
放射性炭素標識イミダゾール誘導体
【課題】脳血管障害等の診断に有用な陽電子断層撮影診断用の放射性リガンドを提供すること。
【解決手段】式(I)に示される、放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩。

(Qは、水素原子又はC1〜C4アルキル基であり、nは、3〜8の整数であり、R1は、水素原子、C1〜C4アルキル基又はハロゲン原子であり、R2は、C1〜C4アルキル基であり、R3は、C1〜C4アルキル基である。ただし、R2又はR3は、11Cを導入したC1〜C4アルキル基である。)
【解決手段】式(I)に示される、放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩。
(Qは、水素原子又はC1〜C4アルキル基であり、nは、3〜8の整数であり、R1は、水素原子、C1〜C4アルキル基又はハロゲン原子であり、R2は、C1〜C4アルキル基であり、R3は、C1〜C4アルキル基である。ただし、R2又はR3は、11Cを導入したC1〜C4アルキル基である。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、放射性炭素標識イミダゾール誘導体及びこれを用いた診断薬に関し、特に、陽電子断層撮影(positron emission tomography;PET)診断用の放射性リガンドに関する。
【背景技術】
【0002】
20-Hydroxyeicosatetraenoic acid(20−HETE)は、アラキドン酸から20−HETE産生酵素であるチトクロームP450 4A又は4Fによって代謝される産物であり、腎臓、肝臓、脳血管組織等の主要臓器で産生されることが知られている。アラキドン酸代謝産物である20−HETEは、上記主要臓器において血管収縮や炎症反応に関与すると考えられており、例えば20−HETEの産生増大は脳血管障害の病態進行を促進する。したがって、その産生酵素(20−HETE産生酵素)の発現量を知ることは脳血管障害等の各種病態の程度や予後を知る上で重要である。
【0003】
近年、癌の診断、心筋や脳の代謝活動の判定などの臨床目的あるいは生体の代謝活動、脳機能の解明などの基礎科学において、陽電子断層撮影PETが広範に利用されるようになっている。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】国際公開第WO2001/032164号パンフレット
【特許文献2】国際公開第WO2001/096309号パンフレット
【特許文献3】国際公開第WO2002/088071号パンフレット
【特許文献4】国際公開第WO2004/024677号パンフレット
【特許文献5】特開2004−010513号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
20−HETE産生酵素に対するPETは、脳血管障害等の診断や研究に有用であると考えられるが、当該PETリガンドの存在は知られていない
20−HETE産生酵素阻害活性を有する化合物が報告されているが(特許文献1〜5)、これらの化合物をPETリガンドとして利用したという報告はない。
【0006】
本発明の目的は、20−HETE産生酵素に結合し、脳血管障害等の診断や研究に有用なPETリガンドを提供することにある。
【課題を解決するための手段】
【0007】
本発明者らは前述した課題を解決する目的で鋭意探索研究した結果、優れた20−HETE産生酵素への親和性を示す放射性同位元素で標識した化合物を見出し、本発明を完成した。
【0008】
すなわち、本発明は、
(1)下記一般式(I)に示される、放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩、
【0009】
【化1】
【0010】
(上記一般式(I)において、
Qは、水素原子又はC1〜C4アルキル基であり、
nは、3〜8の整数であり、
R1は、水素原子、C1〜C4アルキル基又はハロゲン原子であり、
R2は、C1〜C4アルキル基であり、
R3は、C1〜C4アルキル基である。
ただし、R2又はR3は、11Cを導入したC1〜C4アルキル基である。)
(2)前記一般式(I)において、Qが水素原子であり、nが6であり、R1が水素原子である、(1)に記載の放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩、又は
(3)(1)又は(2)に記載の放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩を含有する、脳血管障害の診断薬である。
【発明の効果】
【0011】
本発明により、20−HETE産生酵素に結合する化合物が見出され、脳、腎臓、肝臓等の各種臓器における20−HETE産生酵素の量を可視的に測定することが可能となった。特に本発明の化合物は、脳組織における集積が顕著であり、脳血管障害等の診断や研究のためのリガンドとして有用である。
【図面の簡単な説明】
【0012】
【図1】ラット脳虚血再灌流後28日にわたる障害半球(虚血側半球)における11C標識化合物Aの集積変化を示す図である。
【図2】11C標識化合物Aの脳内における集積に対する過剰量非標識化合物A投与の影響を示す図である。
【発明を実施するための形態】
【0013】
本発明において、「C1〜C4アルキル基」すなわち「炭素数1〜4のアルキル基」とは、直鎖状又は分岐鎖状のアルキル基を示し、例えばメチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、1−メチルプロピル基などである。
【0014】
本発明において、「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子又はヨウ素原子である。
【0015】
本発明において、「脳血管障害」とは、脳梗塞、脳出血、クモ膜下出血、もやもや病、慢性硬膜下血腫等の障害を意味し、本発明の化合物は、例えば脳梗塞において認められる虚血再灌流障害の診断に特に有用である。
【0016】
本発明における化合物(I)及びその製薬学的に許容される塩は、特許文献5に記載の方法と同様に製造される化合物を用い、下記反応式で示される方法で製造することができる。式中、Q、R1、R2、R3は、前記と同義である。
【0017】
なお、以下の製造方法の説明において、「不活性溶媒」とは、例えばベンゼン、トルエン、キシレン、ピリジン等の芳香族系溶媒;
ヘキサン、ペンタン、シクロヘキサン等の炭化水素系溶媒;
ジクロロメタン、クロロホルム、1,2−ジクロロエタン、四塩化炭素等のハロゲン化炭化水素系溶媒;
テトラヒドロフラン、ジエチルエーテル、1,2−ジメトキシエタン、1,4−ジオキサン等のエーテル系溶媒;
酢酸エチル、酢酸イソプロピル等のエステル系溶媒;
メタノール、エタノール、イソプロピルアルコール、tert−ブチルアルコール、エチレングリコール等のアルコール系溶媒;
アセトン、メチルエチルケトン等のケトン系溶媒;
N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ヘキサメチルリン酸トリアミド等のアミド系溶媒;
ジメチルスルホキシド等のスルホキシド系溶媒;
アセトニトリル、プロピオニトリル等のニトリル系溶媒;及び
水であり、並びにこれらの均一系及び不均一系混合溶媒等である。これらの不活性溶媒は当業者に公知である種々の反応条件に応じて適宜選択される。
【0018】
「塩基」とは、例えば水素化ナトリウム、水素化カリウム等のアルカリ金属又はアルカリ土類金属の水素化物;
ナトリウムアミド、リチウムジイソプロピルアミド(LDA)、リチウム 2,2,6,6−テトラメチルピペリジド(LTMP)、リチウムヘキサメチルジシラジド(LHMDS)、カリウムヘキサメチルジシラジド(KHMDS)等のアルカリ金属又はアルカリ土類金属のアミド;
ナトリウムメトキシド、ナトリウムエトキシド、カリウム tert−ブトキシド等のアルカリ金属又はアルカリ土類金属のアルコキシド;
ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム等のアルキルリチウム;
水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化バリウム等のアルカリ金属又はアルカリ土類金属の水酸化物;
炭酸ナトリウム、炭酸カリウム、炭酸セシウム等のアルカリ金属又はアルカリ土類金属の炭酸塩;
炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属又はアルカリ土類金属の炭酸水素塩;
フッ化カリウム、フッ化セシウム等のアルカリ金属又はアルカリ土類金属のフッ化物;
トリエチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン、N,N−ジメチルアニリン、1,8−ジアザビシクロ[5.4.0]ウンデク−7−エン(DBU)等のアミン;
ピリジン、N,N−ジメチル−4−アミノピリジン(DMAP)、2,6−ルチジン、イミダゾール等の塩基性複素環化合物等である。これらの塩基は当業者に公知である種々の反応条件に応じて適宜選択される。
【0019】
また、「酸」とは、例えば塩酸、臭化水素酸、硫酸、硝酸、リン酸等の無機酸;
p−トルエンスルホン酸、メタンスルホン酸、トリフルオロ酢酸(TFA)、ギ酸、酢酸等の有機酸;
三フッ化ホウ素ジエチルエーテル錯体、三臭化ホウ素、トリメチルシリルヨージド、塩化亜鉛(II)、塩化アルミニウム(III)、塩化チタン(IV)等のルイス酸である。これらの酸は当業者に公知である種々の反応条件に応じて適宜選択される。
【0020】
[製造法]
本発明における化合物(I)は、例えば以下のスキーム1に示される方法で製造することができる。
(スキーム1)
【0021】
【化2】
【0022】
上記スキーム1における式中、Xは、ハロゲン原子を示す。また、式中、Q、R1、R2、R3及びnは、前記と同義である。
また、Bocはtert−ブトキシカルボニル基であり、Acはアセチル基である。
【0023】
以下、スキーム1中の各工程をさらに具体的に説明する。
工程1:不活性溶媒中、塩基存在下又は非存在下、化合物(1)をアセチル化剤とのアセチル化反応に供することにより、化合物(2)を得ることができる。ここで、アセチル化剤とは、例えば無水酢酸、アセチルクロリド等である。
【0024】
工程2:不活性溶媒中、塩基存在下又は非存在下、添加剤存在下又は非存在下、化合物(2)をハロゲン化アルキルとのアルキル化反応に供することにより、化合物(3)を得ることができる。ここで、ハロゲン化アルキルは、例えばヨウ化メチル等である。
【0025】
工程3:化合物(3)のアセチル基を当業者に公知である種々の有機合成手法[プロテクティブ グループス イン オーガニック シンセシス(Protective Groups in Organic Synthesis)第4版、ジョン ウィリー アンド サンズ(John Wiley & Sons, INC.)参照]を用いて除去することにより、化合物(4)を得ることができる。
【0026】
工程4:不活性溶媒中、化合物(4)と化合物(5)との光延反応に供することにより、化合物(6)を得ることができる。光延反応とは、例えばトリフェニルホスフィン或いはトリブチルホスフィン等の有機リン化合物及び例えばアゾジカルボン酸ジエチル(DEAD)、アゾジカルボン酸ジイソプロピル(DIAD)或いはN,N,N',N'−テトラメチルアゾジカルボキサミド(TMAD)等のアゾ化合物を用いる反応、又は、例えばシアノメチレントリブチルホスホラン(CMBP)等のリンイリド試薬を用いる反応である[ケミカル レビューズ(Chemical Reviews)2009年、第109巻、p.2551−2651参照]。化合物(5)は、市販化合物、公知化合物又は当業者に公知である種々の有機合成手法を用いて市販化合物又は公知化合物より合成した化合物を用いることができる。
【0027】
工程5:化合物(6)のtert−ブトキシカルボニル基を当業者に公知である種々の有機合成手法[プロテクティブ グループス イン オーガニック シンセシス(Protective Groups in Organic Synthesis)第4版、ジョン ウィリー アンド サンズ(John Wiley & Sons, INC.)参照]を用いて除去することにより、化合物(7)を得ることができる。
【0028】
工程6:不活性溶媒中、塩基存在下又は非存在下、添加剤存在下又は非存在下、化合物(7)を11Cを導入したハロゲン化アルキルとのアルキル化反応に供することにより、本発明化合物(I)を得ることができる。
【0029】
以下、実施例と試験例を挙げて本発明を更に詳しく説明する。
【実施例】
【0030】
本発明は下記の実施例によって更に詳細に説明されるが、これら実施例は本発明を限定するものではなく、また、本発明の範囲を逸脱しない範囲で変化させてもよい。
実施例中で言及する「SNAP Cartridge KP-Sil」とはバイオタージ社製のパックドカラムである。また、「Purif-Pack SI30」とは昭光サイエンティフィック社製のパックドカラムである。NHシリカゲルとは富士シリシア社製のクロマトレックスNHシリカゲルである。「室温」とは5〜35℃を意味する。
【0031】
実施例中記載の各機器データは以下の測定機器で測定した。
MSスペクトル:島津製作所社製LCMS−2010EV
【0032】
実施例中で使用した略語を以下に示す。
APCI(大気圧化学イオン化)、DMF(N,N−ジメチルホルムアミド)、ESI(エレクトロスプレーイオン化)、MS(質量分析)、THF(テトラヒドロフラン)、v/v(容量/容量)。
【0033】
(参考例1)
6−[4−(1H−イミダゾル−1−イル)フェノキシ]−N,N'−ジメチルヘキサン−1−アミン 2塩酸塩(以下、非標識化合物A)の合成
特許文献5に記載の方法に従い合成した。
ESI/APCI MS m/z 288 [M+H]+.
【0034】
(実施例1)
11C標識 6−[4−(1H−イミダゾル−1−イル)フェノキシ]−N,N'−ジメチルヘキサン−1−アミン(以下、11C標識化合物A)の合成
【0035】
工程1−1:
tert−ブチル(6−ヒドロキシへキシル)カーバメート(15.7g)のピリジン(47mL)溶液に、氷冷下、無水酢酸(47mL)を加え、室温で19時間撹拌した。反応混合物を氷冷し、水(100mL)を加え、10分間撹拌した。酢酸エチル(300mL)で抽出し、有機層を水(150mL×1、100mL×1)及び飽和食塩水(100mL)で順次洗浄した。有機層を無水MgSO4で乾燥して減圧下、濃縮し、未精製の6−[(tert−ブトキシカルボニル)アミノ]ヘキシルアセテート(17.7g、無色油状物質)を得た。
ESI/APCI MS m/z 282 [M+Na]+.
【0036】
工程1−2:
工程1−1で得られた化合物(17.7g)のDMF(200mL)溶液に、氷冷下、60%NaH(4.08g)を少しずつ加え、同温で30分間撹拌した。氷冷下、ヨウ化メチル(5.10mL)を滴下し、室温で3時間撹拌した。反応混合物を氷冷し、飽和NH4Cl水溶液(250mL)を加え、酢酸エチル(450mL)で抽出した。有機層を飽和食塩水(400mL×3)で洗浄し、無水MgSO4で乾燥して減圧下、濃縮した。残渣をカラムクロマトグラフィー(SNAP Cartridge KP-Sil、移動相:ヘキサン/酢酸エチル=90/10〜70/30;v/v)及び(Purif-Pack SI30、移動相:ヘキサン/酢酸エチル=90/10〜70/30;v/v)×2で精製し、6−[(tert−ブトキシカルボニル)(メチル)アミノ]ヘキシルアセテート(6.71g、無色油状物質)を得た。
ESI/APCI MS m/z 296 [M+Na]+.
【0037】
工程1−3:
工程1−2で得られた化合物(6.70g)のMeOH(67mL)溶液に、K2CO3(340mg)を加え、室温で1.5時間撹拌した。減圧下、MeOHを濃縮し、酢酸エチル(100mL)を加え、水(20mL)及び飽和食塩水(20mL)で洗浄し、無水MgSO4で乾燥して減圧下、濃縮した。残渣をカラムクロマトグラフィー(SNAP Cartridge KP-Sil、移動相:ヘキサン/酢酸エチル=70/30〜50/50;v/v)で精製した。後処理・精製過程での生成物を再びMeOH(60mL)溶液に、K2CO3(340mg)を加え、室温で2時間撹拌した。反応混合物に水(20mL)及び飽和NH4Cl水溶液(20mL)を加え中和し、減圧下、MeOHを濃縮した。CHCl3(200mL)で抽出後、飽和食塩水(30mL)で洗浄し、無水MgSO4で乾燥して減圧下、濃縮した。残渣をカラムクロマトグラフィー(SNAP Cartridge KP-Sil、移動相:ヘキサン/酢酸エチル=70/30〜50/50;v/v)で精製し、先のカラム精製で得られた目的物と一緒にし、tert−ブチル (6−ヒドロキシへキシル)メチルカーバメート(4.67g、無色油状物質)を得た。
ESI/APCI MS m/z 254 [M+Na]+.
【0038】
工程1−4:
工程1−3で得られた化合物(2.32g)、4−(1H−イミダゾル−1−イル)フェノール(2.41g)及びトリフェニルホスフィン(3.92g)のTHF(25mL)懸濁液に、氷冷下、40%アゾジカルボン酸ジイソプロピル/トルエン溶液(7.58g)を滴下し、室温で66時間撹拌した。反応混合物にNHシリカゲルを加え、減圧下、濃縮した。残渣をカラムクロマトグラフィー(NHシリカゲル、移動相:ヘキサン/酢酸エチル=50/50;v/v)、(Purif-Pack SI30、移動相:ヘキサン/酢酸エチル=50/50〜0/100;v/v)及び(NHシリカゲル、移動相:ヘキサン/酢酸エチル=50/50;v/v)で精製し、tert−ブチル{6−[4−(1H−イミダゾル−1−イル)フェノキシ]へキシル}メチルカーバメート(1.39g、無色油状物質)を得た。
ESI/APCI MS m/z 374 [M+H]+.
【0039】
工程1−5:
工程1−4で得られた化合物(1.38g)の酢酸エチル(5mL)溶液に、4M塩酸/酢酸エチル(5mL)を加え、室温で1.5時間撹拌した。反応混合物に2M NaOH水溶液を加え中和し、水(10mL)を加え、酢酸エチル(70mL×2)及びCHCl3(70mL)で抽出した。水層を減圧下、濃縮し、CHCl3(70mL)を加え、室温で12時間撹拌した。不溶物を濾取し、濾液と先の抽出で得られた有機層を一緒にし、無水MgSO4で乾燥して減圧下、濃縮した。残渣をカラムクロマトグラフィー(NHシリカゲル、移動相:酢酸エチル〜CHCl3/MeOH=95/5;v/v)及び再結晶(ヘキサン/酢酸エチル)で順次精製し、6−[4−(1H−イミダゾル−1−イル)フェノキシ]−N−メチルへキサン−1−アミン(366mg、無色固体)を得た。
ESI/APCI MS m/z 274 [M+H]+.
【0040】
工程1−6:
標識用合成装置として、理化学研究所分子イメージング科学研究センターに設置してある標準型標識用合成装置を用いた。化学薬品は市販のものをそのまま用いた。脱水N,N−ジメチルホルムアミド(DMF)(ナカライ社製)、HPLC用アセトニトリル(ナカライ社製)及び酢酸アンモニウム(東京化成社製)を使用した。
11C核の製造には、住友重機械工業社製サイクロトロンCYPRIS HM−12Sを使用し、14N(p,α)11Cの核反応により製造した。[11C]ヨウ化メチルの合成には、専用の標識用合成装置を用いて、11CO2ガスを出発物質として、11CO2→11CH3OH→11CH3Iの順に変換して合成した。
一方、工程1−5で得られた化合物(0.5mg、1.8μmol)のDMF溶液(0.2mL)を標識用合成装置の反応容器に準備し、室温に設置した([11C]ヨウ化メチルを吹き込む10−20分前に設置した)。続いて、本反応溶液に[11C]ヨウ化メチルを60−80mL/minのガス流量で吹き込み、90℃で5分間加熱した。得られた反応溶液を0.5mLの洗浄液(アセトニトリル:30mM酢酸アンモニウム=23:77溶液)で希釈し、分取HPLCを用いて11C標識化合物Aを分取し、分取したフラクションを、エバポレーターを用いて減圧濃縮した。濃縮液は臨床用投与溶液(生理食塩水:3mL)を用いて希釈し、無菌バイヤルに入れた。本溶液の一部(20μL)を分析HPLCに供して、目的化合物の同定、純度検定、及び比放射能の算出を行った。なお、11C標識化合物Aの同定は非標識化合物Aを用いて行った。
11C標識化合物Aの総放射能:1.5〜2.5GBq、合成時間:40分、放射化学純度:99%以上、化学的純度:99%以上、比放射能:60〜100GBq/μmol
分取条件:分取用カラムはナカライ社製MS−II 10mm×250mm、ガードカラムはナカライ社製MS−II 10mm×20mmを使用した。流量は6mL/minで、移動層は30mM CH3COONH4:CH3CN(5%H2O添加)=77:23を使用した。UV検出波長254nm及びγ線検出器で測定した結果、11C標識化合物Aの保持時間は約10分であった。
【0041】
<試験例>
(試験方法)
ラット脳虚血モデルは、栓子法(intraluminal filament method)を用いた60分間の一過性中大脳動脈閉塞(t−MCAO)により作製した。虚血再灌流後3〜28日に、11C標識化合物A(放射比活性40GBq/μmol)は、1.5%isoflurane麻酔下のラットに対して埋め込まれたカニューレを通して尾静脈に注入された(200MBq/kg;1.5μg/kg)。Dynamic PETスキャンは、11C標識化合物A注入直後から小動物用PETスキャナー(microPET focus220)を用いて90分間実施された。11C標識化合物Aの結合能(binding potential)は、小脳を参照領域としてmultilinear reference-tissue model 2(MRTM2)(PMOD Technology, version 3.0)により計算された。
【0042】
(試験結果)
正常ラットで、11C標識化合物Aは腎臓、肝臓等の20−HETE産生酵素が豊富に存在する臓器において高い集積を示した。当該化合物は脳においても高い集積を示した。
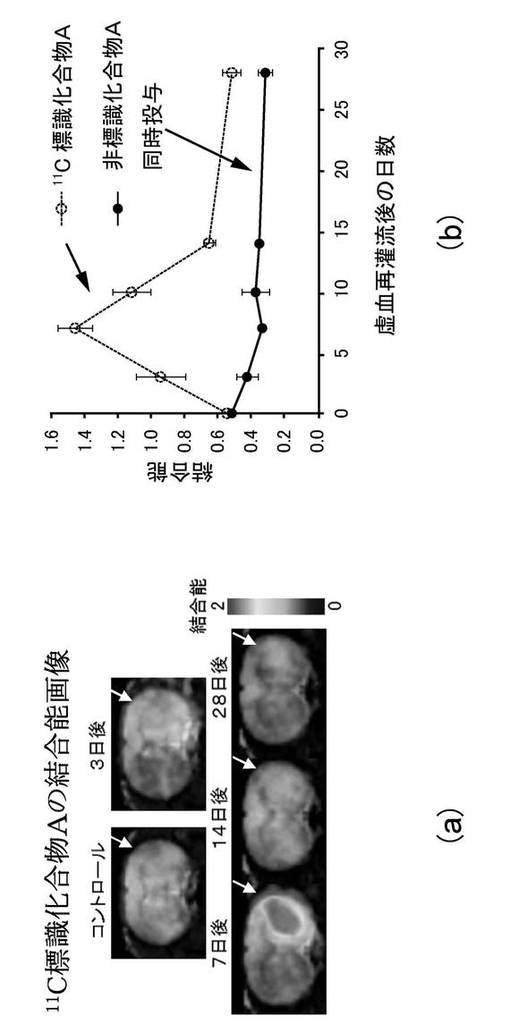
図1(a)及び図1(b)は、ラット脳虚血再灌流後28日にわたる障害半球(虚血側半球)における11C標識化合物Aの集積変化(PETスキャン結果)を示す図である。図1(a)中の白抜き矢印は、障害半球(虚血側半球)を差し示している。
11C標識化合物Aを用いたPETスキャンにより、ラット脳虚血再灌流後28日にわたる20−HETE産生酵素の変動をモニターしたところ、図1(a)及び図1(b)に示したように、虚血再灌流後7日及び10日に障害半球(虚血側半球)において対照半球(非虚血側半球)よりも11C標識化合物Aの高い集積が認められた。当該集積は、過剰量の非標識化合物Aを同時投与(10mg/kg)することによって消失した(図1(a)、図1(b)、図2(a)及び図2(b))。
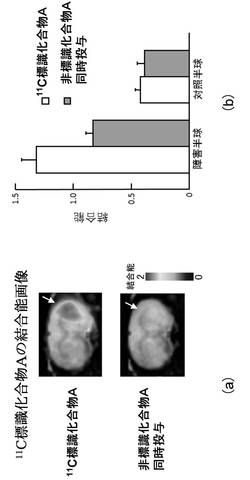
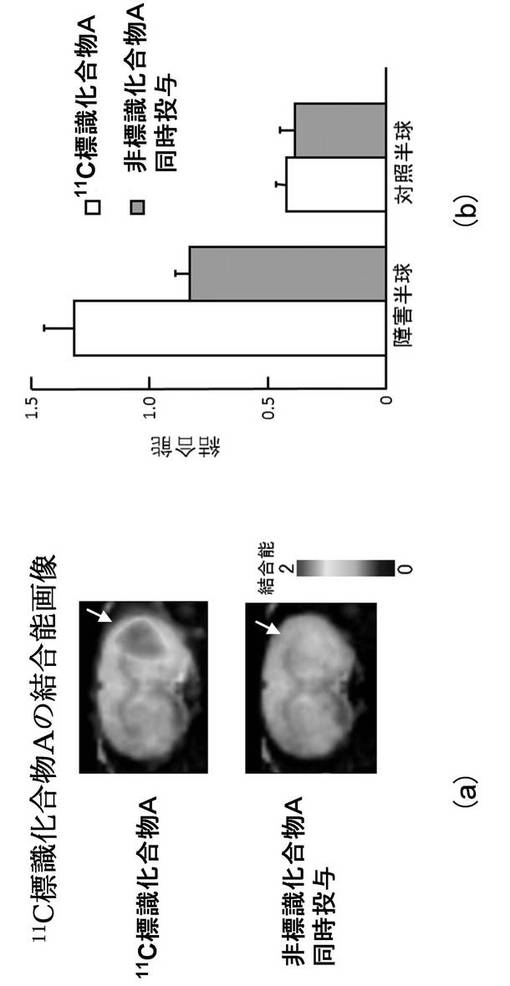
図2(a)及び図2(b)は、11C標識化合物Aの脳内における集積に対する過剰量非標識化合物A投与の影響を示す図である。図2(a)中の白抜き矢印は、障害半球(虚血側半球)を差し示している。
このように、11C標識化合物Aは、ラット脳虚血障害時の20−HETE産生酵素量の時間依存的な変化について定量的な情報を提供した。
【産業上の利用可能性】
【0043】
本発明の化合物は、脳血管障害等の早期診断に有用なPET用の放射性リガンドとして利用可能である。
【技術分野】
【0001】
本発明は、放射性炭素標識イミダゾール誘導体及びこれを用いた診断薬に関し、特に、陽電子断層撮影(positron emission tomography;PET)診断用の放射性リガンドに関する。
【背景技術】
【0002】
20-Hydroxyeicosatetraenoic acid(20−HETE)は、アラキドン酸から20−HETE産生酵素であるチトクロームP450 4A又は4Fによって代謝される産物であり、腎臓、肝臓、脳血管組織等の主要臓器で産生されることが知られている。アラキドン酸代謝産物である20−HETEは、上記主要臓器において血管収縮や炎症反応に関与すると考えられており、例えば20−HETEの産生増大は脳血管障害の病態進行を促進する。したがって、その産生酵素(20−HETE産生酵素)の発現量を知ることは脳血管障害等の各種病態の程度や予後を知る上で重要である。
【0003】
近年、癌の診断、心筋や脳の代謝活動の判定などの臨床目的あるいは生体の代謝活動、脳機能の解明などの基礎科学において、陽電子断層撮影PETが広範に利用されるようになっている。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】国際公開第WO2001/032164号パンフレット
【特許文献2】国際公開第WO2001/096309号パンフレット
【特許文献3】国際公開第WO2002/088071号パンフレット
【特許文献4】国際公開第WO2004/024677号パンフレット
【特許文献5】特開2004−010513号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
20−HETE産生酵素に対するPETは、脳血管障害等の診断や研究に有用であると考えられるが、当該PETリガンドの存在は知られていない
20−HETE産生酵素阻害活性を有する化合物が報告されているが(特許文献1〜5)、これらの化合物をPETリガンドとして利用したという報告はない。
【0006】
本発明の目的は、20−HETE産生酵素に結合し、脳血管障害等の診断や研究に有用なPETリガンドを提供することにある。
【課題を解決するための手段】
【0007】
本発明者らは前述した課題を解決する目的で鋭意探索研究した結果、優れた20−HETE産生酵素への親和性を示す放射性同位元素で標識した化合物を見出し、本発明を完成した。
【0008】
すなわち、本発明は、
(1)下記一般式(I)に示される、放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩、
【0009】
【化1】
【0010】
(上記一般式(I)において、
Qは、水素原子又はC1〜C4アルキル基であり、
nは、3〜8の整数であり、
R1は、水素原子、C1〜C4アルキル基又はハロゲン原子であり、
R2は、C1〜C4アルキル基であり、
R3は、C1〜C4アルキル基である。
ただし、R2又はR3は、11Cを導入したC1〜C4アルキル基である。)
(2)前記一般式(I)において、Qが水素原子であり、nが6であり、R1が水素原子である、(1)に記載の放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩、又は
(3)(1)又は(2)に記載の放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩を含有する、脳血管障害の診断薬である。
【発明の効果】
【0011】
本発明により、20−HETE産生酵素に結合する化合物が見出され、脳、腎臓、肝臓等の各種臓器における20−HETE産生酵素の量を可視的に測定することが可能となった。特に本発明の化合物は、脳組織における集積が顕著であり、脳血管障害等の診断や研究のためのリガンドとして有用である。
【図面の簡単な説明】
【0012】
【図1】ラット脳虚血再灌流後28日にわたる障害半球(虚血側半球)における11C標識化合物Aの集積変化を示す図である。
【図2】11C標識化合物Aの脳内における集積に対する過剰量非標識化合物A投与の影響を示す図である。
【発明を実施するための形態】
【0013】
本発明において、「C1〜C4アルキル基」すなわち「炭素数1〜4のアルキル基」とは、直鎖状又は分岐鎖状のアルキル基を示し、例えばメチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、1−メチルプロピル基などである。
【0014】
本発明において、「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子又はヨウ素原子である。
【0015】
本発明において、「脳血管障害」とは、脳梗塞、脳出血、クモ膜下出血、もやもや病、慢性硬膜下血腫等の障害を意味し、本発明の化合物は、例えば脳梗塞において認められる虚血再灌流障害の診断に特に有用である。
【0016】
本発明における化合物(I)及びその製薬学的に許容される塩は、特許文献5に記載の方法と同様に製造される化合物を用い、下記反応式で示される方法で製造することができる。式中、Q、R1、R2、R3は、前記と同義である。
【0017】
なお、以下の製造方法の説明において、「不活性溶媒」とは、例えばベンゼン、トルエン、キシレン、ピリジン等の芳香族系溶媒;
ヘキサン、ペンタン、シクロヘキサン等の炭化水素系溶媒;
ジクロロメタン、クロロホルム、1,2−ジクロロエタン、四塩化炭素等のハロゲン化炭化水素系溶媒;
テトラヒドロフラン、ジエチルエーテル、1,2−ジメトキシエタン、1,4−ジオキサン等のエーテル系溶媒;
酢酸エチル、酢酸イソプロピル等のエステル系溶媒;
メタノール、エタノール、イソプロピルアルコール、tert−ブチルアルコール、エチレングリコール等のアルコール系溶媒;
アセトン、メチルエチルケトン等のケトン系溶媒;
N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ヘキサメチルリン酸トリアミド等のアミド系溶媒;
ジメチルスルホキシド等のスルホキシド系溶媒;
アセトニトリル、プロピオニトリル等のニトリル系溶媒;及び
水であり、並びにこれらの均一系及び不均一系混合溶媒等である。これらの不活性溶媒は当業者に公知である種々の反応条件に応じて適宜選択される。
【0018】
「塩基」とは、例えば水素化ナトリウム、水素化カリウム等のアルカリ金属又はアルカリ土類金属の水素化物;
ナトリウムアミド、リチウムジイソプロピルアミド(LDA)、リチウム 2,2,6,6−テトラメチルピペリジド(LTMP)、リチウムヘキサメチルジシラジド(LHMDS)、カリウムヘキサメチルジシラジド(KHMDS)等のアルカリ金属又はアルカリ土類金属のアミド;
ナトリウムメトキシド、ナトリウムエトキシド、カリウム tert−ブトキシド等のアルカリ金属又はアルカリ土類金属のアルコキシド;
ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、メチルリチウム等のアルキルリチウム;
水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化バリウム等のアルカリ金属又はアルカリ土類金属の水酸化物;
炭酸ナトリウム、炭酸カリウム、炭酸セシウム等のアルカリ金属又はアルカリ土類金属の炭酸塩;
炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属又はアルカリ土類金属の炭酸水素塩;
フッ化カリウム、フッ化セシウム等のアルカリ金属又はアルカリ土類金属のフッ化物;
トリエチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン、N,N−ジメチルアニリン、1,8−ジアザビシクロ[5.4.0]ウンデク−7−エン(DBU)等のアミン;
ピリジン、N,N−ジメチル−4−アミノピリジン(DMAP)、2,6−ルチジン、イミダゾール等の塩基性複素環化合物等である。これらの塩基は当業者に公知である種々の反応条件に応じて適宜選択される。
【0019】
また、「酸」とは、例えば塩酸、臭化水素酸、硫酸、硝酸、リン酸等の無機酸;
p−トルエンスルホン酸、メタンスルホン酸、トリフルオロ酢酸(TFA)、ギ酸、酢酸等の有機酸;
三フッ化ホウ素ジエチルエーテル錯体、三臭化ホウ素、トリメチルシリルヨージド、塩化亜鉛(II)、塩化アルミニウム(III)、塩化チタン(IV)等のルイス酸である。これらの酸は当業者に公知である種々の反応条件に応じて適宜選択される。
【0020】
[製造法]
本発明における化合物(I)は、例えば以下のスキーム1に示される方法で製造することができる。
(スキーム1)
【0021】
【化2】
【0022】
上記スキーム1における式中、Xは、ハロゲン原子を示す。また、式中、Q、R1、R2、R3及びnは、前記と同義である。
また、Bocはtert−ブトキシカルボニル基であり、Acはアセチル基である。
【0023】
以下、スキーム1中の各工程をさらに具体的に説明する。
工程1:不活性溶媒中、塩基存在下又は非存在下、化合物(1)をアセチル化剤とのアセチル化反応に供することにより、化合物(2)を得ることができる。ここで、アセチル化剤とは、例えば無水酢酸、アセチルクロリド等である。
【0024】
工程2:不活性溶媒中、塩基存在下又は非存在下、添加剤存在下又は非存在下、化合物(2)をハロゲン化アルキルとのアルキル化反応に供することにより、化合物(3)を得ることができる。ここで、ハロゲン化アルキルは、例えばヨウ化メチル等である。
【0025】
工程3:化合物(3)のアセチル基を当業者に公知である種々の有機合成手法[プロテクティブ グループス イン オーガニック シンセシス(Protective Groups in Organic Synthesis)第4版、ジョン ウィリー アンド サンズ(John Wiley & Sons, INC.)参照]を用いて除去することにより、化合物(4)を得ることができる。
【0026】
工程4:不活性溶媒中、化合物(4)と化合物(5)との光延反応に供することにより、化合物(6)を得ることができる。光延反応とは、例えばトリフェニルホスフィン或いはトリブチルホスフィン等の有機リン化合物及び例えばアゾジカルボン酸ジエチル(DEAD)、アゾジカルボン酸ジイソプロピル(DIAD)或いはN,N,N',N'−テトラメチルアゾジカルボキサミド(TMAD)等のアゾ化合物を用いる反応、又は、例えばシアノメチレントリブチルホスホラン(CMBP)等のリンイリド試薬を用いる反応である[ケミカル レビューズ(Chemical Reviews)2009年、第109巻、p.2551−2651参照]。化合物(5)は、市販化合物、公知化合物又は当業者に公知である種々の有機合成手法を用いて市販化合物又は公知化合物より合成した化合物を用いることができる。
【0027】
工程5:化合物(6)のtert−ブトキシカルボニル基を当業者に公知である種々の有機合成手法[プロテクティブ グループス イン オーガニック シンセシス(Protective Groups in Organic Synthesis)第4版、ジョン ウィリー アンド サンズ(John Wiley & Sons, INC.)参照]を用いて除去することにより、化合物(7)を得ることができる。
【0028】
工程6:不活性溶媒中、塩基存在下又は非存在下、添加剤存在下又は非存在下、化合物(7)を11Cを導入したハロゲン化アルキルとのアルキル化反応に供することにより、本発明化合物(I)を得ることができる。
【0029】
以下、実施例と試験例を挙げて本発明を更に詳しく説明する。
【実施例】
【0030】
本発明は下記の実施例によって更に詳細に説明されるが、これら実施例は本発明を限定するものではなく、また、本発明の範囲を逸脱しない範囲で変化させてもよい。
実施例中で言及する「SNAP Cartridge KP-Sil」とはバイオタージ社製のパックドカラムである。また、「Purif-Pack SI30」とは昭光サイエンティフィック社製のパックドカラムである。NHシリカゲルとは富士シリシア社製のクロマトレックスNHシリカゲルである。「室温」とは5〜35℃を意味する。
【0031】
実施例中記載の各機器データは以下の測定機器で測定した。
MSスペクトル:島津製作所社製LCMS−2010EV
【0032】
実施例中で使用した略語を以下に示す。
APCI(大気圧化学イオン化)、DMF(N,N−ジメチルホルムアミド)、ESI(エレクトロスプレーイオン化)、MS(質量分析)、THF(テトラヒドロフラン)、v/v(容量/容量)。
【0033】
(参考例1)
6−[4−(1H−イミダゾル−1−イル)フェノキシ]−N,N'−ジメチルヘキサン−1−アミン 2塩酸塩(以下、非標識化合物A)の合成
特許文献5に記載の方法に従い合成した。
ESI/APCI MS m/z 288 [M+H]+.
【0034】
(実施例1)
11C標識 6−[4−(1H−イミダゾル−1−イル)フェノキシ]−N,N'−ジメチルヘキサン−1−アミン(以下、11C標識化合物A)の合成
【0035】
工程1−1:
tert−ブチル(6−ヒドロキシへキシル)カーバメート(15.7g)のピリジン(47mL)溶液に、氷冷下、無水酢酸(47mL)を加え、室温で19時間撹拌した。反応混合物を氷冷し、水(100mL)を加え、10分間撹拌した。酢酸エチル(300mL)で抽出し、有機層を水(150mL×1、100mL×1)及び飽和食塩水(100mL)で順次洗浄した。有機層を無水MgSO4で乾燥して減圧下、濃縮し、未精製の6−[(tert−ブトキシカルボニル)アミノ]ヘキシルアセテート(17.7g、無色油状物質)を得た。
ESI/APCI MS m/z 282 [M+Na]+.
【0036】
工程1−2:
工程1−1で得られた化合物(17.7g)のDMF(200mL)溶液に、氷冷下、60%NaH(4.08g)を少しずつ加え、同温で30分間撹拌した。氷冷下、ヨウ化メチル(5.10mL)を滴下し、室温で3時間撹拌した。反応混合物を氷冷し、飽和NH4Cl水溶液(250mL)を加え、酢酸エチル(450mL)で抽出した。有機層を飽和食塩水(400mL×3)で洗浄し、無水MgSO4で乾燥して減圧下、濃縮した。残渣をカラムクロマトグラフィー(SNAP Cartridge KP-Sil、移動相:ヘキサン/酢酸エチル=90/10〜70/30;v/v)及び(Purif-Pack SI30、移動相:ヘキサン/酢酸エチル=90/10〜70/30;v/v)×2で精製し、6−[(tert−ブトキシカルボニル)(メチル)アミノ]ヘキシルアセテート(6.71g、無色油状物質)を得た。
ESI/APCI MS m/z 296 [M+Na]+.
【0037】
工程1−3:
工程1−2で得られた化合物(6.70g)のMeOH(67mL)溶液に、K2CO3(340mg)を加え、室温で1.5時間撹拌した。減圧下、MeOHを濃縮し、酢酸エチル(100mL)を加え、水(20mL)及び飽和食塩水(20mL)で洗浄し、無水MgSO4で乾燥して減圧下、濃縮した。残渣をカラムクロマトグラフィー(SNAP Cartridge KP-Sil、移動相:ヘキサン/酢酸エチル=70/30〜50/50;v/v)で精製した。後処理・精製過程での生成物を再びMeOH(60mL)溶液に、K2CO3(340mg)を加え、室温で2時間撹拌した。反応混合物に水(20mL)及び飽和NH4Cl水溶液(20mL)を加え中和し、減圧下、MeOHを濃縮した。CHCl3(200mL)で抽出後、飽和食塩水(30mL)で洗浄し、無水MgSO4で乾燥して減圧下、濃縮した。残渣をカラムクロマトグラフィー(SNAP Cartridge KP-Sil、移動相:ヘキサン/酢酸エチル=70/30〜50/50;v/v)で精製し、先のカラム精製で得られた目的物と一緒にし、tert−ブチル (6−ヒドロキシへキシル)メチルカーバメート(4.67g、無色油状物質)を得た。
ESI/APCI MS m/z 254 [M+Na]+.
【0038】
工程1−4:
工程1−3で得られた化合物(2.32g)、4−(1H−イミダゾル−1−イル)フェノール(2.41g)及びトリフェニルホスフィン(3.92g)のTHF(25mL)懸濁液に、氷冷下、40%アゾジカルボン酸ジイソプロピル/トルエン溶液(7.58g)を滴下し、室温で66時間撹拌した。反応混合物にNHシリカゲルを加え、減圧下、濃縮した。残渣をカラムクロマトグラフィー(NHシリカゲル、移動相:ヘキサン/酢酸エチル=50/50;v/v)、(Purif-Pack SI30、移動相:ヘキサン/酢酸エチル=50/50〜0/100;v/v)及び(NHシリカゲル、移動相:ヘキサン/酢酸エチル=50/50;v/v)で精製し、tert−ブチル{6−[4−(1H−イミダゾル−1−イル)フェノキシ]へキシル}メチルカーバメート(1.39g、無色油状物質)を得た。
ESI/APCI MS m/z 374 [M+H]+.
【0039】
工程1−5:
工程1−4で得られた化合物(1.38g)の酢酸エチル(5mL)溶液に、4M塩酸/酢酸エチル(5mL)を加え、室温で1.5時間撹拌した。反応混合物に2M NaOH水溶液を加え中和し、水(10mL)を加え、酢酸エチル(70mL×2)及びCHCl3(70mL)で抽出した。水層を減圧下、濃縮し、CHCl3(70mL)を加え、室温で12時間撹拌した。不溶物を濾取し、濾液と先の抽出で得られた有機層を一緒にし、無水MgSO4で乾燥して減圧下、濃縮した。残渣をカラムクロマトグラフィー(NHシリカゲル、移動相:酢酸エチル〜CHCl3/MeOH=95/5;v/v)及び再結晶(ヘキサン/酢酸エチル)で順次精製し、6−[4−(1H−イミダゾル−1−イル)フェノキシ]−N−メチルへキサン−1−アミン(366mg、無色固体)を得た。
ESI/APCI MS m/z 274 [M+H]+.
【0040】
工程1−6:
標識用合成装置として、理化学研究所分子イメージング科学研究センターに設置してある標準型標識用合成装置を用いた。化学薬品は市販のものをそのまま用いた。脱水N,N−ジメチルホルムアミド(DMF)(ナカライ社製)、HPLC用アセトニトリル(ナカライ社製)及び酢酸アンモニウム(東京化成社製)を使用した。
11C核の製造には、住友重機械工業社製サイクロトロンCYPRIS HM−12Sを使用し、14N(p,α)11Cの核反応により製造した。[11C]ヨウ化メチルの合成には、専用の標識用合成装置を用いて、11CO2ガスを出発物質として、11CO2→11CH3OH→11CH3Iの順に変換して合成した。
一方、工程1−5で得られた化合物(0.5mg、1.8μmol)のDMF溶液(0.2mL)を標識用合成装置の反応容器に準備し、室温に設置した([11C]ヨウ化メチルを吹き込む10−20分前に設置した)。続いて、本反応溶液に[11C]ヨウ化メチルを60−80mL/minのガス流量で吹き込み、90℃で5分間加熱した。得られた反応溶液を0.5mLの洗浄液(アセトニトリル:30mM酢酸アンモニウム=23:77溶液)で希釈し、分取HPLCを用いて11C標識化合物Aを分取し、分取したフラクションを、エバポレーターを用いて減圧濃縮した。濃縮液は臨床用投与溶液(生理食塩水:3mL)を用いて希釈し、無菌バイヤルに入れた。本溶液の一部(20μL)を分析HPLCに供して、目的化合物の同定、純度検定、及び比放射能の算出を行った。なお、11C標識化合物Aの同定は非標識化合物Aを用いて行った。
11C標識化合物Aの総放射能:1.5〜2.5GBq、合成時間:40分、放射化学純度:99%以上、化学的純度:99%以上、比放射能:60〜100GBq/μmol
分取条件:分取用カラムはナカライ社製MS−II 10mm×250mm、ガードカラムはナカライ社製MS−II 10mm×20mmを使用した。流量は6mL/minで、移動層は30mM CH3COONH4:CH3CN(5%H2O添加)=77:23を使用した。UV検出波長254nm及びγ線検出器で測定した結果、11C標識化合物Aの保持時間は約10分であった。
【0041】
<試験例>
(試験方法)
ラット脳虚血モデルは、栓子法(intraluminal filament method)を用いた60分間の一過性中大脳動脈閉塞(t−MCAO)により作製した。虚血再灌流後3〜28日に、11C標識化合物A(放射比活性40GBq/μmol)は、1.5%isoflurane麻酔下のラットに対して埋め込まれたカニューレを通して尾静脈に注入された(200MBq/kg;1.5μg/kg)。Dynamic PETスキャンは、11C標識化合物A注入直後から小動物用PETスキャナー(microPET focus220)を用いて90分間実施された。11C標識化合物Aの結合能(binding potential)は、小脳を参照領域としてmultilinear reference-tissue model 2(MRTM2)(PMOD Technology, version 3.0)により計算された。
【0042】
(試験結果)
正常ラットで、11C標識化合物Aは腎臓、肝臓等の20−HETE産生酵素が豊富に存在する臓器において高い集積を示した。当該化合物は脳においても高い集積を示した。
図1(a)及び図1(b)は、ラット脳虚血再灌流後28日にわたる障害半球(虚血側半球)における11C標識化合物Aの集積変化(PETスキャン結果)を示す図である。図1(a)中の白抜き矢印は、障害半球(虚血側半球)を差し示している。
11C標識化合物Aを用いたPETスキャンにより、ラット脳虚血再灌流後28日にわたる20−HETE産生酵素の変動をモニターしたところ、図1(a)及び図1(b)に示したように、虚血再灌流後7日及び10日に障害半球(虚血側半球)において対照半球(非虚血側半球)よりも11C標識化合物Aの高い集積が認められた。当該集積は、過剰量の非標識化合物Aを同時投与(10mg/kg)することによって消失した(図1(a)、図1(b)、図2(a)及び図2(b))。
図2(a)及び図2(b)は、11C標識化合物Aの脳内における集積に対する過剰量非標識化合物A投与の影響を示す図である。図2(a)中の白抜き矢印は、障害半球(虚血側半球)を差し示している。
このように、11C標識化合物Aは、ラット脳虚血障害時の20−HETE産生酵素量の時間依存的な変化について定量的な情報を提供した。
【産業上の利用可能性】
【0043】
本発明の化合物は、脳血管障害等の早期診断に有用なPET用の放射性リガンドとして利用可能である。
【特許請求の範囲】
【請求項1】
下記一般式(I)に示される、放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩。
【化1】
(上記一般式(I)において、
Qは、水素原子又はC1〜C4アルキル基であり、
nは、3〜8の整数であり、
R1は、水素原子、C1〜C4アルキル基又はハロゲン原子であり、
R2は、C1〜C4アルキル基であり、
R3は、C1〜C4アルキル基である。
ただし、R2又はR3は、11Cを導入したC1〜C4アルキル基である。)
【請求項2】
前記一般式(I)において、Qが水素原子であり、nが6であり、R1が水素原子である、請求項1に記載の放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩。
【請求項3】
請求項1又は2に記載の放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩を含有する、脳血管障害の診断薬。
【請求項1】
下記一般式(I)に示される、放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩。
【化1】
(上記一般式(I)において、
Qは、水素原子又はC1〜C4アルキル基であり、
nは、3〜8の整数であり、
R1は、水素原子、C1〜C4アルキル基又はハロゲン原子であり、
R2は、C1〜C4アルキル基であり、
R3は、C1〜C4アルキル基である。
ただし、R2又はR3は、11Cを導入したC1〜C4アルキル基である。)
【請求項2】
前記一般式(I)において、Qが水素原子であり、nが6であり、R1が水素原子である、請求項1に記載の放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩。
【請求項3】
請求項1又は2に記載の放射性炭素標識イミダゾール誘導体又はその製薬学的に許容される塩を含有する、脳血管障害の診断薬。
【図1】

【図2】
【図2】
【公開番号】特開2012−211112(P2012−211112A)
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願番号】特願2011−78160(P2011−78160)
【出願日】平成23年3月31日(2011.3.31)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、文部科学省、科学技術試験研究委託事業、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(000002819)大正製薬株式会社 (437)
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【Fターム(参考)】
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願日】平成23年3月31日(2011.3.31)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、文部科学省、科学技術試験研究委託事業、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(000002819)大正製薬株式会社 (437)
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【Fターム(参考)】
[ Back to top ]