
新規なぺプチド類
【課題】ノルアドレナリン、セロトニン、ドパミン、グルタミン酸及びグリシンなどの神経伝達物質のニューロンのアミン輸送因子の阻害剤として有用な新規ぺプチド類及びそれらの誘導体の取得と利用法の開発。
【解決手段】円錐巻貝から単離されたχ−コノトキシン・ぺプチドは、天然産のぺプチドであってもよく、合成され、又組替え体でもよく、その誘導体であってもよい。該ペプチドはニューロンのアミノ酸輸送因子を阻害する能力を有する。該ペプチドの全部一部又は一部をコードする核酸分子、該ペプチドに対する抗体、及びそれらの使用及びそれらを含む治療方法。
【解決手段】円錐巻貝から単離されたχ−コノトキシン・ぺプチドは、天然産のぺプチドであってもよく、合成され、又組替え体でもよく、その誘導体であってもよい。該ペプチドはニューロンのアミノ酸輸送因子を阻害する能力を有する。該ペプチドの全部一部又は一部をコードする核酸分子、該ペプチドに対する抗体、及びそれらの使用及びそれらを含む治療方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はノルアドレナリン、セロトニン、ドパミン、グルタミン酸及びグリシンなどの神経伝達物質のニューロンのアミン輸送因子の阻害剤として有用な新規ぺプチド類及びそれらの誘導体に関する。本発明はこれらのぺプチド類を含む医薬組成物、これらのぺプチド類の活性類似体を発見するのに有用な核酸プローブ、ニューロンのノルアドレナリン輸送因子阻害活性を有する化合物を発見するための検定法、及び失禁、心臓血管状態及び気分変調などの状態、これらに限定されないが、の予防又は治療におけるこれらのぺプチド類の使用にも関する。
【背景技術】
【0002】
コナス属(円錐巻貝)の海洋性巻貝類は、自己の餌を捕るために精巧な生化学的戦法を用いる。魚、虫又は他の軟体動物いずれかの捕食者として、この円錐巻貝はその餌に生物活性を有する小ぺプチド類のカクテルを含む毒を注入する。これらの毒性分子は、コノトキシン類(conotoxins)と呼ばれるが、様々な受容体及びイオンチャンネル類を標的とすることにより神経伝達を妨害する。どの単一のコナス種から得られる毒も100種より多くの異なるぺプチド類を含んでいる。このコノトキシン類はその生理学的標的に基づいてクラス分けされている。今日までに、10のクラスが記述されてきた。ω−コノトキシンクラスのぺプチド類は電圧感受性のCa2+チャンネルを標的としこれを遮断して、神経伝達物質の放出を阻害する。α−コノトキシン類とΨ−コノトキシン類はニコチン性ACh受容体を標的としこれをブロックして神経節遮断及び神経筋遮断を惹き起こす。μ−コノトキシンクラスのぺプチド類は電圧感受性のNa+チャンネル類を遮断して筋肉及び神経の活動電位を阻害するように作用する。δ−コノトキシン類は電圧感受性のNa+チャンネル類の不活性化を標的としこれを遅延させてニューロンの興奮性を高める。κ−コノトキシンクラスのぺプチド類は電圧感受性のK+チャンネル類を標的としこれを遮断する。これらもニューロンの興奮性の向上を惹起する。コノプレッシン類はバソプレッシン受容体アンタゴニストであり、そしてコナントキン類はNMDA受容体アンタゴニストである。より最近、電圧感受性の非特異的カチオンチャンネルを標的とする新たなγ−コノトキシンクラスの原型、及び5HT3受容体に拮抗する新たなσ−コノトキシンクラスの原型が記述された。
【発明の概要】
【0003】
χ−コノトキシンクラスと以下に呼ばれる、コノトキシンの新しいクラスが存在することがここに発見されるに至った。このクラスはニューロンのアミン輸送因子を阻害する能力を持つことを特徴とするものである。
【0004】
神経伝達物質の再吸収を阻害する化合物は尿失禁、排尿筋不安定性及び間質性膀胱炎などの尿道下方障害の治療に有用であることが見出されてきた。このような化合物の一つは「イミプラミン」であり、これはノルアドレナリン再吸収の阻害に加え、カルシウムチャンネルの遮断に悪影響を及ぼすこと、そして抗コリン作用性の、局部麻酔薬的活性及び他の幾つかの効果を示すことが示されてきた。ノルアドレナリン再吸収を阻害できる他の化合物については、米国特許第5,441,985号に記載されている。これらの化合物はイミプラミンに比べて低い抗コリン作用性効果を有するといわれる。
【0005】
本発明のぺプチド類の場合には、神経伝達物質再吸収のこの阻害は、放出されたノルアドレナリンをシナプスからニューロンに迅速に戻して浄化する働きをするノルアドレナリン輸送因子などのニューロンの神経伝達物質輸送因子を選択的に阻害することにより達成される。
【0006】
本発明のぺプチド類はアミン輸送因子を阻害する活性を有する最初のぺプチド類である。今日までに特性決定された他のコノトキシン・ぺプチド類はすべてイオンチャンネル類又は細胞表面上の受容体類を標的とする。
【図面の簡単な説明】
【0007】
【図1】図1は二等分されたラット前立腺精管の1回の超最大パルス(55V,1ms)での電場刺激による収縮の時間経過に及ぼすχ−MrIAの典型的な効果を示すグラフである。χ−MrIA(30nM〜3μM)はその器官浴に半対数単位刻みで累積的に添加した。
【図2】図2は外因性α1アドレナリン受容体アゴニストに対するラット精巣上体精管の二等分された部分の収縮性応答に及ぼすχ−MrIA,−MrIAの効果のグラフである。(a)はχ−MrIA,−MrIAの不存在(○)及び1μM(△)又は3μM(□)の存在の下でのノルアドレナリンに対する、log(濃度)−応答曲線、(b)は3μMのχ−MrIAの存在下(□)及び非存在下(○)におけるメトキサミンに対するlog(濃度)−応答曲線。(a)及び(b)における各データ点は4〜5の個体の組織調製物についての観察の平均値±SEMを表す。一部の誤差棒は記号によって曖昧になっている。
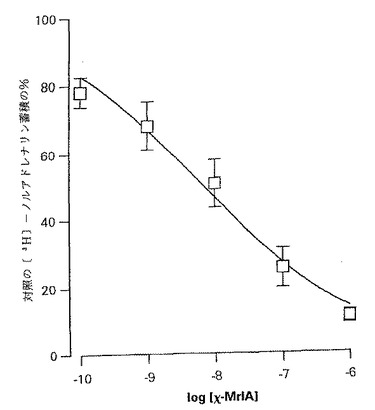
【図3】図3はヒトニューロンのノルアドレナリン輸送因子のcDNAクローンでトランスフェクトされたチャイニーズハムスター卵巣(CHO)細胞による[3H] −ノルアドレナリンのデシプラミン感受性蓄積のχ−MrIAによる阻害を示すグラフである。[3H] −ノルアドレナリンの蓄積はχ−MrIAの非存在下における細胞の取込みの百分率として表す。データ点は4回の別々の実験の観察の平均値±SEMを表す。
【図4】図4はイモガイ(coneshell)毒のぺプチド配列の誘導を表す図式的表示である。プライマーAP1+CHI−1Bを用いる5’RACE・PCRにより5’UTRとリーダーぺプチド配列を得る。このリーダーぺプチド配列を用いて次にχ−コノトキシン類に特異的なPCRプライマーを作成する。プライマーCHI−1A+ANCHORを用いる3’UTRにより、残りの成熟ぺプチド配列及び3’UTR配列の誘導を完成した。
【発明を実施するための形態】
【0008】
本発明の一つの側面によれば、ニューロンのアミン輸送因子を阻害する能力を有する単離され、合成され又は組換えで得られたχ−コノトキシン・ぺプチドが提供される。
このニューロンのアミン輸送因子はニューロンのノルアドレナリン輸送因子であることが好ましい。
【0009】
このχ−コノトキシン・ぺプチドは円錐巻貝から単離された天然産のぺプチドであってもよく、又はその誘導体であってもよい。
該χ−コノトキシン・ぺプチドはχ−MrIA若しくはχ−MrIB、又はそれらの誘導体であることが好ましい。χ−MrIA及びχ−MrIBは軟体動物狩猟性の円錐巻貝であるコナス・マルモレウス(Conus marmoreus)の毒から単離されうる。これらは、共に長さが13アミノ酸残基のぺプチドであり、2個のジスルフィド結合を含んでいる。これらのぺプチドはニコチン性ACh受容体アンタゴニストとして作用するα−コノトキシンクラスのメンバーに最も相同性を示す。
【0010】
χ−MrIA及びχ−MrIBのアミノ酸配列は以下のとおりである。
χ−MrIA NGVCCGYKLCHOC 配列番号:1
χ−MrIB VGVCCGYKLCHOC 配列番号:2
そのC末端は遊離の酸でもアミド化されていてもよい。
【0011】
上記の配列において、「O」は4−ヒドロキシプロリン(Hyp)を指す。このアミノ酸残基はコードされたぺプチドの翻訳後修飾から得られ、従ってそのヌクレオチド配列には直接コードされていない。
【0012】
このχ−コノトキシン・ぺプチドはニューロンのノルアドレナリン輸送因子の選択的阻害剤であることが好ましい。「選択的」及び「選択的に」という用語は、本明細書で用いるとき、ニューロンのノルアドレナリン輸送因子の阻害剤としての該ぺプチドの活性がα1−アドレナリン受容体におけるどの活性よりもかなり大きいことを意味する。
【0013】
米国特許第5,441,985号は、抗コリン作用性効果を殆ど持たないノルアドレナリン再吸収阻害剤は下部尿道障害の治療にとりわけ有用であることを示す。χ−MrIAも検出可能な抗コリン作用性効果を持たないことが見出された。
【0014】
従って、本発明の好ましい実施態様では、ニューロンのノルアドレナリン輸送因子を選択的に阻害する能力を有し、かつ、抗コリン作用性効果を持たないか殆ど持たない、単離され、合成され又は組換えで得られたχ−コノトキシン・ぺプチドが提供される。
【0015】
χ−MrIAはナトリウムチャンネル遮断剤としての活性もドパミン輸送因子阻害剤としての活性も持たないことも見出された。他のノルアドレナリン輸送因子阻害剤が普通に共有しているこれらの付加的な薬理学的活性がχ−MrIA及び本発明の好ましいぺプチドに欠如していることは、これらのぺプチドを有用な薬理学的道具とする。
【0016】
本発明のこのχ−コノトキシン・ぺプチド類は、χ−MrIA及びχ−MrIBなどの天然に生ずるぺプチド類であってもよく、天然に生ずるぺプチド類の誘導体であってもよい。
【0017】
天然に生ずるχ−コノトキシン・ぺプチド類、例えば、χ−MrIA及びχ−MrIBとの関連で本明細書で用いるとき、「誘導体」という用語は、天然に生ずるぺプチド類とは一つ以上のアミノ酸の欠失、付加、置換又は側鎖の修飾により異なっているぺプチドを指す。ニューロンのノルアドレナリン輸送因子を阻害する能力を持たないこのような誘導体は本発明の範囲内には入らない。
【0018】
置換はアミノ酸が異種の天然型アミノ酸残基又は非天然型アミノ酸残基で置換されるアミノ酸の変更を包含する。このような置換は、ポリぺプチドに含まれるアミノ酸残基が極性、側鎖の官能基又はサイズに関して類似の特性を持つ別の天然型アミノ酸で置換される場合、例えばSer←→Thr←→Pro←→Hyp←→Gly←→Ala、Val←→Ile←→Leu、His←→Lys←→Arg、Asn←→Gln←→Asp←→Glu又はPhe←→Trp←→Tyrの場合には、「同類置換」として分類されうる。一部の非天然型アミノ酸が天然型アミノ酸を置換するのに適当であることもあることは理解されるべきである。例えば、オルニチン、ホモアルギニン及びジメチルリジンはHis、Arg及びLysに関連する。
【0019】
本発明により包摂される置換は「非同類置換」であってもよい。この場合は、ポリぺプチド中に存在するアミノ酸残基が異なる基を持つ天然アミノ酸などの異なる特性を持つアミノ酸で置換され(例えば、荷電を持ったアミノ酸又は疎水性アミノ酸がアラニンで置換されること)、また、天然アミノ酸が非天然型アミノ酸で置換される。
【0020】
アミノ酸置換は通常1残基の置換であるが、かたまりの又は分散した複数の残基の置換でもよい。
アミノ酸置換は同類置換が好ましい。
【0021】
付加は一つ以上の天然型又は非天然型のアミノ酸残基の付加を含む。欠失は一つ以上のアミノ酸残基の欠失を含む。
上述のように本発明はアミノ酸の一つ以上が側鎖の修飾を受けたぺプチドを含む。本発明が予定する側鎖の修飾の例としては、アルデヒドとの反応の後NaBH4での還元による還元的アルキル化、メチルアセチミデートでのアミディネーション(amidination)、無水酢酸によるアシル化、シアネートによるアミノ基のカルバモイル化、2,4,6−トリニトロベンゼンスルホン酸(TNBS)によるアミノ基のトリニトロベンジル化、無水コハク酸及び無水テトラヒドロフタル酸によるアミノ基のアシル化、及びピリドキサル−5−リン酸で処理した後NaBH4での還元によるリジンのピリドキシル化等によるアミノ基の修飾が挙げられる。
【0022】
アルギニン残基のグアニジン基は2,3−ブタンジオン、フェニルグリオキサル及びグリオキサル等の試薬との複素環縮合産物の形成により修飾されうる。
カルボキシル基はO−アシルイソウレア形成を経てカルボジイミド活性化の後誘導体化により、例えば対応するアミドに誘導することにより修飾されうる。
【0023】
スルフヒドリル基は、ヨード酢酸又はヨードアセトアミドでのカルボキシメチル化、システイン酸への過ギ酸酸化、他のチオール化合物との混合ジスルフィドの形成、マレイミド、無水マレイン酸又は他の置換マレイミドとの反応、4−クロロメルクリ安息香酸、4−クロロメルクリフェニルスルホン酸、フェニルメルクリクロリド、2−クロロメルクリ−4−ニトロフェノール及び他の水銀化合物を用いる水銀誘導体の形成、アルカリ性pHでのシアン酸塩によるカルバモイル化などの方法により修飾しうる。システイン残基の修飾はいずれも、必要なジスルフィド結合を形成する該ぺプチドの能力に悪影響を与えてはならない。該ぺプチドが一つ以上のジスルフィド結合の代わりにジセレン結合を形成するようにシステインのスルフヒドリル基をセレン等価体と置換することも可能である。
【0024】
トリプトファン残基は、例えば、N−ブロモスクシンイミドでの酸化又は2−ヒドロキシ−5−ニトロベンジルブロミド若しくはスルフェニルハライドによるインドール環のアルキル化により修飾しうる。他方、チロシン残基はテトラニトロメタンでの硝酸化により変化させて3−ニトロチロシン誘導体を形成させうる。
【0025】
ヒスチジン残基のイミダゾール環の修飾は、ヨード酢酸誘導体によるアルキル化又はジエチルピロカーボネートによるN−カルベトキシル化により達成しうる。
プロリン残基は、例えば、4−位におけるヒドロキシル化により修飾しうる。
【0026】
修飾された側鎖を持つアミノ酸及び他の非天然型アミノ酸の一部のリストを表1に示す。
【0027】
【表1a】

【0028】
【表1b】
【0029】
【表1c】
【0030】
これらのタイプの修飾は、個体に投与されるとき又は診断用試薬として使用されるとき該ぺプチドを安定化するのに重要でありうる。
本発明が予定する他の誘導体としては、全くグリコシル化されていない分子から変更されたグリコシル化分子までのある範囲のグリコシル化変異型が挙げられる。変更されたグリコシル化パターンは異なる宿主細胞における組換え分子の発現から生じうる。
【0031】
本発明のχ−コノトキシン類は通常そのC末端でアミド化されているが、遊離のカルボキシル末端又はC末端における他の修飾を有する化合物も本発明の範囲内にあると考えられる。このぺプチド類はそのC末端がアミド化されているか又は遊離のカルボキシルを有することが好ましい。
【0032】
天然産のχ−コノトキシン・ぺプチド類の誘導体はCys残基及び特徴的なジスルフィド結合様式を保持していることが好ましい。誘導体には付加的なCys残基が含まれうる、ただしそれらがジスルフィド結合の形成の間保護されている場合である。
【0033】
修飾により天然産のχ−コノトキシン・ぺプチド類の誘導体を形成する場合、活性な天然産ぺプチド類のアミノ酸配列を比較して、あるとすれば、どの残基が活性ぺプチド間で保存されているかを決定することが有用である。これらの保存された残基の置換は、禁止されるわけではないが、保存されていない残基の置換よりは好ましくない。
【0034】
Alaで一つ以上の残基を置換している誘導体は薬理団(pharmacophore)を同定するために使用することができる。一時に一つ又は二つのアミノ酸だけがAlaで置換されることが好ましい。この薬理学的クラスのぺプチドのその受容体への結合に関与する相互作用のタイプをより正確に規定するのを助けるため、荷電を持った残基、極性の残基又は疎水性の残基がそれぞれ置換されているさらなる新たなぺプチド類を作ることができる。荷電が逆転したり、又は極性の残基が疎水性の残基と置換したりする非同類置換は、結合に関与する残基をそれ以上に同定することができる。これらのぺプチドはすべて、効力の改善又は選択性の増大を示す潜在能力を有する。非天然型アミノ酸の変更も、効力、選択性、及び/又は安定性を改善するために含められよう。
【0035】
露出されている残基は受容体結合に関与する可能性が最も高く、そして体系的に置換することができる。効力及び/又は選択性の改善を目指して追加的な結合相互作用を獲得するため、より長い側鎖型又は非同類置換を用いて、結合に関与する残基及び薬理団の直ぐ周囲にある残基を変えることは、特に強調される。MrIA又はMrIBの環の大きさや尾を減少させたり拡大したりすると、活性はさらに変化する。
【0036】
MrIA又はMrIBは一つの尾(残基1〜3)と二つの環(残基6〜9及び残基11〜12)から構成されることに注目されるが、χ−コノトキシン・ぺプチド類及び本発明の誘導体はアミノ酸及びジスルフィド結合のこの特定の配列を持つものに限定されない。他の配列も可能であり、その結果得られるぺプチドが必要な活性を有するならば、そのぺプチドは本発明の範囲に入る。該ぺプチド類は少なくとも二つのシステイン残基及び少なくとも一つのジスルフィド結合を持つことが好ましく、又は四つのシステイン残基と二つのジスルフィド結合を持つことがさらに好ましい。
【0037】
これらのぺプチド類のジスルフィド結合の連結はA−B/C−D、A−C/B−D又はA−D/B−Cでありうるが、後者がMrIA及びMrIBにとって好ましい。A、B、C及びDはそれぞれジスルフィド結合の形成に関与する第一、第二、第三及び第四Cys残基を指す。
【0038】
これらのぺプチド類に標識を付け、同じ部位に作用する新たな分子を同定するための結合検定法を確立するために使用することもできる。例えば、MrIA又はMrIBの標識化リガンドには、Tyr又は他の適当な残基を介してトリチウムを含ませ又は放射活性ヨウ素若しくは類似のものを付着させることができよう。各ぺプチドの全体にわたるTyrを調査すれば、該Tyrの組み込みに適する位置を決定できる。組織ホモジネート又は化合物若しくは混合物により発現された輸送因子に対するこのような標識ぺプチド類の結合の阻害から、この部位において活性な新たなぺプチド、ヒト組織を含む哺乳類の血清及び神経及び筋肉組織中に存在するぺプチドを含む新たなぺプチドの同定が可能となるであろう。この検定法はMrIA及びMrIBと同じ部位にも作用する非ぺプチド分子であって
、これらのぺプチドの経口的に活性な形として有用性を持ちうるものの同定をも可能とする。標識ぺプチド類はさらに種々の組織にわたるぺプチド結合の位置を同定するためのオートラジオグラフによる研究を可能とする。
【0039】
これらの配列の部分を使用してESTRデータベースを調査し、哺乳類において同様に作用する内因性リガンドを同定するために使用できる関連配列情報を含むぺプチド類又はタンパク質類を哺乳類で同定することができる。
【0040】
本発明のχ−コノトキシン類は標準的なぺプチド合成の後の酸化的ジスルフィド結合形成の方法により調製しうる。例えば、その線状ぺプチドは、シュノルツァーら(1992)が記述したように、BOC化学を用いる固相法により合成しうる。脱保護及び固体支持体からの切断の後、還元型ぺプチドを調製用クロマトグラフィーにより精製する。この精製された還元型ぺプチドを、例えば実施例2で述べるように、緩衝系で酸化する。この酸化型ぺプチドを調製用クロマトグラフィーを用いて精製した。
【0041】
コノトキシン類の合成を記述する参考文献としては、サトーら、リューら及びWO91/07980が挙げられる。
χ−コノトキシン類は組換えDNA技術を用いて調製することもできる。所望のぺプチド配列をコードするヌクレオチド配列を適当なベクター中に挿入し、適当な発現系でタンパク質を発現させる。場合によっては、この発現されたぺプチドのさらなる化学修飾、例えばC末端のアミド化、が適当でありうる。一部の状況の下では、ぺプチド発現後の化学的工程として、発現したぺプチドの酸化的結合形成を行うことが望ましい。これには折り畳まれていないぺプチドを得るため還元的工程を先行させてもよい。当業者はぺプチドの還元及び酸化に適する条件を容易に決定しうる。
【0042】
本発明はさらに、上述のχ−コノトキシン・ぺプチドをコードするヌクレオチド配列又はコードする配列に相補的な配列を含む単離された核酸分子を提供する。
本発明の更なる側面において、χ−コノトキシン・ぺプチドの全部又は一部をコードするヌクレオチド配列又はコードする配列に相補的な配列を含む核酸プローブが提供される。
【0043】
特に好ましい実施態様では、該核酸プローブは配列番号:1又は配列番号:2に示す配列をコードするヌクレオチド配列又はコードする配列に相補的な配列を含む。
本明細書で用いるとき、「プローブ」という言及には、増幅に用いられるプライマー又は直接ハイブリダイゼーションに使用するためのプローブに対する言及が含まれる。
【0044】
本発明のさらに別の側面は、本発明のχ−コノトキシン・ぺプチド類に対する抗体に関する。このような抗体はモノクローナルでもポリクローナルでもよく、標準的技法を用いて該ぺプチドに対して天然に生ずる抗体から選択してもよく、又は該ぺプチドに対し特異的に形成させてもよい。後者の場合、該ぺプチドはまず担体分子と混合する必要がありうる。本発明の抗体は治療薬又は診断薬として特に有用である。
【0045】
この点に関して、特異的な抗体を本発明のぺプチド類をスクリーニングするためにを使用することができる。そのような検定のための技法は当分野で良く知られており、例えば、サンドイッチ検定法やELISA法が含まれる。ぺプチドのレベルについての知識はある種の治療計画をモニターするのに重要でありうる。
【0046】
当分野で知られた技法を用いて抗イディオタイプ抗体を調製することも可能である。これらの抗イディオタイプ抗体及び治療薬としてのその使用は本発明の更なる側面を表す。
本発明の核酸分子はDNAであってもRNAであってもよい。該核酸分子がDNA型であるときは、それはゲノムDNA又はcDNAでありうる。本発明の核酸分子のRNA型は一般にmRNAである。
【0047】
本発明の核酸分子は通常単離された形であるが、それらはベクター分子、とりわけ発現ベクター分子などの他の遺伝子分子に組み込まれ、又は連結され、又は他の方法で融合され又は関係付けられていてもよい。ベクター及び発現ベクターは一般に複製可能であり、そして適用可能であれば、原核細胞又は真核細胞の一方又は両方で発現可能である。原核細胞には大腸菌、バチルス・スピーシーズ、及びシュードモナス・スピーシーズが含まれることが好ましい。好ましい真核細胞としては、酵母細胞、糸状菌細胞、哺乳類細胞及び昆虫細胞が挙げられる。
【0048】
従って、本発明の別の側面は、ベクター部分と本発明のぺプチドをコードすることができる遺伝子とを含む遺伝子構築物を予定する。
該遺伝子構築物の遺伝子部分は、プロモーターが適当な細胞中で該遺伝子部分の発現を指示することができるように該ベクター上の該プロモーターに機能的に連結されていることが好ましい。
【0049】
本発明は、このような遺伝子構築物及びそれを含む原核細胞又は真核細胞にまで及ぶ。
他の分子中にその活性を工学的に組み込むために、場合によっては余分の機能を持つ新たな分子を作成するために、他のコノトキシン類又は付加的に他のぺプチド若しくはタンパク質とMrIAなどのχ−コノトキシン類とのキメラを作成することができる。これはその薬理団を含むこれらのぺプチド配列の部分(単数又は複数)を用いて行うことが好ましいであろう。この薬理団が断続的である場合、薬理団を構成する複数の部分が受容体に結合できるよう該新たな構築物中に配置されるべきである。他のコノトキシン類とのキメラは、追加されたCys残基及び追加されたジスルフィド結合を含んでもよい。
【0050】
一つの活性クラス内のコノトキシン・ぺプチド類が、それぞれのシステイン残基の間のぺプチド環を持つ類似のジスルフィド結合パターンを有することは普通である。χ−MrIA及びχ−MrIBの場合、ジスルフィド結合は第一と第四、第二と第三のシステイン残基を連結する。このパターンはα−コノトキシン・ぺプチド類で観察された結合パターンとは異なる。この相違にもかかわらず、χ−コノトキシン・ぺプチドの環をα−コノトキシンなどの別のぺプチド由来の配列を含む環で置換することによりキメラ誘導体を調製しうる。
【0051】
本発明は他のぺプチド類に結合したχ−コノトキシン・ぺプチド類と同様にχ−コノトキシン・ぺプチドのダイマー、トリマー等をも含む。
本発明のχ−コノトキシン・ぺプチド類は10〜30アミノ酸を持つことが好ましく、11〜20アミノ酸を持つことがより好ましい。
【0052】
天然に生ずるχ−コノトキシン・ぺプチド類の完全な遺伝子配列は、クローニング及びDNA配列決定と併用した5’RACEと3’RACEの組合せ戦術を用いて明らかにしうる。
【0053】
本発明のχ−コノトキシン・ぺプチド類はニューロンのノルアドレナリン輸送因子を阻害する活性を持つ。従って、本発明はニューロンのノルアドレナリン輸送因子の阻害剤としてのχ−コノトキシン・ぺプチド類の使用、及びニューロンのノルアドレナリン輸送因子の阻害がそれに関する有効な治療と関係がある病気又は状態の治療又は予防における使用を提供する。薬理学的作用物質のこのような活性は、排尿系若しくは心臓血管系の病気若しくは状態又は気分変調の予防又は治療、又は苦痛若しくは炎症の治療又は制御における活性と関係がある。
【0054】
従って、本発明は、排尿若しくは心臓血管の状態若しくは病気又は気分変調の治療又は予防、又は苦痛若しくは炎症の治療又は制御のための方法であって、ニューロンのノルアドレナリン輸送因子を阻害する能力を持つ単離され、合成され、又は組換えで得られたχ−コノトキシン・ぺプチドの有効量を哺乳類に投与する工程を含む方法を提供する。
【0055】
排尿系の病気若しくは状態の例としては、尿失禁及び排便失調が挙げられる。心臓血管の病気若しくは状態の例には、種々の起源の不整脈及び冠不全が含まれる。気分変調の例としては、うつ病、不安、及び喫煙などの切望が含まれる。苦痛の例としては、慢性の苦痛、神経障害性の苦痛又は炎症性の苦痛が挙げられる。
【0056】
該哺乳類は、予防的な意味で該ぺプチドが投与されることもありうるが、このような治療を必要とするものであることが好ましい。
本発明はニューロンのノルアドレナリン輸送因子を阻害する能力を持つ単離され、合成され又は組換えで得られたχ−コノトキシン・ぺプチドと、薬学的に許容しうる担体又は希釈剤を含む組成物をも提供する。
【0057】
該組成物は薬学的組成物の形であることが好ましい。
また、排尿又は心臓血管の状態若しくは病気、又は気分変調の治療又は予防用、又は苦痛若しくは炎症の治療若しくは制御用の医薬の製造における、ニューロンのノルアドレナリン輸送因子を阻害する能力を有する単離され、合成され又は組換えで得られたχ−コノトキシン・ぺプチドの使用も提供される。
【0058】
ノルアドレナリン輸送因子は神経細胞により発現されるだけでなく、胎盤、肺の内皮細胞及び子宮などの他の組織でも発現されることも注目される。本発明のぺプチド類はこれらのノルアドレナリン輸送因子の阻害にも有効であり、これらの輸送因子が絡んでいる状態を治療する際に有用でありうる。
【0059】
当業者は容易に理解するように、投与の経路及び薬学的に許容しうる担体の性質は該状態の性質及び治療される哺乳類に左右される。具体的な担体又は送達系の選択、及び投与経路の選択は当業者により容易に決定され得ると思われる。該活性ぺプチドを含む任意の製剤の調製においては、該ぺプチドの活性が工程中に破壊されないように、かつ、該ぺプチドが破壊されずに作用部位まで到達できるように注意が払われるべきである。状況によっては、例えば、ミクロカプセル化などの当分野で知られた手段により該ぺプチドを保護することが必要であることもある。同様に、選ばれる投与経路は該ぺプチドがその作用部位に到達するようなものであるべきである。
【0060】
注射可能な用途に適切な薬学的剤形には、滅菌注射可能な溶液又は分散液、及び滅菌注射可能溶液の即時調製用の滅菌粉末が含まれる。これらは製造及び貯蔵の条件下で安定であるべきであり、酸化及び細菌や黴などの微生物の汚染作用に対して保護されうる。
【0061】
当分野の熟練者は常法的アプローチで本発明のぺプチド類又は修飾されたぺプチド類のための適切な処方を容易に決定しうる。好ましいpH範囲及び、例えば抗酸化剤などの適当な賦形剤の特定は当分野では機械的作業である(例えば、クリーランドら,1993を参照)。緩衝系は望みの範囲のpH値を得るために機械的に用いられ、例えば、酢酸塩、クエン酸塩、乳酸塩及びコハク酸塩などのカルボン酸緩衝液が含まれる。このような処方には、BHT又はビタミンEなどのフェノール性化合物、メチオニン又は亜硫酸塩などの還元剤、及びEDTAなどの金属キレート剤を含む多様な抗酸化剤が利用可能である。
【0062】
注射可能な溶液又は分散液のための溶媒又は分散媒としては、活性ぺプチドのための任意の従来の溶媒又は担体系が含まれ、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール及び液状ポリエチレングリコールなど)、それらの適当な混合液、及び植物油が含まれる。例えば、レシチンなどの被覆剤の使用により、分散液の場合には要求される粒子サイズの維持により、そして界面活性剤の使用により、適当な流動性を維持できる。微生物の作用の防止は、必要ならば、例えば、パラベン、塩化ブタノール、フェノール、ソルビン酸、チメロサール等の種々の抗細菌剤及び抗真菌剤を含めることにより可能となる。多くの場合、重量オスモル濃度を調整するための物質、例えば、糖類又は塩化ナトリウムを含むことが好ましい。注射用の処方は血液と等張であるこ
とが好ましい。注射可能な組成物の長期吸収性は、例えば、モノステアリン酸アルミニウム及びゼラチンなどの吸収を遅延する薬剤を組成物に含めることによりもたらされ得る。注射可能な使用に適切な薬学的剤形は、静脈内、筋肉内、大脳内、鞘内、硬膜外の注射又は注入を含む任意の適切な経路により送達されうる。
【0063】
注射可能な滅菌溶液は、必要量の活性化合物を、上に列挙したような種々の他の成分と適切な溶媒中で混合し、必要ならば、その後濾過滅菌することにより調製される。一般に、分散液は、基本的な分散媒と上に列挙したものから選択された必要な他の成分とを含む滅菌ビークル中に種々の滅菌した活性成分を混合することにより調製される。注射可能な滅菌溶液調製用の滅菌粉末の場合、調製の好ましい方法は、活性成分に任意の追加の望みの成分を加え予め滅菌濾過した溶液を真空乾燥又は凍結乾燥したものである。
【0064】
該活性成分が適切に保護されている場合、該成分は、例えば、不活性希釈剤若しくは同化しうる食用担体とともに経口投与してもよく、又は硬い殻若しくは軟い殻のゼラチンカプセルに封入してもよく、錠剤に圧縮してもよく、常食の食物と直接混合してもよい。経口治療投与では、該活性化合物は賦形剤と混合してもよく、摂取可能な錠剤、バッカル錠、トローチ、カプセル、エリキシル、懸濁液、シロップ、オブラート剤等の剤形で用いてもよい。このような組成物及び調製物は少なくとも1重量%の活性化合物を含むことが好ましい。もちろん、組成物及び調製物の百分率は様々であり、その単位剤形の重量の約5%から約80%の間にあるのが都合がよい。このような治療に有用な組成物における活性化合物の量は、適切な用量が得られるような量である。
【0065】
錠剤、トローチ、丸薬、カプセル等は以下に列挙するような成分も含みうる。ゴム、アラビアゴム、コーンスターチ又はゼラチン等の結合剤、リン酸二カルシウムなどの賦形剤、コーンスターチ、ポテトスターチ、アルギン酸等の崩壊剤、ステアリン酸マグネシウム塩などの滑剤、及びスクロース、ラクトース又はサッカリンなどの甘味料が添加されてもよく、あるいはペパーミント、冬緑油、又はさくらんぼ風味などの香味料が添加されてもよい。投与単位剤形がカプセルの場合、上記タイプの物質に加えて液体の担体を含んでもよい。種々の他の物質が被覆剤として又はその他の方法で投与単位の物理的形態を改変するために存在してもよい。例えば、錠剤、丸剤、又はカプセルがセラック、糖又はその両方で被覆されてもよい。シロップ又はエリキシルは活性化合物、甘味剤としてのスクロース、保存剤としてのメチルパラベン及びプロピルパラベン、さくらんぼ又はオレンジ風味の色素及び香味料を含んでもよい。もちろん、いかなる投与単位剤形を調製するのに用いられるいずれの物質も、その使用量において薬学的に純粋であり実質的に無毒性であるべきである。さらに、これらの活性化合物は放出制御調製物及び放出制御処方に組込まれてもよい。
【0066】
本発明は、投与、例えば、クリーム、ローション、ジェル等の局所的適用、又は、吸入若しくは鼻腔内送達に適した組成物、例えば、溶液若しくは乾燥粉末にふさわしい任意の他の剤形にも及ぶ。
【0067】
静脈内、大脳内、鞘内若しくは硬膜外の送達に適する剤形などの非経口投与剤形は好ましい。
薬学的に許容しうる担体及び/又は希釈剤には、任意の及び全ての溶媒、分散媒、被覆剤、抗細菌剤及び抗真菌剤、等張剤、及び吸収遅延剤等が含まれる。薬学的に活性な物質のためのこのような媒体及び薬剤の使用は当分野では周知である。通常の媒体及び薬剤はいずれも、活性成分と配合禁忌であるときに限りそれを除いて、治療用組成物にそれらを使用することが意図される。補足的活性成分もまた該組成物に組み込まれ得る。
【0068】
投与の容易性及び服用量の均一性のため、非経口組成物を投与単位剤形に製剤化することは特に利点がある。本明細書で使用されるとき、投与単位剤形とは、治療される哺乳類の対象にとって一回の投薬に適した物理的に個別の単位を指し、各単位は必要とされる薬学的担体とともに所望の治療効果を生じるように計算され予め測られた量の活性物質を含む。本発明の新規な投与単位剤形の仕様は、(a)活性物質のユニークな特性及び達成される具体的な治療効果ならびに(b)本明細書で詳細に開示されるような身体の健康が損なわれた病気状態を有する生物の患者における病気の治療のためにこのような活性物質を調合する技術に固有の制限によって左右され、直接これらに依存する。
【0069】
この主要な活性成分は、簡便かつ効果的な投与のために投与単位剤形中に適切な薬学的に許容しうる担体とともに有効な量で調合される。単位投与剤形は、例えば、0.25μgから約2000mgまでの範囲の量で主要な活性成分を含むことができる。割合で表わすと、この活性化合物は一般的に1mlの担体に対して約0.25μgから約200mgまで存在する。補足的活性成分を含む組成物の場合、一回投薬量は前記成分の通常の一日服用量及び投与の方法を参照して決定される。
【0070】
添付の図面及び実施例を参照してここに本発明を記述するが、以下の記述の具体性が本発明についてのこれまでの記述の一般性にとって代わるものではないことが理解されるべきである。
【実施例】
【0071】
薬物
この実施例及び以下の実施例に用いられる薬物には、デシプラミン塩酸塩(シグマ)、インドメタシン(シグマ)、メトキサミン塩酸塩(シグマ)、(−)−ノルアドレナリン二酒石酸塩(シグマ)、[3H] −1−ノルアドレナリン(比活性2200Ci/mM、ニューイングランド・ヌクレアー、ボストン,MA(U.S.A.))、テトロドトキシン(シグマ)、ヨヒンビン塩酸塩(シグマ)が含まれる。
統計的分析
以下の実施例のデータは4〜6回の実験の平均値及び標準誤差として表される。EC50値の計算のためのシグモイド曲線−適合は、ソフトウエア・パッケージ・イゴール・プロ(ウエイブメトリクス)を用いて非線型回帰により行った。平均値の間の差はソフトウエア・パッケージ・プリズム(グラフパド)を用いスチューデントの両側tテストにより評価した。P<0.05という値は統計的に有意の差を示すものとされた。
【0072】
実施例1
ラットの精管の調製
雄ウイスターラット(250〜350g)を頭部を打って殺し、その精管を切除した。各組織を二等分した精巣上体部分と前立腺部分に分けた。この組織部分を5mlの器官浴中に載せ0.5gの張力を掛けた。この浴溶液は次の組成、即ち、NaCl,119mM、KCl,4.7mM、MgSO4,1.17mM、KH2PO4,1.18mM、NaHCO3,25.0mM、グルコース,5.5mM、CaCl2,2.5mM、EDTA,0.026mMを含んでいた。この浴溶液を37℃に維持し、O2中5%v/vCO2を吹き込んだ。実験の開始前にこの組織調製物を少なくとも45分間平衡化させた。アイソメトリック・フォース・トランスデューサー(ナルコ・バイオ・システムF−60)により収縮を記録し、チャート・バージョン3.5.4/sソフトウエア及びマックラボ/8s・データ・アクイジション・システム(ADインストルーメンツ)を用いて200Hzのサンプリング頻度でパワー・マッキントッシュ・コンピュータに記録した。
【0073】
この二等分した前立腺セグメントを用いて、交感神経系神経伝達により媒介される平滑筋の電気誘発収縮に及ぼすχ−MrIAの効果を試験した。この組織調製物をプラチナ刺激電極で広げた。電場刺激(EFS)は、グラスS44スティミュレーターにより発生させた1ms持続の1個の55Vパルスを用い3分間隔で行った。この収縮はテトロドトキシン(0.1μM)で阻止することができた。このことはこの収縮が神経により媒介されていることを示す。ペプチドは、半対数単位増加で濃度を増加させながら器官浴中に累積的に添加された。各投与量は電気刺激への応答に対する先の投与量の効果が定常レベルに達した直後に適用された。
交感神経系神経伝達に及ぼすχ−MrIAの効果
ラットの前立腺精管の二等分された部分は電場刺激に応答して2相性の収縮を示した。この調製物では、該2相性収縮の二つの成分は時間的に十分に分離された。該応答の第一の部分は主要な応答であり、電気パルスの送達後200msでほぼ最高レベルに達した。該収縮の第二の相は刺激後500msでほぼピークに達した。χ−MrIAの薬理学的活性を同定するための本発明者らの試みは電場刺激されたラット精管におけるその効果の研究で始まった。電場刺激への該調製物の応答に及ぼすχ−MrIAの効果を図1に示す。このコノトキシン(30nM〜3μM)は該収縮の第二相を増加するように作用した。この効果は濃度依存性であることが見出された。χ−MrIAの存在下で得られたトレースから対照の応答を差し引くことにより、該収縮の第二の成分のみの該ペプチドによる特異的増強が明らかとなる。第二の成分を特異的に増強するように作用するχ−MrIAの濃度−応答曲線も構成することができる。
【0074】
電気誘発応答に及ぼすχ−MrIAの作用は高度に特異的であり、該2相性応答の第二の成分のみを増強する。該収縮のこの後の相はノルアドレナリンによって媒介されることが認められており、プラゾシン及び他のα1−アドレナリン受容体アンタゴニストにより選択的に阻害される。該収縮の第一の相は放出されたATPによる接合部後のP2Xプリン受容体の活性化によるものであるが、同様には増強されなかった。該収縮のノルアドレナリン作用性成分の大きさは、交感神経系神経興奮により放出されるノルアドレナリンの量、接合部後のα1−アドレナリン受容体の密度及びそれらのエフェクター系への結合、そしてノルアドレナリンがシナプスから除去される速度により調節される。
【0075】
シナプス前のα2アドレナリン受容体における拮抗は、放出されたノルアドレナリンによる負のフィードバック系の活性化の阻止により、交換神経からのノルアドレナリンの電気誘発放出を増強することが良く認識されている。しかしながら、α2アドレナリン受容体拮抗はχ−MrIAの作用機構ではあり得ない。χ−MrIAと異なり、ヨヒンビンやイダゾキサンなどのα2アドレナリン受容体アンタゴニストはラット精管の収縮のプリン性成分とノルアドレナリン作用性成分を平等に増強するように作用する。さらに、1回のパルスへの応答は、一続きの刺激への応答とは対照的に、これらの受容体部位には刺激の時にアゴニストが存在しないであろうから、この負のフィードバック機構による調節を受けない。従って、ヨヒンビン(1μM)はこの検定では誘発応答に対する効果を持たない
。
【0076】
実施例2
α1−アドレナリン受容体アゴニストに対する応答に及ぼすχ−MrIAの効果を試験するための調製物
ラットの精管は、二等分した精巣上体部分を電気的に刺激しなかったことを除き、上記のように使用した。これらの調製物は、χ−MrIAの存在下及び非存在下におけるノルアドレナリン及びメトキサミンへの濃度応答曲線を求めるために代わりに使用した。この組織では、ノルアドレナリン及びメトキサミンは接合部後のα1−アドレナリン受容体の活性化を介して平滑筋の収縮を惹き起こす。1μM又は3μMいずれかの濃度のχ−MrIAを器官浴に適用し、該組織と20分間平衡化した後ノルアドレナリン又はメトキサミンを累積的に添加した。χ−MrIAを適用しなかった反対側の組織セグメントを対照として用い、調製物当たり一つの濃度応答曲線を求めた。
【0077】
α1−アドレナリン受容体アゴニストへの応答に及ぼすχ−MrIAの効果χ−MrIAの作用が神経伝達物質放出の上流で起こるか下流で起こるかを決定することが、外因的に適用したノルアドレナリンへの応答に及ぼす該ペプチドの効果を調べることにより、可能となった。χ−MrIAは浴に適用されたノルアドレナリンの効力を増強させたので、該コノトキシンは神経の貯蔵からのノルアドレナリンの放出を増大させることによるのではなく、ノルアドレナリンへの応答を増強することにより作用すると本発明者らは結論できる。この増強はα1−アドレナリン受容体応答の増加又はノルアドレナリンの作用の終結の障害の結果として起こりうるであろう。これらのうちのいずれがχ−MrIAの作用機構であったかを確認するため、α1−アドレナリン受容体アゴニストであるメトキ
サミンを使用した。このα1−アドレナリン受容体アゴニストは、それがニューロンのノルアドレナリン輸送因子の基質でないという点でノルアドレナリンとは異なる。この輸送因子は、神経末端内への取り込みによりノルアドレナリン及び他のカテコラアミンをシナプスから消去するように働き、交感神経系で刺激された組織のアドレナリン受容体でのノルアドレナリンの作用を終結させるための主要な機構を表す。メトキサミンはこの機構による除去を受けないから、輸送因子の阻害はその作用の効力を増強しない。
【0078】
二つのα1−アドレナリン受容体アゴニストに対するラット精巣上体精管の二等分されたセグメントの応答に及ぼすχ−MrIAの効果を研究した。χ−MrIAの存在下又は非存在下でのノルアドレナリンに対するlog(濃度)−応答曲線を図2aに示す。1μMの濃度で、χ−MrIAはノルアドレナリンに対する該組織の感受性を増加するように作用し、濃度−応答曲線を左側へシフトさせた。観察された効力の程度は3μMのχ−MrIAでの実験でより大きかった。該コノトキシンのいずれの濃度もノルアドレナリンに対する該組織の最大応答を変更しなかった。χ−MrIAは3μMの濃度でメトキサミンに対する濃度応答曲線に影響を及ぼさなかった(図2b)。ノルアドレナリンへの応答に及ぼすその効果とは対照的に、χ−MrIAがメトキサミンの作用を強めないという観察
は、χ−MrIAがニューロンのノルアドレナリン輸送因子の阻害剤であるということと矛盾しない。
【0079】
メトキサミンへの濃度応答曲線に対するχ−MrIAの効果の欠如は、χ−MrIAがα1−アドレナリン受容体における効果を持たないことをも証明する。これは該曲線の右側への平行移動として明らかであろう。このことはχ−MrIAをノルアドレナリン輸送因子の他の一部の阻害剤、とりわけ抗鬱薬として用いられるものと区別する。多くの好鬱薬の治療標的はニューロンのノルアドレナリン輸送因子である。しかしながら、これらの化合物の多くは、とりわけ三環系抗鬱薬、及びより低い程度ではあるが通常の三環系抗鬱薬とは構造的に関係のない一部のより新しい薬剤は、α1−アドレナリン受容体及びムスカリン様のACh受容体などの他の部位で作用することが認められている。
【0080】
実施例3
モルモットの回腸
一晩絶食させた雄モルモット(285〜425g)の頭を打って殺し、放血した。約1.5cm長の回腸部分を除去し、浴溶液で満たした注射筒を用い温和に洗浄して腸内容物を洗浄した。この調製物を下記の組成の浴溶液、即ち、NaCl,136.9mM、KCl,2.68mM、CaCl2,1.84mM、MgCl2,1.03mM、グルコース,5.55mM、NaHCO3,11.9mM、及びKH2PO4,0.45mM、を37℃まで温め、O2中5%v/vCO2を吹き込んだもの、を含む5mlの器官浴中に置いた。安定なベースラインを作成するため、インドメタシン(10μM)を浴溶液中に含めた。該組織を1.0gの張力を掛けて設置し、実験の開始前40分間平衡化させた。ついで、ニコチンの用量(4μM)を、応答が一致すると観察されるまで15分間隔で適用した。次に、別の用量のニコチンを適用した25分後にχ−MrIA(3μM)を添加した。ニコチンに対するこの応答を、10Hzのサンプリング速度で、アイソメトリックに測定しそしてデジタル化した。
モルモット回腸のニコチンへの応答に及ぼすχ−MrIAの効果
χ−MrIA(3μM)はニコチンに対する回腸部分の応答には有意の効果を及ぼさなかった。該コノトキシンの非存在下では、χ−MrIAが存在した場合の4.07±0.80gと比べ、平均応答は3.83±0.76gであった(p>0.1、対合tテスト,n=4)。α−コノトキシン類は、ニューロンサブタイプ又は筋肉サブタイプいずれかのニコチン性ACh受容体をブロックする。モルモット回腸の単離された断片を用いてχ−MrIAがニューロンのニコチン性ACh受容体を標的としないことが証明された。収縮性応答がムスカリン系受容体の活性化に依存するため、該コノトキシンがムスカリンACh受容体アンタゴニストとしても作用したとするならば、モルモット回腸のニコチンへの応答はχ−MrIAの存在下で減衰したであろうことが予測されよう。この調製物では、ACh及び接合部後の受容体を活性化する種々の他の神経伝達物質、のニコチン誘発放出はχ−MrIAによって影響されなかった。従って、そして多くの他の輸送因子阻害剤とは対照的に、χ−MrIAは抗ムスカリン性活性を欠いている。
【0081】
実施例4
マウスの横隔膜神経−片側横隔膜調製物
雄クアケンブッシュマウス(20〜30g)を頸部転移により殺した。左及び右の片側横隔膜を横隔膜神経を付着させたまま切除した。各片側横隔膜の基部を二つの平行プラチナ刺激電極の間に配置し、横隔膜神経は電場刺激のための二つの小さなプラチナループを介して配置した。該調製物をO2中5%v/vCO2を吹き込んだ5mlの器官浴中で37℃でインキュベートした。この浴溶液の組成は、NaCl,135.0mM、KCl,5.0mM、CaCl2,2.0mM、MgCl2,1.0mM、グルコース,11.0mM、NaHCO3,15.0mM、及びKH2PO4,1.0mMであった。該組織を1.0gの静止張力を掛けて配置した。平衡化のため少なくとも30分間放置した後、該筋肉には2ms持続の30Vパルスを、該神経には0.2ms持続で3Vパルスをそれぞれ用いて、10s間隔で直接刺激及び間接刺激を交互に行った。直接的及び間接的に誘発される収縮に及ぼす3μMの濃度でのχ−MrIAの1回用量の効果を測定した。応答をデジタル化し、精管調製物で述べたように記録した。
【0082】
マウス横隔膜神経−片側横隔膜の電気刺激への応答に及ぼすχ−MrIAの効果横隔膜神経の電場刺激により誘発される収縮、又は直接の筋肉刺激により誘発される収縮は、3μMのχ−MrIA(n=4)により影響されなかった。χ−MrIAはマウスの横隔膜神経−片側横隔膜調製物では筋肉のニコチン性ACh受容体をブロックしない。骨格筋又は運動神経におけるχ−MrIAの活性のこの欠如により、χ−MrIAは、今日までに特性決定された、麻痺性の毒であり従って餌捕捉に明確な役割を持つ、コノトキシン・ペプチド類の大部分とは区別される。
【0083】
実施例5
[3H]−ノルアドレナリンの細胞による取り込み
チャイニーズ・ハムスター卵巣(CHO)細胞を24穴プレート(ファルコン)中、10%v/vウシ胎児血清中で増殖させた。60〜70%コンフルエンスに達したとき、該細胞を、ヒトニューロンのノルアドレナリン輸送因子の全長cDNA(パチョルシークら,(1991) Nature, 350, 350-4)を組み込んでいる発現ベクター(pcDNA3,インビトロゲン)で一時的にトランスフェクトした(リポフェクタミン,ギブコ)。このニューロンのノルアドレナリン輸送因子のcDNAクローンを使用した(ボルム・インスティチュート,ポートランド,OR,USA)。細胞の取り込み研究はトランスフェクションの36時間後に行った。該CHO細胞を、NaCl,157mM、KCl,2.7mM、NaH2PO4,11.8mM、MgCl2,1.0mM、及びCaCl2,0.1mM、pH7.4を含む輸送緩衝液でまず洗浄した。次いで、該細胞を50nMの[3H]−ノルアドレナリン(必要なときは非標識ノルアドレナリンを補足した)及び100μMアスコルビン酸を含む輸送緩衝液と共にインキュベートした。適当なときにχ−MrIA(0.1nM〜1μM)又はデシプラミン(10μM)も含めた。室温で20分後に、該細胞を氷冷リン酸緩衝化食塩水で素早く洗浄した後、0.1%v/vトリトン−X中で溶解した。この細胞溶解物を液体シンチレーション計測にかけ、その放射活性レベルを測定した。さらに、該細胞溶解物の部分標本を用いてタンパク質濃度を測定した(バイオラド・DC・プロテインアッセイ)。該ノルアドレナリン輸送因子による[3H]−ノルアドレナリンの特異的取り込みをデシプラミン(10μM)感受性の成分として定義した。
[3H]−ノルアドレナリンの細胞内蓄積に及ぼすχ−MrIAの効果
ヒトニューロンのノルアドレナリン輸送因子を発現するCHO細胞中へのノルアドレナリンの蓄積はデシプラミン(10μM)による対照量の0.5%未満にまで減少した。このことは該蓄積が殆ど全てクローニングされた輸送因子を介する特異的取り込みによるものであったことを証明する。このノルアドレナリン輸送因子は細胞取り込み研究において該コノトキシンの標的であることが確認された。χ−MrIA(0.1nM〜1μM)は放射標識ノルアドレナリンの蓄積を濃度−依存形式で阻害し(図3)、logIC50値は−8.17±0.0275(n=4)であった。該蓄積を50%だけ阻害するために必要なχ−MrIAの濃度は約7nMであることが見出された。この濃度はデシプラミンが同じ効果を達成するために必要な濃度よりもほぼ1オーダー低い量である。
【0084】
コカインとχ−MrIAは両方とも天然に生ずる化合物であるが、それらは全く似ていない。コカインはコカ植物の葉から抽出されるアルカロイドであるのに対し、χ−MrIAは動物遺伝子が直接コードするぺプチドである。該取り込み輸送因子におけるその効果に加え、コカインは強力な局部麻酔特性を有することが知られている。これはナトリウムチャンネルとカリウムチャンネルの両方の遮断による。検定のいずれにおいてもχ−MrIAの局部麻酔活性についての証拠は見出されなかった。χ−MrIAは単独では精管の正常な張りに対する収縮効果も弛緩効果も持たないことが見出された。同様な研究により、χ−コノトキシンはドパミン輸送因子を阻害しないことが明らかにされた。
【0085】
実施例6
トリチウム化したマジンドールのノルアドレナリン輸送因子への結合を、該輸送因子タンパク質を発現する細胞中で測定した(実施例5を参照)。トリチウム化したマジンドールの結合に及ぼす10-6から10-9のχ−MrIAの影響を測定した。χ−MrIAはトリチウム化したマジンドールの結合には効果を及ぼさなかった。このことは、それがデシプラミン、マジンドール及びコカインなどの従来のノルアドレナリン輸送因子阻害剤とは異なる部位に非競争的に作用することを示す。
【0086】
実施例7
χ−コノトキシン・ぺプチド類の遺伝子配列の誘導
クローニング及びDNA配列決定と併用した5’RACE(Random Amplification of cDNA Ends、cDNA末端の無作為増幅)及び3’RACE組合せ戦術を用いて、該χ−MrIAの完全な遺伝子配列を単離した。
5’RACE
その成熟ぺプチド配列からオリゴヌクレオチドプライマーCH1−1Bを設計した。このオリゴヌクレオチドと該ぺプチドとの関係は以下のとおりであり、該オリゴヌクレオチドの配列と共に示す。
χ−MrIA - NGVCCGYKLCHPC 配列番号:3
CHI−1B 5'- CANGGRTGRCANARYTTRTA -3'
配列番号:4
AP1 5'- CCATCCTAATACGACTCACTATAGGGC-3'
配列番号:5
(上記において、N=A/C/G/T、R=A/G、Y=C/T)
該オリゴヌクレオチドCH1−1BをAP1オリゴヌクレオチドと組合せ、イモガイ(coneshell)毒素管から単離されたmRNA由来のcDNA鋳型を用いて、ポリメラーゼ連鎖反応(PCR)を行った。そのPCR産物はMrIA遺伝子の5’領域を表すが、これを単離し、精製し、細菌ベクター中にクローニングし、配列決定した。MrIAの遺伝子配列はシー.マルモレウス(C. marmoreus)から得た(図4)。
3’RACE
該遺伝子の5’領域のDNA配列を用いて、MrIA配列及び他の密接に関連するぺプチド由来の配列を検出できるオリゴヌクレオチドを設計した。該遺伝子配列との関連での該オリゴヌクレオチドの位置決めは図4に示す。オリゴヌクレオチドCH1−1AをANCHORオリゴヌクレオチドと結合させてPCRに使用し、該遺伝子のリーダーぺプチド、成熟ぺプチド及び3’非翻訳領域(3’UTR)に相当するDNA断片を作成した。シー.マルモレウス由来の毒素管cDNA鋳型のPCRにより、MrIAぺプチドに相当するDNA断片を作成した。
【0087】
これらのオリゴヌクレオチド類のDNA配列は以下のとおりである。
CHI−1A 5'- ACAGGCAGAATGCGCTGTCTCCC -3'
配列番号:6
ANCHOR 5'- AACTGGAAGAATTCGCGGCCGCAGGAAT -3'
配列番号:7
χ−MrIAの完全配列
5’RACEと3’RACEを用いて作成したχ−MrIAの遺伝子配列は該遺伝子の重複断片類を表す。これらの断片類を組合せ、該遺伝子に対する共通配列を作成した。この共通配列は該遺伝子の完全cDNAであり、5’UTR、リーダーぺプチド、成熟ぺプチド及び3’UTRを含む。このχ−MrIAのリーダーぺプチドオリゴヌクレオチド配列及び成熟ぺプチドオリゴヌクレオチド配列を配列番号:8に示し、一方、リーダーぺプチドアミノ酸配列及び成熟ぺプチドアミノ酸配列は配列番号:9に示す。
【0088】
【化1】
【0089】
以下の本明細書及び請求の範囲を通じて、文脈が別義を要求する場合を除き、用語「含む(comprise)」及びその変化「含む(comprises)及び(comprising)」は、述べられた主体若しくは工程又は主体群若しくは工程群の含有を意味するが、任意の他の主体若しくは工程又は主体群若しくは工程群の排除を意味するものではないと理解されるべきである。
【0090】
当業者は本明細書に記述された発明が具体的に記述されたもの以外の変更及び修飾を受け易いことを認めるはずである。本発明はこのような変更及び修飾をすべて包含するものと解すべきである。本発明は本明細書で個々に若しくは集合的に言及し又は示したすべての工程、特徴、組成物及び化合物、及び該工程又は特徴の任意の組合せ及びすべての組合せをも包含する。
【技術分野】
【0001】
本発明はノルアドレナリン、セロトニン、ドパミン、グルタミン酸及びグリシンなどの神経伝達物質のニューロンのアミン輸送因子の阻害剤として有用な新規ぺプチド類及びそれらの誘導体に関する。本発明はこれらのぺプチド類を含む医薬組成物、これらのぺプチド類の活性類似体を発見するのに有用な核酸プローブ、ニューロンのノルアドレナリン輸送因子阻害活性を有する化合物を発見するための検定法、及び失禁、心臓血管状態及び気分変調などの状態、これらに限定されないが、の予防又は治療におけるこれらのぺプチド類の使用にも関する。
【背景技術】
【0002】
コナス属(円錐巻貝)の海洋性巻貝類は、自己の餌を捕るために精巧な生化学的戦法を用いる。魚、虫又は他の軟体動物いずれかの捕食者として、この円錐巻貝はその餌に生物活性を有する小ぺプチド類のカクテルを含む毒を注入する。これらの毒性分子は、コノトキシン類(conotoxins)と呼ばれるが、様々な受容体及びイオンチャンネル類を標的とすることにより神経伝達を妨害する。どの単一のコナス種から得られる毒も100種より多くの異なるぺプチド類を含んでいる。このコノトキシン類はその生理学的標的に基づいてクラス分けされている。今日までに、10のクラスが記述されてきた。ω−コノトキシンクラスのぺプチド類は電圧感受性のCa2+チャンネルを標的としこれを遮断して、神経伝達物質の放出を阻害する。α−コノトキシン類とΨ−コノトキシン類はニコチン性ACh受容体を標的としこれをブロックして神経節遮断及び神経筋遮断を惹き起こす。μ−コノトキシンクラスのぺプチド類は電圧感受性のNa+チャンネル類を遮断して筋肉及び神経の活動電位を阻害するように作用する。δ−コノトキシン類は電圧感受性のNa+チャンネル類の不活性化を標的としこれを遅延させてニューロンの興奮性を高める。κ−コノトキシンクラスのぺプチド類は電圧感受性のK+チャンネル類を標的としこれを遮断する。これらもニューロンの興奮性の向上を惹起する。コノプレッシン類はバソプレッシン受容体アンタゴニストであり、そしてコナントキン類はNMDA受容体アンタゴニストである。より最近、電圧感受性の非特異的カチオンチャンネルを標的とする新たなγ−コノトキシンクラスの原型、及び5HT3受容体に拮抗する新たなσ−コノトキシンクラスの原型が記述された。
【発明の概要】
【0003】
χ−コノトキシンクラスと以下に呼ばれる、コノトキシンの新しいクラスが存在することがここに発見されるに至った。このクラスはニューロンのアミン輸送因子を阻害する能力を持つことを特徴とするものである。
【0004】
神経伝達物質の再吸収を阻害する化合物は尿失禁、排尿筋不安定性及び間質性膀胱炎などの尿道下方障害の治療に有用であることが見出されてきた。このような化合物の一つは「イミプラミン」であり、これはノルアドレナリン再吸収の阻害に加え、カルシウムチャンネルの遮断に悪影響を及ぼすこと、そして抗コリン作用性の、局部麻酔薬的活性及び他の幾つかの効果を示すことが示されてきた。ノルアドレナリン再吸収を阻害できる他の化合物については、米国特許第5,441,985号に記載されている。これらの化合物はイミプラミンに比べて低い抗コリン作用性効果を有するといわれる。
【0005】
本発明のぺプチド類の場合には、神経伝達物質再吸収のこの阻害は、放出されたノルアドレナリンをシナプスからニューロンに迅速に戻して浄化する働きをするノルアドレナリン輸送因子などのニューロンの神経伝達物質輸送因子を選択的に阻害することにより達成される。
【0006】
本発明のぺプチド類はアミン輸送因子を阻害する活性を有する最初のぺプチド類である。今日までに特性決定された他のコノトキシン・ぺプチド類はすべてイオンチャンネル類又は細胞表面上の受容体類を標的とする。
【図面の簡単な説明】
【0007】
【図1】図1は二等分されたラット前立腺精管の1回の超最大パルス(55V,1ms)での電場刺激による収縮の時間経過に及ぼすχ−MrIAの典型的な効果を示すグラフである。χ−MrIA(30nM〜3μM)はその器官浴に半対数単位刻みで累積的に添加した。
【図2】図2は外因性α1アドレナリン受容体アゴニストに対するラット精巣上体精管の二等分された部分の収縮性応答に及ぼすχ−MrIA,−MrIAの効果のグラフである。(a)はχ−MrIA,−MrIAの不存在(○)及び1μM(△)又は3μM(□)の存在の下でのノルアドレナリンに対する、log(濃度)−応答曲線、(b)は3μMのχ−MrIAの存在下(□)及び非存在下(○)におけるメトキサミンに対するlog(濃度)−応答曲線。(a)及び(b)における各データ点は4〜5の個体の組織調製物についての観察の平均値±SEMを表す。一部の誤差棒は記号によって曖昧になっている。
【図3】図3はヒトニューロンのノルアドレナリン輸送因子のcDNAクローンでトランスフェクトされたチャイニーズハムスター卵巣(CHO)細胞による[3H] −ノルアドレナリンのデシプラミン感受性蓄積のχ−MrIAによる阻害を示すグラフである。[3H] −ノルアドレナリンの蓄積はχ−MrIAの非存在下における細胞の取込みの百分率として表す。データ点は4回の別々の実験の観察の平均値±SEMを表す。
【図4】図4はイモガイ(coneshell)毒のぺプチド配列の誘導を表す図式的表示である。プライマーAP1+CHI−1Bを用いる5’RACE・PCRにより5’UTRとリーダーぺプチド配列を得る。このリーダーぺプチド配列を用いて次にχ−コノトキシン類に特異的なPCRプライマーを作成する。プライマーCHI−1A+ANCHORを用いる3’UTRにより、残りの成熟ぺプチド配列及び3’UTR配列の誘導を完成した。
【発明を実施するための形態】
【0008】
本発明の一つの側面によれば、ニューロンのアミン輸送因子を阻害する能力を有する単離され、合成され又は組換えで得られたχ−コノトキシン・ぺプチドが提供される。
このニューロンのアミン輸送因子はニューロンのノルアドレナリン輸送因子であることが好ましい。
【0009】
このχ−コノトキシン・ぺプチドは円錐巻貝から単離された天然産のぺプチドであってもよく、又はその誘導体であってもよい。
該χ−コノトキシン・ぺプチドはχ−MrIA若しくはχ−MrIB、又はそれらの誘導体であることが好ましい。χ−MrIA及びχ−MrIBは軟体動物狩猟性の円錐巻貝であるコナス・マルモレウス(Conus marmoreus)の毒から単離されうる。これらは、共に長さが13アミノ酸残基のぺプチドであり、2個のジスルフィド結合を含んでいる。これらのぺプチドはニコチン性ACh受容体アンタゴニストとして作用するα−コノトキシンクラスのメンバーに最も相同性を示す。
【0010】
χ−MrIA及びχ−MrIBのアミノ酸配列は以下のとおりである。
χ−MrIA NGVCCGYKLCHOC 配列番号:1
χ−MrIB VGVCCGYKLCHOC 配列番号:2
そのC末端は遊離の酸でもアミド化されていてもよい。
【0011】
上記の配列において、「O」は4−ヒドロキシプロリン(Hyp)を指す。このアミノ酸残基はコードされたぺプチドの翻訳後修飾から得られ、従ってそのヌクレオチド配列には直接コードされていない。
【0012】
このχ−コノトキシン・ぺプチドはニューロンのノルアドレナリン輸送因子の選択的阻害剤であることが好ましい。「選択的」及び「選択的に」という用語は、本明細書で用いるとき、ニューロンのノルアドレナリン輸送因子の阻害剤としての該ぺプチドの活性がα1−アドレナリン受容体におけるどの活性よりもかなり大きいことを意味する。
【0013】
米国特許第5,441,985号は、抗コリン作用性効果を殆ど持たないノルアドレナリン再吸収阻害剤は下部尿道障害の治療にとりわけ有用であることを示す。χ−MrIAも検出可能な抗コリン作用性効果を持たないことが見出された。
【0014】
従って、本発明の好ましい実施態様では、ニューロンのノルアドレナリン輸送因子を選択的に阻害する能力を有し、かつ、抗コリン作用性効果を持たないか殆ど持たない、単離され、合成され又は組換えで得られたχ−コノトキシン・ぺプチドが提供される。
【0015】
χ−MrIAはナトリウムチャンネル遮断剤としての活性もドパミン輸送因子阻害剤としての活性も持たないことも見出された。他のノルアドレナリン輸送因子阻害剤が普通に共有しているこれらの付加的な薬理学的活性がχ−MrIA及び本発明の好ましいぺプチドに欠如していることは、これらのぺプチドを有用な薬理学的道具とする。
【0016】
本発明のこのχ−コノトキシン・ぺプチド類は、χ−MrIA及びχ−MrIBなどの天然に生ずるぺプチド類であってもよく、天然に生ずるぺプチド類の誘導体であってもよい。
【0017】
天然に生ずるχ−コノトキシン・ぺプチド類、例えば、χ−MrIA及びχ−MrIBとの関連で本明細書で用いるとき、「誘導体」という用語は、天然に生ずるぺプチド類とは一つ以上のアミノ酸の欠失、付加、置換又は側鎖の修飾により異なっているぺプチドを指す。ニューロンのノルアドレナリン輸送因子を阻害する能力を持たないこのような誘導体は本発明の範囲内には入らない。
【0018】
置換はアミノ酸が異種の天然型アミノ酸残基又は非天然型アミノ酸残基で置換されるアミノ酸の変更を包含する。このような置換は、ポリぺプチドに含まれるアミノ酸残基が極性、側鎖の官能基又はサイズに関して類似の特性を持つ別の天然型アミノ酸で置換される場合、例えばSer←→Thr←→Pro←→Hyp←→Gly←→Ala、Val←→Ile←→Leu、His←→Lys←→Arg、Asn←→Gln←→Asp←→Glu又はPhe←→Trp←→Tyrの場合には、「同類置換」として分類されうる。一部の非天然型アミノ酸が天然型アミノ酸を置換するのに適当であることもあることは理解されるべきである。例えば、オルニチン、ホモアルギニン及びジメチルリジンはHis、Arg及びLysに関連する。
【0019】
本発明により包摂される置換は「非同類置換」であってもよい。この場合は、ポリぺプチド中に存在するアミノ酸残基が異なる基を持つ天然アミノ酸などの異なる特性を持つアミノ酸で置換され(例えば、荷電を持ったアミノ酸又は疎水性アミノ酸がアラニンで置換されること)、また、天然アミノ酸が非天然型アミノ酸で置換される。
【0020】
アミノ酸置換は通常1残基の置換であるが、かたまりの又は分散した複数の残基の置換でもよい。
アミノ酸置換は同類置換が好ましい。
【0021】
付加は一つ以上の天然型又は非天然型のアミノ酸残基の付加を含む。欠失は一つ以上のアミノ酸残基の欠失を含む。
上述のように本発明はアミノ酸の一つ以上が側鎖の修飾を受けたぺプチドを含む。本発明が予定する側鎖の修飾の例としては、アルデヒドとの反応の後NaBH4での還元による還元的アルキル化、メチルアセチミデートでのアミディネーション(amidination)、無水酢酸によるアシル化、シアネートによるアミノ基のカルバモイル化、2,4,6−トリニトロベンゼンスルホン酸(TNBS)によるアミノ基のトリニトロベンジル化、無水コハク酸及び無水テトラヒドロフタル酸によるアミノ基のアシル化、及びピリドキサル−5−リン酸で処理した後NaBH4での還元によるリジンのピリドキシル化等によるアミノ基の修飾が挙げられる。
【0022】
アルギニン残基のグアニジン基は2,3−ブタンジオン、フェニルグリオキサル及びグリオキサル等の試薬との複素環縮合産物の形成により修飾されうる。
カルボキシル基はO−アシルイソウレア形成を経てカルボジイミド活性化の後誘導体化により、例えば対応するアミドに誘導することにより修飾されうる。
【0023】
スルフヒドリル基は、ヨード酢酸又はヨードアセトアミドでのカルボキシメチル化、システイン酸への過ギ酸酸化、他のチオール化合物との混合ジスルフィドの形成、マレイミド、無水マレイン酸又は他の置換マレイミドとの反応、4−クロロメルクリ安息香酸、4−クロロメルクリフェニルスルホン酸、フェニルメルクリクロリド、2−クロロメルクリ−4−ニトロフェノール及び他の水銀化合物を用いる水銀誘導体の形成、アルカリ性pHでのシアン酸塩によるカルバモイル化などの方法により修飾しうる。システイン残基の修飾はいずれも、必要なジスルフィド結合を形成する該ぺプチドの能力に悪影響を与えてはならない。該ぺプチドが一つ以上のジスルフィド結合の代わりにジセレン結合を形成するようにシステインのスルフヒドリル基をセレン等価体と置換することも可能である。
【0024】
トリプトファン残基は、例えば、N−ブロモスクシンイミドでの酸化又は2−ヒドロキシ−5−ニトロベンジルブロミド若しくはスルフェニルハライドによるインドール環のアルキル化により修飾しうる。他方、チロシン残基はテトラニトロメタンでの硝酸化により変化させて3−ニトロチロシン誘導体を形成させうる。
【0025】
ヒスチジン残基のイミダゾール環の修飾は、ヨード酢酸誘導体によるアルキル化又はジエチルピロカーボネートによるN−カルベトキシル化により達成しうる。
プロリン残基は、例えば、4−位におけるヒドロキシル化により修飾しうる。
【0026】
修飾された側鎖を持つアミノ酸及び他の非天然型アミノ酸の一部のリストを表1に示す。
【0027】
【表1a】
【0028】
【表1b】
【0029】
【表1c】
【0030】
これらのタイプの修飾は、個体に投与されるとき又は診断用試薬として使用されるとき該ぺプチドを安定化するのに重要でありうる。
本発明が予定する他の誘導体としては、全くグリコシル化されていない分子から変更されたグリコシル化分子までのある範囲のグリコシル化変異型が挙げられる。変更されたグリコシル化パターンは異なる宿主細胞における組換え分子の発現から生じうる。
【0031】
本発明のχ−コノトキシン類は通常そのC末端でアミド化されているが、遊離のカルボキシル末端又はC末端における他の修飾を有する化合物も本発明の範囲内にあると考えられる。このぺプチド類はそのC末端がアミド化されているか又は遊離のカルボキシルを有することが好ましい。
【0032】
天然産のχ−コノトキシン・ぺプチド類の誘導体はCys残基及び特徴的なジスルフィド結合様式を保持していることが好ましい。誘導体には付加的なCys残基が含まれうる、ただしそれらがジスルフィド結合の形成の間保護されている場合である。
【0033】
修飾により天然産のχ−コノトキシン・ぺプチド類の誘導体を形成する場合、活性な天然産ぺプチド類のアミノ酸配列を比較して、あるとすれば、どの残基が活性ぺプチド間で保存されているかを決定することが有用である。これらの保存された残基の置換は、禁止されるわけではないが、保存されていない残基の置換よりは好ましくない。
【0034】
Alaで一つ以上の残基を置換している誘導体は薬理団(pharmacophore)を同定するために使用することができる。一時に一つ又は二つのアミノ酸だけがAlaで置換されることが好ましい。この薬理学的クラスのぺプチドのその受容体への結合に関与する相互作用のタイプをより正確に規定するのを助けるため、荷電を持った残基、極性の残基又は疎水性の残基がそれぞれ置換されているさらなる新たなぺプチド類を作ることができる。荷電が逆転したり、又は極性の残基が疎水性の残基と置換したりする非同類置換は、結合に関与する残基をそれ以上に同定することができる。これらのぺプチドはすべて、効力の改善又は選択性の増大を示す潜在能力を有する。非天然型アミノ酸の変更も、効力、選択性、及び/又は安定性を改善するために含められよう。
【0035】
露出されている残基は受容体結合に関与する可能性が最も高く、そして体系的に置換することができる。効力及び/又は選択性の改善を目指して追加的な結合相互作用を獲得するため、より長い側鎖型又は非同類置換を用いて、結合に関与する残基及び薬理団の直ぐ周囲にある残基を変えることは、特に強調される。MrIA又はMrIBの環の大きさや尾を減少させたり拡大したりすると、活性はさらに変化する。
【0036】
MrIA又はMrIBは一つの尾(残基1〜3)と二つの環(残基6〜9及び残基11〜12)から構成されることに注目されるが、χ−コノトキシン・ぺプチド類及び本発明の誘導体はアミノ酸及びジスルフィド結合のこの特定の配列を持つものに限定されない。他の配列も可能であり、その結果得られるぺプチドが必要な活性を有するならば、そのぺプチドは本発明の範囲に入る。該ぺプチド類は少なくとも二つのシステイン残基及び少なくとも一つのジスルフィド結合を持つことが好ましく、又は四つのシステイン残基と二つのジスルフィド結合を持つことがさらに好ましい。
【0037】
これらのぺプチド類のジスルフィド結合の連結はA−B/C−D、A−C/B−D又はA−D/B−Cでありうるが、後者がMrIA及びMrIBにとって好ましい。A、B、C及びDはそれぞれジスルフィド結合の形成に関与する第一、第二、第三及び第四Cys残基を指す。
【0038】
これらのぺプチド類に標識を付け、同じ部位に作用する新たな分子を同定するための結合検定法を確立するために使用することもできる。例えば、MrIA又はMrIBの標識化リガンドには、Tyr又は他の適当な残基を介してトリチウムを含ませ又は放射活性ヨウ素若しくは類似のものを付着させることができよう。各ぺプチドの全体にわたるTyrを調査すれば、該Tyrの組み込みに適する位置を決定できる。組織ホモジネート又は化合物若しくは混合物により発現された輸送因子に対するこのような標識ぺプチド類の結合の阻害から、この部位において活性な新たなぺプチド、ヒト組織を含む哺乳類の血清及び神経及び筋肉組織中に存在するぺプチドを含む新たなぺプチドの同定が可能となるであろう。この検定法はMrIA及びMrIBと同じ部位にも作用する非ぺプチド分子であって
、これらのぺプチドの経口的に活性な形として有用性を持ちうるものの同定をも可能とする。標識ぺプチド類はさらに種々の組織にわたるぺプチド結合の位置を同定するためのオートラジオグラフによる研究を可能とする。
【0039】
これらの配列の部分を使用してESTRデータベースを調査し、哺乳類において同様に作用する内因性リガンドを同定するために使用できる関連配列情報を含むぺプチド類又はタンパク質類を哺乳類で同定することができる。
【0040】
本発明のχ−コノトキシン類は標準的なぺプチド合成の後の酸化的ジスルフィド結合形成の方法により調製しうる。例えば、その線状ぺプチドは、シュノルツァーら(1992)が記述したように、BOC化学を用いる固相法により合成しうる。脱保護及び固体支持体からの切断の後、還元型ぺプチドを調製用クロマトグラフィーにより精製する。この精製された還元型ぺプチドを、例えば実施例2で述べるように、緩衝系で酸化する。この酸化型ぺプチドを調製用クロマトグラフィーを用いて精製した。
【0041】
コノトキシン類の合成を記述する参考文献としては、サトーら、リューら及びWO91/07980が挙げられる。
χ−コノトキシン類は組換えDNA技術を用いて調製することもできる。所望のぺプチド配列をコードするヌクレオチド配列を適当なベクター中に挿入し、適当な発現系でタンパク質を発現させる。場合によっては、この発現されたぺプチドのさらなる化学修飾、例えばC末端のアミド化、が適当でありうる。一部の状況の下では、ぺプチド発現後の化学的工程として、発現したぺプチドの酸化的結合形成を行うことが望ましい。これには折り畳まれていないぺプチドを得るため還元的工程を先行させてもよい。当業者はぺプチドの還元及び酸化に適する条件を容易に決定しうる。
【0042】
本発明はさらに、上述のχ−コノトキシン・ぺプチドをコードするヌクレオチド配列又はコードする配列に相補的な配列を含む単離された核酸分子を提供する。
本発明の更なる側面において、χ−コノトキシン・ぺプチドの全部又は一部をコードするヌクレオチド配列又はコードする配列に相補的な配列を含む核酸プローブが提供される。
【0043】
特に好ましい実施態様では、該核酸プローブは配列番号:1又は配列番号:2に示す配列をコードするヌクレオチド配列又はコードする配列に相補的な配列を含む。
本明細書で用いるとき、「プローブ」という言及には、増幅に用いられるプライマー又は直接ハイブリダイゼーションに使用するためのプローブに対する言及が含まれる。
【0044】
本発明のさらに別の側面は、本発明のχ−コノトキシン・ぺプチド類に対する抗体に関する。このような抗体はモノクローナルでもポリクローナルでもよく、標準的技法を用いて該ぺプチドに対して天然に生ずる抗体から選択してもよく、又は該ぺプチドに対し特異的に形成させてもよい。後者の場合、該ぺプチドはまず担体分子と混合する必要がありうる。本発明の抗体は治療薬又は診断薬として特に有用である。
【0045】
この点に関して、特異的な抗体を本発明のぺプチド類をスクリーニングするためにを使用することができる。そのような検定のための技法は当分野で良く知られており、例えば、サンドイッチ検定法やELISA法が含まれる。ぺプチドのレベルについての知識はある種の治療計画をモニターするのに重要でありうる。
【0046】
当分野で知られた技法を用いて抗イディオタイプ抗体を調製することも可能である。これらの抗イディオタイプ抗体及び治療薬としてのその使用は本発明の更なる側面を表す。
本発明の核酸分子はDNAであってもRNAであってもよい。該核酸分子がDNA型であるときは、それはゲノムDNA又はcDNAでありうる。本発明の核酸分子のRNA型は一般にmRNAである。
【0047】
本発明の核酸分子は通常単離された形であるが、それらはベクター分子、とりわけ発現ベクター分子などの他の遺伝子分子に組み込まれ、又は連結され、又は他の方法で融合され又は関係付けられていてもよい。ベクター及び発現ベクターは一般に複製可能であり、そして適用可能であれば、原核細胞又は真核細胞の一方又は両方で発現可能である。原核細胞には大腸菌、バチルス・スピーシーズ、及びシュードモナス・スピーシーズが含まれることが好ましい。好ましい真核細胞としては、酵母細胞、糸状菌細胞、哺乳類細胞及び昆虫細胞が挙げられる。
【0048】
従って、本発明の別の側面は、ベクター部分と本発明のぺプチドをコードすることができる遺伝子とを含む遺伝子構築物を予定する。
該遺伝子構築物の遺伝子部分は、プロモーターが適当な細胞中で該遺伝子部分の発現を指示することができるように該ベクター上の該プロモーターに機能的に連結されていることが好ましい。
【0049】
本発明は、このような遺伝子構築物及びそれを含む原核細胞又は真核細胞にまで及ぶ。
他の分子中にその活性を工学的に組み込むために、場合によっては余分の機能を持つ新たな分子を作成するために、他のコノトキシン類又は付加的に他のぺプチド若しくはタンパク質とMrIAなどのχ−コノトキシン類とのキメラを作成することができる。これはその薬理団を含むこれらのぺプチド配列の部分(単数又は複数)を用いて行うことが好ましいであろう。この薬理団が断続的である場合、薬理団を構成する複数の部分が受容体に結合できるよう該新たな構築物中に配置されるべきである。他のコノトキシン類とのキメラは、追加されたCys残基及び追加されたジスルフィド結合を含んでもよい。
【0050】
一つの活性クラス内のコノトキシン・ぺプチド類が、それぞれのシステイン残基の間のぺプチド環を持つ類似のジスルフィド結合パターンを有することは普通である。χ−MrIA及びχ−MrIBの場合、ジスルフィド結合は第一と第四、第二と第三のシステイン残基を連結する。このパターンはα−コノトキシン・ぺプチド類で観察された結合パターンとは異なる。この相違にもかかわらず、χ−コノトキシン・ぺプチドの環をα−コノトキシンなどの別のぺプチド由来の配列を含む環で置換することによりキメラ誘導体を調製しうる。
【0051】
本発明は他のぺプチド類に結合したχ−コノトキシン・ぺプチド類と同様にχ−コノトキシン・ぺプチドのダイマー、トリマー等をも含む。
本発明のχ−コノトキシン・ぺプチド類は10〜30アミノ酸を持つことが好ましく、11〜20アミノ酸を持つことがより好ましい。
【0052】
天然に生ずるχ−コノトキシン・ぺプチド類の完全な遺伝子配列は、クローニング及びDNA配列決定と併用した5’RACEと3’RACEの組合せ戦術を用いて明らかにしうる。
【0053】
本発明のχ−コノトキシン・ぺプチド類はニューロンのノルアドレナリン輸送因子を阻害する活性を持つ。従って、本発明はニューロンのノルアドレナリン輸送因子の阻害剤としてのχ−コノトキシン・ぺプチド類の使用、及びニューロンのノルアドレナリン輸送因子の阻害がそれに関する有効な治療と関係がある病気又は状態の治療又は予防における使用を提供する。薬理学的作用物質のこのような活性は、排尿系若しくは心臓血管系の病気若しくは状態又は気分変調の予防又は治療、又は苦痛若しくは炎症の治療又は制御における活性と関係がある。
【0054】
従って、本発明は、排尿若しくは心臓血管の状態若しくは病気又は気分変調の治療又は予防、又は苦痛若しくは炎症の治療又は制御のための方法であって、ニューロンのノルアドレナリン輸送因子を阻害する能力を持つ単離され、合成され、又は組換えで得られたχ−コノトキシン・ぺプチドの有効量を哺乳類に投与する工程を含む方法を提供する。
【0055】
排尿系の病気若しくは状態の例としては、尿失禁及び排便失調が挙げられる。心臓血管の病気若しくは状態の例には、種々の起源の不整脈及び冠不全が含まれる。気分変調の例としては、うつ病、不安、及び喫煙などの切望が含まれる。苦痛の例としては、慢性の苦痛、神経障害性の苦痛又は炎症性の苦痛が挙げられる。
【0056】
該哺乳類は、予防的な意味で該ぺプチドが投与されることもありうるが、このような治療を必要とするものであることが好ましい。
本発明はニューロンのノルアドレナリン輸送因子を阻害する能力を持つ単離され、合成され又は組換えで得られたχ−コノトキシン・ぺプチドと、薬学的に許容しうる担体又は希釈剤を含む組成物をも提供する。
【0057】
該組成物は薬学的組成物の形であることが好ましい。
また、排尿又は心臓血管の状態若しくは病気、又は気分変調の治療又は予防用、又は苦痛若しくは炎症の治療若しくは制御用の医薬の製造における、ニューロンのノルアドレナリン輸送因子を阻害する能力を有する単離され、合成され又は組換えで得られたχ−コノトキシン・ぺプチドの使用も提供される。
【0058】
ノルアドレナリン輸送因子は神経細胞により発現されるだけでなく、胎盤、肺の内皮細胞及び子宮などの他の組織でも発現されることも注目される。本発明のぺプチド類はこれらのノルアドレナリン輸送因子の阻害にも有効であり、これらの輸送因子が絡んでいる状態を治療する際に有用でありうる。
【0059】
当業者は容易に理解するように、投与の経路及び薬学的に許容しうる担体の性質は該状態の性質及び治療される哺乳類に左右される。具体的な担体又は送達系の選択、及び投与経路の選択は当業者により容易に決定され得ると思われる。該活性ぺプチドを含む任意の製剤の調製においては、該ぺプチドの活性が工程中に破壊されないように、かつ、該ぺプチドが破壊されずに作用部位まで到達できるように注意が払われるべきである。状況によっては、例えば、ミクロカプセル化などの当分野で知られた手段により該ぺプチドを保護することが必要であることもある。同様に、選ばれる投与経路は該ぺプチドがその作用部位に到達するようなものであるべきである。
【0060】
注射可能な用途に適切な薬学的剤形には、滅菌注射可能な溶液又は分散液、及び滅菌注射可能溶液の即時調製用の滅菌粉末が含まれる。これらは製造及び貯蔵の条件下で安定であるべきであり、酸化及び細菌や黴などの微生物の汚染作用に対して保護されうる。
【0061】
当分野の熟練者は常法的アプローチで本発明のぺプチド類又は修飾されたぺプチド類のための適切な処方を容易に決定しうる。好ましいpH範囲及び、例えば抗酸化剤などの適当な賦形剤の特定は当分野では機械的作業である(例えば、クリーランドら,1993を参照)。緩衝系は望みの範囲のpH値を得るために機械的に用いられ、例えば、酢酸塩、クエン酸塩、乳酸塩及びコハク酸塩などのカルボン酸緩衝液が含まれる。このような処方には、BHT又はビタミンEなどのフェノール性化合物、メチオニン又は亜硫酸塩などの還元剤、及びEDTAなどの金属キレート剤を含む多様な抗酸化剤が利用可能である。
【0062】
注射可能な溶液又は分散液のための溶媒又は分散媒としては、活性ぺプチドのための任意の従来の溶媒又は担体系が含まれ、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール及び液状ポリエチレングリコールなど)、それらの適当な混合液、及び植物油が含まれる。例えば、レシチンなどの被覆剤の使用により、分散液の場合には要求される粒子サイズの維持により、そして界面活性剤の使用により、適当な流動性を維持できる。微生物の作用の防止は、必要ならば、例えば、パラベン、塩化ブタノール、フェノール、ソルビン酸、チメロサール等の種々の抗細菌剤及び抗真菌剤を含めることにより可能となる。多くの場合、重量オスモル濃度を調整するための物質、例えば、糖類又は塩化ナトリウムを含むことが好ましい。注射用の処方は血液と等張であるこ
とが好ましい。注射可能な組成物の長期吸収性は、例えば、モノステアリン酸アルミニウム及びゼラチンなどの吸収を遅延する薬剤を組成物に含めることによりもたらされ得る。注射可能な使用に適切な薬学的剤形は、静脈内、筋肉内、大脳内、鞘内、硬膜外の注射又は注入を含む任意の適切な経路により送達されうる。
【0063】
注射可能な滅菌溶液は、必要量の活性化合物を、上に列挙したような種々の他の成分と適切な溶媒中で混合し、必要ならば、その後濾過滅菌することにより調製される。一般に、分散液は、基本的な分散媒と上に列挙したものから選択された必要な他の成分とを含む滅菌ビークル中に種々の滅菌した活性成分を混合することにより調製される。注射可能な滅菌溶液調製用の滅菌粉末の場合、調製の好ましい方法は、活性成分に任意の追加の望みの成分を加え予め滅菌濾過した溶液を真空乾燥又は凍結乾燥したものである。
【0064】
該活性成分が適切に保護されている場合、該成分は、例えば、不活性希釈剤若しくは同化しうる食用担体とともに経口投与してもよく、又は硬い殻若しくは軟い殻のゼラチンカプセルに封入してもよく、錠剤に圧縮してもよく、常食の食物と直接混合してもよい。経口治療投与では、該活性化合物は賦形剤と混合してもよく、摂取可能な錠剤、バッカル錠、トローチ、カプセル、エリキシル、懸濁液、シロップ、オブラート剤等の剤形で用いてもよい。このような組成物及び調製物は少なくとも1重量%の活性化合物を含むことが好ましい。もちろん、組成物及び調製物の百分率は様々であり、その単位剤形の重量の約5%から約80%の間にあるのが都合がよい。このような治療に有用な組成物における活性化合物の量は、適切な用量が得られるような量である。
【0065】
錠剤、トローチ、丸薬、カプセル等は以下に列挙するような成分も含みうる。ゴム、アラビアゴム、コーンスターチ又はゼラチン等の結合剤、リン酸二カルシウムなどの賦形剤、コーンスターチ、ポテトスターチ、アルギン酸等の崩壊剤、ステアリン酸マグネシウム塩などの滑剤、及びスクロース、ラクトース又はサッカリンなどの甘味料が添加されてもよく、あるいはペパーミント、冬緑油、又はさくらんぼ風味などの香味料が添加されてもよい。投与単位剤形がカプセルの場合、上記タイプの物質に加えて液体の担体を含んでもよい。種々の他の物質が被覆剤として又はその他の方法で投与単位の物理的形態を改変するために存在してもよい。例えば、錠剤、丸剤、又はカプセルがセラック、糖又はその両方で被覆されてもよい。シロップ又はエリキシルは活性化合物、甘味剤としてのスクロース、保存剤としてのメチルパラベン及びプロピルパラベン、さくらんぼ又はオレンジ風味の色素及び香味料を含んでもよい。もちろん、いかなる投与単位剤形を調製するのに用いられるいずれの物質も、その使用量において薬学的に純粋であり実質的に無毒性であるべきである。さらに、これらの活性化合物は放出制御調製物及び放出制御処方に組込まれてもよい。
【0066】
本発明は、投与、例えば、クリーム、ローション、ジェル等の局所的適用、又は、吸入若しくは鼻腔内送達に適した組成物、例えば、溶液若しくは乾燥粉末にふさわしい任意の他の剤形にも及ぶ。
【0067】
静脈内、大脳内、鞘内若しくは硬膜外の送達に適する剤形などの非経口投与剤形は好ましい。
薬学的に許容しうる担体及び/又は希釈剤には、任意の及び全ての溶媒、分散媒、被覆剤、抗細菌剤及び抗真菌剤、等張剤、及び吸収遅延剤等が含まれる。薬学的に活性な物質のためのこのような媒体及び薬剤の使用は当分野では周知である。通常の媒体及び薬剤はいずれも、活性成分と配合禁忌であるときに限りそれを除いて、治療用組成物にそれらを使用することが意図される。補足的活性成分もまた該組成物に組み込まれ得る。
【0068】
投与の容易性及び服用量の均一性のため、非経口組成物を投与単位剤形に製剤化することは特に利点がある。本明細書で使用されるとき、投与単位剤形とは、治療される哺乳類の対象にとって一回の投薬に適した物理的に個別の単位を指し、各単位は必要とされる薬学的担体とともに所望の治療効果を生じるように計算され予め測られた量の活性物質を含む。本発明の新規な投与単位剤形の仕様は、(a)活性物質のユニークな特性及び達成される具体的な治療効果ならびに(b)本明細書で詳細に開示されるような身体の健康が損なわれた病気状態を有する生物の患者における病気の治療のためにこのような活性物質を調合する技術に固有の制限によって左右され、直接これらに依存する。
【0069】
この主要な活性成分は、簡便かつ効果的な投与のために投与単位剤形中に適切な薬学的に許容しうる担体とともに有効な量で調合される。単位投与剤形は、例えば、0.25μgから約2000mgまでの範囲の量で主要な活性成分を含むことができる。割合で表わすと、この活性化合物は一般的に1mlの担体に対して約0.25μgから約200mgまで存在する。補足的活性成分を含む組成物の場合、一回投薬量は前記成分の通常の一日服用量及び投与の方法を参照して決定される。
【0070】
添付の図面及び実施例を参照してここに本発明を記述するが、以下の記述の具体性が本発明についてのこれまでの記述の一般性にとって代わるものではないことが理解されるべきである。
【実施例】
【0071】
薬物
この実施例及び以下の実施例に用いられる薬物には、デシプラミン塩酸塩(シグマ)、インドメタシン(シグマ)、メトキサミン塩酸塩(シグマ)、(−)−ノルアドレナリン二酒石酸塩(シグマ)、[3H] −1−ノルアドレナリン(比活性2200Ci/mM、ニューイングランド・ヌクレアー、ボストン,MA(U.S.A.))、テトロドトキシン(シグマ)、ヨヒンビン塩酸塩(シグマ)が含まれる。
統計的分析
以下の実施例のデータは4〜6回の実験の平均値及び標準誤差として表される。EC50値の計算のためのシグモイド曲線−適合は、ソフトウエア・パッケージ・イゴール・プロ(ウエイブメトリクス)を用いて非線型回帰により行った。平均値の間の差はソフトウエア・パッケージ・プリズム(グラフパド)を用いスチューデントの両側tテストにより評価した。P<0.05という値は統計的に有意の差を示すものとされた。
【0072】
実施例1
ラットの精管の調製
雄ウイスターラット(250〜350g)を頭部を打って殺し、その精管を切除した。各組織を二等分した精巣上体部分と前立腺部分に分けた。この組織部分を5mlの器官浴中に載せ0.5gの張力を掛けた。この浴溶液は次の組成、即ち、NaCl,119mM、KCl,4.7mM、MgSO4,1.17mM、KH2PO4,1.18mM、NaHCO3,25.0mM、グルコース,5.5mM、CaCl2,2.5mM、EDTA,0.026mMを含んでいた。この浴溶液を37℃に維持し、O2中5%v/vCO2を吹き込んだ。実験の開始前にこの組織調製物を少なくとも45分間平衡化させた。アイソメトリック・フォース・トランスデューサー(ナルコ・バイオ・システムF−60)により収縮を記録し、チャート・バージョン3.5.4/sソフトウエア及びマックラボ/8s・データ・アクイジション・システム(ADインストルーメンツ)を用いて200Hzのサンプリング頻度でパワー・マッキントッシュ・コンピュータに記録した。
【0073】
この二等分した前立腺セグメントを用いて、交感神経系神経伝達により媒介される平滑筋の電気誘発収縮に及ぼすχ−MrIAの効果を試験した。この組織調製物をプラチナ刺激電極で広げた。電場刺激(EFS)は、グラスS44スティミュレーターにより発生させた1ms持続の1個の55Vパルスを用い3分間隔で行った。この収縮はテトロドトキシン(0.1μM)で阻止することができた。このことはこの収縮が神経により媒介されていることを示す。ペプチドは、半対数単位増加で濃度を増加させながら器官浴中に累積的に添加された。各投与量は電気刺激への応答に対する先の投与量の効果が定常レベルに達した直後に適用された。
交感神経系神経伝達に及ぼすχ−MrIAの効果
ラットの前立腺精管の二等分された部分は電場刺激に応答して2相性の収縮を示した。この調製物では、該2相性収縮の二つの成分は時間的に十分に分離された。該応答の第一の部分は主要な応答であり、電気パルスの送達後200msでほぼ最高レベルに達した。該収縮の第二の相は刺激後500msでほぼピークに達した。χ−MrIAの薬理学的活性を同定するための本発明者らの試みは電場刺激されたラット精管におけるその効果の研究で始まった。電場刺激への該調製物の応答に及ぼすχ−MrIAの効果を図1に示す。このコノトキシン(30nM〜3μM)は該収縮の第二相を増加するように作用した。この効果は濃度依存性であることが見出された。χ−MrIAの存在下で得られたトレースから対照の応答を差し引くことにより、該収縮の第二の成分のみの該ペプチドによる特異的増強が明らかとなる。第二の成分を特異的に増強するように作用するχ−MrIAの濃度−応答曲線も構成することができる。
【0074】
電気誘発応答に及ぼすχ−MrIAの作用は高度に特異的であり、該2相性応答の第二の成分のみを増強する。該収縮のこの後の相はノルアドレナリンによって媒介されることが認められており、プラゾシン及び他のα1−アドレナリン受容体アンタゴニストにより選択的に阻害される。該収縮の第一の相は放出されたATPによる接合部後のP2Xプリン受容体の活性化によるものであるが、同様には増強されなかった。該収縮のノルアドレナリン作用性成分の大きさは、交感神経系神経興奮により放出されるノルアドレナリンの量、接合部後のα1−アドレナリン受容体の密度及びそれらのエフェクター系への結合、そしてノルアドレナリンがシナプスから除去される速度により調節される。
【0075】
シナプス前のα2アドレナリン受容体における拮抗は、放出されたノルアドレナリンによる負のフィードバック系の活性化の阻止により、交換神経からのノルアドレナリンの電気誘発放出を増強することが良く認識されている。しかしながら、α2アドレナリン受容体拮抗はχ−MrIAの作用機構ではあり得ない。χ−MrIAと異なり、ヨヒンビンやイダゾキサンなどのα2アドレナリン受容体アンタゴニストはラット精管の収縮のプリン性成分とノルアドレナリン作用性成分を平等に増強するように作用する。さらに、1回のパルスへの応答は、一続きの刺激への応答とは対照的に、これらの受容体部位には刺激の時にアゴニストが存在しないであろうから、この負のフィードバック機構による調節を受けない。従って、ヨヒンビン(1μM)はこの検定では誘発応答に対する効果を持たない
。
【0076】
実施例2
α1−アドレナリン受容体アゴニストに対する応答に及ぼすχ−MrIAの効果を試験するための調製物
ラットの精管は、二等分した精巣上体部分を電気的に刺激しなかったことを除き、上記のように使用した。これらの調製物は、χ−MrIAの存在下及び非存在下におけるノルアドレナリン及びメトキサミンへの濃度応答曲線を求めるために代わりに使用した。この組織では、ノルアドレナリン及びメトキサミンは接合部後のα1−アドレナリン受容体の活性化を介して平滑筋の収縮を惹き起こす。1μM又は3μMいずれかの濃度のχ−MrIAを器官浴に適用し、該組織と20分間平衡化した後ノルアドレナリン又はメトキサミンを累積的に添加した。χ−MrIAを適用しなかった反対側の組織セグメントを対照として用い、調製物当たり一つの濃度応答曲線を求めた。
【0077】
α1−アドレナリン受容体アゴニストへの応答に及ぼすχ−MrIAの効果χ−MrIAの作用が神経伝達物質放出の上流で起こるか下流で起こるかを決定することが、外因的に適用したノルアドレナリンへの応答に及ぼす該ペプチドの効果を調べることにより、可能となった。χ−MrIAは浴に適用されたノルアドレナリンの効力を増強させたので、該コノトキシンは神経の貯蔵からのノルアドレナリンの放出を増大させることによるのではなく、ノルアドレナリンへの応答を増強することにより作用すると本発明者らは結論できる。この増強はα1−アドレナリン受容体応答の増加又はノルアドレナリンの作用の終結の障害の結果として起こりうるであろう。これらのうちのいずれがχ−MrIAの作用機構であったかを確認するため、α1−アドレナリン受容体アゴニストであるメトキ
サミンを使用した。このα1−アドレナリン受容体アゴニストは、それがニューロンのノルアドレナリン輸送因子の基質でないという点でノルアドレナリンとは異なる。この輸送因子は、神経末端内への取り込みによりノルアドレナリン及び他のカテコラアミンをシナプスから消去するように働き、交感神経系で刺激された組織のアドレナリン受容体でのノルアドレナリンの作用を終結させるための主要な機構を表す。メトキサミンはこの機構による除去を受けないから、輸送因子の阻害はその作用の効力を増強しない。
【0078】
二つのα1−アドレナリン受容体アゴニストに対するラット精巣上体精管の二等分されたセグメントの応答に及ぼすχ−MrIAの効果を研究した。χ−MrIAの存在下又は非存在下でのノルアドレナリンに対するlog(濃度)−応答曲線を図2aに示す。1μMの濃度で、χ−MrIAはノルアドレナリンに対する該組織の感受性を増加するように作用し、濃度−応答曲線を左側へシフトさせた。観察された効力の程度は3μMのχ−MrIAでの実験でより大きかった。該コノトキシンのいずれの濃度もノルアドレナリンに対する該組織の最大応答を変更しなかった。χ−MrIAは3μMの濃度でメトキサミンに対する濃度応答曲線に影響を及ぼさなかった(図2b)。ノルアドレナリンへの応答に及ぼすその効果とは対照的に、χ−MrIAがメトキサミンの作用を強めないという観察
は、χ−MrIAがニューロンのノルアドレナリン輸送因子の阻害剤であるということと矛盾しない。
【0079】
メトキサミンへの濃度応答曲線に対するχ−MrIAの効果の欠如は、χ−MrIAがα1−アドレナリン受容体における効果を持たないことをも証明する。これは該曲線の右側への平行移動として明らかであろう。このことはχ−MrIAをノルアドレナリン輸送因子の他の一部の阻害剤、とりわけ抗鬱薬として用いられるものと区別する。多くの好鬱薬の治療標的はニューロンのノルアドレナリン輸送因子である。しかしながら、これらの化合物の多くは、とりわけ三環系抗鬱薬、及びより低い程度ではあるが通常の三環系抗鬱薬とは構造的に関係のない一部のより新しい薬剤は、α1−アドレナリン受容体及びムスカリン様のACh受容体などの他の部位で作用することが認められている。
【0080】
実施例3
モルモットの回腸
一晩絶食させた雄モルモット(285〜425g)の頭を打って殺し、放血した。約1.5cm長の回腸部分を除去し、浴溶液で満たした注射筒を用い温和に洗浄して腸内容物を洗浄した。この調製物を下記の組成の浴溶液、即ち、NaCl,136.9mM、KCl,2.68mM、CaCl2,1.84mM、MgCl2,1.03mM、グルコース,5.55mM、NaHCO3,11.9mM、及びKH2PO4,0.45mM、を37℃まで温め、O2中5%v/vCO2を吹き込んだもの、を含む5mlの器官浴中に置いた。安定なベースラインを作成するため、インドメタシン(10μM)を浴溶液中に含めた。該組織を1.0gの張力を掛けて設置し、実験の開始前40分間平衡化させた。ついで、ニコチンの用量(4μM)を、応答が一致すると観察されるまで15分間隔で適用した。次に、別の用量のニコチンを適用した25分後にχ−MrIA(3μM)を添加した。ニコチンに対するこの応答を、10Hzのサンプリング速度で、アイソメトリックに測定しそしてデジタル化した。
モルモット回腸のニコチンへの応答に及ぼすχ−MrIAの効果
χ−MrIA(3μM)はニコチンに対する回腸部分の応答には有意の効果を及ぼさなかった。該コノトキシンの非存在下では、χ−MrIAが存在した場合の4.07±0.80gと比べ、平均応答は3.83±0.76gであった(p>0.1、対合tテスト,n=4)。α−コノトキシン類は、ニューロンサブタイプ又は筋肉サブタイプいずれかのニコチン性ACh受容体をブロックする。モルモット回腸の単離された断片を用いてχ−MrIAがニューロンのニコチン性ACh受容体を標的としないことが証明された。収縮性応答がムスカリン系受容体の活性化に依存するため、該コノトキシンがムスカリンACh受容体アンタゴニストとしても作用したとするならば、モルモット回腸のニコチンへの応答はχ−MrIAの存在下で減衰したであろうことが予測されよう。この調製物では、ACh及び接合部後の受容体を活性化する種々の他の神経伝達物質、のニコチン誘発放出はχ−MrIAによって影響されなかった。従って、そして多くの他の輸送因子阻害剤とは対照的に、χ−MrIAは抗ムスカリン性活性を欠いている。
【0081】
実施例4
マウスの横隔膜神経−片側横隔膜調製物
雄クアケンブッシュマウス(20〜30g)を頸部転移により殺した。左及び右の片側横隔膜を横隔膜神経を付着させたまま切除した。各片側横隔膜の基部を二つの平行プラチナ刺激電極の間に配置し、横隔膜神経は電場刺激のための二つの小さなプラチナループを介して配置した。該調製物をO2中5%v/vCO2を吹き込んだ5mlの器官浴中で37℃でインキュベートした。この浴溶液の組成は、NaCl,135.0mM、KCl,5.0mM、CaCl2,2.0mM、MgCl2,1.0mM、グルコース,11.0mM、NaHCO3,15.0mM、及びKH2PO4,1.0mMであった。該組織を1.0gの静止張力を掛けて配置した。平衡化のため少なくとも30分間放置した後、該筋肉には2ms持続の30Vパルスを、該神経には0.2ms持続で3Vパルスをそれぞれ用いて、10s間隔で直接刺激及び間接刺激を交互に行った。直接的及び間接的に誘発される収縮に及ぼす3μMの濃度でのχ−MrIAの1回用量の効果を測定した。応答をデジタル化し、精管調製物で述べたように記録した。
【0082】
マウス横隔膜神経−片側横隔膜の電気刺激への応答に及ぼすχ−MrIAの効果横隔膜神経の電場刺激により誘発される収縮、又は直接の筋肉刺激により誘発される収縮は、3μMのχ−MrIA(n=4)により影響されなかった。χ−MrIAはマウスの横隔膜神経−片側横隔膜調製物では筋肉のニコチン性ACh受容体をブロックしない。骨格筋又は運動神経におけるχ−MrIAの活性のこの欠如により、χ−MrIAは、今日までに特性決定された、麻痺性の毒であり従って餌捕捉に明確な役割を持つ、コノトキシン・ペプチド類の大部分とは区別される。
【0083】
実施例5
[3H]−ノルアドレナリンの細胞による取り込み
チャイニーズ・ハムスター卵巣(CHO)細胞を24穴プレート(ファルコン)中、10%v/vウシ胎児血清中で増殖させた。60〜70%コンフルエンスに達したとき、該細胞を、ヒトニューロンのノルアドレナリン輸送因子の全長cDNA(パチョルシークら,(1991) Nature, 350, 350-4)を組み込んでいる発現ベクター(pcDNA3,インビトロゲン)で一時的にトランスフェクトした(リポフェクタミン,ギブコ)。このニューロンのノルアドレナリン輸送因子のcDNAクローンを使用した(ボルム・インスティチュート,ポートランド,OR,USA)。細胞の取り込み研究はトランスフェクションの36時間後に行った。該CHO細胞を、NaCl,157mM、KCl,2.7mM、NaH2PO4,11.8mM、MgCl2,1.0mM、及びCaCl2,0.1mM、pH7.4を含む輸送緩衝液でまず洗浄した。次いで、該細胞を50nMの[3H]−ノルアドレナリン(必要なときは非標識ノルアドレナリンを補足した)及び100μMアスコルビン酸を含む輸送緩衝液と共にインキュベートした。適当なときにχ−MrIA(0.1nM〜1μM)又はデシプラミン(10μM)も含めた。室温で20分後に、該細胞を氷冷リン酸緩衝化食塩水で素早く洗浄した後、0.1%v/vトリトン−X中で溶解した。この細胞溶解物を液体シンチレーション計測にかけ、その放射活性レベルを測定した。さらに、該細胞溶解物の部分標本を用いてタンパク質濃度を測定した(バイオラド・DC・プロテインアッセイ)。該ノルアドレナリン輸送因子による[3H]−ノルアドレナリンの特異的取り込みをデシプラミン(10μM)感受性の成分として定義した。
[3H]−ノルアドレナリンの細胞内蓄積に及ぼすχ−MrIAの効果
ヒトニューロンのノルアドレナリン輸送因子を発現するCHO細胞中へのノルアドレナリンの蓄積はデシプラミン(10μM)による対照量の0.5%未満にまで減少した。このことは該蓄積が殆ど全てクローニングされた輸送因子を介する特異的取り込みによるものであったことを証明する。このノルアドレナリン輸送因子は細胞取り込み研究において該コノトキシンの標的であることが確認された。χ−MrIA(0.1nM〜1μM)は放射標識ノルアドレナリンの蓄積を濃度−依存形式で阻害し(図3)、logIC50値は−8.17±0.0275(n=4)であった。該蓄積を50%だけ阻害するために必要なχ−MrIAの濃度は約7nMであることが見出された。この濃度はデシプラミンが同じ効果を達成するために必要な濃度よりもほぼ1オーダー低い量である。
【0084】
コカインとχ−MrIAは両方とも天然に生ずる化合物であるが、それらは全く似ていない。コカインはコカ植物の葉から抽出されるアルカロイドであるのに対し、χ−MrIAは動物遺伝子が直接コードするぺプチドである。該取り込み輸送因子におけるその効果に加え、コカインは強力な局部麻酔特性を有することが知られている。これはナトリウムチャンネルとカリウムチャンネルの両方の遮断による。検定のいずれにおいてもχ−MrIAの局部麻酔活性についての証拠は見出されなかった。χ−MrIAは単独では精管の正常な張りに対する収縮効果も弛緩効果も持たないことが見出された。同様な研究により、χ−コノトキシンはドパミン輸送因子を阻害しないことが明らかにされた。
【0085】
実施例6
トリチウム化したマジンドールのノルアドレナリン輸送因子への結合を、該輸送因子タンパク質を発現する細胞中で測定した(実施例5を参照)。トリチウム化したマジンドールの結合に及ぼす10-6から10-9のχ−MrIAの影響を測定した。χ−MrIAはトリチウム化したマジンドールの結合には効果を及ぼさなかった。このことは、それがデシプラミン、マジンドール及びコカインなどの従来のノルアドレナリン輸送因子阻害剤とは異なる部位に非競争的に作用することを示す。
【0086】
実施例7
χ−コノトキシン・ぺプチド類の遺伝子配列の誘導
クローニング及びDNA配列決定と併用した5’RACE(Random Amplification of cDNA Ends、cDNA末端の無作為増幅)及び3’RACE組合せ戦術を用いて、該χ−MrIAの完全な遺伝子配列を単離した。
5’RACE
その成熟ぺプチド配列からオリゴヌクレオチドプライマーCH1−1Bを設計した。このオリゴヌクレオチドと該ぺプチドとの関係は以下のとおりであり、該オリゴヌクレオチドの配列と共に示す。
χ−MrIA - NGVCCGYKLCHPC 配列番号:3
CHI−1B 5'- CANGGRTGRCANARYTTRTA -3'
配列番号:4
AP1 5'- CCATCCTAATACGACTCACTATAGGGC-3'
配列番号:5
(上記において、N=A/C/G/T、R=A/G、Y=C/T)
該オリゴヌクレオチドCH1−1BをAP1オリゴヌクレオチドと組合せ、イモガイ(coneshell)毒素管から単離されたmRNA由来のcDNA鋳型を用いて、ポリメラーゼ連鎖反応(PCR)を行った。そのPCR産物はMrIA遺伝子の5’領域を表すが、これを単離し、精製し、細菌ベクター中にクローニングし、配列決定した。MrIAの遺伝子配列はシー.マルモレウス(C. marmoreus)から得た(図4)。
3’RACE
該遺伝子の5’領域のDNA配列を用いて、MrIA配列及び他の密接に関連するぺプチド由来の配列を検出できるオリゴヌクレオチドを設計した。該遺伝子配列との関連での該オリゴヌクレオチドの位置決めは図4に示す。オリゴヌクレオチドCH1−1AをANCHORオリゴヌクレオチドと結合させてPCRに使用し、該遺伝子のリーダーぺプチド、成熟ぺプチド及び3’非翻訳領域(3’UTR)に相当するDNA断片を作成した。シー.マルモレウス由来の毒素管cDNA鋳型のPCRにより、MrIAぺプチドに相当するDNA断片を作成した。
【0087】
これらのオリゴヌクレオチド類のDNA配列は以下のとおりである。
CHI−1A 5'- ACAGGCAGAATGCGCTGTCTCCC -3'
配列番号:6
ANCHOR 5'- AACTGGAAGAATTCGCGGCCGCAGGAAT -3'
配列番号:7
χ−MrIAの完全配列
5’RACEと3’RACEを用いて作成したχ−MrIAの遺伝子配列は該遺伝子の重複断片類を表す。これらの断片類を組合せ、該遺伝子に対する共通配列を作成した。この共通配列は該遺伝子の完全cDNAであり、5’UTR、リーダーぺプチド、成熟ぺプチド及び3’UTRを含む。このχ−MrIAのリーダーぺプチドオリゴヌクレオチド配列及び成熟ぺプチドオリゴヌクレオチド配列を配列番号:8に示し、一方、リーダーぺプチドアミノ酸配列及び成熟ぺプチドアミノ酸配列は配列番号:9に示す。
【0088】
【化1】
【0089】
以下の本明細書及び請求の範囲を通じて、文脈が別義を要求する場合を除き、用語「含む(comprise)」及びその変化「含む(comprises)及び(comprising)」は、述べられた主体若しくは工程又は主体群若しくは工程群の含有を意味するが、任意の他の主体若しくは工程又は主体群若しくは工程群の排除を意味するものではないと理解されるべきである。
【0090】
当業者は本明細書に記述された発明が具体的に記述されたもの以外の変更及び修飾を受け易いことを認めるはずである。本発明はこのような変更及び修飾をすべて包含するものと解すべきである。本発明は本明細書で個々に若しくは集合的に言及し又は示したすべての工程、特徴、組成物及び化合物、及び該工程又は特徴の任意の組合せ及びすべての組合せをも包含する。
【特許請求の範囲】
【請求項1】
下記の配列
NGVCCGYKLCHOC 配列番号:1
及び VGVCCGYKLCHOC 配列番号:2
から選択され、二以上のアミノ酸の付加および/または同類置換を受けた配列を持ち、ニューロンのアミン輸送因子を阻害する能力を有する、単離されまたは合成されたχ−コノトキシン・ぺプチド。
【請求項2】
前記配列が13〜30のアミノ酸を持つ、請求項1記載のχ−コノトキシン・ぺプチド。
【請求項3】
ニューロンのノルアドレナリン輸送因子を阻害する能力を有する請求項1または請求項2記載のχ−コノトキシン・ぺプチド。
【請求項4】
ニューロンのノルアドレナリン輸送因子の選択的阻害剤である請求項3記載のχコノトキシン・ぺプチド。
【請求項5】
抗コリン作用性の効果を持たないか又は殆ど持たない請求項3記載のχ−コノトキシン・ぺプチド。
【請求項6】
ナトリウムチャンネル・ブロッカーとしての活性を持たないか殆ど持たない請求項3記載のχ−コノトキシン・ぺプチド。
【請求項7】
ドパミン輸送因子の阻害剤としての活性を持たないか殆ど持たない請求項3記載のχ−コノトキシン・ぺプチド。
【請求項8】
4個のシステイン残基と2個のジスルフィド結合を持つ請求項1または請求項2記載のχ−コノトキシン・ぺプチド。
【請求項9】
該ジスルフィド結合の連結がA−D/B−Cであり、A、B、C及びDがそれぞれ第一、第二、第三及び第四システイン残基である、請求項8記載のχ−コノトキシン・ぺプチド。
【請求項10】
ニューロンのノルアドレナリン輸送因子の阻害剤としてのある分子の活性を試験するための受容体結合検定における、請求項1または請求項2記載のχ−コノトキシン・ぺプチドの使用。
【請求項11】
請求項1〜請求項9いずれか1項に記載のχ−コノトキシン・ペプチドをコードするヌクレオチド配列又はコードする配列に相補的な配列を含む単離された核酸分子。
【請求項12】
請求項1または請求項2記載のχ−コノトキシン・ぺプチドに対するモノクローナル抗体又はポリクローナル抗体。
【請求項13】
ベクター部分と請求項1または請求項2記載のχ−コノトキシン・ぺプチドをコードすることができる核酸を含む遺伝子構築物。
【請求項14】
排尿若しくは心臓血管の状態若しくは病気又は気分変調の治療若しくは予防、又は苦痛若しくは炎症の治療若しくは制御のための医薬の製造における、請求項1〜請求項9いずれか1項に記載のχ−コノトキシン・ぺプチドの使用。
【請求項15】
該排尿システムの病気又は状態が尿失禁又は排便失調である請求項14記載の使用。
【請求項16】
該心臓血管の病気又は状態が不整脈又は冠不全である請求項14記載の方法。
【請求項17】
該気分変調がうつ病、不安又は喫煙の切望である請求項14記載の方法。
【請求項18】
該苦痛が慢性の苦痛、神経障害性の苦痛又は炎症性の苦痛である請求項14記載の方法。
【請求項19】
請求項1〜請求項9いずれか1項に記載のχ−コノトキシン・ぺプチド、及び薬学的に許容しうる担体又は希釈剤を含む組成物。
【請求項20】
医薬組成物である請求項19記載の組成物。
【請求項21】
ニューロンのノルアドレナリン輸送因子を阻害するための請求項3記載のぺプチドの使用。
【請求項22】
ニューロンのノルアドレナリン輸送因子の阻害が有効な治療又は予防と関連がある病気又は状態を治療又は予防するための医薬の製造における、請求項3記載のχ−コノトキシン・ぺプチドの使用。
【請求項1】
下記の配列
NGVCCGYKLCHOC 配列番号:1
及び VGVCCGYKLCHOC 配列番号:2
から選択され、二以上のアミノ酸の付加および/または同類置換を受けた配列を持ち、ニューロンのアミン輸送因子を阻害する能力を有する、単離されまたは合成されたχ−コノトキシン・ぺプチド。
【請求項2】
前記配列が13〜30のアミノ酸を持つ、請求項1記載のχ−コノトキシン・ぺプチド。
【請求項3】
ニューロンのノルアドレナリン輸送因子を阻害する能力を有する請求項1または請求項2記載のχ−コノトキシン・ぺプチド。
【請求項4】
ニューロンのノルアドレナリン輸送因子の選択的阻害剤である請求項3記載のχコノトキシン・ぺプチド。
【請求項5】
抗コリン作用性の効果を持たないか又は殆ど持たない請求項3記載のχ−コノトキシン・ぺプチド。
【請求項6】
ナトリウムチャンネル・ブロッカーとしての活性を持たないか殆ど持たない請求項3記載のχ−コノトキシン・ぺプチド。
【請求項7】
ドパミン輸送因子の阻害剤としての活性を持たないか殆ど持たない請求項3記載のχ−コノトキシン・ぺプチド。
【請求項8】
4個のシステイン残基と2個のジスルフィド結合を持つ請求項1または請求項2記載のχ−コノトキシン・ぺプチド。
【請求項9】
該ジスルフィド結合の連結がA−D/B−Cであり、A、B、C及びDがそれぞれ第一、第二、第三及び第四システイン残基である、請求項8記載のχ−コノトキシン・ぺプチド。
【請求項10】
ニューロンのノルアドレナリン輸送因子の阻害剤としてのある分子の活性を試験するための受容体結合検定における、請求項1または請求項2記載のχ−コノトキシン・ぺプチドの使用。
【請求項11】
請求項1〜請求項9いずれか1項に記載のχ−コノトキシン・ペプチドをコードするヌクレオチド配列又はコードする配列に相補的な配列を含む単離された核酸分子。
【請求項12】
請求項1または請求項2記載のχ−コノトキシン・ぺプチドに対するモノクローナル抗体又はポリクローナル抗体。
【請求項13】
ベクター部分と請求項1または請求項2記載のχ−コノトキシン・ぺプチドをコードすることができる核酸を含む遺伝子構築物。
【請求項14】
排尿若しくは心臓血管の状態若しくは病気又は気分変調の治療若しくは予防、又は苦痛若しくは炎症の治療若しくは制御のための医薬の製造における、請求項1〜請求項9いずれか1項に記載のχ−コノトキシン・ぺプチドの使用。
【請求項15】
該排尿システムの病気又は状態が尿失禁又は排便失調である請求項14記載の使用。
【請求項16】
該心臓血管の病気又は状態が不整脈又は冠不全である請求項14記載の方法。
【請求項17】
該気分変調がうつ病、不安又は喫煙の切望である請求項14記載の方法。
【請求項18】
該苦痛が慢性の苦痛、神経障害性の苦痛又は炎症性の苦痛である請求項14記載の方法。
【請求項19】
請求項1〜請求項9いずれか1項に記載のχ−コノトキシン・ぺプチド、及び薬学的に許容しうる担体又は希釈剤を含む組成物。
【請求項20】
医薬組成物である請求項19記載の組成物。
【請求項21】
ニューロンのノルアドレナリン輸送因子を阻害するための請求項3記載のぺプチドの使用。
【請求項22】
ニューロンのノルアドレナリン輸送因子の阻害が有効な治療又は予防と関連がある病気又は状態を治療又は予防するための医薬の製造における、請求項3記載のχ−コノトキシン・ぺプチドの使用。
【図1】


【図2】

【図3】
【図4】

【図2】
【図3】
【図4】
【公開番号】特開2010−46088(P2010−46088A)
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願番号】特願2009−245436(P2009−245436)
【出願日】平成21年10月26日(2009.10.26)
【分割の表示】特願2000−574555(P2000−574555)の分割
【原出願日】平成11年10月1日(1999.10.1)
【出願人】(506411900)ゼノム リミテッド (1)
【Fターム(参考)】
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願日】平成21年10月26日(2009.10.26)
【分割の表示】特願2000−574555(P2000−574555)の分割
【原出願日】平成11年10月1日(1999.10.1)
【出願人】(506411900)ゼノム リミテッド (1)
【Fターム(参考)】
[ Back to top ]