
新規なグルココルチコイド受容体アゴニスト
本発明は、式(I)の新規なグルココルチコイド受容体アゴニストならびにそれを調製するための方法および中間体に関する。本発明はまた、これらの化合物を含有する医薬組成物、1種または複数の他の治療剤とのその組合せ、さらに、いくつかの炎症性およびアレルギー性疾患、障害および状態を治療するためのその使用に関する。
【化1】

【化1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なグルココルチコイド受容体アゴニストならびに薬学的に許容できるその塩または前記グルココルチコイド受容体アゴニストもしくは塩の薬学的に許容できる溶媒和物、それを調製する方法および中間体に関する。本発明はまた、これらの化合物を含有する医薬組成物、1種または複数の他の治療剤とのその組合せ、さらに、いくつかの炎症性およびアレルギー性疾患、障害および状態を治療するためのその使用に関する。
【背景技術】
【0002】
グルココルチコイド受容体アゴニストは、幅広い炎症性および免疫障害を治療するために不可欠である強力な抗炎症薬である。治療に組み込まれた最初の化合物は、天然のコルチコステロイドヒドロコルチゾンに由来した。核分子の初めの構造的修飾は、ミネラルコルチコイド受容体を上回るグルココルチコイドに対する選択性の向上を目的としていた。構造と活性との関係がさらによく理解されたことに基づき、次世代の化合物は、より高い受容体親和性を、したがってより高い効果を示した。局所塗布されるグルココルチコイドでは、薬物標的化により、例えば、コルチコステロイド製剤の吸入または皮膚塗布により、さらなる進歩が達成された。最近の開発は、代謝不安定性な官能基を活性分子に組み込んで、局所塗布後の全身曝露を最小化することにより、副作用を可能な限り低減することに焦点を当てていた。治療標的組織に対する高い親和性は、標的への効果および作用時間を高め、体循環への再分布を遅くすることにより標的外への全身作用を制限する特性と認識された。
【0003】
グルココルチコイド受容体アゴニストは、炎症性およびアレルギー性状態、例えば、喘息、閉塞性気道疾患、鼻炎、炎症性腸疾患、乾癬、湿疹などの管理で使用される。既に市販されているグルココルチコイドの例には、以下のものが包含される。
【0004】
【化1】
【0005】
これらの化合物は、幅広い細胞種類において、グルココルチコイド受容体に結合し、それを活性化する。活性化された受容体は、核中のグルココルチコイド応答要素に結合して、鍵となる調節機能を有する遺伝子の転写を活性化または阻害する。詳細には、これらの化合物は、好酸球および好中球などの炎症性白血球が炎症部位へと動員されるのを妨げ、また、白血球および組織細胞からの炎症性媒介物質の形成および放出を阻害することにより、炎症性疾患において有効である。
【0006】
最初のコルチコステロイドが市販されて以来、例えば、下式のWO05/028495に記載されている化合物など、様々な構造を有する数多くのコルチコステロイドが提案されている:
【0007】
【化2】
[式中、
【0008】
【化3】
は、二重結合であってよく、R1およびR2は、Fであってよく、R3は、OHであってよく、R4はHであってよく、R5は、フェニルにより置換されていてもよいC5〜10アリールであってよく、ここで、前記フェニルは、アルキル、アルコキシまたはハロゲンにより置換されていてもよい]。
【発明の概要】
【発明が解決しようとする課題】
【0009】
しかしながら、例えば、効果、治療指数、薬物動態、薬物/薬物相互作用および/または副作用に関して、最も適した薬理学的プロファイルを有するであろう改良されたグルココルチコイド受容体アゴニストが未だに必要とされている。本明細書では、式(I)の化合物:
【0010】
【化4】
[式中、R1は、
【0011】
【化5】
からなる群から選択され、ここで、*は、フェニル環の炭素へのR1の結合点を示している]
または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物を提供する。
【課題を解決するための手段】
【0012】
第1の実施形態では、式(Ia)のグルココルチコイド受容体アゴニストのサブグループ、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物が好ましい
【0013】
【化6】
[式中、R1は、
【0014】
【化7】
からなる群から選択され、ここで、*は、フェニル環の炭素へのR1の結合点を示している]。
【発明を実施するための形態】
【0015】
したがって本発明は、下記の好ましい化合物に及ぶ:
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−ベンジルフェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルチオ)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a、10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−8−(4−{[(4−ヒドロキシフェニル)チオ]メチル}フェニル)−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a、10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルスルフィニル)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a、10b,11,12−ドデカヒドロ−2H−ナフト−[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;および
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}フェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b、9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン。
【0016】
本発明によるさらなる好ましいグルココルチコイド受容体アゴニストは、(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルチオ)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オンである。
【0017】
本発明による他の好ましいグルココルチコイド受容体アゴニストは、(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−8−(4−{[(4−ヒドロキシフェニル)チオ]メチル}フェニル)−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オンである。
【0018】
本発明によるまだ他の好ましいグルココルチコイド受容体アゴニストは、(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}フェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オンである。
【0019】
本発明による式(I)の化合物は、様々な方法で、下記に例示されている方法などによる慣用の手順を使用して調製することができ、ここで、R1は、別段に述べられていない限り、式(I)の化合物に関して既に定義された通りである。しかし、当業者であれば、他の経路も同等に実施可能であり得ることを理解するであろう。
【0020】
式(I)の化合物は、下記の通りスキーム1に従って調製することができる:
【0021】
【化8】
スキーム1では、式(I)の化合物は、式(II)の化合物を式(IIa)の適切なアルデヒドと、または式(IIb)の適切なアルデヒド同等物と反応させることにより調製することができる。簡便には、過剰のアルデヒドもしくはアルデヒド同等物または化学量論的量のアルデヒドもしくはアルデヒド同等物を、アルキルスルホン酸(例えば、トリフルオロメタンスルホン酸)などの酸の存在下に、適切な溶媒(例えば、アセトニトリル、エチレングリコールジメチルエーテル、1,4−ジオキサンまたはジクロロメタン)の存在下に、任意選択で乾燥剤(硫酸マグネシウムまたは硫酸ナトリウムなど)の存在下に、周囲温度または低温で使用することにより、反応を行う。
【0022】
式(II)の化合物は、文献(例えば、Fried,J.、US3,177,231(1965))で知られている方法により式(III)の化合物を反応させるか、またはホウフッ化水素酸水溶液で周囲温度または40℃などの高温で処理することにより調製することができる。
【0023】
別法では、R1が式:
【0024】
【化9】
である式(I)の化合物はまた、R1が式:
【0025】
【化10】
である式(I)の化合物を適切な酸化剤と反応させることにより調製することができる。簡便には、やや過剰の過酸化水素などの酸化剤を、適切な溶媒(例えば、ヘキサフルオロイソプロパノール)の存在下、周囲温度または低温で使用することにより、この反応を行う(Tet.Lett.、39、3141〜3144、J.P.Begueら、1998参照)。
【0026】
式(I)の化合物はまた、式(III)の化合物を式(IIa)の適切なアルデヒドと、または式(IIb)の適切なアルデヒド同等物と反応させることにより調製することができる。簡便には、過剰のアルデヒドもしくはアルデヒド同等物または化学量論的量のアルデヒドもしくはアルデヒド同等物を、酸(例えば、トリフルオロメタンスルホン酸または過塩素酸など)の存在下、適切な溶媒(例えば、アセトニトリルまたは1,4−ジオキサンなど)の存在下、任意選択で添加剤(例えば、砂など)の存在下、周囲温度または低温で使用することにより、反応を行う。
【0027】
式(I)の化合物はまた、R2およびR3がホルミルと定義される式(IV)の化合物を式(IIa)の適切なアルデヒドと、または式(IIb)の適切なアルデヒド同等物と、文献(例えばWO2005/028495)で知られている方法により反応させることにより調製することができる。
【0028】
R2およびR3がホルミルと定義される式(IV)の化合物は、式(III)の化合物を、文献(例えばWO2005/028495)で知られている方法により反応させることにより調製することができる。
【0029】
式(III)の化合物は、市販されている。
【0030】
本発明では、「適切なアルデヒド」は、式(IIa)のアルデヒドを意味する:
【0031】
【化11】
[式中、R1は、式(I)の化合物で既に定義された通りである]。言い換えると、本発明による適切なアルデヒドは、
式(Va)の4−ベンジルベンズアルデヒド
【0032】
【化12】
式(VIa)の4−{[3−(メチルチオ)フェニル]チオ}ベンズアルデヒド
【0033】
【化13】
式(VIIa)の4−{[(4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド
【0034】
【化14】
式(VIIIa)の4−{[3−(メチルスルフィニル)フェニル]チオ}ベンズアルデヒド:
【0035】
【化15】
または
式(IXa)の4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド
【0036】
【化16】
からなる群から選択される。
【0037】
別法では、「適切なアルデヒド同等物」は、式(IIb)の化合物を意味し:
【0038】
【化17】
これはまた、「ビスルフィト付加生成物」とも称され、ここで、R1は、式(I)の化合物で既に定義された通りである。言い換えると、本発明による適切なアルデヒド同等物は、
式(Vb)のナトリウムヒドロキシル(ベンジルフェニル)メタンスルホネート
【0039】
【化18】
【0040】
(VIb)のナトリウムヒドロキシ(4−{[3−(メチルチオ)フェニル]チオ}フェニル)メタンスルホネート
【0041】
【化19】
式(VIIb)のナトリウムヒドロキシル(4−{[(4−ヒドロキシフェニル)チオ]メチル}フェニル)メタンスルホネート
【0042】
【化20】
式(VIIIb)のナトリウムヒドロキシ(4−{[4−(メチルスルフィニル)フェニル]チオ}フェニル)メタンスルホネート
【0043】
【化21】
または
式(IXb)のナトリウム(4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}フェニル)(ヒドロキシ)メタンスルホネート
【0044】
【化22】
からなる群から選択される。
【0045】
上記で述べられたアルデヒドまたはアルデヒド同等物は、市販されているか、または当業者によく知られている慣用の手順に従って容易に調製することができる。
【0046】
本明細書で上記された式(I)の化合物の調製方法のいくつかのステップでは、反応することが望ましくない反応する可能性のある官能基を保護し、最終的には前記保護基を分離する必要のあることがある。このような場合、任意の相容性な保護基を使用することができる。詳細には、T.W.GREENE(Protective Groups in Organic Synthesis、A.Wiley−Interscience Publication、1981)またはP.J.Kocienski(Protecting groups、Georg Thieme Verlag、1994)により記載された方法などの保護および脱保護の方法を使用することができる。
【0047】
上記の反応および前述の方法で使用される新規な出発物質の調製は全て、慣用的であり、その実施または調製のための適切な試薬および反応条件、さらに、所望の生成物を単離するための手順は、文献手順ならびにその例および調製を参照して、当業者によく知られている。
【0048】
また、式(I)の化合物、さらに、それを調製するための中間体は、例えば、結晶化またはクロマトグラフィーなどの様々なよく知られている方法により精製することができる。
【0049】
式(I)の化合物の薬学的に許容できる塩には、その塩基塩が包含される。適切な塩基塩は、非毒性の塩を形成する塩基から形成される。例には、アルミニウム、アルギニン、ベンザチン、カルシウム、コリン、ジエチルアミン、ジオラミン、グリシン、リシン、マグネシウム、メグルミン、オラミン、カリウム、ナトリウム、トロメタミンおよび亜鉛塩が包含される。
【0050】
式(I)の化合物の薬学的に許容できる塩にはまた、その酸塩が包含されることもある。また、酸および塩基の半塩、例えば、半硫酸塩および半カルシウム塩を形成することもできる。
【0051】
適切な塩についての総説に関しては、StahlおよびWermuthによる「Handbook of Pharmaceutical Salts:Properties,Selection,and Use」(Wiley−VCH、2002)参照。
【0052】
式(I)の化合物の薬学的に許容できる塩は、3種の方法のうちの1種または複数により調製することができる:
(i)式(I)の化合物を所望の酸または塩基と反応させることによる方法、
(ii)所望の酸または塩基を使用して、式(I)の化合物の適切な前駆体から酸または塩基不安定性保護基を除去するか、または適切な環式前駆体、例えば、ラクトンまたはラクタムを開環する方法、または
(iii)適切な酸もしくは塩基との反応により、または適切なイオン交換カラムを用いて、式(I)の化合物の1種の塩を他の塩に変換する方法。
【0053】
3種の反応を全て典型的には、溶液で実施する。生じた塩を沈殿させて、濾過により集めるか、または溶媒を蒸発させることにより回収することができる。生じた塩の電離度は、完全な電離から、ほぼ非電離まで変動してよい。
【0054】
本発明の化合物は、完全な非晶質から完全な結晶までの範囲の固体状態の連続で存在し得る。「非晶質」との用語は、その物質が、分子レベルで長距離秩序を欠いていて、温度に応じて固体または液体の物理的特性を示し得る状態を指す。典型的には、このような物質は、特有のX線回折パターンを示さず、固体の特性を示しながらも、液体としてより形式的には記載される。加熱すると、固体特性から液体特性への変化が生じ、これは、状態変化、典型的には二次変化により特徴づけられる(「ガラス遷移」)。「結晶」との用語は、その物質が、分子レベルで規則的に配列している内部構造を有し、規定のピークを有する特有のX線回折パターンを示す固相を指す。このような物質はまた、十分に加熱されると、液体の特性を示すが、固体から液体への変化は、相変化、典型的には一次変化により特徴づけられる(「融点」)。
【0055】
本発明の化合物およびその塩はまた、非溶媒和形態および溶媒和形態で存在し得る。「溶媒和物」との用語は本明細書では、本発明の化合物および1個または複数の薬学的に許容できる溶媒分子、例えば、エタノールを含む分子複合体を記載するために使用されている。「水和物」との用語は、前記溶媒が水である場合に使用される。
【0056】
有機水和物に関して現在認められている分類体系は、孤立サイト、チャネルまたは金属イオン配位水和物を定義する分類体系である。K.R.Morrisによる「Polymorphism in Pharmaceutical Solids」(H.G.Brittain編、Marcel Dekker、1995)参照。孤立サイト水和物は、その水分子が、有機分子の介在により、相互の直接的な接触から孤立している水和物である。チャネル水和物では、水分子は、格子チャネル内に存在し、他の水分子に隣接している。金属イオン配位水和物では、水分子は、金属イオンに結合している。
【0057】
溶媒または水が密に結合していると、複合体は、湿度とは独立に、十分に定義される化学量論を有するはずである。しかしながら、チャネル溶媒和物および吸湿性化合物においてのように、溶媒または水の結合が弱い場合、水/溶媒含分は、湿度および乾燥状態に左右される。このようなケースでは、非化学量論が標準となる。
【0058】
また、薬物および少なくとも1種の他の成分が、化学量論的量または非化学量論的量で存在している多成分複合体(塩および溶媒和物以外)も、本発明の範囲内に包含される。このタイプの複合体には、包接化合物(薬物−ホスト包接複合体)および共結晶が包含される。後者は典型的には、非共有結合相互作用を介して相互に結合している中性分子成分同士の結晶複合体と定義されるが、中性分子と塩との複合体であってもよい。溶融結晶化、溶媒からの再結晶化または成分同士の物理的粉砕により、共結晶を調製することができる(O.AlmarssonおよびM.J.ZaworotkoによるChem Commun、17、1889〜1896(2004)参照)。多成分複合体の一般的な総説に関しては、HaleblianによるJ.Pharm.Sci、64(8)、1269〜1288(1975年8月)参照。
【0059】
本発明の化合物はまた、適切な条件に掛けると、中間状態(中間相または液晶)でも存在し得る。中間状態は、真の結晶状態と真の液体状態(溶融または溶液)との中間である。温度変化の結果として生じる液晶性は、「サーモトロピック」と記載され、水または他の溶媒などの第2の成分を加えると生じる液晶性は、「リオトロピック」と記載される。リオトロピック中間相を形成する可能性のある化合物は、「両親媒性」と記載され、イオン性(−COO−Na+、−COO−K+または−SO3−Na+など)または非イオン性(−N−N+(CH3)3など)極性ヘッド基を持つ分子からなる。さらなる情報に関しては、N.H.HartshorneおよびA.Stuartによる「Crystals and the Polarizing Microscope」、4th Edition(Edward Arnold、1970)参照。
【0060】
後記では、本発明の化合物に関する言及は全て、その塩、溶媒和物、多成分複合体および液晶ならびにその塩の溶媒和物、多成分複合体および液晶に対する言及を包含する。
【0061】
本発明の化合物には、後記で定義される通りのその全ての多形および晶癖、そのプロドラッグおよび異性体(光学、幾何および互変異性体を包含)ならびに同位体標識された式(I)の化合物を包含する、上記で定義された通りの式(I)の化合物が包含される。
【0062】
前記のように、本発明の化合物のいわゆる「プロドラッグ」もまた、本発明の範囲内である。それ自体は薬理活性をほとんど有さないか、有さないことのある式(I)の化合物のある種の誘導体は、体内または体上に投与されると、例えば、加水分解により変換されて、所望の活性を有する式(I)の化合物になり得る。このような誘導体が、「プロドラッグ」と称される。プロドラッグの使用に関するさらなる情報は、「Pro−drugs as Novel Delivery Systems」、Vol.14、ACS Symposium Series(T.HiguchiおよびW.Stella)および「Bioreversible Carriers in Drug Design」、Pergamon Press、1987(E.B.Roche編、American Pharmaceutical Association)で見ることができる。
【0063】
例えば、式(I)の化合物中に存在する適切な官能基を、例えば、H.Bundgaardによる「Design of Prodrugs」(Elsevier、1985)に記載されている通りに、当業者に「プロ部分」として知られているある種の部分で置き換えることにより、本発明によるプロドラッグを製造することができる。
【0064】
本発明によるプロドラッグのいくつかの例には、式(I)の化合物がアルコール官能基(−OH)を含有する場合には、そのエーテルが包含され、例えば、式(I)の化合物のアルコール官能基の水素が(C1〜C6)アルカノイルオキシメチルにより置き換えられている化合物が包含される。
【0065】
上述の例による代替基のさらなる例および他のプロドラッグタイプの例は、上述の参考文献で見ることができる。
【0066】
さらに、ある種の式(I)の化合物はそれ自体、他の式(I)の化合物のプロドラッグとして作用し得る。
【0067】
また、式(I)の化合物の代謝産物、即ち、薬物が投与されるとin vivoで形成される化合物も、本発明の範囲内に包含される。本発明による代謝産物のいくつかの例には:
(i)式(I)の化合物がメチル基を含有する場合、そのヒドロキシメチル誘導体(−CH3→−CH2OH)、
(ii)式(I)の化合物がフェニル部分を含有する場合、そのフェノール誘導体(−Ph→−PhOH)、および
(iii)式(I)の化合物がスルフィドを含有する場合、そのスルホキシド誘導体(−SPh→−S(O)Ph)
が包含される。
【0068】
1個または複数の不斉炭素原子を含有する式(I)の化合物は、2種以上の立体異性体として存在し得る。構造異性体が、低エネルギー障壁を介して相互転換可能である場合、互変異性(「tautomerism」)が生じ得る。これは、例えばイミノ、ケトまたはオキシム基を含有する式(I)の化合物では、プロトン互変異性の形態を、または芳香族部分を含有する化合物ではいわゆる原子価互変異性の形態をとり得る。したがって、単一化合物が、1種を超える異性で存在し得る。1種を超える異性を示す化合物およびそれらの1種または複数の混合物を包含する式Iの化合物の立体異性体、幾何異性体および互変異性形態全てが、本発明の範囲内に包含される。また、対イオンが光学活性である酸付加塩もしくは塩基塩、例えば、d−乳酸塩もしくはl−リシンまたはラセミ体、例えばdl−酒石酸塩もしくはdl−アルギニンが包含される。
【0069】
個々の鏡像異性体を調製/単離するための慣用の技術には、適切な光学的に純粋な前駆体からのキラル合成または例えば、キラル高圧液体クロマトグラフィー(HPLC)を使用してのラセミ体(または塩もしくは誘導体のラセミ体)の分割が包含される。
【0070】
別法では、ラセミ体(またはラセミ前駆体)を適切な光学的に活性な化合物、例えば、アルコールと、または式(I)の化合物が酸性または塩基性部分を含有する場合には、1−フェニルエチルアミンまたは酒石酸などの塩基または酸と反応させることができる。生じたジアステレオ異性体の混合物を、クロマトグラフィーおよび/または分別結晶化により分離し、そのジアステレオ異性体の一方または両方を、当業者によく知られている手段により対応する純粋な1種または複数の鏡像異性体に変換することもできる。
【0071】
クロマトグラフィー、典型的にはHPLCを不斉樹脂上で、炭化水素、典型的には、イソプロパノール0から50体積%、典型的には2%から20体積%およびアルキルアミン0から5体積%、典型的にはジエチルアミン0.1体積%を含有するヘプタンまたはヘキサンからなる移動相と共に使用すると、本発明のキラル化合物(およびそのキラル前駆体)を鏡像異性的に濃縮された形態で得ることができる。溶離液を濃縮すると、濃縮混合物が得られる。
【0072】
任意のラセミ体が結晶化する場合、2種の異なるタイプの結晶が可能である。第1のタイプは、両方の鏡像異性体を等モル量で含有する結晶の1種の均一な形態が生じる前記のラセミ化合物(真のラセミ化合物)である。第2のタイプは、それぞれ単一の鏡像異性体を含む2種の形態の結晶が等モル量で生じるラセミ混合物または複合体である。
【0073】
ラセミ混合物中に存在する結晶形態の両方が、同一の物理的特性を有する一方で、これらは、真のラセミ化合物に対して異なる物理的特性を有することがある。ラセミ混合物は、当業者に知られている慣用の技術により分離することができる。例えば、E.L.ElielおよびS.H.Wilenによる「Stereochemistry of Organic Compounds」(Wiley、1994)参照。
【0074】
本発明は、1個または複数の原子が、同じ原子番号を有するが、自然で優勢な原子質量または質量数とは異なる原子質量または質量数を有する原子に置き換えられている薬学的に許容できる同位体標識された式(I)の化合物全てを包含する。
【0075】
本発明の化合物中に包含されるのに適している同位体の例には、2Hおよび3Hなどの水素、11C、13Cおよび14Cなどの炭素、36Clなどの塩素、18Fなどのフッ素、123Iおよび125Iなどのヨウ素、13Nおよび15Nなどの窒素、15O、17Oおよび18Oなどの酸素、32Pなどのリンならびに35Sなどの硫黄の同位体が包含される。
【0076】
ある種の同位体標識された式(I)の化合物、例えば、放射性同位体を導入されているものは、薬物および/または基質組織分布研究で有用である。放射性同位体のトリチウム、即ち3Hおよび炭素−14、即ち14Cは、導入の容易さおよび検出の迅速な手段である点において、この目的のために特に有用である。
【0077】
ジュウテリウム、即ち2Hなどの重同位体での置換は、より大きな代謝安定性、例えば、高いin vivo半減期または低い用量要求から生じるある種の治療的利点をもたらし得るので、場合によっては好ましいことがある。
【0078】
11C、18F、15Oおよび13Nなどの陽電子放出同位体での置換は、基質受容体占有率を調べるための陽電子放出断層撮影法(PET)研究において有用であり得る。
【0079】
当業者に知られている慣用の技術により、または前に使用されていた非標識試薬の代わりに適切な同位体標識試薬を使用する添付の実施例および調製に記載のプロセスと同様のプロセスにより、同位体標識された式(I)の化合物を通常は調製することができる。
【0080】
本発明による薬学的に許容できる溶媒和物には、結晶化の溶媒が同位体置換されている、例えば、D2O、d6−アセトン、d6−DMSOであるものが包含される。
【0081】
提示されている適応症を治療するために最も適切な剤形および投与経路を選択するために、溶解性および溶解安定性(pH全体で)、透過性などのその生物薬学的特性に関して、式(I)の化合物を評価すべきである。
【0082】
薬学的使用を意図されている本発明の化合物は、結晶または非晶質生成物として投与することができる。これらは、沈殿、結晶化、凍結乾燥、噴霧乾燥または蒸発乾燥などの方法により、例えば、固体プラグ、粉末またはフィルムとして得ることができる。マイクロ波または高周波乾燥を、この目的のために使用することができる。
【0083】
これらは、単独で、または1種もしくは複数の他の本発明の化合物と組み合わせて、または1種もしくは複数の他の薬物と組み合わせて(またはその任意の組合せとして)投与することができる。通常、これらは、1種または複数の薬学的に許容できる賦形剤と共に製剤として投与される。「賦形剤」との用語は本明細書では、例えば、希釈剤、担体および補助剤などの1種または複数の本発明の化合物以外の任意の成分を記載するために使用される。賦形剤の選択は、特定の投与方法、溶解性および安定性に対する賦形剤の作用ならびに剤形の性質などの要因に大きく左右される。
【0084】
本発明の化合物を送達するために適した医薬組成物およびその調製方法は、当業者には容易に分かるであろう。このような組成物およびその調製方法は、例えば、「Remington’s Pharmaceutical Sciences」、19th Edition(Mack Publishing Company、1995)で見ることができる。
【0085】
本発明の化合物は、経口で投与することができる。経口投与は、化合物が胃腸管に入るような嚥下および/または化合物が口から直接、血流に入る頬、舌もしくは舌下投与を伴ってよい。
【0086】
経口投与に適している製剤には、錠剤などの固体、半固体および液体系;多粒子もしくはナノ粒子、液体または粉末を含有する軟質または硬質カプセル;ロゼンジ(液体充填を包含);チューイング剤;ゲル;急速分散剤形;フィルム;卵形剤(ovule);スプレーならびに頬/粘膜接着パッチが包含される。
【0087】
液体製剤には、懸濁剤、液剤、シロップおよびエリキシルが包含される。このような製剤を、軟質または硬質カプセル(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース製)中の充填剤として使用することもでき、典型的には、担体、例えば、水、エタノール、ポリエチレングリコール、プロピレングリコール、メチルセルロースまたは適切なオイルならびに1種または複数の乳化剤および/または懸濁化剤を含む。液体製剤はまた、固体、例えば、サシェからの再構成により調製することができる。
【0088】
本発明の化合物はまた、LiangおよびChenによる「Expert Opinion in Therapeutic Patents」、11(6)、981〜986(2001)に記載されているものなどの急速溶解、急速崩壊剤形で使用することができる。
【0089】
錠剤剤形では、用量に応じて、薬物は、剤形の1重量%から80重量%、より典型的には剤形の5重量%から60重量%を構成していてよい。薬物の他に、錠剤は通常、崩壊剤を含有する。崩壊剤の例には、デンプングリコール酸ナトリウム、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、メチルセルロース、微結晶性セルロース、低級アルキル置換ヒドロキシプロピルセルロース、デンプン、α化デンプンおよびアルギン酸ナトリウムが包含される。通常、崩壊剤は、剤形の1重量%から25重量%、好ましくは5重量%から20重量%を構成している。
【0090】
結合剤を通常は使用して、錠剤製剤に粘着性を付与する。適切な結合剤には、微結晶性セルロース、ゼラチン、糖、ポリエチレングリコール、天然および合成ゴム、ポリビニルピロリドン、α化デンプン、ヒドロキシプロピルセルロースならびにヒドロキシプロピルメチルセルロースが包含される。錠剤はまた、ラクトース(一水和物、噴霧乾燥一水和物、無水物など)、マンニトール、キシリトール、デキストロース、スクロース、ソルビトール、微結晶性セルロース、デンプンおよび二塩基性リン酸カルシウム二水和物などの希釈剤を含有してよい。
【0091】
錠剤はまた、ラウリル硫酸ナトリウムおよびポリソルベート80などの界面活性剤ならびに二酸化ケイ素およびタルクなどの流動促進剤を含んでもよい。存在する場合には、界面活性剤は、錠剤の0.2重量%から5重量%を構成してよく、流動促進剤は、錠剤の0.2重量%から1重量%を構成してよい。
【0092】
また、錠剤は通常、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、ステアリルフマル酸ナトリウムおよびステアリン酸マグネシウムとラウリル硫酸ナトリウムとの混合物などの滑剤を含有する。滑剤は通常、錠剤の0.25重量%から10重量%、好ましくは0.5重量%から3重量%の量を構成する。
【0093】
他の可能な成分には、抗酸化剤、着色剤、香料、防腐剤および矯味剤が包含される。
【0094】
例示的な錠剤は、薬物約80%まで、結合剤約10重量%から約90重量%、希釈剤約0重量%から約85重量%、崩壊剤約2重量%から約10重量%および滑剤約0.25重量%から約10重量%を含有する。
【0095】
錠剤ブレンドを、直接か、またはローラーにより圧縮して、錠剤を形成することができる。別法では、錠剤ブレンドまたは一部のブレンドを湿潤、乾燥もしくは溶融顆粒化するか、溶融凝固させるか、または押し出し、その後に錠剤化することができる。最終製剤は、1つまたは複数の層を含んでよく、コーティングされているか、またはコーティングされてなくてよい。さらに、カプセル封入されていてよい。
【0096】
錠剤の製剤は、H.LiebermanおよびL.Lachmanによる「Pharmaceutical Dosage Forms:Tablets,Vol.1」(Marcel Dekker、New.York、1980)で検討されている。
【0097】
ヒトまたは動物使用のための摂取可能な口腔フィルムは典型的には、柔軟な水溶性または水膨潤性薄膜剤形であり、これは、迅速に溶解するか、粘膜接着性であってよく、典型的には、式Iの化合物、フィルム形成ポリマー、結合剤、溶媒、湿潤剤、可塑剤、安定剤または乳化剤、粘度調節剤および溶媒を含む。製剤のうちのいくつかの成分は、1つを超える機能を果たすことがある。
【0098】
式(I)の化合物は、水溶性または不溶性であってよい。水溶性化合物は典型的には、溶質1重量%から80重量%、より典型的には20重量%から50重量%を含む。溶解性の低い化合物は、より高い割合の組成、典型的には、溶質88重量%までを含んでよい。別法では、式(I)の化合物は多粒子ビーズの形態であってよい。
【0099】
フィルム形成ポリマーは、天然多糖類、タンパク質または合成親水コロイドからなる群から選択されてよく、典型的には、0.01から99重量%の範囲、より典型的には、30から80重量%の範囲で存在する。
【0100】
他の可能な成分には、抗酸化剤、着色剤、香料および香増強剤、防腐剤、唾液刺激剤、冷却剤、補助溶媒(油を包含する)、緩和剤、増量剤、消泡剤、界面活性剤および矯味剤が包含される。
【0101】
本発明によるフィルムは典型的には、剥離可能なバッキング支持体または紙にコーティングされた薄い水性フィルムを蒸発乾燥させることにより調製される。これは、乾燥オーブンまたはトンネル、典型的には組み合わされたコーティングドライヤーで、または凍結乾燥もしくは真空化により行うことができる。
【0102】
経口投与のための固体製剤を、即時および/または変更放出であるように製剤することもできる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出およびプログラム放出が包含される。
【0103】
本発明の目的に適している変更放出製剤は、米国特許第6,106,864号に記載されている。高エネルギー分散液および浸透性のコーティングされた粒子などの他の適切な放出技術の詳細は、Vermaらによる「Pharmaceutical Technology On−line」、25(2)、1〜14(2001)で見ることができる。調節放出を達成するためにチューインガムを使用することは、WO00/35298に記載されている。
【0104】
本発明の化合物はまた、血流中、筋肉中または内臓に直接投与することもできる。非経口投与に適している手段には、静脈内、動脈内、腹腔内、クモ膜下、心室内、尿管内、胸骨内、頭蓋内、筋肉内、滑液包内および皮下が包含される。非経口投与のための適切なデバイスには、針(微細針を包含する)注射器、無針注射器および点滴技術が包含される。
【0105】
非経口製剤は典型的には、塩、炭水化物および緩衝剤(好ましくはpH3から9に)などの賦形剤を含有してよい水溶液であるが、いくつかの用途では、これらをより適切に、無菌非水溶液として、または無菌の発熱物質不含水などの適切な媒体と共に使用される乾燥形態として製剤することができる。
【0106】
例えば、凍結乾燥による無菌条件下での非経口製剤の調製は、当業者によく知られている標準的な製薬技術を使用して容易に達成することができる。
【0107】
非経口溶液を調製する際に使用される式(I)の化合物の溶解性は、溶解性増強剤を導入するなどの適切な製剤技術を使用することにより高めることができる。
【0108】
非経口投与のための製剤は、即時および/または変更放出であるように製剤することができる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出およびプログラム放出が包含される。したがって本発明の化合物を、懸濁剤として、または活性化合物の変更放出をもたらす移植デポーとして投与するための固体、半固体もしくはチキソトロピー液として製剤することができる。このような製剤の例には、薬物コーティングされたステントならびに薬物負荷されたポリ(dl−乳酸−コグリコール酸)(PGLA)微小球を含む半固体および懸濁液が包含される。
【0109】
本発明の化合物はまた、皮膚または粘膜に局所、皮膚(皮内)または経皮で投与することもできる。この目的のための典型的な製剤には、ゲル、ヒドロゲル、ローション、液剤、クリーム、軟膏、散布剤、包帯、フォーム剤、フィルム剤、皮膚パッチ、ウェハ、インプラント、スポンジ、繊維、帯具およびマイクロエマルションが包含される。リポソームもまた使用することができる。典型的な担体には、アルコール、水、鉱油、流動ワセリン、白色ワセリン、グリセリン、ポリエチレングリコールおよびプロピレングリコールが包含される。透過増強剤を導入することもできる。例えば、FinninおよびMorganによるJ.Pharm.Sci、88(10)、955〜958(1999年10月)参照。
【0110】
局所投与の他の手段には、電気穿孔法、イオン導入法、音波泳動法、音泳動法および微細針または無針(例えば、Powderject(商標)、Bioject(商標)など)注射による送達が包含される。
【0111】
局所投与のための製剤は、即時および/または変更放出であるように製剤することができる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出およびプログラム放出が包含される。
【0112】
本発明の化合物はまた、鼻腔内または吸入により、典型的には乾燥粉末の形態(単独で、混合物として、例えば、ラクトースとの乾燥ブレンドで、または混合成分粒子として、例えば、ホスファチジルコリンなどのリン脂質と混合して)で、乾燥粉末吸入器から、エアロゾルスプレーとして、加圧容器、ポンプ、スプレー、噴霧器(好ましくは、微細な霧を生じさせるために電磁流体力学を使用する噴霧器)またはネブライザーから、1,1,1,2−テトラフルオロエタンまたは1,1,1,2,3,3,3−ヘプタフルオロプロパンなどの適切な噴射剤を使用して、もしくは使用せずに、または点鼻薬として投与することができる。鼻腔内使用では、粉末は、生体接着剤、例えば、キトサンまたはシクロデキストリンを含んでもよい。
【0113】
加圧容器、ポンプ、スプレー、噴霧器またはネブライザーは、例えば、エタノール、エタノール水溶液または活性剤の分散、可溶化もしくはその放出の延長のために適している別の薬剤、溶媒としての噴射剤およびトリオレイン酸ソルビタン、オレイン酸もしくはオリゴ乳酸などの任意選択の界面活性剤を含む本発明の化合物の溶液または懸濁液を含有する。
【0114】
乾燥粉末または懸濁液製剤で使用する前に、薬物生成物を、吸入により送達するために適したサイズ(典型的には5ミクロン未満)まで超微粉砕する。これは、スパイラルジェット粉砕、流動床ジェット粉砕、ナノ粒子を形成するための臨界液体処理、高圧均一化または噴霧乾燥などの任意の適切な粉砕方法により達成することができる。
【0115】
吸入器または注入器で使用するためのカプセル(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース製)、ブリスターおよびカートリッジを、本発明の化合物、ラクトースまたはデンプンなどの適切な粉末基剤およびl−ロイシン、マンニトールまたはステアリン酸マグネシウムなどの性能改良剤の粉末混合物を含有するように製剤することができる。ラクトースは、無水であってよいか、または一水和物の形態であってよいが、後者が好ましい。他の適切な賦形剤には、デキストラン、グルコース、マルトース、ソルビトール、キシリトール、フルクトース、スクロースおよびトレハロースが包含される。
【0116】
微細な霧を発生させるために電磁流体力学を使用する噴霧器で使用するために適している液剤は、動作1回当たり本発明の化合物1μgから20mgを含有してよく、その動作体積は、1μlから100μlまで変動してよい。典型的な製剤は、式Iの化合物、プロピレングリコール、無菌水、エタノールおよび塩化ナトリウムを含んでよい。プロピレングリコールの代わりに使用することができる別の溶媒には、グリセロールおよびポリエチレングリコールが包含される。
【0117】
メントールおよびレボメントールなどの適切な香料またはサッカリンもしくはサッカリンナトリウムなどの甘味料を、吸入/鼻腔内投与を意図されている本発明の製剤に加えることができる。
【0118】
吸入/鼻腔内投与のための製剤は、例えば、PGLAを使用して、即時および/または変更放出であるように製剤することができる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出およびプログラム放出が包含される。
【0119】
乾燥粉末吸入器およびエアロゾルの場合、投与単位は、計測量を送達するバルブ手段により決定される。本発明による単位は典型的には、式(I)の化合物0.001mgから10mgを含有する計測量または「パフ」を投与するように設計される。全1日用量は典型的には、0.001mgから40mgの範囲であり、これを単回用量で、またはより通常は、1日を通して分けた用量として投与することができる。
【0120】
本発明の化合物は、直腸または膣で、例えば、坐剤、ペッサリまたは浣腸剤の形態で投与することができる。カカオバターは、慣用的な坐剤基剤であるが、様々な代替物を適切に使用することができる。
【0121】
直腸/膣投与のための製剤を、即時および/または変更放出であるように製剤することができる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出およびプログラム放出が包含される。
【0122】
本発明の化合物はまた、眼または耳に、典型的には等張性pH調節無菌食塩水中の超微粉砕された懸濁液または溶液の液滴の形態で直接投与することもできる。眼および耳投与に適している他の製剤には、軟膏、ゲル、生分解性(例えば、吸収性ゲルスポンジ、コラーゲン)および非生分解性(例えば、シリコーン)インプラント、ウェハ、レンズならびに粒子またはニオソームもしくはリポソームなどの小胞系が包含される。架橋ポリアクリル酸、ポリビニルアルコール、ヒアルロン酸、セルロース系ポリマー、例えば、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロースもしくはメチルセルロースまたはヘテロ多糖ポリマー、例えば、ゲランゴムなどのポリマーを、塩化ベンザルコニウムなどの防腐剤と共に導入することができる。このような製剤はまた、イオン泳動法により送達することができる。眼/耳投与のための製剤は、即時および/または変更放出であるように製剤することができる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出またはプログラム放出が包含される。
【0123】
本発明による式(I)の化合物は、鼻、吸入および局所投与に特に適している。
【0124】
前記の投与方法のいずれかで使用するために、本発明の化合物を、シクロデキストリンおよび適切なその誘導体などの溶解性高分子成分またはポリエチレングリコール−含有ポリマーと組み合わせて、その溶解性、溶解速度、矯味、生物学的利用率および/または安定性を改良することができる。
【0125】
例えば、薬物−シクロデキストリン複合体は通常、多くの剤形および投与経路に有用であることが判明している。包接複合体と非包接複合体の両方を使用することができる。薬物との直接的な複合化の代わりに、シクロデキストリンを補助的添加剤、即ち、担体、希釈剤または可溶化剤として使用することができる。これらの目的のために最も一般的に使用されるのは、アルファ−、ベータ−およびガンマ−シクロデキストリンであり、この例は、国際特許出願WO91/11172、WO94/02518およびWO98/55148で見ることができる。
【0126】
例えば、特定の疾患または状態を治療する目的で、活性化合物の組合せを投与することが望ましい場合、そのうちの少なくとも1種が本発明による化合物を含有する2種以上の医薬組成物を簡便に、それらの組成物を同時投与するために適しているキットの形態に組み合わせることができることも、本発明の範囲内である。
【0127】
したがって、本発明のキットは、そのうちの少なくとも1種が本発明による式(I)の化合物を含有する2種以上の別々の医薬組成物と、容器、別々のボトルまたは別々のフォイルパケットなどの前記の組成物を別々に保持するための手段とを含む。このようなキットの例は、錠剤、カプセルなどを包装するために使用される通常のブリスターパックである。
【0128】
本発明のキットは、別の剤形、例えば経口と非経口を投与するために、別々の組成物を別々の投与間隔で投与するために、または別々の組成物を相互に用量決定するために特に適している。服薬遵守を補助するために、キットは典型的には、投与指示書を含み、いわゆる記憶補助体と共に提供され得る。
【0129】
ヒト患者に投与するためには、本発明の化合物の全1日用量は典型的には、勿論、投与方式に応じて0.001mgから5000mgの範囲、好ましくは0.01mgから1000mgの範囲である。例えば、経口投与または静脈内、筋肉内、関節内もしくは関節周囲投与は、0.01mgから1000mg、好ましくは0.01mgから100mgの全1日用量を必要とし得る。全1日用量を単回または分割用量で投与することができ、医師の裁量で、本明細書に示されている典型的な範囲外に該当してもよい。
【0130】
これらの投与は、約60kgから70kgの体重を有する平均的なヒト対象に基づく。医師であれば、乳児および高齢者などのこの範囲外に体重が該当する対象での用量を容易に決定することができるであろう。疑問を回避するために、「治療」に関する本明細書での言及は、治癒的、緩和的および予防的治療に関する言及を包含する。
【0131】
式(I)の化合物は、グルココルチコイド受容体と相互作用することができ、したがって、下記でさらに記載するような幅広い治療用途を有する。それというのも、哺乳動物全ての生理においてグルココルチコイド受容体は必須の役割を果たすためである。
【0132】
したがって、本発明は、グルココルチコイド受容体が関与している疾患、障害および状態の治療または予防で使用するための式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物に関する。本発明はさらに、グルココルチコイド受容体が関与している疾患、障害および状態を治療するための医薬品を製造するための式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物の使用に関する。本発明はまたさらに、ヒトを包含する哺乳動物をグルココルチコイド受容体アゴニストで治療する方法に関し、この方法は、前記哺乳動物を有効量の式(I)の化合物で、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物で治療することを包含する。
【0133】
このような疾患、障害および状態の例には、湿疹、乾癬、皮膚炎、心因性掻痒症および超過敏反応などの皮膚疾患;鼻炎、副鼻腔炎、喘息、鼻ポリープ、慢性閉塞性肺疾患(COPD)および線維症などの鼻、喉および肺の炎症状態;炎症性腸疾患、クローン病および潰瘍性大腸炎などの腸の炎症性疾患;関節リウマチなどの自己免疫疾患;多発性硬化症および播種性エリテマトーデス;非感染性炎症(結膜炎)などの眼状態が包含される。化合物はまた、癌(例えば、膠腫および前立腺癌)、後天性免疫不全症候群、変形性関節症、敗血症性ショック、移植片拒絶、肺気腫(特に、COPD患者)、虚血後病変、肺高血圧、急性呼吸窮迫症候群、冠動脈血管形成術後の再狭窄の予防、スティーヴェンズ−ジョンソン症候群、HELLP症候群(重症子癇前症の異型)、肺炎、慢性活動性肝炎、血液学的障害、腎疾患および急性脊髄損傷に適用し得る。
【0134】
より特には、本発明による化合物は、湿疹、乾癬、皮膚炎、心因性掻痒症および超過敏反応などの皮膚疾患;鼻炎、副鼻腔炎、喘息、鼻ポリープ、慢性閉塞性肺疾患(COPD)および線維症などの鼻、喉および肺の炎症状態;炎症性腸疾患、クローン病および潰瘍性大腸炎などの腸の炎症性疾患;ならびに関節リウマチなどの自己免疫疾患;ならびに結膜炎などの眼状態を治療するために有用である。
【0135】
より特に、本発明はまた、
あらゆるタイプ、病因または病原の皮膚疾患、詳細には、湿疹、乾癬、アレルギー性皮膚炎、神経皮膚炎、心因性掻痒症および超過敏反応;
非感染性眼炎症(結膜炎)などの眼状態;
あらゆるタイプ、病因または病原の季節性アレルギー性鼻炎もしくは通年性アレルギー性鼻炎または副鼻腔炎、詳細には、化膿性または非化膿性副鼻腔炎、急性または慢性副鼻腔炎および篩骨蜂巣炎、前頭洞炎、上顎洞炎または蝶形骨洞炎からなる群から選択されるメンバーである副鼻腔炎;
あらゆるタイプ、病因または病原の喘息、詳細には、アトピー性喘息、非アトピー性喘息、アレルギー性喘息、アトピー性気管支IgE媒介喘息、気管支喘息、本態性喘息、真性喘息、病態生理学的障害に起因する内因性喘息、環境因子に起因する外因性喘息、未知または不顕性原因の本態性喘息、非アトピー性喘息、気管支炎性喘息、肺気腫性喘息、運動誘発喘息、アレルゲン誘発喘息、冷気誘発喘息、職業性喘息、細菌、真菌、原虫またはウイルス感染に起因する感染性喘息、非アレルギー性喘息、初発喘息、喘鳴乳児症候群および細気管支炎からなる群から選択されるメンバーである喘息、
あらゆるタイプ、病因または病原の閉塞性または炎症性気道疾患、詳細には、慢性好酸球性肺炎、慢性閉塞性肺疾患(COPD)、COPDを随伴する、または随伴しない慢性気管支炎、肺気腫または呼吸窮迫を包含するCOPD、不可逆性進行性気道閉塞を特徴とするCOPD、成人呼吸窮迫症候群(ARDS)、他の薬物療法に起因する気道過反応性の再燃および肺高血圧症を随伴する気道疾患からなる群から選択されるメンバーである閉塞性または炎症性気道疾患、
あらゆるタイプ、病因または病原の鼻ポリープ;
あらゆるタイプ、病因または病原の線維症、詳細には、炎症性気道疾患を随伴する肺線維症;
あらゆるタイプ、病因または病原の腸の炎症性疾患、詳細には、潰瘍性大腸炎およびクローン病;
あらゆるタイプ、病因または病原の自己免疫疾患、詳細には、関節リウマチ、多発性硬化症および播種性エリテマトーデス
からなる群から選択される疾患、障害および状態を治療する際に使用するための式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物に関する。
【0136】
なおより特には、本発明による化合物は、喘息、COPD、アレルギー性鼻炎、鼻ポリープ、クローン病、湿疹および乾癬を治療するためにより特には有用である。
【0137】
本発明の他の実施形態では、本発明の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物はまた、患者に同時投与して、これらに限られないが、(i)気管支収縮、(ii)炎症、(iii)アレルギー、(iv)組織破壊、(v)息切れ、咳などの徴候および症状を包含する病態生理学的関連疾患プロセスの治療などのいくつかの特に望ましい治療最終結果を得るための1種または複数の追加的な治療剤との組合せとして使用することができる。第2またはそれ以上の追加的な治療剤はまた、式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物、または当分野で知られている1種または複数のグルココルチコイド受容体アゴニストであってもよい。より典型的には、第2またはそれ以上の治療剤は、異なる群の治療剤から選択される。
【0138】
本明細書で使用される場合、本発明の化合物および1種または複数の他の治療剤に関する「同時投与」、「同時投与される」および「〜と組み合わせて」との用語は、下記を意味し、下記に関し、さらに、下記を包含することとする:
各成分を実質的に同時に患者に放出する単一剤形に、各成分を一緒に製剤する場合であって、式(I)の化合物および治療剤のこのような組合せを、治療を必要とする患者に同時に投与すること、
患者によって実質的に同時に摂取されて、前記各成分が実質的に同時に前記患者に放出される別々の剤形に、各成分を相互に別々に製剤する場合であって、式(I)の化合物および治療剤のこのような組合せを、治療を必要とする患者に実質的に同時に投与すること、
患者によって、各投与間に有意の時間的間隔を伴う連続する時間に摂取されて、前記各成分が実質的に異なる時間に前記患者に放出される別々の剤形に、各成分を相互に別々に製剤する場合であって、式(I)の化合物および治療剤のこのような組合せを、治療を必要とする患者に連続して投与すること、および
各成分を制御して放出する単一剤形に、各成分を一緒に製剤し、その上、それらを患者が同じか異なる時点で同時に、連続しておよび/または重複して投与する場合であって、式(I)の化合物および治療剤のこのような組合せを、治療を必要とする患者に連続して投与すること
(ここで、各部を、同じか異なる経路により投与することができる)。
【0139】
本発明の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物と組み合わせて使用することができる他の治療剤の適切な例には、決してこれらには限られないが、
(a)5−リポキシゲナーゼ(5−LO)阻害剤または5−リポキシゲナーゼ活性化タンパク質(FLAP)アンタゴニスト、
(b)LTB4、LTC4、LTD4およびLTE4のアンタゴニストを包含するロイコトリエンアンタゴニスト(LTRA)、
(c)ロイコトリエンC4シンターゼの阻害剤、
(d)H1、H3およびH4アンタゴニストを包含するヒスタミン受容体アンタゴニスト、
(e)うっ血除去用途のためのα1−およびα2−アドレノセプターアゴニスト血管収縮交感神経様作動薬、
(f)PDE阻害剤、例えば、PDE3、PDE4およびPDE5阻害剤、
(g)テオフィリン、
(h)クロモグリク酸ナトリウム、
(i)非選択的および選択的COX−1またはCOX−2阻害剤の両方であるCOX阻害剤(NSAID)、
(j)プロスタグランジン受容体アンタゴニストおよびhPGDSなどのプロスタグランジンシンターゼの阻害剤、
(k)ムスカリン様M3受容体アンタゴニストまたは抗コリン作動薬、
(l)β2−アドレノセプターアゴニスト、
(m)例えば、IgE、IL3、IL4、IL9、IL10、IL13、IL17A、GMCSFおよびその受容体などの内生炎症促進性実体に対して活性なモノクローナル抗体、
(n)抗腫瘍壊死因子(抗TNF−α)剤、
(o)VLA−4アンタゴニストを包含する接着分子阻害剤、
(p)キニン−B1−およびB2−受容体アンタゴニスト、
(q)IgE経路の阻害剤およびシクロスポリンを包含する免疫抑制剤、
(r)例えば、MMP9およびMMP12などのマトリックスメタロプロテアーゼ(MMP)の阻害剤、
(s)タキキニンNK1、NK2およびNK3受容体アンタゴニスト、
(t)エラスターゼ阻害剤、特に、好中球エラスターゼ阻害剤などのプロテアーゼ阻害剤、
(u)アデノシンA2a受容体アゴニストおよびA2bアンタゴニスト、
(v)ウロキナーゼの阻害剤、
(w)D2アゴニストなどのドーパミン受容体に作用する化合物、
(x)IKK阻害剤などのNFκβ経路の調節剤、
(y)p38MAPキナーゼ、PI3キナーゼ、JAKキナーゼ、sykキナーゼ、EGFR、MK−2、fynキナーゼまたはITKなどのサイトカインシグナル伝達経路の調節剤、
(z)粘液溶解薬または鎮咳薬として分類することができる薬剤、
(aa)例えば、マコリド(macolide)類似体およびPI3KδまたはAKT1、2、3の阻害剤などの吸入コルチコステロイドに対する応答を増強または再増感する薬剤、
(bb)気道でコロニー形成し得る微生物に対して有効な抗生物質および抗ウイルス剤、
(cc)HDAC活性化剤、
(dd)CXCR1、CXCR2およびCXCR3アンタゴニスト、
(ee)インテグリンアンタゴニスト、
(ff)ケモカインおよびケモカイン受容体アンタゴニスト、
(gg)上皮性ナトリウムチャネル(ENaC)遮断剤または上皮性ナトリウムチャネル(ENaC)阻害剤、
(hh)CRACイオンチャネル遮断剤またはCRAC阻害剤、
(ii)P2Y2アゴニストおよび他のヌクレオチド受容体アゴニスト、
(jj)P2X7アンタゴニスト、
(kk)VAP1の阻害剤、
(ll)トロンボキサンの阻害剤、
(mm)ナイアシン、および
(nn)VLAM、ICAMおよびELAMを包含する接着因子
が包含される。
【0140】
本発明では、式(I)の化合物と、
例えば、イプラトロピウム塩、即ち、臭化物、チオトロピウム塩、即ち、臭化物、オキシトロピウム塩、即ち、臭化物、トロスピウム塩、アクリジニウム塩、ペレンゼピンおよびテレンゼピンを包含するムスカリン様M3受容体アゴニストまたは抗コリン作動薬、
例えば、エフェドリン、アドレナリン、イソプレナリン、メタプロテレノール、フェニレフリン、フェニルプロパノールアミン、ピルブテロール、レプロテロール、リミテロール、イソエタリン、トロブテロール、カルモテロール、アルブテロール、テルブタリン、バムブテロール、フェノテロール、サルブタモール、ツロブテロール ホルモテロール、サルメテロールならびにWO05/080313、WO05/080324、WO05/092840およびWO2007/010356に記載されているアゴニストを包含するβ2−アドレノセプターアゴニスト;
PDE4阻害剤、詳細には、吸入PDE4阻害剤、
テオフィリン、
H1およびH3アンタゴニスト、例えば、ロラタジンおよびメタピリレンを包含するヒスタミン受容体アンタゴニストまたは
アデノシンA2a受容体アゴニスト、例えば、WO01/94368に記載されているもの
との組合せが好ましい。
【0141】
好ましい態様では、本発明の化合物は、β2−アドレノセプターアゴニストおよび抗コリン作動薬から選択される他の治療剤と組み合わせることができる。他の好ましい態様には、本発明による化合物と、β2−アドレノセプターアゴニストおよび抗コリン作動薬との3種組合せが包含される。
【0142】
下記の非限定的な実施例により、本発明を説明する:
【図面の簡単な説明】
【0143】
【図1】実施例1のDSCサーモグラムである。
【図2】実施例1のPXRDパターンである。
【図3】実施例2のDSCサーモグラムである。
【図4】実施例2のPXRDパターンである。
【図5】実施例5のDSCサーモグラムである。
【図6】実施例5のPXRDパターンである。
【0144】
プロトコル
下記の全ての実施例で、下記の実験条件を使用した:
【0145】
示差走査熱分析(DSC)
示差走査熱分析を、TA Instrument Q1000 DSCを使用し、リッドを備えたアルミニウムパンで行った。試料約3mgを、1分当たり20℃で、試料に応じて10℃から250℃または10℃から300℃または20℃から300℃の範囲にわたって、窒素ガスパージを伴って加熱した。
【0146】
粉末X線回折方法(PXRD)
自動試料チェンジャー、シータ−シータゴニオメーター、自動ビーム広がりスリットおよびPSD Vantec−1検出器を備えたBruker−AXS Ltd.D4粉末X線回折計を使用して、粉末X線回折パターンを決定した。低バックグラウンドキャビティシリコンウェハに試料量をマウントすることにより、試料を分析のために調製した。試料を回転させ、その間、40kV/30mAで運転されるX線管を用いて、銅K−アルファ1X線(波長=1.5406Å)を照射した。2°から55°の2シータ範囲にわたって0.018°ステップ当たり0.2秒カウントに設定された連続モードで運転するゴニオメーターを用いて、分析を行った。
【実施例】
【0147】
調製1
(6α,11β,16α)−6,9−ジフルオロ−11,16,17,21−テトラヒドロキシプレグナ−1,4−ジエン−3,20−ジオン
【0148】
【化23】
(4bR,6bS,9aR,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a,8,8−テトラメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン(10.3g、22.76mmol、市販)を48%ホウフッ化水素酸水溶液(100mL)に懸濁させ、生じた懸濁液を周囲温度、窒素雰囲気下で7時間撹拌した。次いで、懸濁液を水(200mL)で希釈し、濾過し、固体を水(500mL)で洗浄した。固体ケークをメタノール(200mL)に懸濁させ、真空濃縮した。生じた固体をtert−ブチル−メチルエーテル(150mL)に懸濁させ、濾過し、tert−ブチルメチルエーテル(200mL)で洗浄すると、表題化合物が白色の固体、収率95%、8.9gとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.82 (s, 3H), 1.46 (s,
3H), 1.31-1.51 (m, 3H), 1.77-1.87 (m, 1H), 2.08-2.33 (m, 3H), 2.37-2.46 (m,
1H), 4.08 (d, 1H), 4.09-4.16 (m, 1H), 4.48 (d, 1H), 4.63 (br, 1H), 4.75 (dd,
1H), 5.35 (d, 1H), 5.51-5.69 (m, 1H), 6.08 (s, 1H), 6.26 (dd, 1H), 7.24 (dd,
1H) ppm.
LRMS (ESI): m/z 411 [M-H]-
【0149】
調製2
4−ベンジルベンズアルデヒド
【0150】
【化24】
臭化ベンジル(41g、240mmol)、4−ホルミルベンゼンボロン酸(28g、186.7mmol)、パラジウムテトラキストリフェニルホスフィン(7.9g、6.84mmol)および炭酸カリウム(84.7g、613mmol)をテトラヒドロフラン(620mL)中で合わせ、80℃で窒素下に8時間加熱した。生じた懸濁液を周囲温度に冷却し、一晩撹拌した。反応混合物を10%クエン酸(50mL)に注ぎ、酢酸エチル(3×50mL)で抽出した。合わせた有機抽出物をブライン(100mL)で洗浄し、乾燥させ(硫酸マグネシウム)、溶媒を真空除去した。生じたオイルをカラムクロマトグラフィーによりシリカゲルで、酢酸エチル:ヘプタン(体積で0:1から1:5へ変化)で溶離して精製すると、表題化合物が無色のオイル、収率83%、30.45gとして得られた。
1H NMR
(400 MHz, CDCl3) δ: 4.06 (s, 2H), 7.38-7.17
(m, 7H), 7.83-7.79 (m, 2H), 9.97 (s, 1H) ppm.
LRMS (ESI): m/z 197 [M+H]+
【0151】
調製3
4−{[3−(メチルチオ)フェニル]チオ}ベンズアルデヒド
【0152】
【化25】
Rumpf,P.、Bull.soc.chim.(1940)、7、pp.632〜4に示されている通りに調製された3−(メチルチオ)ベンゼンチオール(19.9g、127.3mmol)のアセトニトリル(60mL)溶液を4−フルオロベンズアルデヒド(13.4mL、127mmol)で、続いて、炭酸カリウム(19.4g、140mmol)で処理した。周囲温度で18時間撹拌した後に、懸濁液を水(200mL)で希釈し、酢酸エチル(3×300mL)で抽出した。合わせた有機抽出物をブライン(2×100mL)で洗浄し、乾燥させ(硫酸マグネシウム)、溶媒を真空除去すると、無色のオイルが得られた。粗製のオイルをフラッシュカラムクロマトグラフィーによりシリカゲルで、ヘプタン:酢酸エチル(体積で1:0から9:1へ変化)で溶離して精製すると、表題化合物が無色のオイル、収率33%、10.8gとして得られた。
1H NMR
(400 MHz, CDCl3) δ: 2.48 (s, 3H), 7.25-7.34
(m, 5H), 7.38 (m, 1H), 7.73-7.75 (d,2H) ppm.
LRMS (API): m/z 261 [M+H]+
【0153】
調製4
4−{[(4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド
【0154】
【化26】
4−ブロモメチルベンズアルデヒド(0.3g、1.5mmol)および4−ヒドロキシチオフェノール(0.2g、1.5mmol)の1,4−ジオキサン(10mL)溶液を脱ガスし、トリエチルアミン(0.44mL、3.11mmol)で処理した。1日撹拌した後に、混合物を水(20mL)で希釈し、酢酸エチル(2×20mL)で抽出した。合わせた有機抽出物をブラインで洗浄し、乾燥させ(硫酸ナトリウム)、溶媒を真空除去した。粗製物質をフラッシュカラムクロマトグラフィーによりシリカゲルで、ヘプタン:酢酸エチル(体積で9:1から0:1へ変化)で溶離して精製すると、表題化合物が固体、収率62%、220mgとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 4.01 (s, 2H), 6.66 (d,
2H), 7.13 (d, 2H), 7.38 (d, 2H), 7.77 (d, 2H), 9.54 (s, 1H), 9.93 (s, 1H) ppm.
LRMS (ESI): m/z 243 [M-H]-
【0155】
調製5
3−クロロ−4−ヒドロキシフェニルチオシアネート
【0156】
【化27】
ヘキサフルオロイソプロパノール(10mL)中のトリメチルシリルイソチオシアネート(24.5g、187mmol)を氷冷された2−クロロフェノールのヘキサフルオロイソプロパノール(30mL)溶液に滴下添加した。反応混合物を10分間撹拌し、次いで、[ビス(トリフルオロアセトキシ)ヨード]ベンゼン(60.2g、140mmol)を滴下添加し、その間、内部温度を5℃未満に維持した。添加が完了した後に、反応混合物を5〜10℃で4時間撹拌し、次いで、真空濃縮すると、黄色の固体が得られた。これを、ジクロロメタン(100mL)に入れ、セライトパッドで濾過した。濾液を真空濃縮すると、黄色のオイルが得られ、これを、フラッシュカラムクロマトグラフィーによりシリカゲルで、ジクロロメタン:ヘプタン(体積で3:7から0:1へ変化)で溶離して精製すると、生成物が黄色の固体、収率30%、5.2gとして得られた。
1H NMR
(400 MHz, CDCl3) δ: 5.99 (bs, 1H), 7.06-7.10
(m, 1H), 7.40-7.43 (m, 1H), 7.59 (s, 1H).
LRMS (ESI): m/z 186 [M+H]+
【0157】
別法では、3−クロロ−4−ヒドロキシフェニルチオシアネートを下記の通り調製した:
臭素(0.40mL、7.78mmol)の酢酸(0.80mL)溶液を、2−クロロフェノール(1.00g、7.78mmol)およびチオシアン酸ナトリウム(2.27g、28.0mmol)の酢酸(6mL)中の懸濁液に滴下添加した。添加の間、内部温度を16℃から25℃に維持した。反応混合物を周囲温度で1時間撹拌した。次いで、水30mLおよび酢酸エチル30mLを反応混合物に加え、これを、celite(登録商標)パッドで濾過した。層を分離し、水性層を酢酸エチル(2×30mL)で抽出した。合わせた有機層を乾燥させ(硫酸マグネシウム)、真空濃縮すると、オレンジ色の半固体が得られた。これを酢酸エチル50mLに入れ、第2のcelite(登録商標)パッドで濾過すると、暗オレンジ色のオイル1.31gが得られた。この物質をさらに精製することなく、調製6で使用した。
【0158】
調製6
2−クロロ−4−メルカプトフェノール
【0159】
【化28】
水素化アルミニウムリチウム(テトラヒドロフラン中1Mの溶液として、71.0ml、71.0mmol)を氷冷された3−クロロ−4−ヒドロキシフェニルチオシアネート(4.20g、22.6mmol)のテトラヒドロフラン(100mL)中の溶液に窒素下に滴下添加した。反応混合物を撹拌し、室温に5時間にわたって加温した。混合物を5℃に冷却し、さらなるガス発生が観察されなくなるまで、テトラヒドロフラン:水の1:1混合物でクエンチした。次いで、1Nの塩酸水溶液(30mL)を加え、混合物を酢酸エチル(2×70mL)で抽出した。合わせた有機層を乾燥させ(硫酸ナトリウム)、真空濃縮すると、無色の結晶質固体、収率100%、3.60gが得られた。
1H NMR
(400 MHz, CDCl3) δ: 3.42 (s, 1H), 5.67, (bs,
1H), 6.89-6.92 (m, 1H), 7.13-7.16 (m, 1H), 7.33 (s, 1H).
LRMS (ESI): m/z 161 [M+H]+
【0160】
調製7
4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド
【0161】
【化29】
トリエチルアミン(5.0mL、35.9mmol)を2−クロロ−4−メルカプトフェノール(3.60g、22.4mmol)および4−ブロモメチルベンズアルデヒド(3.93g、19.7mmol)のジオキサン(150mL)中の溶液に室温、窒素下で滴下添加した。反応混合物を室温で20時間撹拌した。水(100mL)を反応混合物に加え、次いでこれを、酢酸エチルおよびブライン(それぞれ200mL)に分配した。水性層を酢酸エチル(100mL)で再抽出した。合わせた有機層を硫酸ナトリウム上で乾燥させ、真空濃縮すると、黄色の固体が得られた。これをアセトニトリル(5mL/g)と共に30分間摩砕することにより精製した。濾過の後に、明黄色の固体が純粋な生成物、収率84%、5.27gとして得られた。
1H NMR
(400 MHz, CDCl3) δ: 3.96 (s, 2H), 5.55 (bs,
1H), 6.82-6.86 (m, 1H), 7.02-7.06 (m, 1H), 7.19-7.23 (m, 1H), 7.24-7.28 (m,
1H), 7.71-7.74 (m, 2H), 9.92 (s, 1H).
LRMS (ESI): m/z 279 [M+H]+
【0162】
調製8
3−(メチルチオ)ベンゼンチオール
【0163】
【化30】
ベンゼン−1,3−ジチオール(50g、0.351mmol)の2−メチルテトラヒドロフラン(375mL、0.351mmol)溶液を硫酸ジメチル(33.3mL、0.351mol)で、続いて、2−メチルテトラヒドロフラン(25mL、連続洗浄として使用)で処理した。生じた溶液を0℃から5℃に冷却し、反応混合物の温度を15℃未満に維持しながら水酸化ナトリウム(2Mの水溶液、210.6mL)を、続いて、2−メチルテトラヒドロフラン(25mL、連続洗浄として使用)を滴下添加し、窒素下、50℃に4時間加熱した。周囲温度まで冷却した後に、生じた溶液をtert−ブチルメチルエーテル(500mL)で処理し、相を分離した。有機相を水酸化ナトリウム(2Mの水溶液、250mL)で抽出し、合わせた水性相を10℃に冷却し、塩酸(6Mの水溶液、500mL)を加え、その間、混合物の温度を25℃未満に維持した。生じた溶液をtert−ブチルメチルエーテル(2×250mL)で抽出し、合わせた有機相を水(500mL)で洗浄し、真空濃縮すると、表題化合物が黄色のオイル、収率78%、42.9gとして得られた。この物質は、HPLCにより、Rumpf(Bull.soc.chim.(1940)、7、pp.632〜4)の方法により調製された物質と同一であることが判明した。
【0164】
調製9
ナトリウムヒドロキシ(4−{[3−(メチルチオ)フェニル]チオ}フェニル)メタンスルホネート
【0165】
【化31】
調製8で調製された3−(メチルチオ)ベンゼンチオール(290g、1.856mol)のアセトニトリル(3L)溶液を窒素で1時間パージし、次いで、4−フルオロベンズアルデヒド(196mL、1.856mol)で、続いて、アセトニトリル(150mL、連続洗浄として使用)で処理した。生じた溶液を1,1’,3,3’−テトラメチルグアニジン(256mL、2.04mol)で、続いて、アセトニトリル(150mL、連続洗浄として使用)で処理し、50℃に窒素下に16時間加熱した。生じた溶液を周囲温度に冷却し、酢酸エチル(3L)で希釈し、塩酸(2Mの水溶液、1.5L)および重炭酸ナトリウム(1Mの水溶液、3L)およびブライン(半飽和、1.5L)で洗浄した。生じた溶液を大気圧で蒸留することにより濃縮して2Lの体積にし、アセトニトリル(3L)で希釈した。生じた溶液を大気圧での蒸留により濃縮して2Lの体積にし、アセトニトリル(3L)で希釈し、大気圧での蒸留により濃縮して3Lの最終体積にした。生じた溶液を周囲温度に冷却し、メタ重亜硫酸ナトリウム(377g、1.982mol)の水(3L)溶液で処理した。周囲温度で48時間撹拌した後に、生じた懸濁液を濾過し、集められた固体を水(2×2.5L)およびアセトニトリル(2×2.5L)で洗浄した。固体をアセトニトリル(2L)に懸濁させ、周囲温度で18時間撹拌し、この時間の後に、懸濁液を濾過し、固体をアセトニトリル(2×1L)で洗浄し、50℃で真空乾燥させると、表題化合物が白色の固体、収率55%、368.7gとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 2.41 (s, 3H), 4.97 (d,
1H), 5.90 (d, 1H), 6.98 (m, H), 7.12 (m, 2H), 7.26 (m, 3H), 7.46 (d, 2H) ppm.
【0166】
(実施例1)
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−ベンジルフェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
【0167】
【化32】
(4bR,6bS,9aR,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a,8,8−テトラメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト−[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン(8g、18mmol)および調製2で得られた4−ベンジルベンズアルデヒド(10.4g、53mmol)を、氷冷され撹拌されている砂(80g)のトルエン(80mL)懸濁液に加えた。70%過塩素酸水溶液(4mL、70mmol)を5分にわたって滴下添加し、次いで、溶液を周囲温度で21時間撹拌した。飽和重炭酸ナトリウム溶液(100mL)を反応混合物に、続いて、酢酸エチル(100mL)を加え、溶液を撹拌し、次いで、濾過した。砂を飽和重炭酸ナトリウム溶液(50mL)で、次いで、酢酸エチル(100mL)で洗浄した。水性層を分離し、酢酸エチル(2×50mL)で抽出し、合わせた有機抽出物をブライン(100mL)で洗浄し、乾燥させ(硫酸マグネシウム)、溶媒を真空除去した。生じた粘稠性の黄色のオイル(10g)をDCM:酢酸エチル(体積で9:1)で希釈すると、固体物質の沈殿が生じ、これを濾過により集めた。乾燥の後に、物質を酢酸エチル:ヘプタン(体積で4:1)から再結晶化させると、白色の結晶物質、収率44%、4.36gが得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.82 (s, 3H), 1.45 (s,
3H), 1.43-1.50 (m, 1H), 1.62-1.71 (m, 3H), 1.97-2.03 (m, 1H), 2.16-2.30 (m,
2H), 2.51-2.65 (m, 1H), 3.87 (s, 2H), 4.12-4.18 (m, 1H), 4.15 (dd, 1H), 4.46
(dd, 1H), 4.91 (d, 1H), 5.02 (t, 1H), 5.40 (s, 1H), 5.45 (d, 1H), 5.51-5.69 (m,
1H), 6.09 (s, 1H), 6.25 (dd, 1H), 7.10-7.24 (m, 8H), 7.29-7.32 (m, 2H) ppm.
LRMS (ESI): m/z 591 [M+H]+
【0168】
1.960mgの試料を、示差走査熱分析(DSC)により、20℃/分で、10℃から300℃の傾斜で分析した。得られたDSCサーモグラムを、図1に、平坦な基線および250.6℃での溶融に対応する鋭い吸熱と共に示す。
【0169】
上記のプロセスにより製造される結晶形はまた、図2の対応する粉末X線回折パターンに示されている特徴を有する。主な特徴的なピークは、8.0、16.0、16.9、24.0および24.2度2θ±0.1度2θにあり、さらに、表1に示す。
【0170】
【表1】
【0171】
(実施例2)
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルチオ)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
【0172】
【化33】
調製1で得られた(6α,11β,16α)−6,9−ジフルオロ−11,16,17,21−テトラヒドロキシプレグナ−1,4−ジエン−3,20−ジオン(6.6g、16mmol)および調製3で得られた4−{[3−(メチルチオ)フェニル]チオ}ベンズアルデヒド(4.5g、17.28mmol)の1,4−ジオキサン(70mL)中の懸濁液を硫酸マグネシウム(10g、83.1mmol)で処理した。懸濁液を水浴中で冷却し、トリフルオロメタンスルホン酸(7.5mL、82mmol)を加えた。室温で24時間撹拌した後に、混合物を水(200mL)で希釈し、酢酸エチル(2×200mL)で抽出した。合わせた有機抽出物を水(200mL)およびブライン(2×150mL)で洗浄し、乾燥させ(硫酸マグネシウム)、溶媒を真空除去した。粗製のゴムをフラッシュカラムクロマトグラフィーによりシリカゲルで、ヘプタン:tert−ブチルメチルエーテル(体積で4:1から1:0へ変化)で、次いで、ヘプタン:酢酸エチル(体積で3:7から0:1へ変化)で溶離して精製すると、黄色の泡が得られた。さらに、フラッシュカラムクロマトグラフィーによりシリカゲルで、ペンタン:酢酸エチル(体積で4:1から2:3へ変化)で溶離すると、黄色の固体が得られた。この固体を2−ブタノンから、次いで、アセトニトリルから再結晶化させると、表題化合物が白色の固体、収率18%、1.93gとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.85 (s, 3H),
1.43-1.53 (m, 1H), 1.48 (s, 3H), 1.65-1.71 (m, 3H), 1.97-2.06 (m, 1H),
2.14-2.31 (m, 2H), 2.41 (s, 3H), 2.55-2.67 (m, 1H), 4.16-4.22 (m, 2H),
4.49-4.55 (dd, 1H), 4.95 (d, 1H), 5.09 (t, 1H), 5.48 (s, 1H), 5.50-5.51 (m,
1H), 5.54-5.79 (m, 1H), 6.10 (s, 1H), 6.26-6.29 (m, 1H), 7.05-7.07 (m, 1H),
7.18-7.20 (m, 2H), 7.23-7.26 (m, 1H), 7.27-7.29 (m, 1H), 7.31-7.33 (d, 2H),
7.41-7.43 (d, 2H) ppm.
LRMS (ESI): m/z 655 [M+H]+
【0173】
別法では、表題化合物を下記の通り調製した:
調製1で得られた(6α,11β,16α)−6,9−ジフルオロ−11,16,17,21−テトラヒドロキシプレグナ−1,4−ジエン−3,20−ジオン(434g、1.050mol)および硫酸マグネシウム(417g、3.47mol)のアセトニトリル(4.34L)中の懸濁液を窒素下に18時間撹拌した。調製9で得られたナトリウムヒドロキシ(4−{[3−(メチルチオ)フェニル]チオ}フェニル)メタンスルホネート(460g、1.26mol)を加え、生じた懸濁液をトリフルオロメタンスルホン酸(443mL、5.01mmol)で処理し、その間、混合物の温度を24℃未満に維持した。周囲温度で75分間撹拌した後に、混合物を酢酸n−ブチル(4.4L)および水(4.4L)で処理し、さらに酢酸n−ブチル(400mL)を連続洗浄として使用して分離器に移した。相を分離し、有機相を水(4.4L)で洗浄し、炭酸水素ナトリウム(10%水溶液、2×2.2L)で洗浄し、水(2.2L)で洗浄した。生じた懸濁液を濾過し、濾液を真空濃縮して、溶媒4.26Lを除去した。残渣を35℃に冷却し、2−ブタノン(4L)で処理し、周囲温度に冷却し、18時間撹拌した。生じた懸濁液を濾過し、固体を2−ブタノン(2×2L)で洗浄した。固体をエタノール(2−ブタノンで変性、8L)に懸濁させ、10分間還流加熱し、周囲温度に冷却した。生じた懸濁液を濾過し、固体をエタノール(2−ブタノンで変性、2×2L)およびアセトニトリル(900mL)で洗浄した。固体をアセトニトリル(2.6L)に懸濁させ、還流加熱し、アセトニトリル(1.3L)で処理し、大気圧での蒸留により濃縮して、溶媒2.7Lを除去した。生じた懸濁液をアセトニトリル(1.75L)で処理し、還流加熱し、周囲温度に冷却した。生じた懸濁液を濾過し、固体をアセトニトリル(2×450mL)で洗浄し、40℃で真空乾燥させると、表題化合物が白色の固体、収率37%、307.3gとして得られた。こうして得られた化合物は、先行する方法により得られた化合物と同一であった。
【0174】
本明細書中、上記された第1の方法に従って得られた生成物のうちの2.847mgの試料を、示差走査熱分析(DSC)により、20℃/分で10から300℃の傾斜で分析した。第1の吸熱事象は、114.5℃で観察され、これは、不純物におそらく対応している。溶融が184.8℃で観察される。対応するサーモグラムを図3に示す。
【0175】
上記の方法により製造された結晶形はまた、図4の対応する粉末X線回折パターンに示されている特徴を有する。主な特徴的なピークは、10.0、16.5、17.0、20.0および25.5度2θ±0.1度2θにあり、さらに、下記の表2に示されている。
【0176】
【表2】
【0177】
(実施例3)
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−8−(4−{[(4−ヒドロキシフェニル)チオ]メチル}フェニル)−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
【0178】
【化34】
調製1で得られた(6α,11β,16α)−6,9−ジフルオロ−11,16,17,21−テトラヒドロキシプレグナ−1,4−ジエン−3,20−ジオン(99.8mg、0.24mmol)および調製4で得られた4−{[(4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド(148mg、0.61mmol)の1,4−ジオキサン(3mL)中の懸濁液を乾燥硫酸マグネシウム(430mg、3.57mmol)およびトリフルオロメタンスルホン酸(43μL、0.49mmol)で処理した。1日撹拌した後に、反応混合物を濾過し、集められた固体を酢酸エチル(20mL)で洗浄した。合わせた濾液を水(100mL)に注ぎ、酢酸エチル(3×20mL)で抽出した。合わせた有機抽出物をブライン(50mL)で洗浄し、乾燥させ(硫酸ナトリウム)、溶媒を真空除去した。粗製物質をフラッシュカラムクロマトグラフィーによりシリカゲルで、ヘプタン:酢酸エチル(体積で3:1から0:1へ変化)で溶離して精製すると、表題化合物が白色の固体、収率14%、21mgとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.85 (s, 3H),1.48 (s,
3H), 1.5 (m, 1H), 1.62-1.73 (m, 3H), 2.01-2.05 (m, 1H), 2.17-2.24 (m, 1H),
2.25-2.31 (m, 1H), 2.55-2.70 (m, 1H), 4.01 (s, 2H), 4.15-4.22 (m, 2H), 4.50
(dd, 1H), 4.94 (d, 1H), 5.05 (t, 1H), 5.43 (s, 2H), 5.48 (m, 1H), 5.55-5.71 (m,
1H), 6.11 (s, 1H), 6.28 (dd, 1H), 6.66 (d, 2H), 7.13 (d, 2H), 7.24 (m, 3H),
7.31 (d, 2H), 9.48 (s, 1H) ppm.
LRMS (ESI): m/z 639 [M+H]+
【0179】
(実施例4)
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルスルフィニル)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
【0180】
【化35】
実施例2で調製された(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルチオ)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2Hナフト[2’,1’:4,5]−インデノ[1,2−d][1,3]ジオキソール−2−オン(979mg、1.50mmol)をヘキサフルオロイソプロパノール(6mL、57.0mmol)に懸濁させ、氷浴中で冷却し、その後、過酸化水素(水中30重量%、203mg、1.79mmol)を滴下添加した。反応を周囲温度で90分間撹拌した。次いで、反応混合物を25%w/vの亜硫酸ナトリウム水溶液(30mL)に注いだ。水性層を酢酸エチル(3×50mL)で抽出し、合わせた有機抽出物を乾燥させ(硫酸ナトリウム)、溶媒を真空除去した。粗製物質をフラッシュカラムクロマトグラフィーによりシリカゲルで、ジクロロメタン:メタノール(体積で9:1)で溶離して精製すると、表題化合物が白色の固体、収率61%、610mgとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.87 (s, 3H), 1.50 (s, 3H),
1.47-1.59 (m, 1H), 1.65-1.78 (m, 3H), 2.01-2.09 (m, 1H), 2.17-2.34 (m, 2H),
2.58-2.71 (m, 1H), 2.74 (m, 3H), 4.18-4.26 (m, 2H), 4.52-4.58 (m, 1H), 4.98 (d,
1H), 5.10-5.13 (m, 1H), 5.52 (s, 1H), 5.53-5.54 (m, 1H), 5.58-5.78 (m, 1H),
6.12 (s, 1H), 6.31 (dd, 1H), 7.27 (d, 1H), 7.41-7.48 (m, 5H), 7.54- 7.64 (m,
3H) ppm.
LRMS (ESI): m/z 671 [M+H]+
【0181】
(実施例5)
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}フェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
【0182】
【化36】
トリフルオロメタンスルホン酸(4.76mL、53.8mmol)を、調製7で得られた4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド(5.25g、18.8mmol)、調製1で得られた(6α,11β,16α)−6,9−ジフルオロ−11,16,17,21−テトラヒドロキシプレグナ−1,4−ジエン−3,20−ジオン(7.40g、17.9mmol)および硫酸マグネシウム(6.82g、53.8mmol)のアセトニトリル(80mL)中の氷冷懸濁液に窒素下に滴下添加した。反応混合物を5〜10℃で4時間撹拌し、次いで、氷水(100mL)に注ぎ、酢酸エチル(3×150mL)で抽出した。合わせた有機層を乾燥させ(硫酸ナトリウム)、真空濃縮すると、茶色の泡が得られた。これを、フラッシュカラムクロマトグラフィー(シリカゲル)により、ジクロロメタン:メタノール(体積で100:0から90:10へ変化)で溶離して精製すると、オレンジ色の泡が得られた。これを、フラッシュカラムクロマトグラフィー(シリカゲル)により、ジクロロメタン:メタノール(体積で100:0から90:10へ変化)で溶離してさらに精製すると、黄色の泡(6.0g)が得られた。これを、温酢酸エチルに入れ、播種して沈殿を誘発した。固体を濾過し、乾燥させると、淡黄色の固体1.18gが得られた。母液を蒸発させ、酢酸エチルに再び入れ、播種して第2の収量1.46gを生じさせた。これを3回繰り返すと、2.40gの最終収量が得られた。3つのバッチ全てを合わせ、温酢酸エチル中で摩砕し、濾過し、乾燥させると、淡黄色の固体3.5g、収率27%が得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.86 (s, 3H), 1.50 (s,
3H), 1.48-1.57 (m, 1H), 1.62-1.77 (m, 3H), 2.01-2.08 (m, 1H), 2.18-2.33 (m,
2H), 2.55-2.72 (m, 1H), 4.09 (s, 2H), 4.16-4.24 (m, 2H), 4.48-4.55 (m, 1H),
4.93-4.97 (m, 1H), 5.45 (s, 1H), 5.50-5.53 (m, 1H), 5.55-5.61 (m, 1/2H),
5.68-5.74 (m, 1/2H), 6.13 (s, 1H), 6.27-6.32 (m, 1H), 6.85-6.88 (s, 1H),
7.10-7.13 (m, 1H), 7.25-7.36 (m, 1H), 10.28 (bs, 1H).
LRMS (ESI): m/z 673 [M]
【0183】
2.769mgの試料を、示差走査熱分析(DSC)により、20℃/分、10℃から300℃の傾斜で分析した。得られたDSCサーモグラムを、図5に、平坦な基線および254℃にピークがある溶融に対応する鋭い吸熱と共に示す。
【0184】
上記のプロセスにより製造される結晶形はまた、図6の対応する粉末X線回折パターンに示されている特徴を有する。主な特徴的なピークは、14.1、16.6、20.0、21.8および25.2度2θ±0.1度2θにあり、さらに、表3に示す。
【0185】
【表3】
【0186】
(実施例6)
in vitro薬理学的活性
式(I)の化合物の薬理学的活性を、グルココルチコイドアゴニスト活性のin vitroアッセイで、ならびにin vivoでの抗炎症活性を予測し得るヒト血液および単離白血球TNF−α放出アッセイで評価した。
【0187】
グルココルチコイド受容体(GR)アゴニスト効果を、MMTV−ルシフェラーゼレポーター構成を安定導入されたヒト軟骨肉腫細胞系SW1353で決定した。SW1353は元々、ヒトGRを発現し、これは、グルココルチコイドアゴニストと結合すると、MMTVプロモーター内のグルココルチコイド応答要素を活性化させて、ルシフェラーゼ遺伝子の発現を駆動する。
【0188】
凍結されたSW1353細胞を、ピルビン酸ナトリウムまたはフェノールレッドを伴わず、2mMのL−グルタミン、1μg/mlのインスリン、2mg/mlのラクトアルブミンヒドロシレート(hydrosylate)および0.5μg/mlのアスコルベートを補足されているDMEM培地中で復活させた。細胞を細胞約5000/ウェル(35μl/ウェル)で384ウェルの透明床組織培養処理プレートに播種した。ステロイド用量−応答希釈をステロイド希釈剤(2.5%(v/v)のDMSOおよび0.05%(v/v)のプルロニック界面活性剤を含有するPBS)中で調製し、5μlを各ウェルに加えた。ステロイド希釈剤を用いて、体積を各ウェル当たり50μlにした。陽性対照ウェルは、1μMのデキサメタゾンを含有した。プレートを37℃、空気/5%CO2雰囲気中、加湿インキュベーター中で約18時間インキュベーションし、その後、Britelite試薬(10μl;Perkin−Elmer)を各ウェルに加えた。各プレートを暗所で2分間インキュベーションし、LJL Biosystems Analystルミノメーターを使用して、発光を定量した。試験化合物でのデータ(デキサメタゾン陽性対照に対するパーセンテージとして表示)を使用して、用量応答曲線を構成し、それから、EC50値を推定した。下記のデータが得られた。
【0189】
【表4】
【0190】
また、in vitroでのヒト白血球に対する化合物の抗炎症活性を、リポ多糖類(LPS)刺激されたヒト全血(WB)および単離ヒト末梢単核細胞(PBMC)からの腫瘍壊死因子−α(TNF−α)放出の阻害を決定することにより評価した。
【0191】
健康で投薬されていない提供者からの末梢静脈血を、エチレンジアミン四酢酸(EDTA)を抗凝血薬として使用して集めた。PBMC調製のために、血液試料を、無菌のリン酸緩衝溶液で1:1で希釈し、次いで、ACCUSPIN(商標)System−Histopaque(登録商標)−1077管(Sigma−Aldrich、St Louis、MO)を使用して分離し、400gで35分間遠心分離した。軟膜細胞をPBS中へ除去し、200gで10分間遠心分離し、PBMCアッセイ緩衝液(ハンクス液、0.28%[w/v]の4−[2−ヒドロキシエチル]−1−ピペラジンエタンスルホン酸[HEPES]、0.01%[w/v]の低内毒素ウシ血清アルブミン[BSA]に再懸濁させた。白血球分画を行い、1ml当たり1×106リンパ球まで、PBMCアッセイ緩衝液中でPBMC希釈した。
【0192】
試験化合物をDMSOに溶かし、PBMCアッセイ緩衝液(最終DMSO濃度1%)中で希釈して、適切な濃度範囲、例えば、0.001nMから10000nMをカバーした。試験化合物溶液の試料または媒体(20μl)を96ウェルの組織培養処理プレート(Corning)に加え、PBMC(160μl)またはWB(160μl)を各ウェルに加えた。アッセイ混合物を37℃、5%CO2を補足された空気雰囲気を含有する加湿インキュベーター中で1時間インキュベーションし、その後、LPS(PBMCでは100ng/mlまたはWBでは1μg/mlを20μl)を加えた。プレートをインキュベーターにさらに18時間戻し、次いで、遠心分離し、その後、上澄みの試料を回収した。試料中のTNF−αを、酵素結合免疫吸着アッセイ(ELISA)(Invitrogenキット no CHC−1754;Invitrogen Carlsbad、CA)を使用し、製造者指示に従って決定した。用量応答曲線を構成し、これから、IC50値を算出した。下記のデータが得られた。
【0193】
【表5】
【0194】
(実施例7)
in vivo薬理学的活性
薬理学的活性は、下記に記載されているものなどの肺炎症のin vivoモデルで評価することができる。この手順の主な目的は、気管を介して肺に直接投与した場合の式(I)の化合物の抗炎症活性を決定することであった。
【0195】
試験化合物を、0.5%(w/v)のTween−80を含有するリン酸緩衝溶液中に溶かすか、微細な懸濁液として調製して、一連の用量レベルを得た。雄のCD Sprague−Dawleyラット(300〜450g)をn=6の研究群に無作為化し、次いで、5%のイソフルランを含む麻酔チャンバー中、O23l/分で短時間麻酔を掛けた。試験化合物製剤または用量媒体(100μl)の1種を、Hamiltonシリンジを使用して麻酔を掛けられたラットそれぞれの気管に直接注射した。次いで、動物を麻酔剤から回復させた。研究設計に応じて、動物に、単回用量の化合物を与えるか、または1日1回、連続4日間処置した。投与の4時間後に(または繰り返し投与研究での最終投与の4時間後に)、超音波ネブライザーならびに最大1回呼吸量および速度(5ml、160ストローク/分)に設定されている小動物齧歯類人工呼吸器に接続されているチャンバー(300×300×450mm)に、ラットを入れた。37℃に予め加温された生理食塩水に溶かされた1mg/mlのLPS(Sigma−Aldrich、L2630)10mlをチャンバーへ噴霧した。15分後に、換気機およびネブライザーを止め、動物をチャンバー内に保持して、霧をさらに15分間呼吸させ、その後、ホームケージに戻した。
【0196】
LPS処置終了の4時間後に、動物を、1ml/kgのPentoject IPを用いて最終的な麻酔に掛けた。気管にカニューレを挿入し、肺を、2.6mMのEDTAを含有するPBS4×2.5mlで洗浄し、洗浄液を集めた。細気管支肺胞洗浄液(BAL)1mlを、40%ウシ血清アルブミン(BSA)125μlに加え、細胞カウントを、Advia 120血液学系(Siemens)を使用して決定した。繰り返し投与研究では、グルココルチコイドアゴニスト曝露に応答して減少することが知られていて、グルココルチコイドアゴニストの全身作用を評価するために使用されているので、体重ならびに副腎および胸腺の重量もまた決定した。一部の繰り返し投与実験では、最終血液試料を各ラットから集め、血清および血漿を調製し、血清中のコルチコステロンおよび血漿中のACTHの濃度を、全身グルココルチコイドアゴニスト作用の追加的なマーカーとして決定した。一部の研究では、知られているグルココルチコイドアゴニストであるプロピオン酸フルチカゾンを、陽性対称としてのラットの別の群に投与した。
【0197】
LPS−誘発肺好中球およびグルココルチコイドアゴニスト全身作用の各マーカーの阻害に関して、別々の用量応答曲線を構成した。半数最大有効量(ED50)値を、近似曲線から推定した。また、グルココルチコイドアゴニスト活性の全身マーカーに対する作用に関するED50値を肺好中球増加の阻害に関するED50で割って、治療指数(TI)の値を決定した。
【0198】
こうして、上記のアッセイで試験された本発明による式(I)の化合物は、下記の表に列挙されている通り、単回用量の投与後に肺好中球増加の阻害に活性を示すことが判明した。
【0199】
【表6】
【技術分野】
【0001】
本発明は、新規なグルココルチコイド受容体アゴニストならびに薬学的に許容できるその塩または前記グルココルチコイド受容体アゴニストもしくは塩の薬学的に許容できる溶媒和物、それを調製する方法および中間体に関する。本発明はまた、これらの化合物を含有する医薬組成物、1種または複数の他の治療剤とのその組合せ、さらに、いくつかの炎症性およびアレルギー性疾患、障害および状態を治療するためのその使用に関する。
【背景技術】
【0002】
グルココルチコイド受容体アゴニストは、幅広い炎症性および免疫障害を治療するために不可欠である強力な抗炎症薬である。治療に組み込まれた最初の化合物は、天然のコルチコステロイドヒドロコルチゾンに由来した。核分子の初めの構造的修飾は、ミネラルコルチコイド受容体を上回るグルココルチコイドに対する選択性の向上を目的としていた。構造と活性との関係がさらによく理解されたことに基づき、次世代の化合物は、より高い受容体親和性を、したがってより高い効果を示した。局所塗布されるグルココルチコイドでは、薬物標的化により、例えば、コルチコステロイド製剤の吸入または皮膚塗布により、さらなる進歩が達成された。最近の開発は、代謝不安定性な官能基を活性分子に組み込んで、局所塗布後の全身曝露を最小化することにより、副作用を可能な限り低減することに焦点を当てていた。治療標的組織に対する高い親和性は、標的への効果および作用時間を高め、体循環への再分布を遅くすることにより標的外への全身作用を制限する特性と認識された。
【0003】
グルココルチコイド受容体アゴニストは、炎症性およびアレルギー性状態、例えば、喘息、閉塞性気道疾患、鼻炎、炎症性腸疾患、乾癬、湿疹などの管理で使用される。既に市販されているグルココルチコイドの例には、以下のものが包含される。
【0004】
【化1】
【0005】
これらの化合物は、幅広い細胞種類において、グルココルチコイド受容体に結合し、それを活性化する。活性化された受容体は、核中のグルココルチコイド応答要素に結合して、鍵となる調節機能を有する遺伝子の転写を活性化または阻害する。詳細には、これらの化合物は、好酸球および好中球などの炎症性白血球が炎症部位へと動員されるのを妨げ、また、白血球および組織細胞からの炎症性媒介物質の形成および放出を阻害することにより、炎症性疾患において有効である。
【0006】
最初のコルチコステロイドが市販されて以来、例えば、下式のWO05/028495に記載されている化合物など、様々な構造を有する数多くのコルチコステロイドが提案されている:
【0007】
【化2】
[式中、
【0008】
【化3】
は、二重結合であってよく、R1およびR2は、Fであってよく、R3は、OHであってよく、R4はHであってよく、R5は、フェニルにより置換されていてもよいC5〜10アリールであってよく、ここで、前記フェニルは、アルキル、アルコキシまたはハロゲンにより置換されていてもよい]。
【発明の概要】
【発明が解決しようとする課題】
【0009】
しかしながら、例えば、効果、治療指数、薬物動態、薬物/薬物相互作用および/または副作用に関して、最も適した薬理学的プロファイルを有するであろう改良されたグルココルチコイド受容体アゴニストが未だに必要とされている。本明細書では、式(I)の化合物:
【0010】
【化4】
[式中、R1は、
【0011】
【化5】
からなる群から選択され、ここで、*は、フェニル環の炭素へのR1の結合点を示している]
または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物を提供する。
【課題を解決するための手段】
【0012】
第1の実施形態では、式(Ia)のグルココルチコイド受容体アゴニストのサブグループ、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物が好ましい
【0013】
【化6】
[式中、R1は、
【0014】
【化7】
からなる群から選択され、ここで、*は、フェニル環の炭素へのR1の結合点を示している]。
【発明を実施するための形態】
【0015】
したがって本発明は、下記の好ましい化合物に及ぶ:
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−ベンジルフェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルチオ)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a、10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−8−(4−{[(4−ヒドロキシフェニル)チオ]メチル}フェニル)−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a、10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルスルフィニル)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a、10b,11,12−ドデカヒドロ−2H−ナフト−[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;および
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}フェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b、9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン。
【0016】
本発明によるさらなる好ましいグルココルチコイド受容体アゴニストは、(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルチオ)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オンである。
【0017】
本発明による他の好ましいグルココルチコイド受容体アゴニストは、(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−8−(4−{[(4−ヒドロキシフェニル)チオ]メチル}フェニル)−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オンである。
【0018】
本発明によるまだ他の好ましいグルココルチコイド受容体アゴニストは、(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}フェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オンである。
【0019】
本発明による式(I)の化合物は、様々な方法で、下記に例示されている方法などによる慣用の手順を使用して調製することができ、ここで、R1は、別段に述べられていない限り、式(I)の化合物に関して既に定義された通りである。しかし、当業者であれば、他の経路も同等に実施可能であり得ることを理解するであろう。
【0020】
式(I)の化合物は、下記の通りスキーム1に従って調製することができる:
【0021】
【化8】
スキーム1では、式(I)の化合物は、式(II)の化合物を式(IIa)の適切なアルデヒドと、または式(IIb)の適切なアルデヒド同等物と反応させることにより調製することができる。簡便には、過剰のアルデヒドもしくはアルデヒド同等物または化学量論的量のアルデヒドもしくはアルデヒド同等物を、アルキルスルホン酸(例えば、トリフルオロメタンスルホン酸)などの酸の存在下に、適切な溶媒(例えば、アセトニトリル、エチレングリコールジメチルエーテル、1,4−ジオキサンまたはジクロロメタン)の存在下に、任意選択で乾燥剤(硫酸マグネシウムまたは硫酸ナトリウムなど)の存在下に、周囲温度または低温で使用することにより、反応を行う。
【0022】
式(II)の化合物は、文献(例えば、Fried,J.、US3,177,231(1965))で知られている方法により式(III)の化合物を反応させるか、またはホウフッ化水素酸水溶液で周囲温度または40℃などの高温で処理することにより調製することができる。
【0023】
別法では、R1が式:
【0024】
【化9】
である式(I)の化合物はまた、R1が式:
【0025】
【化10】
である式(I)の化合物を適切な酸化剤と反応させることにより調製することができる。簡便には、やや過剰の過酸化水素などの酸化剤を、適切な溶媒(例えば、ヘキサフルオロイソプロパノール)の存在下、周囲温度または低温で使用することにより、この反応を行う(Tet.Lett.、39、3141〜3144、J.P.Begueら、1998参照)。
【0026】
式(I)の化合物はまた、式(III)の化合物を式(IIa)の適切なアルデヒドと、または式(IIb)の適切なアルデヒド同等物と反応させることにより調製することができる。簡便には、過剰のアルデヒドもしくはアルデヒド同等物または化学量論的量のアルデヒドもしくはアルデヒド同等物を、酸(例えば、トリフルオロメタンスルホン酸または過塩素酸など)の存在下、適切な溶媒(例えば、アセトニトリルまたは1,4−ジオキサンなど)の存在下、任意選択で添加剤(例えば、砂など)の存在下、周囲温度または低温で使用することにより、反応を行う。
【0027】
式(I)の化合物はまた、R2およびR3がホルミルと定義される式(IV)の化合物を式(IIa)の適切なアルデヒドと、または式(IIb)の適切なアルデヒド同等物と、文献(例えばWO2005/028495)で知られている方法により反応させることにより調製することができる。
【0028】
R2およびR3がホルミルと定義される式(IV)の化合物は、式(III)の化合物を、文献(例えばWO2005/028495)で知られている方法により反応させることにより調製することができる。
【0029】
式(III)の化合物は、市販されている。
【0030】
本発明では、「適切なアルデヒド」は、式(IIa)のアルデヒドを意味する:
【0031】
【化11】
[式中、R1は、式(I)の化合物で既に定義された通りである]。言い換えると、本発明による適切なアルデヒドは、
式(Va)の4−ベンジルベンズアルデヒド
【0032】
【化12】
式(VIa)の4−{[3−(メチルチオ)フェニル]チオ}ベンズアルデヒド
【0033】
【化13】
式(VIIa)の4−{[(4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド
【0034】
【化14】
式(VIIIa)の4−{[3−(メチルスルフィニル)フェニル]チオ}ベンズアルデヒド:
【0035】
【化15】
または
式(IXa)の4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド
【0036】
【化16】
からなる群から選択される。
【0037】
別法では、「適切なアルデヒド同等物」は、式(IIb)の化合物を意味し:
【0038】
【化17】
これはまた、「ビスルフィト付加生成物」とも称され、ここで、R1は、式(I)の化合物で既に定義された通りである。言い換えると、本発明による適切なアルデヒド同等物は、
式(Vb)のナトリウムヒドロキシル(ベンジルフェニル)メタンスルホネート
【0039】
【化18】
【0040】
(VIb)のナトリウムヒドロキシ(4−{[3−(メチルチオ)フェニル]チオ}フェニル)メタンスルホネート
【0041】
【化19】
式(VIIb)のナトリウムヒドロキシル(4−{[(4−ヒドロキシフェニル)チオ]メチル}フェニル)メタンスルホネート
【0042】
【化20】
式(VIIIb)のナトリウムヒドロキシ(4−{[4−(メチルスルフィニル)フェニル]チオ}フェニル)メタンスルホネート
【0043】
【化21】
または
式(IXb)のナトリウム(4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}フェニル)(ヒドロキシ)メタンスルホネート
【0044】
【化22】
からなる群から選択される。
【0045】
上記で述べられたアルデヒドまたはアルデヒド同等物は、市販されているか、または当業者によく知られている慣用の手順に従って容易に調製することができる。
【0046】
本明細書で上記された式(I)の化合物の調製方法のいくつかのステップでは、反応することが望ましくない反応する可能性のある官能基を保護し、最終的には前記保護基を分離する必要のあることがある。このような場合、任意の相容性な保護基を使用することができる。詳細には、T.W.GREENE(Protective Groups in Organic Synthesis、A.Wiley−Interscience Publication、1981)またはP.J.Kocienski(Protecting groups、Georg Thieme Verlag、1994)により記載された方法などの保護および脱保護の方法を使用することができる。
【0047】
上記の反応および前述の方法で使用される新規な出発物質の調製は全て、慣用的であり、その実施または調製のための適切な試薬および反応条件、さらに、所望の生成物を単離するための手順は、文献手順ならびにその例および調製を参照して、当業者によく知られている。
【0048】
また、式(I)の化合物、さらに、それを調製するための中間体は、例えば、結晶化またはクロマトグラフィーなどの様々なよく知られている方法により精製することができる。
【0049】
式(I)の化合物の薬学的に許容できる塩には、その塩基塩が包含される。適切な塩基塩は、非毒性の塩を形成する塩基から形成される。例には、アルミニウム、アルギニン、ベンザチン、カルシウム、コリン、ジエチルアミン、ジオラミン、グリシン、リシン、マグネシウム、メグルミン、オラミン、カリウム、ナトリウム、トロメタミンおよび亜鉛塩が包含される。
【0050】
式(I)の化合物の薬学的に許容できる塩にはまた、その酸塩が包含されることもある。また、酸および塩基の半塩、例えば、半硫酸塩および半カルシウム塩を形成することもできる。
【0051】
適切な塩についての総説に関しては、StahlおよびWermuthによる「Handbook of Pharmaceutical Salts:Properties,Selection,and Use」(Wiley−VCH、2002)参照。
【0052】
式(I)の化合物の薬学的に許容できる塩は、3種の方法のうちの1種または複数により調製することができる:
(i)式(I)の化合物を所望の酸または塩基と反応させることによる方法、
(ii)所望の酸または塩基を使用して、式(I)の化合物の適切な前駆体から酸または塩基不安定性保護基を除去するか、または適切な環式前駆体、例えば、ラクトンまたはラクタムを開環する方法、または
(iii)適切な酸もしくは塩基との反応により、または適切なイオン交換カラムを用いて、式(I)の化合物の1種の塩を他の塩に変換する方法。
【0053】
3種の反応を全て典型的には、溶液で実施する。生じた塩を沈殿させて、濾過により集めるか、または溶媒を蒸発させることにより回収することができる。生じた塩の電離度は、完全な電離から、ほぼ非電離まで変動してよい。
【0054】
本発明の化合物は、完全な非晶質から完全な結晶までの範囲の固体状態の連続で存在し得る。「非晶質」との用語は、その物質が、分子レベルで長距離秩序を欠いていて、温度に応じて固体または液体の物理的特性を示し得る状態を指す。典型的には、このような物質は、特有のX線回折パターンを示さず、固体の特性を示しながらも、液体としてより形式的には記載される。加熱すると、固体特性から液体特性への変化が生じ、これは、状態変化、典型的には二次変化により特徴づけられる(「ガラス遷移」)。「結晶」との用語は、その物質が、分子レベルで規則的に配列している内部構造を有し、規定のピークを有する特有のX線回折パターンを示す固相を指す。このような物質はまた、十分に加熱されると、液体の特性を示すが、固体から液体への変化は、相変化、典型的には一次変化により特徴づけられる(「融点」)。
【0055】
本発明の化合物およびその塩はまた、非溶媒和形態および溶媒和形態で存在し得る。「溶媒和物」との用語は本明細書では、本発明の化合物および1個または複数の薬学的に許容できる溶媒分子、例えば、エタノールを含む分子複合体を記載するために使用されている。「水和物」との用語は、前記溶媒が水である場合に使用される。
【0056】
有機水和物に関して現在認められている分類体系は、孤立サイト、チャネルまたは金属イオン配位水和物を定義する分類体系である。K.R.Morrisによる「Polymorphism in Pharmaceutical Solids」(H.G.Brittain編、Marcel Dekker、1995)参照。孤立サイト水和物は、その水分子が、有機分子の介在により、相互の直接的な接触から孤立している水和物である。チャネル水和物では、水分子は、格子チャネル内に存在し、他の水分子に隣接している。金属イオン配位水和物では、水分子は、金属イオンに結合している。
【0057】
溶媒または水が密に結合していると、複合体は、湿度とは独立に、十分に定義される化学量論を有するはずである。しかしながら、チャネル溶媒和物および吸湿性化合物においてのように、溶媒または水の結合が弱い場合、水/溶媒含分は、湿度および乾燥状態に左右される。このようなケースでは、非化学量論が標準となる。
【0058】
また、薬物および少なくとも1種の他の成分が、化学量論的量または非化学量論的量で存在している多成分複合体(塩および溶媒和物以外)も、本発明の範囲内に包含される。このタイプの複合体には、包接化合物(薬物−ホスト包接複合体)および共結晶が包含される。後者は典型的には、非共有結合相互作用を介して相互に結合している中性分子成分同士の結晶複合体と定義されるが、中性分子と塩との複合体であってもよい。溶融結晶化、溶媒からの再結晶化または成分同士の物理的粉砕により、共結晶を調製することができる(O.AlmarssonおよびM.J.ZaworotkoによるChem Commun、17、1889〜1896(2004)参照)。多成分複合体の一般的な総説に関しては、HaleblianによるJ.Pharm.Sci、64(8)、1269〜1288(1975年8月)参照。
【0059】
本発明の化合物はまた、適切な条件に掛けると、中間状態(中間相または液晶)でも存在し得る。中間状態は、真の結晶状態と真の液体状態(溶融または溶液)との中間である。温度変化の結果として生じる液晶性は、「サーモトロピック」と記載され、水または他の溶媒などの第2の成分を加えると生じる液晶性は、「リオトロピック」と記載される。リオトロピック中間相を形成する可能性のある化合物は、「両親媒性」と記載され、イオン性(−COO−Na+、−COO−K+または−SO3−Na+など)または非イオン性(−N−N+(CH3)3など)極性ヘッド基を持つ分子からなる。さらなる情報に関しては、N.H.HartshorneおよびA.Stuartによる「Crystals and the Polarizing Microscope」、4th Edition(Edward Arnold、1970)参照。
【0060】
後記では、本発明の化合物に関する言及は全て、その塩、溶媒和物、多成分複合体および液晶ならびにその塩の溶媒和物、多成分複合体および液晶に対する言及を包含する。
【0061】
本発明の化合物には、後記で定義される通りのその全ての多形および晶癖、そのプロドラッグおよび異性体(光学、幾何および互変異性体を包含)ならびに同位体標識された式(I)の化合物を包含する、上記で定義された通りの式(I)の化合物が包含される。
【0062】
前記のように、本発明の化合物のいわゆる「プロドラッグ」もまた、本発明の範囲内である。それ自体は薬理活性をほとんど有さないか、有さないことのある式(I)の化合物のある種の誘導体は、体内または体上に投与されると、例えば、加水分解により変換されて、所望の活性を有する式(I)の化合物になり得る。このような誘導体が、「プロドラッグ」と称される。プロドラッグの使用に関するさらなる情報は、「Pro−drugs as Novel Delivery Systems」、Vol.14、ACS Symposium Series(T.HiguchiおよびW.Stella)および「Bioreversible Carriers in Drug Design」、Pergamon Press、1987(E.B.Roche編、American Pharmaceutical Association)で見ることができる。
【0063】
例えば、式(I)の化合物中に存在する適切な官能基を、例えば、H.Bundgaardによる「Design of Prodrugs」(Elsevier、1985)に記載されている通りに、当業者に「プロ部分」として知られているある種の部分で置き換えることにより、本発明によるプロドラッグを製造することができる。
【0064】
本発明によるプロドラッグのいくつかの例には、式(I)の化合物がアルコール官能基(−OH)を含有する場合には、そのエーテルが包含され、例えば、式(I)の化合物のアルコール官能基の水素が(C1〜C6)アルカノイルオキシメチルにより置き換えられている化合物が包含される。
【0065】
上述の例による代替基のさらなる例および他のプロドラッグタイプの例は、上述の参考文献で見ることができる。
【0066】
さらに、ある種の式(I)の化合物はそれ自体、他の式(I)の化合物のプロドラッグとして作用し得る。
【0067】
また、式(I)の化合物の代謝産物、即ち、薬物が投与されるとin vivoで形成される化合物も、本発明の範囲内に包含される。本発明による代謝産物のいくつかの例には:
(i)式(I)の化合物がメチル基を含有する場合、そのヒドロキシメチル誘導体(−CH3→−CH2OH)、
(ii)式(I)の化合物がフェニル部分を含有する場合、そのフェノール誘導体(−Ph→−PhOH)、および
(iii)式(I)の化合物がスルフィドを含有する場合、そのスルホキシド誘導体(−SPh→−S(O)Ph)
が包含される。
【0068】
1個または複数の不斉炭素原子を含有する式(I)の化合物は、2種以上の立体異性体として存在し得る。構造異性体が、低エネルギー障壁を介して相互転換可能である場合、互変異性(「tautomerism」)が生じ得る。これは、例えばイミノ、ケトまたはオキシム基を含有する式(I)の化合物では、プロトン互変異性の形態を、または芳香族部分を含有する化合物ではいわゆる原子価互変異性の形態をとり得る。したがって、単一化合物が、1種を超える異性で存在し得る。1種を超える異性を示す化合物およびそれらの1種または複数の混合物を包含する式Iの化合物の立体異性体、幾何異性体および互変異性形態全てが、本発明の範囲内に包含される。また、対イオンが光学活性である酸付加塩もしくは塩基塩、例えば、d−乳酸塩もしくはl−リシンまたはラセミ体、例えばdl−酒石酸塩もしくはdl−アルギニンが包含される。
【0069】
個々の鏡像異性体を調製/単離するための慣用の技術には、適切な光学的に純粋な前駆体からのキラル合成または例えば、キラル高圧液体クロマトグラフィー(HPLC)を使用してのラセミ体(または塩もしくは誘導体のラセミ体)の分割が包含される。
【0070】
別法では、ラセミ体(またはラセミ前駆体)を適切な光学的に活性な化合物、例えば、アルコールと、または式(I)の化合物が酸性または塩基性部分を含有する場合には、1−フェニルエチルアミンまたは酒石酸などの塩基または酸と反応させることができる。生じたジアステレオ異性体の混合物を、クロマトグラフィーおよび/または分別結晶化により分離し、そのジアステレオ異性体の一方または両方を、当業者によく知られている手段により対応する純粋な1種または複数の鏡像異性体に変換することもできる。
【0071】
クロマトグラフィー、典型的にはHPLCを不斉樹脂上で、炭化水素、典型的には、イソプロパノール0から50体積%、典型的には2%から20体積%およびアルキルアミン0から5体積%、典型的にはジエチルアミン0.1体積%を含有するヘプタンまたはヘキサンからなる移動相と共に使用すると、本発明のキラル化合物(およびそのキラル前駆体)を鏡像異性的に濃縮された形態で得ることができる。溶離液を濃縮すると、濃縮混合物が得られる。
【0072】
任意のラセミ体が結晶化する場合、2種の異なるタイプの結晶が可能である。第1のタイプは、両方の鏡像異性体を等モル量で含有する結晶の1種の均一な形態が生じる前記のラセミ化合物(真のラセミ化合物)である。第2のタイプは、それぞれ単一の鏡像異性体を含む2種の形態の結晶が等モル量で生じるラセミ混合物または複合体である。
【0073】
ラセミ混合物中に存在する結晶形態の両方が、同一の物理的特性を有する一方で、これらは、真のラセミ化合物に対して異なる物理的特性を有することがある。ラセミ混合物は、当業者に知られている慣用の技術により分離することができる。例えば、E.L.ElielおよびS.H.Wilenによる「Stereochemistry of Organic Compounds」(Wiley、1994)参照。
【0074】
本発明は、1個または複数の原子が、同じ原子番号を有するが、自然で優勢な原子質量または質量数とは異なる原子質量または質量数を有する原子に置き換えられている薬学的に許容できる同位体標識された式(I)の化合物全てを包含する。
【0075】
本発明の化合物中に包含されるのに適している同位体の例には、2Hおよび3Hなどの水素、11C、13Cおよび14Cなどの炭素、36Clなどの塩素、18Fなどのフッ素、123Iおよび125Iなどのヨウ素、13Nおよび15Nなどの窒素、15O、17Oおよび18Oなどの酸素、32Pなどのリンならびに35Sなどの硫黄の同位体が包含される。
【0076】
ある種の同位体標識された式(I)の化合物、例えば、放射性同位体を導入されているものは、薬物および/または基質組織分布研究で有用である。放射性同位体のトリチウム、即ち3Hおよび炭素−14、即ち14Cは、導入の容易さおよび検出の迅速な手段である点において、この目的のために特に有用である。
【0077】
ジュウテリウム、即ち2Hなどの重同位体での置換は、より大きな代謝安定性、例えば、高いin vivo半減期または低い用量要求から生じるある種の治療的利点をもたらし得るので、場合によっては好ましいことがある。
【0078】
11C、18F、15Oおよび13Nなどの陽電子放出同位体での置換は、基質受容体占有率を調べるための陽電子放出断層撮影法(PET)研究において有用であり得る。
【0079】
当業者に知られている慣用の技術により、または前に使用されていた非標識試薬の代わりに適切な同位体標識試薬を使用する添付の実施例および調製に記載のプロセスと同様のプロセスにより、同位体標識された式(I)の化合物を通常は調製することができる。
【0080】
本発明による薬学的に許容できる溶媒和物には、結晶化の溶媒が同位体置換されている、例えば、D2O、d6−アセトン、d6−DMSOであるものが包含される。
【0081】
提示されている適応症を治療するために最も適切な剤形および投与経路を選択するために、溶解性および溶解安定性(pH全体で)、透過性などのその生物薬学的特性に関して、式(I)の化合物を評価すべきである。
【0082】
薬学的使用を意図されている本発明の化合物は、結晶または非晶質生成物として投与することができる。これらは、沈殿、結晶化、凍結乾燥、噴霧乾燥または蒸発乾燥などの方法により、例えば、固体プラグ、粉末またはフィルムとして得ることができる。マイクロ波または高周波乾燥を、この目的のために使用することができる。
【0083】
これらは、単独で、または1種もしくは複数の他の本発明の化合物と組み合わせて、または1種もしくは複数の他の薬物と組み合わせて(またはその任意の組合せとして)投与することができる。通常、これらは、1種または複数の薬学的に許容できる賦形剤と共に製剤として投与される。「賦形剤」との用語は本明細書では、例えば、希釈剤、担体および補助剤などの1種または複数の本発明の化合物以外の任意の成分を記載するために使用される。賦形剤の選択は、特定の投与方法、溶解性および安定性に対する賦形剤の作用ならびに剤形の性質などの要因に大きく左右される。
【0084】
本発明の化合物を送達するために適した医薬組成物およびその調製方法は、当業者には容易に分かるであろう。このような組成物およびその調製方法は、例えば、「Remington’s Pharmaceutical Sciences」、19th Edition(Mack Publishing Company、1995)で見ることができる。
【0085】
本発明の化合物は、経口で投与することができる。経口投与は、化合物が胃腸管に入るような嚥下および/または化合物が口から直接、血流に入る頬、舌もしくは舌下投与を伴ってよい。
【0086】
経口投与に適している製剤には、錠剤などの固体、半固体および液体系;多粒子もしくはナノ粒子、液体または粉末を含有する軟質または硬質カプセル;ロゼンジ(液体充填を包含);チューイング剤;ゲル;急速分散剤形;フィルム;卵形剤(ovule);スプレーならびに頬/粘膜接着パッチが包含される。
【0087】
液体製剤には、懸濁剤、液剤、シロップおよびエリキシルが包含される。このような製剤を、軟質または硬質カプセル(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース製)中の充填剤として使用することもでき、典型的には、担体、例えば、水、エタノール、ポリエチレングリコール、プロピレングリコール、メチルセルロースまたは適切なオイルならびに1種または複数の乳化剤および/または懸濁化剤を含む。液体製剤はまた、固体、例えば、サシェからの再構成により調製することができる。
【0088】
本発明の化合物はまた、LiangおよびChenによる「Expert Opinion in Therapeutic Patents」、11(6)、981〜986(2001)に記載されているものなどの急速溶解、急速崩壊剤形で使用することができる。
【0089】
錠剤剤形では、用量に応じて、薬物は、剤形の1重量%から80重量%、より典型的には剤形の5重量%から60重量%を構成していてよい。薬物の他に、錠剤は通常、崩壊剤を含有する。崩壊剤の例には、デンプングリコール酸ナトリウム、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、メチルセルロース、微結晶性セルロース、低級アルキル置換ヒドロキシプロピルセルロース、デンプン、α化デンプンおよびアルギン酸ナトリウムが包含される。通常、崩壊剤は、剤形の1重量%から25重量%、好ましくは5重量%から20重量%を構成している。
【0090】
結合剤を通常は使用して、錠剤製剤に粘着性を付与する。適切な結合剤には、微結晶性セルロース、ゼラチン、糖、ポリエチレングリコール、天然および合成ゴム、ポリビニルピロリドン、α化デンプン、ヒドロキシプロピルセルロースならびにヒドロキシプロピルメチルセルロースが包含される。錠剤はまた、ラクトース(一水和物、噴霧乾燥一水和物、無水物など)、マンニトール、キシリトール、デキストロース、スクロース、ソルビトール、微結晶性セルロース、デンプンおよび二塩基性リン酸カルシウム二水和物などの希釈剤を含有してよい。
【0091】
錠剤はまた、ラウリル硫酸ナトリウムおよびポリソルベート80などの界面活性剤ならびに二酸化ケイ素およびタルクなどの流動促進剤を含んでもよい。存在する場合には、界面活性剤は、錠剤の0.2重量%から5重量%を構成してよく、流動促進剤は、錠剤の0.2重量%から1重量%を構成してよい。
【0092】
また、錠剤は通常、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、ステアリルフマル酸ナトリウムおよびステアリン酸マグネシウムとラウリル硫酸ナトリウムとの混合物などの滑剤を含有する。滑剤は通常、錠剤の0.25重量%から10重量%、好ましくは0.5重量%から3重量%の量を構成する。
【0093】
他の可能な成分には、抗酸化剤、着色剤、香料、防腐剤および矯味剤が包含される。
【0094】
例示的な錠剤は、薬物約80%まで、結合剤約10重量%から約90重量%、希釈剤約0重量%から約85重量%、崩壊剤約2重量%から約10重量%および滑剤約0.25重量%から約10重量%を含有する。
【0095】
錠剤ブレンドを、直接か、またはローラーにより圧縮して、錠剤を形成することができる。別法では、錠剤ブレンドまたは一部のブレンドを湿潤、乾燥もしくは溶融顆粒化するか、溶融凝固させるか、または押し出し、その後に錠剤化することができる。最終製剤は、1つまたは複数の層を含んでよく、コーティングされているか、またはコーティングされてなくてよい。さらに、カプセル封入されていてよい。
【0096】
錠剤の製剤は、H.LiebermanおよびL.Lachmanによる「Pharmaceutical Dosage Forms:Tablets,Vol.1」(Marcel Dekker、New.York、1980)で検討されている。
【0097】
ヒトまたは動物使用のための摂取可能な口腔フィルムは典型的には、柔軟な水溶性または水膨潤性薄膜剤形であり、これは、迅速に溶解するか、粘膜接着性であってよく、典型的には、式Iの化合物、フィルム形成ポリマー、結合剤、溶媒、湿潤剤、可塑剤、安定剤または乳化剤、粘度調節剤および溶媒を含む。製剤のうちのいくつかの成分は、1つを超える機能を果たすことがある。
【0098】
式(I)の化合物は、水溶性または不溶性であってよい。水溶性化合物は典型的には、溶質1重量%から80重量%、より典型的には20重量%から50重量%を含む。溶解性の低い化合物は、より高い割合の組成、典型的には、溶質88重量%までを含んでよい。別法では、式(I)の化合物は多粒子ビーズの形態であってよい。
【0099】
フィルム形成ポリマーは、天然多糖類、タンパク質または合成親水コロイドからなる群から選択されてよく、典型的には、0.01から99重量%の範囲、より典型的には、30から80重量%の範囲で存在する。
【0100】
他の可能な成分には、抗酸化剤、着色剤、香料および香増強剤、防腐剤、唾液刺激剤、冷却剤、補助溶媒(油を包含する)、緩和剤、増量剤、消泡剤、界面活性剤および矯味剤が包含される。
【0101】
本発明によるフィルムは典型的には、剥離可能なバッキング支持体または紙にコーティングされた薄い水性フィルムを蒸発乾燥させることにより調製される。これは、乾燥オーブンまたはトンネル、典型的には組み合わされたコーティングドライヤーで、または凍結乾燥もしくは真空化により行うことができる。
【0102】
経口投与のための固体製剤を、即時および/または変更放出であるように製剤することもできる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出およびプログラム放出が包含される。
【0103】
本発明の目的に適している変更放出製剤は、米国特許第6,106,864号に記載されている。高エネルギー分散液および浸透性のコーティングされた粒子などの他の適切な放出技術の詳細は、Vermaらによる「Pharmaceutical Technology On−line」、25(2)、1〜14(2001)で見ることができる。調節放出を達成するためにチューインガムを使用することは、WO00/35298に記載されている。
【0104】
本発明の化合物はまた、血流中、筋肉中または内臓に直接投与することもできる。非経口投与に適している手段には、静脈内、動脈内、腹腔内、クモ膜下、心室内、尿管内、胸骨内、頭蓋内、筋肉内、滑液包内および皮下が包含される。非経口投与のための適切なデバイスには、針(微細針を包含する)注射器、無針注射器および点滴技術が包含される。
【0105】
非経口製剤は典型的には、塩、炭水化物および緩衝剤(好ましくはpH3から9に)などの賦形剤を含有してよい水溶液であるが、いくつかの用途では、これらをより適切に、無菌非水溶液として、または無菌の発熱物質不含水などの適切な媒体と共に使用される乾燥形態として製剤することができる。
【0106】
例えば、凍結乾燥による無菌条件下での非経口製剤の調製は、当業者によく知られている標準的な製薬技術を使用して容易に達成することができる。
【0107】
非経口溶液を調製する際に使用される式(I)の化合物の溶解性は、溶解性増強剤を導入するなどの適切な製剤技術を使用することにより高めることができる。
【0108】
非経口投与のための製剤は、即時および/または変更放出であるように製剤することができる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出およびプログラム放出が包含される。したがって本発明の化合物を、懸濁剤として、または活性化合物の変更放出をもたらす移植デポーとして投与するための固体、半固体もしくはチキソトロピー液として製剤することができる。このような製剤の例には、薬物コーティングされたステントならびに薬物負荷されたポリ(dl−乳酸−コグリコール酸)(PGLA)微小球を含む半固体および懸濁液が包含される。
【0109】
本発明の化合物はまた、皮膚または粘膜に局所、皮膚(皮内)または経皮で投与することもできる。この目的のための典型的な製剤には、ゲル、ヒドロゲル、ローション、液剤、クリーム、軟膏、散布剤、包帯、フォーム剤、フィルム剤、皮膚パッチ、ウェハ、インプラント、スポンジ、繊維、帯具およびマイクロエマルションが包含される。リポソームもまた使用することができる。典型的な担体には、アルコール、水、鉱油、流動ワセリン、白色ワセリン、グリセリン、ポリエチレングリコールおよびプロピレングリコールが包含される。透過増強剤を導入することもできる。例えば、FinninおよびMorganによるJ.Pharm.Sci、88(10)、955〜958(1999年10月)参照。
【0110】
局所投与の他の手段には、電気穿孔法、イオン導入法、音波泳動法、音泳動法および微細針または無針(例えば、Powderject(商標)、Bioject(商標)など)注射による送達が包含される。
【0111】
局所投与のための製剤は、即時および/または変更放出であるように製剤することができる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出およびプログラム放出が包含される。
【0112】
本発明の化合物はまた、鼻腔内または吸入により、典型的には乾燥粉末の形態(単独で、混合物として、例えば、ラクトースとの乾燥ブレンドで、または混合成分粒子として、例えば、ホスファチジルコリンなどのリン脂質と混合して)で、乾燥粉末吸入器から、エアロゾルスプレーとして、加圧容器、ポンプ、スプレー、噴霧器(好ましくは、微細な霧を生じさせるために電磁流体力学を使用する噴霧器)またはネブライザーから、1,1,1,2−テトラフルオロエタンまたは1,1,1,2,3,3,3−ヘプタフルオロプロパンなどの適切な噴射剤を使用して、もしくは使用せずに、または点鼻薬として投与することができる。鼻腔内使用では、粉末は、生体接着剤、例えば、キトサンまたはシクロデキストリンを含んでもよい。
【0113】
加圧容器、ポンプ、スプレー、噴霧器またはネブライザーは、例えば、エタノール、エタノール水溶液または活性剤の分散、可溶化もしくはその放出の延長のために適している別の薬剤、溶媒としての噴射剤およびトリオレイン酸ソルビタン、オレイン酸もしくはオリゴ乳酸などの任意選択の界面活性剤を含む本発明の化合物の溶液または懸濁液を含有する。
【0114】
乾燥粉末または懸濁液製剤で使用する前に、薬物生成物を、吸入により送達するために適したサイズ(典型的には5ミクロン未満)まで超微粉砕する。これは、スパイラルジェット粉砕、流動床ジェット粉砕、ナノ粒子を形成するための臨界液体処理、高圧均一化または噴霧乾燥などの任意の適切な粉砕方法により達成することができる。
【0115】
吸入器または注入器で使用するためのカプセル(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース製)、ブリスターおよびカートリッジを、本発明の化合物、ラクトースまたはデンプンなどの適切な粉末基剤およびl−ロイシン、マンニトールまたはステアリン酸マグネシウムなどの性能改良剤の粉末混合物を含有するように製剤することができる。ラクトースは、無水であってよいか、または一水和物の形態であってよいが、後者が好ましい。他の適切な賦形剤には、デキストラン、グルコース、マルトース、ソルビトール、キシリトール、フルクトース、スクロースおよびトレハロースが包含される。
【0116】
微細な霧を発生させるために電磁流体力学を使用する噴霧器で使用するために適している液剤は、動作1回当たり本発明の化合物1μgから20mgを含有してよく、その動作体積は、1μlから100μlまで変動してよい。典型的な製剤は、式Iの化合物、プロピレングリコール、無菌水、エタノールおよび塩化ナトリウムを含んでよい。プロピレングリコールの代わりに使用することができる別の溶媒には、グリセロールおよびポリエチレングリコールが包含される。
【0117】
メントールおよびレボメントールなどの適切な香料またはサッカリンもしくはサッカリンナトリウムなどの甘味料を、吸入/鼻腔内投与を意図されている本発明の製剤に加えることができる。
【0118】
吸入/鼻腔内投与のための製剤は、例えば、PGLAを使用して、即時および/または変更放出であるように製剤することができる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出およびプログラム放出が包含される。
【0119】
乾燥粉末吸入器およびエアロゾルの場合、投与単位は、計測量を送達するバルブ手段により決定される。本発明による単位は典型的には、式(I)の化合物0.001mgから10mgを含有する計測量または「パフ」を投与するように設計される。全1日用量は典型的には、0.001mgから40mgの範囲であり、これを単回用量で、またはより通常は、1日を通して分けた用量として投与することができる。
【0120】
本発明の化合物は、直腸または膣で、例えば、坐剤、ペッサリまたは浣腸剤の形態で投与することができる。カカオバターは、慣用的な坐剤基剤であるが、様々な代替物を適切に使用することができる。
【0121】
直腸/膣投与のための製剤を、即時および/または変更放出であるように製剤することができる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出およびプログラム放出が包含される。
【0122】
本発明の化合物はまた、眼または耳に、典型的には等張性pH調節無菌食塩水中の超微粉砕された懸濁液または溶液の液滴の形態で直接投与することもできる。眼および耳投与に適している他の製剤には、軟膏、ゲル、生分解性(例えば、吸収性ゲルスポンジ、コラーゲン)および非生分解性(例えば、シリコーン)インプラント、ウェハ、レンズならびに粒子またはニオソームもしくはリポソームなどの小胞系が包含される。架橋ポリアクリル酸、ポリビニルアルコール、ヒアルロン酸、セルロース系ポリマー、例えば、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロースもしくはメチルセルロースまたはヘテロ多糖ポリマー、例えば、ゲランゴムなどのポリマーを、塩化ベンザルコニウムなどの防腐剤と共に導入することができる。このような製剤はまた、イオン泳動法により送達することができる。眼/耳投与のための製剤は、即時および/または変更放出であるように製剤することができる。変更放出製剤には、遅延放出、持続放出、パルス放出、調節放出、ターゲット放出またはプログラム放出が包含される。
【0123】
本発明による式(I)の化合物は、鼻、吸入および局所投与に特に適している。
【0124】
前記の投与方法のいずれかで使用するために、本発明の化合物を、シクロデキストリンおよび適切なその誘導体などの溶解性高分子成分またはポリエチレングリコール−含有ポリマーと組み合わせて、その溶解性、溶解速度、矯味、生物学的利用率および/または安定性を改良することができる。
【0125】
例えば、薬物−シクロデキストリン複合体は通常、多くの剤形および投与経路に有用であることが判明している。包接複合体と非包接複合体の両方を使用することができる。薬物との直接的な複合化の代わりに、シクロデキストリンを補助的添加剤、即ち、担体、希釈剤または可溶化剤として使用することができる。これらの目的のために最も一般的に使用されるのは、アルファ−、ベータ−およびガンマ−シクロデキストリンであり、この例は、国際特許出願WO91/11172、WO94/02518およびWO98/55148で見ることができる。
【0126】
例えば、特定の疾患または状態を治療する目的で、活性化合物の組合せを投与することが望ましい場合、そのうちの少なくとも1種が本発明による化合物を含有する2種以上の医薬組成物を簡便に、それらの組成物を同時投与するために適しているキットの形態に組み合わせることができることも、本発明の範囲内である。
【0127】
したがって、本発明のキットは、そのうちの少なくとも1種が本発明による式(I)の化合物を含有する2種以上の別々の医薬組成物と、容器、別々のボトルまたは別々のフォイルパケットなどの前記の組成物を別々に保持するための手段とを含む。このようなキットの例は、錠剤、カプセルなどを包装するために使用される通常のブリスターパックである。
【0128】
本発明のキットは、別の剤形、例えば経口と非経口を投与するために、別々の組成物を別々の投与間隔で投与するために、または別々の組成物を相互に用量決定するために特に適している。服薬遵守を補助するために、キットは典型的には、投与指示書を含み、いわゆる記憶補助体と共に提供され得る。
【0129】
ヒト患者に投与するためには、本発明の化合物の全1日用量は典型的には、勿論、投与方式に応じて0.001mgから5000mgの範囲、好ましくは0.01mgから1000mgの範囲である。例えば、経口投与または静脈内、筋肉内、関節内もしくは関節周囲投与は、0.01mgから1000mg、好ましくは0.01mgから100mgの全1日用量を必要とし得る。全1日用量を単回または分割用量で投与することができ、医師の裁量で、本明細書に示されている典型的な範囲外に該当してもよい。
【0130】
これらの投与は、約60kgから70kgの体重を有する平均的なヒト対象に基づく。医師であれば、乳児および高齢者などのこの範囲外に体重が該当する対象での用量を容易に決定することができるであろう。疑問を回避するために、「治療」に関する本明細書での言及は、治癒的、緩和的および予防的治療に関する言及を包含する。
【0131】
式(I)の化合物は、グルココルチコイド受容体と相互作用することができ、したがって、下記でさらに記載するような幅広い治療用途を有する。それというのも、哺乳動物全ての生理においてグルココルチコイド受容体は必須の役割を果たすためである。
【0132】
したがって、本発明は、グルココルチコイド受容体が関与している疾患、障害および状態の治療または予防で使用するための式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物に関する。本発明はさらに、グルココルチコイド受容体が関与している疾患、障害および状態を治療するための医薬品を製造するための式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物の使用に関する。本発明はまたさらに、ヒトを包含する哺乳動物をグルココルチコイド受容体アゴニストで治療する方法に関し、この方法は、前記哺乳動物を有効量の式(I)の化合物で、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物で治療することを包含する。
【0133】
このような疾患、障害および状態の例には、湿疹、乾癬、皮膚炎、心因性掻痒症および超過敏反応などの皮膚疾患;鼻炎、副鼻腔炎、喘息、鼻ポリープ、慢性閉塞性肺疾患(COPD)および線維症などの鼻、喉および肺の炎症状態;炎症性腸疾患、クローン病および潰瘍性大腸炎などの腸の炎症性疾患;関節リウマチなどの自己免疫疾患;多発性硬化症および播種性エリテマトーデス;非感染性炎症(結膜炎)などの眼状態が包含される。化合物はまた、癌(例えば、膠腫および前立腺癌)、後天性免疫不全症候群、変形性関節症、敗血症性ショック、移植片拒絶、肺気腫(特に、COPD患者)、虚血後病変、肺高血圧、急性呼吸窮迫症候群、冠動脈血管形成術後の再狭窄の予防、スティーヴェンズ−ジョンソン症候群、HELLP症候群(重症子癇前症の異型)、肺炎、慢性活動性肝炎、血液学的障害、腎疾患および急性脊髄損傷に適用し得る。
【0134】
より特には、本発明による化合物は、湿疹、乾癬、皮膚炎、心因性掻痒症および超過敏反応などの皮膚疾患;鼻炎、副鼻腔炎、喘息、鼻ポリープ、慢性閉塞性肺疾患(COPD)および線維症などの鼻、喉および肺の炎症状態;炎症性腸疾患、クローン病および潰瘍性大腸炎などの腸の炎症性疾患;ならびに関節リウマチなどの自己免疫疾患;ならびに結膜炎などの眼状態を治療するために有用である。
【0135】
より特に、本発明はまた、
あらゆるタイプ、病因または病原の皮膚疾患、詳細には、湿疹、乾癬、アレルギー性皮膚炎、神経皮膚炎、心因性掻痒症および超過敏反応;
非感染性眼炎症(結膜炎)などの眼状態;
あらゆるタイプ、病因または病原の季節性アレルギー性鼻炎もしくは通年性アレルギー性鼻炎または副鼻腔炎、詳細には、化膿性または非化膿性副鼻腔炎、急性または慢性副鼻腔炎および篩骨蜂巣炎、前頭洞炎、上顎洞炎または蝶形骨洞炎からなる群から選択されるメンバーである副鼻腔炎;
あらゆるタイプ、病因または病原の喘息、詳細には、アトピー性喘息、非アトピー性喘息、アレルギー性喘息、アトピー性気管支IgE媒介喘息、気管支喘息、本態性喘息、真性喘息、病態生理学的障害に起因する内因性喘息、環境因子に起因する外因性喘息、未知または不顕性原因の本態性喘息、非アトピー性喘息、気管支炎性喘息、肺気腫性喘息、運動誘発喘息、アレルゲン誘発喘息、冷気誘発喘息、職業性喘息、細菌、真菌、原虫またはウイルス感染に起因する感染性喘息、非アレルギー性喘息、初発喘息、喘鳴乳児症候群および細気管支炎からなる群から選択されるメンバーである喘息、
あらゆるタイプ、病因または病原の閉塞性または炎症性気道疾患、詳細には、慢性好酸球性肺炎、慢性閉塞性肺疾患(COPD)、COPDを随伴する、または随伴しない慢性気管支炎、肺気腫または呼吸窮迫を包含するCOPD、不可逆性進行性気道閉塞を特徴とするCOPD、成人呼吸窮迫症候群(ARDS)、他の薬物療法に起因する気道過反応性の再燃および肺高血圧症を随伴する気道疾患からなる群から選択されるメンバーである閉塞性または炎症性気道疾患、
あらゆるタイプ、病因または病原の鼻ポリープ;
あらゆるタイプ、病因または病原の線維症、詳細には、炎症性気道疾患を随伴する肺線維症;
あらゆるタイプ、病因または病原の腸の炎症性疾患、詳細には、潰瘍性大腸炎およびクローン病;
あらゆるタイプ、病因または病原の自己免疫疾患、詳細には、関節リウマチ、多発性硬化症および播種性エリテマトーデス
からなる群から選択される疾患、障害および状態を治療する際に使用するための式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物に関する。
【0136】
なおより特には、本発明による化合物は、喘息、COPD、アレルギー性鼻炎、鼻ポリープ、クローン病、湿疹および乾癬を治療するためにより特には有用である。
【0137】
本発明の他の実施形態では、本発明の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物はまた、患者に同時投与して、これらに限られないが、(i)気管支収縮、(ii)炎症、(iii)アレルギー、(iv)組織破壊、(v)息切れ、咳などの徴候および症状を包含する病態生理学的関連疾患プロセスの治療などのいくつかの特に望ましい治療最終結果を得るための1種または複数の追加的な治療剤との組合せとして使用することができる。第2またはそれ以上の追加的な治療剤はまた、式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物、または当分野で知られている1種または複数のグルココルチコイド受容体アゴニストであってもよい。より典型的には、第2またはそれ以上の治療剤は、異なる群の治療剤から選択される。
【0138】
本明細書で使用される場合、本発明の化合物および1種または複数の他の治療剤に関する「同時投与」、「同時投与される」および「〜と組み合わせて」との用語は、下記を意味し、下記に関し、さらに、下記を包含することとする:
各成分を実質的に同時に患者に放出する単一剤形に、各成分を一緒に製剤する場合であって、式(I)の化合物および治療剤のこのような組合せを、治療を必要とする患者に同時に投与すること、
患者によって実質的に同時に摂取されて、前記各成分が実質的に同時に前記患者に放出される別々の剤形に、各成分を相互に別々に製剤する場合であって、式(I)の化合物および治療剤のこのような組合せを、治療を必要とする患者に実質的に同時に投与すること、
患者によって、各投与間に有意の時間的間隔を伴う連続する時間に摂取されて、前記各成分が実質的に異なる時間に前記患者に放出される別々の剤形に、各成分を相互に別々に製剤する場合であって、式(I)の化合物および治療剤のこのような組合せを、治療を必要とする患者に連続して投与すること、および
各成分を制御して放出する単一剤形に、各成分を一緒に製剤し、その上、それらを患者が同じか異なる時点で同時に、連続しておよび/または重複して投与する場合であって、式(I)の化合物および治療剤のこのような組合せを、治療を必要とする患者に連続して投与すること
(ここで、各部を、同じか異なる経路により投与することができる)。
【0139】
本発明の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物と組み合わせて使用することができる他の治療剤の適切な例には、決してこれらには限られないが、
(a)5−リポキシゲナーゼ(5−LO)阻害剤または5−リポキシゲナーゼ活性化タンパク質(FLAP)アンタゴニスト、
(b)LTB4、LTC4、LTD4およびLTE4のアンタゴニストを包含するロイコトリエンアンタゴニスト(LTRA)、
(c)ロイコトリエンC4シンターゼの阻害剤、
(d)H1、H3およびH4アンタゴニストを包含するヒスタミン受容体アンタゴニスト、
(e)うっ血除去用途のためのα1−およびα2−アドレノセプターアゴニスト血管収縮交感神経様作動薬、
(f)PDE阻害剤、例えば、PDE3、PDE4およびPDE5阻害剤、
(g)テオフィリン、
(h)クロモグリク酸ナトリウム、
(i)非選択的および選択的COX−1またはCOX−2阻害剤の両方であるCOX阻害剤(NSAID)、
(j)プロスタグランジン受容体アンタゴニストおよびhPGDSなどのプロスタグランジンシンターゼの阻害剤、
(k)ムスカリン様M3受容体アンタゴニストまたは抗コリン作動薬、
(l)β2−アドレノセプターアゴニスト、
(m)例えば、IgE、IL3、IL4、IL9、IL10、IL13、IL17A、GMCSFおよびその受容体などの内生炎症促進性実体に対して活性なモノクローナル抗体、
(n)抗腫瘍壊死因子(抗TNF−α)剤、
(o)VLA−4アンタゴニストを包含する接着分子阻害剤、
(p)キニン−B1−およびB2−受容体アンタゴニスト、
(q)IgE経路の阻害剤およびシクロスポリンを包含する免疫抑制剤、
(r)例えば、MMP9およびMMP12などのマトリックスメタロプロテアーゼ(MMP)の阻害剤、
(s)タキキニンNK1、NK2およびNK3受容体アンタゴニスト、
(t)エラスターゼ阻害剤、特に、好中球エラスターゼ阻害剤などのプロテアーゼ阻害剤、
(u)アデノシンA2a受容体アゴニストおよびA2bアンタゴニスト、
(v)ウロキナーゼの阻害剤、
(w)D2アゴニストなどのドーパミン受容体に作用する化合物、
(x)IKK阻害剤などのNFκβ経路の調節剤、
(y)p38MAPキナーゼ、PI3キナーゼ、JAKキナーゼ、sykキナーゼ、EGFR、MK−2、fynキナーゼまたはITKなどのサイトカインシグナル伝達経路の調節剤、
(z)粘液溶解薬または鎮咳薬として分類することができる薬剤、
(aa)例えば、マコリド(macolide)類似体およびPI3KδまたはAKT1、2、3の阻害剤などの吸入コルチコステロイドに対する応答を増強または再増感する薬剤、
(bb)気道でコロニー形成し得る微生物に対して有効な抗生物質および抗ウイルス剤、
(cc)HDAC活性化剤、
(dd)CXCR1、CXCR2およびCXCR3アンタゴニスト、
(ee)インテグリンアンタゴニスト、
(ff)ケモカインおよびケモカイン受容体アンタゴニスト、
(gg)上皮性ナトリウムチャネル(ENaC)遮断剤または上皮性ナトリウムチャネル(ENaC)阻害剤、
(hh)CRACイオンチャネル遮断剤またはCRAC阻害剤、
(ii)P2Y2アゴニストおよび他のヌクレオチド受容体アゴニスト、
(jj)P2X7アンタゴニスト、
(kk)VAP1の阻害剤、
(ll)トロンボキサンの阻害剤、
(mm)ナイアシン、および
(nn)VLAM、ICAMおよびELAMを包含する接着因子
が包含される。
【0140】
本発明では、式(I)の化合物と、
例えば、イプラトロピウム塩、即ち、臭化物、チオトロピウム塩、即ち、臭化物、オキシトロピウム塩、即ち、臭化物、トロスピウム塩、アクリジニウム塩、ペレンゼピンおよびテレンゼピンを包含するムスカリン様M3受容体アゴニストまたは抗コリン作動薬、
例えば、エフェドリン、アドレナリン、イソプレナリン、メタプロテレノール、フェニレフリン、フェニルプロパノールアミン、ピルブテロール、レプロテロール、リミテロール、イソエタリン、トロブテロール、カルモテロール、アルブテロール、テルブタリン、バムブテロール、フェノテロール、サルブタモール、ツロブテロール ホルモテロール、サルメテロールならびにWO05/080313、WO05/080324、WO05/092840およびWO2007/010356に記載されているアゴニストを包含するβ2−アドレノセプターアゴニスト;
PDE4阻害剤、詳細には、吸入PDE4阻害剤、
テオフィリン、
H1およびH3アンタゴニスト、例えば、ロラタジンおよびメタピリレンを包含するヒスタミン受容体アンタゴニストまたは
アデノシンA2a受容体アゴニスト、例えば、WO01/94368に記載されているもの
との組合せが好ましい。
【0141】
好ましい態様では、本発明の化合物は、β2−アドレノセプターアゴニストおよび抗コリン作動薬から選択される他の治療剤と組み合わせることができる。他の好ましい態様には、本発明による化合物と、β2−アドレノセプターアゴニストおよび抗コリン作動薬との3種組合せが包含される。
【0142】
下記の非限定的な実施例により、本発明を説明する:
【図面の簡単な説明】
【0143】
【図1】実施例1のDSCサーモグラムである。
【図2】実施例1のPXRDパターンである。
【図3】実施例2のDSCサーモグラムである。
【図4】実施例2のPXRDパターンである。
【図5】実施例5のDSCサーモグラムである。
【図6】実施例5のPXRDパターンである。
【0144】
プロトコル
下記の全ての実施例で、下記の実験条件を使用した:
【0145】
示差走査熱分析(DSC)
示差走査熱分析を、TA Instrument Q1000 DSCを使用し、リッドを備えたアルミニウムパンで行った。試料約3mgを、1分当たり20℃で、試料に応じて10℃から250℃または10℃から300℃または20℃から300℃の範囲にわたって、窒素ガスパージを伴って加熱した。
【0146】
粉末X線回折方法(PXRD)
自動試料チェンジャー、シータ−シータゴニオメーター、自動ビーム広がりスリットおよびPSD Vantec−1検出器を備えたBruker−AXS Ltd.D4粉末X線回折計を使用して、粉末X線回折パターンを決定した。低バックグラウンドキャビティシリコンウェハに試料量をマウントすることにより、試料を分析のために調製した。試料を回転させ、その間、40kV/30mAで運転されるX線管を用いて、銅K−アルファ1X線(波長=1.5406Å)を照射した。2°から55°の2シータ範囲にわたって0.018°ステップ当たり0.2秒カウントに設定された連続モードで運転するゴニオメーターを用いて、分析を行った。
【実施例】
【0147】
調製1
(6α,11β,16α)−6,9−ジフルオロ−11,16,17,21−テトラヒドロキシプレグナ−1,4−ジエン−3,20−ジオン
【0148】
【化23】
(4bR,6bS,9aR,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a,8,8−テトラメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン(10.3g、22.76mmol、市販)を48%ホウフッ化水素酸水溶液(100mL)に懸濁させ、生じた懸濁液を周囲温度、窒素雰囲気下で7時間撹拌した。次いで、懸濁液を水(200mL)で希釈し、濾過し、固体を水(500mL)で洗浄した。固体ケークをメタノール(200mL)に懸濁させ、真空濃縮した。生じた固体をtert−ブチル−メチルエーテル(150mL)に懸濁させ、濾過し、tert−ブチルメチルエーテル(200mL)で洗浄すると、表題化合物が白色の固体、収率95%、8.9gとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.82 (s, 3H), 1.46 (s,
3H), 1.31-1.51 (m, 3H), 1.77-1.87 (m, 1H), 2.08-2.33 (m, 3H), 2.37-2.46 (m,
1H), 4.08 (d, 1H), 4.09-4.16 (m, 1H), 4.48 (d, 1H), 4.63 (br, 1H), 4.75 (dd,
1H), 5.35 (d, 1H), 5.51-5.69 (m, 1H), 6.08 (s, 1H), 6.26 (dd, 1H), 7.24 (dd,
1H) ppm.
LRMS (ESI): m/z 411 [M-H]-
【0149】
調製2
4−ベンジルベンズアルデヒド
【0150】
【化24】
臭化ベンジル(41g、240mmol)、4−ホルミルベンゼンボロン酸(28g、186.7mmol)、パラジウムテトラキストリフェニルホスフィン(7.9g、6.84mmol)および炭酸カリウム(84.7g、613mmol)をテトラヒドロフラン(620mL)中で合わせ、80℃で窒素下に8時間加熱した。生じた懸濁液を周囲温度に冷却し、一晩撹拌した。反応混合物を10%クエン酸(50mL)に注ぎ、酢酸エチル(3×50mL)で抽出した。合わせた有機抽出物をブライン(100mL)で洗浄し、乾燥させ(硫酸マグネシウム)、溶媒を真空除去した。生じたオイルをカラムクロマトグラフィーによりシリカゲルで、酢酸エチル:ヘプタン(体積で0:1から1:5へ変化)で溶離して精製すると、表題化合物が無色のオイル、収率83%、30.45gとして得られた。
1H NMR
(400 MHz, CDCl3) δ: 4.06 (s, 2H), 7.38-7.17
(m, 7H), 7.83-7.79 (m, 2H), 9.97 (s, 1H) ppm.
LRMS (ESI): m/z 197 [M+H]+
【0151】
調製3
4−{[3−(メチルチオ)フェニル]チオ}ベンズアルデヒド
【0152】
【化25】
Rumpf,P.、Bull.soc.chim.(1940)、7、pp.632〜4に示されている通りに調製された3−(メチルチオ)ベンゼンチオール(19.9g、127.3mmol)のアセトニトリル(60mL)溶液を4−フルオロベンズアルデヒド(13.4mL、127mmol)で、続いて、炭酸カリウム(19.4g、140mmol)で処理した。周囲温度で18時間撹拌した後に、懸濁液を水(200mL)で希釈し、酢酸エチル(3×300mL)で抽出した。合わせた有機抽出物をブライン(2×100mL)で洗浄し、乾燥させ(硫酸マグネシウム)、溶媒を真空除去すると、無色のオイルが得られた。粗製のオイルをフラッシュカラムクロマトグラフィーによりシリカゲルで、ヘプタン:酢酸エチル(体積で1:0から9:1へ変化)で溶離して精製すると、表題化合物が無色のオイル、収率33%、10.8gとして得られた。
1H NMR
(400 MHz, CDCl3) δ: 2.48 (s, 3H), 7.25-7.34
(m, 5H), 7.38 (m, 1H), 7.73-7.75 (d,2H) ppm.
LRMS (API): m/z 261 [M+H]+
【0153】
調製4
4−{[(4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド
【0154】
【化26】
4−ブロモメチルベンズアルデヒド(0.3g、1.5mmol)および4−ヒドロキシチオフェノール(0.2g、1.5mmol)の1,4−ジオキサン(10mL)溶液を脱ガスし、トリエチルアミン(0.44mL、3.11mmol)で処理した。1日撹拌した後に、混合物を水(20mL)で希釈し、酢酸エチル(2×20mL)で抽出した。合わせた有機抽出物をブラインで洗浄し、乾燥させ(硫酸ナトリウム)、溶媒を真空除去した。粗製物質をフラッシュカラムクロマトグラフィーによりシリカゲルで、ヘプタン:酢酸エチル(体積で9:1から0:1へ変化)で溶離して精製すると、表題化合物が固体、収率62%、220mgとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 4.01 (s, 2H), 6.66 (d,
2H), 7.13 (d, 2H), 7.38 (d, 2H), 7.77 (d, 2H), 9.54 (s, 1H), 9.93 (s, 1H) ppm.
LRMS (ESI): m/z 243 [M-H]-
【0155】
調製5
3−クロロ−4−ヒドロキシフェニルチオシアネート
【0156】
【化27】
ヘキサフルオロイソプロパノール(10mL)中のトリメチルシリルイソチオシアネート(24.5g、187mmol)を氷冷された2−クロロフェノールのヘキサフルオロイソプロパノール(30mL)溶液に滴下添加した。反応混合物を10分間撹拌し、次いで、[ビス(トリフルオロアセトキシ)ヨード]ベンゼン(60.2g、140mmol)を滴下添加し、その間、内部温度を5℃未満に維持した。添加が完了した後に、反応混合物を5〜10℃で4時間撹拌し、次いで、真空濃縮すると、黄色の固体が得られた。これを、ジクロロメタン(100mL)に入れ、セライトパッドで濾過した。濾液を真空濃縮すると、黄色のオイルが得られ、これを、フラッシュカラムクロマトグラフィーによりシリカゲルで、ジクロロメタン:ヘプタン(体積で3:7から0:1へ変化)で溶離して精製すると、生成物が黄色の固体、収率30%、5.2gとして得られた。
1H NMR
(400 MHz, CDCl3) δ: 5.99 (bs, 1H), 7.06-7.10
(m, 1H), 7.40-7.43 (m, 1H), 7.59 (s, 1H).
LRMS (ESI): m/z 186 [M+H]+
【0157】
別法では、3−クロロ−4−ヒドロキシフェニルチオシアネートを下記の通り調製した:
臭素(0.40mL、7.78mmol)の酢酸(0.80mL)溶液を、2−クロロフェノール(1.00g、7.78mmol)およびチオシアン酸ナトリウム(2.27g、28.0mmol)の酢酸(6mL)中の懸濁液に滴下添加した。添加の間、内部温度を16℃から25℃に維持した。反応混合物を周囲温度で1時間撹拌した。次いで、水30mLおよび酢酸エチル30mLを反応混合物に加え、これを、celite(登録商標)パッドで濾過した。層を分離し、水性層を酢酸エチル(2×30mL)で抽出した。合わせた有機層を乾燥させ(硫酸マグネシウム)、真空濃縮すると、オレンジ色の半固体が得られた。これを酢酸エチル50mLに入れ、第2のcelite(登録商標)パッドで濾過すると、暗オレンジ色のオイル1.31gが得られた。この物質をさらに精製することなく、調製6で使用した。
【0158】
調製6
2−クロロ−4−メルカプトフェノール
【0159】
【化28】
水素化アルミニウムリチウム(テトラヒドロフラン中1Mの溶液として、71.0ml、71.0mmol)を氷冷された3−クロロ−4−ヒドロキシフェニルチオシアネート(4.20g、22.6mmol)のテトラヒドロフラン(100mL)中の溶液に窒素下に滴下添加した。反応混合物を撹拌し、室温に5時間にわたって加温した。混合物を5℃に冷却し、さらなるガス発生が観察されなくなるまで、テトラヒドロフラン:水の1:1混合物でクエンチした。次いで、1Nの塩酸水溶液(30mL)を加え、混合物を酢酸エチル(2×70mL)で抽出した。合わせた有機層を乾燥させ(硫酸ナトリウム)、真空濃縮すると、無色の結晶質固体、収率100%、3.60gが得られた。
1H NMR
(400 MHz, CDCl3) δ: 3.42 (s, 1H), 5.67, (bs,
1H), 6.89-6.92 (m, 1H), 7.13-7.16 (m, 1H), 7.33 (s, 1H).
LRMS (ESI): m/z 161 [M+H]+
【0160】
調製7
4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド
【0161】
【化29】
トリエチルアミン(5.0mL、35.9mmol)を2−クロロ−4−メルカプトフェノール(3.60g、22.4mmol)および4−ブロモメチルベンズアルデヒド(3.93g、19.7mmol)のジオキサン(150mL)中の溶液に室温、窒素下で滴下添加した。反応混合物を室温で20時間撹拌した。水(100mL)を反応混合物に加え、次いでこれを、酢酸エチルおよびブライン(それぞれ200mL)に分配した。水性層を酢酸エチル(100mL)で再抽出した。合わせた有機層を硫酸ナトリウム上で乾燥させ、真空濃縮すると、黄色の固体が得られた。これをアセトニトリル(5mL/g)と共に30分間摩砕することにより精製した。濾過の後に、明黄色の固体が純粋な生成物、収率84%、5.27gとして得られた。
1H NMR
(400 MHz, CDCl3) δ: 3.96 (s, 2H), 5.55 (bs,
1H), 6.82-6.86 (m, 1H), 7.02-7.06 (m, 1H), 7.19-7.23 (m, 1H), 7.24-7.28 (m,
1H), 7.71-7.74 (m, 2H), 9.92 (s, 1H).
LRMS (ESI): m/z 279 [M+H]+
【0162】
調製8
3−(メチルチオ)ベンゼンチオール
【0163】
【化30】
ベンゼン−1,3−ジチオール(50g、0.351mmol)の2−メチルテトラヒドロフラン(375mL、0.351mmol)溶液を硫酸ジメチル(33.3mL、0.351mol)で、続いて、2−メチルテトラヒドロフラン(25mL、連続洗浄として使用)で処理した。生じた溶液を0℃から5℃に冷却し、反応混合物の温度を15℃未満に維持しながら水酸化ナトリウム(2Mの水溶液、210.6mL)を、続いて、2−メチルテトラヒドロフラン(25mL、連続洗浄として使用)を滴下添加し、窒素下、50℃に4時間加熱した。周囲温度まで冷却した後に、生じた溶液をtert−ブチルメチルエーテル(500mL)で処理し、相を分離した。有機相を水酸化ナトリウム(2Mの水溶液、250mL)で抽出し、合わせた水性相を10℃に冷却し、塩酸(6Mの水溶液、500mL)を加え、その間、混合物の温度を25℃未満に維持した。生じた溶液をtert−ブチルメチルエーテル(2×250mL)で抽出し、合わせた有機相を水(500mL)で洗浄し、真空濃縮すると、表題化合物が黄色のオイル、収率78%、42.9gとして得られた。この物質は、HPLCにより、Rumpf(Bull.soc.chim.(1940)、7、pp.632〜4)の方法により調製された物質と同一であることが判明した。
【0164】
調製9
ナトリウムヒドロキシ(4−{[3−(メチルチオ)フェニル]チオ}フェニル)メタンスルホネート
【0165】
【化31】
調製8で調製された3−(メチルチオ)ベンゼンチオール(290g、1.856mol)のアセトニトリル(3L)溶液を窒素で1時間パージし、次いで、4−フルオロベンズアルデヒド(196mL、1.856mol)で、続いて、アセトニトリル(150mL、連続洗浄として使用)で処理した。生じた溶液を1,1’,3,3’−テトラメチルグアニジン(256mL、2.04mol)で、続いて、アセトニトリル(150mL、連続洗浄として使用)で処理し、50℃に窒素下に16時間加熱した。生じた溶液を周囲温度に冷却し、酢酸エチル(3L)で希釈し、塩酸(2Mの水溶液、1.5L)および重炭酸ナトリウム(1Mの水溶液、3L)およびブライン(半飽和、1.5L)で洗浄した。生じた溶液を大気圧で蒸留することにより濃縮して2Lの体積にし、アセトニトリル(3L)で希釈した。生じた溶液を大気圧での蒸留により濃縮して2Lの体積にし、アセトニトリル(3L)で希釈し、大気圧での蒸留により濃縮して3Lの最終体積にした。生じた溶液を周囲温度に冷却し、メタ重亜硫酸ナトリウム(377g、1.982mol)の水(3L)溶液で処理した。周囲温度で48時間撹拌した後に、生じた懸濁液を濾過し、集められた固体を水(2×2.5L)およびアセトニトリル(2×2.5L)で洗浄した。固体をアセトニトリル(2L)に懸濁させ、周囲温度で18時間撹拌し、この時間の後に、懸濁液を濾過し、固体をアセトニトリル(2×1L)で洗浄し、50℃で真空乾燥させると、表題化合物が白色の固体、収率55%、368.7gとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 2.41 (s, 3H), 4.97 (d,
1H), 5.90 (d, 1H), 6.98 (m, H), 7.12 (m, 2H), 7.26 (m, 3H), 7.46 (d, 2H) ppm.
【0166】
(実施例1)
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−ベンジルフェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
【0167】
【化32】
(4bR,6bS,9aR,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a,8,8−テトラメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト−[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン(8g、18mmol)および調製2で得られた4−ベンジルベンズアルデヒド(10.4g、53mmol)を、氷冷され撹拌されている砂(80g)のトルエン(80mL)懸濁液に加えた。70%過塩素酸水溶液(4mL、70mmol)を5分にわたって滴下添加し、次いで、溶液を周囲温度で21時間撹拌した。飽和重炭酸ナトリウム溶液(100mL)を反応混合物に、続いて、酢酸エチル(100mL)を加え、溶液を撹拌し、次いで、濾過した。砂を飽和重炭酸ナトリウム溶液(50mL)で、次いで、酢酸エチル(100mL)で洗浄した。水性層を分離し、酢酸エチル(2×50mL)で抽出し、合わせた有機抽出物をブライン(100mL)で洗浄し、乾燥させ(硫酸マグネシウム)、溶媒を真空除去した。生じた粘稠性の黄色のオイル(10g)をDCM:酢酸エチル(体積で9:1)で希釈すると、固体物質の沈殿が生じ、これを濾過により集めた。乾燥の後に、物質を酢酸エチル:ヘプタン(体積で4:1)から再結晶化させると、白色の結晶物質、収率44%、4.36gが得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.82 (s, 3H), 1.45 (s,
3H), 1.43-1.50 (m, 1H), 1.62-1.71 (m, 3H), 1.97-2.03 (m, 1H), 2.16-2.30 (m,
2H), 2.51-2.65 (m, 1H), 3.87 (s, 2H), 4.12-4.18 (m, 1H), 4.15 (dd, 1H), 4.46
(dd, 1H), 4.91 (d, 1H), 5.02 (t, 1H), 5.40 (s, 1H), 5.45 (d, 1H), 5.51-5.69 (m,
1H), 6.09 (s, 1H), 6.25 (dd, 1H), 7.10-7.24 (m, 8H), 7.29-7.32 (m, 2H) ppm.
LRMS (ESI): m/z 591 [M+H]+
【0168】
1.960mgの試料を、示差走査熱分析(DSC)により、20℃/分で、10℃から300℃の傾斜で分析した。得られたDSCサーモグラムを、図1に、平坦な基線および250.6℃での溶融に対応する鋭い吸熱と共に示す。
【0169】
上記のプロセスにより製造される結晶形はまた、図2の対応する粉末X線回折パターンに示されている特徴を有する。主な特徴的なピークは、8.0、16.0、16.9、24.0および24.2度2θ±0.1度2θにあり、さらに、表1に示す。
【0170】
【表1】
【0171】
(実施例2)
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルチオ)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
【0172】
【化33】
調製1で得られた(6α,11β,16α)−6,9−ジフルオロ−11,16,17,21−テトラヒドロキシプレグナ−1,4−ジエン−3,20−ジオン(6.6g、16mmol)および調製3で得られた4−{[3−(メチルチオ)フェニル]チオ}ベンズアルデヒド(4.5g、17.28mmol)の1,4−ジオキサン(70mL)中の懸濁液を硫酸マグネシウム(10g、83.1mmol)で処理した。懸濁液を水浴中で冷却し、トリフルオロメタンスルホン酸(7.5mL、82mmol)を加えた。室温で24時間撹拌した後に、混合物を水(200mL)で希釈し、酢酸エチル(2×200mL)で抽出した。合わせた有機抽出物を水(200mL)およびブライン(2×150mL)で洗浄し、乾燥させ(硫酸マグネシウム)、溶媒を真空除去した。粗製のゴムをフラッシュカラムクロマトグラフィーによりシリカゲルで、ヘプタン:tert−ブチルメチルエーテル(体積で4:1から1:0へ変化)で、次いで、ヘプタン:酢酸エチル(体積で3:7から0:1へ変化)で溶離して精製すると、黄色の泡が得られた。さらに、フラッシュカラムクロマトグラフィーによりシリカゲルで、ペンタン:酢酸エチル(体積で4:1から2:3へ変化)で溶離すると、黄色の固体が得られた。この固体を2−ブタノンから、次いで、アセトニトリルから再結晶化させると、表題化合物が白色の固体、収率18%、1.93gとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.85 (s, 3H),
1.43-1.53 (m, 1H), 1.48 (s, 3H), 1.65-1.71 (m, 3H), 1.97-2.06 (m, 1H),
2.14-2.31 (m, 2H), 2.41 (s, 3H), 2.55-2.67 (m, 1H), 4.16-4.22 (m, 2H),
4.49-4.55 (dd, 1H), 4.95 (d, 1H), 5.09 (t, 1H), 5.48 (s, 1H), 5.50-5.51 (m,
1H), 5.54-5.79 (m, 1H), 6.10 (s, 1H), 6.26-6.29 (m, 1H), 7.05-7.07 (m, 1H),
7.18-7.20 (m, 2H), 7.23-7.26 (m, 1H), 7.27-7.29 (m, 1H), 7.31-7.33 (d, 2H),
7.41-7.43 (d, 2H) ppm.
LRMS (ESI): m/z 655 [M+H]+
【0173】
別法では、表題化合物を下記の通り調製した:
調製1で得られた(6α,11β,16α)−6,9−ジフルオロ−11,16,17,21−テトラヒドロキシプレグナ−1,4−ジエン−3,20−ジオン(434g、1.050mol)および硫酸マグネシウム(417g、3.47mol)のアセトニトリル(4.34L)中の懸濁液を窒素下に18時間撹拌した。調製9で得られたナトリウムヒドロキシ(4−{[3−(メチルチオ)フェニル]チオ}フェニル)メタンスルホネート(460g、1.26mol)を加え、生じた懸濁液をトリフルオロメタンスルホン酸(443mL、5.01mmol)で処理し、その間、混合物の温度を24℃未満に維持した。周囲温度で75分間撹拌した後に、混合物を酢酸n−ブチル(4.4L)および水(4.4L)で処理し、さらに酢酸n−ブチル(400mL)を連続洗浄として使用して分離器に移した。相を分離し、有機相を水(4.4L)で洗浄し、炭酸水素ナトリウム(10%水溶液、2×2.2L)で洗浄し、水(2.2L)で洗浄した。生じた懸濁液を濾過し、濾液を真空濃縮して、溶媒4.26Lを除去した。残渣を35℃に冷却し、2−ブタノン(4L)で処理し、周囲温度に冷却し、18時間撹拌した。生じた懸濁液を濾過し、固体を2−ブタノン(2×2L)で洗浄した。固体をエタノール(2−ブタノンで変性、8L)に懸濁させ、10分間還流加熱し、周囲温度に冷却した。生じた懸濁液を濾過し、固体をエタノール(2−ブタノンで変性、2×2L)およびアセトニトリル(900mL)で洗浄した。固体をアセトニトリル(2.6L)に懸濁させ、還流加熱し、アセトニトリル(1.3L)で処理し、大気圧での蒸留により濃縮して、溶媒2.7Lを除去した。生じた懸濁液をアセトニトリル(1.75L)で処理し、還流加熱し、周囲温度に冷却した。生じた懸濁液を濾過し、固体をアセトニトリル(2×450mL)で洗浄し、40℃で真空乾燥させると、表題化合物が白色の固体、収率37%、307.3gとして得られた。こうして得られた化合物は、先行する方法により得られた化合物と同一であった。
【0174】
本明細書中、上記された第1の方法に従って得られた生成物のうちの2.847mgの試料を、示差走査熱分析(DSC)により、20℃/分で10から300℃の傾斜で分析した。第1の吸熱事象は、114.5℃で観察され、これは、不純物におそらく対応している。溶融が184.8℃で観察される。対応するサーモグラムを図3に示す。
【0175】
上記の方法により製造された結晶形はまた、図4の対応する粉末X線回折パターンに示されている特徴を有する。主な特徴的なピークは、10.0、16.5、17.0、20.0および25.5度2θ±0.1度2θにあり、さらに、下記の表2に示されている。
【0176】
【表2】
【0177】
(実施例3)
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−8−(4−{[(4−ヒドロキシフェニル)チオ]メチル}フェニル)−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
【0178】
【化34】
調製1で得られた(6α,11β,16α)−6,9−ジフルオロ−11,16,17,21−テトラヒドロキシプレグナ−1,4−ジエン−3,20−ジオン(99.8mg、0.24mmol)および調製4で得られた4−{[(4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド(148mg、0.61mmol)の1,4−ジオキサン(3mL)中の懸濁液を乾燥硫酸マグネシウム(430mg、3.57mmol)およびトリフルオロメタンスルホン酸(43μL、0.49mmol)で処理した。1日撹拌した後に、反応混合物を濾過し、集められた固体を酢酸エチル(20mL)で洗浄した。合わせた濾液を水(100mL)に注ぎ、酢酸エチル(3×20mL)で抽出した。合わせた有機抽出物をブライン(50mL)で洗浄し、乾燥させ(硫酸ナトリウム)、溶媒を真空除去した。粗製物質をフラッシュカラムクロマトグラフィーによりシリカゲルで、ヘプタン:酢酸エチル(体積で3:1から0:1へ変化)で溶離して精製すると、表題化合物が白色の固体、収率14%、21mgとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.85 (s, 3H),1.48 (s,
3H), 1.5 (m, 1H), 1.62-1.73 (m, 3H), 2.01-2.05 (m, 1H), 2.17-2.24 (m, 1H),
2.25-2.31 (m, 1H), 2.55-2.70 (m, 1H), 4.01 (s, 2H), 4.15-4.22 (m, 2H), 4.50
(dd, 1H), 4.94 (d, 1H), 5.05 (t, 1H), 5.43 (s, 2H), 5.48 (m, 1H), 5.55-5.71 (m,
1H), 6.11 (s, 1H), 6.28 (dd, 1H), 6.66 (d, 2H), 7.13 (d, 2H), 7.24 (m, 3H),
7.31 (d, 2H), 9.48 (s, 1H) ppm.
LRMS (ESI): m/z 639 [M+H]+
【0179】
(実施例4)
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルスルフィニル)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
【0180】
【化35】
実施例2で調製された(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルチオ)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2Hナフト[2’,1’:4,5]−インデノ[1,2−d][1,3]ジオキソール−2−オン(979mg、1.50mmol)をヘキサフルオロイソプロパノール(6mL、57.0mmol)に懸濁させ、氷浴中で冷却し、その後、過酸化水素(水中30重量%、203mg、1.79mmol)を滴下添加した。反応を周囲温度で90分間撹拌した。次いで、反応混合物を25%w/vの亜硫酸ナトリウム水溶液(30mL)に注いだ。水性層を酢酸エチル(3×50mL)で抽出し、合わせた有機抽出物を乾燥させ(硫酸ナトリウム)、溶媒を真空除去した。粗製物質をフラッシュカラムクロマトグラフィーによりシリカゲルで、ジクロロメタン:メタノール(体積で9:1)で溶離して精製すると、表題化合物が白色の固体、収率61%、610mgとして得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.87 (s, 3H), 1.50 (s, 3H),
1.47-1.59 (m, 1H), 1.65-1.78 (m, 3H), 2.01-2.09 (m, 1H), 2.17-2.34 (m, 2H),
2.58-2.71 (m, 1H), 2.74 (m, 3H), 4.18-4.26 (m, 2H), 4.52-4.58 (m, 1H), 4.98 (d,
1H), 5.10-5.13 (m, 1H), 5.52 (s, 1H), 5.53-5.54 (m, 1H), 5.58-5.78 (m, 1H),
6.12 (s, 1H), 6.31 (dd, 1H), 7.27 (d, 1H), 7.41-7.48 (m, 5H), 7.54- 7.64 (m,
3H) ppm.
LRMS (ESI): m/z 671 [M+H]+
【0181】
(実施例5)
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}フェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
【0182】
【化36】
トリフルオロメタンスルホン酸(4.76mL、53.8mmol)を、調製7で得られた4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}ベンズアルデヒド(5.25g、18.8mmol)、調製1で得られた(6α,11β,16α)−6,9−ジフルオロ−11,16,17,21−テトラヒドロキシプレグナ−1,4−ジエン−3,20−ジオン(7.40g、17.9mmol)および硫酸マグネシウム(6.82g、53.8mmol)のアセトニトリル(80mL)中の氷冷懸濁液に窒素下に滴下添加した。反応混合物を5〜10℃で4時間撹拌し、次いで、氷水(100mL)に注ぎ、酢酸エチル(3×150mL)で抽出した。合わせた有機層を乾燥させ(硫酸ナトリウム)、真空濃縮すると、茶色の泡が得られた。これを、フラッシュカラムクロマトグラフィー(シリカゲル)により、ジクロロメタン:メタノール(体積で100:0から90:10へ変化)で溶離して精製すると、オレンジ色の泡が得られた。これを、フラッシュカラムクロマトグラフィー(シリカゲル)により、ジクロロメタン:メタノール(体積で100:0から90:10へ変化)で溶離してさらに精製すると、黄色の泡(6.0g)が得られた。これを、温酢酸エチルに入れ、播種して沈殿を誘発した。固体を濾過し、乾燥させると、淡黄色の固体1.18gが得られた。母液を蒸発させ、酢酸エチルに再び入れ、播種して第2の収量1.46gを生じさせた。これを3回繰り返すと、2.40gの最終収量が得られた。3つのバッチ全てを合わせ、温酢酸エチル中で摩砕し、濾過し、乾燥させると、淡黄色の固体3.5g、収率27%が得られた。
1H NMR
(400 MHz, DMSO-d6) δ: 0.86 (s, 3H), 1.50 (s,
3H), 1.48-1.57 (m, 1H), 1.62-1.77 (m, 3H), 2.01-2.08 (m, 1H), 2.18-2.33 (m,
2H), 2.55-2.72 (m, 1H), 4.09 (s, 2H), 4.16-4.24 (m, 2H), 4.48-4.55 (m, 1H),
4.93-4.97 (m, 1H), 5.45 (s, 1H), 5.50-5.53 (m, 1H), 5.55-5.61 (m, 1/2H),
5.68-5.74 (m, 1/2H), 6.13 (s, 1H), 6.27-6.32 (m, 1H), 6.85-6.88 (s, 1H),
7.10-7.13 (m, 1H), 7.25-7.36 (m, 1H), 10.28 (bs, 1H).
LRMS (ESI): m/z 673 [M]
【0183】
2.769mgの試料を、示差走査熱分析(DSC)により、20℃/分、10℃から300℃の傾斜で分析した。得られたDSCサーモグラムを、図5に、平坦な基線および254℃にピークがある溶融に対応する鋭い吸熱と共に示す。
【0184】
上記のプロセスにより製造される結晶形はまた、図6の対応する粉末X線回折パターンに示されている特徴を有する。主な特徴的なピークは、14.1、16.6、20.0、21.8および25.2度2θ±0.1度2θにあり、さらに、表3に示す。
【0185】
【表3】
【0186】
(実施例6)
in vitro薬理学的活性
式(I)の化合物の薬理学的活性を、グルココルチコイドアゴニスト活性のin vitroアッセイで、ならびにin vivoでの抗炎症活性を予測し得るヒト血液および単離白血球TNF−α放出アッセイで評価した。
【0187】
グルココルチコイド受容体(GR)アゴニスト効果を、MMTV−ルシフェラーゼレポーター構成を安定導入されたヒト軟骨肉腫細胞系SW1353で決定した。SW1353は元々、ヒトGRを発現し、これは、グルココルチコイドアゴニストと結合すると、MMTVプロモーター内のグルココルチコイド応答要素を活性化させて、ルシフェラーゼ遺伝子の発現を駆動する。
【0188】
凍結されたSW1353細胞を、ピルビン酸ナトリウムまたはフェノールレッドを伴わず、2mMのL−グルタミン、1μg/mlのインスリン、2mg/mlのラクトアルブミンヒドロシレート(hydrosylate)および0.5μg/mlのアスコルベートを補足されているDMEM培地中で復活させた。細胞を細胞約5000/ウェル(35μl/ウェル)で384ウェルの透明床組織培養処理プレートに播種した。ステロイド用量−応答希釈をステロイド希釈剤(2.5%(v/v)のDMSOおよび0.05%(v/v)のプルロニック界面活性剤を含有するPBS)中で調製し、5μlを各ウェルに加えた。ステロイド希釈剤を用いて、体積を各ウェル当たり50μlにした。陽性対照ウェルは、1μMのデキサメタゾンを含有した。プレートを37℃、空気/5%CO2雰囲気中、加湿インキュベーター中で約18時間インキュベーションし、その後、Britelite試薬(10μl;Perkin−Elmer)を各ウェルに加えた。各プレートを暗所で2分間インキュベーションし、LJL Biosystems Analystルミノメーターを使用して、発光を定量した。試験化合物でのデータ(デキサメタゾン陽性対照に対するパーセンテージとして表示)を使用して、用量応答曲線を構成し、それから、EC50値を推定した。下記のデータが得られた。
【0189】
【表4】
【0190】
また、in vitroでのヒト白血球に対する化合物の抗炎症活性を、リポ多糖類(LPS)刺激されたヒト全血(WB)および単離ヒト末梢単核細胞(PBMC)からの腫瘍壊死因子−α(TNF−α)放出の阻害を決定することにより評価した。
【0191】
健康で投薬されていない提供者からの末梢静脈血を、エチレンジアミン四酢酸(EDTA)を抗凝血薬として使用して集めた。PBMC調製のために、血液試料を、無菌のリン酸緩衝溶液で1:1で希釈し、次いで、ACCUSPIN(商標)System−Histopaque(登録商標)−1077管(Sigma−Aldrich、St Louis、MO)を使用して分離し、400gで35分間遠心分離した。軟膜細胞をPBS中へ除去し、200gで10分間遠心分離し、PBMCアッセイ緩衝液(ハンクス液、0.28%[w/v]の4−[2−ヒドロキシエチル]−1−ピペラジンエタンスルホン酸[HEPES]、0.01%[w/v]の低内毒素ウシ血清アルブミン[BSA]に再懸濁させた。白血球分画を行い、1ml当たり1×106リンパ球まで、PBMCアッセイ緩衝液中でPBMC希釈した。
【0192】
試験化合物をDMSOに溶かし、PBMCアッセイ緩衝液(最終DMSO濃度1%)中で希釈して、適切な濃度範囲、例えば、0.001nMから10000nMをカバーした。試験化合物溶液の試料または媒体(20μl)を96ウェルの組織培養処理プレート(Corning)に加え、PBMC(160μl)またはWB(160μl)を各ウェルに加えた。アッセイ混合物を37℃、5%CO2を補足された空気雰囲気を含有する加湿インキュベーター中で1時間インキュベーションし、その後、LPS(PBMCでは100ng/mlまたはWBでは1μg/mlを20μl)を加えた。プレートをインキュベーターにさらに18時間戻し、次いで、遠心分離し、その後、上澄みの試料を回収した。試料中のTNF−αを、酵素結合免疫吸着アッセイ(ELISA)(Invitrogenキット no CHC−1754;Invitrogen Carlsbad、CA)を使用し、製造者指示に従って決定した。用量応答曲線を構成し、これから、IC50値を算出した。下記のデータが得られた。
【0193】
【表5】
【0194】
(実施例7)
in vivo薬理学的活性
薬理学的活性は、下記に記載されているものなどの肺炎症のin vivoモデルで評価することができる。この手順の主な目的は、気管を介して肺に直接投与した場合の式(I)の化合物の抗炎症活性を決定することであった。
【0195】
試験化合物を、0.5%(w/v)のTween−80を含有するリン酸緩衝溶液中に溶かすか、微細な懸濁液として調製して、一連の用量レベルを得た。雄のCD Sprague−Dawleyラット(300〜450g)をn=6の研究群に無作為化し、次いで、5%のイソフルランを含む麻酔チャンバー中、O23l/分で短時間麻酔を掛けた。試験化合物製剤または用量媒体(100μl)の1種を、Hamiltonシリンジを使用して麻酔を掛けられたラットそれぞれの気管に直接注射した。次いで、動物を麻酔剤から回復させた。研究設計に応じて、動物に、単回用量の化合物を与えるか、または1日1回、連続4日間処置した。投与の4時間後に(または繰り返し投与研究での最終投与の4時間後に)、超音波ネブライザーならびに最大1回呼吸量および速度(5ml、160ストローク/分)に設定されている小動物齧歯類人工呼吸器に接続されているチャンバー(300×300×450mm)に、ラットを入れた。37℃に予め加温された生理食塩水に溶かされた1mg/mlのLPS(Sigma−Aldrich、L2630)10mlをチャンバーへ噴霧した。15分後に、換気機およびネブライザーを止め、動物をチャンバー内に保持して、霧をさらに15分間呼吸させ、その後、ホームケージに戻した。
【0196】
LPS処置終了の4時間後に、動物を、1ml/kgのPentoject IPを用いて最終的な麻酔に掛けた。気管にカニューレを挿入し、肺を、2.6mMのEDTAを含有するPBS4×2.5mlで洗浄し、洗浄液を集めた。細気管支肺胞洗浄液(BAL)1mlを、40%ウシ血清アルブミン(BSA)125μlに加え、細胞カウントを、Advia 120血液学系(Siemens)を使用して決定した。繰り返し投与研究では、グルココルチコイドアゴニスト曝露に応答して減少することが知られていて、グルココルチコイドアゴニストの全身作用を評価するために使用されているので、体重ならびに副腎および胸腺の重量もまた決定した。一部の繰り返し投与実験では、最終血液試料を各ラットから集め、血清および血漿を調製し、血清中のコルチコステロンおよび血漿中のACTHの濃度を、全身グルココルチコイドアゴニスト作用の追加的なマーカーとして決定した。一部の研究では、知られているグルココルチコイドアゴニストであるプロピオン酸フルチカゾンを、陽性対称としてのラットの別の群に投与した。
【0197】
LPS−誘発肺好中球およびグルココルチコイドアゴニスト全身作用の各マーカーの阻害に関して、別々の用量応答曲線を構成した。半数最大有効量(ED50)値を、近似曲線から推定した。また、グルココルチコイドアゴニスト活性の全身マーカーに対する作用に関するED50値を肺好中球増加の阻害に関するED50で割って、治療指数(TI)の値を決定した。
【0198】
こうして、上記のアッセイで試験された本発明による式(I)の化合物は、下記の表に列挙されている通り、単回用量の投与後に肺好中球増加の阻害に活性を示すことが判明した。
【0199】
【表6】
【特許請求の範囲】
【請求項1】
式(I)の化合物:
【化1】
[式中、R1は、
【化2】
からなる群から選択され、ここで、*は、フェニル環の炭素へのR1の結合点を示している]
または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物。
【請求項2】
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−ベンジルフェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルチオ)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−8−(4−{[(4−ヒドロキシフェニル)チオ]メチル}フェニル)−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルスルフィニル)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a、10b,11,12−ドデカヒドロ−2H−ナフト−[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;および
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}フェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b、9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
からなる群から選択される、請求項1に記載の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物。
【請求項3】
有効量の請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物と、1種または複数の薬学的に許容できる賦形剤とを含む医薬組成物。
【請求項4】
医薬品として使用するための、請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物。
【請求項5】
グルココルチコイド受容体が関与している疾患、障害および状態を治療する際に使用するための、請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物。
【請求項6】
湿疹、乾癬、皮膚炎、心因性掻痒症および超過敏反応などの皮膚疾患;鼻炎、副鼻腔炎、喘息、鼻ポリープ、慢性閉塞性肺疾患(COPD)および線維症などの鼻、喉および肺の炎症状態;炎症性腸疾患、クローン病および潰瘍性大腸炎などの腸の炎症性疾患;ならびに関節リウマチなどの自己免疫疾患;ならびに結膜炎などの眼状態からなる群から選択される疾患、障害および状態を治療する際に使用するための、請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物。
【請求項7】
グルココルチコイド受容体が関与している疾患、障害および状態を治療するための薬物を製造するための、請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物の使用。
【請求項8】
請求項6に記載の群から選択される疾患、障害および状態を治療するための薬物を製造するための、請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物の使用。
【請求項9】
ヒトを包含する哺乳動物をグルココルチコイド受容体アゴニストで治療する方法であって、前記哺乳動物を有効量の請求項1または2のいずれか一項に記載の式(I)の化合物で、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物で治療することを包含する方法。
【請求項10】
請求項1または2のいずれか一項に記載の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物と、
(a)5−リポキシゲナーゼ(5−LO)阻害剤または5−リポキシゲナーゼ活性化タンパク質(FLAP)アンタゴニスト、
(b)LTB4、LTC4、LTD4およびLTE4のアンタゴニストを包含するロイコトリエンアンタゴニスト(LTRA)、
(c)ロイコトリエンC4シンターゼの阻害剤、
(d)H1、H3およびH4アンタゴニストを包含するヒスタミン受容体アンタゴニスト、
(e)うっ血除去用途のためのα1−およびα2−アドレノセプターアゴニスト血管収縮交感神経様作動薬、
(f)PDE阻害剤、例えば、PDE3、PDE4およびPDE5阻害剤、
(g)テオフィリン、
(h)クロモグリク酸ナトリウム、
(i)非選択的および選択的COX−1またはCOX−2阻害剤の両方であるCOX阻害剤(NSAID)、
(j)プロスタグランジン受容体アンタゴニストおよびhPGDSなどのプロスタグランジンシンターゼの阻害剤、
(k)ムスカリン様M3受容体アンタゴニストまたは抗コリン作動薬、
(l)β2−アドレノセプターアゴニスト、
(m)例えば、IgE、IL3、IL4、IL9、IL10、IL13、IL17A、GMCSFおよびその受容体などの内生炎症促進性実体に対して活性なモノクローナル抗体、
(n)抗腫瘍壊死因子(抗TNF−α)剤、
(o)VLA−4アンタゴニストを包含する接着分子阻害剤、
(p)キニン−B1−およびB2−受容体アンタゴニスト、
(q)IgE経路の阻害剤およびシクロスポリンを包含する免疫抑制剤、
(r)例えば、MMP9およびMMP12などのマトリックスメタロプロテアーゼ(MMP)の阻害剤、
(s)タキキニンNK1、NK2およびNK3受容体アンタゴニスト、
(t)エラスターゼ阻害剤、特に、好中球エラスターゼ阻害剤などのプロテアーゼ阻害剤、
(u)アデノシンA2a受容体アゴニストおよびA2bアンタゴニスト、
(v)ウロキナーゼの阻害剤、
(w)D2アゴニストなどのドーパミン受容体に作用する化合物、
(x)IKK阻害剤などのNFκβ経路の調節剤、
(y)p38MAPキナーゼ、PI3キナーゼ、JAKキナーゼ、sykキナーゼ、EGFR、MK−2、fynキナーゼまたはITKなどのサイトカインシグナル伝達経路の調節剤、
(z)粘液溶解薬または鎮咳薬として分類することができる薬剤、
(aa)例えば、マコリド類似体およびPI3KδまたはAKT1、2、3の阻害剤などの吸入コルチコステロイドに対する応答を増強または再増感する薬剤、
(bb)気道でコロニー形成し得る微生物に対して有効な抗生物質および抗ウイルス剤、
(cc)HDAC活性化剤、
(dd)CXCR1、CXCR2およびCXCR3アンタゴニスト、
(ee)インテグリンアンタゴニスト、
(ff)ケモカインおよびケモカイン受容体アンタゴニスト、
(gg)上皮性ナトリウムチャネル(ENaC)遮断剤または上皮性ナトリウムチャネル(ENaC)阻害剤、
(hh)CRACイオンチャネル遮断剤またはCRAC阻害剤、
(ii)P2Y2アゴニストおよび他のヌクレオチド受容体アゴニスト、
(jj)P2X7アンタゴニスト、
(kk)VAP1の阻害剤、
(ll)トロンボキサンの阻害剤、
(mm)ナイアシン、および
(nn)VLAM、ICAMおよびELAMを包含する接着因子
から選択される他の治療剤との組合せ。
【請求項11】
他の治療活性剤をさらに含む、請求項3に記載の医薬組成物。
【請求項12】
前記他の治療活性剤がβ2−アドレノセプターアゴニストである、請求項11に記載の医薬組成物。
【請求項13】
前記他の治療活性剤が抗コリン作動薬である、請求項11に記載の医薬組成物。
【請求項14】
β2−アドレノセプターアゴニストおよび抗コリン作動薬をさらに含む、請求項3に記載の医薬組成物。
【請求項15】
請求項1または2のいずれか一項に記載の式(I)の化合物を調製する方法であって、式(II)の化合物:
【化3】
または式(III)の化合物:
【化4】
または式(IV)の化合物:
【化5】
[式中、R2およびR3はホルミルである]
を式(IIa)の適切なアルデヒド:
【化6】
または式(IIb)の適切なアルデヒド同等物:
【化7】
[式中、R1は、請求項1に記載されている通りである]
と反応させるステップを含む方法。
【請求項16】
R1が式:
【化8】
である請求項1または2のいずれか一項に記載の式(I)の化合物を調製する方法であって、R1が式:
【化9】
である請求項1または2のいずれか一項に記載の式(I)の化合物を酸化剤と反応させるステップを含む方法。
【請求項1】
式(I)の化合物:
【化1】
[式中、R1は、
【化2】
からなる群から選択され、ここで、*は、フェニル環の炭素へのR1の結合点を示している]
または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物。
【請求項2】
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−ベンジルフェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルチオ)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−8−(4−{[(4−ヒドロキシフェニル)チオ]メチル}フェニル)−4a,6a−ジメチル−4a,4b,5,6,6a,6b,9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−8−(4−{[3−(メチルスルフィニル)フェニル]チオ}フェニル)−4a,4b,5,6,6a,6b,9a,10,10a、10b,11,12−ドデカヒドロ−2H−ナフト−[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン;および
(4aS,4bR,5S,6aS,6bS,8R,9aR,10aS,10bS,12S)−8−(4−{[(3−クロロ−4−ヒドロキシフェニル)チオ]メチル}フェニル)−4b,12−ジフルオロ−6b−グリコロイル−5−ヒドロキシ−4a,6a−ジメチル−4a,4b,5,6,6a,6b、9a,10,10a,10b,11,12−ドデカヒドロ−2H−ナフト[2’,1’:4,5]インデノ[1,2−d][1,3]ジオキソール−2−オン
からなる群から選択される、請求項1に記載の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物。
【請求項3】
有効量の請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物と、1種または複数の薬学的に許容できる賦形剤とを含む医薬組成物。
【請求項4】
医薬品として使用するための、請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物。
【請求項5】
グルココルチコイド受容体が関与している疾患、障害および状態を治療する際に使用するための、請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物。
【請求項6】
湿疹、乾癬、皮膚炎、心因性掻痒症および超過敏反応などの皮膚疾患;鼻炎、副鼻腔炎、喘息、鼻ポリープ、慢性閉塞性肺疾患(COPD)および線維症などの鼻、喉および肺の炎症状態;炎症性腸疾患、クローン病および潰瘍性大腸炎などの腸の炎症性疾患;ならびに関節リウマチなどの自己免疫疾患;ならびに結膜炎などの眼状態からなる群から選択される疾患、障害および状態を治療する際に使用するための、請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物。
【請求項7】
グルココルチコイド受容体が関与している疾患、障害および状態を治療するための薬物を製造するための、請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物の使用。
【請求項8】
請求項6に記載の群から選択される疾患、障害および状態を治療するための薬物を製造するための、請求項1または2のいずれか一項に記載の式(I)の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物の使用。
【請求項9】
ヒトを包含する哺乳動物をグルココルチコイド受容体アゴニストで治療する方法であって、前記哺乳動物を有効量の請求項1または2のいずれか一項に記載の式(I)の化合物で、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物で治療することを包含する方法。
【請求項10】
請求項1または2のいずれか一項に記載の化合物、または薬学的に許容できるその塩または前記化合物もしくは塩の薬学的に許容できる溶媒和物と、
(a)5−リポキシゲナーゼ(5−LO)阻害剤または5−リポキシゲナーゼ活性化タンパク質(FLAP)アンタゴニスト、
(b)LTB4、LTC4、LTD4およびLTE4のアンタゴニストを包含するロイコトリエンアンタゴニスト(LTRA)、
(c)ロイコトリエンC4シンターゼの阻害剤、
(d)H1、H3およびH4アンタゴニストを包含するヒスタミン受容体アンタゴニスト、
(e)うっ血除去用途のためのα1−およびα2−アドレノセプターアゴニスト血管収縮交感神経様作動薬、
(f)PDE阻害剤、例えば、PDE3、PDE4およびPDE5阻害剤、
(g)テオフィリン、
(h)クロモグリク酸ナトリウム、
(i)非選択的および選択的COX−1またはCOX−2阻害剤の両方であるCOX阻害剤(NSAID)、
(j)プロスタグランジン受容体アンタゴニストおよびhPGDSなどのプロスタグランジンシンターゼの阻害剤、
(k)ムスカリン様M3受容体アンタゴニストまたは抗コリン作動薬、
(l)β2−アドレノセプターアゴニスト、
(m)例えば、IgE、IL3、IL4、IL9、IL10、IL13、IL17A、GMCSFおよびその受容体などの内生炎症促進性実体に対して活性なモノクローナル抗体、
(n)抗腫瘍壊死因子(抗TNF−α)剤、
(o)VLA−4アンタゴニストを包含する接着分子阻害剤、
(p)キニン−B1−およびB2−受容体アンタゴニスト、
(q)IgE経路の阻害剤およびシクロスポリンを包含する免疫抑制剤、
(r)例えば、MMP9およびMMP12などのマトリックスメタロプロテアーゼ(MMP)の阻害剤、
(s)タキキニンNK1、NK2およびNK3受容体アンタゴニスト、
(t)エラスターゼ阻害剤、特に、好中球エラスターゼ阻害剤などのプロテアーゼ阻害剤、
(u)アデノシンA2a受容体アゴニストおよびA2bアンタゴニスト、
(v)ウロキナーゼの阻害剤、
(w)D2アゴニストなどのドーパミン受容体に作用する化合物、
(x)IKK阻害剤などのNFκβ経路の調節剤、
(y)p38MAPキナーゼ、PI3キナーゼ、JAKキナーゼ、sykキナーゼ、EGFR、MK−2、fynキナーゼまたはITKなどのサイトカインシグナル伝達経路の調節剤、
(z)粘液溶解薬または鎮咳薬として分類することができる薬剤、
(aa)例えば、マコリド類似体およびPI3KδまたはAKT1、2、3の阻害剤などの吸入コルチコステロイドに対する応答を増強または再増感する薬剤、
(bb)気道でコロニー形成し得る微生物に対して有効な抗生物質および抗ウイルス剤、
(cc)HDAC活性化剤、
(dd)CXCR1、CXCR2およびCXCR3アンタゴニスト、
(ee)インテグリンアンタゴニスト、
(ff)ケモカインおよびケモカイン受容体アンタゴニスト、
(gg)上皮性ナトリウムチャネル(ENaC)遮断剤または上皮性ナトリウムチャネル(ENaC)阻害剤、
(hh)CRACイオンチャネル遮断剤またはCRAC阻害剤、
(ii)P2Y2アゴニストおよび他のヌクレオチド受容体アゴニスト、
(jj)P2X7アンタゴニスト、
(kk)VAP1の阻害剤、
(ll)トロンボキサンの阻害剤、
(mm)ナイアシン、および
(nn)VLAM、ICAMおよびELAMを包含する接着因子
から選択される他の治療剤との組合せ。
【請求項11】
他の治療活性剤をさらに含む、請求項3に記載の医薬組成物。
【請求項12】
前記他の治療活性剤がβ2−アドレノセプターアゴニストである、請求項11に記載の医薬組成物。
【請求項13】
前記他の治療活性剤が抗コリン作動薬である、請求項11に記載の医薬組成物。
【請求項14】
β2−アドレノセプターアゴニストおよび抗コリン作動薬をさらに含む、請求項3に記載の医薬組成物。
【請求項15】
請求項1または2のいずれか一項に記載の式(I)の化合物を調製する方法であって、式(II)の化合物:
【化3】
または式(III)の化合物:
【化4】
または式(IV)の化合物:
【化5】
[式中、R2およびR3はホルミルである]
を式(IIa)の適切なアルデヒド:
【化6】
または式(IIb)の適切なアルデヒド同等物:
【化7】
[式中、R1は、請求項1に記載されている通りである]
と反応させるステップを含む方法。
【請求項16】
R1が式:
【化8】
である請求項1または2のいずれか一項に記載の式(I)の化合物を調製する方法であって、R1が式:
【化9】
である請求項1または2のいずれか一項に記載の式(I)の化合物を酸化剤と反応させるステップを含む方法。
【図1】

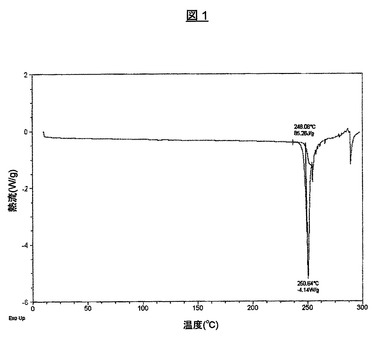
【図2】
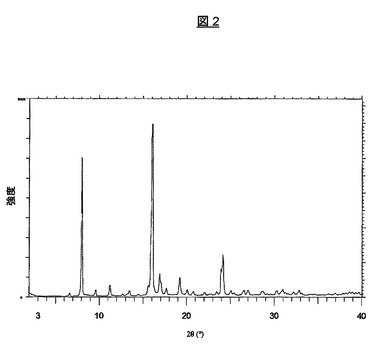
【図3】
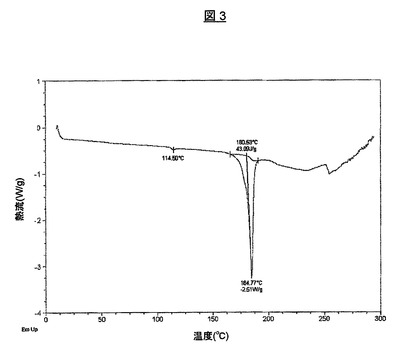
【図4】
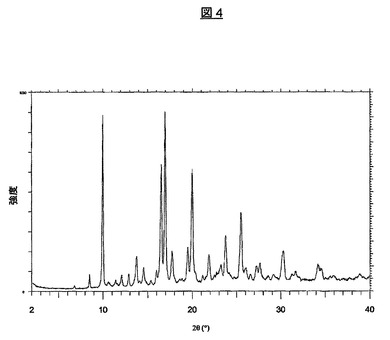
【図5】
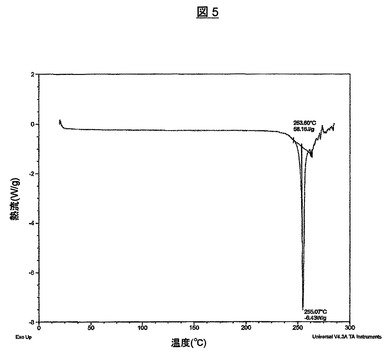
【図6】
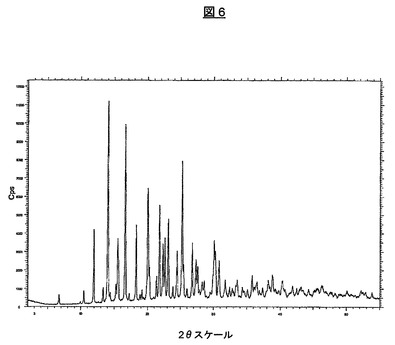
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2011−505350(P2011−505350A)
【公表日】平成23年2月24日(2011.2.24)
【国際特許分類】
【出願番号】特願2010−535483(P2010−535483)
【出願日】平成20年11月17日(2008.11.17)
【国際出願番号】PCT/IB2008/054801
【国際公開番号】WO2009/069032
【国際公開日】平成21年6月4日(2009.6.4)
【出願人】(597014501)ファイザー・リミテッド (107)
【氏名又は名称原語表記】Pfizer Limited
【住所又は居所原語表記】Ramsgate Road, Sandwich, Kent, England
【Fターム(参考)】
【公表日】平成23年2月24日(2011.2.24)
【国際特許分類】
【出願日】平成20年11月17日(2008.11.17)
【国際出願番号】PCT/IB2008/054801
【国際公開番号】WO2009/069032
【国際公開日】平成21年6月4日(2009.6.4)
【出願人】(597014501)ファイザー・リミテッド (107)
【氏名又は名称原語表記】Pfizer Limited
【住所又は居所原語表記】Ramsgate Road, Sandwich, Kent, England
【Fターム(参考)】
[ Back to top ]