
新規なコレステロールエステラーゼの製造方法
【課題】広範囲の脂肪酸エステルを加水分解する性質があり、安定性が高く、至適pHおよびpH安定性の範囲が広く、比活性が高いコレステロールエステラーゼを効率よく製造する方法の提供。
【解決手段】Burkholderia stabilis(FERM P−21014株)を培養し、コレステロールエステラーゼを得ることを特徴とするコレステロールエステラーゼの製造方法。
【解決手段】Burkholderia stabilis(FERM P−21014株)を培養し、コレステロールエステラーゼを得ることを特徴とするコレステロールエステラーゼの製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なコレステロールエステラーゼの製造方法に関する。
【背景技術】
【0002】
コレステロールは人体の各臓器および各器官に広く分布している。高コレステロール血症は、動脈硬化、心筋梗塞等と関連し、低コレステロール血症は悪液質、甲状腺機能昂進等と関連するので、特に血中コレステロール濃度の把握は臨床医学的に重要である。
従来、血中コレステロールを測定する酵素法は、コレステロールオキシダーゼを用いた方法(例えば、非特許文献1参照)と、コレステロールデヒドロゲナーゼを用いた方法(例えば特許文献1又は特許文献2参照)が報告されている。
血中コレステロールとしては、遊離コレステロールと脂肪酸コレステロールエステルが存在する。コレステロールオキシダーゼ(以下CODと略する場合がある)とコレステロールデヒドロゲナーゼ(以下CDHと略する場合がある)は、遊離のコレステロールのみに作用して酸化或いは還元するため、酵素法を用いて血中コレステロールを測定する場合には、コレステロールエステラーゼ(以下CEと略する場合がある)でコレステロールエステルを加水分解してコレステロールを遊離させなければならない。
CEは、これまでにシュードモナス属(特許文献3〜6など参照)、ストレプトミセス属(特許文献7参照)、ノカルディア属(特許文献8参照)、カワラタケ(特許文献9参照)、スエヒロタケ(特許文献10参照)、ブルクホルデリア属(特許文献11参照)、キサントモナス属(特許文献11参照)などを由来とするものが知られている。
CEを、血中コレステロールの測定などの臨床診断試薬用酵素として利用する場合、CEが以下の性質を持つと有利である。
1)広範囲の脂肪酸エステルを加水分解する性質。
2)液体中での高い保存安定性。
3)広い至適pHと高いpH安定性。
4)微生物の単位培養当たりの酵素生産量が大きい事とともに、単位タンパク質当たりの酵素活性(比活性)が高い事。
これらの性質が有利な理由は、順に、
1)被験対象液中、例えば血中のコレステロールエステルの脂肪酸成分は、リノール酸、オレイン酸、及びパルミチン酸を中心として、他にもアラキドン酸、パルミトレイン酸、ステアリン酸、及びミリスチン酸などの広範な種類が含まれており、これらを加水分解する必要がある為。
2)臨床診断試薬は液状で長期間保存できると利便性が高い為。
3)臨床診断試薬組成の検討に際して、原料や試薬組成条件の選択の幅が広がる為。
4)経済性が良い為。特に、比活性が高い性質は製造上、例えば、イ)不要な混在物質の割合を減らす事が容易である、ロ)カラムクロマトグラフィーに用いる樹脂が少なくて済み精製が容易である、ハ)遺伝子組み換え体にした場合、高い酵素の発現量が期待できる、など有利である。
例えば、非特許文献2には、シュードモナス属エルギノーサ由来CEの比活性は8.5U/mgとの報告があり、特許文献6には、シュードモナス属フルオレッセンス由来CEは0.101U/mg、シュードモナス属ストゥッツェリー属由来CEの比活性は796U/mg以上との報告があり、特許文献5には、シュードモナス属由来CEは0.24U/mgとの報告がある一方で非特許文献3には、15.2U/mgとの報告がある。又、特許文献7には、ストレプトミセス属由来CEの比活性は5U/mgとの報告がある一方で非特許文献4には、78.8U/mgとの報告があり、特許文献9には、カワラタケ由来CEの比活性は5.51U/mgとの報告があり、特許文献10には、スエヒロタケ由来CEの比活性は2.5U/mgとの報告があり、特許文献11には、ブルクホルデリア属由来CEの比活性は51U/mgとの報告があり、特許文献12には、キサントモナス属由来CEの比活性は85.6U/mgとの報告がある。
これらのうち、比活性が比較的高いストレプトミセス属由来CEは培養力価が0.05U/ml以下である(非特許文献4)ために高い経済性があるとは言えない。キサントモナス属由来CEは、キサントモナス属細菌は斑点細菌病、黒腐病、又は褐班細菌病などの植物病原細菌である場合が多い事を考慮すれば容易に産業利用できるとは言い難い。シュードモナス属ストゥッツェリー属由来CEは35℃から失活が認められ、45℃で約40%失活する(特許文献6)という産業利用しづらい性質を有する。
すなわち、従来のCEはその性質上或いは生産上何らかの問題がある。
【特許文献1】特公平2−49720号公報
【特許文献2】特公平5−176797号公報
【特許文献3】特開昭50−157588号公報
【特許文献4】特開昭56−42586号公報
【特許文献5】特開昭52−7483 号公報
【特許文献6】特開平9−251号公報
【特許文献7】特開昭53−109992号公報
【特許文献8】特開昭57−43686号公報
【特許文献9】特開昭55−114288号公報
【特許文献10】特開昭53−9391号公報
【特許文献11】特開平10−127278号公報
【特許文献12】特開平11−56355号公報
【非特許文献1】Charles C. Allain、 Lucy S. Poon、 Cicely S. G. Chan、 W. Richmond、 C. Fu、Enzymatic Determination of Total Serum Cholesterol、Clin. Chem.、20巻、470頁、1974年
【非特許文献2】Akio SUGIHARA、Yuji SHIMADA、Atsuo NOMURA、Tadamasa TERAI、Masaki IMAYASU、Yusuke NAGAI、Toshihiro NAGAO、Yomi WATANABE、Yoshio TOMINAGA、Purification and Characterization of a Novel Cholesterol Esterase from Pseudomonas aeruginosa, with Its Application to Cleaning Lipid-stained Contact Lenses、Bioscience, Biotechnology, and Biochemistry、66巻、2347頁、2002年
【非特許文献3】Noriyuki Doukyu、Rikizo Aono、Purification of Extracellular Cholesterol Oxidase with High Activity in the Presence of Organic Solvents from Pseudomonas sp. Strain ST-200、Applied and Environmental Microbiology、64巻、1929頁、1998年
【非特許文献4】Hongyu Xiang、Naoki Takaya、Takayuki Hoshino、Novel Cholesterol Esterase Secreted by Streptomyces Persists during Aqueous Long-Term Storage、Journal of Bioscience and Bioengineering、101巻、19頁、2006年
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明の課題は、1)広範囲の脂肪酸エステルを加水分解する性質があり、2)液体中での保存安定性が高く、3)至適pHおよびpH安定性の範囲が広く、且つ4)比活性が高いコレステロールエステラーゼを効率よく製造する方法を提供する事にある。
【課題を解決するための手段】
【0004】
本発明者らは、上記の課題を解決するために鋭意努力した結果、土壌より分離したBurkholderia stabilis(FERM P−21014株)から生産されるコレステロールエステラーゼが、所望の性質を有する事を見出した。さらに、その効率的な工業的規模の製造方法を確立して本発明を完成に至った。即ち、本発明は、Burkholderia stabilisを培養し、その培養物から広範囲の脂肪酸エステルを加水分解可能な、安定性が高く、至適pHおよびpH安定性の範囲が広く、比活性が高いCEを採取する事を特徴とするCEの製造方法に関し、具体的には以下に関する。
〔1〕Burkholderia stabilisを培養し、培養物から下記特性を有するコレステロールエステラーゼを得ることを特徴とするコレステロールエステラーゼの製造方法。
(1)作用
1分子の水の存在下、1分子のコレステロールエステルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
〔2〕Burkholderia stabilis が、Burkholderia stabilis FERM P−21014株である上記〔1〕に記載の製造方法。
〔3〕コレステロールエステラーゼが、配列表配列番号1のアミノ酸配列或いは配列番号1のアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を有することを特徴とする上記〔1〕又は〔2〕に記載のコレステロールエステラーゼの製造方法。
〔4〕配列表配列番号1のアミノ酸配列或いは配列番号1のアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列をコードする塩基配列からなるDNAを宿主に移入し、該宿主を培養し、該培養物から下記特性を有するコレステロールエステラーゼを得ることを特徴とするコレステロールエステラーゼの製造方法。
(1)作用
1分子の水の存在下、1分子のコレステロールエステルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
〔5〕上記〔1〕から〔4〕のいずれかに記載の製造方法により得られるコレステロールエステラーゼ。
〔6〕下記(a)又は(b)のコレステロールエステラーゼをコードするDNAを含有する組み換えベクター。
(a)配列表配列番号1に示すアミノ酸配列を有するコレステロールエステラーゼ。
(b)配列表配列番号1に示すアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を有し、かつ下記特性を有するコレステロールエステラーゼ。
(1)作用
1分子の水の存在下、1分子のコレステロールエスルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
〔7〕上記〔6〕に記載の組み換えベクターを含む形質転換体。
〔8〕Burkholderia stabilisFERM P−21014株。
〔9〕下記特性を有するコレステロールエステラーゼを製造するための、Burkholderia stabilis(FERM P−21014)株の使用。
(1)作用
1分子の水の存在下、1分子のコレステロールエスルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
【発明の効果】
【0005】
本発明により、1)広範囲の脂肪酸エステルを加水分解する性質があり、2)安定性が高く、3)至適pHおよびpH安定性の範囲が広く、且つ4)比活性が高いコレステロールエステラーゼを効率よく製造する方法を提供することができる。
【発明を実施するための最良の形態】
【0006】
Burkholderia stabilisの菌学的性質を以下に示す。+は陽性、−は陰性、wは反応弱い、を示す。
1.形態学的性質
培養条件: Nutrient agar培地 30℃
細胞の形態: 桿菌(0.7〜0.8×1.5〜2.0μm)
細胞の多様性の有無:−
運動性(鞭毛の着生状態):+(極毛)
胞子の有無:−
2.培養的性質
(a)培養条件: Nutrient agar培地 30℃
色:淡黄色
光沢:+
色素産生:−
(b)培養条件: Nutrient broth培地 30℃
表面発育の有無:−
培地の混濁の有無:+
(c)培養条件: ゼラチン穿刺培養 30℃
生育状態:+
ゼラチン液化:−
(d)培養条件: リトマスミルク 30℃
凝固:−
液化:−
3.生理学的性質
グラム染色性:−
硝酸塩の還元:+
脱窒反応:−
MRテスト:−
VPテスト:−
インドール産生:−
硫化水素の生成:−
デンプンの加水分解:−
クエン酸の利用(Koser):+
クエン酸の利用(Christensen):+
無機窒素源の利用 硝酸塩:+w
無機窒素源の利用 アンモニウム塩:+
ウレアーゼ活性:−
カタラーゼ:+
オキシダーゼ:−
生育の範囲pH 5:+
生育の範囲pH 8:+
生育の範囲pH 9:+
生育の範囲温度20℃:+
生育の範囲温度25℃:+
生育の範囲温度37℃:+
生育の範囲温度45℃:−
嫌気的生育性:−
O−Fテスト(酸化/発酵):+/−
4.糖類からの酸産生/ガス産生
L−アラビノース:+/−
D−グルコース:+/−
D−フラクトース:+w/−
マルトース:+/−
ラクトース:+/−
D−ソルビトール:−/−
イノシトール:+w/−
D−キシロース:+/−
D−マンノース:−/−
D−ガラクトース:+/−
シュークロース:+/−
トレハロース:+/−
D−マンニトール:+w/−
グリセリン:−/−
5.その他の生理学的性質
β−ガラクトシダーゼ活性:+
アルギニンジヒドロラーゼ活性:−
リジンデカルボキシラーゼ活性:−
トリプトファンデアミナーゼ活性:−
ゼラチナーゼ活性:−
【0007】
BLASTを用いた細菌基準株データベースに対する相同性検索の結果、本菌株の16SrDNA塩基配列はBurkholderia由来の16SrDNAに高い相同性を示し、Burkholderia stabilis LMG14294株の16SrDNAに対し相同率99.9%、Burkholderia pyrrocinia LMG14191株の16SrDNAに対し相同率99.7%の相同性を示した。GenBank/DDBJ/EMBLに対する相同性検索の結果においても、Burkholderia stabilis LMG14294株の16SrDNAに対し相同率99.9%の相同性を示した。また、本菌株の16SrDNAと細菌基準株データベースに対する相同性検索上位30株の16SrDNAをもちいて行った簡易分子系統樹解析の結果、本菌株はBurkholderia stabilisの16SrDNAとほぼ同一の系統枝を形成し、Burkholderia pyrrociniaにも近縁である事が示された。
【0008】
本菌株は、硝酸塩を還元し、インドールを産生せず、ウレアーゼ活性を示さず、β−ガラクトシダーゼ活性を示した。さらに、42℃で生育せず、リパーゼ活性およびレシチナーゼ活性を示し、リジンデカルボキシラーゼおよびオルニチンデカルボキシラーゼ活性を示さず、クリステンセンのクエン酸を利用した。また、グリセリンを酸化せず、アラビノース等を酸化した。これらの性状は16SrDNA塩基配列において近縁性が示唆されたBurkholderia stabilisおよびBurkholderia pyrrociniaと類似するが、β−ガラクトシダーゼ活性を示す点においてBurkholderia stabilisと異なり、硝酸塩を還元する点および硫化水素を産生しない点においてBurkholderia pyrrociniaと異なった。
【0009】
以上の事から、本菌株はBurkholderia属に含まれ、既知種ではBurkholderia stabilisおよびBurkholderia pyrrociniaに近縁であるが、16SrDNA塩基配列解析および生理生化学性状試験の結果からはBurkholderia pyrrociniaよりBurkholderia stabilisにより近縁であるために本菌株はBurkholderia stabilisと同定した。
このBurkholderia stabilisは、独立行政法人産業技術総合研究所特許性物寄託センターに平成18年8月31日付けで寄託し、受託番号FERM P−21014を得た。
【0010】
本発明のBurkholderia stabilisの培養条件はその栄養生理的性質を考慮して培養条件を選択すれば良く、通常多くの場合は、液体培養で行うが、工業的には深部通気撹拌培養を行うのが有利である。培地の栄養源としては、通常の栄養培地を使用でき、炭素源、窒素源、無機塩類等を適当に含有するものであれば、天然培地、合成培地のいずれも使用できる。
炭素源としてはグルコース、グリセロール、シュークロース、糖蜜、澱粉などの糖質、アルコール類、有機酸類、オリーブ油、大豆油等の油脂、及びこれらの組み合わせを用いることができるが、オレイン酸とミルクカゼインの組み合わせが好ましい。窒素源としてはコーンスティープリカー(CSL)、大豆粉、ペプトン、肉エキス、酵母エキス等の有機態窒素、硫安、硝酸アンモニウム、尿素などの無機態窒素を用いることができるが、酵母エキスが好ましい。無機塩類としては、食塩、塩化カリウム、硫酸マグネシウム、リン酸第一カリウム、リン酸第二カリウム、硫酸第一鉄などが使用できるが、塩化カリウム、硫酸マグネシウム、リン酸第一カリウムの組み合わせが好ましい。更に生育を促進し、酵素生産能を高めるためにエマルゲン、トリトン、ツイーン、スパンなどの適当な界面活性剤を添加してもよい。
【0011】
培養温度は、微生物が発育し、CEを生産する範囲で適宜変更し得るが、本発明のBurkholderia stabilisの場合、好ましくは、10から42℃程度、さらに好ましくは25から38℃程度である。培養時間は、条件によって多少異なるが、CEが最高収量に達する時期を見計らって適当な時期に培養を終了すればよく、本発明のBurkholderia stabilisの場合、通常は20から56時間程度である。
【0012】
培地pHは、菌が発育し、CEを生産する範囲で適宜変更し得るが、本発明のBurkholderia stabilisの場合、好ましくはpH6から8程度である。
【0013】
また、本発明のCEを製造するに当たっては、本発明のBurkholderia stabilisを栄養培地で培養して菌体内、培養液中またはその両方にCEを産生せしめることができるが、菌体内に産生させる製造方法が好ましい。
通常微生物由来のCEやリパーゼなどのエステラーゼ類は菌体外に分泌される分泌型の酵素である。これは、エステラーゼ活性により自らの細胞膜を分解する事を防ぐためと考えられる。したがって、特許文献5〜7、9〜12、及び非特許文献2〜4には全て菌体外に分泌されたCEを培養液中からCEを精製し製造する方法が記載されている。
また、CEを菌体内と菌体外に同時に産生する例も報告されているが(特許文献3〜4)、これらも菌体内より菌体外の方がCEの生産量が多い為、菌体外に分泌されたCEを培養液中から精製し製造する方法が記載されている。
【0014】
本発明のBurkholderia stabilisは栄養培地で培養して菌体内に蓄積したCEを精製して製造する事ができるが、菌体外に分泌されたCEを培養液中から精製し製造する方法に比べて次のような利点がある。
1)培地成分がCEの製造に影響を与えない。
2)培養液を濃縮するための減圧濃縮装置や濾過濃縮装置などの特別な設備が必要ない。
3)CEを培養液中から沈澱分離するために培養スケールに見合った大量の硫安、アセトン、またはアルコールの使用の必要がない。
本発明のBurkholderia stabilisを栄養培地で培養して菌体内にCEを産生させた場合、培養終了後、得られた培養物を濾過または遠心分離などの手段により菌体を採集し、次いでこの菌体を機械的方法、リゾチームなどの酵素的方法、またはアセトンや界面活性剤を用いた化学的な方法で破壊し、又、必要に応じてEDTA及び/または適当な界面活性剤やアセトンなどを添加して本発明のCEの抽出液を得る。
【0015】
本発明のBurkholderia stabilisを栄養培地で培養して菌体外にCEを産生させた場合、培養液にアセトン、硫安、エタノール、又、必要に応じてEDTA及び/または適当な界面活性剤などを添加して本発明のCEを濃縮するか、もしくは濃縮する事なく精製する。
【0016】
菌体内から得られた本発明のCEの抽出液または菌体外のCEは硫安分画、アセトン分画、アルコール分画、ゲル濾過、アフィニティークロマトグラフィー等の吸着クロマトグラフィー、イオン交換クロマトグラフィーにより処理して、純度の良い本発明のCEを得ることができる。
【0017】
本発明のCEは、比活性が高いため、経済性に優れ製造上有利である。好ましい比活性はコレステロールエステルを基質とした場合100U/mg以上であるが、更に好ましくは120U/mg以上で、最適には150U/mg以上である。なお、比活性はCEの精製純度によって変動する場合がある事は言うまでもない。
以下に、本発明のCEの理化学的性質を示す。尚、本発明の実験に使用した試薬類は、特に断らない限り、和光純薬工業社製、国産化学社製、シグマアルドリッチ社製など市販で容易に入手できるものを使用した。また、組換えDNA実験酵素試薬(制限酵素など)、ベクターDNA、キット類は特に指摘しない限りタカラバイオ株式会社より購入したものである。
【0018】
<作用>
本発明のCEの作用は(式1)の通りであり、1分子の水の存在下、1分子のコレステロールエステルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
コレステロールエステル + H2O → コレステロール + 脂肪酸 (式1)
この作用を示す酵素は、ステロールエステラーゼ(EC 3.1.1.13)に分類される。ステロールエステラーゼの作用は(式2)の通りであり、1分子の水の存在下、1分子のステロールエステルを1分子のステロールと1分子の脂肪酸に加水分解する。
ステロールエステル + H2O → ステロール + 脂肪酸 (式2)
【0019】
<酵素活性測定法>
反応試薬混合液組成
0.2M リン酸カリウム緩衝液 pH6.8 0.60ml
0.35% 4−アミノアンチピリン溶液 0.30ml
0.2% フェノール溶液 0.30ml
100U/ml ペルオキシダーゼ溶液 0.30ml
3% トリトンX−100溶液 0.30ml
10U/ml コレステロールオキシダーゼ溶液 0.60ml
基質溶液(仔牛血清液) 0.30ml
精製水 0.30ml
小試験管に上記組成の反応試薬混合液を3.0ml量り、37℃で予備加温する。10分後適当に希釈したCE溶液を0.05ml加えて混和し37℃で反応を開始する。反応開始後493nmにおける吸光度を測定して直線的に反応している1分間当たりの吸光度変化を求める。求められた吸光度変化をAs/min、CE溶液の代わりに精製水を用いた盲検をAb/minとして、酵素活性(U/ml)は(式3)で算出する。
酵素活性(U/ml)=[{(As/min−Ab/min)/12}×1/2]×3.05/0.05 (式3)
【0020】
<タンパク質濃度測定法>
タンパク質濃度はバイオラッド社のプロテインアッセイキットを用いて使用説明書記載の方法に従って測定し、ウシ血清アルブミン(以下、BSAと略称する)をスタンダードとして算出した。
【0021】
<熱安定性>
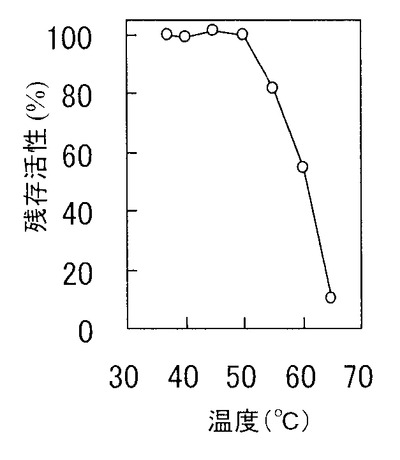
酵素を0.1mg/mlになるように20mMトリス塩酸緩衝液(pH8.0)中に溶解し、各温度で10分間熱処理した後のCEの残存活性を前記酵素活性測定法に従って測定した。図1に示すとおり、本発明のCEは55℃、10分間の熱処理で、80%以上の活性を保持する。
【0022】
<至適pH>
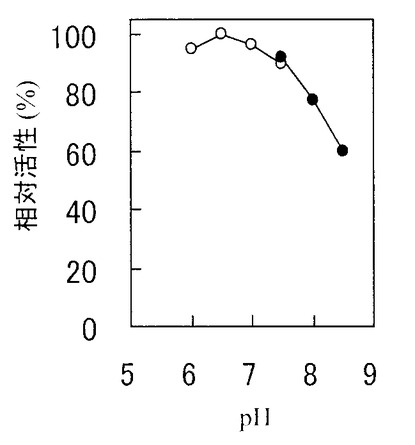
上記酵素活性測定法の緩衝液をリン酸カリウム緩衝液(pH6.0から8.0、図中○印)とトリス塩酸緩衝液(pH7.5から8.5、図中●印)として酵素活性を求めた。最大活性を100%とした相対活性を図2に示した。本発明のCEに対する至適pHはpH6.5±0.5であった。
【0023】
<pH安定性>
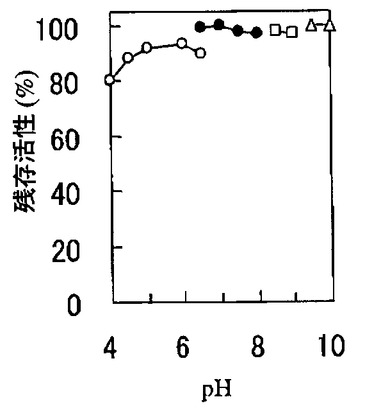
酵素を0.1mg/mlになるように100mMの3,3−ジメチルグルタル酸NaOH緩衝液(pH4.0から6.5、図中○印)、リン酸カリウム緩衝液(pH6.5から8.0、図中●印)、トリス塩酸緩衝液(pH8.5から9.0、図中□印)、グリシンNaOH緩衝液(pH9.0から10.0、図中△印)に溶解して37℃で60分間加温処理した後のCEの残存活性を前記酵素活性測定法に従って測定した。図3に示すとおり、本発明のCEは37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
【0024】
<分子量>
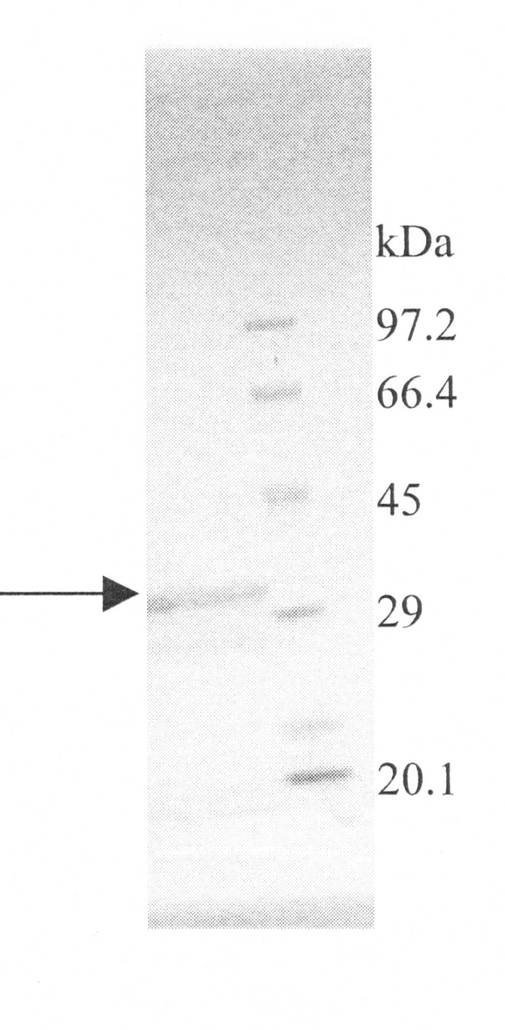
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000であった。
【0025】
<分子量>
Sephadex G−100による分子量が、31000±3000であった。
【0026】
<等電点>
キャリアーアンホラインを用いた電気泳動法にてpH4.25±0.5であった。
【0027】
<基質特異性>
上記酵素活性測定法にて、表1に示した基質に対する相対活性を、コレステロールオレートを100%として測定した。
【0028】
【表1】

【0029】
<Km>
上記酵素活性測定法にて、Km値の1/10倍から10倍の間で5つ以上の異なる濃度になるように基質濃度を調製して、ラインウェーバー・バーク逆数プロットにより見かけのKm値を算出した。その結果、コレステロールリノレートに対して1.0〜1.5mMであった。仔牛血清液を用いた場合0.5〜1.5mMであった。なお、仔牛血清液のロット差は考慮していない。
【0030】
<Burkholderia stabilis FERM P−21014のマウス毒力試験>
(材料と方法)
1.マウス:Slc:ICR系、4週齢、雄、体重約20gを用いた。白血球減少マウスモデルはサイクロフォスファマイド(CY)150mg/kg、100mg/kgをそれぞれ感染の4日前、1日前に投与した。
2.菌液:Nutrient broth培地にて培養したBurkholderia stabilis FERM P−21014(表中、B.stabilisと示す)を2500rpm5分間で遠心集菌し、PBSで懸濁して用いた。対照菌株はPseudomonas aeruginosa 0844株(表中、P.aeruginosaと示す。バイオセーフティレベル2)を用いた。
3.試験菌接種
第一試験群:正常マウスに5%ムチンを加えたPBS懸濁菌液を腹腔内接種した。
第二試験群:CY処置白血球減少マウスにPBS懸濁菌液を腹腔内接種した。
第三試験群:CY処置白血球減少マウスにPBS懸濁菌液を静脈内接種した。
4.判定:菌接種5日後までの死亡数によった。
(結果)
結果を表2に示す。
【0031】
【表2】
【0032】
Burkholderia stabilis FERM P−21014の正常マウスに対する毒力は殆ど認められなかった。白血球減少マウスに対する毒力は認められるがPseudomonas aeruginosa 0844株に比べて弱かった。日和見感染症菌であるPseudomonas aeruginosa 0844株と比較してBurkholderia stabilis FERM P−21014の毒力は弱いと考えられた。
【0033】
<比活性>
上記酵素活性測定法にて、色々なエステラーゼの比活性を比較した。結果を表3に示す。
【0034】
【表3】
【0035】
<アミノ酸配列>
本発明におけるBurkholderia stabilis FERM P−21014のCEを構成するアミノ酸配列は、具体的には、配列表配列番号1のアミノ酸配列の1から360で表されるが、配列番号1のアミノ酸配列の1から360で表されるアミノ酸配列からなるポリペプチドによる酵素活性発現と同様の効果を発現する、配列番号1のアミノ酸配列の1から360のアミノ酸配列の一部から実質的になるアミノ酸配列や、酵素活性発現に関与しない一部のアミノ酸の配列を変異、欠損または付加したもの、及びその均等物も含まれる。
付加するアミノ酸残基としてはシグナルペプチドまたはT7タグ、Hisタグ、Sタグ、Trxタグ、CBDタグ、DsbAタグ、GSTタグ、Nusタグなど通常用いられるタグも挙げられる。これらは構造遺伝子のN末端側或いはC末端側に単独、或いは複数付加する事ができる。
【0036】
<塩基配列>
配列表配列番号1の塩基配列1〜1080で表されるDNAは、そのN末端側およびC末端側のアミノ酸残基またはポリペプチド残基を含めたアミノ酸配列の各アミノ酸に対応する一連のコドンのうちいずれか1個のコドンからなるDNAであれば良い。また、配列番号1のアミノ酸配列の1から360で表されるアミノ酸配列からなるポリペプチドによる酵素活性発現と同様の効果を発現する、配列番号1のアミノ酸配列の1から360のアミノ酸配列の一部から実質的になるアミノ酸配列や酵素活性発現に関与しない一部のアミノ酸の配列を変異等により欠失、置換もしくは付加したもの、及びその均等物をコードするDNAも、そのN末端側およびC末端側のアミノ酸残基またはポリペプチド残基を含めたアミノ酸配列の各アミノ酸に対応する一連のコドンのうちいずれか1個のコドンからなるDNAであれば良い。
DNAの供与体である微生物としては、好ましくはBurkholderia stabilis FERM P−21014が挙げられるが、CEの機能や製造上の利点を向上するために、他の微生物由来のDNAや合成オリゴヌクレオチドを構造遺伝子の5’末端側或いは3’末端側に単独、或いは複数付加する事ができる。
【0037】
<ベクター>
本発明におけるBurkholderia stabilis FERM P−21014のCEをコードするDNAを組み込むベクターとしては、宿主微生物体内で自律的に増殖しうるファージまたはプラスミドから遺伝子組み換え用として構築されたものが適しており、ファージベクターとしては、例えば、エシェリヒア・コリに属する微生物を宿主微生物とする場合にはλgt・λC、λgt・λBなどが使用できる。
また、プラスミドベクターとしては、例えば、エシェリヒア・コリを宿主微生物とする場合には、プラスミドpET−3a、pET−11a、pET−32aなどのpETベクター(Novagen)またはpBR322、pBR325、pACYC184、pUC12、pUC13、pUC18、pUC19、pUC118、pINI、BluescriptKS+、枯草菌を宿主とする場合にはpWH1520、pUB110、pKH300PLK、放線菌を宿主とする場合にはpIJ680、pIJ702、酵母、特にサッカロマイセス・セレビジアエを宿主とする場合にはYRp7、pYC1、YEp13などが使用できる。他にもロドコッカス属やシュードモナス属に移入できるベクターも使用できる。
本発明の組み換えベクターは、上記ベクターに配列表配列番号1に示すアミノ酸配列を有するCE或いは該アミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を有し、かつ同様の酵素活性を有するCEをコードするDNAを組み込むことによって得ることができ、この組み換えベクターを下記に示す宿主に移入して発現させることによって本発明の形質転換体を得ることができる。
【0038】
<宿主>
プラスミドを移入する宿主微生物としては、組み換えDNAが安定かつ自律的に増殖可能であればよく、例えば宿主微生物がエシェリヒア・コリに属する微生物の場合、エシェリヒア・コリBL21、エシェリヒア・コリ BL21(DE3)、エシェリヒア・コリ BL21trxB、エシェリヒア・コリ Rosetta(DE3)、エシェリヒア・コリ Rosetta、エシェリヒア・コリ Rosetta(DE3)pLysS、エシェリヒア・コリ Rosetta(DE3)pLacl、エシェリヒア・コリ RosettaBlue、エシェリヒア・コリRosetta−gami、エシェリヒア・コリOrigami、エシェリヒア・コリ Origami、エシェリヒア・コリ Tuner、エシェリヒア・コリ DH1、エシェリヒア・コリ JM109、エシェリヒア・コリ W3110、エシェリヒア・コリC600などが利用できる。
また、宿主微生物がバチラス属に属する微生物の場合、バチラス・サチリス、バチラス・メガテリウムなど、放線菌に属する微生物の場合、ストレプトマイセス・リビダンスTK24など、サッカロマイセス・セルビシエに属する微生物の場合、サッカロマイセス・セルビシエ INVSC1などが使用できる。他にもロドコッカス属やシュードモナス属も宿主微生物として用いる事ができる。
また、ファージベクターを移入する宿主微生物としては、組み換えDNAが安定かつ自律的に増殖可能であればよく、例えば宿主微生物がエシェリヒア・コリに属する微生物の場合、エシェリヒア・コリDH1、エシェリヒア・コリ W3110、エシェリヒア・コリC600などが利用できる。
【実施例】
【0039】
本発明を実施例に基づいて説明するが、本発明の範囲は以下の例に限定されることはない。なお、実施例中、常法に従い、と記述した遺伝子操作技術は、例えばマニアティスらの方法(Maniatis,T.,et al.Molecular Cloning.Cold Spring Harbor Laboratory 1982,1989)や、市販の各種酵素、キット類に添付された手順に従えば実施できるものである。
【0040】
[実施例1]
<培養1>
容量500mlの三角フラスコに、前培養用培地としてLB液体培地100mlを入れて、蒸気加圧滅菌した後、Burkholderia stabilis FERM P−21014株を1白金耳接種した。次いで28℃で24時間振とう培養することにより、前培養液を得た。容量2リットルのジャーに、ビール酵母エキス3%、グリセロール3%、CSL2%、塩化ナトリウム0.5%、リン酸二カリウム0.2%及びトライトンX−100 0.5%からなる液体培地を入れ、pHを7に調製した後121℃で20分間蒸気加圧滅菌した後、上記の前培養液を2ml接種し、28℃、600rpmで36時間通気培養を行った。培養後、遠心分離して菌体を得た。6基培養を行ったところ、CEの培養力価は4〜6U/mlであった。
【0041】
[実施例2]
<培養2>
容量500mlの三角フラスコに、前培養用培地としてLB液体培地100mlを入れて、蒸気加圧滅菌した後、Burkholderia stabilis FERM P−21014株を1白金耳接種した。次いで28℃で24時間振とう培養することにより、前培養液を得た。容量2リットルのジャーに、大豆油2%、スキムミルク1%、ビール酵母エキス0.5%、塩化カリウム0.1%、硫酸マグネシウム0.05%からなる液体培地を入れ、pHを6.7に調製した後113℃で30分間蒸気加圧滅菌した後、上記の前培養液を2ml接種し、28℃、600rpmで36時間通気培養を行った。培養後、遠心分離して菌体を得た。6基培養を行ったところ、CEの培養力価は8〜10U/mlであった。
【0042】
[実施例3]
<培養3>
容量500mlの三角フラスコに、前培養用培地としてLB液体培地100mlを入れて、蒸気加圧滅菌した後、Burkholderia stabilis FERM P−21014株を1白金耳接種した。次いで28℃で24時間振とう培養することにより、前培養液を得た。容量2リットルのジャーに、オレイン酸2%、ミルクカゼイン1%、ビール酵母エキス0.5%、塩化カリウム0.1%、硫酸マグネシウム0.05%からなる液体培地を入れ、113℃で30分間蒸気加圧滅菌した後、上記の前培養液を2ml接種し、28℃、600rpmで30時間通気培養を行った。培養後、遠心分離して菌体を得た。6基培養を行ったところ、CEの培養力価は10〜12U/mlであった。
【0043】
[実施例4]
<培養4>
容量500mlの三角フラスコに、前培養用培地としてLB液体培地100mlを入れて、蒸気加圧滅菌した後、Burkholderia stabilis FERM P−21014株を1白金耳接種した。次いで28℃で24時間振とう培養することにより、前培養液を得た。容量2リットルのジャーに、ビール酵母エキス3%、ソルビトール1.5%、グリセロール1.5%からなる液体培地を入れ、113℃で30分間蒸気加圧滅菌した後、上記の前培養液を2ml接種し、28℃、600rpmで30時間通気培養を行った。培養後、遠心分離して菌体を得た。6基培養を行ったところ、CEの培養力価は1〜2U/mlであった。
【0044】
[実施例5]
<培養5>
容量500mlの三角フラスコに、前培養用培地としてLB液体培地100mlを入れて、蒸気加圧滅菌した後、Burkholderia stabilis FERM P−21014株を1白金耳接種した。次いで28℃で24時間振とう培養することにより、前培養液を得た。容量2リットルのジャーに、ビール酵母エキス3%、オリーブ油1.5%、グリセロール1.5%からなる液体培地を入れ、113℃で30分間蒸気加圧滅菌した後、上記の前培養液を2ml接種し、28℃、600rpmで30時間通気培養を行った。培養後、遠心分離して菌体を得た。6基培養を行ったところ、CEの培養力価は5〜7U/mlであった。
【0045】
[実施例6]
<精製1>
実施例1〜3で得られたそれぞれの菌体に0.1%リゾチーム、5mMのEDTA、1%トリトンX−100からなる可溶化液1L(pH8)で37℃3時間可溶化した後、5Lのアセトンを加えて5℃で6時間静置した。生じた沈殿を遠心分離により集めて、10mMトリス塩酸緩衝液(pH7.8)に溶解した。次に、得られた溶液に硫安を最終濃度35%になるように加えて室温で6時間静置した。生じた沈殿を遠心分離により集めて、10mMトリス塩酸緩衝液(pH7.8)に溶解した。次に、得られた溶液を10mMリン酸カリウム緩衝液pH7.5で平衡化したG−25(GEヘルスケアバイオサイエンス社製)で脱塩した後50℃で15分間加熱処理し、生じた沈殿を遠心分離により除去して精製酵素を得た。各精製収率は50%であった。得られた精製CEの比活性はいずれも150〜160U/mgであった。
【0046】
[実施例7]
<精製2>
実施例1〜3で得られた菌体を10%トリトンX−100で45℃17時間可溶化した後、5Lのアセトンを加えて5℃で6時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液に硫安を最終濃度35%になるように加えて室温で6時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液を10mMリン酸カリウム緩衝液pH7.5で平衡化したG−25(GEヘルスケアバイオサイエンス社製)で脱塩した後50℃で15分間加熱処理し、生じた沈殿を遠心分離により除去して精製酵素を得た。精製収率は40%であった。得られた精製CEの比活性は150〜170U/mgであった。
【0047】
[実施例8]
<精製3>
実施例1〜3で得られた菌体に5Lのアセトンを加えて室温で17時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液に硫安を最終濃度35%になるように加えて室温で6時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液を10mMリン酸カリウム緩衝液pH7.5で平衡化したG−25(GEヘルスケアバイオサイエンス社製)で脱塩した後50℃で15分間加熱処理し、生じた沈殿を遠心分離により除去して精製酵素を得た。精製収率は60%であった。得られた精製CEの比活性は160〜180U/mgであった。
【0048】
[実施例9]
<精製4>
実施例1〜3で得られた菌体に5Lのアセトンを加えて室温で17時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液を10mMのトリス−塩酸緩衝液(pH8.5)で平衡化したDEAE sep.FF(GEヘルスケアバイオサイエンス社製)に吸着させた。10mMのトリス−塩酸緩衝液(pH8.5)で充分に洗浄した後、0及び0.5Mの塩化カリウムを含む10mMのトリス−塩酸緩衝液(pH8.5)を用いたリニアグラジェントにて溶出した。活性画分に最終濃度15%になるように硫安を添加し、15%の硫安を含む10mMリン酸カリウム緩衝液pH7.5で平衡化したPhenyl sep.FF(GEヘルスケアバイオサイエンス社製)に吸着して15及び0%の硫安を含む10mMリン酸カリウム緩衝液pH7.5を用いたリニアグラジェントにて溶出した。活性画分は10mMリン酸カリウム緩衝液pH7.5で平衡化したG−25で脱塩した後、50℃で15分間加熱処理し、生じた沈殿を遠心分離により除去して精製酵素を得た。精製収率は20%であった。得られた精製CEの比活性は170〜190U/mgであった。
【0049】
[実施例10]
<精製5>
実施例1〜3で得られた菌体に0.1%リゾチーム、5mMのEDTA、1%トリトンX−100からなる可溶化液1L(pH8)で37℃17時間可溶化した後、5Lのアセトンを加えて5℃で17時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液を10mMのトリス−塩酸緩衝液(pH8.5)で平衡化したDEAE sep.FFに吸着させた。10mMのトリス−塩酸緩衝液(pH8.5)で充分に洗浄した後、0及び0.5Mの塩化カリウムを含む10mMのトリス−塩酸緩衝液(pH8.5)を用いたリニアグラジェントにて溶出した。活性画分に最終濃度15%になるように硫安を添加し、15%の硫安を含む10mMリン酸カリウム緩衝液pH7.5で平衡化したPhenyl sep.FFに吸着して15及び0%の硫安を含む10mMリン酸カリウム緩衝液pH7.5を用いたリニアグラジェントにて溶出した。活性画分は10mMリン酸カリウム緩衝液pH7.5で平衡化したG−25で脱塩した後、50℃で15分間加熱処理し、生じた沈殿を遠心分離により除去して精製酵素を得た。精製収率は10%であった。得られた精製CEの比活性は170〜190U/mgであった。
【0050】
[実施例11]
<DNAの抽出>
実施例1〜3で得られた菌体の一部を50mMのトリス−塩酸(pH8.0)、50mMのEDTA、15%シュークロースを含む1mg/mlリゾチーム溶液で37℃、10分処理した後、SDSを最終濃度0.25%になるよう添加して菌体を溶解した。さらに等量のフェノール/クロロホルム=1:1混合液を加え、30分攪拌した後、12000rpmで15分遠心分離処理をして水層を回収した。回収した水層に10分の1量の3Mの酢酸ナトリウム(pH5.5)を混合後、2倍量のエタノールを静かに重層し、ゲノムDNAをガラス棒に巻き付かせて分離した。分離したゲノムDNAを、10mMトリス−塩酸(pH8.0)、1mMのEDTA水溶液(TEバッファー)20mlに溶解し、20mg/mlのRNaseAを200μl加え、37℃で1時間保温し、混在しているRNAを分解した。次いで、等量のフェノール/クロロホルム混合液を加え、前記と同様に処理して、水層を分取した。分取した水層に10分の1量の3Mの酢酸ナトリウム(pH5.5)と2倍量のエタノールを加えて前記の方法でもう一度ゲノムDNAを分離した。この染色体を50mlのTE(10mMのトリス−塩酸緩衝液(pH8.0)、1mMのEDTA(pH8.0))に溶解し、TE飽和のフェノールとクロロホルムの1対1混和液20mlを加え、全体を懸濁した後、同様の遠心分離を繰り返し、上層を再び別の容器に移した。この分離した上層20mlに3Mの酢酸ナトリウム緩衝液(pH5.5)2mlとエタノール50mlを加え、撹拌後−70℃で5分間冷却した後、遠心分離(2000G、4℃、15分)し、沈澱した染色体を75%エタノールで洗い、減圧乾燥した。以上の操作によりBurkholderia stabilisのDNA標品約1mgを得た。
【0051】
[実施例12]
<本発明の酵素の部分アミノ酸配列決定>
1Mのウレア pH8に溶解した実施例10で得た本発明の酵素1mgを、アミノペプチダーゼLys−CもしくはAsp−Nで断片化した(37℃17時間)。断片は定法に従い0.1%のトリフルオロ酢酸を含むアセトニトリル−水系の逆相カラムクロマトグラフィーで分離精製しエドマン分解法にて部分アミノ酸配列を決定した。
【0052】
[実施例13]
<本発明の酵素遺伝子の部分断片の分離>
本発明のCEの遺伝子を分離するために、データベースに登録されている既知のCE遺伝子のDNA配列を元に配列番号2および3のDNA配列を有するPCR用DNAプライマーを設計し、外部機関(BEX社)に依頼して合成した。このDNAプライマーを用いて実施例11で調製したBurkholderia stabilisのDNA標品を鋳型としてPCR反応を行い、約500bpのDNA断片を取得した。このDNA断片の塩基配列を外部機関(BMR社)に依頼して決定したところ、実施例12で決定した部分アミノ酸配列の一部をコードする配列を含むことが確認され、本発明のCE遺伝子の一部であることを確認した。
【0053】
[実施例14]
<放射性DNAプローブの作製>
実施例13で取得したCE遺伝子の一部を含むDNA断片を鋳型として370kBq(キロベクレル)の[α−32P]dCTP(NEN社製)存在下、BcaBEST Labeling Kitを用いてランダムプライマー法によりラジオアイソトープ32Pを取り込ませた放射性オリゴヌクレオチドプローブを作製した。
【0054】
[実施例15]
<本発明の酵素遺伝子含有DNAフラグメントの検定>
実施例11で調製したBurkholderia stabilisのDNA標品(10μg)を各種制限酵素で切断し、1.5%アガロースゲル(タカラバイオ社、AgaroseH14(商品名)、40mMのTris−酢酸緩衝液(pH7.4)、2mMのEDTA)で150V、1.5時間電気泳動し、常法に従ってサザンブロッティングを行い、アガロースゲルからナイロンメンブレン(PALL社製:バイオダインA)にDNAを移行させた。
このメンブレンを風乾後、1平方cmあたり0.05mlのハイブリダイゼーション溶液(0.1%のフィコール、0.1%のポリビニルピロリドン、0.1%のBSA(シグマ社製)、0.75Mの塩化ナトリウム、75mMのクエン酸ナトリウム、50mMのリン酸3ナトリウム、0.1%のドデシル硫酸ナトリウム、250μg/mlのサケ精子DNA(フナコシ社製)、30%のホルムアミド)に浸し、42℃で2時間プレハイブリダイゼーション処理を行った。処理終了後、ハイブリダイゼーション溶液を新しいものに交換し、実施例14で作成した放射性DNAプローブを74kBq添加し、同じく42℃でハイブリダイゼーション処理を1晩行った。ハイブリダイゼーション後、メンブレンを100平方cm当り50mlの洗浄液(75mMの塩化ナトリウム、7.5mMのクエン酸ナトリウム、0.1%のSDS)で50℃下、10分洗った後、メンブレンを自然乾燥した。この乾燥したメンブレンをX線フィルム(富士写真フィルム社製 New RXO−H)に重ね、遮光下、−70℃で4時間オートラジオグラフィーを行った。
オートラジオグラフィー終了後、フィルムを現像し、各制限酵素による切断染色体が示すポジティブバンドのサイズを観察した。
その結果、EcoRVによる切断により約6kbのDNAフラグメント上に本発明のCE遺伝子が含有されることが明らかとなり、EcoRVで切断した染色体DNAの6kbフラグメントから遺伝子ライブラリーを作成することとした。
【0055】
[実施例16]
<遺伝子ライブラリーの作成>
実施例11で調製したBurkholderia stabilisのDNA標品10μgを制限酵素EcoRVで切断し、常法に従い約6kbのDNAフラグメントを分離した。このDNAフラグメントを、制限酵素SmaIで切断しアルカリフォスファターゼ(以下BAPと略称)1uで切断末端を脱リン酸化した1μgのpUC119と、DNA Ligation Kitで連結させた。これを用いて、常法に従ってコンピテント細胞としたエシェリヒア・コリ・JM109(東洋紡績社販売)をトランスフォーメーションし、50μg/mlアンピシリン含有LB(バクトトリプトン(DIFCO社製)10g/l、酵母エキス(DIFCO社製)5g/l、NaCl 10g/l)1.5%寒天平板培地にて一夜培養し、アンピシリン耐性コロニーを得て遺伝子ライブラリーとした。
【0056】
[実施例17]
<本発明の酵素遺伝子含有クローンのスクリーニング>
実施例16で得られた遺伝子ライブラリーを、ナイロンメンブレン(PALL社製:バイオダインA)にレプリカし、このメンブレンに添付のマニュアルに従って菌体のDNAを固定した。
このDNAを固定したメンブレンを実施例15に示したハイブリダイゼーション溶液(メンブレン1平方cm当たり20μl)に浸し、42℃で2時間プレハイブリダイゼーション処理を行った。処理終了後、ハイブリダイゼーション溶液を新しいものに交換し、実施例14で作成した放射性DNAプローブを74kBq添加し、同じく42℃でハイブリダイゼーション処理を1晩行った。ハイブリダイゼーション後、メンブレンを実施例15に示した50℃の洗浄液(メンブレン100平方cm当り50ml)で10分洗った後、自然乾燥した。この乾燥したメンブレンをX線フィルム(富士写真フィルム社製 New RXO−H)に重ね、遮光下、−70℃で4時間オートラジオグラフィーを行った。
オートラジオグラフィー終了後、フィルムを現像し、ポジティブシグナルを示すコロニーを確認した。
【0057】
[実施例18]
<組み換えプラスミドの抽出と酵素遺伝子塩基配列の決定>
実施例17で選ばれたポジティブシグナルを示すコロニーを50μg/mlのアンピシリン含有LB液体培地1.5mlに植菌し37℃で16時間振盪培養した後、常法に従ってプラスミドを抽出した。このプラスミドに挿入された染色体断片の塩基配列を外部機関(BMR社)に依頼して解析したところ、実施例12で決定した本発明のCEの部分アミノ酸配列をコードする構造遺伝子領域の全領域が確認された。この構造遺伝子の塩基配列を決定して本発明の酵素遺伝子の塩基配列とした。本発明の酵素遺伝子の塩基配列とそのコードするアミノ酸配列を配列番号1に示した。
【産業上の利用可能性】
【0058】
本発明によれば、広範囲の脂肪酸エステルを加水分解する性質があり、安定性が高く、至適pHおよびpH安定性の範囲が広く、比活性が高いCEを効率よく製造する方法を提供する事ができる。
【図面の簡単な説明】
【0059】
【図1】本発明によるコレステロールエステラーゼの熱安定性を示す図である。
【図2】本発明によるコレステロールエステラーゼの至適pHを示す図である。
【図3】本発明によるコレステロールエステラーゼのpH安定性を示す図である。
【図4】本発明によるコレステロールエステラーゼのSDS−PAGEである。
【技術分野】
【0001】
本発明は、新規なコレステロールエステラーゼの製造方法に関する。
【背景技術】
【0002】
コレステロールは人体の各臓器および各器官に広く分布している。高コレステロール血症は、動脈硬化、心筋梗塞等と関連し、低コレステロール血症は悪液質、甲状腺機能昂進等と関連するので、特に血中コレステロール濃度の把握は臨床医学的に重要である。
従来、血中コレステロールを測定する酵素法は、コレステロールオキシダーゼを用いた方法(例えば、非特許文献1参照)と、コレステロールデヒドロゲナーゼを用いた方法(例えば特許文献1又は特許文献2参照)が報告されている。
血中コレステロールとしては、遊離コレステロールと脂肪酸コレステロールエステルが存在する。コレステロールオキシダーゼ(以下CODと略する場合がある)とコレステロールデヒドロゲナーゼ(以下CDHと略する場合がある)は、遊離のコレステロールのみに作用して酸化或いは還元するため、酵素法を用いて血中コレステロールを測定する場合には、コレステロールエステラーゼ(以下CEと略する場合がある)でコレステロールエステルを加水分解してコレステロールを遊離させなければならない。
CEは、これまでにシュードモナス属(特許文献3〜6など参照)、ストレプトミセス属(特許文献7参照)、ノカルディア属(特許文献8参照)、カワラタケ(特許文献9参照)、スエヒロタケ(特許文献10参照)、ブルクホルデリア属(特許文献11参照)、キサントモナス属(特許文献11参照)などを由来とするものが知られている。
CEを、血中コレステロールの測定などの臨床診断試薬用酵素として利用する場合、CEが以下の性質を持つと有利である。
1)広範囲の脂肪酸エステルを加水分解する性質。
2)液体中での高い保存安定性。
3)広い至適pHと高いpH安定性。
4)微生物の単位培養当たりの酵素生産量が大きい事とともに、単位タンパク質当たりの酵素活性(比活性)が高い事。
これらの性質が有利な理由は、順に、
1)被験対象液中、例えば血中のコレステロールエステルの脂肪酸成分は、リノール酸、オレイン酸、及びパルミチン酸を中心として、他にもアラキドン酸、パルミトレイン酸、ステアリン酸、及びミリスチン酸などの広範な種類が含まれており、これらを加水分解する必要がある為。
2)臨床診断試薬は液状で長期間保存できると利便性が高い為。
3)臨床診断試薬組成の検討に際して、原料や試薬組成条件の選択の幅が広がる為。
4)経済性が良い為。特に、比活性が高い性質は製造上、例えば、イ)不要な混在物質の割合を減らす事が容易である、ロ)カラムクロマトグラフィーに用いる樹脂が少なくて済み精製が容易である、ハ)遺伝子組み換え体にした場合、高い酵素の発現量が期待できる、など有利である。
例えば、非特許文献2には、シュードモナス属エルギノーサ由来CEの比活性は8.5U/mgとの報告があり、特許文献6には、シュードモナス属フルオレッセンス由来CEは0.101U/mg、シュードモナス属ストゥッツェリー属由来CEの比活性は796U/mg以上との報告があり、特許文献5には、シュードモナス属由来CEは0.24U/mgとの報告がある一方で非特許文献3には、15.2U/mgとの報告がある。又、特許文献7には、ストレプトミセス属由来CEの比活性は5U/mgとの報告がある一方で非特許文献4には、78.8U/mgとの報告があり、特許文献9には、カワラタケ由来CEの比活性は5.51U/mgとの報告があり、特許文献10には、スエヒロタケ由来CEの比活性は2.5U/mgとの報告があり、特許文献11には、ブルクホルデリア属由来CEの比活性は51U/mgとの報告があり、特許文献12には、キサントモナス属由来CEの比活性は85.6U/mgとの報告がある。
これらのうち、比活性が比較的高いストレプトミセス属由来CEは培養力価が0.05U/ml以下である(非特許文献4)ために高い経済性があるとは言えない。キサントモナス属由来CEは、キサントモナス属細菌は斑点細菌病、黒腐病、又は褐班細菌病などの植物病原細菌である場合が多い事を考慮すれば容易に産業利用できるとは言い難い。シュードモナス属ストゥッツェリー属由来CEは35℃から失活が認められ、45℃で約40%失活する(特許文献6)という産業利用しづらい性質を有する。
すなわち、従来のCEはその性質上或いは生産上何らかの問題がある。
【特許文献1】特公平2−49720号公報
【特許文献2】特公平5−176797号公報
【特許文献3】特開昭50−157588号公報
【特許文献4】特開昭56−42586号公報
【特許文献5】特開昭52−7483 号公報
【特許文献6】特開平9−251号公報
【特許文献7】特開昭53−109992号公報
【特許文献8】特開昭57−43686号公報
【特許文献9】特開昭55−114288号公報
【特許文献10】特開昭53−9391号公報
【特許文献11】特開平10−127278号公報
【特許文献12】特開平11−56355号公報
【非特許文献1】Charles C. Allain、 Lucy S. Poon、 Cicely S. G. Chan、 W. Richmond、 C. Fu、Enzymatic Determination of Total Serum Cholesterol、Clin. Chem.、20巻、470頁、1974年
【非特許文献2】Akio SUGIHARA、Yuji SHIMADA、Atsuo NOMURA、Tadamasa TERAI、Masaki IMAYASU、Yusuke NAGAI、Toshihiro NAGAO、Yomi WATANABE、Yoshio TOMINAGA、Purification and Characterization of a Novel Cholesterol Esterase from Pseudomonas aeruginosa, with Its Application to Cleaning Lipid-stained Contact Lenses、Bioscience, Biotechnology, and Biochemistry、66巻、2347頁、2002年
【非特許文献3】Noriyuki Doukyu、Rikizo Aono、Purification of Extracellular Cholesterol Oxidase with High Activity in the Presence of Organic Solvents from Pseudomonas sp. Strain ST-200、Applied and Environmental Microbiology、64巻、1929頁、1998年
【非特許文献4】Hongyu Xiang、Naoki Takaya、Takayuki Hoshino、Novel Cholesterol Esterase Secreted by Streptomyces Persists during Aqueous Long-Term Storage、Journal of Bioscience and Bioengineering、101巻、19頁、2006年
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明の課題は、1)広範囲の脂肪酸エステルを加水分解する性質があり、2)液体中での保存安定性が高く、3)至適pHおよびpH安定性の範囲が広く、且つ4)比活性が高いコレステロールエステラーゼを効率よく製造する方法を提供する事にある。
【課題を解決するための手段】
【0004】
本発明者らは、上記の課題を解決するために鋭意努力した結果、土壌より分離したBurkholderia stabilis(FERM P−21014株)から生産されるコレステロールエステラーゼが、所望の性質を有する事を見出した。さらに、その効率的な工業的規模の製造方法を確立して本発明を完成に至った。即ち、本発明は、Burkholderia stabilisを培養し、その培養物から広範囲の脂肪酸エステルを加水分解可能な、安定性が高く、至適pHおよびpH安定性の範囲が広く、比活性が高いCEを採取する事を特徴とするCEの製造方法に関し、具体的には以下に関する。
〔1〕Burkholderia stabilisを培養し、培養物から下記特性を有するコレステロールエステラーゼを得ることを特徴とするコレステロールエステラーゼの製造方法。
(1)作用
1分子の水の存在下、1分子のコレステロールエステルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
〔2〕Burkholderia stabilis が、Burkholderia stabilis FERM P−21014株である上記〔1〕に記載の製造方法。
〔3〕コレステロールエステラーゼが、配列表配列番号1のアミノ酸配列或いは配列番号1のアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を有することを特徴とする上記〔1〕又は〔2〕に記載のコレステロールエステラーゼの製造方法。
〔4〕配列表配列番号1のアミノ酸配列或いは配列番号1のアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列をコードする塩基配列からなるDNAを宿主に移入し、該宿主を培養し、該培養物から下記特性を有するコレステロールエステラーゼを得ることを特徴とするコレステロールエステラーゼの製造方法。
(1)作用
1分子の水の存在下、1分子のコレステロールエステルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
〔5〕上記〔1〕から〔4〕のいずれかに記載の製造方法により得られるコレステロールエステラーゼ。
〔6〕下記(a)又は(b)のコレステロールエステラーゼをコードするDNAを含有する組み換えベクター。
(a)配列表配列番号1に示すアミノ酸配列を有するコレステロールエステラーゼ。
(b)配列表配列番号1に示すアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を有し、かつ下記特性を有するコレステロールエステラーゼ。
(1)作用
1分子の水の存在下、1分子のコレステロールエスルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
〔7〕上記〔6〕に記載の組み換えベクターを含む形質転換体。
〔8〕Burkholderia stabilisFERM P−21014株。
〔9〕下記特性を有するコレステロールエステラーゼを製造するための、Burkholderia stabilis(FERM P−21014)株の使用。
(1)作用
1分子の水の存在下、1分子のコレステロールエスルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
【発明の効果】
【0005】
本発明により、1)広範囲の脂肪酸エステルを加水分解する性質があり、2)安定性が高く、3)至適pHおよびpH安定性の範囲が広く、且つ4)比活性が高いコレステロールエステラーゼを効率よく製造する方法を提供することができる。
【発明を実施するための最良の形態】
【0006】
Burkholderia stabilisの菌学的性質を以下に示す。+は陽性、−は陰性、wは反応弱い、を示す。
1.形態学的性質
培養条件: Nutrient agar培地 30℃
細胞の形態: 桿菌(0.7〜0.8×1.5〜2.0μm)
細胞の多様性の有無:−
運動性(鞭毛の着生状態):+(極毛)
胞子の有無:−
2.培養的性質
(a)培養条件: Nutrient agar培地 30℃
色:淡黄色
光沢:+
色素産生:−
(b)培養条件: Nutrient broth培地 30℃
表面発育の有無:−
培地の混濁の有無:+
(c)培養条件: ゼラチン穿刺培養 30℃
生育状態:+
ゼラチン液化:−
(d)培養条件: リトマスミルク 30℃
凝固:−
液化:−
3.生理学的性質
グラム染色性:−
硝酸塩の還元:+
脱窒反応:−
MRテスト:−
VPテスト:−
インドール産生:−
硫化水素の生成:−
デンプンの加水分解:−
クエン酸の利用(Koser):+
クエン酸の利用(Christensen):+
無機窒素源の利用 硝酸塩:+w
無機窒素源の利用 アンモニウム塩:+
ウレアーゼ活性:−
カタラーゼ:+
オキシダーゼ:−
生育の範囲pH 5:+
生育の範囲pH 8:+
生育の範囲pH 9:+
生育の範囲温度20℃:+
生育の範囲温度25℃:+
生育の範囲温度37℃:+
生育の範囲温度45℃:−
嫌気的生育性:−
O−Fテスト(酸化/発酵):+/−
4.糖類からの酸産生/ガス産生
L−アラビノース:+/−
D−グルコース:+/−
D−フラクトース:+w/−
マルトース:+/−
ラクトース:+/−
D−ソルビトール:−/−
イノシトール:+w/−
D−キシロース:+/−
D−マンノース:−/−
D−ガラクトース:+/−
シュークロース:+/−
トレハロース:+/−
D−マンニトール:+w/−
グリセリン:−/−
5.その他の生理学的性質
β−ガラクトシダーゼ活性:+
アルギニンジヒドロラーゼ活性:−
リジンデカルボキシラーゼ活性:−
トリプトファンデアミナーゼ活性:−
ゼラチナーゼ活性:−
【0007】
BLASTを用いた細菌基準株データベースに対する相同性検索の結果、本菌株の16SrDNA塩基配列はBurkholderia由来の16SrDNAに高い相同性を示し、Burkholderia stabilis LMG14294株の16SrDNAに対し相同率99.9%、Burkholderia pyrrocinia LMG14191株の16SrDNAに対し相同率99.7%の相同性を示した。GenBank/DDBJ/EMBLに対する相同性検索の結果においても、Burkholderia stabilis LMG14294株の16SrDNAに対し相同率99.9%の相同性を示した。また、本菌株の16SrDNAと細菌基準株データベースに対する相同性検索上位30株の16SrDNAをもちいて行った簡易分子系統樹解析の結果、本菌株はBurkholderia stabilisの16SrDNAとほぼ同一の系統枝を形成し、Burkholderia pyrrociniaにも近縁である事が示された。
【0008】
本菌株は、硝酸塩を還元し、インドールを産生せず、ウレアーゼ活性を示さず、β−ガラクトシダーゼ活性を示した。さらに、42℃で生育せず、リパーゼ活性およびレシチナーゼ活性を示し、リジンデカルボキシラーゼおよびオルニチンデカルボキシラーゼ活性を示さず、クリステンセンのクエン酸を利用した。また、グリセリンを酸化せず、アラビノース等を酸化した。これらの性状は16SrDNA塩基配列において近縁性が示唆されたBurkholderia stabilisおよびBurkholderia pyrrociniaと類似するが、β−ガラクトシダーゼ活性を示す点においてBurkholderia stabilisと異なり、硝酸塩を還元する点および硫化水素を産生しない点においてBurkholderia pyrrociniaと異なった。
【0009】
以上の事から、本菌株はBurkholderia属に含まれ、既知種ではBurkholderia stabilisおよびBurkholderia pyrrociniaに近縁であるが、16SrDNA塩基配列解析および生理生化学性状試験の結果からはBurkholderia pyrrociniaよりBurkholderia stabilisにより近縁であるために本菌株はBurkholderia stabilisと同定した。
このBurkholderia stabilisは、独立行政法人産業技術総合研究所特許性物寄託センターに平成18年8月31日付けで寄託し、受託番号FERM P−21014を得た。
【0010】
本発明のBurkholderia stabilisの培養条件はその栄養生理的性質を考慮して培養条件を選択すれば良く、通常多くの場合は、液体培養で行うが、工業的には深部通気撹拌培養を行うのが有利である。培地の栄養源としては、通常の栄養培地を使用でき、炭素源、窒素源、無機塩類等を適当に含有するものであれば、天然培地、合成培地のいずれも使用できる。
炭素源としてはグルコース、グリセロール、シュークロース、糖蜜、澱粉などの糖質、アルコール類、有機酸類、オリーブ油、大豆油等の油脂、及びこれらの組み合わせを用いることができるが、オレイン酸とミルクカゼインの組み合わせが好ましい。窒素源としてはコーンスティープリカー(CSL)、大豆粉、ペプトン、肉エキス、酵母エキス等の有機態窒素、硫安、硝酸アンモニウム、尿素などの無機態窒素を用いることができるが、酵母エキスが好ましい。無機塩類としては、食塩、塩化カリウム、硫酸マグネシウム、リン酸第一カリウム、リン酸第二カリウム、硫酸第一鉄などが使用できるが、塩化カリウム、硫酸マグネシウム、リン酸第一カリウムの組み合わせが好ましい。更に生育を促進し、酵素生産能を高めるためにエマルゲン、トリトン、ツイーン、スパンなどの適当な界面活性剤を添加してもよい。
【0011】
培養温度は、微生物が発育し、CEを生産する範囲で適宜変更し得るが、本発明のBurkholderia stabilisの場合、好ましくは、10から42℃程度、さらに好ましくは25から38℃程度である。培養時間は、条件によって多少異なるが、CEが最高収量に達する時期を見計らって適当な時期に培養を終了すればよく、本発明のBurkholderia stabilisの場合、通常は20から56時間程度である。
【0012】
培地pHは、菌が発育し、CEを生産する範囲で適宜変更し得るが、本発明のBurkholderia stabilisの場合、好ましくはpH6から8程度である。
【0013】
また、本発明のCEを製造するに当たっては、本発明のBurkholderia stabilisを栄養培地で培養して菌体内、培養液中またはその両方にCEを産生せしめることができるが、菌体内に産生させる製造方法が好ましい。
通常微生物由来のCEやリパーゼなどのエステラーゼ類は菌体外に分泌される分泌型の酵素である。これは、エステラーゼ活性により自らの細胞膜を分解する事を防ぐためと考えられる。したがって、特許文献5〜7、9〜12、及び非特許文献2〜4には全て菌体外に分泌されたCEを培養液中からCEを精製し製造する方法が記載されている。
また、CEを菌体内と菌体外に同時に産生する例も報告されているが(特許文献3〜4)、これらも菌体内より菌体外の方がCEの生産量が多い為、菌体外に分泌されたCEを培養液中から精製し製造する方法が記載されている。
【0014】
本発明のBurkholderia stabilisは栄養培地で培養して菌体内に蓄積したCEを精製して製造する事ができるが、菌体外に分泌されたCEを培養液中から精製し製造する方法に比べて次のような利点がある。
1)培地成分がCEの製造に影響を与えない。
2)培養液を濃縮するための減圧濃縮装置や濾過濃縮装置などの特別な設備が必要ない。
3)CEを培養液中から沈澱分離するために培養スケールに見合った大量の硫安、アセトン、またはアルコールの使用の必要がない。
本発明のBurkholderia stabilisを栄養培地で培養して菌体内にCEを産生させた場合、培養終了後、得られた培養物を濾過または遠心分離などの手段により菌体を採集し、次いでこの菌体を機械的方法、リゾチームなどの酵素的方法、またはアセトンや界面活性剤を用いた化学的な方法で破壊し、又、必要に応じてEDTA及び/または適当な界面活性剤やアセトンなどを添加して本発明のCEの抽出液を得る。
【0015】
本発明のBurkholderia stabilisを栄養培地で培養して菌体外にCEを産生させた場合、培養液にアセトン、硫安、エタノール、又、必要に応じてEDTA及び/または適当な界面活性剤などを添加して本発明のCEを濃縮するか、もしくは濃縮する事なく精製する。
【0016】
菌体内から得られた本発明のCEの抽出液または菌体外のCEは硫安分画、アセトン分画、アルコール分画、ゲル濾過、アフィニティークロマトグラフィー等の吸着クロマトグラフィー、イオン交換クロマトグラフィーにより処理して、純度の良い本発明のCEを得ることができる。
【0017】
本発明のCEは、比活性が高いため、経済性に優れ製造上有利である。好ましい比活性はコレステロールエステルを基質とした場合100U/mg以上であるが、更に好ましくは120U/mg以上で、最適には150U/mg以上である。なお、比活性はCEの精製純度によって変動する場合がある事は言うまでもない。
以下に、本発明のCEの理化学的性質を示す。尚、本発明の実験に使用した試薬類は、特に断らない限り、和光純薬工業社製、国産化学社製、シグマアルドリッチ社製など市販で容易に入手できるものを使用した。また、組換えDNA実験酵素試薬(制限酵素など)、ベクターDNA、キット類は特に指摘しない限りタカラバイオ株式会社より購入したものである。
【0018】
<作用>
本発明のCEの作用は(式1)の通りであり、1分子の水の存在下、1分子のコレステロールエステルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
コレステロールエステル + H2O → コレステロール + 脂肪酸 (式1)
この作用を示す酵素は、ステロールエステラーゼ(EC 3.1.1.13)に分類される。ステロールエステラーゼの作用は(式2)の通りであり、1分子の水の存在下、1分子のステロールエステルを1分子のステロールと1分子の脂肪酸に加水分解する。
ステロールエステル + H2O → ステロール + 脂肪酸 (式2)
【0019】
<酵素活性測定法>
反応試薬混合液組成
0.2M リン酸カリウム緩衝液 pH6.8 0.60ml
0.35% 4−アミノアンチピリン溶液 0.30ml
0.2% フェノール溶液 0.30ml
100U/ml ペルオキシダーゼ溶液 0.30ml
3% トリトンX−100溶液 0.30ml
10U/ml コレステロールオキシダーゼ溶液 0.60ml
基質溶液(仔牛血清液) 0.30ml
精製水 0.30ml
小試験管に上記組成の反応試薬混合液を3.0ml量り、37℃で予備加温する。10分後適当に希釈したCE溶液を0.05ml加えて混和し37℃で反応を開始する。反応開始後493nmにおける吸光度を測定して直線的に反応している1分間当たりの吸光度変化を求める。求められた吸光度変化をAs/min、CE溶液の代わりに精製水を用いた盲検をAb/minとして、酵素活性(U/ml)は(式3)で算出する。
酵素活性(U/ml)=[{(As/min−Ab/min)/12}×1/2]×3.05/0.05 (式3)
【0020】
<タンパク質濃度測定法>
タンパク質濃度はバイオラッド社のプロテインアッセイキットを用いて使用説明書記載の方法に従って測定し、ウシ血清アルブミン(以下、BSAと略称する)をスタンダードとして算出した。
【0021】
<熱安定性>
酵素を0.1mg/mlになるように20mMトリス塩酸緩衝液(pH8.0)中に溶解し、各温度で10分間熱処理した後のCEの残存活性を前記酵素活性測定法に従って測定した。図1に示すとおり、本発明のCEは55℃、10分間の熱処理で、80%以上の活性を保持する。
【0022】
<至適pH>
上記酵素活性測定法の緩衝液をリン酸カリウム緩衝液(pH6.0から8.0、図中○印)とトリス塩酸緩衝液(pH7.5から8.5、図中●印)として酵素活性を求めた。最大活性を100%とした相対活性を図2に示した。本発明のCEに対する至適pHはpH6.5±0.5であった。
【0023】
<pH安定性>
酵素を0.1mg/mlになるように100mMの3,3−ジメチルグルタル酸NaOH緩衝液(pH4.0から6.5、図中○印)、リン酸カリウム緩衝液(pH6.5から8.0、図中●印)、トリス塩酸緩衝液(pH8.5から9.0、図中□印)、グリシンNaOH緩衝液(pH9.0から10.0、図中△印)に溶解して37℃で60分間加温処理した後のCEの残存活性を前記酵素活性測定法に従って測定した。図3に示すとおり、本発明のCEは37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
【0024】
<分子量>
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000であった。
【0025】
<分子量>
Sephadex G−100による分子量が、31000±3000であった。
【0026】
<等電点>
キャリアーアンホラインを用いた電気泳動法にてpH4.25±0.5であった。
【0027】
<基質特異性>
上記酵素活性測定法にて、表1に示した基質に対する相対活性を、コレステロールオレートを100%として測定した。
【0028】
【表1】
【0029】
<Km>
上記酵素活性測定法にて、Km値の1/10倍から10倍の間で5つ以上の異なる濃度になるように基質濃度を調製して、ラインウェーバー・バーク逆数プロットにより見かけのKm値を算出した。その結果、コレステロールリノレートに対して1.0〜1.5mMであった。仔牛血清液を用いた場合0.5〜1.5mMであった。なお、仔牛血清液のロット差は考慮していない。
【0030】
<Burkholderia stabilis FERM P−21014のマウス毒力試験>
(材料と方法)
1.マウス:Slc:ICR系、4週齢、雄、体重約20gを用いた。白血球減少マウスモデルはサイクロフォスファマイド(CY)150mg/kg、100mg/kgをそれぞれ感染の4日前、1日前に投与した。
2.菌液:Nutrient broth培地にて培養したBurkholderia stabilis FERM P−21014(表中、B.stabilisと示す)を2500rpm5分間で遠心集菌し、PBSで懸濁して用いた。対照菌株はPseudomonas aeruginosa 0844株(表中、P.aeruginosaと示す。バイオセーフティレベル2)を用いた。
3.試験菌接種
第一試験群:正常マウスに5%ムチンを加えたPBS懸濁菌液を腹腔内接種した。
第二試験群:CY処置白血球減少マウスにPBS懸濁菌液を腹腔内接種した。
第三試験群:CY処置白血球減少マウスにPBS懸濁菌液を静脈内接種した。
4.判定:菌接種5日後までの死亡数によった。
(結果)
結果を表2に示す。
【0031】
【表2】
【0032】
Burkholderia stabilis FERM P−21014の正常マウスに対する毒力は殆ど認められなかった。白血球減少マウスに対する毒力は認められるがPseudomonas aeruginosa 0844株に比べて弱かった。日和見感染症菌であるPseudomonas aeruginosa 0844株と比較してBurkholderia stabilis FERM P−21014の毒力は弱いと考えられた。
【0033】
<比活性>
上記酵素活性測定法にて、色々なエステラーゼの比活性を比較した。結果を表3に示す。
【0034】
【表3】
【0035】
<アミノ酸配列>
本発明におけるBurkholderia stabilis FERM P−21014のCEを構成するアミノ酸配列は、具体的には、配列表配列番号1のアミノ酸配列の1から360で表されるが、配列番号1のアミノ酸配列の1から360で表されるアミノ酸配列からなるポリペプチドによる酵素活性発現と同様の効果を発現する、配列番号1のアミノ酸配列の1から360のアミノ酸配列の一部から実質的になるアミノ酸配列や、酵素活性発現に関与しない一部のアミノ酸の配列を変異、欠損または付加したもの、及びその均等物も含まれる。
付加するアミノ酸残基としてはシグナルペプチドまたはT7タグ、Hisタグ、Sタグ、Trxタグ、CBDタグ、DsbAタグ、GSTタグ、Nusタグなど通常用いられるタグも挙げられる。これらは構造遺伝子のN末端側或いはC末端側に単独、或いは複数付加する事ができる。
【0036】
<塩基配列>
配列表配列番号1の塩基配列1〜1080で表されるDNAは、そのN末端側およびC末端側のアミノ酸残基またはポリペプチド残基を含めたアミノ酸配列の各アミノ酸に対応する一連のコドンのうちいずれか1個のコドンからなるDNAであれば良い。また、配列番号1のアミノ酸配列の1から360で表されるアミノ酸配列からなるポリペプチドによる酵素活性発現と同様の効果を発現する、配列番号1のアミノ酸配列の1から360のアミノ酸配列の一部から実質的になるアミノ酸配列や酵素活性発現に関与しない一部のアミノ酸の配列を変異等により欠失、置換もしくは付加したもの、及びその均等物をコードするDNAも、そのN末端側およびC末端側のアミノ酸残基またはポリペプチド残基を含めたアミノ酸配列の各アミノ酸に対応する一連のコドンのうちいずれか1個のコドンからなるDNAであれば良い。
DNAの供与体である微生物としては、好ましくはBurkholderia stabilis FERM P−21014が挙げられるが、CEの機能や製造上の利点を向上するために、他の微生物由来のDNAや合成オリゴヌクレオチドを構造遺伝子の5’末端側或いは3’末端側に単独、或いは複数付加する事ができる。
【0037】
<ベクター>
本発明におけるBurkholderia stabilis FERM P−21014のCEをコードするDNAを組み込むベクターとしては、宿主微生物体内で自律的に増殖しうるファージまたはプラスミドから遺伝子組み換え用として構築されたものが適しており、ファージベクターとしては、例えば、エシェリヒア・コリに属する微生物を宿主微生物とする場合にはλgt・λC、λgt・λBなどが使用できる。
また、プラスミドベクターとしては、例えば、エシェリヒア・コリを宿主微生物とする場合には、プラスミドpET−3a、pET−11a、pET−32aなどのpETベクター(Novagen)またはpBR322、pBR325、pACYC184、pUC12、pUC13、pUC18、pUC19、pUC118、pINI、BluescriptKS+、枯草菌を宿主とする場合にはpWH1520、pUB110、pKH300PLK、放線菌を宿主とする場合にはpIJ680、pIJ702、酵母、特にサッカロマイセス・セレビジアエを宿主とする場合にはYRp7、pYC1、YEp13などが使用できる。他にもロドコッカス属やシュードモナス属に移入できるベクターも使用できる。
本発明の組み換えベクターは、上記ベクターに配列表配列番号1に示すアミノ酸配列を有するCE或いは該アミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を有し、かつ同様の酵素活性を有するCEをコードするDNAを組み込むことによって得ることができ、この組み換えベクターを下記に示す宿主に移入して発現させることによって本発明の形質転換体を得ることができる。
【0038】
<宿主>
プラスミドを移入する宿主微生物としては、組み換えDNAが安定かつ自律的に増殖可能であればよく、例えば宿主微生物がエシェリヒア・コリに属する微生物の場合、エシェリヒア・コリBL21、エシェリヒア・コリ BL21(DE3)、エシェリヒア・コリ BL21trxB、エシェリヒア・コリ Rosetta(DE3)、エシェリヒア・コリ Rosetta、エシェリヒア・コリ Rosetta(DE3)pLysS、エシェリヒア・コリ Rosetta(DE3)pLacl、エシェリヒア・コリ RosettaBlue、エシェリヒア・コリRosetta−gami、エシェリヒア・コリOrigami、エシェリヒア・コリ Origami、エシェリヒア・コリ Tuner、エシェリヒア・コリ DH1、エシェリヒア・コリ JM109、エシェリヒア・コリ W3110、エシェリヒア・コリC600などが利用できる。
また、宿主微生物がバチラス属に属する微生物の場合、バチラス・サチリス、バチラス・メガテリウムなど、放線菌に属する微生物の場合、ストレプトマイセス・リビダンスTK24など、サッカロマイセス・セルビシエに属する微生物の場合、サッカロマイセス・セルビシエ INVSC1などが使用できる。他にもロドコッカス属やシュードモナス属も宿主微生物として用いる事ができる。
また、ファージベクターを移入する宿主微生物としては、組み換えDNAが安定かつ自律的に増殖可能であればよく、例えば宿主微生物がエシェリヒア・コリに属する微生物の場合、エシェリヒア・コリDH1、エシェリヒア・コリ W3110、エシェリヒア・コリC600などが利用できる。
【実施例】
【0039】
本発明を実施例に基づいて説明するが、本発明の範囲は以下の例に限定されることはない。なお、実施例中、常法に従い、と記述した遺伝子操作技術は、例えばマニアティスらの方法(Maniatis,T.,et al.Molecular Cloning.Cold Spring Harbor Laboratory 1982,1989)や、市販の各種酵素、キット類に添付された手順に従えば実施できるものである。
【0040】
[実施例1]
<培養1>
容量500mlの三角フラスコに、前培養用培地としてLB液体培地100mlを入れて、蒸気加圧滅菌した後、Burkholderia stabilis FERM P−21014株を1白金耳接種した。次いで28℃で24時間振とう培養することにより、前培養液を得た。容量2リットルのジャーに、ビール酵母エキス3%、グリセロール3%、CSL2%、塩化ナトリウム0.5%、リン酸二カリウム0.2%及びトライトンX−100 0.5%からなる液体培地を入れ、pHを7に調製した後121℃で20分間蒸気加圧滅菌した後、上記の前培養液を2ml接種し、28℃、600rpmで36時間通気培養を行った。培養後、遠心分離して菌体を得た。6基培養を行ったところ、CEの培養力価は4〜6U/mlであった。
【0041】
[実施例2]
<培養2>
容量500mlの三角フラスコに、前培養用培地としてLB液体培地100mlを入れて、蒸気加圧滅菌した後、Burkholderia stabilis FERM P−21014株を1白金耳接種した。次いで28℃で24時間振とう培養することにより、前培養液を得た。容量2リットルのジャーに、大豆油2%、スキムミルク1%、ビール酵母エキス0.5%、塩化カリウム0.1%、硫酸マグネシウム0.05%からなる液体培地を入れ、pHを6.7に調製した後113℃で30分間蒸気加圧滅菌した後、上記の前培養液を2ml接種し、28℃、600rpmで36時間通気培養を行った。培養後、遠心分離して菌体を得た。6基培養を行ったところ、CEの培養力価は8〜10U/mlであった。
【0042】
[実施例3]
<培養3>
容量500mlの三角フラスコに、前培養用培地としてLB液体培地100mlを入れて、蒸気加圧滅菌した後、Burkholderia stabilis FERM P−21014株を1白金耳接種した。次いで28℃で24時間振とう培養することにより、前培養液を得た。容量2リットルのジャーに、オレイン酸2%、ミルクカゼイン1%、ビール酵母エキス0.5%、塩化カリウム0.1%、硫酸マグネシウム0.05%からなる液体培地を入れ、113℃で30分間蒸気加圧滅菌した後、上記の前培養液を2ml接種し、28℃、600rpmで30時間通気培養を行った。培養後、遠心分離して菌体を得た。6基培養を行ったところ、CEの培養力価は10〜12U/mlであった。
【0043】
[実施例4]
<培養4>
容量500mlの三角フラスコに、前培養用培地としてLB液体培地100mlを入れて、蒸気加圧滅菌した後、Burkholderia stabilis FERM P−21014株を1白金耳接種した。次いで28℃で24時間振とう培養することにより、前培養液を得た。容量2リットルのジャーに、ビール酵母エキス3%、ソルビトール1.5%、グリセロール1.5%からなる液体培地を入れ、113℃で30分間蒸気加圧滅菌した後、上記の前培養液を2ml接種し、28℃、600rpmで30時間通気培養を行った。培養後、遠心分離して菌体を得た。6基培養を行ったところ、CEの培養力価は1〜2U/mlであった。
【0044】
[実施例5]
<培養5>
容量500mlの三角フラスコに、前培養用培地としてLB液体培地100mlを入れて、蒸気加圧滅菌した後、Burkholderia stabilis FERM P−21014株を1白金耳接種した。次いで28℃で24時間振とう培養することにより、前培養液を得た。容量2リットルのジャーに、ビール酵母エキス3%、オリーブ油1.5%、グリセロール1.5%からなる液体培地を入れ、113℃で30分間蒸気加圧滅菌した後、上記の前培養液を2ml接種し、28℃、600rpmで30時間通気培養を行った。培養後、遠心分離して菌体を得た。6基培養を行ったところ、CEの培養力価は5〜7U/mlであった。
【0045】
[実施例6]
<精製1>
実施例1〜3で得られたそれぞれの菌体に0.1%リゾチーム、5mMのEDTA、1%トリトンX−100からなる可溶化液1L(pH8)で37℃3時間可溶化した後、5Lのアセトンを加えて5℃で6時間静置した。生じた沈殿を遠心分離により集めて、10mMトリス塩酸緩衝液(pH7.8)に溶解した。次に、得られた溶液に硫安を最終濃度35%になるように加えて室温で6時間静置した。生じた沈殿を遠心分離により集めて、10mMトリス塩酸緩衝液(pH7.8)に溶解した。次に、得られた溶液を10mMリン酸カリウム緩衝液pH7.5で平衡化したG−25(GEヘルスケアバイオサイエンス社製)で脱塩した後50℃で15分間加熱処理し、生じた沈殿を遠心分離により除去して精製酵素を得た。各精製収率は50%であった。得られた精製CEの比活性はいずれも150〜160U/mgであった。
【0046】
[実施例7]
<精製2>
実施例1〜3で得られた菌体を10%トリトンX−100で45℃17時間可溶化した後、5Lのアセトンを加えて5℃で6時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液に硫安を最終濃度35%になるように加えて室温で6時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液を10mMリン酸カリウム緩衝液pH7.5で平衡化したG−25(GEヘルスケアバイオサイエンス社製)で脱塩した後50℃で15分間加熱処理し、生じた沈殿を遠心分離により除去して精製酵素を得た。精製収率は40%であった。得られた精製CEの比活性は150〜170U/mgであった。
【0047】
[実施例8]
<精製3>
実施例1〜3で得られた菌体に5Lのアセトンを加えて室温で17時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液に硫安を最終濃度35%になるように加えて室温で6時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液を10mMリン酸カリウム緩衝液pH7.5で平衡化したG−25(GEヘルスケアバイオサイエンス社製)で脱塩した後50℃で15分間加熱処理し、生じた沈殿を遠心分離により除去して精製酵素を得た。精製収率は60%であった。得られた精製CEの比活性は160〜180U/mgであった。
【0048】
[実施例9]
<精製4>
実施例1〜3で得られた菌体に5Lのアセトンを加えて室温で17時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液を10mMのトリス−塩酸緩衝液(pH8.5)で平衡化したDEAE sep.FF(GEヘルスケアバイオサイエンス社製)に吸着させた。10mMのトリス−塩酸緩衝液(pH8.5)で充分に洗浄した後、0及び0.5Mの塩化カリウムを含む10mMのトリス−塩酸緩衝液(pH8.5)を用いたリニアグラジェントにて溶出した。活性画分に最終濃度15%になるように硫安を添加し、15%の硫安を含む10mMリン酸カリウム緩衝液pH7.5で平衡化したPhenyl sep.FF(GEヘルスケアバイオサイエンス社製)に吸着して15及び0%の硫安を含む10mMリン酸カリウム緩衝液pH7.5を用いたリニアグラジェントにて溶出した。活性画分は10mMリン酸カリウム緩衝液pH7.5で平衡化したG−25で脱塩した後、50℃で15分間加熱処理し、生じた沈殿を遠心分離により除去して精製酵素を得た。精製収率は20%であった。得られた精製CEの比活性は170〜190U/mgであった。
【0049】
[実施例10]
<精製5>
実施例1〜3で得られた菌体に0.1%リゾチーム、5mMのEDTA、1%トリトンX−100からなる可溶化液1L(pH8)で37℃17時間可溶化した後、5Lのアセトンを加えて5℃で17時間静置した。生じた沈殿を遠心分離により集めて、10mMリン酸カリウム緩衝液pH7.5に溶解した。次に、得られた溶液を10mMのトリス−塩酸緩衝液(pH8.5)で平衡化したDEAE sep.FFに吸着させた。10mMのトリス−塩酸緩衝液(pH8.5)で充分に洗浄した後、0及び0.5Mの塩化カリウムを含む10mMのトリス−塩酸緩衝液(pH8.5)を用いたリニアグラジェントにて溶出した。活性画分に最終濃度15%になるように硫安を添加し、15%の硫安を含む10mMリン酸カリウム緩衝液pH7.5で平衡化したPhenyl sep.FFに吸着して15及び0%の硫安を含む10mMリン酸カリウム緩衝液pH7.5を用いたリニアグラジェントにて溶出した。活性画分は10mMリン酸カリウム緩衝液pH7.5で平衡化したG−25で脱塩した後、50℃で15分間加熱処理し、生じた沈殿を遠心分離により除去して精製酵素を得た。精製収率は10%であった。得られた精製CEの比活性は170〜190U/mgであった。
【0050】
[実施例11]
<DNAの抽出>
実施例1〜3で得られた菌体の一部を50mMのトリス−塩酸(pH8.0)、50mMのEDTA、15%シュークロースを含む1mg/mlリゾチーム溶液で37℃、10分処理した後、SDSを最終濃度0.25%になるよう添加して菌体を溶解した。さらに等量のフェノール/クロロホルム=1:1混合液を加え、30分攪拌した後、12000rpmで15分遠心分離処理をして水層を回収した。回収した水層に10分の1量の3Mの酢酸ナトリウム(pH5.5)を混合後、2倍量のエタノールを静かに重層し、ゲノムDNAをガラス棒に巻き付かせて分離した。分離したゲノムDNAを、10mMトリス−塩酸(pH8.0)、1mMのEDTA水溶液(TEバッファー)20mlに溶解し、20mg/mlのRNaseAを200μl加え、37℃で1時間保温し、混在しているRNAを分解した。次いで、等量のフェノール/クロロホルム混合液を加え、前記と同様に処理して、水層を分取した。分取した水層に10分の1量の3Mの酢酸ナトリウム(pH5.5)と2倍量のエタノールを加えて前記の方法でもう一度ゲノムDNAを分離した。この染色体を50mlのTE(10mMのトリス−塩酸緩衝液(pH8.0)、1mMのEDTA(pH8.0))に溶解し、TE飽和のフェノールとクロロホルムの1対1混和液20mlを加え、全体を懸濁した後、同様の遠心分離を繰り返し、上層を再び別の容器に移した。この分離した上層20mlに3Mの酢酸ナトリウム緩衝液(pH5.5)2mlとエタノール50mlを加え、撹拌後−70℃で5分間冷却した後、遠心分離(2000G、4℃、15分)し、沈澱した染色体を75%エタノールで洗い、減圧乾燥した。以上の操作によりBurkholderia stabilisのDNA標品約1mgを得た。
【0051】
[実施例12]
<本発明の酵素の部分アミノ酸配列決定>
1Mのウレア pH8に溶解した実施例10で得た本発明の酵素1mgを、アミノペプチダーゼLys−CもしくはAsp−Nで断片化した(37℃17時間)。断片は定法に従い0.1%のトリフルオロ酢酸を含むアセトニトリル−水系の逆相カラムクロマトグラフィーで分離精製しエドマン分解法にて部分アミノ酸配列を決定した。
【0052】
[実施例13]
<本発明の酵素遺伝子の部分断片の分離>
本発明のCEの遺伝子を分離するために、データベースに登録されている既知のCE遺伝子のDNA配列を元に配列番号2および3のDNA配列を有するPCR用DNAプライマーを設計し、外部機関(BEX社)に依頼して合成した。このDNAプライマーを用いて実施例11で調製したBurkholderia stabilisのDNA標品を鋳型としてPCR反応を行い、約500bpのDNA断片を取得した。このDNA断片の塩基配列を外部機関(BMR社)に依頼して決定したところ、実施例12で決定した部分アミノ酸配列の一部をコードする配列を含むことが確認され、本発明のCE遺伝子の一部であることを確認した。
【0053】
[実施例14]
<放射性DNAプローブの作製>
実施例13で取得したCE遺伝子の一部を含むDNA断片を鋳型として370kBq(キロベクレル)の[α−32P]dCTP(NEN社製)存在下、BcaBEST Labeling Kitを用いてランダムプライマー法によりラジオアイソトープ32Pを取り込ませた放射性オリゴヌクレオチドプローブを作製した。
【0054】
[実施例15]
<本発明の酵素遺伝子含有DNAフラグメントの検定>
実施例11で調製したBurkholderia stabilisのDNA標品(10μg)を各種制限酵素で切断し、1.5%アガロースゲル(タカラバイオ社、AgaroseH14(商品名)、40mMのTris−酢酸緩衝液(pH7.4)、2mMのEDTA)で150V、1.5時間電気泳動し、常法に従ってサザンブロッティングを行い、アガロースゲルからナイロンメンブレン(PALL社製:バイオダインA)にDNAを移行させた。
このメンブレンを風乾後、1平方cmあたり0.05mlのハイブリダイゼーション溶液(0.1%のフィコール、0.1%のポリビニルピロリドン、0.1%のBSA(シグマ社製)、0.75Mの塩化ナトリウム、75mMのクエン酸ナトリウム、50mMのリン酸3ナトリウム、0.1%のドデシル硫酸ナトリウム、250μg/mlのサケ精子DNA(フナコシ社製)、30%のホルムアミド)に浸し、42℃で2時間プレハイブリダイゼーション処理を行った。処理終了後、ハイブリダイゼーション溶液を新しいものに交換し、実施例14で作成した放射性DNAプローブを74kBq添加し、同じく42℃でハイブリダイゼーション処理を1晩行った。ハイブリダイゼーション後、メンブレンを100平方cm当り50mlの洗浄液(75mMの塩化ナトリウム、7.5mMのクエン酸ナトリウム、0.1%のSDS)で50℃下、10分洗った後、メンブレンを自然乾燥した。この乾燥したメンブレンをX線フィルム(富士写真フィルム社製 New RXO−H)に重ね、遮光下、−70℃で4時間オートラジオグラフィーを行った。
オートラジオグラフィー終了後、フィルムを現像し、各制限酵素による切断染色体が示すポジティブバンドのサイズを観察した。
その結果、EcoRVによる切断により約6kbのDNAフラグメント上に本発明のCE遺伝子が含有されることが明らかとなり、EcoRVで切断した染色体DNAの6kbフラグメントから遺伝子ライブラリーを作成することとした。
【0055】
[実施例16]
<遺伝子ライブラリーの作成>
実施例11で調製したBurkholderia stabilisのDNA標品10μgを制限酵素EcoRVで切断し、常法に従い約6kbのDNAフラグメントを分離した。このDNAフラグメントを、制限酵素SmaIで切断しアルカリフォスファターゼ(以下BAPと略称)1uで切断末端を脱リン酸化した1μgのpUC119と、DNA Ligation Kitで連結させた。これを用いて、常法に従ってコンピテント細胞としたエシェリヒア・コリ・JM109(東洋紡績社販売)をトランスフォーメーションし、50μg/mlアンピシリン含有LB(バクトトリプトン(DIFCO社製)10g/l、酵母エキス(DIFCO社製)5g/l、NaCl 10g/l)1.5%寒天平板培地にて一夜培養し、アンピシリン耐性コロニーを得て遺伝子ライブラリーとした。
【0056】
[実施例17]
<本発明の酵素遺伝子含有クローンのスクリーニング>
実施例16で得られた遺伝子ライブラリーを、ナイロンメンブレン(PALL社製:バイオダインA)にレプリカし、このメンブレンに添付のマニュアルに従って菌体のDNAを固定した。
このDNAを固定したメンブレンを実施例15に示したハイブリダイゼーション溶液(メンブレン1平方cm当たり20μl)に浸し、42℃で2時間プレハイブリダイゼーション処理を行った。処理終了後、ハイブリダイゼーション溶液を新しいものに交換し、実施例14で作成した放射性DNAプローブを74kBq添加し、同じく42℃でハイブリダイゼーション処理を1晩行った。ハイブリダイゼーション後、メンブレンを実施例15に示した50℃の洗浄液(メンブレン100平方cm当り50ml)で10分洗った後、自然乾燥した。この乾燥したメンブレンをX線フィルム(富士写真フィルム社製 New RXO−H)に重ね、遮光下、−70℃で4時間オートラジオグラフィーを行った。
オートラジオグラフィー終了後、フィルムを現像し、ポジティブシグナルを示すコロニーを確認した。
【0057】
[実施例18]
<組み換えプラスミドの抽出と酵素遺伝子塩基配列の決定>
実施例17で選ばれたポジティブシグナルを示すコロニーを50μg/mlのアンピシリン含有LB液体培地1.5mlに植菌し37℃で16時間振盪培養した後、常法に従ってプラスミドを抽出した。このプラスミドに挿入された染色体断片の塩基配列を外部機関(BMR社)に依頼して解析したところ、実施例12で決定した本発明のCEの部分アミノ酸配列をコードする構造遺伝子領域の全領域が確認された。この構造遺伝子の塩基配列を決定して本発明の酵素遺伝子の塩基配列とした。本発明の酵素遺伝子の塩基配列とそのコードするアミノ酸配列を配列番号1に示した。
【産業上の利用可能性】
【0058】
本発明によれば、広範囲の脂肪酸エステルを加水分解する性質があり、安定性が高く、至適pHおよびpH安定性の範囲が広く、比活性が高いCEを効率よく製造する方法を提供する事ができる。
【図面の簡単な説明】
【0059】
【図1】本発明によるコレステロールエステラーゼの熱安定性を示す図である。
【図2】本発明によるコレステロールエステラーゼの至適pHを示す図である。
【図3】本発明によるコレステロールエステラーゼのpH安定性を示す図である。
【図4】本発明によるコレステロールエステラーゼのSDS−PAGEである。
【特許請求の範囲】
【請求項1】
Burkholderia stabilisを培養し、培養物から下記特性を有するコレステロールエステラーゼを得ることを特徴とするコレステロールエステラーゼの製造方法。
(1)作用
1分子の水の存在下、1分子のコレステロールエステルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
【請求項2】
Burkholderia stabilis が、Burkholderia stabilis FERM P−21014株である請求項1に記載の製造方法。
【請求項3】
コレステロールエステラーゼが、配列表配列番号1のアミノ酸配列或いは配列番号1のアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を有することを特徴とする請求項1又は2に記載のコレステロールエステラーゼの製造方法。
【請求項4】
配列表配列番号1のアミノ酸配列或いは配列番号1のアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列をコードする塩基配列からなるDNAを宿主に移入し、該宿主を培養し、該培養物から下記特性を有するコレステロールエステラーゼを得ることを特徴とするコレステロールエステラーゼの製造方法。
(1)作用
1分子の水の存在下、1分子のコレステロールエステルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
【請求項5】
請求項1から4のいずれかに記載の製造方法により得られるコレステロールエステラーゼ。
【請求項6】
下記(a)又は(b)のコレステロールエステラーゼをコードするDNAを含有する組み換えベクター。
(a)配列表配列番号1に示すアミノ酸配列を有するコレステロールエステラーゼ。
(b)配列表配列番号1に示すアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を有し、かつ下記特性を有するコレステロールエステラーゼ。
(1)作用
1分子の水の存在下、1分子のコレステロールエスルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
【請求項7】
請求項6に記載の組み換えベクターを含む形質転換体。
【請求項8】
Burkholderia stabilis FERM P−21014株。
【請求項9】
下記特性を有するコレステロールエステラーゼを製造するための、Burkholderia stabilis(FERM P−21014)株の使用。
(1)作用
1分子の水の存在下、1分子のコレステロールエスルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
【請求項1】
Burkholderia stabilisを培養し、培養物から下記特性を有するコレステロールエステラーゼを得ることを特徴とするコレステロールエステラーゼの製造方法。
(1)作用
1分子の水の存在下、1分子のコレステロールエステルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
【請求項2】
Burkholderia stabilis が、Burkholderia stabilis FERM P−21014株である請求項1に記載の製造方法。
【請求項3】
コレステロールエステラーゼが、配列表配列番号1のアミノ酸配列或いは配列番号1のアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を有することを特徴とする請求項1又は2に記載のコレステロールエステラーゼの製造方法。
【請求項4】
配列表配列番号1のアミノ酸配列或いは配列番号1のアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列をコードする塩基配列からなるDNAを宿主に移入し、該宿主を培養し、該培養物から下記特性を有するコレステロールエステラーゼを得ることを特徴とするコレステロールエステラーゼの製造方法。
(1)作用
1分子の水の存在下、1分子のコレステロールエステルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
【請求項5】
請求項1から4のいずれかに記載の製造方法により得られるコレステロールエステラーゼ。
【請求項6】
下記(a)又は(b)のコレステロールエステラーゼをコードするDNAを含有する組み換えベクター。
(a)配列表配列番号1に示すアミノ酸配列を有するコレステロールエステラーゼ。
(b)配列表配列番号1に示すアミノ酸配列から1又は数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を有し、かつ下記特性を有するコレステロールエステラーゼ。
(1)作用
1分子の水の存在下、1分子のコレステロールエスルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
【請求項7】
請求項6に記載の組み換えベクターを含む形質転換体。
【請求項8】
Burkholderia stabilis FERM P−21014株。
【請求項9】
下記特性を有するコレステロールエステラーゼを製造するための、Burkholderia stabilis(FERM P−21014)株の使用。
(1)作用
1分子の水の存在下、1分子のコレステロールエスルを1分子のコレステロールと1分子の脂肪酸に加水分解する。
(2)熱安定性
55℃、10分間の熱処理で、80%以上の活性を保持する。
(3)比活性
コレステロールエステルを基質とした場合、100U/mg以上。
(4)至適pH
pH6.5±0.5。
(5)pH安定性
37℃、60分間でpH4から10の範囲で80%以上の活性を保持する。
(6)分子量
SDSポリアクリルアミドゲル電気泳動法による分子量が、29500±3000。
(7)分子量
Sephadex G−100による分子量が、31000±3000。
(8)等電点
pH4.25±0.5。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2008−86277(P2008−86277A)
【公開日】平成20年4月17日(2008.4.17)
【国際特許分類】
【出願番号】特願2006−272414(P2006−272414)
【出願日】平成18年10月4日(2006.10.4)
【出願人】(303046299)旭化成ファーマ株式会社 (105)
【Fターム(参考)】
【公開日】平成20年4月17日(2008.4.17)
【国際特許分類】
【出願日】平成18年10月4日(2006.10.4)
【出願人】(303046299)旭化成ファーマ株式会社 (105)
【Fターム(参考)】
[ Back to top ]