
新規なシタグリプチンの塩
本発明は、新規の薬学的に許容されるシタグリプチンの塩、これらの調製のための方法およびこれらを含む医薬組成物に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規の薬学的に許容されるシタグリプチンの塩、これらの調製のための方法およびこれらを含む医薬組成物に関する。
【背景技術】
【0002】
化合物、(2R)−4−オキソ−4−[3−(トリフルオロメチル)−5,6−ジヒドロ[1,2,4]−トリアゾロ[4,3−a]ピラジン−7(8H)−イル]−1−(2,4,5−トリフルオロフェニル)ブタン−2−アミン、別名シタグリプチン(化1)は、ジペプチジルペプチダーゼ−IV(DPP−IV)の阻害薬として作用することが示されている。
【0003】
【化1】

【0004】
WO2003/004498は、総じて言えば、糖尿病、特に2型糖尿病などの、ジペプチジルペプチダーゼ−IV酵素が関係している疾患の治療または予防において有用であるジペプチジルペプチダーゼ−IVの阻害薬に関するものであり、具体的にはシタグリプチンを開示している。全体を通して、薬学的に許容される非毒性の酸から調製される塩など、一般的なクラスの化合物の塩の例が挙げられている。WO2003/004498は、特定の結晶形の塩の調製および特性に関しては何も記載していない。
【0005】
WO2005/003135は、ジペプチジルペプチダーゼ−IV阻害薬であるシタグリプチンのリン酸二水素塩およびその結晶性水和物、特に、結晶性一水和物を記載している。WO2005/003135には、シタグリプチンリン酸二水素塩および結晶性水和物は、処理、取扱い、および投与の容易さなどの医薬組成物の調製における利点を有すると記載されている。特に、これらの塩は、ストレス、高い温度および湿度に対する安定性などの改善された物理的および化学的安定性、ならびに、溶解度および溶解速度などの改善された物理化学的特性を示す。
【0006】
WO2005/020920は、シタグリプチンのリン酸二水素塩の結晶性無水物I型、II型およびIII型ならびに溶媒和物を記載している。
【0007】
WO2005/030127は、シタグリプチンのリン酸二水素塩の新規な結晶性無水物IV型を記載している。
【0008】
WO2006/033848は、非晶性のシタグリプチンリン酸二水素塩を記載している。
【0009】
WO2005/072530は、結晶性のシタグリプチンの塩酸、ベンゼンスルホン酸、p−トルエンスルホン酸、10−カンファースルホン酸および酒石酸の塩ならびにこれらの水和物を記載している。
【0010】
WO2007/035198は、シタグリプチンのドデシル硫酸塩、特に、その結晶性無水物形を記載している。
【0011】
塩は、作用機構に基づいて、その主要な薬学的活性を変更することなく、母化合物の物理的および生物学的特徴をしばしば改善する。
【0012】
それゆえ、改善された物理的および/または化学的特性を有するシタグリプチンの新規な塩を得る必要性が継続して存在する。本発明は、医薬製剤の機能を決定する活性薬剤成分の本質的な特性としての、水または水性媒体への向上した溶解度を有する、シタグリプチンの新規な塩を提供することによりその必要性を満たす。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】国際公開第2003/004498号
【特許文献2】国際公開第2005/003135号
【特許文献3】国際公開第2005/020920号
【特許文献4】国際公開第2005/030127号
【特許文献5】国際公開第2006/033848号
【特許文献6】国際公開第2005/072530号
【特許文献7】国際公開第2007/035198号
【発明の概要】
【課題を解決するための手段】
【0014】
本発明は、主要な態様および好ましい実施形態を含む以下の各項を提供する。これらは、それぞれ単独でおよび組合せで、特に、上記の目的を解決することに寄与し、結果として追加の利点を提供する。
【0015】
(1)D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、エタンスルホン酸、シュウ酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸からなる群から選択される薬学的に許容される酸とのシタグリプチンの塩、ならびに前記塩の水和物および溶媒和物。
【0016】
(2)薬学的に許容される酸が、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、L−マンデル酸およびD−マンデル酸からなる群から選択される、(1)項に記載のシタグリプチンの塩、ならびに前記塩の水和物および溶媒和物。
【0017】
(3)シタグリプチンD−グルクロン酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、(1)項に記載のシタグリプチンの塩。
【0018】
(4)結晶形であり、以下の特徴的な反射角2θ:5.1±0.2°、12.7±0.2°、15.4±0.2°、17.1±0.2°、19.5±0.2°、21.5±0.2°、22.5±0.2°、26.2±0.2°および26.9±0.2°を含む粉末X線回折パターンを有する、前記項に記載のシタグリプチンD−グルクロン酸塩。
【0019】
(5)シタグリプチングルタル酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、(1)項に記載のシタグリプチンの塩。
【0020】
(6)結晶形であり、以下の特徴的な反射角2θ:6.4±0.2°、8.0±0.2°、12.9±0.2°、15.4±0.2°、17.6±0.2°、20.8±0.2°、23.0±0.2°、24.7±0.2°、25.4±0.2°および26.6±0.2°を含む粉末X線回折パターンを有する、(5)項に記載のシタグリプチングルタル酸塩。
【0021】
(7)シタグリプチン硫酸水素塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、(1)項に記載のシタグリプチンの塩。
【0022】
(8)結晶形であり、以下の特徴的な反射角2θ:4.7±0.2°、14.2±0.2°、15.4±0.2°、18.1±0.2°、19.3±0.2°、22.0±0.2°、23.3±0.2°、24.4±0.2°、26.3±0.2°および26.8±0.2°を含む粉末X線回折パターンを有する、(7)項に記載のシタグリプチン硫酸水素塩。
【0023】
(9)シタグリプチンL−乳酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、(1)項に記載のシタグリプチンの塩。
【0024】
(10)結晶形であり、以下の特徴的な反射角2θ:6.4±0.2°、7.9±0.2°、10.5±0.2°、17.8±0.2°、20.3±0.2°、21.5±0.2°、23.8±0.2°、24.5±0.2°、25.7±0.2°および27.3±0.2°を含む粉末X線回折パターンを有する、項(9)に記載のシタグリプチンL−乳酸塩。
【0025】
(11)シタグリプチンシュウ酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、(1)項に記載のシタグリプチンの塩。
【0026】
(12)結晶形であり、以下の特徴的な反射角2θ:8.4±0.2°、11.2±0.2°、17.0±0.2°、17.5±0.2°、18.4±0.2°、20.9±0.2°、23.9±0.2°、25.4±0.2°、27.0±0.2°および27.9±0.2°を含む粉末X線回折パターンを有する、(11)項に記載のシタグリプチンシュウ酸塩。
【0027】
(13)a)シタグリプチン(塩基)ならびにD−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸からなる群から選択される薬学的に許容される酸を含む混合物を準備するステップ、
b)得られたシタグリプチンの塩を単離するステップ
を含む、(1)から(12)項のいずれか一項に記載のシタグリプチンの塩の調製方法。
【0028】
(14)前記混合物が、シタグリプチン塩基および選択された薬学的に許容される酸を液体媒体中に溶解した後、前記シタグリプチンの塩が形成されるまで、溶液を場合によって攪拌しながら、40℃より低い温度、好ましくは、30℃より低い温度に維持することによって準備され、得られたシタグリプチンの塩が、温度を40℃以上に上昇させることなく、液体媒体から単離される、(13)項に記載の方法。
【0029】
(15)溶解後、塩が形成されるまで、前記混合物を約20から25℃の温度に保つ、(13)または(14)項に記載の方法。
【0030】
(16)(1)から(12)項のいずれか一項に記載のシタグリプチンの塩を含む医薬組成物。
【0031】
(17)前記シタグリプチンの塩が、シタグリプチンD−グルクロン酸塩である、(16)項に記載の医薬組成物。
【0032】
(18)前記シタグリプチンの塩が、シタグリプチングルタル酸塩である、(16)項に記載の医薬組成物。
【0033】
(19)前記シタグリプチンの塩が、シタグリプチン硫酸水素塩である、(16)項に記載の医薬組成物。
【0034】
(20)前記シタグリプチンの塩が、シタグリプチンL−乳酸塩である、(16)項に記載の医薬組成物。
【0035】
(21)前記シタグリプチンの塩が、シタグリプチンシュウ酸塩である、(16)項に記載の医薬組成物。
【0036】
(22)哺乳類における2型糖尿病の治療処置に使用するための、(16)から(21)項のいずれか一項に記載の医薬組成物。
【0037】
個々の塩の存在および特性は、本質的に予測できないものであることは強調せねばならない。したがって、数多くの酸が代替品として試験可能であるが、もしあるとすれば、どの酸が、医薬組成物中に含有させるのに適切な物理的および/または化学的特性を有するシタグリプチンの塩を提供し得るか、およびもし提供し得るとすれば、どの酸が適切な投与量または投与形態を提供し得るかを、当業者が予測するのは不可能である。例えば、活性化合物は、原則的には、錠剤またはカプセル中に、中性の形態でまたはマグネシウム塩で使用し得るが、一方で非経口使用のためにはナトリウム塩が好ましいことがある。シタグリプチンは、好ましくは、リン酸水素塩として使用されるが、リン酸塩はカルシウムおよびマグネシウムイオンにより容易に沈殿するため、カルシウムまたはマグネシウムを含む賦形剤が、生体利用性に影響を与えることがあることは周知である。さらに、カルシウムに富んだ食物を同時に摂取すると不規則な生体挙動を引き起こし得る。
【0038】
したがって、中性、酸性またはアルカリ性の水性媒体中への適切な溶解度、技術的に重要な有機溶媒への溶解度、水/脂質分配係数、電荷付与性(electrochargeability)、保存安定性、熱安定性、水および酸素に対する不活性、吸湿性、結晶型、粒径と表面、溶解プロファイル、賦形剤および組合わされる活性成分との相溶性、または最終投与形態の設計に対する特別な特性などの有利な物理化学的および生体動態学的特性を有し得る、新規のシタグリプチンの塩を得る必要性が存在する。特定の塩が、精製および不純物除去の容易さなどの、技術上有効な特性を有し得る。確かに、生理学的条件下での良好な溶解性は、特に、即時放出の経口の最終投与形態のために最も重要な特性の1つである。
【0039】
驚くべきことに、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸の群から選択される薬学的に許容されるイオンを用いると、特定の物理化学的特性を有するシタグリプチンの十分に規定された塩が形成されることが見出された。理論に固執するわけではないが、特別に選択されたイオンは、シタグリプチン構造のアミド基に結合しているベータアミン基と好ましい結合を形成し、それにより対応するシタグリプチンの塩ならびにその水和物および溶媒和物の分解を制御し、さらに水または水性媒体への迅速な溶解を可能とすると仮定し得る。
【0040】
高い水溶性および高い安定性の両者の点に関する特定の物理化学的特性の観点から、本発明の好ましい塩は、シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩およびシタグリプチンシュウ酸塩、ならびに前記塩の水和物および溶媒和物である。結晶形であるそれぞれの塩はまた、特徴的なX線回折パターンを有する固体状態を与える。
【0041】
特に、薬学的に許容される酸が、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、L−マンデル酸およびD−マンデル酸(ならびに前記塩の水和物および溶媒和物)からなる群から選択される場合は、例えば、シタグリプチンリン酸二水素塩と比較して、良好な水溶解性および高い熱安定性の両方よい全体的な特性を得ることができる。さらに、生理学的にどこにでも見られる典型的なイオンの特性も有することは、そのような塩の薬学的受容性にとって好ましい。
【0042】
具体的に言うと、本明細書に開示した塩の安定性、特に熱的安定性は、例えば、処理、取扱い、保存等の条件下での分解生成物の発生傾向を制御することに関係している。
【0043】
本発明に記載の、特に好ましい塩(シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩およびシタグリプチンシュウ酸塩)は、安定であり、水への非常に良好な溶解性を示す。さらに、そのような塩は、取扱いおよび処理が容易であり、それゆえ様々の薬学的投与形態の製造に適している。一方で熱的安定性を保ちながら、水への優れた溶解性を有することは、得られる医薬組成物にとって価値ある有用な機能を確保する。
【0044】
以下に、本発明を、添付図面を参照しながら好ましい実施形態および実施例によってより詳細に記載するが、これらの実施形態、実施例および図面は、例示の目的のためにのみ提供するのであり、いかなる意味でも本発明を制限するものではない。
【図面の簡単な説明】
【0045】
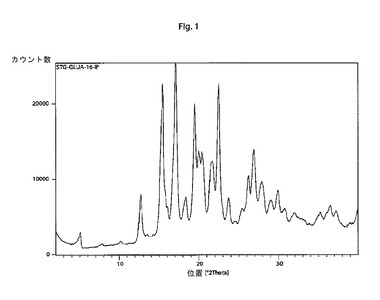
【図1】本発明の結晶性シタグリプチンD−グルクロン酸塩の特徴的なX線回折パターンである。
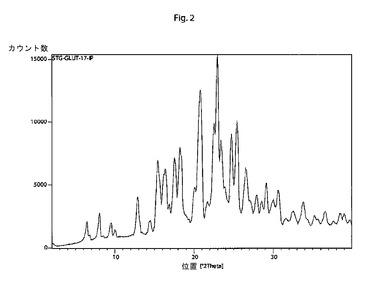
【図2】本発明の結晶性シタグリプチングルタル酸塩の特徴的なX線回折パターンである。
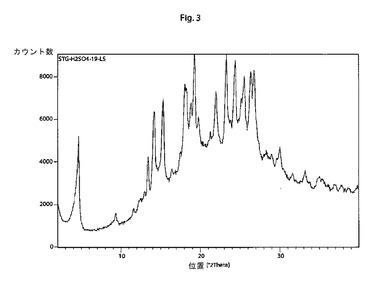
【図3】本発明の結晶性シタグリプチン硫酸水素塩の特徴的なX線回折パターンである。
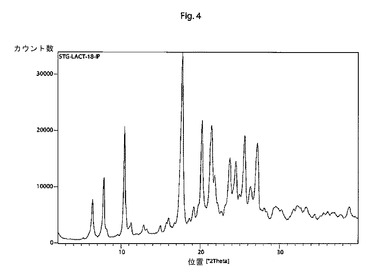
【図4】本発明の結晶性シタグリプチンL−乳酸塩の特徴的なX線回折パターンである。
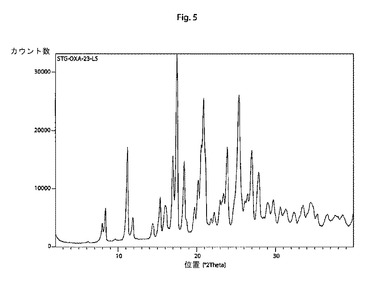
【図5】本発明の結晶性シタグリプチンシュウ酸塩の特徴的なX線回折パターンである。
【発明を実施するための形態】
【0046】
本発明は、酸が、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸からなる群から選択されるシタグリプチンの新規な酸塩、および前記酸塩の水和物ならびに溶媒和物に関する。
【0047】
好ましくは、薬学的に許容される酸は、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、L−マンデル酸およびD−マンデル酸および前記酸塩の水和物ならびに溶媒和物からなる群から選択される。本発明に記載の、特に好ましい塩は、シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩およびシタグリプチンシュウ酸塩である。
【0048】
ある態様では、本発明は、シタグリプチンD−グルクロン酸塩またはその水和物もしくは溶媒和物に関する。
【0049】
他の態様では、本発明は、結晶性シタグリプチンD−グルクロン酸塩に関する。
【0050】
他の態様では、本発明は、以下の特徴的な反射角2θ:5.1±0.2°、12.7±0.2°、15.4±0.2°、17.1±0.2°、19.5±0.2°、21.5±0.2°、22.5±0.2°、26.2±0.2°および26.9±0.2°を含む粉末X線回折パターンを有する結晶性シタグリプチンD−グルクロン酸塩に関する。
【0051】
さらに他の態様では、本発明は、非晶形のシタグリプチンD−グルクロン酸塩に関する。
【0052】
他の態様では、本発明は、シタグリプチングルタル酸塩またはその水和物もしくは溶媒和物に関する。
【0053】
他の態様では、本発明は、結晶性シタグリプチングルタル酸塩に関する。
【0054】
他の態様では、本発明は、以下の特徴的な反射角2θ:6.4±0.2°、8.0±0.2°、12.9±0.2°、15.4±0.2°、17.6±0.2°、20.8±0.2°、23.0±0.2°、24.7±0.2°、25.4±0.2°および26.6±0.2°を含む粉末X線回折パターンを有する結晶性シタグリプチングルタル酸塩に関する。
【0055】
さらに他の態様では、本発明は、非晶形のシタグリプチングルタル酸塩に関する。
【0056】
他の態様では、本発明は、シタグリプチン硫酸水素塩またはその水和物もしくは溶媒和物に関する。
【0057】
他の態様では、本発明は、結晶性シタグリプチン硫酸水素塩に関する。
【0058】
他の態様では、本発明は、以下の特徴的な反射角2θ:4.7±0.2°、14.2±0.2°、15.4±0.2°、18.1±0.2°、19.3±0.2°、22.0±0.2°、23.3±0.2°、24.4±0.2°、26.3±0.2°および26.8±0.2°を含む粉末X線回折パターンを有する結晶性シタグリプチン硫酸水素塩に関する。
【0059】
さらに他の態様では、本発明は、非晶形のシタグリプチン硫酸水素塩に関する。
【0060】
他の態様では、本発明は、シタグリプチンL−乳酸塩またはその水和物もしくは溶媒和物に関する。
【0061】
他の態様では、本発明は、結晶性シタグリプチンL−乳酸塩に関する。
【0062】
他の態様では、本発明は、以下の特徴的な反射角2θ:6.4±0.2°、7.9±0.2°、10.5±0.2°、17.8±0.2°、20.3±0.2°、21.5±0.2°、23.8±0.2°、24.5±0.2°、25.7±0.2°および27.3±0.2°を含む粉末X線回折パターンを有する結晶性シタグリプチンL−乳酸塩に関する。
【0063】
さらに他の態様では、本発明は、非晶形のシタグリプチンL−乳酸塩に関する。
【0064】
他の態様では、本発明は、シタグリプチンシュウ酸塩またはその水和物もしくは溶媒和物に関する。
【0065】
他の態様では、本発明は、結晶性シタグリプチンシュウ酸塩に関する。
【0066】
他の態様では、本発明は、以下の特徴的な反射角2θ:8.4±0.2°、11.2±0.2°、17.0±0.2°、17.5±0.2°、18.4±0.2°、20.9±0.2°、23.9±0.2°、25.4±0.2°、27.0±0.2°および27.9±0.2°を含む粉末X線回折パターンを有する結晶性シタグリプチンシュウ酸塩に関する。
【0067】
さらに他の態様では、本発明は、非晶形のシタグリプチンシュウ酸塩に関する。
【0068】
他の態様では、本発明は、それぞれ、単一の溶媒または溶媒混合物からなる適切な溶媒系中に、シタグリプチン塩基とD−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸の群から選択される薬学的に許容される酸との混合物を準備するステップ、ならびに沈殿、固体塩の濾過、蒸発、噴霧乾燥または当業界に公知の他の通常の技法により、得られたシタグリプチンの塩を単離するステップにより、シタグリプチンとD−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸の群から選択される薬学的に許容される酸との塩を調製するための方法に関する。
【0069】
適切な溶媒は、アルコール、ケトン、ニトリル、エステルおよび水またはこれらの混合物から選択される溶媒、好ましくは、アセトン、C1−C4のアルコール(好ましくは、メタノール、エタノールおよびイソプロピルアルコール(iPrOH)から選択される)、アセトニトリルおよび水またはこれらの混合物から選択される溶媒である。
【0070】
薬学的に許容される酸は、自然の状態でまたは溶液状態で、シタグリプチン塩基の溶液中へ添加され得る。
【0071】
薬学的に許容される酸は、好ましくは、シタグリプチン塩基と等モル比で添加され、または酸が過剰で用いられる。
【0072】
シタグリプチン塩基と薬学的に許容される酸との混合物を含む溶媒系の温度は、室温から溶媒系の沸点までである。シタグリプチン塩基および選択された有機酸が液体媒体に溶解された後、得られた溶液を、好ましくは、40℃より低い温度、より好ましくは、30℃より低い温度、適切には20から25℃などの室温付近に、塩が形成されるまで保持される。そのような低温条件に注意を払うことは、分解生成物の発生を危険にさらすことなく、安定な塩の形態を得ることおよび保持することに役立つ。より高温では、溶液中に、本発明の塩と共にかなりの量の分解または分裂生成物が発生することが見出された。
【0073】
シタグリプチンの塩は、反応溶液から沈殿させることによって単離または回収することができる。沈殿は、溶媒に応じて自然に生じ得る。もしくは、特に反応混合物の初期温度が高い場合は、反応混合物の温度を下げることによって沈殿を引き起こすこともできる。沈殿はまた、好ましくは減圧下で反応溶液の容積を減少させることによって引き起こすこともでき、溶媒を完全に蒸発させることによって引き起こすこともできる。さらに、貧溶媒、例えば、水、エーテルおよび炭化水素を添加することによって沈殿を行ってもよい。または、種晶を添加することによって沈殿を開始してもよい。
【0074】
本発明の1つの態様では、シタグリプチンの塩は、アルコール、ケトン、ニトリル、エステルおよび水またはこれらの混合物から選択される溶媒、好ましくは、アセトン、C1−C4のアルコール、アセトニトリルおよび水から選択される溶媒中のシタグリプチンの溶液に、薬学的に許容される酸を、自然の状態または溶液状態で添加し、場合によって、混合物を加熱して溶液を得、冷却して調製する。加熱した溶液から得られる攪拌混合物を、40℃未満、好ましくは約20から25℃などの室温以下に(場合によって、種晶を加えながら)冷却し、場合によって溶媒の一部を蒸発させて溶液を濃縮させた後、40℃未満、好ましくは、−10から25℃の間の適切な温度にて、得られた溶液を長時間放置することで、塩が沈殿する。
【0075】
別の方法として、出発成分の一方または両方の懸濁液中で再沈殿させることによって、塩が形成し、または、好ましくは、水、エーテルおよび炭化水素から、最も好ましくは、水およびジエチルエーテルから選択される貧溶媒を添加して沈殿させることによって、塩が形成する。イソプロピルアルコール(iPrOH)も貧溶媒として用い得る。
【0076】
本発明の他の態様では、シタグリプチンの塩は、薬学的に許容される酸を、自然の状態または溶解状態で、シタグリプチン塩基を含む低級アルコール、好ましくは、メタノール、エタノールまたはイソプロピルアルコール(iPrOH)の溶液に添加し、次いで溶媒を完全にまたは一部蒸発させて調製する。
【0077】
本発明の他の態様では、シタグリプチンD−グルクロン酸塩、シタグリプチンL−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩、シタグリプチンD−乳酸塩、シタグリプチンシュウ酸塩、シタグリプチンエタンスルホン酸塩、シタグリプチン酢酸塩、シタグリプチンL−マンデル酸塩、シタグリプチンD−マンデル酸塩、シタグリプチンカプリン酸塩、シタグリプチン安息香酸塩、シタグリプチン馬尿酸塩、シタグリプチントランス−桂皮酸塩、シタグリプチンマロン酸塩、シタグリプチンクエン酸塩、シタグリプチン1−ヒドロキシ−2−ナフトエ酸塩、シタグリプチンクロトン酸塩、またはシタグリプチンアスコルビン酸塩は、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸またはアスコルビン酸を、固体状態でまたは溶液状態で、アルコール、ケトン、ニトリル、エステルおよび水またはこれらの混合物から選択される溶媒、好ましくは、アセトン、C1−C4のアルコール、アセトニトリルおよび水からから選択される溶媒中のシタグリプチンの溶液に添加し、場合によって、混合物を加熱して溶液を得、冷却して調製する。加熱した溶液から得られる攪拌混合物を、40℃未満、好ましくは約20から25℃などの室温以下に(場合によって、種晶を加えながら)冷却し、場合によって溶媒の一部を蒸発させて溶液を濃縮させた後、40℃未満、好ましくは−10から25℃の間の適切な温度にて、得られた溶液を長時間放置することで、塩が沈殿する。別の方法として、出発成分の一方または両方の懸濁液中で再沈殿させることによって、塩が形成し、または、好ましくは、水、エーテルおよび炭化水素から選択される貧溶媒を添加して沈殿させることによって、塩が形成する。イソプロピルアルコール(iPrOH)も貧溶媒として用い得る。
【0078】
1つの好ましい例としては、シタグリプチン塩基をメタノールに溶解させる。固体状態のD−グルクロン酸を、攪拌下、典型的には約20から25℃などの室温付近で、好ましくは、シタグリプチン塩基に等モル比でまたはわずかに過剰で、シタグリプチン塩基の溶液中に添加する。均一な溶液が形成されたら、イソプロピルアルコール(iPrOH)を、攪拌下、典型的には室温付近で、徐々に添加する。次いで、分散液を6から24時間、好ましくは、約12時間、ほぼ室温で攪拌する。得られた結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集め、場合によってはイソプロピルアルコール(iPrOH)で洗浄する。
【0079】
そのような手順に従って調製されたシタグリプチンD−グルクロン酸塩は、以下の特徴的な反射角2θ:5.1±0.2°、12.7±0.2°、15.4±0.2°、17.1±0.2°、19.5±0.2°、21.5±0.2°、22.5±0.2°、26.2±0.2°および26.9±0.2°を含む粉末X線回折パターンを示す。
【0080】
そのような手順に従って調製されたシタグリプチンD−グルクロン酸塩は、約126から129℃の融点を示す。
【0081】
他の好ましい例では、シタグリプチン塩基をアセトニトリルに溶解させる。好ましくは、アセトニトリル中のグルタル酸の溶液を、シタグリプチン塩基の溶液に、攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で滴加する。その後、溶液混合物を、透明な溶液が形成されるまで還流温度に加熱し、次いで、好ましくは、攪拌下で室温付近まで徐冷する。溶液を冷却している間に、場合によっては種晶を加える。得られた結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0082】
前記種晶は、シタグリプチン遊離塩基をアセトニトリルに溶解して調製する。好ましくは、アセトニトリル中のグルタル酸溶液を、シタグリプチン塩基の溶液に、攪拌下、典型的には室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比で滴加し、次いで溶媒を蒸発させる。
【0083】
そのような手順に従って調製されたシタグリプチングルタル酸塩は、以下の特徴的な反射角2θ:6.4±0.2°、8.0±0.2°、12.9±0.2°、15.4±0.2°、17.6±0.2°、20.8±0.2°、23.0±0.2°、24.7±0.2°、25.4±0.2°および26.6±0.2°を含む粉末X線回折パターンを示す。
【0084】
そのような手順に従って調製されたシタグリプチングルタル酸塩は、約112から118℃の融点を示す。
【0085】
他の好ましい例では、シタグリプチン塩基をアセトニトリルに溶解させる。好ましくは、アセトニトリル中の硫酸の溶液(好ましくは、95から97%の濃硫酸を用いる)を、シタグリプチン塩基の溶液に、攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で滴加する。その後、溶液混合物を、好ましくは、約2時間、典型的には室温付近の温度で攪拌する。次いで、溶媒を蒸発させ、形成された固体を、好ましくは、メチルt−ブチルエーテル(tBuOMe)中に、場合によっては、攪拌下、典型的には室温付近の温度で、好ましくは、約2時間分散させる。得られた結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0086】
そのような手順に従って調製されたシタグリプチン硫酸水素塩は、以下の特徴的な反射角2θ:4.7±0.2°、14.2±0.2°、15.4±0.2°、18.1±0.2°、19.3±0.2°、22.0±0.2°、23.3±0.2°、24.4±0.2°、26.3±0.2°および26.8±0.2°を含む粉末X線回折パターンを示す。
【0087】
そのような手順に従って調製されたシタグリプチン硫酸水素塩は、約184から190℃の融点を示す。
【0088】
他の好ましい例では、シタグリプチン塩基を、好ましくは、それぞれ約1:5の容積比のメタノールおよびイソプロピルアルコール(iPrOH)の混合物に溶解させる。L−乳酸、好ましくは、濃度99%の濃乳酸を用いるが、シタグリプチン塩基の溶液に、典型的には約20から25℃などの室温付近の温度で、場合によっては攪拌下で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で滴加する。その後、種晶を加える。次いで、得られた分散液を6から24時間、好ましくは、約12時間室温付近で攪拌する。得られた結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0089】
前記種晶は、好ましくは、それぞれ約1:5の容積比のメタノールおよびイソプロピルアルコール(iPrOH)の混合物にシタグリプチン遊離塩基を溶解させて調製する。L−乳酸、好ましくは、濃度99%の濃乳酸を用いるが、シタグリプチン塩基の溶液に、典型的には室温付近の温度で、場合によっては攪拌下で、好ましくは、シタグリプチン塩基に対して等モル比で滴加し、次いで溶媒を蒸発させる。
【0090】
そのような手順に従って調製されたシタグリプチンL−乳酸塩は、以下の特徴的な反射角2θ:6.4±0.2°、7.9±0.2°、10.5±0.2°、17.8±0.2°、20.3±0.2°、21.5±0.2°、23.8±0.2°、24.5±0.2°、25.7±0.2°および27.3±0.2°を含む粉末X線回折パターンを示す。
【0091】
そのような手順に従って調製されたシタグリプチンL−乳酸塩は、約150から151℃の融点を示す。
【0092】
他の好ましい例では、シタグリプチン塩基をエタノールに溶解させる。シュウ酸を固体状態でシタグリプチン塩基の溶液に、攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で添加する。溶液を沈殿が形成されるまで攪拌する。得られた結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0093】
そのような手順に従って調製されたシタグリプチンシュウ酸塩は、以下の特徴的な反射角2θ:8.4±0.2°、11.2±0.2°、17.0±0.2°、17.5±0.2°、18.4±0.2°、20.9±0.2°、23.9±0.2°、25.4±0.2°、27.0±0.2°および27.9±0.2°を含む粉末X線回折パターンを示す。
【0094】
そのような手順に従って調製されたシタグリプチンシュウ酸塩は、約189から190℃の融点を示す。
【0095】
他の好ましい例では、シタグリプチン塩基を、好ましくはそれぞれ約1:6の容積比の、メタノールおよびイソプロピルアルコール(iPrOH)の混合物に溶解させる。エタンスルホン酸の水溶液、好ましくは、エタンスルホン酸の0.2M溶液を、シタグリプチン塩基の溶液に、攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で滴加する。その後、溶液混合物を好ましくは、約2時間、典型的には室温付近の温度で攪拌する。得られたシタグリプチンエタンスルホン酸塩の結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0096】
そのような手順に従って調製されたシタグリプチンエタンスルホン酸塩は、約199から202℃の融点を示す。
【0097】
他の好ましい例では、シタグリプチン塩基をメタノールに溶解させる。酢酸、好ましくは100%の濃酢酸を、シタグリプチン塩基の溶液に、攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で滴加する。その後、溶媒を蒸発させて形成した固体を、好ましくは、イソプロピルアルコール(iPrOH)中に分散し、次いで残渣全体が溶解するまで70℃付近まで加熱する。次いで、得られた溶液を、好ましくは、−5℃未満の温度で、6から24時間、好ましくは、約12時間冷凍室に入れる。得られたシタグリプチン酢酸塩の結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0098】
そのような手順に従って調製されたシタグリプチン酢酸塩は、約113から117℃の融点を示す。
【0099】
他の好ましい例では、シタグリプチン塩基をアセトニトリルに溶解させる。カプリン酸を固体状態で、シタグリプチン塩基の溶液に、典型的には約20から25℃などの室温付近の温度で、場合によっては攪拌下で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で添加する。得られた溶液を約1時間攪拌し、次いで溶媒を蒸発させる。固体のシタグリプチンカプリン酸塩を、場合によってはさらに6から24時間、好ましくは、約12時間、30℃付近で減圧下で乾燥する。
【0100】
そのような手順に従って調製されたシタグリプチンカプリン酸塩は、約93から97℃の融点を示す。
【0101】
さらに他の好ましい例では、シタグリプチン塩基をエタノールに溶解させる。L−マンデル酸を固体状態で、シタグリプチン塩基の溶液に、場合によっては攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で添加する。次いで溶媒を蒸発させ、シタグリプチンL−マンデル酸塩を白色粉末として得る。
【0102】
そのような手順に従って調製されたシタグリプチンL−マンデル酸塩は、約166から170℃の融点を示す。
【0103】
本発明の他の態様は、本発明の薬学的に許容される酸とのシタグリプチンの塩、すなわち、シタグリプチンD−グルクロン酸塩、シタグリプチンL−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩、シタグリプチンD−乳酸塩、シタグリプチンシュウ酸塩、シタグリプチンエタンスルホン酸塩、シタグリプチン酢酸塩、シタグリプチンL−マンデル酸塩、シタグリプチンD−マンデル酸塩、シタグリプチンカプリン酸塩、シタグリプチン安息香酸塩、シタグリプチン馬尿酸塩、シタグリプチントランス−桂皮酸塩、シタグリプチンマロン酸塩、シタグリプチンクエン酸塩、シタグリプチン1−ヒドロキシ−2−ナフトエ酸塩、シタグリプチンクロトン酸塩またはシタグリプチンアスコルビン酸塩、好ましくは、シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩またはシタグリプチンシュウ酸塩の治療有効量を、1種以上の薬学的に許容される担体または他の賦形剤とともに単回投与形態で投与するための医薬組成物である。
【0104】
本発明のシタグリプチンの塩の治療有効量は、シタグリプチン塩基として計算された場合、5から200mg、好ましくは、10から150mg、さらに好ましくは、25から100mgの範囲の塩の量である。
【0105】
本発明に従う薬学的に許容される塩は、例えば、錠剤、カプセル、ペレット、粒剤および坐薬の形態またはこれらの組合せ形態に具現化され得る。本発明に従う医薬組成物は、本発明のシタグリプチンの塩を、即時放出または制御放出するのに適し得る。固体の医薬組成物は、例えば、ペレット化を容易にするまたは分解もしくは吸収を制御する目的で被覆され得る。
【0106】
薬学的に許容される賦形剤は、結合剤、希釈剤、崩壊剤、安定化剤、保存剤、潤滑剤、芳香剤、着香剤、甘味剤および医薬技術の分野で公知の他の賦形剤からなる群から選択され得る。好ましくは、担体および賦形剤は、ラクトース、微小結晶性セルロース、セルロース誘導体、(例えば、ヒドロキシプロピルセルロース、クロスカルメロースナトリウム)、ポリアクリレート、炭酸カルシウム、でんぷん、コロイド状二酸化珪素、無水2塩基性リン酸カルシウム、デンプングリコール酸ナトリウム、タルク、ステアリン酸マグネシウム、ナトリウムステアリルフマラート、マンニトール、ポリビニルピロリドン、ポリエチレングリコールおよび医薬技術の分野で公知の他の賦形剤からなる群から選択され得る。
【0107】
適宜、本発明の医薬組成物は、シタグリプチンの塩に加えて1種以上の追加の薬学的活性成分を含む組合せの製品であり得る。好ましくは、1種以上の追加の薬学的活性成分は、インシュリン感受性剤、インシュリン、インシュリン模倣薬、スルホニル尿素、α‐グルコシダーゼ阻害薬、グルカゴン受容体アンタゴニスト、GLP−1、GLP−1類縁体、GLP−1模倣薬、GLP−1受容体アゴニスト、GIP、GIP模倣薬、PACAP、PACAP模倣薬、PACAP受容体アゴニスト、コレステロール低下剤、PPARδアゴニスト、抗肥満化合物、回腸胆汁酸輸送阻害薬、炎症状態での使用を意図した薬剤、抗高血圧薬、グルコキナーゼ活性薬(GKAs)、11β−ヒドロキシステロイドデヒドロキナーゼ1型阻害薬、コレステリルエステル輸送タンパク(CETP)阻害薬およびフラクトース1,6−ビスホスファターゼ阻害薬からなる群から選択される。
【0108】
最も好ましい追加の薬学的に活性な成分は、メトフォルミンおよび/またはその薬学的に許容される塩である。
【0109】
本発明に記載の医薬組成物は、医薬技術の分野に公知の方法により調製され得る。しかしながら、本明細書に開示されているシタグリプチンの塩の温度感受性の観点からは、本発明に記載の医薬組成物の調製方法の好ましい実施形態では、最終的な所望の医薬組成物を得るためのシタグリプチンの塩の処理を含むすべてのステップが、40℃、好ましくは、30℃より低い温度で実施される。このことは、特に、溶液状態または湿った状態で実施されるステップに当てはまる。結果として、各シタグリプチンの塩は、安定性を保持することができ、分解生成物をより少なく生成することができる。安定性およびより少ない分解生成物の生成は、本明細書に開示されている各融点によって規定することができる。
【0110】
本発明の他の態様は、本発明に記載のシタグリプチンの塩、すなわち、シタグリプチンD−グルクロン酸塩、シタグリプチンL−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩、シタグリプチンD−乳酸塩、シタグリプチンシュウ酸塩、シタグリプチンエタンスルホン酸塩、シタグリプチン酢酸塩、シタグリプチンL−マンデル酸塩、シタグリプチンD−マンデル酸塩、シタグリプチンカプリン酸塩、シタグリプチン安息香酸塩、シタグリプチン馬尿酸塩、シタグリプチントランス−桂皮酸塩、シタグリプチンマロン酸塩、シタグリプチンクエン酸塩、シタグリプチン1−ヒドロキシ−2−ナフトエ酸塩、シタグリプチンクロトン酸塩またはシタグリプチンアスコルビン酸塩、好ましくは、シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩またはシタグリプチンシュウ酸塩の有効量を用いることによる薬剤を使用しての、哺乳類におけるDPP−IV阻害薬が適用される臨床状態の治療および/または予防、特に、2型糖尿病、高血糖症、インシュリン抵抗性、および肥満の治療のための方法である。
【0111】
他の態様では、本発明は、哺乳類におけるDPP−IV阻害薬が適用される臨床状態の治療および/または予防、特に、2型糖尿病、高血糖症、インシュリン抵抗性、および肥満の治療のための薬剤の製造のための、本発明に記載のシタグリプチンの塩、すなわち、シタグリプチンD−グルクロン酸塩、シタグリプチンL−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩、シタグリプチンD−乳酸塩、シタグリプチンシュウ酸塩、シタグリプチンエタンスルホン酸塩、シタグリプチン酢酸塩、シタグリプチンL−マンデル酸塩、シタグリプチンD−マンデル酸塩、シタグリプチンカプリン酸塩、シタグリプチン安息香酸塩、シタグリプチン馬尿酸塩、シタグリプチントランス−桂皮酸塩、シタグリプチンマロン酸塩、シタグリプチンクエン酸塩、シタグリプチン1−ヒドロキシ−2−ナフトエ酸塩、シタグリプチンクロトン酸塩またはシタグリプチンアスコルビン酸塩、好ましくは、シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩またはシタグリプチンシュウ酸塩の使用に関する。
【実施例】
【0112】
実験の手順
【0113】
【表1】
【0114】
水への溶解度は、Avantium Technologies社のCrystal 16(商標)を用いて測定した。
【0115】
シタグリプチンリン酸塩は、WO2005/003135に記載の手順に従って調製した。
【0116】
シタグリプチン遊離塩基は、WO2003/004498に記載の手順に従って調製した。
【0117】
(実施例1)
(シタグリプチンD−グルクロン酸塩)
100mlのフラスコ中で、シタグリプチン遊離塩基(1.20g)およびD−グルクロン酸(604mg)を、メタノール(8ml)および水(3ml)に攪拌下溶解させた。均一な溶液が形成されたら、室温で、攪拌しながら、iPrOH(イソプロピルアルコール)(50ml)を徐々に添加した。白色沈殿が生じたら、分散液を室温で12時間攪拌した。形成された結晶を多孔性セラミックフィルターを通す吸引濾過を用いて集め、10mlのiPrOHで洗浄し、白色粉末(1.64g、92%)を得た。融点:126−129℃。
【0118】
(実施例2)
(シタグリプチングルタル酸塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(1.01g)をアセトニトリル(10ml)に溶解させた。次いで、グルタル酸(344mg)を含む、アセトニトリル(7ml)の溶液を攪拌下滴加した。添加後、沈殿が粘着性の残渣と共に出現した。次いで、透明な溶液が得られるまで、混合物を還流温度まで加熱し、種晶を加えて攪拌しながら室温まで徐冷した。形成された結晶を多孔性セラミックフィルターを通す吸引濾過を用いて集め、白色粉末(1.20g、90%)を得た。融点:112−118℃。
【0119】
実施例2で用いた種晶は、シタグリプチン遊離塩基(1.01g)をアセトニトリル(10ml)に溶解し、グルタル酸(344mg)を含む、アセトニトリル(7ml)を攪拌下滴加し、次いで溶媒を蒸発させて調製した。
【0120】
(実施例3)
(シタグリプチン硫酸水素塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(505mg)をアセトニトリル(10ml)に溶解させた。この溶液に、H2SO4(66.4μl、濃度95−97%)を含む、アセトニトリル(5ml)の溶液を滴加し、2時間以上攪拌した。濾過を試みたが成功しなかったので、溶液を蒸発乾固させ、形成した固体を、メチルt−ブチルエーテル(tBuOMe)(50 ml)中に、攪拌下、室温で2時間かけて分散した。形成した分散液を多孔性セラミックフィルターを通して濾過し、白色結晶を集めた(592mg。95%)。融点:184−190℃。
【0121】
(実施例4)
(シタグリプチンL−乳酸塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(1.20g)をメタノール(2ml)およびiPrOH(10ml)に溶解させた。次いで、L−乳酸(243μl、濃度99%)を滴加した。種晶を加えると、沈殿が始まった。分散液を室温で12時間攪拌した。形成された結晶を多孔性セラミックフィルターを通す吸引濾過を用いて集め、白色粉末(571mg、35%)を得た。融点:150−151℃。
【0122】
実施例4で用いた種晶は、シタグリプチン遊離塩基(1.20g)をメタノール(2ml)およびiPrOH(10ml)に溶解し、L−乳酸(243μl、濃度99%)を攪拌しながら滴加し、次いで溶媒を蒸発させて調製した。
【0123】
(実施例5)
(シタグリプチンシュウ酸塩)
エタノール(15ml)に溶解させたシタグリプチン遊離塩基(1.05g)の溶液に、シュウ酸(0.23g)を添加し、溶液を室温で沈殿が形成されるまで攪拌した。結晶(1.05g、85%)を濾過により集めた。融点:189−190℃。
【0124】
(実施例6)
(シタグリプチンエタンスルホン酸塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(303mg)をメタノール(1.5ml)およびiPrOH(10ml)に溶解させた。次いで、エタンスルホン酸の0.2M溶液(3.72ml)を滴加し、室温で2時間、攪拌した。形成された結晶を多孔性セラミックフィルターを通す吸引濾過を用いて集め、白色粉末(272mg、71%)を得た。融点:199−202℃。
【0125】
(実施例7)
(シタグリプチン酢酸塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(500mg)を3mlのメタノール中に溶解し、酢酸(81.7μl、濃度100%)を攪拌下、室温で滴加した。溶媒を蒸発させ、9mlのiPrOHをフラスコに添加した。残渣の全体が溶解するまで、約70℃までフラスコを加熱し、次いで12時間、冷凍室に入れた。形成された結晶を多孔性セラミックフィルターを通す吸引濾過を用いて白色粉末(505mg、88%)として集めた。融点:113−117℃。
【0126】
(実施例8)
(シタグリプチンカプリン酸塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(307mg)をアセトニトリル(10ml)中に溶解し、カプリン酸(130mg)を添加した。室温で1時間、反応混合物を攪拌した後、溶媒を蒸発させた。固体残渣を一晩、さらに乾燥(5mBarの減圧下、30℃)した。白色粉末(437mg)を集めた。融点:93−97℃。
【0127】
(実施例9)
(シタグリプチンL−マンデル酸塩)
シタグリプチン遊離塩基(0.54g)およびL−マンデル酸(0.20g)をエタノール(20ml)に溶解させた。溶液を蒸発させて白色粉末(0.70g、95%)を得た。融点:166−170℃。
【0128】
シタグリプチンL−リンゴ酸塩、シタグリプチンクエン酸塩、シタグリプチンマロン酸塩、シタグリプチンD−マンデル酸塩、シタグリプチン安息香酸塩およびシタグリプチン1−ヒドロキシ−2−ナフトエ酸塩などの他のシタグリプチンの塩は、実施例1から9に同様の手順に従って調製した。
【0129】
分析の方法
X線粉末回折法
粉末X線回折(XRD)パターンを得るための条件:粉末X線回折パターンは、X’Celerator検出器を有するPhilips X’Pert PRO 回折装置を用いて、CuKa線(45kVおよび40mAで操作している管)を用い、Bragg−Brentano(反射)配置で、当業界に公知の方法により得た。データは、0.033°2θのステップで、2から40°2θまで記録し、測定時間は、ステップ当り50秒である。可変分散および抗散乱スリットを、照射サンプル長を12mmに保つために用いた。
【技術分野】
【0001】
本発明は、新規の薬学的に許容されるシタグリプチンの塩、これらの調製のための方法およびこれらを含む医薬組成物に関する。
【背景技術】
【0002】
化合物、(2R)−4−オキソ−4−[3−(トリフルオロメチル)−5,6−ジヒドロ[1,2,4]−トリアゾロ[4,3−a]ピラジン−7(8H)−イル]−1−(2,4,5−トリフルオロフェニル)ブタン−2−アミン、別名シタグリプチン(化1)は、ジペプチジルペプチダーゼ−IV(DPP−IV)の阻害薬として作用することが示されている。
【0003】
【化1】
【0004】
WO2003/004498は、総じて言えば、糖尿病、特に2型糖尿病などの、ジペプチジルペプチダーゼ−IV酵素が関係している疾患の治療または予防において有用であるジペプチジルペプチダーゼ−IVの阻害薬に関するものであり、具体的にはシタグリプチンを開示している。全体を通して、薬学的に許容される非毒性の酸から調製される塩など、一般的なクラスの化合物の塩の例が挙げられている。WO2003/004498は、特定の結晶形の塩の調製および特性に関しては何も記載していない。
【0005】
WO2005/003135は、ジペプチジルペプチダーゼ−IV阻害薬であるシタグリプチンのリン酸二水素塩およびその結晶性水和物、特に、結晶性一水和物を記載している。WO2005/003135には、シタグリプチンリン酸二水素塩および結晶性水和物は、処理、取扱い、および投与の容易さなどの医薬組成物の調製における利点を有すると記載されている。特に、これらの塩は、ストレス、高い温度および湿度に対する安定性などの改善された物理的および化学的安定性、ならびに、溶解度および溶解速度などの改善された物理化学的特性を示す。
【0006】
WO2005/020920は、シタグリプチンのリン酸二水素塩の結晶性無水物I型、II型およびIII型ならびに溶媒和物を記載している。
【0007】
WO2005/030127は、シタグリプチンのリン酸二水素塩の新規な結晶性無水物IV型を記載している。
【0008】
WO2006/033848は、非晶性のシタグリプチンリン酸二水素塩を記載している。
【0009】
WO2005/072530は、結晶性のシタグリプチンの塩酸、ベンゼンスルホン酸、p−トルエンスルホン酸、10−カンファースルホン酸および酒石酸の塩ならびにこれらの水和物を記載している。
【0010】
WO2007/035198は、シタグリプチンのドデシル硫酸塩、特に、その結晶性無水物形を記載している。
【0011】
塩は、作用機構に基づいて、その主要な薬学的活性を変更することなく、母化合物の物理的および生物学的特徴をしばしば改善する。
【0012】
それゆえ、改善された物理的および/または化学的特性を有するシタグリプチンの新規な塩を得る必要性が継続して存在する。本発明は、医薬製剤の機能を決定する活性薬剤成分の本質的な特性としての、水または水性媒体への向上した溶解度を有する、シタグリプチンの新規な塩を提供することによりその必要性を満たす。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】国際公開第2003/004498号
【特許文献2】国際公開第2005/003135号
【特許文献3】国際公開第2005/020920号
【特許文献4】国際公開第2005/030127号
【特許文献5】国際公開第2006/033848号
【特許文献6】国際公開第2005/072530号
【特許文献7】国際公開第2007/035198号
【発明の概要】
【課題を解決するための手段】
【0014】
本発明は、主要な態様および好ましい実施形態を含む以下の各項を提供する。これらは、それぞれ単独でおよび組合せで、特に、上記の目的を解決することに寄与し、結果として追加の利点を提供する。
【0015】
(1)D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、エタンスルホン酸、シュウ酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸からなる群から選択される薬学的に許容される酸とのシタグリプチンの塩、ならびに前記塩の水和物および溶媒和物。
【0016】
(2)薬学的に許容される酸が、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、L−マンデル酸およびD−マンデル酸からなる群から選択される、(1)項に記載のシタグリプチンの塩、ならびに前記塩の水和物および溶媒和物。
【0017】
(3)シタグリプチンD−グルクロン酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、(1)項に記載のシタグリプチンの塩。
【0018】
(4)結晶形であり、以下の特徴的な反射角2θ:5.1±0.2°、12.7±0.2°、15.4±0.2°、17.1±0.2°、19.5±0.2°、21.5±0.2°、22.5±0.2°、26.2±0.2°および26.9±0.2°を含む粉末X線回折パターンを有する、前記項に記載のシタグリプチンD−グルクロン酸塩。
【0019】
(5)シタグリプチングルタル酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、(1)項に記載のシタグリプチンの塩。
【0020】
(6)結晶形であり、以下の特徴的な反射角2θ:6.4±0.2°、8.0±0.2°、12.9±0.2°、15.4±0.2°、17.6±0.2°、20.8±0.2°、23.0±0.2°、24.7±0.2°、25.4±0.2°および26.6±0.2°を含む粉末X線回折パターンを有する、(5)項に記載のシタグリプチングルタル酸塩。
【0021】
(7)シタグリプチン硫酸水素塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、(1)項に記載のシタグリプチンの塩。
【0022】
(8)結晶形であり、以下の特徴的な反射角2θ:4.7±0.2°、14.2±0.2°、15.4±0.2°、18.1±0.2°、19.3±0.2°、22.0±0.2°、23.3±0.2°、24.4±0.2°、26.3±0.2°および26.8±0.2°を含む粉末X線回折パターンを有する、(7)項に記載のシタグリプチン硫酸水素塩。
【0023】
(9)シタグリプチンL−乳酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、(1)項に記載のシタグリプチンの塩。
【0024】
(10)結晶形であり、以下の特徴的な反射角2θ:6.4±0.2°、7.9±0.2°、10.5±0.2°、17.8±0.2°、20.3±0.2°、21.5±0.2°、23.8±0.2°、24.5±0.2°、25.7±0.2°および27.3±0.2°を含む粉末X線回折パターンを有する、項(9)に記載のシタグリプチンL−乳酸塩。
【0025】
(11)シタグリプチンシュウ酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、(1)項に記載のシタグリプチンの塩。
【0026】
(12)結晶形であり、以下の特徴的な反射角2θ:8.4±0.2°、11.2±0.2°、17.0±0.2°、17.5±0.2°、18.4±0.2°、20.9±0.2°、23.9±0.2°、25.4±0.2°、27.0±0.2°および27.9±0.2°を含む粉末X線回折パターンを有する、(11)項に記載のシタグリプチンシュウ酸塩。
【0027】
(13)a)シタグリプチン(塩基)ならびにD−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸からなる群から選択される薬学的に許容される酸を含む混合物を準備するステップ、
b)得られたシタグリプチンの塩を単離するステップ
を含む、(1)から(12)項のいずれか一項に記載のシタグリプチンの塩の調製方法。
【0028】
(14)前記混合物が、シタグリプチン塩基および選択された薬学的に許容される酸を液体媒体中に溶解した後、前記シタグリプチンの塩が形成されるまで、溶液を場合によって攪拌しながら、40℃より低い温度、好ましくは、30℃より低い温度に維持することによって準備され、得られたシタグリプチンの塩が、温度を40℃以上に上昇させることなく、液体媒体から単離される、(13)項に記載の方法。
【0029】
(15)溶解後、塩が形成されるまで、前記混合物を約20から25℃の温度に保つ、(13)または(14)項に記載の方法。
【0030】
(16)(1)から(12)項のいずれか一項に記載のシタグリプチンの塩を含む医薬組成物。
【0031】
(17)前記シタグリプチンの塩が、シタグリプチンD−グルクロン酸塩である、(16)項に記載の医薬組成物。
【0032】
(18)前記シタグリプチンの塩が、シタグリプチングルタル酸塩である、(16)項に記載の医薬組成物。
【0033】
(19)前記シタグリプチンの塩が、シタグリプチン硫酸水素塩である、(16)項に記載の医薬組成物。
【0034】
(20)前記シタグリプチンの塩が、シタグリプチンL−乳酸塩である、(16)項に記載の医薬組成物。
【0035】
(21)前記シタグリプチンの塩が、シタグリプチンシュウ酸塩である、(16)項に記載の医薬組成物。
【0036】
(22)哺乳類における2型糖尿病の治療処置に使用するための、(16)から(21)項のいずれか一項に記載の医薬組成物。
【0037】
個々の塩の存在および特性は、本質的に予測できないものであることは強調せねばならない。したがって、数多くの酸が代替品として試験可能であるが、もしあるとすれば、どの酸が、医薬組成物中に含有させるのに適切な物理的および/または化学的特性を有するシタグリプチンの塩を提供し得るか、およびもし提供し得るとすれば、どの酸が適切な投与量または投与形態を提供し得るかを、当業者が予測するのは不可能である。例えば、活性化合物は、原則的には、錠剤またはカプセル中に、中性の形態でまたはマグネシウム塩で使用し得るが、一方で非経口使用のためにはナトリウム塩が好ましいことがある。シタグリプチンは、好ましくは、リン酸水素塩として使用されるが、リン酸塩はカルシウムおよびマグネシウムイオンにより容易に沈殿するため、カルシウムまたはマグネシウムを含む賦形剤が、生体利用性に影響を与えることがあることは周知である。さらに、カルシウムに富んだ食物を同時に摂取すると不規則な生体挙動を引き起こし得る。
【0038】
したがって、中性、酸性またはアルカリ性の水性媒体中への適切な溶解度、技術的に重要な有機溶媒への溶解度、水/脂質分配係数、電荷付与性(electrochargeability)、保存安定性、熱安定性、水および酸素に対する不活性、吸湿性、結晶型、粒径と表面、溶解プロファイル、賦形剤および組合わされる活性成分との相溶性、または最終投与形態の設計に対する特別な特性などの有利な物理化学的および生体動態学的特性を有し得る、新規のシタグリプチンの塩を得る必要性が存在する。特定の塩が、精製および不純物除去の容易さなどの、技術上有効な特性を有し得る。確かに、生理学的条件下での良好な溶解性は、特に、即時放出の経口の最終投与形態のために最も重要な特性の1つである。
【0039】
驚くべきことに、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸の群から選択される薬学的に許容されるイオンを用いると、特定の物理化学的特性を有するシタグリプチンの十分に規定された塩が形成されることが見出された。理論に固執するわけではないが、特別に選択されたイオンは、シタグリプチン構造のアミド基に結合しているベータアミン基と好ましい結合を形成し、それにより対応するシタグリプチンの塩ならびにその水和物および溶媒和物の分解を制御し、さらに水または水性媒体への迅速な溶解を可能とすると仮定し得る。
【0040】
高い水溶性および高い安定性の両者の点に関する特定の物理化学的特性の観点から、本発明の好ましい塩は、シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩およびシタグリプチンシュウ酸塩、ならびに前記塩の水和物および溶媒和物である。結晶形であるそれぞれの塩はまた、特徴的なX線回折パターンを有する固体状態を与える。
【0041】
特に、薬学的に許容される酸が、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、L−マンデル酸およびD−マンデル酸(ならびに前記塩の水和物および溶媒和物)からなる群から選択される場合は、例えば、シタグリプチンリン酸二水素塩と比較して、良好な水溶解性および高い熱安定性の両方よい全体的な特性を得ることができる。さらに、生理学的にどこにでも見られる典型的なイオンの特性も有することは、そのような塩の薬学的受容性にとって好ましい。
【0042】
具体的に言うと、本明細書に開示した塩の安定性、特に熱的安定性は、例えば、処理、取扱い、保存等の条件下での分解生成物の発生傾向を制御することに関係している。
【0043】
本発明に記載の、特に好ましい塩(シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩およびシタグリプチンシュウ酸塩)は、安定であり、水への非常に良好な溶解性を示す。さらに、そのような塩は、取扱いおよび処理が容易であり、それゆえ様々の薬学的投与形態の製造に適している。一方で熱的安定性を保ちながら、水への優れた溶解性を有することは、得られる医薬組成物にとって価値ある有用な機能を確保する。
【0044】
以下に、本発明を、添付図面を参照しながら好ましい実施形態および実施例によってより詳細に記載するが、これらの実施形態、実施例および図面は、例示の目的のためにのみ提供するのであり、いかなる意味でも本発明を制限するものではない。
【図面の簡単な説明】
【0045】
【図1】本発明の結晶性シタグリプチンD−グルクロン酸塩の特徴的なX線回折パターンである。
【図2】本発明の結晶性シタグリプチングルタル酸塩の特徴的なX線回折パターンである。
【図3】本発明の結晶性シタグリプチン硫酸水素塩の特徴的なX線回折パターンである。
【図4】本発明の結晶性シタグリプチンL−乳酸塩の特徴的なX線回折パターンである。
【図5】本発明の結晶性シタグリプチンシュウ酸塩の特徴的なX線回折パターンである。
【発明を実施するための形態】
【0046】
本発明は、酸が、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸からなる群から選択されるシタグリプチンの新規な酸塩、および前記酸塩の水和物ならびに溶媒和物に関する。
【0047】
好ましくは、薬学的に許容される酸は、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、L−マンデル酸およびD−マンデル酸および前記酸塩の水和物ならびに溶媒和物からなる群から選択される。本発明に記載の、特に好ましい塩は、シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩およびシタグリプチンシュウ酸塩である。
【0048】
ある態様では、本発明は、シタグリプチンD−グルクロン酸塩またはその水和物もしくは溶媒和物に関する。
【0049】
他の態様では、本発明は、結晶性シタグリプチンD−グルクロン酸塩に関する。
【0050】
他の態様では、本発明は、以下の特徴的な反射角2θ:5.1±0.2°、12.7±0.2°、15.4±0.2°、17.1±0.2°、19.5±0.2°、21.5±0.2°、22.5±0.2°、26.2±0.2°および26.9±0.2°を含む粉末X線回折パターンを有する結晶性シタグリプチンD−グルクロン酸塩に関する。
【0051】
さらに他の態様では、本発明は、非晶形のシタグリプチンD−グルクロン酸塩に関する。
【0052】
他の態様では、本発明は、シタグリプチングルタル酸塩またはその水和物もしくは溶媒和物に関する。
【0053】
他の態様では、本発明は、結晶性シタグリプチングルタル酸塩に関する。
【0054】
他の態様では、本発明は、以下の特徴的な反射角2θ:6.4±0.2°、8.0±0.2°、12.9±0.2°、15.4±0.2°、17.6±0.2°、20.8±0.2°、23.0±0.2°、24.7±0.2°、25.4±0.2°および26.6±0.2°を含む粉末X線回折パターンを有する結晶性シタグリプチングルタル酸塩に関する。
【0055】
さらに他の態様では、本発明は、非晶形のシタグリプチングルタル酸塩に関する。
【0056】
他の態様では、本発明は、シタグリプチン硫酸水素塩またはその水和物もしくは溶媒和物に関する。
【0057】
他の態様では、本発明は、結晶性シタグリプチン硫酸水素塩に関する。
【0058】
他の態様では、本発明は、以下の特徴的な反射角2θ:4.7±0.2°、14.2±0.2°、15.4±0.2°、18.1±0.2°、19.3±0.2°、22.0±0.2°、23.3±0.2°、24.4±0.2°、26.3±0.2°および26.8±0.2°を含む粉末X線回折パターンを有する結晶性シタグリプチン硫酸水素塩に関する。
【0059】
さらに他の態様では、本発明は、非晶形のシタグリプチン硫酸水素塩に関する。
【0060】
他の態様では、本発明は、シタグリプチンL−乳酸塩またはその水和物もしくは溶媒和物に関する。
【0061】
他の態様では、本発明は、結晶性シタグリプチンL−乳酸塩に関する。
【0062】
他の態様では、本発明は、以下の特徴的な反射角2θ:6.4±0.2°、7.9±0.2°、10.5±0.2°、17.8±0.2°、20.3±0.2°、21.5±0.2°、23.8±0.2°、24.5±0.2°、25.7±0.2°および27.3±0.2°を含む粉末X線回折パターンを有する結晶性シタグリプチンL−乳酸塩に関する。
【0063】
さらに他の態様では、本発明は、非晶形のシタグリプチンL−乳酸塩に関する。
【0064】
他の態様では、本発明は、シタグリプチンシュウ酸塩またはその水和物もしくは溶媒和物に関する。
【0065】
他の態様では、本発明は、結晶性シタグリプチンシュウ酸塩に関する。
【0066】
他の態様では、本発明は、以下の特徴的な反射角2θ:8.4±0.2°、11.2±0.2°、17.0±0.2°、17.5±0.2°、18.4±0.2°、20.9±0.2°、23.9±0.2°、25.4±0.2°、27.0±0.2°および27.9±0.2°を含む粉末X線回折パターンを有する結晶性シタグリプチンシュウ酸塩に関する。
【0067】
さらに他の態様では、本発明は、非晶形のシタグリプチンシュウ酸塩に関する。
【0068】
他の態様では、本発明は、それぞれ、単一の溶媒または溶媒混合物からなる適切な溶媒系中に、シタグリプチン塩基とD−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸の群から選択される薬学的に許容される酸との混合物を準備するステップ、ならびに沈殿、固体塩の濾過、蒸発、噴霧乾燥または当業界に公知の他の通常の技法により、得られたシタグリプチンの塩を単離するステップにより、シタグリプチンとD−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸の群から選択される薬学的に許容される酸との塩を調製するための方法に関する。
【0069】
適切な溶媒は、アルコール、ケトン、ニトリル、エステルおよび水またはこれらの混合物から選択される溶媒、好ましくは、アセトン、C1−C4のアルコール(好ましくは、メタノール、エタノールおよびイソプロピルアルコール(iPrOH)から選択される)、アセトニトリルおよび水またはこれらの混合物から選択される溶媒である。
【0070】
薬学的に許容される酸は、自然の状態でまたは溶液状態で、シタグリプチン塩基の溶液中へ添加され得る。
【0071】
薬学的に許容される酸は、好ましくは、シタグリプチン塩基と等モル比で添加され、または酸が過剰で用いられる。
【0072】
シタグリプチン塩基と薬学的に許容される酸との混合物を含む溶媒系の温度は、室温から溶媒系の沸点までである。シタグリプチン塩基および選択された有機酸が液体媒体に溶解された後、得られた溶液を、好ましくは、40℃より低い温度、より好ましくは、30℃より低い温度、適切には20から25℃などの室温付近に、塩が形成されるまで保持される。そのような低温条件に注意を払うことは、分解生成物の発生を危険にさらすことなく、安定な塩の形態を得ることおよび保持することに役立つ。より高温では、溶液中に、本発明の塩と共にかなりの量の分解または分裂生成物が発生することが見出された。
【0073】
シタグリプチンの塩は、反応溶液から沈殿させることによって単離または回収することができる。沈殿は、溶媒に応じて自然に生じ得る。もしくは、特に反応混合物の初期温度が高い場合は、反応混合物の温度を下げることによって沈殿を引き起こすこともできる。沈殿はまた、好ましくは減圧下で反応溶液の容積を減少させることによって引き起こすこともでき、溶媒を完全に蒸発させることによって引き起こすこともできる。さらに、貧溶媒、例えば、水、エーテルおよび炭化水素を添加することによって沈殿を行ってもよい。または、種晶を添加することによって沈殿を開始してもよい。
【0074】
本発明の1つの態様では、シタグリプチンの塩は、アルコール、ケトン、ニトリル、エステルおよび水またはこれらの混合物から選択される溶媒、好ましくは、アセトン、C1−C4のアルコール、アセトニトリルおよび水から選択される溶媒中のシタグリプチンの溶液に、薬学的に許容される酸を、自然の状態または溶液状態で添加し、場合によって、混合物を加熱して溶液を得、冷却して調製する。加熱した溶液から得られる攪拌混合物を、40℃未満、好ましくは約20から25℃などの室温以下に(場合によって、種晶を加えながら)冷却し、場合によって溶媒の一部を蒸発させて溶液を濃縮させた後、40℃未満、好ましくは、−10から25℃の間の適切な温度にて、得られた溶液を長時間放置することで、塩が沈殿する。
【0075】
別の方法として、出発成分の一方または両方の懸濁液中で再沈殿させることによって、塩が形成し、または、好ましくは、水、エーテルおよび炭化水素から、最も好ましくは、水およびジエチルエーテルから選択される貧溶媒を添加して沈殿させることによって、塩が形成する。イソプロピルアルコール(iPrOH)も貧溶媒として用い得る。
【0076】
本発明の他の態様では、シタグリプチンの塩は、薬学的に許容される酸を、自然の状態または溶解状態で、シタグリプチン塩基を含む低級アルコール、好ましくは、メタノール、エタノールまたはイソプロピルアルコール(iPrOH)の溶液に添加し、次いで溶媒を完全にまたは一部蒸発させて調製する。
【0077】
本発明の他の態様では、シタグリプチンD−グルクロン酸塩、シタグリプチンL−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩、シタグリプチンD−乳酸塩、シタグリプチンシュウ酸塩、シタグリプチンエタンスルホン酸塩、シタグリプチン酢酸塩、シタグリプチンL−マンデル酸塩、シタグリプチンD−マンデル酸塩、シタグリプチンカプリン酸塩、シタグリプチン安息香酸塩、シタグリプチン馬尿酸塩、シタグリプチントランス−桂皮酸塩、シタグリプチンマロン酸塩、シタグリプチンクエン酸塩、シタグリプチン1−ヒドロキシ−2−ナフトエ酸塩、シタグリプチンクロトン酸塩、またはシタグリプチンアスコルビン酸塩は、D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、エタンスルホン酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸またはアスコルビン酸を、固体状態でまたは溶液状態で、アルコール、ケトン、ニトリル、エステルおよび水またはこれらの混合物から選択される溶媒、好ましくは、アセトン、C1−C4のアルコール、アセトニトリルおよび水からから選択される溶媒中のシタグリプチンの溶液に添加し、場合によって、混合物を加熱して溶液を得、冷却して調製する。加熱した溶液から得られる攪拌混合物を、40℃未満、好ましくは約20から25℃などの室温以下に(場合によって、種晶を加えながら)冷却し、場合によって溶媒の一部を蒸発させて溶液を濃縮させた後、40℃未満、好ましくは−10から25℃の間の適切な温度にて、得られた溶液を長時間放置することで、塩が沈殿する。別の方法として、出発成分の一方または両方の懸濁液中で再沈殿させることによって、塩が形成し、または、好ましくは、水、エーテルおよび炭化水素から選択される貧溶媒を添加して沈殿させることによって、塩が形成する。イソプロピルアルコール(iPrOH)も貧溶媒として用い得る。
【0078】
1つの好ましい例としては、シタグリプチン塩基をメタノールに溶解させる。固体状態のD−グルクロン酸を、攪拌下、典型的には約20から25℃などの室温付近で、好ましくは、シタグリプチン塩基に等モル比でまたはわずかに過剰で、シタグリプチン塩基の溶液中に添加する。均一な溶液が形成されたら、イソプロピルアルコール(iPrOH)を、攪拌下、典型的には室温付近で、徐々に添加する。次いで、分散液を6から24時間、好ましくは、約12時間、ほぼ室温で攪拌する。得られた結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集め、場合によってはイソプロピルアルコール(iPrOH)で洗浄する。
【0079】
そのような手順に従って調製されたシタグリプチンD−グルクロン酸塩は、以下の特徴的な反射角2θ:5.1±0.2°、12.7±0.2°、15.4±0.2°、17.1±0.2°、19.5±0.2°、21.5±0.2°、22.5±0.2°、26.2±0.2°および26.9±0.2°を含む粉末X線回折パターンを示す。
【0080】
そのような手順に従って調製されたシタグリプチンD−グルクロン酸塩は、約126から129℃の融点を示す。
【0081】
他の好ましい例では、シタグリプチン塩基をアセトニトリルに溶解させる。好ましくは、アセトニトリル中のグルタル酸の溶液を、シタグリプチン塩基の溶液に、攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で滴加する。その後、溶液混合物を、透明な溶液が形成されるまで還流温度に加熱し、次いで、好ましくは、攪拌下で室温付近まで徐冷する。溶液を冷却している間に、場合によっては種晶を加える。得られた結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0082】
前記種晶は、シタグリプチン遊離塩基をアセトニトリルに溶解して調製する。好ましくは、アセトニトリル中のグルタル酸溶液を、シタグリプチン塩基の溶液に、攪拌下、典型的には室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比で滴加し、次いで溶媒を蒸発させる。
【0083】
そのような手順に従って調製されたシタグリプチングルタル酸塩は、以下の特徴的な反射角2θ:6.4±0.2°、8.0±0.2°、12.9±0.2°、15.4±0.2°、17.6±0.2°、20.8±0.2°、23.0±0.2°、24.7±0.2°、25.4±0.2°および26.6±0.2°を含む粉末X線回折パターンを示す。
【0084】
そのような手順に従って調製されたシタグリプチングルタル酸塩は、約112から118℃の融点を示す。
【0085】
他の好ましい例では、シタグリプチン塩基をアセトニトリルに溶解させる。好ましくは、アセトニトリル中の硫酸の溶液(好ましくは、95から97%の濃硫酸を用いる)を、シタグリプチン塩基の溶液に、攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で滴加する。その後、溶液混合物を、好ましくは、約2時間、典型的には室温付近の温度で攪拌する。次いで、溶媒を蒸発させ、形成された固体を、好ましくは、メチルt−ブチルエーテル(tBuOMe)中に、場合によっては、攪拌下、典型的には室温付近の温度で、好ましくは、約2時間分散させる。得られた結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0086】
そのような手順に従って調製されたシタグリプチン硫酸水素塩は、以下の特徴的な反射角2θ:4.7±0.2°、14.2±0.2°、15.4±0.2°、18.1±0.2°、19.3±0.2°、22.0±0.2°、23.3±0.2°、24.4±0.2°、26.3±0.2°および26.8±0.2°を含む粉末X線回折パターンを示す。
【0087】
そのような手順に従って調製されたシタグリプチン硫酸水素塩は、約184から190℃の融点を示す。
【0088】
他の好ましい例では、シタグリプチン塩基を、好ましくは、それぞれ約1:5の容積比のメタノールおよびイソプロピルアルコール(iPrOH)の混合物に溶解させる。L−乳酸、好ましくは、濃度99%の濃乳酸を用いるが、シタグリプチン塩基の溶液に、典型的には約20から25℃などの室温付近の温度で、場合によっては攪拌下で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で滴加する。その後、種晶を加える。次いで、得られた分散液を6から24時間、好ましくは、約12時間室温付近で攪拌する。得られた結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0089】
前記種晶は、好ましくは、それぞれ約1:5の容積比のメタノールおよびイソプロピルアルコール(iPrOH)の混合物にシタグリプチン遊離塩基を溶解させて調製する。L−乳酸、好ましくは、濃度99%の濃乳酸を用いるが、シタグリプチン塩基の溶液に、典型的には室温付近の温度で、場合によっては攪拌下で、好ましくは、シタグリプチン塩基に対して等モル比で滴加し、次いで溶媒を蒸発させる。
【0090】
そのような手順に従って調製されたシタグリプチンL−乳酸塩は、以下の特徴的な反射角2θ:6.4±0.2°、7.9±0.2°、10.5±0.2°、17.8±0.2°、20.3±0.2°、21.5±0.2°、23.8±0.2°、24.5±0.2°、25.7±0.2°および27.3±0.2°を含む粉末X線回折パターンを示す。
【0091】
そのような手順に従って調製されたシタグリプチンL−乳酸塩は、約150から151℃の融点を示す。
【0092】
他の好ましい例では、シタグリプチン塩基をエタノールに溶解させる。シュウ酸を固体状態でシタグリプチン塩基の溶液に、攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で添加する。溶液を沈殿が形成されるまで攪拌する。得られた結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0093】
そのような手順に従って調製されたシタグリプチンシュウ酸塩は、以下の特徴的な反射角2θ:8.4±0.2°、11.2±0.2°、17.0±0.2°、17.5±0.2°、18.4±0.2°、20.9±0.2°、23.9±0.2°、25.4±0.2°、27.0±0.2°および27.9±0.2°を含む粉末X線回折パターンを示す。
【0094】
そのような手順に従って調製されたシタグリプチンシュウ酸塩は、約189から190℃の融点を示す。
【0095】
他の好ましい例では、シタグリプチン塩基を、好ましくはそれぞれ約1:6の容積比の、メタノールおよびイソプロピルアルコール(iPrOH)の混合物に溶解させる。エタンスルホン酸の水溶液、好ましくは、エタンスルホン酸の0.2M溶液を、シタグリプチン塩基の溶液に、攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で滴加する。その後、溶液混合物を好ましくは、約2時間、典型的には室温付近の温度で攪拌する。得られたシタグリプチンエタンスルホン酸塩の結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0096】
そのような手順に従って調製されたシタグリプチンエタンスルホン酸塩は、約199から202℃の融点を示す。
【0097】
他の好ましい例では、シタグリプチン塩基をメタノールに溶解させる。酢酸、好ましくは100%の濃酢酸を、シタグリプチン塩基の溶液に、攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で滴加する。その後、溶媒を蒸発させて形成した固体を、好ましくは、イソプロピルアルコール(iPrOH)中に分散し、次いで残渣全体が溶解するまで70℃付近まで加熱する。次いで、得られた溶液を、好ましくは、−5℃未満の温度で、6から24時間、好ましくは、約12時間冷凍室に入れる。得られたシタグリプチン酢酸塩の結晶を、好ましくは、多孔性セラミックフィルターを通す吸引濾過を用いて集める。
【0098】
そのような手順に従って調製されたシタグリプチン酢酸塩は、約113から117℃の融点を示す。
【0099】
他の好ましい例では、シタグリプチン塩基をアセトニトリルに溶解させる。カプリン酸を固体状態で、シタグリプチン塩基の溶液に、典型的には約20から25℃などの室温付近の温度で、場合によっては攪拌下で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で添加する。得られた溶液を約1時間攪拌し、次いで溶媒を蒸発させる。固体のシタグリプチンカプリン酸塩を、場合によってはさらに6から24時間、好ましくは、約12時間、30℃付近で減圧下で乾燥する。
【0100】
そのような手順に従って調製されたシタグリプチンカプリン酸塩は、約93から97℃の融点を示す。
【0101】
さらに他の好ましい例では、シタグリプチン塩基をエタノールに溶解させる。L−マンデル酸を固体状態で、シタグリプチン塩基の溶液に、場合によっては攪拌下、典型的には約20から25℃などの室温付近の温度で、好ましくは、シタグリプチン塩基に対して等モル比でまたはわずかに過剰で添加する。次いで溶媒を蒸発させ、シタグリプチンL−マンデル酸塩を白色粉末として得る。
【0102】
そのような手順に従って調製されたシタグリプチンL−マンデル酸塩は、約166から170℃の融点を示す。
【0103】
本発明の他の態様は、本発明の薬学的に許容される酸とのシタグリプチンの塩、すなわち、シタグリプチンD−グルクロン酸塩、シタグリプチンL−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩、シタグリプチンD−乳酸塩、シタグリプチンシュウ酸塩、シタグリプチンエタンスルホン酸塩、シタグリプチン酢酸塩、シタグリプチンL−マンデル酸塩、シタグリプチンD−マンデル酸塩、シタグリプチンカプリン酸塩、シタグリプチン安息香酸塩、シタグリプチン馬尿酸塩、シタグリプチントランス−桂皮酸塩、シタグリプチンマロン酸塩、シタグリプチンクエン酸塩、シタグリプチン1−ヒドロキシ−2−ナフトエ酸塩、シタグリプチンクロトン酸塩またはシタグリプチンアスコルビン酸塩、好ましくは、シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩またはシタグリプチンシュウ酸塩の治療有効量を、1種以上の薬学的に許容される担体または他の賦形剤とともに単回投与形態で投与するための医薬組成物である。
【0104】
本発明のシタグリプチンの塩の治療有効量は、シタグリプチン塩基として計算された場合、5から200mg、好ましくは、10から150mg、さらに好ましくは、25から100mgの範囲の塩の量である。
【0105】
本発明に従う薬学的に許容される塩は、例えば、錠剤、カプセル、ペレット、粒剤および坐薬の形態またはこれらの組合せ形態に具現化され得る。本発明に従う医薬組成物は、本発明のシタグリプチンの塩を、即時放出または制御放出するのに適し得る。固体の医薬組成物は、例えば、ペレット化を容易にするまたは分解もしくは吸収を制御する目的で被覆され得る。
【0106】
薬学的に許容される賦形剤は、結合剤、希釈剤、崩壊剤、安定化剤、保存剤、潤滑剤、芳香剤、着香剤、甘味剤および医薬技術の分野で公知の他の賦形剤からなる群から選択され得る。好ましくは、担体および賦形剤は、ラクトース、微小結晶性セルロース、セルロース誘導体、(例えば、ヒドロキシプロピルセルロース、クロスカルメロースナトリウム)、ポリアクリレート、炭酸カルシウム、でんぷん、コロイド状二酸化珪素、無水2塩基性リン酸カルシウム、デンプングリコール酸ナトリウム、タルク、ステアリン酸マグネシウム、ナトリウムステアリルフマラート、マンニトール、ポリビニルピロリドン、ポリエチレングリコールおよび医薬技術の分野で公知の他の賦形剤からなる群から選択され得る。
【0107】
適宜、本発明の医薬組成物は、シタグリプチンの塩に加えて1種以上の追加の薬学的活性成分を含む組合せの製品であり得る。好ましくは、1種以上の追加の薬学的活性成分は、インシュリン感受性剤、インシュリン、インシュリン模倣薬、スルホニル尿素、α‐グルコシダーゼ阻害薬、グルカゴン受容体アンタゴニスト、GLP−1、GLP−1類縁体、GLP−1模倣薬、GLP−1受容体アゴニスト、GIP、GIP模倣薬、PACAP、PACAP模倣薬、PACAP受容体アゴニスト、コレステロール低下剤、PPARδアゴニスト、抗肥満化合物、回腸胆汁酸輸送阻害薬、炎症状態での使用を意図した薬剤、抗高血圧薬、グルコキナーゼ活性薬(GKAs)、11β−ヒドロキシステロイドデヒドロキナーゼ1型阻害薬、コレステリルエステル輸送タンパク(CETP)阻害薬およびフラクトース1,6−ビスホスファターゼ阻害薬からなる群から選択される。
【0108】
最も好ましい追加の薬学的に活性な成分は、メトフォルミンおよび/またはその薬学的に許容される塩である。
【0109】
本発明に記載の医薬組成物は、医薬技術の分野に公知の方法により調製され得る。しかしながら、本明細書に開示されているシタグリプチンの塩の温度感受性の観点からは、本発明に記載の医薬組成物の調製方法の好ましい実施形態では、最終的な所望の医薬組成物を得るためのシタグリプチンの塩の処理を含むすべてのステップが、40℃、好ましくは、30℃より低い温度で実施される。このことは、特に、溶液状態または湿った状態で実施されるステップに当てはまる。結果として、各シタグリプチンの塩は、安定性を保持することができ、分解生成物をより少なく生成することができる。安定性およびより少ない分解生成物の生成は、本明細書に開示されている各融点によって規定することができる。
【0110】
本発明の他の態様は、本発明に記載のシタグリプチンの塩、すなわち、シタグリプチンD−グルクロン酸塩、シタグリプチンL−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩、シタグリプチンD−乳酸塩、シタグリプチンシュウ酸塩、シタグリプチンエタンスルホン酸塩、シタグリプチン酢酸塩、シタグリプチンL−マンデル酸塩、シタグリプチンD−マンデル酸塩、シタグリプチンカプリン酸塩、シタグリプチン安息香酸塩、シタグリプチン馬尿酸塩、シタグリプチントランス−桂皮酸塩、シタグリプチンマロン酸塩、シタグリプチンクエン酸塩、シタグリプチン1−ヒドロキシ−2−ナフトエ酸塩、シタグリプチンクロトン酸塩またはシタグリプチンアスコルビン酸塩、好ましくは、シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩またはシタグリプチンシュウ酸塩の有効量を用いることによる薬剤を使用しての、哺乳類におけるDPP−IV阻害薬が適用される臨床状態の治療および/または予防、特に、2型糖尿病、高血糖症、インシュリン抵抗性、および肥満の治療のための方法である。
【0111】
他の態様では、本発明は、哺乳類におけるDPP−IV阻害薬が適用される臨床状態の治療および/または予防、特に、2型糖尿病、高血糖症、インシュリン抵抗性、および肥満の治療のための薬剤の製造のための、本発明に記載のシタグリプチンの塩、すなわち、シタグリプチンD−グルクロン酸塩、シタグリプチンL−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩、シタグリプチンD−乳酸塩、シタグリプチンシュウ酸塩、シタグリプチンエタンスルホン酸塩、シタグリプチン酢酸塩、シタグリプチンL−マンデル酸塩、シタグリプチンD−マンデル酸塩、シタグリプチンカプリン酸塩、シタグリプチン安息香酸塩、シタグリプチン馬尿酸塩、シタグリプチントランス−桂皮酸塩、シタグリプチンマロン酸塩、シタグリプチンクエン酸塩、シタグリプチン1−ヒドロキシ−2−ナフトエ酸塩、シタグリプチンクロトン酸塩またはシタグリプチンアスコルビン酸塩、好ましくは、シタグリプチンD−グルクロン酸塩、シタグリプチングルタル酸塩、シタグリプチン硫酸水素塩、シタグリプチンL−乳酸塩またはシタグリプチンシュウ酸塩の使用に関する。
【実施例】
【0112】
実験の手順
【0113】
【表1】
【0114】
水への溶解度は、Avantium Technologies社のCrystal 16(商標)を用いて測定した。
【0115】
シタグリプチンリン酸塩は、WO2005/003135に記載の手順に従って調製した。
【0116】
シタグリプチン遊離塩基は、WO2003/004498に記載の手順に従って調製した。
【0117】
(実施例1)
(シタグリプチンD−グルクロン酸塩)
100mlのフラスコ中で、シタグリプチン遊離塩基(1.20g)およびD−グルクロン酸(604mg)を、メタノール(8ml)および水(3ml)に攪拌下溶解させた。均一な溶液が形成されたら、室温で、攪拌しながら、iPrOH(イソプロピルアルコール)(50ml)を徐々に添加した。白色沈殿が生じたら、分散液を室温で12時間攪拌した。形成された結晶を多孔性セラミックフィルターを通す吸引濾過を用いて集め、10mlのiPrOHで洗浄し、白色粉末(1.64g、92%)を得た。融点:126−129℃。
【0118】
(実施例2)
(シタグリプチングルタル酸塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(1.01g)をアセトニトリル(10ml)に溶解させた。次いで、グルタル酸(344mg)を含む、アセトニトリル(7ml)の溶液を攪拌下滴加した。添加後、沈殿が粘着性の残渣と共に出現した。次いで、透明な溶液が得られるまで、混合物を還流温度まで加熱し、種晶を加えて攪拌しながら室温まで徐冷した。形成された結晶を多孔性セラミックフィルターを通す吸引濾過を用いて集め、白色粉末(1.20g、90%)を得た。融点:112−118℃。
【0119】
実施例2で用いた種晶は、シタグリプチン遊離塩基(1.01g)をアセトニトリル(10ml)に溶解し、グルタル酸(344mg)を含む、アセトニトリル(7ml)を攪拌下滴加し、次いで溶媒を蒸発させて調製した。
【0120】
(実施例3)
(シタグリプチン硫酸水素塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(505mg)をアセトニトリル(10ml)に溶解させた。この溶液に、H2SO4(66.4μl、濃度95−97%)を含む、アセトニトリル(5ml)の溶液を滴加し、2時間以上攪拌した。濾過を試みたが成功しなかったので、溶液を蒸発乾固させ、形成した固体を、メチルt−ブチルエーテル(tBuOMe)(50 ml)中に、攪拌下、室温で2時間かけて分散した。形成した分散液を多孔性セラミックフィルターを通して濾過し、白色結晶を集めた(592mg。95%)。融点:184−190℃。
【0121】
(実施例4)
(シタグリプチンL−乳酸塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(1.20g)をメタノール(2ml)およびiPrOH(10ml)に溶解させた。次いで、L−乳酸(243μl、濃度99%)を滴加した。種晶を加えると、沈殿が始まった。分散液を室温で12時間攪拌した。形成された結晶を多孔性セラミックフィルターを通す吸引濾過を用いて集め、白色粉末(571mg、35%)を得た。融点:150−151℃。
【0122】
実施例4で用いた種晶は、シタグリプチン遊離塩基(1.20g)をメタノール(2ml)およびiPrOH(10ml)に溶解し、L−乳酸(243μl、濃度99%)を攪拌しながら滴加し、次いで溶媒を蒸発させて調製した。
【0123】
(実施例5)
(シタグリプチンシュウ酸塩)
エタノール(15ml)に溶解させたシタグリプチン遊離塩基(1.05g)の溶液に、シュウ酸(0.23g)を添加し、溶液を室温で沈殿が形成されるまで攪拌した。結晶(1.05g、85%)を濾過により集めた。融点:189−190℃。
【0124】
(実施例6)
(シタグリプチンエタンスルホン酸塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(303mg)をメタノール(1.5ml)およびiPrOH(10ml)に溶解させた。次いで、エタンスルホン酸の0.2M溶液(3.72ml)を滴加し、室温で2時間、攪拌した。形成された結晶を多孔性セラミックフィルターを通す吸引濾過を用いて集め、白色粉末(272mg、71%)を得た。融点:199−202℃。
【0125】
(実施例7)
(シタグリプチン酢酸塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(500mg)を3mlのメタノール中に溶解し、酢酸(81.7μl、濃度100%)を攪拌下、室温で滴加した。溶媒を蒸発させ、9mlのiPrOHをフラスコに添加した。残渣の全体が溶解するまで、約70℃までフラスコを加熱し、次いで12時間、冷凍室に入れた。形成された結晶を多孔性セラミックフィルターを通す吸引濾過を用いて白色粉末(505mg、88%)として集めた。融点:113−117℃。
【0126】
(実施例8)
(シタグリプチンカプリン酸塩)
50mlのフラスコ中で、シタグリプチン遊離塩基(307mg)をアセトニトリル(10ml)中に溶解し、カプリン酸(130mg)を添加した。室温で1時間、反応混合物を攪拌した後、溶媒を蒸発させた。固体残渣を一晩、さらに乾燥(5mBarの減圧下、30℃)した。白色粉末(437mg)を集めた。融点:93−97℃。
【0127】
(実施例9)
(シタグリプチンL−マンデル酸塩)
シタグリプチン遊離塩基(0.54g)およびL−マンデル酸(0.20g)をエタノール(20ml)に溶解させた。溶液を蒸発させて白色粉末(0.70g、95%)を得た。融点:166−170℃。
【0128】
シタグリプチンL−リンゴ酸塩、シタグリプチンクエン酸塩、シタグリプチンマロン酸塩、シタグリプチンD−マンデル酸塩、シタグリプチン安息香酸塩およびシタグリプチン1−ヒドロキシ−2−ナフトエ酸塩などの他のシタグリプチンの塩は、実施例1から9に同様の手順に従って調製した。
【0129】
分析の方法
X線粉末回折法
粉末X線回折(XRD)パターンを得るための条件:粉末X線回折パターンは、X’Celerator検出器を有するPhilips X’Pert PRO 回折装置を用いて、CuKa線(45kVおよび40mAで操作している管)を用い、Bragg−Brentano(反射)配置で、当業界に公知の方法により得た。データは、0.033°2θのステップで、2から40°2θまで記録し、測定時間は、ステップ当り50秒である。可変分散および抗散乱スリットを、照射サンプル長を12mmに保つために用いた。
【特許請求の範囲】
【請求項1】
D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、L−マンデル酸およびD−マンデル酸からなる群から選択される薬学的に許容される酸とのシタグリプチンの塩、および前記塩の水和物ならびに溶媒和物。
【請求項2】
薬学的に許容される酸が、D−グルクロン酸、L−グルクロン酸、シュウ酸、L−乳酸およびD−乳酸からなる群から選択される、請求項1に記載のシタグリプチンの塩。
【請求項3】
シタグリプチンD−グルクロン酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、請求項1に記載のシタグリプチンの塩。
【請求項4】
結晶形であり、以下の特徴的な反射角2θ:5.1±0.2°、12.7±0.2°、15.4±0.2°、17.1±0.2°、19.5±0.2°、21.5±0.2°、22.5±0.2°、26.2±0.2°および26.9±0.2°を含む粉末X線回折パターンを有する、請求項3に記載のシタグリプチンD−グルクロン酸塩。
【請求項5】
シタグリプチングルタル酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、請求項1に記載のシタグリプチンの塩。
【請求項6】
結晶形であり、以下の特徴的な反射角2θ:6.4±0.2°、8.0±0.2°、12.9±0.2°、15.4±0.2°、17.6±0.2°、20.8±0.2°、23.0±0.2°、24.7±0.2°、25.4±0.2°および26.6±0.2°を含む粉末X線回折パターンを有する、請求項5に記載のシタグリプチングルタル酸塩。
【請求項7】
シタグリプチンL−乳酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、請求項1に記載のシタグリプチンの塩。
【請求項8】
結晶形であり、以下の特徴的な反射角2θ:6.4±0.2°、7.9±0.2°、10.5±0.2°、17.8±0.2°、20.3±0.2°、21.5±0.2°、23.8±0.2°、24.5±0.2°、25.7±0.2°および27.3±0.2°を含む粉末X線回折パターンを有する、請求項7に記載の結晶シタグリプチンL−乳酸塩。
【請求項9】
シタグリプチンシュウ酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、請求項1に記載のシタグリプチンの塩。
【請求項10】
結晶形であり、以下の特徴的な反射角2θ:8.4±0.2°、11.2±0.2°、17.0±0.2°、17.5±0.2°、18.4±0.2°、20.9±0.2°、23.9±0.2°、25.4±0.2°、27.0±0.2°および27.9±0.2°を含む粉末X線回折パターンを有する、請求項10に記載のシタグリプチンシュウ酸塩。
【請求項11】
D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、エタンスルホン酸、シュウ酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸からなる群から選択される薬学的に許容される酸とのシタグリプチンの塩、および前記塩の水和物ならびに溶媒和物。
【請求項12】
a)シタグリプチン(塩基)および選択された薬学的に許容される酸を含む混合物を準備するステップ、および
b)得られたシタグリプチンの塩を単離するステップ
を含み、
前記混合物は、シタグリプチン塩基および選択された薬学的に許容される酸を液体媒体中に溶解した後、前記シタグリプチンの塩が形成されるまで、溶液を場合によって攪拌しながら、40℃より低い温度、好ましくは、30℃より低い温度に維持することによって形成され、得られたシタグリプチンの塩は、温度を40℃以上に上昇させることなく、液体媒体から単離される、請求項1から11のいずれか一項に記載のシタグリプチンの塩の調製のための方法。
【請求項13】
ステップa)が、調製される塩に対して特異的になるよう選択された溶媒中に、シタグリプチン塩基を溶解するステップ、および選択された薬学的に許容される酸を、好ましくは、固体状態で、得られた溶液中に添加するステップを含む、請求項12に記載の方法。
【請求項14】
グルコロン酸塩を調製する場合は、メタノールを用い、
グルタル酸塩を調製する場合は、アセトニトリルを用い、
硫酸塩を調製する場合は、アセトニトリルを用い、
乳酸塩を調製する場合は、メタノールとイソプロピルアルコールとの混合物を用い、
シュウ酸塩を調製する場合は、エタノールを用い、
酢酸塩を調製する場合は、メタノールを用い、
カプリン酸塩を調製する場合は、アセトニトリルを用い、および
マンデル酸塩を調製する場合は、エタノールを用いており、
それぞれ選択された薬学的に許容される酸を添加する前に、シタグリプチン塩基をそれぞれ溶解する、請求項13に記載の方法。
【請求項15】
溶解後、塩が形成されるまで、前記混合物を約20から25℃の温度に保つ、請求項12に記載の方法。
【請求項16】
請求項1から11のいずれか一項に記載のシタグリプチンの塩を含む医薬組成物。
【請求項17】
前記シタグリプチンの塩が、シタグリプチンD−グルクロン酸塩、シタグリプチンL−グルクロン酸塩、シタグリプチンL−乳酸塩、シタグリプチンD−乳酸塩、またはシタグリプチンシュウ酸塩である、請求項16に記載の医薬組成物。
【請求項18】
前記シタグリプチンの塩が、シタグリプチンD−グルクロン酸塩またはシタグリプチンL−グルクロン酸塩である、請求項17に記載の医薬組成物。
【請求項19】
哺乳類における2型糖尿病の治療処置に使用するための、請求項16から18のいずれか一項に記載の医薬組成物。
【請求項1】
D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、シュウ酸、L−マンデル酸およびD−マンデル酸からなる群から選択される薬学的に許容される酸とのシタグリプチンの塩、および前記塩の水和物ならびに溶媒和物。
【請求項2】
薬学的に許容される酸が、D−グルクロン酸、L−グルクロン酸、シュウ酸、L−乳酸およびD−乳酸からなる群から選択される、請求項1に記載のシタグリプチンの塩。
【請求項3】
シタグリプチンD−グルクロン酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、請求項1に記載のシタグリプチンの塩。
【請求項4】
結晶形であり、以下の特徴的な反射角2θ:5.1±0.2°、12.7±0.2°、15.4±0.2°、17.1±0.2°、19.5±0.2°、21.5±0.2°、22.5±0.2°、26.2±0.2°および26.9±0.2°を含む粉末X線回折パターンを有する、請求項3に記載のシタグリプチンD−グルクロン酸塩。
【請求項5】
シタグリプチングルタル酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、請求項1に記載のシタグリプチンの塩。
【請求項6】
結晶形であり、以下の特徴的な反射角2θ:6.4±0.2°、8.0±0.2°、12.9±0.2°、15.4±0.2°、17.6±0.2°、20.8±0.2°、23.0±0.2°、24.7±0.2°、25.4±0.2°および26.6±0.2°を含む粉末X線回折パターンを有する、請求項5に記載のシタグリプチングルタル酸塩。
【請求項7】
シタグリプチンL−乳酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、請求項1に記載のシタグリプチンの塩。
【請求項8】
結晶形であり、以下の特徴的な反射角2θ:6.4±0.2°、7.9±0.2°、10.5±0.2°、17.8±0.2°、20.3±0.2°、21.5±0.2°、23.8±0.2°、24.5±0.2°、25.7±0.2°および27.3±0.2°を含む粉末X線回折パターンを有する、請求項7に記載の結晶シタグリプチンL−乳酸塩。
【請求項9】
シタグリプチンシュウ酸塩、またはその水和物もしくは溶媒和物であり、場合によって、結晶形または非晶形である、請求項1に記載のシタグリプチンの塩。
【請求項10】
結晶形であり、以下の特徴的な反射角2θ:8.4±0.2°、11.2±0.2°、17.0±0.2°、17.5±0.2°、18.4±0.2°、20.9±0.2°、23.9±0.2°、25.4±0.2°、27.0±0.2°および27.9±0.2°を含む粉末X線回折パターンを有する、請求項10に記載のシタグリプチンシュウ酸塩。
【請求項11】
D−グルクロン酸、L−グルクロン酸、グルタル酸、硫酸、L−乳酸、D−乳酸、エタンスルホン酸、シュウ酸、酢酸、L−マンデル酸、D−マンデル酸、カプリン酸、安息香酸、馬尿酸、トランス−桂皮酸、マロン酸、クエン酸、1−ヒドロキシ−2−ナフトエ酸、クロトン酸およびアスコルビン酸からなる群から選択される薬学的に許容される酸とのシタグリプチンの塩、および前記塩の水和物ならびに溶媒和物。
【請求項12】
a)シタグリプチン(塩基)および選択された薬学的に許容される酸を含む混合物を準備するステップ、および
b)得られたシタグリプチンの塩を単離するステップ
を含み、
前記混合物は、シタグリプチン塩基および選択された薬学的に許容される酸を液体媒体中に溶解した後、前記シタグリプチンの塩が形成されるまで、溶液を場合によって攪拌しながら、40℃より低い温度、好ましくは、30℃より低い温度に維持することによって形成され、得られたシタグリプチンの塩は、温度を40℃以上に上昇させることなく、液体媒体から単離される、請求項1から11のいずれか一項に記載のシタグリプチンの塩の調製のための方法。
【請求項13】
ステップa)が、調製される塩に対して特異的になるよう選択された溶媒中に、シタグリプチン塩基を溶解するステップ、および選択された薬学的に許容される酸を、好ましくは、固体状態で、得られた溶液中に添加するステップを含む、請求項12に記載の方法。
【請求項14】
グルコロン酸塩を調製する場合は、メタノールを用い、
グルタル酸塩を調製する場合は、アセトニトリルを用い、
硫酸塩を調製する場合は、アセトニトリルを用い、
乳酸塩を調製する場合は、メタノールとイソプロピルアルコールとの混合物を用い、
シュウ酸塩を調製する場合は、エタノールを用い、
酢酸塩を調製する場合は、メタノールを用い、
カプリン酸塩を調製する場合は、アセトニトリルを用い、および
マンデル酸塩を調製する場合は、エタノールを用いており、
それぞれ選択された薬学的に許容される酸を添加する前に、シタグリプチン塩基をそれぞれ溶解する、請求項13に記載の方法。
【請求項15】
溶解後、塩が形成されるまで、前記混合物を約20から25℃の温度に保つ、請求項12に記載の方法。
【請求項16】
請求項1から11のいずれか一項に記載のシタグリプチンの塩を含む医薬組成物。
【請求項17】
前記シタグリプチンの塩が、シタグリプチンD−グルクロン酸塩、シタグリプチンL−グルクロン酸塩、シタグリプチンL−乳酸塩、シタグリプチンD−乳酸塩、またはシタグリプチンシュウ酸塩である、請求項16に記載の医薬組成物。
【請求項18】
前記シタグリプチンの塩が、シタグリプチンD−グルクロン酸塩またはシタグリプチンL−グルクロン酸塩である、請求項17に記載の医薬組成物。
【請求項19】
哺乳類における2型糖尿病の治療処置に使用するための、請求項16から18のいずれか一項に記載の医薬組成物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2012−517456(P2012−517456A)
【公表日】平成24年8月2日(2012.8.2)
【国際特許分類】
【出願番号】特願2011−549546(P2011−549546)
【出願日】平成22年2月10日(2010.2.10)
【国際出願番号】PCT/EP2010/051661
【国際公開番号】WO2010/092090
【国際公開日】平成22年8月19日(2010.8.19)
【出願人】(504359293)レツク・フアーマシユーテイカルズ・デー・デー (60)
【Fターム(参考)】
【公表日】平成24年8月2日(2012.8.2)
【国際特許分類】
【出願日】平成22年2月10日(2010.2.10)
【国際出願番号】PCT/EP2010/051661
【国際公開番号】WO2010/092090
【国際公開日】平成22年8月19日(2010.8.19)
【出願人】(504359293)レツク・フアーマシユーテイカルズ・デー・デー (60)
【Fターム(参考)】
[ Back to top ]