
新規なネオマイシンホスホトランスフェラーゼ遺伝子及び高生産組換え細胞の選抜方法
【課題】本発明は新規改変ネオマイシンホスホトランスフェラーゼ遺伝子及び高生産性組換え細胞についての選抜方法を提供する。
【解決手段】特定の改変ネオマイシンホスホトランスフェラーゼ遺伝子を選抜可能なマーカーとして使用する。本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子はアミノ酸位置91、182、198、227、240又は261において野生型遺伝子とは異なるアミノ酸をコードする変異体であることが好ましい。より具体的には、本発明のネオマイシンホスホトランスフェラーゼ遺伝子は変異体Glu182Gly、Glu182Asp、Trp91Ala、Val198Gly、Asp227Ala、Asp227Val、Asp227Gly、Asp261Asn、Asp261Gly又はPhe240Ileである。
【解決手段】特定の改変ネオマイシンホスホトランスフェラーゼ遺伝子を選抜可能なマーカーとして使用する。本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子はアミノ酸位置91、182、198、227、240又は261において野生型遺伝子とは異なるアミノ酸をコードする変異体であることが好ましい。より具体的には、本発明のネオマイシンホスホトランスフェラーゼ遺伝子は変異体Glu182Gly、Glu182Asp、Trp91Ala、Val198Gly、Asp227Ala、Asp227Val、Asp227Gly、Asp261Asn、Asp261Gly又はPhe240Ileである。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は新規改変ネオマイシンホスホトランスフェラーゼ遺伝子及び高生産性組換え細胞の選抜方法におけるそれらの使用に関する。それ故、本発明はまた好ましくは異種プロモーターに機能し得る形で連結された対象遺伝子と組み合わされた、改変ネオマイシンホスホトランスフェラーゼ遺伝子を含む新規発現ベクターに関する。更に、本発明は相当する高生産性組換え細胞を使用する異種遺伝子産物の調製方法に関する。
【背景技術】
【0002】
哺乳動物細胞は複雑な生物医薬タンパク質の生成に好ましい宿主細胞である。何とならば、翻訳後に起こる修飾が機能上及び薬物速度論上の両方でヒトと共存性があるからである。主な妥当な細胞型はハイブリドーマ、ミエローマCHO(チャイニーズハムスター卵巣)細胞及びBHK(ベビーハムスター腎臓)細胞である。宿主細胞の培養は無血清かつ無タンパク質生成条件下で次第に行なわれるようになっている。これらの理由はコスト低減、組換えタンパク質の精製における低減された妨害及び病原体(例えば、プリオン及びウイルス)の導入の可能性の低下である。宿主細胞としてのCHO細胞の使用が一層広まりつつある。何とならば、これらの細胞が無血清かつ無タンパク質培地中の懸濁増殖に適しており、かつ規制当局により安全な生産細胞と見なされ、受け入れられているからである。
対象異種遺伝子(GOI)を発現する安定な哺乳動物細胞株を生成するために、異種遺伝子は一般にトランスフェクションにより選抜可能なマーカー遺伝子、例えば、ネオマイシンホスホトランスフェラーゼ遺伝子(NPT)と一緒に所望の細胞株に挿入される。異種遺伝子及び選抜可能なマーカー遺伝子は一つの個々のベクター又は別々の同時トランスフェクションされたベクターから開始して宿主細胞中で発現し得る。トランスフェクションの2〜3日後に、トランスフェクションされた細胞が選択薬剤、例えば、ネオマイシンホスホトランスフェラーゼ遺伝子(NPT遺伝子)を使用する場合にはG418を含む培地に移され、これらの選抜条件下で数週にわたって培養される。外来性DNAを組み込んだ耐性細胞が出現してこれを単離でき、所望の遺伝子産物(GOIの)の発現について研究し得る。
【0003】
所望のタンパク質の高発現を有する細胞株を樹立する際の重大な問題が、宿主細胞ゲノム中の転写活性又は不活性遺伝子座への組換えベクターのランダムかつ方向性のない(undirected)組込みから生じる。結果として、異種遺伝子の全く異なる発現率を有する細胞の集団が得られ、細胞の生産性は一般に正規分布に従う。それ故、対象異種遺伝子の非常に高い発現を有する細胞クローンを同定するために、多数のクローンを検査し、試験することが必要であり、これは時間を浪費し、労力集中的であり、高価である。それ故、トランスフェクションに使用されるベクター系の改良は、好適な選抜戦略によりトランスフェクションされた細胞集団中の高生産細胞の比率を増大しようと試み、それによりクローン同定に関係する出費及び作業を低減しようとした。このような発現系の開発が本発明の主題である。
アミノグリコシド-3'-ホスホトランスフェラーゼII酵素(ネオマイシンホスホトランスフェラーゼ)(EC27195)(その遺伝子はエシェリキア・コリ中のトランスポゾン5に付随する)が幾つかの生物(例えば、バクテリア、酵母、植物及び哺乳動物細胞)中の選抜可能なマーカーとして使用される。この酵素は末端リン酸をATPからアミノヘキソース環Iの3'ヒドロキシル基に転移することにより抗生物質を不活性化することにより、種々のアミノグリコシド抗生物質、例えば、ネオマイシン、カナマイシン及びG418に対する耐性を与える。野生型ネオマイシンホスホトランスフェラーゼに加えて、低下されたホスホトランスフェラーゼ活性ひいてはバクテリア(Blazquezら, 1991; Kocabiyikら, 1992; Yenofskyら, 1990)及びタバコからの葉の切片(Yenofskyら, 1990)におけるアミノグリコシド抗生物質に対する低下された耐性を有する幾つかの変異体が知られている。
これらの変異体の一種(Glu182Asp)が胚性幹細胞を選抜するためのマーカーとして使用され、ネオマイシンホスホトランスフェラーゼ遺伝子が標的相同組換え(遺伝子ターゲッティング)(Hansonら, 1995)によりc-myc遺伝子に組み込まれた。著者らはそれら自体を遺伝子ターゲッティングのための改変酵素の使用に限定している。
【0004】
特許出願WO 99/53046には生産に妥当な哺乳動物細胞中の改変ネオマイシンホスホトランスフェラーゼ遺伝子(Asp261Asn)の発現が記載されている。著者らは哺乳動物細胞中の対象遺伝子の発現のための非クローニング方法を記載している。プロモーター要素、対象遺伝子、及びIRES(“内部リボソームエントリー部位”)要素と結合された選抜可能なマーカーをコードする三つの個々のDNAフラグメントによる細胞の同時トランスフェクションにより、細胞を選択圧下で計画的に増殖させることが可能であり、その場合、全ての三つのDNAフラグメントが機能性2シストロン転写単位として組み合わされる(プロモーター-対象遺伝子-IRES-ネオマイシンホスホトランスフェラーゼ遺伝子)。これらの要素の組合せはトランスフェクションされた細胞中のみで生じ、その結果、いくつかの細胞のみが要素の正しい配置を示す。更に、増幅可能な選抜可能マーカーを使用した遺伝子増幅後に、高生産性クローンが生成し得ない。反復した選抜及び遺伝子増幅後に、生成された細胞は細胞当り1日当りせいぜい6pg(6pg/細胞/日)のタンパク質しか示さなかった。
刊行物のいずれもが一種以上の対象遺伝子そしてまた低下された抗生物質耐性を有する改変ネオマイシンホスホトランスフェラーゼ遺伝子の両方のための一種以上の完全な機能性転写単位を含む哺乳動物細胞のための高発現ベクター系(これは組換え生物医薬タンパク質を調製するために高生産性細胞を開発することを可能にする)の調製に特別な適性を有する改変ネオマイシンホスホトランスフェラーゼ遺伝子を開示していない。WO 99/53046に記載されたDNA構築物はジヒドロ葉酸還元酵素(DHFR)の遺伝子に機能し得る形で連結された無プロモーターネオマイシン遺伝子のみを含む。
それ故、特に生物医薬方法のための相当する高発現ベクター系の開発に、利用できる好適な改変ネオマイシンホスホトランスフェラーゼ遺伝子をつくるという需要がある。それ故、本発明の課題は相当する新規改変ネオマイシンホスホトランスフェラーゼ遺伝子、改変ネオマイシンホスホトランスフェラーゼ遺伝子及び異種プロモーターに機能し得る形で連結された対象遺伝子を含む発現ベクター、高生産性組換え細胞、好ましくは哺乳動物細胞の選抜方法、並びに異種遺伝子産物の生産方法を提供することであった。
驚くことに、本発明の範囲内で、高生産性細胞の選抜についてのそれらの特別な適性を特徴とする新規な高度に選抜的なネオマイシンホスホトランスフェラーゼ遺伝子を生成し、同定することが可能であった。
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は新規改変ネオマイシンホスホトランスフェラーゼ遺伝子を提供する。
【課題を解決するための手段】
【0006】
驚くことに、同時に組み込まれた対象遺伝子の高発現率を有するトランスフェクションされた哺乳動物細胞の濃縮が以下に記載される改変ネオマイシンホスホトランスフェラーゼ遺伝子を選抜可能なマーカーとして使用することにより達成し得ることがわかった。選抜可能なマーカーとしての野生型ネオマイシンホスホトランスフェラーゼの使用と較べて、本発明の新規ネオマイシンホスホトランスフェラーゼ遺伝子の一つによるトランスフェクション後に、細胞が1.4〜14.6倍に増大されたタンパク質(抗体)の生産性を示した。
本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子はアミノ酸位置91、182、198、227、240又は261において野生型遺伝子とは異なるアミノ酸をコードする変異体であることが好ましい。好ましい実施態様において、本発明のネオマイシンホスホトランスフェラーゼ遺伝子は変異体Glu182Gly、Glu182Asp、Trp91Ala、Val198Gly、Asp227Ala、Asp227Val、Asp227Gly、Asp261Asn、Asp261Gly又はPhe240Ileである。高生産性哺乳動物細胞を選抜するために、変異体Trp91Ala、Asp227Val、Asp261Asn、Asp261Gly及びPhe240Ileを使用することが特に好適と判明し、一方、変異体Asp227Val及びAsp261Glyは順に最高の生産性を有する細胞クローンを与え、それ故、特に好ましい。
【0007】
高生産性細胞が異種プロモーターに機能し得る形で連結された対象異種遺伝子及び本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子を含む真核細胞発現ベクターの使用により得られた。発現ベクターはその他の調節要素、例えば、一種以上のプロモーターに機能し得る形で連結された一種以上のエンハンサーを含むことが好ましい。また、好ましくは内部リボソームエントリー部位(IRES)(これは異種プロモーターの制御下で、蛍光タンパク質をコードする遺伝子及び対象タンパク質/産物をコードする遺伝子の2シストロン性発現を可能にする)を介して、対象遺伝子及び異種プロモーターに機能し得る形で連結される蛍光タンパク質の遺伝子を更に含む発現ベクターが好ましい。注目する異種遺伝子がユビキチン/S27aプロモーターの制御下にある発現ベクターが特に好適である。
また、本発明は対象遺伝子に代えてこのような遺伝子をとり込むための、即ち、制限エンドヌクレアーゼのための多重認識配列による配列選択のための多重クローニング部位を含む発現ベクターに関する。
別の局面において、本発明は本発明の上記改変ネオマイシンホスホトランスフェラーゼ遺伝子の一つを含む組換え哺乳動物細胞に関する。加えて、本発明は本発明の発現ベクターの一つでトランスフェクションされた組換え哺乳動物細胞に関する。これらは組換えげっ歯類細胞であることが好ましく、その中で組換えハムスター細胞、例えば、CHO細胞又はBHK細胞が特に好ましい。別の好ましい実施態様において、前記組換え細胞は増幅可能な選抜可能マーカーの遺伝子、例えば、ジヒドロ葉酸還元酵素(DHFR)の遺伝子で更にトランスフェクションされる。
【0008】
また、本発明は(i) 哺乳動物細胞のプール(集団)を改変ネオマイシンホスホトランスフェラーゼ(これはわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5〜26%の活性及び/又は上記改変の一つを有する)の遺伝子でトランスフェクションすること、(ii)前記哺乳動物細胞を前記改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、および、(iii)前記哺乳動物細胞を前記哺乳動物細胞の増殖に選択的に作用し、かつネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、を特徴とする、改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現する組換え哺乳動物細胞の濃縮方法に関する。
また、本発明は(i) 哺乳動物細胞のプールを少なくとも一つの対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ(これはわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5〜26%の活性及び/又は上記改変の一つを示す)の遺伝子でトランスフェクションすること、(ii)前記細胞を一以上の対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、(iii) 前記哺乳動物細胞を前記哺乳動物細胞の増殖に選択的に作用し、かつ改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、および、(iv)一以上の対象タンパク質を前記哺乳動物細胞又は培養上清から得ることを特徴とする、組換え哺乳動物細胞中の少なくとも一つの対象遺伝子の発現方法に関する。
更に、本発明は(i) 組換え哺乳動物細胞を本発明の発現ベクター(これは注目する遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子に加えて蛍光タンパク質をコードする)でトランスフェクションすること、(ii)前記哺乳動物細胞を一以上の対象遺伝子、蛍光タンパク質をコードする遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、(iii) 前記哺乳動物細胞を前記哺乳動物細胞の増殖に選択的に作用し、かつネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、および、(iv)前記哺乳動物細胞をフローサイトメトリー分析によりソーティングすることを特徴とする、少なくとも一つの対象異種遺伝子を発現する組換え哺乳動物細胞を得ること、および選抜する方法に関する。
哺乳動物細胞が増幅可能な選抜可能マーカー遺伝子、例えば、DHFR遺伝子で更にトランスフェクションされた場合、哺乳動物細胞を増幅可能な選抜可能マーカー遺伝子も発現される条件下で培養すること、及び培地に増幅可能な選抜可能マーカー遺伝子の増幅をもたらす選択薬剤を添加することが可能である。
本発明の方法は懸濁培養増殖に適している哺乳動物細胞、即ち、懸濁培養で培養される哺乳動物細胞を用いて行なわれることが好ましい。その他の実施態様は哺乳動物細胞、好ましくは懸濁液中の増殖に適している哺乳動物細胞が無血清条件下で培養される方法に関する。
【発明を実施するための最良の形態】
【0009】
発明の詳細な記載
アミノ酸位置に関する下記の情報は夫々の場合に配列番号1を有する野生型ネオマイシンホスホトランスフェラーゼ遺伝子によりコードされるアミノ酸の位置に関する。“改変ネオマイシンホスホトランスフェラーゼ遺伝子”はネオマイシンホスホトランスフェラーゼ活性を有するポリペプチドをコードする核酸を意味し、そのポリペプチドは明細書に更に充分に記載されるアミノ酸位置(これらは配列番号2を有する野生型タンパク質に相同である)の少なくとも一つでその野生型タンパク質とは異なるアミノ酸を有する。この状況で、“相同”という用語は変異を有する配列領域が所謂通常の“アライメント”アルゴリズム、例えば、“BLAST”(Altschul, S.F., Gish, W., Miller, W., Myers, E.W.&Lipman, D.J. (1990)“基本的局所アライメント検索ツール” J. Mol. Biol. 215:403-410; Gish, W.&States, D.J. (1993)“データベース類似性検索によるタンパク質コーディング領域の同定” Nature Genet. 3:266-272; Madden, T.L., Tatusov, R.L.&Zhang, J. (1996)“ネットワークBLASTサーバーの適用” Meth. Enzymol. 266:131-141; Zhang, J.&Madden, T.L. (1997)“パワーBLAST:インタラクチブ又は自動化配列分析及び注釈のための新しいネットワークBLAST適用”Genome Res. 7:649-656; Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller,及びDavid J. Lipman (1997),“ギャップ付きBLAST及びPSI-BLAST:タンパク質データベース検索プログラムの新しい作成”Nucleic Acids Res. 25:3389-3402)を使用して基準配列、この場合には配列番号2の野生型ネオマイシンホスホトランスフェラーゼの配列と対応させ得ることを意味する。配列はそれらがそれらの配列順序で対応する場合に対応し、通常の“アライメント”アルゴリズムを使用して同定し得る。
【0010】
本発明は新規改変ネオマイシンホスホトランスフェラーゼ遺伝子及び異種遺伝子産物、好ましくは生物医薬上妥当なポリペプチド又はタンパク質の高発現を可能にする哺乳動物細胞を調製し、選抜する方法を提供する。本発明の方法は、対象遺伝子に加えてトランスフェクションされた細胞にトランスフェクションされなかった細胞に対し選択的な利点を与える本発明のネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞の選抜に主として基づいている。驚くことに、本明細書に記載された本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子(mNPT遺伝子)の使用が野生型ネオマイシンホスホトランスフェラーゼ遺伝子(wtNPT遺伝子)に対する実質的な選択的利点を有することがわかった。特に、これはwtNPTと較べて低い酵素活性を有する変異体の使用に関する。
【0011】
本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子
wtNPTの酵素活性のわずかに1〜80%、好ましくはわずかに1〜60%を有するNPTをコードする改変NPT遺伝子を使用することが特に好適と判明した。好ましいNPT変異体はwtNPTの酵素活性のわずかに1〜30%を有するものであり、wtNPTの酵素活性のわずかに1.5〜26%を有するものが特に好ましい。NPTの酵素活性は、例えば、実施例4に記載され、方法5として示されるドットアッセイで測定し得る。
野生型ネオマイシンホスホトランスフェラーゼという用語はアミノグリコシド-3'-ホスホトランスフェラーゼII酵素(EC 2.7.1.95)をコードするネオマイシンホスホトランスフェラーゼ遺伝子を表し、その酵素の遺伝子は、に天然にはエシェリキア・コリ中のトランスポゾン5と関連しており、例えば、配列番号2に示されるアミノ酸配列を含み、又は配列番号1に示されるヌクレオチド配列によりコードされる。この酵素はアミノヘキソース環Iの3'ヒドロキシル基へのATPの末端リン酸の転移により抗生物質を不活性化することにより、種々のアミノグリコシド抗生物質、例えば、ネオマイシン、カナマイシン及びG418に対する耐性を与える。また、wtNPTという用語は配列番号1によりコードされるNPTに匹敵する酵素活性を有する全てのNPTを表す。これは特にATPから基質への末端ホスフェートの転移を触媒作用する酵素活性中心が同じ又はほぼ同じコンホメーションで存在し(Shawら, 1993; Honら, 1997; Burkら, 2001)、こうして配列番号2のアミノ酸配列を含む酵素に匹敵する酵素活性を有するこれらのNPTを含む。wtNPTはそれが配列番号2により特定されるNPTにより示される酵素活性の約81〜150%、好ましくは90〜120%を示す場合に匹敵する酵素活性を有し、その活性は実施例4に記載され、方法5と称されるドットアッセイで測定し得る。
【0012】
wtNPTと較べての酵素活性の低下がアミノ酸配列の改変、例えば、少なくとも一種又はそれ以上のアミノ酸の置換、挿入又は欠失に基づく変異体が基本的に好ましい。欠失変異体、挿入変異体及び置換変異体は“部位特異的変異導入”及び/又は“PCRをベースとする変異導入技術”により生成し得る。好適な方法が、例えば、Lottspeich及びZorbas(1998)(その他の文献とともに36.1章)により記載されている。
驚くことに、ネオマイシンホスホトランスフェラーゼ変異体が選抜可能なマーカーとして使用される場合(この場合、少なくともアミノ酸位置91のアミノ酸トリプトファン、アミノ酸位置182のアミノ酸グルタミン酸、アミノ酸位置198のアミノ酸バリン、アミノ酸位置227のアミノ酸アスパラギン酸、アミノ酸位置261のアミノ酸アスパラギン酸又はアミノ酸位置240のアミノ酸フェニルアラニンがwtNPTと較べて変化されていた)、注目する同時組込み遺伝子について高い発現率を有するトランスフェクションされた哺乳動物細胞の特に有効な濃縮を達成することが可能であることがわかった。それ故、位置91、182、198、227及び/又は240のアミノ酸に影響する変異体が好ましい。置換変異体、即ち、野生型中のこの位置に生じるアミノ酸が別のアミノ酸により置換された変異体が特に有利である。相当するアミノ酸の変化がwt-NPTと較べて1〜80%まで、好ましくは1〜60%まで、更に好ましくは1.5〜30%まで、最も好ましくは1.5%〜26%までの酵素活性の低下をもたらす相当する置換変異体が更に好ましい。wt-NPTと較べた酵素活性がわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5%〜26%であるようにアミノ酸91、227、261及び/又は240がそれに従って改変された改変NPT遺伝子が特に好ましい。アミノ酸位置227のアミノ酸が改変NPTの酵素活性がwt-NPTと較べて26%以下、好ましくは1〜20%、更に好ましくは1〜16%であるような形態で改変された置換変異体が最も好ましい。
【0013】
本発明の別の実施態様によれば、有利な変異体は、wtNPTとの比較により、アミノ酸位置91、182又は227においてグリシン、アラニン、バリン、ロイシン、イソロイシン、フェニルアラニン又はチロシンをコードする変異体である。更に、アミノ酸位置182のグルタミン酸はまたアスパラギン酸、アスパラギン、グルタミン又はあらゆるその他の好ましくは負に荷電されていてもよいアミノ酸により置換されていてもよい。また、wtNPTとの比較により、アミノ酸位置198においてグリシン、アラニン、ロイシン、イソロイシン、フェニルアラニン、チロシン又はトリプトファンをコードする改変NPT遺伝子が好ましい。また、wtNPTとの比較により、アミノ酸位置261においてグリシン、アラニン、ロイシン、イソロイシン、フェニルアラニン、チロシン、トリプトファン、アスパラギン、グルタミン又はアスパラギン酸をコードする改変NPT遺伝子が好ましい。特に、選抜可能なマーカーとして変異体Glu182Gly、Glu182Asp、Trp91Ala、Val198Gly、Asp227Ala、Asp227Val、Asp227Gly、Asp261Gly、Asp261Asn及びPhe240Ileを用いて、同時組込みされる対象遺伝子の高発現率を有するトランスフェクションされた哺乳動物細胞の濃縮を得ることが可能であることがわかり、これらの変異体が特に好ましいという結果が得られた。変異体Asp227Val、Asp227Gly、Asp261Gly、Asp261Asn、Phe240Ile及びTrp91Alaが更に好ましい。何とならば、最良の濃縮率がそれらを使用して得られるからである。変異体Asp227Valが特に好ましい。
【0014】
対照的に、Asp190Gly変異体及びAsp208Gly変異体は無血清培養条件下でトランスフェクションされたCHO-DG44細胞の選抜に不適なマーカーであることが判明した。これらの変異体(Asp190Gly、Asp208Gly)のおそらく大いに低下された酵素機能の結果として、わずかにいくつかの細胞が選抜期後に得られ、さらにそれらは増殖及び生命力が更にひどく損なわれていた。
野生型を基準として位置182及び227のアミノ酸はアミノグリコシド-3'-ホスホトランスフェラーゼのC末端領域中の三つの保存モチーフの外に位置される非保存アミノ酸である。位置91のアミノ酸はまた非保存アミノ酸に属し、アミノグリコシド-3'-ホスホトランスフェラーゼのN末端領域中の保存モチーフの一つの外に位置される。対照的に、位置198及び240のアミノ酸はNPTのC末端領域中の保存アミノ酸であるが、それにもかかわらず保存モチーフの外にある。対照的に、位置261のアミノ酸はC末端領域の第三の保存モチーフ中の保存アミノ酸である(Shawら, 1993; Honら, 1997; Burkら, 2001)。
選抜可能なマーカーとしてのwtNPTの使用と較べて、Glu182Gly、Glu182Asp及びVal198Gly変異体の場合の細胞は1.4-2.4倍に増大された生産性を示し、Asp227Gly変異体の場合には生産性が1.6-4.1倍に増大され、Asp227Ala又はTrp91Ala変異体の場合には生産性が2.2倍又は4倍に増大され、Phe240Ile又はAsp261Asn変異体の場合には生産性が5.7倍又は7.3倍に増大され、またAsp261Gly又はAsp227Val変異体の場合にはそれが9.3倍又は14.6倍に一層増大された。マルチ鎖タンパク質(抗体)を発現するために、同時トランスフェクションが行なわれた。二つのタンパク質鎖はそれら自身のベクターにより夫々発現され、一つのベクターはNPT遺伝子を更にコードし、一方、別のベクターは増幅可能で選抜可能なジヒドロ葉酸還元酵素遺伝子をコードする。
【0015】
このように、本発明は(i)哺乳動物細胞のプールを野生型ネオマイシンホスホトランスフェラーゼの活性のわずかに1〜80%、好ましくは1〜60%、更に好ましくは1.5〜30%、最も好ましくは1.5〜26%及び/又は本明細書に記載された改変の一つを有する改変ネオマイシンホスホトランスフェラーゼの遺伝子でトランスフェクションすること、(ii)前記哺乳動物細胞を改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、および、(iii) 前記哺乳動物細胞を哺乳動物細胞の増殖に選択的に作用し、かつネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、を特徴とする、改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現する組換え哺乳動物細胞の濃縮方法に関する。
特に使用される改変NPT遺伝子が、野生型遺伝子との比較して、アミノ酸位置91のアラニン、アミノ酸位置182のグリシン又はアスパラギン酸、アミノ酸位置198のグリシン、アミノ酸位置227のアラニン、グリシン又はバリン、アミノ酸位置261のグリシン又はアスパラギン或いはアミノ酸位置240のイソロイシンをコードする改変NPTをコードする場合、この出願に更に詳しく記載された改変NPT遺伝子を使用する相当する方法が特に好ましい。野生型遺伝子との比較により、アミノ酸位置227のバリン及び/又はアミノ酸位置261のグリシン及び/又はアスパラギンをコードするNPT遺伝子が更に好ましい。野生型遺伝子との比較により、アミノ酸位置240でイソロイシン又はアミノ酸位置227でバリンをコードするこれらのNPT遺伝子が特に好ましく、野生型遺伝子と較べてアミノ酸位置227でバリンをコードするNPT遺伝子が特に好ましい。当然、本発明はまた改変NPT遺伝子及び相当するアミノ酸交換の組み合わせを含む本発明の改変NPT遺伝子の使用を含む。
更に、本発明は(i) 異種プロモーターに機能し得る形で連結された注目する異種遺伝子及び(ii)野生型ネオマイシンホスホトランスフェラーゼと較べて低い酵素活性を有するネオマイシンホスホトランスフェラーゼをコードする本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子を含む真核細胞発現ベクターに関する。本発明の目的において、“低い”又は“より低い”酵素活性はwtNPTの酵素活性のせいぜい80%、好ましくは1〜80%、更に好ましくはわずかに1〜60%に相当する酵素活性を意味する。本発明の一実施態様によれば、“より低い酵素活性”は野生型ネオマイシンホスホトランスフェラーゼと較べて1〜30%、好ましくは1.5〜26%の酵素活性を表す。
【0016】
好ましい発現ベクターはwtNPTの酵素活性のわずかに1〜80%、好ましくはわずかに1〜60%を有する改変NPTをコードする改変NPT遺伝子を含む。また、wtNPTの酵素活性のわずかに1〜30%を有する変異体をコードする改変NPT遺伝子を有する発現ベクターが好ましい。wtNPTの酵素活性のわずかに1.5〜26%を有する変異体をコードする改変NPT遺伝子を含むこれらの発現ベクターが特に好ましく、その活性は実施例4に記載され、方法5と称されるドットアッセイで測定される。
本発明の別の実施態様において、発現ベクターは、wtNPTと較べて、アミノ酸位置Trp91、Glu182、Val198、Asp227、Phe240又は位置Asp261で改変された改変NPTの遺伝子を含む。この意味で、位置Trp91、Glu182、Val198、Asp227、Phe240又はAsp261で改変され、wtNPTの酵素活性のわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5〜26%を有するNPT変異体が好ましい。アミノ酸Trp91、Glu182又はAsp227が相当する位置でグリシン、アラニン、バリン、ロイシン、イソロイシン、フェニルアラニン又はチロシンにより夫々置換されることが好ましい。位置182のグルタミン酸がまたアスパラギン酸、アスパラギン、グルタミン又は別の好ましくは負に荷電されていてもよいアミノ酸により置換されることが好ましい。また、wtNPTと較べてアミノ酸位置198でグリシン、アラニン、ロイシン、イソロイシン、フェニルアラニン、チロシン又はトリプトファンをコードする改変NPT遺伝子が好ましい。加えて、wtNPTと較べてアミノ酸位置240でグリシン、アラニン、バリン、イソロイシン、チロシン又はトリプトファンをコードする改変NPT遺伝子が好ましい。また、wtNPTと較べて、アミノ酸261でグリシン、アラニン、ロイシン、イソロイシン、フェニルアラニン、チロシン、トリプトファン、アスパラギン、グルタミン又はアスパラギン酸をコードする改変NPT遺伝子が好ましい。位置227のアスパラギン酸がグリシン、アラニン、バリン、ロイシン又はイソロイシンにより置換され、位置261のアスパラギン酸がアラニン、バリン、ロイシン、イソロイシン又はグルタミン、特にグリシン又はアスパラギンにより置換される変異体を使用することが特に好ましい。
【0017】
Glu182Gly変異体、Glu182Asp変異体、Trp91Ala変異体、Val198Gly変異体、Asp227Ala変異体、Asp227Val変異体、Asp227Gly変異体、Asp261Gly変異体、Asp261Asn変異体又はPhe240Ile変異体をコードする改変NPT遺伝子を含む発現ベクターが特に好ましく、これはGlu182Gly変異体の場合には配列番号4のアミノ酸配列を含み、Glu182Asp変異体の場合には配列番号20のアミノ酸配列を含み、Trp91Ala変異体の場合には配列番号6のアミノ酸配列を含み、Val198Gly変異体の場合には配列番号8のアミノ酸配列を含み、Asp227Ala変異体の場合には配列番号10のアミノ酸配列を含み、Asp227Val変異体の場合には配列番号12のアミノ酸配列を含み、Asp227Gly変異体の場合には配列番号22のアミノ酸配列を含み、Asp261Gly変異体の場合には配列番号14のアミノ酸配列を含み、Asp261Asn変異体の場合には配列番号16のアミノ酸配列を含み、またPhe240Ile変異体の場合には配列番号18のアミノ酸配列を含む。特に発現ベクターが配列番号12、配列番号22、配列番号14、配列番号16、配列番号18もしくは配列番号6に示されたアミノ酸配列を含む場合、又はそれが配列番号11、配列番号21、配列番号13、配列番号15、配列番号17もしくは配列番号5に示された核酸配列によりコードされ、もしくはそれを含む場合、Asp227Val変異体、Asp227Gly変異体、Asp261Gly変異体、Asp261Asn変異体、Phe240Ile変異体又はTrp91Ala変異体を使用する発現ベクターが最も好ましい。
加えて、本発明はwtNPTと較べてアミノ酸位置Trp91、Val198位置又はPhe240位置にwtアミノ酸とは異なるアミノ酸をコードする改変ネオマイシンホスホトランスフェラーゼ遺伝子及びそれらの遺伝子産物を初めて提供する。特に、本発明はwtNPTと較べて低下された酵素活性を有するTrp91変異体、Val198変異体又はPhe240変異体を初めて提供する。ここに記載され、本発明の範囲内で利用できるようにされた改変NPTはアミノ酸位置91でアラニン、位置198でグリシン、また位置240でイソロイシンをコードすることが好ましい。更に、本発明は、wtNPTと較べて、位置182でグリシン、位置227でアラニン又はバリン、また位置261でグリシンをコードするNPT変異体を最初に提供する。遺伝子及び遺伝子産物(酵素)の両方が本発明の範囲内で初めて提供される。この状況で、本発明は配列番号4、配列番号6、配列番号8、配列番号10、配列番号12、配列番号14及び配列番号18のアミノ酸配列を有する改変NPTを初めて提供する。更に、本発明は配列番号3、配列番号5、配列番号7、配列番号9、配列番号11、配列番号13、及び配列番号17のDNA配列を有する改変NPT遺伝子を提供する。
【0018】
対象遺伝子
本発明の発現ベクター中に含まれる対象遺伝子は対象産物をコードするあらゆる長さのヌクレオチド配列を含む。遺伝子産物又は“対象産物”は一般にタンパク質、ポリペプチド、ペプチド又はこれらのフラグメントもしくは誘導体である。しかしながら、それはまたRNA又はアンチセンスRNAであってもよい。対象とする遺伝子はその完全長、短くされた形態、融合遺伝子として、又は標識遺伝子として存在してもよい。それはゲノムDNA又は好ましくはcDNA或いは融合の相当するフラグメントであってもよい。対象遺伝子は天然の遺伝子配列であってもよく、又はそれは変異されてもよく、もしくはそれ以外に改変されてもよい。このような改変として、特別な宿主細胞に適合するためのコドン最適化及びヒト化が挙げられる。対象遺伝子は、例えば、分泌されたポリペプチド、細胞質ポリペプチド、核配置されたポリペプチド、膜結合されたポリペプチド又は細胞表面結合されたポリペプチドをコードしてもよい。
“ヌクレオチド配列”又は“核酸配列”という用語はオリゴヌクレオチド、ヌクレオチド、ポリヌクレオチド及びこれらのフラグメントだけでなく、一本鎖又は二本鎖として生じ、遺伝子のコーディング鎖又は非コーディング鎖に相当し得るゲノム起源又は合成起源のDNA又はRNAを示す。核酸配列は通常の技術、例えば、部位特異的変異導入又はPCR媒介変異導入(例えば、Sambrookら, 1989又はAusubelら, 1994に記載される)を使用して改変されてもよい。
【0019】
“コードする”とは核酸中のヌクレオチドの特定配列、例えば、染色体又はmRNA中の遺伝子が、生物学的プロセスにおいてその他のポリマー及び巨大分子、例えば、rRNA、tRNA、mRNA、その他のRNA分子、cDNA又はポリペプチドの合成のためのマトリックスとして作用する性質又は能力を意味する。それ故、所望のタンパク質が細胞又は別の生物系中でmRNAの転写、続いて翻訳により生成される場合、その遺伝子はタンパク質をコードする。ヌクレオチド配列がmRNA配列と同じである、通常配列データバンク、例えば、EMBL又はGenBankに示されるコーディング鎖、そしてまた転写のためのマトリックスとして作用する遺伝子又はcDNAの非コーディング鎖のいずれも産物又はタンパク質をコードすると称されることがある。また、タンパク質をコードする核酸には、縮重遺伝子コードに基づいてヌクレオチド配列の異なる順序を有するがタンパク質の同じアミノ酸配列をもたらす核酸も含まれる。タンパク質をコードする核酸配列はまたイントロンを含んでもよい。
cDNAという用語は遺伝子から生じたmRNA又はその他のRNAからの逆転写および第二DNAストランドの合成により調製されるデオキシリボ核酸を表す。cDNAが二本鎖DNA分子として存在する場合、それはコーディングストランド及び非コーディングストランドの両方を含む。
イントロンという用語はあらゆる長さの非コーディングヌクレオチド配列を表す。それらは多くの真核生物遺伝子中に天然に存在し、スプライシングとして知られている方法により既に転写されたmRNA前駆体から排除される。これは成功裏のタンパク質合成のために正しいリーディングフレームを有するプロセッシングされた成熟mRNAを生じるように5'末端及び3'末端におけるイントロンの正確な切除及び得られるmRNA末端の正確な結合を必要とする。このスプライシング過程に関与するスプライスドナー部位及びスプライスアクセプター部位の多く、即ち、エキソン-イントロン境界又はイントロン-エキソン境界に直接配置された配列が、今までに特性決定されている。総覧について、Ohshimaら, 1987を参照のこと。
【0020】
対象とするタンパク質/産物
生物医薬上重要なタンパク質/ポリペプチドとして、例えば、抗体、酵素、サイトカイン、リンホカイン、付着分子、受容体及びこれらの誘導体又はフラグメントが挙げれるが、これらに限定されない。一般に、アゴニストもしくはアンタゴニストとして作用し、かつ/又は治療もしくは診断上の適用を有する全てのポリペプチドが有益である。
“ポリペプチド”という用語はアミノ酸配列又はタンパク質について使用され、あらゆる長さのアミノ酸のポリマーを表す。この用語はまたグリコシル化、リン酸化、アセチル化又はタンパク質プロセシングの如き反応により翻訳後に修飾されたタンパク質を含む。ポリペプチドの構造は、例えば、その生物学的活性を保持しながらアミノ酸の置換、欠失又は挿入及びその他のタンパク質との融合により改変されてもよい。こうして、“ポリペプチド”という用語はまた、例えば、免疫グロブリン成分、例えば、Fc成分、及び成長因子、例えば、インターロイキンからなる融合タンパク質を含む。
治療タンパク質の例はインスリン、インスリン様成長因子、ヒト成長ホルモン(hGH)及びその他の成長因子、組織プラスミノーゲンアクチベーター(tPA)、エリスロポエチン(EPO)、サイトカイン、例えば、インターロイキン(IL)、例えば、IL-1、IL-2、IL-3、IL-4、IL-5、IL-6、IL-7、IL-8、IL-9、IL-10、IL-11、IL-12、IL-13、IL-14、IL-15、IL-16、IL-17、IL-18、インターフェロン(IFN)-α、-β、-γ、-ω又は-タウ、腫瘍壊死因子(TNF)、例えば、TNF-α、β又はγ、TRAIL、G-CSF、GM-CSF、M-CSF、MCP-1及びVEGFである。その他の例はモノクローナル抗体、ポリクローナル抗体、多重特異性抗体及び一本鎖抗体並びにこれらのフラグメント、例えば、Fab、Fab'、F(ab')2、Fc及びFc'フラグメント、軽鎖(L鎖)免疫グロブリン及び重鎖(H鎖)免疫グロブリン並びにこれらの定常領域、可変領域又は超可変領域だけでなく、Fvフラグメント及びFdフラグメントである(Chamovら, 1999)。抗体はヒト起源であっても非ヒト起源のものであってもよい。また、ヒト化抗体及びキメラ抗体が可能である。
【0021】
Fabフラグメント(フラグメント抗原結合=Fab)は隣接定常領域により一緒に保持される両方の鎖の可変領域からなる。それらは、例えば、通常の抗体からパパインの如きプロテアーゼで処理することにより、又はDNAクローニングにより産生されてもよい。その他の抗体フラグメントはペプシンによるタンパク質分解消化により産生し得るF(Ab')2フラグメントである。
遺伝子クローニングにより、重鎖の可変領域(VH)及び軽鎖の可変領域(VL)のみからなる短くされた抗体フラグメントを調製することが可能である。これらはFvフラグメント(可変フラグメント=可変部分のフラグメント)として知られている。定常鎖のシステイン基を介しての共有結合がこれらのFvフラグメントでは可能ではないので、それらはしばしば或る種のその他の方法により安定化される。この目的のために、重鎖及び軽鎖の可変領域が約10〜30アミノ酸、好ましくは15アミノ酸の短いペプチドフラグメントによりしばしば一緒に結合される。これはVH及びVLがペプチドリンカーにより一緒に結合される単一ポリペプチド鎖を生じる。このような抗体フラグメントもまた一本鎖Fvフラグメント(scFv)と称される。scFv抗体の例が知られており、例えば、Hustonら, 1988に記載されている。
過去何年かに、種々の戦略が多量体のscFv誘導体を産生するために開発されている。その目的は、改良された薬物速度論的性質及び増大された結合活性を有する組換え抗体を産生するためである。scFvフラグメントの多量体化を得るために、それらが多量体化ドメインを有する融合タンパク質として産生される。多量体化ドメインは、例えば、IgGのCH3領域又はらせん構造(“コイルドコイル構造”)、例えば、ロイシンジッパードメインであってもよい。その他の戦略では、scFvフラグメントのVH領域とVL領域の間の相互作用が多量体化に使用される(例えば、ジアボディ、トリボディ及びペンタボディ)。
【0022】
ジアボディ(diabody)(二量体抗体)という用語は当業界で2価のホモ二量体scFv誘導体を表すのに使用される。scFv分子中のペプチドリンカーを5〜10アミノ酸に短くすることはVH/VL鎖を重ねることによりホモ二量体の生成をもたらす。ジアボディは挿入されたジスルフィド架橋により更に安定化されてもよい。ジアボディの例が文献、例えば、Perisicら, 1994に見られる。
ミニボディという用語は当業界で2価のホモ二量体scFv誘導体を表すのに使用される。それは二量体化領域として免疫グロブリン、好ましくはIgG、最も好ましくはIgG1のCH3領域を含む融合タンパク質からなる。これはまたIgGのヒンジ領域及びリンカー領域によりscFvフラグメントを連結する。このようなミニボディの例がHuら, 1996により記載されている。
トリアボディという用語は当業界で3価のホモ三量体のscFv誘導体を表すのに使用される(Korttら, 1997)。リンカー配列を使用しないVH VLの直接融合が三量体の生成をもたらす。
2価、3価又は4価の構造を有するミニボディとして当業界で知られているフラグメントはまたscFvフラグメントの誘導体である。多量体化は二量体、三量体又は四量体のコイルドコイル構造により得られる(Packら, 1993及び1995; Lovejoyら, 1993)。
【0023】
蛍光タンパク質をコードする遺伝子
別の実施態様において、本発明の発現ベクターは、好ましくは対象とする遺伝子に機能し得る形で連結された、蛍光タンパク質をコードする遺伝子を含む。両方の遺伝子は対象とするタンパク質/産物及び蛍光タンパク質が2シストロン性mRNAによりコードされるように単一異種プロモーターの制御下で転写されることが好ましい。これは蛍光タンパク質の発現率により、対象とするタンパク質/産物を多量に生成する細胞を同定することを可能にする。
蛍光タンパク質は、例えば、緑色、青緑色、青色、黄色又はその他の着色蛍光タンパク質であってもよい。一つの特別な例はオワンクラゲ(Aequorea victoria)又はウミシイタケ(Renilla reniformis)から得られた緑色蛍光タンパク質(GFP)及びそれらから発生された変異体である。例えば、Bennetら, 1998; Chalfieら, 1994; WO 01/04306及びその中に引用された文献を参照のこと。
その他の蛍光タンパク質及びそれらをコードする遺伝子がWO 00/34318、WO 00/34326、WO 00/34526及びWO 01/27150(これらは参考として本明細書に含まれる)に記載されている。これらの蛍光タンパク質は種花虫綱の非バイオルミネセント生物、例えば、アネモニア・マジャノ(Anemonia majano)、ツツウミヅタ(Clavularia)種、スナギンチャク(Zoanthus)種I、スナギンチャク種II、ディスコゾーマ・ストリアタ(Discosoma striata)、ディスコゾーマ種“レッド”、ディスコゾーマ種“グリーン”、ディスコゾーマ種“マゼンタ”、ヘビイソギンチャク(Anemonia sulcata)の蛍光体である。
本発明に従って使用される蛍光タンパク質は野生型タンパク質に加えて天然の、又は遺伝子操作された変異体及び変異体、これらのフラグメント、誘導体又は変異体(これらは、例えば、その他のタンパク質又はペプチドと融合された)を含む。使用される変異は、例えば、励起スペクトル又は発光スペクトル、発色団の形成、タンパク質の吸光係数又は安定性を変化し得る。更に、哺乳動物細胞又はその他の種中の発現がコドン最適化により改良し得る。本発明によれば、蛍光タンパク質はまた選抜可能なマーカー、好ましくは増幅可能な選抜可能マーカー、例えば、ジヒドロ葉酸還元酵素(DHFR)との融合に使用されてもよい。
蛍光タンパク質により放出された蛍光はタンパク質を、例えば、蛍光活性化セルソーター(FACS)によるスルーフローサイトメトリー又は蛍光顕微鏡により検出することを可能にする。
【0024】
その他の調節要素
発現ベクターは対象とする遺伝子そしてまた好ましくは蛍光タンパク質の遺伝子の発現を可能にする少なくとも一つの異種プロモーターを含む。
プロモーターという用語はそれと機能機能可能に連結された遺伝子又は配列の転写を可能にし、制御するポリヌクレオチド配列を表す。プロモーターはRNAポリメラーゼを結合するための認識配列及び転写のための開始部位(転写開始部位)を含む。或る種の細胞型又は宿主細胞中で所望の配列を発現するために、好適な機能性プロモーターが選ばれる必要がある。当業者は構成的プロモーター、誘導性プロモーター及び抑制プロモーターを含む、種々の源からの種々のプロモーターを良く知っているであろう。それらはデータバンク、例えば、GenBankに寄託され、別々の要素或いは市販又は個々の源からのポリヌクレオチド配列内でクローン化された要素として得られてもよい。誘導性プロモーターでは、プロモーターの活性がシグナルに応答して低下又は増大され得る。誘導性プロモーターの一つの例はテトラサイクリン(tet)プロモーターである。これはテトラサイクリン調節トランスアクチベータータンパク質(tTA)により誘導し得るテトラサイクリンオペレーター配列(tetO)を含む。テトラサイクリンの存在下で、tetOへのtTAの結合が抑制される。その他の誘導性プロモーターの例はjun、fos、メタロチオネイン及び熱ショックプロモーターである(また、Sambrookら, 1989; Gossenら, 1994を参照のこと)。
真核生物中の高発現に特に適しているプロモーターの中に、例えば、ハムスターのユビキチン/S27aプロモーター(WO 97/15664)、SV40初期プロモーター、アデノウイルス主要後期プロモーター、マウスメタロチオネイン-Iプロモーター、ラウス肉腫ウイルスの長末端リピート領域、ヒトサイトメガロウイルスの初期プロモーターがある。その他の異種哺乳動物プロモーターの例はアクチン、免疫グロブリン又は一以上の熱ショックプロモーターである。
【0025】
相当する異種プロモーターは発現カセット中の転写活性を増大/調節するためにその他の調節配列に機能し得る形で連結し得る。
例えば、プロモーターは転写活性を増大するためにエンハンサー配列に機能可能に連結することができる。このために、一以上のエンハンサー及び/又はエンハンサー配列の幾つかのコピー、例えば、CMV又はSV40エンハンサーを使用することもできる。それ故、本発明の発現ベクターは、別の実施態様において、一以上のエンハンサー/エンハンサー配列、好ましくはCMV又はSV40エンハンサーを含む。
エンハンサーという用語はcis配置でプロモーターの活性に作用し、こうしてこのプロモーターに機能可能に連結された遺伝子の転写を刺激するポリヌクレオチド配列を表す。プロモーターと違って、エンハンサーの効果は位置及び配向とは独立であり、それ故、それらはイントロン内又は更にはコーディング領域内の、転写単位の前又は後に位置し得る。エンハンサーは転写単位の直ぐの付近及びプロモーターからのかなりの距離の両方に位置されてもよい。また、プロモーターと物理的かつ機能的な重なりを有することが可能である。当業者は独立の要素又はポリヌクレオチド配列(例えば、ATCCに寄託され、又は市販の源及び個々の源からの)内でクローン化された要素として利用できる種々の源からの幾つかのエンハンサー(データバンク、例えば、GenBankに寄託されたもの、例えば、SV40エンハンサー、CMVエンハンサー、ポリオーマエンハンサー、アデノウイルスエンハンサー)を知っているであろう。また、幾つかのプロモーター配列はエンハンサー配列、例えば、頻繁に使用されるCMVプロモーターを含む。ヒトCMVエンハンサーはこれまでに同定された最強のエンハンサーの一つである。誘導エンハンサーの一例はメタロチオネインエンハンサーであり、これはグルココルチコイド又は重金属により刺激し得る。
別の可能な改変は、例えば、多重Sp1結合部位の導入である。プロモーター配列は転写活性の制御/調節を可能にする調節配列と組み合わされてもよい。こうして、プロモーターは抑制性/誘導性にされ得る。これは、例えば、転写因子をアップレギュレート又はダウンレギュレートするための結合部位である配列に連結することにより行ない得る。例えば、上記転写因子Sp1は転写活性に正の効果を有する。別の例はアクチベータータンパク質AP1のための結合部位であり、これは転写に正及び負の両方で作用し得る。AP1の活性はあらゆる種類の因子、例えば、成長因子、サイトカイン及び血清により調節し得る(Faisstら, 1992及びその中の文献)。転写効率はまた一つ、二つ、三つ又はそれ以上の塩基の変異(置換、挿入又は欠失)によりプロモーター配列を変化させることにより増大させることができ、次いでレポーター遺伝子アッセイで、プロモーター活性が増大したか否かを測定することができる。
【0026】
基本的に、付加的な調節要素は異種プロモーター、エンハンサー、終結シグナル及びポリアデニル化シグナル並びにその他の発現調節要素を含む。誘導調節配列及び構成的調節配列の両方が種々の細胞型について知られている。
“転写調節要素”は一般に発現すべき遺伝子の上流のプロモーター、転写開始部位及び終結部位並びにポリアデニル化シグナルを含む。
“転写開始部位”という用語は一次転写産物、即ち、mRNA前駆体中にとり込まれる第一核酸に相当する構築物中の核酸を表す。転写開始部位はプロモーター配列と重なっていることもある。
“転写終結部位”という用語は、通常対象とする遺伝子又は転写されるべきである遺伝子部分の3'末端にあってRNAポリメラーゼによる転写の終結をもたらすヌクレオチド配列を表す。
“ポリアデニル化シグナル”は真核生物mRNAの3'末端にある特定部位における開裂及び開裂された3'末端における約100-200アデニンヌクレオチドの配列(ポリAテール)の転写後のとり込みを生じるシグナル配列である。ポリアデニル化シグナルは開裂部位の約10-30ヌクレオチド上流の配列AATAAA及び下流に位置される配列を含む。種々のポリアデニル化要素、例えば、tk ポリA、SV40後期及び初期ポリA又はBGHポリAが知られている(例えば、米国特許第5,112,458号に記載されている)。
本発明の好ましい実施態様において、夫々の転写単位はプロモーター又はプロモーター/エンハンサー要素、対象とする遺伝子及び/又はマーカー遺伝子だけでなく、転写終結要素を有する。別の好ましい実施態様において、転写単位は二つの更なる翻訳調節単位を含む。
【0027】
“翻訳調節要素”は、発現すべき夫々のポリペプチドについて、翻訳開始部位(AUG)、停止コドン及びポリAシグナルを含む。最適の発現のために、転写レベル又は発現レベルで発現に影響し得る潜在的に望ましくない付加的な翻訳開始コドン又はその他の配列を排除するために、発現される核酸配列の5'-及び/又は3'-未翻訳領域を除去、追加又は変化することが推奨されるかもしれない。発現を促進するために、リボソームコンセンサス結合部位がまた開始コドンの直ぐ上流に挿入されてもよい。分泌されるポリペプチドを生成するために、対象とする遺伝子は合成されたポリペプチドをER膜に輸送するシグナル前駆体ペプチドをコードするシグナル配列を通常含む。シグナル配列は分泌されるタンパク質のアミノ末端に常にではないがしばしば位置され、そのタンパク質がER膜で濾過された後にシグナルペプチダーゼにより切断される。遺伝子配列はそれ自体のシグナル配列を必ずではないが通常含むであろう。天然シグナル配列が存在しない場合、異種シグナル配列が既知の様式で導入されてもよい。この種の多くのシグナル配列が当業者に知られており、配列データバンク、例えば、GenBank及びEMBLに寄託されている。
本発明の一つの重要な調節要素は内部リボソームエントリー部位(IRES)である。IRES要素は5'-末端メチルグアノシニウムキャップ(CAP構造)及び上流遺伝子とは独立に翻訳開始を機能的に活性化する配列を含み、動物細胞中で単一転写産物からの二つのシストロン(オープンリーディングフレーム)の翻訳を可能にする。IRES要素は直ぐ下流に位置されるオープンリーディングフレームの翻訳のための独立のリボソーム侵入部位を与える。マルチシストロン性であり得る細菌mRNA(即ち、それはmRNAにより交互に翻訳される多くの異なるポリペプチド又は産物をコードし得る)とは対照的に、動物細胞からのmRNAの大半はモノシストロン性であり、唯一のタンパク質又は産物をコードする。真核生物細胞におけるマルチシストロン性転写産物の場合、その翻訳は最も近い上流にあった翻訳開始部位から開始され、最初の停止コドンにより停止され、その後に転写産物がリボソームから放出されるであろう。こうして、mRNAによりコードされた最初のポリペプチド又は産物のみが翻訳中に生成されるであろう。対照的に、転写産物中の第二の、又はその後のオープンリーディングフレームに機能し得る形で連結されるIRES要素を含むマルチシストロン性転写産物はその下流に位置されたオープンリーディングフレームのその後の翻訳を可能にし、その結果、同じ転写産物によりコードされた2以上のポリペプチド又は産物が真核生物細胞中で生成される。
【0028】
IRES要素は種々の長さ及び種々の起源のものであってもよく、例えば、脳心筋炎ウイルス(EMCV)又はその他のピコルナウイルスに由来してもよい。種々のIRES配列及びベクターの構築物中のそれらの使用が文献に記載されており、例えば、Pelletierら, 1988; Jangら, 1989; Daviesら, 1992; Adamら, 1991; Morganら, 1992; Sugimotoら, 1994; Rameshら, 1996; Mosserら, 1997を参照のこと。
下流に位置される遺伝子配列はIRES要素の3'末端に機能し得る形で連結され、即ち、遺伝子の発現が影響されず、もしくはほんの限界的に影響され、又は意図される目的に充分な発現を有するように間隔が選ばれる。充分な発現のためのIRES要素とその下流に位置される遺伝子の開始コドンの間の最適の許される距離は間隔を変化させ、レポーター遺伝子アッセイを使用して発現率を間隔の関数として測定することにより簡単な実験により決定し得る。
記載された手段により、異種遺伝子産物の発現に大いに有益である最適発現カセットを得ることが可能である。それ故、一つ以上のこのような手段により得られた発現カセットは本発明の更なる主題である。
【0029】
ハムスター-ユビキチン/S27aプロモーター
別の実施態様において、本発明の発現ベクターは、好ましくは対象とする遺伝子に機能し得る形で連結され、更に好ましくは対象とする遺伝子及び蛍光タンパク質をコードする遺伝子に機能し得る形で連結された、ハムスターのユビキチン/S27aプロモーターを含む。
ハムスターのユビキチン/S27aプロモーターはWO 97/15664に記載されている強力な同種プロモーターである。このようなプロモーターは下記の特徴の少なくとも一つを有することが好ましい:GCに富む配列領域、Sp1結合部位、ポリピリミジン要素、TATAボックスの不在。Sp1結合部位を有するが、TATAボックスを有しないプロモーターが特に好ましい。また、構成的に活性化され、特に血清含有細胞培養条件、低血清細胞培養条件及び無血清細胞培養条件下で同等に活性であるプロモーターが好ましい。別の実施態様において、それは誘導性プロモーター、特に血清の除去により活性化されるプロモーターである。
特に有利な実施態様はWO 97/15664の図5に含まれるようなヌクレオチド配列を有するプロモーターである。図5の位置-161から-45までの配列を含むプロモーター配列が特に好ましい。
本特許明細書の実施例に使用されるプロモーターは配列表の配列番号55の位置1923から2406までの配列を有するDNA分子を夫々含む。この配列はWO 97/15664の図5からのフラグメント-372〜+111に相当し、好ましいプロモーターを表し、即ち、好ましいプロモーターはこの配列領域を含むべきである。別の好適なプロモーターフラグメントは位置2134から2406まで(WO 97/15664の図5中の-161から+111までに相当する)の配列を含む。位置2251から2406までの配列のみを含むプロモーターは最早機能性ではない(WO 97/15664の図5中の位置-45から+111までに相当する)。プロモーター配列を位置2134から出発して5'方向に延長することが可能である。
また、完全ハムスターユビキチン/S27aプロモーター配列の機能性部分断片だけでなく、例えば、置換、挿入又は欠失により改変されたその部分断片の完全配列の機能性変異体/変異体を使用することが可能である。相当する部分断片、変異体又は変異体が以下にまた“改変プロモーター”と称される。
【0030】
改変プロモーターは、必要によりその他の調節要素と組み合わされてもよく、配列番号55に示されたヌクレオチド配列の位置1923から2406まで(WO 97/15664の図5からの-372から+111まで)のプロモーターフラグメントの転写活性に相当する転写活性を有することが好ましい。改変プロモーターはそれが比較レポーター遺伝子アッセイで1923〜2406フラグメント(-372〜+111フラグメント)の活性の少なくとも50%、好ましくは少なくとも80%、更に好ましくは少なくとも90%、最も好ましくは少なくとも100%を有する転写活性を有する場合に本発明の目的に有益であると判明する。少なくとも80%、好ましくは少なくとも85%、好ましくは少なくとも90%、更に好ましくは少なくとも95%、最も好ましくは少なくとも97%のハムスターユビキチン/S27aプロモーターの野生型配列配列番号55に対する最小配列相同性を有し、比較レポーター遺伝子アッセイで相当するプロモーター活性を有する改変プロモーターが特に好ましい。
相当する比較レポーター遺伝子アッセイで、基準配列を含む試験すべきプロモーターフラグメントが、無プロモーターレポーター遺伝子、例えば、ルシフェラーゼ、分泌アルカリ性ホスファターゼ又は緑色蛍光タンパク質(GFP)をコードする無プロモーターレポーター遺伝子の前にクローン化される。続いて、これらの構築物(プロモーター配列+レポーター遺伝子)がトランスフェクションにより試験細胞、例えば、CHO-DG44に導入され、当該プロモーターフラグメントによるレポーター遺伝子発現の誘導がレポーター遺伝子のタンパク質含量を測定することにより測定される。相当する試験が、例えば、Ausubelら, Current Protocols in Mokecular Biology, 1994(最新)に見られる。
ハムスターユビキチン/S27aプロモーター及び改変プロモーター(これはまた、例えば、5'未翻訳領域又はその選ばれたフラグメント、及びコーディング領域だけでなく、ユビキチン/S27a遺伝子の3'-未翻訳領域又はその選ばれたフラグメントを含んでもよい)のプロモーター配列は、例えば、Sambrookら, 1989; Ausubelら, 1994に記載されたような種々の通常の方法を使用してWO 97/15664に記載された配列の知識により当業者によれば得ることが出来る。WO 97/15664に記載された配列から出発して、例えば、好適なフラグメントが選択され、このフラクションの配列を含むオリゴヌクレオチドプローブが化学合成されてもよい。この種のプローブは、例えば、ハムスターゲノムのライブラリーからのハイブリダイゼーションにより、ユビキチン/S27a遺伝子又はその5'未翻訳領域もしくはその他のフラグメントをクローン化するのに使用することができる。上記レポーター遺伝子アッセイを使用して、当業者は大いに努力しなくてもプロモーター活性フラグメントを同定し、それらを本発明の目的に使用する状態に置かれている。5'未翻訳領域又はその特別なフラグメントはゲノムDNA又はゲノムライブラリーからの相当するプライマーによるPCR増幅により容易に得られる。5'未翻訳領域のフラグメントはまた一層大きいDNAフラグメントからの制限エキソヌクレアーゼIII消化により得ることもできる。このようなDNA分子はまた化学合成することもでき、又はライゲーションにより化学合成されたフラグメントから生成することもできる。
【0031】
欠失変異体、挿入変異体及び置換変異体は“部位特異的変異導入”及び/又は“PCRをベースとする変異導入技術”により生成することができる。対応する方法が、例えば、Lottspeich及びZorbas 1998 36.1章に、更なる文献とともに記載されている。
ハムスターユビキチン/S27a遺伝子の5'未翻訳領域又はハムスターユビキチンS27a遺伝子のS27a部分もしくは3'-未翻訳領域からのプローブによる交差ハイブリダイゼーションにより、その他の種、好ましくは哺乳動物種の相当する相同遺伝子から好適なプロモーター配列を同定し、単離することがまた可能である。好適な技術が、例えば、Lottspeich及びZorbas 1998 23章に記載されている。遺伝子はそれらのヌクレオチド配列がそれが相同である遺伝子のヌクレオチド配列に対し少なくとも70%、好ましくは少なくとも80%、好ましくは少なくとも90%、更に好ましくは少なくとも95%、最も好ましくは少なくとも97%の一致を示す場合に本発明の目的において“相同”である。
上記手段を使用して、異種遺伝子産物の発現に高度に有益である最適化発現カセットを得ることが可能である。それ故、一つ以上のこのような手段により得られた発現カセットは本発明の更なる対象である。
【0032】
本発明の発現ベクターの調製
本発明の発現ベクターは理論上、例えば、Sambrookら(1989)により記載されたような、当業界で知られている通常の方法により調製することができる。また、Sambrookはベクターの機能性成分、例えば、好適なプロモーター(ハムスターユビキチン/S27aプロモーターに加えて)、エンハンサー、終結シグナル及びポリアデニル化シグナル、抗生物質耐性遺伝子、選抜可能なマーカー、複製開始点並びにスプライシングシグナルを記載している。それらを生成するのに通常のクローニングベクター、例えば、プラスミド、バクテリオファージ、ファージミド、コスミド又はウイルスベクター、例えば、バキュロウイルス、レトロウイルス、アデノウイルス、アデノ関連ウイルス及び単純ヘルペスウイルスだけでなく、人工染色体/ミニ染色体を使用してもよい。真核細胞発現ベクターは典型的にはまた原核生物配列、例えば、複製起点及び細菌中のベクターの複製及び選抜を可能にする抗生物質耐性遺伝子を含む。ポリヌクレオチド配列の導入のための多重クローニング部位を含む幾つかの真核細胞発現ベクターが知られており、幾つかが種々の会社、例えば、ストラタジーン(ラ・ジョラ、CA、USA);インビトロジェン(カールスバッド、CA、USA);プロメガ(マジソン、WI、USA)又はBDバイオサイエンシズ・クロンテク(パロ・アルト、CA、USA)から商業的に入手できる。
【0033】
異種プロモーター、対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子そして必要により蛍光タンパク質をコードする遺伝子、付加的な調節要素、例えば、内部リボソームエントリー部位(IRES)、エンハンサー又はポリアデニル化シグナルは、当業者に良く知られている様式で発現ベクターに導入される。本発明の発現ベクターは、最小限でも、異種プロモーター、対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子を含む。発現ベクターはまた蛍光タンパク質をコードする遺伝子を含むことが好ましい。本発明によれば、ユビキチン/S27aプロモーターを異種プロモーターとして使用することが特に好ましい。異種プロモーター、好ましくはユビキチン/S27aプロモーター、対象とする遺伝子及び蛍光タンパク質をコードする遺伝子が一緒に機能可能に連結され、又は機能可能に連結され、ネオマイシンホスホトランスフェラーゼ遺伝子が同じ転写単位又は別々の転写単位中に位置される発現ベクターが特に好ましい。
本記載の範囲内で、“機能可能な連結”又は“機能可能に連結された”という用語は2以上の核酸配列又は部分配列がそれらの意図された機能を奏し得るように位置されている2以上の核酸配列又は部分配列を表す。例えば、プロモーター/エンハンサーはそれがシス位置で連結された遺伝子配列の転写を調節又は調節することができる場合にコーディング遺伝子配列に機能的に連結されている。必ずではないが、一般に、機能的に連結されたDNA配列は一緒に接近しており、そして二つのコーディング遺伝子配列が連結される場合又は分泌シグナル配列が連結される場合には、同じリーディングフレーム中にある。機能可能に連結されたプロモーターは一般にコーディング遺伝子配列の上流に配置されるが、それは必ずしも近くにある必要はない。エンハンサーは、それらが遺伝子配列の転写を補助する限り、いずれかの近くにある必要はない。この目的のために、それらは遺伝子配列の上流及び下流の両方に、おそらくそれから若干の距離にあってもよい。ポリアデニル化部位は転写がコーディング配列を介してポリアデニル化シグナルに進行するような様式でそれが遺伝子配列の3'末端に位置される場合に遺伝子配列に機能可能に連結される。連結は通常の組換え方法に従って、例えば、PCR技術、好適な制限切断部位におけるライゲーション又はスプライシングにより起こり得る。好適な制限切断部位が利用できない場合、合成オリゴヌクレオチドリンカー又はアダプターがそれ自体知られている様式で使用されてもよい。本発明によれば、好ましくは、機能可能な連結はイントロン配列を介して起こらない。
記載された実施態様の一つにおいて、異種プロモーター、好ましくはユビキチン/S27aプロモーター、対象遺伝子及び蛍光タンパク質をコードする遺伝子が一緒に機能可能に連結される。これは、例えば、対象遺伝子及び蛍光タンパク質をコードする遺伝子の両方が同じ異種プロモーターから出発して発現されることを意味する。
【0034】
特に好ましい実施態様において、機能可能な連結はIRES要素を介して起こり、その結果、両方の遺伝子から2シストロン性mRNAが合成される。本発明の発現ベクターは一以上のプロモーターに機能的に作用するエンハンサー要素を更に含んでもよい。異種プロモーター、好ましくはユビキチン/S27aプロモーター又はその改変形態がエンハンサー要素、例えば、SV40エンハンサー又はCMVエンハンサー要素に連結される発現ベクターが特に好ましい。
基本的には、発現ベクター内の遺伝子の発現は一以上の転写単位から出発して起こってもよい。転写単位という用語は転写すべき一以上の遺伝子を含む領域と定義される。転写単位内の遺伝子はこのような単位内の全ての遺伝子が同じプロモーター又はプロモーター/エンハンサーの転写制御下にあるような様式で互いに機能し得る形で連結される。遺伝子のこの転写連結の結果として、一種より多いタンパク質又は産物が転写単位から転写でき、こうして発現し得る。夫々の転写単位はその中に含まれる遺伝子配列の転写及び翻訳に必要である調節要素を含む。夫々の転写単位は同じ又は異なる調節要素を含見える。IRES要素又はイントロンが転写単位内の遺伝子の機能可能な連結に使用されてもよい。
発現ベクターは対象遺伝子、改変NPT遺伝子及び必要により蛍光タンパク質をコードする遺伝子を発現するための単一転写単位を含んでもよい。また、これらの遺伝子はまた二以上の転写単位中に配置されてもよい。転写単位内の遺伝子の種々の組み合わせが可能である。本発明の別の実施態様において、一つ、二つ又はそれ以上の転写単位からなる一より多い発現ベクターが同時トランスフェクション又はあらゆる所望の順序の連続トランスフェクションにより宿主細胞に挿入されてもよい。転写単位の適切な発現が確実にされることを条件として、夫々のベクターについての調節要素及び遺伝子のあらゆる組み合わせが選択し得る。必要であれば、その他の調節要素及び遺伝子、例えば、対象とする付加的な遺伝子又は選抜可能なマーカーが発現ベクターに配置されてもよい。
【0035】
それ故、対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼをコードする遺伝子を含む本発明の発現ベクターは両方の遺伝子を一つの転写単位中に又は二つの別々の転写単位中に含んでもよい。夫々の転写単位は一以上の遺伝子産物を転写し、発現し得る。両方の遺伝子が一つの転写単位中に含まれる場合、それらは同じプロモーター又はプロモーター/エンハンサーの制御下にあり、IRES要素が全ての成分の機能可能な連結を確実にするのに使用されることが好ましい。改変ネオマイシンホスホトランスフェラーゼをコードする遺伝子及び対象遺伝子が二つの別々の転写単位中に含まれる場合、それらは同じ又は異なるプロモーター/エンハンサーの制御下にあってもよい。しかしながら、改変NPT遺伝子については、より弱い異種プロモーター、例えば、SV40初期プロモーターが使用されることが好ましく、エンハンサーが使用されないことが好ましい。二つの別々の転写単位を有する発現ベクターが本発明の範囲内で好ましい。一つの(2シストロンの)転写単位は対象遺伝子、および、場合により蛍光タンパク質をコードする遺伝子、を含み、一方、別の転写単位は改変NPT遺伝子を含む。夫々の転写単位はポリAシグナル、好ましくはBGHポリA又はSV40ポリAをコードする配列により3'末端で特定されていることが好ましい。
また、本発明によれば、対象遺伝子に代えて、制限エンドヌクレアーゼの認識配列を介した対象遺伝子のクローニングを可能にするマルチクローニング部位のみを有する発現ベクターも好ましい。全ての種類の制限エンドヌクレアーゼだけでなく、関連制限エンドヌクレアーゼのための多くの認識配列が従来技術により知られている。認識配列として少なくとも6のヌクレオチドからなる配列が使用されることが好ましい。好適な認識配列のリストが、例えば、Sambrookら, 1989に見られる。
【0036】
宿主細胞
本発明の発現ベクターによるトランスフェクションのために、真核生物宿主細胞、好ましくは哺乳動物細胞、更に特別にはげっ歯類細胞、例えば、マウス細胞株、ラット細胞株及びハムスター細胞株が使用される。本発明の発現ベクターによる相当する細胞の成功裏のトランスフェクションは形質転換され、遺伝子改変された、組換え細胞又はトランスジェニック細胞をもたらし、これらもまた本発明の主題である。
本発明の目的に好ましい宿主細胞はハムスター細胞、例えば、BHK21細胞、BHK TK- CHO細胞、CHO-K1細胞、CHO-DUKX細胞、CHO-DUKX B1細胞及びCHO-DG44細胞又はこれらの細胞株の誘導体/子孫である。CHO-DG44細胞、CHO-DUKX細胞、CHO-K1細胞及びBHK21細胞、特にCHO-DG44細胞及びCHO-DUKX細胞が特に好ましい。また、マウスからのミエローマ細胞、好ましくはNS0細胞及びSp2/0細胞及びこれらの細胞株の誘導体/子孫も好適である。
本発明に従って使用し得るハムスター細胞及びマウス細胞の例が下記の表1に示される。しかしながら、これらの細胞の誘導体及び子孫、ヒト、マウス、ラット、サル、げっ歯類の細胞株(これらに限定されない)を含む他の哺乳動物細胞、又は酵母細胞、昆虫細胞及び植物細胞を含む(これらに限定されない)真核細胞を、生物医薬タンパク質の生成のための宿主細胞として使用することもできる。
【0037】
表1. ハムスター生産細胞株及びマウス生産細胞株

【0038】
本発明のポリヌクレオチド又は発現ベクターの一つによる真核生物宿主細胞のトランスフェクションは通常の方法(Sambrookら, 1989; Ausubelら, 1994)により行なわれる。トランスフェクションの好適な方法として、例えば、リポソーム媒介トランスフェクション、リン酸カルシウム共沈、エレクトロポレーション、ポリカチオン(例えば、DEAEデキストラン)媒介トランスフェクション、プロトプラスト融合、マイクロインジェクション及びウイルス感染が挙げられる。本発明によれば、安定的トランスフェクションが行なわれることが好ましく、この場合、構築物が宿主細胞のゲノム又は人工染色体/ミニ染色体に組み込まれ、又は宿主細胞中に安定な様式でエピソームにより含まれる。最適トランスフェクション頻度及び当該宿主中の異種遺伝子の発現を与えるトランスフェクション方法が好ましい。定義によれば、宿主細胞に挿入されるいかなる配列又はいかなる遺伝子も宿主細胞に関して“異種配列”又は“異種遺伝子”と称される。これはたとえ導入すべき配列又は導入すべき遺伝子が宿主細胞の内在性配列又は内在性遺伝子と同じであるとしても適用される。例えば、ハムスター宿主細胞に導入されたハムスターアクチン遺伝子はこの定義により異種遺伝子である。
【0039】
本発明によれば、本発明の発現ベクターの一つでトランスフェクションされた組換え哺乳動物細胞、好ましくはげっ歯類細胞、最も好ましくはハムスター細胞、例えば、CHO細胞又はBHK細胞が好ましい。
ヘテロマータンパク質、例えば、モノクローナル抗体(mAb)の組換え生成において、好適な宿主細胞のトランスフェクションは理論的に二つの異なる方法により行ない得る。この種のmAbは幾つかのサブユニット、重鎖及び軽鎖を含む。これらのサブユニットをコードする遺伝子は単一プラスミド上の独立又はマルチシストロンの転写単位中に収容されてもよく、これにより宿主細胞がその後にトランスフェクションされる。これは宿主細胞のゲノムへの組込み後に遺伝子の化学量論的提示を確保することを意図している。しかしながら、独立の転写単位の場合、異なるタンパク質をコードするmRNAが同じ安定性並びに転写効率及び翻訳効率を示すことがここで確実にされる必要がある。第二の場合、遺伝子の発現は単一プロモーターによりマルチシストロン転写単位内で起こり、単一の転写産物が生成される。IRES要素を使用することにより、遺伝子の高度に有効な内部翻訳開始が第二シストロン及びその後のシストロン中で得られる。しかしながら、これらのシストロンに関する発現率は第一シストロンの発現率よりも低い(所謂“キャップ”依存性前開始複合体による第1シストロンの翻訳開始は、IRES依存性翻訳開始よりもずっと有効である)。シストロンの真に等モルの発現を得るために、例えば、付加的なシストロン間の要素が導入されてもよく、これがIRES要素と連係して一様な発現率を確実にする(WO 94/05785)。
【0040】
幾つかの異種タンパク質を同時に生成する別の可能な方法(これが本発明によれば好ましい)は同時トランスフェクションであり、この場合、遺伝子が異なる発現ベクターに別々に組み込まれる。これは互いの遺伝子及び遺伝子産物の或る比率が調節でき、それによりmRNA安定性並びに転写及び翻訳の効率の差を調整するという利点を有する。加えて、発現ベクターがそれらの小さいサイズのためにより安定であり、クローニング中及びトランスフェクション中の両方で取り扱い易い。
それ故、本発明の一つの特別な実施態様において、宿主細胞が対象とする一以上のその他のタンパク質をコードする遺伝子を有する一以上のベクターで更にトランスフェクションされ、好ましくは同時トランスフェクションされる。同時トランスフェクションに使用されるその他の一以上のベクターは、例えば、同じプロモーター/エンハンサー組み合わせの制御下で対象とするその他の一以上のタンパク質及び少なくとも一つの選抜可能なマーカー、例えば、ジヒドロ葉酸還元酵素をコードする。
本発明によれば、無血清条件下、場合により動物タンパク質/ペプチドを含まない培地中で、宿主細胞が樹立され、適合され、培養されることが好ましい。商業的に得られる培地の例として、ハムF12(シグマ、ダイゼンホッフェン、DE)、RPMI-1640(シグマ)、ダルベッコ改良イーグル培地(DMEM;シグマ)、最小必須培地(MEM;シグマ)、イスコフ改良ダルベッコ培地(IMDM;シグマ)、CD-CHO(インビトロゲン、カールスバッド、CA、USA)、CHO-S-SFMII(インビトロゲン)、無血清CHO培地(シグマ)及び無タンパク質CHO培地(シグマ)が挙げられる。これらの培地の夫々は、場合により種々の化合物、例えば、ホルモン及び/又はその他の成長因子(例えば、インスリン、トランスフェリン、表皮成長因子、インスリン様成長因子)、塩(例えば、塩化ナトリウム、カルシウム、マグネシウム、リン酸塩)、緩衝剤(例えば、HEPES)、ヌクレオシド(例えば、アデノシン、チミジン)、グルタミン、グルコース又はその他の等価な栄養素、抗生物質及び/又は微量要素を添加してもよい。無血清培地が本発明によれば好ましいが、宿主細胞はまた好適な量の血清と混合された培地を使用して培養され、続いてタンパク質が生成されてもよい。一以上の選抜可能なマーカー遺伝子を発現する遺伝子改変細胞を選抜するために、一以上の選択薬剤が培地に添加される。
【0041】
“選択薬剤”という用語は選抜可能なマーカー遺伝子に関する欠損を有する宿主細胞の増殖又は生存に影響する物質を表す。本発明の範囲内で、ジェネティシン(G418)が改変ネオマイシンホスホトランスフェラーゼ遺伝子を有する異種宿主細胞の選抜のための培地添加剤として使用されることが好ましい。培地1ml当り100〜800μgのG418濃度が使用されることが好ましく、培地1ml当り300〜400μgが最も好ましい。宿主細胞が幾つかの発現ベクターでトランスフェクションされる場合、例えば、対象とする幾つかの遺伝子が宿主細胞に別々に導入される場合、それらは一般に異なる選抜可能なマーカー遺伝子を有する。
選抜可能なマーカー遺伝子は培地への相当する選択薬剤の添加によりこの遺伝子を含む細胞の特異的な選抜を可能にする遺伝子である。例示として、抗生物質耐性遺伝子を陽性の選抜可能なマーカーとして使用することができる。この遺伝子で形質転換された細胞のみが相当する抗生物質の存在下で増殖することができ、従って、選抜される。一方、形質転換されなかった細胞は、これらの選抜条件下で増殖又は生存することができない。陽性、陰性及び2機能性の選抜可能マーカーがある。陽性の選抜可能マーカーは耐性を選択薬剤に与えることにより、又は宿主細胞中の代謝もしくは異化代謝欠陥を相殺することにより形質転換細胞の選抜ひいては濃縮を可能にする。対照的に、選抜可能マーカーの遺伝子を受けた細胞は陰性の選抜可能マーカーにより選択的に排除し得る。これの例は単純ヘルペスウイルスのチミジンキナーゼ遺伝子であり、アシクロビル又はガンシクロビルの同時添加による細胞中のその発現はその排除をもたらす。本発明に使用される選抜可能マーカー(増幅可能な選抜可能マーカーを含む)として、遺伝子改変された変異体及び変異体、フラグメント、機能性均等物、誘導体、同族体及びその他のタンパク質又はペプチドとの融合が挙げられるが、選抜可能マーカーがその選抜特性を保持することを条件とする。このような誘導体は選択的であると考えられる領域又はドメイン中のアミノ酸配列にかなりの相同性を示す。2機能性(陽性/陰性)マーカーを含む多数の選抜可能なマーカー遺伝子が文献に記載されている(例えば、WO 92/08796及びWO 94/28143を参照のこと)。真核生物細胞中で通常使用される選抜可能なマーカーの例として、アミノグリコシドホスホトランスフェラーゼ(APH)、ハイグロマイシンホスホトランスフェラーゼ(HYG)、ジヒドロ葉酸還元酵素(DHFR)、チミジンキナーゼ(TK)、グルタミンシンセターゼ、アスパラギンシンセターゼの遺伝子並びにネオマイシン(G418)、プロマイシン、ヒスチジノールD、ベロマイシン、フレオマイシン及びゼオシンに対する耐性を与える遺伝子が挙げられる。
また、形質転換細胞を蛍光活性化セルソーティング(FACS)により選抜することが可能である。このために、細菌のβ-ガラクトシダーゼ、細胞表面マーカー又は蛍光タンパク質を形質転換細胞の選抜に使用することができる(例えば、緑色蛍光タンパク質(GFP)並びにオワンクラゲ及びウミシイタケ又はその他の種からのその変異体;赤色の蛍光タンパク質及びその他の色で蛍光を発するタンパク質並びに非バイオルミネセント生物、例えば、ディスコゾーマ種(Discosoma sp.)、ミナミウメボシイソギンチャク種(Anemonia sp.)、ツツウミヅタ種(Clavularia sp.)、スナギンチャク種(Zoanthus sp.)からのそれらの変異体)。
【0042】
遺伝子発現及び高生産性宿主細胞の選抜
遺伝子発現という用語は宿主細胞中の異種遺伝子配列の転写及び/翻訳に関する。発現率は一般に宿主細胞中に存在する相当するmRNAの量に基づいて、又は対象遺伝子によりコードされる生成される遺伝子産物の量に基づいて測定し得る。選択されたヌクレオチド配列の転写により生成されるmRNAの量は、例えば、ノーザンブロットハイブリダイゼーション、リボヌクレアーゼ-RNA-保護、細胞RNAのin situハイブリダイゼーション又はPCR方法(Sambrookら, 1989; Ausubelら, 1994)により測定し得る。選択されたヌクレオチド配列によりコードされるタンパク質はまた種々の方法、例えば、ELISA、ウェスタンブロット、ラジオイムノアッセイ、免疫沈殿、タンパク質の生物学的活性の検出又はタンパク質の免疫染色、続いてFACS分析(Sambrookら, 1989; Ausubelら, 1994)により測定し得る。
【0043】
“高発現レベル(又は率)、高発現、増大された発現又は高生産性”という用語は宿主細胞に導入される異種配列、例えば、治療タンパク質をコードする遺伝子の長く持続する、充分に高い発現又は合成を表す。本発明の細胞が、ここに記載された本発明の方法の一つにより培養され、この細胞が遺伝子増幅無しに、1日当りおよそ0.5pg(0.5pg/細胞/日)より少なくとも多い所望の遺伝子産物を生じる場合に、増大され、もしくは高い発現又は高発現レベルもしくは率或いは高生産性が存在する。また、本発明の細胞が、事前の遺伝子増幅無しで1日当りおよそ1.0pg(1.0pg/細胞/日)より少なくとも多い所望の遺伝子産物を生じる場合に、増大され、もしくは高い発現又は高発現もしくは率或いは高生産性が存在する。特に本発明の細胞が事前の遺伝子増幅無しに1日当りおよそ1.5pg(1.5pg/細胞/日)より少なくとも多い所望の遺伝子産物を生じる場合に、増大され、もしくは高い発現又は高発現レベルもしくは率或いは高生産性が存在する。特に本発明の細胞が事前の遺伝子増幅無しに1日当りおよそ2.0pg(2.0pg/細胞/日)より少なくとも多い所望の遺伝子産物を生じる場合に、増大され、もしくは高い発現又は高発現レベルもしくは率或いは高生産性が存在する。特に、本発明の細胞が事前の遺伝子増幅無しで1日当りおよそ3.0pg(3.0pg/細胞/日)より少なくとも多い所望の遺伝子産物を生じる場合に,増大され、もしくは高い発現又は特に高い発現レベルもしくは率或いは特に高い生産性が存在する。簡単な遺伝子増幅工程により、例えば、以下に記載されるようなDHFR/MTX増幅系を使用して、生産性が少なくとも2〜10倍に増大でき、従って、“高発現”、“増大された発現”又は“高生産性”という用語は、遺伝子増幅工程にかけられた細胞に関しては、この細胞が少なくとも1日当りおよそ5pg(5pg/細胞/日)より多く、好ましくは少なくともおよそ10pg/細胞/日より多く、更に好ましくは少なくともおよそ15pg/細胞/日より多く、更に好ましくは少なくともおよそ20pg/細胞/日より多く、又は少なくともおよそ30pg/細胞/日より多い所望の遺伝子産物を生じる場合に使用される。
【0044】
高発現もしくは増大された発現、高生産性又は高発現レベルもしくは率は本発明の発現ベクターの一つを使用すること、および本発明の方法の一つの使用、の両方により得られる。
例えば、対象遺伝子及び改変NPT遺伝子の同時発現により、異種遺伝子を高度に発現する細胞を選抜し、同定することが可能である。wtNPTと較べて、改変NPTは対象とする異種遺伝子の高発現とともに安定にトランスフェクションされた宿主細胞の一層有効な選抜を可能にする。
従って、本発明は(i) 哺乳動物細胞のプールを少なくとも一つの対象遺伝子及び野生型ネオマイシンホスホトランスフェラーゼと比較してその活性のわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5〜26%を有する改変ネオマイシンホスホトランスフェラーゼの一つの遺伝子でトランスフェクションすること、(ii)前記対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で前記細胞を培養し、(iii) 前記哺乳動物細胞を少なくとも一種の選択薬剤、好ましくはG418(これは哺乳動物細胞の増殖に選択的に作用し、かつ改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える)の存在下で培養すること、および(iv)一以上の対象タンパク質を前記哺乳動物細胞又は培養上清から得ることを特徴とする、組換え哺乳動物細胞中の少なくとも一つの対象遺伝子の発現方法に関する。本発明の発現ベクターでトランスフェクションされた組換え哺乳動物細胞が使用されることが好ましい。
また、本発明は(i) 哺乳動物細胞のプールを少なくとも一つの対象遺伝子及び野生型ネオマイシンホスホトランスフェラーゼとの比較によりその活性のわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5〜26%を有する改変ネオマイシンホスホトランスフェラーゼの遺伝子でトランスフェクションすること、(ii)前記哺乳動物細胞を前記対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、(iii) 前記哺乳動物細胞を少なくとも一種の選択薬剤、好ましくはG418(これは哺乳動物細胞の増殖に選択的に作用し、かつ改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える)の存在下で培養することを特徴とする、少なくとも一つの対象遺伝子を発現する組換え哺乳動物細胞の選抜方法に関する。
【0045】
少なくとも一つの対象遺伝子を発現させる方法及び対応する対象遺伝子を発現する組換え細胞を選抜する方法が特に好ましいのは、本出願に更に詳しく記載されるNPT遺伝子が使用される場合、特にアミノ酸位置182においてグリシン又はアスパラギン酸、アミノ酸位置91においてアラニン、アミノ酸位置198においてグリシン、アミノ酸位置227においてグリシン又はバリン、アミノ酸位置261においてグリシン又はアスパラギン或いはアミノ酸位置240においてイソロイシンをコードする、野生型遺伝子と比較して改変されたNPT遺伝子が使用される場合である。特にAsp227Val変異体、Asp227Gly変異体、Asp261Gly変異体、Asp261Asn変異体、Phe240Ile変異体又はTrp91Ala変異体を使用する場合が好ましい。一般に、この特許明細書に記載された本発明の全ての改変ネオマイシンホスホトランスフェラーゼ遺伝子がこのような方法に好適である。好ましいネオマイシンホスホトランスフェラーゼ遺伝子については、改変ネオマイシンホスホトランスフェラーゼ遺伝子に関する節を参照のこと。
対象とする遺伝子及び改変NPT遺伝子を発現する細胞の選抜は、例えば、G418を選択薬剤として添加することにより行なわれる。しかしながら、その他のアミノグリコシド抗生物質、例えば、ネオマイシン又はカナマイシンを使用することも可能である。本発明の細胞は培地1ml当り200〜800μgのG418中で培養され、選抜されることが好ましい。培地1ml当り300〜700μgのG418を添加することが特に好ましいと判明した。培地1ml当りおよそ400μgのG418の添加が最も好ましい実施態様である。このような方法を使用して、特に高い発現率を有する組換え細胞を選抜することが可能である。wtNPTの使用との比較により、選抜可能なマーカーとしての培地1ml当り400μgのG418による選抜後に、細胞はGlu182Gly変異体、Glu182Asp変異体及びVal198Gly変異体の場合に1.4-2.4倍、Asp227Gly変異体の場合に1.6-4.1倍、Asp227Ala変異体又はTrp91Ala変異体の場合に2.2又は4倍、Phe240Ile変異体又はAsp261Asn変異体の場合に5.7又は7.3倍更にはAsp261Gly変異体又はAsp227Val変異体の場合に9.3又は14.6倍に増大された生産性を示した。種々の改変NPT遺伝子に関する比生産能が図6に示される。
【0046】
相当する方法は、付加的な選抜可能なマーカーとして、一種以上の蛍光タンパク質(例えば、GFP)又は細胞表面マーカーを含む組換え宿主細胞のFACS補助選抜と組み合わされてもよい。増大された発現を得るその他の方法、及び異なる方法の組み合わせがまた使用されてもよく、例えば、(人工)転写因子の使用、内在性又は異種遺伝子配列をアップレギュレートするための天然又は合成の薬剤による細胞の処理、mRNA又はタンパク質の安定性(半減期)を改良すること、mRNA翻訳の開始を改良すること、エピソームプラスミドの使用により遺伝子量を増大すること(複製起点としてウイルス配列、例えば、SV40、ポリオーマ、アデノウイルス、EBV又はBPVの使用に基づく)、増幅促進配列(Hemannら, 1994)の使用又はDNAコンカテマー(Monacoら, 1996)をベースとするin vitro増幅系に基づいている。
対象遺伝子及び蛍光タンパク質をコードする遺伝子の共役した転写は選抜可能なマーカーとしての改変NPT遺伝子の使用と連係して特に有効と判明した。得られる2シストロン性mRNAは対象タンパク質/産物及び蛍光タンパク質の両方を発現する。対象とするタンパク質及び蛍光タンパク質の発現のこのカップリングに基づいて、本発明によれば発現された蛍光タンパク質により、例えば、蛍光活性化セルソーティング装置(FACS)を使用してソーティングすることにより高生産性組換え宿主細胞を同定し、単離することが容易に可能である。
高い生命力及び所望の遺伝子産物の増大された発現率を示す組換え宿主細胞の選抜は多段階の工程である。本発明の発現ベクターでトランスフェクションされた(又は場合により例えば別のベクターで同時トランスフェクションされてもよい)宿主細胞は、例えば、選択薬剤、例えば、培地1ml当り100μg、200μg、400μg、600μg、800μg又はそれ以上のG418の濃度のG418の存在下の培養により、改変NPTを発現する細胞の選抜を可能にする条件下で培養される。次いで相当する細胞が蛍光タンパク質の最高発現率を示す細胞/細胞集団を同定し、ソーティングするために、対象遺伝子に結合された、蛍光タンパク質をコードする遺伝子の発現について少なくとも調べられる。蛍光タンパク質の最高発現率を有する細胞の10-20%に属する細胞のみがソーティングされ、更に培養されることが好ましい。実際には、これは蛍光細胞の最も明るい10%がソーティングされ、更に培養されることを意味する。それ故、細胞混合物の蛍光細胞の最も明るい5%、好ましくは最も明るい3%又は更には最も明るい1%がソーティングされ、複製し得る。特に好ましい実施態様において、蛍光細胞の最も明るい0.5%又は最も明るい0.1%のみがソーティングされ、複製される。
【0047】
選抜工程は細胞プールについて、又は前ソーティングされた細胞プール/細胞クローンを使用して行うことができる。一つ以上、好ましくは二つ以上、特に三つ以上のソーティング工程が行なわれてもよく、個々のソーティング工程の間で細胞が特定の時間、例えば、プールの場合にはおよそ2週にわたって培養され、複製されてもよい。図11及び12は、例えば、変異体Asp227Glyについて遺伝子増幅工程を用いて、また用いないでFACSに基づくソーティング後の比生産能を示す。
従って、本発明は(i) 組換え哺乳動物細胞を本発明の発現ベクターでトランスフェクションすること、(ii)トランスフェクションされた細胞を一以上の対象遺伝子、蛍光タンパク質をコードする遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、(iii) 前記哺乳動物細胞を前記哺乳動物細胞の増殖に選択的に作用し、かつ前記改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、および、(iv)蛍光遺伝子の特に高い発現を示す前記哺乳動物細胞をフローサイトメトリー分析によりソーティングすること、を特徴とする、少なくとも一つの対象異種遺伝子を発現する組換え哺乳動物細胞を得、選抜する方法に関する。所望により、工程(ii)〜(iv)が工程(iv)で得られた細胞を用いて1回又は数回繰り返されてもよい。
ソーティングされた哺乳動物細胞が、付加的な遺伝子増幅工程を用いないで、1日当りかつ細胞当り0.5pg(0.5pg/細胞/日)より多く、好ましくは1pg/細胞/日より大きく、更に好ましくは2pg/細胞/日より大きく、更に好ましくは3pg/細胞/日より大きく、更に好ましくは4pg/細胞/日より大きく、例えば、5、6、8、9、10pg/細胞/日より大きく、15、20、25pg/細胞/日より大きい等の所望の一以上の遺伝子産物の平均比生産能を有することを特徴とする方法が好ましい。上記のように、これらの細胞の生産性は、例えば、DHFR/MTX系を使用する簡単な遺伝子増幅工程により少なくとも2〜10倍に増大し得る。これは、例えば、NTP変異体Asp227Glyを使用する選抜について図12に示されている。比生産能は20〜25pg/細胞/日であった。
【0048】
また、本発明によれば、好適にソーティングされた細胞が複製され、対象とするコードされた遺伝子産物を調製するのに使用される方法も好ましい。これのために、選抜された高生産性細胞が無血清培地中で、好ましくは対象遺伝子の発現を可能にする条件下で懸濁培養される。対象タンパク質/産物は細胞培地から分泌された遺伝子産物として得られることが好ましい。しかしながら、タンパク質が分泌シグナルなしで発現される場合、遺伝子産物はまた細胞溶解産物から単離することもできる。その他の組換えタンパク質及び宿主細胞タンパク質を実質的に含まない純粋な均一産物を得るために、通常の精製操作が行なわれる。最初に、細胞及び細胞破砕物が培地又は溶解産物から除去される。次いで所望の遺伝子産物から、例えば、イムノアフィニティーカラム及びイオン交換カラムによる分別、エタノール沈殿、逆相HPLC又はセファデックス、シリカ又はDEAEの如き陽イオン交換樹脂によるクロマトグラフィーにより、夾雑可溶性タンパク質、ポリペプチド及び核酸が除かれる。組換え宿主細胞により発現された異種タンパク質の精製をもたらす方法が当業者に知られており、文献、例えば、Harrisら, 1995及びScopes 1988に記載されている。
【0049】
増幅可能な選抜可能マーカー遺伝子
加えて、本発明の細胞はまた必要によりそれらが増幅可能な選抜可能遺伝子の増幅をもたらす選択薬剤の存在下で培養される、一以上の遺伝子増幅工程にかけられてもよい。この工程は蛍光タンパク質を発現し、好ましくはFACS(好ましくはここに記載された方法の一つにおいて)により1回又は数回予備ソーティングされた細胞及び未だソーティングされていない細胞のどちらで行うことも出来る。
前提条件は宿主細胞が増幅可能な選抜可能マーカーをコードする遺伝子で更にトランスフェクションされることである。増幅可能な選抜可能マーカーをコードする遺伝子は本発明の発現ベクターの一つに存在するか、又は別のベクターにより宿主細胞に導入されることが推考される。
増幅可能な選抜可能マーカー遺伝子は或る培養条件下の真核生物細胞の増殖に必要とされる酵素を通常コードする。例えば、増幅可能な選抜可能なマーカー遺伝子はジヒドロ葉酸還元酵素(DHFR)をコードしてもよい。この場合、その遺伝子はそれでトランスフェクションされた宿主細胞が選択薬剤メトトレキセート(MTX)の存在下で培養される場合に増幅される。
下記の表2はその他の増幅可能な選抜可能なマーカー遺伝子及び本発明に従って使用し得る関連する選択薬剤の例を示し、これらはKaufman著Methods in Enzymology, 185:537-566 (1990)による総覧に記載されている。
【0050】
表2. 増幅可能な選抜可能なマーカー遺伝子
【0051】
表2続き
【0052】
本発明によれば、使用される増幅可能な選抜可能マーカー遺伝子はDHFRの機能を有するポリペプチド、例えば、DHFR又は蛍光タンパク質及びDHFRからの融合タンパク質をコードする遺伝子であることが好ましい。DHFRはプリンの生合成に必要である。DHFR遺伝子を欠いている細胞はプリン欠損培地中で増殖し得ない。それ故、DHFR遺伝子は無プリン培地中で培養される細胞中で選抜し、遺伝子を増幅するのに有益な選抜可能なマーカーである。DHFR遺伝子と連係して使用される選択培地はメトトレキセート(MTX)である。
それ故、本発明は下記の工程:(i) 対象タンパク質/産物、改変ネオマイシンホスホトランスフェラーゼ及びDHFRを少なくともコードする遺伝子による宿主細胞のトランスフェクション工程、(ii)種々の遺伝子の発現を可能にする条件下の前記細胞の培養工程、及び(iii)少なくとも増幅可能な選抜可能なマーカー遺伝子の増幅を可能にする選択薬剤、例えば、メトトレキセートの存在下で前記細胞を培養することによる同時組込み遺伝子の増幅工程、を含む、組換え哺乳動物細胞を調製および選抜する方法を含む。トランスフェクションされた細胞は血清の不在下で増大する濃度のMTXを添加してヒポキサンチン/チミジンを含まない培地中で培養されることが好ましい。最初の増幅工程におけるMTXの濃度は少なくとも5nMであることが好ましい。しかしながら、MTXの濃度はまた少なくとも20nM又は100nMであってもよく、段階的に1μMまで増大し得る。個々の場合には、1μMより大きい濃度、例えば、2μMが使用されてもよい。
【0053】
相当する細胞が更に蛍光タンパク質の遺伝子で形質転換される場合、これらの細胞は蛍光活性化細胞ソーティング装置(FACS)を使用して同定され、ソーティングされ、次いで遺伝子増幅工程において少なくとも20nM、好ましくは50nM又は100nMのMTXの存在下で培養することができる。このようにして、生産性を細胞当り1日当り実質的に20pg以上、好ましくは21以上、22以上、23以上、24以上、25以上等、30以上、35以上、40以上等の遺伝子産物まで増大させることが可能である。宿主細胞は少なくとも対象遺伝子及び増幅可能な選抜可能マーカー遺伝子のコピー数を増大するために一以上の遺伝子増幅工程にかけられてもよい。本発明によれば、得られる高生産性は、ネオマイシン、カナマイシン及びG418のようなアミノグリコシド抗生物質に対するネオマイシンホスホトランスフェラーゼ媒介耐性による有効な予備選抜と関連している。それ故、必要とされる遺伝子増幅工程の数を減少し、例えば、単一遺伝子増幅のみを行なうことが可能である。
更なる実施態様において、こうしてまた、本発明は少なくとも一つの対象異種遺伝子を発現する組換え哺乳動物細胞を得る方法及び選抜する方法に関するものであり、(i) 組換え哺乳動物細胞を本発明の発現ベクター及び増幅可能な選抜可能マーカー遺伝子でトランスフェクションすること、(ii)前記哺乳動物細胞を一以上の対象遺伝子、改変ネオマイシンホスホトランスフェラーゼ遺伝子及び蛍光タンパク質をコードする遺伝子の発現を可能にする条件下で培養すること、(iii) 前記哺乳動物細胞を前記哺乳動物細胞の増殖に選択的に作用し、かつネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、(iv)蛍光タンパク質の高発現を示す前記哺乳動物細胞をフローサイトメトリー分析によりソーティングすること、(v) ソーティングされた前記細胞を前記増幅可能な選抜可能なマーカー遺伝子が発現される条件下で培養すること、および(vi)選択薬剤を前記増幅可能な選抜可能なマーカー遺伝子の増幅をもたらす培地に添加すること、を特徴とする。
【0054】
本発明において記載された改変ネオマイシンホスホトランスフェラーゼ遺伝子が使用される相当する方法が特に好ましい。また、単一の増幅工程が行なわれる方法が好ましい。また、細胞当り1日当り20pg以上、好ましくは21以上、22以上、23以上、25以上等、30以上、35以上、40以上等の所望の一以上の遺伝子産物の平均比生産能を示す組換え哺乳動物細胞をもたらす相当する方法が好ましい。
哺乳動物細胞、好ましくはマウスミエローマ細胞及びハムスター細胞が、増幅可能な選抜可能マーカーとしてのDHFRの使用に好ましい宿主細胞である。細胞株CHO-DUKX(ATCC CRL-9096)及びCHO-GD44(Urlaubら, 1983)が特に好ましい。何とならば、変異の結果としてそれら自体のDHFR活性を有しないからである。自身が内因性DHFR活性を有するその他のタイプの細胞中でDHFR誘導増幅を同様に使用することができるように、トランスフェクション過程でメトトレキセートに対する低下された感受性を有するタンパク質をコードする変異DHFR遺伝子(Simonsonら, 1983; Wiglerら, 1980; Haberら, 1982)を使用することが可能である。
DHFRマーカーはDHFR陰性細胞、例えば、CHO-DG44又はCHO-DUKXを使用する場合に選抜及びその後の増幅に特に適している。何とならば、これらの細胞は内因性DHFRを発現せず、それ故、無プリン培地中で増殖しないからである。従って、DHFR遺伝子が支配的な選抜可能マーカーとしてここで使用することができ、形質転換細胞がヒポキサンチン/チミジンを含まない培地中で選抜される。
本発明が幾つかの非限定実施例を参照して以下に更に充分に記載される。
【0055】
略号
Ala (=A) アラニン
AP: アルカリ性ホスファターゼ
Asn (=N): アスパラギン
Asp (=D): アスパラギン酸
bp: 塩基対
BSA: ウシ血清アルブミン
CHO: チャイニーズハムスター卵巣
dhfr: ジヒドロ葉酸還元酵素
DMSO: ジメチルスルホキシド
ELISA: 酵素結合免疫吸着検定法
FACS: 蛍光活性化セルソーター
FITC: フルオレセイン-イソチオシアネート
GFP: 緑色蛍光タンパク質
Glu (=E): グルタミン酸
Gly (=G): グリシン
HBSS: ハンクス平衡塩溶液
HT: ヒポキサンチン/チミジン
Ile (=I): イソロイシン
IRES: 内部リボソームエントリー部位
kb: キロベース
mAb: モノクローナル抗体
MCP-1: 単球走化性タンパク質1
MTX: メトトレキセート
MW: 平均値
NPT: ネオマイシンホスホトランスフェラーゼ
PCR: ポリメラーゼ連鎖反応
PBS: リン酸緩衝生理食塩水
Phe (=F): フェニルアラニン
Trp (=W): トリプトファン
Val (=V): バリン
WT: 野生型
【0056】
方法
1. 細胞培養及びトランスフェクション
細胞CHO-DG44/dhfr-/-(Urlaubら, 1983)を細胞培養フラスコ中で37℃で湿った大気、5%のCO2中でヒポキサンチン及びチミジン(インビトロゲンGmbH、カールスルーヘ、DE)を添加した無血清CHO-S-SFMII培地中で懸濁細胞として恒常的に培養した。細胞カウント及び生命力をCASY1セルカウンター(シェルフェ・システム、DE)又はトリプタンブルー染色により測定し、次いで1-3x105/mlの濃度で播き、2-3日毎に繰り返した。
リポフェクタミン・プラス試薬(インビトロゲンGmbH)をCHO-DG44のトランスフェクションに使用した。夫々のトランスフェクション混合物について、合計1μgのプラスミド-DNA、4μLのリポフェクタミン及び6μLのプラス試薬を製造業者の指示に従って一緒に混合し、200μLの体積でHT添加CHO-S-SFMII培地0.8ml中の6x105の指数増殖しているCHO-DG44細胞に添加した。細胞インキュベーター中で37℃で3時間のインキュベーション後に、HT添加CHO-S-SFMII培地2mlを添加した。NPTをベースとする選抜のために、細胞をトランスフェクションの2日後にG418(インビトロゲン)を含むHT添加CHO-S-SFMII培地に移し、3〜4日毎に培地を交換した。原則として、400μg/mlのG418を選抜のために添加し、また幾つかの実験シリーズでは、濃度を200μg/mlに低下し、又は500、600もしくは800μg/mlに上昇させた。同時トランスフェクション(一つの発現ベクターはDHFRを含み、別の発現ベクターはネオマイシンホスホトランスフェラーゼ選抜可能なマーカーを含む)の場合のDHFR及びNPTをベースとする選抜において、細胞をトランスフェクションの2日後にヒポキサンチン及びチミジンを添加しないCHO-S-SFMII培地に移し、またG418(インビトロゲン)を400μg/mlの濃度で培地に添加した。
組込み異種遺伝子のDHFRをベースとする遺伝子増幅をHTを含まないCHO-S-SFMII培地への5-2000nMの濃度の選択薬剤MTX(シグマ、ダイゼンホッフェン、DE)の添加により得ることができる。
【0057】
2. 発現ベクター
発現を分析するために、pAD-CMVベクター(Wernerら, 1998)をベースとし、CMVエンハンサー/ハムスターユビキチン/S27aプロモーター(WO 97/15664)の組み合わせによる異種遺伝子の構成的発現を媒介する真核細胞発現ベクターを使用した。基本ベクターpBIDは増幅可能な選抜可能なマーカーとして作用するdhfr-ミニジーン(例えば、EP-0-393-438を参照のこと)を含むが、ベクターpBINではそのdhfr-ミニジーンがネオマイシンホスホトランスフェラーゼ耐性遺伝子により置換されていた(図1)。この目的のために、SV40初期プロモーター及びTK-ポリアデニル化シグナルを含む、選抜可能なマーカーネオマイシンホスホトランスフェラーゼを、1640 bp Bsu36Iフラグメントとして商業的プラスミドpBK-CMV(ストラタゲン、ラ・ジョラ、CA、USA)から単離した。そのフラグメントの末端をクレノー-DNA-ポリメラーゼでフィルイン(fill in)する反応後に、フラグメントをベクターpBIDの3750bp Bsu36I/StuIフラグメントとつなぎ、これをまたクレノー-DNA-ポリメラーゼで処理した。
2シストロン性基本ベクターpBIDG(図1)中、IRES-GFP遺伝子領域はベクターpIRES2-EGFP(クロンテク、パロ・アルト、CA、USA)から単離され、ベクターpBID中でCMVエンハンサー/プロモーターの制御下におかれ、その結果、プロモーター領域とIRES-要素の間の多重クローニング部位が保持されている。下記の手順を使用した。プラスミドpIRES2-EGFPが鋳型として作用するPCR変異導入では、一方で、IRES配列内のHindIII切断部位AAGCTTを変異導入プライマーの使用により配列ATGCTTに変換し、除去した。他方で、IRES配列の5'末端に相補性を有するプライマーによりXbaI切断部位を挿入し、又はGFP配列の3'末端に相補性を有するプライマーによりSpeI切断部位を導入した。得られたPCRフラグメント(これは完全IRES及びGFP配列を含む)を、XbaI及びSpeIで消化し、ベクターpBIDのマルチクローニング部位の3'末端の単一XbaI切断部位にクローン化した。同じ方法で、ベクターpIRES2-EGFPからのIRES-GFP遺伝子領域をベクターpBIN中のCMVエンハンサー/ハムスターユビキチン/S27aプロモーターの制御下においた。これにより2シストロン性基本ベクターpBINGが生された(図1)。
ヒトMCP-1 cDNA(Yoshimuraら, 1989)を0.3kb HindIII/EcoRIフラグメントとしてベクターpBINの相当する切断部位にクローン化して、ベクターpBIN-MCP1を生じた(図2A)。
モノクローナルヒト化IgG2抗体を発現するために、重鎖をベクターpBID又はBamHI及びHindIIIで消化されたpBIDGに1.5kb BamHI/HindIIIフラグメントとしてクローン化して、ベクターpBID-HC又はpBIDG-HCを得た(図2B)。一方で、軽鎖をベクターpBIN又はpBINGの相当する切断部位に0.7kb BamHI/HindIIIフラグメントとしてクローン化して、ベクターpBIN-LC又はpBING-LCを生じた(図2B)。
【0058】
3. FACS
フローサイトメトリー分析及びソーティングをクールター・エピックス・アルタ装置で行なった。このFACSは488nmの励起波長を有するヘリウム-アルゴンレーザーに適合している。蛍光タンパク質に適した波長で蛍光強度を吸収し、取り付けられたソフトウェア、クールター・エクスポ32によって処理される。ソーティングは8000-10000イベント/秒の速度で通常行なう。懸濁された細胞を遠心分離し(180xgで5分)、HBSS中1-1.5x107/mlの細胞濃度に調節することができる。次いで細胞をそれらの蛍光タンパク質シグナルに従ってソーティングすることができる。細胞を培地を予め含む試験管に取り、次いで遠心分離し、ソーティングされた細胞の数に応じて、好適な培養容器に接種し、又はマイクロタイタープレートに直接入れる。
4. ELISA
安定にトランスフェクションされたCHO-DG44細胞の上清中のMCP-1力価を製造業者の指示(BDバイオサイエンシズ・ファーミンゲン、ハイデルベルグ、DE)に従ってOptEIAヒトMCP-1セットキットを使用してELISAにより定量した。
安定にトランスフェクションされたCHO-DG44細胞の上清中のIgG2 mAbを標準的な操作(Current Protocols in Molecular Biology, Ausubelら, 1994, 最新)に従って、一方で、ヤギ抗ヒトIgG Fcフラグメント(ダイアノバ、ハンブルグ、DE)また他方でAP-結合ヤギ抗ヒトカッパ軽鎖抗体(シグマ)を使用してELISAにより定量した。精製IgG2抗体を標準として使用した。生産性(pg/細胞/日)を式pg/((Ct-Co)t/ln(Ct-Co))(式中、Co及びCtは夫々播種時及び回収時の細胞カウントであり、かつtは培養時間である)により計算した。
【0059】
5. NPT酵素活性を測定するためのドットアッセイ
細胞抽出物をDuchら, 1990の方法に従って調製するために、6x106の細胞をPBSで2回洗浄し、次いで抽出緩衝液(0.135M Tris-HCl pH 6.8、20%のグリセロール、4mMジチオスレイトール)600μL中で再懸濁させた。ドライアイス又は水の浴中の凍結及び解凍の4サイクル後に、細胞破砕物を遠心分離により除去し、上清をその後の酵素アッセイに使用した。細胞抽出物中のタンパク質濃度をBIO-RADタンパク質アッセイ(Bio-RadラボラトリイズGmbH、ミュンヘン、DE)を使用して、標準タンパク質としてBSAを用いてブラッドフォードアッセイにより測定した(Current Protocols in Molecular Biology, Ausubelら, 1994, 最新)。NPT酵素活性を測定するために、ドットアッセイをPlattら, 1987のプロトコルに基づいて行なった。これのために、タンパク質5μg、2.5μg及び1.25μgを抽出緩衝液で20μLの最終体積に調節し、トランスフェクションしなかったCHO-DG44細胞からの細胞抽出物で5μgの合計タンパク質含量まで足した。アッセイ緩衝液(67mM Tris-HCl pH 7.1、42mM MgCl2、400mM NH4Cl)200μL±40μg/mlのG418±5μCiの〔γ-33P〕-ATP/ml(NEN)の添加後に、抽出物を27℃で135分間インキュベートした。次いで抽出物を96ウェル真空マニホルド(シュライヘル&シュル、ダッセル、DE)中でワットマン3MM紙の一層、P81ホスホセルロース膜(ワットマン・ラボラトリイ部門、マイドストーン、英国)及びニトロセルロース膜(シュライヘル&シュル)のサンドイッチで濾過した。タンパク質キナーゼによりリン酸化されたタンパク質及び非リン酸化タンパク質がニトロセルロースに結合し、一方、リン酸化G418がニトロセルロースを通過し、ホスホセルロースに結合する。脱イオン水で3回洗浄した後、膜を装置から除去し、再度水で洗浄し、次いで空気乾燥させた。ホスホイメージャー(モレキュラー・ダイナミクス、クレフェルド、DE)を使用して、放射能シグナルを定量した。
【0060】
6. ノーザンブロット分析
全RNAを製造業者の指示(インビトロゲンGmbH、カルスルーヘ、DE)に従ってTRIZOL試薬を用いて細胞から単離し、ゲル電気泳動によるRNA30μgの分離及びハイボンドN+ナイロン膜(アマシャム・バイオサイエンシズ、フライブルグ、DE)への転写をグリオキサール/DMSO変性RNAに関する標準操作(Current Protocols in Molecular Biology, Ausubelら, 1994, 最新)に従って行なった。ジーンイメージズCDP-スター検出キット(アマシャム・バイオサイエンシズ)によるその後の非放射性ハイブリダイゼーションに使用したプローブはジーンイメージズ・ランダム・プライム標識キット(アマシャム・バイオサイエンシズ、フライブルグ、DE)で製造業者の指示に従ってFITC-dUTP標識された、PCR産物(これはNPT遺伝子のコーディング領域を含む)であった。
【0061】
7. ドットブロット分析
DNA単離キットを製造業者の指示(細胞及び組織用のDNA単離キット;ロシェ・ダイアグノスチクスGmbH、マンハイム、DE)に従って使用して、ゲノムDNAを細胞から単離した。種々の量のDNA(10μg、5μg、2.5μg、1.25μg、0.63μg及び0.32μg)をアルカリ性緩衝液中で96ウェル真空マニホルド(シュライヘル&シュル、ダッセル、DE)を使用して通常の方法(Ausubelら, 1994)によりハイボンドN+ナイロン膜(アマシャム・バイオサイエンシズ、フライブルグ、DE)で濾過した。トランスフェクションしなかったCHO-DG44細胞を陰性対照として使用した。プラスミドpBIN-LCを標準として使用した(320pg、160pg、80pg、40pg、20pg、10pg、5pg、2.5pg)。ジーンイメージズCDP-スター検出キット(アマシャム・バイオサイエンシズ)によるその後の非放射性ハイブリダイゼーションに使用したプローブは、製造業者の指示に従ってジーンイメージズランダムプライム標識キット(アマシャム・バイオサイエンシズ、フライブルグ、DE)によりFITC-dUTP標識した、NPT遺伝子のコーディング領域を含むPCR産物とした。イメージマスターVDS-CL(アマシャム・バイオサイエンシズ)を使用して、ケミルミネセンスシグナルを定量した。次いでタイトレーションしたプラスミドDNAのシグナル強さから得られた標準シリーズを使用して、細胞中のnpt遺伝子のコピー数を測定した。アボガドロ定数を使用してプラスミド分子の数を計算し、CHO細胞のDNA含量は約5pgであるとした。
【実施例1】
【0062】
実施例1.ネオマイシンホスホトランスフェラーゼの変異導入
NPT変異体Glu182Gly(配列番号3)、Trp91Ala(配列番号5)、Val198Gly(配列番号7)、Asp227Ala(配列番号9)、Asp227Val(配列番号11)、Asp261Gly(配列番号13)、Asp261Asn(配列番号15)、Phe240Ile(配列番号17)、Glu182Asp(配列番号19)、Asp227Gly(配列番号21)、Asp190Gly(配列番号23)及びAsp208Gly(配列番号25)を調製するのに必要とされる野生型NPT-遺伝子中の塩基置換を、PCRにより変異導入プライマーを使用して行なった(図3)。ベクターpBIN(図1)又はpBK-CMV(ストラタゲン、ラ・ジョラ、USA)をPCR変異導入のための鋳型として使用した。最初に変異体の5'又は3'部分を別々のPCR操作で調製した。変異体Glu182Gly、Glu182Asp、Trp91Ala、Asp190Gly、Val198Gly、Asp208Gly及びAsp227Glyを調製するために、Neofor5(配列番号27)と関連する変異導入リバース(rev)プライマー又はNeorev5(配列番号28)と関連する変異導入フォワード(for)プライマーからなるプライマーの組み合わせを増幅に使用した:プライマーの組合せは、
−NPT変異体Glu182Gly(配列番号3)の場合には、Neofor5(配列番号27)とE182Grev(配列番号32)又はNeorev5(配列番号28)とE182Gfor(配列番号31)から;
−NPT変異体Glu182Asp(配列番号19)の場合には、Neofor5(配列番号27)とE182Drev(配列番号48)又はNeorev5(配列番号28)とE182Dfor(配列番号47)から;
−NPT変異体Trp91Ala(配列番号5)の場合には、Neofor5(配列番号27)とW91Arev(配列番号34)又はNeorev5(配列番号28)とW91Afor(配列番号33)から;
−NPT変異体Val198Gly(配列番号7)の場合には、Neofor5(配列番号27)とV198Grev(配列番号36)又はNeorev5(配列番号28)とV198Gfor(配列番号35)から;
−NPT変異体Asp190Gly(配列番号23)の場合には、Neofor5(配列番号27)とD190Grev(配列番号50)又はNeorev5(配列番号28)とD190Gfor(配列番号49)から;
−NPT変異体Asp208Gly(配列番号25)の場合には、Neofor5(配列番号27)とD208Grev(配列番号52)又はNeorev5(配列番号28)とD208Gfor(配列番号51)から;
−NPT変異体Asp227Gly(配列番号21)の場合には、Neofor5(配列番号27)とD227Grev(配列番号54)又はNeorev5(配列番号28)とD227Gfor(配列番号52)からなる。
変異体Asp227Ala、Asp227Val、Asp261Gly、Asp261Asn及びPhe240Ileを調製するために、Neofor2(配列番号29)と関連する変異導入リバース(rev)プライマー又はIC49(配列番号30)と関連する変異導入フォワード(for)プライマーからなるプライマーの組み合わせを増幅に使用した:プライマーの組合せは、
−NPT変異体Asp227Ala(配列番号9)の場合には、Neofor2(配列番号29)とD227Arev(配列番号38)又はIC49(配列番号30)とD227Afor(配列番号37)から;
−NPT変異体Asp227Val(配列番号11)の場合には、Neofor2(配列番号29)とD227Vrev(配列番号40)又はIC49(配列番号30)とD227Vfor(配列番号39)から;
−NPT変異体Asp261Gly(配列番号13)の場合には、Neofor2(配列番号29)とD261Grev(配列番号42)又はIC49(配列番号30)とD261Gfor(配列番号41)から;
−NPT変異体Asp261Asn(配列番号15)の場合には、Neofor2(配列番号29)とD261Nrev(配列番号44)又はIC49(配列番号30)とD261Nfor(配列番号43)から;
−NPT変異体Phe240Ile(配列番号17)の場合には、Neofor2(配列番号29)とF240Irev(配列番号46)又はIC49(配列番号30)とF240Ifor(配列番号45)からなる。
【0063】
次いで当該変異体の5'部分のコーディング鎖及び3'部分の相補鎖を変異導入プライマー配列により形成された重なり領域中のハイブリダイゼーションにより組み合わせ、一本鎖領域をフィルインし、全生成物をプライマーNeofor5(配列番号27)とNeorev5(配列番号28)又はNeofor2(配列番号29)とIC49(配列番号30)によるPCRで再度増幅した。これらのPCR産物をStuI/RsrII(変異体Glu182Gly、Trp91Ala及びVal198GlyのNeofor5/Neorev5 PCR産物)、StuI/BstBI(変異体Glu182Asp、Asp190Gly、Asp208Gly及びAsp227GlyのNeofor5/Neorev5 PCR産物)又はDraIII/RsrII(変異体Asp227Ala、Asp227Val、Asp261Gly、Asp261Asn及びPhe240IleのNeofor2/IC49 PCR産物)で消化した。次いでベクターpBIN-LC(図2B)又はpBK-CMV(ストラタジーン、ラ・ジョラ、USA)中で、野生型NPT配列の部分をStuI/RsrII消化、DraIII/RsrII消化又はStuI/BstBI消化により除去し、PCR産物の相当するフラグメントにより置換した。相補鎖及びコーディング鎖の両方のシーケンシングにより、種々の変異体中の所望の塩基置換を確認し、残りのDNA配列が野生型NPT配列に一致することを確かめた。このようにして、発現ベクターpBIN1-LC、pBIN2-LC、pBIN3-LC、pBIN4-LC、pBIN5-LC、pBIN6-LC、pBIN7-LC及びpBIN8-LCを生成した。これらはNPT変異体Glu182Gly、Trp91Ala、Val198Gly、Asp227Ala、Asp227Val、Asp261Gly、Asp261Asn又はPhe240Ileを含む(図2B)。
【0064】
残りのNPT変異体を改変pBK-CMVからの1640 bp Bsu36Iフラグメントとして単離し、フラグメント末端をクレノーDNAポリメラーゼでフィルインし、ベクターpBIDの3750 bp Bsu36I/StuIフラグメントとつなぎ、これをまたクレノーDNAポリメラーゼで処理した。このようにして、発現ベクターpKS-N5、pKS-N6、pKS-N7及びpKS-N8を生成し、これらは夫々NPT変異体Glu182Asp、Asp190Gly、Asp208Gly及びAsp227Glyを含んでいた。次いでヒトMCP-1 cDNAを0.3 kb HindIII/EcoRIフラグメント(図2A)としてこれらの発現ベクターにクローン化し、又はヒト化IgG2抗体のH鎖を0.7 kb HindIII/BamHIフラグメント(図2B)としてこれらの発現ベクターにクローン化した。
ネオマイシンホスホトランスフェラーゼに挿入された変異は一方で保存ドメイン、例えば、モチーフ1、2及び3(Shawら, 1993)に隣接するより保存的な(Val198Gly、Phe240Ile)又はあまり保存的でない(Trp91Ala、Glu182Gly、Glu182Asp、Asp227Ala、Asp227Val、Asp227Gly)アミノ酸の置換である。他方で、変異は保存モチーフ1(Asp190Gly)、2(Asp208Gly)又は3(Asp261Gly、Asp261Asn)内に位置され、保存アミノ酸に関する。
【実施例2】
【0065】
実施例2.安定的にトランスフェクションされたMCP-1発現細胞の選抜に対するNPT変異の影響
MCPをCHO細胞中の一本鎖タンパク質の発現の例として使用した。これのために、CHO-DG44をpKS-N5-MCP1、pKS-N6-MCP1、pKS-N7-MCP1、pKS-N8-MCP1又はpBIN-MCP1でトランスフェクションした(図2A)。二つの二重調製を行なった。トランスフェクションの2日後に、細胞を96ウェル-プレートに接種し(2000細胞/ウェル)、HT添加CHO-S-SFMII培地中で400μg/mlのG418で選抜した。pBIN-MCP1でトランスフェクションされた細胞の場合、選抜をまた800μg/mlのG418で並行して行なった。得られた細胞集団を24ウェルプレートを経て6ウェルプレートに継代した。選抜期中でさえも、種々のトランスフェクション混合物間で相違を検出することができた。選抜がNPT野生型遺伝子(配列番号1)で行なわれた細胞集団とは対照的に、変異されたNPTでトランスフェクションされたこれらの細胞集団では、より少ない細胞しかG418による初期の選抜に耐えなかった。それ故、これらの細胞集団を約4日後まで24ウェルプレートに移すことができなかった。また、pKS-N6-MCP1及びpKS-N7-MCP1でトランスフェクションされた混合物では、安定にトランスフェクションされた細胞はG418の400μg/mlの濃度で全く選抜することができなかった。おそらく、変異Asp190Gly及びAsp208Glyを有するNPT変異体では酵素機能がひどく損なわれ、充分なG418分子が不活化されることができず安定にトランスフェクションされた細胞の増殖ができないのであろう。実際、G418濃度を200μg/mlに低下させた場合、いくらかの細胞が最初の選抜期に耐えたが、それらは全て増殖及び生命力がひどく損なわれており、変異体Asp208Glyの場合のいくつかの例外は別として、エクスパンジョンが可能ではなかった。
変異体Glu182Asp及びAsp227Gly又はNPT野生型でトランスフェクションされた細胞から、18プールを6ウェル中で4回の継代にわたって培養し(混合物1及び2の夫々9プール)、生成されたMCP-1の濃度を細胞培養上清中でELISAにより測定した。NPT変異体が選抜可能なマーカーとして使用された細胞プールは、400又は更には800μg/mlのG418によりNPT野生型を用いて選抜が行なわれた細胞プールよりも平均で50%−57%(Glu182Asp変異体)又は57%−65%(Asp227Gly変異体)高い生産性を示した(図5)。このように、NPT変異体を選抜可能マーカーとして使用することにより、トランスフェクションされた細胞集団中の高生産性細胞の比率が実際に増大し得た。
【実施例3】
【0066】
実施例3.安定にトランスフェクションされたmAb発現細胞の選抜に関するNPT変異の影響 同時トランスフェクションでは、CHO-DG44細胞を最初にプラスミド組み合わせpBIDG-HC/pBIN-LC(NPT野生型)、pBIDG-HC/pKS-N5-LC(Glu182Asp NPT変異体)又はpBIDG-HC/pKS-N8-LC(Asp227Gly NPT変異体)でトランスフェクションした(図2B)。使用したベクター配置では、ヒト化IgG抗体の二つのタンパク質鎖はそれら自体のベクター(これは別々の転写単位中のDHFR又はネオマイシン選抜可能なマーカーを更にコードする)により夫々発現される。産物遺伝子の発現はCMV-エンハンサー/ハムスターユビキチン/S27aプロモーターにより媒介される。しかしながら、例えば、CMVエンハンサー/プロモーター、SV40エンハンサー/ハムスターユビキチン/S27aプロモーター又はその他のプロモーター組み合わせを使用して、同等なデータを得ることも出来る。
全てにおいて、プラスミド組み合わせ当り6プールを使用して、トランスフェクションの4シリーズを夫々の場合に行なった。選抜がNPT野生型遺伝子を用いて行なわれた細胞集団とは対照的に、変異されたNPTでトランスフェクションされたこれらの細胞集団では、より少ない細胞がG418による初期の選抜に耐えた。付加的な400μg/mlのG418によるHTを含まないCHO-S-SFMII培地中のトランスフェクションされた細胞集団の2〜3週の選抜後に、細胞培養上清中の抗体力価を幾つかの実験(6−8)にわたってELISAにより測定した。選抜可能なマーカーとしてのNPT野生型遺伝子の使用との比較により、Glu182Asp変異体で選抜された細胞は夫々平均で86%及び77%の生産性及び力価の増大を示し、Asp227Gly変異体で選抜された細胞は更に夫々126%及び107%の生産性及び力価の増大を示した。このように、低下された酵素活性を有するNPT変異体を使用することにより、2倍程度まで高い基本的生産性を有する細胞を選択的に濃縮することが可能であった。
【0067】
別のトランスフェクションシリーズでは、選抜に関するG418の異なる濃度の影響を試験した。400、500及び600μg/mlのG418をトランスフェクションされた細胞プール、夫々の場合に3プールの選抜に使用した。高濃度では、有意に少ない細胞が細胞集団(選抜がNPT野生型遺伝子を用いて行なわれた)中で選抜に耐え、その効果はAsp227Gly変異体で最大であった。しかしながら、得られたトランスフェクションされた細胞集団は増殖及び生命力の低下を示さなかった。しかしながら、トランスフェクションに使用したプラスミド組み合わせ内で得られた生産性と力価の間で有意差は検出できなかった。しかし、ここでさえも、NPT変異体により選抜された細胞は、平均して最高の生産性を再度有しており、Asp227Gly変異体によりもたらされる生産性はNPT野生型のそれよりも4倍高く、続いてGlu182Asp変異体が2.4倍高い生産性を有していた(図6A)。
次いで同時トランスフェクションでは、CHO-DG44細胞をプラスミドの組み合わせpBIDG-HC/pBIN-LC(NPT野生型)、pBIDG-HC/pBIN1-LC(Glu182Gly NPT変異体)、pBIDG-HC/pBIN2-LC(Trp91Ala NPT変異体)、pBIDG-HC/pBIN3-LC(Val198Gly NPT変異体)、pBIDG-HC/pBIN4-LC(Asp227Ala)、pBIDG-HC/pBIN5-LC (Asp227Val NPT変異体)、pBIDG-HC/pBIN6-LC (Asp261Gly NPT変異体)、pBIDG-HC/pBIN7-LC (Asp261Asn NPT変異体)又はpBIDG-HC/pBIN8-LC (Phe240Ile NPT変異体)でトランスフェクションした(図2B)。使用したベクター配置では、再度モノクローナルヒト化IgG抗体の二つのタンパク質鎖はそれ自体のベクター(これは更にまた別々の転写単位中のDHFR又はネオマイシン選抜可能なマーカーをコードする)により夫々発現される。
【0068】
夫々のプラスミド組み合わせについて、5プールをトランスフェクションした。選抜がNPT野生型遺伝子を用いて行なわれた細胞集団とは対照的に、変異されたNPTでトランスフェクションされた少ない細胞が細胞集団中でG418による初期の選抜に耐えた。400μg/mlのG418を添加したHTを含まないCHO-S-SFMII培地中のトランスフェクションされた細胞プールの2〜3週の選抜後に、細胞培養上清中の抗体力価を6回の実験にわたってELISAにより測定した。図6Bは試験中にプールから測定された力価及び生産性の平均を示す。選抜可能なマーカーとしてのNPT野生型遺伝子の使用と較べて、NPT変異体で選抜された全ての細胞プールが夫々平均で1.4−14.6倍及び1.4−10.8倍の生産性及び力価の増大を示した(図6B)。高い基本的生産性を有する細胞の最良の選択的濃縮がこうしてNPT変異体Asp227Val及びAsp261Glyで得られ、夫々14.6倍及び9.3倍の平均生産性の増大を有していた。
ベクターpBIDG-HCは別の選抜可能なマーカー、GFPを含む。GFPはIRES要素を介して重鎖に転写的に連結される。それ故、標的タンパク質の発現と選抜可能なマーカーGFPとの間に得られる相関関係はFACS分析で測定されたGFP蛍光に基づいてトランスフェクションされた細胞集団における発現のレベル及び発現レベルの分布を迅速に評価することを可能にする。G418を添加したHTを含まないCHO-S-SFMII培地中のトランスフェクションされた細胞集団の2〜3週の選抜後に、GFP蛍光をFACS分析で測定した(図7)。GFP蛍光シグナルは実際にモノクローナルIgG2抗体について得られた力価データと相関関係があった。また、NPT変異体Asp227Val、Asp261Gly、Asp161Asn及びPhe240Ileで選抜されたプールは高いGFP蛍光を有する細胞の高比率を有し、NPT変異体Trp91Ala、Asp227Ala、Asp227Gly、Gly182Asp、Glu182Gly及びVal198Glyで選抜された細胞がそれに続いた。
選択薬剤メトトレキセート(MTX)を培地に添加することにより、dhfr媒介遺伝子増幅を誘導することにより細胞の生産性を更に増大することが可能であった。こうして、例えば、100nMのMTXによる簡単な増幅工程後に、プラスミドの組み合わせpBIDG-HC/pBIN5-LC (NPT変異体Asp227Val)、pBIDG-HC/pBIN6-LC (NPT変異体Asp261Gly)、pBIDG-HC/pBIN7-LC (NPT変異体Asp261Asn)及びpBIDG-HC/pBIN8-LC (NPT変異体Phe240Ile)による同時トランスフェクションにより得られた細胞プール中の比生産能は2〜4倍に増大でき、プールに依存して、4〜14pg/細胞/日の生産性を得ることができた。図8は、例として、pBIDG-HC/pBIN5-LC (NPT変異体Asp227Val)の同時トランスフェクションにより得られた細胞プールについて、MTXの添加(100nMのMTX、続いて500nMのMTX)により得られた27pg/細胞/日への生産性の増大を示す。
【実施例4】
【0069】
実施例4.NPT酵素活性の測定及び比較
NPT変異体の酵素活性をNPT野生型のそれと比較するために、ドットアッセイをPlattら, 1987の操作に基づいて行なって細胞抽出物中のNPT活性を測定し、例としてNPT変異体Glu182Asp及びAsp227Glyについて図9Aに示した。細胞抽出物をNPT野生型遺伝子(配列番号1)又はNPT変異体Glu182Gly(配列番号3)、Glu182Asp(配列番号19)、Trp91Ala(配列番号5)、Asp190Gly(配列番号23)、Val198Gly(配列番号7)、Asp208Gly(配列番号25)、Asp227Ala(配列番号9)、Asp227Val(配列番号11)、Asp227Gly(配列番号21)、Asp261Gly(配列番号13)、Asp261Asn(配列番号15)又はPhe240Ile(配列番号17)でトランスフェクションされ選抜された二つの異なるmAb発現細胞プールから調製した。トランスフェクションしなかったCHO-DG44細胞からの細胞抽出物を陰性対照として使用した。
NPT変異体の酵素活性はNPT野生型と較べて有意に低下した。平均で、NPT変異体は野生型酵素活性のわずかに1.5%〜62%を有し、3.1%及び1.5%を有するNPT変異体Asp261Gly及びAsp261Asnは最低の残留活性を有し、また61.9%及び53.2%を有するNPT変異体Val198Gly及びTrp91Alaは最高の残留活性を有する(図9B)。ホスホセルロース上で得られたシグナルはNPT酵素活性により生じたG418のリン酸化に特異的であった。NPT基質G418をアッセイ緩衝液に添加しない場合、ホスホセルロース上で活性は検出できなかった。ニトロセルロース膜上で得られたシグナル(これらは細胞抽出物中のタンパク質キナーゼによりリン酸化されたタンパク質から生じる)を、適用した同じ量のサンプルについての内部対照として使用した。
NPT野生型と較べてNPT変異体の低下した酵素活性は低下した遺伝子発現のためではあり得なかった。逆に、全RNAに関するノーザンブロット分析により、NPT野生型でトランスフェクションされ、高いNPT酵素活性を示す細胞プールが、NPT変異体でトランスフェクションされた細胞プールよりも少ないRNAを発現することが示された(図10)。唯一の例外はNPT変異体Glu182Glyでトランスフェクションされ、匹敵する量のNPT-mRNAを発現する細胞であった。これらのトランスフェクションされた細胞集団からのゲノムDNAについて行なわれたドットブロット分析では、NPT変異体の高発現が遺伝子量効果及び/又はより転写的に活性なゲノム領域への外来性DNAの組込みにより得られたことが示された(図10)。例えば、NPT変異体Trp91Alaで選抜された細胞では、遺伝子量効果がプール1で優性であり、一方、プール2では、組込み効果が支配的である。このようにして、低下した酵素活性を有するマーカーが選抜に使用されたトランスフェクション細胞は、同じ選択圧のもとで、選択薬剤に対する低下した耐性を相殺するのに充分なマーカータンパク質を合成することができる。
【実施例5】
【0070】
実施例5.GFPをベースとするFACSソーティングによるmAbの高発現を有する細胞の単離
同時トランスフェクションでは、CHO-DG44細胞をモノクローナルヒト化IgG2抗体をコードする、プラスミドの組み合わせpBID-HC及びpBING-LCでトランスフェクションした(図2B)。使用されたベクター配置において、抗体の二つのタンパク質鎖はそれら自体のベクター(これはまた別々の転写単位中でDHFR又は改変ネオマイシンホスホトランスフェラーゼ選抜可能なマーカー(Asp227Gly変異体;配列番号21)を更にコードする)により夫々発現される。加えて、ベクターpBING-LCは別の選抜可能なマーカー、GFPを含む。ベクターpBID-HC/pBING-LCによるCHO-DG44の同時トランスフェクションにおける、IRES要素によるGFP及び軽鎖の発現の転写連鎖の結果として、高いGFP含量を有する細胞を連続FACSソーティングにより純粋に選択することにより、モノクローナル抗体の高発現を有する細胞を単離することが短時間内で可能であった。全てにおいて、8の別々の細胞プールをトランスフェクションし、これらから400μg/mlのG418を添加したHTを含まないCHO-S-SFMII培地中の最初の2〜3週の選抜後に、安定にトランスフェクションされた細胞集団を得た。全ての8プールの力価及び生産性を幾つかの実験(7−8)にわたってELISAにより測定した。平均で、力価は約1.4mg/Lであり、生産性は1日当り約1.3pg/細胞であった。その後の連続のFACSをベースとするソーティングのために、プール5及び8を選抜し、プール5は最高の生産性を有し、またプール8は全てのプールの平均に相当する生産性を有していた。夫々の工程において、最高のGFP蛍光を有する細胞の5%をFACSによりソーティングし、更にプール中で培養した。このソーティングを夫々のソーティング間で約2週の培養期間で全部で6回まで行なった。
【0071】
二つのタンパク質鎖はそれら自体のベクターによりそれぞれ発現され、GFPとの転写カップリングの結果として、GFPをベースとするFACSソーティングの際に軽鎖の発現について選抜することしかできなかったが、驚くことに、mAb生産性とGFP蛍光の間に良い相関関係があることがわかった(図13)。生産性はFACSをベースとするソーティング単独により9.5pg/細胞/日に増大させることができた(図11)。また、ハムスタープロモーターをCMV-エンハンサーに代えてSV40-エンハンサーに機能可能に連結した場合に、匹敵するデータを得た。その後の単一のMTX増幅工程により、最初のソーティング工程のプール5及び8から出発して、100nMのMTXを選択培地に添加することにより、プールの生産性を平均20pg/細胞/日以上に増大させることが可能であった(図12)。蛍光タンパク質の高発現レベルは細胞増殖及び生命力にどんな負の効果をも有していなかった。遺伝子増幅中に、細胞の増殖性はまた、特にMTXが高濃度で添加される場合に、MTXの添加によりひどく負に影響される。しかしながら、予備ソーティングされた細胞プールは100nMのMTXの実に高い初期用量に応答してかなり強い特性を示した。それらは極めて良好に選抜期に耐え、即ち、わずかに2週後に、高生命力及び良好な増殖速度を有する細胞集団を得た。
加えて、高生産性細胞を選抜するための開発時間が通常の段階的遺伝子増幅戦略(これは一般にMTXの増大する添加とともに4増幅段階を含む)と較べて、約6週に短縮された。これは低下された酵素活性を有する改変NPT選抜可能なマーカーを使用することにより得られた、対象遺伝子の増大された発現を有するトランスフェクションされた細胞の濃縮、続いてその後の遺伝子増幅工程とともにGFPをベースとするFACSソーティングの組み合わされた使用により得られた。
【0072】
参考文献
Adam, M.A. et al., J Virol 1991, 65, 4985 - 4990
Altschul, S.F. et al., Nucleic Acids Res. 1997, 25, 3389 - 3402
Altschul, S.F. et al., J Mol Biol 1990, 215, 403 - 410
Ausubel, F.M. et al., Current Protocols in molecular biology. New York: Greene Publishing Associates and Wiley-Interscience. 1994 (updated)
Blazques, J. et al., Molecular Microbiology 1991, 5(6), 1511 1518
Bennett, R.P. et al., BioTechniques 1998, 24, 478 - 482
Burk, D.L. et al., Biochemistry 2001, 40 (30), 8756 - 8764
Chalfie, M. et al., Science 1994, 263, 802 - 805
Chamov, S.M. et al., Antibody Fusion Proteins, Wiley-Liss Inc., 1999
Davies, M.V. et al., J Virol 1992, 66, 1924 - 1932
Faisst, S. et al., Nucleic Acids Research 1992, 20, 3 26
Gish, W. & States, D.J., Nature Genet. 1993, 3, 266 - 272
Gossen, M. et al., Curr Opi Biotech 1994, 5, 516 - 520
Duch, M. et al., Gene 1990, 95, 285 - 288
Hanson, K.D. et al., Mol Cell Biol 1995, 15(1), 45 51
Haber, D.A. et al., Somatic Cell Genetics 1982, 8, 499 - 508
Harris et al., Protein Purification: A Practical Approach, Pickwood and Hames, eds., IRL Press, 1995
Hemann, C. et al., DNA Cell Biol 1994, 13 (4), 437 - 445
Hon, W. et al., Cell 1997, 89, 887 - 895
Hu, S. et al., Cancer Res. 1996, 56 (13), 3055 - 3061
Huston, C. et al., Proc Natl Acad Sci USA 1988, 85 (16), 5879 - 5883
Jang, S.K. et al., J Virol 1989, 63, 1651 1660
Kaufman, R.J., Methods in Enzymology 1990, 185, 537 - 566
Kocabiyik, S. et al., Biochem Biophys Res Commun 1992, 185(3), 925 931
Kortt, A.A. et al., Protein Engineering 1997, 10 (4), 423 - 433
Lottspeich F. and Zorbas H. eds., Bioanalytic, Spektrum Akad. Verl., 1998
Lovejoy, B. et al., Science 1993, 259, 1288 - 1293
Madden, T.L. et al., Meth. Enzymol. 1996, 266, 131 - 141;
Monaco, L. et al., Gene 1996, 180, 145 15
Morgan, R.A. et al., Nucleic Acids Research 1992, 20, 1293 - 1299
Mosser, D.D. et al., BioTechniques 1997, 22, 150 - 161
Ohshima, Y. et al., J Mol Biol 1987, 195, 247 - 259
Pack, P. et al., Biotechnology 1993, 11, 1271 1277
Pack, P. et al., J Mol Biol 1995, 246 (11), 28 - 34
Pelletier, J. et al., Nature 1988, 334, 320 325
Perisic, O. et al., Structure 1994, 2, 1217 - 1226
Platt, S.G. et al., Analyt Biochem 1987, 162, 529 - 535
Ramesh, N. et al., Nucleic Acids Research 1996, 24, 2697 2700
Sambrook, J., Fritsch, E.F. & Maniatis, T.,Molecular Cloning: A Laboratory Manual Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, 1989
Scopes, R., Protein Purification, Springer Verlag, 1988
Shaw, K.J. et al., Microbiological Reviews 1993, 57(1), 138 - 163
Simonson, C.C. et al., Proc Natl Acad Sci USA 1983, 80, 2495 - 2499
Sugimoto et al., Biotechnology 1994, 12, 694 - 698
Yenofsky, R.L. et al., Proc Natl Acad Sci USA 1990, 87, 3435 3439
Yoshimura, T. et al., FEBS LETTERS 1989, 244(2), 487 - 493
Urlaub, G. et al., Cell 1983, 33, 405 412
Werner, R.G. et al., Arzneim.-Forsch./Drug.Res. 1998, 48, 870 - 880
Wigler, M. et al., Proc Natl Acad Sci USA 1980, 77, 3567 - 3570
Zhang, J. & Madden, T.L. Genome Res. 1997, 7, 649 - 656;
【図面の簡単な説明】
【0073】
【図1】CHO-DG44細胞中で組換えタンパク質を発現するのに使用した基本ベクターの模式図を示す。“P/E”はCMVエンハンサーとハムスター-ユビキチン/S27aプロモーターの組み合わせであり、“P”自体はプロモーター要素を示し、かつ“T”は転写の終結シグナルであり、これは転写されたmRNAのポリアデニル化に必要とされる。夫々の転写単位内の転写開始の位置及び方向が矢印により示される。異種遺伝子のクローニングのために、制限エンドヌクレアーゼのための多重切断部位(マルチクローニング部位-MCS)を含む配列領域がプロモーター要素の後に挿入されている。増幅可能な選抜可能マーカージヒドロ葉酸還元酵素は“dhfr”と略記され、また選抜可能なマーカーネオマイシンホスホトランスフェラーゼは“npt”(npt野生型又はnpt変異体)と略記される。脳心筋炎ウイルスに由来する“IRES”要素は2シストロン性転写単位内で内部リボソームエントリー部位として作用し、続く緑色蛍光タンパク質“GFP”の翻訳を可能にする。
【図2A】一本鎖タンパク質(図2A)をコードし、CHO-DG44細胞をトランスフェクションするのに使用される真核細胞発現ベクターの図解を示す。“P/E”はCMVエンハンサーとハムスターユビキチン/S27aプロモーターの組み合わせであり、“P”それ自体はプロモーター要素であり、かつ“T”は転写されたmRNAのポリアデニル化に必要とされる転写の終結シグナルである。夫々の転写単位内の転写開始の位置及び方向が矢印により示される。増幅可能な選抜可能マーカージヒドロ葉酸還元酵素は“dhfr”と略記され、また選抜可能なマーカーネオマイシンホスホトランスフェラーゼは“npt”と略記される。NPT変異体E182G(配列番号3)、E182D(配列番号19)、W91A(配列番号5)、D190G(配列番号23)、V198G(配列番号7)、D208G(配列番号25)、D227A(配列番号9)、D227V(配列番号11)、D227G(配列番号21)、D261G(配列番号13)、D261N(配列番号15)及びF240I(配列番号17)は示された位置に改変アミノ酸(1文字コードで示される)をもたらす点変異を含む。脳心筋炎ウイルスに由来する“IRES”要素は2シストロン性転写単位内で内部リボソームエントリー部位として作用し、その後の緑色蛍光タンパク質“GFP”の翻訳を可能にする。“MCP-1”は単球走化性タンパク質1をコードし、一方、“HC”及び“LC”はヒト化モノクローナルIgG2抗体の重鎖及び軽鎖を夫々コードする。
【図2B】モノクローナル抗体のサブユニット(図2B)をコードし、CHO-DG44細胞をトランスフェクションするのに使用される真核細胞発現ベクターの図解を示す。“P/E”はCMVエンハンサーとハムスターユビキチン/S27aプロモーターの組み合わせであり、“P”それ自体はプロモーター要素であり、かつ“T”は転写されたmRNAのポリアデニル化に必要とされる転写の終結シグナルである。夫々の転写単位内の転写開始の位置及び方向が矢印により示される。増幅可能な選抜可能マーカージヒドロ葉酸還元酵素は“dhfr”と略記され、また選抜可能なマーカーネオマイシンホスホトランスフェラーゼは“npt”と略記される。NPT変異体E182G(配列番号3)、E182D(配列番号19)、W91A(配列番号5)、D190G(配列番号23)、V198G(配列番号7)、D208G(配列番号25)、D227A(配列番号9)、D227V(配列番号11)、D227G(配列番号21)、D261G(配列番号13)、D261N(配列番号15)及びF240I(配列番号17)は示された位置に改変アミノ酸(1文字コードで示される)をもたらす点変異を含む。脳心筋炎ウイルスに由来する“IRES”要素は2シストロン性転写単位内で内部リボソームエントリー部位として作用し、その後の緑色蛍光タンパク質“GFP”の翻訳を可能にする。“MCP-1”は単球走化性タンパク質1をコードし、一方、“HC”及び“LC”はヒト化モノクローナルIgG2抗体の重鎖及び軽鎖を夫々コードする。
【図3】変異導入プライマーによるPCRにより点変異が挿入されたネオマイシンホスホトランスフェラーゼ(npt)遺伝子の配列の部分を示す。大文字はnptコーディング領域のヌクレオチド配列を示し、一方、小文字は隣接非コーディングヌクレオチド配列を示す。ヌクレオチド配列(3文字コード)から予測されたアミノ酸配列をコーディングヌクレオチド配列の上に示してある。矢印は使用されたプライマーの方向、長さ及び位置を示し、実線の矢印は変異導入フォワードプライマーを示し、破線は変異導入リバースプライマーを示し、点線はnpt遺伝子又は変異部位の夫々の上流に位置されたプライマーNeofor5(配列番号27)又はNeofor2(配列番号29)を示し、また点-ダッシュ線はnpt遺伝子又は変異部位の夫々の下流に位置されたプライマーNeorev5(配列番号28)又はIC49(配列番号30)を示す。野生型配列に対して交換されたヌクレオチドが矢印の上下で強調されている。
【図4】保存ドメイン及びNPTアミノ酸配列内の挿入されたNPT変異の位置を示す。種々のアミノグリコシド改変酵素間の配列相同性に基づいて、種々の保存ドメインがNPTタンパク質配列内で同定された(灰色で示される)。酵素のC末端領域中の三つのモチーフは特別な機能を明らかに有する。モチーフ1及び2はおそらくATP触媒作用又はヌクレオチド結合における末端リン酸の触媒的転移に関係し、一方、モチーフ3はATP加水分解及び/又は酵素-アミノグリコシド複合体におけるコンホメーションの変化に機能を有すると考えられる。アミノグリコシド改変酵素の少なくとも70%中に表れるアミノ酸を太字で強調してある。下線を施したアミノ酸はそれらの類似性に基づいて同じグループに帰属され、アミノグリコシド改変酵素の少なくとも70%中に表れるものである。アステリスクでマークしたアミノ酸は変異部位の位置を示す。
【図5】安定にトランスフェクションされたMCP-1発現細胞の選抜に対するNPT変異の影響を示す。このために、CHO-DG44細胞をベクターpBIN-MCP1、pKS-N5-MCP1及びpKS-N8-MCP1(図2A)でトランスフェクションした。これらは選抜可能なマーカーとしてNPT野生型(WT)又はNPT変異体Glu182Asp及びAsp227Glyを含んでいた。安定にトランスフェクションされた細胞を選抜するために、400μg/ml又は800μg/mlのG418を選択薬剤として培地に添加した。生成された組換えタンパク質MCP-1の細胞培養上清中の濃度をELISAにより測定し、細胞当り1日当りの比生産能を計算した。バーは6ウェル皿中の6種の培養からの18プールの比生産能又は力価の平均を表す。
【図6A】安定にトランスフェクションされたmAb発現細胞の選抜に関するNPT変異の影響を調べた。このために、CHO-DG44細胞をプラスミド組み合わせpBIDG-HC/pBIN-LC(NPT野生型)、pBIDG-HC/pKS-N5-LC(NPT変異体Glu182Asp)もしくはpBIDG-HC/pKS-N8-LC(NPT変異体Asp227Gly)(図6A)(これらは選抜可能なマーカーとして使用されたNPT遺伝子(野生型又は変異体)のみを互いに異にする)でトランスフェクションした。産生された組換えモノクローナルIgG2抗体の細胞培養上清中の濃度をELISAにより測定し、細胞当り1日当りの比生産能を計算した。全てにおいて、5〜9プールを夫々のベクター組み合わせについてセットアップした。バーは75cm2のフラスコ中の6種の培養実験からの試験中の全てのプールの比生産能又は力価の平均を表す。相対力価又は相対比生産能を計算するために、NPT野生型遺伝子で選抜されたプールの平均を1とした。
【図6B】安定にトランスフェクションされたmAb発現細胞の選抜に関するNPT変異の影響を調べた。このために、CHO-DG44細胞をプラスミド組み合わせpBIDG-HC/pBIN-LC(NPT野生型)、pBIDG-HC/pBIN1-LC(NPT変異体Glu182Gly)、pBIDG-HC/pBIN2-LC(NPT変異体Trp91Ala)、pBIDG-HC/pBIN3-LC(NPT変異体Val198G)、pBIDG-HC/pBIN4-LC(NPT変異体Asp227Ala)、pBIDG-HC/pBIN5-LC(NPT変異体Asp227Val)、pBIDG-HC/pBIN6-LC(NPT変異体Asp261Gly)、pBIDG-HC/pBIN7-LC(NPT変異体Asp261Asn)もしくはpBIDG-HC/pBIN8-LC(NPT変異体Phe240Ile)(図6B)(これらは選抜可能なマーカーとして使用されたNPT遺伝子(野生型又は変異体)のみを互いに異にする)でトランスフェクションした。産生された組換えモノクローナルIgG2抗体の細胞培養上清中の濃度をELISAにより測定し、細胞当り1日当りの比生産能を計算した。全てにおいて、5〜9プールを夫々のベクター組み合わせについてセットアップした。バーは75cm2のフラスコ中の6種の培養実験からの試験中の全てのプールの比生産能又は力価の平均を表す。相対力価又は相対比生産能を計算するために、NPT野生型遺伝子で選抜されたプールの平均を1とした。
【図7】本発明のNPT変異体を選抜可能なマーカーとして使用することによる、トランスフェクションされた細胞プール中の高GFP発現を有する細胞の濃縮を示す。このために、CHO-DG44細胞をプラスミド組み合わせpBIDG-HC/pBIN-LC(NPT野生型)、pBIDG-HC/pKS-N5-LC(NPT変異体Glu182Asp)、pBIDG-HC/pKS-N8-LC(NPT変異体Asp227Gly)、pBIDG-HC/pBIN1-LC(NPT変異体Glu182Gly)、pBIDG-HC/pBIN2-LC(NPT変異体Trp91Ala)、pBIDG-HC/pBIN3-LC(NPT変異体Val198G)、pBIDG-HC/pBIN4-LC(NPT変異体Asp227Ala)、pBIDG-HC/pBIN5-LC(NPT変異体Asp227Val)、pBIDG-HC/pBIN6-LC(NPT変異体Asp261Gly)、pBIDG-HC/pBIN7-LC(NPT変異体Asp261Asn)又はpBIDG-HC/pBIN8-LC(NPT変異体Phe240Ile)(夫々の場合に5〜9プール)(これらは選抜可能なマーカーとして使用されたNPT遺伝子(野生型又は変異体)のみを互いに異にする)でトランスフェクションした。更に、pBIDGベクターはまたマーカー遺伝子としてGFPを含んでいた。G418を添加したHTを含まない培地中のトランスフェクションされた細胞プールの2〜3週の選抜後に、GFP蛍光をFACS分析により測定した。陰性対照として使用されたトランスフェクションされなかったCHO-DG44細胞を除く、夫々のグラフは同じプラスミド組み合わせでトランスフェクションされたプールからの平均GFP蛍光を表す。
【図8】ベクターの組み合わせpBIDG-HC/pBIN-LC(野生型)又はpBIDG-HC/pBIN5-LC(NPT変異体D227V)によるCHO-DG44のトランスフェクションから得られた細胞プールをその例とするdhfr媒介遺伝子増幅により得られたmAb生産性の増大を示す。G418の存在下のヒポキサンチン/チミジンを含まないCHO-S-SFMII培地中の最初の選抜後に、dhfr媒介遺伝子増幅を培地への100nM次いで500nMのMTXの添加により行なった。プールの細胞培養上清中のmAbの濃度をELISAにより測定し、細胞当り1日当りの比生産能(pg/細胞*日)を計算した。夫々のデータ点は75cm2のフラスコ中の6回の培養実験の平均を表す。
【図9A】ドットアッセイにおける、NPT野生型と較べた本発明のNPT変異体の酵素活性を示す。このために、mAbを発現する2種の異なる細胞プール(プール1及び2)(これらはNPT野生型遺伝子(配列番号1)又はNPT変異体E182G(配列番号3)、E182D(配列番号19)、W91A(配列番号5)、V198G(配列番号7)、D227A(配列番号9)、D227V(配列番号11)、D227G(配列番号21)、D261G(配列番号13)、D261N(配列番号15)及びF240I(配列番号17)Glu182Asp又はAsp227Glyでトランスフェクションし、選抜した)から細胞抽出物を調製した。トランスフェクションしなかったCHO-DG44細胞を陰性対照として使用した。G418をリン酸化アッセイで基質として使用した。抽出物を96ウェル真空マニホルド中でP81ホスホセルロースとニトロセルロース膜のサンドイッチで濾過した。タンパク質キナーゼによりリン酸化されたタンパク質そしてまたリン酸化されなかったタンパク質はニトロセルロースに結合し、一方、リン酸化G418及びリン酸化されなかったG418はニトロセルロースを通過し、ホスホセルロースに結合する。
【図9B】ドットアッセイにおける、NPT野生型と較べた本発明のNPT変異体の酵素活性を示す。このために、mAbを発現する2種の異なる細胞プール(プール1及び2)(これらはNPT野生型遺伝子(配列番号1)又はNPT変異体E182G(配列番号3)、E182D(配列番号19)、W91A(配列番号5)、V198G(配列番号7)、D227A(配列番号9)、D227V(配列番号11)、D227G(配列番号21)、D261G(配列番号13)、D261N(配列番号15)及びF240I(配列番号17)Glu182Asp又はAsp227Glyでトランスフェクションし、選抜した)から細胞抽出物を調製した。トランスフェクションしなかったCHO-DG44細胞を陰性対照として使用した。G418をリン酸化アッセイで基質として使用した。抽出物を96ウェル真空マニホルド中でP81ホスホセルロースとニトロセルロース膜のサンドイッチで濾過した。タンパク質キナーゼによりリン酸化されたタンパク質そしてまたリン酸化されなかったタンパク質はニトロセルロースに結合し、一方、リン酸化G418及びリン酸化されなかったG418はニトロセルロースを通過し、ホスホセルロースに結合する。放射性シグナルを検出し、ホスホイメージャーを使用して定量した。5μgの抽出物で得られたシグナルを使用して酵素活性%を計算した。酵素活性%はmAbを発現する2種の細胞プールからのNPT変異体の平均を表し、野生型NPTの酵素活性を100%とする。
【図10】トランスフェクションされた細胞プール中のNPT発現及びNPT遺伝子コピーの数のノーザンブロット分析を示す。このために、NPT野生型遺伝子(配列番号1)又はNPT変異体E182G(配列番号3)、E182D(配列番号19)、W91A(配列番号5)、V198G(配列番号7)、D227A(配列番号9)、D227V(配列番号11)、D227G(配列番号21)、D261G(配列番号13)、D261N(配列番号15)及びF240I(配列番号17)でトランスフェクションして選抜し、mAbを発現する2種の異なる細胞プールから全RNAを調製した。トランスフェクションしなかったCHO-DG44細胞を陰性対照として使用した。RNA30μgをNPT遺伝子のコーディング領域を含むFITC-dUTP標識PCR産物とハイブリダイズさせた。全てのトランスフェクション細胞において、約1.3kbの特異的な単一のNPT転写産物が検出された。ドットブロット分析でnpt遺伝子コピー数を測定するために、ゲノムDNAをmAbを発現する上記細胞プールから単離した。 10μg、5μg、2.5μg、1.25μg、0.63μg及び0.32μgのゲノムDNAをNPT遺伝子のコーディング領域を含むFITC-dUTP標識PCR産物とハイブリダイズした。トランスフェクションしなかったCHO-DG44細胞を陰性対照として使用した。プラスミドpBIN-LCを標準として使用した(320pg、160pg、80pg、40pg、20pg、10pg、5pg、2.5pg)。タイトレーションしたプラスミド-DNAについて測定されたシグナル強さから、測定された標準シリーズを使用して、細胞プール中のnpt遺伝子のコピー数を計算した。
【図11】二つの細胞プール(細胞プール5及び8)を例として、FACSを使用するGFPをベースとする選抜によるmAb高発現細胞プールの単離を示す。ベクターpBID-HC及びpBING-LCによる同時トランスフェクションから得られた細胞プールをGFPをベースとする連続FACSソーティングにかけた。プールの細胞培養上清中のIgG2抗体の濃度を夫々のソーティング工程後にELISAにより測定し、細胞当り1日当りの比生産能(pg/細胞*日)を計算した。全てにおいて、6回のソーティングを行ない、夫々の場合に最高のGFP蛍光を有する細胞の5%をソーティングした。夫々のデータ点は75cm2のフラスコ中の少なくとも6回の培養実験の平均を表す。
【図12】細胞プール5及び8(図11を参照のこと)を例として、GFPをベースとする選抜をMTX増幅工程と組み合わせることにより得られたmAb生産性の増大を示す。ベクターpBID-HC及びpBING-LCによるCHO-DG44の同時トランスフェクションの2週後に、最高のGFP蛍光を有する細胞の5%をプール5及び8からソーティングした。次いで100nMのメトトレキセート(MTX)を培地に添加することにより、dhfr媒介遺伝子増幅を行なった。プールの細胞培養上清中のmAbの濃度をELISAにより測定し、細胞当り1日当りの比生産能(pg/細胞*日)を計算した。夫々のデータ点は75cm2のフラスコ中の少なくとも6回の培養実験の平均を表す。
【図13】細胞プール5及び8(図11を参照のこと)を例として、抗体生産性とGFP蛍光の間の相関関係を示す。CHO-DG44をベクター組み合わせpBID-HC及びpBING-LCでトランスフェクションすることにより、これらの細胞プールを得た。それらを連続のGFPをベースとするFACSソーティングにかけ、最高のGFP蛍光を有する細胞の5%をソーティングした。プールの細胞培養上清中のIgG2抗体の濃度を夫々のソーティング工程後にELISAにより測定し、細胞当り1日当りの比生産能(pg/細胞*日)を計算した。夫々のデータ点は75cm2のフラスコ中の少なくとも6回の培養実験の平均を表す。
【技術分野】
【0001】
本発明は新規改変ネオマイシンホスホトランスフェラーゼ遺伝子及び高生産性組換え細胞の選抜方法におけるそれらの使用に関する。それ故、本発明はまた好ましくは異種プロモーターに機能し得る形で連結された対象遺伝子と組み合わされた、改変ネオマイシンホスホトランスフェラーゼ遺伝子を含む新規発現ベクターに関する。更に、本発明は相当する高生産性組換え細胞を使用する異種遺伝子産物の調製方法に関する。
【背景技術】
【0002】
哺乳動物細胞は複雑な生物医薬タンパク質の生成に好ましい宿主細胞である。何とならば、翻訳後に起こる修飾が機能上及び薬物速度論上の両方でヒトと共存性があるからである。主な妥当な細胞型はハイブリドーマ、ミエローマCHO(チャイニーズハムスター卵巣)細胞及びBHK(ベビーハムスター腎臓)細胞である。宿主細胞の培養は無血清かつ無タンパク質生成条件下で次第に行なわれるようになっている。これらの理由はコスト低減、組換えタンパク質の精製における低減された妨害及び病原体(例えば、プリオン及びウイルス)の導入の可能性の低下である。宿主細胞としてのCHO細胞の使用が一層広まりつつある。何とならば、これらの細胞が無血清かつ無タンパク質培地中の懸濁増殖に適しており、かつ規制当局により安全な生産細胞と見なされ、受け入れられているからである。
対象異種遺伝子(GOI)を発現する安定な哺乳動物細胞株を生成するために、異種遺伝子は一般にトランスフェクションにより選抜可能なマーカー遺伝子、例えば、ネオマイシンホスホトランスフェラーゼ遺伝子(NPT)と一緒に所望の細胞株に挿入される。異種遺伝子及び選抜可能なマーカー遺伝子は一つの個々のベクター又は別々の同時トランスフェクションされたベクターから開始して宿主細胞中で発現し得る。トランスフェクションの2〜3日後に、トランスフェクションされた細胞が選択薬剤、例えば、ネオマイシンホスホトランスフェラーゼ遺伝子(NPT遺伝子)を使用する場合にはG418を含む培地に移され、これらの選抜条件下で数週にわたって培養される。外来性DNAを組み込んだ耐性細胞が出現してこれを単離でき、所望の遺伝子産物(GOIの)の発現について研究し得る。
【0003】
所望のタンパク質の高発現を有する細胞株を樹立する際の重大な問題が、宿主細胞ゲノム中の転写活性又は不活性遺伝子座への組換えベクターのランダムかつ方向性のない(undirected)組込みから生じる。結果として、異種遺伝子の全く異なる発現率を有する細胞の集団が得られ、細胞の生産性は一般に正規分布に従う。それ故、対象異種遺伝子の非常に高い発現を有する細胞クローンを同定するために、多数のクローンを検査し、試験することが必要であり、これは時間を浪費し、労力集中的であり、高価である。それ故、トランスフェクションに使用されるベクター系の改良は、好適な選抜戦略によりトランスフェクションされた細胞集団中の高生産細胞の比率を増大しようと試み、それによりクローン同定に関係する出費及び作業を低減しようとした。このような発現系の開発が本発明の主題である。
アミノグリコシド-3'-ホスホトランスフェラーゼII酵素(ネオマイシンホスホトランスフェラーゼ)(EC27195)(その遺伝子はエシェリキア・コリ中のトランスポゾン5に付随する)が幾つかの生物(例えば、バクテリア、酵母、植物及び哺乳動物細胞)中の選抜可能なマーカーとして使用される。この酵素は末端リン酸をATPからアミノヘキソース環Iの3'ヒドロキシル基に転移することにより抗生物質を不活性化することにより、種々のアミノグリコシド抗生物質、例えば、ネオマイシン、カナマイシン及びG418に対する耐性を与える。野生型ネオマイシンホスホトランスフェラーゼに加えて、低下されたホスホトランスフェラーゼ活性ひいてはバクテリア(Blazquezら, 1991; Kocabiyikら, 1992; Yenofskyら, 1990)及びタバコからの葉の切片(Yenofskyら, 1990)におけるアミノグリコシド抗生物質に対する低下された耐性を有する幾つかの変異体が知られている。
これらの変異体の一種(Glu182Asp)が胚性幹細胞を選抜するためのマーカーとして使用され、ネオマイシンホスホトランスフェラーゼ遺伝子が標的相同組換え(遺伝子ターゲッティング)(Hansonら, 1995)によりc-myc遺伝子に組み込まれた。著者らはそれら自体を遺伝子ターゲッティングのための改変酵素の使用に限定している。
【0004】
特許出願WO 99/53046には生産に妥当な哺乳動物細胞中の改変ネオマイシンホスホトランスフェラーゼ遺伝子(Asp261Asn)の発現が記載されている。著者らは哺乳動物細胞中の対象遺伝子の発現のための非クローニング方法を記載している。プロモーター要素、対象遺伝子、及びIRES(“内部リボソームエントリー部位”)要素と結合された選抜可能なマーカーをコードする三つの個々のDNAフラグメントによる細胞の同時トランスフェクションにより、細胞を選択圧下で計画的に増殖させることが可能であり、その場合、全ての三つのDNAフラグメントが機能性2シストロン転写単位として組み合わされる(プロモーター-対象遺伝子-IRES-ネオマイシンホスホトランスフェラーゼ遺伝子)。これらの要素の組合せはトランスフェクションされた細胞中のみで生じ、その結果、いくつかの細胞のみが要素の正しい配置を示す。更に、増幅可能な選抜可能マーカーを使用した遺伝子増幅後に、高生産性クローンが生成し得ない。反復した選抜及び遺伝子増幅後に、生成された細胞は細胞当り1日当りせいぜい6pg(6pg/細胞/日)のタンパク質しか示さなかった。
刊行物のいずれもが一種以上の対象遺伝子そしてまた低下された抗生物質耐性を有する改変ネオマイシンホスホトランスフェラーゼ遺伝子の両方のための一種以上の完全な機能性転写単位を含む哺乳動物細胞のための高発現ベクター系(これは組換え生物医薬タンパク質を調製するために高生産性細胞を開発することを可能にする)の調製に特別な適性を有する改変ネオマイシンホスホトランスフェラーゼ遺伝子を開示していない。WO 99/53046に記載されたDNA構築物はジヒドロ葉酸還元酵素(DHFR)の遺伝子に機能し得る形で連結された無プロモーターネオマイシン遺伝子のみを含む。
それ故、特に生物医薬方法のための相当する高発現ベクター系の開発に、利用できる好適な改変ネオマイシンホスホトランスフェラーゼ遺伝子をつくるという需要がある。それ故、本発明の課題は相当する新規改変ネオマイシンホスホトランスフェラーゼ遺伝子、改変ネオマイシンホスホトランスフェラーゼ遺伝子及び異種プロモーターに機能し得る形で連結された対象遺伝子を含む発現ベクター、高生産性組換え細胞、好ましくは哺乳動物細胞の選抜方法、並びに異種遺伝子産物の生産方法を提供することであった。
驚くことに、本発明の範囲内で、高生産性細胞の選抜についてのそれらの特別な適性を特徴とする新規な高度に選抜的なネオマイシンホスホトランスフェラーゼ遺伝子を生成し、同定することが可能であった。
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は新規改変ネオマイシンホスホトランスフェラーゼ遺伝子を提供する。
【課題を解決するための手段】
【0006】
驚くことに、同時に組み込まれた対象遺伝子の高発現率を有するトランスフェクションされた哺乳動物細胞の濃縮が以下に記載される改変ネオマイシンホスホトランスフェラーゼ遺伝子を選抜可能なマーカーとして使用することにより達成し得ることがわかった。選抜可能なマーカーとしての野生型ネオマイシンホスホトランスフェラーゼの使用と較べて、本発明の新規ネオマイシンホスホトランスフェラーゼ遺伝子の一つによるトランスフェクション後に、細胞が1.4〜14.6倍に増大されたタンパク質(抗体)の生産性を示した。
本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子はアミノ酸位置91、182、198、227、240又は261において野生型遺伝子とは異なるアミノ酸をコードする変異体であることが好ましい。好ましい実施態様において、本発明のネオマイシンホスホトランスフェラーゼ遺伝子は変異体Glu182Gly、Glu182Asp、Trp91Ala、Val198Gly、Asp227Ala、Asp227Val、Asp227Gly、Asp261Asn、Asp261Gly又はPhe240Ileである。高生産性哺乳動物細胞を選抜するために、変異体Trp91Ala、Asp227Val、Asp261Asn、Asp261Gly及びPhe240Ileを使用することが特に好適と判明し、一方、変異体Asp227Val及びAsp261Glyは順に最高の生産性を有する細胞クローンを与え、それ故、特に好ましい。
【0007】
高生産性細胞が異種プロモーターに機能し得る形で連結された対象異種遺伝子及び本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子を含む真核細胞発現ベクターの使用により得られた。発現ベクターはその他の調節要素、例えば、一種以上のプロモーターに機能し得る形で連結された一種以上のエンハンサーを含むことが好ましい。また、好ましくは内部リボソームエントリー部位(IRES)(これは異種プロモーターの制御下で、蛍光タンパク質をコードする遺伝子及び対象タンパク質/産物をコードする遺伝子の2シストロン性発現を可能にする)を介して、対象遺伝子及び異種プロモーターに機能し得る形で連結される蛍光タンパク質の遺伝子を更に含む発現ベクターが好ましい。注目する異種遺伝子がユビキチン/S27aプロモーターの制御下にある発現ベクターが特に好適である。
また、本発明は対象遺伝子に代えてこのような遺伝子をとり込むための、即ち、制限エンドヌクレアーゼのための多重認識配列による配列選択のための多重クローニング部位を含む発現ベクターに関する。
別の局面において、本発明は本発明の上記改変ネオマイシンホスホトランスフェラーゼ遺伝子の一つを含む組換え哺乳動物細胞に関する。加えて、本発明は本発明の発現ベクターの一つでトランスフェクションされた組換え哺乳動物細胞に関する。これらは組換えげっ歯類細胞であることが好ましく、その中で組換えハムスター細胞、例えば、CHO細胞又はBHK細胞が特に好ましい。別の好ましい実施態様において、前記組換え細胞は増幅可能な選抜可能マーカーの遺伝子、例えば、ジヒドロ葉酸還元酵素(DHFR)の遺伝子で更にトランスフェクションされる。
【0008】
また、本発明は(i) 哺乳動物細胞のプール(集団)を改変ネオマイシンホスホトランスフェラーゼ(これはわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5〜26%の活性及び/又は上記改変の一つを有する)の遺伝子でトランスフェクションすること、(ii)前記哺乳動物細胞を前記改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、および、(iii)前記哺乳動物細胞を前記哺乳動物細胞の増殖に選択的に作用し、かつネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、を特徴とする、改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現する組換え哺乳動物細胞の濃縮方法に関する。
また、本発明は(i) 哺乳動物細胞のプールを少なくとも一つの対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ(これはわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5〜26%の活性及び/又は上記改変の一つを示す)の遺伝子でトランスフェクションすること、(ii)前記細胞を一以上の対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、(iii) 前記哺乳動物細胞を前記哺乳動物細胞の増殖に選択的に作用し、かつ改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、および、(iv)一以上の対象タンパク質を前記哺乳動物細胞又は培養上清から得ることを特徴とする、組換え哺乳動物細胞中の少なくとも一つの対象遺伝子の発現方法に関する。
更に、本発明は(i) 組換え哺乳動物細胞を本発明の発現ベクター(これは注目する遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子に加えて蛍光タンパク質をコードする)でトランスフェクションすること、(ii)前記哺乳動物細胞を一以上の対象遺伝子、蛍光タンパク質をコードする遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、(iii) 前記哺乳動物細胞を前記哺乳動物細胞の増殖に選択的に作用し、かつネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、および、(iv)前記哺乳動物細胞をフローサイトメトリー分析によりソーティングすることを特徴とする、少なくとも一つの対象異種遺伝子を発現する組換え哺乳動物細胞を得ること、および選抜する方法に関する。
哺乳動物細胞が増幅可能な選抜可能マーカー遺伝子、例えば、DHFR遺伝子で更にトランスフェクションされた場合、哺乳動物細胞を増幅可能な選抜可能マーカー遺伝子も発現される条件下で培養すること、及び培地に増幅可能な選抜可能マーカー遺伝子の増幅をもたらす選択薬剤を添加することが可能である。
本発明の方法は懸濁培養増殖に適している哺乳動物細胞、即ち、懸濁培養で培養される哺乳動物細胞を用いて行なわれることが好ましい。その他の実施態様は哺乳動物細胞、好ましくは懸濁液中の増殖に適している哺乳動物細胞が無血清条件下で培養される方法に関する。
【発明を実施するための最良の形態】
【0009】
発明の詳細な記載
アミノ酸位置に関する下記の情報は夫々の場合に配列番号1を有する野生型ネオマイシンホスホトランスフェラーゼ遺伝子によりコードされるアミノ酸の位置に関する。“改変ネオマイシンホスホトランスフェラーゼ遺伝子”はネオマイシンホスホトランスフェラーゼ活性を有するポリペプチドをコードする核酸を意味し、そのポリペプチドは明細書に更に充分に記載されるアミノ酸位置(これらは配列番号2を有する野生型タンパク質に相同である)の少なくとも一つでその野生型タンパク質とは異なるアミノ酸を有する。この状況で、“相同”という用語は変異を有する配列領域が所謂通常の“アライメント”アルゴリズム、例えば、“BLAST”(Altschul, S.F., Gish, W., Miller, W., Myers, E.W.&Lipman, D.J. (1990)“基本的局所アライメント検索ツール” J. Mol. Biol. 215:403-410; Gish, W.&States, D.J. (1993)“データベース類似性検索によるタンパク質コーディング領域の同定” Nature Genet. 3:266-272; Madden, T.L., Tatusov, R.L.&Zhang, J. (1996)“ネットワークBLASTサーバーの適用” Meth. Enzymol. 266:131-141; Zhang, J.&Madden, T.L. (1997)“パワーBLAST:インタラクチブ又は自動化配列分析及び注釈のための新しいネットワークBLAST適用”Genome Res. 7:649-656; Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller,及びDavid J. Lipman (1997),“ギャップ付きBLAST及びPSI-BLAST:タンパク質データベース検索プログラムの新しい作成”Nucleic Acids Res. 25:3389-3402)を使用して基準配列、この場合には配列番号2の野生型ネオマイシンホスホトランスフェラーゼの配列と対応させ得ることを意味する。配列はそれらがそれらの配列順序で対応する場合に対応し、通常の“アライメント”アルゴリズムを使用して同定し得る。
【0010】
本発明は新規改変ネオマイシンホスホトランスフェラーゼ遺伝子及び異種遺伝子産物、好ましくは生物医薬上妥当なポリペプチド又はタンパク質の高発現を可能にする哺乳動物細胞を調製し、選抜する方法を提供する。本発明の方法は、対象遺伝子に加えてトランスフェクションされた細胞にトランスフェクションされなかった細胞に対し選択的な利点を与える本発明のネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞の選抜に主として基づいている。驚くことに、本明細書に記載された本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子(mNPT遺伝子)の使用が野生型ネオマイシンホスホトランスフェラーゼ遺伝子(wtNPT遺伝子)に対する実質的な選択的利点を有することがわかった。特に、これはwtNPTと較べて低い酵素活性を有する変異体の使用に関する。
【0011】
本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子
wtNPTの酵素活性のわずかに1〜80%、好ましくはわずかに1〜60%を有するNPTをコードする改変NPT遺伝子を使用することが特に好適と判明した。好ましいNPT変異体はwtNPTの酵素活性のわずかに1〜30%を有するものであり、wtNPTの酵素活性のわずかに1.5〜26%を有するものが特に好ましい。NPTの酵素活性は、例えば、実施例4に記載され、方法5として示されるドットアッセイで測定し得る。
野生型ネオマイシンホスホトランスフェラーゼという用語はアミノグリコシド-3'-ホスホトランスフェラーゼII酵素(EC 2.7.1.95)をコードするネオマイシンホスホトランスフェラーゼ遺伝子を表し、その酵素の遺伝子は、に天然にはエシェリキア・コリ中のトランスポゾン5と関連しており、例えば、配列番号2に示されるアミノ酸配列を含み、又は配列番号1に示されるヌクレオチド配列によりコードされる。この酵素はアミノヘキソース環Iの3'ヒドロキシル基へのATPの末端リン酸の転移により抗生物質を不活性化することにより、種々のアミノグリコシド抗生物質、例えば、ネオマイシン、カナマイシン及びG418に対する耐性を与える。また、wtNPTという用語は配列番号1によりコードされるNPTに匹敵する酵素活性を有する全てのNPTを表す。これは特にATPから基質への末端ホスフェートの転移を触媒作用する酵素活性中心が同じ又はほぼ同じコンホメーションで存在し(Shawら, 1993; Honら, 1997; Burkら, 2001)、こうして配列番号2のアミノ酸配列を含む酵素に匹敵する酵素活性を有するこれらのNPTを含む。wtNPTはそれが配列番号2により特定されるNPTにより示される酵素活性の約81〜150%、好ましくは90〜120%を示す場合に匹敵する酵素活性を有し、その活性は実施例4に記載され、方法5と称されるドットアッセイで測定し得る。
【0012】
wtNPTと較べての酵素活性の低下がアミノ酸配列の改変、例えば、少なくとも一種又はそれ以上のアミノ酸の置換、挿入又は欠失に基づく変異体が基本的に好ましい。欠失変異体、挿入変異体及び置換変異体は“部位特異的変異導入”及び/又は“PCRをベースとする変異導入技術”により生成し得る。好適な方法が、例えば、Lottspeich及びZorbas(1998)(その他の文献とともに36.1章)により記載されている。
驚くことに、ネオマイシンホスホトランスフェラーゼ変異体が選抜可能なマーカーとして使用される場合(この場合、少なくともアミノ酸位置91のアミノ酸トリプトファン、アミノ酸位置182のアミノ酸グルタミン酸、アミノ酸位置198のアミノ酸バリン、アミノ酸位置227のアミノ酸アスパラギン酸、アミノ酸位置261のアミノ酸アスパラギン酸又はアミノ酸位置240のアミノ酸フェニルアラニンがwtNPTと較べて変化されていた)、注目する同時組込み遺伝子について高い発現率を有するトランスフェクションされた哺乳動物細胞の特に有効な濃縮を達成することが可能であることがわかった。それ故、位置91、182、198、227及び/又は240のアミノ酸に影響する変異体が好ましい。置換変異体、即ち、野生型中のこの位置に生じるアミノ酸が別のアミノ酸により置換された変異体が特に有利である。相当するアミノ酸の変化がwt-NPTと較べて1〜80%まで、好ましくは1〜60%まで、更に好ましくは1.5〜30%まで、最も好ましくは1.5%〜26%までの酵素活性の低下をもたらす相当する置換変異体が更に好ましい。wt-NPTと較べた酵素活性がわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5%〜26%であるようにアミノ酸91、227、261及び/又は240がそれに従って改変された改変NPT遺伝子が特に好ましい。アミノ酸位置227のアミノ酸が改変NPTの酵素活性がwt-NPTと較べて26%以下、好ましくは1〜20%、更に好ましくは1〜16%であるような形態で改変された置換変異体が最も好ましい。
【0013】
本発明の別の実施態様によれば、有利な変異体は、wtNPTとの比較により、アミノ酸位置91、182又は227においてグリシン、アラニン、バリン、ロイシン、イソロイシン、フェニルアラニン又はチロシンをコードする変異体である。更に、アミノ酸位置182のグルタミン酸はまたアスパラギン酸、アスパラギン、グルタミン又はあらゆるその他の好ましくは負に荷電されていてもよいアミノ酸により置換されていてもよい。また、wtNPTとの比較により、アミノ酸位置198においてグリシン、アラニン、ロイシン、イソロイシン、フェニルアラニン、チロシン又はトリプトファンをコードする改変NPT遺伝子が好ましい。また、wtNPTとの比較により、アミノ酸位置261においてグリシン、アラニン、ロイシン、イソロイシン、フェニルアラニン、チロシン、トリプトファン、アスパラギン、グルタミン又はアスパラギン酸をコードする改変NPT遺伝子が好ましい。特に、選抜可能なマーカーとして変異体Glu182Gly、Glu182Asp、Trp91Ala、Val198Gly、Asp227Ala、Asp227Val、Asp227Gly、Asp261Gly、Asp261Asn及びPhe240Ileを用いて、同時組込みされる対象遺伝子の高発現率を有するトランスフェクションされた哺乳動物細胞の濃縮を得ることが可能であることがわかり、これらの変異体が特に好ましいという結果が得られた。変異体Asp227Val、Asp227Gly、Asp261Gly、Asp261Asn、Phe240Ile及びTrp91Alaが更に好ましい。何とならば、最良の濃縮率がそれらを使用して得られるからである。変異体Asp227Valが特に好ましい。
【0014】
対照的に、Asp190Gly変異体及びAsp208Gly変異体は無血清培養条件下でトランスフェクションされたCHO-DG44細胞の選抜に不適なマーカーであることが判明した。これらの変異体(Asp190Gly、Asp208Gly)のおそらく大いに低下された酵素機能の結果として、わずかにいくつかの細胞が選抜期後に得られ、さらにそれらは増殖及び生命力が更にひどく損なわれていた。
野生型を基準として位置182及び227のアミノ酸はアミノグリコシド-3'-ホスホトランスフェラーゼのC末端領域中の三つの保存モチーフの外に位置される非保存アミノ酸である。位置91のアミノ酸はまた非保存アミノ酸に属し、アミノグリコシド-3'-ホスホトランスフェラーゼのN末端領域中の保存モチーフの一つの外に位置される。対照的に、位置198及び240のアミノ酸はNPTのC末端領域中の保存アミノ酸であるが、それにもかかわらず保存モチーフの外にある。対照的に、位置261のアミノ酸はC末端領域の第三の保存モチーフ中の保存アミノ酸である(Shawら, 1993; Honら, 1997; Burkら, 2001)。
選抜可能なマーカーとしてのwtNPTの使用と較べて、Glu182Gly、Glu182Asp及びVal198Gly変異体の場合の細胞は1.4-2.4倍に増大された生産性を示し、Asp227Gly変異体の場合には生産性が1.6-4.1倍に増大され、Asp227Ala又はTrp91Ala変異体の場合には生産性が2.2倍又は4倍に増大され、Phe240Ile又はAsp261Asn変異体の場合には生産性が5.7倍又は7.3倍に増大され、またAsp261Gly又はAsp227Val変異体の場合にはそれが9.3倍又は14.6倍に一層増大された。マルチ鎖タンパク質(抗体)を発現するために、同時トランスフェクションが行なわれた。二つのタンパク質鎖はそれら自身のベクターにより夫々発現され、一つのベクターはNPT遺伝子を更にコードし、一方、別のベクターは増幅可能で選抜可能なジヒドロ葉酸還元酵素遺伝子をコードする。
【0015】
このように、本発明は(i)哺乳動物細胞のプールを野生型ネオマイシンホスホトランスフェラーゼの活性のわずかに1〜80%、好ましくは1〜60%、更に好ましくは1.5〜30%、最も好ましくは1.5〜26%及び/又は本明細書に記載された改変の一つを有する改変ネオマイシンホスホトランスフェラーゼの遺伝子でトランスフェクションすること、(ii)前記哺乳動物細胞を改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、および、(iii) 前記哺乳動物細胞を哺乳動物細胞の増殖に選択的に作用し、かつネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、を特徴とする、改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現する組換え哺乳動物細胞の濃縮方法に関する。
特に使用される改変NPT遺伝子が、野生型遺伝子との比較して、アミノ酸位置91のアラニン、アミノ酸位置182のグリシン又はアスパラギン酸、アミノ酸位置198のグリシン、アミノ酸位置227のアラニン、グリシン又はバリン、アミノ酸位置261のグリシン又はアスパラギン或いはアミノ酸位置240のイソロイシンをコードする改変NPTをコードする場合、この出願に更に詳しく記載された改変NPT遺伝子を使用する相当する方法が特に好ましい。野生型遺伝子との比較により、アミノ酸位置227のバリン及び/又はアミノ酸位置261のグリシン及び/又はアスパラギンをコードするNPT遺伝子が更に好ましい。野生型遺伝子との比較により、アミノ酸位置240でイソロイシン又はアミノ酸位置227でバリンをコードするこれらのNPT遺伝子が特に好ましく、野生型遺伝子と較べてアミノ酸位置227でバリンをコードするNPT遺伝子が特に好ましい。当然、本発明はまた改変NPT遺伝子及び相当するアミノ酸交換の組み合わせを含む本発明の改変NPT遺伝子の使用を含む。
更に、本発明は(i) 異種プロモーターに機能し得る形で連結された注目する異種遺伝子及び(ii)野生型ネオマイシンホスホトランスフェラーゼと較べて低い酵素活性を有するネオマイシンホスホトランスフェラーゼをコードする本発明の改変ネオマイシンホスホトランスフェラーゼ遺伝子を含む真核細胞発現ベクターに関する。本発明の目的において、“低い”又は“より低い”酵素活性はwtNPTの酵素活性のせいぜい80%、好ましくは1〜80%、更に好ましくはわずかに1〜60%に相当する酵素活性を意味する。本発明の一実施態様によれば、“より低い酵素活性”は野生型ネオマイシンホスホトランスフェラーゼと較べて1〜30%、好ましくは1.5〜26%の酵素活性を表す。
【0016】
好ましい発現ベクターはwtNPTの酵素活性のわずかに1〜80%、好ましくはわずかに1〜60%を有する改変NPTをコードする改変NPT遺伝子を含む。また、wtNPTの酵素活性のわずかに1〜30%を有する変異体をコードする改変NPT遺伝子を有する発現ベクターが好ましい。wtNPTの酵素活性のわずかに1.5〜26%を有する変異体をコードする改変NPT遺伝子を含むこれらの発現ベクターが特に好ましく、その活性は実施例4に記載され、方法5と称されるドットアッセイで測定される。
本発明の別の実施態様において、発現ベクターは、wtNPTと較べて、アミノ酸位置Trp91、Glu182、Val198、Asp227、Phe240又は位置Asp261で改変された改変NPTの遺伝子を含む。この意味で、位置Trp91、Glu182、Val198、Asp227、Phe240又はAsp261で改変され、wtNPTの酵素活性のわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5〜26%を有するNPT変異体が好ましい。アミノ酸Trp91、Glu182又はAsp227が相当する位置でグリシン、アラニン、バリン、ロイシン、イソロイシン、フェニルアラニン又はチロシンにより夫々置換されることが好ましい。位置182のグルタミン酸がまたアスパラギン酸、アスパラギン、グルタミン又は別の好ましくは負に荷電されていてもよいアミノ酸により置換されることが好ましい。また、wtNPTと較べてアミノ酸位置198でグリシン、アラニン、ロイシン、イソロイシン、フェニルアラニン、チロシン又はトリプトファンをコードする改変NPT遺伝子が好ましい。加えて、wtNPTと較べてアミノ酸位置240でグリシン、アラニン、バリン、イソロイシン、チロシン又はトリプトファンをコードする改変NPT遺伝子が好ましい。また、wtNPTと較べて、アミノ酸261でグリシン、アラニン、ロイシン、イソロイシン、フェニルアラニン、チロシン、トリプトファン、アスパラギン、グルタミン又はアスパラギン酸をコードする改変NPT遺伝子が好ましい。位置227のアスパラギン酸がグリシン、アラニン、バリン、ロイシン又はイソロイシンにより置換され、位置261のアスパラギン酸がアラニン、バリン、ロイシン、イソロイシン又はグルタミン、特にグリシン又はアスパラギンにより置換される変異体を使用することが特に好ましい。
【0017】
Glu182Gly変異体、Glu182Asp変異体、Trp91Ala変異体、Val198Gly変異体、Asp227Ala変異体、Asp227Val変異体、Asp227Gly変異体、Asp261Gly変異体、Asp261Asn変異体又はPhe240Ile変異体をコードする改変NPT遺伝子を含む発現ベクターが特に好ましく、これはGlu182Gly変異体の場合には配列番号4のアミノ酸配列を含み、Glu182Asp変異体の場合には配列番号20のアミノ酸配列を含み、Trp91Ala変異体の場合には配列番号6のアミノ酸配列を含み、Val198Gly変異体の場合には配列番号8のアミノ酸配列を含み、Asp227Ala変異体の場合には配列番号10のアミノ酸配列を含み、Asp227Val変異体の場合には配列番号12のアミノ酸配列を含み、Asp227Gly変異体の場合には配列番号22のアミノ酸配列を含み、Asp261Gly変異体の場合には配列番号14のアミノ酸配列を含み、Asp261Asn変異体の場合には配列番号16のアミノ酸配列を含み、またPhe240Ile変異体の場合には配列番号18のアミノ酸配列を含む。特に発現ベクターが配列番号12、配列番号22、配列番号14、配列番号16、配列番号18もしくは配列番号6に示されたアミノ酸配列を含む場合、又はそれが配列番号11、配列番号21、配列番号13、配列番号15、配列番号17もしくは配列番号5に示された核酸配列によりコードされ、もしくはそれを含む場合、Asp227Val変異体、Asp227Gly変異体、Asp261Gly変異体、Asp261Asn変異体、Phe240Ile変異体又はTrp91Ala変異体を使用する発現ベクターが最も好ましい。
加えて、本発明はwtNPTと較べてアミノ酸位置Trp91、Val198位置又はPhe240位置にwtアミノ酸とは異なるアミノ酸をコードする改変ネオマイシンホスホトランスフェラーゼ遺伝子及びそれらの遺伝子産物を初めて提供する。特に、本発明はwtNPTと較べて低下された酵素活性を有するTrp91変異体、Val198変異体又はPhe240変異体を初めて提供する。ここに記載され、本発明の範囲内で利用できるようにされた改変NPTはアミノ酸位置91でアラニン、位置198でグリシン、また位置240でイソロイシンをコードすることが好ましい。更に、本発明は、wtNPTと較べて、位置182でグリシン、位置227でアラニン又はバリン、また位置261でグリシンをコードするNPT変異体を最初に提供する。遺伝子及び遺伝子産物(酵素)の両方が本発明の範囲内で初めて提供される。この状況で、本発明は配列番号4、配列番号6、配列番号8、配列番号10、配列番号12、配列番号14及び配列番号18のアミノ酸配列を有する改変NPTを初めて提供する。更に、本発明は配列番号3、配列番号5、配列番号7、配列番号9、配列番号11、配列番号13、及び配列番号17のDNA配列を有する改変NPT遺伝子を提供する。
【0018】
対象遺伝子
本発明の発現ベクター中に含まれる対象遺伝子は対象産物をコードするあらゆる長さのヌクレオチド配列を含む。遺伝子産物又は“対象産物”は一般にタンパク質、ポリペプチド、ペプチド又はこれらのフラグメントもしくは誘導体である。しかしながら、それはまたRNA又はアンチセンスRNAであってもよい。対象とする遺伝子はその完全長、短くされた形態、融合遺伝子として、又は標識遺伝子として存在してもよい。それはゲノムDNA又は好ましくはcDNA或いは融合の相当するフラグメントであってもよい。対象遺伝子は天然の遺伝子配列であってもよく、又はそれは変異されてもよく、もしくはそれ以外に改変されてもよい。このような改変として、特別な宿主細胞に適合するためのコドン最適化及びヒト化が挙げられる。対象遺伝子は、例えば、分泌されたポリペプチド、細胞質ポリペプチド、核配置されたポリペプチド、膜結合されたポリペプチド又は細胞表面結合されたポリペプチドをコードしてもよい。
“ヌクレオチド配列”又は“核酸配列”という用語はオリゴヌクレオチド、ヌクレオチド、ポリヌクレオチド及びこれらのフラグメントだけでなく、一本鎖又は二本鎖として生じ、遺伝子のコーディング鎖又は非コーディング鎖に相当し得るゲノム起源又は合成起源のDNA又はRNAを示す。核酸配列は通常の技術、例えば、部位特異的変異導入又はPCR媒介変異導入(例えば、Sambrookら, 1989又はAusubelら, 1994に記載される)を使用して改変されてもよい。
【0019】
“コードする”とは核酸中のヌクレオチドの特定配列、例えば、染色体又はmRNA中の遺伝子が、生物学的プロセスにおいてその他のポリマー及び巨大分子、例えば、rRNA、tRNA、mRNA、その他のRNA分子、cDNA又はポリペプチドの合成のためのマトリックスとして作用する性質又は能力を意味する。それ故、所望のタンパク質が細胞又は別の生物系中でmRNAの転写、続いて翻訳により生成される場合、その遺伝子はタンパク質をコードする。ヌクレオチド配列がmRNA配列と同じである、通常配列データバンク、例えば、EMBL又はGenBankに示されるコーディング鎖、そしてまた転写のためのマトリックスとして作用する遺伝子又はcDNAの非コーディング鎖のいずれも産物又はタンパク質をコードすると称されることがある。また、タンパク質をコードする核酸には、縮重遺伝子コードに基づいてヌクレオチド配列の異なる順序を有するがタンパク質の同じアミノ酸配列をもたらす核酸も含まれる。タンパク質をコードする核酸配列はまたイントロンを含んでもよい。
cDNAという用語は遺伝子から生じたmRNA又はその他のRNAからの逆転写および第二DNAストランドの合成により調製されるデオキシリボ核酸を表す。cDNAが二本鎖DNA分子として存在する場合、それはコーディングストランド及び非コーディングストランドの両方を含む。
イントロンという用語はあらゆる長さの非コーディングヌクレオチド配列を表す。それらは多くの真核生物遺伝子中に天然に存在し、スプライシングとして知られている方法により既に転写されたmRNA前駆体から排除される。これは成功裏のタンパク質合成のために正しいリーディングフレームを有するプロセッシングされた成熟mRNAを生じるように5'末端及び3'末端におけるイントロンの正確な切除及び得られるmRNA末端の正確な結合を必要とする。このスプライシング過程に関与するスプライスドナー部位及びスプライスアクセプター部位の多く、即ち、エキソン-イントロン境界又はイントロン-エキソン境界に直接配置された配列が、今までに特性決定されている。総覧について、Ohshimaら, 1987を参照のこと。
【0020】
対象とするタンパク質/産物
生物医薬上重要なタンパク質/ポリペプチドとして、例えば、抗体、酵素、サイトカイン、リンホカイン、付着分子、受容体及びこれらの誘導体又はフラグメントが挙げれるが、これらに限定されない。一般に、アゴニストもしくはアンタゴニストとして作用し、かつ/又は治療もしくは診断上の適用を有する全てのポリペプチドが有益である。
“ポリペプチド”という用語はアミノ酸配列又はタンパク質について使用され、あらゆる長さのアミノ酸のポリマーを表す。この用語はまたグリコシル化、リン酸化、アセチル化又はタンパク質プロセシングの如き反応により翻訳後に修飾されたタンパク質を含む。ポリペプチドの構造は、例えば、その生物学的活性を保持しながらアミノ酸の置換、欠失又は挿入及びその他のタンパク質との融合により改変されてもよい。こうして、“ポリペプチド”という用語はまた、例えば、免疫グロブリン成分、例えば、Fc成分、及び成長因子、例えば、インターロイキンからなる融合タンパク質を含む。
治療タンパク質の例はインスリン、インスリン様成長因子、ヒト成長ホルモン(hGH)及びその他の成長因子、組織プラスミノーゲンアクチベーター(tPA)、エリスロポエチン(EPO)、サイトカイン、例えば、インターロイキン(IL)、例えば、IL-1、IL-2、IL-3、IL-4、IL-5、IL-6、IL-7、IL-8、IL-9、IL-10、IL-11、IL-12、IL-13、IL-14、IL-15、IL-16、IL-17、IL-18、インターフェロン(IFN)-α、-β、-γ、-ω又は-タウ、腫瘍壊死因子(TNF)、例えば、TNF-α、β又はγ、TRAIL、G-CSF、GM-CSF、M-CSF、MCP-1及びVEGFである。その他の例はモノクローナル抗体、ポリクローナル抗体、多重特異性抗体及び一本鎖抗体並びにこれらのフラグメント、例えば、Fab、Fab'、F(ab')2、Fc及びFc'フラグメント、軽鎖(L鎖)免疫グロブリン及び重鎖(H鎖)免疫グロブリン並びにこれらの定常領域、可変領域又は超可変領域だけでなく、Fvフラグメント及びFdフラグメントである(Chamovら, 1999)。抗体はヒト起源であっても非ヒト起源のものであってもよい。また、ヒト化抗体及びキメラ抗体が可能である。
【0021】
Fabフラグメント(フラグメント抗原結合=Fab)は隣接定常領域により一緒に保持される両方の鎖の可変領域からなる。それらは、例えば、通常の抗体からパパインの如きプロテアーゼで処理することにより、又はDNAクローニングにより産生されてもよい。その他の抗体フラグメントはペプシンによるタンパク質分解消化により産生し得るF(Ab')2フラグメントである。
遺伝子クローニングにより、重鎖の可変領域(VH)及び軽鎖の可変領域(VL)のみからなる短くされた抗体フラグメントを調製することが可能である。これらはFvフラグメント(可変フラグメント=可変部分のフラグメント)として知られている。定常鎖のシステイン基を介しての共有結合がこれらのFvフラグメントでは可能ではないので、それらはしばしば或る種のその他の方法により安定化される。この目的のために、重鎖及び軽鎖の可変領域が約10〜30アミノ酸、好ましくは15アミノ酸の短いペプチドフラグメントによりしばしば一緒に結合される。これはVH及びVLがペプチドリンカーにより一緒に結合される単一ポリペプチド鎖を生じる。このような抗体フラグメントもまた一本鎖Fvフラグメント(scFv)と称される。scFv抗体の例が知られており、例えば、Hustonら, 1988に記載されている。
過去何年かに、種々の戦略が多量体のscFv誘導体を産生するために開発されている。その目的は、改良された薬物速度論的性質及び増大された結合活性を有する組換え抗体を産生するためである。scFvフラグメントの多量体化を得るために、それらが多量体化ドメインを有する融合タンパク質として産生される。多量体化ドメインは、例えば、IgGのCH3領域又はらせん構造(“コイルドコイル構造”)、例えば、ロイシンジッパードメインであってもよい。その他の戦略では、scFvフラグメントのVH領域とVL領域の間の相互作用が多量体化に使用される(例えば、ジアボディ、トリボディ及びペンタボディ)。
【0022】
ジアボディ(diabody)(二量体抗体)という用語は当業界で2価のホモ二量体scFv誘導体を表すのに使用される。scFv分子中のペプチドリンカーを5〜10アミノ酸に短くすることはVH/VL鎖を重ねることによりホモ二量体の生成をもたらす。ジアボディは挿入されたジスルフィド架橋により更に安定化されてもよい。ジアボディの例が文献、例えば、Perisicら, 1994に見られる。
ミニボディという用語は当業界で2価のホモ二量体scFv誘導体を表すのに使用される。それは二量体化領域として免疫グロブリン、好ましくはIgG、最も好ましくはIgG1のCH3領域を含む融合タンパク質からなる。これはまたIgGのヒンジ領域及びリンカー領域によりscFvフラグメントを連結する。このようなミニボディの例がHuら, 1996により記載されている。
トリアボディという用語は当業界で3価のホモ三量体のscFv誘導体を表すのに使用される(Korttら, 1997)。リンカー配列を使用しないVH VLの直接融合が三量体の生成をもたらす。
2価、3価又は4価の構造を有するミニボディとして当業界で知られているフラグメントはまたscFvフラグメントの誘導体である。多量体化は二量体、三量体又は四量体のコイルドコイル構造により得られる(Packら, 1993及び1995; Lovejoyら, 1993)。
【0023】
蛍光タンパク質をコードする遺伝子
別の実施態様において、本発明の発現ベクターは、好ましくは対象とする遺伝子に機能し得る形で連結された、蛍光タンパク質をコードする遺伝子を含む。両方の遺伝子は対象とするタンパク質/産物及び蛍光タンパク質が2シストロン性mRNAによりコードされるように単一異種プロモーターの制御下で転写されることが好ましい。これは蛍光タンパク質の発現率により、対象とするタンパク質/産物を多量に生成する細胞を同定することを可能にする。
蛍光タンパク質は、例えば、緑色、青緑色、青色、黄色又はその他の着色蛍光タンパク質であってもよい。一つの特別な例はオワンクラゲ(Aequorea victoria)又はウミシイタケ(Renilla reniformis)から得られた緑色蛍光タンパク質(GFP)及びそれらから発生された変異体である。例えば、Bennetら, 1998; Chalfieら, 1994; WO 01/04306及びその中に引用された文献を参照のこと。
その他の蛍光タンパク質及びそれらをコードする遺伝子がWO 00/34318、WO 00/34326、WO 00/34526及びWO 01/27150(これらは参考として本明細書に含まれる)に記載されている。これらの蛍光タンパク質は種花虫綱の非バイオルミネセント生物、例えば、アネモニア・マジャノ(Anemonia majano)、ツツウミヅタ(Clavularia)種、スナギンチャク(Zoanthus)種I、スナギンチャク種II、ディスコゾーマ・ストリアタ(Discosoma striata)、ディスコゾーマ種“レッド”、ディスコゾーマ種“グリーン”、ディスコゾーマ種“マゼンタ”、ヘビイソギンチャク(Anemonia sulcata)の蛍光体である。
本発明に従って使用される蛍光タンパク質は野生型タンパク質に加えて天然の、又は遺伝子操作された変異体及び変異体、これらのフラグメント、誘導体又は変異体(これらは、例えば、その他のタンパク質又はペプチドと融合された)を含む。使用される変異は、例えば、励起スペクトル又は発光スペクトル、発色団の形成、タンパク質の吸光係数又は安定性を変化し得る。更に、哺乳動物細胞又はその他の種中の発現がコドン最適化により改良し得る。本発明によれば、蛍光タンパク質はまた選抜可能なマーカー、好ましくは増幅可能な選抜可能マーカー、例えば、ジヒドロ葉酸還元酵素(DHFR)との融合に使用されてもよい。
蛍光タンパク質により放出された蛍光はタンパク質を、例えば、蛍光活性化セルソーター(FACS)によるスルーフローサイトメトリー又は蛍光顕微鏡により検出することを可能にする。
【0024】
その他の調節要素
発現ベクターは対象とする遺伝子そしてまた好ましくは蛍光タンパク質の遺伝子の発現を可能にする少なくとも一つの異種プロモーターを含む。
プロモーターという用語はそれと機能機能可能に連結された遺伝子又は配列の転写を可能にし、制御するポリヌクレオチド配列を表す。プロモーターはRNAポリメラーゼを結合するための認識配列及び転写のための開始部位(転写開始部位)を含む。或る種の細胞型又は宿主細胞中で所望の配列を発現するために、好適な機能性プロモーターが選ばれる必要がある。当業者は構成的プロモーター、誘導性プロモーター及び抑制プロモーターを含む、種々の源からの種々のプロモーターを良く知っているであろう。それらはデータバンク、例えば、GenBankに寄託され、別々の要素或いは市販又は個々の源からのポリヌクレオチド配列内でクローン化された要素として得られてもよい。誘導性プロモーターでは、プロモーターの活性がシグナルに応答して低下又は増大され得る。誘導性プロモーターの一つの例はテトラサイクリン(tet)プロモーターである。これはテトラサイクリン調節トランスアクチベータータンパク質(tTA)により誘導し得るテトラサイクリンオペレーター配列(tetO)を含む。テトラサイクリンの存在下で、tetOへのtTAの結合が抑制される。その他の誘導性プロモーターの例はjun、fos、メタロチオネイン及び熱ショックプロモーターである(また、Sambrookら, 1989; Gossenら, 1994を参照のこと)。
真核生物中の高発現に特に適しているプロモーターの中に、例えば、ハムスターのユビキチン/S27aプロモーター(WO 97/15664)、SV40初期プロモーター、アデノウイルス主要後期プロモーター、マウスメタロチオネイン-Iプロモーター、ラウス肉腫ウイルスの長末端リピート領域、ヒトサイトメガロウイルスの初期プロモーターがある。その他の異種哺乳動物プロモーターの例はアクチン、免疫グロブリン又は一以上の熱ショックプロモーターである。
【0025】
相当する異種プロモーターは発現カセット中の転写活性を増大/調節するためにその他の調節配列に機能し得る形で連結し得る。
例えば、プロモーターは転写活性を増大するためにエンハンサー配列に機能可能に連結することができる。このために、一以上のエンハンサー及び/又はエンハンサー配列の幾つかのコピー、例えば、CMV又はSV40エンハンサーを使用することもできる。それ故、本発明の発現ベクターは、別の実施態様において、一以上のエンハンサー/エンハンサー配列、好ましくはCMV又はSV40エンハンサーを含む。
エンハンサーという用語はcis配置でプロモーターの活性に作用し、こうしてこのプロモーターに機能可能に連結された遺伝子の転写を刺激するポリヌクレオチド配列を表す。プロモーターと違って、エンハンサーの効果は位置及び配向とは独立であり、それ故、それらはイントロン内又は更にはコーディング領域内の、転写単位の前又は後に位置し得る。エンハンサーは転写単位の直ぐの付近及びプロモーターからのかなりの距離の両方に位置されてもよい。また、プロモーターと物理的かつ機能的な重なりを有することが可能である。当業者は独立の要素又はポリヌクレオチド配列(例えば、ATCCに寄託され、又は市販の源及び個々の源からの)内でクローン化された要素として利用できる種々の源からの幾つかのエンハンサー(データバンク、例えば、GenBankに寄託されたもの、例えば、SV40エンハンサー、CMVエンハンサー、ポリオーマエンハンサー、アデノウイルスエンハンサー)を知っているであろう。また、幾つかのプロモーター配列はエンハンサー配列、例えば、頻繁に使用されるCMVプロモーターを含む。ヒトCMVエンハンサーはこれまでに同定された最強のエンハンサーの一つである。誘導エンハンサーの一例はメタロチオネインエンハンサーであり、これはグルココルチコイド又は重金属により刺激し得る。
別の可能な改変は、例えば、多重Sp1結合部位の導入である。プロモーター配列は転写活性の制御/調節を可能にする調節配列と組み合わされてもよい。こうして、プロモーターは抑制性/誘導性にされ得る。これは、例えば、転写因子をアップレギュレート又はダウンレギュレートするための結合部位である配列に連結することにより行ない得る。例えば、上記転写因子Sp1は転写活性に正の効果を有する。別の例はアクチベータータンパク質AP1のための結合部位であり、これは転写に正及び負の両方で作用し得る。AP1の活性はあらゆる種類の因子、例えば、成長因子、サイトカイン及び血清により調節し得る(Faisstら, 1992及びその中の文献)。転写効率はまた一つ、二つ、三つ又はそれ以上の塩基の変異(置換、挿入又は欠失)によりプロモーター配列を変化させることにより増大させることができ、次いでレポーター遺伝子アッセイで、プロモーター活性が増大したか否かを測定することができる。
【0026】
基本的に、付加的な調節要素は異種プロモーター、エンハンサー、終結シグナル及びポリアデニル化シグナル並びにその他の発現調節要素を含む。誘導調節配列及び構成的調節配列の両方が種々の細胞型について知られている。
“転写調節要素”は一般に発現すべき遺伝子の上流のプロモーター、転写開始部位及び終結部位並びにポリアデニル化シグナルを含む。
“転写開始部位”という用語は一次転写産物、即ち、mRNA前駆体中にとり込まれる第一核酸に相当する構築物中の核酸を表す。転写開始部位はプロモーター配列と重なっていることもある。
“転写終結部位”という用語は、通常対象とする遺伝子又は転写されるべきである遺伝子部分の3'末端にあってRNAポリメラーゼによる転写の終結をもたらすヌクレオチド配列を表す。
“ポリアデニル化シグナル”は真核生物mRNAの3'末端にある特定部位における開裂及び開裂された3'末端における約100-200アデニンヌクレオチドの配列(ポリAテール)の転写後のとり込みを生じるシグナル配列である。ポリアデニル化シグナルは開裂部位の約10-30ヌクレオチド上流の配列AATAAA及び下流に位置される配列を含む。種々のポリアデニル化要素、例えば、tk ポリA、SV40後期及び初期ポリA又はBGHポリAが知られている(例えば、米国特許第5,112,458号に記載されている)。
本発明の好ましい実施態様において、夫々の転写単位はプロモーター又はプロモーター/エンハンサー要素、対象とする遺伝子及び/又はマーカー遺伝子だけでなく、転写終結要素を有する。別の好ましい実施態様において、転写単位は二つの更なる翻訳調節単位を含む。
【0027】
“翻訳調節要素”は、発現すべき夫々のポリペプチドについて、翻訳開始部位(AUG)、停止コドン及びポリAシグナルを含む。最適の発現のために、転写レベル又は発現レベルで発現に影響し得る潜在的に望ましくない付加的な翻訳開始コドン又はその他の配列を排除するために、発現される核酸配列の5'-及び/又は3'-未翻訳領域を除去、追加又は変化することが推奨されるかもしれない。発現を促進するために、リボソームコンセンサス結合部位がまた開始コドンの直ぐ上流に挿入されてもよい。分泌されるポリペプチドを生成するために、対象とする遺伝子は合成されたポリペプチドをER膜に輸送するシグナル前駆体ペプチドをコードするシグナル配列を通常含む。シグナル配列は分泌されるタンパク質のアミノ末端に常にではないがしばしば位置され、そのタンパク質がER膜で濾過された後にシグナルペプチダーゼにより切断される。遺伝子配列はそれ自体のシグナル配列を必ずではないが通常含むであろう。天然シグナル配列が存在しない場合、異種シグナル配列が既知の様式で導入されてもよい。この種の多くのシグナル配列が当業者に知られており、配列データバンク、例えば、GenBank及びEMBLに寄託されている。
本発明の一つの重要な調節要素は内部リボソームエントリー部位(IRES)である。IRES要素は5'-末端メチルグアノシニウムキャップ(CAP構造)及び上流遺伝子とは独立に翻訳開始を機能的に活性化する配列を含み、動物細胞中で単一転写産物からの二つのシストロン(オープンリーディングフレーム)の翻訳を可能にする。IRES要素は直ぐ下流に位置されるオープンリーディングフレームの翻訳のための独立のリボソーム侵入部位を与える。マルチシストロン性であり得る細菌mRNA(即ち、それはmRNAにより交互に翻訳される多くの異なるポリペプチド又は産物をコードし得る)とは対照的に、動物細胞からのmRNAの大半はモノシストロン性であり、唯一のタンパク質又は産物をコードする。真核生物細胞におけるマルチシストロン性転写産物の場合、その翻訳は最も近い上流にあった翻訳開始部位から開始され、最初の停止コドンにより停止され、その後に転写産物がリボソームから放出されるであろう。こうして、mRNAによりコードされた最初のポリペプチド又は産物のみが翻訳中に生成されるであろう。対照的に、転写産物中の第二の、又はその後のオープンリーディングフレームに機能し得る形で連結されるIRES要素を含むマルチシストロン性転写産物はその下流に位置されたオープンリーディングフレームのその後の翻訳を可能にし、その結果、同じ転写産物によりコードされた2以上のポリペプチド又は産物が真核生物細胞中で生成される。
【0028】
IRES要素は種々の長さ及び種々の起源のものであってもよく、例えば、脳心筋炎ウイルス(EMCV)又はその他のピコルナウイルスに由来してもよい。種々のIRES配列及びベクターの構築物中のそれらの使用が文献に記載されており、例えば、Pelletierら, 1988; Jangら, 1989; Daviesら, 1992; Adamら, 1991; Morganら, 1992; Sugimotoら, 1994; Rameshら, 1996; Mosserら, 1997を参照のこと。
下流に位置される遺伝子配列はIRES要素の3'末端に機能し得る形で連結され、即ち、遺伝子の発現が影響されず、もしくはほんの限界的に影響され、又は意図される目的に充分な発現を有するように間隔が選ばれる。充分な発現のためのIRES要素とその下流に位置される遺伝子の開始コドンの間の最適の許される距離は間隔を変化させ、レポーター遺伝子アッセイを使用して発現率を間隔の関数として測定することにより簡単な実験により決定し得る。
記載された手段により、異種遺伝子産物の発現に大いに有益である最適発現カセットを得ることが可能である。それ故、一つ以上のこのような手段により得られた発現カセットは本発明の更なる主題である。
【0029】
ハムスター-ユビキチン/S27aプロモーター
別の実施態様において、本発明の発現ベクターは、好ましくは対象とする遺伝子に機能し得る形で連結され、更に好ましくは対象とする遺伝子及び蛍光タンパク質をコードする遺伝子に機能し得る形で連結された、ハムスターのユビキチン/S27aプロモーターを含む。
ハムスターのユビキチン/S27aプロモーターはWO 97/15664に記載されている強力な同種プロモーターである。このようなプロモーターは下記の特徴の少なくとも一つを有することが好ましい:GCに富む配列領域、Sp1結合部位、ポリピリミジン要素、TATAボックスの不在。Sp1結合部位を有するが、TATAボックスを有しないプロモーターが特に好ましい。また、構成的に活性化され、特に血清含有細胞培養条件、低血清細胞培養条件及び無血清細胞培養条件下で同等に活性であるプロモーターが好ましい。別の実施態様において、それは誘導性プロモーター、特に血清の除去により活性化されるプロモーターである。
特に有利な実施態様はWO 97/15664の図5に含まれるようなヌクレオチド配列を有するプロモーターである。図5の位置-161から-45までの配列を含むプロモーター配列が特に好ましい。
本特許明細書の実施例に使用されるプロモーターは配列表の配列番号55の位置1923から2406までの配列を有するDNA分子を夫々含む。この配列はWO 97/15664の図5からのフラグメント-372〜+111に相当し、好ましいプロモーターを表し、即ち、好ましいプロモーターはこの配列領域を含むべきである。別の好適なプロモーターフラグメントは位置2134から2406まで(WO 97/15664の図5中の-161から+111までに相当する)の配列を含む。位置2251から2406までの配列のみを含むプロモーターは最早機能性ではない(WO 97/15664の図5中の位置-45から+111までに相当する)。プロモーター配列を位置2134から出発して5'方向に延長することが可能である。
また、完全ハムスターユビキチン/S27aプロモーター配列の機能性部分断片だけでなく、例えば、置換、挿入又は欠失により改変されたその部分断片の完全配列の機能性変異体/変異体を使用することが可能である。相当する部分断片、変異体又は変異体が以下にまた“改変プロモーター”と称される。
【0030】
改変プロモーターは、必要によりその他の調節要素と組み合わされてもよく、配列番号55に示されたヌクレオチド配列の位置1923から2406まで(WO 97/15664の図5からの-372から+111まで)のプロモーターフラグメントの転写活性に相当する転写活性を有することが好ましい。改変プロモーターはそれが比較レポーター遺伝子アッセイで1923〜2406フラグメント(-372〜+111フラグメント)の活性の少なくとも50%、好ましくは少なくとも80%、更に好ましくは少なくとも90%、最も好ましくは少なくとも100%を有する転写活性を有する場合に本発明の目的に有益であると判明する。少なくとも80%、好ましくは少なくとも85%、好ましくは少なくとも90%、更に好ましくは少なくとも95%、最も好ましくは少なくとも97%のハムスターユビキチン/S27aプロモーターの野生型配列配列番号55に対する最小配列相同性を有し、比較レポーター遺伝子アッセイで相当するプロモーター活性を有する改変プロモーターが特に好ましい。
相当する比較レポーター遺伝子アッセイで、基準配列を含む試験すべきプロモーターフラグメントが、無プロモーターレポーター遺伝子、例えば、ルシフェラーゼ、分泌アルカリ性ホスファターゼ又は緑色蛍光タンパク質(GFP)をコードする無プロモーターレポーター遺伝子の前にクローン化される。続いて、これらの構築物(プロモーター配列+レポーター遺伝子)がトランスフェクションにより試験細胞、例えば、CHO-DG44に導入され、当該プロモーターフラグメントによるレポーター遺伝子発現の誘導がレポーター遺伝子のタンパク質含量を測定することにより測定される。相当する試験が、例えば、Ausubelら, Current Protocols in Mokecular Biology, 1994(最新)に見られる。
ハムスターユビキチン/S27aプロモーター及び改変プロモーター(これはまた、例えば、5'未翻訳領域又はその選ばれたフラグメント、及びコーディング領域だけでなく、ユビキチン/S27a遺伝子の3'-未翻訳領域又はその選ばれたフラグメントを含んでもよい)のプロモーター配列は、例えば、Sambrookら, 1989; Ausubelら, 1994に記載されたような種々の通常の方法を使用してWO 97/15664に記載された配列の知識により当業者によれば得ることが出来る。WO 97/15664に記載された配列から出発して、例えば、好適なフラグメントが選択され、このフラクションの配列を含むオリゴヌクレオチドプローブが化学合成されてもよい。この種のプローブは、例えば、ハムスターゲノムのライブラリーからのハイブリダイゼーションにより、ユビキチン/S27a遺伝子又はその5'未翻訳領域もしくはその他のフラグメントをクローン化するのに使用することができる。上記レポーター遺伝子アッセイを使用して、当業者は大いに努力しなくてもプロモーター活性フラグメントを同定し、それらを本発明の目的に使用する状態に置かれている。5'未翻訳領域又はその特別なフラグメントはゲノムDNA又はゲノムライブラリーからの相当するプライマーによるPCR増幅により容易に得られる。5'未翻訳領域のフラグメントはまた一層大きいDNAフラグメントからの制限エキソヌクレアーゼIII消化により得ることもできる。このようなDNA分子はまた化学合成することもでき、又はライゲーションにより化学合成されたフラグメントから生成することもできる。
【0031】
欠失変異体、挿入変異体及び置換変異体は“部位特異的変異導入”及び/又は“PCRをベースとする変異導入技術”により生成することができる。対応する方法が、例えば、Lottspeich及びZorbas 1998 36.1章に、更なる文献とともに記載されている。
ハムスターユビキチン/S27a遺伝子の5'未翻訳領域又はハムスターユビキチンS27a遺伝子のS27a部分もしくは3'-未翻訳領域からのプローブによる交差ハイブリダイゼーションにより、その他の種、好ましくは哺乳動物種の相当する相同遺伝子から好適なプロモーター配列を同定し、単離することがまた可能である。好適な技術が、例えば、Lottspeich及びZorbas 1998 23章に記載されている。遺伝子はそれらのヌクレオチド配列がそれが相同である遺伝子のヌクレオチド配列に対し少なくとも70%、好ましくは少なくとも80%、好ましくは少なくとも90%、更に好ましくは少なくとも95%、最も好ましくは少なくとも97%の一致を示す場合に本発明の目的において“相同”である。
上記手段を使用して、異種遺伝子産物の発現に高度に有益である最適化発現カセットを得ることが可能である。それ故、一つ以上のこのような手段により得られた発現カセットは本発明の更なる対象である。
【0032】
本発明の発現ベクターの調製
本発明の発現ベクターは理論上、例えば、Sambrookら(1989)により記載されたような、当業界で知られている通常の方法により調製することができる。また、Sambrookはベクターの機能性成分、例えば、好適なプロモーター(ハムスターユビキチン/S27aプロモーターに加えて)、エンハンサー、終結シグナル及びポリアデニル化シグナル、抗生物質耐性遺伝子、選抜可能なマーカー、複製開始点並びにスプライシングシグナルを記載している。それらを生成するのに通常のクローニングベクター、例えば、プラスミド、バクテリオファージ、ファージミド、コスミド又はウイルスベクター、例えば、バキュロウイルス、レトロウイルス、アデノウイルス、アデノ関連ウイルス及び単純ヘルペスウイルスだけでなく、人工染色体/ミニ染色体を使用してもよい。真核細胞発現ベクターは典型的にはまた原核生物配列、例えば、複製起点及び細菌中のベクターの複製及び選抜を可能にする抗生物質耐性遺伝子を含む。ポリヌクレオチド配列の導入のための多重クローニング部位を含む幾つかの真核細胞発現ベクターが知られており、幾つかが種々の会社、例えば、ストラタジーン(ラ・ジョラ、CA、USA);インビトロジェン(カールスバッド、CA、USA);プロメガ(マジソン、WI、USA)又はBDバイオサイエンシズ・クロンテク(パロ・アルト、CA、USA)から商業的に入手できる。
【0033】
異種プロモーター、対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子そして必要により蛍光タンパク質をコードする遺伝子、付加的な調節要素、例えば、内部リボソームエントリー部位(IRES)、エンハンサー又はポリアデニル化シグナルは、当業者に良く知られている様式で発現ベクターに導入される。本発明の発現ベクターは、最小限でも、異種プロモーター、対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子を含む。発現ベクターはまた蛍光タンパク質をコードする遺伝子を含むことが好ましい。本発明によれば、ユビキチン/S27aプロモーターを異種プロモーターとして使用することが特に好ましい。異種プロモーター、好ましくはユビキチン/S27aプロモーター、対象とする遺伝子及び蛍光タンパク質をコードする遺伝子が一緒に機能可能に連結され、又は機能可能に連結され、ネオマイシンホスホトランスフェラーゼ遺伝子が同じ転写単位又は別々の転写単位中に位置される発現ベクターが特に好ましい。
本記載の範囲内で、“機能可能な連結”又は“機能可能に連結された”という用語は2以上の核酸配列又は部分配列がそれらの意図された機能を奏し得るように位置されている2以上の核酸配列又は部分配列を表す。例えば、プロモーター/エンハンサーはそれがシス位置で連結された遺伝子配列の転写を調節又は調節することができる場合にコーディング遺伝子配列に機能的に連結されている。必ずではないが、一般に、機能的に連結されたDNA配列は一緒に接近しており、そして二つのコーディング遺伝子配列が連結される場合又は分泌シグナル配列が連結される場合には、同じリーディングフレーム中にある。機能可能に連結されたプロモーターは一般にコーディング遺伝子配列の上流に配置されるが、それは必ずしも近くにある必要はない。エンハンサーは、それらが遺伝子配列の転写を補助する限り、いずれかの近くにある必要はない。この目的のために、それらは遺伝子配列の上流及び下流の両方に、おそらくそれから若干の距離にあってもよい。ポリアデニル化部位は転写がコーディング配列を介してポリアデニル化シグナルに進行するような様式でそれが遺伝子配列の3'末端に位置される場合に遺伝子配列に機能可能に連結される。連結は通常の組換え方法に従って、例えば、PCR技術、好適な制限切断部位におけるライゲーション又はスプライシングにより起こり得る。好適な制限切断部位が利用できない場合、合成オリゴヌクレオチドリンカー又はアダプターがそれ自体知られている様式で使用されてもよい。本発明によれば、好ましくは、機能可能な連結はイントロン配列を介して起こらない。
記載された実施態様の一つにおいて、異種プロモーター、好ましくはユビキチン/S27aプロモーター、対象遺伝子及び蛍光タンパク質をコードする遺伝子が一緒に機能可能に連結される。これは、例えば、対象遺伝子及び蛍光タンパク質をコードする遺伝子の両方が同じ異種プロモーターから出発して発現されることを意味する。
【0034】
特に好ましい実施態様において、機能可能な連結はIRES要素を介して起こり、その結果、両方の遺伝子から2シストロン性mRNAが合成される。本発明の発現ベクターは一以上のプロモーターに機能的に作用するエンハンサー要素を更に含んでもよい。異種プロモーター、好ましくはユビキチン/S27aプロモーター又はその改変形態がエンハンサー要素、例えば、SV40エンハンサー又はCMVエンハンサー要素に連結される発現ベクターが特に好ましい。
基本的には、発現ベクター内の遺伝子の発現は一以上の転写単位から出発して起こってもよい。転写単位という用語は転写すべき一以上の遺伝子を含む領域と定義される。転写単位内の遺伝子はこのような単位内の全ての遺伝子が同じプロモーター又はプロモーター/エンハンサーの転写制御下にあるような様式で互いに機能し得る形で連結される。遺伝子のこの転写連結の結果として、一種より多いタンパク質又は産物が転写単位から転写でき、こうして発現し得る。夫々の転写単位はその中に含まれる遺伝子配列の転写及び翻訳に必要である調節要素を含む。夫々の転写単位は同じ又は異なる調節要素を含見える。IRES要素又はイントロンが転写単位内の遺伝子の機能可能な連結に使用されてもよい。
発現ベクターは対象遺伝子、改変NPT遺伝子及び必要により蛍光タンパク質をコードする遺伝子を発現するための単一転写単位を含んでもよい。また、これらの遺伝子はまた二以上の転写単位中に配置されてもよい。転写単位内の遺伝子の種々の組み合わせが可能である。本発明の別の実施態様において、一つ、二つ又はそれ以上の転写単位からなる一より多い発現ベクターが同時トランスフェクション又はあらゆる所望の順序の連続トランスフェクションにより宿主細胞に挿入されてもよい。転写単位の適切な発現が確実にされることを条件として、夫々のベクターについての調節要素及び遺伝子のあらゆる組み合わせが選択し得る。必要であれば、その他の調節要素及び遺伝子、例えば、対象とする付加的な遺伝子又は選抜可能なマーカーが発現ベクターに配置されてもよい。
【0035】
それ故、対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼをコードする遺伝子を含む本発明の発現ベクターは両方の遺伝子を一つの転写単位中に又は二つの別々の転写単位中に含んでもよい。夫々の転写単位は一以上の遺伝子産物を転写し、発現し得る。両方の遺伝子が一つの転写単位中に含まれる場合、それらは同じプロモーター又はプロモーター/エンハンサーの制御下にあり、IRES要素が全ての成分の機能可能な連結を確実にするのに使用されることが好ましい。改変ネオマイシンホスホトランスフェラーゼをコードする遺伝子及び対象遺伝子が二つの別々の転写単位中に含まれる場合、それらは同じ又は異なるプロモーター/エンハンサーの制御下にあってもよい。しかしながら、改変NPT遺伝子については、より弱い異種プロモーター、例えば、SV40初期プロモーターが使用されることが好ましく、エンハンサーが使用されないことが好ましい。二つの別々の転写単位を有する発現ベクターが本発明の範囲内で好ましい。一つの(2シストロンの)転写単位は対象遺伝子、および、場合により蛍光タンパク質をコードする遺伝子、を含み、一方、別の転写単位は改変NPT遺伝子を含む。夫々の転写単位はポリAシグナル、好ましくはBGHポリA又はSV40ポリAをコードする配列により3'末端で特定されていることが好ましい。
また、本発明によれば、対象遺伝子に代えて、制限エンドヌクレアーゼの認識配列を介した対象遺伝子のクローニングを可能にするマルチクローニング部位のみを有する発現ベクターも好ましい。全ての種類の制限エンドヌクレアーゼだけでなく、関連制限エンドヌクレアーゼのための多くの認識配列が従来技術により知られている。認識配列として少なくとも6のヌクレオチドからなる配列が使用されることが好ましい。好適な認識配列のリストが、例えば、Sambrookら, 1989に見られる。
【0036】
宿主細胞
本発明の発現ベクターによるトランスフェクションのために、真核生物宿主細胞、好ましくは哺乳動物細胞、更に特別にはげっ歯類細胞、例えば、マウス細胞株、ラット細胞株及びハムスター細胞株が使用される。本発明の発現ベクターによる相当する細胞の成功裏のトランスフェクションは形質転換され、遺伝子改変された、組換え細胞又はトランスジェニック細胞をもたらし、これらもまた本発明の主題である。
本発明の目的に好ましい宿主細胞はハムスター細胞、例えば、BHK21細胞、BHK TK- CHO細胞、CHO-K1細胞、CHO-DUKX細胞、CHO-DUKX B1細胞及びCHO-DG44細胞又はこれらの細胞株の誘導体/子孫である。CHO-DG44細胞、CHO-DUKX細胞、CHO-K1細胞及びBHK21細胞、特にCHO-DG44細胞及びCHO-DUKX細胞が特に好ましい。また、マウスからのミエローマ細胞、好ましくはNS0細胞及びSp2/0細胞及びこれらの細胞株の誘導体/子孫も好適である。
本発明に従って使用し得るハムスター細胞及びマウス細胞の例が下記の表1に示される。しかしながら、これらの細胞の誘導体及び子孫、ヒト、マウス、ラット、サル、げっ歯類の細胞株(これらに限定されない)を含む他の哺乳動物細胞、又は酵母細胞、昆虫細胞及び植物細胞を含む(これらに限定されない)真核細胞を、生物医薬タンパク質の生成のための宿主細胞として使用することもできる。
【0037】
表1. ハムスター生産細胞株及びマウス生産細胞株
【0038】
本発明のポリヌクレオチド又は発現ベクターの一つによる真核生物宿主細胞のトランスフェクションは通常の方法(Sambrookら, 1989; Ausubelら, 1994)により行なわれる。トランスフェクションの好適な方法として、例えば、リポソーム媒介トランスフェクション、リン酸カルシウム共沈、エレクトロポレーション、ポリカチオン(例えば、DEAEデキストラン)媒介トランスフェクション、プロトプラスト融合、マイクロインジェクション及びウイルス感染が挙げられる。本発明によれば、安定的トランスフェクションが行なわれることが好ましく、この場合、構築物が宿主細胞のゲノム又は人工染色体/ミニ染色体に組み込まれ、又は宿主細胞中に安定な様式でエピソームにより含まれる。最適トランスフェクション頻度及び当該宿主中の異種遺伝子の発現を与えるトランスフェクション方法が好ましい。定義によれば、宿主細胞に挿入されるいかなる配列又はいかなる遺伝子も宿主細胞に関して“異種配列”又は“異種遺伝子”と称される。これはたとえ導入すべき配列又は導入すべき遺伝子が宿主細胞の内在性配列又は内在性遺伝子と同じであるとしても適用される。例えば、ハムスター宿主細胞に導入されたハムスターアクチン遺伝子はこの定義により異種遺伝子である。
【0039】
本発明によれば、本発明の発現ベクターの一つでトランスフェクションされた組換え哺乳動物細胞、好ましくはげっ歯類細胞、最も好ましくはハムスター細胞、例えば、CHO細胞又はBHK細胞が好ましい。
ヘテロマータンパク質、例えば、モノクローナル抗体(mAb)の組換え生成において、好適な宿主細胞のトランスフェクションは理論的に二つの異なる方法により行ない得る。この種のmAbは幾つかのサブユニット、重鎖及び軽鎖を含む。これらのサブユニットをコードする遺伝子は単一プラスミド上の独立又はマルチシストロンの転写単位中に収容されてもよく、これにより宿主細胞がその後にトランスフェクションされる。これは宿主細胞のゲノムへの組込み後に遺伝子の化学量論的提示を確保することを意図している。しかしながら、独立の転写単位の場合、異なるタンパク質をコードするmRNAが同じ安定性並びに転写効率及び翻訳効率を示すことがここで確実にされる必要がある。第二の場合、遺伝子の発現は単一プロモーターによりマルチシストロン転写単位内で起こり、単一の転写産物が生成される。IRES要素を使用することにより、遺伝子の高度に有効な内部翻訳開始が第二シストロン及びその後のシストロン中で得られる。しかしながら、これらのシストロンに関する発現率は第一シストロンの発現率よりも低い(所謂“キャップ”依存性前開始複合体による第1シストロンの翻訳開始は、IRES依存性翻訳開始よりもずっと有効である)。シストロンの真に等モルの発現を得るために、例えば、付加的なシストロン間の要素が導入されてもよく、これがIRES要素と連係して一様な発現率を確実にする(WO 94/05785)。
【0040】
幾つかの異種タンパク質を同時に生成する別の可能な方法(これが本発明によれば好ましい)は同時トランスフェクションであり、この場合、遺伝子が異なる発現ベクターに別々に組み込まれる。これは互いの遺伝子及び遺伝子産物の或る比率が調節でき、それによりmRNA安定性並びに転写及び翻訳の効率の差を調整するという利点を有する。加えて、発現ベクターがそれらの小さいサイズのためにより安定であり、クローニング中及びトランスフェクション中の両方で取り扱い易い。
それ故、本発明の一つの特別な実施態様において、宿主細胞が対象とする一以上のその他のタンパク質をコードする遺伝子を有する一以上のベクターで更にトランスフェクションされ、好ましくは同時トランスフェクションされる。同時トランスフェクションに使用されるその他の一以上のベクターは、例えば、同じプロモーター/エンハンサー組み合わせの制御下で対象とするその他の一以上のタンパク質及び少なくとも一つの選抜可能なマーカー、例えば、ジヒドロ葉酸還元酵素をコードする。
本発明によれば、無血清条件下、場合により動物タンパク質/ペプチドを含まない培地中で、宿主細胞が樹立され、適合され、培養されることが好ましい。商業的に得られる培地の例として、ハムF12(シグマ、ダイゼンホッフェン、DE)、RPMI-1640(シグマ)、ダルベッコ改良イーグル培地(DMEM;シグマ)、最小必須培地(MEM;シグマ)、イスコフ改良ダルベッコ培地(IMDM;シグマ)、CD-CHO(インビトロゲン、カールスバッド、CA、USA)、CHO-S-SFMII(インビトロゲン)、無血清CHO培地(シグマ)及び無タンパク質CHO培地(シグマ)が挙げられる。これらの培地の夫々は、場合により種々の化合物、例えば、ホルモン及び/又はその他の成長因子(例えば、インスリン、トランスフェリン、表皮成長因子、インスリン様成長因子)、塩(例えば、塩化ナトリウム、カルシウム、マグネシウム、リン酸塩)、緩衝剤(例えば、HEPES)、ヌクレオシド(例えば、アデノシン、チミジン)、グルタミン、グルコース又はその他の等価な栄養素、抗生物質及び/又は微量要素を添加してもよい。無血清培地が本発明によれば好ましいが、宿主細胞はまた好適な量の血清と混合された培地を使用して培養され、続いてタンパク質が生成されてもよい。一以上の選抜可能なマーカー遺伝子を発現する遺伝子改変細胞を選抜するために、一以上の選択薬剤が培地に添加される。
【0041】
“選択薬剤”という用語は選抜可能なマーカー遺伝子に関する欠損を有する宿主細胞の増殖又は生存に影響する物質を表す。本発明の範囲内で、ジェネティシン(G418)が改変ネオマイシンホスホトランスフェラーゼ遺伝子を有する異種宿主細胞の選抜のための培地添加剤として使用されることが好ましい。培地1ml当り100〜800μgのG418濃度が使用されることが好ましく、培地1ml当り300〜400μgが最も好ましい。宿主細胞が幾つかの発現ベクターでトランスフェクションされる場合、例えば、対象とする幾つかの遺伝子が宿主細胞に別々に導入される場合、それらは一般に異なる選抜可能なマーカー遺伝子を有する。
選抜可能なマーカー遺伝子は培地への相当する選択薬剤の添加によりこの遺伝子を含む細胞の特異的な選抜を可能にする遺伝子である。例示として、抗生物質耐性遺伝子を陽性の選抜可能なマーカーとして使用することができる。この遺伝子で形質転換された細胞のみが相当する抗生物質の存在下で増殖することができ、従って、選抜される。一方、形質転換されなかった細胞は、これらの選抜条件下で増殖又は生存することができない。陽性、陰性及び2機能性の選抜可能マーカーがある。陽性の選抜可能マーカーは耐性を選択薬剤に与えることにより、又は宿主細胞中の代謝もしくは異化代謝欠陥を相殺することにより形質転換細胞の選抜ひいては濃縮を可能にする。対照的に、選抜可能マーカーの遺伝子を受けた細胞は陰性の選抜可能マーカーにより選択的に排除し得る。これの例は単純ヘルペスウイルスのチミジンキナーゼ遺伝子であり、アシクロビル又はガンシクロビルの同時添加による細胞中のその発現はその排除をもたらす。本発明に使用される選抜可能マーカー(増幅可能な選抜可能マーカーを含む)として、遺伝子改変された変異体及び変異体、フラグメント、機能性均等物、誘導体、同族体及びその他のタンパク質又はペプチドとの融合が挙げられるが、選抜可能マーカーがその選抜特性を保持することを条件とする。このような誘導体は選択的であると考えられる領域又はドメイン中のアミノ酸配列にかなりの相同性を示す。2機能性(陽性/陰性)マーカーを含む多数の選抜可能なマーカー遺伝子が文献に記載されている(例えば、WO 92/08796及びWO 94/28143を参照のこと)。真核生物細胞中で通常使用される選抜可能なマーカーの例として、アミノグリコシドホスホトランスフェラーゼ(APH)、ハイグロマイシンホスホトランスフェラーゼ(HYG)、ジヒドロ葉酸還元酵素(DHFR)、チミジンキナーゼ(TK)、グルタミンシンセターゼ、アスパラギンシンセターゼの遺伝子並びにネオマイシン(G418)、プロマイシン、ヒスチジノールD、ベロマイシン、フレオマイシン及びゼオシンに対する耐性を与える遺伝子が挙げられる。
また、形質転換細胞を蛍光活性化セルソーティング(FACS)により選抜することが可能である。このために、細菌のβ-ガラクトシダーゼ、細胞表面マーカー又は蛍光タンパク質を形質転換細胞の選抜に使用することができる(例えば、緑色蛍光タンパク質(GFP)並びにオワンクラゲ及びウミシイタケ又はその他の種からのその変異体;赤色の蛍光タンパク質及びその他の色で蛍光を発するタンパク質並びに非バイオルミネセント生物、例えば、ディスコゾーマ種(Discosoma sp.)、ミナミウメボシイソギンチャク種(Anemonia sp.)、ツツウミヅタ種(Clavularia sp.)、スナギンチャク種(Zoanthus sp.)からのそれらの変異体)。
【0042】
遺伝子発現及び高生産性宿主細胞の選抜
遺伝子発現という用語は宿主細胞中の異種遺伝子配列の転写及び/翻訳に関する。発現率は一般に宿主細胞中に存在する相当するmRNAの量に基づいて、又は対象遺伝子によりコードされる生成される遺伝子産物の量に基づいて測定し得る。選択されたヌクレオチド配列の転写により生成されるmRNAの量は、例えば、ノーザンブロットハイブリダイゼーション、リボヌクレアーゼ-RNA-保護、細胞RNAのin situハイブリダイゼーション又はPCR方法(Sambrookら, 1989; Ausubelら, 1994)により測定し得る。選択されたヌクレオチド配列によりコードされるタンパク質はまた種々の方法、例えば、ELISA、ウェスタンブロット、ラジオイムノアッセイ、免疫沈殿、タンパク質の生物学的活性の検出又はタンパク質の免疫染色、続いてFACS分析(Sambrookら, 1989; Ausubelら, 1994)により測定し得る。
【0043】
“高発現レベル(又は率)、高発現、増大された発現又は高生産性”という用語は宿主細胞に導入される異種配列、例えば、治療タンパク質をコードする遺伝子の長く持続する、充分に高い発現又は合成を表す。本発明の細胞が、ここに記載された本発明の方法の一つにより培養され、この細胞が遺伝子増幅無しに、1日当りおよそ0.5pg(0.5pg/細胞/日)より少なくとも多い所望の遺伝子産物を生じる場合に、増大され、もしくは高い発現又は高発現レベルもしくは率或いは高生産性が存在する。また、本発明の細胞が、事前の遺伝子増幅無しで1日当りおよそ1.0pg(1.0pg/細胞/日)より少なくとも多い所望の遺伝子産物を生じる場合に、増大され、もしくは高い発現又は高発現もしくは率或いは高生産性が存在する。特に本発明の細胞が事前の遺伝子増幅無しに1日当りおよそ1.5pg(1.5pg/細胞/日)より少なくとも多い所望の遺伝子産物を生じる場合に、増大され、もしくは高い発現又は高発現レベルもしくは率或いは高生産性が存在する。特に本発明の細胞が事前の遺伝子増幅無しに1日当りおよそ2.0pg(2.0pg/細胞/日)より少なくとも多い所望の遺伝子産物を生じる場合に、増大され、もしくは高い発現又は高発現レベルもしくは率或いは高生産性が存在する。特に、本発明の細胞が事前の遺伝子増幅無しで1日当りおよそ3.0pg(3.0pg/細胞/日)より少なくとも多い所望の遺伝子産物を生じる場合に,増大され、もしくは高い発現又は特に高い発現レベルもしくは率或いは特に高い生産性が存在する。簡単な遺伝子増幅工程により、例えば、以下に記載されるようなDHFR/MTX増幅系を使用して、生産性が少なくとも2〜10倍に増大でき、従って、“高発現”、“増大された発現”又は“高生産性”という用語は、遺伝子増幅工程にかけられた細胞に関しては、この細胞が少なくとも1日当りおよそ5pg(5pg/細胞/日)より多く、好ましくは少なくともおよそ10pg/細胞/日より多く、更に好ましくは少なくともおよそ15pg/細胞/日より多く、更に好ましくは少なくともおよそ20pg/細胞/日より多く、又は少なくともおよそ30pg/細胞/日より多い所望の遺伝子産物を生じる場合に使用される。
【0044】
高発現もしくは増大された発現、高生産性又は高発現レベルもしくは率は本発明の発現ベクターの一つを使用すること、および本発明の方法の一つの使用、の両方により得られる。
例えば、対象遺伝子及び改変NPT遺伝子の同時発現により、異種遺伝子を高度に発現する細胞を選抜し、同定することが可能である。wtNPTと較べて、改変NPTは対象とする異種遺伝子の高発現とともに安定にトランスフェクションされた宿主細胞の一層有効な選抜を可能にする。
従って、本発明は(i) 哺乳動物細胞のプールを少なくとも一つの対象遺伝子及び野生型ネオマイシンホスホトランスフェラーゼと比較してその活性のわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5〜26%を有する改変ネオマイシンホスホトランスフェラーゼの一つの遺伝子でトランスフェクションすること、(ii)前記対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で前記細胞を培養し、(iii) 前記哺乳動物細胞を少なくとも一種の選択薬剤、好ましくはG418(これは哺乳動物細胞の増殖に選択的に作用し、かつ改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える)の存在下で培養すること、および(iv)一以上の対象タンパク質を前記哺乳動物細胞又は培養上清から得ることを特徴とする、組換え哺乳動物細胞中の少なくとも一つの対象遺伝子の発現方法に関する。本発明の発現ベクターでトランスフェクションされた組換え哺乳動物細胞が使用されることが好ましい。
また、本発明は(i) 哺乳動物細胞のプールを少なくとも一つの対象遺伝子及び野生型ネオマイシンホスホトランスフェラーゼとの比較によりその活性のわずかに1〜80%、好ましくはわずかに1〜60%、更に好ましくはわずかに1.5〜30%、最も好ましくはわずかに1.5〜26%を有する改変ネオマイシンホスホトランスフェラーゼの遺伝子でトランスフェクションすること、(ii)前記哺乳動物細胞を前記対象遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、(iii) 前記哺乳動物細胞を少なくとも一種の選択薬剤、好ましくはG418(これは哺乳動物細胞の増殖に選択的に作用し、かつ改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える)の存在下で培養することを特徴とする、少なくとも一つの対象遺伝子を発現する組換え哺乳動物細胞の選抜方法に関する。
【0045】
少なくとも一つの対象遺伝子を発現させる方法及び対応する対象遺伝子を発現する組換え細胞を選抜する方法が特に好ましいのは、本出願に更に詳しく記載されるNPT遺伝子が使用される場合、特にアミノ酸位置182においてグリシン又はアスパラギン酸、アミノ酸位置91においてアラニン、アミノ酸位置198においてグリシン、アミノ酸位置227においてグリシン又はバリン、アミノ酸位置261においてグリシン又はアスパラギン或いはアミノ酸位置240においてイソロイシンをコードする、野生型遺伝子と比較して改変されたNPT遺伝子が使用される場合である。特にAsp227Val変異体、Asp227Gly変異体、Asp261Gly変異体、Asp261Asn変異体、Phe240Ile変異体又はTrp91Ala変異体を使用する場合が好ましい。一般に、この特許明細書に記載された本発明の全ての改変ネオマイシンホスホトランスフェラーゼ遺伝子がこのような方法に好適である。好ましいネオマイシンホスホトランスフェラーゼ遺伝子については、改変ネオマイシンホスホトランスフェラーゼ遺伝子に関する節を参照のこと。
対象とする遺伝子及び改変NPT遺伝子を発現する細胞の選抜は、例えば、G418を選択薬剤として添加することにより行なわれる。しかしながら、その他のアミノグリコシド抗生物質、例えば、ネオマイシン又はカナマイシンを使用することも可能である。本発明の細胞は培地1ml当り200〜800μgのG418中で培養され、選抜されることが好ましい。培地1ml当り300〜700μgのG418を添加することが特に好ましいと判明した。培地1ml当りおよそ400μgのG418の添加が最も好ましい実施態様である。このような方法を使用して、特に高い発現率を有する組換え細胞を選抜することが可能である。wtNPTの使用との比較により、選抜可能なマーカーとしての培地1ml当り400μgのG418による選抜後に、細胞はGlu182Gly変異体、Glu182Asp変異体及びVal198Gly変異体の場合に1.4-2.4倍、Asp227Gly変異体の場合に1.6-4.1倍、Asp227Ala変異体又はTrp91Ala変異体の場合に2.2又は4倍、Phe240Ile変異体又はAsp261Asn変異体の場合に5.7又は7.3倍更にはAsp261Gly変異体又はAsp227Val変異体の場合に9.3又は14.6倍に増大された生産性を示した。種々の改変NPT遺伝子に関する比生産能が図6に示される。
【0046】
相当する方法は、付加的な選抜可能なマーカーとして、一種以上の蛍光タンパク質(例えば、GFP)又は細胞表面マーカーを含む組換え宿主細胞のFACS補助選抜と組み合わされてもよい。増大された発現を得るその他の方法、及び異なる方法の組み合わせがまた使用されてもよく、例えば、(人工)転写因子の使用、内在性又は異種遺伝子配列をアップレギュレートするための天然又は合成の薬剤による細胞の処理、mRNA又はタンパク質の安定性(半減期)を改良すること、mRNA翻訳の開始を改良すること、エピソームプラスミドの使用により遺伝子量を増大すること(複製起点としてウイルス配列、例えば、SV40、ポリオーマ、アデノウイルス、EBV又はBPVの使用に基づく)、増幅促進配列(Hemannら, 1994)の使用又はDNAコンカテマー(Monacoら, 1996)をベースとするin vitro増幅系に基づいている。
対象遺伝子及び蛍光タンパク質をコードする遺伝子の共役した転写は選抜可能なマーカーとしての改変NPT遺伝子の使用と連係して特に有効と判明した。得られる2シストロン性mRNAは対象タンパク質/産物及び蛍光タンパク質の両方を発現する。対象とするタンパク質及び蛍光タンパク質の発現のこのカップリングに基づいて、本発明によれば発現された蛍光タンパク質により、例えば、蛍光活性化セルソーティング装置(FACS)を使用してソーティングすることにより高生産性組換え宿主細胞を同定し、単離することが容易に可能である。
高い生命力及び所望の遺伝子産物の増大された発現率を示す組換え宿主細胞の選抜は多段階の工程である。本発明の発現ベクターでトランスフェクションされた(又は場合により例えば別のベクターで同時トランスフェクションされてもよい)宿主細胞は、例えば、選択薬剤、例えば、培地1ml当り100μg、200μg、400μg、600μg、800μg又はそれ以上のG418の濃度のG418の存在下の培養により、改変NPTを発現する細胞の選抜を可能にする条件下で培養される。次いで相当する細胞が蛍光タンパク質の最高発現率を示す細胞/細胞集団を同定し、ソーティングするために、対象遺伝子に結合された、蛍光タンパク質をコードする遺伝子の発現について少なくとも調べられる。蛍光タンパク質の最高発現率を有する細胞の10-20%に属する細胞のみがソーティングされ、更に培養されることが好ましい。実際には、これは蛍光細胞の最も明るい10%がソーティングされ、更に培養されることを意味する。それ故、細胞混合物の蛍光細胞の最も明るい5%、好ましくは最も明るい3%又は更には最も明るい1%がソーティングされ、複製し得る。特に好ましい実施態様において、蛍光細胞の最も明るい0.5%又は最も明るい0.1%のみがソーティングされ、複製される。
【0047】
選抜工程は細胞プールについて、又は前ソーティングされた細胞プール/細胞クローンを使用して行うことができる。一つ以上、好ましくは二つ以上、特に三つ以上のソーティング工程が行なわれてもよく、個々のソーティング工程の間で細胞が特定の時間、例えば、プールの場合にはおよそ2週にわたって培養され、複製されてもよい。図11及び12は、例えば、変異体Asp227Glyについて遺伝子増幅工程を用いて、また用いないでFACSに基づくソーティング後の比生産能を示す。
従って、本発明は(i) 組換え哺乳動物細胞を本発明の発現ベクターでトランスフェクションすること、(ii)トランスフェクションされた細胞を一以上の対象遺伝子、蛍光タンパク質をコードする遺伝子及び改変ネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能にする条件下で培養すること、(iii) 前記哺乳動物細胞を前記哺乳動物細胞の増殖に選択的に作用し、かつ前記改変ネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、および、(iv)蛍光遺伝子の特に高い発現を示す前記哺乳動物細胞をフローサイトメトリー分析によりソーティングすること、を特徴とする、少なくとも一つの対象異種遺伝子を発現する組換え哺乳動物細胞を得、選抜する方法に関する。所望により、工程(ii)〜(iv)が工程(iv)で得られた細胞を用いて1回又は数回繰り返されてもよい。
ソーティングされた哺乳動物細胞が、付加的な遺伝子増幅工程を用いないで、1日当りかつ細胞当り0.5pg(0.5pg/細胞/日)より多く、好ましくは1pg/細胞/日より大きく、更に好ましくは2pg/細胞/日より大きく、更に好ましくは3pg/細胞/日より大きく、更に好ましくは4pg/細胞/日より大きく、例えば、5、6、8、9、10pg/細胞/日より大きく、15、20、25pg/細胞/日より大きい等の所望の一以上の遺伝子産物の平均比生産能を有することを特徴とする方法が好ましい。上記のように、これらの細胞の生産性は、例えば、DHFR/MTX系を使用する簡単な遺伝子増幅工程により少なくとも2〜10倍に増大し得る。これは、例えば、NTP変異体Asp227Glyを使用する選抜について図12に示されている。比生産能は20〜25pg/細胞/日であった。
【0048】
また、本発明によれば、好適にソーティングされた細胞が複製され、対象とするコードされた遺伝子産物を調製するのに使用される方法も好ましい。これのために、選抜された高生産性細胞が無血清培地中で、好ましくは対象遺伝子の発現を可能にする条件下で懸濁培養される。対象タンパク質/産物は細胞培地から分泌された遺伝子産物として得られることが好ましい。しかしながら、タンパク質が分泌シグナルなしで発現される場合、遺伝子産物はまた細胞溶解産物から単離することもできる。その他の組換えタンパク質及び宿主細胞タンパク質を実質的に含まない純粋な均一産物を得るために、通常の精製操作が行なわれる。最初に、細胞及び細胞破砕物が培地又は溶解産物から除去される。次いで所望の遺伝子産物から、例えば、イムノアフィニティーカラム及びイオン交換カラムによる分別、エタノール沈殿、逆相HPLC又はセファデックス、シリカ又はDEAEの如き陽イオン交換樹脂によるクロマトグラフィーにより、夾雑可溶性タンパク質、ポリペプチド及び核酸が除かれる。組換え宿主細胞により発現された異種タンパク質の精製をもたらす方法が当業者に知られており、文献、例えば、Harrisら, 1995及びScopes 1988に記載されている。
【0049】
増幅可能な選抜可能マーカー遺伝子
加えて、本発明の細胞はまた必要によりそれらが増幅可能な選抜可能遺伝子の増幅をもたらす選択薬剤の存在下で培養される、一以上の遺伝子増幅工程にかけられてもよい。この工程は蛍光タンパク質を発現し、好ましくはFACS(好ましくはここに記載された方法の一つにおいて)により1回又は数回予備ソーティングされた細胞及び未だソーティングされていない細胞のどちらで行うことも出来る。
前提条件は宿主細胞が増幅可能な選抜可能マーカーをコードする遺伝子で更にトランスフェクションされることである。増幅可能な選抜可能マーカーをコードする遺伝子は本発明の発現ベクターの一つに存在するか、又は別のベクターにより宿主細胞に導入されることが推考される。
増幅可能な選抜可能マーカー遺伝子は或る培養条件下の真核生物細胞の増殖に必要とされる酵素を通常コードする。例えば、増幅可能な選抜可能なマーカー遺伝子はジヒドロ葉酸還元酵素(DHFR)をコードしてもよい。この場合、その遺伝子はそれでトランスフェクションされた宿主細胞が選択薬剤メトトレキセート(MTX)の存在下で培養される場合に増幅される。
下記の表2はその他の増幅可能な選抜可能なマーカー遺伝子及び本発明に従って使用し得る関連する選択薬剤の例を示し、これらはKaufman著Methods in Enzymology, 185:537-566 (1990)による総覧に記載されている。
【0050】
表2. 増幅可能な選抜可能なマーカー遺伝子
【0051】
表2続き
【0052】
本発明によれば、使用される増幅可能な選抜可能マーカー遺伝子はDHFRの機能を有するポリペプチド、例えば、DHFR又は蛍光タンパク質及びDHFRからの融合タンパク質をコードする遺伝子であることが好ましい。DHFRはプリンの生合成に必要である。DHFR遺伝子を欠いている細胞はプリン欠損培地中で増殖し得ない。それ故、DHFR遺伝子は無プリン培地中で培養される細胞中で選抜し、遺伝子を増幅するのに有益な選抜可能なマーカーである。DHFR遺伝子と連係して使用される選択培地はメトトレキセート(MTX)である。
それ故、本発明は下記の工程:(i) 対象タンパク質/産物、改変ネオマイシンホスホトランスフェラーゼ及びDHFRを少なくともコードする遺伝子による宿主細胞のトランスフェクション工程、(ii)種々の遺伝子の発現を可能にする条件下の前記細胞の培養工程、及び(iii)少なくとも増幅可能な選抜可能なマーカー遺伝子の増幅を可能にする選択薬剤、例えば、メトトレキセートの存在下で前記細胞を培養することによる同時組込み遺伝子の増幅工程、を含む、組換え哺乳動物細胞を調製および選抜する方法を含む。トランスフェクションされた細胞は血清の不在下で増大する濃度のMTXを添加してヒポキサンチン/チミジンを含まない培地中で培養されることが好ましい。最初の増幅工程におけるMTXの濃度は少なくとも5nMであることが好ましい。しかしながら、MTXの濃度はまた少なくとも20nM又は100nMであってもよく、段階的に1μMまで増大し得る。個々の場合には、1μMより大きい濃度、例えば、2μMが使用されてもよい。
【0053】
相当する細胞が更に蛍光タンパク質の遺伝子で形質転換される場合、これらの細胞は蛍光活性化細胞ソーティング装置(FACS)を使用して同定され、ソーティングされ、次いで遺伝子増幅工程において少なくとも20nM、好ましくは50nM又は100nMのMTXの存在下で培養することができる。このようにして、生産性を細胞当り1日当り実質的に20pg以上、好ましくは21以上、22以上、23以上、24以上、25以上等、30以上、35以上、40以上等の遺伝子産物まで増大させることが可能である。宿主細胞は少なくとも対象遺伝子及び増幅可能な選抜可能マーカー遺伝子のコピー数を増大するために一以上の遺伝子増幅工程にかけられてもよい。本発明によれば、得られる高生産性は、ネオマイシン、カナマイシン及びG418のようなアミノグリコシド抗生物質に対するネオマイシンホスホトランスフェラーゼ媒介耐性による有効な予備選抜と関連している。それ故、必要とされる遺伝子増幅工程の数を減少し、例えば、単一遺伝子増幅のみを行なうことが可能である。
更なる実施態様において、こうしてまた、本発明は少なくとも一つの対象異種遺伝子を発現する組換え哺乳動物細胞を得る方法及び選抜する方法に関するものであり、(i) 組換え哺乳動物細胞を本発明の発現ベクター及び増幅可能な選抜可能マーカー遺伝子でトランスフェクションすること、(ii)前記哺乳動物細胞を一以上の対象遺伝子、改変ネオマイシンホスホトランスフェラーゼ遺伝子及び蛍光タンパク質をコードする遺伝子の発現を可能にする条件下で培養すること、(iii) 前記哺乳動物細胞を前記哺乳動物細胞の増殖に選択的に作用し、かつネオマイシンホスホトランスフェラーゼ遺伝子を発現するこれらの細胞の増殖に優先性を与える少なくとも一種の選択薬剤の存在下で培養すること、(iv)蛍光タンパク質の高発現を示す前記哺乳動物細胞をフローサイトメトリー分析によりソーティングすること、(v) ソーティングされた前記細胞を前記増幅可能な選抜可能なマーカー遺伝子が発現される条件下で培養すること、および(vi)選択薬剤を前記増幅可能な選抜可能なマーカー遺伝子の増幅をもたらす培地に添加すること、を特徴とする。
【0054】
本発明において記載された改変ネオマイシンホスホトランスフェラーゼ遺伝子が使用される相当する方法が特に好ましい。また、単一の増幅工程が行なわれる方法が好ましい。また、細胞当り1日当り20pg以上、好ましくは21以上、22以上、23以上、25以上等、30以上、35以上、40以上等の所望の一以上の遺伝子産物の平均比生産能を示す組換え哺乳動物細胞をもたらす相当する方法が好ましい。
哺乳動物細胞、好ましくはマウスミエローマ細胞及びハムスター細胞が、増幅可能な選抜可能マーカーとしてのDHFRの使用に好ましい宿主細胞である。細胞株CHO-DUKX(ATCC CRL-9096)及びCHO-GD44(Urlaubら, 1983)が特に好ましい。何とならば、変異の結果としてそれら自体のDHFR活性を有しないからである。自身が内因性DHFR活性を有するその他のタイプの細胞中でDHFR誘導増幅を同様に使用することができるように、トランスフェクション過程でメトトレキセートに対する低下された感受性を有するタンパク質をコードする変異DHFR遺伝子(Simonsonら, 1983; Wiglerら, 1980; Haberら, 1982)を使用することが可能である。
DHFRマーカーはDHFR陰性細胞、例えば、CHO-DG44又はCHO-DUKXを使用する場合に選抜及びその後の増幅に特に適している。何とならば、これらの細胞は内因性DHFRを発現せず、それ故、無プリン培地中で増殖しないからである。従って、DHFR遺伝子が支配的な選抜可能マーカーとしてここで使用することができ、形質転換細胞がヒポキサンチン/チミジンを含まない培地中で選抜される。
本発明が幾つかの非限定実施例を参照して以下に更に充分に記載される。
【0055】
略号
Ala (=A) アラニン
AP: アルカリ性ホスファターゼ
Asn (=N): アスパラギン
Asp (=D): アスパラギン酸
bp: 塩基対
BSA: ウシ血清アルブミン
CHO: チャイニーズハムスター卵巣
dhfr: ジヒドロ葉酸還元酵素
DMSO: ジメチルスルホキシド
ELISA: 酵素結合免疫吸着検定法
FACS: 蛍光活性化セルソーター
FITC: フルオレセイン-イソチオシアネート
GFP: 緑色蛍光タンパク質
Glu (=E): グルタミン酸
Gly (=G): グリシン
HBSS: ハンクス平衡塩溶液
HT: ヒポキサンチン/チミジン
Ile (=I): イソロイシン
IRES: 内部リボソームエントリー部位
kb: キロベース
mAb: モノクローナル抗体
MCP-1: 単球走化性タンパク質1
MTX: メトトレキセート
MW: 平均値
NPT: ネオマイシンホスホトランスフェラーゼ
PCR: ポリメラーゼ連鎖反応
PBS: リン酸緩衝生理食塩水
Phe (=F): フェニルアラニン
Trp (=W): トリプトファン
Val (=V): バリン
WT: 野生型
【0056】
方法
1. 細胞培養及びトランスフェクション
細胞CHO-DG44/dhfr-/-(Urlaubら, 1983)を細胞培養フラスコ中で37℃で湿った大気、5%のCO2中でヒポキサンチン及びチミジン(インビトロゲンGmbH、カールスルーヘ、DE)を添加した無血清CHO-S-SFMII培地中で懸濁細胞として恒常的に培養した。細胞カウント及び生命力をCASY1セルカウンター(シェルフェ・システム、DE)又はトリプタンブルー染色により測定し、次いで1-3x105/mlの濃度で播き、2-3日毎に繰り返した。
リポフェクタミン・プラス試薬(インビトロゲンGmbH)をCHO-DG44のトランスフェクションに使用した。夫々のトランスフェクション混合物について、合計1μgのプラスミド-DNA、4μLのリポフェクタミン及び6μLのプラス試薬を製造業者の指示に従って一緒に混合し、200μLの体積でHT添加CHO-S-SFMII培地0.8ml中の6x105の指数増殖しているCHO-DG44細胞に添加した。細胞インキュベーター中で37℃で3時間のインキュベーション後に、HT添加CHO-S-SFMII培地2mlを添加した。NPTをベースとする選抜のために、細胞をトランスフェクションの2日後にG418(インビトロゲン)を含むHT添加CHO-S-SFMII培地に移し、3〜4日毎に培地を交換した。原則として、400μg/mlのG418を選抜のために添加し、また幾つかの実験シリーズでは、濃度を200μg/mlに低下し、又は500、600もしくは800μg/mlに上昇させた。同時トランスフェクション(一つの発現ベクターはDHFRを含み、別の発現ベクターはネオマイシンホスホトランスフェラーゼ選抜可能なマーカーを含む)の場合のDHFR及びNPTをベースとする選抜において、細胞をトランスフェクションの2日後にヒポキサンチン及びチミジンを添加しないCHO-S-SFMII培地に移し、またG418(インビトロゲン)を400μg/mlの濃度で培地に添加した。
組込み異種遺伝子のDHFRをベースとする遺伝子増幅をHTを含まないCHO-S-SFMII培地への5-2000nMの濃度の選択薬剤MTX(シグマ、ダイゼンホッフェン、DE)の添加により得ることができる。
【0057】
2. 発現ベクター
発現を分析するために、pAD-CMVベクター(Wernerら, 1998)をベースとし、CMVエンハンサー/ハムスターユビキチン/S27aプロモーター(WO 97/15664)の組み合わせによる異種遺伝子の構成的発現を媒介する真核細胞発現ベクターを使用した。基本ベクターpBIDは増幅可能な選抜可能なマーカーとして作用するdhfr-ミニジーン(例えば、EP-0-393-438を参照のこと)を含むが、ベクターpBINではそのdhfr-ミニジーンがネオマイシンホスホトランスフェラーゼ耐性遺伝子により置換されていた(図1)。この目的のために、SV40初期プロモーター及びTK-ポリアデニル化シグナルを含む、選抜可能なマーカーネオマイシンホスホトランスフェラーゼを、1640 bp Bsu36Iフラグメントとして商業的プラスミドpBK-CMV(ストラタゲン、ラ・ジョラ、CA、USA)から単離した。そのフラグメントの末端をクレノー-DNA-ポリメラーゼでフィルイン(fill in)する反応後に、フラグメントをベクターpBIDの3750bp Bsu36I/StuIフラグメントとつなぎ、これをまたクレノー-DNA-ポリメラーゼで処理した。
2シストロン性基本ベクターpBIDG(図1)中、IRES-GFP遺伝子領域はベクターpIRES2-EGFP(クロンテク、パロ・アルト、CA、USA)から単離され、ベクターpBID中でCMVエンハンサー/プロモーターの制御下におかれ、その結果、プロモーター領域とIRES-要素の間の多重クローニング部位が保持されている。下記の手順を使用した。プラスミドpIRES2-EGFPが鋳型として作用するPCR変異導入では、一方で、IRES配列内のHindIII切断部位AAGCTTを変異導入プライマーの使用により配列ATGCTTに変換し、除去した。他方で、IRES配列の5'末端に相補性を有するプライマーによりXbaI切断部位を挿入し、又はGFP配列の3'末端に相補性を有するプライマーによりSpeI切断部位を導入した。得られたPCRフラグメント(これは完全IRES及びGFP配列を含む)を、XbaI及びSpeIで消化し、ベクターpBIDのマルチクローニング部位の3'末端の単一XbaI切断部位にクローン化した。同じ方法で、ベクターpIRES2-EGFPからのIRES-GFP遺伝子領域をベクターpBIN中のCMVエンハンサー/ハムスターユビキチン/S27aプロモーターの制御下においた。これにより2シストロン性基本ベクターpBINGが生された(図1)。
ヒトMCP-1 cDNA(Yoshimuraら, 1989)を0.3kb HindIII/EcoRIフラグメントとしてベクターpBINの相当する切断部位にクローン化して、ベクターpBIN-MCP1を生じた(図2A)。
モノクローナルヒト化IgG2抗体を発現するために、重鎖をベクターpBID又はBamHI及びHindIIIで消化されたpBIDGに1.5kb BamHI/HindIIIフラグメントとしてクローン化して、ベクターpBID-HC又はpBIDG-HCを得た(図2B)。一方で、軽鎖をベクターpBIN又はpBINGの相当する切断部位に0.7kb BamHI/HindIIIフラグメントとしてクローン化して、ベクターpBIN-LC又はpBING-LCを生じた(図2B)。
【0058】
3. FACS
フローサイトメトリー分析及びソーティングをクールター・エピックス・アルタ装置で行なった。このFACSは488nmの励起波長を有するヘリウム-アルゴンレーザーに適合している。蛍光タンパク質に適した波長で蛍光強度を吸収し、取り付けられたソフトウェア、クールター・エクスポ32によって処理される。ソーティングは8000-10000イベント/秒の速度で通常行なう。懸濁された細胞を遠心分離し(180xgで5分)、HBSS中1-1.5x107/mlの細胞濃度に調節することができる。次いで細胞をそれらの蛍光タンパク質シグナルに従ってソーティングすることができる。細胞を培地を予め含む試験管に取り、次いで遠心分離し、ソーティングされた細胞の数に応じて、好適な培養容器に接種し、又はマイクロタイタープレートに直接入れる。
4. ELISA
安定にトランスフェクションされたCHO-DG44細胞の上清中のMCP-1力価を製造業者の指示(BDバイオサイエンシズ・ファーミンゲン、ハイデルベルグ、DE)に従ってOptEIAヒトMCP-1セットキットを使用してELISAにより定量した。
安定にトランスフェクションされたCHO-DG44細胞の上清中のIgG2 mAbを標準的な操作(Current Protocols in Molecular Biology, Ausubelら, 1994, 最新)に従って、一方で、ヤギ抗ヒトIgG Fcフラグメント(ダイアノバ、ハンブルグ、DE)また他方でAP-結合ヤギ抗ヒトカッパ軽鎖抗体(シグマ)を使用してELISAにより定量した。精製IgG2抗体を標準として使用した。生産性(pg/細胞/日)を式pg/((Ct-Co)t/ln(Ct-Co))(式中、Co及びCtは夫々播種時及び回収時の細胞カウントであり、かつtは培養時間である)により計算した。
【0059】
5. NPT酵素活性を測定するためのドットアッセイ
細胞抽出物をDuchら, 1990の方法に従って調製するために、6x106の細胞をPBSで2回洗浄し、次いで抽出緩衝液(0.135M Tris-HCl pH 6.8、20%のグリセロール、4mMジチオスレイトール)600μL中で再懸濁させた。ドライアイス又は水の浴中の凍結及び解凍の4サイクル後に、細胞破砕物を遠心分離により除去し、上清をその後の酵素アッセイに使用した。細胞抽出物中のタンパク質濃度をBIO-RADタンパク質アッセイ(Bio-RadラボラトリイズGmbH、ミュンヘン、DE)を使用して、標準タンパク質としてBSAを用いてブラッドフォードアッセイにより測定した(Current Protocols in Molecular Biology, Ausubelら, 1994, 最新)。NPT酵素活性を測定するために、ドットアッセイをPlattら, 1987のプロトコルに基づいて行なった。これのために、タンパク質5μg、2.5μg及び1.25μgを抽出緩衝液で20μLの最終体積に調節し、トランスフェクションしなかったCHO-DG44細胞からの細胞抽出物で5μgの合計タンパク質含量まで足した。アッセイ緩衝液(67mM Tris-HCl pH 7.1、42mM MgCl2、400mM NH4Cl)200μL±40μg/mlのG418±5μCiの〔γ-33P〕-ATP/ml(NEN)の添加後に、抽出物を27℃で135分間インキュベートした。次いで抽出物を96ウェル真空マニホルド(シュライヘル&シュル、ダッセル、DE)中でワットマン3MM紙の一層、P81ホスホセルロース膜(ワットマン・ラボラトリイ部門、マイドストーン、英国)及びニトロセルロース膜(シュライヘル&シュル)のサンドイッチで濾過した。タンパク質キナーゼによりリン酸化されたタンパク質及び非リン酸化タンパク質がニトロセルロースに結合し、一方、リン酸化G418がニトロセルロースを通過し、ホスホセルロースに結合する。脱イオン水で3回洗浄した後、膜を装置から除去し、再度水で洗浄し、次いで空気乾燥させた。ホスホイメージャー(モレキュラー・ダイナミクス、クレフェルド、DE)を使用して、放射能シグナルを定量した。
【0060】
6. ノーザンブロット分析
全RNAを製造業者の指示(インビトロゲンGmbH、カルスルーヘ、DE)に従ってTRIZOL試薬を用いて細胞から単離し、ゲル電気泳動によるRNA30μgの分離及びハイボンドN+ナイロン膜(アマシャム・バイオサイエンシズ、フライブルグ、DE)への転写をグリオキサール/DMSO変性RNAに関する標準操作(Current Protocols in Molecular Biology, Ausubelら, 1994, 最新)に従って行なった。ジーンイメージズCDP-スター検出キット(アマシャム・バイオサイエンシズ)によるその後の非放射性ハイブリダイゼーションに使用したプローブはジーンイメージズ・ランダム・プライム標識キット(アマシャム・バイオサイエンシズ、フライブルグ、DE)で製造業者の指示に従ってFITC-dUTP標識された、PCR産物(これはNPT遺伝子のコーディング領域を含む)であった。
【0061】
7. ドットブロット分析
DNA単離キットを製造業者の指示(細胞及び組織用のDNA単離キット;ロシェ・ダイアグノスチクスGmbH、マンハイム、DE)に従って使用して、ゲノムDNAを細胞から単離した。種々の量のDNA(10μg、5μg、2.5μg、1.25μg、0.63μg及び0.32μg)をアルカリ性緩衝液中で96ウェル真空マニホルド(シュライヘル&シュル、ダッセル、DE)を使用して通常の方法(Ausubelら, 1994)によりハイボンドN+ナイロン膜(アマシャム・バイオサイエンシズ、フライブルグ、DE)で濾過した。トランスフェクションしなかったCHO-DG44細胞を陰性対照として使用した。プラスミドpBIN-LCを標準として使用した(320pg、160pg、80pg、40pg、20pg、10pg、5pg、2.5pg)。ジーンイメージズCDP-スター検出キット(アマシャム・バイオサイエンシズ)によるその後の非放射性ハイブリダイゼーションに使用したプローブは、製造業者の指示に従ってジーンイメージズランダムプライム標識キット(アマシャム・バイオサイエンシズ、フライブルグ、DE)によりFITC-dUTP標識した、NPT遺伝子のコーディング領域を含むPCR産物とした。イメージマスターVDS-CL(アマシャム・バイオサイエンシズ)を使用して、ケミルミネセンスシグナルを定量した。次いでタイトレーションしたプラスミドDNAのシグナル強さから得られた標準シリーズを使用して、細胞中のnpt遺伝子のコピー数を測定した。アボガドロ定数を使用してプラスミド分子の数を計算し、CHO細胞のDNA含量は約5pgであるとした。
【実施例1】
【0062】
実施例1.ネオマイシンホスホトランスフェラーゼの変異導入
NPT変異体Glu182Gly(配列番号3)、Trp91Ala(配列番号5)、Val198Gly(配列番号7)、Asp227Ala(配列番号9)、Asp227Val(配列番号11)、Asp261Gly(配列番号13)、Asp261Asn(配列番号15)、Phe240Ile(配列番号17)、Glu182Asp(配列番号19)、Asp227Gly(配列番号21)、Asp190Gly(配列番号23)及びAsp208Gly(配列番号25)を調製するのに必要とされる野生型NPT-遺伝子中の塩基置換を、PCRにより変異導入プライマーを使用して行なった(図3)。ベクターpBIN(図1)又はpBK-CMV(ストラタゲン、ラ・ジョラ、USA)をPCR変異導入のための鋳型として使用した。最初に変異体の5'又は3'部分を別々のPCR操作で調製した。変異体Glu182Gly、Glu182Asp、Trp91Ala、Asp190Gly、Val198Gly、Asp208Gly及びAsp227Glyを調製するために、Neofor5(配列番号27)と関連する変異導入リバース(rev)プライマー又はNeorev5(配列番号28)と関連する変異導入フォワード(for)プライマーからなるプライマーの組み合わせを増幅に使用した:プライマーの組合せは、
−NPT変異体Glu182Gly(配列番号3)の場合には、Neofor5(配列番号27)とE182Grev(配列番号32)又はNeorev5(配列番号28)とE182Gfor(配列番号31)から;
−NPT変異体Glu182Asp(配列番号19)の場合には、Neofor5(配列番号27)とE182Drev(配列番号48)又はNeorev5(配列番号28)とE182Dfor(配列番号47)から;
−NPT変異体Trp91Ala(配列番号5)の場合には、Neofor5(配列番号27)とW91Arev(配列番号34)又はNeorev5(配列番号28)とW91Afor(配列番号33)から;
−NPT変異体Val198Gly(配列番号7)の場合には、Neofor5(配列番号27)とV198Grev(配列番号36)又はNeorev5(配列番号28)とV198Gfor(配列番号35)から;
−NPT変異体Asp190Gly(配列番号23)の場合には、Neofor5(配列番号27)とD190Grev(配列番号50)又はNeorev5(配列番号28)とD190Gfor(配列番号49)から;
−NPT変異体Asp208Gly(配列番号25)の場合には、Neofor5(配列番号27)とD208Grev(配列番号52)又はNeorev5(配列番号28)とD208Gfor(配列番号51)から;
−NPT変異体Asp227Gly(配列番号21)の場合には、Neofor5(配列番号27)とD227Grev(配列番号54)又はNeorev5(配列番号28)とD227Gfor(配列番号52)からなる。
変異体Asp227Ala、Asp227Val、Asp261Gly、Asp261Asn及びPhe240Ileを調製するために、Neofor2(配列番号29)と関連する変異導入リバース(rev)プライマー又はIC49(配列番号30)と関連する変異導入フォワード(for)プライマーからなるプライマーの組み合わせを増幅に使用した:プライマーの組合せは、
−NPT変異体Asp227Ala(配列番号9)の場合には、Neofor2(配列番号29)とD227Arev(配列番号38)又はIC49(配列番号30)とD227Afor(配列番号37)から;
−NPT変異体Asp227Val(配列番号11)の場合には、Neofor2(配列番号29)とD227Vrev(配列番号40)又はIC49(配列番号30)とD227Vfor(配列番号39)から;
−NPT変異体Asp261Gly(配列番号13)の場合には、Neofor2(配列番号29)とD261Grev(配列番号42)又はIC49(配列番号30)とD261Gfor(配列番号41)から;
−NPT変異体Asp261Asn(配列番号15)の場合には、Neofor2(配列番号29)とD261Nrev(配列番号44)又はIC49(配列番号30)とD261Nfor(配列番号43)から;
−NPT変異体Phe240Ile(配列番号17)の場合には、Neofor2(配列番号29)とF240Irev(配列番号46)又はIC49(配列番号30)とF240Ifor(配列番号45)からなる。
【0063】
次いで当該変異体の5'部分のコーディング鎖及び3'部分の相補鎖を変異導入プライマー配列により形成された重なり領域中のハイブリダイゼーションにより組み合わせ、一本鎖領域をフィルインし、全生成物をプライマーNeofor5(配列番号27)とNeorev5(配列番号28)又はNeofor2(配列番号29)とIC49(配列番号30)によるPCRで再度増幅した。これらのPCR産物をStuI/RsrII(変異体Glu182Gly、Trp91Ala及びVal198GlyのNeofor5/Neorev5 PCR産物)、StuI/BstBI(変異体Glu182Asp、Asp190Gly、Asp208Gly及びAsp227GlyのNeofor5/Neorev5 PCR産物)又はDraIII/RsrII(変異体Asp227Ala、Asp227Val、Asp261Gly、Asp261Asn及びPhe240IleのNeofor2/IC49 PCR産物)で消化した。次いでベクターpBIN-LC(図2B)又はpBK-CMV(ストラタジーン、ラ・ジョラ、USA)中で、野生型NPT配列の部分をStuI/RsrII消化、DraIII/RsrII消化又はStuI/BstBI消化により除去し、PCR産物の相当するフラグメントにより置換した。相補鎖及びコーディング鎖の両方のシーケンシングにより、種々の変異体中の所望の塩基置換を確認し、残りのDNA配列が野生型NPT配列に一致することを確かめた。このようにして、発現ベクターpBIN1-LC、pBIN2-LC、pBIN3-LC、pBIN4-LC、pBIN5-LC、pBIN6-LC、pBIN7-LC及びpBIN8-LCを生成した。これらはNPT変異体Glu182Gly、Trp91Ala、Val198Gly、Asp227Ala、Asp227Val、Asp261Gly、Asp261Asn又はPhe240Ileを含む(図2B)。
【0064】
残りのNPT変異体を改変pBK-CMVからの1640 bp Bsu36Iフラグメントとして単離し、フラグメント末端をクレノーDNAポリメラーゼでフィルインし、ベクターpBIDの3750 bp Bsu36I/StuIフラグメントとつなぎ、これをまたクレノーDNAポリメラーゼで処理した。このようにして、発現ベクターpKS-N5、pKS-N6、pKS-N7及びpKS-N8を生成し、これらは夫々NPT変異体Glu182Asp、Asp190Gly、Asp208Gly及びAsp227Glyを含んでいた。次いでヒトMCP-1 cDNAを0.3 kb HindIII/EcoRIフラグメント(図2A)としてこれらの発現ベクターにクローン化し、又はヒト化IgG2抗体のH鎖を0.7 kb HindIII/BamHIフラグメント(図2B)としてこれらの発現ベクターにクローン化した。
ネオマイシンホスホトランスフェラーゼに挿入された変異は一方で保存ドメイン、例えば、モチーフ1、2及び3(Shawら, 1993)に隣接するより保存的な(Val198Gly、Phe240Ile)又はあまり保存的でない(Trp91Ala、Glu182Gly、Glu182Asp、Asp227Ala、Asp227Val、Asp227Gly)アミノ酸の置換である。他方で、変異は保存モチーフ1(Asp190Gly)、2(Asp208Gly)又は3(Asp261Gly、Asp261Asn)内に位置され、保存アミノ酸に関する。
【実施例2】
【0065】
実施例2.安定的にトランスフェクションされたMCP-1発現細胞の選抜に対するNPT変異の影響
MCPをCHO細胞中の一本鎖タンパク質の発現の例として使用した。これのために、CHO-DG44をpKS-N5-MCP1、pKS-N6-MCP1、pKS-N7-MCP1、pKS-N8-MCP1又はpBIN-MCP1でトランスフェクションした(図2A)。二つの二重調製を行なった。トランスフェクションの2日後に、細胞を96ウェル-プレートに接種し(2000細胞/ウェル)、HT添加CHO-S-SFMII培地中で400μg/mlのG418で選抜した。pBIN-MCP1でトランスフェクションされた細胞の場合、選抜をまた800μg/mlのG418で並行して行なった。得られた細胞集団を24ウェルプレートを経て6ウェルプレートに継代した。選抜期中でさえも、種々のトランスフェクション混合物間で相違を検出することができた。選抜がNPT野生型遺伝子(配列番号1)で行なわれた細胞集団とは対照的に、変異されたNPTでトランスフェクションされたこれらの細胞集団では、より少ない細胞しかG418による初期の選抜に耐えなかった。それ故、これらの細胞集団を約4日後まで24ウェルプレートに移すことができなかった。また、pKS-N6-MCP1及びpKS-N7-MCP1でトランスフェクションされた混合物では、安定にトランスフェクションされた細胞はG418の400μg/mlの濃度で全く選抜することができなかった。おそらく、変異Asp190Gly及びAsp208Glyを有するNPT変異体では酵素機能がひどく損なわれ、充分なG418分子が不活化されることができず安定にトランスフェクションされた細胞の増殖ができないのであろう。実際、G418濃度を200μg/mlに低下させた場合、いくらかの細胞が最初の選抜期に耐えたが、それらは全て増殖及び生命力がひどく損なわれており、変異体Asp208Glyの場合のいくつかの例外は別として、エクスパンジョンが可能ではなかった。
変異体Glu182Asp及びAsp227Gly又はNPT野生型でトランスフェクションされた細胞から、18プールを6ウェル中で4回の継代にわたって培養し(混合物1及び2の夫々9プール)、生成されたMCP-1の濃度を細胞培養上清中でELISAにより測定した。NPT変異体が選抜可能なマーカーとして使用された細胞プールは、400又は更には800μg/mlのG418によりNPT野生型を用いて選抜が行なわれた細胞プールよりも平均で50%−57%(Glu182Asp変異体)又は57%−65%(Asp227Gly変異体)高い生産性を示した(図5)。このように、NPT変異体を選抜可能マーカーとして使用することにより、トランスフェクションされた細胞集団中の高生産性細胞の比率が実際に増大し得た。
【実施例3】
【0066】
実施例3.安定にトランスフェクションされたmAb発現細胞の選抜に関するNPT変異の影響 同時トランスフェクションでは、CHO-DG44細胞を最初にプラスミド組み合わせpBIDG-HC/pBIN-LC(NPT野生型)、pBIDG-HC/pKS-N5-LC(Glu182Asp NPT変異体)又はpBIDG-HC/pKS-N8-LC(Asp227Gly NPT変異体)でトランスフェクションした(図2B)。使用したベクター配置では、ヒト化IgG抗体の二つのタンパク質鎖はそれら自体のベクター(これは別々の転写単位中のDHFR又はネオマイシン選抜可能なマーカーを更にコードする)により夫々発現される。産物遺伝子の発現はCMV-エンハンサー/ハムスターユビキチン/S27aプロモーターにより媒介される。しかしながら、例えば、CMVエンハンサー/プロモーター、SV40エンハンサー/ハムスターユビキチン/S27aプロモーター又はその他のプロモーター組み合わせを使用して、同等なデータを得ることも出来る。
全てにおいて、プラスミド組み合わせ当り6プールを使用して、トランスフェクションの4シリーズを夫々の場合に行なった。選抜がNPT野生型遺伝子を用いて行なわれた細胞集団とは対照的に、変異されたNPTでトランスフェクションされたこれらの細胞集団では、より少ない細胞がG418による初期の選抜に耐えた。付加的な400μg/mlのG418によるHTを含まないCHO-S-SFMII培地中のトランスフェクションされた細胞集団の2〜3週の選抜後に、細胞培養上清中の抗体力価を幾つかの実験(6−8)にわたってELISAにより測定した。選抜可能なマーカーとしてのNPT野生型遺伝子の使用との比較により、Glu182Asp変異体で選抜された細胞は夫々平均で86%及び77%の生産性及び力価の増大を示し、Asp227Gly変異体で選抜された細胞は更に夫々126%及び107%の生産性及び力価の増大を示した。このように、低下された酵素活性を有するNPT変異体を使用することにより、2倍程度まで高い基本的生産性を有する細胞を選択的に濃縮することが可能であった。
【0067】
別のトランスフェクションシリーズでは、選抜に関するG418の異なる濃度の影響を試験した。400、500及び600μg/mlのG418をトランスフェクションされた細胞プール、夫々の場合に3プールの選抜に使用した。高濃度では、有意に少ない細胞が細胞集団(選抜がNPT野生型遺伝子を用いて行なわれた)中で選抜に耐え、その効果はAsp227Gly変異体で最大であった。しかしながら、得られたトランスフェクションされた細胞集団は増殖及び生命力の低下を示さなかった。しかしながら、トランスフェクションに使用したプラスミド組み合わせ内で得られた生産性と力価の間で有意差は検出できなかった。しかし、ここでさえも、NPT変異体により選抜された細胞は、平均して最高の生産性を再度有しており、Asp227Gly変異体によりもたらされる生産性はNPT野生型のそれよりも4倍高く、続いてGlu182Asp変異体が2.4倍高い生産性を有していた(図6A)。
次いで同時トランスフェクションでは、CHO-DG44細胞をプラスミドの組み合わせpBIDG-HC/pBIN-LC(NPT野生型)、pBIDG-HC/pBIN1-LC(Glu182Gly NPT変異体)、pBIDG-HC/pBIN2-LC(Trp91Ala NPT変異体)、pBIDG-HC/pBIN3-LC(Val198Gly NPT変異体)、pBIDG-HC/pBIN4-LC(Asp227Ala)、pBIDG-HC/pBIN5-LC (Asp227Val NPT変異体)、pBIDG-HC/pBIN6-LC (Asp261Gly NPT変異体)、pBIDG-HC/pBIN7-LC (Asp261Asn NPT変異体)又はpBIDG-HC/pBIN8-LC (Phe240Ile NPT変異体)でトランスフェクションした(図2B)。使用したベクター配置では、再度モノクローナルヒト化IgG抗体の二つのタンパク質鎖はそれ自体のベクター(これは更にまた別々の転写単位中のDHFR又はネオマイシン選抜可能なマーカーをコードする)により夫々発現される。
【0068】
夫々のプラスミド組み合わせについて、5プールをトランスフェクションした。選抜がNPT野生型遺伝子を用いて行なわれた細胞集団とは対照的に、変異されたNPTでトランスフェクションされた少ない細胞が細胞集団中でG418による初期の選抜に耐えた。400μg/mlのG418を添加したHTを含まないCHO-S-SFMII培地中のトランスフェクションされた細胞プールの2〜3週の選抜後に、細胞培養上清中の抗体力価を6回の実験にわたってELISAにより測定した。図6Bは試験中にプールから測定された力価及び生産性の平均を示す。選抜可能なマーカーとしてのNPT野生型遺伝子の使用と較べて、NPT変異体で選抜された全ての細胞プールが夫々平均で1.4−14.6倍及び1.4−10.8倍の生産性及び力価の増大を示した(図6B)。高い基本的生産性を有する細胞の最良の選択的濃縮がこうしてNPT変異体Asp227Val及びAsp261Glyで得られ、夫々14.6倍及び9.3倍の平均生産性の増大を有していた。
ベクターpBIDG-HCは別の選抜可能なマーカー、GFPを含む。GFPはIRES要素を介して重鎖に転写的に連結される。それ故、標的タンパク質の発現と選抜可能なマーカーGFPとの間に得られる相関関係はFACS分析で測定されたGFP蛍光に基づいてトランスフェクションされた細胞集団における発現のレベル及び発現レベルの分布を迅速に評価することを可能にする。G418を添加したHTを含まないCHO-S-SFMII培地中のトランスフェクションされた細胞集団の2〜3週の選抜後に、GFP蛍光をFACS分析で測定した(図7)。GFP蛍光シグナルは実際にモノクローナルIgG2抗体について得られた力価データと相関関係があった。また、NPT変異体Asp227Val、Asp261Gly、Asp161Asn及びPhe240Ileで選抜されたプールは高いGFP蛍光を有する細胞の高比率を有し、NPT変異体Trp91Ala、Asp227Ala、Asp227Gly、Gly182Asp、Glu182Gly及びVal198Glyで選抜された細胞がそれに続いた。
選択薬剤メトトレキセート(MTX)を培地に添加することにより、dhfr媒介遺伝子増幅を誘導することにより細胞の生産性を更に増大することが可能であった。こうして、例えば、100nMのMTXによる簡単な増幅工程後に、プラスミドの組み合わせpBIDG-HC/pBIN5-LC (NPT変異体Asp227Val)、pBIDG-HC/pBIN6-LC (NPT変異体Asp261Gly)、pBIDG-HC/pBIN7-LC (NPT変異体Asp261Asn)及びpBIDG-HC/pBIN8-LC (NPT変異体Phe240Ile)による同時トランスフェクションにより得られた細胞プール中の比生産能は2〜4倍に増大でき、プールに依存して、4〜14pg/細胞/日の生産性を得ることができた。図8は、例として、pBIDG-HC/pBIN5-LC (NPT変異体Asp227Val)の同時トランスフェクションにより得られた細胞プールについて、MTXの添加(100nMのMTX、続いて500nMのMTX)により得られた27pg/細胞/日への生産性の増大を示す。
【実施例4】
【0069】
実施例4.NPT酵素活性の測定及び比較
NPT変異体の酵素活性をNPT野生型のそれと比較するために、ドットアッセイをPlattら, 1987の操作に基づいて行なって細胞抽出物中のNPT活性を測定し、例としてNPT変異体Glu182Asp及びAsp227Glyについて図9Aに示した。細胞抽出物をNPT野生型遺伝子(配列番号1)又はNPT変異体Glu182Gly(配列番号3)、Glu182Asp(配列番号19)、Trp91Ala(配列番号5)、Asp190Gly(配列番号23)、Val198Gly(配列番号7)、Asp208Gly(配列番号25)、Asp227Ala(配列番号9)、Asp227Val(配列番号11)、Asp227Gly(配列番号21)、Asp261Gly(配列番号13)、Asp261Asn(配列番号15)又はPhe240Ile(配列番号17)でトランスフェクションされ選抜された二つの異なるmAb発現細胞プールから調製した。トランスフェクションしなかったCHO-DG44細胞からの細胞抽出物を陰性対照として使用した。
NPT変異体の酵素活性はNPT野生型と較べて有意に低下した。平均で、NPT変異体は野生型酵素活性のわずかに1.5%〜62%を有し、3.1%及び1.5%を有するNPT変異体Asp261Gly及びAsp261Asnは最低の残留活性を有し、また61.9%及び53.2%を有するNPT変異体Val198Gly及びTrp91Alaは最高の残留活性を有する(図9B)。ホスホセルロース上で得られたシグナルはNPT酵素活性により生じたG418のリン酸化に特異的であった。NPT基質G418をアッセイ緩衝液に添加しない場合、ホスホセルロース上で活性は検出できなかった。ニトロセルロース膜上で得られたシグナル(これらは細胞抽出物中のタンパク質キナーゼによりリン酸化されたタンパク質から生じる)を、適用した同じ量のサンプルについての内部対照として使用した。
NPT野生型と較べてNPT変異体の低下した酵素活性は低下した遺伝子発現のためではあり得なかった。逆に、全RNAに関するノーザンブロット分析により、NPT野生型でトランスフェクションされ、高いNPT酵素活性を示す細胞プールが、NPT変異体でトランスフェクションされた細胞プールよりも少ないRNAを発現することが示された(図10)。唯一の例外はNPT変異体Glu182Glyでトランスフェクションされ、匹敵する量のNPT-mRNAを発現する細胞であった。これらのトランスフェクションされた細胞集団からのゲノムDNAについて行なわれたドットブロット分析では、NPT変異体の高発現が遺伝子量効果及び/又はより転写的に活性なゲノム領域への外来性DNAの組込みにより得られたことが示された(図10)。例えば、NPT変異体Trp91Alaで選抜された細胞では、遺伝子量効果がプール1で優性であり、一方、プール2では、組込み効果が支配的である。このようにして、低下した酵素活性を有するマーカーが選抜に使用されたトランスフェクション細胞は、同じ選択圧のもとで、選択薬剤に対する低下した耐性を相殺するのに充分なマーカータンパク質を合成することができる。
【実施例5】
【0070】
実施例5.GFPをベースとするFACSソーティングによるmAbの高発現を有する細胞の単離
同時トランスフェクションでは、CHO-DG44細胞をモノクローナルヒト化IgG2抗体をコードする、プラスミドの組み合わせpBID-HC及びpBING-LCでトランスフェクションした(図2B)。使用されたベクター配置において、抗体の二つのタンパク質鎖はそれら自体のベクター(これはまた別々の転写単位中でDHFR又は改変ネオマイシンホスホトランスフェラーゼ選抜可能なマーカー(Asp227Gly変異体;配列番号21)を更にコードする)により夫々発現される。加えて、ベクターpBING-LCは別の選抜可能なマーカー、GFPを含む。ベクターpBID-HC/pBING-LCによるCHO-DG44の同時トランスフェクションにおける、IRES要素によるGFP及び軽鎖の発現の転写連鎖の結果として、高いGFP含量を有する細胞を連続FACSソーティングにより純粋に選択することにより、モノクローナル抗体の高発現を有する細胞を単離することが短時間内で可能であった。全てにおいて、8の別々の細胞プールをトランスフェクションし、これらから400μg/mlのG418を添加したHTを含まないCHO-S-SFMII培地中の最初の2〜3週の選抜後に、安定にトランスフェクションされた細胞集団を得た。全ての8プールの力価及び生産性を幾つかの実験(7−8)にわたってELISAにより測定した。平均で、力価は約1.4mg/Lであり、生産性は1日当り約1.3pg/細胞であった。その後の連続のFACSをベースとするソーティングのために、プール5及び8を選抜し、プール5は最高の生産性を有し、またプール8は全てのプールの平均に相当する生産性を有していた。夫々の工程において、最高のGFP蛍光を有する細胞の5%をFACSによりソーティングし、更にプール中で培養した。このソーティングを夫々のソーティング間で約2週の培養期間で全部で6回まで行なった。
【0071】
二つのタンパク質鎖はそれら自体のベクターによりそれぞれ発現され、GFPとの転写カップリングの結果として、GFPをベースとするFACSソーティングの際に軽鎖の発現について選抜することしかできなかったが、驚くことに、mAb生産性とGFP蛍光の間に良い相関関係があることがわかった(図13)。生産性はFACSをベースとするソーティング単独により9.5pg/細胞/日に増大させることができた(図11)。また、ハムスタープロモーターをCMV-エンハンサーに代えてSV40-エンハンサーに機能可能に連結した場合に、匹敵するデータを得た。その後の単一のMTX増幅工程により、最初のソーティング工程のプール5及び8から出発して、100nMのMTXを選択培地に添加することにより、プールの生産性を平均20pg/細胞/日以上に増大させることが可能であった(図12)。蛍光タンパク質の高発現レベルは細胞増殖及び生命力にどんな負の効果をも有していなかった。遺伝子増幅中に、細胞の増殖性はまた、特にMTXが高濃度で添加される場合に、MTXの添加によりひどく負に影響される。しかしながら、予備ソーティングされた細胞プールは100nMのMTXの実に高い初期用量に応答してかなり強い特性を示した。それらは極めて良好に選抜期に耐え、即ち、わずかに2週後に、高生命力及び良好な増殖速度を有する細胞集団を得た。
加えて、高生産性細胞を選抜するための開発時間が通常の段階的遺伝子増幅戦略(これは一般にMTXの増大する添加とともに4増幅段階を含む)と較べて、約6週に短縮された。これは低下された酵素活性を有する改変NPT選抜可能なマーカーを使用することにより得られた、対象遺伝子の増大された発現を有するトランスフェクションされた細胞の濃縮、続いてその後の遺伝子増幅工程とともにGFPをベースとするFACSソーティングの組み合わされた使用により得られた。
【0072】
参考文献
Adam, M.A. et al., J Virol 1991, 65, 4985 - 4990
Altschul, S.F. et al., Nucleic Acids Res. 1997, 25, 3389 - 3402
Altschul, S.F. et al., J Mol Biol 1990, 215, 403 - 410
Ausubel, F.M. et al., Current Protocols in molecular biology. New York: Greene Publishing Associates and Wiley-Interscience. 1994 (updated)
Blazques, J. et al., Molecular Microbiology 1991, 5(6), 1511 1518
Bennett, R.P. et al., BioTechniques 1998, 24, 478 - 482
Burk, D.L. et al., Biochemistry 2001, 40 (30), 8756 - 8764
Chalfie, M. et al., Science 1994, 263, 802 - 805
Chamov, S.M. et al., Antibody Fusion Proteins, Wiley-Liss Inc., 1999
Davies, M.V. et al., J Virol 1992, 66, 1924 - 1932
Faisst, S. et al., Nucleic Acids Research 1992, 20, 3 26
Gish, W. & States, D.J., Nature Genet. 1993, 3, 266 - 272
Gossen, M. et al., Curr Opi Biotech 1994, 5, 516 - 520
Duch, M. et al., Gene 1990, 95, 285 - 288
Hanson, K.D. et al., Mol Cell Biol 1995, 15(1), 45 51
Haber, D.A. et al., Somatic Cell Genetics 1982, 8, 499 - 508
Harris et al., Protein Purification: A Practical Approach, Pickwood and Hames, eds., IRL Press, 1995
Hemann, C. et al., DNA Cell Biol 1994, 13 (4), 437 - 445
Hon, W. et al., Cell 1997, 89, 887 - 895
Hu, S. et al., Cancer Res. 1996, 56 (13), 3055 - 3061
Huston, C. et al., Proc Natl Acad Sci USA 1988, 85 (16), 5879 - 5883
Jang, S.K. et al., J Virol 1989, 63, 1651 1660
Kaufman, R.J., Methods in Enzymology 1990, 185, 537 - 566
Kocabiyik, S. et al., Biochem Biophys Res Commun 1992, 185(3), 925 931
Kortt, A.A. et al., Protein Engineering 1997, 10 (4), 423 - 433
Lottspeich F. and Zorbas H. eds., Bioanalytic, Spektrum Akad. Verl., 1998
Lovejoy, B. et al., Science 1993, 259, 1288 - 1293
Madden, T.L. et al., Meth. Enzymol. 1996, 266, 131 - 141;
Monaco, L. et al., Gene 1996, 180, 145 15
Morgan, R.A. et al., Nucleic Acids Research 1992, 20, 1293 - 1299
Mosser, D.D. et al., BioTechniques 1997, 22, 150 - 161
Ohshima, Y. et al., J Mol Biol 1987, 195, 247 - 259
Pack, P. et al., Biotechnology 1993, 11, 1271 1277
Pack, P. et al., J Mol Biol 1995, 246 (11), 28 - 34
Pelletier, J. et al., Nature 1988, 334, 320 325
Perisic, O. et al., Structure 1994, 2, 1217 - 1226
Platt, S.G. et al., Analyt Biochem 1987, 162, 529 - 535
Ramesh, N. et al., Nucleic Acids Research 1996, 24, 2697 2700
Sambrook, J., Fritsch, E.F. & Maniatis, T.,Molecular Cloning: A Laboratory Manual Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, 1989
Scopes, R., Protein Purification, Springer Verlag, 1988
Shaw, K.J. et al., Microbiological Reviews 1993, 57(1), 138 - 163
Simonson, C.C. et al., Proc Natl Acad Sci USA 1983, 80, 2495 - 2499
Sugimoto et al., Biotechnology 1994, 12, 694 - 698
Yenofsky, R.L. et al., Proc Natl Acad Sci USA 1990, 87, 3435 3439
Yoshimura, T. et al., FEBS LETTERS 1989, 244(2), 487 - 493
Urlaub, G. et al., Cell 1983, 33, 405 412
Werner, R.G. et al., Arzneim.-Forsch./Drug.Res. 1998, 48, 870 - 880
Wigler, M. et al., Proc Natl Acad Sci USA 1980, 77, 3567 - 3570
Zhang, J. & Madden, T.L. Genome Res. 1997, 7, 649 - 656;
【図面の簡単な説明】
【0073】
【図1】CHO-DG44細胞中で組換えタンパク質を発現するのに使用した基本ベクターの模式図を示す。“P/E”はCMVエンハンサーとハムスター-ユビキチン/S27aプロモーターの組み合わせであり、“P”自体はプロモーター要素を示し、かつ“T”は転写の終結シグナルであり、これは転写されたmRNAのポリアデニル化に必要とされる。夫々の転写単位内の転写開始の位置及び方向が矢印により示される。異種遺伝子のクローニングのために、制限エンドヌクレアーゼのための多重切断部位(マルチクローニング部位-MCS)を含む配列領域がプロモーター要素の後に挿入されている。増幅可能な選抜可能マーカージヒドロ葉酸還元酵素は“dhfr”と略記され、また選抜可能なマーカーネオマイシンホスホトランスフェラーゼは“npt”(npt野生型又はnpt変異体)と略記される。脳心筋炎ウイルスに由来する“IRES”要素は2シストロン性転写単位内で内部リボソームエントリー部位として作用し、続く緑色蛍光タンパク質“GFP”の翻訳を可能にする。
【図2A】一本鎖タンパク質(図2A)をコードし、CHO-DG44細胞をトランスフェクションするのに使用される真核細胞発現ベクターの図解を示す。“P/E”はCMVエンハンサーとハムスターユビキチン/S27aプロモーターの組み合わせであり、“P”それ自体はプロモーター要素であり、かつ“T”は転写されたmRNAのポリアデニル化に必要とされる転写の終結シグナルである。夫々の転写単位内の転写開始の位置及び方向が矢印により示される。増幅可能な選抜可能マーカージヒドロ葉酸還元酵素は“dhfr”と略記され、また選抜可能なマーカーネオマイシンホスホトランスフェラーゼは“npt”と略記される。NPT変異体E182G(配列番号3)、E182D(配列番号19)、W91A(配列番号5)、D190G(配列番号23)、V198G(配列番号7)、D208G(配列番号25)、D227A(配列番号9)、D227V(配列番号11)、D227G(配列番号21)、D261G(配列番号13)、D261N(配列番号15)及びF240I(配列番号17)は示された位置に改変アミノ酸(1文字コードで示される)をもたらす点変異を含む。脳心筋炎ウイルスに由来する“IRES”要素は2シストロン性転写単位内で内部リボソームエントリー部位として作用し、その後の緑色蛍光タンパク質“GFP”の翻訳を可能にする。“MCP-1”は単球走化性タンパク質1をコードし、一方、“HC”及び“LC”はヒト化モノクローナルIgG2抗体の重鎖及び軽鎖を夫々コードする。
【図2B】モノクローナル抗体のサブユニット(図2B)をコードし、CHO-DG44細胞をトランスフェクションするのに使用される真核細胞発現ベクターの図解を示す。“P/E”はCMVエンハンサーとハムスターユビキチン/S27aプロモーターの組み合わせであり、“P”それ自体はプロモーター要素であり、かつ“T”は転写されたmRNAのポリアデニル化に必要とされる転写の終結シグナルである。夫々の転写単位内の転写開始の位置及び方向が矢印により示される。増幅可能な選抜可能マーカージヒドロ葉酸還元酵素は“dhfr”と略記され、また選抜可能なマーカーネオマイシンホスホトランスフェラーゼは“npt”と略記される。NPT変異体E182G(配列番号3)、E182D(配列番号19)、W91A(配列番号5)、D190G(配列番号23)、V198G(配列番号7)、D208G(配列番号25)、D227A(配列番号9)、D227V(配列番号11)、D227G(配列番号21)、D261G(配列番号13)、D261N(配列番号15)及びF240I(配列番号17)は示された位置に改変アミノ酸(1文字コードで示される)をもたらす点変異を含む。脳心筋炎ウイルスに由来する“IRES”要素は2シストロン性転写単位内で内部リボソームエントリー部位として作用し、その後の緑色蛍光タンパク質“GFP”の翻訳を可能にする。“MCP-1”は単球走化性タンパク質1をコードし、一方、“HC”及び“LC”はヒト化モノクローナルIgG2抗体の重鎖及び軽鎖を夫々コードする。
【図3】変異導入プライマーによるPCRにより点変異が挿入されたネオマイシンホスホトランスフェラーゼ(npt)遺伝子の配列の部分を示す。大文字はnptコーディング領域のヌクレオチド配列を示し、一方、小文字は隣接非コーディングヌクレオチド配列を示す。ヌクレオチド配列(3文字コード)から予測されたアミノ酸配列をコーディングヌクレオチド配列の上に示してある。矢印は使用されたプライマーの方向、長さ及び位置を示し、実線の矢印は変異導入フォワードプライマーを示し、破線は変異導入リバースプライマーを示し、点線はnpt遺伝子又は変異部位の夫々の上流に位置されたプライマーNeofor5(配列番号27)又はNeofor2(配列番号29)を示し、また点-ダッシュ線はnpt遺伝子又は変異部位の夫々の下流に位置されたプライマーNeorev5(配列番号28)又はIC49(配列番号30)を示す。野生型配列に対して交換されたヌクレオチドが矢印の上下で強調されている。
【図4】保存ドメイン及びNPTアミノ酸配列内の挿入されたNPT変異の位置を示す。種々のアミノグリコシド改変酵素間の配列相同性に基づいて、種々の保存ドメインがNPTタンパク質配列内で同定された(灰色で示される)。酵素のC末端領域中の三つのモチーフは特別な機能を明らかに有する。モチーフ1及び2はおそらくATP触媒作用又はヌクレオチド結合における末端リン酸の触媒的転移に関係し、一方、モチーフ3はATP加水分解及び/又は酵素-アミノグリコシド複合体におけるコンホメーションの変化に機能を有すると考えられる。アミノグリコシド改変酵素の少なくとも70%中に表れるアミノ酸を太字で強調してある。下線を施したアミノ酸はそれらの類似性に基づいて同じグループに帰属され、アミノグリコシド改変酵素の少なくとも70%中に表れるものである。アステリスクでマークしたアミノ酸は変異部位の位置を示す。
【図5】安定にトランスフェクションされたMCP-1発現細胞の選抜に対するNPT変異の影響を示す。このために、CHO-DG44細胞をベクターpBIN-MCP1、pKS-N5-MCP1及びpKS-N8-MCP1(図2A)でトランスフェクションした。これらは選抜可能なマーカーとしてNPT野生型(WT)又はNPT変異体Glu182Asp及びAsp227Glyを含んでいた。安定にトランスフェクションされた細胞を選抜するために、400μg/ml又は800μg/mlのG418を選択薬剤として培地に添加した。生成された組換えタンパク質MCP-1の細胞培養上清中の濃度をELISAにより測定し、細胞当り1日当りの比生産能を計算した。バーは6ウェル皿中の6種の培養からの18プールの比生産能又は力価の平均を表す。
【図6A】安定にトランスフェクションされたmAb発現細胞の選抜に関するNPT変異の影響を調べた。このために、CHO-DG44細胞をプラスミド組み合わせpBIDG-HC/pBIN-LC(NPT野生型)、pBIDG-HC/pKS-N5-LC(NPT変異体Glu182Asp)もしくはpBIDG-HC/pKS-N8-LC(NPT変異体Asp227Gly)(図6A)(これらは選抜可能なマーカーとして使用されたNPT遺伝子(野生型又は変異体)のみを互いに異にする)でトランスフェクションした。産生された組換えモノクローナルIgG2抗体の細胞培養上清中の濃度をELISAにより測定し、細胞当り1日当りの比生産能を計算した。全てにおいて、5〜9プールを夫々のベクター組み合わせについてセットアップした。バーは75cm2のフラスコ中の6種の培養実験からの試験中の全てのプールの比生産能又は力価の平均を表す。相対力価又は相対比生産能を計算するために、NPT野生型遺伝子で選抜されたプールの平均を1とした。
【図6B】安定にトランスフェクションされたmAb発現細胞の選抜に関するNPT変異の影響を調べた。このために、CHO-DG44細胞をプラスミド組み合わせpBIDG-HC/pBIN-LC(NPT野生型)、pBIDG-HC/pBIN1-LC(NPT変異体Glu182Gly)、pBIDG-HC/pBIN2-LC(NPT変異体Trp91Ala)、pBIDG-HC/pBIN3-LC(NPT変異体Val198G)、pBIDG-HC/pBIN4-LC(NPT変異体Asp227Ala)、pBIDG-HC/pBIN5-LC(NPT変異体Asp227Val)、pBIDG-HC/pBIN6-LC(NPT変異体Asp261Gly)、pBIDG-HC/pBIN7-LC(NPT変異体Asp261Asn)もしくはpBIDG-HC/pBIN8-LC(NPT変異体Phe240Ile)(図6B)(これらは選抜可能なマーカーとして使用されたNPT遺伝子(野生型又は変異体)のみを互いに異にする)でトランスフェクションした。産生された組換えモノクローナルIgG2抗体の細胞培養上清中の濃度をELISAにより測定し、細胞当り1日当りの比生産能を計算した。全てにおいて、5〜9プールを夫々のベクター組み合わせについてセットアップした。バーは75cm2のフラスコ中の6種の培養実験からの試験中の全てのプールの比生産能又は力価の平均を表す。相対力価又は相対比生産能を計算するために、NPT野生型遺伝子で選抜されたプールの平均を1とした。
【図7】本発明のNPT変異体を選抜可能なマーカーとして使用することによる、トランスフェクションされた細胞プール中の高GFP発現を有する細胞の濃縮を示す。このために、CHO-DG44細胞をプラスミド組み合わせpBIDG-HC/pBIN-LC(NPT野生型)、pBIDG-HC/pKS-N5-LC(NPT変異体Glu182Asp)、pBIDG-HC/pKS-N8-LC(NPT変異体Asp227Gly)、pBIDG-HC/pBIN1-LC(NPT変異体Glu182Gly)、pBIDG-HC/pBIN2-LC(NPT変異体Trp91Ala)、pBIDG-HC/pBIN3-LC(NPT変異体Val198G)、pBIDG-HC/pBIN4-LC(NPT変異体Asp227Ala)、pBIDG-HC/pBIN5-LC(NPT変異体Asp227Val)、pBIDG-HC/pBIN6-LC(NPT変異体Asp261Gly)、pBIDG-HC/pBIN7-LC(NPT変異体Asp261Asn)又はpBIDG-HC/pBIN8-LC(NPT変異体Phe240Ile)(夫々の場合に5〜9プール)(これらは選抜可能なマーカーとして使用されたNPT遺伝子(野生型又は変異体)のみを互いに異にする)でトランスフェクションした。更に、pBIDGベクターはまたマーカー遺伝子としてGFPを含んでいた。G418を添加したHTを含まない培地中のトランスフェクションされた細胞プールの2〜3週の選抜後に、GFP蛍光をFACS分析により測定した。陰性対照として使用されたトランスフェクションされなかったCHO-DG44細胞を除く、夫々のグラフは同じプラスミド組み合わせでトランスフェクションされたプールからの平均GFP蛍光を表す。
【図8】ベクターの組み合わせpBIDG-HC/pBIN-LC(野生型)又はpBIDG-HC/pBIN5-LC(NPT変異体D227V)によるCHO-DG44のトランスフェクションから得られた細胞プールをその例とするdhfr媒介遺伝子増幅により得られたmAb生産性の増大を示す。G418の存在下のヒポキサンチン/チミジンを含まないCHO-S-SFMII培地中の最初の選抜後に、dhfr媒介遺伝子増幅を培地への100nM次いで500nMのMTXの添加により行なった。プールの細胞培養上清中のmAbの濃度をELISAにより測定し、細胞当り1日当りの比生産能(pg/細胞*日)を計算した。夫々のデータ点は75cm2のフラスコ中の6回の培養実験の平均を表す。
【図9A】ドットアッセイにおける、NPT野生型と較べた本発明のNPT変異体の酵素活性を示す。このために、mAbを発現する2種の異なる細胞プール(プール1及び2)(これらはNPT野生型遺伝子(配列番号1)又はNPT変異体E182G(配列番号3)、E182D(配列番号19)、W91A(配列番号5)、V198G(配列番号7)、D227A(配列番号9)、D227V(配列番号11)、D227G(配列番号21)、D261G(配列番号13)、D261N(配列番号15)及びF240I(配列番号17)Glu182Asp又はAsp227Glyでトランスフェクションし、選抜した)から細胞抽出物を調製した。トランスフェクションしなかったCHO-DG44細胞を陰性対照として使用した。G418をリン酸化アッセイで基質として使用した。抽出物を96ウェル真空マニホルド中でP81ホスホセルロースとニトロセルロース膜のサンドイッチで濾過した。タンパク質キナーゼによりリン酸化されたタンパク質そしてまたリン酸化されなかったタンパク質はニトロセルロースに結合し、一方、リン酸化G418及びリン酸化されなかったG418はニトロセルロースを通過し、ホスホセルロースに結合する。
【図9B】ドットアッセイにおける、NPT野生型と較べた本発明のNPT変異体の酵素活性を示す。このために、mAbを発現する2種の異なる細胞プール(プール1及び2)(これらはNPT野生型遺伝子(配列番号1)又はNPT変異体E182G(配列番号3)、E182D(配列番号19)、W91A(配列番号5)、V198G(配列番号7)、D227A(配列番号9)、D227V(配列番号11)、D227G(配列番号21)、D261G(配列番号13)、D261N(配列番号15)及びF240I(配列番号17)Glu182Asp又はAsp227Glyでトランスフェクションし、選抜した)から細胞抽出物を調製した。トランスフェクションしなかったCHO-DG44細胞を陰性対照として使用した。G418をリン酸化アッセイで基質として使用した。抽出物を96ウェル真空マニホルド中でP81ホスホセルロースとニトロセルロース膜のサンドイッチで濾過した。タンパク質キナーゼによりリン酸化されたタンパク質そしてまたリン酸化されなかったタンパク質はニトロセルロースに結合し、一方、リン酸化G418及びリン酸化されなかったG418はニトロセルロースを通過し、ホスホセルロースに結合する。放射性シグナルを検出し、ホスホイメージャーを使用して定量した。5μgの抽出物で得られたシグナルを使用して酵素活性%を計算した。酵素活性%はmAbを発現する2種の細胞プールからのNPT変異体の平均を表し、野生型NPTの酵素活性を100%とする。
【図10】トランスフェクションされた細胞プール中のNPT発現及びNPT遺伝子コピーの数のノーザンブロット分析を示す。このために、NPT野生型遺伝子(配列番号1)又はNPT変異体E182G(配列番号3)、E182D(配列番号19)、W91A(配列番号5)、V198G(配列番号7)、D227A(配列番号9)、D227V(配列番号11)、D227G(配列番号21)、D261G(配列番号13)、D261N(配列番号15)及びF240I(配列番号17)でトランスフェクションして選抜し、mAbを発現する2種の異なる細胞プールから全RNAを調製した。トランスフェクションしなかったCHO-DG44細胞を陰性対照として使用した。RNA30μgをNPT遺伝子のコーディング領域を含むFITC-dUTP標識PCR産物とハイブリダイズさせた。全てのトランスフェクション細胞において、約1.3kbの特異的な単一のNPT転写産物が検出された。ドットブロット分析でnpt遺伝子コピー数を測定するために、ゲノムDNAをmAbを発現する上記細胞プールから単離した。 10μg、5μg、2.5μg、1.25μg、0.63μg及び0.32μgのゲノムDNAをNPT遺伝子のコーディング領域を含むFITC-dUTP標識PCR産物とハイブリダイズした。トランスフェクションしなかったCHO-DG44細胞を陰性対照として使用した。プラスミドpBIN-LCを標準として使用した(320pg、160pg、80pg、40pg、20pg、10pg、5pg、2.5pg)。タイトレーションしたプラスミド-DNAについて測定されたシグナル強さから、測定された標準シリーズを使用して、細胞プール中のnpt遺伝子のコピー数を計算した。
【図11】二つの細胞プール(細胞プール5及び8)を例として、FACSを使用するGFPをベースとする選抜によるmAb高発現細胞プールの単離を示す。ベクターpBID-HC及びpBING-LCによる同時トランスフェクションから得られた細胞プールをGFPをベースとする連続FACSソーティングにかけた。プールの細胞培養上清中のIgG2抗体の濃度を夫々のソーティング工程後にELISAにより測定し、細胞当り1日当りの比生産能(pg/細胞*日)を計算した。全てにおいて、6回のソーティングを行ない、夫々の場合に最高のGFP蛍光を有する細胞の5%をソーティングした。夫々のデータ点は75cm2のフラスコ中の少なくとも6回の培養実験の平均を表す。
【図12】細胞プール5及び8(図11を参照のこと)を例として、GFPをベースとする選抜をMTX増幅工程と組み合わせることにより得られたmAb生産性の増大を示す。ベクターpBID-HC及びpBING-LCによるCHO-DG44の同時トランスフェクションの2週後に、最高のGFP蛍光を有する細胞の5%をプール5及び8からソーティングした。次いで100nMのメトトレキセート(MTX)を培地に添加することにより、dhfr媒介遺伝子増幅を行なった。プールの細胞培養上清中のmAbの濃度をELISAにより測定し、細胞当り1日当りの比生産能(pg/細胞*日)を計算した。夫々のデータ点は75cm2のフラスコ中の少なくとも6回の培養実験の平均を表す。
【図13】細胞プール5及び8(図11を参照のこと)を例として、抗体生産性とGFP蛍光の間の相関関係を示す。CHO-DG44をベクター組み合わせpBID-HC及びpBING-LCでトランスフェクションすることにより、これらの細胞プールを得た。それらを連続のGFPをベースとするFACSソーティングにかけ、最高のGFP蛍光を有する細胞の5%をソーティングした。プールの細胞培養上清中のIgG2抗体の濃度を夫々のソーティング工程後にELISAにより測定し、細胞当り1日当りの比生産能(pg/細胞*日)を計算した。夫々のデータ点は75cm2のフラスコ中の少なくとも6回の培養実験の平均を表す。
【特許請求の範囲】
【請求項1】
i)アミノ酸位置91番、ii) アミノ酸位置198番、iii)アミノ酸位置240番、またはiv)i)〜iii)の組合せ、において野生型ネオマイシンホスホトランスフェラーゼ遺伝子がコートするネオマイシンホスホトランスフェラーゼと異なるアミノ酸をコードする、野生型ネオマイシンホスホトランスフェラーゼ遺伝子に対して改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項2】
改変されたネオマイシンホスホトランスフェラーゼ遺伝子によってコードされるネオマイシンホスホトランスフェラーゼが野生型ネオマイシンホスホトランスフェラーゼ遺伝子よりも低い酵素活性を有する、請求項1記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項3】
i)アミノ酸位置91にアラニン、ii)アミノ酸位置198にグリシン、iii)アミノ酸位置240にイソロイシン、またはiv)i)〜iii)の組合せ、をコードする、請求項1または2記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項4】
配列番号6、配列番号8、または配列番号18記載のアミノ酸配列を有するポリペプチドをコードする、請求項1〜3のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項5】
配列番号5、配列番号7または配列番号17記載の配列を含む、または前記配列からなる、請求項1〜4のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項6】
i)アミノ酸位置182にグリシン、ii)アミノ酸位置227にアラニン若しくはバリン、iii)アミノ酸位置261にグリシン、またはiv)i〜iii)の組合せ、をコードする、野生型ネオマイシンホスホトランスフェラーゼの遺伝子に比較して改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項7】
配列番号4、配列番号10、配列番号12または配列番号14記載のアミノ酸配列を有するポリペプチドをコードする、請求項6記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項8】
配列番号3、配列番号9、配列番号11または配列番号13記載の配列を含む、または前記配列からなる請求項6または7記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項9】
請求項1〜8いずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子によってコードされる改変されたネオマイシンホスホトランスフェラーゼ。
【請求項10】
請求項1〜8いずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子を含む真核細胞発現ベクター。
【請求項11】
異種プロモーターに機能的に連結した対象異種遺伝子、および野生型ネオマイシンホスホトランスフェラーゼよりも低い酵素活性を有するネオマイシンホスホトランスフェラーゼをコードする改変されたネオマイシンホスホトランスフェラーゼ遺伝子を含む真核細胞発現ベクター。
【請求項12】
以下の特徴を有する請求項11記載の発現ベクター:
a)改変されたネオマイシンホスホトランスフェラーゼ遺伝子が請求項1〜5のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子である、または、
b)改変されたネオマイシンホスホトランスフェラーゼ遺伝子が、i)アミノ酸位置182ii)アミノ酸位置227、またはiii)i)およびiiの組合せ、において野生型ネオマイシンホスホトランスフェラーゼ遺伝子の対応する位置のアミノ酸と異なるアミノ酸をコードする、または、
c)改変されたネオマイシンホスホトランスフェラーゼ遺伝子が、アミノ酸位置261においてグリシンをコードする。
【請求項13】
野生型オマイシンホスホトランスフェラーゼ遺伝子に対して改変されたネオマイシンホスホトランスフェラーゼ遺伝子が、i)アミノ酸位置182においてグリシンまたはアスパラギン酸、ii)アミノ酸位置227においてアラニン、グリシン、またはバリン、またはiii)i)およびii)の組合せ、をコードする請求項12記載の発現ベクター。
【請求項14】
改変されたネオマイシンホスホトランスフェラーゼ遺伝子が配列番号4、配列番号6、配列番号8、配列番号10、配列番号12、配列番号14、配列番号18、配列番号20、または配列番号22記載のアミノ酸配列を有するタンパク質をコードする、請求項12記載の発現ベクター。
【請求項15】
1以上のプロモーターに機能的に連結した1以上のエンハンサーを含む、請求項12〜14のいずれか1項記載の発現ベクター。
【請求項16】
エンハンサーがCMVまたはSV40のエンハンサーである、請求項15記載の発現ベクター。
【請求項17】
ハムスターユビキチン/S27aプロモーターを含む、請求項11〜16のいずれか1項記載の発現ベクター。
【請求項18】
異種遺伝子がハムスターユビキチン/S27aプロモーターの制御下にある、請求項17記載の発現ベクター。
【請求項19】
蛍光タンパク質の遺伝子をさらに含み、場合により前記蛍光タンパク質の遺伝子が異種遺伝子および異種プロモーターに機能的に連結しているまたは連結されるであろう、請求項11〜18のいずれか1項記載の発現ベクター。
【請求項20】
蛍光タンパク質をコードする遺伝子、および、対象タンパク質/産物をコードする遺伝子の二シストロン性発現を異種プロモーターの制御下で可能とする内部リボソームエントリー部位(IRES)を更に含む、請求項19記載の発現ベクター。
【請求項21】
蛍光タンパク質をコードする遺伝子、および改変されたネオマイシンホスホトランスフェラーゼ遺伝子が単一の、または、別個の2つの転写単位中に位置している、請求項19または20記載の発現ベクター。
【請求項22】
請求項1〜8のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子を含む哺乳動物細胞。
【請求項23】
請求項10〜18のいずれか1項記載の発現ベクターでトランスフェクションされた哺乳動物細胞。
【請求項24】
請求項19〜21のいずれか1項記載の発現ベクターでトランスフェクションされた哺乳動物細胞。
【請求項25】
さらに増幅可能な選抜可能マーカー遺伝子でトランスフェクションされた、請求項22〜24のいずれか1項記載の哺乳動物細胞。
【請求項26】
増幅可能な選抜可能マーカーがジヒドロ葉酸還元酵素(DHFR)である、請求項25記載の哺乳動物細胞。
【請求項27】
げっ歯類細胞である、請求項22〜26のいずれか1項記載の哺乳動物細胞。
【請求項28】
げっ歯類細胞がCHOまたはBHK細胞である、請求項27記載の哺乳動物細胞。
【請求項29】
以下を特徴とする、哺乳動物細胞を濃縮する方法:
(i)哺乳動物の集団を請求項1〜8のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子、またはE182D、D227GまたはD261Nネオマイシンホスホトランスフェラーゼ変異体をコードする改変されたネオマイシンホスホトランスフェラーゼ遺伝子でトランスフェクションすること;
(ii)前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能とする条件下で前記哺乳動物細胞を培養すること;および、
(iii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること。
【請求項30】
以下を特徴とする、少なくとも1つの対象異種遺伝子を発現する細胞を取得および選抜する方法:
(i)哺乳動物の集団を、少なくとも1つの対象遺伝子、および、請求項1〜8のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子またはE128D、D227G若しくはD261Nネオマイシンホスホトランスフェラーゼ変異体をコードする改変されたネオマイシンホスホトランスフェラーゼ遺伝子でトランスフェクションすること;
(ii)前記対象遺伝子の発現、および前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現、を可能とする条件下で前記哺乳動物細胞を培養すること;および、
(iii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること。
【請求項31】
更に哺乳動物細胞を増幅可能な選抜可能マーカー遺伝子でトランスフェクションすること、および、選抜した前記哺乳動物細胞を好ましくは少なくとも1つの遺伝子増幅工程に供することを含む請求項30記載の方法であって、前記増幅可能な選抜可能マーカー遺伝子が好ましくはDHFRであり前記遺伝子増幅が好ましくはメトトレキセートの添加によって行われる前記方法。
【請求項32】
以下を特徴とする、少なくとも1つの対象異種遺伝子を発現する細胞を取得および選抜する方法:
(i)請求項19〜21記載の発現ベクターで哺乳動物細胞をトランスフェクションすること;
(ii)前記哺乳動物細胞を対象遺伝子の発現、蛍光タンパク質をコードする遺伝子の発現、および改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現、を可能とする条件下で培養すること;
(iii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること;および、
(iv)前記哺乳動物細胞をフローサイトメトリー分析によってソーティングすること。
【請求項33】
哺更に乳動物細胞を増幅可能な選抜可能マーカー遺伝子でトランスフェクションすること、および、フローサイトメトリー分析によってソーティングした細胞を少なくとも1つの遺伝子増幅工程に供することを含む請求項32記載の方法であって、増幅可能な前記選抜可能マーカー遺伝子が好ましくはDHFRであり前記遺伝子増幅が好ましくはメトトレキセートによって行われる前記方法。
【請求項34】
以下を特徴とする、少なくとも一つの対象タンパク質を組換え細胞で生産する方法:
(i)哺乳動物の集団を、少なくとも1つの対象遺伝子、および、請求項1〜8のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子またはE128D、D227G若しくはD261Nネオマイシンホスホトランスフェラーゼ変異体をコードする改変されたネオマイシンホスホトランスフェラーゼ遺伝子でトランスフェクションすること;
(ii)前記対象遺伝子の発現、および前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現、を可能とする条件下で前記哺乳動物細胞を培養すること;
(iii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること;および、
(iv)前記哺乳動物細胞または前記培養の培養上清から前記対象タンパク質を回収すること。
【請求項35】
以下を特徴とする、少なくとも一つの対象タンパク質を組換え細胞で生産する方法:
(i)請求項19〜21記載の発現ベクターで哺乳動物細胞をトランスフェクションすること;
(ii)前記哺乳動物細胞を対象遺伝子の発現、蛍光タンパク質をコードする遺伝子の発現、および改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現、を可能とする条件下で培養すること;
(iii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること;
(iv)前記哺乳動物細胞をフローサイトメトリー分析によってソーティングすること;および、
(v)前記哺乳動物細胞または前記培養の培養上清から対象タンパク質を回収すること。
【請求項36】
以下を特徴とする、少なくとも一つの対象タンパク質を生産する方法:
(i)対象遺伝子の発現、改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現、および増幅可能選抜可能マーカー遺伝子の発現、を可能とする条件下で、請求項25または26記載の哺乳動物を培養すること;
(ii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること;
(iii)前記選抜した哺乳動物を少なくとも一つの遺伝子増幅工程に供すること;および、
(iv)前記哺乳動物細胞または前記培養の培養上清から対象タンパク質を回収すること。
【請求項37】
哺乳動物細胞をヘテロマータンパク質/産物をコードする少なくとも2つの対象遺伝子でトランスフェクションすること、および、
(i)前記ヘテロマータンパク質/産物のサブユニットの発現が可能となる条件下で前記哺乳動物細胞を培養すること、および、
(ii)前記ヘテロマータンパク質/産物を前記培養の培養物または培地から単離すること、
を特徴とする、請求項34〜36のいずれか1項記載の方法。
【請求項38】
ソーティングされた細胞が、細胞あたり、一日あたり所望の遺伝子産物の5pgを超える平均比生産能を示す、請求項34〜37のいずれか1項記載の方法。
【請求項39】
分類された細胞が、細胞あたり、一日あたり所望の遺伝子産物の20pgを超える平均比生産能を示す、請求項36または37記載の方法。
【請求項40】
哺乳動物細胞がげっ歯類細胞である、請求項29〜39のいずれか1項記載の方法。
【請求項41】
げっ歯類細胞がCHOまたはBHK細胞である、請求項40記載の方法。
【請求項42】
哺乳動物細胞が懸濁培養される、請求項29〜41記載の方法。
【請求項43】
哺乳動物細胞が無血清培養される、請求項29〜42記載の方法。
【請求項1】
i)アミノ酸位置91番、ii) アミノ酸位置198番、iii)アミノ酸位置240番、またはiv)i)〜iii)の組合せ、において野生型ネオマイシンホスホトランスフェラーゼ遺伝子がコートするネオマイシンホスホトランスフェラーゼと異なるアミノ酸をコードする、野生型ネオマイシンホスホトランスフェラーゼ遺伝子に対して改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項2】
改変されたネオマイシンホスホトランスフェラーゼ遺伝子によってコードされるネオマイシンホスホトランスフェラーゼが野生型ネオマイシンホスホトランスフェラーゼ遺伝子よりも低い酵素活性を有する、請求項1記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項3】
i)アミノ酸位置91にアラニン、ii)アミノ酸位置198にグリシン、iii)アミノ酸位置240にイソロイシン、またはiv)i)〜iii)の組合せ、をコードする、請求項1または2記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項4】
配列番号6、配列番号8、または配列番号18記載のアミノ酸配列を有するポリペプチドをコードする、請求項1〜3のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項5】
配列番号5、配列番号7または配列番号17記載の配列を含む、または前記配列からなる、請求項1〜4のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項6】
i)アミノ酸位置182にグリシン、ii)アミノ酸位置227にアラニン若しくはバリン、iii)アミノ酸位置261にグリシン、またはiv)i〜iii)の組合せ、をコードする、野生型ネオマイシンホスホトランスフェラーゼの遺伝子に比較して改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項7】
配列番号4、配列番号10、配列番号12または配列番号14記載のアミノ酸配列を有するポリペプチドをコードする、請求項6記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項8】
配列番号3、配列番号9、配列番号11または配列番号13記載の配列を含む、または前記配列からなる請求項6または7記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子。
【請求項9】
請求項1〜8いずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子によってコードされる改変されたネオマイシンホスホトランスフェラーゼ。
【請求項10】
請求項1〜8いずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子を含む真核細胞発現ベクター。
【請求項11】
異種プロモーターに機能的に連結した対象異種遺伝子、および野生型ネオマイシンホスホトランスフェラーゼよりも低い酵素活性を有するネオマイシンホスホトランスフェラーゼをコードする改変されたネオマイシンホスホトランスフェラーゼ遺伝子を含む真核細胞発現ベクター。
【請求項12】
以下の特徴を有する請求項11記載の発現ベクター:
a)改変されたネオマイシンホスホトランスフェラーゼ遺伝子が請求項1〜5のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子である、または、
b)改変されたネオマイシンホスホトランスフェラーゼ遺伝子が、i)アミノ酸位置182ii)アミノ酸位置227、またはiii)i)およびiiの組合せ、において野生型ネオマイシンホスホトランスフェラーゼ遺伝子の対応する位置のアミノ酸と異なるアミノ酸をコードする、または、
c)改変されたネオマイシンホスホトランスフェラーゼ遺伝子が、アミノ酸位置261においてグリシンをコードする。
【請求項13】
野生型オマイシンホスホトランスフェラーゼ遺伝子に対して改変されたネオマイシンホスホトランスフェラーゼ遺伝子が、i)アミノ酸位置182においてグリシンまたはアスパラギン酸、ii)アミノ酸位置227においてアラニン、グリシン、またはバリン、またはiii)i)およびii)の組合せ、をコードする請求項12記載の発現ベクター。
【請求項14】
改変されたネオマイシンホスホトランスフェラーゼ遺伝子が配列番号4、配列番号6、配列番号8、配列番号10、配列番号12、配列番号14、配列番号18、配列番号20、または配列番号22記載のアミノ酸配列を有するタンパク質をコードする、請求項12記載の発現ベクター。
【請求項15】
1以上のプロモーターに機能的に連結した1以上のエンハンサーを含む、請求項12〜14のいずれか1項記載の発現ベクター。
【請求項16】
エンハンサーがCMVまたはSV40のエンハンサーである、請求項15記載の発現ベクター。
【請求項17】
ハムスターユビキチン/S27aプロモーターを含む、請求項11〜16のいずれか1項記載の発現ベクター。
【請求項18】
異種遺伝子がハムスターユビキチン/S27aプロモーターの制御下にある、請求項17記載の発現ベクター。
【請求項19】
蛍光タンパク質の遺伝子をさらに含み、場合により前記蛍光タンパク質の遺伝子が異種遺伝子および異種プロモーターに機能的に連結しているまたは連結されるであろう、請求項11〜18のいずれか1項記載の発現ベクター。
【請求項20】
蛍光タンパク質をコードする遺伝子、および、対象タンパク質/産物をコードする遺伝子の二シストロン性発現を異種プロモーターの制御下で可能とする内部リボソームエントリー部位(IRES)を更に含む、請求項19記載の発現ベクター。
【請求項21】
蛍光タンパク質をコードする遺伝子、および改変されたネオマイシンホスホトランスフェラーゼ遺伝子が単一の、または、別個の2つの転写単位中に位置している、請求項19または20記載の発現ベクター。
【請求項22】
請求項1〜8のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子を含む哺乳動物細胞。
【請求項23】
請求項10〜18のいずれか1項記載の発現ベクターでトランスフェクションされた哺乳動物細胞。
【請求項24】
請求項19〜21のいずれか1項記載の発現ベクターでトランスフェクションされた哺乳動物細胞。
【請求項25】
さらに増幅可能な選抜可能マーカー遺伝子でトランスフェクションされた、請求項22〜24のいずれか1項記載の哺乳動物細胞。
【請求項26】
増幅可能な選抜可能マーカーがジヒドロ葉酸還元酵素(DHFR)である、請求項25記載の哺乳動物細胞。
【請求項27】
げっ歯類細胞である、請求項22〜26のいずれか1項記載の哺乳動物細胞。
【請求項28】
げっ歯類細胞がCHOまたはBHK細胞である、請求項27記載の哺乳動物細胞。
【請求項29】
以下を特徴とする、哺乳動物細胞を濃縮する方法:
(i)哺乳動物の集団を請求項1〜8のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子、またはE182D、D227GまたはD261Nネオマイシンホスホトランスフェラーゼ変異体をコードする改変されたネオマイシンホスホトランスフェラーゼ遺伝子でトランスフェクションすること;
(ii)前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現を可能とする条件下で前記哺乳動物細胞を培養すること;および、
(iii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること。
【請求項30】
以下を特徴とする、少なくとも1つの対象異種遺伝子を発現する細胞を取得および選抜する方法:
(i)哺乳動物の集団を、少なくとも1つの対象遺伝子、および、請求項1〜8のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子またはE128D、D227G若しくはD261Nネオマイシンホスホトランスフェラーゼ変異体をコードする改変されたネオマイシンホスホトランスフェラーゼ遺伝子でトランスフェクションすること;
(ii)前記対象遺伝子の発現、および前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現、を可能とする条件下で前記哺乳動物細胞を培養すること;および、
(iii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること。
【請求項31】
更に哺乳動物細胞を増幅可能な選抜可能マーカー遺伝子でトランスフェクションすること、および、選抜した前記哺乳動物細胞を好ましくは少なくとも1つの遺伝子増幅工程に供することを含む請求項30記載の方法であって、前記増幅可能な選抜可能マーカー遺伝子が好ましくはDHFRであり前記遺伝子増幅が好ましくはメトトレキセートの添加によって行われる前記方法。
【請求項32】
以下を特徴とする、少なくとも1つの対象異種遺伝子を発現する細胞を取得および選抜する方法:
(i)請求項19〜21記載の発現ベクターで哺乳動物細胞をトランスフェクションすること;
(ii)前記哺乳動物細胞を対象遺伝子の発現、蛍光タンパク質をコードする遺伝子の発現、および改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現、を可能とする条件下で培養すること;
(iii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること;および、
(iv)前記哺乳動物細胞をフローサイトメトリー分析によってソーティングすること。
【請求項33】
哺更に乳動物細胞を増幅可能な選抜可能マーカー遺伝子でトランスフェクションすること、および、フローサイトメトリー分析によってソーティングした細胞を少なくとも1つの遺伝子増幅工程に供することを含む請求項32記載の方法であって、増幅可能な前記選抜可能マーカー遺伝子が好ましくはDHFRであり前記遺伝子増幅が好ましくはメトトレキセートによって行われる前記方法。
【請求項34】
以下を特徴とする、少なくとも一つの対象タンパク質を組換え細胞で生産する方法:
(i)哺乳動物の集団を、少なくとも1つの対象遺伝子、および、請求項1〜8のいずれか1項記載の改変されたネオマイシンホスホトランスフェラーゼ遺伝子またはE128D、D227G若しくはD261Nネオマイシンホスホトランスフェラーゼ変異体をコードする改変されたネオマイシンホスホトランスフェラーゼ遺伝子でトランスフェクションすること;
(ii)前記対象遺伝子の発現、および前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現、を可能とする条件下で前記哺乳動物細胞を培養すること;
(iii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること;および、
(iv)前記哺乳動物細胞または前記培養の培養上清から前記対象タンパク質を回収すること。
【請求項35】
以下を特徴とする、少なくとも一つの対象タンパク質を組換え細胞で生産する方法:
(i)請求項19〜21記載の発現ベクターで哺乳動物細胞をトランスフェクションすること;
(ii)前記哺乳動物細胞を対象遺伝子の発現、蛍光タンパク質をコードする遺伝子の発現、および改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現、を可能とする条件下で培養すること;
(iii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること;
(iv)前記哺乳動物細胞をフローサイトメトリー分析によってソーティングすること;および、
(v)前記哺乳動物細胞または前記培養の培養上清から対象タンパク質を回収すること。
【請求項36】
以下を特徴とする、少なくとも一つの対象タンパク質を生産する方法:
(i)対象遺伝子の発現、改変されたネオマイシンホスホトランスフェラーゼ遺伝子の発現、および増幅可能選抜可能マーカー遺伝子の発現、を可能とする条件下で、請求項25または26記載の哺乳動物を培養すること;
(ii)前記哺乳動物細胞の増殖に対して選択的に作用する少なくとも1種の選択試薬の存在下で前記哺乳動物細胞を培養すること、および、前記改変されたネオマイシンホスホトランスフェラーゼ遺伝子を発現する細胞を優先的に増殖させること;
(iii)前記選抜した哺乳動物を少なくとも一つの遺伝子増幅工程に供すること;および、
(iv)前記哺乳動物細胞または前記培養の培養上清から対象タンパク質を回収すること。
【請求項37】
哺乳動物細胞をヘテロマータンパク質/産物をコードする少なくとも2つの対象遺伝子でトランスフェクションすること、および、
(i)前記ヘテロマータンパク質/産物のサブユニットの発現が可能となる条件下で前記哺乳動物細胞を培養すること、および、
(ii)前記ヘテロマータンパク質/産物を前記培養の培養物または培地から単離すること、
を特徴とする、請求項34〜36のいずれか1項記載の方法。
【請求項38】
ソーティングされた細胞が、細胞あたり、一日あたり所望の遺伝子産物の5pgを超える平均比生産能を示す、請求項34〜37のいずれか1項記載の方法。
【請求項39】
分類された細胞が、細胞あたり、一日あたり所望の遺伝子産物の20pgを超える平均比生産能を示す、請求項36または37記載の方法。
【請求項40】
哺乳動物細胞がげっ歯類細胞である、請求項29〜39のいずれか1項記載の方法。
【請求項41】
げっ歯類細胞がCHOまたはBHK細胞である、請求項40記載の方法。
【請求項42】
哺乳動物細胞が懸濁培養される、請求項29〜41記載の方法。
【請求項43】
哺乳動物細胞が無血清培養される、請求項29〜42記載の方法。
【図1】

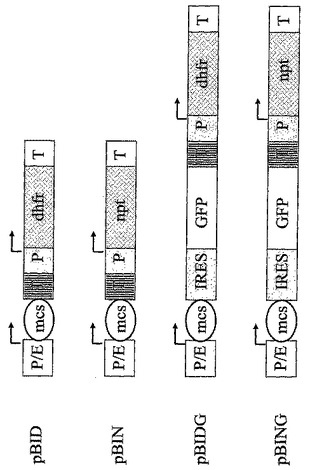
【図2A】
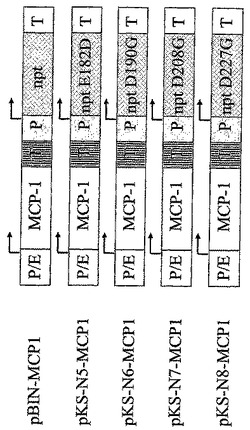
【図2B】
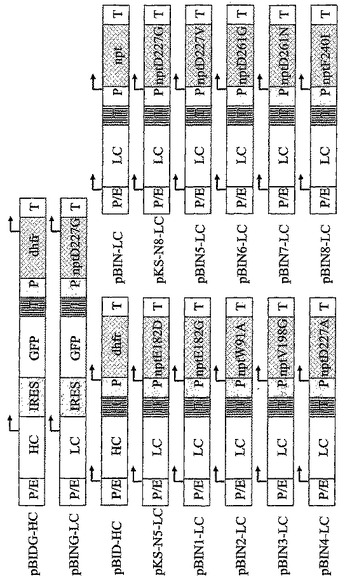
【図3】

【図4】
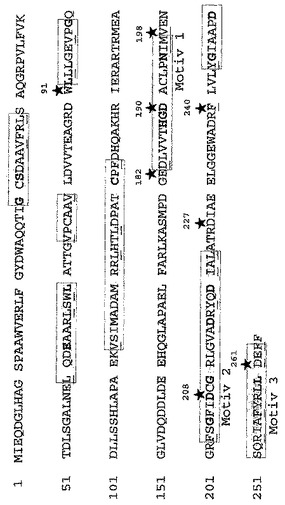
【図5】
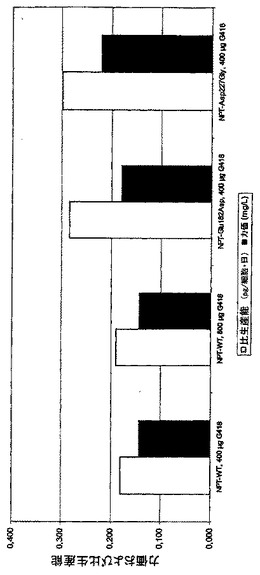
【図6A】
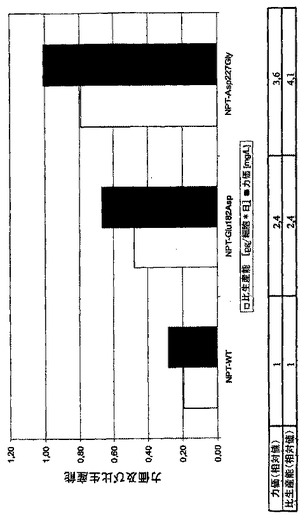
【図6B】
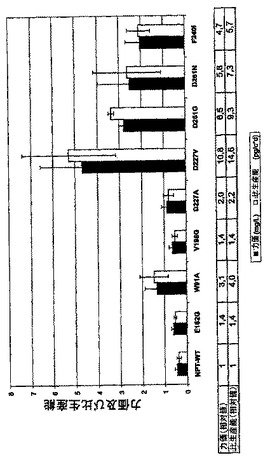
【図7】
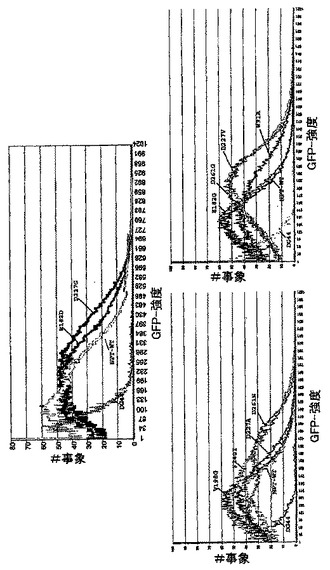
【図8】
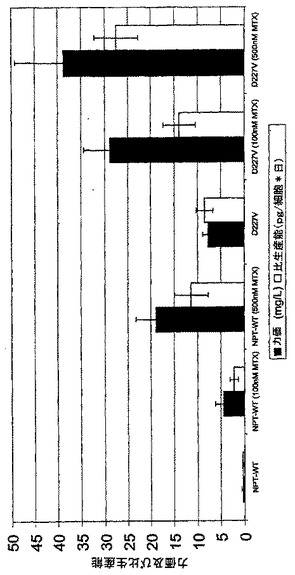
【図9A】
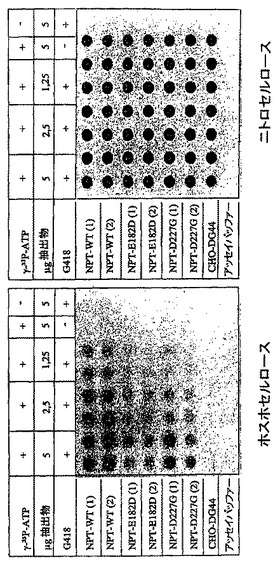
【図9B】

【図10】
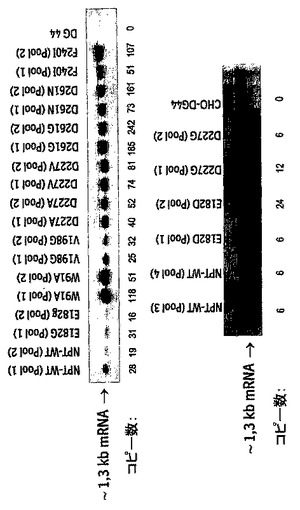
【図11】
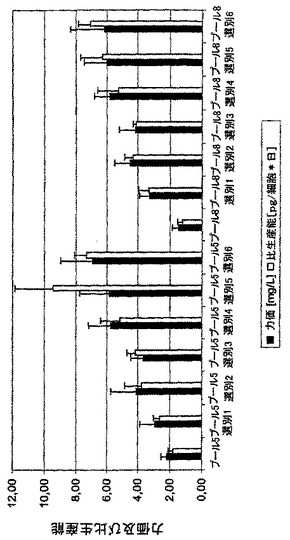
【図12】
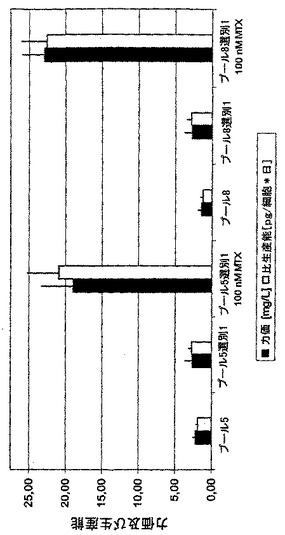
【図13】
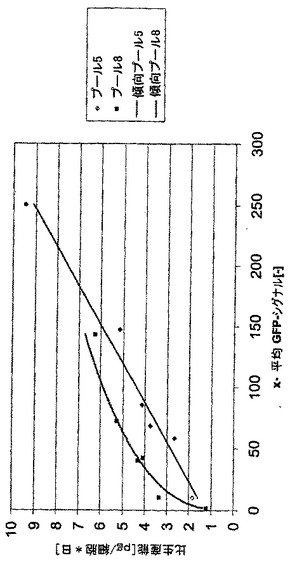
【図2A】
【図2B】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図7】
【図8】
【図9A】
【図9B】
【図10】
【図11】
【図12】
【図13】
【公開番号】特開2009−50279(P2009−50279A)
【公開日】平成21年3月12日(2009.3.12)
【国際特許分類】
【出願番号】特願2008−306392(P2008−306392)
【出願日】平成20年12月1日(2008.12.1)
【分割の表示】特願2004−570678(P2004−570678)の分割
【原出願日】平成15年11月25日(2003.11.25)
【出願人】(503137975)ベーリンガー インゲルハイム ファルマ ゲゼルシャフト ミット ベシュレンクテル ハフツング ウント コンパニー コマンディトゲゼルシャフト (129)
【Fターム(参考)】
【公開日】平成21年3月12日(2009.3.12)
【国際特許分類】
【出願日】平成20年12月1日(2008.12.1)
【分割の表示】特願2004−570678(P2004−570678)の分割
【原出願日】平成15年11月25日(2003.11.25)
【出願人】(503137975)ベーリンガー インゲルハイム ファルマ ゲゼルシャフト ミット ベシュレンクテル ハフツング ウント コンパニー コマンディトゲゼルシャフト (129)
【Fターム(参考)】
[ Back to top ]