
新規な塩および医学的使用
【課題】
より有効で、かつ/または忍容性のより良好な痛風のための新規な治療を提供すること。
【解決手段】
本発明は、高尿酸血症または痛風などの、高血中尿酸レベルに関連する疾患を治療するための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩を提供する。別の一態様において本発明は、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩を提供する。
より有効で、かつ/または忍容性のより良好な痛風のための新規な治療を提供すること。
【解決手段】
本発明は、高尿酸血症または痛風などの、高血中尿酸レベルに関連する疾患を治療するための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩を提供する。別の一態様において本発明は、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドの新規な医学的使用、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドの改良された薬学的に許容できる塩、およびその組成物に関する。
【背景技術】
【0002】
化合物4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドは、電位開口型ナトリウムチャネル(Nav)阻害剤、より具体的にはNav1.7阻害剤であり、その内容の全体が参照により本明細書に組み込まれている国際特許出願公開WO2010/079443号の実施例788として開示されている。その化合物は、Nav1.7阻害剤として、広範囲の障害、特に疼痛の治療に潜在的に有用であり、疼痛には、急性疼痛;慢性疼痛;神経障害性疼痛;炎症性疼痛;内臓疼痛;術後疼痛を含む侵害受容性疼痛;ならびに癌性疼痛、背部および口腔顔面疼痛を含む、内臓、胃腸管、頭蓋構造、筋骨格系、脊椎、泌尿生殖器系、心臓血管系、およびCNSに関連する混合型の疼痛タイプが挙げられる。
【0003】
尿酸は、ヒトにおけるプリン代謝の最終生成物である。ヒトでは、他の多くの動物と異なり、尿酸はそれ以上分解されないが、主に(70%)尿中に排出され、残りの30%は糞便に排泄される。高尿酸血症は、尿酸の過剰産生または低下した排泄として定義されており、血清尿酸(sUA)の過剰産生もしくは排泄低下、または両方の組合せとして生じる可能性がある。症例の約90%では尿酸の腎性排泄低下が高尿酸血症の主要な原因であり、過剰産生を原因とするものは10%未満である。sUA濃度が6.8mg/dLよりも上昇すると、尿酸が尿酸一ナトリウムなどの塩の形態で結晶化し、これらの結晶が関節中、腱上、および周辺組織中に沈着する。これらの結晶(痛風結節として知られている)は、局所的な免疫介在性炎症反応を引き起こし、痛風となる。痛風のリスクは、sUAレベルが上昇するにつれて増加する。
【0004】
症例の約90%では尿酸の腎性排泄低下が高尿酸血症の主要な原因であり、過剰産生を原因とするものは10%未満である。痛風のリスクは、尿酸濃度が上昇するにつれて増加する。
【0005】
痛風は様々な形で現れる可能性がある疼痛状態であるが、最も一般的なのは、足の親指、踵部、膝、手首、および指にしばしば生じる急性の炎症性関節炎(発赤、圧痛、発熱、関節腫脹)の反復性発作である。
【0006】
痛風は、尿酸結晶の炎症および疼痛の原因および影響の両方を低減させるために薬剤で治療される。
【0007】
痛風に関連する疼痛は、一般に、非ステロイド性抗炎症薬(NSAID)、コルヒチン、およびステロイドなどの疼痛用薬および抗炎症薬で治療される。痛風の原因の治療には、sUAレベルを低下させる薬剤を使用することができる。これらには、次の薬剤が挙げられる:尿酸産生を引き起こす酵素を阻害する薬剤、例えば、キサンチン酸化酵素阻害剤(例えば、アロプリノール、フェブキソスタット、またはチソプリン)またはプリンヌクレオシドホスホリラーゼ(PNP)阻害剤(例えば、ウロデシン)など;尿酸を代謝させる薬剤、例えば、ウリカーゼとしても知られている尿酸酸化酵素(例えば、ペグロチカーゼ)など;または尿中での尿酸の排泄を増加させる薬剤(尿酸排泄薬)、尿酸排泄薬には、ベンズヨーダロン、イソブロミンジオン、プロベネシッドおよびスルフィンピラゾン、およびURAT−1阻害剤(例えば、ベンズブロマロン)などの、血液中への尿酸の腎再吸収に関与する輸送体を阻害する薬剤がある。
【0008】
URAT−1はまた、溶質キャリアファミリー22(有機アニオン/カチオン輸送体)、メンバー12として知られており、遺伝子SLC22A12によってコードされている。ヒト遺伝子の分析から、SLC22A12遺伝子の多型体が血清尿酸の変化に直接関連することが実証されている。したがって、URAT−1などの尿酸輸送阻害剤は、痛風の治療に有効である。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際特許出願公開WO2010/079443号
【特許文献2】米国特許第6,106,864号
【特許文献3】国際特許出願公開WO00/35298号
【特許文献4】国際特許出願公開WO91/11172号
【特許文献5】国際特許出願公開WO94/02518号
【特許文献6】国際特許出願公開WO98/55148号
【非特許文献】
【0010】
【非特許文献1】「Remington’s Pharmaceutical Sciences」、第19版(Mack Publishing Company、1995)
【非特許文献2】LiangおよびChenによるExpert Opinion in Therapeutic Patents、11(6)、981〜986(2001)
【非特許文献3】H.LiebermanおよびL.Lachmanによる「Pharmaceutical Dosage Forms:Tablets」、第1巻(Marcel Dekker、New York、1980)
【非特許文献4】Vermaらによる「Pharmaceutical Technology On−line」、25(2)、1〜14(2001)
【非特許文献5】FinninおよびMorganによるJ Pharm Sci、88(10)、955〜958(1999年10月)
【発明の概要】
【発明が解決しようとする課題】
【0011】
より有効で、かつ/または忍容性のより良好な痛風のための新規な治療を提供することが継続して必要とされている。
【課題を解決するための手段】
【0012】
驚くべきことに、今回、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドが血中尿酸レベルを低下させることが分かった。本明細書に示すように、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドは、URAT−1の阻害剤である。この尿酸低下効果は、表5〜9ならびに図7および8のデータを用いて、より詳細に以下に論じている。これらのデータは、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドおよびそのトシル酸塩から調製した経口分散剤を使用して得られた。
【0013】
したがって、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドは、高尿酸血症(例えば尿路結石)に関連する腎障害を含む高尿酸血症;ならびに痛風結節および痛風性関節炎を含む痛風などの、高血中尿酸レベルに関連する疾患の治療に有用である。また、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドは、URAT−1阻害剤が必要される疾患の治療にも有用であるということになる。
【0014】
第1の態様では、本発明は、高血中尿酸レベルに関連する疾患を治療するための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩を提供する。
【図面の簡単な説明】
【0015】
【発明を実施するための形態】
【0016】
一実施形態では、高血中尿酸レベルに関連する疾患は、高尿酸血症である。
【0017】
別の一実施形態では、高血中尿酸レベルに関連する疾患は、痛風である。
【0018】
別の一態様では、本発明は、URAT−1阻害剤の適応がある疾患を治療するための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩を提供する。
【0019】
別の一態様では、本発明は、高血中尿酸レベルに関連する疾患を治療するための医薬の製造のための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩の使用を提供する。
【0020】
別の一態様では、本発明は、URAT−1阻害剤の適応がある疾患を治療するための医薬の製造のための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩の使用を提供する。
【0021】
別の一態様では、本発明は、高血中尿酸レベルに関連する疾患を治療する方法であって、有効量の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩を投与するステップを含む方法を提供する。
【0022】
別の一態様では、本発明は、URAT−1阻害剤の適応がある疾患を治療する方法であって、有効量の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩を投与するステップを含む方法を提供する。
【0023】
驚くべきことに、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩は、薬学的に許容できる製剤の調製に特に適するいくつかの予想外の性質を有することが分かった。このトシル酸塩は、特に製剤化および保存に関して、遊離塩基を上回る化学安定性の向上を示している。これはまた、遊離塩基よりも良好な固形安定性を与える結晶形を形成することができる。驚くべきことに、このトシル酸塩は、分離に関して他の塩よりも大きな安定性を示し、また、良好な水溶性を示した。
【0024】
したがって、別の一態様では、本発明は、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩を提供する。
【0025】
一実施形態では、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩は、結晶性固体である。
【0026】
別の実施形態では、結晶性固体は、CuKα1X線放射(波長=1.5406Å)の使用による、下記の表4および4aで定義する一連のピークから選択される特徴的2シータ(2θ)ピークのうちの3、4、5、または6個を示す粉末X線回折(PXRD)パターンを特徴とする。
【0027】
別の実施形態では、結晶性固体は、CuKα1X線放射(波長=1.5406Å)の使用による、9.0、9.3、10.0、10.7、11.6、12.5、12.9、13.2、13.8、14.4、16.0、16.6、17.5、17.8、18.1、21.4、および23.4°(±0.2°2θ)からなる群から、より好ましくは、9.0、9.3、10.0、10.7、11.6、12.9、13.2、16.0、16.6、17.5、17.8、18.1、21.4、および23.4°(±0.2°2θ)からなる群から、最も好ましくは、11.6、12.9、16.0、17.5、17.8、18.1°(±0.2°2θ)からなる群から選択される特徴的2θピークのうちの任意の3、4、5、または6個を示すPXRDパターンを特徴とする。
【0028】
別の実施形態では、結晶性固体は、CuKα1X線放射(波長=1.540562Å)の使用による、9.0、10.7、16.0、21.4、および23.4°(±0.1°2θ)の主な2θピークを示す粉末X線回折(PXRD)パターンを特徴とする。
【0029】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩は、一般に、1種または複数の薬学的に許容できる賦形剤と併せた製剤として投与される。本明細書では、用語「賦形剤」は、前述のベンゼンスルホンアミド以外の任意の成分の説明に使用される。賦形剤の選択は、特定の投与様式、溶解性および安定性に対する賦形剤の影響、ならびに剤形の性質などの要因に大きく依存する。
【0030】
別の一態様では、本発明は、1種または複数の薬学的に許容できる賦形剤と一緒に4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩を含む医薬組成物を提供する。
【0031】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩の送達に適した医薬組成物、およびそれらの調製方法は、当業者には容易に明らかであろう。こうした組成物および方法は、例えば、「Remington’s Pharmaceutical Sciences」、第19版(Mack Publishing Company、1995)に見ることができる。
【0032】
適切な投与様式としては、経口、非経口、局所、吸入/鼻腔内、直腸/膣内、および眼/耳経由の投与が挙げられる。
【0033】
前述の投与様式に適した製剤は、即時および/または調節放出になるように製剤化することができる。調節放出製剤としては、遅延、持続、パルス、制御、標的、および計画放出が挙げられる。
【0034】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩は、経口投与することができる。経口投与には、薬物が胃腸管に入るような嚥下、または薬物が口から直接血流に入る頬側もしくは舌下投与を使用することができる。経口投与に適した製剤としては、錠剤、微粒子剤、液剤、または散剤を含有するカプセル剤、ロゼンジ剤(液体入りを含む)、咀嚼剤(chew)、多粒子剤およびナノ粒子剤、ゲル剤、固溶体剤、リポソーム剤、フィルム剤、腔坐剤(ovule)、噴霧剤などの固形製剤、液体製剤、ならびに頬/粘膜付着性パッチ剤が挙げられる。
【0035】
液体製剤としては、懸濁剤、液剤、シロップ剤、およびエリキシル剤が挙げられる。こうした製剤は、軟質または硬質カプセル剤における充填剤として使用することができ、一般に、担体、例えば、水、エタノール、ポリエチレングリコール、プロピレングリコール、メチルセルロース、または適切な油、ならびに1種または複数の乳化剤および/または懸濁化剤を含む。液体製剤はまた、例えば、サシェからの固体の再構成によって調製することができる。
【0036】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩はまた、LiangおよびChenによるExpert Opinion in Therapeutic Patents、11(6)、981〜986(2001)に記載されている剤形などの速溶解性、速崩壊性剤形において使用することができる。
【0037】
錠剤剤形の場合、用量に応じて、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩、すなわち、薬物は、剤形の1重量%〜80重量%、より典型的には、剤形の5重量%〜60重量%とすることができる。この薬物に加えて、錠剤は、一般に崩壊剤を含有する。崩壊剤の例としては、デンプングリコール酸ナトリウム、カルボキシルメチルセルロースナトリウム、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、メチルセルロース、微結晶性セルロース、低級アルキル置換ヒドロキシプロピルセルロース、デンプン、アルファ化デンプン、およびアルギン酸ナトリウムが挙げられる。一般に、崩壊剤は、剤形の1重量%〜25重量%、好ましくは、5重量%〜20重量%を占める。
【0038】
結合剤は、一般に、錠剤製剤に凝集性を付与するのに使用される。適切な結合剤としては、微結晶性セルロース、ゼラチン、糖、ポリエチレングリコール、天然および合成ゴム、ポリビニルピロリドン、アルファ化デンプン、ヒドロキシプロピルセルロース、ならびにヒドロキシプロピルメチルセルロースが挙げられる。錠剤はまた、ラクトース(一水和物、噴霧乾燥させた一水和物、無水など)、マンニトール、キシリトール、デキストロース、スクロース、ソルビトール、微結晶性セルロース、デンプン、および第二リン酸カルシウム二水和物などの希釈剤を含有することができる。
【0039】
錠剤はまた、ラウリル硫酸ナトリウムおよびポリソルベート80などの界面活性剤、ならびに二酸化ケイ素およびタルクなどの流動促進剤を含んでいてもよい。存在する場合には、界面活性剤は、錠剤の0.2重量%〜5重量%を占め、流動促進剤は、錠剤の0.2重量%〜1重量%を占めることができる。
【0040】
錠剤はまた、一般に、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、ステアリルフマル酸ナトリウム、およびステアリン酸マグネシウムとラウリル硫酸ナトリウムとの混合物などの滑沢剤を含有する。滑沢剤は、一般に、錠剤の0.25重量%〜10重量%、好ましくは、0.5重量%〜3重量%を占める。他の可能な成分としては、酸化防止剤、着色剤、香味剤、保存剤、および風味マスキング剤が挙げられる。
【0041】
例示的な錠剤は、薬物を約80%まで、結合剤を約10重量%〜約90重量%、希釈剤を約0重量%〜約85重量%、崩壊剤を約2重量%〜約10重量%、滑沢剤を約0.25重量%〜約10重量%含有する。錠剤ブレンドを、直接、またはローラーで圧縮して、錠剤を作製することができる。あるいは、錠剤ブレンドまたはブレンドの一部を、湿式、乾式、もしくは融解式粒状化、融解凍結化、または押出成形化した後、錠剤化することができる。最終製剤は、1層または複数層で構成することができ、コーティングされていても、コーティングされていなくてもよく、さらにカプセル化されていてもよい。錠剤の製剤化は、H.LiebermanおよびL.Lachmanによる「Pharmaceutical Dosage Forms:Tablets」、第1巻(Marcel Dekker、New York、1980)に論じられている。
【0042】
本発明の目的に適した調節放出製剤は、米国特許第6,106,864号に記載されている。高エネルギー分散剤ならびに浸透および被覆粒子剤などの他の適切な放出技術の詳細は、Vermaらによる「Pharmaceutical Technology On−line」、25(2)、1〜14(2001)に見られる。制御放出を実現するためのチューインガムの使用は、国際特許出願公開WO00/35298号に記載されている。
【0043】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩はまた、血流中、筋肉中、または内部器官中に直接投与することができる。非経口投与に適した手段としては、静脈内、動脈内、腹腔内、くも膜下腔内、脳室内、尿道内、胸骨内、頭蓋内、筋肉内、および皮下が挙げられる。非経口投与に適したデバイスとしては、有針(顕微針を含む)注射器、無針注射器、および注入技術が挙げられる。
【0044】
非経口製剤は、一般に、塩、炭水化物、および緩衝剤(好ましくはpHが3〜9)などの賦形剤を含有することができる水溶液であるが、いくつかの用途では、これらは、より適切には、滅菌非水溶液として、または乾燥形態として製剤化して、滅菌した発熱物質を含まない水などの適切なビヒクルと共に使用することができる。
【0045】
滅菌条件下、例えば凍結乾燥による非経口製剤の調製は、当業者によく知られている標準的製薬技術を使用して容易に実施することができる。
【0046】
非経口溶液の調製において使用する4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩の溶解性は、溶解促進剤を組み込むなど、適切な製剤技術を使用することで増大させることができる。非経口投与用製剤は、即時および/または調節放出になるように製剤化することができる。調節放出製剤としては、遅延、持続、パルス、制御、標的、および計画放出が挙げられる。
【0047】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩はまた、皮膚または粘膜に局所的に、すなわち皮膚的または経皮的に投与することができる。この目的のための典型的な製剤としては、ゲル剤、ヒドロゲル剤、ローション剤、液剤、クリーム剤、軟膏剤、散布剤、包帯剤、フォーム剤、フィルム剤、皮膚パッチ剤、ウエハー剤、インプラント剤、スポンジ剤、ファイバー剤、絆創膏剤、およびマイクロエマルジョン剤が挙げられる。また、リポソーム剤も使用することができる。典型的な担体としては、アルコール、水、鉱物油、流動ワセリン、白色ワセリン、グリセリン、ポリエチレングリコール、およびプロピレングリコールが挙げられる。浸透促進剤を組み込むことができ、例えば、FinninおよびMorganによるJ Pharm Sci、88(10)、955〜958(1999年10月)を参照されたい。
【0048】
局所投与の他の手段としては、エレクトロポレーション、イオントフォレーシス、フォノフォレーシス、ソノフォレーシス、および顕微針または無針(例えば、Powderject(商標)、Bioject(商標)など)注射による送達が挙げられる。
【0049】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩はまた、一般に、乾燥粉末用吸入器からの乾燥粉末(単独で、例えば、ラクトースとの乾燥ブレンドでの混合物として、または、例えば、ホスファチジルコリンなどのリン脂質と混合した混合成分粒子として)の形態で、または1,1,1,2−テトラフルオロエタンもしくは1,1,1,2,3,3,3−ヘプタフルオロプロパンなどの適切な噴霧剤の使用の有無にかかわらず加圧容器、ポンプ、スプレー、アトマイザー(好ましくは細かい霧を発生する電気流体力学法を使用するアトマイザー)、もしくはネブライザーからのエアロゾルスプレーとして、鼻腔内にまたは吸入によって投与することができる。鼻腔内使用では、粉末は、生体接着剤、例えば、キトサンまたはシクロデキストリンを含むことができる。
【0050】
加圧容器、ポンプ、スプレー、アトマイザー、またはネブライザーは、例えば、エタノール、エタノール水溶液、または有効物の分散、可溶化、もしくは拡張放出に適した代替試剤、溶媒としての噴霧剤(複数可)、およびトリオレイン酸ソルビタン、オレイン酸、またはオリゴ乳酸などの任意の界面活性剤を含む本発明の化合物(複数可)の溶液または懸濁液を含有する。
【0051】
乾燥粉末または懸濁製剤での使用の前に、薬物製品を、吸入による送達に適した大きさに微小化する(一般に5ミクロン未満)。これは、スパイラルジェットミル法、フルイドベッドジェットミル法、ナノ粒子を形成するための超臨界流体プロセス法、高圧ホモジナイズ法、または噴霧乾燥法などの任意の適切な粉砕法によって実現することができる。
【0052】
吸入器または注入器において使用されるカプセル剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース製)、ブリスター剤、およびカートリッジ剤は、薬物製品と、ラクトースまたはデンプンなどの適切な粉末基剤と、l−ロイシン、マンニトール、またはステアリン酸マグネシウムなどの性能調節剤との粉末混合物を含有するように製剤化することができる。ラクトースは、無水でも一水和物の形態でもよく、好ましくは後者である。他の適切な賦形剤としては、デキストラン、グルコース、マルトース、ソルビトール、キシリトール、フルクトース、スクロース、およびトレハロースが挙げられる。
【0053】
吸入/鼻腔内投与を目的とする本発明のこれらの製剤に、メントールおよびレボメントールなどの適切な香料、またはサッカリンもしくはサッカリンナトリウムなどの甘味料を添加することができる。
【0054】
乾燥粉末用吸入器およびエアロゾルの場合、投薬単位は、計量した量を送達するバルブによって決定される。本発明による単位は、一般に、リスト(I)の化合物を1μg〜100mg含有する計量済み用量または「パフ(puff)」を投与するように構成されている。総1日用量は、典型的には、1μg〜200mgの範囲であり、これは、単回用量で、または、より一般には、1日を通しての分割用量として投与することができる。
【0055】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩はまた、典型的には、等張のpH調整した滅菌生理食塩水において微細化された懸濁液または溶液の液滴の形態で、眼または耳に直接投与することができる。眼および耳への投与に適した他の製剤としては、軟膏剤、生分解性(例えば、吸収性ゲルスポンジ、コラーゲン)および非生分解性(例えば、シリコーン)のインプラント剤、ウエハー剤、レンズ、ならびにニオソームまたはリポソームなどの粒子または小胞系が挙げられる。架橋ポリアクリル酸、ポリビニルアルコール、ヒアルロン酸などのポリマー、セルロースポリマー、例えば、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、もしくはメチルセルロース、またはヘテロ多糖ポリマー、例えばジェランガムを、塩化ベンザルコニウムなどの保存剤と一緒に組み込むことができる。こうした製剤はまた、イオントフォレーシスによって送達することができる。
【0056】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩は、前述の投与様式のいずれかで使用するために、シクロデキストリンおよび適切なその誘導体またはポリエチレングリコール含有ポリマーなどの可溶な巨大分子体と組み合わせて、これらの溶解性、溶出速度、風味マスキング、生体利用能、および/または安定性を向上させることができる。
【0057】
薬物−シクロデキストリン複合体は、例えば、ほとんどの剤形および投与経路にとって一般に有用であることが分かっている。包接複合体と非包接複合体のどちらも使用することができる。薬物との直接複合体化の代わりに、シクロデキストリンを、補助添加剤として、すなわち、担体、希釈剤、または可溶化剤として使用することができる。これらの目的には、α、β、およびγ−シクロデキストリンが最も一般的に使用されており、これらの例は、国際特許出願公開WO91/11172号、同WO94/02518号、および同WO98/55148号に見ることができる。
【0058】
ヒト患者への投与では、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩の総1日用量は、典型的には、1mg〜10g、例えば10mg〜1gなど、例えば25mg〜500mgの範囲にあるが、当然ながら投与様式および効果に依存する。総1日用量は、単回用量または分割用量で投与することができ、特定の患者の年齢、体重、および応答に応じて、医師の裁量で本明細書に示される典型的な範囲から外れる可能性がある。
【0059】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩は、痛風の治療のために、別の1種の薬理学的に有効な化合物、または別の2種以上の薬理学的に有効な化合物と有用に組み合わせることができる。こうした組合せから、患者のコンプライアンス、投薬の容易さ、および相乗活性を含めた著しい利益がもたらされる可能性がある。
【0060】
下記の組合せでは、本発明の化合物は、別の治療剤または薬剤と組み合わせて、同時に、逐次的に、または別々に投与することができる。
【0061】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド、またはトシル酸塩などの薬学的に許容できるその塩は、以下から選択される1種または複数の薬剤と組み合わせて投与することができる:
・抗炎症薬、例えば、NSAID(例えば、セレコキシブ)、コルヒチン、またはステロイドなど;
・キサンチン酸化酵素阻害剤(例えば、アロプリノール、フェブキソスタット、またはチソプリン)、またはプリンヌクレオシドホスホリラーゼ(PNP)阻害剤(例えば、ウロデシン);
・ウリカーゼ(例えば、ペグロチカーゼまたはラスブリカーゼ);または
・尿酸排泄薬、例えば、ベンズヨーダロン、イソブロミンジオン、プロベネシッドおよびスルフィンピラゾン、またはURAT−1阻害剤(例えば、ベンズブロマロン)などの、血液中への尿酸の腎再吸収に関与する輸送体を阻害する薬剤など。
【0062】
本明細書における治療に関するすべての言及には、治癒的、姑息的、および予防的治療があることを理解されたい。
【0063】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドは、類似構造の化合物を調製するために当技術分野でよく知られている任意の方法によって、詳細には、国際特許出願公開WO2010/079443号に記載の実施例788などに詳述されている特定の方法によって調製することができる。
【0064】
本発明を以下の非限定的な実施例により説明する。
【実施例1】
【0065】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の調製
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド(国際特許出願公開WO2010/079443号の実施例788、36.75g、73.45mmol)を含む酢酸エチル(20mL/g、735mL)に、メタノール(3mL/g、110.25mL)を添加し、混合物を50℃に加熱した。p−トルエンスルホン酸一水和物(13.27g、69.77mmol)のメタノール(2mL/g、73.50mL)溶液を、反応混合物に滴下漏斗で6分間かけて添加し、続いて、メタノール(1mL/g、36.75mL)をさらに添加した。反応混合物を室温に冷却し、真空下でろ過し、固体を酢酸エチル:メタノール(9:1、2×37mL)で洗浄した。固体を50℃の真空下で一晩乾燥させて、表題化合物を易流動性の灰色がかった白色固体(43.99g、65.41mmol、89%)として得た。
【0066】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩の分光分析の詳細を以下に示す。
【0067】
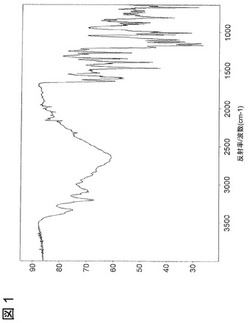
赤外線(IR)分光法
赤外吸収スペクトルは、単一反射の減衰全反射法(ATR)を使用して記録した。スペクトルは、ThermoNicolet Avatar 360 FT IR分光計およびSmart Golden Gate(商標)付属品を使用して、4cm−1分解で得た。この手法は試料調製が不要であった。スペクトルは図1に示している。
【0068】
質量分析法(MS)
フルスキャン質量スペクトルは、図2および図3に示しており、エレクトロスプレーの正(ES+)および負(ES−)イオン化によってそれぞれ得た。データは、エレクトロスプレー源を取り付けたBruker MaXis Quadrupole Time of Flight質量分析計を使用して記録した。内部較正はギ酸ナトリウム溶液を使用して行い、質量範囲m/z113〜m/z997に関して0.2mDa(ES+)および0.3mDa(ES−)の最大質量偏差が観測された。
【0069】
ES+ならびにES−データに関する精密質量測定、理論的モノアイソトピック質量、ならびに観測される付加体およびフラグメントイオンの分子式は、表1および2にそれぞれ示している。対応する質量スペクトルは、図2および3にそれぞれ示している。
【0070】
【表1】

【0071】
【表2】
【0072】
核磁気共鳴(NMR)分光法
プロトン(1H)NMRスペクトルは、液体でDMSOd6において得た。データは、599.77MHzでプロトンにチューニングされた三重共鳴低温プローブを装備したBruker AVANCE III 600MHz NMR分光計において30℃で得た。スペクトルは、DMSOd5(2.50ppm)を基準とした。
【0073】
図4に示す、下記の構造に関して標識した1H NMRスペクトルは、12個の芳香族プロトンおよび3個の脂肪族(1個のCH3基)プロトンの存在を明らかにしている。1Hの化学シフト帰属は、表3に要約している。
【0074】
【化1】
【0075】
【表3】
【0076】
紫外線/可視(UV/Vis)分光測光法
UV/可視スペクトルは、Hitachi U−3000分光光度計を使用して、メタノールにおいて1.09mg/100mLの濃度で得た。それを図5に示している。2個のλ最大が281nmおよび240nmに観察されている。
【0077】
PXRDによる4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の特性分析
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の粉末X線回折パターンを、自動試料交換器、θ−θゴニオメータジオメトリ、自動ビーム発散スリット、およびPSD Vantec−1検出器を取り付けたBruker−AXS Ltd.のD4 ENDEAVOR粉末X線回折計を使用して決定した。試料を0.5mmのキャビティを有する低バックグラウンドシリコンウエハー標本マウント上に載置して分析準備をした。X線管を35kV/40mAで操作して、標本を、銅のKα1X線(波長=1.5406Å)で照射しながら回転させた。分析は、データ収集に関して、室温で2°〜55°の2θ範囲にわたって0.018°ステップサイズにつき0.2秒の連続モード設定とした。ピーク検索は、閾値および幅のパラメータをそれぞれ1および0.3に設定したものを使用して、Bruker−AXSから発売されたEvaソフトウェアを用いて行った。機器の較正は、コランダム標準品(NIST:SRM 1976 XRD平板強度標準)を使用して確認した。
【0078】
機器、試料、および試料調製の違いにより、本明細書では、ピーク値は、ピーク値のばらつきから推定して報告している。これは、ピーク値に固有の偏差のために、固体状態の化学技術分野において普通に行われている。粉末X線回折での2θX軸値の典型的なばらつきは、+または−0.2°2θ程度である。
【0079】
ピーク強度のばらつきは、個々の結晶が外部X線源に対して試料容器中でどのように配向しているかの結果である(「優先配向」として知られている)。この配向効果は、結晶に関する構造情報を提供するものではない。
【0080】
さらに、本発明の結晶材料が薬学的賦形剤などの追加成分と混合されるか、またはそれらで希釈される場合、上述の特徴的ピークの強度が変わるであろうことも当業者なら理解されよう。このため、諸成分の混合物の特徴的ピークの検出を可能にするために、上述のPXRD法をわずかに最適化しなければならないことを当業者なら理解されよう。この最適化には、より強力なX線源(波長=1.5406Å)、わずかに異なるステップサイズ、またはわずかに異なるステップ時間の使用が挙げられる。
【0081】
また、異なる波長を使用して測定すると、ブラッグの式nλ=2d sinθに従って異なるシフトが得られることを当業者なら理解されよう。代替波長の使用によって生じるこうしたさらなるPXRDパターンは、本発明の結晶材料のPXRDパターンの代替表現と考えられ、したがって、本発明の範囲内に含まれる。
【0082】
PXRDパターンは、図6に示している。主な2θピーク位置および相対強度は、表4および4aに列挙している。
【0083】
【表4】
【0084】
【表5】
【実施例2】
【0085】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド噴霧乾燥拡散(SDD)の調製
テトラヒドロフラン(安定剤不含、14.5kg)および水(0.76kg)を、上部マウントミキサーを装備したステンレス鋼槽に加えた。次いで、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド(742.4g)を溶液に添加し、固体がすべて完全に溶解するまで、少なくとも1時間混合した。(酢酸/コハク酸)ヒドロキシプロピルメチルセルロース(中級顆粒、1338.4g)を溶液に添加し、完全に溶解するまで混合した。次いで、下表の条件を使用して、溶液を窒素ガス下で噴霧乾燥させた。
【0086】
【表6】
【0087】
次いで、得られた4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのSDDを、対流トレー乾燥機において40℃/50%相対湿度(RH)で最低6時間トレー乾燥させ、続いて、RHを40℃/75%まで上昇させ、さらに最低25時間トレー乾燥させた。
【0088】
SDDは、必要となるまで2〜8℃で保存した。
【実施例3】
【0089】
経口投与用分散剤の調製:
(a)4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩を使用
メチルセルロースビヒクル(0.5%w/v)を以下のように調製した。ビーカーにおいて潅注水(water for irrigation)(600mL)を80℃〜90℃に加熱した。メチルセルロース(5g)粉末を、粉末が完全に分散するまで撹拌しながら添加した。次いで、分散物を氷浴へ移動させ、冷精製水(400mL)を添加しながら素早く冷却させて、透明な溶液を得た。
【0090】
必要量の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩(10mg〜2400mg)を適切な大きさのガラス製褐色投薬瓶に秤量し、ある量のビヒクル(0.5%(w/v)メチルセルロース)を添加して、経口投与用分散剤を調製した。添加したビヒクル量は、薬物濃度が範囲0.6〜50mg/mLになるように、用量に応じて決定した:範囲10mg〜30mg未満の薬物用量では15mL;範囲30mg〜2400mgの薬物用量では50mL。
【0091】
分散剤を2〜8℃で保存し、72時間以内に投薬容器から直接投与した。投与後、ガラス製投薬瓶をほぼ等量の飲料水で2回すすいで、投薬量を含めた全投与量が240mLになるようにした。
【0092】
(b)4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのSDDを使用
メチルセルロースビヒクル0.5%(w/v)を、上述の実施例3(a)で詳述した手順を使用して調製した。
【0093】
必要量の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドSDDを適切な大きさのガラス製褐色投薬瓶に秤量し、ある量のビヒクル(0.5%(w/v)メチルセルロース)を添加して、経口投与用分散剤を調製した。添加したビヒクル量は、薬物濃度が範囲0.6〜50mg/mLになるように、用量に応じて決定した:用量範囲10mg〜2400mgでは20mL〜100mL。
【0094】
分散剤を2〜8℃で保存し、72時間以内に投薬容器から直接投与した。投与後、ガラス製投薬瓶をほぼ等量の飲料水で2回すすいで、投薬量を含めた全投与量が240mLになるようにした。
【0095】
生物学的活性
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドおよびトシル酸塩などの薬学的に許容できるその塩が血中尿酸レベルを低下させる能力について、次の実験で実証した。尿酸測定は、市販の比色測定法キット(Beckman Coulter)を使用して行った。
【実施例4】
【0096】
単回用量研究:6コホートの健常な対象者における二重盲検無作為化プラセボ対照クロスオーバー研究。
範囲が10mg〜2400mgの4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドSDD(「SDD」)経口投与用分散剤の単回用量を調査した。さらに、範囲が200mg〜1000mgの4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩(「TS」)経口投与用分散剤の単回用量も調査した。
【0097】
以下の用量を調査し、すべての用量を絶食状態で与えたが、例外として、SDD分散剤200mgおよび1000mgの場合は、絶食状態と高脂肪食後(「摂食」)の両方で与えた。
コホート1:SDD10mg、SDD100mg、SDD300mg、TS200mg、プラセボ
コホート2:SDD30mg、SDD300mg、SDD(摂食)200mg、プラセボ
コホート3:SDD100mg、SDD200mg、SDD300mg、プラセボ
コホート4:SDD450mg、SDD600mg、SDD800mg、SDD1000mg、プラセボ
コホート5:TS600mg、TS1000mg、SDD(摂食)1000mg
コホート6:SDD1250mg、SDD1600mg、SDD2000mg、SSD2400mg、プラセボ
【0098】
合計61人の対象者(すべて男性)が参加し、全員がSDDまたはTS経口投与用分散剤の投与を少なくとも1回受けた。
【0099】
用量群ごとの尿酸レベルの平均データを以下のように示している。
・SDD絶食 表5
・SDD摂食および絶食 表6
・TS 表7
【0100】
【表7】
【0101】
【表8】
【0102】
【表9】
【0103】
投与の48時間後に、血液中の尿酸の用量依存的な低下が認められた。通常、10〜1000mgの用量では、値は正常範囲(3.5〜7.2mg/dL)内にあったが、1250〜2400mgの用量では、尿酸レベルの低下はより著しく、対象者の少なくとも半数が、投与48時間後で正常下限(LNN)値よりも低くなった。すべての投与前値および追跡値は、LNNよりも高かった。投与48時間後にLNNをやや下回る1人の対象者を除くプラセボの全対象者(n=42)が、正常範囲内の尿酸値となった。
【実施例5】
【0104】
多回用量研究:健常な対象者における二重盲検無作為化プラセボ対照研究。
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドSDD経口投与用分散剤の100mg1日2回(BID)、300mgBID、および600mgBIDの多回経口用量、ならびにプラセボを14日間調査した。対象者は、朝の投与前の一晩および晩の投与前の少なくとも2時間は絶食した。投与後の少なくとも2時間は食物を控えた。
【0105】
合計30人の対象者(すべて男性)を研究に登録し、27人の対象者が研究を完了した(3人の対象者は、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド600mgBIDでの治療中に有害事象が生じたので研究を止めた)。
【0106】
用量群ごとの尿酸レベルの平均データを表8および図7に示している。
【0107】
【表10】
【0108】
血液中の尿酸の用量依存的な低下は、4日目に明らかになり(最初の投与後評価)、最低平均値は4または7日目に生じた。8人中5人の対象者が、7日目に、100mgBID(低尿酸の対象者の実範囲は2.6〜3.4mg/dLであり、1人の対象者はベースラインの限界より低く、実値は2.7mg/dL)および300mg(低尿酸の対象者の実範囲は2.2〜3.4mg/dL)で尿酸値がLLNより低くなった。全対象者が、600mgBID投与の4および7日目に尿酸値がLNNより低くなった(実値は<1.5〜3.0mg/dL)。値は継続投与したにもかかわらず10および14日目に通常増加したが、1人を除く全対象者が16日目に正常範囲に戻った(最終投与の2日後)。残りの対象者は、ベースライン時での尿酸値が最低(4.7mg/dL)であり、継続値は、2.2mg/dL(4日目)、1.5mg/dL未満(7日目)、1.6mg/dL(10日目)、3.1mg/dL(14日目)、および追跡時4.9mg/dLであった。プラセボ投与を受けた全対象者(n=6)は、全時点で正常範囲内の尿酸濃度であった。
【実施例6】
【0109】
多回用量研究:健常な対象者および高齢の対象者における二重盲検無作為化プラセボ対照研究。
以下のように、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドTS経口投与用分散剤の多回経口用量、ならびにプラセボを14日間調査した。
・健常な対象者、300mg1日2回(BID)
・健常な対象者、450mgBID、および
・高齢の対象者、300mgBID。
【0110】
対象者は、朝の投与前の一晩および晩の投与前の少なくとも2時間は絶食した。投与後の少なくとも2時間は食物を控えた。
【0111】
合計49人の対象者を研究に登録し、そのうちの39人が4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドTS投与を受け、10人がプラセボ投与を受けた。
【0112】
用量群ごとの平均血中尿酸レベルおよび尿排泄データを表9に示している。図8は、尿中に排泄された尿酸のパーセントを示している。
【0113】
【表11】
【0114】
血液中の尿酸濃度は、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドの投与後に低下した。全対象者が、1日目では血中尿酸値がLLNよりも高かった。プラセボ投与を受けた対象者は、研究期間を通して血中尿酸濃度がLLNよりも高かった。450mgBID投与を受けた対象者では、血中尿酸濃度の中央値は3日目にLLNよりも低くなり、6日目には、全コホート(300mgおよび450mgBID)の中央値がLLNよりも低くなった。全対象者が、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド投与を中止して2日以内(すなわち16日目までに)に、血中尿酸濃度がLLNよりも高く戻った。
【0115】
1日目、次いで、6、14、および16日目の投与前の24時間にわたって採取された尿中の尿酸も測定した。尿中尿酸の排泄率パーセントを計算し、線形混合効果モデルで分析した。これらのデータの要約は、図8に示している。データは、尿中尿酸の排泄率が、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドの投与中には増加し、16日目までにベースラインに戻ることを示唆している。
【実施例7】
【0116】
URAT−1阻害活性
URAT−1輸送体(tranporter)の阻害剤としての4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドの効力を、以下のように決定した。
【0117】
HEK293細胞を、熱不活性化ウシ胎児血清(10%v/v)、100U/mLのペニシリン、および100μg/mLのストレプトマイシンを補充した、L−GlutaMax(1L当たりグルコース4.5g)を含むダルベッコの改変イーグル培地(DMEM)からなる培地で成長させた。HEK細胞を、約95%空気/5%CO2で約37℃の加湿インキュベーターにおいて75cm2組織培養フラスコで常法通り培養した。ほぼコンフルエントなHEK細胞培養物をトリプシン処理により採取し、培地に再懸濁させ、このプロセスを週に1または2回繰り返して、使用に十分な細胞を得た。
【0118】
取込み実験では、HEK293細胞を、ポリ−D−リシンでコーティングした24ウェルプレートにウェル当たり4×105細胞の密度で播種した。約5%CO2の空気が入っている加湿インキュベーターにおいて、細胞を約37℃で1日間培養した。その後、リポフェクタミン(Lipofectamine)2000試薬を使用して、細胞をpcDNA3.1/hygro/URAT1(HEK−URAT1細胞)またはpcDNA3.1/hygro(HEK対照細胞)でトランスフェクトした。約5%CO2の空気が入っている加湿インキュベーターにおいて約37℃で約24時間後、細胞を実験に使用した。
【0119】
トランスフェクトした1日後、培地をウェルから除去し、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド(0〜30μM)の非存在下および存在下で、細胞を塩素不含インキュベーション培地(125mMのグルコン酸Na、4.8mMのグルコン酸K、1.3mMのグルコン酸Ca、1.2mMのKH2PO4、1.2mMのMgSO4、5.6mMのD−グルコース、25mMのHEPES、pH7.4)0.2mLと共に約37℃で15分間プレインキュベートした。その後、インキュベーション培地を除去し、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド(0〜30μM)の非存在下および存在下で、[14C]尿酸(20μM)を含有する塩素不含インキュベーション培地0.2mLを添加し、これをトリプリケートで行った。細胞を約37℃で2分間インキュベートした。インキュベーションの終わりに、培地を吸引し、単層を氷冷インキュベーション培地1mLで素早く2回すすいだ。続いて、細胞を0.5NのNaOH0.5mLで可溶化し、各ウェルの一定量の細胞溶解物試料をシンチレーションバイアルに収集した。[14C]尿酸の濃度を液体シンチレーション計数器(LSC)で決定した。[14C]尿酸輸送の阻害はまた、よく知られている阻害剤ベンズブロマロン(30μM)の存在下で決定した。最終的な有機溶媒は、1%(v/v)未満であった。
【0120】
可溶化したHEK細胞のタンパク質含有量は、タンパク質標準物としてウシ血清アルブミン(BSA)(濃度域0〜1mg/mL)を含むバイオ−ラッドブラッドフォード(Bio−Rad Bradford)試薬を使用して、ブラッドフォード法により決定した。BSA溶液または可溶化した細胞を、希釈した染料試薬濃縮物(Bio−Rad製)と混合した。室温で10分間インキュベートした後、595nmで吸光度を測定した。
【0121】
細胞溶解物試料中の放射活性量を、液体シンチレーション計数器(LSC)により決定した。液体シンチラント(Hionic Fluor(商標))を全試料に添加し、放射活性を、QuantaSmart(商標)ソフトウェアを使用して、TriCarb3100TR液体シンチレーションカウンタのLSCにより決定し、これらのカウントはすべて、tSIE/AEC(自動効率補正つき外部標準変換スペクトル指数(transformed Spectral Index of external standards coupled to Automatic Efficiency Correction))を使用して、DPMに変換した。機器の較正手順は、試験機関で確立されている。試料はすべて少なくとも2分間カウントした。バックグラウンド値は、試料を含まない液体シンチラントを使用して、各試料系列で測定した。HEK細胞中の[14C]−尿酸の蓄積(pmol/mgタンパク質)を計算し、IC50値を、50%輸送阻害に必要な阻害剤の濃度と定義し、ヒル(Hill)式を用いるグラフパッドプリズム(GraphPad Prism)バージョン4.00を使用して計算した。
【0122】
データ
上述の方法で測定した4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドによる尿酸取込みの阻害は、基準化合物ベンズブロマロンに対して正規化すると、3.54μMである。
【図1】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【技術分野】
【0001】
本発明は、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドの新規な医学的使用、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドの改良された薬学的に許容できる塩、およびその組成物に関する。
【背景技術】
【0002】
化合物4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドは、電位開口型ナトリウムチャネル(Nav)阻害剤、より具体的にはNav1.7阻害剤であり、その内容の全体が参照により本明細書に組み込まれている国際特許出願公開WO2010/079443号の実施例788として開示されている。その化合物は、Nav1.7阻害剤として、広範囲の障害、特に疼痛の治療に潜在的に有用であり、疼痛には、急性疼痛;慢性疼痛;神経障害性疼痛;炎症性疼痛;内臓疼痛;術後疼痛を含む侵害受容性疼痛;ならびに癌性疼痛、背部および口腔顔面疼痛を含む、内臓、胃腸管、頭蓋構造、筋骨格系、脊椎、泌尿生殖器系、心臓血管系、およびCNSに関連する混合型の疼痛タイプが挙げられる。
【0003】
尿酸は、ヒトにおけるプリン代謝の最終生成物である。ヒトでは、他の多くの動物と異なり、尿酸はそれ以上分解されないが、主に(70%)尿中に排出され、残りの30%は糞便に排泄される。高尿酸血症は、尿酸の過剰産生または低下した排泄として定義されており、血清尿酸(sUA)の過剰産生もしくは排泄低下、または両方の組合せとして生じる可能性がある。症例の約90%では尿酸の腎性排泄低下が高尿酸血症の主要な原因であり、過剰産生を原因とするものは10%未満である。sUA濃度が6.8mg/dLよりも上昇すると、尿酸が尿酸一ナトリウムなどの塩の形態で結晶化し、これらの結晶が関節中、腱上、および周辺組織中に沈着する。これらの結晶(痛風結節として知られている)は、局所的な免疫介在性炎症反応を引き起こし、痛風となる。痛風のリスクは、sUAレベルが上昇するにつれて増加する。
【0004】
症例の約90%では尿酸の腎性排泄低下が高尿酸血症の主要な原因であり、過剰産生を原因とするものは10%未満である。痛風のリスクは、尿酸濃度が上昇するにつれて増加する。
【0005】
痛風は様々な形で現れる可能性がある疼痛状態であるが、最も一般的なのは、足の親指、踵部、膝、手首、および指にしばしば生じる急性の炎症性関節炎(発赤、圧痛、発熱、関節腫脹)の反復性発作である。
【0006】
痛風は、尿酸結晶の炎症および疼痛の原因および影響の両方を低減させるために薬剤で治療される。
【0007】
痛風に関連する疼痛は、一般に、非ステロイド性抗炎症薬(NSAID)、コルヒチン、およびステロイドなどの疼痛用薬および抗炎症薬で治療される。痛風の原因の治療には、sUAレベルを低下させる薬剤を使用することができる。これらには、次の薬剤が挙げられる:尿酸産生を引き起こす酵素を阻害する薬剤、例えば、キサンチン酸化酵素阻害剤(例えば、アロプリノール、フェブキソスタット、またはチソプリン)またはプリンヌクレオシドホスホリラーゼ(PNP)阻害剤(例えば、ウロデシン)など;尿酸を代謝させる薬剤、例えば、ウリカーゼとしても知られている尿酸酸化酵素(例えば、ペグロチカーゼ)など;または尿中での尿酸の排泄を増加させる薬剤(尿酸排泄薬)、尿酸排泄薬には、ベンズヨーダロン、イソブロミンジオン、プロベネシッドおよびスルフィンピラゾン、およびURAT−1阻害剤(例えば、ベンズブロマロン)などの、血液中への尿酸の腎再吸収に関与する輸送体を阻害する薬剤がある。
【0008】
URAT−1はまた、溶質キャリアファミリー22(有機アニオン/カチオン輸送体)、メンバー12として知られており、遺伝子SLC22A12によってコードされている。ヒト遺伝子の分析から、SLC22A12遺伝子の多型体が血清尿酸の変化に直接関連することが実証されている。したがって、URAT−1などの尿酸輸送阻害剤は、痛風の治療に有効である。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際特許出願公開WO2010/079443号
【特許文献2】米国特許第6,106,864号
【特許文献3】国際特許出願公開WO00/35298号
【特許文献4】国際特許出願公開WO91/11172号
【特許文献5】国際特許出願公開WO94/02518号
【特許文献6】国際特許出願公開WO98/55148号
【非特許文献】
【0010】
【非特許文献1】「Remington’s Pharmaceutical Sciences」、第19版(Mack Publishing Company、1995)
【非特許文献2】LiangおよびChenによるExpert Opinion in Therapeutic Patents、11(6)、981〜986(2001)
【非特許文献3】H.LiebermanおよびL.Lachmanによる「Pharmaceutical Dosage Forms:Tablets」、第1巻(Marcel Dekker、New York、1980)
【非特許文献4】Vermaらによる「Pharmaceutical Technology On−line」、25(2)、1〜14(2001)
【非特許文献5】FinninおよびMorganによるJ Pharm Sci、88(10)、955〜958(1999年10月)
【発明の概要】
【発明が解決しようとする課題】
【0011】
より有効で、かつ/または忍容性のより良好な痛風のための新規な治療を提供することが継続して必要とされている。
【課題を解決するための手段】
【0012】
驚くべきことに、今回、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドが血中尿酸レベルを低下させることが分かった。本明細書に示すように、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドは、URAT−1の阻害剤である。この尿酸低下効果は、表5〜9ならびに図7および8のデータを用いて、より詳細に以下に論じている。これらのデータは、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドおよびそのトシル酸塩から調製した経口分散剤を使用して得られた。
【0013】
したがって、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドは、高尿酸血症(例えば尿路結石)に関連する腎障害を含む高尿酸血症;ならびに痛風結節および痛風性関節炎を含む痛風などの、高血中尿酸レベルに関連する疾患の治療に有用である。また、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドは、URAT−1阻害剤が必要される疾患の治療にも有用であるということになる。
【0014】
第1の態様では、本発明は、高血中尿酸レベルに関連する疾患を治療するための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩を提供する。
【図面の簡単な説明】
【0015】
【発明を実施するための形態】
【0016】
一実施形態では、高血中尿酸レベルに関連する疾患は、高尿酸血症である。
【0017】
別の一実施形態では、高血中尿酸レベルに関連する疾患は、痛風である。
【0018】
別の一態様では、本発明は、URAT−1阻害剤の適応がある疾患を治療するための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩を提供する。
【0019】
別の一態様では、本発明は、高血中尿酸レベルに関連する疾患を治療するための医薬の製造のための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩の使用を提供する。
【0020】
別の一態様では、本発明は、URAT−1阻害剤の適応がある疾患を治療するための医薬の製造のための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩の使用を提供する。
【0021】
別の一態様では、本発明は、高血中尿酸レベルに関連する疾患を治療する方法であって、有効量の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩を投与するステップを含む方法を提供する。
【0022】
別の一態様では、本発明は、URAT−1阻害剤の適応がある疾患を治療する方法であって、有効量の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩を投与するステップを含む方法を提供する。
【0023】
驚くべきことに、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩は、薬学的に許容できる製剤の調製に特に適するいくつかの予想外の性質を有することが分かった。このトシル酸塩は、特に製剤化および保存に関して、遊離塩基を上回る化学安定性の向上を示している。これはまた、遊離塩基よりも良好な固形安定性を与える結晶形を形成することができる。驚くべきことに、このトシル酸塩は、分離に関して他の塩よりも大きな安定性を示し、また、良好な水溶性を示した。
【0024】
したがって、別の一態様では、本発明は、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩を提供する。
【0025】
一実施形態では、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩は、結晶性固体である。
【0026】
別の実施形態では、結晶性固体は、CuKα1X線放射(波長=1.5406Å)の使用による、下記の表4および4aで定義する一連のピークから選択される特徴的2シータ(2θ)ピークのうちの3、4、5、または6個を示す粉末X線回折(PXRD)パターンを特徴とする。
【0027】
別の実施形態では、結晶性固体は、CuKα1X線放射(波長=1.5406Å)の使用による、9.0、9.3、10.0、10.7、11.6、12.5、12.9、13.2、13.8、14.4、16.0、16.6、17.5、17.8、18.1、21.4、および23.4°(±0.2°2θ)からなる群から、より好ましくは、9.0、9.3、10.0、10.7、11.6、12.9、13.2、16.0、16.6、17.5、17.8、18.1、21.4、および23.4°(±0.2°2θ)からなる群から、最も好ましくは、11.6、12.9、16.0、17.5、17.8、18.1°(±0.2°2θ)からなる群から選択される特徴的2θピークのうちの任意の3、4、5、または6個を示すPXRDパターンを特徴とする。
【0028】
別の実施形態では、結晶性固体は、CuKα1X線放射(波長=1.540562Å)の使用による、9.0、10.7、16.0、21.4、および23.4°(±0.1°2θ)の主な2θピークを示す粉末X線回折(PXRD)パターンを特徴とする。
【0029】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩は、一般に、1種または複数の薬学的に許容できる賦形剤と併せた製剤として投与される。本明細書では、用語「賦形剤」は、前述のベンゼンスルホンアミド以外の任意の成分の説明に使用される。賦形剤の選択は、特定の投与様式、溶解性および安定性に対する賦形剤の影響、ならびに剤形の性質などの要因に大きく依存する。
【0030】
別の一態様では、本発明は、1種または複数の薬学的に許容できる賦形剤と一緒に4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩を含む医薬組成物を提供する。
【0031】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩の送達に適した医薬組成物、およびそれらの調製方法は、当業者には容易に明らかであろう。こうした組成物および方法は、例えば、「Remington’s Pharmaceutical Sciences」、第19版(Mack Publishing Company、1995)に見ることができる。
【0032】
適切な投与様式としては、経口、非経口、局所、吸入/鼻腔内、直腸/膣内、および眼/耳経由の投与が挙げられる。
【0033】
前述の投与様式に適した製剤は、即時および/または調節放出になるように製剤化することができる。調節放出製剤としては、遅延、持続、パルス、制御、標的、および計画放出が挙げられる。
【0034】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩は、経口投与することができる。経口投与には、薬物が胃腸管に入るような嚥下、または薬物が口から直接血流に入る頬側もしくは舌下投与を使用することができる。経口投与に適した製剤としては、錠剤、微粒子剤、液剤、または散剤を含有するカプセル剤、ロゼンジ剤(液体入りを含む)、咀嚼剤(chew)、多粒子剤およびナノ粒子剤、ゲル剤、固溶体剤、リポソーム剤、フィルム剤、腔坐剤(ovule)、噴霧剤などの固形製剤、液体製剤、ならびに頬/粘膜付着性パッチ剤が挙げられる。
【0035】
液体製剤としては、懸濁剤、液剤、シロップ剤、およびエリキシル剤が挙げられる。こうした製剤は、軟質または硬質カプセル剤における充填剤として使用することができ、一般に、担体、例えば、水、エタノール、ポリエチレングリコール、プロピレングリコール、メチルセルロース、または適切な油、ならびに1種または複数の乳化剤および/または懸濁化剤を含む。液体製剤はまた、例えば、サシェからの固体の再構成によって調製することができる。
【0036】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩はまた、LiangおよびChenによるExpert Opinion in Therapeutic Patents、11(6)、981〜986(2001)に記載されている剤形などの速溶解性、速崩壊性剤形において使用することができる。
【0037】
錠剤剤形の場合、用量に応じて、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩、すなわち、薬物は、剤形の1重量%〜80重量%、より典型的には、剤形の5重量%〜60重量%とすることができる。この薬物に加えて、錠剤は、一般に崩壊剤を含有する。崩壊剤の例としては、デンプングリコール酸ナトリウム、カルボキシルメチルセルロースナトリウム、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、メチルセルロース、微結晶性セルロース、低級アルキル置換ヒドロキシプロピルセルロース、デンプン、アルファ化デンプン、およびアルギン酸ナトリウムが挙げられる。一般に、崩壊剤は、剤形の1重量%〜25重量%、好ましくは、5重量%〜20重量%を占める。
【0038】
結合剤は、一般に、錠剤製剤に凝集性を付与するのに使用される。適切な結合剤としては、微結晶性セルロース、ゼラチン、糖、ポリエチレングリコール、天然および合成ゴム、ポリビニルピロリドン、アルファ化デンプン、ヒドロキシプロピルセルロース、ならびにヒドロキシプロピルメチルセルロースが挙げられる。錠剤はまた、ラクトース(一水和物、噴霧乾燥させた一水和物、無水など)、マンニトール、キシリトール、デキストロース、スクロース、ソルビトール、微結晶性セルロース、デンプン、および第二リン酸カルシウム二水和物などの希釈剤を含有することができる。
【0039】
錠剤はまた、ラウリル硫酸ナトリウムおよびポリソルベート80などの界面活性剤、ならびに二酸化ケイ素およびタルクなどの流動促進剤を含んでいてもよい。存在する場合には、界面活性剤は、錠剤の0.2重量%〜5重量%を占め、流動促進剤は、錠剤の0.2重量%〜1重量%を占めることができる。
【0040】
錠剤はまた、一般に、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、ステアリルフマル酸ナトリウム、およびステアリン酸マグネシウムとラウリル硫酸ナトリウムとの混合物などの滑沢剤を含有する。滑沢剤は、一般に、錠剤の0.25重量%〜10重量%、好ましくは、0.5重量%〜3重量%を占める。他の可能な成分としては、酸化防止剤、着色剤、香味剤、保存剤、および風味マスキング剤が挙げられる。
【0041】
例示的な錠剤は、薬物を約80%まで、結合剤を約10重量%〜約90重量%、希釈剤を約0重量%〜約85重量%、崩壊剤を約2重量%〜約10重量%、滑沢剤を約0.25重量%〜約10重量%含有する。錠剤ブレンドを、直接、またはローラーで圧縮して、錠剤を作製することができる。あるいは、錠剤ブレンドまたはブレンドの一部を、湿式、乾式、もしくは融解式粒状化、融解凍結化、または押出成形化した後、錠剤化することができる。最終製剤は、1層または複数層で構成することができ、コーティングされていても、コーティングされていなくてもよく、さらにカプセル化されていてもよい。錠剤の製剤化は、H.LiebermanおよびL.Lachmanによる「Pharmaceutical Dosage Forms:Tablets」、第1巻(Marcel Dekker、New York、1980)に論じられている。
【0042】
本発明の目的に適した調節放出製剤は、米国特許第6,106,864号に記載されている。高エネルギー分散剤ならびに浸透および被覆粒子剤などの他の適切な放出技術の詳細は、Vermaらによる「Pharmaceutical Technology On−line」、25(2)、1〜14(2001)に見られる。制御放出を実現するためのチューインガムの使用は、国際特許出願公開WO00/35298号に記載されている。
【0043】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩はまた、血流中、筋肉中、または内部器官中に直接投与することができる。非経口投与に適した手段としては、静脈内、動脈内、腹腔内、くも膜下腔内、脳室内、尿道内、胸骨内、頭蓋内、筋肉内、および皮下が挙げられる。非経口投与に適したデバイスとしては、有針(顕微針を含む)注射器、無針注射器、および注入技術が挙げられる。
【0044】
非経口製剤は、一般に、塩、炭水化物、および緩衝剤(好ましくはpHが3〜9)などの賦形剤を含有することができる水溶液であるが、いくつかの用途では、これらは、より適切には、滅菌非水溶液として、または乾燥形態として製剤化して、滅菌した発熱物質を含まない水などの適切なビヒクルと共に使用することができる。
【0045】
滅菌条件下、例えば凍結乾燥による非経口製剤の調製は、当業者によく知られている標準的製薬技術を使用して容易に実施することができる。
【0046】
非経口溶液の調製において使用する4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩の溶解性は、溶解促進剤を組み込むなど、適切な製剤技術を使用することで増大させることができる。非経口投与用製剤は、即時および/または調節放出になるように製剤化することができる。調節放出製剤としては、遅延、持続、パルス、制御、標的、および計画放出が挙げられる。
【0047】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩はまた、皮膚または粘膜に局所的に、すなわち皮膚的または経皮的に投与することができる。この目的のための典型的な製剤としては、ゲル剤、ヒドロゲル剤、ローション剤、液剤、クリーム剤、軟膏剤、散布剤、包帯剤、フォーム剤、フィルム剤、皮膚パッチ剤、ウエハー剤、インプラント剤、スポンジ剤、ファイバー剤、絆創膏剤、およびマイクロエマルジョン剤が挙げられる。また、リポソーム剤も使用することができる。典型的な担体としては、アルコール、水、鉱物油、流動ワセリン、白色ワセリン、グリセリン、ポリエチレングリコール、およびプロピレングリコールが挙げられる。浸透促進剤を組み込むことができ、例えば、FinninおよびMorganによるJ Pharm Sci、88(10)、955〜958(1999年10月)を参照されたい。
【0048】
局所投与の他の手段としては、エレクトロポレーション、イオントフォレーシス、フォノフォレーシス、ソノフォレーシス、および顕微針または無針(例えば、Powderject(商標)、Bioject(商標)など)注射による送達が挙げられる。
【0049】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩はまた、一般に、乾燥粉末用吸入器からの乾燥粉末(単独で、例えば、ラクトースとの乾燥ブレンドでの混合物として、または、例えば、ホスファチジルコリンなどのリン脂質と混合した混合成分粒子として)の形態で、または1,1,1,2−テトラフルオロエタンもしくは1,1,1,2,3,3,3−ヘプタフルオロプロパンなどの適切な噴霧剤の使用の有無にかかわらず加圧容器、ポンプ、スプレー、アトマイザー(好ましくは細かい霧を発生する電気流体力学法を使用するアトマイザー)、もしくはネブライザーからのエアロゾルスプレーとして、鼻腔内にまたは吸入によって投与することができる。鼻腔内使用では、粉末は、生体接着剤、例えば、キトサンまたはシクロデキストリンを含むことができる。
【0050】
加圧容器、ポンプ、スプレー、アトマイザー、またはネブライザーは、例えば、エタノール、エタノール水溶液、または有効物の分散、可溶化、もしくは拡張放出に適した代替試剤、溶媒としての噴霧剤(複数可)、およびトリオレイン酸ソルビタン、オレイン酸、またはオリゴ乳酸などの任意の界面活性剤を含む本発明の化合物(複数可)の溶液または懸濁液を含有する。
【0051】
乾燥粉末または懸濁製剤での使用の前に、薬物製品を、吸入による送達に適した大きさに微小化する(一般に5ミクロン未満)。これは、スパイラルジェットミル法、フルイドベッドジェットミル法、ナノ粒子を形成するための超臨界流体プロセス法、高圧ホモジナイズ法、または噴霧乾燥法などの任意の適切な粉砕法によって実現することができる。
【0052】
吸入器または注入器において使用されるカプセル剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース製)、ブリスター剤、およびカートリッジ剤は、薬物製品と、ラクトースまたはデンプンなどの適切な粉末基剤と、l−ロイシン、マンニトール、またはステアリン酸マグネシウムなどの性能調節剤との粉末混合物を含有するように製剤化することができる。ラクトースは、無水でも一水和物の形態でもよく、好ましくは後者である。他の適切な賦形剤としては、デキストラン、グルコース、マルトース、ソルビトール、キシリトール、フルクトース、スクロース、およびトレハロースが挙げられる。
【0053】
吸入/鼻腔内投与を目的とする本発明のこれらの製剤に、メントールおよびレボメントールなどの適切な香料、またはサッカリンもしくはサッカリンナトリウムなどの甘味料を添加することができる。
【0054】
乾燥粉末用吸入器およびエアロゾルの場合、投薬単位は、計量した量を送達するバルブによって決定される。本発明による単位は、一般に、リスト(I)の化合物を1μg〜100mg含有する計量済み用量または「パフ(puff)」を投与するように構成されている。総1日用量は、典型的には、1μg〜200mgの範囲であり、これは、単回用量で、または、より一般には、1日を通しての分割用量として投与することができる。
【0055】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩はまた、典型的には、等張のpH調整した滅菌生理食塩水において微細化された懸濁液または溶液の液滴の形態で、眼または耳に直接投与することができる。眼および耳への投与に適した他の製剤としては、軟膏剤、生分解性(例えば、吸収性ゲルスポンジ、コラーゲン)および非生分解性(例えば、シリコーン)のインプラント剤、ウエハー剤、レンズ、ならびにニオソームまたはリポソームなどの粒子または小胞系が挙げられる。架橋ポリアクリル酸、ポリビニルアルコール、ヒアルロン酸などのポリマー、セルロースポリマー、例えば、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、もしくはメチルセルロース、またはヘテロ多糖ポリマー、例えばジェランガムを、塩化ベンザルコニウムなどの保存剤と一緒に組み込むことができる。こうした製剤はまた、イオントフォレーシスによって送達することができる。
【0056】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩は、前述の投与様式のいずれかで使用するために、シクロデキストリンおよび適切なその誘導体またはポリエチレングリコール含有ポリマーなどの可溶な巨大分子体と組み合わせて、これらの溶解性、溶出速度、風味マスキング、生体利用能、および/または安定性を向上させることができる。
【0057】
薬物−シクロデキストリン複合体は、例えば、ほとんどの剤形および投与経路にとって一般に有用であることが分かっている。包接複合体と非包接複合体のどちらも使用することができる。薬物との直接複合体化の代わりに、シクロデキストリンを、補助添加剤として、すなわち、担体、希釈剤、または可溶化剤として使用することができる。これらの目的には、α、β、およびγ−シクロデキストリンが最も一般的に使用されており、これらの例は、国際特許出願公開WO91/11172号、同WO94/02518号、および同WO98/55148号に見ることができる。
【0058】
ヒト患者への投与では、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩の総1日用量は、典型的には、1mg〜10g、例えば10mg〜1gなど、例えば25mg〜500mgの範囲にあるが、当然ながら投与様式および効果に依存する。総1日用量は、単回用量または分割用量で投与することができ、特定の患者の年齢、体重、および応答に応じて、医師の裁量で本明細書に示される典型的な範囲から外れる可能性がある。
【0059】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたはトシル酸塩などの薬学的に許容できるその塩は、痛風の治療のために、別の1種の薬理学的に有効な化合物、または別の2種以上の薬理学的に有効な化合物と有用に組み合わせることができる。こうした組合せから、患者のコンプライアンス、投薬の容易さ、および相乗活性を含めた著しい利益がもたらされる可能性がある。
【0060】
下記の組合せでは、本発明の化合物は、別の治療剤または薬剤と組み合わせて、同時に、逐次的に、または別々に投与することができる。
【0061】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド、またはトシル酸塩などの薬学的に許容できるその塩は、以下から選択される1種または複数の薬剤と組み合わせて投与することができる:
・抗炎症薬、例えば、NSAID(例えば、セレコキシブ)、コルヒチン、またはステロイドなど;
・キサンチン酸化酵素阻害剤(例えば、アロプリノール、フェブキソスタット、またはチソプリン)、またはプリンヌクレオシドホスホリラーゼ(PNP)阻害剤(例えば、ウロデシン);
・ウリカーゼ(例えば、ペグロチカーゼまたはラスブリカーゼ);または
・尿酸排泄薬、例えば、ベンズヨーダロン、イソブロミンジオン、プロベネシッドおよびスルフィンピラゾン、またはURAT−1阻害剤(例えば、ベンズブロマロン)などの、血液中への尿酸の腎再吸収に関与する輸送体を阻害する薬剤など。
【0062】
本明細書における治療に関するすべての言及には、治癒的、姑息的、および予防的治療があることを理解されたい。
【0063】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドは、類似構造の化合物を調製するために当技術分野でよく知られている任意の方法によって、詳細には、国際特許出願公開WO2010/079443号に記載の実施例788などに詳述されている特定の方法によって調製することができる。
【0064】
本発明を以下の非限定的な実施例により説明する。
【実施例1】
【0065】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の調製
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド(国際特許出願公開WO2010/079443号の実施例788、36.75g、73.45mmol)を含む酢酸エチル(20mL/g、735mL)に、メタノール(3mL/g、110.25mL)を添加し、混合物を50℃に加熱した。p−トルエンスルホン酸一水和物(13.27g、69.77mmol)のメタノール(2mL/g、73.50mL)溶液を、反応混合物に滴下漏斗で6分間かけて添加し、続いて、メタノール(1mL/g、36.75mL)をさらに添加した。反応混合物を室温に冷却し、真空下でろ過し、固体を酢酸エチル:メタノール(9:1、2×37mL)で洗浄した。固体を50℃の真空下で一晩乾燥させて、表題化合物を易流動性の灰色がかった白色固体(43.99g、65.41mmol、89%)として得た。
【0066】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩の分光分析の詳細を以下に示す。
【0067】
赤外線(IR)分光法
赤外吸収スペクトルは、単一反射の減衰全反射法(ATR)を使用して記録した。スペクトルは、ThermoNicolet Avatar 360 FT IR分光計およびSmart Golden Gate(商標)付属品を使用して、4cm−1分解で得た。この手法は試料調製が不要であった。スペクトルは図1に示している。
【0068】
質量分析法(MS)
フルスキャン質量スペクトルは、図2および図3に示しており、エレクトロスプレーの正(ES+)および負(ES−)イオン化によってそれぞれ得た。データは、エレクトロスプレー源を取り付けたBruker MaXis Quadrupole Time of Flight質量分析計を使用して記録した。内部較正はギ酸ナトリウム溶液を使用して行い、質量範囲m/z113〜m/z997に関して0.2mDa(ES+)および0.3mDa(ES−)の最大質量偏差が観測された。
【0069】
ES+ならびにES−データに関する精密質量測定、理論的モノアイソトピック質量、ならびに観測される付加体およびフラグメントイオンの分子式は、表1および2にそれぞれ示している。対応する質量スペクトルは、図2および3にそれぞれ示している。
【0070】
【表1】
【0071】
【表2】
【0072】
核磁気共鳴(NMR)分光法
プロトン(1H)NMRスペクトルは、液体でDMSOd6において得た。データは、599.77MHzでプロトンにチューニングされた三重共鳴低温プローブを装備したBruker AVANCE III 600MHz NMR分光計において30℃で得た。スペクトルは、DMSOd5(2.50ppm)を基準とした。
【0073】
図4に示す、下記の構造に関して標識した1H NMRスペクトルは、12個の芳香族プロトンおよび3個の脂肪族(1個のCH3基)プロトンの存在を明らかにしている。1Hの化学シフト帰属は、表3に要約している。
【0074】
【化1】
【0075】
【表3】
【0076】
紫外線/可視(UV/Vis)分光測光法
UV/可視スペクトルは、Hitachi U−3000分光光度計を使用して、メタノールにおいて1.09mg/100mLの濃度で得た。それを図5に示している。2個のλ最大が281nmおよび240nmに観察されている。
【0077】
PXRDによる4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の特性分析
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の粉末X線回折パターンを、自動試料交換器、θ−θゴニオメータジオメトリ、自動ビーム発散スリット、およびPSD Vantec−1検出器を取り付けたBruker−AXS Ltd.のD4 ENDEAVOR粉末X線回折計を使用して決定した。試料を0.5mmのキャビティを有する低バックグラウンドシリコンウエハー標本マウント上に載置して分析準備をした。X線管を35kV/40mAで操作して、標本を、銅のKα1X線(波長=1.5406Å)で照射しながら回転させた。分析は、データ収集に関して、室温で2°〜55°の2θ範囲にわたって0.018°ステップサイズにつき0.2秒の連続モード設定とした。ピーク検索は、閾値および幅のパラメータをそれぞれ1および0.3に設定したものを使用して、Bruker−AXSから発売されたEvaソフトウェアを用いて行った。機器の較正は、コランダム標準品(NIST:SRM 1976 XRD平板強度標準)を使用して確認した。
【0078】
機器、試料、および試料調製の違いにより、本明細書では、ピーク値は、ピーク値のばらつきから推定して報告している。これは、ピーク値に固有の偏差のために、固体状態の化学技術分野において普通に行われている。粉末X線回折での2θX軸値の典型的なばらつきは、+または−0.2°2θ程度である。
【0079】
ピーク強度のばらつきは、個々の結晶が外部X線源に対して試料容器中でどのように配向しているかの結果である(「優先配向」として知られている)。この配向効果は、結晶に関する構造情報を提供するものではない。
【0080】
さらに、本発明の結晶材料が薬学的賦形剤などの追加成分と混合されるか、またはそれらで希釈される場合、上述の特徴的ピークの強度が変わるであろうことも当業者なら理解されよう。このため、諸成分の混合物の特徴的ピークの検出を可能にするために、上述のPXRD法をわずかに最適化しなければならないことを当業者なら理解されよう。この最適化には、より強力なX線源(波長=1.5406Å)、わずかに異なるステップサイズ、またはわずかに異なるステップ時間の使用が挙げられる。
【0081】
また、異なる波長を使用して測定すると、ブラッグの式nλ=2d sinθに従って異なるシフトが得られることを当業者なら理解されよう。代替波長の使用によって生じるこうしたさらなるPXRDパターンは、本発明の結晶材料のPXRDパターンの代替表現と考えられ、したがって、本発明の範囲内に含まれる。
【0082】
PXRDパターンは、図6に示している。主な2θピーク位置および相対強度は、表4および4aに列挙している。
【0083】
【表4】
【0084】
【表5】
【実施例2】
【0085】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド噴霧乾燥拡散(SDD)の調製
テトラヒドロフラン(安定剤不含、14.5kg)および水(0.76kg)を、上部マウントミキサーを装備したステンレス鋼槽に加えた。次いで、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド(742.4g)を溶液に添加し、固体がすべて完全に溶解するまで、少なくとも1時間混合した。(酢酸/コハク酸)ヒドロキシプロピルメチルセルロース(中級顆粒、1338.4g)を溶液に添加し、完全に溶解するまで混合した。次いで、下表の条件を使用して、溶液を窒素ガス下で噴霧乾燥させた。
【0086】
【表6】
【0087】
次いで、得られた4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのSDDを、対流トレー乾燥機において40℃/50%相対湿度(RH)で最低6時間トレー乾燥させ、続いて、RHを40℃/75%まで上昇させ、さらに最低25時間トレー乾燥させた。
【0088】
SDDは、必要となるまで2〜8℃で保存した。
【実施例3】
【0089】
経口投与用分散剤の調製:
(a)4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのトシル酸塩を使用
メチルセルロースビヒクル(0.5%w/v)を以下のように調製した。ビーカーにおいて潅注水(water for irrigation)(600mL)を80℃〜90℃に加熱した。メチルセルロース(5g)粉末を、粉末が完全に分散するまで撹拌しながら添加した。次いで、分散物を氷浴へ移動させ、冷精製水(400mL)を添加しながら素早く冷却させて、透明な溶液を得た。
【0090】
必要量の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩(10mg〜2400mg)を適切な大きさのガラス製褐色投薬瓶に秤量し、ある量のビヒクル(0.5%(w/v)メチルセルロース)を添加して、経口投与用分散剤を調製した。添加したビヒクル量は、薬物濃度が範囲0.6〜50mg/mLになるように、用量に応じて決定した:範囲10mg〜30mg未満の薬物用量では15mL;範囲30mg〜2400mgの薬物用量では50mL。
【0091】
分散剤を2〜8℃で保存し、72時間以内に投薬容器から直接投与した。投与後、ガラス製投薬瓶をほぼ等量の飲料水で2回すすいで、投薬量を含めた全投与量が240mLになるようにした。
【0092】
(b)4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドのSDDを使用
メチルセルロースビヒクル0.5%(w/v)を、上述の実施例3(a)で詳述した手順を使用して調製した。
【0093】
必要量の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドSDDを適切な大きさのガラス製褐色投薬瓶に秤量し、ある量のビヒクル(0.5%(w/v)メチルセルロース)を添加して、経口投与用分散剤を調製した。添加したビヒクル量は、薬物濃度が範囲0.6〜50mg/mLになるように、用量に応じて決定した:用量範囲10mg〜2400mgでは20mL〜100mL。
【0094】
分散剤を2〜8℃で保存し、72時間以内に投薬容器から直接投与した。投与後、ガラス製投薬瓶をほぼ等量の飲料水で2回すすいで、投薬量を含めた全投与量が240mLになるようにした。
【0095】
生物学的活性
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドおよびトシル酸塩などの薬学的に許容できるその塩が血中尿酸レベルを低下させる能力について、次の実験で実証した。尿酸測定は、市販の比色測定法キット(Beckman Coulter)を使用して行った。
【実施例4】
【0096】
単回用量研究:6コホートの健常な対象者における二重盲検無作為化プラセボ対照クロスオーバー研究。
範囲が10mg〜2400mgの4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドSDD(「SDD」)経口投与用分散剤の単回用量を調査した。さらに、範囲が200mg〜1000mgの4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩(「TS」)経口投与用分散剤の単回用量も調査した。
【0097】
以下の用量を調査し、すべての用量を絶食状態で与えたが、例外として、SDD分散剤200mgおよび1000mgの場合は、絶食状態と高脂肪食後(「摂食」)の両方で与えた。
コホート1:SDD10mg、SDD100mg、SDD300mg、TS200mg、プラセボ
コホート2:SDD30mg、SDD300mg、SDD(摂食)200mg、プラセボ
コホート3:SDD100mg、SDD200mg、SDD300mg、プラセボ
コホート4:SDD450mg、SDD600mg、SDD800mg、SDD1000mg、プラセボ
コホート5:TS600mg、TS1000mg、SDD(摂食)1000mg
コホート6:SDD1250mg、SDD1600mg、SDD2000mg、SSD2400mg、プラセボ
【0098】
合計61人の対象者(すべて男性)が参加し、全員がSDDまたはTS経口投与用分散剤の投与を少なくとも1回受けた。
【0099】
用量群ごとの尿酸レベルの平均データを以下のように示している。
・SDD絶食 表5
・SDD摂食および絶食 表6
・TS 表7
【0100】
【表7】
【0101】
【表8】
【0102】
【表9】
【0103】
投与の48時間後に、血液中の尿酸の用量依存的な低下が認められた。通常、10〜1000mgの用量では、値は正常範囲(3.5〜7.2mg/dL)内にあったが、1250〜2400mgの用量では、尿酸レベルの低下はより著しく、対象者の少なくとも半数が、投与48時間後で正常下限(LNN)値よりも低くなった。すべての投与前値および追跡値は、LNNよりも高かった。投与48時間後にLNNをやや下回る1人の対象者を除くプラセボの全対象者(n=42)が、正常範囲内の尿酸値となった。
【実施例5】
【0104】
多回用量研究:健常な対象者における二重盲検無作為化プラセボ対照研究。
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドSDD経口投与用分散剤の100mg1日2回(BID)、300mgBID、および600mgBIDの多回経口用量、ならびにプラセボを14日間調査した。対象者は、朝の投与前の一晩および晩の投与前の少なくとも2時間は絶食した。投与後の少なくとも2時間は食物を控えた。
【0105】
合計30人の対象者(すべて男性)を研究に登録し、27人の対象者が研究を完了した(3人の対象者は、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド600mgBIDでの治療中に有害事象が生じたので研究を止めた)。
【0106】
用量群ごとの尿酸レベルの平均データを表8および図7に示している。
【0107】
【表10】
【0108】
血液中の尿酸の用量依存的な低下は、4日目に明らかになり(最初の投与後評価)、最低平均値は4または7日目に生じた。8人中5人の対象者が、7日目に、100mgBID(低尿酸の対象者の実範囲は2.6〜3.4mg/dLであり、1人の対象者はベースラインの限界より低く、実値は2.7mg/dL)および300mg(低尿酸の対象者の実範囲は2.2〜3.4mg/dL)で尿酸値がLLNより低くなった。全対象者が、600mgBID投与の4および7日目に尿酸値がLNNより低くなった(実値は<1.5〜3.0mg/dL)。値は継続投与したにもかかわらず10および14日目に通常増加したが、1人を除く全対象者が16日目に正常範囲に戻った(最終投与の2日後)。残りの対象者は、ベースライン時での尿酸値が最低(4.7mg/dL)であり、継続値は、2.2mg/dL(4日目)、1.5mg/dL未満(7日目)、1.6mg/dL(10日目)、3.1mg/dL(14日目)、および追跡時4.9mg/dLであった。プラセボ投与を受けた全対象者(n=6)は、全時点で正常範囲内の尿酸濃度であった。
【実施例6】
【0109】
多回用量研究:健常な対象者および高齢の対象者における二重盲検無作為化プラセボ対照研究。
以下のように、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドTS経口投与用分散剤の多回経口用量、ならびにプラセボを14日間調査した。
・健常な対象者、300mg1日2回(BID)
・健常な対象者、450mgBID、および
・高齢の対象者、300mgBID。
【0110】
対象者は、朝の投与前の一晩および晩の投与前の少なくとも2時間は絶食した。投与後の少なくとも2時間は食物を控えた。
【0111】
合計49人の対象者を研究に登録し、そのうちの39人が4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドTS投与を受け、10人がプラセボ投与を受けた。
【0112】
用量群ごとの平均血中尿酸レベルおよび尿排泄データを表9に示している。図8は、尿中に排泄された尿酸のパーセントを示している。
【0113】
【表11】
【0114】
血液中の尿酸濃度は、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドの投与後に低下した。全対象者が、1日目では血中尿酸値がLLNよりも高かった。プラセボ投与を受けた対象者は、研究期間を通して血中尿酸濃度がLLNよりも高かった。450mgBID投与を受けた対象者では、血中尿酸濃度の中央値は3日目にLLNよりも低くなり、6日目には、全コホート(300mgおよび450mgBID)の中央値がLLNよりも低くなった。全対象者が、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド投与を中止して2日以内(すなわち16日目までに)に、血中尿酸濃度がLLNよりも高く戻った。
【0115】
1日目、次いで、6、14、および16日目の投与前の24時間にわたって採取された尿中の尿酸も測定した。尿中尿酸の排泄率パーセントを計算し、線形混合効果モデルで分析した。これらのデータの要約は、図8に示している。データは、尿中尿酸の排泄率が、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドの投与中には増加し、16日目までにベースラインに戻ることを示唆している。
【実施例7】
【0116】
URAT−1阻害活性
URAT−1輸送体(tranporter)の阻害剤としての4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドの効力を、以下のように決定した。
【0117】
HEK293細胞を、熱不活性化ウシ胎児血清(10%v/v)、100U/mLのペニシリン、および100μg/mLのストレプトマイシンを補充した、L−GlutaMax(1L当たりグルコース4.5g)を含むダルベッコの改変イーグル培地(DMEM)からなる培地で成長させた。HEK細胞を、約95%空気/5%CO2で約37℃の加湿インキュベーターにおいて75cm2組織培養フラスコで常法通り培養した。ほぼコンフルエントなHEK細胞培養物をトリプシン処理により採取し、培地に再懸濁させ、このプロセスを週に1または2回繰り返して、使用に十分な細胞を得た。
【0118】
取込み実験では、HEK293細胞を、ポリ−D−リシンでコーティングした24ウェルプレートにウェル当たり4×105細胞の密度で播種した。約5%CO2の空気が入っている加湿インキュベーターにおいて、細胞を約37℃で1日間培養した。その後、リポフェクタミン(Lipofectamine)2000試薬を使用して、細胞をpcDNA3.1/hygro/URAT1(HEK−URAT1細胞)またはpcDNA3.1/hygro(HEK対照細胞)でトランスフェクトした。約5%CO2の空気が入っている加湿インキュベーターにおいて約37℃で約24時間後、細胞を実験に使用した。
【0119】
トランスフェクトした1日後、培地をウェルから除去し、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド(0〜30μM)の非存在下および存在下で、細胞を塩素不含インキュベーション培地(125mMのグルコン酸Na、4.8mMのグルコン酸K、1.3mMのグルコン酸Ca、1.2mMのKH2PO4、1.2mMのMgSO4、5.6mMのD−グルコース、25mMのHEPES、pH7.4)0.2mLと共に約37℃で15分間プレインキュベートした。その後、インキュベーション培地を除去し、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミド(0〜30μM)の非存在下および存在下で、[14C]尿酸(20μM)を含有する塩素不含インキュベーション培地0.2mLを添加し、これをトリプリケートで行った。細胞を約37℃で2分間インキュベートした。インキュベーションの終わりに、培地を吸引し、単層を氷冷インキュベーション培地1mLで素早く2回すすいだ。続いて、細胞を0.5NのNaOH0.5mLで可溶化し、各ウェルの一定量の細胞溶解物試料をシンチレーションバイアルに収集した。[14C]尿酸の濃度を液体シンチレーション計数器(LSC)で決定した。[14C]尿酸輸送の阻害はまた、よく知られている阻害剤ベンズブロマロン(30μM)の存在下で決定した。最終的な有機溶媒は、1%(v/v)未満であった。
【0120】
可溶化したHEK細胞のタンパク質含有量は、タンパク質標準物としてウシ血清アルブミン(BSA)(濃度域0〜1mg/mL)を含むバイオ−ラッドブラッドフォード(Bio−Rad Bradford)試薬を使用して、ブラッドフォード法により決定した。BSA溶液または可溶化した細胞を、希釈した染料試薬濃縮物(Bio−Rad製)と混合した。室温で10分間インキュベートした後、595nmで吸光度を測定した。
【0121】
細胞溶解物試料中の放射活性量を、液体シンチレーション計数器(LSC)により決定した。液体シンチラント(Hionic Fluor(商標))を全試料に添加し、放射活性を、QuantaSmart(商標)ソフトウェアを使用して、TriCarb3100TR液体シンチレーションカウンタのLSCにより決定し、これらのカウントはすべて、tSIE/AEC(自動効率補正つき外部標準変換スペクトル指数(transformed Spectral Index of external standards coupled to Automatic Efficiency Correction))を使用して、DPMに変換した。機器の較正手順は、試験機関で確立されている。試料はすべて少なくとも2分間カウントした。バックグラウンド値は、試料を含まない液体シンチラントを使用して、各試料系列で測定した。HEK細胞中の[14C]−尿酸の蓄積(pmol/mgタンパク質)を計算し、IC50値を、50%輸送阻害に必要な阻害剤の濃度と定義し、ヒル(Hill)式を用いるグラフパッドプリズム(GraphPad Prism)バージョン4.00を使用して計算した。
【0122】
データ
上述の方法で測定した4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドによる尿酸取込みの阻害は、基準化合物ベンズブロマロンに対して正規化すると、3.54μMである。
【図1】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【特許請求の範囲】
【請求項1】
高血中尿酸レベルに関連する疾患を治療するための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩。
【請求項2】
高血中尿酸レベルに関連する疾患が高尿酸血症である、請求項1に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩。
【請求項3】
高血中尿酸レベルに関連する疾患が痛風である、請求項1に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩。
【請求項4】
高血中尿酸レベルに関連する疾患を治療するための医薬の製造のための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩の使用。
【請求項5】
高血中尿酸レベルに関連する疾患が高尿酸血症である、請求項4に記載の使用。
【請求項6】
高血中尿酸レベルに関連する疾患が痛風である、請求項4に記載の使用。
【請求項7】
高血中尿酸レベルに関連する疾患を治療する方法であって、こうした治療を必要とする対象への、治療有効量の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩の投与を含む方法。
【請求項8】
高血中尿酸レベルに関連する疾患を治療するための、1種または複数の追加の薬学的に活性な薬剤と組み合わせた、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩。
【請求項9】
1種または複数の追加の薬学的に活性な薬剤が、
・抗炎症薬、例えば、NSAID(例えば、セレコキシブ)、コルヒチン、またはステロイドなど;
・キサンチン酸化酵素阻害剤(例えば、アロプリノール、フェブキソスタット、またはチソプリン)、またはプリンヌクレオシドホスホリラーゼ(PNP)阻害剤(例えば、ウロデシン);
・ウリカーゼ(例えば、ペグロチカーゼまたはラスブリカーゼ);または
・尿酸排泄薬、例えば、ベンズヨーダロン、イソブロミンジオン、プロベネシッドおよびスルフィンピラゾン、またはURAT−1阻害剤(例えば、ベンズブロマロン)などの、血液中への尿酸の腎再吸収に関与する輸送体を阻害する薬剤など
から選択される、請求項8に記載の組合せ。
【請求項10】
高血中尿酸レベルに関連する疾患が高尿酸血症である、請求項8または9のいずれか一項に記載の組合せ。
【請求項11】
高血中尿酸レベルに関連する疾患が痛風である、請求項8または9のいずれか一項に記載の方法。
【請求項12】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項13】
結晶性固体である、請求項12に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項14】
CuKα1X線放射(波長=1.5406Å)の使用による、9.0、9.3、10.0、10.7、11.6、12.5、12.9、13.2、13.8、14.4、16.0、16.6、17.5、17.8、18.1、21.4、および23.4°(±0.2°2θ)からなる群から選択される3、4、5、または6個の特徴的2シータ(2θ)ピークのうちのいずれかを示す粉末X線回折パターン(PXRD)パターンを特徴とする、請求項13に記載の結晶性固体。
【請求項15】
請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩と薬学的に許容できる担体とを含む医薬組成物。
【請求項16】
錠剤またはカプセル剤の形態である、請求項15に記載の医薬組成物。
【請求項17】
医薬として使用するための、請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項18】
NaV1.7阻害剤の適応がある障害の治療において使用するための、請求項17に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項19】
NaV1.7阻害剤の適応がある障害が、疼痛、好ましくは、神経障害性疼痛、侵害受容性疼痛、または炎症性疼痛である、請求項18に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項20】
高血中尿酸レベルに関連する疾患の治療において使用するための、請求項17に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項21】
疼痛、好ましくは、神経障害性疼痛、侵害受容性疼痛、または炎症性疼痛を治療するための医薬の製造のための、請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の使用。
【請求項22】
高血中尿酸レベルに関連する疾患を治療するための医薬の製造のための、請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の使用。
【請求項23】
疼痛、好ましくは、神経障害性疼痛、侵害受容性疼痛、または炎症性疼痛を治療する方法であって、こうした治療を必要とする対象への、治療有効量の請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の投与を含む方法。
【請求項24】
高血中尿酸レベルに関連する疾患を治療する方法であって、こうした治療を必要とする対象への、治療有効量の請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の投与を含む方法。
【請求項1】
高血中尿酸レベルに関連する疾患を治療するための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩。
【請求項2】
高血中尿酸レベルに関連する疾患が高尿酸血症である、請求項1に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩。
【請求項3】
高血中尿酸レベルに関連する疾患が痛風である、請求項1に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩。
【請求項4】
高血中尿酸レベルに関連する疾患を治療するための医薬の製造のための4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩の使用。
【請求項5】
高血中尿酸レベルに関連する疾患が高尿酸血症である、請求項4に記載の使用。
【請求項6】
高血中尿酸レベルに関連する疾患が痛風である、請求項4に記載の使用。
【請求項7】
高血中尿酸レベルに関連する疾患を治療する方法であって、こうした治療を必要とする対象への、治療有効量の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩の投与を含む方法。
【請求項8】
高血中尿酸レベルに関連する疾患を治療するための、1種または複数の追加の薬学的に活性な薬剤と組み合わせた、4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドまたは薬学的に許容できるその塩。
【請求項9】
1種または複数の追加の薬学的に活性な薬剤が、
・抗炎症薬、例えば、NSAID(例えば、セレコキシブ)、コルヒチン、またはステロイドなど;
・キサンチン酸化酵素阻害剤(例えば、アロプリノール、フェブキソスタット、またはチソプリン)、またはプリンヌクレオシドホスホリラーゼ(PNP)阻害剤(例えば、ウロデシン);
・ウリカーゼ(例えば、ペグロチカーゼまたはラスブリカーゼ);または
・尿酸排泄薬、例えば、ベンズヨーダロン、イソブロミンジオン、プロベネシッドおよびスルフィンピラゾン、またはURAT−1阻害剤(例えば、ベンズブロマロン)などの、血液中への尿酸の腎再吸収に関与する輸送体を阻害する薬剤など
から選択される、請求項8に記載の組合せ。
【請求項10】
高血中尿酸レベルに関連する疾患が高尿酸血症である、請求項8または9のいずれか一項に記載の組合せ。
【請求項11】
高血中尿酸レベルに関連する疾患が痛風である、請求項8または9のいずれか一項に記載の方法。
【請求項12】
4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項13】
結晶性固体である、請求項12に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項14】
CuKα1X線放射(波長=1.5406Å)の使用による、9.0、9.3、10.0、10.7、11.6、12.5、12.9、13.2、13.8、14.4、16.0、16.6、17.5、17.8、18.1、21.4、および23.4°(±0.2°2θ)からなる群から選択される3、4、5、または6個の特徴的2シータ(2θ)ピークのうちのいずれかを示す粉末X線回折パターン(PXRD)パターンを特徴とする、請求項13に記載の結晶性固体。
【請求項15】
請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩と薬学的に許容できる担体とを含む医薬組成物。
【請求項16】
錠剤またはカプセル剤の形態である、請求項15に記載の医薬組成物。
【請求項17】
医薬として使用するための、請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項18】
NaV1.7阻害剤の適応がある障害の治療において使用するための、請求項17に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項19】
NaV1.7阻害剤の適応がある障害が、疼痛、好ましくは、神経障害性疼痛、侵害受容性疼痛、または炎症性疼痛である、請求項18に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項20】
高血中尿酸レベルに関連する疾患の治療において使用するための、請求項17に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩。
【請求項21】
疼痛、好ましくは、神経障害性疼痛、侵害受容性疼痛、または炎症性疼痛を治療するための医薬の製造のための、請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の使用。
【請求項22】
高血中尿酸レベルに関連する疾患を治療するための医薬の製造のための、請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の使用。
【請求項23】
疼痛、好ましくは、神経障害性疼痛、侵害受容性疼痛、または炎症性疼痛を治療する方法であって、こうした治療を必要とする対象への、治療有効量の請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の投与を含む方法。
【請求項24】
高血中尿酸レベルに関連する疾患を治療する方法であって、こうした治療を必要とする対象への、治療有効量の請求項12から14のいずれか一項に記載の4−[2−(5−アミノ−1H−ピラゾール−4−イル)−4−クロロフェノキシ]−5−クロロ−2−フルオロ−N−(1,3−チアゾール−4−イル)ベンゼンスルホンアミドトシル酸塩の投与を含む方法。
【公開番号】特開2013−87119(P2013−87119A)
【公開日】平成25年5月13日(2013.5.13)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−229438(P2012−229438)
【出願日】平成24年10月17日(2012.10.17)
【出願人】(597014501)ファイザー・リミテッド (107)
【氏名又は名称原語表記】Pfizer Limited
【住所又は居所原語表記】Ramsgate Road, Sandwich, Kent, England
【Fターム(参考)】
【公開日】平成25年5月13日(2013.5.13)
【国際特許分類】
【出願番号】特願2012−229438(P2012−229438)
【出願日】平成24年10月17日(2012.10.17)
【出願人】(597014501)ファイザー・リミテッド (107)
【氏名又は名称原語表記】Pfizer Limited
【住所又は居所原語表記】Ramsgate Road, Sandwich, Kent, England
【Fターム(参考)】
[ Back to top ]