
新規な機能性ペプチド核酸およびその製法
【課題】コストパフォーマンスに優れ、かつ機能性分子を超高速に導入することができる、機能性PNAの新規合成方法およびそれに用いる化合物の提供。
【解決手段】機能性PNAオリゴマーを製造する方法であって、保護基によって保護されたアデニン、グアニン、シトシンまたはチミンを有するPNAモノマーユニットを、Fmoc−ω−アミノ酸−BocPNA−OHと反応させてPNAオリゴマーを合成した後、該PNAオリゴマーに遊離カルボン酸を有する機能性分子を導入し、さらに前記保護基の脱保護を行うことによって、機能性PNAオリゴマーを製造することを含む特徴とする前記方法、該方法によって合成される化合物および前駆体的PNAモノマーユニットとして機能するFmoc−ω−アミノ酸−BocPNA−OH。
【解決手段】機能性PNAオリゴマーを製造する方法であって、保護基によって保護されたアデニン、グアニン、シトシンまたはチミンを有するPNAモノマーユニットを、Fmoc−ω−アミノ酸−BocPNA−OHと反応させてPNAオリゴマーを合成した後、該PNAオリゴマーに遊離カルボン酸を有する機能性分子を導入し、さらに前記保護基の脱保護を行うことによって、機能性PNAオリゴマーを製造することを含む特徴とする前記方法、該方法によって合成される化合物および前駆体的PNAモノマーユニットとして機能するFmoc−ω−アミノ酸−BocPNA−OH。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、機能性ペプチド核酸モノマーの新規な製造方法、該製造方法によって製造された機能性ペプチド核酸オリゴマーおよびその中間体に関する。より詳細には、前駆体的PNAモノマーユニットをPNAオリゴマーに導入した後に1種または2種以上の機能性分子をポスト合成的に導入することを特徴とする、前記製造方法に関する。
【背景技術】
【0002】
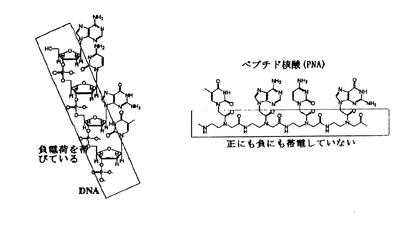
核酸は生物の遺伝情報を司るDNAおよびRNAである。これに対して、ペプチド核酸(PNA)とは、核酸の糖リン酸骨格をN−(2−アミノエチル)グリシン骨格に変換した修飾核酸である(図1)。DNA/RNAの糖リン酸骨格は中性条件で負電荷を帯びていて相補鎖間の静電的な反発があるが、PNAの背骨構造はもともと電荷を持たないので静電的な反発がない。そのためPNAは従来の核酸と比較して、高い二重鎖形成能をもち、高い塩基配列認識能を持つ。さらにPNAは生体内ヌクレアーゼ・プロテアーゼに対し非常に安定で分解されないので、アンチセンス分子として遺伝子治療に応用することが検討されている。
【0003】
従来のDNAを媒体にしていた技術をPNA化することにより、これまで克服できなかったDNAの欠点を補うことが可能となった。例えば、遺伝情報の体系的な解析を高速に且つ大量に行うための「DNAマイクロアレイ技術」および塩基配列を特異的に認識したことを蛍光発光により検出できるプローブとして最近開発された「モレキュラービーコン」に応用することが可能である。これらはいずれも酵素耐性に乏しいDNAを媒体とするため、これらの技術を用いるに際しては厳密なサンプリングが要求される。この要求を満たすことが、前記の技術を高度化する上での鍵となっている。
【0004】
一方PNAは酵素に対し完全な耐性を持つので、DNAマイクロアレイ技術およびモレキュラービーコンにおいてPNAをDNAに代用することによって、前記技術の欠点が克服され、さらに長所が引き出されるものと期待されている。DNAマイクロアレイ技術およびモレキュラービーコン以外にもPNA化することにより発展が期待される分野は数多いが、それらにおいてはPNAの効率的な機能化、すなわちPNAモノマーへの機能性分子の効率的な導入による新規なPNAモノマーの設計が必要である。
【0005】
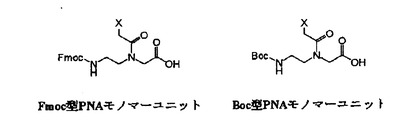
PNAオリゴマーの合成方法には通常の固相ペプチド合成法を用いるので、PNAモノマーユニットをPNAの背骨構造によって分類すると、Fmoc型PNAモノマーユニットとtBoc型PNAモノマーユニットの2種類が含まれる(図2)。
【0006】
Fmoc型PNAモノマーユニットの合成方法は既に確立されており、しかもそのオリゴマーの合成は一般的なDNA自動合成機によって可能であるため、下記のルート
【0007】
【化1】

【0008】
によって、少量スケールでの合成が可能となっている。
【0009】
当初PNAには下記のようなtBoc型PNAモノマーユニット
【0010】
【化2】
【0011】
が採用され、その後より効率のよい合成方法
【0012】
【化3】
【0013】
が確立された。しかし、前述したように取り扱いが容易なFmoc型が開発されたため、tBoc型の使用頻度は減少している。
【0014】
しかし、グアニン・チミン・シトシン・アデニン4種類の核酸塩基以外の機能性分子を導入する際、例えば光機能性分子を導入する際には、導入する機能性分子がアルカリ条件に不安定な場合が多いので、アルカリ条件を使用しないtBoc型PNA背骨構造の有用性は高い。「t−ブトキシカルボニルアミノエチルアミン及びアミノ酸誘導体の製造方法」に関しては、本発明者らが特願2000−268638として既に特許出願中である。
【0015】
これ以外にも、光機能性オリゴPNAのモノマーユニットの合成例は過去に5例が知られている。これら全てが上記ルートを用いているが、その収率については記載がないか、または極めて低いものでしかない(Peter E. Nielsen, GeraldHaaiman, Anne B. Eldrup PCT Int. Appl. (1998) WO 985295 A1 19981126, T. A. Tran, R.-H. Mattern, B. A. Morgan (1999) J. Pept. Res, 53, 134-145,Jesper Lohse et al. (1997) Bioconjugate Chem., 8, 503-509, Hans-georg Batz, Henrik Frydenlund Hansen, et al. Pct Int. Appl. (1998) WO 9837232A2 19980827, Bruce Armitage, Troels Koch, et al. (1998) Nucleic Acid Res., 26, 715-720)。また、用いられる化合物の構造がアルカリ性条件に比較的安定であることが特徴的であるため、アルカリ性条件に不安定な発色団が付くと、前記従来法と類似の方法、すなわち下記ルートA
【0016】
【化4】
【0017】
【化5】
【0018】
では効率良く合成できないと予想された。
したがって、一般に光機能性分子等の機能性分子は高価な場合が多いため、より合目的的な機能性PNAの合成方法、すなわち、1)機能性PNAモノマーユニットの設計における、機能性分子のPNA背骨構造への効率的な導入、2)コストパフォーマンスを考えた合成ルート、および3)遺伝子診断薬としての応用へ適応させるための、これらの機能性分子を超高速に導入する方法が探求された。
【0019】
上記課題に鑑み、本発明者らは、機能性PNAモノマーの新規製造方法として、下記ルートB
【0020】
【化6】
【0021】
に示すように、PNA背骨構造にt−ブトキシカルボニルアミノエチルアミン誘導体6を用いて1のペンタフルオロフェニル基を含む活性エステル体5と縮合してほぼ定量的に光機能性PNAモノマー4を合成する方法を見出した。
【0022】
また、本発明者らは、機能性PNAモノマーを合成する別法として、PNA背骨構造に上記t−ブトキシカルボニルアミノエチルアミン誘導体6の代わりにベンジルオキシカルボニル−ω−アミノ酸誘導体を用いる方法(ルートC)を見出した。これらの方法については、既に特許出願がなされている。
【0023】
したがって、最終的に機能性PNAを合成するための方法として、上記ルートBおよびルートCのいずれかを用いる方法によって機能性PNAモノマーを合成した後に、それらを重合する方法が工業的にも確立されつつある。すなわち、現在までの機能性PNAの合成法によってPNAプローブとして用いられる機能性PNAを工業的に大量合成することは可能になりつつある。
【0024】
一方、コストパフォーマンスの向上および機能性分子を超高速に導入することを目的とした、機能性PNAを合成方法の改良もなされている。例えば、前記機能性PNAモノマーユニットを用いる方法とは異なるアプローチとして、下記の前駆体的PNAモノマーユニットを利用することによって、ポスト合成的に機能性分子をPNAオリゴマーに導入する方法が報告されている(Oliver Seitz; Tetrahedron Letters 1999, 40, 4161-4164.)。
【0025】
【化7】
【0026】
当該方法は、前記前駆体的PNAモノマーユニットをPNAオリゴマーに導入した後、さらに機能性分子を導入することによって機能性PNAを合成するものである。しかし、当該方法においては、導入できる機能性分子の種類が限定される等の欠点がある。
【0027】
例えば、下図に示すように、市販されている光機能性分子のsuccinimideエステルを導入することはできず、導入するためにはFmoc-Gly等のリンカーをまず導入する必要があるが、結果として上記化合物は使用しにくいものになっている。
【0028】
【化8】
【0029】
また、細胞中に導入するためのに蛍光プローブとして、これまでDNAオリゴマー・RNAオリゴマー・PNAオリゴマーが利用されているが、これらを細胞中に導入するためには、当然ながら細胞膜を通過させなければならない。しかし、細胞膜は膜表面が負電荷を帯びているため、元々負に帯電しているDNA/RNAオリゴマーを導入するのは非常に困難である。
【0030】
一方PNAオリゴマーは電気的に中性であるが、膜透過しにくいという結果が得られている。したがって、PNAオリゴマーを細胞内に導入するに際しては、膜表面を前処理してその導入をしやすくしたり、あるいはトランスフェクション試薬を用いて導入せざるを得ないのが現状である。
【0031】
しかし、そのような処理を施してPNAオリゴマーを導入した場合においてプローブの機能が発揮されたとしても、本来生体が示す挙動を正確に表現していることは必ずしも保証されない。しかも、これは細胞1個の場合であり、多細胞(個体)での利用に至っては到底不可能である。このような現状および観点から、膜透過性機能を有する蛍光PNAプローブの開発が有用であると考えられている。
【0032】
なお、膜透過性機能を有する蛍光PNAプローブは既に存在する。例えば、1)膜透過性機能を有するオリゴペプチドをPNAに結合させたもの、2)膜透過性機能を有するリン脂質をPNAに結合させたものが挙げられる。しかしながら、これらは膜透過した後細胞内においてプロテアーゼ等の酵素によりPNA以外の部分が分解され、細胞内に滞留してしまうことが予想される。このことは、ターゲットを捕捉出来なかった過剰なPNAプローブが膜透過性機能を失い、その後の洗浄過程で細胞外に出にくくなることにつながるため、本来細胞が持っている遺伝子発現系を正確に表現できないことを意味する。
【発明の開示】
【発明が解決しようとする課題】
【0033】
したがって、本発明は、コストパフォーマンスに優れ、かつ機能性分子を超高速に導入することができる、機能性PNAの新規合成方法およびそれに用いる化合物、ならびに新規機能性PNAを提供することを目的とする。
【課題を解決するための手段】
【0034】
上記課題に鑑み研究を重ねた結果、本発明者らは、前駆体的PNAモノマーユニットの構造を最適化することによって、驚くべきことに、従来法における前記課題が克服され、かつ極めて広範にわたる機能性PNAを合成できることを見出し、本発明を完成するに至った。
【0035】
すなわち、本発明は、機能性PNAオリゴマーを製造する方法であって、保護基によって保護されたアデニン、グアニン、シトシンまたはチミンを有するPNAモノマーユニットを、Fmoc−ω−アミノ酸−BocPNA−OHと反応させてPNAオリゴマーを合成した後、該PNAオリゴマーに遊離カルボン酸を有する機能性分子を導入し、さらに前記保護基の脱保護を行うことを含む、前記方法に関する。
【0036】
また、本発明は、Fmoc−ω−アミノ酸−BocPNA−OHが、Fmoc−ω−アミノ酸ペンタフルオロフェニルエステルとBocPNA−OHとの反応によって製造されたものであることを特徴とする、前記方法に関する。さらに、本発明は、Fmoc−ω−アミノ酸ペンタフルオロフェニルエステルが、Fmoc−ω−アミノ酸とペンタフルオロフェノールとの反応によって製造されたものであることを特徴とする、前記方法に関する。
【0037】
さらにまた、本発明は、機能性分子を導入した後に、さらに別異の機能性分子の導入を行うことを特徴とする、前記方法に関する。またさらに、本発明は、導入される機能性分子が、光機能性分子、膜透過性機能分子、臓器選択性機能分子、殺菌性機能分子および分子認識性機能分子から選択されることを特徴とする、、前記方法に関する。また、本発明は、導入される機能性分子が、光機能性分子および膜透過性機能分子を含むことを特徴とする、前記方法に関する。そして、本発明は、光機能性分子が、FITC、ROX、TAMRAまたはDabcylであり、膜透過性機能分子が水溶性アミノ酸であることを特徴とする、前記方法に関する。
【0038】
また、本発明は、アデニン、グアニン、シトシンまたはチミンを保護する保護基が、Z基であることを特徴とする、前記方法に関する。
【0039】
さらに、本発明は、PNAオリゴマーの合成が、tBoc法用固相担体を用いたPNA鎖における縮合・伸長を含むことを特徴とする、前記方法に関する。またさらに、本発明は、tBoc法用固相担体がMBHAであることを特徴とする、前記方法に関する。さらにまた、本発明は、遊離カルボン酸を有する機能性分子の導入が、Fmoc基をピペリジン処理によって選択的に脱保護して得られた1級アミノ基との脱水縮合によって行われることを特徴とする、前記方法に関する。
【0040】
また、本発明は、Fmoc−ω−アミノ酸−BocPNA−OHが、下記一般式(I)
【0041】
【化9】
【0042】
(式中、nは1〜15までの整数を表す)で表される化合物であることを特徴とする、請求項1〜7のいずれかに記載の方法。
また、本発明は、下記a)〜d):
a)Fmoc−ω−アミノ酸ペンタフルオロフェニルエステルを製造する工程において、Fmoc−ω−アミノ酸とペンタフルオロフェノールとを反応させること;
b)Fmoc−ω−アミノ酸−BocPNA−OHを製造する工程において、Fmoc−ω−アミノ酸ペンタフルオロフェニルエステルをBocPNA−OHと反応させることによってFmoc−ω−アミノ基をBocPNA−OHに導入すること;
c)Fmoc−ω−アミノ酸−BocPNA−OHからPNAオリゴマーを製造する工程において、PNAモノマーユニットを、Fmoc−ω−アミノ酸−BocPNA−OHと反応させてPNAオリゴマーを製造すること;および
d)前記PNAオリゴマーから機能性PNAオリゴマーを製造する工程において、PNAオリゴマーへの機能性分子の導入が、Fmoc基をピペリジン処理によって選択的に脱保護して得られた1級アミノ基との脱水縮合によって行われる行うこと、の1または2以上を含むことを特徴とする、前記方法に関する。
そして、本発明は、下記一般式(I)
【0043】
【化10】
【0044】
(式中、nは1〜15までの整数を表す)で表される化合物に関する。
【0045】
そしてさらに、本発明は、一般式(I)
【0046】
【化11】
【0047】
(式中、nは1〜15までの整数を表す)で表される化合物を製造する方法であって、Fmoc−ω−アミノ酸をペンタフルオロフェノールと反応させ、その反応物をBocPNA−OHと反応させることによってFmoc−ω−アミノ基を導入することを特徴とする、前記方法に関する。
【0048】
そして、本発明は、下記一般式(II)
【0049】
【化12】
【0050】
(式中、Bは、互いに独立し、同一または異なって、アデニン、グアニン、シトシンまたはチミンであり、Rは、互いに独立し、同一または異なって、Fmoc基または機能性カルボン酸誘導体であり、R1は、水素原子または機能性カルボン酸誘導体であり、a〜hは0〜10の整数であり、X1〜X3、Y1、Y2およびZ1〜Z5はいずれも0以上の整数であり、X1+X2+X3≧0であり、Y1+Y2>0であり、Z1+Z2+Z3+Z4+Z5≧0である。ただし、X1+X2+X3およびZ1+Z2+Z3+Z4+Z5が同時に0であることはなく、X1+X2+X3=0の場合、R1は機能性カルボン酸誘導体である。)で表される化合物に関する。
【0051】
さらに、本発明は、Z1+Z2+Z3+Z4+Z5=0であり、R1が水素原子である、前記化合物に関する。
【0052】
また、本発明は、Rがメチルレッドのカルボン酸誘導体を含むことを特徴とする、前記化合物に関する。
【0053】
さらに、本発明は、X1+X2+X3=9であり、Y1+Y2=1であることを特徴とする、前記化合物に関する。
【0054】
また、本発明は、X1=3、X2=6であり、Y1=1であることを特徴とする、前記化合物に関する。
【0055】
さらに、本発明は、RまたはR1が細胞膜透過性機能分子誘導体であることを特徴とする、前記化合物に関する。
【0056】
さらにまた、本発明は、さらにR1が機能性カルボン酸誘導体であることを特徴とする、前記化合物に関する。
【0057】
また、本発明は、X1=Z1=1であることを特徴とする、前記化合物に関する。
【0058】
さらに、本発明は、Y1≧2であり、Z2=1であることを特徴とする、前記化合物に関する。
【0059】
またさらに、本発明は、a≦6であり、b≦4であり、f≦6であることを特徴とする、前記化合物に関する。
【0060】
また、本発明は、R1が光機能性カルボン酸誘導体であることを特徴とする、前記化合物に関する。
さらに、本発明は、下記一般式(III)
【0061】
【化13】
【0062】
(式中、nは1〜15の整数を表す)で表されることを特徴とする化合物に関する。
【0063】
さらに、本発明は、下記一般式(III)
【0064】
【化14】
【0065】
(式中、nは1〜15の整数を表す)で表される化合物を製造する方法であって、Fmoc−ω−アミノ酸とペンタフルオロフェノールとを反応させることを特徴とする、前記方法に関する。
【0066】
本発明は、PNA背骨構造にFmoc−ω−アミノ酸を導入した前駆体的PNAモノマーユニット、すなわちFmoc−ω−アミノ酸−BocPNA−OHをPNAオリゴマーに導入した後、機能性分子をポスト合成的に導入することにより、ほぼ定量的に光機能性PNAオリゴマーを合成できることに成功したものである。
【0067】
上記特徴により、本発明の製造方法においては、導入する機能性分子として市販のsuccinimideエステルを使用する必要がなく、カルボン酸を有する化合物であれば問題なく利用でき且つ定量的に導入できる。そのため、本発明による製造方法はコストパフォーマンスに極めて優れている。
【0068】
また、前記前駆体的PNAモノマーユニットを機能性PNAオリゴマーに導入した後にレジンを分割することにより、それぞれのレジンに異なった機能性分子を導入することができる。したがって、本発明による製造方法によれば、極めて高速な機能性PNAオリゴマーの合成手法を開発することが可能となる。
【0069】
本発明の方法によって効率的な合成が可能になる機能性PNAオリゴマーの例として、下記一般式(II)
【0070】
【化15】
【0071】
(式中、Bは、互いに独立し、同一または異なって、アデニン、グアニン、シトシンまたはチミンであり、Rは、互いに独立し、同一または異なって、Fmoc基または機能性カルボン酸誘導体であり、R1は、水素原子または機能性カルボン酸誘導体であり、a〜hは0〜10の整数であり、X1〜X3、Y1、Y2およびZ1〜Z5はいずれも0以上の整数であり、X1+X2+X3≧0であり、Y1+Y2>0であり、Z1+Z2+Z3+Z4+Z5≧0である。ただし、X1+X2+X3およびZ1+Z2+Z3+Z4+Z5が同時に0であることはなく、X1+X2+X3=0の場合、R1は機能性カルボン酸誘導体である。)で表される化合物において、Z1+Z2+Z3+Z4+Z5=0であり、R1が水素原子である化合物を挙げることができる。
【0072】
本発明によれば、前記一般式(II)で表される化合物において同一または異なる機能性分子を、任意の複数の位置に導入することも可能となる。すなわち、前記前駆体的PNAモノマーユニットを用いてPNAオリゴマーを導入した後、ピペリジン処理と機能性分子のポスト合成的導入を一括して行うことによるものであるが、これはPNAオリゴマーの細胞膜透過機能を向上させるアンテナペディアを高速に設計する上で、欠かせないものである。この点においても、本発明による方法は極めて優れたものである。
【0073】
このように製造される化合物の例として、前記一般式(II)において、Z1+Z2+Z3+Z4+Z5>0であり、Rが細胞膜透過性分子誘導体であり、R1が機能性カルボン酸誘導体であるものが挙げられる。
【0074】
このプローブは大きく蛍光標識領域・細胞膜透過性機能領域・分子認識領域の3つに分けることができ、それぞれをリンカー部位(Z1〜Z5の添字付きで表される部分)を介して結合させた形をしている。
【0075】
蛍光標識化合物は市販のものも、既に本発明者らがPCT出願を行った新規蛍光標識化PNAモノマーユニットも用いることができる。
【0076】
分子認識部位は市販のPNAユニットを用いて合成する。このものの特徴は、既に国内特許出願を行っている一般式(I)で表される新規PNAユニットを膜透過性機能領域部に用いていることである。該一般式(I)で表される新規PNAユニットは機能性分子をポスト合成的に導入するために開発された前駆体ユニットであり、これを複数個並べて導入した後、前記したように同一機能性分子を一括導入できることを特徴としている。
【0077】
したがって、本発明によれば、光機能性分子に限定されることない、多種多様な機能性分子を、PNA中に容易かつ極めて効率的に導入することができるようになる。
【0078】
このような機能性分子として、Naphthalimide型、Flavin型、Dabcyl型、Biotin型、FAM型、Rhodamine型、TAMRA型、ROX型、HABA型、Pyrene型、Coumarine型等の光機能性モノマーユニット、膜透過機能性分子、臓器選択機能性分子、殺菌機能性分子および分子認識機能性分子等が挙げられる。
【0079】
すなわち、本発明における「機能性」の語は、光機能性のみならず、膜透過性、臓器選択性、殺菌機能性および分子認識機能性等を含む、ある特定の修飾を行うことによって化合物に新たに付与される種々の機能の全てを意味するものである。
【0080】
さらに、本発明における「機能性PNA」の語は、PNAモノマー同士が2−(N−アミノエチル)グリシン骨格によって直接結合したもののみならず、その間にリンカーとしての炭化水素鎖等を含むものも意味するものである。
【発明を実施するための最良の形態】
【0081】
ここで、本発明による方法の特徴を更に詳細に説明する。
本発明によるオリゴPNAを合成するルートは、典型的には、下図
【0082】
【化16】
【0083】
に示すとおりである。
まず、必要に応じて、下図
【0084】
【化17】
【0085】
に示すように、Fmoc−ω−アミノ酸とペンタフルオロフェノール(PfpOH)とを反応させて得られるFmoc−ω−アミノ酸ペンタフルオロフェニルエステル(Fmoc−ω−アミノ酸−OPfp)とから、Fmoc−ω−アミノ酸−BocPNA−OHを合成する。
【0086】
以後の工程において用いる該Fmoc−ω−アミノ酸−OPfpの溶液を得るには、DMFなどの有機溶媒、またはアセトン等と水とを含む水溶性溶媒したものいずれも好適に用いることができる。前記水溶性溶媒を用いた場合には、精製等の後処理の面におけるメリットを有するものである。
【0087】
前記Fmoc−ω−アミノ酸−OPfpは、Fmoc−ω−アミノ酸とPfpOHをDMF溶液中にてDCCを加えて反応させることによって、例えば下記式(III)
【0088】
【化18】
【0089】
(式中、nは1〜15の整数を表す)で表されるものが得られる。
次いで、これにBocPNA−OHのDMF溶液にジイソプロピルエチルアミンとともに添加し、Fmoc−ω−アミノ酸−BocPNA−OHを得る。Fmoc−ω−アミノ酸−BocPNA−OHは、PNAモノマーユニットの前駆体として機能するため、前駆体的PNAモノマーユニットと呼ぶことができる。
【0090】
式(I)において、nとしては1〜15までの整数を適宜選択できるが、nの値が大きい方が、ハイブリッド形成時の立体的反発(あるいは障害)を軽減する点において好ましい。
次に、下図
【0091】
【化19】
【0092】
に示すように、前駆体的PNAモノマーユニットを用いて、オリゴマーIaを合成する。具体的には、Z基(N−ベンジルオキシカルボニル基)等で保護されたアデニン、グアニン、シトシンまたはチミンを有するPNAモノマーユニットを、前駆体的PNAモノマーユニットと反応させ、tBoc法用固相担体を用いてPNA鎖を逐次縮合・伸長せしめる。
【0093】
PNA鎖の縮合においては、予めtBoc基を脱離しておく必要があるが、その方法に制限はなく、一般的な方法が用いられる。それに続く縮合には、HATU、HBTUおよびBOP等の一般的な縮合剤が用いられる。また、固相担体に関しては、tBoc法用のものであれば特に制限はないが、特にMBHAが好適に用いられる。
【0094】
次に、下図
【0095】
【化20】
【0096】
に示すように、ピペリジン処理によってFmoc基を選択的に脱保護してアミノ基とし、Ibを得て、さらに、下図
【0097】
【化21】
【0098】
に示すように、該Ibの前記アミノ基に遊離カルボン酸を有する機能性分子を脱水縮合してIcを得る。前記カルボン酸として特に制限はないが、反応性の点においては脂肪族カルボン酸が芳香族カルボン酸を上回るため、脂肪族カルボン酸を用いると製造の効率が高く好ましい。
【0099】
また、ピペリジン処理によるFmoc基の脱保護は、ある程度の時間をかけることによって好適に行われる。特に、20〜40分が好適であり、最も好適には30分であった。
【0100】
縮合剤の種類に特に制限はなく、前記PNA鎖の縮合と同様に、HATU、HBTUおよびBOP等の一般的な縮合剤が用いられる。
【0101】
なお、機能性分子の導入は、Fmoc−ω−アミノ酸−BocPNA−OHを縮合した後、直ちに行ってもよく(第1法)、あるいは、Fmoc−ω−アミノ酸−BocPNA−OHを含む全てのPNAモノマーユニットを逐次縮合した後に行ってもよい(第2法)。
最後に、下図
【0102】
【化22】
【0103】
に示すように、担体レジンからの切り出しとZ基の脱保護を同時に行うことによって、目的とするPNAオリゴマーIdを得る。
【0104】
切り出しおよび脱保護は、Fmoc基の脱保護の後に行われる限りにおいてはその条件に特に制限はない。例えば、TFA/TFMSA/p-cresol/Thioanisole=60/25/10/10のような一般的な条件において好適に行われる。
【0105】
上記のように、本発明による方法においては、従来の機能性モノマー合成に用いる活性エステル化の合成を要する方法とは異なり、機能性分子をそのまま利用できる。また一旦Iaを合成した後に種々の機能性分子が導入可能であるため、従来困難であった高速かつ多様な並列PNAプローブ合成が可能である。
【0106】
Fmoc−ω−アミノ酸−BocPNA−OHとPNA鎖を有する分子との反応を含む本発明による方法によれば、下記一般式(II)
【0107】
【化23】
【0108】
(式中、Bは、互いに独立し、同一または異なって、アデニン、グアニン、シトシンまたはチミンであり、Rは、互いに独立し、同一または異なって、Fmoc基または機能性カルボン酸誘導体であり、R1は、水素原子または機能性カルボン酸誘導体であり、a〜hは0〜10の整数であり、X1〜X3、Y1、Y2およびZ1〜Z5はいずれも0以上の整数であり、X1+X2+X3≧0であり、Y1+Y2>0であり、Z1+Z2+Z3+Z4+Z5≧0である。ただし、X1+X2+X3およびZ1+Z2+Z3+Z4+Z5が同時に0であることはなく、X1+X2+X3=0の場合、R1は機能性カルボン酸誘導体である。)に示す化合物等が好適に合成される。
【0109】
本発明の方法によって、式(II)で表される化合物として、例えば、Rがメチルレッドのカルボン酸誘導体を含むもの、さらにX1+X2+X3=9であり、Y1+Y2=1であるもの、またはX1=3、X2=6であり、Y1=1であるもの等が、特に好適に合成される。
【0110】
また、前記一般式(II)で表される化合物において、複数の機能性分子が導入されたものとして、例えば、RまたはR1が細胞膜透過性機能分子誘導体であるものが好適に合成される。このような化合物は、典型的には、Rが細胞膜透過性機能分子誘導体等であり、R1が光機能性分子等の機能性カルボン酸誘導体等であるもの、すなわち、末端部を含む複数部位に機能性分子が導入され、それらによって複数の機能が付与された化合物である。このような化合物は、例えば下記のように模式化することができる。
【0111】
【化24】
【0112】
このような化合物は、例えば前記一般式(II)において、X1=Z1=Z2=1であり、かつY1≧2である化合物である。このような化合物は合成のしやすさおよび合成コストの面等において好適である。
【0113】
上記化合物において、a、bおよびfはそれぞれ0〜10の整数であれば特に限定されないが、例えばa≦6であり、b≦4であり、f≦6であるものであっても、合成上および実用上のいずれにおいても支障はない。
【0114】
リンカー部位を導入することによって、個々の機能性部位および塩基配列認識領域の干渉を防ぎ、分子の機能をより確実なものにすることができる。本明細書におけるPNA、PNAモノマーおよびPNAオリゴマーの語には、リンカー部位をその末端および/または内部に含むものも包含される。これらの部位または領域間の相互干渉を防ぐための部位としては、前記リンカー部位のみならず、一般式(I)におけるf〜hを、所望に応じて選択することによっても可能である。
【0115】
リンカー部位を構成する基としては、直鎖状または分枝状の炭化水素およびそれらのエーテル体等が挙げられるが、直鎖状炭化水素基は導入の容易さおよびコストなどの面から好適であり、特に炭素数1〜6の直鎖状炭化水素基が好適である。また、エーテル体は、その汎用性において好適である。
【0116】
前記複数の機能性分子が導入された化合物は、例えばKoch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; Orum, H. J. PeptideRes. 1997, 49, 80-88.を利用して好適に合成される。塩基配列認識部位は、市販の各種PNAモノマーを用いて固相合成によりオリゴマー化することができる。リンカー部位には、市販のBoc−7−アミノヘプタン酸、Boc−6−アミノカプロン酸等を用いることができる。
【0117】
1の機能性分子として光機能性分子を導入すれば、蛍光標識することが可能であり、かつ他の機能も有する化合物を合成することができる。このような蛍光標識部位として、市販のFITC、ROX、TAMRAまたはDabcyl等の市販の活性エステル型蛍光標識化合物を用いて、多様な蛍光発光波長を選択することが可能であるが、導入される蛍光標識化合物はこれらに限定されるものではない。
【0118】
本発明の化合物に導入し得る他の機能性の例としては、膜透過性機能が挙げられる。このような膜透過性機能部位は、前回特許前記一般式(I)で表される化合物を用いることにより、同様に導入することができる。膜透過性を向上させることができる機能性分子としてアルギニンが挙げられるが、リシンおよびセリン等の他の水溶性アミノ酸も好適に用いることができる。
【0119】
また、Fmocアミノ酸ユニットを利用することによって、複数個のアミノ酸を導入することも可能である。その合成例は実施例20および21にも示した。しかしながら、上記2化合物は本発明による膜透過性機能を有する蛍光PNAプローブのモデル化合物であり、本発明はこれらに限定されるものではない。
【0120】
これらのプローブの特徴は、「全てPNA型になっているので、完全な酵素耐性を有すること」である。すなわち、これまでの膜透過性機能を有するプローブは、PNAと膜透過性機能を有するペプチド鎖あるいはリン脂質を共有結合させたものが主流であったが、これらの既知のプローブは優れた膜透過性機能を有するものの、一旦細胞内に入ると酵素群によりペプチド鎖あるいはリン脂質が分解されることが予想される。したがって、これらは、ターゲットを認識していない分解を受けたプローブを洗浄過程で完全に取り除くことができないという欠点を有する。
【0121】
これに対して、今回設計したプローブは、細胞内においても酵素分解を受けないため、ターゲットを認識していないプローブは洗浄過程で完全に取り除かれるため、正確な遺伝子発現量の定量を可能とするものである。なお、これらの機能性を有する化合物以外にも、ラクトースやトリスエックス等の臓器選択機能性分子、タナチンやセクロピン等の殺菌機能性分子およびビオローゲン等の分子認識機能性分子等も、本発明によれば制限なく導入することが可能であり、そのような化合物を、大量に低コストで実用に供することが可能になる。
【実施例】
【0122】
以下に実施例を用いて本発明をさらに詳細に説明するが、本発明の範囲はこれに限られるものではない。
【0123】
(実施例1)Fmoc-Gly-BocPNA-OHの合成(1)
Fmoc-Gly-OH (891 mg, 3.0 mmol) とPfpOH (754 mg, 4.5 mmol)のDMF溶液(12mL)にDCC (845 mg, 4.5 mmol)を氷冷下加え、この反応液を0℃で30分次いで室温で15時間撹拌した。反応液を濾過し濾液を減圧濃縮し、残渣をBocPNA-OH (436 mg, 2.0 mmol)のDMF溶液(16 mL)にdiisopropylethylamine (445 μL, 2.6 mmol) を加え、室温で15時間撹拌した。これを減圧濃縮し残渣をシリカゲルカラムクロマト法(0-50% MeOH/CH2Cl2)により精製しFmoc-Gly-BocPNA-OH (121 mg, 12%)を得た。1H NMR (DMSO-d6)δ 7.88 (d, J = 7.0 Hz, 2 H), 7.72 (d, J = 7.0 Hz, 2 H), 7.62 (brt) and 7.56 (brt) (1 H), 7.41 (t, J = 7.0 Hz, 2 H), 7.33 (t, J = 7.0 Hz, 2 H), 7.18 (m, 2 H), 6.85 (brt) and 6.79 (brt) (1 H), 4.35 - 4.15 (m, 3 H), 4.05 - 3.85 (m, 3 H), 3.77 (m, 1 H), 3.40 - 3.25 (m, 2 H), 3.10 (m) and 3.03 (s) (2 H), 1.37 (brs, 9 H); 13C NMR (DMSO-d6) δ172.2 (d), 169.10 (d), 156.34 (d), 155.58 (d), 143.83, 140.66, 127.58, 127.04, 125.24, 120.04, 77.77 (d), 65.71, 47.34 (d), 46.72, 46.65 (d), 29.23 (d), 28.14 (d); FABMS m/z 498 [(M+H)+].
【0124】
(実施例2) Fmoc-C7-OPfpの合成
Fmoc-C7-OH(381.9 mg,1.0 mmol)とPfpOH(349.7 mg、1.9 mmol)のDMF溶液(2.5mL)にDCC(392.0 mg,1.9 mmol)を氷冷下で加え、この反応液を0 ℃で30分次いで室温で一晩攪拌した。反応終了後DCUreaを濾別して濾液を減圧濃縮し、残渣をシリカゲルカラムクロマト法(CH2Cl2)により精製した後、Hexaneで再結晶し白色粉末としてFmoc-C7-OPfp (537.5 mg,98%)を得た。1H-NMR (CDCl3)δ7.76(d,J= 7.6 Hz, 2 H),7.59 (d,J = 7.6 Hz, 2 H),7.40 (t,J = 7.4 Hz, 2 H),7.31 (t,J = 7.4 Hz, 2 H),4.70 - 4.73 (brt,1 H),4.47 - 4.40 (brd,2 H),4.22 (t,J = 6.42 Hz, 1 H),3.20 (q,J = 5.94 Hz, 2 H),2.66 (t,J = 7.38 Hz, 2 H),1.80 - 1.75 (m,2 H),1.55 - 1.50 (m,2 H),1.45 - 1.34 (m,6 H); 13C-NMR (CDCl3)δ 169.44, 156.43, 143.98, 141.96(m), 141.29, 140.23, 138.67(m), 136.99(m), 127.60, 126.96, 124.97, 119.91, 66.49, 55.73,47.29, 41.34(d), 34.89, 33.22, 29.85, 28.70, 26.42, 25.43, 24.60 ;HRMS(FAB+) calcd. for C29H27F5NO4[(M+H)+] 547.5131 observed 548.1861.
【0125】
(実施例3) Fmoc-Gly-BocPNA-OHの合成(2)
アセトン(6.0 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-Gly-OPfp (240.9 mg,0.52 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で6時間攪拌した。氷冷した1 N塩酸で冷却した反応溶液をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ溶液を濃縮し、シリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)及びLH-20(MeOH)で精製した。この後、塩化メチレンに溶かし、減圧濃縮しアモルファスパウダーとしてFmoc-Gly-BocPNA-OH (157.3 mg,80%)を得た。
【0126】
(実施例4)Fmoc-β-Ala-BocPNA-OHの合成(1)
Fmoc-β-Ala-OH (311 mg, 1.0 mmol)とPfpOH (334 mg, 1.75 mmol)のDMF溶液(2.5 mL)にDCC (288 mg, 1.4 mmol)を氷冷下加え、この反応液を0℃で30分次いで室温で15時間撹拌した。反応液を濾過し濾液を減圧濃縮し、残渣をシリカゲルカラムクロマト法(CH2Cl2)により精製した。これをHexaneとCH2Cl2で再結晶し、白色粉末としてFmoc-β-Ala-OPfp (429 mg, 90%) を得た。Fmoc-β-Ala-OPfp(100 mg, 0.21 mmol)とBocPNA-OH (41 mg, 0.19 mmol)のDMF溶液(2 mL)にdiisopropylethylamine (36 μL, 0.21 mmol) を加え、室温で15時間撹拌した。これを減圧濃縮し、残渣をシリカゲルカラムクロマト法(0-10% MeOH/CH2Cl2)により精製しFmoc-β-Ala-BocPNA-OH (41 mg, 42%)を得た。1H NMR (DMSO-d6)δ 7.88 (d, J = 7.4 Hz, 2 H), 7.68 (d, J = 7.4 Hz, 2 H), 7.41 (t, J= 7.3 Hz, 2 H), 7.33 (t, J = 7.3 Hz, 2 H), 7.18 (m, 2 H), 6.83 (brt) and6.72 (brt) (2 H), 4.3 - 4.2 (m, 4 H), 4.05 - 3.9 (m, 3 H), 3.33 (brt) and 3.29 (brt) (2 H), 3.19 (m, 2 H), 3.07 (brq) and 3.02 (brq) (2 H), 1.36 (brs, 9 H); 13C NMR (DMSO-d6) δ171.20 (d), 170.85 (d), 155.93, 155.56, 143.87, 140.69, 127.55, 127.01, 125.09, 120.05, 77.73 (d), 65.30 (d),59.69, 47.35 (d), 46.68, 46.49 (d), 37.99 (d), 36.72 (d), 28.14 (d).
【0127】
(実施例5)Fmoc-β-Ala-BocPNA-OHの合成(2)
アセトン(1.25 mL)と水(1.25 mL)の混合溶液にNaHCO3(92.4 mg,1.1 mmol)を加え、Fmoc-β-Ala-OPfp (476.0 mg,1.0 mmol)とBocPNA-OH (87.3 mg,0.55 mmol)を溶解し、室温で6時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、反応液に1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ溶液を濃縮し、シリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。この後、塩化メチレンに溶かし、減圧濃縮しアモルファスパウダーとしてFmoc-β-Ala-BocPNA-OH (225.9 mg,80%)を得た。
【0128】
(実施例6)Fmoc-GABA-BocPNA-OHの合成(1)
Fmoc-GABA-OPfp (100 mg, 0.20 mmol)とBocPNA-OH (40 mg, 0.18 mmol)のDMF溶液(2 mL)にdiisopropylethylamine (34 μL, 0.20 mmol) を加え、室温で15時間撹拌した。これを減圧濃縮し、残渣をシリカゲルカラムクロマト法(0-20% MeOH/CH2Cl2)により精製しFmoc-GABA-BocPNA-OH (43 mg, 45%)を得た。1H NMR (DMSO-d6)δ 7.88 (d, J = 7.4 Hz, 2 H), 7.68 (d, J = 7.4 Hz, 2 H),7.41 (t, J = 7.4 Hz, 2 H), 7.33 (t, J = 7.4 Hz, 2 H), 7.29 (m, 1 H), 6.82 (brt) and 6.71 (brt) (1 H), 4.3 - 4.2 (m, 4 H), 4.05 - 3.9 (m, 3 H),3.35 - 3.25 (m, 2 H), 3.1 - 2.95 (m, 4 H), 1.36 (brs, 9 H); 13C NMR (DMSO-d6) δ172.2 (d), 171.5 (d), 156.03, 155.60 (d), 143.89, 140.68, 127.54, 127.00, 125.50, 120.04, 77.70 (d), 65.19, 54.84, 47.89 (d), 46.97 (d), 46.72, 38.20 (d), 29.23 (d), 28.14 (d), 24.98 (d).
【0129】
(実施例7)Fmoc-GABA-OPfpの合成
Fmoc-GABA-OH (325 mg, 1.0 mmol)とPfpOH (221 mg, 1.2 mmol)のDMF溶液(2.5mL)にDCC (248 mg, 1.2 mmol)を氷冷下加え、この反応液を0℃で30分次いで室温で15時間撹拌した。反応液を濾過し濾液を減圧濃縮し、残渣をシリカゲルカラムクロマト法(CH2Cl2)により精製し、白色粉末としてFmoc-GABA-OPfp (463mg, 94%) を得た。1H NMR (CDCl3)δ7.77 (d, J = 7.5 Hz, 2 H), 7.59 (d, J =7.5 Hz, 2 H), 7.40 (t, J = 7.5 Hz, 2 H), 7.31 (t, J = 7.5 Hz, 2 H), 4.85 (brs, 1 H), 4.45 (d, J = 6.3 Hz, 2 H), 4.21 (t, J = 6.3 Hz, 2 H), 3.32(d, J = 6.5 Hz, 2 H), 2.71 (t, J = 6.5 Hz, 2 H), 1.98 (t, J = 6.5 Hz, 2H); 13C NMR(CDCl3)δ169.02, 156.49, 143.83, 141.87 (m), 141.28, 140.23 (m), 138.61 (m), 136.94 (m), 127.62, 127.46, 124.89, 119.88, 66.52, 47.25, 39.92, 30.42, 25.03.
【0130】
(実施例8) Fmoc-GABA-BocPNA-OHの合成(2)
アセトン(5.0 mL)と水(1.0 mL)の混合溶液にNaHCO3 (67.2 mg,0.8 mmol)を加え、Fmoc-GABA-OPfp (255.5 mg,0.52 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で8時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、反応液に1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ溶液を濃縮し、シリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮しアモルファスパウダーとしてFmoc-GABA-BocPNA-OH (175.9 mg,84%)を得た。
【0131】
(実施例9) Fmoc-C4-BocPNA-OHの合成
アセトン(4.0 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C4-Opfp (323.5 mg,0.64 mmol)とBocPNA-OH(87.3 mg,0.4 mmol)を溶解し、室温で12時間攪拌した。氷冷した1 N塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ、シリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮しアモルファスパウダーとしてFmoc-C4-BocPNA-OH (190.7 mg,88%)を得た。1H-NMR(CDCl3) δ7.76(d,J = 6.7 Hz, 2 H),6.96 (mi) and 6.66 (ma) (brd, J = 6.7 Hz, 2 H), 7.41 - 7.37(m, 2 H), 7.32 - 7.28(m,2 H), 7.14 (ma) and 6.68 (mi) (m, 1 H), 5.54 (ma) and 5.43 (mi) (brt, 1 H), 4.45(mi) and 4.37(ma) (m, 2 H), 4.24 - 4.21 (m, 1 H),4.08 - 3.95 (m,2 H),3.54 - 3.48 (m,2 H),3.29 - 3.11 (m, 4 H),2.43 - 2.25 (m, 2 H),1.70 - 1.29 (m,13 H); 13C-NMR (CDCl3) δ 174.39,173.07,171.95,157.51,156.79(d),156.11,144.06(d),141.16,127.46(d),126.90(d),119.77(d), 81.42,79.67,66.39(d),53.35,49.46(d),49.17,48.60,47.15(d), 40.89,40.32(d),38.68,31.87,31.41,29.59,29.11(d),28.28,21.77(d); HRMS (FAB+) calcd. for C29H37N3O7[(M+H)+] 539.2632 observed 540.2707.
【0132】
(実施例10) Fmoc-C5-BocPNA-OHの合成
アセトン(7.5 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C5-OPfp (311.0 mg,0.6 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で24時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ溶液を濃縮し、シリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮し白色のアモルファスパウダーとして、Fmoc-C5-BocPNA-OH (198.0 mg,90%)を得た。1H-NMR (DMSO-d6)δ 7.88(d,J = 7.4 Hz, 2 H),7.68 (d, J =7.2 Hz,2 H),7.41 (t,J = 7.4 Hz, 2 H),7.32 (t,J = 7.4 Hz, 2 H),7.22(brt,1 H),6.81 (ma) and 6.67 (mi) (brt, 1 H),4.33 (mi) and 4.29 (ma)(brd,2 H),4.20 (t,J = 7.1 Hz, 1 H),4.08 (mi) and 3.90 (ma) (brs,2H),3.09 - 2.94 (m,4 H),2.30 (ma) and2.14 (mi) (brt,2 H),1.51 - 1.45(m,2 H),1.41 - 1.31 (brs,11 H),1.29-1.21 (m,8 H); 13C-NMR (CDCl3)δ175.1(d),172.27(d),157.20(t),156.63,144.43(d),141.69,128.07,127.45,125.40(d),120.35,81.71,80.00,67.36(d),50.42,49.81(d),48.90(d),47.82(d),41.77,41.16,40.64,39.19,33.12,32.75,29.45(d),28.81,26.59,24.97,24.70; HRMS (FAB+) calcd. for C30H39N3O7[(M+H)+] 553.2788 observed 554.2873.
【0133】
(実施例11) Fmoc-C6-BocPNA-OHの合成
アセトン(6.0 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C6-OPfp (331.9 mg,0.6 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で24時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ、溶液を濃縮しシリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮し白色のアモルファスパウダーとしてFmoc-C6-BocPNA-OH (197.0 mg,87%)を得た。1H-NMR (DMSO-d6)δ7.88 (d,J =7.7 Hz, 2 H),7.68 (ma) and 7.63 (mi) (brd,J = 7.4 Hz,2 H),7.40 (t,J = 7.4 Hz,2 H), 7.32 (t, J =7.4 Hz,2 H), 7.22 (brt, 1 H), 6.79 (ma) and 6.66 (mi) (brt, 1 H), 4.39(mi) and 4.29 (ma) (brt, 2 H), 4.20 (t, J = 6.7 Hz,1 H) 4.08 (mi) and 3.91 (ma) (brs, 2 H), 3.10 - 2.97 (m, 4 H),2.31 (ma) and 2.15 (mi) (brt,2 H),1.50 - 1.47(m,2 H),1.41 - 1.36(m,11 H),1.28-1.24(brd,6 H);13C-NMR (CDCl3)δ175.23(d),172.41(d),157.11(d),156.60,144.34(d),141.69,128.07,127.45,125.40(d),120.35,81.68,80.00(d),67.72,67.50(d),53.87,50.77,50.14(d),48.90,47.82(d),41.29(d),41.36,40.69,39.18,33.19,32.96,30.00,29.11,28.81,26.75(d),25.27(d),24.80(d); HRMS(FAB+) calcd. for C29H41N3O7[(M+H)+] 567.2945 observed 568.3027.
【0134】
(実施例12) Fmoc-C7-BocPNA-OHの合成
アセトン(7.0 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C7-OPfp (328.5 mg,0.6 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で24時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ、溶液を濃縮しシリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮し白色のアモルファスパウダーとしてFmoc-C7-BocPNA-OH (196.1 mg,84%)を得た。1H-NMR (DMSO-d6);δ7.88(d,J = 7.7 Hz,2 H),7.68 (ma) and 7.63 (mi) (brd,J = 7.4 Hz,2 H),7.40 (t,J = 7.4 Hz,2 H),7.32 (t,J =7.4 Hz,2 H),7.22 (brt,1 H),6.79 (ma) and 6.79 (mi) (brt,1 H),4.39(mi) and 4.29(ma) (brd,J = 6.9 Hz,2 H),4.05 (t,J = 6.7 Hz,1 H),4.08 (mi) and 3.91 (ma) (brs,2 H),3.12 - 2.95 (m,4 H),2.31 (mi) and 2.15 (ma) (brt,2 H),1.50 - 1.47 (m,2 H),1.42 - 1.34 (m,11 H),1.25 (brd,2 H); 13C-NMR (CDCl3) δ 174.72,172.19,156.52,156.05,143.78(d),141.14,127.51, 126.89, 124.86(d), 119.79, 79.43(d),66.80(d),53.33,50.19,49.20, 48.50,47.14(d),41.18(d), 38.60,32.28(d),29.60, 28.83,28.26, 26.27(d),24.68(d),21.77(d); HRMS (FAB+) calcd. for C32H43N3O7[(M+H)+] 581.3101 observed 582.3171.
【0135】
(実施例13) Fmoc-C10-BocPNA-OH
アセトン(7.0 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C10-OPfp (353.7 mg,0.6 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で24時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ、溶液を濃縮しシリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮し白色のアモルファスパウダーとしてFmoc-C10-BocPNA-OH (218.5 mg,88%)を得た。1H-NMR (CDCl3)δ9.60 (brs,1 H),7.73 (d,J = 7.6 Hz,2 H),7.58 (d,J = 6.8 Hz,2 H),7.37 (t,J = 6.8 Hz,2 H),7.29 (t,J = 7.2Hz,2 H),5.52 (ma) and 5.35 (mi) (brd,1 H),5.00 (s,1 H),4.45 (mi)and 4.40 (ma) (brd,J = 6.4 Hz,2 H),4.23 - 4.22 (m,1 H),4.09 (mi) and 4.04 (ma) (brs,2 H),3.57 - 3.46 (m,2 H),3.29 - 3.03 (m,4 H),1.66- 1.58 (brs,2 H),1.52 - 1.37 (m,11 H),1.33 - 1.20 (brs,12 H); 13C-NMR (CDCl3)δ174.5(d),172.43(d),171.64(d),157.83(d),156.98,156.03,143.78(d),141.17,127.52,126.90,124.86(d),119.70(d),81.08,79.43(d),67.26,66.43,50.14,49.29,48.145(d),47.17(t),41.47,40.99,40.16,38.63,32.90,32.43(d),29.55(d),29.22(m),28.28,26.56(d),24.98,24.75; HRMS (FAB+) calcd. for C35H49N3O7[(M+H)+] 623.3571 observed 624.3643.
【0136】
(実施例14)Fmoc-C11-OPfpの合成
Fmoc-C11-OH (437.5 mg,2.0 mmol)とPfpOH(276.6 mg,3.0 mmol)のDMF溶液(2.5 mL)にDCC(309.5 mg,3.0 mmol)を氷冷下で加え、この反応液を0 ℃で30分次いで室温で18時間攪拌した。反応終了後DCUreaを濾別して濾液を減圧濃縮し、残渣をシリカゲルカラムクロマト法(CH2Cl2)により精製した後、Hexaneで再結晶し白色粉末としてFmoc-C11-OPfp (575.6 mg,96%)を得た。1H-NMR (CDCl3)δ7.79(d,J = 7.6 Hz,2 H),7.63 (d,J = 7.2 Hz, 2 H),7.43 (t,J =7.6 Hz, 2 H),7.34 (t,J = 7.2 Hz, 2 H),4.86 (brt,1 H),4.47 (mi) and 4.44 (ma)(brd,2 H),4.25 (t,1 H),3.22 (q,J = 6.1 Hz, 2 H),2.68 (t,J = 7.2 Hz, 2 H),1.80 (m,2 H),1.56 - 1.52 (m,2 H),1.47 - 1.42 (m,2 H),1.39- 1.30 (m,12 H); HRMS (FAB+) calcd. for C33H34F5NO4[(M+H)+]603.6194 observed 604.2490.
【0137】
(実施例15) Fmoc-C11-BocPNA-OH
アセトン(10 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C11-OPfp (362.2 mg,0.6 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で48時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ、溶液を濃縮しシリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮し白色のアモルファスパウダーとしてFmoc-C11-BocPNA-OH (227.6 mg,89%)を得た。1H-NMR (CDCl3);δ9.62 (brs,1 H),7.74 (d,J = 7.6 Hz,2 H),7.57 (d,J = 7.5 Hz,2 H),7.37 (t,J = 7.1 Hz,2 H),7.28 (t,J = 6.8Hz,2 H),5.53 (ma) and 5.35 (mi) (brs,H),5.00 (brt,1 H),4.43 (mi)and 4.37 (ma) (brd,J = 6.4 Hz,2 H),4.22 - 4.19 (m,1 H),4.06 (mi) and 4.01 (ma) (brs,2 H),3.51 - 3.44 (m,2 H),3.23 - 3.08 (m,4 H),2.36(ma) and 2.21 (mi) (brt,J = 7.0 Hz,2 H),1.69 - 1.58 (brs,2 H),1.52- 1.40 (m,11 H),1.29 - 1.25 (brd,14 H); 13C-NMR (CDCl3)δ175.45(d),172.428(d),157.91(d),157.01,156.57,144.33(d),141.72,128.05,127.43,125.40(d),120.35,81.63,80.00(d),67.77,66.97,50.67,49.84,49.20(d),47.72(t),42.10,41.53,40.70,39.18,33.51,33.07,30.032,29.83(m),28.82,27.00(d),25.44,25.35; HRMS (FAB+) calcd. for C36H51N3O7[(M+H)+] 637.3727 observed 638.3794.
【0138】
(実施例16)PNA oligo 1(H2N-G-A-T-pMR-G-A-C-G-C-CONH2)の合成(第1法)
tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; φrum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、固相担体MBHA(50 mg)に各種PNAモノマーユニット(チミン7.7 mg、シトシン10.1 mg、アデニン10.6 mg、グアニン10.9 mg、Fmoc-Gly-BocPNA-OH 10.0 mg;各20 mmol)と縮合剤HBTU(7.6 mg、20 mmol)とDIEA(7.0 μL、20 mmol)を用いて逐次伸長反応を行った。この間、Fmoc-Gly-BocPNA-OHを縮合した際には、次のチミジンPNAモノマーユニットと縮合する前に、まずピペリジン処理(20% piperidine in DMF 3 mL、室温30分)してFmoc基を脱保護して、次いで光機能性分子のカルボン酸誘導体としてp-Methyl Red-OH(10.8 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(13.9 μL、40 mmol)を用いて縮合し目的の位置に導入した。その後、チミジン、アデニン、グアニンのPNAモノマーユニットを逐次縮合したあと、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しとZ基の脱保護を同時に行い、後処理して、目的とするPNA oligo 1を得た。UV λmax (H2O) 303, 548 (nm)。
【0139】
(実施例17)PNA oligo 1の合成(第2法)
tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; φrum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、固相担体MBHA(50 mg)に各種PNAモノマーユニット(チミン7.7 mg、シトシン10.1 mg、アデニン10.6 mg、グアニン10.9 mg、Fmoc-Gly-BocPNA-OH 10.0 mg;各20 mmol)と縮合剤HBTU(7.6 mg、20 mmol)とDIEA(7.0 μL、20 mmol)を用いて逐次伸長反応を行った。全てのモノマーユニットを逐次縮合した後、ピペリジン処理(20% piperidine in DMF、室温30分)してFmoc-Gly残基を脱保護して、次いで光機能性分子のカルボン酸誘導体としてp-Methyl Red-OH(10.8 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(13.9 μL、40 mmol)を用いて縮合し目的の位置に導入した。その後、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しとZ基の脱保護を同時に行い、後処理して、目的とするPNA oligo 1を得た。UV λmax (H2O) 302, 550 (nm)。
【0140】
(実施例18)PNA oligo 2(H2N-G-A-T-mMR-G-A-C-G-C-CONH2)の合成(第2法)
tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; φrum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、固相担体MBHA(50 mg)に各種PNAモノマーユニット(チミン7.7 mg、シトシン10.1 mg、アデニン10.6 mg、グアニン10.9 mg、Fmoc-Gly-BocPNA-OH 10.0 mg;各20 mmol)と縮合剤HBTU(7.6 mg、20 mmol)とDIEA(7.0 μL、20 mmol)を用いて逐次伸長反応を行った。全てのモノマーユニットを逐次縮合した後、ピペリジン処理(20% piperidine in DMF、室温30分)してFmoc-Gly残基を脱保護して、次いで光機能性分子のカルボン酸誘導体としてm-Methyl Red-OH(10.8 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(13.9 μL、40 mmol)を用いて縮合し目的の位置に導入した。その後、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しとZ基の脱保護を同時に行い、後処理して、目的とするPNA oligo 2を得た。UVλmax (H2O) 308, 570 (nm)。
【0141】
(実施例19)PNA oligo 3(H2N-G-A-T-oMR-G-A-C-G-C-CONH2)の合成(第2法)
tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; φrum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、固相担体MBHA(50 mg)に各種PNAモノマーユニット(チミン7.7 mg、シトシン10.1 mg、アデニン10.6 mg、グアニン10.9 mg、Fmoc-Gly-BocPNA-OH 10.0 mg;各20 mmol)と縮合剤HBTU(7.6 mg、20 mmol)とDIEA(7.0 μL、20 mmol)を用いて逐次伸長反応を行った。全てのモノマーユニットを逐次縮合した後、ピペリジン処理(20% piperidine in DMF、室温30分)してFmoc-Gly残基を脱保護して、次いで光機能性分子のカルボン酸誘導体としてo-Methyl Red-OH(10.8 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(13.9 μL、40 mmol)を用いて縮合し目的の位置に導入した。その後、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しとZ基の脱保護を同時に行い、後処理して、目的とするPNA oligo 1を得た。UV λmax (H2O) 302, 561 (nm)。
【0142】
(実施例20)膜透過性機能を有する蛍光PNAプローブ(1)の合成
【0143】
【化25】
【0144】
前記第2法を用いた。
標準的tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; Orum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、まず、固相担体MBHA(50 mg)にチミンPNAモノマーユニット(7.7 mg、20 mmol)、縮合剤HBTU(7.6 mg、20 mmol)とDIEA(3.5 mL、20 mmol)を用いて逐次伸長反応を行った(塩基配列認識領域の設計)。次いで、リンカー用ω-アミノ酸Boc-7-aminoheptanoic Acid(5.2 mg、20 mmol)、Fmoc-Ahx-BocPNA-OH(10.0 mg、20 mmol)と再度Boc-7-aminoheptanoic Acidを、縮合剤HBTU(7.6 mg、20 mmol)とDIEA(3.5 mL、20 mmol)を用いて順次縮合させた(リンカー部位と膜透過性機能領域の設計)。全てのユニットを逐次縮合した後、ピペリジン処理(20% piperidine in DMF、室温3分)してFmoc基を脱保護した。次いで、機能性カルボン酸誘導体としてFmoc-Arg(Mts)-OH(23.1 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(7.0 mL、40 mmol)を用いて縮合し目的の位置に機能性分子を導入した(膜透過性機能の導入)。TFA処理(95% TFA/5% m-cresol)によりBoc基を脱保護した後、FITC(9.3 mg,25 mmol)をDIEA(17.4 mL, 100 mmol)存在下、室温で12時間振盪し蛍光標識化した(蛍光標識部位の設計)。最後にピペリジン処理(20% piperidine in DMF、室温3分)して残るFmoc基を脱保護した後、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しを行い、後処理して、目的物を得た。MALDI-TOF MS: calcd. 2096.26 (M + H+), found 2096.36.
【0145】
(実施例21)膜透過性機能を有する蛍光PNAプローブ(2)の合成
【0146】
【化26】
【0147】
標準的tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; Orum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、まず、固相担体MBHA(50 mg)にチミンPNAモノマーユニット(7.7 mg、20 mmol)、縮合剤HBTU(7.6 mg、20 mmol)とDIEA(3.5 mL、20 mmol)を用いて逐次伸長反応を行った(塩基配列認識領域の設計)。
【0148】
次いで、リンカー用ω-アミノ酸Boc-7-aminoheptanoic Acid(5.2 mg、20 mmol)、Fmoc-Ahx-BocPNA-OH(10.0 mg、20 mmol)と再度Boc-7-aminoheptanoic Acidを、縮合剤HBTU(7.6 mg、20 mmol)とDIEA(3.5 mL、20 mmol)を用いて順次縮合させた(リンカー部位と膜透過性機能領域の設計)。全てのユニットを逐次縮合した後、ピペリジン処理(20% piperidine in DMF、室温3分)してFmoc基を脱保護した。次いで、機能性カルボン酸誘導体としてFmoc-Arg(Mts)-OH(23.1 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(7.0 mL、40 mmol)を用いて縮合し目的の位置に機能性分子を導入した(膜透過性機能の導入)。
【0149】
これをピペリジン処理(20% piperidine in DMF、室温3分)してFmoc基を脱保護した後、再度Fmoc-Arg(Mts)-OH(23.1 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(7.0 mL、40 mmol)を用いて縮合し目的の位置に機能性分子を導入した(膜透過性機能の追加導入)。TFA処理(95% TFA/5% m-cresol)によりBoc基を脱保護した後、FITC(9.3 mg, 25 mmol)をDIEA(17.4 mL, 100 mmol)存在下、室温で12時間振盪し蛍光標識化した(蛍光標識部位の設計)。
【0150】
最後にピペリジン処理(20% piperidine in DMF、室温3分)して残るFmoc基を脱保護した後、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しを行い、後処理して、目的物を得た。MALDI-TOF MS: calcd. 2252.44 (M + H+), found 2252.33.
[発明の効果]
本発明によれば、光機能性分子に限定されることない多種多様な機能性分子を、PNA中に容易に導入することができ、多種多様な機能性分子をPNA中に容易かつ効率的に導入することができ、遺伝子治療などに用いられる種々のPNAの構築が可能になる。
【図面の簡単な説明】
【0151】
【図1】DNAとPNAの構造および荷電の状況の違いを表す図である。
【図2】2種類のPNAモノマーユニットの構造を表す図である。
【技術分野】
【0001】
本発明は、機能性ペプチド核酸モノマーの新規な製造方法、該製造方法によって製造された機能性ペプチド核酸オリゴマーおよびその中間体に関する。より詳細には、前駆体的PNAモノマーユニットをPNAオリゴマーに導入した後に1種または2種以上の機能性分子をポスト合成的に導入することを特徴とする、前記製造方法に関する。
【背景技術】
【0002】
核酸は生物の遺伝情報を司るDNAおよびRNAである。これに対して、ペプチド核酸(PNA)とは、核酸の糖リン酸骨格をN−(2−アミノエチル)グリシン骨格に変換した修飾核酸である(図1)。DNA/RNAの糖リン酸骨格は中性条件で負電荷を帯びていて相補鎖間の静電的な反発があるが、PNAの背骨構造はもともと電荷を持たないので静電的な反発がない。そのためPNAは従来の核酸と比較して、高い二重鎖形成能をもち、高い塩基配列認識能を持つ。さらにPNAは生体内ヌクレアーゼ・プロテアーゼに対し非常に安定で分解されないので、アンチセンス分子として遺伝子治療に応用することが検討されている。
【0003】
従来のDNAを媒体にしていた技術をPNA化することにより、これまで克服できなかったDNAの欠点を補うことが可能となった。例えば、遺伝情報の体系的な解析を高速に且つ大量に行うための「DNAマイクロアレイ技術」および塩基配列を特異的に認識したことを蛍光発光により検出できるプローブとして最近開発された「モレキュラービーコン」に応用することが可能である。これらはいずれも酵素耐性に乏しいDNAを媒体とするため、これらの技術を用いるに際しては厳密なサンプリングが要求される。この要求を満たすことが、前記の技術を高度化する上での鍵となっている。
【0004】
一方PNAは酵素に対し完全な耐性を持つので、DNAマイクロアレイ技術およびモレキュラービーコンにおいてPNAをDNAに代用することによって、前記技術の欠点が克服され、さらに長所が引き出されるものと期待されている。DNAマイクロアレイ技術およびモレキュラービーコン以外にもPNA化することにより発展が期待される分野は数多いが、それらにおいてはPNAの効率的な機能化、すなわちPNAモノマーへの機能性分子の効率的な導入による新規なPNAモノマーの設計が必要である。
【0005】
PNAオリゴマーの合成方法には通常の固相ペプチド合成法を用いるので、PNAモノマーユニットをPNAの背骨構造によって分類すると、Fmoc型PNAモノマーユニットとtBoc型PNAモノマーユニットの2種類が含まれる(図2)。
【0006】
Fmoc型PNAモノマーユニットの合成方法は既に確立されており、しかもそのオリゴマーの合成は一般的なDNA自動合成機によって可能であるため、下記のルート
【0007】
【化1】
【0008】
によって、少量スケールでの合成が可能となっている。
【0009】
当初PNAには下記のようなtBoc型PNAモノマーユニット
【0010】
【化2】
【0011】
が採用され、その後より効率のよい合成方法
【0012】
【化3】
【0013】
が確立された。しかし、前述したように取り扱いが容易なFmoc型が開発されたため、tBoc型の使用頻度は減少している。
【0014】
しかし、グアニン・チミン・シトシン・アデニン4種類の核酸塩基以外の機能性分子を導入する際、例えば光機能性分子を導入する際には、導入する機能性分子がアルカリ条件に不安定な場合が多いので、アルカリ条件を使用しないtBoc型PNA背骨構造の有用性は高い。「t−ブトキシカルボニルアミノエチルアミン及びアミノ酸誘導体の製造方法」に関しては、本発明者らが特願2000−268638として既に特許出願中である。
【0015】
これ以外にも、光機能性オリゴPNAのモノマーユニットの合成例は過去に5例が知られている。これら全てが上記ルートを用いているが、その収率については記載がないか、または極めて低いものでしかない(Peter E. Nielsen, GeraldHaaiman, Anne B. Eldrup PCT Int. Appl. (1998) WO 985295 A1 19981126, T. A. Tran, R.-H. Mattern, B. A. Morgan (1999) J. Pept. Res, 53, 134-145,Jesper Lohse et al. (1997) Bioconjugate Chem., 8, 503-509, Hans-georg Batz, Henrik Frydenlund Hansen, et al. Pct Int. Appl. (1998) WO 9837232A2 19980827, Bruce Armitage, Troels Koch, et al. (1998) Nucleic Acid Res., 26, 715-720)。また、用いられる化合物の構造がアルカリ性条件に比較的安定であることが特徴的であるため、アルカリ性条件に不安定な発色団が付くと、前記従来法と類似の方法、すなわち下記ルートA
【0016】
【化4】
【0017】
【化5】
【0018】
では効率良く合成できないと予想された。
したがって、一般に光機能性分子等の機能性分子は高価な場合が多いため、より合目的的な機能性PNAの合成方法、すなわち、1)機能性PNAモノマーユニットの設計における、機能性分子のPNA背骨構造への効率的な導入、2)コストパフォーマンスを考えた合成ルート、および3)遺伝子診断薬としての応用へ適応させるための、これらの機能性分子を超高速に導入する方法が探求された。
【0019】
上記課題に鑑み、本発明者らは、機能性PNAモノマーの新規製造方法として、下記ルートB
【0020】
【化6】
【0021】
に示すように、PNA背骨構造にt−ブトキシカルボニルアミノエチルアミン誘導体6を用いて1のペンタフルオロフェニル基を含む活性エステル体5と縮合してほぼ定量的に光機能性PNAモノマー4を合成する方法を見出した。
【0022】
また、本発明者らは、機能性PNAモノマーを合成する別法として、PNA背骨構造に上記t−ブトキシカルボニルアミノエチルアミン誘導体6の代わりにベンジルオキシカルボニル−ω−アミノ酸誘導体を用いる方法(ルートC)を見出した。これらの方法については、既に特許出願がなされている。
【0023】
したがって、最終的に機能性PNAを合成するための方法として、上記ルートBおよびルートCのいずれかを用いる方法によって機能性PNAモノマーを合成した後に、それらを重合する方法が工業的にも確立されつつある。すなわち、現在までの機能性PNAの合成法によってPNAプローブとして用いられる機能性PNAを工業的に大量合成することは可能になりつつある。
【0024】
一方、コストパフォーマンスの向上および機能性分子を超高速に導入することを目的とした、機能性PNAを合成方法の改良もなされている。例えば、前記機能性PNAモノマーユニットを用いる方法とは異なるアプローチとして、下記の前駆体的PNAモノマーユニットを利用することによって、ポスト合成的に機能性分子をPNAオリゴマーに導入する方法が報告されている(Oliver Seitz; Tetrahedron Letters 1999, 40, 4161-4164.)。
【0025】
【化7】
【0026】
当該方法は、前記前駆体的PNAモノマーユニットをPNAオリゴマーに導入した後、さらに機能性分子を導入することによって機能性PNAを合成するものである。しかし、当該方法においては、導入できる機能性分子の種類が限定される等の欠点がある。
【0027】
例えば、下図に示すように、市販されている光機能性分子のsuccinimideエステルを導入することはできず、導入するためにはFmoc-Gly等のリンカーをまず導入する必要があるが、結果として上記化合物は使用しにくいものになっている。
【0028】
【化8】
【0029】
また、細胞中に導入するためのに蛍光プローブとして、これまでDNAオリゴマー・RNAオリゴマー・PNAオリゴマーが利用されているが、これらを細胞中に導入するためには、当然ながら細胞膜を通過させなければならない。しかし、細胞膜は膜表面が負電荷を帯びているため、元々負に帯電しているDNA/RNAオリゴマーを導入するのは非常に困難である。
【0030】
一方PNAオリゴマーは電気的に中性であるが、膜透過しにくいという結果が得られている。したがって、PNAオリゴマーを細胞内に導入するに際しては、膜表面を前処理してその導入をしやすくしたり、あるいはトランスフェクション試薬を用いて導入せざるを得ないのが現状である。
【0031】
しかし、そのような処理を施してPNAオリゴマーを導入した場合においてプローブの機能が発揮されたとしても、本来生体が示す挙動を正確に表現していることは必ずしも保証されない。しかも、これは細胞1個の場合であり、多細胞(個体)での利用に至っては到底不可能である。このような現状および観点から、膜透過性機能を有する蛍光PNAプローブの開発が有用であると考えられている。
【0032】
なお、膜透過性機能を有する蛍光PNAプローブは既に存在する。例えば、1)膜透過性機能を有するオリゴペプチドをPNAに結合させたもの、2)膜透過性機能を有するリン脂質をPNAに結合させたものが挙げられる。しかしながら、これらは膜透過した後細胞内においてプロテアーゼ等の酵素によりPNA以外の部分が分解され、細胞内に滞留してしまうことが予想される。このことは、ターゲットを捕捉出来なかった過剰なPNAプローブが膜透過性機能を失い、その後の洗浄過程で細胞外に出にくくなることにつながるため、本来細胞が持っている遺伝子発現系を正確に表現できないことを意味する。
【発明の開示】
【発明が解決しようとする課題】
【0033】
したがって、本発明は、コストパフォーマンスに優れ、かつ機能性分子を超高速に導入することができる、機能性PNAの新規合成方法およびそれに用いる化合物、ならびに新規機能性PNAを提供することを目的とする。
【課題を解決するための手段】
【0034】
上記課題に鑑み研究を重ねた結果、本発明者らは、前駆体的PNAモノマーユニットの構造を最適化することによって、驚くべきことに、従来法における前記課題が克服され、かつ極めて広範にわたる機能性PNAを合成できることを見出し、本発明を完成するに至った。
【0035】
すなわち、本発明は、機能性PNAオリゴマーを製造する方法であって、保護基によって保護されたアデニン、グアニン、シトシンまたはチミンを有するPNAモノマーユニットを、Fmoc−ω−アミノ酸−BocPNA−OHと反応させてPNAオリゴマーを合成した後、該PNAオリゴマーに遊離カルボン酸を有する機能性分子を導入し、さらに前記保護基の脱保護を行うことを含む、前記方法に関する。
【0036】
また、本発明は、Fmoc−ω−アミノ酸−BocPNA−OHが、Fmoc−ω−アミノ酸ペンタフルオロフェニルエステルとBocPNA−OHとの反応によって製造されたものであることを特徴とする、前記方法に関する。さらに、本発明は、Fmoc−ω−アミノ酸ペンタフルオロフェニルエステルが、Fmoc−ω−アミノ酸とペンタフルオロフェノールとの反応によって製造されたものであることを特徴とする、前記方法に関する。
【0037】
さらにまた、本発明は、機能性分子を導入した後に、さらに別異の機能性分子の導入を行うことを特徴とする、前記方法に関する。またさらに、本発明は、導入される機能性分子が、光機能性分子、膜透過性機能分子、臓器選択性機能分子、殺菌性機能分子および分子認識性機能分子から選択されることを特徴とする、、前記方法に関する。また、本発明は、導入される機能性分子が、光機能性分子および膜透過性機能分子を含むことを特徴とする、前記方法に関する。そして、本発明は、光機能性分子が、FITC、ROX、TAMRAまたはDabcylであり、膜透過性機能分子が水溶性アミノ酸であることを特徴とする、前記方法に関する。
【0038】
また、本発明は、アデニン、グアニン、シトシンまたはチミンを保護する保護基が、Z基であることを特徴とする、前記方法に関する。
【0039】
さらに、本発明は、PNAオリゴマーの合成が、tBoc法用固相担体を用いたPNA鎖における縮合・伸長を含むことを特徴とする、前記方法に関する。またさらに、本発明は、tBoc法用固相担体がMBHAであることを特徴とする、前記方法に関する。さらにまた、本発明は、遊離カルボン酸を有する機能性分子の導入が、Fmoc基をピペリジン処理によって選択的に脱保護して得られた1級アミノ基との脱水縮合によって行われることを特徴とする、前記方法に関する。
【0040】
また、本発明は、Fmoc−ω−アミノ酸−BocPNA−OHが、下記一般式(I)
【0041】
【化9】
【0042】
(式中、nは1〜15までの整数を表す)で表される化合物であることを特徴とする、請求項1〜7のいずれかに記載の方法。
また、本発明は、下記a)〜d):
a)Fmoc−ω−アミノ酸ペンタフルオロフェニルエステルを製造する工程において、Fmoc−ω−アミノ酸とペンタフルオロフェノールとを反応させること;
b)Fmoc−ω−アミノ酸−BocPNA−OHを製造する工程において、Fmoc−ω−アミノ酸ペンタフルオロフェニルエステルをBocPNA−OHと反応させることによってFmoc−ω−アミノ基をBocPNA−OHに導入すること;
c)Fmoc−ω−アミノ酸−BocPNA−OHからPNAオリゴマーを製造する工程において、PNAモノマーユニットを、Fmoc−ω−アミノ酸−BocPNA−OHと反応させてPNAオリゴマーを製造すること;および
d)前記PNAオリゴマーから機能性PNAオリゴマーを製造する工程において、PNAオリゴマーへの機能性分子の導入が、Fmoc基をピペリジン処理によって選択的に脱保護して得られた1級アミノ基との脱水縮合によって行われる行うこと、の1または2以上を含むことを特徴とする、前記方法に関する。
そして、本発明は、下記一般式(I)
【0043】
【化10】
【0044】
(式中、nは1〜15までの整数を表す)で表される化合物に関する。
【0045】
そしてさらに、本発明は、一般式(I)
【0046】
【化11】
【0047】
(式中、nは1〜15までの整数を表す)で表される化合物を製造する方法であって、Fmoc−ω−アミノ酸をペンタフルオロフェノールと反応させ、その反応物をBocPNA−OHと反応させることによってFmoc−ω−アミノ基を導入することを特徴とする、前記方法に関する。
【0048】
そして、本発明は、下記一般式(II)
【0049】
【化12】
【0050】
(式中、Bは、互いに独立し、同一または異なって、アデニン、グアニン、シトシンまたはチミンであり、Rは、互いに独立し、同一または異なって、Fmoc基または機能性カルボン酸誘導体であり、R1は、水素原子または機能性カルボン酸誘導体であり、a〜hは0〜10の整数であり、X1〜X3、Y1、Y2およびZ1〜Z5はいずれも0以上の整数であり、X1+X2+X3≧0であり、Y1+Y2>0であり、Z1+Z2+Z3+Z4+Z5≧0である。ただし、X1+X2+X3およびZ1+Z2+Z3+Z4+Z5が同時に0であることはなく、X1+X2+X3=0の場合、R1は機能性カルボン酸誘導体である。)で表される化合物に関する。
【0051】
さらに、本発明は、Z1+Z2+Z3+Z4+Z5=0であり、R1が水素原子である、前記化合物に関する。
【0052】
また、本発明は、Rがメチルレッドのカルボン酸誘導体を含むことを特徴とする、前記化合物に関する。
【0053】
さらに、本発明は、X1+X2+X3=9であり、Y1+Y2=1であることを特徴とする、前記化合物に関する。
【0054】
また、本発明は、X1=3、X2=6であり、Y1=1であることを特徴とする、前記化合物に関する。
【0055】
さらに、本発明は、RまたはR1が細胞膜透過性機能分子誘導体であることを特徴とする、前記化合物に関する。
【0056】
さらにまた、本発明は、さらにR1が機能性カルボン酸誘導体であることを特徴とする、前記化合物に関する。
【0057】
また、本発明は、X1=Z1=1であることを特徴とする、前記化合物に関する。
【0058】
さらに、本発明は、Y1≧2であり、Z2=1であることを特徴とする、前記化合物に関する。
【0059】
またさらに、本発明は、a≦6であり、b≦4であり、f≦6であることを特徴とする、前記化合物に関する。
【0060】
また、本発明は、R1が光機能性カルボン酸誘導体であることを特徴とする、前記化合物に関する。
さらに、本発明は、下記一般式(III)
【0061】
【化13】
【0062】
(式中、nは1〜15の整数を表す)で表されることを特徴とする化合物に関する。
【0063】
さらに、本発明は、下記一般式(III)
【0064】
【化14】
【0065】
(式中、nは1〜15の整数を表す)で表される化合物を製造する方法であって、Fmoc−ω−アミノ酸とペンタフルオロフェノールとを反応させることを特徴とする、前記方法に関する。
【0066】
本発明は、PNA背骨構造にFmoc−ω−アミノ酸を導入した前駆体的PNAモノマーユニット、すなわちFmoc−ω−アミノ酸−BocPNA−OHをPNAオリゴマーに導入した後、機能性分子をポスト合成的に導入することにより、ほぼ定量的に光機能性PNAオリゴマーを合成できることに成功したものである。
【0067】
上記特徴により、本発明の製造方法においては、導入する機能性分子として市販のsuccinimideエステルを使用する必要がなく、カルボン酸を有する化合物であれば問題なく利用でき且つ定量的に導入できる。そのため、本発明による製造方法はコストパフォーマンスに極めて優れている。
【0068】
また、前記前駆体的PNAモノマーユニットを機能性PNAオリゴマーに導入した後にレジンを分割することにより、それぞれのレジンに異なった機能性分子を導入することができる。したがって、本発明による製造方法によれば、極めて高速な機能性PNAオリゴマーの合成手法を開発することが可能となる。
【0069】
本発明の方法によって効率的な合成が可能になる機能性PNAオリゴマーの例として、下記一般式(II)
【0070】
【化15】
【0071】
(式中、Bは、互いに独立し、同一または異なって、アデニン、グアニン、シトシンまたはチミンであり、Rは、互いに独立し、同一または異なって、Fmoc基または機能性カルボン酸誘導体であり、R1は、水素原子または機能性カルボン酸誘導体であり、a〜hは0〜10の整数であり、X1〜X3、Y1、Y2およびZ1〜Z5はいずれも0以上の整数であり、X1+X2+X3≧0であり、Y1+Y2>0であり、Z1+Z2+Z3+Z4+Z5≧0である。ただし、X1+X2+X3およびZ1+Z2+Z3+Z4+Z5が同時に0であることはなく、X1+X2+X3=0の場合、R1は機能性カルボン酸誘導体である。)で表される化合物において、Z1+Z2+Z3+Z4+Z5=0であり、R1が水素原子である化合物を挙げることができる。
【0072】
本発明によれば、前記一般式(II)で表される化合物において同一または異なる機能性分子を、任意の複数の位置に導入することも可能となる。すなわち、前記前駆体的PNAモノマーユニットを用いてPNAオリゴマーを導入した後、ピペリジン処理と機能性分子のポスト合成的導入を一括して行うことによるものであるが、これはPNAオリゴマーの細胞膜透過機能を向上させるアンテナペディアを高速に設計する上で、欠かせないものである。この点においても、本発明による方法は極めて優れたものである。
【0073】
このように製造される化合物の例として、前記一般式(II)において、Z1+Z2+Z3+Z4+Z5>0であり、Rが細胞膜透過性分子誘導体であり、R1が機能性カルボン酸誘導体であるものが挙げられる。
【0074】
このプローブは大きく蛍光標識領域・細胞膜透過性機能領域・分子認識領域の3つに分けることができ、それぞれをリンカー部位(Z1〜Z5の添字付きで表される部分)を介して結合させた形をしている。
【0075】
蛍光標識化合物は市販のものも、既に本発明者らがPCT出願を行った新規蛍光標識化PNAモノマーユニットも用いることができる。
【0076】
分子認識部位は市販のPNAユニットを用いて合成する。このものの特徴は、既に国内特許出願を行っている一般式(I)で表される新規PNAユニットを膜透過性機能領域部に用いていることである。該一般式(I)で表される新規PNAユニットは機能性分子をポスト合成的に導入するために開発された前駆体ユニットであり、これを複数個並べて導入した後、前記したように同一機能性分子を一括導入できることを特徴としている。
【0077】
したがって、本発明によれば、光機能性分子に限定されることない、多種多様な機能性分子を、PNA中に容易かつ極めて効率的に導入することができるようになる。
【0078】
このような機能性分子として、Naphthalimide型、Flavin型、Dabcyl型、Biotin型、FAM型、Rhodamine型、TAMRA型、ROX型、HABA型、Pyrene型、Coumarine型等の光機能性モノマーユニット、膜透過機能性分子、臓器選択機能性分子、殺菌機能性分子および分子認識機能性分子等が挙げられる。
【0079】
すなわち、本発明における「機能性」の語は、光機能性のみならず、膜透過性、臓器選択性、殺菌機能性および分子認識機能性等を含む、ある特定の修飾を行うことによって化合物に新たに付与される種々の機能の全てを意味するものである。
【0080】
さらに、本発明における「機能性PNA」の語は、PNAモノマー同士が2−(N−アミノエチル)グリシン骨格によって直接結合したもののみならず、その間にリンカーとしての炭化水素鎖等を含むものも意味するものである。
【発明を実施するための最良の形態】
【0081】
ここで、本発明による方法の特徴を更に詳細に説明する。
本発明によるオリゴPNAを合成するルートは、典型的には、下図
【0082】
【化16】
【0083】
に示すとおりである。
まず、必要に応じて、下図
【0084】
【化17】
【0085】
に示すように、Fmoc−ω−アミノ酸とペンタフルオロフェノール(PfpOH)とを反応させて得られるFmoc−ω−アミノ酸ペンタフルオロフェニルエステル(Fmoc−ω−アミノ酸−OPfp)とから、Fmoc−ω−アミノ酸−BocPNA−OHを合成する。
【0086】
以後の工程において用いる該Fmoc−ω−アミノ酸−OPfpの溶液を得るには、DMFなどの有機溶媒、またはアセトン等と水とを含む水溶性溶媒したものいずれも好適に用いることができる。前記水溶性溶媒を用いた場合には、精製等の後処理の面におけるメリットを有するものである。
【0087】
前記Fmoc−ω−アミノ酸−OPfpは、Fmoc−ω−アミノ酸とPfpOHをDMF溶液中にてDCCを加えて反応させることによって、例えば下記式(III)
【0088】
【化18】
【0089】
(式中、nは1〜15の整数を表す)で表されるものが得られる。
次いで、これにBocPNA−OHのDMF溶液にジイソプロピルエチルアミンとともに添加し、Fmoc−ω−アミノ酸−BocPNA−OHを得る。Fmoc−ω−アミノ酸−BocPNA−OHは、PNAモノマーユニットの前駆体として機能するため、前駆体的PNAモノマーユニットと呼ぶことができる。
【0090】
式(I)において、nとしては1〜15までの整数を適宜選択できるが、nの値が大きい方が、ハイブリッド形成時の立体的反発(あるいは障害)を軽減する点において好ましい。
次に、下図
【0091】
【化19】
【0092】
に示すように、前駆体的PNAモノマーユニットを用いて、オリゴマーIaを合成する。具体的には、Z基(N−ベンジルオキシカルボニル基)等で保護されたアデニン、グアニン、シトシンまたはチミンを有するPNAモノマーユニットを、前駆体的PNAモノマーユニットと反応させ、tBoc法用固相担体を用いてPNA鎖を逐次縮合・伸長せしめる。
【0093】
PNA鎖の縮合においては、予めtBoc基を脱離しておく必要があるが、その方法に制限はなく、一般的な方法が用いられる。それに続く縮合には、HATU、HBTUおよびBOP等の一般的な縮合剤が用いられる。また、固相担体に関しては、tBoc法用のものであれば特に制限はないが、特にMBHAが好適に用いられる。
【0094】
次に、下図
【0095】
【化20】
【0096】
に示すように、ピペリジン処理によってFmoc基を選択的に脱保護してアミノ基とし、Ibを得て、さらに、下図
【0097】
【化21】
【0098】
に示すように、該Ibの前記アミノ基に遊離カルボン酸を有する機能性分子を脱水縮合してIcを得る。前記カルボン酸として特に制限はないが、反応性の点においては脂肪族カルボン酸が芳香族カルボン酸を上回るため、脂肪族カルボン酸を用いると製造の効率が高く好ましい。
【0099】
また、ピペリジン処理によるFmoc基の脱保護は、ある程度の時間をかけることによって好適に行われる。特に、20〜40分が好適であり、最も好適には30分であった。
【0100】
縮合剤の種類に特に制限はなく、前記PNA鎖の縮合と同様に、HATU、HBTUおよびBOP等の一般的な縮合剤が用いられる。
【0101】
なお、機能性分子の導入は、Fmoc−ω−アミノ酸−BocPNA−OHを縮合した後、直ちに行ってもよく(第1法)、あるいは、Fmoc−ω−アミノ酸−BocPNA−OHを含む全てのPNAモノマーユニットを逐次縮合した後に行ってもよい(第2法)。
最後に、下図
【0102】
【化22】
【0103】
に示すように、担体レジンからの切り出しとZ基の脱保護を同時に行うことによって、目的とするPNAオリゴマーIdを得る。
【0104】
切り出しおよび脱保護は、Fmoc基の脱保護の後に行われる限りにおいてはその条件に特に制限はない。例えば、TFA/TFMSA/p-cresol/Thioanisole=60/25/10/10のような一般的な条件において好適に行われる。
【0105】
上記のように、本発明による方法においては、従来の機能性モノマー合成に用いる活性エステル化の合成を要する方法とは異なり、機能性分子をそのまま利用できる。また一旦Iaを合成した後に種々の機能性分子が導入可能であるため、従来困難であった高速かつ多様な並列PNAプローブ合成が可能である。
【0106】
Fmoc−ω−アミノ酸−BocPNA−OHとPNA鎖を有する分子との反応を含む本発明による方法によれば、下記一般式(II)
【0107】
【化23】
【0108】
(式中、Bは、互いに独立し、同一または異なって、アデニン、グアニン、シトシンまたはチミンであり、Rは、互いに独立し、同一または異なって、Fmoc基または機能性カルボン酸誘導体であり、R1は、水素原子または機能性カルボン酸誘導体であり、a〜hは0〜10の整数であり、X1〜X3、Y1、Y2およびZ1〜Z5はいずれも0以上の整数であり、X1+X2+X3≧0であり、Y1+Y2>0であり、Z1+Z2+Z3+Z4+Z5≧0である。ただし、X1+X2+X3およびZ1+Z2+Z3+Z4+Z5が同時に0であることはなく、X1+X2+X3=0の場合、R1は機能性カルボン酸誘導体である。)に示す化合物等が好適に合成される。
【0109】
本発明の方法によって、式(II)で表される化合物として、例えば、Rがメチルレッドのカルボン酸誘導体を含むもの、さらにX1+X2+X3=9であり、Y1+Y2=1であるもの、またはX1=3、X2=6であり、Y1=1であるもの等が、特に好適に合成される。
【0110】
また、前記一般式(II)で表される化合物において、複数の機能性分子が導入されたものとして、例えば、RまたはR1が細胞膜透過性機能分子誘導体であるものが好適に合成される。このような化合物は、典型的には、Rが細胞膜透過性機能分子誘導体等であり、R1が光機能性分子等の機能性カルボン酸誘導体等であるもの、すなわち、末端部を含む複数部位に機能性分子が導入され、それらによって複数の機能が付与された化合物である。このような化合物は、例えば下記のように模式化することができる。
【0111】
【化24】
【0112】
このような化合物は、例えば前記一般式(II)において、X1=Z1=Z2=1であり、かつY1≧2である化合物である。このような化合物は合成のしやすさおよび合成コストの面等において好適である。
【0113】
上記化合物において、a、bおよびfはそれぞれ0〜10の整数であれば特に限定されないが、例えばa≦6であり、b≦4であり、f≦6であるものであっても、合成上および実用上のいずれにおいても支障はない。
【0114】
リンカー部位を導入することによって、個々の機能性部位および塩基配列認識領域の干渉を防ぎ、分子の機能をより確実なものにすることができる。本明細書におけるPNA、PNAモノマーおよびPNAオリゴマーの語には、リンカー部位をその末端および/または内部に含むものも包含される。これらの部位または領域間の相互干渉を防ぐための部位としては、前記リンカー部位のみならず、一般式(I)におけるf〜hを、所望に応じて選択することによっても可能である。
【0115】
リンカー部位を構成する基としては、直鎖状または分枝状の炭化水素およびそれらのエーテル体等が挙げられるが、直鎖状炭化水素基は導入の容易さおよびコストなどの面から好適であり、特に炭素数1〜6の直鎖状炭化水素基が好適である。また、エーテル体は、その汎用性において好適である。
【0116】
前記複数の機能性分子が導入された化合物は、例えばKoch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; Orum, H. J. PeptideRes. 1997, 49, 80-88.を利用して好適に合成される。塩基配列認識部位は、市販の各種PNAモノマーを用いて固相合成によりオリゴマー化することができる。リンカー部位には、市販のBoc−7−アミノヘプタン酸、Boc−6−アミノカプロン酸等を用いることができる。
【0117】
1の機能性分子として光機能性分子を導入すれば、蛍光標識することが可能であり、かつ他の機能も有する化合物を合成することができる。このような蛍光標識部位として、市販のFITC、ROX、TAMRAまたはDabcyl等の市販の活性エステル型蛍光標識化合物を用いて、多様な蛍光発光波長を選択することが可能であるが、導入される蛍光標識化合物はこれらに限定されるものではない。
【0118】
本発明の化合物に導入し得る他の機能性の例としては、膜透過性機能が挙げられる。このような膜透過性機能部位は、前回特許前記一般式(I)で表される化合物を用いることにより、同様に導入することができる。膜透過性を向上させることができる機能性分子としてアルギニンが挙げられるが、リシンおよびセリン等の他の水溶性アミノ酸も好適に用いることができる。
【0119】
また、Fmocアミノ酸ユニットを利用することによって、複数個のアミノ酸を導入することも可能である。その合成例は実施例20および21にも示した。しかしながら、上記2化合物は本発明による膜透過性機能を有する蛍光PNAプローブのモデル化合物であり、本発明はこれらに限定されるものではない。
【0120】
これらのプローブの特徴は、「全てPNA型になっているので、完全な酵素耐性を有すること」である。すなわち、これまでの膜透過性機能を有するプローブは、PNAと膜透過性機能を有するペプチド鎖あるいはリン脂質を共有結合させたものが主流であったが、これらの既知のプローブは優れた膜透過性機能を有するものの、一旦細胞内に入ると酵素群によりペプチド鎖あるいはリン脂質が分解されることが予想される。したがって、これらは、ターゲットを認識していない分解を受けたプローブを洗浄過程で完全に取り除くことができないという欠点を有する。
【0121】
これに対して、今回設計したプローブは、細胞内においても酵素分解を受けないため、ターゲットを認識していないプローブは洗浄過程で完全に取り除かれるため、正確な遺伝子発現量の定量を可能とするものである。なお、これらの機能性を有する化合物以外にも、ラクトースやトリスエックス等の臓器選択機能性分子、タナチンやセクロピン等の殺菌機能性分子およびビオローゲン等の分子認識機能性分子等も、本発明によれば制限なく導入することが可能であり、そのような化合物を、大量に低コストで実用に供することが可能になる。
【実施例】
【0122】
以下に実施例を用いて本発明をさらに詳細に説明するが、本発明の範囲はこれに限られるものではない。
【0123】
(実施例1)Fmoc-Gly-BocPNA-OHの合成(1)
Fmoc-Gly-OH (891 mg, 3.0 mmol) とPfpOH (754 mg, 4.5 mmol)のDMF溶液(12mL)にDCC (845 mg, 4.5 mmol)を氷冷下加え、この反応液を0℃で30分次いで室温で15時間撹拌した。反応液を濾過し濾液を減圧濃縮し、残渣をBocPNA-OH (436 mg, 2.0 mmol)のDMF溶液(16 mL)にdiisopropylethylamine (445 μL, 2.6 mmol) を加え、室温で15時間撹拌した。これを減圧濃縮し残渣をシリカゲルカラムクロマト法(0-50% MeOH/CH2Cl2)により精製しFmoc-Gly-BocPNA-OH (121 mg, 12%)を得た。1H NMR (DMSO-d6)δ 7.88 (d, J = 7.0 Hz, 2 H), 7.72 (d, J = 7.0 Hz, 2 H), 7.62 (brt) and 7.56 (brt) (1 H), 7.41 (t, J = 7.0 Hz, 2 H), 7.33 (t, J = 7.0 Hz, 2 H), 7.18 (m, 2 H), 6.85 (brt) and 6.79 (brt) (1 H), 4.35 - 4.15 (m, 3 H), 4.05 - 3.85 (m, 3 H), 3.77 (m, 1 H), 3.40 - 3.25 (m, 2 H), 3.10 (m) and 3.03 (s) (2 H), 1.37 (brs, 9 H); 13C NMR (DMSO-d6) δ172.2 (d), 169.10 (d), 156.34 (d), 155.58 (d), 143.83, 140.66, 127.58, 127.04, 125.24, 120.04, 77.77 (d), 65.71, 47.34 (d), 46.72, 46.65 (d), 29.23 (d), 28.14 (d); FABMS m/z 498 [(M+H)+].
【0124】
(実施例2) Fmoc-C7-OPfpの合成
Fmoc-C7-OH(381.9 mg,1.0 mmol)とPfpOH(349.7 mg、1.9 mmol)のDMF溶液(2.5mL)にDCC(392.0 mg,1.9 mmol)を氷冷下で加え、この反応液を0 ℃で30分次いで室温で一晩攪拌した。反応終了後DCUreaを濾別して濾液を減圧濃縮し、残渣をシリカゲルカラムクロマト法(CH2Cl2)により精製した後、Hexaneで再結晶し白色粉末としてFmoc-C7-OPfp (537.5 mg,98%)を得た。1H-NMR (CDCl3)δ7.76(d,J= 7.6 Hz, 2 H),7.59 (d,J = 7.6 Hz, 2 H),7.40 (t,J = 7.4 Hz, 2 H),7.31 (t,J = 7.4 Hz, 2 H),4.70 - 4.73 (brt,1 H),4.47 - 4.40 (brd,2 H),4.22 (t,J = 6.42 Hz, 1 H),3.20 (q,J = 5.94 Hz, 2 H),2.66 (t,J = 7.38 Hz, 2 H),1.80 - 1.75 (m,2 H),1.55 - 1.50 (m,2 H),1.45 - 1.34 (m,6 H); 13C-NMR (CDCl3)δ 169.44, 156.43, 143.98, 141.96(m), 141.29, 140.23, 138.67(m), 136.99(m), 127.60, 126.96, 124.97, 119.91, 66.49, 55.73,47.29, 41.34(d), 34.89, 33.22, 29.85, 28.70, 26.42, 25.43, 24.60 ;HRMS(FAB+) calcd. for C29H27F5NO4[(M+H)+] 547.5131 observed 548.1861.
【0125】
(実施例3) Fmoc-Gly-BocPNA-OHの合成(2)
アセトン(6.0 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-Gly-OPfp (240.9 mg,0.52 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で6時間攪拌した。氷冷した1 N塩酸で冷却した反応溶液をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ溶液を濃縮し、シリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)及びLH-20(MeOH)で精製した。この後、塩化メチレンに溶かし、減圧濃縮しアモルファスパウダーとしてFmoc-Gly-BocPNA-OH (157.3 mg,80%)を得た。
【0126】
(実施例4)Fmoc-β-Ala-BocPNA-OHの合成(1)
Fmoc-β-Ala-OH (311 mg, 1.0 mmol)とPfpOH (334 mg, 1.75 mmol)のDMF溶液(2.5 mL)にDCC (288 mg, 1.4 mmol)を氷冷下加え、この反応液を0℃で30分次いで室温で15時間撹拌した。反応液を濾過し濾液を減圧濃縮し、残渣をシリカゲルカラムクロマト法(CH2Cl2)により精製した。これをHexaneとCH2Cl2で再結晶し、白色粉末としてFmoc-β-Ala-OPfp (429 mg, 90%) を得た。Fmoc-β-Ala-OPfp(100 mg, 0.21 mmol)とBocPNA-OH (41 mg, 0.19 mmol)のDMF溶液(2 mL)にdiisopropylethylamine (36 μL, 0.21 mmol) を加え、室温で15時間撹拌した。これを減圧濃縮し、残渣をシリカゲルカラムクロマト法(0-10% MeOH/CH2Cl2)により精製しFmoc-β-Ala-BocPNA-OH (41 mg, 42%)を得た。1H NMR (DMSO-d6)δ 7.88 (d, J = 7.4 Hz, 2 H), 7.68 (d, J = 7.4 Hz, 2 H), 7.41 (t, J= 7.3 Hz, 2 H), 7.33 (t, J = 7.3 Hz, 2 H), 7.18 (m, 2 H), 6.83 (brt) and6.72 (brt) (2 H), 4.3 - 4.2 (m, 4 H), 4.05 - 3.9 (m, 3 H), 3.33 (brt) and 3.29 (brt) (2 H), 3.19 (m, 2 H), 3.07 (brq) and 3.02 (brq) (2 H), 1.36 (brs, 9 H); 13C NMR (DMSO-d6) δ171.20 (d), 170.85 (d), 155.93, 155.56, 143.87, 140.69, 127.55, 127.01, 125.09, 120.05, 77.73 (d), 65.30 (d),59.69, 47.35 (d), 46.68, 46.49 (d), 37.99 (d), 36.72 (d), 28.14 (d).
【0127】
(実施例5)Fmoc-β-Ala-BocPNA-OHの合成(2)
アセトン(1.25 mL)と水(1.25 mL)の混合溶液にNaHCO3(92.4 mg,1.1 mmol)を加え、Fmoc-β-Ala-OPfp (476.0 mg,1.0 mmol)とBocPNA-OH (87.3 mg,0.55 mmol)を溶解し、室温で6時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、反応液に1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ溶液を濃縮し、シリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。この後、塩化メチレンに溶かし、減圧濃縮しアモルファスパウダーとしてFmoc-β-Ala-BocPNA-OH (225.9 mg,80%)を得た。
【0128】
(実施例6)Fmoc-GABA-BocPNA-OHの合成(1)
Fmoc-GABA-OPfp (100 mg, 0.20 mmol)とBocPNA-OH (40 mg, 0.18 mmol)のDMF溶液(2 mL)にdiisopropylethylamine (34 μL, 0.20 mmol) を加え、室温で15時間撹拌した。これを減圧濃縮し、残渣をシリカゲルカラムクロマト法(0-20% MeOH/CH2Cl2)により精製しFmoc-GABA-BocPNA-OH (43 mg, 45%)を得た。1H NMR (DMSO-d6)δ 7.88 (d, J = 7.4 Hz, 2 H), 7.68 (d, J = 7.4 Hz, 2 H),7.41 (t, J = 7.4 Hz, 2 H), 7.33 (t, J = 7.4 Hz, 2 H), 7.29 (m, 1 H), 6.82 (brt) and 6.71 (brt) (1 H), 4.3 - 4.2 (m, 4 H), 4.05 - 3.9 (m, 3 H),3.35 - 3.25 (m, 2 H), 3.1 - 2.95 (m, 4 H), 1.36 (brs, 9 H); 13C NMR (DMSO-d6) δ172.2 (d), 171.5 (d), 156.03, 155.60 (d), 143.89, 140.68, 127.54, 127.00, 125.50, 120.04, 77.70 (d), 65.19, 54.84, 47.89 (d), 46.97 (d), 46.72, 38.20 (d), 29.23 (d), 28.14 (d), 24.98 (d).
【0129】
(実施例7)Fmoc-GABA-OPfpの合成
Fmoc-GABA-OH (325 mg, 1.0 mmol)とPfpOH (221 mg, 1.2 mmol)のDMF溶液(2.5mL)にDCC (248 mg, 1.2 mmol)を氷冷下加え、この反応液を0℃で30分次いで室温で15時間撹拌した。反応液を濾過し濾液を減圧濃縮し、残渣をシリカゲルカラムクロマト法(CH2Cl2)により精製し、白色粉末としてFmoc-GABA-OPfp (463mg, 94%) を得た。1H NMR (CDCl3)δ7.77 (d, J = 7.5 Hz, 2 H), 7.59 (d, J =7.5 Hz, 2 H), 7.40 (t, J = 7.5 Hz, 2 H), 7.31 (t, J = 7.5 Hz, 2 H), 4.85 (brs, 1 H), 4.45 (d, J = 6.3 Hz, 2 H), 4.21 (t, J = 6.3 Hz, 2 H), 3.32(d, J = 6.5 Hz, 2 H), 2.71 (t, J = 6.5 Hz, 2 H), 1.98 (t, J = 6.5 Hz, 2H); 13C NMR(CDCl3)δ169.02, 156.49, 143.83, 141.87 (m), 141.28, 140.23 (m), 138.61 (m), 136.94 (m), 127.62, 127.46, 124.89, 119.88, 66.52, 47.25, 39.92, 30.42, 25.03.
【0130】
(実施例8) Fmoc-GABA-BocPNA-OHの合成(2)
アセトン(5.0 mL)と水(1.0 mL)の混合溶液にNaHCO3 (67.2 mg,0.8 mmol)を加え、Fmoc-GABA-OPfp (255.5 mg,0.52 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で8時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、反応液に1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ溶液を濃縮し、シリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮しアモルファスパウダーとしてFmoc-GABA-BocPNA-OH (175.9 mg,84%)を得た。
【0131】
(実施例9) Fmoc-C4-BocPNA-OHの合成
アセトン(4.0 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C4-Opfp (323.5 mg,0.64 mmol)とBocPNA-OH(87.3 mg,0.4 mmol)を溶解し、室温で12時間攪拌した。氷冷した1 N塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ、シリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮しアモルファスパウダーとしてFmoc-C4-BocPNA-OH (190.7 mg,88%)を得た。1H-NMR(CDCl3) δ7.76(d,J = 6.7 Hz, 2 H),6.96 (mi) and 6.66 (ma) (brd, J = 6.7 Hz, 2 H), 7.41 - 7.37(m, 2 H), 7.32 - 7.28(m,2 H), 7.14 (ma) and 6.68 (mi) (m, 1 H), 5.54 (ma) and 5.43 (mi) (brt, 1 H), 4.45(mi) and 4.37(ma) (m, 2 H), 4.24 - 4.21 (m, 1 H),4.08 - 3.95 (m,2 H),3.54 - 3.48 (m,2 H),3.29 - 3.11 (m, 4 H),2.43 - 2.25 (m, 2 H),1.70 - 1.29 (m,13 H); 13C-NMR (CDCl3) δ 174.39,173.07,171.95,157.51,156.79(d),156.11,144.06(d),141.16,127.46(d),126.90(d),119.77(d), 81.42,79.67,66.39(d),53.35,49.46(d),49.17,48.60,47.15(d), 40.89,40.32(d),38.68,31.87,31.41,29.59,29.11(d),28.28,21.77(d); HRMS (FAB+) calcd. for C29H37N3O7[(M+H)+] 539.2632 observed 540.2707.
【0132】
(実施例10) Fmoc-C5-BocPNA-OHの合成
アセトン(7.5 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C5-OPfp (311.0 mg,0.6 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で24時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ溶液を濃縮し、シリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮し白色のアモルファスパウダーとして、Fmoc-C5-BocPNA-OH (198.0 mg,90%)を得た。1H-NMR (DMSO-d6)δ 7.88(d,J = 7.4 Hz, 2 H),7.68 (d, J =7.2 Hz,2 H),7.41 (t,J = 7.4 Hz, 2 H),7.32 (t,J = 7.4 Hz, 2 H),7.22(brt,1 H),6.81 (ma) and 6.67 (mi) (brt, 1 H),4.33 (mi) and 4.29 (ma)(brd,2 H),4.20 (t,J = 7.1 Hz, 1 H),4.08 (mi) and 3.90 (ma) (brs,2H),3.09 - 2.94 (m,4 H),2.30 (ma) and2.14 (mi) (brt,2 H),1.51 - 1.45(m,2 H),1.41 - 1.31 (brs,11 H),1.29-1.21 (m,8 H); 13C-NMR (CDCl3)δ175.1(d),172.27(d),157.20(t),156.63,144.43(d),141.69,128.07,127.45,125.40(d),120.35,81.71,80.00,67.36(d),50.42,49.81(d),48.90(d),47.82(d),41.77,41.16,40.64,39.19,33.12,32.75,29.45(d),28.81,26.59,24.97,24.70; HRMS (FAB+) calcd. for C30H39N3O7[(M+H)+] 553.2788 observed 554.2873.
【0133】
(実施例11) Fmoc-C6-BocPNA-OHの合成
アセトン(6.0 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C6-OPfp (331.9 mg,0.6 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で24時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ、溶液を濃縮しシリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮し白色のアモルファスパウダーとしてFmoc-C6-BocPNA-OH (197.0 mg,87%)を得た。1H-NMR (DMSO-d6)δ7.88 (d,J =7.7 Hz, 2 H),7.68 (ma) and 7.63 (mi) (brd,J = 7.4 Hz,2 H),7.40 (t,J = 7.4 Hz,2 H), 7.32 (t, J =7.4 Hz,2 H), 7.22 (brt, 1 H), 6.79 (ma) and 6.66 (mi) (brt, 1 H), 4.39(mi) and 4.29 (ma) (brt, 2 H), 4.20 (t, J = 6.7 Hz,1 H) 4.08 (mi) and 3.91 (ma) (brs, 2 H), 3.10 - 2.97 (m, 4 H),2.31 (ma) and 2.15 (mi) (brt,2 H),1.50 - 1.47(m,2 H),1.41 - 1.36(m,11 H),1.28-1.24(brd,6 H);13C-NMR (CDCl3)δ175.23(d),172.41(d),157.11(d),156.60,144.34(d),141.69,128.07,127.45,125.40(d),120.35,81.68,80.00(d),67.72,67.50(d),53.87,50.77,50.14(d),48.90,47.82(d),41.29(d),41.36,40.69,39.18,33.19,32.96,30.00,29.11,28.81,26.75(d),25.27(d),24.80(d); HRMS(FAB+) calcd. for C29H41N3O7[(M+H)+] 567.2945 observed 568.3027.
【0134】
(実施例12) Fmoc-C7-BocPNA-OHの合成
アセトン(7.0 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C7-OPfp (328.5 mg,0.6 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で24時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ、溶液を濃縮しシリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮し白色のアモルファスパウダーとしてFmoc-C7-BocPNA-OH (196.1 mg,84%)を得た。1H-NMR (DMSO-d6);δ7.88(d,J = 7.7 Hz,2 H),7.68 (ma) and 7.63 (mi) (brd,J = 7.4 Hz,2 H),7.40 (t,J = 7.4 Hz,2 H),7.32 (t,J =7.4 Hz,2 H),7.22 (brt,1 H),6.79 (ma) and 6.79 (mi) (brt,1 H),4.39(mi) and 4.29(ma) (brd,J = 6.9 Hz,2 H),4.05 (t,J = 6.7 Hz,1 H),4.08 (mi) and 3.91 (ma) (brs,2 H),3.12 - 2.95 (m,4 H),2.31 (mi) and 2.15 (ma) (brt,2 H),1.50 - 1.47 (m,2 H),1.42 - 1.34 (m,11 H),1.25 (brd,2 H); 13C-NMR (CDCl3) δ 174.72,172.19,156.52,156.05,143.78(d),141.14,127.51, 126.89, 124.86(d), 119.79, 79.43(d),66.80(d),53.33,50.19,49.20, 48.50,47.14(d),41.18(d), 38.60,32.28(d),29.60, 28.83,28.26, 26.27(d),24.68(d),21.77(d); HRMS (FAB+) calcd. for C32H43N3O7[(M+H)+] 581.3101 observed 582.3171.
【0135】
(実施例13) Fmoc-C10-BocPNA-OH
アセトン(7.0 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C10-OPfp (353.7 mg,0.6 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で24時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ、溶液を濃縮しシリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮し白色のアモルファスパウダーとしてFmoc-C10-BocPNA-OH (218.5 mg,88%)を得た。1H-NMR (CDCl3)δ9.60 (brs,1 H),7.73 (d,J = 7.6 Hz,2 H),7.58 (d,J = 6.8 Hz,2 H),7.37 (t,J = 6.8 Hz,2 H),7.29 (t,J = 7.2Hz,2 H),5.52 (ma) and 5.35 (mi) (brd,1 H),5.00 (s,1 H),4.45 (mi)and 4.40 (ma) (brd,J = 6.4 Hz,2 H),4.23 - 4.22 (m,1 H),4.09 (mi) and 4.04 (ma) (brs,2 H),3.57 - 3.46 (m,2 H),3.29 - 3.03 (m,4 H),1.66- 1.58 (brs,2 H),1.52 - 1.37 (m,11 H),1.33 - 1.20 (brs,12 H); 13C-NMR (CDCl3)δ174.5(d),172.43(d),171.64(d),157.83(d),156.98,156.03,143.78(d),141.17,127.52,126.90,124.86(d),119.70(d),81.08,79.43(d),67.26,66.43,50.14,49.29,48.145(d),47.17(t),41.47,40.99,40.16,38.63,32.90,32.43(d),29.55(d),29.22(m),28.28,26.56(d),24.98,24.75; HRMS (FAB+) calcd. for C35H49N3O7[(M+H)+] 623.3571 observed 624.3643.
【0136】
(実施例14)Fmoc-C11-OPfpの合成
Fmoc-C11-OH (437.5 mg,2.0 mmol)とPfpOH(276.6 mg,3.0 mmol)のDMF溶液(2.5 mL)にDCC(309.5 mg,3.0 mmol)を氷冷下で加え、この反応液を0 ℃で30分次いで室温で18時間攪拌した。反応終了後DCUreaを濾別して濾液を減圧濃縮し、残渣をシリカゲルカラムクロマト法(CH2Cl2)により精製した後、Hexaneで再結晶し白色粉末としてFmoc-C11-OPfp (575.6 mg,96%)を得た。1H-NMR (CDCl3)δ7.79(d,J = 7.6 Hz,2 H),7.63 (d,J = 7.2 Hz, 2 H),7.43 (t,J =7.6 Hz, 2 H),7.34 (t,J = 7.2 Hz, 2 H),4.86 (brt,1 H),4.47 (mi) and 4.44 (ma)(brd,2 H),4.25 (t,1 H),3.22 (q,J = 6.1 Hz, 2 H),2.68 (t,J = 7.2 Hz, 2 H),1.80 (m,2 H),1.56 - 1.52 (m,2 H),1.47 - 1.42 (m,2 H),1.39- 1.30 (m,12 H); HRMS (FAB+) calcd. for C33H34F5NO4[(M+H)+]603.6194 observed 604.2490.
【0137】
(実施例15) Fmoc-C11-BocPNA-OH
アセトン(10 mL)と水(1.0 mL)の混合溶液にNaHCO3(67.2 mg,0.8 mmol)を加え、Fmoc-C11-OPfp (362.2 mg,0.6 mmol)とBocPNA-OH (87.3 mg,0.4 mmol)を溶解し、室温で48時間攪拌した。氷冷した1 N 塩酸で溶液(0 ℃)をpH 3.0とし、さらに1%クエン酸水溶液を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄をした。有機層を無水硫酸マグネシウムで乾燥させ、溶液を濃縮しシリカゲルカラムクロマト法(1-5% MeOH/CH2Cl2)で精製した。塩化メチレンに溶かした後、減圧濃縮し白色のアモルファスパウダーとしてFmoc-C11-BocPNA-OH (227.6 mg,89%)を得た。1H-NMR (CDCl3);δ9.62 (brs,1 H),7.74 (d,J = 7.6 Hz,2 H),7.57 (d,J = 7.5 Hz,2 H),7.37 (t,J = 7.1 Hz,2 H),7.28 (t,J = 6.8Hz,2 H),5.53 (ma) and 5.35 (mi) (brs,H),5.00 (brt,1 H),4.43 (mi)and 4.37 (ma) (brd,J = 6.4 Hz,2 H),4.22 - 4.19 (m,1 H),4.06 (mi) and 4.01 (ma) (brs,2 H),3.51 - 3.44 (m,2 H),3.23 - 3.08 (m,4 H),2.36(ma) and 2.21 (mi) (brt,J = 7.0 Hz,2 H),1.69 - 1.58 (brs,2 H),1.52- 1.40 (m,11 H),1.29 - 1.25 (brd,14 H); 13C-NMR (CDCl3)δ175.45(d),172.428(d),157.91(d),157.01,156.57,144.33(d),141.72,128.05,127.43,125.40(d),120.35,81.63,80.00(d),67.77,66.97,50.67,49.84,49.20(d),47.72(t),42.10,41.53,40.70,39.18,33.51,33.07,30.032,29.83(m),28.82,27.00(d),25.44,25.35; HRMS (FAB+) calcd. for C36H51N3O7[(M+H)+] 637.3727 observed 638.3794.
【0138】
(実施例16)PNA oligo 1(H2N-G-A-T-pMR-G-A-C-G-C-CONH2)の合成(第1法)
tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; φrum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、固相担体MBHA(50 mg)に各種PNAモノマーユニット(チミン7.7 mg、シトシン10.1 mg、アデニン10.6 mg、グアニン10.9 mg、Fmoc-Gly-BocPNA-OH 10.0 mg;各20 mmol)と縮合剤HBTU(7.6 mg、20 mmol)とDIEA(7.0 μL、20 mmol)を用いて逐次伸長反応を行った。この間、Fmoc-Gly-BocPNA-OHを縮合した際には、次のチミジンPNAモノマーユニットと縮合する前に、まずピペリジン処理(20% piperidine in DMF 3 mL、室温30分)してFmoc基を脱保護して、次いで光機能性分子のカルボン酸誘導体としてp-Methyl Red-OH(10.8 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(13.9 μL、40 mmol)を用いて縮合し目的の位置に導入した。その後、チミジン、アデニン、グアニンのPNAモノマーユニットを逐次縮合したあと、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しとZ基の脱保護を同時に行い、後処理して、目的とするPNA oligo 1を得た。UV λmax (H2O) 303, 548 (nm)。
【0139】
(実施例17)PNA oligo 1の合成(第2法)
tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; φrum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、固相担体MBHA(50 mg)に各種PNAモノマーユニット(チミン7.7 mg、シトシン10.1 mg、アデニン10.6 mg、グアニン10.9 mg、Fmoc-Gly-BocPNA-OH 10.0 mg;各20 mmol)と縮合剤HBTU(7.6 mg、20 mmol)とDIEA(7.0 μL、20 mmol)を用いて逐次伸長反応を行った。全てのモノマーユニットを逐次縮合した後、ピペリジン処理(20% piperidine in DMF、室温30分)してFmoc-Gly残基を脱保護して、次いで光機能性分子のカルボン酸誘導体としてp-Methyl Red-OH(10.8 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(13.9 μL、40 mmol)を用いて縮合し目的の位置に導入した。その後、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しとZ基の脱保護を同時に行い、後処理して、目的とするPNA oligo 1を得た。UV λmax (H2O) 302, 550 (nm)。
【0140】
(実施例18)PNA oligo 2(H2N-G-A-T-mMR-G-A-C-G-C-CONH2)の合成(第2法)
tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; φrum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、固相担体MBHA(50 mg)に各種PNAモノマーユニット(チミン7.7 mg、シトシン10.1 mg、アデニン10.6 mg、グアニン10.9 mg、Fmoc-Gly-BocPNA-OH 10.0 mg;各20 mmol)と縮合剤HBTU(7.6 mg、20 mmol)とDIEA(7.0 μL、20 mmol)を用いて逐次伸長反応を行った。全てのモノマーユニットを逐次縮合した後、ピペリジン処理(20% piperidine in DMF、室温30分)してFmoc-Gly残基を脱保護して、次いで光機能性分子のカルボン酸誘導体としてm-Methyl Red-OH(10.8 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(13.9 μL、40 mmol)を用いて縮合し目的の位置に導入した。その後、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しとZ基の脱保護を同時に行い、後処理して、目的とするPNA oligo 2を得た。UVλmax (H2O) 308, 570 (nm)。
【0141】
(実施例19)PNA oligo 3(H2N-G-A-T-oMR-G-A-C-G-C-CONH2)の合成(第2法)
tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; φrum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、固相担体MBHA(50 mg)に各種PNAモノマーユニット(チミン7.7 mg、シトシン10.1 mg、アデニン10.6 mg、グアニン10.9 mg、Fmoc-Gly-BocPNA-OH 10.0 mg;各20 mmol)と縮合剤HBTU(7.6 mg、20 mmol)とDIEA(7.0 μL、20 mmol)を用いて逐次伸長反応を行った。全てのモノマーユニットを逐次縮合した後、ピペリジン処理(20% piperidine in DMF、室温30分)してFmoc-Gly残基を脱保護して、次いで光機能性分子のカルボン酸誘導体としてo-Methyl Red-OH(10.8 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(13.9 μL、40 mmol)を用いて縮合し目的の位置に導入した。その後、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しとZ基の脱保護を同時に行い、後処理して、目的とするPNA oligo 1を得た。UV λmax (H2O) 302, 561 (nm)。
【0142】
(実施例20)膜透過性機能を有する蛍光PNAプローブ(1)の合成
【0143】
【化25】
【0144】
前記第2法を用いた。
標準的tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; Orum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、まず、固相担体MBHA(50 mg)にチミンPNAモノマーユニット(7.7 mg、20 mmol)、縮合剤HBTU(7.6 mg、20 mmol)とDIEA(3.5 mL、20 mmol)を用いて逐次伸長反応を行った(塩基配列認識領域の設計)。次いで、リンカー用ω-アミノ酸Boc-7-aminoheptanoic Acid(5.2 mg、20 mmol)、Fmoc-Ahx-BocPNA-OH(10.0 mg、20 mmol)と再度Boc-7-aminoheptanoic Acidを、縮合剤HBTU(7.6 mg、20 mmol)とDIEA(3.5 mL、20 mmol)を用いて順次縮合させた(リンカー部位と膜透過性機能領域の設計)。全てのユニットを逐次縮合した後、ピペリジン処理(20% piperidine in DMF、室温3分)してFmoc基を脱保護した。次いで、機能性カルボン酸誘導体としてFmoc-Arg(Mts)-OH(23.1 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(7.0 mL、40 mmol)を用いて縮合し目的の位置に機能性分子を導入した(膜透過性機能の導入)。TFA処理(95% TFA/5% m-cresol)によりBoc基を脱保護した後、FITC(9.3 mg,25 mmol)をDIEA(17.4 mL, 100 mmol)存在下、室温で12時間振盪し蛍光標識化した(蛍光標識部位の設計)。最後にピペリジン処理(20% piperidine in DMF、室温3分)して残るFmoc基を脱保護した後、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しを行い、後処理して、目的物を得た。MALDI-TOF MS: calcd. 2096.26 (M + H+), found 2096.36.
【0145】
(実施例21)膜透過性機能を有する蛍光PNAプローブ(2)の合成
【0146】
【化26】
【0147】
標準的tBoc法(cf. Koch, T.; Hansen, H.F.; Andersen, P.; Larsen, T.; Batz, H.G.; Otteson, K.; Orum, H. J. Peptide Res. 1997, 49, 80-88.)に従い、まず、固相担体MBHA(50 mg)にチミンPNAモノマーユニット(7.7 mg、20 mmol)、縮合剤HBTU(7.6 mg、20 mmol)とDIEA(3.5 mL、20 mmol)を用いて逐次伸長反応を行った(塩基配列認識領域の設計)。
【0148】
次いで、リンカー用ω-アミノ酸Boc-7-aminoheptanoic Acid(5.2 mg、20 mmol)、Fmoc-Ahx-BocPNA-OH(10.0 mg、20 mmol)と再度Boc-7-aminoheptanoic Acidを、縮合剤HBTU(7.6 mg、20 mmol)とDIEA(3.5 mL、20 mmol)を用いて順次縮合させた(リンカー部位と膜透過性機能領域の設計)。全てのユニットを逐次縮合した後、ピペリジン処理(20% piperidine in DMF、室温3分)してFmoc基を脱保護した。次いで、機能性カルボン酸誘導体としてFmoc-Arg(Mts)-OH(23.1 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(7.0 mL、40 mmol)を用いて縮合し目的の位置に機能性分子を導入した(膜透過性機能の導入)。
【0149】
これをピペリジン処理(20% piperidine in DMF、室温3分)してFmoc基を脱保護した後、再度Fmoc-Arg(Mts)-OH(23.1 mg、40 mmol)を縮合剤HBTU(15.2 mg、40 mmol)とDIEA(7.0 mL、40 mmol)を用いて縮合し目的の位置に機能性分子を導入した(膜透過性機能の追加導入)。TFA処理(95% TFA/5% m-cresol)によりBoc基を脱保護した後、FITC(9.3 mg, 25 mmol)をDIEA(17.4 mL, 100 mmol)存在下、室温で12時間振盪し蛍光標識化した(蛍光標識部位の設計)。
【0150】
最後にピペリジン処理(20% piperidine in DMF、室温3分)して残るFmoc基を脱保護した後、常法(TFA/TFMSA/p-cresol/thioanisol=60/25/10/10)により固相担体からの切り出しを行い、後処理して、目的物を得た。MALDI-TOF MS: calcd. 2252.44 (M + H+), found 2252.33.
[発明の効果]
本発明によれば、光機能性分子に限定されることない多種多様な機能性分子を、PNA中に容易に導入することができ、多種多様な機能性分子をPNA中に容易かつ効率的に導入することができ、遺伝子治療などに用いられる種々のPNAの構築が可能になる。
【図面の簡単な説明】
【0151】
【図1】DNAとPNAの構造および荷電の状況の違いを表す図である。
【図2】2種類のPNAモノマーユニットの構造を表す図である。
【特許請求の範囲】
【請求項1】
下記一般式(I)
【化1】
(式中、Bは、互いに独立し、同一または異なっている、アデニン、グアニン、シトシンまたはチミンであり、Rは、互いに独立し、同一または異なって、機能性カルボン酸誘導体であり、R1は、水素原子または機能性カルボン酸誘導体であり、a〜hは0〜10の整数であり、X1〜X3、Y1、Y2およびZ1〜Z5はいずれも0以上の整数であり、X1+X2+X3≧0であり、Y1+Y2>0であり、Z1+Z2+Z3+Z4+Z5≧0である。ただし、X1+X2+X3およびZ1+Z2+Z3+Z4+Z5が同時に0であることはなく、X1+X2+X3=0の場合、R1は機能性カルボン酸誘導体である。)で表され、機能性カルボン酸誘導体が、細胞膜透過性機能分子の誘導体、又は光活性機能分子の誘導体、又は臓器選択機能性分子の誘導体、又は殺菌機能性分子の誘導体、又は分子認識機能性分子の誘導体の何れかであることに基づくPNAオリゴマー誘導体化合物。
【請求項2】
膜透過性機能分子が水溶性アミノ酸であることを特徴とする請求項1記載のPNAオリゴマー誘導体化合物。
【請求項3】
水溶性アミノ酸が、アルギニン、リジン、セリンの何れかであることを特徴とする請求項2記載のPNAオリゴマー誘導体化合物。
【請求項4】
光活性機能分子が活性エステル型蛍光標識化合物であることを特徴とする請求項1記載のPNAオリゴマー誘導体化合物。
【請求項5】
活性エステル型蛍光標識化合物が、FITC、ROX、TAMRA、Dabcylの何れかであることを特徴とする請求項4記載のPNAオリゴマー誘導体化合物。
【請求項6】
臓器選択機能性分子が、ラクトース、トリスエックスの何れかであることを特徴とする請求項1記載のPNAオリゴマー誘導体化合物。
【請求項7】
分子認識機能性分子が、ビオローゲンであることを特徴とする請求項1記載のPNAオリゴマー誘導体化合物。
【請求項8】
細胞膜透過性機能分子の誘導体、及び光活性機能分子の誘導体の双方を有していることを特徴とする請求項1記載のPNAオリゴマー誘導体化合物。
【請求項9】
X1=Z1=1であることを特徴とする、請求項8に記載のPNAオリゴマー誘導体化合物。
【請求項10】
Y1≧2であり、Z2=1であることを特徴とする、請求項8、9に記載のPNAオリゴマー誘導体化合物。
【請求項11】
a≦6であり、b≦4であり、f≦6であることを特徴とする、請求項8〜10のいずれ
かに記載のPNAオリゴマー誘導体化合物。
【請求項12】
R1が光機能性カルボン酸誘導体であることを特徴とする、請求項8〜11のいずれかに記載のPNAオリゴマー誘導体化合物。
【請求項13】
下記一般式(II)
【化2】
(式中、Bは、互いに独立し、同一または異なって、アデニン、グアニン、シトシンまた
はチミンであり、Rは、互いに独立し、同一または異なって、機能性カルボン酸誘導体であり、R1は、水素原子または機能性カルボン酸誘導体であり、a〜hは0〜10の整数であり、X1〜X3、Y1、Y2およびZ1〜Z5はいずれも0以上の整数であり、X1+X2+X3≧0であり、Y1+Y2>0であり、Z1+Z2+Z3+Z4+Z5≧0である。ただし、X1+X2+X3およびZ1+Z2+Z3+Z4+Z5が同時に0であることはなく、X1+X2+X3=0の場合、R1は機能性カルボン酸誘導体である。)で表され、機能性カルボン酸誘導体が水溶性アミノ酸の誘導体、活性エステル型蛍光標識化合物の誘導体の少なくとも何れか一方であることに基づく化合物。
【請求項14】
水溶性アミノ酸が、アルギニン、リジン、セリンの何れかであることを特徴とする、請求項13に記載の化合物。
【請求項15】
活性エステル型蛍光標識化合物がFITC、ROX、TAMRA、Dabcylの何れかであることを特徴とする請求項13に記載の化合物。
【請求項16】
機能性カルボン酸誘導体として、水溶性アミノ酸の誘導体、及び活性エステル型蛍光標識化合物の誘導体の双方を採用することによって膜透過性機能を有する蛍光PNAプローブ機能を有している請求項13に記載の化合物。
【請求項17】
水溶性アミノ酸がアルギニンであり、活性エステル型蛍光標識化合物がFITCであることを特徴とする請求項16に記載の化合物。
【請求項18】
X1=Z1=1であることを特徴とする、請求項16に記載の化合物。
【請求項19】
Y1≧2であり、Z2=1であることを特徴とする、請求項16、17のいずれかに記載の化合物。
【請求項20】
a≦6であり、b≦4であり、f ≦6であることを特徴とする、請求項16〜19のいずれかに記載の化合物。
【請求項21】
R1が光機能性カルボン酸誘導体であることを特徴とする、請求項16〜19のいずれかに記載の化合物。
【請求項1】
下記一般式(I)
【化1】
(式中、Bは、互いに独立し、同一または異なっている、アデニン、グアニン、シトシンまたはチミンであり、Rは、互いに独立し、同一または異なって、機能性カルボン酸誘導体であり、R1は、水素原子または機能性カルボン酸誘導体であり、a〜hは0〜10の整数であり、X1〜X3、Y1、Y2およびZ1〜Z5はいずれも0以上の整数であり、X1+X2+X3≧0であり、Y1+Y2>0であり、Z1+Z2+Z3+Z4+Z5≧0である。ただし、X1+X2+X3およびZ1+Z2+Z3+Z4+Z5が同時に0であることはなく、X1+X2+X3=0の場合、R1は機能性カルボン酸誘導体である。)で表され、機能性カルボン酸誘導体が、細胞膜透過性機能分子の誘導体、又は光活性機能分子の誘導体、又は臓器選択機能性分子の誘導体、又は殺菌機能性分子の誘導体、又は分子認識機能性分子の誘導体の何れかであることに基づくPNAオリゴマー誘導体化合物。
【請求項2】
膜透過性機能分子が水溶性アミノ酸であることを特徴とする請求項1記載のPNAオリゴマー誘導体化合物。
【請求項3】
水溶性アミノ酸が、アルギニン、リジン、セリンの何れかであることを特徴とする請求項2記載のPNAオリゴマー誘導体化合物。
【請求項4】
光活性機能分子が活性エステル型蛍光標識化合物であることを特徴とする請求項1記載のPNAオリゴマー誘導体化合物。
【請求項5】
活性エステル型蛍光標識化合物が、FITC、ROX、TAMRA、Dabcylの何れかであることを特徴とする請求項4記載のPNAオリゴマー誘導体化合物。
【請求項6】
臓器選択機能性分子が、ラクトース、トリスエックスの何れかであることを特徴とする請求項1記載のPNAオリゴマー誘導体化合物。
【請求項7】
分子認識機能性分子が、ビオローゲンであることを特徴とする請求項1記載のPNAオリゴマー誘導体化合物。
【請求項8】
細胞膜透過性機能分子の誘導体、及び光活性機能分子の誘導体の双方を有していることを特徴とする請求項1記載のPNAオリゴマー誘導体化合物。
【請求項9】
X1=Z1=1であることを特徴とする、請求項8に記載のPNAオリゴマー誘導体化合物。
【請求項10】
Y1≧2であり、Z2=1であることを特徴とする、請求項8、9に記載のPNAオリゴマー誘導体化合物。
【請求項11】
a≦6であり、b≦4であり、f≦6であることを特徴とする、請求項8〜10のいずれ
かに記載のPNAオリゴマー誘導体化合物。
【請求項12】
R1が光機能性カルボン酸誘導体であることを特徴とする、請求項8〜11のいずれかに記載のPNAオリゴマー誘導体化合物。
【請求項13】
下記一般式(II)
【化2】
(式中、Bは、互いに独立し、同一または異なって、アデニン、グアニン、シトシンまた
はチミンであり、Rは、互いに独立し、同一または異なって、機能性カルボン酸誘導体であり、R1は、水素原子または機能性カルボン酸誘導体であり、a〜hは0〜10の整数であり、X1〜X3、Y1、Y2およびZ1〜Z5はいずれも0以上の整数であり、X1+X2+X3≧0であり、Y1+Y2>0であり、Z1+Z2+Z3+Z4+Z5≧0である。ただし、X1+X2+X3およびZ1+Z2+Z3+Z4+Z5が同時に0であることはなく、X1+X2+X3=0の場合、R1は機能性カルボン酸誘導体である。)で表され、機能性カルボン酸誘導体が水溶性アミノ酸の誘導体、活性エステル型蛍光標識化合物の誘導体の少なくとも何れか一方であることに基づく化合物。
【請求項14】
水溶性アミノ酸が、アルギニン、リジン、セリンの何れかであることを特徴とする、請求項13に記載の化合物。
【請求項15】
活性エステル型蛍光標識化合物がFITC、ROX、TAMRA、Dabcylの何れかであることを特徴とする請求項13に記載の化合物。
【請求項16】
機能性カルボン酸誘導体として、水溶性アミノ酸の誘導体、及び活性エステル型蛍光標識化合物の誘導体の双方を採用することによって膜透過性機能を有する蛍光PNAプローブ機能を有している請求項13に記載の化合物。
【請求項17】
水溶性アミノ酸がアルギニンであり、活性エステル型蛍光標識化合物がFITCであることを特徴とする請求項16に記載の化合物。
【請求項18】
X1=Z1=1であることを特徴とする、請求項16に記載の化合物。
【請求項19】
Y1≧2であり、Z2=1であることを特徴とする、請求項16、17のいずれかに記載の化合物。
【請求項20】
a≦6であり、b≦4であり、f ≦6であることを特徴とする、請求項16〜19のいずれかに記載の化合物。
【請求項21】
R1が光機能性カルボン酸誘導体であることを特徴とする、請求項16〜19のいずれかに記載の化合物。
【図1】

【図2】
【図2】
【公開番号】特開2007−275071(P2007−275071A)
【公開日】平成19年10月25日(2007.10.25)
【国際特許分類】
【出願番号】特願2007−141136(P2007−141136)
【出願日】平成19年5月28日(2007.5.28)
【分割の表示】特願2002−121667(P2002−121667)の分割
【原出願日】平成14年4月24日(2002.4.24)
【出願人】(503280961)株式会社クレディアジャパン (10)
【Fターム(参考)】
【公開日】平成19年10月25日(2007.10.25)
【国際特許分類】
【出願日】平成19年5月28日(2007.5.28)
【分割の表示】特願2002−121667(P2002−121667)の分割
【原出願日】平成14年4月24日(2002.4.24)
【出願人】(503280961)株式会社クレディアジャパン (10)
【Fターム(参考)】
[ Back to top ]