
新規な脂肪酸誘導体
【課題】薬剤および農薬等の多くの生物学的活性化合物の作用を化学的誘導体化によって改良する方法を提供する。
【解決手段】分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含む、薬剤および農薬等の生物学的活性化合物の性質を、これら官能基の1つ以上を式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、COOCH2R、および−SCH2Rであらわされるものから選択される親油性基で置換することによって改良する。ここでRは、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である。
【解決手段】分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含む、薬剤および農薬等の生物学的活性化合物の性質を、これら官能基の1つ以上を式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、COOCH2R、および−SCH2Rであらわされるものから選択される親油性基で置換することによって改良する。ここでRは、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は生物学的活性化合物に関するものであり、薬剤および農薬等の多くの生物学的活性化合物の作用を化学的誘導体化によって改良する方法を提供する。
【背景技術】
【0002】
医学界では患者の作用部位への薬剤運搬効果の研究、改良に多大の関心がもたれている。本研究は主として薬剤の腸から血流への吸収に焦点を当てている。但しその他の生物学的障壁を通過する運搬も、多くの疾患、例えば癌、感染症、炎症、CNS疾患等の治療に必要な治療効果を得るために重要な役割を演ずることは多い。細胞膜経由輸送は或る治療化合物の最適効果達成を妨害する主原因であることが多い。
【0003】
最近数十年にわたり、悪性および感染性疾患の治療における薬剤耐性がますます増えており、今や重大な臨床的問題とみなされている。薬剤耐性の発現は多くのメカニズムによって起こるが、微生物および細胞が毒性化合物を有毒レベル以下にまで除去する正常メカニズムがトリガーされることによることが非常に多い。一例は、癌細胞における多剤耐性(MDR)の発現である。この場合、MDRは、細胞が有毒化合物の非常に効率的な流出を実現する細胞膜タンパク質ポンプに関係することがよくある。臨床的立場、すなわち細胞増殖抑制剤で腫瘍を治療する場合には、最も強力なタンパク質ポンプを有する細胞が優先的に生き残り、これらの細胞は増殖して、種々の薬剤による治療に耐性を有する新しい腫瘍になるかも知れない。同様な作用メカニズムは、その他の治療領域、例えば抗マラリヤ剤による治療等で見られる不十分な効果の原因になるかも知れない。
【0004】
臨床における耐性メカニズムを回避することを試みる幾つかの方法が知られている。例えばベラパミル等のCa2+チャンネルブロッカーまたはシクロスポリンのような免疫調節剤との併用が試みられた。しかし顕著な改善はこれまで報告されていない。
【0005】
文献には、化合物類を脂肪酸と結合させて化学的結合誘導体または物理的混合物のどちらかを形成することによって、治療化合物の治療指数、バイオアベイラビリティー、膜通過性、器官標的化などを改善する幾つかの提案がある。
【0006】
例えば、欧州特許出願第393920号は、長鎖(C16以上)アシル基で誘導体化した抗ウィルス性ヌクレオシドおよびヌクレオシド類似体が親化合物より有効であることを開示している。これら分子の脂肪酸部分は、γ−リノレン酸またはリノール酸等のポリ不飽和脂肪酸から形成されることが記載されている。
【0007】
米国特許出願第3920630号は、2、2'−無水−アラシチジンおよびその5'−O−アシレート類が、抗ウィルス薬として、アラシチジンそのものと同様な全般的生物学的効果および治療効果を有することを教示している。特に化合物2、2'−無水−5'−オレイル−アラシチジンについて記載されている。
【0008】
欧州特許出願第56265号は、アラビノ−フラノシル−チミン(Ara T)と1−17炭素原子を有する飽和酸とのエステル類を開示している。
【0009】
PCT/WO90/00555からは、特に燐酸基を介してヌクレオシドのペントース基の5'−位置に結合した脂質誘導体が公知である。この誘導体化の目的は、ヌクレオシド類をより親油性にして、リポソームがそれらを含むことができるようにすることである。リポソームはHIVウィルスを担持することが知られている細胞であるマクロファージおよび単球に優先的に取り込まれる。そこで標的化効果が得られると記載されている。
【0010】
ヌクレオシド類似体の抗ウィルスおよび抗癌活性は、投与薬剤の細胞内燐酸化に直接関係している。この生化学的変換は通常はウィルスおよび/または細胞内酵素によって行われる。この効果を改善するために、WO96/25421は比較的短い鎖(C14以下)を有する飽和または不飽和脂肪酸によるヌクレオシドの燐脂質誘導体を開示している。
【0011】
その他のクラスの薬物学的物質の特性を、脂肪酸による誘導体化によって改良する方法も研究されている。
【0012】
例えばWO96/22303は、数種のカテゴリーの治療化合物(コルチコステロン、オピオイド、およびオピオイド拮抗薬、抗ウィルス性ヌクレオシド類、シクロスポリン類および関連シクロペプチド類、葉酸拮抗薬、カテコールアミン前駆体およびカテコールアミン類、およびカルボン酸基を含むアルキル化剤)の薬物動態プロフィールおよびデリバリーモードを、トロメタミンまたはエタノールアミン誘導体等のリンカー/スペーサー基を使用して脂肪酸の1ないし3個のアシル誘導体とこれらとを結合することによって変え得ることを教示している。パルミチン酸は好適脂肪酸である。
【0013】
数種のNSAIDsの親油性プロドラッグはブンドガード(H.Bundgaard)ら(International Journal of Pharmaceutics、43巻、101−110ページ、1988)およびシャンバグ(V.R.Shanbhag)ら(Journal of Pharmaceutical Sciences、149、81巻、2号、1992年2月)から公知である。プロドラッグ的観点に加えて、GI(胃腸)刺激が減少することが報告されている。EP−A−0195570は、ガンマリノレン酸およびジホモ−ガンマ−リノレン酸とNSAIDsとの組み合わせが連続的に摂取した場合のNSAIDsによる副作用を軽減することを示唆している。
【0014】
米国特許第5,284,876号は、CNS疾患治療において経口プロドラッグとしてドーパミンのドコサヘキサエン酸アミドを使用することを教示している。
【0015】
皮膚および経口投与どちらでもいわゆる浸透促進剤として用いられる脂肪酸/脂肪酸誘導体を含む物理的混合物がPCT/US94/02880およびPCT/SE96/00122から公知である。
【発明の概要】
【発明が解決しようとする課題】
【0016】
上記のように、これら先行提案の多くは抗ウィルス性ヌクレオシドおよびヌクレオシド類似体の脂肪酸誘導体に関係する。実際、或る種のポリ不飽和脂肪酸がウィルスを攻撃することは以前から知られているから、こうなることは驚くにあたらない。EP−A−0642525において、我々自身、ヌクレオシドおよびヌクレオシド誘導体がオレイン酸(cis−9−オクタデセン酸)、エライジン酸(trans−9−オクタデセン酸)、cis−11−エイコセン酸またはtrans−11−エイコセン酸と反応して対応する5'−O−モノエステルを形成することによって上記ヌクレオシドおよびヌクレオシド誘導体の抗ウィルス効果が著しく高まることを教示した。我々は、これら4種類のモノ不飽和、ω−9 C18またはC20脂肪酸から得られる好都合な効果が脂肪酸誘導体化によって一般に得られるものより優れていることを示した。
【課題を解決するための手段】
【0017】
今や驚くべきことに、本発明により多くの異なる生物学的活性化合物の性質を ω−9 C18またはC20モノ不飽和脂肪酸による誘導体化によって好都合に変え得ることを我々は見いだした。こうして本発明は、例えば多くの薬剤および農薬の価値を高めるために広く、かつ簡単に利用できる方法を提供する。
【0018】
概して本発明は、一面では分子構造中にアルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸およびカルボン酸エステル基から選択される1つ以上の官能基を含む、ヌクレオシドまたはヌクレオシド誘導体以外の生物学的活性化合物の親油性誘導体を提供する。上記親油性誘導体は前記生物学的活性化合物の少なくとも一つの前記官能基が、下記の式であらわされるものから選択される親油性基によって置換された分子構造によって、特徴づけられる:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2RN−CONHCH2Rおよび−SCH2R。前記式中、Rはcis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニルおよびtrans−10−ノナデセニルから選択される親油性部分である。
【図面の簡単な説明】
【0019】
【図1】図1は肉芽腫の液体重量を示す。リポソーム投与対照動物の肉芽腫の平均液体含有量は62.69mgであった。減少は全投与群に認められた(ナプロキセン12%、ナプロキセンオレイルエーテル9%、ナプロキセンオレイルエステル14%およびナプロキセンオレイルアミド22%)。ナプロキセンオレイルアミドで得られた結果が特に顕著であった。
【図2】図2は肉芽腫組織乾燥重量を示す。リポソーム投与対照動物の肉芽腫の組織乾燥重量は14.36mgであった。ナプロキセンは組織乾燥重量に影響を与えないように見えた。残る投与群では減少が認められた(ナプロキセンオレイルエーテル16%、ナプロキセンオレイルエステル12%およびナプロキセンオレイルアミド38%)。ここでもナプロキセンオレイルアミドで得られる減少が最大であった。
【図3】図3はマウスの皮下に2週間埋め込まれたコットン−ラップド軟骨のグリコサミノグリカン含有量を示す。埋め込まれなかった対照軟骨は平均グリコサミノグリカン1168mgであった。リポソーム投与対照動物に2週間移植するとグリコサミノグリカンは60%減少した。ナプロキセンオレイルエーテル投与動物から取り出したインプラントを除き、残る投与群のインプラントはリポソーム投与対照群のそれらより少ないグリコサミノグリカンを含む傾向があった(ナプロキセン16%、ナプロキセンオレイルエステル12%およびナプロキセンオレイルアミド11%)。
【図4】図4はマウスの皮下に2週間埋め込まれたコットン−ラップド軟骨のヒドロキシプロリン含有量を示す。埋め込まれなかった対照軟骨は平均ヒドロキシプロリン含有量329mgであった。リポソーム投与対照動物に2週間移植するとヒドロキシプロリンは19%減少した。ナプロキセンオレイルエーテル投与動物から取り出したインプラントを除き、残る投与群からのインプラントはリポソーム投与対照群のそれらより少ないヒドロキシプロリンを含む傾向があった(ナプロキセン12%、ナプロキセンオレイルエステル8%、ナプロキセンオレイルアミド3%)。
【図5】誘導体の効果は明らかで、驚くべきものである。図5からわかるように、プレドニゾロンがチモサン刺激−細胞の活性に与える影響は6時間目の化学ルミネッセンスのわずかな減少として認められるだけである。プレドニゾロンエライデートでは、炎症細胞の活性は投与後48時間にわたって対照に比較して減少する。その効果はプレドニゾロンそのものの効果より明らかに大きく、持続する。
【図6】誘導体類の効果は明らかで驚くべきものである。図6からわかるように、特に好ましい脂肪酸の一つの例として、プレドニゾロン−エライデートの効果は最大であった。その他の脂肪酸誘導体では化学ルミネッセンスはほんのわずかな影響を受けただけである。プレドニゾロン−エライデートでは炎症細胞の活性は投与後48時間にわたり、対照および残りの脂肪酸誘導体に比較して減少する。
【図7】図7に示すように、腹腔洗浄液中の細胞数は実質的に減少し、この減少は投与後48時間まで明らかであった。
【図8】細胞を分別すると、腹腔洗浄液中のマクロファージの数に主な影響が出ることがわかった。
【図9】プレドニゾロンではその効果は遥かに少なく、6時間および12時間目に認められただけである。ベタメタゾンとベタメタゾン−エライデートとの比較でも、腹腔洗浄液中のマクロファージの数の測定で、さほど明らかではないが、同様な効果が認められた。
【図10】気道反応性。
【図11】細胞を2×105/mlの懸濁液にして、20μMの薬剤にさらした。種々の時間に部分を取り、氷冷PBSですすぎ、その後フローサイトメーターを通過させた。図11にはドキソルビシンおよびドキソルビシン誘導体の時間の関数としての蓄積が見られる。細胞系COR−L123/P(ヒト大肺細胞)およびその耐性細胞系COR−L123/Rではドキソルビシン誘導体(ドキソルビシン−エライジン−アミドおよびドキソルビシン−オレイル−カルバメート)濃度はほぼ同じであった、一方ドキソルビシン濃度は耐性細胞系では遥かに低かった。
【図12】細胞を上記のようにダウノルビシン、ダウノルビシン−エライジンアミドおよびダウノルビシン−オレインカルバメートにさらした。薬剤濃度は10μMに減らした。耐性および非耐性細胞への取り込みによる蛍光は誘導体に関しては2種類の細胞で多かれ少なかれ同じである。
【図13】ダウノルビシンそのものではこれらの細胞内への取り込みによる蛍光は2種類の細胞で差があった。
【発明を実施するための形態】
【0020】
本発明の好適一実施態様において、治療化合物をn−9 C18またはC20モノ不飽和脂肪酸で誘導体化することによって、上記治療化合物の生物学的効果は改善される。以下に、下記の群から選択される薬剤類に本発明を適用することに関して詳細に論議する:
1.抗癌剤;
2.抗炎症剤;
−NSAIDs
−副腎皮質ステロイド類
3.抗生物質およびその他の抗菌剤;
4.抗寄生虫薬;
5.CNS薬;
6.心臓血管薬;
7.抗凝固剤。
【0021】
しかし本発明は、薬物学的に活性であり、分子構造中にn−9 C18またはC20モノ不飽和脂肪酸と結合できる官能基を1つ以上含むいかなる化合物にも広く応用できる。例えば、本発明を用いて、鎮痛剤、殺真菌薬、高脂血症薬、制吐剤および診断薬等の医学的化合物の生物学的効果を改善することもできる。
【0022】
本発明による治療的活性化合物の親油性誘導体は、熟練せる当業者には公知の一般的方法によって、薬物学的に容認される担体および賦形薬と共に処方される。用量の割合は親薬剤のそれと関係する。但し本発明の親油性誘導体が親薬剤の効果を著しく増強する場合にはその用量を通常レベルより減らすことができる。
【0023】
本発明の好都合な効果は十分確立された薬剤類で証明されたとはいえ、同様な改良はまだ開発途上にあるその他の薬剤類でも示されるかも知れないと考えられる。すなわち、本発明の親油性誘導体で認められた改良特性の仮定的説明は一般的に適用されるものであって、治療的効果の特別のメカニズムに限られるものではない。
【0024】
本発明による治療的活性化合物の親油性誘導体の幾つかが示す、我々が発見した特に貴重な特性は、それらが薬剤耐性を克服することである。理論によって束縛されるものではないが、本発明の親油性誘導体は何らかの方法で膜タンパク質ポンプと相互作用し、それらの細胞からの活性(毒性)化合物の除去が阻止され、その結果活性化合物の濃度を長期間治療的有効濃度に保つことが可能となる。少なくとも本発明は、本発明による親薬剤およびその親油性誘導体の同時投与によって薬剤耐性効果を克服する可能性にも通じる。親薬剤とその親油性誘導体は容易に投与できるように、同じ製剤中に含まれるのが好適である。但し若干の場合には親薬剤と親油性誘導体とを別々の投与型に含めるのも好ましい。親薬剤の用量に対する親油性誘導体の用量は適切な試験によって決められるが、一般には、重量で1:1から1000:1までの範囲である。
【0025】
前にも述べたように、本発明は普通は薬剤だけでなく、生物学的活性を有するいかなる化合物にも適用できる。
【0026】
生物学的活性化合物の別の経済的重要な群は、農業および園芸に用いられる生成物、例えば殺虫剤、殺真菌薬および除草剤等である。農薬は構造および作用モード共に非常に種々様々である。例えば、幾つかのよく知られた取り込み経路がある;例えば植物は活性化合物を根系から、または直接植物の葉または幹から摂取することができ、一方殺虫剤は、害虫が襲う植物を介して、または直接接触によって取り込まれる。本発明による農薬の親油性誘導体は植物によっても昆虫およびその他の害虫によっても取り込まれやすい特性を有することが判明している。その上、本発明の誘導体類は、薬剤耐性のように問題になりつつある殺虫剤耐性の克服にも役立つ。
【0027】
本発明によって好都合に誘導体化できる生物学的活性化合物のその他の群は、食物および食品添加物、例えば保存料、香料およびスパイス等を含む。
【0028】
本発明の親油性誘導体は、親薬剤またはその他の生物学的活性化合物分子を、炭素原子18または20の炭素鎖を有するcis−またはtrans−n−9モノ不飽和脂肪酸、脂肪酸アルコールまたは脂肪アミンと、またはこのような脂肪酸、脂肪アルコールまたは脂肪アミンの反応性誘導体、例えば酸塩化物、反応性エステル類、ハロゲン化物などと反応させることによって合成される。n−9とは、脂質部分のC末端から数えて9および10の位置が不飽和であることを示す。このため、使用できる脂肪酸類(およびそれらから誘導されたアルコールおよびアミン類)はcis−9−オクタデセン酸(オレイン酸)、trans−9−オクタデセン酸(エライジン酸)、cis−11−エイコセン酸およびtrans−11−エイコセン酸である。
【0029】
親生物学的活性化合物と脂肪酸、脂肪アルコールまたは脂肪アミン化合物とのカップリング反応は、熟練せる当業者に公知の種々の方法によって達成できる。親分子中に2つ以上の誘導体化可能の官能基が存在する場合、カップリング段階において保護基または改良合成法を用いて必要な選択を行うことができる。一般的に、反応の進行を薄層クロマトグラフィー(TLC)および適した溶媒系を用いて追跡することができる。TLCによって確認して反応が完了したとき、生成物を有機溶媒で抽出し、クロマトグラフィーおよび/または適切な溶媒系からの再結晶によって精製するのが一般的である。親出発原料にヒドロキシル、アミノ、チオールまたはカルボキシル基が1つより多く存在する場合は、アルキル化またはアシル化化合物の混合物が生成するかも知れない。その後個々のモノ−またはポリ−誘導体化合物は例えばクロマトグラフィーによって分離することができる。
【0030】
カップリング反応は一段階で行われることが多く、親油性誘導体は普通は良い安定性を有する結晶として回収できる。このことは最終的医薬品を上首尾にガレノス処理するために役立つ。
【0031】
本発明によって用いられる調製法は以下に示す反応スキーム、並びに本明細書の後部で述べる実施例によって説明される。
【0032】
ここで本発明を数種類の薬剤についてさらに詳細に説明する。
【0033】
抗炎症剤
リウマチ性関節炎、骨関節炎、ベクテリエフ症候群、全身性紅斑性狼瘡(SLE)、喘息、通風等の重大な疾患の多くは炎症性反応を引き起こす異常免疫応答の結果である。炎症性プロセスは、例えば抗原−抗体相互作用、感染性作用物質、虚血等の多数の刺激によって誘起される一連の事象を含む。肉眼的レベルでは、その反応は紅斑、浮腫、敏感(痛覚過敏)、および疼痛という臨床症状を伴うのが普通である。炎症性疾患は主として3種類の薬、すなわちNSAIDs(アスピリン様薬剤と呼ぶこともある)、免疫抑制剤(例えばメトトレキセート、シクロホスファミドおよび最近ではシクロスポリン)および副腎皮質ステロイド類(ヒドロコーチゾン、プレドニゾロン、等)で治療される。その治療は主として疾患初期の疼痛および/または強度を抑制する。現在の治療法は高用量および/または長い治療期間によるひどい副作用によって制限されることが多い。
【0034】
可逆的気道閉塞−喘息−は呼吸疾患の最も一般的なものである。気管支過反応性の程度は普通は副腎皮質ステロイドおよび/または気管支拡張剤の定期的吸入によってコントロールまたは軽減される。大部分の治療法は平均して2−3時間の治療効果をもたらす。大部分の呼吸器エアゾール類では、吸収は経腸または経口投与による吸収とほぼ等しい。抗炎症効果および免疫抑制効果による治療は一時的である。長期治療のためには副作用を抑制した最小量を用いるべきである。
【0035】
重症喘息発作ではメチルプレドニゾロン琥珀酸ナトリウムを静脈注射し、その後10日間経口投与する。喘息の急性悪化は経口コルチコステロイドの短期間コースで治療することが多い。気管支喘息を治療する治療法に吸入コルチコステロイドを含めるやり方が最近明らかに増加している。ベクロメタゾン・ジプロピオネート、トランシノロンアセトニドまたはフルニゾリドは経口コルチコステロイドコースの期間を短縮し、或いは完全に経口コルチコステロイドの代わりとなる。これらの薬剤を推奨用量で用いる際にはより少ない副腎機能抑制が認められる。
【0036】
慢性喘息の局所治療におけるステロイド使用は吸入器によって便利に行われる。これは全身的投与のひどい副作用のリスクを制限する。局所投与後の速やか且つ選択的作用を確実にするためには、気管支の内皮細胞上の受容体への付加および前記受容体との相互作用が必須である。本発明の脂肪酸誘導体は局所投与から得られる利点をより一層改善することができる。気道炎症は致命的喘息発作の顕著な特徴と見なされ、軽度の喘息発作の気管支生検でも同様な変化が見いだされる。重症喘息発作は炎症細胞、主として肺胞マクロファージの気道への多量流入と関係がある。この状態はウィルスによって誘起される気道過剰反応性の外観に非常に似ている。マクロファージは反応性酸素種を放出することができ、それらは肺抵抗を高め、ヒスタミンを遊離させる。一般には炎症細胞、特にマクロファージの抑制を測定するモデルを用いて、可能性のある喘息薬の効果を評価することができる。化学ルミネッセンスを反応性酸素種の放出の尺度として用いることができる。下に示すように、炎症細胞、主にマクロファージのラット腹腔内への流入を本発明の副腎皮質ステロイド誘導体によって減らすことができる。刺激時の炎症細胞の活性も減少し、誘導体投与後、親薬剤投与後よりもより長期間にわたってその減少が認められる。それら誘導体は投与後最低48時間は有効である。この長時間にわたる活性は喘息治療において非常に好都合である。
【0037】
天然ホルモン類はin vivoでは非常に速やかに分解することが多いため、注射しない限り治療効果はほとんど得られないことが多い。これらの分子または天然化合物に類似した合成類似体を本発明の脂肪酸と結合することによって薬物動態挙動を変え、治療効果を改善することができる。これは全身投与および局所投与両方に言えることである。
【0038】
本発明によって誘導体化し得る副腎皮質ステロイド類およびその他の喘息薬の例は次のものを含める:
【0039】
【化1】

【0040】
【化2】
【0041】
炎症性疾患の治療に最も一般に用いられる薬剤はNSAIDsである。よく使用されるこの群の製品はいろいろあり、主なものはナプロキセン、ジクロフェナック(ボルタレン)、ピロキシカム(フェルデン)およびサリチル酸誘導体である。NSAIDsは抗炎症性、鎮痛性および解熱剤であるが、それらの主要な臨床的用途は炎症性疾患の治療である。NSAIDsはこれらの疾患による痛みおよび炎症の症状緩和をもたらすが、ひどい発症中の組織の病的損傷の進行は止めない。今日知られているNSAIDs製剤のなかで肉芽腫性組織の生成を明らかに軽減するものはない。肉芽腫性液含有量の減少は認められるとはいえ、その効果は肉芽腫性固体物質含有量の同時減少としては反映されない。これら薬剤の主な作用モードはプロスタグランジン生合成の阻止(シクロオキシゲナーゼの阻害)である。各薬剤の分布および薬物動態特性は薬剤活性に重要な影響を有する。これも、同じ化学ファミリーであっても、異なるNSAIDs薬剤に対しては各患者の応答性に大きなばらつきが起きる理由であると考えられる。例えば、異なるプロピオン酸誘導体に対する耐性に大きいばらつきがあることが報告されている。
【0042】
NSAIDsの主な特性はそれらのシクロオキシゲナーゼ阻止能力であり、したがってPGG2およびPGH2およびこれらから誘導される全てのエイコサノイド類(PGI2、TXB2、PGE2等)の生合成を阻止する。他方NSAIDsがリポキシゲナーゼを(少なくとも同程度に)阻止することは知られておらず、そのためロイコトリエン類(LTB4およびLTC4)の合成には影響しない。プロスタグランジンPGI2およびPGE2は炎症過程に重要な役割を演ずる。それらは浮腫をおこし、多分血管透過性を高める。PGI2は炎症性疾患に関連する痛みの主な要因である。ロイコトリエン類は炎症発作の第二および第三相における重要なメディエータである。そしてNSAIDsはリポキシゲナーゼを治療的に役立つ程度には阻止しないから、それらは炎症性疾患の変性部には影響しない。
【0043】
NSAIDsには副作用があり、それらは時には重症になり得る。最も一般的な副作用は胃潰瘍または腸潰瘍の誘発傾向であり、したがって痛み、吐気、胸焼けおよび時には出血および貧血をおこす。これらの効果はプロスタグランジン生合成の阻止と相関するものである。PGI2およびTXB2が存在しないため、血小板は凝集能力を失い、それはそれで出血時間を長引かせる。多くの症例で、NSAIDsがリウマチ性疾患の進行には好ましい効果を与えないことは明らかであり、状況によっては疾患プロセスを悪化さえすることを示唆する証拠がある。これは軟骨分解の増強による重度の基質喪失としてあらわれる。
【0044】
その他の副作用、例えば塩および水分の貯留、高カリウム血症および腎血流減少等もプロスタグランジン合成阻害と関連し、治療を不可能にすることがある。若干の患者においてアナフィラキシーショックをおこすこともあるアスピリン過敏症では、アスピリン様薬剤による治療はできない。
【0045】
親NSAIDsは、非ステロイド性抗炎症剤として分類され、アルコール、エーテル、フェノール、アミノ(第一、第二または第三)、アミド、チオール、カルボン酸およびカルボキシルエステル基から選択される1つ以上の誘導体化可能基を有するいかなる化合物でもよい。このクラスの現在公知のNSAIDsは下記の化合物を含む:
【0046】
【化3】
【0047】
【化4】
【0048】
【化5】
【0049】
【化6】
【0050】
【化7】
【0051】
【化8】
【0052】
【化9】
【0053】
上に示したように、既知NSAIDsの多くは上に定義した種類の誘導体化可能基を一つより多く含む。これらの場合、これらの官能基の1つ以上が本発明による親油性基によって置換することができ、2つ以上の親油性基がある場合、これらは同じ親油性基でも異なる親油性基でもよい。
【0054】
本発明の親油性抗炎症剤誘導体は親薬剤と炭素原子18または20の鎖長を有するcisまたはtrans n−9モノ不飽和脂肪酸、脂肪アルコールまたは脂肪アミンとの反応、またはこのような脂肪酸、脂肪アルコールまたは脂肪アミンの反応性誘導体、例えば酸塩化物、反応性エステル、ハロゲン化物およびメシレート等との反応によって合成される。n−9とは、脂質部分のC末端から数えて9および10位置の間に不飽和があることを示す。例えば使用し得る脂肪酸(およびそれらから誘導されるアルコール類およびアミン類)はcis−9−オクタデセン酸(オレイン酸)、trans−9−オクタデセン酸(エライジン酸)、cis−11−エイコセン酸およびtrans−11−エイコセン酸である。
【0055】
親薬剤と脂肪酸、脂肪アルコールまたは脂肪アミン化合物との間のカップリング反応は熟練せる当業者には公知の種々の方法によって達成できる。親薬剤に2つ以上の誘導化可能の官能基が存在する際には、保護基または改良合成法を用いてカップリング段階に必要な選択性を得ることができる。
【0056】
一般的に、反応の進行は薄層クロマトグラフィー(TLC)および適した溶媒系を用いて追跡することができる。TLCによって確認して反応が完了したときに、生成物を有機溶媒で抽出し、クロマトグラフィーおよび/または適切な溶媒系からの再結晶によって精製するのが一般的である。NSAIDs出発原料にヒドロキシル、アミノ、チオールまたはカルボキシル基が1つより多く存在する場合は、アルキル化またはアシル化化合物の混合物が生成するかも知れない。その場合は個々のモノ−またはポリ−誘導体化合物を例えばクロマトグラフィーによって分離することができる。
【0057】
本発明によって用いられる合成法は下に与えられる反応スキーム並びに本明細書の後部に与えられる実施例によって説明される。
【0058】
第一の反応スキームはサリチル酸の誘導体化を説明する。
【0059】
【化10】
【0060】
スキーム1
サリチル酸ナトリウムを脂肪アルコールメシレート(R'−OMS)で処理すると、サリチル酸エステル(I)が生成する。この反応の変法はサリチル酸エステル−2−エーテル(II)を与える。ここでエステルおよびエーテル中の炭化水素残基は同じである。この生成物は、サリチル酸エチル(IV)をアルキル化してエチルサリチレート−2−エーテル(V)を生成し、その後そのエチルエステルの加水分解によってサリチル酸−2−エーテル(III)を得るという方法でより好適に得られる。
【0061】
これらの方法の組み合わせにより、式IIであらわされる、エステル置換基およびエーテル置換基が異なる二付加物の合成が可能となる。
【0062】
第二の反応スキームはナプロキセンの誘導体化を説明する。
【0063】
【化11】
【0064】
スキーム2
ナプロキセンエステル(VII)またはアミド(VIII)誘導体を、N,N '−ジシクロヘキシルカルボジイミド(DCC)またはO−(1H−ベンゾトリアゾール−1−イル)−N,N,N',N'−テトラメチルウロニウムテトラフルオロボレート(TBTU)のようなカップリング試薬を用いてナプロキセン(VI)と対応するアルコールまたはアミン(R'−OHまたはR'−NH2)から合成する。
【0065】
長鎖エーテル同族体(XII)はナプロキセン(VI)から、最初に芳香族6−メチルエーテルを脱メチル化して生成物(IX)を与え、その後プロピオン酸側鎖をエステル化する(X)方法で合成される。フェノール部分のアルキル化(XI)およびエチル−エステルの加水分解は生成物(XII)を与えた。これらの方法の組み合わせは、エーテルおよびエステルまたはアミドの炭化水素残基が異なるかまたは同じである二付加物を与えることができる。
【0066】
第三の反応スキームはピロキシカムの誘導体化を説明する。
【0067】
【化12】
【0068】
スキーム3
ピロキシカムエステル(XIV)をピロキシカム(XIII)および対応する脂肪酸塩化物(R'COC1)から合成する。ピロキシカムのアミド窒素のアシル化が可能であり、少量のN−アシル化生成物並びにジアシル化生成物が分離される。主要生成物(XIV)の同定は改良NMR法によって行われる。
【0069】
第四の反応スキームはジクロフェナックの誘導体化を説明する。
【0070】
【化13】
スキーム4
ジクロフェナックエステル(XVI)またはアミド(VXII)はジクロフェナック(XV)および対応するアルコールまたはアミン(R−OHまたはR−NH2)から、DCCまたはTBTUのようなカップリング試薬を用いて合成される。異性体アミド(XVIII)は対応する脂肪酸R'−COOHとXVから、カップリング試薬としてTBTUを用いて作ることができる。
【0071】
第五反応スキームは、ベタメタゾン(XIX)およびプレドニゾロン(XX)の誘導体化を説明する。多くのステロイドには第一、第二および第三アルコール機能があり、それら全てをエステルに変換することができる。しかし適度に良い選択性があり、第一アルコールはカップリング試薬としてDCCを用いて、或いは脂肪酸塩化物の直接使用によってエステル化される。
【0072】
【化14】
【0073】
スキーム5
本発明のチオ誘導体類は上記反応スキームによって示されるものと類似の方法によって合成される。
【0074】
本発明による特異的親油性抗炎症剤誘導体の製法はこの後の実施例によって説明され、実施例6および7は中間体化合物の製法を説明する。
【実施例】
【0075】
実施例1
2−ヒドロキシ安息香酸−(cis−9'−オクタデセニル)エステル
無水N,N'−ジメチルホルムアミド40ml中水素化ナトリウム(60%)(0.21g、5.25×10-3mol)の懸濁液に、2−ヒドロキシ安息香酸(サリチル酸)(0.726g、5.25×10-3mol)を加え、混合物をN2 下で80℃で1時間撹拌した。cis−9−オクタデセノール−メシレート(1 .82g、5.25×10-3mol)を加え、22時間撹拌し続けた。冷やした反応混合物を濃縮し、残渣を100mlクロロホルムに溶解した。有機相を水で洗い、炭酸水素ナトリウムおよびブラインで希釈した。乾燥した相を蒸発乾固し、粗生成物をシリカゲルカラムで精製した。その際溶出系としてヘキサン中5%エーテルを用いた。均質フラクションを蒸発すると表題化合物1.3g(64%)が得られた。
【0076】
【化15】
【0077】
実施例2
2−(cis−9'−オクタデセノキシ)−エチル−ベンゾエート
無水N,N−ジメチルホルムアミド40ml中水素化ナトリウム(60%)(0.206g、5.15×10-3mol)の懸濁液に2−ヒドロキシ−エチル−ベンゾエート(0.86g、5.15×10-3mol)を加え、その混合物をN2下で80℃で1時間撹拌した。cis−9−オクタデセノール−メシレート(1. 78g、5.15×10-3mol)を加え、40時間撹拌し続けた。冷やした反応混合物を高真空で蒸発し、残留物をクロロホルムおよび水で処理した。乾燥した有機相を濃縮し、粗生成物をシリカゲルカラムで精製した。その際ヘキサン中5%エーテルで溶出した。均質フラクションを集めると表題化合物1.11g(52%)が得られた。
【0078】
【化16】
【0079】
実施例3
2−(cis−9'−オクタデセノキシ)−安息香酸
エタノール25mlおよび水50ml中2−(cis−9'−オクタデセノキシ)−エチル−ベンゾエート(1.11g、2.66×10-3mol)の懸濁液に水酸化リチウム(2.0g)を加え、反応混合物を90℃で6時間撹拌した。エタノールを蒸発し、クロロホルム100mlを加えた。5N HClを注意深く添加してpH7に調節した。有機相を水で洗った。溶媒除去後、生成物をシリカゲルカラムで精製した。その際溶出系としてクロロホルム中2%メタノールを用いた。表題化合物1.0g(96%)が得られた。
【0080】
【化17】
【0081】
実施例4
1−(p−クロロベンゾイル)−5−メトキシ−2−メチルインドール−3−酢酸−(cis−9'−オクタデセニル)アミド
無水N,N−ジメチルホルムアミド6ml中1−(p−クロロベンゾイル)−5−メトキシ−2−メチルインドール−3−酢酸(インドメタシン)(0.56g、1.56×10-3mol)およびTBTU(0.51g、1.56×10-3mol)の溶液に、N,N−ジイソプロピルエチラミン(0.53ml、3.12 ×10-3mol)を加え、反応混合物をN2下で室温で30分間撹拌した。無水N,N−ジメチルホルムアミド6m1中cis−9−オクタデセニル−アミン(0.42g、1.56×10-3mol)の溶液を加え、撹拌を3時間続けた。溶媒を高真空で蒸発し、残留物をクロロホルムと水とに分割した。乾燥した有機相を濃縮し、生成物をシリカゲルカラムで精製した。その際クロロホルム中2%エタノールを溶媒系として用いた。均質フラクションを蒸発すると若干のDMFを含む表題化合物が1.05g得られた。その生成物をエーテルに溶解し、水で洗い、有機相を乾燥し、蒸発すると表題化合物0.88g(92%)が得られた。
【0082】
【化18】
実施例5
S(+)−2−(6−メトキシ−2−ナフチル)プロピオン酸−(cis−9' −オクタデセニル)−アミド(ナプロキセンオレイルアミド)
無水N,N−ジメチルホルムアミド20ml中ナプロキセン(1.65g、7. 15×10-3mol)およびTBTU(2.30g、7.15×10-3mol)の溶液に、N,N−ジイソプロピルエチラミン(2.45ml)14.3×10-3mol)を加え、反応混合物をN2下で室温で30分間撹拌した。無水N,N−ジメチルホルムアミド25ml中1−アミノ−cis−9−オクタデセン(1.91g、7.15×10-3mol)の溶液を加え、3時間撹拌を続けた。溶媒を高真空で蒸発し、残留物をクロロホルムと水との間に分割した。乾燥した有機相を濃縮し、生成物をシリカゲルカラムで精製した。その際溶出系としてクロロホルム中3%メタノールを用いた。均質フラクションを蒸発すると表題化合物2.77g(81%)が得られた。
【0083】
【化19】
【0084】
実施例6
S(+)−2−(6−ヒドロキシ−2−ナフチル)プロピオン酸
十分撹拌した無水N,N−ジメチルホルムアミド中水素化ナトリウム(60%)(12.9g、0.336mol)懸濁液に、N,N−ジメチルホルムアミド300ml中エタンチオール(24.3ml、0.328mol)溶液を滴下した。N,N−ジメチルホルムアミド150ml中ナプロキセン(15g、0.065mol)溶液をゆっくり加え、反応混合物を150℃で3時間加熱した。澄明溶液を冷却し、3.5N HClでpHを調節した(2−3)。溶媒を高真空で蒸発し、残留物をエーテル150mlと水90mlとの混合物で処理した。固体沈殿物を濾去し、濾液を濃縮した。残留物をクロロホルム90mlと水90mlとの混合物で処理し、冷蔵庫に24時間保存した。白色沈殿物を濾去し、洗い、乾燥すると、表題化合物10.1g(72%)が得られた。
【0085】
【化20】
【0086】
実施例7
S(+)−2−(6−ヒドロキシ−2−ナフチル)プロピオン酸−エチルエステル
無水エタノール1200ml中S(+)−2−(6−ヒドロキシ−2−ナフチル)プロピオン酸(5.0g、23×10-3mol)溶液にp−トルエン−スルホン酸(0.2g)を加え、反応混合物を24時間還流加熱した。冷やした混合物を固体NaHCO3部分と共に撹拌した。その溶液を濾過し、溶媒を蒸発した。残留物をクロロホルムに溶解し、水で洗った。有機相を濃縮し、粗生成物をシリカゲルカラムでクロロホルム中2%メタノールで溶出した。均質フラクションは表題化合物4.8g(80%)を与えた。
【0087】
【化21】
【0088】
実施例8
S(+)−2−(6−[cis−9'−オクタデセノキシ]−2−ナフチル)−プロピオン酸−エチルエステル
無水N,N−ジメチルホルムアミド350ml中水素化ナトリウム(60%)(0.47g、11.8×10-3mol)懸濁液にS(+)−2−(6−ヒドロキシ−2−ナフチル)プロピオン酸−エチルエステルを加え、反応混合物をN2下で室温で2時間撹拌した。N,N−ジメチルホルムアミド5ml中cis−9−オクタデセノール−メシレート(3.91g、10.7×10-3mol)溶液を加え、撹拌を48時間続けた。溶媒を高真空で蒸発し、残留物をクロロホルムと水で処理した。乾燥した有機相を濃縮し、粗生成物をシリカゲルカラムで精製した。その際クロロホルムで溶出した。均質フラクションは表題化合物2.93g(56%)を与えた。
【0089】
【化22】
【0090】
実施例9
S(+)−2−(6−[cis−9'−オクタデセノキシ]−2−ナフチル)−プロピオン酸(ナプロキセンオレイルエーテル)
テトラヒドロフラン115mlおよび1M NaOH25ml中S(+)−2−(6−[cis−9'−オクタデセノキシ]−2−ナフチル)−プロピオン酸−エチルエステル(3.79g、7.67×10-3mol)溶液を室温で10日間撹拌した。1M HCl17mlを加え、溶媒を蒸発した。残留物をクロロホルムおよび水に取り、1M HClでpHを1に調節した。有機相を水で洗い、乾燥し(MgSO4)、濃縮すると表題化合物3.25g(94%)が得られた。
【0091】
【化23】
【0092】
実施例10
4−O−(trans−9'−オクタデセノイル)−2−メチル−N(2−ピリジル)−2H−1,2−ベンゾチアジン−3−カルボキサミド−1,1−ジオキシド
無水N,N−ジメチルホルムアミド25ml中4−ヒドロキシ−2−メチル−N[2−ピリジル]−2H,1,2−ベンゾチアジン−3−カルボキサミド−1,1−ジオキシド(ピロキシカム)溶液に、ジクロロメタン20ml中trans−9−オクタデセノイルクロリド(2.2g、7.53×10-3mol)溶液2mlを加え、反応混合物をN2下で室温で撹拌した。残りの酸塩化物溶液を2mlづつ2時間間隔で加えた。合計80時間の反応時間後、溶媒を高真空で蒸発した。残留物をエーテル200mlに溶解し、水と少量のNaHCO3(水溶液)で洗った。乾燥した(MgSO4)有機相を濃縮し、粗生成物をシリカゲルカラムで精製した。その際エチルアセテート/ヘキサン(40:60)で溶出した。均質フラクションを集め、蒸発すると、固体物質3.56gが得られ、それをペンタン/エーテル中で還流した。冷やした混合物を一晩4℃に保った。固体物質を濾去し、ペンタンで洗い、乾燥すると表題化合物3.5g(78%)が得られた。
【0093】
【化24】
【0094】
実施例11
[2−(2,6−ジクロロフェニル)アミノ]ベンゼン酢酸)−(cis−9' −オクタデセニル)−エステル
ジクロロメタン15mlおよびN,N−ジメチルホルムアミド3ml中(2−[2、6−ジクロロフェニル])−アミノ]ベンゼン酢酸ナトリウム(ジクロフェナック)(0.48g、1.6×10-3mol)溶液に、酢酸(0.09ml、1.6×10-3mol)、cis−9−オクタデシン−1−01(0.42g、1.6×10-3mol)、4−ジメチルーアミノピリジン(DMAP)(50mg)およびDCC(0.34g、1.7×10-3mol)を加え、反応混合物を0 ℃で6時間、室温で48時間撹拌した。白色沈殿を濾去し、ジクロロメタンで洗った。有機相を水で洗い、乾燥し(MgSO4)、濃縮し、シリカゲルカラムで精製した。その際エチルアセテート/ヘキサン(40:60)で溶出した。均質フラクションは表題化合物0.45g(53%)を無色液体として与えた。
【0095】
【化25】
【0096】
実施例12
4−O−(cis−11'−エイコサノイル)−2−メチル−N(2−ピリジル)−2H−1,2−ベンゾチアジン−3−カルボキサミド−1,1−ジオキシド
無水N,N−ジメチルホルムアミド3ml中4−ヒドロキシ−2−メチル−N[2−ピリジル]−2H−1,2−ベンゾチアジン−3−カルボキサミド−1,1−ジオキシド(ピロキシカム)(0.3g、0.990×10-3mol)溶液にジクロロメタン2.5ml中cis−11−エイコサノイルクロリド(0.29g、0.90×10-3mol)溶液1.5mlを加えた。残りの酸塩化物溶液を2時間後に加えた。合計80時間の反応時間後に溶媒を高真空で蒸発した。残留物をエーテル40mlに溶解し、水および少量のNaHCO3(水溶液)で洗った。乾燥した有機相を濃縮し、粗生成物をシリカゲルガラムで精製した。その際エチルアセテート/ヘキサン(40:60)で溶出した。均質フラクションを集め、蒸発すると表題化合物0.42g(75%)が得られた。
【0097】
【化26】
【0098】
実施例13
S(+)−2−(6−メトキシ−2−ナフチル)プロピオン酸−cis−9'−オクタデセニル−エステル
ジクロロメタン10ml中S(+)−2−(6−メトキシ−2−ナフチル)プロピオン酸(ナプロキセン)(0.15g、0.65mmol)溶液に、cis−9−オクタデセノール(0.18g、0.67mmol)、DCC(0.13g、0.67mmol)、4−ジメチルアミノピリジン(DMAP)(20mg)を加え、反応混合物をN2下、室温で3時間撹拌した。生成した白色沈殿を濾去し、ジクロロメタンで洗った。 溶媒を蒸発し、生成物をシリカゲルカラムで精製した。その際溶出液としてジクロロメタンを用いた。均質フラクションは表題化合物0.25g(80%)を与えた。
【0099】
【化27】
【0100】
実施例14
11β,17α,21−トリヒドロキシプレグナ−1,4−ジエン−3、20−ジオン−21−エライデート
無水ジオキサン200mlおよびピリジン6.5ml中11β、17α、21−トリヒドロキシプレグナ−1、4−ジエン−3、20−ジオン(プレドニゾロン)溶液に、エライジン酸塩化物(8.0g、26.6mmol)を加え、反応混合物を10℃で3時間撹拌した。少量のメタノールを加え、溶媒を高真空で蒸発した。残留物をエーテルと水とに分配した。有機相を酒石酸(水溶液)、NaHCO3(水溶液)および水で洗った。乾燥した有機相を濃縮し、生成物をシリカゲルカラムで精製した。その際溶出系としてヘプタン/エチルアセテート/メタノール(64:32:4)を用いた。均質フラクションを蒸発すると表題化合物9.18g(90%)が得られた。
【0101】
【化28】
【0102】
実施例15
9−フルオロ−11β,17,21−トリヒドロキシ−16β−メチルプレグナ−1,4−ジエン−3,20−ジオン−21−エライデート
無水ジオキサン40mlおよびピリジン1ml中9−フルオロ−11β,17,21−トリヒドロキシ−16β−メチルプレグナ−1,4−ジエン−3,20−ジオン(ベタメタゾン)(0.9g、2.3mmol)の懸濁液に、エライジン酸塩化物(1.13g、3.03mmol)を加え、反応混合物を周囲温度で48時間撹拌した。少量のメタノールを加え、溶媒を高真空で蒸発した。残留物をエーテルと水とに分配した。有機相を酒石酸(水溶液)、NaHCO3(水溶液)および水で洗った。乾燥した有機相を濃縮し、生成物をシリカゲルカラムで精製した。その際溶出系としてヘプタン/エチルアセテート/メタノール(64:32:4)を用いた。不純フラクションを再精製し、均質フラクションを蒸発すると表題化合物1.02g(65%)が得られた。
【0103】
【化29】
【0104】
NSAIDsおよびその他の抗炎症剤が一般に処方される状態を治療する場合、本発明の親油性誘導体を経腸的または非経口的に全身投与することができる。
【0105】
好適形で経腸投与する場合、本発明の化合物はソフトまたはハードゼラチンカプセル、錠剤、顆粒、粒または粉末、糖衣錠、シロップ、懸濁液または溶液として処方することができる。
【0106】
非経口的投与の場合、本発明の化合物を注射または注入溶液、懸濁液または乳濁液として調製するのが適切である。
【0107】
本発明の医薬組成物は一般的方法によって調製される。例えばそれら製剤は不活性または薬力学的活性な添加物を含むことができる。錠剤または顆粒等は一般的な結合剤、増量剤、担体物質または希釈剤を含むことができる。液体製剤は例えば滅菌溶液の形で存在する。
【0108】
カプセルは活性成分以外に増量剤または濃化剤を含むことができる。さらに香味改良添加物並びに、保存料、安定剤、保水剤および乳化剤等の普通用いられる物質、浸透圧を変える塩類、緩衝剤およびその他の添加物等も含んでよい。
【0109】
所望ならば、本発明の化合物の薬剤は抗酸化剤、例えばトコフェロール、N−メチル−トコフェラミン、ブチル化ヒドロキシアニソール、アスコルビン酸またはブチル化ヒドロキシトルエン等を含むことができる。
【0110】
本発明による化合物の用量は治療すべき疾患の性質およびその進行段階によって、使用法および使用経路並びに患者の要求によって変化する。一般に全身投与する際の成人1日量は約0.1−100mg/kg体重/日、好適には0.5−30mg/kg/日である。
【0111】
本発明はさらに、炎症性、疼痛誘発および/または発熱状態の治療法において、そのような治療を必要とするヒト患者に本発明の少なくとも1種類の化合物を投与することを含む治療法に関係する。
【0112】
現在好まれる本発明の抗炎症薬親油性誘導体は、親薬剤がナプロキセンであるものである。特に、我々はナプロキセンオレイルエーテル、ナプロキセンオレイルエステルおよびナプロキセンオレイルアミドがナプロキセンそのものよりも改善された抗炎症効果を示すことを見いだした。動物in vivoモデルにおいて、これらの誘導体は肉芽腫液体含有量に関して、炎症の第一相に対して改善された効果を示した。さらに驚くべき効果は、特にナプロキセンオレイルアミド投与で肉芽腫組織乾燥重量が減少したことである。これは、既知NSAIDsによる治療では得られなかった組織損傷の減少を意味する。この効果の大きさは、治療量のステロイドによってのみ期待できるものであった。NSAIDsの重症副作用である軟骨分解の減少も認められた。
【0113】
これらの所見、例えば肉芽腫乾燥重量の減少によってわかる直接的改善効果、および軟骨分解の減少等の組み合わせはナプロキセン誘導体の治療指数を有意に改善する。これらの誘導体で治療した動物は、親化合物で治療した動物よりもかなり小さい侵襲性を示した。これは、これら誘導体の消化器副作用の誘発力がより小さいことを強く示唆する。
【0114】
理論によって束縛されるものではないが、この高まった抗炎症効果は、これら誘導体の親油性性質のために細胞による取り込みが増加するためであり、またはナプロキセンの活性とは全く異なる活性によるものであると考えることができる。ナプロキセンに付加した脂肪酸末端は反応性酸素種(ROS)の掃去剤(これは多くのメカニズムによって抗炎症性であり得る)として作用するらしい。例えば、組織損傷はROS感受性プロテアーゼーインヒビターの保護によって阻止されるし、タンパク質に対する酸化的損傷によって生成する内因性抗原類の形成は防止され、ヒアルロン酸を解重合から保護することによって血管由来因子の生成を阻止する。
【0115】
移植軟骨では、ナプロキセン様のその他のNSAIDsはプロテオグリカンおよびコラーゲン喪失を増加する傾向がある。それに対して、本発明のナプロキセン誘導体は軟骨からのプロテオグリカンまたはコラーゲンの喪失を高める傾向を示さない。シクロオキシゲナーゼ阻害は軟骨に対するNSAIDsの有害作用の原因であると考えられ、ナプロキセン誘導体はナプロキセンと共にこの酵素を阻止し得ることから、ナプロキセン誘導体のより大きいサイズおよび親油性特性が軟骨基質からナプロキセン誘導体を排除することが示唆される。
【0116】
これらナプロキセン誘導体の高められた抗炎症効果を明らかにする実験をここに詳細に述べる。
【0117】
生物学的効果
使用した肉芽腫誘起性軟骨分解のin vivoモデルは、滅菌コットンに包んだラット大腿骨頭軟骨をマウスの背中の皮下に埋め込んだものである。そのコットンは明白なT細胞が関係した肉芽腫性反応を誘発し、それは移植軟骨からの基質化合物の喪失に通ずる。可能性のある薬剤の抗関節炎効果を試験する手段として、このモデルは幾つかの明白な長所を有する。このモデルは軟骨基質の喪失を確認するための定量的生化学的終点を有する慢性糜爛性疾患を含む。抗炎症性活性はコットン肉芽腫の湿潤および乾燥重量から判断でき、軟骨保護効果は移植軟骨のグリコサミノグリカンおよびヒドロキシプロリン含有量(それぞれプロテオグリカンおよびコラーゲンを示す)から測定できる。肉芽腫は分離しており、必要に応じて摘出し、種々のメディエータまたは酵素を評価することができる。
【0118】
雌TOマウス(21±4g)群(n=10)においてコットンで包んだラット大腿骨頭軟骨を皮下に移植した。2週間目にインプラントを除去した。コットンは肉芽腫様反応を誘発し、同時にプロテオグリカンを移植軟骨から遊離させた。等モル量のナプロキセン(30mg/kg)およびナプロキセン誘導体(60mg/kg)を毎日経口投与し、肉芽腫発生に対する影響および軟骨プロテオグリカン含有量に与える影響を評価した。それら化合物はリポソームとして処方され、空のリポソームを賦形薬コントロールとして用いた。
【0119】
15mg/mlリポソーム組成物は次のように調製した:グリセロール/滅菌水緩衝液中で脂質誘導体(DMSO中)とレシチン(エタノール中)を1:1(w/w)で混合し、その後透析して溶媒類を除去する。誘導体化しないNSAIDs化合物の7.5mg/mlリポソーム組成物は、グリセロール/滅菌水中で空のリポソームに上記化合物を加えることによって調製した。
【0120】
結果をマン−ホィットニーを用いるINSTATで分析した。p値はタイ(ties)を補正した。p<0.05を有意とした。
【0121】
試験した化合物
ナプロキセン(VI)、ナプロキセンオレイルエーテル(XII)、ナプロキセンオレイルエステル(VII)およびナプロキセンオレイルアミド(VIII)。化合物XII、VIIおよびVIIIのR'はcis−CH2(CH2)7CH=CH(CH2)7 CH3である。
【0122】
このモデルでナプロキセンで得られたこれらの結果は、文献に報告されたナプロキセンおよびその他のNSAIDsを用いた同様な研究から判明した結果に匹敵する。
【0123】
薬剤投与を対照と比較した場合、肉芽腫の液体含有量は減少した、そしてその減少はナプロキセンオレイルアミドの場合には有意であることが判明した。組織乾燥重量はナプロキセンによって影響を受けないように見えたが、親油性有意は減少をおこすように見え、それはここでもナプロキセンオレイルアミドの場合に有意であった。これらの顕著な所見は、ナプロキセン誘導体がナプロキセンそのものに比し、軟骨分解を減らすことを強く示唆する。これは実際に確認された。なぜならばナプロキセンがリポソーム対照および親油性誘導体に比較して移植軟骨からのプロテオグリカン喪失を増加することがわかったからである。同じ所見はヒドロキシプロリン含有量として評価した軟骨分解にも反映された。但しナプロキセン投与動物からのインプラントは比較的少ないコラーゲンをもつ傾向があったが、投与群の間に統計的有意差はなかった。
【0124】
その上、ナプロキセン投与動物は侵襲的挙動を示し、その結果10インプラントのうち4インプラントを失った。リポソーム投与群またはナプロキセン誘導体投与群では同様な侵襲的挙動の結果としてインプラントが失われることはなかった。これはナプロキセン誘導体が親NSAIDsよりもよく耐えられたことを示唆する。
【0125】
上記の実験は、本発明によって誘導体化することによりナプロキセンの生物学的特性がかなり改善されることを示すものである。
【0126】
プレドニゾロンおよびベタメタゾンおよびそれらの誘導体のラット腹腔内単球/ マクロファージに与える影響
雄ラットの腹腔内に0時に10mg/ml試験化合物を4ml、または賦形薬のみを注射した。投与後6、12、25、48および72時間目に腹腔を生理的食塩液40mlで洗浄し、分離した細胞を洗い、数え、分別した。それらの細胞をオプソニン結合−チモサン(zymosan)、N−ホルミル−L−ロイシル−L−フェニルアラニン(fMLP)またはホルボール−12−ミリステート13−アセテート(PMA)で刺激し、細胞の活性を化学ルミネッセンス生成によって1時間にわたり測定した。
【0127】
誘導体の効果は明らかで、驚くべきものである。図5からわかるように、プレドニゾロンがチモサン刺激−細胞の活性に与える影響は6時間目の化学ルミネッセンスのわずかな減少として認められるだけである。プレドニゾロンエライデートでは、炎症細胞の活性は投与後48時間にわたって対照に比較して減少する。その効果はプレドニゾロンそのものの効果より明らかに大きく、持続する。
【0128】
選択したプレドニゾロン誘導体の効果をさらに研究するために別の実験系列を行った。この実験ではプレドニゾロンの7種類の脂肪酸エステルの抗炎症効果を比較した。
【0129】
雄ラットに試験化合物10mg/mlの或る用量、または賦形薬のみを0時に腹腔内注射した。投与後48時間に腹腔を洗浄し、分離した細胞を洗い、数え、分別した。細胞をその後オプソニン結合−チモサン、N−ホルミル−L−ロイシル−L−フェニルアラニン(fMLP)またはホルボール12−ミリステート13−アセテート(PMA)で刺激し、細胞活性を化学ルミネッセンス生成によって1時間にわたり測定した。
【0130】
誘導体類の効果は明らかで驚くべきものである。図6からわかるように、特に好ましい脂肪酸の一つの例として、プレドニゾロン−エライデートの効果は最大であった。その他の脂肪酸誘導体では化学ルミネッセンスはほんのわずかな影響を受けただけである。プレドニゾロン−エライデートでは炎症細胞の活性は投与後48時間にわたり、対照および残りの脂肪酸誘導体に比較して減少する。
【0131】
図7に示すように、腹腔洗浄液中の細胞数は実質的に減少し、この減少は投与後48時間まで明らかであった。
【0132】
細胞を分別すると、腹腔洗浄液中のマクロファージの数に主な影響が出ることがわかった。これは図8に示される。プレドニゾロンではその効果は遥かに少なく、6時間および12時間目に認められただけである。ベタメタゾンとベタメタゾン−エライデートとの比較でも、腹腔洗浄液中のマクロファージの数の測定で、さほど明らかではないが、同様な効果が認められた(図9)。
【0133】
プレドニゾロン−エライジン酸エステルの直接的抗喘息効果を研究するために、試験化合物を気道過剰反応モデルで評価した。
【0134】
ラットの内毒素誘起性気道変化に与えるプレドニゾロンエライジン酸の影響
急性炎症性気道変化のラットモデルにおいて、動物をエアゾール化内毒素(LPS)にさらす。これは90分以内に気管および気管支に広汎性好中性炎症をおこし、同時に気管支肺胞液中の好中球数の増加および喘息の重要な特徴である気道反応性上昇をおこす。10匹の雄F344ラットをチェンバー内で30分間100μg/ml LPSにさらした。エアゾールにさらす12時間前と4時間前に動物にプレドニゾロンまたはプレドニゾロン誘導体、3mg/kgを点滴注入した。エアゾール被曝終了後90分に、5−ヒドロキシトリプタミンに対する気道反応性の測定および気道炎症の評価のために動物の準備を行った。肺抵抗の最低50%増加が認められるまで、5HTを5分ごとに静脈注射に加えた。そして肺抵抗を50%増加するのに必要な5HT量、PC50RL、を計算した。プレドニゾロンも誘導体も気管支肺胞液中の炎症細胞数に影響を与えなかった。気道反応性は図10に示すように、プレドニゾロン−エライジン酸エステルによって驚くべく高程度の影響を受けた。これは喘息治療において非常に重要であるかも知れない。
【0135】
抗癌剤
効果的な癌化学療法治療は若干の重大な障害のために制限される。そのうちの幾つかは完全にまたは一部克服できるかも知れない。作用を確実により特異的にするあらゆる薬剤の変形は、患者にとって直接的利益となる。最も重要な要求は、問題の腫瘍が提供される治療に対して感受性を有することである。これは治療薬の種類および作用メカニズムに大きく依存し、実際の治療を開始する前の生検/分離−腫瘍細胞のin vitro実験で評価することができる。腫瘍を数種の薬剤に感受性にし得る方法も知られている。
【0136】
化学療法薬剤はそれらの性質として細胞に毒性を有する。悪性腫瘍細胞がその薬剤に比較的敏感である限り、好ましい状況となる。もしもその薬剤が腫瘍組織/腫瘍細胞に蓄積する傾向があれば、治療ポテンシャルはさらに改善される。治療指数をさらに改善するためには器官標的化が重要な要因である。一次腫瘍、特に初期、または別の腫瘍型から転移した一次腫瘍は、肝臓、脾臓、肺、脳等の選ばれた組織に限られることが非常に多い。薬剤の性質、その処方、または投与法がその薬剤を選択した組織に向かわせるならば、非常に選択的な腫瘍根絶が達成されるかも知れない。
【0137】
本発明の好適抗癌性誘導体は以下に述べる試験で証明されるように、改善された治療指数を有する。
【0138】
親抗癌化合物は、悪性腫瘍の治療に有用な特性を有する群に分類でき、アルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸およびカルボン酸エステル基から選択される1つ以上の誘導化可能基を有する化合物ならばいかなる化合物でもよい。本発明によって誘導体化できる現在使用できる抗癌剤の若干の例は次のものを含む:
【0139】
【化30】
【0140】
【化31】
【0141】
【化32】
【0142】
【化33】
【0143】
【化34】
【0144】
上記のように、公知の抗癌剤の多くは上に定義した種類の誘導化可能基を1つより多く含む。これらの場合、これら官能基の1つ以上を本発明による親油性基によって置換することができる。そして2つ以上の親油性基が存在する場合、これらは同じ基でも異なる基でもよい。
【0145】
本発明の親油性抗癌性誘導体は既述の一般的製法によって合成することができる。
【0146】
例えば、以下の反応スキームはドキソルビシン(XXI)およびダウノルビシン(XXII)からのアミド類およびカルバメート類の形成を説明する。親化合物(類)のアミノ基を、脂肪酸(RCOOH)または脂肪アルコール(R'OH)から作られるアシル−チアゾリジン−2−チオンまたはアルキルオキシ−カルボニル−チアゾリジン−2−チオン試薬と反応させて、アミドまたはカルバメートに選択的に誘導することができる。
【0147】
【化35】
【0148】
スキーム6
次に記すスキーム7も2つの抗癌性アルキル化剤クロラムブシル(XXIII)およびメルファラン(XXIV)の誘導体化を説明するものである。一官能価クロラムブシルは多数の方法によってエステル化され、またはアミドに変換できる。しかし二官能価メルファランは自己縮合または環形成反応等の多くの副反応を受けることがある。未保護メルファランにおいては、DCCまたはTBTU等のカップリング試薬の使用の有用性は限られているが、アミン官能基はアシル−チアゾリジン−2−チオン試薬によって、対応するアミドに都合よく変換できる。
【0149】
【化36】
【0150】
スキーム7
本発明による特異的抗癌性誘導体の製法は以下の実施例によって説明される。実施例18および21は中間体の製法に関係する。
【0151】
実施例16
クロラムブシル−オレイル エステル
ジクロロメタン70ml中4−[p−[ビス(2−クロルエチル)アミノ]−フェニル]酪酸(クロラムブシル)(0.966g、3.18mmol)およびオレイルアルコール(01893g、3.33mmol)の溶液に、DCC(0.72g、3.5mmol)およびN,N−ジメチル−アミノ−ピリジン(DMAP)(25mg)を加え、反応混合物を周囲温度で12時間撹拌した。固体沈殿を濾去し、残留物をCH2Cl250mlに溶解し、水で洗った。有機相にエーテル25mlを加え、固体沈殿を濾去した。濾液を蒸発し、残留物をシリカゲルカラムで精製した。この際CH2Cl2を溶出液として用いた。均質フラクションを蒸発すると表題化合物1.0g(55%)が得られた。
【0152】
【化37】
【0153】
実施例17
エライジン酸メルファランアミド
DMF24ml、水4mlおよびトリエチルアミン4ml中、L−3−[p−[ビス(2−クロルエチル)アミノ]−フェニル]アラニン(メルファラン)(0.603g、1.98mmol)の溶液に、DMF12ml中3−チアゾリジン−2−チオン−エライジルアミド(0.617g、1.61mmol)を加えた。反応混合物を暗所で室温で1.5時間撹拌した。溶媒を高真空で蒸発し、残留物をクロロホルム100mlに溶解し、pH5.5の水で洗った。有機相をAgNO3(水溶液)、水(pH5.5)および飽和NaCl(水溶液)で洗った。その有機相を蒸発すると表題化合物0.84g(75%)が得られた。
【0154】
【化38】
【0155】
実施例18
3−エライドイル−1,3−チアゾリジン−2−チオン
ジクロロメタン(20ml)中エライジル酸(2.0g)7.1mmol)、DMAP(86mg、0.7mmol)、1,3−チアゾリジン−2−チオン(1. 0g、8.4mmol)およびDCC(1.7g、8.2mmol)の混合物をN2 下で0℃で1時間撹拌し、その後周囲温度でさらに5時間撹拌した。追加のDCC(41mg、0.2mmol)を加え、反応物を同温度で2時間撹拌した。処理後フラッシュクロマトグラフィー(SiO2;四塩化炭素−クロロホルム1:0、1:1、0:1)を行うと、表題化合物2.56g(94%)が黄色ワックス様固体として得られた。
【0156】
【化39】
【0157】
実施例19
エライジン酸ダウノルビシンアミド
塩酸ダウノルビシン(250mg、0.44mmol)および3−エライドイル−1,3−チアゾリジン−2−チオン(実施例18、400mg、1.04mmol)をTHF(20ml)と、炭酸ナトリウム(0.12M NaHCO3、0.8M Na2CO3)で緩衝したブライン(4M NaCl 20ml)との間に分配した。混合物を暗所でN2下で周囲温度で4時間激しく撹拌した。相を分離し、水相をエーテルで抽出した(3×10ml)。合一した有機相を硝酸ナトリウム水溶液で洗った(3×10ml 2M)。存在する1、3−チアゾリジン−2−チオンを除去するために、ピリジン(1.0ml)を加え、エーテル相を、硝酸銀(0.2M)を含む硝酸ナトリウム(2M)水溶液(2×3ml)と共に激しく振とうした。各処理後、混合物をセライトを通して濾過し、エーテル(20ml)ですすいだ。エーテル相を硝酸ナトリウム水溶液(5ml 2M)およびブライン(5ml)で洗い、最後に乾燥した(MgSO4)。粗生成物をピリジン(0.2%w/w)で予備調製したシリカゲルカラムで精製した。溶出液としてクロロホルム中0.2%ピリジンおよび0.6%メタノールを用いた。表題化合物332mg(95%)が暗赤色粉末として得られた。
【0158】
【化40】
【0159】
実施例20
エライジン酸ドキソルビシンアミド
塩酸ドキソルビシン(400mg、0.69mmol)を上記のように、THF(35ml)および緩衝ブライン(35ml)中、3−エライドイル−1,3−チアゾリジン−2−チオン(実施例18、400mg、1.04mmol)で周囲温度で10分間処理した。反応を完了するために追加のアミド化試薬(実施例18:100mg、0.26mmol)が必要であった。6時間後、相を分離し、水相をTHFで抽出した。合一した有機相をブラインで洗い、乾燥した(MgSO4)。実施例19に記載したフラッシュクロマトグラフィーによって精製すると表題化合物440mg(79%)が暗赤色結晶として得られた(mp.115−116℃)。
【0160】
【化41】
【0161】
実施例21
3−(cis−9−オクタデセン−1−オキシカルボニル)−1,3−チアゾリジン−2−チオン
上記化合物は実質的に、エチル同族体のためのチェン(Chen)およびヤング( Yang)の方法によって合成した。N2下で0℃に保持したクロロホルム(乾燥、エタノール−フリー;15ml)中2−チオキソ−3−チアゾリジンカルボニルクロリド2(1.6g、8.6mmol)およびTEA(1.3ml、9.3mmol)の撹拌溶液に、オレイルアルコール(cis−9−オクタデセン−1−オール;2.8g、10.4mmol)を10分間以内に加えた。その混合物を同温度で80分間撹拌し、氷水(5ml)を加えた。塩酸(0.5ml 1M)を滴下して水相のpHを6に調節した。標準的に処理し、その後フラッシュクロマトグラフィー(SiO2:ヘキサン−クロロホルム1:1、1:2、1:3、0:1)を行うと、黄色油1.78g(50%)が得られた。
【0162】
【化42】
【0163】
実施例22
ダウノルビシン オレイル カルバメート、[N−(cis−9−オクタデセン−1−オキシカルボニル)ダウノルビシン]
塩酸ダウノルビシン(250mg、0.44mmol)を3−(cis−9−オクタデセン−1−オキシカルボニル)−1,3−チアゾリジン−2−チオン(実施例21、550mg、1.33mmol)と共に実施例19においてアミド同族体について記載したように27時間処理した。実施例20に記載したようにTHFで抽出した後得られた粗生成物をエーテル(40ml)に溶解した。存在する1,3−チアゾリジン−2−チオンを請求項19の方法によって除去した。粗生成物をピリジン(0.2%w/w)で予備調製したシリカゲルガラムで精製した。その際ベンゼン中0.2%ピリジンおよび0−10%メタノールで溶出した。表題化合物321mg(87%)が暗赤色粉末として得られた。
【0164】
【化43】
【0165】
実施例23
ドキソルビシン オレイル カルバメート、[N−(cis−9−オクタデセン−1−オキシカルボニル)ドキソルビシン]
塩酸ドキソルビシン(250mg、0.43mmol)を3−(cis−9−オクタデセン−1−オキシカルボニル)−1,3−チアゾリジン−2−チオン(実施例21:700mg、1.69mmol)と共に実施例19においてアミド同族体について記載したように69時間処理した。THFを減圧下で除去し、生成した懸濁液を水(20ml)とピリジン−クロロホルム1:4(25ml)に分配した。未溶解の物質を、有機相に加えたピリジン(15ml合一)で処理した。揮発物は減圧下で蒸発によって除去した。残留残渣(1.1g)を硝酸銀水溶液(0.6ml 1M)を含む酢酸エチルで20−30℃で20分間超音波処理した。生成した懸濁液をセライトを通して濾過し、酢酸エチル(20ml合一)ですすいだ。超音波処理サイクルをさらに硝酸銀(0.4ml 1M)で繰り返した。合一した有機相をブライン(5ml)で洗い、乾燥した(MgSO4)。回転蒸発器で酢酸エチルを蒸発することによって得た粗生成物(0.61g)を実施例22に記載のようにしてフラッシュクロマトグラフィーによって精製すると、表題化合物208mg(58%)が暗赤色ガラスとして得られる。
【0166】
【化44】
【0167】
実施例24
タキソール−2'−エライデート
タキソール(25mg、0.029mmol)を先ず最初に、ピリジン(3×1ml)への溶解および減圧下における蒸発を繰り返すことによって乾燥した。それからピリジン(1ml)に溶解すると、澄明な無色溶液が得られ、それにN−(3−ジメチルアミノプロピル)−N'−エチルカルボジイミド塩酸(9mg、0.05mmol)、DMAP(2mg、0.02mmol)、エライジン酸(10mg、0.035mmol)および無水MgSO4(3mg)を混合物として加えた。反応物をN2下で室温で48時間撹拌した。ピリジンを減圧下で蒸発して除去し、残留物をDCM(25ml)に溶解した。有機相をその後乾燥し(MgSO4)、濾過し、減圧下で蒸発すると白色固体が得られる。それをフラッシュクロマトグラフィー(SiO2;ジエチルエーテルヘキサン1:1ないし1:0勾配溶出)によって精製すると表題化合物が白色固体(25mg、77%)として得られた。
【0168】
【化45】
【0169】
ネズミ皮下ADJ/PC6形質細胞腫およびそのシスプラチン抵抗サブラインにおけるメルファラン−エライジンアミドおよびクロラムブシル−オレイルエステルのin vivo抗腫瘍効果
不飽和度の異なるクロラムブシルおよびクロラムブシル−脂肪酸結合物のヒトリンパ腫および正常ヒト末梢血リンパ球の細胞毒性がアネル(A.Anel)らのBioc hemical Pharmacology、40巻、6号、1193−1200ページ、1990、に記載されている。クロラムブシル−アラキドン酸およびクロラムブシル−ドコサヘキサエン酸のリンパ腫細胞に対する毒性はクロラムブシルまたは遊離脂肪酸の個々の毒性ポテンシャルと等しいかまたはより高い。これに対し、オレイン酸およびエライジン酸を例とする本発明の脂肪酸誘導体は親薬剤のみに比し遥かに毒性が小さい。これは下記の実験によって示される。
【0170】
ネズミ充実性ADJ/PC6形質細胞腫および、シスプラチンおよびその他のアルキル化剤に対する耐性があるということから選択されたそのサブラインを体重20−25gのBALB/C雌マウスに1mm3腫瘍断片として皮下に移植した。メルファランまたはメルファランエライジンアミド、またはクロラムブシルまたはクロラムブシル−オレイルエステルを腫瘍移植後20日目に腹腔内に単回投与した。腫瘍を30日目に切除し、対照および治療群の重量を比較した。活性はED90として測定した抗腫瘍効果と比較した薬剤毒性測定値、LD50(mg/kg)、に基づいて決められた。ED90は、対照と比較して腫瘍質量を90%減少させるのに必要な量(mg/kg)である。
【0171】
表1からわかるように、メルファラン−エライジンアミドおよびクロラムブシルオレイルエステルどちらのLD50(mg/kg)も、それぞれメルファランおよびクロラムブシルのそれらと比較して遥かに多量であった。これは毒性の減少を意味する。
【0172】
【表1】
【0173】
メルファラン−エライジン酸アミドでは感受性−およびシスプラチン耐性腫瘍どちらでもED90に達したが、メルファランはシスプラチン耐性腫瘍では全く活性を示さなかった。メルファラン−エライジン酸アミドのED90は60mg/kgであった。
【0174】
多剤耐性を有する、または有しない細胞内へのドキソルビシンおよびドキソルビシン誘導体の細胞内蓄積
腫瘍細胞は長期間化学療法後、抗癌剤に耐性を示すことがある。薬剤耐性の一つの形は、多剤耐性(MDR)である。その場合細胞はビンカアルカロイド類、アントラサイクリン類、アクチノマイシンDおよびコルヒチン等の種々の薬に交差耐性を示す。MDR表現型はP−糖タンパク質と呼ばれる特殊の群の膜通過性糖タンパク質(P−gp)の過剰発現と関係している。P−gpはエネルギー依存性薬剤排出ポンプとして機能するように見える。P−糖タンパク質は抗癌剤を細胞外に活発に排出することによって抗癌剤の細胞内濃度をその有効濃度より下に下げることができる。カルシウムチャンネル−ブロッカーであるベラパミルは、抗腫瘍薬の細胞内濃度を高めることによってMDRを元に戻すことができる。ジヒドロピリジンおよびピリジン同族体、カルモジュリン阻止物質、合成イソプレノイド類、向リソソーム剤、ビスベンジルイソキノリン−アルカロイド、キニジン、キナクリン、リドカイン、フェノキサジン、アミオダロンおよびシクロスポリンAは、細胞に同時投与した際、またはin vivoに同時投与される際にMDRを変化させる薬のその他の例である。まだ実験的レベルであるが、癌治療においては耐性変更剤の使用が一般的になりつつある。
【0175】
これらの高度に生体内活性である化合物はそれら自体問題がないわけではない。軽度ないし重度の生命を脅かす副作用が認められるため、in vitroで認められる非常に有望な結果を臨床的に得ることは不可能である。
【0176】
これらの作用物質の大部分はカチオン性且つ親油性である。親油性は耐性モジュレータにとって好ましい特徴である。リポソーム封入ドキソルビシンについてその多剤耐性克服効果を試験した。細胞内薬剤濃度はドキソルビシンのリポソームを用いると2倍になった(Cancer chemotherapy and Pharumacology、1991、28巻:259−265ページ)。リポソームのみでもMDRに影響するかも知れない(リポソームによって誘起される多剤耐性細胞内への薬剤の蓄積の増加、Cancer Research、52巻、3241−3245、1992)、(カルジオリピン、ホスファチジルイノシトール、ジオレイルホスファチジン酸のリポソーム)。
【0177】
細胞を2×105/mlの懸濁液にして、20μMの薬剤にさらした。種々の時間に部分を取り、氷冷PBSですすぎ、その後フローサイトメーターを通過させた。図11にはドキソルビシンおよびドキソルビシン誘導体の時間の関数としての蓄積が見られる。細胞系COR−L123/P(ヒト大肺細胞)およびその耐性細胞系COR−L123/Rではドキソルビシン誘導体(ドキソルビシン−エライジン−アミドおよびドキソルビシン−オレイル−カルバメート)濃度はほぼ同じであった、一方ドキソルビシン濃度は耐性細胞系では遥かに低かった。
【0178】
多剤耐性を有するまたは有しない細胞内へのダウノルビシンおよびダウノルビシ ン誘導体の細胞内蓄積
細胞を上記のようにダウノルビシン、ダウノルビシン−エライジンアミドおよびダウノルビシン−オレインカルバメートにさらした。薬剤濃度は10μMに減らした。耐性および非耐性細胞への取り込みによる蛍光は誘導体に関しては2種類の細胞で多かれ少なかれ同じであり(図12)、ダウノルビシンそのものではこれらの細胞内への取り込みによる蛍光は2種類の細胞で差があった(図13)。
【0179】
ドキソルビシン−エライジン酸アミドの同時投与によるドキソルビシンに対する 細胞の感受性増加
MTTアッセイを用いて化合物の毒性を調べた。アッセイ前の6日間、細胞を化合物にさらした。使用した細胞系はPgPを過剰発現するH69/LX4、ヒト小肺細胞系であった。その細胞系はドキソルビシン−エライジン酸アミドのみに高度に耐性であり、ドキソルビシン−エライジン酸アミドのIC50値は>50μMである。しかし驚くべきことに、ドキソルビシン−エライジン−アミドを5μM濃度でドキソルビシンと同時に投与すると、上記細胞系のドキソルビシン感受性はIC50=0.4μMからIC50=0.08μMに高まる。20μMドキソルビシン−エライジン−アミドの添加はドキソルビシン感受性をIC500.04=μMに高める。表2の結果は、この誘導体が細胞の耐性メカニズムと相互作用し、ドキソルビシンそのものの効果を強め、そして感受性細胞系のレベルにまで回復することができることを示す。
【0180】
【表2】
【0181】
上記図11−13に示したように、アントラサイクリン類ドキソルビシンおよびダウノルビシンの脂肪酸誘導体はドキソ耐性細胞系のMDRメカニズムに対して調節作用を有する。耐性細胞系に親薬剤と同時投与する際、親薬剤に対する感受性が再び感受性細胞系と同じ桁になる。このMDRモジュレータに対するこのアプローチは好都合である。なぜならば同時投与した薬剤はまさに活性化合物の誘導体であり、それはin vivoで加水分解されて活性薬剤と無毒性脂肪酸残基を遊離するからである。
【0182】
抗菌剤
この治療分野には非常に種々様々の薬剤が含まれる。
【0183】
抗菌剤の最も重要な群は多分ペニシリンであるが、薬剤耐性がますます深刻になるにつれ、細菌感染症を治療するための代替治療薬に焦点が当てられてきた。他の作用メカニズムを有する他の薬剤で治療されるとはいえ、マイコバクテリウムおよび原虫によって起きる疾患の治療においても、細菌感染症の克服に重要である因子類の幾つか、例えば細胞内取り込み、組織分布、耐性メカニズムの回避等は同様に重要である。
【0184】
この分野で使用される全ての薬剤は標的感染症に対して非常に良い〜中程度の効果を有する。この領域では通常の新薬開発の他に、より良い経口バイオアベイラビリティを有する誘導体、すなわち純粋プロドラッグ類の開発に焦点が当てられており、この研究は薬剤耐性問題にはほとんど影響をもたない。
【0185】
抗生物質の臨床的効果はその抗菌活性で決まるだけでなく、その薬物学的および薬物動態的特性によっても決まる。プロドラッグは抗生物質の安定性および溶解度を高めるために、そして親化合物の経口吸収、組織浸透性および作用持続時間を改良するために用いられている。経口投与後の血清中濃度の上昇は抗生物質の組織濃度の改善につながる。
【0186】
ペニシリンおよびその他の関連β−ラクタム抗生物質領域では、単純アルキルエステル類は安定過ぎてプロドラッグとして使用できないとしばしば言われている。好適プロドラッグは1ないし3個のメチレンリンカーまたはメトキシカルボニル アルキルエステルを有するdoubleエステルである。これらの側鎖改変は経口バイオアベイラビリティの改良を容易にし、これらの誘導体は生体内で十分に不安定で、血流中の内因性または微生物由来酵素による加水分解性代謝によって活性薬剤を遊離する。これらのペニシリン−プロドラッグは薬剤耐性状態にほとんど影響を与えない。ペニシリンおよび密接に関係する同族体に対して活性な、最も優勢で、合理的によく特徴づけられた耐性メカニズムは、細菌が獲得した、加水分解酵素β−ラクタマーゼを産生する能力である。その酵素は細胞内−および細胞外両方で見いだされる。それはその薬が実際の細菌に到達する前にすでに血流中で破壊され得ることを意味する。その他の種類の薬剤耐性は純粋な排除メカニズムによる。このメカニズムによって薬剤は細菌またはその他の微生物に入ることを阻止される。本発明の脂質誘導体は血流中ではさほど容易には加水分解されず、そのため血流中でその薬剤誘導体はよりよく循環することができる。特に、新規脂質誘導体の高い細胞内輸送および付加的効果が排除メカニズムを克服し、活性薬剤が細胞/細菌のその他のコンパートメントにおいて放出され、そこではそれは加水分解酵素には無関係にその作用をあらわすことができると考えられる。
【0187】
本発明によって誘導体化できる抗生物質およびその他の抗菌剤の若干の例は以下の化合物を含む:
【0188】
【化46】
【0189】
【化47】
【0190】
【化48】
【0191】
上に図示したように、抗菌剤は誘導化可能基を1つより多く含むことができる。これらの場合にはこれら官能基の1つ以上が本発明によって親油性基で置換でき、2つ以上の親油性基がある場合は同じ親油性基でも異なる親油性基でもよい。
【0192】
本発明の親油性抗菌化合物は既述のように同じ一般的方法によって合成できる。しかし、数種のペニシリン誘導体の選択的および効率的誘導体化は、例えば親薬剤に複数の官能基(−OH2−NH−および−NH2)が存在する等、種々の要因により、βラクタム環の開裂、化合物のその他の再構成または分解によって複雑になるかも知れない。そのため保護基および種々の試薬系を使用して、スキーム8のアンピシリン(XXV)で示されるように、選択的誘導体化を容易にすることができる。
【0193】
第一アミノ基はアシル−チアゾリジン−2−チオンによって選択的に脂肪酸アンピシリンアミド(XXVI)に変換できる。同じアミノ官能基はベンザルデヒドでシッフ塩基(XXVII)として保護される。そのカルボン酸をセシウム塩に変形し、さらに脂肪臭化物(RBr)と反応させる。軽度酸性加水分解によりアミノ基が再形成され、アンピシリン−脂肪酸エステル(XXVIII)が生成する。
【0194】
【化49】
【0195】
スキーム8
三官能価抗結核薬パラ−アミノ−サリチル酸、PAS(XXIX)、の選択的誘導体化がスキーム9に示される。PASそのものは多くの反応条件下では不安定で、自己縮合および二付加物形成が起こり得る。カルボン酸をそのセシウム塩に変形することができ、さらにメシレートとの反応によって対応するエステルに変形できる。脂肪酸塩化物は主としてアミノ基と反応し、対応するアミドを与える。フェノール基に対する反応から生ずる生成物は微塩基性加水分解により除去することができる。
【0196】
【化50】
【0197】
スキーム9
本発明による特異的抗菌化合物の製法を以下に示す。
【0198】
実施例25
パラ−アミノ−サリチル酸エライジルエステル
無水DMF50ml中パラ−アミノ−サリチル酸(PAS)(0.47g、3. 1mmol)および炭酸セシウム(0.98g、3.0mmol)の懸濁液にエライジルメシレート(1.0g、2.9mmol)を加え、反応混合物を周囲温度で60時間、35℃で48時間撹拌した。反応混合物をエーテルと水で抽出し、有機相をNaHCO3(水溶液)および水で洗った。溶媒を留去し、残留物をシリカゲルカラムで精製した。溶出系としてヘプタン/CH2Cl2/AcOH/MeOH(85:15:2:2)を用いた。生成物含有フラクションを蒸発すると表題化合物0.6g(51%)が得られた。
【0199】
【化51】
【0200】
実施例26
4−(エライドアミド)−サリチル酸
無水THF100ml中PAS(2.6g、17mmol)の溶液にTHF20ml中エライジン酸塩化物(5.11g、17mmol)の溶液を0℃で滴下した。反応混合物を周囲温度で24時間撹拌した。さらにエライジン酸塩化物(1.0g)を加えた。溶媒を高真空で蒸発し、残留物をエーテルと水との間に分配した。有機相を酒石酸(水溶液)および水で洗った。溶媒を留去し、残留物を水5mlを加えたメタノール100mlに溶解した。固体沈殿を濾去し、エタノールから再結晶すると粗材料3.9gが得られた。この材料1.5gを、1M NaOH 11ml加えたエタノール100mlに溶解した。混合物を周囲温度で2時間撹拌し、0.5M酒石酸(水溶液)30mlで酸性にした。固体沈殿を水で洗い、エーテルに溶解し、有機相を酒石酸(水溶液)で洗った。溶媒を留去し、残留物を無水クロロホルム×2から蒸発すると表題化合物1.3g(87%)が得られた。
【0201】
【化52】
【0202】
実施例27
エライジン酸クロラムフェニコールエステル
無水DMF10mlおよびピリジン2ml中D−threo−2、2'−ジクロロ−N−[β−ヒドロキシ−α−(ヒドロキシメチル)]−p−ニトロフェネチルアセタミド(0.20g、0.62mmol)の溶液に、DMF3ml中エライジル酸塩化物(0.19g、0.62mmol)の溶液を加えた。反応混合物を周囲温度で12時間撹拌した。溶媒を高真空で蒸発し、残留物を酢酸エチルと水との間に分配した。有機相を濃縮し、粗生成物をシリカケルカラムで、溶出系として酢酸エチル/ヘキサン(1:1)を用いて精製した。均質フラクションを蒸発すると表題化合物0.14g(39%)が得られた。
【0203】
【化53】
【0204】
実施例28
オキサシリン−オレイル エステル
水76ml中(5−メチル−3−フェニル−4−イソキサゾリル)ペニシリン(オキサシリン)ナトリウム塩(1.0g、2.4mmol)の溶液に、0.2MHCl(水溶液)13.1mlを加え、その混合物を蒸発した。残留物をメタノール50mlおよび水5mlに溶解した。Cs2CO3の20%溶液(水溶液)をpH7になるまで加えた。混合物を蒸発乾固した。
DMF50ml中残留物の溶液にオレイルブロミド(0.78g、2.4mmol)を加え、反応混合物を周囲温度で72時間撹拌した。溶媒を高真空中で蒸発し、残留物を水およびクロロホルムで抽出した。有機相を濃縮し、粗生成物をシリカゲルカラムで、溶媒系として酢酸エチル/ヘキサン(40:60)を用いて精製した。均質フラクションを蒸発すると表題化合物0.69g(45%)が得られた。
【0205】
【化54】
【0206】
実施例29
エライジン酸アンピシリンアミド
アセトニトリル10ml中D−(−)−(α−アミノベンジル)ペニシリン(アンピシリン)(0.10g、0.29mmol)の溶液に、3−チアゾリジン−2−チオン−エライジルアミドおよびDBU(0.043ml、0.29mmol)溶液を加え、二相反応混合物を周囲温度で72時間激しく撹拌した。溶媒を留去し、残留物を酢酸エチルと飽和塩化ナトリウム(水溶液)との間に分配した。粗生成物を分離し、メタノールに再溶解した。混合物を蒸発乾固すると、表題化合物0.1g(55%)が得られた。
【0207】
【化55】
【0208】
実施例30
アンピシリン−オレイルエステル
DMF30ml中アンピシリン(1.21g、3.5mmol)および炭酸水素カリウム(0.35g、3.5mmol)の懸濁液に、ベンザルデヒド(0.92g、8.7mmol)を加え、反応混合物を0℃で4時間撹拌した。炭酸水素カリウム(0.35g、3.5mmol)およびオレイルブロミド(1.21g、3. 7mmol)を加え、0℃で2時間、周囲温度で12時間撹拌を続けた。溶媒を高真空で蒸発し、残留物を酢酸エチルおよび冷水(0℃)で抽出した。有機相を蒸発すると黄色シロップ2.46gが得られた。
粗生成物をアセトニトリルに溶解し、1M HCl(水溶液)をpH=2になるまで加えた。水30mlを加え、アセトニトリルを留去した。生成物を酢酸エチルおよびジクロロメタンで抽出した。合一した有機相を蒸発し、粗生成物をシリカゲルカラムで溶媒系として酢酸エチル中1%トリエチルアミンを用いて精製した。均質フラクションを蒸発すると表題化合物0.8g(38%)が得られた。
【0209】
【化56】
【0210】
抗寄生虫薬
寄生虫感染はヒト医学および獣医学における重大問題である。寄生虫は通常、食物/水を介して、または昆虫が刺すことによってホストに侵入する。寄生虫は腸管(上皮細胞層)または血流に見いだされる、この場合赤血球または、その他の、肺または脳等の標的器官が感染するかも知れない。寄生虫はホストの細胞間および細胞内両方に見いだされる。原虫について言うと、寄生虫の生活環には数段階あることがよくあり、全ての段階が治療されるわけではない。
【0211】
ヒトにおける最も主要な寄生虫感染症はマラリアであり、一方動物、特に鳥(家禽)では腸感染、コクシジューム症が主な問題である。未治療状態では糞便は胞子を含み、それは動物または新たな人々の再感染に導く(他の種属とのクロスオーバーも)。
【0212】
活性薬剤を寄生虫そのものに効率的に運搬し、または例えばマラリアおよびコクシジューム症では、寄生虫感染細胞内に運搬することが重要である。
【0213】
本発明の親油性抗寄生虫誘導体は既述の一般的製法によって合成できる。
【0214】
例えば、反応スキーム10は抗マラリア薬ヒドロキシ−クロロキン(XXX)のアシル化を示す。反応は第一OH基に完全に選択的である。
【0215】
【化57】
【0216】
スキーム10
生物学的効果
選択されたヒドロキシクロロキノン誘導体の高められた抗マラリア効果を示す実験を次に示す。
【0217】
ヒドロキシクロロキノン−エライジン酸エステルのマウス−マラリアに与える効果
NK65系薬剤感受性P.bergheiの4日間試験をスイス アルビノ雌マウスで行った。ヒドロキシクロロキノン−エライデート0.063、0.25、1.0および4mg/kgおよびヒドロキシクロロキノン0.094、0.395、1.5および6mg/kgを3匹づつのマウス群に4日間腹腔内投与した。寄生虫接種物107感染細胞を0日目に静脈内投与し、その日のうちに薬剤を投与した。動物にその後3日間投与し、5日目に尾血液フィルムを作り、寄生虫血を検査した。モル−ベースで、この4日間試験においてエライジン酸誘導体はエライジン酸そのものよりも2.5−3倍効果が高かった。これらの知見はヒトにおけるマラリア治療に非常に重要である。
【0218】
【表3】
【0219】
ヒドロキシクロロキノン硫酸またはヒドロキシクロロキノンエライデートでマウス血液中のP.bergheiマラリア寄生虫を50%、90%および99%減少させる有効量がこの表に示される。
【0220】
本発明によって誘導体化できる抗寄生虫薬の例は下記を含む:
【0221】
【化58】
【0222】
マラリアは最も広くはびこっている寄生虫病であり、推定マラリア発生率は臨床症例が年間200万−500万台である。抗マラリア薬に対する獲得耐性は大きな問題となりつつある。
【0223】
ヒトは、普通、感染した雌アノフェレス蚊に刺されると、注入された(胞子虫の)種虫に感染する。その寄生虫は速やかに循環を出て、肝実質細胞に入り、そこで増殖し、組織shizontsに発達する。薬剤が肝臓内の形質胞体状の組織に作用するならば、それら薬剤は原因予防に用いられる。本発明の脂質誘導体は肝組織に親和性を有するから、これらの化合物はマラリア病の組織型により有効であり、P.vivaxおよびP.ovaleの肝臓型を根絶するかも知れない。これらの型は生き残り、後に増殖し、最初の感染の数カ月ないし数年後に赤血球感染を再発させる。
【0224】
P.falciparumはヒトマラリアの症例の85%以上を占める。P.falciparumの耐性種は薬剤を十分に高い濃度まで蓄積しない。Ca2チャンネルブロッカーはクロロキンのような薬に対する感受性を一部回復する。多数の化学的に関係ない薬に対する交差耐性は、悪性腫瘍に見られる多剤耐性と同様である。
【0225】
脂肪酸はそれら自体が抗マラリア効果を有するらしい(クルグリアク(Krugli ak)ら、実験寄生虫学81巻、97−105ページ、1995)。オレイン酸、エライジン酸、リノレイン酸のような脂肪酸がPlasmodium vinckei petteriまたはPlasmodium yoelii nigeriensisに感染したマウスにおける寄生虫血の発生を阻止した。
【0226】
しかし、ヒトにおいて同様な効果を得るのに必要な細胞内濃度は、非現実的多量の脂肪酸の摂取を必要とする。
【0227】
ここでも本発明の実施態様に基づき、抗マラリア薬誘導体が驚くほど効率的に高濃度で細胞内寄生虫に運搬され、薬剤耐性メカニズムを克服することさえできる。
【0228】
本発明の抗マラリア誘導体の製法を示す実施例を以下に記す:
【0229】
実施例31
7−クロロ−4−[4−[エチル(2−エライドイルオキシエチル)アミノ]1−メチルブチルアミノ]−キノリン
ジクロロメタン30ml中7−クロロ−4−[4−[エチル(2−ヒドロキシエチル)アミノ]1−メチルブチルアミノ]キノリン(ヒドロキシクロロキン)(3.17g、9.4mmol)の溶液に、エライジン酸塩化物(2.82g、9. 4mmol)を加え、反応混合物を周囲温度で48時間撹拌した。少量のメタノールを加え、溶媒を高真空で蒸発した。残留物をシリカゲルカラムで反復精製した(第一行程:クロロホルム/メタノール9:1、第二行程:クロロホルム/メタノール95:5)。均質フラクションを蒸発すると表題化合物1.18g(21%)が得られる。
【0230】
【化59】
【0231】
本発明によって誘導体化できるその他の領域内の薬の例をさらに幾つか挙げる。
【0232】
中枢神経系薬
【0233】
【化60】
【0234】
心臓血管薬
【0235】
【化61】
【0236】
下記の一般的反応スキームは、本発明によるワルファリンおよびセロケンの誘導体の製法を示す。
【0237】
スキーム11は抗凝固剤ワルファリン(XXXI)のアシル化を示す。
【0238】
【化62】
【0239】
スキーム11
セロケン(XXXII)のヒドロキシ基の選択的アシル化はアミノ官能基の存在によって複雑になる。アミノ官能基をBOC誘導体として好適に保護し、脂肪酸塩化物の使用によってOH基を変形する。これらの反応はスキーム12に示される。
【0240】
【化63】
【0241】
スキーム12
反応スキーム11の特殊な実施例を次に示す。
【0242】
実施例32
3−(α−アセトニルベンジル)−4−エライドイルオキシクマリン
無水ジオキサン120mlおよびピリジン25ml中3−(α−アセトニルベンジル)−4−ヒドロキシクマリン(ワルファリン)(3.70g、12mmol)の溶液にエライジン酸塩化物(3.60g、12mmol)を加え、反応混合物を周囲温度で4時間撹拌した。溶媒を高真空で蒸発した。残留物をエーテルと水との間に分配した。有機相を酒石酸(水溶液)、NaHCO3および水で洗った。乾燥した有機相を濃縮し、生成物をシリカゲルカラムで、溶出系としてヘプタン/酢酸エチル(6:1)を用いて精製した。不純なフラクションを再精製し、均質フラクションを蒸発すると表題化合物が淡黄色油として5.1g(70%)得られる。
【0243】
【化64】
【0244】
上述のように、本発明はヒトまたは動物医学に有用な化合物だけでなく、あらゆる種類の生物学的活性化合物に一般的に適用できる。特に、アルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸およびカルボン酸エステル基から選択される1つ以上の官能基を有する農薬を本発明によって誘導体化できる。このような農薬および園芸用化学薬品の例は次のものを含む:
【0245】
【化65】
【0246】
【化66】
【0247】
【化67】
【0248】
【化68】
【0249】
【化69】
【技術分野】
【0001】
本発明は生物学的活性化合物に関するものであり、薬剤および農薬等の多くの生物学的活性化合物の作用を化学的誘導体化によって改良する方法を提供する。
【背景技術】
【0002】
医学界では患者の作用部位への薬剤運搬効果の研究、改良に多大の関心がもたれている。本研究は主として薬剤の腸から血流への吸収に焦点を当てている。但しその他の生物学的障壁を通過する運搬も、多くの疾患、例えば癌、感染症、炎症、CNS疾患等の治療に必要な治療効果を得るために重要な役割を演ずることは多い。細胞膜経由輸送は或る治療化合物の最適効果達成を妨害する主原因であることが多い。
【0003】
最近数十年にわたり、悪性および感染性疾患の治療における薬剤耐性がますます増えており、今や重大な臨床的問題とみなされている。薬剤耐性の発現は多くのメカニズムによって起こるが、微生物および細胞が毒性化合物を有毒レベル以下にまで除去する正常メカニズムがトリガーされることによることが非常に多い。一例は、癌細胞における多剤耐性(MDR)の発現である。この場合、MDRは、細胞が有毒化合物の非常に効率的な流出を実現する細胞膜タンパク質ポンプに関係することがよくある。臨床的立場、すなわち細胞増殖抑制剤で腫瘍を治療する場合には、最も強力なタンパク質ポンプを有する細胞が優先的に生き残り、これらの細胞は増殖して、種々の薬剤による治療に耐性を有する新しい腫瘍になるかも知れない。同様な作用メカニズムは、その他の治療領域、例えば抗マラリヤ剤による治療等で見られる不十分な効果の原因になるかも知れない。
【0004】
臨床における耐性メカニズムを回避することを試みる幾つかの方法が知られている。例えばベラパミル等のCa2+チャンネルブロッカーまたはシクロスポリンのような免疫調節剤との併用が試みられた。しかし顕著な改善はこれまで報告されていない。
【0005】
文献には、化合物類を脂肪酸と結合させて化学的結合誘導体または物理的混合物のどちらかを形成することによって、治療化合物の治療指数、バイオアベイラビリティー、膜通過性、器官標的化などを改善する幾つかの提案がある。
【0006】
例えば、欧州特許出願第393920号は、長鎖(C16以上)アシル基で誘導体化した抗ウィルス性ヌクレオシドおよびヌクレオシド類似体が親化合物より有効であることを開示している。これら分子の脂肪酸部分は、γ−リノレン酸またはリノール酸等のポリ不飽和脂肪酸から形成されることが記載されている。
【0007】
米国特許出願第3920630号は、2、2'−無水−アラシチジンおよびその5'−O−アシレート類が、抗ウィルス薬として、アラシチジンそのものと同様な全般的生物学的効果および治療効果を有することを教示している。特に化合物2、2'−無水−5'−オレイル−アラシチジンについて記載されている。
【0008】
欧州特許出願第56265号は、アラビノ−フラノシル−チミン(Ara T)と1−17炭素原子を有する飽和酸とのエステル類を開示している。
【0009】
PCT/WO90/00555からは、特に燐酸基を介してヌクレオシドのペントース基の5'−位置に結合した脂質誘導体が公知である。この誘導体化の目的は、ヌクレオシド類をより親油性にして、リポソームがそれらを含むことができるようにすることである。リポソームはHIVウィルスを担持することが知られている細胞であるマクロファージおよび単球に優先的に取り込まれる。そこで標的化効果が得られると記載されている。
【0010】
ヌクレオシド類似体の抗ウィルスおよび抗癌活性は、投与薬剤の細胞内燐酸化に直接関係している。この生化学的変換は通常はウィルスおよび/または細胞内酵素によって行われる。この効果を改善するために、WO96/25421は比較的短い鎖(C14以下)を有する飽和または不飽和脂肪酸によるヌクレオシドの燐脂質誘導体を開示している。
【0011】
その他のクラスの薬物学的物質の特性を、脂肪酸による誘導体化によって改良する方法も研究されている。
【0012】
例えばWO96/22303は、数種のカテゴリーの治療化合物(コルチコステロン、オピオイド、およびオピオイド拮抗薬、抗ウィルス性ヌクレオシド類、シクロスポリン類および関連シクロペプチド類、葉酸拮抗薬、カテコールアミン前駆体およびカテコールアミン類、およびカルボン酸基を含むアルキル化剤)の薬物動態プロフィールおよびデリバリーモードを、トロメタミンまたはエタノールアミン誘導体等のリンカー/スペーサー基を使用して脂肪酸の1ないし3個のアシル誘導体とこれらとを結合することによって変え得ることを教示している。パルミチン酸は好適脂肪酸である。
【0013】
数種のNSAIDsの親油性プロドラッグはブンドガード(H.Bundgaard)ら(International Journal of Pharmaceutics、43巻、101−110ページ、1988)およびシャンバグ(V.R.Shanbhag)ら(Journal of Pharmaceutical Sciences、149、81巻、2号、1992年2月)から公知である。プロドラッグ的観点に加えて、GI(胃腸)刺激が減少することが報告されている。EP−A−0195570は、ガンマリノレン酸およびジホモ−ガンマ−リノレン酸とNSAIDsとの組み合わせが連続的に摂取した場合のNSAIDsによる副作用を軽減することを示唆している。
【0014】
米国特許第5,284,876号は、CNS疾患治療において経口プロドラッグとしてドーパミンのドコサヘキサエン酸アミドを使用することを教示している。
【0015】
皮膚および経口投与どちらでもいわゆる浸透促進剤として用いられる脂肪酸/脂肪酸誘導体を含む物理的混合物がPCT/US94/02880およびPCT/SE96/00122から公知である。
【発明の概要】
【発明が解決しようとする課題】
【0016】
上記のように、これら先行提案の多くは抗ウィルス性ヌクレオシドおよびヌクレオシド類似体の脂肪酸誘導体に関係する。実際、或る種のポリ不飽和脂肪酸がウィルスを攻撃することは以前から知られているから、こうなることは驚くにあたらない。EP−A−0642525において、我々自身、ヌクレオシドおよびヌクレオシド誘導体がオレイン酸(cis−9−オクタデセン酸)、エライジン酸(trans−9−オクタデセン酸)、cis−11−エイコセン酸またはtrans−11−エイコセン酸と反応して対応する5'−O−モノエステルを形成することによって上記ヌクレオシドおよびヌクレオシド誘導体の抗ウィルス効果が著しく高まることを教示した。我々は、これら4種類のモノ不飽和、ω−9 C18またはC20脂肪酸から得られる好都合な効果が脂肪酸誘導体化によって一般に得られるものより優れていることを示した。
【課題を解決するための手段】
【0017】
今や驚くべきことに、本発明により多くの異なる生物学的活性化合物の性質を ω−9 C18またはC20モノ不飽和脂肪酸による誘導体化によって好都合に変え得ることを我々は見いだした。こうして本発明は、例えば多くの薬剤および農薬の価値を高めるために広く、かつ簡単に利用できる方法を提供する。
【0018】
概して本発明は、一面では分子構造中にアルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸およびカルボン酸エステル基から選択される1つ以上の官能基を含む、ヌクレオシドまたはヌクレオシド誘導体以外の生物学的活性化合物の親油性誘導体を提供する。上記親油性誘導体は前記生物学的活性化合物の少なくとも一つの前記官能基が、下記の式であらわされるものから選択される親油性基によって置換された分子構造によって、特徴づけられる:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2RN−CONHCH2Rおよび−SCH2R。前記式中、Rはcis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニルおよびtrans−10−ノナデセニルから選択される親油性部分である。
【図面の簡単な説明】
【0019】
【図1】図1は肉芽腫の液体重量を示す。リポソーム投与対照動物の肉芽腫の平均液体含有量は62.69mgであった。減少は全投与群に認められた(ナプロキセン12%、ナプロキセンオレイルエーテル9%、ナプロキセンオレイルエステル14%およびナプロキセンオレイルアミド22%)。ナプロキセンオレイルアミドで得られた結果が特に顕著であった。
【図2】図2は肉芽腫組織乾燥重量を示す。リポソーム投与対照動物の肉芽腫の組織乾燥重量は14.36mgであった。ナプロキセンは組織乾燥重量に影響を与えないように見えた。残る投与群では減少が認められた(ナプロキセンオレイルエーテル16%、ナプロキセンオレイルエステル12%およびナプロキセンオレイルアミド38%)。ここでもナプロキセンオレイルアミドで得られる減少が最大であった。
【図3】図3はマウスの皮下に2週間埋め込まれたコットン−ラップド軟骨のグリコサミノグリカン含有量を示す。埋め込まれなかった対照軟骨は平均グリコサミノグリカン1168mgであった。リポソーム投与対照動物に2週間移植するとグリコサミノグリカンは60%減少した。ナプロキセンオレイルエーテル投与動物から取り出したインプラントを除き、残る投与群のインプラントはリポソーム投与対照群のそれらより少ないグリコサミノグリカンを含む傾向があった(ナプロキセン16%、ナプロキセンオレイルエステル12%およびナプロキセンオレイルアミド11%)。
【図4】図4はマウスの皮下に2週間埋め込まれたコットン−ラップド軟骨のヒドロキシプロリン含有量を示す。埋め込まれなかった対照軟骨は平均ヒドロキシプロリン含有量329mgであった。リポソーム投与対照動物に2週間移植するとヒドロキシプロリンは19%減少した。ナプロキセンオレイルエーテル投与動物から取り出したインプラントを除き、残る投与群からのインプラントはリポソーム投与対照群のそれらより少ないヒドロキシプロリンを含む傾向があった(ナプロキセン12%、ナプロキセンオレイルエステル8%、ナプロキセンオレイルアミド3%)。
【図5】誘導体の効果は明らかで、驚くべきものである。図5からわかるように、プレドニゾロンがチモサン刺激−細胞の活性に与える影響は6時間目の化学ルミネッセンスのわずかな減少として認められるだけである。プレドニゾロンエライデートでは、炎症細胞の活性は投与後48時間にわたって対照に比較して減少する。その効果はプレドニゾロンそのものの効果より明らかに大きく、持続する。
【図6】誘導体類の効果は明らかで驚くべきものである。図6からわかるように、特に好ましい脂肪酸の一つの例として、プレドニゾロン−エライデートの効果は最大であった。その他の脂肪酸誘導体では化学ルミネッセンスはほんのわずかな影響を受けただけである。プレドニゾロン−エライデートでは炎症細胞の活性は投与後48時間にわたり、対照および残りの脂肪酸誘導体に比較して減少する。
【図7】図7に示すように、腹腔洗浄液中の細胞数は実質的に減少し、この減少は投与後48時間まで明らかであった。
【図8】細胞を分別すると、腹腔洗浄液中のマクロファージの数に主な影響が出ることがわかった。
【図9】プレドニゾロンではその効果は遥かに少なく、6時間および12時間目に認められただけである。ベタメタゾンとベタメタゾン−エライデートとの比較でも、腹腔洗浄液中のマクロファージの数の測定で、さほど明らかではないが、同様な効果が認められた。
【図10】気道反応性。
【図11】細胞を2×105/mlの懸濁液にして、20μMの薬剤にさらした。種々の時間に部分を取り、氷冷PBSですすぎ、その後フローサイトメーターを通過させた。図11にはドキソルビシンおよびドキソルビシン誘導体の時間の関数としての蓄積が見られる。細胞系COR−L123/P(ヒト大肺細胞)およびその耐性細胞系COR−L123/Rではドキソルビシン誘導体(ドキソルビシン−エライジン−アミドおよびドキソルビシン−オレイル−カルバメート)濃度はほぼ同じであった、一方ドキソルビシン濃度は耐性細胞系では遥かに低かった。
【図12】細胞を上記のようにダウノルビシン、ダウノルビシン−エライジンアミドおよびダウノルビシン−オレインカルバメートにさらした。薬剤濃度は10μMに減らした。耐性および非耐性細胞への取り込みによる蛍光は誘導体に関しては2種類の細胞で多かれ少なかれ同じである。
【図13】ダウノルビシンそのものではこれらの細胞内への取り込みによる蛍光は2種類の細胞で差があった。
【発明を実施するための形態】
【0020】
本発明の好適一実施態様において、治療化合物をn−9 C18またはC20モノ不飽和脂肪酸で誘導体化することによって、上記治療化合物の生物学的効果は改善される。以下に、下記の群から選択される薬剤類に本発明を適用することに関して詳細に論議する:
1.抗癌剤;
2.抗炎症剤;
−NSAIDs
−副腎皮質ステロイド類
3.抗生物質およびその他の抗菌剤;
4.抗寄生虫薬;
5.CNS薬;
6.心臓血管薬;
7.抗凝固剤。
【0021】
しかし本発明は、薬物学的に活性であり、分子構造中にn−9 C18またはC20モノ不飽和脂肪酸と結合できる官能基を1つ以上含むいかなる化合物にも広く応用できる。例えば、本発明を用いて、鎮痛剤、殺真菌薬、高脂血症薬、制吐剤および診断薬等の医学的化合物の生物学的効果を改善することもできる。
【0022】
本発明による治療的活性化合物の親油性誘導体は、熟練せる当業者には公知の一般的方法によって、薬物学的に容認される担体および賦形薬と共に処方される。用量の割合は親薬剤のそれと関係する。但し本発明の親油性誘導体が親薬剤の効果を著しく増強する場合にはその用量を通常レベルより減らすことができる。
【0023】
本発明の好都合な効果は十分確立された薬剤類で証明されたとはいえ、同様な改良はまだ開発途上にあるその他の薬剤類でも示されるかも知れないと考えられる。すなわち、本発明の親油性誘導体で認められた改良特性の仮定的説明は一般的に適用されるものであって、治療的効果の特別のメカニズムに限られるものではない。
【0024】
本発明による治療的活性化合物の親油性誘導体の幾つかが示す、我々が発見した特に貴重な特性は、それらが薬剤耐性を克服することである。理論によって束縛されるものではないが、本発明の親油性誘導体は何らかの方法で膜タンパク質ポンプと相互作用し、それらの細胞からの活性(毒性)化合物の除去が阻止され、その結果活性化合物の濃度を長期間治療的有効濃度に保つことが可能となる。少なくとも本発明は、本発明による親薬剤およびその親油性誘導体の同時投与によって薬剤耐性効果を克服する可能性にも通じる。親薬剤とその親油性誘導体は容易に投与できるように、同じ製剤中に含まれるのが好適である。但し若干の場合には親薬剤と親油性誘導体とを別々の投与型に含めるのも好ましい。親薬剤の用量に対する親油性誘導体の用量は適切な試験によって決められるが、一般には、重量で1:1から1000:1までの範囲である。
【0025】
前にも述べたように、本発明は普通は薬剤だけでなく、生物学的活性を有するいかなる化合物にも適用できる。
【0026】
生物学的活性化合物の別の経済的重要な群は、農業および園芸に用いられる生成物、例えば殺虫剤、殺真菌薬および除草剤等である。農薬は構造および作用モード共に非常に種々様々である。例えば、幾つかのよく知られた取り込み経路がある;例えば植物は活性化合物を根系から、または直接植物の葉または幹から摂取することができ、一方殺虫剤は、害虫が襲う植物を介して、または直接接触によって取り込まれる。本発明による農薬の親油性誘導体は植物によっても昆虫およびその他の害虫によっても取り込まれやすい特性を有することが判明している。その上、本発明の誘導体類は、薬剤耐性のように問題になりつつある殺虫剤耐性の克服にも役立つ。
【0027】
本発明によって好都合に誘導体化できる生物学的活性化合物のその他の群は、食物および食品添加物、例えば保存料、香料およびスパイス等を含む。
【0028】
本発明の親油性誘導体は、親薬剤またはその他の生物学的活性化合物分子を、炭素原子18または20の炭素鎖を有するcis−またはtrans−n−9モノ不飽和脂肪酸、脂肪酸アルコールまたは脂肪アミンと、またはこのような脂肪酸、脂肪アルコールまたは脂肪アミンの反応性誘導体、例えば酸塩化物、反応性エステル類、ハロゲン化物などと反応させることによって合成される。n−9とは、脂質部分のC末端から数えて9および10の位置が不飽和であることを示す。このため、使用できる脂肪酸類(およびそれらから誘導されたアルコールおよびアミン類)はcis−9−オクタデセン酸(オレイン酸)、trans−9−オクタデセン酸(エライジン酸)、cis−11−エイコセン酸およびtrans−11−エイコセン酸である。
【0029】
親生物学的活性化合物と脂肪酸、脂肪アルコールまたは脂肪アミン化合物とのカップリング反応は、熟練せる当業者に公知の種々の方法によって達成できる。親分子中に2つ以上の誘導体化可能の官能基が存在する場合、カップリング段階において保護基または改良合成法を用いて必要な選択を行うことができる。一般的に、反応の進行を薄層クロマトグラフィー(TLC)および適した溶媒系を用いて追跡することができる。TLCによって確認して反応が完了したとき、生成物を有機溶媒で抽出し、クロマトグラフィーおよび/または適切な溶媒系からの再結晶によって精製するのが一般的である。親出発原料にヒドロキシル、アミノ、チオールまたはカルボキシル基が1つより多く存在する場合は、アルキル化またはアシル化化合物の混合物が生成するかも知れない。その後個々のモノ−またはポリ−誘導体化合物は例えばクロマトグラフィーによって分離することができる。
【0030】
カップリング反応は一段階で行われることが多く、親油性誘導体は普通は良い安定性を有する結晶として回収できる。このことは最終的医薬品を上首尾にガレノス処理するために役立つ。
【0031】
本発明によって用いられる調製法は以下に示す反応スキーム、並びに本明細書の後部で述べる実施例によって説明される。
【0032】
ここで本発明を数種類の薬剤についてさらに詳細に説明する。
【0033】
抗炎症剤
リウマチ性関節炎、骨関節炎、ベクテリエフ症候群、全身性紅斑性狼瘡(SLE)、喘息、通風等の重大な疾患の多くは炎症性反応を引き起こす異常免疫応答の結果である。炎症性プロセスは、例えば抗原−抗体相互作用、感染性作用物質、虚血等の多数の刺激によって誘起される一連の事象を含む。肉眼的レベルでは、その反応は紅斑、浮腫、敏感(痛覚過敏)、および疼痛という臨床症状を伴うのが普通である。炎症性疾患は主として3種類の薬、すなわちNSAIDs(アスピリン様薬剤と呼ぶこともある)、免疫抑制剤(例えばメトトレキセート、シクロホスファミドおよび最近ではシクロスポリン)および副腎皮質ステロイド類(ヒドロコーチゾン、プレドニゾロン、等)で治療される。その治療は主として疾患初期の疼痛および/または強度を抑制する。現在の治療法は高用量および/または長い治療期間によるひどい副作用によって制限されることが多い。
【0034】
可逆的気道閉塞−喘息−は呼吸疾患の最も一般的なものである。気管支過反応性の程度は普通は副腎皮質ステロイドおよび/または気管支拡張剤の定期的吸入によってコントロールまたは軽減される。大部分の治療法は平均して2−3時間の治療効果をもたらす。大部分の呼吸器エアゾール類では、吸収は経腸または経口投与による吸収とほぼ等しい。抗炎症効果および免疫抑制効果による治療は一時的である。長期治療のためには副作用を抑制した最小量を用いるべきである。
【0035】
重症喘息発作ではメチルプレドニゾロン琥珀酸ナトリウムを静脈注射し、その後10日間経口投与する。喘息の急性悪化は経口コルチコステロイドの短期間コースで治療することが多い。気管支喘息を治療する治療法に吸入コルチコステロイドを含めるやり方が最近明らかに増加している。ベクロメタゾン・ジプロピオネート、トランシノロンアセトニドまたはフルニゾリドは経口コルチコステロイドコースの期間を短縮し、或いは完全に経口コルチコステロイドの代わりとなる。これらの薬剤を推奨用量で用いる際にはより少ない副腎機能抑制が認められる。
【0036】
慢性喘息の局所治療におけるステロイド使用は吸入器によって便利に行われる。これは全身的投与のひどい副作用のリスクを制限する。局所投与後の速やか且つ選択的作用を確実にするためには、気管支の内皮細胞上の受容体への付加および前記受容体との相互作用が必須である。本発明の脂肪酸誘導体は局所投与から得られる利点をより一層改善することができる。気道炎症は致命的喘息発作の顕著な特徴と見なされ、軽度の喘息発作の気管支生検でも同様な変化が見いだされる。重症喘息発作は炎症細胞、主として肺胞マクロファージの気道への多量流入と関係がある。この状態はウィルスによって誘起される気道過剰反応性の外観に非常に似ている。マクロファージは反応性酸素種を放出することができ、それらは肺抵抗を高め、ヒスタミンを遊離させる。一般には炎症細胞、特にマクロファージの抑制を測定するモデルを用いて、可能性のある喘息薬の効果を評価することができる。化学ルミネッセンスを反応性酸素種の放出の尺度として用いることができる。下に示すように、炎症細胞、主にマクロファージのラット腹腔内への流入を本発明の副腎皮質ステロイド誘導体によって減らすことができる。刺激時の炎症細胞の活性も減少し、誘導体投与後、親薬剤投与後よりもより長期間にわたってその減少が認められる。それら誘導体は投与後最低48時間は有効である。この長時間にわたる活性は喘息治療において非常に好都合である。
【0037】
天然ホルモン類はin vivoでは非常に速やかに分解することが多いため、注射しない限り治療効果はほとんど得られないことが多い。これらの分子または天然化合物に類似した合成類似体を本発明の脂肪酸と結合することによって薬物動態挙動を変え、治療効果を改善することができる。これは全身投与および局所投与両方に言えることである。
【0038】
本発明によって誘導体化し得る副腎皮質ステロイド類およびその他の喘息薬の例は次のものを含める:
【0039】
【化1】
【0040】
【化2】
【0041】
炎症性疾患の治療に最も一般に用いられる薬剤はNSAIDsである。よく使用されるこの群の製品はいろいろあり、主なものはナプロキセン、ジクロフェナック(ボルタレン)、ピロキシカム(フェルデン)およびサリチル酸誘導体である。NSAIDsは抗炎症性、鎮痛性および解熱剤であるが、それらの主要な臨床的用途は炎症性疾患の治療である。NSAIDsはこれらの疾患による痛みおよび炎症の症状緩和をもたらすが、ひどい発症中の組織の病的損傷の進行は止めない。今日知られているNSAIDs製剤のなかで肉芽腫性組織の生成を明らかに軽減するものはない。肉芽腫性液含有量の減少は認められるとはいえ、その効果は肉芽腫性固体物質含有量の同時減少としては反映されない。これら薬剤の主な作用モードはプロスタグランジン生合成の阻止(シクロオキシゲナーゼの阻害)である。各薬剤の分布および薬物動態特性は薬剤活性に重要な影響を有する。これも、同じ化学ファミリーであっても、異なるNSAIDs薬剤に対しては各患者の応答性に大きなばらつきが起きる理由であると考えられる。例えば、異なるプロピオン酸誘導体に対する耐性に大きいばらつきがあることが報告されている。
【0042】
NSAIDsの主な特性はそれらのシクロオキシゲナーゼ阻止能力であり、したがってPGG2およびPGH2およびこれらから誘導される全てのエイコサノイド類(PGI2、TXB2、PGE2等)の生合成を阻止する。他方NSAIDsがリポキシゲナーゼを(少なくとも同程度に)阻止することは知られておらず、そのためロイコトリエン類(LTB4およびLTC4)の合成には影響しない。プロスタグランジンPGI2およびPGE2は炎症過程に重要な役割を演ずる。それらは浮腫をおこし、多分血管透過性を高める。PGI2は炎症性疾患に関連する痛みの主な要因である。ロイコトリエン類は炎症発作の第二および第三相における重要なメディエータである。そしてNSAIDsはリポキシゲナーゼを治療的に役立つ程度には阻止しないから、それらは炎症性疾患の変性部には影響しない。
【0043】
NSAIDsには副作用があり、それらは時には重症になり得る。最も一般的な副作用は胃潰瘍または腸潰瘍の誘発傾向であり、したがって痛み、吐気、胸焼けおよび時には出血および貧血をおこす。これらの効果はプロスタグランジン生合成の阻止と相関するものである。PGI2およびTXB2が存在しないため、血小板は凝集能力を失い、それはそれで出血時間を長引かせる。多くの症例で、NSAIDsがリウマチ性疾患の進行には好ましい効果を与えないことは明らかであり、状況によっては疾患プロセスを悪化さえすることを示唆する証拠がある。これは軟骨分解の増強による重度の基質喪失としてあらわれる。
【0044】
その他の副作用、例えば塩および水分の貯留、高カリウム血症および腎血流減少等もプロスタグランジン合成阻害と関連し、治療を不可能にすることがある。若干の患者においてアナフィラキシーショックをおこすこともあるアスピリン過敏症では、アスピリン様薬剤による治療はできない。
【0045】
親NSAIDsは、非ステロイド性抗炎症剤として分類され、アルコール、エーテル、フェノール、アミノ(第一、第二または第三)、アミド、チオール、カルボン酸およびカルボキシルエステル基から選択される1つ以上の誘導体化可能基を有するいかなる化合物でもよい。このクラスの現在公知のNSAIDsは下記の化合物を含む:
【0046】
【化3】
【0047】
【化4】
【0048】
【化5】
【0049】
【化6】
【0050】
【化7】
【0051】
【化8】
【0052】
【化9】
【0053】
上に示したように、既知NSAIDsの多くは上に定義した種類の誘導体化可能基を一つより多く含む。これらの場合、これらの官能基の1つ以上が本発明による親油性基によって置換することができ、2つ以上の親油性基がある場合、これらは同じ親油性基でも異なる親油性基でもよい。
【0054】
本発明の親油性抗炎症剤誘導体は親薬剤と炭素原子18または20の鎖長を有するcisまたはtrans n−9モノ不飽和脂肪酸、脂肪アルコールまたは脂肪アミンとの反応、またはこのような脂肪酸、脂肪アルコールまたは脂肪アミンの反応性誘導体、例えば酸塩化物、反応性エステル、ハロゲン化物およびメシレート等との反応によって合成される。n−9とは、脂質部分のC末端から数えて9および10位置の間に不飽和があることを示す。例えば使用し得る脂肪酸(およびそれらから誘導されるアルコール類およびアミン類)はcis−9−オクタデセン酸(オレイン酸)、trans−9−オクタデセン酸(エライジン酸)、cis−11−エイコセン酸およびtrans−11−エイコセン酸である。
【0055】
親薬剤と脂肪酸、脂肪アルコールまたは脂肪アミン化合物との間のカップリング反応は熟練せる当業者には公知の種々の方法によって達成できる。親薬剤に2つ以上の誘導化可能の官能基が存在する際には、保護基または改良合成法を用いてカップリング段階に必要な選択性を得ることができる。
【0056】
一般的に、反応の進行は薄層クロマトグラフィー(TLC)および適した溶媒系を用いて追跡することができる。TLCによって確認して反応が完了したときに、生成物を有機溶媒で抽出し、クロマトグラフィーおよび/または適切な溶媒系からの再結晶によって精製するのが一般的である。NSAIDs出発原料にヒドロキシル、アミノ、チオールまたはカルボキシル基が1つより多く存在する場合は、アルキル化またはアシル化化合物の混合物が生成するかも知れない。その場合は個々のモノ−またはポリ−誘導体化合物を例えばクロマトグラフィーによって分離することができる。
【0057】
本発明によって用いられる合成法は下に与えられる反応スキーム並びに本明細書の後部に与えられる実施例によって説明される。
【0058】
第一の反応スキームはサリチル酸の誘導体化を説明する。
【0059】
【化10】
【0060】
スキーム1
サリチル酸ナトリウムを脂肪アルコールメシレート(R'−OMS)で処理すると、サリチル酸エステル(I)が生成する。この反応の変法はサリチル酸エステル−2−エーテル(II)を与える。ここでエステルおよびエーテル中の炭化水素残基は同じである。この生成物は、サリチル酸エチル(IV)をアルキル化してエチルサリチレート−2−エーテル(V)を生成し、その後そのエチルエステルの加水分解によってサリチル酸−2−エーテル(III)を得るという方法でより好適に得られる。
【0061】
これらの方法の組み合わせにより、式IIであらわされる、エステル置換基およびエーテル置換基が異なる二付加物の合成が可能となる。
【0062】
第二の反応スキームはナプロキセンの誘導体化を説明する。
【0063】
【化11】
【0064】
スキーム2
ナプロキセンエステル(VII)またはアミド(VIII)誘導体を、N,N '−ジシクロヘキシルカルボジイミド(DCC)またはO−(1H−ベンゾトリアゾール−1−イル)−N,N,N',N'−テトラメチルウロニウムテトラフルオロボレート(TBTU)のようなカップリング試薬を用いてナプロキセン(VI)と対応するアルコールまたはアミン(R'−OHまたはR'−NH2)から合成する。
【0065】
長鎖エーテル同族体(XII)はナプロキセン(VI)から、最初に芳香族6−メチルエーテルを脱メチル化して生成物(IX)を与え、その後プロピオン酸側鎖をエステル化する(X)方法で合成される。フェノール部分のアルキル化(XI)およびエチル−エステルの加水分解は生成物(XII)を与えた。これらの方法の組み合わせは、エーテルおよびエステルまたはアミドの炭化水素残基が異なるかまたは同じである二付加物を与えることができる。
【0066】
第三の反応スキームはピロキシカムの誘導体化を説明する。
【0067】
【化12】
【0068】
スキーム3
ピロキシカムエステル(XIV)をピロキシカム(XIII)および対応する脂肪酸塩化物(R'COC1)から合成する。ピロキシカムのアミド窒素のアシル化が可能であり、少量のN−アシル化生成物並びにジアシル化生成物が分離される。主要生成物(XIV)の同定は改良NMR法によって行われる。
【0069】
第四の反応スキームはジクロフェナックの誘導体化を説明する。
【0070】
【化13】
スキーム4
ジクロフェナックエステル(XVI)またはアミド(VXII)はジクロフェナック(XV)および対応するアルコールまたはアミン(R−OHまたはR−NH2)から、DCCまたはTBTUのようなカップリング試薬を用いて合成される。異性体アミド(XVIII)は対応する脂肪酸R'−COOHとXVから、カップリング試薬としてTBTUを用いて作ることができる。
【0071】
第五反応スキームは、ベタメタゾン(XIX)およびプレドニゾロン(XX)の誘導体化を説明する。多くのステロイドには第一、第二および第三アルコール機能があり、それら全てをエステルに変換することができる。しかし適度に良い選択性があり、第一アルコールはカップリング試薬としてDCCを用いて、或いは脂肪酸塩化物の直接使用によってエステル化される。
【0072】
【化14】
【0073】
スキーム5
本発明のチオ誘導体類は上記反応スキームによって示されるものと類似の方法によって合成される。
【0074】
本発明による特異的親油性抗炎症剤誘導体の製法はこの後の実施例によって説明され、実施例6および7は中間体化合物の製法を説明する。
【実施例】
【0075】
実施例1
2−ヒドロキシ安息香酸−(cis−9'−オクタデセニル)エステル
無水N,N'−ジメチルホルムアミド40ml中水素化ナトリウム(60%)(0.21g、5.25×10-3mol)の懸濁液に、2−ヒドロキシ安息香酸(サリチル酸)(0.726g、5.25×10-3mol)を加え、混合物をN2 下で80℃で1時間撹拌した。cis−9−オクタデセノール−メシレート(1 .82g、5.25×10-3mol)を加え、22時間撹拌し続けた。冷やした反応混合物を濃縮し、残渣を100mlクロロホルムに溶解した。有機相を水で洗い、炭酸水素ナトリウムおよびブラインで希釈した。乾燥した相を蒸発乾固し、粗生成物をシリカゲルカラムで精製した。その際溶出系としてヘキサン中5%エーテルを用いた。均質フラクションを蒸発すると表題化合物1.3g(64%)が得られた。
【0076】
【化15】
【0077】
実施例2
2−(cis−9'−オクタデセノキシ)−エチル−ベンゾエート
無水N,N−ジメチルホルムアミド40ml中水素化ナトリウム(60%)(0.206g、5.15×10-3mol)の懸濁液に2−ヒドロキシ−エチル−ベンゾエート(0.86g、5.15×10-3mol)を加え、その混合物をN2下で80℃で1時間撹拌した。cis−9−オクタデセノール−メシレート(1. 78g、5.15×10-3mol)を加え、40時間撹拌し続けた。冷やした反応混合物を高真空で蒸発し、残留物をクロロホルムおよび水で処理した。乾燥した有機相を濃縮し、粗生成物をシリカゲルカラムで精製した。その際ヘキサン中5%エーテルで溶出した。均質フラクションを集めると表題化合物1.11g(52%)が得られた。
【0078】
【化16】
【0079】
実施例3
2−(cis−9'−オクタデセノキシ)−安息香酸
エタノール25mlおよび水50ml中2−(cis−9'−オクタデセノキシ)−エチル−ベンゾエート(1.11g、2.66×10-3mol)の懸濁液に水酸化リチウム(2.0g)を加え、反応混合物を90℃で6時間撹拌した。エタノールを蒸発し、クロロホルム100mlを加えた。5N HClを注意深く添加してpH7に調節した。有機相を水で洗った。溶媒除去後、生成物をシリカゲルカラムで精製した。その際溶出系としてクロロホルム中2%メタノールを用いた。表題化合物1.0g(96%)が得られた。
【0080】
【化17】
【0081】
実施例4
1−(p−クロロベンゾイル)−5−メトキシ−2−メチルインドール−3−酢酸−(cis−9'−オクタデセニル)アミド
無水N,N−ジメチルホルムアミド6ml中1−(p−クロロベンゾイル)−5−メトキシ−2−メチルインドール−3−酢酸(インドメタシン)(0.56g、1.56×10-3mol)およびTBTU(0.51g、1.56×10-3mol)の溶液に、N,N−ジイソプロピルエチラミン(0.53ml、3.12 ×10-3mol)を加え、反応混合物をN2下で室温で30分間撹拌した。無水N,N−ジメチルホルムアミド6m1中cis−9−オクタデセニル−アミン(0.42g、1.56×10-3mol)の溶液を加え、撹拌を3時間続けた。溶媒を高真空で蒸発し、残留物をクロロホルムと水とに分割した。乾燥した有機相を濃縮し、生成物をシリカゲルカラムで精製した。その際クロロホルム中2%エタノールを溶媒系として用いた。均質フラクションを蒸発すると若干のDMFを含む表題化合物が1.05g得られた。その生成物をエーテルに溶解し、水で洗い、有機相を乾燥し、蒸発すると表題化合物0.88g(92%)が得られた。
【0082】
【化18】
実施例5
S(+)−2−(6−メトキシ−2−ナフチル)プロピオン酸−(cis−9' −オクタデセニル)−アミド(ナプロキセンオレイルアミド)
無水N,N−ジメチルホルムアミド20ml中ナプロキセン(1.65g、7. 15×10-3mol)およびTBTU(2.30g、7.15×10-3mol)の溶液に、N,N−ジイソプロピルエチラミン(2.45ml)14.3×10-3mol)を加え、反応混合物をN2下で室温で30分間撹拌した。無水N,N−ジメチルホルムアミド25ml中1−アミノ−cis−9−オクタデセン(1.91g、7.15×10-3mol)の溶液を加え、3時間撹拌を続けた。溶媒を高真空で蒸発し、残留物をクロロホルムと水との間に分割した。乾燥した有機相を濃縮し、生成物をシリカゲルカラムで精製した。その際溶出系としてクロロホルム中3%メタノールを用いた。均質フラクションを蒸発すると表題化合物2.77g(81%)が得られた。
【0083】
【化19】
【0084】
実施例6
S(+)−2−(6−ヒドロキシ−2−ナフチル)プロピオン酸
十分撹拌した無水N,N−ジメチルホルムアミド中水素化ナトリウム(60%)(12.9g、0.336mol)懸濁液に、N,N−ジメチルホルムアミド300ml中エタンチオール(24.3ml、0.328mol)溶液を滴下した。N,N−ジメチルホルムアミド150ml中ナプロキセン(15g、0.065mol)溶液をゆっくり加え、反応混合物を150℃で3時間加熱した。澄明溶液を冷却し、3.5N HClでpHを調節した(2−3)。溶媒を高真空で蒸発し、残留物をエーテル150mlと水90mlとの混合物で処理した。固体沈殿物を濾去し、濾液を濃縮した。残留物をクロロホルム90mlと水90mlとの混合物で処理し、冷蔵庫に24時間保存した。白色沈殿物を濾去し、洗い、乾燥すると、表題化合物10.1g(72%)が得られた。
【0085】
【化20】
【0086】
実施例7
S(+)−2−(6−ヒドロキシ−2−ナフチル)プロピオン酸−エチルエステル
無水エタノール1200ml中S(+)−2−(6−ヒドロキシ−2−ナフチル)プロピオン酸(5.0g、23×10-3mol)溶液にp−トルエン−スルホン酸(0.2g)を加え、反応混合物を24時間還流加熱した。冷やした混合物を固体NaHCO3部分と共に撹拌した。その溶液を濾過し、溶媒を蒸発した。残留物をクロロホルムに溶解し、水で洗った。有機相を濃縮し、粗生成物をシリカゲルカラムでクロロホルム中2%メタノールで溶出した。均質フラクションは表題化合物4.8g(80%)を与えた。
【0087】
【化21】
【0088】
実施例8
S(+)−2−(6−[cis−9'−オクタデセノキシ]−2−ナフチル)−プロピオン酸−エチルエステル
無水N,N−ジメチルホルムアミド350ml中水素化ナトリウム(60%)(0.47g、11.8×10-3mol)懸濁液にS(+)−2−(6−ヒドロキシ−2−ナフチル)プロピオン酸−エチルエステルを加え、反応混合物をN2下で室温で2時間撹拌した。N,N−ジメチルホルムアミド5ml中cis−9−オクタデセノール−メシレート(3.91g、10.7×10-3mol)溶液を加え、撹拌を48時間続けた。溶媒を高真空で蒸発し、残留物をクロロホルムと水で処理した。乾燥した有機相を濃縮し、粗生成物をシリカゲルカラムで精製した。その際クロロホルムで溶出した。均質フラクションは表題化合物2.93g(56%)を与えた。
【0089】
【化22】
【0090】
実施例9
S(+)−2−(6−[cis−9'−オクタデセノキシ]−2−ナフチル)−プロピオン酸(ナプロキセンオレイルエーテル)
テトラヒドロフラン115mlおよび1M NaOH25ml中S(+)−2−(6−[cis−9'−オクタデセノキシ]−2−ナフチル)−プロピオン酸−エチルエステル(3.79g、7.67×10-3mol)溶液を室温で10日間撹拌した。1M HCl17mlを加え、溶媒を蒸発した。残留物をクロロホルムおよび水に取り、1M HClでpHを1に調節した。有機相を水で洗い、乾燥し(MgSO4)、濃縮すると表題化合物3.25g(94%)が得られた。
【0091】
【化23】
【0092】
実施例10
4−O−(trans−9'−オクタデセノイル)−2−メチル−N(2−ピリジル)−2H−1,2−ベンゾチアジン−3−カルボキサミド−1,1−ジオキシド
無水N,N−ジメチルホルムアミド25ml中4−ヒドロキシ−2−メチル−N[2−ピリジル]−2H,1,2−ベンゾチアジン−3−カルボキサミド−1,1−ジオキシド(ピロキシカム)溶液に、ジクロロメタン20ml中trans−9−オクタデセノイルクロリド(2.2g、7.53×10-3mol)溶液2mlを加え、反応混合物をN2下で室温で撹拌した。残りの酸塩化物溶液を2mlづつ2時間間隔で加えた。合計80時間の反応時間後、溶媒を高真空で蒸発した。残留物をエーテル200mlに溶解し、水と少量のNaHCO3(水溶液)で洗った。乾燥した(MgSO4)有機相を濃縮し、粗生成物をシリカゲルカラムで精製した。その際エチルアセテート/ヘキサン(40:60)で溶出した。均質フラクションを集め、蒸発すると、固体物質3.56gが得られ、それをペンタン/エーテル中で還流した。冷やした混合物を一晩4℃に保った。固体物質を濾去し、ペンタンで洗い、乾燥すると表題化合物3.5g(78%)が得られた。
【0093】
【化24】
【0094】
実施例11
[2−(2,6−ジクロロフェニル)アミノ]ベンゼン酢酸)−(cis−9' −オクタデセニル)−エステル
ジクロロメタン15mlおよびN,N−ジメチルホルムアミド3ml中(2−[2、6−ジクロロフェニル])−アミノ]ベンゼン酢酸ナトリウム(ジクロフェナック)(0.48g、1.6×10-3mol)溶液に、酢酸(0.09ml、1.6×10-3mol)、cis−9−オクタデシン−1−01(0.42g、1.6×10-3mol)、4−ジメチルーアミノピリジン(DMAP)(50mg)およびDCC(0.34g、1.7×10-3mol)を加え、反応混合物を0 ℃で6時間、室温で48時間撹拌した。白色沈殿を濾去し、ジクロロメタンで洗った。有機相を水で洗い、乾燥し(MgSO4)、濃縮し、シリカゲルカラムで精製した。その際エチルアセテート/ヘキサン(40:60)で溶出した。均質フラクションは表題化合物0.45g(53%)を無色液体として与えた。
【0095】
【化25】
【0096】
実施例12
4−O−(cis−11'−エイコサノイル)−2−メチル−N(2−ピリジル)−2H−1,2−ベンゾチアジン−3−カルボキサミド−1,1−ジオキシド
無水N,N−ジメチルホルムアミド3ml中4−ヒドロキシ−2−メチル−N[2−ピリジル]−2H−1,2−ベンゾチアジン−3−カルボキサミド−1,1−ジオキシド(ピロキシカム)(0.3g、0.990×10-3mol)溶液にジクロロメタン2.5ml中cis−11−エイコサノイルクロリド(0.29g、0.90×10-3mol)溶液1.5mlを加えた。残りの酸塩化物溶液を2時間後に加えた。合計80時間の反応時間後に溶媒を高真空で蒸発した。残留物をエーテル40mlに溶解し、水および少量のNaHCO3(水溶液)で洗った。乾燥した有機相を濃縮し、粗生成物をシリカゲルガラムで精製した。その際エチルアセテート/ヘキサン(40:60)で溶出した。均質フラクションを集め、蒸発すると表題化合物0.42g(75%)が得られた。
【0097】
【化26】
【0098】
実施例13
S(+)−2−(6−メトキシ−2−ナフチル)プロピオン酸−cis−9'−オクタデセニル−エステル
ジクロロメタン10ml中S(+)−2−(6−メトキシ−2−ナフチル)プロピオン酸(ナプロキセン)(0.15g、0.65mmol)溶液に、cis−9−オクタデセノール(0.18g、0.67mmol)、DCC(0.13g、0.67mmol)、4−ジメチルアミノピリジン(DMAP)(20mg)を加え、反応混合物をN2下、室温で3時間撹拌した。生成した白色沈殿を濾去し、ジクロロメタンで洗った。 溶媒を蒸発し、生成物をシリカゲルカラムで精製した。その際溶出液としてジクロロメタンを用いた。均質フラクションは表題化合物0.25g(80%)を与えた。
【0099】
【化27】
【0100】
実施例14
11β,17α,21−トリヒドロキシプレグナ−1,4−ジエン−3、20−ジオン−21−エライデート
無水ジオキサン200mlおよびピリジン6.5ml中11β、17α、21−トリヒドロキシプレグナ−1、4−ジエン−3、20−ジオン(プレドニゾロン)溶液に、エライジン酸塩化物(8.0g、26.6mmol)を加え、反応混合物を10℃で3時間撹拌した。少量のメタノールを加え、溶媒を高真空で蒸発した。残留物をエーテルと水とに分配した。有機相を酒石酸(水溶液)、NaHCO3(水溶液)および水で洗った。乾燥した有機相を濃縮し、生成物をシリカゲルカラムで精製した。その際溶出系としてヘプタン/エチルアセテート/メタノール(64:32:4)を用いた。均質フラクションを蒸発すると表題化合物9.18g(90%)が得られた。
【0101】
【化28】
【0102】
実施例15
9−フルオロ−11β,17,21−トリヒドロキシ−16β−メチルプレグナ−1,4−ジエン−3,20−ジオン−21−エライデート
無水ジオキサン40mlおよびピリジン1ml中9−フルオロ−11β,17,21−トリヒドロキシ−16β−メチルプレグナ−1,4−ジエン−3,20−ジオン(ベタメタゾン)(0.9g、2.3mmol)の懸濁液に、エライジン酸塩化物(1.13g、3.03mmol)を加え、反応混合物を周囲温度で48時間撹拌した。少量のメタノールを加え、溶媒を高真空で蒸発した。残留物をエーテルと水とに分配した。有機相を酒石酸(水溶液)、NaHCO3(水溶液)および水で洗った。乾燥した有機相を濃縮し、生成物をシリカゲルカラムで精製した。その際溶出系としてヘプタン/エチルアセテート/メタノール(64:32:4)を用いた。不純フラクションを再精製し、均質フラクションを蒸発すると表題化合物1.02g(65%)が得られた。
【0103】
【化29】
【0104】
NSAIDsおよびその他の抗炎症剤が一般に処方される状態を治療する場合、本発明の親油性誘導体を経腸的または非経口的に全身投与することができる。
【0105】
好適形で経腸投与する場合、本発明の化合物はソフトまたはハードゼラチンカプセル、錠剤、顆粒、粒または粉末、糖衣錠、シロップ、懸濁液または溶液として処方することができる。
【0106】
非経口的投与の場合、本発明の化合物を注射または注入溶液、懸濁液または乳濁液として調製するのが適切である。
【0107】
本発明の医薬組成物は一般的方法によって調製される。例えばそれら製剤は不活性または薬力学的活性な添加物を含むことができる。錠剤または顆粒等は一般的な結合剤、増量剤、担体物質または希釈剤を含むことができる。液体製剤は例えば滅菌溶液の形で存在する。
【0108】
カプセルは活性成分以外に増量剤または濃化剤を含むことができる。さらに香味改良添加物並びに、保存料、安定剤、保水剤および乳化剤等の普通用いられる物質、浸透圧を変える塩類、緩衝剤およびその他の添加物等も含んでよい。
【0109】
所望ならば、本発明の化合物の薬剤は抗酸化剤、例えばトコフェロール、N−メチル−トコフェラミン、ブチル化ヒドロキシアニソール、アスコルビン酸またはブチル化ヒドロキシトルエン等を含むことができる。
【0110】
本発明による化合物の用量は治療すべき疾患の性質およびその進行段階によって、使用法および使用経路並びに患者の要求によって変化する。一般に全身投与する際の成人1日量は約0.1−100mg/kg体重/日、好適には0.5−30mg/kg/日である。
【0111】
本発明はさらに、炎症性、疼痛誘発および/または発熱状態の治療法において、そのような治療を必要とするヒト患者に本発明の少なくとも1種類の化合物を投与することを含む治療法に関係する。
【0112】
現在好まれる本発明の抗炎症薬親油性誘導体は、親薬剤がナプロキセンであるものである。特に、我々はナプロキセンオレイルエーテル、ナプロキセンオレイルエステルおよびナプロキセンオレイルアミドがナプロキセンそのものよりも改善された抗炎症効果を示すことを見いだした。動物in vivoモデルにおいて、これらの誘導体は肉芽腫液体含有量に関して、炎症の第一相に対して改善された効果を示した。さらに驚くべき効果は、特にナプロキセンオレイルアミド投与で肉芽腫組織乾燥重量が減少したことである。これは、既知NSAIDsによる治療では得られなかった組織損傷の減少を意味する。この効果の大きさは、治療量のステロイドによってのみ期待できるものであった。NSAIDsの重症副作用である軟骨分解の減少も認められた。
【0113】
これらの所見、例えば肉芽腫乾燥重量の減少によってわかる直接的改善効果、および軟骨分解の減少等の組み合わせはナプロキセン誘導体の治療指数を有意に改善する。これらの誘導体で治療した動物は、親化合物で治療した動物よりもかなり小さい侵襲性を示した。これは、これら誘導体の消化器副作用の誘発力がより小さいことを強く示唆する。
【0114】
理論によって束縛されるものではないが、この高まった抗炎症効果は、これら誘導体の親油性性質のために細胞による取り込みが増加するためであり、またはナプロキセンの活性とは全く異なる活性によるものであると考えることができる。ナプロキセンに付加した脂肪酸末端は反応性酸素種(ROS)の掃去剤(これは多くのメカニズムによって抗炎症性であり得る)として作用するらしい。例えば、組織損傷はROS感受性プロテアーゼーインヒビターの保護によって阻止されるし、タンパク質に対する酸化的損傷によって生成する内因性抗原類の形成は防止され、ヒアルロン酸を解重合から保護することによって血管由来因子の生成を阻止する。
【0115】
移植軟骨では、ナプロキセン様のその他のNSAIDsはプロテオグリカンおよびコラーゲン喪失を増加する傾向がある。それに対して、本発明のナプロキセン誘導体は軟骨からのプロテオグリカンまたはコラーゲンの喪失を高める傾向を示さない。シクロオキシゲナーゼ阻害は軟骨に対するNSAIDsの有害作用の原因であると考えられ、ナプロキセン誘導体はナプロキセンと共にこの酵素を阻止し得ることから、ナプロキセン誘導体のより大きいサイズおよび親油性特性が軟骨基質からナプロキセン誘導体を排除することが示唆される。
【0116】
これらナプロキセン誘導体の高められた抗炎症効果を明らかにする実験をここに詳細に述べる。
【0117】
生物学的効果
使用した肉芽腫誘起性軟骨分解のin vivoモデルは、滅菌コットンに包んだラット大腿骨頭軟骨をマウスの背中の皮下に埋め込んだものである。そのコットンは明白なT細胞が関係した肉芽腫性反応を誘発し、それは移植軟骨からの基質化合物の喪失に通ずる。可能性のある薬剤の抗関節炎効果を試験する手段として、このモデルは幾つかの明白な長所を有する。このモデルは軟骨基質の喪失を確認するための定量的生化学的終点を有する慢性糜爛性疾患を含む。抗炎症性活性はコットン肉芽腫の湿潤および乾燥重量から判断でき、軟骨保護効果は移植軟骨のグリコサミノグリカンおよびヒドロキシプロリン含有量(それぞれプロテオグリカンおよびコラーゲンを示す)から測定できる。肉芽腫は分離しており、必要に応じて摘出し、種々のメディエータまたは酵素を評価することができる。
【0118】
雌TOマウス(21±4g)群(n=10)においてコットンで包んだラット大腿骨頭軟骨を皮下に移植した。2週間目にインプラントを除去した。コットンは肉芽腫様反応を誘発し、同時にプロテオグリカンを移植軟骨から遊離させた。等モル量のナプロキセン(30mg/kg)およびナプロキセン誘導体(60mg/kg)を毎日経口投与し、肉芽腫発生に対する影響および軟骨プロテオグリカン含有量に与える影響を評価した。それら化合物はリポソームとして処方され、空のリポソームを賦形薬コントロールとして用いた。
【0119】
15mg/mlリポソーム組成物は次のように調製した:グリセロール/滅菌水緩衝液中で脂質誘導体(DMSO中)とレシチン(エタノール中)を1:1(w/w)で混合し、その後透析して溶媒類を除去する。誘導体化しないNSAIDs化合物の7.5mg/mlリポソーム組成物は、グリセロール/滅菌水中で空のリポソームに上記化合物を加えることによって調製した。
【0120】
結果をマン−ホィットニーを用いるINSTATで分析した。p値はタイ(ties)を補正した。p<0.05を有意とした。
【0121】
試験した化合物
ナプロキセン(VI)、ナプロキセンオレイルエーテル(XII)、ナプロキセンオレイルエステル(VII)およびナプロキセンオレイルアミド(VIII)。化合物XII、VIIおよびVIIIのR'はcis−CH2(CH2)7CH=CH(CH2)7 CH3である。
【0122】
このモデルでナプロキセンで得られたこれらの結果は、文献に報告されたナプロキセンおよびその他のNSAIDsを用いた同様な研究から判明した結果に匹敵する。
【0123】
薬剤投与を対照と比較した場合、肉芽腫の液体含有量は減少した、そしてその減少はナプロキセンオレイルアミドの場合には有意であることが判明した。組織乾燥重量はナプロキセンによって影響を受けないように見えたが、親油性有意は減少をおこすように見え、それはここでもナプロキセンオレイルアミドの場合に有意であった。これらの顕著な所見は、ナプロキセン誘導体がナプロキセンそのものに比し、軟骨分解を減らすことを強く示唆する。これは実際に確認された。なぜならばナプロキセンがリポソーム対照および親油性誘導体に比較して移植軟骨からのプロテオグリカン喪失を増加することがわかったからである。同じ所見はヒドロキシプロリン含有量として評価した軟骨分解にも反映された。但しナプロキセン投与動物からのインプラントは比較的少ないコラーゲンをもつ傾向があったが、投与群の間に統計的有意差はなかった。
【0124】
その上、ナプロキセン投与動物は侵襲的挙動を示し、その結果10インプラントのうち4インプラントを失った。リポソーム投与群またはナプロキセン誘導体投与群では同様な侵襲的挙動の結果としてインプラントが失われることはなかった。これはナプロキセン誘導体が親NSAIDsよりもよく耐えられたことを示唆する。
【0125】
上記の実験は、本発明によって誘導体化することによりナプロキセンの生物学的特性がかなり改善されることを示すものである。
【0126】
プレドニゾロンおよびベタメタゾンおよびそれらの誘導体のラット腹腔内単球/ マクロファージに与える影響
雄ラットの腹腔内に0時に10mg/ml試験化合物を4ml、または賦形薬のみを注射した。投与後6、12、25、48および72時間目に腹腔を生理的食塩液40mlで洗浄し、分離した細胞を洗い、数え、分別した。それらの細胞をオプソニン結合−チモサン(zymosan)、N−ホルミル−L−ロイシル−L−フェニルアラニン(fMLP)またはホルボール−12−ミリステート13−アセテート(PMA)で刺激し、細胞の活性を化学ルミネッセンス生成によって1時間にわたり測定した。
【0127】
誘導体の効果は明らかで、驚くべきものである。図5からわかるように、プレドニゾロンがチモサン刺激−細胞の活性に与える影響は6時間目の化学ルミネッセンスのわずかな減少として認められるだけである。プレドニゾロンエライデートでは、炎症細胞の活性は投与後48時間にわたって対照に比較して減少する。その効果はプレドニゾロンそのものの効果より明らかに大きく、持続する。
【0128】
選択したプレドニゾロン誘導体の効果をさらに研究するために別の実験系列を行った。この実験ではプレドニゾロンの7種類の脂肪酸エステルの抗炎症効果を比較した。
【0129】
雄ラットに試験化合物10mg/mlの或る用量、または賦形薬のみを0時に腹腔内注射した。投与後48時間に腹腔を洗浄し、分離した細胞を洗い、数え、分別した。細胞をその後オプソニン結合−チモサン、N−ホルミル−L−ロイシル−L−フェニルアラニン(fMLP)またはホルボール12−ミリステート13−アセテート(PMA)で刺激し、細胞活性を化学ルミネッセンス生成によって1時間にわたり測定した。
【0130】
誘導体類の効果は明らかで驚くべきものである。図6からわかるように、特に好ましい脂肪酸の一つの例として、プレドニゾロン−エライデートの効果は最大であった。その他の脂肪酸誘導体では化学ルミネッセンスはほんのわずかな影響を受けただけである。プレドニゾロン−エライデートでは炎症細胞の活性は投与後48時間にわたり、対照および残りの脂肪酸誘導体に比較して減少する。
【0131】
図7に示すように、腹腔洗浄液中の細胞数は実質的に減少し、この減少は投与後48時間まで明らかであった。
【0132】
細胞を分別すると、腹腔洗浄液中のマクロファージの数に主な影響が出ることがわかった。これは図8に示される。プレドニゾロンではその効果は遥かに少なく、6時間および12時間目に認められただけである。ベタメタゾンとベタメタゾン−エライデートとの比較でも、腹腔洗浄液中のマクロファージの数の測定で、さほど明らかではないが、同様な効果が認められた(図9)。
【0133】
プレドニゾロン−エライジン酸エステルの直接的抗喘息効果を研究するために、試験化合物を気道過剰反応モデルで評価した。
【0134】
ラットの内毒素誘起性気道変化に与えるプレドニゾロンエライジン酸の影響
急性炎症性気道変化のラットモデルにおいて、動物をエアゾール化内毒素(LPS)にさらす。これは90分以内に気管および気管支に広汎性好中性炎症をおこし、同時に気管支肺胞液中の好中球数の増加および喘息の重要な特徴である気道反応性上昇をおこす。10匹の雄F344ラットをチェンバー内で30分間100μg/ml LPSにさらした。エアゾールにさらす12時間前と4時間前に動物にプレドニゾロンまたはプレドニゾロン誘導体、3mg/kgを点滴注入した。エアゾール被曝終了後90分に、5−ヒドロキシトリプタミンに対する気道反応性の測定および気道炎症の評価のために動物の準備を行った。肺抵抗の最低50%増加が認められるまで、5HTを5分ごとに静脈注射に加えた。そして肺抵抗を50%増加するのに必要な5HT量、PC50RL、を計算した。プレドニゾロンも誘導体も気管支肺胞液中の炎症細胞数に影響を与えなかった。気道反応性は図10に示すように、プレドニゾロン−エライジン酸エステルによって驚くべく高程度の影響を受けた。これは喘息治療において非常に重要であるかも知れない。
【0135】
抗癌剤
効果的な癌化学療法治療は若干の重大な障害のために制限される。そのうちの幾つかは完全にまたは一部克服できるかも知れない。作用を確実により特異的にするあらゆる薬剤の変形は、患者にとって直接的利益となる。最も重要な要求は、問題の腫瘍が提供される治療に対して感受性を有することである。これは治療薬の種類および作用メカニズムに大きく依存し、実際の治療を開始する前の生検/分離−腫瘍細胞のin vitro実験で評価することができる。腫瘍を数種の薬剤に感受性にし得る方法も知られている。
【0136】
化学療法薬剤はそれらの性質として細胞に毒性を有する。悪性腫瘍細胞がその薬剤に比較的敏感である限り、好ましい状況となる。もしもその薬剤が腫瘍組織/腫瘍細胞に蓄積する傾向があれば、治療ポテンシャルはさらに改善される。治療指数をさらに改善するためには器官標的化が重要な要因である。一次腫瘍、特に初期、または別の腫瘍型から転移した一次腫瘍は、肝臓、脾臓、肺、脳等の選ばれた組織に限られることが非常に多い。薬剤の性質、その処方、または投与法がその薬剤を選択した組織に向かわせるならば、非常に選択的な腫瘍根絶が達成されるかも知れない。
【0137】
本発明の好適抗癌性誘導体は以下に述べる試験で証明されるように、改善された治療指数を有する。
【0138】
親抗癌化合物は、悪性腫瘍の治療に有用な特性を有する群に分類でき、アルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸およびカルボン酸エステル基から選択される1つ以上の誘導化可能基を有する化合物ならばいかなる化合物でもよい。本発明によって誘導体化できる現在使用できる抗癌剤の若干の例は次のものを含む:
【0139】
【化30】
【0140】
【化31】
【0141】
【化32】
【0142】
【化33】
【0143】
【化34】
【0144】
上記のように、公知の抗癌剤の多くは上に定義した種類の誘導化可能基を1つより多く含む。これらの場合、これら官能基の1つ以上を本発明による親油性基によって置換することができる。そして2つ以上の親油性基が存在する場合、これらは同じ基でも異なる基でもよい。
【0145】
本発明の親油性抗癌性誘導体は既述の一般的製法によって合成することができる。
【0146】
例えば、以下の反応スキームはドキソルビシン(XXI)およびダウノルビシン(XXII)からのアミド類およびカルバメート類の形成を説明する。親化合物(類)のアミノ基を、脂肪酸(RCOOH)または脂肪アルコール(R'OH)から作られるアシル−チアゾリジン−2−チオンまたはアルキルオキシ−カルボニル−チアゾリジン−2−チオン試薬と反応させて、アミドまたはカルバメートに選択的に誘導することができる。
【0147】
【化35】
【0148】
スキーム6
次に記すスキーム7も2つの抗癌性アルキル化剤クロラムブシル(XXIII)およびメルファラン(XXIV)の誘導体化を説明するものである。一官能価クロラムブシルは多数の方法によってエステル化され、またはアミドに変換できる。しかし二官能価メルファランは自己縮合または環形成反応等の多くの副反応を受けることがある。未保護メルファランにおいては、DCCまたはTBTU等のカップリング試薬の使用の有用性は限られているが、アミン官能基はアシル−チアゾリジン−2−チオン試薬によって、対応するアミドに都合よく変換できる。
【0149】
【化36】
【0150】
スキーム7
本発明による特異的抗癌性誘導体の製法は以下の実施例によって説明される。実施例18および21は中間体の製法に関係する。
【0151】
実施例16
クロラムブシル−オレイル エステル
ジクロロメタン70ml中4−[p−[ビス(2−クロルエチル)アミノ]−フェニル]酪酸(クロラムブシル)(0.966g、3.18mmol)およびオレイルアルコール(01893g、3.33mmol)の溶液に、DCC(0.72g、3.5mmol)およびN,N−ジメチル−アミノ−ピリジン(DMAP)(25mg)を加え、反応混合物を周囲温度で12時間撹拌した。固体沈殿を濾去し、残留物をCH2Cl250mlに溶解し、水で洗った。有機相にエーテル25mlを加え、固体沈殿を濾去した。濾液を蒸発し、残留物をシリカゲルカラムで精製した。この際CH2Cl2を溶出液として用いた。均質フラクションを蒸発すると表題化合物1.0g(55%)が得られた。
【0152】
【化37】
【0153】
実施例17
エライジン酸メルファランアミド
DMF24ml、水4mlおよびトリエチルアミン4ml中、L−3−[p−[ビス(2−クロルエチル)アミノ]−フェニル]アラニン(メルファラン)(0.603g、1.98mmol)の溶液に、DMF12ml中3−チアゾリジン−2−チオン−エライジルアミド(0.617g、1.61mmol)を加えた。反応混合物を暗所で室温で1.5時間撹拌した。溶媒を高真空で蒸発し、残留物をクロロホルム100mlに溶解し、pH5.5の水で洗った。有機相をAgNO3(水溶液)、水(pH5.5)および飽和NaCl(水溶液)で洗った。その有機相を蒸発すると表題化合物0.84g(75%)が得られた。
【0154】
【化38】
【0155】
実施例18
3−エライドイル−1,3−チアゾリジン−2−チオン
ジクロロメタン(20ml)中エライジル酸(2.0g)7.1mmol)、DMAP(86mg、0.7mmol)、1,3−チアゾリジン−2−チオン(1. 0g、8.4mmol)およびDCC(1.7g、8.2mmol)の混合物をN2 下で0℃で1時間撹拌し、その後周囲温度でさらに5時間撹拌した。追加のDCC(41mg、0.2mmol)を加え、反応物を同温度で2時間撹拌した。処理後フラッシュクロマトグラフィー(SiO2;四塩化炭素−クロロホルム1:0、1:1、0:1)を行うと、表題化合物2.56g(94%)が黄色ワックス様固体として得られた。
【0156】
【化39】
【0157】
実施例19
エライジン酸ダウノルビシンアミド
塩酸ダウノルビシン(250mg、0.44mmol)および3−エライドイル−1,3−チアゾリジン−2−チオン(実施例18、400mg、1.04mmol)をTHF(20ml)と、炭酸ナトリウム(0.12M NaHCO3、0.8M Na2CO3)で緩衝したブライン(4M NaCl 20ml)との間に分配した。混合物を暗所でN2下で周囲温度で4時間激しく撹拌した。相を分離し、水相をエーテルで抽出した(3×10ml)。合一した有機相を硝酸ナトリウム水溶液で洗った(3×10ml 2M)。存在する1、3−チアゾリジン−2−チオンを除去するために、ピリジン(1.0ml)を加え、エーテル相を、硝酸銀(0.2M)を含む硝酸ナトリウム(2M)水溶液(2×3ml)と共に激しく振とうした。各処理後、混合物をセライトを通して濾過し、エーテル(20ml)ですすいだ。エーテル相を硝酸ナトリウム水溶液(5ml 2M)およびブライン(5ml)で洗い、最後に乾燥した(MgSO4)。粗生成物をピリジン(0.2%w/w)で予備調製したシリカゲルカラムで精製した。溶出液としてクロロホルム中0.2%ピリジンおよび0.6%メタノールを用いた。表題化合物332mg(95%)が暗赤色粉末として得られた。
【0158】
【化40】
【0159】
実施例20
エライジン酸ドキソルビシンアミド
塩酸ドキソルビシン(400mg、0.69mmol)を上記のように、THF(35ml)および緩衝ブライン(35ml)中、3−エライドイル−1,3−チアゾリジン−2−チオン(実施例18、400mg、1.04mmol)で周囲温度で10分間処理した。反応を完了するために追加のアミド化試薬(実施例18:100mg、0.26mmol)が必要であった。6時間後、相を分離し、水相をTHFで抽出した。合一した有機相をブラインで洗い、乾燥した(MgSO4)。実施例19に記載したフラッシュクロマトグラフィーによって精製すると表題化合物440mg(79%)が暗赤色結晶として得られた(mp.115−116℃)。
【0160】
【化41】
【0161】
実施例21
3−(cis−9−オクタデセン−1−オキシカルボニル)−1,3−チアゾリジン−2−チオン
上記化合物は実質的に、エチル同族体のためのチェン(Chen)およびヤング( Yang)の方法によって合成した。N2下で0℃に保持したクロロホルム(乾燥、エタノール−フリー;15ml)中2−チオキソ−3−チアゾリジンカルボニルクロリド2(1.6g、8.6mmol)およびTEA(1.3ml、9.3mmol)の撹拌溶液に、オレイルアルコール(cis−9−オクタデセン−1−オール;2.8g、10.4mmol)を10分間以内に加えた。その混合物を同温度で80分間撹拌し、氷水(5ml)を加えた。塩酸(0.5ml 1M)を滴下して水相のpHを6に調節した。標準的に処理し、その後フラッシュクロマトグラフィー(SiO2:ヘキサン−クロロホルム1:1、1:2、1:3、0:1)を行うと、黄色油1.78g(50%)が得られた。
【0162】
【化42】
【0163】
実施例22
ダウノルビシン オレイル カルバメート、[N−(cis−9−オクタデセン−1−オキシカルボニル)ダウノルビシン]
塩酸ダウノルビシン(250mg、0.44mmol)を3−(cis−9−オクタデセン−1−オキシカルボニル)−1,3−チアゾリジン−2−チオン(実施例21、550mg、1.33mmol)と共に実施例19においてアミド同族体について記載したように27時間処理した。実施例20に記載したようにTHFで抽出した後得られた粗生成物をエーテル(40ml)に溶解した。存在する1,3−チアゾリジン−2−チオンを請求項19の方法によって除去した。粗生成物をピリジン(0.2%w/w)で予備調製したシリカゲルガラムで精製した。その際ベンゼン中0.2%ピリジンおよび0−10%メタノールで溶出した。表題化合物321mg(87%)が暗赤色粉末として得られた。
【0164】
【化43】
【0165】
実施例23
ドキソルビシン オレイル カルバメート、[N−(cis−9−オクタデセン−1−オキシカルボニル)ドキソルビシン]
塩酸ドキソルビシン(250mg、0.43mmol)を3−(cis−9−オクタデセン−1−オキシカルボニル)−1,3−チアゾリジン−2−チオン(実施例21:700mg、1.69mmol)と共に実施例19においてアミド同族体について記載したように69時間処理した。THFを減圧下で除去し、生成した懸濁液を水(20ml)とピリジン−クロロホルム1:4(25ml)に分配した。未溶解の物質を、有機相に加えたピリジン(15ml合一)で処理した。揮発物は減圧下で蒸発によって除去した。残留残渣(1.1g)を硝酸銀水溶液(0.6ml 1M)を含む酢酸エチルで20−30℃で20分間超音波処理した。生成した懸濁液をセライトを通して濾過し、酢酸エチル(20ml合一)ですすいだ。超音波処理サイクルをさらに硝酸銀(0.4ml 1M)で繰り返した。合一した有機相をブライン(5ml)で洗い、乾燥した(MgSO4)。回転蒸発器で酢酸エチルを蒸発することによって得た粗生成物(0.61g)を実施例22に記載のようにしてフラッシュクロマトグラフィーによって精製すると、表題化合物208mg(58%)が暗赤色ガラスとして得られる。
【0166】
【化44】
【0167】
実施例24
タキソール−2'−エライデート
タキソール(25mg、0.029mmol)を先ず最初に、ピリジン(3×1ml)への溶解および減圧下における蒸発を繰り返すことによって乾燥した。それからピリジン(1ml)に溶解すると、澄明な無色溶液が得られ、それにN−(3−ジメチルアミノプロピル)−N'−エチルカルボジイミド塩酸(9mg、0.05mmol)、DMAP(2mg、0.02mmol)、エライジン酸(10mg、0.035mmol)および無水MgSO4(3mg)を混合物として加えた。反応物をN2下で室温で48時間撹拌した。ピリジンを減圧下で蒸発して除去し、残留物をDCM(25ml)に溶解した。有機相をその後乾燥し(MgSO4)、濾過し、減圧下で蒸発すると白色固体が得られる。それをフラッシュクロマトグラフィー(SiO2;ジエチルエーテルヘキサン1:1ないし1:0勾配溶出)によって精製すると表題化合物が白色固体(25mg、77%)として得られた。
【0168】
【化45】
【0169】
ネズミ皮下ADJ/PC6形質細胞腫およびそのシスプラチン抵抗サブラインにおけるメルファラン−エライジンアミドおよびクロラムブシル−オレイルエステルのin vivo抗腫瘍効果
不飽和度の異なるクロラムブシルおよびクロラムブシル−脂肪酸結合物のヒトリンパ腫および正常ヒト末梢血リンパ球の細胞毒性がアネル(A.Anel)らのBioc hemical Pharmacology、40巻、6号、1193−1200ページ、1990、に記載されている。クロラムブシル−アラキドン酸およびクロラムブシル−ドコサヘキサエン酸のリンパ腫細胞に対する毒性はクロラムブシルまたは遊離脂肪酸の個々の毒性ポテンシャルと等しいかまたはより高い。これに対し、オレイン酸およびエライジン酸を例とする本発明の脂肪酸誘導体は親薬剤のみに比し遥かに毒性が小さい。これは下記の実験によって示される。
【0170】
ネズミ充実性ADJ/PC6形質細胞腫および、シスプラチンおよびその他のアルキル化剤に対する耐性があるということから選択されたそのサブラインを体重20−25gのBALB/C雌マウスに1mm3腫瘍断片として皮下に移植した。メルファランまたはメルファランエライジンアミド、またはクロラムブシルまたはクロラムブシル−オレイルエステルを腫瘍移植後20日目に腹腔内に単回投与した。腫瘍を30日目に切除し、対照および治療群の重量を比較した。活性はED90として測定した抗腫瘍効果と比較した薬剤毒性測定値、LD50(mg/kg)、に基づいて決められた。ED90は、対照と比較して腫瘍質量を90%減少させるのに必要な量(mg/kg)である。
【0171】
表1からわかるように、メルファラン−エライジンアミドおよびクロラムブシルオレイルエステルどちらのLD50(mg/kg)も、それぞれメルファランおよびクロラムブシルのそれらと比較して遥かに多量であった。これは毒性の減少を意味する。
【0172】
【表1】
【0173】
メルファラン−エライジン酸アミドでは感受性−およびシスプラチン耐性腫瘍どちらでもED90に達したが、メルファランはシスプラチン耐性腫瘍では全く活性を示さなかった。メルファラン−エライジン酸アミドのED90は60mg/kgであった。
【0174】
多剤耐性を有する、または有しない細胞内へのドキソルビシンおよびドキソルビシン誘導体の細胞内蓄積
腫瘍細胞は長期間化学療法後、抗癌剤に耐性を示すことがある。薬剤耐性の一つの形は、多剤耐性(MDR)である。その場合細胞はビンカアルカロイド類、アントラサイクリン類、アクチノマイシンDおよびコルヒチン等の種々の薬に交差耐性を示す。MDR表現型はP−糖タンパク質と呼ばれる特殊の群の膜通過性糖タンパク質(P−gp)の過剰発現と関係している。P−gpはエネルギー依存性薬剤排出ポンプとして機能するように見える。P−糖タンパク質は抗癌剤を細胞外に活発に排出することによって抗癌剤の細胞内濃度をその有効濃度より下に下げることができる。カルシウムチャンネル−ブロッカーであるベラパミルは、抗腫瘍薬の細胞内濃度を高めることによってMDRを元に戻すことができる。ジヒドロピリジンおよびピリジン同族体、カルモジュリン阻止物質、合成イソプレノイド類、向リソソーム剤、ビスベンジルイソキノリン−アルカロイド、キニジン、キナクリン、リドカイン、フェノキサジン、アミオダロンおよびシクロスポリンAは、細胞に同時投与した際、またはin vivoに同時投与される際にMDRを変化させる薬のその他の例である。まだ実験的レベルであるが、癌治療においては耐性変更剤の使用が一般的になりつつある。
【0175】
これらの高度に生体内活性である化合物はそれら自体問題がないわけではない。軽度ないし重度の生命を脅かす副作用が認められるため、in vitroで認められる非常に有望な結果を臨床的に得ることは不可能である。
【0176】
これらの作用物質の大部分はカチオン性且つ親油性である。親油性は耐性モジュレータにとって好ましい特徴である。リポソーム封入ドキソルビシンについてその多剤耐性克服効果を試験した。細胞内薬剤濃度はドキソルビシンのリポソームを用いると2倍になった(Cancer chemotherapy and Pharumacology、1991、28巻:259−265ページ)。リポソームのみでもMDRに影響するかも知れない(リポソームによって誘起される多剤耐性細胞内への薬剤の蓄積の増加、Cancer Research、52巻、3241−3245、1992)、(カルジオリピン、ホスファチジルイノシトール、ジオレイルホスファチジン酸のリポソーム)。
【0177】
細胞を2×105/mlの懸濁液にして、20μMの薬剤にさらした。種々の時間に部分を取り、氷冷PBSですすぎ、その後フローサイトメーターを通過させた。図11にはドキソルビシンおよびドキソルビシン誘導体の時間の関数としての蓄積が見られる。細胞系COR−L123/P(ヒト大肺細胞)およびその耐性細胞系COR−L123/Rではドキソルビシン誘導体(ドキソルビシン−エライジン−アミドおよびドキソルビシン−オレイル−カルバメート)濃度はほぼ同じであった、一方ドキソルビシン濃度は耐性細胞系では遥かに低かった。
【0178】
多剤耐性を有するまたは有しない細胞内へのダウノルビシンおよびダウノルビシ ン誘導体の細胞内蓄積
細胞を上記のようにダウノルビシン、ダウノルビシン−エライジンアミドおよびダウノルビシン−オレインカルバメートにさらした。薬剤濃度は10μMに減らした。耐性および非耐性細胞への取り込みによる蛍光は誘導体に関しては2種類の細胞で多かれ少なかれ同じであり(図12)、ダウノルビシンそのものではこれらの細胞内への取り込みによる蛍光は2種類の細胞で差があった(図13)。
【0179】
ドキソルビシン−エライジン酸アミドの同時投与によるドキソルビシンに対する 細胞の感受性増加
MTTアッセイを用いて化合物の毒性を調べた。アッセイ前の6日間、細胞を化合物にさらした。使用した細胞系はPgPを過剰発現するH69/LX4、ヒト小肺細胞系であった。その細胞系はドキソルビシン−エライジン酸アミドのみに高度に耐性であり、ドキソルビシン−エライジン酸アミドのIC50値は>50μMである。しかし驚くべきことに、ドキソルビシン−エライジン−アミドを5μM濃度でドキソルビシンと同時に投与すると、上記細胞系のドキソルビシン感受性はIC50=0.4μMからIC50=0.08μMに高まる。20μMドキソルビシン−エライジン−アミドの添加はドキソルビシン感受性をIC500.04=μMに高める。表2の結果は、この誘導体が細胞の耐性メカニズムと相互作用し、ドキソルビシンそのものの効果を強め、そして感受性細胞系のレベルにまで回復することができることを示す。
【0180】
【表2】
【0181】
上記図11−13に示したように、アントラサイクリン類ドキソルビシンおよびダウノルビシンの脂肪酸誘導体はドキソ耐性細胞系のMDRメカニズムに対して調節作用を有する。耐性細胞系に親薬剤と同時投与する際、親薬剤に対する感受性が再び感受性細胞系と同じ桁になる。このMDRモジュレータに対するこのアプローチは好都合である。なぜならば同時投与した薬剤はまさに活性化合物の誘導体であり、それはin vivoで加水分解されて活性薬剤と無毒性脂肪酸残基を遊離するからである。
【0182】
抗菌剤
この治療分野には非常に種々様々の薬剤が含まれる。
【0183】
抗菌剤の最も重要な群は多分ペニシリンであるが、薬剤耐性がますます深刻になるにつれ、細菌感染症を治療するための代替治療薬に焦点が当てられてきた。他の作用メカニズムを有する他の薬剤で治療されるとはいえ、マイコバクテリウムおよび原虫によって起きる疾患の治療においても、細菌感染症の克服に重要である因子類の幾つか、例えば細胞内取り込み、組織分布、耐性メカニズムの回避等は同様に重要である。
【0184】
この分野で使用される全ての薬剤は標的感染症に対して非常に良い〜中程度の効果を有する。この領域では通常の新薬開発の他に、より良い経口バイオアベイラビリティを有する誘導体、すなわち純粋プロドラッグ類の開発に焦点が当てられており、この研究は薬剤耐性問題にはほとんど影響をもたない。
【0185】
抗生物質の臨床的効果はその抗菌活性で決まるだけでなく、その薬物学的および薬物動態的特性によっても決まる。プロドラッグは抗生物質の安定性および溶解度を高めるために、そして親化合物の経口吸収、組織浸透性および作用持続時間を改良するために用いられている。経口投与後の血清中濃度の上昇は抗生物質の組織濃度の改善につながる。
【0186】
ペニシリンおよびその他の関連β−ラクタム抗生物質領域では、単純アルキルエステル類は安定過ぎてプロドラッグとして使用できないとしばしば言われている。好適プロドラッグは1ないし3個のメチレンリンカーまたはメトキシカルボニル アルキルエステルを有するdoubleエステルである。これらの側鎖改変は経口バイオアベイラビリティの改良を容易にし、これらの誘導体は生体内で十分に不安定で、血流中の内因性または微生物由来酵素による加水分解性代謝によって活性薬剤を遊離する。これらのペニシリン−プロドラッグは薬剤耐性状態にほとんど影響を与えない。ペニシリンおよび密接に関係する同族体に対して活性な、最も優勢で、合理的によく特徴づけられた耐性メカニズムは、細菌が獲得した、加水分解酵素β−ラクタマーゼを産生する能力である。その酵素は細胞内−および細胞外両方で見いだされる。それはその薬が実際の細菌に到達する前にすでに血流中で破壊され得ることを意味する。その他の種類の薬剤耐性は純粋な排除メカニズムによる。このメカニズムによって薬剤は細菌またはその他の微生物に入ることを阻止される。本発明の脂質誘導体は血流中ではさほど容易には加水分解されず、そのため血流中でその薬剤誘導体はよりよく循環することができる。特に、新規脂質誘導体の高い細胞内輸送および付加的効果が排除メカニズムを克服し、活性薬剤が細胞/細菌のその他のコンパートメントにおいて放出され、そこではそれは加水分解酵素には無関係にその作用をあらわすことができると考えられる。
【0187】
本発明によって誘導体化できる抗生物質およびその他の抗菌剤の若干の例は以下の化合物を含む:
【0188】
【化46】
【0189】
【化47】
【0190】
【化48】
【0191】
上に図示したように、抗菌剤は誘導化可能基を1つより多く含むことができる。これらの場合にはこれら官能基の1つ以上が本発明によって親油性基で置換でき、2つ以上の親油性基がある場合は同じ親油性基でも異なる親油性基でもよい。
【0192】
本発明の親油性抗菌化合物は既述のように同じ一般的方法によって合成できる。しかし、数種のペニシリン誘導体の選択的および効率的誘導体化は、例えば親薬剤に複数の官能基(−OH2−NH−および−NH2)が存在する等、種々の要因により、βラクタム環の開裂、化合物のその他の再構成または分解によって複雑になるかも知れない。そのため保護基および種々の試薬系を使用して、スキーム8のアンピシリン(XXV)で示されるように、選択的誘導体化を容易にすることができる。
【0193】
第一アミノ基はアシル−チアゾリジン−2−チオンによって選択的に脂肪酸アンピシリンアミド(XXVI)に変換できる。同じアミノ官能基はベンザルデヒドでシッフ塩基(XXVII)として保護される。そのカルボン酸をセシウム塩に変形し、さらに脂肪臭化物(RBr)と反応させる。軽度酸性加水分解によりアミノ基が再形成され、アンピシリン−脂肪酸エステル(XXVIII)が生成する。
【0194】
【化49】
【0195】
スキーム8
三官能価抗結核薬パラ−アミノ−サリチル酸、PAS(XXIX)、の選択的誘導体化がスキーム9に示される。PASそのものは多くの反応条件下では不安定で、自己縮合および二付加物形成が起こり得る。カルボン酸をそのセシウム塩に変形することができ、さらにメシレートとの反応によって対応するエステルに変形できる。脂肪酸塩化物は主としてアミノ基と反応し、対応するアミドを与える。フェノール基に対する反応から生ずる生成物は微塩基性加水分解により除去することができる。
【0196】
【化50】
【0197】
スキーム9
本発明による特異的抗菌化合物の製法を以下に示す。
【0198】
実施例25
パラ−アミノ−サリチル酸エライジルエステル
無水DMF50ml中パラ−アミノ−サリチル酸(PAS)(0.47g、3. 1mmol)および炭酸セシウム(0.98g、3.0mmol)の懸濁液にエライジルメシレート(1.0g、2.9mmol)を加え、反応混合物を周囲温度で60時間、35℃で48時間撹拌した。反応混合物をエーテルと水で抽出し、有機相をNaHCO3(水溶液)および水で洗った。溶媒を留去し、残留物をシリカゲルカラムで精製した。溶出系としてヘプタン/CH2Cl2/AcOH/MeOH(85:15:2:2)を用いた。生成物含有フラクションを蒸発すると表題化合物0.6g(51%)が得られた。
【0199】
【化51】
【0200】
実施例26
4−(エライドアミド)−サリチル酸
無水THF100ml中PAS(2.6g、17mmol)の溶液にTHF20ml中エライジン酸塩化物(5.11g、17mmol)の溶液を0℃で滴下した。反応混合物を周囲温度で24時間撹拌した。さらにエライジン酸塩化物(1.0g)を加えた。溶媒を高真空で蒸発し、残留物をエーテルと水との間に分配した。有機相を酒石酸(水溶液)および水で洗った。溶媒を留去し、残留物を水5mlを加えたメタノール100mlに溶解した。固体沈殿を濾去し、エタノールから再結晶すると粗材料3.9gが得られた。この材料1.5gを、1M NaOH 11ml加えたエタノール100mlに溶解した。混合物を周囲温度で2時間撹拌し、0.5M酒石酸(水溶液)30mlで酸性にした。固体沈殿を水で洗い、エーテルに溶解し、有機相を酒石酸(水溶液)で洗った。溶媒を留去し、残留物を無水クロロホルム×2から蒸発すると表題化合物1.3g(87%)が得られた。
【0201】
【化52】
【0202】
実施例27
エライジン酸クロラムフェニコールエステル
無水DMF10mlおよびピリジン2ml中D−threo−2、2'−ジクロロ−N−[β−ヒドロキシ−α−(ヒドロキシメチル)]−p−ニトロフェネチルアセタミド(0.20g、0.62mmol)の溶液に、DMF3ml中エライジル酸塩化物(0.19g、0.62mmol)の溶液を加えた。反応混合物を周囲温度で12時間撹拌した。溶媒を高真空で蒸発し、残留物を酢酸エチルと水との間に分配した。有機相を濃縮し、粗生成物をシリカケルカラムで、溶出系として酢酸エチル/ヘキサン(1:1)を用いて精製した。均質フラクションを蒸発すると表題化合物0.14g(39%)が得られた。
【0203】
【化53】
【0204】
実施例28
オキサシリン−オレイル エステル
水76ml中(5−メチル−3−フェニル−4−イソキサゾリル)ペニシリン(オキサシリン)ナトリウム塩(1.0g、2.4mmol)の溶液に、0.2MHCl(水溶液)13.1mlを加え、その混合物を蒸発した。残留物をメタノール50mlおよび水5mlに溶解した。Cs2CO3の20%溶液(水溶液)をpH7になるまで加えた。混合物を蒸発乾固した。
DMF50ml中残留物の溶液にオレイルブロミド(0.78g、2.4mmol)を加え、反応混合物を周囲温度で72時間撹拌した。溶媒を高真空中で蒸発し、残留物を水およびクロロホルムで抽出した。有機相を濃縮し、粗生成物をシリカゲルカラムで、溶媒系として酢酸エチル/ヘキサン(40:60)を用いて精製した。均質フラクションを蒸発すると表題化合物0.69g(45%)が得られた。
【0205】
【化54】
【0206】
実施例29
エライジン酸アンピシリンアミド
アセトニトリル10ml中D−(−)−(α−アミノベンジル)ペニシリン(アンピシリン)(0.10g、0.29mmol)の溶液に、3−チアゾリジン−2−チオン−エライジルアミドおよびDBU(0.043ml、0.29mmol)溶液を加え、二相反応混合物を周囲温度で72時間激しく撹拌した。溶媒を留去し、残留物を酢酸エチルと飽和塩化ナトリウム(水溶液)との間に分配した。粗生成物を分離し、メタノールに再溶解した。混合物を蒸発乾固すると、表題化合物0.1g(55%)が得られた。
【0207】
【化55】
【0208】
実施例30
アンピシリン−オレイルエステル
DMF30ml中アンピシリン(1.21g、3.5mmol)および炭酸水素カリウム(0.35g、3.5mmol)の懸濁液に、ベンザルデヒド(0.92g、8.7mmol)を加え、反応混合物を0℃で4時間撹拌した。炭酸水素カリウム(0.35g、3.5mmol)およびオレイルブロミド(1.21g、3. 7mmol)を加え、0℃で2時間、周囲温度で12時間撹拌を続けた。溶媒を高真空で蒸発し、残留物を酢酸エチルおよび冷水(0℃)で抽出した。有機相を蒸発すると黄色シロップ2.46gが得られた。
粗生成物をアセトニトリルに溶解し、1M HCl(水溶液)をpH=2になるまで加えた。水30mlを加え、アセトニトリルを留去した。生成物を酢酸エチルおよびジクロロメタンで抽出した。合一した有機相を蒸発し、粗生成物をシリカゲルカラムで溶媒系として酢酸エチル中1%トリエチルアミンを用いて精製した。均質フラクションを蒸発すると表題化合物0.8g(38%)が得られた。
【0209】
【化56】
【0210】
抗寄生虫薬
寄生虫感染はヒト医学および獣医学における重大問題である。寄生虫は通常、食物/水を介して、または昆虫が刺すことによってホストに侵入する。寄生虫は腸管(上皮細胞層)または血流に見いだされる、この場合赤血球または、その他の、肺または脳等の標的器官が感染するかも知れない。寄生虫はホストの細胞間および細胞内両方に見いだされる。原虫について言うと、寄生虫の生活環には数段階あることがよくあり、全ての段階が治療されるわけではない。
【0211】
ヒトにおける最も主要な寄生虫感染症はマラリアであり、一方動物、特に鳥(家禽)では腸感染、コクシジューム症が主な問題である。未治療状態では糞便は胞子を含み、それは動物または新たな人々の再感染に導く(他の種属とのクロスオーバーも)。
【0212】
活性薬剤を寄生虫そのものに効率的に運搬し、または例えばマラリアおよびコクシジューム症では、寄生虫感染細胞内に運搬することが重要である。
【0213】
本発明の親油性抗寄生虫誘導体は既述の一般的製法によって合成できる。
【0214】
例えば、反応スキーム10は抗マラリア薬ヒドロキシ−クロロキン(XXX)のアシル化を示す。反応は第一OH基に完全に選択的である。
【0215】
【化57】
【0216】
スキーム10
生物学的効果
選択されたヒドロキシクロロキノン誘導体の高められた抗マラリア効果を示す実験を次に示す。
【0217】
ヒドロキシクロロキノン−エライジン酸エステルのマウス−マラリアに与える効果
NK65系薬剤感受性P.bergheiの4日間試験をスイス アルビノ雌マウスで行った。ヒドロキシクロロキノン−エライデート0.063、0.25、1.0および4mg/kgおよびヒドロキシクロロキノン0.094、0.395、1.5および6mg/kgを3匹づつのマウス群に4日間腹腔内投与した。寄生虫接種物107感染細胞を0日目に静脈内投与し、その日のうちに薬剤を投与した。動物にその後3日間投与し、5日目に尾血液フィルムを作り、寄生虫血を検査した。モル−ベースで、この4日間試験においてエライジン酸誘導体はエライジン酸そのものよりも2.5−3倍効果が高かった。これらの知見はヒトにおけるマラリア治療に非常に重要である。
【0218】
【表3】
【0219】
ヒドロキシクロロキノン硫酸またはヒドロキシクロロキノンエライデートでマウス血液中のP.bergheiマラリア寄生虫を50%、90%および99%減少させる有効量がこの表に示される。
【0220】
本発明によって誘導体化できる抗寄生虫薬の例は下記を含む:
【0221】
【化58】
【0222】
マラリアは最も広くはびこっている寄生虫病であり、推定マラリア発生率は臨床症例が年間200万−500万台である。抗マラリア薬に対する獲得耐性は大きな問題となりつつある。
【0223】
ヒトは、普通、感染した雌アノフェレス蚊に刺されると、注入された(胞子虫の)種虫に感染する。その寄生虫は速やかに循環を出て、肝実質細胞に入り、そこで増殖し、組織shizontsに発達する。薬剤が肝臓内の形質胞体状の組織に作用するならば、それら薬剤は原因予防に用いられる。本発明の脂質誘導体は肝組織に親和性を有するから、これらの化合物はマラリア病の組織型により有効であり、P.vivaxおよびP.ovaleの肝臓型を根絶するかも知れない。これらの型は生き残り、後に増殖し、最初の感染の数カ月ないし数年後に赤血球感染を再発させる。
【0224】
P.falciparumはヒトマラリアの症例の85%以上を占める。P.falciparumの耐性種は薬剤を十分に高い濃度まで蓄積しない。Ca2チャンネルブロッカーはクロロキンのような薬に対する感受性を一部回復する。多数の化学的に関係ない薬に対する交差耐性は、悪性腫瘍に見られる多剤耐性と同様である。
【0225】
脂肪酸はそれら自体が抗マラリア効果を有するらしい(クルグリアク(Krugli ak)ら、実験寄生虫学81巻、97−105ページ、1995)。オレイン酸、エライジン酸、リノレイン酸のような脂肪酸がPlasmodium vinckei petteriまたはPlasmodium yoelii nigeriensisに感染したマウスにおける寄生虫血の発生を阻止した。
【0226】
しかし、ヒトにおいて同様な効果を得るのに必要な細胞内濃度は、非現実的多量の脂肪酸の摂取を必要とする。
【0227】
ここでも本発明の実施態様に基づき、抗マラリア薬誘導体が驚くほど効率的に高濃度で細胞内寄生虫に運搬され、薬剤耐性メカニズムを克服することさえできる。
【0228】
本発明の抗マラリア誘導体の製法を示す実施例を以下に記す:
【0229】
実施例31
7−クロロ−4−[4−[エチル(2−エライドイルオキシエチル)アミノ]1−メチルブチルアミノ]−キノリン
ジクロロメタン30ml中7−クロロ−4−[4−[エチル(2−ヒドロキシエチル)アミノ]1−メチルブチルアミノ]キノリン(ヒドロキシクロロキン)(3.17g、9.4mmol)の溶液に、エライジン酸塩化物(2.82g、9. 4mmol)を加え、反応混合物を周囲温度で48時間撹拌した。少量のメタノールを加え、溶媒を高真空で蒸発した。残留物をシリカゲルカラムで反復精製した(第一行程:クロロホルム/メタノール9:1、第二行程:クロロホルム/メタノール95:5)。均質フラクションを蒸発すると表題化合物1.18g(21%)が得られる。
【0230】
【化59】
【0231】
本発明によって誘導体化できるその他の領域内の薬の例をさらに幾つか挙げる。
【0232】
中枢神経系薬
【0233】
【化60】
【0234】
心臓血管薬
【0235】
【化61】
【0236】
下記の一般的反応スキームは、本発明によるワルファリンおよびセロケンの誘導体の製法を示す。
【0237】
スキーム11は抗凝固剤ワルファリン(XXXI)のアシル化を示す。
【0238】
【化62】
【0239】
スキーム11
セロケン(XXXII)のヒドロキシ基の選択的アシル化はアミノ官能基の存在によって複雑になる。アミノ官能基をBOC誘導体として好適に保護し、脂肪酸塩化物の使用によってOH基を変形する。これらの反応はスキーム12に示される。
【0240】
【化63】
【0241】
スキーム12
反応スキーム11の特殊な実施例を次に示す。
【0242】
実施例32
3−(α−アセトニルベンジル)−4−エライドイルオキシクマリン
無水ジオキサン120mlおよびピリジン25ml中3−(α−アセトニルベンジル)−4−ヒドロキシクマリン(ワルファリン)(3.70g、12mmol)の溶液にエライジン酸塩化物(3.60g、12mmol)を加え、反応混合物を周囲温度で4時間撹拌した。溶媒を高真空で蒸発した。残留物をエーテルと水との間に分配した。有機相を酒石酸(水溶液)、NaHCO3および水で洗った。乾燥した有機相を濃縮し、生成物をシリカゲルカラムで、溶出系としてヘプタン/酢酸エチル(6:1)を用いて精製した。不純なフラクションを再精製し、均質フラクションを蒸発すると表題化合物が淡黄色油として5.1g(70%)得られる。
【0243】
【化64】
【0244】
上述のように、本発明はヒトまたは動物医学に有用な化合物だけでなく、あらゆる種類の生物学的活性化合物に一般的に適用できる。特に、アルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸およびカルボン酸エステル基から選択される1つ以上の官能基を有する農薬を本発明によって誘導体化できる。このような農薬および園芸用化学薬品の例は次のものを含む:
【0245】
【化65】
【0246】
【化66】
【0247】
【化67】
【0248】
【化68】
【0249】
【化69】
【特許請求の範囲】
【請求項1】
分子構造中に、アルコール、エーテル、フェニル、およびアミノ基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、コーチゾン、フルオシノロン、フルドロコーチゾン、ヒドロコーチゾン、メチルプレドニゾロン、プレドニゾロン、トリアムシノロン、パラメタゾン、プレドニゾン、およびベクロメタゾンから選択された副腎皮質ステロイドの親油性誘導体。
【請求項2】
11β,17α,21−トリヒドロキシプレグナ−1,4−ジエン−3,20−ジオン−21−エライデート。
【請求項3】
9−フルオロ−11β,17,21−トリヒドロキシ−16β−メチルプレグナ−1,4−ジエン−3,20−ジオン−21−エライデート。
【請求項4】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、アセメタシン、アルクロフェナック、アンフェナック、アスピリン、ベンダザック、ベノリレート、ベノキサプロフェン、ブクロキシ酸、ブフェキサマク、ブマジゾン、ブチブフェン、カルプロフェン、シンメタシン、クリダナク、クロメタシン、クロリパク、ジフルニザル、エプロチノール、エトドラック、エトフェナメート、フェルビナク、フェンブフェン、フェンクロフェナク、フェンクロラク、フェンドサル、フェノプロフェン、フェンチアザク、フェルフェナミン酸、フルルビプロフェン、グラフェニン、イブフェナク、イブプロフェン、インドメタシン、イソフェゾラク、イソキセパク、ケトプロフェン、ケトロラック、ロナゾラック、メクロフェナム酸、メファナミン酸、メチアジン酸、ナブメトン、ニフルム酸、オルシプレナリン、オキサメタシン、オキサプロジン、ピラゾラク、プロチジン酸、スリンダク、スルガム、テニダプ、テノキシカム、チアラミド、チノリジン、トルフェナム酸、トルメチン、およびゾメピラックから選択された非ステロイド抗炎症薬(NSAID)の親油性誘導体。
【請求項5】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、メゲストロール、メドロキシプロゲステロン、ヘキセストロール、トリロスタン、アミノグルテチミド、エピチオスタノール、カルステロン、ポドフィリン酸2−エチルヒドラジド、ピラルビシン、ドキソルビシン、ダウノルビシン、タキソール、モピダモル、ミトキサントロン、ロニダミン、エトポシド、エフロルニチン、デフォサミド、トリメトレキセート、メトトレキセート、デオプテリン、チオグアニン、チアミプレン、メルカプトプリン、ダカルバジン、ニムスチン、クロロゾトシン、メルファラン、エストラムスチン、シクロホスファミド、クロラムブシル、およびトリメチロールメラミンから選択された抗癌剤の親油性誘導体。
【請求項6】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、オキサシリン、アンピシリン、アモキシシリン、セファレキシン、セファロチン、セファロスポリン、ドキシサイクリン、クロラムフェニコール、エタムブトール、シプロフロキサシン、エンロフロキサシン、ジフロキサシン、およびダノフロキサシンから選択された抗菌剤の親油性誘導体。
【請求項7】
前記抗菌剤が、クロラムフェニコール、オキサシリン、およびアンピシリンから選択される請求の範囲第6項記載の親油性誘導体。
【請求項8】
化合物:エライジン酸アンピシリンアミド。
【請求項9】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、アモジアキン、ヒドロキシクロロキン、メフロキン、メパクリン、デコキナート、ゾアレン、下記の式の化合物:
【化1】
および下記の式の化合物:
【化2】
から選択された抗寄生虫薬の親油性誘導体。
【請求項10】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、カプトプリル、エナラプリル、ブニトロール、セロケン、ラベタロール、およびセロケンから選択された心臓血管薬の親油性誘導体。
【請求項11】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、アミノトリアゾール、アシュラム、ベナゾリン、ブロモフェノキシム、ブロモキシニル、2,4−D、DICAMBA、ジクロブトゥラゾール、ジノテルブ、フルアジホップ、メコプロップ、ピクロラム、スルホメスロン、メタミドフォス、トリクロロフォン、アンシミドール、ホルモジン、シクロヘキシミド、ヒメキサゾール、およびエチリモールから選択された農薬の親油性誘導体。
【請求項1】
分子構造中に、アルコール、エーテル、フェニル、およびアミノ基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、コーチゾン、フルオシノロン、フルドロコーチゾン、ヒドロコーチゾン、メチルプレドニゾロン、プレドニゾロン、トリアムシノロン、パラメタゾン、プレドニゾン、およびベクロメタゾンから選択された副腎皮質ステロイドの親油性誘導体。
【請求項2】
11β,17α,21−トリヒドロキシプレグナ−1,4−ジエン−3,20−ジオン−21−エライデート。
【請求項3】
9−フルオロ−11β,17,21−トリヒドロキシ−16β−メチルプレグナ−1,4−ジエン−3,20−ジオン−21−エライデート。
【請求項4】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、アセメタシン、アルクロフェナック、アンフェナック、アスピリン、ベンダザック、ベノリレート、ベノキサプロフェン、ブクロキシ酸、ブフェキサマク、ブマジゾン、ブチブフェン、カルプロフェン、シンメタシン、クリダナク、クロメタシン、クロリパク、ジフルニザル、エプロチノール、エトドラック、エトフェナメート、フェルビナク、フェンブフェン、フェンクロフェナク、フェンクロラク、フェンドサル、フェノプロフェン、フェンチアザク、フェルフェナミン酸、フルルビプロフェン、グラフェニン、イブフェナク、イブプロフェン、インドメタシン、イソフェゾラク、イソキセパク、ケトプロフェン、ケトロラック、ロナゾラック、メクロフェナム酸、メファナミン酸、メチアジン酸、ナブメトン、ニフルム酸、オルシプレナリン、オキサメタシン、オキサプロジン、ピラゾラク、プロチジン酸、スリンダク、スルガム、テニダプ、テノキシカム、チアラミド、チノリジン、トルフェナム酸、トルメチン、およびゾメピラックから選択された非ステロイド抗炎症薬(NSAID)の親油性誘導体。
【請求項5】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、メゲストロール、メドロキシプロゲステロン、ヘキセストロール、トリロスタン、アミノグルテチミド、エピチオスタノール、カルステロン、ポドフィリン酸2−エチルヒドラジド、ピラルビシン、ドキソルビシン、ダウノルビシン、タキソール、モピダモル、ミトキサントロン、ロニダミン、エトポシド、エフロルニチン、デフォサミド、トリメトレキセート、メトトレキセート、デオプテリン、チオグアニン、チアミプレン、メルカプトプリン、ダカルバジン、ニムスチン、クロロゾトシン、メルファラン、エストラムスチン、シクロホスファミド、クロラムブシル、およびトリメチロールメラミンから選択された抗癌剤の親油性誘導体。
【請求項6】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、オキサシリン、アンピシリン、アモキシシリン、セファレキシン、セファロチン、セファロスポリン、ドキシサイクリン、クロラムフェニコール、エタムブトール、シプロフロキサシン、エンロフロキサシン、ジフロキサシン、およびダノフロキサシンから選択された抗菌剤の親油性誘導体。
【請求項7】
前記抗菌剤が、クロラムフェニコール、オキサシリン、およびアンピシリンから選択される請求の範囲第6項記載の親油性誘導体。
【請求項8】
化合物:エライジン酸アンピシリンアミド。
【請求項9】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、アモジアキン、ヒドロキシクロロキン、メフロキン、メパクリン、デコキナート、ゾアレン、下記の式の化合物:
【化1】
および下記の式の化合物:
【化2】
から選択された抗寄生虫薬の親油性誘導体。
【請求項10】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、チオール、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、カプトプリル、エナラプリル、ブニトロール、セロケン、ラベタロール、およびセロケンから選択された心臓血管薬の親油性誘導体。
【請求項11】
分子構造中に、アルコール、エーテル、フェニル、アミノ、アミド、カルボン酸、およびカルボン酸エステル基から選択される1つ以上の官能基を含み、少なくとも1つの前記官能基が、式:RCOO−、RCONH−、RCOS−、RCH2O−、RCH2NH−、−COOCH2R、−CONHCH2R、および−SCH2Rから選択される親油性基によって置換されており、前記式中Rが、cis−8−ヘプタデセニル、trans−8−ヘプタデセニル、cis−10−ノナデセニル、およびtrans−10−ノナデセニルから選択される親油性部分である、アミノトリアゾール、アシュラム、ベナゾリン、ブロモフェノキシム、ブロモキシニル、2,4−D、DICAMBA、ジクロブトゥラゾール、ジノテルブ、フルアジホップ、メコプロップ、ピクロラム、スルホメスロン、メタミドフォス、トリクロロフォン、アンシミドール、ホルモジン、シクロヘキシミド、ヒメキサゾール、およびエチリモールから選択された農薬の親油性誘導体。
【図1】

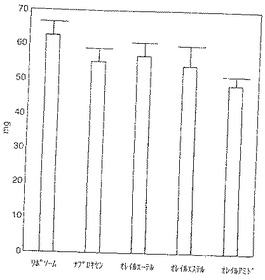
【図2】
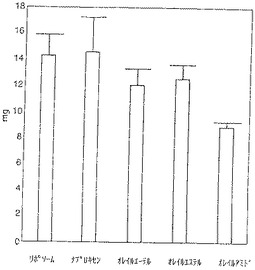
【図3】
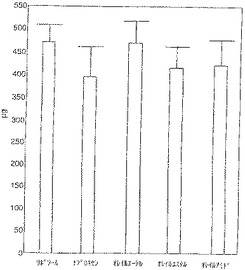
【図4】
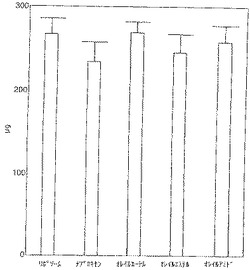
【図5】
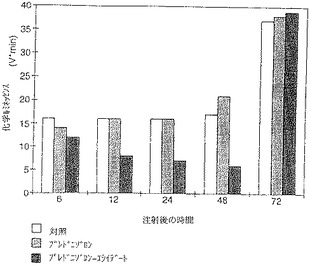
【図6】
【図7】
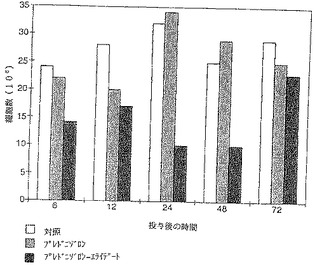
【図8】
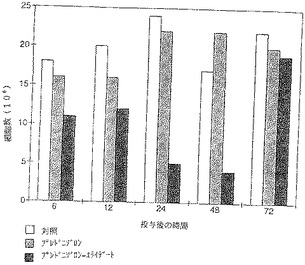
【図9】
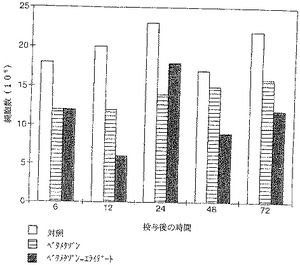
【図10】

【図11】
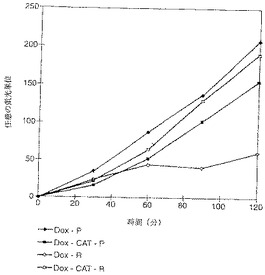
【図12】
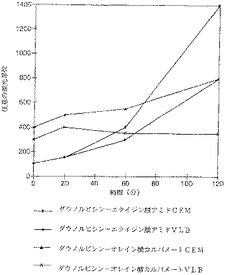
【図13】
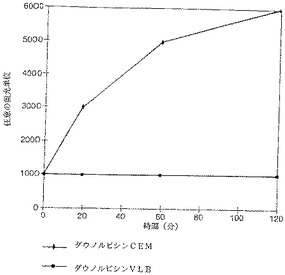
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公開番号】特開2009−280583(P2009−280583A)
【公開日】平成21年12月3日(2009.12.3)
【国際特許分類】
【出願番号】特願2009−141714(P2009−141714)
【出願日】平成21年6月12日(2009.6.12)
【分割の表示】特願平10−531863の分割
【原出願日】平成10年1月23日(1998.1.23)
【出願人】(507311957)
【氏名又は名称原語表記】CLAVIS PHARMA ASA
【住所又は居所原語表記】Parkveien 55, 0256 Oslo, Norway
【Fターム(参考)】
【公開日】平成21年12月3日(2009.12.3)
【国際特許分類】
【出願日】平成21年6月12日(2009.6.12)
【分割の表示】特願平10−531863の分割
【原出願日】平成10年1月23日(1998.1.23)
【出願人】(507311957)
【氏名又は名称原語表記】CLAVIS PHARMA ASA
【住所又は居所原語表記】Parkveien 55, 0256 Oslo, Norway
【Fターム(参考)】
[ Back to top ]