
新規な葉酸依存性酵素阻害剤
【課題】本来の基質が葉酸又は葉酸誘導体(葉酸塩)である酵素を阻害し、癌のような疾患の治療に用いられる新規化合物の提供。
【解決手段】下式の化合物、またはその製薬上許容されうる塩に関する:

(式中、Zは、OまたはSであり;nは、1〜3であり;R3は、CH2CH2COOH等であり;R4は、水素原子、COOH等、Aはプテリジン環等)。
【解決手段】下式の化合物、またはその製薬上許容されうる塩に関する:
(式中、Zは、OまたはSであり;nは、1〜3であり;R3は、CH2CH2COOH等であり;R4は、水素原子、COOH等、Aはプテリジン環等)。
【発明の詳細な説明】
【技術分野】
【0001】
明細書
本発明は、新規な一連の化合物に関する。特に本発明は、本来の基質が葉酸または葉酸誘導体(葉酸塩)である酵素を阻害し、癌のような疾患の治療に用いられうる新規な化合物に関する。
【背景技術】
【0002】
癌細胞は他の多くの細胞よりも急速に複製することから、より多くのヌクレオチド(デオキシリボ核酸(DNA)およびリボ核酸(RNA)の前駆体)を必要とする。全ての細胞が常に剰余のヌクレオチド(アデノシン三リン酸(ATP)を除く)を貯蔵しているわけではないため、ヌクレオチドはDNAおよびRNAの合成の間、継続的に合成される必要がある。従って、癌細胞の複製はヌクレオチド生合成の阻害に対して健常な細胞の複製よりも感受性が高くなりがちであり、このため、かような阻害が可能な治療剤が関心を集めつつある。
【0003】
ヌクレオチドは、基本的な代謝前駆体からデノボ経路を経て、あるいは、核酸分解の生成物である遊離塩基およびヌクレオシドが再利用されるサルベージ経路を経て、生物学的に合成されうる。新規な化学治療剤の探索に関して主に関心を集めているのは、デノボ経路である。
【0004】
ヌクレオチドである2’−デオキシアデノシン−5’−一リン酸(dAMP)および2’−デオキシグアノシン−5’−一リン酸(dGMP)はともに、イノシン一リン酸(IMP)から新たに合成され、このIMPは5−ホスホリボシル−1−ピロリン酸(PPRP)由来である。このPPRPとIMPとの間の生合成経路に関与している2つの酵素は、GARトランスホルミラーゼおよびAICARトランスホルミラーゼである。以下に示すように、GARトランスホルミラーゼは、N10−ホルミルテトラヒドロ葉酸を用いてグリシンアミドリボヌクレオチド(GAR)をホルミルグリシンアミドリボヌクレオチド(FGAR)に変換する。一方、AICARトランスホルミラーゼは、同じ化合物を用いて5−アミノイミダゾール−4−カルボキサミドリボヌクレオチド(AICAR)をN−ホルミルアミノイミダゾール−4−カルボキサミドリボヌクレオチド(FAICAR)に変換する。
【0005】
【化1】
【0006】
これに対し、ヌクレオチドである2’−デオキシチミジン−5’−一リン酸(dTMP)は、2’−デオキシウリジン−5’−一リン酸(dUMP)からデノボ合成により製造される。これは、酵素であるチミジル酸シンターゼにより触媒される変換反応である。この変換反応において、N5,N10−メチレン−テトラヒドロ葉酸は7,8−ジヒドロ葉酸へと還元される。そして、N5,N10−メチレン−テトラヒドロ葉酸は、酵素であるジヒドロ葉酸レダクターゼ(DHFR)およびセリンヒドロキシメチルトランスフェラーゼを用いてテトラヒドロ葉酸を経由して再生される。これらのプロセスを以下に図示する。
【0007】
【化2】
【0008】
これらのメカニズムの阻害が、癌の治療に利用されている。例えば、米国特許第2,512,572号には、「葉酸拮抗薬」に分類され、本来の基質よりも約100倍高い親和性で結合してジヒドロ葉酸レダクターゼを競合的に阻害し、dTMPの合成に必須のテトラヒドロ葉酸の再生を阻害することによりDNA合成を阻害する、強力な化学治療剤であるメトトレキサートを含む多くの置換プテリジン類が開示されている。これにより、癌細胞においていわゆる「チミン欠乏死」が誘導される。メトトレキサートはまた、程度は小さいものの、GARトランスホルミラーゼ、AICARトランスホルミラーゼおよびチミジル酸シンターゼをも阻害する。メトトレキサートおよび他の関連する抗葉酸剤の構造を以下に示す。
【0009】
【表1】
【0010】
米国特許第4,684,653号には、式:
【0011】
【化3】
【0012】
式中、R1は、OHまたはNH2であり、R3は、水素原子、メチル基またはエチル基である、
の化合物およびその対応する5,6,7,8−テトラヒドロ誘導体が開示されている。これらの化合物は、葉酸およびその代謝誘導体を基質として利用する1または2以上の酵素に対して効果を有することが開示されている。
【0013】
米国特許第5,344,932号には、式:
【0014】
【化4】
【0015】
式中、R5は、水素原子またはNH2であり、R4は、水素原子またはメトキシ基であり、R2は、水素原子または製薬上許容されうるカチオンである、
のグルタミン酸誘導体が開示されており、これらの化合物が、葉酸およびその代謝誘導体を基質として利用する1または2以上の酵素に対して阻害効果を有することが開示されている。
【0016】
米国特許4,077,957号には:
【0017】
【化5】
【0018】
を含む種々のプテリジン化合物の合成方法が開示されている。
【0019】
かような化合物は、癌治療のための新たな治療戦略の開発に有用であることが判明しているが、効果が小さい、ある患者ではかような薬物に対してもともと耐性がある、または後天的に耐性となる、毒性や好ましくない副作用があるといった、使用に関連した多くの問題が依然として存在する。従って、癌の治療に用いられることができ、上述した問題の1または2以上を解消しうる代替化合物に対する要求は依然として残っている。
【発明の開示】
【0020】
従って、本発明の第1の形態によれば、式Iの化合物、またはその製薬上許容されうる塩:
【0021】
【化6】
【0022】
式中、
Zは、OまたはSであり;
nは、1〜3であり;
R3は、−CO2R8、−C(O)SR8、−C(O)NHR8、−C(S)OR8、−C(S)SR8、−C(S)NHR8、−C(NH)SR8、または−C(NH)NHR8(ここで、R8は水素原子もしくはアルキル基である)であり;
R4は、水素原子、−CH2R5または−CH2CH2R5(ここで、R5は、それぞれ独立してR3の定義のいずれかを有する)であり;
Bは、−NR2−、−CH2NR2−、−CH2CH2NR2−、−CH2CHR7−または−CH2O−(ここで、R2は、水素原子またはC1−3のアルキル基、アルケニル基もしくはアルキニル基であり、R7は、水素原子またはC1−3のアルキル基もしくはアルコキシ基である)であり;
Aは:
【0023】
【化7】
【0024】
式中、
R1は、−NH2または−OHであり;
CおよびDは、それぞれ独立して、1または2以上のヘテロ原子を含んでもよい、5員または6員の、置換されたまたは非置換の、芳香族環または非芳香族環であり、Cは任意の可能な位置で基Bと結合している、
である、
が提供される。
【0025】
本発明の第2の形態によれば、治療に使用される、本発明の第1の形態の化合物が提供される。
【0026】
本発明の第3の形態によれば、本発明の第1または第2の形態の化合物を含む医薬組成物が提供される。
【0027】
本発明の第4の形態によれば、葉酸または葉酸誘導体に依存性の酵素の阻害に応答性の症状の治療に用いられる医薬の製造のための、本発明の第1または第2の形態の化合物の使用が提供される。
【0028】
本発明の第5の形態によれば、癌の治療に用いられる医薬の製造のための、本発明の第1または第2の形態の化合物の使用が提供される。
【0029】
本発明の第6の形態によれば、以下の段階:
(a)式IIの化合物:
【0030】
【化8】
【0031】
式中、Aは上記と同様の定義であり、mは、0、1または2であり、Xは脱離基である、
を、式IIIの化合物:
【0032】
【化9】
【0033】
式中、Z、n、R2、R3およびR4は上記と同様の定義である、
と反応させる段階;
(b)式IVの化合物:
【0034】
【化10】
【0035】
式中、Aは上記と同様の定義であり、Xは脱離基である、
を、式Vの化合物:
【0036】
【化11】
【0037】
式中、Z、n、R3およびR4は上記と同様の定義である、
と反応させる段階;あるいは、
(c)以下の化合物VIまたはVII:
【0038】
【化12】
【0039】
式中、A、Z、n、R3、R4およびR7は上記と同様の定義であり、いずれの場合においても、Yは独立してハロゲン原子である、
の一方を、対応する有機金属試薬に変換させ、前記有機金属試薬を化合物VIまたはVIIの他方と反応させる段階、
を有する、本発明の第1または第2の形態の化合物の製造方法が提供される。
【0040】
本発明の第7の形態によれば、式II−Aの化合物:
【0041】
【化13】
【0042】
式中、Xは、Cl、BrまたはIである、
を、式III−Aの化合物:
【0043】
【化14】
【0044】
と反応させる段階を有する、式I−A:
【0045】
【化15】
【0046】
式中、n、R1、R3およびR4は、上記と同様の定義である、
を有する、本発明の第1または第2の形態の化合物またはその製薬上許容されうる塩の製造方法が提供される。
【0047】
本発明の第8の形態によれば、上記で定義され、R4が−CH2R5または−CH2CH2R5である場合を除く、式III、VまたはVIIの化合物が提供される。
【0048】
本発明の種々の形態のいずれかの発明の好ましい実施形態を以下に記載し、または従属項において規定する。
【0049】
本発明の種々の形態の全てにおいて、C*とマークされた炭素は不斉炭素であってもよく(R4が水素原子ではない場合)、この場合、式Iの化合物は、ラセミ体の形態で存在しても、常法によりその(+)または(−)光学異性体に分離されてもよいことは当然である。また、いくつかの化合物では、他のキラル中心が存在して1または2以上の別の光学異性体のペアを生じてもよい。例えば、Bが−CH2CHR7−であり、R7がC1−3のアルキル基またはアルコキシ基である化合物においては、第2のキラル中心が存在する。かようなラセミ体の形態または光学異性体の形態は全て、本発明の技術的範囲に含まれるものである。さらに、式Iの化合物は、1または2以上の互変異性体の形態で存在してもよいことは理解されるであろうし、これらの形態のそれぞれもまた、本発明の技術的範囲に含まれるものである。
【0050】
以下により詳細に記載される通り、式Iの化合物は葉酸の構造類似体であり、インビトロでメトトレキサートに匹敵するレベルで、ジヒドロ葉酸レダクターゼ(DHFR)のような葉酸または葉酸誘導体(葉酸塩)に依存性の酵素の阻害剤としての作用を有することが判明したのである。式Iの化合物はまた、動物モデルにおいてインビボでも腫瘍の増殖を阻害する活性を有することが判明している。ここではメカニズムの詳細は割愛するが、後者の活性は、DHFRの競合的拮抗薬として作用する当該化合物の活性に起因するかもしれないことが予想される。式Iの化合物は、癌や、葉酸または葉酸誘導体に依存性の酵素の阻害に応答性の症状の治療に用いられうる。
【0051】
式Iの好ましい化合物は、以下の条件の1または2以上を満たす:
ZがOである;
nが1である;
R3が−CO2R8であり、R4が−CH2CH2CO2R8である;
R8が、水素原子、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基またはtert−ブチル基であり、好ましくは水素原子、メチル基またはエチル基であり、より好ましくは水素原子である;
Bが、−CH2NR2−、−CH2CHR7−または−CH2O−であり、好ましくは−CH2NR2−である;
R2が、水素原子、メチル基、エチル基または−CH2C≡CHであり、好ましくは水素原子である;
R7が、水素原子、メチル基、エチル基またはメトキシ基であり、好ましくは水素原子である。
【0052】
また、式III、VまたはVIIの好ましい化合物は、上述したZ、n、R2、R3、R4および/またはR7の1または2以上の好ましい規定を有する。
【0053】
式I、II、IVまたはVIの基Aにおいて、Dは、好ましくは5員のヘテロ芳香族環である。好ましくは、Aは:
【0054】
【化16】
【0055】
である。
【0056】
基Aにおいて、Cは、以下の基(隣接する環および基Bに結合する点を示す)の1つであってもよい:
【0057】
【化17】
【0058】
式中、Xは、CHまたはNであり、かつ、YがCであり、R6が水素原子、メチル基、エチル基またはHCOであるか、YがNであり、R6が孤立電子対である、のいずれかである。好ましい実施形態において、XおよびYはともにNであり、R6は孤立電子対である。
【0059】
特に好ましい基Aは、天然のプテリジン類および他のへテロ環塩基に極めて類似した、以下の構造を有する:
【0060】
【化18】
【0061】
特に関心を集めているのは、以下の2つのA基である:
【0062】
【化19】
【0063】
上述した2つのA基を有し、Bが−CH2NR2−であり、R2が、水素原子、メチル基、エチル基または−CH2C≡CHであり、ZがOであり、nが1であり、R3が−CO2R8であり、好ましくは加水分解可能な任意のエステル基である、式Iの化合物が特に好ましい。この群の化合物の個別の例としては、以下のものが挙げられる:
【0064】
【化20】
【0065】
式Iの化合物は、容易に入手可能で安価な出発物質から、本発明の方法により製造されうる。例えば、Bが−NR2−、−CH2NR2−または−CH2CH2NR2−である場合に、式Iの化合物は、式IIの化合物を式IIIの化合物とカップリングさせることにより製造されうる。脱離基Xは、一般的には塩素、臭素またはヨウ素などのハロゲンであり、特には臭素またはヨウ素である。この反応は、好ましくはジメチルホルムアミド(DMF)またはジメチルアセトアミド(DMAc)などの双極性非プロトン性溶媒中で行われうる。フッ化カリウムなどの塩基性触媒が用いられてもよく、これにより第3級アミンまたは重炭酸ナトリウムと比べて高い収率が得られる。必要であれば、上記の反応前に、本技術分野において公知の適切な保護基を用いて反応性の高い基を保護し、後に、常法により脱保護してもよい。例えば、R3が水素原子であり、R4が−CH2CH2CO2Hである場合、これらの酸基は、例えばメチルエステル基により保護され、その後にエタノール中での水酸化ナトリウムを用いたアルカリ加水分解などの公知の手法により脱保護され、氷酢酸などの酸の添加により沈殿しうる。従って、式IIおよび式IIIの化合物のいずれかまたは双方が保護された形態であるこれらの化合物の反応は、本発明の方法に包含されることが理解されうるであろう。
【0066】
Bが−CH2O−である場合において、式Iの化合物は、例えば、式IVの化合物を、ウィリアムソンエーテル型反応により式Vの化合物とカップリングさせることにより製造されうる。この反応において、式Vの化合物は一般的に、式IVの化合物との反応前に、例えばNaHなどの塩基を用いてアロキシド(aroxide)イオンの形態に変換される。Xは、任意の適切な脱離基であればよく、特にはハロゲン原子である。
【0067】
Bが−CH2CHR7−である場合において、式Iの化合物は、例えば、式VIの化合物を、任意の公知の炭素−炭素結合形成反応により式VIIの化合物とカップリングさせることにより製造されることができ、特にこれらの反応は、グリニャール試薬やリチウム化合物または銅−リチウム化合物などの有機金属試薬の使用または生成を伴う。例えば、式VIIの化合物を、対応するグリニャール試薬またはリチウム銅酸化物試薬に変換し、式VIの化合物と反応させてもよい。あるいは、式VIの化合物を、対応するグリニャール試薬またはリチウム銅酸化物試薬に変換し、式VIIの化合物と反応させてもよい。上述した通り、反応性の置換基のための適切な保護基は、本技術分野の当業者に周知であろう。
【0068】
中間体II〜VIIは、従来の手法により製造されうる。一例を挙げると、式III、VまたはVIIの化合物は、式:
【0069】
【化21】
【0070】
の化合物を、式:
【0071】
【化22】
【0072】
式中、B’は、−NHR2、−OHまたは−CHYR7であり、Xは脱離基である、
の化合物と、塩基の存在下で反応させることにより製造されうる。これに続き、必要であれば適切な保護基を用いて、加水分解および脱炭酸によりシアノ基を除去してもよい。
【0073】
式Iの化合物は、葉酸、特に葉酸の代謝誘導体を基質として利用する1または2以上の酵素に対し、阻害作用を有する。これらの酵素としては、GARトランスホルミラーゼ、AICARトランスホルミラーゼ、ジヒドロ葉酸レダクターゼおよびチミジル酸シンターゼが挙げられる。前記化合物は、ジヒドロ葉酸レダクターゼの阻害剤として特に活性であるように思われる。前記化合物は、単独でまたは組み合わせて、絨毛癌、白血病、女性乳房の腺癌、頭部および頸部の表層の癌、扁平上皮癌または小細胞癌、および種々のリンパ肉腫などの、従来はメトトレキサートで治療されてきた新生物を治療するために用いられうる。理論に拘束されるわけではないが、式Iの化合物の修飾されたケトメチレン性またはチオケトメチレン性の側鎖により、メトトレキサートに比べて腎毒性が低減されるものと考えられる。前記ケトメチレン性またはチオケトメチレン性の基の不安定性はより低いため、加水分解による不活性化は最小限であり、これにより半減期は長くなる。また、式Iの化合物においては、従来技術と比較して物理化学的な性質が改善されている。
【0074】
上述したように、癌細胞は健常な細胞よりも急速に複製するためにより多くのヌクレオチドを必要とすることから、本発明の化合物における主要な関心は、癌の治療における使用に関する。しかしながら、急速に増殖する細胞は全て、ヌクレオチドに対して類似の高い要求性を示すことから、本発明の化合物の治療上の有用性はこれに限られない。例えば、メトトレキサートは乾癬および菌状息肉腫の治療、および子宮外妊娠の患者における流産の誘導に用いられてきた。メトトレキサートはまた、慢性関節リウマチの治療にも用いられてきた。ただし、この例における作用のメカニズムは完全には判明していない。
【0075】
本発明の化合物は、新生物に罹患して治療を必要としている哺乳動物(好ましくは、ヒト)に対し、単独で、または他の抗癌剤もしくはステロイドなどの他の治療剤と組み合わせて、経口または好ましくは非経口のいずれかで投与されうる。非経口の投与経路としては、筋肉内、髄腔内、静脈内または動脈内が挙げられる。
【実施例】
【0076】
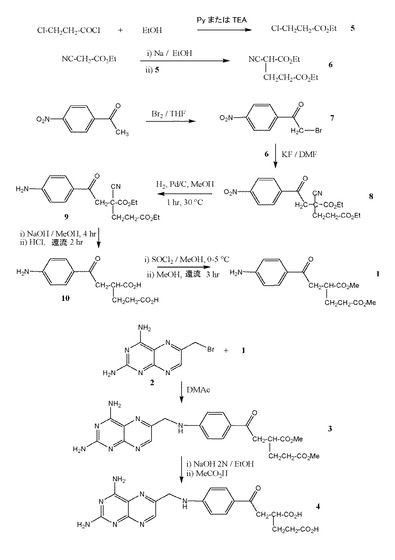
本発明がより完全に理解されるように、添付の図面を参照しながら実施例により本発明を説明する。ここで、以下の実施例1および2に詳細に記載されるように、図1は本発明による2つの化合物(化合物3および4)の全合成の模式図である。
【0077】
以下の実施例は本発明を例示するためのものであり、本発明をいかようにも制限するものではない。
【0078】
実施例1
化合物3の合成
出発物質の入手源
3−クロロプロパノイルクロライドおよびエチルシアノアセテートは、シグマ−アルドリッチ カンパニー リミテッド(ジ オールド ブリックヤード、ニュー ロード、ジリンガム、ドルセット SP8 4XT、イギリス)より購入し、または常法により合成した。α−ブロモ−p−ニトロ−アセトフェノン(7)は、テトラヒドロフラン(THF)中で臭素を用いたp−ニトロアセトフェノンの臭素化により得た。2,4−ジアミノ−6−ブロモメチルプテリジン(2)は、常法により得た(例えば、米国特許第4,077,957号および米国特許第4,224,446号を参照)。
【0079】
段階A:
化合物6の合成
ピリジンまたはトリエチルアセテートの存在下で、エタノールを用いて3−クロロプロパノイルクロライドをエステル化して、3−クロロプロピオン酸エチル(5)を製造した。L.Ruzicka et.al.,Helv.Chim.Acta 17,183−200(1934),CA 28:2584,またはKoelsch,C.F.,J.Am.Chem.65,2458−9(1943)の手法に従い、3−クロロプロピオン酸エチル(5)をエチルシアノアセテートと縮合させて、α−シアノグルタル酸ジエチル(6)を形成した。1H−NMRにより予想される構造を確認した。GC:純度97%。
【0080】
段階B:
化合物8の合成
0〜5℃にて、175g(0.71mmol)のα−ブロモ−p−ニトロ−アセトフェノン(7)を、175g(0.82mmol)のα−シアノグルタル酸ジエチル(6)および175g(3mmol)のKFの500mL DMF中の懸濁液に分割して添加した。この反応を薄層クロマトグラフィ(TLC)によりモニターした。4時間後、0.1%の酢酸を含有するpH5の水2Lに反応混合物を懸濁させた。水をデカンテーションした後、水(2×750mL)で粘着性の沈殿物を洗浄し、次いでメタノール300mLで練和した。結晶化が完了したら、沈殿物を濾過し、過剰のメタノールおよびエーテルで連続的に洗浄し、融点92.1℃の黄色固体である、化合物8を210g得た(収率68%)。シリカゲルでのクロマトグラフィ(ベンゼン:シクロヘキサン:エタノール=50:50:5)後、生成物の融点は99.7℃であった。
【0081】
シリカゲルプレート上でのTLC(ベンゼン:エタノール:シクロヘキサン:石油エーテル:酢酸=5:1:3:10:0.1)ではRf(保持因子)0.38の単一のスポットが確認された。HPLC:純度97%。
【0082】
段階C:
化合物9の合成
30g(0.08mmol)の化合物(8)をメタノール400mLに溶解させ、6gの20%Pd/C触媒の存在下、室温にて水素化フラスコ中で水素化した。理論体積の水素(c.6200mL;0.28mmol)が1時間で吸収された(TLCコントロール)。白金触媒を濾過し、メタノールを蒸発させた。得られた粗生成物を真空乾燥により固化させて、黄色固体である化合物(9)を27.6g得て(収率99%)、これをさらに精製することなく後述する未精製の化合物(10)への変換に用いた。純度はTLC分析に使用可能な値であった。TLC(クロロホルム:メタノール=4:1)ではRf0.5の単一のスポットが確認された(4−ジメチルアミノベンズアルデヒドとの特徴的な反応)。塩酸中で還流後、塩酸塩を単離した。LC−MSおよび1H−NMRにより、予想される構造を確認した。HPLC:純度99%。
【0083】
段階D:
化合物10の合成
メタノール1000mL中の52.2g(0.15mmol)の中間体(9)の溶液を調製した。6N NaOH188mLを、室温にて1時間かけて滴下し、その溶液を12時間静置した。次いで、水300mLでこの反応混合物を希釈し、高真空下で濃縮した。残渣に37%塩酸700mLを添加し、得られた混合物を加熱して4時間還流した。
【0084】
メタノール1.5Lで、得られた混合物を希釈し、塩化ナトリウムの沈殿を濾過により除去した。この濾液を段階Eにおいて用いた。希釈前に、懸濁液を濾過し、過剰の水、アセトンおよびエーテルで連続的に沈殿物を洗浄することにより少量の二塩基酸(10)を単離した。TLC(クロロホルム:メタノール=4:1)ではRf0.26の単一のスポットが確認された。
【0085】
段階E:
化合物1の合成
段階Dにおいて得られたジカルボン酸(10)のメタノール溶液を0〜5℃に冷却し、塩化チオニル100mLを滴下した。反応混合物を還流下で3時間撹拌し、次いで室温まで冷却し、溶媒を蒸発留去した。得られた沈殿物を濾過し、エーテルで洗浄して、融点115〜116℃の固体である化合物1を27g得た(収率63%)。
【0086】
テトラヒドロフランからの再結晶後、116〜117℃の融点を有する化合物1の白色結晶17.5gを得た。TLC(クロロホルム:メタノール=4:1)ではRf0.73の単一のスポットが確認された。UVスペクトル:234、319nm(メタノール)。1H−NMRスペクトル:2.0(2H,m,CH2CH2COOCH3),2.5(2H,t,CH2CH2COOCH3),3.1(2H,m,COCH2),3.5(1H,m,COCH2CH),3.75(6H,s,COOCH3),7.6−8.0(4H,m,CH原子)。HPLC:純度99%
段階F:
N−[4−[[(2,4−ジアミノ−6−プテリジニル)メチル]アミノ]ベンゾイル]シュードグルタミン酸エステル(化合物3)の合成
7g(27.4mmol)の2,4−ジアミノ−6−ブロモメチルプテリジン(2)および7g(23.4mmol)のN−[(4−メチルアミノ)ベンゾイル]シュードグルタミン酸ジメチル(1)の70mLのN,N−ジメチルアセトアミド中での混合物を70℃にて30分間撹拌し、次いで室温にて一晩、遮光下で静置し、次いで100℃に10分間再度加熱した。この反応をTLCにより制御した。冷却後、酢酸でpH4に酸性化された水(1000mL)に反応混合物を注いだ。生成した黄褐色の沈殿物を濾過し、水で3回洗浄し、空気乾燥させた。2.6gの橙黄色の生成物(3)を得た(融点200〜210℃)。10%NaHCO3で濾液を処理し、生成した沈殿物を同様に分離して、ジメチルエステル(3)の第2の画分を得た(2g)。合計収率:36%。TLC(クロロホルム:メタノール=4:1)ではRf0.48の単一のスポットが確認された。UVスペクトル:210、242、332(0.1N塩酸);238、335(メタノール)。
【0087】
実施例2
N−[4−[[(2,4−ジアミノ−6−プテリジニル)メチル]アミノ]ベンゾイル]シュードグルタミン酸(化合物4)の合成
1g(2.1mmol)のジメチルエステル(3)を、2N NaOH10mLおよびエタノール25mLの溶液に分割して添加し、混合物を室温にて4時間撹拌した。生成した沈殿物を濾過し、蒸留水に溶解させた。このアルカリ溶液を活性炭で処理し、濾過し、10%酢酸でpHを4.5に調整した。沈殿物を濾過し、pH4.5の水、次いでアセトンで洗浄して、化合物4を0.8g得た(収率85%)。茶色の固体である生成物を、分取HPTLC(高速薄層クロマトグラフィ)により精製した。CH3CN−H2O−NH4OH(50:50:5)で溶離した後、pH8のNaOH溶液100mLでシリカゲルから二塩基酸を抽出した。凍結乾燥により水分を除去した。TLC(CH3CN:H2O:NH4OH=7:2:1)ではRf0.80の単一のスポットが確認された。質量スペクトル:m/z 120(M+,100%)。IRスペクトル(KBr):1651(COCH2)、1594(C=C)、1563、1403(C=O酸)、1176(C−O)、823(CH)。UVスペクトル:242、332nm(0.1N塩酸);232、259、325nm(0.1N NaOH);229、262、318(メタノール)。1H−NMR:1.6(3H,m,CH−CH2)、2.2(2H,t,CH2CH2COOH)、2.9(2H,m,COCH2)、4.6(2H,s,CH2NH)、6.8−7.8(4H,m,CH原子)、9(1H,s,7−CH)。HPLC:純度97%。
【0088】
式Iの他の化合物は、上述した手法を適宜適応させることにより、製造されうる。例えば、中間体(1)および類似体化合物は、ホルムアルデヒドおよびシアノ水素化ホウ素ナトリウムとの反応により、対応するN−メチル誘導体に変換されうる。また、中間体(6)は、常法に従ってシアノ酢酸エチルをアクリル酸エチルと反応させることにより、製造されうる。
【0089】
実施例3
インビトロでのDHFRの阻害
式Iの化合物のインビトロでのジヒドロ葉酸レダクターゼ(DHFR)阻害能を、標準的なDHFR酵素阻害アッセイを用いて測定した。DHFR酵素についてはラット肝から精製し、または、大腸菌での組換え発現により製造された市販のDHFRを用いた。酵素活性のアッセイは、50mM N−トリス(ヒドロキシメチル)メチル−2−アミノエタンスルホン酸(pH7.0)、1mM EDTA、75μM 2−メルカプトエタノール、0.1%ウシ血清アルブミン、20μMジヒドロ葉酸および100μM NADPHを含有する溶液の340nmでのUV吸光度の変化を、37℃にてモニターすることにより行った。ジヒドロ葉酸を添加することにより、反応が開始した。各阻害剤の力価を2回ずつ測定し、平均のDHFR活性を阻害剤濃度に対してプロットして、IC50値を得た。IC50(化合物)/IC50(MTX)の比を相対IC50と定義し、化合物438および化合物497についての(5つのうちの)1つの代表的な実験のメトトレキサート(MTX)と比較した結果を表1に示す。得られた結果を全体的に見ると、試験した式Iの化合物の多くがインビトロでMTXに類似の活性を有していることが示唆される。
【0090】
【表2】
【0091】
注:化合物438は、R3(−CH2CH2CO2H)がHに置換されていること以外は、図1の化合物4と同一の構造を有する。
【0092】
実施例4
インビトロでの細胞毒性
薬剤添加後の72時間までの異なる時点での細胞の生存率を測定することにより、多数の腫瘍細胞株(CCRF−CEM、HepG2、HeLa、KB、L1210、A549およびCOLO205)に対する式1の化合物の細胞毒性を評価し、メトトレキサートおよびペメトレキセド(アリムタ(登録商標)として入手)の対応する細胞毒性と比較した。式Iの化合物は、試験した全ての細胞株の増殖に対して強力な阻害効果を示し、最も強い阻害効果はL1210細胞株に対するものであった。メトトレキサートおよびペメトレキセドと比較すると、式Iの化合物は全ての細胞株に対して類似かより強い阻害効果を示し、細胞毒性の発現がより速い場合もあった。さらに、Bが−CH2NH−である式Iの化合物が特に強い細胞毒性を示すことが判明した。
【0093】
ヒポキサンチンなどのプリン類やアミノイミダゾールカルボキサミドを(非常に高濃度まで)添加しても、細胞毒性は元に戻らなかったが、ロイコボリンを添加すると細胞毒性が元に戻った。これは、細胞毒性が葉酸に関連したメカニズムへの拮抗によるものであることを示す。DHFRが主要な標的である提案された作用メカニズムと一致するように、チミジンの添加では高濃度の場合にのみ、式Iの化合物により誘導された細胞毒性は元に戻った。これらの効果によれば、プリンのデノボ合成が特異的に阻害され、メトトレキサートの場合よりは顕著であるものの、チミジル酸サイクルはあまり大きく阻害されないことが示される。式Iの化合物はまた、メトトレキサートに匹敵する濃度範囲でグリシンアミドリボヌクレオチドトランスフェラーゼをも阻害した。
【0094】
実施例5
動物モデルにおけるインビボでの腫瘍阻害
式Iの化合物のマウスにおける腫瘍増殖阻害能を、以下のようにして試験した。DBA/2マウス(8匹のマウス/処理の群)の腋窩部に、5×106個のL1210細胞を皮下注射した。生理食塩水単独、または式Iの化合物を含有する生理食塩水を腹腔内投与した後、所定の時間に(生理食塩水のみを投与された)コントロールの腫瘍の長さおよび幅を測定し、試験化合物を投与された動物の値と比較して、阻害百分率を算出した。1日1回6日間の試験化合物含有生理食塩水の腹腔内投与により、60%で腫瘍が消失して長期間生存に至った(腫瘍重量がゼロ)。生理食塩水処理したコントロール動物および化合物4(0.5mg/kg)を投与した動物の生存期間の中央値は、それぞれ6.7日および15.6日であった。化合物の経口投与では高用量の阻害剤が必要とされ、それほど顕著ではないが依然として有意な腫瘍重量の減少および25%の長期生存が、食塩水処理のコントロール群と比較すると見られた。
【0095】
式Iの化合物はまた、癌腫W256(TGI=28%)に対してもインビボで活性を示した。これはおそらく、他の抗葉酸剤と比較して高い溶解性とこれに起因する腫瘍への受動輸送による。ウォーカー(Walker)−256ラット腫瘍モデルにおいて、生理食塩水処理したコントロール動物および化合物4(1mg/kg)を腹腔内投与した動物の生存期間の中央値は、それぞれ22.5日および46.3日超であった。
【0096】
以下の異なる処理計画により腫瘍の進展を評価した。その結果を表2に示す。
【0097】
【表3】
【図面の簡単な説明】
【0098】
【図1】本発明による2つの化合物(化合物3および4)の全合成の模式図である。
【技術分野】
【0001】
明細書
本発明は、新規な一連の化合物に関する。特に本発明は、本来の基質が葉酸または葉酸誘導体(葉酸塩)である酵素を阻害し、癌のような疾患の治療に用いられうる新規な化合物に関する。
【背景技術】
【0002】
癌細胞は他の多くの細胞よりも急速に複製することから、より多くのヌクレオチド(デオキシリボ核酸(DNA)およびリボ核酸(RNA)の前駆体)を必要とする。全ての細胞が常に剰余のヌクレオチド(アデノシン三リン酸(ATP)を除く)を貯蔵しているわけではないため、ヌクレオチドはDNAおよびRNAの合成の間、継続的に合成される必要がある。従って、癌細胞の複製はヌクレオチド生合成の阻害に対して健常な細胞の複製よりも感受性が高くなりがちであり、このため、かような阻害が可能な治療剤が関心を集めつつある。
【0003】
ヌクレオチドは、基本的な代謝前駆体からデノボ経路を経て、あるいは、核酸分解の生成物である遊離塩基およびヌクレオシドが再利用されるサルベージ経路を経て、生物学的に合成されうる。新規な化学治療剤の探索に関して主に関心を集めているのは、デノボ経路である。
【0004】
ヌクレオチドである2’−デオキシアデノシン−5’−一リン酸(dAMP)および2’−デオキシグアノシン−5’−一リン酸(dGMP)はともに、イノシン一リン酸(IMP)から新たに合成され、このIMPは5−ホスホリボシル−1−ピロリン酸(PPRP)由来である。このPPRPとIMPとの間の生合成経路に関与している2つの酵素は、GARトランスホルミラーゼおよびAICARトランスホルミラーゼである。以下に示すように、GARトランスホルミラーゼは、N10−ホルミルテトラヒドロ葉酸を用いてグリシンアミドリボヌクレオチド(GAR)をホルミルグリシンアミドリボヌクレオチド(FGAR)に変換する。一方、AICARトランスホルミラーゼは、同じ化合物を用いて5−アミノイミダゾール−4−カルボキサミドリボヌクレオチド(AICAR)をN−ホルミルアミノイミダゾール−4−カルボキサミドリボヌクレオチド(FAICAR)に変換する。
【0005】
【化1】
【0006】
これに対し、ヌクレオチドである2’−デオキシチミジン−5’−一リン酸(dTMP)は、2’−デオキシウリジン−5’−一リン酸(dUMP)からデノボ合成により製造される。これは、酵素であるチミジル酸シンターゼにより触媒される変換反応である。この変換反応において、N5,N10−メチレン−テトラヒドロ葉酸は7,8−ジヒドロ葉酸へと還元される。そして、N5,N10−メチレン−テトラヒドロ葉酸は、酵素であるジヒドロ葉酸レダクターゼ(DHFR)およびセリンヒドロキシメチルトランスフェラーゼを用いてテトラヒドロ葉酸を経由して再生される。これらのプロセスを以下に図示する。
【0007】
【化2】
【0008】
これらのメカニズムの阻害が、癌の治療に利用されている。例えば、米国特許第2,512,572号には、「葉酸拮抗薬」に分類され、本来の基質よりも約100倍高い親和性で結合してジヒドロ葉酸レダクターゼを競合的に阻害し、dTMPの合成に必須のテトラヒドロ葉酸の再生を阻害することによりDNA合成を阻害する、強力な化学治療剤であるメトトレキサートを含む多くの置換プテリジン類が開示されている。これにより、癌細胞においていわゆる「チミン欠乏死」が誘導される。メトトレキサートはまた、程度は小さいものの、GARトランスホルミラーゼ、AICARトランスホルミラーゼおよびチミジル酸シンターゼをも阻害する。メトトレキサートおよび他の関連する抗葉酸剤の構造を以下に示す。
【0009】
【表1】
【0010】
米国特許第4,684,653号には、式:
【0011】
【化3】
【0012】
式中、R1は、OHまたはNH2であり、R3は、水素原子、メチル基またはエチル基である、
の化合物およびその対応する5,6,7,8−テトラヒドロ誘導体が開示されている。これらの化合物は、葉酸およびその代謝誘導体を基質として利用する1または2以上の酵素に対して効果を有することが開示されている。
【0013】
米国特許第5,344,932号には、式:
【0014】
【化4】
【0015】
式中、R5は、水素原子またはNH2であり、R4は、水素原子またはメトキシ基であり、R2は、水素原子または製薬上許容されうるカチオンである、
のグルタミン酸誘導体が開示されており、これらの化合物が、葉酸およびその代謝誘導体を基質として利用する1または2以上の酵素に対して阻害効果を有することが開示されている。
【0016】
米国特許4,077,957号には:
【0017】
【化5】
【0018】
を含む種々のプテリジン化合物の合成方法が開示されている。
【0019】
かような化合物は、癌治療のための新たな治療戦略の開発に有用であることが判明しているが、効果が小さい、ある患者ではかような薬物に対してもともと耐性がある、または後天的に耐性となる、毒性や好ましくない副作用があるといった、使用に関連した多くの問題が依然として存在する。従って、癌の治療に用いられることができ、上述した問題の1または2以上を解消しうる代替化合物に対する要求は依然として残っている。
【発明の開示】
【0020】
従って、本発明の第1の形態によれば、式Iの化合物、またはその製薬上許容されうる塩:
【0021】
【化6】
【0022】
式中、
Zは、OまたはSであり;
nは、1〜3であり;
R3は、−CO2R8、−C(O)SR8、−C(O)NHR8、−C(S)OR8、−C(S)SR8、−C(S)NHR8、−C(NH)SR8、または−C(NH)NHR8(ここで、R8は水素原子もしくはアルキル基である)であり;
R4は、水素原子、−CH2R5または−CH2CH2R5(ここで、R5は、それぞれ独立してR3の定義のいずれかを有する)であり;
Bは、−NR2−、−CH2NR2−、−CH2CH2NR2−、−CH2CHR7−または−CH2O−(ここで、R2は、水素原子またはC1−3のアルキル基、アルケニル基もしくはアルキニル基であり、R7は、水素原子またはC1−3のアルキル基もしくはアルコキシ基である)であり;
Aは:
【0023】
【化7】
【0024】
式中、
R1は、−NH2または−OHであり;
CおよびDは、それぞれ独立して、1または2以上のヘテロ原子を含んでもよい、5員または6員の、置換されたまたは非置換の、芳香族環または非芳香族環であり、Cは任意の可能な位置で基Bと結合している、
である、
が提供される。
【0025】
本発明の第2の形態によれば、治療に使用される、本発明の第1の形態の化合物が提供される。
【0026】
本発明の第3の形態によれば、本発明の第1または第2の形態の化合物を含む医薬組成物が提供される。
【0027】
本発明の第4の形態によれば、葉酸または葉酸誘導体に依存性の酵素の阻害に応答性の症状の治療に用いられる医薬の製造のための、本発明の第1または第2の形態の化合物の使用が提供される。
【0028】
本発明の第5の形態によれば、癌の治療に用いられる医薬の製造のための、本発明の第1または第2の形態の化合物の使用が提供される。
【0029】
本発明の第6の形態によれば、以下の段階:
(a)式IIの化合物:
【0030】
【化8】
【0031】
式中、Aは上記と同様の定義であり、mは、0、1または2であり、Xは脱離基である、
を、式IIIの化合物:
【0032】
【化9】
【0033】
式中、Z、n、R2、R3およびR4は上記と同様の定義である、
と反応させる段階;
(b)式IVの化合物:
【0034】
【化10】
【0035】
式中、Aは上記と同様の定義であり、Xは脱離基である、
を、式Vの化合物:
【0036】
【化11】
【0037】
式中、Z、n、R3およびR4は上記と同様の定義である、
と反応させる段階;あるいは、
(c)以下の化合物VIまたはVII:
【0038】
【化12】
【0039】
式中、A、Z、n、R3、R4およびR7は上記と同様の定義であり、いずれの場合においても、Yは独立してハロゲン原子である、
の一方を、対応する有機金属試薬に変換させ、前記有機金属試薬を化合物VIまたはVIIの他方と反応させる段階、
を有する、本発明の第1または第2の形態の化合物の製造方法が提供される。
【0040】
本発明の第7の形態によれば、式II−Aの化合物:
【0041】
【化13】
【0042】
式中、Xは、Cl、BrまたはIである、
を、式III−Aの化合物:
【0043】
【化14】
【0044】
と反応させる段階を有する、式I−A:
【0045】
【化15】
【0046】
式中、n、R1、R3およびR4は、上記と同様の定義である、
を有する、本発明の第1または第2の形態の化合物またはその製薬上許容されうる塩の製造方法が提供される。
【0047】
本発明の第8の形態によれば、上記で定義され、R4が−CH2R5または−CH2CH2R5である場合を除く、式III、VまたはVIIの化合物が提供される。
【0048】
本発明の種々の形態のいずれかの発明の好ましい実施形態を以下に記載し、または従属項において規定する。
【0049】
本発明の種々の形態の全てにおいて、C*とマークされた炭素は不斉炭素であってもよく(R4が水素原子ではない場合)、この場合、式Iの化合物は、ラセミ体の形態で存在しても、常法によりその(+)または(−)光学異性体に分離されてもよいことは当然である。また、いくつかの化合物では、他のキラル中心が存在して1または2以上の別の光学異性体のペアを生じてもよい。例えば、Bが−CH2CHR7−であり、R7がC1−3のアルキル基またはアルコキシ基である化合物においては、第2のキラル中心が存在する。かようなラセミ体の形態または光学異性体の形態は全て、本発明の技術的範囲に含まれるものである。さらに、式Iの化合物は、1または2以上の互変異性体の形態で存在してもよいことは理解されるであろうし、これらの形態のそれぞれもまた、本発明の技術的範囲に含まれるものである。
【0050】
以下により詳細に記載される通り、式Iの化合物は葉酸の構造類似体であり、インビトロでメトトレキサートに匹敵するレベルで、ジヒドロ葉酸レダクターゼ(DHFR)のような葉酸または葉酸誘導体(葉酸塩)に依存性の酵素の阻害剤としての作用を有することが判明したのである。式Iの化合物はまた、動物モデルにおいてインビボでも腫瘍の増殖を阻害する活性を有することが判明している。ここではメカニズムの詳細は割愛するが、後者の活性は、DHFRの競合的拮抗薬として作用する当該化合物の活性に起因するかもしれないことが予想される。式Iの化合物は、癌や、葉酸または葉酸誘導体に依存性の酵素の阻害に応答性の症状の治療に用いられうる。
【0051】
式Iの好ましい化合物は、以下の条件の1または2以上を満たす:
ZがOである;
nが1である;
R3が−CO2R8であり、R4が−CH2CH2CO2R8である;
R8が、水素原子、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基またはtert−ブチル基であり、好ましくは水素原子、メチル基またはエチル基であり、より好ましくは水素原子である;
Bが、−CH2NR2−、−CH2CHR7−または−CH2O−であり、好ましくは−CH2NR2−である;
R2が、水素原子、メチル基、エチル基または−CH2C≡CHであり、好ましくは水素原子である;
R7が、水素原子、メチル基、エチル基またはメトキシ基であり、好ましくは水素原子である。
【0052】
また、式III、VまたはVIIの好ましい化合物は、上述したZ、n、R2、R3、R4および/またはR7の1または2以上の好ましい規定を有する。
【0053】
式I、II、IVまたはVIの基Aにおいて、Dは、好ましくは5員のヘテロ芳香族環である。好ましくは、Aは:
【0054】
【化16】
【0055】
である。
【0056】
基Aにおいて、Cは、以下の基(隣接する環および基Bに結合する点を示す)の1つであってもよい:
【0057】
【化17】
【0058】
式中、Xは、CHまたはNであり、かつ、YがCであり、R6が水素原子、メチル基、エチル基またはHCOであるか、YがNであり、R6が孤立電子対である、のいずれかである。好ましい実施形態において、XおよびYはともにNであり、R6は孤立電子対である。
【0059】
特に好ましい基Aは、天然のプテリジン類および他のへテロ環塩基に極めて類似した、以下の構造を有する:
【0060】
【化18】
【0061】
特に関心を集めているのは、以下の2つのA基である:
【0062】
【化19】
【0063】
上述した2つのA基を有し、Bが−CH2NR2−であり、R2が、水素原子、メチル基、エチル基または−CH2C≡CHであり、ZがOであり、nが1であり、R3が−CO2R8であり、好ましくは加水分解可能な任意のエステル基である、式Iの化合物が特に好ましい。この群の化合物の個別の例としては、以下のものが挙げられる:
【0064】
【化20】
【0065】
式Iの化合物は、容易に入手可能で安価な出発物質から、本発明の方法により製造されうる。例えば、Bが−NR2−、−CH2NR2−または−CH2CH2NR2−である場合に、式Iの化合物は、式IIの化合物を式IIIの化合物とカップリングさせることにより製造されうる。脱離基Xは、一般的には塩素、臭素またはヨウ素などのハロゲンであり、特には臭素またはヨウ素である。この反応は、好ましくはジメチルホルムアミド(DMF)またはジメチルアセトアミド(DMAc)などの双極性非プロトン性溶媒中で行われうる。フッ化カリウムなどの塩基性触媒が用いられてもよく、これにより第3級アミンまたは重炭酸ナトリウムと比べて高い収率が得られる。必要であれば、上記の反応前に、本技術分野において公知の適切な保護基を用いて反応性の高い基を保護し、後に、常法により脱保護してもよい。例えば、R3が水素原子であり、R4が−CH2CH2CO2Hである場合、これらの酸基は、例えばメチルエステル基により保護され、その後にエタノール中での水酸化ナトリウムを用いたアルカリ加水分解などの公知の手法により脱保護され、氷酢酸などの酸の添加により沈殿しうる。従って、式IIおよび式IIIの化合物のいずれかまたは双方が保護された形態であるこれらの化合物の反応は、本発明の方法に包含されることが理解されうるであろう。
【0066】
Bが−CH2O−である場合において、式Iの化合物は、例えば、式IVの化合物を、ウィリアムソンエーテル型反応により式Vの化合物とカップリングさせることにより製造されうる。この反応において、式Vの化合物は一般的に、式IVの化合物との反応前に、例えばNaHなどの塩基を用いてアロキシド(aroxide)イオンの形態に変換される。Xは、任意の適切な脱離基であればよく、特にはハロゲン原子である。
【0067】
Bが−CH2CHR7−である場合において、式Iの化合物は、例えば、式VIの化合物を、任意の公知の炭素−炭素結合形成反応により式VIIの化合物とカップリングさせることにより製造されることができ、特にこれらの反応は、グリニャール試薬やリチウム化合物または銅−リチウム化合物などの有機金属試薬の使用または生成を伴う。例えば、式VIIの化合物を、対応するグリニャール試薬またはリチウム銅酸化物試薬に変換し、式VIの化合物と反応させてもよい。あるいは、式VIの化合物を、対応するグリニャール試薬またはリチウム銅酸化物試薬に変換し、式VIIの化合物と反応させてもよい。上述した通り、反応性の置換基のための適切な保護基は、本技術分野の当業者に周知であろう。
【0068】
中間体II〜VIIは、従来の手法により製造されうる。一例を挙げると、式III、VまたはVIIの化合物は、式:
【0069】
【化21】
【0070】
の化合物を、式:
【0071】
【化22】
【0072】
式中、B’は、−NHR2、−OHまたは−CHYR7であり、Xは脱離基である、
の化合物と、塩基の存在下で反応させることにより製造されうる。これに続き、必要であれば適切な保護基を用いて、加水分解および脱炭酸によりシアノ基を除去してもよい。
【0073】
式Iの化合物は、葉酸、特に葉酸の代謝誘導体を基質として利用する1または2以上の酵素に対し、阻害作用を有する。これらの酵素としては、GARトランスホルミラーゼ、AICARトランスホルミラーゼ、ジヒドロ葉酸レダクターゼおよびチミジル酸シンターゼが挙げられる。前記化合物は、ジヒドロ葉酸レダクターゼの阻害剤として特に活性であるように思われる。前記化合物は、単独でまたは組み合わせて、絨毛癌、白血病、女性乳房の腺癌、頭部および頸部の表層の癌、扁平上皮癌または小細胞癌、および種々のリンパ肉腫などの、従来はメトトレキサートで治療されてきた新生物を治療するために用いられうる。理論に拘束されるわけではないが、式Iの化合物の修飾されたケトメチレン性またはチオケトメチレン性の側鎖により、メトトレキサートに比べて腎毒性が低減されるものと考えられる。前記ケトメチレン性またはチオケトメチレン性の基の不安定性はより低いため、加水分解による不活性化は最小限であり、これにより半減期は長くなる。また、式Iの化合物においては、従来技術と比較して物理化学的な性質が改善されている。
【0074】
上述したように、癌細胞は健常な細胞よりも急速に複製するためにより多くのヌクレオチドを必要とすることから、本発明の化合物における主要な関心は、癌の治療における使用に関する。しかしながら、急速に増殖する細胞は全て、ヌクレオチドに対して類似の高い要求性を示すことから、本発明の化合物の治療上の有用性はこれに限られない。例えば、メトトレキサートは乾癬および菌状息肉腫の治療、および子宮外妊娠の患者における流産の誘導に用いられてきた。メトトレキサートはまた、慢性関節リウマチの治療にも用いられてきた。ただし、この例における作用のメカニズムは完全には判明していない。
【0075】
本発明の化合物は、新生物に罹患して治療を必要としている哺乳動物(好ましくは、ヒト)に対し、単独で、または他の抗癌剤もしくはステロイドなどの他の治療剤と組み合わせて、経口または好ましくは非経口のいずれかで投与されうる。非経口の投与経路としては、筋肉内、髄腔内、静脈内または動脈内が挙げられる。
【実施例】
【0076】
本発明がより完全に理解されるように、添付の図面を参照しながら実施例により本発明を説明する。ここで、以下の実施例1および2に詳細に記載されるように、図1は本発明による2つの化合物(化合物3および4)の全合成の模式図である。
【0077】
以下の実施例は本発明を例示するためのものであり、本発明をいかようにも制限するものではない。
【0078】
実施例1
化合物3の合成
出発物質の入手源
3−クロロプロパノイルクロライドおよびエチルシアノアセテートは、シグマ−アルドリッチ カンパニー リミテッド(ジ オールド ブリックヤード、ニュー ロード、ジリンガム、ドルセット SP8 4XT、イギリス)より購入し、または常法により合成した。α−ブロモ−p−ニトロ−アセトフェノン(7)は、テトラヒドロフラン(THF)中で臭素を用いたp−ニトロアセトフェノンの臭素化により得た。2,4−ジアミノ−6−ブロモメチルプテリジン(2)は、常法により得た(例えば、米国特許第4,077,957号および米国特許第4,224,446号を参照)。
【0079】
段階A:
化合物6の合成
ピリジンまたはトリエチルアセテートの存在下で、エタノールを用いて3−クロロプロパノイルクロライドをエステル化して、3−クロロプロピオン酸エチル(5)を製造した。L.Ruzicka et.al.,Helv.Chim.Acta 17,183−200(1934),CA 28:2584,またはKoelsch,C.F.,J.Am.Chem.65,2458−9(1943)の手法に従い、3−クロロプロピオン酸エチル(5)をエチルシアノアセテートと縮合させて、α−シアノグルタル酸ジエチル(6)を形成した。1H−NMRにより予想される構造を確認した。GC:純度97%。
【0080】
段階B:
化合物8の合成
0〜5℃にて、175g(0.71mmol)のα−ブロモ−p−ニトロ−アセトフェノン(7)を、175g(0.82mmol)のα−シアノグルタル酸ジエチル(6)および175g(3mmol)のKFの500mL DMF中の懸濁液に分割して添加した。この反応を薄層クロマトグラフィ(TLC)によりモニターした。4時間後、0.1%の酢酸を含有するpH5の水2Lに反応混合物を懸濁させた。水をデカンテーションした後、水(2×750mL)で粘着性の沈殿物を洗浄し、次いでメタノール300mLで練和した。結晶化が完了したら、沈殿物を濾過し、過剰のメタノールおよびエーテルで連続的に洗浄し、融点92.1℃の黄色固体である、化合物8を210g得た(収率68%)。シリカゲルでのクロマトグラフィ(ベンゼン:シクロヘキサン:エタノール=50:50:5)後、生成物の融点は99.7℃であった。
【0081】
シリカゲルプレート上でのTLC(ベンゼン:エタノール:シクロヘキサン:石油エーテル:酢酸=5:1:3:10:0.1)ではRf(保持因子)0.38の単一のスポットが確認された。HPLC:純度97%。
【0082】
段階C:
化合物9の合成
30g(0.08mmol)の化合物(8)をメタノール400mLに溶解させ、6gの20%Pd/C触媒の存在下、室温にて水素化フラスコ中で水素化した。理論体積の水素(c.6200mL;0.28mmol)が1時間で吸収された(TLCコントロール)。白金触媒を濾過し、メタノールを蒸発させた。得られた粗生成物を真空乾燥により固化させて、黄色固体である化合物(9)を27.6g得て(収率99%)、これをさらに精製することなく後述する未精製の化合物(10)への変換に用いた。純度はTLC分析に使用可能な値であった。TLC(クロロホルム:メタノール=4:1)ではRf0.5の単一のスポットが確認された(4−ジメチルアミノベンズアルデヒドとの特徴的な反応)。塩酸中で還流後、塩酸塩を単離した。LC−MSおよび1H−NMRにより、予想される構造を確認した。HPLC:純度99%。
【0083】
段階D:
化合物10の合成
メタノール1000mL中の52.2g(0.15mmol)の中間体(9)の溶液を調製した。6N NaOH188mLを、室温にて1時間かけて滴下し、その溶液を12時間静置した。次いで、水300mLでこの反応混合物を希釈し、高真空下で濃縮した。残渣に37%塩酸700mLを添加し、得られた混合物を加熱して4時間還流した。
【0084】
メタノール1.5Lで、得られた混合物を希釈し、塩化ナトリウムの沈殿を濾過により除去した。この濾液を段階Eにおいて用いた。希釈前に、懸濁液を濾過し、過剰の水、アセトンおよびエーテルで連続的に沈殿物を洗浄することにより少量の二塩基酸(10)を単離した。TLC(クロロホルム:メタノール=4:1)ではRf0.26の単一のスポットが確認された。
【0085】
段階E:
化合物1の合成
段階Dにおいて得られたジカルボン酸(10)のメタノール溶液を0〜5℃に冷却し、塩化チオニル100mLを滴下した。反応混合物を還流下で3時間撹拌し、次いで室温まで冷却し、溶媒を蒸発留去した。得られた沈殿物を濾過し、エーテルで洗浄して、融点115〜116℃の固体である化合物1を27g得た(収率63%)。
【0086】
テトラヒドロフランからの再結晶後、116〜117℃の融点を有する化合物1の白色結晶17.5gを得た。TLC(クロロホルム:メタノール=4:1)ではRf0.73の単一のスポットが確認された。UVスペクトル:234、319nm(メタノール)。1H−NMRスペクトル:2.0(2H,m,CH2CH2COOCH3),2.5(2H,t,CH2CH2COOCH3),3.1(2H,m,COCH2),3.5(1H,m,COCH2CH),3.75(6H,s,COOCH3),7.6−8.0(4H,m,CH原子)。HPLC:純度99%
段階F:
N−[4−[[(2,4−ジアミノ−6−プテリジニル)メチル]アミノ]ベンゾイル]シュードグルタミン酸エステル(化合物3)の合成
7g(27.4mmol)の2,4−ジアミノ−6−ブロモメチルプテリジン(2)および7g(23.4mmol)のN−[(4−メチルアミノ)ベンゾイル]シュードグルタミン酸ジメチル(1)の70mLのN,N−ジメチルアセトアミド中での混合物を70℃にて30分間撹拌し、次いで室温にて一晩、遮光下で静置し、次いで100℃に10分間再度加熱した。この反応をTLCにより制御した。冷却後、酢酸でpH4に酸性化された水(1000mL)に反応混合物を注いだ。生成した黄褐色の沈殿物を濾過し、水で3回洗浄し、空気乾燥させた。2.6gの橙黄色の生成物(3)を得た(融点200〜210℃)。10%NaHCO3で濾液を処理し、生成した沈殿物を同様に分離して、ジメチルエステル(3)の第2の画分を得た(2g)。合計収率:36%。TLC(クロロホルム:メタノール=4:1)ではRf0.48の単一のスポットが確認された。UVスペクトル:210、242、332(0.1N塩酸);238、335(メタノール)。
【0087】
実施例2
N−[4−[[(2,4−ジアミノ−6−プテリジニル)メチル]アミノ]ベンゾイル]シュードグルタミン酸(化合物4)の合成
1g(2.1mmol)のジメチルエステル(3)を、2N NaOH10mLおよびエタノール25mLの溶液に分割して添加し、混合物を室温にて4時間撹拌した。生成した沈殿物を濾過し、蒸留水に溶解させた。このアルカリ溶液を活性炭で処理し、濾過し、10%酢酸でpHを4.5に調整した。沈殿物を濾過し、pH4.5の水、次いでアセトンで洗浄して、化合物4を0.8g得た(収率85%)。茶色の固体である生成物を、分取HPTLC(高速薄層クロマトグラフィ)により精製した。CH3CN−H2O−NH4OH(50:50:5)で溶離した後、pH8のNaOH溶液100mLでシリカゲルから二塩基酸を抽出した。凍結乾燥により水分を除去した。TLC(CH3CN:H2O:NH4OH=7:2:1)ではRf0.80の単一のスポットが確認された。質量スペクトル:m/z 120(M+,100%)。IRスペクトル(KBr):1651(COCH2)、1594(C=C)、1563、1403(C=O酸)、1176(C−O)、823(CH)。UVスペクトル:242、332nm(0.1N塩酸);232、259、325nm(0.1N NaOH);229、262、318(メタノール)。1H−NMR:1.6(3H,m,CH−CH2)、2.2(2H,t,CH2CH2COOH)、2.9(2H,m,COCH2)、4.6(2H,s,CH2NH)、6.8−7.8(4H,m,CH原子)、9(1H,s,7−CH)。HPLC:純度97%。
【0088】
式Iの他の化合物は、上述した手法を適宜適応させることにより、製造されうる。例えば、中間体(1)および類似体化合物は、ホルムアルデヒドおよびシアノ水素化ホウ素ナトリウムとの反応により、対応するN−メチル誘導体に変換されうる。また、中間体(6)は、常法に従ってシアノ酢酸エチルをアクリル酸エチルと反応させることにより、製造されうる。
【0089】
実施例3
インビトロでのDHFRの阻害
式Iの化合物のインビトロでのジヒドロ葉酸レダクターゼ(DHFR)阻害能を、標準的なDHFR酵素阻害アッセイを用いて測定した。DHFR酵素についてはラット肝から精製し、または、大腸菌での組換え発現により製造された市販のDHFRを用いた。酵素活性のアッセイは、50mM N−トリス(ヒドロキシメチル)メチル−2−アミノエタンスルホン酸(pH7.0)、1mM EDTA、75μM 2−メルカプトエタノール、0.1%ウシ血清アルブミン、20μMジヒドロ葉酸および100μM NADPHを含有する溶液の340nmでのUV吸光度の変化を、37℃にてモニターすることにより行った。ジヒドロ葉酸を添加することにより、反応が開始した。各阻害剤の力価を2回ずつ測定し、平均のDHFR活性を阻害剤濃度に対してプロットして、IC50値を得た。IC50(化合物)/IC50(MTX)の比を相対IC50と定義し、化合物438および化合物497についての(5つのうちの)1つの代表的な実験のメトトレキサート(MTX)と比較した結果を表1に示す。得られた結果を全体的に見ると、試験した式Iの化合物の多くがインビトロでMTXに類似の活性を有していることが示唆される。
【0090】
【表2】
【0091】
注:化合物438は、R3(−CH2CH2CO2H)がHに置換されていること以外は、図1の化合物4と同一の構造を有する。
【0092】
実施例4
インビトロでの細胞毒性
薬剤添加後の72時間までの異なる時点での細胞の生存率を測定することにより、多数の腫瘍細胞株(CCRF−CEM、HepG2、HeLa、KB、L1210、A549およびCOLO205)に対する式1の化合物の細胞毒性を評価し、メトトレキサートおよびペメトレキセド(アリムタ(登録商標)として入手)の対応する細胞毒性と比較した。式Iの化合物は、試験した全ての細胞株の増殖に対して強力な阻害効果を示し、最も強い阻害効果はL1210細胞株に対するものであった。メトトレキサートおよびペメトレキセドと比較すると、式Iの化合物は全ての細胞株に対して類似かより強い阻害効果を示し、細胞毒性の発現がより速い場合もあった。さらに、Bが−CH2NH−である式Iの化合物が特に強い細胞毒性を示すことが判明した。
【0093】
ヒポキサンチンなどのプリン類やアミノイミダゾールカルボキサミドを(非常に高濃度まで)添加しても、細胞毒性は元に戻らなかったが、ロイコボリンを添加すると細胞毒性が元に戻った。これは、細胞毒性が葉酸に関連したメカニズムへの拮抗によるものであることを示す。DHFRが主要な標的である提案された作用メカニズムと一致するように、チミジンの添加では高濃度の場合にのみ、式Iの化合物により誘導された細胞毒性は元に戻った。これらの効果によれば、プリンのデノボ合成が特異的に阻害され、メトトレキサートの場合よりは顕著であるものの、チミジル酸サイクルはあまり大きく阻害されないことが示される。式Iの化合物はまた、メトトレキサートに匹敵する濃度範囲でグリシンアミドリボヌクレオチドトランスフェラーゼをも阻害した。
【0094】
実施例5
動物モデルにおけるインビボでの腫瘍阻害
式Iの化合物のマウスにおける腫瘍増殖阻害能を、以下のようにして試験した。DBA/2マウス(8匹のマウス/処理の群)の腋窩部に、5×106個のL1210細胞を皮下注射した。生理食塩水単独、または式Iの化合物を含有する生理食塩水を腹腔内投与した後、所定の時間に(生理食塩水のみを投与された)コントロールの腫瘍の長さおよび幅を測定し、試験化合物を投与された動物の値と比較して、阻害百分率を算出した。1日1回6日間の試験化合物含有生理食塩水の腹腔内投与により、60%で腫瘍が消失して長期間生存に至った(腫瘍重量がゼロ)。生理食塩水処理したコントロール動物および化合物4(0.5mg/kg)を投与した動物の生存期間の中央値は、それぞれ6.7日および15.6日であった。化合物の経口投与では高用量の阻害剤が必要とされ、それほど顕著ではないが依然として有意な腫瘍重量の減少および25%の長期生存が、食塩水処理のコントロール群と比較すると見られた。
【0095】
式Iの化合物はまた、癌腫W256(TGI=28%)に対してもインビボで活性を示した。これはおそらく、他の抗葉酸剤と比較して高い溶解性とこれに起因する腫瘍への受動輸送による。ウォーカー(Walker)−256ラット腫瘍モデルにおいて、生理食塩水処理したコントロール動物および化合物4(1mg/kg)を腹腔内投与した動物の生存期間の中央値は、それぞれ22.5日および46.3日超であった。
【0096】
以下の異なる処理計画により腫瘍の進展を評価した。その結果を表2に示す。
【0097】
【表3】
【図面の簡単な説明】
【0098】
【図1】本発明による2つの化合物(化合物3および4)の全合成の模式図である。
【特許請求の範囲】
【請求項1】
式Iの化合物、またはその製薬上許容されうる塩:
【化1】
式中、
Zは、OまたはSであり;
nは、1〜3であり;
R3は、−CO2R8、−C(O)SR8、−C(O)NHR8、−C(S)OR8、−C(S)SR8、−C(S)NHR8、−C(NH)SR8、または−C(NH)NHR8(ここで、R8は水素原子もしくはアルキル基である)であり;
R4は、水素原子、−CH2R5または−CH2CH2R5(ここで、R5は、それぞれ独立してR3の定義のいずれかを有する)であり;
Bは、−NR2−、−CH2NR2−、−CH2CH2NR2−、−CH2CHR7−または−CH2O−(ここで、R2は、水素原子またはC1−3のアルキル基、アルケニル基もしくはアルキニル基であり、R7は、水素原子またはC1−3のアルキル基もしくはアルコキシ基である)であり;
Aは:
【化2】
式中、
R1は、−NH2または−OHであり;
CおよびDは、それぞれ独立して、1または2以上のヘテロ原子を含んでもよい、5員または6員の、置換されたまたは非置換の、芳香族環または非芳香族環であり、Cは任意の可能な位置で基Bと結合している、
である。
【請求項2】
ZがOである、請求項1に記載の化合物。
【請求項3】
nが1である、請求項1または2に記載の化合物。
【請求項4】
R8が、水素原子、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基またはtert−ブチル基である、請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
R3が−CO2R8であり、R4が−CH2CH2CO2R8である、請求項1〜4のいずれか1項に記載の化合物。
【請求項6】
R8が、水素原子、メチル基またはエチル基である、請求項5に記載の化合物。
【請求項7】
Bが、−CH2NR2−、−CH2CHR7−または−CH2O−である、請求項1〜6のいずれか1項に記載の化合物。
【請求項8】
Bが−CH2NR2−である、請求項7に記載の化合物。
【請求項9】
R2が、水素原子、メチル基、エチル基または−CH2C≡CHである、請求項1〜8のいずれか1項に記載の化合物。
【請求項10】
R7が、水素原子、メチル基、エチル基またはメトキシ基である、請求項1〜9のいずれか1項に記載の化合物。
【請求項11】
Dが5員の芳香族環である、請求項1〜10のいずれか1項に記載の化合物。
【請求項12】
Aが:
【化3】
である、請求項11に記載の化合物。
【請求項13】
Cが:
【化4】
式中、Xは、CHまたはNであり、かつ、
YがCであり、R6が水素原子、メチル基、エチル基またはHCOであるか、
YがNであり、R6が孤立電子対である、のいずれかである、
である、請求項1〜12のいずれか1項に記載の化合物。
【請求項14】
Aが、式A−iまたは式A−iiで表される、請求項13に記載の化合物。
【化5】
【請求項15】
Aが、式A−i−1または式A−ii−1で表される、請求項14に記載の化合物。
【化6】
【請求項16】
Bが−CH2NR2−であり、R2が、水素原子、メチル基、エチル基または−CH2C≡CHであり、ZがOであり、nが1であり、R3が−CO2R8である、請求項15に記載の化合物。
【請求項17】
Bが、−CH2NR2−、−CH2CH2−、−CH2CHCH3−または−CH2O−であり、R2が水素原子、メチル基、エチル基または−CH2C≡CHであり、ZがOであり、nが1であり、R3が−CO2R8であり、R4が、水素原子または−CH2CH2CO2R8であり、R8が、それぞれ独立して水素原子、メチル基またはエチル基であり、AがA−iでありYがCである場合に、R6は水素原子であり、AがA−iiである場合にR1は−OHである、請求項14に記載の化合物。
【請求項18】
治療に使用される、請求項1〜17のいずれか1項に記載の化合物。
【請求項19】
請求項1〜18のいずれか1項に記載の化合物を含む、医薬組成物。
【請求項20】
葉酸または葉酸誘導体に依存性の酵素の阻害に応答性の症状の治療に用いられる医薬の製造のための、請求項1〜19のいずれか1項に記載の化合物の使用。
【請求項21】
癌の治療に用いられる医薬の製造のための、請求項1〜19のいずれか1項に記載の化合物の使用。
【請求項22】
以下の段階:
(a)式IIの化合物:
【化7】
式中、Aは請求項1と同様の定義であり、mは、0、1または2であり、Xは脱離基である、
を、式IIIの化合物:
【化8】
式中、Z、n、R2、R3およびR4は請求項1と同様の定義である、
と反応させる段階;
(b)式IVの化合物:
【化9】
式中、Aは請求項1と同様の定義であり、Xは脱離基である、
を、式Vの化合物:
【化10】
式中、Z、n、R3およびR4は請求項1と同様の定義である、
と反応させる段階;あるいは、
(c)以下の化合物VIまたはVII:
【化11】
式中、A、Z、n、R3、R4およびR7は請求項1と同様の定義であり、いずれの場合においても、Yは独立してハロゲン原子である、
の一方を、対応する有機金属試薬に変換させ、前記有機金属試薬を化合物VIまたはVIIの他方と反応させる段階、
を有する、請求項1に記載の化合物の製造方法。
【請求項23】
式II−Aの化合物:
【化12】
式中、Xは、Cl、BrまたはIである、
を、式III−Aの化合物:
【化13】
と反応させる段階を有する、式I−A:
【化14】
式中、n、R1、R3およびR4は、請求項1と同様の定義である、
を有する、請求項1に記載の化合物またはその製薬上許容されうる塩の製造方法。
【請求項24】
請求項22において定義され、R4が−CH2R5または−CH2CH2R5である、式III、VまたはVIIの化合物。
【請求項1】
式Iの化合物、またはその製薬上許容されうる塩:
【化1】
式中、
Zは、OまたはSであり;
nは、1〜3であり;
R3は、−CO2R8、−C(O)SR8、−C(O)NHR8、−C(S)OR8、−C(S)SR8、−C(S)NHR8、−C(NH)SR8、または−C(NH)NHR8(ここで、R8は水素原子もしくはアルキル基である)であり;
R4は、水素原子、−CH2R5または−CH2CH2R5(ここで、R5は、それぞれ独立してR3の定義のいずれかを有する)であり;
Bは、−NR2−、−CH2NR2−、−CH2CH2NR2−、−CH2CHR7−または−CH2O−(ここで、R2は、水素原子またはC1−3のアルキル基、アルケニル基もしくはアルキニル基であり、R7は、水素原子またはC1−3のアルキル基もしくはアルコキシ基である)であり;
Aは:
【化2】
式中、
R1は、−NH2または−OHであり;
CおよびDは、それぞれ独立して、1または2以上のヘテロ原子を含んでもよい、5員または6員の、置換されたまたは非置換の、芳香族環または非芳香族環であり、Cは任意の可能な位置で基Bと結合している、
である。
【請求項2】
ZがOである、請求項1に記載の化合物。
【請求項3】
nが1である、請求項1または2に記載の化合物。
【請求項4】
R8が、水素原子、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基またはtert−ブチル基である、請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
R3が−CO2R8であり、R4が−CH2CH2CO2R8である、請求項1〜4のいずれか1項に記載の化合物。
【請求項6】
R8が、水素原子、メチル基またはエチル基である、請求項5に記載の化合物。
【請求項7】
Bが、−CH2NR2−、−CH2CHR7−または−CH2O−である、請求項1〜6のいずれか1項に記載の化合物。
【請求項8】
Bが−CH2NR2−である、請求項7に記載の化合物。
【請求項9】
R2が、水素原子、メチル基、エチル基または−CH2C≡CHである、請求項1〜8のいずれか1項に記載の化合物。
【請求項10】
R7が、水素原子、メチル基、エチル基またはメトキシ基である、請求項1〜9のいずれか1項に記載の化合物。
【請求項11】
Dが5員の芳香族環である、請求項1〜10のいずれか1項に記載の化合物。
【請求項12】
Aが:
【化3】
である、請求項11に記載の化合物。
【請求項13】
Cが:
【化4】
式中、Xは、CHまたはNであり、かつ、
YがCであり、R6が水素原子、メチル基、エチル基またはHCOであるか、
YがNであり、R6が孤立電子対である、のいずれかである、
である、請求項1〜12のいずれか1項に記載の化合物。
【請求項14】
Aが、式A−iまたは式A−iiで表される、請求項13に記載の化合物。
【化5】
【請求項15】
Aが、式A−i−1または式A−ii−1で表される、請求項14に記載の化合物。
【化6】
【請求項16】
Bが−CH2NR2−であり、R2が、水素原子、メチル基、エチル基または−CH2C≡CHであり、ZがOであり、nが1であり、R3が−CO2R8である、請求項15に記載の化合物。
【請求項17】
Bが、−CH2NR2−、−CH2CH2−、−CH2CHCH3−または−CH2O−であり、R2が水素原子、メチル基、エチル基または−CH2C≡CHであり、ZがOであり、nが1であり、R3が−CO2R8であり、R4が、水素原子または−CH2CH2CO2R8であり、R8が、それぞれ独立して水素原子、メチル基またはエチル基であり、AがA−iでありYがCである場合に、R6は水素原子であり、AがA−iiである場合にR1は−OHである、請求項14に記載の化合物。
【請求項18】
治療に使用される、請求項1〜17のいずれか1項に記載の化合物。
【請求項19】
請求項1〜18のいずれか1項に記載の化合物を含む、医薬組成物。
【請求項20】
葉酸または葉酸誘導体に依存性の酵素の阻害に応答性の症状の治療に用いられる医薬の製造のための、請求項1〜19のいずれか1項に記載の化合物の使用。
【請求項21】
癌の治療に用いられる医薬の製造のための、請求項1〜19のいずれか1項に記載の化合物の使用。
【請求項22】
以下の段階:
(a)式IIの化合物:
【化7】
式中、Aは請求項1と同様の定義であり、mは、0、1または2であり、Xは脱離基である、
を、式IIIの化合物:
【化8】
式中、Z、n、R2、R3およびR4は請求項1と同様の定義である、
と反応させる段階;
(b)式IVの化合物:
【化9】
式中、Aは請求項1と同様の定義であり、Xは脱離基である、
を、式Vの化合物:
【化10】
式中、Z、n、R3およびR4は請求項1と同様の定義である、
と反応させる段階;あるいは、
(c)以下の化合物VIまたはVII:
【化11】
式中、A、Z、n、R3、R4およびR7は請求項1と同様の定義であり、いずれの場合においても、Yは独立してハロゲン原子である、
の一方を、対応する有機金属試薬に変換させ、前記有機金属試薬を化合物VIまたはVIIの他方と反応させる段階、
を有する、請求項1に記載の化合物の製造方法。
【請求項23】
式II−Aの化合物:
【化12】
式中、Xは、Cl、BrまたはIである、
を、式III−Aの化合物:
【化13】
と反応させる段階を有する、式I−A:
【化14】
式中、n、R1、R3およびR4は、請求項1と同様の定義である、
を有する、請求項1に記載の化合物またはその製薬上許容されうる塩の製造方法。
【請求項24】
請求項22において定義され、R4が−CH2R5または−CH2CH2R5である、式III、VまたはVIIの化合物。
【図1】

【公開番号】特開2009−114192(P2009−114192A)
【公開日】平成21年5月28日(2009.5.28)
【国際特許分類】
【出願番号】特願2008−293362(P2008−293362)
【出願日】平成20年11月17日(2008.11.17)
【分割の表示】特願2007−531773(P2007−531773)の分割
【原出願日】平成18年8月16日(2006.8.16)
【出願人】(507087041)
【Fターム(参考)】
【公開日】平成21年5月28日(2009.5.28)
【国際特許分類】
【出願日】平成20年11月17日(2008.11.17)
【分割の表示】特願2007−531773(P2007−531773)の分割
【原出願日】平成18年8月16日(2006.8.16)
【出願人】(507087041)
【Fターム(参考)】
[ Back to top ]