
新規な蛍光プローブ及びキナーゼ阻害剤の新規スクリーニング方法
【課題】キナーゼ阻害剤のスクリーニング方法およびその検出を可能にする新規なベンゾオキサジアゾール蛍光プローブの提供。
【解決手段】式(I)
【化1】

(式中、Zは、キナーゼ阻害活性を有する化合物を表わし、Lは、結合、置換されてもよいアミノ基、カルボニル基、置換されてもよい炭化水素基、置換されてもよいアルキルカルバモイル基、置換されてもよいアシルアミノ基、スルフィド基、置換されてもよいアルコキシ基を表し、Xは、結合、置換されてもよいアミノ基、置換されてもよいヒドラジノ基、チオカルボニルアミノ基を表し、R1はニトロ基、ジメチルスルファモイル基を表す。)
【解決手段】式(I)
【化1】
(式中、Zは、キナーゼ阻害活性を有する化合物を表わし、Lは、結合、置換されてもよいアミノ基、カルボニル基、置換されてもよい炭化水素基、置換されてもよいアルキルカルバモイル基、置換されてもよいアシルアミノ基、スルフィド基、置換されてもよいアルコキシ基を表し、Xは、結合、置換されてもよいアミノ基、置換されてもよいヒドラジノ基、チオカルボニルアミノ基を表し、R1はニトロ基、ジメチルスルファモイル基を表す。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、薬剤を開発するための技術分野に属し、特に、医薬品となり得るキナーゼ阻害剤をスクリーニングする方法およびその検出を可能にする新規なベンゾオキサジアゾール蛍光プローブに関する。
【背景技術】
【0002】
プロテインキナーゼは、自己または他のタンパク質上のチロシン、セリンあるいはスレオニンをリン酸化することにより細胞増殖、細胞間コミュニケーション、細胞生存等に関与するシグナルを伝えており、これらのリン酸化シグナルは正常細胞の増殖、生存等に重要な役割を果たしている。多くの疾患は、何らかの原因で正常に働かなくなったプロテインキナーゼを介したリン酸化シグナル、つまり異常な細胞応答に関連していることが知られている(非特許文献1参照)。このような疾患としては、自己免疫疾患、炎症性疾患、骨疾患、代謝性疾患、神経学的疾患及び神経変性疾患、癌、心臓血管の疾患、アレルギー及び喘息、アルツハイマー病、ならびにホルモン関連の疾患が挙げられる。したがって、これらの疾患に関与するキナーゼを阻害する化合物はこれらの疾病などに対する予防/治療薬になると考えられ、新しいキナーゼ阻害剤の開発が日夜続けられている(非特許文献2、3参照)。
【0003】
キナーゼ阻害剤の効率的な開発のためには、対象となるキナーゼに対して、阻害剤の候補となり得る化合物群をスクリーニングすることのできる評価系を確立することが必要である。しかしながら、キナーゼはヒトの生体内に500種類以上存在しているとされており(非特許文献4参照)、対象となるキナーゼ以外を阻害しないような化合物を見出すことが、副作用の少ない薬剤を開発するうえで非常に重要である。これまでに報告されているキナーゼ阻害剤は、キナーゼに共通の基質であるATPの同じ部位に結合するものがほとんどであるが、この結合部位は同じATPを結合するようにできていることから容易に類推できるように非常に構造類似性が高く、選択的なキナーゼ阻害剤を得ることは容易ではない(非特許文献5参照)。
【0004】
生体内ではキナーゼ活性は精密に調節されており、キナーゼ自身もキナーゼによるリン酸化によってon/off(活性型と不活性型)の調節を受けており、その立体構造が大きく変化するとされている。つまり同じキナーゼでも、活性型か不活性型かのどちらを阻害するかで選択性に違いが出てくることが知られている(非特許文献6参照)。
【0005】
現在、報告されているほとんどのキナーゼ阻害剤は活性型キナーゼを阻害するものであるが、前述のとおり活性型キナーゼのATP結合部位の構造は、極めて高い類似性を持つため、そこに結合する阻害剤で高い選択性を出すのは難しいとされている。
【0006】
一方、不活性型キナーゼに結合する阻害剤は、活性化ループの構造の変化によって出現する新たな領域との相互作用を利用できるため非常に高い選択性を示す場合が多い。つまり、活性型キナーゼの立体構造は類似しているが、不活性型キナーゼでは必ずしもそうではないということである。また不活性型キナーゼは競合基質であるATPとの親和性も低下しており、不活性型キナーゼに結合する物質には、生体内ではより強力な生理作用が期待できる。しかしながら、これまで報告されている不活性型キナーゼに結合する阻害剤は、ほとんどが偶然発見されたものであり、活性のないキナーゼを用いて化合物のスクリーニングを実施するのは非常に難しいとされている。
【0007】
ところで、キナーゼを阻害する化合物をスクリーニングする方法としては、活性型キナーゼの酵素活性阻害を指標とした評価系が最も一般的である(非特許文献7参照)。競合結合測定法(competitive binding assay)により、プローブとして用いられる標準阻害剤に対する拮抗阻害の評価系も用いられることもあるが(例えば、LanthaScreen(商標) Eu Kinase Binding Assay、インビトロジェン社)、拮抗阻害を検出するためにはキナーゼタンパク自体を化学的あるいは生物的に修飾する必要があり、さらに2次試薬も添加する必要があるため、バックグラウンドシグナルの上昇や操作が煩雑にならざるを得ない。
【0008】
通常、蛍光プローブを合成する場合には、生化学分野でよく用いられているフルオレセインやその誘導体などの蛍光色素が用いられている。しかしながら、これらの蛍光色素は、それ自身が強い蛍光を発するため、測定時には標的タンパク質と結合していないプローブを取り除くか、あるいは、先に述べたように標的タンパクの修飾や2次試薬の添加などが必要となる。一方、蛍光検出−HPLCなどによく用いられている蛍光ラベル化試薬、ベンゾオキサジアゾール誘導体は、有機溶媒中など疎水性環境で非常に強い蛍光を発するが、例えば水中ではほぼ無蛍光となることが知られており(例えば、東京化成工業のWebページ、HPLC用ラベル化剤、http://www.tokyokasei.co.jp/product/analytical−chem/A007.shtmlなどを参照)、この性質を利用した、タンパク質内部の疎水性環境を探るような蛍光プローブの例がいくつか報告されている(例えば非特許文献8、9などを参照)。
【0009】
ちなみに、特許文献としては、国際公開第2003/097667号、国際公開第2003/089665号等にキナーゼ阻害剤をスクリーニングする方法が知られているが(特許文献1、特許文献2)、これらの方法は先に述べたように満足とは限らない。また、国際公開第2004/085618号に不活性型キナーゼを標的としたスクリーニング方法について記載されているが(特許文献3)、どのようにして評価するのか、またスクリーニング方法の実証結果など全く示されていない。国際公開第2005/033330号には、当業界で通常用いられているフルオレセイン誘導体を蛍光色素として利用した蛍光偏光法によるアッセイ方法について開示されているが(特許文献4)、不活性型キナーゼのスクリーニングに関する記載は全くなされていない。国際公開第2008/033834号、国際公開第2008/033854号には、ラジオアイソトープや蛍光色素でラベル化したBTK(Bruton’s tyrosine kinase)阻害剤がバイオアッセイにも使用できることが記載されているものの、ラベル化化合物およびバイオアッセイに関しては全く検討されていない(特許文献5、特許文献6)。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Hunter,T.,Cell,(1995),80,225−236
【非特許文献2】Cohen,P.,Nat.Rev.Drug Discov.,(2002),1,309−315
【非特許文献3】Shchemelinin,I.,et al.,Folia Biol(Praha),(2006),52,81−100
【非特許文献4】Manning,G.,et al.,Science,(2002),298,1912−1934
【非特許文献5】Zhang,J.,et al.,Nat.Rev.Cancer,(2009),9,28−39
【非特許文献6】Liu,Y.,et al.,Nat.Chem.Biol.,(2006),2,358−364
【非特許文献7】Comley,J.,Drug Discovery World,Fall 2004,45−56
【非特許文献8】Kenner,R.A., et al.,Biochemistry,(1971),10,4433−4440
【非特許文献9】Rasmussen,S.G.、et al.,J.Biol.Chem.,(2001)、276、4717−4723
【特許文献】
【0011】
【特許文献1】WO2003/097667号
【特許文献2】WO2003/089665号
【特許文献3】WO2004/085618号
【特許文献4】WO2005/033330号
【特許文献5】WO2008/033834号
【特許文献6】WO2008/033854号
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明の目的は、活性型ないし不活性型キナーゼに結合する薬剤を簡便且つ確実にスクリーニングすることのできる新しいスクリーニング方法、およびその検出を可能にする新規なベンゾオキサジアゾール蛍光プローブを提供することにある。
【課題を解決するための手段】
【0013】
本発明者は、前述のとおりベンゾオキサジアゾール誘導体が環境に依存して蛍光強度を変化させる性質に着目し、この性質を利用することにより上述の目的を達成し得る本発明を案出したものである。かくして、本発明に従えば、活性型ないし不活性型キナーゼに結合する薬剤のスクリーニングが簡便に可能となり、キナーゼ阻害剤にベンゾオキサジアゾール誘導体を結合させた蛍光プローブとキナーゼの混合物に被験物質を添加し、その蛍光強度の変化が観察されたときに、被験物質をキナーゼ阻害剤として選択することを特徴とする方法が提供される。
【発明の効果】
【0014】
本発明のスクリーニング方法および新規なベンゾオキサジアゾール蛍光プローブを用いれば、活性型キナーゼのみならず、不活性型キナーゼに結合する薬剤を簡便且つ確実にスクリーニングすることができ、従来の競合結合測定法において必要だった標的酵素の修飾および2次試薬が不要となる。かくして、本発明は、キナーゼ阻害剤の効率的なスクリーニングを通じて、自己免疫疾患、炎症性疾患、骨疾患、代謝性疾患、神経学的疾患及び神経変性疾患、癌、心臓血管の疾患、アレルギー及び喘息、アルツハイマー病、ならびにホルモン関連の疾患などに対する予防・治療薬の開発に資するものである。
【図面の簡単な説明】
【0015】
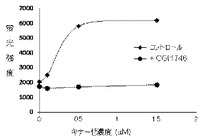
【図1】本発明の蛍光プローブが脱リン酸化(不活性型)BTKに結合することで、水溶液中と比較して有意に高く酵素濃度依存的に蛍光を発することを示す。また、キナーゼ阻害剤の添加により蛍光強度が低下することを示す。
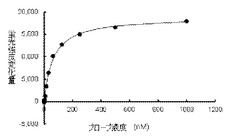
【図2】脱リン酸化(不活性型)BTKと結合することで、本発明の蛍光プローブの蛍光が濃度依存的に増加することを示す。
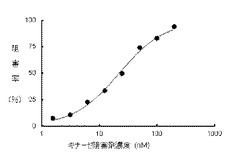
【図3】本発明の蛍光プローブを使用して、試験化合物を種々の希釈で脱リン酸化(不活性型)BTK結合活性を指標にしてスクリーニングする場合に得られた蛍光変化結果を示す。
【発明を実施するための最良の形態】
【0016】
本発明者らは、酵素活性がない、もしくは非常に弱い不活性型キナーゼを用いて、その基質結合部位に拮抗的に結合する化合物を見つけるために、競合結合測定法に注目して研究を行った。このとき、通常、生化学分野でよく用いられるフルオレセインなどの蛍光色素とは異なる性質を有するベンゾオキサジアゾール誘導体が、キナーゼの結合度に応じて蛍光強度が変化することにより、2次試薬等を必要としない非常に簡便な競合阻害試験に利用できるものと考え、本発明を導き出したものである。
本発明は、以下の(1)〜(7)によって達成される。
(1)下式(I)を有する新規なベンゾオキサジアゾール蛍光プローブ。
【0017】
【化1】
【0018】
(式中、Zは、キナーゼ阻害活性を有する化合物を表わし、Lは、結合、置換されてもよいアミノ基、カルボニル基、置換されてもよい炭化水素基、置換されてもよいアルキルカルバモイル基、置換されてもよいアシルアミノ基、スルフィド基、置換されてもよいアルコキシ基を表し、Xは、結合、置換されてもよいアミノ基、置換されてもよいヒドラジノ基、チオカルボニルアミノ基を表し、R1はニトロ基、ジメチルスルファモイル基を表す。)
(2)Zが、p38MAPK阻害活性を有する化合物である、(1)に記載の蛍光プローブ。
(3)Zが、下式(Ia)を有する化合物である、(1)または(2)に記載の蛍光プローブ。
【0019】
【化2】
(式中、R2は置換されてもよいアルコキシ基を表す。)
(4)Zが、BTK阻害活性を有する化合物である、(1)に記載の蛍光プローブ。
(5)Zが、下式(Ib)を有する化合物である、(1)または(4)に記載の蛍光プローブ。
【0020】
【化3】
(6)(1)から(5)の何れか1つに記載の蛍光プローブを使用することを特徴とするキナーゼ阻害剤をスクリーニングする方法。
(7)不活性型キナーゼを標的とした、(6)に記載のスクリーニング方法。
【0021】
以下、本発明に従うキナーゼ阻害剤のスクリーニング法において用いられる蛍光プローブの作製法、ならびに当該蛍光プローブを用いるスクリーニング法に従って本発明の実施の形態を説明する。但し、以下に示す具体的な実施の形態は例示のためのものであり、本発明の実施の形態はこれに限定されるものではない。
【0022】
本発明の新しいキナーゼ阻害剤のスクリーニング法の実施を可能にする新規なベンゾオキサジアゾール蛍光プローブは、下式(I)で示される化合物である。
【0023】
【化4】
【0024】
(式中、Zは、キナーゼ阻害活性を有する化合物を表わし、Lは、結合、置換されてもよいアミノ基、カルボニル基、置換されてもよい炭化水素基、置換されてもよいアルキルカルバモイル基、置換されてもよいアシルアミノ基、スルフィド基、置換されてもよいアルコキシ基を表し、Xは、結合、置換されてもよいアミノ基、置換されてもよいヒドラジノ基、チオカルボニルアミノ基を表し、R1はニトロ基、ジメチルスルファモイル基を表す。)
【0025】
尚、上記で「結合」とは、“ZとX”、“Lとベンゾオキサジアゾール基”、あるいは“Zとベンゾオキサジアゾール基”が直接結合した場合を意味する。
【0026】
前記式(I)において、キナーゼ阻害活性を有する化合物としては、標的とする活性型あるいは不活性型キナーゼの、触媒ドメインのATP結合部位、基質結合部位もしくはアロステリック部位に可逆的に結合する化合物であれば、新規化合物や公知化合物の何れであってもよいが、このような性質を有する化合物としてはいくつかのものが既に知られており、これらを使用することができる。
例えば、ATP結合部位に結合して活性型キナーゼを阻害するものとしては、スタウロスポリン、スニチニブ(N−(2−diethylaminoethyl)−5−[(Z)−(5−fluoro−2−oxo−1H−indol−3−ylidene)methyl]−2,4−dimethyl−1H−pyrrole−3−carboxamide)、ダサチニブ(N−(2−chloro−6−methylphenyl)−2−[[6−[4−(2−hydroxyethyl)−1−piperazinyl]−2−methyl−4−pyrimidinyl]amino]−5−thiazolecarboxamide monohydrate)などのキナーゼ阻害薬もしくはそれらの誘導体などが挙げられる。
【0027】
不活性型キナーゼを標的とする場合は、不活性型キナーゼにより強く結合することが知られている化合物、具体的には、不活性型BTKを阻害するN−[3−[4,5−Dihydro−4−methyl−6−[[4−(4−morpholinylcarbonyl)phenyl]amino]−5−oxo−2−pyrazinyl]−2−methylphenyl]−4−(tert−butyl)benzamide(CGI1746)に代表される上記[化3]の部分構造をもつ誘導体、不活性型p38MAPKを阻害するdoramapimodに代表される上記[化2]の部分構造をもつ誘導体、不活性型ABLを阻害するイマチニブ、ニロチニブもしくはそれらの誘導体などが挙げられる。
【0028】
また、アロステリック部位に結合してキナーゼ活性を阻害することが知られているものとしては、MAPKキナーゼのMEKを阻害する2−(2−Chloro−4−iodo−phenylamino)−N−cyclopropylmethoxy−3,4−difluoro−benzamide(PD−184352,CI−1040)もしくはそれらの誘導体などが挙げられる。
前記式(I)において、「置換されてもよい炭化水素基」の「炭化水素」部分としては、例えば、
a)炭素数1から6の直鎖状、あるいは分岐鎖状のアルキル基(例えば、メチル、エチル、イソプロピル、tert−ブチル、ヘキシル等)、
b)炭素数1から6の直鎖状、あるいは分岐鎖状のアルケニル基(例えば、ビニル、アリル、イソプロペニル、2−ブテニル等)、
c)炭素数2から6のアルキニル基(例えば、エチニル、プロパルギル、2−ブチニル等)、
d)炭素数3から8のシクロアルキル基(例えば、シクロプロピル、シクロペンチル、シクロヘキシル、シクロヘプチル等)、
e)炭素数3から8のシクロアルケニル基(例えば、シクロヘキセニル、シクロヘプテニル等)、
【0029】
f)アラルキル基、アラルキル基のアリール部分としては、炭素数6から14のアリールが挙げられ(例えば、フェニル、ナフチル、インデニル等)、アラルキル基のアルキレン部分としては、前記アルキル基から水素原子を1つ除いたものと同義である、
などが挙げられる。
【0030】
「置換されてもよい炭化水素基」の「置換基」としては、置換されてもよいアミノ基、置換されてもよいアルコキシ基、置換されてよいカルバモイル基、置換されてもよいアシルアミノ基、置換されてもよいスルフィド基等を挙げることができる。
【0031】
「置換されてもよいアミノ基」としては、置換もしくは非置換の炭素数1から6の直鎖状、分枝状もしくは環状のアルキル基を有するアミノ基または直鎖状、分枝状もしくは環状のアルキル基の炭素原子がヘテロ原子に置換されていてもよいアミノ基を表し、具体的にはメチルアミノ基、ピロリジノ基、ピペリジノ基、ピペラジノ基、モルホリノ基、アニリノ基などが挙げられる。
【0032】
「置換されてもよいアルキルカルバモイル基」としては、置換もしくは非置換の炭素数1から6の直鎖状、分枝状もしくは環状のアルキル基を有するアルキルカルバモイル基又は直鎖状、分枝状もしくは環状のアルキル基の炭素原子がヘテロ原子に置換されていてもよいアルキルカルバモイル基を表す。
【0033】
「置換されてもよいアシルアミノ基」としては、置換もしくは非置換の炭素数1から6の直鎖状、分枝状もしくは環状のアルキル基を有するアルキルカルボニルアミノ基又は直鎖状、分枝状もしくは環状のアルキル基の炭素原子がヘテロ原子に置換されていてもよいアルキルカルボニルアミノ基を表す。
【0034】
「置換されてもよいアルコキシ基」としては、置換もしくは非置換の炭素数1から6の直鎖状、分枝状もしくは環状のアルキル基を有するアルコキシ基又は直鎖状、分枝状もしくは環状のアルキル基の炭素原子がヘテロ原子に置換されていてもよいアルコキシ基を表す。
【0035】
「置換されてもよいヒドラジノ基」としては、置換もしくは非置換の炭素数1から6の直鎖状、分枝状もしくは環状のアルキル基を有するヒドラジノ基または直鎖状、分枝状もしくは環状のアルキル基の炭素原子がヘテロ原子に置換されていてもよいヒドラジノ基を表す。
【0036】
ベンゾオキサジアゾール基を、キナーゼ阻害活性を有する化合物に結合するためのリンカーである式(I)のLおよびXとしては、市販で入手可能なベンゾオキサジアゾール誘導体を蛍光標識色素として結合するのに使用し得る当業界で知られているあらゆる基であってもよい。典型的には、この実施態様において、LおよびXは、ベンゾオキサジアゾール誘導体の反応性官能基であるフッ素、塩素などのハロゲンまたはイソチオシアネート基と、キナーゼ阻害活性を有する化合物の適当な位置に導入したアミン性官能基との反応により生成されてもよい。
【0037】
ベンゾオキサジアゾール基は、Zのキナーゼ阻害活性を保持しつつ、キナーゼに結合すると蛍光が変化するように適宜、選ばれたLおよびXを介してZと結合を形成する。
【0038】
本発明の蛍光プローブ(I)は、例えば、置換基の種類によって、異性体が存在する場合がある。本明細書において、それらの異性体の一形態のみの化学構造で記載することがあるが、本発明には、構造上生じ得るすべての異性体(幾何異性体、光学異性体、互変異性体など)も含有し、異性体単体、またはそれらの混合物も含有する。
【0039】
また、本発明の蛍光プローブ(I)は、その水溶性などの物性を調節するために、塩にして用いる場合があり、本発明には、化学性質上、生じ得るすべての塩も含有する。
本発明の蛍光プローブ(I)の具体的な化合物としては、以下の化合物が挙げられる。
【0040】
1−(3−(t‐ブチル)−1−(4−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)フェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア
【0041】
1−(3−(t‐ブチル)−1−(3−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)フェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア
【0042】
N−(4−(3−(t‐ブチル)−5−(3−(ナフタレン−1−イル)ウレイド)−1H−ピラゾロ−1−イル)フェニル)−3−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)プロパンアミド
【0043】
3−(3−(t‐ブチル)−5−(3−(ナフタレン−1−イル)ウレイド)−1H−ピラゾロ−1−イル)−N−(2−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)エチル)ベンズアミド
【0044】
4−(3−(t‐ブチル)−5−(3−(ナフタレン−1−イル)ウレイド)−1H−ピラゾロ−1−イル)−N−(2−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)エチル)ベンズアミド
【0045】
4−(3−(t‐ブチル)−5−(3−(4−(2−モルホリノエトキシ)ナフタレン−1−イル)ウレイド)−1H−ピラゾロ−1−イル)−N−(2−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)エチル)ベンズアミド
【0046】
1−(3−(t‐ブチル)−1−(4−(4−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)1‐ブチン−1−イル)フェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア
【0047】
1−(3−(t‐ブチル)−1−(4−(4−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)ブチル)フェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア
【0048】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−(4−(7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)ピペラジン−1−カルボニル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0049】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−((2−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)エチル)カルバモイル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0050】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−(2−(((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)メチル)モルホリン−4−カルボニル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0051】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−((2−(2−(2−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)エトキシ)エトキシ)エチル)カルバモイル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0052】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−(4−(2−(メチル(7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)アセチル)ピペラジン−1−カルボニル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0053】
4−(t‐ブチル)−N−(3−(6−((4−(4−(7−(N,N−ジメチルスルファモイル)ベンゾ[C][1,2,5]オキサジアゾール−4−イル)ピペラジン−1−カルボニル)フェニル)アミノ)−4−メチル−5−オキソ−4,5−ジヒドロピラジン−2−イル)−2−メチルフェニル)ベンズアミド
【0054】
本発明の蛍光プローブ(I)は、例えば以下の方法によって製造することができる。なお、以下に示した製造法において、定義した基が実施方法の条件下で変化するか、または方法を実施するのに不適切な場合、有機合成化学で通常用いられる方法、例えば、官能基の保護、脱保護[T.W.Greene,Protective Groups in Organic Synthesis 3rd Edition,John Wiley&Sons,Inc.,1999]等の手段を付すことにより容易に製造することができる。また、必要に応じて置換基導入等の反応工程の順序を変えることもできる。
スキーム1
【0055】
【化5】
【0056】
スキーム1において、R1、L、XおよびZは先に定義されたとおりであり、
ベンゾオキサジアゾール誘導体化試薬(III)は、蛍光ラベル化試薬として種々のAを官能基として有する誘導体が市販されており(例えば、東京化成工業株式会社などから入手可能)、官能基Aは化合物(II)と反応後、Xに変換される。もし、ZがリンカーLもしくはベンゾオキサジアゾール誘導体化試薬(III)と反応しうる適当な官能基を適当な位置に有しないときには、その前駆体化合物や、有機合成化学で通常用いられる方法を実施することにより、所望の位置に所望の官能基を有する誘導体を合成し、その誘導体を用いてスキーム1の反応を実施してもよい。また、リンカーLを介して、ベンゾオキサジアゾール誘導体化試薬(III)でZをラベル化するときは、アミノ基、カルボキシル基、水酸基、チオール基など、ベンゾオキサジアゾール誘導体化試薬(III)との反応に適当な官能基を適当な位置に有しているL’を、必要であれば官能基の保護、脱保護を繰り返して、当業界で知られている通常の有機合成の条件などを用いてZに導入し、得られた化合物(II)を適当なベンゾキサジアゾール誘導体化試薬(III)と反応させることにより、式(I)の所望の化合物を得ることができる。
ベンゾオキサジアゾール誘導体化試薬(III)は、例えば、
【0057】
アミノ基やメルカプト基のラベル化試薬として、AがFやClなどのハロゲン原子であるNBD−Cl、NBD−F、イソチオシアネート基であるNBD−NCS、
【0058】
カルボキシル基のラベル化試薬として、AがピペラジンであるNBD−PZ、DBD−PZ、2−アミノエチルアミノ基であるDBD−ED、N−ヒドラジノカルボニルメチル−N−メチルアミノ基であるNBD−CO−Hz
などが市販で入手可能である。
【0059】
また、カルボニル基のラベル化には、Aがヒドラジノ基であるNBD−HやDBD−H、水酸基のラベル化としては、AがN−クロロホルミルメチル−N−メチルアミノ基であるDBD−COClなどが市販されている。
また、本発明のスクリーニング方法は、上記方法により得られた本発明の蛍光プローブとキナーゼの混合物に被験物質を添加し、その蛍光強度の変化(低下)が観察されたときに、被験物質をキナーゼ阻害剤として選択することを特徴とする。
【実施例】
【0060】
以下に実施例を挙げて本発明をさらに具体的に説明するが、これらの実施例により本発明が限定されるものではない。
【0061】
化合物の同定は水素核磁気共鳴スペクトル(1H−NMR)及びマススペクトル(MS)により行った。1H−NMRは、特に指示のないかぎりは400MHzで測定されたものであり、また化合物及び測定条件によっては交換性水素が明瞭に観測されない場合がある。質量分析は液体クロマトグラフ質量分析計(LCMS)システムを用い、以下に示したA法もしくはB法にて測定した。
【0062】
A法:LCMS−2010Aシステム(島津製作所)を使用し、質量分析はエレクトロスプレー(ESI)法により測定した。分離カラムはCadenza CD‐C18(50×2mm)(インタクト社製)を用いた。溶出には10mMギ酸水溶液(A液)および10mMギ酸メタノール溶液(B液)によるグラジェント法を用い、溶出条件は、流速0.5mL/分、0分〜0.5分(B液:5%(v/v))、0.5分〜2.5分(B液:5〜95%(v/v)直線グラジェント)、2.5分〜4分(B液:95%(v/v))とした。
【0063】
B法:液体クロマトグラフ質量分析計システム(micromass ZQマス検出器、日本ウォーターズ社製)を使用し、質量分析はエレクトロスプレー(ESI)法により測定した。分離カラムはUnison US‐C18(50×4.6mm)(インタクト社製)を用いた。溶出には10mMギ酸水溶液(A液)および10mMギ酸メタノール溶液(B液)によるグラジェント法を用い、溶出条件は、溶出条件は、流速2mL/分、0分〜0.5分(B液:5%(v/v))、0.5分〜3分(B液:10〜90%(v/v)直線グラジェント)、3分〜4分(B液:90%(v/v))とした。
実施例1から8は、本発明の蛍光プローブ(I)のうち、Zがp38MAPK阻害活性を有する化合物である場合を例として、具体的に説明する。
実施例1
【0064】
1−(3−(t‐ブチル)−1−(4−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)フェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア
【0065】
【化6】
(第1工程)
【0066】
4−ニトロフェニルヒドラジン(766mg)、ピバロイルアセトニトリル(626mg)をエタノール(12mL)に溶解し、濃塩酸(2.5mL)を加え90℃で24時間撹拌した。反応混合物を減圧濃縮し、得られた粗生成物をエタノールで懸濁洗浄し、3−(t‐ブチル)−1−(4−ニトロフェニル)−1H−ピラゾロ−5−アミン塩酸塩1.31gを得た。
(第2工程)
【0067】
3−(t‐ブチル)−1−(4−ニトロフェニル)−1H−ピラゾロ−5−アミン塩酸塩(297mg)にジクロロメタン(10mL)、トリエチルアミン(167μL)、1−ナフチルイソシアネート(158μL)を加え80℃で18時間撹拌した。反応混合物を減圧濃縮し、得られた残渣に水(10mL)を加え、酢酸エチル(50mL×2)で抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して1−(3−(t‐ブチル)−1−(4−ニトロフェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア145mgを得た。
(第3工程)
【0068】
1−(3−(t‐ブチル)−1−(4−ニトロフェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア(145mg)をエタノール(7mL)に溶解し、ギ酸アンモニウム(132mg)、触媒量の10%パラジウム−炭素を加え90℃で3時間還流した。不溶物をセライトろ過した後、ろ液を減圧濃縮した。得られた残渣をフラッシュクロマトグラフィーで精製し、1−(1−(4−アミノフェニル)−3−(t‐ブチル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア81mgを得た。
(第4工程)
【0069】
1−(1−(4−アミノフェニル)−3−(t‐ブチル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア(21mg)をエタノール(7mL)に溶解し、4‐フルオロ‐7‐ニトロ-2,1,3‐ベンゾオキサジアゾール(10mg)を加え室温で18時間撹拌した。析出した固体をろ取し、ジエチルエーテルで洗浄後、無水硫酸ナトリウムで乾燥させて表題化合物22mgを得た。LCMS:HPLC保持時間3.53分(A法)、m/z563(MH+)。
【0070】
1H-NMR (400 MHz, DMSO-d6) δ ppm 1.31 (s, 9H), 6.44 (s, 1H), 6.85 (d, J=8.78 Hz, 1H), 7.47 (t, J=7.91 Hz, 1H), 7.51 - 7.61 (m, 2H), 7.62 - 7.77 (m, 5H), 7.87 - 7.98 (m, 2H), 8.04 (d, J=8.28 Hz, 1H), 8.57 (d, J=8.78 Hz, 1H), 8.90 (s, 1H), 9.06 (s, 1H), 11.18 (s, 1H).
【0071】
下記実施例2から7の化合物は、実施例1に記載の方法もしくは類似の方法に従い、適当な試薬および有機合成化学で通常用いられる方法により製造した。表中のMethodはLCMSの分析方法、RTはLCMSにおけるHPLC保持時間、MSはLCMSにおけるマススペクトル分析データ(特に指示のない限りポジティブイオンモードで測定されたMH+)を示す。
【0072】
【表1】
また、実施例9から14は、本発明の蛍光プローブ(I)のうち、ZがBTK阻害活性を有する化合物である場合を例として、具体的に説明する。
参考例1
【0073】
4−((6−(3−(4−(t‐ブチル)ベンズアミド)−2−メチルフェニル)−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)安息香酸
【0074】
【化7】
(第1工程)
【0075】
文献J.Med.Chem.Vol.48、p.1901(2005年)に記載の方法で調製した3,5−ジブロモ−1−メチルピラジン−2(1H)−オン(593mg)のジメチルアセトアミド溶液(3.7mL)に4‐アミノ安息香酸エチル(475mg)を加え、マイクロウェーブ合成装置を用いて105℃で20時間反応させた。析出した固体をろ取し、酢酸エチルで洗浄後、乾燥させてエチル 4−((6−ブロモ−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート620mgを得た。
【0076】
1H-NMR (400 MHz, CDCl3) δppm 1.39 (t, J=7.15 Hz, 3H), 3.55 (s, 3H), 4.37 (q, J=7.28 Hz, 2H), 6.83 (s, 1H), 7.82 (d, J=8.78 Hz, 2H), 8.06 (d,J=8.78 Hz, 2H), 8.45 (s, 1H).
(第2工程)
【0077】
エチル 4−((6−ブロモ−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート(870mg)の1,4−ジオキサン溶液(7.6mL)に3‐アミノ‐2‐メチルフェニルボロン酸ピナコールエステル(749mg)、テトラキス(トリフェニルホスフィン)パラジウム(0)(400mg)、2.4M炭酸ナトリウム水溶液(1.5mL)を加え、105℃で60時間撹拌した。不溶物をセライトろ過した後、ろ液にジクロロメタン(50mL)を加え、2M塩酸(50mL)で抽出した。水層に2M水酸化ナトリウム水溶液(60mL)を加えてアルカリ性にした。析出した固体をろ取し、水で洗浄後、乾燥させてエチル 4−(6−(3−アミノ−2−メチルフェニル)−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート588mgを得た。
(第3工程)
【0078】
エチル 4−(6−(3−アミノ−2−メチルフェニル)−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート(588mg)のジクロロメタン溶液溶液(7.8mL)にピリジン(0.14mL)、4‐t‐ブチルベンゾイルクロリド(0.455mL)を加え、室温で4時間撹拌した。反応混合物をクロロホルム(100mL)で希釈し、1M水酸化ナトリウム水溶液、水、飽和食塩水で順次洗浄した。得られた有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。得られた残渣をフラッシュクロマトグラフィーで精製し、エチル 4−((6−(3−(4−(t‐ブチル)ベンズアミド)−2−メチルフェニル)−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート534mgを得た。
(第4工程)
【0079】
エチル 4−((6−(3−(4−(t‐ブチル)ベンズアミド)−2−メチルフェニル)−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート(400mg)のテトラヒドロフラン溶液(7mL)に1M水酸化ナトリウム水溶液(7mL)、エタノール(7mL)を加え、80℃で3時間撹拌した。反応混合物に1M塩酸(7mL)、水(50mL)を加え、酢酸エチル(20mL×3)で抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して表題化合物377mgを得た。
【0080】
1H-NMR (400 MHz, DMSO-d6) δ ppm 1.33 (s, 9H), 2.30 (s, 3H), 3.58 (s, 3H), 7.25 - 7.36 (m, 3H), 7.40 (dd, J=6.53, 2.51 Hz, 1H), 7.56 (d, J=8.28 Hz, 2H), 7.85 (d, J=8.78 Hz, 2H), 7.96 (d, J=8.53 Hz, 2H), 8.14 (d, J=8.78 Hz, 2H), 9.59 (s, 1H), 9.88 (s, 1H), 12.58 (s, 1H).
実施例9
【0081】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−(4−(7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)ピペラジン−1−カルボニル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0082】
【化8】
【0083】
参考例1で得られた化合物(50mg)のテトラヒドロフラン溶液(1.6mL)に1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(56mg)、1−ヒドロキシベンゾトリアゾール(45mg)を加え、室温で1時間撹拌したのち、4‐ニトロ‐7‐ピペラジノ‐2,1,3‐ベンゾオキサジアゾール(36.6mg)のジメチルホルムアミド(0.3mL)溶液を加え、室温でさらに18時間撹拌した。反応混合物にクロロホルム(50mL)を加え、1M塩酸、水、飽和重曹水、及び飽和食塩水で順次洗浄した。得られた有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し表題化合物63mgを得た。LCMS:HPLC保持時間3.72分(B法)、m/z742(MH+)。
【0084】
1H-NMR (400 MHz, DMSO-d6) δ ppm 1.31 (s, 9H), 2.30 (s, 3H), 3.58 (s, 3H), 3.80 (br. s., 4H), 4.22 (br. s., 4H), 6.60 (d, J=9.29 Hz, 1H), 7.24 - 7.41 (m, 4H), 7.44 (d, J=8.53 Hz, 2H), 7.53 (d, J=8.53 Hz, 2H), 7.94 (d, J=8.53 Hz, 2H), 8.12 (d, J=8.78 Hz, 2H), 8.52 (d, J=9.03 Hz, 1H), 9.50 (s, 1H), 9.89 (s, 1H).
【0085】
下記実施例10から13に示す化合物は、実施例9に記載の方法もしくは類似の方法に従い、適当な試薬および有機合成化学で通常用いられる方法により製造した。表中のMethodはLCMSの分析方法、RTはLCMSにおけるHPLC保持時間、MSはLCMSにおけるマススペクトル分析データ(特に指示のない限りポジティブイオンモードで測定されたMH+)を示す。
【0086】
【表2】
実施例14
【0087】
4−(t‐ブチル)−N−(3−(6−((4−(4−(7−(N,N−ジメチルスルファモイル)ベンゾ[C][1,2,5]オキサジアゾール−4−イル)ピペラジン−1−カルボニル)フェニル)アミノ)−4−メチル−5−オキソ−4,5−ジヒドロピラジン−2−イル)−2−メチルフェニル)ベンズアミド
【0088】
【化9】
【0089】
実施例9に記載の手順に従って、4‐ニトロ‐7‐ピペラジノ‐2,1,3‐ベンゾオキサジアゾールの替わりに4‐(N,N‐ジメチルアミノスルホニル)‐7‐ピペラジノ‐2,1,3‐ベンゾオキサジアゾール(27mg)を用い、反応・処理することにより表題化合物47mgを得た。
LCMS:HPLC保持時間3.72分(B法)、m/z804(MH+)。
【0090】
1H-NMR (400 MHz, DMSO-d6) δ ppm 1.31 (s, 9H), 2.30 (s, 3H), 2.65 - 2.80 (m, 6H), 3.58 (s, 3H), 3.75 (br. s., 4H), 3.96 (br. s., 4H), 6.60 (d, J=8.28 Hz, 1H), 7.25 - 7.34 (m, 3H), 7.38 (dd, J=6.90, 2.38 Hz, 1H), 7.43 (d, J=8.53 Hz, 2H), 7.54 (d, J=8.53 Hz, 2H), 7.86 (d, J=8.03 Hz, 1H), 7.94 (d, J=8.28 Hz, 2H), 8.12(d, J=8.78 Hz, 2H), 9.50 (s, 1H), 9.90 (s, 1H).
実施例15
リン酸化(活性型)および脱リン酸化(不活性型)BTKに対するキナーゼ活性阻害試験
【0091】
リン酸化(活性型)BTKは、ビオチン化BTK蛋白質BTN−BTK(カルナバイオサイエンス社製)酵素溶液にATP、MgCl2をそれぞれ3mM、10mMとなるように添加し、4℃で一晩反応させた後、10DG Desalting Columnを用いてバッファー交換を行い、未反応のATPを除去することにより得た。
【0092】
脱リン酸化(不活性型)BTKは、BTN−BTK酵素溶液にλ protein phosphatase(New England BioLabs社製、Code No.P0753S)とMnCl2をそれぞれ10U/μg、2mMとなるように添加し、4℃で一晩反応させた後、抗DYKDDDDK−tag抗体アガロースゲルクロマトグラフィーによりλ protein phosphataseを除去したのち、10DG Desalting Columnを用いてバッファー交換を行い得た。
【0093】
キナーゼ活性の測定は、MSAアッセイキット(QuickScout Screening Assist(商標)kit、カルナバイオサイエンス社製)を用いて行った。キナーゼ反応の基質は、キット付属のFITC標識SRCtideペプチドを用いた。アッセイバッファー[20mM HEPES、0.01%TritonX−100(商標)、2mM dithiothreitol、pH7.5]を用い、基質(4μM)、MgCl2(20mM)、ATP(100μM、リン酸化BTK用もしくは200μM、脱リン酸化BTK用)となるように調整し、基質混合液を作成した。またキナーゼを0.6nMとなるようアッセイバッファーで希釈して酵素溶液を調製した。被験化合物の10mM DMSO溶液から、10濃度(0.00003mM、0.0001mM、0.0003mM、0.001mM、0.003mM、0.01mM、0.03mM、0.1mM、0.3mM、1mM)にDMSOでさらに希釈し、それぞれをアッセイバッファーで25倍希釈して、薬物溶液を調整した(4%DMSO溶液)。
【0094】
薬物溶液もしくは溶媒溶液(コントロール)(4%DMSO−アッセイバッファー)5μL、基質混合液5μL、および酵素溶液10μLをポリプロピレン製384穴プレートのウェル中で混合し、1時間室温で反応させた後、60μLのキット付属のターミネーションバッファーを添加し反応を停止させた。反応阻害率は、LabChip EZ ReaderIIシステム(Caliper Life Sciences社製)を用い、アッセイキットのプロトコールに従って測定した。
【0095】
FITC標識SRCtideペプチド(キナーゼ反応基質:Substrate),及び、そのリン酸化物(Product)の、蛍光強度のピークの高さをそれぞれSおよびPとし、被験化合物の阻害率(%)を次の式に従って算出した。
またブランクとして酵素溶液の代わりにアッセイバッファーを添加したものを測定した。
被験化合物のキナーゼ阻害率(%)は、次の式に従って算出した。
キナーゼ阻害率(%)=(1−(C−A)/(B−A))×100
ただし、AはブランクのP/(P+S)、BはコントロールのP/(P+S)、Cは被験化合物ウェルのP/(P+S)を示す。
また、IC50値は、阻害率と被験化合物濃度(対数)の回帰分析により算出した。
【0096】
表3に示すとおり、実施例9から11の本発明の蛍光プローブは、蛍光標識していないCGI1746(公知の不活性型キナーゼに結合する化合物)と同様に脱リン酸化BTKのキナーゼ活性を強く阻害することが判明した。
【0097】
【表3】
実施例16
標的キナーゼへの結合による蛍光プローブの蛍光強度変化の測定
【0098】
本発明の蛍光プローブ(実施例9)の1mM DMSO溶液をアッセイバッファー[50mM HEPES、0.01%Brij−35(商標)、1mM EGTA、2mM dithiothreitol、10mM MgCl2、pH7.5]で希釈し、1μMの蛍光プローブ溶液を調整した。同様にして競合化合物(CGI1746)の1mM DMSO溶液から100μMの競合化合物溶液を調整した。また標的キナーゼとして、脱リン酸化(不活性型)BTKを0.2、1、3μMとなるようアッセイバッファーで希釈して酵素溶液を調製した。ポリスチレン製384穴プレートの各ウェルに、酵素溶液と蛍光プローブ溶液を等量ずつ加え、室温で1時間インキュベートした。ブランクとして、酵素溶液の代わりにアッセイバッファーを添加したものを測定した。また、競合試験として、最終濃度5μMとなるように上記競合化合物溶液を加えて調整した反応溶液も測定した。各ウェルの蛍光強度をSynergyH1マイクロプレートリーダー(Biotek社製)で測定した(測定波長:励起485nm/蛍光550nm)。
【0099】
結果を図1に示す。図1に示されるように標的キナーゼである脱リン酸化(不活性型)BTKの存在により濃度依存的に蛍光プローブの蛍光強度の増加が認められる。また、競合化合物であるキナーゼ阻害剤CGI1746の添加により蛍光強度が低下することから、この系は結合部位特異的に起こり、また可逆的なことが理解される。
実施例17
脱リン酸化(不活性型)BTKを用いた蛍光プローブの結合活性測定
【0100】
蛍光プローブ(実施例9)の10mM DMSO溶液から、2mMを最高濃度として公比2でDMSO希釈した13濃度のDMSO溶液を、さらに実施例16記載のアッセイバッファーで1000倍希釈して、蛍光プローブ溶液を調整した(0.1%DMSO溶液)。同様にして競合化合物(CGI1746)の1mM DMSO溶液から実施例16記載のアッセイバッファーを用いて40μMの競合化合物溶液を調整した。また脱リン酸化(不活性型)BTKを120nMとなるようアッセイバッファーで希釈して酵素溶液を調製した。蛍光プローブ溶液40μL、コントロール溶液(4%DMSO−アッセイバッファー)20μL、および酵素溶液20μLをポリプロピレン製384穴プレートのウェル中で混合し、1時間室温でインキュベートした後、各ウェルの蛍光強度をSynergyH1マイクロプレートリーダーで測定した(測定波長:励起485nm/蛍光550nm)。またコントロール溶液の代わりに競合化合物溶液を加えたウェルの蛍光強度も測定し、これをバックグラウンドとして測定値から差し引き、蛍光強度変化量(ΔRFU)とした。各蛍光プローブ濃度における蛍光強度変化量(ΔRFU)を蛍光プローブ化合物濃度に対してプロットし、飽和曲線を作成し、GraphPad Prism(商標)ソフトウェア(GraphPad Software社製)を用いて、蛍光プローブの乖離定数(Kd)を算出した。
図2に測定結果を示す。脱リン酸化(不活性型)BTKに対する蛍光プローブ(実施例9)のKd値は63nMと測定された。
【0101】
この結果は、本発明の蛍光プローブが、不活性型BTKから非常に解離し易く、競合する不活性型キナーゼを標的とする候補化合物のスクリーニングに有用であることを示している。
実施例18
脱リン酸化(不活性型)BTKを用いた競合結合測定試験
【0102】
本発明の蛍光プローブ(実施例9)の1mM DMSO溶液を実施例16記載のアッセイバッファーで希釈し、100nMの蛍光プローブ溶液を調整した。また脱リン酸化(不活性型)BTKを100nMとなるようアッセイバッファーで希釈して酵素溶液を調製した。被験化合物(CGI1746)の10mM DMSO溶液から、20μMを最高濃度として公比2でDMSO希釈した8濃度のDMSO溶液を、さらに実施例16記載のアッセイバッファーで25倍希釈して、薬物溶液を調整した(4%DMSO溶液)。薬物溶液20μLと酵素溶液20μLをポリスチレン製384穴プレートのウェル中で混合し、15分後、蛍光プローブ溶液(40μL)を添加した。室温で2時間インキュベートした後、各ウェルの蛍光強度をSynergyH1マイクロプレートリーダーで測定した(測定波長:励起485nm/蛍光550nm)。蛍光強度とキナーゼ阻害剤濃度をプロットしIC50値を算出した。
【0103】
結果を図3に示す。脱リン酸化(不活性型)BTKと蛍光プローブ(実施例9)に対するCGI1746の競合阻害活性(IC50値)は22nMと測定された。
【0104】
つまり、本発明の蛍光プローブを用いて、既知の不活性型BTKを阻害する化合物(CGI1746)の阻害活性を決定できたことは、本発明の蛍光プローブが、不活性型キナーゼのスクリーニングに有効であることを示している。
【技術分野】
【0001】
本発明は、薬剤を開発するための技術分野に属し、特に、医薬品となり得るキナーゼ阻害剤をスクリーニングする方法およびその検出を可能にする新規なベンゾオキサジアゾール蛍光プローブに関する。
【背景技術】
【0002】
プロテインキナーゼは、自己または他のタンパク質上のチロシン、セリンあるいはスレオニンをリン酸化することにより細胞増殖、細胞間コミュニケーション、細胞生存等に関与するシグナルを伝えており、これらのリン酸化シグナルは正常細胞の増殖、生存等に重要な役割を果たしている。多くの疾患は、何らかの原因で正常に働かなくなったプロテインキナーゼを介したリン酸化シグナル、つまり異常な細胞応答に関連していることが知られている(非特許文献1参照)。このような疾患としては、自己免疫疾患、炎症性疾患、骨疾患、代謝性疾患、神経学的疾患及び神経変性疾患、癌、心臓血管の疾患、アレルギー及び喘息、アルツハイマー病、ならびにホルモン関連の疾患が挙げられる。したがって、これらの疾患に関与するキナーゼを阻害する化合物はこれらの疾病などに対する予防/治療薬になると考えられ、新しいキナーゼ阻害剤の開発が日夜続けられている(非特許文献2、3参照)。
【0003】
キナーゼ阻害剤の効率的な開発のためには、対象となるキナーゼに対して、阻害剤の候補となり得る化合物群をスクリーニングすることのできる評価系を確立することが必要である。しかしながら、キナーゼはヒトの生体内に500種類以上存在しているとされており(非特許文献4参照)、対象となるキナーゼ以外を阻害しないような化合物を見出すことが、副作用の少ない薬剤を開発するうえで非常に重要である。これまでに報告されているキナーゼ阻害剤は、キナーゼに共通の基質であるATPの同じ部位に結合するものがほとんどであるが、この結合部位は同じATPを結合するようにできていることから容易に類推できるように非常に構造類似性が高く、選択的なキナーゼ阻害剤を得ることは容易ではない(非特許文献5参照)。
【0004】
生体内ではキナーゼ活性は精密に調節されており、キナーゼ自身もキナーゼによるリン酸化によってon/off(活性型と不活性型)の調節を受けており、その立体構造が大きく変化するとされている。つまり同じキナーゼでも、活性型か不活性型かのどちらを阻害するかで選択性に違いが出てくることが知られている(非特許文献6参照)。
【0005】
現在、報告されているほとんどのキナーゼ阻害剤は活性型キナーゼを阻害するものであるが、前述のとおり活性型キナーゼのATP結合部位の構造は、極めて高い類似性を持つため、そこに結合する阻害剤で高い選択性を出すのは難しいとされている。
【0006】
一方、不活性型キナーゼに結合する阻害剤は、活性化ループの構造の変化によって出現する新たな領域との相互作用を利用できるため非常に高い選択性を示す場合が多い。つまり、活性型キナーゼの立体構造は類似しているが、不活性型キナーゼでは必ずしもそうではないということである。また不活性型キナーゼは競合基質であるATPとの親和性も低下しており、不活性型キナーゼに結合する物質には、生体内ではより強力な生理作用が期待できる。しかしながら、これまで報告されている不活性型キナーゼに結合する阻害剤は、ほとんどが偶然発見されたものであり、活性のないキナーゼを用いて化合物のスクリーニングを実施するのは非常に難しいとされている。
【0007】
ところで、キナーゼを阻害する化合物をスクリーニングする方法としては、活性型キナーゼの酵素活性阻害を指標とした評価系が最も一般的である(非特許文献7参照)。競合結合測定法(competitive binding assay)により、プローブとして用いられる標準阻害剤に対する拮抗阻害の評価系も用いられることもあるが(例えば、LanthaScreen(商標) Eu Kinase Binding Assay、インビトロジェン社)、拮抗阻害を検出するためにはキナーゼタンパク自体を化学的あるいは生物的に修飾する必要があり、さらに2次試薬も添加する必要があるため、バックグラウンドシグナルの上昇や操作が煩雑にならざるを得ない。
【0008】
通常、蛍光プローブを合成する場合には、生化学分野でよく用いられているフルオレセインやその誘導体などの蛍光色素が用いられている。しかしながら、これらの蛍光色素は、それ自身が強い蛍光を発するため、測定時には標的タンパク質と結合していないプローブを取り除くか、あるいは、先に述べたように標的タンパクの修飾や2次試薬の添加などが必要となる。一方、蛍光検出−HPLCなどによく用いられている蛍光ラベル化試薬、ベンゾオキサジアゾール誘導体は、有機溶媒中など疎水性環境で非常に強い蛍光を発するが、例えば水中ではほぼ無蛍光となることが知られており(例えば、東京化成工業のWebページ、HPLC用ラベル化剤、http://www.tokyokasei.co.jp/product/analytical−chem/A007.shtmlなどを参照)、この性質を利用した、タンパク質内部の疎水性環境を探るような蛍光プローブの例がいくつか報告されている(例えば非特許文献8、9などを参照)。
【0009】
ちなみに、特許文献としては、国際公開第2003/097667号、国際公開第2003/089665号等にキナーゼ阻害剤をスクリーニングする方法が知られているが(特許文献1、特許文献2)、これらの方法は先に述べたように満足とは限らない。また、国際公開第2004/085618号に不活性型キナーゼを標的としたスクリーニング方法について記載されているが(特許文献3)、どのようにして評価するのか、またスクリーニング方法の実証結果など全く示されていない。国際公開第2005/033330号には、当業界で通常用いられているフルオレセイン誘導体を蛍光色素として利用した蛍光偏光法によるアッセイ方法について開示されているが(特許文献4)、不活性型キナーゼのスクリーニングに関する記載は全くなされていない。国際公開第2008/033834号、国際公開第2008/033854号には、ラジオアイソトープや蛍光色素でラベル化したBTK(Bruton’s tyrosine kinase)阻害剤がバイオアッセイにも使用できることが記載されているものの、ラベル化化合物およびバイオアッセイに関しては全く検討されていない(特許文献5、特許文献6)。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Hunter,T.,Cell,(1995),80,225−236
【非特許文献2】Cohen,P.,Nat.Rev.Drug Discov.,(2002),1,309−315
【非特許文献3】Shchemelinin,I.,et al.,Folia Biol(Praha),(2006),52,81−100
【非特許文献4】Manning,G.,et al.,Science,(2002),298,1912−1934
【非特許文献5】Zhang,J.,et al.,Nat.Rev.Cancer,(2009),9,28−39
【非特許文献6】Liu,Y.,et al.,Nat.Chem.Biol.,(2006),2,358−364
【非特許文献7】Comley,J.,Drug Discovery World,Fall 2004,45−56
【非特許文献8】Kenner,R.A., et al.,Biochemistry,(1971),10,4433−4440
【非特許文献9】Rasmussen,S.G.、et al.,J.Biol.Chem.,(2001)、276、4717−4723
【特許文献】
【0011】
【特許文献1】WO2003/097667号
【特許文献2】WO2003/089665号
【特許文献3】WO2004/085618号
【特許文献4】WO2005/033330号
【特許文献5】WO2008/033834号
【特許文献6】WO2008/033854号
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明の目的は、活性型ないし不活性型キナーゼに結合する薬剤を簡便且つ確実にスクリーニングすることのできる新しいスクリーニング方法、およびその検出を可能にする新規なベンゾオキサジアゾール蛍光プローブを提供することにある。
【課題を解決するための手段】
【0013】
本発明者は、前述のとおりベンゾオキサジアゾール誘導体が環境に依存して蛍光強度を変化させる性質に着目し、この性質を利用することにより上述の目的を達成し得る本発明を案出したものである。かくして、本発明に従えば、活性型ないし不活性型キナーゼに結合する薬剤のスクリーニングが簡便に可能となり、キナーゼ阻害剤にベンゾオキサジアゾール誘導体を結合させた蛍光プローブとキナーゼの混合物に被験物質を添加し、その蛍光強度の変化が観察されたときに、被験物質をキナーゼ阻害剤として選択することを特徴とする方法が提供される。
【発明の効果】
【0014】
本発明のスクリーニング方法および新規なベンゾオキサジアゾール蛍光プローブを用いれば、活性型キナーゼのみならず、不活性型キナーゼに結合する薬剤を簡便且つ確実にスクリーニングすることができ、従来の競合結合測定法において必要だった標的酵素の修飾および2次試薬が不要となる。かくして、本発明は、キナーゼ阻害剤の効率的なスクリーニングを通じて、自己免疫疾患、炎症性疾患、骨疾患、代謝性疾患、神経学的疾患及び神経変性疾患、癌、心臓血管の疾患、アレルギー及び喘息、アルツハイマー病、ならびにホルモン関連の疾患などに対する予防・治療薬の開発に資するものである。
【図面の簡単な説明】
【0015】
【図1】本発明の蛍光プローブが脱リン酸化(不活性型)BTKに結合することで、水溶液中と比較して有意に高く酵素濃度依存的に蛍光を発することを示す。また、キナーゼ阻害剤の添加により蛍光強度が低下することを示す。
【図2】脱リン酸化(不活性型)BTKと結合することで、本発明の蛍光プローブの蛍光が濃度依存的に増加することを示す。
【図3】本発明の蛍光プローブを使用して、試験化合物を種々の希釈で脱リン酸化(不活性型)BTK結合活性を指標にしてスクリーニングする場合に得られた蛍光変化結果を示す。
【発明を実施するための最良の形態】
【0016】
本発明者らは、酵素活性がない、もしくは非常に弱い不活性型キナーゼを用いて、その基質結合部位に拮抗的に結合する化合物を見つけるために、競合結合測定法に注目して研究を行った。このとき、通常、生化学分野でよく用いられるフルオレセインなどの蛍光色素とは異なる性質を有するベンゾオキサジアゾール誘導体が、キナーゼの結合度に応じて蛍光強度が変化することにより、2次試薬等を必要としない非常に簡便な競合阻害試験に利用できるものと考え、本発明を導き出したものである。
本発明は、以下の(1)〜(7)によって達成される。
(1)下式(I)を有する新規なベンゾオキサジアゾール蛍光プローブ。
【0017】
【化1】
【0018】
(式中、Zは、キナーゼ阻害活性を有する化合物を表わし、Lは、結合、置換されてもよいアミノ基、カルボニル基、置換されてもよい炭化水素基、置換されてもよいアルキルカルバモイル基、置換されてもよいアシルアミノ基、スルフィド基、置換されてもよいアルコキシ基を表し、Xは、結合、置換されてもよいアミノ基、置換されてもよいヒドラジノ基、チオカルボニルアミノ基を表し、R1はニトロ基、ジメチルスルファモイル基を表す。)
(2)Zが、p38MAPK阻害活性を有する化合物である、(1)に記載の蛍光プローブ。
(3)Zが、下式(Ia)を有する化合物である、(1)または(2)に記載の蛍光プローブ。
【0019】
【化2】
(式中、R2は置換されてもよいアルコキシ基を表す。)
(4)Zが、BTK阻害活性を有する化合物である、(1)に記載の蛍光プローブ。
(5)Zが、下式(Ib)を有する化合物である、(1)または(4)に記載の蛍光プローブ。
【0020】
【化3】
(6)(1)から(5)の何れか1つに記載の蛍光プローブを使用することを特徴とするキナーゼ阻害剤をスクリーニングする方法。
(7)不活性型キナーゼを標的とした、(6)に記載のスクリーニング方法。
【0021】
以下、本発明に従うキナーゼ阻害剤のスクリーニング法において用いられる蛍光プローブの作製法、ならびに当該蛍光プローブを用いるスクリーニング法に従って本発明の実施の形態を説明する。但し、以下に示す具体的な実施の形態は例示のためのものであり、本発明の実施の形態はこれに限定されるものではない。
【0022】
本発明の新しいキナーゼ阻害剤のスクリーニング法の実施を可能にする新規なベンゾオキサジアゾール蛍光プローブは、下式(I)で示される化合物である。
【0023】
【化4】
【0024】
(式中、Zは、キナーゼ阻害活性を有する化合物を表わし、Lは、結合、置換されてもよいアミノ基、カルボニル基、置換されてもよい炭化水素基、置換されてもよいアルキルカルバモイル基、置換されてもよいアシルアミノ基、スルフィド基、置換されてもよいアルコキシ基を表し、Xは、結合、置換されてもよいアミノ基、置換されてもよいヒドラジノ基、チオカルボニルアミノ基を表し、R1はニトロ基、ジメチルスルファモイル基を表す。)
【0025】
尚、上記で「結合」とは、“ZとX”、“Lとベンゾオキサジアゾール基”、あるいは“Zとベンゾオキサジアゾール基”が直接結合した場合を意味する。
【0026】
前記式(I)において、キナーゼ阻害活性を有する化合物としては、標的とする活性型あるいは不活性型キナーゼの、触媒ドメインのATP結合部位、基質結合部位もしくはアロステリック部位に可逆的に結合する化合物であれば、新規化合物や公知化合物の何れであってもよいが、このような性質を有する化合物としてはいくつかのものが既に知られており、これらを使用することができる。
例えば、ATP結合部位に結合して活性型キナーゼを阻害するものとしては、スタウロスポリン、スニチニブ(N−(2−diethylaminoethyl)−5−[(Z)−(5−fluoro−2−oxo−1H−indol−3−ylidene)methyl]−2,4−dimethyl−1H−pyrrole−3−carboxamide)、ダサチニブ(N−(2−chloro−6−methylphenyl)−2−[[6−[4−(2−hydroxyethyl)−1−piperazinyl]−2−methyl−4−pyrimidinyl]amino]−5−thiazolecarboxamide monohydrate)などのキナーゼ阻害薬もしくはそれらの誘導体などが挙げられる。
【0027】
不活性型キナーゼを標的とする場合は、不活性型キナーゼにより強く結合することが知られている化合物、具体的には、不活性型BTKを阻害するN−[3−[4,5−Dihydro−4−methyl−6−[[4−(4−morpholinylcarbonyl)phenyl]amino]−5−oxo−2−pyrazinyl]−2−methylphenyl]−4−(tert−butyl)benzamide(CGI1746)に代表される上記[化3]の部分構造をもつ誘導体、不活性型p38MAPKを阻害するdoramapimodに代表される上記[化2]の部分構造をもつ誘導体、不活性型ABLを阻害するイマチニブ、ニロチニブもしくはそれらの誘導体などが挙げられる。
【0028】
また、アロステリック部位に結合してキナーゼ活性を阻害することが知られているものとしては、MAPKキナーゼのMEKを阻害する2−(2−Chloro−4−iodo−phenylamino)−N−cyclopropylmethoxy−3,4−difluoro−benzamide(PD−184352,CI−1040)もしくはそれらの誘導体などが挙げられる。
前記式(I)において、「置換されてもよい炭化水素基」の「炭化水素」部分としては、例えば、
a)炭素数1から6の直鎖状、あるいは分岐鎖状のアルキル基(例えば、メチル、エチル、イソプロピル、tert−ブチル、ヘキシル等)、
b)炭素数1から6の直鎖状、あるいは分岐鎖状のアルケニル基(例えば、ビニル、アリル、イソプロペニル、2−ブテニル等)、
c)炭素数2から6のアルキニル基(例えば、エチニル、プロパルギル、2−ブチニル等)、
d)炭素数3から8のシクロアルキル基(例えば、シクロプロピル、シクロペンチル、シクロヘキシル、シクロヘプチル等)、
e)炭素数3から8のシクロアルケニル基(例えば、シクロヘキセニル、シクロヘプテニル等)、
【0029】
f)アラルキル基、アラルキル基のアリール部分としては、炭素数6から14のアリールが挙げられ(例えば、フェニル、ナフチル、インデニル等)、アラルキル基のアルキレン部分としては、前記アルキル基から水素原子を1つ除いたものと同義である、
などが挙げられる。
【0030】
「置換されてもよい炭化水素基」の「置換基」としては、置換されてもよいアミノ基、置換されてもよいアルコキシ基、置換されてよいカルバモイル基、置換されてもよいアシルアミノ基、置換されてもよいスルフィド基等を挙げることができる。
【0031】
「置換されてもよいアミノ基」としては、置換もしくは非置換の炭素数1から6の直鎖状、分枝状もしくは環状のアルキル基を有するアミノ基または直鎖状、分枝状もしくは環状のアルキル基の炭素原子がヘテロ原子に置換されていてもよいアミノ基を表し、具体的にはメチルアミノ基、ピロリジノ基、ピペリジノ基、ピペラジノ基、モルホリノ基、アニリノ基などが挙げられる。
【0032】
「置換されてもよいアルキルカルバモイル基」としては、置換もしくは非置換の炭素数1から6の直鎖状、分枝状もしくは環状のアルキル基を有するアルキルカルバモイル基又は直鎖状、分枝状もしくは環状のアルキル基の炭素原子がヘテロ原子に置換されていてもよいアルキルカルバモイル基を表す。
【0033】
「置換されてもよいアシルアミノ基」としては、置換もしくは非置換の炭素数1から6の直鎖状、分枝状もしくは環状のアルキル基を有するアルキルカルボニルアミノ基又は直鎖状、分枝状もしくは環状のアルキル基の炭素原子がヘテロ原子に置換されていてもよいアルキルカルボニルアミノ基を表す。
【0034】
「置換されてもよいアルコキシ基」としては、置換もしくは非置換の炭素数1から6の直鎖状、分枝状もしくは環状のアルキル基を有するアルコキシ基又は直鎖状、分枝状もしくは環状のアルキル基の炭素原子がヘテロ原子に置換されていてもよいアルコキシ基を表す。
【0035】
「置換されてもよいヒドラジノ基」としては、置換もしくは非置換の炭素数1から6の直鎖状、分枝状もしくは環状のアルキル基を有するヒドラジノ基または直鎖状、分枝状もしくは環状のアルキル基の炭素原子がヘテロ原子に置換されていてもよいヒドラジノ基を表す。
【0036】
ベンゾオキサジアゾール基を、キナーゼ阻害活性を有する化合物に結合するためのリンカーである式(I)のLおよびXとしては、市販で入手可能なベンゾオキサジアゾール誘導体を蛍光標識色素として結合するのに使用し得る当業界で知られているあらゆる基であってもよい。典型的には、この実施態様において、LおよびXは、ベンゾオキサジアゾール誘導体の反応性官能基であるフッ素、塩素などのハロゲンまたはイソチオシアネート基と、キナーゼ阻害活性を有する化合物の適当な位置に導入したアミン性官能基との反応により生成されてもよい。
【0037】
ベンゾオキサジアゾール基は、Zのキナーゼ阻害活性を保持しつつ、キナーゼに結合すると蛍光が変化するように適宜、選ばれたLおよびXを介してZと結合を形成する。
【0038】
本発明の蛍光プローブ(I)は、例えば、置換基の種類によって、異性体が存在する場合がある。本明細書において、それらの異性体の一形態のみの化学構造で記載することがあるが、本発明には、構造上生じ得るすべての異性体(幾何異性体、光学異性体、互変異性体など)も含有し、異性体単体、またはそれらの混合物も含有する。
【0039】
また、本発明の蛍光プローブ(I)は、その水溶性などの物性を調節するために、塩にして用いる場合があり、本発明には、化学性質上、生じ得るすべての塩も含有する。
本発明の蛍光プローブ(I)の具体的な化合物としては、以下の化合物が挙げられる。
【0040】
1−(3−(t‐ブチル)−1−(4−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)フェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア
【0041】
1−(3−(t‐ブチル)−1−(3−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)フェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア
【0042】
N−(4−(3−(t‐ブチル)−5−(3−(ナフタレン−1−イル)ウレイド)−1H−ピラゾロ−1−イル)フェニル)−3−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)プロパンアミド
【0043】
3−(3−(t‐ブチル)−5−(3−(ナフタレン−1−イル)ウレイド)−1H−ピラゾロ−1−イル)−N−(2−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)エチル)ベンズアミド
【0044】
4−(3−(t‐ブチル)−5−(3−(ナフタレン−1−イル)ウレイド)−1H−ピラゾロ−1−イル)−N−(2−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)エチル)ベンズアミド
【0045】
4−(3−(t‐ブチル)−5−(3−(4−(2−モルホリノエトキシ)ナフタレン−1−イル)ウレイド)−1H−ピラゾロ−1−イル)−N−(2−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)エチル)ベンズアミド
【0046】
1−(3−(t‐ブチル)−1−(4−(4−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)1‐ブチン−1−イル)フェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア
【0047】
1−(3−(t‐ブチル)−1−(4−(4−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)ブチル)フェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア
【0048】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−(4−(7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)ピペラジン−1−カルボニル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0049】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−((2−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)エチル)カルバモイル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0050】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−(2−(((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)メチル)モルホリン−4−カルボニル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0051】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−((2−(2−(2−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)エトキシ)エトキシ)エチル)カルバモイル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0052】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−(4−(2−(メチル(7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)アセチル)ピペラジン−1−カルボニル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0053】
4−(t‐ブチル)−N−(3−(6−((4−(4−(7−(N,N−ジメチルスルファモイル)ベンゾ[C][1,2,5]オキサジアゾール−4−イル)ピペラジン−1−カルボニル)フェニル)アミノ)−4−メチル−5−オキソ−4,5−ジヒドロピラジン−2−イル)−2−メチルフェニル)ベンズアミド
【0054】
本発明の蛍光プローブ(I)は、例えば以下の方法によって製造することができる。なお、以下に示した製造法において、定義した基が実施方法の条件下で変化するか、または方法を実施するのに不適切な場合、有機合成化学で通常用いられる方法、例えば、官能基の保護、脱保護[T.W.Greene,Protective Groups in Organic Synthesis 3rd Edition,John Wiley&Sons,Inc.,1999]等の手段を付すことにより容易に製造することができる。また、必要に応じて置換基導入等の反応工程の順序を変えることもできる。
スキーム1
【0055】
【化5】
【0056】
スキーム1において、R1、L、XおよびZは先に定義されたとおりであり、
ベンゾオキサジアゾール誘導体化試薬(III)は、蛍光ラベル化試薬として種々のAを官能基として有する誘導体が市販されており(例えば、東京化成工業株式会社などから入手可能)、官能基Aは化合物(II)と反応後、Xに変換される。もし、ZがリンカーLもしくはベンゾオキサジアゾール誘導体化試薬(III)と反応しうる適当な官能基を適当な位置に有しないときには、その前駆体化合物や、有機合成化学で通常用いられる方法を実施することにより、所望の位置に所望の官能基を有する誘導体を合成し、その誘導体を用いてスキーム1の反応を実施してもよい。また、リンカーLを介して、ベンゾオキサジアゾール誘導体化試薬(III)でZをラベル化するときは、アミノ基、カルボキシル基、水酸基、チオール基など、ベンゾオキサジアゾール誘導体化試薬(III)との反応に適当な官能基を適当な位置に有しているL’を、必要であれば官能基の保護、脱保護を繰り返して、当業界で知られている通常の有機合成の条件などを用いてZに導入し、得られた化合物(II)を適当なベンゾキサジアゾール誘導体化試薬(III)と反応させることにより、式(I)の所望の化合物を得ることができる。
ベンゾオキサジアゾール誘導体化試薬(III)は、例えば、
【0057】
アミノ基やメルカプト基のラベル化試薬として、AがFやClなどのハロゲン原子であるNBD−Cl、NBD−F、イソチオシアネート基であるNBD−NCS、
【0058】
カルボキシル基のラベル化試薬として、AがピペラジンであるNBD−PZ、DBD−PZ、2−アミノエチルアミノ基であるDBD−ED、N−ヒドラジノカルボニルメチル−N−メチルアミノ基であるNBD−CO−Hz
などが市販で入手可能である。
【0059】
また、カルボニル基のラベル化には、Aがヒドラジノ基であるNBD−HやDBD−H、水酸基のラベル化としては、AがN−クロロホルミルメチル−N−メチルアミノ基であるDBD−COClなどが市販されている。
また、本発明のスクリーニング方法は、上記方法により得られた本発明の蛍光プローブとキナーゼの混合物に被験物質を添加し、その蛍光強度の変化(低下)が観察されたときに、被験物質をキナーゼ阻害剤として選択することを特徴とする。
【実施例】
【0060】
以下に実施例を挙げて本発明をさらに具体的に説明するが、これらの実施例により本発明が限定されるものではない。
【0061】
化合物の同定は水素核磁気共鳴スペクトル(1H−NMR)及びマススペクトル(MS)により行った。1H−NMRは、特に指示のないかぎりは400MHzで測定されたものであり、また化合物及び測定条件によっては交換性水素が明瞭に観測されない場合がある。質量分析は液体クロマトグラフ質量分析計(LCMS)システムを用い、以下に示したA法もしくはB法にて測定した。
【0062】
A法:LCMS−2010Aシステム(島津製作所)を使用し、質量分析はエレクトロスプレー(ESI)法により測定した。分離カラムはCadenza CD‐C18(50×2mm)(インタクト社製)を用いた。溶出には10mMギ酸水溶液(A液)および10mMギ酸メタノール溶液(B液)によるグラジェント法を用い、溶出条件は、流速0.5mL/分、0分〜0.5分(B液:5%(v/v))、0.5分〜2.5分(B液:5〜95%(v/v)直線グラジェント)、2.5分〜4分(B液:95%(v/v))とした。
【0063】
B法:液体クロマトグラフ質量分析計システム(micromass ZQマス検出器、日本ウォーターズ社製)を使用し、質量分析はエレクトロスプレー(ESI)法により測定した。分離カラムはUnison US‐C18(50×4.6mm)(インタクト社製)を用いた。溶出には10mMギ酸水溶液(A液)および10mMギ酸メタノール溶液(B液)によるグラジェント法を用い、溶出条件は、溶出条件は、流速2mL/分、0分〜0.5分(B液:5%(v/v))、0.5分〜3分(B液:10〜90%(v/v)直線グラジェント)、3分〜4分(B液:90%(v/v))とした。
実施例1から8は、本発明の蛍光プローブ(I)のうち、Zがp38MAPK阻害活性を有する化合物である場合を例として、具体的に説明する。
実施例1
【0064】
1−(3−(t‐ブチル)−1−(4−((7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)アミノ)フェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア
【0065】
【化6】
(第1工程)
【0066】
4−ニトロフェニルヒドラジン(766mg)、ピバロイルアセトニトリル(626mg)をエタノール(12mL)に溶解し、濃塩酸(2.5mL)を加え90℃で24時間撹拌した。反応混合物を減圧濃縮し、得られた粗生成物をエタノールで懸濁洗浄し、3−(t‐ブチル)−1−(4−ニトロフェニル)−1H−ピラゾロ−5−アミン塩酸塩1.31gを得た。
(第2工程)
【0067】
3−(t‐ブチル)−1−(4−ニトロフェニル)−1H−ピラゾロ−5−アミン塩酸塩(297mg)にジクロロメタン(10mL)、トリエチルアミン(167μL)、1−ナフチルイソシアネート(158μL)を加え80℃で18時間撹拌した。反応混合物を減圧濃縮し、得られた残渣に水(10mL)を加え、酢酸エチル(50mL×2)で抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して1−(3−(t‐ブチル)−1−(4−ニトロフェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア145mgを得た。
(第3工程)
【0068】
1−(3−(t‐ブチル)−1−(4−ニトロフェニル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア(145mg)をエタノール(7mL)に溶解し、ギ酸アンモニウム(132mg)、触媒量の10%パラジウム−炭素を加え90℃で3時間還流した。不溶物をセライトろ過した後、ろ液を減圧濃縮した。得られた残渣をフラッシュクロマトグラフィーで精製し、1−(1−(4−アミノフェニル)−3−(t‐ブチル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア81mgを得た。
(第4工程)
【0069】
1−(1−(4−アミノフェニル)−3−(t‐ブチル)−1H−ピラゾロ−5−イル)−3−(ナフタレン−1−イル)ウレア(21mg)をエタノール(7mL)に溶解し、4‐フルオロ‐7‐ニトロ-2,1,3‐ベンゾオキサジアゾール(10mg)を加え室温で18時間撹拌した。析出した固体をろ取し、ジエチルエーテルで洗浄後、無水硫酸ナトリウムで乾燥させて表題化合物22mgを得た。LCMS:HPLC保持時間3.53分(A法)、m/z563(MH+)。
【0070】
1H-NMR (400 MHz, DMSO-d6) δ ppm 1.31 (s, 9H), 6.44 (s, 1H), 6.85 (d, J=8.78 Hz, 1H), 7.47 (t, J=7.91 Hz, 1H), 7.51 - 7.61 (m, 2H), 7.62 - 7.77 (m, 5H), 7.87 - 7.98 (m, 2H), 8.04 (d, J=8.28 Hz, 1H), 8.57 (d, J=8.78 Hz, 1H), 8.90 (s, 1H), 9.06 (s, 1H), 11.18 (s, 1H).
【0071】
下記実施例2から7の化合物は、実施例1に記載の方法もしくは類似の方法に従い、適当な試薬および有機合成化学で通常用いられる方法により製造した。表中のMethodはLCMSの分析方法、RTはLCMSにおけるHPLC保持時間、MSはLCMSにおけるマススペクトル分析データ(特に指示のない限りポジティブイオンモードで測定されたMH+)を示す。
【0072】
【表1】
また、実施例9から14は、本発明の蛍光プローブ(I)のうち、ZがBTK阻害活性を有する化合物である場合を例として、具体的に説明する。
参考例1
【0073】
4−((6−(3−(4−(t‐ブチル)ベンズアミド)−2−メチルフェニル)−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)安息香酸
【0074】
【化7】
(第1工程)
【0075】
文献J.Med.Chem.Vol.48、p.1901(2005年)に記載の方法で調製した3,5−ジブロモ−1−メチルピラジン−2(1H)−オン(593mg)のジメチルアセトアミド溶液(3.7mL)に4‐アミノ安息香酸エチル(475mg)を加え、マイクロウェーブ合成装置を用いて105℃で20時間反応させた。析出した固体をろ取し、酢酸エチルで洗浄後、乾燥させてエチル 4−((6−ブロモ−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート620mgを得た。
【0076】
1H-NMR (400 MHz, CDCl3) δppm 1.39 (t, J=7.15 Hz, 3H), 3.55 (s, 3H), 4.37 (q, J=7.28 Hz, 2H), 6.83 (s, 1H), 7.82 (d, J=8.78 Hz, 2H), 8.06 (d,J=8.78 Hz, 2H), 8.45 (s, 1H).
(第2工程)
【0077】
エチル 4−((6−ブロモ−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート(870mg)の1,4−ジオキサン溶液(7.6mL)に3‐アミノ‐2‐メチルフェニルボロン酸ピナコールエステル(749mg)、テトラキス(トリフェニルホスフィン)パラジウム(0)(400mg)、2.4M炭酸ナトリウム水溶液(1.5mL)を加え、105℃で60時間撹拌した。不溶物をセライトろ過した後、ろ液にジクロロメタン(50mL)を加え、2M塩酸(50mL)で抽出した。水層に2M水酸化ナトリウム水溶液(60mL)を加えてアルカリ性にした。析出した固体をろ取し、水で洗浄後、乾燥させてエチル 4−(6−(3−アミノ−2−メチルフェニル)−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート588mgを得た。
(第3工程)
【0078】
エチル 4−(6−(3−アミノ−2−メチルフェニル)−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート(588mg)のジクロロメタン溶液溶液(7.8mL)にピリジン(0.14mL)、4‐t‐ブチルベンゾイルクロリド(0.455mL)を加え、室温で4時間撹拌した。反応混合物をクロロホルム(100mL)で希釈し、1M水酸化ナトリウム水溶液、水、飽和食塩水で順次洗浄した。得られた有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。得られた残渣をフラッシュクロマトグラフィーで精製し、エチル 4−((6−(3−(4−(t‐ブチル)ベンズアミド)−2−メチルフェニル)−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート534mgを得た。
(第4工程)
【0079】
エチル 4−((6−(3−(4−(t‐ブチル)ベンズアミド)−2−メチルフェニル)−4−メチル−3−オキソ−3,4−ジヒドロピラジン−2−イル)アミノ)ベンゾエート(400mg)のテトラヒドロフラン溶液(7mL)に1M水酸化ナトリウム水溶液(7mL)、エタノール(7mL)を加え、80℃で3時間撹拌した。反応混合物に1M塩酸(7mL)、水(50mL)を加え、酢酸エチル(20mL×3)で抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して表題化合物377mgを得た。
【0080】
1H-NMR (400 MHz, DMSO-d6) δ ppm 1.33 (s, 9H), 2.30 (s, 3H), 3.58 (s, 3H), 7.25 - 7.36 (m, 3H), 7.40 (dd, J=6.53, 2.51 Hz, 1H), 7.56 (d, J=8.28 Hz, 2H), 7.85 (d, J=8.78 Hz, 2H), 7.96 (d, J=8.53 Hz, 2H), 8.14 (d, J=8.78 Hz, 2H), 9.59 (s, 1H), 9.88 (s, 1H), 12.58 (s, 1H).
実施例9
【0081】
4−(t‐ブチル)−N−(2−メチル−3−(4−メチル−6−((4−(4−(7−ニトロベンゾ[c][1,2,5]オキサジアゾール−4−イル)ピペラジン−1−カルボニル)フェニル)アミノ)−5−オキソ−4,5−ジヒドロピラジン−2−イル)フェニル)ベンズアミド
【0082】
【化8】
【0083】
参考例1で得られた化合物(50mg)のテトラヒドロフラン溶液(1.6mL)に1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(56mg)、1−ヒドロキシベンゾトリアゾール(45mg)を加え、室温で1時間撹拌したのち、4‐ニトロ‐7‐ピペラジノ‐2,1,3‐ベンゾオキサジアゾール(36.6mg)のジメチルホルムアミド(0.3mL)溶液を加え、室温でさらに18時間撹拌した。反応混合物にクロロホルム(50mL)を加え、1M塩酸、水、飽和重曹水、及び飽和食塩水で順次洗浄した。得られた有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し表題化合物63mgを得た。LCMS:HPLC保持時間3.72分(B法)、m/z742(MH+)。
【0084】
1H-NMR (400 MHz, DMSO-d6) δ ppm 1.31 (s, 9H), 2.30 (s, 3H), 3.58 (s, 3H), 3.80 (br. s., 4H), 4.22 (br. s., 4H), 6.60 (d, J=9.29 Hz, 1H), 7.24 - 7.41 (m, 4H), 7.44 (d, J=8.53 Hz, 2H), 7.53 (d, J=8.53 Hz, 2H), 7.94 (d, J=8.53 Hz, 2H), 8.12 (d, J=8.78 Hz, 2H), 8.52 (d, J=9.03 Hz, 1H), 9.50 (s, 1H), 9.89 (s, 1H).
【0085】
下記実施例10から13に示す化合物は、実施例9に記載の方法もしくは類似の方法に従い、適当な試薬および有機合成化学で通常用いられる方法により製造した。表中のMethodはLCMSの分析方法、RTはLCMSにおけるHPLC保持時間、MSはLCMSにおけるマススペクトル分析データ(特に指示のない限りポジティブイオンモードで測定されたMH+)を示す。
【0086】
【表2】
実施例14
【0087】
4−(t‐ブチル)−N−(3−(6−((4−(4−(7−(N,N−ジメチルスルファモイル)ベンゾ[C][1,2,5]オキサジアゾール−4−イル)ピペラジン−1−カルボニル)フェニル)アミノ)−4−メチル−5−オキソ−4,5−ジヒドロピラジン−2−イル)−2−メチルフェニル)ベンズアミド
【0088】
【化9】
【0089】
実施例9に記載の手順に従って、4‐ニトロ‐7‐ピペラジノ‐2,1,3‐ベンゾオキサジアゾールの替わりに4‐(N,N‐ジメチルアミノスルホニル)‐7‐ピペラジノ‐2,1,3‐ベンゾオキサジアゾール(27mg)を用い、反応・処理することにより表題化合物47mgを得た。
LCMS:HPLC保持時間3.72分(B法)、m/z804(MH+)。
【0090】
1H-NMR (400 MHz, DMSO-d6) δ ppm 1.31 (s, 9H), 2.30 (s, 3H), 2.65 - 2.80 (m, 6H), 3.58 (s, 3H), 3.75 (br. s., 4H), 3.96 (br. s., 4H), 6.60 (d, J=8.28 Hz, 1H), 7.25 - 7.34 (m, 3H), 7.38 (dd, J=6.90, 2.38 Hz, 1H), 7.43 (d, J=8.53 Hz, 2H), 7.54 (d, J=8.53 Hz, 2H), 7.86 (d, J=8.03 Hz, 1H), 7.94 (d, J=8.28 Hz, 2H), 8.12(d, J=8.78 Hz, 2H), 9.50 (s, 1H), 9.90 (s, 1H).
実施例15
リン酸化(活性型)および脱リン酸化(不活性型)BTKに対するキナーゼ活性阻害試験
【0091】
リン酸化(活性型)BTKは、ビオチン化BTK蛋白質BTN−BTK(カルナバイオサイエンス社製)酵素溶液にATP、MgCl2をそれぞれ3mM、10mMとなるように添加し、4℃で一晩反応させた後、10DG Desalting Columnを用いてバッファー交換を行い、未反応のATPを除去することにより得た。
【0092】
脱リン酸化(不活性型)BTKは、BTN−BTK酵素溶液にλ protein phosphatase(New England BioLabs社製、Code No.P0753S)とMnCl2をそれぞれ10U/μg、2mMとなるように添加し、4℃で一晩反応させた後、抗DYKDDDDK−tag抗体アガロースゲルクロマトグラフィーによりλ protein phosphataseを除去したのち、10DG Desalting Columnを用いてバッファー交換を行い得た。
【0093】
キナーゼ活性の測定は、MSAアッセイキット(QuickScout Screening Assist(商標)kit、カルナバイオサイエンス社製)を用いて行った。キナーゼ反応の基質は、キット付属のFITC標識SRCtideペプチドを用いた。アッセイバッファー[20mM HEPES、0.01%TritonX−100(商標)、2mM dithiothreitol、pH7.5]を用い、基質(4μM)、MgCl2(20mM)、ATP(100μM、リン酸化BTK用もしくは200μM、脱リン酸化BTK用)となるように調整し、基質混合液を作成した。またキナーゼを0.6nMとなるようアッセイバッファーで希釈して酵素溶液を調製した。被験化合物の10mM DMSO溶液から、10濃度(0.00003mM、0.0001mM、0.0003mM、0.001mM、0.003mM、0.01mM、0.03mM、0.1mM、0.3mM、1mM)にDMSOでさらに希釈し、それぞれをアッセイバッファーで25倍希釈して、薬物溶液を調整した(4%DMSO溶液)。
【0094】
薬物溶液もしくは溶媒溶液(コントロール)(4%DMSO−アッセイバッファー)5μL、基質混合液5μL、および酵素溶液10μLをポリプロピレン製384穴プレートのウェル中で混合し、1時間室温で反応させた後、60μLのキット付属のターミネーションバッファーを添加し反応を停止させた。反応阻害率は、LabChip EZ ReaderIIシステム(Caliper Life Sciences社製)を用い、アッセイキットのプロトコールに従って測定した。
【0095】
FITC標識SRCtideペプチド(キナーゼ反応基質:Substrate),及び、そのリン酸化物(Product)の、蛍光強度のピークの高さをそれぞれSおよびPとし、被験化合物の阻害率(%)を次の式に従って算出した。
またブランクとして酵素溶液の代わりにアッセイバッファーを添加したものを測定した。
被験化合物のキナーゼ阻害率(%)は、次の式に従って算出した。
キナーゼ阻害率(%)=(1−(C−A)/(B−A))×100
ただし、AはブランクのP/(P+S)、BはコントロールのP/(P+S)、Cは被験化合物ウェルのP/(P+S)を示す。
また、IC50値は、阻害率と被験化合物濃度(対数)の回帰分析により算出した。
【0096】
表3に示すとおり、実施例9から11の本発明の蛍光プローブは、蛍光標識していないCGI1746(公知の不活性型キナーゼに結合する化合物)と同様に脱リン酸化BTKのキナーゼ活性を強く阻害することが判明した。
【0097】
【表3】
実施例16
標的キナーゼへの結合による蛍光プローブの蛍光強度変化の測定
【0098】
本発明の蛍光プローブ(実施例9)の1mM DMSO溶液をアッセイバッファー[50mM HEPES、0.01%Brij−35(商標)、1mM EGTA、2mM dithiothreitol、10mM MgCl2、pH7.5]で希釈し、1μMの蛍光プローブ溶液を調整した。同様にして競合化合物(CGI1746)の1mM DMSO溶液から100μMの競合化合物溶液を調整した。また標的キナーゼとして、脱リン酸化(不活性型)BTKを0.2、1、3μMとなるようアッセイバッファーで希釈して酵素溶液を調製した。ポリスチレン製384穴プレートの各ウェルに、酵素溶液と蛍光プローブ溶液を等量ずつ加え、室温で1時間インキュベートした。ブランクとして、酵素溶液の代わりにアッセイバッファーを添加したものを測定した。また、競合試験として、最終濃度5μMとなるように上記競合化合物溶液を加えて調整した反応溶液も測定した。各ウェルの蛍光強度をSynergyH1マイクロプレートリーダー(Biotek社製)で測定した(測定波長:励起485nm/蛍光550nm)。
【0099】
結果を図1に示す。図1に示されるように標的キナーゼである脱リン酸化(不活性型)BTKの存在により濃度依存的に蛍光プローブの蛍光強度の増加が認められる。また、競合化合物であるキナーゼ阻害剤CGI1746の添加により蛍光強度が低下することから、この系は結合部位特異的に起こり、また可逆的なことが理解される。
実施例17
脱リン酸化(不活性型)BTKを用いた蛍光プローブの結合活性測定
【0100】
蛍光プローブ(実施例9)の10mM DMSO溶液から、2mMを最高濃度として公比2でDMSO希釈した13濃度のDMSO溶液を、さらに実施例16記載のアッセイバッファーで1000倍希釈して、蛍光プローブ溶液を調整した(0.1%DMSO溶液)。同様にして競合化合物(CGI1746)の1mM DMSO溶液から実施例16記載のアッセイバッファーを用いて40μMの競合化合物溶液を調整した。また脱リン酸化(不活性型)BTKを120nMとなるようアッセイバッファーで希釈して酵素溶液を調製した。蛍光プローブ溶液40μL、コントロール溶液(4%DMSO−アッセイバッファー)20μL、および酵素溶液20μLをポリプロピレン製384穴プレートのウェル中で混合し、1時間室温でインキュベートした後、各ウェルの蛍光強度をSynergyH1マイクロプレートリーダーで測定した(測定波長:励起485nm/蛍光550nm)。またコントロール溶液の代わりに競合化合物溶液を加えたウェルの蛍光強度も測定し、これをバックグラウンドとして測定値から差し引き、蛍光強度変化量(ΔRFU)とした。各蛍光プローブ濃度における蛍光強度変化量(ΔRFU)を蛍光プローブ化合物濃度に対してプロットし、飽和曲線を作成し、GraphPad Prism(商標)ソフトウェア(GraphPad Software社製)を用いて、蛍光プローブの乖離定数(Kd)を算出した。
図2に測定結果を示す。脱リン酸化(不活性型)BTKに対する蛍光プローブ(実施例9)のKd値は63nMと測定された。
【0101】
この結果は、本発明の蛍光プローブが、不活性型BTKから非常に解離し易く、競合する不活性型キナーゼを標的とする候補化合物のスクリーニングに有用であることを示している。
実施例18
脱リン酸化(不活性型)BTKを用いた競合結合測定試験
【0102】
本発明の蛍光プローブ(実施例9)の1mM DMSO溶液を実施例16記載のアッセイバッファーで希釈し、100nMの蛍光プローブ溶液を調整した。また脱リン酸化(不活性型)BTKを100nMとなるようアッセイバッファーで希釈して酵素溶液を調製した。被験化合物(CGI1746)の10mM DMSO溶液から、20μMを最高濃度として公比2でDMSO希釈した8濃度のDMSO溶液を、さらに実施例16記載のアッセイバッファーで25倍希釈して、薬物溶液を調整した(4%DMSO溶液)。薬物溶液20μLと酵素溶液20μLをポリスチレン製384穴プレートのウェル中で混合し、15分後、蛍光プローブ溶液(40μL)を添加した。室温で2時間インキュベートした後、各ウェルの蛍光強度をSynergyH1マイクロプレートリーダーで測定した(測定波長:励起485nm/蛍光550nm)。蛍光強度とキナーゼ阻害剤濃度をプロットしIC50値を算出した。
【0103】
結果を図3に示す。脱リン酸化(不活性型)BTKと蛍光プローブ(実施例9)に対するCGI1746の競合阻害活性(IC50値)は22nMと測定された。
【0104】
つまり、本発明の蛍光プローブを用いて、既知の不活性型BTKを阻害する化合物(CGI1746)の阻害活性を決定できたことは、本発明の蛍光プローブが、不活性型キナーゼのスクリーニングに有効であることを示している。
【特許請求の範囲】
【請求項1】
下式(I)を有する新規なベンゾオキサジアゾール蛍光プローブ。
【化1】
(式中、Zは、キナーゼ阻害活性を有する化合物を表わし、Lは、結合、置換されてもよいアミノ基、カルボニル基、置換されてもよい炭化水素基、置換されてもよいアルキルカルバモイル基、置換されてもよいアシルアミノ基、スルフィド基、置換されてもよいアルコキシ基を表し、Xは、結合、置換されてもよいアミノ基、置換されてもよいヒドラジノ基、チオカルボニルアミノ基を表し、R1はニトロ基、ジメチルスルファモイル基を表す。)
【請求項2】
Zが、p38MAPK阻害活性を有する化合物である、請求項1に記載の蛍光プローブ。
【請求項3】
Zが、下式(Ia)を有する化合物である、請求項1または2に記載の蛍光プローブ。
【化2】
(式中、R2は置換されてもよいアルコキシ基を表す。)
【請求項4】
Zが、BTK阻害活性を有する化合物である、請求項1に記載の蛍光プローブ。
【請求項5】
Zが、下式(Ib)を有する化合物である、請求項1または4に記載の蛍光プローブ。
【化3】
【請求項6】
請求項1から5の何れか1項に記載の蛍光プローブを使用することを特徴とするキナーゼ阻害剤をスクリーニングする方法。
【請求項7】
不活性型キナーゼを標的とした、請求項6に記載のスクリーニング方法。
【請求項1】
下式(I)を有する新規なベンゾオキサジアゾール蛍光プローブ。
【化1】
(式中、Zは、キナーゼ阻害活性を有する化合物を表わし、Lは、結合、置換されてもよいアミノ基、カルボニル基、置換されてもよい炭化水素基、置換されてもよいアルキルカルバモイル基、置換されてもよいアシルアミノ基、スルフィド基、置換されてもよいアルコキシ基を表し、Xは、結合、置換されてもよいアミノ基、置換されてもよいヒドラジノ基、チオカルボニルアミノ基を表し、R1はニトロ基、ジメチルスルファモイル基を表す。)
【請求項2】
Zが、p38MAPK阻害活性を有する化合物である、請求項1に記載の蛍光プローブ。
【請求項3】
Zが、下式(Ia)を有する化合物である、請求項1または2に記載の蛍光プローブ。
【化2】
(式中、R2は置換されてもよいアルコキシ基を表す。)
【請求項4】
Zが、BTK阻害活性を有する化合物である、請求項1に記載の蛍光プローブ。
【請求項5】
Zが、下式(Ib)を有する化合物である、請求項1または4に記載の蛍光プローブ。
【化3】
【請求項6】
請求項1から5の何れか1項に記載の蛍光プローブを使用することを特徴とするキナーゼ阻害剤をスクリーニングする方法。
【請求項7】
不活性型キナーゼを標的とした、請求項6に記載のスクリーニング方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公開番号】特開2013−104687(P2013−104687A)
【公開日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願番号】特願2011−246695(P2011−246695)
【出願日】平成23年11月10日(2011.11.10)
【出願人】(503286424)カルナバイオサイエンス株式会社 (4)
【Fターム(参考)】
【公開日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願日】平成23年11月10日(2011.11.10)
【出願人】(503286424)カルナバイオサイエンス株式会社 (4)
【Fターム(参考)】
[ Back to top ]