
新規なC−17−ヘテロアリールステロイドCYP17阻害剤/抗アンドロゲン:合成、インビトロ生物活性、薬物動態および抗腫瘍活性
【課題】ヒトCYP17酵素の阻害剤であり、そして野生タイプおよび変異アンドロゲン受容体(AR)の両方のアンタゴニストであり、ヒト前立腺癌の処置に使用できる化合物の提供。
【解決手段】下記式(I)で表される3β−ヒドロキシ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン化合物。

【解決手段】下記式(I)で表される3β−ヒドロキシ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン化合物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新しい化学的実在物、特にステロイドC−17ベンゾアゾール、ピリミジノアゾール(アザベンゾアゾール)およびジアジンを提供する。本発明はまた、これらベンゾアゾール、ピリミジノアゾールおよびジアジンの合成方法を提供する。一具体例において、ベンゾアゾールまたはピリミジノアゾールの合成方法は、3β−アセトキシ−17−クロロ−16−ホルミルアンドロスタ−5,16−ジエンまたはその類縁体と、そしてベンゾアゾールまたはピリミジノアゾール求核試薬の求核性ビニル“付加−脱離”置換反応のステップを含んでいる。他の具体例において、ジアジンの合成方法は、17−ヨードアンドロスタ−5,6−ジエン−3β−オールまたはその類縁体のトリブチルスタニルジアジンとのパラジウム触媒交差カップリング反応を含んでいる。
【0002】
本発明の化合物は、ヒトCYP17酵素の強力な阻害剤であり、また野生タイプおよび変異株アンドロゲンレセプタ−(AR)の強力な拮抗剤である。最も強力なCYP17阻害剤は、3β−ヒドロキシ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン(5、コード名VN/124−1)、3β−ヒドロキシ−17−(51−ピリミジン)アンドロスタ−5,16−ジエン(15)、および17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−6,16−ジエン−3−オン(6)であり、それらのIC50値はそれぞれ300,500および915nMである。化合物5,6,14および15は、変異株およびLNCaPARおよび野生タイプARへの3H−R1881(メチルトリエノロン、安定な合成アンドロゲン)の結合防止に有効であったが、しかし後者へは2.2ないし5倍高い結合効率を有している。化合物5および6は,強力な純粋AR拮抗剤であることを示した。細胞成育研究は、化合物5および6は低いマイクロモル範囲(すなわち<10μM)のIC50値でDHT−刺激LNCaPおよびLAPC4前立腺癌細胞の成育を阻止する。それらの阻止強度はカソデックスに匹敵するが、しかしフルタミドより著しくすぐれている。マウスにおける化合物5および6の薬物動態が検討された。5および6の50mg/kgの腹腔内投与後、30ないし60分後それぞれ16.82および5.15ng/mlのピーク血漿レベルが出現し、両方の化合物は血漿から速やかに排出され(それぞれ終末半減期44.17および39.93分)、そして8時間目にどちらも検出されなかった。注目すべきことに、化合物5は一時的に17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−3−オンと同定された代謝物へ急速に変換された。インビボでテストする時、5はアンドロゲン依存性LAPC4ヒト前立腺癌キセノグラフトの成育の阻害に非常に有効であることが証明されたが、6は無効であった。化合物5(50mg/kg 1日2回)は、対照と比較して平均最終腫瘍体積の93.8%減少(P=0.00065)をもたらし、そして去勢よりも有意に効果的であった。我々の知る限り、これはアンドロゲン依存性前立腺腫瘍成育の抑制において去勢より有意にもっと効果的な抗ホルモン剤(アンドロゲン合成の阻害剤(CYP17阻害剤)/抗アンドロゲン)の最初の例である。これらの印象的抗癌性に鑑み、化合物5その他はヒト前立腺癌の処置に使用することができる。
【背景技術】
【0003】
前立腺癌(PCA)は最も普通の悪性腫瘍であり、そして全世界において年令関連癌死亡の原因である。肺癌は別にし、PCAは男性において最も普通の癌の形であり、そしてアメリカ人男性の第2の癌死因である。2004年においてアメリカ合衆国では、前立腺癌の推定230,000症例が診断され、そして約23,000人男性がこの病気で死亡するであろうといわれている(Jemal et al.Cancer Statics,2004.CA Cancer J.Clin.2004,54,8−29)。1992−1999の期間、アフリカ系男性アメリカ人の平均年間PCA症例はコーカサス系男性よりも59%高く、そして平均年間死亡率はコーカサス系男性の2倍以上であった(American Cancer Society−Cancer Facts and Figures 2003)。アンドロゲンはPCAの発生、成育および進行において重要な役割を果す(McConnell,J.D.,“Physiological basis of endocrine therapy for prostatic Cancer”,Urol.Clin.North Am.1991,18:1−13)。この点に関し2つの最も重要なアンドロゲンはテストステロン(T)およびジヒドテストステロン(DHT)である。睾丸はTの約90%を合成し、そして残りは副腎によって合成される。Tは主として前立腺に局在する酵素ステロイド5α−レダクターゼによってもっと強力なアンドロゲンDHTへ変換される(Bruggins et al.“The conversion of testosterone to 5α−androstan−17β−ol−3−one by rat prostate in vivo and in vitro”,J.Biol.Chem.,1968,243,2012−2021)。Huggins et al.は1941年に進行および転移PCAのための療法としてアンドロゲン奪取を導入した(Huggins et al.“Studies on prostatic cancer:2.The effect of prostate gland”,Arch.Surg.,1994,43,209−212)。その後アンドロゲン奪取療法はPCA患者の多数セッティングにおいて最も有益な応答を産むことが示された(Denmeade et al.“A history of prostate cancer treatment”,Nature Rev.Cancer,2002,2:389−396)。睾丸摘除術(外科的またはGnRHアゴニストでの医学的)は大部分の前立腺癌患者のための標準的処置オプションとして残っている。医学的および外科的睾丸摘除術は睾丸によるアンドロゲン生産を減らすかまたは排除するが、しかし副腎中のアンドロゲン合成には影響しない。睾丸摘除術と、副腎アンドロゲンの作用を阻害する抗アンドロゲン剤との併用療法はPCA患者の生存を有意に延長するといういくつかの研究が報告された(Crawford et al.“A controlled trial of lenprolide with and without flutamide in prostatic carcinoma”,N.Eng.J.Med.,1989,321,419−424;Crawford et al.“Tratment of newly diagnosed state D2 prostate cancer with lefnprolide and flutamide or leuprolide alone,phase III;intergroup study 0036”,J.Urol,1992,147;417A;Denis L.,“Role of maximal androgen blockade in advanced prostate cancer”,Prostate,1994,5(Suppl.),17s−22s)。Mohlerらによる最近の特色ある論文(Mohler et al,“The androgen axis in recurrent prostate cancer”,Clin.Cancer Res.2004,10,440−448)において、TおよびDHTはアンドロゲン受容体を活性化するのに十分なレベルにおいて再発性PCA組織内に出現することが明らかに証明された。加えて、同系PCAキセノグラフトモデルのマイクロアレイに基いたプロファイリングを使用して、Sawyerらは(Chen et al,“Molecular determinant of resistance to antiandrogen therapy”,Nat.Med.2004,10,33−39)、アンドロゲン受容体mRNAの適度の増加が抗アンドロゲン療法に対する抵抗性の発展に一貫して関連する唯一の変化であることを発見した。睾丸、副腎および他の組織中のアンドロゲン合成を阻害する強力なそして特異性化合物はPCAの処置のためにもっと効果的であり得る(Njar,V.C.O.;Brodie,A.M.H.,“Inhibitors of 17α−hydroxylase−C17,20−lyase(CYP17):Potent Agents for the treatment of prostate cancer,”Current Pharm.Design,1999,5:163−180)。
【0004】
睾丸および副腎において、Tの生合成における最後のステップは、逐次的に作用しそして両方とも単一の酵素チトクロームP450モノオキシゲナーゼ17α−ヒドロキシラーゼ/17,20−リアーゼ(CYP17)によって触媒される、二つのキー反応を含んでいる(Hall,D.F.,“Cytochrome P−450 C21SCC:one enzyme with two actions:Hydroxylase and lyase”,J.Steroid Biochem.Molec.Biol,1991,40,527−532)。抗カビ剤として、そしてP450酵素阻害のためにケトコナゲールも適度なCYP17阻害剤であり、そしてPCAの処置に臨床的に使用された(Trachtenberg et al,“Ketoconazole:A novel and rapid treatment for advanced prostatic cancer”,J.Urol.,1983,130,152−153)。処理の注意深いスケジュールはさもなければホルモン抵抗性癌患者において長期応答を発生させることができることが報告された(Muscato et al,“Optimal dosing of ketoconazole and hydrocortisone leads to long responses in hormone refractory prostate cancer”,Proc.Am.Assoc.Cancer Res.,1994,13:22(Abstract))。さらにケトコナゾールはフルタミド脱退にもかかわらず進行したPCA患者において活性を保有することが発見された(Small et al,“Ketoconazole retains activity in advanced prostatic cancer patients with progression despite flutamide withdrawal”,J.Urol.,1997,157,1204−1207)。今ではケトコナゾールは肝臓毒性および他の副作用のために使用が取り止められているけれども、このことはCYP17のもっと強力なそして選択的阻害剤はこの病気の処置において進んだ段階でもそしてホルモン抵抗性らしい一部の患者においてさえも有用な剤を提供し得ることを示唆する。
【0005】
CYP17の種々の強力なステロイドおよび非ステロイド阻害剤が報告されており、そしてあるものはゲッ歯類モデルにおいてテストステロイド生産の強力な阻害剤であることが示された(上出、Njar and Brodie)。最近Jarmanらは彼らの最も強力なCYP17阻害剤アビラテロンの前立腺癌患者におけるホルモンインパクトを記載した(O’Donnell et al,“Hormonal Impact of the 17α−Hydroxylase/C17,20−lyase inhibitors abiraterone acetate(CB7630)in patients with prostate cancer”,Br.J.Cancer,2004,90:2317−2325)。我々の強力なCYP17阻害剤のいくつかも5α−レダクターゼを阻害し、および/または強力な抗腫瘍活性を有する強力な抗アンドロゲン剤であることを示した。(上出Njar and Brodie,and Long et al,“Antiadrogenic effects of novel androgen synthesis inhibitors on hormone−dependent prostate cencer”,Cancer Res,2000,60,6630−6640)。本発明の背景のさらなる例証は、米国特許Nos.5,994,335;6,200,965;および6,444,683である。
【発明の概要】
【課題を解決するための手段】
【0006】
我々は一連の強力なCYP17阻害剤、17−ベンゾアゾール、17−ピリミジノアゾールおよび17−ジアジンを発見した(例えば以下に記載するように他の構造にも類似して適用することができる化合物の製造例のためのスキーム1および2を見よ)。これらC−17ヘテロアリールステロイドの製造のための刺激は、ベンズイミダゾール、ベンゾトリアゾール、ピリミジノアゾール、およびジアジン構造、いわゆる恩恵享受構造(privileged structure)を新しい分子へ導入する我々の願望に基いている(Nicolaou et al,“Natural product−like combinational libraries based on privileged structures.1.General principles and solid−phase synthesis of benzopyrans”,J.Am.Chem.Soc.2000,122,9939−9953)“Privileged strncture”なる術語は、多種類の無関係分子標的と相互作用することができる構造モチーフを記載するためにEvansらにより初めて導入された(J.Med,Chem.Soc.2000,122,9939−9953)。これらの枠組、特にベンズイミダゾール枠組は、それらの多岐にわたる生物学的活性のポートフオリオのため、および多種類の有用な薬物の部分構造として医薬化学において広く注目されている(Nicolaou et al.前出)。
【0007】
本発明のC−17ヘテロアリールステロイド化合物は以下の一般式Iを有する。
【0008】
【化1】
【0009】
式中、
ABC環構造は、任意に置換されてもよいステロイドまたはその類縁体のA,BおよびC環部分であり;
16,17位間の結合(実線と点線)は二重結合か、または化合物が17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−3−オンである時には単結合であり;そして
Xは任意に置換されてもよいベンズイミダゾール、ベンゾトリアゾール、ピリミジノイミダゾール(プリン)、ピリミジノトリアゾールまたはジアジンであり、ベンズイミダゾール、ベンゾトリアゾールおよびピリミドイミダゾール基は5員環の窒素原子を介してステロイド残基へ結合しており、そしてジアジン基はジアジン環の炭素原子を介してステロイド残基へ結合している。
【0010】
これらの化合物の薬学的に許容し得る塩も本発明に含まれる。
【0011】
ABC環構造のための任意の置換基は、一以上のアルキルおよびハロゲン化アルキル(好ましくはC1−6);環構造に直接結合した二重結合を含むアルケニルおよびハロゲン化アルケニル(好ましくはC1−6);ハロゲン;アミノ;アミノアルキレン;ヒドロキシイミノ;およびヒドロキシを含む。さらに任意に、ABC環構造の隣接する水素を除去し、隣接する炭素原子間を追加の結合によって置換し、環構造中のこれら炭素間に二重結合を形成してもよい。ABC環構造上の好ましい任意の置換基は環構造の10位および/または13位のメチル基である。
【0012】
ベンズイミダゾール、ベンゾトリアゾール、ピリミジノイミダゾール、ピリミジノトリアゾール、またはジアジン構造のための任意の置換基は、ハロゲン、アミノ、アミノアルキレン、ヒドロキシ、−SH,−S−アルキル、アルキルおよびハロゲン化アルキル(好ましくはC1−6)を含む。これらの任意の置換基はベンズイミダゾール、ベンゾトリアゾール、ピリミジノイミダゾール、ピリミジノトリアゾールまたはジアジン構造の環炭素原子上にあるであろう。
【0013】
ベンズイミダゾール、ベンゾトリアゾール、ピリミジノイミダゾール、ピリミジノトリアゾールおよびジアジン構造はそれぞれ以下の式を有する。
【0014】
【化2】
【0015】
式中の*はステロイド残基への結合を示す。
【0016】
好ましい一具体例においては、ABC環構造は、C−17ヘテロアリール置換基への結合に隣接するD環と共有する炭素、すなわち13位においてアルキル、特にメチル置換を除いて無置換のC環を有する。
【0017】
好ましい他の具体例においては、ABC環構造のA,BおよびC環は3β−ヒドロキシアンドロスタ−5,16−ジエンまたは3−オキソアンドロスタ−5,16−ジエンに基づく慣用構造を有する。しかし他の具体例においては、AおよびB環は以下の構造1−25の一つを有する。
【0018】
【化3】
【0019】
以下に1−25のAB環を有し、そしてXがベンズイミダゾールであるCおよびD環が慣用である化合物の化学名をリストする。
【0020】
化合物1:3β−ヒドロキシ−3α−メチル−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物2:3β−フルオロ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物3:3β−クロロ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物4:3β−ブロモ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物5:3β−ヨード−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物6:3β−アミノ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物7:17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−3,5,16−トリエン;
化合物8:17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−2,4,16−トリエン;
化合物9:17−(1H−ベンズイミダゾール−1−イル)−3−メチレンアンドロスタ−5,16−トリエン;
化合物10:17−(1H−ベンズイミダゾール−1−イル)−3−メチレンアンドロスタ−4,16−トリエン;
化合物11:3,3−ジフルオロ−17−(1H−イミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物12:3,3−ジフルオロ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−4,16−ジエン;
化合物13:17−(1H−ベンズイミダゾール−1−イル)−3−メチレンアンドロスタ−2,4,16−トリエン;
化合物14:17−(1H−ベンズイミダゾール−1−イル)−3−メチレンアンドロスタ−2,4,5,16−テトラエン;
化合物15:3,3−ジフルオロ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−2,4,16−トリエン;
化合物16:3,3−ジフルオロ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−2,4,6,16−テトラエン;
化合物17:3−ヒドロキシイミノ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物18:3−ヒドロキシイミノ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−4,16−ジエン;
化合物19:3−ヒドロキシイミノ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−2,4,16−トリエン;
化合物20:3−ヒドロキシイミノ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−2,4,6,16−テトラエン;
化合物21:3−ヒドロキシ−17−(1H−ベンズイミダゾール−1−イル)エストラ−1,3,5(10),16−テトラエン;
化合物22:3−フルオロ−17−(1H−ベンズイミダゾール−1−イル)エストラ−1,3,5(10),16−テトラエン;
化合物23:3−クロロ−17−(1H−ベンズイミダゾール−1−イル)エストラ−1,3,5(10),16−テトラエン;
化合物24:3−ブロモ−17−(1H−ベンズイミダゾール−1−イル)エストラ−1,3,5(10),16−トリエン;
化合物25:3−ヨード−17−(1H−ベンズイミダゾール−1−イル)エストラ−1,3,5(10),16−テトラエン
【0021】
ヘテロアリール環のための任意の置換基Xの例は以下の構造26−40によって示され、ここでXはベンズイミダゾールである。Xが置換されたベンズイミダゾール、ピリミジノイミダゾール、ピリミジノトリアゾール、ピラジンまたはピリミジンである類似化合物も企図される。
【0022】
【化4】
【0023】
ヘテロアリール環のための任意の置換基Xの他の例は以下の構造41−46によって示され、ここでXは置換されたC−17−アザベンズイミダゾール(すなわちピリミジノイミダゾールまたはプリン)である。Xが置換されたベンズイミダゾール、ベンゾトリアゾール、ピリミジノトリアゾール、ピラジンまたはピリミジンである類似化合物も企図される。
【0024】
【化5】
【0025】
特に好ましい化合物は以下の構造M5,M6,M9およびM10の化合物である。
【0026】
【化6】
【0027】
CYP17およびステロイド5α−レダクターゼと比較したこれら化合物の阻害活性、アンドロゲン受容体への結合およびトランス活性化、および二つのヒト前立腺癌細胞株LNCaPおよびLAPC−4に対する抗増殖効果が研究された。化合物5および6の薬物動態がマウスで評価され、そしてヒトLAPC−4前立腺癌に対するインビボ抗腫瘍活性もマウスで評価された。我々の知る限り、化合物15を除いてここに記載したすべての化合物は新規化合物を代表する(Haidar et al,“Novel steroidal pyrimidyl inhibitors of P450 17(17α−hydroxylase/C17−20−lyase)”,Arch.Pharm.Med.Chem.2001,334,373−374;and Haidar et al,“Effects of novel 17α−hydroxylase/C17−20−ylase(P450 17,CYP17)inhibitors on androgen synthesis in vivo”,J.Steroid Biochem.Molec.Biol.,2003,84,555−562)。
【0028】
17−ベンゾアゾールおよび17−ジアジンの製造はそれぞれスキーム1および2に要約されている。これらの方法はここに記載した他の類縁体へも同様に適用することができる。
【0029】
我々の17−ベンゾアゾール合成のキー中間体である、3β−アセトキシ−17−クロロ−16−ホルミルアンドロスタ−5,16−ジエン(2)は、(1)から以前記載された我々の日常的操作によって得られた(Njar et al,“Nucleophilic Vinylic“addition−elimination”substitntion reaction of 3β−acetoxy−17−chloro−16−formylandrosta−5,16−diene:A novel and general route to 17−substituted−△16−steroids.Part I.Synthesis of novel 17−azolyl−△16−steroids;inhibitors of 17a−hydoxylase/17,20−lyase(P45017a)”,Bioorg.Med.Chem.Lett.1996,6,2777−2782;and“Novel 17−azolyl steroids;potent inhibitors of cytochrome P450 17a−hydroxylase/17,20−lyase(P45017a):Potential agents for the treatment of prostate cancer”,J.Med.Chem.1998,41,902−912)。DMF中K2CO3の存在下2のベンズイミダゾールでの約80℃における処理は殆ど定量的収率で3β−アセトキシ−17−1H−ベンズイミダゾール3を与えた。化合物3は還流ベンゾニトリル中で活性炭上の10%パラジウムでスムースに脱ホルミル化され、93%で化合物4を与えた。この化合物の加水分解は必要な3β−ヒドロキシ−17−ベンズイミダゾール5を与えた。5のOppenauer酸化変法は対応する△4−3−オキソ類縁体6を与えた。
【0030】
DMF中約80℃におけるK2CO3存在下のベンゾトリアゾールと2との反応は、すぐれた収率で所望の3β−アセトキシ−17−ベンゾ−1H−1,2,3−トリアゾール7bと、そして約5%収率で2H−1,2,3−トリアゾール位置異性体を与えた。これの2位置異性体はシリカゲル上のフラッシュカラムクロマトグラフィー(FCC)によって容易に分離され、そしてそれらのそれぞれのプロトンNMRスペクトルによって容易に同定された。このように対称2H−1,2,3−トリアゾール7aの4個の芳香族プロトンはδ7.43,7.45,7.88および7.90のダブレットの2対として現れ、他方非対称1H−1,2,3−トリアゾール7bの4個の芳香族プロトンは、δ7.46(2H)のマルチプレットおよびδ7.57(1H)と8.15(1H)におけるダブレットとして現れた。加えて、7aの16−CHOプロトンは、7bのδ9.59に比較してδ10.66へ著しくシフトした。還流キシレン中のRh(1,3−ビス(ジフェニルホスフィ))プロパン)2+Cl−触媒〔Rh(dppp)2+Cl−〕のその場の生成による脱ホルミル化は化合物8を与え、3β−アセトキシ基の加水分解の後、我々は標的とする3β−ヒドロキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−5,16−ジエン(9)を90%収率で得た。9の酸化は好収率で10を与えた。
【0031】
17−ジアジン(17−ジアジン14および17−ピリミジン15)の合成は容易に入手し得るデヒドロエピアンドロステロン(11,スキーム2)から始め、この化合物を以前Potter et alらが記載したようにヒドラジンヒドラートとヒドラジンサルフェートでの処理により対応する17−ヒドラゾンへ変換した(Potter et al,“A convenient,large−scale synthesis of abiraterone acetate〔3β−acetoxy−17(3−pyridyl)androsta−5,16−diene〕,a potent new drug for the treatment of prostate cancer,Org.Prep.Proc.Int.1997,29,123−128)。1,1,3,3−テトラメチルグアニジンの存在下ヨウ素での12の処理はすぐれた収率でビニル17−ヨウダイド12を与えた。(2−トリブチルスタニル)ピラジンまたは(5−トリブチルスタニル)ピリミジンでの13のパラジウム触媒交差カップリング反応(Choshi et al,“Total Synthesis of Grossularines−1 and−2,”J.Org.Chem.1995,60,5899−5904)は、それぞれ3β−ヒドロキシ−17−(2−ピラジル)アンドロスタ−5,16−ジエン(14,15%)と、3β−ヒドロキシ−17−(5−ピリミジル)アンドロスタ−5,16−ジエン(15,10%)を与えるように進行した。これら二つの交差カップリング反応の低収率は採用した反応条件下でのスタニルジアジン試薬の不安定性のためであり得る。標的化合物14および15の構造はそれらのプロトンNMRスペクトルに容易に同定された。14中の17−ピラジン部分の3個の非均等プロトンはδ8.35,8.48および8.70における3シングレットとして現われ、15中の17−ピリミジン部分の3個のプロトンは2個の均等プロトンがδ8.73においてシングレットとして現われ、1個のプロトンはδ9.07に現われた。さらに14および15の17−ジアジン基は、前駆体△16−17−イオダイド13の16−プロトンに関してそれらそれぞれの16−オレフィン性プロトンの化学的シフトに異なる影響を発揮する。14中の16−Hはδ6.77においてシングレットとして現われ、13中の16−H(δ6.14)に比較して著しく脱シールドされ、15中の16−Hは13と同様にδ6.11に現われた。上で示したように化合物15はHaidarらによって以前に報告されたが、これはそこで記載された操作とは異なる操作で合成された。
【0032】
新規化合物の代表的サンプルが次に以後のセクションに詳細に記載した広範なインビトロおよびインビボ研究にかけられた。
【0033】
本発明は、それを必要とする対象へ本発明に従った化合物の有効量を投与することを含む、前立腺癌または前立腺肥大を処理する方法にも関する。用語“処置”は慣用的に、例えば前立腺病に関連する一以上の症状を撲滅、緩和、減少、寛解、改善等の目的のため対象の管理またはケアの意味で使用される。処置することができる前立腺病の例は、例えば前立腺肥大(BPH)および前立腺癌(例えば前立腺悪性腫瘍)を含む。
【0034】
投与の特定の投与量および頻度は、特定の活性化合物の活性度、その代謝安定性および作用の長さ、排泄速度、投与モードおよび時間、対象の年令、体重、健康状態、性別、食事等、および前立腺癌または肥大の重篤度を含む種々のファクターに依存して変動し得る。化合物のどのような有効量、例えば毎日約1mgないし500mg、もっと特定すれば毎日約50mgないし150mgを投与することができる。化合物は、例えば経口、非経口、腹腔内、外用、経皮(例えば標準的パッチを使用して)、眼内、経鼻、局所、エアロゾル、スプレー、吸入のような非経口、皮下、静脈内、筋肉内、バッカル、舌下、経直腸、動脈内および硬膜下腔内を含む、どのようなルートによるどのような形によっても投与することができる。本発明の化合物は単独で、または例えば適当な薬剤組成物を製造するための生理学的に許容し得る担体のような活性または不活性成分との組合せで投与することができる。
【0035】
ここで引用したすべての出願、特許および発表、および2005年3月2日に出願した米国仮出願No.60/657,390の全体の開示を参照としてここに取入れる。
【0036】
さらに考究することなく、以上の説明を利用して当業者は本発明をその全範囲において利用できるものと信じられる。それ故以下の好ましい特定の具体例は単に例証と解すべきであり、開示の残りの限定ではない。
【0037】
以上および以下の実施例において、すべての温度は未補正の摂氏で表され、そしてすべての部およびパーセントは特記しない限り重量による。
【図面の簡単な説明】
【0038】
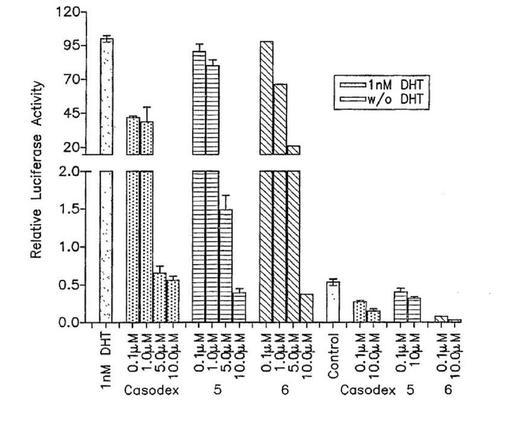
【図1】LNCaP−ARR2−1u前立腺癌細胞中のLNCaP−ARを介して仲介されるルシフェラーゼの転写活性に対する5,6およびカソデックスの効果。ステロイド不含倍地中の細胞はビヒクルか、または1nM DHTありまたはなしの5かまたはカソデックスの増大する濃度で18時間処理された。次に細胞は材料および方法に記載されたようにルシフェラーゼ活性についてアッセイされた。棒は、別々の3実験からの3系列ウエル中の平均光ユニット〔秒あたりのカウント(CPS)/単位タンパク、すなわち相対的ルシフェラーゼ活性〕によって表される。
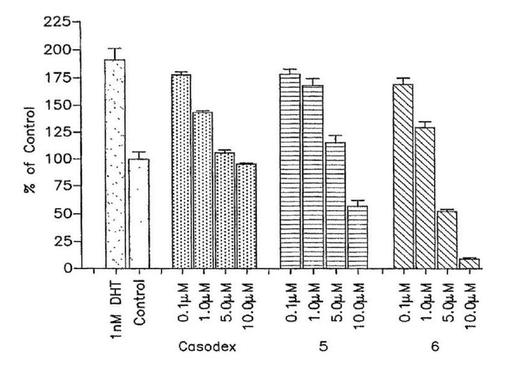
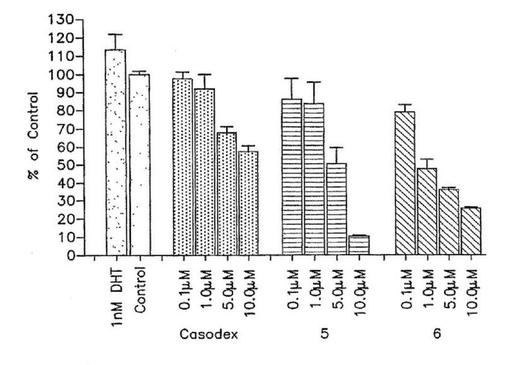
【図2a】(a)LNCaPおよび(b)LAPC4前立腺癌細胞発育に対する5,6およびカソデックスの効果。細胞はプレート前ステロイド不含倍地中で生育された。次に材料および方法に記載されたように3系列ウエルが5,6またはカソデックスおよびDHTの増大する濃度で同時処理された。処理7日後の発育阻害のパーセント(対照と比較して)がWST−1アッセイを使用して決定された。結果は3系列で実施された3実験の平均値および標準偏差を表す。
【図2b】(a)LNCaPおよび(b)LAPC4前立腺癌細胞発育に対する5,6およびカソデックスの効果。細胞はプレート前ステロイド不含倍地中で生育された。次に材料および方法に記載されたように3系列ウエルが5,6またはカソデックスおよびDHTの増大する濃度で同時処理された。処理7日後の発育阻害のパーセント(対照と比較して)がWST−1アッセイを使用して決定された。結果は3系列で実施された3実験の平均値および標準偏差を表す。
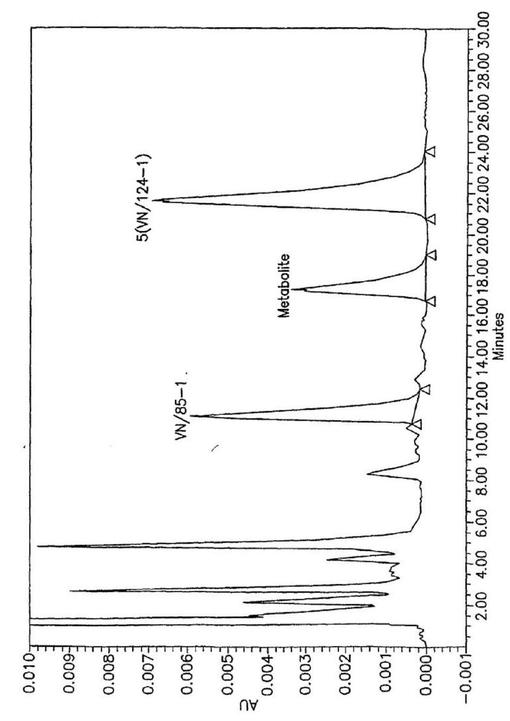
【図3】5,6(内部標準)およびマウス血漿から抽出した代謝産物の典型的HPLCクロマトグラフィーチャート。16,代謝産物および5の保留時間はそれぞれ11.5,17.3および21.6分であった。
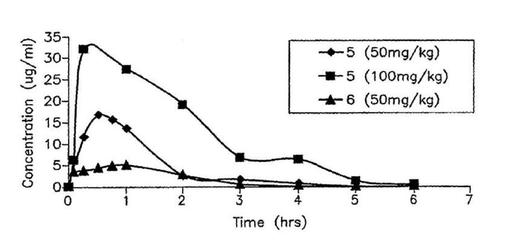
【図4】雄SCIDマウスへの1回の皮下ボーラス投与量の投与後の5および6の薬物動態プロフィル。各データポイントはマウス3匹から得た平均血漿濃度を表す。標準偏差(示さず)は、平均値の±5〜8%であった。
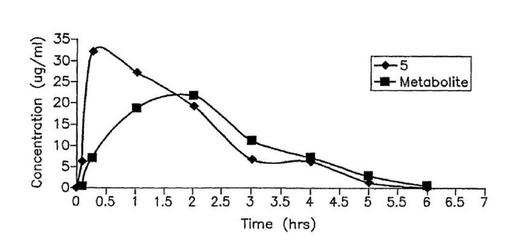
【図5】雄マウスへの1回の皮下ボーラス投与(100mg/kg体重)後の5および代謝産物の薬物動態プロフィル。
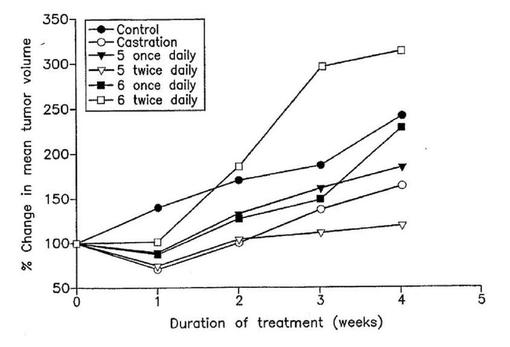
【図6】雄SCIDマウスにおけるLAPC4前立腺腫瘍の発育に対する5,6および睾丸除去術のインビボ抗腫瘍活性。LAPC4腫瘍を有するマウス5匹のグループが5(0.15mmol/kg/dayまたは0.30mmol/kg/day)で処置された。処置28日後腫瘍体積が測定された。腫瘍体積の標準偏差(示さず)は平均値の±10〜12%であった。
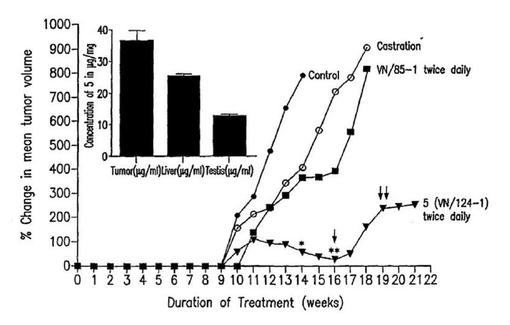
【図7】雄SCIDマウスにおけるLAPC4前立腺腫瘍の形成および発育に対する5,16および睾丸除去術の効果。3×107個のLAPC4細胞がCIDマウスの背面脇に皮下注入された。マウスの1グループは去勢された。マウスの他のグループはビヒクルか、または5(0.15mmol/kg 1日2回)または16(0.15mmol/kg 1日2回)を投与された。5または16による毎日の処置は細胞接種1日後に開始された。毎週腫瘍体積が測定され、腫瘍体積の変化のパーセントが処置16週後に決定された。*は、14週において対照、去勢および16に対する5の有意差(それぞれP=0.00065,0.05および0.0097)を示す。**は、16週において去勢および16に対する5の有意差(それぞれP=0.047および0.0047)を示す。↓および↓↓:5の減らした投与量期間
【発明を実施するための形態】
【0039】
(実施例)
生物学的研究
CYP17阻害研究:CYP17阻害アッセイは我々の以前報告した操作に従って実施される。そこでは酵素源としてインタクトなチトクロームP450c17−発現E.coliが使用される(Grigoryev et al,“Cytochrome P450c17−expressing Escherichia coli as a first−step screening system for 17α−hydroxylase−C17,20−lyase inhibitors”,Anal.Biochem.1999,267,319−330;and“Effects of new 17α−hydroxylase/C17,20−lyase inhibitors on LNCaP prostate cancer cell growth in vitro and invivo”,Br.J.Cancer,1999,81,622−630)。化合物のIC50値は投与量応答曲線から決定され、そして表1にリストされている。ケトコナゾール、アビラテロン(臨床試験中のCYP17阻害剤(前出O’Donnell)チャート1)、および3β−ヒドロキシ−17−(1H−イミダゾール−1−イル)アンドロスタ−5,16−ジエン(VN/85−1,化合物16,チャート1,最も強力なCYP17阻害剤だと信じられている(Njar et al,Current Pharm.Design,1999,5,163−180))のIC50値も比較のため同じアッセイシステムにおいて決定される。新しい17−複素環化合物のいくつかはIC50値300−915nMでCYP17の強力な阻害を示す。ベンゾイミダゾール5および6は、ベンゾトリアゾール9および10よりも4ないし6倍強力である。この結果は17−複素環の電子的性格が阻害活性に影響することを示唆する。さらに△5−3β−オール官能を有する化合物5および9は、△4−3−オン官能を有する対応する類縁体6および10よりもそれぞれ少なくとも3倍強力である。これらの結果は簡単な17−アゾールCYP17阻害剤についての我々の以前の結果と対照的である。それらの阻害剤シリーズにおいては、△5−3β−オールアゾールと対応する△4−3−オンアゾールの間には阻害強度に目立った差はない(Njar et al,J.Med.Chem.1998,41,902−912,前出)。可能性ある説明は、よりバルキーなベンゾアゾールは酵素の活性部位に異なって結合し、3位における部分の相互作用が結合に対して重要になって来ることである。一部のP450チトクロームのヘム成分に対する基質または阻害リガンドの結合はUV−vis差分光分析(Jefcoat C.R.,“Measurement of substrate and inhibitor binding to microsomal cytochrome P450 by optical difference spetroscopy”,Methods Enzymol.1978,52,258−279)を用いて検討される。このアプローチは我々が以前発表した標準的操作に従って拡張される(Njar et al,Bioorg.Med.Chem.Lett,1996,6,2777−2782;and J.Med.Chem,1998,41,902−912)。化合物5および9は各自タイプII差スペクトルを誘発し、低スピン鉄の生成を伴なってステロイドの窒素(ベンゾイミダゾールまたはベンゾトリアゾールのN−3)のCYP17のヘムへの配置を指示する。5および9(426nM)との酵素コンプレックスのソーレー最大のためのピーク位置はCYPシステムに対する窒素リガンドの結合についての利用できるデータと一致し、そして我々の他の17−アゾリルCYP17阻害剤でのデータとも一致する(Njar et al,Bioorg.Med.Chem.Lett.1996,6,2776−2782;and J.Med.Chem.1998,41,902−912)。ベンゾアゾール窒素とCYP17のヘム鉄との相互作用は、5および9の結合親和力が17−イミダゾール基を有するより小さい立体を要求する16と同じであるため17位におけるバルクトラランスを示唆する。
【0040】
テストした2つの17−ジアジンのうち、500nMのIC50値を持つ17−ピリミジン15は17−ピラジン(IC50=3810nM)よりも約8倍強力である。ベンゾアゾール場合のように、この結果は17−複素環の電子的性格が阻害活性に影響することを示唆する。最後に、ケトコナゾールおよびアビラテロンについて同じアッセイにおけるIC50値が評価される(表1)。このシリーズにおいて最も強力である17−ベンズイミダゾール5は、これら化合物よりもCYP17阻害においてそれぞれ約4倍および約5倍の改善を示すが、しかし16より強力ではない。
【0041】
インビトロにおけるヒト5α−レダクターゼイソエンザイムタイプ1および2の阻害:ある種のCYP13阻害剤はヒト5α−レダクターゼ酵素を阻害することができるとの以前の発見を基にして、我々はCYP17阻害剤の新しいシリーズを簡略に評価した。化合物5,6,9,10および参照としてのフィナステライドの阻害活性は、Hartman et al,“Synthesis and evaluation of 2’−substituted 4−(4’−carboxy−or 4’−carboxy methyl benzylidene)−N−acylpiperidines:Highly potent and in vivo active steroid 5α−reductase type 2 inhibitors”,J.Med.Chem.2002,45,3406−3417に記載されているように、DU−145細胞株(ヒトタイプ1酵素)およびヒトBPH組織のホモジネート(ヒトタイプ2酵素)を使用して決定される。いくつかの化合物についてIC50値または濃度10μMにおける阻害パーセント値が表1に提供されている。化合物6のみがタイプ1および2酵素の両方に強力な阻害を示すが(それぞれIC50値=770および480nM)、それはフィナステライド(それぞれIC50値=60および2nM)よりも数倍弱い。
【0042】
LNCaPおよびPC−3Aアンドロゲン受容体結合アッセイ:以前我々は我々のCYP17阻害剤のいくつかは変異および野生タイプARに対し強力な抗アンドロゲンであることを証明したので(Long et al.and Njar et al,J.Med Chem.1998,41,902−912前出)、CYP17阻害剤のこのシリーズがこれら受容体へ結合する能力を評価することに関心があった。AR競合は、変異ARを発現するアンドロゲン感受性LNCaP細胞、および野生タイプAR(PC−3ARと命名された)で安定して形質転換されたアンドロゲン非依存性PC−3細胞中の標識したR1881(〔3H〕−R1881)を用いて決定される。化合物5,6,14および15はナノモル濃度範囲において投与量応答態様でARの両方のタイプへの結合に対して標識したR1881と効果的に競合する(数値示さず)。それぞれIC50値384,242,336および374nMを持つ化合物5,6,14および15(表1)対野生タイプARは、臨床的に使用される抗アンドロゲンであるフルタミド(IC50=10,985nM)よりも29倍ないし45倍強力である。表1に示すように、5および6の変異ARに対する結合親和力は現在使用されている抗アンドロゲンであるカソデックスに匹敵するが、しかし再びフルタミドよりすぐれている。しかしながらフルタミドの生物学的活性はもっと強力なAR拮抗剤である代謝物ヒドロキシフルタミドから主として誘導される。
【0043】
LNCaP変異AR仲介転写に対する剤の効果:次に我々は化合物5および6はARアンタゴニストかまたアゴニストとして作用しているのかを質問した。アンドロゲン−調節転写活性化について研究は、プロバシンルシフェラーゼレポーター構築物AARZ−Luc(ルシラーゼ活性アッセイ)で過渡的に形質転換したLNCaP細胞中で実施される(Kim et al,“Synergism of cytoplasmic Rinase in IL6−induced ligand−independent activation of androgen receptor in prostate cancer cells”,Oncogene,2004,23:1838−1844;and Zhang et al,“A Small composite probasin promoter confers high levels of prostate−specifec gene expression through regulation by androgens and glucocorticoids in Vitro and in Vivo”,Endocrinology,2000,141:4698−4710)。化合物5,6またはカソデックスは各自0.1および10.0μMにおいてルシフェラーゼ活性に対して効果を持たないが、ルシフェラーゼ発現は18時間の1.0nM DHTでの処理後に約99.6倍増大される(図1)。さらに1.0nM DHTへの曝露によって誘発されたルシフェラーゼ発現は5,6およびカソデックスにより濃度依存態様でそして同様な態様で減少する(図1)。合わせてこれらの結果は、カソデックスと同様に化合物5および6はARアゴニストまたは部分的アゴニスト活性を持たず、そして強力な純粋なアンドロゲンアンタゴニストと考えることができることを示唆する。野生タイプARを発現するPC−3AR/LV細胞で化合物をテストしなかったが、これらも同じ態様に挙動するらしい。我々は以前我々のCYP17阻害剤のいくつかはフルタミドよりもカソデックスにもっと匹敵したことを示し(Long et al,前出)、そしてこのことはこれら新しい化合物でもそのとおりであるらしい。一般に、我々の新規化合物は両方のARタイプと強力に相互作用し、化合物は野生タイプまたは変異ARを発現する腫瘍を有する患者、または増幅したAR発現を有する患者の処置のために有用であり得ることを指示する。
【0044】
インビトロにおいてベンゾアゾールのLNCaPおよびLAPC−4前立腺癌細胞生育に対する効果:1nM DHTによって刺激された変異LNCaP細胞の増殖を阻害する化合物5および6の能力が調べられる。DHTのこの濃度は、ビヒクル処理細胞に比較して約2倍LNCaP細胞増殖を刺激した(図2A)。図2Aに示すように、化合物5および6は、各自それぞれIC50値6.0および1.8μMで投与量応答様式でDHT−誘発LNCaP細胞増殖を阻止する。カソデックスが陽性対照として使用され、そしてDHT−誘発LNCaP細胞増殖の同様な阻害を示す(図2A,IC50=8.6μM)。10nM DHTでのアンドロゲン感受性LAPC4前立腺癌細胞株の処理は、驚くべきことに細胞増殖を有意に誘発しない(図2B)。他の研究者も、アンドロゲンに対するLAPC4細胞の応答はLNCaP細胞において観察される程顕著でないことを報告している(Thompson et al,“Androgen antagonist activity by the antioxidant moiety of vitamin E,2,2,5,7,8−pentamethyl −6−chromanol in human prostate cancer cells”,Molec.Cancer Thera.2003,2,797−803)。しかしながら、化合物5,6およびカソデックスは各自LNCaPと同様にこの細胞株の投与量応答阻害を示す(図2B)。LAPC4細胞増殖の阻害強度の順序は6>5>カソデックスであり、IC50値はそれぞれ1.0,3.2および10μMである。合わせてこれらの結果は、5および6は上に記載したそれらのアンドロゲン受容体結合および活性化性質に相関して、細胞増殖の刺激においてDHTの作用をブロックするように作用していることを示唆する。化合物5および6は今日まで記載された最も強力な抗アンドロゲンである。
【0045】
5および6の薬物動態学および5の代謝:
化合物5および6についての雄性SCIDマウス中の薬物動態学的性質が他の阻害剤についての最近記載された操作に従って研究される(Nnane et al,“Pharmacokinetic profile of 3β−hydroxy −17−(1H−1,2,3−triazol−1−yl)androsta−5,16−diene(VN/87−1),a potent androgen synthesis inhibitor in mice”,J.Steroid Biochem.Molec.Biol.2001,71,145−152;and Handratta et al,“Potent CYP17 inhibitors:improved syntheses,pharmacokinetics and anti−tumor activity in the LNCaP human prostate cancer model”,J.Steroid Biochem.Molec.Biol.2004,92,155−165)。結果は表2および図3−5に要約されている。逆相HPLCにおいて5(保留時間=21.6min)は内部標準(16,rt=11.5min)、代謝産物(rt=17.3min)およびマウス血漿中の他の内因性化合物から良く解像される(図3)。5に対する較正曲線は直線で、そしてその検出限界は100ng/mlである。HPLCアッセイがバリデートされ、そしてマウス血漿中の5のモニターのために使用される。
【0046】
皮下投与後、5の血漿濃度は平均半減期約44.17分と、排出速度定数56.5min−1をもって指数的に低下する。化合物5は全身循環から1986.14ml/h/kgの速度で排除され、そして投与8時間後検出されなかった。5の皮下投与後の血漿濃度に基づく計算した非コンパートメント薬物動態学的パラメータが表2に示されている。雄性SCIDマウスへの5の皮下投与(50および100mg/kg)後の血漿濃度一時間曲線が図4に示されている。5の皮下投与後、マウス中の観察された血漿濃度は投与30.0分でピークレベルに達する。化合物5は皮下部位から良く吸収され、そして皮下投与後の血漿濃度対時間プロフィルの曲線下の面積AUCは、投与量を50から100mg/kgへ変えた時投与量に比例して増加する。さらに、排除半減期および平均滞留時間は、5の投与量を50から100mg/kgへ変える時比較的コンスタントである(表1)。これらの結果は5の薬物動態プロフィルは投与量依存性であることを指示する。
【0047】
図5は、有意義な量の極性代謝産物(保留時間17.3min、図3を見よ)が5から生成し、そしてインビボ薬物動態学的研究の間血漿中に存在することを示す。この代謝産物の最高量は投与約2時間後に達した67.72%である。この代謝産物は化合物6と同じ保留時間を示す。この代謝産物は一時的にLC−MSと同定され、その分子質量(m/z391=M+H+)は3−オキソ−△5,16−テトラヒドロ化合物5(すなわち、17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−3−オン)の構造と一致する。この代謝産物は、5から3β−OH→3−オキソの酸化、次いで△5および△16二重結合の還元(レダクターゼ)を経て生成し得る。類似の代謝産物は、雄マウス中の密接に関連するステロイド17−イミダゾールにおいて(3β−OH→3−オキソ、次いで△5二重結合の異性体化の結果として生成した)以前に同定された(Handratta et al,前出)。
【0048】
5,すなわち17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−3−オンの主要な代謝産物はトランス−アンドロステロンから合成することができる。スキーム3を見よ。これら類似の活性を以っていることが予期される。
【0049】
マウス中の6のインビボ薬物動態は、比較的低いCmaxおよび著しく高い排出速度(図4および表2)のため、化合物5と似ていない。加えて、我々は、化合物5についての我々の観察とは対照的に、血漿中に化合物6の代謝産物を検出しなかった。
【0050】
SCIDマウスに生育させたLAPC4キセノグラフトに対する5および6の効果:印象的な多数のインビトロ生物学活性、すなわちCYP17の強力な阻害、強い抗増殖前立腺癌細胞活性および抗アンドロゲン活性を基にして、5および6がアンドロゲン依存性LAPC4ヒト前立腺癌キセノグラフトモデルにおいてインビボ抗腫瘍有効性研究のために選択される。
【0051】
第1の実験において、SCIDマウスのよく確立されたLAPC4前立腺癌腫瘍の生育に対する化合物5および6の効果が決定され、そして参照処置として去勢が使用された。腫瘍保有マウスは5または6の2通りの投与(0.15mmol/kg 1日1回または0.15mmol/kg 1日2回)を受けるように割当てられた(n=5/グループ)。腫瘍体積が毎週測定され、そしてビヒクルを受けているか去勢マウスの対照と比較される。
【0052】
去勢は対照と比較して最終腫瘍体積の55%減少へ導く(図6)。5の0.15mmol/kg 1日1回および0.15mmol/kg 1日2回は、ビヒクル処理対照動物の腫瘍に比較してそれぞれ平均最終腫瘍体積の41%および86.5%減少をもたらした(図6)。5で処理したマウスのすぐれた腫瘍発育阻害とは対照的に、化合物6で処理したマウスは低投与量において効果がないか、または対照と比較して腫瘍発育の刺激を示す(図6)。この化合物はインビトロにおいてPCA細胞生育の阻害において非常に有効であり、そして高度に抗アンドロゲン性であったから(図1を見よ)、化合物6がインビボにおいてLAPCA腫瘍細胞の発育を阻害することができないことは特に失望的である。5および6のインビボ抗腫瘍有効性の高度に有意な不均衡な二つの化合物の薬物動態学的性質の差に安易に帰せしめることはできない。これら二つの密接に関連した化合物のインビボ抗腫瘍有効性の劇的な差に含まれる理由は現時点では未知である。しかしながら、6が動物においてアンドロゲン受容体の強力なアゴニストであり得る代謝産物へ変換され、そのため腫瘍生育刺激を生ぜしめることへ帰せしめることができる。この研究の間、すべてのマウスは週1回体重測定した。すべての処理グループの体重は僅かに増加し、そして対照グループで観察されたのと類似であった。すべての動物は健康に見え、そして有害効果は観察されず、化合物は有意な毒性を持たないことを示唆する。
【0053】
第2のインビボ実験は、SCIDマウスに生育しているLAPC4前立腺癌細胞の成育を阻害する5および以前強力なCYP17阻害剤/抗アンドロゲンと同定された16(Grogoriyer et al.and Njar et al.J.Med.Chem,1998,41,902−912前出)の能力をテストし、そして去勢が参照処置として使用される。この実験において処理はマウスがホルモン依存性LAPC4細胞を皮下接種され、そして去勢されるかまたは5または16を1日2回皮下注射された日から始まる。図7は治療21週間の間の腫瘍の出現およびサイズに対する種々の処置の効果を示す。
【0054】
すべての他のグループは、触知可能なそして測定可能な腫瘍へ11週に発達した16で処置したグループ(0.5mmol/kg 1日2回)を除いて、治療の10週において触知可能で測定可能な腫瘍へ発展する。対照マウス中の総腫瘍体積は、大きい腫瘍のため、マウスが犠牲にされる時処置の14週に亘って8倍増加する。このように他のグループの腫瘍体積は処置の14週において対照グループに匹敵する。去勢したマウスの腫瘍体積はたった4.1倍(対照に比較して約50%減少)増加し、16で処置したマウスで観察された3.7倍増加(対照に比較して53.8%減少)に似ている。5で処置したマウスにおいては(0.15mmol/kg 1日2回)、腫瘍体積はたった0.5倍だけ増加し、これは対照マウスに対して93.8%減少を表す(P=0.00065)。16週において、化合物5で処置した動物の平均腫瘍体積は、測定し得る腫瘍が出現する時の10週におけるそれらの平均腫瘍体積よりも低い(殆ど無視し得るかまたは静止している)ことが見られる。さらに、5は、それぞれP=0.005および0.05において16または去勢に比較して腫瘍に対して有意な阻害効果を生ぜしめる。一般に、対照、去勢および化合物16処置マウスの腫瘍は急速に成長するが、他方5処置マウスの腫瘍は非常に遅く、そして二段階で成長する(図7)。化合物5は最も効果的な剤であり、そして腫瘍成長の阻害において去勢よりも有意にもっと効果的である。16はCYP17阻害において5より6倍強力であるが、5はすぐれたインビボ抗腫瘍活性を発揮することは興味深い。この現象に責任ある理由は現時点では未知であるが、しかし一部は5のより良い薬物動態学性質であろう。
【0055】
体眠している化合物5処理前立腺癌細胞(図7,16週を見よ)が5のもっと低い投与量で生育できるかどうかを決定するため、その投与量を16〜19週から0.15mmol/kg 週3回(投与量の78.6%低減)へ減らした。化合物の減らした投与量でのこの処置の期間、腫瘍は発育を回復した(図7)。この3週間の間の後、通常の投与量での薬物処置へ復帰し、そして腫瘍発育は遅くなり、プラトーへ達する。これらのデータはこの処理の細胞静止的性格を示唆し、そして抗腫瘍効果を達成するために連続的投与の必要性を推論させる。
【0056】
実験の終りにおいて、腫瘍および5−処置マウス中の5のレベルが決定される。最終投与1時間後の腫瘍、睾丸、および肝臓内のHPLCによる5のレベル(図7中の挿入部分)が決定される。興味あることに、代謝産物の少量(5に対して約15%)が肝臓組織内にのみ検出される。この代謝産物は血漿中に観察された代謝産物(前を見よ)と同じ保留時間を持つ。5の39.0±8.4μg/mg組織の最高濃度が皮下腫瘍において計測される。肝臓および睾丸中の濃度はより低いがしかし検出可能である。腫瘍中の5のレベルは血漿中で測定されたレベルよりも有意に高い。これは実験期間を通して化合物の蓄積の結果であり得る。このように5による腫瘍成長の阻害は一部は腫瘍キセノグラフト中のより高い濃度が前立腺癌細胞に細胞毒性/細胞静止効果を発揮するものと説明することができる。前立腺癌細胞のケトコナゾール(適度なCYP17阻害剤)の可能な直接の細胞毒効果を示唆する証拠があることを言及しなければならない。加えて、睾丸中の5の蓄積は動物内のテストステロン合成の阻害を可能となし得る。
【0057】
LAPC4はアンドロゲン依存性であることが良く確立されているけれども、これらの細胞はアンドロゲン非依存性になることができ、そしてそうなることで患者中の前立腺癌の発展を真似する適当なモデルを代表する(Chen et al.上出、およびKline et al.“Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice”,Nat.Med.1997,3,402−408)。図7に示すように、我々はこの現象を繰り返すことができる。さらに我々の結果は、16による処理または去勢は一定期間(アンドロゲン依存フェーズ)腫瘍成長を効果的に抑止するが、しかし腫瘍がインタクトな対照マウスにおけるのと全く同様に急速に成長するから、その後は非効果的になった(多分アンドロゲン非依存フェーズの結果)ことを示している。5で処理したマウス中の腫瘍の成長は処理期間全体を通じて強力に抑制される。このことは、5はアンドロゲン非依存性前立腺癌に効果を有することを示唆する。この化合物による処置はLAPC4腫瘍細胞を長期間アンドロゲン依存性に留め、それ故抗アンドロゲン治療に応答性とすることを可能することがもっともらしい。
【0058】
進んだそして再発したPCA中のARの上方調節および関与を証明する最近の研究(Mohler et al.and Chen et al.前出)は、PCAを処置するための薬物の開発のための標的としてアンドロゲン受容体に関心を再び呼び戻した(Tindall et al,“Symposium on androgen action in prostate cancer”,Cancer Res.2004,64,7178−7180)。その潜在的性質のため、5はすぐれた候補になり得る。
【0059】
結論:
データは、△16ステロイドのC17置換基をCYP17の強力な阻害剤および強力なARアンタゴニストを製造するために修飾する我々の以前の着想を補強する。17−ベンズイミダゾール5および6はCYP17のヘム鉄に配位することを示し、これはそれらの酵素阻害活性、ARアンタゴニズム、および前立腺癌細胞成長に一部責任ある性質である。驚くべきことに、これら化合物はそれらの抗腫瘍活性において非常に異なっており、5はLAPC4腫瘍キセノグラフトの成長において著しく抑制を生ぜしめ、対照的に6(0.15mmol/kg 1日2回)は腫瘍成長を促進する。本研究は、5はヒト前立腺成長の強力な阻害剤であり、そして去勢よりも著しく効果的である説得力ある証拠を提供する。これは前立腺癌腫瘍に対して去勢よりももっと効果的であるという点においてインビボ抗腫瘍効果を示すCYP17阻害剤/抗アンドロゲン剤の最初の例である。これらの印象的な生物学的活性は5をしてヒトの前立腺癌の処置のための可能性ある薬物としてさらなる開発のための強力な候補にならしめる。ベンズイミダゾール基を含んでいる化合物5のすぐれた抗腫瘍活性はベンズイミダゾールをして好ましい基とならしめる。しかしながら、上に議論した5の類縁体は関連した活性を持つことが予期され、そして本発明に含まれる。
【0060】
実験の部
化学:一般的操作およびテクニックは以前の報告と同じである(Njar et al.J.Med.Chem.1998,41,902−912)。赤外スペクトルはCHCl3溶液を使用してパーキンエルマー1600FTIR分光光度計で記録される。高解像度質量スペクトル(HRMS)は3−テルサフィネガンFTMS−2000FF質量分析計、EIモード(Ohio State University,Department of Chemistry)で測定される。キー標的化合物の純度基準として、我々は化合物均質性を指示するHPLCクロマトグラフィーを伴った高解像度質量スペクトルデータを提供した。低解像度質量スペクトル(LRMS)はフィネガンLCR−MSで測定される。融点はFischer Johns融点装置で測定され、そして補正されない。デヒドロエピアンドロステロンおよびデヒドロエピアンドロステロンアセテートはAldrich Milwaukee,WIから購入した。トリブチルスタニルピリミジンおよび2−トリブチルスタニルピラジンはFrontier Scientific,Inc.,Logan,UTから購入した。
【0061】
3β−アセトキシ−17−クロロ−16−ホルミルアンドロスタ−5,16−ジエン(2):この化合物は3β−アセトキシアンドロスト−5−エン−17−オン(1)から以前記載し、スペクトルおよび分析データを提供したように製造した(Njar et al,J.Med.Chem.1998,41,902−912)。
【0062】
3β−アセトキシ−17−(1H−ベンズイミダゾール−1−イル)−16−ホルミルアンドロスタ−5,16−ジエン(3):乾燥DMF(20ml)中の3β−アセトキシ−17−クロロ−16−ホルミルアンドスタ−5,16−ジエン(2,2.5g,6.65mmol)と、ベンズイミダゾール(2.35g,19.9mmol)と、そしてK2CO3(2.76g,23.9mmol)の混合物をアルゴン下1.5時間約80℃においてかきまぜる。室温へ冷却後、反応混合物を氷水(250ml)へ注ぎ、生成する沈澱を濾過し、水で洗い、乾燥して粗製の汚い白色固体(約2.9g)を得る。FCC(石油エーテル/EtOAc/Et3N=6:4:0.3)は純粋な化合物3を2.3g(88.7%)を与える。
【0063】
【化7】
【0064】
3β−アセトキシ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン(4):
乾燥ベンゾニトリル(10ml)中の3β−アセトキシ−17−(1H−イミダゾール−1−イル)−16−ホルミルアンドロスタ−5,16−ジエン(3,2.04g,4.45mmol)の溶液を活性炭上の10%パラジウム(1.02g,3の50重量%)の存在下5時間還流した。室温へ冷却後、触媒をセライトパッドで濾過して除いた。濾液を蒸発し、残渣をFCC(石油エーテル/EtOA/Et3N=7.5:3:0.5)によって精製した。純粋な化合物4を1.41g(73.8%)を与えた。
【0065】
【化8】
【0066】
3β−ヒドロキシ−17−(1H−イミダゾール−1−イル)アンドロスタ−5,6−ジエン(5):アセテート4(1.3g,3.02mmol)を不活性Ar雰囲気下メタノール(20ml)に溶かし、生成した溶液を10%メタノール性KOH(8ml)で処理した。混合物を室温で1.5時間かきまぜ,減圧下約40℃で10ml体積へ濃縮した。この溶液へ氷水(300ml)を注ぎ、生成する白色沈澱を濾過し、水洗し、乾燥した。EtOAc/MeOHからの結晶化は5(1.10g,94%)を与えた。
【0067】
【化9】
【0068】
17−(1H−ベンゾイミダゾール−1−イル)アンドスタ−4,16−ジエン−3−オン(6):化合物5(660mg,1.70mmol),1−メチル−4−ピペリドン(2.5ml)およびトルエン(4ml)の混合物から約10mlを留去した。次にアルミニウムイソプロポキサイド(521mg,2.55mmol)を加え、混合物をAr下4時間還流した。冷後混合物をEtOAc(50ml)で希釈し、5%NaHCO3(×3)および食塩水(×2)で次々に洗い、乾燥(Na2SO4)した。溶媒を蒸発し、粗生成物をFCC(CH2Cl2/EtOAc=25:1)で精製し、標題化合物6(544mg,82%)を得た。
【0069】
【化10】
【0070】
3β−アセトキシ−17−クロロ−16−ホルミルアンドロスタ−4,16−ジエン(2)と、ベンゾ−1H−1,2,3−トリアゾールおよびK2CO3との反応:3β−アセトキシ−17−(ベンゾ−2H−トリアゾール−2−イル)−16−ホルミルアンドロスタ−5,16−ジエン(7a)および3β−アセトキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)−16−ホルミルアンドロスタ−5,16−ジエン(7b):乾燥DMF(20ml)中の化合物2(2.5g,6.65mmol)、ベンゾトリアゾール(2.35g,19.9mmol)およびK2CO3(2.76g,23.9mmol)の混合物をAr下45分約80℃においてかきまぜた。室温へ冷却後、反応混合物を氷水(250ml)へ注ぎ、生成する沈澱を濾過し、水で洗い、乾燥して粗製の汚れた白色固体を得た。FCC(石油エーテル/EtOAc=4:1)は最初にマイナーな生成物として3β−アセトキシ−17−(ベンゾ−2H−1,2,3−トリアゾール−2−イル)−16−ホルミルアンドスタ−5,16−ジエン(7a,0.3g,9.8%)を与えた。
【0071】
【化11】
【0072】
同じ溶媒系でのさらなる溶出は主要生成物、3β−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)−16−ホルミルアンドスタ−5,16−ジエン(7b,2.3g,75.4%)を与えた。
【0073】
【化12】
【0074】
3β−アセトキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−5,16−ジエン(8):乾燥キシレン(40ml)中のビス(トリフェニルホスフィン)ロジウム(I)カルボニルクロライド(303mg,0.438mmol)と、1,3−ビス(ジフェニルホスフィノ)プロパン(394mg,0.954mmol)の混合物をAr下80℃で15分間黄色沈澱が生成するまで攪拌した。化合物7b(1.71g,3.72mmol)を加え、混合物をAr下18時間還流し、減圧下濃縮した。粗生成物はFCC(石油エーテル/EtOAc/Et3N=8.9:1.0:0.1)は純粋な化合物8の1.2g(74.7%)を与えた。
【0075】
【化13】
【0076】
3β−ヒドロキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−5,16−ジエン(9):3β−アセトキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−5,16−ジエン(8,700mg,1.62mmol)を用いて、化合物5について記載した方法を追従した。EtOAc/MeOHからの再結晶は標題化合物9(600mg,95%)を与えた。
【0077】
【化14】
【0078】
17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−4,16−ジエン−3−オン(10):β−ヒドロキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−5,16−ジエン(9,500mg,1.28mmol)を使用し、化合物6について記載した方法に従った。粗生成物のFCC(CH2Cl2/EtOH=50:1)による精製は標題化合物10(420mg,84.4%)を与えた。
【0079】
【化15】
【0080】
デヒドロエピアンドロステロン−17−ヒドラゾン(12):デヒドロエピアンドロステロン(11,3.5g,12.2mmol)をエタノール(60ml)に溶解し、得られた溶液をヒドラジンヒドラート(2.37ml,0.049mmol)で、次に水0.25ml中の硫酸ヒドラジン(7.9mg,0.061mmol)の溶液で処理した。混合物を室温で12時間かきまぜ、次に氷水中へ注いだ。生成する沈澱を濾取し、水洗し、乾燥し、標題化合物12の白色結晶を得た。
【0081】
【化16】
【0082】
17−ヨードアンドロスタ−5,16−ジエン−3β−オール(13):乾燥THF(144ml)および乾燥EtOH(72ml)のヨウ素(12.16g,0.0203mol)のかきまぜ溶液を氷欲で0℃に冷却し、そして溶液を1,1,3,3−テトラメチルグアニジン(6.72ml,6.27g,0.054mol)で処理した。THF(81ml)中の化合物12(3.0mg,9.9mmol)の溶液をヨウ素溶液へ反応温度を0℃に保って2時間を要して滴下した。次に反応混合物を真空下濃縮し、氷欲で冷却し、真空下室温で乾燥し、黄色固体(13,3.65g,92.4%)を得た。
【0083】
【化17】
【0084】
3β−ヒドロキシ−17−(2−ピラジル)−アンドロスタ−5,16−ジエン(14):乾燥DMF(10ml)中の17−ヨードアンドロスタ−5,6−ジエン−3β−オール(13,6.5g,1.257mmol)の溶液と、テトラキス(トリフェニルホスフェート)パラジウム(Pd(PPh3)4)(71.6mg,0.062mmol)と、(2−トリブチルスタニル)ピラジン(774.6mg,2.099mmol)の混合物を120℃で20時間加熱した。冷後混合物を冷水(50ml)で希釈し、EtOAc(30ml×3)で抽出した。合併したEtOAc抽出液を食塩水および水で洗い、Na2SO4上で乾燥し、濃縮して褐色固体を得た。この粗生成物をカラムクロマトグラフィー(FCC,石油エーテル/EtOAc/Et3N=3:2:0.15)によって精製し、14(66mg,15%)を得た。
【0085】
【化18】
【0086】
3β−ヒドロキシ−17−(5−ピリミジル)−アンドロスタ−5,6−ジエン(15):乾燥DMF 10mlに溶解した(5−トリブチルスタニル)ピリミジン(1.0g,2.710mmol)と、(Pd(PPh3)4)(92.88mg,0.0804mmol)を使用し、14について上で記載した13(0.645g,1.623mmol)の反応と、続いてのFCC(石油エーテル/EtOAc/Et3N)=3:2:0.15)による精製は、3β−ヒドロキシ−17−(5−ピリミジル)−アンドロスタ−5,16−ジエン(15,44mg,10%)を与えた。
【0087】
【化19】
【0088】
CYP17のインビトアッセイ:化合物のインビトロCYP17阻害活性は、酵素源としてインタクトなP450c17発現E.coli(Grigoryev,前出)を利用する我々の急速酢酸放出アッセイ(AARA)を使用して評価される。これは〔21−3H〕−17α−ヒドロキシプレグネノロンを基質として使用することを含み、そしてCYP17活性は基質のC−21側鎖の分裂の間生成したトリチウム化酢酸の量によって測定される。これは、この分野の研究者(Grigoryev,前出)によって使用されたHPLC分析操作に匹敵する。IC50値は適切の範囲にわたって阻害%対阻害剤濃度に関するプロットから直接得られる。各化合物は最大5濃度においてテストされる。アッセイは三系列で行われ、そしてIC50値は三系列実験の平均値として報告される。標準差は平均値の±5%である。
【0089】
ヒト5α−レダクターゼタイプ1および2アッセイ:化合物および参照としてのフィナステライドの阻害活性は、Hartmannらによって記載された操作(Picard et al,“Synthesis and evaluation of 2’−substituted 4−(4’−carboxy−methylbenzylidene)−N−acylpiperidines:Highly potent and in vivo active steroid 5α−reductase type 2 inhibitors”,J.Med,Chem.2002,45,3406−3417)に従い、DU145細胞株(ヒトタイプ1酵素のため)およびヒト前立腺ホモジネート(タイプ2酵素のためのBPH組織)を使用して決定される。10μMの濃度における阻害パーセントか、またはもっと強力な化合物の場合、IC50値が決定される。
【0090】
競合アンドロゲン受容体(AR)結合およびルシフェラーゼアッセイ:AR結合/競合アッセイ:24ウエルマルチウエルディッシュウエルをポリ−L−リジン(0.05mg/ml)で5分間コートし、乾燥し、無菌蒸留水でリンスし、2時間乾燥する。LNCaP ARおよび野生タイプARへのR1881結合の動力学を決定するため、LNCaPおよびPC3AR細胞をステロイド不含倍地中の24ウエルディッシュにプレートし、結合を許容する。翌日倍地を、0.1%BSAを補給し、〔3H〕R1881(0.01−10nM)を含有し、そして非特異的結合を決定するため冷DHTの200倍過剰が存在または不存在で、そしてプロゲステロンおよびグルココルチコイド受容体を飽和させるための1μMトリアムシノロンを含む血清不含、ステロイド不含RPM1で置換する。37℃において2時間のインキュベーション期間後、細胞を氷冷DPBSで2回洗い、そして0.5%SDSおよび20%グリセロールを含有するDPBS中に可溶化する。抽出物を除去し、そして細胞結合放射活性をシンチレーションカウンター中でカウントする。Graphpad Prismソフトウエアを用いて非直線回帰によってKdおよびBmax決定を含むデータを解析する。両方の細胞株中のARを殆ど飽和させるのに必要な濃度を確立し、そして受容体から〔3H〕R1881(5.0nM)を移動させるテスト化合物(0.1nM−10μM)の能力を上に記載したように決定する。各化合物のIC50値はGraphpad Prismソフトウエア(GraphPad Software,Inc,San Diego,CA)での非線形回帰によって決定される。
【0091】
ルシフェラーゼトランス活性化アッセイ:転写活性化アッセイは、少しの修飾を加えて前出のKimらにより記載された方法によって実施される。プロバシンルシフェラーゼレポーター構築物ARR2−Lucは、Vanderbilt University Medical CenterのDr.R.MatsusiRから好意に提供された最小プロバシンプロモーターARR2(Endocrinology 2000,141,4698−4710)をPGL3−エンハンサーベクター(Promegd)のポリクローナルリンカー領域に挿入することによって発生させる。概略すると、ポリ−L−リジンをコートした24−ウエルプレートに発育させたLNCaP細胞が5%活性炭処理FBS(Hyclon)を含有するフェノールレッド不含RPMI1640倍地中のARR2−Lucで形質転換される。形質転換24時間後、細胞はDHTありまたはなしの新鮮なフェノールレッド不含血清不含RPMI1640倍地および阻害剤と18時間インキュベートされる。ルシフェラーゼ活性は製造者の指示書(Promega)に従って二重ルシフェラーゼアッセイを使用して3系列で測定される。結果は誘発倍率、すなわち対照ルシフェラーゼ活性で除した処理細胞の相対的ルシフェラーゼ活性で表される。
【0092】
細胞培養および生存性:LNCaP細胞は10%FBSおよび1%ペニシリン/ストレプトマイシン溶液を補給したRPMI1640倍地中で発育させる。細胞増殖に対する新規化合物の効果を決定するため、細胞は実験開始前3日間ステロイド不含培地へ移される。ステロイド不含培地は、5%デキストランコートした、活性炭処理血清と、そして1%ペニシリン/ストレプトマイシン溶液を補給したフェノールレッドフ含RPMIからなっていた。発育研究は細胞(3×104)を24ウエルマルチウエルデイッシュ(Corning Inc.,Corning,NY)中にプレートすることによって実施される。24時間の結合期間後、培地を吸引し、そしてビヒクルまたはDHTの指示された濃度(1nM)および化合物(0.1μM−10μM)を含有するステロイド不含培地で置換される。対照ウエルはビヒクル(エタノール)で処理される。この培地は3日毎に交換され、そして生存細胞の数が7日目にWST−1〔4−〔3−(4−ヨードフェニク)−2−(4−ニトロフェニル)−2H−5−テトラゾリオ〕−1,3−ベンゼンジスルホネート〕アッセイによって比較される。上述の時間細胞のインキュベーション後、10%WST−1溶液が各ウエルへ加えられ、そして37℃で3時間インキュベートされる。インキュベーション後、プレートを僅かに振とうし、そして直ちに査定型マルチウエル分光分析計で450nmにおいて読み取られる。すべての結果は3ウエルの最小値の平均値で表される。追加の対照は細胞なしの培地のみからなる。
【0093】
薬物動態学的研究:すべての動物実験は、メリーランド大学医学部の動物ケア委員会のガイドラインおよび承認に従って実施される。NCI,FredericR,MD,USAから得た体重20〜22gの雄SCIDマウス(8〜10週令)は約25℃,50%相対湿度および12時間明サイクルおよび2時間暗サイクルにコントロールされた環境に維持され、そして食物および水に自由にアクセスすることが許容される。化合物5および6は水中の40%β−シクロデキストリン中に処方され、1回の皮下投与量が与えられる。薬物投与6時間までの種々の時間において動物が犠牲にされ、そして軽いハロタン(Ayerst,New York,NY,USA)麻酔のもとで心臓穿刺によって血液が収集された。
HPLC分析:ステロイドおよび適切な内部標準のクロマトグラフィー分離および定量は、以前記載されたように、ペリクルC18で包装されたWatersガードカートリッジによって保護されたWaters Novapak C18カラム(3.9×150mm)上のWaters Novapak C18カラム上の逆相HPLC法によって達成される。概略すると、この研究に使用されるHPLCシステムは、Waters溶媒送達システム、Waters717plusオートサンプラーおよび242.7nmで作動するWaters996発光ダイオード列デテクターと組合せたWatersコントローラー(Milford,MA)よりなる。移動相組成は水/MeOH/CH3CN(35:35:30 v/v/v+Et3N 200mlおよび移動相1000mlあたりNH4OAc 0.77g)であり、流速は1.0ml/minである。HPLC分析は環境温度で実施され、そしてデータ取得およびマネージメントはWatersミレニアムクロマトグラフィーマネージャーで達成される。
【0094】
サンプル調製:マウス血漿(200μl),5または6およびVN185−1(内部標準、100μg/mlの10μl)を含んでいる試験管がボルテッスミキサーを用いてジエチルエーテル(2×2ml)で3分間抽出され、そして3000gにおいて5分間遠心される。ゆるやかな空気流のもとで有機層が蒸発乾固される。残渣は移動相の部分標本(100μl)中に再構成され、HPLC分析前に0.2μmテフロンフィルターを用いて濾過される。
【0095】
較正曲線およびHPLCアッセイバリデーション:血漿および組織中の5および血漿中の6のための較正曲線は、0.1−100.0μg/mlの最終濃度を与えるように、未処置動物からの血漿(200μl)および組織調製物(200μl)を含んでいる抽出チューブ(2系列)中へ化合物の変化する量をスパイクすることによって構築される。適切なブランク抽出チューブも調製され、そして内部標準の部分標本が最終濃度5μg/mlを与えるように各抽出チューブへ加えられる。この較正サンプルは上に記載したサンプル調製操作を受ける。再構成した抽出液を部分標本(50μl)がHPLCシステムへ注入され、そして各検体のピーク面積の内部標準のピーク面積に対する比が5または6の濃度に対してプロットされる。アッセイの精密度および正確性はブランク血漿中の阻害剤の既知濃度の範囲から決定され、そしてHPL操作を受ける。研究は別々に3回繰り返される。
【0096】
データ解析:薬物動態計算は以前記載したとおり実施される。非コンパートメント薬物動態計算はWinNOnlin(Scientific Consulting Inc.)を用いて実施される。Windowsバージョン1.0のためのSigmastat上の変動のワンウエイ解析が異なる処理グループを95%信頼性レベルにおいて比較するために使用される。Bonferroni post−hocテストが有意性の決定のために使用される。0.05以下のP−値が統計学的に有意と考えられる。
【0097】
インビボ抗腫瘍研究(LAPC−4前立腺癌キセノグラフト):すべての動物実験はメリーランド大学医学部の動物ケア委員会のガイドラインおよび承認に従って実施される。Nationasl Cancer Institute−Frederick Cancer Research and Development Center(Frederick,MD)から購入した4〜6週令の重篤結合免疫不全(SCID)マウスが光および湿度の制御された条件下の病原体不存在環境に収容され、そして食物および水への自由なアクセスが許容される。腫瘍は実質的に以前記載されたとおりにマウスに皮下で接種されたLAPC4細胞から発達される。LAPC4細胞は80%コンフルエントまで15%FBSプラス1%PSおよび10nmDHTを加えたIMEM中に生育される。細胞はDPHS中に掻き取られ、遠心によって集められ、そして3×107細胞/mlにおいてMatrigel中に再懸濁(10mg/ml)される。マウスは各脇腹の一部位に細胞懸濁液100μlを皮下注射される。腫瘍はキャリパーで毎週計測され、そして腫瘍体積が一式:4/3πr12×r2(r1<r2)によって計算される。
【0098】
第1の実験において、LAPC4腫瘍は接種後8〜10週間発育が許容される。匹敵する腫瘍体積を有する5匹のマウスのグループは、去勢されるか、または5および6で処置される(0.15mmol/kg 1日1回および0.15mmol/kg 1日2回,9a.m.および5p.m.)。マウスはメトキシフルオラン麻酔のもとで去勢される。化合物5および6は食塩水中の0.3%ヒドロキシプロピルセルロース溶液中17.2mg/mlに調製され、そしてマウスは毎日皮下注射を受けた。対照および去勢マウスはビヒクルのみで処置されるか。腫瘍は処置の4週間毎週計測され、そして腫瘍体積が計算される。処置期間の終りに動物はハロタン麻酔のもとに犠牲にされ、そして腫瘍が摘出され、計量されそして−80℃で貯蔵される。動物はまた毎週計量され、そして一般的健康状態および処置による可能性ある毒性についてモニターされる。
【0099】
第2の実験においては、マウスはLAPC4細胞を接種され、そして各群5匹の4グループに分割される。対照および去勢グループはビヒクルのみを受け、他の2グループはVN/85−1(0.15mmol/kg 1日2回,9a.m.および3p.m.)か、または5(0.15mmol/kg 1日2回,9a.m.および3p.m.)を受ける。これら処置はLAPC4細胞接種1日後から開始され、対照グリープには14週、19週(VN/85−1および去勢グループ)および5処置グループには21週続けられ、そして腫瘍が測定され、上に記載したように処理される。
【0100】
腫瘍、肝臓および睾丸中の5(VN/124−1)レベルの測定:VN/124−1処置グループ中の動物を最後のVN/124−1投与1時間後に犠牲にし、そして腫瘍、肝臓および睾丸を収穫し、液体窒素中にスナップ凍結する。組織サンプルはリン酸バッファー(pH=7.4,0.5ml/mg組織)中にホモジナイズする。ホモジテイズした組織(200μl)を内部標準VN/85−1(100μg/mlストック溶液から10μl)でスパイクし、次にボルテックスミキサーに3分間かけることによりEt2O(2×2ml)で抽出し、次いで3000gで5分間遠心する。Et2O抽出液をゆるやかな空気流の下で蒸発乾固する。残渣をHPLC移動相100μl中に再構成し、0.2μmテフロンフィルターで濾過し、次に上に記載したようにHPLCにより分析される。
【0101】
以上の実施例は、以上の実施例において使用された反応剤および/または作業条件を一般的にまたは特定的に記載した本発明の反応剤および/または作業条件をもって置換することによって同様の成功度で繰り返すことができる。
【0102】
以上の説明から、当業者は本発明の本質特徴を容易に確かめることができ、そしてその精神および範囲を逸脱することなく種々の用途および条件に適合するように本発明の変更および修飾をなすことができる。
【0103】
表1:新規17−ヘテロアリール化合物のCYP17および5α−レダクターゼ活性およびアンドロゲン受容体結合
【0104】
【表1】
【0105】
a 我々は以前VN/81−1の合成を報告した(Njar et al,前出)。アビラテロンはPotterら(A convenient,lerge−scalesyn thesis of a biraterone acetate〔3β−acetoxy−17(3−pyridyl)androsta−5,16−diene〕,a potent new drug for the treatment of prostate cancer.Org.Prep.Proc.Int.1997,29,123−128)によって記載されたように合成した。
b IC50は酵素活性を50%阻害するのに要する阻害剤の濃度,CYP17については各自2系列、5α−レダクターゼおよびAR結合については3系列。
c IC50はアンドロゲン受容体から〔3H〕R1881を50%移動させるのに要する化合物の濃度。
d タイプ1酵素を発現する前立腺腫瘍細胞株(DV−145);基質:(5nM〔1β3H〕アンドロステンジオン。
e BPH組織からの酵素(タイプ2酵素),タンパク125μg,基質:20nM〔1β,2β−3H〕テストステロン。
f ni=10μMまで阻害なし、−=測定せず
【0106】
表2:5(50および100mg/kg)および6(50mg/kg)の皮下投与後の薬物動態
【0107】
【表2】
【0108】
a 値は平均±S.E.で表現される。n=5
【0109】
スキームの簡単な説明
【0110】
【化20】
【0111】
チャート1:アベラテロンおよびVN/85−1(16)の構造
【0112】
【化21】
【0113】
スキーム1:17−ベンゾアゾール化合物(5,6,9および10)の合成
【0114】
【化22】
【0115】
スキーム2:17−ジアジン化合物(14および15)の合成
【0116】
【化23】
【0117】
スキーム3:VNLG/81を含む、トランスアンドロステロンの代謝産物の合成
【技術分野】
【0001】
本発明は、新しい化学的実在物、特にステロイドC−17ベンゾアゾール、ピリミジノアゾール(アザベンゾアゾール)およびジアジンを提供する。本発明はまた、これらベンゾアゾール、ピリミジノアゾールおよびジアジンの合成方法を提供する。一具体例において、ベンゾアゾールまたはピリミジノアゾールの合成方法は、3β−アセトキシ−17−クロロ−16−ホルミルアンドロスタ−5,16−ジエンまたはその類縁体と、そしてベンゾアゾールまたはピリミジノアゾール求核試薬の求核性ビニル“付加−脱離”置換反応のステップを含んでいる。他の具体例において、ジアジンの合成方法は、17−ヨードアンドロスタ−5,6−ジエン−3β−オールまたはその類縁体のトリブチルスタニルジアジンとのパラジウム触媒交差カップリング反応を含んでいる。
【0002】
本発明の化合物は、ヒトCYP17酵素の強力な阻害剤であり、また野生タイプおよび変異株アンドロゲンレセプタ−(AR)の強力な拮抗剤である。最も強力なCYP17阻害剤は、3β−ヒドロキシ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン(5、コード名VN/124−1)、3β−ヒドロキシ−17−(51−ピリミジン)アンドロスタ−5,16−ジエン(15)、および17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−6,16−ジエン−3−オン(6)であり、それらのIC50値はそれぞれ300,500および915nMである。化合物5,6,14および15は、変異株およびLNCaPARおよび野生タイプARへの3H−R1881(メチルトリエノロン、安定な合成アンドロゲン)の結合防止に有効であったが、しかし後者へは2.2ないし5倍高い結合効率を有している。化合物5および6は,強力な純粋AR拮抗剤であることを示した。細胞成育研究は、化合物5および6は低いマイクロモル範囲(すなわち<10μM)のIC50値でDHT−刺激LNCaPおよびLAPC4前立腺癌細胞の成育を阻止する。それらの阻止強度はカソデックスに匹敵するが、しかしフルタミドより著しくすぐれている。マウスにおける化合物5および6の薬物動態が検討された。5および6の50mg/kgの腹腔内投与後、30ないし60分後それぞれ16.82および5.15ng/mlのピーク血漿レベルが出現し、両方の化合物は血漿から速やかに排出され(それぞれ終末半減期44.17および39.93分)、そして8時間目にどちらも検出されなかった。注目すべきことに、化合物5は一時的に17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−3−オンと同定された代謝物へ急速に変換された。インビボでテストする時、5はアンドロゲン依存性LAPC4ヒト前立腺癌キセノグラフトの成育の阻害に非常に有効であることが証明されたが、6は無効であった。化合物5(50mg/kg 1日2回)は、対照と比較して平均最終腫瘍体積の93.8%減少(P=0.00065)をもたらし、そして去勢よりも有意に効果的であった。我々の知る限り、これはアンドロゲン依存性前立腺腫瘍成育の抑制において去勢より有意にもっと効果的な抗ホルモン剤(アンドロゲン合成の阻害剤(CYP17阻害剤)/抗アンドロゲン)の最初の例である。これらの印象的抗癌性に鑑み、化合物5その他はヒト前立腺癌の処置に使用することができる。
【背景技術】
【0003】
前立腺癌(PCA)は最も普通の悪性腫瘍であり、そして全世界において年令関連癌死亡の原因である。肺癌は別にし、PCAは男性において最も普通の癌の形であり、そしてアメリカ人男性の第2の癌死因である。2004年においてアメリカ合衆国では、前立腺癌の推定230,000症例が診断され、そして約23,000人男性がこの病気で死亡するであろうといわれている(Jemal et al.Cancer Statics,2004.CA Cancer J.Clin.2004,54,8−29)。1992−1999の期間、アフリカ系男性アメリカ人の平均年間PCA症例はコーカサス系男性よりも59%高く、そして平均年間死亡率はコーカサス系男性の2倍以上であった(American Cancer Society−Cancer Facts and Figures 2003)。アンドロゲンはPCAの発生、成育および進行において重要な役割を果す(McConnell,J.D.,“Physiological basis of endocrine therapy for prostatic Cancer”,Urol.Clin.North Am.1991,18:1−13)。この点に関し2つの最も重要なアンドロゲンはテストステロン(T)およびジヒドテストステロン(DHT)である。睾丸はTの約90%を合成し、そして残りは副腎によって合成される。Tは主として前立腺に局在する酵素ステロイド5α−レダクターゼによってもっと強力なアンドロゲンDHTへ変換される(Bruggins et al.“The conversion of testosterone to 5α−androstan−17β−ol−3−one by rat prostate in vivo and in vitro”,J.Biol.Chem.,1968,243,2012−2021)。Huggins et al.は1941年に進行および転移PCAのための療法としてアンドロゲン奪取を導入した(Huggins et al.“Studies on prostatic cancer:2.The effect of prostate gland”,Arch.Surg.,1994,43,209−212)。その後アンドロゲン奪取療法はPCA患者の多数セッティングにおいて最も有益な応答を産むことが示された(Denmeade et al.“A history of prostate cancer treatment”,Nature Rev.Cancer,2002,2:389−396)。睾丸摘除術(外科的またはGnRHアゴニストでの医学的)は大部分の前立腺癌患者のための標準的処置オプションとして残っている。医学的および外科的睾丸摘除術は睾丸によるアンドロゲン生産を減らすかまたは排除するが、しかし副腎中のアンドロゲン合成には影響しない。睾丸摘除術と、副腎アンドロゲンの作用を阻害する抗アンドロゲン剤との併用療法はPCA患者の生存を有意に延長するといういくつかの研究が報告された(Crawford et al.“A controlled trial of lenprolide with and without flutamide in prostatic carcinoma”,N.Eng.J.Med.,1989,321,419−424;Crawford et al.“Tratment of newly diagnosed state D2 prostate cancer with lefnprolide and flutamide or leuprolide alone,phase III;intergroup study 0036”,J.Urol,1992,147;417A;Denis L.,“Role of maximal androgen blockade in advanced prostate cancer”,Prostate,1994,5(Suppl.),17s−22s)。Mohlerらによる最近の特色ある論文(Mohler et al,“The androgen axis in recurrent prostate cancer”,Clin.Cancer Res.2004,10,440−448)において、TおよびDHTはアンドロゲン受容体を活性化するのに十分なレベルにおいて再発性PCA組織内に出現することが明らかに証明された。加えて、同系PCAキセノグラフトモデルのマイクロアレイに基いたプロファイリングを使用して、Sawyerらは(Chen et al,“Molecular determinant of resistance to antiandrogen therapy”,Nat.Med.2004,10,33−39)、アンドロゲン受容体mRNAの適度の増加が抗アンドロゲン療法に対する抵抗性の発展に一貫して関連する唯一の変化であることを発見した。睾丸、副腎および他の組織中のアンドロゲン合成を阻害する強力なそして特異性化合物はPCAの処置のためにもっと効果的であり得る(Njar,V.C.O.;Brodie,A.M.H.,“Inhibitors of 17α−hydroxylase−C17,20−lyase(CYP17):Potent Agents for the treatment of prostate cancer,”Current Pharm.Design,1999,5:163−180)。
【0004】
睾丸および副腎において、Tの生合成における最後のステップは、逐次的に作用しそして両方とも単一の酵素チトクロームP450モノオキシゲナーゼ17α−ヒドロキシラーゼ/17,20−リアーゼ(CYP17)によって触媒される、二つのキー反応を含んでいる(Hall,D.F.,“Cytochrome P−450 C21SCC:one enzyme with two actions:Hydroxylase and lyase”,J.Steroid Biochem.Molec.Biol,1991,40,527−532)。抗カビ剤として、そしてP450酵素阻害のためにケトコナゲールも適度なCYP17阻害剤であり、そしてPCAの処置に臨床的に使用された(Trachtenberg et al,“Ketoconazole:A novel and rapid treatment for advanced prostatic cancer”,J.Urol.,1983,130,152−153)。処理の注意深いスケジュールはさもなければホルモン抵抗性癌患者において長期応答を発生させることができることが報告された(Muscato et al,“Optimal dosing of ketoconazole and hydrocortisone leads to long responses in hormone refractory prostate cancer”,Proc.Am.Assoc.Cancer Res.,1994,13:22(Abstract))。さらにケトコナゾールはフルタミド脱退にもかかわらず進行したPCA患者において活性を保有することが発見された(Small et al,“Ketoconazole retains activity in advanced prostatic cancer patients with progression despite flutamide withdrawal”,J.Urol.,1997,157,1204−1207)。今ではケトコナゾールは肝臓毒性および他の副作用のために使用が取り止められているけれども、このことはCYP17のもっと強力なそして選択的阻害剤はこの病気の処置において進んだ段階でもそしてホルモン抵抗性らしい一部の患者においてさえも有用な剤を提供し得ることを示唆する。
【0005】
CYP17の種々の強力なステロイドおよび非ステロイド阻害剤が報告されており、そしてあるものはゲッ歯類モデルにおいてテストステロイド生産の強力な阻害剤であることが示された(上出、Njar and Brodie)。最近Jarmanらは彼らの最も強力なCYP17阻害剤アビラテロンの前立腺癌患者におけるホルモンインパクトを記載した(O’Donnell et al,“Hormonal Impact of the 17α−Hydroxylase/C17,20−lyase inhibitors abiraterone acetate(CB7630)in patients with prostate cancer”,Br.J.Cancer,2004,90:2317−2325)。我々の強力なCYP17阻害剤のいくつかも5α−レダクターゼを阻害し、および/または強力な抗腫瘍活性を有する強力な抗アンドロゲン剤であることを示した。(上出Njar and Brodie,and Long et al,“Antiadrogenic effects of novel androgen synthesis inhibitors on hormone−dependent prostate cencer”,Cancer Res,2000,60,6630−6640)。本発明の背景のさらなる例証は、米国特許Nos.5,994,335;6,200,965;および6,444,683である。
【発明の概要】
【課題を解決するための手段】
【0006】
我々は一連の強力なCYP17阻害剤、17−ベンゾアゾール、17−ピリミジノアゾールおよび17−ジアジンを発見した(例えば以下に記載するように他の構造にも類似して適用することができる化合物の製造例のためのスキーム1および2を見よ)。これらC−17ヘテロアリールステロイドの製造のための刺激は、ベンズイミダゾール、ベンゾトリアゾール、ピリミジノアゾール、およびジアジン構造、いわゆる恩恵享受構造(privileged structure)を新しい分子へ導入する我々の願望に基いている(Nicolaou et al,“Natural product−like combinational libraries based on privileged structures.1.General principles and solid−phase synthesis of benzopyrans”,J.Am.Chem.Soc.2000,122,9939−9953)“Privileged strncture”なる術語は、多種類の無関係分子標的と相互作用することができる構造モチーフを記載するためにEvansらにより初めて導入された(J.Med,Chem.Soc.2000,122,9939−9953)。これらの枠組、特にベンズイミダゾール枠組は、それらの多岐にわたる生物学的活性のポートフオリオのため、および多種類の有用な薬物の部分構造として医薬化学において広く注目されている(Nicolaou et al.前出)。
【0007】
本発明のC−17ヘテロアリールステロイド化合物は以下の一般式Iを有する。
【0008】
【化1】
【0009】
式中、
ABC環構造は、任意に置換されてもよいステロイドまたはその類縁体のA,BおよびC環部分であり;
16,17位間の結合(実線と点線)は二重結合か、または化合物が17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−3−オンである時には単結合であり;そして
Xは任意に置換されてもよいベンズイミダゾール、ベンゾトリアゾール、ピリミジノイミダゾール(プリン)、ピリミジノトリアゾールまたはジアジンであり、ベンズイミダゾール、ベンゾトリアゾールおよびピリミドイミダゾール基は5員環の窒素原子を介してステロイド残基へ結合しており、そしてジアジン基はジアジン環の炭素原子を介してステロイド残基へ結合している。
【0010】
これらの化合物の薬学的に許容し得る塩も本発明に含まれる。
【0011】
ABC環構造のための任意の置換基は、一以上のアルキルおよびハロゲン化アルキル(好ましくはC1−6);環構造に直接結合した二重結合を含むアルケニルおよびハロゲン化アルケニル(好ましくはC1−6);ハロゲン;アミノ;アミノアルキレン;ヒドロキシイミノ;およびヒドロキシを含む。さらに任意に、ABC環構造の隣接する水素を除去し、隣接する炭素原子間を追加の結合によって置換し、環構造中のこれら炭素間に二重結合を形成してもよい。ABC環構造上の好ましい任意の置換基は環構造の10位および/または13位のメチル基である。
【0012】
ベンズイミダゾール、ベンゾトリアゾール、ピリミジノイミダゾール、ピリミジノトリアゾール、またはジアジン構造のための任意の置換基は、ハロゲン、アミノ、アミノアルキレン、ヒドロキシ、−SH,−S−アルキル、アルキルおよびハロゲン化アルキル(好ましくはC1−6)を含む。これらの任意の置換基はベンズイミダゾール、ベンゾトリアゾール、ピリミジノイミダゾール、ピリミジノトリアゾールまたはジアジン構造の環炭素原子上にあるであろう。
【0013】
ベンズイミダゾール、ベンゾトリアゾール、ピリミジノイミダゾール、ピリミジノトリアゾールおよびジアジン構造はそれぞれ以下の式を有する。
【0014】
【化2】
【0015】
式中の*はステロイド残基への結合を示す。
【0016】
好ましい一具体例においては、ABC環構造は、C−17ヘテロアリール置換基への結合に隣接するD環と共有する炭素、すなわち13位においてアルキル、特にメチル置換を除いて無置換のC環を有する。
【0017】
好ましい他の具体例においては、ABC環構造のA,BおよびC環は3β−ヒドロキシアンドロスタ−5,16−ジエンまたは3−オキソアンドロスタ−5,16−ジエンに基づく慣用構造を有する。しかし他の具体例においては、AおよびB環は以下の構造1−25の一つを有する。
【0018】
【化3】
【0019】
以下に1−25のAB環を有し、そしてXがベンズイミダゾールであるCおよびD環が慣用である化合物の化学名をリストする。
【0020】
化合物1:3β−ヒドロキシ−3α−メチル−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物2:3β−フルオロ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物3:3β−クロロ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物4:3β−ブロモ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物5:3β−ヨード−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物6:3β−アミノ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物7:17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−3,5,16−トリエン;
化合物8:17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−2,4,16−トリエン;
化合物9:17−(1H−ベンズイミダゾール−1−イル)−3−メチレンアンドロスタ−5,16−トリエン;
化合物10:17−(1H−ベンズイミダゾール−1−イル)−3−メチレンアンドロスタ−4,16−トリエン;
化合物11:3,3−ジフルオロ−17−(1H−イミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物12:3,3−ジフルオロ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−4,16−ジエン;
化合物13:17−(1H−ベンズイミダゾール−1−イル)−3−メチレンアンドロスタ−2,4,16−トリエン;
化合物14:17−(1H−ベンズイミダゾール−1−イル)−3−メチレンアンドロスタ−2,4,5,16−テトラエン;
化合物15:3,3−ジフルオロ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−2,4,16−トリエン;
化合物16:3,3−ジフルオロ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−2,4,6,16−テトラエン;
化合物17:3−ヒドロキシイミノ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン;
化合物18:3−ヒドロキシイミノ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−4,16−ジエン;
化合物19:3−ヒドロキシイミノ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−2,4,16−トリエン;
化合物20:3−ヒドロキシイミノ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−2,4,6,16−テトラエン;
化合物21:3−ヒドロキシ−17−(1H−ベンズイミダゾール−1−イル)エストラ−1,3,5(10),16−テトラエン;
化合物22:3−フルオロ−17−(1H−ベンズイミダゾール−1−イル)エストラ−1,3,5(10),16−テトラエン;
化合物23:3−クロロ−17−(1H−ベンズイミダゾール−1−イル)エストラ−1,3,5(10),16−テトラエン;
化合物24:3−ブロモ−17−(1H−ベンズイミダゾール−1−イル)エストラ−1,3,5(10),16−トリエン;
化合物25:3−ヨード−17−(1H−ベンズイミダゾール−1−イル)エストラ−1,3,5(10),16−テトラエン
【0021】
ヘテロアリール環のための任意の置換基Xの例は以下の構造26−40によって示され、ここでXはベンズイミダゾールである。Xが置換されたベンズイミダゾール、ピリミジノイミダゾール、ピリミジノトリアゾール、ピラジンまたはピリミジンである類似化合物も企図される。
【0022】
【化4】
【0023】
ヘテロアリール環のための任意の置換基Xの他の例は以下の構造41−46によって示され、ここでXは置換されたC−17−アザベンズイミダゾール(すなわちピリミジノイミダゾールまたはプリン)である。Xが置換されたベンズイミダゾール、ベンゾトリアゾール、ピリミジノトリアゾール、ピラジンまたはピリミジンである類似化合物も企図される。
【0024】
【化5】
【0025】
特に好ましい化合物は以下の構造M5,M6,M9およびM10の化合物である。
【0026】
【化6】
【0027】
CYP17およびステロイド5α−レダクターゼと比較したこれら化合物の阻害活性、アンドロゲン受容体への結合およびトランス活性化、および二つのヒト前立腺癌細胞株LNCaPおよびLAPC−4に対する抗増殖効果が研究された。化合物5および6の薬物動態がマウスで評価され、そしてヒトLAPC−4前立腺癌に対するインビボ抗腫瘍活性もマウスで評価された。我々の知る限り、化合物15を除いてここに記載したすべての化合物は新規化合物を代表する(Haidar et al,“Novel steroidal pyrimidyl inhibitors of P450 17(17α−hydroxylase/C17−20−lyase)”,Arch.Pharm.Med.Chem.2001,334,373−374;and Haidar et al,“Effects of novel 17α−hydroxylase/C17−20−ylase(P450 17,CYP17)inhibitors on androgen synthesis in vivo”,J.Steroid Biochem.Molec.Biol.,2003,84,555−562)。
【0028】
17−ベンゾアゾールおよび17−ジアジンの製造はそれぞれスキーム1および2に要約されている。これらの方法はここに記載した他の類縁体へも同様に適用することができる。
【0029】
我々の17−ベンゾアゾール合成のキー中間体である、3β−アセトキシ−17−クロロ−16−ホルミルアンドロスタ−5,16−ジエン(2)は、(1)から以前記載された我々の日常的操作によって得られた(Njar et al,“Nucleophilic Vinylic“addition−elimination”substitntion reaction of 3β−acetoxy−17−chloro−16−formylandrosta−5,16−diene:A novel and general route to 17−substituted−△16−steroids.Part I.Synthesis of novel 17−azolyl−△16−steroids;inhibitors of 17a−hydoxylase/17,20−lyase(P45017a)”,Bioorg.Med.Chem.Lett.1996,6,2777−2782;and“Novel 17−azolyl steroids;potent inhibitors of cytochrome P450 17a−hydroxylase/17,20−lyase(P45017a):Potential agents for the treatment of prostate cancer”,J.Med.Chem.1998,41,902−912)。DMF中K2CO3の存在下2のベンズイミダゾールでの約80℃における処理は殆ど定量的収率で3β−アセトキシ−17−1H−ベンズイミダゾール3を与えた。化合物3は還流ベンゾニトリル中で活性炭上の10%パラジウムでスムースに脱ホルミル化され、93%で化合物4を与えた。この化合物の加水分解は必要な3β−ヒドロキシ−17−ベンズイミダゾール5を与えた。5のOppenauer酸化変法は対応する△4−3−オキソ類縁体6を与えた。
【0030】
DMF中約80℃におけるK2CO3存在下のベンゾトリアゾールと2との反応は、すぐれた収率で所望の3β−アセトキシ−17−ベンゾ−1H−1,2,3−トリアゾール7bと、そして約5%収率で2H−1,2,3−トリアゾール位置異性体を与えた。これの2位置異性体はシリカゲル上のフラッシュカラムクロマトグラフィー(FCC)によって容易に分離され、そしてそれらのそれぞれのプロトンNMRスペクトルによって容易に同定された。このように対称2H−1,2,3−トリアゾール7aの4個の芳香族プロトンはδ7.43,7.45,7.88および7.90のダブレットの2対として現れ、他方非対称1H−1,2,3−トリアゾール7bの4個の芳香族プロトンは、δ7.46(2H)のマルチプレットおよびδ7.57(1H)と8.15(1H)におけるダブレットとして現れた。加えて、7aの16−CHOプロトンは、7bのδ9.59に比較してδ10.66へ著しくシフトした。還流キシレン中のRh(1,3−ビス(ジフェニルホスフィ))プロパン)2+Cl−触媒〔Rh(dppp)2+Cl−〕のその場の生成による脱ホルミル化は化合物8を与え、3β−アセトキシ基の加水分解の後、我々は標的とする3β−ヒドロキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−5,16−ジエン(9)を90%収率で得た。9の酸化は好収率で10を与えた。
【0031】
17−ジアジン(17−ジアジン14および17−ピリミジン15)の合成は容易に入手し得るデヒドロエピアンドロステロン(11,スキーム2)から始め、この化合物を以前Potter et alらが記載したようにヒドラジンヒドラートとヒドラジンサルフェートでの処理により対応する17−ヒドラゾンへ変換した(Potter et al,“A convenient,large−scale synthesis of abiraterone acetate〔3β−acetoxy−17(3−pyridyl)androsta−5,16−diene〕,a potent new drug for the treatment of prostate cancer,Org.Prep.Proc.Int.1997,29,123−128)。1,1,3,3−テトラメチルグアニジンの存在下ヨウ素での12の処理はすぐれた収率でビニル17−ヨウダイド12を与えた。(2−トリブチルスタニル)ピラジンまたは(5−トリブチルスタニル)ピリミジンでの13のパラジウム触媒交差カップリング反応(Choshi et al,“Total Synthesis of Grossularines−1 and−2,”J.Org.Chem.1995,60,5899−5904)は、それぞれ3β−ヒドロキシ−17−(2−ピラジル)アンドロスタ−5,16−ジエン(14,15%)と、3β−ヒドロキシ−17−(5−ピリミジル)アンドロスタ−5,16−ジエン(15,10%)を与えるように進行した。これら二つの交差カップリング反応の低収率は採用した反応条件下でのスタニルジアジン試薬の不安定性のためであり得る。標的化合物14および15の構造はそれらのプロトンNMRスペクトルに容易に同定された。14中の17−ピラジン部分の3個の非均等プロトンはδ8.35,8.48および8.70における3シングレットとして現われ、15中の17−ピリミジン部分の3個のプロトンは2個の均等プロトンがδ8.73においてシングレットとして現われ、1個のプロトンはδ9.07に現われた。さらに14および15の17−ジアジン基は、前駆体△16−17−イオダイド13の16−プロトンに関してそれらそれぞれの16−オレフィン性プロトンの化学的シフトに異なる影響を発揮する。14中の16−Hはδ6.77においてシングレットとして現われ、13中の16−H(δ6.14)に比較して著しく脱シールドされ、15中の16−Hは13と同様にδ6.11に現われた。上で示したように化合物15はHaidarらによって以前に報告されたが、これはそこで記載された操作とは異なる操作で合成された。
【0032】
新規化合物の代表的サンプルが次に以後のセクションに詳細に記載した広範なインビトロおよびインビボ研究にかけられた。
【0033】
本発明は、それを必要とする対象へ本発明に従った化合物の有効量を投与することを含む、前立腺癌または前立腺肥大を処理する方法にも関する。用語“処置”は慣用的に、例えば前立腺病に関連する一以上の症状を撲滅、緩和、減少、寛解、改善等の目的のため対象の管理またはケアの意味で使用される。処置することができる前立腺病の例は、例えば前立腺肥大(BPH)および前立腺癌(例えば前立腺悪性腫瘍)を含む。
【0034】
投与の特定の投与量および頻度は、特定の活性化合物の活性度、その代謝安定性および作用の長さ、排泄速度、投与モードおよび時間、対象の年令、体重、健康状態、性別、食事等、および前立腺癌または肥大の重篤度を含む種々のファクターに依存して変動し得る。化合物のどのような有効量、例えば毎日約1mgないし500mg、もっと特定すれば毎日約50mgないし150mgを投与することができる。化合物は、例えば経口、非経口、腹腔内、外用、経皮(例えば標準的パッチを使用して)、眼内、経鼻、局所、エアロゾル、スプレー、吸入のような非経口、皮下、静脈内、筋肉内、バッカル、舌下、経直腸、動脈内および硬膜下腔内を含む、どのようなルートによるどのような形によっても投与することができる。本発明の化合物は単独で、または例えば適当な薬剤組成物を製造するための生理学的に許容し得る担体のような活性または不活性成分との組合せで投与することができる。
【0035】
ここで引用したすべての出願、特許および発表、および2005年3月2日に出願した米国仮出願No.60/657,390の全体の開示を参照としてここに取入れる。
【0036】
さらに考究することなく、以上の説明を利用して当業者は本発明をその全範囲において利用できるものと信じられる。それ故以下の好ましい特定の具体例は単に例証と解すべきであり、開示の残りの限定ではない。
【0037】
以上および以下の実施例において、すべての温度は未補正の摂氏で表され、そしてすべての部およびパーセントは特記しない限り重量による。
【図面の簡単な説明】
【0038】
【図1】LNCaP−ARR2−1u前立腺癌細胞中のLNCaP−ARを介して仲介されるルシフェラーゼの転写活性に対する5,6およびカソデックスの効果。ステロイド不含倍地中の細胞はビヒクルか、または1nM DHTありまたはなしの5かまたはカソデックスの増大する濃度で18時間処理された。次に細胞は材料および方法に記載されたようにルシフェラーゼ活性についてアッセイされた。棒は、別々の3実験からの3系列ウエル中の平均光ユニット〔秒あたりのカウント(CPS)/単位タンパク、すなわち相対的ルシフェラーゼ活性〕によって表される。
【図2a】(a)LNCaPおよび(b)LAPC4前立腺癌細胞発育に対する5,6およびカソデックスの効果。細胞はプレート前ステロイド不含倍地中で生育された。次に材料および方法に記載されたように3系列ウエルが5,6またはカソデックスおよびDHTの増大する濃度で同時処理された。処理7日後の発育阻害のパーセント(対照と比較して)がWST−1アッセイを使用して決定された。結果は3系列で実施された3実験の平均値および標準偏差を表す。
【図2b】(a)LNCaPおよび(b)LAPC4前立腺癌細胞発育に対する5,6およびカソデックスの効果。細胞はプレート前ステロイド不含倍地中で生育された。次に材料および方法に記載されたように3系列ウエルが5,6またはカソデックスおよびDHTの増大する濃度で同時処理された。処理7日後の発育阻害のパーセント(対照と比較して)がWST−1アッセイを使用して決定された。結果は3系列で実施された3実験の平均値および標準偏差を表す。
【図3】5,6(内部標準)およびマウス血漿から抽出した代謝産物の典型的HPLCクロマトグラフィーチャート。16,代謝産物および5の保留時間はそれぞれ11.5,17.3および21.6分であった。
【図4】雄SCIDマウスへの1回の皮下ボーラス投与量の投与後の5および6の薬物動態プロフィル。各データポイントはマウス3匹から得た平均血漿濃度を表す。標準偏差(示さず)は、平均値の±5〜8%であった。
【図5】雄マウスへの1回の皮下ボーラス投与(100mg/kg体重)後の5および代謝産物の薬物動態プロフィル。
【図6】雄SCIDマウスにおけるLAPC4前立腺腫瘍の発育に対する5,6および睾丸除去術のインビボ抗腫瘍活性。LAPC4腫瘍を有するマウス5匹のグループが5(0.15mmol/kg/dayまたは0.30mmol/kg/day)で処置された。処置28日後腫瘍体積が測定された。腫瘍体積の標準偏差(示さず)は平均値の±10〜12%であった。
【図7】雄SCIDマウスにおけるLAPC4前立腺腫瘍の形成および発育に対する5,16および睾丸除去術の効果。3×107個のLAPC4細胞がCIDマウスの背面脇に皮下注入された。マウスの1グループは去勢された。マウスの他のグループはビヒクルか、または5(0.15mmol/kg 1日2回)または16(0.15mmol/kg 1日2回)を投与された。5または16による毎日の処置は細胞接種1日後に開始された。毎週腫瘍体積が測定され、腫瘍体積の変化のパーセントが処置16週後に決定された。*は、14週において対照、去勢および16に対する5の有意差(それぞれP=0.00065,0.05および0.0097)を示す。**は、16週において去勢および16に対する5の有意差(それぞれP=0.047および0.0047)を示す。↓および↓↓:5の減らした投与量期間
【発明を実施するための形態】
【0039】
(実施例)
生物学的研究
CYP17阻害研究:CYP17阻害アッセイは我々の以前報告した操作に従って実施される。そこでは酵素源としてインタクトなチトクロームP450c17−発現E.coliが使用される(Grigoryev et al,“Cytochrome P450c17−expressing Escherichia coli as a first−step screening system for 17α−hydroxylase−C17,20−lyase inhibitors”,Anal.Biochem.1999,267,319−330;and“Effects of new 17α−hydroxylase/C17,20−lyase inhibitors on LNCaP prostate cancer cell growth in vitro and invivo”,Br.J.Cancer,1999,81,622−630)。化合物のIC50値は投与量応答曲線から決定され、そして表1にリストされている。ケトコナゾール、アビラテロン(臨床試験中のCYP17阻害剤(前出O’Donnell)チャート1)、および3β−ヒドロキシ−17−(1H−イミダゾール−1−イル)アンドロスタ−5,16−ジエン(VN/85−1,化合物16,チャート1,最も強力なCYP17阻害剤だと信じられている(Njar et al,Current Pharm.Design,1999,5,163−180))のIC50値も比較のため同じアッセイシステムにおいて決定される。新しい17−複素環化合物のいくつかはIC50値300−915nMでCYP17の強力な阻害を示す。ベンゾイミダゾール5および6は、ベンゾトリアゾール9および10よりも4ないし6倍強力である。この結果は17−複素環の電子的性格が阻害活性に影響することを示唆する。さらに△5−3β−オール官能を有する化合物5および9は、△4−3−オン官能を有する対応する類縁体6および10よりもそれぞれ少なくとも3倍強力である。これらの結果は簡単な17−アゾールCYP17阻害剤についての我々の以前の結果と対照的である。それらの阻害剤シリーズにおいては、△5−3β−オールアゾールと対応する△4−3−オンアゾールの間には阻害強度に目立った差はない(Njar et al,J.Med.Chem.1998,41,902−912,前出)。可能性ある説明は、よりバルキーなベンゾアゾールは酵素の活性部位に異なって結合し、3位における部分の相互作用が結合に対して重要になって来ることである。一部のP450チトクロームのヘム成分に対する基質または阻害リガンドの結合はUV−vis差分光分析(Jefcoat C.R.,“Measurement of substrate and inhibitor binding to microsomal cytochrome P450 by optical difference spetroscopy”,Methods Enzymol.1978,52,258−279)を用いて検討される。このアプローチは我々が以前発表した標準的操作に従って拡張される(Njar et al,Bioorg.Med.Chem.Lett,1996,6,2777−2782;and J.Med.Chem,1998,41,902−912)。化合物5および9は各自タイプII差スペクトルを誘発し、低スピン鉄の生成を伴なってステロイドの窒素(ベンゾイミダゾールまたはベンゾトリアゾールのN−3)のCYP17のヘムへの配置を指示する。5および9(426nM)との酵素コンプレックスのソーレー最大のためのピーク位置はCYPシステムに対する窒素リガンドの結合についての利用できるデータと一致し、そして我々の他の17−アゾリルCYP17阻害剤でのデータとも一致する(Njar et al,Bioorg.Med.Chem.Lett.1996,6,2776−2782;and J.Med.Chem.1998,41,902−912)。ベンゾアゾール窒素とCYP17のヘム鉄との相互作用は、5および9の結合親和力が17−イミダゾール基を有するより小さい立体を要求する16と同じであるため17位におけるバルクトラランスを示唆する。
【0040】
テストした2つの17−ジアジンのうち、500nMのIC50値を持つ17−ピリミジン15は17−ピラジン(IC50=3810nM)よりも約8倍強力である。ベンゾアゾール場合のように、この結果は17−複素環の電子的性格が阻害活性に影響することを示唆する。最後に、ケトコナゾールおよびアビラテロンについて同じアッセイにおけるIC50値が評価される(表1)。このシリーズにおいて最も強力である17−ベンズイミダゾール5は、これら化合物よりもCYP17阻害においてそれぞれ約4倍および約5倍の改善を示すが、しかし16より強力ではない。
【0041】
インビトロにおけるヒト5α−レダクターゼイソエンザイムタイプ1および2の阻害:ある種のCYP13阻害剤はヒト5α−レダクターゼ酵素を阻害することができるとの以前の発見を基にして、我々はCYP17阻害剤の新しいシリーズを簡略に評価した。化合物5,6,9,10および参照としてのフィナステライドの阻害活性は、Hartman et al,“Synthesis and evaluation of 2’−substituted 4−(4’−carboxy−or 4’−carboxy methyl benzylidene)−N−acylpiperidines:Highly potent and in vivo active steroid 5α−reductase type 2 inhibitors”,J.Med.Chem.2002,45,3406−3417に記載されているように、DU−145細胞株(ヒトタイプ1酵素)およびヒトBPH組織のホモジネート(ヒトタイプ2酵素)を使用して決定される。いくつかの化合物についてIC50値または濃度10μMにおける阻害パーセント値が表1に提供されている。化合物6のみがタイプ1および2酵素の両方に強力な阻害を示すが(それぞれIC50値=770および480nM)、それはフィナステライド(それぞれIC50値=60および2nM)よりも数倍弱い。
【0042】
LNCaPおよびPC−3Aアンドロゲン受容体結合アッセイ:以前我々は我々のCYP17阻害剤のいくつかは変異および野生タイプARに対し強力な抗アンドロゲンであることを証明したので(Long et al.and Njar et al,J.Med Chem.1998,41,902−912前出)、CYP17阻害剤のこのシリーズがこれら受容体へ結合する能力を評価することに関心があった。AR競合は、変異ARを発現するアンドロゲン感受性LNCaP細胞、および野生タイプAR(PC−3ARと命名された)で安定して形質転換されたアンドロゲン非依存性PC−3細胞中の標識したR1881(〔3H〕−R1881)を用いて決定される。化合物5,6,14および15はナノモル濃度範囲において投与量応答態様でARの両方のタイプへの結合に対して標識したR1881と効果的に競合する(数値示さず)。それぞれIC50値384,242,336および374nMを持つ化合物5,6,14および15(表1)対野生タイプARは、臨床的に使用される抗アンドロゲンであるフルタミド(IC50=10,985nM)よりも29倍ないし45倍強力である。表1に示すように、5および6の変異ARに対する結合親和力は現在使用されている抗アンドロゲンであるカソデックスに匹敵するが、しかし再びフルタミドよりすぐれている。しかしながらフルタミドの生物学的活性はもっと強力なAR拮抗剤である代謝物ヒドロキシフルタミドから主として誘導される。
【0043】
LNCaP変異AR仲介転写に対する剤の効果:次に我々は化合物5および6はARアンタゴニストかまたアゴニストとして作用しているのかを質問した。アンドロゲン−調節転写活性化について研究は、プロバシンルシフェラーゼレポーター構築物AARZ−Luc(ルシラーゼ活性アッセイ)で過渡的に形質転換したLNCaP細胞中で実施される(Kim et al,“Synergism of cytoplasmic Rinase in IL6−induced ligand−independent activation of androgen receptor in prostate cancer cells”,Oncogene,2004,23:1838−1844;and Zhang et al,“A Small composite probasin promoter confers high levels of prostate−specifec gene expression through regulation by androgens and glucocorticoids in Vitro and in Vivo”,Endocrinology,2000,141:4698−4710)。化合物5,6またはカソデックスは各自0.1および10.0μMにおいてルシフェラーゼ活性に対して効果を持たないが、ルシフェラーゼ発現は18時間の1.0nM DHTでの処理後に約99.6倍増大される(図1)。さらに1.0nM DHTへの曝露によって誘発されたルシフェラーゼ発現は5,6およびカソデックスにより濃度依存態様でそして同様な態様で減少する(図1)。合わせてこれらの結果は、カソデックスと同様に化合物5および6はARアゴニストまたは部分的アゴニスト活性を持たず、そして強力な純粋なアンドロゲンアンタゴニストと考えることができることを示唆する。野生タイプARを発現するPC−3AR/LV細胞で化合物をテストしなかったが、これらも同じ態様に挙動するらしい。我々は以前我々のCYP17阻害剤のいくつかはフルタミドよりもカソデックスにもっと匹敵したことを示し(Long et al,前出)、そしてこのことはこれら新しい化合物でもそのとおりであるらしい。一般に、我々の新規化合物は両方のARタイプと強力に相互作用し、化合物は野生タイプまたは変異ARを発現する腫瘍を有する患者、または増幅したAR発現を有する患者の処置のために有用であり得ることを指示する。
【0044】
インビトロにおいてベンゾアゾールのLNCaPおよびLAPC−4前立腺癌細胞生育に対する効果:1nM DHTによって刺激された変異LNCaP細胞の増殖を阻害する化合物5および6の能力が調べられる。DHTのこの濃度は、ビヒクル処理細胞に比較して約2倍LNCaP細胞増殖を刺激した(図2A)。図2Aに示すように、化合物5および6は、各自それぞれIC50値6.0および1.8μMで投与量応答様式でDHT−誘発LNCaP細胞増殖を阻止する。カソデックスが陽性対照として使用され、そしてDHT−誘発LNCaP細胞増殖の同様な阻害を示す(図2A,IC50=8.6μM)。10nM DHTでのアンドロゲン感受性LAPC4前立腺癌細胞株の処理は、驚くべきことに細胞増殖を有意に誘発しない(図2B)。他の研究者も、アンドロゲンに対するLAPC4細胞の応答はLNCaP細胞において観察される程顕著でないことを報告している(Thompson et al,“Androgen antagonist activity by the antioxidant moiety of vitamin E,2,2,5,7,8−pentamethyl −6−chromanol in human prostate cancer cells”,Molec.Cancer Thera.2003,2,797−803)。しかしながら、化合物5,6およびカソデックスは各自LNCaPと同様にこの細胞株の投与量応答阻害を示す(図2B)。LAPC4細胞増殖の阻害強度の順序は6>5>カソデックスであり、IC50値はそれぞれ1.0,3.2および10μMである。合わせてこれらの結果は、5および6は上に記載したそれらのアンドロゲン受容体結合および活性化性質に相関して、細胞増殖の刺激においてDHTの作用をブロックするように作用していることを示唆する。化合物5および6は今日まで記載された最も強力な抗アンドロゲンである。
【0045】
5および6の薬物動態学および5の代謝:
化合物5および6についての雄性SCIDマウス中の薬物動態学的性質が他の阻害剤についての最近記載された操作に従って研究される(Nnane et al,“Pharmacokinetic profile of 3β−hydroxy −17−(1H−1,2,3−triazol−1−yl)androsta−5,16−diene(VN/87−1),a potent androgen synthesis inhibitor in mice”,J.Steroid Biochem.Molec.Biol.2001,71,145−152;and Handratta et al,“Potent CYP17 inhibitors:improved syntheses,pharmacokinetics and anti−tumor activity in the LNCaP human prostate cancer model”,J.Steroid Biochem.Molec.Biol.2004,92,155−165)。結果は表2および図3−5に要約されている。逆相HPLCにおいて5(保留時間=21.6min)は内部標準(16,rt=11.5min)、代謝産物(rt=17.3min)およびマウス血漿中の他の内因性化合物から良く解像される(図3)。5に対する較正曲線は直線で、そしてその検出限界は100ng/mlである。HPLCアッセイがバリデートされ、そしてマウス血漿中の5のモニターのために使用される。
【0046】
皮下投与後、5の血漿濃度は平均半減期約44.17分と、排出速度定数56.5min−1をもって指数的に低下する。化合物5は全身循環から1986.14ml/h/kgの速度で排除され、そして投与8時間後検出されなかった。5の皮下投与後の血漿濃度に基づく計算した非コンパートメント薬物動態学的パラメータが表2に示されている。雄性SCIDマウスへの5の皮下投与(50および100mg/kg)後の血漿濃度一時間曲線が図4に示されている。5の皮下投与後、マウス中の観察された血漿濃度は投与30.0分でピークレベルに達する。化合物5は皮下部位から良く吸収され、そして皮下投与後の血漿濃度対時間プロフィルの曲線下の面積AUCは、投与量を50から100mg/kgへ変えた時投与量に比例して増加する。さらに、排除半減期および平均滞留時間は、5の投与量を50から100mg/kgへ変える時比較的コンスタントである(表1)。これらの結果は5の薬物動態プロフィルは投与量依存性であることを指示する。
【0047】
図5は、有意義な量の極性代謝産物(保留時間17.3min、図3を見よ)が5から生成し、そしてインビボ薬物動態学的研究の間血漿中に存在することを示す。この代謝産物の最高量は投与約2時間後に達した67.72%である。この代謝産物は化合物6と同じ保留時間を示す。この代謝産物は一時的にLC−MSと同定され、その分子質量(m/z391=M+H+)は3−オキソ−△5,16−テトラヒドロ化合物5(すなわち、17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−3−オン)の構造と一致する。この代謝産物は、5から3β−OH→3−オキソの酸化、次いで△5および△16二重結合の還元(レダクターゼ)を経て生成し得る。類似の代謝産物は、雄マウス中の密接に関連するステロイド17−イミダゾールにおいて(3β−OH→3−オキソ、次いで△5二重結合の異性体化の結果として生成した)以前に同定された(Handratta et al,前出)。
【0048】
5,すなわち17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−3−オンの主要な代謝産物はトランス−アンドロステロンから合成することができる。スキーム3を見よ。これら類似の活性を以っていることが予期される。
【0049】
マウス中の6のインビボ薬物動態は、比較的低いCmaxおよび著しく高い排出速度(図4および表2)のため、化合物5と似ていない。加えて、我々は、化合物5についての我々の観察とは対照的に、血漿中に化合物6の代謝産物を検出しなかった。
【0050】
SCIDマウスに生育させたLAPC4キセノグラフトに対する5および6の効果:印象的な多数のインビトロ生物学活性、すなわちCYP17の強力な阻害、強い抗増殖前立腺癌細胞活性および抗アンドロゲン活性を基にして、5および6がアンドロゲン依存性LAPC4ヒト前立腺癌キセノグラフトモデルにおいてインビボ抗腫瘍有効性研究のために選択される。
【0051】
第1の実験において、SCIDマウスのよく確立されたLAPC4前立腺癌腫瘍の生育に対する化合物5および6の効果が決定され、そして参照処置として去勢が使用された。腫瘍保有マウスは5または6の2通りの投与(0.15mmol/kg 1日1回または0.15mmol/kg 1日2回)を受けるように割当てられた(n=5/グループ)。腫瘍体積が毎週測定され、そしてビヒクルを受けているか去勢マウスの対照と比較される。
【0052】
去勢は対照と比較して最終腫瘍体積の55%減少へ導く(図6)。5の0.15mmol/kg 1日1回および0.15mmol/kg 1日2回は、ビヒクル処理対照動物の腫瘍に比較してそれぞれ平均最終腫瘍体積の41%および86.5%減少をもたらした(図6)。5で処理したマウスのすぐれた腫瘍発育阻害とは対照的に、化合物6で処理したマウスは低投与量において効果がないか、または対照と比較して腫瘍発育の刺激を示す(図6)。この化合物はインビトロにおいてPCA細胞生育の阻害において非常に有効であり、そして高度に抗アンドロゲン性であったから(図1を見よ)、化合物6がインビボにおいてLAPCA腫瘍細胞の発育を阻害することができないことは特に失望的である。5および6のインビボ抗腫瘍有効性の高度に有意な不均衡な二つの化合物の薬物動態学的性質の差に安易に帰せしめることはできない。これら二つの密接に関連した化合物のインビボ抗腫瘍有効性の劇的な差に含まれる理由は現時点では未知である。しかしながら、6が動物においてアンドロゲン受容体の強力なアゴニストであり得る代謝産物へ変換され、そのため腫瘍生育刺激を生ぜしめることへ帰せしめることができる。この研究の間、すべてのマウスは週1回体重測定した。すべての処理グループの体重は僅かに増加し、そして対照グループで観察されたのと類似であった。すべての動物は健康に見え、そして有害効果は観察されず、化合物は有意な毒性を持たないことを示唆する。
【0053】
第2のインビボ実験は、SCIDマウスに生育しているLAPC4前立腺癌細胞の成育を阻害する5および以前強力なCYP17阻害剤/抗アンドロゲンと同定された16(Grogoriyer et al.and Njar et al.J.Med.Chem,1998,41,902−912前出)の能力をテストし、そして去勢が参照処置として使用される。この実験において処理はマウスがホルモン依存性LAPC4細胞を皮下接種され、そして去勢されるかまたは5または16を1日2回皮下注射された日から始まる。図7は治療21週間の間の腫瘍の出現およびサイズに対する種々の処置の効果を示す。
【0054】
すべての他のグループは、触知可能なそして測定可能な腫瘍へ11週に発達した16で処置したグループ(0.5mmol/kg 1日2回)を除いて、治療の10週において触知可能で測定可能な腫瘍へ発展する。対照マウス中の総腫瘍体積は、大きい腫瘍のため、マウスが犠牲にされる時処置の14週に亘って8倍増加する。このように他のグループの腫瘍体積は処置の14週において対照グループに匹敵する。去勢したマウスの腫瘍体積はたった4.1倍(対照に比較して約50%減少)増加し、16で処置したマウスで観察された3.7倍増加(対照に比較して53.8%減少)に似ている。5で処置したマウスにおいては(0.15mmol/kg 1日2回)、腫瘍体積はたった0.5倍だけ増加し、これは対照マウスに対して93.8%減少を表す(P=0.00065)。16週において、化合物5で処置した動物の平均腫瘍体積は、測定し得る腫瘍が出現する時の10週におけるそれらの平均腫瘍体積よりも低い(殆ど無視し得るかまたは静止している)ことが見られる。さらに、5は、それぞれP=0.005および0.05において16または去勢に比較して腫瘍に対して有意な阻害効果を生ぜしめる。一般に、対照、去勢および化合物16処置マウスの腫瘍は急速に成長するが、他方5処置マウスの腫瘍は非常に遅く、そして二段階で成長する(図7)。化合物5は最も効果的な剤であり、そして腫瘍成長の阻害において去勢よりも有意にもっと効果的である。16はCYP17阻害において5より6倍強力であるが、5はすぐれたインビボ抗腫瘍活性を発揮することは興味深い。この現象に責任ある理由は現時点では未知であるが、しかし一部は5のより良い薬物動態学性質であろう。
【0055】
体眠している化合物5処理前立腺癌細胞(図7,16週を見よ)が5のもっと低い投与量で生育できるかどうかを決定するため、その投与量を16〜19週から0.15mmol/kg 週3回(投与量の78.6%低減)へ減らした。化合物の減らした投与量でのこの処置の期間、腫瘍は発育を回復した(図7)。この3週間の間の後、通常の投与量での薬物処置へ復帰し、そして腫瘍発育は遅くなり、プラトーへ達する。これらのデータはこの処理の細胞静止的性格を示唆し、そして抗腫瘍効果を達成するために連続的投与の必要性を推論させる。
【0056】
実験の終りにおいて、腫瘍および5−処置マウス中の5のレベルが決定される。最終投与1時間後の腫瘍、睾丸、および肝臓内のHPLCによる5のレベル(図7中の挿入部分)が決定される。興味あることに、代謝産物の少量(5に対して約15%)が肝臓組織内にのみ検出される。この代謝産物は血漿中に観察された代謝産物(前を見よ)と同じ保留時間を持つ。5の39.0±8.4μg/mg組織の最高濃度が皮下腫瘍において計測される。肝臓および睾丸中の濃度はより低いがしかし検出可能である。腫瘍中の5のレベルは血漿中で測定されたレベルよりも有意に高い。これは実験期間を通して化合物の蓄積の結果であり得る。このように5による腫瘍成長の阻害は一部は腫瘍キセノグラフト中のより高い濃度が前立腺癌細胞に細胞毒性/細胞静止効果を発揮するものと説明することができる。前立腺癌細胞のケトコナゾール(適度なCYP17阻害剤)の可能な直接の細胞毒効果を示唆する証拠があることを言及しなければならない。加えて、睾丸中の5の蓄積は動物内のテストステロン合成の阻害を可能となし得る。
【0057】
LAPC4はアンドロゲン依存性であることが良く確立されているけれども、これらの細胞はアンドロゲン非依存性になることができ、そしてそうなることで患者中の前立腺癌の発展を真似する適当なモデルを代表する(Chen et al.上出、およびKline et al.“Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice”,Nat.Med.1997,3,402−408)。図7に示すように、我々はこの現象を繰り返すことができる。さらに我々の結果は、16による処理または去勢は一定期間(アンドロゲン依存フェーズ)腫瘍成長を効果的に抑止するが、しかし腫瘍がインタクトな対照マウスにおけるのと全く同様に急速に成長するから、その後は非効果的になった(多分アンドロゲン非依存フェーズの結果)ことを示している。5で処理したマウス中の腫瘍の成長は処理期間全体を通じて強力に抑制される。このことは、5はアンドロゲン非依存性前立腺癌に効果を有することを示唆する。この化合物による処置はLAPC4腫瘍細胞を長期間アンドロゲン依存性に留め、それ故抗アンドロゲン治療に応答性とすることを可能することがもっともらしい。
【0058】
進んだそして再発したPCA中のARの上方調節および関与を証明する最近の研究(Mohler et al.and Chen et al.前出)は、PCAを処置するための薬物の開発のための標的としてアンドロゲン受容体に関心を再び呼び戻した(Tindall et al,“Symposium on androgen action in prostate cancer”,Cancer Res.2004,64,7178−7180)。その潜在的性質のため、5はすぐれた候補になり得る。
【0059】
結論:
データは、△16ステロイドのC17置換基をCYP17の強力な阻害剤および強力なARアンタゴニストを製造するために修飾する我々の以前の着想を補強する。17−ベンズイミダゾール5および6はCYP17のヘム鉄に配位することを示し、これはそれらの酵素阻害活性、ARアンタゴニズム、および前立腺癌細胞成長に一部責任ある性質である。驚くべきことに、これら化合物はそれらの抗腫瘍活性において非常に異なっており、5はLAPC4腫瘍キセノグラフトの成長において著しく抑制を生ぜしめ、対照的に6(0.15mmol/kg 1日2回)は腫瘍成長を促進する。本研究は、5はヒト前立腺成長の強力な阻害剤であり、そして去勢よりも著しく効果的である説得力ある証拠を提供する。これは前立腺癌腫瘍に対して去勢よりももっと効果的であるという点においてインビボ抗腫瘍効果を示すCYP17阻害剤/抗アンドロゲン剤の最初の例である。これらの印象的な生物学的活性は5をしてヒトの前立腺癌の処置のための可能性ある薬物としてさらなる開発のための強力な候補にならしめる。ベンズイミダゾール基を含んでいる化合物5のすぐれた抗腫瘍活性はベンズイミダゾールをして好ましい基とならしめる。しかしながら、上に議論した5の類縁体は関連した活性を持つことが予期され、そして本発明に含まれる。
【0060】
実験の部
化学:一般的操作およびテクニックは以前の報告と同じである(Njar et al.J.Med.Chem.1998,41,902−912)。赤外スペクトルはCHCl3溶液を使用してパーキンエルマー1600FTIR分光光度計で記録される。高解像度質量スペクトル(HRMS)は3−テルサフィネガンFTMS−2000FF質量分析計、EIモード(Ohio State University,Department of Chemistry)で測定される。キー標的化合物の純度基準として、我々は化合物均質性を指示するHPLCクロマトグラフィーを伴った高解像度質量スペクトルデータを提供した。低解像度質量スペクトル(LRMS)はフィネガンLCR−MSで測定される。融点はFischer Johns融点装置で測定され、そして補正されない。デヒドロエピアンドロステロンおよびデヒドロエピアンドロステロンアセテートはAldrich Milwaukee,WIから購入した。トリブチルスタニルピリミジンおよび2−トリブチルスタニルピラジンはFrontier Scientific,Inc.,Logan,UTから購入した。
【0061】
3β−アセトキシ−17−クロロ−16−ホルミルアンドロスタ−5,16−ジエン(2):この化合物は3β−アセトキシアンドロスト−5−エン−17−オン(1)から以前記載し、スペクトルおよび分析データを提供したように製造した(Njar et al,J.Med.Chem.1998,41,902−912)。
【0062】
3β−アセトキシ−17−(1H−ベンズイミダゾール−1−イル)−16−ホルミルアンドロスタ−5,16−ジエン(3):乾燥DMF(20ml)中の3β−アセトキシ−17−クロロ−16−ホルミルアンドスタ−5,16−ジエン(2,2.5g,6.65mmol)と、ベンズイミダゾール(2.35g,19.9mmol)と、そしてK2CO3(2.76g,23.9mmol)の混合物をアルゴン下1.5時間約80℃においてかきまぜる。室温へ冷却後、反応混合物を氷水(250ml)へ注ぎ、生成する沈澱を濾過し、水で洗い、乾燥して粗製の汚い白色固体(約2.9g)を得る。FCC(石油エーテル/EtOAc/Et3N=6:4:0.3)は純粋な化合物3を2.3g(88.7%)を与える。
【0063】
【化7】
【0064】
3β−アセトキシ−17−(1H−ベンズイミダゾール−1−イル)アンドロスタ−5,16−ジエン(4):
乾燥ベンゾニトリル(10ml)中の3β−アセトキシ−17−(1H−イミダゾール−1−イル)−16−ホルミルアンドロスタ−5,16−ジエン(3,2.04g,4.45mmol)の溶液を活性炭上の10%パラジウム(1.02g,3の50重量%)の存在下5時間還流した。室温へ冷却後、触媒をセライトパッドで濾過して除いた。濾液を蒸発し、残渣をFCC(石油エーテル/EtOA/Et3N=7.5:3:0.5)によって精製した。純粋な化合物4を1.41g(73.8%)を与えた。
【0065】
【化8】
【0066】
3β−ヒドロキシ−17−(1H−イミダゾール−1−イル)アンドロスタ−5,6−ジエン(5):アセテート4(1.3g,3.02mmol)を不活性Ar雰囲気下メタノール(20ml)に溶かし、生成した溶液を10%メタノール性KOH(8ml)で処理した。混合物を室温で1.5時間かきまぜ,減圧下約40℃で10ml体積へ濃縮した。この溶液へ氷水(300ml)を注ぎ、生成する白色沈澱を濾過し、水洗し、乾燥した。EtOAc/MeOHからの結晶化は5(1.10g,94%)を与えた。
【0067】
【化9】
【0068】
17−(1H−ベンゾイミダゾール−1−イル)アンドスタ−4,16−ジエン−3−オン(6):化合物5(660mg,1.70mmol),1−メチル−4−ピペリドン(2.5ml)およびトルエン(4ml)の混合物から約10mlを留去した。次にアルミニウムイソプロポキサイド(521mg,2.55mmol)を加え、混合物をAr下4時間還流した。冷後混合物をEtOAc(50ml)で希釈し、5%NaHCO3(×3)および食塩水(×2)で次々に洗い、乾燥(Na2SO4)した。溶媒を蒸発し、粗生成物をFCC(CH2Cl2/EtOAc=25:1)で精製し、標題化合物6(544mg,82%)を得た。
【0069】
【化10】
【0070】
3β−アセトキシ−17−クロロ−16−ホルミルアンドロスタ−4,16−ジエン(2)と、ベンゾ−1H−1,2,3−トリアゾールおよびK2CO3との反応:3β−アセトキシ−17−(ベンゾ−2H−トリアゾール−2−イル)−16−ホルミルアンドロスタ−5,16−ジエン(7a)および3β−アセトキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)−16−ホルミルアンドロスタ−5,16−ジエン(7b):乾燥DMF(20ml)中の化合物2(2.5g,6.65mmol)、ベンゾトリアゾール(2.35g,19.9mmol)およびK2CO3(2.76g,23.9mmol)の混合物をAr下45分約80℃においてかきまぜた。室温へ冷却後、反応混合物を氷水(250ml)へ注ぎ、生成する沈澱を濾過し、水で洗い、乾燥して粗製の汚れた白色固体を得た。FCC(石油エーテル/EtOAc=4:1)は最初にマイナーな生成物として3β−アセトキシ−17−(ベンゾ−2H−1,2,3−トリアゾール−2−イル)−16−ホルミルアンドスタ−5,16−ジエン(7a,0.3g,9.8%)を与えた。
【0071】
【化11】
【0072】
同じ溶媒系でのさらなる溶出は主要生成物、3β−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)−16−ホルミルアンドスタ−5,16−ジエン(7b,2.3g,75.4%)を与えた。
【0073】
【化12】
【0074】
3β−アセトキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−5,16−ジエン(8):乾燥キシレン(40ml)中のビス(トリフェニルホスフィン)ロジウム(I)カルボニルクロライド(303mg,0.438mmol)と、1,3−ビス(ジフェニルホスフィノ)プロパン(394mg,0.954mmol)の混合物をAr下80℃で15分間黄色沈澱が生成するまで攪拌した。化合物7b(1.71g,3.72mmol)を加え、混合物をAr下18時間還流し、減圧下濃縮した。粗生成物はFCC(石油エーテル/EtOAc/Et3N=8.9:1.0:0.1)は純粋な化合物8の1.2g(74.7%)を与えた。
【0075】
【化13】
【0076】
3β−ヒドロキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−5,16−ジエン(9):3β−アセトキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−5,16−ジエン(8,700mg,1.62mmol)を用いて、化合物5について記載した方法を追従した。EtOAc/MeOHからの再結晶は標題化合物9(600mg,95%)を与えた。
【0077】
【化14】
【0078】
17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−4,16−ジエン−3−オン(10):β−ヒドロキシ−17−(ベンゾ−1H−1,2,3−トリアゾール−1−イル)アンドロスタ−5,16−ジエン(9,500mg,1.28mmol)を使用し、化合物6について記載した方法に従った。粗生成物のFCC(CH2Cl2/EtOH=50:1)による精製は標題化合物10(420mg,84.4%)を与えた。
【0079】
【化15】
【0080】
デヒドロエピアンドロステロン−17−ヒドラゾン(12):デヒドロエピアンドロステロン(11,3.5g,12.2mmol)をエタノール(60ml)に溶解し、得られた溶液をヒドラジンヒドラート(2.37ml,0.049mmol)で、次に水0.25ml中の硫酸ヒドラジン(7.9mg,0.061mmol)の溶液で処理した。混合物を室温で12時間かきまぜ、次に氷水中へ注いだ。生成する沈澱を濾取し、水洗し、乾燥し、標題化合物12の白色結晶を得た。
【0081】
【化16】
【0082】
17−ヨードアンドロスタ−5,16−ジエン−3β−オール(13):乾燥THF(144ml)および乾燥EtOH(72ml)のヨウ素(12.16g,0.0203mol)のかきまぜ溶液を氷欲で0℃に冷却し、そして溶液を1,1,3,3−テトラメチルグアニジン(6.72ml,6.27g,0.054mol)で処理した。THF(81ml)中の化合物12(3.0mg,9.9mmol)の溶液をヨウ素溶液へ反応温度を0℃に保って2時間を要して滴下した。次に反応混合物を真空下濃縮し、氷欲で冷却し、真空下室温で乾燥し、黄色固体(13,3.65g,92.4%)を得た。
【0083】
【化17】
【0084】
3β−ヒドロキシ−17−(2−ピラジル)−アンドロスタ−5,16−ジエン(14):乾燥DMF(10ml)中の17−ヨードアンドロスタ−5,6−ジエン−3β−オール(13,6.5g,1.257mmol)の溶液と、テトラキス(トリフェニルホスフェート)パラジウム(Pd(PPh3)4)(71.6mg,0.062mmol)と、(2−トリブチルスタニル)ピラジン(774.6mg,2.099mmol)の混合物を120℃で20時間加熱した。冷後混合物を冷水(50ml)で希釈し、EtOAc(30ml×3)で抽出した。合併したEtOAc抽出液を食塩水および水で洗い、Na2SO4上で乾燥し、濃縮して褐色固体を得た。この粗生成物をカラムクロマトグラフィー(FCC,石油エーテル/EtOAc/Et3N=3:2:0.15)によって精製し、14(66mg,15%)を得た。
【0085】
【化18】
【0086】
3β−ヒドロキシ−17−(5−ピリミジル)−アンドロスタ−5,6−ジエン(15):乾燥DMF 10mlに溶解した(5−トリブチルスタニル)ピリミジン(1.0g,2.710mmol)と、(Pd(PPh3)4)(92.88mg,0.0804mmol)を使用し、14について上で記載した13(0.645g,1.623mmol)の反応と、続いてのFCC(石油エーテル/EtOAc/Et3N)=3:2:0.15)による精製は、3β−ヒドロキシ−17−(5−ピリミジル)−アンドロスタ−5,16−ジエン(15,44mg,10%)を与えた。
【0087】
【化19】
【0088】
CYP17のインビトアッセイ:化合物のインビトロCYP17阻害活性は、酵素源としてインタクトなP450c17発現E.coli(Grigoryev,前出)を利用する我々の急速酢酸放出アッセイ(AARA)を使用して評価される。これは〔21−3H〕−17α−ヒドロキシプレグネノロンを基質として使用することを含み、そしてCYP17活性は基質のC−21側鎖の分裂の間生成したトリチウム化酢酸の量によって測定される。これは、この分野の研究者(Grigoryev,前出)によって使用されたHPLC分析操作に匹敵する。IC50値は適切の範囲にわたって阻害%対阻害剤濃度に関するプロットから直接得られる。各化合物は最大5濃度においてテストされる。アッセイは三系列で行われ、そしてIC50値は三系列実験の平均値として報告される。標準差は平均値の±5%である。
【0089】
ヒト5α−レダクターゼタイプ1および2アッセイ:化合物および参照としてのフィナステライドの阻害活性は、Hartmannらによって記載された操作(Picard et al,“Synthesis and evaluation of 2’−substituted 4−(4’−carboxy−methylbenzylidene)−N−acylpiperidines:Highly potent and in vivo active steroid 5α−reductase type 2 inhibitors”,J.Med,Chem.2002,45,3406−3417)に従い、DU145細胞株(ヒトタイプ1酵素のため)およびヒト前立腺ホモジネート(タイプ2酵素のためのBPH組織)を使用して決定される。10μMの濃度における阻害パーセントか、またはもっと強力な化合物の場合、IC50値が決定される。
【0090】
競合アンドロゲン受容体(AR)結合およびルシフェラーゼアッセイ:AR結合/競合アッセイ:24ウエルマルチウエルディッシュウエルをポリ−L−リジン(0.05mg/ml)で5分間コートし、乾燥し、無菌蒸留水でリンスし、2時間乾燥する。LNCaP ARおよび野生タイプARへのR1881結合の動力学を決定するため、LNCaPおよびPC3AR細胞をステロイド不含倍地中の24ウエルディッシュにプレートし、結合を許容する。翌日倍地を、0.1%BSAを補給し、〔3H〕R1881(0.01−10nM)を含有し、そして非特異的結合を決定するため冷DHTの200倍過剰が存在または不存在で、そしてプロゲステロンおよびグルココルチコイド受容体を飽和させるための1μMトリアムシノロンを含む血清不含、ステロイド不含RPM1で置換する。37℃において2時間のインキュベーション期間後、細胞を氷冷DPBSで2回洗い、そして0.5%SDSおよび20%グリセロールを含有するDPBS中に可溶化する。抽出物を除去し、そして細胞結合放射活性をシンチレーションカウンター中でカウントする。Graphpad Prismソフトウエアを用いて非直線回帰によってKdおよびBmax決定を含むデータを解析する。両方の細胞株中のARを殆ど飽和させるのに必要な濃度を確立し、そして受容体から〔3H〕R1881(5.0nM)を移動させるテスト化合物(0.1nM−10μM)の能力を上に記載したように決定する。各化合物のIC50値はGraphpad Prismソフトウエア(GraphPad Software,Inc,San Diego,CA)での非線形回帰によって決定される。
【0091】
ルシフェラーゼトランス活性化アッセイ:転写活性化アッセイは、少しの修飾を加えて前出のKimらにより記載された方法によって実施される。プロバシンルシフェラーゼレポーター構築物ARR2−Lucは、Vanderbilt University Medical CenterのDr.R.MatsusiRから好意に提供された最小プロバシンプロモーターARR2(Endocrinology 2000,141,4698−4710)をPGL3−エンハンサーベクター(Promegd)のポリクローナルリンカー領域に挿入することによって発生させる。概略すると、ポリ−L−リジンをコートした24−ウエルプレートに発育させたLNCaP細胞が5%活性炭処理FBS(Hyclon)を含有するフェノールレッド不含RPMI1640倍地中のARR2−Lucで形質転換される。形質転換24時間後、細胞はDHTありまたはなしの新鮮なフェノールレッド不含血清不含RPMI1640倍地および阻害剤と18時間インキュベートされる。ルシフェラーゼ活性は製造者の指示書(Promega)に従って二重ルシフェラーゼアッセイを使用して3系列で測定される。結果は誘発倍率、すなわち対照ルシフェラーゼ活性で除した処理細胞の相対的ルシフェラーゼ活性で表される。
【0092】
細胞培養および生存性:LNCaP細胞は10%FBSおよび1%ペニシリン/ストレプトマイシン溶液を補給したRPMI1640倍地中で発育させる。細胞増殖に対する新規化合物の効果を決定するため、細胞は実験開始前3日間ステロイド不含培地へ移される。ステロイド不含培地は、5%デキストランコートした、活性炭処理血清と、そして1%ペニシリン/ストレプトマイシン溶液を補給したフェノールレッドフ含RPMIからなっていた。発育研究は細胞(3×104)を24ウエルマルチウエルデイッシュ(Corning Inc.,Corning,NY)中にプレートすることによって実施される。24時間の結合期間後、培地を吸引し、そしてビヒクルまたはDHTの指示された濃度(1nM)および化合物(0.1μM−10μM)を含有するステロイド不含培地で置換される。対照ウエルはビヒクル(エタノール)で処理される。この培地は3日毎に交換され、そして生存細胞の数が7日目にWST−1〔4−〔3−(4−ヨードフェニク)−2−(4−ニトロフェニル)−2H−5−テトラゾリオ〕−1,3−ベンゼンジスルホネート〕アッセイによって比較される。上述の時間細胞のインキュベーション後、10%WST−1溶液が各ウエルへ加えられ、そして37℃で3時間インキュベートされる。インキュベーション後、プレートを僅かに振とうし、そして直ちに査定型マルチウエル分光分析計で450nmにおいて読み取られる。すべての結果は3ウエルの最小値の平均値で表される。追加の対照は細胞なしの培地のみからなる。
【0093】
薬物動態学的研究:すべての動物実験は、メリーランド大学医学部の動物ケア委員会のガイドラインおよび承認に従って実施される。NCI,FredericR,MD,USAから得た体重20〜22gの雄SCIDマウス(8〜10週令)は約25℃,50%相対湿度および12時間明サイクルおよび2時間暗サイクルにコントロールされた環境に維持され、そして食物および水に自由にアクセスすることが許容される。化合物5および6は水中の40%β−シクロデキストリン中に処方され、1回の皮下投与量が与えられる。薬物投与6時間までの種々の時間において動物が犠牲にされ、そして軽いハロタン(Ayerst,New York,NY,USA)麻酔のもとで心臓穿刺によって血液が収集された。
HPLC分析:ステロイドおよび適切な内部標準のクロマトグラフィー分離および定量は、以前記載されたように、ペリクルC18で包装されたWatersガードカートリッジによって保護されたWaters Novapak C18カラム(3.9×150mm)上のWaters Novapak C18カラム上の逆相HPLC法によって達成される。概略すると、この研究に使用されるHPLCシステムは、Waters溶媒送達システム、Waters717plusオートサンプラーおよび242.7nmで作動するWaters996発光ダイオード列デテクターと組合せたWatersコントローラー(Milford,MA)よりなる。移動相組成は水/MeOH/CH3CN(35:35:30 v/v/v+Et3N 200mlおよび移動相1000mlあたりNH4OAc 0.77g)であり、流速は1.0ml/minである。HPLC分析は環境温度で実施され、そしてデータ取得およびマネージメントはWatersミレニアムクロマトグラフィーマネージャーで達成される。
【0094】
サンプル調製:マウス血漿(200μl),5または6およびVN185−1(内部標準、100μg/mlの10μl)を含んでいる試験管がボルテッスミキサーを用いてジエチルエーテル(2×2ml)で3分間抽出され、そして3000gにおいて5分間遠心される。ゆるやかな空気流のもとで有機層が蒸発乾固される。残渣は移動相の部分標本(100μl)中に再構成され、HPLC分析前に0.2μmテフロンフィルターを用いて濾過される。
【0095】
較正曲線およびHPLCアッセイバリデーション:血漿および組織中の5および血漿中の6のための較正曲線は、0.1−100.0μg/mlの最終濃度を与えるように、未処置動物からの血漿(200μl)および組織調製物(200μl)を含んでいる抽出チューブ(2系列)中へ化合物の変化する量をスパイクすることによって構築される。適切なブランク抽出チューブも調製され、そして内部標準の部分標本が最終濃度5μg/mlを与えるように各抽出チューブへ加えられる。この較正サンプルは上に記載したサンプル調製操作を受ける。再構成した抽出液を部分標本(50μl)がHPLCシステムへ注入され、そして各検体のピーク面積の内部標準のピーク面積に対する比が5または6の濃度に対してプロットされる。アッセイの精密度および正確性はブランク血漿中の阻害剤の既知濃度の範囲から決定され、そしてHPL操作を受ける。研究は別々に3回繰り返される。
【0096】
データ解析:薬物動態計算は以前記載したとおり実施される。非コンパートメント薬物動態計算はWinNOnlin(Scientific Consulting Inc.)を用いて実施される。Windowsバージョン1.0のためのSigmastat上の変動のワンウエイ解析が異なる処理グループを95%信頼性レベルにおいて比較するために使用される。Bonferroni post−hocテストが有意性の決定のために使用される。0.05以下のP−値が統計学的に有意と考えられる。
【0097】
インビボ抗腫瘍研究(LAPC−4前立腺癌キセノグラフト):すべての動物実験はメリーランド大学医学部の動物ケア委員会のガイドラインおよび承認に従って実施される。Nationasl Cancer Institute−Frederick Cancer Research and Development Center(Frederick,MD)から購入した4〜6週令の重篤結合免疫不全(SCID)マウスが光および湿度の制御された条件下の病原体不存在環境に収容され、そして食物および水への自由なアクセスが許容される。腫瘍は実質的に以前記載されたとおりにマウスに皮下で接種されたLAPC4細胞から発達される。LAPC4細胞は80%コンフルエントまで15%FBSプラス1%PSおよび10nmDHTを加えたIMEM中に生育される。細胞はDPHS中に掻き取られ、遠心によって集められ、そして3×107細胞/mlにおいてMatrigel中に再懸濁(10mg/ml)される。マウスは各脇腹の一部位に細胞懸濁液100μlを皮下注射される。腫瘍はキャリパーで毎週計測され、そして腫瘍体積が一式:4/3πr12×r2(r1<r2)によって計算される。
【0098】
第1の実験において、LAPC4腫瘍は接種後8〜10週間発育が許容される。匹敵する腫瘍体積を有する5匹のマウスのグループは、去勢されるか、または5および6で処置される(0.15mmol/kg 1日1回および0.15mmol/kg 1日2回,9a.m.および5p.m.)。マウスはメトキシフルオラン麻酔のもとで去勢される。化合物5および6は食塩水中の0.3%ヒドロキシプロピルセルロース溶液中17.2mg/mlに調製され、そしてマウスは毎日皮下注射を受けた。対照および去勢マウスはビヒクルのみで処置されるか。腫瘍は処置の4週間毎週計測され、そして腫瘍体積が計算される。処置期間の終りに動物はハロタン麻酔のもとに犠牲にされ、そして腫瘍が摘出され、計量されそして−80℃で貯蔵される。動物はまた毎週計量され、そして一般的健康状態および処置による可能性ある毒性についてモニターされる。
【0099】
第2の実験においては、マウスはLAPC4細胞を接種され、そして各群5匹の4グループに分割される。対照および去勢グループはビヒクルのみを受け、他の2グループはVN/85−1(0.15mmol/kg 1日2回,9a.m.および3p.m.)か、または5(0.15mmol/kg 1日2回,9a.m.および3p.m.)を受ける。これら処置はLAPC4細胞接種1日後から開始され、対照グリープには14週、19週(VN/85−1および去勢グループ)および5処置グループには21週続けられ、そして腫瘍が測定され、上に記載したように処理される。
【0100】
腫瘍、肝臓および睾丸中の5(VN/124−1)レベルの測定:VN/124−1処置グループ中の動物を最後のVN/124−1投与1時間後に犠牲にし、そして腫瘍、肝臓および睾丸を収穫し、液体窒素中にスナップ凍結する。組織サンプルはリン酸バッファー(pH=7.4,0.5ml/mg組織)中にホモジナイズする。ホモジテイズした組織(200μl)を内部標準VN/85−1(100μg/mlストック溶液から10μl)でスパイクし、次にボルテックスミキサーに3分間かけることによりEt2O(2×2ml)で抽出し、次いで3000gで5分間遠心する。Et2O抽出液をゆるやかな空気流の下で蒸発乾固する。残渣をHPLC移動相100μl中に再構成し、0.2μmテフロンフィルターで濾過し、次に上に記載したようにHPLCにより分析される。
【0101】
以上の実施例は、以上の実施例において使用された反応剤および/または作業条件を一般的にまたは特定的に記載した本発明の反応剤および/または作業条件をもって置換することによって同様の成功度で繰り返すことができる。
【0102】
以上の説明から、当業者は本発明の本質特徴を容易に確かめることができ、そしてその精神および範囲を逸脱することなく種々の用途および条件に適合するように本発明の変更および修飾をなすことができる。
【0103】
表1:新規17−ヘテロアリール化合物のCYP17および5α−レダクターゼ活性およびアンドロゲン受容体結合
【0104】
【表1】
【0105】
a 我々は以前VN/81−1の合成を報告した(Njar et al,前出)。アビラテロンはPotterら(A convenient,lerge−scalesyn thesis of a biraterone acetate〔3β−acetoxy−17(3−pyridyl)androsta−5,16−diene〕,a potent new drug for the treatment of prostate cancer.Org.Prep.Proc.Int.1997,29,123−128)によって記載されたように合成した。
b IC50は酵素活性を50%阻害するのに要する阻害剤の濃度,CYP17については各自2系列、5α−レダクターゼおよびAR結合については3系列。
c IC50はアンドロゲン受容体から〔3H〕R1881を50%移動させるのに要する化合物の濃度。
d タイプ1酵素を発現する前立腺腫瘍細胞株(DV−145);基質:(5nM〔1β3H〕アンドロステンジオン。
e BPH組織からの酵素(タイプ2酵素),タンパク125μg,基質:20nM〔1β,2β−3H〕テストステロン。
f ni=10μMまで阻害なし、−=測定せず
【0106】
表2:5(50および100mg/kg)および6(50mg/kg)の皮下投与後の薬物動態
【0107】
【表2】
【0108】
a 値は平均±S.E.で表現される。n=5
【0109】
スキームの簡単な説明
【0110】
【化20】
【0111】
チャート1:アベラテロンおよびVN/85−1(16)の構造
【0112】
【化21】
【0113】
スキーム1:17−ベンゾアゾール化合物(5,6,9および10)の合成
【0114】
【化22】
【0115】
スキーム2:17−ジアジン化合物(14および15)の合成
【0116】
【化23】
【0117】
スキーム3:VNLG/81を含む、トランスアンドロステロンの代謝産物の合成
【特許請求の範囲】
【請求項1】
前立腺疾患を処置する際に使用するための式1(化1)の化合物、又はその薬学的に許容し得る塩。
【化1】
【請求項2】
前記前立腺疾患が前立腺癌又は前立腺肥大症であることを特徴とする請求項1に記載の化合物。
【請求項3】
式1(化1)の化合物又はその薬学的に許容し得る塩が任意の他の活性成分と組み合わせられることを特徴とする、前立腺疾患の処置において使用するための請求項1又は2に記載の化合物。
【化1】
【請求項4】
式1(化1)の化合物又はその薬学的に許容し得る塩が任意の他の活性成分と組み合わせられることを特徴とする、前立腺疾患の処置における使用。
【化1】
【請求項1】
前立腺疾患を処置する際に使用するための式1(化1)の化合物、又はその薬学的に許容し得る塩。
【化1】
【請求項2】
前記前立腺疾患が前立腺癌又は前立腺肥大症であることを特徴とする請求項1に記載の化合物。
【請求項3】
式1(化1)の化合物又はその薬学的に許容し得る塩が任意の他の活性成分と組み合わせられることを特徴とする、前立腺疾患の処置において使用するための請求項1又は2に記載の化合物。
【化1】
【請求項4】
式1(化1)の化合物又はその薬学的に許容し得る塩が任意の他の活性成分と組み合わせられることを特徴とする、前立腺疾患の処置における使用。
【化1】
【図1】

【図2a】
【図2b】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2a】
【図2b】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−255026(P2012−255026A)
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−196525(P2012−196525)
【出願日】平成24年9月6日(2012.9.6)
【分割の表示】特願2007−558143(P2007−558143)の分割
【原出願日】平成18年3月2日(2006.3.2)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
2.WINDOWS
【出願人】(507293321)ユニバーシティ オブ メリーランド,ボルティモア (2)
【Fターム(参考)】
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願日】平成24年9月6日(2012.9.6)
【分割の表示】特願2007−558143(P2007−558143)の分割
【原出願日】平成18年3月2日(2006.3.2)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
2.WINDOWS
【出願人】(507293321)ユニバーシティ オブ メリーランド,ボルティモア (2)
【Fターム(参考)】
[ Back to top ]