
新規のペプチド、その調製プロセス、及びその使用
本発明は、一般式(I)によるペプチド、

並びに、その薬剤的に許容可能な塩、エステル、及び薬剤的に許容可能なプロドラッグに関する。ここで、
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、である。
更に、本発明は、それらを含む医薬調製物及びキット、それらを使用したスクリーニング及び単離方法、並びに医薬調製物の製造におけるそれらの使用に関する。
並びに、その薬剤的に許容可能な塩、エステル、及び薬剤的に許容可能なプロドラッグに関する。ここで、
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、である。
更に、本発明は、それらを含む医薬調製物及びキット、それらを使用したスクリーニング及び単離方法、並びに医薬調製物の製造におけるそれらの使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規のペプチド、特にオリゴペプチドに関し、更に、こうしたペプチドの生産のための方法と、医薬の生産におけるこうしたペプチドの使用とに関する。
【背景技術】
【0002】
補体系は、ヒト及び動物生命体の先天性免疫における最も重要な構成要素の1つである。補体系は、免疫系一般と同様に、侵入病原体及び変性した宿主構造(例えば、アポトーシス細胞)を認識、標識、及び除去する能力を有する。補体系は、先天性免疫系の一部として、病原微生物に対する生命体の第1の防御線の1つを形成するが、幾つかの点において、適応(後天性)免疫系にも関連しており、先天性免疫及び適応免疫メカニズム間の言わば橋渡しをしている(Walport 2001a; Walport 2001b; Morgan 2005)。補体系は、約30のタンパク質成分から成るネットワークであり、こうした成分は、血漿中において、可溶性の形態として見られ、更には、細胞の表面に付着した受容体及びモジュレータ(例えば、阻害因子)の形態でも見られる。当該系の主成分は、セリンプロテアーゼチモーゲンであり、厳密に定められた順序でカスケードの形で相互に活性化し合う。活性化プロテアーゼの特定の基質は、チオエステル結合を含むタンパク質である(補体系の成分C4及びC3)。これらの基質が活性化プロテアーゼにより切断される時、反応性チオエステル基が分子の表面に露出した状態となり、これにより、当該切断された分子を、攻撃される細胞の表面に付着させることが可能となる。結果として、こうした細胞は標識され、免疫系により認識可能となる。
【0003】
補体系の生物学的機能は、極めて多様且つ複雑であり、今日に至るまで全ての詳細については探求がなされていない。最も重要な機能の1つは、直接的な細胞毒性活性であり、補体系の終結的要素をなす成分から形成された膜侵襲複合体(MAC)により誘発される。MAPは、異物と認識された細胞の膜に穿孔し、溶解をもたらすことで、こうした細胞を破壊する。
【0004】
補体系の他の重要な機能には、細胞表面に定着する活性補体成分(例えば、C1q、MBL、C4b、C3b)が白血球(例えば、マクロファージ)による貪食を促進する際のオプソニン化がある。こうした白血球は、破壊するべき細胞を飲み込む。
【0005】
更に、補体系の炎症開始における役割も、特に重要である。補体活性化中に放出された切断産物は、白血球に対するその化学走性刺激作用(chemotactic stimulating effects)により、炎症プロセスを開始させる(Mollnes 2002)。
【0006】
補体系の成分は、適切な信号(例えば、異質細胞、病原体の侵入)により補体カスケードの活性化が誘発されるまでは、不活性(チモーゲン)型として血漿中に存在する。補体系の正常な活性は、免疫ホメオスタシスを維持する観点から重要である。異常な活性低下と無制御な活性過剰とは、共に重篤な疾患の発生、又は既存の疾患の悪化をもたらす恐れがある(Szebeni 2004)。
【0007】
補体系は、古典経路、レクチン経路、及び副経路という3種類の経路で活性化され得る。古典経路の第1のステップにおいて、C1複合体は、活性化因子、すなわち異物と認識された生物学的構造の表面に結合する。C1複合体は、認識タンパク質分子(C1q)と、それに結合するセリンプロテアーゼ(C1r、C1s)とから成る超分子複合体である(Arlaud 2002)。まず、C1q分子が、免疫複合体、アポトーシス細胞、C反応性タンパク質、及び他の活性化因子構造と結合する。C1q分子が活性化因子と結合した結果として、C1複合体に存在するセリンプロテアーゼチモーゲンは徐々に活性化される。四量体C1s−C1r−C1r−C1sにおいて、まず、C1rチモーゲンが自己活性化し、その後、活性C1r分子が、C1s分子を切断及び活性化する。活性C1sは、補体系のC4及びC2成分を切断し、その切断産物は、C3転換酵素(convertase)複合体(C4bC2a)の前駆体となる。C3転換酵素は、C3成分を分割し、C5転換酵素(C4bC2aC3b)へ変換する。C5転換酵素は、C5を切断し、その後、補体系の活性化は、3種類全ての経路の特徴である終結期に達する(MACの形成)。
【0008】
補体系の別の経路であるレクチン経路の活性化は、古典経路のものと非常に似ている(Fujita 2004)。しかしながら、この場合、MBL(「マンノース結合レクチン」)及びフィコリン(H、L、及びM型)という幾つかの異なる種類の認識分子が関与する。こうした分子は、微生物表面上の糖鎖構造に結合する。認識分子の結合に続いて、MASP−2(「MBL関連セリンプロテアーゼ」−2)チモーゲンの自己活性化が生じる。活性MASP−2は、C4及びC2成分を切断し、古典経路の過程において既に説明したC3転換酵素複合体の形成をもたらし、これ以降、プロセスは上述したように続く。
【0009】
副経路は、C3成分の切断と、異物と認識された生物学的構造の表面への固定とにより開始される(Harboe 2008)。当該切断によって形成されたC3b成分は、微生物の細胞膜に結合した場合には、同時に、B因子(C3bB)と呼ばれるセリンプロテアーゼのチモーゲン型にも結合し、B因子は、活性型で血中に存在するD因子による切断により活性化される。このように形成されたC3bBb複合体は、副経路のC3転換酵素であり、更なるC3b分子により補完された後、C5転換酵素に変化する。副経路は、C3成分(C3w)の緩やかな加水分解により、自発的に独立して誘発され得るが、古典経路又はレクチン経路の何れかがC3切断の時点まで到達した場合、副経路は、その効果を著しく増幅させる。
【0010】
上述した経路のうち、最近になって発見され最も理解が進んでいないが、本発明の観点からは最も重要である、レクチン経路について、更に詳細に説明する。数種類のプロテアーゼ及び非触媒タンパク質が、幾つかの異なる形態で存在する認識分子(重合の度合いが異なるMBL及びフィコリン)に結合する。MASP−2は、それ自体でも補体カスケードを開始する能力を有するが(Ambrus 2003; Gal 2005)、この酵素は、MASP−1に比べ少ない量(0.5μg/ml)で存在している。存在する量がより多いMASP−1プロテアーゼ(7μg/ml)の生理的機能は、未だ完全には探求されていない。
【0011】
MASP−1は、それ自体では補体カスケードを開始できないが(C2のみ切断可能でありC4は切断できない)、その活性は、幾つかの点でMASP−2の活性を補っており、そのため、活性MASP−1は、レクチン経路の作用を増幅及び完了させるために必要となり得る。MASP−1はトロンビンにある程度類似したプロテアーゼであり、血液中において、2つの主要なタンパク質分解カスケード系である補体系と血液凝固系との間の橋渡しをすることを示す幾つかの兆候がある(Hajela 2002; Krarup 2008)。
【0012】
MASP−1及びMASP−2の遺伝子は共に、選択的スプライシング産物を有する。MAp19(sMAP)タンパク質は、MASP−2遺伝子から生産され、MASP−2の最初の2つのドメイン(CUB1−EGF)を含む。MASP−3 mRNAは、MASP−1遺伝子から転写される。MASP−3の最初の5つのドメインは、MASP−1のドメインと同じだが、それらはセリンプロテアーゼドメインにおいて異なる。MASP−3は、合成基質に対するタンパク質分解活性が低く、その天然基質は知られていない。また、他の初期プロテアーゼとは異なり、C1阻害剤分子との複合体を形成しない。恐らく、MAp19及びMASP−3の両方が存在することは、タンパク質分解に関して不活性であるこれらのタンパク質が認識分子上の結合部位について活性MASP−2及びMASP−1酵素と競合することから、レクチン経路の活性化に抗することとなる。
【0013】
上述したように、ヒト又は動物生命体における補体系の異常は、疾患を発生させる恐れがある。補体系の無制御な活性化は、自己組織の損傷、及び炎症又は自己免疫状態の発生をもたらす恐れがある(Beinrohr 2008)。こうした状態の1つは、虚血再灌流(以降、IR)障害であり、組織への酸素供給が何らかの理由(例えば、血管閉塞)により一時的に制限又は中断され(虚血)、血液循環の回復(再灌流)後、細胞破壊が始まる際に生じる。再灌流中、補体系は、虚血細胞を変性した自己細胞として認識し、これらを除去するために炎症反応を開始する。この現象は、心筋梗塞及び脳卒中後に生じる組織損傷の原因の一部であり、冠動脈バイパス形成手術及び臓器移植術中に合併症を引き起こす場合もある(Markiewski 2007)。レクチン経路は、IR障害の発生において恐らく一定の役割を果たす。この理由から、レクチン経路の意図的な抑制は、IR障害の程度及び結果を低減し得る。関節リウマチ(以降、RA)の場合にも、RA中に関節に蓄積されるグリコシル化状態が変化したIgG−G0抗体型とMBLが結合するため、レクチン経路は、活性状態となる場合がある。補体系の無制御活性は、様々な神経変性疾患(例えば、アルツハイマー病、ハンチントン病、パーキンソン病、多発性硬化症)の発生及び維持においても役割を果たし、加齢性黄斑変性症(AMD)の発病における主な要因の1つでもある(Bora 2008)。後者の臨床像は、先進工業国における加齢による視力喪失の全症例の半分に関与する。補体系は、自己免疫性腎炎(糸球体腎炎)の形態の1つと、更に別の自己免疫疾患、即ちSLE(全身性エリテマトーデス)にも関連し得る。
【0014】
補体系を初期ステップ中に阻害する場合、一般的な免疫抑制を誘発することなく、特定の活性化経路の効率的且つ選択的な阻害が可能となる。MASP−1及びMASP−2酵素を阻害することにより、レクチン経路を選択的に遮断すること(例えば、上述した疾患において)が可能となり、免疫複合体の除去に関与する古典経路は、これにより影響されず、機能を続ける。
【0015】
C1r、C1s、MASP−1、MASP−2、及びMASP−3酵素は、同じドメイン構造を有する酵素ファミリを形成する(Gal 2007)。タンパク質分解活性に関与するトリプシン様セリンプロテアーゼ(SP)ドメインの前に、5つの非触媒ドメインがある。分子のN末端部分を形成する3つのドメインCUB1−EGF−CUB2(CUB=C1r/C1s、ウニUegf及び骨形成タンパク質−1、EGF=上皮成長因子)は、分子の二量体化(MASP−1及びMASP−2の両方の場合)に関与すると共に、分子との相互作用、例えば、認識分子との結合に関与する。
【0016】
分子のC末端CCP1−CCP2−SPフラグメント(CCP=補体制御タンパク質)は、その触媒性に関して、分子全体と同等である。補体プロテアーゼの特性の1つは、基質特異性が非常に狭く、ほんの少数のタンパク質基質の明確に定められたペプチド結合だけを切断する能力を有することである。CCPモジュール及びSPドメインは、共に、この精密な特異性に寄与する。
【0017】
SPドメインは、セリンプロテアーゼの特徴である活性中心と、基質結合ポケットと、オキシアニオン穴(oxyanion hole)とを含む。8個の表面ループ領域は、様々なプロテアーゼにおいて立体配座が大きく異なり、サブサイト特異性を定める上で決定的な役割を果たす。
【0018】
CCPモジュールは、一方において、触媒領域の構造を安定化させ、他方において、大きなタンパク質基質に対する結合部位を含む。トリプシン様セリンプロテアーゼを阻害するために一般に使用される小分子化合物(例えば、ベンズアミジン、NPGB、FUT−175)は、補体プロテアーゼの活性も阻害するが(Schwertz 2008)、この阻害は、十分に選択的ではなく、血漿中の他のセリンプロテアーゼ、例えば、血液凝固酵素、カリクレイン等の不活性化にも及ぶ。
【0019】
補体系の天然阻害剤としては唯一既知のものであるC1阻害タンパク質は、血中を循環し、セルピンファミリに属するものであるが、比較的広い特異性を有する。
【0020】
レクチン経路を効率的且つ選択的に阻害する能力を有する化合物又は天然阻害タンパク質は、最新技術においても知られていない。
【先行技術文献】
【非特許文献】
【0021】
【非特許文献1】Ambrus, G., Gal, P., Kojima, M., Szilagyi, K., Balczer, J., Antal, J., Graf, L., Laich, A., Moffatt, B., Schwaeble, W., Sim, R.B. and Zavodszky, P. (2003) Natural substrates and inhibitors of mannan-binding lectin-associated serine protease-1 and -2: a study on recombinant catalytic fragments. J. Immunol. 170, 1374-1382.
【非特許文献2】Arlaud, G.J., Gaboriaud, C., Thielens, N.M., Budayova-Spano, M., Rossi, V. and Fontecilla-Camps, J.C. (2002) Structural biology of the C1 complex of complement unveils the mechanisms of its activation and proteolytic activity. Mol. Immunol. 39, 383-394
【非特許文献3】Atherton, E.; Sheppard, R.C. (1989). Solid Phase peptide synthesis: a practical approach. Oxford, England: IRL Press. ISBN 0199630674.
【非特許文献4】Beinrohr, L., Dobo, J., Zavodszky, P. and Gal, P. (2008) C1, MBL-MASPs and C1-inhibitor: novel approaches for targeting complement-mediated inflammation. Trends in Molecular Medicine doi:10.1016/j.molmed.2008.09.009
【非特許文献5】Bora, N.S., Jha, P. and Bora, P.S. (2008) The role of complement in ocular pathology. Semin. Immunopathol. 30, 85-95
【非特許文献6】Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. "WebLogo: A sequence logo generator", Genome Research, 14:1188-1190
【非特許文献7】Dobo, J., Harmat, V., Sebestyen, E., Beinrohr, L., Zavodszky, P. & Gal, P. (2008). Purification, crystallization and preliminary X-ray analysis of human mannose-binding lectin-associated serine protease-1 (MASP-1) catalytic region. Acta Crystallogr Sect F Struct Biol Cryst Commun 64, 781-4.
【非特許文献8】Empie, M. W. & Laskowski, M., Jr. (1982). Thermodynamics and kinetics of single residue replacements in avian ovomucoid third domains: effect on inhibitor interactions with serine proteinases. Biochemistry 21, 2274-84.
【非特許文献9】Fujita, T., Matsushita, M. and Endo, Y. (2004) The lectin-complement pathway - its role in innate immunity and evolution. Immunol. Rev. 198, 185-202
【非特許文献10】Gal, P., Barna, L., Kocsis, A. and Zavodszky P. (2007) Serine proteases of the classical and lectin pathways: Similarities and differences Immunobiol. 212, 267-277
【非特許文献11】Gal, P., Harmat, V., Kocsis, A., Bian, T., Barna, L., Ambrus, G., Vegh, B., Balczer, J., Sim, R.B., Naray-Szabo, G., Zavodszky, P. (2005) A true autoactivating enzyme. Structural insights into mannose-binding lectin-associated serine protease-2 activation. J. Biol. Chem. 280, 33435-33444.
【非特許文献12】Hajela, K., Kojima, M., Ambrus, G., Wong, K.H.N., Moffatt, B.E., Fergula, J., Hajela, S., Gal, P., Sim, R.B. (2002) The biological functions of MBL-associated serine proteases (MASPs). Immunbiol. 205, 467-475.
【非特許文献13】Harboe, M and Mollnes, T.E. (2008) The alternative complement pathway revisited. J. Cell. Mol. Med. 12, 1074-1084
【非特許文献14】Harmat, V., Gal, P., Kardos, J., Szilagyi, K., Ambrus, G., Vegh, B., Naray-Szabo, G. & Zavodszky, P. (2004). The structure of MBL-associated serine protease-2 reveals that identical substrate specificities of C1s and MASP-2 are realized through different sets of enzyme-substrate interactions. J Mol Biol 342, 1533-46.
【非特許文献15】Korsinczky, M. L., Schirra, H. J., Rosengren, K. J., West, J., Condie, B. A., Otvos, L., Anderson, M. A. & Craik, D. J. (2001). Solution structures by 1H NMR of the novel cyclic trypsin inhibitor SFTI-1 from sunflower seeds and an acyclic permutant. J Mol Biol 311, 579-91.
【非特許文献16】Krarup, A, Gulla, K.C., Gal, P. Hajela, K. and Sim R.B. (2008) The action of MBL associated serine protease 1 (MASP1) on factor XIII and fibrinogen. Biochim. Biophys. Acta. 1784, 1294-1300.
【非特許文献17】Kunkel, T. A., Bebenek, K. & Mcclary, J. (1991). Efficient Site-Directed Mutagenesis Using Uracil-Containing DNA. Methods in Enzymology 204, 125-139.
【非特許文献18】Laskowski, M., Jr. & Kato, I. (1980). Protein inhibitors of proteinases. Annu Rev Biochem 49, 593-626.
【非特許文献19】Luckett, S., Garcia, R. S., Barker, J. J., Konarev, A. V., Shewry, P. R., Clarke, A. R. & Brady, R. L. (1999). High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J Mol Biol 290, 525-33.
【非特許文献20】Malik, Z., Amir, S., Pal, G., Buzas, Z., Varallyay, E., Antal, J., Szilagyi, Z., Vekey, K., Asboth, B., Patthy, A. & Graf, L. (1999). Proteinase inhibitors from desert locust, Schistocerca gregaria: engineering of both P-1 and P-1 ' residues converts a potent chymotrypsin inhibitor to a potent trypsin. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology 1434, 143-150.
【非特許文献21】Markiewski, M.M. and Lambris, J.D. (2007) The role of complement in inflammatory diseases - from behind the scenes into the spotlight. Am J. Pathol. 171, 715-727.
【非特許文献22】Mollnes, T.E., Song, W.C. and Lambris, J.D. (2002) Complement in inflammatory tissue damage and disease. Trends Immunol. 23, 61-64
【非特許文献23】Morgan, B.P., Marchbank, K.J., Longhi, M.P., Harris, C.L. and Gallimore, A.M. (2005) Complement: central to innate immunity and bridging to adaptive responses. Immunol. Lett. 97, 171-179
【非特許文献24】Mulvenna, J. P., Foley, F. M. & Craik, D. J. (2005). Discovery, structural determination, and putative processing of the precursor protein that produces the cyclic trypsin inhibitor sunflower trypsin inhibitor 1. J Biol Chem 280, 32245-53.
【非特許文献25】Schneider TD, Stephens RM. 1990. "Sequence Logos: A New Way to Display Consensus Sequences." Nucleic Acids Res. 18:6097-6100
【非特許文献26】Schwertz H, Carter JM, Russ M, Schubert S, Schlitt A, Buerke U, Schmidt M, Hillen H, Werdan K, Buerke M. (2008) Serine protease inhibitor nafamostat given before reperfusion reduces inflammatory myocardial injury by complement and neutrophil inhibition. J Cardiovasc Pharmacol. 2008 Aug;52(2):151-60.
【非特許文献27】Smith, G. P. (1985). Filamentous Fusion Phage - Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 228, 1315-1317.
【非特許文献28】Szebeni, J. ed. (2004) The complement system. Novel roles in health and disease Kluwer Academic Publishers, ISBN 1-4020-8055-7.
【非特許文献29】Szenthe, B., Patthy, A., Gaspari, Z., Kekesi, A. K., Graf, L. & Pal, G. (2007). When the surface tells what lies beneath: Combinatorial phage-display mutagenesis reveals complex networks of surface-core interactions in the pacifastin protease inhibitor family. Journal of Molecular Biology 370, 63-79.
【非特許文献30】Walport, M.J. (2001a) Complement. First of two parts. N. Eng. J. Med. 344, 1058-1066.
【非特許文献31】Walport, M.J. (2001b) Complement. Second of two parts. N. Eng. J. Med. 344, 1140-1144.
【発明の概要】
【発明が解決しようとする課題】
【0022】
レクチン経路を含む補体系の阻害は、補体系の異常の結果として生じるヒト及び動物の疾患に対抗するための効率的なツールとなり得る。しかしながら、現在、こうした疾患と闘うために、補体系、主にレクチン経路を望ましい範囲で阻害できる化合物として利用できるものは無い。上で詳細に説明したように、MASP−1及びMASP−2酵素を阻害することによりレクチン経路を選択的に阻害し得る。
【0023】
この理由から、本発明者らは、MASP−1及び/又はMASP−2酵素を阻害することにより補体系のレクチン経路を選択的に阻害できる化合物の開発を目標に定めた。
【課題を解決するための手段】
【0024】
驚いたことに、我々は、以下の一般式(I)によるペプチドが上述した目的に適していることを発見した。
ここで、
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、Wである。
【0025】
上記によれば、本発明は、一般式(I)によるペプチド、その塩、エステル、及び薬剤的に許容可能なプロドラッグに関する。
【0026】
特に好ましくは、本発明は、以下の配列を有するペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、
GICSRSLPPICIPD(SEQ ID NO 3)、
GVCSRSLPPICWPD(SEQ ID NO 4)、
GMCSRSYPPVCIPD(SEQ ID NO 5)、
GYCSRSIPPVCIPD(SEQ ID NO 6)、
GWCSRSYPPVCIPD(SEQ ID NO 7)、及び
配列GICSRSLPPICIPD(SEQ ID NO 3)を有するペプチドの環状のもの、
及びその塩又はエステルに関する。
【0027】
最も好ましくは、本発明は、配列GYCSRSYPPVCIPD(SEQ ID NO 2)及びGICSRSLPPICIPD(SEQ ID NO 3)を有するペプチド、その塩及びエステルに関する。
【0028】
更に、本発明は、一般式(I)による少なくとも1つのペプチド、その塩、エステル、又はプロドラッグ、及び少なくとも1つの他の添加物を含有する医薬調製物に関する。この添加物は、好ましくは、活性薬剤の制御された放出を確保するマトリクスである。
【0029】
本発明は、特に、以下の配列を有する少なくとも1つのペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、
GICSRSLPPICIPD(SEQ ID NO 3)、
GVCSRSLPPICWPD(SEQ ID NO 4)、
GMCSRSYPPVCIPD(SEQ ID NO 5)、
GYCSRSIPPVCIPD(SEQ ID NO 6)、
GWCSRSYPPVCIPD(SEQ ID NO 7)、
配列GICSRSLPPICIPD(SEQ ID NO 3)を有するペプチドの環状のもの、
及び/又はその薬剤的に許容可能な塩もしくはエステル、を含有する医薬調製物に関する。
特に好ましくは、本発明による医薬調製物は、配列GYCSRSYPPVCIPD及びGICSRSLPPICIPDを有するペプチド、及び/又は、その薬剤的に許容可能な塩及び/又はエステルを含有する。
【0030】
本発明は、更に、一般式(I)による少なくとも1つのペプチド、その塩又はエステルを含むキットに関する。
【0031】
本発明は、更に、MASP酵素を阻害する可能性がある化合物のスクリーニング手順に関し、その過程においては、本発明による標識ペプチドを、MASPを含有する溶液に添加し、試験対象の1つ以上の化合物を含有する溶液をそれに添加し、放出された有標ペプチドの量を測定する。これに関して、MASP酵素は、好ましくは、MASP−1又はMASP−2酵素である。
【0032】
本発明は、更に、補体系の阻害により治療可能な疾患を治療することに適した医薬調製物の製造における、一般式(I)によるペプチド、及びその薬剤的に許容可能な塩又はエステルの使用に関する。これによれば、疾患は、好ましくは以下の群から選択され得る:炎症性及び自己免疫疾患、特に好ましくは、虚血再灌流障害、関節リウマチ、神経変性疾患、加齢性黄斑変性症、糸球体腎炎、全身性エリテマトーデス、及び補体活性化関連仮性アレルギ。
【0033】
本発明は、更に、MASP酵素を単離するための手順に関し、その過程においては、一般式(I)による1つ以上の固定化ペプチドを有する担体を、MASP酵素を含有する溶液に接触させ、調製物を洗浄する。これに関して、MASP酵素は、好ましくは、MASP−1又はMASP−2酵素である。
【0034】
上述した本発明によるペプチドには、MASP−1及びMASP−2酵素の両方を阻害するものと、MASP−2酵素のみを阻害しMASP−1酵素を阻害しないものとがある。しかしながら、こうした本発明によるペプチドは、MASP酵素と密接に関係するトロンビンを、非常に高濃度でなければ阻害せず、一般には、トリプシンもほんの僅かにしか阻害しない。
【図面の簡単な説明】
【0035】
【図1】ファージディスプレイ法の概略図である。
【図2】アガロースゲル上で行った、実施例1.1.3.2に記載のダイジェスチョンの結果を確認する図である(レーン1は、ダイジェストされたpMal−p2X lacIq遺伝子を示し、レーン2は、ダイジェストされたpBlueKS_NheI_Nsiベクタを示す)。
【図3】実施例1.1.4.3に記載のライゲーション及び形質転換に使用したベクタ及びインサートを検査して濃度を確認する試験の結果を示す図である。
【図4】実施例2.2.2に記載のライゲーション試験に関連して調製したゲルの写真を示す図である。
【図5】得られた配列の配列ロゴ図であり 図5aは、MASP−2から選択されMASP−2に特異的な配列に関する配列図であり、 図5bは、MASP−2から選択されたが、MASP−1も認識する配列に関する配列図であり、 図5cは、MASP−1から選択されたが、MASP−2も認識する配列に関する配列図である。
【図6】血液凝固に対する本発明によるペプチドの影響に関する用量関連試験結果を示す図であり、 図6aは、トロンビンを血漿に添加することにより血漿凝固(フィブリン形成)を誘発する過程を有する、トロンビン時間の測定実験を示す図であり、 図6bは、組織因子を血漿に添加することにより血漿凝固(フィブリン形成)を誘発する過程を有する、プロトロンビン時間の測定実験を示す図であり、 図6cは、血液凝固のいわゆる「接触活性化(contact activated)」又は「内因性(intrinsic)」経路を模倣する、トロンボプラスチン時間の測定実験を示す図である。
【図7】3つの補体活性化経路に対する本発明によるペプチドの影響を示す図であり、 図7aは、選択的「S」ペプチドの影響を示す図であり、 図7bは、非選択的「NS」ペプチドの影響を示す図である。
【発明を実施するための形態】
【0036】
本発明は、MASP−1及びMASP−2酵素(又はMASP−2酵素のみ)を選択的に阻害するペプチド及びペプチド誘導体に関する。
【0037】
本発明は、更に、記載した配列と配列的に類似し、その生物学的活性も記載した配列と比較して類似する、アミノ酸配列に関する。対象となるペプチドの生物学的機能を変えること無く、特定の側鎖改変又はアミノ酸置換を実施し得るということは、当業者には明白である。こうした改変は、アミノ酸側鎖の相対的類似、例えば、大きさ、電荷、疎水性、親水性等における類似に基づくものであり得る。こうした変更の目的は、酵素分解に対するペプチドの安定性の増加、或いは特定の薬物動態パラメータの改善である場合がある。
【0038】
本発明の保護の範囲は、更に、検出性を確保する要素(例えば、蛍光基、放射性原子等)が統合されたペプチドを含む。
【0039】
更に、本発明の保護の範囲には、幾つかの追加のアミノ酸を、N末端、C末端、又は両末端に含むペプチドが含まれるが、これは当該追加アミノ酸が元の配列の生物学的活性を大きく損なわないことを条件とする。端部に位置するこうした追加のアミノ酸の目的は、固定化を促進すること、他の試薬との連結の可能性を確保すること、溶解度、吸収、その他の特性に影響を与えることであり得る。
【0040】
配列内のアミノ酸側鎖は、IUPACの推奨する方法を使用して表示した(Nomenclature of α-Amino Acids, Recommendations, 1974 - Biochemistry, 14(2), 1975)。
【0041】
本発明は、更に、本発明による一般式(I)によるペプチドの薬剤的に許容可能な塩に関する。これは、ヒト又は動物の組織と接触させた時に、不要な度合いの毒性、炎症、アレルギ症状、又はこれらに類似する現象を引き起こさない塩を意味する。酸付加塩の非限定的な例として、以下が挙げられる:酢酸塩、クエン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、酪酸塩、ジグルコン酸塩, ヘミ硫酸塩、フマル酸塩、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、乳酸塩、マレイン酸塩、メタンスルホン酸塩、シュウ酸塩、プロピオン酸塩、コハク酸塩、酒石酸塩、リン酸塩、グルタミン酸塩。塩基付加塩の非限定的な例として、以下のものに基づく塩が挙げられる:アルカリ金属及びアルカリ土類金属(リチウム、カリウム、ナトリウム、カルシウム、マグネシウム、アルミニウム)、第4アンモニウム塩、アミンカチオン(メチルアミン、エチルアミン、ジエチルアミン等)。
【0042】
本発明に関して、プロドラッグとは、インビボで本発明によるペプチドへ転換される化合物である。転換は、例えば、血液中で酵素加水分解によって起こり得る。
【0043】
本発明によるペプチドは、適切な生物学的効果を達成するために1つ以上の添加物が必要となる医薬調製物に使用することができる。こうした製剤は、例えば、当業者に周知である、活性薬剤の制御された放出を確保するマトリクスと組み合わせた医薬調製物であり得る。一般に、活性薬剤の制御された放出を確保するマトリクスは、適切な組織(例えば、血漿)に入った時に、例えば、酵素又は酸塩基加水分解の過程によって分解する重合体である(例えば、ポリラクチド、ポリグリコライド)。
【0044】
本発明による医薬調製物では、希釈剤、充填剤、pH調節剤、溶解促進物質、着色添加物、抗酸化剤、保存料、等張剤等、最新技術水準において公知の他の添加物を使用することが可能である。こうした添加物は、最新技術水準において公知である。
【0045】
好ましくは、本発明による医薬調製物は、非経口(静脈内、筋肉内、皮下)投与を介して生命体内に入れることができる。これを考慮すると、好適な医薬組成物は、水性又は非水性の溶液、分散液、懸濁液、乳液、或いは、使用直前に上述した流体の何れかに変化させることが可能な固形(例えば、粉末)製剤にしてよい。こうした流体において、適切な媒体、担体、希釈剤、又は溶媒は、例えば、水、エタノール、様々なポリオール(例えば、グリセリン、プロピレングリコール、ポリエチレングリコール、及びこれらに類似する物質)、カルボキシメチルセルロース、様々な(植物)油、有機エステル、及びこれら全ての物質の混合物であり得る。
【0046】
本発明による医薬調製物の好ましい剤形には、例えば、タブレット、粉末、顆粒、座薬、注射液、シロップ等が含まれる。
【0047】
投与量は、疾患の種類、患者の性別、年齢、体重、及び、疾患の重篤性に応じて決まる。経口投与の場合、活性薬剤について、好ましい1日あたりの量は、例えば、0.01mgから1gの間であり、非経口投与(例えば、静脈内に投与される製剤)の場合、好ましい1日あたりの量は、例えば、0.001mgから100mgの間であり得る。
【0048】
更に、当該医薬調製物は、最新技術において公知であるリポソーム又はマイクロカプセルの形で使用することができる。本発明によるペプチドはまた、遺伝子治療の最先端の手段により標的生物に入れることもできる。
【0049】
所望の医療効果を達成するために、MASP−1又はMASP−2を選択的に阻害する活性薬剤が必要である場合には、本発明による一般式(I)によるペプチドから、選択的阻害ペプチドを選択することが好ましい。例えば、MASP−2酵素を選択的に阻害する本発明によるペプチドは、配列GYCSRSYPPVCIPD(SEQ ID NO 2)を有するペプチドにしてよく、一方、MASP−1酵素を選択的に阻害する本発明によるペプチドは、配列GICSRSLPPICIPD(SEQ ID NO 3)を有するペプチドにしてよい。特定の治療目標を達成するためには、配列GICSRSLPPICIPD(SEQ ID NO 3)を有する本発明による環状ペプチド等、MASP−1及びMASP−2の両方を阻害するペプチドを使用することが好ましい場合もある。
【0050】
本発明によるペプチドは、様々なMASP酵素の測定又は位置特定を(何れかのMASP酵素に特異的な形で、或いはMASP−1及びMASP−2酵素の両方に同時に特異的な形で)行うために使用可能な様々なキットにおいて、好ましく使用することができる。こうした使用は、競合的及び非競合的試験、ラジオイムノアッセイ、生物発光及び化学発光試験、蛍光定量試験、酵素結合アッセイ(例えば、ELISA)、免疫細胞化学アッセイ等にまで及び得る。
【0051】
本発明によれば、キットは、例えば競合的結合アッセイによって、MASP酵素の潜在的阻害剤を検査することに適したものが特に好ましい。こうしたキットの支援により、本発明によるペプチドをMASP酵素からどのくらい引き離すことができるかについて、潜在的阻害剤の能力を測定することができる。これを検出するためには、本発明によるペプチドは、何らかの形(例えば、蛍光基又は放射性原子の組み込み)により標識する必要がある。
【0052】
本発明によるキットは、更に、溶液及び試薬を調製するために必要な他の溶液、ツール、及び出発物質と、使用説明書とを含み得る。
【0053】
一般式(I)による本発明による化合物(ペプチド)は、更に、MASP酵素を阻害する可能性のある化合物をスクリーニングするために使用することができる。こうしたスクリーニング手順の過程において、一般式(I)によるペプチドは、後の時点での検出性を確保するために、標識(蛍光、放射性等)された形態で使用される。こうしたペプチドを含有する調製物を、MASP酵素を含有する溶液に添加し、その過程において、ペプチドはMASP酵素に結合する。適切なインキュベーション時間の後、試験対象の化合物/化合物群を含有する溶液を、当該調製物に添加し、その後、更にインキュベーション時間を設ける。MASP酵素に結合する化合物は(試験化合物が部分的又は完全にペプチドと同じ部位で酵素表面に結合するか、或いは別の場所だが、その結合により、ペプチドと結合する能力を喪失させるような形でMASP酵素の立体配座が変更される場合)、その阻害能力の範囲でMASP分子から標識ペプチドを引き離す。引き離されたペプチドの濃度は、ペプチド分子上で使用した(蛍光又は放射性)標識を検出するのに適した任意の方法を用いて決定することができる。インキュベーション時間、洗浄条件、検出方法、他のパラメータは、当業者に知られるやり方で最適化することができる。本発明によるスクリーニング手順は、高スループットスクリーニング(HTS)手順においても使用することができる。
【0054】
本発明によるペプチドは、まず第一に、疾患の治療において使用可能であり、その場合、補体系の働きの阻害は、好ましい効果を有する。結果として、本発明は、更に、こうした疾患の治療用の医薬の生産におけるペプチドの使用にも関連する。上で詳細に説明したように、こうした疾患は、第一に、特定の炎症性疾患及び自己免疫疾患であり、特に、以下の疾患である:虚血再灌流障害、関節リウマチ、神経変性疾患(例えば、アルツハイマー病、ハンチントン病、パーキンソン病、多発性硬化症)、加齢性黄斑変性症、糸球体腎炎、全身性エリテマトーデス。
【0055】
本発明による化合物は、更に、MASPタンパク質を単離するために使用でき、それは、ペプチドを固定化し、このように作製した調製物をMASP酵素の含有が推定される溶液に接触させることによって行われる。この溶液が実際にMASP酵素を含有する場合には、当該酵素は固定化ペプチドにより結合される。この手順は、分析及び調製の両方の目的に適したものになり得る。所定のペプチドのMASP酵素上での結合のジオメトリが知られていない場合、この手順においては、一つのペプチドを幾つかの方向から固定して使用するか、或いは複数のペプチドを使用してでも、適切な結合を確保するべきである。MASP酵素を含有する溶液は、純粋なタンパク質溶液、様々な程度まで精製した抽出物、組織調製物等であり得る。
【0056】
ファージディスプレイ
本発明によるペプチドは、ファージディスプレイ法を用いて開発した。
【0057】
ファージシスプレイ法は、誘導性(directed)インビトロ進化の実現に適しており、最先端の手順(Smith 1985)の主要ステップは、図1において確認することができる。この手順では、進化に関与するタンパク質の遺伝子が、バクテリオファージ外被タンパク質遺伝子に連結される。これにより、バクテリオファージが形成される時、融合タンパク質が生成され、ファージの表面に組み込まれた状態となる。ファージ粒子は、当該外来タンパク質の遺伝子を内部に保有しており、その表面には、当該外来タンパク質を提示している。当該タンパク質及びその遺伝子は、ファージを介して物理的に結び付けられる。我々は、誘導性タンパク質進化のために、タンパク質をコードする遺伝子のコドンを、慎重に決めた様式で改変する。合成オリゴヌクレオチドの混合物に基づくコンビネーション的変異導入を用いて、多数のコドンを同時に変更することが可能である。変異の位置と位置毎の変異多様性(variability)は同時に決定される。
【0058】
数十億の変異体を含むDNAライブラリを作製してバクテリアに入れた後、ファージタンパク質ライブラリが作製される。各ファージは、1種類のタンパク質変異体のみを提示し、この変異体の遺伝子のみを保有する。個々の変異体は、試験者により選択された(通常は表面に結合された)特定の標的分子と結合する能力に基づいて、親和性クロマトグラフィ及びその類似法により、互いに分離することができる。それと同時に、単純なタンパク質親和性クロマトグラフィとは異なり、このように選択されたファージタンパク質変異体は2つの重要な特性を有する。第一に、増殖する能力を有しているという点であり、第二に、コード遺伝子をファージ粒子に包んで保有しているという点である。
【0059】
この進化の間においては、変異体が個別に調べられるのではなく、何十億もの実験が実際に同時に行われるのである。結合した変異体は、増殖させ、選択-増殖のサイクルを何回か繰り返した後、機能的変異体が豊富に含まれた集団が得られる。この集団から、タンパク質が依然としてファージ上に提示された状態で、個々のクローンを機能試験において検査する。当該試験によって適切とされたファージタンパク質変異体は、物理的に連結した遺伝子の配列決定をすることにより同定される。個別の測定に加えて、適度に大きな数の機能選択クローンの配列を分析することにより、どのようなアミノ酸配列がその機能を達成することを可能とするかについても明らかになる。これにより、実際の実験に基づくデータベースが作成され、配列-機能アルゴリズムを発展させることが可能となる。これに基づいて最善とされた変異体は、独立したタンパク質として生産され、更に正確な追加試験において検査される。
【0060】
ライブラリの作製
SFTI(ヒマワリトリプシン阻害剤)分子は、トリプシン阻害活性を有しており、以下の配列を有する14アミノ酸ペプチドである:GRCTKSIPPICFPD(SEQ ID NO 1)。自然界においては、即ち、植物のヒマワリでは、これは環状の形態で形成されるため、ここでN末端として示したグリシンと、C末端として示したアスパラギン酸とは、ペプチド結合により連結される。2個のシステインは、互いにジスルフィド架橋を形成する。ジスルフィド架橋が完全であるならば、上に示した線状形態であっても強力なトリプシン阻害剤となることが、インビトロ試験により実証されている(Korsinczky, 2001)。SFTI分子の他の特別な特徴は、酵素と相互作用する遙かに大きなボーマン−バーク阻害剤の分子部分と構造的に事実上同一であることである(Luckett 1999; Korsinczky, 2001; Mulvenna 2005)。ボーマン−バーク阻害剤で保存されておりSFTI分子と同一の部分に下線を付す:GRCTKSIPPICFPD。全ての下線部分は、1つ(位置4のトレオニン)を除き、ライブラリを作製する際に維持された。
【0061】
ライブラリを設計する際には、以下のランダム化スキームを使用した:
ここでも、下線を付しているのは、構造的な理由から改変させなかった位置である。位置「O」では、20個全ての天然アミノ酸が許容されたが、位置P1では、スキーム(R/K)により示した2つの塩基性アミノ酸のみが許容された。イタリック体の部分は、我々の当初の予想に基づいて、プロテアーゼと接触することはないと推定されたため、改変しなかった。
【0062】
ファージディスプレイ中に高親和性結合分子を選択可能とするためには、提示される結合分子が、ファージ当たり少ないコピー数で、理想的には1個の単一コピー(一価ファージディスプレイ)で存在することが重要となる。これにより、幾つかの固定標的分子に同時に結合することにより見た目の上で高親和性結合(アビディティ)となるものを回避することができる。このため、上述したSFTIライブラリは、ファージタンパク質p8に連結して発現させた時にファージ当たり1個の単一コピーとして出現することが実証されているキモトリプシン阻害剤分子(Szenthe 2007)と融合させた状態で発現させた。これは、我々の予備実験において、MASP酵素を阻害せず、MASP酵素と結合もしないことが実証されたSchistocerca Gregariaキモトリプシン阻害剤(SGCI)である(Malik, 1999)。
【0063】
SFTIライブラリのエレメントとSGCI分子との間には、更に、タグとライブラリのエレメントとの間で適切な距離が維持されるようなペプチドリンクを使用して、モノクローナル抗体により認識可能な直線状エピトープタグを挿入した。これは、いわゆる「フラッグタグ」であり、2つの目的を果たす。一方の目的は、ファージ表面上でのライブラリの提示を容易に明らかにできるようにすることである。他方の目的は、当該タグに対する抗体を使用することによるコントロール選択の結果として得られたクローンの配列決定をすることにより、特定の標的酵素、即ち、MASP1及びMASP2が欠如した状態ではいかなる配列のクローンが得られるかを明らかにすることである。このようにして、酵素に対して行われた選択の結果を、当該抗体に基づいて選択された群と比較することにより、他の何らかの影響(例えば、より効率的な生産)の結果ではなく真に酵素との結合に起因し得る、典型的な位置依存的アミノ酸嗜好性(preferences)を明らかにすることができる。
【実施例】
【0064】
以下、本発明を実施例に基づいて詳細に説明するが、しかしながら、実施例は、本発明を限定する例と見做すべきではない。
【0065】
実施例を通して、ファージミド系を構築する上で可能な方法(実施例1)、ライブラリの調製(実施例2)、ファージの選択(実施例3)、及び結果(実施例4)を示す。実施例5では、ペプチド合成及び関連する分析試験を説明する。
【0066】
実施例1:ファージミド系の構築
1.1. ファージミドベクタの構築
最初のステップとして、入手可能な市販のベクタから出発して、独自のファージミドベクタを構築した。このために、新たに制限エンドヌクレアーゼ切断部位を形成する必要があり、これはクンケル式変異誘発を使用して実現した(Kunkel, 1991)。
【0067】
1.1.1. ウラシル含有一本鎖クンケルテンプレートの調製
1.1.1.1. 形質転換
0.5μl pBluescript II KS(−)ファージミド(Stratagene、cat#212208、51.1μg/μl、2961bp)
8μl KCM溶液[0.5M KCl、0.15M CaCl2、0.25M MgCl]
31.5μl USP蒸留水
40μl CJ236 K12 E.coliコンピテント細胞
形質転換体を、氷上で20分間、その後、室温で10分間インキュベートした。体積の10倍量(800μl)のLB培地を加え、30分間に渡り、37℃、200rpmで振盪した。100μlの量を一晩、37℃で、LB−アンピシリンプレート[LB、100μg/mlアンピシリン]上において培養した。
【0068】
1.1.1.2. 感染
翌日、コロニを2mlの培地[LB、100μg/mlアンピシリン、30μg/mlクロラムフェニコール]に接種し、37℃で一晩、200rpmで振盪しながらインキュベートした。次に、一晩培養した培養物2μlを、上述したものと同じ組成の培地2mlに接種し、37℃で200rpmで振盪しながら6時間培養した。その後、30μl M13KO7ヘルパーファージ(NEB、cat#N0315S)に感染させ、200rpmで振盪しながら、37℃の状態で40分間インキュベートした。当該出発培養物の全てを、30ml[2YT、100μg/mlアンピシリン、30μg/mlクロラムフェニコール]培地へ移した。ファージは、培養物を200rpmで振盪しながら、37℃で、16乃至18時間に渡り一晩培養することにより生産した。翌朝、培養物を4℃で10分間、8,000で遠心した。上清を清潔なチューブに移し、体積の1/5の量(6ml)の溶液[2.5M NaCl、20%PEG−8000]を加え室温で20分間インキュベートした後、ファージを溶液から沈殿させた。沈殿物を4℃で20分間、10,000rpmで遠心し、上清をピペットで取り除いた。沈殿物を、800μlのPBS緩衝液において溶解した。
【0069】
一本鎖プラスミドは、Qiaprep Spin M13キット(Qiagen、cat#27704)を使用して、キットに付属のレシピによりファージから取得し、100μlの10倍希釈EB緩衝液によりカラムから溶出させた。産物の濃度は、35倍希釈において260nmで確認した(ssDNS OD260nm=1=33ng/μl)。上述した手順の結果として得られた一本鎖ウラシル含有pKSファージミドベクタの濃度は、407μg/mlであった。
【0070】
1.1.2. クンケル式変異誘発を使用した切断部位Nsi及びNheIの導入
1.1.2.1. オリゴのリン酸化
変異プライマ:
Blue_NheI_in_779(36mer、SEQ ID NO 8):
5'-cgcaattaaccctcagctagcggaacaaaagctggg-3'
Blue_NsiI_in_1089(36mer、SEQ ID NO 9):
5'-ccgcctttgagtgagatgcatccgctcgccgcagcc-3'
・2μl 10×濃縮TM緩衝液[0.5 M Tris−HCl、0.1M MgCl2、pH7.5]
・2μl 10mM ATP
・1μl 100mM DTT
・1μl T4ポリヌクレオチドキナーゼ(Fermentas、10u/μl)
・36ng Blue_NheIプライマ(4μl)/36ng Blue_Nsiプライマ(3.5μl)
・10μl USP蒸留水/10.5μl USP蒸留水
別個の2つのプライマについての2つのリン酸化反応を、共に体積20μl中に添加し、45分間37℃でインキュベートした。
【0071】
1.1.2.2. オリゴヌクレオチドのハイブリダイゼーション
テンプレート:プライマの割合は、体積25μl中のモル比が1:3となるように設定した。
・2.5μl一本鎖クンケルテンプレート(1μg)
・2μlリン酸化Blue_NheIプライマ
・2μlリン酸化Blue_Nsiプライマ
・2.5μl 10×濃縮TM緩衝液
・16μl USP蒸留水
反応混合物は、90℃の水槽内で1分間加熱した後、直ちに50℃のサーモスタット内へ移して更に3分間置いた。短時間遠心し、氷中に置いた。
【0072】
1.1.2.3. 二本鎖産物の調製、精製、ダイジェスチョン
オリゴヌクレオチドのハイブリダイゼーション後、第2DNA合成により、二本鎖産物をインビトロで生産した。二本鎖産物の一方の鎖はウラシルを含む当初のクンケルテンプレートであり、プライマの延長により形成された、変異を有する他方の鎖は、ウラシルを含んでいなかった。
・1μl 10mM ATP
・1μl 25mM dNTP
・1.5μl 100mM DTT
・0.6μl T4リガーゼ(NEB、400u/μl)
・0.3μl T7ポリメラーゼ(Fermentas、10u/μl)
反応混合物は、14℃で一晩インキュベートした。混合物全体を1%アガロースゲル上で泳動させ、Qiaquick Gel Extractionキット(Qiagen、cat#28704)によりレシピに従って分離及び精製した。産物は、30μl EB緩衝液で溶出させ、上述したレシピによりE.coli XL1 Blueコンピテント細胞に形質転換した。これらの細胞は、ウラシルを含む鎖を分解するため、3mlの培養物中で培養したバクテリアでは、ウラシルを含まない変異鎖の複製により増殖したベクタのクローンが主に存在する。二本鎖ベクタは、Mini Plus Plasmid DNA Extraction System(Viogen、cat#GF2001)キットを使用して、50μl EB緩衝液中に単離した。
遺伝手術の次のステップとして、新たに導入した切断部位において産物を25μl中でダイジェストする。
・20μlベクタミニプレップ
・2.5μl 10×濃縮Y Tango緩衝液(Fermentas)
・1.25μl USP蒸留水
・0.50μl NheI(Fermentas、10u/ml)
・0.75μl Nsi(Promega、10 u/ml)
ダイジェスチョンは、37℃で一晩行った。産物は、2%アガロースゲル上において、電気泳動を使用して確認し、その後、上述した方法を使用してダイジェスト化プラスミドをゲルから単離した後、キットにより精製した。このようにして入手したベクタの名前は、pBlueKS_NheI_Nsiである。
【0073】
1.1.3 lacIq遺伝子の追加
1.1.3.1 PCR
lacIq遺伝子及びマルトース結合タンパク質(MBP)シグナル配列を、pMal−p2Xベクタ(NEB、cat# N8077S、200μg/ml)からPCRを使用して分離した。
プライマ:
pMal_lac_forward(SEQ ID NO 10):
5'-gtcagtatgcatccgacaccatcgaatggtg-3'
pMal_NheI_rev(SEQ ID NO 11):
5'-gtcagtgctagcgccgaggcggaaaacatcatcg-3'
・5μl 10×濃縮Pfu緩衝液
・0.4μl 25mM dNTPs
・10μl 25mM MgSO4
・0.5μl pMal−p2Xテンプレート
・0.5μl 5μM pMal_lac_forwardプライマ
・0.5μl 5μM pMal_NheI_revプライマ
・1μl Pfuポリメラーゼ(Fermentas、2.5Wu/μl)
・36.5μl USP蒸留水
PCR中に用いたプログラム:
95℃ 180s
95℃ 45s
65℃ 45s
72℃ 240s
72℃ 480s
ステップ2乃至4は、20回繰り返した。
【0074】
1.1.3.2 ダイジェスチョン
産物を、説明書に従ってGenElute PCR Clean Upキット(Sigma、cat#NA1020)を使用して精製し、制限酵素により37℃で一晩ダイジェストし、ライゲーションに必要な粘着末端(sticky ends)を利用可能とした。
・20μl PCR産物(lacIq遺伝子)
・2.5μl 10×濃縮Y Tango緩衝液(Fermentas)
・1μl Nsi酵素(=AvaIII、Fermentas、10u/μl)
・0.5μl NheI酵素
ダイジェストしたPCR産物は、上述したようにキットにより精製し、事前に調製、ダイジェスト、精製したファージミドベクタと共に、1%アガロースジェル上で確認した。結果は、図2に示しており、レーン1は、ダイジェストされたpMal−p2X lacIq遺伝子に対応し、レーン2は、ダイジェストされたpBlueKS_NheI_Nsiベクタに対応する。
【0075】
1.1.3.3. ライゲーション
2μlダイジェスト済みpBlueKS_NheI_Nsiベクタ
6μlダイジェスト済みpMal−p2X lacIq遺伝子
1μl 10×倍濃縮T4リガーゼ緩衝液
1μl T4リガーゼ(Fermentas、1 Weiss u/μl)
ライゲーションは、室温で2時間に渡り実行した。ライゲーション産物は、上述したように40μlコンピテントE.coli XL1 Blue細胞に形質転換した。100μlの形質転換産物[LB、100μg/mlアンピシリン]を寒天プレートに塗布し、37℃で一晩インキュベートした。発生したコロニから、ミニプレップ培養物を接種し、Viogenキットを使用してプラスミドを単離した。ライゲーションは、37℃での1時間の制限消化により確認した。EcoRI酵素に対しては、添加したlacIq遺伝子の内部においてのみ切断部位が存在した。
・3.5μlミニプレップ産物
・1μl 10×EcoRI緩衝液
・0.26μl EcoRI酵素(Fermentas、10u/μl)
・5.24μl USP蒸留水
1%アガロースゲルに基づいて、ダイジェスチョンが発生したこと、すなわちライゲーションが成功したことが確認できる。新規のファージミドベクタの名前は、pBlueKS_NheI_Nsi_lacIqである。
【0076】
1.1.4. エピトープタグ及びSGCI部の導入
1.1.4.1. PCR
エピトープタグとして使用されるフラッグタグのアミノ酸配列はDYKDDDDK(SEQ ID NO 12)である。SGCI部は、外被タンパク質p8に融合させ、エピトープタグは、SGCIのN末端に融合させた。上述したように、SGCIの存在により1価での発現が確保されるため、1個のファージは、最大で1個のライブラリ構成ペプチドをその表面に提示する。
プライマ:
pGP8−Tag−NheI(SEQ ID NO 13):
5'-gtcagtgctagcatcggattataaagacgatgac-3'
P8−XbaI−rev(SEQ ID NO 14):
5'-gtcagttctagattattagcttgctttcgaggtg-3'
・5μl 10×濃縮Pfu緩衝液
・8μl 25mM MgSO4
・0.4μl 25mM dNTPs
・2μlテンプレート:pGP8−Tag−SGCIベクタ(以前の構築物)
・0.5μl 5μM pGP8−Tag−NheIプライマ
・0.5μl 5μM P8−XbaI−revプライマ
・1μl Pfuポリメラーゼ(Fermentas、2.5u/μl)
・36.2μl USP蒸留水
PCR中に用いたプログラム:
95℃ 180s
95℃ 45s
60℃ 45s
72℃ 60s
72℃ 480s
ステップ2乃至4は、25回繰り返した。
PCR産物は、Sigma GenElute PCR Clean Upキットを使用して、レシピに従って精製した。
【0077】
1.1.4.2. 制限消化
pBlueKS_NheI_Nsi_lacIqベクタを、37℃で2時間に渡り制限酵素によりダイジェストし、フラッグタグ−SGCI部のライゲーションを可能にした。
・2.5μl pBlueKS_NheI_Nsi_lacIqミニプレップ
・3.5μl 10×濃縮Tango緩衝液
・1.5μl XbaI(Fermentas、10u/μl)
・1.5μl NheI(Fermentas、10u/μl)
・3.5μl USP蒸留水
産物を1%アガロースゲルから単離し、Viogen Gel−Mキットにより精製し、45μlの水に溶出させた。その後、産物をアルカリホスファターゼにより、37℃で45分間処理した。
・ゲルから単離した43μlダイジェスト済みpBlueKS_NheI_Nsi_lacIqベクタ
・1μlエビアルカリホスファターゼ(SAP、Fermentas、1u/μl)
・5μl 10×濃縮SAP緩衝液
ホスファターゼは、65℃で15分間加熱し不活性化した。
【0078】
1.1.4.3. ライゲーション及び形質転換
反応混合物を調製する前に、ベクタ及びインサートを1.8%アガロースゲルで泳動し、濃度を確認した。結果を図3に示す。
【0079】
図3において、それぞれのレーンは以下を意味する:
1. 6μl 1kb DNAラダー(Fermentas)
2. フラッグタグ−SGCI−p8 PCR産物
3. ダイジェスト済み精製pBlueKS_NheI_Nsi_lacIqベクタ
【0080】
ライゲーションのために、反応混合物と対照産物とを、室温で90分間インキュベートした。
・2μl pBlueKS_NheI_Nsi_lacIqベクタ
・7μl フラッグタグ−SGCI−p8 PCR産物
・1μl 10×濃縮T4リガーゼ緩衝液
・1μl T4リガーゼ(Fermentas、1Wu/μl)
ライゲーションした産物は、上述したようにコンピテントE.coli XL1 Blue細胞に形質転換し、塗布して37℃で一晩培養した。
【0081】
それぞれ3mlである10アリコートの培地に、個別のバクテリアコロニを接種した後、一晩培養した液体培養物を調製し、そこから二本鎖プラスミドを単離した。XbaI及びNheI酵素により生成した粘着端部は、互いに対しても適合するため、挿入が適切な方向で実現された場合のものを、10個のクローンからDNA配列決定により単離した。Big Dye Terminator v3.1 cycle Sequencing Kit(Applied Biosystems、cat#4336917)システムをPCR反応に使用した。配列決定は、BIOMI Kft.(Godollo)により実行された。確認した10個の試料から、2つの良好な挿入が見つかった。新たなベクタの名前は、pKS−Tag−SGCI−p8である。
【0082】
1.1.5. Ser−Glyアダプタの挿入
1価での発現のために、以下の機能ユニットの配列を作製した:ライブラリ構成要素−Ser/Gly/リンカー−フラッグタグ−SGCI−p8。この目的のために、pKS−Tag−SGCI−p8ベクタがNheI及びXhoI酵素により切断され、このステップの結果として、元のフラッグタグが削除された。その後、ベクタを、適切なNheI及びXhoI粘着端部を伴うGly−Serリンカー(GGSGGSGG、SEQ ID NO 15)及びフラッグタグを含むアダプタにライゲートした。ライゲーションを確認するためにBamHI切断部位をフラッグタグの内部に形成しておいた。この酵素は、適切にライゲートしたベクタを2つの部位で分割し、そこで形成された産物は159塩基対の長さを有し、アガロースゲル電気泳動を使用して検出可能となる。
・20μl pKS−Tag−SGCI−p8ベクタミニプレップ
・3μl 10×Y Tango緩衝液
・2μl XhoI(Fermentas、10u/μl)
・5μl USP蒸留水
ベクタは、37℃で2時間ダイジェストし、与えられた条件はXhoIにとって至適条件ではなかったことから、その後、0.8%アガロースゲル上で、ダイジェスチョンが完了したかどうかを確認した。1μl NheI酵素をこれに添加し、37度で1時間インキュベートした。産物は、Viogen Gel−Mキットによりアガロースゲルから単離した。
リンカ及びフラッグタグを含むアダプタは、ダイジェスト済みベクタと共に環化させた。
アダプタ:
Ser−Gly_forward(SEQ ID NO 16):
5'-ctagctggcgggtcgggtggatccggtggcgattataaagacgatgatgacaaac-3'
Ser−Gly_reverse(SEQ ID NO 17):
5'-tcgagtttgtcatcatcgtctttataatcgccaccggatccacccgacccgccag-3'
・15μlダイジェスト済みpKS−Tag−SGCI−p8ベクタ
・2.8μl 1.3ng/μl Ser−Gly_forwardプライマ
・1.7ml 2.2ng/μl Ser−Gly_reverseプライマ
反応混合物は、90℃で1分間、その後、50℃で3分間インキュベートし、短時間遠心し、氷上に置いた。ライゲーションのため、以下のものをこれに添加した:
・2.2μl 10倍濃縮T4リガーゼ緩衝液
・1μl T4リガーゼ(Fermentas、1 Weiss u/μl)
ライゲーションは、16℃で一晩に渡って行った。コンピテントE.coli XL1 Blue細胞は、上述したように形質転換し、その後、形質転換産物[LB、100mg/ml]をプレートに塗布した。コロニから、スタータを一晩接種し、Viogen Mini−Mキットにより、ミニプレッププラスミドを説明書に従い精製した。得られた試料を、Big Dye Terminator v3.1 cycle Sequencingキットを使用して、DNA配列決定により確認し、PCR産物は、BIOMI Kft.(Godollo、ハンガリ)により処理された。
【0083】
以下では、このようにして調製されたファージミドに基づいてライブラリを作製しており、その名前はpKS−SG−Tag−SGCI−p8である。
【0084】
実施例2:ファージライブラリの調製
配列決定により確認したpKS−SG−Tag−SGCI−p8ベクタは、DNAライブラリを作製するためのテンプレートの役割を果たし、DNAライブラリは、縮重(degenerated)ライブラリオリゴ及びベクタ特異的オリゴをプライマとして使用することにより、ポリメラーゼ連鎖反応(PCR)を使用して作製した。このようにして作製されたPCR産物を、pKS−SG−Tag−SGCI−p8ベクタに挿入した。
【0085】
2.1. PCR
2.1.1. ライブラリオリゴ
上述したように、ライブラリを計画する際には、以下のランダム化スキームを使用した:
SFTIライブラリは、6箇所の選択位置(「O」位置)が完全にランダム化され、即ち、20個全てのアミノ酸の発生が許容され、位置P1では、アルギニン及びリジンのみが許容される(「R/K」位置)ように調製された。縮重オリゴヌクレオチドに関するIUPACコードを使用すると、ライブラリのオリゴヌクレオチド配列は、次のようになる(SEQ ID NO 19):
ペプチドをコードする部分には下線を付しており、ランダム化したコドンは、太字で示している。
【0086】
2.1.2 DNAライブラリの調製
ライブラリは、PCRを使用して調製され、ここで一方のオリゴは、挿入されるべきライブラリ構成要素を保有し、他方のオリゴは、ユニバーサル外部プライマである。反応混合液の総量は300μlであったが、6本のPCRチューブに分割した。
・30μl 10×濃縮Taq緩衝液
・36μl 25mM MgCl2
・2.4μl 25mM dNTP
・15μl 13μM SFTIライブラリオリゴヌクレオチド
・22μl 10μM pVIII 3’プライマ
・9μl(450ng)pKS−SG−Tag−SGCI−p8テンプレート
・180.6μl USP蒸留水
・5μl Taqポリメラーゼ(Fermentas、5u/μl)
プログラム:
1. 95℃ 60s
2. 95℃ 30s
3. 50℃ 30s
4. 72℃ 60s
5. 72℃ 120s
ステップ2乃至4は、15回繰り返した.
PCR産物は、1.5%アガロースゲル上で確認し、ExoI酵素によりダイジェストして、プライマを除去した。チューブ当たり1μlのExoI酵素と共に、37℃で45分間インキュベートし、その後、80℃で酵素を不活性化した。ホモ二重鎖を増幅するため、短い重合サイクルを挿入し、プライマは、一般に使用される外部プライマとした。
pVIII_3’(SEQ ID NO 18):
5'-gctagttattgctcagcggtggcttgctttcgaggtgaatttc-3'
各チューブに以下を添加した:
・2.5μl 2.5mM dNTP
・1μl 100μM pVIII_3’プライマ
・0.8μl Taqポリメラーゼ(Fermentas、5u/μl)
プログラムは、以前のPCRの場合と同じだが、2サイクルのみを実行した。
産物を1.5%アガロースゲル上で再度確認し、ExoI酵素によりダイジェストし、6本のPCRチューブの内容物を、3本のカラム上で、Sigma PCRクリーンアップキットによりレシピに従って精製した。溶出は、10倍に希釈したEB緩衝液中において、52μl/カラムの量で行った。
【0087】
2.2 pKS−SG−Tag−SGCI−p8ファージミドベクタへのDNAライブラリの挿入
2.2.1 ダイジェスチョン
ベクタと、インサートの役割を果たすDNAライブラリとは、2段階でダイジェストし、最初に、NheI酵素により切断を行った。DNAライブラリのダイジェスチョン中に分離した不要部分は、ほぼ完全に産物と同じサイズであったため、反応混合物から除去できなかった。この断片がベクタに入り込むのを防ぐために、ダイジェスチョンの第1の段階にはSacI酵素を更に添加した。これにより、不要部分の末端近くで、精製により除去可能な小さなフラグメントが分離され、残された大きな断片は、SacIの粘着末端を有しておりライゲーションできない。インキュベーションは、37℃で8時間及び一晩実行した。
・93μl pKS−SG−Tag−SGCI−p8ベクタ(40μg)
・15μl 10×Y Tango緩衝液
・4μl NheI酵素(Fermentas、10u/μl)
・38μl USP蒸留水
(V=150μl)
・35μl DNSライブラリPCR産物
・15μl 10×Y Tango緩衝液
・4μl NheI酵素(Fermentas、10u/μl)
・4μl SacI酵素(Fermentas、10u/μl)
・38μl USP蒸留水
(V=150μl)
続いて、他方の粘着末端を生成するAcc651(=KpnI)酵素を2倍量で添加した。Tango緩衝液の濃度も2倍とした。
ダイジェスト済みpKS−SG−Tag−SGCI−p8ベクタに対して:
・8μl Acc651(Fermentas、10u/μl)
・19.8μl 10×濃縮Tango緩衝液
ダイジェスト済みDNAライブラリに対して:
・8μl Acc651(Fermentas、10u/μl)
・11μl 10×濃縮Y Tango緩衝液
【0088】
2.2.2 ライゲーション
まず、両方のダイジェスチョン産物をゲルから分離した。ベクタは、0.8%アガロースゲルから分離し、6個のポケットに分割して、Viogen Gel−Mキットを使用して6本のカラム上で精製した。DNAライブラリは、1.8%ゲルから分離し、3本のカラム上で精製した(図4)。図4に示したゲル画像の線は、以下を意味する:
1. 1μl 100bp DNAラダー
2. 1μl精製DNAライブラリ
3. 1μl精製ベクタ
4. 5μl 1kb DNAラダー
【0089】
全ての試料をライゲーションに使用し、6本のチューブに分割して、16℃で18時間インキュベートした:
・210ml精製pKS−SG−Tag−SGCI−p8ベクタ
・100ml精製SFTI DNAライブラリ
・2ml T4リガーゼ(NEB、400,000ul/ml)
・35ml USP蒸留水
産物は、Qiagen Gel Eluteキットにより精製したが、ゲルから分離したのではなく、カラム上で精製のみを行った。溶出は、2×60μl USP蒸留水において行った。
【0090】
2.3 エレクトロポレーション、ファージライブラリの増殖
ライブラリは、エレクトロポレーションによりスーパコンピテント細胞へ導入した。我々の目的は、プラスミドを可能な限り多くの細胞に導入し、ライブラリに108乃至109片が含まれるようにすることである。
USP蒸留水中にあるため無塩状態であるDNAライブラリを、2×350mlのスーパコンピテント細胞に添加した。操作は、直径0.2cmのキュベット内において、以下のプロトコルに従って行った:2.5kV、200ohm、25μF。
エレクトロポレーション後、細胞を2×25mlのSOC培地へ慎重に移し、30分間、100rpm、37℃でインキュベートした後、試料を取り出し、段階希釈を行い、[LB]、[LB、100μg/mlアンピシリン]、及び[LB、10μg/mlテトラサイクリン]プレート上に滴下し、37℃で一晩培養した。非エレクトロポレーション対照産物と、水と共にエレクトロポレーションした対照産物とについても、同じ手順に従った。試料を取り出した後、2×25mlの培養物に2×250μlのM13KO7ヘルパーファージを感染させ、37℃で30分間、220rpmで振盪した後、産物全体を接種した。2×250 ml[2YT、100μg/mlアンピシリン、30μg/mlカナマイシン]培養物を2つの2リットルフラスコにおいて、37℃、220rpmで18時間培養した。
【0091】
タイトレーションに基づくと、我々のライブラリは、1.2×109の変異体を含んでいた。
【0092】
実施例3:ファージの選択
以下の例では、MASP−1及びMASP−2標的酵素に対する、上述した例により構築されたライブラリの選択を示す。
【0093】
3.1 標的酵素
ヒトMASP標的は、セリンプロテアーゼ(SP)ドメインと、2つの補体制御タンパク質ドメイン(CCP−1、−2)から成る(Gal 2007)。これらは組換えフラグメント産物であり、分子全体の触媒活性を有する。タンパク質は、封入体(inclusion body)の形態で生産し、ここから、生物活性を有する立体配座を再生(renaturation)により取得した。精製は、アニオン及びカチオンの交換分離により行った。タンパク質の活性は、溶液中、及び、ELISAプレートに結合させた形態で試験した。生産については、別の研究において詳細に説明されている(Ambrus 2003)。
選択において使用した標的のデータ:
MASP−1 CCP1−CCP2−SP:Mw=45478Da、cstock=0.58g/l(以降、MASP−1)
MASP−2 CCP1−CCP2−SP:Mw=44017Da、cstock=0.45g/l(以降、MASP−2)
抗フラッグタグ抗体:cstock=4g/l、(Sigma、マウスにおいて生成したモノクローナル抗フラッグM2抗体、cat# F3165)
【0094】
3.2 選択の工程
3.2.1 ファージの単離
第2.3章において説明した操作の終わりに、ファージを2×250mlの培養物において18時間に渡り作製した。選択の第1の工程では、これらを単離し、ライブラリを直ちにディスプレイのために使用できるようにした。
細胞培養物を、4℃、8,000rpmで、10分間遠心した。バクテリオファージを含む上清を清潔な遠心チューブに注ぎ入れ、その体積の1/5の沈殿剤を添加した[2.5M NaCl、20%PEG−8000]。沈殿は、室温で20分間行った。その後、4℃、10,000rpmで、15分間再度遠心した。上清を捨て、再度短時間遠心し、残液をピペットで取り除いた。白色のファージ沈殿物を25ml[PBS、5mg/ml BSA、0.05%Tween−20]緩衝液に溶解した。細胞片の可能性があるものを除去するために、再度遠心し、上清を清潔なチューブに移した。
【0095】
3.2.2.第1の選択サイクル
a)固定化:標的分子を96ウェルNunc Maxisorp ELISAプレート(cat#442404)上に固定化した。固定化操作中、MASP−1及び−2の濃度は、20μg/mlで、抗フラッグタグ抗体の濃度は2μg/mlとした。タンパク質を固定化緩衝液[200mM Na2CO3、pH9.4]において希釈し、100μlをウェルに配置した。固定化の時間は、タンパク質毎に最適化した。MASP−1は、110rev/minで混合しながら室温で60分間インキュベートし、抗体は、30分間インキュベートし、MASP−2は、4℃で一晩インキュベートした。第1の選択サイクルでは、標的タンパク質当たり12ウェルを使用した。1列おきに空の列を残した。陰性対照として、固定化バッファのみを1列に配置した。この列は、その後、標的タンパク質で覆ったものと同じ方法で処理した。
b)ブロッキング:固定化溶液を取り除き、200μl/wellのブロッキング緩衝液[PBS、5mg/ml BSA]をプレートに入れた。室温で少なくとも1時間、150rev/minで混合しながらインキュベートした。
c)洗浄:ELISAプレートを、1lの洗浄緩衝液[PBS、0.05%Tween−20]を使用して4回洗浄した。
d)選択:上述したように単離したライブラリのファージを、ピペットにより、各ウェルに100μlずつの量で入れた。室温で、110rev/minで混合しながら2.5時間インキュベートした。
e)E.coli XL1 Blue培養物:選択の実施中に、XLI Blue細胞を、前もって新たに取り出したプレートから、接種ループを使用して2×30ml[2YT、10μg/mlテトラサイクリン]の培地に接種した。これらの細胞は、後の時点で、標的タンパク質から溶出したファージに感染させる。感染の際、細胞は、指数関数的増殖の段階にある必要がある。OD600nmが約0.3乃至0.5の培養物が必要であり、これは、37℃、220rpmで、2乃至3時間培養することにより得られた。
f)洗浄:ELISAプレートを、3リットルの洗浄緩衝液を使用して12回洗浄した。
g)溶出:溶出は、100μl/wellの100mM HCl溶液を使用して行った。酸を加えて、5分間振盪し、各ウェルから、1ウェルずつ順に取り出した。個々の標的タンパク質から溶出したファージは、12×15μlの1M Trisベース緩衝液を事前に入れたチューブに収集して、ファージを含有する酸性溶液を素早く中和した。チューブは、直ちに混合し氷上に載置した。
h)感染:指数関数的増殖の段階にあるXL1 Blue培養物4.5mlを、試験管に入れ、標的タンパク質から溶出したファージ溶液500μlにより感染させた。MASP−1及びMASP−2、並びに抗体及び陰性対照物質(negative control substance)からそれぞれ溶出したファージにより、合計4つの感染を行った。培養物は、37℃、220rpmで、30分間インキュベートした。
i)タイトレーション:各感染培養物から20μlの試料を取り出し、2YT培地により体積の10倍に希釈し、更なる10×希釈により段階希釈物の調製を行った。各段階から、10μl[LB、100μg/mlアンピシリン]をプレートに滴下し、一晩37℃で培養した。
j)ヘルパーファージによる感染:試料採取の直後に、50μlのM13KO7ヘルパーファージを試験管内の各培養物に添加し、更に30分間インキュベートした。
k)全ての感染培養物を3×200ml[2YT、100μg/mlアンピシリン、30μg/mlカナマイシン]培地に移し、220rpmで混合しながら、37℃で18時間インキュベートした。対照物質は、タイトレーションのためにのみ必要であったため、更なる処理は行わなかった。
l)濃縮(Enrichment):翌朝、タイトレーションをチェックすると、1回のみの選択サイクル後に、対照物質と比較して大きな差が検出できた。抗体から溶出したファージの数は、バックグラウンドから溶出したファージの数よりも4桁も多く、MASPの場合、その差は、1乃至1.5桁であった。
【0096】
3.2.3. 第2の選択サイクル
このサイクルでは、第1の選択サイクルの場合と同じ工程を繰り返したが、ブロッキング緩衝液及び洗浄緩衝液中では、2mg/mlカゼイン(Pierce、cat#37528)をBSAの代わりに使用した。この変更により、BSAに結合するファージの増殖を回避することができる。この工程において、各標的タンパク質は、それぞれ対照を伴い(12ウェル)、前のサイクルにおいて溶出及び増殖させたファージを、各標的タンパク質に配置した。
18時間かけて産生されたファージを上述したように単離したが、最終的に、10mlの無菌PBS緩衝液に溶解した。ファージ溶液の濃度を268nmで測定し、その後、[PBS、2mg/mlカゼイン、0.05%Tween−20]緩衝液により希釈し、それぞれが均一なOD268値0.5を有するようにして、この状態で、導入の工程において使用した。第2の選択サイクル後、新たな2.7mlの指数関数的増殖中XL1 Blue細胞を300μlの溶出ファージにより感染させた。6種類全てのケース(標的タンパク質3種+対照物質3種)でタイトレーションを実行した後、更にヘルパーファージに感染させた培養物を30ml[2YT、100μg/mlアンピシリン、30μg/mlカナマイシン]培地内へ移した。
第2の選択サイクル後、抗フラッグタグ抗体について104倍の濃縮、MASP−1について10倍の濃縮、MASP−2について20倍の濃縮が得られた。
【0097】
3.2.4 第3の選択サイクル
第2のサイクルの場合と全て同じように実行し、カゼインも緩衝液中に維持した。単離後、ファージを2.8mlの無菌PBSに溶解し、提示のために、OD268が約0.5となるように希釈した。
第3の選択サイクル後、対照物質と比較して、非常に大きな濃縮値が得られた。差は、抗フラッグタグ抗体について105倍、両方のMASPについて104倍となった。
【0098】
3.3. ファージELISAアッセイを用いた個別クローンの試験
この試験では、選択された個々のクローンの内どれほどの割合が、バックグラウンドシグナルを示さずに標的タンパク質に結合能を有するかを調べた。
a)感染:MASP−1及びMASP−2の場合、選択サイクル2及び3からの溶出ファージ10μlを、指数関数的段階にあるXL1 Blue培養物90μlに添加した。30分間、220rpmで混合しながら37℃でインキュベートし、その後、20μlの量を取り出し、180μlの2YT培地をこれに添加した。この10倍希釈を更に2回繰り返した。各希釈段階から、100μlを[LB、100μg/mlアンピシリン]プレートに塗布し、37℃で一晩培養した。第1の選択サイクルにおいて抗フラッグタグ抗体から溶出したファージはまず希釈し、この後でのみ、細胞を感染させた。これは、抗体はELISAプレートの表面に遥かに容易に固定され、従って遙かに多くのファージが溶出したためである。高いファージ濃度のため、1個の細胞が複数のファージに感染し、混合した不可解な配列が生じるリスクがある。
b)注入:いわゆる「シングルルース」チューブ内で、500μlの培地[2YT、100μg/mlアンピシリン、50μl M13KO7ヘルパーファージ]中へ、個々のコロニを接種した。これらのチューブは、96ウェルELISAプレートの配置と同様に配置されており、個別に動くため、プレートインキュベータにおいて、300rev/minで混合しながら37℃で少量の培養物を生産することに適している。
c)固定化:MASP−1及びMASP−2タンパク質は濃度0.01μg/μlにおいて、抗フラッグタグ抗体は濃度1μg/mlにおいて、体積100μl/ウェルで、選択に関して上述したように、Nunc ELISA Maxisorpプレート上で固定化した。各クローンは、それぞれの標的タンパク質と、バックグラウンドと、抗フラッグタグ抗体とに対して試験した。
d)18時間後、チューブをプレート遠心機において、2,500rpmで10分間、4℃で遠心し、上清をピペットで清潔なチューブに移した。ELISA後、残留上清を2時間65℃で加熱した。この後、−20℃での保存が可能となり、配列決定に使用可能となる。
e)ブロッキング:液体を固定化試料から取り除き、200μl/ウェルの[PBS、2mg/mlカゼイン]ブロッキング緩衝液を各ウェル内に入れた。150rev/minで混合しながら、少なくとも1時間、室温でインキュベートした。
f)洗浄:1リットルの洗浄緩衝液を使用して、プレートを4回洗浄した。
g)ファージの付与:上述したように作製及び単離したファージを、[PBS、2mg/mlカゼイン、0.05%Tween−20]緩衝液を使用して2倍に希釈し、100μlをウェル内に配置した。同じクローンから、試料をピペットで合計3つのウェルに入れた。110rev/minで混合しながら、室温で1時間、インキュベートした。
h)洗浄:1.5リットルの洗浄緩衝液を使用して、プレートを6回洗浄した。
i)抗M13抗体:[PBS、2mg/mlカゼイン、0.05%Tween−20]緩衝液により10,000倍に希釈したHRPコンジュゲート・モノクローナル抗M13抗体(Amersham、cat#27−9421−01)100μlをウェルに配置し、110rev/minで混合しながら、室温で30分間インキュベートした。
j)洗浄:1.5リットルの洗浄緩衝液を使用して、プレートを6回洗浄した後、PBSにより2回洗浄した。
k)発色:USP蒸留水により2倍量に希釈した1−Step Ultra TMB−ELISA基質(Pierce、cat#34028)100μlを各ウェル内に配置し、少しの間、振盪し、50μlの1M HClを各ウェルに添加することで反応を停止させた。
l)読み取り:BioTrak II(Amersham)プレート読み取り光度計を使用して、吸光度を450nmにおいて測定した。
バックグラウンドの強度が低く、それぞれの標的タンパク質に対して少なくとも3倍以上強いシグナルを提示していた場合、ファージ上清からサンプルを取って、DNA配列決定用の試料を調製した。上清2μlを使用し、Big Dye Terminator v3.1 cycle Sequencing Kit(Applied Biosystems、cat#4336917)システムをPCR反応に使用した。これはBIOMI Kft.(Godollo)により実行された。
配列を解釈したところ、MASPの場合、第3のサイクルでは、個別の配列が僅かしか発見されず、僅かな種類のみが濃縮されていたため、第2の選択サイクルから更にクローンを選択及び試験する必要があることが判明した。我々の目的は、標的タンパク質のアミノ酸嗜好性に関するパターンを構築できるように、可能な限り多様な多数の配列を収集することであった。
【0099】
実施例4:結果
この実施例では、実施例1−3において説明した試験の結果、即ち、得られた配列について説明する。
【0100】
MASP−1より溶出したファージから、ELISAを使用して、32のクローンを試験し、最終的に9種類の個別の配列を見出した。MASP−2の場合、80のELISAポイントから21種類の個別の配列が得られ、抗フラッグタグ抗体の場合、72の試験クローンから57種類の解釈可能な配列が得られた。
【0101】
結果を解釈する際には、ディスプレイバイアスの影響を考慮する必要があった。このための一方法は、コドンの標準化であるが、これは、DNAライブラリを構築するために使用したNNKコドンでは、個々のアミノ酸について同じ頻度が保証されないためである。他方、更に現実的なアプローチとしては、前記抗体に基づいて選択された配列のデータによる標準化である。理論的に可能な全ての配列タイプが、ファージの表面上で提示可能となる訳ではない。なぜなら、それらの一部は実現可能な構造を発生させなかったり、或いは、ファージにとって負荷が大きすぎるものであったりするからである。しかしながら、抗体から取得した配列は現実に発生した配列であり、これらは、標的タンパク質に対して実行した選択の初期ステップにおいて存在していたものであるため、MASPに特異的な形態は、これらに基づいて得られた。
【0102】
データ標準化後、我々は、インターネット上でアクセス可能なWebLogo(http://weblogo.berkeley.edu/logo.cgi; Crooks 2004 and Schneider 1990)の支援により、当該配列に関する配列ロゴ図を作成した。我々は、個々の位置においてどのアミノ酸が好まれているか、また、MASP−1由来かMASP−2由来かに応じて、互いにどのくらい異なるかを調べた。更に、我々のデータを、フレームとしての役割を果たす野性型SFTIの配列と比較した。SFTIはウシトリプシンに対してナノモーラー(nM)以下の親和性で作用する阻害剤である。
個々のクローンは、上述したELISA系において、バックグラウンドとして使用したBSAと、標的として使用したそれぞれのタンパク質とについて調査し、更に、他方のMASP分子についても、交差反応の可能性を確認するために調査した。結果に基づいて、配列は、3つの群に分類することができる:
a)MASP−2から選択され、これに特異的な配列、
b)MASP−2から選択されたが、MASP−1も認識する配列、及び
c)MASP−1から選択されたが、MASP−2も認識する配列。
【0103】
MASP−1のみを特異的に認識する群は発見できなかった。2つの非選択的な群(b及びc)は、どちらのMASP標的に対して選択されたかに関係なく、非常に類似する傾向を示していた。配列ロゴ図の横軸では、個別位置の番号が確認でき、部位P1は、位置5に対応する。配列ロゴ図は、図5に示しており、ここで、図5の番号(5a、5b、5c)は、a)、b)、c)と分類した群の配列ロゴ図にそのままの順番で対応している。各位置において、ロゴのカラム高さは、その要素(我々の場合、20種類の異なるアミノ酸)の発生がどのくらい均等かを示す。この発生が均等でないほど、カラムは高くなる。完全に均等な分布の場合(20種類全てのアミノ酸が5%の割合で発生する場合)、高さはゼロになる。最高値は、1種類のみの要素(アミノ酸)が発生した場合に起こる。カラム内では、個々のアミノ酸は、発生頻度に基づいて配置され、最も頻度の高いものが一番上になる。アミノ酸を示す文字の高さは、その位置における相対的な発生頻度に比例している(例えば、発生頻度50%の場合、カラムの高さの半分となる)。カラーの図の場合、一般に、類似する化学特性を有するアミノ酸は、同一又は類似する色で図示されるが、本特許明細書の図では、異なる灰色の陰影を用いている。
【0104】
ロゴ図の支援により、我々は、選択的及び非選択的な群のコンセンサス配列を決定し、選択に由来するクローンの名前に基づいて、M2−6E及びM2−4Gペプチドと命名し、その活性を反映した名前は、「S」ペプチド(選択的(selective)のS)又は「NS」ペプチド(非選択的(non−selective)のNS)とした(以下参照)。
MASP−2選択的M2−6Eクローン(SEQ ID NO 2):
「S」ペプチド GYCSRSYPPVCIPD
非選択的M2−4Gクローン(SEQ ID NO 3):
「NS」ペプチド GICSRSLPPICIPD
【0105】
上述したペプチドと、その点突然変異体及び環状変異体を、固相ペプチド合成により生成した。合成及びペプチド分析試験は、実施例5において説明する。
【0106】
実施例5:ペプチド合成及び分析
5.1. ペプチドの調製、再生、及び品質検査
ペプチドは、標準的なFmoc(N−(9−フルオレニル)メトキシカルボニル)手順(Atherton 1989)を使用した固相ペプチド合成により生成した。担体からの分離と、保護基の同時除去とは、1,2−エタンジチオール、チオアニソール、水、及びフェノールがラジカル捕獲剤として存在する状態で、TFA(トリフルオロ酢酸)法を使用して実行した。ほぼ乾燥するまで溶液が蒸発した後、低温のジエチルエーテルを使用して、産物を沈殿させた。沈殿物を水に溶解した後、揮発性成分を凍結乾燥により除去した。再生のため、即ち、ペプチドの2本のシステイニル側鎖間にジスルフィド架橋を形成するために、凍結乾燥産物を、濃度0.1mg/mlで水に溶解した。継続的なエアリングに加えて溶液を混合することにより、酸化を実行し、pH値は、N,N−ジイソプロピル−エチルアミンを加えることによりアルカリ値(8乃至9)に維持した。完全な酸化の達成は、逆相HPLC手順及び質量分析法を用いて試験した。95%超の均質な形態での酸化産物の単離も、逆相HPLC手順により実行した。
M2−4Gペプチドの場合、環状形態でも生成され、この場合、線状のもののN及びC末端間に、ペプチド結合を形成させた。環化は、下記のように実行した。ペプチド合成を、2−ClTrt(2−クロロトリチル)樹脂上で行い、1%TFAを含有するDCM(ジクロロメタン)溶液を使用して、ここからペプチドを分離した。こうした条件下では、側鎖保護基は、ペプチド上に留まる。逆相HPCL手順を用いた分離ペプチドの精製後、線状ペプチドを、ペプチドの最終濃度が0.1mMになる量のDMF(ジメチルホルムアミド)を使用して溶解した。その後、1.1当量のHATU(1−[ビス(ジメチルアミノ)メチレン]−1H−1,2,3−トリアゾロ[4,5−b]ピリジン−3−オキシドヘキサフルオロリン酸)と、3.0当量のDIPEA(ジイソプロピルエチルアミン)を、これに添加した。溶液を30分間室温で混合した後、逆相HPCL手順及び質量分析法を用いて環化の効率を試験した。環化の完了後、試料を蒸発させ、調製的逆相HPLC手順を用いて、ペプチドを精製した。
【0107】
各分離ペプチドについて、品質コントロールは、質量分析手順を用いて実行した。質量分析法による分析は、HP1100型HPLC−ESI−MSシステムにより、10mMギ酸アンモニウムpH3.5溶液を使用して、フローインジェクション法により行った。機器は、以下のパラメータに設定した。乾燥ガス及び噴霧(atomizing)ガスは、共に窒素とし、乾燥ガスの流量は、10l/分、温度は300℃とした。噴霧ガスの圧力は、210kPa、キャピラリ電圧は、3500Vとした。全イオン電流(TIC)クロマトグラムは、300乃至2000質量/電荷の範囲において正イオンの設定で作成された。質量データは、Agilent ChemStationソフトウェアにより評価した。
作製した個々の阻害剤の名前、配列、及び質量データを下表1に示す。
【0108】
【表1】
表1:化学合成により製造した、本発明による幾つかのペプチド阻害剤の理論的及び実測分子量
【0109】
表1に示した配列において、ライブラリ構築中にランダム化した位置には下線を付しており、野性型SFTIとアミノ酸が異なる位置は、太字で示している。
【0110】
5.2.合成ペプチド基質による定数Kiの決定
ペプチドの阻害能力を、まず、MASP酵素とトリプシンとについて測定した。MASP酵素に対して最も有望な阻害データを示した2つのペプチド(以下参照)のみ、トロンビンについても阻害能力を測定した。
【0111】
5.2.1.MASP酵素による測定
測定に使用した合成基質は、Z−L−Lys−SBzl塩酸塩(Sigma、C3647)であり、ここから10mMストックソリューションを調製した。反応は、1mlの量で、室温において、[20mM HEPES、145mM NaCl、5mM CaCl2、0.05%Triton−X100]から成る緩衝液中で行った。酵素により切断された基質は、溶液内に2倍超過で存在するジチオジピリジン補助基質(Aldrithiol−4、Sigma、cat#143057)との反応に入る。このようにして形成された発色基の放出を、分光光度計により324nmでモニタした。段階希釈を合成ペプチドから調製し、これに酵素を添加し、室温で1時間インキュベートした。基質の濃度及び測定時間の長さは、酵素が基質の10%未満を消費する条件となるように選択した。測定の過程では、強力結合(tight-binding)阻害剤の解析用に開発された測定方法を使用した(Empie, 1982)。反応の初期段階で示された直線の傾斜は、阻害されていない酵素反応の場合に得られた傾斜により標準化され、酵素の量で乗じた。この結果として、遊離酵素の濃度を求め、阻害剤の濃度の関数として示し、次の等式1に従って表した。
ここで、Eは、遊離(非阻害)酵素濃度、E0は、初期酵素濃度である。MASP−1 MASP−2濃度は、C1阻害剤による滴定により決定した。結果は、平行して行われた複数の測定の平均として計算した。結果を、5.3.の表2にまとめる。
【0112】
5.3. トリプシン及びトロンビンに関する測定
2つのコンセンサスペプチド、即ち、M2−6E及びM2−4Gが、最も有望なMASP−2及びMASP−1阻害剤であることがわかったため、トリプシン及びトロンビンに対する阻害能力に関してこれらを当初のSFTI分子と比較することにより、その解析を続けた。トリプシン阻害の測定には、上述した測定条件を使用し、従って、トリプシンの活性は、Z−L−Lys−SBzl塩酸塩基質において、阻害ペプチド濃度の関数として測定した。評価は、上述したように行った。
【0113】
MASP酵素は、その生理的機能を血液中で遂行するため、ペプチドを使用することについての可能性は、血清中の他のプロテアーゼの活性に対してどのような影響を有するかにより決まる。我々は、血液凝固の中心的な酵素であるトロンビンを、同様の条件下で、但しZ−Gly−Pro−Arg−pNa基質を用いて、調査した。当該p−ニトロアニリドは、補助基質を必要とせず、産物の形成を、分光光度計において405nmで直接モニタすることができる。細いキュベット内の測定体積を350μlとし、基質の濃度は、505μMとした。トロンビンは、異なる阻害剤濃度で、20分間室温でインキュベートした。トロンビンの量は、活性部位滴定法を使用して決定した。評価は、上述したように行った。結果を下表2にまとめる。
【0114】
【表2】
表2:個々の阻害剤の酵素阻害を要約する表。表示した配列において、下線及び太字は、表1と同じ意味を有する。
【0115】
特に記載がない場合、阻害剤は、開鎖(open chain)を有する。記号「NG」は、使用した中で最も高い阻害剤濃度をもってしても阻害が測定できなかったことを意味する。記号「−」は、その酵素/阻害剤対に関して測定が実行されなかったことを意味する。
【0116】
データに基づくと、選択的ペプチド(M2−6E、SEQ ID NO 2)は、MASP−2を選択的に阻害し、MASP−1に対する活性はなく、トリプシンに対してはその活性は4桁も低く、トロンビン阻害剤としての能力も非常に低いと言える。これと対照的に、非選択的ペプチド(M2−4G、環状、SEQ ID NO 3)は、遙かに一般的な阻害剤の特徴を示す。このペプチドは、4種類全てのプロテアーゼを阻害し、トリプシンに対しては、野性型SFTI−1よりも遙かに弱い。トロンビン阻害剤としては劣っているが、野性型と比較すると、親和性が向上している。
【0117】
5.4. 血液凝固に対するペプチドの作用
健康な個体から採取した血漿を用いて、血液凝固の測定を行った。静脈穿刺により取得し、クエン酸ナトリウム(3.8%wt/vol)により処理した血液から、血漿を遠心(2000g、15分間、Jouan CR412遠心機)により単離した。
【0118】
血液凝固の外因経路を試験するプロトロンビン時間(PT)を、Sysmex CA−500(Sysmex、日本)自動システムにおいてInnovin試薬(Dale Behring、ドイツ、マーブルク)を使用して測定した。血液凝固の内因経路を試験する活性化部分トロンボプラスチン時間(APTT)と、トロンビンの働きを直接試験するトロンビン時間(TT)とを、Coag−A−Mate MAX(BioMerieux、フランス)アナライザにおいて、TriniClot試薬(Trinity Biotech、アイルランド、ウィックロ)及びReanal試薬(Reanal Finechemical、ハンガリ)を使用して測定した。
【0119】
血液凝固に対するペプチドの影響を調査するために、我々は用量依存性を測定した。結果を図6のグラフに示す。各図において、破線間の領域は、その測定との関連における正常範囲を示す。縦座標には、時間を秒単位で定め、横座標には、阻害剤濃度の対数を、μM単位で示す。
【0120】
図6aは、トロンビン時間を測定するための実験を示しており、その過程では、血漿にトロンビンを添加することにより血漿凝固(フィブリン形成)を開始させた。外部から添加したトロンビンの影響を、ペプチドの濃度を増加させつつ阻害し(横座標)、凝固に必要な時間を測定する(縦座標)。図6bは、プロトロンビン時間を測定するための実験を示しており、その過程では、血漿に組織因子を添加することにより血漿凝固(フィブリン形成)を開始させ、その結果、第VII因子の活性化により、トロンビンを活性化するプロトロンビナーゼ複合体が幾つかのステップにおいて形成される。この実験では、外傷(血管損傷)の結果として活性化される血液凝固の外因経路を模倣している。組織因子により開始されるプロテアーゼカスケードの構成要素を、ペプチドの濃度を増加させつつ阻害し(横座標)、凝固に必要な時間を測定する(縦座標)。図6cは、例えば、血液中のコラーゲンの発生により生理的に開始される、血液凝固のいわゆる「接触活性又は内因性」経路を模倣した、活性トロンボプラスチン時間を測定するための実験を示す。実験では、コラーゲンの代わりに、異なる大表面(large-surface)素材、例えばカオリン粉末を添加することにより実現する。この結果として、活性化第XII因子を介して、やはりプロテアーゼカスケードが開始され、その結果として、トロンビンを活性化するプロトロンビナーゼ複合体が形成される。このプロテアーゼカスケードの構成要素を、ペプチドの濃度を増加させつつ阻害し(横座標)、凝固に必要な時間を測定する(縦座標)。
【0121】
3種類全ての測定について、選択的「S」ペプチドは、濃度が200μMであっても正常領域近くに留まっており、従って、MASP阻害という側面で問題となるような濃度において凝固を阻害しなかった。これとは対照的に、非選択的“NS”ペプチドは、200μMの場合に、極端な測定値に達し、これは、血液凝固を有意に阻害したことを意味する。前章で説明したデータでは、「NS」ペプチドがトロンビンを10μMのKi値で阻害することが実証されており、このことだけでも、当該試験において示された作用が説明される。血液凝固の最後のステップにおいて、トロンビンは、フィブリノゲンを分解し、これによりフィブリンに基づく凝塊を形成する酵素である。そのため、トロンビンを阻害することは、それだけで血液凝固を効率的に阻害するのに十分となる。このため、上述した血液凝固試験に基づき、比較的好適にトロンビンを阻害する「NS」ペプチドが、血液凝固カスケード中で機能的観点においてトロンビンに先行する他の血液凝固因子(例えば、VIIA、IXa、Xa、XIa、XIIa)を同じく阻害するか否かを判断することはできない。その一方で、3種類全ての試験において実証された選択的「S」ペプチドの血液凝固に対する作用の弱さは、このペプチドは、カスケードの初期成分に対しても強力な阻害剤にはなり得ないことを示している。
【0122】
5.5.本発明によるペプチドの3つの補体活性化経路に対する作用
詳細に上述してきたように、補体系は、3つの経路により活性化可能であり、同じ単一の終点につながる。3つの活性化経路は、古典経路、レクチン経路、及び副経路を含む。MASPは、レクチン経路の初期段階の酵素であるため、本発明によるMASP阻害剤が、レクチン経路、他の2つの活性化経路、及び3経路が出会った後の共通段階に対して、どのような作用を有するかを知ることは重要である。
【0123】
測定のため、我々は、補体経路の選択的測定用に開発された、いわゆるWIELISAキット(Euro−Diagnostica AB、COMPL300)を、キットに付属の使用説明書に基づいて使用した。測定の指針は、3つの活性化経路に従って3種類の測定条件を使用し、現在検査中の補体活性経路は機能可能な状態にすると同時に他の2つの経路は不活性であるようにすることである。その一方で、測定において検出する産物は、経路選択的な成分ではなく、3つ活性経路の共通部分の最終要素であるC5−9複合体である。
【0124】
測定のために、血液試料を1時間室温でインキュベートし、その後、遠心して、血清を小分けにして−80℃で保存した。血清を、処方に従って所定の補体経路用の緩衝液により希釈し、20分間室温でインキュベートし、ペプチドの段階希釈をこれに添加し、20分間室温でインキュベートし、その後、ピペットで、特別なELISAプレートの適切なウェルに移し入れた。続いて、洗浄、インキュベート、及び抗体の添加を使用説明書に従って実行した。基質と共に20分間インキュベートした後、分光光度計において450nmでデータを読み取った。各測定点において平行実験を行い、100%の活性は、阻害剤を含まない血清により表された。測定は、同じプレート上で同時に、溶解した1つの同じ血清試料を用いて、実行した。
【0125】
当該測定により、「S」ペプチド及び「NS」ペプチドが共に補体系のレクチン経路の効率的且つ特異的な阻害剤であるという極めて重要な結果が得られた。この結果は、我々の現在の知識によればレクチン経路の開始に関与する酵素であるMASP−2酵素を、両方のペプチドが非常に効率的に阻害するという、前述の結果と一致するものである。
【0126】
多数のセリンプロテアーゼが補体系において作用しており、その一部は、MASP酵素に非常に似ている。これにもかかわらず、「S」ペプチドも「NS」ペプチドも、古典経路および副経路を阻害しなかった。
【0127】
古典経路及び副経路の測定の過程では、本発明によるペプチドの存在は、最終的なC5−9複合体の形成を阻害しなかったことから、本発明のペプチドが補体系の共通部分のプロテアーゼを阻害しないことは確実であり、従って、レクチン経路の阻害は、実際に、レクチン経路の開始時点において、すなわちMASP酵素のレベルにおいて生じたものである。WIELISA測定の過程で得られたIC50データは、合成基質に基づくMASP−2阻害の測定の過程で得られたki値に比べて約30倍、60倍高いことは指摘するに値する。これには以下の説明が可能である:阻害ペプチドは、MASP−2酵素と直接、基質結合部位において結合し、この結合は、当該同じ酵素表面と小さな合成基質との比較的弱い相互作用に対して上手く競合する。しかしながら、生理学的基質は、プロテアーゼドメインに位置した基質結合部位に加え、他の表面(エキソサイト)を介した結合を形成することも可能であり、これらは小さな合成基質よりも高い親和性で酵素と結合する。この高い親和性のため、阻害ペプチドは、バランスを酵素基質複合体から酵素阻害剤複合体の方へ移行させるために、より高い濃度で使用する必要がある。
【技術分野】
【0001】
本発明は、新規のペプチド、特にオリゴペプチドに関し、更に、こうしたペプチドの生産のための方法と、医薬の生産におけるこうしたペプチドの使用とに関する。
【背景技術】
【0002】
補体系は、ヒト及び動物生命体の先天性免疫における最も重要な構成要素の1つである。補体系は、免疫系一般と同様に、侵入病原体及び変性した宿主構造(例えば、アポトーシス細胞)を認識、標識、及び除去する能力を有する。補体系は、先天性免疫系の一部として、病原微生物に対する生命体の第1の防御線の1つを形成するが、幾つかの点において、適応(後天性)免疫系にも関連しており、先天性免疫及び適応免疫メカニズム間の言わば橋渡しをしている(Walport 2001a; Walport 2001b; Morgan 2005)。補体系は、約30のタンパク質成分から成るネットワークであり、こうした成分は、血漿中において、可溶性の形態として見られ、更には、細胞の表面に付着した受容体及びモジュレータ(例えば、阻害因子)の形態でも見られる。当該系の主成分は、セリンプロテアーゼチモーゲンであり、厳密に定められた順序でカスケードの形で相互に活性化し合う。活性化プロテアーゼの特定の基質は、チオエステル結合を含むタンパク質である(補体系の成分C4及びC3)。これらの基質が活性化プロテアーゼにより切断される時、反応性チオエステル基が分子の表面に露出した状態となり、これにより、当該切断された分子を、攻撃される細胞の表面に付着させることが可能となる。結果として、こうした細胞は標識され、免疫系により認識可能となる。
【0003】
補体系の生物学的機能は、極めて多様且つ複雑であり、今日に至るまで全ての詳細については探求がなされていない。最も重要な機能の1つは、直接的な細胞毒性活性であり、補体系の終結的要素をなす成分から形成された膜侵襲複合体(MAC)により誘発される。MAPは、異物と認識された細胞の膜に穿孔し、溶解をもたらすことで、こうした細胞を破壊する。
【0004】
補体系の他の重要な機能には、細胞表面に定着する活性補体成分(例えば、C1q、MBL、C4b、C3b)が白血球(例えば、マクロファージ)による貪食を促進する際のオプソニン化がある。こうした白血球は、破壊するべき細胞を飲み込む。
【0005】
更に、補体系の炎症開始における役割も、特に重要である。補体活性化中に放出された切断産物は、白血球に対するその化学走性刺激作用(chemotactic stimulating effects)により、炎症プロセスを開始させる(Mollnes 2002)。
【0006】
補体系の成分は、適切な信号(例えば、異質細胞、病原体の侵入)により補体カスケードの活性化が誘発されるまでは、不活性(チモーゲン)型として血漿中に存在する。補体系の正常な活性は、免疫ホメオスタシスを維持する観点から重要である。異常な活性低下と無制御な活性過剰とは、共に重篤な疾患の発生、又は既存の疾患の悪化をもたらす恐れがある(Szebeni 2004)。
【0007】
補体系は、古典経路、レクチン経路、及び副経路という3種類の経路で活性化され得る。古典経路の第1のステップにおいて、C1複合体は、活性化因子、すなわち異物と認識された生物学的構造の表面に結合する。C1複合体は、認識タンパク質分子(C1q)と、それに結合するセリンプロテアーゼ(C1r、C1s)とから成る超分子複合体である(Arlaud 2002)。まず、C1q分子が、免疫複合体、アポトーシス細胞、C反応性タンパク質、及び他の活性化因子構造と結合する。C1q分子が活性化因子と結合した結果として、C1複合体に存在するセリンプロテアーゼチモーゲンは徐々に活性化される。四量体C1s−C1r−C1r−C1sにおいて、まず、C1rチモーゲンが自己活性化し、その後、活性C1r分子が、C1s分子を切断及び活性化する。活性C1sは、補体系のC4及びC2成分を切断し、その切断産物は、C3転換酵素(convertase)複合体(C4bC2a)の前駆体となる。C3転換酵素は、C3成分を分割し、C5転換酵素(C4bC2aC3b)へ変換する。C5転換酵素は、C5を切断し、その後、補体系の活性化は、3種類全ての経路の特徴である終結期に達する(MACの形成)。
【0008】
補体系の別の経路であるレクチン経路の活性化は、古典経路のものと非常に似ている(Fujita 2004)。しかしながら、この場合、MBL(「マンノース結合レクチン」)及びフィコリン(H、L、及びM型)という幾つかの異なる種類の認識分子が関与する。こうした分子は、微生物表面上の糖鎖構造に結合する。認識分子の結合に続いて、MASP−2(「MBL関連セリンプロテアーゼ」−2)チモーゲンの自己活性化が生じる。活性MASP−2は、C4及びC2成分を切断し、古典経路の過程において既に説明したC3転換酵素複合体の形成をもたらし、これ以降、プロセスは上述したように続く。
【0009】
副経路は、C3成分の切断と、異物と認識された生物学的構造の表面への固定とにより開始される(Harboe 2008)。当該切断によって形成されたC3b成分は、微生物の細胞膜に結合した場合には、同時に、B因子(C3bB)と呼ばれるセリンプロテアーゼのチモーゲン型にも結合し、B因子は、活性型で血中に存在するD因子による切断により活性化される。このように形成されたC3bBb複合体は、副経路のC3転換酵素であり、更なるC3b分子により補完された後、C5転換酵素に変化する。副経路は、C3成分(C3w)の緩やかな加水分解により、自発的に独立して誘発され得るが、古典経路又はレクチン経路の何れかがC3切断の時点まで到達した場合、副経路は、その効果を著しく増幅させる。
【0010】
上述した経路のうち、最近になって発見され最も理解が進んでいないが、本発明の観点からは最も重要である、レクチン経路について、更に詳細に説明する。数種類のプロテアーゼ及び非触媒タンパク質が、幾つかの異なる形態で存在する認識分子(重合の度合いが異なるMBL及びフィコリン)に結合する。MASP−2は、それ自体でも補体カスケードを開始する能力を有するが(Ambrus 2003; Gal 2005)、この酵素は、MASP−1に比べ少ない量(0.5μg/ml)で存在している。存在する量がより多いMASP−1プロテアーゼ(7μg/ml)の生理的機能は、未だ完全には探求されていない。
【0011】
MASP−1は、それ自体では補体カスケードを開始できないが(C2のみ切断可能でありC4は切断できない)、その活性は、幾つかの点でMASP−2の活性を補っており、そのため、活性MASP−1は、レクチン経路の作用を増幅及び完了させるために必要となり得る。MASP−1はトロンビンにある程度類似したプロテアーゼであり、血液中において、2つの主要なタンパク質分解カスケード系である補体系と血液凝固系との間の橋渡しをすることを示す幾つかの兆候がある(Hajela 2002; Krarup 2008)。
【0012】
MASP−1及びMASP−2の遺伝子は共に、選択的スプライシング産物を有する。MAp19(sMAP)タンパク質は、MASP−2遺伝子から生産され、MASP−2の最初の2つのドメイン(CUB1−EGF)を含む。MASP−3 mRNAは、MASP−1遺伝子から転写される。MASP−3の最初の5つのドメインは、MASP−1のドメインと同じだが、それらはセリンプロテアーゼドメインにおいて異なる。MASP−3は、合成基質に対するタンパク質分解活性が低く、その天然基質は知られていない。また、他の初期プロテアーゼとは異なり、C1阻害剤分子との複合体を形成しない。恐らく、MAp19及びMASP−3の両方が存在することは、タンパク質分解に関して不活性であるこれらのタンパク質が認識分子上の結合部位について活性MASP−2及びMASP−1酵素と競合することから、レクチン経路の活性化に抗することとなる。
【0013】
上述したように、ヒト又は動物生命体における補体系の異常は、疾患を発生させる恐れがある。補体系の無制御な活性化は、自己組織の損傷、及び炎症又は自己免疫状態の発生をもたらす恐れがある(Beinrohr 2008)。こうした状態の1つは、虚血再灌流(以降、IR)障害であり、組織への酸素供給が何らかの理由(例えば、血管閉塞)により一時的に制限又は中断され(虚血)、血液循環の回復(再灌流)後、細胞破壊が始まる際に生じる。再灌流中、補体系は、虚血細胞を変性した自己細胞として認識し、これらを除去するために炎症反応を開始する。この現象は、心筋梗塞及び脳卒中後に生じる組織損傷の原因の一部であり、冠動脈バイパス形成手術及び臓器移植術中に合併症を引き起こす場合もある(Markiewski 2007)。レクチン経路は、IR障害の発生において恐らく一定の役割を果たす。この理由から、レクチン経路の意図的な抑制は、IR障害の程度及び結果を低減し得る。関節リウマチ(以降、RA)の場合にも、RA中に関節に蓄積されるグリコシル化状態が変化したIgG−G0抗体型とMBLが結合するため、レクチン経路は、活性状態となる場合がある。補体系の無制御活性は、様々な神経変性疾患(例えば、アルツハイマー病、ハンチントン病、パーキンソン病、多発性硬化症)の発生及び維持においても役割を果たし、加齢性黄斑変性症(AMD)の発病における主な要因の1つでもある(Bora 2008)。後者の臨床像は、先進工業国における加齢による視力喪失の全症例の半分に関与する。補体系は、自己免疫性腎炎(糸球体腎炎)の形態の1つと、更に別の自己免疫疾患、即ちSLE(全身性エリテマトーデス)にも関連し得る。
【0014】
補体系を初期ステップ中に阻害する場合、一般的な免疫抑制を誘発することなく、特定の活性化経路の効率的且つ選択的な阻害が可能となる。MASP−1及びMASP−2酵素を阻害することにより、レクチン経路を選択的に遮断すること(例えば、上述した疾患において)が可能となり、免疫複合体の除去に関与する古典経路は、これにより影響されず、機能を続ける。
【0015】
C1r、C1s、MASP−1、MASP−2、及びMASP−3酵素は、同じドメイン構造を有する酵素ファミリを形成する(Gal 2007)。タンパク質分解活性に関与するトリプシン様セリンプロテアーゼ(SP)ドメインの前に、5つの非触媒ドメインがある。分子のN末端部分を形成する3つのドメインCUB1−EGF−CUB2(CUB=C1r/C1s、ウニUegf及び骨形成タンパク質−1、EGF=上皮成長因子)は、分子の二量体化(MASP−1及びMASP−2の両方の場合)に関与すると共に、分子との相互作用、例えば、認識分子との結合に関与する。
【0016】
分子のC末端CCP1−CCP2−SPフラグメント(CCP=補体制御タンパク質)は、その触媒性に関して、分子全体と同等である。補体プロテアーゼの特性の1つは、基質特異性が非常に狭く、ほんの少数のタンパク質基質の明確に定められたペプチド結合だけを切断する能力を有することである。CCPモジュール及びSPドメインは、共に、この精密な特異性に寄与する。
【0017】
SPドメインは、セリンプロテアーゼの特徴である活性中心と、基質結合ポケットと、オキシアニオン穴(oxyanion hole)とを含む。8個の表面ループ領域は、様々なプロテアーゼにおいて立体配座が大きく異なり、サブサイト特異性を定める上で決定的な役割を果たす。
【0018】
CCPモジュールは、一方において、触媒領域の構造を安定化させ、他方において、大きなタンパク質基質に対する結合部位を含む。トリプシン様セリンプロテアーゼを阻害するために一般に使用される小分子化合物(例えば、ベンズアミジン、NPGB、FUT−175)は、補体プロテアーゼの活性も阻害するが(Schwertz 2008)、この阻害は、十分に選択的ではなく、血漿中の他のセリンプロテアーゼ、例えば、血液凝固酵素、カリクレイン等の不活性化にも及ぶ。
【0019】
補体系の天然阻害剤としては唯一既知のものであるC1阻害タンパク質は、血中を循環し、セルピンファミリに属するものであるが、比較的広い特異性を有する。
【0020】
レクチン経路を効率的且つ選択的に阻害する能力を有する化合物又は天然阻害タンパク質は、最新技術においても知られていない。
【先行技術文献】
【非特許文献】
【0021】
【非特許文献1】Ambrus, G., Gal, P., Kojima, M., Szilagyi, K., Balczer, J., Antal, J., Graf, L., Laich, A., Moffatt, B., Schwaeble, W., Sim, R.B. and Zavodszky, P. (2003) Natural substrates and inhibitors of mannan-binding lectin-associated serine protease-1 and -2: a study on recombinant catalytic fragments. J. Immunol. 170, 1374-1382.
【非特許文献2】Arlaud, G.J., Gaboriaud, C., Thielens, N.M., Budayova-Spano, M., Rossi, V. and Fontecilla-Camps, J.C. (2002) Structural biology of the C1 complex of complement unveils the mechanisms of its activation and proteolytic activity. Mol. Immunol. 39, 383-394
【非特許文献3】Atherton, E.; Sheppard, R.C. (1989). Solid Phase peptide synthesis: a practical approach. Oxford, England: IRL Press. ISBN 0199630674.
【非特許文献4】Beinrohr, L., Dobo, J., Zavodszky, P. and Gal, P. (2008) C1, MBL-MASPs and C1-inhibitor: novel approaches for targeting complement-mediated inflammation. Trends in Molecular Medicine doi:10.1016/j.molmed.2008.09.009
【非特許文献5】Bora, N.S., Jha, P. and Bora, P.S. (2008) The role of complement in ocular pathology. Semin. Immunopathol. 30, 85-95
【非特許文献6】Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. "WebLogo: A sequence logo generator", Genome Research, 14:1188-1190
【非特許文献7】Dobo, J., Harmat, V., Sebestyen, E., Beinrohr, L., Zavodszky, P. & Gal, P. (2008). Purification, crystallization and preliminary X-ray analysis of human mannose-binding lectin-associated serine protease-1 (MASP-1) catalytic region. Acta Crystallogr Sect F Struct Biol Cryst Commun 64, 781-4.
【非特許文献8】Empie, M. W. & Laskowski, M., Jr. (1982). Thermodynamics and kinetics of single residue replacements in avian ovomucoid third domains: effect on inhibitor interactions with serine proteinases. Biochemistry 21, 2274-84.
【非特許文献9】Fujita, T., Matsushita, M. and Endo, Y. (2004) The lectin-complement pathway - its role in innate immunity and evolution. Immunol. Rev. 198, 185-202
【非特許文献10】Gal, P., Barna, L., Kocsis, A. and Zavodszky P. (2007) Serine proteases of the classical and lectin pathways: Similarities and differences Immunobiol. 212, 267-277
【非特許文献11】Gal, P., Harmat, V., Kocsis, A., Bian, T., Barna, L., Ambrus, G., Vegh, B., Balczer, J., Sim, R.B., Naray-Szabo, G., Zavodszky, P. (2005) A true autoactivating enzyme. Structural insights into mannose-binding lectin-associated serine protease-2 activation. J. Biol. Chem. 280, 33435-33444.
【非特許文献12】Hajela, K., Kojima, M., Ambrus, G., Wong, K.H.N., Moffatt, B.E., Fergula, J., Hajela, S., Gal, P., Sim, R.B. (2002) The biological functions of MBL-associated serine proteases (MASPs). Immunbiol. 205, 467-475.
【非特許文献13】Harboe, M and Mollnes, T.E. (2008) The alternative complement pathway revisited. J. Cell. Mol. Med. 12, 1074-1084
【非特許文献14】Harmat, V., Gal, P., Kardos, J., Szilagyi, K., Ambrus, G., Vegh, B., Naray-Szabo, G. & Zavodszky, P. (2004). The structure of MBL-associated serine protease-2 reveals that identical substrate specificities of C1s and MASP-2 are realized through different sets of enzyme-substrate interactions. J Mol Biol 342, 1533-46.
【非特許文献15】Korsinczky, M. L., Schirra, H. J., Rosengren, K. J., West, J., Condie, B. A., Otvos, L., Anderson, M. A. & Craik, D. J. (2001). Solution structures by 1H NMR of the novel cyclic trypsin inhibitor SFTI-1 from sunflower seeds and an acyclic permutant. J Mol Biol 311, 579-91.
【非特許文献16】Krarup, A, Gulla, K.C., Gal, P. Hajela, K. and Sim R.B. (2008) The action of MBL associated serine protease 1 (MASP1) on factor XIII and fibrinogen. Biochim. Biophys. Acta. 1784, 1294-1300.
【非特許文献17】Kunkel, T. A., Bebenek, K. & Mcclary, J. (1991). Efficient Site-Directed Mutagenesis Using Uracil-Containing DNA. Methods in Enzymology 204, 125-139.
【非特許文献18】Laskowski, M., Jr. & Kato, I. (1980). Protein inhibitors of proteinases. Annu Rev Biochem 49, 593-626.
【非特許文献19】Luckett, S., Garcia, R. S., Barker, J. J., Konarev, A. V., Shewry, P. R., Clarke, A. R. & Brady, R. L. (1999). High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J Mol Biol 290, 525-33.
【非特許文献20】Malik, Z., Amir, S., Pal, G., Buzas, Z., Varallyay, E., Antal, J., Szilagyi, Z., Vekey, K., Asboth, B., Patthy, A. & Graf, L. (1999). Proteinase inhibitors from desert locust, Schistocerca gregaria: engineering of both P-1 and P-1 ' residues converts a potent chymotrypsin inhibitor to a potent trypsin. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology 1434, 143-150.
【非特許文献21】Markiewski, M.M. and Lambris, J.D. (2007) The role of complement in inflammatory diseases - from behind the scenes into the spotlight. Am J. Pathol. 171, 715-727.
【非特許文献22】Mollnes, T.E., Song, W.C. and Lambris, J.D. (2002) Complement in inflammatory tissue damage and disease. Trends Immunol. 23, 61-64
【非特許文献23】Morgan, B.P., Marchbank, K.J., Longhi, M.P., Harris, C.L. and Gallimore, A.M. (2005) Complement: central to innate immunity and bridging to adaptive responses. Immunol. Lett. 97, 171-179
【非特許文献24】Mulvenna, J. P., Foley, F. M. & Craik, D. J. (2005). Discovery, structural determination, and putative processing of the precursor protein that produces the cyclic trypsin inhibitor sunflower trypsin inhibitor 1. J Biol Chem 280, 32245-53.
【非特許文献25】Schneider TD, Stephens RM. 1990. "Sequence Logos: A New Way to Display Consensus Sequences." Nucleic Acids Res. 18:6097-6100
【非特許文献26】Schwertz H, Carter JM, Russ M, Schubert S, Schlitt A, Buerke U, Schmidt M, Hillen H, Werdan K, Buerke M. (2008) Serine protease inhibitor nafamostat given before reperfusion reduces inflammatory myocardial injury by complement and neutrophil inhibition. J Cardiovasc Pharmacol. 2008 Aug;52(2):151-60.
【非特許文献27】Smith, G. P. (1985). Filamentous Fusion Phage - Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 228, 1315-1317.
【非特許文献28】Szebeni, J. ed. (2004) The complement system. Novel roles in health and disease Kluwer Academic Publishers, ISBN 1-4020-8055-7.
【非特許文献29】Szenthe, B., Patthy, A., Gaspari, Z., Kekesi, A. K., Graf, L. & Pal, G. (2007). When the surface tells what lies beneath: Combinatorial phage-display mutagenesis reveals complex networks of surface-core interactions in the pacifastin protease inhibitor family. Journal of Molecular Biology 370, 63-79.
【非特許文献30】Walport, M.J. (2001a) Complement. First of two parts. N. Eng. J. Med. 344, 1058-1066.
【非特許文献31】Walport, M.J. (2001b) Complement. Second of two parts. N. Eng. J. Med. 344, 1140-1144.
【発明の概要】
【発明が解決しようとする課題】
【0022】
レクチン経路を含む補体系の阻害は、補体系の異常の結果として生じるヒト及び動物の疾患に対抗するための効率的なツールとなり得る。しかしながら、現在、こうした疾患と闘うために、補体系、主にレクチン経路を望ましい範囲で阻害できる化合物として利用できるものは無い。上で詳細に説明したように、MASP−1及びMASP−2酵素を阻害することによりレクチン経路を選択的に阻害し得る。
【0023】
この理由から、本発明者らは、MASP−1及び/又はMASP−2酵素を阻害することにより補体系のレクチン経路を選択的に阻害できる化合物の開発を目標に定めた。
【課題を解決するための手段】
【0024】
驚いたことに、我々は、以下の一般式(I)によるペプチドが上述した目的に適していることを発見した。
ここで、
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、Wである。
【0025】
上記によれば、本発明は、一般式(I)によるペプチド、その塩、エステル、及び薬剤的に許容可能なプロドラッグに関する。
【0026】
特に好ましくは、本発明は、以下の配列を有するペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、
GICSRSLPPICIPD(SEQ ID NO 3)、
GVCSRSLPPICWPD(SEQ ID NO 4)、
GMCSRSYPPVCIPD(SEQ ID NO 5)、
GYCSRSIPPVCIPD(SEQ ID NO 6)、
GWCSRSYPPVCIPD(SEQ ID NO 7)、及び
配列GICSRSLPPICIPD(SEQ ID NO 3)を有するペプチドの環状のもの、
及びその塩又はエステルに関する。
【0027】
最も好ましくは、本発明は、配列GYCSRSYPPVCIPD(SEQ ID NO 2)及びGICSRSLPPICIPD(SEQ ID NO 3)を有するペプチド、その塩及びエステルに関する。
【0028】
更に、本発明は、一般式(I)による少なくとも1つのペプチド、その塩、エステル、又はプロドラッグ、及び少なくとも1つの他の添加物を含有する医薬調製物に関する。この添加物は、好ましくは、活性薬剤の制御された放出を確保するマトリクスである。
【0029】
本発明は、特に、以下の配列を有する少なくとも1つのペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、
GICSRSLPPICIPD(SEQ ID NO 3)、
GVCSRSLPPICWPD(SEQ ID NO 4)、
GMCSRSYPPVCIPD(SEQ ID NO 5)、
GYCSRSIPPVCIPD(SEQ ID NO 6)、
GWCSRSYPPVCIPD(SEQ ID NO 7)、
配列GICSRSLPPICIPD(SEQ ID NO 3)を有するペプチドの環状のもの、
及び/又はその薬剤的に許容可能な塩もしくはエステル、を含有する医薬調製物に関する。
特に好ましくは、本発明による医薬調製物は、配列GYCSRSYPPVCIPD及びGICSRSLPPICIPDを有するペプチド、及び/又は、その薬剤的に許容可能な塩及び/又はエステルを含有する。
【0030】
本発明は、更に、一般式(I)による少なくとも1つのペプチド、その塩又はエステルを含むキットに関する。
【0031】
本発明は、更に、MASP酵素を阻害する可能性がある化合物のスクリーニング手順に関し、その過程においては、本発明による標識ペプチドを、MASPを含有する溶液に添加し、試験対象の1つ以上の化合物を含有する溶液をそれに添加し、放出された有標ペプチドの量を測定する。これに関して、MASP酵素は、好ましくは、MASP−1又はMASP−2酵素である。
【0032】
本発明は、更に、補体系の阻害により治療可能な疾患を治療することに適した医薬調製物の製造における、一般式(I)によるペプチド、及びその薬剤的に許容可能な塩又はエステルの使用に関する。これによれば、疾患は、好ましくは以下の群から選択され得る:炎症性及び自己免疫疾患、特に好ましくは、虚血再灌流障害、関節リウマチ、神経変性疾患、加齢性黄斑変性症、糸球体腎炎、全身性エリテマトーデス、及び補体活性化関連仮性アレルギ。
【0033】
本発明は、更に、MASP酵素を単離するための手順に関し、その過程においては、一般式(I)による1つ以上の固定化ペプチドを有する担体を、MASP酵素を含有する溶液に接触させ、調製物を洗浄する。これに関して、MASP酵素は、好ましくは、MASP−1又はMASP−2酵素である。
【0034】
上述した本発明によるペプチドには、MASP−1及びMASP−2酵素の両方を阻害するものと、MASP−2酵素のみを阻害しMASP−1酵素を阻害しないものとがある。しかしながら、こうした本発明によるペプチドは、MASP酵素と密接に関係するトロンビンを、非常に高濃度でなければ阻害せず、一般には、トリプシンもほんの僅かにしか阻害しない。
【図面の簡単な説明】
【0035】
【図1】ファージディスプレイ法の概略図である。
【図2】アガロースゲル上で行った、実施例1.1.3.2に記載のダイジェスチョンの結果を確認する図である(レーン1は、ダイジェストされたpMal−p2X lacIq遺伝子を示し、レーン2は、ダイジェストされたpBlueKS_NheI_Nsiベクタを示す)。
【図3】実施例1.1.4.3に記載のライゲーション及び形質転換に使用したベクタ及びインサートを検査して濃度を確認する試験の結果を示す図である。
【図4】実施例2.2.2に記載のライゲーション試験に関連して調製したゲルの写真を示す図である。
【図5】得られた配列の配列ロゴ図であり 図5aは、MASP−2から選択されMASP−2に特異的な配列に関する配列図であり、 図5bは、MASP−2から選択されたが、MASP−1も認識する配列に関する配列図であり、 図5cは、MASP−1から選択されたが、MASP−2も認識する配列に関する配列図である。
【図6】血液凝固に対する本発明によるペプチドの影響に関する用量関連試験結果を示す図であり、 図6aは、トロンビンを血漿に添加することにより血漿凝固(フィブリン形成)を誘発する過程を有する、トロンビン時間の測定実験を示す図であり、 図6bは、組織因子を血漿に添加することにより血漿凝固(フィブリン形成)を誘発する過程を有する、プロトロンビン時間の測定実験を示す図であり、 図6cは、血液凝固のいわゆる「接触活性化(contact activated)」又は「内因性(intrinsic)」経路を模倣する、トロンボプラスチン時間の測定実験を示す図である。
【図7】3つの補体活性化経路に対する本発明によるペプチドの影響を示す図であり、 図7aは、選択的「S」ペプチドの影響を示す図であり、 図7bは、非選択的「NS」ペプチドの影響を示す図である。
【発明を実施するための形態】
【0036】
本発明は、MASP−1及びMASP−2酵素(又はMASP−2酵素のみ)を選択的に阻害するペプチド及びペプチド誘導体に関する。
【0037】
本発明は、更に、記載した配列と配列的に類似し、その生物学的活性も記載した配列と比較して類似する、アミノ酸配列に関する。対象となるペプチドの生物学的機能を変えること無く、特定の側鎖改変又はアミノ酸置換を実施し得るということは、当業者には明白である。こうした改変は、アミノ酸側鎖の相対的類似、例えば、大きさ、電荷、疎水性、親水性等における類似に基づくものであり得る。こうした変更の目的は、酵素分解に対するペプチドの安定性の増加、或いは特定の薬物動態パラメータの改善である場合がある。
【0038】
本発明の保護の範囲は、更に、検出性を確保する要素(例えば、蛍光基、放射性原子等)が統合されたペプチドを含む。
【0039】
更に、本発明の保護の範囲には、幾つかの追加のアミノ酸を、N末端、C末端、又は両末端に含むペプチドが含まれるが、これは当該追加アミノ酸が元の配列の生物学的活性を大きく損なわないことを条件とする。端部に位置するこうした追加のアミノ酸の目的は、固定化を促進すること、他の試薬との連結の可能性を確保すること、溶解度、吸収、その他の特性に影響を与えることであり得る。
【0040】
配列内のアミノ酸側鎖は、IUPACの推奨する方法を使用して表示した(Nomenclature of α-Amino Acids, Recommendations, 1974 - Biochemistry, 14(2), 1975)。
【0041】
本発明は、更に、本発明による一般式(I)によるペプチドの薬剤的に許容可能な塩に関する。これは、ヒト又は動物の組織と接触させた時に、不要な度合いの毒性、炎症、アレルギ症状、又はこれらに類似する現象を引き起こさない塩を意味する。酸付加塩の非限定的な例として、以下が挙げられる:酢酸塩、クエン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、酪酸塩、ジグルコン酸塩, ヘミ硫酸塩、フマル酸塩、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、乳酸塩、マレイン酸塩、メタンスルホン酸塩、シュウ酸塩、プロピオン酸塩、コハク酸塩、酒石酸塩、リン酸塩、グルタミン酸塩。塩基付加塩の非限定的な例として、以下のものに基づく塩が挙げられる:アルカリ金属及びアルカリ土類金属(リチウム、カリウム、ナトリウム、カルシウム、マグネシウム、アルミニウム)、第4アンモニウム塩、アミンカチオン(メチルアミン、エチルアミン、ジエチルアミン等)。
【0042】
本発明に関して、プロドラッグとは、インビボで本発明によるペプチドへ転換される化合物である。転換は、例えば、血液中で酵素加水分解によって起こり得る。
【0043】
本発明によるペプチドは、適切な生物学的効果を達成するために1つ以上の添加物が必要となる医薬調製物に使用することができる。こうした製剤は、例えば、当業者に周知である、活性薬剤の制御された放出を確保するマトリクスと組み合わせた医薬調製物であり得る。一般に、活性薬剤の制御された放出を確保するマトリクスは、適切な組織(例えば、血漿)に入った時に、例えば、酵素又は酸塩基加水分解の過程によって分解する重合体である(例えば、ポリラクチド、ポリグリコライド)。
【0044】
本発明による医薬調製物では、希釈剤、充填剤、pH調節剤、溶解促進物質、着色添加物、抗酸化剤、保存料、等張剤等、最新技術水準において公知の他の添加物を使用することが可能である。こうした添加物は、最新技術水準において公知である。
【0045】
好ましくは、本発明による医薬調製物は、非経口(静脈内、筋肉内、皮下)投与を介して生命体内に入れることができる。これを考慮すると、好適な医薬組成物は、水性又は非水性の溶液、分散液、懸濁液、乳液、或いは、使用直前に上述した流体の何れかに変化させることが可能な固形(例えば、粉末)製剤にしてよい。こうした流体において、適切な媒体、担体、希釈剤、又は溶媒は、例えば、水、エタノール、様々なポリオール(例えば、グリセリン、プロピレングリコール、ポリエチレングリコール、及びこれらに類似する物質)、カルボキシメチルセルロース、様々な(植物)油、有機エステル、及びこれら全ての物質の混合物であり得る。
【0046】
本発明による医薬調製物の好ましい剤形には、例えば、タブレット、粉末、顆粒、座薬、注射液、シロップ等が含まれる。
【0047】
投与量は、疾患の種類、患者の性別、年齢、体重、及び、疾患の重篤性に応じて決まる。経口投与の場合、活性薬剤について、好ましい1日あたりの量は、例えば、0.01mgから1gの間であり、非経口投与(例えば、静脈内に投与される製剤)の場合、好ましい1日あたりの量は、例えば、0.001mgから100mgの間であり得る。
【0048】
更に、当該医薬調製物は、最新技術において公知であるリポソーム又はマイクロカプセルの形で使用することができる。本発明によるペプチドはまた、遺伝子治療の最先端の手段により標的生物に入れることもできる。
【0049】
所望の医療効果を達成するために、MASP−1又はMASP−2を選択的に阻害する活性薬剤が必要である場合には、本発明による一般式(I)によるペプチドから、選択的阻害ペプチドを選択することが好ましい。例えば、MASP−2酵素を選択的に阻害する本発明によるペプチドは、配列GYCSRSYPPVCIPD(SEQ ID NO 2)を有するペプチドにしてよく、一方、MASP−1酵素を選択的に阻害する本発明によるペプチドは、配列GICSRSLPPICIPD(SEQ ID NO 3)を有するペプチドにしてよい。特定の治療目標を達成するためには、配列GICSRSLPPICIPD(SEQ ID NO 3)を有する本発明による環状ペプチド等、MASP−1及びMASP−2の両方を阻害するペプチドを使用することが好ましい場合もある。
【0050】
本発明によるペプチドは、様々なMASP酵素の測定又は位置特定を(何れかのMASP酵素に特異的な形で、或いはMASP−1及びMASP−2酵素の両方に同時に特異的な形で)行うために使用可能な様々なキットにおいて、好ましく使用することができる。こうした使用は、競合的及び非競合的試験、ラジオイムノアッセイ、生物発光及び化学発光試験、蛍光定量試験、酵素結合アッセイ(例えば、ELISA)、免疫細胞化学アッセイ等にまで及び得る。
【0051】
本発明によれば、キットは、例えば競合的結合アッセイによって、MASP酵素の潜在的阻害剤を検査することに適したものが特に好ましい。こうしたキットの支援により、本発明によるペプチドをMASP酵素からどのくらい引き離すことができるかについて、潜在的阻害剤の能力を測定することができる。これを検出するためには、本発明によるペプチドは、何らかの形(例えば、蛍光基又は放射性原子の組み込み)により標識する必要がある。
【0052】
本発明によるキットは、更に、溶液及び試薬を調製するために必要な他の溶液、ツール、及び出発物質と、使用説明書とを含み得る。
【0053】
一般式(I)による本発明による化合物(ペプチド)は、更に、MASP酵素を阻害する可能性のある化合物をスクリーニングするために使用することができる。こうしたスクリーニング手順の過程において、一般式(I)によるペプチドは、後の時点での検出性を確保するために、標識(蛍光、放射性等)された形態で使用される。こうしたペプチドを含有する調製物を、MASP酵素を含有する溶液に添加し、その過程において、ペプチドはMASP酵素に結合する。適切なインキュベーション時間の後、試験対象の化合物/化合物群を含有する溶液を、当該調製物に添加し、その後、更にインキュベーション時間を設ける。MASP酵素に結合する化合物は(試験化合物が部分的又は完全にペプチドと同じ部位で酵素表面に結合するか、或いは別の場所だが、その結合により、ペプチドと結合する能力を喪失させるような形でMASP酵素の立体配座が変更される場合)、その阻害能力の範囲でMASP分子から標識ペプチドを引き離す。引き離されたペプチドの濃度は、ペプチド分子上で使用した(蛍光又は放射性)標識を検出するのに適した任意の方法を用いて決定することができる。インキュベーション時間、洗浄条件、検出方法、他のパラメータは、当業者に知られるやり方で最適化することができる。本発明によるスクリーニング手順は、高スループットスクリーニング(HTS)手順においても使用することができる。
【0054】
本発明によるペプチドは、まず第一に、疾患の治療において使用可能であり、その場合、補体系の働きの阻害は、好ましい効果を有する。結果として、本発明は、更に、こうした疾患の治療用の医薬の生産におけるペプチドの使用にも関連する。上で詳細に説明したように、こうした疾患は、第一に、特定の炎症性疾患及び自己免疫疾患であり、特に、以下の疾患である:虚血再灌流障害、関節リウマチ、神経変性疾患(例えば、アルツハイマー病、ハンチントン病、パーキンソン病、多発性硬化症)、加齢性黄斑変性症、糸球体腎炎、全身性エリテマトーデス。
【0055】
本発明による化合物は、更に、MASPタンパク質を単離するために使用でき、それは、ペプチドを固定化し、このように作製した調製物をMASP酵素の含有が推定される溶液に接触させることによって行われる。この溶液が実際にMASP酵素を含有する場合には、当該酵素は固定化ペプチドにより結合される。この手順は、分析及び調製の両方の目的に適したものになり得る。所定のペプチドのMASP酵素上での結合のジオメトリが知られていない場合、この手順においては、一つのペプチドを幾つかの方向から固定して使用するか、或いは複数のペプチドを使用してでも、適切な結合を確保するべきである。MASP酵素を含有する溶液は、純粋なタンパク質溶液、様々な程度まで精製した抽出物、組織調製物等であり得る。
【0056】
ファージディスプレイ
本発明によるペプチドは、ファージディスプレイ法を用いて開発した。
【0057】
ファージシスプレイ法は、誘導性(directed)インビトロ進化の実現に適しており、最先端の手順(Smith 1985)の主要ステップは、図1において確認することができる。この手順では、進化に関与するタンパク質の遺伝子が、バクテリオファージ外被タンパク質遺伝子に連結される。これにより、バクテリオファージが形成される時、融合タンパク質が生成され、ファージの表面に組み込まれた状態となる。ファージ粒子は、当該外来タンパク質の遺伝子を内部に保有しており、その表面には、当該外来タンパク質を提示している。当該タンパク質及びその遺伝子は、ファージを介して物理的に結び付けられる。我々は、誘導性タンパク質進化のために、タンパク質をコードする遺伝子のコドンを、慎重に決めた様式で改変する。合成オリゴヌクレオチドの混合物に基づくコンビネーション的変異導入を用いて、多数のコドンを同時に変更することが可能である。変異の位置と位置毎の変異多様性(variability)は同時に決定される。
【0058】
数十億の変異体を含むDNAライブラリを作製してバクテリアに入れた後、ファージタンパク質ライブラリが作製される。各ファージは、1種類のタンパク質変異体のみを提示し、この変異体の遺伝子のみを保有する。個々の変異体は、試験者により選択された(通常は表面に結合された)特定の標的分子と結合する能力に基づいて、親和性クロマトグラフィ及びその類似法により、互いに分離することができる。それと同時に、単純なタンパク質親和性クロマトグラフィとは異なり、このように選択されたファージタンパク質変異体は2つの重要な特性を有する。第一に、増殖する能力を有しているという点であり、第二に、コード遺伝子をファージ粒子に包んで保有しているという点である。
【0059】
この進化の間においては、変異体が個別に調べられるのではなく、何十億もの実験が実際に同時に行われるのである。結合した変異体は、増殖させ、選択-増殖のサイクルを何回か繰り返した後、機能的変異体が豊富に含まれた集団が得られる。この集団から、タンパク質が依然としてファージ上に提示された状態で、個々のクローンを機能試験において検査する。当該試験によって適切とされたファージタンパク質変異体は、物理的に連結した遺伝子の配列決定をすることにより同定される。個別の測定に加えて、適度に大きな数の機能選択クローンの配列を分析することにより、どのようなアミノ酸配列がその機能を達成することを可能とするかについても明らかになる。これにより、実際の実験に基づくデータベースが作成され、配列-機能アルゴリズムを発展させることが可能となる。これに基づいて最善とされた変異体は、独立したタンパク質として生産され、更に正確な追加試験において検査される。
【0060】
ライブラリの作製
SFTI(ヒマワリトリプシン阻害剤)分子は、トリプシン阻害活性を有しており、以下の配列を有する14アミノ酸ペプチドである:GRCTKSIPPICFPD(SEQ ID NO 1)。自然界においては、即ち、植物のヒマワリでは、これは環状の形態で形成されるため、ここでN末端として示したグリシンと、C末端として示したアスパラギン酸とは、ペプチド結合により連結される。2個のシステインは、互いにジスルフィド架橋を形成する。ジスルフィド架橋が完全であるならば、上に示した線状形態であっても強力なトリプシン阻害剤となることが、インビトロ試験により実証されている(Korsinczky, 2001)。SFTI分子の他の特別な特徴は、酵素と相互作用する遙かに大きなボーマン−バーク阻害剤の分子部分と構造的に事実上同一であることである(Luckett 1999; Korsinczky, 2001; Mulvenna 2005)。ボーマン−バーク阻害剤で保存されておりSFTI分子と同一の部分に下線を付す:GRCTKSIPPICFPD。全ての下線部分は、1つ(位置4のトレオニン)を除き、ライブラリを作製する際に維持された。
【0061】
ライブラリを設計する際には、以下のランダム化スキームを使用した:
ここでも、下線を付しているのは、構造的な理由から改変させなかった位置である。位置「O」では、20個全ての天然アミノ酸が許容されたが、位置P1では、スキーム(R/K)により示した2つの塩基性アミノ酸のみが許容された。イタリック体の部分は、我々の当初の予想に基づいて、プロテアーゼと接触することはないと推定されたため、改変しなかった。
【0062】
ファージディスプレイ中に高親和性結合分子を選択可能とするためには、提示される結合分子が、ファージ当たり少ないコピー数で、理想的には1個の単一コピー(一価ファージディスプレイ)で存在することが重要となる。これにより、幾つかの固定標的分子に同時に結合することにより見た目の上で高親和性結合(アビディティ)となるものを回避することができる。このため、上述したSFTIライブラリは、ファージタンパク質p8に連結して発現させた時にファージ当たり1個の単一コピーとして出現することが実証されているキモトリプシン阻害剤分子(Szenthe 2007)と融合させた状態で発現させた。これは、我々の予備実験において、MASP酵素を阻害せず、MASP酵素と結合もしないことが実証されたSchistocerca Gregariaキモトリプシン阻害剤(SGCI)である(Malik, 1999)。
【0063】
SFTIライブラリのエレメントとSGCI分子との間には、更に、タグとライブラリのエレメントとの間で適切な距離が維持されるようなペプチドリンクを使用して、モノクローナル抗体により認識可能な直線状エピトープタグを挿入した。これは、いわゆる「フラッグタグ」であり、2つの目的を果たす。一方の目的は、ファージ表面上でのライブラリの提示を容易に明らかにできるようにすることである。他方の目的は、当該タグに対する抗体を使用することによるコントロール選択の結果として得られたクローンの配列決定をすることにより、特定の標的酵素、即ち、MASP1及びMASP2が欠如した状態ではいかなる配列のクローンが得られるかを明らかにすることである。このようにして、酵素に対して行われた選択の結果を、当該抗体に基づいて選択された群と比較することにより、他の何らかの影響(例えば、より効率的な生産)の結果ではなく真に酵素との結合に起因し得る、典型的な位置依存的アミノ酸嗜好性(preferences)を明らかにすることができる。
【実施例】
【0064】
以下、本発明を実施例に基づいて詳細に説明するが、しかしながら、実施例は、本発明を限定する例と見做すべきではない。
【0065】
実施例を通して、ファージミド系を構築する上で可能な方法(実施例1)、ライブラリの調製(実施例2)、ファージの選択(実施例3)、及び結果(実施例4)を示す。実施例5では、ペプチド合成及び関連する分析試験を説明する。
【0066】
実施例1:ファージミド系の構築
1.1. ファージミドベクタの構築
最初のステップとして、入手可能な市販のベクタから出発して、独自のファージミドベクタを構築した。このために、新たに制限エンドヌクレアーゼ切断部位を形成する必要があり、これはクンケル式変異誘発を使用して実現した(Kunkel, 1991)。
【0067】
1.1.1. ウラシル含有一本鎖クンケルテンプレートの調製
1.1.1.1. 形質転換
0.5μl pBluescript II KS(−)ファージミド(Stratagene、cat#212208、51.1μg/μl、2961bp)
8μl KCM溶液[0.5M KCl、0.15M CaCl2、0.25M MgCl]
31.5μl USP蒸留水
40μl CJ236 K12 E.coliコンピテント細胞
形質転換体を、氷上で20分間、その後、室温で10分間インキュベートした。体積の10倍量(800μl)のLB培地を加え、30分間に渡り、37℃、200rpmで振盪した。100μlの量を一晩、37℃で、LB−アンピシリンプレート[LB、100μg/mlアンピシリン]上において培養した。
【0068】
1.1.1.2. 感染
翌日、コロニを2mlの培地[LB、100μg/mlアンピシリン、30μg/mlクロラムフェニコール]に接種し、37℃で一晩、200rpmで振盪しながらインキュベートした。次に、一晩培養した培養物2μlを、上述したものと同じ組成の培地2mlに接種し、37℃で200rpmで振盪しながら6時間培養した。その後、30μl M13KO7ヘルパーファージ(NEB、cat#N0315S)に感染させ、200rpmで振盪しながら、37℃の状態で40分間インキュベートした。当該出発培養物の全てを、30ml[2YT、100μg/mlアンピシリン、30μg/mlクロラムフェニコール]培地へ移した。ファージは、培養物を200rpmで振盪しながら、37℃で、16乃至18時間に渡り一晩培養することにより生産した。翌朝、培養物を4℃で10分間、8,000で遠心した。上清を清潔なチューブに移し、体積の1/5の量(6ml)の溶液[2.5M NaCl、20%PEG−8000]を加え室温で20分間インキュベートした後、ファージを溶液から沈殿させた。沈殿物を4℃で20分間、10,000rpmで遠心し、上清をピペットで取り除いた。沈殿物を、800μlのPBS緩衝液において溶解した。
【0069】
一本鎖プラスミドは、Qiaprep Spin M13キット(Qiagen、cat#27704)を使用して、キットに付属のレシピによりファージから取得し、100μlの10倍希釈EB緩衝液によりカラムから溶出させた。産物の濃度は、35倍希釈において260nmで確認した(ssDNS OD260nm=1=33ng/μl)。上述した手順の結果として得られた一本鎖ウラシル含有pKSファージミドベクタの濃度は、407μg/mlであった。
【0070】
1.1.2. クンケル式変異誘発を使用した切断部位Nsi及びNheIの導入
1.1.2.1. オリゴのリン酸化
変異プライマ:
Blue_NheI_in_779(36mer、SEQ ID NO 8):
5'-cgcaattaaccctcagctagcggaacaaaagctggg-3'
Blue_NsiI_in_1089(36mer、SEQ ID NO 9):
5'-ccgcctttgagtgagatgcatccgctcgccgcagcc-3'
・2μl 10×濃縮TM緩衝液[0.5 M Tris−HCl、0.1M MgCl2、pH7.5]
・2μl 10mM ATP
・1μl 100mM DTT
・1μl T4ポリヌクレオチドキナーゼ(Fermentas、10u/μl)
・36ng Blue_NheIプライマ(4μl)/36ng Blue_Nsiプライマ(3.5μl)
・10μl USP蒸留水/10.5μl USP蒸留水
別個の2つのプライマについての2つのリン酸化反応を、共に体積20μl中に添加し、45分間37℃でインキュベートした。
【0071】
1.1.2.2. オリゴヌクレオチドのハイブリダイゼーション
テンプレート:プライマの割合は、体積25μl中のモル比が1:3となるように設定した。
・2.5μl一本鎖クンケルテンプレート(1μg)
・2μlリン酸化Blue_NheIプライマ
・2μlリン酸化Blue_Nsiプライマ
・2.5μl 10×濃縮TM緩衝液
・16μl USP蒸留水
反応混合物は、90℃の水槽内で1分間加熱した後、直ちに50℃のサーモスタット内へ移して更に3分間置いた。短時間遠心し、氷中に置いた。
【0072】
1.1.2.3. 二本鎖産物の調製、精製、ダイジェスチョン
オリゴヌクレオチドのハイブリダイゼーション後、第2DNA合成により、二本鎖産物をインビトロで生産した。二本鎖産物の一方の鎖はウラシルを含む当初のクンケルテンプレートであり、プライマの延長により形成された、変異を有する他方の鎖は、ウラシルを含んでいなかった。
・1μl 10mM ATP
・1μl 25mM dNTP
・1.5μl 100mM DTT
・0.6μl T4リガーゼ(NEB、400u/μl)
・0.3μl T7ポリメラーゼ(Fermentas、10u/μl)
反応混合物は、14℃で一晩インキュベートした。混合物全体を1%アガロースゲル上で泳動させ、Qiaquick Gel Extractionキット(Qiagen、cat#28704)によりレシピに従って分離及び精製した。産物は、30μl EB緩衝液で溶出させ、上述したレシピによりE.coli XL1 Blueコンピテント細胞に形質転換した。これらの細胞は、ウラシルを含む鎖を分解するため、3mlの培養物中で培養したバクテリアでは、ウラシルを含まない変異鎖の複製により増殖したベクタのクローンが主に存在する。二本鎖ベクタは、Mini Plus Plasmid DNA Extraction System(Viogen、cat#GF2001)キットを使用して、50μl EB緩衝液中に単離した。
遺伝手術の次のステップとして、新たに導入した切断部位において産物を25μl中でダイジェストする。
・20μlベクタミニプレップ
・2.5μl 10×濃縮Y Tango緩衝液(Fermentas)
・1.25μl USP蒸留水
・0.50μl NheI(Fermentas、10u/ml)
・0.75μl Nsi(Promega、10 u/ml)
ダイジェスチョンは、37℃で一晩行った。産物は、2%アガロースゲル上において、電気泳動を使用して確認し、その後、上述した方法を使用してダイジェスト化プラスミドをゲルから単離した後、キットにより精製した。このようにして入手したベクタの名前は、pBlueKS_NheI_Nsiである。
【0073】
1.1.3 lacIq遺伝子の追加
1.1.3.1 PCR
lacIq遺伝子及びマルトース結合タンパク質(MBP)シグナル配列を、pMal−p2Xベクタ(NEB、cat# N8077S、200μg/ml)からPCRを使用して分離した。
プライマ:
pMal_lac_forward(SEQ ID NO 10):
5'-gtcagtatgcatccgacaccatcgaatggtg-3'
pMal_NheI_rev(SEQ ID NO 11):
5'-gtcagtgctagcgccgaggcggaaaacatcatcg-3'
・5μl 10×濃縮Pfu緩衝液
・0.4μl 25mM dNTPs
・10μl 25mM MgSO4
・0.5μl pMal−p2Xテンプレート
・0.5μl 5μM pMal_lac_forwardプライマ
・0.5μl 5μM pMal_NheI_revプライマ
・1μl Pfuポリメラーゼ(Fermentas、2.5Wu/μl)
・36.5μl USP蒸留水
PCR中に用いたプログラム:
95℃ 180s
95℃ 45s
65℃ 45s
72℃ 240s
72℃ 480s
ステップ2乃至4は、20回繰り返した。
【0074】
1.1.3.2 ダイジェスチョン
産物を、説明書に従ってGenElute PCR Clean Upキット(Sigma、cat#NA1020)を使用して精製し、制限酵素により37℃で一晩ダイジェストし、ライゲーションに必要な粘着末端(sticky ends)を利用可能とした。
・20μl PCR産物(lacIq遺伝子)
・2.5μl 10×濃縮Y Tango緩衝液(Fermentas)
・1μl Nsi酵素(=AvaIII、Fermentas、10u/μl)
・0.5μl NheI酵素
ダイジェストしたPCR産物は、上述したようにキットにより精製し、事前に調製、ダイジェスト、精製したファージミドベクタと共に、1%アガロースジェル上で確認した。結果は、図2に示しており、レーン1は、ダイジェストされたpMal−p2X lacIq遺伝子に対応し、レーン2は、ダイジェストされたpBlueKS_NheI_Nsiベクタに対応する。
【0075】
1.1.3.3. ライゲーション
2μlダイジェスト済みpBlueKS_NheI_Nsiベクタ
6μlダイジェスト済みpMal−p2X lacIq遺伝子
1μl 10×倍濃縮T4リガーゼ緩衝液
1μl T4リガーゼ(Fermentas、1 Weiss u/μl)
ライゲーションは、室温で2時間に渡り実行した。ライゲーション産物は、上述したように40μlコンピテントE.coli XL1 Blue細胞に形質転換した。100μlの形質転換産物[LB、100μg/mlアンピシリン]を寒天プレートに塗布し、37℃で一晩インキュベートした。発生したコロニから、ミニプレップ培養物を接種し、Viogenキットを使用してプラスミドを単離した。ライゲーションは、37℃での1時間の制限消化により確認した。EcoRI酵素に対しては、添加したlacIq遺伝子の内部においてのみ切断部位が存在した。
・3.5μlミニプレップ産物
・1μl 10×EcoRI緩衝液
・0.26μl EcoRI酵素(Fermentas、10u/μl)
・5.24μl USP蒸留水
1%アガロースゲルに基づいて、ダイジェスチョンが発生したこと、すなわちライゲーションが成功したことが確認できる。新規のファージミドベクタの名前は、pBlueKS_NheI_Nsi_lacIqである。
【0076】
1.1.4. エピトープタグ及びSGCI部の導入
1.1.4.1. PCR
エピトープタグとして使用されるフラッグタグのアミノ酸配列はDYKDDDDK(SEQ ID NO 12)である。SGCI部は、外被タンパク質p8に融合させ、エピトープタグは、SGCIのN末端に融合させた。上述したように、SGCIの存在により1価での発現が確保されるため、1個のファージは、最大で1個のライブラリ構成ペプチドをその表面に提示する。
プライマ:
pGP8−Tag−NheI(SEQ ID NO 13):
5'-gtcagtgctagcatcggattataaagacgatgac-3'
P8−XbaI−rev(SEQ ID NO 14):
5'-gtcagttctagattattagcttgctttcgaggtg-3'
・5μl 10×濃縮Pfu緩衝液
・8μl 25mM MgSO4
・0.4μl 25mM dNTPs
・2μlテンプレート:pGP8−Tag−SGCIベクタ(以前の構築物)
・0.5μl 5μM pGP8−Tag−NheIプライマ
・0.5μl 5μM P8−XbaI−revプライマ
・1μl Pfuポリメラーゼ(Fermentas、2.5u/μl)
・36.2μl USP蒸留水
PCR中に用いたプログラム:
95℃ 180s
95℃ 45s
60℃ 45s
72℃ 60s
72℃ 480s
ステップ2乃至4は、25回繰り返した。
PCR産物は、Sigma GenElute PCR Clean Upキットを使用して、レシピに従って精製した。
【0077】
1.1.4.2. 制限消化
pBlueKS_NheI_Nsi_lacIqベクタを、37℃で2時間に渡り制限酵素によりダイジェストし、フラッグタグ−SGCI部のライゲーションを可能にした。
・2.5μl pBlueKS_NheI_Nsi_lacIqミニプレップ
・3.5μl 10×濃縮Tango緩衝液
・1.5μl XbaI(Fermentas、10u/μl)
・1.5μl NheI(Fermentas、10u/μl)
・3.5μl USP蒸留水
産物を1%アガロースゲルから単離し、Viogen Gel−Mキットにより精製し、45μlの水に溶出させた。その後、産物をアルカリホスファターゼにより、37℃で45分間処理した。
・ゲルから単離した43μlダイジェスト済みpBlueKS_NheI_Nsi_lacIqベクタ
・1μlエビアルカリホスファターゼ(SAP、Fermentas、1u/μl)
・5μl 10×濃縮SAP緩衝液
ホスファターゼは、65℃で15分間加熱し不活性化した。
【0078】
1.1.4.3. ライゲーション及び形質転換
反応混合物を調製する前に、ベクタ及びインサートを1.8%アガロースゲルで泳動し、濃度を確認した。結果を図3に示す。
【0079】
図3において、それぞれのレーンは以下を意味する:
1. 6μl 1kb DNAラダー(Fermentas)
2. フラッグタグ−SGCI−p8 PCR産物
3. ダイジェスト済み精製pBlueKS_NheI_Nsi_lacIqベクタ
【0080】
ライゲーションのために、反応混合物と対照産物とを、室温で90分間インキュベートした。
・2μl pBlueKS_NheI_Nsi_lacIqベクタ
・7μl フラッグタグ−SGCI−p8 PCR産物
・1μl 10×濃縮T4リガーゼ緩衝液
・1μl T4リガーゼ(Fermentas、1Wu/μl)
ライゲーションした産物は、上述したようにコンピテントE.coli XL1 Blue細胞に形質転換し、塗布して37℃で一晩培養した。
【0081】
それぞれ3mlである10アリコートの培地に、個別のバクテリアコロニを接種した後、一晩培養した液体培養物を調製し、そこから二本鎖プラスミドを単離した。XbaI及びNheI酵素により生成した粘着端部は、互いに対しても適合するため、挿入が適切な方向で実現された場合のものを、10個のクローンからDNA配列決定により単離した。Big Dye Terminator v3.1 cycle Sequencing Kit(Applied Biosystems、cat#4336917)システムをPCR反応に使用した。配列決定は、BIOMI Kft.(Godollo)により実行された。確認した10個の試料から、2つの良好な挿入が見つかった。新たなベクタの名前は、pKS−Tag−SGCI−p8である。
【0082】
1.1.5. Ser−Glyアダプタの挿入
1価での発現のために、以下の機能ユニットの配列を作製した:ライブラリ構成要素−Ser/Gly/リンカー−フラッグタグ−SGCI−p8。この目的のために、pKS−Tag−SGCI−p8ベクタがNheI及びXhoI酵素により切断され、このステップの結果として、元のフラッグタグが削除された。その後、ベクタを、適切なNheI及びXhoI粘着端部を伴うGly−Serリンカー(GGSGGSGG、SEQ ID NO 15)及びフラッグタグを含むアダプタにライゲートした。ライゲーションを確認するためにBamHI切断部位をフラッグタグの内部に形成しておいた。この酵素は、適切にライゲートしたベクタを2つの部位で分割し、そこで形成された産物は159塩基対の長さを有し、アガロースゲル電気泳動を使用して検出可能となる。
・20μl pKS−Tag−SGCI−p8ベクタミニプレップ
・3μl 10×Y Tango緩衝液
・2μl XhoI(Fermentas、10u/μl)
・5μl USP蒸留水
ベクタは、37℃で2時間ダイジェストし、与えられた条件はXhoIにとって至適条件ではなかったことから、その後、0.8%アガロースゲル上で、ダイジェスチョンが完了したかどうかを確認した。1μl NheI酵素をこれに添加し、37度で1時間インキュベートした。産物は、Viogen Gel−Mキットによりアガロースゲルから単離した。
リンカ及びフラッグタグを含むアダプタは、ダイジェスト済みベクタと共に環化させた。
アダプタ:
Ser−Gly_forward(SEQ ID NO 16):
5'-ctagctggcgggtcgggtggatccggtggcgattataaagacgatgatgacaaac-3'
Ser−Gly_reverse(SEQ ID NO 17):
5'-tcgagtttgtcatcatcgtctttataatcgccaccggatccacccgacccgccag-3'
・15μlダイジェスト済みpKS−Tag−SGCI−p8ベクタ
・2.8μl 1.3ng/μl Ser−Gly_forwardプライマ
・1.7ml 2.2ng/μl Ser−Gly_reverseプライマ
反応混合物は、90℃で1分間、その後、50℃で3分間インキュベートし、短時間遠心し、氷上に置いた。ライゲーションのため、以下のものをこれに添加した:
・2.2μl 10倍濃縮T4リガーゼ緩衝液
・1μl T4リガーゼ(Fermentas、1 Weiss u/μl)
ライゲーションは、16℃で一晩に渡って行った。コンピテントE.coli XL1 Blue細胞は、上述したように形質転換し、その後、形質転換産物[LB、100mg/ml]をプレートに塗布した。コロニから、スタータを一晩接種し、Viogen Mini−Mキットにより、ミニプレッププラスミドを説明書に従い精製した。得られた試料を、Big Dye Terminator v3.1 cycle Sequencingキットを使用して、DNA配列決定により確認し、PCR産物は、BIOMI Kft.(Godollo、ハンガリ)により処理された。
【0083】
以下では、このようにして調製されたファージミドに基づいてライブラリを作製しており、その名前はpKS−SG−Tag−SGCI−p8である。
【0084】
実施例2:ファージライブラリの調製
配列決定により確認したpKS−SG−Tag−SGCI−p8ベクタは、DNAライブラリを作製するためのテンプレートの役割を果たし、DNAライブラリは、縮重(degenerated)ライブラリオリゴ及びベクタ特異的オリゴをプライマとして使用することにより、ポリメラーゼ連鎖反応(PCR)を使用して作製した。このようにして作製されたPCR産物を、pKS−SG−Tag−SGCI−p8ベクタに挿入した。
【0085】
2.1. PCR
2.1.1. ライブラリオリゴ
上述したように、ライブラリを計画する際には、以下のランダム化スキームを使用した:
SFTIライブラリは、6箇所の選択位置(「O」位置)が完全にランダム化され、即ち、20個全てのアミノ酸の発生が許容され、位置P1では、アルギニン及びリジンのみが許容される(「R/K」位置)ように調製された。縮重オリゴヌクレオチドに関するIUPACコードを使用すると、ライブラリのオリゴヌクレオチド配列は、次のようになる(SEQ ID NO 19):
ペプチドをコードする部分には下線を付しており、ランダム化したコドンは、太字で示している。
【0086】
2.1.2 DNAライブラリの調製
ライブラリは、PCRを使用して調製され、ここで一方のオリゴは、挿入されるべきライブラリ構成要素を保有し、他方のオリゴは、ユニバーサル外部プライマである。反応混合液の総量は300μlであったが、6本のPCRチューブに分割した。
・30μl 10×濃縮Taq緩衝液
・36μl 25mM MgCl2
・2.4μl 25mM dNTP
・15μl 13μM SFTIライブラリオリゴヌクレオチド
・22μl 10μM pVIII 3’プライマ
・9μl(450ng)pKS−SG−Tag−SGCI−p8テンプレート
・180.6μl USP蒸留水
・5μl Taqポリメラーゼ(Fermentas、5u/μl)
プログラム:
1. 95℃ 60s
2. 95℃ 30s
3. 50℃ 30s
4. 72℃ 60s
5. 72℃ 120s
ステップ2乃至4は、15回繰り返した.
PCR産物は、1.5%アガロースゲル上で確認し、ExoI酵素によりダイジェストして、プライマを除去した。チューブ当たり1μlのExoI酵素と共に、37℃で45分間インキュベートし、その後、80℃で酵素を不活性化した。ホモ二重鎖を増幅するため、短い重合サイクルを挿入し、プライマは、一般に使用される外部プライマとした。
pVIII_3’(SEQ ID NO 18):
5'-gctagttattgctcagcggtggcttgctttcgaggtgaatttc-3'
各チューブに以下を添加した:
・2.5μl 2.5mM dNTP
・1μl 100μM pVIII_3’プライマ
・0.8μl Taqポリメラーゼ(Fermentas、5u/μl)
プログラムは、以前のPCRの場合と同じだが、2サイクルのみを実行した。
産物を1.5%アガロースゲル上で再度確認し、ExoI酵素によりダイジェストし、6本のPCRチューブの内容物を、3本のカラム上で、Sigma PCRクリーンアップキットによりレシピに従って精製した。溶出は、10倍に希釈したEB緩衝液中において、52μl/カラムの量で行った。
【0087】
2.2 pKS−SG−Tag−SGCI−p8ファージミドベクタへのDNAライブラリの挿入
2.2.1 ダイジェスチョン
ベクタと、インサートの役割を果たすDNAライブラリとは、2段階でダイジェストし、最初に、NheI酵素により切断を行った。DNAライブラリのダイジェスチョン中に分離した不要部分は、ほぼ完全に産物と同じサイズであったため、反応混合物から除去できなかった。この断片がベクタに入り込むのを防ぐために、ダイジェスチョンの第1の段階にはSacI酵素を更に添加した。これにより、不要部分の末端近くで、精製により除去可能な小さなフラグメントが分離され、残された大きな断片は、SacIの粘着末端を有しておりライゲーションできない。インキュベーションは、37℃で8時間及び一晩実行した。
・93μl pKS−SG−Tag−SGCI−p8ベクタ(40μg)
・15μl 10×Y Tango緩衝液
・4μl NheI酵素(Fermentas、10u/μl)
・38μl USP蒸留水
(V=150μl)
・35μl DNSライブラリPCR産物
・15μl 10×Y Tango緩衝液
・4μl NheI酵素(Fermentas、10u/μl)
・4μl SacI酵素(Fermentas、10u/μl)
・38μl USP蒸留水
(V=150μl)
続いて、他方の粘着末端を生成するAcc651(=KpnI)酵素を2倍量で添加した。Tango緩衝液の濃度も2倍とした。
ダイジェスト済みpKS−SG−Tag−SGCI−p8ベクタに対して:
・8μl Acc651(Fermentas、10u/μl)
・19.8μl 10×濃縮Tango緩衝液
ダイジェスト済みDNAライブラリに対して:
・8μl Acc651(Fermentas、10u/μl)
・11μl 10×濃縮Y Tango緩衝液
【0088】
2.2.2 ライゲーション
まず、両方のダイジェスチョン産物をゲルから分離した。ベクタは、0.8%アガロースゲルから分離し、6個のポケットに分割して、Viogen Gel−Mキットを使用して6本のカラム上で精製した。DNAライブラリは、1.8%ゲルから分離し、3本のカラム上で精製した(図4)。図4に示したゲル画像の線は、以下を意味する:
1. 1μl 100bp DNAラダー
2. 1μl精製DNAライブラリ
3. 1μl精製ベクタ
4. 5μl 1kb DNAラダー
【0089】
全ての試料をライゲーションに使用し、6本のチューブに分割して、16℃で18時間インキュベートした:
・210ml精製pKS−SG−Tag−SGCI−p8ベクタ
・100ml精製SFTI DNAライブラリ
・2ml T4リガーゼ(NEB、400,000ul/ml)
・35ml USP蒸留水
産物は、Qiagen Gel Eluteキットにより精製したが、ゲルから分離したのではなく、カラム上で精製のみを行った。溶出は、2×60μl USP蒸留水において行った。
【0090】
2.3 エレクトロポレーション、ファージライブラリの増殖
ライブラリは、エレクトロポレーションによりスーパコンピテント細胞へ導入した。我々の目的は、プラスミドを可能な限り多くの細胞に導入し、ライブラリに108乃至109片が含まれるようにすることである。
USP蒸留水中にあるため無塩状態であるDNAライブラリを、2×350mlのスーパコンピテント細胞に添加した。操作は、直径0.2cmのキュベット内において、以下のプロトコルに従って行った:2.5kV、200ohm、25μF。
エレクトロポレーション後、細胞を2×25mlのSOC培地へ慎重に移し、30分間、100rpm、37℃でインキュベートした後、試料を取り出し、段階希釈を行い、[LB]、[LB、100μg/mlアンピシリン]、及び[LB、10μg/mlテトラサイクリン]プレート上に滴下し、37℃で一晩培養した。非エレクトロポレーション対照産物と、水と共にエレクトロポレーションした対照産物とについても、同じ手順に従った。試料を取り出した後、2×25mlの培養物に2×250μlのM13KO7ヘルパーファージを感染させ、37℃で30分間、220rpmで振盪した後、産物全体を接種した。2×250 ml[2YT、100μg/mlアンピシリン、30μg/mlカナマイシン]培養物を2つの2リットルフラスコにおいて、37℃、220rpmで18時間培養した。
【0091】
タイトレーションに基づくと、我々のライブラリは、1.2×109の変異体を含んでいた。
【0092】
実施例3:ファージの選択
以下の例では、MASP−1及びMASP−2標的酵素に対する、上述した例により構築されたライブラリの選択を示す。
【0093】
3.1 標的酵素
ヒトMASP標的は、セリンプロテアーゼ(SP)ドメインと、2つの補体制御タンパク質ドメイン(CCP−1、−2)から成る(Gal 2007)。これらは組換えフラグメント産物であり、分子全体の触媒活性を有する。タンパク質は、封入体(inclusion body)の形態で生産し、ここから、生物活性を有する立体配座を再生(renaturation)により取得した。精製は、アニオン及びカチオンの交換分離により行った。タンパク質の活性は、溶液中、及び、ELISAプレートに結合させた形態で試験した。生産については、別の研究において詳細に説明されている(Ambrus 2003)。
選択において使用した標的のデータ:
MASP−1 CCP1−CCP2−SP:Mw=45478Da、cstock=0.58g/l(以降、MASP−1)
MASP−2 CCP1−CCP2−SP:Mw=44017Da、cstock=0.45g/l(以降、MASP−2)
抗フラッグタグ抗体:cstock=4g/l、(Sigma、マウスにおいて生成したモノクローナル抗フラッグM2抗体、cat# F3165)
【0094】
3.2 選択の工程
3.2.1 ファージの単離
第2.3章において説明した操作の終わりに、ファージを2×250mlの培養物において18時間に渡り作製した。選択の第1の工程では、これらを単離し、ライブラリを直ちにディスプレイのために使用できるようにした。
細胞培養物を、4℃、8,000rpmで、10分間遠心した。バクテリオファージを含む上清を清潔な遠心チューブに注ぎ入れ、その体積の1/5の沈殿剤を添加した[2.5M NaCl、20%PEG−8000]。沈殿は、室温で20分間行った。その後、4℃、10,000rpmで、15分間再度遠心した。上清を捨て、再度短時間遠心し、残液をピペットで取り除いた。白色のファージ沈殿物を25ml[PBS、5mg/ml BSA、0.05%Tween−20]緩衝液に溶解した。細胞片の可能性があるものを除去するために、再度遠心し、上清を清潔なチューブに移した。
【0095】
3.2.2.第1の選択サイクル
a)固定化:標的分子を96ウェルNunc Maxisorp ELISAプレート(cat#442404)上に固定化した。固定化操作中、MASP−1及び−2の濃度は、20μg/mlで、抗フラッグタグ抗体の濃度は2μg/mlとした。タンパク質を固定化緩衝液[200mM Na2CO3、pH9.4]において希釈し、100μlをウェルに配置した。固定化の時間は、タンパク質毎に最適化した。MASP−1は、110rev/minで混合しながら室温で60分間インキュベートし、抗体は、30分間インキュベートし、MASP−2は、4℃で一晩インキュベートした。第1の選択サイクルでは、標的タンパク質当たり12ウェルを使用した。1列おきに空の列を残した。陰性対照として、固定化バッファのみを1列に配置した。この列は、その後、標的タンパク質で覆ったものと同じ方法で処理した。
b)ブロッキング:固定化溶液を取り除き、200μl/wellのブロッキング緩衝液[PBS、5mg/ml BSA]をプレートに入れた。室温で少なくとも1時間、150rev/minで混合しながらインキュベートした。
c)洗浄:ELISAプレートを、1lの洗浄緩衝液[PBS、0.05%Tween−20]を使用して4回洗浄した。
d)選択:上述したように単離したライブラリのファージを、ピペットにより、各ウェルに100μlずつの量で入れた。室温で、110rev/minで混合しながら2.5時間インキュベートした。
e)E.coli XL1 Blue培養物:選択の実施中に、XLI Blue細胞を、前もって新たに取り出したプレートから、接種ループを使用して2×30ml[2YT、10μg/mlテトラサイクリン]の培地に接種した。これらの細胞は、後の時点で、標的タンパク質から溶出したファージに感染させる。感染の際、細胞は、指数関数的増殖の段階にある必要がある。OD600nmが約0.3乃至0.5の培養物が必要であり、これは、37℃、220rpmで、2乃至3時間培養することにより得られた。
f)洗浄:ELISAプレートを、3リットルの洗浄緩衝液を使用して12回洗浄した。
g)溶出:溶出は、100μl/wellの100mM HCl溶液を使用して行った。酸を加えて、5分間振盪し、各ウェルから、1ウェルずつ順に取り出した。個々の標的タンパク質から溶出したファージは、12×15μlの1M Trisベース緩衝液を事前に入れたチューブに収集して、ファージを含有する酸性溶液を素早く中和した。チューブは、直ちに混合し氷上に載置した。
h)感染:指数関数的増殖の段階にあるXL1 Blue培養物4.5mlを、試験管に入れ、標的タンパク質から溶出したファージ溶液500μlにより感染させた。MASP−1及びMASP−2、並びに抗体及び陰性対照物質(negative control substance)からそれぞれ溶出したファージにより、合計4つの感染を行った。培養物は、37℃、220rpmで、30分間インキュベートした。
i)タイトレーション:各感染培養物から20μlの試料を取り出し、2YT培地により体積の10倍に希釈し、更なる10×希釈により段階希釈物の調製を行った。各段階から、10μl[LB、100μg/mlアンピシリン]をプレートに滴下し、一晩37℃で培養した。
j)ヘルパーファージによる感染:試料採取の直後に、50μlのM13KO7ヘルパーファージを試験管内の各培養物に添加し、更に30分間インキュベートした。
k)全ての感染培養物を3×200ml[2YT、100μg/mlアンピシリン、30μg/mlカナマイシン]培地に移し、220rpmで混合しながら、37℃で18時間インキュベートした。対照物質は、タイトレーションのためにのみ必要であったため、更なる処理は行わなかった。
l)濃縮(Enrichment):翌朝、タイトレーションをチェックすると、1回のみの選択サイクル後に、対照物質と比較して大きな差が検出できた。抗体から溶出したファージの数は、バックグラウンドから溶出したファージの数よりも4桁も多く、MASPの場合、その差は、1乃至1.5桁であった。
【0096】
3.2.3. 第2の選択サイクル
このサイクルでは、第1の選択サイクルの場合と同じ工程を繰り返したが、ブロッキング緩衝液及び洗浄緩衝液中では、2mg/mlカゼイン(Pierce、cat#37528)をBSAの代わりに使用した。この変更により、BSAに結合するファージの増殖を回避することができる。この工程において、各標的タンパク質は、それぞれ対照を伴い(12ウェル)、前のサイクルにおいて溶出及び増殖させたファージを、各標的タンパク質に配置した。
18時間かけて産生されたファージを上述したように単離したが、最終的に、10mlの無菌PBS緩衝液に溶解した。ファージ溶液の濃度を268nmで測定し、その後、[PBS、2mg/mlカゼイン、0.05%Tween−20]緩衝液により希釈し、それぞれが均一なOD268値0.5を有するようにして、この状態で、導入の工程において使用した。第2の選択サイクル後、新たな2.7mlの指数関数的増殖中XL1 Blue細胞を300μlの溶出ファージにより感染させた。6種類全てのケース(標的タンパク質3種+対照物質3種)でタイトレーションを実行した後、更にヘルパーファージに感染させた培養物を30ml[2YT、100μg/mlアンピシリン、30μg/mlカナマイシン]培地内へ移した。
第2の選択サイクル後、抗フラッグタグ抗体について104倍の濃縮、MASP−1について10倍の濃縮、MASP−2について20倍の濃縮が得られた。
【0097】
3.2.4 第3の選択サイクル
第2のサイクルの場合と全て同じように実行し、カゼインも緩衝液中に維持した。単離後、ファージを2.8mlの無菌PBSに溶解し、提示のために、OD268が約0.5となるように希釈した。
第3の選択サイクル後、対照物質と比較して、非常に大きな濃縮値が得られた。差は、抗フラッグタグ抗体について105倍、両方のMASPについて104倍となった。
【0098】
3.3. ファージELISAアッセイを用いた個別クローンの試験
この試験では、選択された個々のクローンの内どれほどの割合が、バックグラウンドシグナルを示さずに標的タンパク質に結合能を有するかを調べた。
a)感染:MASP−1及びMASP−2の場合、選択サイクル2及び3からの溶出ファージ10μlを、指数関数的段階にあるXL1 Blue培養物90μlに添加した。30分間、220rpmで混合しながら37℃でインキュベートし、その後、20μlの量を取り出し、180μlの2YT培地をこれに添加した。この10倍希釈を更に2回繰り返した。各希釈段階から、100μlを[LB、100μg/mlアンピシリン]プレートに塗布し、37℃で一晩培養した。第1の選択サイクルにおいて抗フラッグタグ抗体から溶出したファージはまず希釈し、この後でのみ、細胞を感染させた。これは、抗体はELISAプレートの表面に遥かに容易に固定され、従って遙かに多くのファージが溶出したためである。高いファージ濃度のため、1個の細胞が複数のファージに感染し、混合した不可解な配列が生じるリスクがある。
b)注入:いわゆる「シングルルース」チューブ内で、500μlの培地[2YT、100μg/mlアンピシリン、50μl M13KO7ヘルパーファージ]中へ、個々のコロニを接種した。これらのチューブは、96ウェルELISAプレートの配置と同様に配置されており、個別に動くため、プレートインキュベータにおいて、300rev/minで混合しながら37℃で少量の培養物を生産することに適している。
c)固定化:MASP−1及びMASP−2タンパク質は濃度0.01μg/μlにおいて、抗フラッグタグ抗体は濃度1μg/mlにおいて、体積100μl/ウェルで、選択に関して上述したように、Nunc ELISA Maxisorpプレート上で固定化した。各クローンは、それぞれの標的タンパク質と、バックグラウンドと、抗フラッグタグ抗体とに対して試験した。
d)18時間後、チューブをプレート遠心機において、2,500rpmで10分間、4℃で遠心し、上清をピペットで清潔なチューブに移した。ELISA後、残留上清を2時間65℃で加熱した。この後、−20℃での保存が可能となり、配列決定に使用可能となる。
e)ブロッキング:液体を固定化試料から取り除き、200μl/ウェルの[PBS、2mg/mlカゼイン]ブロッキング緩衝液を各ウェル内に入れた。150rev/minで混合しながら、少なくとも1時間、室温でインキュベートした。
f)洗浄:1リットルの洗浄緩衝液を使用して、プレートを4回洗浄した。
g)ファージの付与:上述したように作製及び単離したファージを、[PBS、2mg/mlカゼイン、0.05%Tween−20]緩衝液を使用して2倍に希釈し、100μlをウェル内に配置した。同じクローンから、試料をピペットで合計3つのウェルに入れた。110rev/minで混合しながら、室温で1時間、インキュベートした。
h)洗浄:1.5リットルの洗浄緩衝液を使用して、プレートを6回洗浄した。
i)抗M13抗体:[PBS、2mg/mlカゼイン、0.05%Tween−20]緩衝液により10,000倍に希釈したHRPコンジュゲート・モノクローナル抗M13抗体(Amersham、cat#27−9421−01)100μlをウェルに配置し、110rev/minで混合しながら、室温で30分間インキュベートした。
j)洗浄:1.5リットルの洗浄緩衝液を使用して、プレートを6回洗浄した後、PBSにより2回洗浄した。
k)発色:USP蒸留水により2倍量に希釈した1−Step Ultra TMB−ELISA基質(Pierce、cat#34028)100μlを各ウェル内に配置し、少しの間、振盪し、50μlの1M HClを各ウェルに添加することで反応を停止させた。
l)読み取り:BioTrak II(Amersham)プレート読み取り光度計を使用して、吸光度を450nmにおいて測定した。
バックグラウンドの強度が低く、それぞれの標的タンパク質に対して少なくとも3倍以上強いシグナルを提示していた場合、ファージ上清からサンプルを取って、DNA配列決定用の試料を調製した。上清2μlを使用し、Big Dye Terminator v3.1 cycle Sequencing Kit(Applied Biosystems、cat#4336917)システムをPCR反応に使用した。これはBIOMI Kft.(Godollo)により実行された。
配列を解釈したところ、MASPの場合、第3のサイクルでは、個別の配列が僅かしか発見されず、僅かな種類のみが濃縮されていたため、第2の選択サイクルから更にクローンを選択及び試験する必要があることが判明した。我々の目的は、標的タンパク質のアミノ酸嗜好性に関するパターンを構築できるように、可能な限り多様な多数の配列を収集することであった。
【0099】
実施例4:結果
この実施例では、実施例1−3において説明した試験の結果、即ち、得られた配列について説明する。
【0100】
MASP−1より溶出したファージから、ELISAを使用して、32のクローンを試験し、最終的に9種類の個別の配列を見出した。MASP−2の場合、80のELISAポイントから21種類の個別の配列が得られ、抗フラッグタグ抗体の場合、72の試験クローンから57種類の解釈可能な配列が得られた。
【0101】
結果を解釈する際には、ディスプレイバイアスの影響を考慮する必要があった。このための一方法は、コドンの標準化であるが、これは、DNAライブラリを構築するために使用したNNKコドンでは、個々のアミノ酸について同じ頻度が保証されないためである。他方、更に現実的なアプローチとしては、前記抗体に基づいて選択された配列のデータによる標準化である。理論的に可能な全ての配列タイプが、ファージの表面上で提示可能となる訳ではない。なぜなら、それらの一部は実現可能な構造を発生させなかったり、或いは、ファージにとって負荷が大きすぎるものであったりするからである。しかしながら、抗体から取得した配列は現実に発生した配列であり、これらは、標的タンパク質に対して実行した選択の初期ステップにおいて存在していたものであるため、MASPに特異的な形態は、これらに基づいて得られた。
【0102】
データ標準化後、我々は、インターネット上でアクセス可能なWebLogo(http://weblogo.berkeley.edu/logo.cgi; Crooks 2004 and Schneider 1990)の支援により、当該配列に関する配列ロゴ図を作成した。我々は、個々の位置においてどのアミノ酸が好まれているか、また、MASP−1由来かMASP−2由来かに応じて、互いにどのくらい異なるかを調べた。更に、我々のデータを、フレームとしての役割を果たす野性型SFTIの配列と比較した。SFTIはウシトリプシンに対してナノモーラー(nM)以下の親和性で作用する阻害剤である。
個々のクローンは、上述したELISA系において、バックグラウンドとして使用したBSAと、標的として使用したそれぞれのタンパク質とについて調査し、更に、他方のMASP分子についても、交差反応の可能性を確認するために調査した。結果に基づいて、配列は、3つの群に分類することができる:
a)MASP−2から選択され、これに特異的な配列、
b)MASP−2から選択されたが、MASP−1も認識する配列、及び
c)MASP−1から選択されたが、MASP−2も認識する配列。
【0103】
MASP−1のみを特異的に認識する群は発見できなかった。2つの非選択的な群(b及びc)は、どちらのMASP標的に対して選択されたかに関係なく、非常に類似する傾向を示していた。配列ロゴ図の横軸では、個別位置の番号が確認でき、部位P1は、位置5に対応する。配列ロゴ図は、図5に示しており、ここで、図5の番号(5a、5b、5c)は、a)、b)、c)と分類した群の配列ロゴ図にそのままの順番で対応している。各位置において、ロゴのカラム高さは、その要素(我々の場合、20種類の異なるアミノ酸)の発生がどのくらい均等かを示す。この発生が均等でないほど、カラムは高くなる。完全に均等な分布の場合(20種類全てのアミノ酸が5%の割合で発生する場合)、高さはゼロになる。最高値は、1種類のみの要素(アミノ酸)が発生した場合に起こる。カラム内では、個々のアミノ酸は、発生頻度に基づいて配置され、最も頻度の高いものが一番上になる。アミノ酸を示す文字の高さは、その位置における相対的な発生頻度に比例している(例えば、発生頻度50%の場合、カラムの高さの半分となる)。カラーの図の場合、一般に、類似する化学特性を有するアミノ酸は、同一又は類似する色で図示されるが、本特許明細書の図では、異なる灰色の陰影を用いている。
【0104】
ロゴ図の支援により、我々は、選択的及び非選択的な群のコンセンサス配列を決定し、選択に由来するクローンの名前に基づいて、M2−6E及びM2−4Gペプチドと命名し、その活性を反映した名前は、「S」ペプチド(選択的(selective)のS)又は「NS」ペプチド(非選択的(non−selective)のNS)とした(以下参照)。
MASP−2選択的M2−6Eクローン(SEQ ID NO 2):
「S」ペプチド GYCSRSYPPVCIPD
非選択的M2−4Gクローン(SEQ ID NO 3):
「NS」ペプチド GICSRSLPPICIPD
【0105】
上述したペプチドと、その点突然変異体及び環状変異体を、固相ペプチド合成により生成した。合成及びペプチド分析試験は、実施例5において説明する。
【0106】
実施例5:ペプチド合成及び分析
5.1. ペプチドの調製、再生、及び品質検査
ペプチドは、標準的なFmoc(N−(9−フルオレニル)メトキシカルボニル)手順(Atherton 1989)を使用した固相ペプチド合成により生成した。担体からの分離と、保護基の同時除去とは、1,2−エタンジチオール、チオアニソール、水、及びフェノールがラジカル捕獲剤として存在する状態で、TFA(トリフルオロ酢酸)法を使用して実行した。ほぼ乾燥するまで溶液が蒸発した後、低温のジエチルエーテルを使用して、産物を沈殿させた。沈殿物を水に溶解した後、揮発性成分を凍結乾燥により除去した。再生のため、即ち、ペプチドの2本のシステイニル側鎖間にジスルフィド架橋を形成するために、凍結乾燥産物を、濃度0.1mg/mlで水に溶解した。継続的なエアリングに加えて溶液を混合することにより、酸化を実行し、pH値は、N,N−ジイソプロピル−エチルアミンを加えることによりアルカリ値(8乃至9)に維持した。完全な酸化の達成は、逆相HPLC手順及び質量分析法を用いて試験した。95%超の均質な形態での酸化産物の単離も、逆相HPLC手順により実行した。
M2−4Gペプチドの場合、環状形態でも生成され、この場合、線状のもののN及びC末端間に、ペプチド結合を形成させた。環化は、下記のように実行した。ペプチド合成を、2−ClTrt(2−クロロトリチル)樹脂上で行い、1%TFAを含有するDCM(ジクロロメタン)溶液を使用して、ここからペプチドを分離した。こうした条件下では、側鎖保護基は、ペプチド上に留まる。逆相HPCL手順を用いた分離ペプチドの精製後、線状ペプチドを、ペプチドの最終濃度が0.1mMになる量のDMF(ジメチルホルムアミド)を使用して溶解した。その後、1.1当量のHATU(1−[ビス(ジメチルアミノ)メチレン]−1H−1,2,3−トリアゾロ[4,5−b]ピリジン−3−オキシドヘキサフルオロリン酸)と、3.0当量のDIPEA(ジイソプロピルエチルアミン)を、これに添加した。溶液を30分間室温で混合した後、逆相HPCL手順及び質量分析法を用いて環化の効率を試験した。環化の完了後、試料を蒸発させ、調製的逆相HPLC手順を用いて、ペプチドを精製した。
【0107】
各分離ペプチドについて、品質コントロールは、質量分析手順を用いて実行した。質量分析法による分析は、HP1100型HPLC−ESI−MSシステムにより、10mMギ酸アンモニウムpH3.5溶液を使用して、フローインジェクション法により行った。機器は、以下のパラメータに設定した。乾燥ガス及び噴霧(atomizing)ガスは、共に窒素とし、乾燥ガスの流量は、10l/分、温度は300℃とした。噴霧ガスの圧力は、210kPa、キャピラリ電圧は、3500Vとした。全イオン電流(TIC)クロマトグラムは、300乃至2000質量/電荷の範囲において正イオンの設定で作成された。質量データは、Agilent ChemStationソフトウェアにより評価した。
作製した個々の阻害剤の名前、配列、及び質量データを下表1に示す。
【0108】
【表1】
表1:化学合成により製造した、本発明による幾つかのペプチド阻害剤の理論的及び実測分子量
【0109】
表1に示した配列において、ライブラリ構築中にランダム化した位置には下線を付しており、野性型SFTIとアミノ酸が異なる位置は、太字で示している。
【0110】
5.2.合成ペプチド基質による定数Kiの決定
ペプチドの阻害能力を、まず、MASP酵素とトリプシンとについて測定した。MASP酵素に対して最も有望な阻害データを示した2つのペプチド(以下参照)のみ、トロンビンについても阻害能力を測定した。
【0111】
5.2.1.MASP酵素による測定
測定に使用した合成基質は、Z−L−Lys−SBzl塩酸塩(Sigma、C3647)であり、ここから10mMストックソリューションを調製した。反応は、1mlの量で、室温において、[20mM HEPES、145mM NaCl、5mM CaCl2、0.05%Triton−X100]から成る緩衝液中で行った。酵素により切断された基質は、溶液内に2倍超過で存在するジチオジピリジン補助基質(Aldrithiol−4、Sigma、cat#143057)との反応に入る。このようにして形成された発色基の放出を、分光光度計により324nmでモニタした。段階希釈を合成ペプチドから調製し、これに酵素を添加し、室温で1時間インキュベートした。基質の濃度及び測定時間の長さは、酵素が基質の10%未満を消費する条件となるように選択した。測定の過程では、強力結合(tight-binding)阻害剤の解析用に開発された測定方法を使用した(Empie, 1982)。反応の初期段階で示された直線の傾斜は、阻害されていない酵素反応の場合に得られた傾斜により標準化され、酵素の量で乗じた。この結果として、遊離酵素の濃度を求め、阻害剤の濃度の関数として示し、次の等式1に従って表した。
ここで、Eは、遊離(非阻害)酵素濃度、E0は、初期酵素濃度である。MASP−1 MASP−2濃度は、C1阻害剤による滴定により決定した。結果は、平行して行われた複数の測定の平均として計算した。結果を、5.3.の表2にまとめる。
【0112】
5.3. トリプシン及びトロンビンに関する測定
2つのコンセンサスペプチド、即ち、M2−6E及びM2−4Gが、最も有望なMASP−2及びMASP−1阻害剤であることがわかったため、トリプシン及びトロンビンに対する阻害能力に関してこれらを当初のSFTI分子と比較することにより、その解析を続けた。トリプシン阻害の測定には、上述した測定条件を使用し、従って、トリプシンの活性は、Z−L−Lys−SBzl塩酸塩基質において、阻害ペプチド濃度の関数として測定した。評価は、上述したように行った。
【0113】
MASP酵素は、その生理的機能を血液中で遂行するため、ペプチドを使用することについての可能性は、血清中の他のプロテアーゼの活性に対してどのような影響を有するかにより決まる。我々は、血液凝固の中心的な酵素であるトロンビンを、同様の条件下で、但しZ−Gly−Pro−Arg−pNa基質を用いて、調査した。当該p−ニトロアニリドは、補助基質を必要とせず、産物の形成を、分光光度計において405nmで直接モニタすることができる。細いキュベット内の測定体積を350μlとし、基質の濃度は、505μMとした。トロンビンは、異なる阻害剤濃度で、20分間室温でインキュベートした。トロンビンの量は、活性部位滴定法を使用して決定した。評価は、上述したように行った。結果を下表2にまとめる。
【0114】
【表2】
表2:個々の阻害剤の酵素阻害を要約する表。表示した配列において、下線及び太字は、表1と同じ意味を有する。
【0115】
特に記載がない場合、阻害剤は、開鎖(open chain)を有する。記号「NG」は、使用した中で最も高い阻害剤濃度をもってしても阻害が測定できなかったことを意味する。記号「−」は、その酵素/阻害剤対に関して測定が実行されなかったことを意味する。
【0116】
データに基づくと、選択的ペプチド(M2−6E、SEQ ID NO 2)は、MASP−2を選択的に阻害し、MASP−1に対する活性はなく、トリプシンに対してはその活性は4桁も低く、トロンビン阻害剤としての能力も非常に低いと言える。これと対照的に、非選択的ペプチド(M2−4G、環状、SEQ ID NO 3)は、遙かに一般的な阻害剤の特徴を示す。このペプチドは、4種類全てのプロテアーゼを阻害し、トリプシンに対しては、野性型SFTI−1よりも遙かに弱い。トロンビン阻害剤としては劣っているが、野性型と比較すると、親和性が向上している。
【0117】
5.4. 血液凝固に対するペプチドの作用
健康な個体から採取した血漿を用いて、血液凝固の測定を行った。静脈穿刺により取得し、クエン酸ナトリウム(3.8%wt/vol)により処理した血液から、血漿を遠心(2000g、15分間、Jouan CR412遠心機)により単離した。
【0118】
血液凝固の外因経路を試験するプロトロンビン時間(PT)を、Sysmex CA−500(Sysmex、日本)自動システムにおいてInnovin試薬(Dale Behring、ドイツ、マーブルク)を使用して測定した。血液凝固の内因経路を試験する活性化部分トロンボプラスチン時間(APTT)と、トロンビンの働きを直接試験するトロンビン時間(TT)とを、Coag−A−Mate MAX(BioMerieux、フランス)アナライザにおいて、TriniClot試薬(Trinity Biotech、アイルランド、ウィックロ)及びReanal試薬(Reanal Finechemical、ハンガリ)を使用して測定した。
【0119】
血液凝固に対するペプチドの影響を調査するために、我々は用量依存性を測定した。結果を図6のグラフに示す。各図において、破線間の領域は、その測定との関連における正常範囲を示す。縦座標には、時間を秒単位で定め、横座標には、阻害剤濃度の対数を、μM単位で示す。
【0120】
図6aは、トロンビン時間を測定するための実験を示しており、その過程では、血漿にトロンビンを添加することにより血漿凝固(フィブリン形成)を開始させた。外部から添加したトロンビンの影響を、ペプチドの濃度を増加させつつ阻害し(横座標)、凝固に必要な時間を測定する(縦座標)。図6bは、プロトロンビン時間を測定するための実験を示しており、その過程では、血漿に組織因子を添加することにより血漿凝固(フィブリン形成)を開始させ、その結果、第VII因子の活性化により、トロンビンを活性化するプロトロンビナーゼ複合体が幾つかのステップにおいて形成される。この実験では、外傷(血管損傷)の結果として活性化される血液凝固の外因経路を模倣している。組織因子により開始されるプロテアーゼカスケードの構成要素を、ペプチドの濃度を増加させつつ阻害し(横座標)、凝固に必要な時間を測定する(縦座標)。図6cは、例えば、血液中のコラーゲンの発生により生理的に開始される、血液凝固のいわゆる「接触活性又は内因性」経路を模倣した、活性トロンボプラスチン時間を測定するための実験を示す。実験では、コラーゲンの代わりに、異なる大表面(large-surface)素材、例えばカオリン粉末を添加することにより実現する。この結果として、活性化第XII因子を介して、やはりプロテアーゼカスケードが開始され、その結果として、トロンビンを活性化するプロトロンビナーゼ複合体が形成される。このプロテアーゼカスケードの構成要素を、ペプチドの濃度を増加させつつ阻害し(横座標)、凝固に必要な時間を測定する(縦座標)。
【0121】
3種類全ての測定について、選択的「S」ペプチドは、濃度が200μMであっても正常領域近くに留まっており、従って、MASP阻害という側面で問題となるような濃度において凝固を阻害しなかった。これとは対照的に、非選択的“NS”ペプチドは、200μMの場合に、極端な測定値に達し、これは、血液凝固を有意に阻害したことを意味する。前章で説明したデータでは、「NS」ペプチドがトロンビンを10μMのKi値で阻害することが実証されており、このことだけでも、当該試験において示された作用が説明される。血液凝固の最後のステップにおいて、トロンビンは、フィブリノゲンを分解し、これによりフィブリンに基づく凝塊を形成する酵素である。そのため、トロンビンを阻害することは、それだけで血液凝固を効率的に阻害するのに十分となる。このため、上述した血液凝固試験に基づき、比較的好適にトロンビンを阻害する「NS」ペプチドが、血液凝固カスケード中で機能的観点においてトロンビンに先行する他の血液凝固因子(例えば、VIIA、IXa、Xa、XIa、XIIa)を同じく阻害するか否かを判断することはできない。その一方で、3種類全ての試験において実証された選択的「S」ペプチドの血液凝固に対する作用の弱さは、このペプチドは、カスケードの初期成分に対しても強力な阻害剤にはなり得ないことを示している。
【0122】
5.5.本発明によるペプチドの3つの補体活性化経路に対する作用
詳細に上述してきたように、補体系は、3つの経路により活性化可能であり、同じ単一の終点につながる。3つの活性化経路は、古典経路、レクチン経路、及び副経路を含む。MASPは、レクチン経路の初期段階の酵素であるため、本発明によるMASP阻害剤が、レクチン経路、他の2つの活性化経路、及び3経路が出会った後の共通段階に対して、どのような作用を有するかを知ることは重要である。
【0123】
測定のため、我々は、補体経路の選択的測定用に開発された、いわゆるWIELISAキット(Euro−Diagnostica AB、COMPL300)を、キットに付属の使用説明書に基づいて使用した。測定の指針は、3つの活性化経路に従って3種類の測定条件を使用し、現在検査中の補体活性経路は機能可能な状態にすると同時に他の2つの経路は不活性であるようにすることである。その一方で、測定において検出する産物は、経路選択的な成分ではなく、3つ活性経路の共通部分の最終要素であるC5−9複合体である。
【0124】
測定のために、血液試料を1時間室温でインキュベートし、その後、遠心して、血清を小分けにして−80℃で保存した。血清を、処方に従って所定の補体経路用の緩衝液により希釈し、20分間室温でインキュベートし、ペプチドの段階希釈をこれに添加し、20分間室温でインキュベートし、その後、ピペットで、特別なELISAプレートの適切なウェルに移し入れた。続いて、洗浄、インキュベート、及び抗体の添加を使用説明書に従って実行した。基質と共に20分間インキュベートした後、分光光度計において450nmでデータを読み取った。各測定点において平行実験を行い、100%の活性は、阻害剤を含まない血清により表された。測定は、同じプレート上で同時に、溶解した1つの同じ血清試料を用いて、実行した。
【0125】
当該測定により、「S」ペプチド及び「NS」ペプチドが共に補体系のレクチン経路の効率的且つ特異的な阻害剤であるという極めて重要な結果が得られた。この結果は、我々の現在の知識によればレクチン経路の開始に関与する酵素であるMASP−2酵素を、両方のペプチドが非常に効率的に阻害するという、前述の結果と一致するものである。
【0126】
多数のセリンプロテアーゼが補体系において作用しており、その一部は、MASP酵素に非常に似ている。これにもかかわらず、「S」ペプチドも「NS」ペプチドも、古典経路および副経路を阻害しなかった。
【0127】
古典経路及び副経路の測定の過程では、本発明によるペプチドの存在は、最終的なC5−9複合体の形成を阻害しなかったことから、本発明のペプチドが補体系の共通部分のプロテアーゼを阻害しないことは確実であり、従って、レクチン経路の阻害は、実際に、レクチン経路の開始時点において、すなわちMASP酵素のレベルにおいて生じたものである。WIELISA測定の過程で得られたIC50データは、合成基質に基づくMASP−2阻害の測定の過程で得られたki値に比べて約30倍、60倍高いことは指摘するに値する。これには以下の説明が可能である:阻害ペプチドは、MASP−2酵素と直接、基質結合部位において結合し、この結合は、当該同じ酵素表面と小さな合成基質との比較的弱い相互作用に対して上手く競合する。しかしながら、生理学的基質は、プロテアーゼドメインに位置した基質結合部位に加え、他の表面(エキソサイト)を介した結合を形成することも可能であり、これらは小さな合成基質よりも高い親和性で酵素と結合する。この高い親和性のため、阻害ペプチドは、バランスを酵素基質複合体から酵素阻害剤複合体の方へ移行させるために、より高い濃度で使用する必要がある。
【特許請求の範囲】
【請求項1】
一般式(I)によるペプチド、並びにその塩、エステル、及び薬剤的に許容可能なプロドラッグ;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項2】
以下の配列を有するペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、
GICSRSLPPICIPD(SEQ ID NO 3)、
GVCSRSLPPICWPD(SEQ ID NO 4)、
GMCSRSYPPVCIPD(SEQ ID NO 5)、
GYCSRSIPPVCIPD(SEQ ID NO 6)、
GWCSRSYPPVCIPD(SEQ ID NO 7)、及び
配列GICSRSLPPICIPD(SEQ ID NO 3)を有する環状ペプチド、
並びにその塩及びエステルから選択される、請求項1に記載のペプチド。
【請求項3】
以下の配列を有するペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、及び
GICSRSLPPICIPD(SEQ ID NO 3)、
並びにその塩及びエステルから選択される、請求項2に記載のペプチド、。
【請求項4】
医薬調製物であって、一般式(I)による少なくとも1つのペプチド、
及び/又は、一般式(I)によるペプチドの薬剤的に許容可能な塩、エステル、又はプロドラッグ、
及び少なくとも1つの他の添加物を含有する医薬調製物;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項5】
少なくとも1つの前記添加物は、制御された活性薬剤の放出を確保するマトリクスである、請求項4に記載の医薬調製物。
【請求項6】
一般式(I)による前記ペプチドは、以下の配列を有するペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、
GICSRSLPPICIPD(SEQ ID NO 3)、
GVCSRSLPPICWPD(SEQ ID NO 4)、
GMCSRSYPPVCIPD(SEQ ID NO 5)、
GYCSRSIPPVCIPD(SEQ ID NO 6)、
GWCSRSYPPVCIPD(SEQ ID NO 7)、及び
配列GICSRSLPPICIPD(SEQ ID NO 3)を有する環状ペプチド、
及び/又は、その薬剤的に許容可能な塩及びエステルから選択される、請求項4又は5に記載の医薬調製物。
【請求項7】
前記ペプチドは、以下の配列を有するペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、及び
GICSRSLPPICIPD(SEQ ID NO 3)、
並びにその薬剤的に許容可能な塩及びエステルから選択される、請求項6に記載の医薬調製物。
【請求項8】
一般式(I)による1つ以上のペプチド、
及び/又は、その塩もしくはエステルを含むキット;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項9】
MASP酵素を阻害する可能性のある化合物をスクリーニングするための方法であって、その過程において、
i)標識された一般式(I)によるペプチド、
及び/又は、その塩もしくはエステルを、MASPを含有する溶液に添加し、
ii)試験対象の1つ以上の化合物を含有する溶液をそこに添加し、
iii)放出された有標ペプチドの量を測定する、方法;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項10】
MASP酵素は、MASP−1又はMASP−2酵素から選択される、請求項9に記載の方法。
【請求項11】
補体系の阻害により治療可能な疾患の治療用の医薬調製物の製造における、一般式(I)によるペプチド、
又はその薬剤的に許容可能な塩もしくはエステルの使用;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項12】
補体系を阻害することにより治療可能な前記疾患は、炎症性疾患及び自己免疫疾患から選択される、請求項11に記載の使用。
【請求項13】
補体系を阻害することにより治療可能な前記疾患は、虚血再灌流障害、関節リウマチ、神経変性疾患、加齢性黄斑変性症、糸球体腎炎、全身性エリテマトーデスから選択される、請求項11に記載の使用。
【請求項14】
補体系を阻害することにより治療可能な前記疾患は、補体活性化関連仮性アレルギである、請求項11に記載の使用。
【請求項15】
MASP酵素を単離するための方法であって、その過程において、
i)一般式(I)によるペプチド、
及び/又は、その塩もしくはエステルを、担体上において固定化し、
ii)このように固定化された前記ペプチドを、MASP酵素を含有する溶液に接触させ、
iii)当該調製物を洗浄する、方法;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項16】
MASP酵素が、MASP−1又はMASP−2酵素から選択される、請求項15に記載の方法。
【請求項1】
一般式(I)によるペプチド、並びにその塩、エステル、及び薬剤的に許容可能なプロドラッグ;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項2】
以下の配列を有するペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、
GICSRSLPPICIPD(SEQ ID NO 3)、
GVCSRSLPPICWPD(SEQ ID NO 4)、
GMCSRSYPPVCIPD(SEQ ID NO 5)、
GYCSRSIPPVCIPD(SEQ ID NO 6)、
GWCSRSYPPVCIPD(SEQ ID NO 7)、及び
配列GICSRSLPPICIPD(SEQ ID NO 3)を有する環状ペプチド、
並びにその塩及びエステルから選択される、請求項1に記載のペプチド。
【請求項3】
以下の配列を有するペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、及び
GICSRSLPPICIPD(SEQ ID NO 3)、
並びにその塩及びエステルから選択される、請求項2に記載のペプチド、。
【請求項4】
医薬調製物であって、一般式(I)による少なくとも1つのペプチド、
及び/又は、一般式(I)によるペプチドの薬剤的に許容可能な塩、エステル、又はプロドラッグ、
及び少なくとも1つの他の添加物を含有する医薬調製物;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項5】
少なくとも1つの前記添加物は、制御された活性薬剤の放出を確保するマトリクスである、請求項4に記載の医薬調製物。
【請求項6】
一般式(I)による前記ペプチドは、以下の配列を有するペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、
GICSRSLPPICIPD(SEQ ID NO 3)、
GVCSRSLPPICWPD(SEQ ID NO 4)、
GMCSRSYPPVCIPD(SEQ ID NO 5)、
GYCSRSIPPVCIPD(SEQ ID NO 6)、
GWCSRSYPPVCIPD(SEQ ID NO 7)、及び
配列GICSRSLPPICIPD(SEQ ID NO 3)を有する環状ペプチド、
及び/又は、その薬剤的に許容可能な塩及びエステルから選択される、請求項4又は5に記載の医薬調製物。
【請求項7】
前記ペプチドは、以下の配列を有するペプチド:
GYCSRSYPPVCIPD(SEQ ID NO 2)、及び
GICSRSLPPICIPD(SEQ ID NO 3)、
並びにその薬剤的に許容可能な塩及びエステルから選択される、請求項6に記載の医薬調製物。
【請求項8】
一般式(I)による1つ以上のペプチド、
及び/又は、その塩もしくはエステルを含むキット;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項9】
MASP酵素を阻害する可能性のある化合物をスクリーニングするための方法であって、その過程において、
i)標識された一般式(I)によるペプチド、
及び/又は、その塩もしくはエステルを、MASPを含有する溶液に添加し、
ii)試験対象の1つ以上の化合物を含有する溶液をそこに添加し、
iii)放出された有標ペプチドの量を測定する、方法;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項10】
MASP酵素は、MASP−1又はMASP−2酵素から選択される、請求項9に記載の方法。
【請求項11】
補体系の阻害により治療可能な疾患の治療用の医薬調製物の製造における、一般式(I)によるペプチド、
又はその薬剤的に許容可能な塩もしくはエステルの使用;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項12】
補体系を阻害することにより治療可能な前記疾患は、炎症性疾患及び自己免疫疾患から選択される、請求項11に記載の使用。
【請求項13】
補体系を阻害することにより治療可能な前記疾患は、虚血再灌流障害、関節リウマチ、神経変性疾患、加齢性黄斑変性症、糸球体腎炎、全身性エリテマトーデスから選択される、請求項11に記載の使用。
【請求項14】
補体系を阻害することにより治療可能な前記疾患は、補体活性化関連仮性アレルギである、請求項11に記載の使用。
【請求項15】
MASP酵素を単離するための方法であって、その過程において、
i)一般式(I)によるペプチド、
及び/又は、その塩もしくはエステルを、担体上において固定化し、
ii)このように固定化された前記ペプチドを、MASP酵素を含有する溶液に接触させ、
iii)当該調製物を洗浄する、方法;
ここで
X1は、Y、M、W、I、V、A、及び
X2は、R、K、及び
X3は、Y、F、I、M、L、E、D、H、及び
X4は、V、I、H、及び
X5は、I、V、Y、F、W、
である。
【請求項16】
MASP酵素が、MASP−1又はMASP−2酵素から選択される、請求項15に記載の方法。
【図3】

【図5a】

【図5b】

【図5c】

【図6a】
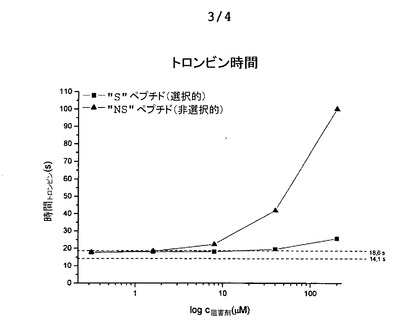
【図6b】
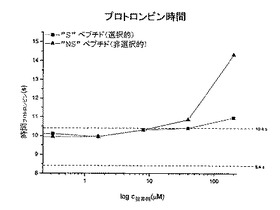
【図6c】
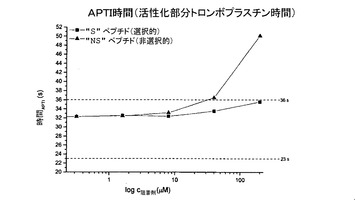
【図7】
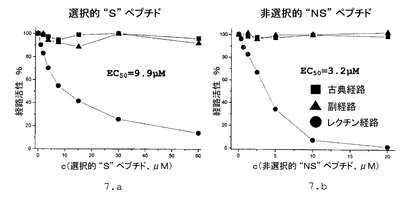
【図1】
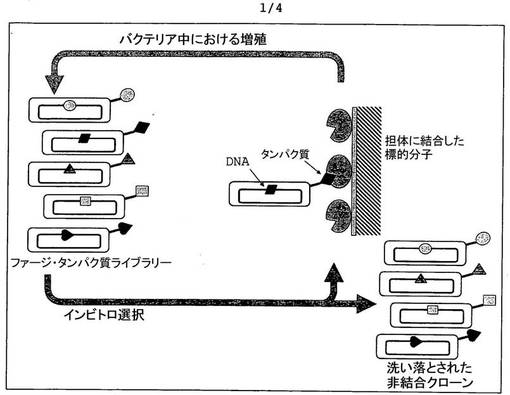
【図2】

【図4】
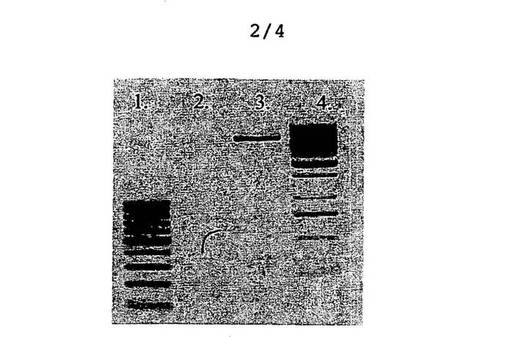
【図5a】
【図5b】
【図5c】
【図6a】
【図6b】
【図6c】
【図7】
【図1】
【図2】
【図4】
【公表番号】特表2012−528138(P2012−528138A)
【公表日】平成24年11月12日(2012.11.12)
【国際特許分類】
【出願番号】特願2012−512461(P2012−512461)
【出願日】平成22年5月25日(2010.5.25)
【国際出願番号】PCT/HU2010/000061
【国際公開番号】WO2010/136831
【国際公開日】平成22年12月2日(2010.12.2)
【出願人】(511286492)
【氏名又は名称原語表記】EOTVOS LORAND TUDOMANYEGYETEM
【出願人】(511286506)
【氏名又は名称原語表記】MAGYAR TUDOMANYOS AKADEMIA SZEGEDI BIOLOGIAI KOZPONT ENZIMOLOGIAI INTEZETE
【Fターム(参考)】
【公表日】平成24年11月12日(2012.11.12)
【国際特許分類】
【出願日】平成22年5月25日(2010.5.25)
【国際出願番号】PCT/HU2010/000061
【国際公開番号】WO2010/136831
【国際公開日】平成22年12月2日(2010.12.2)
【出願人】(511286492)
【氏名又は名称原語表記】EOTVOS LORAND TUDOMANYEGYETEM
【出願人】(511286506)
【氏名又は名称原語表記】MAGYAR TUDOMANYOS AKADEMIA SZEGEDI BIOLOGIAI KOZPONT ENZIMOLOGIAI INTEZETE
【Fターム(参考)】
[ Back to top ]