
新規アルコールアルデヒド脱水素酵素
【課題】アルコールおよび/またはアルデヒド脱水素酵素活性を有する新規組換え酵素調製物を提供する。
【解決手段】グルコノバクターに存在する固有のアルコールおよび/またはアルデヒド脱水素酵素をコードする遺伝子をクローン化した。該遺伝子の特定配列により同定されるポリペプチドを組み合わせたキメラ組換え酵素、並びに一つまたはそれ以上のアミノ酸残基の付加、挿入、欠失および/または置換を含むポリペプチドより成るアルコールおよび/またはアルデヒド脱水素酵素調製物。更に、該調製物をL-ソルボースおよび/またはD-ソルビトールに作用させることにより、2−ケトーL-ギュロン酸を調製し、これを既知の方法でL-アスコルビン酸に変換する製造方法。
【解決手段】グルコノバクターに存在する固有のアルコールおよび/またはアルデヒド脱水素酵素をコードする遺伝子をクローン化した。該遺伝子の特定配列により同定されるポリペプチドを組み合わせたキメラ組換え酵素、並びに一つまたはそれ以上のアミノ酸残基の付加、挿入、欠失および/または置換を含むポリペプチドより成るアルコールおよび/またはアルデヒド脱水素酵素調製物。更に、該調製物をL-ソルボースおよび/またはD-ソルビトールに作用させることにより、2−ケトーL-ギュロン酸を調製し、これを既知の方法でL-アスコルビン酸に変換する製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アルコールおよび/またはアルデヒド脱水素酵素活性を有するアルコール/アルデヒド脱水素酵素(以下AADHと称する)の組換え酵素調製物に関する。本発明はまた、AADHをコードする新規組換えDNA分子、該DNAを含む組換え発現ベクター、該組換えDNA分子および/または該組換え発現ベクターを含有する組換え生物に関する。更に本発明は、AADHの組換え酵素調製物の製造方法および該組換え酵素調製物を利用するアルデヒド、カルボン酸、ケトン、特に2-ケト-L-ギュロン酸(以下2KGAと称する)の製造方法および該組換え生物を利用するアルデヒド、カルボン酸、ケトン、特に2KGAの製造方法に関する。
【背景技術】
【0002】
2KGAはL-アスコルビン酸(ビタミンC)生産の重要な中間体である。数多くの微生物がD-ソルビトールまたはL-ソルボースから2KGAを生産することが知られている。特公昭51-40154(1976)はアセトバクター(Acetobacter)属、バクテリウム(Bacterium)属またはシュードモナス(Pseudomonas)属の微生物によるD-ソルビトールからの2KGA生産を開示している。「Acta Micorobiologica Sinica 21巻 (2号)185〜191頁 (1981)」によれば、2KGAはL-ソルボースから微生物の混合培養物、特にシュードモナスストリアータ(Pseudomonasstriata)とグルコノバクター オキシダンス(Gluconobacteroxydans)の混合培養により生産することが可能である。ヨーロッパ特許出願公開第0221 707号は共存微生物存在および非存在下でのシュードグルコノバクターサッカロケトゲネス(Pseudogluconobactersaccharoketogenes)によるL-ソルボースからの2KGA生産を開示している。ヨーロッパ特許出願公開第0278 447号はDSM番号4025(グルコノバクターオキシダンス)および他のDSM番号4026(バチルス メガテリウム株、Bacillusmegaterium)を含む混合培養物によるL-ソルボースからの2KGA製造方法を開示している。ヨーロッパ特許出願公開第88116156号はG.オキシダンスDSM番号4025によるL-ソルボースからの2KGA製造方法を開示している。
【0003】
G.オキシダンスDSM番号4025からAADHが精製され、アルコール、アルデヒドの酸化を触媒するという性質が調べられた。すなわちAADHは、アルコールから対応するアルデヒドおよびケトンを、アルデヒドからカルボン酸を生産することができた(朝倉、星野、ヨーロッパ特許出願公開第606621号)。更に、特に本AADHはL-ソルボースからL-ソルボソンを経由して2KGAへの酸化を触媒した。AADHの精製標品の物理化学的性質は、次の通りである:1)至適pH:約7.0〜9.02)至適温度:約20℃〜40℃3)分子量:135,000 +/- 5,000ダルトン(分子量64,500 +/- 2,000のアルファサブユニットと分子量62,500 +/- 2,000のベータサブユニットのどの組み合わせでもよい2つのサブユニットを含む)
4)基質特異性:L-ソルボース、L-ソルボソン、D-ソルビトール、D-グルコース、D-マンニトール、D-フルクトース、DL-グリセルアルデヒド、エタノール、1-プロパノール、1-ブタノール、1-ペンタノール、1-ヘキサノール、1-ヘプタノール、2-プロパノール、2-ブタノール、プロピオンアルデヒド、PEG1000、PEG2000、PEG4000、PEG6000およびポリビニルアルコールを含む一級および二級アルコールとアルデヒドに活性を有する5)補欠分子族:ピロロキノリンキノン6)等電点:約4.4。
【0004】
該AADHをコードする遺伝子をクローン化できれば、大量のAADH組換え酵素調製物の生産、ならびに種々のアルデヒド、ケトンおよびカルボン酸、特に2KGAの生産が可能な組換え生物を作成するのに有用である。しかし、今日までかかる遺伝子類のクローニングに関する報告はない。
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、アルコールおよび/またはアルデヒド脱水素酵素活性を有するAADHの新規組換え酵素調製物を提供することを課題とする。また、本発明は、AADHをコードする新規組換えDNA分子、該DNAを含む組換え発現ベクター、該DNAおよび/または組換え発現ベクターを有する組換え生物、組換えAADHの製造方法、組換えAADHまたは組換え生物を利用するアルデヒド、カルボン酸、ケトン、特に2KGAの製造方法を提供することを課題とする。
より詳しくは、本発明の一面は配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチド、並びに配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドの間のキメラ組換え酵素、並びに一つまたはそれ以上のアミノ酸残基の付加、挿入、欠失および/または置換を含む上記で同定されるポリペプチドの機能的誘導体からなる群より選択され、該酵素ポリペプチドがアルコールおよび/またはアルデヒド脱水素酵素活性を有する、一つまたはそれ以上の酵素ポリペプチドを含むアルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物に関する。
【0006】
かかる同等の機能を有する誘導体は、当技術分野において周知の化学的ペプチド合成、または本明細書に開示されているような当技術分野において周知の方法により、たとえばサムブルック(Sambrook)ら(分子クローニング(Molecular Cloning), Cold Spring Harbour Laboratory Press, New York,USA,第2版、1989)により開示されているようなDNA配列に基づいた組換えDNA技術、のいずれの方法でも作成することが可能である。蛋白質やペプチド中のアミノ酸をその分子の活性を概して変えずに置換することは、当技術分野において周知であり、たとえばH.ニューラス(Neurath) R. L.ヒル(Hill)により「蛋白質(The Proteins)」(Academic Press, New York, 1979, 特に14頁の図6参照)に述べられている。最も通常起こる置換は:Ala/Ser、Val/Ile、Asp/Glu、Thr/Ser、Ala/Gly、Ala/Thr、Ser/Asn、Ala/Val、Ser/Gly、Tyr/Phe、Ala/Pro、Lys/Arg、Asp/Asn、Leu/Ile、Leu/Val、Ala/Glu、Asp/Glyおよびこれらの逆である。
【0007】
本発明の他の一面は、配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチド、並びに配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドの間のキメラ組換え酵素、並びに一つまたはそれ以上のアミノ酸残基の付加、挿入、欠失および/または置換を含む上記で同定されるポリペプチドの機能的誘導体からなる群より選択され、該酵素ポリペプチドがアルコールおよび/またはアルデヒド脱水素酵素活性を有する、少なくとも一つの酵素ポリペプチドをコードする組換えDNA分子に関する。
【0008】
さらに本発明は、たとえば本配列表に開示されているようなアルコールおよび/またはアルデヒド脱水素酵素活性を有するポリペプチドをコードするDNA配列、およびそれらの相補鎖、またはこれらの配列を含むDNA配列、該配列またはそれらから誘導される断片と標準的条件下でハイブリダイズするDNA配列、および遺伝子コードの縮退という理由により上記DNA配列と標準的条件下でハイブリダイズはしないが全く同じアミノ酸配列を有するポリペプチドをコードするDNA配列に関する。
【0009】
ハイブリダイゼーションの「標準的条件」とは、特異的ハイブリダイゼーションシグナルを検出するのに当業者により一般的に使用され、かつ例えばサムブルック(Sambrook)ら(分子クローニング(Molecular Cloning), Cold Spring Harbour Laboratory Press, New York,USA, 第2版、1989)により述べられている条件、好ましくは、当業者に公知のサムブルック(Sambrook)らにより述べられたいわゆるストリンジェントハイブリダイゼーション-非ストリンジェント洗浄条件、より好ましくはいわゆるストリンジェントハイブリダイゼーション-ストリンジェント洗浄条件を意味する。さらに当技術分野で周知の方法を使用し本明細書に開示されているDNA配列に基づいて設計したプライマーを用いてポリメラーゼ連鎖反応(PCR)により作成できるDNA配列を提供することもまた本発明の目的である。本発明のDNA配列はまたヨーロッパ特許出願第747 483号に記載されているように合成することも可能であると理解される。
【0010】
本発明の更なる一面は、上記で定義される一つまたはそれ以上の組換えDNA分子を有する組換え発現ベクターおよび上述の組換え発現ベクターおよび/または、一つまたはそれ以上のDNA分子を染色体上に有する組換え生物に関する。
【0011】
本発明の更なる一面は、適当な培地で上述の組換え生物を培養し、該組換え酵素調製物を回収することを含む、上記で定義されるアルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物の製造方法に関する。
【0012】
本発明の他の一面は、上記で定義される組換え生物を使用しての基質を生産物に変換することを含むアルデヒド、ケトン、またはカルボン酸を対応する基質から生産する製造方法に関する。
【0013】
本発明の更に他の一面は、L-ソルボースおよび/またはD-ソルビトールを含有する適当な培地中での上記に定義する組換え生物の醗酵を含む2-ケト-L-ギュロン酸の製造方法に関する。
【0014】
本発明の他の一面は、本発明の組換え酵素調製物を含有する反応混合液をインキュベートすることを含むアルデヒド、ケトンまたはカルボン酸を対応する基質から生産する製造方法に関する。
【0015】
本発明の更に他の一面は、上記に定義する組換え酵素調製物とL-ソルボースおよび/またはD-ソルビトールを含有する反応混合液をインキュベートすることを含む2-ケト-L-ギュロン酸の製造方法に関する。
【0016】
上述のように2-ケト-L-ギュロン酸の製造方法に効果的であり、該製造方法により得られる2-ケト-L-ギュロン酸が当技術分野で既知の方法によりビタミンC(L-アスコルビン酸)に変換されることを特徴とする2-ケト-L-ギュロン酸からのビタミンCの製造方法を提供することも本発明の課題である。
【課題を解決するための手段】
【0017】
本発明に記述されているAADH遺伝子は上述のように種々のアルコールおよびアルデヒドの酸化を触媒することが可能なAADH酵素をコードしている。具体的には、グルコノバクターに存在する固有のAADH酵素遺伝子をクローン化し、発現させた。当業者であれば、本発明の知見に基づき、グルコノバクターに加えて他のAADH酵素遺伝子の供給源も見出すことが可能である。
【0018】
特定の好ましいグルコノバクターオキシダンス株は、DSM番号4025の下、「Deutsche Sammlung vonMikroorganismen in Gottingen(ドイツ)」に寄託されている。
【0019】
更に、本菌株の継代培養株は、寄託番号FERM BP-3812として、日本工業技術院生命工学工業技術研究所に寄託されている。ヨーロッパ特許出願公開第0278477号に本菌株の性質が開示されている。
【0020】
本発明で利用されるAADH遺伝子および組換え微生物は以下の工程で取得できる:(1)コロニーハイブリダイゼーション、プラークハイブリダイゼーション、PCRクローニング、ウェスタンブロット分析、サザンブロットハイブリダイゼーション等の手法による染色体DNAからのAADH遺伝子のクローニング(2)該AADH遺伝子の常法による塩基配列決定およびAADH遺伝子を含有しそれを効率良く発現する組換え発現ベクターの構築(3)形質転換、形質導入、接合伝達およびエレクトロポレーションによる組換え発現ベクターまたは染色体上に組換えAADH遺伝子を有する組換え微生物の構築。
【0021】
本発明の上記見地に応用可能な材料および技術を以下のように詳細に例示する。
【0022】
全染色体DNAは当業者に周知の方法(マーマー(Marmaur) J., J. Mol. Biol. 3巻、208頁、1961)により精製できる。次に目的遺伝子を有する菌株の遺伝子ライブラリーは上記染色体DNAおよび以下に詳細に述べられるベクターを用い構築可能である。AADHをコードする遺伝子は、以下の方法で染色体DNAからプラスミドまたはファージどちらのベクターにもクローン化可能である。
【0023】
1)精製酵素の部分アミノ酸配列を決定し、その配列情報に従ってオリゴヌクレオチドを合成し、遺伝子ライブラリーから目的の遺伝子をサザンブロットハイブリダイゼーション、コロニーハイブリダイゼーション、またはプラークハイブリダイゼーションにより選択する。
【0024】
2)上述のように合成したオリゴヌクレオチドをプライマーとして用いるポリメラーゼ連鎖反応(PCR)により所望の遺伝子の部分配列を増幅し、そのPCR生成物をプローブとして用い遺伝子ライブラリーから目的の完全長の遺伝子をサザンブロットハイブリダイゼーション、コロニーハイブリダイゼーション、またはプラークハイブリダイゼーションにより選択する。
【0025】
3)たとえば「Methods in Enzymology、73巻、46頁、1981」に述べられているような前述の方法で所望の酵素蛋白質に対する抗体を調製し、ウェスタンブロット分析を含む免疫学的分析により所望のポリペプチドを発現するクローンを選択する。
【0026】
4)所望の酵素の一つに対する相同体(homologs)のアミノ酸配列を整列し、相同体間で共通のアミノ酸配列を選択し、その共通配列をコードするオリゴヌクレオチドを合成し、そのオリゴヌクレオチドをプライマーとして用いるPCRにより所望の遺伝子部分配列を増幅し、上記2)の方法で述べた完全配列を選択する。
【0027】
所望の遺伝子の塩基配列はM13ファージを用いるダイデオキシチェインターミネーション法(サンガー(Sanger) F.ら、Proc. Natl. Acad. Sci. USA、74巻、5463〜5467頁、1977)のような公知の方法で決定できる。
【0028】
必要に応じて、このように決定された塩基配列情報を(使用コドンを考慮して)利用することにより、進化的に分岐したアルコールおよび/またはアルデヒド脱水素酵素をコードする遺伝子を、該塩基配列から推定されるアミノ酸配列に従って合成されたプローブを用いるコロニーハイブリダイゼーションまたはサザンブロットハイブリダイゼーションにより、または該情報により合成されたプライマーを用いるポリメラーゼ連鎖反応(PCR)により、異種の生物からも単離可能である。
【0029】
所望の遺伝子、すなわち本発明のDNA配列を効率良く発現するために、種々のプロモーターを使用することが可能である。たとえば、該遺伝子自身のプロモーターや、Tn5(D. E. Berg, C. M. Berg, 1983, Bio/Technology 1巻、417〜435頁)のカナマイシン耐性遺伝子、pBR322のアンピシリン耐性遺伝子のような抗生物質耐性遺伝子のプロモーター、大腸菌(Escherichia coli)のベータガラクトシダーゼ遺伝子のプロモーター(lac)、trp、tac、trcプロモーター、ラムダファージのプロモーターおよび大腸菌、シュードモナスプチダ(Pseudomonasputida)、アセトバクター キシリナム(Acetobacter xylinum)、アセトバクター パスチュリアヌス(Acetobacter pasteurianus)、アセトバクターアセチ(Acetobacter aceti)、アセトバクター ハンゼニイ(Acetobacter hansenii)およびグルコノバクターオキシダンス等の細菌を含む微生物、哺乳動物細胞および植物細胞を含む宿主中で機能しうるいずれのプロモーターも使用可能である。
【0030】
さらに遺伝情報をコードする配列が導入される宿主細胞中で機能可能なシャインダルガノ(SD)配列(たとえばAGGAGG等で、宿主細胞中で機能しうる天然および合成配列も含む)および転写終結因子(宿主細胞中で機能しうる天然および合成配列を含む逆位反復配列)のような他の制御因子も上述のプロモーターとともに使用することができる。
【0031】
ペリプラズムに局在するポリペプチド(AADH)の発現のためには、通常15〜50アミノ酸残基を含み全体的に疎水性であるシグナルペプチドが不可欠である。シグナルペプチドをコードするDNAは宿主細胞中で機能しうるものであればどのような天然配列でも合成配列でもよい。
【0032】
多様な宿主/クローニングベクターの組み合わせを二重鎖DNAのクローニングに使用できる。クローニングベクターは一般的に複製起点、制御因子、マルチクローニング部位を含むクローニング部位、およびアンピシリン、テトラサイクリン、カナマイシン、ストレプトマイシン、ゲンタマイシン、スペクチノマイシン等の抗生物質耐性遺伝子のような選択マーカーを含むプラスミドまたはファージである。
【0033】
大腸菌中での本発明のDNA配列の発現のための好ましいベクターは、pBR322またはpUC18、pBluescriptIIを含むその誘導体、pACYC177、pACYC184(J. Bacteriol.、134巻、1141〜1156頁、1978)およびその誘導体、ならびにRK2、RSF1010のような広宿主域プラスミドから誘導されるベクターのような大腸菌中で通常使用されるどのようなベクターからも選択できる。G.オキシダンスDSM番号4025を含むグルコノバクターおよびP.プチダ中での本発明のDNA配列の発現のための好ましいベクターは、大腸菌のような好ましいクローニング宿主と同様、グルコノバクターおよび/またはP.プチダ中で複製できるどのようなベクターからも選択できる。好ましいベクターは、pVK102のようなコスミドベクターとその誘導体、およびRSF1010とその誘導体、ならびにグルコノバクターで機能する複製起点および大腸菌で機能するもう一つの複製起点を有するベクターのような広宿主域ベクターである。ベクターのコピー数および安定性はクローン化遺伝子の安定で効率よい発現のため、およびクローン化遺伝子を有する宿主細胞の効率よい培養のために注意深く考慮すべきである。Tn5のような転位因子を含むDNA分子も、本発明のDNA配列を好ましい宿主に、特に染色体上に、導入するベクターとして使用可能である。本発明の所望のDNA配列と共に、好ましい宿主から単離されたどのようなDNAを含むDNA分子も、好ましい宿主に、特に染色体上に、本発明の所望のDNA配列を導入するのに有用である。そのようなDNA分子は形質転換、形質導入、接合伝達またはエレクトロポレーションにより好ましい宿主に導入することが可能である。
【0034】
使用可能な宿主としては、微生物、哺乳動物細胞および植物細胞等が挙げられる。好ましい微生物としては、大腸菌、P.プチダ、A.キシリナム、A.パスチュリアヌス、A.アセチ、A.ハンゼニイ、G.オキシダンスおよび組換えAADHを生産可能ないずれのグラム陰性細菌でもよい細菌が挙げられる。該微生物の機能的同等株、継代培養株、変異株および変種も本発明に使用することが可能である。好ましい株は、大腸菌 K12株およびその誘導株、P.プチダまたはG.オキシダンス DSM番号4025株である。
【0035】
機能的なAADHをコードする本発明のDNA配列を用い、当技術分野で周知の方法を用い上述の宿主中で機能することが可能なプロモーターやリボゾーム結合部位のような制御領域を有する適当なベクターに連結することにより、発現プラスミドを産出する。そのような組換え発現ベクターの構造を具体的に図1、2、4および10に示す。
【0036】
組換え発現ベクターを有する組換え微生物を構築するためには、形質転換、形質導入、接合伝達(フィリップゲンハート(Philipp Genhardt)ら、「一般的分子細菌学に関する方法(Methods for general and molecular bacteriology)、14および15章、American Society forMicrobiology、1994)およびエレクトロポレーションを含む種々の遺伝子導入方法を使用することが可能である。組換え体を作成する方法としては、分子生物学の分野で周知の方法の中から選択して使用できる。通常の形質転換系は、大腸菌、シュードモナスおよびアセトバクターにおいて使用できる。形質導入系は、大腸菌に使用できる。接合伝達系は、グラム陽性菌および大腸菌、P.プチダおよびG.オキシダンスを含むグラム陰性菌において広く使用できる。好ましい接合伝達方法は、国際公開公報89/06688号に開示されている。接合伝達は、液体培地中または固体培地表面において起こりうる。好ましい受容菌は、適当な組換え発現ベクターを用い活性を有するAADHを生産しうる大腸菌、P.プチダおよびG.オキシダンスから選択される。2KGA生産のための好ましい受容菌は、G.オキシダンスDSM番号4025である。接合伝達の受容菌には、通常選択マーカーが付与される。たとえばナリディキシン酸またはリファンピシンに対する耐性が通常選択される。
【0037】
本発明で提供されるAADHは、アルコールおよび/またはアルデヒドの酸化を触媒し、該当する基質からアルデヒド、ケトンまたはカルボン酸を生産できる。更に具体的には、本発明で提供されるAADHは、L-ソルボースを酸化してL-ソルボソンを経由し2KGAを生成する反応および/またはD-ソルビトールを酸化してL-ソルボースを生成する反応を触媒することができる。更に具体的には、本発明で提供されるAADHは、アミノ酸配列が配列番号5、6、7および8で各々示される酵素A、酵素 A'、酵素 A"および酵素 Bを含んでいる。
【0038】
塩基配列が配列番号1、2、3および4で各々示される遺伝子で、その遺伝子がアミノ酸配列が配列番号5、6、7および8で各々示されるポリペプチドをコードする酵素 A、酵素 A'、酵素 A"および酵素 B遺伝子は、G.オキシダンス株DSM番号4025から取得することができる。
【0039】
本発明で提供される酵素 A、酵素 A'、酵素 A"および酵素 Bを含むAADHは、別個に、適当な生物を培養し、その細胞を破砕し、その破砕された細胞の無細胞抽出物、好ましくは微生物の可溶性画分から単離精製することにより調製できる。
【0040】
本発明で提供される組換え生物は、好気的条件下適当な栄養分を補足した水性培地中で培養することができる。該培養はpH約4.0から9.0の範囲、好ましくはpH約6.0から8.0の範囲で行うことができる。培養期間は、使用するpH、温度、栄養培地に応じて変わるが、通常2から5日が好ましい結果をもたらす。培養を行うのに好ましい温度範囲は約13℃から45℃、好ましくは18℃から42℃である。
【0041】
培養培地として通常必要な栄養分は、同化可能な炭素源、消化可能な窒素源、無機物質、ビタミン類、微量元素および他の生育促進因子などである。同化可能な炭素源としては、グリセロール、D-グルコース、D-マンニトール、D-フルクトース、D-アラビトール、D-ソルビトールおよびL-ソルボースなどが使用できる。
【0042】
酵母エキス、肉エキス、ペプトン、カゼイン、コーンスティープリカー、尿素、アミノ酸、硝酸塩、アンモニウム塩などの種々の有機または無機物質を窒素源として使用できる。無機物質としては、硫酸マグネシウム、リン酸カリウム、塩化第一鉄、塩化第二鉄、炭酸カルシウムなどが使用できる。
【0043】
本発明に係る組換え酵素調製物においては、[1] 配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチド、並びに配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドの間のキメラ組換え酵素、並びに一つまたはそれ以上のアミノ酸残基の付加、挿入、欠失および/または置換を含む上記で同定されるポリペプチドの機能的誘導体からなる群より選択され、該酵素ポリペプチドがアルコールおよび/またはアルデヒド脱水素酵素活性を有する、一つまたはそれ以上の酵素ポリペプチドを含む、アルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物であることを特徴とする。
【0044】
また、本発明に係る組換え酵素調製物においては、[2] ポリペプチドが、酵素A/B1、酵素 A/B2、酵素 A/B3、酵素 B/A1、酵素 B/A2、酵素 B/A3、酵素 sA2、酵素 sA21、酵素 sA22、酵素 sBおよびそれらの機能的誘導体のキメラ酵素である、前記[1]に記載の組換え酵素調製物であることを特徴とする。
【0045】
また、本発明に係る組換え酵素調製物においては、[3] 酵素ポリペプチドが、ホモ二量体および/またはヘテロ二量体の形態で存在する、前記[1]または[2]に記載の組換え酵素調製物であることを特徴とする。
【0046】
また、本発明に係るDNA分子においては、[4] 上記[1]または[2]で定義されるポリペプチドをコードするDNA分子であることを特徴とする。
【0047】
また、本発明に係るDNA分子においては、[5] 直鎖状もしくは環状DNAまたは染色体上での挿入DNA断片のいずれかの形態で存在する、前記[4]に記載DNA分子であることを特徴とする。
【0048】
また、本発明に係る組換え発現ベクターにおいては、[6] 上記[4]または[5]のいずれかで定義される一つまたはそれ以上のDNA分子を含む、組換え発現ベクターであることを特徴とする。
【0049】
また、本発明に係る組換え発現ベクターにおいては、[7] DNA分子が、一つまたはそれ以上の遺伝子制御配列に機能的に結合しており、適当な宿主細胞において上記[1]〜[3]のいずれかに記載の酵素ポリペプチドを発現可能である、前記[6]に記載の組換え発現ベクターであることを特徴とする。
【0050】
また、本発明に係る組換え発現ベクターにおいては、[8] pSSA102R、pSSA'101R、pSSA"102、pSSB103R、pSSAp-B、pSSA/B101R、pSSA/B102R、pSSA/B103R、pSSB/A101R、pSSB/A102R、pSSB/A103R、pSSsA2、pSSsA21、pSSsA22およびpSSsBからなる群より選択される、前記[7]に記載の組換え発現ベクターであることを特徴とする。
【0051】
また、本発明に係る組換え生物においては、[9] 上記[6]〜[8]のいずれかに記載の組換え発現ベクターまたは上記[4]もしくは[5]のいずれかで定義される一つまたはそれ以上のDNA分子を有する、組換え生物であることを特徴とする。
【0052】
また、本発明に係る組換え生物においては、[10] 宿主細胞が、微生物、哺乳動物細胞および植物細胞からなる群より選択される、前記[9]に記載の組換え生物であることを特徴とする。
【0053】
また、本発明に係る組換え生物においては、[11] 宿主細胞が、大腸菌(Escherichia coli)、シュードモナスプチダ(Pseudomonasputida)、アセトバクターキシリナム(Acetobacter xylinum)、アセトバクターパスチュリアヌス(Acetobacterpasteurianus)、アセトバクター アセチ(Acetobacter aceti)、アセトバクター ハンゼニイ(Acetobacter hansenii)およびグルコノバクターオキシダンス(Gluconobacteroxydans)等の細菌からなる群より選択される微生物である、前記[9]または[10]に記載の組換え生物であることを特徴とする。
【0054】
また、本発明に係る組換え生物においては、[12] 宿主細胞が、グルコノバクター オキシダンス[DSM番号4025]である、前記[11]に記載の組換え生物であることを特徴とする。
【0055】
また、本発明に係る組換え酵素調製物の製造方法においては、[13] 上記[10]〜[12]のいずれかで定義される組換え生物を適当な培地で培養し、組換え酵素を回収することを含む、前記[1]、[2]または[3]で定義されるアルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物の製造方法であることを特徴とする。
【0056】
また、本発明に係るアルデヒド、ケトンまたはカルボン酸生成物の対応する基質からの製造方法においては、[14] 上記[10]〜[12]のいずれかで定義される組換え生物の生化学的反応により基質を生産物に変換することを含む、アルデヒド、ケトンまたはカルボン酸生成物を対応する基質から製造する方法であることを特徴とする。
【0057】
また、本発明に係るL-ソルボースおよび/またはD-ソルビトールからの2-ケト-L-ギュロン酸の製造方法においては、[15] 上記[10]〜[12]のいずれかで定義される組換え生物の生化学的反応によりL-ソルボースおよび/またはD-ソルビトールを2-ケト-L-ギュロン酸に変換することを含む、L-ソルボースおよび/またはD-ソルビトールから2-ケト-L-ギュロン酸を製造する方法であることを特徴とする。
【0058】
また、本発明に係るアルデヒド、ケトン、またはカルボン酸生成物の対応する基質からの製造方法においては、[16] 上記[1]〜[3]のいずれかで定義される組換え酵素調製物および基質を含有する反応混合液をインキュベートすることを含む、アルデヒド、ケトンまたはカルボン酸生成物を対応する基質から製造する方法であることを特徴とする。
【0059】
また、本発明に係る2-ケト-L-ギュロン酸の製造方法においては、[17] 上記[1]〜[3]のいずれかで定義される組換え酵素調製物とL-ソルボースおよび/またはD-ソルビトールとを含有する反応混合液をインキュベートすることを含む、2-ケト-L-ギュロン酸の製造方法であることを特徴とする。
【0060】
また、本発明に係る2-ケト-L-ギュロン酸からのL-アスコルビン酸の製造方法においては、[18] 上記[15]または[17]に記載の製造方法が達成され、該製造方法により得られる2-ケト-L-ギュロン酸が、当技術分野で既知の方法によりL-アスコルビン酸に変換されることを特徴とする、2-ケト-L-ギュロン酸からのL-アスコルビン酸の製造方法であることを特徴とする。
【発明の効果】
【0061】
アルコールおよび/またはアルデヒド脱水素酵素活性を有するアルコールおよび/またはアルデヒド脱水素酵素遺伝子をクローン化したことにより、組換えDNA技術を用いて、クローン化した遺伝子を含むDNA分子、該DNA分子を含む組換え発現ベクター、該DNA分子または該発現ベクターを含む組換え体を作成することが可能となり、また、これらを用いてクローン化した遺伝子を発現させることにより、種々のアルデヒド、ケトン、カルボン酸、特に2-ケト-L-ギュロン酸、の生成に有用なアルコールおよび/またはアルデヒド脱水素酵素の組み換え調製物を製造することが可能となった。
【発明を実施するための最良の形態】
【0062】
以下に特にP.プチダから得られた精製組換えAADH酵素の性質およびその生産方法を示す。
【0063】
(1)酵素活性本発明のAADHは以下の反応式に従い電子受容体存在下D-ソルビトール、L-ソルボースおよびL-ソルボソンを含むアルコールおよびアルデヒドの酸化を触媒する。
アルコール+電子受容体 → アルデヒド+還元型電子受容体アルコール+電子受容体 → ケトン+還元型電子受容体アルデヒド+電子受容体 → カルボン酸+還元型電子受容体糖アルコール+電子受容体 → アルドース+還元型電子受容体糖アルコール+電子受容体 → ケトース+還元型電子受容体アルデヒドケトース+電子受容体 → ケトカルボン酸+還元型電子受容体カルボン酸+電子受容体 → ケトカルボン酸+還元型電子受容体酵素は分子酸素を受容体として用いない。受容体として、2、6-ジクロロフェノールインドフェノール(DCIP)、フェナジンメソサルフェート(PMS)、ウェスターブルー、フェリサイアナイド、補酵素Qまたはチトクロームcが使用できる。
【0064】
酵素活性1単位は1分間あたり1μモルのDCIPの還元を触媒する酵素量と定義した。DCIPのpH 8.0における分子吸光係数は15 mM-1とした。標準反応混合液(1.0 ml)は0.1 mM DCIP、1 mM PMS、2〜125mM 基質、50 mMTris-リンゴ酸-NaOH緩衝液(pH 8.0)および10 μlの酵素液を含む。対照としては上記の組成から基質を除いたものを用いた。
【0065】
(2) AADHの性質a) 酵素反応の基質特異性と生成物酵素 A、酵素 A'、酵素 A"および酵素 Bは8種の基質、n-プロパノール、イソプロパノール、D-グルコース、D-ソルビトール、L-ソルボソン、D-マンニトール、L-ソルボースおよびD-フルクトース、を用いた上記(1)におけるそれらの基質特異性によって特徴づけられる。結果を表1に示した。
【0066】
【表1】

【0067】
酵素 Bはn-プロパノールおよびイソプロパノールに対して比較的低い反応性を示したが、D-グルコースおよびD-マンニトールに対して高い反応性を示した。酵素 A、A'およびA"は、D-グルコースおよびD-マンニトールに対して低い反応性を示したが、n-プロパノールおよびイソプロパノールに対して高い反応性を示した。これら三酵素は酵素 A'がL-ソルボースおよびD-フルクトースに対してきわめて低い反応性を示した以外には、類似した基質特異性パターンを示した。
【0068】
酵素 A、酵素 A'、酵素 A"または酵素 Bを用いた反応における基質からの生成物は、既知物質を用いた薄層クロマトグラフィー(TLC)および/または高速液体クロマトグラフィー(HPLC)により分析された。酵素 A、酵素 A'、および酵素 A"(以後Aグループとする)はD-ソルビトール、L-ソルボース、L-ソルボソン、D-マンニトールおよびD-フルクトースをそれぞれL-ギュロースを伴うD-グルコース、2KGAを伴うL-ソルボソン、2KGA、D-マンノース、および2-ケト-D-グルコン酸(2KD)に変換した。酵素 B(以後Bグループとする)はD-グルコース、D-ソルビトール、L-ソルボソン、D-マンニトール、L-イドース、グリセロール、D-グルコン酸、D-マンノン酸をそれぞれD-グルコン酸、L-ソルボース、2KGA、D-フルクトース、L-イドン酸、ジヒドロキシアセトン、5-ケト-D-グルコン酸および5-ケト-D-マンノン酸に変換した。L-ソルボソンに対する反応性における類似性から、D-グルコソンは上記すべてのAADHにより2KDに変換されうる。事実、Aグループ酵素はその直接生成物がD-グルコソンと考えられるD-フルクトースから2KDを生成した。これらすべての酵素はD-ソルビトールやD-マンニトールのような糖アルコールを含むアルコール、ならびにD-グルコースのようなアルドース、およびL-ソルボソンのようなケトースを含むアルデヒドの両者に活性を示した。
【0069】
b) 至適pH表2に示すように、すべての酵素はそれらの至適点をpH8.0〜8.5に有する。酵素 A"とBは、酵素 AとA'に比較して低pH側に比較的広い至適pH範囲を持つ。
【0070】
【表2】
【0071】
c) pH安定性酵素 A、酵素 A'、酵素 A"および酵素 Bを様々なpH値の緩衝液中3時間、25℃でインキュベートした後にその残存活性を測定し、pH8.0でインキュベートしなかった値に対する相対活性として表示した。
【0072】
表3に示すように酵素 A、A、A" およびBはpH 6から9の範囲で安定であった。
【0073】
【表3】
【0074】
d)温度安定性表4に各酵素の4、20、30、40、50および60℃での5分間処理後の残存活性を示す。
【0075】
【表4】
【0076】
e)金属イオンおよび阻害剤の影響種々の金属及び阻害剤により酵素を処理した後の残存活性を表5に示す。MgCl2とCaCl2はほぼ酵素に対して無影響であったが、他の金属イオン、特にCuCl2はその反応性に強い影響をおよぼした。EGTAおよびEDTAは顕著に酵素 A、A'およびA"を阻害した。しかし、酵素 BはAグループ酵素群よりもEGTAとEDTAによる阻害が少なかった。
【0077】
【表5】
【0078】
f)分子量とサブユニットP.プチダ接合伝達体から精製された酵素 A、A'、A"およびBは、ドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS-PAGE)での計測によると、分子量約64,000、62,500、62,500および60,000のそれぞれの単一種ユニットを含む。酵素 A、A'、A"およびB遺伝子/DNA配列類を同一宿主内で発現させた場合、酵素A、A'、A"およびBのユニット間のいかなる組み合わせでもヘテロ二量体を形成しうる。
【0079】
g) N-末端アミノ酸配列成熟酵素 AおよびBのN-末端配列を以下に示す。
酵素 A :Gln-Val-Thr-Pro-Val-Thr----酵素 A" :ブロック化されたN-末端残基酵素 B :Gln-Val-Thr-Pro-Ile-Thr-Asp-Glu-Leu-Leu-Ala----成熟酵素 A'のN-末端は標品の純度不足のため決定されていない。
【0080】
(3) AADHの生産細胞は遠心分離または濾過により発酵培養液より回収される。回収された細胞を緩衝溶液中に懸濁し、ホモジナイザー、超音波または溶菌酵素処理などによって破砕し細胞の破砕溶液を得る。
【0081】
AADHは破砕された細胞の無細胞抽出液から、好ましくは微生物の可溶性画分から硫安沈殿、透析、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィーおよびアフィニティークロマトグラフィーの様な一般的蛋白質精製法により、単離精製される。
【0082】
(4)酵素反応酵素反応はTris-HCl緩衝液、燐酸緩衝液等の緩衝液中で、例えばDCIP、PMS、ウェスターブルー、フェリサイアナイド、補酵素Q、チトクロームc等の電子受容体存在下、約10℃から約50℃、好ましくは20℃から40℃、の温度においてpH値約6.0から約9.0においてで実施される。反応混合液中の基質の濃度は他の反応条件により変化させうるが、一般的に約1〜200g/l、最も好ましくは1〜100 g/lである。
【0083】
本酵素反応には、AADHは適当な担体を用いる固定化状態においても使用できる。当技術分野において一般に知られているいずれの酵素固定化法も使用可能である。例えば、酵素を官能基を有する樹脂膜、顆粒などに直接結合させてもよく、または官能基を有する架橋化合物、例えばグルタルアルデヒドを介して樹脂に結合させてもよい。
【0084】
本発明で提供されるポリペプチドには、酵素 A、酵素 A'、酵素 A"および酵素Bを含むAADH遺伝子から調製される誘導体、および遺伝子コドンの縮退から生ずる遺伝子相同体またはAADH遺伝子に相当な相同性のある天然、合成または組換え体のいずれの遺伝子配列から調製される関連誘導体も含まれる。本誘導体は、配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドと同等の機能を有し、それが一つまたはそれ以上のアミノ酸残基の付加、欠失および/または置換を含み、その該酵素ポリペプチドがアルコールおよび/またはアルデヒド脱水素酵素活性を有する変異体でもよい。これらの変異遺伝子は、AADH遺伝子を紫外線、エックス線、ガンマ線を照射することにより、または亜硝酸、N-メチル-N'-ニトロ-N-ニトロソグアニジン(NTG)または他の適当な変異剤で処理することにより、または自然変異を起こした株を分離することにより、または当技術分野において周知の標準的インビトロ変異処理をすることにより作成することが可能である。
【0085】
AADHポリペプチドの誘導体には、また配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドの間のキメラ組換え酵素が含まれる。このキメラ体は、図6に示されるように制限酵素とT4-リガーゼを用い、遺伝子間で共通の制限酵素部位で本発明のDNA配列の2つまたはそれ以上のDNA配列を試験管内で組換えることにより、または図8に示されるように二遺伝子間で共通の塩基配列の位置での2つのAADH遺伝子間の生体内組換えにより調製することができる。
【0086】
AADHポリペプチドの誘導体には、またAADHポリペプチドのアミノ末端、カルボキシル末端および/または内部に、ポリペプチドが付加されたポリペプチドも含まれる。カルボキシル末端にG.オキシダンスDSM番号4025のチトクロムcポリペプチド(17〜18kDa)が融合した酵素 B、酵素 A/B25および酵素 A/B3は、D-ソルビトールからL-ソルボースへの変換において実施例4に述べられている酵素Bと同様のAADH活性を示し、L-ソルボースから2KGAへの変換において実施例14に述べられている酵素 A/B25および酵素 A/B3と同様のAADH活性を示した。このように本発明で提供されるAADHに、AADH活性を維持したまま、かなり長いポリペプチドを付加または挿入することが可能である。
【0087】
上述のAADHポリペプチドの誘導体は、所望の基質特異性、基質への高親和性、阻害物質への低親和性、温度および/またはpHに対する高安定性、および高触媒速度のような好ましい性質を示す可能性が高い。以下の実施例に述べられているように、該誘導体は目的の生産物の生産性を改良することが可能であると思われる。
【0088】
本発明の酵素ポリペプチドは、通常二量体の形態で生産される。このような二量体には、酵素 A、A'、A"、またはBもしくはキメラを含むそれらの誘導体のホモ二量体および上述の二つの異なった酵素ポリペプチドを含むヘテロ二量体が含まれている。本発明の組換え酵素調製物はまた、一つまたはそれ以上の該ホモ二量体および/またはヘテロ二量体を含んでいる。
【0089】
本発明で提供される組換え生物は、アルコールおよび/またはアルデヒド脱水素酵素活性を有するAADHの組換え酵素調製物の生産に非常に有用である。該組換え生物は、該組換え酵素調製物を利用することによる、および該組換え生物を利用することによるアルデヒド、カルボン酸、ケトン、特に2KGAの生産にもまた有用である。
【0090】
該組換え生物を用いる2KGAの生産は、上述の培地および培養条件を用いる発酵により実施され得る。2KGAの生産は、上述の組換え生物を大腸菌、P.プチダ、B.メガテリウムのような共存生物とともに用いても行い得る。
【実施例】
【0091】
[実施例1] AADH遺伝子のクローニング(1)G.オキシダンスDSM番号4025のゲノムライブラリーの構築染色体DNAは以下のように調製した。G.オキシダンスDSM番号4025は、20 mlのNS2培地[5.0% D-マンニトール、0.25% MgSO4・7H2O、1.75% コーンスティープリカー(corn steep liquor: CSL)、5.0% パン酵母 (オリエンタル酵母社製)、0.5%CaCO3、0.5% 尿素(別滅菌)、2.0% 寒天(滅菌前pH7.0)を含む寒天平板上で27℃で3日間培養した。この寒天平板から細胞を集め、10 mlの1 mM EDTAを含む10mM Tris-HCl(pH8.0)緩衝液で洗浄し、5 mlの20 mM EDTAを含む10mM Tris-HCl(pH8.0)緩衝液に懸濁した。この細胞懸濁液を終濃度400μg/mlのリゾチーム(シグマケミカルズ社製(Sigma Chemicals))で37℃、30分間処理し、次にプロナーゼ(400単位)で37℃、30分間処理し、1%SDS で37℃、1時間処理した。染色体DNAは、マニアティス(Maniatis)らにより述べられている方法「分子クローニング:研究室マニュアル(Molecular cloning:a laboratory manual) Cold Spring Harbor Laboratory, Cold Spring Harbor, N. Y.,(1982)」に従いフェノールとRNase A(ベーリンガーマンハイム社製(Boeheringer Mannheim))で処理した。染色体DNA(200 μg)は、168単位のSalI(ベーリンガーマンハイム社製(Boeheringer Mannheim))で37℃、5〜90分間処理した。この処理で得られた15〜35 kbの部分消化断片を分取アガロースゲル電気泳動(アガロース0.7%)で単離した。単離は目的断片を含むゲル断片を切り出し、その中のDNAをゲルから40 mM Tris-酢酸と2 mM EDTAを含むTAE緩衝液中へ電気的に溶出した。このようにして40 μgの目的DNAが得られた。並行して、8 μgのコスミドベクターpVK102(ATCC 37158)をSalIで完全消化し、牛大腸アルカリフォスファターゼ(ベーリンガーマンハイム社製(Boeheringer Mannheim))で供給会社の指示に従い処理した。0.4 μgのpVK102と15〜35kbのSalI断片(0.2〜2 μg)をライゲーションキット(宝酒造社製)を用い26℃で10分間処理した。結合したDNAを今度は、供給会社(アマーシャム社(Amersham))の指示した方法に従って、インビトロパッケージング(結合したDNAをファージコート蛋白質の部品と混合する)に使用した。このようにして生じたファージ顆粒を大腸菌 ED8767[ミューレイ(Murray), N. E., W. J. ブランマー(Brammer)およびK. ミューレイ(Murray), Mol. Gen. Genet.、150巻、53〜61頁、(1977)]に感染させた。約3,000個のカナマイシン耐性(Kmr)テトラサイクリン感受性(Tcs)のコロニーが得られ、テストした24コロニー全てが挿入DNAを有していた。その平均鎖長は26.5 kbであった。もう一つの55,000クローンを含むG.オキシダンスDSM番号4025のコスミドライブラリーは、上述の方法とほぼ同様にしてEcoRIで部分消化したG.オキシダンスDSM番号4025の染色体DNAをコスミドpVK100のEcoRI部位に挿入して構築した。テストした24コロニー全てが平均鎖長27 kbの挿入断片を有していた。
【0092】
これら2種類の大腸菌 ED8767中に作成されたコスミドライブラリーを、大腸菌 ED8767ライブラリーから抽出された組換えプラスミドDNA混合物を用いて、大腸菌 S17-1[Tra+, Bio/Technology、1巻、784〜791頁、(1983)]へ移した。約4,000株の大腸菌 S17-1のKmr形質転換株を爪楊枝で拾い上げ、各々別個に50μg/mlのカナマイシンを添加した10 g/lのバクトトリプトン(ディフコ社製(Difco))、5g/lの酵母エキス(ディフコ社(Difco))、5 g/lのNaClを含むLB培地100 μlを含むマイクロタイタープレート中で37℃で培養した後、15%グリセロール存在下、-80℃で大腸菌 S17-1中のコスミドライブラリーとして保存した。
【0093】
この様に、G.オキシダンス DSM番号4025のSalI-およびEcoRI-コスミドライブラリーが大腸菌S17-1中に作成された。このライブラリーから各々1,400クローンずつが、別々に大腸菌 S17-1からP.プチダ ATCC21812へ接合伝達により次のように移された。-80℃でマイクロタイタープレート中に保存していた1,400培養物を融解し、各々の穴に100 μlの新鮮なLB培地を含むマイクロタイタープレートにプレート移し換えカートリッジ(ナンク社製(Nunc))を用いて移し、37℃で一晩培養した。ナリディキシン酸耐性(Nalr)を付与されたP.プチダ ATCC21812は、100mlの2.5%のマンニトール、0.5%の酵母エキス(ディフコラボラトリー(DifcoLaboratories))、0.3%バクトトリプトン(ディフコ(Difco))を含むMB培地中、30℃で一晩培養した。このP.プチダの培養液50 μlをコスミドライブラリーの培養液を含む1,400の穴各々に添加した。この1,400の細胞混合液を、プレート移し換えカートリッジを用いて5%フルクトース、1% 酵母エキス (ディフコ(Difco))、1%ポリペプトン(大五栄養社製)、1.8% 寒天を含むFB寒天培地表面に置いたニトロセルロースフィルター上にスポットし、27℃で一晩培養した。ナリディキシン酸は、遺伝子供与体である大腸菌から接合伝達体を選択するために用いた。フィルター上に生育した細胞は、別々に50 μg/mlのナリディキシン酸および50 μg/mlのカナマイシンを含有するMB寒天培地(以下MNK寒天平板と称する)上に移植し、接合伝達体を選択するために27℃で4日間培養した。この結果得られたコロニーを上述のようにMNK寒天平板に移植して精製した。このように1,400株のP.プチダ接合伝達体[G.オキシダンス DSM番号4025のP.プチダ中での遺伝子ライブラリー]を調製した。
【0094】
(2)G.オキシダンス DSM番号4025のAADH遺伝子クローンの免疫学的スクリーニングまず、MNK寒天平板上に維持されている350株の接合伝達体(SalIライブラリー175株、EcoRIライブラリー175株)を別々に5 mlのMNK培地を含む試験管中で培養した。その各々1.5 mlの培養液から細胞を集菌し、以下のようにウェスタンブロット解析のための処理に供した。細胞は、50 μlのLaemmli緩衝液[62.5 mM Tris-HCl(pH6.8)、10% グリセロール、5% メルカプトエタノール、2% SDS]に懸濁した。この細胞懸濁液を3分間煮沸し、その10 μlの細胞溶菌液をSDS-PAGEに供した。その結果得られた蛋白質バンドを次に40V、200 mAで16時間操作される電気ブロット装置(マリソル社製)を用いて、20% メタノールを含む2.5 mM Tris-19.2 mM グリシン緩衝液(pH8.6)中で16時間ニトロセルロースフィルターに電気的に移した。そのフィルターは、次に20 mM Tris(pH7.5)、500 mM NaClを含むTBS緩衝液中の3%ゼラチンの中で1時間処理した。このフィルターを20mM Tris(pH7.5)、500 mM NaCl、0.05% Tween 20を含むTTBS緩衝液で軽く洗浄した後、1% ゼラチンを含むTTBS緩衝液中で1/500に希釈した抗AADH抗体を含む一次抗体で1時間処理した。この抗AADH抗体は、G.オキシダンス DSM番号4025から精製したAADH蛋白質をインコンプリートアジュバントと混合し、その混合物を白兎に2週間間隔で2度インジェクトし、2度目のインジェクトの1週間後全採血し、その血清画分を抗AADH抗体として調製した。次に、このフィルターをTTBS緩衝液中で5分間ずつ2度洗浄し、1% ゼラチンを含むTTBS緩衝液中で1/3,000に希釈した二次抗体を含む二次抗体(山羊-抗兎IgG-西洋わさびパーオキシダーゼコンジュゲート)溶液で1時間処理した。そのフィルターをTTBS緩衝液中で2度洗浄し、TBS緩衝液中で1度洗浄した後、Konica Immunostaining HRP Kit IS-50B(コニカ社製)を用い、青色バンドが観察されるまで供給会社の推奨する方法に従いフィルターを発色試薬に浸漬した。実際のスクリーニングには、ウェスタンブロットの一次スクリーニングのために5つの細胞溶菌液を混合したものを1レーンにのせた。70個の混合液の中で14個の試料が陽性バンドを示した。その9つの試料が分子量約64,000の免疫反応性を有する蛋白質を有していたが、この中の2つの試料のシグナルは弱かった。別の1試料は分子量約60,000の残りの4試料は分子量約55,000の免疫反応性を有する蛋白質を有していた。
【0095】
分子量約64,000に強いシグナルを示した7個の混合試料について、その混合試料中の陽性クローンを同定するためにその個々の試料をウェスタンブロットの二次スクリーニングに供した。1混合試料あたり、1陽性クローンが同定された。得られた7クローンの有するプラスミドを各々p6E10、p16C8、p16F4、p17E8、p1E2、p24D4およびp26C3と命名した。制限酵素解析により、p6E10、p16C8、p16F4およびp17E8の4プラスミドは同じDNA領域を含み、他の3プラスミドは前者の4プラスミドとは異なる領域を含むことが明らかになった。
【0096】
(3)コロニーブロットおよびサザンブロットハイブリダイゼーションによるコスミドライブラリーからのAADH遺伝子のスクリーニング上述の免疫スクリーニングで得られた遺伝子以外のAADH遺伝子を検索するために、G.オキシダンス DSM番号4025の大腸菌 ED8767中で構築された全コスミドライブラリー(SalI-およびEcoRI-ライブラリー)を、p24D4の0.9 kb SalI断片とのコロニーおよびサザンブロットハイブリダイゼーションによってスクリーニングした。この0.9 kb SalI 断片とは、G.オキシダンス DSM番号4025から精製された天然AADH酵素の一つの内部アミノ酸配列、MetMetValThrAsnValAspValGlnMetSerThrGlu、に従って合成されたオリゴヌクレオチドプローブATGATGGT(GATC)AC(GATC)AA(TC)GT とハイブリダイゼーションしたDNA断片である。その内部アミノ酸配列は、天然AADH酵素消化後、自動ガス層シークエンサー470A(アプライドバイオシステム社製(Applied Biosystems))を用いて決定されたものである。上記コスミドライブラリーの細胞を適当に希釈し、LK寒天平板に塗抹した。生じたコロニーは、ナイロンフィルターに転写した後、32P-標識0.9 kb SalI断片とのハイブリダイゼーションにより解析した。約1%のコロニーが陽性シグナルを示した。SalIライブラリーから41株、EcoRIライブラリーから20株を選択し、それらを制限酵素解析後、サザンブロット解析に供した。このスクリーニングで以下のように6種類の異なるAADH遺伝子関連DNA領域を単離した。4種類は既に単離していた領域でp24D4、p1E2、p26C3、p17E8が有しており、2種類は新規でpSS31およびpSS53と命名された2種類の別個のプラスミドが有していた。残りのプラスミドpSS33は、p24D4およびpSS31が有している2領域の両方を有していた。
【0097】
(4)AADHクローンの免疫学的および酵素学的解析p24D4、p1E2、p26C3、pSS31およびp17E8を有するP.プチダの細胞溶菌液のウェスタンブロット解析の結果、これらの5クローンは各々分子量約64,000、62,500、62,500、60,000および62,000の蛋白質をコードすることが示された。プラスミドpSS33は、分子量約64,000および60,000の2種類の免疫反応性を有する蛋白質をコードしていた。しかしpSS53は、いかなる免疫反応性を有する蛋白質もコードしていなかった。
【0098】
各々のクローンの酵素活性(無細胞抽出液、可溶画分および膜画分)を光学分析により測定した。各々のクローンの細胞を5 mlのMB培地を含む試験管に植菌し、30℃で24時間培養した。その培養液を200 mlの新鮮なMB培地を含む500 ml容のフラスコに移し、回転フラスコ培養機上で30℃で24時間培養した。得られた細胞を6,000 x g 10分間遠心分離により集菌した後、40 mlの冷緩衝液(50 mM Tris-HCl、pH7.5、5 mM MgCl2、0.5 mM フェニルメチルスルフォニルフルオリド)で洗浄し、同緩衝液に懸濁して5 mlあたり1 g 湿重量細胞濃度の細胞懸濁液を調製した。この細胞懸濁液は、フレンチプレス細胞破砕機で二回処理(1,500 kg/cm2)し、得られたホモジネートを6,000 x g 10分間遠心分離し、細胞破片を除去した。ここで得られた無細胞抽出液(CFE)を100,000 x g 60分間遠心分離した。この上清とペレットを細胞質画分および膜画分として各々集め、以下のPMS-DCIP分析に供した。酵素反応混合液(1.0 ml)には、100 μM DCIP、1 mM PMS、50 mM Tris-リンゴ酸-NaOH緩衝液(pH8.0)、基質および酵素(10 μl)が含まれる。基質依存の600 nmでのDCIPの吸光度の減少を、コントロン社製(Kontron)の吸光度計UVIKON810を用いて25℃で測定した。表6はクローンの無細胞抽出液および可溶画分の酵素活性のレベルを示している。基質特異性によれば、各々のプラスミドにコードされている酵素は大きく三つのグループ、A、B、Cグループに分類された。AグループはL-ソルボース、D-ソルビトール、1-プロパノールの酸化を触媒し、BグループはD-グルコースおよびD-ソルビトールの酸化を触媒するが、Cグループは使用した基質に対し明瞭に検出できる活性を示さなかった。Aグループには三種類のタイプA、A'、A"(各々のプラスミドが有するDNAの物理地図によりお互い区別される)が存在していた。B、Cグループは各々染色体DNAの一領域から由来する一タイプの蛋白質のみから構成されていた。
【0099】
【表6】
【0100】
[実施例2] 塩基配列決定酵素 A、酵素 A'、酵素 A"および酵素 B遺伝子の塩基配列は、各々プラスミドp24D4、p1E2、p26C3およびpSS31を用いてM13mp18およびM13mp19(ベーリンガーマンハイム社製(Boehringer Mannheim))を用いるジデオキシチェーン終結法により行った。各々の遺伝子について一つのオープンリーディングフレーム(open reading frame、ORF)が見つかった。これら4遺伝子の塩基配列を、配列表中に配列番号:1、配列番号:2、配列番号:3、配列番号:4として示す。またそれら塩基配列から推定されるアミノ酸配列を配列番号:5、配列番号:6、配列番号:7、配列番号:8に示す。酵素 A、酵素 A'、酵素 A"および酵素 B遺伝子のORFは、各々1737、1737、1734および1737 bp長であり、23アミノ酸残基のシグナル配列を含む579、579、578および579アミノ酸残基をコードしている。酵素 A、酵素 A'、酵素A"および酵素 B間のホモロジーを表7に示す。
【0101】
【表7】
【0102】
図5は成熟型酵素 A、酵素 B酵素のアミノ酸配列を相互に比較できるように整列(アライメント)表示している。
【0103】
酵素 A、酵素 A'、酵素 A"および酵素 Bのホモロジー検索の結果、A.アセチ(T.イノウエら、J. Bacteriol.、171巻、3115〜3122頁)またはA.ポリオキソゲネス(Acetobacter polyoxogenes、T.タマキら、B. B. A.、1088巻、292〜300頁)のアルコール脱水素酵素およびパラコッカスデニトリフィカンス(Paracoccusdenitrificans、N.ハームス(Harms)ら、J. Bacteriol.、169巻、3966〜3975頁)、メチロバクテリウム オーガノフィラム(Methylobacterium organophilum、S.M.マクリン(Machlin)ら、J. Bacteriol.、170巻、4739〜4747頁)またはメチロバクテリウム エクストーケンス(Methylobacteriumextorquens、D.J.アンダーソン(Anderson)ら、Gene、90巻、171〜176頁)のメタノール脱水素酵素を含む幾つかのキノ蛋白質(quino-proteins)とは、むしろ低いホモロジー(いずれのポリペプチド間においても26〜31%の範囲のホモロジー)を示した。
【0104】
[実施例3] AADH遺伝子のサブクローニング酵素 A遺伝子は、最初pVK100のEcoRI切断部位に約25 kbの挿入断片を有するコスミドクローンp24D4としてクローン化された。次にこの酵素 A遺伝子を、酵素A遺伝子カセットとして使用するためにサブクローン化した。酵素 A遺伝子のORFとその上流下流に約500bpの非コード領域を有する2.7 kbのEcoRV断片を、p24D4からM13mp18中に単離された3.4 kb NruI断片から切り出しpUC18のHindIII切断部位にHindIIIリンカー (CAAGCTTG)を用いて結合した。その結果得られたプラスミドをpSSA202と命名した。次に、酵素 A遺伝子カセット(2.7kb HindIII画分)をpVK102のHindIII切断部位に挿入してpSSA102Rを得た。このプラスミドpSSA102Rを実施例1-(1)に述べる接合伝達方法によりナリディキシン酸耐性のP.プチダ[ATCC21812]に導入した。このpSSA102Rを有するP.プチダの接合伝達体は、50 μg/ml ナリディキシン酸および10 μg/ml テトラサイクリンを含有するMB寒天培地(MNT寒天培地)上で選択され、ミニ休止菌体反応に供された。20 g/lのL-ソルボース、3 g/lのNaCl、10 g/lのCaCO3および爪楊枝でMNT寒天培地から集菌した細胞を含む反応混合液(100 μl)を室温で24時間、静かに振とうさせた。この反応混合液をTLCで分析して、その生産物が2KGAであると同定した。一方宿主であるナリディキシン酸耐性のP.プチダ[ATCC21812]を用いた同様の休止菌体反応では、2KGAは観察されなかった。
【0105】
酵素 B遺伝子は、最初pVK102のSalI切断部位に約30 kbの挿入断片を有するコスミドクローンpSS31としてクローン化された。この酵素 B遺伝子を6.5 kbのBglII 断片としてpVK101(ATCC37157)のBglII切断部位にサブクローン化してpSSB102を得た。次に酵素 B遺伝子をカセットとして使用するためにさらにサブクローン化した。まず6.5 kbのBglII断片をpUC18のBamHI切断部位にクローン化し、pSSB202を得た。次にpSSB202から2.3 kb XhoII断片を切り出した。この2.3 kb XhoII断片は、酵素 BのORFの他に、120bpの5'-非コード領域および500 bpの3'-非コード領域を含む。この2.3 kb XhoII断片をクレノウフラグメント(Klenowfragment)での処理により接着末端を平滑化し、HindIIIリンカーを用いてpUC18のHindIII切断部位にクローン化してpSSB203を得た。この酵素 B遺伝子カセット(2.3 kbHindIII断片)をpVK102のHindIII切断部位に挿入してpSSB103Rを作成した。このプラスミドpSSB103Rを接合伝達方法によりナリディキシン酸耐性のP.プチダ[ATCC21812]に導入した。このpSSB103Rを有するP.プチダの接合伝達体は、MNT寒天培地上で選択され、ミニ休止菌体反応に供された。このpSSB103Rを有するP.プチダの接合伝達体は休止菌体反応において酵素 B活性(D-ソルビトールからL-ソルボースを生成)を示した。酵素 B遺伝子カセットの塩基配列を決定したところ、上記XhoII断片は、XhoII-XhoII断片ではなくてXhoII-XhoI断片であることが判明した。おそらくXhoIがXhoII酵素標品に混入していたものと思われた。
【0106】
酵素 A'および酵素 A"遺伝子は、最初pVK102のSalI切断部位に約30 kbの挿入断片を持つp1E2およびp26C3のコスミドクローンとして得られ、更に基本的に上述のようにサブクローン化をおこなった。3.5 kb XhoII断片中の酵素 A'遺伝子をpVK102のBglII切断部位にサブクローン化しpSSA'101Rを得た。2.7 kb EcoRV断片中の酵素 A"遺伝子は、まずM13mp19にサブクローン化した後pVK102のHindIIIおよびBglII切断部位の間に再サブクローン化しpSSA"102を得た。
【0107】
[実施例4] P.プチダ接合伝達体からのAADHの単離と性質(1) 微生物の培養酵素 A、A'、A"およびBの遺伝子を含むpVK102コスミドベクター(それぞれpSSA102R、p1E2、p26C3およびpSSB103R)を持つP.プチダ[ATCC21812]を抗生物質存在下MB培地で培養した。抗生物質は以下に示すように添加した。pSSA102R(酵素 A)およびpSSB103R(酵素 B)に対しては5 μg/mlテトラサイクリンを、p1E2(酵素 A')およびp26C3(酵素 A")に対しては25 μg/ml カナマイシンを添加した。各々の抗生物質を含んだMB寒天平板培地より、細胞を5 mlの各々の抗生物質を含んだMB培地を含んだ10本の試験管に植菌し30℃で振とう培養した。2日培養後、細胞を100 mlの同培地を含む500 ml エーレンマイヤーフラスコ10本に移植し30℃で振とう培養した。1日培養後、以上の種培養を合わせて18Lの培地を含んだ30 L ジャーファーメンター(丸菱製)に移植し攪拌回転数300rpm、通気量1.0 vvm、30℃で18時間培養した。細胞を6,000 x g、10分の遠心操作で回収し、5 mM CaCl2、1 mM MgCl2、0.2 M NaCl、2.5% スクロース及び0.5 mM PMSFを含む1.5 Lの25 mM Tris-HCl緩衝液(pH 7.5)で1度洗浄し使用時まで-20℃で保存した。結果として湿重量で約150 gの菌体を得た。
【0108】
(2)クローン化された酵素 A、A'、A"およびBの精製酵素 A、A'、A"およびBの精製はほぼ同様のスケールで同一の方法で行った。すべての操作は特に記載のない場合4〜10℃で行った。酵素 A、A'、A"およびBの酵素活性の測定は精製段階を通してそれぞれ、L-ソルボース、n-プロパノール、n-プロパノールおよびD-グルコースを基質として、実施例1に述べられた分光学的測定法により行った。細胞(8〜10g全蛋白質を含む湿重量で約100gの菌体)を融解し、約200mlの25 mMTris-HCl緩衝液(pH8.0)中に懸濁し、フレンチプレス(1500 kg/cm2)を2回通過させることにより破砕した。DNAによる溶液の粘性を軽減するため、DNAase及びMgCl2をおのおの0.01 mg/ml及び1 mMの終濃度で溶液に添加した。細胞残骸は6,000 x g、10分の遠心操作で除去した。得られた上清を25 mM Tris-HCl緩衝液(pH 8.0)で240 ml容に調整し、不溶性膜画分を除去するため100,000 x g、90分間遠心処理した。可溶性上清をTris緩衝液で240 ml容に調整し、ピロロキノリンキノン(PQQ)及びCaCl2をそれぞれ終濃度12.5 μM及び5 mMで添加した後、溶液を室温下、15分間激しく攪拌した。上記のように調整された可溶性画分を硫安により分画した。硫安飽和35〜60%画分を沈殿させ100 mlの5 mM CaCl2及び5% スクロースを含む25 mM Tris-HCl緩衝液(pH 8.0)で再懸濁した後、PQQを再び12.5 μM終濃度添加した。酵素溶液を1000 mlの同緩衝液(PQQ非添加)で終夜透析した。固形ポリエチレングリコール#6000を透析終了液に緩やかな攪拌下徐々に添加した。30分間攪拌後、沈殿物を10,000 x g、20分間の遠心操作で除去し、上清を上記の同緩衝液で200ml容に調整した。
【0109】
上記のように調製した酵素溶液を以下の三つのクロマトグラフィー操作で精製した。
【0110】
第1段階:DEAE-トヨパール 650M粗酵素液を5 mM CaCl2および5% スクロースを含む25 mM Tris-HCl緩衝液(pH 8.0)で平衡化したDEAE-トヨパール 650Mカラム(2.5 x 40 cm)に供した。カラムを400 mlの同緩衝液で洗浄後、酵素の溶出を2000 ml同緩衝液中の0〜0.5M NaCl直線濃度勾配を用い流速150 ml/時で行った。酵素活性画分を回収しNaClを含まない同緩衝液で2倍容に希釈した。
【0111】
第2段階:Q-セファロース(ファーストフロー)
酵素溶液をNaClを含まない同緩衝液で平衡化したQ-セファロース(ファーストフロー)カラム(1.5 x 20 cm)に供した。カラムを200 mlの0.2 M NaClを含む同緩衝液で洗浄後、酵素の溶出を600 ml同緩衝液中の0.2〜0.6 M NaCl直線濃度勾配を用い流速50 ml/時で行った。酵素活性画分を回収し窒素ガス下、限界濾過膜(アミコン社製、PM-30)を用い2.5 ml容に濃縮した。
【0112】
第3段階:セファクリルS-300HR(ゲル濾過)
濃縮された酵素を5 mM CaCl2、5% スクロースおよび0.2 M NaClを含む25 mM HEPES緩衝液(pH 7.5)で平衡化したセファクリルS-300HR(2.5 x 100 cm)でゲル濾過分画した。カラムは同緩衝液で流速20 ml/時で展開した。酵素活性画分を回収し上記と同様の限界濾過膜を用い1 ml以下に濃縮した後、-80℃で保存した。HEPES緩衝液中濃縮された酵素は-80℃下少なくとも2ヵ月は安定であった。
【0113】
結果として、26.0 mgの酵素 A、0.35mgの酵素 A'、0.41 mgの酵素 A"および5.0 mgの酵素 Bを得た。
【0114】
(3)酵素 A、A'、A"およびBの性質a) 分子量およびサブユニット酵素 A、A'、A"およびBは同一条件下のセファクリルS-300HRによるゲル濾過において同一位置に溶出された。酵素の分子量は分子量標準蛋白質(SDS-PAGE スタンダード、低分子用、バイオラド社製(Bio-Rad))との比較により約135,000と推定された。酵素 A、A'、A"およびBはSDS-PAGE分析においてそれぞれ64,000、62,500、62,500および60,000の分子量を有する均一な単一バンドを示した。酵素A、A'、A"およびBのバンド全ては抗-AADHウサギ血清を用いたウェスタンブロッティング分析により検出された。従って、全酵素は2つの同一サブユニットを含むホモ二量体であると結論づけられた。
【0115】
b) N-末端アミノ酸配列とアミノ酸組成成熟酵素 A、A"およびBのN-末端アミノ酸配列をエドマン法[Acta Chem.Scand.、4巻、283〜293頁 (1950年)]による自動気相シーケンサー(470A,アプライドバイオシステム社製(AppliedBiosystems))を用いて分析した。酵素 A'のN-末端は標品の純度不足のため決定されていない。以下に結果を示す。
酵素 A :Gln-Val-Thr-Pro-Val-Thr----酵素 A" :ブロック化されたN-末端残基酵素 B :Gln-Val-Thr-Pro-Ile-Thr-Asp-Glu-Leu-Leu-Ala----決定された酵素 A及びBの配列は配列番号:5と配列番号:8に記載された塩基配列より推定される配列(24番目の残基より始まる)と同一であった。この結果は酵素の初めの23残基がシグナル配列であることを示す。酵素 A及びBとの類似性から酵素 A'およびA"の初めの23残基もシグナル配列であると推定される。
【0116】
酵素 Aのアミノ酸組成を決定した。蛋白質は6 N 塩酸で110℃、24時間または4M メタンスルホン酸(過ギ酸酸化後)で115℃、24時間処理により加水分解した。アミノ酸分析はコントロン社製(Kontron)のアミノ酸分析機(ニンヒドリン型)で行った。分析結果を酵素 AのDNA配列より推定されるアミノ酸組成と比較した。結果は精製された酵素 Aが明確に酵素 A遺伝子の産物であることを示した。
【0117】
c) 基質特異性酵素 A、A'、A"およびBを上述の8つの基質、n-プロパノール、イソプロパノール、D-グルコース、D-ソルビトール、L-ソルボソン、D-マンニトール、L-ソルボース及びD-フルクトースを用いたPMS-DCIP分析におけるそれらの基質特異性で性格づけした。結果を表1に示す。
【0118】
d) 物理化学的性質酵素 A(L-ソルボース脱水素酵素活性により)、酵素 A'(n-プロパノール脱水素酵素活性により)、酵素A"(L-ソルボース脱水素酵素活性により)および酵素 B(D-ソルビトール脱水素酵素活性により)の物理化学試験、至適pH、pH安定性および温度安定性をPMS-DCIP分析により行った。
【0119】
表2は酵素の至適pHの結果を要約したものである。酵素活性は様々なpHの緩衝液を用いてPMS-DCIP分光学法分析により測定した。緩衝液は50 mM Tris-リンゴ酸-NaOH緩衝液をpH 6.0、6.5、7.0、7.5、8.0および8.5用に、50 mM Glycine-NaOH緩衝液をpH 9.0および9.5用に用いた。各pH 6.0、6.5、7.0、7.5、8.0、8.5、9.0および9.5におけるDCIPの分子吸光係数はそれぞれ10.8、13.2、14.5、14.9、15.0、15.1、15.1および15.1とした。すべての酵素はその至適点をpH 8.0〜8.5に示した。酵素 A"とBは酵素 AとA'に比較して低pH側に比較的広い至適pH範囲を有していた。
【0120】
表3は酵素のpH安定性の結果を示す。酵素(約0.01 mg/ml)を5% スクロース、0.2 M NaClおよび5 mM CaCl2を含む50 mM 各緩衝液中で3時間インキュベートしPMS-DCIP分光学法分析により測定した。緩衝液はpH 4と5の酢酸ソーダ、pH 6、7および8のTris-リンゴ酸-NaOHおよびpH 9と10のGlycine-NaOH各緩衝液を用いた。表中の値はpH 8.0でインキュベートしなかった値に対する相対活性として表示した。各酵素に対して基質として酵素 A及びA"には125 mM L-ソルボース、酵素 A'には50 mM n-プロパノール、酵素 Bには125 mM D-ソルビトールを用いた。酵素 A、A'、A"およびBのpH-安定性はほぼ同様で、pH 6から9の範囲で安定であった。
【0121】
表4は酵素の温度安定性試験の結果を示す。5% スクロース、0.2 M NaCl及び5mM CaCl2を含む25 mM HEPES緩衝液(pH7.5)中で酵素(約0.05 mg/ml)を表に示した温度(4〜60℃)で5分間インキュベート後、氷冷しPMS-DCIP分光学法分析により測定した。残存活性を4℃でインキュベートした値に対する相対活性で表示した。各酵素に対する基質として酵素 AおよびA"には125 mM L-ソルボース、酵素 A'には50 mM n-プロパノール、酵素 Bには125 mM D-ソルビトールを用いた。40℃、5分間処理の酵素の場合、残存活性は酵素 Aで20%、酵素 A'、A"およびBで70〜85%であった。
【0122】
e) 阻害剤5% スクロースを含む25 mM HEPES緩衝液(pH7.5)中で酵素(約0.05 mg/ml)を金属または阻害剤とともに30分間、25℃でインキュベートした。残存活性を実施例1に記載したPMS-DCIP分光学法分析で測定した。残存活性は対照に対する相対活性で表わしている。酵素に対する金属イオンの影響を表5に記載した。MgCl2とCaCl2はほぼ酵素に対して無影響であったが、他の金属イオン、特にCuCl2、は強い影響を及ぼした。酵素に対する阻害剤の影響も表5に記載した。EGTAとEDTAは酵素 A、A'及びBを強く阻害した。しかし、酵素 Bに対するEDTA及びEGTAによる阻害はAグループ酵素に対する阻害よりも軽度であった。
【0123】
[実施例5] 大腸菌中での酵素 Bの効率的生産酵素 Bのシグナルペプチド領域を大腸菌のマルトース結合蛋白質(malE)のシグナルペプチドと以下のように置換した。2種のオリゴヌクレオチド(配列番号:9および配列番号:10)をアプライドバイオシステム社製(Applied Biosystems)の381A DNA合成機を用いて合成し、アニール化しアミノ酸配列(配列番号:11)、MetLysIleLysThrGlyAlaArgIleLeuAlaLeuSerAlaLeuThrThrMetMetPheSerAlaSerAlaLeuAla(Gln)をコードする2本鎖DNA断片を生成させ、この2本鎖DNA断片をT4ポリヌクレオチドキナーゼで処理した [J. Biol.Chem.、259巻、10606〜10613頁 (1984)]。pSSB203(実施例3 参照)を制限酵素SphIで切断後、T4DNAポリメラーゼで処理しBstPIで切断した。得られた元来のシグナル配列および成熟酵素Bの一番めのアミノ酸残基(Gln)をコードする領域のない酵素 B遺伝子を有する1.72 kb DNA断片を、アガロースゲル電気泳動を行った後アガロースゲルから単離した。大腸菌の発現ベクター、pTrc99A(ファーマシア社製(Pharmacia))を制限酵素NcoI(ATG開始コドンの位置)およびSmaIで切断し、上記2DNA断片と結合した。この結果得られたプラスミドをpTrcMal-EnzBと命名し、大腸菌 JM109を形質転換するのに用いた。得られた形質転換体を、100μg/mlのアンピシリンを添加したLB600 mlを含む2 L容のフラスコ2本中で28℃で生育させ、細胞濃度が約1.5 OD600に達した時に、IPTGを0.1mMになるよう添加し、さらに3〜4時間培養を続けた。得られた細胞は、25℃、10分間の遠心分離(4,000 x g)により集菌し、25℃で500 mlの20%シュークロースを含む30 mMTris-HCl(pH8.0)緩衝液に懸濁した。この細胞懸濁液に終濃度1 mMのEDTAを添加し、25℃でゆっくり振とうした後、細胞を4℃で15分間遠心分離(8,000 x g)にかけた。得た細胞を500 mlの氷冷した5 mM MgSO4に再懸濁し、4℃で5分間ゆっくり振とうした。この細胞懸濁液を4℃で10分間遠心分離(8,000 x g)にかけ、その上清を冷浸透圧ショック抽出液として得た。この抽出液には、SDS-PAGE分析で50〜60%以上の純度を有する酵素 B蛋白質(分子量60,000)が含まれていた。この上清にまず終濃度20 mMになるようにTris-HCl(pH8.0)を添加し、終濃度10 mM EDTAを添加後25℃10分間インキュベートし、次に終濃度20 mMのCaCl2を添加後10分間インキュベートし、最後に終濃度25 μMのPQQを添加後10分間インキュベートした。この上清に酵素の安定化のため終濃度20 mMのα-メチル-D-グルコシド(競合阻害剤)を加えた。酵素 Bはつぎの2種類のクロマトグラフィーにより完全精製された。まず上記上清を1 mM CaCl2および20 mM α-メチル-D-グルコシドを含む20 mM Tris-HCl(pH8.0)緩衝液で平衡化したQ-セファロースカラム(1.6 x 12 cm)上に供し、酵素 Bを600mlの0〜0.4 M NaCl直線勾配を持つ同緩衝液で溶出した。約0.25 MNaClの濃度で溶出された赤色蛋白質ピークを集め、Centricon-30(アミコン社製(Amicon))により約0.5 mlにまで濃縮した。最終的に、酵素 Bを0.2M NaCl、1 mMCaCl2、および20 mM α-メチル-D-グルコシドを含む20 mM HEPES(pH7.8)緩衝液を用い、セファクリルS-300HRカラムを通過させた。分子量135,000ダルトンの付近に溶出される赤色蛋白質ピークを最終精製酵素 Bとして集めた。結果として約8mgの精製酵素 Bを1.2リッターの大腸菌培養液から得た。
【0124】
[実施例6] 宿主-ベクター系G.オキシダンス[DSM番号4025]のための宿主-ベクター系を広宿主域コスミドpVK102を使用する接合伝達系を用いて確立した。最初は、ナリディキシン酸耐性を有するG.オキシダンス[DSM番号4025]から唯一株の接合伝達体が分離された。新たな宿主GOS2は、上記pVK102を有するG.オキシダンス[DSM番号4025]の接合伝達体からpVK102を除去することにより単離された。二番目の宿主GOS2Rは、DNA供与体である大腸菌から接合伝達体を簡便に選択できるようにリファンピシン(100μg/ml)耐性をGOS2に付与して誘導した。GOS2RへのプラスミドpVK102の導入頻度は、受容菌あたり10-3〜10-4個の接合伝達体が得られるものであった。しかし、GOS2Rの2KGA生産性はG.オキシダンスDSM番号4025より約10%低かった。三番目の宿主GORS6-35は、G.オキシダンス DSM番号4025からリファンピシン耐性、高2KGA生産性、比較的高い遺伝子導入適格性を有する株を接合伝達、プラスミド除去、2KGA醗酵を含む一連の実験を通して選択することにより得られた。
【0125】
(1)GOS2の単離G.オキシダンス[DSM番号4025]にナリディキシン酸耐性を以下のように付与した。G.オキシダンス[DSM番号4025]の細胞をTrypticase Soy Broth(BBL、BectonDickinson Microbiology Systems 社製)(以下Tと略す)寒天培地に50 μg/mlのナリディキシン酸を含む寒天培地(以下TNと略す)上に塗抹し、この塗抹平板を27℃で5日間培養した。その結果得られたコロニーを再度同上TN寒天培地に塗抹し、ナリディキシン酸耐性G.オキシダンス[DSM番号4025]、GONを得た。広宿主域コスミドpVK102(Kmr、Tcr)を、pVK102を保持する大腸菌からGONへ以下のように三者接合伝達により導入した。ヘルパー株であるpRK2013を保持する大腸菌およびpVK102を保持するDNA供与体の大腸菌は、50 μg/mlのカナマイシンを添加したLB培地中で37℃一晩培養した。これらの培養液をカナマイシンを添加した新鮮なLB培地に移植し、さらに5〜6時間培養した。受容菌GONは、TN液体培地中で30℃一晩培養した。大腸菌およびGON株は別々に遠心分離し、各々等量でもとの十分の一量のT培地に再懸濁した。各々の細胞懸濁液100 μlを混合し、その混合液の30μlをNS2寒天平板の表面上に置いたニトロセルロースフィルター上にスポットした。接合伝達体の選択は、50 μg/mlのナリディキシン酸および50 μg/mlのカナマイシンを含むT寒天培地(TNK寒天培地)上で行われた。数個のコロニーが選択平板上で得られたが、同時に多くの大腸菌自然変異体(Nalr、Kmr)も出現した。これら接合伝達体の候補株からプラスミドおよび染色体DNAを調製し、コントロールのpVK102とG.オキシダンス[DSM番号4025]の染色体DNAと、制限酵素解析およびサザンブロットハイブリダイゼーションにより比較した。その結果、一株のpVK102を有するG.オキシダンス[DSM番号4025]、GON8-1の分離を確認した。GON8-1から調製されたプラスミドDNAはpVK102DNAと同一であり、大腸菌中で複製できた。またGON8-1の染色体DNAは、G.オキシダンスDSM番号4025の染色体と同一であった。
【0126】
高い接合伝達頻度を有する宿主として使用できる株を分離するために、上記接合伝達体GON8-1からプラスミドpVK102を以下のように除去した。GON8-1を抗生物質非存在下、Tブロス中で30℃で2日間培養し、その培養液を新鮮なTブロスに2%の割合で移植した。このような培養を3回繰り返し得られた細胞をT寒天平板に塗抹し、27℃で4日間培養した。得られたコロニーをKms株を分離するためにTNKおよびTN寒天平板上に拾い上げた。このKms株の一株はGOS2と命名され、サザンブロットハイブリダイゼーションによりpVK102のいかなる領域をも含まないことが確認された。次に、pVK102を接合伝達によりGOS2に導入した。この株はG.オキシダンス[DSM番号4025]よりも102〜103高い遺伝子導入適格性 (すなわち10-5〜10-6接合伝達体/受容体)を示した。
【0127】
(2)GOS2のリファンピシン耐性変異株、GOS2Rの単離GOS2細胞を20〜100μg/mlのリファンピシンを含むT寒天平板上を繰り返し移植することで、GOS2からのリファンピシン耐性(Rifr)変異株を単離した。得たRifr株の一つをGOS2Rと命名した。GOS2R株は非常に高い遺伝子導入適格性を示し、TRK寒天(100 μg/mlのリファンピシンおよび50 μg/mlのカナマイシンを含むT寒天培地)およびTRT寒天(100 μg/mlのリファンピシンおよび3 μg/mlのテトラサイクリンを含むT寒天培地)上でそれぞれ受容菌あたり各々10-2〜10-3および10-4の接合伝達体がGOS2R株から得られた。
【0128】
GOS2RのL-ソルボースからの2KGA生産性を、G.オキシダンス[DSM番号4025]の2KGA生産性と以下のように比較した。NS2寒天培地上で維持されている細胞を5 mlの種培養培地[8% L-ソルボース(別滅菌)、0.05%グリセロール、0.25 %MgSO4・7H2O、1.75% コーンスティープリカー(corn steep liquor)、5.0% パン酵母、1.5% CaCO3および0.5% 尿素(別滅菌)(滅菌前pH7.0)]に植菌し、30℃で24時間培養した。得られた種培養液5 mlを50 mlの生産培地PMS10[10% L-ソルボース(別滅菌)、0.05% グリセロール、0.25 % MgSO4・7H2O、3% コーンスティープリカー、6.25% パン酵母、1.5% CaCO3および1.6% 尿素(別滅菌)(滅菌前pH7.5)]を含む500mlのエルレンマイヤーフラスコに植菌し、30℃で4日間振とう培養(180 rpm)した。2KGAの定量は、高速液体クロマトグラフィー分析により行った。GOS2RおよびG.オキシダンス[DSM番号4025]は、各々87.3および97.3 g/lの2KGAを生産した。
【0129】
(3)2KGA高生産性を有するGORS6-35の単離L-ソルボースからの2KGA生産性が、G.オキシダンス[DSM番号4025]と同じである株中で、AADH遺伝子のセルフクローニングの評価を行うために、新たな宿主を以下のように作成した。すなわち、(1)200 μg/mlのリファンピシンに対する耐性を付与する、(2)pVK102を導入後、除去する、(3)L-ソルボースからの2KGA高生産株を選択することににより作成した。このようにして得られたGORS6-35は次の二つの性質:(1)親株のG.オキシダンス[DSM番号4025]とほぼ同等の2KGA生産性(10% L-ソルボースから約100 g/lの2KGA生産)および(2)受容菌あたり10-6〜10-7の接合伝達体を与える遺伝子導入適格性を示した。
【0130】
[実施例7] プロモーター置換酵素 B遺伝子の構築G.オキシダンスDSM番号4025株の無細胞抽出液をSDS-ポリアクリルアミドゲル電気泳動に供し、そのゲルをクマシーブリリアントブルー(Coomassie BrilliantBlue)R-250で染色した結果、酵素 Aは上記菌株細胞内において発現量の最も高い蛋白質の一つであることが判明したので、酵素 A遺伝子のプロモーター(PA)はG.オキシダンス DSM番号4025株の中で強力なものと思われる。このPAおよび、G.オキシダンス DSM番号4025株においてカナマイシン耐性を示しうるTn5のカナマイシン耐性遺伝子のプロモーター(PTn5)を、図10に示すように酵素 B遺伝子のSD配列を含む構造遺伝子に連結した。
【0131】
酵素 B遺伝子を含む2.3 kb HindIII断片をM13 mp18に挿入し、得られたファージDNAをT7-GEN(登録商標)インビトロ変異誘発キット(In vitro Mutagenesis Kit(東洋紡社製))を用いて、供給会社の推奨する方法に従って部位特異的変異導入を行った(図9)。酵素 B遺伝子の上流に、酵素 Bのプロモーターの代わりに種々のプロモーターを導入するために、SD配列の上流にBamHI部位を作成した。変異プライマーGTTAGCGCGGTGGATCCCCATTGGAGG(BamHI部位を含む27塩基、配列番号:12)は、アプライドバイオシステム社製(Applied Biosystems)の381A DNA合成機を用いて合成した。結果として得られるBamHI-HindIII断片は、酵素 Bのプロモーター(PB)を含まず、酵素 BのSDおよび構造遺伝子を有している。
【0132】
酵素 A遺伝子のプロモーター(PA)は、HindIIIおよびBamHI部位を付加したプライマーを用いたPCR法によりクローン化した。PCR反応は、GeneAmp(登録商標)DNA増幅試薬キット(DNA Amplification Reagent Kit(宝酒造社製))を用い、サーマルサイクラー、Zymoreactor II(アトー社製)により行った。反応条件は、酵素添加前処理(94℃、5分)、30サイクルの変性段階(94℃、1分)、会合段階(60℃、1分)、合成段階(72℃、1分)に、後処理(72℃、5分)で行った。鋳型のDNAは、プラスミドpSSA202(pUC18に酵素 A遺伝子を含む2.7 kbのHindIII断片をクローン化したもの)を用いた。反応液の組成は、添付のバファーに対して、200μMのdNTP等モル混液、1 μMの各プライマー、1 ngの鋳型DNAおよび2.5単位のAmpliTaq(登録商標)DNAポリメラーゼとした。結果としてSD配列の上流の300 bpの断片が増幅され、この断片をpUC18のHindIIIとBamHI 部位の間にクローン化し配列に利用した。得られたクローンについては、PCRのミスインコーポレーションによる変異が導入されていないことを塩基配列決定により確認した。
【0133】
カナマイシン耐性遺伝子のプロモーター(PTn5)は、プラスミドpNeo (ファーマシア社製(Pharmacia))からまずHindIII-PstI断片として単離し、pUC18のマルチクローニング部位にクローン化し、PTn5をHindIII-BamHI断片として切り出した。
【0134】
PAおよびPTn5プロモーターを含むHindIII-BamHI断片を、PBプロモーターを除いた酵素 Bの構造遺伝子を含むBamHI-HindIII 断片とともにpUC18のHindIII部位に導入した。得られたプラスミドより調製したHindIII断片をpVK100にクローン化し、pSSAp-BおよびpSSPTn5-Bを作成し、実施例6記載の接合伝達法によってGOS2Rに導入した。
【0135】
[実施例8] フラスコ培養でのGOS2R接合伝達株による2KGA生産(1)酵素 A遺伝子増幅接合伝達体の単独菌フラスコ培養におけるL-ソルボースからの2KGA生産酵素 A発現プラスミドpSSA102RおよびベクタープラスミドpVK102を実施例6記載の接合伝達法によりGOS2Rへ導入した。得られた接合伝達体は30 μg/mlのテトラサイクリンを含むNS2寒天平板培地で維持し、L-ソルボースからの2KGA発酵に供した。接合伝達体細胞を実施例6に示すように種培地5mlに接種し、30℃で24時間培養した。得られた種培養液5mlを、実施例6記載のPMS10生産培地またはPMS12生産培地[12 % L-ソルボース(別滅菌)、0.05% グリセロール、0.25% MgSO4・7H2O、3% コーンスティープリカー(CSL)、10% パン酵母、1.5% CaCO3および2% 尿素(別滅菌)(pHは滅菌前に7.5に調整)]50 mlの入った500 ml-エルレンマイヤーフラスコに接種した。培養は、30℃、180rpmで振とうし、4日ないし5日行った。結果として、GOS2R(pSSA102R)およびGOS2R(pVK102)は、10 %のL-ソルボースからは、4日間でそれぞれ92.2、89.1 g/lの2KGAを生産し、12 %のL-ソルボースからは、5日間でそれぞれ105.7、99.9 g/lの2KGAを生産した。
【0136】
(2)GOS2R(pSSB103R)の単独菌フラスコ培養におけるD-ソルビトールからの2KGA生産酵素 B発現プラスミドpSSB103RおよびベクタープラスミドpVK102を実施例6記載の接合伝達法によりGOS2Rに導入した。得られた接合伝達体は30 μg/mlのテトラサイクリンを含むNS2寒天平板培地で維持し、D-ソルビトールからの2KGA発酵に供した。接合伝達体細胞を8% D-ソルビトール、0.05% グリセロール、0.25% MgSO4・7H2O、1.75% コーンスティープリカー(CSL)、5.0% パン酵母、1.5% CaCO3および0.5% 尿素(別滅菌)(pHは滅菌前に7.0に調整)を含む種培地5 mlに接種し、30℃で24時間培養した。得られた種培養液5 mlを、表8に示す3種の生産培地50 mlの入った500 ml-エルレンマイヤーフラスコに接種し、3日間30℃で振とう培養した(180rpm)。結果として、GOS2R(pSSB103R)は、およそ61.5、71.5および73.0 g/lの2KGAを8%、10%および12%のD-ソルビトールからそれぞれ生産した。一方GOS2R(pVK102)は、それぞれ19.5、25.4および30.2 g/lの2KGAを生産した。
【0137】
【表8】
【0138】
滅菌前pH 7.5に調整*:別滅菌。
【0139】
(3)GOS2R(pSSAp-B)およびGOS2R(pSSPTn5-B)単独菌フラスコ培養におけるD-ソルビトールからの2KGA生産GOS2R(pSSAp-B)、GOS2R(pSSPTn5-B)、GOS2R(pSSB103R)およびGOS2R(pVK100)の細胞を、実施例8の(2)に示したように、エルレンマイヤーフラスコでPMSL10生産培地を用いて30℃で3日間培養した。生成した2KGA量は、表9に示すとおりである。
【0140】
【表9】
【0141】
[実施例9] 単独菌による3Lジャー培養でのD-ソルビトールからの2KGA生産GOS2R(pSSB103R)による単独菌発酵実施例8の(2)に示したように試験管で調製した種培養液5mlずつを、同じ種培地50 mlの入った500 ml-エルレンマイヤーフラスコ4本に接種し、30℃で24時間振とう培養した(180rpm)。得られた種培養液200 mlを、3 mlの消泡剤を添加したPMSL10生産培地1800 mlを含む3Lジャーファーメンターに接種した。ファーメンターは、30℃、700 rpm、0.5 vvmで操作した。D-ソルビトールは以下の2通りの方法で流加した。(1)24時間目から30時間目までの6時間で、50% D-ソルビトール液200 mlを流加、または(2)24時間目から32.3時間目までの8.3時間で、50% D-ソルビトール液280 mlを流加。結果として、(1)および(2)の方法の流加培養でそれぞれ、99.0、103.4 g/lの2KGAを51時間で生産した。
【0142】
[実施例10] 酵素 B遺伝子増幅GOS2Rと大腸菌とのフラスコでの混合培養によるD-ソルビトールからの2KGA生産(1)B.メガテリウム、大腸菌、P.プチダとの混合培養系発酵生育因子供給体として、B.メガテリウム[DSM番号4026]、大腸菌 HB101およびP.プチダ[ATCC21812]を、0.3% 酵母エキス(Difco)、0.3% 肉エキス(極東製薬)、3% コーンスティープリカー、1% ポリペプトン(極東)、0.1% 尿素、0.1%KH2PO4、0.02% MgSO4・7H2O、2% L-ソルボース、0.1% CaCO3(滅菌前にpH7.1に調整)を含む種培養培地150 mlで、24時間それぞれ37、37、30℃で培養した。GOS2R(pSSB103R)株は、実施例8の(2)に示すように、5 mlの種培養培地を入れた2本の試験管で24時間30℃で培養した。GOS2R(pSSB103R)の種培養液4 mlと生育因子供給菌の種培養液3.5 mlを、8% D-ソルビトール、0.01% MgSO4o7H2O、1% コーンスティープリカー、0.1% KH2PO4、0.6% CaCO3、1.5%尿素(別滅菌)および消泡剤(フラスコあたり1滴)(滅菌前にpH7.0に調整)を含む混合培養用生産培地50 mlを含む500 ml-エルレンマイヤーフラスコに接種し、30℃で46.5時間振とう培養した。結果として、B.メガテリウムDSM番号4026、大腸菌 HB101およびP.プチダ[ATCC21812]との混合培養において、それぞれ49.9、54.1、31.3 g/lの2KGAを生産した。
【0143】
(2)GOS2R(pSSAp-B)と大腸菌とのフラスコでの混合培養系発酵GOS2R(pSSAp-B)と大腸菌との混合培養を、大腸菌の種培養培地における2% L-ソルボースを2% D-ソルビトールに変えた以外は上記の方法と同じ様式で行った。結果として、GOS2R(pSSAp-B)は10 %のD-ソルビトールから73.7 g/lの2KGAを48.5時間で生産した。
【0144】
[実施例11] 組換え体のAADHによる2KGAの生産1.7 mg/mlの精製酵素 A(実施例4によって精製)、50 mM Tris-HCl、pH7.5、5mM CaCl2、8 mg/ml ウシ血清アルブミン(BSA)、1 mM PMS、20 μg/ml PQQおよび4% L-ソルボースを含む反応液を、20時間ゆるやかに振とうしながら30℃でインキュベートしたところ、約2 g/lの2KGA(TLCで定量)が生成した。
【0145】
また、各々2.4 mg/mlの精製酵素 Aおよび酵素 B(実施例4によって精製)、50mMTris-HCl (pH7.5)、5 mM CaCl2、8 mg/ml BSA、1 mM PMS、20 μg/ml PQQおよび2 % D-ソルビトールを含む反応液を、20時間ゆるやかに振とうしながら30℃でインキュベートしたところ、0.25 g/lの2KGA(HPLCで定量)および約5 g/lのL-ソルボース(TLCで定量)が生成した。
【0146】
[実施例12] アルコールからアルデヒド、アルコールまたはカルボン酸からケトン、アルデヒドからカルボン酸の生産様々な基質を用いて、精製酵素 Aまたは酵素 Bによる酵素反応を実施例11に示したように行った。生産物はTLCおよび/またはHPLCにより同定し、結果を表10に示した。
【0147】
【表10】
【0148】
酵素 AはD-フルクトースを2KDに変換したことから、D-フルクトースからの中間体としてD-グルコソンを生成していると考えられる。
【0149】
[実施例13] P.プチダの接合伝達体による2KGAおよびL-ソルボースの生産1% CaCO3、0.3% NaCl、1 mM PMS、5 μg/ml PQQ、2% L-ソルボースおよびOD600ユニット10の、pSSA102RまたはpVK100を保有するナリディキシン酸耐性(Nalr)P.プチダ[ATCC21812]を含む休止菌体混液(2 ml)を30℃で緩やかに振とうしながら17時間インキュベートした。結果として、pSSA102RないしpVK100を保有するNalr P.プチダ[ATCC21812]は、それぞれ18.9、0.0 g/lの2KGAを生成した。
【0150】
また、1 % CaCO3、0.3 % NaCl、1 mM PMS、5 μg/ml PQQ、2 % D-ソルビトールおよびOD600ユニット10のpSSB103RまたはpVK100を保有するNalr P.プチダ[ATCC21812]を含む休止菌体混液(2 ml)を30℃で緩やかに振とうしながら17時間インキュベートした。結果として、pSSB103RまたはpVK100を保有するNalr P.プチダ[ATCC21812]は、それぞれ7.8または0.0 g/lのL-ソルボースを生成した。
【0151】
[実施例14] キメラAADH酵素の作成とその性質(1) キメラAADH酵素の作成AADH酵素の基質特異性を変えるために、酵素 Aと酵素 B間の種々のキメラ酵素を作成した。
【0152】
1)図2は方策I(制限酵素切断-連結法)によって作成したキメラ遺伝子の構造を示している。両遺伝子で共通の制限酵素切断部位、AvaI(酵素 A 遺伝子の塩基番号603)、EcoRI(塩基番号1084)、SalI(塩基番号1470)(図7)をその作成のために使用した。まず、酵素 Aおよび酵素 Bの遺伝子カセット(それぞれ2.7 kb、2.3 kbのHindIII断片)を、この順番で同じ向きにpUC18にクローン化して、プラスミドpSSAB201を作成した。また、酵素 B、酵素 A遺伝子の順で、同様にカセットをpUC18に導入し、プラスミドpSSBA201を得た(図3)。次に、これらのプラスミドを各制限酵素で部分切断したのち連結して大腸菌JM109を形質転換した。得られたアンピシリン耐性形質転換体からプラスミドを分析し、酵素A遺伝子を頭とするキメラ遺伝子カセットの場合には2.7 kb、酵素 Bを頭とするキメラ遺伝子カセットの場合には2.3 kbの期待される構造を有するHindIII断片を与えるものを選んだ。このようにして作成したキメラ遺伝子のカセットをpVK102のHindIII部位に導入し、それぞれ酵素 A/B1、酵素 A/B2、酵素 A/B3、酵素 B/A1、酵素 B/A2、および酵素 B/A3をコードする遺伝子をもつプラスミドpSSA/B101R、pSSA/B102R、pSSA/B103R、pSSB/A101R、pSSB/A102RおよびpSSB/A103Rを図2に示すように作成した。これら6つのプラスミドは実施例1に示したように接合伝達法により、Nalr P.プチダに導入した。
【0153】
2)図8は方策IIによるキメラ遺伝子の作成の概略を示している。方策IIでは、AADH酵素の基質特異性を変えるのに、任意の位置で組換えたキメラが作成される細胞内相同組換え法を採用している。この方法の原理は以下のようである。
(i)選択マーカーを持つ一つのプラスミド上に、2つの相同性のある遺伝子を直列に並べ,(ii)2つの遺伝子の間に存在する制限酵素部位で切断し、直鎖状になったプラスミドを用いて、野性型recAを有する大腸菌を形質転換し、(iii)2つの遺伝子間の様々な位置で、組換えた結果環化したDNAを保有する形質転換体を、選択マーカーを指標に選択する、というものである。図8に示すように、2種の制限酵素を用いて、プラスミドpSSAB201およびpSSBA201[pUC18上に酵素 Aおよび酵素 B両遺伝子を有するプラスミド(図3)]を直鎖状とし、大腸菌 JM101(recA+)株を形質転換した。形質転換体は1μgのDNAあたり101〜102得られた。不当な組換えを起こしたものを、DNA鎖長を指標に除去した結果、正しい組換え体の比率は30%であった。酵素 A遺伝子のC末端側約3分の2を失う形のXhoI-BalI断片を用いると、効率よくN末端側3分の1以内で組換えたキメラが得られた。次に、得られた組換え体を特定の3つのSmaI、SphI、SalIおよびBalI切断部位(図7)によって区切られる組換え位置グループに分類した。このようにして作成した様々なキメラ遺伝子をHindIII断片のカセットとしてpVK100にクローン化し、Nalr P.プチダに接合伝達法により導入した。
【0154】
(2)キメラAADH酵素の性質1) 制限酵素切断-連結法により得られたキメラの性質NalrP.プチダで発現したキメラ酵素の酵素学的性質を、実施例1に記載したように接合伝達体細胞の可溶性画分を用いて調べた。図11に示すように8種類の基質をこの評価に用いた。酵素 A/B1および酵素 A/B3は、酵素 Aに比べて発現レベルが低いものの、酵素 A型の基質特異性を示した。一方、酵素 B/A1、酵素B/A2、酵素 B/A3は、酵素 Aに由来する領域が増加するとともに、n-プロパノールに対する活性(酵素 A型)が高まるものの、酵素 B型の基質特異性を示した。酵素 B/A1遺伝子の発現レベルは野生型の酵素 B遺伝子よりも高く約2倍であった。制限酵素切断-連結法によって得たキメラ酵素の性質から、酵素 Aまたは酵素 BにおけるN末側3分の1が基本的に基質特異性を決定していると結論された。
【0155】
2)相同組換え法によって得られたキメラの性質上記のように、この方法により得られたキメラの中で、図7に示されるSmaI2とSalI切断部位の間で組換えた18のキメラ酵素のうち7つが好ましい基質特異性を示した。この7つのキメラ酵素は、D-ソルビトールからはL-ソルボースを生成し、酵素 Aで生成されるD-グルコースを生じなかった。そしてL-ソルボースからは、酵素 Aと同様に2KGAを生成した。キメラ遺伝子の組換え位置は、実施例2に示すように塩基配列の決定により確定した。大まかな構造が「N末側9分の2が酵素 A、残りのC末側9分の7が酵素 B」となっているこのタイプのキメラ酵素は、酵素 superA型として分類した。その結果、酵素 superA型に属する7つのキメラ酵素の組換え位置は3種類であった。すなわち、酵素 A/B21(酵素 Aの第1〜128残基に加えて、酵素 Bの第129〜556残基を含むキメラ酵素)、酵素 A/B22(酵素 Aの第1〜125残基に加えて、酵素 Bの第126〜556残基を含むキメラ酵素)および酵素 A/B25(酵素 Aの第1〜135残基に加えて、酵素 Bの第136〜556残基を含むキメラ酵素)である。酵素 A/B21、酵素 A/B22、または酵素 A/B25遺伝子を発現するP.プチダ接合伝達体は、D-ソルビトールをD-グルコースには変換せずにL-ソルボースに変換した。その他に得られたキメラ酵素酵素 A/B31(酵素 Aの第1〜95残基に加えて、酵素 Bの第96〜556残基を含むキメラ酵素)は、D-ソルビトールを効率よくL-ソルボースに変換したが、L-ソルボースを2KGAには変換しなかった。すなわち、このキメラ酵素は酵素 B型の活性を示した。上述したキメラの発現レベルは、野性型の酵素 Bに比べて高かったが、これは酵素 B遺伝子が、酵素 A遺伝子には見られない「まれなコドン(rare codon)」を多く含むためと思われる。尚、このコドン解析は、プログラム「Codon Preference」(Wisconsin Sequence Analysis Package(登録商標)、Genetics Computer Group)により行った。
【0156】
(3)キメラ遺伝子におけるコドン使用の改良好ましいコドン使用という点においてキメラ酵素(酵素A/B21、酵素 A/B22、酵素 A/B25および酵素 A/B31)遺伝子をさらに改良するために、それらの酵素 B残基からの配列を有するC末側の3分の2の領域を、酵素 A遺伝子残基のC末側の3分の2と置き換えた。酵素 A/B21、酵素 A/B22、酵素 A/B25および酵素 A/B31遺伝子を用いて、それぞれ以下の4つの新しいキメラ遺伝子を構築した。すなわち、酵素 sA21(酵素 Aの第1〜128残基、酵素 Bの第129〜180残基、そして酵素 Aの第180〜556残基を含むキメラ酵素)遺伝子、酵素 sA22(酵素 Aの第1〜125残基、酵素 Bの第126〜180残基、そして酵素 Aの第180〜556残基を含むキメラ酵素)遺伝子、酵素 sA2(酵素 Aの第1〜135残基、酵素 Bの第136〜180残基、そして酵素 Aの第180〜556残基を含むキメラ酵素)遺伝子、および酵素 sB(酵素 Aの第1〜95残基、酵素 Bの第96〜180残基、そして酵素 Aの第180〜556残基を含むキメラ酵素)遺伝子である(図4)。酵素 sA2および酵素 sB遺伝子の置換実験の実際は、それぞれpUC18に酵素 sA遺伝子と酵素 B/A1遺伝子を並べた形でもつプラスミドおよびpUC18に酵素 A/B31遺伝子と酵素 B/A1遺伝子を並べた形でもつプラスミドを、AvaIによって部分切断後連結し、大腸菌 JM109を形質転換した後、形質転換体のプラスミドの構造を制限酵素切断様式で解析するとともに、塩基配列を決定して期待される組み換え部位のAvaI切断部位で置換されていることを確認した。酵素 sA21および酵素 sA22遺伝子作成のための置換実験は、酵素 sA2のN末端部をコードするHindIII-SspI断片を、それぞれ酵素 A/B21および酵素 A/B22遺伝子の組換え位置を含んでいるHindIII-SspI断片と置換することにより行った(図4)。
【0157】
(4)キメラ酵素の速度論的解析表11および表12に酵素 sA2および酵素 sBの速度論的性質を、それぞれ酵素 Aおよび酵素 Bと比較してまとめた。
【0158】
【表11】
【0159】
酵素 sA2および酵素 AによるL-ソルボースからの生成物は2KGAであった。D-ソルビトールからの生成物は、酵素 sA2では微量のD-グルコースを伴うもののL-ソルボースであり、酵素 AではD-グルコースのみであった。このように、酵素 sA2はD-ソルビトールからの2KGA生産に望ましい性質、すなわち、酵素 A同様のL-ソルボースから2KGAを生成するためのL-ソルボース/L-ソルボソン脱水素酵素活性、および酵素 B同様のD-ソルビトールからL-ソルボースを生成するD-ソルビトール脱水素酵素活性を示した。
【0160】
【表12】
【0161】
酵素 Bと比較して、酵素 sBはD-ソルビトールに対する親和性が高く、またD-ソルビトールからL-ソルボースへの変換に対して阻害剤となるD-ソルビトールの酸化生成物であるL-ソルボースに対する親和性は低くなっていた。
【0162】
[実施例15] キメラAADH酵素を増幅したGOS2R誘導株によるD-ソルビトールからの2KGA生産酵素 sA2および酵素 sBを評価するにあたり、GOBΔKおよびGOI13株を作成した。GOBΔKは、自殺ベクターpSUP201[アンピシリン耐性(Ampr)、クロラムフェニコール耐性(Cmr)、mob+、pBR325誘導体、Bio/Technology, 1巻、784〜791頁、1983]を使ってGOS2R株に対して酵素 B遺伝子全体を欠失させ、代わりにプラスミドpUC4K[4.1 kb、Kmr、Ampr、ファーマシア社製(Pharmacia)、ヴィエラ(Viera), J.およびメッシング(Messing), J.、Gene 19巻、259頁、(1982)]から単離した1.28kb Kmr 遺伝子カセットを挿入することにより作成した。
【0163】
GOI13はGOBΔKからさらに野性型酵素 A遺伝子を酵素 sB遺伝子に置換し、野性型酵素 A"遺伝子を、自殺ベクターpSUP202[Ampr、Cmr、Tcr、mob+、pBR325誘導体、Bio/Technology、1巻、784〜791頁、(1983)]を用いてゲンタマイシン耐性(Gmr)遺伝子カセットと置換することによって欠失させて作成した。Gmr遺伝子カセットは、DNA断片Tn5-GM[ササガワら、Gene 56巻、 283〜288頁、(1987)]を鋳型としたPCR増幅法により、PstI切断部位を両端に付加するように設計し、得られたPCR産物をpUC4KのPstI切断部位に挿入することによってpUC8Gとして得た。Gmr遺伝子は、pUC8GからEcoRI、BamHI、SalIまたはPstIで切断することによって単離することが可能である。
【0164】
(1)2KGA生産における酵素 sA2増幅の効果プラスミドpSSsA2(pVK100に酵素 sA2遺伝子を含む2.7 kb HindIIIカセットを導入したもの)および対照となるプラスミドpSSA102R(pVK102に酵素 A遺伝子を含む2.7 kb HindIIIカセットを導入したもの)を実施例6記載の接合伝達法によってGOI13に導入した。得られた接合伝達体を実施例8に示すように、PMSL10培地を用いて30℃で4日間培養した。pSSsA2およびpSSA102Rを有するGOI13は、それぞれ66.3および38.5 g/lの2KGAと、それぞれ8.4 および25.9 g/lの2KD(D-ソルビトールからの2KGA生産において、D-ソルビトールからD-グルコースおよびD-グルコン酸を経て生成する副産物)を生成した。
【0165】
(2)プラスミドpSSsA21およびpSSsA22[pVK100に、それぞれ酵素 sA21および酵素 sA22遺伝子を含む2.7 kb HindIIIカセットを導入したもの(図4)]を実施例6記載の接合伝達法によってGOI13に導入した。得られた接合伝達体を実施例8に示すように、PMSL10培地を用いて30℃で4日間培養した。pSSsA21およびpSSsA22を有するGOI13は、それぞれ66.8および77.4g/lの2KGAと、それぞれ0.3および0.4 g/lの2KDを生成した。
【0166】
(3)2KGA生産における酵素 sBの効果プラスミドpSSsB[pVK100に酵素 sB遺伝子を含む2.7 kb HindIIIカセットを導入したもの(図4)]および対照となるプラスミドpSSB103R(pVK102に酵素 B遺伝子を含む2.3 kb 酵素 B遺伝子を導入したもの)を接合伝達法によってGOBΔKに導入した。pSSsBを有するGOBΔK、pSSB103Rを有するGOBΔKおよびGOBΔKを実施例8の(2)に記載したようにPMSL8培地で培養したところ、それぞれ52.0、46.8、および1.1 g/lの2KGAと、それぞれ6.9、9.3、32.3 g/lの2KDを生成した。
【0167】
GOI13(野性型の酵素 B、酵素 Aおよび酵素 A"を欠失し、染色体DNA上に1コピーの酵素 sB遺伝子を有する)をPMSL10培地で2日間培養したところ、79.3 g/lのL-ソルボースを生成した。
【図面の簡単な説明】
【0168】
【図1】本発明の組換え酵素 Aまたは酵素 Bをコードする組換えDNA分子を各々有する組換え発現ベクターの構造の概略図である。
【図2】本発明のキメラ酵素をコードする組換えDNA分子を各々有する組換え発現ベクターの構造の概略図である。
【図3】相同組換え法によるキメラの構築のための直列に並ぶ酵素 Aおよび酵素 Bの構造遺伝子を含有する組換えDNA分子を有する材料プラスミドの構造の概略図である。
【図4】好ましいコドンを用いたキメラ酵素 sA2、酵素 sA21、酵素 sA22または酵素 sBを各々コードする組換え発現ベクターの構造の概略図である。酵素 sA2は「酵素 Aの第1〜第135残基と酵素 Bの第136〜第180残基と酵素 Aの第180〜第556残基」を含む構造を有する。酵素 sA21は「酵素 Aの第1〜第128残基と酵素 Bの第129〜第180残基と酵素 Aの第180〜第556残基」を含む構造を有する。酵素 sA22は「酵素 Aの第1〜第125残基と酵素 Bの第126〜第180残基と酵素 Aの第180〜第556残基」を含む構造を有する。酵素 sBは「酵素 Aの第1〜第95残基と酵素 Bの第96〜第180残基と酵素 Aの第180〜第556残基」を含む構造を有する。上述の番号は各々の成熟(mature)酵素のアミノ酸残基の番号である。図中、*1、*2、*3、*4および*5は塩基配列におけるAvaI切断部位に相当する組換え部位を示し、各々成熟型酵素 Aのアミノ酸残基番号135、128、125、95および180を示す。
【図5】成熟酵素 Aと酵素 Bのアミノ酸配列の並列図である。図中、*で示した領域をコードする塩基配列は、図2に示されるキメラ遺伝子を構築するために用いた、AvaI、EcoRIおよびSalI切断部位を表す。
【図6】本発明のキメラ酵素をコードする組換え遺伝子の構築法を図示したものである。
【図7】酵素 A遺伝子と酵素 B遺伝子の制限酵素地図を図示している。図中、*は図2および図6に示したキメラ遺伝子を作成するのに用いたAvaI、EcoRIおよびSalI切断部位を表す。
【図8】二つのAADH遺伝子中の共通塩基配列における生体内相同組換えによるキメラ遺伝子の構築法を図示している。
【図9】酵素 B遺伝子の上流にBamHI部位を導入するための部位特異的変異法を示す。
【図10】酵素 B遺伝子のプロモーターの置換方法を図示している。
【図11】本発明のキメラ酵素の基質特異性のグラフを示す。図中、酵素活性は(1)では、n-プロパノールに対する、(2)ではD-グルコースに対する相対活性として表した。酵素 A/B2はシュードモナス プチダにおける発現レベルが低かったため測定対象から除外した。
【技術分野】
【0001】
本発明は、アルコールおよび/またはアルデヒド脱水素酵素活性を有するアルコール/アルデヒド脱水素酵素(以下AADHと称する)の組換え酵素調製物に関する。本発明はまた、AADHをコードする新規組換えDNA分子、該DNAを含む組換え発現ベクター、該組換えDNA分子および/または該組換え発現ベクターを含有する組換え生物に関する。更に本発明は、AADHの組換え酵素調製物の製造方法および該組換え酵素調製物を利用するアルデヒド、カルボン酸、ケトン、特に2-ケト-L-ギュロン酸(以下2KGAと称する)の製造方法および該組換え生物を利用するアルデヒド、カルボン酸、ケトン、特に2KGAの製造方法に関する。
【背景技術】
【0002】
2KGAはL-アスコルビン酸(ビタミンC)生産の重要な中間体である。数多くの微生物がD-ソルビトールまたはL-ソルボースから2KGAを生産することが知られている。特公昭51-40154(1976)はアセトバクター(Acetobacter)属、バクテリウム(Bacterium)属またはシュードモナス(Pseudomonas)属の微生物によるD-ソルビトールからの2KGA生産を開示している。「Acta Micorobiologica Sinica 21巻 (2号)185〜191頁 (1981)」によれば、2KGAはL-ソルボースから微生物の混合培養物、特にシュードモナスストリアータ(Pseudomonasstriata)とグルコノバクター オキシダンス(Gluconobacteroxydans)の混合培養により生産することが可能である。ヨーロッパ特許出願公開第0221 707号は共存微生物存在および非存在下でのシュードグルコノバクターサッカロケトゲネス(Pseudogluconobactersaccharoketogenes)によるL-ソルボースからの2KGA生産を開示している。ヨーロッパ特許出願公開第0278 447号はDSM番号4025(グルコノバクターオキシダンス)および他のDSM番号4026(バチルス メガテリウム株、Bacillusmegaterium)を含む混合培養物によるL-ソルボースからの2KGA製造方法を開示している。ヨーロッパ特許出願公開第88116156号はG.オキシダンスDSM番号4025によるL-ソルボースからの2KGA製造方法を開示している。
【0003】
G.オキシダンスDSM番号4025からAADHが精製され、アルコール、アルデヒドの酸化を触媒するという性質が調べられた。すなわちAADHは、アルコールから対応するアルデヒドおよびケトンを、アルデヒドからカルボン酸を生産することができた(朝倉、星野、ヨーロッパ特許出願公開第606621号)。更に、特に本AADHはL-ソルボースからL-ソルボソンを経由して2KGAへの酸化を触媒した。AADHの精製標品の物理化学的性質は、次の通りである:1)至適pH:約7.0〜9.02)至適温度:約20℃〜40℃3)分子量:135,000 +/- 5,000ダルトン(分子量64,500 +/- 2,000のアルファサブユニットと分子量62,500 +/- 2,000のベータサブユニットのどの組み合わせでもよい2つのサブユニットを含む)
4)基質特異性:L-ソルボース、L-ソルボソン、D-ソルビトール、D-グルコース、D-マンニトール、D-フルクトース、DL-グリセルアルデヒド、エタノール、1-プロパノール、1-ブタノール、1-ペンタノール、1-ヘキサノール、1-ヘプタノール、2-プロパノール、2-ブタノール、プロピオンアルデヒド、PEG1000、PEG2000、PEG4000、PEG6000およびポリビニルアルコールを含む一級および二級アルコールとアルデヒドに活性を有する5)補欠分子族:ピロロキノリンキノン6)等電点:約4.4。
【0004】
該AADHをコードする遺伝子をクローン化できれば、大量のAADH組換え酵素調製物の生産、ならびに種々のアルデヒド、ケトンおよびカルボン酸、特に2KGAの生産が可能な組換え生物を作成するのに有用である。しかし、今日までかかる遺伝子類のクローニングに関する報告はない。
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、アルコールおよび/またはアルデヒド脱水素酵素活性を有するAADHの新規組換え酵素調製物を提供することを課題とする。また、本発明は、AADHをコードする新規組換えDNA分子、該DNAを含む組換え発現ベクター、該DNAおよび/または組換え発現ベクターを有する組換え生物、組換えAADHの製造方法、組換えAADHまたは組換え生物を利用するアルデヒド、カルボン酸、ケトン、特に2KGAの製造方法を提供することを課題とする。
より詳しくは、本発明の一面は配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチド、並びに配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドの間のキメラ組換え酵素、並びに一つまたはそれ以上のアミノ酸残基の付加、挿入、欠失および/または置換を含む上記で同定されるポリペプチドの機能的誘導体からなる群より選択され、該酵素ポリペプチドがアルコールおよび/またはアルデヒド脱水素酵素活性を有する、一つまたはそれ以上の酵素ポリペプチドを含むアルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物に関する。
【0006】
かかる同等の機能を有する誘導体は、当技術分野において周知の化学的ペプチド合成、または本明細書に開示されているような当技術分野において周知の方法により、たとえばサムブルック(Sambrook)ら(分子クローニング(Molecular Cloning), Cold Spring Harbour Laboratory Press, New York,USA,第2版、1989)により開示されているようなDNA配列に基づいた組換えDNA技術、のいずれの方法でも作成することが可能である。蛋白質やペプチド中のアミノ酸をその分子の活性を概して変えずに置換することは、当技術分野において周知であり、たとえばH.ニューラス(Neurath) R. L.ヒル(Hill)により「蛋白質(The Proteins)」(Academic Press, New York, 1979, 特に14頁の図6参照)に述べられている。最も通常起こる置換は:Ala/Ser、Val/Ile、Asp/Glu、Thr/Ser、Ala/Gly、Ala/Thr、Ser/Asn、Ala/Val、Ser/Gly、Tyr/Phe、Ala/Pro、Lys/Arg、Asp/Asn、Leu/Ile、Leu/Val、Ala/Glu、Asp/Glyおよびこれらの逆である。
【0007】
本発明の他の一面は、配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチド、並びに配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドの間のキメラ組換え酵素、並びに一つまたはそれ以上のアミノ酸残基の付加、挿入、欠失および/または置換を含む上記で同定されるポリペプチドの機能的誘導体からなる群より選択され、該酵素ポリペプチドがアルコールおよび/またはアルデヒド脱水素酵素活性を有する、少なくとも一つの酵素ポリペプチドをコードする組換えDNA分子に関する。
【0008】
さらに本発明は、たとえば本配列表に開示されているようなアルコールおよび/またはアルデヒド脱水素酵素活性を有するポリペプチドをコードするDNA配列、およびそれらの相補鎖、またはこれらの配列を含むDNA配列、該配列またはそれらから誘導される断片と標準的条件下でハイブリダイズするDNA配列、および遺伝子コードの縮退という理由により上記DNA配列と標準的条件下でハイブリダイズはしないが全く同じアミノ酸配列を有するポリペプチドをコードするDNA配列に関する。
【0009】
ハイブリダイゼーションの「標準的条件」とは、特異的ハイブリダイゼーションシグナルを検出するのに当業者により一般的に使用され、かつ例えばサムブルック(Sambrook)ら(分子クローニング(Molecular Cloning), Cold Spring Harbour Laboratory Press, New York,USA, 第2版、1989)により述べられている条件、好ましくは、当業者に公知のサムブルック(Sambrook)らにより述べられたいわゆるストリンジェントハイブリダイゼーション-非ストリンジェント洗浄条件、より好ましくはいわゆるストリンジェントハイブリダイゼーション-ストリンジェント洗浄条件を意味する。さらに当技術分野で周知の方法を使用し本明細書に開示されているDNA配列に基づいて設計したプライマーを用いてポリメラーゼ連鎖反応(PCR)により作成できるDNA配列を提供することもまた本発明の目的である。本発明のDNA配列はまたヨーロッパ特許出願第747 483号に記載されているように合成することも可能であると理解される。
【0010】
本発明の更なる一面は、上記で定義される一つまたはそれ以上の組換えDNA分子を有する組換え発現ベクターおよび上述の組換え発現ベクターおよび/または、一つまたはそれ以上のDNA分子を染色体上に有する組換え生物に関する。
【0011】
本発明の更なる一面は、適当な培地で上述の組換え生物を培養し、該組換え酵素調製物を回収することを含む、上記で定義されるアルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物の製造方法に関する。
【0012】
本発明の他の一面は、上記で定義される組換え生物を使用しての基質を生産物に変換することを含むアルデヒド、ケトン、またはカルボン酸を対応する基質から生産する製造方法に関する。
【0013】
本発明の更に他の一面は、L-ソルボースおよび/またはD-ソルビトールを含有する適当な培地中での上記に定義する組換え生物の醗酵を含む2-ケト-L-ギュロン酸の製造方法に関する。
【0014】
本発明の他の一面は、本発明の組換え酵素調製物を含有する反応混合液をインキュベートすることを含むアルデヒド、ケトンまたはカルボン酸を対応する基質から生産する製造方法に関する。
【0015】
本発明の更に他の一面は、上記に定義する組換え酵素調製物とL-ソルボースおよび/またはD-ソルビトールを含有する反応混合液をインキュベートすることを含む2-ケト-L-ギュロン酸の製造方法に関する。
【0016】
上述のように2-ケト-L-ギュロン酸の製造方法に効果的であり、該製造方法により得られる2-ケト-L-ギュロン酸が当技術分野で既知の方法によりビタミンC(L-アスコルビン酸)に変換されることを特徴とする2-ケト-L-ギュロン酸からのビタミンCの製造方法を提供することも本発明の課題である。
【課題を解決するための手段】
【0017】
本発明に記述されているAADH遺伝子は上述のように種々のアルコールおよびアルデヒドの酸化を触媒することが可能なAADH酵素をコードしている。具体的には、グルコノバクターに存在する固有のAADH酵素遺伝子をクローン化し、発現させた。当業者であれば、本発明の知見に基づき、グルコノバクターに加えて他のAADH酵素遺伝子の供給源も見出すことが可能である。
【0018】
特定の好ましいグルコノバクターオキシダンス株は、DSM番号4025の下、「Deutsche Sammlung vonMikroorganismen in Gottingen(ドイツ)」に寄託されている。
【0019】
更に、本菌株の継代培養株は、寄託番号FERM BP-3812として、日本工業技術院生命工学工業技術研究所に寄託されている。ヨーロッパ特許出願公開第0278477号に本菌株の性質が開示されている。
【0020】
本発明で利用されるAADH遺伝子および組換え微生物は以下の工程で取得できる:(1)コロニーハイブリダイゼーション、プラークハイブリダイゼーション、PCRクローニング、ウェスタンブロット分析、サザンブロットハイブリダイゼーション等の手法による染色体DNAからのAADH遺伝子のクローニング(2)該AADH遺伝子の常法による塩基配列決定およびAADH遺伝子を含有しそれを効率良く発現する組換え発現ベクターの構築(3)形質転換、形質導入、接合伝達およびエレクトロポレーションによる組換え発現ベクターまたは染色体上に組換えAADH遺伝子を有する組換え微生物の構築。
【0021】
本発明の上記見地に応用可能な材料および技術を以下のように詳細に例示する。
【0022】
全染色体DNAは当業者に周知の方法(マーマー(Marmaur) J., J. Mol. Biol. 3巻、208頁、1961)により精製できる。次に目的遺伝子を有する菌株の遺伝子ライブラリーは上記染色体DNAおよび以下に詳細に述べられるベクターを用い構築可能である。AADHをコードする遺伝子は、以下の方法で染色体DNAからプラスミドまたはファージどちらのベクターにもクローン化可能である。
【0023】
1)精製酵素の部分アミノ酸配列を決定し、その配列情報に従ってオリゴヌクレオチドを合成し、遺伝子ライブラリーから目的の遺伝子をサザンブロットハイブリダイゼーション、コロニーハイブリダイゼーション、またはプラークハイブリダイゼーションにより選択する。
【0024】
2)上述のように合成したオリゴヌクレオチドをプライマーとして用いるポリメラーゼ連鎖反応(PCR)により所望の遺伝子の部分配列を増幅し、そのPCR生成物をプローブとして用い遺伝子ライブラリーから目的の完全長の遺伝子をサザンブロットハイブリダイゼーション、コロニーハイブリダイゼーション、またはプラークハイブリダイゼーションにより選択する。
【0025】
3)たとえば「Methods in Enzymology、73巻、46頁、1981」に述べられているような前述の方法で所望の酵素蛋白質に対する抗体を調製し、ウェスタンブロット分析を含む免疫学的分析により所望のポリペプチドを発現するクローンを選択する。
【0026】
4)所望の酵素の一つに対する相同体(homologs)のアミノ酸配列を整列し、相同体間で共通のアミノ酸配列を選択し、その共通配列をコードするオリゴヌクレオチドを合成し、そのオリゴヌクレオチドをプライマーとして用いるPCRにより所望の遺伝子部分配列を増幅し、上記2)の方法で述べた完全配列を選択する。
【0027】
所望の遺伝子の塩基配列はM13ファージを用いるダイデオキシチェインターミネーション法(サンガー(Sanger) F.ら、Proc. Natl. Acad. Sci. USA、74巻、5463〜5467頁、1977)のような公知の方法で決定できる。
【0028】
必要に応じて、このように決定された塩基配列情報を(使用コドンを考慮して)利用することにより、進化的に分岐したアルコールおよび/またはアルデヒド脱水素酵素をコードする遺伝子を、該塩基配列から推定されるアミノ酸配列に従って合成されたプローブを用いるコロニーハイブリダイゼーションまたはサザンブロットハイブリダイゼーションにより、または該情報により合成されたプライマーを用いるポリメラーゼ連鎖反応(PCR)により、異種の生物からも単離可能である。
【0029】
所望の遺伝子、すなわち本発明のDNA配列を効率良く発現するために、種々のプロモーターを使用することが可能である。たとえば、該遺伝子自身のプロモーターや、Tn5(D. E. Berg, C. M. Berg, 1983, Bio/Technology 1巻、417〜435頁)のカナマイシン耐性遺伝子、pBR322のアンピシリン耐性遺伝子のような抗生物質耐性遺伝子のプロモーター、大腸菌(Escherichia coli)のベータガラクトシダーゼ遺伝子のプロモーター(lac)、trp、tac、trcプロモーター、ラムダファージのプロモーターおよび大腸菌、シュードモナスプチダ(Pseudomonasputida)、アセトバクター キシリナム(Acetobacter xylinum)、アセトバクター パスチュリアヌス(Acetobacter pasteurianus)、アセトバクターアセチ(Acetobacter aceti)、アセトバクター ハンゼニイ(Acetobacter hansenii)およびグルコノバクターオキシダンス等の細菌を含む微生物、哺乳動物細胞および植物細胞を含む宿主中で機能しうるいずれのプロモーターも使用可能である。
【0030】
さらに遺伝情報をコードする配列が導入される宿主細胞中で機能可能なシャインダルガノ(SD)配列(たとえばAGGAGG等で、宿主細胞中で機能しうる天然および合成配列も含む)および転写終結因子(宿主細胞中で機能しうる天然および合成配列を含む逆位反復配列)のような他の制御因子も上述のプロモーターとともに使用することができる。
【0031】
ペリプラズムに局在するポリペプチド(AADH)の発現のためには、通常15〜50アミノ酸残基を含み全体的に疎水性であるシグナルペプチドが不可欠である。シグナルペプチドをコードするDNAは宿主細胞中で機能しうるものであればどのような天然配列でも合成配列でもよい。
【0032】
多様な宿主/クローニングベクターの組み合わせを二重鎖DNAのクローニングに使用できる。クローニングベクターは一般的に複製起点、制御因子、マルチクローニング部位を含むクローニング部位、およびアンピシリン、テトラサイクリン、カナマイシン、ストレプトマイシン、ゲンタマイシン、スペクチノマイシン等の抗生物質耐性遺伝子のような選択マーカーを含むプラスミドまたはファージである。
【0033】
大腸菌中での本発明のDNA配列の発現のための好ましいベクターは、pBR322またはpUC18、pBluescriptIIを含むその誘導体、pACYC177、pACYC184(J. Bacteriol.、134巻、1141〜1156頁、1978)およびその誘導体、ならびにRK2、RSF1010のような広宿主域プラスミドから誘導されるベクターのような大腸菌中で通常使用されるどのようなベクターからも選択できる。G.オキシダンスDSM番号4025を含むグルコノバクターおよびP.プチダ中での本発明のDNA配列の発現のための好ましいベクターは、大腸菌のような好ましいクローニング宿主と同様、グルコノバクターおよび/またはP.プチダ中で複製できるどのようなベクターからも選択できる。好ましいベクターは、pVK102のようなコスミドベクターとその誘導体、およびRSF1010とその誘導体、ならびにグルコノバクターで機能する複製起点および大腸菌で機能するもう一つの複製起点を有するベクターのような広宿主域ベクターである。ベクターのコピー数および安定性はクローン化遺伝子の安定で効率よい発現のため、およびクローン化遺伝子を有する宿主細胞の効率よい培養のために注意深く考慮すべきである。Tn5のような転位因子を含むDNA分子も、本発明のDNA配列を好ましい宿主に、特に染色体上に、導入するベクターとして使用可能である。本発明の所望のDNA配列と共に、好ましい宿主から単離されたどのようなDNAを含むDNA分子も、好ましい宿主に、特に染色体上に、本発明の所望のDNA配列を導入するのに有用である。そのようなDNA分子は形質転換、形質導入、接合伝達またはエレクトロポレーションにより好ましい宿主に導入することが可能である。
【0034】
使用可能な宿主としては、微生物、哺乳動物細胞および植物細胞等が挙げられる。好ましい微生物としては、大腸菌、P.プチダ、A.キシリナム、A.パスチュリアヌス、A.アセチ、A.ハンゼニイ、G.オキシダンスおよび組換えAADHを生産可能ないずれのグラム陰性細菌でもよい細菌が挙げられる。該微生物の機能的同等株、継代培養株、変異株および変種も本発明に使用することが可能である。好ましい株は、大腸菌 K12株およびその誘導株、P.プチダまたはG.オキシダンス DSM番号4025株である。
【0035】
機能的なAADHをコードする本発明のDNA配列を用い、当技術分野で周知の方法を用い上述の宿主中で機能することが可能なプロモーターやリボゾーム結合部位のような制御領域を有する適当なベクターに連結することにより、発現プラスミドを産出する。そのような組換え発現ベクターの構造を具体的に図1、2、4および10に示す。
【0036】
組換え発現ベクターを有する組換え微生物を構築するためには、形質転換、形質導入、接合伝達(フィリップゲンハート(Philipp Genhardt)ら、「一般的分子細菌学に関する方法(Methods for general and molecular bacteriology)、14および15章、American Society forMicrobiology、1994)およびエレクトロポレーションを含む種々の遺伝子導入方法を使用することが可能である。組換え体を作成する方法としては、分子生物学の分野で周知の方法の中から選択して使用できる。通常の形質転換系は、大腸菌、シュードモナスおよびアセトバクターにおいて使用できる。形質導入系は、大腸菌に使用できる。接合伝達系は、グラム陽性菌および大腸菌、P.プチダおよびG.オキシダンスを含むグラム陰性菌において広く使用できる。好ましい接合伝達方法は、国際公開公報89/06688号に開示されている。接合伝達は、液体培地中または固体培地表面において起こりうる。好ましい受容菌は、適当な組換え発現ベクターを用い活性を有するAADHを生産しうる大腸菌、P.プチダおよびG.オキシダンスから選択される。2KGA生産のための好ましい受容菌は、G.オキシダンスDSM番号4025である。接合伝達の受容菌には、通常選択マーカーが付与される。たとえばナリディキシン酸またはリファンピシンに対する耐性が通常選択される。
【0037】
本発明で提供されるAADHは、アルコールおよび/またはアルデヒドの酸化を触媒し、該当する基質からアルデヒド、ケトンまたはカルボン酸を生産できる。更に具体的には、本発明で提供されるAADHは、L-ソルボースを酸化してL-ソルボソンを経由し2KGAを生成する反応および/またはD-ソルビトールを酸化してL-ソルボースを生成する反応を触媒することができる。更に具体的には、本発明で提供されるAADHは、アミノ酸配列が配列番号5、6、7および8で各々示される酵素A、酵素 A'、酵素 A"および酵素 Bを含んでいる。
【0038】
塩基配列が配列番号1、2、3および4で各々示される遺伝子で、その遺伝子がアミノ酸配列が配列番号5、6、7および8で各々示されるポリペプチドをコードする酵素 A、酵素 A'、酵素 A"および酵素 B遺伝子は、G.オキシダンス株DSM番号4025から取得することができる。
【0039】
本発明で提供される酵素 A、酵素 A'、酵素 A"および酵素 Bを含むAADHは、別個に、適当な生物を培養し、その細胞を破砕し、その破砕された細胞の無細胞抽出物、好ましくは微生物の可溶性画分から単離精製することにより調製できる。
【0040】
本発明で提供される組換え生物は、好気的条件下適当な栄養分を補足した水性培地中で培養することができる。該培養はpH約4.0から9.0の範囲、好ましくはpH約6.0から8.0の範囲で行うことができる。培養期間は、使用するpH、温度、栄養培地に応じて変わるが、通常2から5日が好ましい結果をもたらす。培養を行うのに好ましい温度範囲は約13℃から45℃、好ましくは18℃から42℃である。
【0041】
培養培地として通常必要な栄養分は、同化可能な炭素源、消化可能な窒素源、無機物質、ビタミン類、微量元素および他の生育促進因子などである。同化可能な炭素源としては、グリセロール、D-グルコース、D-マンニトール、D-フルクトース、D-アラビトール、D-ソルビトールおよびL-ソルボースなどが使用できる。
【0042】
酵母エキス、肉エキス、ペプトン、カゼイン、コーンスティープリカー、尿素、アミノ酸、硝酸塩、アンモニウム塩などの種々の有機または無機物質を窒素源として使用できる。無機物質としては、硫酸マグネシウム、リン酸カリウム、塩化第一鉄、塩化第二鉄、炭酸カルシウムなどが使用できる。
【0043】
本発明に係る組換え酵素調製物においては、[1] 配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチド、並びに配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドの間のキメラ組換え酵素、並びに一つまたはそれ以上のアミノ酸残基の付加、挿入、欠失および/または置換を含む上記で同定されるポリペプチドの機能的誘導体からなる群より選択され、該酵素ポリペプチドがアルコールおよび/またはアルデヒド脱水素酵素活性を有する、一つまたはそれ以上の酵素ポリペプチドを含む、アルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物であることを特徴とする。
【0044】
また、本発明に係る組換え酵素調製物においては、[2] ポリペプチドが、酵素A/B1、酵素 A/B2、酵素 A/B3、酵素 B/A1、酵素 B/A2、酵素 B/A3、酵素 sA2、酵素 sA21、酵素 sA22、酵素 sBおよびそれらの機能的誘導体のキメラ酵素である、前記[1]に記載の組換え酵素調製物であることを特徴とする。
【0045】
また、本発明に係る組換え酵素調製物においては、[3] 酵素ポリペプチドが、ホモ二量体および/またはヘテロ二量体の形態で存在する、前記[1]または[2]に記載の組換え酵素調製物であることを特徴とする。
【0046】
また、本発明に係るDNA分子においては、[4] 上記[1]または[2]で定義されるポリペプチドをコードするDNA分子であることを特徴とする。
【0047】
また、本発明に係るDNA分子においては、[5] 直鎖状もしくは環状DNAまたは染色体上での挿入DNA断片のいずれかの形態で存在する、前記[4]に記載DNA分子であることを特徴とする。
【0048】
また、本発明に係る組換え発現ベクターにおいては、[6] 上記[4]または[5]のいずれかで定義される一つまたはそれ以上のDNA分子を含む、組換え発現ベクターであることを特徴とする。
【0049】
また、本発明に係る組換え発現ベクターにおいては、[7] DNA分子が、一つまたはそれ以上の遺伝子制御配列に機能的に結合しており、適当な宿主細胞において上記[1]〜[3]のいずれかに記載の酵素ポリペプチドを発現可能である、前記[6]に記載の組換え発現ベクターであることを特徴とする。
【0050】
また、本発明に係る組換え発現ベクターにおいては、[8] pSSA102R、pSSA'101R、pSSA"102、pSSB103R、pSSAp-B、pSSA/B101R、pSSA/B102R、pSSA/B103R、pSSB/A101R、pSSB/A102R、pSSB/A103R、pSSsA2、pSSsA21、pSSsA22およびpSSsBからなる群より選択される、前記[7]に記載の組換え発現ベクターであることを特徴とする。
【0051】
また、本発明に係る組換え生物においては、[9] 上記[6]〜[8]のいずれかに記載の組換え発現ベクターまたは上記[4]もしくは[5]のいずれかで定義される一つまたはそれ以上のDNA分子を有する、組換え生物であることを特徴とする。
【0052】
また、本発明に係る組換え生物においては、[10] 宿主細胞が、微生物、哺乳動物細胞および植物細胞からなる群より選択される、前記[9]に記載の組換え生物であることを特徴とする。
【0053】
また、本発明に係る組換え生物においては、[11] 宿主細胞が、大腸菌(Escherichia coli)、シュードモナスプチダ(Pseudomonasputida)、アセトバクターキシリナム(Acetobacter xylinum)、アセトバクターパスチュリアヌス(Acetobacterpasteurianus)、アセトバクター アセチ(Acetobacter aceti)、アセトバクター ハンゼニイ(Acetobacter hansenii)およびグルコノバクターオキシダンス(Gluconobacteroxydans)等の細菌からなる群より選択される微生物である、前記[9]または[10]に記載の組換え生物であることを特徴とする。
【0054】
また、本発明に係る組換え生物においては、[12] 宿主細胞が、グルコノバクター オキシダンス[DSM番号4025]である、前記[11]に記載の組換え生物であることを特徴とする。
【0055】
また、本発明に係る組換え酵素調製物の製造方法においては、[13] 上記[10]〜[12]のいずれかで定義される組換え生物を適当な培地で培養し、組換え酵素を回収することを含む、前記[1]、[2]または[3]で定義されるアルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物の製造方法であることを特徴とする。
【0056】
また、本発明に係るアルデヒド、ケトンまたはカルボン酸生成物の対応する基質からの製造方法においては、[14] 上記[10]〜[12]のいずれかで定義される組換え生物の生化学的反応により基質を生産物に変換することを含む、アルデヒド、ケトンまたはカルボン酸生成物を対応する基質から製造する方法であることを特徴とする。
【0057】
また、本発明に係るL-ソルボースおよび/またはD-ソルビトールからの2-ケト-L-ギュロン酸の製造方法においては、[15] 上記[10]〜[12]のいずれかで定義される組換え生物の生化学的反応によりL-ソルボースおよび/またはD-ソルビトールを2-ケト-L-ギュロン酸に変換することを含む、L-ソルボースおよび/またはD-ソルビトールから2-ケト-L-ギュロン酸を製造する方法であることを特徴とする。
【0058】
また、本発明に係るアルデヒド、ケトン、またはカルボン酸生成物の対応する基質からの製造方法においては、[16] 上記[1]〜[3]のいずれかで定義される組換え酵素調製物および基質を含有する反応混合液をインキュベートすることを含む、アルデヒド、ケトンまたはカルボン酸生成物を対応する基質から製造する方法であることを特徴とする。
【0059】
また、本発明に係る2-ケト-L-ギュロン酸の製造方法においては、[17] 上記[1]〜[3]のいずれかで定義される組換え酵素調製物とL-ソルボースおよび/またはD-ソルビトールとを含有する反応混合液をインキュベートすることを含む、2-ケト-L-ギュロン酸の製造方法であることを特徴とする。
【0060】
また、本発明に係る2-ケト-L-ギュロン酸からのL-アスコルビン酸の製造方法においては、[18] 上記[15]または[17]に記載の製造方法が達成され、該製造方法により得られる2-ケト-L-ギュロン酸が、当技術分野で既知の方法によりL-アスコルビン酸に変換されることを特徴とする、2-ケト-L-ギュロン酸からのL-アスコルビン酸の製造方法であることを特徴とする。
【発明の効果】
【0061】
アルコールおよび/またはアルデヒド脱水素酵素活性を有するアルコールおよび/またはアルデヒド脱水素酵素遺伝子をクローン化したことにより、組換えDNA技術を用いて、クローン化した遺伝子を含むDNA分子、該DNA分子を含む組換え発現ベクター、該DNA分子または該発現ベクターを含む組換え体を作成することが可能となり、また、これらを用いてクローン化した遺伝子を発現させることにより、種々のアルデヒド、ケトン、カルボン酸、特に2-ケト-L-ギュロン酸、の生成に有用なアルコールおよび/またはアルデヒド脱水素酵素の組み換え調製物を製造することが可能となった。
【発明を実施するための最良の形態】
【0062】
以下に特にP.プチダから得られた精製組換えAADH酵素の性質およびその生産方法を示す。
【0063】
(1)酵素活性本発明のAADHは以下の反応式に従い電子受容体存在下D-ソルビトール、L-ソルボースおよびL-ソルボソンを含むアルコールおよびアルデヒドの酸化を触媒する。
アルコール+電子受容体 → アルデヒド+還元型電子受容体アルコール+電子受容体 → ケトン+還元型電子受容体アルデヒド+電子受容体 → カルボン酸+還元型電子受容体糖アルコール+電子受容体 → アルドース+還元型電子受容体糖アルコール+電子受容体 → ケトース+還元型電子受容体アルデヒドケトース+電子受容体 → ケトカルボン酸+還元型電子受容体カルボン酸+電子受容体 → ケトカルボン酸+還元型電子受容体酵素は分子酸素を受容体として用いない。受容体として、2、6-ジクロロフェノールインドフェノール(DCIP)、フェナジンメソサルフェート(PMS)、ウェスターブルー、フェリサイアナイド、補酵素Qまたはチトクロームcが使用できる。
【0064】
酵素活性1単位は1分間あたり1μモルのDCIPの還元を触媒する酵素量と定義した。DCIPのpH 8.0における分子吸光係数は15 mM-1とした。標準反応混合液(1.0 ml)は0.1 mM DCIP、1 mM PMS、2〜125mM 基質、50 mMTris-リンゴ酸-NaOH緩衝液(pH 8.0)および10 μlの酵素液を含む。対照としては上記の組成から基質を除いたものを用いた。
【0065】
(2) AADHの性質a) 酵素反応の基質特異性と生成物酵素 A、酵素 A'、酵素 A"および酵素 Bは8種の基質、n-プロパノール、イソプロパノール、D-グルコース、D-ソルビトール、L-ソルボソン、D-マンニトール、L-ソルボースおよびD-フルクトース、を用いた上記(1)におけるそれらの基質特異性によって特徴づけられる。結果を表1に示した。
【0066】
【表1】
【0067】
酵素 Bはn-プロパノールおよびイソプロパノールに対して比較的低い反応性を示したが、D-グルコースおよびD-マンニトールに対して高い反応性を示した。酵素 A、A'およびA"は、D-グルコースおよびD-マンニトールに対して低い反応性を示したが、n-プロパノールおよびイソプロパノールに対して高い反応性を示した。これら三酵素は酵素 A'がL-ソルボースおよびD-フルクトースに対してきわめて低い反応性を示した以外には、類似した基質特異性パターンを示した。
【0068】
酵素 A、酵素 A'、酵素 A"または酵素 Bを用いた反応における基質からの生成物は、既知物質を用いた薄層クロマトグラフィー(TLC)および/または高速液体クロマトグラフィー(HPLC)により分析された。酵素 A、酵素 A'、および酵素 A"(以後Aグループとする)はD-ソルビトール、L-ソルボース、L-ソルボソン、D-マンニトールおよびD-フルクトースをそれぞれL-ギュロースを伴うD-グルコース、2KGAを伴うL-ソルボソン、2KGA、D-マンノース、および2-ケト-D-グルコン酸(2KD)に変換した。酵素 B(以後Bグループとする)はD-グルコース、D-ソルビトール、L-ソルボソン、D-マンニトール、L-イドース、グリセロール、D-グルコン酸、D-マンノン酸をそれぞれD-グルコン酸、L-ソルボース、2KGA、D-フルクトース、L-イドン酸、ジヒドロキシアセトン、5-ケト-D-グルコン酸および5-ケト-D-マンノン酸に変換した。L-ソルボソンに対する反応性における類似性から、D-グルコソンは上記すべてのAADHにより2KDに変換されうる。事実、Aグループ酵素はその直接生成物がD-グルコソンと考えられるD-フルクトースから2KDを生成した。これらすべての酵素はD-ソルビトールやD-マンニトールのような糖アルコールを含むアルコール、ならびにD-グルコースのようなアルドース、およびL-ソルボソンのようなケトースを含むアルデヒドの両者に活性を示した。
【0069】
b) 至適pH表2に示すように、すべての酵素はそれらの至適点をpH8.0〜8.5に有する。酵素 A"とBは、酵素 AとA'に比較して低pH側に比較的広い至適pH範囲を持つ。
【0070】
【表2】
【0071】
c) pH安定性酵素 A、酵素 A'、酵素 A"および酵素 Bを様々なpH値の緩衝液中3時間、25℃でインキュベートした後にその残存活性を測定し、pH8.0でインキュベートしなかった値に対する相対活性として表示した。
【0072】
表3に示すように酵素 A、A、A" およびBはpH 6から9の範囲で安定であった。
【0073】
【表3】
【0074】
d)温度安定性表4に各酵素の4、20、30、40、50および60℃での5分間処理後の残存活性を示す。
【0075】
【表4】
【0076】
e)金属イオンおよび阻害剤の影響種々の金属及び阻害剤により酵素を処理した後の残存活性を表5に示す。MgCl2とCaCl2はほぼ酵素に対して無影響であったが、他の金属イオン、特にCuCl2はその反応性に強い影響をおよぼした。EGTAおよびEDTAは顕著に酵素 A、A'およびA"を阻害した。しかし、酵素 BはAグループ酵素群よりもEGTAとEDTAによる阻害が少なかった。
【0077】
【表5】
【0078】
f)分子量とサブユニットP.プチダ接合伝達体から精製された酵素 A、A'、A"およびBは、ドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS-PAGE)での計測によると、分子量約64,000、62,500、62,500および60,000のそれぞれの単一種ユニットを含む。酵素 A、A'、A"およびB遺伝子/DNA配列類を同一宿主内で発現させた場合、酵素A、A'、A"およびBのユニット間のいかなる組み合わせでもヘテロ二量体を形成しうる。
【0079】
g) N-末端アミノ酸配列成熟酵素 AおよびBのN-末端配列を以下に示す。
酵素 A :Gln-Val-Thr-Pro-Val-Thr----酵素 A" :ブロック化されたN-末端残基酵素 B :Gln-Val-Thr-Pro-Ile-Thr-Asp-Glu-Leu-Leu-Ala----成熟酵素 A'のN-末端は標品の純度不足のため決定されていない。
【0080】
(3) AADHの生産細胞は遠心分離または濾過により発酵培養液より回収される。回収された細胞を緩衝溶液中に懸濁し、ホモジナイザー、超音波または溶菌酵素処理などによって破砕し細胞の破砕溶液を得る。
【0081】
AADHは破砕された細胞の無細胞抽出液から、好ましくは微生物の可溶性画分から硫安沈殿、透析、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィーおよびアフィニティークロマトグラフィーの様な一般的蛋白質精製法により、単離精製される。
【0082】
(4)酵素反応酵素反応はTris-HCl緩衝液、燐酸緩衝液等の緩衝液中で、例えばDCIP、PMS、ウェスターブルー、フェリサイアナイド、補酵素Q、チトクロームc等の電子受容体存在下、約10℃から約50℃、好ましくは20℃から40℃、の温度においてpH値約6.0から約9.0においてで実施される。反応混合液中の基質の濃度は他の反応条件により変化させうるが、一般的に約1〜200g/l、最も好ましくは1〜100 g/lである。
【0083】
本酵素反応には、AADHは適当な担体を用いる固定化状態においても使用できる。当技術分野において一般に知られているいずれの酵素固定化法も使用可能である。例えば、酵素を官能基を有する樹脂膜、顆粒などに直接結合させてもよく、または官能基を有する架橋化合物、例えばグルタルアルデヒドを介して樹脂に結合させてもよい。
【0084】
本発明で提供されるポリペプチドには、酵素 A、酵素 A'、酵素 A"および酵素Bを含むAADH遺伝子から調製される誘導体、および遺伝子コドンの縮退から生ずる遺伝子相同体またはAADH遺伝子に相当な相同性のある天然、合成または組換え体のいずれの遺伝子配列から調製される関連誘導体も含まれる。本誘導体は、配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドと同等の機能を有し、それが一つまたはそれ以上のアミノ酸残基の付加、欠失および/または置換を含み、その該酵素ポリペプチドがアルコールおよび/またはアルデヒド脱水素酵素活性を有する変異体でもよい。これらの変異遺伝子は、AADH遺伝子を紫外線、エックス線、ガンマ線を照射することにより、または亜硝酸、N-メチル-N'-ニトロ-N-ニトロソグアニジン(NTG)または他の適当な変異剤で処理することにより、または自然変異を起こした株を分離することにより、または当技術分野において周知の標準的インビトロ変異処理をすることにより作成することが可能である。
【0085】
AADHポリペプチドの誘導体には、また配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドの間のキメラ組換え酵素が含まれる。このキメラ体は、図6に示されるように制限酵素とT4-リガーゼを用い、遺伝子間で共通の制限酵素部位で本発明のDNA配列の2つまたはそれ以上のDNA配列を試験管内で組換えることにより、または図8に示されるように二遺伝子間で共通の塩基配列の位置での2つのAADH遺伝子間の生体内組換えにより調製することができる。
【0086】
AADHポリペプチドの誘導体には、またAADHポリペプチドのアミノ末端、カルボキシル末端および/または内部に、ポリペプチドが付加されたポリペプチドも含まれる。カルボキシル末端にG.オキシダンスDSM番号4025のチトクロムcポリペプチド(17〜18kDa)が融合した酵素 B、酵素 A/B25および酵素 A/B3は、D-ソルビトールからL-ソルボースへの変換において実施例4に述べられている酵素Bと同様のAADH活性を示し、L-ソルボースから2KGAへの変換において実施例14に述べられている酵素 A/B25および酵素 A/B3と同様のAADH活性を示した。このように本発明で提供されるAADHに、AADH活性を維持したまま、かなり長いポリペプチドを付加または挿入することが可能である。
【0087】
上述のAADHポリペプチドの誘導体は、所望の基質特異性、基質への高親和性、阻害物質への低親和性、温度および/またはpHに対する高安定性、および高触媒速度のような好ましい性質を示す可能性が高い。以下の実施例に述べられているように、該誘導体は目的の生産物の生産性を改良することが可能であると思われる。
【0088】
本発明の酵素ポリペプチドは、通常二量体の形態で生産される。このような二量体には、酵素 A、A'、A"、またはBもしくはキメラを含むそれらの誘導体のホモ二量体および上述の二つの異なった酵素ポリペプチドを含むヘテロ二量体が含まれている。本発明の組換え酵素調製物はまた、一つまたはそれ以上の該ホモ二量体および/またはヘテロ二量体を含んでいる。
【0089】
本発明で提供される組換え生物は、アルコールおよび/またはアルデヒド脱水素酵素活性を有するAADHの組換え酵素調製物の生産に非常に有用である。該組換え生物は、該組換え酵素調製物を利用することによる、および該組換え生物を利用することによるアルデヒド、カルボン酸、ケトン、特に2KGAの生産にもまた有用である。
【0090】
該組換え生物を用いる2KGAの生産は、上述の培地および培養条件を用いる発酵により実施され得る。2KGAの生産は、上述の組換え生物を大腸菌、P.プチダ、B.メガテリウムのような共存生物とともに用いても行い得る。
【実施例】
【0091】
[実施例1] AADH遺伝子のクローニング(1)G.オキシダンスDSM番号4025のゲノムライブラリーの構築染色体DNAは以下のように調製した。G.オキシダンスDSM番号4025は、20 mlのNS2培地[5.0% D-マンニトール、0.25% MgSO4・7H2O、1.75% コーンスティープリカー(corn steep liquor: CSL)、5.0% パン酵母 (オリエンタル酵母社製)、0.5%CaCO3、0.5% 尿素(別滅菌)、2.0% 寒天(滅菌前pH7.0)を含む寒天平板上で27℃で3日間培養した。この寒天平板から細胞を集め、10 mlの1 mM EDTAを含む10mM Tris-HCl(pH8.0)緩衝液で洗浄し、5 mlの20 mM EDTAを含む10mM Tris-HCl(pH8.0)緩衝液に懸濁した。この細胞懸濁液を終濃度400μg/mlのリゾチーム(シグマケミカルズ社製(Sigma Chemicals))で37℃、30分間処理し、次にプロナーゼ(400単位)で37℃、30分間処理し、1%SDS で37℃、1時間処理した。染色体DNAは、マニアティス(Maniatis)らにより述べられている方法「分子クローニング:研究室マニュアル(Molecular cloning:a laboratory manual) Cold Spring Harbor Laboratory, Cold Spring Harbor, N. Y.,(1982)」に従いフェノールとRNase A(ベーリンガーマンハイム社製(Boeheringer Mannheim))で処理した。染色体DNA(200 μg)は、168単位のSalI(ベーリンガーマンハイム社製(Boeheringer Mannheim))で37℃、5〜90分間処理した。この処理で得られた15〜35 kbの部分消化断片を分取アガロースゲル電気泳動(アガロース0.7%)で単離した。単離は目的断片を含むゲル断片を切り出し、その中のDNAをゲルから40 mM Tris-酢酸と2 mM EDTAを含むTAE緩衝液中へ電気的に溶出した。このようにして40 μgの目的DNAが得られた。並行して、8 μgのコスミドベクターpVK102(ATCC 37158)をSalIで完全消化し、牛大腸アルカリフォスファターゼ(ベーリンガーマンハイム社製(Boeheringer Mannheim))で供給会社の指示に従い処理した。0.4 μgのpVK102と15〜35kbのSalI断片(0.2〜2 μg)をライゲーションキット(宝酒造社製)を用い26℃で10分間処理した。結合したDNAを今度は、供給会社(アマーシャム社(Amersham))の指示した方法に従って、インビトロパッケージング(結合したDNAをファージコート蛋白質の部品と混合する)に使用した。このようにして生じたファージ顆粒を大腸菌 ED8767[ミューレイ(Murray), N. E., W. J. ブランマー(Brammer)およびK. ミューレイ(Murray), Mol. Gen. Genet.、150巻、53〜61頁、(1977)]に感染させた。約3,000個のカナマイシン耐性(Kmr)テトラサイクリン感受性(Tcs)のコロニーが得られ、テストした24コロニー全てが挿入DNAを有していた。その平均鎖長は26.5 kbであった。もう一つの55,000クローンを含むG.オキシダンスDSM番号4025のコスミドライブラリーは、上述の方法とほぼ同様にしてEcoRIで部分消化したG.オキシダンスDSM番号4025の染色体DNAをコスミドpVK100のEcoRI部位に挿入して構築した。テストした24コロニー全てが平均鎖長27 kbの挿入断片を有していた。
【0092】
これら2種類の大腸菌 ED8767中に作成されたコスミドライブラリーを、大腸菌 ED8767ライブラリーから抽出された組換えプラスミドDNA混合物を用いて、大腸菌 S17-1[Tra+, Bio/Technology、1巻、784〜791頁、(1983)]へ移した。約4,000株の大腸菌 S17-1のKmr形質転換株を爪楊枝で拾い上げ、各々別個に50μg/mlのカナマイシンを添加した10 g/lのバクトトリプトン(ディフコ社製(Difco))、5g/lの酵母エキス(ディフコ社(Difco))、5 g/lのNaClを含むLB培地100 μlを含むマイクロタイタープレート中で37℃で培養した後、15%グリセロール存在下、-80℃で大腸菌 S17-1中のコスミドライブラリーとして保存した。
【0093】
この様に、G.オキシダンス DSM番号4025のSalI-およびEcoRI-コスミドライブラリーが大腸菌S17-1中に作成された。このライブラリーから各々1,400クローンずつが、別々に大腸菌 S17-1からP.プチダ ATCC21812へ接合伝達により次のように移された。-80℃でマイクロタイタープレート中に保存していた1,400培養物を融解し、各々の穴に100 μlの新鮮なLB培地を含むマイクロタイタープレートにプレート移し換えカートリッジ(ナンク社製(Nunc))を用いて移し、37℃で一晩培養した。ナリディキシン酸耐性(Nalr)を付与されたP.プチダ ATCC21812は、100mlの2.5%のマンニトール、0.5%の酵母エキス(ディフコラボラトリー(DifcoLaboratories))、0.3%バクトトリプトン(ディフコ(Difco))を含むMB培地中、30℃で一晩培養した。このP.プチダの培養液50 μlをコスミドライブラリーの培養液を含む1,400の穴各々に添加した。この1,400の細胞混合液を、プレート移し換えカートリッジを用いて5%フルクトース、1% 酵母エキス (ディフコ(Difco))、1%ポリペプトン(大五栄養社製)、1.8% 寒天を含むFB寒天培地表面に置いたニトロセルロースフィルター上にスポットし、27℃で一晩培養した。ナリディキシン酸は、遺伝子供与体である大腸菌から接合伝達体を選択するために用いた。フィルター上に生育した細胞は、別々に50 μg/mlのナリディキシン酸および50 μg/mlのカナマイシンを含有するMB寒天培地(以下MNK寒天平板と称する)上に移植し、接合伝達体を選択するために27℃で4日間培養した。この結果得られたコロニーを上述のようにMNK寒天平板に移植して精製した。このように1,400株のP.プチダ接合伝達体[G.オキシダンス DSM番号4025のP.プチダ中での遺伝子ライブラリー]を調製した。
【0094】
(2)G.オキシダンス DSM番号4025のAADH遺伝子クローンの免疫学的スクリーニングまず、MNK寒天平板上に維持されている350株の接合伝達体(SalIライブラリー175株、EcoRIライブラリー175株)を別々に5 mlのMNK培地を含む試験管中で培養した。その各々1.5 mlの培養液から細胞を集菌し、以下のようにウェスタンブロット解析のための処理に供した。細胞は、50 μlのLaemmli緩衝液[62.5 mM Tris-HCl(pH6.8)、10% グリセロール、5% メルカプトエタノール、2% SDS]に懸濁した。この細胞懸濁液を3分間煮沸し、その10 μlの細胞溶菌液をSDS-PAGEに供した。その結果得られた蛋白質バンドを次に40V、200 mAで16時間操作される電気ブロット装置(マリソル社製)を用いて、20% メタノールを含む2.5 mM Tris-19.2 mM グリシン緩衝液(pH8.6)中で16時間ニトロセルロースフィルターに電気的に移した。そのフィルターは、次に20 mM Tris(pH7.5)、500 mM NaClを含むTBS緩衝液中の3%ゼラチンの中で1時間処理した。このフィルターを20mM Tris(pH7.5)、500 mM NaCl、0.05% Tween 20を含むTTBS緩衝液で軽く洗浄した後、1% ゼラチンを含むTTBS緩衝液中で1/500に希釈した抗AADH抗体を含む一次抗体で1時間処理した。この抗AADH抗体は、G.オキシダンス DSM番号4025から精製したAADH蛋白質をインコンプリートアジュバントと混合し、その混合物を白兎に2週間間隔で2度インジェクトし、2度目のインジェクトの1週間後全採血し、その血清画分を抗AADH抗体として調製した。次に、このフィルターをTTBS緩衝液中で5分間ずつ2度洗浄し、1% ゼラチンを含むTTBS緩衝液中で1/3,000に希釈した二次抗体を含む二次抗体(山羊-抗兎IgG-西洋わさびパーオキシダーゼコンジュゲート)溶液で1時間処理した。そのフィルターをTTBS緩衝液中で2度洗浄し、TBS緩衝液中で1度洗浄した後、Konica Immunostaining HRP Kit IS-50B(コニカ社製)を用い、青色バンドが観察されるまで供給会社の推奨する方法に従いフィルターを発色試薬に浸漬した。実際のスクリーニングには、ウェスタンブロットの一次スクリーニングのために5つの細胞溶菌液を混合したものを1レーンにのせた。70個の混合液の中で14個の試料が陽性バンドを示した。その9つの試料が分子量約64,000の免疫反応性を有する蛋白質を有していたが、この中の2つの試料のシグナルは弱かった。別の1試料は分子量約60,000の残りの4試料は分子量約55,000の免疫反応性を有する蛋白質を有していた。
【0095】
分子量約64,000に強いシグナルを示した7個の混合試料について、その混合試料中の陽性クローンを同定するためにその個々の試料をウェスタンブロットの二次スクリーニングに供した。1混合試料あたり、1陽性クローンが同定された。得られた7クローンの有するプラスミドを各々p6E10、p16C8、p16F4、p17E8、p1E2、p24D4およびp26C3と命名した。制限酵素解析により、p6E10、p16C8、p16F4およびp17E8の4プラスミドは同じDNA領域を含み、他の3プラスミドは前者の4プラスミドとは異なる領域を含むことが明らかになった。
【0096】
(3)コロニーブロットおよびサザンブロットハイブリダイゼーションによるコスミドライブラリーからのAADH遺伝子のスクリーニング上述の免疫スクリーニングで得られた遺伝子以外のAADH遺伝子を検索するために、G.オキシダンス DSM番号4025の大腸菌 ED8767中で構築された全コスミドライブラリー(SalI-およびEcoRI-ライブラリー)を、p24D4の0.9 kb SalI断片とのコロニーおよびサザンブロットハイブリダイゼーションによってスクリーニングした。この0.9 kb SalI 断片とは、G.オキシダンス DSM番号4025から精製された天然AADH酵素の一つの内部アミノ酸配列、MetMetValThrAsnValAspValGlnMetSerThrGlu、に従って合成されたオリゴヌクレオチドプローブATGATGGT(GATC)AC(GATC)AA(TC)GT とハイブリダイゼーションしたDNA断片である。その内部アミノ酸配列は、天然AADH酵素消化後、自動ガス層シークエンサー470A(アプライドバイオシステム社製(Applied Biosystems))を用いて決定されたものである。上記コスミドライブラリーの細胞を適当に希釈し、LK寒天平板に塗抹した。生じたコロニーは、ナイロンフィルターに転写した後、32P-標識0.9 kb SalI断片とのハイブリダイゼーションにより解析した。約1%のコロニーが陽性シグナルを示した。SalIライブラリーから41株、EcoRIライブラリーから20株を選択し、それらを制限酵素解析後、サザンブロット解析に供した。このスクリーニングで以下のように6種類の異なるAADH遺伝子関連DNA領域を単離した。4種類は既に単離していた領域でp24D4、p1E2、p26C3、p17E8が有しており、2種類は新規でpSS31およびpSS53と命名された2種類の別個のプラスミドが有していた。残りのプラスミドpSS33は、p24D4およびpSS31が有している2領域の両方を有していた。
【0097】
(4)AADHクローンの免疫学的および酵素学的解析p24D4、p1E2、p26C3、pSS31およびp17E8を有するP.プチダの細胞溶菌液のウェスタンブロット解析の結果、これらの5クローンは各々分子量約64,000、62,500、62,500、60,000および62,000の蛋白質をコードすることが示された。プラスミドpSS33は、分子量約64,000および60,000の2種類の免疫反応性を有する蛋白質をコードしていた。しかしpSS53は、いかなる免疫反応性を有する蛋白質もコードしていなかった。
【0098】
各々のクローンの酵素活性(無細胞抽出液、可溶画分および膜画分)を光学分析により測定した。各々のクローンの細胞を5 mlのMB培地を含む試験管に植菌し、30℃で24時間培養した。その培養液を200 mlの新鮮なMB培地を含む500 ml容のフラスコに移し、回転フラスコ培養機上で30℃で24時間培養した。得られた細胞を6,000 x g 10分間遠心分離により集菌した後、40 mlの冷緩衝液(50 mM Tris-HCl、pH7.5、5 mM MgCl2、0.5 mM フェニルメチルスルフォニルフルオリド)で洗浄し、同緩衝液に懸濁して5 mlあたり1 g 湿重量細胞濃度の細胞懸濁液を調製した。この細胞懸濁液は、フレンチプレス細胞破砕機で二回処理(1,500 kg/cm2)し、得られたホモジネートを6,000 x g 10分間遠心分離し、細胞破片を除去した。ここで得られた無細胞抽出液(CFE)を100,000 x g 60分間遠心分離した。この上清とペレットを細胞質画分および膜画分として各々集め、以下のPMS-DCIP分析に供した。酵素反応混合液(1.0 ml)には、100 μM DCIP、1 mM PMS、50 mM Tris-リンゴ酸-NaOH緩衝液(pH8.0)、基質および酵素(10 μl)が含まれる。基質依存の600 nmでのDCIPの吸光度の減少を、コントロン社製(Kontron)の吸光度計UVIKON810を用いて25℃で測定した。表6はクローンの無細胞抽出液および可溶画分の酵素活性のレベルを示している。基質特異性によれば、各々のプラスミドにコードされている酵素は大きく三つのグループ、A、B、Cグループに分類された。AグループはL-ソルボース、D-ソルビトール、1-プロパノールの酸化を触媒し、BグループはD-グルコースおよびD-ソルビトールの酸化を触媒するが、Cグループは使用した基質に対し明瞭に検出できる活性を示さなかった。Aグループには三種類のタイプA、A'、A"(各々のプラスミドが有するDNAの物理地図によりお互い区別される)が存在していた。B、Cグループは各々染色体DNAの一領域から由来する一タイプの蛋白質のみから構成されていた。
【0099】
【表6】
【0100】
[実施例2] 塩基配列決定酵素 A、酵素 A'、酵素 A"および酵素 B遺伝子の塩基配列は、各々プラスミドp24D4、p1E2、p26C3およびpSS31を用いてM13mp18およびM13mp19(ベーリンガーマンハイム社製(Boehringer Mannheim))を用いるジデオキシチェーン終結法により行った。各々の遺伝子について一つのオープンリーディングフレーム(open reading frame、ORF)が見つかった。これら4遺伝子の塩基配列を、配列表中に配列番号:1、配列番号:2、配列番号:3、配列番号:4として示す。またそれら塩基配列から推定されるアミノ酸配列を配列番号:5、配列番号:6、配列番号:7、配列番号:8に示す。酵素 A、酵素 A'、酵素 A"および酵素 B遺伝子のORFは、各々1737、1737、1734および1737 bp長であり、23アミノ酸残基のシグナル配列を含む579、579、578および579アミノ酸残基をコードしている。酵素 A、酵素 A'、酵素A"および酵素 B間のホモロジーを表7に示す。
【0101】
【表7】
【0102】
図5は成熟型酵素 A、酵素 B酵素のアミノ酸配列を相互に比較できるように整列(アライメント)表示している。
【0103】
酵素 A、酵素 A'、酵素 A"および酵素 Bのホモロジー検索の結果、A.アセチ(T.イノウエら、J. Bacteriol.、171巻、3115〜3122頁)またはA.ポリオキソゲネス(Acetobacter polyoxogenes、T.タマキら、B. B. A.、1088巻、292〜300頁)のアルコール脱水素酵素およびパラコッカスデニトリフィカンス(Paracoccusdenitrificans、N.ハームス(Harms)ら、J. Bacteriol.、169巻、3966〜3975頁)、メチロバクテリウム オーガノフィラム(Methylobacterium organophilum、S.M.マクリン(Machlin)ら、J. Bacteriol.、170巻、4739〜4747頁)またはメチロバクテリウム エクストーケンス(Methylobacteriumextorquens、D.J.アンダーソン(Anderson)ら、Gene、90巻、171〜176頁)のメタノール脱水素酵素を含む幾つかのキノ蛋白質(quino-proteins)とは、むしろ低いホモロジー(いずれのポリペプチド間においても26〜31%の範囲のホモロジー)を示した。
【0104】
[実施例3] AADH遺伝子のサブクローニング酵素 A遺伝子は、最初pVK100のEcoRI切断部位に約25 kbの挿入断片を有するコスミドクローンp24D4としてクローン化された。次にこの酵素 A遺伝子を、酵素A遺伝子カセットとして使用するためにサブクローン化した。酵素 A遺伝子のORFとその上流下流に約500bpの非コード領域を有する2.7 kbのEcoRV断片を、p24D4からM13mp18中に単離された3.4 kb NruI断片から切り出しpUC18のHindIII切断部位にHindIIIリンカー (CAAGCTTG)を用いて結合した。その結果得られたプラスミドをpSSA202と命名した。次に、酵素 A遺伝子カセット(2.7kb HindIII画分)をpVK102のHindIII切断部位に挿入してpSSA102Rを得た。このプラスミドpSSA102Rを実施例1-(1)に述べる接合伝達方法によりナリディキシン酸耐性のP.プチダ[ATCC21812]に導入した。このpSSA102Rを有するP.プチダの接合伝達体は、50 μg/ml ナリディキシン酸および10 μg/ml テトラサイクリンを含有するMB寒天培地(MNT寒天培地)上で選択され、ミニ休止菌体反応に供された。20 g/lのL-ソルボース、3 g/lのNaCl、10 g/lのCaCO3および爪楊枝でMNT寒天培地から集菌した細胞を含む反応混合液(100 μl)を室温で24時間、静かに振とうさせた。この反応混合液をTLCで分析して、その生産物が2KGAであると同定した。一方宿主であるナリディキシン酸耐性のP.プチダ[ATCC21812]を用いた同様の休止菌体反応では、2KGAは観察されなかった。
【0105】
酵素 B遺伝子は、最初pVK102のSalI切断部位に約30 kbの挿入断片を有するコスミドクローンpSS31としてクローン化された。この酵素 B遺伝子を6.5 kbのBglII 断片としてpVK101(ATCC37157)のBglII切断部位にサブクローン化してpSSB102を得た。次に酵素 B遺伝子をカセットとして使用するためにさらにサブクローン化した。まず6.5 kbのBglII断片をpUC18のBamHI切断部位にクローン化し、pSSB202を得た。次にpSSB202から2.3 kb XhoII断片を切り出した。この2.3 kb XhoII断片は、酵素 BのORFの他に、120bpの5'-非コード領域および500 bpの3'-非コード領域を含む。この2.3 kb XhoII断片をクレノウフラグメント(Klenowfragment)での処理により接着末端を平滑化し、HindIIIリンカーを用いてpUC18のHindIII切断部位にクローン化してpSSB203を得た。この酵素 B遺伝子カセット(2.3 kbHindIII断片)をpVK102のHindIII切断部位に挿入してpSSB103Rを作成した。このプラスミドpSSB103Rを接合伝達方法によりナリディキシン酸耐性のP.プチダ[ATCC21812]に導入した。このpSSB103Rを有するP.プチダの接合伝達体は、MNT寒天培地上で選択され、ミニ休止菌体反応に供された。このpSSB103Rを有するP.プチダの接合伝達体は休止菌体反応において酵素 B活性(D-ソルビトールからL-ソルボースを生成)を示した。酵素 B遺伝子カセットの塩基配列を決定したところ、上記XhoII断片は、XhoII-XhoII断片ではなくてXhoII-XhoI断片であることが判明した。おそらくXhoIがXhoII酵素標品に混入していたものと思われた。
【0106】
酵素 A'および酵素 A"遺伝子は、最初pVK102のSalI切断部位に約30 kbの挿入断片を持つp1E2およびp26C3のコスミドクローンとして得られ、更に基本的に上述のようにサブクローン化をおこなった。3.5 kb XhoII断片中の酵素 A'遺伝子をpVK102のBglII切断部位にサブクローン化しpSSA'101Rを得た。2.7 kb EcoRV断片中の酵素 A"遺伝子は、まずM13mp19にサブクローン化した後pVK102のHindIIIおよびBglII切断部位の間に再サブクローン化しpSSA"102を得た。
【0107】
[実施例4] P.プチダ接合伝達体からのAADHの単離と性質(1) 微生物の培養酵素 A、A'、A"およびBの遺伝子を含むpVK102コスミドベクター(それぞれpSSA102R、p1E2、p26C3およびpSSB103R)を持つP.プチダ[ATCC21812]を抗生物質存在下MB培地で培養した。抗生物質は以下に示すように添加した。pSSA102R(酵素 A)およびpSSB103R(酵素 B)に対しては5 μg/mlテトラサイクリンを、p1E2(酵素 A')およびp26C3(酵素 A")に対しては25 μg/ml カナマイシンを添加した。各々の抗生物質を含んだMB寒天平板培地より、細胞を5 mlの各々の抗生物質を含んだMB培地を含んだ10本の試験管に植菌し30℃で振とう培養した。2日培養後、細胞を100 mlの同培地を含む500 ml エーレンマイヤーフラスコ10本に移植し30℃で振とう培養した。1日培養後、以上の種培養を合わせて18Lの培地を含んだ30 L ジャーファーメンター(丸菱製)に移植し攪拌回転数300rpm、通気量1.0 vvm、30℃で18時間培養した。細胞を6,000 x g、10分の遠心操作で回収し、5 mM CaCl2、1 mM MgCl2、0.2 M NaCl、2.5% スクロース及び0.5 mM PMSFを含む1.5 Lの25 mM Tris-HCl緩衝液(pH 7.5)で1度洗浄し使用時まで-20℃で保存した。結果として湿重量で約150 gの菌体を得た。
【0108】
(2)クローン化された酵素 A、A'、A"およびBの精製酵素 A、A'、A"およびBの精製はほぼ同様のスケールで同一の方法で行った。すべての操作は特に記載のない場合4〜10℃で行った。酵素 A、A'、A"およびBの酵素活性の測定は精製段階を通してそれぞれ、L-ソルボース、n-プロパノール、n-プロパノールおよびD-グルコースを基質として、実施例1に述べられた分光学的測定法により行った。細胞(8〜10g全蛋白質を含む湿重量で約100gの菌体)を融解し、約200mlの25 mMTris-HCl緩衝液(pH8.0)中に懸濁し、フレンチプレス(1500 kg/cm2)を2回通過させることにより破砕した。DNAによる溶液の粘性を軽減するため、DNAase及びMgCl2をおのおの0.01 mg/ml及び1 mMの終濃度で溶液に添加した。細胞残骸は6,000 x g、10分の遠心操作で除去した。得られた上清を25 mM Tris-HCl緩衝液(pH 8.0)で240 ml容に調整し、不溶性膜画分を除去するため100,000 x g、90分間遠心処理した。可溶性上清をTris緩衝液で240 ml容に調整し、ピロロキノリンキノン(PQQ)及びCaCl2をそれぞれ終濃度12.5 μM及び5 mMで添加した後、溶液を室温下、15分間激しく攪拌した。上記のように調整された可溶性画分を硫安により分画した。硫安飽和35〜60%画分を沈殿させ100 mlの5 mM CaCl2及び5% スクロースを含む25 mM Tris-HCl緩衝液(pH 8.0)で再懸濁した後、PQQを再び12.5 μM終濃度添加した。酵素溶液を1000 mlの同緩衝液(PQQ非添加)で終夜透析した。固形ポリエチレングリコール#6000を透析終了液に緩やかな攪拌下徐々に添加した。30分間攪拌後、沈殿物を10,000 x g、20分間の遠心操作で除去し、上清を上記の同緩衝液で200ml容に調整した。
【0109】
上記のように調製した酵素溶液を以下の三つのクロマトグラフィー操作で精製した。
【0110】
第1段階:DEAE-トヨパール 650M粗酵素液を5 mM CaCl2および5% スクロースを含む25 mM Tris-HCl緩衝液(pH 8.0)で平衡化したDEAE-トヨパール 650Mカラム(2.5 x 40 cm)に供した。カラムを400 mlの同緩衝液で洗浄後、酵素の溶出を2000 ml同緩衝液中の0〜0.5M NaCl直線濃度勾配を用い流速150 ml/時で行った。酵素活性画分を回収しNaClを含まない同緩衝液で2倍容に希釈した。
【0111】
第2段階:Q-セファロース(ファーストフロー)
酵素溶液をNaClを含まない同緩衝液で平衡化したQ-セファロース(ファーストフロー)カラム(1.5 x 20 cm)に供した。カラムを200 mlの0.2 M NaClを含む同緩衝液で洗浄後、酵素の溶出を600 ml同緩衝液中の0.2〜0.6 M NaCl直線濃度勾配を用い流速50 ml/時で行った。酵素活性画分を回収し窒素ガス下、限界濾過膜(アミコン社製、PM-30)を用い2.5 ml容に濃縮した。
【0112】
第3段階:セファクリルS-300HR(ゲル濾過)
濃縮された酵素を5 mM CaCl2、5% スクロースおよび0.2 M NaClを含む25 mM HEPES緩衝液(pH 7.5)で平衡化したセファクリルS-300HR(2.5 x 100 cm)でゲル濾過分画した。カラムは同緩衝液で流速20 ml/時で展開した。酵素活性画分を回収し上記と同様の限界濾過膜を用い1 ml以下に濃縮した後、-80℃で保存した。HEPES緩衝液中濃縮された酵素は-80℃下少なくとも2ヵ月は安定であった。
【0113】
結果として、26.0 mgの酵素 A、0.35mgの酵素 A'、0.41 mgの酵素 A"および5.0 mgの酵素 Bを得た。
【0114】
(3)酵素 A、A'、A"およびBの性質a) 分子量およびサブユニット酵素 A、A'、A"およびBは同一条件下のセファクリルS-300HRによるゲル濾過において同一位置に溶出された。酵素の分子量は分子量標準蛋白質(SDS-PAGE スタンダード、低分子用、バイオラド社製(Bio-Rad))との比較により約135,000と推定された。酵素 A、A'、A"およびBはSDS-PAGE分析においてそれぞれ64,000、62,500、62,500および60,000の分子量を有する均一な単一バンドを示した。酵素A、A'、A"およびBのバンド全ては抗-AADHウサギ血清を用いたウェスタンブロッティング分析により検出された。従って、全酵素は2つの同一サブユニットを含むホモ二量体であると結論づけられた。
【0115】
b) N-末端アミノ酸配列とアミノ酸組成成熟酵素 A、A"およびBのN-末端アミノ酸配列をエドマン法[Acta Chem.Scand.、4巻、283〜293頁 (1950年)]による自動気相シーケンサー(470A,アプライドバイオシステム社製(AppliedBiosystems))を用いて分析した。酵素 A'のN-末端は標品の純度不足のため決定されていない。以下に結果を示す。
酵素 A :Gln-Val-Thr-Pro-Val-Thr----酵素 A" :ブロック化されたN-末端残基酵素 B :Gln-Val-Thr-Pro-Ile-Thr-Asp-Glu-Leu-Leu-Ala----決定された酵素 A及びBの配列は配列番号:5と配列番号:8に記載された塩基配列より推定される配列(24番目の残基より始まる)と同一であった。この結果は酵素の初めの23残基がシグナル配列であることを示す。酵素 A及びBとの類似性から酵素 A'およびA"の初めの23残基もシグナル配列であると推定される。
【0116】
酵素 Aのアミノ酸組成を決定した。蛋白質は6 N 塩酸で110℃、24時間または4M メタンスルホン酸(過ギ酸酸化後)で115℃、24時間処理により加水分解した。アミノ酸分析はコントロン社製(Kontron)のアミノ酸分析機(ニンヒドリン型)で行った。分析結果を酵素 AのDNA配列より推定されるアミノ酸組成と比較した。結果は精製された酵素 Aが明確に酵素 A遺伝子の産物であることを示した。
【0117】
c) 基質特異性酵素 A、A'、A"およびBを上述の8つの基質、n-プロパノール、イソプロパノール、D-グルコース、D-ソルビトール、L-ソルボソン、D-マンニトール、L-ソルボース及びD-フルクトースを用いたPMS-DCIP分析におけるそれらの基質特異性で性格づけした。結果を表1に示す。
【0118】
d) 物理化学的性質酵素 A(L-ソルボース脱水素酵素活性により)、酵素 A'(n-プロパノール脱水素酵素活性により)、酵素A"(L-ソルボース脱水素酵素活性により)および酵素 B(D-ソルビトール脱水素酵素活性により)の物理化学試験、至適pH、pH安定性および温度安定性をPMS-DCIP分析により行った。
【0119】
表2は酵素の至適pHの結果を要約したものである。酵素活性は様々なpHの緩衝液を用いてPMS-DCIP分光学法分析により測定した。緩衝液は50 mM Tris-リンゴ酸-NaOH緩衝液をpH 6.0、6.5、7.0、7.5、8.0および8.5用に、50 mM Glycine-NaOH緩衝液をpH 9.0および9.5用に用いた。各pH 6.0、6.5、7.0、7.5、8.0、8.5、9.0および9.5におけるDCIPの分子吸光係数はそれぞれ10.8、13.2、14.5、14.9、15.0、15.1、15.1および15.1とした。すべての酵素はその至適点をpH 8.0〜8.5に示した。酵素 A"とBは酵素 AとA'に比較して低pH側に比較的広い至適pH範囲を有していた。
【0120】
表3は酵素のpH安定性の結果を示す。酵素(約0.01 mg/ml)を5% スクロース、0.2 M NaClおよび5 mM CaCl2を含む50 mM 各緩衝液中で3時間インキュベートしPMS-DCIP分光学法分析により測定した。緩衝液はpH 4と5の酢酸ソーダ、pH 6、7および8のTris-リンゴ酸-NaOHおよびpH 9と10のGlycine-NaOH各緩衝液を用いた。表中の値はpH 8.0でインキュベートしなかった値に対する相対活性として表示した。各酵素に対して基質として酵素 A及びA"には125 mM L-ソルボース、酵素 A'には50 mM n-プロパノール、酵素 Bには125 mM D-ソルビトールを用いた。酵素 A、A'、A"およびBのpH-安定性はほぼ同様で、pH 6から9の範囲で安定であった。
【0121】
表4は酵素の温度安定性試験の結果を示す。5% スクロース、0.2 M NaCl及び5mM CaCl2を含む25 mM HEPES緩衝液(pH7.5)中で酵素(約0.05 mg/ml)を表に示した温度(4〜60℃)で5分間インキュベート後、氷冷しPMS-DCIP分光学法分析により測定した。残存活性を4℃でインキュベートした値に対する相対活性で表示した。各酵素に対する基質として酵素 AおよびA"には125 mM L-ソルボース、酵素 A'には50 mM n-プロパノール、酵素 Bには125 mM D-ソルビトールを用いた。40℃、5分間処理の酵素の場合、残存活性は酵素 Aで20%、酵素 A'、A"およびBで70〜85%であった。
【0122】
e) 阻害剤5% スクロースを含む25 mM HEPES緩衝液(pH7.5)中で酵素(約0.05 mg/ml)を金属または阻害剤とともに30分間、25℃でインキュベートした。残存活性を実施例1に記載したPMS-DCIP分光学法分析で測定した。残存活性は対照に対する相対活性で表わしている。酵素に対する金属イオンの影響を表5に記載した。MgCl2とCaCl2はほぼ酵素に対して無影響であったが、他の金属イオン、特にCuCl2、は強い影響を及ぼした。酵素に対する阻害剤の影響も表5に記載した。EGTAとEDTAは酵素 A、A'及びBを強く阻害した。しかし、酵素 Bに対するEDTA及びEGTAによる阻害はAグループ酵素に対する阻害よりも軽度であった。
【0123】
[実施例5] 大腸菌中での酵素 Bの効率的生産酵素 Bのシグナルペプチド領域を大腸菌のマルトース結合蛋白質(malE)のシグナルペプチドと以下のように置換した。2種のオリゴヌクレオチド(配列番号:9および配列番号:10)をアプライドバイオシステム社製(Applied Biosystems)の381A DNA合成機を用いて合成し、アニール化しアミノ酸配列(配列番号:11)、MetLysIleLysThrGlyAlaArgIleLeuAlaLeuSerAlaLeuThrThrMetMetPheSerAlaSerAlaLeuAla(Gln)をコードする2本鎖DNA断片を生成させ、この2本鎖DNA断片をT4ポリヌクレオチドキナーゼで処理した [J. Biol.Chem.、259巻、10606〜10613頁 (1984)]。pSSB203(実施例3 参照)を制限酵素SphIで切断後、T4DNAポリメラーゼで処理しBstPIで切断した。得られた元来のシグナル配列および成熟酵素Bの一番めのアミノ酸残基(Gln)をコードする領域のない酵素 B遺伝子を有する1.72 kb DNA断片を、アガロースゲル電気泳動を行った後アガロースゲルから単離した。大腸菌の発現ベクター、pTrc99A(ファーマシア社製(Pharmacia))を制限酵素NcoI(ATG開始コドンの位置)およびSmaIで切断し、上記2DNA断片と結合した。この結果得られたプラスミドをpTrcMal-EnzBと命名し、大腸菌 JM109を形質転換するのに用いた。得られた形質転換体を、100μg/mlのアンピシリンを添加したLB600 mlを含む2 L容のフラスコ2本中で28℃で生育させ、細胞濃度が約1.5 OD600に達した時に、IPTGを0.1mMになるよう添加し、さらに3〜4時間培養を続けた。得られた細胞は、25℃、10分間の遠心分離(4,000 x g)により集菌し、25℃で500 mlの20%シュークロースを含む30 mMTris-HCl(pH8.0)緩衝液に懸濁した。この細胞懸濁液に終濃度1 mMのEDTAを添加し、25℃でゆっくり振とうした後、細胞を4℃で15分間遠心分離(8,000 x g)にかけた。得た細胞を500 mlの氷冷した5 mM MgSO4に再懸濁し、4℃で5分間ゆっくり振とうした。この細胞懸濁液を4℃で10分間遠心分離(8,000 x g)にかけ、その上清を冷浸透圧ショック抽出液として得た。この抽出液には、SDS-PAGE分析で50〜60%以上の純度を有する酵素 B蛋白質(分子量60,000)が含まれていた。この上清にまず終濃度20 mMになるようにTris-HCl(pH8.0)を添加し、終濃度10 mM EDTAを添加後25℃10分間インキュベートし、次に終濃度20 mMのCaCl2を添加後10分間インキュベートし、最後に終濃度25 μMのPQQを添加後10分間インキュベートした。この上清に酵素の安定化のため終濃度20 mMのα-メチル-D-グルコシド(競合阻害剤)を加えた。酵素 Bはつぎの2種類のクロマトグラフィーにより完全精製された。まず上記上清を1 mM CaCl2および20 mM α-メチル-D-グルコシドを含む20 mM Tris-HCl(pH8.0)緩衝液で平衡化したQ-セファロースカラム(1.6 x 12 cm)上に供し、酵素 Bを600mlの0〜0.4 M NaCl直線勾配を持つ同緩衝液で溶出した。約0.25 MNaClの濃度で溶出された赤色蛋白質ピークを集め、Centricon-30(アミコン社製(Amicon))により約0.5 mlにまで濃縮した。最終的に、酵素 Bを0.2M NaCl、1 mMCaCl2、および20 mM α-メチル-D-グルコシドを含む20 mM HEPES(pH7.8)緩衝液を用い、セファクリルS-300HRカラムを通過させた。分子量135,000ダルトンの付近に溶出される赤色蛋白質ピークを最終精製酵素 Bとして集めた。結果として約8mgの精製酵素 Bを1.2リッターの大腸菌培養液から得た。
【0124】
[実施例6] 宿主-ベクター系G.オキシダンス[DSM番号4025]のための宿主-ベクター系を広宿主域コスミドpVK102を使用する接合伝達系を用いて確立した。最初は、ナリディキシン酸耐性を有するG.オキシダンス[DSM番号4025]から唯一株の接合伝達体が分離された。新たな宿主GOS2は、上記pVK102を有するG.オキシダンス[DSM番号4025]の接合伝達体からpVK102を除去することにより単離された。二番目の宿主GOS2Rは、DNA供与体である大腸菌から接合伝達体を簡便に選択できるようにリファンピシン(100μg/ml)耐性をGOS2に付与して誘導した。GOS2RへのプラスミドpVK102の導入頻度は、受容菌あたり10-3〜10-4個の接合伝達体が得られるものであった。しかし、GOS2Rの2KGA生産性はG.オキシダンスDSM番号4025より約10%低かった。三番目の宿主GORS6-35は、G.オキシダンス DSM番号4025からリファンピシン耐性、高2KGA生産性、比較的高い遺伝子導入適格性を有する株を接合伝達、プラスミド除去、2KGA醗酵を含む一連の実験を通して選択することにより得られた。
【0125】
(1)GOS2の単離G.オキシダンス[DSM番号4025]にナリディキシン酸耐性を以下のように付与した。G.オキシダンス[DSM番号4025]の細胞をTrypticase Soy Broth(BBL、BectonDickinson Microbiology Systems 社製)(以下Tと略す)寒天培地に50 μg/mlのナリディキシン酸を含む寒天培地(以下TNと略す)上に塗抹し、この塗抹平板を27℃で5日間培養した。その結果得られたコロニーを再度同上TN寒天培地に塗抹し、ナリディキシン酸耐性G.オキシダンス[DSM番号4025]、GONを得た。広宿主域コスミドpVK102(Kmr、Tcr)を、pVK102を保持する大腸菌からGONへ以下のように三者接合伝達により導入した。ヘルパー株であるpRK2013を保持する大腸菌およびpVK102を保持するDNA供与体の大腸菌は、50 μg/mlのカナマイシンを添加したLB培地中で37℃一晩培養した。これらの培養液をカナマイシンを添加した新鮮なLB培地に移植し、さらに5〜6時間培養した。受容菌GONは、TN液体培地中で30℃一晩培養した。大腸菌およびGON株は別々に遠心分離し、各々等量でもとの十分の一量のT培地に再懸濁した。各々の細胞懸濁液100 μlを混合し、その混合液の30μlをNS2寒天平板の表面上に置いたニトロセルロースフィルター上にスポットした。接合伝達体の選択は、50 μg/mlのナリディキシン酸および50 μg/mlのカナマイシンを含むT寒天培地(TNK寒天培地)上で行われた。数個のコロニーが選択平板上で得られたが、同時に多くの大腸菌自然変異体(Nalr、Kmr)も出現した。これら接合伝達体の候補株からプラスミドおよび染色体DNAを調製し、コントロールのpVK102とG.オキシダンス[DSM番号4025]の染色体DNAと、制限酵素解析およびサザンブロットハイブリダイゼーションにより比較した。その結果、一株のpVK102を有するG.オキシダンス[DSM番号4025]、GON8-1の分離を確認した。GON8-1から調製されたプラスミドDNAはpVK102DNAと同一であり、大腸菌中で複製できた。またGON8-1の染色体DNAは、G.オキシダンスDSM番号4025の染色体と同一であった。
【0126】
高い接合伝達頻度を有する宿主として使用できる株を分離するために、上記接合伝達体GON8-1からプラスミドpVK102を以下のように除去した。GON8-1を抗生物質非存在下、Tブロス中で30℃で2日間培養し、その培養液を新鮮なTブロスに2%の割合で移植した。このような培養を3回繰り返し得られた細胞をT寒天平板に塗抹し、27℃で4日間培養した。得られたコロニーをKms株を分離するためにTNKおよびTN寒天平板上に拾い上げた。このKms株の一株はGOS2と命名され、サザンブロットハイブリダイゼーションによりpVK102のいかなる領域をも含まないことが確認された。次に、pVK102を接合伝達によりGOS2に導入した。この株はG.オキシダンス[DSM番号4025]よりも102〜103高い遺伝子導入適格性 (すなわち10-5〜10-6接合伝達体/受容体)を示した。
【0127】
(2)GOS2のリファンピシン耐性変異株、GOS2Rの単離GOS2細胞を20〜100μg/mlのリファンピシンを含むT寒天平板上を繰り返し移植することで、GOS2からのリファンピシン耐性(Rifr)変異株を単離した。得たRifr株の一つをGOS2Rと命名した。GOS2R株は非常に高い遺伝子導入適格性を示し、TRK寒天(100 μg/mlのリファンピシンおよび50 μg/mlのカナマイシンを含むT寒天培地)およびTRT寒天(100 μg/mlのリファンピシンおよび3 μg/mlのテトラサイクリンを含むT寒天培地)上でそれぞれ受容菌あたり各々10-2〜10-3および10-4の接合伝達体がGOS2R株から得られた。
【0128】
GOS2RのL-ソルボースからの2KGA生産性を、G.オキシダンス[DSM番号4025]の2KGA生産性と以下のように比較した。NS2寒天培地上で維持されている細胞を5 mlの種培養培地[8% L-ソルボース(別滅菌)、0.05%グリセロール、0.25 %MgSO4・7H2O、1.75% コーンスティープリカー(corn steep liquor)、5.0% パン酵母、1.5% CaCO3および0.5% 尿素(別滅菌)(滅菌前pH7.0)]に植菌し、30℃で24時間培養した。得られた種培養液5 mlを50 mlの生産培地PMS10[10% L-ソルボース(別滅菌)、0.05% グリセロール、0.25 % MgSO4・7H2O、3% コーンスティープリカー、6.25% パン酵母、1.5% CaCO3および1.6% 尿素(別滅菌)(滅菌前pH7.5)]を含む500mlのエルレンマイヤーフラスコに植菌し、30℃で4日間振とう培養(180 rpm)した。2KGAの定量は、高速液体クロマトグラフィー分析により行った。GOS2RおよびG.オキシダンス[DSM番号4025]は、各々87.3および97.3 g/lの2KGAを生産した。
【0129】
(3)2KGA高生産性を有するGORS6-35の単離L-ソルボースからの2KGA生産性が、G.オキシダンス[DSM番号4025]と同じである株中で、AADH遺伝子のセルフクローニングの評価を行うために、新たな宿主を以下のように作成した。すなわち、(1)200 μg/mlのリファンピシンに対する耐性を付与する、(2)pVK102を導入後、除去する、(3)L-ソルボースからの2KGA高生産株を選択することににより作成した。このようにして得られたGORS6-35は次の二つの性質:(1)親株のG.オキシダンス[DSM番号4025]とほぼ同等の2KGA生産性(10% L-ソルボースから約100 g/lの2KGA生産)および(2)受容菌あたり10-6〜10-7の接合伝達体を与える遺伝子導入適格性を示した。
【0130】
[実施例7] プロモーター置換酵素 B遺伝子の構築G.オキシダンスDSM番号4025株の無細胞抽出液をSDS-ポリアクリルアミドゲル電気泳動に供し、そのゲルをクマシーブリリアントブルー(Coomassie BrilliantBlue)R-250で染色した結果、酵素 Aは上記菌株細胞内において発現量の最も高い蛋白質の一つであることが判明したので、酵素 A遺伝子のプロモーター(PA)はG.オキシダンス DSM番号4025株の中で強力なものと思われる。このPAおよび、G.オキシダンス DSM番号4025株においてカナマイシン耐性を示しうるTn5のカナマイシン耐性遺伝子のプロモーター(PTn5)を、図10に示すように酵素 B遺伝子のSD配列を含む構造遺伝子に連結した。
【0131】
酵素 B遺伝子を含む2.3 kb HindIII断片をM13 mp18に挿入し、得られたファージDNAをT7-GEN(登録商標)インビトロ変異誘発キット(In vitro Mutagenesis Kit(東洋紡社製))を用いて、供給会社の推奨する方法に従って部位特異的変異導入を行った(図9)。酵素 B遺伝子の上流に、酵素 Bのプロモーターの代わりに種々のプロモーターを導入するために、SD配列の上流にBamHI部位を作成した。変異プライマーGTTAGCGCGGTGGATCCCCATTGGAGG(BamHI部位を含む27塩基、配列番号:12)は、アプライドバイオシステム社製(Applied Biosystems)の381A DNA合成機を用いて合成した。結果として得られるBamHI-HindIII断片は、酵素 Bのプロモーター(PB)を含まず、酵素 BのSDおよび構造遺伝子を有している。
【0132】
酵素 A遺伝子のプロモーター(PA)は、HindIIIおよびBamHI部位を付加したプライマーを用いたPCR法によりクローン化した。PCR反応は、GeneAmp(登録商標)DNA増幅試薬キット(DNA Amplification Reagent Kit(宝酒造社製))を用い、サーマルサイクラー、Zymoreactor II(アトー社製)により行った。反応条件は、酵素添加前処理(94℃、5分)、30サイクルの変性段階(94℃、1分)、会合段階(60℃、1分)、合成段階(72℃、1分)に、後処理(72℃、5分)で行った。鋳型のDNAは、プラスミドpSSA202(pUC18に酵素 A遺伝子を含む2.7 kbのHindIII断片をクローン化したもの)を用いた。反応液の組成は、添付のバファーに対して、200μMのdNTP等モル混液、1 μMの各プライマー、1 ngの鋳型DNAおよび2.5単位のAmpliTaq(登録商標)DNAポリメラーゼとした。結果としてSD配列の上流の300 bpの断片が増幅され、この断片をpUC18のHindIIIとBamHI 部位の間にクローン化し配列に利用した。得られたクローンについては、PCRのミスインコーポレーションによる変異が導入されていないことを塩基配列決定により確認した。
【0133】
カナマイシン耐性遺伝子のプロモーター(PTn5)は、プラスミドpNeo (ファーマシア社製(Pharmacia))からまずHindIII-PstI断片として単離し、pUC18のマルチクローニング部位にクローン化し、PTn5をHindIII-BamHI断片として切り出した。
【0134】
PAおよびPTn5プロモーターを含むHindIII-BamHI断片を、PBプロモーターを除いた酵素 Bの構造遺伝子を含むBamHI-HindIII 断片とともにpUC18のHindIII部位に導入した。得られたプラスミドより調製したHindIII断片をpVK100にクローン化し、pSSAp-BおよびpSSPTn5-Bを作成し、実施例6記載の接合伝達法によってGOS2Rに導入した。
【0135】
[実施例8] フラスコ培養でのGOS2R接合伝達株による2KGA生産(1)酵素 A遺伝子増幅接合伝達体の単独菌フラスコ培養におけるL-ソルボースからの2KGA生産酵素 A発現プラスミドpSSA102RおよびベクタープラスミドpVK102を実施例6記載の接合伝達法によりGOS2Rへ導入した。得られた接合伝達体は30 μg/mlのテトラサイクリンを含むNS2寒天平板培地で維持し、L-ソルボースからの2KGA発酵に供した。接合伝達体細胞を実施例6に示すように種培地5mlに接種し、30℃で24時間培養した。得られた種培養液5mlを、実施例6記載のPMS10生産培地またはPMS12生産培地[12 % L-ソルボース(別滅菌)、0.05% グリセロール、0.25% MgSO4・7H2O、3% コーンスティープリカー(CSL)、10% パン酵母、1.5% CaCO3および2% 尿素(別滅菌)(pHは滅菌前に7.5に調整)]50 mlの入った500 ml-エルレンマイヤーフラスコに接種した。培養は、30℃、180rpmで振とうし、4日ないし5日行った。結果として、GOS2R(pSSA102R)およびGOS2R(pVK102)は、10 %のL-ソルボースからは、4日間でそれぞれ92.2、89.1 g/lの2KGAを生産し、12 %のL-ソルボースからは、5日間でそれぞれ105.7、99.9 g/lの2KGAを生産した。
【0136】
(2)GOS2R(pSSB103R)の単独菌フラスコ培養におけるD-ソルビトールからの2KGA生産酵素 B発現プラスミドpSSB103RおよびベクタープラスミドpVK102を実施例6記載の接合伝達法によりGOS2Rに導入した。得られた接合伝達体は30 μg/mlのテトラサイクリンを含むNS2寒天平板培地で維持し、D-ソルビトールからの2KGA発酵に供した。接合伝達体細胞を8% D-ソルビトール、0.05% グリセロール、0.25% MgSO4・7H2O、1.75% コーンスティープリカー(CSL)、5.0% パン酵母、1.5% CaCO3および0.5% 尿素(別滅菌)(pHは滅菌前に7.0に調整)を含む種培地5 mlに接種し、30℃で24時間培養した。得られた種培養液5 mlを、表8に示す3種の生産培地50 mlの入った500 ml-エルレンマイヤーフラスコに接種し、3日間30℃で振とう培養した(180rpm)。結果として、GOS2R(pSSB103R)は、およそ61.5、71.5および73.0 g/lの2KGAを8%、10%および12%のD-ソルビトールからそれぞれ生産した。一方GOS2R(pVK102)は、それぞれ19.5、25.4および30.2 g/lの2KGAを生産した。
【0137】
【表8】
【0138】
滅菌前pH 7.5に調整*:別滅菌。
【0139】
(3)GOS2R(pSSAp-B)およびGOS2R(pSSPTn5-B)単独菌フラスコ培養におけるD-ソルビトールからの2KGA生産GOS2R(pSSAp-B)、GOS2R(pSSPTn5-B)、GOS2R(pSSB103R)およびGOS2R(pVK100)の細胞を、実施例8の(2)に示したように、エルレンマイヤーフラスコでPMSL10生産培地を用いて30℃で3日間培養した。生成した2KGA量は、表9に示すとおりである。
【0140】
【表9】
【0141】
[実施例9] 単独菌による3Lジャー培養でのD-ソルビトールからの2KGA生産GOS2R(pSSB103R)による単独菌発酵実施例8の(2)に示したように試験管で調製した種培養液5mlずつを、同じ種培地50 mlの入った500 ml-エルレンマイヤーフラスコ4本に接種し、30℃で24時間振とう培養した(180rpm)。得られた種培養液200 mlを、3 mlの消泡剤を添加したPMSL10生産培地1800 mlを含む3Lジャーファーメンターに接種した。ファーメンターは、30℃、700 rpm、0.5 vvmで操作した。D-ソルビトールは以下の2通りの方法で流加した。(1)24時間目から30時間目までの6時間で、50% D-ソルビトール液200 mlを流加、または(2)24時間目から32.3時間目までの8.3時間で、50% D-ソルビトール液280 mlを流加。結果として、(1)および(2)の方法の流加培養でそれぞれ、99.0、103.4 g/lの2KGAを51時間で生産した。
【0142】
[実施例10] 酵素 B遺伝子増幅GOS2Rと大腸菌とのフラスコでの混合培養によるD-ソルビトールからの2KGA生産(1)B.メガテリウム、大腸菌、P.プチダとの混合培養系発酵生育因子供給体として、B.メガテリウム[DSM番号4026]、大腸菌 HB101およびP.プチダ[ATCC21812]を、0.3% 酵母エキス(Difco)、0.3% 肉エキス(極東製薬)、3% コーンスティープリカー、1% ポリペプトン(極東)、0.1% 尿素、0.1%KH2PO4、0.02% MgSO4・7H2O、2% L-ソルボース、0.1% CaCO3(滅菌前にpH7.1に調整)を含む種培養培地150 mlで、24時間それぞれ37、37、30℃で培養した。GOS2R(pSSB103R)株は、実施例8の(2)に示すように、5 mlの種培養培地を入れた2本の試験管で24時間30℃で培養した。GOS2R(pSSB103R)の種培養液4 mlと生育因子供給菌の種培養液3.5 mlを、8% D-ソルビトール、0.01% MgSO4o7H2O、1% コーンスティープリカー、0.1% KH2PO4、0.6% CaCO3、1.5%尿素(別滅菌)および消泡剤(フラスコあたり1滴)(滅菌前にpH7.0に調整)を含む混合培養用生産培地50 mlを含む500 ml-エルレンマイヤーフラスコに接種し、30℃で46.5時間振とう培養した。結果として、B.メガテリウムDSM番号4026、大腸菌 HB101およびP.プチダ[ATCC21812]との混合培養において、それぞれ49.9、54.1、31.3 g/lの2KGAを生産した。
【0143】
(2)GOS2R(pSSAp-B)と大腸菌とのフラスコでの混合培養系発酵GOS2R(pSSAp-B)と大腸菌との混合培養を、大腸菌の種培養培地における2% L-ソルボースを2% D-ソルビトールに変えた以外は上記の方法と同じ様式で行った。結果として、GOS2R(pSSAp-B)は10 %のD-ソルビトールから73.7 g/lの2KGAを48.5時間で生産した。
【0144】
[実施例11] 組換え体のAADHによる2KGAの生産1.7 mg/mlの精製酵素 A(実施例4によって精製)、50 mM Tris-HCl、pH7.5、5mM CaCl2、8 mg/ml ウシ血清アルブミン(BSA)、1 mM PMS、20 μg/ml PQQおよび4% L-ソルボースを含む反応液を、20時間ゆるやかに振とうしながら30℃でインキュベートしたところ、約2 g/lの2KGA(TLCで定量)が生成した。
【0145】
また、各々2.4 mg/mlの精製酵素 Aおよび酵素 B(実施例4によって精製)、50mMTris-HCl (pH7.5)、5 mM CaCl2、8 mg/ml BSA、1 mM PMS、20 μg/ml PQQおよび2 % D-ソルビトールを含む反応液を、20時間ゆるやかに振とうしながら30℃でインキュベートしたところ、0.25 g/lの2KGA(HPLCで定量)および約5 g/lのL-ソルボース(TLCで定量)が生成した。
【0146】
[実施例12] アルコールからアルデヒド、アルコールまたはカルボン酸からケトン、アルデヒドからカルボン酸の生産様々な基質を用いて、精製酵素 Aまたは酵素 Bによる酵素反応を実施例11に示したように行った。生産物はTLCおよび/またはHPLCにより同定し、結果を表10に示した。
【0147】
【表10】
【0148】
酵素 AはD-フルクトースを2KDに変換したことから、D-フルクトースからの中間体としてD-グルコソンを生成していると考えられる。
【0149】
[実施例13] P.プチダの接合伝達体による2KGAおよびL-ソルボースの生産1% CaCO3、0.3% NaCl、1 mM PMS、5 μg/ml PQQ、2% L-ソルボースおよびOD600ユニット10の、pSSA102RまたはpVK100を保有するナリディキシン酸耐性(Nalr)P.プチダ[ATCC21812]を含む休止菌体混液(2 ml)を30℃で緩やかに振とうしながら17時間インキュベートした。結果として、pSSA102RないしpVK100を保有するNalr P.プチダ[ATCC21812]は、それぞれ18.9、0.0 g/lの2KGAを生成した。
【0150】
また、1 % CaCO3、0.3 % NaCl、1 mM PMS、5 μg/ml PQQ、2 % D-ソルビトールおよびOD600ユニット10のpSSB103RまたはpVK100を保有するNalr P.プチダ[ATCC21812]を含む休止菌体混液(2 ml)を30℃で緩やかに振とうしながら17時間インキュベートした。結果として、pSSB103RまたはpVK100を保有するNalr P.プチダ[ATCC21812]は、それぞれ7.8または0.0 g/lのL-ソルボースを生成した。
【0151】
[実施例14] キメラAADH酵素の作成とその性質(1) キメラAADH酵素の作成AADH酵素の基質特異性を変えるために、酵素 Aと酵素 B間の種々のキメラ酵素を作成した。
【0152】
1)図2は方策I(制限酵素切断-連結法)によって作成したキメラ遺伝子の構造を示している。両遺伝子で共通の制限酵素切断部位、AvaI(酵素 A 遺伝子の塩基番号603)、EcoRI(塩基番号1084)、SalI(塩基番号1470)(図7)をその作成のために使用した。まず、酵素 Aおよび酵素 Bの遺伝子カセット(それぞれ2.7 kb、2.3 kbのHindIII断片)を、この順番で同じ向きにpUC18にクローン化して、プラスミドpSSAB201を作成した。また、酵素 B、酵素 A遺伝子の順で、同様にカセットをpUC18に導入し、プラスミドpSSBA201を得た(図3)。次に、これらのプラスミドを各制限酵素で部分切断したのち連結して大腸菌JM109を形質転換した。得られたアンピシリン耐性形質転換体からプラスミドを分析し、酵素A遺伝子を頭とするキメラ遺伝子カセットの場合には2.7 kb、酵素 Bを頭とするキメラ遺伝子カセットの場合には2.3 kbの期待される構造を有するHindIII断片を与えるものを選んだ。このようにして作成したキメラ遺伝子のカセットをpVK102のHindIII部位に導入し、それぞれ酵素 A/B1、酵素 A/B2、酵素 A/B3、酵素 B/A1、酵素 B/A2、および酵素 B/A3をコードする遺伝子をもつプラスミドpSSA/B101R、pSSA/B102R、pSSA/B103R、pSSB/A101R、pSSB/A102RおよびpSSB/A103Rを図2に示すように作成した。これら6つのプラスミドは実施例1に示したように接合伝達法により、Nalr P.プチダに導入した。
【0153】
2)図8は方策IIによるキメラ遺伝子の作成の概略を示している。方策IIでは、AADH酵素の基質特異性を変えるのに、任意の位置で組換えたキメラが作成される細胞内相同組換え法を採用している。この方法の原理は以下のようである。
(i)選択マーカーを持つ一つのプラスミド上に、2つの相同性のある遺伝子を直列に並べ,(ii)2つの遺伝子の間に存在する制限酵素部位で切断し、直鎖状になったプラスミドを用いて、野性型recAを有する大腸菌を形質転換し、(iii)2つの遺伝子間の様々な位置で、組換えた結果環化したDNAを保有する形質転換体を、選択マーカーを指標に選択する、というものである。図8に示すように、2種の制限酵素を用いて、プラスミドpSSAB201およびpSSBA201[pUC18上に酵素 Aおよび酵素 B両遺伝子を有するプラスミド(図3)]を直鎖状とし、大腸菌 JM101(recA+)株を形質転換した。形質転換体は1μgのDNAあたり101〜102得られた。不当な組換えを起こしたものを、DNA鎖長を指標に除去した結果、正しい組換え体の比率は30%であった。酵素 A遺伝子のC末端側約3分の2を失う形のXhoI-BalI断片を用いると、効率よくN末端側3分の1以内で組換えたキメラが得られた。次に、得られた組換え体を特定の3つのSmaI、SphI、SalIおよびBalI切断部位(図7)によって区切られる組換え位置グループに分類した。このようにして作成した様々なキメラ遺伝子をHindIII断片のカセットとしてpVK100にクローン化し、Nalr P.プチダに接合伝達法により導入した。
【0154】
(2)キメラAADH酵素の性質1) 制限酵素切断-連結法により得られたキメラの性質NalrP.プチダで発現したキメラ酵素の酵素学的性質を、実施例1に記載したように接合伝達体細胞の可溶性画分を用いて調べた。図11に示すように8種類の基質をこの評価に用いた。酵素 A/B1および酵素 A/B3は、酵素 Aに比べて発現レベルが低いものの、酵素 A型の基質特異性を示した。一方、酵素 B/A1、酵素B/A2、酵素 B/A3は、酵素 Aに由来する領域が増加するとともに、n-プロパノールに対する活性(酵素 A型)が高まるものの、酵素 B型の基質特異性を示した。酵素 B/A1遺伝子の発現レベルは野生型の酵素 B遺伝子よりも高く約2倍であった。制限酵素切断-連結法によって得たキメラ酵素の性質から、酵素 Aまたは酵素 BにおけるN末側3分の1が基本的に基質特異性を決定していると結論された。
【0155】
2)相同組換え法によって得られたキメラの性質上記のように、この方法により得られたキメラの中で、図7に示されるSmaI2とSalI切断部位の間で組換えた18のキメラ酵素のうち7つが好ましい基質特異性を示した。この7つのキメラ酵素は、D-ソルビトールからはL-ソルボースを生成し、酵素 Aで生成されるD-グルコースを生じなかった。そしてL-ソルボースからは、酵素 Aと同様に2KGAを生成した。キメラ遺伝子の組換え位置は、実施例2に示すように塩基配列の決定により確定した。大まかな構造が「N末側9分の2が酵素 A、残りのC末側9分の7が酵素 B」となっているこのタイプのキメラ酵素は、酵素 superA型として分類した。その結果、酵素 superA型に属する7つのキメラ酵素の組換え位置は3種類であった。すなわち、酵素 A/B21(酵素 Aの第1〜128残基に加えて、酵素 Bの第129〜556残基を含むキメラ酵素)、酵素 A/B22(酵素 Aの第1〜125残基に加えて、酵素 Bの第126〜556残基を含むキメラ酵素)および酵素 A/B25(酵素 Aの第1〜135残基に加えて、酵素 Bの第136〜556残基を含むキメラ酵素)である。酵素 A/B21、酵素 A/B22、または酵素 A/B25遺伝子を発現するP.プチダ接合伝達体は、D-ソルビトールをD-グルコースには変換せずにL-ソルボースに変換した。その他に得られたキメラ酵素酵素 A/B31(酵素 Aの第1〜95残基に加えて、酵素 Bの第96〜556残基を含むキメラ酵素)は、D-ソルビトールを効率よくL-ソルボースに変換したが、L-ソルボースを2KGAには変換しなかった。すなわち、このキメラ酵素は酵素 B型の活性を示した。上述したキメラの発現レベルは、野性型の酵素 Bに比べて高かったが、これは酵素 B遺伝子が、酵素 A遺伝子には見られない「まれなコドン(rare codon)」を多く含むためと思われる。尚、このコドン解析は、プログラム「Codon Preference」(Wisconsin Sequence Analysis Package(登録商標)、Genetics Computer Group)により行った。
【0156】
(3)キメラ遺伝子におけるコドン使用の改良好ましいコドン使用という点においてキメラ酵素(酵素A/B21、酵素 A/B22、酵素 A/B25および酵素 A/B31)遺伝子をさらに改良するために、それらの酵素 B残基からの配列を有するC末側の3分の2の領域を、酵素 A遺伝子残基のC末側の3分の2と置き換えた。酵素 A/B21、酵素 A/B22、酵素 A/B25および酵素 A/B31遺伝子を用いて、それぞれ以下の4つの新しいキメラ遺伝子を構築した。すなわち、酵素 sA21(酵素 Aの第1〜128残基、酵素 Bの第129〜180残基、そして酵素 Aの第180〜556残基を含むキメラ酵素)遺伝子、酵素 sA22(酵素 Aの第1〜125残基、酵素 Bの第126〜180残基、そして酵素 Aの第180〜556残基を含むキメラ酵素)遺伝子、酵素 sA2(酵素 Aの第1〜135残基、酵素 Bの第136〜180残基、そして酵素 Aの第180〜556残基を含むキメラ酵素)遺伝子、および酵素 sB(酵素 Aの第1〜95残基、酵素 Bの第96〜180残基、そして酵素 Aの第180〜556残基を含むキメラ酵素)遺伝子である(図4)。酵素 sA2および酵素 sB遺伝子の置換実験の実際は、それぞれpUC18に酵素 sA遺伝子と酵素 B/A1遺伝子を並べた形でもつプラスミドおよびpUC18に酵素 A/B31遺伝子と酵素 B/A1遺伝子を並べた形でもつプラスミドを、AvaIによって部分切断後連結し、大腸菌 JM109を形質転換した後、形質転換体のプラスミドの構造を制限酵素切断様式で解析するとともに、塩基配列を決定して期待される組み換え部位のAvaI切断部位で置換されていることを確認した。酵素 sA21および酵素 sA22遺伝子作成のための置換実験は、酵素 sA2のN末端部をコードするHindIII-SspI断片を、それぞれ酵素 A/B21および酵素 A/B22遺伝子の組換え位置を含んでいるHindIII-SspI断片と置換することにより行った(図4)。
【0157】
(4)キメラ酵素の速度論的解析表11および表12に酵素 sA2および酵素 sBの速度論的性質を、それぞれ酵素 Aおよび酵素 Bと比較してまとめた。
【0158】
【表11】
【0159】
酵素 sA2および酵素 AによるL-ソルボースからの生成物は2KGAであった。D-ソルビトールからの生成物は、酵素 sA2では微量のD-グルコースを伴うもののL-ソルボースであり、酵素 AではD-グルコースのみであった。このように、酵素 sA2はD-ソルビトールからの2KGA生産に望ましい性質、すなわち、酵素 A同様のL-ソルボースから2KGAを生成するためのL-ソルボース/L-ソルボソン脱水素酵素活性、および酵素 B同様のD-ソルビトールからL-ソルボースを生成するD-ソルビトール脱水素酵素活性を示した。
【0160】
【表12】
【0161】
酵素 Bと比較して、酵素 sBはD-ソルビトールに対する親和性が高く、またD-ソルビトールからL-ソルボースへの変換に対して阻害剤となるD-ソルビトールの酸化生成物であるL-ソルボースに対する親和性は低くなっていた。
【0162】
[実施例15] キメラAADH酵素を増幅したGOS2R誘導株によるD-ソルビトールからの2KGA生産酵素 sA2および酵素 sBを評価するにあたり、GOBΔKおよびGOI13株を作成した。GOBΔKは、自殺ベクターpSUP201[アンピシリン耐性(Ampr)、クロラムフェニコール耐性(Cmr)、mob+、pBR325誘導体、Bio/Technology, 1巻、784〜791頁、1983]を使ってGOS2R株に対して酵素 B遺伝子全体を欠失させ、代わりにプラスミドpUC4K[4.1 kb、Kmr、Ampr、ファーマシア社製(Pharmacia)、ヴィエラ(Viera), J.およびメッシング(Messing), J.、Gene 19巻、259頁、(1982)]から単離した1.28kb Kmr 遺伝子カセットを挿入することにより作成した。
【0163】
GOI13はGOBΔKからさらに野性型酵素 A遺伝子を酵素 sB遺伝子に置換し、野性型酵素 A"遺伝子を、自殺ベクターpSUP202[Ampr、Cmr、Tcr、mob+、pBR325誘導体、Bio/Technology、1巻、784〜791頁、(1983)]を用いてゲンタマイシン耐性(Gmr)遺伝子カセットと置換することによって欠失させて作成した。Gmr遺伝子カセットは、DNA断片Tn5-GM[ササガワら、Gene 56巻、 283〜288頁、(1987)]を鋳型としたPCR増幅法により、PstI切断部位を両端に付加するように設計し、得られたPCR産物をpUC4KのPstI切断部位に挿入することによってpUC8Gとして得た。Gmr遺伝子は、pUC8GからEcoRI、BamHI、SalIまたはPstIで切断することによって単離することが可能である。
【0164】
(1)2KGA生産における酵素 sA2増幅の効果プラスミドpSSsA2(pVK100に酵素 sA2遺伝子を含む2.7 kb HindIIIカセットを導入したもの)および対照となるプラスミドpSSA102R(pVK102に酵素 A遺伝子を含む2.7 kb HindIIIカセットを導入したもの)を実施例6記載の接合伝達法によってGOI13に導入した。得られた接合伝達体を実施例8に示すように、PMSL10培地を用いて30℃で4日間培養した。pSSsA2およびpSSA102Rを有するGOI13は、それぞれ66.3および38.5 g/lの2KGAと、それぞれ8.4 および25.9 g/lの2KD(D-ソルビトールからの2KGA生産において、D-ソルビトールからD-グルコースおよびD-グルコン酸を経て生成する副産物)を生成した。
【0165】
(2)プラスミドpSSsA21およびpSSsA22[pVK100に、それぞれ酵素 sA21および酵素 sA22遺伝子を含む2.7 kb HindIIIカセットを導入したもの(図4)]を実施例6記載の接合伝達法によってGOI13に導入した。得られた接合伝達体を実施例8に示すように、PMSL10培地を用いて30℃で4日間培養した。pSSsA21およびpSSsA22を有するGOI13は、それぞれ66.8および77.4g/lの2KGAと、それぞれ0.3および0.4 g/lの2KDを生成した。
【0166】
(3)2KGA生産における酵素 sBの効果プラスミドpSSsB[pVK100に酵素 sB遺伝子を含む2.7 kb HindIIIカセットを導入したもの(図4)]および対照となるプラスミドpSSB103R(pVK102に酵素 B遺伝子を含む2.3 kb 酵素 B遺伝子を導入したもの)を接合伝達法によってGOBΔKに導入した。pSSsBを有するGOBΔK、pSSB103Rを有するGOBΔKおよびGOBΔKを実施例8の(2)に記載したようにPMSL8培地で培養したところ、それぞれ52.0、46.8、および1.1 g/lの2KGAと、それぞれ6.9、9.3、32.3 g/lの2KDを生成した。
【0167】
GOI13(野性型の酵素 B、酵素 Aおよび酵素 A"を欠失し、染色体DNA上に1コピーの酵素 sB遺伝子を有する)をPMSL10培地で2日間培養したところ、79.3 g/lのL-ソルボースを生成した。
【図面の簡単な説明】
【0168】
【図1】本発明の組換え酵素 Aまたは酵素 Bをコードする組換えDNA分子を各々有する組換え発現ベクターの構造の概略図である。
【図2】本発明のキメラ酵素をコードする組換えDNA分子を各々有する組換え発現ベクターの構造の概略図である。
【図3】相同組換え法によるキメラの構築のための直列に並ぶ酵素 Aおよび酵素 Bの構造遺伝子を含有する組換えDNA分子を有する材料プラスミドの構造の概略図である。
【図4】好ましいコドンを用いたキメラ酵素 sA2、酵素 sA21、酵素 sA22または酵素 sBを各々コードする組換え発現ベクターの構造の概略図である。酵素 sA2は「酵素 Aの第1〜第135残基と酵素 Bの第136〜第180残基と酵素 Aの第180〜第556残基」を含む構造を有する。酵素 sA21は「酵素 Aの第1〜第128残基と酵素 Bの第129〜第180残基と酵素 Aの第180〜第556残基」を含む構造を有する。酵素 sA22は「酵素 Aの第1〜第125残基と酵素 Bの第126〜第180残基と酵素 Aの第180〜第556残基」を含む構造を有する。酵素 sBは「酵素 Aの第1〜第95残基と酵素 Bの第96〜第180残基と酵素 Aの第180〜第556残基」を含む構造を有する。上述の番号は各々の成熟(mature)酵素のアミノ酸残基の番号である。図中、*1、*2、*3、*4および*5は塩基配列におけるAvaI切断部位に相当する組換え部位を示し、各々成熟型酵素 Aのアミノ酸残基番号135、128、125、95および180を示す。
【図5】成熟酵素 Aと酵素 Bのアミノ酸配列の並列図である。図中、*で示した領域をコードする塩基配列は、図2に示されるキメラ遺伝子を構築するために用いた、AvaI、EcoRIおよびSalI切断部位を表す。
【図6】本発明のキメラ酵素をコードする組換え遺伝子の構築法を図示したものである。
【図7】酵素 A遺伝子と酵素 B遺伝子の制限酵素地図を図示している。図中、*は図2および図6に示したキメラ遺伝子を作成するのに用いたAvaI、EcoRIおよびSalI切断部位を表す。
【図8】二つのAADH遺伝子中の共通塩基配列における生体内相同組換えによるキメラ遺伝子の構築法を図示している。
【図9】酵素 B遺伝子の上流にBamHI部位を導入するための部位特異的変異法を示す。
【図10】酵素 B遺伝子のプロモーターの置換方法を図示している。
【図11】本発明のキメラ酵素の基質特異性のグラフを示す。図中、酵素活性は(1)では、n-プロパノールに対する、(2)ではD-グルコースに対する相対活性として表した。酵素 A/B2はシュードモナス プチダにおける発現レベルが低かったため測定対象から除外した。
【特許請求の範囲】
【請求項1】
配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチド、並びに配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドの間のキメラ組換え酵素、並びに一つまたはそれ以上のアミノ酸残基の付加、挿入、欠失および/または置換を含む上記で同定されるポリペプチドの機能的誘導体からなる群より選択され、該酵素ポリペプチドがアルコールおよび/またはアルデヒド脱水素酵素活性を有する、一つまたはそれ以上の酵素ポリペプチドを含む、アルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物。
【請求項2】
ポリペプチドが、酵素 A/B1、酵素 A/B2、酵素 A/B3、酵素B/A1、酵素 B/A2、酵素 B/A3、酵素 sA2、酵素 sA21、酵素 sA22、酵素 sBおよびそれらの機能的誘導体のキメラ酵素である、請求項1記載の組換え酵素調製物。
【請求項3】
酵素ポリペプチドが、ホモ二量体および/またはヘテロ二量体の形態で存在する、請求項1または2記載の組換え酵素調製物。
【請求項4】
請求項1または2で定義されるポリペプチドをコードするDNA分子。
【請求項5】
直鎖状もしくは環状DNAまたは染色体上での挿入DNA断片のいずれかの形態で存在する、請求項4記載のDNA分子。
【請求項6】
請求項4または5のいずれか1項で定義される一つまたはそれ以上のDNA分子を含む、組換え発現ベクター。
【請求項7】
DNA分子が、一つまたはそれ以上の遺伝子制御配列に機能的に結合しており、適当な宿主細胞において請求項1〜3のいずれか1項に記載の酵素ポリペプチドを発現可能である、請求項6記載の組換え発現ベクター。
【請求項8】
pSSA102R、pSSA'101R、pSSA"102、pSSB103R、pSSAp-B、pSSA/B101R、pSSA/B102R、pSSA/B103R、pSSB/A101R、pSSB/A102R、pSSB/A103R、pSSsA2、pSSsA21、pSSsA22およびpSSsBからなる群より選択される、請求項7記載の組換え発現ベクター。
【請求項9】
請求項6〜8のいずれか1項に記載の組換え発現ベクターまたは請求項4もしくは5のいずれか1項で定義される一つまたはそれ以上のDNA分子を有する組換え生物。
【請求項10】
宿主細胞が、微生物、哺乳動物細胞および植物細胞からなる群より選択される、請求項9記載の組換え生物。
【請求項11】
宿主細胞が、大腸菌(Escherichia coli)、シュードモナスプチダ(Pseudomonas putida)、アセトバクターキシリナム(Acetobacterxylinum)、アセトバクター パスチュリアヌス(Acetobacterpasteurianus)、アセトバクター アセチ(Acetobacter aceti)、アセトバクター ハンゼニイ(Acetobacter hansenii)およびグルコノバクターオキシダンス(Gluconobacteroxydans)等の細菌類からなる群より選択される微生物である、請求項9または10記載の組換え生物。
【請求項12】
宿主細胞が、グルコノバクターオキシダンス[DSM番号4025]である、請求項11記載の組換え生物。
【請求項13】
請求項10〜12のいずれか1項で定義される組換え生物を適当な培地で培養し、組換え酵素を回収することを含む、請求項1、2または3で定義されるアルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物の製造方法。
【請求項14】
請求項10〜12のいずれか1項で定義される組換え生物の生化学的反応により基質を生産物に変換することを含む、アルデヒド、ケトンまたはカルボン酸生成物を対応する基質から製造する方法。
【請求項15】
請求項10〜12のいずれか1項で定義される組換え生物の生化学的反応によりL-ソルボースおよび/またはD-ソルビトールを2-ケト-L-ギュロン酸に変換することを含む、L-ソルボースおよび/またはD-ソルビトールから2-ケト-L-ギュロン酸を製造する方法。
【請求項16】
請求項1〜3のいずれか1項で定義される組換え酵素調製物および基質を含有する反応混合液をインキュベートすることを含む、アルデヒド、ケトン、またはカルボン酸生成物を対応する基質から製造する方法。
【請求項17】
請求項1〜3のいずれか1項で定義される組換え酵素調製物とL-ソルボースおよび/またはD-ソルビトールとを含有する反応混合液をインキュベートすることを含む、2-ケト-L-ギュロン酸の製造方法。
【請求項18】
請求項15または17記載の製造方法が達成され、該製造方法により得られる2-ケト-L-ギュロン酸が、当技術分野で既知の方法によりL-アスコルビン酸に変換されることを特徴とする、2-ケト-L-ギュロン酸からのL-アスコルビン酸の製造方法。
【請求項1】
配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチド、並びに配列番号:5、配列番号:6、配列番号:7および配列番号:8により同定されるポリペプチドの間のキメラ組換え酵素、並びに一つまたはそれ以上のアミノ酸残基の付加、挿入、欠失および/または置換を含む上記で同定されるポリペプチドの機能的誘導体からなる群より選択され、該酵素ポリペプチドがアルコールおよび/またはアルデヒド脱水素酵素活性を有する、一つまたはそれ以上の酵素ポリペプチドを含む、アルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物。
【請求項2】
ポリペプチドが、酵素 A/B1、酵素 A/B2、酵素 A/B3、酵素B/A1、酵素 B/A2、酵素 B/A3、酵素 sA2、酵素 sA21、酵素 sA22、酵素 sBおよびそれらの機能的誘導体のキメラ酵素である、請求項1記載の組換え酵素調製物。
【請求項3】
酵素ポリペプチドが、ホモ二量体および/またはヘテロ二量体の形態で存在する、請求項1または2記載の組換え酵素調製物。
【請求項4】
請求項1または2で定義されるポリペプチドをコードするDNA分子。
【請求項5】
直鎖状もしくは環状DNAまたは染色体上での挿入DNA断片のいずれかの形態で存在する、請求項4記載のDNA分子。
【請求項6】
請求項4または5のいずれか1項で定義される一つまたはそれ以上のDNA分子を含む、組換え発現ベクター。
【請求項7】
DNA分子が、一つまたはそれ以上の遺伝子制御配列に機能的に結合しており、適当な宿主細胞において請求項1〜3のいずれか1項に記載の酵素ポリペプチドを発現可能である、請求項6記載の組換え発現ベクター。
【請求項8】
pSSA102R、pSSA'101R、pSSA"102、pSSB103R、pSSAp-B、pSSA/B101R、pSSA/B102R、pSSA/B103R、pSSB/A101R、pSSB/A102R、pSSB/A103R、pSSsA2、pSSsA21、pSSsA22およびpSSsBからなる群より選択される、請求項7記載の組換え発現ベクター。
【請求項9】
請求項6〜8のいずれか1項に記載の組換え発現ベクターまたは請求項4もしくは5のいずれか1項で定義される一つまたはそれ以上のDNA分子を有する組換え生物。
【請求項10】
宿主細胞が、微生物、哺乳動物細胞および植物細胞からなる群より選択される、請求項9記載の組換え生物。
【請求項11】
宿主細胞が、大腸菌(Escherichia coli)、シュードモナスプチダ(Pseudomonas putida)、アセトバクターキシリナム(Acetobacterxylinum)、アセトバクター パスチュリアヌス(Acetobacterpasteurianus)、アセトバクター アセチ(Acetobacter aceti)、アセトバクター ハンゼニイ(Acetobacter hansenii)およびグルコノバクターオキシダンス(Gluconobacteroxydans)等の細菌類からなる群より選択される微生物である、請求項9または10記載の組換え生物。
【請求項12】
宿主細胞が、グルコノバクターオキシダンス[DSM番号4025]である、請求項11記載の組換え生物。
【請求項13】
請求項10〜12のいずれか1項で定義される組換え生物を適当な培地で培養し、組換え酵素を回収することを含む、請求項1、2または3で定義されるアルコールおよび/またはアルデヒド脱水素酵素活性を有する組換え酵素調製物の製造方法。
【請求項14】
請求項10〜12のいずれか1項で定義される組換え生物の生化学的反応により基質を生産物に変換することを含む、アルデヒド、ケトンまたはカルボン酸生成物を対応する基質から製造する方法。
【請求項15】
請求項10〜12のいずれか1項で定義される組換え生物の生化学的反応によりL-ソルボースおよび/またはD-ソルビトールを2-ケト-L-ギュロン酸に変換することを含む、L-ソルボースおよび/またはD-ソルビトールから2-ケト-L-ギュロン酸を製造する方法。
【請求項16】
請求項1〜3のいずれか1項で定義される組換え酵素調製物および基質を含有する反応混合液をインキュベートすることを含む、アルデヒド、ケトン、またはカルボン酸生成物を対応する基質から製造する方法。
【請求項17】
請求項1〜3のいずれか1項で定義される組換え酵素調製物とL-ソルボースおよび/またはD-ソルビトールとを含有する反応混合液をインキュベートすることを含む、2-ケト-L-ギュロン酸の製造方法。
【請求項18】
請求項15または17記載の製造方法が達成され、該製造方法により得られる2-ケト-L-ギュロン酸が、当技術分野で既知の方法によりL-アスコルビン酸に変換されることを特徴とする、2-ケト-L-ギュロン酸からのL-アスコルビン酸の製造方法。
【図1】

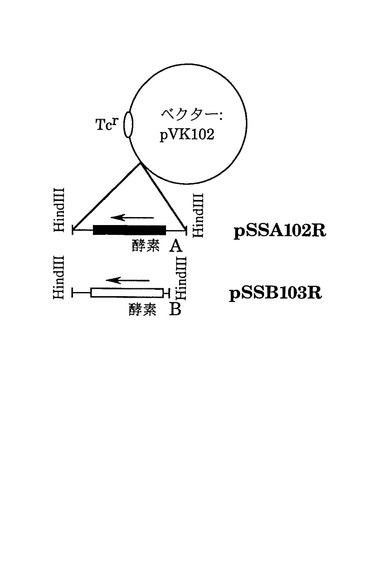
【図2】
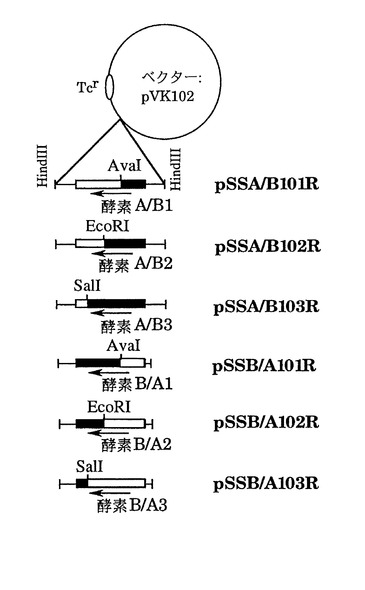
【図3】
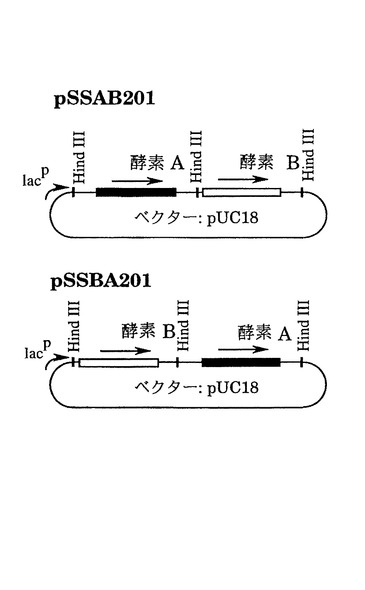
【図4】

【図5】

【図6】
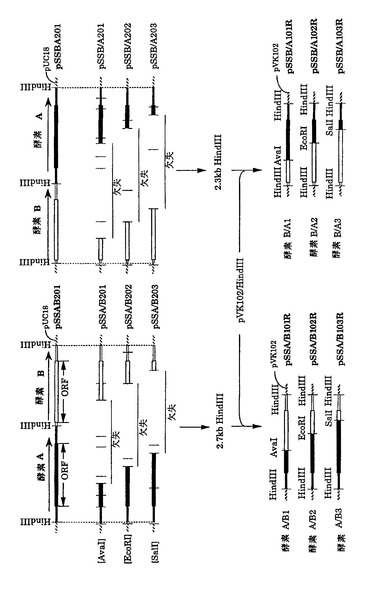
【図7】

【図8】
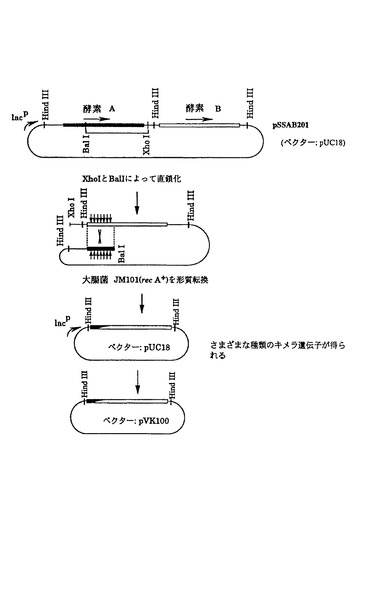
【図9】
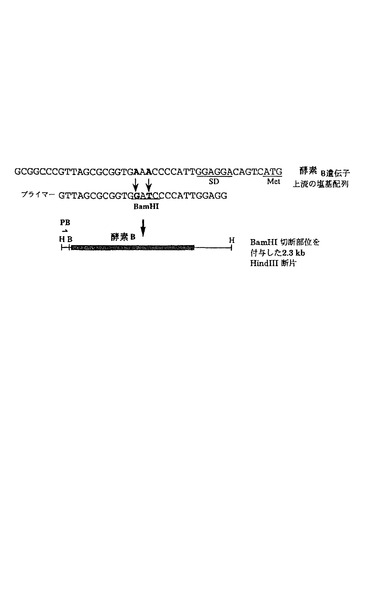
【図10】
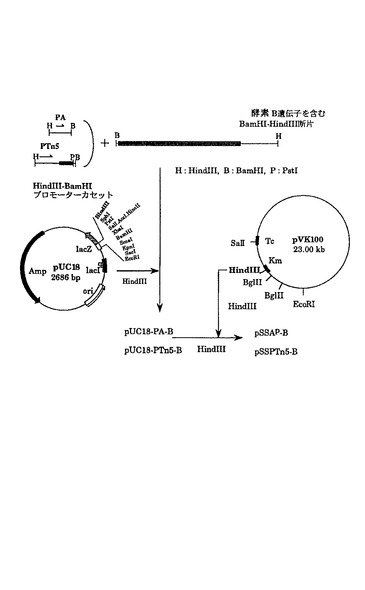
【図11】
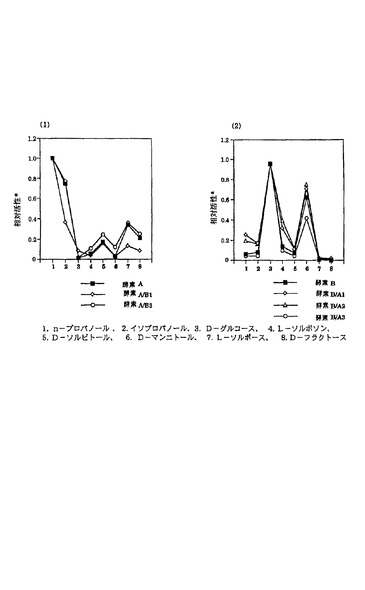
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公開番号】特開2007−275070(P2007−275070A)
【公開日】平成19年10月25日(2007.10.25)
【国際特許分類】
【出願番号】特願2007−134731(P2007−134731)
【出願日】平成19年5月21日(2007.5.21)
【分割の表示】特願平9−273790の分割
【原出願日】平成9年9月19日(1997.9.19)
【出願人】(503220392)ディーエスエム アイピー アセッツ ビー.ブイ. (873)
【Fターム(参考)】
【公開日】平成19年10月25日(2007.10.25)
【国際特許分類】
【出願日】平成19年5月21日(2007.5.21)
【分割の表示】特願平9−273790の分割
【原出願日】平成9年9月19日(1997.9.19)
【出願人】(503220392)ディーエスエム アイピー アセッツ ビー.ブイ. (873)
【Fターム(参考)】
[ Back to top ]