
新規イメージング法
本発明は、被検体において線維形成の識別を容易にするのに有用な方法に関する。本発明の方法は、肝臓の線維形成を診断するための方法の一部として応用する場合に特に有用である。本発明は、被検体において線維形成を識別するための方法に使用するための化合物も用意する。本発明の別の態様は、被検体において線維形成を識別するための方法に使用するための医薬品の調製に使用するための化合物である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、インビボイメージング法、特に、ある種の公知のインビボイメージング剤の新規用途に関する。本発明の好ましいインビボイメージング法は単光子放射断層撮影(SPECT)及び陽電子放射断層撮影(PET)である。
【背景技術】
【0002】
慢性ウイルス性肝炎、非アルコール性脂肪性肝炎(NASH)、寄生虫血症、先天性代謝異常及び飲酒による中毒性障害を始めとする肝線維症を引き起こす可能性のある傷害は地理的に広く分布し、有病率は高い。こうした要因はすべて、肝硬変と潜在的に肝癌を招く線維症が、依然として世界中での主な病因及び死因の一つであることを意味している。英国だけをとっても、肝疾患は現在5番目に多い死因であり、その発症率は増加しつつある(Iredale 2003 BMJ Vol. 327 pp 143−147)。
【0003】
非アルコール性脂肪性肝疾患(NAFLD)とNASHの2種類の脂肪肝疾患が存在する。米国の人口の約24%がNAFLDをもつと考えられており、これは低い頻度でNASHへと進行する。NAFLDはメタボリックシンドロームに関連しており、肥満、高脂血症、高血圧及びII型糖尿病と関係がある。米国では約4700万人がメタボリックシンドロームをもっていると考えられている。米国の人口のうち推定860万人がNASHを有していて、線維症及び肝硬変に罹患するおそれがあり、NASH患者の20〜28%が10年間で肝硬変を発症すると考えられている。NAFLDはごく一般的で、NASHへ、ひいては肝硬変へと進行しかねないNAFLDの範囲の重症度の低い端を表す。肝線維症はNASHから肝硬変へと進行するリスクの指標である。
【0004】
肝線維症の検出に現在用いられているアプローチは幾つかの重大な短所をもつ。コラーゲン沈着のパターンの組織学的分析による肝生検は、肝疾患ステージ及び肝線維症を評価するための最も信頼できる基準とみなされている。しかし、この方法には、若干の有病率、偶発的な死亡率、高いコスト、サンプリング誤差及び線維症の程度を分類する際の肝臓病理学者間での高い観察者間変動を伴う。生検での肝臓のサンプリングは、評価すべき肝臓の5万分の1にすぎず、ステージ診断の誤りを招くおそれがある。さらに、コラーゲンは繊維組織のマーカーであり、コラーゲンは活動性線維症の後期ステージにおいても、疾患プロセスが解消しつつある場所でも見つかるので、コラーゲンは活動性疾患の理想的標的ではない。現在、肝線維症を非侵襲的方法で有効に特徴付けてモニターすることができる手段はない。そのため、早期治療介入して肝線維症の進行を遅らせ停止させるのに悪影響がある。さらに、病気の進行を適宜モニターするには、3〜5年毎に繰返し生検を行うことが推奨されている。肝線維症の検出に利用できる血液検査の価値が限られているのは、線維症の程度又は線維症と肝硬変との区別に用いることができないからである。現在、NASHとNAFLDを区別し、或いはNASHにおける線維症を十分に定量化し特徴付けるのに利用できる方法はない。
【0005】
肝星細胞(HSC)は、肝臓における主要なフィブロコンピテント(fibrocompetent)細胞と広くみなされている。進行性肝線維症中に、HSCは活性化して増殖するが、線維症の解消中には、肝臓瘢痕の分解と同時に起こる広範囲のHSCアポトーシスがある。線維症のプロセスのこの進行性ステージは、線維形成と呼ばれる。活性化HSC上のインテグリン発現の上方制御が報告されている(Zhou et al J. Biol. Chem. 2004; 279(23): 23996−24006)。HSCの活性化は、線維形成及び、続く肝線維症の開始及び進行に不可欠である。従って、HSC活性化のマーカーは、線維形成のイメージングにとっての標的機会である。この事に関して、最近になって顕著になってきたプロセスのマーカーはインテグリンである(Zhou et al 2004 J. Biol. Chem.; 279: 23996−24006; Patsenker et al 2007 J. Hepatol.; 46(5): 878−887; Zhou et al 2006 J. Biol. Chem.; 281: 39757−39765; Carloni et al 1996 Gastroenterology: 110: 1127−36)。
【0006】
インビボイメージング用途のための適当に標識されたインテグリン結合剤の使用についてこれまで記載されている。
【0007】
国際公開第2004/020435号は、αvインテグリン又はそのサブタイプの発現に起因する細胞増殖の病理学的な上方制御又は調整不全を伴う疾患の治療に有用であるインテグリン結合性ピペリジニル化合物を開示している。国際公開第2004/020435号は、発明の化合物が、インビボイメージングに適している部分とコンジュゲートし、非侵襲的腫瘍イメージング剤として使用できることも開示している。
【0008】
国際公開第2007/088041号は、ある疾患の治療及び/又は予防に有用である、あるクラスの小分子インテグリン結合性化合物を開示しており、その疾患は好ましくはαvβ1インテグリンにより仲介される疾患である。肝線維症は、国際公開第2007/088041号の化合物が使用される特定の疾患として包含されている。治療及び予防に加えて、国際公開第2007/088041号には、開示された化合物がインビボイメージング部分を含んでいてインビボイメージングに使用することができることが教示している。
【0009】
国際公開第2003/006491号、国際公開第2005/012335号及び国際公開第2005/123767号は、血管新生に関係する受容体に結合するRGDペプチド系の化合物に関し、受容体は、インテグリンを包含する。開示されている化合物は、抗新生物剤又はインビボイメージング部分のいずれかを含み、血管新生に関係する疾患の治療及びインビボイメージングに有用であると教示されている。
【0010】
国際公開第2006/054904号は、細胞外マトリックス(ECM)をターゲティングする造影剤としてのインビボイメージング部分で標識されたRGDペプチドの使用を開示している。この造影剤は、肝線維症を始めとするコラーゲンの過剰な形成に関連する疾患を診断及びモニターする際に有用であると言われている。しかし、放射標識されたインテグリン結合性RGDペプチドの排泄が主に肝胆道系を介して起きることが知られていることから、肝線維症のインビボイメージングにRGDペプチドを使用することへの阻害要因がある(Haubner 1999 J. Nuc. Med.; 40: 1061−71)。さらに、上述のように、疾患プロセスが解消しつつある場合並びに活動性線維症の後期ステージの両方でコラーゲンが見いだされることがあるため、コラーゲンは、活動性線維症の理想的マーカーではない。治療レジメンの用途が、より適切であるかもしれず、肝硬変が発症する前の、例えば、NASHにおける線維症の早期ステージで臨床的により有効である可能性も高い、より早期の活動性ステージを標的とすることがより有利であろう。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】国際公開第2007/148074号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0012】
そこで、肝線維症の早期状態(「線維形成」とも呼ばれる)を識別し、それによって、最も効果的に治療することができるステージで疾患プロセスへ介入するための方法が必要である。
【課題を解決するための手段】
【0013】
本発明は、被検体の肝臓における線維形成の識別を容易にするのに有用な方法に関する。本発明は、被検体の肝臓における線維形成を識別するための方法に使用するための化合物も用意する。本発明の別の態様は、被検体の肝臓における線維形成を識別するための方法に使用するための医薬品の調製に使用するための化合物である。本発明は、RGDペプチド系の化合物が、活性化肝星細胞(HSC)を検出し、それによって、肝線維症の早期診断に有用な方法を用意するために効果的に使用することができることを例証する。
【図面の簡単な説明】
【0014】
【図1】活性化ヒト肝星細胞とEA−Hy926膜の両方と特異的に結合する化合物6を示す図である。Kiは、EA−Hy926膜アッセイで約10nMと決定され、EC50は、LX−2細胞アッセイで約1nMと決定された。LX−2アッセイにおける結合の特異性は、非放射性の化合物6による化合物6結合の特異的阻害によって示された。低親和性スクランブル陰性対照は、インテグリンとも活性化星細胞ともにも結合しない。
【図2】活性化ヒト肝星細胞とEA−Hy926膜の両方と特異的に結合する化合物6を示す図である。Kiは、EA−Hy926膜アッセイで約10nMと決定され、EC50は、LX−2細胞アッセイで約1nMと決定された。LX−2アッセイにおける結合の特異性は、非放射性の化合物6による化合物6結合の特異的阻害によって示された。低親和性スクランブル陰性対照は、インテグリンとも活性化星細胞とも結合しない。
【図3】活性化肝星細胞膜とEA−Hy926膜の両方と特異的に結合する化合物3を示す図である。Kiは、観察されたように、EA−Hy926膜アッセイで約1nMと決定され、星細胞膜アッセイで59nMと決定された。化合物8は、親和性において化合物3に続くように見え、Kiは、活性化星細胞膜アッセイで56nMと決定され、EA−Hy926膜アッセイで6nMと決定された。陰性対照スクランブルRGDペプチドは、どちらの細胞タイプともほとんど結合を示さない。
【図4】活性化肝星細胞膜とEA−Hy926膜の両方と特異的に結合する化合物3を示す図である。Kiは、観察されたように、EA−Hy926膜アッセイで約1nMと決定され、星細胞膜アッセイで59nMと決定された。化合物8は、親和性において化合物3に続くように見え、Kiは、活性化星細胞膜アッセイで56nMと決定され、EA−Hy926膜アッセイで6nMと決定された。陰性対照スクランブルRGDペプチドは、どちらの細胞タイプともほとんど結合を示さない。
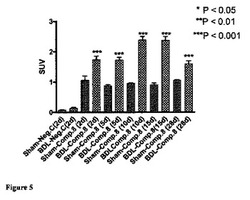
【図5】肝臓取込みが観察されない陰性対照スクランブルRGDペプチドと比較した、胆管結紮(BDL)ラットからの肝臓による化合物8の特異的取込みを例証する図である。BDL肝臓取込みは、線維形成の程度にも比例しており、最大取込みは、線維形成がその最高レベルである術後10及び15日目に観察された。
【図6】陰性対照化合物(RGDスクランブルペプチド)と対比した、BDLラット肝臓中へのRGDペプチドの特異的取込み例示する図である。この実験からのデータは、化合物3と化合物8が共に、BDLとシャムの間の取込みの有意差が観察されない陰性対照と比較して、注射から1時間後にシャム動物と比較して、BDLの肝臓中に有意に保持されることを示した。
【図7】術後15日目のBDLにおける有意なαvインテグリン上方制御を示す図である。
【図8】図8は、図7との関連して、術後の全時点におけるBDL動物の肝臓におけるαvインテグリン発現と化合物取込みの相関関係を例証している。
【発明を実施するための形態】
【0015】
一つの態様では、本発明は、被検体の肝臓における線維形成の存在、場所及び/又は量を決定する方法に用いられる式Iの化合物であって、該方法は、
(i)検出可能な量の式Iの化合物を投与しておいた被検体を準備する段階と、
(ii)肝臓中の線維形成性組織に式Iの化合物を結合させる段階と、
(iii)式Iの化合物から放出される信号をインビボイメージング法で検出する段階と、
(iv)信号の場所及び/又は量を表す画像を形成する段階
とを含んでおり、式Iの化合物は以下の通り定義される。
【0016】
【化1】

【0017】
式中、
Gはグリシンを表し、
Dはアスパラギン酸を表し、
X1は、アスパラギン酸、グルタミン酸、リジン、ホモリジンもしくはC3〜6ジアミノアルカン酸又はそれらの誘導体から選択されるアミノ酸を表し、
X2及びX4は独立に側鎖同士が連結して環化架橋を形成するアミノ酸残基(例えばジスルフィド又はチオエーテル結合を形成するシステイン又はホモシステイン)又は環化架橋を形成し得る他のアミノ酸(例えばアスパラギン酸及びリジン)を表し、
X3はアルギニン、N−メチルアルギニン又はアルギニン模倣体を表し、
X5はチロシン、フェニルアラニン、3−ヨード−チロシン C4〜6シクロアルキルアラニン、ナフチルアラニン又はそれらの誘導体を表し、
X6はX6をC(=O)基に連結するチオエーテル結合又はチオアセタール結合のいずれかを形成するチオール含有アミノ酸を表し、
W1及びW2は独立に任意要素としてのリンカー基であって、W1が存在するときはX1のアミノ酸側鎖部分に連結し、W2が存在するときはX6のカルボキシ基に連結し、
Z1及びZ2は、Z1とZ2の少なくとも一方がイメージング部分であることを条件として、独立にイメージング部分、糖部分、有機色素基又は水素である。
【0018】
式Iの化合物について、
X1は好ましくはリジンである。
X2及びX4は好ましくは独立にシステイン又はホモシステインであり、最も好ましくは共にシステインである。
X3は好ましくはアルギニンである。
X5は好ましくはシクロヘキシルアラニン、フェニルアラニン又は3−ヨード−チロシンであり、最も好ましくはフェニルアラニンである。
X6は好ましくはシステイン又はホモシステインであり、最も好ましくはシステインである。
【0019】
式Iの好ましい実施形態では、
X1はリジンであり、
X2及びX4は独立にシステイン又はホモシステインであり、
X3はアルギニンであり、
X5ハフェニルアラニン又は3−ヨード−チロシンであり、
X6ハシステイン又はホモシステインである。
【0020】
式Iの最も好ましい実施形態では、
X1はリジンであり、
X2及びX4は共にシステインであり、
X3はアルギニンであり、
X5はフェニルアラニンであり、
X6はシステインである。
【0021】
本発明において、「線維形成」という用語は、具体的に、とりわけ肝星細胞(HSC)が活性化され、インテグリンを発現する、線維症の活動性の進行ステージをいう。HSCは、肝臓における主要なフィブロコンピテント細胞と広くみなされている。線維形成中に、HSCは活性化して増殖するが、線維症の解消中には、肝臓瘢痕の分解と同時に起こる広範囲のHSCアポトーシスがある。さらに、線維症中に、コラーゲンなどの細胞外マトリックス(ECM)成分の沈着が起きることはない。従って、ECM成分の存在は、線維症の後期ステージ及び線維症の解消の特徴である。従って、線維形成中の疾患プロセスを標的にすることは、治療の用途が最も適切である活動性疾患のより良い指標を用意する。
【0022】
本発明において、「アミノ酸」という用語は、アミノ基、カルボキシル基、水素原子及びアミノ酸側鎖基からなり、これらがすべて(αアミノ酸の場合)α炭素と呼ばれる1つの炭素に結合したものをいう。アミノ酸としては、特に限定されないが、天然アミノ酸が挙げられる。天然アミノ酸は、天然タンパク質のアミノ酸単位が誘導されるものであり、当業者に周知である。本明細書でアミノ酸に関して用いられる「それらの誘導体」という用語は、側鎖が天然アミノ酸の側鎖の誘導体であるものをいう(“Amino Acid Derivatives” 1999 Oxford University Press, Barrett, Ed.参照)。
【0023】
「アミノ酸模倣体」という用語は、天然アミノ酸の合成類似体であってアイソスターであるもの、つまり天然化合物の立体及び電子構造を模倣して設計されたものを意味する。かかるアイソスターは当業者に周知であり、特に限定されないが、デプシペプチド、レトロインベルソペプチド、チオアミド、シクロアルカン又は1,5−二置換テトラゾールなどが挙げられる(M. Goodman, Biopolymers, 24, 137, (1985)参照)。
【0024】
「環化架橋」という用語は、架橋を導入できる官能基をもつアミノ酸同士又はアミノ酸と−(CH2)O−もしくは−(CH2)O−C6H4−基の組合せをいう。oは1〜10の正の整数を表す。好ましい例は、ジスルフィド、−(CH2)4−カルバ架橋のようなジスルフィド模倣体、チオアセタール架橋、チオエーテル架橋(シスタチオン又はランチオニン)、エステル及びエーテルを含有する架橋、並びにアミド架橋である。好ましくは、1つの架橋はジスルフィド結合を形成し、第二の架橋はチオエーテル(スルフィド)結合からなる。例えば、環化架橋がX2とX4のような2つのアミノ酸で形成されている場合、システイン又はホモシステインの側鎖がシステイン、ホモシステイン、セリン、スレオニン又はアルデヒド含有アミノ酸の側鎖と連結して環化架橋を形成する。
【0025】
「アルギニン模倣体」という用語は、アミノ酸模倣体に関する上記の定義と同様に通り、天然アルギニンの合成類似体であるアイソスターである。
【0026】
式Iの化合物のペプチドは、あらゆる公知の化学合成法で合成できるが、特に有用な方法は自動ペプチド合成装置を用いたMerrifieldの固相法である(J. Am. Chem. Soc., 85: 2149 (1964))。合成法の標準的手順は、E.Atherton & R.C.Sheppard, “Solid phase peptide synthesis: a practical approach, 1989, IRL Press, Oxfordに記載されている。
【0027】
酸不安定リンカー基を有する合成樹脂を用いて、これに、C末端を保護した所望のアミノ酸残基をアミド結合の形成によって結合させる。例えば、(ジメトキシフェニル−アミノメチル)−フェノキシ誘導リンカーを有するいわゆるRinkアミドAM樹脂を用いることができる(Rink, H. (1987), Tetrahedron Lett. 30, p.3787)。この樹脂からペプチドを酸分解で開裂させると、ペプチドアミドが得られる。別法として、O−ビス−(アミノエチル)エチレングリコールトリチル樹脂(K. Barlos et al (1988), Liebigs Ann. Chem, p.1079)を用いると、酸分解開裂によって第一級アミンハンドルを有するペプチドを得ることができる。
【0028】
イメージング部分での標識は好適には「前駆体化合物」によって実施されるが、前駆体化合物は、式Iの化合物の誘導体であって、適当な化学的形態の1以上のイメージング部分との化学反応が部位特異的に起こり、最小限の段階数(理想的には一段階)で実施でき、しかも多大な精製を行わずに(理想的にはそれ以上精製しなくても)所望の式Iの化合物が得られるように設計される。かかる前駆体化合物は合成品であり、良好な化学的純度で得ることができる。前駆体化合物は、任意には、式Iの化合物の官能基に対する保護基を含んでいてもよい。
【0029】
「保護基」という用語は、不都合な化学反応は阻害又は抑制するが、分子の残りの部分を修飾しない十分穏和な条件下で当該官能基から脱離させることができる十分な反応性をもつように設計された基を意味する。脱保護後に、所望の生成物が得られる。保護基は当業者に周知であり、好適には、アミン基については、Boc(Bocはtert−ブチルオキシカルボニルである。)、Fmoc(Fmocはフルオレニルメトキシカルボニルである。)、トリフルオロアセチル、アリルオキシカルボニル、Dde[すなわち、1−(4,4−ジメチル−2,6−ジオキソシクロヘキシリデン)エチル]又はNpys(すなわち、3−ニトロ−2−ピリジンスルフェニル)から、カルボキシル基については、メチルエステル、tert−ブチルエステル又はベンジルエステルから選択される。
ヒドロキシ基に対する適当な保護基は、メチル、エチル又はtert−ブチル、アルコキシメチル又はアルコキシエチル、ベンジル、アセチル、ベンゾイル、トリチル(Trt)或いはテトラブチルジメチルシリルのようなトリアルキルシリルである。チオール基に対する適当な保護基は、トリチル及び4−メトキシベンジルである。その他の保護基の使用については、‘Protective Groups in Organic Synthesis’,Theorodora W.Greene and Peter G.M.Wuts(Third Edition、John Wiley & Sons,1999)に記載されている。
【0030】
本発明の「リンカー基」は式−(L)n−の基である。
式中、
各Lは独立に−C(=O)−、−CR′2−、−CR′=CR′−、−C≡C−、−CR′2CO2−、−CO2CR′2−、−NR′−、−NR′CO−、−CONR′−、−NR′(C=O)NR′−、−NR′(C=S)NR′−、−SO2NR′−、−NR′SO2−、−CR′2OCR′2−、−CR′2SCR′2−、−CR′2NR′ CR′2−、C4-8シクロヘテロアルキレン基、C4-8シクロアルキレン基、C5-12アリーレン基、C3-12ヘテロアリーレン基、アミノ酸、ポリアルキレングリコール、ポリ乳酸又はポリグリコール酸部分であり、
nは1〜15の整数であり、
各R′基は独立にH又はC1-10アルキル、C3-10アルキルアリール、C2-10アルコキシアルキル、C1-10ヒドロキシアルキル、C1-10フルオロアルキルであるか、或いは2以上のR′基がそれらに結合した原子と共に炭素環、複素環式、飽和又は不飽和環を形成したものである。
【0031】
ただし、リンカー基は100個以下の原子鎖、好ましくは50個以下の原子鎖である。リンカー基は、最も好ましくは10〜50個の原子鎖、特に好ましくは、10〜30個の原子鎖である。好ましいL基は、−C(=O)−、−CH2−、−NH−、−NHC(=O)−、−C(=O)NH−、−CH2−O−CH2−及びアミノ酸である。
【0032】
好ましくは、リンカーはバイオモディファイアー基としての機能する。「バイオモディファイヤー基」は式Iの化合物の薬物動態及び血中クリアランス速度を変化させる機能をもつ。適当なバイオモディファイヤー基の一例は、単分散PEG構成単位を1〜10単位含むものである。また、バイオモディファイアー基は1〜10個のアミノ酸残基も表すものであってもよい。バイオモディファイアー基の好ましいアミノ酸残基はリジン及びグルタミン酸のような荷電アミノ酸、又はシステイン酸及びホスホノアラニンのような荷電非天然アミノ酸である。さらに、アミノ酸グリシン、アスパラギン酸及びセリンを含んでいてもよい。好ましい実施形態では、バイオモディファイアー基は単分散PEG様構造の式(II)の17−アミノ−5−オキソ−6−アザ−3,9,12,15−テトラオキサヘプタデカン酸を含む表す。
【0033】
【化2】
【0034】
式中、mは1〜10の整数であり、C末端単位はアミド基である。バイオモディファイアー基は、化合物の薬物動態及び血中クリアランス速度を変化させる機能をもつ。本発明におけるバイオモディファイアーの機能は、組織中への取込みを減少させ、腎臓からの排泄を増大させ、もってバックグラウンド干渉を低減して優れたインビボ画像を与える。バイオモディファイアー基は、グルタル酸及び/又はコハク酸及び/又はポリエチレングリコール系単位及び/又は上記の式IIの単位から誘導される部分を表すものであってもよい。リンカー基の性状は、式Iの化合物の標的受容体に対する親和性を損なうべきではない。さらに、リンカー基は、式Iの化合物のバックグラウンド肝臓取込みを増加させるべきではないが、これは、過度に大きなポリエチレングリコール系単位を使用した場合に起きりかねない。
【0035】
Z1又はZ2のいずれかが糖部分である場合、それらもバイオモディファイアー基として機能し得る。「糖部分」は炭水化物基でり、通常は多価アルコールのアルデヒド又はケトン誘導体である。糖部分は、フルクトース又はグルコースのような単量体(単糖)であってもよいし、糖が2つ結合して二糖を形成したものであってもよい。二糖には、グルコースとフルクトースとで構成されたスクロースのような糖がある。糖という用語には、置換及び非置換糖並びに糖誘導体が包含する。好ましくは、糖は、グルコース、グルコサミン、ガラクトース、ガラクトサミン、マンノース、ラクトース、フコース及びそれらの誘導体、例えばシアル酸、グルコサミンの誘導体から選択される。糖は好ましくはα又はβである。糖は、特に、マンノピラノシド又はガラクトースピラノシドであってもよい。糖のヒドロキシル基は、例えば1以上のアセチル基で保護してもよい。糖部分は好ましくはN−アセチル化される。かかる糖の好ましい例としては、N−アセチルガラクトサミン、シアル酸、ノイラミン酸、N−アセチルガラクトース及びN−アセチルグルコサミンが挙げられる。
【0036】
「有機色素基」は、紫外乃至近赤外域の波長を有する電磁スペクトルの光と相互作用する有機色素であればよい。好ましい有機色素基としては、広範に非局在化した電子系を有する基が挙げられる。最も好ましい有機色素基はシアニン染料(CyDye(商標))である。シアニン色素は、奇数個の炭素原子が交互に炭素−炭素単結合と炭素−炭素多重(好ましくは二重)結合とで連結したポリエン連鎖であって、両端にアミノ基を有していてその一方が第四級化されているものとして定義される化合物である。シアニン及び類似のアリール−リンカー−アリール発色団は、任意にはペンダント置換基又は縮合環置換基を有する。シアニン染料及びその合成に関する一般的な説明は、米国特許第6048982号、同第5268486号及び欧州特許第1037947号に記載されている。本発明では、シアニン色素は好ましくはカルバシアニン、オキサシアニン、チアシアニン及びアザシアニンからなる群から選択される。
【0037】
本明細書に記載した方法の「検出」段階は、式Iの「イメージング部分」から放出される信号を、信号の検出器によって検出することを含む。この検出段階は、信号データの取得として理解することもできる。本発明での使用に適したイメージング部分から放出される信号の例は、(i)γ線のように人体の外部で検出できるもの、又は(ii)術中用に設計された放射線検出器のように、インビボ用に設計された検出器の使用によって検出できるものである。本明細書に記載された方法の「形成」段階は、コンピューターによって実施され、取得した信号データに再構成アルゴリズムを適用してデータセットを得る。このデータセットを操作して、被検体の関心領域を示す画像を形成する。
【0038】
本発明の化合物のイメージング部分は、好ましくは以下の(i)〜(iv)から選択される。
(i)放射性金属イオン、
(ii)γ線放出型放射性ハロゲン、
(iii)陽電子放出型放射性非金属、及び
(iv)常磁性金属イオン。
【0039】
イメージング部分が放射性金属イオンつまり放射性金属である場合、好適な放射性金属は、64Cu、48V、52Fe、55Co、94mTc又は68Gaのような陽電子放射体、或いは99mTc、111In、113mIn又は67Gaのようなγ線放射体である。好ましい放射性金属は99mTc、64Cu、68Ga及び111Inである。最も好ましい放射性金属はγ放射体、特に99mTcである。
【0040】
イメージング部分が常磁性金属イオンである場合、かかる金属イオンの好適なものとして、Gd(III)、Mn(II)、Cu(II)、Cr(III)、Fe(III)、Co(II)、Er(II)、Ni(II)、Eu(III)又はDy(III)が挙げられる。好ましい常磁性金属イオンはGd(III)、Mn(II)及びFe(III)であり、Gd(III)が特に好ましい。
【0041】
式Iの化合物のイメージング部分が金属イオンである場合、イメージング部分は好ましくは金属イオンと合成配位子との金属錯体として存在する。「金属鎖体」という用語は、金属イオンと1以上の配位子との配位鎖体を意味する。金属鎖体は「キレート交換耐性」、つまり金属の配位部位に対する他の潜在的な競合配位子との配位子交換を容易に起こさないものであるのが極めて好ましい。潜在的な競合配位子としては、インビトロ標品中の他の賦形剤(製剤に使用される例えば放射線防護剤又は抗菌保存剤)又は生体の内在性化合物(例えばグルタチオン、トランスフェリン又は血漿タンパク質)がある。「合成」という用語は、その通常の意味、つまり天然資源(例えば哺乳類の身体)から分離されたものではなく、人造のものを意味する。かかる化合物は、その製造並びに不純物プロファイルを十分に制御できるという利点がある。
【0042】
キレート交換耐性の金属鎖体を形成する本発明の使用に適した配位子としては、(金属ドナー原子同士が炭素原子又は非配位ヘテロ原子の非配位骨格で連結されて)五又は六員キレート環が形成されるように2〜6、好ましくは2〜4個の金属ドナー原子が配列したキレート剤、或いはイソニトリル、ホスフィン又はジアゼニドのように金属イオンに強く結合するドナー原子を含む単座配位子が挙げられる。キレート剤の一部として金属によく結合するドナー原子の例は、アミン、チオール、アミド、オキシム及びホスフィンである。ホスフィン類は強固な金属鎖体を形成し、単座又は二座ホスフィンであっても適当な金属鎖体を形成する。イソニトリル及びジアゼニドの線状構造は、それらをキレート剤に導入するのが容易ではないので、通例、単座配位子として使用される。適当なイソニトリルの例としては、tert−ブチルイソニトリルのような単純なアルキルイソニトリル及びMIBI(すなわち、1−イソシアノ−2−メトキシ−2−メチルプロパン)のようなエーテル置換イソニトリルが挙げられる。適当なホスフィンの例としては、テトロホスミン及び単座ホスフィン類、例えばトリス(3−メトキシプロピル)ホスフィンが挙げられる。適当なジアゼニドの例としては、配位子のHYNIC系配位子、すなわちヒドラジン置換ピリジン又はニコチンアミドが挙げられる。
【0043】
金属イオンがテクネチウムである場合、キレート交換耐性の金属鎖体を形成するのに適したキレート剤の例としては、特に限定されないが、以下の(i)〜(v)が挙げられる。
(i)ジアミンジオキシム;
(ii)チオールトリアミドドナーセットを有するN3S配位子、例えばMAG3(メルカプトアセチルトリグリシン)及び関連配位子、又はジアミドピリジンチオールドナーセットを有するもの、例えばPica;
(iii)ジアミンジチオールドナーセットを有するN2S2配位子、例えばBAT又はECD(すなわちエチルシステイネート二量体)又はアミドアミンジチオールドナーセットを有するもの、例えばMAMA;
(iv)テトラミン、アミドトリアミン又はジアミンジアミンドナーセットを有する開環又はマクロ環状配位子であるN4配位子、例えばサイクラム、モノオキシサイクラム又はジオキシサイクラム;或いは
(v)ジアミンジフェノールドナーセットを有するN2O2配位子。
【0044】
イメージング部分がテクネチウムである場合、本発明の好ましいキレート剤はジアミンジオキシム及びテトラアミンであり、それらの好ましいものについて以下で詳しく説明する。
【0045】
好ましいジアミンジオキシムは、次の式VIIのものである。
【0046】
【化3】
【0047】
式中、
E1〜E6は各々独立にR*基であり、
各R*はH、C1-10アルキル、C3-10アルキルアリール、C2-10アルコキシアルキル、C1-10ヒドロキシアルキル、C1-10フルオロアルキル、C2-10カルボキシアルキル又はC1-10アミノアルキルであるか、或いは2以上のR*基がそれらと結合した原子と共に飽和又は不飽和炭素環又は複素環を形成するもので、1以上のR*基がCBPと結合しており、
Q′は式−(J′)e−の架橋基であり、eは3、4又は5であり、各J′は独立に−O−、−NR*−又は−C(R*)2−であるが、−(J′)f−が、−O−又は−NR*−であるJ′基を最大1個しか含まないことを条件とする。
【0048】
好ましいQ′基は以下のものである。
Q′=−(CH2)(CHR*)(CH2)−、すなわちプロピレンアミンオキシムつまりPnAO誘導体、
Q′=−(CH2)2(CHR*)(CH2)2−、すなわちペンチレンアミンオキシムつまりPentAO誘導体、
Q′=−(CH2)2NR*(CH2)2−。
【0049】
E1〜E6は、好ましくは、C1-3アルキル、アルキルアリール、アルコキシアルキル、ヒドロキシアルキル、フルオロアルキル、カルボキシアルキル、アミノアルキルから選択される。最も好ましくは、各E1〜E6基はCH3である。
【0050】
ジアミンジオキシムは、好ましくは、E1もしくはE6のR*基又はQ′部分のR*基で結合している。最も好ましくは、Q′部分のR*基で結合する。Q′部分のR*基で結合している場合、R*基は好ましくは橋頭位である。この場合、Q′は、好ましくは、−(CH2)(CHR*)(CH2)−、−(CH2)2(CHR*)(CH2)2−、又は−(CH2)2NR*(CH2)2−であり、最も好ましくは−(CH2)2(CHR*)(CH2)2−である。特に好ましい二官能性ジアミンジオキシムキレート剤は、次の式VIIaのものである。
【0051】
【化4】
【0052】
式中、E7〜E20は各々独立に上記で定義したR*基であり、
GはN又はCR*であり、
Y′は式Iのペプチド部分との連結点である。
【0053】
式VIIaの好ましいキレート剤は、次の式VIIbのものである。
【0054】
【化5】
【0055】
式中、Gは上記で定義した通りであり、好ましくはCHである。キレートIの調製方法は、国際公開第03/006070号に開示されている。
【0056】
好ましいテトラアミンキレート剤は、次の式VIIIのものである。
【0057】
【化6】
【0058】
式中、
Y″は式Iの残りの部分との連結点であり、
E21〜E26は上記で定義したR*基である。
【0059】
最も好ましいテトラアミンキレートは式VIIIaであり、
【0060】
【化7】
【0061】
式中、
Y″は上記で定義した通りである。式VIIIaのキレートの合成方法は国際公開第06/008496号に開示されている。
【0062】
上述の配位子は、テクネチウム(例えば94mTc又は99mTc)の錯体の形成に特に適しており、Jurisson et al, Chem.Rev.,99,2205−2218(1999)に詳細に記載されている。この配位子は、銅(64Cu又は67Cu)、バナジウム(例えば48V)、鉄(例えば52Fe)又はコバルト(例えば55Co)のような他の金属にも有用である。
【0063】
その他の適当な配位子については、インジウム、イットリウム及びガドリニウム、特に大環状アミノカルボキシレート及びアミノホスホン酸配位子に特に適した配位子を始めとして、Sandozの国際公開第91/01144号に記載されている。ガドリニウムの非イオン性(すなわち中性)の金属錯体を形成する配位子は公知であり、米国特許第4885363号に記載されている。
かかるドナー原子を有する好適なキレート剤の例としては、1、4、7、10−テトラアザシクロデカン−1、4、7、10−テトラ酢酸(DOTA)及びジエチレントリアミンペンタ酢酸(DTPA)が挙げられる。ガドリニウムに対して特に好ましいのは、DTPA、エチレンジアミン四酢酸(EDTA)、トリエチレンテトラアミン六酢酸(TTHA)、1,4,7,10−テトラアザシクロドデカン−1,4,7,10−四酢酸(DOTA)、10−(2−ヒドロキシプロピル)−1,4,7,10−テトラアザシクロドデカン−1,4,7−三酢酸(DO3A)及びこれらの誘導体を始めとするキレートである。
【0064】
イメージング部分が、金属錯体の一部として存在する金属イオンである場合、リンカー基が付随して存在するのが好ましい。こうした場合のリンカー基の役割は、金属の配位で得られる比較的嵩高い金属錯体を、ペプチドの活性部位から遠ざけることによって、例えば、基質の結合に支障をきたさないようにすることである。これは、嵩高い基が活性部位から遠ざかる自由度をもつようにするための柔軟性(例えば単純なアルキル鎖)及び/又は金属錯体を活性部位から遠ざけるシクロアルキル系もしくはアリール系スペーサーのような剛直性の組合せによって達成することができる。こうしたキレートに関して好ましいリンカー基は、原子数2〜10、最も好ましくは原子数2〜5の骨格鎖を含むもので、原子数2又は3のものが特に好ましい。原子数2の最短のリンカー基骨格鎖であっても、ペプチドからキレーターが十分に離隔して相互反応が最小限になるという利点が得られる。さらに、ペプチドは、金属イオンへのキレーターの配位に有効に拮抗できなくなる。こうして、ペプチドの生物学的ターゲティング特性とキレーターの金属錯形成能が共に保持される。なお、金属錯体がペプチドにそれらの結合が血中で容易に代謝されないように結合していることが強く望まれる。かかる代謝によって、標識ペプチドがインビボで所望の標的部位に達する前に、イメージング金属錯体が開裂してしまうからである。そこで、ペプチドは、好ましくは、容易には代謝されない結合を含むリンカー基を介して金属錯体に共有結合させる。かかる好適な結合は、炭素−炭素結合、アミド結合、尿素もしくはチオ尿素結合又はエーテル結合である。
【0065】
アルキレン基又はアリーレン基のような非ペプチド系リンカー基は、式Iのペプチドと有意の水素結合がないため、リンカーがペプチドと相互作用しないという利点を有する。好ましいアルキレンスペーサー基は−(CH2)q−であり、式中、qは2〜5の整数である。好ましくは、qは2又は3である。好ましいアリーレンスペーサーは次の式IXのものである。
【0066】
【化8】
【0067】
式中、a及びbは各々独立に0、1又は2である。
【0068】
好ましいリンカー基は−CH2CH2−(L)p−であり、Lは上記で定義した通りであり、pは0〜3の整数である。最も好ましくは、−(L)p−は−CO−又は−NR−である。式VIIbについて、GがNであって、−(L)p−が−NH−である場合、この組合せは市販の対称中間生成物N(CH2CH2NH2)3から誘導できるという追加の利点がある。
【0069】
イメージング用金属がテクネチウムの場合、通常のテクネチウム出発原料は過テクネチウム酸塩、すなわちTcO4-つまり酸化状態がTc(VII)のテクネチウムである。過テクネチウム酸塩自体は金属錯体を形成しにくいので、テクネチウム錯体の製造に際しては、テクネチウムの酸化状態を低酸化状態(Tc(I)〜Tc(V))に還元することによって錯形成を促進するため、第一スズイオンのような適当な還元剤を添加する必要がある。溶媒は有機溶媒でも、水性溶媒でも、それらの混合物でもよい。溶媒が有機溶媒を含む場合、有機溶媒は好ましくはエタノール又はDMSOのような生体適合性溶媒である。好ましくは、溶媒は水性溶媒であり、最も好ましくは等張塩類溶液である。
【0070】
イメージング部分がγ線放出型放射性ハロゲンである場合、放射性ハロゲンは好適には123I、131I又は77Brから選択される。125Iは、外部からのインビボイメージング用のイメージング部分としての使用には適していないので、特に除外してある。インビボイメージングに好ましいγ線放出型放射性ハロゲンは123Iである。
【0071】
イメージング部分が放射性ヨウ素である場合、式Iの化合物は、求電子又は求核ヨウ素化或いは標識アルデヒド又はケトンとの縮合を起こすような誘導体を含む前駆体化合物によって得ることができる。前者に属するものの例としては、以下の(a)〜(c)が挙げられる。
(a)トリアルキルスタンナン(例えばトリメチルスタンニル又はトリブチルスタンニル)、トリアルキルシラン(例えばトリメチルシリル)又は有機ホウ素化合物(例えば、ボロン酸エステル又はオルガノトリフルオロボレート)のような有機金属誘導体、
(b)ハロゲン交換のための非放射性臭化アルキル、或いは求核ヨウ素化のためのアルキルトシレート、メシレート又はトリフレート、
(c)求電子ヨウ素化用の活性化芳香族環(例えばフェノール)及び求核ヨウ素化用の活性化芳香族環(例えばアリールヨードニウム、アリールジアゾニウム、アリールトリアルキルアンモニウム塩又はニトロアリール誘導体)。
【0072】
かかる前駆体として好適なものは、ヨウ化又は臭化アリールのような非放射性ハロゲン原子(放射性ヨウ素交換を可能とするため)、有機金属前駆体化合物(例えばトリアルキルスズ、トリアルキルシリル又は有機ホウ素化合物)、トリアゼンのような有機前駆体、或いはヨードニウム塩のような求核置換反応のための良好な脱離基を含む。放射性ヨウ素化では、前駆体は好ましくは有機金属前駆体化合物を含み、最も好ましくはトリアルキルスズを含む。
【0073】
前駆体及び放射性ヨウ素を有機分子に導入する方法は、Bolton, J.Lab.Comp.Radiopharm.,45,485−528(2002)に記載されている。適当なボロン酸エステル有機ホウ素化合物及びその製造方法は、Kabalaka et al, Nucl.Med.Biol.,29、841−843(2002)及び30,369−373(2003)に記載されている。適当なオルガノトリフルオロホウ化物及びその製造方法は、Kabalaka et al, Nucl.Med.Biol.,31、935−938(2004)に記載されている。
【0074】
放射性ヨウ素を結合させることのできるアリール基の例としては、以下のものがある。
【0075】
【化9】
【0076】
これらはいずれも、芳香族環での放射性ヨウ素置換が容易な置換基を含んでいる。チロシン残基では、その内在フェノール基を利用して放射性ヨウ素化を実施できる。
【0077】
放射性ヨウ素を含む他の置換基は、例えば以下のような放射性ハロゲン交換による直接ヨウ素化によって合成することができる。
【0078】
【化10】
【0079】
飽和脂肪族系に結合したヨウ素原子はインビボで代謝され易く、放射性ヨウ素が失われ易いことが知られているので、放射性ヨウ素原子は好ましくは芳香族環(ベンゼン環など)又はビニル基に直接共有結合で結合させる。
【0080】
イメージング部分が陽電子放出型放射性非金属である場合、かかる陽電子放射体の好適なものとして、11C、13N、15O、17F、18F、75Br、76Br又は124Iが挙げられる。好ましい陽電子放出型放射性非金属は11C、13N、18F及び124Iであり、特に好ましくは11C及び18Fであり、最も好ましくは18Fである。
【0081】
放射性フッ素化は、臭化アルキル、アルキルメシレート又はアルキルトシレートのような良好な脱離基を有する前駆体化合物の適当な化学基と18F−フッ化物との反応を用いた直接標識法で実施できる。18Fは、18F(CH2)3OH反応体を用いたN−ハロアセチル基のアルキル化によって導入することもでき、−NH(CO)CH2O(CH2)318F誘導体が得られる。アリール系については、アリールジアゾニウム塩、アリールニトロ化合物又はアリール第四級アンモニウム塩からの18F−フッ化物求核置換が、アリール−18F誘導体への好適な経路である。
【0082】
本発明の18F−標識化合物は、18Fフルオロジアルキルアミンの形成後、18Fフルオロジアルキルアミンを、例えば塩素、P(O)Ph3又は活性化エステルを含む前駆体と反応させてアミドを形成することによって得ることができる。
【0083】
ペプチドの放射性フッ素化に特に適した別の放射性フッ素化法は、国際公開第03/080544号に記載されており、チオールカップリングを利用する。以下の置換基のいずれかを含む前駆体化合物を式Xの化合物と反応させて、それぞれ式(Xa)又は(Xb)の放射性フッ素化イメージング剤を生成させる。
【0084】
【化11】
【0085】
18F−XX−SH (X)
式中、YXは式−(LY)w−のリンカーであって、適宜1〜6のヘテロ原子を含んでいてもよく(LYはLについて上記で定義した通りであり、yは1〜10である。)、
XXは、式−(LX)x−のリンカーであって、適宜1〜10のヘテロ原子を含んでいてもよく(LXはLについて上記で定義した通りであり、xは1〜30である。)、
*は化合物の残りの部分との連結点である。
【0086】
【化12】
【0087】
式中、XX、YX及び*は、上記で定義した通りである。
【0088】
ペプチドの放射性フッ素化に特に適した追加の方法は、国際公開第04/080492号に記載されており、アミノキシカップリングを利用する。放射性フッ素化は、式(XI)の前駆体化合物を式(XIa)の化合物と反応させるか、或いは式(XII)の前駆体化合物を式(XIIa)と反応させることによって実施され、それぞれ式(XIII)又は(XIV)のコンジュゲートが得られる。
【0089】
【化13】
【0090】
式中、XXI及びXXIIは、リンカー基−(LXI)z−であって、適宜1〜6のヘテロ原子を含んでいてもよい(LXIは、Lについて上記で定義した通りであり、zは1〜10である。)。
R1は、アルデヒド部分、ケトン部分、アセタールのような保護アルデヒド、ケタールのような保護ケトン、或いは酸化剤を用いてアルデヒド又はケトンへと迅速かつ効率的に酸化できるジオール又はN−末端セリン残基のような官能基である。
R2は、水性緩衝液のような穏和な条件下でR1と部位特異的に反応して安定なコンジュゲートを与える官能基である。R2は、第一級アミン、第二級アミン、ヒドロキシルアミン、ヒドラジン、ヒドラジド、アミノキシ、フェニルヒドラジン、セミカルバジド又はチオセミカルバジドのようなアンモニア誘導体であってもよく、好ましくは、ヒドラジン、ヒドラジド又はアミノキシ基である。
R3は、R4と部位特異的に反応する官能基である。R3は、第一級アミン、第二級アミン、ヒドロキシルアミン、ヒドラジン、ヒドラジド、アミノキシ、フェニルヒドラジン、セミカルバジド又はチオセミカルバジドなどのアンモニア誘導体であってもよく、好ましくは、ヒドラジン、ヒドラジド又はアミノキシ基である。
R4は、アルデヒド部分、ケトン部分、アセタールのような保護アルデヒド、ケタールのような保護ケトン、或いは酸化剤を用いてアルデヒド又はケトンへと迅速かつ効率的に酸化できるジオール又はN−末端セリン残基のような官能基である。
【0091】
【化14】
【0092】
式中、Wは−CO−NH−、−NH−、−O−、−NHCONH−又は−NHCSNH−であり、好ましくは−CO−NH−、−NH−又は−O−であり、YはH、C1〜6アルキル又はC5〜6アリール置換基であり、XXI、XXII及び*は上記で定義した通りである。
【0093】
18F−標識誘導体の合成経路についてのさらに詳しい内容は、Bolton,J.Lab.Comp.Radiopharm.,45,485−528(2002)に記載されている。
【0094】
好ましいイメージング部分は、生体に投与した後、単光子放射断層撮影(SPECT)、陽電子放射断層撮影(PET)及び磁気共鳴イメージング(MRI)などの手段によって外部から非侵襲的に検出することができるものである。最も好ましいイメージング部分は、放射性、特に、放射性金属イオン、γ線放出型放射性ハロゲン及び陽電子放出型放射性非金属、特に、SPECT又はPETを用いたイメージングに適したもの、例えば、99mTc、123I、11C及び18Fである。
【0095】
好ましい一実施形態では、W1及びW2は共にリンカー基を表し、Z1は有機色素基を表し、Z2はイメージング部分を表す。これらの化合物及びその調製方法は、国際公開第2006/054904号に記載されている。最も好ましくは、Z2は放射性イメージング部分である。これらの好ましい化合物の例としては、以下のものが挙げられる。
【0096】
【化15】
【0097】
【化16】
【0098】
化合物1〜4の調製方法は、国際公開第2006/054904号に詳述されている。
【0099】
別の好ましい実施形態では、W1はリンカー基を表し、Z1はイメージング部分を表し、W2は任意要素としてのリンカー基を表し、Z2は水素である。これらの化合物及びその調製方法は、国際公開第2005/012335号に記載されている。これらの好ましい化合物の例としては、以下のものが挙げられる。
【0100】
【化17】
【0101】
化合物5及び6の調製方法は、国際公開第2005/012335号に詳述されている。
【0102】
別の好ましい実施形態では、W1は任意要素としてのリンカー基を表し、Z1は水素を表し、W2はリンカー基を表し、Z2はイメージング部分を表す。これらの化合物及びその調製方法は、国際公開第2003/006491号に記載されている。これらの好ましい化合物の例としては、以下のものが挙げられる。
【0103】
【化18】
【0104】
【化19】
【0105】
化合物7〜10の調製方法は、国際公開第2003/006491号に詳述されている。
【0106】
化合物1〜4及び7〜10について、別法として、ジアミンジオキシムキレート部分は、化合物1a〜4a及び7a〜10aを形成するためにテトラアミンキレート部分で置き換えられていることがさらに好ましい。
【0107】
【化20】
【0108】
【化21】
【0109】
【化22】
【0110】
【化23】
【0111】
化合物1a〜10aの調製方法は、化合物1〜10のジアミンジオキシムキレートの代わりに化合物1a〜10aのテトラアミンキレートを用いる点を除いて、化合物1〜10の方法と類似している。テトラアミンキレートの連結は、Bocで保護された分子種との通常のペプチドカップリングを用いて達成される。
【0112】
生体分布を改善することを目的として、すなわち、主にバックグラウンド肝臓取込み低減するために、多くの追加の修飾化合物を合成した。化合物11〜15の合成の詳細を、以下の実施例8〜12に示す。化合物11は、ジアミンジオキシムキレートからテトラアミンキレートに変える効果を評価するために設計した。化合物12では、システイン酸基を加えた。化合物13は、ペプチドのN−末端側に追加のPEG部分を有する。化合物14は、ペプチドのC−末端側に追加のPEG部分を有する。化合物15では、テトラアミンキレートを、多くのグルタミン酸残基に加えて使用した。
【0113】
本明細書に記載されている方法は、検出可能な量の式Iの化合物を投与しておいた被検体を「準備」することから始める。本発明の方法の目的は、診断上有用な画像の用意である。従って、式Iの化合物の被検体への投与は、画像の形成を容易にするために必要な予備段階であると理解することができる。好ましくは、被検体は、哺乳類、最も好ましくはヒトである。最も好ましくは、被検体は、インビボでのインタクトな哺乳類の身体である。従って、好ましい実施形態では、式Iの化合物は、生体適合性担体と共に、哺乳類への投与に適した形態の化合物を含む医薬組成物として投与されてきた。投与の好ましい経路は、血管内投与である。代替実施形態では、検出可能な量の式Iの化合物の投与は、本方法の一部として行われる。
【0114】
用意する段階に続いて検出段階に先立って、式Iの化合物を、被検体における任意の線維形成性組織に結合させる。例えば、被検体がインタクトな哺乳類である場合、式Iの化合物は、哺乳類の身体を動的に進み、その中の様々な組織と接触する。化合物が、任意の線維形成性組織に接触するとすぐに、線維形成性組織からの化合物のクリアランスが、非線維形成性組織からより時間がかかるように、特異的相互作用が起きる。線維形成性組織と特異的に結合している化合物の検出が、線維形成性組織と結合している化合物と非線維形成性組織に結合している化合物の比の結果として可能になるある時点に到達する。理想的なそのような比は、2:1以上である。
【0115】
「生体適合性担体」とは、式Iの化合物を懸濁又は溶解できる流体、特に液体であって、組成物が生理学的に認容できるもの、つまり毒性も耐え難い不快感も伴わずに哺乳類の身体に投与することができるものである。生体適合性担体は好適には注射可能な担体液であり、例えば、発熱物質を含まない注射用の滅菌水、食塩液のような水溶液(これは注射用の最終製剤が等張性又は非低張性となるように調整するのに都合がよい)、1種以上の張度調節物質(例えば血漿陽イオンと生体適合性対イオンとの塩)、糖(例えばグルコース又はスクロース)、糖アルコール(例えばソルビトール又はマンニトール)、グリコール(例えばグリセロール)その他の非イオン性ポリオール材料(例えばポリエチレングリコール、プロピレングリコールなど)の水溶液である。生体適合性担体は、エタノールのような生体適合性の有機溶媒を含んでいてもよい。かかる有機溶媒は、親油性の高い化合物又は製剤の可溶化に有用である。好ましくは、生体適合性担体は発熱物質を含まない注射用水、等張塩類溶液又はエタノール水溶液である。静脈内注射用の生体適合性担体のpHは好適には4.0〜10.5の範囲内である。
【0116】
かかる医薬組成物は、好適には、無菌状態を維持したまま皮下注射針で一回又は複数回穿刺するのに適したシール(例えばクリンプオン式セプタムシール蓋)を備えた容器に入れて供給される。かかる容器には、1回又は複数回分の患者用量を入れることができる。好ましい多用量容器は、複数回分の患者用量を収容した単一バルクバイアル(例えば容積10〜30cm3のもの)からなり、臨床症状に応じて製剤の有効期間中様々な時間間隔で1回分の患者用量を臨床グレードのシリンジに吸引することができる。プレフィルドシリンジは1回分の患者用量つまり「単位用量」を収容するように設計され、そのため好ましくは使い捨て又はその他臨床用に適したシリンジである。医薬組成物が放射性医薬組成物の場合、プレフィルドシリンジは、適宜、オペレーターを放射能被曝から保護するためのシリンジシールドを備えていてもよい。かかる適当な放射性医薬品シリンジシールドは当技術分野で公知であり、好ましくは鉛又はタングステンからなる。
【0117】
医薬組成物は、キットから調製することができる。或いは、医薬組成物は、所望の滅菌生成物が得られるような無菌製造条件下で製造してもよい。医薬組成物を非滅菌条件下で調製した後、例えばγ線照射、オートクレーブ処理、乾熱又は化学的処理(例えばエチレンオキサイドでの処理)を用いて最終的に滅菌してもよい。
【0118】
本発明の造影剤に関して上述した通り、放射性医薬組成物に対して最も好ましい本発明の放射性造影基は99mTc、123I、11C及び18Fである。
【0119】
本発明の別の態様では、式Iの化合物は、被検体の器官又は身体領域における線維形成の存在、場所及び/又は量を決定するための医薬品の調製に用いることができる。式Iの好ましい及び最も好ましい実施形態、器官又は身体領域、被検体及び投与方法は、上記で定義した通りである。
【0120】
本発明は、活性化HSCの存在、場所及び/又は量を評価するのに有用であり、線維形成の指標を与える。これが特に有利であるのは、線維形成性組織が、線維性組織よりも早期活動性疾患の優れたマーカーであり、繊維性組織は、疾患プロセスが解消しつつある場所にも存在するからである。従って、疾患プロセスの識別を、治療の実施が最も有効であるステージで行うことができる。
【0121】
これらの利点を、下記の非限定的な実施例で例証する。
【0122】
実施例の簡単な説明
実施例1は、化合物2が、インビトロで活性化ヒトHSCと特異的に結合することを例証する。
【0123】
実施例2は、ラット胆管結紮モデル及び関連する偽手術モデルを準備するのに使用される方法、並びに組織病理学的検証について記載する。
【0124】
実施例3は、化合物4の取込みと線維形成の相関関係を例証する。線維形成のマーカーは、術後15日目で(実施例2に記載されている)ラット胆管結紮(BDL)モデルにおいて高く、ラットBDLモデルにおける線維形成性肝臓中への化合物4の取込みは、術後15日目で最も高い。
【0125】
実施例4は、BDL線維形成性肝臓との化合物4の結合が、非放射性の化合物4により特異的に阻害されることを例証する。
【0126】
実施例5は、化合物4の肝臓取込みとαvインテグリンの肝臓発現の相関関係を例証する。
【0127】
実施例6は、BDL線維形成性肝臓との化合物8の結合が、非放射性の化合物8により特異的に阻害されることを例証する。
【0128】
実施例7は、αv発現と化合物8の取込みの相関関係を例証する。
【0129】
実施例8〜12は、化合物11〜15の合成について記載する。
【0130】
【表1】
【実施例】
【0131】
実施例1:活性化ヒトHSCへTの化合物6の結合
1(i) EA−Hy926細胞から調製した膜との結合
化合物6の阻害定数を、報文に記載された膜結合アッセイを使用して測定した(Indrevoll et al, Bioorg & Med Chem Lett, 2006, 16, 6190−6193)。
【0132】
簡潔に述べると、ヒト内皮腺癌細胞系EA−Hy926からの膜を調製し、精製した膜画分についてKdを計算した。次いで、競合的結合アッセイを確立し、非放射性の化合物6についての阻害定数を測定した。125I−エキスタチンを標識リガンドとして使用し、非放射性のエキスタチンを参照標準として使用した。
【0133】
非放射性の試験化合物(非放射性のエキスタチン又は非放射性の化合物6のいずれか)の合計16通りの希釈液を調製し、125I−エキスタチンと膜の組合せと混合してから37℃で1時間インキュベーションした。数回の洗浄後、結合した材料を、Skatronマイクロハーベスターを使用してフィルター上に収集した。最後に、フィルタースポットを切り取り、Packard γ−カウンター中でカウントした。
【0134】
図1は、EA−Hy926膜上の非放射性の化合物6及びエキスタチンと対比した125I−エキスタチンの結合を図示している。
【0135】
1(ii) 活性化ヒト肝星細胞との結合
活性化ヒト肝星細胞LX−2(Mount Sinai School of Medicine(米国ニューヨーク)のScott L.Friedman教授から提供されたもの)を、10%FBS、ペニシリン、ストレプトマイシン及びグルタミンを含有するダルベッコ変法イーグル培地中でコンフルエンスまで12ウェルプレート(Nunc)において培養した。細胞を、50mM Tris、pH7,4、150mM NaCl、5mM MnCl2、1mM CaCl2及び0,01% BSAを含有する冷緩衝液中で2回洗浄した。細胞を、攪拌しながら4℃で60分間、様々な濃度の非放射性の化合物と一緒に、痕跡量の標識合物(0.1nM 125I化合物又は0.1nM 125I−エキスタチンのいずれか)を含有する緩衝液中でさらにインキュベートした。インキュベーション後、非結合材料を、冷緩衝液で3回細胞を洗浄することにより除去した。細胞を、0.1M NaOHを添加することにより壁から引き離し、チューブに移し、γ−カウンター(Packard)中でカウントした。図2は、観察された活性値を図示している。
【0136】
実施例2:化合物3及び化合物8の活性化ラットHSCとの結合
2(i) EA−Hy926細胞から調製した膜との結合
化合物3及び化合物8についての阻害定数を、実施例1に記載されている方法を使用して決定した。図3は、EA−Hy926膜上の非放射性の化合物3、化合物8及びエキスタチンと対比した125I−エキスタチンの結合を図示している。
【0137】
2(ii) 細胞及び培養
活性化ラット肝星細胞系(不死化)は、University of OsloのTrond Berg教授から、そのご好意によって入手した。細胞を、10%FBS、グルタミン及びペニシリン/ストレプトマイシンを含有するダルベッコ変法イーグル培地中でコンフルエンスまで150mmの培養フラスコにおいて培養した。
【0138】
2(iii) 膜の調製
細胞を、氷冷PBS、pH7.4中で2回洗浄し、PBS10mlを添加し、ラバーポリスマンでこそぎ落とし、氷上の50mlバイアルに移した。別のPBS10mlをフラスコに添加し、こそぎ落とし、第一の細胞と混合した。細胞を4℃で2000rpmで10分間遠心分離した。上清を流し捨て、ペレットをPBS3ml中に再懸濁した。すべてのペレットを1つのチューブ中に混合し、上記のように遠心分離し、最終ペレットを直ちに−70℃で冷凍した。
【0139】
細胞ペレットを、50mM Tris−HCl、5mM MgCl2、1mM EGTA、pH7,4と、さらに、プロテアーゼ阻害剤、10ug/mlロイペプチン、10ug/mlペプスタチン、200ug/mlバシトラシン、0.5ug/mlアプロチニン及び100uM PMSFを含有する氷冷ホモジネーション緩衝液(細胞の10倍量、例えば、ペレット2gにホモジネーション緩衝液20ml)中に再懸濁した。細胞を、Dounceホモジナイザー、ペストルB中で10ストローク3回により氷上でホモジナイズした。ホモジネートを、4℃で2100rpmで5分間遠心分離し、上清を流し捨てた。ペレットを、ホモジネーション緩衝液10ml中に再懸濁し、10ストローク2回で再びホモジナイズした後、10分間2100rpmで遠心分離した。この上清を第一の上清と混合し、JA−17ローター付きBeckman Coulter Centrifuge中、16500rpm(29000×g)で遠心分離した。ペレットを、ホモジネーション緩衝液20mL中に再懸濁し、遠心分離段階を繰り返した。
【0140】
上清を流し捨て、ペレットを、10mM Hepes、135mM NaCl、4,8mM KCl、1,7mM MgSO4、2,5mM CaCl2、1.0mM NaH2PO4、pH7,4を含有する結合緩衝液3ml中に再懸濁した。タンパク質測定のためのサンプルを取り除いた後、結合緩衝液さらに3mlを添加した。300ulのアリコートを、直ちに−80℃で冷凍した。
【0141】
2(iv) タンパク質測定
十分に混合した膜調製物100ulを、1:10に希釈し、段階希釈液及びタンパク質含量を、BCA Protein Assay Reagentキット(Pierce No.23225)中で取扱説明書に従って測定した。
【0142】
2(v) 星細胞膜上のインテグリンとの結合を試験する実験設計
実験を96ウェルプレート中で準備した。各ウェルに、緩衝液(50mM Tris、pH7,4、150mM NaCl、5mM MnCl2、1mM CaCl2、0,01% BSA)60ul、非放射性の化合物3、化合物8又は非放射性のエキスタチン20ul、125−Iエキスタチン20ul及び膜溶液(ウェル当たり膜約1ugに相当するように緩衝液中で1:30に希釈した)50ulを添加し、攪拌しながら60分間37℃でインキュベートした。インキュベーション後、非結合材料を、Skatron細胞ハーベスター中でPBSにより洗い流し、結合材料をフィルタースポット中に濃縮した。このフィルタースポットを切り取り、チューブに添加し、最終的に、γ−カウンター中でカウントした。フィルターは、4時間以上水中0.3%PEI中で予浸した。図4は、得られた結果を示している。
【0143】
実施例3:胆管結紮(BDL)及びシャム動物
3(i) 動物モデルの準備
非近交系雄性Sprague Dawleyラット(180〜200g;Charles River)を、すべての胆管結紮(BDL)及びシャム試験に使用した。6日の馴化後、ラットを、2つの群(BDL群及びシャム群)に分けた。
【0144】
BDL動物については、腹部を剪毛し、ベタジン溶液と、続いて、皮下(s.c.)のカルプロフェン5mg/kg及びs.c.のブプロノルフィン(bupronorphine)5mg/kgで消毒し、イソフルラン麻酔下で、正中開腹術を実施し、総胆管の場所を見つけた。胆管を二重結紮し、第一の結紮は肝管の結合部の間で行い、第二の結紮は、膵管の入口の上で行った。
【0145】
第二の群(シャム動物)の腹部を剪毛し、ベタジン溶液と、続いて、s.c.のカルプロフェン5mg/kg及びs.c.のブプロノルフィン5mg/kgで消毒した。動物は、胆管を操作し、縫合糸を胆管の下に通す偽手術を受けた。
【0146】
閉じる前に、食塩水2〜3mlを、各動物の腹膜中に投与した。筋膜及び皮膚を閉じ、動物にs.c.のメタクロプロミド(metaclopromide)2mg/kg、s.c.のバイトリル(Baytril)5mg/kg、及びs.c.の食塩水約2mlを投与した。カルプロフェン(5mg/kg)を、必要に応じて、続く2、3日間与えた。動物を、実験の期間中厳重にモニターした。
【0147】
3(ii) 試験化合物の投与及び生体分布
術後の適切な日に、BDL及びシャム動物を取り出し、イソフルラン麻酔下に置き、次いで、各動物に、尾静脈を介して静脈内(i.v.)に0.3ml(約3MBq)を注射した。試験品目の注射後の適切な時点で、各動物をイソフルランで再麻酔し、頸椎脱臼により屠殺して秤量し、バーコード走査システムを介して重量を記録した。各動物を解剖し、下記の器官及び組織を摘出し、BASILカウンタープロトコル40又は手動カウンティングを使用してカウントした。
【0148】
骨* 筋肉*
血液* 腎臓
膀胱及び尿(B/U) 肺
肝臓* 脾臓
胃及び内容物 小腸及び大腸(SI及びLI)
心臓 甲状腺
皮膚* 屠殺体
注射部位
*秤量したサンプル
全器官(例えば、肝臓)で記録された活性を、バックグラウンド放射能及び放射性崩壊に対して補正し、放射能の生体分布を、式Iを参照して計算した。
【0149】
【数1】
【0150】
秤量した組織サンプル(例えば、血液)における注射した放射能に占める割合を計算すると、式2から、全組織における%i.d.を得た。
【0151】
【数2】
【0152】
【表2】
【0153】
3(iii) 組織病理学的評価
各動物の肝臓からの切片を、室温で10%緩衝中性ホルマリン溶液中で固定し、パラフィンに包埋し、5μmの薄い切片を調製した。切片を、胆管過形成、壊死及び炎症の評価についてはMayer’s Hematoxylin and Eosin(H&E)、及び線維症については報文に記載された標準染色プロトコル(Gomori 1950 Am. J. Clin. Pathol. 20:661−663)を使用するGomori’sトリクロム染色(コラーゲン染色)で染色した。
【0154】
肝臓損傷の組織学的グレーディングを、各病変の範囲及び重症度を評価し、次いで、病変を以下の通り、すなわち、病変なし=0、最小=1、軽度=2、中等度=3、顕著=4、重度=5と0〜5に点数化することにより半定量的に評価した。組織の組織形態学的評価を、術後の日数と、手順(すなわち、シャム対胆管結紮)の両方に関してブラインドベースで実施した。全データを追加の病理学者がピアレビューした。病理学的分析は、光学顕微鏡により実施した。切片を、40倍、100倍又は400倍対物レンズを使用して明視野照明下で観察した。
【0155】
表4は、組織病理学的評価を要約している。
【0156】
【表3】
【0157】
提示されているデータは、線維形成に関係する特異的取込み示す分子を識別するための肝線維症の適当なモデルとしての、特に、15日目における、このモデルの使用を支持している。
【0158】
実施例4:シャムとラットBDLモデル(線維形成のマーカーが高いときの15日目)における線維形成性肝臓への化合物8の取込量の対比
BDL及びシャムモデルを、実施例3に記載されているように準備した。化合物8の取込み、以下の通り、
【0159】
【数3】
【0160】
屠殺体における放射能の割合を、解剖後の残留屠殺体における秒当たりカウントを参照し、屠殺体中に残っているサンプリング組織に対して補正することにより計算した。身体組成の生物学的変動は、組織値の推定におけるわずかな不正確さと、従って、屠殺体値の過度の補正又は補正不足をもたらしたかも知れない。屠殺体値が過度に補正された場合、これは、負の値をもたらすこともあった。
【0161】
図5は、術後2、5、10、15、28日目のうちの1日における化合物8、及び術後2日目のスクランブル陰性対照(以下の構造)の尾静脈注射の1時間のBDL動物及び偽手術動物の肝臓におけるSUVとして表される放射能の保持を要約している。
【0162】
【化24】
【0163】
結果は、試験されたすべての時点における偽手術動物と比較してBDLにおける放射能の有意な保持を例証しており、放射能の最高保持は、術後10及び15日目にBDL動物からの肝臓において観察された。術後のいずれの時点においても偽手術動物における肝臓取込みに有意差はなかった。
【0164】
実施例5:BDLモデルにおける術後15日目に、化合物3及び化合物8は、シャム動物及び陰性対照との対比で、線維形成肝臓に取込まれる
BDL及びシャム動物モデルを、実施例3に記載されているように準備した。術後15日目に、化合物3、化合物8又は陰性対照のうちの1つを各動物に投与し、肝臓取込み実施例3に記載されているように評価した。
【0165】
陰性対照の構造は以下の通りであった。
【0166】
【化25】
【0167】
図6は、偽手術動物と比較して、BDLモデルにおける化合物3及び8の有意に高い肝臓取込み図示している。
【0168】
実施例6:BDL線維形成性肝臓との化合物8の結合は、非放射性の化合物8により特異的に阻害される
放射能の生体分布を、1:100及び1:10000倍過剰の非放射性の化合物8の添加前と添加後の両方で、化合物8約3MBqの尾静脈を介する静脈内注射から1時間後のBDL及び偽手術雄性Sprague Dawleyラット(実施例3に記載されている手順)において術後15日目に検討した。試験品目の注射後の適切な時点で、各動物をイソフルランで再麻酔し、頸椎脱臼により屠殺して秤量した。各動物を解剖し、器官及び組織(以下の表5及び6を参照)を摘出し、BASILカウンタープロトコル40又は手動カウンティングを使用してカウントした。
【0169】
BDL動物における注射後1時間における活性の蓄積(%i.d.)の主要部位を以下の表5に要約する。
【0170】
【表4】
【0171】
シャム動物における注射後1時間における活性の蓄積(%i.d.)の主要部位を以下の表6に要約する。
【0172】
【表5】
【0173】
実施例7:15日目のBDLにおける化合物8の取込み及びα−v上方制御
BDL及びシャム動物モデルを、実施例3に記載されているように準備した。術後2、15及び28日目の各々で、化合物8約3MBqを動物に投与し、肝臓取込み実施例3に記載されているように評価した。
【0174】
動物の肝臓におけるαvインテグリンの発現を、BDL及び偽手術動物のホモジナイズした肝臓において測定した。術後2、15及び28日目の各々で、動物を屠殺し、それらの肝臓を摘出し、−70℃で保存した。
【0175】
予め秤量した肝臓を、−70℃フリーザーから取り出し、少量の液体窒素の存在下で予冷した乳鉢及び乳棒中に入れた。肝臓を、ほぼ均一な粉末になるまで粉砕した(肝臓の粉砕中に、少量の液体窒素を加え、組織を常に冷凍状態に保った)。粉砕した肝臓を、予冷した医薬測定器具に入れ、さらに使用するまで−70℃フリーザーに入れた。冷凍した粉砕組織1gを、1mM EDTA、0.25Mスクロースを含有する氷冷RIPA緩衝液(Sigma;150mM NaCl、1.0% Igepal CA−630(界面活性剤)、0.5%デオキシコール酸ナトリウム(陰イオン性界面活性剤)、0.1% SDS、50mM Tris、pH8.0)10ml中に直接加え、プロテアーゼ阻害剤カクテル(Sigma)(溶解緩衝液ml当たりカクテル10μl)を新たに加えた。混合物を、4℃でシャープブレードホモジナイザーでホモジナイズし、30分間湿った氷上に保持した。
【0176】
ホモジネートを、予冷したガラス組織Dounce中に移し、乳棒の3回の上下ストロークを4℃で適用した。次いで、組織ホモジネートを、予冷した遠心分離管中に移し、30000rpm及び4℃で30分間遠心分離した。上清を取り出し(全細胞ライセート)、湿った氷上に置いた。相対的タンパク質濃度を、製造者の取扱説明書に従ってBSA標準品を含む市販のタンパク質アッセイキット(Pierce、USA)を使用して各サンプルにおいて決定した(BSAアリコートは、ホモジネーションのために使用された変法RIPA緩衝液中で調製すべきである)。各サンプルからの上清を、H2O中で1:50〜1:200へさらに希釈し、タンパク質濃度を、99マルチウェルPlateリーダー(iEMS)を使用して決定した。各サンプルを、1mlアリコートに分け、さらなる使用まで−70℃で保存した。
【0177】
抗α−vインテグリン捕捉抗体(BD;カタログ番号:611013)を、結合溶液(0.05Mトリス、0.138M NaCl及び0.0027M KCl)中で適切な濃度に希釈し、50μl又は100μlのいずれかを、高タンパク質結合ELISAプレート(Nunc−Immuno(商標)プレート96ウェルプレート、MaxiSorp)の各ウェルに添加した。使用される抗体の量は、200ng/ウェルとした。プレートを密封し、37℃で1時間又は4℃で一晩インキュベートした。プレートを空にして、マイクロタイタープレート上のタンパク質結合のための残った部位を、ブロッキング緩衝液(ウェル当たり0.02%アジ化ナトリウム約300μlを含む3% BSA/TBS)と共にインキュベートすることにより飽和させた。プレートを密封し、37℃で1時間インキュベートし、次いで、TBS/0.01% Tween−20で3回洗浄した。ニートの肝臓ホモジネート上清及び/又は異なる濃度の精製α−vインテグリン標準品を、ブロッキング緩衝液中で調製し、コーティングしたプレート中に三つ組みで添加し(ウェル当たり100μl)、次いで、密封し、37℃で2時間インキュベートした。プレートを、TBS/0.05% Tween−20で4回洗浄し、ブロッキング緩衝液中で希釈された第二の抗インテグリン検出抗体(Chemicon AB1930)をウェルに添加し、37℃で1時間インキュベートした。プレートを、TBS/0.05% Tween−20で4回洗浄し、検出抗体に特異的な二次抗体アルカリホスファターゼコンジュゲート(Sigma;カタログ番号:A7539)をウェルに添加し、37℃で1時間インキュベートした。プレートを、TBS/0.05% Tween−20で4回洗浄し、NPP基質溶液100μlを各ウェルに添加し、室温で2時間又は4℃で一晩インキュベートした。加水分解は、視覚的検査により定性的に又はマイクロタイタープレートリーダーで定量的にモニターした。NPPの加水分解は、推奨インキュベーション時間が経過した後に黄色に見え、標的波長(405nm)における光学密度を、ELISAプレートリーダー(Spectra−Max Plus)で測定した。加水分解は、5M水酸化ナトリウムNaOH50μlを添加することにより停止させた。
【0178】
定量的結果については、未知サンプルの信号を、標準曲線の信号と比較した。統計分析は、GraphPad PRISM、Version4.0を使用して実施した。群間差は、ノンパラメトリックな一元配置ANOVAにより分析した。すべての統計分析において、0.05未満の確率値(P<0.05)を有意と見なした。
【0179】
図7及び図8は、シャム動物と対比したBDL動物におけるαv発現を図示しており、αv発現と化合物8の取込みの相関関係を例証している。
【0180】
実施例8:化合物11の合成
【0181】
【化26】
【0182】
Boc−テトラアミン−N−ヒドロキシスクシンイミドエステル(国際公開第2006/008496号)(36mg、0.050mmol)を、10分間DMF(0.5mL)中のHOAt(1.4mg、0.010mmol)及びNMM(17μL、0.15mmol)で予め活性化し、次いで、DMF(0.5mL)中のCys2−6;c[CH2CO−Lys−Cys−Arg−Gly−Asp−Cys−Phe−Cys]−PEG(4)−ジグリコロイル−NH2(国際公開第03/006491号)(12.6mg、0.010mmol)の溶液に加えた。反応混合物を、3日間室温で撹拌し、次いで、真空中で濃縮した。Boc保護基を、90分間TFA/水/トリイソプロピルシラン(95:2.5:2.5、10mL)溶液の添加により除去した。混合物を濃縮し、粗生成物をエーテルから沈殿させ、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;40分間勾配10〜20%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、凍結乾燥後に10.4mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜20%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=2.02分、m/z1458.7(MH)+は、構造を支持した。
【0183】
99mTcによる標識は、窒素でパージしたP46バイアルに、下記の、MeOH中のIGF前駆体1又は2 100μg、Na2CO3/NaHCO3緩衝液(pH9.2)0.5ml、Drytec(商標)99mTc発生剤からのTcO4-0.5ml、SnCl2/MDP溶液(N2でパージした食塩水100ml中にSnCl210.2mg及びメチレンジホスホン酸101mgを含有する)0.1mlを加えることにより行った。
【0184】
実施例9:化合物12の合成
【0185】
【化27】
【0186】
合成は、0.3mmolスケールで(ローディング量0.432g、0.71mmol/g)RinkアミドAM樹脂上で実施した。最初の3つのカップリング段階は、手動窒素バブラー装置中で行った。樹脂上のFmoc基は、標準プロトコル(DMF中20%ピペリジン)により切断した。Fmoc−Cys(トリチル)−OH(702mg、1.20mmol)を、標準カップリング試薬HATU及びDIEAによりDMF中の樹脂にカップリングさせた。カップリングの終了は、標準Kaiser試験によりチェックした。Fmoc切断後、カップリングを繰り返し、第二のシステインを導入した。樹脂を、ジクロロメタン/TFA/トリイソプロピルシラン(94:5:1、10mL)溶液中に懸濁した。3分後、黄色の溶液を流し出した。この段階を6回繰り返し、側鎖チオール機能の完全な脱保護を確保した。樹脂を、ジクロロメタンで洗浄し、乾燥した(30分間窒素流)。20%ギ酸(18mL)と35%過酸化水素(2mL)の混合物を、室温で1時間放置し、0℃まで冷却した。樹脂を、過ギ酸溶液のアリコート(2×4mL)で洗浄し、次いで、緩やかに攪拌しながら5℃で12時間過ギ酸溶液(5mL)中に保持した。樹脂のアリコートを切断し(TFA/水/トリイソプロピルシラン、95:2.5:2.5)、LC−MS(カラムPhenomenex Luna C18(2)50×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配5〜50%B;流速0.3mL/分、214nm及び254nmにおけるUV検出、ESI−MS)により分析すると、tR=2.48分、m/z564.3(MNa)+は、構造を支持した。
【0187】
上記の樹脂のFmoc除去(標準プロトコル)後、Fmoc−アミノ−PEG(4)−ジグリコール酸(796mg、1.50mmol)を、標準カップリング試薬(HATU及びDIEA)を使用してDMF中の樹脂にカップリングさせた。6時間後、アリコートを切断し(前の段落に記載されている)、LC−MS(カラムPhenomenex Luna C18(2)50×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配5〜50%B;流速0.3mL/分、214nm及び254nmにおけるUV検出、ESI−MS)により分析すると、tR=3.28分、m/z832.3(MH)+は、完全な変換を示した。
【0188】
すべての後続アミノ酸を、上記からの樹脂上約0.25mmol上でSlowMocカップリングプロトコルを使用してAB1433A自動ペプチド合成機を使用してカップリングさせると、H−Lys(Boc)−Cys(tBu)−Arg(Pmc)−Gly−Asp(OtBu)−Cys(tBu)−Phe−Cys(Trt)−PEG4−ジグリコロイル−CyA−CyA−Rinkアミド樹脂が得られた。
【0189】
ジクロロメタン(10mL)中のクロロ酢酸(142mg、1.50mmol)及びDCC(155mg、0.75mmol)の溶液を、15分間室温で撹拌し、次いで、濾過し、濃縮した。残渣をDMFに取り、上記のペプチド樹脂に加えた。ペプチドを、2時間TFA/水/トリイソプロピルシラン溶液(95:2.5:2.5、10mL)を使用して樹脂から切断した。溶液を濃縮し、粗ペプチドをエーテルから沈殿させ、真空中で乾燥すると、340mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)50×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配0〜50%B;流速0.3mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析、tR=3.09分、m/z1710.6(MH)+は、クロロアセチル化ペプチドを支持した。
【0190】
50%アセトニトリル(64mL)中のペプチド(68mg)の溶液に、pHが8.5になるまで2.5%アンモニアを加えた。混合物を16時間室温で撹拌した。LC−MS分析は、完全なチオエーテル環化を支持した。アセトニトリルを減圧下で除去し、水溶液を凍結乾燥した。単離した材料を、TFA中DMSOの5%溶液(100mL)に取り、60分間室温で撹拌した。混合物を濃縮し、粗生成物を、エーテルからの沈殿により単離し、続く、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配5〜20%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、固体材料48mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)50×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配0〜30%B;流速0.3mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=3.18分、m/z1560.5(MH)+は、ジスルフィド架橋形成を支持した。
【0191】
NMP(1.5mL)中のペプチド(5mg)、キレートI−グルタリル テトラフルオロチオフェノールエステル(国際公開第03/006491号)(5mg)及びDIEA(2.7μl)の部分的に溶けた混合物を、一晩40℃で加熱した。混合物を、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;40分間勾配0〜30%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、純粋な生成物2.9mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配0〜30%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=3.83分、m/z1000.7(MH2)2+は、構造を支持した。
【0192】
99mTcによる標識は、国際公開第2003/006491号に記載の通り実施した。
【0193】
実施例10:化合物13の合成
【0194】
【化28】
【0195】
Boc−PEG(6)−ジグリコール酸(50mg、0.10mmol)を、標準カップリング試薬(HATU及びNMM)を使用し、DMF中のCys2−6;c[CH2CO−Lys−Cys−Arg−Gly−Asp−Cys−Phe−Cys]−PEG(4)−ジグリコロイル−NH2(国際公開第03/006491号)(25.2mg、0.020mmol)にカップリングさせた。反応混合物を、一晩撹拌し、真空中で濃縮した。Boc保護基を、室温で45分間TFA/水/トリイソプロピルシラン(95:2.5:2.5、10mL)の添加により除去した。混合物を濃縮し、続いて、粗材料をエーテルから沈殿させた。生成物を、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;40分間勾配10〜30%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、5.5mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜25%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=3.01分、m/z1636.0(MH)+は、生成物を支持した。
【0196】
キレートI−グルタル酸(国際公開第03/006491号)(15.6mg、0.034mmol)を、標準カップリング試薬(PyAOP(7−アザベンゾトリアゾール−1−イルオキシ−トリス−(ピロリジノ)ホスホニウムヘキサフルオロホスフェート)及びNMM)を使用して、DMF中のペプチド(5.5mg、0.0034mmol)にカップリングさせた。4時間後、混合物を、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;40分間勾配15〜30%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、3.8mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配15〜30%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=2.13分、m/z1039.0(MH2)2+は、構造を支持した。
【0197】
99mTcによる標識は、国際公開第2003/006491号に記載の通り実施した。
【0198】
実施例11:化合物14の合成
【0199】
【化29】
【0200】
合成は、0.80mmolスケールでRink Amide MBHA樹脂(ローディング量0.72mmol/g)上で手動窒素バブラー装置中で行った。樹脂上のFmoc基は、標準プロトコルにより切断した。Fmoc−PEG(4)−ジグリコール酸(850mg、1.6mmol)を、DMF中で標準カップリング試薬(PyAOP及びNMM)を使用して樹脂にカップリングさせた。コンジュゲーション反応と、続く、Fmoc脱保護を、5つのPEG(4)−ジグリコロイル基が樹脂上に導入されるまで繰り返した。
【0201】
Fmoc−Cys(Trt)−OH(1.0mmol)を、DMF/ジクロロメタン(1:1、6mL)中で標準カップリング試薬(PyAOP及びNMM)を使用して、上記の樹脂(0.2mmol)にカップリングさせた。2.5時間後、樹脂のアリコートを切断し(上記)、LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜80%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)により分析すると、tR=2.3分、m/z1794.1(MH)+は、完全な変換を示した。
【0202】
すべての後続アミノ酸を、上記からの樹脂上約0.1mmol上でSlowMocカップリングプロトコルを使用し、AB1433A自動ペプチド合成機を使用してカップリングさせると、H−Lys(Boc)−Cys(tBu)−Arg(Pbf)−Gly−Asp(OtBu)−Cys(tBu)−Phe−Cys(Trt)−(PEG(4)−ジグリコロイル)5−Rinkアミド樹脂が得られた。
【0203】
クロロ酢酸無水物(2.00mmol)を、1時間ジクロロメタン(10mL)中のクロロ酢酸(378mg、4.00mmol)及びDCC(412mg、2.00mmol)の溶液を撹拌し、続いて、濾過することによって合成した。溶液を濃縮し、残渣をDMF(3mL)に取り、上記のペプチド樹脂に加えた。NMM(220μL、2.00mmol)を加え、混合物を3時間放置した。樹脂のアリコートを切断し(上記)、LC−MSにより分析すると、完全な変換を支持した。ペプチドを、3時間TFA/水/トリイソプロピルシラン溶液(95:2.5:2.5、10mL)の添加により樹脂から切断した。溶液を濃縮した。
【0204】
上記からの残渣を、水(100mL)に取り、エーテルで抽出した。水溶液にアセトニトリル(100mL)を加え、pHを、希アンモニアの添加により8に調整した。反応物を一晩放置した。アセトニトリルを蒸発させ、粗生成物を凍結乾燥により単離すると、154mgが得られた。LC−MSによる分析は、完全なチオエーテル環化を支持した。
【0205】
上記からの単離材料を、TFA中DMSOの5%溶液(80mL)に取り、60分間室温で撹拌した。混合物を濃縮し、粗生成物を、エーテルからの沈殿により単離し、続いて、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配15〜25%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、純粋な材料5mgが得られた。LC−MSによる分析は、ジスルフィド架橋形成を支持した。
【0206】
キレートI−グルタル酸(国際公開第03/006491号)(9mg、0.02mmol)を、一晩DMF中で標準カップリング試薬(PyAOP及びDIEA)を使用して上記のペプチドにカップリングさせた。生成物を、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配10〜30%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、1.3mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜60%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=1.8分、m/z1430.1(MH2)2+は、構造を支持した。
【0207】
99mTcによる標識は、国際公開第2003/006491号に記載の通り実施した。
【0208】
実施例12:化合物15の合成
【0209】
【化30】
【0210】
ペプチド配列H−Lys(Boc)−Cys(tBu)−Arg(Pbf)−Gly−Asp(OtBu)−Cys(tBu)−Phe−Cys(Trt)−Gly−[Glu(OtBu)]5−NH2を、SlowMocカップリング方法を使用してRinkアミドMBHA樹脂(0.1mmol)上でABI433A自動ペプチド合成機で組み立てた。
【0211】
ジクロロメタン(10mL)中のクロロ酢酸(378mg、4.00mmol)及びDCC(412mg、2.00mmol)の溶液を、1時間室温で撹拌し、次いで、濾過し、濃縮した。残渣をDMFに取り、上記のペプチド樹脂に加えた。NMM(220μl、2.00mmol)を加え、反応物を2時間放置した。樹脂のアリコートを切断し(上記)、LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配20〜30%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)により分析すると、tR=2.2分、m/z1820.7(MH)+は、完全な変換を支持した。ペプチドを、2時間TFA/水/トリイソプロピルシラン溶液(95:2.5:2.5、10mL)の添加により樹脂から切断した。溶液を濃縮し、粗生成物を、エーテルからの沈殿により単離し、続いて、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配20〜40%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製した。半純粋な生成物を凍結乾燥により単離し、50%アセトニトリル/水(6mL)に取った。pHを、希アンモニアの添加により8に調整し、反応物を一晩撹拌した。アセトニトリルを減圧下で蒸発させ、生成物を凍結乾燥により単離すると、14.8mgが得られた。LC−MS[カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜60%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=2.0分、m/z1785.0(MH)+は、完全なチオエーテル環化を支持した。
【0212】
上記の単離ペプチドを、TFA中DMSOの5%溶液(8mL)に取り、15分間室温で撹拌した。混合物を濃縮し、粗生成物を、エーテルから沈殿させ、続いて、調製用HPLC(カラムPhenomenex Luna C18(2)250×10mm、10μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配10〜20%B;流速5mL/分、214nm及び254nmにおけるUV検出)により精製すると、純粋な材料2mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜60%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=1.0分、m/z1670.8(MH)+は、ペプチドを支持した。
【0213】
DMF中の上記からのペプチド(2mg、1μmol)及びBoc−テトラアミン−N−ヒドロキシスクシンイミドエステル(国際公開第2006/008496号)(8.6mg、0.012mmol)の溶液に、DIEA(3μL、0.02mmol)を加えた。反応混合物を一晩撹拌し、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配20〜80%B;流速10mL/分、214nm及び254nmにおけるUV検出)による精製にかけた。純粋な生成物を凍結乾燥し、TFAとジクロロメタンの混合物(1:1、2mL)に溶かした。2時間後、溶媒を蒸発させ、残渣をアセトニトリル/水に取り、凍結乾燥すると、純粋な生成物1.0mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配0〜20%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=4.2分、m/z935.5(MH2)2+は、構造を支持した。
【0214】
99mTcによる標識は、上記の実施例8において化合物11について記載されているように行った。
【技術分野】
【0001】
本発明は、インビボイメージング法、特に、ある種の公知のインビボイメージング剤の新規用途に関する。本発明の好ましいインビボイメージング法は単光子放射断層撮影(SPECT)及び陽電子放射断層撮影(PET)である。
【背景技術】
【0002】
慢性ウイルス性肝炎、非アルコール性脂肪性肝炎(NASH)、寄生虫血症、先天性代謝異常及び飲酒による中毒性障害を始めとする肝線維症を引き起こす可能性のある傷害は地理的に広く分布し、有病率は高い。こうした要因はすべて、肝硬変と潜在的に肝癌を招く線維症が、依然として世界中での主な病因及び死因の一つであることを意味している。英国だけをとっても、肝疾患は現在5番目に多い死因であり、その発症率は増加しつつある(Iredale 2003 BMJ Vol. 327 pp 143−147)。
【0003】
非アルコール性脂肪性肝疾患(NAFLD)とNASHの2種類の脂肪肝疾患が存在する。米国の人口の約24%がNAFLDをもつと考えられており、これは低い頻度でNASHへと進行する。NAFLDはメタボリックシンドロームに関連しており、肥満、高脂血症、高血圧及びII型糖尿病と関係がある。米国では約4700万人がメタボリックシンドロームをもっていると考えられている。米国の人口のうち推定860万人がNASHを有していて、線維症及び肝硬変に罹患するおそれがあり、NASH患者の20〜28%が10年間で肝硬変を発症すると考えられている。NAFLDはごく一般的で、NASHへ、ひいては肝硬変へと進行しかねないNAFLDの範囲の重症度の低い端を表す。肝線維症はNASHから肝硬変へと進行するリスクの指標である。
【0004】
肝線維症の検出に現在用いられているアプローチは幾つかの重大な短所をもつ。コラーゲン沈着のパターンの組織学的分析による肝生検は、肝疾患ステージ及び肝線維症を評価するための最も信頼できる基準とみなされている。しかし、この方法には、若干の有病率、偶発的な死亡率、高いコスト、サンプリング誤差及び線維症の程度を分類する際の肝臓病理学者間での高い観察者間変動を伴う。生検での肝臓のサンプリングは、評価すべき肝臓の5万分の1にすぎず、ステージ診断の誤りを招くおそれがある。さらに、コラーゲンは繊維組織のマーカーであり、コラーゲンは活動性線維症の後期ステージにおいても、疾患プロセスが解消しつつある場所でも見つかるので、コラーゲンは活動性疾患の理想的標的ではない。現在、肝線維症を非侵襲的方法で有効に特徴付けてモニターすることができる手段はない。そのため、早期治療介入して肝線維症の進行を遅らせ停止させるのに悪影響がある。さらに、病気の進行を適宜モニターするには、3〜5年毎に繰返し生検を行うことが推奨されている。肝線維症の検出に利用できる血液検査の価値が限られているのは、線維症の程度又は線維症と肝硬変との区別に用いることができないからである。現在、NASHとNAFLDを区別し、或いはNASHにおける線維症を十分に定量化し特徴付けるのに利用できる方法はない。
【0005】
肝星細胞(HSC)は、肝臓における主要なフィブロコンピテント(fibrocompetent)細胞と広くみなされている。進行性肝線維症中に、HSCは活性化して増殖するが、線維症の解消中には、肝臓瘢痕の分解と同時に起こる広範囲のHSCアポトーシスがある。線維症のプロセスのこの進行性ステージは、線維形成と呼ばれる。活性化HSC上のインテグリン発現の上方制御が報告されている(Zhou et al J. Biol. Chem. 2004; 279(23): 23996−24006)。HSCの活性化は、線維形成及び、続く肝線維症の開始及び進行に不可欠である。従って、HSC活性化のマーカーは、線維形成のイメージングにとっての標的機会である。この事に関して、最近になって顕著になってきたプロセスのマーカーはインテグリンである(Zhou et al 2004 J. Biol. Chem.; 279: 23996−24006; Patsenker et al 2007 J. Hepatol.; 46(5): 878−887; Zhou et al 2006 J. Biol. Chem.; 281: 39757−39765; Carloni et al 1996 Gastroenterology: 110: 1127−36)。
【0006】
インビボイメージング用途のための適当に標識されたインテグリン結合剤の使用についてこれまで記載されている。
【0007】
国際公開第2004/020435号は、αvインテグリン又はそのサブタイプの発現に起因する細胞増殖の病理学的な上方制御又は調整不全を伴う疾患の治療に有用であるインテグリン結合性ピペリジニル化合物を開示している。国際公開第2004/020435号は、発明の化合物が、インビボイメージングに適している部分とコンジュゲートし、非侵襲的腫瘍イメージング剤として使用できることも開示している。
【0008】
国際公開第2007/088041号は、ある疾患の治療及び/又は予防に有用である、あるクラスの小分子インテグリン結合性化合物を開示しており、その疾患は好ましくはαvβ1インテグリンにより仲介される疾患である。肝線維症は、国際公開第2007/088041号の化合物が使用される特定の疾患として包含されている。治療及び予防に加えて、国際公開第2007/088041号には、開示された化合物がインビボイメージング部分を含んでいてインビボイメージングに使用することができることが教示している。
【0009】
国際公開第2003/006491号、国際公開第2005/012335号及び国際公開第2005/123767号は、血管新生に関係する受容体に結合するRGDペプチド系の化合物に関し、受容体は、インテグリンを包含する。開示されている化合物は、抗新生物剤又はインビボイメージング部分のいずれかを含み、血管新生に関係する疾患の治療及びインビボイメージングに有用であると教示されている。
【0010】
国際公開第2006/054904号は、細胞外マトリックス(ECM)をターゲティングする造影剤としてのインビボイメージング部分で標識されたRGDペプチドの使用を開示している。この造影剤は、肝線維症を始めとするコラーゲンの過剰な形成に関連する疾患を診断及びモニターする際に有用であると言われている。しかし、放射標識されたインテグリン結合性RGDペプチドの排泄が主に肝胆道系を介して起きることが知られていることから、肝線維症のインビボイメージングにRGDペプチドを使用することへの阻害要因がある(Haubner 1999 J. Nuc. Med.; 40: 1061−71)。さらに、上述のように、疾患プロセスが解消しつつある場合並びに活動性線維症の後期ステージの両方でコラーゲンが見いだされることがあるため、コラーゲンは、活動性線維症の理想的マーカーではない。治療レジメンの用途が、より適切であるかもしれず、肝硬変が発症する前の、例えば、NASHにおける線維症の早期ステージで臨床的により有効である可能性も高い、より早期の活動性ステージを標的とすることがより有利であろう。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】国際公開第2007/148074号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0012】
そこで、肝線維症の早期状態(「線維形成」とも呼ばれる)を識別し、それによって、最も効果的に治療することができるステージで疾患プロセスへ介入するための方法が必要である。
【課題を解決するための手段】
【0013】
本発明は、被検体の肝臓における線維形成の識別を容易にするのに有用な方法に関する。本発明は、被検体の肝臓における線維形成を識別するための方法に使用するための化合物も用意する。本発明の別の態様は、被検体の肝臓における線維形成を識別するための方法に使用するための医薬品の調製に使用するための化合物である。本発明は、RGDペプチド系の化合物が、活性化肝星細胞(HSC)を検出し、それによって、肝線維症の早期診断に有用な方法を用意するために効果的に使用することができることを例証する。
【図面の簡単な説明】
【0014】
【図1】活性化ヒト肝星細胞とEA−Hy926膜の両方と特異的に結合する化合物6を示す図である。Kiは、EA−Hy926膜アッセイで約10nMと決定され、EC50は、LX−2細胞アッセイで約1nMと決定された。LX−2アッセイにおける結合の特異性は、非放射性の化合物6による化合物6結合の特異的阻害によって示された。低親和性スクランブル陰性対照は、インテグリンとも活性化星細胞ともにも結合しない。
【図2】活性化ヒト肝星細胞とEA−Hy926膜の両方と特異的に結合する化合物6を示す図である。Kiは、EA−Hy926膜アッセイで約10nMと決定され、EC50は、LX−2細胞アッセイで約1nMと決定された。LX−2アッセイにおける結合の特異性は、非放射性の化合物6による化合物6結合の特異的阻害によって示された。低親和性スクランブル陰性対照は、インテグリンとも活性化星細胞とも結合しない。
【図3】活性化肝星細胞膜とEA−Hy926膜の両方と特異的に結合する化合物3を示す図である。Kiは、観察されたように、EA−Hy926膜アッセイで約1nMと決定され、星細胞膜アッセイで59nMと決定された。化合物8は、親和性において化合物3に続くように見え、Kiは、活性化星細胞膜アッセイで56nMと決定され、EA−Hy926膜アッセイで6nMと決定された。陰性対照スクランブルRGDペプチドは、どちらの細胞タイプともほとんど結合を示さない。
【図4】活性化肝星細胞膜とEA−Hy926膜の両方と特異的に結合する化合物3を示す図である。Kiは、観察されたように、EA−Hy926膜アッセイで約1nMと決定され、星細胞膜アッセイで59nMと決定された。化合物8は、親和性において化合物3に続くように見え、Kiは、活性化星細胞膜アッセイで56nMと決定され、EA−Hy926膜アッセイで6nMと決定された。陰性対照スクランブルRGDペプチドは、どちらの細胞タイプともほとんど結合を示さない。
【図5】肝臓取込みが観察されない陰性対照スクランブルRGDペプチドと比較した、胆管結紮(BDL)ラットからの肝臓による化合物8の特異的取込みを例証する図である。BDL肝臓取込みは、線維形成の程度にも比例しており、最大取込みは、線維形成がその最高レベルである術後10及び15日目に観察された。
【図6】陰性対照化合物(RGDスクランブルペプチド)と対比した、BDLラット肝臓中へのRGDペプチドの特異的取込み例示する図である。この実験からのデータは、化合物3と化合物8が共に、BDLとシャムの間の取込みの有意差が観察されない陰性対照と比較して、注射から1時間後にシャム動物と比較して、BDLの肝臓中に有意に保持されることを示した。
【図7】術後15日目のBDLにおける有意なαvインテグリン上方制御を示す図である。
【図8】図8は、図7との関連して、術後の全時点におけるBDL動物の肝臓におけるαvインテグリン発現と化合物取込みの相関関係を例証している。
【発明を実施するための形態】
【0015】
一つの態様では、本発明は、被検体の肝臓における線維形成の存在、場所及び/又は量を決定する方法に用いられる式Iの化合物であって、該方法は、
(i)検出可能な量の式Iの化合物を投与しておいた被検体を準備する段階と、
(ii)肝臓中の線維形成性組織に式Iの化合物を結合させる段階と、
(iii)式Iの化合物から放出される信号をインビボイメージング法で検出する段階と、
(iv)信号の場所及び/又は量を表す画像を形成する段階
とを含んでおり、式Iの化合物は以下の通り定義される。
【0016】
【化1】
【0017】
式中、
Gはグリシンを表し、
Dはアスパラギン酸を表し、
X1は、アスパラギン酸、グルタミン酸、リジン、ホモリジンもしくはC3〜6ジアミノアルカン酸又はそれらの誘導体から選択されるアミノ酸を表し、
X2及びX4は独立に側鎖同士が連結して環化架橋を形成するアミノ酸残基(例えばジスルフィド又はチオエーテル結合を形成するシステイン又はホモシステイン)又は環化架橋を形成し得る他のアミノ酸(例えばアスパラギン酸及びリジン)を表し、
X3はアルギニン、N−メチルアルギニン又はアルギニン模倣体を表し、
X5はチロシン、フェニルアラニン、3−ヨード−チロシン C4〜6シクロアルキルアラニン、ナフチルアラニン又はそれらの誘導体を表し、
X6はX6をC(=O)基に連結するチオエーテル結合又はチオアセタール結合のいずれかを形成するチオール含有アミノ酸を表し、
W1及びW2は独立に任意要素としてのリンカー基であって、W1が存在するときはX1のアミノ酸側鎖部分に連結し、W2が存在するときはX6のカルボキシ基に連結し、
Z1及びZ2は、Z1とZ2の少なくとも一方がイメージング部分であることを条件として、独立にイメージング部分、糖部分、有機色素基又は水素である。
【0018】
式Iの化合物について、
X1は好ましくはリジンである。
X2及びX4は好ましくは独立にシステイン又はホモシステインであり、最も好ましくは共にシステインである。
X3は好ましくはアルギニンである。
X5は好ましくはシクロヘキシルアラニン、フェニルアラニン又は3−ヨード−チロシンであり、最も好ましくはフェニルアラニンである。
X6は好ましくはシステイン又はホモシステインであり、最も好ましくはシステインである。
【0019】
式Iの好ましい実施形態では、
X1はリジンであり、
X2及びX4は独立にシステイン又はホモシステインであり、
X3はアルギニンであり、
X5ハフェニルアラニン又は3−ヨード−チロシンであり、
X6ハシステイン又はホモシステインである。
【0020】
式Iの最も好ましい実施形態では、
X1はリジンであり、
X2及びX4は共にシステインであり、
X3はアルギニンであり、
X5はフェニルアラニンであり、
X6はシステインである。
【0021】
本発明において、「線維形成」という用語は、具体的に、とりわけ肝星細胞(HSC)が活性化され、インテグリンを発現する、線維症の活動性の進行ステージをいう。HSCは、肝臓における主要なフィブロコンピテント細胞と広くみなされている。線維形成中に、HSCは活性化して増殖するが、線維症の解消中には、肝臓瘢痕の分解と同時に起こる広範囲のHSCアポトーシスがある。さらに、線維症中に、コラーゲンなどの細胞外マトリックス(ECM)成分の沈着が起きることはない。従って、ECM成分の存在は、線維症の後期ステージ及び線維症の解消の特徴である。従って、線維形成中の疾患プロセスを標的にすることは、治療の用途が最も適切である活動性疾患のより良い指標を用意する。
【0022】
本発明において、「アミノ酸」という用語は、アミノ基、カルボキシル基、水素原子及びアミノ酸側鎖基からなり、これらがすべて(αアミノ酸の場合)α炭素と呼ばれる1つの炭素に結合したものをいう。アミノ酸としては、特に限定されないが、天然アミノ酸が挙げられる。天然アミノ酸は、天然タンパク質のアミノ酸単位が誘導されるものであり、当業者に周知である。本明細書でアミノ酸に関して用いられる「それらの誘導体」という用語は、側鎖が天然アミノ酸の側鎖の誘導体であるものをいう(“Amino Acid Derivatives” 1999 Oxford University Press, Barrett, Ed.参照)。
【0023】
「アミノ酸模倣体」という用語は、天然アミノ酸の合成類似体であってアイソスターであるもの、つまり天然化合物の立体及び電子構造を模倣して設計されたものを意味する。かかるアイソスターは当業者に周知であり、特に限定されないが、デプシペプチド、レトロインベルソペプチド、チオアミド、シクロアルカン又は1,5−二置換テトラゾールなどが挙げられる(M. Goodman, Biopolymers, 24, 137, (1985)参照)。
【0024】
「環化架橋」という用語は、架橋を導入できる官能基をもつアミノ酸同士又はアミノ酸と−(CH2)O−もしくは−(CH2)O−C6H4−基の組合せをいう。oは1〜10の正の整数を表す。好ましい例は、ジスルフィド、−(CH2)4−カルバ架橋のようなジスルフィド模倣体、チオアセタール架橋、チオエーテル架橋(シスタチオン又はランチオニン)、エステル及びエーテルを含有する架橋、並びにアミド架橋である。好ましくは、1つの架橋はジスルフィド結合を形成し、第二の架橋はチオエーテル(スルフィド)結合からなる。例えば、環化架橋がX2とX4のような2つのアミノ酸で形成されている場合、システイン又はホモシステインの側鎖がシステイン、ホモシステイン、セリン、スレオニン又はアルデヒド含有アミノ酸の側鎖と連結して環化架橋を形成する。
【0025】
「アルギニン模倣体」という用語は、アミノ酸模倣体に関する上記の定義と同様に通り、天然アルギニンの合成類似体であるアイソスターである。
【0026】
式Iの化合物のペプチドは、あらゆる公知の化学合成法で合成できるが、特に有用な方法は自動ペプチド合成装置を用いたMerrifieldの固相法である(J. Am. Chem. Soc., 85: 2149 (1964))。合成法の標準的手順は、E.Atherton & R.C.Sheppard, “Solid phase peptide synthesis: a practical approach, 1989, IRL Press, Oxfordに記載されている。
【0027】
酸不安定リンカー基を有する合成樹脂を用いて、これに、C末端を保護した所望のアミノ酸残基をアミド結合の形成によって結合させる。例えば、(ジメトキシフェニル−アミノメチル)−フェノキシ誘導リンカーを有するいわゆるRinkアミドAM樹脂を用いることができる(Rink, H. (1987), Tetrahedron Lett. 30, p.3787)。この樹脂からペプチドを酸分解で開裂させると、ペプチドアミドが得られる。別法として、O−ビス−(アミノエチル)エチレングリコールトリチル樹脂(K. Barlos et al (1988), Liebigs Ann. Chem, p.1079)を用いると、酸分解開裂によって第一級アミンハンドルを有するペプチドを得ることができる。
【0028】
イメージング部分での標識は好適には「前駆体化合物」によって実施されるが、前駆体化合物は、式Iの化合物の誘導体であって、適当な化学的形態の1以上のイメージング部分との化学反応が部位特異的に起こり、最小限の段階数(理想的には一段階)で実施でき、しかも多大な精製を行わずに(理想的にはそれ以上精製しなくても)所望の式Iの化合物が得られるように設計される。かかる前駆体化合物は合成品であり、良好な化学的純度で得ることができる。前駆体化合物は、任意には、式Iの化合物の官能基に対する保護基を含んでいてもよい。
【0029】
「保護基」という用語は、不都合な化学反応は阻害又は抑制するが、分子の残りの部分を修飾しない十分穏和な条件下で当該官能基から脱離させることができる十分な反応性をもつように設計された基を意味する。脱保護後に、所望の生成物が得られる。保護基は当業者に周知であり、好適には、アミン基については、Boc(Bocはtert−ブチルオキシカルボニルである。)、Fmoc(Fmocはフルオレニルメトキシカルボニルである。)、トリフルオロアセチル、アリルオキシカルボニル、Dde[すなわち、1−(4,4−ジメチル−2,6−ジオキソシクロヘキシリデン)エチル]又はNpys(すなわち、3−ニトロ−2−ピリジンスルフェニル)から、カルボキシル基については、メチルエステル、tert−ブチルエステル又はベンジルエステルから選択される。
ヒドロキシ基に対する適当な保護基は、メチル、エチル又はtert−ブチル、アルコキシメチル又はアルコキシエチル、ベンジル、アセチル、ベンゾイル、トリチル(Trt)或いはテトラブチルジメチルシリルのようなトリアルキルシリルである。チオール基に対する適当な保護基は、トリチル及び4−メトキシベンジルである。その他の保護基の使用については、‘Protective Groups in Organic Synthesis’,Theorodora W.Greene and Peter G.M.Wuts(Third Edition、John Wiley & Sons,1999)に記載されている。
【0030】
本発明の「リンカー基」は式−(L)n−の基である。
式中、
各Lは独立に−C(=O)−、−CR′2−、−CR′=CR′−、−C≡C−、−CR′2CO2−、−CO2CR′2−、−NR′−、−NR′CO−、−CONR′−、−NR′(C=O)NR′−、−NR′(C=S)NR′−、−SO2NR′−、−NR′SO2−、−CR′2OCR′2−、−CR′2SCR′2−、−CR′2NR′ CR′2−、C4-8シクロヘテロアルキレン基、C4-8シクロアルキレン基、C5-12アリーレン基、C3-12ヘテロアリーレン基、アミノ酸、ポリアルキレングリコール、ポリ乳酸又はポリグリコール酸部分であり、
nは1〜15の整数であり、
各R′基は独立にH又はC1-10アルキル、C3-10アルキルアリール、C2-10アルコキシアルキル、C1-10ヒドロキシアルキル、C1-10フルオロアルキルであるか、或いは2以上のR′基がそれらに結合した原子と共に炭素環、複素環式、飽和又は不飽和環を形成したものである。
【0031】
ただし、リンカー基は100個以下の原子鎖、好ましくは50個以下の原子鎖である。リンカー基は、最も好ましくは10〜50個の原子鎖、特に好ましくは、10〜30個の原子鎖である。好ましいL基は、−C(=O)−、−CH2−、−NH−、−NHC(=O)−、−C(=O)NH−、−CH2−O−CH2−及びアミノ酸である。
【0032】
好ましくは、リンカーはバイオモディファイアー基としての機能する。「バイオモディファイヤー基」は式Iの化合物の薬物動態及び血中クリアランス速度を変化させる機能をもつ。適当なバイオモディファイヤー基の一例は、単分散PEG構成単位を1〜10単位含むものである。また、バイオモディファイアー基は1〜10個のアミノ酸残基も表すものであってもよい。バイオモディファイアー基の好ましいアミノ酸残基はリジン及びグルタミン酸のような荷電アミノ酸、又はシステイン酸及びホスホノアラニンのような荷電非天然アミノ酸である。さらに、アミノ酸グリシン、アスパラギン酸及びセリンを含んでいてもよい。好ましい実施形態では、バイオモディファイアー基は単分散PEG様構造の式(II)の17−アミノ−5−オキソ−6−アザ−3,9,12,15−テトラオキサヘプタデカン酸を含む表す。
【0033】
【化2】
【0034】
式中、mは1〜10の整数であり、C末端単位はアミド基である。バイオモディファイアー基は、化合物の薬物動態及び血中クリアランス速度を変化させる機能をもつ。本発明におけるバイオモディファイアーの機能は、組織中への取込みを減少させ、腎臓からの排泄を増大させ、もってバックグラウンド干渉を低減して優れたインビボ画像を与える。バイオモディファイアー基は、グルタル酸及び/又はコハク酸及び/又はポリエチレングリコール系単位及び/又は上記の式IIの単位から誘導される部分を表すものであってもよい。リンカー基の性状は、式Iの化合物の標的受容体に対する親和性を損なうべきではない。さらに、リンカー基は、式Iの化合物のバックグラウンド肝臓取込みを増加させるべきではないが、これは、過度に大きなポリエチレングリコール系単位を使用した場合に起きりかねない。
【0035】
Z1又はZ2のいずれかが糖部分である場合、それらもバイオモディファイアー基として機能し得る。「糖部分」は炭水化物基でり、通常は多価アルコールのアルデヒド又はケトン誘導体である。糖部分は、フルクトース又はグルコースのような単量体(単糖)であってもよいし、糖が2つ結合して二糖を形成したものであってもよい。二糖には、グルコースとフルクトースとで構成されたスクロースのような糖がある。糖という用語には、置換及び非置換糖並びに糖誘導体が包含する。好ましくは、糖は、グルコース、グルコサミン、ガラクトース、ガラクトサミン、マンノース、ラクトース、フコース及びそれらの誘導体、例えばシアル酸、グルコサミンの誘導体から選択される。糖は好ましくはα又はβである。糖は、特に、マンノピラノシド又はガラクトースピラノシドであってもよい。糖のヒドロキシル基は、例えば1以上のアセチル基で保護してもよい。糖部分は好ましくはN−アセチル化される。かかる糖の好ましい例としては、N−アセチルガラクトサミン、シアル酸、ノイラミン酸、N−アセチルガラクトース及びN−アセチルグルコサミンが挙げられる。
【0036】
「有機色素基」は、紫外乃至近赤外域の波長を有する電磁スペクトルの光と相互作用する有機色素であればよい。好ましい有機色素基としては、広範に非局在化した電子系を有する基が挙げられる。最も好ましい有機色素基はシアニン染料(CyDye(商標))である。シアニン色素は、奇数個の炭素原子が交互に炭素−炭素単結合と炭素−炭素多重(好ましくは二重)結合とで連結したポリエン連鎖であって、両端にアミノ基を有していてその一方が第四級化されているものとして定義される化合物である。シアニン及び類似のアリール−リンカー−アリール発色団は、任意にはペンダント置換基又は縮合環置換基を有する。シアニン染料及びその合成に関する一般的な説明は、米国特許第6048982号、同第5268486号及び欧州特許第1037947号に記載されている。本発明では、シアニン色素は好ましくはカルバシアニン、オキサシアニン、チアシアニン及びアザシアニンからなる群から選択される。
【0037】
本明細書に記載した方法の「検出」段階は、式Iの「イメージング部分」から放出される信号を、信号の検出器によって検出することを含む。この検出段階は、信号データの取得として理解することもできる。本発明での使用に適したイメージング部分から放出される信号の例は、(i)γ線のように人体の外部で検出できるもの、又は(ii)術中用に設計された放射線検出器のように、インビボ用に設計された検出器の使用によって検出できるものである。本明細書に記載された方法の「形成」段階は、コンピューターによって実施され、取得した信号データに再構成アルゴリズムを適用してデータセットを得る。このデータセットを操作して、被検体の関心領域を示す画像を形成する。
【0038】
本発明の化合物のイメージング部分は、好ましくは以下の(i)〜(iv)から選択される。
(i)放射性金属イオン、
(ii)γ線放出型放射性ハロゲン、
(iii)陽電子放出型放射性非金属、及び
(iv)常磁性金属イオン。
【0039】
イメージング部分が放射性金属イオンつまり放射性金属である場合、好適な放射性金属は、64Cu、48V、52Fe、55Co、94mTc又は68Gaのような陽電子放射体、或いは99mTc、111In、113mIn又は67Gaのようなγ線放射体である。好ましい放射性金属は99mTc、64Cu、68Ga及び111Inである。最も好ましい放射性金属はγ放射体、特に99mTcである。
【0040】
イメージング部分が常磁性金属イオンである場合、かかる金属イオンの好適なものとして、Gd(III)、Mn(II)、Cu(II)、Cr(III)、Fe(III)、Co(II)、Er(II)、Ni(II)、Eu(III)又はDy(III)が挙げられる。好ましい常磁性金属イオンはGd(III)、Mn(II)及びFe(III)であり、Gd(III)が特に好ましい。
【0041】
式Iの化合物のイメージング部分が金属イオンである場合、イメージング部分は好ましくは金属イオンと合成配位子との金属錯体として存在する。「金属鎖体」という用語は、金属イオンと1以上の配位子との配位鎖体を意味する。金属鎖体は「キレート交換耐性」、つまり金属の配位部位に対する他の潜在的な競合配位子との配位子交換を容易に起こさないものであるのが極めて好ましい。潜在的な競合配位子としては、インビトロ標品中の他の賦形剤(製剤に使用される例えば放射線防護剤又は抗菌保存剤)又は生体の内在性化合物(例えばグルタチオン、トランスフェリン又は血漿タンパク質)がある。「合成」という用語は、その通常の意味、つまり天然資源(例えば哺乳類の身体)から分離されたものではなく、人造のものを意味する。かかる化合物は、その製造並びに不純物プロファイルを十分に制御できるという利点がある。
【0042】
キレート交換耐性の金属鎖体を形成する本発明の使用に適した配位子としては、(金属ドナー原子同士が炭素原子又は非配位ヘテロ原子の非配位骨格で連結されて)五又は六員キレート環が形成されるように2〜6、好ましくは2〜4個の金属ドナー原子が配列したキレート剤、或いはイソニトリル、ホスフィン又はジアゼニドのように金属イオンに強く結合するドナー原子を含む単座配位子が挙げられる。キレート剤の一部として金属によく結合するドナー原子の例は、アミン、チオール、アミド、オキシム及びホスフィンである。ホスフィン類は強固な金属鎖体を形成し、単座又は二座ホスフィンであっても適当な金属鎖体を形成する。イソニトリル及びジアゼニドの線状構造は、それらをキレート剤に導入するのが容易ではないので、通例、単座配位子として使用される。適当なイソニトリルの例としては、tert−ブチルイソニトリルのような単純なアルキルイソニトリル及びMIBI(すなわち、1−イソシアノ−2−メトキシ−2−メチルプロパン)のようなエーテル置換イソニトリルが挙げられる。適当なホスフィンの例としては、テトロホスミン及び単座ホスフィン類、例えばトリス(3−メトキシプロピル)ホスフィンが挙げられる。適当なジアゼニドの例としては、配位子のHYNIC系配位子、すなわちヒドラジン置換ピリジン又はニコチンアミドが挙げられる。
【0043】
金属イオンがテクネチウムである場合、キレート交換耐性の金属鎖体を形成するのに適したキレート剤の例としては、特に限定されないが、以下の(i)〜(v)が挙げられる。
(i)ジアミンジオキシム;
(ii)チオールトリアミドドナーセットを有するN3S配位子、例えばMAG3(メルカプトアセチルトリグリシン)及び関連配位子、又はジアミドピリジンチオールドナーセットを有するもの、例えばPica;
(iii)ジアミンジチオールドナーセットを有するN2S2配位子、例えばBAT又はECD(すなわちエチルシステイネート二量体)又はアミドアミンジチオールドナーセットを有するもの、例えばMAMA;
(iv)テトラミン、アミドトリアミン又はジアミンジアミンドナーセットを有する開環又はマクロ環状配位子であるN4配位子、例えばサイクラム、モノオキシサイクラム又はジオキシサイクラム;或いは
(v)ジアミンジフェノールドナーセットを有するN2O2配位子。
【0044】
イメージング部分がテクネチウムである場合、本発明の好ましいキレート剤はジアミンジオキシム及びテトラアミンであり、それらの好ましいものについて以下で詳しく説明する。
【0045】
好ましいジアミンジオキシムは、次の式VIIのものである。
【0046】
【化3】
【0047】
式中、
E1〜E6は各々独立にR*基であり、
各R*はH、C1-10アルキル、C3-10アルキルアリール、C2-10アルコキシアルキル、C1-10ヒドロキシアルキル、C1-10フルオロアルキル、C2-10カルボキシアルキル又はC1-10アミノアルキルであるか、或いは2以上のR*基がそれらと結合した原子と共に飽和又は不飽和炭素環又は複素環を形成するもので、1以上のR*基がCBPと結合しており、
Q′は式−(J′)e−の架橋基であり、eは3、4又は5であり、各J′は独立に−O−、−NR*−又は−C(R*)2−であるが、−(J′)f−が、−O−又は−NR*−であるJ′基を最大1個しか含まないことを条件とする。
【0048】
好ましいQ′基は以下のものである。
Q′=−(CH2)(CHR*)(CH2)−、すなわちプロピレンアミンオキシムつまりPnAO誘導体、
Q′=−(CH2)2(CHR*)(CH2)2−、すなわちペンチレンアミンオキシムつまりPentAO誘導体、
Q′=−(CH2)2NR*(CH2)2−。
【0049】
E1〜E6は、好ましくは、C1-3アルキル、アルキルアリール、アルコキシアルキル、ヒドロキシアルキル、フルオロアルキル、カルボキシアルキル、アミノアルキルから選択される。最も好ましくは、各E1〜E6基はCH3である。
【0050】
ジアミンジオキシムは、好ましくは、E1もしくはE6のR*基又はQ′部分のR*基で結合している。最も好ましくは、Q′部分のR*基で結合する。Q′部分のR*基で結合している場合、R*基は好ましくは橋頭位である。この場合、Q′は、好ましくは、−(CH2)(CHR*)(CH2)−、−(CH2)2(CHR*)(CH2)2−、又は−(CH2)2NR*(CH2)2−であり、最も好ましくは−(CH2)2(CHR*)(CH2)2−である。特に好ましい二官能性ジアミンジオキシムキレート剤は、次の式VIIaのものである。
【0051】
【化4】
【0052】
式中、E7〜E20は各々独立に上記で定義したR*基であり、
GはN又はCR*であり、
Y′は式Iのペプチド部分との連結点である。
【0053】
式VIIaの好ましいキレート剤は、次の式VIIbのものである。
【0054】
【化5】
【0055】
式中、Gは上記で定義した通りであり、好ましくはCHである。キレートIの調製方法は、国際公開第03/006070号に開示されている。
【0056】
好ましいテトラアミンキレート剤は、次の式VIIIのものである。
【0057】
【化6】
【0058】
式中、
Y″は式Iの残りの部分との連結点であり、
E21〜E26は上記で定義したR*基である。
【0059】
最も好ましいテトラアミンキレートは式VIIIaであり、
【0060】
【化7】
【0061】
式中、
Y″は上記で定義した通りである。式VIIIaのキレートの合成方法は国際公開第06/008496号に開示されている。
【0062】
上述の配位子は、テクネチウム(例えば94mTc又は99mTc)の錯体の形成に特に適しており、Jurisson et al, Chem.Rev.,99,2205−2218(1999)に詳細に記載されている。この配位子は、銅(64Cu又は67Cu)、バナジウム(例えば48V)、鉄(例えば52Fe)又はコバルト(例えば55Co)のような他の金属にも有用である。
【0063】
その他の適当な配位子については、インジウム、イットリウム及びガドリニウム、特に大環状アミノカルボキシレート及びアミノホスホン酸配位子に特に適した配位子を始めとして、Sandozの国際公開第91/01144号に記載されている。ガドリニウムの非イオン性(すなわち中性)の金属錯体を形成する配位子は公知であり、米国特許第4885363号に記載されている。
かかるドナー原子を有する好適なキレート剤の例としては、1、4、7、10−テトラアザシクロデカン−1、4、7、10−テトラ酢酸(DOTA)及びジエチレントリアミンペンタ酢酸(DTPA)が挙げられる。ガドリニウムに対して特に好ましいのは、DTPA、エチレンジアミン四酢酸(EDTA)、トリエチレンテトラアミン六酢酸(TTHA)、1,4,7,10−テトラアザシクロドデカン−1,4,7,10−四酢酸(DOTA)、10−(2−ヒドロキシプロピル)−1,4,7,10−テトラアザシクロドデカン−1,4,7−三酢酸(DO3A)及びこれらの誘導体を始めとするキレートである。
【0064】
イメージング部分が、金属錯体の一部として存在する金属イオンである場合、リンカー基が付随して存在するのが好ましい。こうした場合のリンカー基の役割は、金属の配位で得られる比較的嵩高い金属錯体を、ペプチドの活性部位から遠ざけることによって、例えば、基質の結合に支障をきたさないようにすることである。これは、嵩高い基が活性部位から遠ざかる自由度をもつようにするための柔軟性(例えば単純なアルキル鎖)及び/又は金属錯体を活性部位から遠ざけるシクロアルキル系もしくはアリール系スペーサーのような剛直性の組合せによって達成することができる。こうしたキレートに関して好ましいリンカー基は、原子数2〜10、最も好ましくは原子数2〜5の骨格鎖を含むもので、原子数2又は3のものが特に好ましい。原子数2の最短のリンカー基骨格鎖であっても、ペプチドからキレーターが十分に離隔して相互反応が最小限になるという利点が得られる。さらに、ペプチドは、金属イオンへのキレーターの配位に有効に拮抗できなくなる。こうして、ペプチドの生物学的ターゲティング特性とキレーターの金属錯形成能が共に保持される。なお、金属錯体がペプチドにそれらの結合が血中で容易に代謝されないように結合していることが強く望まれる。かかる代謝によって、標識ペプチドがインビボで所望の標的部位に達する前に、イメージング金属錯体が開裂してしまうからである。そこで、ペプチドは、好ましくは、容易には代謝されない結合を含むリンカー基を介して金属錯体に共有結合させる。かかる好適な結合は、炭素−炭素結合、アミド結合、尿素もしくはチオ尿素結合又はエーテル結合である。
【0065】
アルキレン基又はアリーレン基のような非ペプチド系リンカー基は、式Iのペプチドと有意の水素結合がないため、リンカーがペプチドと相互作用しないという利点を有する。好ましいアルキレンスペーサー基は−(CH2)q−であり、式中、qは2〜5の整数である。好ましくは、qは2又は3である。好ましいアリーレンスペーサーは次の式IXのものである。
【0066】
【化8】
【0067】
式中、a及びbは各々独立に0、1又は2である。
【0068】
好ましいリンカー基は−CH2CH2−(L)p−であり、Lは上記で定義した通りであり、pは0〜3の整数である。最も好ましくは、−(L)p−は−CO−又は−NR−である。式VIIbについて、GがNであって、−(L)p−が−NH−である場合、この組合せは市販の対称中間生成物N(CH2CH2NH2)3から誘導できるという追加の利点がある。
【0069】
イメージング用金属がテクネチウムの場合、通常のテクネチウム出発原料は過テクネチウム酸塩、すなわちTcO4-つまり酸化状態がTc(VII)のテクネチウムである。過テクネチウム酸塩自体は金属錯体を形成しにくいので、テクネチウム錯体の製造に際しては、テクネチウムの酸化状態を低酸化状態(Tc(I)〜Tc(V))に還元することによって錯形成を促進するため、第一スズイオンのような適当な還元剤を添加する必要がある。溶媒は有機溶媒でも、水性溶媒でも、それらの混合物でもよい。溶媒が有機溶媒を含む場合、有機溶媒は好ましくはエタノール又はDMSOのような生体適合性溶媒である。好ましくは、溶媒は水性溶媒であり、最も好ましくは等張塩類溶液である。
【0070】
イメージング部分がγ線放出型放射性ハロゲンである場合、放射性ハロゲンは好適には123I、131I又は77Brから選択される。125Iは、外部からのインビボイメージング用のイメージング部分としての使用には適していないので、特に除外してある。インビボイメージングに好ましいγ線放出型放射性ハロゲンは123Iである。
【0071】
イメージング部分が放射性ヨウ素である場合、式Iの化合物は、求電子又は求核ヨウ素化或いは標識アルデヒド又はケトンとの縮合を起こすような誘導体を含む前駆体化合物によって得ることができる。前者に属するものの例としては、以下の(a)〜(c)が挙げられる。
(a)トリアルキルスタンナン(例えばトリメチルスタンニル又はトリブチルスタンニル)、トリアルキルシラン(例えばトリメチルシリル)又は有機ホウ素化合物(例えば、ボロン酸エステル又はオルガノトリフルオロボレート)のような有機金属誘導体、
(b)ハロゲン交換のための非放射性臭化アルキル、或いは求核ヨウ素化のためのアルキルトシレート、メシレート又はトリフレート、
(c)求電子ヨウ素化用の活性化芳香族環(例えばフェノール)及び求核ヨウ素化用の活性化芳香族環(例えばアリールヨードニウム、アリールジアゾニウム、アリールトリアルキルアンモニウム塩又はニトロアリール誘導体)。
【0072】
かかる前駆体として好適なものは、ヨウ化又は臭化アリールのような非放射性ハロゲン原子(放射性ヨウ素交換を可能とするため)、有機金属前駆体化合物(例えばトリアルキルスズ、トリアルキルシリル又は有機ホウ素化合物)、トリアゼンのような有機前駆体、或いはヨードニウム塩のような求核置換反応のための良好な脱離基を含む。放射性ヨウ素化では、前駆体は好ましくは有機金属前駆体化合物を含み、最も好ましくはトリアルキルスズを含む。
【0073】
前駆体及び放射性ヨウ素を有機分子に導入する方法は、Bolton, J.Lab.Comp.Radiopharm.,45,485−528(2002)に記載されている。適当なボロン酸エステル有機ホウ素化合物及びその製造方法は、Kabalaka et al, Nucl.Med.Biol.,29、841−843(2002)及び30,369−373(2003)に記載されている。適当なオルガノトリフルオロホウ化物及びその製造方法は、Kabalaka et al, Nucl.Med.Biol.,31、935−938(2004)に記載されている。
【0074】
放射性ヨウ素を結合させることのできるアリール基の例としては、以下のものがある。
【0075】
【化9】
【0076】
これらはいずれも、芳香族環での放射性ヨウ素置換が容易な置換基を含んでいる。チロシン残基では、その内在フェノール基を利用して放射性ヨウ素化を実施できる。
【0077】
放射性ヨウ素を含む他の置換基は、例えば以下のような放射性ハロゲン交換による直接ヨウ素化によって合成することができる。
【0078】
【化10】
【0079】
飽和脂肪族系に結合したヨウ素原子はインビボで代謝され易く、放射性ヨウ素が失われ易いことが知られているので、放射性ヨウ素原子は好ましくは芳香族環(ベンゼン環など)又はビニル基に直接共有結合で結合させる。
【0080】
イメージング部分が陽電子放出型放射性非金属である場合、かかる陽電子放射体の好適なものとして、11C、13N、15O、17F、18F、75Br、76Br又は124Iが挙げられる。好ましい陽電子放出型放射性非金属は11C、13N、18F及び124Iであり、特に好ましくは11C及び18Fであり、最も好ましくは18Fである。
【0081】
放射性フッ素化は、臭化アルキル、アルキルメシレート又はアルキルトシレートのような良好な脱離基を有する前駆体化合物の適当な化学基と18F−フッ化物との反応を用いた直接標識法で実施できる。18Fは、18F(CH2)3OH反応体を用いたN−ハロアセチル基のアルキル化によって導入することもでき、−NH(CO)CH2O(CH2)318F誘導体が得られる。アリール系については、アリールジアゾニウム塩、アリールニトロ化合物又はアリール第四級アンモニウム塩からの18F−フッ化物求核置換が、アリール−18F誘導体への好適な経路である。
【0082】
本発明の18F−標識化合物は、18Fフルオロジアルキルアミンの形成後、18Fフルオロジアルキルアミンを、例えば塩素、P(O)Ph3又は活性化エステルを含む前駆体と反応させてアミドを形成することによって得ることができる。
【0083】
ペプチドの放射性フッ素化に特に適した別の放射性フッ素化法は、国際公開第03/080544号に記載されており、チオールカップリングを利用する。以下の置換基のいずれかを含む前駆体化合物を式Xの化合物と反応させて、それぞれ式(Xa)又は(Xb)の放射性フッ素化イメージング剤を生成させる。
【0084】
【化11】
【0085】
18F−XX−SH (X)
式中、YXは式−(LY)w−のリンカーであって、適宜1〜6のヘテロ原子を含んでいてもよく(LYはLについて上記で定義した通りであり、yは1〜10である。)、
XXは、式−(LX)x−のリンカーであって、適宜1〜10のヘテロ原子を含んでいてもよく(LXはLについて上記で定義した通りであり、xは1〜30である。)、
*は化合物の残りの部分との連結点である。
【0086】
【化12】
【0087】
式中、XX、YX及び*は、上記で定義した通りである。
【0088】
ペプチドの放射性フッ素化に特に適した追加の方法は、国際公開第04/080492号に記載されており、アミノキシカップリングを利用する。放射性フッ素化は、式(XI)の前駆体化合物を式(XIa)の化合物と反応させるか、或いは式(XII)の前駆体化合物を式(XIIa)と反応させることによって実施され、それぞれ式(XIII)又は(XIV)のコンジュゲートが得られる。
【0089】
【化13】
【0090】
式中、XXI及びXXIIは、リンカー基−(LXI)z−であって、適宜1〜6のヘテロ原子を含んでいてもよい(LXIは、Lについて上記で定義した通りであり、zは1〜10である。)。
R1は、アルデヒド部分、ケトン部分、アセタールのような保護アルデヒド、ケタールのような保護ケトン、或いは酸化剤を用いてアルデヒド又はケトンへと迅速かつ効率的に酸化できるジオール又はN−末端セリン残基のような官能基である。
R2は、水性緩衝液のような穏和な条件下でR1と部位特異的に反応して安定なコンジュゲートを与える官能基である。R2は、第一級アミン、第二級アミン、ヒドロキシルアミン、ヒドラジン、ヒドラジド、アミノキシ、フェニルヒドラジン、セミカルバジド又はチオセミカルバジドのようなアンモニア誘導体であってもよく、好ましくは、ヒドラジン、ヒドラジド又はアミノキシ基である。
R3は、R4と部位特異的に反応する官能基である。R3は、第一級アミン、第二級アミン、ヒドロキシルアミン、ヒドラジン、ヒドラジド、アミノキシ、フェニルヒドラジン、セミカルバジド又はチオセミカルバジドなどのアンモニア誘導体であってもよく、好ましくは、ヒドラジン、ヒドラジド又はアミノキシ基である。
R4は、アルデヒド部分、ケトン部分、アセタールのような保護アルデヒド、ケタールのような保護ケトン、或いは酸化剤を用いてアルデヒド又はケトンへと迅速かつ効率的に酸化できるジオール又はN−末端セリン残基のような官能基である。
【0091】
【化14】
【0092】
式中、Wは−CO−NH−、−NH−、−O−、−NHCONH−又は−NHCSNH−であり、好ましくは−CO−NH−、−NH−又は−O−であり、YはH、C1〜6アルキル又はC5〜6アリール置換基であり、XXI、XXII及び*は上記で定義した通りである。
【0093】
18F−標識誘導体の合成経路についてのさらに詳しい内容は、Bolton,J.Lab.Comp.Radiopharm.,45,485−528(2002)に記載されている。
【0094】
好ましいイメージング部分は、生体に投与した後、単光子放射断層撮影(SPECT)、陽電子放射断層撮影(PET)及び磁気共鳴イメージング(MRI)などの手段によって外部から非侵襲的に検出することができるものである。最も好ましいイメージング部分は、放射性、特に、放射性金属イオン、γ線放出型放射性ハロゲン及び陽電子放出型放射性非金属、特に、SPECT又はPETを用いたイメージングに適したもの、例えば、99mTc、123I、11C及び18Fである。
【0095】
好ましい一実施形態では、W1及びW2は共にリンカー基を表し、Z1は有機色素基を表し、Z2はイメージング部分を表す。これらの化合物及びその調製方法は、国際公開第2006/054904号に記載されている。最も好ましくは、Z2は放射性イメージング部分である。これらの好ましい化合物の例としては、以下のものが挙げられる。
【0096】
【化15】
【0097】
【化16】
【0098】
化合物1〜4の調製方法は、国際公開第2006/054904号に詳述されている。
【0099】
別の好ましい実施形態では、W1はリンカー基を表し、Z1はイメージング部分を表し、W2は任意要素としてのリンカー基を表し、Z2は水素である。これらの化合物及びその調製方法は、国際公開第2005/012335号に記載されている。これらの好ましい化合物の例としては、以下のものが挙げられる。
【0100】
【化17】
【0101】
化合物5及び6の調製方法は、国際公開第2005/012335号に詳述されている。
【0102】
別の好ましい実施形態では、W1は任意要素としてのリンカー基を表し、Z1は水素を表し、W2はリンカー基を表し、Z2はイメージング部分を表す。これらの化合物及びその調製方法は、国際公開第2003/006491号に記載されている。これらの好ましい化合物の例としては、以下のものが挙げられる。
【0103】
【化18】
【0104】
【化19】
【0105】
化合物7〜10の調製方法は、国際公開第2003/006491号に詳述されている。
【0106】
化合物1〜4及び7〜10について、別法として、ジアミンジオキシムキレート部分は、化合物1a〜4a及び7a〜10aを形成するためにテトラアミンキレート部分で置き換えられていることがさらに好ましい。
【0107】
【化20】
【0108】
【化21】
【0109】
【化22】
【0110】
【化23】
【0111】
化合物1a〜10aの調製方法は、化合物1〜10のジアミンジオキシムキレートの代わりに化合物1a〜10aのテトラアミンキレートを用いる点を除いて、化合物1〜10の方法と類似している。テトラアミンキレートの連結は、Bocで保護された分子種との通常のペプチドカップリングを用いて達成される。
【0112】
生体分布を改善することを目的として、すなわち、主にバックグラウンド肝臓取込み低減するために、多くの追加の修飾化合物を合成した。化合物11〜15の合成の詳細を、以下の実施例8〜12に示す。化合物11は、ジアミンジオキシムキレートからテトラアミンキレートに変える効果を評価するために設計した。化合物12では、システイン酸基を加えた。化合物13は、ペプチドのN−末端側に追加のPEG部分を有する。化合物14は、ペプチドのC−末端側に追加のPEG部分を有する。化合物15では、テトラアミンキレートを、多くのグルタミン酸残基に加えて使用した。
【0113】
本明細書に記載されている方法は、検出可能な量の式Iの化合物を投与しておいた被検体を「準備」することから始める。本発明の方法の目的は、診断上有用な画像の用意である。従って、式Iの化合物の被検体への投与は、画像の形成を容易にするために必要な予備段階であると理解することができる。好ましくは、被検体は、哺乳類、最も好ましくはヒトである。最も好ましくは、被検体は、インビボでのインタクトな哺乳類の身体である。従って、好ましい実施形態では、式Iの化合物は、生体適合性担体と共に、哺乳類への投与に適した形態の化合物を含む医薬組成物として投与されてきた。投与の好ましい経路は、血管内投与である。代替実施形態では、検出可能な量の式Iの化合物の投与は、本方法の一部として行われる。
【0114】
用意する段階に続いて検出段階に先立って、式Iの化合物を、被検体における任意の線維形成性組織に結合させる。例えば、被検体がインタクトな哺乳類である場合、式Iの化合物は、哺乳類の身体を動的に進み、その中の様々な組織と接触する。化合物が、任意の線維形成性組織に接触するとすぐに、線維形成性組織からの化合物のクリアランスが、非線維形成性組織からより時間がかかるように、特異的相互作用が起きる。線維形成性組織と特異的に結合している化合物の検出が、線維形成性組織と結合している化合物と非線維形成性組織に結合している化合物の比の結果として可能になるある時点に到達する。理想的なそのような比は、2:1以上である。
【0115】
「生体適合性担体」とは、式Iの化合物を懸濁又は溶解できる流体、特に液体であって、組成物が生理学的に認容できるもの、つまり毒性も耐え難い不快感も伴わずに哺乳類の身体に投与することができるものである。生体適合性担体は好適には注射可能な担体液であり、例えば、発熱物質を含まない注射用の滅菌水、食塩液のような水溶液(これは注射用の最終製剤が等張性又は非低張性となるように調整するのに都合がよい)、1種以上の張度調節物質(例えば血漿陽イオンと生体適合性対イオンとの塩)、糖(例えばグルコース又はスクロース)、糖アルコール(例えばソルビトール又はマンニトール)、グリコール(例えばグリセロール)その他の非イオン性ポリオール材料(例えばポリエチレングリコール、プロピレングリコールなど)の水溶液である。生体適合性担体は、エタノールのような生体適合性の有機溶媒を含んでいてもよい。かかる有機溶媒は、親油性の高い化合物又は製剤の可溶化に有用である。好ましくは、生体適合性担体は発熱物質を含まない注射用水、等張塩類溶液又はエタノール水溶液である。静脈内注射用の生体適合性担体のpHは好適には4.0〜10.5の範囲内である。
【0116】
かかる医薬組成物は、好適には、無菌状態を維持したまま皮下注射針で一回又は複数回穿刺するのに適したシール(例えばクリンプオン式セプタムシール蓋)を備えた容器に入れて供給される。かかる容器には、1回又は複数回分の患者用量を入れることができる。好ましい多用量容器は、複数回分の患者用量を収容した単一バルクバイアル(例えば容積10〜30cm3のもの)からなり、臨床症状に応じて製剤の有効期間中様々な時間間隔で1回分の患者用量を臨床グレードのシリンジに吸引することができる。プレフィルドシリンジは1回分の患者用量つまり「単位用量」を収容するように設計され、そのため好ましくは使い捨て又はその他臨床用に適したシリンジである。医薬組成物が放射性医薬組成物の場合、プレフィルドシリンジは、適宜、オペレーターを放射能被曝から保護するためのシリンジシールドを備えていてもよい。かかる適当な放射性医薬品シリンジシールドは当技術分野で公知であり、好ましくは鉛又はタングステンからなる。
【0117】
医薬組成物は、キットから調製することができる。或いは、医薬組成物は、所望の滅菌生成物が得られるような無菌製造条件下で製造してもよい。医薬組成物を非滅菌条件下で調製した後、例えばγ線照射、オートクレーブ処理、乾熱又は化学的処理(例えばエチレンオキサイドでの処理)を用いて最終的に滅菌してもよい。
【0118】
本発明の造影剤に関して上述した通り、放射性医薬組成物に対して最も好ましい本発明の放射性造影基は99mTc、123I、11C及び18Fである。
【0119】
本発明の別の態様では、式Iの化合物は、被検体の器官又は身体領域における線維形成の存在、場所及び/又は量を決定するための医薬品の調製に用いることができる。式Iの好ましい及び最も好ましい実施形態、器官又は身体領域、被検体及び投与方法は、上記で定義した通りである。
【0120】
本発明は、活性化HSCの存在、場所及び/又は量を評価するのに有用であり、線維形成の指標を与える。これが特に有利であるのは、線維形成性組織が、線維性組織よりも早期活動性疾患の優れたマーカーであり、繊維性組織は、疾患プロセスが解消しつつある場所にも存在するからである。従って、疾患プロセスの識別を、治療の実施が最も有効であるステージで行うことができる。
【0121】
これらの利点を、下記の非限定的な実施例で例証する。
【0122】
実施例の簡単な説明
実施例1は、化合物2が、インビトロで活性化ヒトHSCと特異的に結合することを例証する。
【0123】
実施例2は、ラット胆管結紮モデル及び関連する偽手術モデルを準備するのに使用される方法、並びに組織病理学的検証について記載する。
【0124】
実施例3は、化合物4の取込みと線維形成の相関関係を例証する。線維形成のマーカーは、術後15日目で(実施例2に記載されている)ラット胆管結紮(BDL)モデルにおいて高く、ラットBDLモデルにおける線維形成性肝臓中への化合物4の取込みは、術後15日目で最も高い。
【0125】
実施例4は、BDL線維形成性肝臓との化合物4の結合が、非放射性の化合物4により特異的に阻害されることを例証する。
【0126】
実施例5は、化合物4の肝臓取込みとαvインテグリンの肝臓発現の相関関係を例証する。
【0127】
実施例6は、BDL線維形成性肝臓との化合物8の結合が、非放射性の化合物8により特異的に阻害されることを例証する。
【0128】
実施例7は、αv発現と化合物8の取込みの相関関係を例証する。
【0129】
実施例8〜12は、化合物11〜15の合成について記載する。
【0130】
【表1】
【実施例】
【0131】
実施例1:活性化ヒトHSCへTの化合物6の結合
1(i) EA−Hy926細胞から調製した膜との結合
化合物6の阻害定数を、報文に記載された膜結合アッセイを使用して測定した(Indrevoll et al, Bioorg & Med Chem Lett, 2006, 16, 6190−6193)。
【0132】
簡潔に述べると、ヒト内皮腺癌細胞系EA−Hy926からの膜を調製し、精製した膜画分についてKdを計算した。次いで、競合的結合アッセイを確立し、非放射性の化合物6についての阻害定数を測定した。125I−エキスタチンを標識リガンドとして使用し、非放射性のエキスタチンを参照標準として使用した。
【0133】
非放射性の試験化合物(非放射性のエキスタチン又は非放射性の化合物6のいずれか)の合計16通りの希釈液を調製し、125I−エキスタチンと膜の組合せと混合してから37℃で1時間インキュベーションした。数回の洗浄後、結合した材料を、Skatronマイクロハーベスターを使用してフィルター上に収集した。最後に、フィルタースポットを切り取り、Packard γ−カウンター中でカウントした。
【0134】
図1は、EA−Hy926膜上の非放射性の化合物6及びエキスタチンと対比した125I−エキスタチンの結合を図示している。
【0135】
1(ii) 活性化ヒト肝星細胞との結合
活性化ヒト肝星細胞LX−2(Mount Sinai School of Medicine(米国ニューヨーク)のScott L.Friedman教授から提供されたもの)を、10%FBS、ペニシリン、ストレプトマイシン及びグルタミンを含有するダルベッコ変法イーグル培地中でコンフルエンスまで12ウェルプレート(Nunc)において培養した。細胞を、50mM Tris、pH7,4、150mM NaCl、5mM MnCl2、1mM CaCl2及び0,01% BSAを含有する冷緩衝液中で2回洗浄した。細胞を、攪拌しながら4℃で60分間、様々な濃度の非放射性の化合物と一緒に、痕跡量の標識合物(0.1nM 125I化合物又は0.1nM 125I−エキスタチンのいずれか)を含有する緩衝液中でさらにインキュベートした。インキュベーション後、非結合材料を、冷緩衝液で3回細胞を洗浄することにより除去した。細胞を、0.1M NaOHを添加することにより壁から引き離し、チューブに移し、γ−カウンター(Packard)中でカウントした。図2は、観察された活性値を図示している。
【0136】
実施例2:化合物3及び化合物8の活性化ラットHSCとの結合
2(i) EA−Hy926細胞から調製した膜との結合
化合物3及び化合物8についての阻害定数を、実施例1に記載されている方法を使用して決定した。図3は、EA−Hy926膜上の非放射性の化合物3、化合物8及びエキスタチンと対比した125I−エキスタチンの結合を図示している。
【0137】
2(ii) 細胞及び培養
活性化ラット肝星細胞系(不死化)は、University of OsloのTrond Berg教授から、そのご好意によって入手した。細胞を、10%FBS、グルタミン及びペニシリン/ストレプトマイシンを含有するダルベッコ変法イーグル培地中でコンフルエンスまで150mmの培養フラスコにおいて培養した。
【0138】
2(iii) 膜の調製
細胞を、氷冷PBS、pH7.4中で2回洗浄し、PBS10mlを添加し、ラバーポリスマンでこそぎ落とし、氷上の50mlバイアルに移した。別のPBS10mlをフラスコに添加し、こそぎ落とし、第一の細胞と混合した。細胞を4℃で2000rpmで10分間遠心分離した。上清を流し捨て、ペレットをPBS3ml中に再懸濁した。すべてのペレットを1つのチューブ中に混合し、上記のように遠心分離し、最終ペレットを直ちに−70℃で冷凍した。
【0139】
細胞ペレットを、50mM Tris−HCl、5mM MgCl2、1mM EGTA、pH7,4と、さらに、プロテアーゼ阻害剤、10ug/mlロイペプチン、10ug/mlペプスタチン、200ug/mlバシトラシン、0.5ug/mlアプロチニン及び100uM PMSFを含有する氷冷ホモジネーション緩衝液(細胞の10倍量、例えば、ペレット2gにホモジネーション緩衝液20ml)中に再懸濁した。細胞を、Dounceホモジナイザー、ペストルB中で10ストローク3回により氷上でホモジナイズした。ホモジネートを、4℃で2100rpmで5分間遠心分離し、上清を流し捨てた。ペレットを、ホモジネーション緩衝液10ml中に再懸濁し、10ストローク2回で再びホモジナイズした後、10分間2100rpmで遠心分離した。この上清を第一の上清と混合し、JA−17ローター付きBeckman Coulter Centrifuge中、16500rpm(29000×g)で遠心分離した。ペレットを、ホモジネーション緩衝液20mL中に再懸濁し、遠心分離段階を繰り返した。
【0140】
上清を流し捨て、ペレットを、10mM Hepes、135mM NaCl、4,8mM KCl、1,7mM MgSO4、2,5mM CaCl2、1.0mM NaH2PO4、pH7,4を含有する結合緩衝液3ml中に再懸濁した。タンパク質測定のためのサンプルを取り除いた後、結合緩衝液さらに3mlを添加した。300ulのアリコートを、直ちに−80℃で冷凍した。
【0141】
2(iv) タンパク質測定
十分に混合した膜調製物100ulを、1:10に希釈し、段階希釈液及びタンパク質含量を、BCA Protein Assay Reagentキット(Pierce No.23225)中で取扱説明書に従って測定した。
【0142】
2(v) 星細胞膜上のインテグリンとの結合を試験する実験設計
実験を96ウェルプレート中で準備した。各ウェルに、緩衝液(50mM Tris、pH7,4、150mM NaCl、5mM MnCl2、1mM CaCl2、0,01% BSA)60ul、非放射性の化合物3、化合物8又は非放射性のエキスタチン20ul、125−Iエキスタチン20ul及び膜溶液(ウェル当たり膜約1ugに相当するように緩衝液中で1:30に希釈した)50ulを添加し、攪拌しながら60分間37℃でインキュベートした。インキュベーション後、非結合材料を、Skatron細胞ハーベスター中でPBSにより洗い流し、結合材料をフィルタースポット中に濃縮した。このフィルタースポットを切り取り、チューブに添加し、最終的に、γ−カウンター中でカウントした。フィルターは、4時間以上水中0.3%PEI中で予浸した。図4は、得られた結果を示している。
【0143】
実施例3:胆管結紮(BDL)及びシャム動物
3(i) 動物モデルの準備
非近交系雄性Sprague Dawleyラット(180〜200g;Charles River)を、すべての胆管結紮(BDL)及びシャム試験に使用した。6日の馴化後、ラットを、2つの群(BDL群及びシャム群)に分けた。
【0144】
BDL動物については、腹部を剪毛し、ベタジン溶液と、続いて、皮下(s.c.)のカルプロフェン5mg/kg及びs.c.のブプロノルフィン(bupronorphine)5mg/kgで消毒し、イソフルラン麻酔下で、正中開腹術を実施し、総胆管の場所を見つけた。胆管を二重結紮し、第一の結紮は肝管の結合部の間で行い、第二の結紮は、膵管の入口の上で行った。
【0145】
第二の群(シャム動物)の腹部を剪毛し、ベタジン溶液と、続いて、s.c.のカルプロフェン5mg/kg及びs.c.のブプロノルフィン5mg/kgで消毒した。動物は、胆管を操作し、縫合糸を胆管の下に通す偽手術を受けた。
【0146】
閉じる前に、食塩水2〜3mlを、各動物の腹膜中に投与した。筋膜及び皮膚を閉じ、動物にs.c.のメタクロプロミド(metaclopromide)2mg/kg、s.c.のバイトリル(Baytril)5mg/kg、及びs.c.の食塩水約2mlを投与した。カルプロフェン(5mg/kg)を、必要に応じて、続く2、3日間与えた。動物を、実験の期間中厳重にモニターした。
【0147】
3(ii) 試験化合物の投与及び生体分布
術後の適切な日に、BDL及びシャム動物を取り出し、イソフルラン麻酔下に置き、次いで、各動物に、尾静脈を介して静脈内(i.v.)に0.3ml(約3MBq)を注射した。試験品目の注射後の適切な時点で、各動物をイソフルランで再麻酔し、頸椎脱臼により屠殺して秤量し、バーコード走査システムを介して重量を記録した。各動物を解剖し、下記の器官及び組織を摘出し、BASILカウンタープロトコル40又は手動カウンティングを使用してカウントした。
【0148】
骨* 筋肉*
血液* 腎臓
膀胱及び尿(B/U) 肺
肝臓* 脾臓
胃及び内容物 小腸及び大腸(SI及びLI)
心臓 甲状腺
皮膚* 屠殺体
注射部位
*秤量したサンプル
全器官(例えば、肝臓)で記録された活性を、バックグラウンド放射能及び放射性崩壊に対して補正し、放射能の生体分布を、式Iを参照して計算した。
【0149】
【数1】
【0150】
秤量した組織サンプル(例えば、血液)における注射した放射能に占める割合を計算すると、式2から、全組織における%i.d.を得た。
【0151】
【数2】
【0152】
【表2】
【0153】
3(iii) 組織病理学的評価
各動物の肝臓からの切片を、室温で10%緩衝中性ホルマリン溶液中で固定し、パラフィンに包埋し、5μmの薄い切片を調製した。切片を、胆管過形成、壊死及び炎症の評価についてはMayer’s Hematoxylin and Eosin(H&E)、及び線維症については報文に記載された標準染色プロトコル(Gomori 1950 Am. J. Clin. Pathol. 20:661−663)を使用するGomori’sトリクロム染色(コラーゲン染色)で染色した。
【0154】
肝臓損傷の組織学的グレーディングを、各病変の範囲及び重症度を評価し、次いで、病変を以下の通り、すなわち、病変なし=0、最小=1、軽度=2、中等度=3、顕著=4、重度=5と0〜5に点数化することにより半定量的に評価した。組織の組織形態学的評価を、術後の日数と、手順(すなわち、シャム対胆管結紮)の両方に関してブラインドベースで実施した。全データを追加の病理学者がピアレビューした。病理学的分析は、光学顕微鏡により実施した。切片を、40倍、100倍又は400倍対物レンズを使用して明視野照明下で観察した。
【0155】
表4は、組織病理学的評価を要約している。
【0156】
【表3】
【0157】
提示されているデータは、線維形成に関係する特異的取込み示す分子を識別するための肝線維症の適当なモデルとしての、特に、15日目における、このモデルの使用を支持している。
【0158】
実施例4:シャムとラットBDLモデル(線維形成のマーカーが高いときの15日目)における線維形成性肝臓への化合物8の取込量の対比
BDL及びシャムモデルを、実施例3に記載されているように準備した。化合物8の取込み、以下の通り、
【0159】
【数3】
【0160】
屠殺体における放射能の割合を、解剖後の残留屠殺体における秒当たりカウントを参照し、屠殺体中に残っているサンプリング組織に対して補正することにより計算した。身体組成の生物学的変動は、組織値の推定におけるわずかな不正確さと、従って、屠殺体値の過度の補正又は補正不足をもたらしたかも知れない。屠殺体値が過度に補正された場合、これは、負の値をもたらすこともあった。
【0161】
図5は、術後2、5、10、15、28日目のうちの1日における化合物8、及び術後2日目のスクランブル陰性対照(以下の構造)の尾静脈注射の1時間のBDL動物及び偽手術動物の肝臓におけるSUVとして表される放射能の保持を要約している。
【0162】
【化24】
【0163】
結果は、試験されたすべての時点における偽手術動物と比較してBDLにおける放射能の有意な保持を例証しており、放射能の最高保持は、術後10及び15日目にBDL動物からの肝臓において観察された。術後のいずれの時点においても偽手術動物における肝臓取込みに有意差はなかった。
【0164】
実施例5:BDLモデルにおける術後15日目に、化合物3及び化合物8は、シャム動物及び陰性対照との対比で、線維形成肝臓に取込まれる
BDL及びシャム動物モデルを、実施例3に記載されているように準備した。術後15日目に、化合物3、化合物8又は陰性対照のうちの1つを各動物に投与し、肝臓取込み実施例3に記載されているように評価した。
【0165】
陰性対照の構造は以下の通りであった。
【0166】
【化25】
【0167】
図6は、偽手術動物と比較して、BDLモデルにおける化合物3及び8の有意に高い肝臓取込み図示している。
【0168】
実施例6:BDL線維形成性肝臓との化合物8の結合は、非放射性の化合物8により特異的に阻害される
放射能の生体分布を、1:100及び1:10000倍過剰の非放射性の化合物8の添加前と添加後の両方で、化合物8約3MBqの尾静脈を介する静脈内注射から1時間後のBDL及び偽手術雄性Sprague Dawleyラット(実施例3に記載されている手順)において術後15日目に検討した。試験品目の注射後の適切な時点で、各動物をイソフルランで再麻酔し、頸椎脱臼により屠殺して秤量した。各動物を解剖し、器官及び組織(以下の表5及び6を参照)を摘出し、BASILカウンタープロトコル40又は手動カウンティングを使用してカウントした。
【0169】
BDL動物における注射後1時間における活性の蓄積(%i.d.)の主要部位を以下の表5に要約する。
【0170】
【表4】
【0171】
シャム動物における注射後1時間における活性の蓄積(%i.d.)の主要部位を以下の表6に要約する。
【0172】
【表5】
【0173】
実施例7:15日目のBDLにおける化合物8の取込み及びα−v上方制御
BDL及びシャム動物モデルを、実施例3に記載されているように準備した。術後2、15及び28日目の各々で、化合物8約3MBqを動物に投与し、肝臓取込み実施例3に記載されているように評価した。
【0174】
動物の肝臓におけるαvインテグリンの発現を、BDL及び偽手術動物のホモジナイズした肝臓において測定した。術後2、15及び28日目の各々で、動物を屠殺し、それらの肝臓を摘出し、−70℃で保存した。
【0175】
予め秤量した肝臓を、−70℃フリーザーから取り出し、少量の液体窒素の存在下で予冷した乳鉢及び乳棒中に入れた。肝臓を、ほぼ均一な粉末になるまで粉砕した(肝臓の粉砕中に、少量の液体窒素を加え、組織を常に冷凍状態に保った)。粉砕した肝臓を、予冷した医薬測定器具に入れ、さらに使用するまで−70℃フリーザーに入れた。冷凍した粉砕組織1gを、1mM EDTA、0.25Mスクロースを含有する氷冷RIPA緩衝液(Sigma;150mM NaCl、1.0% Igepal CA−630(界面活性剤)、0.5%デオキシコール酸ナトリウム(陰イオン性界面活性剤)、0.1% SDS、50mM Tris、pH8.0)10ml中に直接加え、プロテアーゼ阻害剤カクテル(Sigma)(溶解緩衝液ml当たりカクテル10μl)を新たに加えた。混合物を、4℃でシャープブレードホモジナイザーでホモジナイズし、30分間湿った氷上に保持した。
【0176】
ホモジネートを、予冷したガラス組織Dounce中に移し、乳棒の3回の上下ストロークを4℃で適用した。次いで、組織ホモジネートを、予冷した遠心分離管中に移し、30000rpm及び4℃で30分間遠心分離した。上清を取り出し(全細胞ライセート)、湿った氷上に置いた。相対的タンパク質濃度を、製造者の取扱説明書に従ってBSA標準品を含む市販のタンパク質アッセイキット(Pierce、USA)を使用して各サンプルにおいて決定した(BSAアリコートは、ホモジネーションのために使用された変法RIPA緩衝液中で調製すべきである)。各サンプルからの上清を、H2O中で1:50〜1:200へさらに希釈し、タンパク質濃度を、99マルチウェルPlateリーダー(iEMS)を使用して決定した。各サンプルを、1mlアリコートに分け、さらなる使用まで−70℃で保存した。
【0177】
抗α−vインテグリン捕捉抗体(BD;カタログ番号:611013)を、結合溶液(0.05Mトリス、0.138M NaCl及び0.0027M KCl)中で適切な濃度に希釈し、50μl又は100μlのいずれかを、高タンパク質結合ELISAプレート(Nunc−Immuno(商標)プレート96ウェルプレート、MaxiSorp)の各ウェルに添加した。使用される抗体の量は、200ng/ウェルとした。プレートを密封し、37℃で1時間又は4℃で一晩インキュベートした。プレートを空にして、マイクロタイタープレート上のタンパク質結合のための残った部位を、ブロッキング緩衝液(ウェル当たり0.02%アジ化ナトリウム約300μlを含む3% BSA/TBS)と共にインキュベートすることにより飽和させた。プレートを密封し、37℃で1時間インキュベートし、次いで、TBS/0.01% Tween−20で3回洗浄した。ニートの肝臓ホモジネート上清及び/又は異なる濃度の精製α−vインテグリン標準品を、ブロッキング緩衝液中で調製し、コーティングしたプレート中に三つ組みで添加し(ウェル当たり100μl)、次いで、密封し、37℃で2時間インキュベートした。プレートを、TBS/0.05% Tween−20で4回洗浄し、ブロッキング緩衝液中で希釈された第二の抗インテグリン検出抗体(Chemicon AB1930)をウェルに添加し、37℃で1時間インキュベートした。プレートを、TBS/0.05% Tween−20で4回洗浄し、検出抗体に特異的な二次抗体アルカリホスファターゼコンジュゲート(Sigma;カタログ番号:A7539)をウェルに添加し、37℃で1時間インキュベートした。プレートを、TBS/0.05% Tween−20で4回洗浄し、NPP基質溶液100μlを各ウェルに添加し、室温で2時間又は4℃で一晩インキュベートした。加水分解は、視覚的検査により定性的に又はマイクロタイタープレートリーダーで定量的にモニターした。NPPの加水分解は、推奨インキュベーション時間が経過した後に黄色に見え、標的波長(405nm)における光学密度を、ELISAプレートリーダー(Spectra−Max Plus)で測定した。加水分解は、5M水酸化ナトリウムNaOH50μlを添加することにより停止させた。
【0178】
定量的結果については、未知サンプルの信号を、標準曲線の信号と比較した。統計分析は、GraphPad PRISM、Version4.0を使用して実施した。群間差は、ノンパラメトリックな一元配置ANOVAにより分析した。すべての統計分析において、0.05未満の確率値(P<0.05)を有意と見なした。
【0179】
図7及び図8は、シャム動物と対比したBDL動物におけるαv発現を図示しており、αv発現と化合物8の取込みの相関関係を例証している。
【0180】
実施例8:化合物11の合成
【0181】
【化26】
【0182】
Boc−テトラアミン−N−ヒドロキシスクシンイミドエステル(国際公開第2006/008496号)(36mg、0.050mmol)を、10分間DMF(0.5mL)中のHOAt(1.4mg、0.010mmol)及びNMM(17μL、0.15mmol)で予め活性化し、次いで、DMF(0.5mL)中のCys2−6;c[CH2CO−Lys−Cys−Arg−Gly−Asp−Cys−Phe−Cys]−PEG(4)−ジグリコロイル−NH2(国際公開第03/006491号)(12.6mg、0.010mmol)の溶液に加えた。反応混合物を、3日間室温で撹拌し、次いで、真空中で濃縮した。Boc保護基を、90分間TFA/水/トリイソプロピルシラン(95:2.5:2.5、10mL)溶液の添加により除去した。混合物を濃縮し、粗生成物をエーテルから沈殿させ、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;40分間勾配10〜20%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、凍結乾燥後に10.4mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜20%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=2.02分、m/z1458.7(MH)+は、構造を支持した。
【0183】
99mTcによる標識は、窒素でパージしたP46バイアルに、下記の、MeOH中のIGF前駆体1又は2 100μg、Na2CO3/NaHCO3緩衝液(pH9.2)0.5ml、Drytec(商標)99mTc発生剤からのTcO4-0.5ml、SnCl2/MDP溶液(N2でパージした食塩水100ml中にSnCl210.2mg及びメチレンジホスホン酸101mgを含有する)0.1mlを加えることにより行った。
【0184】
実施例9:化合物12の合成
【0185】
【化27】
【0186】
合成は、0.3mmolスケールで(ローディング量0.432g、0.71mmol/g)RinkアミドAM樹脂上で実施した。最初の3つのカップリング段階は、手動窒素バブラー装置中で行った。樹脂上のFmoc基は、標準プロトコル(DMF中20%ピペリジン)により切断した。Fmoc−Cys(トリチル)−OH(702mg、1.20mmol)を、標準カップリング試薬HATU及びDIEAによりDMF中の樹脂にカップリングさせた。カップリングの終了は、標準Kaiser試験によりチェックした。Fmoc切断後、カップリングを繰り返し、第二のシステインを導入した。樹脂を、ジクロロメタン/TFA/トリイソプロピルシラン(94:5:1、10mL)溶液中に懸濁した。3分後、黄色の溶液を流し出した。この段階を6回繰り返し、側鎖チオール機能の完全な脱保護を確保した。樹脂を、ジクロロメタンで洗浄し、乾燥した(30分間窒素流)。20%ギ酸(18mL)と35%過酸化水素(2mL)の混合物を、室温で1時間放置し、0℃まで冷却した。樹脂を、過ギ酸溶液のアリコート(2×4mL)で洗浄し、次いで、緩やかに攪拌しながら5℃で12時間過ギ酸溶液(5mL)中に保持した。樹脂のアリコートを切断し(TFA/水/トリイソプロピルシラン、95:2.5:2.5)、LC−MS(カラムPhenomenex Luna C18(2)50×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配5〜50%B;流速0.3mL/分、214nm及び254nmにおけるUV検出、ESI−MS)により分析すると、tR=2.48分、m/z564.3(MNa)+は、構造を支持した。
【0187】
上記の樹脂のFmoc除去(標準プロトコル)後、Fmoc−アミノ−PEG(4)−ジグリコール酸(796mg、1.50mmol)を、標準カップリング試薬(HATU及びDIEA)を使用してDMF中の樹脂にカップリングさせた。6時間後、アリコートを切断し(前の段落に記載されている)、LC−MS(カラムPhenomenex Luna C18(2)50×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配5〜50%B;流速0.3mL/分、214nm及び254nmにおけるUV検出、ESI−MS)により分析すると、tR=3.28分、m/z832.3(MH)+は、完全な変換を示した。
【0188】
すべての後続アミノ酸を、上記からの樹脂上約0.25mmol上でSlowMocカップリングプロトコルを使用してAB1433A自動ペプチド合成機を使用してカップリングさせると、H−Lys(Boc)−Cys(tBu)−Arg(Pmc)−Gly−Asp(OtBu)−Cys(tBu)−Phe−Cys(Trt)−PEG4−ジグリコロイル−CyA−CyA−Rinkアミド樹脂が得られた。
【0189】
ジクロロメタン(10mL)中のクロロ酢酸(142mg、1.50mmol)及びDCC(155mg、0.75mmol)の溶液を、15分間室温で撹拌し、次いで、濾過し、濃縮した。残渣をDMFに取り、上記のペプチド樹脂に加えた。ペプチドを、2時間TFA/水/トリイソプロピルシラン溶液(95:2.5:2.5、10mL)を使用して樹脂から切断した。溶液を濃縮し、粗ペプチドをエーテルから沈殿させ、真空中で乾燥すると、340mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)50×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配0〜50%B;流速0.3mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析、tR=3.09分、m/z1710.6(MH)+は、クロロアセチル化ペプチドを支持した。
【0190】
50%アセトニトリル(64mL)中のペプチド(68mg)の溶液に、pHが8.5になるまで2.5%アンモニアを加えた。混合物を16時間室温で撹拌した。LC−MS分析は、完全なチオエーテル環化を支持した。アセトニトリルを減圧下で除去し、水溶液を凍結乾燥した。単離した材料を、TFA中DMSOの5%溶液(100mL)に取り、60分間室温で撹拌した。混合物を濃縮し、粗生成物を、エーテルからの沈殿により単離し、続く、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配5〜20%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、固体材料48mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)50×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配0〜30%B;流速0.3mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=3.18分、m/z1560.5(MH)+は、ジスルフィド架橋形成を支持した。
【0191】
NMP(1.5mL)中のペプチド(5mg)、キレートI−グルタリル テトラフルオロチオフェノールエステル(国際公開第03/006491号)(5mg)及びDIEA(2.7μl)の部分的に溶けた混合物を、一晩40℃で加熱した。混合物を、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;40分間勾配0〜30%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、純粋な生成物2.9mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配0〜30%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=3.83分、m/z1000.7(MH2)2+は、構造を支持した。
【0192】
99mTcによる標識は、国際公開第2003/006491号に記載の通り実施した。
【0193】
実施例10:化合物13の合成
【0194】
【化28】
【0195】
Boc−PEG(6)−ジグリコール酸(50mg、0.10mmol)を、標準カップリング試薬(HATU及びNMM)を使用し、DMF中のCys2−6;c[CH2CO−Lys−Cys−Arg−Gly−Asp−Cys−Phe−Cys]−PEG(4)−ジグリコロイル−NH2(国際公開第03/006491号)(25.2mg、0.020mmol)にカップリングさせた。反応混合物を、一晩撹拌し、真空中で濃縮した。Boc保護基を、室温で45分間TFA/水/トリイソプロピルシラン(95:2.5:2.5、10mL)の添加により除去した。混合物を濃縮し、続いて、粗材料をエーテルから沈殿させた。生成物を、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;40分間勾配10〜30%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、5.5mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜25%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=3.01分、m/z1636.0(MH)+は、生成物を支持した。
【0196】
キレートI−グルタル酸(国際公開第03/006491号)(15.6mg、0.034mmol)を、標準カップリング試薬(PyAOP(7−アザベンゾトリアゾール−1−イルオキシ−トリス−(ピロリジノ)ホスホニウムヘキサフルオロホスフェート)及びNMM)を使用して、DMF中のペプチド(5.5mg、0.0034mmol)にカップリングさせた。4時間後、混合物を、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;40分間勾配15〜30%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、3.8mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配15〜30%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=2.13分、m/z1039.0(MH2)2+は、構造を支持した。
【0197】
99mTcによる標識は、国際公開第2003/006491号に記載の通り実施した。
【0198】
実施例11:化合物14の合成
【0199】
【化29】
【0200】
合成は、0.80mmolスケールでRink Amide MBHA樹脂(ローディング量0.72mmol/g)上で手動窒素バブラー装置中で行った。樹脂上のFmoc基は、標準プロトコルにより切断した。Fmoc−PEG(4)−ジグリコール酸(850mg、1.6mmol)を、DMF中で標準カップリング試薬(PyAOP及びNMM)を使用して樹脂にカップリングさせた。コンジュゲーション反応と、続く、Fmoc脱保護を、5つのPEG(4)−ジグリコロイル基が樹脂上に導入されるまで繰り返した。
【0201】
Fmoc−Cys(Trt)−OH(1.0mmol)を、DMF/ジクロロメタン(1:1、6mL)中で標準カップリング試薬(PyAOP及びNMM)を使用して、上記の樹脂(0.2mmol)にカップリングさせた。2.5時間後、樹脂のアリコートを切断し(上記)、LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜80%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)により分析すると、tR=2.3分、m/z1794.1(MH)+は、完全な変換を示した。
【0202】
すべての後続アミノ酸を、上記からの樹脂上約0.1mmol上でSlowMocカップリングプロトコルを使用し、AB1433A自動ペプチド合成機を使用してカップリングさせると、H−Lys(Boc)−Cys(tBu)−Arg(Pbf)−Gly−Asp(OtBu)−Cys(tBu)−Phe−Cys(Trt)−(PEG(4)−ジグリコロイル)5−Rinkアミド樹脂が得られた。
【0203】
クロロ酢酸無水物(2.00mmol)を、1時間ジクロロメタン(10mL)中のクロロ酢酸(378mg、4.00mmol)及びDCC(412mg、2.00mmol)の溶液を撹拌し、続いて、濾過することによって合成した。溶液を濃縮し、残渣をDMF(3mL)に取り、上記のペプチド樹脂に加えた。NMM(220μL、2.00mmol)を加え、混合物を3時間放置した。樹脂のアリコートを切断し(上記)、LC−MSにより分析すると、完全な変換を支持した。ペプチドを、3時間TFA/水/トリイソプロピルシラン溶液(95:2.5:2.5、10mL)の添加により樹脂から切断した。溶液を濃縮した。
【0204】
上記からの残渣を、水(100mL)に取り、エーテルで抽出した。水溶液にアセトニトリル(100mL)を加え、pHを、希アンモニアの添加により8に調整した。反応物を一晩放置した。アセトニトリルを蒸発させ、粗生成物を凍結乾燥により単離すると、154mgが得られた。LC−MSによる分析は、完全なチオエーテル環化を支持した。
【0205】
上記からの単離材料を、TFA中DMSOの5%溶液(80mL)に取り、60分間室温で撹拌した。混合物を濃縮し、粗生成物を、エーテルからの沈殿により単離し、続いて、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配15〜25%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、純粋な材料5mgが得られた。LC−MSによる分析は、ジスルフィド架橋形成を支持した。
【0206】
キレートI−グルタル酸(国際公開第03/006491号)(9mg、0.02mmol)を、一晩DMF中で標準カップリング試薬(PyAOP及びDIEA)を使用して上記のペプチドにカップリングさせた。生成物を、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配10〜30%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製すると、1.3mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜60%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=1.8分、m/z1430.1(MH2)2+は、構造を支持した。
【0207】
99mTcによる標識は、国際公開第2003/006491号に記載の通り実施した。
【0208】
実施例12:化合物15の合成
【0209】
【化30】
【0210】
ペプチド配列H−Lys(Boc)−Cys(tBu)−Arg(Pbf)−Gly−Asp(OtBu)−Cys(tBu)−Phe−Cys(Trt)−Gly−[Glu(OtBu)]5−NH2を、SlowMocカップリング方法を使用してRinkアミドMBHA樹脂(0.1mmol)上でABI433A自動ペプチド合成機で組み立てた。
【0211】
ジクロロメタン(10mL)中のクロロ酢酸(378mg、4.00mmol)及びDCC(412mg、2.00mmol)の溶液を、1時間室温で撹拌し、次いで、濾過し、濃縮した。残渣をDMFに取り、上記のペプチド樹脂に加えた。NMM(220μl、2.00mmol)を加え、反応物を2時間放置した。樹脂のアリコートを切断し(上記)、LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配20〜30%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)により分析すると、tR=2.2分、m/z1820.7(MH)+は、完全な変換を支持した。ペプチドを、2時間TFA/水/トリイソプロピルシラン溶液(95:2.5:2.5、10mL)の添加により樹脂から切断した。溶液を濃縮し、粗生成物を、エーテルからの沈殿により単離し、続いて、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配20〜40%B;流速10mL/分、214nm及び254nmにおけるUV検出)により精製した。半純粋な生成物を凍結乾燥により単離し、50%アセトニトリル/水(6mL)に取った。pHを、希アンモニアの添加により8に調整し、反応物を一晩撹拌した。アセトニトリルを減圧下で蒸発させ、生成物を凍結乾燥により単離すると、14.8mgが得られた。LC−MS[カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜60%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=2.0分、m/z1785.0(MH)+は、完全なチオエーテル環化を支持した。
【0212】
上記の単離ペプチドを、TFA中DMSOの5%溶液(8mL)に取り、15分間室温で撹拌した。混合物を濃縮し、粗生成物を、エーテルから沈殿させ、続いて、調製用HPLC(カラムPhenomenex Luna C18(2)250×10mm、10μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配10〜20%B;流速5mL/分、214nm及び254nmにおけるUV検出)により精製すると、純粋な材料2mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配10〜60%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=1.0分、m/z1670.8(MH)+は、ペプチドを支持した。
【0213】
DMF中の上記からのペプチド(2mg、1μmol)及びBoc−テトラアミン−N−ヒドロキシスクシンイミドエステル(国際公開第2006/008496号)(8.6mg、0.012mmol)の溶液に、DIEA(3μL、0.02mmol)を加えた。反応混合物を一晩撹拌し、調製用HPLC(カラムPhenomenex Luna C18(2)250×21.2mm、5μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;60分間勾配20〜80%B;流速10mL/分、214nm及び254nmにおけるUV検出)による精製にかけた。純粋な生成物を凍結乾燥し、TFAとジクロロメタンの混合物(1:1、2mL)に溶かした。2時間後、溶媒を蒸発させ、残渣をアセトニトリル/水に取り、凍結乾燥すると、純粋な生成物1.0mgが得られた。LC−MS(カラムPhenomenex Luna C18(2)20×2mm、3μm、溶媒:A=水/0.1% TFA及びB=アセトニトリル/0.1% TFA;5分間勾配0〜20%B;流速0.6mL/分、214nm及び254nmにおけるUV検出、ESI−MS)による分析tR=4.2分、m/z935.5(MH2)2+は、構造を支持した。
【0214】
99mTcによる標識は、上記の実施例8において化合物11について記載されているように行った。
【特許請求の範囲】
【請求項1】
被検体の肝臓における線維形成の存在、場所及び/又は量を決定する方法に用いられる式Iの化合物であって、該方法が、
(i)検出可能な量の式Iの化合物を投与しておいた被検体を準備する段階と、
(ii)肝臓中の線維形成性組織に式Iの化合物を結合させる段階と、
(iii)式Iの化合物から放出される信号をインビボイメージング法で検出する段階と、
(iv)信号の場所及び/又は量を表す画像を形成する段階
とを含んでおり、式Iの化合物は以下の通り定義される化合物。
【化1】
式中、
Gはグリシンを表し、
Dはアスパラギン酸を表し、
X1は、アスパラギン酸、グルタミン酸、リジン、ホモリジンもしくはC3〜6ジアミノアルカン酸又はそれらの誘導体から選択されるアミノ酸を表し、
X2及びX4は独立に側鎖同士が連結して環化架橋を形成するアミノ酸残基(例えばジスルフィド又はチオエーテル結合を形成するシステイン又はホモシステイン)又は環化架橋を形成し得る他のアミノ酸(例えばアスパラギン酸及びリジン)を表し、
X3はアルギニン、N−メチルアルギニン又はアルギニン模倣体を表し、
X5はチロシン、フェニルアラニン、3−ヨード−チロシン C4〜6シクロアルキルアラニン、ナフチルアラニン又はそれらの誘導体を表し、
X6はX6をC(=O)基に連結するチオエーテル結合又はチオアセタール結合のいずれかを形成するチオール含有アミノ酸を表し、
W1及びW2は独立に任意要素としてのリンカー基であって、W1が存在するときはX1のアミノ酸側鎖部分に連結し、W2が存在するときはX6のカルボキシ基に連結し、
Z1及びZ2は、Z1とZ2の少なくとも一方がイメージング部分であることを条件として、独立にイメージング部分、糖部分、有機色素基又は水素である。
【請求項2】
X1がリシンである、請求項1記載の化合物。
【請求項3】
X2及びX4が独立にシステイン又はホモシステインである、請求項1又は請求項2記載の化合物。
【請求項4】
X3がアルギニンである、請求項1乃至請求項3のいずれか1項記載の化合物。
【請求項5】
X5がシクロヘキシルアラニン、フェニルアラニン又は3−ヨード−チロシンである、請求項1乃至請求項4のいずれか1項記載の化合物。
【請求項6】
X6がシステイン又はホモシステインである、請求項1乃至請求項5のいずれか1項記載の化合物。
【請求項7】
X1がリシンであり、X2及びX4が共にシステインであり、X3がアルギニンであり、X5がフェニルアラニンであり、X6がシステインである、請求項1乃至請求項6のいずれか1項記載の化合物。
【請求項8】
W1及びW2の少なくとも一方が存在していて、リンカー基が式−(L)n−の基である、請求項1乃至請求項7のいずれか1項記載の化合物。
式中
各Lは独立に−C(=O)−、−CR′2−、−CR′=CR′−、−C≡C−、−CR′2CO2−、−CO2CR′2−、−NR′−、−NR′CO−、−CONR′−、−NR′(C=O)NR′−、−NR′(C=S)NR′−、−SO2NR′−、−NR′SO2−、−CR′2OCR′2−、−CR′2SCR′2−、−CR′2NR′CR′2−、C4-8シクロヘテロアルキレン基、C4-8シクロアルキレン基、C5-12アリーレン基、C3-12ヘテロアリーレン基、アミノ酸、ポリアルキレングリコール、ポリ乳酸又はポリグリコール酸部分であり、
nは1〜15の整数であり、
各R′基は独立にH又はC1-10アルキル、C3-10アルキルアリール、C2-10アルコキシアルキル、C1-10ヒドロキシアルキル、C1-10フルオロアルキルであるか、或いは2以上のR′基がそれらに結合した原子と共に炭素環、複素環式、飽和又は不飽和環を形成したものである。
【請求項9】
W1及びW2の少なくとも一方が存在していて、リンカー基が1〜10単位の単分散PEG構成単位を表す、請求項1乃至請求項8のいずれか1項記載の化合物。
【請求項10】
W1及びW2の少なくとも一方が存在していて、リンカー基が1〜10のアミノ酸残基を表す、請求項1乃至請求項8のいずれか1項記載の化合物。
【請求項11】
Z1及びZ2の一方が糖部分である、請求項1乃至請求項8のいずれか1項記載の化合物。
【請求項12】
Z1及びZ2の一方が有機色素基である、請求項1乃至請求項8のいずれか1項記載の化合物。
【請求項13】
前記イメージング部分が以下の(i)〜(iv)から選択される、請求項1乃至請求項12のいずれか1項記載の化合物。
(i)放射性金属イオン、
(ii)γ線放出型放射性ハロゲン、
(iii)陽電子放出型放射性非金属、及び
(iv)常磁性金属イオン。
【請求項14】
前記イメージング部分が前記(i)〜(iii)から選択される、請求項13記載の化合物。
【請求項15】
前記イメージング部分が99mTc、123I、11C及び18Fから選択される、請求項14記載の化合物。
【請求項16】
生体適合性担体と共に、哺乳類への投与に適した形態の医薬組成物として調製される、請求項1乃至請求項15のいずれか1項記載の化合物。
【請求項17】
被検体がインビボでのインタクトな哺乳類の身体である、請求項1記載の方法に使用するための請求項16記載の化合物。
【請求項18】
被検体がヒトである、請求項1記載の方法に使用するための請求項16記載の化合物。
【請求項19】
被検体の器官又は身体領域における線維形成の存在、場所及び/又は量を決定するための医薬品の調製に使用される、請求項1乃至請求項16のいずれか1項記載の式Iの化合物。
【請求項1】
被検体の肝臓における線維形成の存在、場所及び/又は量を決定する方法に用いられる式Iの化合物であって、該方法が、
(i)検出可能な量の式Iの化合物を投与しておいた被検体を準備する段階と、
(ii)肝臓中の線維形成性組織に式Iの化合物を結合させる段階と、
(iii)式Iの化合物から放出される信号をインビボイメージング法で検出する段階と、
(iv)信号の場所及び/又は量を表す画像を形成する段階
とを含んでおり、式Iの化合物は以下の通り定義される化合物。
【化1】
式中、
Gはグリシンを表し、
Dはアスパラギン酸を表し、
X1は、アスパラギン酸、グルタミン酸、リジン、ホモリジンもしくはC3〜6ジアミノアルカン酸又はそれらの誘導体から選択されるアミノ酸を表し、
X2及びX4は独立に側鎖同士が連結して環化架橋を形成するアミノ酸残基(例えばジスルフィド又はチオエーテル結合を形成するシステイン又はホモシステイン)又は環化架橋を形成し得る他のアミノ酸(例えばアスパラギン酸及びリジン)を表し、
X3はアルギニン、N−メチルアルギニン又はアルギニン模倣体を表し、
X5はチロシン、フェニルアラニン、3−ヨード−チロシン C4〜6シクロアルキルアラニン、ナフチルアラニン又はそれらの誘導体を表し、
X6はX6をC(=O)基に連結するチオエーテル結合又はチオアセタール結合のいずれかを形成するチオール含有アミノ酸を表し、
W1及びW2は独立に任意要素としてのリンカー基であって、W1が存在するときはX1のアミノ酸側鎖部分に連結し、W2が存在するときはX6のカルボキシ基に連結し、
Z1及びZ2は、Z1とZ2の少なくとも一方がイメージング部分であることを条件として、独立にイメージング部分、糖部分、有機色素基又は水素である。
【請求項2】
X1がリシンである、請求項1記載の化合物。
【請求項3】
X2及びX4が独立にシステイン又はホモシステインである、請求項1又は請求項2記載の化合物。
【請求項4】
X3がアルギニンである、請求項1乃至請求項3のいずれか1項記載の化合物。
【請求項5】
X5がシクロヘキシルアラニン、フェニルアラニン又は3−ヨード−チロシンである、請求項1乃至請求項4のいずれか1項記載の化合物。
【請求項6】
X6がシステイン又はホモシステインである、請求項1乃至請求項5のいずれか1項記載の化合物。
【請求項7】
X1がリシンであり、X2及びX4が共にシステインであり、X3がアルギニンであり、X5がフェニルアラニンであり、X6がシステインである、請求項1乃至請求項6のいずれか1項記載の化合物。
【請求項8】
W1及びW2の少なくとも一方が存在していて、リンカー基が式−(L)n−の基である、請求項1乃至請求項7のいずれか1項記載の化合物。
式中
各Lは独立に−C(=O)−、−CR′2−、−CR′=CR′−、−C≡C−、−CR′2CO2−、−CO2CR′2−、−NR′−、−NR′CO−、−CONR′−、−NR′(C=O)NR′−、−NR′(C=S)NR′−、−SO2NR′−、−NR′SO2−、−CR′2OCR′2−、−CR′2SCR′2−、−CR′2NR′CR′2−、C4-8シクロヘテロアルキレン基、C4-8シクロアルキレン基、C5-12アリーレン基、C3-12ヘテロアリーレン基、アミノ酸、ポリアルキレングリコール、ポリ乳酸又はポリグリコール酸部分であり、
nは1〜15の整数であり、
各R′基は独立にH又はC1-10アルキル、C3-10アルキルアリール、C2-10アルコキシアルキル、C1-10ヒドロキシアルキル、C1-10フルオロアルキルであるか、或いは2以上のR′基がそれらに結合した原子と共に炭素環、複素環式、飽和又は不飽和環を形成したものである。
【請求項9】
W1及びW2の少なくとも一方が存在していて、リンカー基が1〜10単位の単分散PEG構成単位を表す、請求項1乃至請求項8のいずれか1項記載の化合物。
【請求項10】
W1及びW2の少なくとも一方が存在していて、リンカー基が1〜10のアミノ酸残基を表す、請求項1乃至請求項8のいずれか1項記載の化合物。
【請求項11】
Z1及びZ2の一方が糖部分である、請求項1乃至請求項8のいずれか1項記載の化合物。
【請求項12】
Z1及びZ2の一方が有機色素基である、請求項1乃至請求項8のいずれか1項記載の化合物。
【請求項13】
前記イメージング部分が以下の(i)〜(iv)から選択される、請求項1乃至請求項12のいずれか1項記載の化合物。
(i)放射性金属イオン、
(ii)γ線放出型放射性ハロゲン、
(iii)陽電子放出型放射性非金属、及び
(iv)常磁性金属イオン。
【請求項14】
前記イメージング部分が前記(i)〜(iii)から選択される、請求項13記載の化合物。
【請求項15】
前記イメージング部分が99mTc、123I、11C及び18Fから選択される、請求項14記載の化合物。
【請求項16】
生体適合性担体と共に、哺乳類への投与に適した形態の医薬組成物として調製される、請求項1乃至請求項15のいずれか1項記載の化合物。
【請求項17】
被検体がインビボでのインタクトな哺乳類の身体である、請求項1記載の方法に使用するための請求項16記載の化合物。
【請求項18】
被検体がヒトである、請求項1記載の方法に使用するための請求項16記載の化合物。
【請求項19】
被検体の器官又は身体領域における線維形成の存在、場所及び/又は量を決定するための医薬品の調製に使用される、請求項1乃至請求項16のいずれか1項記載の式Iの化合物。
【図1】

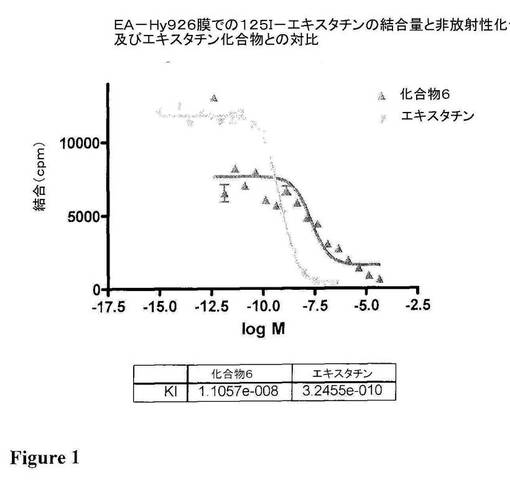
【図2】
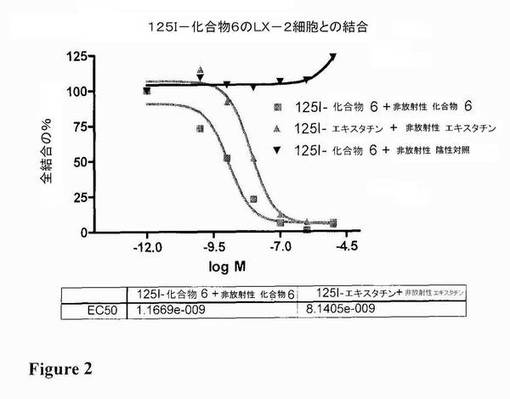
【図3】
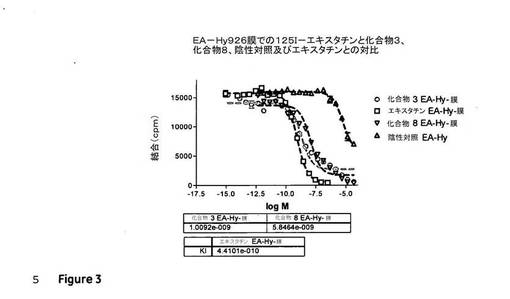
【図4】
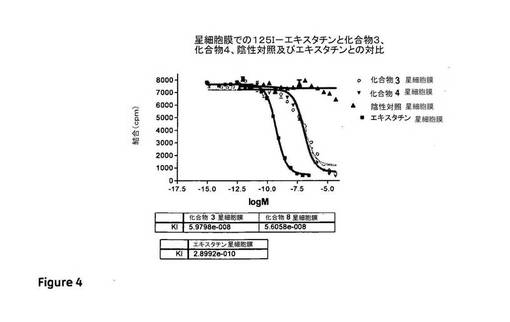
【図5】
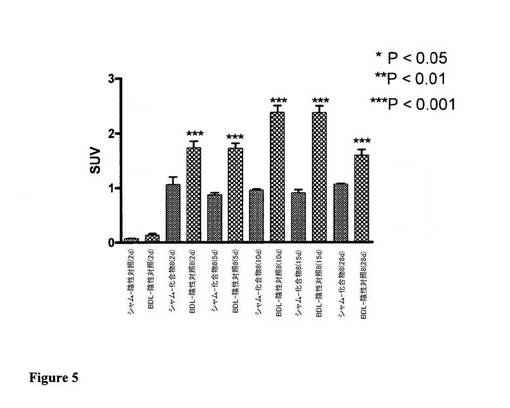
【図6】
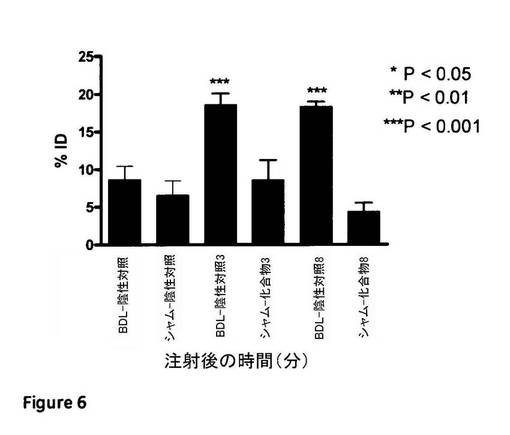
【図7】
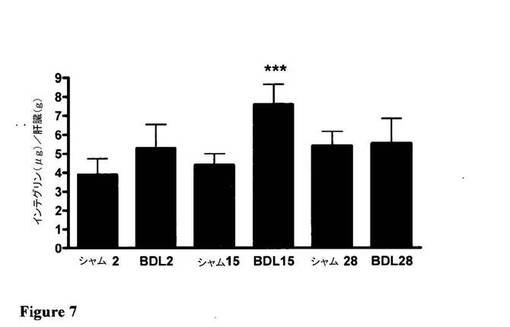
【図8】
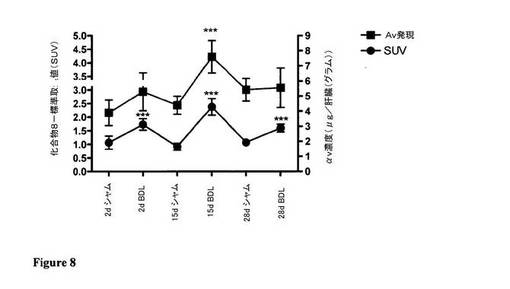
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2011−506273(P2011−506273A)
【公表日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願番号】特願2010−534461(P2010−534461)
【出願日】平成20年11月19日(2008.11.19)
【国際出願番号】PCT/EP2008/065824
【国際公開番号】WO2009/071444
【国際公開日】平成21年6月11日(2009.6.11)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
【公表日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願日】平成20年11月19日(2008.11.19)
【国際出願番号】PCT/EP2008/065824
【国際公開番号】WO2009/071444
【国際公開日】平成21年6月11日(2009.6.11)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
[ Back to top ]