
新規サルビアノール酸化合物L、その調製方法及び使用
新規サルビアノール酸L化合物、その調製方法、上記サルビアノール酸Lを含有する医薬組成物及び心臓・脳血管疾患を治療するための薬剤を調製するためのその使用が開示される。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、伝統的な漢方薬分野、より具体的には新種のサルビアノール酸化合物に関する。
【背景技術】
【0002】
サルビア・ミルチオルヒザ・ブゲ(Salvia miltiorrhiza Bge.)(ファム・ラビアタエ(Fam. Labiatae))の乾燥根である丹参(Radix Salviae Miltiorrhizae:ラディックス・サルビアエ・ミルチオルヒザエ)(中国生薬、以下「タンジン」と称する)は、苦味があり、少し冷たい感じがし、うっ血を除去することによって疼痛を止める機能を伴って心臓及び肝臓のチャネルに作用し、血流を活性化させて、また心臓を清浄にすることによって不穏状態を軽減する。一連の現在の薬理学的研究がタンジンに対して行われており、タンジンが冠状動脈を拡張させ、微小循環を改善させて、心臓を保護する効果を有し、また血小板凝集を抑制及び除去し、無酸素耐性の身体能力、並びに抗肝炎、抗腫瘍及び抗ウイルス等の活性を高めることが可能であることを示している。
【0003】
2001年に、中国医学科学院 北京協和医科大学の薬物研究所(Institute of Materia Madica, Chinese Academy of Medical Sciences & Peking Union Medical College)のL.N. Li et al.(非特許文献1)は、サルビアノール酸A、B、C、D、E、F、G、H、I、J、リトスペルミン酸、ロスマリン酸及びイソサルビアノール酸C等を含むタンジン又は同属の植物から単離されるフェノール酸誘導体の13個の水溶性生理活性成分が存在することを報告した。さらに、これらの成分の薬理学的作用もまた開示されている。
【0004】
Rena. Kasimu et al.(非特許文献2)は、サルビアノール酸Kの化学構造について報告した。
【0005】
外国の研究者らもまた、タンジンの水溶性生理活性成分に関して研究してきた。1999年には、ジョージ・ワシントン大学(George Washington University)は、抗HIVインテグラーゼ及び他のウイルスに対する13個のサルビアノール酸誘導体の効果に関して米国特許を出願して、最終的に付与されている。これらは全て、タンジンが、多大な潜在性を持ち、かつ開発する価値のある医療用植物資源であることを示唆した。
【0006】
本発明の上記サルビアノール酸Lは、大量のスクリーニングのプロセスにおいてタンジン中で見出された、まさに新規の化合物である。これまで、この化合物に関連する構造及び薬理学的効果はいまだに報告されていない。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Bulletin of Medical Research, 2001, Vol 30(7)
【非特許文献2】Journal of Xinjiang Medical University, 2002, Vol. 25(3)
【発明の概要】
【課題を解決するための手段】
【0008】
本発明の目的は、サルビアノール酸Lの新規化合物を提供することである。
【0009】
本発明のさらなる目的は、サルビアノール酸Lを含む医薬組成物を提供することである。
【0010】
本発明の別の目的は、サルビアノール酸Lを調製する方法を提供することである。
【0011】
本発明の別の目的は、心臓血管疾患を治療するための薬剤の調製におけるサルビアノール酸Lの使用を提供することである。
【図面の簡単な説明】
【0012】
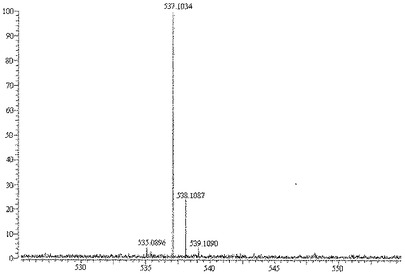
【図1】サルビアノール酸Lの高分解能質量スペクトログラムを示す図である。
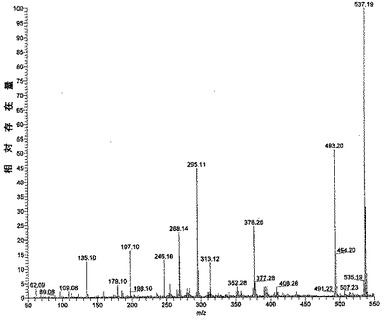
【図2】サルビアノール酸Lのエレクトロスプレーイオン化質量スペクトログラムを示す図である。
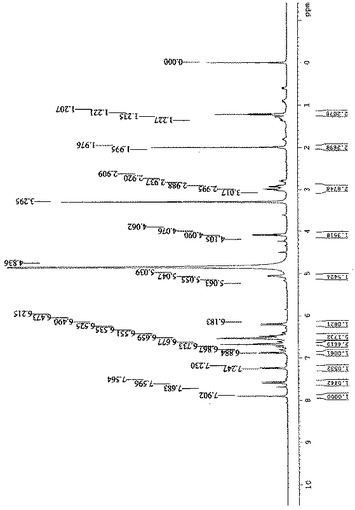
【図3】CD3ODを使用することによる500MHzでのサルビアノール酸Lの1H−NMRダイアグラムを示す図である。
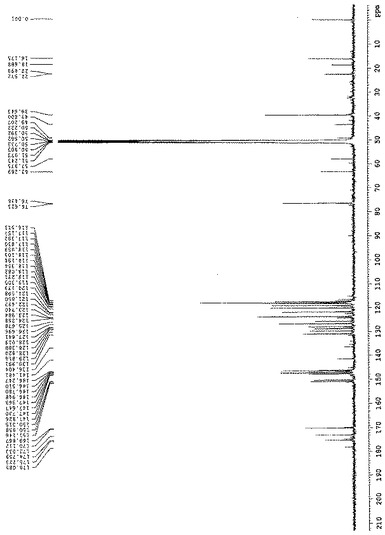
【図4】CD3ODを使用することによる125MHzでのサルビアノール酸Lの13C−NMRダイアグラムを示す図である。
【図5】CD3ODを使用することによる125MHzでのサルビアノール酸LのDEPTダイアグラムを示す図である。
【図6】CD3ODを使用することによる500MHzでのサルビアノール酸LのgCOSYダイアグラムを示す図である。
【図7】CD3ODを使用することによる500MHzでのサルビアノール酸LのgHMBCダイアグラムを示す図である。
【図8】CD3ODを使用することによる500MHzでのサルビアノール酸LのgHMQCダイアグラムを示す図である。
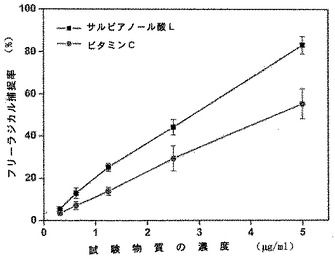
【図9】試験物質のフリーラジカルを捕捉する能力を示す図である。
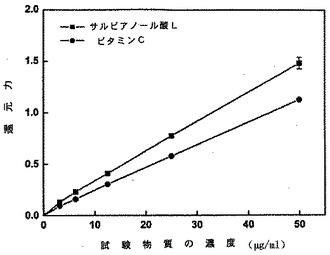
【図10】サルビアノール酸とビタミンCとの間での還元力の比較を示す図である。
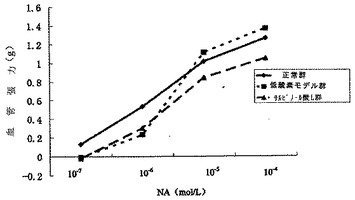
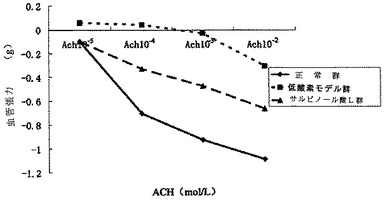
【図11】血管収縮に対するサルビアノール酸Lの凍結乾燥粉末の効果を示す図である。
【図12】血管拡張に対するサルビアノール酸Lの凍結乾燥粉末の効果を示す図である。
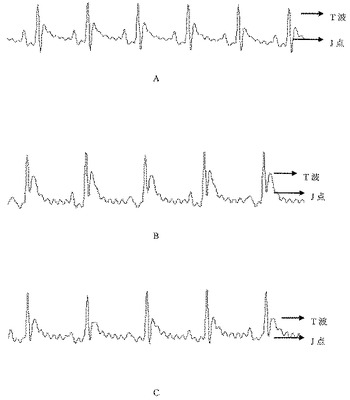
【図13】ピツイトリンで下垂体を処理した後の心電図(ECG)を示す図であり、ここでA)は、モデル対照群から得られる正常なECGであり、B)は、ピツイトリンを投与した15秒後のモデル対照群から得られるものであり、C)は、ピツイトリンを投与した30秒後のモデル対照群から得られるものである。
【0013】
本発明は、下記のように一般式(I)で表わされる新規化合物、その薬学的に許容可能な塩、溶媒和物及び加水分解可能なエステルに関する:
【化1】
【発明を実施するための形態】
【0014】
本発明によれば、フェノール酸の新規化合物の構造は、物理化学的特性、高分解能質量分析法(QFT−ESI)、エレクトロスプレーイオン化質量分析法(ESI−MS)、1H−NMR、13C−NMR、DEPT、gCOSY、gHMBC及びgHMQCにより特定された。
【0015】
本発明の化合物は、淡い帯黄色粉末である。
【0016】
本発明による化合物は、FeCl3による薄層クロマトグラフィ(TLC)発色反応で陽性の結果を示し、本発明による化合物がフェノール系化合物であり得ることを示唆した。
【0017】
m/z 537.1034で擬分子イオンピークを示した高分解能質量分析法(QFT−ESI)により、分子式は、不飽和度Ω 17を有するC27H22O12であることが確認された。
【0018】
ESI−MSでは、m/z 537での本発明による化合物の分子イオンピークは、まず8’’−カルボキシル基を容易に損失することができ(−44)、その結果m/z 493でフラグメントイオンピークを形成し(サルビアノール酸Aの分子イオンピークと同じ構造を有する)、次にサルビアノール酸Aのフラグメンテーション規則性に従ってm/z 313、m/z 295で2つのフラグメントイオンピークを形成する。
【0019】
本発明によれば、サルビアノール酸Aのフラグメンテーション規則性は下記の通りに提示される:
【化2】
【0020】
m/z 493、m/z 313、m/z 295での主なフラグメントイオンピークが、その質量スペクトルにおけるサルビアノール酸Aの主なイオンピークであることは明らかである。したがって、本発明の化合物は、サルビアノール酸Aの骨格構造と同じ骨格構造を有する。
【0021】
プロトン核磁気共鳴(1H−NMR)スペクトルは、δ 5.09(1H,dd,J=8.0,4.5Hz)での酸素に結合されたメテニルプロトンの1個のシグナル、δ 6.88(1H,d,J=8.5Hz)、δ 7.25(1H,d,J=8.5Hz)、δ 7.59(1H,d,J=16.0Hz)、δ 6.22(1H,d,J=16.0Hz)、δ 6.68(1H,s)、δ 6.55(2H,d,J=8.0Hz)、δ 6.58(1H,d,J=2.0Hz)、δ 6.69(1H,d,J=8.0Hz)、δ 6.54(1H,dd,J=8.5,2.0Hz)、δ 7.92(1H,s)での芳香族プロトンの11個のシグナル、δ 3.01(2H,ddd,J=14.0,8.0,4.5Hz)での脂肪族プロトンの2個のシグナルを示す。
【0022】
炭素−13核磁気共鳴(13C−NMR)スペクトルは、δ 39.6での1個の脂肪族炭素シグナル、δ 76.4での酸素に結合されたメテニル炭素の1個のシグナル、δ 170.1、δ 173.0、δ 175.1でのカルボニル炭素の3個のシグナル、及びδ 117.4、δ 117.8、δ 117.8、δ 118.2、δ 119.2、δ 120.2、δ 121.7、δ 123.7、δ 125.7、δ 126.6、δ 128.0、δ 128.8、δ 129.9、δ 130.9、δ 146.2、δ 146.5、δ 146.9、δ 147.4、δ 147.7、δ 147.8、δ 150.3、δ 150.9での二重結合炭素の22個のシグナルを含む27個の炭素シグナルを示す。
【0023】
DEPTスペクトルは、分子中に1×CH2、12×CH及び14×Cが存在することを示す。
【0024】
13C−NMRスペクトルにより提供される情報とともに、1H−NMRスペクトルにおける芳香族プロトンの化学シフト及び相互結合を踏まえて、本発明の化合物は、2つの1,3,4−三置換ベンゼン環、1つの1,2,3,4−四置換ベンゼン環、1つのトランス型二重結合及び1つの一置換二重結合を有するとみなされる。これらは全て、丹参由来のサルビアノール酸の化合物の分光分析法の特徴と一致する。
【0025】
上記の結果として、本発明の化合物は最初に、構造的に丹参における報告されたサルビアノール酸化合物に対して類似性を示すフェノール酸化合物である可能性が高いと推論することができた。
【化3】
【0026】
従来技術及び関連スペクトル研究と比較して、1H−NMRがサルビアノール酸Aでは、2対のトランス型二重結合プロトンを示したのに対して、本発明の化合物では、1対だけのトランス型二重結合プロトン及び1つの一置換二重結合プロトンを示した以外は、本発明の化合物はサルビアノール酸Aと類似したスペクトル特性を有することが見出されており、また13C−NMRは、本発明の化合物ではサルビアノール酸Aのカルボニル炭素シグナルよりも1つ多いカルボニル炭素シグナルが存在する一方で、C−7’’及びC−8’’は、それぞれ8ppm及び6ppmだけ低磁場へシフトされることを示す。結果として、本発明の化合物と、サルビアノール酸Aとの差は、C−7’’又はC−8’’がカルボキシル基で置換されていることである。
【0027】
C−7’’及びC−8’’の置換をさらに確認するために、本発明の化合物の2D−NMR研究を行って、そのHMBCスペクトルの結果により、H−7’’とC−9’’、H−7’’とC−2’’、H−7’’とC−2及びH−7’’とC−6’’との間にロングレンジカップリングが存在することが示された。したがって、本発明の化合物ではC−8’’がカルボキシル基により置換されていると推測することができた。
【0028】
したがって、従来技術と比較して、本発明の化合物は、新規のサルビアノール酸化合物であり、これを「サルビアノール酸L」と称する。
【化4】
【0029】
実際に、抽出のプロセス中に本発明の化合物で起こる立体配置及び立体配座の変化に起因して、対応する変化がそのスペクトルデータで起こるが、立体配置変化及び立体配座変化により生じる各種異性体は、本発明の保護範囲の範疇にある。
【0030】
通常の専門知識及び従来技術によれば、本発明のサルビアノール酸Lはまた、その薬学的に許容可能な塩又は溶媒和物の形態で使用することができる。本発明によるサルビアノール酸Lの上記薬学的に許容可能な塩は、従来の塩形成方法により生成される、無機塩基又は有機塩基から生成される従来の薬学的に許容可能な塩を包含する。塩の好適な例としては、ナトリウム塩、カリウム塩、リチウム塩、マグネシウム塩、アルミニウム塩、カルシウム塩、亜鉛塩又はN,N’−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、N−メチルグルコサミン、プロカイン及びベルベリンと反応させることによって生成される塩が挙げられる。以下で記載するサルビアノール酸Lには、式(I)で表わされるサルビアノール酸L、並びにその薬学的に許容可能な塩、溶媒和物及び加水分解可能なエステルが含まれている。
【0031】
本発明の上記サルビアノール酸Lは、医薬組成物の形態で適切に投与され、従来、1種類又は複数種類の薬学的に許容可能なキャリア或いは賦形剤とともに使用することができる。さらに、可能であれば、本発明の上記サルビアノール酸Lは、原薬、好ましくは医薬調製物として直接使用される活性成分として投与することができる。他の成分との適合性及び患者に対する安全性の観点から、キャリアは薬学的に許容可能でなくてはならない。
【0032】
したがって、本発明は、他の治療学的成分及び/又は予防学的成分を伴って又は伴わずに、本発明のサルビアノール酸L及び1種類又は複数種類の薬学的に許容可能なキャリアを含むサルビアノール酸Lの医薬調製物を提供する。これらの調製物は、経口的に、非経口的に(皮下(例えば、注射又はリザーバ型錠剤)、皮内、クモ膜下腔内、筋内(例えば、リザーバ型)及び静脈内を含む)、直腸的に、及び局所的に(例えば、舌下)投与することができる。しかしながら、最も望ましい投与形態は、患者の疾患に依存する。
【0033】
上記医薬調製物は単位調製物であり得る。また、上記医薬調製物は、薬学分野で既知の任意の方法により調製することができる。これらの方法は全て、本発明のサルビアノール酸Lを、1種類又は複数種類の補助剤成分を構成するキャリアと組み合わせる工程を包含する。一般的に言えば、本発明の上記調製物は下記の通りに製造される:本発明のサルビアノール酸Lを液体若しくは粉砕した固体キャリア又はその混合物と一様かつ密に組み合わせて、その結果半製品が得られ、必要に応じて、次に上記半製品を所望の調製物へ形成させる。
【0034】
通常、一連の標準的な薬学的技術を使用して、サルビアノール酸L及び医薬キャリアを利用することによって本発明の医薬組成物を得ることができる。この技術には、混合、造粒及び加圧成型が含まれる。当業者には既知であるように、薬学的に許容可能なキャリア又は希釈剤の特徴及び形態は、下記要素に依存する:混合される活性成分の量、投与経路及び他の既知の要素。本明細書中では上記薬学的に許容可能なキャリアは、組成物とともに投与することができる有機キャリア又は無機キャリアの全種、例えば固体調製物に使用される賦形剤、潤滑剤、結合剤、崩壊剤及びコーティング剤、又は着色剤及び甘味剤のような医薬添加物を指す。上記医薬キャリアは、マンニトール又はソルビトールのような糖アルコール、ピロ亜硫酸ナトリウム、亜硫酸水素ナトリウム、チオ硫酸ナトリウム、塩酸システイン、チオグリコール酸、メチオニン、ビタミンC、二ナトリウムEDTA、EDTAカルシウムナトリウム、一価アルカリ金属の炭酸塩、酢酸塩、リン酸塩又はそれらの水溶液、塩酸、酢酸、硫酸、リン酸、アミノ酸、塩化ナトリウム、塩化カリウム、乳酸ナトリウム、キシリトール、マルトース、グルコース、フルクトース、デキストラン、グリシン、デンプン、スクロース、ラクトース、マンニトール、ケイ素誘導体、セルロース及びその誘導体、アルギン酸塩、ゼラチン、ポリビニルピロリドン(PVP)、グリセロール、ツイーン−80、寒天、炭酸カルシウム、炭酸水素カルシウム、界面活性剤、PEG、シクロデキストリン、β−シクロデキストリン、リン脂質材料、カオリン、タルク粉末、ステアリン酸カルシウム、ステアリン酸マグネシウム等からなる群から選択される。
【0035】
上述の医薬組成物は、錠剤(例えば、糖衣錠、フィルムコート錠及び腸溶錠);カプセル(例えば、硬質カプセル及び軟質カプセル);経口溶液;バッカル錠;顆粒;沸騰水中に溶解させた後に摂取する顆粒;丸剤;粉末;ペースト;ペレット;懸濁液;散剤;リカー;注射剤;坐剤;ペースト(例えば、軟膏及び硬膏);クリーム;スプレー剤;ドロップ及びパッチを含む任意の薬学的に許容可能な投薬形態へ配合され得る。好ましくは、調製物は、カプセル、錠剤、経口溶液、顆粒、丸剤、粉末、ペレット及びペーストのような経口投薬形態で、並びに注射可能粉末、注射剤及び輸液等のような注射の形態で存在する。最も好ましくは、調製物は錠剤の形態である。
【0036】
これらの望ましい調製物の中でも、上記経口調製物は、一般的に使用される賦形剤、結合剤、増量剤、希釈剤、錠剤加圧成型剤、潤滑剤、崩壊剤、着色剤、風味剤及び湿潤剤を含有することができ、必要に応じて、錠剤はコーティングすることができる。
【0037】
上記賦形剤の好ましい例としては、ラクトース、D−マンニトール、D−ソルビトール、デンプン(例えば、α−デンプン)、デキストリン、結晶性セルロース、低置換ヒドロキシプロピルセルロース、ナトリウムカルボキシメチルセルロース、アラビアゴム、アミロペクチン、軽質無水ケイ酸、合成ケイ酸アルミニウム又はケイ酸アルミニウムマグネシウム等が挙げられる。
【0038】
上記潤滑剤の好ましい例としては、ステアリン酸マグネシウム、ステアリン酸カルシウム、タルカム粉末及びシリカゲル等が挙げられる。
【0039】
上記結合剤の好ましい例としては、α−デンプン、スクロース、ゼラチン、アラビアゴム、メチルセルロース、カルボキシメチルセルロース、ナトリウムカルボキシメチルセルロース、結晶性セルロース、糖、D−マンニトール、トレハロース、デキストリン、アミロペクチン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ピロリドン等が挙げられる。
【0040】
上記崩壊剤の好ましい例としては、ラクトース、糖、デンプン、カルボキシメチルセルロース、カルシウムカルボキシメチルセルロース、アミノアルキルナトリウム、ナトリウムカルボキシメチルデンプン、軽質無水ケイ酸、低置換ヒドロキシプロピルセルロース等が挙げられる。
【0041】
上記コーティング剤の好ましい例としては、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、エチルセルロース、カルボキシメチルセルロース、ポリビニルアルコール等が挙げられる。
【0042】
上記着色剤の好ましい例としては、水溶性食用タルトラジン色素(食用赤色2号及び3号、食用黄色4号及び5号、食用青色1号及び2号のような食用色素)、水不溶性レーキ顔料(例えば、上述の水溶性食用タルトラジン色素のアンモニウム塩)及び天然色素(例えば、β−カロテン、クロロフィル及びベンガラ)等が挙げられる。
【0043】
上記甘味剤の好ましい例としては、サッカリンナトリウム、グリシルレチン酸、アスパルテーム及びステビオシド等が挙げられる。
【0044】
錠剤を調製する従来方法は、本発明のサルビアノール酸Lを1種類又は複数種類の薬学的に許容可能な賦形剤と組み合わせること、及び続いて加圧成型すること又は成形することを含む。
【0045】
さらに、本発明のサルビアノール酸Lはまた、経口液体調製物、例えば、水溶性又は油溶性の懸濁液、溶液、エマルジョン、シロップ等に配合することもできる。本発明のサルビアノール酸Lはまた、乾燥製品へ調製し、使用前に水又は他の好適なキャリアと再ブレンドすることができる。この種の液体調製物は、沈殿防止剤(例えば、ソルビトールシロップ、メチルセルロース、グルコース/シロップ、ゼラチン、ヒドロキシエチルセルロース、カルボキシメチルセルロース、ステアリン酸アルミニウムゲル又は硬化食用脂);乳化剤(例えば、レシチン、モノオレイン酸ソルビタン又はアラビアゴム);非水性キャリア(食用油を含む)(例えば、扁桃油、分留ヤシ油、バター性エステル、プロピレングリコール又はエタノール);並びに防腐剤(例えば、メチルパラベン、ニパソール及びソルビン酸)を含む従来の添加物を含有する。
【0046】
非経口的に投与される調製物としては、水性及び非水性の滅菌注射剤が挙げられ、任意にこれらの調製物は、抗酸化剤、緩衝剤、静菌剤及び等張剤等を含有する。また、非経口的に投与される調製剤としては、水性及び非水性の滅菌懸濁液が挙げられ、任意にこれらの調製物は、沈殿防止剤及び増粘剤を含有する。上記調製物は、単回用量又は複数回用量容器(例えば、密封アンプル及びバイアル)で保存することができ、これは、凍結乾燥条件下で保管することができ、使用前に滅菌液体キャリア、例えば注射用の水で再溶解させることができる。
【0047】
直腸投与される調製物は、従来の坐剤基剤(例えば、ココアバター、ステアリン酸又は他のグリセリド若しくはエチレングリコール)を含有する坐剤であり得る。
【0048】
口腔で局所的に投与される調製物、例えばバッカル調製物又は舌下用調製物は、トローチ(ここで、活性成分は、スクロース及びアラビアゴムのような風味付けされた基剤中に包埋される)、同様に香錠(ここで、活性成分は、ゼラチン及びグリセロール、又はスクロース及びアラビアゴムのような基剤中に包埋される)が挙げられる。
【0049】
本発明のサルビアノール酸Lは、リザーバ型調製物へ配合され、かかる持続放出調製物は、移植(例えば、皮下移植又は筋内移植)又は筋内注射により投与することができる。したがって、本発明のサルビアノール酸Lは、好適なポリマー、疎水性材料(例えば、許容可能な油中のエマルジョン)若しくはイオン交換樹脂を用いて調製することができ、又は難溶性誘導体、例えば難溶性塩へと調製することができる。
【0050】
通常の専門知識及び従来技術によれば、本発明に関連した医療効果は、或る特定の疾患又は症状の予防並びに治療を包含する。本発明のサルビアノール酸Lの治療上有効な量は、疾患の特性及び患者個人の状態に依存するか、又は医師の助言に従う。概して、成人に関する治療上有効な量は、0.02mg/日〜5000mg/日、好ましくは1mg/日〜1500mg/日の範囲内である。上述するように、この量は、単回投与量又は複数回用量(これは、適切な間隔で、例えば1日2回、1日3回、1日4回又はそれ以上で患者に摂取される)であり得る。本発明の上記調製物は、錠剤及びカプセルに関しては0.1wt%〜99wt%、好ましくは30wt%〜95wt%の活性成分、及び液体調製物に関しては3wt%〜50wt%の活性成分を含む。
【0051】
本発明は下記のように実施する:
a)抽出:丹参生薬又は丹参及び他の生薬の混合物を水で抽出して、アルコールを添加し沈殿させて上清を得て、続いて上清を濃縮して、抽出物を得る工程、
b)分離:工程a)の抽出物を水中に溶解させて、多孔質吸収性樹脂上に適用させて、続いて樹脂を水で溶出させて、溶出液を得て、溶出液を酸性にして、酸性にした溶出液を再び多孔質吸収性樹脂上に適用し、樹脂を酸性水溶液で洗浄して、不純物を除去して、続いて樹脂をエタノールで溶出させて、エタノール溶出液を得て、エタノール溶出液を濃縮して、抽出物を得る工程、
c)精製:乾燥方法を使用することにより工程b)の抽出物をシリカゲルカラム上に適用し、クロロホルム、メタノール及びギ酸の移動相で定組成溶出させて、溶出液を収集して、TLCにより溶出プロセス全体をモニタリングして、特徴的に類似した溶出液を組み合わせて、サルビアノール酸Lを得る工程。
【0052】
工程a)では、上記丹参生薬又は丹参及び他の生薬の混合物を煎じ片にスライスするか、顆粒又は粉末へと粉砕させ、好ましくは煎じ片にスライスすることができる。好ましくは、丹参の根が、丹参生薬として使用される。上記他の生薬は、丹参と適合性である当業者に既知の中国生薬、好ましくはラディックス・ノトギンセング(Radix Notoginseng)、ラディックス・アストラガリ(Radix Astragali)及び/又はラディックス・ポリゴニ・ムルチフロリ(Radix Polygoni Multiflori)を指す。
【0053】
工程a)では、上記水抽出は下記の通り:生薬を、生薬の容積の4倍〜8倍の水で、好ましくは生薬の容積の4倍の水で、1.5時間〜3.5時間、好ましくは2時間煎じて、濾過して、薬物残渣を、薬物残渣の容積の3倍〜6倍の水で1時間〜3時間、好ましくは薬物残渣の容積の3倍の水で1時間煎じて、濾過して、濾液を組み合わせて、濾液を濃縮して、相対密度1.11〜1.28(80℃)、好ましくは1.2(80℃)を有する抽出物を得ることである。より簡単に単離されるようにフェノール酸物質を塩化するために、アルカリ水溶液が上記水抽出工程で使用されることが好ましく、好ましくは上記アルカリは、重炭酸ナトリウム、炭酸ナトリウム、水酸化ナトリウム、重炭酸カリウム、炭酸カリウム及び水酸化カリウム、より好ましくは重炭酸ナトリウム又は水酸化ナトリウムからなる群から選択される少なくとも1つである。上記アルカリ水溶液は、0.30%〜0.68%の濃度での重炭酸ナトリウム水溶液、又は0.0025‰〜0.004‰の濃度での水酸化ナトリウム水溶液、好ましくは0.45%の濃度での重炭酸ナトリウム水溶液である。
【0054】
工程a)では、上記アルコール沈殿は下記の通り:エタノールの含有量が65%〜70%(25℃)、好ましくは70%になるまで95%エタノールを抽出物に添加し、沈殿させて、12時間〜36時間、好ましくは24時間静置して、減圧条件下でエタノールを回収することによって上清を濃縮して、相対密度1.30〜1.38(60℃)、好ましくは1.37(60℃)を有する抽出物を得ることである。
【0055】
脂溶性不純物をより良好に排除するために、アルコール抽出が、水抽出工程の前に実施されることが好ましい。アルコール抽出工程では、生薬の容積の5倍〜8倍の50%〜95%エタノールで2回、各回毎に1時間〜2時間煎じて、濾過して、エタノール抽出溶液を廃棄して、上述の水抽出として薬物残渣を抽出する。
【0056】
工程b)では、上記多孔質樹脂カラムは、無極性又は微極性の樹脂、例えば、AB−8、HPD450、HPD700、D101、D4020又はX5、好ましくはAB−8であり得る。生薬対多孔質吸収性樹脂の重量比は、5:1〜1:1、好ましくは4:1である。樹脂カラムは、総容積の8倍〜15倍、好ましくは総容積の12倍の水で洗浄し、それにより水溶出液が得られる。
【0057】
塩酸を、水溶出液のpH値を2.2〜3.5、好ましくは3.0へ調整するために水溶出液に添加する。
【0058】
上記酸性溶出液を、再度多孔質吸収性樹脂上へ適用して、生薬対多孔質吸収性樹脂の重量比は5:1〜1:1、好ましくは4:1であり、カラムを、溶出液がほぼ無色になるまで、pH値2.2〜3.5、好ましくは3.0を有する塩酸で洗浄する。
【0059】
さらに、3倍〜8倍の50%〜95%エタノール、好ましくは4倍の95%エタノールを使用して、カラムを洗浄し、溶出液を濃縮して、アルコール臭を伴わない抽出物を得る。
【0060】
工程c)では、工程b)で濃縮された抽出物を有機溶媒、好ましくはメタノールで溶解し、クロマトグラフィシリカゲルと混合して、好ましくは添加される200メッシュ〜300メッシュのクロマトグラフィシリカゲルの重量は、抽出物の重量に等しい。十分に混合されたサンプルを、十分に充填されたシリカゲルカラム上に置き、好ましくは充填されるシリカゲルは、200メッシュ〜300メッシュのシリカゲルであり、カラムをクロロホルム:メタノール:ギ酸(容積比は90:10:3〜40:10:0.5である)、好ましくはクロロホルム:メタノール:ギ酸(容積比は85:15:3である)の移動相で溶出させる。上記溶出は、定組成溶出(溶出液の比は不変である)又は勾配溶出(溶出液の比は、時間の経過とともに変化する)であり得る。ここで、上記勾配溶出は、当該技術分野における常識を使用することによって、収集される物質の極性に従って調整することができ、例えば、溶出液の極性が徐々に増大した。溶出プロセスを正確にモニタリングするために、クロロホルム:メタノール:ギ酸(容積比は50:10:2である)の展開溶媒を用いたTLCが好ましい。
【0061】
特徴的に類似した溶出液を組み合わせて、サルビアノール酸Lが得られる。
【0062】
より良好な分離効果を達成するために、分取用液体クロマトグラフィを分離ツールとして使用することができる。例えば、サルビアノール酸Lは、下記分離条件で調製される:Waters製のDelta prep 4000半分取用液体クロマトグラフィ、カラム:Aglient製のZorbax XDB−C18(21.2mm×150mm、5μm)、移動相:アセトニトリル:0.1%ギ酸水溶液(15:85)、流速:20ml/分、検出波長:280nm。
【0063】
薬力学的試験で示されるように、サルビアノール酸Lのフリーラジカルを捕捉する(scavenging)能力は、ビタミンCの能力よりもはるかに高い(表3、図9を参照)。さらに、本発明のサルビアノール酸Lの還元力は、ビタミンCの還元力よりも高い(図10を参照)。本発明のサルビアノール酸Lは、抗酸化及びフリーラジカル捕捉の活性を保有する。結果として、本発明のサルビアノール酸Lは、フリーラジカルを捕捉する活性及び予防的抗酸化機能を有する薬へ調製することができる。
【0064】
さらに、本発明はまた、心臓血管疾患を治療するための薬の調製における上記サルビアノール酸Lの使用に関する。上記心臓血管疾患が、低酸素誘導性血管拡張機能不全、酸素欠乏、グルコース欠乏及び過酸化状態により引き起こされるin vitroでのニューロン障害、並びに急性心筋虚血からなる群から選択される少なくとも1つの疾患である。
【0065】
本発明の薬力学的試験で示されるように、サルビアノール酸Lの凍結乾燥粉末は、ノルエピネフリンの血管収縮曲線の或る特定の右側シフトを引き起こし得るが、有意な差はない。サルビアノール酸Lの凍結乾燥粉末は、3つのAch濃度(10-5mol/L、10-4mol/L、10-3mol/L)で無酸素性血管輪に対して血管拡張効果を有意に増強した(P<0.05)。サルビアノール酸Lが、低酸素により引き起こされる血管拡張機能不全を改善させるのに重要な役割を果たすことが示される(表7〜表8、及び図11〜図12を参照)。
【0066】
本発明のサルビアノール酸Lは、虚血及び低酸素症により引き起こされる血管内皮傷害の寛解、血管内皮過形成の促進、虚血及び低酸素により引き起こされる心筋細胞傷害の改善、アテローム性動脈硬化症に対する抵抗性、血小板凝縮の阻害及び血栓形成に対する抵抗性を含む心臓血管系に対する広範な薬力学的効果を有する。さらに、上記サルビアノール酸Lは、冠状動脈を拡張させる効果、冠血流を増大させる効果及び脳虚血により引き起こされる傷害を防止する効果を有する。
【0067】
本発明の薬力学的試験で示されるように、本発明のサルビアノール酸Lは、酸素欠乏、グルコース欠乏及び過酸化水素により引き起こされるin vitroでの神経系細胞傷害に対して有意な改善効果を有し、細胞生存率を高めることができ、酸素欠乏、グルコース欠乏及び過酸化状態から神経細胞を保護する機能を有する(表12〜表15を参照)。さらに、本発明のサルビアノール酸Lは、急性心筋虚血を治療する効果を有する(表16〜表17を参照)。
【実施例】
【0068】
抗酸化及びフリーラジカル捕捉に対する本発明のサルビアノール酸Lの好適な効果はさらに、下記の具体的な実験データにより示される。
【0069】
別記されない限り、本発明で言及される単位%及び‰は重量比を表す。
【0070】
実施例1 サルビアノール酸Lの調製
タンジン煎じ片を、抽出器中に入れた。生薬の容積の4倍の水(0.45%重炭酸ナトリウムを含有する)を抽出器へ加えて、2時間煎じて、濾過した。薬物残渣は、薬物残渣の容積の3倍の水で1時間煎じ続けて、濾過して、濾液を組み合わせて、濃縮して、相対密度1.2(80℃)を有する抽出物を得た。最終エタノール含有量が70%(25℃)になるまで95%エタノールを抽出物に添加して、沈殿を実施して、12時間以上の間、静置させた。エタノールを減圧条件下で回収して、相対密度1.37(60℃)を有する抽出物を得た。
【0071】
上述の抽出物を水で溶解させた後、AB−8多孔質吸収性樹脂カラム上に適用させて、カラムを総容積の12倍の水で溶出させて、水溶出液を得た。水溶出液のpHは、塩酸でpH3.0に調整した。再び、酸性にした水溶出液をAB−8多孔質吸収性樹脂カラム上へ適用させた。pH値3.0を有する酸性水溶液を使用して、溶出液がほぼ無色になるまで、カラムを洗浄した。さらに、総容積の4倍の容積を有する95%エタノールを使用して、溶出させて、溶出液を得た後、溶出液を濃縮して、アルコール臭を伴わない粘度の高い抽出物を得た。
【0072】
得られた抽出物をメタノールで溶解させて、ここで200メッシュ〜300メッシュのクロマトグラフィシリカゲルを添加して、混合して、添加されるクロマトグラフィシリカゲルの重量は、抽出物の重量に等しい。混合サンプルを十分に充填されたシリカゲルカラム上に置き、カラムをクロロホルム:メタノール:ギ酸(容積比は85:15:3である)の移動相で溶出させる。TLCを使用して、全溶出プロセスをモニタリングして、特徴的に類似した溶出液を組み合わせて、サルビアノール酸Lを得た。
【0073】
高分解能質量分析法(QFT−ESI)を使用することによって、擬分子イオンピークは[M−H]+m/z 537.1034であった。
【0074】
【表1】
【0075】
DEPTスペクトルは、分子中に1×CH2、12×CH及び14×Cが存在することを示した。
【0076】
実施例2 サルビアノール酸Lの調製
タンジン及びサンシチ(Sanqi)の煎じ片を、抽出器中に入れた。生薬の容積の6倍の水(0.45%重炭酸ナトリウムを含有する)を抽出器へ加えて、3時間煎じて、濾過した。薬物残渣は、薬物残渣の容積の5倍の水で2時間煎じ続けて、濾過して、濾液を組み合わせて、濃縮して、相対密度1.25(80℃)を有する抽出物を得た。最終エタノール含有量が68%(25℃)になるまで95%エタノールを抽出物に添加して、沈殿を実施して、12時間以上の間、静置させた。エタノールを減圧条件下で回収して、相対密度1.32(60℃)を有する抽出物を得た。
【0077】
上述の抽出物を水で溶解させた後、AB−8多孔質吸収性樹脂カラム上に適用させて、カラムを総容積の12倍の水で溶出させて、水溶出液を得た。水溶出液のpHは、塩酸でpH2.5に調整した。再び、酸性にした水溶出液をAB−8多孔質吸収性樹脂カラム上へ適用させた。pH値3.0を有する酸性水溶液を使用して、溶出液がほぼ無色になるまで、カラムを洗浄した。さらに、総容積の5倍の95%エタノールを使用して、溶出させて、溶出液を得た後、溶出液を濃縮して、アルコール臭を伴わない粘度の高い抽出物を得た。
【0078】
得られた抽出物をメタノールで溶解させて、ここで200メッシュ〜300メッシュのクロマトグラフィシリカゲルを添加して、混合して、添加されるクロマトグラフィシリカゲルの重量は、抽出物の重量に等しい。混合サンプルを十分に充填されたシリカゲルカラム上に置き、カラムをクロロホルム:メタノール:ギ酸(容積比は85:15:3である)の移動相で溶出させる。TLCを使用して、全溶出プロセスをモニタリングして、特徴的に類似した溶出液を組み合わせて、サルビアノール酸Lを得た。
【0079】
高分解能質量分析法(QFT−ESI)を使用することによって、擬分子イオンピークは[M−H]+m/z 537.1027であった。
【0080】
【表2】
【0081】
DEPTスペクトルは、分子中に1×CH2、12×CH及び14×Cが存在することを示した。
【0082】
実施例3 サルビアノール酸Lの調製
タンジン煎じ片を抽出器に入れた。生薬の容積の6倍の85%エタノールを抽出器に加えて2回、各回毎に2時間煎じて、濾過した。エタノール抽出溶液は廃棄した。
【0083】
薬物残渣を、薬物残渣の容積の4倍の水(0.45%重炭酸ナトリウムを含有する)で2時間煎じて、濾過し、薬物残渣を、薬物残渣の容積の3倍の水で1時間、煎じ続けて、濾過した。濾液を組み合わせて、濃縮して、相対密度1.2(80℃)を有する抽出物を得た。最終エタノール含有量が70%(25℃)になるまで95%エタノールを抽出物に添加して、沈殿を実施して、12時間以上の間、静置させた。エタノールを減圧条件下で回収して、相対密度1.37(60℃)を有する抽出物を得た。
【0084】
上述で得られた抽出物を水で溶解させた後、AB−8多孔質吸収性樹脂カラム上に適用させて、カラムを総容積の12倍の水で溶出させて、水溶出液を得た。水溶出液のpHは、塩酸でpH3.0に調整した。再び、酸性にした水溶出液をAB−8多孔質吸収性樹脂カラム上へ適用させた。pH値3.0を有する酸性水溶液を使用して、溶出液がほぼ無色になるまで、カラムを洗浄した。さらに、総容積の4倍の95%エタノールを使用して、溶出させて、溶出液を得て、溶出液を濃縮して、アルコール臭を伴わない粘度の高い抽出物を得た。
【0085】
得られた抽出物をメタノールで溶解させて、ここで200メッシュ〜300メッシュのクロマトグラフィシリカゲルを添加して、混合して、添加されるクロマトグラフィシリカゲルの重量は、抽出物の重量に等しい。混合サンプルを十分に充填されたシリカゲルカラム上に置き、カラムをクロロホルム:メタノール:ギ酸(容積比は85:15:3である)の移動相で溶出させる。TLCを使用して、全溶出プロセスをモニタリングして、特徴的に類似した溶出液を組み合わせて、サルビアノール酸Lを得た。
【0086】
実施例4 サルビアノール酸Lの錠剤の調製
配合:
サルビアノール酸L 100g
微結晶性セルロース 50g
ラクトース 50g
デンプン 51g
ナトリウムカルボキシメチルデンプン 12g
5% PVP無水エタノール 適量
ステアリン酸マグネシウム 3g
【0087】
上記配合物を1000個の錠剤に調製した。
【0088】
調製プロセス:
1.造粒
サルビアノール酸L及び配合中に列挙した他の補助剤を、それぞれ100メッシュの篩に通して篩過した。配合投与量に従って、サルビアノール酸L、微結晶性セルロース、デンプン及びナトリウムカルボキシメチルデンプンを、同等に漸増する方法を用いることによって十分ブレンドした。適量の5% PVP無水エタノールを使用して、軟質材料を製造して、14メッシュの篩を用いて造粒して、50℃〜60℃で1時間乾燥させた。配合投与量に従ってステアリン酸マグネシウムを添加して、14メッシュの篩で顆粒を篩過した。
【0089】
2.錠剤の加圧成型
得られた顆粒を、特定のダイアモンド形状のパンチで加圧成型して、錠剤を調製した。
【0090】
実施例5 サルビアノール酸Lのカプセルの調製
配合:
サルビアノール酸 100g
デンプン 200g
ナトリウムカルボキシメチルデンプン 12g
5% PVP無水エタノール 適量
ステアリン酸マグネシウム 3g
【0091】
上記配合物を1000個のカプセルに調製した。
【0092】
調製プロセス:
1.造粒
サルビアノール酸L及び配合中に列挙した他の補助剤を、それぞれ100メッシュの篩に通して篩過した。配合投与量に従って、サルビアノール酸L、デンプン及びナトリウムカルボキシメチルデンプンを、同等に漸増する方法に従って十分ブレンドした。適量の5% PVP無水エタノールを使用して、軟質材料を製造して、14メッシュの篩を用いて造粒して、50℃〜60℃で1時間乾燥させた。配合投与量に従ってステアリン酸マグネシウムを添加して、14メッシュの篩で顆粒を篩過した。
【0093】
2.封入
得られた顆粒をカプセルへ充填した。
【0094】
実施例6 サルビアノール酸Lの注射剤の調製
配合:
サルビアノール酸L 100g
マンニトール 100g
注射用の水 2500mlまで
【0095】
上記配合物を1000ユニットに調製した。
【0096】
調製プロセス:
サルビアノール酸Lを取り出して、注射用の水1000mlで溶解させて、一様に攪拌した。マンニトールを、注射用の水500mlで溶解させて、上述のサルビアノール酸L溶液に添加して、一様に攪拌して、そこへ活性炭0.5gを添加して、不変温度で20分間攪拌して濾過した。濾液のpHを4.5〜5.0へ調整して、注射用の水で2500mlまで希釈して、滅菌濾過して、別個に充填して製品を得た。
【0097】
実施例7 サルビアノール酸Lの凍結乾燥粉末の調製
配合:
サルビアノール酸L 100g
マンニトール 100g
注射用の水 2000ml
【0098】
上記配合物を1000ユニットに調製した。
【0099】
調製プロセス:
サルビアノール酸L及びマンニトールを秤量して、攪拌することによって注射用の水1500mlで溶解させて、そこへ20分間攪拌することにより脱色のために活性炭0.5gを添加して、溶液を微小空洞フィルターフィルム(0.45μm)に通して濾過して、炭素を除去して、注射用の水で2000mlまで希釈した。得られた溶液を滅菌濾過して、別個に充填して、凍結乾燥させて、製品を得た。
【0100】
薬力学的実施例
薬力学的実施例1 フサルビアノール酸Lのフリーラジカル捕捉反応
フリーラジカルは、活性の高い物質の一種であると考えられている。フリーラジカルは、細胞の代謝プロセス中に順次生成され得る。それらの直接的又は間接的な酸化効果に起因して、フリーラジカルは、生理学的プロセス及び病理学的プロセスに広く関与することが示されている。過剰量のフリーラジカルの存在下で、フリーラジカルは常に、酸化により身体中の高分子(例えば、核酸、タンパク質、糖類及び脂質等)を攻撃する。酸化によるこれらの物質の変性(架橋及び破壊)により、フリーラジカルは細胞構造及び機能に損傷を引き起こして、身体の組織破壊及び変性変化をもたらす。多くの研究により示されているように、フリーラジカルは、多数の疾患の病理学的プロセスに寄与しており、心臓血管疾患、幾つかのがん、老人性白内障及び黄斑変性症、幾つかの炎症及び多様なタイプのニューロン疾患のような多くの疾患を引き起こす。
【0101】
化学構造分析は、サルビアノール酸化合物がフェノール性ヒドロキシル基の供与体であり、それらの抗酸化活性に対する構造基盤を有することを示す。この研究では、サルビアノール酸Lのフリーラジカル捕捉活性を観察するのに、1,1−ジフェニル−2−ピクリル−ヒドラジル(DPPH)フリーラジカル捕捉反応モデルを使用している。
【0102】
1.試薬及び装置
95%を超える純度を有するサルビアノール酸Lは、Tianjin Tasly Group Academyにより提供され、実施例1の方法に従って調製した。
【0103】
ビタミンC及びDPPHは、SIGMA Inc.から購入した。
【0104】
紫外線分光光度計(UV−1800)は、Beijing Rayleigh Analytical Instrument Co., Ltd.から購入した。
【0105】
2.実験方法
総反応容積は2mlであった。80%メタノール(v/v)中の種々の濃度でのサンプル溶液1mlを、100μMのDPPHメタノール溶液に添加して、一様に混合して、暗所で25℃で20分間溶液を反応させた。反応溶液の吸光度を517nmで測定した。この研究では、ビタミンCを陽性対照とみなした。フリーラジカル捕捉率を下記方程式に従って算出した:
フリーラジカル捕捉率(%)=[1−Aサンプル/A対照)/A対照]×100%
(式中、Aサンプルは、試験サンプルの吸光度を意味し、A対照は、ブランク対照の吸光度を意味する)。
【0106】
3.実験結果
表3及び図9は、種々の濃度でのサルビアノール酸L及びビタミンCのDPPHフリーラジカル捕捉率を示す。サルビアノール酸Lは、ビタミンCのフリーラジカル捕捉率よりもはるかに高いフリーラジカル捕捉率を有していた。
【0107】
【表3】
【0108】
薬力学的実施例2 サルビアノール酸Lの還元力の測定
或る程度までは、予防的抗酸化に関する潜在能力は、薬物の還元力により表わされる。本発明のサルビアノール酸Lの還元力に関して研究が行われた。
【0109】
1.試薬及び装置
95%を超える純度を有するサルビアノール酸Lは、Tianjin Tasly Group Academyにより提供され、実施例1の方法に従って調製した。
【0110】
分析上純粋なフェリシアン化カリウムは、Tianjin No.1 Chemical Reagent Factoryから購入した。
【0111】
分析上純粋なトリクロロ酢酸は、Sinopharm Chemical Reagent Co., Ltd.から購入した。
【0112】
分析上純粋な塩化第二鉄は、Tianjin Fengchuan Chemical Reagent Science and Technology Co., Ltd.から購入した。
【0113】
ビタミンCは、SIGMA Inc.から購入した。
【0114】
紫外線分光光度計(UV−1800)は、Beijing Rayleigh Analytical Instrument Co., Ltd.から購入した。
【0115】
冷却遠心機(Z323K)は、HEMMLE(ドイツ)から購入した。
【0116】
2.実験方法
種々の濃度のサルビアノール酸L及び1.0%フェリシアン化カリウム溶液を含有する200mM リン酸緩衝液(pH6.8)0.5mlを、それぞれ吸い取って、水浴(50℃)で20分間加熱した後に氷浴上で冷却した。トリクロロ酢酸溶液(10%)0.5mlを添加して、1000g/分で10分間遠心分離した。得られた上清1.0mlを採取して、そこへ蒸留水1.0ml及び塩化第二鉄溶液(0.1%)0.2mlを添加して、さらに10分間静置させて、吸光度を700nmで測定した。同時に、ブランク実験を実行した。ビタミンCは、強力な還元物質であり、この研究において陽性対照として役割を果たす。サンプルの還元力は、試験サンプルの吸光度から、ブランク対照の吸光度を差し引くことにより表わされる。したがって、吸光度が高いほど、還元力が強力であることを意味する。
【0117】
3.実験結果
図10に示されるように、両方の物質が濃度依存的な吸光度を有し、サルビアノール酸Lの還元力は、ビタミンCの還元力よりもはるかに強力であった。
【0118】
下記薬力学的実験例3〜5で使用されるNo.1抽出物及びNo.2抽出物の成分の決定並びに調製
実験で使用される材料は全て、Tianjin Tasly Group Academy TCM Instituteにより提供された。No.1抽出物の含有量は、生薬 6.825g/g(No.1抽出物)であり、No.2抽出物は、生薬 4.162g/g(No.2抽出物)であった。
【0119】
調製プロセス
No.1抽出物の調製プロセス:
89.8wt%の丹参(中国名:タンジン)及び9.6wt%のラディックス・ノトギンセング(中国名:サンシチ)の混合物を水(0.45%重炭酸ナトリウムを含有する)で2回(5倍の水で2時間、また4倍の水で1時間)、順次抽出した。95%エタノール(v/v)を使用して、還流を用いて水抽出溶液を濃縮した。エタノール抽出溶液中の最終エタノール含有量が70%になるまで、エタノール沈殿を実施した。さらに一晩静置した後、上清を採取して、濃縮して、No.1抽出物を得た。
【0120】
No.2抽出物の調製プロセス:
89.8wt%の丹参(中国名:タンジン)及び9.6wt%のラディックス・ノトギンセング(中国名:サンシチ)の混合物を水で2回(5倍の水で2時間、また4倍の水で1時間)、順次抽出した。95%エタノール(v/v)を使用して、還流を用いて水抽出溶液を濃縮した。エタノール抽出溶液中の最終エタノール含有量が70%になるまで、エタノール沈殿を実施した。さらに一晩静置した後、上清を採取して、濃縮して、No.2抽出物を得た。
【0121】
サルビアノール酸Lを本発明の実施例1の方法により調製した。
【0122】
検出方法
分析条件は下記の通りであった:Waters製の2695 HPLC、Agilent製のZorbax SB−C18(4.6mm×250mm、5μm)クロマトグラフィカラム、0.02%リン酸水溶液を移動相Aとして使用し、0.02%リン酸を含有する80%(v/v)アセトニトリル溶液を移動相Bとして使用し、勾配溶出は、下記表4に従って実施し、流速は1ml/分であり、検出波長は280nmであり、カラム温度は30℃であり、記録時間は50分であった。
【0123】
【表4】
【0124】
No.1抽出物及びNo.2抽出物中の各成分の含有量を、下記表5及び表6に提示した。
【0125】
【表5】
【0126】
【表6】
【0127】
薬力学的実施例3 単離ラット胸部大動脈に対するサルビアノール酸Lの凍結乾燥粉末の効果
実験材料
1.試験材料及び試薬:サルビアノール酸Lの凍結乾燥粉末は、Tianjin Tasly Group Academy TCM Instituteにより提供された。クエン酸ノルエピネフリン(NA)及びアセチルコリン(ACH)はSigma Inc.から購入して、バッチ番号は、1377511及び44908131であった。クレブス溶液を調製するための原材料には、塩化カリウム、塩化ナトリウム、リン酸二水素カリウム、重炭酸ナトリウム、硫酸マグネシウム、グルコース及び塩化カルシウムが含まれていた。
【0128】
2.主な装置:MedLab(登録商標)単離組織トラフ及びMedlab−U/8C獲得システムは、Nanjing Medease Science and Technology Co., Ltd.により製造された。他の装置としては、張力変換器、デジタル制御超恒温槽SC−15、化学天秤、浄水器及び酸素ボンベが挙げられた。
【0129】
3.実験動物:適正な体重のSDラット(雄又は雌)は、証明書番号SCXK(Jing)2007−0001とともにBeijing Vital River Laboratory Animal Technology Co., Ltd.から提供された。ラットは全て、動物給餌室で室温20℃〜25℃でラット特別食(Beijing Keaoxieli Diet CO., Ltd.により製造)及び水道水が与えられ、12時間照射した。
【0130】
実験方法
1.投与用量の設計
サルビアノール酸Lの凍結乾燥粉末の用量は、他のサルビアノール酸の薬力学的実験に基づいて確認された。この研究では、用量は、0.1mg/mlであった。
クレブス溶液(mol/L):NaCl(120)、NaHCO3(25)、KH2PO4(1.2)、MgSO4(1.2)、KCl(4.5)、CaCl2(1.25)、C6H12O6(グルコース11.1)。
KCl:各時点でKCl(3mol/L)100μlを添加した(最終濃度は60mmol/L)。
NA:10-4mol/L(最終濃度は10-6mol/L)は、総計4つの勾配で希釈した。
ACH:10-3mol/L(最終濃度は10-5mol/L)は、総計4つの勾配で希釈した。
【0131】
2.グループ分け
ラットには、その日に薬物の調製に従って、無作為に群に配置した自由食を与えた。各群には8匹のラットが存在し、各ラットから入手可能な4つの血管輪データが存在することを確認した。この研究では、ラットを3つの群:正常群、低酸素モデル群及びサルビアノール酸L+低酸素モデル群に分けた。
【0132】
3.実験方法
SDラットには、その日に薬物の調製に従って、無作為に群に配置した自由食を与えた。各群には8匹のラットが存在した。ラットを頸椎の脱臼により屠殺して、迅速に開胸して、胸部大動脈を取り出した。0℃で、胸部大動脈を、酸素を吹き込んだクレブス溶液へ入れて、ここで結合組織を除去して、胸部大動脈を、直径約2mmを有する血管輪にして、血管輪を、一定温度37℃で分離浴トラフ中で慎重に付した。酸素をトラフへ吹き込み、そこへ張力変換器及びマルチチャネル生理学的記録計を接続させた。基礎張力は2gであり、血管輪は45min-1時間平衡化させて、クレブス溶液を15分間隔で交換した。平衡化させた後、血管輪を塩化カリウム溶液で20分間前処理して、溶出させた。15分の平衡化後、血管輪を再び塩化カリウム溶液でもう1回前処理して、生理学的に極値の血管収縮を達成した。次に、種々の勾配レベル(10-7mol/L、10-6mol/L、10-5mol/L、10-4mol/L)を踏まえてNAを添加して、血管収縮を観察した。ピーク値に到達すると、値は定常期で安定化した。続いて、種々の勾配レベル(10-5mol/L、10-4mol/L、10-3mol/L、10-2mol/L)を踏まえてACHを添加して、血管拡張を観察した。NA及びACHを添加するプロセス中は、クレブス溶液は交換することができない。
【0133】
低酸素モデル群では、酸素の供給は、塩化カリウム溶液により2回前処理した後に、20分間懸濁した。同時に、サルビアノール酸Lの凍結乾燥粉末又はクレブス溶液(等量で)を添加して、一緒に浸して、その後種々の勾配レベルを踏まえてNA及びACHを添加した。クレブス溶液は、低酸素の始まりから、ACHの最終濃度勾配の添加の終わりまで交換することができない。
【0134】
結果は、t検定を使用することにより統計学的に解析した。
【0135】
実験結果
1.血管収縮に対する効果
結果で示されるように、サルビアノール酸Lの乾燥粉末は、本実験条件下では正常群と比較して血管収縮に対して有意な効果が見られず、血管−張力曲線の明らかな右側シフトが見られた。データは表7に見られた。
【0136】
【表7】
【0137】
血管収縮に対するサルビアノール酸Lの凍結乾燥粉末の効果は図11に見られた。
【0138】
2.血管拡張に対する効果
この結果で示されるように、正常群と比較して、血管拡張は、本実験条件下では低酸素モデル群における4つのACH勾配レベルで明らかに減衰した(P<0.01)のに対して、サルビアノール酸L群及び正常群において有意な差は見られなかった。低酸素モデル群と比較して、血管拡張は、サルビアノール酸L群における3つのACH勾配レベルで明らかに増強した(P<0.05)。このことにより、サルビアノール酸L群が、低酸素誘導性血管拡張の機能不全を有意に改善させることができることが示唆された。データは表8に見られた。
【0139】
【表8】
【0140】
血管拡張に対するサルビアノール酸Lの凍結乾燥粉末の効果は図12に見られた。
【0141】
実験的な結論:
サルビアノール酸Lの凍結乾燥粉末は、或る程度はNAの血管収縮曲線の右側シフトを引き起こすことに対して効果があったが、有意な差は観察されなかった。20分間の低酸素は、モデル群においてACHにより引き起こされる血管輪の勾配拡張において有意な低下を引き起こす可能性があり(P<0.01)、拡張機能障害が発生した。対比して、サルビアノール酸Lの凍結乾燥粉末は、ACHの3つの勾配レベル(10-5mol/L、10-4mol/L、10-3mol/L)で無酸素血管輪の拡張において有意な増強を示した(p<0.05)。サルビアノール酸Lが、低酸素により引き起こされる血管拡張機能障害に対して有意な改善効果を有することが確認された。
【0142】
注意点に関する論述:
1.クレブス溶液を調製する際、混濁を防止するために他の物質を全て添加するまで、塩化カルシウム及びグルコースは添加しなかった。クレブス溶液は綿状沈殿を回避するため、長時間、室温で維持することができない。最終的に、クレブス溶液は要時調製した。
【0143】
2.心臓大動脈は、できる限り早く、氷浴中に取り出して、装置により引き起こされる血管輪への傷害を低減させるべきである。心臓大動脈は、血管活性の低減を防止する目的で、血管アーチに十分に密接して取り出されるべきである。
【0144】
3.酸素は、小泡の形態で排気され、特大の泡が、張力変換器に影響を与える場合があり、データの歪みをもたらすため、できるだけ小さくなければならない。
【0145】
薬力学的実施例4 in vitroでの神経細胞に対するサルビアノール酸Lの凍結乾燥粉末及びその抽出物の保護効果
実験材料
1.主な装置:スーパークリーンベンチは、Antai Cleaning Equipment Inc.により製造され、定温CO2インキュベータは、Heraeus(ドイツ)から購入し、ELISA読取計は、BIO-RAD Inc.(米国)から購入し、平坦振とう台は、Jiangsu Guangming Experimental Apparatus Manufacturerで購入し、倒立生物顕微鏡は、オリンパス株式会社(日本)から購入した。
【0146】
2.主な試薬:DMEM高グルコース培地及びDMEMグルコース非含有培地は、GIBCOにより調製され、トリプシンは、SIGMAから購入し、ウシ胎児血清は、PAAから購入し、MTT及びDMSOは、Sigmaから購入し、LDH試験キットは、Nanjing Jiangcheng Bioengineering Instituteから購入した。
【0147】
3.使い捨て材料:96ウェル細胞培養マイクロプレートは、CORNINGにより調製された。
4.細胞染色:PC12。
【0148】
実験方法:
1.MTT法
a.MTTを、各ウェル中に20μlで96ウェルマイクロプレートに添加して、インキュベータ中で4時間反応させた。
b.上清を廃棄して、続いて各ウェル中でDMSO 150μlを添加して、平坦振とう台上で10分間振とうさせた。
c.各ウェルの吸光度を波長570nmでELISA読取計により測定して、細胞生存率を算出した。
細胞生存率%=(薬物投与群のOD値/陰性対照群のOD値)×100%
【0149】
2.LDH活性の測定
測定実験は、Nanjing Jiangcheng Bioengineering Instituteにより提供されるLDH試験キットの仕様に従って実行した。詳細な工程は表9に提示した。
【0150】
【表9】
【0151】
LDH活性(U/L)=(ODU−ODC)/(ODS−ODB)×CS×N×1000
(式中、ODUは、試験サンプルの吸光度を表し、ODCは、対照サンプルの吸光度を表し、ODBは、ブランクの吸光度を表し、ODSは、標準溶液の吸光度を表し、CSは、2mmol/Lの標準濃度を表し、Nは、測定前のサンプルの希釈倍数を表す)。
【0152】
実験結果:
1.過酸化水素で損傷させたモデルの確立
a.良好な条件での指数増殖期にあるPC12細胞を、PBSで2回洗浄した後に、0.25%トリプシン消化溶液を添加して、37℃で約1分間消化を実施した。この反応は、血清含有培養培地の添加により終了させて、遠心分離して再懸濁させて、続いて細胞を計数して、2×104細胞/ml〜4×104細胞/mlの細胞密度を有する懸濁液を調製した。
b.得られた細胞懸濁液を、各ウェル中に180μlで96ウェルマイクロプレートへ接種して(n=3)、定温CO2インキュベータ中で37℃で24時間インキュベートした。
c.グループ分け及び処理:ブランク対照群(PBS)、溶媒対照群(DMSO)、モデル群(H2O2)及び陽性対照群(エダラボン)の4つの群が存在した。
ブランク対照群:PBSの添加のみ。
溶媒対照群:0.1%DMSOの添加。
モデル群:過酸化水素(H2O2)の濃度はそれぞれ、0.25mM、0.5mM及び1mMあり、反応時間は1時間であった。
陽性対照群:エダラボン(2μg/ml)を陽性薬物として添加して、続いて6時間前処理して、そこへ0.5mM過酸化水素を添加して、1時間損傷させて、新たに調製したDMEM+10%FBS培養培地(ウェル1つ当たり200μl)と交換した。
d.細胞活性をMTT法により測定した。
【0153】
【表10】
【0154】
表10で示されるように、0.5mM H2O2で1時間処理した後に、PC12細胞生存率は40%であり、阻害率は60%であった。過酸化水素で損傷させたモデルは、0.5mM H2O2で1時間処理したPC12細胞であった。
【0155】
2.酸素−グルコース欠乏(OGD)モデルの確立
a.良好な条件での指数増殖期にあるPC12細胞を、PBSで2回洗浄した後に、0.25%トリプシン消化溶液を添加して、37℃で約1分間消化を実施した。この反応は、血清含有培養培地の添加により終了させて、遠心分離して再懸濁させて、続いて細胞を計数して、2×104細胞/ml〜4×104細胞/mlの細胞密度を有する懸濁液を調製した。
b.得られた細胞懸濁液を、各ウェル中に180μlで96ウェルマイクロプレートへ接種して(n=3)、定温CO2インキュベータ中で37℃で24時間インキュベートした。
c.グループ分け及び処理:ブランク対照群(酸素正常状態+0.1%DMSO)、モデル群(OGD+0.1%DMSO、酸素−グルコース欠乏)及び陽性対照群(エダラボン)の3つの群が存在した。
モデル群:培養マイクロプレート中の細胞を、グルコース非含有DMEM培地を用いて培養し、これを低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移した。インキュベーションの期間後に、測定を実行した。
陽性対照群:エダラボン(2μg/ml)を陽性薬物として使用した。薬物を添加して、6時間前処理した後、グルコース非含有培地(ウェル1つ当たり180μl)と交換した。薬物を再び添加して、低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移した。インキュベーションの期間後に、測定を実行した。
d.細胞活性はMTT法により測定した。
【0156】
【表11】
【0157】
表11に示されるように、酸素−グルコース欠乏で損傷させたPC12細胞の生存率は、ちょうど42%であり、阻害率は58%であった。したがって、この研究での酸素−グルコース欠乏モデルは、細胞を培養するように選択されたグルコース非含有DMEMであり、これを低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移した。インキュベーションの期間後に、測定を実行した。
【0158】
3.H2O2で損傷させたPC12細胞の細胞生存率に対する薬物の効果
a.良好な条件での指数増殖期にあるPC12細胞を、PBSで2回洗浄した後に、0.25%トリプシン消化溶液を添加して、37℃で約1分間消化を実施した。この反応は、血清含有培養培地の添加により終了させて、遠心分離して再懸濁させて、続いて細胞を計数して、2×104細胞/ml〜4×104細胞/mlの細胞密度を有する懸濁液を調製した。
b.得られた細胞懸濁液を、各ウェル中に180μlで96ウェルマイクロプレートへ接種して(n=3)、定温CO2インキュベータ中で37℃で24時間インキュベートした。
c.グループ分け及び処理:ブランク対照群(PBS)、溶媒対照群(DMSO又は酢酸エチル)、モデル群(H2O2)、陽性対照群(エダラボン)及び薬物処理群の5つの群が存在した。
モデル群:0.5mM過酸化水素(H2O2)を使用して、1時間処理した。
陽性対照群:陽性薬物として使用されるエダラボン(2μg/ml)を細胞に添加して、6時間前処理して、そこへ0.5mM過酸化水素を添加して、1時間損傷させて、新たに調製したDMEM+10%FBS培地と交換した。
薬物処理群:細胞を培養マイクロプレートへ接種して、まず種々の濃度での種々の試験薬物を添加して、ウェル1つ当たり20μlで6時間前処理して、そこへ0.5mM H2O2を添加して、1時間損傷させて、続いて新たに調製したDMEM+10%FBS培養培地と交換した。
d.LDH活性の測定のために、上清を収集した(ウェル1つ当たり20μl)。
e.細胞マイクロプレートにおける細胞の活性をMTT法により測定した。
【0159】
【表12】
【0160】
薬物濃度はそれぞれ、0.1%、0.01%及び0.001%でDMSOを用いて調製し、これらは、相当する濃度を有する溶媒対照群と比較するものとする。ここで、薬物処理群はモデル群(0.5mM H2O2+EtOAc)と比較し、モデル群(H2O2+EtOAc)は溶媒対照群(EtOAc)と比較した。
【0161】
【表13】
【0162】
ここで、薬物処理群はモデル群(0.5mM H2O2+EtOAc)と比較した一方で、モデル群(H2O2+EtOAc)は溶媒対照群(EtOAc)と比較した。
【0163】
4.酸素−グルコース欠乏のPC12細胞の生存率に対する薬物の効果
a.良好な条件での指数増殖期にあるPC12細胞を、PBSで2回洗浄した後に、0.25%トリプシン消化溶液を添加して、37℃で約1分間消化を実施した。この反応は、血清含有培養培地の添加により終了させて、遠心分離して再懸濁させて、続いて細胞を計数して、2×104細胞/ml〜4×104細胞/mlの細胞密度を有する懸濁液を調製した。
b.得られた細胞懸濁液を、各ウェル中に180μlで96ウェルマイクロプレートへ接種して(n=3)、定温CO2インキュベータ中で37℃で24時間インキュベートした。
c.グループ分け及び処理:ブランク対照群(酸素正常状態+0.1%DMSO)、モデル群(OGD+DMSO、酸素−グルコース欠乏)、陽性対照群(エダラボン)及び薬物処理群の4つの群が存在した。
モデル群:マイクロプレート中の細胞用の培養培地を、グルコース非含有DMEMへ変更し、低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移して、一晩培養した。
陽性対照群:エダラボン(2μg/ml)を陽性薬物として使用した。薬物を添加して、6時間前処理した後、培養培地を、グルコース非含有DMEM培地(ウェル1つ当たり180μl)と交換した。薬物を再び添加して、低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移して、一晩培養した。
薬物処理群:種々の濃度での薬物を添加して、6時間前処理して、培養培地をグルコース非含有DMEM培地(ウェル1つ当たり180μl)へ変更した。薬物を再び添加して、低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移して、一晩培養した。
d.翌日に、得られた上清をウェル1つ当たり20μl収集して、これをLDH活性の測定に使用した。
e.細胞の活性をMTT法により測定した。
【0164】
【表14】
【0165】
ここで、薬物処理群をブランク対照群(OGD+EtOAc)と比較し、モデル群(OGD+EtOAc)をブランク対照群(酸素正常状態+DMSO)と比較した。
【0166】
f.LDH活性
【0167】
【表15】
【0168】
ここで、薬物処理群をブランク対照群(OGD+EtOAc)と比較した一方で、モデル群(OGD+EtOAc)をブランク対照群(酸素正常状態+EtOAc)と比較した。
【0169】
結論:
実験の結果:0.02μg/mlの投与量でサルビアノール酸Lの凍結乾燥粉末を用いて処理した場合、H2O2で損傷させたPC12細胞の細胞生存率は47%(P<0.05)であったのに対して、0.2μg/mlの投与量では、LDH活性は474(P<0.05)であった。0.02μg/ml、0.2μg/ml及び2μg/mlの投与量でのサルビアノール酸LのNo.1抽出物に関しては、LDH活性はそれぞれ、483(P<0.01)、416(P<0.01)及び465(P<0.05)であったのに対して、0.2μg/ml及び2μg/mlの投与量でのサルビアノール酸LのNo.2抽出物に関しては、LDH活性はそれぞれ、407(P<0.01)及び488(P<0.01)であり、モデル群と比較して、ともにLDH活性を低減させる効果を有していた。
【0170】
0.02μg/ml及び0.2μg/mlの投与量でサルビアノール酸Lの凍結乾燥粉末を用いて処理した場合、OGD細胞の細胞生存率はそれぞれ、48%(P<0.01)及び37%(P<0.05)であった。0.2μg/ml及び2μg/mlの投与量でのサルビアノール酸LのNo.1抽出物に関して、生存率はそれぞれ、40%(P<0.01)及び42%(P<0.01)であったのに対して、0.02μg/ml、0.2μg/ml及び2μg/mlの投与量でのサルビアノール酸LのNo.2抽出物については、生存率はそれぞれ、47%(P<0.01)、47%(P<0.01)及び41%(P<0.05)であった。2μg/mlの投与量でサルビアノール酸Lの凍結乾燥粉末を用いて処理した場合、LDH活性は40(P<0.05)であった。さらに、2μg/mlの投与量でのサルビアノール酸LのNo.1抽出物に関して、LDH活性は31(P<0.01)であったのに対して、0.2μg/mlの投与量でのサルビアノール酸LのNo.2抽出物については、LDH活性は31(P<0.05)であった。
【0171】
実験で示されるように、サルビアノール酸Lの凍結乾燥粉末は、OGD又はH2O2により引き起こされるin vitroでの神経障害に対して有意な改善効果を有していただけでなく、細胞の生存率も増加させた。したがって、サルビアノール酸Lは、酸素欠乏、グルコース欠乏及び過酸化の条件において神経細胞を保護する機能を有することが確認された。
【0172】
薬力学的実施例5 ラットにおける実験的急性心筋虚血に対するサルビアノール酸Lの凍結乾燥粉末及び抽出物の保護効果
実験材料:
1.試験材料及び試薬:ピツイトリン(Pit)注射剤は、Nanjing Xinbai Pharmaceutical Co., Ltd.により製造され、バッチ番号は070302であった。生理食塩水は、Tianjin Tian’an Pharmaceutical Co., Ltd.により製造され、バッチ番号は200605241(仕様:500ml/瓶)であった。
【0173】
2.主な装置:MedLab(登録商標)8チャネル生理学的記録計は、Nanjing Medease Science and Technology Co., Ltd.により製造された。
【0174】
3.動物:SDラット(適正な体重の雄又は雌)は、証明書番号SCXK(Jing)2007−0001とともにBeijing Vital River Laboratory Animal Technology Co., Ltd.により提供された。ラットには全て、20℃〜25℃の室温での動物給餌室中でラット特別食(Beijing Keaoxieli Diet Co., Ltd.により製造)及び水道水が与えられ、12時間照射した。
【0175】
実験方法
1.投与用量の設計
No.1抽出物の含有量は、生薬6.825g/gであり、No.2抽出物の含有量は、生薬4.162g/gであった。
【0176】
No.1抽出物及びNo.2抽出物の両方に関して、高用量及び低用量:それぞれ生薬1.086g/kg及び生薬0.543g/kgの2つの群が存在した。生薬の用量変換に従って、高用量No.1抽出物中のサルビアノール酸Lの凍結乾燥粉末の投与用量は4.67mg/kgであり、低用量群では2.33mg/kgであった。サルビアノール酸Lは、No.2抽出物では見出されなかった。
【0177】
サルビアノール酸の凍結乾燥粉末の投与用量は10.0mg/kg及び5.0mg/kgであった。
【0178】
2.グループ分け
2.1 動物のスクリーニング
正式の実験の前に、ラットに尾側静脈を介してピツイトリン(Pit)(1U/kg)を注射した。正常ECG及び注射の5分後のECGを記録して、J点の上昇及びT波異常を観察した。注射前に異常ECGが見られた動物又はPitに対して非感受性である動物は拒絶した。
【0179】
2.2 動物のグループ分け
望ましいラットを7群に分けた:(1)モデル対照群、(2)タンジンのNo.1抽出物低用量群(A群)、(3)タンジンのNo.1抽出物高用量群(B群)、(4)タンジンのNo.2抽出物低用量群(C群)、(5)タンジンのNo.2抽出物高用量群(D群)、(6)サルビアノール酸Lの凍結乾燥粉末低用量群(E群)及び(7)サルビアノール酸Lの凍結乾燥粉末高用量群(F群)。
【0180】
3.実験方法
SDラット(半分が雄で、半分が雌)を、無作為に群に分けた(各群中に動物8匹)。処理群中のラットを、種々のサンプルの水性懸濁液を毎日投与したのに対して、モデル対照群中のラットは、等量の生理食塩水を投与した。動物は全て、7日間連続して投与された。最終投与の40分後に、ラットに麻酔して、リードII正常ECGを記録するためのデバイスに接続した。ピツイトリン(Pit)を、尾側静脈を介して1U/kg(体重)の投与量で一定速度で約10秒以内に注射した。ECG変化を、投与の0秒後、5秒後、10秒後、15秒後、30秒後、45秒後、1分後、2分後、3分後、4分後、5分後、10分後及び15分後に記録した。各群のPitの注射前と注射後との間の差、並びに処理群とモデル対照群との差を比較して、J点及びT波の変化を分析して、データをt検定により解析した。
【0181】
実験結果
1.J点に対する効果
結果で示されるように、モデル対照群と比較して、F群(サルビアノール酸Lの凍結乾燥粉末高用量群)におけるECGのJ点の上昇度は、ピツイトリンにより引き起こされる急性心筋虚血において15秒、30秒及び45秒でより小さく、その差は、本実験条件下では統計学的有意性を有していた(P<0.05)。モデル対照群と比較して、B群(タンジンのNo.1抽出物高用量群)におけるECGのJ点の上昇度は15秒でより小さく、その差は統計学的有意性を有していた(P<0.05)。しかしながら、モデル対照群と比較して、他の群は、各時点で有意な差を示さなかった。データは表16に見られた。
【0182】
【表16】
【0183】
2.T波に対する効果
結果で示されるように、モデル対照群と比較して、15秒及び30秒でのF群(サルビアノール酸Lの凍結乾燥粉末高用量群)のECGのT波の上昇度はより小さく、その差は、本実験条件下では統計学的有意性を有していた(P<0.05)。同様に、モデル対照群と比較して、15秒でのB群(タンジンのNo.1抽出物高用量群)におけるECGのT波の上昇度はより小さく、その差は統計学的有意性を有していた(P<0.05)。しかしながら、モデル対照群と比較して、他の群は、各時点で有意な差を示さなかった。データは表17に見られた。
【0184】
【表17】
【0185】
結論:
モデル対照群と比較して、F群(サルビアノール酸Lの凍結乾燥粉末高用量群)におけるECGのJ点及びT波の上昇度は、15秒及び30秒でより小さく、その差は統計学的有意性を有していた(P<0.05)。
【0186】
モデル対照群と比較して、15秒でのJ点及びT波はともに、B群(タンジンのNo.1抽出物高用量群)において有意に減少する(P<0.05)。
【0187】
モデル対照群と比較して、他の群は、各時点でJ点及びT波において有意な減少を示さなかった。
【0188】
結果で示されるように、この研究の下では、サルビアノール酸Lの凍結乾燥粉末(10mg/kg)及び4.67mg/kgの濃度でのサルビアノール酸Lを含有するNo.1抽出物は、抗急性心筋虚血の効果を有していたが、サルビアノール酸Lを含有しないNo.2抽出物における実験的投与量の下では、抗急性心筋虚血の効果は観察されなかった。
【0189】
注意点に関する論述:
1.J点の定義:QRS波群の終点とSTセグメントとの組合せ点。
【0190】
2.ピツイトリンの冠状血管に対する収縮効果に起因して、ピツイトリンの静脈内注射は、正常ラットにおいて急性心筋虚血を誘導することができ、ECGにおけるJ点及びT波の両方の明らかな上昇をもたらす。薬物処理群においてJ点のシフトが有意に回復し、T波は、試験薬物が投与された後に徐々に正常レベルへ減少しており、これにより、薬物が、冠状血管に対するピツイトリンの収縮効果により誘導される急性心筋虚血に対して拮抗作用を有することが示唆された。I期異常(0秒〜45秒以内でピツイトリンにより誘導される)に対してもII期異常(45秒〜15分以内でピツイトリンにより誘導される)に対しても治療上の効果を有する薬物は通常、抗心筋虚血の効果を有すると考えられた。
【0191】
3.実験中、同じバッチ番号のピツイトリンを使用して、実験結果に対する薬物の効力単位の影響を回避すべきである。ピツイトリンは、2時間を超える間隔で注射して、薬物耐性を回避すべきである。好ましくは、選択した動物を1日おきに使用する。
【技術分野】
【0001】
本発明は、伝統的な漢方薬分野、より具体的には新種のサルビアノール酸化合物に関する。
【背景技術】
【0002】
サルビア・ミルチオルヒザ・ブゲ(Salvia miltiorrhiza Bge.)(ファム・ラビアタエ(Fam. Labiatae))の乾燥根である丹参(Radix Salviae Miltiorrhizae:ラディックス・サルビアエ・ミルチオルヒザエ)(中国生薬、以下「タンジン」と称する)は、苦味があり、少し冷たい感じがし、うっ血を除去することによって疼痛を止める機能を伴って心臓及び肝臓のチャネルに作用し、血流を活性化させて、また心臓を清浄にすることによって不穏状態を軽減する。一連の現在の薬理学的研究がタンジンに対して行われており、タンジンが冠状動脈を拡張させ、微小循環を改善させて、心臓を保護する効果を有し、また血小板凝集を抑制及び除去し、無酸素耐性の身体能力、並びに抗肝炎、抗腫瘍及び抗ウイルス等の活性を高めることが可能であることを示している。
【0003】
2001年に、中国医学科学院 北京協和医科大学の薬物研究所(Institute of Materia Madica, Chinese Academy of Medical Sciences & Peking Union Medical College)のL.N. Li et al.(非特許文献1)は、サルビアノール酸A、B、C、D、E、F、G、H、I、J、リトスペルミン酸、ロスマリン酸及びイソサルビアノール酸C等を含むタンジン又は同属の植物から単離されるフェノール酸誘導体の13個の水溶性生理活性成分が存在することを報告した。さらに、これらの成分の薬理学的作用もまた開示されている。
【0004】
Rena. Kasimu et al.(非特許文献2)は、サルビアノール酸Kの化学構造について報告した。
【0005】
外国の研究者らもまた、タンジンの水溶性生理活性成分に関して研究してきた。1999年には、ジョージ・ワシントン大学(George Washington University)は、抗HIVインテグラーゼ及び他のウイルスに対する13個のサルビアノール酸誘導体の効果に関して米国特許を出願して、最終的に付与されている。これらは全て、タンジンが、多大な潜在性を持ち、かつ開発する価値のある医療用植物資源であることを示唆した。
【0006】
本発明の上記サルビアノール酸Lは、大量のスクリーニングのプロセスにおいてタンジン中で見出された、まさに新規の化合物である。これまで、この化合物に関連する構造及び薬理学的効果はいまだに報告されていない。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Bulletin of Medical Research, 2001, Vol 30(7)
【非特許文献2】Journal of Xinjiang Medical University, 2002, Vol. 25(3)
【発明の概要】
【課題を解決するための手段】
【0008】
本発明の目的は、サルビアノール酸Lの新規化合物を提供することである。
【0009】
本発明のさらなる目的は、サルビアノール酸Lを含む医薬組成物を提供することである。
【0010】
本発明の別の目的は、サルビアノール酸Lを調製する方法を提供することである。
【0011】
本発明の別の目的は、心臓血管疾患を治療するための薬剤の調製におけるサルビアノール酸Lの使用を提供することである。
【図面の簡単な説明】
【0012】
【図1】サルビアノール酸Lの高分解能質量スペクトログラムを示す図である。
【図2】サルビアノール酸Lのエレクトロスプレーイオン化質量スペクトログラムを示す図である。
【図3】CD3ODを使用することによる500MHzでのサルビアノール酸Lの1H−NMRダイアグラムを示す図である。
【図4】CD3ODを使用することによる125MHzでのサルビアノール酸Lの13C−NMRダイアグラムを示す図である。
【図5】CD3ODを使用することによる125MHzでのサルビアノール酸LのDEPTダイアグラムを示す図である。
【図6】CD3ODを使用することによる500MHzでのサルビアノール酸LのgCOSYダイアグラムを示す図である。
【図7】CD3ODを使用することによる500MHzでのサルビアノール酸LのgHMBCダイアグラムを示す図である。
【図8】CD3ODを使用することによる500MHzでのサルビアノール酸LのgHMQCダイアグラムを示す図である。
【図9】試験物質のフリーラジカルを捕捉する能力を示す図である。
【図10】サルビアノール酸とビタミンCとの間での還元力の比較を示す図である。
【図11】血管収縮に対するサルビアノール酸Lの凍結乾燥粉末の効果を示す図である。
【図12】血管拡張に対するサルビアノール酸Lの凍結乾燥粉末の効果を示す図である。
【図13】ピツイトリンで下垂体を処理した後の心電図(ECG)を示す図であり、ここでA)は、モデル対照群から得られる正常なECGであり、B)は、ピツイトリンを投与した15秒後のモデル対照群から得られるものであり、C)は、ピツイトリンを投与した30秒後のモデル対照群から得られるものである。
【0013】
本発明は、下記のように一般式(I)で表わされる新規化合物、その薬学的に許容可能な塩、溶媒和物及び加水分解可能なエステルに関する:
【化1】
【発明を実施するための形態】
【0014】
本発明によれば、フェノール酸の新規化合物の構造は、物理化学的特性、高分解能質量分析法(QFT−ESI)、エレクトロスプレーイオン化質量分析法(ESI−MS)、1H−NMR、13C−NMR、DEPT、gCOSY、gHMBC及びgHMQCにより特定された。
【0015】
本発明の化合物は、淡い帯黄色粉末である。
【0016】
本発明による化合物は、FeCl3による薄層クロマトグラフィ(TLC)発色反応で陽性の結果を示し、本発明による化合物がフェノール系化合物であり得ることを示唆した。
【0017】
m/z 537.1034で擬分子イオンピークを示した高分解能質量分析法(QFT−ESI)により、分子式は、不飽和度Ω 17を有するC27H22O12であることが確認された。
【0018】
ESI−MSでは、m/z 537での本発明による化合物の分子イオンピークは、まず8’’−カルボキシル基を容易に損失することができ(−44)、その結果m/z 493でフラグメントイオンピークを形成し(サルビアノール酸Aの分子イオンピークと同じ構造を有する)、次にサルビアノール酸Aのフラグメンテーション規則性に従ってm/z 313、m/z 295で2つのフラグメントイオンピークを形成する。
【0019】
本発明によれば、サルビアノール酸Aのフラグメンテーション規則性は下記の通りに提示される:
【化2】
【0020】
m/z 493、m/z 313、m/z 295での主なフラグメントイオンピークが、その質量スペクトルにおけるサルビアノール酸Aの主なイオンピークであることは明らかである。したがって、本発明の化合物は、サルビアノール酸Aの骨格構造と同じ骨格構造を有する。
【0021】
プロトン核磁気共鳴(1H−NMR)スペクトルは、δ 5.09(1H,dd,J=8.0,4.5Hz)での酸素に結合されたメテニルプロトンの1個のシグナル、δ 6.88(1H,d,J=8.5Hz)、δ 7.25(1H,d,J=8.5Hz)、δ 7.59(1H,d,J=16.0Hz)、δ 6.22(1H,d,J=16.0Hz)、δ 6.68(1H,s)、δ 6.55(2H,d,J=8.0Hz)、δ 6.58(1H,d,J=2.0Hz)、δ 6.69(1H,d,J=8.0Hz)、δ 6.54(1H,dd,J=8.5,2.0Hz)、δ 7.92(1H,s)での芳香族プロトンの11個のシグナル、δ 3.01(2H,ddd,J=14.0,8.0,4.5Hz)での脂肪族プロトンの2個のシグナルを示す。
【0022】
炭素−13核磁気共鳴(13C−NMR)スペクトルは、δ 39.6での1個の脂肪族炭素シグナル、δ 76.4での酸素に結合されたメテニル炭素の1個のシグナル、δ 170.1、δ 173.0、δ 175.1でのカルボニル炭素の3個のシグナル、及びδ 117.4、δ 117.8、δ 117.8、δ 118.2、δ 119.2、δ 120.2、δ 121.7、δ 123.7、δ 125.7、δ 126.6、δ 128.0、δ 128.8、δ 129.9、δ 130.9、δ 146.2、δ 146.5、δ 146.9、δ 147.4、δ 147.7、δ 147.8、δ 150.3、δ 150.9での二重結合炭素の22個のシグナルを含む27個の炭素シグナルを示す。
【0023】
DEPTスペクトルは、分子中に1×CH2、12×CH及び14×Cが存在することを示す。
【0024】
13C−NMRスペクトルにより提供される情報とともに、1H−NMRスペクトルにおける芳香族プロトンの化学シフト及び相互結合を踏まえて、本発明の化合物は、2つの1,3,4−三置換ベンゼン環、1つの1,2,3,4−四置換ベンゼン環、1つのトランス型二重結合及び1つの一置換二重結合を有するとみなされる。これらは全て、丹参由来のサルビアノール酸の化合物の分光分析法の特徴と一致する。
【0025】
上記の結果として、本発明の化合物は最初に、構造的に丹参における報告されたサルビアノール酸化合物に対して類似性を示すフェノール酸化合物である可能性が高いと推論することができた。
【化3】
【0026】
従来技術及び関連スペクトル研究と比較して、1H−NMRがサルビアノール酸Aでは、2対のトランス型二重結合プロトンを示したのに対して、本発明の化合物では、1対だけのトランス型二重結合プロトン及び1つの一置換二重結合プロトンを示した以外は、本発明の化合物はサルビアノール酸Aと類似したスペクトル特性を有することが見出されており、また13C−NMRは、本発明の化合物ではサルビアノール酸Aのカルボニル炭素シグナルよりも1つ多いカルボニル炭素シグナルが存在する一方で、C−7’’及びC−8’’は、それぞれ8ppm及び6ppmだけ低磁場へシフトされることを示す。結果として、本発明の化合物と、サルビアノール酸Aとの差は、C−7’’又はC−8’’がカルボキシル基で置換されていることである。
【0027】
C−7’’及びC−8’’の置換をさらに確認するために、本発明の化合物の2D−NMR研究を行って、そのHMBCスペクトルの結果により、H−7’’とC−9’’、H−7’’とC−2’’、H−7’’とC−2及びH−7’’とC−6’’との間にロングレンジカップリングが存在することが示された。したがって、本発明の化合物ではC−8’’がカルボキシル基により置換されていると推測することができた。
【0028】
したがって、従来技術と比較して、本発明の化合物は、新規のサルビアノール酸化合物であり、これを「サルビアノール酸L」と称する。
【化4】
【0029】
実際に、抽出のプロセス中に本発明の化合物で起こる立体配置及び立体配座の変化に起因して、対応する変化がそのスペクトルデータで起こるが、立体配置変化及び立体配座変化により生じる各種異性体は、本発明の保護範囲の範疇にある。
【0030】
通常の専門知識及び従来技術によれば、本発明のサルビアノール酸Lはまた、その薬学的に許容可能な塩又は溶媒和物の形態で使用することができる。本発明によるサルビアノール酸Lの上記薬学的に許容可能な塩は、従来の塩形成方法により生成される、無機塩基又は有機塩基から生成される従来の薬学的に許容可能な塩を包含する。塩の好適な例としては、ナトリウム塩、カリウム塩、リチウム塩、マグネシウム塩、アルミニウム塩、カルシウム塩、亜鉛塩又はN,N’−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、N−メチルグルコサミン、プロカイン及びベルベリンと反応させることによって生成される塩が挙げられる。以下で記載するサルビアノール酸Lには、式(I)で表わされるサルビアノール酸L、並びにその薬学的に許容可能な塩、溶媒和物及び加水分解可能なエステルが含まれている。
【0031】
本発明の上記サルビアノール酸Lは、医薬組成物の形態で適切に投与され、従来、1種類又は複数種類の薬学的に許容可能なキャリア或いは賦形剤とともに使用することができる。さらに、可能であれば、本発明の上記サルビアノール酸Lは、原薬、好ましくは医薬調製物として直接使用される活性成分として投与することができる。他の成分との適合性及び患者に対する安全性の観点から、キャリアは薬学的に許容可能でなくてはならない。
【0032】
したがって、本発明は、他の治療学的成分及び/又は予防学的成分を伴って又は伴わずに、本発明のサルビアノール酸L及び1種類又は複数種類の薬学的に許容可能なキャリアを含むサルビアノール酸Lの医薬調製物を提供する。これらの調製物は、経口的に、非経口的に(皮下(例えば、注射又はリザーバ型錠剤)、皮内、クモ膜下腔内、筋内(例えば、リザーバ型)及び静脈内を含む)、直腸的に、及び局所的に(例えば、舌下)投与することができる。しかしながら、最も望ましい投与形態は、患者の疾患に依存する。
【0033】
上記医薬調製物は単位調製物であり得る。また、上記医薬調製物は、薬学分野で既知の任意の方法により調製することができる。これらの方法は全て、本発明のサルビアノール酸Lを、1種類又は複数種類の補助剤成分を構成するキャリアと組み合わせる工程を包含する。一般的に言えば、本発明の上記調製物は下記の通りに製造される:本発明のサルビアノール酸Lを液体若しくは粉砕した固体キャリア又はその混合物と一様かつ密に組み合わせて、その結果半製品が得られ、必要に応じて、次に上記半製品を所望の調製物へ形成させる。
【0034】
通常、一連の標準的な薬学的技術を使用して、サルビアノール酸L及び医薬キャリアを利用することによって本発明の医薬組成物を得ることができる。この技術には、混合、造粒及び加圧成型が含まれる。当業者には既知であるように、薬学的に許容可能なキャリア又は希釈剤の特徴及び形態は、下記要素に依存する:混合される活性成分の量、投与経路及び他の既知の要素。本明細書中では上記薬学的に許容可能なキャリアは、組成物とともに投与することができる有機キャリア又は無機キャリアの全種、例えば固体調製物に使用される賦形剤、潤滑剤、結合剤、崩壊剤及びコーティング剤、又は着色剤及び甘味剤のような医薬添加物を指す。上記医薬キャリアは、マンニトール又はソルビトールのような糖アルコール、ピロ亜硫酸ナトリウム、亜硫酸水素ナトリウム、チオ硫酸ナトリウム、塩酸システイン、チオグリコール酸、メチオニン、ビタミンC、二ナトリウムEDTA、EDTAカルシウムナトリウム、一価アルカリ金属の炭酸塩、酢酸塩、リン酸塩又はそれらの水溶液、塩酸、酢酸、硫酸、リン酸、アミノ酸、塩化ナトリウム、塩化カリウム、乳酸ナトリウム、キシリトール、マルトース、グルコース、フルクトース、デキストラン、グリシン、デンプン、スクロース、ラクトース、マンニトール、ケイ素誘導体、セルロース及びその誘導体、アルギン酸塩、ゼラチン、ポリビニルピロリドン(PVP)、グリセロール、ツイーン−80、寒天、炭酸カルシウム、炭酸水素カルシウム、界面活性剤、PEG、シクロデキストリン、β−シクロデキストリン、リン脂質材料、カオリン、タルク粉末、ステアリン酸カルシウム、ステアリン酸マグネシウム等からなる群から選択される。
【0035】
上述の医薬組成物は、錠剤(例えば、糖衣錠、フィルムコート錠及び腸溶錠);カプセル(例えば、硬質カプセル及び軟質カプセル);経口溶液;バッカル錠;顆粒;沸騰水中に溶解させた後に摂取する顆粒;丸剤;粉末;ペースト;ペレット;懸濁液;散剤;リカー;注射剤;坐剤;ペースト(例えば、軟膏及び硬膏);クリーム;スプレー剤;ドロップ及びパッチを含む任意の薬学的に許容可能な投薬形態へ配合され得る。好ましくは、調製物は、カプセル、錠剤、経口溶液、顆粒、丸剤、粉末、ペレット及びペーストのような経口投薬形態で、並びに注射可能粉末、注射剤及び輸液等のような注射の形態で存在する。最も好ましくは、調製物は錠剤の形態である。
【0036】
これらの望ましい調製物の中でも、上記経口調製物は、一般的に使用される賦形剤、結合剤、増量剤、希釈剤、錠剤加圧成型剤、潤滑剤、崩壊剤、着色剤、風味剤及び湿潤剤を含有することができ、必要に応じて、錠剤はコーティングすることができる。
【0037】
上記賦形剤の好ましい例としては、ラクトース、D−マンニトール、D−ソルビトール、デンプン(例えば、α−デンプン)、デキストリン、結晶性セルロース、低置換ヒドロキシプロピルセルロース、ナトリウムカルボキシメチルセルロース、アラビアゴム、アミロペクチン、軽質無水ケイ酸、合成ケイ酸アルミニウム又はケイ酸アルミニウムマグネシウム等が挙げられる。
【0038】
上記潤滑剤の好ましい例としては、ステアリン酸マグネシウム、ステアリン酸カルシウム、タルカム粉末及びシリカゲル等が挙げられる。
【0039】
上記結合剤の好ましい例としては、α−デンプン、スクロース、ゼラチン、アラビアゴム、メチルセルロース、カルボキシメチルセルロース、ナトリウムカルボキシメチルセルロース、結晶性セルロース、糖、D−マンニトール、トレハロース、デキストリン、アミロペクチン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ピロリドン等が挙げられる。
【0040】
上記崩壊剤の好ましい例としては、ラクトース、糖、デンプン、カルボキシメチルセルロース、カルシウムカルボキシメチルセルロース、アミノアルキルナトリウム、ナトリウムカルボキシメチルデンプン、軽質無水ケイ酸、低置換ヒドロキシプロピルセルロース等が挙げられる。
【0041】
上記コーティング剤の好ましい例としては、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、エチルセルロース、カルボキシメチルセルロース、ポリビニルアルコール等が挙げられる。
【0042】
上記着色剤の好ましい例としては、水溶性食用タルトラジン色素(食用赤色2号及び3号、食用黄色4号及び5号、食用青色1号及び2号のような食用色素)、水不溶性レーキ顔料(例えば、上述の水溶性食用タルトラジン色素のアンモニウム塩)及び天然色素(例えば、β−カロテン、クロロフィル及びベンガラ)等が挙げられる。
【0043】
上記甘味剤の好ましい例としては、サッカリンナトリウム、グリシルレチン酸、アスパルテーム及びステビオシド等が挙げられる。
【0044】
錠剤を調製する従来方法は、本発明のサルビアノール酸Lを1種類又は複数種類の薬学的に許容可能な賦形剤と組み合わせること、及び続いて加圧成型すること又は成形することを含む。
【0045】
さらに、本発明のサルビアノール酸Lはまた、経口液体調製物、例えば、水溶性又は油溶性の懸濁液、溶液、エマルジョン、シロップ等に配合することもできる。本発明のサルビアノール酸Lはまた、乾燥製品へ調製し、使用前に水又は他の好適なキャリアと再ブレンドすることができる。この種の液体調製物は、沈殿防止剤(例えば、ソルビトールシロップ、メチルセルロース、グルコース/シロップ、ゼラチン、ヒドロキシエチルセルロース、カルボキシメチルセルロース、ステアリン酸アルミニウムゲル又は硬化食用脂);乳化剤(例えば、レシチン、モノオレイン酸ソルビタン又はアラビアゴム);非水性キャリア(食用油を含む)(例えば、扁桃油、分留ヤシ油、バター性エステル、プロピレングリコール又はエタノール);並びに防腐剤(例えば、メチルパラベン、ニパソール及びソルビン酸)を含む従来の添加物を含有する。
【0046】
非経口的に投与される調製物としては、水性及び非水性の滅菌注射剤が挙げられ、任意にこれらの調製物は、抗酸化剤、緩衝剤、静菌剤及び等張剤等を含有する。また、非経口的に投与される調製剤としては、水性及び非水性の滅菌懸濁液が挙げられ、任意にこれらの調製物は、沈殿防止剤及び増粘剤を含有する。上記調製物は、単回用量又は複数回用量容器(例えば、密封アンプル及びバイアル)で保存することができ、これは、凍結乾燥条件下で保管することができ、使用前に滅菌液体キャリア、例えば注射用の水で再溶解させることができる。
【0047】
直腸投与される調製物は、従来の坐剤基剤(例えば、ココアバター、ステアリン酸又は他のグリセリド若しくはエチレングリコール)を含有する坐剤であり得る。
【0048】
口腔で局所的に投与される調製物、例えばバッカル調製物又は舌下用調製物は、トローチ(ここで、活性成分は、スクロース及びアラビアゴムのような風味付けされた基剤中に包埋される)、同様に香錠(ここで、活性成分は、ゼラチン及びグリセロール、又はスクロース及びアラビアゴムのような基剤中に包埋される)が挙げられる。
【0049】
本発明のサルビアノール酸Lは、リザーバ型調製物へ配合され、かかる持続放出調製物は、移植(例えば、皮下移植又は筋内移植)又は筋内注射により投与することができる。したがって、本発明のサルビアノール酸Lは、好適なポリマー、疎水性材料(例えば、許容可能な油中のエマルジョン)若しくはイオン交換樹脂を用いて調製することができ、又は難溶性誘導体、例えば難溶性塩へと調製することができる。
【0050】
通常の専門知識及び従来技術によれば、本発明に関連した医療効果は、或る特定の疾患又は症状の予防並びに治療を包含する。本発明のサルビアノール酸Lの治療上有効な量は、疾患の特性及び患者個人の状態に依存するか、又は医師の助言に従う。概して、成人に関する治療上有効な量は、0.02mg/日〜5000mg/日、好ましくは1mg/日〜1500mg/日の範囲内である。上述するように、この量は、単回投与量又は複数回用量(これは、適切な間隔で、例えば1日2回、1日3回、1日4回又はそれ以上で患者に摂取される)であり得る。本発明の上記調製物は、錠剤及びカプセルに関しては0.1wt%〜99wt%、好ましくは30wt%〜95wt%の活性成分、及び液体調製物に関しては3wt%〜50wt%の活性成分を含む。
【0051】
本発明は下記のように実施する:
a)抽出:丹参生薬又は丹参及び他の生薬の混合物を水で抽出して、アルコールを添加し沈殿させて上清を得て、続いて上清を濃縮して、抽出物を得る工程、
b)分離:工程a)の抽出物を水中に溶解させて、多孔質吸収性樹脂上に適用させて、続いて樹脂を水で溶出させて、溶出液を得て、溶出液を酸性にして、酸性にした溶出液を再び多孔質吸収性樹脂上に適用し、樹脂を酸性水溶液で洗浄して、不純物を除去して、続いて樹脂をエタノールで溶出させて、エタノール溶出液を得て、エタノール溶出液を濃縮して、抽出物を得る工程、
c)精製:乾燥方法を使用することにより工程b)の抽出物をシリカゲルカラム上に適用し、クロロホルム、メタノール及びギ酸の移動相で定組成溶出させて、溶出液を収集して、TLCにより溶出プロセス全体をモニタリングして、特徴的に類似した溶出液を組み合わせて、サルビアノール酸Lを得る工程。
【0052】
工程a)では、上記丹参生薬又は丹参及び他の生薬の混合物を煎じ片にスライスするか、顆粒又は粉末へと粉砕させ、好ましくは煎じ片にスライスすることができる。好ましくは、丹参の根が、丹参生薬として使用される。上記他の生薬は、丹参と適合性である当業者に既知の中国生薬、好ましくはラディックス・ノトギンセング(Radix Notoginseng)、ラディックス・アストラガリ(Radix Astragali)及び/又はラディックス・ポリゴニ・ムルチフロリ(Radix Polygoni Multiflori)を指す。
【0053】
工程a)では、上記水抽出は下記の通り:生薬を、生薬の容積の4倍〜8倍の水で、好ましくは生薬の容積の4倍の水で、1.5時間〜3.5時間、好ましくは2時間煎じて、濾過して、薬物残渣を、薬物残渣の容積の3倍〜6倍の水で1時間〜3時間、好ましくは薬物残渣の容積の3倍の水で1時間煎じて、濾過して、濾液を組み合わせて、濾液を濃縮して、相対密度1.11〜1.28(80℃)、好ましくは1.2(80℃)を有する抽出物を得ることである。より簡単に単離されるようにフェノール酸物質を塩化するために、アルカリ水溶液が上記水抽出工程で使用されることが好ましく、好ましくは上記アルカリは、重炭酸ナトリウム、炭酸ナトリウム、水酸化ナトリウム、重炭酸カリウム、炭酸カリウム及び水酸化カリウム、より好ましくは重炭酸ナトリウム又は水酸化ナトリウムからなる群から選択される少なくとも1つである。上記アルカリ水溶液は、0.30%〜0.68%の濃度での重炭酸ナトリウム水溶液、又は0.0025‰〜0.004‰の濃度での水酸化ナトリウム水溶液、好ましくは0.45%の濃度での重炭酸ナトリウム水溶液である。
【0054】
工程a)では、上記アルコール沈殿は下記の通り:エタノールの含有量が65%〜70%(25℃)、好ましくは70%になるまで95%エタノールを抽出物に添加し、沈殿させて、12時間〜36時間、好ましくは24時間静置して、減圧条件下でエタノールを回収することによって上清を濃縮して、相対密度1.30〜1.38(60℃)、好ましくは1.37(60℃)を有する抽出物を得ることである。
【0055】
脂溶性不純物をより良好に排除するために、アルコール抽出が、水抽出工程の前に実施されることが好ましい。アルコール抽出工程では、生薬の容積の5倍〜8倍の50%〜95%エタノールで2回、各回毎に1時間〜2時間煎じて、濾過して、エタノール抽出溶液を廃棄して、上述の水抽出として薬物残渣を抽出する。
【0056】
工程b)では、上記多孔質樹脂カラムは、無極性又は微極性の樹脂、例えば、AB−8、HPD450、HPD700、D101、D4020又はX5、好ましくはAB−8であり得る。生薬対多孔質吸収性樹脂の重量比は、5:1〜1:1、好ましくは4:1である。樹脂カラムは、総容積の8倍〜15倍、好ましくは総容積の12倍の水で洗浄し、それにより水溶出液が得られる。
【0057】
塩酸を、水溶出液のpH値を2.2〜3.5、好ましくは3.0へ調整するために水溶出液に添加する。
【0058】
上記酸性溶出液を、再度多孔質吸収性樹脂上へ適用して、生薬対多孔質吸収性樹脂の重量比は5:1〜1:1、好ましくは4:1であり、カラムを、溶出液がほぼ無色になるまで、pH値2.2〜3.5、好ましくは3.0を有する塩酸で洗浄する。
【0059】
さらに、3倍〜8倍の50%〜95%エタノール、好ましくは4倍の95%エタノールを使用して、カラムを洗浄し、溶出液を濃縮して、アルコール臭を伴わない抽出物を得る。
【0060】
工程c)では、工程b)で濃縮された抽出物を有機溶媒、好ましくはメタノールで溶解し、クロマトグラフィシリカゲルと混合して、好ましくは添加される200メッシュ〜300メッシュのクロマトグラフィシリカゲルの重量は、抽出物の重量に等しい。十分に混合されたサンプルを、十分に充填されたシリカゲルカラム上に置き、好ましくは充填されるシリカゲルは、200メッシュ〜300メッシュのシリカゲルであり、カラムをクロロホルム:メタノール:ギ酸(容積比は90:10:3〜40:10:0.5である)、好ましくはクロロホルム:メタノール:ギ酸(容積比は85:15:3である)の移動相で溶出させる。上記溶出は、定組成溶出(溶出液の比は不変である)又は勾配溶出(溶出液の比は、時間の経過とともに変化する)であり得る。ここで、上記勾配溶出は、当該技術分野における常識を使用することによって、収集される物質の極性に従って調整することができ、例えば、溶出液の極性が徐々に増大した。溶出プロセスを正確にモニタリングするために、クロロホルム:メタノール:ギ酸(容積比は50:10:2である)の展開溶媒を用いたTLCが好ましい。
【0061】
特徴的に類似した溶出液を組み合わせて、サルビアノール酸Lが得られる。
【0062】
より良好な分離効果を達成するために、分取用液体クロマトグラフィを分離ツールとして使用することができる。例えば、サルビアノール酸Lは、下記分離条件で調製される:Waters製のDelta prep 4000半分取用液体クロマトグラフィ、カラム:Aglient製のZorbax XDB−C18(21.2mm×150mm、5μm)、移動相:アセトニトリル:0.1%ギ酸水溶液(15:85)、流速:20ml/分、検出波長:280nm。
【0063】
薬力学的試験で示されるように、サルビアノール酸Lのフリーラジカルを捕捉する(scavenging)能力は、ビタミンCの能力よりもはるかに高い(表3、図9を参照)。さらに、本発明のサルビアノール酸Lの還元力は、ビタミンCの還元力よりも高い(図10を参照)。本発明のサルビアノール酸Lは、抗酸化及びフリーラジカル捕捉の活性を保有する。結果として、本発明のサルビアノール酸Lは、フリーラジカルを捕捉する活性及び予防的抗酸化機能を有する薬へ調製することができる。
【0064】
さらに、本発明はまた、心臓血管疾患を治療するための薬の調製における上記サルビアノール酸Lの使用に関する。上記心臓血管疾患が、低酸素誘導性血管拡張機能不全、酸素欠乏、グルコース欠乏及び過酸化状態により引き起こされるin vitroでのニューロン障害、並びに急性心筋虚血からなる群から選択される少なくとも1つの疾患である。
【0065】
本発明の薬力学的試験で示されるように、サルビアノール酸Lの凍結乾燥粉末は、ノルエピネフリンの血管収縮曲線の或る特定の右側シフトを引き起こし得るが、有意な差はない。サルビアノール酸Lの凍結乾燥粉末は、3つのAch濃度(10-5mol/L、10-4mol/L、10-3mol/L)で無酸素性血管輪に対して血管拡張効果を有意に増強した(P<0.05)。サルビアノール酸Lが、低酸素により引き起こされる血管拡張機能不全を改善させるのに重要な役割を果たすことが示される(表7〜表8、及び図11〜図12を参照)。
【0066】
本発明のサルビアノール酸Lは、虚血及び低酸素症により引き起こされる血管内皮傷害の寛解、血管内皮過形成の促進、虚血及び低酸素により引き起こされる心筋細胞傷害の改善、アテローム性動脈硬化症に対する抵抗性、血小板凝縮の阻害及び血栓形成に対する抵抗性を含む心臓血管系に対する広範な薬力学的効果を有する。さらに、上記サルビアノール酸Lは、冠状動脈を拡張させる効果、冠血流を増大させる効果及び脳虚血により引き起こされる傷害を防止する効果を有する。
【0067】
本発明の薬力学的試験で示されるように、本発明のサルビアノール酸Lは、酸素欠乏、グルコース欠乏及び過酸化水素により引き起こされるin vitroでの神経系細胞傷害に対して有意な改善効果を有し、細胞生存率を高めることができ、酸素欠乏、グルコース欠乏及び過酸化状態から神経細胞を保護する機能を有する(表12〜表15を参照)。さらに、本発明のサルビアノール酸Lは、急性心筋虚血を治療する効果を有する(表16〜表17を参照)。
【実施例】
【0068】
抗酸化及びフリーラジカル捕捉に対する本発明のサルビアノール酸Lの好適な効果はさらに、下記の具体的な実験データにより示される。
【0069】
別記されない限り、本発明で言及される単位%及び‰は重量比を表す。
【0070】
実施例1 サルビアノール酸Lの調製
タンジン煎じ片を、抽出器中に入れた。生薬の容積の4倍の水(0.45%重炭酸ナトリウムを含有する)を抽出器へ加えて、2時間煎じて、濾過した。薬物残渣は、薬物残渣の容積の3倍の水で1時間煎じ続けて、濾過して、濾液を組み合わせて、濃縮して、相対密度1.2(80℃)を有する抽出物を得た。最終エタノール含有量が70%(25℃)になるまで95%エタノールを抽出物に添加して、沈殿を実施して、12時間以上の間、静置させた。エタノールを減圧条件下で回収して、相対密度1.37(60℃)を有する抽出物を得た。
【0071】
上述の抽出物を水で溶解させた後、AB−8多孔質吸収性樹脂カラム上に適用させて、カラムを総容積の12倍の水で溶出させて、水溶出液を得た。水溶出液のpHは、塩酸でpH3.0に調整した。再び、酸性にした水溶出液をAB−8多孔質吸収性樹脂カラム上へ適用させた。pH値3.0を有する酸性水溶液を使用して、溶出液がほぼ無色になるまで、カラムを洗浄した。さらに、総容積の4倍の容積を有する95%エタノールを使用して、溶出させて、溶出液を得た後、溶出液を濃縮して、アルコール臭を伴わない粘度の高い抽出物を得た。
【0072】
得られた抽出物をメタノールで溶解させて、ここで200メッシュ〜300メッシュのクロマトグラフィシリカゲルを添加して、混合して、添加されるクロマトグラフィシリカゲルの重量は、抽出物の重量に等しい。混合サンプルを十分に充填されたシリカゲルカラム上に置き、カラムをクロロホルム:メタノール:ギ酸(容積比は85:15:3である)の移動相で溶出させる。TLCを使用して、全溶出プロセスをモニタリングして、特徴的に類似した溶出液を組み合わせて、サルビアノール酸Lを得た。
【0073】
高分解能質量分析法(QFT−ESI)を使用することによって、擬分子イオンピークは[M−H]+m/z 537.1034であった。
【0074】
【表1】
【0075】
DEPTスペクトルは、分子中に1×CH2、12×CH及び14×Cが存在することを示した。
【0076】
実施例2 サルビアノール酸Lの調製
タンジン及びサンシチ(Sanqi)の煎じ片を、抽出器中に入れた。生薬の容積の6倍の水(0.45%重炭酸ナトリウムを含有する)を抽出器へ加えて、3時間煎じて、濾過した。薬物残渣は、薬物残渣の容積の5倍の水で2時間煎じ続けて、濾過して、濾液を組み合わせて、濃縮して、相対密度1.25(80℃)を有する抽出物を得た。最終エタノール含有量が68%(25℃)になるまで95%エタノールを抽出物に添加して、沈殿を実施して、12時間以上の間、静置させた。エタノールを減圧条件下で回収して、相対密度1.32(60℃)を有する抽出物を得た。
【0077】
上述の抽出物を水で溶解させた後、AB−8多孔質吸収性樹脂カラム上に適用させて、カラムを総容積の12倍の水で溶出させて、水溶出液を得た。水溶出液のpHは、塩酸でpH2.5に調整した。再び、酸性にした水溶出液をAB−8多孔質吸収性樹脂カラム上へ適用させた。pH値3.0を有する酸性水溶液を使用して、溶出液がほぼ無色になるまで、カラムを洗浄した。さらに、総容積の5倍の95%エタノールを使用して、溶出させて、溶出液を得た後、溶出液を濃縮して、アルコール臭を伴わない粘度の高い抽出物を得た。
【0078】
得られた抽出物をメタノールで溶解させて、ここで200メッシュ〜300メッシュのクロマトグラフィシリカゲルを添加して、混合して、添加されるクロマトグラフィシリカゲルの重量は、抽出物の重量に等しい。混合サンプルを十分に充填されたシリカゲルカラム上に置き、カラムをクロロホルム:メタノール:ギ酸(容積比は85:15:3である)の移動相で溶出させる。TLCを使用して、全溶出プロセスをモニタリングして、特徴的に類似した溶出液を組み合わせて、サルビアノール酸Lを得た。
【0079】
高分解能質量分析法(QFT−ESI)を使用することによって、擬分子イオンピークは[M−H]+m/z 537.1027であった。
【0080】
【表2】
【0081】
DEPTスペクトルは、分子中に1×CH2、12×CH及び14×Cが存在することを示した。
【0082】
実施例3 サルビアノール酸Lの調製
タンジン煎じ片を抽出器に入れた。生薬の容積の6倍の85%エタノールを抽出器に加えて2回、各回毎に2時間煎じて、濾過した。エタノール抽出溶液は廃棄した。
【0083】
薬物残渣を、薬物残渣の容積の4倍の水(0.45%重炭酸ナトリウムを含有する)で2時間煎じて、濾過し、薬物残渣を、薬物残渣の容積の3倍の水で1時間、煎じ続けて、濾過した。濾液を組み合わせて、濃縮して、相対密度1.2(80℃)を有する抽出物を得た。最終エタノール含有量が70%(25℃)になるまで95%エタノールを抽出物に添加して、沈殿を実施して、12時間以上の間、静置させた。エタノールを減圧条件下で回収して、相対密度1.37(60℃)を有する抽出物を得た。
【0084】
上述で得られた抽出物を水で溶解させた後、AB−8多孔質吸収性樹脂カラム上に適用させて、カラムを総容積の12倍の水で溶出させて、水溶出液を得た。水溶出液のpHは、塩酸でpH3.0に調整した。再び、酸性にした水溶出液をAB−8多孔質吸収性樹脂カラム上へ適用させた。pH値3.0を有する酸性水溶液を使用して、溶出液がほぼ無色になるまで、カラムを洗浄した。さらに、総容積の4倍の95%エタノールを使用して、溶出させて、溶出液を得て、溶出液を濃縮して、アルコール臭を伴わない粘度の高い抽出物を得た。
【0085】
得られた抽出物をメタノールで溶解させて、ここで200メッシュ〜300メッシュのクロマトグラフィシリカゲルを添加して、混合して、添加されるクロマトグラフィシリカゲルの重量は、抽出物の重量に等しい。混合サンプルを十分に充填されたシリカゲルカラム上に置き、カラムをクロロホルム:メタノール:ギ酸(容積比は85:15:3である)の移動相で溶出させる。TLCを使用して、全溶出プロセスをモニタリングして、特徴的に類似した溶出液を組み合わせて、サルビアノール酸Lを得た。
【0086】
実施例4 サルビアノール酸Lの錠剤の調製
配合:
サルビアノール酸L 100g
微結晶性セルロース 50g
ラクトース 50g
デンプン 51g
ナトリウムカルボキシメチルデンプン 12g
5% PVP無水エタノール 適量
ステアリン酸マグネシウム 3g
【0087】
上記配合物を1000個の錠剤に調製した。
【0088】
調製プロセス:
1.造粒
サルビアノール酸L及び配合中に列挙した他の補助剤を、それぞれ100メッシュの篩に通して篩過した。配合投与量に従って、サルビアノール酸L、微結晶性セルロース、デンプン及びナトリウムカルボキシメチルデンプンを、同等に漸増する方法を用いることによって十分ブレンドした。適量の5% PVP無水エタノールを使用して、軟質材料を製造して、14メッシュの篩を用いて造粒して、50℃〜60℃で1時間乾燥させた。配合投与量に従ってステアリン酸マグネシウムを添加して、14メッシュの篩で顆粒を篩過した。
【0089】
2.錠剤の加圧成型
得られた顆粒を、特定のダイアモンド形状のパンチで加圧成型して、錠剤を調製した。
【0090】
実施例5 サルビアノール酸Lのカプセルの調製
配合:
サルビアノール酸 100g
デンプン 200g
ナトリウムカルボキシメチルデンプン 12g
5% PVP無水エタノール 適量
ステアリン酸マグネシウム 3g
【0091】
上記配合物を1000個のカプセルに調製した。
【0092】
調製プロセス:
1.造粒
サルビアノール酸L及び配合中に列挙した他の補助剤を、それぞれ100メッシュの篩に通して篩過した。配合投与量に従って、サルビアノール酸L、デンプン及びナトリウムカルボキシメチルデンプンを、同等に漸増する方法に従って十分ブレンドした。適量の5% PVP無水エタノールを使用して、軟質材料を製造して、14メッシュの篩を用いて造粒して、50℃〜60℃で1時間乾燥させた。配合投与量に従ってステアリン酸マグネシウムを添加して、14メッシュの篩で顆粒を篩過した。
【0093】
2.封入
得られた顆粒をカプセルへ充填した。
【0094】
実施例6 サルビアノール酸Lの注射剤の調製
配合:
サルビアノール酸L 100g
マンニトール 100g
注射用の水 2500mlまで
【0095】
上記配合物を1000ユニットに調製した。
【0096】
調製プロセス:
サルビアノール酸Lを取り出して、注射用の水1000mlで溶解させて、一様に攪拌した。マンニトールを、注射用の水500mlで溶解させて、上述のサルビアノール酸L溶液に添加して、一様に攪拌して、そこへ活性炭0.5gを添加して、不変温度で20分間攪拌して濾過した。濾液のpHを4.5〜5.0へ調整して、注射用の水で2500mlまで希釈して、滅菌濾過して、別個に充填して製品を得た。
【0097】
実施例7 サルビアノール酸Lの凍結乾燥粉末の調製
配合:
サルビアノール酸L 100g
マンニトール 100g
注射用の水 2000ml
【0098】
上記配合物を1000ユニットに調製した。
【0099】
調製プロセス:
サルビアノール酸L及びマンニトールを秤量して、攪拌することによって注射用の水1500mlで溶解させて、そこへ20分間攪拌することにより脱色のために活性炭0.5gを添加して、溶液を微小空洞フィルターフィルム(0.45μm)に通して濾過して、炭素を除去して、注射用の水で2000mlまで希釈した。得られた溶液を滅菌濾過して、別個に充填して、凍結乾燥させて、製品を得た。
【0100】
薬力学的実施例
薬力学的実施例1 フサルビアノール酸Lのフリーラジカル捕捉反応
フリーラジカルは、活性の高い物質の一種であると考えられている。フリーラジカルは、細胞の代謝プロセス中に順次生成され得る。それらの直接的又は間接的な酸化効果に起因して、フリーラジカルは、生理学的プロセス及び病理学的プロセスに広く関与することが示されている。過剰量のフリーラジカルの存在下で、フリーラジカルは常に、酸化により身体中の高分子(例えば、核酸、タンパク質、糖類及び脂質等)を攻撃する。酸化によるこれらの物質の変性(架橋及び破壊)により、フリーラジカルは細胞構造及び機能に損傷を引き起こして、身体の組織破壊及び変性変化をもたらす。多くの研究により示されているように、フリーラジカルは、多数の疾患の病理学的プロセスに寄与しており、心臓血管疾患、幾つかのがん、老人性白内障及び黄斑変性症、幾つかの炎症及び多様なタイプのニューロン疾患のような多くの疾患を引き起こす。
【0101】
化学構造分析は、サルビアノール酸化合物がフェノール性ヒドロキシル基の供与体であり、それらの抗酸化活性に対する構造基盤を有することを示す。この研究では、サルビアノール酸Lのフリーラジカル捕捉活性を観察するのに、1,1−ジフェニル−2−ピクリル−ヒドラジル(DPPH)フリーラジカル捕捉反応モデルを使用している。
【0102】
1.試薬及び装置
95%を超える純度を有するサルビアノール酸Lは、Tianjin Tasly Group Academyにより提供され、実施例1の方法に従って調製した。
【0103】
ビタミンC及びDPPHは、SIGMA Inc.から購入した。
【0104】
紫外線分光光度計(UV−1800)は、Beijing Rayleigh Analytical Instrument Co., Ltd.から購入した。
【0105】
2.実験方法
総反応容積は2mlであった。80%メタノール(v/v)中の種々の濃度でのサンプル溶液1mlを、100μMのDPPHメタノール溶液に添加して、一様に混合して、暗所で25℃で20分間溶液を反応させた。反応溶液の吸光度を517nmで測定した。この研究では、ビタミンCを陽性対照とみなした。フリーラジカル捕捉率を下記方程式に従って算出した:
フリーラジカル捕捉率(%)=[1−Aサンプル/A対照)/A対照]×100%
(式中、Aサンプルは、試験サンプルの吸光度を意味し、A対照は、ブランク対照の吸光度を意味する)。
【0106】
3.実験結果
表3及び図9は、種々の濃度でのサルビアノール酸L及びビタミンCのDPPHフリーラジカル捕捉率を示す。サルビアノール酸Lは、ビタミンCのフリーラジカル捕捉率よりもはるかに高いフリーラジカル捕捉率を有していた。
【0107】
【表3】
【0108】
薬力学的実施例2 サルビアノール酸Lの還元力の測定
或る程度までは、予防的抗酸化に関する潜在能力は、薬物の還元力により表わされる。本発明のサルビアノール酸Lの還元力に関して研究が行われた。
【0109】
1.試薬及び装置
95%を超える純度を有するサルビアノール酸Lは、Tianjin Tasly Group Academyにより提供され、実施例1の方法に従って調製した。
【0110】
分析上純粋なフェリシアン化カリウムは、Tianjin No.1 Chemical Reagent Factoryから購入した。
【0111】
分析上純粋なトリクロロ酢酸は、Sinopharm Chemical Reagent Co., Ltd.から購入した。
【0112】
分析上純粋な塩化第二鉄は、Tianjin Fengchuan Chemical Reagent Science and Technology Co., Ltd.から購入した。
【0113】
ビタミンCは、SIGMA Inc.から購入した。
【0114】
紫外線分光光度計(UV−1800)は、Beijing Rayleigh Analytical Instrument Co., Ltd.から購入した。
【0115】
冷却遠心機(Z323K)は、HEMMLE(ドイツ)から購入した。
【0116】
2.実験方法
種々の濃度のサルビアノール酸L及び1.0%フェリシアン化カリウム溶液を含有する200mM リン酸緩衝液(pH6.8)0.5mlを、それぞれ吸い取って、水浴(50℃)で20分間加熱した後に氷浴上で冷却した。トリクロロ酢酸溶液(10%)0.5mlを添加して、1000g/分で10分間遠心分離した。得られた上清1.0mlを採取して、そこへ蒸留水1.0ml及び塩化第二鉄溶液(0.1%)0.2mlを添加して、さらに10分間静置させて、吸光度を700nmで測定した。同時に、ブランク実験を実行した。ビタミンCは、強力な還元物質であり、この研究において陽性対照として役割を果たす。サンプルの還元力は、試験サンプルの吸光度から、ブランク対照の吸光度を差し引くことにより表わされる。したがって、吸光度が高いほど、還元力が強力であることを意味する。
【0117】
3.実験結果
図10に示されるように、両方の物質が濃度依存的な吸光度を有し、サルビアノール酸Lの還元力は、ビタミンCの還元力よりもはるかに強力であった。
【0118】
下記薬力学的実験例3〜5で使用されるNo.1抽出物及びNo.2抽出物の成分の決定並びに調製
実験で使用される材料は全て、Tianjin Tasly Group Academy TCM Instituteにより提供された。No.1抽出物の含有量は、生薬 6.825g/g(No.1抽出物)であり、No.2抽出物は、生薬 4.162g/g(No.2抽出物)であった。
【0119】
調製プロセス
No.1抽出物の調製プロセス:
89.8wt%の丹参(中国名:タンジン)及び9.6wt%のラディックス・ノトギンセング(中国名:サンシチ)の混合物を水(0.45%重炭酸ナトリウムを含有する)で2回(5倍の水で2時間、また4倍の水で1時間)、順次抽出した。95%エタノール(v/v)を使用して、還流を用いて水抽出溶液を濃縮した。エタノール抽出溶液中の最終エタノール含有量が70%になるまで、エタノール沈殿を実施した。さらに一晩静置した後、上清を採取して、濃縮して、No.1抽出物を得た。
【0120】
No.2抽出物の調製プロセス:
89.8wt%の丹参(中国名:タンジン)及び9.6wt%のラディックス・ノトギンセング(中国名:サンシチ)の混合物を水で2回(5倍の水で2時間、また4倍の水で1時間)、順次抽出した。95%エタノール(v/v)を使用して、還流を用いて水抽出溶液を濃縮した。エタノール抽出溶液中の最終エタノール含有量が70%になるまで、エタノール沈殿を実施した。さらに一晩静置した後、上清を採取して、濃縮して、No.2抽出物を得た。
【0121】
サルビアノール酸Lを本発明の実施例1の方法により調製した。
【0122】
検出方法
分析条件は下記の通りであった:Waters製の2695 HPLC、Agilent製のZorbax SB−C18(4.6mm×250mm、5μm)クロマトグラフィカラム、0.02%リン酸水溶液を移動相Aとして使用し、0.02%リン酸を含有する80%(v/v)アセトニトリル溶液を移動相Bとして使用し、勾配溶出は、下記表4に従って実施し、流速は1ml/分であり、検出波長は280nmであり、カラム温度は30℃であり、記録時間は50分であった。
【0123】
【表4】
【0124】
No.1抽出物及びNo.2抽出物中の各成分の含有量を、下記表5及び表6に提示した。
【0125】
【表5】
【0126】
【表6】
【0127】
薬力学的実施例3 単離ラット胸部大動脈に対するサルビアノール酸Lの凍結乾燥粉末の効果
実験材料
1.試験材料及び試薬:サルビアノール酸Lの凍結乾燥粉末は、Tianjin Tasly Group Academy TCM Instituteにより提供された。クエン酸ノルエピネフリン(NA)及びアセチルコリン(ACH)はSigma Inc.から購入して、バッチ番号は、1377511及び44908131であった。クレブス溶液を調製するための原材料には、塩化カリウム、塩化ナトリウム、リン酸二水素カリウム、重炭酸ナトリウム、硫酸マグネシウム、グルコース及び塩化カルシウムが含まれていた。
【0128】
2.主な装置:MedLab(登録商標)単離組織トラフ及びMedlab−U/8C獲得システムは、Nanjing Medease Science and Technology Co., Ltd.により製造された。他の装置としては、張力変換器、デジタル制御超恒温槽SC−15、化学天秤、浄水器及び酸素ボンベが挙げられた。
【0129】
3.実験動物:適正な体重のSDラット(雄又は雌)は、証明書番号SCXK(Jing)2007−0001とともにBeijing Vital River Laboratory Animal Technology Co., Ltd.から提供された。ラットは全て、動物給餌室で室温20℃〜25℃でラット特別食(Beijing Keaoxieli Diet CO., Ltd.により製造)及び水道水が与えられ、12時間照射した。
【0130】
実験方法
1.投与用量の設計
サルビアノール酸Lの凍結乾燥粉末の用量は、他のサルビアノール酸の薬力学的実験に基づいて確認された。この研究では、用量は、0.1mg/mlであった。
クレブス溶液(mol/L):NaCl(120)、NaHCO3(25)、KH2PO4(1.2)、MgSO4(1.2)、KCl(4.5)、CaCl2(1.25)、C6H12O6(グルコース11.1)。
KCl:各時点でKCl(3mol/L)100μlを添加した(最終濃度は60mmol/L)。
NA:10-4mol/L(最終濃度は10-6mol/L)は、総計4つの勾配で希釈した。
ACH:10-3mol/L(最終濃度は10-5mol/L)は、総計4つの勾配で希釈した。
【0131】
2.グループ分け
ラットには、その日に薬物の調製に従って、無作為に群に配置した自由食を与えた。各群には8匹のラットが存在し、各ラットから入手可能な4つの血管輪データが存在することを確認した。この研究では、ラットを3つの群:正常群、低酸素モデル群及びサルビアノール酸L+低酸素モデル群に分けた。
【0132】
3.実験方法
SDラットには、その日に薬物の調製に従って、無作為に群に配置した自由食を与えた。各群には8匹のラットが存在した。ラットを頸椎の脱臼により屠殺して、迅速に開胸して、胸部大動脈を取り出した。0℃で、胸部大動脈を、酸素を吹き込んだクレブス溶液へ入れて、ここで結合組織を除去して、胸部大動脈を、直径約2mmを有する血管輪にして、血管輪を、一定温度37℃で分離浴トラフ中で慎重に付した。酸素をトラフへ吹き込み、そこへ張力変換器及びマルチチャネル生理学的記録計を接続させた。基礎張力は2gであり、血管輪は45min-1時間平衡化させて、クレブス溶液を15分間隔で交換した。平衡化させた後、血管輪を塩化カリウム溶液で20分間前処理して、溶出させた。15分の平衡化後、血管輪を再び塩化カリウム溶液でもう1回前処理して、生理学的に極値の血管収縮を達成した。次に、種々の勾配レベル(10-7mol/L、10-6mol/L、10-5mol/L、10-4mol/L)を踏まえてNAを添加して、血管収縮を観察した。ピーク値に到達すると、値は定常期で安定化した。続いて、種々の勾配レベル(10-5mol/L、10-4mol/L、10-3mol/L、10-2mol/L)を踏まえてACHを添加して、血管拡張を観察した。NA及びACHを添加するプロセス中は、クレブス溶液は交換することができない。
【0133】
低酸素モデル群では、酸素の供給は、塩化カリウム溶液により2回前処理した後に、20分間懸濁した。同時に、サルビアノール酸Lの凍結乾燥粉末又はクレブス溶液(等量で)を添加して、一緒に浸して、その後種々の勾配レベルを踏まえてNA及びACHを添加した。クレブス溶液は、低酸素の始まりから、ACHの最終濃度勾配の添加の終わりまで交換することができない。
【0134】
結果は、t検定を使用することにより統計学的に解析した。
【0135】
実験結果
1.血管収縮に対する効果
結果で示されるように、サルビアノール酸Lの乾燥粉末は、本実験条件下では正常群と比較して血管収縮に対して有意な効果が見られず、血管−張力曲線の明らかな右側シフトが見られた。データは表7に見られた。
【0136】
【表7】
【0137】
血管収縮に対するサルビアノール酸Lの凍結乾燥粉末の効果は図11に見られた。
【0138】
2.血管拡張に対する効果
この結果で示されるように、正常群と比較して、血管拡張は、本実験条件下では低酸素モデル群における4つのACH勾配レベルで明らかに減衰した(P<0.01)のに対して、サルビアノール酸L群及び正常群において有意な差は見られなかった。低酸素モデル群と比較して、血管拡張は、サルビアノール酸L群における3つのACH勾配レベルで明らかに増強した(P<0.05)。このことにより、サルビアノール酸L群が、低酸素誘導性血管拡張の機能不全を有意に改善させることができることが示唆された。データは表8に見られた。
【0139】
【表8】
【0140】
血管拡張に対するサルビアノール酸Lの凍結乾燥粉末の効果は図12に見られた。
【0141】
実験的な結論:
サルビアノール酸Lの凍結乾燥粉末は、或る程度はNAの血管収縮曲線の右側シフトを引き起こすことに対して効果があったが、有意な差は観察されなかった。20分間の低酸素は、モデル群においてACHにより引き起こされる血管輪の勾配拡張において有意な低下を引き起こす可能性があり(P<0.01)、拡張機能障害が発生した。対比して、サルビアノール酸Lの凍結乾燥粉末は、ACHの3つの勾配レベル(10-5mol/L、10-4mol/L、10-3mol/L)で無酸素血管輪の拡張において有意な増強を示した(p<0.05)。サルビアノール酸Lが、低酸素により引き起こされる血管拡張機能障害に対して有意な改善効果を有することが確認された。
【0142】
注意点に関する論述:
1.クレブス溶液を調製する際、混濁を防止するために他の物質を全て添加するまで、塩化カルシウム及びグルコースは添加しなかった。クレブス溶液は綿状沈殿を回避するため、長時間、室温で維持することができない。最終的に、クレブス溶液は要時調製した。
【0143】
2.心臓大動脈は、できる限り早く、氷浴中に取り出して、装置により引き起こされる血管輪への傷害を低減させるべきである。心臓大動脈は、血管活性の低減を防止する目的で、血管アーチに十分に密接して取り出されるべきである。
【0144】
3.酸素は、小泡の形態で排気され、特大の泡が、張力変換器に影響を与える場合があり、データの歪みをもたらすため、できるだけ小さくなければならない。
【0145】
薬力学的実施例4 in vitroでの神経細胞に対するサルビアノール酸Lの凍結乾燥粉末及びその抽出物の保護効果
実験材料
1.主な装置:スーパークリーンベンチは、Antai Cleaning Equipment Inc.により製造され、定温CO2インキュベータは、Heraeus(ドイツ)から購入し、ELISA読取計は、BIO-RAD Inc.(米国)から購入し、平坦振とう台は、Jiangsu Guangming Experimental Apparatus Manufacturerで購入し、倒立生物顕微鏡は、オリンパス株式会社(日本)から購入した。
【0146】
2.主な試薬:DMEM高グルコース培地及びDMEMグルコース非含有培地は、GIBCOにより調製され、トリプシンは、SIGMAから購入し、ウシ胎児血清は、PAAから購入し、MTT及びDMSOは、Sigmaから購入し、LDH試験キットは、Nanjing Jiangcheng Bioengineering Instituteから購入した。
【0147】
3.使い捨て材料:96ウェル細胞培養マイクロプレートは、CORNINGにより調製された。
4.細胞染色:PC12。
【0148】
実験方法:
1.MTT法
a.MTTを、各ウェル中に20μlで96ウェルマイクロプレートに添加して、インキュベータ中で4時間反応させた。
b.上清を廃棄して、続いて各ウェル中でDMSO 150μlを添加して、平坦振とう台上で10分間振とうさせた。
c.各ウェルの吸光度を波長570nmでELISA読取計により測定して、細胞生存率を算出した。
細胞生存率%=(薬物投与群のOD値/陰性対照群のOD値)×100%
【0149】
2.LDH活性の測定
測定実験は、Nanjing Jiangcheng Bioengineering Instituteにより提供されるLDH試験キットの仕様に従って実行した。詳細な工程は表9に提示した。
【0150】
【表9】
【0151】
LDH活性(U/L)=(ODU−ODC)/(ODS−ODB)×CS×N×1000
(式中、ODUは、試験サンプルの吸光度を表し、ODCは、対照サンプルの吸光度を表し、ODBは、ブランクの吸光度を表し、ODSは、標準溶液の吸光度を表し、CSは、2mmol/Lの標準濃度を表し、Nは、測定前のサンプルの希釈倍数を表す)。
【0152】
実験結果:
1.過酸化水素で損傷させたモデルの確立
a.良好な条件での指数増殖期にあるPC12細胞を、PBSで2回洗浄した後に、0.25%トリプシン消化溶液を添加して、37℃で約1分間消化を実施した。この反応は、血清含有培養培地の添加により終了させて、遠心分離して再懸濁させて、続いて細胞を計数して、2×104細胞/ml〜4×104細胞/mlの細胞密度を有する懸濁液を調製した。
b.得られた細胞懸濁液を、各ウェル中に180μlで96ウェルマイクロプレートへ接種して(n=3)、定温CO2インキュベータ中で37℃で24時間インキュベートした。
c.グループ分け及び処理:ブランク対照群(PBS)、溶媒対照群(DMSO)、モデル群(H2O2)及び陽性対照群(エダラボン)の4つの群が存在した。
ブランク対照群:PBSの添加のみ。
溶媒対照群:0.1%DMSOの添加。
モデル群:過酸化水素(H2O2)の濃度はそれぞれ、0.25mM、0.5mM及び1mMあり、反応時間は1時間であった。
陽性対照群:エダラボン(2μg/ml)を陽性薬物として添加して、続いて6時間前処理して、そこへ0.5mM過酸化水素を添加して、1時間損傷させて、新たに調製したDMEM+10%FBS培養培地(ウェル1つ当たり200μl)と交換した。
d.細胞活性をMTT法により測定した。
【0153】
【表10】
【0154】
表10で示されるように、0.5mM H2O2で1時間処理した後に、PC12細胞生存率は40%であり、阻害率は60%であった。過酸化水素で損傷させたモデルは、0.5mM H2O2で1時間処理したPC12細胞であった。
【0155】
2.酸素−グルコース欠乏(OGD)モデルの確立
a.良好な条件での指数増殖期にあるPC12細胞を、PBSで2回洗浄した後に、0.25%トリプシン消化溶液を添加して、37℃で約1分間消化を実施した。この反応は、血清含有培養培地の添加により終了させて、遠心分離して再懸濁させて、続いて細胞を計数して、2×104細胞/ml〜4×104細胞/mlの細胞密度を有する懸濁液を調製した。
b.得られた細胞懸濁液を、各ウェル中に180μlで96ウェルマイクロプレートへ接種して(n=3)、定温CO2インキュベータ中で37℃で24時間インキュベートした。
c.グループ分け及び処理:ブランク対照群(酸素正常状態+0.1%DMSO)、モデル群(OGD+0.1%DMSO、酸素−グルコース欠乏)及び陽性対照群(エダラボン)の3つの群が存在した。
モデル群:培養マイクロプレート中の細胞を、グルコース非含有DMEM培地を用いて培養し、これを低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移した。インキュベーションの期間後に、測定を実行した。
陽性対照群:エダラボン(2μg/ml)を陽性薬物として使用した。薬物を添加して、6時間前処理した後、グルコース非含有培地(ウェル1つ当たり180μl)と交換した。薬物を再び添加して、低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移した。インキュベーションの期間後に、測定を実行した。
d.細胞活性はMTT法により測定した。
【0156】
【表11】
【0157】
表11に示されるように、酸素−グルコース欠乏で損傷させたPC12細胞の生存率は、ちょうど42%であり、阻害率は58%であった。したがって、この研究での酸素−グルコース欠乏モデルは、細胞を培養するように選択されたグルコース非含有DMEMであり、これを低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移した。インキュベーションの期間後に、測定を実行した。
【0158】
3.H2O2で損傷させたPC12細胞の細胞生存率に対する薬物の効果
a.良好な条件での指数増殖期にあるPC12細胞を、PBSで2回洗浄した後に、0.25%トリプシン消化溶液を添加して、37℃で約1分間消化を実施した。この反応は、血清含有培養培地の添加により終了させて、遠心分離して再懸濁させて、続いて細胞を計数して、2×104細胞/ml〜4×104細胞/mlの細胞密度を有する懸濁液を調製した。
b.得られた細胞懸濁液を、各ウェル中に180μlで96ウェルマイクロプレートへ接種して(n=3)、定温CO2インキュベータ中で37℃で24時間インキュベートした。
c.グループ分け及び処理:ブランク対照群(PBS)、溶媒対照群(DMSO又は酢酸エチル)、モデル群(H2O2)、陽性対照群(エダラボン)及び薬物処理群の5つの群が存在した。
モデル群:0.5mM過酸化水素(H2O2)を使用して、1時間処理した。
陽性対照群:陽性薬物として使用されるエダラボン(2μg/ml)を細胞に添加して、6時間前処理して、そこへ0.5mM過酸化水素を添加して、1時間損傷させて、新たに調製したDMEM+10%FBS培地と交換した。
薬物処理群:細胞を培養マイクロプレートへ接種して、まず種々の濃度での種々の試験薬物を添加して、ウェル1つ当たり20μlで6時間前処理して、そこへ0.5mM H2O2を添加して、1時間損傷させて、続いて新たに調製したDMEM+10%FBS培養培地と交換した。
d.LDH活性の測定のために、上清を収集した(ウェル1つ当たり20μl)。
e.細胞マイクロプレートにおける細胞の活性をMTT法により測定した。
【0159】
【表12】
【0160】
薬物濃度はそれぞれ、0.1%、0.01%及び0.001%でDMSOを用いて調製し、これらは、相当する濃度を有する溶媒対照群と比較するものとする。ここで、薬物処理群はモデル群(0.5mM H2O2+EtOAc)と比較し、モデル群(H2O2+EtOAc)は溶媒対照群(EtOAc)と比較した。
【0161】
【表13】
【0162】
ここで、薬物処理群はモデル群(0.5mM H2O2+EtOAc)と比較した一方で、モデル群(H2O2+EtOAc)は溶媒対照群(EtOAc)と比較した。
【0163】
4.酸素−グルコース欠乏のPC12細胞の生存率に対する薬物の効果
a.良好な条件での指数増殖期にあるPC12細胞を、PBSで2回洗浄した後に、0.25%トリプシン消化溶液を添加して、37℃で約1分間消化を実施した。この反応は、血清含有培養培地の添加により終了させて、遠心分離して再懸濁させて、続いて細胞を計数して、2×104細胞/ml〜4×104細胞/mlの細胞密度を有する懸濁液を調製した。
b.得られた細胞懸濁液を、各ウェル中に180μlで96ウェルマイクロプレートへ接種して(n=3)、定温CO2インキュベータ中で37℃で24時間インキュベートした。
c.グループ分け及び処理:ブランク対照群(酸素正常状態+0.1%DMSO)、モデル群(OGD+DMSO、酸素−グルコース欠乏)、陽性対照群(エダラボン)及び薬物処理群の4つの群が存在した。
モデル群:マイクロプレート中の細胞用の培養培地を、グルコース非含有DMEMへ変更し、低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移して、一晩培養した。
陽性対照群:エダラボン(2μg/ml)を陽性薬物として使用した。薬物を添加して、6時間前処理した後、培養培地を、グルコース非含有DMEM培地(ウェル1つ当たり180μl)と交換した。薬物を再び添加して、低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移して、一晩培養した。
薬物処理群:種々の濃度での薬物を添加して、6時間前処理して、培養培地をグルコース非含有DMEM培地(ウェル1つ当たり180μl)へ変更した。薬物を再び添加して、低酸素チャンバ中に入れて、O2%が2.6未満になったら0.5時間、時間を数え始めて、続いて日常的なインキュベータへ移して、一晩培養した。
d.翌日に、得られた上清をウェル1つ当たり20μl収集して、これをLDH活性の測定に使用した。
e.細胞の活性をMTT法により測定した。
【0164】
【表14】
【0165】
ここで、薬物処理群をブランク対照群(OGD+EtOAc)と比較し、モデル群(OGD+EtOAc)をブランク対照群(酸素正常状態+DMSO)と比較した。
【0166】
f.LDH活性
【0167】
【表15】
【0168】
ここで、薬物処理群をブランク対照群(OGD+EtOAc)と比較した一方で、モデル群(OGD+EtOAc)をブランク対照群(酸素正常状態+EtOAc)と比較した。
【0169】
結論:
実験の結果:0.02μg/mlの投与量でサルビアノール酸Lの凍結乾燥粉末を用いて処理した場合、H2O2で損傷させたPC12細胞の細胞生存率は47%(P<0.05)であったのに対して、0.2μg/mlの投与量では、LDH活性は474(P<0.05)であった。0.02μg/ml、0.2μg/ml及び2μg/mlの投与量でのサルビアノール酸LのNo.1抽出物に関しては、LDH活性はそれぞれ、483(P<0.01)、416(P<0.01)及び465(P<0.05)であったのに対して、0.2μg/ml及び2μg/mlの投与量でのサルビアノール酸LのNo.2抽出物に関しては、LDH活性はそれぞれ、407(P<0.01)及び488(P<0.01)であり、モデル群と比較して、ともにLDH活性を低減させる効果を有していた。
【0170】
0.02μg/ml及び0.2μg/mlの投与量でサルビアノール酸Lの凍結乾燥粉末を用いて処理した場合、OGD細胞の細胞生存率はそれぞれ、48%(P<0.01)及び37%(P<0.05)であった。0.2μg/ml及び2μg/mlの投与量でのサルビアノール酸LのNo.1抽出物に関して、生存率はそれぞれ、40%(P<0.01)及び42%(P<0.01)であったのに対して、0.02μg/ml、0.2μg/ml及び2μg/mlの投与量でのサルビアノール酸LのNo.2抽出物については、生存率はそれぞれ、47%(P<0.01)、47%(P<0.01)及び41%(P<0.05)であった。2μg/mlの投与量でサルビアノール酸Lの凍結乾燥粉末を用いて処理した場合、LDH活性は40(P<0.05)であった。さらに、2μg/mlの投与量でのサルビアノール酸LのNo.1抽出物に関して、LDH活性は31(P<0.01)であったのに対して、0.2μg/mlの投与量でのサルビアノール酸LのNo.2抽出物については、LDH活性は31(P<0.05)であった。
【0171】
実験で示されるように、サルビアノール酸Lの凍結乾燥粉末は、OGD又はH2O2により引き起こされるin vitroでの神経障害に対して有意な改善効果を有していただけでなく、細胞の生存率も増加させた。したがって、サルビアノール酸Lは、酸素欠乏、グルコース欠乏及び過酸化の条件において神経細胞を保護する機能を有することが確認された。
【0172】
薬力学的実施例5 ラットにおける実験的急性心筋虚血に対するサルビアノール酸Lの凍結乾燥粉末及び抽出物の保護効果
実験材料:
1.試験材料及び試薬:ピツイトリン(Pit)注射剤は、Nanjing Xinbai Pharmaceutical Co., Ltd.により製造され、バッチ番号は070302であった。生理食塩水は、Tianjin Tian’an Pharmaceutical Co., Ltd.により製造され、バッチ番号は200605241(仕様:500ml/瓶)であった。
【0173】
2.主な装置:MedLab(登録商標)8チャネル生理学的記録計は、Nanjing Medease Science and Technology Co., Ltd.により製造された。
【0174】
3.動物:SDラット(適正な体重の雄又は雌)は、証明書番号SCXK(Jing)2007−0001とともにBeijing Vital River Laboratory Animal Technology Co., Ltd.により提供された。ラットには全て、20℃〜25℃の室温での動物給餌室中でラット特別食(Beijing Keaoxieli Diet Co., Ltd.により製造)及び水道水が与えられ、12時間照射した。
【0175】
実験方法
1.投与用量の設計
No.1抽出物の含有量は、生薬6.825g/gであり、No.2抽出物の含有量は、生薬4.162g/gであった。
【0176】
No.1抽出物及びNo.2抽出物の両方に関して、高用量及び低用量:それぞれ生薬1.086g/kg及び生薬0.543g/kgの2つの群が存在した。生薬の用量変換に従って、高用量No.1抽出物中のサルビアノール酸Lの凍結乾燥粉末の投与用量は4.67mg/kgであり、低用量群では2.33mg/kgであった。サルビアノール酸Lは、No.2抽出物では見出されなかった。
【0177】
サルビアノール酸の凍結乾燥粉末の投与用量は10.0mg/kg及び5.0mg/kgであった。
【0178】
2.グループ分け
2.1 動物のスクリーニング
正式の実験の前に、ラットに尾側静脈を介してピツイトリン(Pit)(1U/kg)を注射した。正常ECG及び注射の5分後のECGを記録して、J点の上昇及びT波異常を観察した。注射前に異常ECGが見られた動物又はPitに対して非感受性である動物は拒絶した。
【0179】
2.2 動物のグループ分け
望ましいラットを7群に分けた:(1)モデル対照群、(2)タンジンのNo.1抽出物低用量群(A群)、(3)タンジンのNo.1抽出物高用量群(B群)、(4)タンジンのNo.2抽出物低用量群(C群)、(5)タンジンのNo.2抽出物高用量群(D群)、(6)サルビアノール酸Lの凍結乾燥粉末低用量群(E群)及び(7)サルビアノール酸Lの凍結乾燥粉末高用量群(F群)。
【0180】
3.実験方法
SDラット(半分が雄で、半分が雌)を、無作為に群に分けた(各群中に動物8匹)。処理群中のラットを、種々のサンプルの水性懸濁液を毎日投与したのに対して、モデル対照群中のラットは、等量の生理食塩水を投与した。動物は全て、7日間連続して投与された。最終投与の40分後に、ラットに麻酔して、リードII正常ECGを記録するためのデバイスに接続した。ピツイトリン(Pit)を、尾側静脈を介して1U/kg(体重)の投与量で一定速度で約10秒以内に注射した。ECG変化を、投与の0秒後、5秒後、10秒後、15秒後、30秒後、45秒後、1分後、2分後、3分後、4分後、5分後、10分後及び15分後に記録した。各群のPitの注射前と注射後との間の差、並びに処理群とモデル対照群との差を比較して、J点及びT波の変化を分析して、データをt検定により解析した。
【0181】
実験結果
1.J点に対する効果
結果で示されるように、モデル対照群と比較して、F群(サルビアノール酸Lの凍結乾燥粉末高用量群)におけるECGのJ点の上昇度は、ピツイトリンにより引き起こされる急性心筋虚血において15秒、30秒及び45秒でより小さく、その差は、本実験条件下では統計学的有意性を有していた(P<0.05)。モデル対照群と比較して、B群(タンジンのNo.1抽出物高用量群)におけるECGのJ点の上昇度は15秒でより小さく、その差は統計学的有意性を有していた(P<0.05)。しかしながら、モデル対照群と比較して、他の群は、各時点で有意な差を示さなかった。データは表16に見られた。
【0182】
【表16】
【0183】
2.T波に対する効果
結果で示されるように、モデル対照群と比較して、15秒及び30秒でのF群(サルビアノール酸Lの凍結乾燥粉末高用量群)のECGのT波の上昇度はより小さく、その差は、本実験条件下では統計学的有意性を有していた(P<0.05)。同様に、モデル対照群と比較して、15秒でのB群(タンジンのNo.1抽出物高用量群)におけるECGのT波の上昇度はより小さく、その差は統計学的有意性を有していた(P<0.05)。しかしながら、モデル対照群と比較して、他の群は、各時点で有意な差を示さなかった。データは表17に見られた。
【0184】
【表17】
【0185】
結論:
モデル対照群と比較して、F群(サルビアノール酸Lの凍結乾燥粉末高用量群)におけるECGのJ点及びT波の上昇度は、15秒及び30秒でより小さく、その差は統計学的有意性を有していた(P<0.05)。
【0186】
モデル対照群と比較して、15秒でのJ点及びT波はともに、B群(タンジンのNo.1抽出物高用量群)において有意に減少する(P<0.05)。
【0187】
モデル対照群と比較して、他の群は、各時点でJ点及びT波において有意な減少を示さなかった。
【0188】
結果で示されるように、この研究の下では、サルビアノール酸Lの凍結乾燥粉末(10mg/kg)及び4.67mg/kgの濃度でのサルビアノール酸Lを含有するNo.1抽出物は、抗急性心筋虚血の効果を有していたが、サルビアノール酸Lを含有しないNo.2抽出物における実験的投与量の下では、抗急性心筋虚血の効果は観察されなかった。
【0189】
注意点に関する論述:
1.J点の定義:QRS波群の終点とSTセグメントとの組合せ点。
【0190】
2.ピツイトリンの冠状血管に対する収縮効果に起因して、ピツイトリンの静脈内注射は、正常ラットにおいて急性心筋虚血を誘導することができ、ECGにおけるJ点及びT波の両方の明らかな上昇をもたらす。薬物処理群においてJ点のシフトが有意に回復し、T波は、試験薬物が投与された後に徐々に正常レベルへ減少しており、これにより、薬物が、冠状血管に対するピツイトリンの収縮効果により誘導される急性心筋虚血に対して拮抗作用を有することが示唆された。I期異常(0秒〜45秒以内でピツイトリンにより誘導される)に対してもII期異常(45秒〜15分以内でピツイトリンにより誘導される)に対しても治療上の効果を有する薬物は通常、抗心筋虚血の効果を有すると考えられた。
【0191】
3.実験中、同じバッチ番号のピツイトリンを使用して、実験結果に対する薬物の効力単位の影響を回避すべきである。ピツイトリンは、2時間を超える間隔で注射して、薬物耐性を回避すべきである。好ましくは、選択した動物を1日おきに使用する。
【特許請求の範囲】
【請求項1】
一般式(I)を有するサルビアノール酸Lの新規化合物、その薬学的に許容可能な塩、溶媒和物及び加水分解可能なエステル:
【化1】
【請求項2】
下記工程:
a)抽出:丹参生薬又は丹参及び他の生薬の混合物を水で抽出して、アルコールを添加し沈殿させて上清を得て、続いて該上清を濃縮して、抽出物を得る工程、
b)分離:前記工程a)の前記抽出物を水中に溶解させて、多孔質吸収性樹脂上に適用し、続いて該樹脂を水で溶出させて、溶出液を得て、該溶出液を酸性にして、該酸性にした溶出液を再び該多孔質吸収性樹脂上に適用し、該樹脂を酸性水溶液で洗浄して、不純物を除去して、続いて該樹脂をエタノールで溶出させて、エタノール溶出液を得て、該エタノール溶出液を濃縮して、抽出物を得る工程、
c)精製:前記工程b)の前記抽出物をシリカゲルカラム上に適用し、クロロホルム、メタノール及びギ酸の移動相で定組成溶出させて、溶出液を収集して、TLCにより溶出プロセス全体をモニタリングして、特徴的に類似した溶出液を組み合わせて、前記サルビアノール酸Lを得る工程
を含む、請求項1に記載のサルビアノール酸Lを調製する方法。
【請求項3】
前記a)において、前記丹参生薬又は丹参及び他の生薬の混合物を煎じ片にスライスし、前記水抽出が下記の通り:
前記生薬を、該生薬の容積の4倍〜8倍の水で1.5時間〜3.5時間煎じて、濾過して、薬物残渣を、該薬物残渣の容積の3倍〜6倍容量の水で1時間〜3時間煎じて、濾過して、濾液を組み合わせて、該濾液を濃縮して、相対密度1.11〜1.28(80℃)を有する抽出物を得る、煎じることであり、前記アルコール沈殿が下記の通り:
エタノールの含有量が65%〜70%になるまで95%エタノールを前記抽出物に添加して、沈殿させて、12時間〜36時間静置して、減圧条件下でエタノールを回収することによって前記上清を濃縮して、相対密度1.30〜1.38(60℃)を有する抽出物を得ることであり、
前記工程b)において、工程(a)の最終精製物を多孔質吸収性樹脂カラム上へ適用し、前記生薬物対該多孔質吸収性樹脂の重量比が5:1〜1:1であり、該樹脂カラムを総容積の8倍〜15倍の水で洗浄して、水溶出液を得て、塩酸を該水溶出液に添加して、そのpH値を2.2〜3.5へ調整して、前記酸性溶出液を再び該多孔質吸収性樹脂上に適用し、前記生薬対該多孔質吸収性樹脂の重量比が5:1〜1:1であり、該カラムを、該溶出液がほぼ無色になるまでpH値2.2〜3.5を有する塩酸で洗浄して、総容積の3倍〜8倍の50%〜90%エタノールを用いて、該カラムを洗浄し、該溶出液を濃縮して、アルコール臭を伴わない抽出物を得て、前記多孔質吸収性樹脂が、AB−8、HPD450、HPD700、D101、D4020又はX5からなる群から選択される多孔質吸収性樹脂の1つであり、
前記工程(c)において、前記工程(b)において濃縮により得られる前記抽出物を、有機溶媒で溶解し、クロマトグラフィシリカゲルと混合して、十分に混合されたサンプルを、十分に充填されたシリカゲルカラム上に置き、該カラムを、90:10:3〜40:10:0.5の容積比を有するクロロホルム:メタノール:ギ酸の移動相で溶出させることを特徴とする、請求項2に記載の方法。
【請求項4】
前記工程(a)において、前記水抽出は下記の通り:
前記生薬を、該生薬の容積の4倍の水で2時間煎じて、濾過して、薬物残渣を、該薬物残渣の容積の3倍の水で1時間煎じて、濾過して、該濾液を組み合わせて、該濾液を濃縮して、相対密度1.2を有する抽出物を得ることであり、前記アルコール沈殿が下記の通り:
エタノールの含有量が70%になるまで95%エタノールを前記抽出物に添加して、沈殿させて、さらに24時間静置して、減圧条件下でエタノールを回収することによって前記上清を濃縮して、相対密度1.37を有する抽出物を得ることであり、
前記工程(b)において、工程(a)の前記最終抽出物を多孔質吸収性樹脂カラム上に適用し、前記生薬対該多孔質吸収性樹脂の重量比が4:1であり、該樹脂カラムを総容積の12倍の水で洗浄して、水溶出液を得て、塩酸を該水溶出液に添加して、そのpH値を3.0へ調整して、該酸性溶出液を再び該多孔質吸収性樹脂カラム上に適用し、前記生薬対該多孔質吸収性樹脂の重量比が4:1であり、該カラムを、該溶出液がほぼ無色になるまでpH値3.0を有する塩酸で洗浄して、総容積の4倍の95%エタノールを用いて、該カラムを洗浄して、該溶出液を濃縮して、アルコール臭を伴わない抽出物を得て、前記多孔質吸収性樹脂がAB−8であり、
前記工程(c)において、前記工程(b)において濃縮により得られる前記抽出物を、メタノールで溶解し、200メッシュ〜300メッシュのクロマトグラフィシリカゲルカラムと混合して、十分に混合されたサンプルを、十分に充填された200メッシュ〜300メッシュのシリカゲルカラム上に置き、前記カラムを、50:10:2の容積比を有するクロロホルム:メタノール:ギ酸の移動相で溶出させることを特徴とする、請求項2に記載の方法。
【請求項5】
アルカリ水溶液を、前記工程(a)における前記水抽出に使用し、該アルカリ水溶液は、重炭酸ナトリウム、炭酸ナトリウム、水酸化ナトリウム、重炭酸カリウム、炭酸カリウム及び水酸化カリウムからなる群から選択される少なくとも1つであることを特徴とする、請求項2〜4のいずれか一項に記載の方法。
【請求項6】
前記アルカリ水溶液が、重炭酸ナトリウム水溶液又は水酸化ナトリウム水溶液であることを特徴とする、請求項5に記載の方法。
【請求項7】
前記アルカリ水溶液が、0.30%〜0.68%の濃度の重炭酸ナトリウム水溶液、又は0.0025‰〜0.004‰の濃度の水酸化ナトリウム水溶液であることを特徴とする、請求項6に記載の方法。
【請求項8】
前記アルカリ水溶液が、0.45%の濃度の重炭酸ナトリウム水溶液であることを特徴とする、請求項7に記載の方法。
【請求項9】
前記工程(a)が、前記水抽出の前にアルコール抽出をさらに含むことを特徴とする、請求項2〜8のいずれか一項に記載の方法。
【請求項10】
前記アルコール抽出が、下記の通り:
前記生薬の容積の5倍〜8倍の50%〜95%エタノールで2回、各回毎に1時間〜2時間煎じて、濾過して、前記エタノール抽出溶液を廃棄して、薬物残渣を水で抽出する、煎じることであることを特徴とする、請求項9に記載の方法。
【請求項11】
請求項1に記載のサルビアノール酸L及び薬学的に許容可能なキャリアを含む医薬組成物。
【請求項12】
心臓血管疾患を治療するための薬剤の調製における請求項1に記載のサルビアノール酸Lの使用。
【請求項13】
前記心臓血管疾患が、低酸素誘導性血管拡張機能不全、酸素欠乏、グルコース欠乏及び過酸化状態により引き起こされるin vitroでのニューロン障害、並びに急性心筋虚血からなる群から選択される少なくとも1つの疾患である、請求項12に記載の使用。
【請求項14】
フリーラジカルを捕捉する活性を有する薬剤の調製における請求項1に記載のサルビアノール酸Lの使用。
【請求項15】
予防的抗酸化機能の活性を有する薬剤の調製における請求項1に記載のサルビアノール酸Lの使用。
【請求項1】
一般式(I)を有するサルビアノール酸Lの新規化合物、その薬学的に許容可能な塩、溶媒和物及び加水分解可能なエステル:
【化1】
【請求項2】
下記工程:
a)抽出:丹参生薬又は丹参及び他の生薬の混合物を水で抽出して、アルコールを添加し沈殿させて上清を得て、続いて該上清を濃縮して、抽出物を得る工程、
b)分離:前記工程a)の前記抽出物を水中に溶解させて、多孔質吸収性樹脂上に適用し、続いて該樹脂を水で溶出させて、溶出液を得て、該溶出液を酸性にして、該酸性にした溶出液を再び該多孔質吸収性樹脂上に適用し、該樹脂を酸性水溶液で洗浄して、不純物を除去して、続いて該樹脂をエタノールで溶出させて、エタノール溶出液を得て、該エタノール溶出液を濃縮して、抽出物を得る工程、
c)精製:前記工程b)の前記抽出物をシリカゲルカラム上に適用し、クロロホルム、メタノール及びギ酸の移動相で定組成溶出させて、溶出液を収集して、TLCにより溶出プロセス全体をモニタリングして、特徴的に類似した溶出液を組み合わせて、前記サルビアノール酸Lを得る工程
を含む、請求項1に記載のサルビアノール酸Lを調製する方法。
【請求項3】
前記a)において、前記丹参生薬又は丹参及び他の生薬の混合物を煎じ片にスライスし、前記水抽出が下記の通り:
前記生薬を、該生薬の容積の4倍〜8倍の水で1.5時間〜3.5時間煎じて、濾過して、薬物残渣を、該薬物残渣の容積の3倍〜6倍容量の水で1時間〜3時間煎じて、濾過して、濾液を組み合わせて、該濾液を濃縮して、相対密度1.11〜1.28(80℃)を有する抽出物を得る、煎じることであり、前記アルコール沈殿が下記の通り:
エタノールの含有量が65%〜70%になるまで95%エタノールを前記抽出物に添加して、沈殿させて、12時間〜36時間静置して、減圧条件下でエタノールを回収することによって前記上清を濃縮して、相対密度1.30〜1.38(60℃)を有する抽出物を得ることであり、
前記工程b)において、工程(a)の最終精製物を多孔質吸収性樹脂カラム上へ適用し、前記生薬物対該多孔質吸収性樹脂の重量比が5:1〜1:1であり、該樹脂カラムを総容積の8倍〜15倍の水で洗浄して、水溶出液を得て、塩酸を該水溶出液に添加して、そのpH値を2.2〜3.5へ調整して、前記酸性溶出液を再び該多孔質吸収性樹脂上に適用し、前記生薬対該多孔質吸収性樹脂の重量比が5:1〜1:1であり、該カラムを、該溶出液がほぼ無色になるまでpH値2.2〜3.5を有する塩酸で洗浄して、総容積の3倍〜8倍の50%〜90%エタノールを用いて、該カラムを洗浄し、該溶出液を濃縮して、アルコール臭を伴わない抽出物を得て、前記多孔質吸収性樹脂が、AB−8、HPD450、HPD700、D101、D4020又はX5からなる群から選択される多孔質吸収性樹脂の1つであり、
前記工程(c)において、前記工程(b)において濃縮により得られる前記抽出物を、有機溶媒で溶解し、クロマトグラフィシリカゲルと混合して、十分に混合されたサンプルを、十分に充填されたシリカゲルカラム上に置き、該カラムを、90:10:3〜40:10:0.5の容積比を有するクロロホルム:メタノール:ギ酸の移動相で溶出させることを特徴とする、請求項2に記載の方法。
【請求項4】
前記工程(a)において、前記水抽出は下記の通り:
前記生薬を、該生薬の容積の4倍の水で2時間煎じて、濾過して、薬物残渣を、該薬物残渣の容積の3倍の水で1時間煎じて、濾過して、該濾液を組み合わせて、該濾液を濃縮して、相対密度1.2を有する抽出物を得ることであり、前記アルコール沈殿が下記の通り:
エタノールの含有量が70%になるまで95%エタノールを前記抽出物に添加して、沈殿させて、さらに24時間静置して、減圧条件下でエタノールを回収することによって前記上清を濃縮して、相対密度1.37を有する抽出物を得ることであり、
前記工程(b)において、工程(a)の前記最終抽出物を多孔質吸収性樹脂カラム上に適用し、前記生薬対該多孔質吸収性樹脂の重量比が4:1であり、該樹脂カラムを総容積の12倍の水で洗浄して、水溶出液を得て、塩酸を該水溶出液に添加して、そのpH値を3.0へ調整して、該酸性溶出液を再び該多孔質吸収性樹脂カラム上に適用し、前記生薬対該多孔質吸収性樹脂の重量比が4:1であり、該カラムを、該溶出液がほぼ無色になるまでpH値3.0を有する塩酸で洗浄して、総容積の4倍の95%エタノールを用いて、該カラムを洗浄して、該溶出液を濃縮して、アルコール臭を伴わない抽出物を得て、前記多孔質吸収性樹脂がAB−8であり、
前記工程(c)において、前記工程(b)において濃縮により得られる前記抽出物を、メタノールで溶解し、200メッシュ〜300メッシュのクロマトグラフィシリカゲルカラムと混合して、十分に混合されたサンプルを、十分に充填された200メッシュ〜300メッシュのシリカゲルカラム上に置き、前記カラムを、50:10:2の容積比を有するクロロホルム:メタノール:ギ酸の移動相で溶出させることを特徴とする、請求項2に記載の方法。
【請求項5】
アルカリ水溶液を、前記工程(a)における前記水抽出に使用し、該アルカリ水溶液は、重炭酸ナトリウム、炭酸ナトリウム、水酸化ナトリウム、重炭酸カリウム、炭酸カリウム及び水酸化カリウムからなる群から選択される少なくとも1つであることを特徴とする、請求項2〜4のいずれか一項に記載の方法。
【請求項6】
前記アルカリ水溶液が、重炭酸ナトリウム水溶液又は水酸化ナトリウム水溶液であることを特徴とする、請求項5に記載の方法。
【請求項7】
前記アルカリ水溶液が、0.30%〜0.68%の濃度の重炭酸ナトリウム水溶液、又は0.0025‰〜0.004‰の濃度の水酸化ナトリウム水溶液であることを特徴とする、請求項6に記載の方法。
【請求項8】
前記アルカリ水溶液が、0.45%の濃度の重炭酸ナトリウム水溶液であることを特徴とする、請求項7に記載の方法。
【請求項9】
前記工程(a)が、前記水抽出の前にアルコール抽出をさらに含むことを特徴とする、請求項2〜8のいずれか一項に記載の方法。
【請求項10】
前記アルコール抽出が、下記の通り:
前記生薬の容積の5倍〜8倍の50%〜95%エタノールで2回、各回毎に1時間〜2時間煎じて、濾過して、前記エタノール抽出溶液を廃棄して、薬物残渣を水で抽出する、煎じることであることを特徴とする、請求項9に記載の方法。
【請求項11】
請求項1に記載のサルビアノール酸L及び薬学的に許容可能なキャリアを含む医薬組成物。
【請求項12】
心臓血管疾患を治療するための薬剤の調製における請求項1に記載のサルビアノール酸Lの使用。
【請求項13】
前記心臓血管疾患が、低酸素誘導性血管拡張機能不全、酸素欠乏、グルコース欠乏及び過酸化状態により引き起こされるin vitroでのニューロン障害、並びに急性心筋虚血からなる群から選択される少なくとも1つの疾患である、請求項12に記載の使用。
【請求項14】
フリーラジカルを捕捉する活性を有する薬剤の調製における請求項1に記載のサルビアノール酸Lの使用。
【請求項15】
予防的抗酸化機能の活性を有する薬剤の調製における請求項1に記載のサルビアノール酸Lの使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公表番号】特表2012−522022(P2012−522022A)
【公表日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願番号】特願2012−502437(P2012−502437)
【出願日】平成22年3月29日(2010.3.29)
【国際出願番号】PCT/CN2010/071388
【国際公開番号】WO2010/111935
【国際公開日】平成22年10月7日(2010.10.7)
【出願人】(511237874)タスリー・ファーマシューティカル・グループ・カンパニー・リミテッド (1)
【氏名又は名称原語表記】TASLY PHARMACEUTICAL GROUP CO., LTD.
【住所又は居所原語表記】Tasly Modern TCM Garden, Pu Jihe East Road No.2, Beichen District, Tianjin 300410, China
【Fターム(参考)】
【公表日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願日】平成22年3月29日(2010.3.29)
【国際出願番号】PCT/CN2010/071388
【国際公開番号】WO2010/111935
【国際公開日】平成22年10月7日(2010.10.7)
【出願人】(511237874)タスリー・ファーマシューティカル・グループ・カンパニー・リミテッド (1)
【氏名又は名称原語表記】TASLY PHARMACEUTICAL GROUP CO., LTD.
【住所又は居所原語表記】Tasly Modern TCM Garden, Pu Jihe East Road No.2, Beichen District, Tianjin 300410, China
【Fターム(参考)】
[ Back to top ]