
新規シロール化合物及びその製造方法
【課題】 青色領域の発光を有する、新規なシロール化合物を提供することである。
【解決手段】 式(I):

R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルであり、
R3は、互いに独立して、水素、ハロゲン又は−SiH(R1)2であるか、あるいは2個のR3は一緒になって、>Si(R1)2(ここで、R1は、互いに独立して、上記と同義である)を形成している)で示されるシロール化合物である。
【解決手段】 式(I):
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルであり、
R3は、互いに独立して、水素、ハロゲン又は−SiH(R1)2であるか、あるいは2個のR3は一緒になって、>Si(R1)2(ここで、R1は、互いに独立して、上記と同義である)を形成している)で示されるシロール化合物である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なシロール化合物及びその製造方法、並びに新規なシロール化合物を用いた有機エレクトロルミネッセンス素子(以下「有機EL素子」という)及び表示装置に関する。
【背景技術】
【0002】
有機EL素子を用いた表示装置において、フルカラー表示を実現するために、青、緑、赤の各色に発光する発光材料に関して、種々の検討がなされている。しかしながら、青色領域の発光には、励起に高いエネルギーを必要とすることもあり、色純度、発光寿命に優れた青色発光材料の開発が待たれているのが実情である。
【0003】
一方、含ケイ素π共役化合物であるシロール化合物は、電子輸送性のよい発光材料として期待されているが、合成上の問題もあり、2,5-ジアリールシロール誘導体、ラダー型ジベンゾシロール、スピロ型ジベンゾシロールといった例が報告されるにとどまっていた(非特許文献1〜3参照)。
【非特許文献1】Uchida et.al., Chem Mater. 2001, 13, 2680-2683
【非特許文献2】Lee et.al., J. Am.Chem.Soc. 2005, 127, 9071-9078
【非特許文献3】Matsuda et.al., M. Org. Lett. 2007, 9, 133-136
【発明の開示】
【発明が解決しようとする課題】
【0004】
本発明の目的は、青色領域の発光を有する、新規なシロール化合物を提供すること、また、その製造方法を提供することである。さらに、本発明の目的は、新規なシロール化合物を用いた有機EL素子及び表示装置を提供することである。
【0005】
本発明者らは、前記課題を解決するために種々検討を重ねた結果、フリーデル・クラフツ型シリル化反応を活用した環化反応により、新規なシロール化合物の合成が可能であること、さらに、この新規なシロール化合物が、青色領域で発光を有し、有機EL素子の青色発光材料として有用であることを見出し、本発明を完成させるに至った。
【課題を解決するための手段】
【0006】
本発明は、式(I):
【化7】
(式中、
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルであり、
R3は、互いに独立して、水素、ハロゲン又は−SiH(R1)2であるか、あるいは2個のR3は一緒になって、>Si(R1)2(ここで、R1は、互いに独立して、上記と同義である)を形成している)で示されるシロール化合物に関する。
【0007】
また、本発明は、式(II):
【化8】
(式中、R1及びR2は、上記と同義である)で示されるシロール化合物、及びその製造方法であって、式(v):
【化9】
(式中、R1及びR2は、請求項1と同義である)で示される化合物を、塩基の存在下で、脱ヒドリド剤を作用させて、所望であれば、精製して、式(II)で示されるシロール化合物を得る工程を含む方法、又は式(via)及び(vib):
【化10】
(式中、R1及びR2は、上記と同義である)で示される化合物の混合物を、塩基存在下で、脱ヒドリド剤で処理した後、所望であれば、精製して、式(II)で示されるシロール化合物を得る工程を含む方法に関する。
【0008】
さらに、式(III):
【化11】
(式中、R1及びR2は、上記と同義である)で示されるシロール化合物、及びその製造方法であって、式(vic)
【化12】
(式中、R1及びR2は、上記と同義である)で示される化合物を、塩基の存在下で、脱ヒドリド剤で処理した後、所望であれば、精製して、式(III)で示される化合物を得る工程を含む方法に関する。
【0009】
さらに、本発明は、一対の電極間に、少なくとも有機発光層を挟持する有機EL素子であって、上記のいずれかのシロール化合物を含有する有機EL素子に関し、シロール化合物を有機発光層に含有するこの有機エレクトロルミネッセンス素子に関する。また、本発明は、上記の有機EL素子を備えた表示装置に関する。
【発明の効果】
【0010】
本発明のシロール化合物は、可視光領域に強い吸収をもたず、物質の透過性が高いことに加え、青色領域に発光を有する。また、本発明のシロール化合物は、トリフェニレンという二次元的に広がったπ共役系にシロール骨格が縮環しているため、フロンティア軌道が分子全体に広く局在化でき、電化を帯びた励起種が安定に存在することができるので、電気化学的にも安定である。このように、本発明のシロール化合物は、有機EL素子の青色発光材料として理想的である。なお、本発明のシロール化合物の製造方法によれば、フリーデル・クラフツ型シリル化反応を活用した環化反応によって、高価な遷移金属系触媒を用いることなく、本発明のシロール化合物を簡便に合成にできるため、製造方法自体も有用性が高い。
【発明を実施するための最良の形態】
【0011】
本発明のシロール化合物は、式(I):
【化13】
【0012】
(式中、
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルであり、
R3は、互いに独立して、水素、ハロゲン又は−SiH(R1)2であるか、あるいは2個のR3は一緒になって、>Si(R1)2(ここで、R1は、互いに独立して、上記の定義のとおりである)を形成している)で示される。
【0013】
R1は、好ましくは、同一である。アルキルとしては、分岐状又は直鎖状のC1−C8アルキルが挙げられ、例えば分岐状又は直鎖状のC1−C6アルキルであり、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチルなどである。アリールとしては、非置換又は置換フェニルが挙げられ、置換フェニルの置換基としては、分岐状又は直鎖状のC1−C4アルキル(例えば、メチル、エチルである)、分岐状又は直鎖状のC1−C4アルコキシ(例えば、メトキシ、エトキシである)が挙げられる。アラルキルとしては、分岐状又は直鎖状のC1−C4アルキルにフェニルが置換したものが挙げられ、具体的にはベンジルが挙げられる。R1は、好ましくは、フェニルである。
【0014】
R2は、好ましくは、同一である。アルキルとしては、分岐状又は直鎖状のC1−C12アルキルが挙げられ、例えば分岐状又は直鎖状のC1−C8アルキルであり、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、分岐状又は直鎖状のペンチル、ヘキシル、ヘプチル、オクチルである。R2は、好ましくは、分岐状又は直鎖状のC3−C6アルキルであり、特に分岐状又は直鎖状のブチルである。
【0015】
R3に関して、ハロゲンとしては、クロロ、ブロモが挙げられ、好ましくはブロモである。R3が、−SiH(R1)2であるか、あるいは2個のR3は一緒になって、>Si(R1)2の場合に関して、R1は、上記のとおりである。
【0016】
好ましくは、2個のR3が一緒になって、>Si(R1)2を形成したシロール化合物であり、式(II):
【0017】
【化14】
【0018】
(式中、
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルである)で示される。例えば、式(11):
【0019】
【化15】
【0020】
で示されるシロール化合物が挙げられる。
【0021】
また、好ましくは、2個のR3がいずれも水素であるシロール化合物であり、式(III):
【化16】
【0022】
(式中、
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルである)で示される。例えば、式(12):
【0023】
【化17】
【0024】
で示されるシロール化合物が挙げられる。
【0025】
本発明のシロール化合物は、フリーデル・クラフツ型シリル化反応を活用した環化反応により、得ることができる。この環化反応は、以下のモデル系により表すことができる。
【0026】
【化18】
【0027】
上記の環化反応は、溶媒中、塩基の存在下で、0℃から溶媒の還流温度までの温度範囲、好ましくは10〜40℃で、環化させようとする化合物に脱ヒドリド剤を作用させることにより行うことができる。
【0028】
脱ヒドリド剤としては、トリチルカチオン(Ph3C+)を発生させるルイス酸が好ましく、例えば、Ph3CB(C6F5)4、Ph3CClO4、Ph3CBPh4、Ph3COSO2CF3などが挙げられる。対アニオンの反応性の低さの点から、Ph3CB(C6F5)4が好ましい。脱ヒドリド剤は、環化させようとする化合物のヒドロシリル基1モルに対して、1〜1.5モルの量で用いることが好ましい。
【0029】
塩基としては、環化におけるプロトン捕捉能の点から、ピリジン及び置換ピリジンが好ましく、2,6−ルチジン、ピリジンなどが挙げられる。中間種であるシリルカチオンへの配位能の低さの点から、2,6−ルチジンが好ましい。塩基は、環化させようとする化合物のヒドロシリル基1モルに対して、1〜1.5モルの量で用いることが好ましい。
【0030】
溶媒は、反応に不活性な有機溶媒であればよく、ジクロロメタン、クロロホルム、二硫化炭素などが挙げられる。
【0031】
具体的には、下記のスキームに基いて、式(II)及び式(III)のシロール化合物を得ることができる。スキーム1は、フリーデル・クラフツ型シリル化反応を活用した環化反応を2段階で行うプロセスであり、スキーム2は、フリーデル・クラフツ型シリル化反応を活用した環化反応を1段階で行うプロセスである。
【0032】
スキーム1:2段階プロセス
【化19】
【0033】
スキーム2:1段階プロセス
【化20】
【0034】
スキーム1、2の出発物質であるトリフェニレン誘導体(i)からのトリブロモ体((iia)及び(iib))を調製する方法は、J.Mater.Chem., 1997、7(4)、601-605に基いて行うことができ、スキーム中の方法は、適宜、変更することができる。
【0035】
スキーム1、2中のブロモをヒドロシリル化する方法は公知であり、例えば、n−ブチルリチウムを用いてリチオ化した後、(R1)2SiCl2を反応させた後、クロロを水素化アルミニウムリチウムで還元するか、リチオ化した後、(R1)2SiH2を反応させることができる。スキーム中の方法は、適宜、変更することができる。
【0036】
スキーム1、2における環化反応は、溶媒中、塩基の存在下で、0℃から溶媒の還流温度までの温度範囲、好ましくは10〜40℃で、環化させようとする化合物に脱ヒドリド剤を作用させることにより行うことができる。脱ヒドリド剤、塩基、溶媒等の条件については、上記のとおりである。脱ヒドリド剤としては、例えば、Ph3CB(C6F5)4、Ph3CClO4、Ph3CBPh4、Ph3COSO2CF3などが挙げられ、環化させようとする化合物のヒドロシリル基1モルに対して、1〜1.5モルの量で用いることが好ましい。また、塩基としては、例えば、2,6−ルチジンなどが挙げられ、環化させようとする化合物のヒドロシリル基1モルに対して、1〜1.5モルの量で用いることが好ましい。溶媒は、反応に不活性な有機溶媒であればよく、ジクロロメタン、クロロホルム、二硫化炭素などが挙げられる。
【0037】
スキーム1、2により得られる式(II)及び(III)のシロール化合物は、クロマトグラフィー等の公知の手段を用いて、それぞれ精製することができる。式(II)の化合物を収率よく得るためには、スキーム1の方が有利である。
【0038】
スキーム2で生成した式(vic)の化合物を、同様に、溶媒中、塩基の存在下で、脱ヒドリド剤を作用させて、式(III)のシロール化合物を得ることができる。下記スキーム3により得られる(III)のシロール化合物は、クロマトグラフィー等の公知の手段を用いて、精製することができる。
【0039】
スキーム3:
【化21】
【0040】
また、本発明は、一対の電極間に、少なくとも有機発光層を挟持する有機エレクトロルミネッセンス素子であって、本発明のシロール化合物を含有する有機エレクトロルミネッセンス素子に関する。本発明のシロール化合物を有機発光層に含有させて、発光材料として使用することが好ましい。有機発光層には、必要に応じて、本発明のシロール化合物に加えて、公知の発光材料、ドーピング材料、正孔注入材料、電子注入材料等を含有することができる。
【0041】
有機EL素子の構成材料、構造、製造方法については、特に限定されず、公知の構成材料、構造、製造方法を使用することができる。
【0042】
本発明の有機EL素子は、フラットパネルディスプレイをはじめとする表示装置に利用することができる。
【実施例】
【0043】
以下に実施例を挙げて本発明をさらに詳細に説明するが、本発明はこれらの実施例に限定されるものではない。
【0044】
実施例1:化合物11及び化合物12の合成例1(スキーム1)
【化22】
【0045】
工程1:化合物13a及び化合物13bの混合物の合成
【化23】
【0046】
2,3,6,7,11,12−ヘキサn−ブトキシトリフェニレン2.33 g (3.53 mmol)の塩化メチレン溶液 (100 mL)に対し、臭素900 μL (17.7 mmol)を室温で滴下した。室温で1.5時間撹拌した後、亜硫酸ナトリウム水溶液で反応を停止し、水層を塩化メチレンで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた暗緑色の液体をシリカゲルカラムクロマトグラフィー(溶媒;へキサン:CHCl3 = 1:1)で分離し、化合物13aと化合物13bの混合物を1.16 g得た(収率32%)。
【0047】
工程2:化合物15の合成
【化24】
【0048】
上記で得られた化合物13aと化合物13bの混合物(500 mg, 0.56 mmol)をTHF(40 mL)に溶解させ、-78℃においてn-BuLiのヘキサン溶液(1.6 M, 700 μL, 1.1 mmol)を滴下した。-78℃で1時間撹拌した後、Ph2SiCl2(1 mL, 4.8 mmol)を滴下し、室温で14時間撹拌した。この反応溶液にLiAlH4(400 mg, 10.5 mmol)を加え、3時間加熱還流した。反応溶液を室温まで冷却し、氷水に注ぎ、水層をクロロホルムで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた淡黄色の液体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:1)で分離し、化合物14を489 mg得た。化合物14は、化合物13a及び13bの混合物において分子中の臭素2個がPh2SiH基で置き換わった化合物と、分離不可能な副生物との混合物であったが、さらに精製することなく次の反応に用いた。
【0049】
化合物14(300 mg, <0.3 mmol)と2,6-ルチジン(70μL, 0.60 mmol)を乾燥塩化メチレン3 mlに溶解させ、これにPh3CB(C6F5)4(553 mg, 0.60 mmol)の乾燥塩化メチレン溶液(3 mL)を滴下し、室温で16時間撹拌した。亜硫酸ナトリウム水溶液を加えて反応を停止し、塩化メチレンで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた緑褐色の液体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 5:4)で分離し、化合物15を133 mg得た。(工程2の収率31%)
【0050】
化合物15:無色の固体。
1H NMR (500 MHz, CDCl3) δ0.67 (t, 3JHH = 7.5 Hz, 6H), 0.71-0.76 (m, 6H), 1.00 (t, 3JHH = 7.5 Hz, 6H), 1.07-1.18 (m, 8H), 1.29-1.50 (m, 8H), 1.52-1.60 (m, 4H), 1.83-1.92 (m, 4H), 3.73-3.84 (m, 8H), 4.07 (t, 3JHH = 7.0 Hz, 2H), 4.19 (t, 3JHH = 7.0 Hz, 2H), 7.28-7.41 (m, 12H), 7.82-7.85 (m, 8H), 9.58 (s, 1H).
【0051】
工程3:化合物16の合成
【化25】
【0052】
上記で得られた化合物15(200 mg, 0.18 mmol)をジエチルエーテル15 mLに溶解させ、-78℃に冷却した。この溶液に-78℃でt-BuLiのペンタン溶液(2.2 M, 170μL, 0.36 mmol)を滴下し、-78℃で30分撹拌した後、Ph2SiH2(300μL, 1.8 mmol)を滴下した。反応溶液を室温で1時間撹拌した後、19時間加熱還流した。反応溶液を室温まで冷却し、水を加えて反応を停止し、水層をジエチルエーテルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた淡黄色の固体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:1)で分離し、化合物16を113 mg得た。(工程3の収率51%)
【0053】
化合物16:無色の固体。
1H NMR (500 MHz, CDCl3) δ0.65-0.73 (m, 15H), 0.82 (t, 3JHH = 7.4 Hz, 3H), 0.93-1.04 (m, 4H), 1.11-1.14 (m, 6H), 1.22-1.32 (m, 6H), 1.34 (sept, 3JHH = 7.4 Hz, 2H), 1.42-1.54 (m, 6H), 3.31 (t, 3JHH = 7.0 Hz, 2H), 3.56 (t, 3JHH = 7.1 Hz, 2H), 3.68 (t, 3JHH = 7.1 Hz, 2H), 3.73 (t, 3JHH = 6.9 Hz, 2H), 3.77 (t, 3JHH = 7.0 Hz, 2H), 3.85 (t, 3JHH = 6.8 Hz, 2H), 5.99 (s, 1H), 7.30-7.42 (m, 19H), 7.69-7.72 (m, 4H), 7.86 (t, 4JHH = 7.7 Hz, 8H).
【0054】
工程4:化合物11、12の合成
【化26】
【0055】
化合物16(110 mg, 0.091 mmol)と2,6-ルチジン(11μL, 0.091 mmol)を脱水塩化メチレン2 mlに溶解させ、これにPh3CB(C6F5)4(84 mg, 0.091 mmol)の脱水塩化メチレン溶液(2 mL)を滴下し、室温で16時間撹拌した。亜硫酸ナトリウム水溶液を加えて反応を停止し、塩化メチレンで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた淡黄色の固体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:1)で分離し、化合物11および化合物12をそれぞれ20 mgおよび28 mg得た。(工程4の収率:化合物11;18%,化合物12;31%)
【0056】
化合物11:無色の固体。
1H NMR (400 MHz, CDCl3) δ0.69 (t, 3JHH = 7.2 Hz, 18H), 1.11 (sext, 3JHH = 7.2 Hz, 12H), 1.45 (quint, 3JHH = 7.2 Hz, 12H), 3.78 (t, 3JHH = 7.2 Hz, 12H), 7.28-7.40 (m, 18H), 7.83 (dd, 3JHH = 7.6 Hz, , 4JHH = 1.6 Hz, 12H).
化合物12:無色の固体。
1H NMR (500 MHz, CDCl3) δ0.68 (t, 3JHH = 7.4 Hz, 6H), 0.74 (t, 3JHH = 7.4 Hz, 6H), 1.01 (t, 3JHH = 7.4 Hz, 6H), 1.09-1.18 (m, 8H), 1.40 (quint, 3JHH = 7.7 Hz, 4H), 1.46 (quint, 3JHH = 7.7 Hz, 4H), 1.56 (sext, 3JHH = 7.4 Hz, 4H), 1.90 (quint, 3JHH = 7.7 Hz, 4H), 3.75-3.81 (m, 8H), 4.19 (t, 3JHH = 6.4 Hz, 4H), 7.31 (t, 3JHH = 7.5 Hz, 8H), 7.37 (tt, 3JHH = 7.5 Hz, 4JHH = 1.4 Hz, 4H), 7.74 (s, 2H), 7.85 (dd, 3JHH = 7.5 Hz, 4JHH = 1.4 Hz, 8H).
【0057】
実施例2:化合物11及び12の合成例2(スキーム2)
【化27】
【0058】
工程1:化合物10a及び10bの合成
実施例1の工程1にしたがって得られた、化合物13aと13bの混合物(2.50 g, 2.79 mmol)をジエチルエーテル(50 mL)に溶解させ、-78℃においてt-BuLiのペンタン溶液(2.2 M, 7.80 mL, 16.7 mmol)を滴下した。-78℃で30分攪拌した後、Ph2SiH2(3 mL, 16.3 mmol)を滴下し、室温で1時間攪拌した後、18時間加熱還流した。反応溶液を室温まで冷却し、水で反応を停止し、水層をクロロホルムで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた褐色の液体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:1)で分離し、化合物10a, 10bと化合物10cをそれぞれ181 mgと673 mg得た。化合物10a, 10bはさらに精製することなく次の反応に用いた。
【0059】
工程2:化合物11及び12の合成
化合物10a, 10bの混合物(122 mg, 0.10 mmol)と2,6-ルチジン(39 μL, 0.33 mmol)を乾燥塩化メチレン2 mlに溶解させ、これにPh3CB(C6F5)4(307 mg, 0.33 mmol)の乾燥塩化メチレン溶液(2 mL)を滴下し、室温で25時間攪拌した。亜硫酸ナトリウム水溶液を加えて反応を停止し、塩化メチレンで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた淡黄色の固体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:1)で分離し、化合物11および化合物12をそれぞれ8 mgおよび48 mg得た。(工程2の収率:化合物11;4%,化合物12;17%)
【0060】
実施例3:化合物12の合成例(スキーム3)
【化28】
【0061】
実施例2の工程1で得られた化合物10c(100 mg, 0.098 mmol)と2,6-ルチジン(25μL, 0.22 mmol)を乾燥塩化メチレン2 mlに溶解させ、これにPh3CB(C6F5)4(198 mg, 0.22 mmol)の乾燥塩化メチレン溶液(2 mL)を滴下し、室温で28時間攪拌した。亜硫酸ナトリウム水溶液を加えて反応を停止し、塩化メチレンで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた淡黄色の固体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:2)で分離し、化合物12を39 mg得た。(収率39%)
【0062】
光学データの測定
化合物11、12について、以下のようにして光学データを測定した。対照として、ジベンゾシロール誘導体:
【化29】
の測定もあわせて行った。測定はすべて室温で行った。結果を図1〜3に示す。
・紫外可視吸収スペクトル:塩化メチレン溶液(濃度は1.6×10-5 M)として測定。
・蛍光スペクトル:塩化メチレン溶液(濃度は1.6×10-5 M)として測定。励起波長は300 nm。
・蛍光スペクトル(固体):粉末サンプルを測定。励起波長は300 nm。
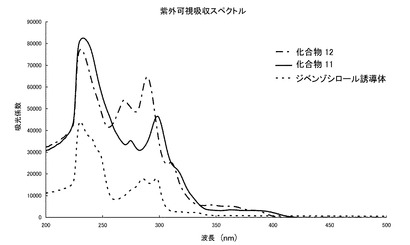
【0063】
図1に示されるように、化合物11、12は、可視領域に強い吸収をもたず、ほぼ無色であることがわかった。
図2に示されるように、化合物11、12は、それぞれ427nm、406nmに発光極大を示す蛍光を発することがわかった。
さらに、図3に示されるように、化合物11、12は、固体状態においても、それぞれ447nm、420nmに発光極大を示す蛍光を発することがわかった。
【0064】
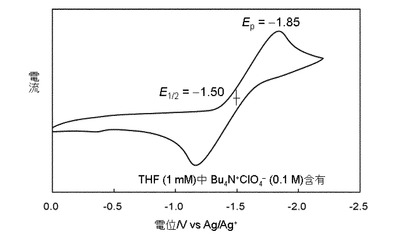
サイクリックボルタンメトリーの測定
化合物12について、以下のようにしてサイクリックボルタンメトリーを測定した。結果を図4に示す。
測定はバッファとしてBu4NClO4(0.1M)を含む12のTHF溶液(1mM)を調製し、銀電極を参照電極とし、挿引速度100mV/sで行った。
【0065】
図4に示されるように、化合物12は可逆的な還元を観測した。シロール誘導体は、還元に対して不安定であり、分解しやすいことが知られているが、化合物12ではπ共役系が二次元的に広がっているために、還元に対する安定性が向上したものと考えられる。
【0066】
本発明のシロール化合物は、可視光領域に強い吸収をもたず、物質の透過性が高いことに加え、青色領域に発光を有し、かつ電気化学的にも安定であるため、有機EL素子の発光材料として理想的である。なお、本発明のシロール化合物の製造方法によれば、フリーデル・クラフツ型シリル化反応を活用した環化反応により、高価な遷移金属系触媒を用いることなく、本発明のシロール化合物を簡便に合成にできるため、製造方法自体も有用性が高い。
【図面の簡単な説明】
【0067】
【図1】紫外可視吸収スペクトルである。
【図2】化合物11、12及び対照化合物の溶液状態での蛍光スペクトルである。
【図3】化合物11、12の固体状態での蛍光スペクトルである。
【図4】化合物12のサイクリックボルタモグラムである。
【技術分野】
【0001】
本発明は、新規なシロール化合物及びその製造方法、並びに新規なシロール化合物を用いた有機エレクトロルミネッセンス素子(以下「有機EL素子」という)及び表示装置に関する。
【背景技術】
【0002】
有機EL素子を用いた表示装置において、フルカラー表示を実現するために、青、緑、赤の各色に発光する発光材料に関して、種々の検討がなされている。しかしながら、青色領域の発光には、励起に高いエネルギーを必要とすることもあり、色純度、発光寿命に優れた青色発光材料の開発が待たれているのが実情である。
【0003】
一方、含ケイ素π共役化合物であるシロール化合物は、電子輸送性のよい発光材料として期待されているが、合成上の問題もあり、2,5-ジアリールシロール誘導体、ラダー型ジベンゾシロール、スピロ型ジベンゾシロールといった例が報告されるにとどまっていた(非特許文献1〜3参照)。
【非特許文献1】Uchida et.al., Chem Mater. 2001, 13, 2680-2683
【非特許文献2】Lee et.al., J. Am.Chem.Soc. 2005, 127, 9071-9078
【非特許文献3】Matsuda et.al., M. Org. Lett. 2007, 9, 133-136
【発明の開示】
【発明が解決しようとする課題】
【0004】
本発明の目的は、青色領域の発光を有する、新規なシロール化合物を提供すること、また、その製造方法を提供することである。さらに、本発明の目的は、新規なシロール化合物を用いた有機EL素子及び表示装置を提供することである。
【0005】
本発明者らは、前記課題を解決するために種々検討を重ねた結果、フリーデル・クラフツ型シリル化反応を活用した環化反応により、新規なシロール化合物の合成が可能であること、さらに、この新規なシロール化合物が、青色領域で発光を有し、有機EL素子の青色発光材料として有用であることを見出し、本発明を完成させるに至った。
【課題を解決するための手段】
【0006】
本発明は、式(I):
【化7】
(式中、
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルであり、
R3は、互いに独立して、水素、ハロゲン又は−SiH(R1)2であるか、あるいは2個のR3は一緒になって、>Si(R1)2(ここで、R1は、互いに独立して、上記と同義である)を形成している)で示されるシロール化合物に関する。
【0007】
また、本発明は、式(II):
【化8】
(式中、R1及びR2は、上記と同義である)で示されるシロール化合物、及びその製造方法であって、式(v):
【化9】
(式中、R1及びR2は、請求項1と同義である)で示される化合物を、塩基の存在下で、脱ヒドリド剤を作用させて、所望であれば、精製して、式(II)で示されるシロール化合物を得る工程を含む方法、又は式(via)及び(vib):
【化10】
(式中、R1及びR2は、上記と同義である)で示される化合物の混合物を、塩基存在下で、脱ヒドリド剤で処理した後、所望であれば、精製して、式(II)で示されるシロール化合物を得る工程を含む方法に関する。
【0008】
さらに、式(III):
【化11】
(式中、R1及びR2は、上記と同義である)で示されるシロール化合物、及びその製造方法であって、式(vic)
【化12】
(式中、R1及びR2は、上記と同義である)で示される化合物を、塩基の存在下で、脱ヒドリド剤で処理した後、所望であれば、精製して、式(III)で示される化合物を得る工程を含む方法に関する。
【0009】
さらに、本発明は、一対の電極間に、少なくとも有機発光層を挟持する有機EL素子であって、上記のいずれかのシロール化合物を含有する有機EL素子に関し、シロール化合物を有機発光層に含有するこの有機エレクトロルミネッセンス素子に関する。また、本発明は、上記の有機EL素子を備えた表示装置に関する。
【発明の効果】
【0010】
本発明のシロール化合物は、可視光領域に強い吸収をもたず、物質の透過性が高いことに加え、青色領域に発光を有する。また、本発明のシロール化合物は、トリフェニレンという二次元的に広がったπ共役系にシロール骨格が縮環しているため、フロンティア軌道が分子全体に広く局在化でき、電化を帯びた励起種が安定に存在することができるので、電気化学的にも安定である。このように、本発明のシロール化合物は、有機EL素子の青色発光材料として理想的である。なお、本発明のシロール化合物の製造方法によれば、フリーデル・クラフツ型シリル化反応を活用した環化反応によって、高価な遷移金属系触媒を用いることなく、本発明のシロール化合物を簡便に合成にできるため、製造方法自体も有用性が高い。
【発明を実施するための最良の形態】
【0011】
本発明のシロール化合物は、式(I):
【化13】
【0012】
(式中、
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルであり、
R3は、互いに独立して、水素、ハロゲン又は−SiH(R1)2であるか、あるいは2個のR3は一緒になって、>Si(R1)2(ここで、R1は、互いに独立して、上記の定義のとおりである)を形成している)で示される。
【0013】
R1は、好ましくは、同一である。アルキルとしては、分岐状又は直鎖状のC1−C8アルキルが挙げられ、例えば分岐状又は直鎖状のC1−C6アルキルであり、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチルなどである。アリールとしては、非置換又は置換フェニルが挙げられ、置換フェニルの置換基としては、分岐状又は直鎖状のC1−C4アルキル(例えば、メチル、エチルである)、分岐状又は直鎖状のC1−C4アルコキシ(例えば、メトキシ、エトキシである)が挙げられる。アラルキルとしては、分岐状又は直鎖状のC1−C4アルキルにフェニルが置換したものが挙げられ、具体的にはベンジルが挙げられる。R1は、好ましくは、フェニルである。
【0014】
R2は、好ましくは、同一である。アルキルとしては、分岐状又は直鎖状のC1−C12アルキルが挙げられ、例えば分岐状又は直鎖状のC1−C8アルキルであり、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、分岐状又は直鎖状のペンチル、ヘキシル、ヘプチル、オクチルである。R2は、好ましくは、分岐状又は直鎖状のC3−C6アルキルであり、特に分岐状又は直鎖状のブチルである。
【0015】
R3に関して、ハロゲンとしては、クロロ、ブロモが挙げられ、好ましくはブロモである。R3が、−SiH(R1)2であるか、あるいは2個のR3は一緒になって、>Si(R1)2の場合に関して、R1は、上記のとおりである。
【0016】
好ましくは、2個のR3が一緒になって、>Si(R1)2を形成したシロール化合物であり、式(II):
【0017】
【化14】
【0018】
(式中、
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルである)で示される。例えば、式(11):
【0019】
【化15】
【0020】
で示されるシロール化合物が挙げられる。
【0021】
また、好ましくは、2個のR3がいずれも水素であるシロール化合物であり、式(III):
【化16】
【0022】
(式中、
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルである)で示される。例えば、式(12):
【0023】
【化17】
【0024】
で示されるシロール化合物が挙げられる。
【0025】
本発明のシロール化合物は、フリーデル・クラフツ型シリル化反応を活用した環化反応により、得ることができる。この環化反応は、以下のモデル系により表すことができる。
【0026】
【化18】
【0027】
上記の環化反応は、溶媒中、塩基の存在下で、0℃から溶媒の還流温度までの温度範囲、好ましくは10〜40℃で、環化させようとする化合物に脱ヒドリド剤を作用させることにより行うことができる。
【0028】
脱ヒドリド剤としては、トリチルカチオン(Ph3C+)を発生させるルイス酸が好ましく、例えば、Ph3CB(C6F5)4、Ph3CClO4、Ph3CBPh4、Ph3COSO2CF3などが挙げられる。対アニオンの反応性の低さの点から、Ph3CB(C6F5)4が好ましい。脱ヒドリド剤は、環化させようとする化合物のヒドロシリル基1モルに対して、1〜1.5モルの量で用いることが好ましい。
【0029】
塩基としては、環化におけるプロトン捕捉能の点から、ピリジン及び置換ピリジンが好ましく、2,6−ルチジン、ピリジンなどが挙げられる。中間種であるシリルカチオンへの配位能の低さの点から、2,6−ルチジンが好ましい。塩基は、環化させようとする化合物のヒドロシリル基1モルに対して、1〜1.5モルの量で用いることが好ましい。
【0030】
溶媒は、反応に不活性な有機溶媒であればよく、ジクロロメタン、クロロホルム、二硫化炭素などが挙げられる。
【0031】
具体的には、下記のスキームに基いて、式(II)及び式(III)のシロール化合物を得ることができる。スキーム1は、フリーデル・クラフツ型シリル化反応を活用した環化反応を2段階で行うプロセスであり、スキーム2は、フリーデル・クラフツ型シリル化反応を活用した環化反応を1段階で行うプロセスである。
【0032】
スキーム1:2段階プロセス
【化19】
【0033】
スキーム2:1段階プロセス
【化20】
【0034】
スキーム1、2の出発物質であるトリフェニレン誘導体(i)からのトリブロモ体((iia)及び(iib))を調製する方法は、J.Mater.Chem., 1997、7(4)、601-605に基いて行うことができ、スキーム中の方法は、適宜、変更することができる。
【0035】
スキーム1、2中のブロモをヒドロシリル化する方法は公知であり、例えば、n−ブチルリチウムを用いてリチオ化した後、(R1)2SiCl2を反応させた後、クロロを水素化アルミニウムリチウムで還元するか、リチオ化した後、(R1)2SiH2を反応させることができる。スキーム中の方法は、適宜、変更することができる。
【0036】
スキーム1、2における環化反応は、溶媒中、塩基の存在下で、0℃から溶媒の還流温度までの温度範囲、好ましくは10〜40℃で、環化させようとする化合物に脱ヒドリド剤を作用させることにより行うことができる。脱ヒドリド剤、塩基、溶媒等の条件については、上記のとおりである。脱ヒドリド剤としては、例えば、Ph3CB(C6F5)4、Ph3CClO4、Ph3CBPh4、Ph3COSO2CF3などが挙げられ、環化させようとする化合物のヒドロシリル基1モルに対して、1〜1.5モルの量で用いることが好ましい。また、塩基としては、例えば、2,6−ルチジンなどが挙げられ、環化させようとする化合物のヒドロシリル基1モルに対して、1〜1.5モルの量で用いることが好ましい。溶媒は、反応に不活性な有機溶媒であればよく、ジクロロメタン、クロロホルム、二硫化炭素などが挙げられる。
【0037】
スキーム1、2により得られる式(II)及び(III)のシロール化合物は、クロマトグラフィー等の公知の手段を用いて、それぞれ精製することができる。式(II)の化合物を収率よく得るためには、スキーム1の方が有利である。
【0038】
スキーム2で生成した式(vic)の化合物を、同様に、溶媒中、塩基の存在下で、脱ヒドリド剤を作用させて、式(III)のシロール化合物を得ることができる。下記スキーム3により得られる(III)のシロール化合物は、クロマトグラフィー等の公知の手段を用いて、精製することができる。
【0039】
スキーム3:
【化21】
【0040】
また、本発明は、一対の電極間に、少なくとも有機発光層を挟持する有機エレクトロルミネッセンス素子であって、本発明のシロール化合物を含有する有機エレクトロルミネッセンス素子に関する。本発明のシロール化合物を有機発光層に含有させて、発光材料として使用することが好ましい。有機発光層には、必要に応じて、本発明のシロール化合物に加えて、公知の発光材料、ドーピング材料、正孔注入材料、電子注入材料等を含有することができる。
【0041】
有機EL素子の構成材料、構造、製造方法については、特に限定されず、公知の構成材料、構造、製造方法を使用することができる。
【0042】
本発明の有機EL素子は、フラットパネルディスプレイをはじめとする表示装置に利用することができる。
【実施例】
【0043】
以下に実施例を挙げて本発明をさらに詳細に説明するが、本発明はこれらの実施例に限定されるものではない。
【0044】
実施例1:化合物11及び化合物12の合成例1(スキーム1)
【化22】
【0045】
工程1:化合物13a及び化合物13bの混合物の合成
【化23】
【0046】
2,3,6,7,11,12−ヘキサn−ブトキシトリフェニレン2.33 g (3.53 mmol)の塩化メチレン溶液 (100 mL)に対し、臭素900 μL (17.7 mmol)を室温で滴下した。室温で1.5時間撹拌した後、亜硫酸ナトリウム水溶液で反応を停止し、水層を塩化メチレンで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた暗緑色の液体をシリカゲルカラムクロマトグラフィー(溶媒;へキサン:CHCl3 = 1:1)で分離し、化合物13aと化合物13bの混合物を1.16 g得た(収率32%)。
【0047】
工程2:化合物15の合成
【化24】
【0048】
上記で得られた化合物13aと化合物13bの混合物(500 mg, 0.56 mmol)をTHF(40 mL)に溶解させ、-78℃においてn-BuLiのヘキサン溶液(1.6 M, 700 μL, 1.1 mmol)を滴下した。-78℃で1時間撹拌した後、Ph2SiCl2(1 mL, 4.8 mmol)を滴下し、室温で14時間撹拌した。この反応溶液にLiAlH4(400 mg, 10.5 mmol)を加え、3時間加熱還流した。反応溶液を室温まで冷却し、氷水に注ぎ、水層をクロロホルムで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた淡黄色の液体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:1)で分離し、化合物14を489 mg得た。化合物14は、化合物13a及び13bの混合物において分子中の臭素2個がPh2SiH基で置き換わった化合物と、分離不可能な副生物との混合物であったが、さらに精製することなく次の反応に用いた。
【0049】
化合物14(300 mg, <0.3 mmol)と2,6-ルチジン(70μL, 0.60 mmol)を乾燥塩化メチレン3 mlに溶解させ、これにPh3CB(C6F5)4(553 mg, 0.60 mmol)の乾燥塩化メチレン溶液(3 mL)を滴下し、室温で16時間撹拌した。亜硫酸ナトリウム水溶液を加えて反応を停止し、塩化メチレンで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた緑褐色の液体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 5:4)で分離し、化合物15を133 mg得た。(工程2の収率31%)
【0050】
化合物15:無色の固体。
1H NMR (500 MHz, CDCl3) δ0.67 (t, 3JHH = 7.5 Hz, 6H), 0.71-0.76 (m, 6H), 1.00 (t, 3JHH = 7.5 Hz, 6H), 1.07-1.18 (m, 8H), 1.29-1.50 (m, 8H), 1.52-1.60 (m, 4H), 1.83-1.92 (m, 4H), 3.73-3.84 (m, 8H), 4.07 (t, 3JHH = 7.0 Hz, 2H), 4.19 (t, 3JHH = 7.0 Hz, 2H), 7.28-7.41 (m, 12H), 7.82-7.85 (m, 8H), 9.58 (s, 1H).
【0051】
工程3:化合物16の合成
【化25】
【0052】
上記で得られた化合物15(200 mg, 0.18 mmol)をジエチルエーテル15 mLに溶解させ、-78℃に冷却した。この溶液に-78℃でt-BuLiのペンタン溶液(2.2 M, 170μL, 0.36 mmol)を滴下し、-78℃で30分撹拌した後、Ph2SiH2(300μL, 1.8 mmol)を滴下した。反応溶液を室温で1時間撹拌した後、19時間加熱還流した。反応溶液を室温まで冷却し、水を加えて反応を停止し、水層をジエチルエーテルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた淡黄色の固体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:1)で分離し、化合物16を113 mg得た。(工程3の収率51%)
【0053】
化合物16:無色の固体。
1H NMR (500 MHz, CDCl3) δ0.65-0.73 (m, 15H), 0.82 (t, 3JHH = 7.4 Hz, 3H), 0.93-1.04 (m, 4H), 1.11-1.14 (m, 6H), 1.22-1.32 (m, 6H), 1.34 (sept, 3JHH = 7.4 Hz, 2H), 1.42-1.54 (m, 6H), 3.31 (t, 3JHH = 7.0 Hz, 2H), 3.56 (t, 3JHH = 7.1 Hz, 2H), 3.68 (t, 3JHH = 7.1 Hz, 2H), 3.73 (t, 3JHH = 6.9 Hz, 2H), 3.77 (t, 3JHH = 7.0 Hz, 2H), 3.85 (t, 3JHH = 6.8 Hz, 2H), 5.99 (s, 1H), 7.30-7.42 (m, 19H), 7.69-7.72 (m, 4H), 7.86 (t, 4JHH = 7.7 Hz, 8H).
【0054】
工程4:化合物11、12の合成
【化26】
【0055】
化合物16(110 mg, 0.091 mmol)と2,6-ルチジン(11μL, 0.091 mmol)を脱水塩化メチレン2 mlに溶解させ、これにPh3CB(C6F5)4(84 mg, 0.091 mmol)の脱水塩化メチレン溶液(2 mL)を滴下し、室温で16時間撹拌した。亜硫酸ナトリウム水溶液を加えて反応を停止し、塩化メチレンで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた淡黄色の固体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:1)で分離し、化合物11および化合物12をそれぞれ20 mgおよび28 mg得た。(工程4の収率:化合物11;18%,化合物12;31%)
【0056】
化合物11:無色の固体。
1H NMR (400 MHz, CDCl3) δ0.69 (t, 3JHH = 7.2 Hz, 18H), 1.11 (sext, 3JHH = 7.2 Hz, 12H), 1.45 (quint, 3JHH = 7.2 Hz, 12H), 3.78 (t, 3JHH = 7.2 Hz, 12H), 7.28-7.40 (m, 18H), 7.83 (dd, 3JHH = 7.6 Hz, , 4JHH = 1.6 Hz, 12H).
化合物12:無色の固体。
1H NMR (500 MHz, CDCl3) δ0.68 (t, 3JHH = 7.4 Hz, 6H), 0.74 (t, 3JHH = 7.4 Hz, 6H), 1.01 (t, 3JHH = 7.4 Hz, 6H), 1.09-1.18 (m, 8H), 1.40 (quint, 3JHH = 7.7 Hz, 4H), 1.46 (quint, 3JHH = 7.7 Hz, 4H), 1.56 (sext, 3JHH = 7.4 Hz, 4H), 1.90 (quint, 3JHH = 7.7 Hz, 4H), 3.75-3.81 (m, 8H), 4.19 (t, 3JHH = 6.4 Hz, 4H), 7.31 (t, 3JHH = 7.5 Hz, 8H), 7.37 (tt, 3JHH = 7.5 Hz, 4JHH = 1.4 Hz, 4H), 7.74 (s, 2H), 7.85 (dd, 3JHH = 7.5 Hz, 4JHH = 1.4 Hz, 8H).
【0057】
実施例2:化合物11及び12の合成例2(スキーム2)
【化27】
【0058】
工程1:化合物10a及び10bの合成
実施例1の工程1にしたがって得られた、化合物13aと13bの混合物(2.50 g, 2.79 mmol)をジエチルエーテル(50 mL)に溶解させ、-78℃においてt-BuLiのペンタン溶液(2.2 M, 7.80 mL, 16.7 mmol)を滴下した。-78℃で30分攪拌した後、Ph2SiH2(3 mL, 16.3 mmol)を滴下し、室温で1時間攪拌した後、18時間加熱還流した。反応溶液を室温まで冷却し、水で反応を停止し、水層をクロロホルムで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた褐色の液体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:1)で分離し、化合物10a, 10bと化合物10cをそれぞれ181 mgと673 mg得た。化合物10a, 10bはさらに精製することなく次の反応に用いた。
【0059】
工程2:化合物11及び12の合成
化合物10a, 10bの混合物(122 mg, 0.10 mmol)と2,6-ルチジン(39 μL, 0.33 mmol)を乾燥塩化メチレン2 mlに溶解させ、これにPh3CB(C6F5)4(307 mg, 0.33 mmol)の乾燥塩化メチレン溶液(2 mL)を滴下し、室温で25時間攪拌した。亜硫酸ナトリウム水溶液を加えて反応を停止し、塩化メチレンで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた淡黄色の固体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:1)で分離し、化合物11および化合物12をそれぞれ8 mgおよび48 mg得た。(工程2の収率:化合物11;4%,化合物12;17%)
【0060】
実施例3:化合物12の合成例(スキーム3)
【化28】
【0061】
実施例2の工程1で得られた化合物10c(100 mg, 0.098 mmol)と2,6-ルチジン(25μL, 0.22 mmol)を乾燥塩化メチレン2 mlに溶解させ、これにPh3CB(C6F5)4(198 mg, 0.22 mmol)の乾燥塩化メチレン溶液(2 mL)を滴下し、室温で28時間攪拌した。亜硫酸ナトリウム水溶液を加えて反応を停止し、塩化メチレンで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムをろ別し、減圧下溶媒を留去した。得られた淡黄色の固体をシリカゲルカラムクロマトグラフィー(溶媒; ヘキサン:CHCl3 = 1:2)で分離し、化合物12を39 mg得た。(収率39%)
【0062】
光学データの測定
化合物11、12について、以下のようにして光学データを測定した。対照として、ジベンゾシロール誘導体:
【化29】
の測定もあわせて行った。測定はすべて室温で行った。結果を図1〜3に示す。
・紫外可視吸収スペクトル:塩化メチレン溶液(濃度は1.6×10-5 M)として測定。
・蛍光スペクトル:塩化メチレン溶液(濃度は1.6×10-5 M)として測定。励起波長は300 nm。
・蛍光スペクトル(固体):粉末サンプルを測定。励起波長は300 nm。
【0063】
図1に示されるように、化合物11、12は、可視領域に強い吸収をもたず、ほぼ無色であることがわかった。
図2に示されるように、化合物11、12は、それぞれ427nm、406nmに発光極大を示す蛍光を発することがわかった。
さらに、図3に示されるように、化合物11、12は、固体状態においても、それぞれ447nm、420nmに発光極大を示す蛍光を発することがわかった。
【0064】
サイクリックボルタンメトリーの測定
化合物12について、以下のようにしてサイクリックボルタンメトリーを測定した。結果を図4に示す。
測定はバッファとしてBu4NClO4(0.1M)を含む12のTHF溶液(1mM)を調製し、銀電極を参照電極とし、挿引速度100mV/sで行った。
【0065】
図4に示されるように、化合物12は可逆的な還元を観測した。シロール誘導体は、還元に対して不安定であり、分解しやすいことが知られているが、化合物12ではπ共役系が二次元的に広がっているために、還元に対する安定性が向上したものと考えられる。
【0066】
本発明のシロール化合物は、可視光領域に強い吸収をもたず、物質の透過性が高いことに加え、青色領域に発光を有し、かつ電気化学的にも安定であるため、有機EL素子の発光材料として理想的である。なお、本発明のシロール化合物の製造方法によれば、フリーデル・クラフツ型シリル化反応を活用した環化反応により、高価な遷移金属系触媒を用いることなく、本発明のシロール化合物を簡便に合成にできるため、製造方法自体も有用性が高い。
【図面の簡単な説明】
【0067】
【図1】紫外可視吸収スペクトルである。
【図2】化合物11、12及び対照化合物の溶液状態での蛍光スペクトルである。
【図3】化合物11、12の固体状態での蛍光スペクトルである。
【図4】化合物12のサイクリックボルタモグラムである。
【特許請求の範囲】
【請求項1】
式(I):
【化1】
(式中、
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルであり、
R3は、互いに独立して、水素、ハロゲン又は−SiH(R1)2であるか、あるいは2個のR3は一緒になって、>Si(R1)2(ここで、R1は、互いに独立して、上記と同義である)を形成している)で示されるシロール化合物。
【請求項2】
式(II):
【化2】
(式中、R1及びR2は、請求項1と同義である)で示される、請求項1記載のシロール化合物。
【請求項3】
式(III):
【化3】
(式中、R1及びR2は、請求項1と同義である)で示される、請求項1記載のシロール化合物。
【請求項4】
請求項2記載の式(II)で示されるシロール化合物の製造方法であって、式(v):
【化4】
(式中、R1及びR2は、請求項1と同義である)で示される化合物を、塩基の存在下で、脱ヒドリド剤を作用させて、所望であれば、精製して、式(II)で示されるシロール化合物を得る工程を含む方法。
【請求項5】
請求項2記載の式(II)で示される化合物の製造方法であって、式(via)及び(vib):
【化5】
(式中、R1及びR2は、請求項1と同義である)で示される化合物の混合物を、塩基存在下で、脱ヒドリド剤で処理した後、所望であれば、精製して、式(II)で示されるシロール化合物を得る工程を含む方法。
【請求項6】
請求項3記載の式(III)で示される化合物の製造方法であって、式(vic)
【化6】
(式中、R1及びR2は、請求項1と同義である)で示される化合物を、塩基の存在下で、脱ヒドリド剤で処理した後、所望であれば、精製して、式(III)で示される化合物を得る工程を含む方法。
【請求項7】
一対の電極間に、少なくとも有機発光層を挟持する有機エレクトロルミネッセンス素子であって、請求項1〜3のいずれか1項記載のシロール化合物を含有する有機エレクトロルミネッセンス素子。
【請求項8】
請求項1〜3のいずれか1項記載のシロール化合物を有機発光層に含有する、請求項7記載の有機エレクトロルミネッセンス素子。
【請求項9】
請求項7又は8記載の有機エレクトロルミネッセンス素子を備えた表示装置。
【請求項1】
式(I):
【化1】
(式中、
R1は、互いに独立して、アルキル、アリール又はアラルキルであり、
R2は、互いに独立して、アルキルであり、
R3は、互いに独立して、水素、ハロゲン又は−SiH(R1)2であるか、あるいは2個のR3は一緒になって、>Si(R1)2(ここで、R1は、互いに独立して、上記と同義である)を形成している)で示されるシロール化合物。
【請求項2】
式(II):
【化2】
(式中、R1及びR2は、請求項1と同義である)で示される、請求項1記載のシロール化合物。
【請求項3】
式(III):
【化3】
(式中、R1及びR2は、請求項1と同義である)で示される、請求項1記載のシロール化合物。
【請求項4】
請求項2記載の式(II)で示されるシロール化合物の製造方法であって、式(v):
【化4】
(式中、R1及びR2は、請求項1と同義である)で示される化合物を、塩基の存在下で、脱ヒドリド剤を作用させて、所望であれば、精製して、式(II)で示されるシロール化合物を得る工程を含む方法。
【請求項5】
請求項2記載の式(II)で示される化合物の製造方法であって、式(via)及び(vib):
【化5】
(式中、R1及びR2は、請求項1と同義である)で示される化合物の混合物を、塩基存在下で、脱ヒドリド剤で処理した後、所望であれば、精製して、式(II)で示されるシロール化合物を得る工程を含む方法。
【請求項6】
請求項3記載の式(III)で示される化合物の製造方法であって、式(vic)
【化6】
(式中、R1及びR2は、請求項1と同義である)で示される化合物を、塩基の存在下で、脱ヒドリド剤で処理した後、所望であれば、精製して、式(III)で示される化合物を得る工程を含む方法。
【請求項7】
一対の電極間に、少なくとも有機発光層を挟持する有機エレクトロルミネッセンス素子であって、請求項1〜3のいずれか1項記載のシロール化合物を含有する有機エレクトロルミネッセンス素子。
【請求項8】
請求項1〜3のいずれか1項記載のシロール化合物を有機発光層に含有する、請求項7記載の有機エレクトロルミネッセンス素子。
【請求項9】
請求項7又は8記載の有機エレクトロルミネッセンス素子を備えた表示装置。
【図1】

【図2】

【図3】

【図4】
【図2】
【図3】
【図4】
【公開番号】特開2010−64976(P2010−64976A)
【公開日】平成22年3月25日(2010.3.25)
【国際特許分類】
【出願番号】特願2008−231979(P2008−231979)
【出願日】平成20年9月10日(2008.9.10)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年3月12日 社団法人日本化学会発行の「日本化学会第88春季年会(2008)講演予稿集II」に発表 平成20年3月26日〜30日 社団法人日本化学会主催の「日本化学会第88春季年会(2008)」において文書をもって発表
【出願人】(504137912)国立大学法人 東京大学 (1,942)
【Fターム(参考)】
【公開日】平成22年3月25日(2010.3.25)
【国際特許分類】
【出願日】平成20年9月10日(2008.9.10)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年3月12日 社団法人日本化学会発行の「日本化学会第88春季年会(2008)講演予稿集II」に発表 平成20年3月26日〜30日 社団法人日本化学会主催の「日本化学会第88春季年会(2008)」において文書をもって発表
【出願人】(504137912)国立大学法人 東京大学 (1,942)
【Fターム(参考)】
[ Back to top ]