
新規タンパク質発現方法
【課題】目的タンパク質を大量に生産する哺乳動物細胞を短期間で作製する技術を提供すること。
【解決手段】哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を動物細胞に導入し、該細胞にメトトレキセートによる遺伝子増幅処理を施すことにより前記課題を解決する。
【解決手段】哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を動物細胞に導入し、該細胞にメトトレキセートによる遺伝子増幅処理を施すことにより前記課題を解決する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、哺乳動物細胞を宿主としてタンパク質を大量に生産する技術に関する。
【背景技術】
【0002】
従来、大腸菌、酵母、動物細胞などを宿主として、有用な物質を生産する技術が開発されており、生産された物質は医薬品や診断試薬など多くの分野で用いられている。生産される物質は主にタンパク質であるが、本来各生物が持っている野生型のタンパク質のみならず、遺伝子組み換え技術を用いて野生型のタンパク質をコードする遺伝子に変異などを導入することで、野生型よりも活性が向上した組み換えタンパク質や、野生型とは異なる機能が付加された組み換えタンパク質も種々生産されている(特許文献1および特許文献2参照)。
【0003】
種々のタンパク質をコードする遺伝子を宿主に導入し発現させてタンパク質を宿主で生産する方法が、タンパク質を工業的に生産する際に一般的に用いられている(以下、生産しようとするタンパク質を「目的タンパク質」、それをコードする遺伝子を「目的タンパク質発現遺伝子」と呼ぶことがある)。前記生産方法で用いられる宿主としては、大腸菌やバチルス属細菌といった細菌、酵母、カビ、昆虫細胞、動物細胞などから適宜選択されるが、従来は取り扱いの容易性や目的タンパク質の生産量が比較的多いという理由から大腸菌が使用されている。しかしながら、大腸菌を宿主として動物由来の目的タンパク質を生産しようとしても目的タンパク質の生産が認められないことや、生産が認められた場合でも、目的タンパク質が不溶化タンパク質として細胞内に蓄積される、または可溶性タンパク質として生産されているが生理活性を有さないタンパク質として生産される場合がある(特許文献3)。
【0004】
一方、酵母、昆虫細胞または動物細胞を宿主として動物由来の目的タンパク質を生産すると、高い確率で生理活性を保持したタンパク質として生産することができる(特許文献2参照)。しかしながら、酵母を宿主とした場合には、付加される糖鎖が哺乳動物細胞由来のものとは異なる場合があり、それが生産された目的タンパク質の活性に影響を及ぼす場合がある(特許文献4)。また、昆虫細胞を宿主とする場合には、目的タンパク質遺伝子を宿主に導入する際にウイルスを使用しなくてはならず、目的タンパク質精製時のウイルス除去に多大な労力を必要とする。これに対して動物細胞を宿主として動物由来の目的タンパク質を生産する場合は、付加される糖鎖はその目的タンパク質を本来修飾しているものとほぼ同様の糖鎖であり、付加される糖鎖が異なることによって生理活性が影響される可能性は低いという利点を有する。また目的タンパク質遺伝子の導入にウイルスを使用する必要がないため、その除去のために多大な労力をかける必要がなく、時間面でも費用面でも有利である。
【0005】
宿主に動物細胞を用いる場合は、前記のような利点を有する一方で、宿主に目的タンパク質発現遺伝子を導入しただけではその生産量が低いという課題がある。そこで、目的タンパク質の生産量を増加させるために、チャイニーズハムスター卵巣(CHO:Chinese Hamster Ovary)細胞等を宿主として、これに目的タンパク質遺伝子とジヒドロ葉酸還元酵素(DHFR:Dihydrofolate reductase)をコードする遺伝子を同時に導入し、DHFRの阻害剤であるメトトレキセート(MTX:Methotrexate)を培養液に加える方法が知られている(非特許文献1)。この方法は、培地中のMTX濃度を段階的に上げることにより、DHFR遺伝子を発現している(DHFRを生産している)細胞を選択し、かつ、DHRF遺伝子とともに目的タンパク質遺伝子を増幅して、結果的に目的タンパク質の生産量を高めるものであるが、操作を完了するまでに6か月以上の時間が必要である。
【0006】
また、哺乳動物複製開始領域(IR:Initiation Region)および核マトリックス結合領域(MAR:Matrix Attachment Region)を宿主に導入することにより、目的タンパク質遺伝子を増幅する方法も知られている(特許文献5)。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開平7−284394号公報(特許2694840号公報)
【特許文献2】特開平7−67665号公報(特許2511251号公報)
【特許文献3】特表2002−531086号公報
【特許文献4】特開平6−277086号公報(特許第3091851号公報)
【特許文献5】特開2003−245083号公報(特許第3755028号公報)
【非特許文献】
【0008】
【非特許文献1】R J Kaufman, L C Wasley, A J Spiliotes, S D Gossels, S A Latt, G R Larsen and R M Kay, "Coamplification and coexpression of human tissue−type plasminogen activator and murine dihydrofolate reductase sequences in Chinese hamster ovary cells", Mol Cell Biol. 1985 July; 5(7): 1750−1759.
【非特許文献2】Shimizu et al., "Amplification of Plasmids Containing a Mammalian Replication Initiation Region Is Mediated by Controllable Conflict between Replication and Transcription", Cancer Research 2003 vol. 63, p5281−5290.
【発明の概要】
【発明が解決しようとする課題】
【0009】
前述したように、動物細胞を宿主として動物由来の目的タンパク質を生産する場合は、生産された目的タンパク質は生理活性を保持しており、精製時のウイルス除去も必要がない。しかし、動物細胞を宿主として使用する場合、目的タンパク質の生産量は大腸菌を宿主として使用する場合と比較して低い。そのため医薬や診断薬に使用することを目的として動物細胞で目的タンパク質を生産するためには、その生産量を高くすることが必要である。また、医薬や診断薬の供給の観点からは、必要な目的タンパク質を短期間で大量に生産することも必要になる。
【0010】
そこで本発明の課題は、目的タンパク質を大量に生産する哺乳動物細胞を短期間で作製する技術を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、哺乳動物複製開始領域(IR)、核マトリックス結合領域(MAR)、ジヒドロ葉酸還元酵素(DHFR)発現遺伝子群および目的タンパク質発現遺伝子群の4つの要素を哺乳動物細胞に導入することにより、従来のDHFRおよびMTXを利用した遺伝子増幅法や、IRおよびMARを利用した遺伝子増幅法の水準はもとより、これらを併用したときに普通に期待される水準よりも著しく早くかつ大量に、目的タンパク質を生産することができることを見出し、本発明を完成させるに至った。
【0012】
すなわち、前記課題を解決するためになされた本発明は、哺乳動物複製開始領域(IR)、核マトリックス結合領域(MAR)、ジヒドロ葉酸還元酵素(DHFR)発現遺伝子群および目的タンパク質発現遺伝子群を含む哺乳動物細胞をメトトレキセート(MTX)処理する工程を含むことを特徴とする、哺乳動物細胞の製造方法を提供する。
【0013】
典型的には、前記メトトレキセート処理する工程は、段階的に濃度を上げながら、メトトレキセートを含有する培地中で前記哺乳動物細胞を培養する複数の段階を含む。
たとえば、前記メトトレキセート処理する工程は、少なくとも、メトトレキセートを5〜25nMの濃度で含有する培地中で前記哺乳動物細胞を培養する第一段階、およびメトトレキセートを第一段階の5〜10倍の濃度で含有する培地中で前記哺乳動物細胞を培養する第二段階を含むことができる。
【0014】
前記哺乳動物細胞の製造方法は、通常さらに、前記哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む哺乳動物細胞を作製するために、あらかじめ、哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む、単一または複数種のベクターを哺乳動物細胞に導入して形質転換する工程を含む。
【0015】
前記哺乳動物複製起点としては、たとえば、c−myc遺伝子座複製起点、ジヒドロ葉酸還元酵素遺伝子座複製起点、またはβ−グロビン遺伝子座複製起点に由来するものが挙げられる。
【0016】
前記マトリックス結合領域としては、たとえば、Igκ遺伝子座マトリックス結合領域、SV40初期領域マトリックス結合領域、またはジヒドロ葉酸還元酵素マトリックス結合領域に由来するものが挙げられる。
【0017】
前記目的タンパク質としては、たとえば、受容体、抗体又は蛍光タンパク質が挙げられる。
上記の哺乳動物細胞の製造方法は、より具体的には、たとえば、以下の順序で行われる(1)から(4)の各工程を含むものである。
(1)哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を動物細胞に導入する工程、
(2)前記導入工程を経た哺乳動物細胞を培養し、目的タンパク質を発現している細胞を選択し単離する工程、
(3)前記単離工程を経た細胞を5〜25nMのメトトレキセートを含む培地に移し、生存可能な細胞を選択する工程、および、
(4)前記選択工程を経た細胞を125〜500nMのメトトレキセートを含む培地に移し、遺伝子が増幅された細胞を選択する工程。
【0018】
そして本発明は、上記の製造方法により得られた哺乳動物細胞を用いて目的タンパク質を製造する方法を提供する。
別の側面において、本発明は、上記のような哺乳動物の製造方法に用いられる、哺乳動物複製開始領域(IR)、核マトリックス結合領域(MAR)、ジヒドロ葉酸還元酵素(DHFR)発現遺伝子群および目的タンパク質発現遺伝子群を含む(が導入された)哺乳動物細胞を提供する。また、本発明は、この哺乳動物細胞にメトトレキセート(MTX)処理をして得られた哺乳動物細胞、すなわち上記の哺乳動物細胞の製造方法により得られた哺乳動物細胞を提供する。さらに本発明は、哺乳動物複製開始領域(IR)、核マトリックス結合領域(MAR)、ジヒドロ葉酸還元酵素(DHFR)発現遺伝子群および目的タンパク質発現遺伝子群を含む単一のベクターを提供する。
【0019】
以下、本発明を詳細に説明する。
IRおよびMARは、両者の組み合わせにより哺乳類細胞内で目的タンパク質発現遺伝子群が増幅し、それが娘細胞へ分離・分配されるものであればどのようなものでもよい。好ましいIRとしては、EBウイルス潜在複製起点(EBV latnt origin)、c−myc遺伝子座複製起点、DHFR遺伝子座複製起点、β−グロビン遺伝子座複製起点等に代表される複製起点に由来するものが挙げられる。好ましいMARとしては、Igκ遺伝子座MAR(AR1)、SV40初期領域MAR、DHFR遺伝子座MAR、等に代表されるMARに由来するものが挙げられる。
【0020】
本発明における「DHFR発現遺伝子群」または「目的タンパク質発現遺伝子群」は、DHFRまたは目的タンパク質をコードする遺伝子を発現してDHFRまたは目的タンパク質を生産するための塩基配列であり、少なくとも宿主で機能するプロモーターおよびその下流に接続されたDHFRまたは目的タンパク質をコードする遺伝子を含むもので、更にその下流にポリアデニレーションシグナルが接続されていてもよい。DHFR発現遺伝子群および目的タンパク質発現遺伝子群を構成するプロモーターは、これらの遺伝子を宿主において発現させることができれば特に制限はない。例えば、CHO細胞、アフリカミドリザル腎繊維芽由来細胞(COS7細胞)、ヒト子宮頸癌由来細胞(Hela細胞)、マウス胎児皮膚由来細胞(NIH3T3)等の哺乳動物由来の細胞を宿主とする場合は、プロモーターとしてSV40初期および後期プロモーター、CMVプロモーター等を使用することが例示できる。
【0021】
宿主細胞は、CHO細胞、COS7細胞、Hela細胞、NIH3T3細胞といった哺乳動物由来の細胞であって、DHFRの生産能力を著しく欠くかまたは欠損した、DHFR欠損株である。例えばCHO細胞のDHFR欠損株は樹立され、極めて容易に入手可能であるし、他の細胞についても、例えばMTXを含む培地で培養を行う等することにより、かかる細胞は選択することが可能である。
【0022】
本発明に係るIR、MAR、DHFR発現遺伝子群および目的タンパク質発現遺伝子群(以下、これら4つの配列または遺伝子群のそれぞれを「要素」と呼ぶことがある)を含む哺乳動物細胞は、例えば、これら4つの要素を含む単一又は複数種のベクターを取り込んで細胞内に保持した状態の哺乳動物細胞や、これら4つの要素を染色体に組み込んで保持した状態の哺乳動物細胞等、種々の形態の細胞を包含する。この哺乳動物細胞は、IR、MAR、DHFR発現遺伝子群および目的タンパク質発現遺伝子群の4つの要素を哺乳動物細胞に導入することにより作製することができる。IR、MARおよび目的タンパク質発現遺伝子群は同時に細胞に導入し(特許文献5)、DHFR発現遺伝子群は、IR、MARおよび目的タンパク質発現遺伝子群の導入に先立って、同時に又はIR、MARおよび目的タンパク発現遺伝子群を導入した後に、細胞に導入する。
【0023】
IR、MARおよび目的タンパク質発現遺伝子群は同一ベクターに載った形態であっても、異なるベクターに載った形態であってもよく、同時に細胞に導入されればそれ以外には特に制限はない。IR、MARおよび目的タンパク質発現遺伝子群を同一のベクターに載せる場合、どの要素が上流であるか等は問わない。異なるベクターに載った形態として、IR、MARおよび目的タンパク質発現遺伝子のいずれか2以上を同一ベクターに載せ、残る遺伝子を他のベクターに載せることや、これら3つの要素を異なる3つのベクターに載せることを例示できる。2つの要素を同一ベクターに載せる場合、どちらの要素が上流であるかは問わない。
【0024】
DHFR発現遺伝子群をIR等と同時に導入する場合、例えば、4つの要素の全てを同一のベクターに載せることや、4つの要素の2または3を同一ベクターにのせ、残る要素を異なるベクターに載せて同時に導入することが例示できる。IR等と同時に導入しない場合、DHFR発現遺伝子群のみを載せたベクターを作製し、使用すればよい。なお、本発明は、目的タンパク質発現遺伝子群の増幅のために上記の哺乳動物細胞をMTX処理することで、目的タンパク質を大量に生産する細胞を提供するものである。従って、上記においてDHFR発現遺伝子群をIR等と同時に導入しない場合、DHFR発現遺伝子群は、MTX処理に先立って導入する。4つの要素が導入され、哺乳動物細胞がこれらを保持している場合には、MTX処理によって目的タンパク質を大量に生産することになる。
【0025】
前記例示した4つの要素を含む単一のベクターを用いると、4つの要素が接続された状態で宿主に保持される確率が高くなり、その結果、MTX処理によってDHFR発現遺伝子群の増幅とともに目的タンパク質発現遺伝子群が増幅される確率も向上できると考えられるため、本発明の態様として好ましい。もっとも、4つの要素のうちの1以上を含む複数種のベクターを用いる場合、例えばIR、MARおよび目的タンパク質発現遺伝子群を含む第1のベクターと、DHFR発現遺伝子群を含む第2のベクターの2種類を用いると各要素が哺乳動物細胞において近接した状態に保持され、MTX処理によってDHFR発現遺伝子群の増幅とともに目的タンパク質発現遺伝子群が増幅される確率において劣る可能性があったとしても、その後の細胞増殖を考慮すると、特に遜色なく目的タンパク質を大量に生産するという本発明の効果を達成することができる。なお、4つの要素を含む単一のベクターについて更に説明すると、4つの要素の位置関係に制限はなく、いずれの要素が上流に位置してもよく、またその転写の向きも制限がない。4つの遺伝子は近接していることが好ましく、例えば実施例で使用したように、各要素を数十から数百塩基程度離間した状態で含むものを例示できる。
【0026】
上記した4つの要素を含むベクターを導入(トランスフェクト)した哺乳動物細細胞を、好ましくはまずそれを培養して目的タンパク質を発現している細胞を選択し単離してから、MTX処理することにより、DHFR発現遺伝子群が導入された細胞を選択することができる。そして培地中のMTX濃度を段階的に上げることによりDHFR発現遺伝子群により産生されるDHFRは阻害されるが、その際にDHFR発現遺伝子群が増幅される。このDHFR発現遺伝子群の増幅の際にDHFR発現遺伝子群に近接して保持された目的タンパク質発現遺伝子群も同時に増幅される。MTX濃度の段階的な上げ方は、一般的には、まず培地中に5〜25nMの濃度で添加し、少なくとももう一段階、必要に応じてさらにもう一段階またはそれ以上の段階にわたって、5〜10倍ずつ濃度を上昇させる方法を用いればよい。各濃度のMTXを添加した培地で、細胞が生育しコロニーを作った段階で、MTX濃度を上げてやればよい。メトトレキセートの濃度を上昇させる段階数、濃度の上昇幅、各段階における培養日数、目的タンパク質の製造に用いる哺乳動物細胞を選別するための最終的な濃度などの条件は、哺乳動物細胞ないし目的タンパク質の製造効率が所望のものとなるよう適宜調整することができるが、本発明の方法を用いれば、全体的な工程期間を従来の遺伝子増幅方法よりも短縮することができる。より具体的な手順としては、例えば、単離した細胞をまず5〜25nMのMTXを含む培地に移して生存可能な細胞を選択し、必要であれば25〜125nMのMTXを含む培地に移して培養する段階を経た後、125〜500nMのMTXを含む培地に移して遺伝子が増幅された細胞を選択することができる。また、各濃度のMTXを添加した培地で生育した細胞を、一度モノクローン化した後、遺伝子増幅数の高い細胞、すなわち目的タンパク質の生産量の多い細胞を単離し、当該細胞に対して更にMTX濃度を上げてもよいし、モノクローン化せずにポリクローンのままMTX濃度を上げてもよい。
【0027】
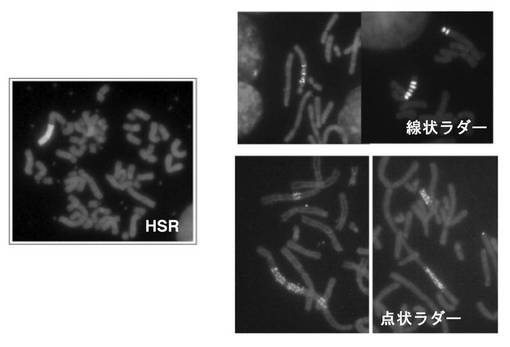
本発明の方法により増幅したDHFR発現遺伝子群および目的タンパク質発現遺伝子群は、染色体外遺伝因子であるDM(Double Minutes)または染色体上のHSR(Homogeneously Staining Resion)いずれかの構造で局在しており、FISHなどの手法を用いて観察することができる。HSRはさらに、均質なHSR、線状ラダー、点状ラダーに分類される(図5参照)。このような遺伝子増幅に関わるDMおよびHSRは、IRおよびMARのみを用いた遺伝子増幅法においても発生し、BFB(Breakage-Fusion-Bridge)cycleにより巨大なHSRが形成されることなどが知られている(たとえば前記非特許文献2参照)。本発明では、従来の遺伝子増幅法よりも早く、より長い点状ラダーHSR(図5参照)が形成されるようになり、これと対応して、より早く、より高いレベルの目的タンパク質の産生が達成される。
【0028】
本発明の目的タンパク質の製造方法には、目的タンパク質の産生量を向上させるための公知の手段をさらに組み合わせてもよい。たとえば、ヒストン脱アセチル化阻害剤(Sodium Butyrateなど)を用いて目的タンパク質発現遺伝子のエピジェネティックなメチル化を阻害することにより、目的タンパク質の産生量を向上させる方法が知られている。本発明の目的タンパク質の製造方法において、IR、MAR、DHFRおよび目的タンパク質発現遺伝子群を導入した動物細胞を培養する際に、ヒストン脱アセチル化阻害剤を添加することにより、目的タンパク質の産生量をより一層向上させることができる。
【0029】
宿主である哺乳動物細胞に遺伝子を導入し形質転換する方法は、ベクターの構成や宿主に応じて適宜選択することができるが、例えばエレクトロポーレーション法、リポフェクション法、リン酸カルシウム法などによるトランスフェクションを例示することができる。たとえば、複数種のベクターを用いる場合は、リポフェクション法が好ましい方法として例示できる。MTX処理を開始する前に行われる、ベクターにより哺乳動物細胞が形質転換されたかどうかの選択は、たとえば、アミノグリコシド3’−ホスホトランスフェラーゼ(カナマイシン抵抗性遺伝子)、ネオマイシンホスホトランスフェラーゼ遺伝子(ネオマイシン抵抗性遺伝子)、ヒグロマイシンBホスホトランスフェラーゼ遺伝子(ヒグロマイシン抵抗性遺伝子)、ブラスティサイジンSデアミナーゼ遺伝子(ブラスティサイジン抵抗性遺伝子)などの薬剤耐性遺伝子、あるいはグルタミンシンセターゼ遺伝子、緑色蛍光タンパク質遺伝子等を選択マーカーとして用いる、公知の一般的な方法に従って行えばよい。あるいは、これらの選択マーカーを用いる代わりに、リボヌクレオチドおよびデオキシリボヌクレオチドを含有しない培地中で哺乳動物細胞(元来DHFR欠損株)を培養することにより、ジヒドロ葉酸レダクターゼ遺伝子(DHFR)が導入された哺乳動物細胞を選択することもできる。
【0030】
形質転換により得られた細胞の培養は、当該細胞に適切な条件で行なえばよい。目的タンパク質発現遺伝子の発現により生産された目的タンパク質は、当該タンパク質が細胞外へ分泌する場合は培養上清から遠心分離などで回収し、適当な方法により精製することができる。生産された目的タンパク質が細胞内に蓄積する場合は、細胞を破砕する等の適当な方法により抽出、精製することができる。
【0031】
本発明の目的タンパク質を大量に生産する方法を用いて発現可能な目的タンパク質は、発現させる目的タンパク質をコードするポリヌクレオチド配列が既知であるものであれば特に限定はなく、例えば、抗体(H鎖およびL鎖)、IL−6(インターロイキン6)などのサイトカイン、エリスロポエチン、インターフェロン、蛍光タンパク質などがあげられる。特に近年注目されている抗体医薬品の製造のために大量に必要とされる、特定の疾患(たとえばがんやリウマチ)に関与する抗原(たとえば細胞表面の受容体)を標的とする抗体は、本発明による生産効率の向上が期待される目的タンパク質である。
【発明の効果】
【0032】
本発明では、IR、MAR、DHFR発現遺伝子群および目的タンパク質発現遺伝子群を動物細胞に導入し、該細胞にMTXによる遺伝子増幅処理を施すことにより、目的タンパク質を大量に生産する哺乳動物細胞を短期間で作製することができる。
【0033】
そして、本発明により医薬品などの有用な目的タンパク質を大量に生産する細胞を作製すると、実施例に示すように、IRおよびMARのみを用いたり、MTXによる遺伝子増幅処理のみを施したりする、従来の目的タンパク質の製造方法と比較して、目的タンパク質の生産量を飛躍的に(たとえば実施例に示すように10倍以上に)増加し、短時間で大量に生産することができる。したがって、従来の目的タンパク質の製造方法と比較し、短時間かつ低コストで目的タンパク質を製造することができる。
【図面の簡単な説明】
【0034】
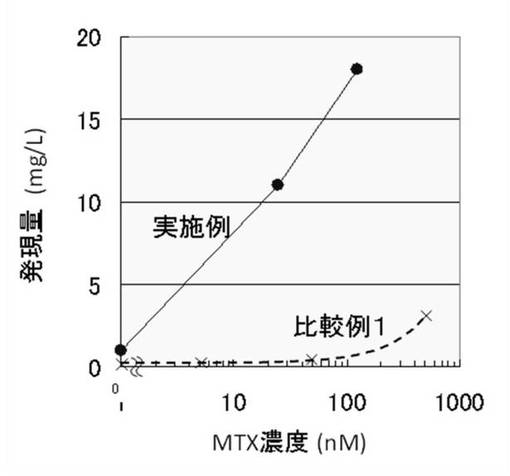
【図1】図1は、実施例1および比較例1−1において、ステップ1から5により培養上清中に発現したFcRの発現量を示すグラフである。
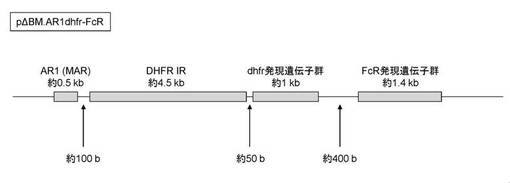
【図2】図2は、実施例1で作製したベクターpΔBM.AR1dhfr−FcRの概略図である。
【図3】図3は、実施例2,比較例2,実施例3,比較例3−1〜3−3で用いたプラスミドA(pΔBN AR1 Psv40−dhfr)、B(pSFV−V Psv40−dhfr)、C(pCMV−d2EGFR)およびD(pMycLH)のマップである。
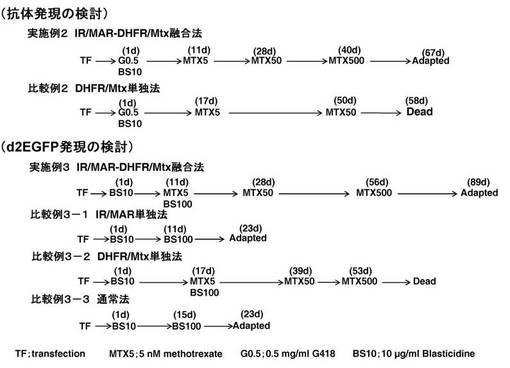
【図4】図4は、実施例2および比較例2(上)、ならびに実施例3および比較例3−1〜3−3(下)の選択法のステップ(形質転換後の日数および使用薬剤)を示す図である。
【図5】図5は、実施例2でFISHにより観察した遺伝子増幅構造の代表例の写真である。
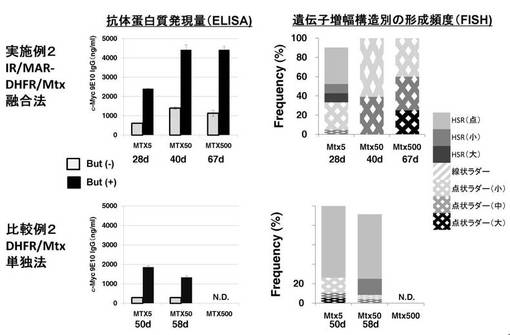
【図6】図6は、実施例2および比較例2における、FISHにより観察した遺伝子増幅構造別の細胞集団中の形成頻度(右)およびELISAにより測定した抗体タンパク質発現量(左)の結果を示すグラフである。図6(左)について、But (-)およびBut (+)は、それぞれSodium Butyrateを添加しなかった場合および添加した場合である。
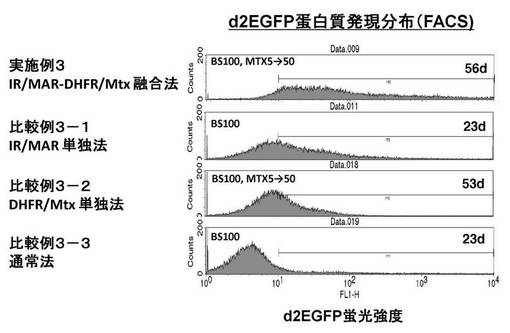
【図7】図7は、実施例3および比較例3−1〜3−3における、FACSで測定したd2EGFPタンパク質発現分布の結果を示すグラフである。
【発明を実施するための形態】
【実施例】
【0035】
以下、本発明をさらに詳細に説明するために実施例を示すが、本発明は実施例に限定されるものではない。
[実施例1]
ステップ1:IR、MAR、DHFR発現遺伝子群およびFcR(可溶性ヒト型FcレセプターFcγRI)発現遺伝子群をコードするポリヌクレオチドを含むベクターの作製
【0036】
(1)pΔB.AR1(非特許文献2)のBamH1−MluIの間に、KpnI、AscI、SalI、SwaI、AsiSI、SbfI、BamHI、MluIからなるマルチクローニングサイトを導入し、pΔBM.AR1.MCSとした。該pΔBM.AR1.MCSを制限酵素BamHIおよびNruIで切断した。両切断末端をT4DNAポリメラーゼにより平滑化し、さらにアルカリフォスファターゼにより脱リン酸化し、pΔBM.AR1/NBb*とした。なお、得られたpΔBM.AR1/NBb*中、IRおよびMARはともにDHFR遺伝子座由来のものである。
【0037】
(2)pECEFcRdhfr(特許文献:特開2009−278948)を制限酵素BamHIで切断し、電気泳動によりDHFR−FcR発現遺伝子群を含んだバンドを切り出した。前記ヌクレオチドをBKLキット(タカラバイオ社製)により平滑化およびリン酸化しdhfr−FcRとした。
【0038】
(3)(1)で調製したpΔBM.AR1/NBb*と(2)で調製したdhfr−FcRを、DNA Ligation Kit<Mighty Mix>(タカラバイオ社製)を用いて連結することで、FcR発現ベクター(pΔBM.AR1dhfr−FcR)を作製した。
【0039】
以上の実施例では、DHFR(dhfr)発現遺伝子群、目的タンパク質(FcR)発現遺伝子群、IR及びMARの4つの要素を同一のベクターに載せているが、作製されたベクターpΔBM.AR1dhfr−FcRの詳細は図2に示した通りである。図2において、MAR(図中ではAR1)は約0.5kb、DHFR遺伝子座由来のIR(図中ではDHFR IR)は約4.6kb、dhfr発現遺伝子群は約1kb、FcR発現遺伝子群は約1.4kbであるが、MARとIRは約100b、IRとdhfr発現遺伝子群は約50b離れているため、これら4つの遺伝子は、全長で約7.5kbとなる。
【0040】
ステップ2:pΔBM.AR1dhfr−FcRのCHO細胞への導入
(1)MEMAlpha培地(インビトロジェン社製)に10%FBS(Fetal Bovine Serum:JRH Biosciences社製)を加えた培地10mLで対数増殖期まで培養したCHO細胞に、ステップ1で作製したFcR発現ベクター(pΔBM.AR1dhfr−FcR)をエレクトロポーレーションシステムGenePulserXcell(バイオラッド社製)を用いて添付の資料に従いトランスフェクションした。
【0041】
(2)(1)の方法でトランスフェクションしたCHO細胞を、リボヌクレオシドおよびデオキシリボヌクレオチド不含のMEMAlpha培地(インビトロジェン社製)に10%透析FBS(Fetal Bovine Serum:biowest社製)を加えた培地10mLで10日間培養した。
【0042】
ステップ3:FcR発現CHO細胞の単離
(1)ステップ2でトランスフェクションしたCHO細胞を、ステップ2−(2)で使用した培地で1細胞/穴になるように希釈し、96穴培養プレート(ファルコン社製)に200マイクロリットルずつ分注した。
【0043】
(2)10日間培養後、ヒトFcRを1μg/穴で固相化したマイクロタイタープレートを用いたELISA(Enzyme−Linked ImmunoSorbent Assay)により培養上清中のFcRの発現量を測定した。その中から、発現量が最も高く、単一コロニーとなっている細胞を選択し単離した。
【0044】
ステップ4:CHO細胞のメトトレキセートによる遺伝子増幅処理および細胞の単離
ステップ3で単離したCHO細胞をステップ2−(2)で使用した培地で対数増殖期まで培養後、5x104細胞を10cmプレート(ファルコン社製)に撒いた。24時間後、25nMメトトレキセート(シグマ社製)を含むステップ2−(2)で使用した培地10mLで15日間培養した。生き残った細胞をステップ3と同様の方法により単離した。さらに、単離した上記CHO細胞を、125nMメトトレキセートを用い上記と同様の方法で、遺伝子増幅処理および細胞の単離を行った。
【0045】
ステップ5:FcR発現量の測定
(1)ステップ4で単離したCHO細胞をステップ2−(2)で使用した培地で対数増殖期後期まで培養後、新たに培地を交換した。3日間培養後、上清を回収した。
【0046】
(2)前記上清を、ヒトFcRを1μg/穴で固相化したマイクロタイタープレートを用いたELISA(Enzyme−Linked ImmunoSorbent Assay)により、上清中に分泌されたFcRの量を測定した。
【0047】
[比較例1−1]
上記ステップ2−(1)において、陰性コントロールとして、ステップ1−(2)で使用したIR(DHFR)およびMAR(AR1)を含まないFcR発現ベクター(pECEFcRdhfr)を(1)と同様の方法によりトランスフェクションするようにしたこと以外は上記実施例1と同様にして、FcR発現量を測定した。
【0048】
[比較例1−2]
上記ステップ4において、25nMメトトレキセートを含まないステップ2−(2)で使用した培地10mLで15日間培養するようにしたこと以外は上記実施例1と同様にして、FcR発現量を測定した。
【0049】
結果
上記実施例1および比較例1−1〜1−2の結果を表1および図1に示す。表1に示す値は、ステップ1から5により培養上清中に発現したFcRの発現量を示す。図1のグラフは、ステップ1から5により培養上清中に発現したFcRの発現量を、実施例1と比較例1−1について示す。
【0050】
IRおよびMARとFcR発現遺伝子を動物細胞に同時に導入し、該細胞にメトトレキセートによる遺伝子増幅処理を施した細胞(実施例)は、FcRが大量に生産されたことがわかる。一方、IRおよびMARをFcR発現遺伝子と同時に導入していない細胞(比較例1−1)は、従来の方法による動物細胞による目的タンパク質発現と同程度の低い発現量であることがわかる。また、一方、IRおよびMARをFcR発現遺伝子と同時に導入したのみで、メトトレキセートによる遺伝子増幅処理を施していない細胞(比較例1−2)も、FcRの発現量は、上記比較例1よりは高いものの上記実施例より著しく低い発現量にとどまった。以上より、IRおよびMARと目的タンパク質発現遺伝子を動物細胞に同時に導入し、該細胞にメトトレキセートによる遺伝子増幅処理を施すことにより、目的タンパク質を大量に生産することが可能であることがわかる。
【0051】
【表1】

[実施例2](抗体発現の検討:IR/MAR−DHFR/MTX融合法)
図3に示す、DHFR(dhfr)タンパク質発現遺伝子群、DHFR遺伝子座由来のIR及びMAR(AR1)を含むプラスミドA(pΔBN AR1 Psv40−dhfr)と、目的タンパク質(抗c−myc抗体のL鎖であるMycLおよび同H鎖であるMycH)発現遺伝子群を含むプラスミドD(pMycLH)を用いて、Lipofectamin2000(インビトロジェン社製)によりCHO−DG44細胞に形質導入した。続いて、図4(実施例2)に示すように、0.5 mg/ml G418および10 μg/ml Blasticidineで選択し(1d)、さらに5 nM methotrexate(11d)、50 nM methotrexate(28d)および500 nM methotrexate(40d)で選択を行った。各ステップの細胞から染色体標本を作製し、導入したプラスミド全長DNAをプローブとしてFISHを行い、遺伝子増幅構造別の形成頻度を調査した。同時に、各ステップの細胞を採取し、5×105cells/mLで96well plateに植え、Sodium Butyrateを添加した場合、添加しなかった場合それぞれについて、3日後に培養上清中の抗体タンパク質発現量をELISA法で決定した。
【0052】
なお、プラスミドAは、pDBN.AR1のBamHI/NruI断片(ヒグロマイシン抵抗性遺伝子を含む)を除き、その位置に、PCRにより増幅したSV40promoterとDHFRcoding配列を挿入することにより作製されたプラスミドであり、ブラストサイジンを用いた選択のためのブラストサイジン耐性遺伝子(Bsr)およびそのプロモーター(SR αPromoter)も含まれている。一方、プラスミドDには、G418を用いた選択のためのカナマイシン耐性遺伝子(Kan)も含まれている。これらの塩基配列は公知である。
【0053】
[比較例2](抗体発現の検討:DHFR/MTX単独法)
前記プラスミドAに代えて、図3に示す、DHFR(dhfr)発現遺伝子群を含むもののIR及びMARを含まないプラスミドB(pSFV−V Psv40−dhfr)を用いるよう変更し、また選択を、図4(比較例2)に示すように、0.5 mg/ml G418および10 μg/ml Blasticidine(1d)、5 nM methotrexate(17d)および50 nM methotrexate(50d)に変更した以外は、実施例2と同様の手順により、FISHによる遺伝子増幅構造別の形成頻度の調査、ならびにELISAによる抗体タンパク質発現量を行った。
【0054】
なお、プラスミドBは、pSFV‐VのBamHI/NruI断片(ヒグロマイシン抵抗性遺伝子を含む)を除き、その位置に、PCRにより増幅したSV40promoterとDHFRcoding配列を挿入することにより作製されたプラスミドであり、ブラストサイジン耐性遺伝子(Bsr)およびそのプロモーター(SR αPromoter)も含まれている。これらの塩基配列は公知である。
【0055】
結果
上記実施例2および比較例2における、FISHにより観察した遺伝子増幅構造の代表例の写真を図5に、遺伝子増幅構造別の形成頻度(FISH)および抗体タンパク質発現量(ELISA)の結果をそれぞれ図6の右および左に示す。従来法(DHFR/MTX単独法)に比べて、本発明の方法(IR/MAR−DHFR/MTX融合法)では、より早く、より長い点状ラダーHSRが形成された。これと対応して、より早く、より高いレベルの抗体産生が検出された。Sodium Butyrateを添加することにより、より一層抗体産生レベルを向上させることができた。
【0056】
[実施例3](d2EGFP発現の検討:IR/MAR−DHFR/MTX融合法)
図3に示す、DHFR(dhfr)発現遺伝子群、IR(DHFR)及びMAR(AR1)含むプラスミドA(pΔBN AR1 Psv40−dhfr)と、目的タンパク質(d2EGFP)発現遺伝子群を含むプラスミドC(pCMV−d2EGFR、N.Shimizu, et al.(2007) J.Cell.Biochem., vol.102, p515-529)を用いて、Lipofectamin2000によりCHO−DG44細胞に形質導入した。続いて、図4(実施例3)に示すように、10 μg/ml Blasticidineで選択し(1d)、さらに5 nM methotrexateおよび100 μg/ml Blasticidine(11d)、50 nM methotrexate(28d)および500 nM methotrexate(56d)で選択を行った。その後、培養した細胞集団のd2EGFPタンパク質発現分布をFACS(fluorescent-activated cell sorter)を用いて測定した。
【0057】
[比較例3−1](d2EGFP発現の検討:IR/MAR単独法)
形質導入したCHO−DG44細胞の選択を、図4(比較例3−1)に示すように、10 μg/ml Blasticidine(1d)および100 μg/ml Blasticidine(11d)に変更した以外は、実施例2と同様の手順により、d2EGFPタンパク質発現分布をFACSを用いて測定した。
【0058】
[比較例3−2](d2EGFP発現の検討:DHFR/MTX単独法)
前記プラスミドAに代えて、図3に示す、DHFR(dhfr)発現遺伝子群を含むもののIR及びMARを含まないプラスミドB(pSFV−V Psv40−dhfr)を用いるよう変更し、また形質導入したCHO−DG44細胞の選択を、図4(比較例3−2)に示すように、10 μg/ml Blasticidine(1d)、5 nM methotrexateおよび100 μg/ml Blasticidine(17d)、50 nM methotrexate(39d)および500 nM methotrexate(53d)に変更した以外は、実施例3と同様の手順により、d2EGFPタンパク質発現分布をFACSを用いて測定した。
【0059】
[比較例3−3](d2EGFP発現の検討:通常法)
前記プラスミドAに代えて、図3に示す、DHFR(dhfr)発現遺伝子群を含むもののIR及びMARを含まないプラスミドB(pSFV−V Psv40−dhfr)を用いるよう変更し、また形質導入したCHO−DG44細胞の選択を図4(比較例3−3)に示すように、10 μg/ml Blasticidine(1d)および100 μg/ml Blasticidine(15d)に変更した以外は、実施例3と同様の手順により、d2EGFPタンパク質発現分布をFACSを用いて測定した。
【0060】
結果
上記実施例3および比較例3−1〜3−3における、FACSにより測定したd2EGFPタンパク質発現分布の結果を図7に示す。従来法(IR/MAR単独法、DHFR/MTX単独法、通常法)に比べて、本発明の方法(IR/MAR−DHFR/MTX融合法)では、d2EGFPタンパク質の発現量の多い細胞が細胞集団中により高頻度で出現することが示されている。
【技術分野】
【0001】
本発明は、哺乳動物細胞を宿主としてタンパク質を大量に生産する技術に関する。
【背景技術】
【0002】
従来、大腸菌、酵母、動物細胞などを宿主として、有用な物質を生産する技術が開発されており、生産された物質は医薬品や診断試薬など多くの分野で用いられている。生産される物質は主にタンパク質であるが、本来各生物が持っている野生型のタンパク質のみならず、遺伝子組み換え技術を用いて野生型のタンパク質をコードする遺伝子に変異などを導入することで、野生型よりも活性が向上した組み換えタンパク質や、野生型とは異なる機能が付加された組み換えタンパク質も種々生産されている(特許文献1および特許文献2参照)。
【0003】
種々のタンパク質をコードする遺伝子を宿主に導入し発現させてタンパク質を宿主で生産する方法が、タンパク質を工業的に生産する際に一般的に用いられている(以下、生産しようとするタンパク質を「目的タンパク質」、それをコードする遺伝子を「目的タンパク質発現遺伝子」と呼ぶことがある)。前記生産方法で用いられる宿主としては、大腸菌やバチルス属細菌といった細菌、酵母、カビ、昆虫細胞、動物細胞などから適宜選択されるが、従来は取り扱いの容易性や目的タンパク質の生産量が比較的多いという理由から大腸菌が使用されている。しかしながら、大腸菌を宿主として動物由来の目的タンパク質を生産しようとしても目的タンパク質の生産が認められないことや、生産が認められた場合でも、目的タンパク質が不溶化タンパク質として細胞内に蓄積される、または可溶性タンパク質として生産されているが生理活性を有さないタンパク質として生産される場合がある(特許文献3)。
【0004】
一方、酵母、昆虫細胞または動物細胞を宿主として動物由来の目的タンパク質を生産すると、高い確率で生理活性を保持したタンパク質として生産することができる(特許文献2参照)。しかしながら、酵母を宿主とした場合には、付加される糖鎖が哺乳動物細胞由来のものとは異なる場合があり、それが生産された目的タンパク質の活性に影響を及ぼす場合がある(特許文献4)。また、昆虫細胞を宿主とする場合には、目的タンパク質遺伝子を宿主に導入する際にウイルスを使用しなくてはならず、目的タンパク質精製時のウイルス除去に多大な労力を必要とする。これに対して動物細胞を宿主として動物由来の目的タンパク質を生産する場合は、付加される糖鎖はその目的タンパク質を本来修飾しているものとほぼ同様の糖鎖であり、付加される糖鎖が異なることによって生理活性が影響される可能性は低いという利点を有する。また目的タンパク質遺伝子の導入にウイルスを使用する必要がないため、その除去のために多大な労力をかける必要がなく、時間面でも費用面でも有利である。
【0005】
宿主に動物細胞を用いる場合は、前記のような利点を有する一方で、宿主に目的タンパク質発現遺伝子を導入しただけではその生産量が低いという課題がある。そこで、目的タンパク質の生産量を増加させるために、チャイニーズハムスター卵巣(CHO:Chinese Hamster Ovary)細胞等を宿主として、これに目的タンパク質遺伝子とジヒドロ葉酸還元酵素(DHFR:Dihydrofolate reductase)をコードする遺伝子を同時に導入し、DHFRの阻害剤であるメトトレキセート(MTX:Methotrexate)を培養液に加える方法が知られている(非特許文献1)。この方法は、培地中のMTX濃度を段階的に上げることにより、DHFR遺伝子を発現している(DHFRを生産している)細胞を選択し、かつ、DHRF遺伝子とともに目的タンパク質遺伝子を増幅して、結果的に目的タンパク質の生産量を高めるものであるが、操作を完了するまでに6か月以上の時間が必要である。
【0006】
また、哺乳動物複製開始領域(IR:Initiation Region)および核マトリックス結合領域(MAR:Matrix Attachment Region)を宿主に導入することにより、目的タンパク質遺伝子を増幅する方法も知られている(特許文献5)。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開平7−284394号公報(特許2694840号公報)
【特許文献2】特開平7−67665号公報(特許2511251号公報)
【特許文献3】特表2002−531086号公報
【特許文献4】特開平6−277086号公報(特許第3091851号公報)
【特許文献5】特開2003−245083号公報(特許第3755028号公報)
【非特許文献】
【0008】
【非特許文献1】R J Kaufman, L C Wasley, A J Spiliotes, S D Gossels, S A Latt, G R Larsen and R M Kay, "Coamplification and coexpression of human tissue−type plasminogen activator and murine dihydrofolate reductase sequences in Chinese hamster ovary cells", Mol Cell Biol. 1985 July; 5(7): 1750−1759.
【非特許文献2】Shimizu et al., "Amplification of Plasmids Containing a Mammalian Replication Initiation Region Is Mediated by Controllable Conflict between Replication and Transcription", Cancer Research 2003 vol. 63, p5281−5290.
【発明の概要】
【発明が解決しようとする課題】
【0009】
前述したように、動物細胞を宿主として動物由来の目的タンパク質を生産する場合は、生産された目的タンパク質は生理活性を保持しており、精製時のウイルス除去も必要がない。しかし、動物細胞を宿主として使用する場合、目的タンパク質の生産量は大腸菌を宿主として使用する場合と比較して低い。そのため医薬や診断薬に使用することを目的として動物細胞で目的タンパク質を生産するためには、その生産量を高くすることが必要である。また、医薬や診断薬の供給の観点からは、必要な目的タンパク質を短期間で大量に生産することも必要になる。
【0010】
そこで本発明の課題は、目的タンパク質を大量に生産する哺乳動物細胞を短期間で作製する技術を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、哺乳動物複製開始領域(IR)、核マトリックス結合領域(MAR)、ジヒドロ葉酸還元酵素(DHFR)発現遺伝子群および目的タンパク質発現遺伝子群の4つの要素を哺乳動物細胞に導入することにより、従来のDHFRおよびMTXを利用した遺伝子増幅法や、IRおよびMARを利用した遺伝子増幅法の水準はもとより、これらを併用したときに普通に期待される水準よりも著しく早くかつ大量に、目的タンパク質を生産することができることを見出し、本発明を完成させるに至った。
【0012】
すなわち、前記課題を解決するためになされた本発明は、哺乳動物複製開始領域(IR)、核マトリックス結合領域(MAR)、ジヒドロ葉酸還元酵素(DHFR)発現遺伝子群および目的タンパク質発現遺伝子群を含む哺乳動物細胞をメトトレキセート(MTX)処理する工程を含むことを特徴とする、哺乳動物細胞の製造方法を提供する。
【0013】
典型的には、前記メトトレキセート処理する工程は、段階的に濃度を上げながら、メトトレキセートを含有する培地中で前記哺乳動物細胞を培養する複数の段階を含む。
たとえば、前記メトトレキセート処理する工程は、少なくとも、メトトレキセートを5〜25nMの濃度で含有する培地中で前記哺乳動物細胞を培養する第一段階、およびメトトレキセートを第一段階の5〜10倍の濃度で含有する培地中で前記哺乳動物細胞を培養する第二段階を含むことができる。
【0014】
前記哺乳動物細胞の製造方法は、通常さらに、前記哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む哺乳動物細胞を作製するために、あらかじめ、哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む、単一または複数種のベクターを哺乳動物細胞に導入して形質転換する工程を含む。
【0015】
前記哺乳動物複製起点としては、たとえば、c−myc遺伝子座複製起点、ジヒドロ葉酸還元酵素遺伝子座複製起点、またはβ−グロビン遺伝子座複製起点に由来するものが挙げられる。
【0016】
前記マトリックス結合領域としては、たとえば、Igκ遺伝子座マトリックス結合領域、SV40初期領域マトリックス結合領域、またはジヒドロ葉酸還元酵素マトリックス結合領域に由来するものが挙げられる。
【0017】
前記目的タンパク質としては、たとえば、受容体、抗体又は蛍光タンパク質が挙げられる。
上記の哺乳動物細胞の製造方法は、より具体的には、たとえば、以下の順序で行われる(1)から(4)の各工程を含むものである。
(1)哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を動物細胞に導入する工程、
(2)前記導入工程を経た哺乳動物細胞を培養し、目的タンパク質を発現している細胞を選択し単離する工程、
(3)前記単離工程を経た細胞を5〜25nMのメトトレキセートを含む培地に移し、生存可能な細胞を選択する工程、および、
(4)前記選択工程を経た細胞を125〜500nMのメトトレキセートを含む培地に移し、遺伝子が増幅された細胞を選択する工程。
【0018】
そして本発明は、上記の製造方法により得られた哺乳動物細胞を用いて目的タンパク質を製造する方法を提供する。
別の側面において、本発明は、上記のような哺乳動物の製造方法に用いられる、哺乳動物複製開始領域(IR)、核マトリックス結合領域(MAR)、ジヒドロ葉酸還元酵素(DHFR)発現遺伝子群および目的タンパク質発現遺伝子群を含む(が導入された)哺乳動物細胞を提供する。また、本発明は、この哺乳動物細胞にメトトレキセート(MTX)処理をして得られた哺乳動物細胞、すなわち上記の哺乳動物細胞の製造方法により得られた哺乳動物細胞を提供する。さらに本発明は、哺乳動物複製開始領域(IR)、核マトリックス結合領域(MAR)、ジヒドロ葉酸還元酵素(DHFR)発現遺伝子群および目的タンパク質発現遺伝子群を含む単一のベクターを提供する。
【0019】
以下、本発明を詳細に説明する。
IRおよびMARは、両者の組み合わせにより哺乳類細胞内で目的タンパク質発現遺伝子群が増幅し、それが娘細胞へ分離・分配されるものであればどのようなものでもよい。好ましいIRとしては、EBウイルス潜在複製起点(EBV latnt origin)、c−myc遺伝子座複製起点、DHFR遺伝子座複製起点、β−グロビン遺伝子座複製起点等に代表される複製起点に由来するものが挙げられる。好ましいMARとしては、Igκ遺伝子座MAR(AR1)、SV40初期領域MAR、DHFR遺伝子座MAR、等に代表されるMARに由来するものが挙げられる。
【0020】
本発明における「DHFR発現遺伝子群」または「目的タンパク質発現遺伝子群」は、DHFRまたは目的タンパク質をコードする遺伝子を発現してDHFRまたは目的タンパク質を生産するための塩基配列であり、少なくとも宿主で機能するプロモーターおよびその下流に接続されたDHFRまたは目的タンパク質をコードする遺伝子を含むもので、更にその下流にポリアデニレーションシグナルが接続されていてもよい。DHFR発現遺伝子群および目的タンパク質発現遺伝子群を構成するプロモーターは、これらの遺伝子を宿主において発現させることができれば特に制限はない。例えば、CHO細胞、アフリカミドリザル腎繊維芽由来細胞(COS7細胞)、ヒト子宮頸癌由来細胞(Hela細胞)、マウス胎児皮膚由来細胞(NIH3T3)等の哺乳動物由来の細胞を宿主とする場合は、プロモーターとしてSV40初期および後期プロモーター、CMVプロモーター等を使用することが例示できる。
【0021】
宿主細胞は、CHO細胞、COS7細胞、Hela細胞、NIH3T3細胞といった哺乳動物由来の細胞であって、DHFRの生産能力を著しく欠くかまたは欠損した、DHFR欠損株である。例えばCHO細胞のDHFR欠損株は樹立され、極めて容易に入手可能であるし、他の細胞についても、例えばMTXを含む培地で培養を行う等することにより、かかる細胞は選択することが可能である。
【0022】
本発明に係るIR、MAR、DHFR発現遺伝子群および目的タンパク質発現遺伝子群(以下、これら4つの配列または遺伝子群のそれぞれを「要素」と呼ぶことがある)を含む哺乳動物細胞は、例えば、これら4つの要素を含む単一又は複数種のベクターを取り込んで細胞内に保持した状態の哺乳動物細胞や、これら4つの要素を染色体に組み込んで保持した状態の哺乳動物細胞等、種々の形態の細胞を包含する。この哺乳動物細胞は、IR、MAR、DHFR発現遺伝子群および目的タンパク質発現遺伝子群の4つの要素を哺乳動物細胞に導入することにより作製することができる。IR、MARおよび目的タンパク質発現遺伝子群は同時に細胞に導入し(特許文献5)、DHFR発現遺伝子群は、IR、MARおよび目的タンパク質発現遺伝子群の導入に先立って、同時に又はIR、MARおよび目的タンパク発現遺伝子群を導入した後に、細胞に導入する。
【0023】
IR、MARおよび目的タンパク質発現遺伝子群は同一ベクターに載った形態であっても、異なるベクターに載った形態であってもよく、同時に細胞に導入されればそれ以外には特に制限はない。IR、MARおよび目的タンパク質発現遺伝子群を同一のベクターに載せる場合、どの要素が上流であるか等は問わない。異なるベクターに載った形態として、IR、MARおよび目的タンパク質発現遺伝子のいずれか2以上を同一ベクターに載せ、残る遺伝子を他のベクターに載せることや、これら3つの要素を異なる3つのベクターに載せることを例示できる。2つの要素を同一ベクターに載せる場合、どちらの要素が上流であるかは問わない。
【0024】
DHFR発現遺伝子群をIR等と同時に導入する場合、例えば、4つの要素の全てを同一のベクターに載せることや、4つの要素の2または3を同一ベクターにのせ、残る要素を異なるベクターに載せて同時に導入することが例示できる。IR等と同時に導入しない場合、DHFR発現遺伝子群のみを載せたベクターを作製し、使用すればよい。なお、本発明は、目的タンパク質発現遺伝子群の増幅のために上記の哺乳動物細胞をMTX処理することで、目的タンパク質を大量に生産する細胞を提供するものである。従って、上記においてDHFR発現遺伝子群をIR等と同時に導入しない場合、DHFR発現遺伝子群は、MTX処理に先立って導入する。4つの要素が導入され、哺乳動物細胞がこれらを保持している場合には、MTX処理によって目的タンパク質を大量に生産することになる。
【0025】
前記例示した4つの要素を含む単一のベクターを用いると、4つの要素が接続された状態で宿主に保持される確率が高くなり、その結果、MTX処理によってDHFR発現遺伝子群の増幅とともに目的タンパク質発現遺伝子群が増幅される確率も向上できると考えられるため、本発明の態様として好ましい。もっとも、4つの要素のうちの1以上を含む複数種のベクターを用いる場合、例えばIR、MARおよび目的タンパク質発現遺伝子群を含む第1のベクターと、DHFR発現遺伝子群を含む第2のベクターの2種類を用いると各要素が哺乳動物細胞において近接した状態に保持され、MTX処理によってDHFR発現遺伝子群の増幅とともに目的タンパク質発現遺伝子群が増幅される確率において劣る可能性があったとしても、その後の細胞増殖を考慮すると、特に遜色なく目的タンパク質を大量に生産するという本発明の効果を達成することができる。なお、4つの要素を含む単一のベクターについて更に説明すると、4つの要素の位置関係に制限はなく、いずれの要素が上流に位置してもよく、またその転写の向きも制限がない。4つの遺伝子は近接していることが好ましく、例えば実施例で使用したように、各要素を数十から数百塩基程度離間した状態で含むものを例示できる。
【0026】
上記した4つの要素を含むベクターを導入(トランスフェクト)した哺乳動物細細胞を、好ましくはまずそれを培養して目的タンパク質を発現している細胞を選択し単離してから、MTX処理することにより、DHFR発現遺伝子群が導入された細胞を選択することができる。そして培地中のMTX濃度を段階的に上げることによりDHFR発現遺伝子群により産生されるDHFRは阻害されるが、その際にDHFR発現遺伝子群が増幅される。このDHFR発現遺伝子群の増幅の際にDHFR発現遺伝子群に近接して保持された目的タンパク質発現遺伝子群も同時に増幅される。MTX濃度の段階的な上げ方は、一般的には、まず培地中に5〜25nMの濃度で添加し、少なくとももう一段階、必要に応じてさらにもう一段階またはそれ以上の段階にわたって、5〜10倍ずつ濃度を上昇させる方法を用いればよい。各濃度のMTXを添加した培地で、細胞が生育しコロニーを作った段階で、MTX濃度を上げてやればよい。メトトレキセートの濃度を上昇させる段階数、濃度の上昇幅、各段階における培養日数、目的タンパク質の製造に用いる哺乳動物細胞を選別するための最終的な濃度などの条件は、哺乳動物細胞ないし目的タンパク質の製造効率が所望のものとなるよう適宜調整することができるが、本発明の方法を用いれば、全体的な工程期間を従来の遺伝子増幅方法よりも短縮することができる。より具体的な手順としては、例えば、単離した細胞をまず5〜25nMのMTXを含む培地に移して生存可能な細胞を選択し、必要であれば25〜125nMのMTXを含む培地に移して培養する段階を経た後、125〜500nMのMTXを含む培地に移して遺伝子が増幅された細胞を選択することができる。また、各濃度のMTXを添加した培地で生育した細胞を、一度モノクローン化した後、遺伝子増幅数の高い細胞、すなわち目的タンパク質の生産量の多い細胞を単離し、当該細胞に対して更にMTX濃度を上げてもよいし、モノクローン化せずにポリクローンのままMTX濃度を上げてもよい。
【0027】
本発明の方法により増幅したDHFR発現遺伝子群および目的タンパク質発現遺伝子群は、染色体外遺伝因子であるDM(Double Minutes)または染色体上のHSR(Homogeneously Staining Resion)いずれかの構造で局在しており、FISHなどの手法を用いて観察することができる。HSRはさらに、均質なHSR、線状ラダー、点状ラダーに分類される(図5参照)。このような遺伝子増幅に関わるDMおよびHSRは、IRおよびMARのみを用いた遺伝子増幅法においても発生し、BFB(Breakage-Fusion-Bridge)cycleにより巨大なHSRが形成されることなどが知られている(たとえば前記非特許文献2参照)。本発明では、従来の遺伝子増幅法よりも早く、より長い点状ラダーHSR(図5参照)が形成されるようになり、これと対応して、より早く、より高いレベルの目的タンパク質の産生が達成される。
【0028】
本発明の目的タンパク質の製造方法には、目的タンパク質の産生量を向上させるための公知の手段をさらに組み合わせてもよい。たとえば、ヒストン脱アセチル化阻害剤(Sodium Butyrateなど)を用いて目的タンパク質発現遺伝子のエピジェネティックなメチル化を阻害することにより、目的タンパク質の産生量を向上させる方法が知られている。本発明の目的タンパク質の製造方法において、IR、MAR、DHFRおよび目的タンパク質発現遺伝子群を導入した動物細胞を培養する際に、ヒストン脱アセチル化阻害剤を添加することにより、目的タンパク質の産生量をより一層向上させることができる。
【0029】
宿主である哺乳動物細胞に遺伝子を導入し形質転換する方法は、ベクターの構成や宿主に応じて適宜選択することができるが、例えばエレクトロポーレーション法、リポフェクション法、リン酸カルシウム法などによるトランスフェクションを例示することができる。たとえば、複数種のベクターを用いる場合は、リポフェクション法が好ましい方法として例示できる。MTX処理を開始する前に行われる、ベクターにより哺乳動物細胞が形質転換されたかどうかの選択は、たとえば、アミノグリコシド3’−ホスホトランスフェラーゼ(カナマイシン抵抗性遺伝子)、ネオマイシンホスホトランスフェラーゼ遺伝子(ネオマイシン抵抗性遺伝子)、ヒグロマイシンBホスホトランスフェラーゼ遺伝子(ヒグロマイシン抵抗性遺伝子)、ブラスティサイジンSデアミナーゼ遺伝子(ブラスティサイジン抵抗性遺伝子)などの薬剤耐性遺伝子、あるいはグルタミンシンセターゼ遺伝子、緑色蛍光タンパク質遺伝子等を選択マーカーとして用いる、公知の一般的な方法に従って行えばよい。あるいは、これらの選択マーカーを用いる代わりに、リボヌクレオチドおよびデオキシリボヌクレオチドを含有しない培地中で哺乳動物細胞(元来DHFR欠損株)を培養することにより、ジヒドロ葉酸レダクターゼ遺伝子(DHFR)が導入された哺乳動物細胞を選択することもできる。
【0030】
形質転換により得られた細胞の培養は、当該細胞に適切な条件で行なえばよい。目的タンパク質発現遺伝子の発現により生産された目的タンパク質は、当該タンパク質が細胞外へ分泌する場合は培養上清から遠心分離などで回収し、適当な方法により精製することができる。生産された目的タンパク質が細胞内に蓄積する場合は、細胞を破砕する等の適当な方法により抽出、精製することができる。
【0031】
本発明の目的タンパク質を大量に生産する方法を用いて発現可能な目的タンパク質は、発現させる目的タンパク質をコードするポリヌクレオチド配列が既知であるものであれば特に限定はなく、例えば、抗体(H鎖およびL鎖)、IL−6(インターロイキン6)などのサイトカイン、エリスロポエチン、インターフェロン、蛍光タンパク質などがあげられる。特に近年注目されている抗体医薬品の製造のために大量に必要とされる、特定の疾患(たとえばがんやリウマチ)に関与する抗原(たとえば細胞表面の受容体)を標的とする抗体は、本発明による生産効率の向上が期待される目的タンパク質である。
【発明の効果】
【0032】
本発明では、IR、MAR、DHFR発現遺伝子群および目的タンパク質発現遺伝子群を動物細胞に導入し、該細胞にMTXによる遺伝子増幅処理を施すことにより、目的タンパク質を大量に生産する哺乳動物細胞を短期間で作製することができる。
【0033】
そして、本発明により医薬品などの有用な目的タンパク質を大量に生産する細胞を作製すると、実施例に示すように、IRおよびMARのみを用いたり、MTXによる遺伝子増幅処理のみを施したりする、従来の目的タンパク質の製造方法と比較して、目的タンパク質の生産量を飛躍的に(たとえば実施例に示すように10倍以上に)増加し、短時間で大量に生産することができる。したがって、従来の目的タンパク質の製造方法と比較し、短時間かつ低コストで目的タンパク質を製造することができる。
【図面の簡単な説明】
【0034】
【図1】図1は、実施例1および比較例1−1において、ステップ1から5により培養上清中に発現したFcRの発現量を示すグラフである。
【図2】図2は、実施例1で作製したベクターpΔBM.AR1dhfr−FcRの概略図である。
【図3】図3は、実施例2,比較例2,実施例3,比較例3−1〜3−3で用いたプラスミドA(pΔBN AR1 Psv40−dhfr)、B(pSFV−V Psv40−dhfr)、C(pCMV−d2EGFR)およびD(pMycLH)のマップである。
【図4】図4は、実施例2および比較例2(上)、ならびに実施例3および比較例3−1〜3−3(下)の選択法のステップ(形質転換後の日数および使用薬剤)を示す図である。
【図5】図5は、実施例2でFISHにより観察した遺伝子増幅構造の代表例の写真である。
【図6】図6は、実施例2および比較例2における、FISHにより観察した遺伝子増幅構造別の細胞集団中の形成頻度(右)およびELISAにより測定した抗体タンパク質発現量(左)の結果を示すグラフである。図6(左)について、But (-)およびBut (+)は、それぞれSodium Butyrateを添加しなかった場合および添加した場合である。
【図7】図7は、実施例3および比較例3−1〜3−3における、FACSで測定したd2EGFPタンパク質発現分布の結果を示すグラフである。
【発明を実施するための形態】
【実施例】
【0035】
以下、本発明をさらに詳細に説明するために実施例を示すが、本発明は実施例に限定されるものではない。
[実施例1]
ステップ1:IR、MAR、DHFR発現遺伝子群およびFcR(可溶性ヒト型FcレセプターFcγRI)発現遺伝子群をコードするポリヌクレオチドを含むベクターの作製
【0036】
(1)pΔB.AR1(非特許文献2)のBamH1−MluIの間に、KpnI、AscI、SalI、SwaI、AsiSI、SbfI、BamHI、MluIからなるマルチクローニングサイトを導入し、pΔBM.AR1.MCSとした。該pΔBM.AR1.MCSを制限酵素BamHIおよびNruIで切断した。両切断末端をT4DNAポリメラーゼにより平滑化し、さらにアルカリフォスファターゼにより脱リン酸化し、pΔBM.AR1/NBb*とした。なお、得られたpΔBM.AR1/NBb*中、IRおよびMARはともにDHFR遺伝子座由来のものである。
【0037】
(2)pECEFcRdhfr(特許文献:特開2009−278948)を制限酵素BamHIで切断し、電気泳動によりDHFR−FcR発現遺伝子群を含んだバンドを切り出した。前記ヌクレオチドをBKLキット(タカラバイオ社製)により平滑化およびリン酸化しdhfr−FcRとした。
【0038】
(3)(1)で調製したpΔBM.AR1/NBb*と(2)で調製したdhfr−FcRを、DNA Ligation Kit<Mighty Mix>(タカラバイオ社製)を用いて連結することで、FcR発現ベクター(pΔBM.AR1dhfr−FcR)を作製した。
【0039】
以上の実施例では、DHFR(dhfr)発現遺伝子群、目的タンパク質(FcR)発現遺伝子群、IR及びMARの4つの要素を同一のベクターに載せているが、作製されたベクターpΔBM.AR1dhfr−FcRの詳細は図2に示した通りである。図2において、MAR(図中ではAR1)は約0.5kb、DHFR遺伝子座由来のIR(図中ではDHFR IR)は約4.6kb、dhfr発現遺伝子群は約1kb、FcR発現遺伝子群は約1.4kbであるが、MARとIRは約100b、IRとdhfr発現遺伝子群は約50b離れているため、これら4つの遺伝子は、全長で約7.5kbとなる。
【0040】
ステップ2:pΔBM.AR1dhfr−FcRのCHO細胞への導入
(1)MEMAlpha培地(インビトロジェン社製)に10%FBS(Fetal Bovine Serum:JRH Biosciences社製)を加えた培地10mLで対数増殖期まで培養したCHO細胞に、ステップ1で作製したFcR発現ベクター(pΔBM.AR1dhfr−FcR)をエレクトロポーレーションシステムGenePulserXcell(バイオラッド社製)を用いて添付の資料に従いトランスフェクションした。
【0041】
(2)(1)の方法でトランスフェクションしたCHO細胞を、リボヌクレオシドおよびデオキシリボヌクレオチド不含のMEMAlpha培地(インビトロジェン社製)に10%透析FBS(Fetal Bovine Serum:biowest社製)を加えた培地10mLで10日間培養した。
【0042】
ステップ3:FcR発現CHO細胞の単離
(1)ステップ2でトランスフェクションしたCHO細胞を、ステップ2−(2)で使用した培地で1細胞/穴になるように希釈し、96穴培養プレート(ファルコン社製)に200マイクロリットルずつ分注した。
【0043】
(2)10日間培養後、ヒトFcRを1μg/穴で固相化したマイクロタイタープレートを用いたELISA(Enzyme−Linked ImmunoSorbent Assay)により培養上清中のFcRの発現量を測定した。その中から、発現量が最も高く、単一コロニーとなっている細胞を選択し単離した。
【0044】
ステップ4:CHO細胞のメトトレキセートによる遺伝子増幅処理および細胞の単離
ステップ3で単離したCHO細胞をステップ2−(2)で使用した培地で対数増殖期まで培養後、5x104細胞を10cmプレート(ファルコン社製)に撒いた。24時間後、25nMメトトレキセート(シグマ社製)を含むステップ2−(2)で使用した培地10mLで15日間培養した。生き残った細胞をステップ3と同様の方法により単離した。さらに、単離した上記CHO細胞を、125nMメトトレキセートを用い上記と同様の方法で、遺伝子増幅処理および細胞の単離を行った。
【0045】
ステップ5:FcR発現量の測定
(1)ステップ4で単離したCHO細胞をステップ2−(2)で使用した培地で対数増殖期後期まで培養後、新たに培地を交換した。3日間培養後、上清を回収した。
【0046】
(2)前記上清を、ヒトFcRを1μg/穴で固相化したマイクロタイタープレートを用いたELISA(Enzyme−Linked ImmunoSorbent Assay)により、上清中に分泌されたFcRの量を測定した。
【0047】
[比較例1−1]
上記ステップ2−(1)において、陰性コントロールとして、ステップ1−(2)で使用したIR(DHFR)およびMAR(AR1)を含まないFcR発現ベクター(pECEFcRdhfr)を(1)と同様の方法によりトランスフェクションするようにしたこと以外は上記実施例1と同様にして、FcR発現量を測定した。
【0048】
[比較例1−2]
上記ステップ4において、25nMメトトレキセートを含まないステップ2−(2)で使用した培地10mLで15日間培養するようにしたこと以外は上記実施例1と同様にして、FcR発現量を測定した。
【0049】
結果
上記実施例1および比較例1−1〜1−2の結果を表1および図1に示す。表1に示す値は、ステップ1から5により培養上清中に発現したFcRの発現量を示す。図1のグラフは、ステップ1から5により培養上清中に発現したFcRの発現量を、実施例1と比較例1−1について示す。
【0050】
IRおよびMARとFcR発現遺伝子を動物細胞に同時に導入し、該細胞にメトトレキセートによる遺伝子増幅処理を施した細胞(実施例)は、FcRが大量に生産されたことがわかる。一方、IRおよびMARをFcR発現遺伝子と同時に導入していない細胞(比較例1−1)は、従来の方法による動物細胞による目的タンパク質発現と同程度の低い発現量であることがわかる。また、一方、IRおよびMARをFcR発現遺伝子と同時に導入したのみで、メトトレキセートによる遺伝子増幅処理を施していない細胞(比較例1−2)も、FcRの発現量は、上記比較例1よりは高いものの上記実施例より著しく低い発現量にとどまった。以上より、IRおよびMARと目的タンパク質発現遺伝子を動物細胞に同時に導入し、該細胞にメトトレキセートによる遺伝子増幅処理を施すことにより、目的タンパク質を大量に生産することが可能であることがわかる。
【0051】
【表1】
[実施例2](抗体発現の検討:IR/MAR−DHFR/MTX融合法)
図3に示す、DHFR(dhfr)タンパク質発現遺伝子群、DHFR遺伝子座由来のIR及びMAR(AR1)を含むプラスミドA(pΔBN AR1 Psv40−dhfr)と、目的タンパク質(抗c−myc抗体のL鎖であるMycLおよび同H鎖であるMycH)発現遺伝子群を含むプラスミドD(pMycLH)を用いて、Lipofectamin2000(インビトロジェン社製)によりCHO−DG44細胞に形質導入した。続いて、図4(実施例2)に示すように、0.5 mg/ml G418および10 μg/ml Blasticidineで選択し(1d)、さらに5 nM methotrexate(11d)、50 nM methotrexate(28d)および500 nM methotrexate(40d)で選択を行った。各ステップの細胞から染色体標本を作製し、導入したプラスミド全長DNAをプローブとしてFISHを行い、遺伝子増幅構造別の形成頻度を調査した。同時に、各ステップの細胞を採取し、5×105cells/mLで96well plateに植え、Sodium Butyrateを添加した場合、添加しなかった場合それぞれについて、3日後に培養上清中の抗体タンパク質発現量をELISA法で決定した。
【0052】
なお、プラスミドAは、pDBN.AR1のBamHI/NruI断片(ヒグロマイシン抵抗性遺伝子を含む)を除き、その位置に、PCRにより増幅したSV40promoterとDHFRcoding配列を挿入することにより作製されたプラスミドであり、ブラストサイジンを用いた選択のためのブラストサイジン耐性遺伝子(Bsr)およびそのプロモーター(SR αPromoter)も含まれている。一方、プラスミドDには、G418を用いた選択のためのカナマイシン耐性遺伝子(Kan)も含まれている。これらの塩基配列は公知である。
【0053】
[比較例2](抗体発現の検討:DHFR/MTX単独法)
前記プラスミドAに代えて、図3に示す、DHFR(dhfr)発現遺伝子群を含むもののIR及びMARを含まないプラスミドB(pSFV−V Psv40−dhfr)を用いるよう変更し、また選択を、図4(比較例2)に示すように、0.5 mg/ml G418および10 μg/ml Blasticidine(1d)、5 nM methotrexate(17d)および50 nM methotrexate(50d)に変更した以外は、実施例2と同様の手順により、FISHによる遺伝子増幅構造別の形成頻度の調査、ならびにELISAによる抗体タンパク質発現量を行った。
【0054】
なお、プラスミドBは、pSFV‐VのBamHI/NruI断片(ヒグロマイシン抵抗性遺伝子を含む)を除き、その位置に、PCRにより増幅したSV40promoterとDHFRcoding配列を挿入することにより作製されたプラスミドであり、ブラストサイジン耐性遺伝子(Bsr)およびそのプロモーター(SR αPromoter)も含まれている。これらの塩基配列は公知である。
【0055】
結果
上記実施例2および比較例2における、FISHにより観察した遺伝子増幅構造の代表例の写真を図5に、遺伝子増幅構造別の形成頻度(FISH)および抗体タンパク質発現量(ELISA)の結果をそれぞれ図6の右および左に示す。従来法(DHFR/MTX単独法)に比べて、本発明の方法(IR/MAR−DHFR/MTX融合法)では、より早く、より長い点状ラダーHSRが形成された。これと対応して、より早く、より高いレベルの抗体産生が検出された。Sodium Butyrateを添加することにより、より一層抗体産生レベルを向上させることができた。
【0056】
[実施例3](d2EGFP発現の検討:IR/MAR−DHFR/MTX融合法)
図3に示す、DHFR(dhfr)発現遺伝子群、IR(DHFR)及びMAR(AR1)含むプラスミドA(pΔBN AR1 Psv40−dhfr)と、目的タンパク質(d2EGFP)発現遺伝子群を含むプラスミドC(pCMV−d2EGFR、N.Shimizu, et al.(2007) J.Cell.Biochem., vol.102, p515-529)を用いて、Lipofectamin2000によりCHO−DG44細胞に形質導入した。続いて、図4(実施例3)に示すように、10 μg/ml Blasticidineで選択し(1d)、さらに5 nM methotrexateおよび100 μg/ml Blasticidine(11d)、50 nM methotrexate(28d)および500 nM methotrexate(56d)で選択を行った。その後、培養した細胞集団のd2EGFPタンパク質発現分布をFACS(fluorescent-activated cell sorter)を用いて測定した。
【0057】
[比較例3−1](d2EGFP発現の検討:IR/MAR単独法)
形質導入したCHO−DG44細胞の選択を、図4(比較例3−1)に示すように、10 μg/ml Blasticidine(1d)および100 μg/ml Blasticidine(11d)に変更した以外は、実施例2と同様の手順により、d2EGFPタンパク質発現分布をFACSを用いて測定した。
【0058】
[比較例3−2](d2EGFP発現の検討:DHFR/MTX単独法)
前記プラスミドAに代えて、図3に示す、DHFR(dhfr)発現遺伝子群を含むもののIR及びMARを含まないプラスミドB(pSFV−V Psv40−dhfr)を用いるよう変更し、また形質導入したCHO−DG44細胞の選択を、図4(比較例3−2)に示すように、10 μg/ml Blasticidine(1d)、5 nM methotrexateおよび100 μg/ml Blasticidine(17d)、50 nM methotrexate(39d)および500 nM methotrexate(53d)に変更した以外は、実施例3と同様の手順により、d2EGFPタンパク質発現分布をFACSを用いて測定した。
【0059】
[比較例3−3](d2EGFP発現の検討:通常法)
前記プラスミドAに代えて、図3に示す、DHFR(dhfr)発現遺伝子群を含むもののIR及びMARを含まないプラスミドB(pSFV−V Psv40−dhfr)を用いるよう変更し、また形質導入したCHO−DG44細胞の選択を図4(比較例3−3)に示すように、10 μg/ml Blasticidine(1d)および100 μg/ml Blasticidine(15d)に変更した以外は、実施例3と同様の手順により、d2EGFPタンパク質発現分布をFACSを用いて測定した。
【0060】
結果
上記実施例3および比較例3−1〜3−3における、FACSにより測定したd2EGFPタンパク質発現分布の結果を図7に示す。従来法(IR/MAR単独法、DHFR/MTX単独法、通常法)に比べて、本発明の方法(IR/MAR−DHFR/MTX融合法)では、d2EGFPタンパク質の発現量の多い細胞が細胞集団中により高頻度で出現することが示されている。
【特許請求の範囲】
【請求項1】
哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む哺乳動物細胞をメトトレキセート処理する工程を含むことを特徴とする、哺乳動物細胞の製造方法。
【請求項2】
前記メトトレキセート処理する工程が、段階的に濃度を上げながら、メトトレキセートを含有する培地中で前記哺乳動物細胞を培養する複数の段階を含む、請求項1に記載の哺乳動物細胞の製造方法。
【請求項3】
前記メトトレキセート処理する工程が、少なくとも、メトトレキセートを5〜25nMの濃度で含有する培地中で前記哺乳動物細胞を培養する第一段階、およびメトトレキセートを第一段階の5〜10倍の濃度で含有する培地中で前記哺乳動物細胞を培養する第二段階を含む、請求項1または2に記載の哺乳動物細胞の製造方法。
【請求項4】
前記哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む哺乳動物細胞を作製するために、あらかじめ、哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む単一のベクターを哺乳動物細胞に導入して形質転換する工程を含む、請求項1〜3のいずれかに記載の哺乳動物細胞の製造方法。
【請求項5】
前記哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む哺乳動物細胞を作製するために、あらかじめ、哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む複数種のベクターを哺乳動物細胞に導入して形質転換する工程を含む、請求項1〜3のいずれかに記載の哺乳動物細胞の製造方法。
【請求項6】
前記哺乳動物複製起点が、c−myc遺伝子座複製起点、ジヒドロ葉酸還元酵素遺伝子座複製起点、またはβ−グロビン遺伝子座複製起点に由来するものである、請求項1〜5のいずれかに記載の哺乳動物細胞の製造方法。
【請求項7】
前記マトリックス結合領域が、Igκ遺伝子座マトリックス結合領域、SV40初期領域マトリックス結合領域、またはジヒドロ葉酸還元酵素マトリックス結合領域に由来するものである、請求項1〜6のいずれかに記載の哺乳動物細胞の製造方法。
【請求項8】
前記目的タンパク質が受容体、抗体又は蛍光タンパク質である、請求項1〜7のいずれかに記載の哺乳動物細胞の製造方法。
【請求項9】
以下の順序で行われる(1)から(4)の各工程を含む、請求項1〜8のいずれかに記載の哺乳動物細胞の製造方法。
(1)哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を動物細胞に導入する工程、
(2)前記導入工程を経た哺乳動物細胞を培養し、目的タンパク質を発現している細胞を選択し単離する工程、
(3)前記単離工程を経た細胞を5〜25nMのメトトレキセートを含む培地に移し、生存可能な細胞を選択する工程、および、
(4)前記選択工程を経た細胞を125〜500nMのメトトレキセートを含む培地に移し、遺伝子が増幅された細胞を選択する工程。
【請求項10】
請求項1〜9のいずれかに記載の製造方法により得られた哺乳動物細胞を用いて目的タンパク質を製造する方法。
【請求項11】
哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む単一のベクター。
【請求項1】
哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む哺乳動物細胞をメトトレキセート処理する工程を含むことを特徴とする、哺乳動物細胞の製造方法。
【請求項2】
前記メトトレキセート処理する工程が、段階的に濃度を上げながら、メトトレキセートを含有する培地中で前記哺乳動物細胞を培養する複数の段階を含む、請求項1に記載の哺乳動物細胞の製造方法。
【請求項3】
前記メトトレキセート処理する工程が、少なくとも、メトトレキセートを5〜25nMの濃度で含有する培地中で前記哺乳動物細胞を培養する第一段階、およびメトトレキセートを第一段階の5〜10倍の濃度で含有する培地中で前記哺乳動物細胞を培養する第二段階を含む、請求項1または2に記載の哺乳動物細胞の製造方法。
【請求項4】
前記哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む哺乳動物細胞を作製するために、あらかじめ、哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む単一のベクターを哺乳動物細胞に導入して形質転換する工程を含む、請求項1〜3のいずれかに記載の哺乳動物細胞の製造方法。
【請求項5】
前記哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む哺乳動物細胞を作製するために、あらかじめ、哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む複数種のベクターを哺乳動物細胞に導入して形質転換する工程を含む、請求項1〜3のいずれかに記載の哺乳動物細胞の製造方法。
【請求項6】
前記哺乳動物複製起点が、c−myc遺伝子座複製起点、ジヒドロ葉酸還元酵素遺伝子座複製起点、またはβ−グロビン遺伝子座複製起点に由来するものである、請求項1〜5のいずれかに記載の哺乳動物細胞の製造方法。
【請求項7】
前記マトリックス結合領域が、Igκ遺伝子座マトリックス結合領域、SV40初期領域マトリックス結合領域、またはジヒドロ葉酸還元酵素マトリックス結合領域に由来するものである、請求項1〜6のいずれかに記載の哺乳動物細胞の製造方法。
【請求項8】
前記目的タンパク質が受容体、抗体又は蛍光タンパク質である、請求項1〜7のいずれかに記載の哺乳動物細胞の製造方法。
【請求項9】
以下の順序で行われる(1)から(4)の各工程を含む、請求項1〜8のいずれかに記載の哺乳動物細胞の製造方法。
(1)哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を動物細胞に導入する工程、
(2)前記導入工程を経た哺乳動物細胞を培養し、目的タンパク質を発現している細胞を選択し単離する工程、
(3)前記単離工程を経た細胞を5〜25nMのメトトレキセートを含む培地に移し、生存可能な細胞を選択する工程、および、
(4)前記選択工程を経た細胞を125〜500nMのメトトレキセートを含む培地に移し、遺伝子が増幅された細胞を選択する工程。
【請求項10】
請求項1〜9のいずれかに記載の製造方法により得られた哺乳動物細胞を用いて目的タンパク質を製造する方法。
【請求項11】
哺乳動物複製開始領域、核マトリックス結合領域、ジヒドロ葉酸還元酵素発現遺伝子群および目的タンパク質発現遺伝子群を含む単一のベクター。
【図1】

【図2】
【図3】

【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−143225(P2012−143225A)
【公開日】平成24年8月2日(2012.8.2)
【国際特許分類】
【出願番号】特願2011−101610(P2011−101610)
【出願日】平成23年4月28日(2011.4.28)
【出願人】(504136568)国立大学法人広島大学 (924)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
【公開日】平成24年8月2日(2012.8.2)
【国際特許分類】
【出願日】平成23年4月28日(2011.4.28)
【出願人】(504136568)国立大学法人広島大学 (924)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
[ Back to top ]