
新規ターミネーターおよびその利用

【課題】糸状菌細胞内でより強力なターミネーターとして機能し得る新規ポリヌクレオチドを提供する。
【解決手段】糸状菌細胞用ターミネーターは、下記の(i)〜(iii)の何れかに示されるポリヌクレオチドを含む。(i)特定の塩基配列からなるポリヌクレオチド。(ii)上記(i)のポリヌクレオチドの部分断片であって、特定の塩基配列の第343位〜第460位を含み、かつ550bp以上のサイズを有するポリヌクレオチド。(iii)上記(i)または(ii)とストリンジェントな条件下でハイブリダイズするポリヌクレオチドであり、かつ糸状菌細胞内でターミネーター活性を有するポリヌクレオチド。
【解決手段】糸状菌細胞用ターミネーターは、下記の(i)〜(iii)の何れかに示されるポリヌクレオチドを含む。(i)特定の塩基配列からなるポリヌクレオチド。(ii)上記(i)のポリヌクレオチドの部分断片であって、特定の塩基配列の第343位〜第460位を含み、かつ550bp以上のサイズを有するポリヌクレオチド。(iii)上記(i)または(ii)とストリンジェントな条件下でハイブリダイズするポリヌクレオチドであり、かつ糸状菌細胞内でターミネーター活性を有するポリヌクレオチド。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は新規なターミネーターおよび当該ターミネーターを用いてタンパク質を効率的に製造するための方法に関する。詳しくは、糸状菌細胞内でターミネーターとして機能し得るポリヌクレオチド、当該ポリヌクレオチドを含むベクター、当該ポリヌクレオチドの上流に任意のプロモーターおよびタンパク質をコードするポリヌクレオチドが連結されてなるベクターを用いたタンパク質の製造方法、当該ポリヌクレオチドの上流に任意のプロモーターおよび目的遺伝子が連結されてなるベクター、および当該ポリヌクレオチドの上流に任意のプロモーターおよび目的遺伝子が連結されてなるベクターを糸状菌細胞に導入することによる目的遺伝子の発現方法等に関するものである。
【背景技術】
【0002】
遺伝子組み換えによる有用タンパク質生産の宿主としては、大腸菌や枯草菌をはじめ酵母、昆虫、植物細胞、動物細胞等数多くの系が開発されており、生産させようとする目的タンパク質によってより適した系が選択されているのが現状である。
【0003】
特に、糸状菌であるアスペルギルス(Aspergillus)属やトリコデルマ(Trichoderma)属は酵素タンパク質を著量に分泌生産する能力に優れているため、工業的な酵素製剤の生産に利用されている。これらの菌株の中には、液体培養を行った際に培養液1リットルあたり20g-protein以上、フスマ培養を行った際に小麦フスマ1kgあたり50g-proteinという高い生産性を示すものもある。
【0004】
一方、異種タンパク質の生産においては、タンパク質のフォールディングや糖鎖付加などの翻訳後修飾が本来の生物と同様に行われることが重要である。糸状菌は大腸菌や酵母より進化的に高等動物に近いため、動物由来異種タンパク質を活性ある形で生産することも期待できる。また、麹菌アスペルギルス・オリゼのように古くから発酵食品の製造に用いられているものが多く、その生産物は安全であるというGRAS(generally regarded as safe)グレードとして認められているため、これらの糸状菌による有用タンパク質生産も認可されやすいと考えられる。
【0005】
アスペルギルス・オリゼをはじめとする糸状菌は一般的に有性生活環が認められず、分生子も多核であるために、古典遺伝学的手法による遺伝解析は容易ではなく、分子生物学的研究も他の微生物に比べて遅れていた。しかし、1980年代後半になって、産業的に重要な糸状菌について形質転換系が確立され、糸状菌の遺伝子工学が発展してきた。このような状況の中、前述した利点にも後押しされ、糸状菌を宿主としたさまざまな有用タンパク質の生産が報告されるようになってきた(例えば、特許文献1、非特許文献1を参照)。また、タンパク質を高発現させるために多くの強力なプロモーターが取得されており、麹菌ではαアミラーゼ遺伝子のプロモーターや(例えば特許文献2を参照)、本発明者らの改良型プロモーター(例えば特許文献3を参照)、スーパーオキサイドジスムターゼ遺伝子のプロモーター(例えば特許文献4を参照)などが使用されその有用性が認められている。
【0006】
さて、遺伝子発現においては、プロモーターによる転写促進に加え、ターミネーターによる効率的な転写終結も重要であると考えられる。植物ではターミネーターとしてシロイヌナズナのHSP18.2遺伝子のターミネーターが単離され、植物発現系における有用性が認められている(例えば非特許文献2を参照)。
【0007】
糸状菌においては、アスペルギルス・オリゼのα-グルコシダーゼ遺伝子AのターミネーターであるT-agdA(例えば非特許文献3を参照)、グルコアミラーゼBのターミネーター(例えば非特許文献4を参照)などがよく使用されている。これらは、各遺伝子の終止コドンの5塩基下流から585塩基まで、および1塩基下流から534塩基までの配列をターミネーターとして利用している。しかし、ターミネーターの配列、構造、機能について詳細に解析した報告はなく、その機構は全く解明されていない。それゆえ、糸状菌宿主においてターミネーターを改善することによりタンパク質の生産量を増大させようとする試みはこれまでなかった。
【0008】
ここで、真核生物で一般に提唱されている転写終結機構について説明する。動物細胞などでは、転写終結部位の近くにAAUAAA という配列があり、これを特定の酵素が認識して、その3’側(下流)約25塩基のところでmRNAを切断している。さらに、mRNAの3’末端に普通200〜400個のA(「ポリ(A)鎖」という)が付加される。このポリ(A)鎖はmRNAの目印で、キャップ構造とともに働いて、翻訳の効率を高めるとされている。また、3’側からの分解を防ぐ働きもある。一方、糸状菌の転写終結機構についてはほとんど解明されておらず、ターミネーター配列と転写終結との関係を定量的に解析した報告はない。もし、糸状菌においてタンパク質生産に利用できるさらに強力なターミネーターを取得できれば、さまざまな有用タンパク質の生産性を向上させることができると考えられる。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特開昭62−272988号公報(公開日:昭和62(1987)年11月27日)
【特許文献2】特開平07−51067号公報(公開日:平成7(1995)年2月28日)
【特許文献3】特開平9−9968号公報(公開日:平成9(1997)年1月14日)
【特許文献4】特開2001−224381号公報(公開日:平成12(2001)年8月21日)
【非特許文献】
【0010】
【非特許文献1】Christensenら、High Level Expression of Recombinant Genes in Aspergillus Oryzae. Nature Biotechnology, 米国, 1988年, 第6巻, p1419−1422
【非特許文献2】真野ら、「ターミネーター領域の改変による導入遺伝子の高発現化」、平成17年度日本生物工学会講演要旨集、P123、社団法人 日本生物工学会、発行日2005年9月25日
【非特許文献3】Minetokiら、Improvement of promoter activity by the introduction of multiple copies of the conserved region III sequence, in volved in the efficient expression of Aspergillus oryzae amylase-encoding genes. Appl. Microbiol. Biotechnol., 米国, 1998年, 第50巻, p459−467
【非特許文献4】Hataら、Nucleotide sequence of an alternative glucoamylase-encoding gene (glaB) expressed in solidstate culture of Aspergillus oryzae. Gene, 米国, 1998年, 第207巻, p127−134
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、糸状菌細胞内でより強力なターミネーターとして機能し得るポリヌクレオチドをアスペルギルス属糸状菌からクローニングし、これをアスペルギルス属等の糸状菌におけるタンパク質の生産に利用することにある。
【課題を解決するための手段】
【0012】
本発明者らは、上記目的を達成するために鋭意検討を行った結果、アスペルギルス・オリゼから新規かつ強力なターミネーターとして機能し得るポリヌクレオチドをクローニングすることに成功し、本発明を完成するに至った。
【0013】
すなわち本発明にかかる糸状菌用ターミネーターは、糸状菌細胞内でターミネーター活性を有し、下記の(i)〜(iii)の何れかに示されるポリヌクレオチドを含むことを特徴とする糸状菌用ターミネーター:
(i)配列番号7に示される塩基配列からなるポリヌクレオチド;
(ii)上記(i)のポリヌクレオチドの部分断片であって、配列番号7に示される塩基配列の第343位〜第460位を含み、かつ550bp以上のサイズを有するポリヌクレオチド;
(iii)上記(i)または(ii)とストリンジェントな条件下でハイブリダイズするポリヌクレオチドであり、かつ糸状菌細胞内でターミネーター活性を有するポリヌクレオチド。
【0014】
また本発明にかかるベクターは上記本発明にかかる糸状菌用ターミネーター、および糸状菌細胞内において機能し得るプロモーターを含むことを特徴としている。
【0015】
また本発明にかかるポリペプチドの生産方法は、上記本発明にかかるベクターのプロモーターの下流であり、かつ糸状菌用ターミネーターの上流に所望のタンパク質をコードする遺伝子が連結されたベクターで、糸状菌を形質転換することを特徴としている。
【0016】
また本発明は、上記本発明にかかる形質転換方法により取得された形質転換体をも包含する。
【0017】
また本発明は、上記本発明にかかる形質転換体を培養する工程を含む、タンパク質の生産方法をも包含する。
【0018】
なお、上記本発明にかかる糸状菌用ターミネーターは全く新規のターミネーターである。また上記本発明にかかる糸状菌用ターミネーターは、アスペルギルス・オリゼのORF予測の終止コドンの下流2000bp以内にORFの転写方向と同じ方向で存在しているものでもなかった。α-グルコシダーゼ遺伝子やグルコアミラーゼB遺伝子のターミネーター等、一般にターミネーターは遺伝子の終止コドンの下流約600bpまでに存在することを考慮すると、今回取得した糸状菌用ターミネーターは、ゲノムデーターベースにおいてターミネーターとして当業者が予測できるDNAではない。また、そのターミネーター活性は、従来公知のT-agdAに比して高い。よって本発明にかかる糸状菌用ターミネーターは、従来技術に対して新規性および進歩性を有することは明白である。
【発明の効果】
【0019】
本発明に係る新規ターミネーターを糸状菌のタンパク質生産系に利用することによって、従来公知の糸状菌のタンパク質生産系に比してタンパク質の生産効率を増大させることが可能となる。
【図面の簡単な説明】
【0020】
【図1】実施例1におけるpSEGA3プラスミドの構築手順を示す図である。
【図2】実施例1において取得されたターミネーター候補DNAのアスペルギルス・オリゼ染色体上の遺伝子座を示す図である。
【図3】実施例1における遺伝子ライブラリーおよびコントロール用ベクターの構築手順を示す図である。
【図4】実施例3において決定された2−5−1ターミネーターの転写終結部位を示す図である。
【図5】実施例2、3における新規ターミネーター導入プラスミドの構築手順を示す図である。
【図6】2−5−1ターミネーターと4種類の断片との関係を示す模式図である。
【発明を実施するための形態】
【0021】
以下、本発明の実施の形態について詳細に説明する。但し、本発明はこれに限定されるものではなく、記述した範囲内で種々の変形を加えた態様で実施できるものである。また、本明細書中に記載された学術文献及び特許文献の全てが、本明細書中において参考として援用される。なお、本明細書において特記しない限り、数値範囲を示す「A〜B」は、「A以上、B以下」であることを示す。
【0022】
(1.糸状菌用ターミネーター)
本発明にかかる糸状菌用ターミネーター(以下「本発明のターミネーター」という)は、アスペルギルス・オリゼのゲノムDNAから見出されたものである。アスペルギルス・オリゼのゲノムの全遺伝子配列の解読は2005年に終了(Nature, 438, 1157-1161 (2005)を参照のこと)し、ポストゲノム期を迎えている。この成果により、有用な遺伝子の探索や生産性の高いプロモーターの取得などが行われている。
【0023】
糸状菌のターミネーターについても予測遺伝子の直後から数百bpの塩基配列をPCR等で取得することが可能となったが、ターミネーターに関しては、翻訳効率の増加やmRNAの安定性への寄与が示唆されているものの、糸状菌において実際にこれらの性能が優れているターミネーターを探索するための知見はほとんど得られていないのが実情である。
【0024】
そこで本発明者らは、アスペルギルス・オリゼのゲノムライブラリーを作製し、ショットガンクローニングを行うことで優れたターミネーターとして機能する塩基配列を取得することを目的として実験を行った。
【0025】
アスペルギルス属糸状菌で自己複製可能なAMA1 (autonomously maintained in Aspergillus) 配列(参考文献:Johnstone, I. L. Microbiol. Sci., 2, 307-311 (1985) )、プロモーターとしてP-enoA(参考文献:Toda, T. et al Curr. Genet. 40, 260-267 (2001))、およびレポーター遺伝子としてイー・コリ由来のβ-グルクロニダーゼ(GUS)をコードする遺伝子(uidA)(参考文献:Jefferson, R. A. et al Proc. Natl. Acad. Sci. USA 83, 8447-8451 (1986))を連結し、高形質転換効率系のターミネーター検索用のプラスミドを構築した。このプラスミドは、AMA1配列を有し、インテグレート型に比べて数百倍の形質転換効率が得られるため、アスペルギルス属糸状菌の遺伝子のショットガンクローニングを可能にする。
【0026】
該プラスミドを用い、アスペルギルス・オリゼ由来の種々のゲノムDNA断片をβ-グルクロニダーゼ遺伝子の下流に公知の方法にてクローニングし、最少培地でβ-グルクロニダーゼを高生産する形質転換体を取得した。最少培地としては、0.2%NaNO3、0.1%K2HPO4、0.05%MgSO4、0.05%KCl、0.001%FeSO4、3%グルコースまたは3%デンプン、pH5.5のツァペックドックス(Czapek-Dox)培地が用いられた。なお、上記「高生産する形質転換体」とは、プロモーターであるP-enoA、レポーター遺伝子であるβ-グルクロニダーゼ遺伝子の下流に、従来までにタンパク質生産に利用されてきたアスペルギルス・オリゼのα-グルコシダーゼA遺伝子のターミネーター(適宜「T-agdA」と表記する)を連結してなるプラスミドで形質転換した時に得られる形質転換体のβ-グルクロニダーゼ活性に比して、アスペルギルス・オリゼのα-グルコシダーゼA遺伝子のターミネーターをアスペルギルス・オリゼ由来の種々のゲノムDNA断片に置換したプラスミドで形質転換されてなる形質転換体のβ-グルクロニダーゼ活性が高いことを意味する。逆に後者のβ-グルクロニダーゼ活性が前者に比して低い場合には、後者の形質転換体は低生産であるといえる。
【0027】
高生産であった形質転換体から公知の方法にてプラスミドDNAを単離し、これらのプラスミドのターミネーター領域のシークエンスを行ったところ、767〜1882bpからなる5種類のDNAの挿入が確認できた。これらのDNAは全てアスペルギルス・オリゼのゲノムデーターベース(独立行政法人製品評価技術基盤機構)上の塩基配列と一致したが、公開されているORF予測の終止コドンの下流2000bp以内にORFの転写方向と同じ方向で存在しているものではなかった。α-グルコシダーゼ遺伝子やグルコアミラーゼB遺伝子のターミネーター等、一般にターミネーターは遺伝子の終止コドンの下流約600bpまでに存在することを考慮すると、今回取得したDNAはいずれの配列も、ゲノムデーターベースにおいてターミネーターとして予測できるDNAではないと考えられる。
【0028】
続いて、上記で取得したDNAをPCRにて増幅し、プロモーターとしてP-No8142(「化学と生物」,2000年,第38巻,第12号,p.831−838)、Hsp12遺伝子の 5’UTR(参考文献「Koda, A. et al Appl. Microbiol. Biotechnol., 70, 333-336 (2006)」)、レポーター遺伝子としてβ-グルクロニダーゼ遺伝子、マーカーとしてniaDを含むベクターのβ-グルクロニダーゼ遺伝子の下流に連結した。これらの発現ベクターを用いて、アスペルギルス・オリゼniaD300(硝酸還元酵素欠損株)を形質転換し、得られた形質転換体のサザン解析を行い、導入ベクターが染色体のniaD座位に1コピーだけ導入された形質転換体を選択した。これらの形質転換体の菌体内β-グルクロニダーゼ比活性を測定することにより、各DNA配列のターミネーターとしての性能を評価した。
【0029】
その結果、配列番号2に示す2−5−1ターミネーターを用いたとき、T-agdA(塩基配列を配列番号1に示す)を導入したコントロールベクターよりもβ-グルクロニダーゼの比活性が1.34倍増大することを見出した。このことにより、ゲノムデーターベースではターミネーターとして予測されていない配列の中に、既知のターミネーターよりも優れたターミネーターとして機能するDNAが存在することが示された。
【0030】
次に、公知の方法にて配列番号2に示す2−5−1ターミネーターのターミネーター活性を保持する領域を特定した。転写終結部位(配列番号2または7に示される塩基配列の第343位〜第460位であると推定)およびその約30bp上流までの塩基を削除しないようにトランケートし、これらをターミネーターとしたプラスミドを作製し、上記と同様の方法で当該プラスミドが1コピー導入されたアスペルギルス・オリゼ形質転換体を取得し、形質転換体の菌体内のβ-グルクロニダーゼ比活性を測定した。
【0031】
その結果、2−5−1ターミネーター(1577bp)の塩基配列の第1−827位までの塩基からなる断片である、2−5−11ターミネーター(827bp、配列番号7);2−5−1ターミネーター(1577bp)の塩基配列の第1−572位までの塩基からなる断片である、2−5−12ターミネーター(572bp、配列番号8);2−5−1ターミネーター(1577bp)の塩基配列の第261−827位までの塩基からなる断片である、2−5−13ターミネーター(567bp、配列番号9)をターミネーターとして用いた場合が、T-agdAをターミネーターとして用いた場合に比して高いβ-グルクロニダーゼ比活性を示した(後出の表4を参照のこと)。つまり、2−5−1ターミネーターおよび2−5−1ターミネーターの断片(2−5−11、2−5−12,2−5−13ターミネーター)は、T-agdAに比して高いターミネーター活性を示すものであることがわかった。
【0032】
一方、2−5−1ターミネーター(1577bp)の塩基配列の第261−572位までの塩基からなる2−5−1の断片である、2−5−14ターミネーター(312bp、配列番号10)をターミネーターとして用いた場合、T-agdAをターミネーターとして用いた場合に比してβ-グルクロニダーゼ比活性が低くなった(後出の表4を参照のこと)。つまり、2−5−14ターミネーターは、T-agdAに比して低いターミネーター活性を示すものであることがわかった。2−5−14ターミネーターは、転写終結部位(配列番号2または7に示される塩基配列の第343位〜第460位であると推定)を含むものであるが、意外にも上記のような結果となった。これはターミネーターとしては、もはや短すぎることが上記結果の原因であると考えられた。
【0033】
図6に2−5−1ターミネーターと、4種類の断片(2−5−11ターミネーター、2−5−12ターミネーター、2−5−13ターミネーター、2−5−14ターミネーター)との関係を示した。図6中の下向き三角は転写終結部位として推定された箇所を示す。2−5−1ターミネーターをトランケートした場合、2−5−11ターミネーター、2−5−12ターミネーター、2−5−13ターミネーターでは、T-agdAよりも高いターミネーター活性を示したが、2−5−14ターミネーターまでトランケートするとターミネーター活性が顕著に低下したことから、(i)配列番号7に示される塩基配列からなる2−5−11ターミネーターを含むポリヌクレオチド、または(ii)上記配列番号7に示される塩基配列からなる2−5−11ターミネーターの部分断片であって、転写終結部位(配列番号7に示される塩基配列の転写終結部位である第343位〜第460位)を含み、かつ550bp以上のサイズを有するポリヌクレオチドを含むポリヌクレオチドが、糸状菌においてターミネーターとして機能し得るということが分かった。
【0034】
また、上記(i)および(ii)のポリヌクレオチドにおいて、1もしくは数個の塩基が欠失、置換もしくは付加された塩基配列であっても、糸状菌細胞内でターミネーター活性を有する限りにおいて本発明にかかる糸状菌用ターミネーターを構成し得る。また、上記(i)または(ii)のポリヌクレオチドと、ストリンジェントな条件下でハイブリダイズできるポリヌクレオチドであって、かつ、糸状菌細胞内でターミネーター活性を有する限りにおいて本発明にかかる糸状菌用ターミネーターを構成し得る。通常、上記(i)または(ii)のポリヌクレオチドと70%以上(好ましくは80%以上、さらに好ましくは90%、最も好ましくは95%以上)の相同性を有するポリヌクレオチドが、本発明にかかる糸状菌用ターミネーターを構成し得る。
【0035】
すなわち本発明のターミネーターは、糸状菌細胞内でターミネーター活性を有し、下記の(i)〜(iii)の何れかに示されるポリヌクレオチドを含むことを特徴としている。
(i)配列番号7に示される塩基配列からなるポリヌクレオチド。
(ii)上記(i)のポリヌクレオチドの部分断片であって、配列番号7に示される塩基配列の第343位〜第460位を含み、かつ550bp以上のサイズを有するポリヌクレオチド。
(iii)上記(i)または(ii)とストリンジェントな条件下でハイブリダイズするポリヌクレオチドであって、糸状菌細胞内でターミネーター活性を有するポリヌクレオチド。
【0036】
上記「ストリンジェントな条件下でハイブリダイズすることができる」とは、あるポリヌクレオチドと、これと相同性の高い他のポリヌクレオチドとの間で、二本鎖を形成させるのに十分な条件であることが意図される。「ストリンジェントな条件」としては、代表的な例として次の条件が挙げられる。すなわち、高イオン濃度下(例えば、6×SSC等)、65℃の温度条件下でハイブリダイズさせた後、低イオン濃度下(例えば、0.1×SSC等)、65℃、30分間の洗浄を行う条件が、「ストリンジェントな条件」の一例として挙げられる。
【0037】
なお、本発明の説明において、「糸状菌細胞内でターミネーター活性を有する」とは、アスペルギルス・オリゼのα-グルコシダーゼA遺伝子のターミネーターT-agdAをターミネーターとして含むプラスミドで糸状菌を形質転換して得られた形質転換体におけるレポーター遺伝子の発現量と、T-agdAの代わりに別のポリヌクレオチドターミネーターとして用いた以外は同一の条件でレポーター遺伝子を発現させた場合の発現量とを比較して、後者が前者と同等以上(つまり、(後者のレポーター発現量)÷(前者のレポーター発現量)≧1.0)であることを意味する。
【0038】
上記「糸状菌」としては、特に限定されるものではないが、例えば、アスペルギルス属糸状菌、ペニシリウム属糸状菌、トリコデルマ属糸状菌、リゾプス属糸状菌、ムコール属糸状菌、モナスカス属糸状菌、フザリウム属糸状菌等を挙げることができる。本発明のターミネーターは、アスペルギルス・オリゼより見出されたものであるため、特にアスペルギルス属糸状菌において奏効し得る。
【0039】
上記「アスペルギルス属糸状菌」としては、例えば、アスペルギルス・ニガー(Aspergillus niger)、アスペルギルス・オリゼ(Aspergillus oryzae)、アスペルギルス・アワモリ(Aspergillus awamori)、アスペルギルス・ウサミ(Aspergillus usami)、アスペルギルス・カワチ(Aspergillus kawachii)、アスペルギルス・ソーヤ(Aspergillus sojae)、アスペルギルス・ニドランス(Aspergillus nidulans)、アスペルギルス・アクレアタス(Aspergillus aculeatus)、アスペルギルス・テレウス(Aspergillus terreus)、アスペルギルス・フォエニシス(Aspergillus phoenicis)等を挙げることができる。
【0040】
また上記「ペニシリウム属糸状菌」としては、例えば、ペニシリウム・カマンベルチ(Penicillium camamberti)、ペニシリウム・ノタトウム(Penicillium notatum)、ペニシリウム・マルチカラー(Penicillium multicolor)、ペニシリウム・パルロゼナム(penicillium purpurogenum)、ペニシリウム・ロックフォルティ(Penicillium roqueforti)等を挙げることができる。
【0041】
また上記「トリコデルマ属糸状菌」としては、例えば、トリコデルマ・リーセイ(Trichoderma reesei)、トリコデルマ・ビリデ(Trichoderma viride)等を挙げることができる。
【0042】
また上記「リゾプス属糸状菌」としては、例えば、リゾプス・オリゼ(Rhizopus oryzae)、リゾプス・ジャポニカス(Rhizopus japonicus)等を挙げることができる。
【0043】
上記「ムコール属糸状菌」としては、例えば、ムコール・エスピー(Mucor sp.)等を挙げることができる。
【0044】
また上記「モナスカス属糸状菌」としては、例えば、モナスカス・パープレウス(Monascus purpureus)等を挙げることができる。
【0045】
また上記「フザリウム属糸状菌」としては、例えば、フザリウム・オキシスポラム(Fusarium oxysporum)等を挙げることができる。
【0046】
また本発明のターミネーターは、糸状菌細胞内でターミネーター活性を有する限りにおいて、上記の(i)〜(iii)の何れかに示されるポリヌクレオチドを含むもので構成されていればよい。つまり本発明のターミネーターは、(i)〜(iii)の何れかに示されるポリヌクレオチドのみからなるものであってもよいし、糸状菌細胞内でターミネーター活性を有する限りにおいてその他の塩基を含むものであってもよい。前者の好適な例としては、配列番号7、8、または9に示されるポリヌクレオチド(つまり2−5−11ターミネーター、2−5−12ターミネーター、2−5−13ターミネーター)が挙げられる。後者の好適な例としては、配列番号2に示される塩基配列からなるポリヌクレオチド(つまり2−5−1ターミネーター)や、配列番号2に示される塩基配列の第827位以降が切断された塩基配列からなるポリヌクレオチドが挙げられ、これらも本願発明のターミネーターに含まれる。
【0047】
(2.本発明のターミネーターの利用)
本発明のターミネーターは、該ターミネーターの上流に糸状菌細胞内で機能し得るプロモーターを連結することによってベクターを構築することができる。上記プロモーターの下流(3’末端側)であり、かつ糸状菌用ターミネーターの上流(5’末端側)に所望のタンパク質(「目的タンパク質」という)をコードする遺伝子(「目的遺伝子」という)を連結し、この目的遺伝子が連結されたベクターでこれらの糸状菌宿主(例えばアスペルギルス属糸状菌等)を形質転換し、それを培養することにより、目的タンパク質を著量生産させることができる。すなわち本発明は、上記ベクター、該ベクターを用いた形質転換方法、該形質転換方法により得られた形質転換体、および該形質転換体を用いたタンパク質の生産方法をも包含する。ベクターを構築するためのDNAの連結、挿入などは、公知の遺伝子工学的手法により行うことができる。
【0048】
(1)本発明のベクター
本発明のベクターを構成する、「糸状菌細胞内において機能し得るプロモーター」としては、糸状菌細胞内で下流に連結されたタンパク質をコードする目的遺伝子を転写する機能を有するポリヌクレオチドであれば特に限定されるものではない。具体的には、α−アミラーゼ(参考文献:Biosci. Biotech. Biochem., 1992年,第56巻,第11号,p1849−1853)、グルコアミラーゼ(参考文献:Current Genetics, 1992年,第22巻,第2号,p85−91)、セルラーゼ、セロビオハイドラーゼ(参考文献:J Biotechnol., 1991年,第17巻,第1号,p35−49)、アセトアミダーゼ(参考文献:Nucleic Acids Res., 1991年,第19巻,第10号,p2655−2660)等の加水分解酵素遺伝子、エノラーゼ(参考文献:Current Genetics, 2001年, p260−267)、3−ホスホグリセレートキナーゼ(参考文献:Mol. Gen. Genet., 1992年,第233巻,第1−2号,p231−40)、グリセルアルデヒド−3−ホスフェートデヒドロゲナーゼ(参考文献:Gene, 1992年,第120巻,第1号,p67−73)、アルコールデヒドロゲナーゼ(参考文献:Gene, 1989年,第79巻,第1号,p119−30)等の解糖系酵素遺伝子、トランスレーションエロンゲーションファクター遺伝子(参考文献:Appl. Microbiol. Biotechnol., 1998年,第50巻,第1号,p85−92)等のプロモーターが挙げられる。好適には、P−No8142プロモーター(参考文献:「化学と生物」,2000年,第38巻,第12号,p.831−838)が用いられる。
【0049】
また本発明のベクターには、以上の他に、所望により当該技術分野で公知の、エンハンサー、スプライシングシグナル、ポリA付加シグナル、選択マーカー、複製起点、目的遺伝子を導入する際に使用する制限酵素認識配列などを付加することができる。また、必要に応じて、目的遺伝子とその他のタンパク質(例えば、グルタチオンSトランスフェラーゼおよびプロテインA)をコードする遺伝子とを連結して本発明のベクターに連結することによって、目的タンパク質をその他のタンパク質との融合タンパク質として発現させることも可能である。このような融合タンパク質は、適当なプロテアーゼを使用して切断することによって、それぞれのタンパク質に分離することができる。
【0050】
また本発明にかかるベクターには、少なくとも1つの選択マーカーが含まれていることが好ましい。このようなマーカー遺伝子としては、一般に糸状菌形質転換体の選択に使用されているマーカーであることが好ましい。例えば、niaD、sC、argB、adeA、ptrA、pyrG等を使用することができる。また、大腸菌等の細菌培養用の薬剤耐性遺伝子(例えば、テトラサイクリン耐性遺伝子、アンピシリン耐性遺伝子等)が含まれていてもよい。上記選択マーカーを用いれば、本発明のベクターが宿主細胞に導入されたか否かを確認することができる。あるいは、目的タンパク質を融合ポリペプチドとして発現させてもよく、例えば、オワンクラゲ由来の緑色蛍光ポリペプチドGFP(Green Fluorescent Protein)をマーカーとして用い、目的タンパク質をGFP融合ポリペプチドとして発現させてもよい。
【0051】
なお、本発明のベクターを構成するポリヌクレオチドの由来は特に限定されるものではないが、本発明を食品等に応用することを考慮すればセルフクローニングを行うことが好ましいといえる。よってセルフクローニングを行う場合、ベクターを構成する各ポリヌクレオチドは、当該ベクターが導入される宿主と同一(同種)の生物である必要がある。つまり、本発明のターミネーターはアスペルギルス・オリゼ由来であるため、ベクターを構成するその他のポリヌクレオチドはアスペルギルス属糸状菌(より好ましくは、アスペルギルス・オリゼ)であることが好ましい。
【0052】
本発明のベクターに含まれ得る制限酵素認識配列としては、ベクター内に1箇所のみ存在し、かつ目的遺伝子の内部に存在しない制限酵素認識配列であれば、特に連結部の制限酵素認識配列として選択することができる。ベクターの汎用性を考慮すると、認識部位の出現頻度が少ない6塩基以上を認識する制限酵素の認識配列とすることが好ましく、8塩基以上を認識する制限酵素の認識配列とすることがより好ましい。6塩基を認識する制限酵素の認識配列としては、例えばGGGCCC(ApaI)、GGATCC(BamHI)、ATCGAT(ClaI)、AAGCTT(HindIII)、CCATGG(NcoI)、CATATG(NdeI)、CACGTG(PmlI)、GCATGC(SphI)、TCTAGA(XbaI)、CTCGAG(XhoI)等が挙げられる。8塩基以上を認識する制限酵素の認識配列としては、例えばGGCGCGCC(AscI)、GCGATCGC(AsiSI)、GGCCGGCC(FseI)、GCGGCCGC(NotI)、TTAATTAA(PacI)、GTTTAAAC(PmeI)、CCTGCAGG(SbfI)、GCCCGGGC(SrfI)、ATTTAAAT(SwaI)等が挙げられる。
【0053】
また本発明にベクターには、相同組み換えを行わせるために必要な宿主糸状菌由来のポリヌクレオチドが含まれていることが好ましい。当該宿主糸状菌由来のポリヌクレオチドの長さは、特に限定されないが、相同組換えの効率を考えると、200bp〜20000bpであることが好ましく、500bp〜10000bpであることがより好ましい。上記宿主糸状菌由来ポリヌクレオチドの長さが200bp以上であれば、相同組換えを効率よく起こさせることができる。また上記宿主糸状菌由来ポリヌクレオチドの長さが20000bp以下であれば、宿主糸状菌の細胞内に効率よく取り込ませることができる。
【0054】
また本発明のベクターには、プロモーターの下流(3’末端側)であり、かつターミネーターの上流(5’末端側)に、目的タンパク質をコードする遺伝子(「目的遺伝子」)が連結されていてもよい。ここで上記目的タンパク質をコードする目的遺伝子としては特に限定するものではないが、発現させることが好ましい有用タンパク質をコードする遺伝子が好ましい。例えば、糸状菌由来の細胞内タンパク質、分泌タンパク質、原核生物由来タンパク質、真核生物由来タンパク質等をコードする遺伝子が挙げられる。具体的には、α−アミラーゼ、グルコアミラーゼ、α−グルコシダーゼ、β−ガラクトシダーゼ、セルラーゼ、キチナーゼ、プロテアーゼ、アミノペプチダーゼ、カルボキシペプチダーゼ、リパーゼ、ホスホリパーゼ、フィターゼ、ヌクレアーゼ、カタラーゼ、グルコースオキシダーゼ、グルコースデヒドロゲナーゼ等の酵素遺伝子や、リボソーム遺伝子、トランスポーター遺伝子、シャペロン遺伝子、転写調節因子遺伝子などの酵素以外のタンパク質をコードする遺伝子からも自由に選択することができ、また機能未知の遺伝子も選択することができる。また、レポーター遺伝子として使用実績の多い大腸菌由来のβ-グルクロニダーゼ遺伝子も目的遺伝子として利用され得る。
【0055】
本発明のベクターには、宿主糸状菌細胞内で目的遺伝子を発現させるための発現単位(プロモーター領域、目的遺伝子のオープンリーディングフレーム、ターミネーター領域を含むポリヌクレオチド鎖)が少なくとも1つ含まれていてもよいが、当該発現単位が複数含まれていてもよい。
【0056】
本発明のベクターを構成する、それぞれのポリヌクレオチドを取得する方法は、特に限定されるものではなく、公知の技術によって取得することができる。例えば、公知の配列情報に基づいて、PCR等の増幅手段を用いる方法によって取得することもできるし、糸状菌の染色体DNAライブラリーからクローニングする方法によって取得することもできる。
【0057】
なおベクターの形状は、環状であっても鎖状であってもよい。
【0058】
(2)本発明の形質転換方法、形質転換体、およびタンパク質の生産方法
本発明の形質転換方法は、本発明のベクターのプロモーターの下流(3'末端側)であり、かつ糸状菌用ターミネーターの上流(5’末端側)に目的タンパク質をコードする遺伝子(目的遺伝子)が連結されているベクターで、糸状菌宿主(例えばアスペルギルス属糸状菌等)を形質転換する方法である。上記ベクターは、本発明のターミネーターを含んでいるため、かかる方法で得られた糸状菌の形質転換体を培養することによって、目的タンパク質を大量に生産させることができる。換言すれば本発明の形質転換方法は、目的タンパク質を高生産し得る糸状菌の作製方法であるとも言える。
【0059】
糸状菌宿主としては、上述の糸状菌が適宜採用され得る。また本発明のベクターを用いた宿主糸状菌の形質転換方法は特に限定されるものではなく、公知の方法が適宜採用される。例えば、カルシウム−PEG法、エレクトロポレーション法、等の公知の方法が形質転換方法として用いることができる。
【0060】
また上記形質転換方法により得られた形質転換体を培養する場合も、糸状菌の培養方法に通常用いられる培地、培養条件を適宜選択することにより行うことができる。形質転換体の培養方法は、固体培養であっても液体培養であってもよい。ここで形質転換体の培養に用いられる培地としては、特に限定されるものではないが、例えばデキストリン−ペプトン培地(2%デキストリン、1%ポリペプトン、0.5% KH2PO4、0.05%MgSO4・7H2O)、Czapek-dox培地(0.2%NaNO3、0.1%K2HPO4、0.05%MgSO4、0.05%KCl、0.001%FeSO4、3%グルコースまたは3%デンプン、pH5.5)、フスマ培地(小麦フスマ、水分含量40%)、等が採用され得る。また培養温度も、形質転換体が生育し得る温度であればよく、適宜最適な温度を検討の上、採用すればよい。形質転換体の培養温度としては、例えば、20〜37℃の範囲が好ましい。
【0061】
上記形質転換体の培養物には目的タンパク質が含まれており、必要に応じて定性また定量分析されたり、培養物から目的タンパク質が単離または精製されたりする。目的タンパク質が菌体外に分泌されている場合、液体培養であれば菌体培養液そのものを回収すればよい。一方、固体培養の場合には適当な緩衝液を用いて培養物から目的タンパク質を含む溶液を抽出して回収すればよい。また菌体内に目的タンパク質が蓄積されている場合は、菌体を公知の手段で破砕し、適当な緩衝液を用いて目的タンパク質を抽出すればよい。なお糸状菌のタンパク質生産系の場合、目的タンパク質は分泌される場合が多い。
【0062】
また、上記のようにして回収した目的タンパク質をさらに精製する場合は、例えば、タンパク質を含む溶液を周知の方法(例えば、硫安沈殿またはエタノール沈殿、酸抽出、陰イオンまたは陽イオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティークロマトグラフィー、ヒドロキシアパタイトクロマトグラフィー、およびレクチンクロマトグラフィー)によって精製すればよい。タンパク質の精製には高速液体クロマトグラフィー(「HPLC」)が好ましく採用され得る。
【0063】
本発明は上述した各実施形態に限定されるものではなく、請求項に示した範囲で種々の変更が可能であり、異なる実施形態にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施形態についても本発明の技術的範囲に含まれる。
【実施例】
【0064】
以下、本発明を実施例により具体的に説明するが、本発明はこれに限定されるものではない。
【0065】
〔実施例1〕アスペルギルス・オリゼ由来新規ターミネーターのクローニング
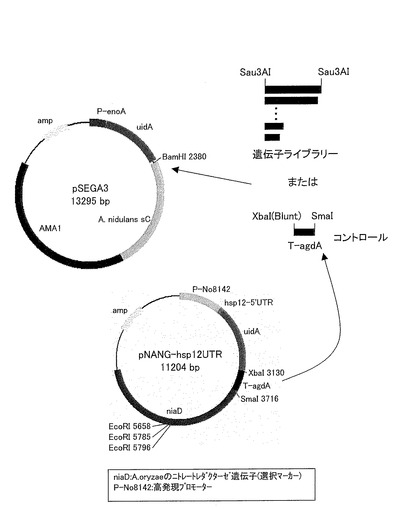
(クローニング用プラスミドpSEGA3の作製)
本実施例において使用するpSEGA3プラスミドの構築手順を、図1に基づいて説明する。図1に示すように、まず、pUC118(タカラバイオ社製)をSapIで消化し、末端をブランチングした後、精製し、アルカリフォスファターゼで脱リン酸化を行った。pUC118のSapI消化物(ブランチング処理済)を再び精製した後、PstIで消化を行い、アガロース電気泳動により、単離および精製を行った。
【0066】
アスペルギルス・ニデランス由来sCマーカー(参考文献:Buxton, F. P. et al Gene, 84, 329-334 (1989))の取得のため、pUsC(参考文献:Yamada, O. et al Biosci. Biotech. Biochem., 61, 1367-1369 (1997))をBspAPI−SmaI消化し、アガロース電気泳動により、単離および精製を行った後に、ブランチングを行い、再び精製した。
【0067】
P-enoA−uidA断片を取得するため、pNGEG-d4(参考文献:Toda, T. et al Curr. Genet. 40, 260-267 (2001))をXbaIで消化し、末端をブランチングした後、精製し、アルカリフォスファターゼで脱リン酸化を行った。再び精製を行った後、PstIで消化を行い、アガロース電気泳動により、P-enoA−uidA断片を単離および精製した。
【0068】
このように調製したpUC118プラスミドと、アスペルギルス・ニデランス由来sCマーカー断片と、P-enoA−uidA断片とをライゲーションし、pSEG3プラスミドを得た。
【0069】
次いで、pSEG3をPstI-DraIIIで消化し、アガロース電気泳動により、単離および精製した後にブランチングを行い、さらに精製した。その後、pSEG3−PstI-DraIII消化物をアルカリフォスファターゼで脱リン酸化を行い、再び精製を行った。
【0070】
アスペルギルス・ニデランス由来AMA1遺伝子断片の取得のため、ARp1(参考文献:Gems, D. H. et al Gene, 98, 61-67 (1991))をHindIIIで処理し、アガロース電気泳動により、単離および精製を行った後に、ブランチングを行い、再び精製した。
【0071】
このように調製したpSEG3プラスミドと、AMA1断片とをライゲーションし、pSEGA3プラスミドを得た。
【0072】
(プラスミドライブラリーの作製)
アスペルギルス・オリゼRIB40(独立行政法人酒類総合研究所より入手)より、ゲノムDNAを取得し、制限酵素Sau3AIで部分分解を行った。アガロース電気泳動を行ってゲノムDNA断片(0.5〜4kb)を回収および精製を行った後に、事前にBamHIで消化および精製しておいたpSEGA3にライゲーションにより挿入した(図3を参照のこと)。
【0073】
また、α-グルコシダーゼターミネーター(T-agdA)をpNAN-hsp12UTR(参考文献:Koda, A. et al Appl. Microbiol. Biotechnol., 70, 333-336 (2006))からXbaI-SmaI処理し、精製後に、ブランチングし、アガロース電気泳動後、単離および精製した後に、事前にBamHIで消化および精製しておいたpSEGA3にライゲーションにより挿入し、コントロール用のプラスミドとした(図3を参照のこと)。
【0074】
(形質転換体の取得とβ−グルクロニダーゼ活性の確認)
プラスミドライブラリーを調製後、アスペルギルス・ニガー NS48株(IFO4343から変異処理により取得したniaD、sC二重欠損株)へ、プロトプラスト-PEG法にて形質転換導入した。スクリーニング用培地として、0.2% 亜硝酸ナトリウムを含有する最少培地(Czapek-dox:0.2%NaNO3、0.1%K2HPO4、0.05%MgSO4、0.05%KCl、0.001%FeSO4、3%グルコースまたは3%デンプン、pH5.5)にβ−グルクロニダーゼが存在するとき青色の色素を生産するX− グルクロナイドを50μg/ml含むプレートに接種し、数日間培養後、青色に発色するコロニーを選択した。
【0075】
T-agdAを挿入したコントロール用プラスミドについても同様に、アスペルギルス・ニガー NS48株へ形質転換導入を行い、形質転換体を取得した。プラスミドライブラリーを導入した形質転換体の中から、コントロール用プラスミドを導入した形質転換体に比べて青色の発色が強い形質転換体をスクリーニングし、候補株5株を選抜した。
【0076】
(候補株からのプラスミドの単離と挿入断片の解析)
β−グルクロニダーゼ高発現であった形質転換体からDNA画分を調製した。当該DNA画分を大腸菌(イー・コリDH5α)を形質転換し、その形質転換体から得られるプラスミドDNAのBamHIサイトに挿入されていたターミネーターとして機能するDNA鎖のシークエンス解析を行った。
【0077】
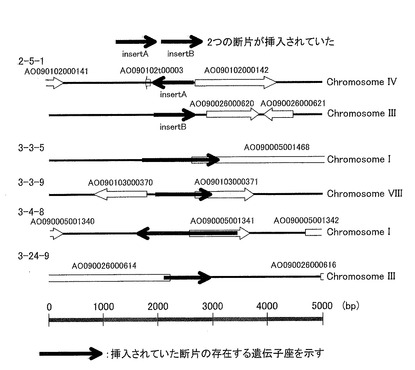
その塩基配列を配列番号2〜6に示す。これらの塩基配列をアスペルギルス・オリゼのゲノムデーターベース(独立行政法人 製品評価技術基盤機構)と照合し、それぞれのDNA鎖が存在する染色体上の座を解析した。図2に、各DNA鎖の染色体上の座を、その近傍のORF予測とともに示した。なお、図2中に示した矢印はORFの転写方向を示している。またORFに記載されているAOに続く番号は、シークエンスされた微生物のゲノムデーターベースに登録されている番号を示しており、AOはアスペルギルス・オリゼに由来するタンパク質をコードする遺伝子であることを意味する。配列番号2〜6に示されるDNA鎖は、いずれもアスペルギルス・オリゼのゲノムデーターベースでORFと予測される塩基配列の直後から2000bp下流以内に、ORFと同じ転写方法で存在しておらず、いずれもターミネーターとして予測され得ない、全く新規なターミネーターであることが判明した。これらを、それぞれ2−5−1ターミネーター(配列番号2)、3−3−5ターミネーター(配列番号3)、3−3−9ターミネーター(配列番号4)、3−4−8ターミネーター(配列番号5)、3−24−9ターミネーター(配列番号6)と称する。
【0078】
なお、配列番号2で示される2−5−1ターミネーターは、異なる2つのDNA(図2中insert Aおよびinsert Bで表記する)から構成されていることが判明した。
【0079】
〔実施例2〕既知のターミネーター(T-agdA)との比較
実施例1で得られたアスペルギルス・オリゼ由来の新規ターミネーターと、従来公知のアスペルギルス・オリゼ由来α−グルコシダーゼ遺伝子のターミネーター(T-agdA)とのターミネーターとしての性能を比較すべく、それぞれのターミネーターを用いたときのタンパク質発現能を、β−グルクロニダーゼ遺伝子をレポーター遺伝子として比較した。
【0080】
比較は、インテグレート型のプラスミドを用いることとして、発明者が以前に作製したpNANG-hsp12UTRを用いることにした。このプラスミドは、高発現プロモーターであるP-No8142、アスペルギルス・オリゼのHsp12遺伝子の5’UTR、およびβ−グルクロニダーゼ遺伝子が連結されており、β−グルクロニダーゼ遺伝子の直後にα−グルコシダーゼ遺伝子のターミネーター(T-agdA)がXbaI-SmaIサイトを利用して導入されている。
【0081】
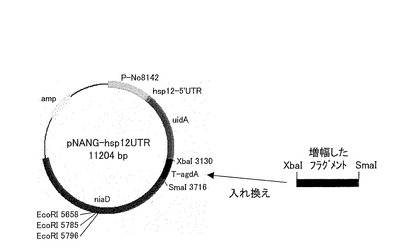
(新規ターミネーター導入プラスミドの作製)
pNANG-hsp12UTR中のα―グルコシダーゼ遺伝子のターミネーターを、新規ターミネーターに置換する手順を図5に示す。図5に示すように、pNANG-hsp12UTRをXbaI−SmaI消化した後、アガロース電気泳動を行って、pNANG-hsp12UTR−XbaI−SmaI消化物を単離および精製した。
【0082】
実施例1にて取得したプラスミドを鋳型にして、配列番号2〜6に示した新規ターミネーターをそれぞれPCR法にて取得した。このとき、それぞれのDNA鎖の5’末端側にXbaIサイトおよび3’末端側にSmaIサイトが付加されるように、両端にGGGが付加されるようにデザインされたプライマーを用いた。PCRに使用したプライマーを表1に示す。
【0083】
【表1】

【0084】
得られた新規ターミネーターをXbaIで消化し、精製した後に、pNANG-hsp12UTRのXbaI−SmaIサイトにライゲーションすることにより、ターミネーターのみが置換されたプラスミドを構築した。
【0085】
ただし、3−4−8ターミネーターはXbaIで切断されるため、3−4−8ターミネーターで置換する場合には以下のようにした。pNANG-hsp12UTRをXbaI消化後にブランチングし、その後SmaIのイソシゾマーであるXmaIにより消化し、さらに精製したプラスミド断片の5’末端側にXbaIサイトに相当するAを含み、3’末端側にSmaIサイトを含むように設計されたプライマーを用いてPCR法にて増幅した。得られたプラスミドをXmaIで消化し、そこに精製した3−4−8ターミネーターのDNA断片を挿入することにより、プラスミドを構築した。
【0086】
なお、β−グルクロニダーゼ遺伝子の終止コドンTGAからXbaIサイトまでATCAACAACの9塩基が挿入されているが、コントロールであるT-agdAがXbaI-SmaIサイトに挿入されているときに著量のβ−グルクロニダーゼが生産されていることから、挿入された9塩基によってターミネーター活性は阻害されていないと考えられる。よって、XbaI-SmaIサイトに任意の新規ターミネーターのDNA断片を導入することで、ターミネーターとしての性能を評価することができると考えられる。
【0087】
(形質転換体の取得とβ−グルクロニダーゼ比活性の測定)
構築したすべてのプラスミドを、同じくプラスミド上に含まれるniaDマーカー遺伝子内のEcoRIサイトで切断して直鎖状にし、アスペルギルス・オリゼniaD300(硝酸還元酵素欠損株)を形質転換した。それぞれの形質転換体からゲノムDNAを抽出し、サザン解析によりゲノム上のniaD遺伝子座にプラスミドが1コピー相同的に導入された形質転換体を得た。これらの形質転換体をデキストリン−ペプトン培地(2%デキストリン、1%ポリペプトン、0.5% KH2PO4、0.05%MgSO4・7H2O)にて培養し、菌体内のβ−グルクロニダーゼ比活性を測定し、それぞれのターミネーターの性能を比較した。その結果を表2に示す。
【0088】
【表2】
【0089】
表2に示すように、α−グルコシダーゼ遺伝子のターミネーター(T-agdA)と比較して、β−グルクロニダーゼ比活性の値に差がみられた。特に、配列番号2に示す2−5−1ターミネーターを利用した場合、β−グルクロニダーゼ比活性の値がT-agdAの1.34倍となっていた。
【0090】
これまでにT-agdAを用いてβ−グルクロニダーゼを生産させた報告(参考文献:Tsuboi, et al Biosci. Biotechnol. Biochem., 69, 206-208 (2005))では、β−グルクロニダーゼが菌体内可溶性タンパク質の約30%をも占めていることが報告されているが、2−5−1ターミネーターを用いることで、β−グルクロニダーゼの生産性をさらに1.34倍増加することができると考えられる。
【0091】
〔実施例3〕2−5−1ターミネーターの短縮化
(3’RACE法による2−5−1ターミネーターの転写終結部位の決定)
2−5−1ターミネーターの転写終結部位を、公知の方法である3’RACE法(Rapid Amplification cDNA Ends)により決定した。具体的な方法を以下に述べる。
【0092】
実施例2で取得した2−5−1ターミネーターを導入した形質転換体を、15mlのデキストリン・ペプトン培地で30℃、2日間振とう培養後、ガラスフィルターで集菌し、滅菌水で洗浄した。この菌体を新しいデキストリン・ペプトン培地15mlに移し、さらに30℃で12時間振とう培養し、同じくガラスフィルターで集菌し滅菌水で洗浄した後、RNAの抽出に用いた。RNAの抽出にはISOGEN(和光純薬社製)を用いた。得られたRNAを用いて、3’-Full RACE core set(タカラバイオ社製)にて逆転写されたクローンを取得し、9クローンのシークエンス解析を行った。
【0093】
決定された転写終結部位を、図4に示す。図4中、転写終結部位を下向き三角で示した。転写終結部位はターミネーターの343〜460bpに存在していた。
【0094】
(2−5−1ターミネーターをトランケートしたターミネーター断片の作製および評価)
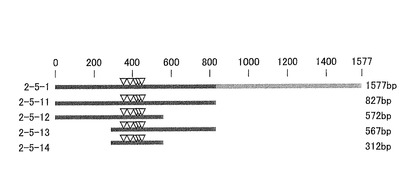
上述の3’RACE法で決定された転写終結部位、およびそれらの転写終結部位より上流30bpまでのヌクレオチド鎖を含む領域を削除しないようにトランケートした2−5−1ターミネーターの断片を4種類作製した。2−5−1ターミネーターと、作製された断片との関係を示す模式図を図6に示した。図6中の下向き三角は、3’RACE法によって決定された2−5−1ターミネーターの転写終結部位に相当する。
【0095】
2−5−11ターミネーター(827bp、配列番号7)は、2−5−1ターミネーター(1577bp)の塩基配列の第1−827位までの塩基からなる2−5−1の断片である。2−5−12ターミネーター(572bp、配列番号8)は、2−5−1ターミネーター(1577bp)の塩基配列の第1−572位までの塩基からなる2−5−1の断片である。2−5−13ターミネーター(567bp、配列番号9)は、2−5−1ターミネーター(1577bp)の塩基配列の第261−827位までの塩基からなる2−5−1の断片である。2−5−14ターミネーター(312bp、配列番号10)は、2−5−1ターミネーター(1577bp)の塩基配列の第261−572位までの塩基からなる2−5−1の断片である。
【0096】
4種類のターミネーターのそれぞれ、またはT-agdAを実施例2と同様の手法で、pNANG-hsp12UTRのXbaI-SmaIサイト内にPCR法にてクローニングした(図5を参照のこと)。PCR法において使用した各プライマーセットを表3に示した。
【0097】
【表3】
【0098】
上記PCR法によって取得した各種ターミネーター断片の塩基配列を配列番号7〜10に示す。これらの各種ターミネーターが挿入されたベクターを用いて、上記の方法でアスペルギルス・オリゼniaD300(硝酸還元酵素欠損株)を形質転換し、該ベクターが相同的に1コピー導入された形質転換体を得た。これらの形質転換体をデキストリン-ペプトン培地にて培養し、菌体内のβ-グルクロニダーゼ比活性を測定し、ターミネーターの性能を比較した。その結果を表4に示す。
【0099】
【表4】
【0100】
表4に示すように、トランケートしたターミネーターのうち、2−5−11ターミネーター、2−5−12ターミネーター、および2−5−13ターミネーターは、従来公知のα−グルコシダーゼ遺伝子のターミネーター(T-agdA)に比較して高いβ-グルクロニダーゼ比活性を示した。また2−5−11ターミネーターおよび2−5−12ターミネーターにいたっては、トランケート前の2−5−1と比較してβ-グルクロニダーゼ比活性が増加した。
【0101】
したがって、2−5−1ターミネーター、並びにその断片である2−5−11ターミネーター、2−5−12ターミネーター、および2−5−13ターミネーターは、糸状菌用のターミネーターとして強力な活性を有するものであることが確認された。
【産業上の利用可能性】
【0102】
本発明は糸状菌細胞内でより強力なターミネーターとして機能し得るポリヌクレオチドを提供する。当該新規ターミネーターを糸状菌のタンパク質生産系に利用することによって、従来公知の糸状菌のタンパク質生産系に比してタンパク質の生産効率を増大させることが可能となる。
【0103】
したがって、本発明は、糸状菌を用いてタンパク質を産生させる、医薬、検査薬、食品等の産業において好適に利用される。
【技術分野】
【0001】
本発明は新規なターミネーターおよび当該ターミネーターを用いてタンパク質を効率的に製造するための方法に関する。詳しくは、糸状菌細胞内でターミネーターとして機能し得るポリヌクレオチド、当該ポリヌクレオチドを含むベクター、当該ポリヌクレオチドの上流に任意のプロモーターおよびタンパク質をコードするポリヌクレオチドが連結されてなるベクターを用いたタンパク質の製造方法、当該ポリヌクレオチドの上流に任意のプロモーターおよび目的遺伝子が連結されてなるベクター、および当該ポリヌクレオチドの上流に任意のプロモーターおよび目的遺伝子が連結されてなるベクターを糸状菌細胞に導入することによる目的遺伝子の発現方法等に関するものである。
【背景技術】
【0002】
遺伝子組み換えによる有用タンパク質生産の宿主としては、大腸菌や枯草菌をはじめ酵母、昆虫、植物細胞、動物細胞等数多くの系が開発されており、生産させようとする目的タンパク質によってより適した系が選択されているのが現状である。
【0003】
特に、糸状菌であるアスペルギルス(Aspergillus)属やトリコデルマ(Trichoderma)属は酵素タンパク質を著量に分泌生産する能力に優れているため、工業的な酵素製剤の生産に利用されている。これらの菌株の中には、液体培養を行った際に培養液1リットルあたり20g-protein以上、フスマ培養を行った際に小麦フスマ1kgあたり50g-proteinという高い生産性を示すものもある。
【0004】
一方、異種タンパク質の生産においては、タンパク質のフォールディングや糖鎖付加などの翻訳後修飾が本来の生物と同様に行われることが重要である。糸状菌は大腸菌や酵母より進化的に高等動物に近いため、動物由来異種タンパク質を活性ある形で生産することも期待できる。また、麹菌アスペルギルス・オリゼのように古くから発酵食品の製造に用いられているものが多く、その生産物は安全であるというGRAS(generally regarded as safe)グレードとして認められているため、これらの糸状菌による有用タンパク質生産も認可されやすいと考えられる。
【0005】
アスペルギルス・オリゼをはじめとする糸状菌は一般的に有性生活環が認められず、分生子も多核であるために、古典遺伝学的手法による遺伝解析は容易ではなく、分子生物学的研究も他の微生物に比べて遅れていた。しかし、1980年代後半になって、産業的に重要な糸状菌について形質転換系が確立され、糸状菌の遺伝子工学が発展してきた。このような状況の中、前述した利点にも後押しされ、糸状菌を宿主としたさまざまな有用タンパク質の生産が報告されるようになってきた(例えば、特許文献1、非特許文献1を参照)。また、タンパク質を高発現させるために多くの強力なプロモーターが取得されており、麹菌ではαアミラーゼ遺伝子のプロモーターや(例えば特許文献2を参照)、本発明者らの改良型プロモーター(例えば特許文献3を参照)、スーパーオキサイドジスムターゼ遺伝子のプロモーター(例えば特許文献4を参照)などが使用されその有用性が認められている。
【0006】
さて、遺伝子発現においては、プロモーターによる転写促進に加え、ターミネーターによる効率的な転写終結も重要であると考えられる。植物ではターミネーターとしてシロイヌナズナのHSP18.2遺伝子のターミネーターが単離され、植物発現系における有用性が認められている(例えば非特許文献2を参照)。
【0007】
糸状菌においては、アスペルギルス・オリゼのα-グルコシダーゼ遺伝子AのターミネーターであるT-agdA(例えば非特許文献3を参照)、グルコアミラーゼBのターミネーター(例えば非特許文献4を参照)などがよく使用されている。これらは、各遺伝子の終止コドンの5塩基下流から585塩基まで、および1塩基下流から534塩基までの配列をターミネーターとして利用している。しかし、ターミネーターの配列、構造、機能について詳細に解析した報告はなく、その機構は全く解明されていない。それゆえ、糸状菌宿主においてターミネーターを改善することによりタンパク質の生産量を増大させようとする試みはこれまでなかった。
【0008】
ここで、真核生物で一般に提唱されている転写終結機構について説明する。動物細胞などでは、転写終結部位の近くにAAUAAA という配列があり、これを特定の酵素が認識して、その3’側(下流)約25塩基のところでmRNAを切断している。さらに、mRNAの3’末端に普通200〜400個のA(「ポリ(A)鎖」という)が付加される。このポリ(A)鎖はmRNAの目印で、キャップ構造とともに働いて、翻訳の効率を高めるとされている。また、3’側からの分解を防ぐ働きもある。一方、糸状菌の転写終結機構についてはほとんど解明されておらず、ターミネーター配列と転写終結との関係を定量的に解析した報告はない。もし、糸状菌においてタンパク質生産に利用できるさらに強力なターミネーターを取得できれば、さまざまな有用タンパク質の生産性を向上させることができると考えられる。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特開昭62−272988号公報(公開日:昭和62(1987)年11月27日)
【特許文献2】特開平07−51067号公報(公開日:平成7(1995)年2月28日)
【特許文献3】特開平9−9968号公報(公開日:平成9(1997)年1月14日)
【特許文献4】特開2001−224381号公報(公開日:平成12(2001)年8月21日)
【非特許文献】
【0010】
【非特許文献1】Christensenら、High Level Expression of Recombinant Genes in Aspergillus Oryzae. Nature Biotechnology, 米国, 1988年, 第6巻, p1419−1422
【非特許文献2】真野ら、「ターミネーター領域の改変による導入遺伝子の高発現化」、平成17年度日本生物工学会講演要旨集、P123、社団法人 日本生物工学会、発行日2005年9月25日
【非特許文献3】Minetokiら、Improvement of promoter activity by the introduction of multiple copies of the conserved region III sequence, in volved in the efficient expression of Aspergillus oryzae amylase-encoding genes. Appl. Microbiol. Biotechnol., 米国, 1998年, 第50巻, p459−467
【非特許文献4】Hataら、Nucleotide sequence of an alternative glucoamylase-encoding gene (glaB) expressed in solidstate culture of Aspergillus oryzae. Gene, 米国, 1998年, 第207巻, p127−134
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、糸状菌細胞内でより強力なターミネーターとして機能し得るポリヌクレオチドをアスペルギルス属糸状菌からクローニングし、これをアスペルギルス属等の糸状菌におけるタンパク質の生産に利用することにある。
【課題を解決するための手段】
【0012】
本発明者らは、上記目的を達成するために鋭意検討を行った結果、アスペルギルス・オリゼから新規かつ強力なターミネーターとして機能し得るポリヌクレオチドをクローニングすることに成功し、本発明を完成するに至った。
【0013】
すなわち本発明にかかる糸状菌用ターミネーターは、糸状菌細胞内でターミネーター活性を有し、下記の(i)〜(iii)の何れかに示されるポリヌクレオチドを含むことを特徴とする糸状菌用ターミネーター:
(i)配列番号7に示される塩基配列からなるポリヌクレオチド;
(ii)上記(i)のポリヌクレオチドの部分断片であって、配列番号7に示される塩基配列の第343位〜第460位を含み、かつ550bp以上のサイズを有するポリヌクレオチド;
(iii)上記(i)または(ii)とストリンジェントな条件下でハイブリダイズするポリヌクレオチドであり、かつ糸状菌細胞内でターミネーター活性を有するポリヌクレオチド。
【0014】
また本発明にかかるベクターは上記本発明にかかる糸状菌用ターミネーター、および糸状菌細胞内において機能し得るプロモーターを含むことを特徴としている。
【0015】
また本発明にかかるポリペプチドの生産方法は、上記本発明にかかるベクターのプロモーターの下流であり、かつ糸状菌用ターミネーターの上流に所望のタンパク質をコードする遺伝子が連結されたベクターで、糸状菌を形質転換することを特徴としている。
【0016】
また本発明は、上記本発明にかかる形質転換方法により取得された形質転換体をも包含する。
【0017】
また本発明は、上記本発明にかかる形質転換体を培養する工程を含む、タンパク質の生産方法をも包含する。
【0018】
なお、上記本発明にかかる糸状菌用ターミネーターは全く新規のターミネーターである。また上記本発明にかかる糸状菌用ターミネーターは、アスペルギルス・オリゼのORF予測の終止コドンの下流2000bp以内にORFの転写方向と同じ方向で存在しているものでもなかった。α-グルコシダーゼ遺伝子やグルコアミラーゼB遺伝子のターミネーター等、一般にターミネーターは遺伝子の終止コドンの下流約600bpまでに存在することを考慮すると、今回取得した糸状菌用ターミネーターは、ゲノムデーターベースにおいてターミネーターとして当業者が予測できるDNAではない。また、そのターミネーター活性は、従来公知のT-agdAに比して高い。よって本発明にかかる糸状菌用ターミネーターは、従来技術に対して新規性および進歩性を有することは明白である。
【発明の効果】
【0019】
本発明に係る新規ターミネーターを糸状菌のタンパク質生産系に利用することによって、従来公知の糸状菌のタンパク質生産系に比してタンパク質の生産効率を増大させることが可能となる。
【図面の簡単な説明】
【0020】
【図1】実施例1におけるpSEGA3プラスミドの構築手順を示す図である。
【図2】実施例1において取得されたターミネーター候補DNAのアスペルギルス・オリゼ染色体上の遺伝子座を示す図である。
【図3】実施例1における遺伝子ライブラリーおよびコントロール用ベクターの構築手順を示す図である。
【図4】実施例3において決定された2−5−1ターミネーターの転写終結部位を示す図である。
【図5】実施例2、3における新規ターミネーター導入プラスミドの構築手順を示す図である。
【図6】2−5−1ターミネーターと4種類の断片との関係を示す模式図である。
【発明を実施するための形態】
【0021】
以下、本発明の実施の形態について詳細に説明する。但し、本発明はこれに限定されるものではなく、記述した範囲内で種々の変形を加えた態様で実施できるものである。また、本明細書中に記載された学術文献及び特許文献の全てが、本明細書中において参考として援用される。なお、本明細書において特記しない限り、数値範囲を示す「A〜B」は、「A以上、B以下」であることを示す。
【0022】
(1.糸状菌用ターミネーター)
本発明にかかる糸状菌用ターミネーター(以下「本発明のターミネーター」という)は、アスペルギルス・オリゼのゲノムDNAから見出されたものである。アスペルギルス・オリゼのゲノムの全遺伝子配列の解読は2005年に終了(Nature, 438, 1157-1161 (2005)を参照のこと)し、ポストゲノム期を迎えている。この成果により、有用な遺伝子の探索や生産性の高いプロモーターの取得などが行われている。
【0023】
糸状菌のターミネーターについても予測遺伝子の直後から数百bpの塩基配列をPCR等で取得することが可能となったが、ターミネーターに関しては、翻訳効率の増加やmRNAの安定性への寄与が示唆されているものの、糸状菌において実際にこれらの性能が優れているターミネーターを探索するための知見はほとんど得られていないのが実情である。
【0024】
そこで本発明者らは、アスペルギルス・オリゼのゲノムライブラリーを作製し、ショットガンクローニングを行うことで優れたターミネーターとして機能する塩基配列を取得することを目的として実験を行った。
【0025】
アスペルギルス属糸状菌で自己複製可能なAMA1 (autonomously maintained in Aspergillus) 配列(参考文献:Johnstone, I. L. Microbiol. Sci., 2, 307-311 (1985) )、プロモーターとしてP-enoA(参考文献:Toda, T. et al Curr. Genet. 40, 260-267 (2001))、およびレポーター遺伝子としてイー・コリ由来のβ-グルクロニダーゼ(GUS)をコードする遺伝子(uidA)(参考文献:Jefferson, R. A. et al Proc. Natl. Acad. Sci. USA 83, 8447-8451 (1986))を連結し、高形質転換効率系のターミネーター検索用のプラスミドを構築した。このプラスミドは、AMA1配列を有し、インテグレート型に比べて数百倍の形質転換効率が得られるため、アスペルギルス属糸状菌の遺伝子のショットガンクローニングを可能にする。
【0026】
該プラスミドを用い、アスペルギルス・オリゼ由来の種々のゲノムDNA断片をβ-グルクロニダーゼ遺伝子の下流に公知の方法にてクローニングし、最少培地でβ-グルクロニダーゼを高生産する形質転換体を取得した。最少培地としては、0.2%NaNO3、0.1%K2HPO4、0.05%MgSO4、0.05%KCl、0.001%FeSO4、3%グルコースまたは3%デンプン、pH5.5のツァペックドックス(Czapek-Dox)培地が用いられた。なお、上記「高生産する形質転換体」とは、プロモーターであるP-enoA、レポーター遺伝子であるβ-グルクロニダーゼ遺伝子の下流に、従来までにタンパク質生産に利用されてきたアスペルギルス・オリゼのα-グルコシダーゼA遺伝子のターミネーター(適宜「T-agdA」と表記する)を連結してなるプラスミドで形質転換した時に得られる形質転換体のβ-グルクロニダーゼ活性に比して、アスペルギルス・オリゼのα-グルコシダーゼA遺伝子のターミネーターをアスペルギルス・オリゼ由来の種々のゲノムDNA断片に置換したプラスミドで形質転換されてなる形質転換体のβ-グルクロニダーゼ活性が高いことを意味する。逆に後者のβ-グルクロニダーゼ活性が前者に比して低い場合には、後者の形質転換体は低生産であるといえる。
【0027】
高生産であった形質転換体から公知の方法にてプラスミドDNAを単離し、これらのプラスミドのターミネーター領域のシークエンスを行ったところ、767〜1882bpからなる5種類のDNAの挿入が確認できた。これらのDNAは全てアスペルギルス・オリゼのゲノムデーターベース(独立行政法人製品評価技術基盤機構)上の塩基配列と一致したが、公開されているORF予測の終止コドンの下流2000bp以内にORFの転写方向と同じ方向で存在しているものではなかった。α-グルコシダーゼ遺伝子やグルコアミラーゼB遺伝子のターミネーター等、一般にターミネーターは遺伝子の終止コドンの下流約600bpまでに存在することを考慮すると、今回取得したDNAはいずれの配列も、ゲノムデーターベースにおいてターミネーターとして予測できるDNAではないと考えられる。
【0028】
続いて、上記で取得したDNAをPCRにて増幅し、プロモーターとしてP-No8142(「化学と生物」,2000年,第38巻,第12号,p.831−838)、Hsp12遺伝子の 5’UTR(参考文献「Koda, A. et al Appl. Microbiol. Biotechnol., 70, 333-336 (2006)」)、レポーター遺伝子としてβ-グルクロニダーゼ遺伝子、マーカーとしてniaDを含むベクターのβ-グルクロニダーゼ遺伝子の下流に連結した。これらの発現ベクターを用いて、アスペルギルス・オリゼniaD300(硝酸還元酵素欠損株)を形質転換し、得られた形質転換体のサザン解析を行い、導入ベクターが染色体のniaD座位に1コピーだけ導入された形質転換体を選択した。これらの形質転換体の菌体内β-グルクロニダーゼ比活性を測定することにより、各DNA配列のターミネーターとしての性能を評価した。
【0029】
その結果、配列番号2に示す2−5−1ターミネーターを用いたとき、T-agdA(塩基配列を配列番号1に示す)を導入したコントロールベクターよりもβ-グルクロニダーゼの比活性が1.34倍増大することを見出した。このことにより、ゲノムデーターベースではターミネーターとして予測されていない配列の中に、既知のターミネーターよりも優れたターミネーターとして機能するDNAが存在することが示された。
【0030】
次に、公知の方法にて配列番号2に示す2−5−1ターミネーターのターミネーター活性を保持する領域を特定した。転写終結部位(配列番号2または7に示される塩基配列の第343位〜第460位であると推定)およびその約30bp上流までの塩基を削除しないようにトランケートし、これらをターミネーターとしたプラスミドを作製し、上記と同様の方法で当該プラスミドが1コピー導入されたアスペルギルス・オリゼ形質転換体を取得し、形質転換体の菌体内のβ-グルクロニダーゼ比活性を測定した。
【0031】
その結果、2−5−1ターミネーター(1577bp)の塩基配列の第1−827位までの塩基からなる断片である、2−5−11ターミネーター(827bp、配列番号7);2−5−1ターミネーター(1577bp)の塩基配列の第1−572位までの塩基からなる断片である、2−5−12ターミネーター(572bp、配列番号8);2−5−1ターミネーター(1577bp)の塩基配列の第261−827位までの塩基からなる断片である、2−5−13ターミネーター(567bp、配列番号9)をターミネーターとして用いた場合が、T-agdAをターミネーターとして用いた場合に比して高いβ-グルクロニダーゼ比活性を示した(後出の表4を参照のこと)。つまり、2−5−1ターミネーターおよび2−5−1ターミネーターの断片(2−5−11、2−5−12,2−5−13ターミネーター)は、T-agdAに比して高いターミネーター活性を示すものであることがわかった。
【0032】
一方、2−5−1ターミネーター(1577bp)の塩基配列の第261−572位までの塩基からなる2−5−1の断片である、2−5−14ターミネーター(312bp、配列番号10)をターミネーターとして用いた場合、T-agdAをターミネーターとして用いた場合に比してβ-グルクロニダーゼ比活性が低くなった(後出の表4を参照のこと)。つまり、2−5−14ターミネーターは、T-agdAに比して低いターミネーター活性を示すものであることがわかった。2−5−14ターミネーターは、転写終結部位(配列番号2または7に示される塩基配列の第343位〜第460位であると推定)を含むものであるが、意外にも上記のような結果となった。これはターミネーターとしては、もはや短すぎることが上記結果の原因であると考えられた。
【0033】
図6に2−5−1ターミネーターと、4種類の断片(2−5−11ターミネーター、2−5−12ターミネーター、2−5−13ターミネーター、2−5−14ターミネーター)との関係を示した。図6中の下向き三角は転写終結部位として推定された箇所を示す。2−5−1ターミネーターをトランケートした場合、2−5−11ターミネーター、2−5−12ターミネーター、2−5−13ターミネーターでは、T-agdAよりも高いターミネーター活性を示したが、2−5−14ターミネーターまでトランケートするとターミネーター活性が顕著に低下したことから、(i)配列番号7に示される塩基配列からなる2−5−11ターミネーターを含むポリヌクレオチド、または(ii)上記配列番号7に示される塩基配列からなる2−5−11ターミネーターの部分断片であって、転写終結部位(配列番号7に示される塩基配列の転写終結部位である第343位〜第460位)を含み、かつ550bp以上のサイズを有するポリヌクレオチドを含むポリヌクレオチドが、糸状菌においてターミネーターとして機能し得るということが分かった。
【0034】
また、上記(i)および(ii)のポリヌクレオチドにおいて、1もしくは数個の塩基が欠失、置換もしくは付加された塩基配列であっても、糸状菌細胞内でターミネーター活性を有する限りにおいて本発明にかかる糸状菌用ターミネーターを構成し得る。また、上記(i)または(ii)のポリヌクレオチドと、ストリンジェントな条件下でハイブリダイズできるポリヌクレオチドであって、かつ、糸状菌細胞内でターミネーター活性を有する限りにおいて本発明にかかる糸状菌用ターミネーターを構成し得る。通常、上記(i)または(ii)のポリヌクレオチドと70%以上(好ましくは80%以上、さらに好ましくは90%、最も好ましくは95%以上)の相同性を有するポリヌクレオチドが、本発明にかかる糸状菌用ターミネーターを構成し得る。
【0035】
すなわち本発明のターミネーターは、糸状菌細胞内でターミネーター活性を有し、下記の(i)〜(iii)の何れかに示されるポリヌクレオチドを含むことを特徴としている。
(i)配列番号7に示される塩基配列からなるポリヌクレオチド。
(ii)上記(i)のポリヌクレオチドの部分断片であって、配列番号7に示される塩基配列の第343位〜第460位を含み、かつ550bp以上のサイズを有するポリヌクレオチド。
(iii)上記(i)または(ii)とストリンジェントな条件下でハイブリダイズするポリヌクレオチドであって、糸状菌細胞内でターミネーター活性を有するポリヌクレオチド。
【0036】
上記「ストリンジェントな条件下でハイブリダイズすることができる」とは、あるポリヌクレオチドと、これと相同性の高い他のポリヌクレオチドとの間で、二本鎖を形成させるのに十分な条件であることが意図される。「ストリンジェントな条件」としては、代表的な例として次の条件が挙げられる。すなわち、高イオン濃度下(例えば、6×SSC等)、65℃の温度条件下でハイブリダイズさせた後、低イオン濃度下(例えば、0.1×SSC等)、65℃、30分間の洗浄を行う条件が、「ストリンジェントな条件」の一例として挙げられる。
【0037】
なお、本発明の説明において、「糸状菌細胞内でターミネーター活性を有する」とは、アスペルギルス・オリゼのα-グルコシダーゼA遺伝子のターミネーターT-agdAをターミネーターとして含むプラスミドで糸状菌を形質転換して得られた形質転換体におけるレポーター遺伝子の発現量と、T-agdAの代わりに別のポリヌクレオチドターミネーターとして用いた以外は同一の条件でレポーター遺伝子を発現させた場合の発現量とを比較して、後者が前者と同等以上(つまり、(後者のレポーター発現量)÷(前者のレポーター発現量)≧1.0)であることを意味する。
【0038】
上記「糸状菌」としては、特に限定されるものではないが、例えば、アスペルギルス属糸状菌、ペニシリウム属糸状菌、トリコデルマ属糸状菌、リゾプス属糸状菌、ムコール属糸状菌、モナスカス属糸状菌、フザリウム属糸状菌等を挙げることができる。本発明のターミネーターは、アスペルギルス・オリゼより見出されたものであるため、特にアスペルギルス属糸状菌において奏効し得る。
【0039】
上記「アスペルギルス属糸状菌」としては、例えば、アスペルギルス・ニガー(Aspergillus niger)、アスペルギルス・オリゼ(Aspergillus oryzae)、アスペルギルス・アワモリ(Aspergillus awamori)、アスペルギルス・ウサミ(Aspergillus usami)、アスペルギルス・カワチ(Aspergillus kawachii)、アスペルギルス・ソーヤ(Aspergillus sojae)、アスペルギルス・ニドランス(Aspergillus nidulans)、アスペルギルス・アクレアタス(Aspergillus aculeatus)、アスペルギルス・テレウス(Aspergillus terreus)、アスペルギルス・フォエニシス(Aspergillus phoenicis)等を挙げることができる。
【0040】
また上記「ペニシリウム属糸状菌」としては、例えば、ペニシリウム・カマンベルチ(Penicillium camamberti)、ペニシリウム・ノタトウム(Penicillium notatum)、ペニシリウム・マルチカラー(Penicillium multicolor)、ペニシリウム・パルロゼナム(penicillium purpurogenum)、ペニシリウム・ロックフォルティ(Penicillium roqueforti)等を挙げることができる。
【0041】
また上記「トリコデルマ属糸状菌」としては、例えば、トリコデルマ・リーセイ(Trichoderma reesei)、トリコデルマ・ビリデ(Trichoderma viride)等を挙げることができる。
【0042】
また上記「リゾプス属糸状菌」としては、例えば、リゾプス・オリゼ(Rhizopus oryzae)、リゾプス・ジャポニカス(Rhizopus japonicus)等を挙げることができる。
【0043】
上記「ムコール属糸状菌」としては、例えば、ムコール・エスピー(Mucor sp.)等を挙げることができる。
【0044】
また上記「モナスカス属糸状菌」としては、例えば、モナスカス・パープレウス(Monascus purpureus)等を挙げることができる。
【0045】
また上記「フザリウム属糸状菌」としては、例えば、フザリウム・オキシスポラム(Fusarium oxysporum)等を挙げることができる。
【0046】
また本発明のターミネーターは、糸状菌細胞内でターミネーター活性を有する限りにおいて、上記の(i)〜(iii)の何れかに示されるポリヌクレオチドを含むもので構成されていればよい。つまり本発明のターミネーターは、(i)〜(iii)の何れかに示されるポリヌクレオチドのみからなるものであってもよいし、糸状菌細胞内でターミネーター活性を有する限りにおいてその他の塩基を含むものであってもよい。前者の好適な例としては、配列番号7、8、または9に示されるポリヌクレオチド(つまり2−5−11ターミネーター、2−5−12ターミネーター、2−5−13ターミネーター)が挙げられる。後者の好適な例としては、配列番号2に示される塩基配列からなるポリヌクレオチド(つまり2−5−1ターミネーター)や、配列番号2に示される塩基配列の第827位以降が切断された塩基配列からなるポリヌクレオチドが挙げられ、これらも本願発明のターミネーターに含まれる。
【0047】
(2.本発明のターミネーターの利用)
本発明のターミネーターは、該ターミネーターの上流に糸状菌細胞内で機能し得るプロモーターを連結することによってベクターを構築することができる。上記プロモーターの下流(3’末端側)であり、かつ糸状菌用ターミネーターの上流(5’末端側)に所望のタンパク質(「目的タンパク質」という)をコードする遺伝子(「目的遺伝子」という)を連結し、この目的遺伝子が連結されたベクターでこれらの糸状菌宿主(例えばアスペルギルス属糸状菌等)を形質転換し、それを培養することにより、目的タンパク質を著量生産させることができる。すなわち本発明は、上記ベクター、該ベクターを用いた形質転換方法、該形質転換方法により得られた形質転換体、および該形質転換体を用いたタンパク質の生産方法をも包含する。ベクターを構築するためのDNAの連結、挿入などは、公知の遺伝子工学的手法により行うことができる。
【0048】
(1)本発明のベクター
本発明のベクターを構成する、「糸状菌細胞内において機能し得るプロモーター」としては、糸状菌細胞内で下流に連結されたタンパク質をコードする目的遺伝子を転写する機能を有するポリヌクレオチドであれば特に限定されるものではない。具体的には、α−アミラーゼ(参考文献:Biosci. Biotech. Biochem., 1992年,第56巻,第11号,p1849−1853)、グルコアミラーゼ(参考文献:Current Genetics, 1992年,第22巻,第2号,p85−91)、セルラーゼ、セロビオハイドラーゼ(参考文献:J Biotechnol., 1991年,第17巻,第1号,p35−49)、アセトアミダーゼ(参考文献:Nucleic Acids Res., 1991年,第19巻,第10号,p2655−2660)等の加水分解酵素遺伝子、エノラーゼ(参考文献:Current Genetics, 2001年, p260−267)、3−ホスホグリセレートキナーゼ(参考文献:Mol. Gen. Genet., 1992年,第233巻,第1−2号,p231−40)、グリセルアルデヒド−3−ホスフェートデヒドロゲナーゼ(参考文献:Gene, 1992年,第120巻,第1号,p67−73)、アルコールデヒドロゲナーゼ(参考文献:Gene, 1989年,第79巻,第1号,p119−30)等の解糖系酵素遺伝子、トランスレーションエロンゲーションファクター遺伝子(参考文献:Appl. Microbiol. Biotechnol., 1998年,第50巻,第1号,p85−92)等のプロモーターが挙げられる。好適には、P−No8142プロモーター(参考文献:「化学と生物」,2000年,第38巻,第12号,p.831−838)が用いられる。
【0049】
また本発明のベクターには、以上の他に、所望により当該技術分野で公知の、エンハンサー、スプライシングシグナル、ポリA付加シグナル、選択マーカー、複製起点、目的遺伝子を導入する際に使用する制限酵素認識配列などを付加することができる。また、必要に応じて、目的遺伝子とその他のタンパク質(例えば、グルタチオンSトランスフェラーゼおよびプロテインA)をコードする遺伝子とを連結して本発明のベクターに連結することによって、目的タンパク質をその他のタンパク質との融合タンパク質として発現させることも可能である。このような融合タンパク質は、適当なプロテアーゼを使用して切断することによって、それぞれのタンパク質に分離することができる。
【0050】
また本発明にかかるベクターには、少なくとも1つの選択マーカーが含まれていることが好ましい。このようなマーカー遺伝子としては、一般に糸状菌形質転換体の選択に使用されているマーカーであることが好ましい。例えば、niaD、sC、argB、adeA、ptrA、pyrG等を使用することができる。また、大腸菌等の細菌培養用の薬剤耐性遺伝子(例えば、テトラサイクリン耐性遺伝子、アンピシリン耐性遺伝子等)が含まれていてもよい。上記選択マーカーを用いれば、本発明のベクターが宿主細胞に導入されたか否かを確認することができる。あるいは、目的タンパク質を融合ポリペプチドとして発現させてもよく、例えば、オワンクラゲ由来の緑色蛍光ポリペプチドGFP(Green Fluorescent Protein)をマーカーとして用い、目的タンパク質をGFP融合ポリペプチドとして発現させてもよい。
【0051】
なお、本発明のベクターを構成するポリヌクレオチドの由来は特に限定されるものではないが、本発明を食品等に応用することを考慮すればセルフクローニングを行うことが好ましいといえる。よってセルフクローニングを行う場合、ベクターを構成する各ポリヌクレオチドは、当該ベクターが導入される宿主と同一(同種)の生物である必要がある。つまり、本発明のターミネーターはアスペルギルス・オリゼ由来であるため、ベクターを構成するその他のポリヌクレオチドはアスペルギルス属糸状菌(より好ましくは、アスペルギルス・オリゼ)であることが好ましい。
【0052】
本発明のベクターに含まれ得る制限酵素認識配列としては、ベクター内に1箇所のみ存在し、かつ目的遺伝子の内部に存在しない制限酵素認識配列であれば、特に連結部の制限酵素認識配列として選択することができる。ベクターの汎用性を考慮すると、認識部位の出現頻度が少ない6塩基以上を認識する制限酵素の認識配列とすることが好ましく、8塩基以上を認識する制限酵素の認識配列とすることがより好ましい。6塩基を認識する制限酵素の認識配列としては、例えばGGGCCC(ApaI)、GGATCC(BamHI)、ATCGAT(ClaI)、AAGCTT(HindIII)、CCATGG(NcoI)、CATATG(NdeI)、CACGTG(PmlI)、GCATGC(SphI)、TCTAGA(XbaI)、CTCGAG(XhoI)等が挙げられる。8塩基以上を認識する制限酵素の認識配列としては、例えばGGCGCGCC(AscI)、GCGATCGC(AsiSI)、GGCCGGCC(FseI)、GCGGCCGC(NotI)、TTAATTAA(PacI)、GTTTAAAC(PmeI)、CCTGCAGG(SbfI)、GCCCGGGC(SrfI)、ATTTAAAT(SwaI)等が挙げられる。
【0053】
また本発明にベクターには、相同組み換えを行わせるために必要な宿主糸状菌由来のポリヌクレオチドが含まれていることが好ましい。当該宿主糸状菌由来のポリヌクレオチドの長さは、特に限定されないが、相同組換えの効率を考えると、200bp〜20000bpであることが好ましく、500bp〜10000bpであることがより好ましい。上記宿主糸状菌由来ポリヌクレオチドの長さが200bp以上であれば、相同組換えを効率よく起こさせることができる。また上記宿主糸状菌由来ポリヌクレオチドの長さが20000bp以下であれば、宿主糸状菌の細胞内に効率よく取り込ませることができる。
【0054】
また本発明のベクターには、プロモーターの下流(3’末端側)であり、かつターミネーターの上流(5’末端側)に、目的タンパク質をコードする遺伝子(「目的遺伝子」)が連結されていてもよい。ここで上記目的タンパク質をコードする目的遺伝子としては特に限定するものではないが、発現させることが好ましい有用タンパク質をコードする遺伝子が好ましい。例えば、糸状菌由来の細胞内タンパク質、分泌タンパク質、原核生物由来タンパク質、真核生物由来タンパク質等をコードする遺伝子が挙げられる。具体的には、α−アミラーゼ、グルコアミラーゼ、α−グルコシダーゼ、β−ガラクトシダーゼ、セルラーゼ、キチナーゼ、プロテアーゼ、アミノペプチダーゼ、カルボキシペプチダーゼ、リパーゼ、ホスホリパーゼ、フィターゼ、ヌクレアーゼ、カタラーゼ、グルコースオキシダーゼ、グルコースデヒドロゲナーゼ等の酵素遺伝子や、リボソーム遺伝子、トランスポーター遺伝子、シャペロン遺伝子、転写調節因子遺伝子などの酵素以外のタンパク質をコードする遺伝子からも自由に選択することができ、また機能未知の遺伝子も選択することができる。また、レポーター遺伝子として使用実績の多い大腸菌由来のβ-グルクロニダーゼ遺伝子も目的遺伝子として利用され得る。
【0055】
本発明のベクターには、宿主糸状菌細胞内で目的遺伝子を発現させるための発現単位(プロモーター領域、目的遺伝子のオープンリーディングフレーム、ターミネーター領域を含むポリヌクレオチド鎖)が少なくとも1つ含まれていてもよいが、当該発現単位が複数含まれていてもよい。
【0056】
本発明のベクターを構成する、それぞれのポリヌクレオチドを取得する方法は、特に限定されるものではなく、公知の技術によって取得することができる。例えば、公知の配列情報に基づいて、PCR等の増幅手段を用いる方法によって取得することもできるし、糸状菌の染色体DNAライブラリーからクローニングする方法によって取得することもできる。
【0057】
なおベクターの形状は、環状であっても鎖状であってもよい。
【0058】
(2)本発明の形質転換方法、形質転換体、およびタンパク質の生産方法
本発明の形質転換方法は、本発明のベクターのプロモーターの下流(3'末端側)であり、かつ糸状菌用ターミネーターの上流(5’末端側)に目的タンパク質をコードする遺伝子(目的遺伝子)が連結されているベクターで、糸状菌宿主(例えばアスペルギルス属糸状菌等)を形質転換する方法である。上記ベクターは、本発明のターミネーターを含んでいるため、かかる方法で得られた糸状菌の形質転換体を培養することによって、目的タンパク質を大量に生産させることができる。換言すれば本発明の形質転換方法は、目的タンパク質を高生産し得る糸状菌の作製方法であるとも言える。
【0059】
糸状菌宿主としては、上述の糸状菌が適宜採用され得る。また本発明のベクターを用いた宿主糸状菌の形質転換方法は特に限定されるものではなく、公知の方法が適宜採用される。例えば、カルシウム−PEG法、エレクトロポレーション法、等の公知の方法が形質転換方法として用いることができる。
【0060】
また上記形質転換方法により得られた形質転換体を培養する場合も、糸状菌の培養方法に通常用いられる培地、培養条件を適宜選択することにより行うことができる。形質転換体の培養方法は、固体培養であっても液体培養であってもよい。ここで形質転換体の培養に用いられる培地としては、特に限定されるものではないが、例えばデキストリン−ペプトン培地(2%デキストリン、1%ポリペプトン、0.5% KH2PO4、0.05%MgSO4・7H2O)、Czapek-dox培地(0.2%NaNO3、0.1%K2HPO4、0.05%MgSO4、0.05%KCl、0.001%FeSO4、3%グルコースまたは3%デンプン、pH5.5)、フスマ培地(小麦フスマ、水分含量40%)、等が採用され得る。また培養温度も、形質転換体が生育し得る温度であればよく、適宜最適な温度を検討の上、採用すればよい。形質転換体の培養温度としては、例えば、20〜37℃の範囲が好ましい。
【0061】
上記形質転換体の培養物には目的タンパク質が含まれており、必要に応じて定性また定量分析されたり、培養物から目的タンパク質が単離または精製されたりする。目的タンパク質が菌体外に分泌されている場合、液体培養であれば菌体培養液そのものを回収すればよい。一方、固体培養の場合には適当な緩衝液を用いて培養物から目的タンパク質を含む溶液を抽出して回収すればよい。また菌体内に目的タンパク質が蓄積されている場合は、菌体を公知の手段で破砕し、適当な緩衝液を用いて目的タンパク質を抽出すればよい。なお糸状菌のタンパク質生産系の場合、目的タンパク質は分泌される場合が多い。
【0062】
また、上記のようにして回収した目的タンパク質をさらに精製する場合は、例えば、タンパク質を含む溶液を周知の方法(例えば、硫安沈殿またはエタノール沈殿、酸抽出、陰イオンまたは陽イオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティークロマトグラフィー、ヒドロキシアパタイトクロマトグラフィー、およびレクチンクロマトグラフィー)によって精製すればよい。タンパク質の精製には高速液体クロマトグラフィー(「HPLC」)が好ましく採用され得る。
【0063】
本発明は上述した各実施形態に限定されるものではなく、請求項に示した範囲で種々の変更が可能であり、異なる実施形態にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施形態についても本発明の技術的範囲に含まれる。
【実施例】
【0064】
以下、本発明を実施例により具体的に説明するが、本発明はこれに限定されるものではない。
【0065】
〔実施例1〕アスペルギルス・オリゼ由来新規ターミネーターのクローニング
(クローニング用プラスミドpSEGA3の作製)
本実施例において使用するpSEGA3プラスミドの構築手順を、図1に基づいて説明する。図1に示すように、まず、pUC118(タカラバイオ社製)をSapIで消化し、末端をブランチングした後、精製し、アルカリフォスファターゼで脱リン酸化を行った。pUC118のSapI消化物(ブランチング処理済)を再び精製した後、PstIで消化を行い、アガロース電気泳動により、単離および精製を行った。
【0066】
アスペルギルス・ニデランス由来sCマーカー(参考文献:Buxton, F. P. et al Gene, 84, 329-334 (1989))の取得のため、pUsC(参考文献:Yamada, O. et al Biosci. Biotech. Biochem., 61, 1367-1369 (1997))をBspAPI−SmaI消化し、アガロース電気泳動により、単離および精製を行った後に、ブランチングを行い、再び精製した。
【0067】
P-enoA−uidA断片を取得するため、pNGEG-d4(参考文献:Toda, T. et al Curr. Genet. 40, 260-267 (2001))をXbaIで消化し、末端をブランチングした後、精製し、アルカリフォスファターゼで脱リン酸化を行った。再び精製を行った後、PstIで消化を行い、アガロース電気泳動により、P-enoA−uidA断片を単離および精製した。
【0068】
このように調製したpUC118プラスミドと、アスペルギルス・ニデランス由来sCマーカー断片と、P-enoA−uidA断片とをライゲーションし、pSEG3プラスミドを得た。
【0069】
次いで、pSEG3をPstI-DraIIIで消化し、アガロース電気泳動により、単離および精製した後にブランチングを行い、さらに精製した。その後、pSEG3−PstI-DraIII消化物をアルカリフォスファターゼで脱リン酸化を行い、再び精製を行った。
【0070】
アスペルギルス・ニデランス由来AMA1遺伝子断片の取得のため、ARp1(参考文献:Gems, D. H. et al Gene, 98, 61-67 (1991))をHindIIIで処理し、アガロース電気泳動により、単離および精製を行った後に、ブランチングを行い、再び精製した。
【0071】
このように調製したpSEG3プラスミドと、AMA1断片とをライゲーションし、pSEGA3プラスミドを得た。
【0072】
(プラスミドライブラリーの作製)
アスペルギルス・オリゼRIB40(独立行政法人酒類総合研究所より入手)より、ゲノムDNAを取得し、制限酵素Sau3AIで部分分解を行った。アガロース電気泳動を行ってゲノムDNA断片(0.5〜4kb)を回収および精製を行った後に、事前にBamHIで消化および精製しておいたpSEGA3にライゲーションにより挿入した(図3を参照のこと)。
【0073】
また、α-グルコシダーゼターミネーター(T-agdA)をpNAN-hsp12UTR(参考文献:Koda, A. et al Appl. Microbiol. Biotechnol., 70, 333-336 (2006))からXbaI-SmaI処理し、精製後に、ブランチングし、アガロース電気泳動後、単離および精製した後に、事前にBamHIで消化および精製しておいたpSEGA3にライゲーションにより挿入し、コントロール用のプラスミドとした(図3を参照のこと)。
【0074】
(形質転換体の取得とβ−グルクロニダーゼ活性の確認)
プラスミドライブラリーを調製後、アスペルギルス・ニガー NS48株(IFO4343から変異処理により取得したniaD、sC二重欠損株)へ、プロトプラスト-PEG法にて形質転換導入した。スクリーニング用培地として、0.2% 亜硝酸ナトリウムを含有する最少培地(Czapek-dox:0.2%NaNO3、0.1%K2HPO4、0.05%MgSO4、0.05%KCl、0.001%FeSO4、3%グルコースまたは3%デンプン、pH5.5)にβ−グルクロニダーゼが存在するとき青色の色素を生産するX− グルクロナイドを50μg/ml含むプレートに接種し、数日間培養後、青色に発色するコロニーを選択した。
【0075】
T-agdAを挿入したコントロール用プラスミドについても同様に、アスペルギルス・ニガー NS48株へ形質転換導入を行い、形質転換体を取得した。プラスミドライブラリーを導入した形質転換体の中から、コントロール用プラスミドを導入した形質転換体に比べて青色の発色が強い形質転換体をスクリーニングし、候補株5株を選抜した。
【0076】
(候補株からのプラスミドの単離と挿入断片の解析)
β−グルクロニダーゼ高発現であった形質転換体からDNA画分を調製した。当該DNA画分を大腸菌(イー・コリDH5α)を形質転換し、その形質転換体から得られるプラスミドDNAのBamHIサイトに挿入されていたターミネーターとして機能するDNA鎖のシークエンス解析を行った。
【0077】
その塩基配列を配列番号2〜6に示す。これらの塩基配列をアスペルギルス・オリゼのゲノムデーターベース(独立行政法人 製品評価技術基盤機構)と照合し、それぞれのDNA鎖が存在する染色体上の座を解析した。図2に、各DNA鎖の染色体上の座を、その近傍のORF予測とともに示した。なお、図2中に示した矢印はORFの転写方向を示している。またORFに記載されているAOに続く番号は、シークエンスされた微生物のゲノムデーターベースに登録されている番号を示しており、AOはアスペルギルス・オリゼに由来するタンパク質をコードする遺伝子であることを意味する。配列番号2〜6に示されるDNA鎖は、いずれもアスペルギルス・オリゼのゲノムデーターベースでORFと予測される塩基配列の直後から2000bp下流以内に、ORFと同じ転写方法で存在しておらず、いずれもターミネーターとして予測され得ない、全く新規なターミネーターであることが判明した。これらを、それぞれ2−5−1ターミネーター(配列番号2)、3−3−5ターミネーター(配列番号3)、3−3−9ターミネーター(配列番号4)、3−4−8ターミネーター(配列番号5)、3−24−9ターミネーター(配列番号6)と称する。
【0078】
なお、配列番号2で示される2−5−1ターミネーターは、異なる2つのDNA(図2中insert Aおよびinsert Bで表記する)から構成されていることが判明した。
【0079】
〔実施例2〕既知のターミネーター(T-agdA)との比較
実施例1で得られたアスペルギルス・オリゼ由来の新規ターミネーターと、従来公知のアスペルギルス・オリゼ由来α−グルコシダーゼ遺伝子のターミネーター(T-agdA)とのターミネーターとしての性能を比較すべく、それぞれのターミネーターを用いたときのタンパク質発現能を、β−グルクロニダーゼ遺伝子をレポーター遺伝子として比較した。
【0080】
比較は、インテグレート型のプラスミドを用いることとして、発明者が以前に作製したpNANG-hsp12UTRを用いることにした。このプラスミドは、高発現プロモーターであるP-No8142、アスペルギルス・オリゼのHsp12遺伝子の5’UTR、およびβ−グルクロニダーゼ遺伝子が連結されており、β−グルクロニダーゼ遺伝子の直後にα−グルコシダーゼ遺伝子のターミネーター(T-agdA)がXbaI-SmaIサイトを利用して導入されている。
【0081】
(新規ターミネーター導入プラスミドの作製)
pNANG-hsp12UTR中のα―グルコシダーゼ遺伝子のターミネーターを、新規ターミネーターに置換する手順を図5に示す。図5に示すように、pNANG-hsp12UTRをXbaI−SmaI消化した後、アガロース電気泳動を行って、pNANG-hsp12UTR−XbaI−SmaI消化物を単離および精製した。
【0082】
実施例1にて取得したプラスミドを鋳型にして、配列番号2〜6に示した新規ターミネーターをそれぞれPCR法にて取得した。このとき、それぞれのDNA鎖の5’末端側にXbaIサイトおよび3’末端側にSmaIサイトが付加されるように、両端にGGGが付加されるようにデザインされたプライマーを用いた。PCRに使用したプライマーを表1に示す。
【0083】
【表1】
【0084】
得られた新規ターミネーターをXbaIで消化し、精製した後に、pNANG-hsp12UTRのXbaI−SmaIサイトにライゲーションすることにより、ターミネーターのみが置換されたプラスミドを構築した。
【0085】
ただし、3−4−8ターミネーターはXbaIで切断されるため、3−4−8ターミネーターで置換する場合には以下のようにした。pNANG-hsp12UTRをXbaI消化後にブランチングし、その後SmaIのイソシゾマーであるXmaIにより消化し、さらに精製したプラスミド断片の5’末端側にXbaIサイトに相当するAを含み、3’末端側にSmaIサイトを含むように設計されたプライマーを用いてPCR法にて増幅した。得られたプラスミドをXmaIで消化し、そこに精製した3−4−8ターミネーターのDNA断片を挿入することにより、プラスミドを構築した。
【0086】
なお、β−グルクロニダーゼ遺伝子の終止コドンTGAからXbaIサイトまでATCAACAACの9塩基が挿入されているが、コントロールであるT-agdAがXbaI-SmaIサイトに挿入されているときに著量のβ−グルクロニダーゼが生産されていることから、挿入された9塩基によってターミネーター活性は阻害されていないと考えられる。よって、XbaI-SmaIサイトに任意の新規ターミネーターのDNA断片を導入することで、ターミネーターとしての性能を評価することができると考えられる。
【0087】
(形質転換体の取得とβ−グルクロニダーゼ比活性の測定)
構築したすべてのプラスミドを、同じくプラスミド上に含まれるniaDマーカー遺伝子内のEcoRIサイトで切断して直鎖状にし、アスペルギルス・オリゼniaD300(硝酸還元酵素欠損株)を形質転換した。それぞれの形質転換体からゲノムDNAを抽出し、サザン解析によりゲノム上のniaD遺伝子座にプラスミドが1コピー相同的に導入された形質転換体を得た。これらの形質転換体をデキストリン−ペプトン培地(2%デキストリン、1%ポリペプトン、0.5% KH2PO4、0.05%MgSO4・7H2O)にて培養し、菌体内のβ−グルクロニダーゼ比活性を測定し、それぞれのターミネーターの性能を比較した。その結果を表2に示す。
【0088】
【表2】
【0089】
表2に示すように、α−グルコシダーゼ遺伝子のターミネーター(T-agdA)と比較して、β−グルクロニダーゼ比活性の値に差がみられた。特に、配列番号2に示す2−5−1ターミネーターを利用した場合、β−グルクロニダーゼ比活性の値がT-agdAの1.34倍となっていた。
【0090】
これまでにT-agdAを用いてβ−グルクロニダーゼを生産させた報告(参考文献:Tsuboi, et al Biosci. Biotechnol. Biochem., 69, 206-208 (2005))では、β−グルクロニダーゼが菌体内可溶性タンパク質の約30%をも占めていることが報告されているが、2−5−1ターミネーターを用いることで、β−グルクロニダーゼの生産性をさらに1.34倍増加することができると考えられる。
【0091】
〔実施例3〕2−5−1ターミネーターの短縮化
(3’RACE法による2−5−1ターミネーターの転写終結部位の決定)
2−5−1ターミネーターの転写終結部位を、公知の方法である3’RACE法(Rapid Amplification cDNA Ends)により決定した。具体的な方法を以下に述べる。
【0092】
実施例2で取得した2−5−1ターミネーターを導入した形質転換体を、15mlのデキストリン・ペプトン培地で30℃、2日間振とう培養後、ガラスフィルターで集菌し、滅菌水で洗浄した。この菌体を新しいデキストリン・ペプトン培地15mlに移し、さらに30℃で12時間振とう培養し、同じくガラスフィルターで集菌し滅菌水で洗浄した後、RNAの抽出に用いた。RNAの抽出にはISOGEN(和光純薬社製)を用いた。得られたRNAを用いて、3’-Full RACE core set(タカラバイオ社製)にて逆転写されたクローンを取得し、9クローンのシークエンス解析を行った。
【0093】
決定された転写終結部位を、図4に示す。図4中、転写終結部位を下向き三角で示した。転写終結部位はターミネーターの343〜460bpに存在していた。
【0094】
(2−5−1ターミネーターをトランケートしたターミネーター断片の作製および評価)
上述の3’RACE法で決定された転写終結部位、およびそれらの転写終結部位より上流30bpまでのヌクレオチド鎖を含む領域を削除しないようにトランケートした2−5−1ターミネーターの断片を4種類作製した。2−5−1ターミネーターと、作製された断片との関係を示す模式図を図6に示した。図6中の下向き三角は、3’RACE法によって決定された2−5−1ターミネーターの転写終結部位に相当する。
【0095】
2−5−11ターミネーター(827bp、配列番号7)は、2−5−1ターミネーター(1577bp)の塩基配列の第1−827位までの塩基からなる2−5−1の断片である。2−5−12ターミネーター(572bp、配列番号8)は、2−5−1ターミネーター(1577bp)の塩基配列の第1−572位までの塩基からなる2−5−1の断片である。2−5−13ターミネーター(567bp、配列番号9)は、2−5−1ターミネーター(1577bp)の塩基配列の第261−827位までの塩基からなる2−5−1の断片である。2−5−14ターミネーター(312bp、配列番号10)は、2−5−1ターミネーター(1577bp)の塩基配列の第261−572位までの塩基からなる2−5−1の断片である。
【0096】
4種類のターミネーターのそれぞれ、またはT-agdAを実施例2と同様の手法で、pNANG-hsp12UTRのXbaI-SmaIサイト内にPCR法にてクローニングした(図5を参照のこと)。PCR法において使用した各プライマーセットを表3に示した。
【0097】
【表3】
【0098】
上記PCR法によって取得した各種ターミネーター断片の塩基配列を配列番号7〜10に示す。これらの各種ターミネーターが挿入されたベクターを用いて、上記の方法でアスペルギルス・オリゼniaD300(硝酸還元酵素欠損株)を形質転換し、該ベクターが相同的に1コピー導入された形質転換体を得た。これらの形質転換体をデキストリン-ペプトン培地にて培養し、菌体内のβ-グルクロニダーゼ比活性を測定し、ターミネーターの性能を比較した。その結果を表4に示す。
【0099】
【表4】
【0100】
表4に示すように、トランケートしたターミネーターのうち、2−5−11ターミネーター、2−5−12ターミネーター、および2−5−13ターミネーターは、従来公知のα−グルコシダーゼ遺伝子のターミネーター(T-agdA)に比較して高いβ-グルクロニダーゼ比活性を示した。また2−5−11ターミネーターおよび2−5−12ターミネーターにいたっては、トランケート前の2−5−1と比較してβ-グルクロニダーゼ比活性が増加した。
【0101】
したがって、2−5−1ターミネーター、並びにその断片である2−5−11ターミネーター、2−5−12ターミネーター、および2−5−13ターミネーターは、糸状菌用のターミネーターとして強力な活性を有するものであることが確認された。
【産業上の利用可能性】
【0102】
本発明は糸状菌細胞内でより強力なターミネーターとして機能し得るポリヌクレオチドを提供する。当該新規ターミネーターを糸状菌のタンパク質生産系に利用することによって、従来公知の糸状菌のタンパク質生産系に比してタンパク質の生産効率を増大させることが可能となる。
【0103】
したがって、本発明は、糸状菌を用いてタンパク質を産生させる、医薬、検査薬、食品等の産業において好適に利用される。
【特許請求の範囲】
【請求項1】
糸状菌細胞内でターミネーター活性を有する糸状菌用ターミネーターであって、下記の(i)〜(iii)の何れかに示されるポリヌクレオチドを含むことを特徴とする糸状菌用ターミネーター:
(i)配列番号7に示される塩基配列からなるポリヌクレオチド;
(ii)上記(i)のポリヌクレオチドの部分断片であって、配列番号7に示される塩基配列の第343位〜第460位を含み、かつ550bp以上のサイズを有するポリヌクレオチド;
(iii)上記(i)または(ii)とストリンジェントな条件下でハイブリダイズするポリヌクレオチドであり、かつ糸状菌細胞内でターミネーター活性を有するポリヌクレオチド。
【請求項2】
請求項1に記載の糸状菌用ターミネーター、および糸状菌細胞内において機能し得るプロモーターを含むことを特徴とするベクター。
【請求項3】
請求項2に記載のベクターのプロモーターの下流であり、かつ糸状菌用ターミネーターの上流に所望のタンパク質をコードする遺伝子が連結されたベクターで、糸状菌を形質転換することを特徴とする形質転換方法。
【請求項4】
請求項3に記載の形質転換方法により取得された形質転換体。
【請求項5】
請求項4に記載の形質転換体を培養する工程を含む、タンパク質の生産方法。
【請求項1】
糸状菌細胞内でターミネーター活性を有する糸状菌用ターミネーターであって、下記の(i)〜(iii)の何れかに示されるポリヌクレオチドを含むことを特徴とする糸状菌用ターミネーター:
(i)配列番号7に示される塩基配列からなるポリヌクレオチド;
(ii)上記(i)のポリヌクレオチドの部分断片であって、配列番号7に示される塩基配列の第343位〜第460位を含み、かつ550bp以上のサイズを有するポリヌクレオチド;
(iii)上記(i)または(ii)とストリンジェントな条件下でハイブリダイズするポリヌクレオチドであり、かつ糸状菌細胞内でターミネーター活性を有するポリヌクレオチド。
【請求項2】
請求項1に記載の糸状菌用ターミネーター、および糸状菌細胞内において機能し得るプロモーターを含むことを特徴とするベクター。
【請求項3】
請求項2に記載のベクターのプロモーターの下流であり、かつ糸状菌用ターミネーターの上流に所望のタンパク質をコードする遺伝子が連結されたベクターで、糸状菌を形質転換することを特徴とする形質転換方法。
【請求項4】
請求項3に記載の形質転換方法により取得された形質転換体。
【請求項5】
請求項4に記載の形質転換体を培養する工程を含む、タンパク質の生産方法。
【図1】


【図2】
【図3】
【図4】

【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2011−167160(P2011−167160A)
【公開日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願番号】特願2010−36288(P2010−36288)
【出願日】平成22年2月22日(2010.2.22)
【出願人】(000204686)大関株式会社 (9)
【Fターム(参考)】
【公開日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願日】平成22年2月22日(2010.2.22)
【出願人】(000204686)大関株式会社 (9)
【Fターム(参考)】
[ Back to top ]