
新規データベース探索様式を用いるタンパク質の特定及び特徴付け
サンプルポリペプチドについて1組の候補ポリペプチドを選択する方法であって、質量分析によって生成されたサンプルポリペプチド断片の質量差に基づいて、候補ポリペプチドを収集する第一の精選、および、サンプルポリペプチドの絶対質量、および前記断片の絶対質量に基づいて、候補ポリペプチドを収集する第二の精選を含む前記方法。

【発明の詳細な説明】
【背景技術】
【0001】
分子生物学の目的の一つは、遺伝子配列によってコードされるタンパク質の構造および生化学的活性を特徴付けることである。タンパク質の構造的特徴付けは、それらのタンパク質が天然の細胞条件下で発現される際の、タンパク質の一次構造(アミノ酸配列)を決定することに相当程度まで依存する。あるタンパク質がmRNAから翻訳された場合、そのタンパク質の一次構造が酵素の作用によって修飾される場合がよくある。こうした修飾としては、アミノ酸残基の側鎖に対する新規活性部分の付加、例えば、セリンに対するリン酸塩の付加、あるいは、タンパク質分解性分断、例えば、イニシエーターまたはシグナル配列の除去が挙げられる。従って、タンパク質の構造的解明は、アミノ酸配列の直線的組織(可変スプライシングおよび多型によって影響される)、および、配列内に生じる可能性のある任意の修飾の存在の両方を含む。
【0002】
この目的を実現するために、プロテオーム研究の一つの大目的は、タンパク質質の上に起こる修飾の詳細を理解することである。このような情報は、タンパク質の生物学的活性を理解するためばかりでなく、ヒト疾患に関連する過程において細胞増殖および分化を調節する薬剤の開発においても極めて重要である。
【0003】
質量分析(MS)は、未知の化合物を特定すること、既知の化合物を定量すること、および、分子の構造を確かめること、これらの目的のために使用される分析技術である。質量分析器は、個々の分子から転換された、複数のイオン質量を測定する装置である。この装置は、それらのイオンの質量対電荷比に基づいて分子量を間接的に測定する。イオン上の電荷は、電子の電荷量の基本単位zで表され、従って、質量対電荷比はm/zで表される。典型的には、質量分析で見られるイオンは、ただ1個の電荷しか持たない(z=1)ので、m/z値は、Daで表した分子量に数値的には等しい。単一荷電イオンでは、m/z比は、特定イオンの質量である。
【0004】
一般に、MSは、サンプルのイオンに、高強度の光子、電子、または中性ガスをぶつけ、結合を破壊し、元の(intact)分子の分子イオンから断片イオンを形成する。MSでは陽イオンおよび陰イオンの両方が生成されるけれども、一方の極性のイオンのみが、特定の装置セットアップによって検出される。気体相サンプルイオンの形成によって、個々のイオンの質量による仕分け、およびそれらの検出が可能となる。サンプルは、固体、液体、または気体であってもよいが、入口から装置の真空室へ入る。静電および/または磁力フィルターを用いて、複数のイオンをそれぞれのm/z比に従ってソートし、これらソートされたイオンは検出器に集束される。検出器において、イオンフラックスは、それに比例する電流に変換される。次に、装置は、これらの電気信号の大きさをm/zの関数として記録し、この情報を質量スペクトラムに変換する。
【0005】
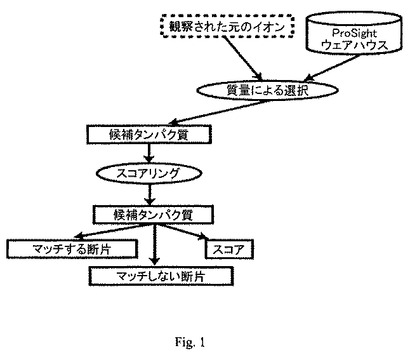
絶対質量探索では、元の質量を、断片イオンの質量と組み合わせて用いることによって、配列データベースからタンパク質を断定的に特定することが可能となる(図1参照)。特定は、指定のデータベースにおいて、元の質量の平均観測値または単一同位元素含有観測値に関するユーザー指定の許容範囲内に含まれる全ての配列を選択することによって実現される。候補単位は、質量をインデックスとするタンパク質データベースから検索されたものであることが好ましい。
【0006】
各候補配列は、観察された断片イオンに基づいて配点(スコア)される。この過程は、各候補配列について、全ての理論的b/yまたはc/z*型断片イオン質量(平均質量または単一同位体質量)を計算し、任意の理論的断片イオンについて、ユーザー指定の許容範囲(絶対的、またはppm)に含まれる、観察された断片イオンの数を数えることを含む。観察された断片イオンの数と、理論的断片イオンに合致する観察された断片イオンの数とを用いて、その特定が偽者である確率を計算する。計算された全てのスコアに考察された候補配列の数を掛け合わせると、確率に基づくスコアが得られる。次に、最低スコア(従って、特定が偽者である確率が最低である)を持つ候補タンパク質を、もっとも候補確率の高いタンパク質と見なす。
【0007】
MSはこれまで、タンパク質のアミノ酸一次配列を決定するために用いられてきた。タンパク質の断片イオンについて観察された質量差を用いて、タンパク質配列の一部のアミノ酸組成が演繹される。十分な数の関連タンパク質断片イオンについてMSデータが利用可能であるならば、これらの配列タグを用いてタンパク質配列を特定することが可能である。
【0008】
MS使用戦略を、プロテオームスケールにおいてタンパク質の修飾を検出する探索の効率と信頼度を向上させるよう現在開発中である。哺乳類ゲノムには、かつて考えられた(非特許文献1)よりもはるかに少ない遺伝子しか存在しないが、各遺伝子について、ヌクレオチド多型、可変RNAスプライシング、RNA編集(RNA editing)、および翻訳後修飾のために、別様のタンパク質形が可能である。修飾によってタンパク質機能を調節することに加えて、環境信号もタンパク質の化学的修飾をもたらす。修飾の検出は、真核細胞における基本的な調節メカニズムおよびヒト疾患の診断を理解する絶好の機会を提供する。
【0009】
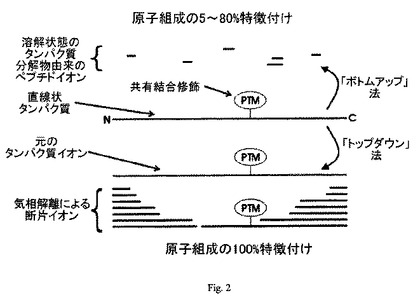
MSによるタンパク質構造決定のもっとも一般的な形は、「ボトムアップ」法の使用を含む。すなわち、最初元のタンパク質を既知の特異性を持つプロテアーゼで消化し、より短いポリペプチド断片を生成する(図2参照)。次に、これらの断片を精製し、MSを用いてその特性を特徴付けする。個々のポリペプチド断片について観察された絶対質量に基づいてアミノ酸組成を類推し、探索アルゴリスムと、既知のタンパク質組成物のデータベースとを用いて、そのタンパク質の正体を演繹することが可能となる。この方法を用い、単一タンパク質について修飾の検出を常套的に行った結果、ほぼ100%の配列をカバーするペプチドマップが生成された(非特許文献2)。しかしながらこの方法でも修飾の特徴付けにギャップを残す可能性がある。なぜなら、プロテアーゼ由来の断片は、別の化学的変化を経過している可能性があり、そのために元のタンパク質について十分にゆとりのある情報を与えない可能性がある。この方法のための探索アルゴリスムは、現在、修飾について、あるタイプの検出およびその位置探査をサポートし、普通に市販もされている(非特許文献3、4、5、および6)。
【0010】
修飾を直接標的する測定技術が現在開発中である。これは、元のタンパク質を、プロテアーゼのトリプシンで消化した時に得られるペプチド断片の分析に基づく。例えば、リン酸化およびグリコシル化の検出は、様々の過程、例えば、修飾を含むポリペプチド断片を単離すること(例えば、修飾されたペプチドの選択的精製)、特異的修飾を検出するためにMSを使用すること(例えば、修飾されたペプチドのマーカーイオンを求めて走査すること)、または、その両方を通じて、検出を強調することによって行われている(非特許文献7、8、9、10、および11)。最後に、二つの生物サンプルから得られたタンパク質の修飾プロフィールの差を検出するのにこれまでボトムアップ法が用いられている(例えば、リン酸プロテオミクス)(非特許文献12、7、8、10、11、13)。これらの技法の内のいくつかは、数百のタンパク質の分析のためにスケールアップされているが、全てのタイプの修飾に通用する汎用的なものはまだない。
【0011】
元のタンパク質の修飾を特定・特徴付けするために、「トップダウン」と呼ばれる別法が開発された(図2参照)。この方法は、先ず、元のタンパク質を断片化するためにタンデム質量分析(MS/MSまたは(MS)n)を用いる。次に、この断片を集め、それに続く断片化と質量測定を繰り返す。従って、このトップダウン法では、元のタンパク質の絶対質量とタンパク質断片イオンの両方が決定される。元のタンパク質がMS分析されるので、構造情報が分析によって誤って失われることがない。そのため、このトップダウン法は、元のタンパク質内に起こった全ての修飾を特定可能とする潜在性を持つ。このトップダウン法を用いて、4種もの多数の生物体から得られた32個のタンパク質について修飾情報が得られた(非特許文献14、15、16、17)。
【0012】
トップダウン法は、全ての修飾に対して通用する汎用的なものである。現在までトップダウン法によって特徴付けされた修飾としては、グリコシル化(非特許文献18、19)、Cysアルキル化(非特許文献20)、ジスルフィド結合形成(非特許文献21)、酸化(非特許文献19)および、リン酸化(非特許文献17)が挙げられる。この方法に対する大きな障害は、タンパク質精製処理における改善(非特許文献22、23)、フーリェ変換MS(FTMS)の自動化(非特許文献24)、4重極FTMSハイブリッド装置の開発(非特許文献25)、および、MS/MSデータによる元のタンパク質の特定に必要なソフトウェアの改善(非特許文献18、17)によって、その高さを減じつつある。しかしながら、データ処理や、修飾を含むタンパク質の完全特性特徴付けのための探索ソフトウェアに関しては依然として大きな障害が存在する。
【0013】
【非特許文献1】Lander et al., “Initial sequencing and analysis of the human genome,” Nature 409:860-921 (2001).
【非特許文献2】Biemann K, Papayannopoulos I. Acc. Chem. Res. 27:370-78 (1994).
【非特許文献3】Clauser KR, Baker P, Burlingame AL. “Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching,” Anal. Chem. 71:2871-82 (1999).
【非特許文献4】Perkins D, Pappin D, Creasy D, Cottrell J. “Probability-based protein identification by searching sequence databases using mass spectroscopy data,” Electrophoresis 20:3351-67 (1999).
【非特許文献5】Wilkins MR, Gasteiger E, Gooley AA, Herbert BR, Molloy MP, Binz PA, Ou K, Sanchez JC, Bairoch A, Williams KL, Hochstrasser DF. “High-throughput mass spectrometric discovery of protein post-translational modifications,” J. Mol. Biol. 289:645-57 (1999)
【非特許文献6】Zhang W, Chait B. “ProFound: an expert system for protein identification using mass spectrometric peptide mapping information,” Anal. Chem. 72:2482-89 (2000).
【非特許文献7】Goshe MB, Conrads TP, Panisko EA, Angell NH, Veenstra TD, Smith RD. “Phosphoprotein isotope-coded affinity tag approach for isolating and quantitating phosphopeptides in proteome-wide analysis,” Anal. Chem. 2001, 73:2578-86 (2001).
【非特許文献8】Oda Y, Nagatus T, Chait BT. “Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome,” Nat. Biotechnol. 19:379-82 (2001)
【非特許文献9】Steen H, Kuster B, Fernamdez M, Pandey A, Mann M. “Detection of tyrosine phosphorylated peptides by precursor ion scanning quadrupole TOF mass spectrometry in positive ion mode,” Anal. Chem. 73:1440-48 (2001).
【非特許文献10】Zhou H, Watts JD, Aebersold R. “A systematic approach to the analysis of protein phosphrylation,” Nat. Biotechnol. 19:375-78 (2001).
【非特許文献11】Ficarro S, McCleland M, Stukenberg P, Burke D, Ross M, Shabanowitz J, Hunt D, White F, “Phosphoproteome analysis by mass spectroscopy and its application to Saccharomyces cerevisiae,” Nat. Biotechnol. 20:301-305 (2002).
【非特許文献12】Oda Y, Huang K, Cross FR, Cowburn D, Chait BJ, “Accurate quantification of protein expression and site-specific phosphorylation,” Proc. Natl. Acad. Sci. U.S.A. 96:6591-96 (1999).
【非特許文献13】Gerber SA, Rush J, Stemann O, Steen H, Kirschner MW, Gygi SP. In: 50th ASMS Conference on Mass Spectrometry and Allied Topics, Orlando, FL, 2002
【非特許文献14】Kelleher NL, Taylor SV, Grannis D, Kinsland C, Chiu HJ, Begley TP, McLafferty FW. “Efficient sequence analysis of the six gene products (7-74 kDa) from the Escherichia coli thiamin biosynthetic operon by tandem high-resolution mass spectrometry,” Protein Sci. 7:1796-1801(1998).
【非特許文献15】Pineda FJ, Lin JS, Fenselau C, Demirev PA, “Testing the significance of microorganism identification by mass spectrometry and proteome database search,” Anal. Chem. 72:3739-44 (2000).
【非特許文献16】Reid GE, Shang H, Hogan JM, Lee GU, McLuckey SA. “Gas-phase concentration, purification, and identification of whole proteins from complex mixtures,” J. Am. Chem. Soc. 124:7353-62 (2002).
【非特許文献17】Meng F, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. “Informatics and multiplexing of intact protein identification in bacteria and the archaea,” Nat. Biotechnol. 19:952-57 (2001).
【非特許文献18】Reid GE, Stephenson JL, McLuckey SA. “Tandem mass spectrometry of ribonuclease A and B: N-linked glycosylation site analysis of whole protein ions,” Anal. Chem. 74:577-83 (2002).
【非特許文献19】Ge Y, ElNagger M, Sze SK, Bin OH, Begley TP, McLafferty FW, Boshoff H, Barry CE, J. Am. Soc. Mass Spectrom. 14:253-61(2003).
【非特許文献20】Kelleher NL, Costello CA, Begley TP, McLafferty FW. J. Am. Soc. Mass Spectrom. 6:981-84 (1995)
【非特許文献21】Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park JH, Begley TP, McLafferty FW. “Top down characterization of larger proteins (45 kDa) by electron capture dissociation and spectrometry,” J. Am. Chem. Soc. 124:672-78 (2002).
【非特許文献22】Kachman MT, Wang H, Schwartz DR, Cho KR, Lubman DM. “A 2-D liquid separations/mass mapping method for interlysate comparison of ovarian cancers,” Anal. Chem. 74:1779-91 (2002).
【非特許文献23】Meng F, Cargile BJ, Patrie SM, Johnson JR, McLoughlin SM, Kelleher NL. “Processing complex mixtures of intact proteins for direct analysis by mass spectrometry,” Anal. Chem. 74:2923-29 (2002).
【非特許文献24】Johnson JR, Meng F, Forbes AJ, Cargile BJ, Kelleher NL. “Fourier-transform mass spectrometry for automated fragmentation and identification of 5-20 kDa proteins in mixtures,” Electrophoresis 23:3217-23 (2002).
【非特許文献25】Belov ME, Nikolaev EN, Anderson GA, Auberry KJ, Harkewicz R, Smith RD. “Electronspray ionization-Fourier tranform ion cyclotron mass spectrometry using ion preselection and external accumulation for ultrahigh sensitivity,” J. Am. Soc. Mass Spectrom. 12:38-48 (2001).
【発明の開示】
【0014】
(本発明の概要)
一つの局面において、本発明は、サンプルポリペプチドについて、質量分析によって生成されたサンプルポリペプチド断片の質量差に基づいて、候補ポリペプチドを収集する第一の精選、および、サンプルポリペプチドの絶対質量および断片の絶対質量に基づいて、候補ポリペプチドを収集する第二の精選を含む方法である。
【0015】
第2の局面において、本発明は、コンピュータによって使用されるコンピュータプログラム製品である。このコンピュータプログラム製品は、サンプルポリペプチドについて1組の候補ポリペプチドを選択するために、コンピュータで読み取り可能な媒体であって、コンピュータで読み取り可能なプログラムコードを有する媒体を含む。コンピュータプログラム製品は、サンプルポリペプチドについて、質量分析によって生成されたサンプルポリペプチドの断片の質量差に基づいて、候補ポリペプチド集合から成る第1精選物、および、サンプルポリペプチドの絶対質量および断片の絶対質量に基づいて、候補ポリペプチド集合から成る第2精選物を含む一組の候補ポリペプチド選択するようにコンピュータに指令するための、コンピュータで読み取り可能なプログラムコードを含む。
【0016】
第3の局面では、本発明は、サンプルポリペプチドについて1組の候補ポリペプチドを選択するシステムであって、質量分析によって生成されたサンプルポリペプチドの断片の質量差に基づいて、候補ポリペプチド集合の第一の精選を実行するための手段、および、サンプルポリペプチドの絶対質量、および質量分析によって生成されたサンプルポリペプチド断片の絶対質量に基づいて、候補ポリペプチド集合の第二の精選を実行するための手段を含む前記システム、および、コンピュータである。
【0017】
(本明細書の用語の定義)
本明細書を通じて、質量分析によって生成される元のポリペプチドの断片を指す場合、「断片」および「断片イオン」という用語は相互交換的に用いられる。
【0018】
「新生ポリペプチド」という用語は、mRNAの最初の翻訳産物を指す。
【0019】
本明細書で用いる「修飾(modification)」という用語は、新生ポリペプチドの一次構造に生じた任意の化学的変化を指す。タンパク質の「修飾」としては、(i) コドン位置における多型であって、タンパク質の一次構造において異なるアミノ酸を生じさせるもの、(ii)可変スプライシングまたはmRNA転写物のRNA編集であって、そのスプライスまたは編集されたmRNAの翻訳時にタンパク質の異なる一次構造を生じさせるもの、および、(iii)翻訳後におけるタンパク質の化学的修飾であって、タンパク質の分子量の変化を生じさせるものが挙げられる。化学的修飾としては、細胞中で天然に見られる翻訳後修飾(例えば、タンパク質分解性分断、タンパク質スプライシング、N-Metおよびシグナル配列排除、リボシル化、リン酸化、アルキル化、ヒドロキシル化、グリコシル化、酸化、還元、ミリスチル化、ビオチニル化、ユビキノン化、ヨード化、ニトロシル化、アミノ化、硫黄付加、ペプチド連結、環化、ヌクレオチド付加、脂肪酸付加、アシル化等)の他に、生物細胞に対して内在的ではない起源から生じる修飾(例えば、環境の変異原、化学的発ガン物質、実験的に誘発された人工的修飾等)が挙げられる。
【0020】
「ショットガン注釈(ショットガン・アノテーション)」という言葉は、ポリペプチドの、あるアミノ酸残基に生じる特定の修飾の記述を指す(例えば、セリンのヒドロキシル基のリン酸化)。典型的には、ショットガン注釈は、ある定義された配列背景の内部で起こる、ポリペプチドのアミノ酸残基の特定の修飾を定義してもよい(例えば、Xを任意のアミノ酸とするRXXS/TXRXなる配列におけるセリンまたはトレオニンのヒドロキシル基のリン酸化)。ショットガン注釈は、指定された修飾体を含むタンパク質形を含むデータベースの拡張をもたらす。ショットガン注釈は、本明細書で「修飾」という用語が使用される意味において、任意のタイプの修飾を含む。
【0021】
「動的に修飾する」という言葉は、探索実行時に、ソフトウェアプログラムまたはデータベースに変化を生じさせることを指す。
【0022】
「動的ショットガン注釈」という言葉は、探索実行時に、データベースのタンパク質構造に対しショットガン注釈を実行することを指す。
【0023】
「拡張」という用語は、より小さい集合に対してショットガン注釈を行った後で、集合に見られるタンパク質形の数の増加を指す。
【0024】
「拡張集合体」という言葉は、より小さい集合に対してショットガン注釈を行った後に得られるタンパク質形の集合を指す。
【0025】
「精選(refining)」という用語は、配列タグモード探索か絶対質量モード探索のいずれかを用いるより大きな集合の問い合わせに続いて得られる集合におけるタンパク質形の数の減少を指す。
【0026】
「精選集合(refined collection)」という言葉は、配列タグモード探索か絶対質量モード探索のいずれかを用いるより大きな集合の問い合わせに続いて得られるタンパク質形の集合を指す。
【0027】
本明細書で用いる「ペプチド」という用語は、D-またはL-アミノ酸から成る単一鎖、あるいは、ペプチド結合で接続されたD-およびL-アミノ酸の混合物から構成される化合物を指す。好ましくは、ペプチドは長さにおいて少なくとも2アミノ酸残基を含み、50アミノ酸未満である。
【0028】
本明細書で用いる「ポリペプチド」は、少なくとも2個のアミノ酸残基から成り、1個以上のペプチド結合を含むポリマーを指す。「ポリペプチド」は、そのポリペプチドがよく定義された立体配置を持つかどうかによらず、ペプチド類およびタンパク質類を含む。好ましくは、ポリペプチドは天然のタンパク質である。
【0029】
本明細書で用いる「タンパク質」という用語は、ペプチド結合で連結された直線的に配されるアミノ酸から構成され、ペプチドと違ってよく定義された立体配置を持つ。タンパク質類は、ペプチド類と違って、50個以上のアミノ酸から成る鎖を含むことが好ましい。本明細書を通じてタンパク質類が好んで使われるが、本発明は全てのポリペプチドに適用可能であることが全体として理解される。
【0030】
「タンパク質形(protein form)」という言葉は、任意の修飾を含む、単一種のポリペプチドまたはタンパク質を指す。従って、単一の遺伝子が、その遺伝子の構造、転写されたmRNA(単数または複数)、および修飾(単複)の性質に応じて、数多くのタンパク質形をコードすることが可能である。
【0031】
「RNAスプライシング」という言葉は、両側に隣接するエキソンRNA配列をホスホジエステル結合連結で接続する一つの任意のRNAにおいて、二つの非隣接ホスホジエステル結合のホスホジエステル結合分断によって、少なくとも1個の介在RNA配列が除去されることを指す。
【0032】
「RNA編集」という言葉は、RNA配列のヌクレオチド組成の変更であって、転写RNAの少なくとも1個の核酸塩基が、異なる水素結合特異性を持つ別の核酸塩基によって置換される変更を指す。このようにして得られた編集RNAは、多型、延長ポリペプチド配列(例えば、終止コドンを除去することによって、または、開始コドンを導入することによって)、または、短縮型ポリペプチド(例えば、終止コドンを導入することによって)をコードしてもよい。
【0033】
「RNA処理」という言葉は、RNA配列の共有的修飾をもたらす任意の反応を指す。「RNA処理」は、RNAスプライシングとRNA編集の両方を含む。
【0034】
「探索モード」という言葉は、ウェアハウス・データベースから候補タンパク質形を特定し、検索する過程を指す。
【0035】
「配列タグ」という言葉は、質量分析によって生成された、ポリペプチドの2本の関連断片の質量差から推論される、ポリペプチド断片の、少なくとも2個の隣接アミノ酸から成る短い末端配列を指す。
【0036】
本明細書でタンパク質と関連して用いる「構造」は、修飾を含む、タンパク質の一次アミノ酸配列を指す。「構造」という用語および「一次構造」という言葉は、本明細書で用いる場合同じ意味を持つ。
【0037】
「ウェアハウス・データベース」という言葉は、2種以上のタンパク質形の集合を指す。
【0038】
(発明の詳細な説明)
本発明は、修飾を含むタンパク質構造を決定するために、ハイブリッド探索モード方法論における発見と、ソフトウェアプラットフォームとを利用する。修飾を含むタンパク質構造を決定するハイブリッド探索モード方法論では、あるサンプルポリペプチドに対する1組の精選候補ポリペプチドを選択するのに、1回の配列タグモード探索と1回以上の絶対質量モード探索の組み合わせが用いられる。この方法および関連するソフトウェアプラットフォームを以下に説明する。
【0039】
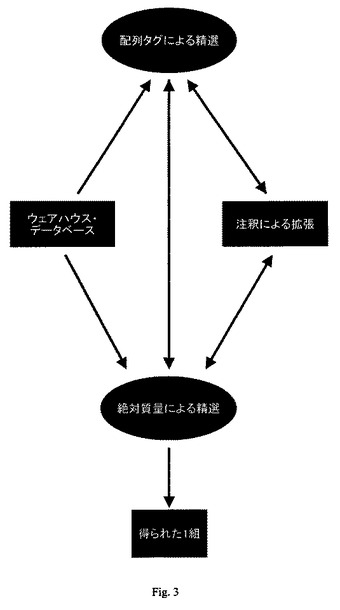
[ハイブリッド探索モード法]
ハイブリッド探索モードは、配列タグ探索の配列特定能力と、絶対質量探索の修飾検出および特徴付け能力とを結合させたものである(図3参照)。このハイブリッド法は、配列タグまたは絶対質量探索プロトコールのいずれかのみを用いた従来法よりも、タンパク質集団を精選するためのより効率的な方法である。ハイブリッド探索では、断片化データおよび1組の候補タンパク質から配列タグが編集される。候補タンパク質は、ウェアハウス・データベースから得られたものであってもよい。次に、タンパク質における各修飾およびその場所の実体が、元のタンパク質イオンおよび断片イオンの質量に注目する絶対質量法を用いて決定される。そのタンパク質形の理論的質量では説明されない質量は、通常、元のタンパク質またはタンパク質断片内に修飾のあるためとされる。
【0040】
タンパク質形のデータベースは、最初は、タンパク質の大集合を含んでいることが好ましい。初期データベースは、注釈のない配列情報を含むことが好ましい。このデータベースは、候補ポリペプチドの初回集合を形成することが好ましい。好ましい実施態様では、配列タグ探索によって、修飾されないポリペプチドから構成される候補タンパク質の集合が精選される。さらに、要すれば任意に、候補タンパク質集合は、修飾を考慮した候補ポリペプチドの注釈によって拡張されてもよい。配列タグ探索の後で、この集合に対して絶対質量モード探索を行って、最後の1組の候補ポリペプチドを獲得するのが好ましい。精選された組がただ一つのタンパク質形しか含んでいない場合、絶対質量探索モードによってそのタンパク質における修飾は一意に特定される。
【0041】
このハイブリッド探索モード法は、必ず1回の配列タグモード探索を行い、その次に、少なくとも1回の絶対質量モード探索を行う。要すれば任意に、絶対質量モード探索を、配列タグモード探索の前に行ってもよい。例えば、このハイブリッド探索モードを用いて「三段階」探索を実行してもよい。この方法では、緩い探索パラメータ(例えば、修飾に関する考慮を最小とするか、正確性許容度を大きくとるか、またはその両方)によって初回の絶対質量断片を用いて候補配列集合を特定し、次に、配列タグモード探索によって候補配列集合を精選する。次に、絶対質量モード探索を実行してその集合をさらに精選することが考えられる。
【0042】
[ソフトウェアプラットフォーム]
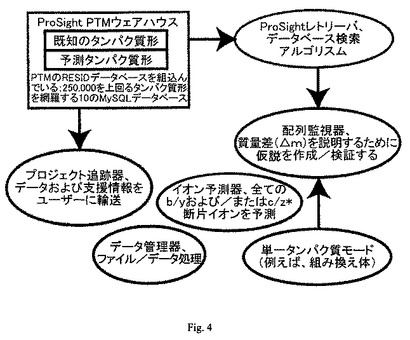
検索アルゴリスム、タンパク質形のウェアハウス・データベース、およびその他のユーティリティを含むコンピュータソフトウェアおよびシステムが記載される(図4参照)。検索アルゴリスムは、観察された断片イオンの絶対質量値に基づくb/yおよびc/z*イオン探索および配列タグ探索をサポートする。タンパク質形のウェアハウス・データベースは、無注釈、および注釈付き修飾情報の両方を含んでもよい。その他のユーティリティとしては、データ管理システム、イオン予測器、データ縮小ツール、および、グラフィック表示インターフェイスツールが挙げられる。
【0043】
[検索アルゴリスム]
検索アルゴリスムは、配列タグ探索モードと絶対質量探索モードとを組み合わせるハイブリッド探索法による、修飾情報を含むタンパク質のトップダウン特定を促進する。図3を参照すると、先ず、元のタンパク質について得られたMSデータと、得られたタンパク質断片イオンとを、タンパク質形のウェアハウス・データベースの配列タグ探索実行にかける。配列タグ探索では、ユーザーは、断片イオンの質量差に基づいてタンパク質の部分配列を決定する。配列タグを生成する際、同じ指定の質量値を持つ複数のアミノ酸(例えば、IleおよびLeu、LysおよびGln)に関するサポートが提供される。一つの実行では、データが含む可能性のある全ての配列タグを表すグラフが生成される。次にこのグラフを分析して、各表示配列タグについて正規の発現を生成する。次に、この部分配列情報を用いて無注釈タンパク質配列のデータベースから候補タンパク質を選択してもよい。要すれば任意に、ユーザーは、手動で編集した配列タグ組を用いて探索を実行してもよい。各候補配列に対し、その配列にマッチする全ての配列タグの長さを掛け合わせることによって計算されるスコアを与える。集束のために、ある指定の許容値よりも高いスコアを持つ配列のみがデータ出力として選択される。
【0044】
探索が配列タグ探索モードで実行される場合、注釈付き配列タグは一般にサポートされない。これは当然である。なぜなら、配列タグが修飾部位と重複することはほとんどあり得ないし、また、注釈付き配列タグの集合に生じる可能性のある全ての修飾を考慮するとなると、データのグラフ表示が複雑になるからである。この制限を用いると、タンパク質データベースに対し強力な直線的探索を実行することが可能となり、検索機能について許容可能な実行測定値が得られる(例えば、検索時間は、通常、実際の探索において3秒未満の実働時間である)。
【0045】
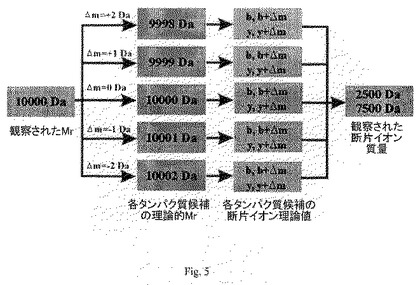
要すれば任意に、デルタMモード(「Δmモード」)と呼ばれる絶対質量探索モードを用いることにより、元のMW入力値とデータベースに収容される理論値との間の質量差を考慮することによって、正体未知の一つの修飾または質量を抱えるタンパク質を探索することが可能となる(図5参照)。元の質量誤差が約±1 Daで探索を実行するとすると、質量正確度の食い違いが生じることがある。またΔm値の正確度も±1 Daであり、断片イオン質量の正確度は数ppmぐらいである。選ばれた入力設定によって、Δmの正確度は変動することがある。
【0046】
[タンパク質形のウェアハウス・データベース]
トップダウン法による特定アルゴリスムでは全て先ず、データベースから候補配列集団を選択する。世界中に公開されるデータベース、例えば、SWISS-PROT、GenBank等ではFASTAファイルとしてタンパク質の無注釈形の利用が可能である。これらのデータベースは、手元の特定のプロジェクトに合わせた、タンパク質形の所望のウェアハウス・データベースの作成が可能となるように配慮されていることがある。FASTAファイルを、ウェアハウス・データベースを埋めることのできるファイルに変換するにはPERLスクリプトを用いるのが好ましい。FASTAファイルを変換中に、FASTAファイルの基本配列には、必要な情報、例えば、平均および単一同位元素含有質量計算値、および配列におけるアミノ酸数が付け加えられる。
【0047】
[ウェアハウス・データベースのショットガン注釈]
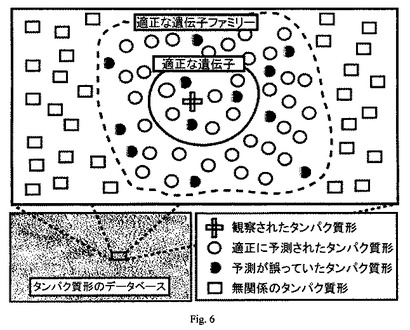
データベースに適切なタンパク質形が見られないためにその特定が妨げられた場合、RESIDの命名法を用いて、注釈付き配列から成るデータ保存体を作成する。RESIDは、既知の修飾タイプに関する有力なデータベースである(Garavelli, JS. “The RESID Database of Protein Modifications: 2003 developments,” Nucleic Acids Res. 31:499-501 (2003))。タンパク質形データベースを保有することによって、異なる配列モチーフの出現によって示される、既知および推測修飾を考慮することが可能になる。この方法は、あるタンパク質形の部分的または完全な特徴付けを、タンパク質形のデータベースから既知のタンパク質を検索することによる、そのタンパク質形の特定内容と結合しようとするものである(図6参照)。
【0048】
データベースで注釈される可能性のある翻訳後修飾事象としては、N-末端アセチル化、シグナルペプチド予測、リン酸化、リポイル化、GPI付着、リボシル化、アルキル化、ヒドロキシル化、グリコシル化、酸化、還元、ミリスチル化、ビオチニル化、ユビキノン化、ニトロシル化、アミノ化、硫黄付加、ペプチド連結、環化、ヌクレオチド付加、脂肪酸付加、アシル化、タンパク質分解性分断等(ポリペプチドについては約150-200種の翻訳後修飾が知られる(Garavelli, JS. “The RESID Database of Protein Modifications: 2003 developments,” Nucleic Acids Res. 31:499-501 (2003))が挙げられる。修飾注釈については、一般公開のデータベース、例えば、SWISS-PROTから、あるいは、ウェアハウス・データベースに修飾注釈を手動で入力することによって獲得することが可能である。
【0049】
各ウェアハウス・データベースは、遺伝子属性、タンパク質形属性、および修飾属性を含む3種の表を持つことが好ましい。遺伝子属性は、遺伝子特定情報と、遺伝子の構造の詳細な記述を含む。タンパク質形属性は、遺伝子特定内容、タンパク質形特定内容、単一同位元素質量、平均質量、アミノ酸数、および、任意の既知の属性、例えば、シグナル配列、開始メチオニン等の属性に対するフラグを含む。修飾属性は、修飾(RESID)特定内容、平均質量、単一同位元素質量、およびRESIDコード属性を含む。
【0050】
ウェアハウス・データベースの主要目的は、検索アルゴリスムからの探索行為を処理することである。検索アルゴリスムは、必ず、質量(平均か、単一同位元素のいずれか)に基づいてウェアハウス・データベースを探索することが好ましい。従って、データベースは質量に基づいて索引されており、対応する配列を速やかに戻し、全体システムの速度を下げないようにしなければならない。タンパク質形の表は、検索アルゴリスムの要求する情報の多くを含む。タンパク質形の表は既に、注釈付き配列および質量の全てを含んでいるのであるから、検索アルゴリスムの探索に対しデータベースから速やかな反応を得ることができる。
【0051】
修飾部位は、タンパク質の遺伝子配列から理論的に予測が可能であるが、注釈データベースを全ての可能な注釈で混雑させるのは多くの場合好ましくない。このような注釈を含めることは、その横断経および検索探索時間延長の観点から取り扱いにくいデータベースを生むことになる。
【0052】
一旦検索アルゴリスムが、配列タグ探索過程に基づいて候補タンパク質の精選集合を特定したら、それらの特定のタンパク質に関する可能な全ての注釈を含む拡張集合の生成が可能である。ウェアハウス・データベースをこのように修飾しても、検索アルゴリスムの性能を危機に陥れることはない。なぜなら、探索行為は、可能なタンパク質形の小集合に限定されるからである。従って、ハイブリッド探索法にウェアハウス・データベースの動的ショットガン注釈付加を含めることも可能である。このタンパク質候補集合が精選されて、候補ポリペプチドおよびその関連修飾から成る最終組が得られたならば、別のサンプルポリペプチドの特性特徴付けが実行される前に、先にウェアハウス・データベースに動的に入力されたショットガン注釈は消去してもよい。
【0053】
イオン予測器は、理論的b/yおよびc/zイオンを予測し、ソフトウェアおよびシステムに含められる。この計算は、ダルトンまたはppmで表した場合の誤差を計算するのに有用である(例えば、実施例1、表1を参照)。
【0054】
[データ縮小ツール]
ソフトウェアおよびシステムには、多数の荷電状態および縮小断片化データにおける水/アンモニア損失による冗長ピークを除去するためのデータ縮小ツールが含まれる。このツールは、獲得MSデータについて、それに検索アルゴリスムを適応する前に、高速分析する場合に有用である。
【0055】
[データベース管理システム]
ウェアハウス・データベースについてはいかなるデータベース管理システムでも使用が可能である。データベース管理システムはMySQLを含むのが好ましい。この、人々の好むデータベースシステムを選ぶのは、それが多くの有用なサポートツールとAPIを持つからであり、システムが一般人にも簡単に手に入るからである。付録に提供されるソフトウェアは、リナックス用の、バージョン11.18頒布3.23.52MySQLを用いている。
【0056】
[グラフィック表示インターフェイスツール]
全ての探索法において、様々なスコアを持つ候補配列の集団が返される。本ソフトウェアおよびシステムには、あらゆる探索法によって得られる候補配列集合を一覧するためのグラフィック表示インターフェイスツールが含まれる。要すれば任意に、このグラフィック表示インターフェイスツールは、本発明の他の特質を含む、局所ワークステーションに組み込まれる。要すれば任意に、グラフィック表示インターフェイスツールは、遠隔サーバーからインターネットを介して入手するデータを一覧できるように適応される。
【0057】
絶対質量モード探索では、ユーザーに、遺伝子記述、配列、配列長、理論的質量、質量差(絶対量およびppm)、マッチするb(またはc)型イオンの数、マッチするy(またはz*)型イオンの数、マッチする断片の合計数、および確率スコア計算値が提示される。次に、ユーザーは、候補タンパク質の集合を列挙された見出しに従って仕分けし、任意の検索配列について断片化詳細を眺める。断片化詳細表示は、ユーザーに対し、その配列にマッチする全ての断片に関する詳細情報を提示する。この画面は、特定されたイオン、観察された質量、理論的質量、単純な質量差(すなわち、例えば、「デルタM」モードによって演繹される質量偏倚が考察される前の)と質量差偏倚(すなわち、「デルタM」モードの場合のような質量偏倚の考察後の)、および、ppmで表した変異差を提示する。このグラフィック表示インターフェイスツールはまた、断片化の詳細を眺めることを可能とする。この詳細は、配列カバー範囲を決定し、断片化パターンの在り処を定めるのに有効な特質であって、適切な特定に対するユーザーの信頼を増すものである。
【0058】
[サポートされているデータベース]
サポートデータベースは、どのような生物体について構成されたものであってもよい。一つの実施態様は、9種の生物体、すなわち、Saccharomyces cerevisiae(酵母)、Escherichia coli(大腸菌)、Arabidopsis thaliana(シロイヌナズナ)、Bacillus subtilis(枯草菌)、Methanococcus jannaschii(古細菌)、Mycoplasma pneumoniae(肺炎マイコプラスマ)、Shewanella oneidensis(シェワネラ・オネイデンシス)、Mus musculus(ハツカネズミ)、およびHomo sapiens(ヒト)を含む生物体のデータベースをサポートする。酵母生物体Saccharomyces cerevisiaeのデータベースは、既知のものと、予測される修飾情報を兼ね備えたもっとも広範な注釈を含む。
【0059】
[データベースのスケール特性]
特に興味あるのは、データベースと探索時間のスケールが修飾情報の増加と共にどのように変化するかである。ある遺伝子と予測修飾は、タンパク質形において、各形が可能な修飾のサブセットを含むために指数関数的となる。従って、n個のタンパク質と、タンパク質当たりm個の可能な加工事象がある場合、一つの実施態様は、O(n2m)個のタンパク質形を含む。検索探索アルゴリスムがO(mlog 2n)で動き、定数は元の許容値に依存するとした場合、絶対質量探索アルゴリスムのスケールはmに対して直線的に変化する。既知および予測タンパク質形から成るデータベースでは、いくつかの修正が適切に予測された場合、観察されたタンパク質形は特定、特徴付けされる可能性がある。公開のタンパク質データベースにおける虚偽情報の増加は、乏しいMS/MSデータに基づく探索を不確かなものにする。一方、探求段階で用いられる修飾情報がより広範で正確になるにつれて、マッチする断片イオン質量の数は増す。
【0060】
[質量分析装置に対するコンピュータインターフェイス]
要すれば任意に、各成分は、コンピュータシステムにおいて質量分析計と相互通信する(in communication with)ように組織されてもよい。一つの実施態様では、コンピュータは局所的ワークステーションである。別の実施態様では、コンピュータは、現場から離れたサーバーである。後の実施態様では、各成分はサーバーに保存され、インターネットによるインターフェイスツールを用いてアクセスされる。質量分析器から生成されたMSデータはコンピュータに伝送され、そこでデータは獲得・保存される。コンピュータの中央処理ユニットは、タンパク質形のウェアハウス・データベースを探索するために、前記好ましい実施態様の一つに従って動作する検索アルゴリスムを用い、獲得されたMSデータの分析を指揮する。オペレータ指定の許容値が、検索アルゴリスムソフトウェアによって提供される選択肢の内から選ばれ、これに基づいて、タンパク質形のウェアハウス・データベースからタンパク質候補を収集し、その後さらに修飾の分析を行うことが可能となる。
【0061】
[医学的応用]
生体内の特定の標的タンパク質における修飾の程度に及ぼす環境シグナルの影響を見分けることが可能である。例えば、ヒトの病態の多くは、修飾、例えばリン酸化によって調整されている。家系内の特定遺伝子の修飾による変化に帰せられる障害は、後生性と診断される。異常な修飾があるかどうかについて特定タンパク質を調査するならば、その他のやり方では既知の遺伝子配列と相関をほとんど示さない病態について新しい洞察が得られる可能性がある。従って、このシステムは、障害や、特定の疾患に罹り易い性質を持つ個人を選び出すための強力なプラットフォームとなる。
【0062】
個々のタンパク質の修飾変化が病気の発生に関与する場合、特定のタンパク質に対する修飾付加または修飾排除を調節する製薬化合物の発見を促進する研究背景に使用可能するようにシステムを構成することも可能である。本明細書に開示される一つの実施態様では、システムは、高処理能力を持つスクリーニング方策の構成成分として設置される。このスクリーニング方策では、ある特定のタンパク質基質の修飾を触媒する修飾活性と連結する酵素を促進または抑制する能力について、候補製薬化合物からなる結合ライブラリーを評価する。タンパク質基質について、MSを用いて修飾の存在(または不在)について尋問する。次に、所望の薬物作用を持つ化合物が、特定の病気に向けた薬剤開発二次プログラムにおいて用いられる。
【0063】
特定タンパク質に対して修飾付加または排除を調節または変調する製薬化合物を評価するために、本システムを臨床背景下に使用可能とするように構成してもよい。一つの実施態様では、患者サンプルについて、特定のタンパク質が薬剤処置に応じて修飾を持つようになったかどうかについて、このシステムを用いて確認することが可能である。例えば、患者サンプルの細胞分解産物から得られる、対象とする標的タンパク質を、均一になるまで精製し、その精製物に対して、本明細書に記載される方法、ソフトウェア、およびシステムに従ってMS/MS分析を行ってもよい。サンプルタンパク質について得られたMSデータと、対応するタンパク質形であって、ウェアハウス・データベースに含まれる、全ての、その天然のショットガン修飾注釈を含めた対応タンパク質形との違いは簡単に獲得されるが、その違いは、治療処方の薬剤活性に関して豊かな情報を提供すると考えられる。
【0064】
本発明を用いることによって、タンパク質の各種修飾を、その発生機構と無関係に、検出することが可能となることは当業者には簡単に察しがつくであろう。例えば、単一タンパク質において、多型の位置、RNAスプライシング、またはmRNAのRNA編集が得られるタンパク質配列におよぼす作用、翻訳後修飾の有無、および、環境誘発性の化学的修飾を特定し、その特性特徴付けするのに本発明を用いてもよい。さらに、ハイブリッドモード探索法によって、理論的に予測されるポリペプチド形と、実際に観測されるポリペプチドの間の質量差を生み出す何らかの生物学的事象またはバイオインフォーマティックな不確かさが検出されることは、通常の熟練度を持つ当業者であれば理解されよう。
【0065】
[ProSight PTM:ソフトウェアと構造]
付録に、本明細書に開示される局面と実施態様を実行するために必要な全てのソフトウェアツール、およびタンパク質形の注釈付きウェアハウス・データベースを提供するコンパクトディスクが含まれている。”ProSight PTM”という名前のこのシステムは好ましい実施態様である。このシステムは、全てインターネット準拠のインターフェイスを持つ4つの主要成分を含む。すなわち、タンパク質データベース(ProSightウェアハウス)、データベース検索アルゴリスム(レトリーバ)、データマネージャー、プロージェクトトラッカー、およびその他のユーティリティである(図4参照、Taylor GK, Kim YB, Forbes AJ, Meng F, McCarthy R, Kelleher NL. “Web and database software for identification of intact proteins using top down mass spectrometry,” Anal. Chem. 75:4081-86 (2003))。
【0066】
スピードが重視される作業、例えば、データベース検索および配点は、iODBCライブラリーを用い、リナックス上においてC++で、オブジェクト指向設計を用いてデータベース結合用に書き込まれる。データ縮小ツールはOCaml(言語発現性のために選ばれる)で書き込まれ、一方、視覚化ツールは、画像変換用GDモジュールを用いてPERLで書き込まれる。
【0067】
絶対質量探索の使用では、ODBCで可動とされる管理システムに、ProSightウェアハウスを起動搭載することを必要とする。インターネット・アプリケーションは、二重プロセッサーAthlon 2200+MP上で走るApache HTTPサーバーによってサポートされるCGIを用いてPERLで書き込まれる。
【実施例】
【0068】
いくつかの実施態様を、S. cerevisiae(酵母)の36-kDaタンパク質と関連する修飾に関するMS/MS分析に注目する特異的具体例と結びつけて開示する。このタンパク質は、後に、グリセルアルデヒドリン酸デヒドロゲナーゼ3型酵素と特定された。Q-FTMSを用いたけれども、どのタイプの質量分析器から得られた元のタンパク質データであっても代替が可能である。手元の特定の応用にとって望ましいように、検索スコアおよび修飾特徴付け率を向上させるため、既知および予想修飾情報を利用するためのデータベース戦略が記載される。
【0069】
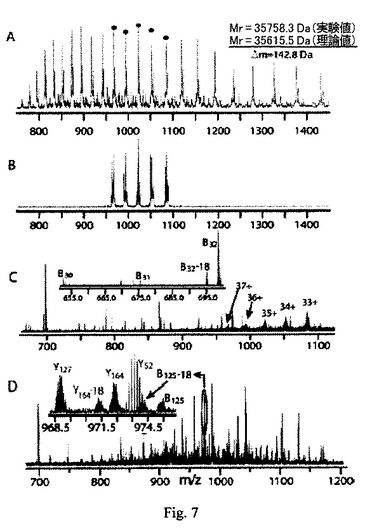
(実施例1:元の酵母タンパク質の自動的トップダウン分析)
35,758.3 DaのMr値を持つ酵母タンパク質が、一つのALS-PAGE/RPLC分画において観察された(図7A)。同じサンプル中に他に3個の成分があった。それらの内一つは、35.8-kDa種に付着したリン酸塩付加物(+98 Da)に対応していた。オンラインの分解アルゴリスムによって、35.8-kDaのタンパク質が摘出され、図7Bに示す5種の荷電状態を選択する適切なSWIFT波形が生成された。IRレーザーを用い、図7CのMS/MSスペクトラムは自動的に生成された。このスペクトラムは、THRASHアルゴリスムによって自動的に検出された、27個の別々の断片イオン質量値に対応する、39個の同位元素分布が観察された。虚偽のピーク(例えば、水損失ピーク)を排除するフィルター通過後、データベース検索用の最終インプットとして20個のイオン質量が用いられた。このタンパク質は、9個のb-型イオンおよび3個のy-型イオンマッチを含む、グリセルアルデヒド-3-リン酸デヒドロゲナーゼ(GAPDH3)であると特定された(表1および2)。この検索のP-スコアは、4 x 10-8であり、これは、この特定内容が偽者事象ではあり得ないことを示す。
【0070】
表1 GAPDH3(配列番号1)のイオン断片化データ
1GAPDH3は331個のアミノ酸を持ち、理論的質量は35,615.5 Da、Δmは142.8 Daである。
【0071】
表2 GAPDH3(配列番号1)1のグラフ表示断片マップ
【0072】
この遺伝子産物(GAPDH3、配列番号1)は、GAPDH遺伝子ファミリーの他のメンバー、GAPDH2(配列番号2)およびGAPDH1(配列番号3)と、それぞれ、96%と80%の相同性を持ちながらはっきりと区別された。これらのデータから、このタンパク質形は、331個のアミノ酸残基の内たった3個が異なるにも拘わらず、ExPASyによって報告された混乱から区別された。さらに、GAPDH3遺伝子産物の観察された分子量は、データベースの配列(開始Met無し)から計算される理論値よりも142 Da大きかった。断片マップによって、この質量差(Δm)はAsp90とAsp192の間に位置づけられ、この配列領域では僅かに2個しかCys残基(Cys149とCys153)はない(表2参照)。
【0073】
次に、このタンパク質形について、手動のQ-FTMS/MSと、超伝導マグネットの外部におけるイオンの衝突解離を用いて尋問したところ、98個の同位元素分布を持つ、図7Dのスペクトラムが得られた。これらのデータを検索アルゴリスムに対するインプットして用いると、+142 DaのΔmは、さらにPro126 Leu154領域に狭められる。これらのデータは、ゲル電気泳動中、2個のCys残基がアクリルアミド(それぞれ+71 Da)によってアルキル化されるという所見と一致する。正確にCys149とCys153に位置づけられたわけではないが、このゲル内修飾にはいくつかの先例があり、PAGE分画における遊離チオールについては予想されている。以上、この全体過程は、トップダウン法による共有的修飾の初期検出を含んでいた。
【0074】
絶対質量検索時間が、スコアされる候補配列の数に直線的に依存するのであれば、元の許容値が小さければ検索時間は短くなる。±2-kDaの許容値を用いて酵母を単純に探索すると、1500個の候補を得るために6秒かかるが、一方、200-Da許容値で同じ探索をすると400 msで終了して200個の候補を得る。ハイブリッド探索は、FASTAファイル入力の数と、考察される配列タグの数に直線的に依存する。5個の配列タグによる探索は4秒で完了する。これまで断片化された酵母タンパク質の内約半分は、観察された断片イオンの絶対質量に基づき検索アルゴリスムによって特定されている。残りについては、20%は、観察された断片イオン同士の間の相対的質量差に基づいて生成される配列タグを通じて特定された。配列タグモードでは、図7Cのデータの自動編集によって、4種のタグ(それぞれ4アミノ酸長で、2個は実際にあり、2個は虚偽のもの)が得られた。配列タグの編集を同じ荷電の断片イオンに限定したところ、適正なタグは僅か二つしか得られなかった。図7Dのデータを用いたところ、荷電状態制限により、8個のタグの内5個は偽者であり(長さは1-4アミノ酸)、6個の内4個は偽者(長さは1-3アミノ酸)であった。
【0075】
(実施例2:修飾活性によって酵素を修飾する化合物のスクリーニング(予測例))
下記の実施例の目的は、修飾活性を示す酵素の機能を、陽性または陰性に修飾する化合物を、結合ライブラリーにおいて特定するための高処理能力方策を概観することである。インビトロ環境における特定例が記載されているけれども、この実施例をインビボ背景に適応させるやり方は簡単に理解される。
【0076】
N-末端ヒスチジンタグ(UpState Biotechnology, Inc., レークプラシッド、ニューヨーク州)を含む、ヒトSrcキナーゼ癌タンパク質の組み換え形を、Srcキナーゼバッファー(100 mM Tris-HCl (pH7.2), 125 mM MgCl2, 25 mM MnCl2, 2 mM EGTA, 500 μM ATP, 0.25 mM オルトバナジン酸ナトリウム、および2 mMジチオスレイトール)に溶解したNi-NTA樹脂でコートした96ウェル皿に固定する。Srcキナーゼバッファーに溶解した試験化合物の添加後、できれば、1ウェル当たり一つの均質化合物、既知の配列を持つSrcタンパク質基質を各ウェルに加える(100 300 μMの濃度で)のが好ましい。これによってリン酸化が可能となる。インキュベーション後、基質を検索し、ProSight PTMシステムを用いてトップダウン質量分析を行う。
【0077】
ある特定の化合物の、Src活性に対する抑制活性は、そのタンパク質の内部にリン酸化チロシン残基と関連する修飾の欠如に基づいて見分けられる。このような化合物は、このトップダウン分析を確認するために、他のアッセイを用いてさらに特性特徴付けするのに好適である。例えば、アッセイに[γ-32P] ATPを用い、P81論文のTCA沈殿アッセイを用いてリン酸化活性を監視してもよい。
【0078】
(実施例3:個人における後生性(epigenetic)障害の検出(予測例))
本実施例の目的は、トップダウン質量分析法を用いて後生性障害と関連する修飾を検出するためのProSight PTMシステムの利便性を明らかにすることである。サンプル組織を、ニワトリ肉腫ウィルスに感染したニワトリを始め、未感染ニワトリから入手する。これらのサンプルをホモジェナイズし、澄明にし、可溶性細胞分解産物を生成する。ニワトリSrcキナーゼの既知のインビボ基質であるγ-カテニンタンパク質を、抗γ-カテニン抗体を用いて細胞分解産物からアフィニティー精製する。次に、この検索されたγ-カテニンサンプルに、トップダウン質量分析およびProSight PTMによる分析を実施する。期待される結果は、正常組織から検索されたγ-カテニンタンパク質は、ProSightウェアハウス・データベースに保存されるタンパク質の正常な修飾プロフィールを表し、それに対し、感染ニワトリから検索されるγ-カテニンタンパク質は、チロシンリン酸化と関連する、追加の修飾を含むことである。
【0079】
(実施例4:実施例1-3の実験手順)
[細胞培養および細胞分解産物の画分]
S. cerevisiae細胞(S288C株)を好気条件下で育成した。約2 gの細胞(湿潤重量)を、2個のプロテアーゼ阻害剤錠剤(Roche Diagnostics、マンハイム、ドイツ)を含む、10 mLの細胞溶解バッファー(25 mM Tris, 1 mM EDTA, 1 mM TCEP, pH7.0, 1 mLのDNAアーゼ添加)に再懸濁した。フレンチプレスにて細胞破壊後、細胞分解産物を、10,000 x g、30分遠心によって澄明にした。次に、この上清を、酸感受性を持つ界面活性剤(ALS)サンプルバッファーと混合し、次に、0.1% SDSの代わりに0.1% ALS-Iを用いるモデル491調整ゲル装置(Bio-Rad)に負荷した。4% Tスタックゲルを用い、12% T分解ゲルを流速0.50 mL/分で移動して溶出した。収集した80個の分画(それぞれ2 mL)の内、2個は、冷却アセトン沈殿、6Mグアニジン塩酸(pH2)によってさらに処理し、逆相液体クロマトグラフィー(RPLC)を用いて分析した。クロマトグラフィーは、標準溶媒(H2O, CH3CN、および0.1% TFA)を用い、15分に渡って直線的勾配を持つ、対称300 C4カラム(4.6 x 50 mm, Waters Inc. ミルフォード、マサチューセッツ州)を用いた。
【0080】
[ESI-Q-FTMS装置設定]
RPLCで分画されたタンパク質は乾燥し、80 μLのESI液(50% ACN, 49% H2O、および1%ギ酸)に再懸濁し、次に、ナノスプレイロボット(Advion BioSciences, Ithaca, NY)に負荷した。これによって、−100 nL/分で5-10 μLのサンプルが直接分析された。本実験で用いられた8.5-T Q-FTMS装置は、別の場所で記載されるように社内で構築したものである。簡単に言うと、先ず、タンパク質イオンが、8本の極に保存され、次に、4重極を通じて移送され、第2の8本極に蓄積され、最終的にICRセルにて分析される。4重極は、質量選択モードでも、「rfのみ」モードでも動作することが可能である。Tclで書かれた自動化スクリプトは、元のタンパク質のスペクトラムを獲得し、次に、オンラインの分解アルゴリスムを呼び出してMr値を計算し、SWIFTが、もっとも多量な5種の荷電状態を単離する。単離された荷電状態に対して5回の走査を行った後、IRレーザーがスイッチオンされ、25回か、50回かいずれかの走査が行われる(0.45秒、75%電源、40-Wレーザー)。図7DのQ-FTMS/MSスペクトラムは、荷電状態が4重極から第2の8本極へ移動する際に、指定の荷電状態の衝突的解離を通じて手動的に獲得されたものである。
【0081】
(参考文献)
Belov ME, Nikolaev EN, Anderson GA, Auberry KJ, Harkewicz R, Smith RD. "Electrospray ionization-Fourier transform ion cyclotron mass spectrometry using ion preselection and external accumulation for ultrahigh sensitivity," J. Am. Soc. Mass Spectrom. 12:38-48 (2001).
Biemann K, Papayannopoulos I. Acc. Chem. Res. 27:370-78 (1994).
Clauser KR, Baker P, Burlingame AL. "Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching," Anal. Chem. 71:2871-82 (1999).
Ficarro S, McCleland M, Stukenberg P, Burke D, Ross M, Shabanowitz J, Hunt D, White F. "Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae," Nat. Biotechnol. 20:301-305 (2002).
Garavelli, JS. "The RESID Database of Protein Modifications: 2003 developments," Nucleic Acids Res. 31:499-501 (2003).
Ge Y, Lawhorn BG, ElNaggar M Strauss E, Park JH, Begley TP, McLafferty FW. "Top down characterization of larger proteins (45 kDa) by electron capture dissociation mass spectrometry," J. Am. Chem. Soc. 124:672-78 (2002).
Ge Y, ElNaggar M, Sze SK, Bin OH, Begley TP, McLafferty FW, Boshoff H, Barry CE. J. Am. Soc. Mass Spectrom. 14:253-61 (2003).
Gerber SA, Rush J, Stemmann O, Steen H, Kirschner MW, Gygi SP. In: 50th ASMS Conference on Mass Spectrometry and Allied Topics, Orlando, FL, 2002.
Goshe MB, Conrads TP, Panisko EA, Angell NH, Veenstra TD, Smith RD. "Phosphoprotein isotope-coded affinity tag approach for isolating and quantitating phosphopeptides in proteome-wide analyses," Anal. Chem. 2001, 73:2578-86 (2001).
Johnson JR, Meng F, Forbes AJ, Cargile BJ, Kelleher NL. "Fourier-transform mass spectrometry for automated fragmentation and identification of 5-20 kDa proteins in mixtures," Electrophoresis 23:3217-23 (2002).
【0082】
Kachman MT Wang H, Schwartz DR, Cho KR, Lubman DM. "A 2-D liquid separations/mass mapping method for interlysate comparison of ovarian cancers," Anal. Chem. 74:1779-91 (2002).
Kelleher NL, Costello CA, Begley TP, McLafferty FW. J. Am. Soc. Mass Spectrom. 6:981-84 (1995).
Kelleher NL, Taylor SV, Grannis D, Kinsland C, Chiu HJ, Begley TP, McLafferty FW. "Efficient sequence analysis of the six gene products (7-74 kDa) from the Escherichia coli thiamin biosynthetic operon by tandem high-resolution mass spectrometry," Protein Sci. 7:1796-1801 (1998).
Lander ES et al. "Initial sequencing and analysis of the human genome," Nature 409:860-921 (2001).
MacCoss MJ McDonald WH, Saraf A, Sadygov R, Clark JM, Tasto JJ, Gould KL, Wolters D, Washburn M, Weiss A Clark JI, Yates JR., III. "Shotgun identification of protein modifications from protein complexes and lens tissue," Proc. Natl. Acad. Sci. U.S.A. 99:7900-7905 (2002).
Meng F, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. "Informatics and multiplexing of intact protein identification in bacteria and the archaea," Nat. Biotechnol. 19:952-57 (2001).
Meng F, Cargile BJ, Patrie SM, Johnson JR, McLoughlin SM, Kelleher NL. "Processing complex mixtures of intact proteins for direct analysis by mass spectrometry," Anal. Chem. 74:2923-29 (2002).
Oda Y, Huang K, Cross FR, Cowburn D, Chait BJ, "Accurate quantitation of protein expression and site-specific phosphorylation," Proc. Natl. Acad. Sci. U.S.A. 96:6591-96 (1999).
Oda Y, Nagasu T, Chait BT. "Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome," Nat. Biotechnol. 19:379-82 (2001).
【0083】
Perkins D, Pappin D, Creasy D, Cottrell J. "Probability-based protein identification by searching sequence databases using mass spectrometry data," Electrophoresis 20:3551-67 (1999).
Pineda FJ, Lin JS, Fenselau C, Demirev PA. "Testing the significance of microorganism identification by mass spectrometry and proteome database search," Anal. Chem. 72:3739-44 (2000).
Reid GE, Shang H, Hogan JM, Lee GU, McLuckey SA. "Gas-phase concentration, purification, and identification of whole proteins from complex mixtures," J. Am. Chem. Soc. 124:7353-62 (2002).
Reid GE, Stephenson JL, McLuckey SA. "Tandem mass spectrometry of ribonuclease A and B: N-linked glycosylation site analysis of whole protein ions," Anal. Chem. 74:577-83 (2002).
Steen H, Kuster B, Fernandez M, Pandey A, Mann M. "Detection of tyrosine phosphorylated peptides by precursor ion scanning quadrupole TOF mass spectrometry in positive ion mode," Anal. Chem. 73:1440-48 (2001).
Taylor GK, Kim YB, Forbes AJ, Meng F, McCarthy R, Kelleher NL "Web and database software for identification of intact proteins using top down mass spectrometry," Anal. Chem. 75:4081-86 (2003).
Wilkins MR, Gasteiger E, Gooley AA, Herbert BR, Molloy MP, Binz PA, Ou K, Sanchez JC, Bairoch A, Williams KL, Hochstrasser DF. "High-throughput mass spectrometric discovery of protein post-translational modifications," J. Mol. Biol. 289:645-57 (1999).
Zhang W, Chait B. "ProFound: an expert system for protein identification using mass spectrometric peptide mapping information," Anal. Chem. 72:2482-89 (2000).
Zhou H, Watts JD, Aebersold R. "A systematic approach to the analysis of protein phosphorylation," Nat. Biotechnol. 19:375-78 (2001).
【図面の簡単な説明】
【0084】
【図1】候補タンパク質を得るためにMSデータについて実施する絶対質量モード探索手順を表すアーキテクチャのフローチャートである。
【図2】MSによるタンパク質の特定と特性特徴付けのための「トップダウン」法および「ボトムアップ」法であって、修飾(例えば、翻訳後修飾(”PTM”))を特定し、位置づける方法を図示する。
【図3】ハイブリッド探索モード法の工程フローチャートを示す。
【図4】検索アルゴリスム(ProSightレトリーバ)、タンパク質形のウェアハウス・データベース(ProSight PTMウェアハウス)、および一次ユーティリティを含むソフトウェアシステムのフローチャートである。
【図5】データベースが「デルタm」モードで探索される実施態様を示す。
【図6】ショットガン注釈付加の模式図を示す。
【図7】S. cerevisiaeから得られたALS-PAGE/RPLC分画に対するMS/MSの例を示す。
【背景技術】
【0001】
分子生物学の目的の一つは、遺伝子配列によってコードされるタンパク質の構造および生化学的活性を特徴付けることである。タンパク質の構造的特徴付けは、それらのタンパク質が天然の細胞条件下で発現される際の、タンパク質の一次構造(アミノ酸配列)を決定することに相当程度まで依存する。あるタンパク質がmRNAから翻訳された場合、そのタンパク質の一次構造が酵素の作用によって修飾される場合がよくある。こうした修飾としては、アミノ酸残基の側鎖に対する新規活性部分の付加、例えば、セリンに対するリン酸塩の付加、あるいは、タンパク質分解性分断、例えば、イニシエーターまたはシグナル配列の除去が挙げられる。従って、タンパク質の構造的解明は、アミノ酸配列の直線的組織(可変スプライシングおよび多型によって影響される)、および、配列内に生じる可能性のある任意の修飾の存在の両方を含む。
【0002】
この目的を実現するために、プロテオーム研究の一つの大目的は、タンパク質質の上に起こる修飾の詳細を理解することである。このような情報は、タンパク質の生物学的活性を理解するためばかりでなく、ヒト疾患に関連する過程において細胞増殖および分化を調節する薬剤の開発においても極めて重要である。
【0003】
質量分析(MS)は、未知の化合物を特定すること、既知の化合物を定量すること、および、分子の構造を確かめること、これらの目的のために使用される分析技術である。質量分析器は、個々の分子から転換された、複数のイオン質量を測定する装置である。この装置は、それらのイオンの質量対電荷比に基づいて分子量を間接的に測定する。イオン上の電荷は、電子の電荷量の基本単位zで表され、従って、質量対電荷比はm/zで表される。典型的には、質量分析で見られるイオンは、ただ1個の電荷しか持たない(z=1)ので、m/z値は、Daで表した分子量に数値的には等しい。単一荷電イオンでは、m/z比は、特定イオンの質量である。
【0004】
一般に、MSは、サンプルのイオンに、高強度の光子、電子、または中性ガスをぶつけ、結合を破壊し、元の(intact)分子の分子イオンから断片イオンを形成する。MSでは陽イオンおよび陰イオンの両方が生成されるけれども、一方の極性のイオンのみが、特定の装置セットアップによって検出される。気体相サンプルイオンの形成によって、個々のイオンの質量による仕分け、およびそれらの検出が可能となる。サンプルは、固体、液体、または気体であってもよいが、入口から装置の真空室へ入る。静電および/または磁力フィルターを用いて、複数のイオンをそれぞれのm/z比に従ってソートし、これらソートされたイオンは検出器に集束される。検出器において、イオンフラックスは、それに比例する電流に変換される。次に、装置は、これらの電気信号の大きさをm/zの関数として記録し、この情報を質量スペクトラムに変換する。
【0005】
絶対質量探索では、元の質量を、断片イオンの質量と組み合わせて用いることによって、配列データベースからタンパク質を断定的に特定することが可能となる(図1参照)。特定は、指定のデータベースにおいて、元の質量の平均観測値または単一同位元素含有観測値に関するユーザー指定の許容範囲内に含まれる全ての配列を選択することによって実現される。候補単位は、質量をインデックスとするタンパク質データベースから検索されたものであることが好ましい。
【0006】
各候補配列は、観察された断片イオンに基づいて配点(スコア)される。この過程は、各候補配列について、全ての理論的b/yまたはc/z*型断片イオン質量(平均質量または単一同位体質量)を計算し、任意の理論的断片イオンについて、ユーザー指定の許容範囲(絶対的、またはppm)に含まれる、観察された断片イオンの数を数えることを含む。観察された断片イオンの数と、理論的断片イオンに合致する観察された断片イオンの数とを用いて、その特定が偽者である確率を計算する。計算された全てのスコアに考察された候補配列の数を掛け合わせると、確率に基づくスコアが得られる。次に、最低スコア(従って、特定が偽者である確率が最低である)を持つ候補タンパク質を、もっとも候補確率の高いタンパク質と見なす。
【0007】
MSはこれまで、タンパク質のアミノ酸一次配列を決定するために用いられてきた。タンパク質の断片イオンについて観察された質量差を用いて、タンパク質配列の一部のアミノ酸組成が演繹される。十分な数の関連タンパク質断片イオンについてMSデータが利用可能であるならば、これらの配列タグを用いてタンパク質配列を特定することが可能である。
【0008】
MS使用戦略を、プロテオームスケールにおいてタンパク質の修飾を検出する探索の効率と信頼度を向上させるよう現在開発中である。哺乳類ゲノムには、かつて考えられた(非特許文献1)よりもはるかに少ない遺伝子しか存在しないが、各遺伝子について、ヌクレオチド多型、可変RNAスプライシング、RNA編集(RNA editing)、および翻訳後修飾のために、別様のタンパク質形が可能である。修飾によってタンパク質機能を調節することに加えて、環境信号もタンパク質の化学的修飾をもたらす。修飾の検出は、真核細胞における基本的な調節メカニズムおよびヒト疾患の診断を理解する絶好の機会を提供する。
【0009】
MSによるタンパク質構造決定のもっとも一般的な形は、「ボトムアップ」法の使用を含む。すなわち、最初元のタンパク質を既知の特異性を持つプロテアーゼで消化し、より短いポリペプチド断片を生成する(図2参照)。次に、これらの断片を精製し、MSを用いてその特性を特徴付けする。個々のポリペプチド断片について観察された絶対質量に基づいてアミノ酸組成を類推し、探索アルゴリスムと、既知のタンパク質組成物のデータベースとを用いて、そのタンパク質の正体を演繹することが可能となる。この方法を用い、単一タンパク質について修飾の検出を常套的に行った結果、ほぼ100%の配列をカバーするペプチドマップが生成された(非特許文献2)。しかしながらこの方法でも修飾の特徴付けにギャップを残す可能性がある。なぜなら、プロテアーゼ由来の断片は、別の化学的変化を経過している可能性があり、そのために元のタンパク質について十分にゆとりのある情報を与えない可能性がある。この方法のための探索アルゴリスムは、現在、修飾について、あるタイプの検出およびその位置探査をサポートし、普通に市販もされている(非特許文献3、4、5、および6)。
【0010】
修飾を直接標的する測定技術が現在開発中である。これは、元のタンパク質を、プロテアーゼのトリプシンで消化した時に得られるペプチド断片の分析に基づく。例えば、リン酸化およびグリコシル化の検出は、様々の過程、例えば、修飾を含むポリペプチド断片を単離すること(例えば、修飾されたペプチドの選択的精製)、特異的修飾を検出するためにMSを使用すること(例えば、修飾されたペプチドのマーカーイオンを求めて走査すること)、または、その両方を通じて、検出を強調することによって行われている(非特許文献7、8、9、10、および11)。最後に、二つの生物サンプルから得られたタンパク質の修飾プロフィールの差を検出するのにこれまでボトムアップ法が用いられている(例えば、リン酸プロテオミクス)(非特許文献12、7、8、10、11、13)。これらの技法の内のいくつかは、数百のタンパク質の分析のためにスケールアップされているが、全てのタイプの修飾に通用する汎用的なものはまだない。
【0011】
元のタンパク質の修飾を特定・特徴付けするために、「トップダウン」と呼ばれる別法が開発された(図2参照)。この方法は、先ず、元のタンパク質を断片化するためにタンデム質量分析(MS/MSまたは(MS)n)を用いる。次に、この断片を集め、それに続く断片化と質量測定を繰り返す。従って、このトップダウン法では、元のタンパク質の絶対質量とタンパク質断片イオンの両方が決定される。元のタンパク質がMS分析されるので、構造情報が分析によって誤って失われることがない。そのため、このトップダウン法は、元のタンパク質内に起こった全ての修飾を特定可能とする潜在性を持つ。このトップダウン法を用いて、4種もの多数の生物体から得られた32個のタンパク質について修飾情報が得られた(非特許文献14、15、16、17)。
【0012】
トップダウン法は、全ての修飾に対して通用する汎用的なものである。現在までトップダウン法によって特徴付けされた修飾としては、グリコシル化(非特許文献18、19)、Cysアルキル化(非特許文献20)、ジスルフィド結合形成(非特許文献21)、酸化(非特許文献19)および、リン酸化(非特許文献17)が挙げられる。この方法に対する大きな障害は、タンパク質精製処理における改善(非特許文献22、23)、フーリェ変換MS(FTMS)の自動化(非特許文献24)、4重極FTMSハイブリッド装置の開発(非特許文献25)、および、MS/MSデータによる元のタンパク質の特定に必要なソフトウェアの改善(非特許文献18、17)によって、その高さを減じつつある。しかしながら、データ処理や、修飾を含むタンパク質の完全特性特徴付けのための探索ソフトウェアに関しては依然として大きな障害が存在する。
【0013】
【非特許文献1】Lander et al., “Initial sequencing and analysis of the human genome,” Nature 409:860-921 (2001).
【非特許文献2】Biemann K, Papayannopoulos I. Acc. Chem. Res. 27:370-78 (1994).
【非特許文献3】Clauser KR, Baker P, Burlingame AL. “Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching,” Anal. Chem. 71:2871-82 (1999).
【非特許文献4】Perkins D, Pappin D, Creasy D, Cottrell J. “Probability-based protein identification by searching sequence databases using mass spectroscopy data,” Electrophoresis 20:3351-67 (1999).
【非特許文献5】Wilkins MR, Gasteiger E, Gooley AA, Herbert BR, Molloy MP, Binz PA, Ou K, Sanchez JC, Bairoch A, Williams KL, Hochstrasser DF. “High-throughput mass spectrometric discovery of protein post-translational modifications,” J. Mol. Biol. 289:645-57 (1999)
【非特許文献6】Zhang W, Chait B. “ProFound: an expert system for protein identification using mass spectrometric peptide mapping information,” Anal. Chem. 72:2482-89 (2000).
【非特許文献7】Goshe MB, Conrads TP, Panisko EA, Angell NH, Veenstra TD, Smith RD. “Phosphoprotein isotope-coded affinity tag approach for isolating and quantitating phosphopeptides in proteome-wide analysis,” Anal. Chem. 2001, 73:2578-86 (2001).
【非特許文献8】Oda Y, Nagatus T, Chait BT. “Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome,” Nat. Biotechnol. 19:379-82 (2001)
【非特許文献9】Steen H, Kuster B, Fernamdez M, Pandey A, Mann M. “Detection of tyrosine phosphorylated peptides by precursor ion scanning quadrupole TOF mass spectrometry in positive ion mode,” Anal. Chem. 73:1440-48 (2001).
【非特許文献10】Zhou H, Watts JD, Aebersold R. “A systematic approach to the analysis of protein phosphrylation,” Nat. Biotechnol. 19:375-78 (2001).
【非特許文献11】Ficarro S, McCleland M, Stukenberg P, Burke D, Ross M, Shabanowitz J, Hunt D, White F, “Phosphoproteome analysis by mass spectroscopy and its application to Saccharomyces cerevisiae,” Nat. Biotechnol. 20:301-305 (2002).
【非特許文献12】Oda Y, Huang K, Cross FR, Cowburn D, Chait BJ, “Accurate quantification of protein expression and site-specific phosphorylation,” Proc. Natl. Acad. Sci. U.S.A. 96:6591-96 (1999).
【非特許文献13】Gerber SA, Rush J, Stemann O, Steen H, Kirschner MW, Gygi SP. In: 50th ASMS Conference on Mass Spectrometry and Allied Topics, Orlando, FL, 2002
【非特許文献14】Kelleher NL, Taylor SV, Grannis D, Kinsland C, Chiu HJ, Begley TP, McLafferty FW. “Efficient sequence analysis of the six gene products (7-74 kDa) from the Escherichia coli thiamin biosynthetic operon by tandem high-resolution mass spectrometry,” Protein Sci. 7:1796-1801(1998).
【非特許文献15】Pineda FJ, Lin JS, Fenselau C, Demirev PA, “Testing the significance of microorganism identification by mass spectrometry and proteome database search,” Anal. Chem. 72:3739-44 (2000).
【非特許文献16】Reid GE, Shang H, Hogan JM, Lee GU, McLuckey SA. “Gas-phase concentration, purification, and identification of whole proteins from complex mixtures,” J. Am. Chem. Soc. 124:7353-62 (2002).
【非特許文献17】Meng F, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. “Informatics and multiplexing of intact protein identification in bacteria and the archaea,” Nat. Biotechnol. 19:952-57 (2001).
【非特許文献18】Reid GE, Stephenson JL, McLuckey SA. “Tandem mass spectrometry of ribonuclease A and B: N-linked glycosylation site analysis of whole protein ions,” Anal. Chem. 74:577-83 (2002).
【非特許文献19】Ge Y, ElNagger M, Sze SK, Bin OH, Begley TP, McLafferty FW, Boshoff H, Barry CE, J. Am. Soc. Mass Spectrom. 14:253-61(2003).
【非特許文献20】Kelleher NL, Costello CA, Begley TP, McLafferty FW. J. Am. Soc. Mass Spectrom. 6:981-84 (1995)
【非特許文献21】Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park JH, Begley TP, McLafferty FW. “Top down characterization of larger proteins (45 kDa) by electron capture dissociation and spectrometry,” J. Am. Chem. Soc. 124:672-78 (2002).
【非特許文献22】Kachman MT, Wang H, Schwartz DR, Cho KR, Lubman DM. “A 2-D liquid separations/mass mapping method for interlysate comparison of ovarian cancers,” Anal. Chem. 74:1779-91 (2002).
【非特許文献23】Meng F, Cargile BJ, Patrie SM, Johnson JR, McLoughlin SM, Kelleher NL. “Processing complex mixtures of intact proteins for direct analysis by mass spectrometry,” Anal. Chem. 74:2923-29 (2002).
【非特許文献24】Johnson JR, Meng F, Forbes AJ, Cargile BJ, Kelleher NL. “Fourier-transform mass spectrometry for automated fragmentation and identification of 5-20 kDa proteins in mixtures,” Electrophoresis 23:3217-23 (2002).
【非特許文献25】Belov ME, Nikolaev EN, Anderson GA, Auberry KJ, Harkewicz R, Smith RD. “Electronspray ionization-Fourier tranform ion cyclotron mass spectrometry using ion preselection and external accumulation for ultrahigh sensitivity,” J. Am. Soc. Mass Spectrom. 12:38-48 (2001).
【発明の開示】
【0014】
(本発明の概要)
一つの局面において、本発明は、サンプルポリペプチドについて、質量分析によって生成されたサンプルポリペプチド断片の質量差に基づいて、候補ポリペプチドを収集する第一の精選、および、サンプルポリペプチドの絶対質量および断片の絶対質量に基づいて、候補ポリペプチドを収集する第二の精選を含む方法である。
【0015】
第2の局面において、本発明は、コンピュータによって使用されるコンピュータプログラム製品である。このコンピュータプログラム製品は、サンプルポリペプチドについて1組の候補ポリペプチドを選択するために、コンピュータで読み取り可能な媒体であって、コンピュータで読み取り可能なプログラムコードを有する媒体を含む。コンピュータプログラム製品は、サンプルポリペプチドについて、質量分析によって生成されたサンプルポリペプチドの断片の質量差に基づいて、候補ポリペプチド集合から成る第1精選物、および、サンプルポリペプチドの絶対質量および断片の絶対質量に基づいて、候補ポリペプチド集合から成る第2精選物を含む一組の候補ポリペプチド選択するようにコンピュータに指令するための、コンピュータで読み取り可能なプログラムコードを含む。
【0016】
第3の局面では、本発明は、サンプルポリペプチドについて1組の候補ポリペプチドを選択するシステムであって、質量分析によって生成されたサンプルポリペプチドの断片の質量差に基づいて、候補ポリペプチド集合の第一の精選を実行するための手段、および、サンプルポリペプチドの絶対質量、および質量分析によって生成されたサンプルポリペプチド断片の絶対質量に基づいて、候補ポリペプチド集合の第二の精選を実行するための手段を含む前記システム、および、コンピュータである。
【0017】
(本明細書の用語の定義)
本明細書を通じて、質量分析によって生成される元のポリペプチドの断片を指す場合、「断片」および「断片イオン」という用語は相互交換的に用いられる。
【0018】
「新生ポリペプチド」という用語は、mRNAの最初の翻訳産物を指す。
【0019】
本明細書で用いる「修飾(modification)」という用語は、新生ポリペプチドの一次構造に生じた任意の化学的変化を指す。タンパク質の「修飾」としては、(i) コドン位置における多型であって、タンパク質の一次構造において異なるアミノ酸を生じさせるもの、(ii)可変スプライシングまたはmRNA転写物のRNA編集であって、そのスプライスまたは編集されたmRNAの翻訳時にタンパク質の異なる一次構造を生じさせるもの、および、(iii)翻訳後におけるタンパク質の化学的修飾であって、タンパク質の分子量の変化を生じさせるものが挙げられる。化学的修飾としては、細胞中で天然に見られる翻訳後修飾(例えば、タンパク質分解性分断、タンパク質スプライシング、N-Metおよびシグナル配列排除、リボシル化、リン酸化、アルキル化、ヒドロキシル化、グリコシル化、酸化、還元、ミリスチル化、ビオチニル化、ユビキノン化、ヨード化、ニトロシル化、アミノ化、硫黄付加、ペプチド連結、環化、ヌクレオチド付加、脂肪酸付加、アシル化等)の他に、生物細胞に対して内在的ではない起源から生じる修飾(例えば、環境の変異原、化学的発ガン物質、実験的に誘発された人工的修飾等)が挙げられる。
【0020】
「ショットガン注釈(ショットガン・アノテーション)」という言葉は、ポリペプチドの、あるアミノ酸残基に生じる特定の修飾の記述を指す(例えば、セリンのヒドロキシル基のリン酸化)。典型的には、ショットガン注釈は、ある定義された配列背景の内部で起こる、ポリペプチドのアミノ酸残基の特定の修飾を定義してもよい(例えば、Xを任意のアミノ酸とするRXXS/TXRXなる配列におけるセリンまたはトレオニンのヒドロキシル基のリン酸化)。ショットガン注釈は、指定された修飾体を含むタンパク質形を含むデータベースの拡張をもたらす。ショットガン注釈は、本明細書で「修飾」という用語が使用される意味において、任意のタイプの修飾を含む。
【0021】
「動的に修飾する」という言葉は、探索実行時に、ソフトウェアプログラムまたはデータベースに変化を生じさせることを指す。
【0022】
「動的ショットガン注釈」という言葉は、探索実行時に、データベースのタンパク質構造に対しショットガン注釈を実行することを指す。
【0023】
「拡張」という用語は、より小さい集合に対してショットガン注釈を行った後で、集合に見られるタンパク質形の数の増加を指す。
【0024】
「拡張集合体」という言葉は、より小さい集合に対してショットガン注釈を行った後に得られるタンパク質形の集合を指す。
【0025】
「精選(refining)」という用語は、配列タグモード探索か絶対質量モード探索のいずれかを用いるより大きな集合の問い合わせに続いて得られる集合におけるタンパク質形の数の減少を指す。
【0026】
「精選集合(refined collection)」という言葉は、配列タグモード探索か絶対質量モード探索のいずれかを用いるより大きな集合の問い合わせに続いて得られるタンパク質形の集合を指す。
【0027】
本明細書で用いる「ペプチド」という用語は、D-またはL-アミノ酸から成る単一鎖、あるいは、ペプチド結合で接続されたD-およびL-アミノ酸の混合物から構成される化合物を指す。好ましくは、ペプチドは長さにおいて少なくとも2アミノ酸残基を含み、50アミノ酸未満である。
【0028】
本明細書で用いる「ポリペプチド」は、少なくとも2個のアミノ酸残基から成り、1個以上のペプチド結合を含むポリマーを指す。「ポリペプチド」は、そのポリペプチドがよく定義された立体配置を持つかどうかによらず、ペプチド類およびタンパク質類を含む。好ましくは、ポリペプチドは天然のタンパク質である。
【0029】
本明細書で用いる「タンパク質」という用語は、ペプチド結合で連結された直線的に配されるアミノ酸から構成され、ペプチドと違ってよく定義された立体配置を持つ。タンパク質類は、ペプチド類と違って、50個以上のアミノ酸から成る鎖を含むことが好ましい。本明細書を通じてタンパク質類が好んで使われるが、本発明は全てのポリペプチドに適用可能であることが全体として理解される。
【0030】
「タンパク質形(protein form)」という言葉は、任意の修飾を含む、単一種のポリペプチドまたはタンパク質を指す。従って、単一の遺伝子が、その遺伝子の構造、転写されたmRNA(単数または複数)、および修飾(単複)の性質に応じて、数多くのタンパク質形をコードすることが可能である。
【0031】
「RNAスプライシング」という言葉は、両側に隣接するエキソンRNA配列をホスホジエステル結合連結で接続する一つの任意のRNAにおいて、二つの非隣接ホスホジエステル結合のホスホジエステル結合分断によって、少なくとも1個の介在RNA配列が除去されることを指す。
【0032】
「RNA編集」という言葉は、RNA配列のヌクレオチド組成の変更であって、転写RNAの少なくとも1個の核酸塩基が、異なる水素結合特異性を持つ別の核酸塩基によって置換される変更を指す。このようにして得られた編集RNAは、多型、延長ポリペプチド配列(例えば、終止コドンを除去することによって、または、開始コドンを導入することによって)、または、短縮型ポリペプチド(例えば、終止コドンを導入することによって)をコードしてもよい。
【0033】
「RNA処理」という言葉は、RNA配列の共有的修飾をもたらす任意の反応を指す。「RNA処理」は、RNAスプライシングとRNA編集の両方を含む。
【0034】
「探索モード」という言葉は、ウェアハウス・データベースから候補タンパク質形を特定し、検索する過程を指す。
【0035】
「配列タグ」という言葉は、質量分析によって生成された、ポリペプチドの2本の関連断片の質量差から推論される、ポリペプチド断片の、少なくとも2個の隣接アミノ酸から成る短い末端配列を指す。
【0036】
本明細書でタンパク質と関連して用いる「構造」は、修飾を含む、タンパク質の一次アミノ酸配列を指す。「構造」という用語および「一次構造」という言葉は、本明細書で用いる場合同じ意味を持つ。
【0037】
「ウェアハウス・データベース」という言葉は、2種以上のタンパク質形の集合を指す。
【0038】
(発明の詳細な説明)
本発明は、修飾を含むタンパク質構造を決定するために、ハイブリッド探索モード方法論における発見と、ソフトウェアプラットフォームとを利用する。修飾を含むタンパク質構造を決定するハイブリッド探索モード方法論では、あるサンプルポリペプチドに対する1組の精選候補ポリペプチドを選択するのに、1回の配列タグモード探索と1回以上の絶対質量モード探索の組み合わせが用いられる。この方法および関連するソフトウェアプラットフォームを以下に説明する。
【0039】
[ハイブリッド探索モード法]
ハイブリッド探索モードは、配列タグ探索の配列特定能力と、絶対質量探索の修飾検出および特徴付け能力とを結合させたものである(図3参照)。このハイブリッド法は、配列タグまたは絶対質量探索プロトコールのいずれかのみを用いた従来法よりも、タンパク質集団を精選するためのより効率的な方法である。ハイブリッド探索では、断片化データおよび1組の候補タンパク質から配列タグが編集される。候補タンパク質は、ウェアハウス・データベースから得られたものであってもよい。次に、タンパク質における各修飾およびその場所の実体が、元のタンパク質イオンおよび断片イオンの質量に注目する絶対質量法を用いて決定される。そのタンパク質形の理論的質量では説明されない質量は、通常、元のタンパク質またはタンパク質断片内に修飾のあるためとされる。
【0040】
タンパク質形のデータベースは、最初は、タンパク質の大集合を含んでいることが好ましい。初期データベースは、注釈のない配列情報を含むことが好ましい。このデータベースは、候補ポリペプチドの初回集合を形成することが好ましい。好ましい実施態様では、配列タグ探索によって、修飾されないポリペプチドから構成される候補タンパク質の集合が精選される。さらに、要すれば任意に、候補タンパク質集合は、修飾を考慮した候補ポリペプチドの注釈によって拡張されてもよい。配列タグ探索の後で、この集合に対して絶対質量モード探索を行って、最後の1組の候補ポリペプチドを獲得するのが好ましい。精選された組がただ一つのタンパク質形しか含んでいない場合、絶対質量探索モードによってそのタンパク質における修飾は一意に特定される。
【0041】
このハイブリッド探索モード法は、必ず1回の配列タグモード探索を行い、その次に、少なくとも1回の絶対質量モード探索を行う。要すれば任意に、絶対質量モード探索を、配列タグモード探索の前に行ってもよい。例えば、このハイブリッド探索モードを用いて「三段階」探索を実行してもよい。この方法では、緩い探索パラメータ(例えば、修飾に関する考慮を最小とするか、正確性許容度を大きくとるか、またはその両方)によって初回の絶対質量断片を用いて候補配列集合を特定し、次に、配列タグモード探索によって候補配列集合を精選する。次に、絶対質量モード探索を実行してその集合をさらに精選することが考えられる。
【0042】
[ソフトウェアプラットフォーム]
検索アルゴリスム、タンパク質形のウェアハウス・データベース、およびその他のユーティリティを含むコンピュータソフトウェアおよびシステムが記載される(図4参照)。検索アルゴリスムは、観察された断片イオンの絶対質量値に基づくb/yおよびc/z*イオン探索および配列タグ探索をサポートする。タンパク質形のウェアハウス・データベースは、無注釈、および注釈付き修飾情報の両方を含んでもよい。その他のユーティリティとしては、データ管理システム、イオン予測器、データ縮小ツール、および、グラフィック表示インターフェイスツールが挙げられる。
【0043】
[検索アルゴリスム]
検索アルゴリスムは、配列タグ探索モードと絶対質量探索モードとを組み合わせるハイブリッド探索法による、修飾情報を含むタンパク質のトップダウン特定を促進する。図3を参照すると、先ず、元のタンパク質について得られたMSデータと、得られたタンパク質断片イオンとを、タンパク質形のウェアハウス・データベースの配列タグ探索実行にかける。配列タグ探索では、ユーザーは、断片イオンの質量差に基づいてタンパク質の部分配列を決定する。配列タグを生成する際、同じ指定の質量値を持つ複数のアミノ酸(例えば、IleおよびLeu、LysおよびGln)に関するサポートが提供される。一つの実行では、データが含む可能性のある全ての配列タグを表すグラフが生成される。次にこのグラフを分析して、各表示配列タグについて正規の発現を生成する。次に、この部分配列情報を用いて無注釈タンパク質配列のデータベースから候補タンパク質を選択してもよい。要すれば任意に、ユーザーは、手動で編集した配列タグ組を用いて探索を実行してもよい。各候補配列に対し、その配列にマッチする全ての配列タグの長さを掛け合わせることによって計算されるスコアを与える。集束のために、ある指定の許容値よりも高いスコアを持つ配列のみがデータ出力として選択される。
【0044】
探索が配列タグ探索モードで実行される場合、注釈付き配列タグは一般にサポートされない。これは当然である。なぜなら、配列タグが修飾部位と重複することはほとんどあり得ないし、また、注釈付き配列タグの集合に生じる可能性のある全ての修飾を考慮するとなると、データのグラフ表示が複雑になるからである。この制限を用いると、タンパク質データベースに対し強力な直線的探索を実行することが可能となり、検索機能について許容可能な実行測定値が得られる(例えば、検索時間は、通常、実際の探索において3秒未満の実働時間である)。
【0045】
要すれば任意に、デルタMモード(「Δmモード」)と呼ばれる絶対質量探索モードを用いることにより、元のMW入力値とデータベースに収容される理論値との間の質量差を考慮することによって、正体未知の一つの修飾または質量を抱えるタンパク質を探索することが可能となる(図5参照)。元の質量誤差が約±1 Daで探索を実行するとすると、質量正確度の食い違いが生じることがある。またΔm値の正確度も±1 Daであり、断片イオン質量の正確度は数ppmぐらいである。選ばれた入力設定によって、Δmの正確度は変動することがある。
【0046】
[タンパク質形のウェアハウス・データベース]
トップダウン法による特定アルゴリスムでは全て先ず、データベースから候補配列集団を選択する。世界中に公開されるデータベース、例えば、SWISS-PROT、GenBank等ではFASTAファイルとしてタンパク質の無注釈形の利用が可能である。これらのデータベースは、手元の特定のプロジェクトに合わせた、タンパク質形の所望のウェアハウス・データベースの作成が可能となるように配慮されていることがある。FASTAファイルを、ウェアハウス・データベースを埋めることのできるファイルに変換するにはPERLスクリプトを用いるのが好ましい。FASTAファイルを変換中に、FASTAファイルの基本配列には、必要な情報、例えば、平均および単一同位元素含有質量計算値、および配列におけるアミノ酸数が付け加えられる。
【0047】
[ウェアハウス・データベースのショットガン注釈]
データベースに適切なタンパク質形が見られないためにその特定が妨げられた場合、RESIDの命名法を用いて、注釈付き配列から成るデータ保存体を作成する。RESIDは、既知の修飾タイプに関する有力なデータベースである(Garavelli, JS. “The RESID Database of Protein Modifications: 2003 developments,” Nucleic Acids Res. 31:499-501 (2003))。タンパク質形データベースを保有することによって、異なる配列モチーフの出現によって示される、既知および推測修飾を考慮することが可能になる。この方法は、あるタンパク質形の部分的または完全な特徴付けを、タンパク質形のデータベースから既知のタンパク質を検索することによる、そのタンパク質形の特定内容と結合しようとするものである(図6参照)。
【0048】
データベースで注釈される可能性のある翻訳後修飾事象としては、N-末端アセチル化、シグナルペプチド予測、リン酸化、リポイル化、GPI付着、リボシル化、アルキル化、ヒドロキシル化、グリコシル化、酸化、還元、ミリスチル化、ビオチニル化、ユビキノン化、ニトロシル化、アミノ化、硫黄付加、ペプチド連結、環化、ヌクレオチド付加、脂肪酸付加、アシル化、タンパク質分解性分断等(ポリペプチドについては約150-200種の翻訳後修飾が知られる(Garavelli, JS. “The RESID Database of Protein Modifications: 2003 developments,” Nucleic Acids Res. 31:499-501 (2003))が挙げられる。修飾注釈については、一般公開のデータベース、例えば、SWISS-PROTから、あるいは、ウェアハウス・データベースに修飾注釈を手動で入力することによって獲得することが可能である。
【0049】
各ウェアハウス・データベースは、遺伝子属性、タンパク質形属性、および修飾属性を含む3種の表を持つことが好ましい。遺伝子属性は、遺伝子特定情報と、遺伝子の構造の詳細な記述を含む。タンパク質形属性は、遺伝子特定内容、タンパク質形特定内容、単一同位元素質量、平均質量、アミノ酸数、および、任意の既知の属性、例えば、シグナル配列、開始メチオニン等の属性に対するフラグを含む。修飾属性は、修飾(RESID)特定内容、平均質量、単一同位元素質量、およびRESIDコード属性を含む。
【0050】
ウェアハウス・データベースの主要目的は、検索アルゴリスムからの探索行為を処理することである。検索アルゴリスムは、必ず、質量(平均か、単一同位元素のいずれか)に基づいてウェアハウス・データベースを探索することが好ましい。従って、データベースは質量に基づいて索引されており、対応する配列を速やかに戻し、全体システムの速度を下げないようにしなければならない。タンパク質形の表は、検索アルゴリスムの要求する情報の多くを含む。タンパク質形の表は既に、注釈付き配列および質量の全てを含んでいるのであるから、検索アルゴリスムの探索に対しデータベースから速やかな反応を得ることができる。
【0051】
修飾部位は、タンパク質の遺伝子配列から理論的に予測が可能であるが、注釈データベースを全ての可能な注釈で混雑させるのは多くの場合好ましくない。このような注釈を含めることは、その横断経および検索探索時間延長の観点から取り扱いにくいデータベースを生むことになる。
【0052】
一旦検索アルゴリスムが、配列タグ探索過程に基づいて候補タンパク質の精選集合を特定したら、それらの特定のタンパク質に関する可能な全ての注釈を含む拡張集合の生成が可能である。ウェアハウス・データベースをこのように修飾しても、検索アルゴリスムの性能を危機に陥れることはない。なぜなら、探索行為は、可能なタンパク質形の小集合に限定されるからである。従って、ハイブリッド探索法にウェアハウス・データベースの動的ショットガン注釈付加を含めることも可能である。このタンパク質候補集合が精選されて、候補ポリペプチドおよびその関連修飾から成る最終組が得られたならば、別のサンプルポリペプチドの特性特徴付けが実行される前に、先にウェアハウス・データベースに動的に入力されたショットガン注釈は消去してもよい。
【0053】
イオン予測器は、理論的b/yおよびc/zイオンを予測し、ソフトウェアおよびシステムに含められる。この計算は、ダルトンまたはppmで表した場合の誤差を計算するのに有用である(例えば、実施例1、表1を参照)。
【0054】
[データ縮小ツール]
ソフトウェアおよびシステムには、多数の荷電状態および縮小断片化データにおける水/アンモニア損失による冗長ピークを除去するためのデータ縮小ツールが含まれる。このツールは、獲得MSデータについて、それに検索アルゴリスムを適応する前に、高速分析する場合に有用である。
【0055】
[データベース管理システム]
ウェアハウス・データベースについてはいかなるデータベース管理システムでも使用が可能である。データベース管理システムはMySQLを含むのが好ましい。この、人々の好むデータベースシステムを選ぶのは、それが多くの有用なサポートツールとAPIを持つからであり、システムが一般人にも簡単に手に入るからである。付録に提供されるソフトウェアは、リナックス用の、バージョン11.18頒布3.23.52MySQLを用いている。
【0056】
[グラフィック表示インターフェイスツール]
全ての探索法において、様々なスコアを持つ候補配列の集団が返される。本ソフトウェアおよびシステムには、あらゆる探索法によって得られる候補配列集合を一覧するためのグラフィック表示インターフェイスツールが含まれる。要すれば任意に、このグラフィック表示インターフェイスツールは、本発明の他の特質を含む、局所ワークステーションに組み込まれる。要すれば任意に、グラフィック表示インターフェイスツールは、遠隔サーバーからインターネットを介して入手するデータを一覧できるように適応される。
【0057】
絶対質量モード探索では、ユーザーに、遺伝子記述、配列、配列長、理論的質量、質量差(絶対量およびppm)、マッチするb(またはc)型イオンの数、マッチするy(またはz*)型イオンの数、マッチする断片の合計数、および確率スコア計算値が提示される。次に、ユーザーは、候補タンパク質の集合を列挙された見出しに従って仕分けし、任意の検索配列について断片化詳細を眺める。断片化詳細表示は、ユーザーに対し、その配列にマッチする全ての断片に関する詳細情報を提示する。この画面は、特定されたイオン、観察された質量、理論的質量、単純な質量差(すなわち、例えば、「デルタM」モードによって演繹される質量偏倚が考察される前の)と質量差偏倚(すなわち、「デルタM」モードの場合のような質量偏倚の考察後の)、および、ppmで表した変異差を提示する。このグラフィック表示インターフェイスツールはまた、断片化の詳細を眺めることを可能とする。この詳細は、配列カバー範囲を決定し、断片化パターンの在り処を定めるのに有効な特質であって、適切な特定に対するユーザーの信頼を増すものである。
【0058】
[サポートされているデータベース]
サポートデータベースは、どのような生物体について構成されたものであってもよい。一つの実施態様は、9種の生物体、すなわち、Saccharomyces cerevisiae(酵母)、Escherichia coli(大腸菌)、Arabidopsis thaliana(シロイヌナズナ)、Bacillus subtilis(枯草菌)、Methanococcus jannaschii(古細菌)、Mycoplasma pneumoniae(肺炎マイコプラスマ)、Shewanella oneidensis(シェワネラ・オネイデンシス)、Mus musculus(ハツカネズミ)、およびHomo sapiens(ヒト)を含む生物体のデータベースをサポートする。酵母生物体Saccharomyces cerevisiaeのデータベースは、既知のものと、予測される修飾情報を兼ね備えたもっとも広範な注釈を含む。
【0059】
[データベースのスケール特性]
特に興味あるのは、データベースと探索時間のスケールが修飾情報の増加と共にどのように変化するかである。ある遺伝子と予測修飾は、タンパク質形において、各形が可能な修飾のサブセットを含むために指数関数的となる。従って、n個のタンパク質と、タンパク質当たりm個の可能な加工事象がある場合、一つの実施態様は、O(n2m)個のタンパク質形を含む。検索探索アルゴリスムがO(mlog 2n)で動き、定数は元の許容値に依存するとした場合、絶対質量探索アルゴリスムのスケールはmに対して直線的に変化する。既知および予測タンパク質形から成るデータベースでは、いくつかの修正が適切に予測された場合、観察されたタンパク質形は特定、特徴付けされる可能性がある。公開のタンパク質データベースにおける虚偽情報の増加は、乏しいMS/MSデータに基づく探索を不確かなものにする。一方、探求段階で用いられる修飾情報がより広範で正確になるにつれて、マッチする断片イオン質量の数は増す。
【0060】
[質量分析装置に対するコンピュータインターフェイス]
要すれば任意に、各成分は、コンピュータシステムにおいて質量分析計と相互通信する(in communication with)ように組織されてもよい。一つの実施態様では、コンピュータは局所的ワークステーションである。別の実施態様では、コンピュータは、現場から離れたサーバーである。後の実施態様では、各成分はサーバーに保存され、インターネットによるインターフェイスツールを用いてアクセスされる。質量分析器から生成されたMSデータはコンピュータに伝送され、そこでデータは獲得・保存される。コンピュータの中央処理ユニットは、タンパク質形のウェアハウス・データベースを探索するために、前記好ましい実施態様の一つに従って動作する検索アルゴリスムを用い、獲得されたMSデータの分析を指揮する。オペレータ指定の許容値が、検索アルゴリスムソフトウェアによって提供される選択肢の内から選ばれ、これに基づいて、タンパク質形のウェアハウス・データベースからタンパク質候補を収集し、その後さらに修飾の分析を行うことが可能となる。
【0061】
[医学的応用]
生体内の特定の標的タンパク質における修飾の程度に及ぼす環境シグナルの影響を見分けることが可能である。例えば、ヒトの病態の多くは、修飾、例えばリン酸化によって調整されている。家系内の特定遺伝子の修飾による変化に帰せられる障害は、後生性と診断される。異常な修飾があるかどうかについて特定タンパク質を調査するならば、その他のやり方では既知の遺伝子配列と相関をほとんど示さない病態について新しい洞察が得られる可能性がある。従って、このシステムは、障害や、特定の疾患に罹り易い性質を持つ個人を選び出すための強力なプラットフォームとなる。
【0062】
個々のタンパク質の修飾変化が病気の発生に関与する場合、特定のタンパク質に対する修飾付加または修飾排除を調節する製薬化合物の発見を促進する研究背景に使用可能するようにシステムを構成することも可能である。本明細書に開示される一つの実施態様では、システムは、高処理能力を持つスクリーニング方策の構成成分として設置される。このスクリーニング方策では、ある特定のタンパク質基質の修飾を触媒する修飾活性と連結する酵素を促進または抑制する能力について、候補製薬化合物からなる結合ライブラリーを評価する。タンパク質基質について、MSを用いて修飾の存在(または不在)について尋問する。次に、所望の薬物作用を持つ化合物が、特定の病気に向けた薬剤開発二次プログラムにおいて用いられる。
【0063】
特定タンパク質に対して修飾付加または排除を調節または変調する製薬化合物を評価するために、本システムを臨床背景下に使用可能とするように構成してもよい。一つの実施態様では、患者サンプルについて、特定のタンパク質が薬剤処置に応じて修飾を持つようになったかどうかについて、このシステムを用いて確認することが可能である。例えば、患者サンプルの細胞分解産物から得られる、対象とする標的タンパク質を、均一になるまで精製し、その精製物に対して、本明細書に記載される方法、ソフトウェア、およびシステムに従ってMS/MS分析を行ってもよい。サンプルタンパク質について得られたMSデータと、対応するタンパク質形であって、ウェアハウス・データベースに含まれる、全ての、その天然のショットガン修飾注釈を含めた対応タンパク質形との違いは簡単に獲得されるが、その違いは、治療処方の薬剤活性に関して豊かな情報を提供すると考えられる。
【0064】
本発明を用いることによって、タンパク質の各種修飾を、その発生機構と無関係に、検出することが可能となることは当業者には簡単に察しがつくであろう。例えば、単一タンパク質において、多型の位置、RNAスプライシング、またはmRNAのRNA編集が得られるタンパク質配列におよぼす作用、翻訳後修飾の有無、および、環境誘発性の化学的修飾を特定し、その特性特徴付けするのに本発明を用いてもよい。さらに、ハイブリッドモード探索法によって、理論的に予測されるポリペプチド形と、実際に観測されるポリペプチドの間の質量差を生み出す何らかの生物学的事象またはバイオインフォーマティックな不確かさが検出されることは、通常の熟練度を持つ当業者であれば理解されよう。
【0065】
[ProSight PTM:ソフトウェアと構造]
付録に、本明細書に開示される局面と実施態様を実行するために必要な全てのソフトウェアツール、およびタンパク質形の注釈付きウェアハウス・データベースを提供するコンパクトディスクが含まれている。”ProSight PTM”という名前のこのシステムは好ましい実施態様である。このシステムは、全てインターネット準拠のインターフェイスを持つ4つの主要成分を含む。すなわち、タンパク質データベース(ProSightウェアハウス)、データベース検索アルゴリスム(レトリーバ)、データマネージャー、プロージェクトトラッカー、およびその他のユーティリティである(図4参照、Taylor GK, Kim YB, Forbes AJ, Meng F, McCarthy R, Kelleher NL. “Web and database software for identification of intact proteins using top down mass spectrometry,” Anal. Chem. 75:4081-86 (2003))。
【0066】
スピードが重視される作業、例えば、データベース検索および配点は、iODBCライブラリーを用い、リナックス上においてC++で、オブジェクト指向設計を用いてデータベース結合用に書き込まれる。データ縮小ツールはOCaml(言語発現性のために選ばれる)で書き込まれ、一方、視覚化ツールは、画像変換用GDモジュールを用いてPERLで書き込まれる。
【0067】
絶対質量探索の使用では、ODBCで可動とされる管理システムに、ProSightウェアハウスを起動搭載することを必要とする。インターネット・アプリケーションは、二重プロセッサーAthlon 2200+MP上で走るApache HTTPサーバーによってサポートされるCGIを用いてPERLで書き込まれる。
【実施例】
【0068】
いくつかの実施態様を、S. cerevisiae(酵母)の36-kDaタンパク質と関連する修飾に関するMS/MS分析に注目する特異的具体例と結びつけて開示する。このタンパク質は、後に、グリセルアルデヒドリン酸デヒドロゲナーゼ3型酵素と特定された。Q-FTMSを用いたけれども、どのタイプの質量分析器から得られた元のタンパク質データであっても代替が可能である。手元の特定の応用にとって望ましいように、検索スコアおよび修飾特徴付け率を向上させるため、既知および予想修飾情報を利用するためのデータベース戦略が記載される。
【0069】
(実施例1:元の酵母タンパク質の自動的トップダウン分析)
35,758.3 DaのMr値を持つ酵母タンパク質が、一つのALS-PAGE/RPLC分画において観察された(図7A)。同じサンプル中に他に3個の成分があった。それらの内一つは、35.8-kDa種に付着したリン酸塩付加物(+98 Da)に対応していた。オンラインの分解アルゴリスムによって、35.8-kDaのタンパク質が摘出され、図7Bに示す5種の荷電状態を選択する適切なSWIFT波形が生成された。IRレーザーを用い、図7CのMS/MSスペクトラムは自動的に生成された。このスペクトラムは、THRASHアルゴリスムによって自動的に検出された、27個の別々の断片イオン質量値に対応する、39個の同位元素分布が観察された。虚偽のピーク(例えば、水損失ピーク)を排除するフィルター通過後、データベース検索用の最終インプットとして20個のイオン質量が用いられた。このタンパク質は、9個のb-型イオンおよび3個のy-型イオンマッチを含む、グリセルアルデヒド-3-リン酸デヒドロゲナーゼ(GAPDH3)であると特定された(表1および2)。この検索のP-スコアは、4 x 10-8であり、これは、この特定内容が偽者事象ではあり得ないことを示す。
【0070】
表1 GAPDH3(配列番号1)のイオン断片化データ
1GAPDH3は331個のアミノ酸を持ち、理論的質量は35,615.5 Da、Δmは142.8 Daである。
【0071】
表2 GAPDH3(配列番号1)1のグラフ表示断片マップ
【0072】
この遺伝子産物(GAPDH3、配列番号1)は、GAPDH遺伝子ファミリーの他のメンバー、GAPDH2(配列番号2)およびGAPDH1(配列番号3)と、それぞれ、96%と80%の相同性を持ちながらはっきりと区別された。これらのデータから、このタンパク質形は、331個のアミノ酸残基の内たった3個が異なるにも拘わらず、ExPASyによって報告された混乱から区別された。さらに、GAPDH3遺伝子産物の観察された分子量は、データベースの配列(開始Met無し)から計算される理論値よりも142 Da大きかった。断片マップによって、この質量差(Δm)はAsp90とAsp192の間に位置づけられ、この配列領域では僅かに2個しかCys残基(Cys149とCys153)はない(表2参照)。
【0073】
次に、このタンパク質形について、手動のQ-FTMS/MSと、超伝導マグネットの外部におけるイオンの衝突解離を用いて尋問したところ、98個の同位元素分布を持つ、図7Dのスペクトラムが得られた。これらのデータを検索アルゴリスムに対するインプットして用いると、+142 DaのΔmは、さらにPro126 Leu154領域に狭められる。これらのデータは、ゲル電気泳動中、2個のCys残基がアクリルアミド(それぞれ+71 Da)によってアルキル化されるという所見と一致する。正確にCys149とCys153に位置づけられたわけではないが、このゲル内修飾にはいくつかの先例があり、PAGE分画における遊離チオールについては予想されている。以上、この全体過程は、トップダウン法による共有的修飾の初期検出を含んでいた。
【0074】
絶対質量検索時間が、スコアされる候補配列の数に直線的に依存するのであれば、元の許容値が小さければ検索時間は短くなる。±2-kDaの許容値を用いて酵母を単純に探索すると、1500個の候補を得るために6秒かかるが、一方、200-Da許容値で同じ探索をすると400 msで終了して200個の候補を得る。ハイブリッド探索は、FASTAファイル入力の数と、考察される配列タグの数に直線的に依存する。5個の配列タグによる探索は4秒で完了する。これまで断片化された酵母タンパク質の内約半分は、観察された断片イオンの絶対質量に基づき検索アルゴリスムによって特定されている。残りについては、20%は、観察された断片イオン同士の間の相対的質量差に基づいて生成される配列タグを通じて特定された。配列タグモードでは、図7Cのデータの自動編集によって、4種のタグ(それぞれ4アミノ酸長で、2個は実際にあり、2個は虚偽のもの)が得られた。配列タグの編集を同じ荷電の断片イオンに限定したところ、適正なタグは僅か二つしか得られなかった。図7Dのデータを用いたところ、荷電状態制限により、8個のタグの内5個は偽者であり(長さは1-4アミノ酸)、6個の内4個は偽者(長さは1-3アミノ酸)であった。
【0075】
(実施例2:修飾活性によって酵素を修飾する化合物のスクリーニング(予測例))
下記の実施例の目的は、修飾活性を示す酵素の機能を、陽性または陰性に修飾する化合物を、結合ライブラリーにおいて特定するための高処理能力方策を概観することである。インビトロ環境における特定例が記載されているけれども、この実施例をインビボ背景に適応させるやり方は簡単に理解される。
【0076】
N-末端ヒスチジンタグ(UpState Biotechnology, Inc., レークプラシッド、ニューヨーク州)を含む、ヒトSrcキナーゼ癌タンパク質の組み換え形を、Srcキナーゼバッファー(100 mM Tris-HCl (pH7.2), 125 mM MgCl2, 25 mM MnCl2, 2 mM EGTA, 500 μM ATP, 0.25 mM オルトバナジン酸ナトリウム、および2 mMジチオスレイトール)に溶解したNi-NTA樹脂でコートした96ウェル皿に固定する。Srcキナーゼバッファーに溶解した試験化合物の添加後、できれば、1ウェル当たり一つの均質化合物、既知の配列を持つSrcタンパク質基質を各ウェルに加える(100 300 μMの濃度で)のが好ましい。これによってリン酸化が可能となる。インキュベーション後、基質を検索し、ProSight PTMシステムを用いてトップダウン質量分析を行う。
【0077】
ある特定の化合物の、Src活性に対する抑制活性は、そのタンパク質の内部にリン酸化チロシン残基と関連する修飾の欠如に基づいて見分けられる。このような化合物は、このトップダウン分析を確認するために、他のアッセイを用いてさらに特性特徴付けするのに好適である。例えば、アッセイに[γ-32P] ATPを用い、P81論文のTCA沈殿アッセイを用いてリン酸化活性を監視してもよい。
【0078】
(実施例3:個人における後生性(epigenetic)障害の検出(予測例))
本実施例の目的は、トップダウン質量分析法を用いて後生性障害と関連する修飾を検出するためのProSight PTMシステムの利便性を明らかにすることである。サンプル組織を、ニワトリ肉腫ウィルスに感染したニワトリを始め、未感染ニワトリから入手する。これらのサンプルをホモジェナイズし、澄明にし、可溶性細胞分解産物を生成する。ニワトリSrcキナーゼの既知のインビボ基質であるγ-カテニンタンパク質を、抗γ-カテニン抗体を用いて細胞分解産物からアフィニティー精製する。次に、この検索されたγ-カテニンサンプルに、トップダウン質量分析およびProSight PTMによる分析を実施する。期待される結果は、正常組織から検索されたγ-カテニンタンパク質は、ProSightウェアハウス・データベースに保存されるタンパク質の正常な修飾プロフィールを表し、それに対し、感染ニワトリから検索されるγ-カテニンタンパク質は、チロシンリン酸化と関連する、追加の修飾を含むことである。
【0079】
(実施例4:実施例1-3の実験手順)
[細胞培養および細胞分解産物の画分]
S. cerevisiae細胞(S288C株)を好気条件下で育成した。約2 gの細胞(湿潤重量)を、2個のプロテアーゼ阻害剤錠剤(Roche Diagnostics、マンハイム、ドイツ)を含む、10 mLの細胞溶解バッファー(25 mM Tris, 1 mM EDTA, 1 mM TCEP, pH7.0, 1 mLのDNAアーゼ添加)に再懸濁した。フレンチプレスにて細胞破壊後、細胞分解産物を、10,000 x g、30分遠心によって澄明にした。次に、この上清を、酸感受性を持つ界面活性剤(ALS)サンプルバッファーと混合し、次に、0.1% SDSの代わりに0.1% ALS-Iを用いるモデル491調整ゲル装置(Bio-Rad)に負荷した。4% Tスタックゲルを用い、12% T分解ゲルを流速0.50 mL/分で移動して溶出した。収集した80個の分画(それぞれ2 mL)の内、2個は、冷却アセトン沈殿、6Mグアニジン塩酸(pH2)によってさらに処理し、逆相液体クロマトグラフィー(RPLC)を用いて分析した。クロマトグラフィーは、標準溶媒(H2O, CH3CN、および0.1% TFA)を用い、15分に渡って直線的勾配を持つ、対称300 C4カラム(4.6 x 50 mm, Waters Inc. ミルフォード、マサチューセッツ州)を用いた。
【0080】
[ESI-Q-FTMS装置設定]
RPLCで分画されたタンパク質は乾燥し、80 μLのESI液(50% ACN, 49% H2O、および1%ギ酸)に再懸濁し、次に、ナノスプレイロボット(Advion BioSciences, Ithaca, NY)に負荷した。これによって、−100 nL/分で5-10 μLのサンプルが直接分析された。本実験で用いられた8.5-T Q-FTMS装置は、別の場所で記載されるように社内で構築したものである。簡単に言うと、先ず、タンパク質イオンが、8本の極に保存され、次に、4重極を通じて移送され、第2の8本極に蓄積され、最終的にICRセルにて分析される。4重極は、質量選択モードでも、「rfのみ」モードでも動作することが可能である。Tclで書かれた自動化スクリプトは、元のタンパク質のスペクトラムを獲得し、次に、オンラインの分解アルゴリスムを呼び出してMr値を計算し、SWIFTが、もっとも多量な5種の荷電状態を単離する。単離された荷電状態に対して5回の走査を行った後、IRレーザーがスイッチオンされ、25回か、50回かいずれかの走査が行われる(0.45秒、75%電源、40-Wレーザー)。図7DのQ-FTMS/MSスペクトラムは、荷電状態が4重極から第2の8本極へ移動する際に、指定の荷電状態の衝突的解離を通じて手動的に獲得されたものである。
【0081】
(参考文献)
Belov ME, Nikolaev EN, Anderson GA, Auberry KJ, Harkewicz R, Smith RD. "Electrospray ionization-Fourier transform ion cyclotron mass spectrometry using ion preselection and external accumulation for ultrahigh sensitivity," J. Am. Soc. Mass Spectrom. 12:38-48 (2001).
Biemann K, Papayannopoulos I. Acc. Chem. Res. 27:370-78 (1994).
Clauser KR, Baker P, Burlingame AL. "Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching," Anal. Chem. 71:2871-82 (1999).
Ficarro S, McCleland M, Stukenberg P, Burke D, Ross M, Shabanowitz J, Hunt D, White F. "Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae," Nat. Biotechnol. 20:301-305 (2002).
Garavelli, JS. "The RESID Database of Protein Modifications: 2003 developments," Nucleic Acids Res. 31:499-501 (2003).
Ge Y, Lawhorn BG, ElNaggar M Strauss E, Park JH, Begley TP, McLafferty FW. "Top down characterization of larger proteins (45 kDa) by electron capture dissociation mass spectrometry," J. Am. Chem. Soc. 124:672-78 (2002).
Ge Y, ElNaggar M, Sze SK, Bin OH, Begley TP, McLafferty FW, Boshoff H, Barry CE. J. Am. Soc. Mass Spectrom. 14:253-61 (2003).
Gerber SA, Rush J, Stemmann O, Steen H, Kirschner MW, Gygi SP. In: 50th ASMS Conference on Mass Spectrometry and Allied Topics, Orlando, FL, 2002.
Goshe MB, Conrads TP, Panisko EA, Angell NH, Veenstra TD, Smith RD. "Phosphoprotein isotope-coded affinity tag approach for isolating and quantitating phosphopeptides in proteome-wide analyses," Anal. Chem. 2001, 73:2578-86 (2001).
Johnson JR, Meng F, Forbes AJ, Cargile BJ, Kelleher NL. "Fourier-transform mass spectrometry for automated fragmentation and identification of 5-20 kDa proteins in mixtures," Electrophoresis 23:3217-23 (2002).
【0082】
Kachman MT Wang H, Schwartz DR, Cho KR, Lubman DM. "A 2-D liquid separations/mass mapping method for interlysate comparison of ovarian cancers," Anal. Chem. 74:1779-91 (2002).
Kelleher NL, Costello CA, Begley TP, McLafferty FW. J. Am. Soc. Mass Spectrom. 6:981-84 (1995).
Kelleher NL, Taylor SV, Grannis D, Kinsland C, Chiu HJ, Begley TP, McLafferty FW. "Efficient sequence analysis of the six gene products (7-74 kDa) from the Escherichia coli thiamin biosynthetic operon by tandem high-resolution mass spectrometry," Protein Sci. 7:1796-1801 (1998).
Lander ES et al. "Initial sequencing and analysis of the human genome," Nature 409:860-921 (2001).
MacCoss MJ McDonald WH, Saraf A, Sadygov R, Clark JM, Tasto JJ, Gould KL, Wolters D, Washburn M, Weiss A Clark JI, Yates JR., III. "Shotgun identification of protein modifications from protein complexes and lens tissue," Proc. Natl. Acad. Sci. U.S.A. 99:7900-7905 (2002).
Meng F, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. "Informatics and multiplexing of intact protein identification in bacteria and the archaea," Nat. Biotechnol. 19:952-57 (2001).
Meng F, Cargile BJ, Patrie SM, Johnson JR, McLoughlin SM, Kelleher NL. "Processing complex mixtures of intact proteins for direct analysis by mass spectrometry," Anal. Chem. 74:2923-29 (2002).
Oda Y, Huang K, Cross FR, Cowburn D, Chait BJ, "Accurate quantitation of protein expression and site-specific phosphorylation," Proc. Natl. Acad. Sci. U.S.A. 96:6591-96 (1999).
Oda Y, Nagasu T, Chait BT. "Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome," Nat. Biotechnol. 19:379-82 (2001).
【0083】
Perkins D, Pappin D, Creasy D, Cottrell J. "Probability-based protein identification by searching sequence databases using mass spectrometry data," Electrophoresis 20:3551-67 (1999).
Pineda FJ, Lin JS, Fenselau C, Demirev PA. "Testing the significance of microorganism identification by mass spectrometry and proteome database search," Anal. Chem. 72:3739-44 (2000).
Reid GE, Shang H, Hogan JM, Lee GU, McLuckey SA. "Gas-phase concentration, purification, and identification of whole proteins from complex mixtures," J. Am. Chem. Soc. 124:7353-62 (2002).
Reid GE, Stephenson JL, McLuckey SA. "Tandem mass spectrometry of ribonuclease A and B: N-linked glycosylation site analysis of whole protein ions," Anal. Chem. 74:577-83 (2002).
Steen H, Kuster B, Fernandez M, Pandey A, Mann M. "Detection of tyrosine phosphorylated peptides by precursor ion scanning quadrupole TOF mass spectrometry in positive ion mode," Anal. Chem. 73:1440-48 (2001).
Taylor GK, Kim YB, Forbes AJ, Meng F, McCarthy R, Kelleher NL "Web and database software for identification of intact proteins using top down mass spectrometry," Anal. Chem. 75:4081-86 (2003).
Wilkins MR, Gasteiger E, Gooley AA, Herbert BR, Molloy MP, Binz PA, Ou K, Sanchez JC, Bairoch A, Williams KL, Hochstrasser DF. "High-throughput mass spectrometric discovery of protein post-translational modifications," J. Mol. Biol. 289:645-57 (1999).
Zhang W, Chait B. "ProFound: an expert system for protein identification using mass spectrometric peptide mapping information," Anal. Chem. 72:2482-89 (2000).
Zhou H, Watts JD, Aebersold R. "A systematic approach to the analysis of protein phosphorylation," Nat. Biotechnol. 19:375-78 (2001).
【図面の簡単な説明】
【0084】
【図1】候補タンパク質を得るためにMSデータについて実施する絶対質量モード探索手順を表すアーキテクチャのフローチャートである。
【図2】MSによるタンパク質の特定と特性特徴付けのための「トップダウン」法および「ボトムアップ」法であって、修飾(例えば、翻訳後修飾(”PTM”))を特定し、位置づける方法を図示する。
【図3】ハイブリッド探索モード法の工程フローチャートを示す。
【図4】検索アルゴリスム(ProSightレトリーバ)、タンパク質形のウェアハウス・データベース(ProSight PTMウェアハウス)、および一次ユーティリティを含むソフトウェアシステムのフローチャートである。
【図5】データベースが「デルタm」モードで探索される実施態様を示す。
【図6】ショットガン注釈付加の模式図を示す。
【図7】S. cerevisiaeから得られたALS-PAGE/RPLC分画に対するMS/MSの例を示す。
【特許請求の範囲】
【請求項1】
サンプルポリペプチドについて1組の候補ポリペプチドを選択する方法であって、
質量分析によって生成されたサンプルポリペプチド断片の質量における差に基づいて、候補ポリペプチドを収集する、第一の精選;及び
サンプルポリペプチドの絶対質量、および前記断片の絶対質量に基づいて、候補ポリペプチドを収集する、第二の精選;を含む前記方法。
【請求項2】
第一の精選が、断片の質量差に基づいて、サンプルポリペプチドの、少なくとも部分的なアミノ酸配列を決定することを含む、請求項1記載の方法。
【請求項3】
サンプルポリペプチドの原形の絶対質量と、サンプルポリペプチド断片の絶対質量とを決定することをさらに含む、請求項2記載の方法。
【請求項4】
精選される集合はウェアハウス・データベースを含み、
サンプルポリペプチドの少なくとも部分的アミノ酸配列に基づいて、ウェアハウス・データベースから候補ポリペプチドを選出することをさらに含むことを特徴とする、請求項2記載の方法。
【請求項5】
サンプルポリペプチドの一次構造を決定する方法であって、
請求項1記載の方法によって1組の候補ポリペプチドを選択すること、
サンプルポリペプチドの絶対質量を、候補ポリペプチドの理論的絶対質量と比較することによって、マッチングの確率スコアを導くこと;および、
マッチングの確率スコアをランク付けすることによって、マッチングの最大確率スコアに基づいて候補ポリペプチドの一つによってサンプルポリペプチドの一次構造を特定すること;
を含む前記方法。
【請求項6】
ウェアハウス・データベースが、ウェアハウス・データベース中の少なくとも一つのポリペプチドについて少なくとも1のショットガン注釈をさらに含む、請求項4記載の方法。
【請求項7】
ショットガン注釈が翻訳後修飾を含む、請求項6記載の方法。
【請求項8】
翻訳後修飾が、リボシル化、リン酸化、アルキル化、ヒドロキシル化、グリコシル化、酸化、還元、ミリスチル化、ビオチニル化、ユビキノン化、ヨード化、ニトロシル化、アミノ化、硫黄付加、環化、ヌクレオチド付加、脂肪酸付加、およびアシル化から成る群から選ばれる少なくとも1の修飾を含む、請求項7記載の方法。
【請求項9】
ウェアハウス・データベースが、コンピュータの電子メモリーに保存されている、請求項4記載の方法。
【請求項10】
ユーザーは、検索アルゴリスムを用いて、電子通信を介してコンピュータにアクセスすることによって、ウェアハウス・データベースから情報を検索することが可能である、請求項9記載の方法。
【請求項11】
検索アルゴリスムが、インターネット・ソフトウェアアプリケーションをさらに含む、請求項10記載の方法。
【請求項12】
ポリペプチド基質を翻訳後に修飾する酵素に対する抑制活性について化合物をスクリーニングする方法であって、
酵素を化合物と接触させて予備混合物を形成すること;
前記予備混合物にポリペプチド基質を加えて、反応混合物を形成すること;
請求項5記載の方法を用いてポリペプチド基質を分析すること;
を含む前記方法。
【請求項13】
酵素による反応を触媒する補助因子の添加をさらに含み、前記補助因子は、ATP、ADP、AMP、GTP、GDP、GMP、CTP、CDP、CMP、UTP、UDPおよびUMPから成る群から選ばれる少なくとも一つの補助因子である、請求項12記載の方法。
【請求項14】
酵素が、固相支持体の上に固定される、請求項12記載の方法。
【請求項15】
コンピュータと共に使用されるコンピュータプログラム製品であって、
サンプルポリペプチドについて1組の候補ポリペプチドを選択するための、コンピュータで読み取り可能なプログラムコードを有するコンピュータで使用可能な媒体;と、
サンプルポリペプチドについて1組の候補ポリペプチドを選択するようコンピュータに指令するためのコンピュータで読み取り可能なプログラムコード;とを含み、
該プログラムコードは、
質量分析によって生成されたサンプルポリペプチド断片の質量差に基づいて、候補ポリペプチドを収集する第一の精選;および、
サンプルポリペプチドの絶対質量、および前記断片の絶対質量に基づいて、候補ポリペプチドを収集する第二の精選;
を含む、前記コンピュータプログラム製品。
【請求項16】
コンピュータで読み取り可能なプログラムコードが、収集の第一の精選を決定するようコンピュータに指令するためのものであり、且つ
前記第一の精選は、断片の質量差に基づいて、サンプルポリペプチドの少なくとも部分的アミノ酸配列を決定することを含む、請求項15記載のコンピュータプログラム。
【請求項17】
サンプルポリペプチドの原形の絶対質量、およびサンプルポリペプチド断片の絶対質量を決定するようコンピュータに指令するためのコンピュータで読み取り可能なプログラムコードをさらに含む、請求項16記載のコンピュータプログラム製品。
【請求項18】
サンプルポリペプチドの少なくとも部分的なアミノ酸配列に基づいて、タンパク質形の集合から候補ポリペプチドを選択するようコンピュータに指令するための、コンピュータで読み取り可能なプログラムコードをさらに含む、請求項16記載のコンピュータプログラム製品。
【請求項19】
請求項1記載の方法によって1組の候補ポリペプチドを選択すること;サンプルポリペプチドの絶対質量を、候補ポリペプチドの理論的絶対質量データと比較することによってマッチングの確率スコアを導くこと;および、マッチングの確率スコアをランク付けすることによって、マッチングの最大確率スコアに基づいて候補ポリペプチドの一つによってサンプルポリペプチドの一次構造を特定すること;をコンピュータに指令して実行させるコンピュータで読み取り可能なプログラムコードをさらに含む、請求項16記載のコンピュータプログラム製品。
【請求項20】
コンピュータ、
タンパク質形のウェアハウス・データベース、および、
一次ユーティリティ
を含むシステムをさらに含む、請求項15記載のコンピュータプログラム製品。
【請求項21】
一次ユーティリティが、データ管理システム、イオン予測器、データ縮小ツール、およびグラフ表示インターフェイスツールから成る群から選ばれる少なくとも一つの要素を含む、請求項20記載のコンピュータプログラム製品。
【請求項22】
ウェアハウス・データベースが、ショットガン注釈をさらに含む、請求項20記載のコンピュータプログラム製品。
【請求項23】
ウェアハウス・データベースが、動的ショットガン注釈をさらに含む、請求項20記載のコンピュータプログラム製品。
【請求項24】
システムがさらに検索アルゴリスムを含み、前記検索アルゴリスムは、絶対質量探索モードと配列タグ探索モードとを含む、請求項20記載のコンピュータプログラム製品。
【請求項25】
絶対質量探索モードが、さらにΔm探索モードを含む、請求項24記載のコンピュータプログラム製品。
【請求項26】
コンピュータと相互通信する質量分析器をさらに含む、請求項20記載のコンピュータプログラム製品。
【請求項27】
コンピュータは、インターネット・ソフトウェアアプリケーションを介してユーザーと相互通信する、請求項20記載のコンピュータプログラム製品。
【請求項28】
コンピュータ;
タンパク質形のウェアハウス・データベース;
ウェアハウス・データベースを探索するための検索アルゴリスム;
データ管理システム;
イオン予測器;
データ縮小ツール;および、
グラフ表示インターフェイスツール;
をさらに含む、請求項20記載のコンピュータプログラム製品。
【請求項29】
サンプルポリペプチドについて1組の候補ポリペプチドを選択するシステムであって、
質量分析によって生成されるサンプルポリペプチド断片の質量差に基づいて、候補ポリペプチド集合の第一の精選を実行するための手段;並びに、
サンプルポリペプチドの絶対質量、および、質量分析によって生成されるサンプルポリペプチド断片の絶対質量に基づいて、候補ポリペプチド集合の第二の精選を実行するための手段;
を含む前記システム。
【請求項30】
コンピュータが質量分析器と相互通信することを特徴とする、請求項29記載のシステム。
【請求項31】
コンピュータは、インターネット・ソフトウェアアプリケーションを介してユーザーと相互通信する、請求項29記載のシステム。
【請求項32】
サンプルポリペプチドについて1組の候補ポリペプチドを選択するためのシステムであって、
請求項15記載のコンピュータプログラム産物、および
コンピュータ
を含む前記システム。
【請求項33】
サンプルポリペプチドおよびサンプルポリペプチド断片の絶対質量に基づいて収集する第三の精選をさらに含み、且つ前記第三の精選が、第一の精選に先立って行われる、請求項1記載の方法。
【請求項1】
サンプルポリペプチドについて1組の候補ポリペプチドを選択する方法であって、
質量分析によって生成されたサンプルポリペプチド断片の質量における差に基づいて、候補ポリペプチドを収集する、第一の精選;及び
サンプルポリペプチドの絶対質量、および前記断片の絶対質量に基づいて、候補ポリペプチドを収集する、第二の精選;を含む前記方法。
【請求項2】
第一の精選が、断片の質量差に基づいて、サンプルポリペプチドの、少なくとも部分的なアミノ酸配列を決定することを含む、請求項1記載の方法。
【請求項3】
サンプルポリペプチドの原形の絶対質量と、サンプルポリペプチド断片の絶対質量とを決定することをさらに含む、請求項2記載の方法。
【請求項4】
精選される集合はウェアハウス・データベースを含み、
サンプルポリペプチドの少なくとも部分的アミノ酸配列に基づいて、ウェアハウス・データベースから候補ポリペプチドを選出することをさらに含むことを特徴とする、請求項2記載の方法。
【請求項5】
サンプルポリペプチドの一次構造を決定する方法であって、
請求項1記載の方法によって1組の候補ポリペプチドを選択すること、
サンプルポリペプチドの絶対質量を、候補ポリペプチドの理論的絶対質量と比較することによって、マッチングの確率スコアを導くこと;および、
マッチングの確率スコアをランク付けすることによって、マッチングの最大確率スコアに基づいて候補ポリペプチドの一つによってサンプルポリペプチドの一次構造を特定すること;
を含む前記方法。
【請求項6】
ウェアハウス・データベースが、ウェアハウス・データベース中の少なくとも一つのポリペプチドについて少なくとも1のショットガン注釈をさらに含む、請求項4記載の方法。
【請求項7】
ショットガン注釈が翻訳後修飾を含む、請求項6記載の方法。
【請求項8】
翻訳後修飾が、リボシル化、リン酸化、アルキル化、ヒドロキシル化、グリコシル化、酸化、還元、ミリスチル化、ビオチニル化、ユビキノン化、ヨード化、ニトロシル化、アミノ化、硫黄付加、環化、ヌクレオチド付加、脂肪酸付加、およびアシル化から成る群から選ばれる少なくとも1の修飾を含む、請求項7記載の方法。
【請求項9】
ウェアハウス・データベースが、コンピュータの電子メモリーに保存されている、請求項4記載の方法。
【請求項10】
ユーザーは、検索アルゴリスムを用いて、電子通信を介してコンピュータにアクセスすることによって、ウェアハウス・データベースから情報を検索することが可能である、請求項9記載の方法。
【請求項11】
検索アルゴリスムが、インターネット・ソフトウェアアプリケーションをさらに含む、請求項10記載の方法。
【請求項12】
ポリペプチド基質を翻訳後に修飾する酵素に対する抑制活性について化合物をスクリーニングする方法であって、
酵素を化合物と接触させて予備混合物を形成すること;
前記予備混合物にポリペプチド基質を加えて、反応混合物を形成すること;
請求項5記載の方法を用いてポリペプチド基質を分析すること;
を含む前記方法。
【請求項13】
酵素による反応を触媒する補助因子の添加をさらに含み、前記補助因子は、ATP、ADP、AMP、GTP、GDP、GMP、CTP、CDP、CMP、UTP、UDPおよびUMPから成る群から選ばれる少なくとも一つの補助因子である、請求項12記載の方法。
【請求項14】
酵素が、固相支持体の上に固定される、請求項12記載の方法。
【請求項15】
コンピュータと共に使用されるコンピュータプログラム製品であって、
サンプルポリペプチドについて1組の候補ポリペプチドを選択するための、コンピュータで読み取り可能なプログラムコードを有するコンピュータで使用可能な媒体;と、
サンプルポリペプチドについて1組の候補ポリペプチドを選択するようコンピュータに指令するためのコンピュータで読み取り可能なプログラムコード;とを含み、
該プログラムコードは、
質量分析によって生成されたサンプルポリペプチド断片の質量差に基づいて、候補ポリペプチドを収集する第一の精選;および、
サンプルポリペプチドの絶対質量、および前記断片の絶対質量に基づいて、候補ポリペプチドを収集する第二の精選;
を含む、前記コンピュータプログラム製品。
【請求項16】
コンピュータで読み取り可能なプログラムコードが、収集の第一の精選を決定するようコンピュータに指令するためのものであり、且つ
前記第一の精選は、断片の質量差に基づいて、サンプルポリペプチドの少なくとも部分的アミノ酸配列を決定することを含む、請求項15記載のコンピュータプログラム。
【請求項17】
サンプルポリペプチドの原形の絶対質量、およびサンプルポリペプチド断片の絶対質量を決定するようコンピュータに指令するためのコンピュータで読み取り可能なプログラムコードをさらに含む、請求項16記載のコンピュータプログラム製品。
【請求項18】
サンプルポリペプチドの少なくとも部分的なアミノ酸配列に基づいて、タンパク質形の集合から候補ポリペプチドを選択するようコンピュータに指令するための、コンピュータで読み取り可能なプログラムコードをさらに含む、請求項16記載のコンピュータプログラム製品。
【請求項19】
請求項1記載の方法によって1組の候補ポリペプチドを選択すること;サンプルポリペプチドの絶対質量を、候補ポリペプチドの理論的絶対質量データと比較することによってマッチングの確率スコアを導くこと;および、マッチングの確率スコアをランク付けすることによって、マッチングの最大確率スコアに基づいて候補ポリペプチドの一つによってサンプルポリペプチドの一次構造を特定すること;をコンピュータに指令して実行させるコンピュータで読み取り可能なプログラムコードをさらに含む、請求項16記載のコンピュータプログラム製品。
【請求項20】
コンピュータ、
タンパク質形のウェアハウス・データベース、および、
一次ユーティリティ
を含むシステムをさらに含む、請求項15記載のコンピュータプログラム製品。
【請求項21】
一次ユーティリティが、データ管理システム、イオン予測器、データ縮小ツール、およびグラフ表示インターフェイスツールから成る群から選ばれる少なくとも一つの要素を含む、請求項20記載のコンピュータプログラム製品。
【請求項22】
ウェアハウス・データベースが、ショットガン注釈をさらに含む、請求項20記載のコンピュータプログラム製品。
【請求項23】
ウェアハウス・データベースが、動的ショットガン注釈をさらに含む、請求項20記載のコンピュータプログラム製品。
【請求項24】
システムがさらに検索アルゴリスムを含み、前記検索アルゴリスムは、絶対質量探索モードと配列タグ探索モードとを含む、請求項20記載のコンピュータプログラム製品。
【請求項25】
絶対質量探索モードが、さらにΔm探索モードを含む、請求項24記載のコンピュータプログラム製品。
【請求項26】
コンピュータと相互通信する質量分析器をさらに含む、請求項20記載のコンピュータプログラム製品。
【請求項27】
コンピュータは、インターネット・ソフトウェアアプリケーションを介してユーザーと相互通信する、請求項20記載のコンピュータプログラム製品。
【請求項28】
コンピュータ;
タンパク質形のウェアハウス・データベース;
ウェアハウス・データベースを探索するための検索アルゴリスム;
データ管理システム;
イオン予測器;
データ縮小ツール;および、
グラフ表示インターフェイスツール;
をさらに含む、請求項20記載のコンピュータプログラム製品。
【請求項29】
サンプルポリペプチドについて1組の候補ポリペプチドを選択するシステムであって、
質量分析によって生成されるサンプルポリペプチド断片の質量差に基づいて、候補ポリペプチド集合の第一の精選を実行するための手段;並びに、
サンプルポリペプチドの絶対質量、および、質量分析によって生成されるサンプルポリペプチド断片の絶対質量に基づいて、候補ポリペプチド集合の第二の精選を実行するための手段;
を含む前記システム。
【請求項30】
コンピュータが質量分析器と相互通信することを特徴とする、請求項29記載のシステム。
【請求項31】
コンピュータは、インターネット・ソフトウェアアプリケーションを介してユーザーと相互通信する、請求項29記載のシステム。
【請求項32】
サンプルポリペプチドについて1組の候補ポリペプチドを選択するためのシステムであって、
請求項15記載のコンピュータプログラム産物、および
コンピュータ
を含む前記システム。
【請求項33】
サンプルポリペプチドおよびサンプルポリペプチド断片の絶対質量に基づいて収集する第三の精選をさらに含み、且つ前記第三の精選が、第一の精選に先立って行われる、請求項1記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2007−531874(P2007−531874A)
【公表日】平成19年11月8日(2007.11.8)
【国際特許分類】
【出願番号】特願2007−502082(P2007−502082)
【出願日】平成17年3月3日(2005.3.3)
【国際出願番号】PCT/US2005/007344
【国際公開番号】WO2005/088303
【国際公開日】平成17年9月22日(2005.9.22)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.リナックス
【出願人】(500033634)ザ・ボード・オブ・トラスティーズ・オブ・ザ・ユニバーシティ・オブ・イリノイ (21)
【氏名又は名称原語表記】THE BOARD OF TRUSTEES OF THE UNIVERSITY OF ILLINOIS
【住所又は居所原語表記】506 South Wright Street, Urbana, IL 61801
【Fターム(参考)】
【公表日】平成19年11月8日(2007.11.8)
【国際特許分類】
【出願日】平成17年3月3日(2005.3.3)
【国際出願番号】PCT/US2005/007344
【国際公開番号】WO2005/088303
【国際公開日】平成17年9月22日(2005.9.22)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.リナックス
【出願人】(500033634)ザ・ボード・オブ・トラスティーズ・オブ・ザ・ユニバーシティ・オブ・イリノイ (21)
【氏名又は名称原語表記】THE BOARD OF TRUSTEES OF THE UNIVERSITY OF ILLINOIS
【住所又は居所原語表記】506 South Wright Street, Urbana, IL 61801
【Fターム(参考)】
[ Back to top ]