
新規ハイドロゲルおよびそれらの使用
本発明は、新規のヒドロゲルならびにこのようなヒドロゲルの作製方法および使用方法を提供する。本発明は、溶液中でのペプチドの自己集合(selfasembly)によって形成され得るヒドロゲルを提供する。このような自己集合は、溶液の1つ以上の特徴の変化によって起こり得る。変化し得る溶液の特徴には、pH、イオン強度、温度、および1つ以上の特定のイオンの濃度が含まれる。さらに、本発明のヒドロゲルを、pH、イオン強度、温度、および1つ以上の特定のイオンの濃度などの1つ以上のヒドロゲルの特徴の変化によって分解(disassemble)することができる。

【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2004年7月28日出願の米国特許出願番号10/900,324号の優先権を主張する。本発明は、NIH助成金番号1P20RR17716−01によって一部支援されている。合衆国政府は、本発明に一定の権利を有し得る。
【背景技術】
【0002】
(発明の背景)
ヒドロゲルは、軟組織工学および骨工学での使用が非常に期待されているクラスの材料である。これらの用途において重要な材料となるヒドロゲルの一般的特徴は、その十分に水和した多孔質構造である。本発明は、既存のテクノロジーでは現在満たされない重要な材料要件を満たす新規のクラスの環境応答性を示すペプチドベースのヒドロゲルを提供する。本発明のヒドロゲルを、種々の細胞型(例えば、線維芽細胞および骨芽細胞)の接着および増殖に適合し、結合組織および骨の生成のための潜在的な組織工学足場となるようにデザインすることができる。組織再生用のヒドロゲルに必要な一連の生物学的性質および材料の性質が求められている。最終的な標的組織型と無関係に、ヒドロゲルは、一般的な一連の生物学的性質を示さなければならない。第1に、この材料は細胞適合性でなければならない。本明細書中で定義される、「細胞適合性」は、所望の細胞に対して細胞傷害性を示してはならないことを意味する。第2に、この材料は生体適合性を示さなければならない。本明細書中で定義される、「生体適合性」は、in vivoに置かれた場合、足場が組織再生に有意な免疫応答および炎症反応を引き起こさず、好ましくは、性分解によって無毒となる種であることを意味する。本発明は、新規の自己集合(self−assembly)法を使用した新規の材料の開発および得られた材料の細胞適合性の評価に関する。
【0003】
いくつかの所望の性質が一見したところ互いに矛盾するので、所望の材料の性質をいずれか1つの材料に包括的に組み込むことは困難である。例えば、理想的なヒドロゲルの形態は、細胞の運動性および栄養素/老廃物の拡散のために多孔性(ナノスケールからマイクロスケールの寸法にわたる)が高い。また、ヒドロゲルは、主に、細胞増殖および最終的に足場を容易に生分解するのに十分な体積を得るためにできるだけ固体材料が少ない水性溶媒から構成されるべきである。しかし、その希薄で多孔質であるにもかかわらず、これらの十分に水和した材料は、機械的に強固でもなければならない。ヒドロゲル化(hydrogelation)過程時に形成される構成分子架橋(化学的および/または物理的)のデザインによってこの明らかな矛盾(希薄な多孔質足場の剛性)に本質的に対処しなければならない。しかし、化学的架橋由来の副生成物が有毒である可能性があり、且つ足場からの除去が困難であるので、化学的架橋の導入は、生物学的に問題があり得る。理想的には、無害の生体適合性の化学的または物理的架橋法を、身体への最終的組み込みのためのin vitroゲル化または生理学的刺激(温度、イオン強度、pHなど)によって架橋の形成が誘発される直接的で迅速なin vivoゲル化のいずれかのために使用すべきである。自己集合を介した材料の形成を開始するための環境要因(trigger)を使用するという考え方が盛んに進められている。例えば、ペプチドの自己集合(およびそれによるゲル化)を、温度および光に感受性を示すリポソームからの塩の放出によって誘発することができることが示されている(Collier,et al.,Journal of the American Chemical Society 2001,123,9463−9464:非特許文献1)。一見したところヒドロゲルの剛性により、予め形成された足場におけるいかなる処理も実行不可能であることがデザインをさらに複雑にしている。例えば、強固な組織工学構築物をin vitroで形成することを望むことができるが、その後に組織再生のために宿主に注入する。恒久的に架橋した強固な網状組織を注入することは不可能である。
【0004】
現在のヒドロゲルテクノロジーは、天然由来の高分子および合成ポリマーの両方を使用する。一般に、天然ポリマーから調製されたヒドロゲルは、好ましい生物学的性質を示すが、所望の材料特性(例えば、低サンプル剛性)を欠き得る。対照的に、所望の材料特性を有する合成ポリマーを設計することができるが、細胞適合性が制限され得る。合成ポリマーの細胞適合性を増加させるための一般的アプローチは、ペプチドエピトープ(例えば、RGDモチーフ)を組み込むことである。しかし、位置特異的に制御された様式での予め形成したポリマーへのこれらのモチーフの組み込みは実現が非常に困難である。結果として、ポリマーの材料特性の制御は問題がある。例えば、細胞接着またはエピトープの接近可能性の制御のために表面上に提示されたエピトープの濃度の制御は困難である。さらに、これらの足場は、その基礎をなす分子網目構造によって微小規模で構造が均一(多孔質ではない)であり、細胞増殖が制限され得る。ゲル網目構造に微小規模で多孔質にするために、これらの系をさらに処理しなければならない(例えば、凍結融解サイクル、粒子浸出、ミクロスフェア焼結、および不織線維(non−woven fiver)形成)。要するに、現在、理想的な組織工学足場の必要な性質を全て首尾よく組み込まれた単一のヒドロゲル系が存在しない。
【0005】
新規のヒドロゲルストラテジーをデザインする機会および必要が存在する。in vitroまたはin vivoで容易に形成することができる強固、多孔質、且つ処理が容易で細胞適合性を示すヒドロゲルが必要である。
【非特許文献1】Collier,et al.,Journal of the American Chemical Society 2001,123,9463−9464
【発明の開示】
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、新規のヒドロゲルを提供する。さらに、本発明は、ヒドロゲル構築のための新規の過程を提供する。本明細書中で使用される、用語「ヒドロゲル」は、好ましくは、大量の水画分をカプセルされ、且つ機械的に自立した希薄な相互連結された足場を意味する。本明細書中で使用される、用語「ヒドロゲル」はまた、水性有機混合物および/または有機溶媒(例えば、DMF、DMSOなど)を含む希薄な相互連結された足場を含む。1つの態様では、本発明は、高性能のヒドロゲル化系を構築するための新規の過程と新規のペプチドとの組み合わせを提供する。1つ以上の環境シグナルまたは刺激(例えば、1つ以上の環境特徴の変化)に反応して二次構造が変化するように新規のペプチド(例えば、MAX1)をデザインした。1つの特定の態様では、本発明のペプチドは水溶液中に存在することができ、ペプチドの二次構造を変化させるために溶液の1つ以上のパラメーターを変化させることができる。特定の実施形態では、溶液のpH、イオン強度、特定のイオンの濃度、および/または温度のうち1つ以上を変化させてペプチドの二次構造を変化させることができる。典型的には、溶液の1つ以上のパラメーターの変化の結果としてペプチドの二次構造が変化した後、ペプチドをより高次の構造(例えば、ヒドロゲル)に集合させる。本発明の別の態様では、環境シグナルは、電磁放射(例えば、光)を含み得る。例えば、本発明のペプチドの構造を変化させることができ、この変化は、電磁放射に供した結果としての一次構造、二次構造、またはその両方の変化であり得る。典型的には、電子放射に曝露した後、本発明のこの態様を使用したペプチドは、所望の二次構造を有することが予想され、より高次の構造(例えば、ヒドロゲル)に自己集合する。
【0007】
1つの態様では、自己集合機構による低粘度の水溶液から強固なヒドロゲル材料(本質的に無限に粘度が変化する)への移行は、好ましくは、各ペプチドの所望の高次構造への折り畳みを前提とする。この分子内折り畳み過程を、所望の環境シグナルのみを用いて起こるように制御することができる。環境シグナルには、生理溶液の条件(37℃、pH7.4、および高塩濃度)が含まれるが、これらに限定されない。生理学的条件下で頑強且つ化学的に無害にゲル化するので、これらのゲルは、主に組織工学分野および創傷治癒分野で見込みがある。
【0008】
1つの特定の実施形態では、1つ以上の環境シグナルに応答してβ−ヘアピン二次構造を採用するようにペプチドをデザインすることができる。典型的には、β−ヘアピン構造の採用後、ペプチドは、より高次の構造(例えば、三次元網目構造、最終的にヒドロゲル)に自己集合する。1つの態様では、ペプチド分子上の側鎖がβ−ヘアピン高次構造中に固有に存在しない限り、自己集合は起こらない。
【0009】
したがって、1つの態様では、本発明は、1つ以上の環境刺激に応答して所望の二次構造を採用するようなペプチドをデザインする過程を提供する。さらに、本発明は、1つ以上の環境刺激に応答してより高次の構造(例えば、ヒドロゲル)を形成するペプチドをデザインする過程を提供する。本発明はまた、このようにして形成されたより高次の構造(例えば、ヒドロゲル)を含む。
【0010】
いくつかの態様では、二次構造の採用およびより高次の構造の形成は関連する。したがって、ペプチドの折り畳みおよび自己集合(例えば、ヒドロゲル)は関連する。本発明のこの態様により、ゲル化が制御される。特に、この態様により、ヒドロゲルの形成速度(すなわち、どれぐらい速くゲルが形成されるか)が制御される。また、自己集合過程の制御により、このようにして形成されたヒドロゲルの物理的特徴(例えば、得られたゲルの剛性)が制御される。1つ以上の環境刺激の適用後に、ヒドロゲルが、約1秒〜約5時間、約1秒〜約4時間、約1秒〜約3時間、約1秒〜約2時間、約1秒〜約1時間、約1秒〜約50分、約1秒〜約40分、約1秒〜約30分、約1秒〜約20分、約1秒〜約15分、約1秒〜約10分、約1秒〜約5分、約1秒〜約2分、約10秒〜約5時間、約10秒〜約4時間、約10秒〜約3時間、約10秒〜約2時間、約10秒〜約1時間、約10秒〜約50分、約10秒〜約40分、約10秒〜約30分、約10秒〜約20分、約10秒〜約15分、約10秒〜約10分、約10秒〜約5分、約10秒〜約2分、約30秒〜約5時間、約30秒〜約4時間、約30秒〜約3時間、約30秒〜約2時間、約30秒〜約1時間、約30秒〜約50分、約30秒〜約40分、約30秒〜約30分、約30秒〜約20分、約30秒〜約15分、約30秒〜約10分、約30秒〜約5分、約30秒〜約2分、約60秒〜約5時間、約60秒〜約4時間、約60秒〜約3時間、約60秒〜約2時間、約60秒〜約1時間、約60秒〜約50分、約60秒〜約40分、約60秒〜約30分、約60秒〜約20分、約60秒〜約15分、約60秒〜約10分、約60秒〜約5分、または約60秒〜約2分で形成することができるように、ペプチドをデザインし、そして/または環境刺激を選択することができる。
【0011】
本発明にしたがって形成されたヒドロゲルは、種々の量の固体材料を有し得る。例えば、約0.1重量%〜約10.0重量%、約0.1重量%〜約9.0重量%、約0.1重量%〜約8.0重量%、約0.1重量%〜約7.0重量%、約0.1重量%〜約6.0重量%、約0.1重量%〜約5.0重量%、約0.1重量%〜約4.0重量%、約0.1重量%〜約3.0重量%、約0.1重量%〜約2.0重量%、約0.1重量%〜約1.0重量%、約0.1重量%〜約0.75重量%、約0.1重量%〜約0.5重量%、約0.1重量%〜約0.25重量%、約0.25重量%〜約10.0重量%、約0.25重量%〜約9.0重量%、約0.25重量%〜約8.0重量%、約0.25重量%〜約7.0重量%、約0.25重量%〜約6.0重量%、約0.25重量%〜約5.0重量%、約0.25重量%〜約4.0重量%、約0.25重量%〜約3.0重量%、約0.25重量%〜約2.0重量%、約0.25重量%〜約1.0重量%、約0.25重量%〜約0.75重量%、約0.25重量%〜約0.5重量%、約0.5重量%〜約10.0重量%、約0.5重量%〜約9.0重量%、約0.5重量%〜約8.0重量%、約0.5重量%〜約7.0重量%、約0.5重量%〜約6.0重量%、約0.5重量%〜約5.0重量%、約0.5重量%〜約4.0重量%、約0.5重量%〜約3.0重量%、約0.5重量%〜約2.0重量%、約0.5重量%〜約1.0重量%、約0.5重量%〜約0.75重量%、約1.0重量%〜約10.0重量%、約1.0重量%〜約9.0重量%、約1.0重量%〜約8.0重量%、約1.0重量%〜約7.0重量%、約1.0重量%〜約6.0重量%、約1.0重量%〜約5.0重量%、約1.0重量%〜約4.0重量%、約1.0重量%〜約3.0重量%、約1.0重量%〜約2.0%、または約1.0重量%〜約1.5重量%のペプチドを含むヒドロゲルを形成することができる。
【0012】
1つの態様では、ペプチドの重量による量およびゲル化速度を変化させて、所望の剛性率(剛性)を有するヒドロゲルを生成することができる。本発明のヒドロゲルは、約1パスカル(Pa)〜約100,000Pa、約1Pa〜約50,000Pa、約1Pa〜約25,000Pa、約1Pa〜約10,000Pa、約1Pa〜約7,500Pa、約1Pa〜約5,000Pa、約1Pa〜約2,500Pa、約1Pa〜約2,000Pa、約1Pa〜約1,500Pa、約1Pa〜約1,000Pa、約1Pa〜約500Pa、約1Pa〜約250Pa、約1Pa〜約100Pa、約100Pa〜約100,000Pa、約100Pa〜約50,000Pa、約100Pa〜約25,000Pa、約100Pa〜約10,000Pa、約100Pa〜約7,500Pa、約100Pa〜約5,000Pa、約100Pa〜約2,500Pa、約100Pa〜約2,000Pa、約100Pa〜約1,500Pa、約100Pa〜約1,000Pa、約100Pa〜約500Pa、または約100Pa〜約250Paの剛性率を有し得る。
【0013】
本発明の1つの態様では、形成されたヒドロゲルを処理することができる。例えば、本発明のヒドロゲルを、動物(例えば、哺乳動物)に注入することができる。本発明のヒドロゲルが自己集合するので、強固なゲルを容易に処理することができる(例えば、シリンジによって注入することができる)一方で、処理の中止直後に再集合/硬化する。関連する態様では、ヒドロゲルを、低粘度溶液からの自己集合によって物理的に形成することができ、したがって、ヒドロゲルを、in vitroまたはin vivoで幾何学的に限定されて生成することができる。
【0014】
いくつかの好ましい実施形態では、本発明のペプチドベースのヒドロゲルは、完全に非細胞傷害性を示し、一般的な哺乳動物細胞(例えば、繊維芽細胞、骨芽細胞)の接着および増殖を促進することもできる。したがって、本発明のヒドロゲルを、細胞培養物中で使用することができる。細胞培養物を、自己集合機構によって三次元でカプセル化することができ、それにより、三次元での細胞付着および増殖が可能になる。いくつかの実施形態では、ヒドロゲルを三次元支持体として使用して、薬学的化合物、ペプチド、タンパク質、および抗体などの治療因子を生成するように操作された細胞株を増殖/維持することができる。ヒドロゲルおよび細胞を含むバイオリアクターによる培地の連続フローにより、化合物の迅速な単離および連続的細胞増殖手段が行われる。
【0015】
いくつかの態様では、本発明のペプチド(例えば、MAX−1および関連するヘアピンペプチド)から調製したヒドロゲルは、グラム陽性細菌およびグラム陰性細菌に対して抗菌挙動を示す。したがって、本発明のヒドロゲルは、臨床的状況(clinical setting)で抗菌性を示し得る。本発明のヒドロゲルのこの特徴により、細胞培養方法と同様に、ヒドロゲルが生きた動物(例えば、ヒトなどの哺乳動物)内におかれる状況でも有用となる。特定の実施形態では、本発明のヒドロゲルを、組織工学のために使用することができる。例えば、所望の量の1つ以上の細胞型を、1つ以上の本発明のペプチドを含む溶液中におくことができる。溶液を含む細胞をヒドロゲルに形成させることができ、この細胞をヒドロゲル全体に分散させることができる。次いで、ヒドロゲルを含む細胞を、例えば、損傷組織と置換するための組織として使用することができる。本発明のヒドロゲルの抗菌特性により、動物に移入された場合に感染防止に役立つ。いくつかの実施形態では、本発明のヒドロゲルを、可逆的にゲル化する(すなわち、1組の条件下でヒドロゲルを形成し、その後に他の条件下で溶液に戻る)ように構築することができる。細胞をヒドロゲル足場に移入し、最終的に溶解して細胞を適所に放出することができるように、これを組織工学への適用の際に使用することができる。抗菌ゲルは、創傷治癒への適用にも有用である。創傷治癒への適用で使用されるヒドロゲルは、ヒドロゲルに加えて治療因子を含み得る。例えば、生理学的条件下(すなわち、創傷との接触)におかれた場合にヒドロゲルが形成されるペプチド溶液は、凝固を促進する薬剤、鎮痛薬、および/または他の治療因子を含み得る。
【0016】
いくつかの態様では、本発明のヒドロゲルを、さらなる至適化のためのさらなる生化学(例えば、増殖因子または細胞接着ペプチドエピトープ)で容易に機能的にすることができる。したがって、本発明のペプチドは、本発明にヒドロゲルの特徴を変化させるペプチドであり得るさらなる成分を含み得る。
【0017】
本発明のヒドロゲルを、環境応答性遮断材料として微小流体(microfluidic)デバイスで使用することができる。例えば、微小流体デバイスのチャネルを、ペプチド水溶液に浸すことができる。所望の位置にチャネル障壁を取り付けるための空間分解能を有するヒドロゲル化を開始することができる。これらの障壁およびダムは、検出装置として作用することができる。溶液がこれらを通過してヒドロゲル溶解(ダム破壊)が起こる場合、初期のチャネルを開口して流動物を検出機に流し、分析物の分析手段が得られる。ヒドロゲル障壁の溶解を使用して、所望の化学転換および反応のための反応成分の混合を開始し、容易にすることもできる。
【0018】
本発明のヒドロゲルを、1つ以上の目的の分析物の検出のためのセンサとして使用することができる。例えば、本発明のペプチド溶液を、分析物を含み得るサンプルと接触させることができる。いくつかの例では、分析物の存在により、溶液のゲル化を誘導し得る。例えば、目的の分析物は金属イオンであってよく、ペプチド溶液の金属イオンとの接触により、溶液がゲル化し得る。他の例では、本発明のヒドロゲルを形成し、次いで、目的の分析物を含み得るサンプルと接触させることができる。分析物の存在により、ヒドロゲルを可溶化することができる。ヒドロゲルの形成または分解を、当業者に周知の標準的技術(例えば、光学的技術)を使用して検出することができる。
【0019】
いくつかの実施形態では、本発明のヒドロゲルを使用して、目的の分子(例えば、生体分子、タンパク質、DNA、RNAなど)の分離のためのマトリクスを調製することができる。ペプチドを、目的の分子の分離に有用な特徴を有するようにデザインすることができる。例えば、目的の分子と特異的に相互作用することができる部分を、ヒドロゲルを作製するために使用されるペプチドにデザインすることができる。特異的に相互作用することができる適切な部分には、エピトープ、リガンド、特異的低分子、および核酸配列などが含まれるが、これらに限定されない。このの型ヒドロゲルを、正の選択様式(すなわち、目的の分子の結合)または負の選択様式(すなわち、夾雑物の結合)のいずれかで使用することができる。この型の実施形態で使用するために、所望の分子を精製するためにヒドロゲルをナノ細孔形態および微小孔形態を制御することが望ましいかもしれない。例えば、本発明のヒドロゲルの孔サイズを、目的の分子の精製を至適化するための使用するペプチドの重量、ゲル化条件、および/またはペプチド構造の変化によって制御することができる。
【0020】
本発明のヒドロゲルを使用して、生きた生物中に存在する材料の耐性および/または接着を改良することができる。例えば、生きた生物中に移植すべき材料(例えば、人工器官、ペースメーカー、支持器(support)など)を、最初に本発明のヒドロゲルでコーティングすることができる。このようなヒドロゲルを、生物組織の移植デバイスへの接着を促進する1つ以上の部分を有するペプチドから作製することができる。このような部分には、接着エピトープなどが含まれ得る。ヒドロゲルはまた、低分子またはエピトープなどの免疫調整(例えば、抑制または刺激)部分を含み得る。
【0021】
別の実施形態では、本発明のヒドロゲルを、水溶液由来の有害金属イオンの改善のために使用することができる。有害金属イオンに結合する官能基を含むペプチドをデザインすることができる。ペプチドを、有害金属イオンを含む溶液に導入し、ペプチドのゲル化を誘導することができる。有害金属をヒドロゲル内に捕捉することができ、ヒドロゲルを、任意の適切な技術(例えば、濾過)によって残りの溶液から分離することができる。任意選択的に、ヒドロゲルを溶解し、有害金属イオンを単離することができる。
【0022】
いくつかの態様では、本発明は、ヒドロゲルの作製方法を提供する。このような方法は、ペプチドを含む溶液を提供し、この溶液の1つ以上の特徴を変化させ、ヒドロゲルが形成されることが必要であり得る。変化する特徴は、その変化の際にヒドロゲルが形成される任意の特徴であり得る。適切な例には、イオン強度、温度、特定のイオンの濃度、およびpHが含まれるが、これらに限定されない。いくつかの実施形態では、溶液の1つ以上の変化させる工程は、溶液を電磁放射と接触させることを含み得る。特定の実施形態では、変化する特徴は、溶液のpHであり得る。いくつかの態様では、溶液の1つ以上の特徴の変化により、塩濃度が約20mM〜約400mMになる。任意の塩(KCl、NaCl、MgCl2、KF、MgSO4)を使用することができる。1つの実施形態では、塩はNaClであり得る。いくつかの実施形態では、溶液は、所望のpH(例えば、pH9未満、約pH6.0〜約pH8.5、約pH7.0〜約pH8.0、または約pH7.4)を有することができ、ヒドロゲル形成の際に同一であっても変化してもよい。
【0023】
いくつかの態様では、本発明は、ヒドロゲルを提供する。このようなヒドロゲルは、ペプチドおよび約20mM〜約400mMの塩を含み得る。上記で考察しているように、任意の塩(例えば、NaCl)を使用することができる。ヒドロゲルを形成することができる任意のペプチド(例えば、MAX1)を使用することができる。
【0024】
1つの態様では、本発明は、ヒドロゲルの作製方法を提供する。このような方法は、ペプチドを含む溶液を動物に注入する工程と、前記溶液が前記動物内でヒドロゲルを形成することとを含み得る。任意の動物(例えば、哺乳動物(ヒトが含まれる))を使用することができる。本発明の態様で使用するための溶液は、いくつの成分を含んでもよい。いくつかの実施形態では、溶液は、1つ以上の治療因子を含み得る。当業者に公知の任意の治療因子を使用することができる。特定の実施形態では、溶液は、低分子、ペプチド、タンパク質、および細胞からなる群から選択される1つ以上の治療因子を含み得る。
【0025】
本発明の別の態様では、本発明は、必要とする動物に治療因子を送達する方法を提供する。このような方法は、治療因子および1つ以上のペプチドを含む溶液を動物に投与する工程と、前記溶液が前記動物内でヒドロゲルを形成することとを含み得る。このような方法を、任意の動物型(ヒトなどの哺乳動物が含まれる)に対して実施することができる。当業者に公知の任意の治療因子型(例えば、低分子、ペプチド、タンパク質、および細胞)を使用することができる。当業者は、治療因子が任意の望ましくない条件を防止および/または改善する任意の薬剤であることを認識している。
【0026】
別の態様では、本発明は、治療因子および1つ以上のペプチドを含むヒドロゲルを動物に投与する工程を含む、必要とする動物に治療因子を送達する方法を提供する。このような方法を、任意の動物型(ヒトなど哺乳動物が含まれる)に対して実施することができる。当業者に公知の任意の治療因子型(例えば、低分子、ペプチド、タンパク質、および細胞)を、使用することができる。
【0027】
別の態様では、本発明は、動物の創傷を処置する方法を提供する。このような方法は、創傷をペプチドを含む溶液と接触させる工程と、前記溶液がヒドロゲルを形成することとを含み得る。本発明のこの態様で使用する溶液は、1つ以上の治療因子をさらに含み得る。この型の方法を、任意の動物型(例えば、哺乳動物(ヒトが含まれる))に対して実施することができる。当業者に公知の任意の治療因子型(例えば、低分子、ペプチド、タンパク質、および細胞)を、使用することができる。
【0028】
別の態様では、本発明は、細胞を増殖させる方法を提供する。このような方法は、細胞を含むヒドロゲルを形成する工程と、細胞生存に適切な条件下で細胞を維持する工程とを含み得る。本発明のこの態様で使用するヒドロゲルは、典型的には、ペプチドを含む。ヒドロゲルを、例えば、ペプチドを含む溶液の1つ以上の特徴の調整によって形成することができる。調整される特徴は、1つ以上のpH、イオン強度、特定のイオンの濃度であり得る。1つの特定の実施形態では、調整される特徴は、イオン強度である。別の特定の実施形態では、調整される特徴はCa2+イオン濃度である。任意の型の細胞(哺乳動物細胞(ヒト細胞が含まれる)などの動物細胞)を、本発明の方法を使用して増殖させることができる。いくつかの特定の実施形態では、細胞は、骨芽細胞または線維芽細胞であり得る。本発明の方法を使用して増殖させるべき細胞は、組換え細胞(すなわち、1つ以上の外因性核酸分子を含み得る)であり得る。このような核酸分子を、細胞のゲノムに組み込み、そして/または染色体外に維持することができる。本発明の方法を使用して増殖させるべき細胞は、目的のタンパク質を発現し得る。目的のタンパク質の例には、抗体が含まれるが、これらに限定されない。
【0029】
別の態様では、本発明は、ヒドロゲルを含むセンサを提供する。本発明のセンサで使用するヒドロゲルは、ヒドロゲルが目的の分析物と接触した場合に変化する1つ以上の特徴を有し得る。目的の分析物は、検出されることが望ましい任意の材料である。いくつかの実施形態では、分析物に応答して変化したヒドロゲルの特徴は剛性である。他の実施形態では、変化した特徴は光学的性質である。光学的特徴には、吸光度、楕円率、および光散乱特性などが含まれるが、これらに限定されない。
【0030】
別の態様では、本発明は、環境条件を検出する方法を提供する。このような方法は、ヒドロゲルを含むセンサを環境条件を代表するサンプルと接触させる工程と、前記ヒドロゲルの1つ以上の特徴を決定する工程とを必要とし得る。典型的には、この型の方法では、ヒドロゲルが目的の分析物と接触した場合に、ヒドロゲルの1つ以上の特徴が変化する。ヒドロゲルの任意の特徴(例えば、剛性および/または光学的性質)を変化させることができる。この型の方法は、ヒドロゲルの特徴を異なる時間に決定した前記ヒドロゲルの同一の特徴と比較する工程も含み得る。
【0031】
別の態様では、本発明は、目的の分子を精製する方法を提供する。このような方法は、目的の分子および1つ以上の夾雑物を含む溶液を、目的の分子がヒドロゲルによって保持される条件下でヒドロゲルに接触させる工程と、ヒドロゲルから前記目的の分子を回収する工程とを含み得る。典型的には、この型の方法では、少なくとも1つの夾雑物がヒドロゲルによって保持されないか、目的の分子よりも低い程度で保持される。目的の分子は、当業者に公知の任意の分子であり得る。目的の分子の例には、タンパク質、核酸分子、および低分子などが含まれる。1つの特定の実施形態では、目的の分子は、抗体であり得る。目的の分子は、治療因子または治療因子の構成要素であり得る。治療因子の構成要素は、治療因子になるように修飾することができる材料である。治療因子の構成要素の1つの例は、治療因子になるように細胞傷害性化合物で共有結合によって修飾することができる抗体である。
【0032】
別の態様では、本発明は、目的の分子を精製する方法であって、目的の分子および1つ以上の夾雑物を含む溶液を、少なくとも1つの夾雑物がヒドロゲルによって保持される条件下でヒドロゲルに接触させる工程と、前記目的の分子を回収する工程とを含む方法を提供する。典型的には、この型の方法では、少なくとも1つの夾雑物がヒドロゲルによって保持されるか、目的の分子よりも高い程度で保持される。目的の分子は、当業者に公知の任意の分子であり得る。目的の分子の例には、タンパク質、核酸分子、および低分子などが含まれる。1つの特定の実施形態では、目的の分子は、抗体であり得る。目的の分子は、治療因子または治療因子の構成要素であり得る。治療因子の構成要素は、治療因子になるように修飾することができる材料である。治療因子の構成要素の1つの例は、治療因子になるように細胞傷害性化合物で共有結合によって修飾することができる抗体である。
【発明を実施するための最良の形態】
【0033】
(発明の詳細な説明)
1つの態様では、本発明は、ゲル足場基礎単位として小ペプチドを使用する自己集合ヒドロゲル化ストラテジーを提供する。ペプチドを、分子内で会合することができるβ−ヘアピン高次構造への分子内折り畳み後にのみ自己集合するようにデザインすることができる。折り畳み事象を、反応のよいヒドロゲル化系を与える環境刺激によって誘発することができる。ペプチドの使用は、化学合成が迅速であることならびにあつらえの材料および生物学的特性のための新規の残基および機能的エピトープの組み込みが可能なことにより有利である。ヒドロゲルの自己集合特性により、外因性架橋剤が必要ない。本明細書中に記載のペプチドベースのヒドロゲルは、強固であるが容易に処理される。このヒドロゲル化ストラテジーにより、潜在的なin vitroおよびin vivo組織工学構築物形成のために使用することができる細胞適合性ゲル化系が得られる。本発明は、ペプチド構造と最終的なヒドロゲルの形態学的、流動学的、および細胞レベルの生物学的性質との間の関係を確立する。以下によってこれを行った。1)ヒドロゲル形成させる折り畳みおよび自己集合および分子のデザインがどのように材料の性質に影響を与えるのかについての基本的理解を得ること。デザインしたペプチド構造と、折り畳み/自己集合過程と、最終的な材料の性質との間の関係の厳密な理解により、あつらえの組織工学特性を有するヒドロゲルを生成することができる。2)適時にペプチド溶液をヒドロゲル化することが可能な活性な分子内折り畳みトリガーのデザインによるヘアピンベースのヒドロゲルの処理可能性の増強。生物学的に適合する合図(例えば、pH、塩濃度、細胞培養培地、光、体温、または特定のイオン濃度(例えば、Ca2+)に応答して折畳まれるペプチドのデザインにより、当業者は、in vitroおよびin vivoの両方での組織工学への適用のためのヒドロゲル化過程をデザインすることができる。3)どのようにしてペプチド構造よび材料の性質がモデル細胞株(例えば、線維芽細胞株および骨芽細胞株)の接着および増殖に影響をあたえるのかを決定すること。β−ヘアピンヒドロゲルのペプチド性および多孔性により、これらの材料が、潜在的な骨および軟組織工学への適用のための細胞適合性の(非細胞傷害性を示し、細胞の接着および増殖を促進する)容易に官能化される基質としての材料の候補となる。ペプチド構造と、ヒドロゲル剛性と、細胞適合性との間に相関関係が確立されている。
【0034】
本発明は、1つの基本的分子デザイン内の1つ以上の上記材料特性を組み入れる。この別のストラテジーは、ヒドロゲルを生成するための以下の4つの基本的デザイン様相(facet)を使用する:1)小さなde novoでデザインされたペプチドを使用してヒドロゲルを調製することができる。2)外因性架橋剤を必要としない純粋な自己集合機構を介してヒドロゲルを構築することができる。3)ペプチドがターゲティングされた高次構造に正確に分子内で折畳まれない限り、自己集合せず、ヒドロゲル骨格形成されないようにペプチドをデザインすることができる。4)この分子内折り畳み事象、およびそれによるヒドロゲル化を、特異的環境刺激によって誘発することができる。
【0035】
図1は、この簡潔な材料構築デザインの根拠の1つの実施形態を示す。正確な溶液条件によってβ−ヘアピン高次構造への分子内折り畳みが決定づけられるまで水溶液中で構築されないように小ペプチドをデザインする。この表面が両親媒性のヘアピンが自己集合されて強固な多孔質のβ−シートリッチなヒドロゲルが得られる。本発明者らは、疎水性に会合した微小繊維内接合部が散在した微小繊維の短いセグメントと一致する自己集合状態のナノ構造(図1中の右側)を提案する(図7Bを参照のこと)。この形態は古典的な微小繊維ベースの集合物が得られる自己会合ペプチドから認められた形態と非常に異なることに留意すべきである。古典的な系において、ペプチドは、積層されたβ−シートリッチな微小繊維と会合する。自己集合過程は、通常は非常に遅く(数時間〜数ヵ月)、ミクロン単位の長さを有し得る微小組織が不可逆的に得られる。例は、十分に開発された成熟した微小繊維の絡みつきの際に形成されるヒドロゲルである。対照的に、本発明者らの提案したペプチド系は、新規の機構を提供し、この機構は、誘発された分子内折り畳みが所望の分子間自己集合前に起こらなければならない。したがって、ヒドロゲル化を、多様な一連の環境トリガーによって開始することができる。ヒドロゲル化事象は、非常に迅速であり(条件によって数秒以内)、必要に応じて、完全に可逆性であるようにデザインすることができる。得られたゲルは、ナノ細孔から微小孔の形態および98%を超える水から構成されているにもかかわらず有意な材料剛性を示すことによって特徴づけられる。
【0036】
複雑な材料を形成するための小ペプチドの実行可能性は、文献に十分に記載されている。ペプチドは、ヘリックスリボン、ナノチューブおよび小胞、表面集合構造、ならびにその他に自己集合することが認められている。小ペプチドからの材料の調製は、これらを迅速に化学合成することができ、新規のアミノ酸残基を組み込むことができるので、有利である。さらに、直交保護ストラテジーの使用により、化学的部分をアミノ酸側鎖と位置選択的に連結してあつらえの機能(例えば、細胞接着)を有する抱合体(conjugate)を得ることが可能である。位置選択性に関して、既存のポリマーの化学修飾に使用される比較的選択性のない方法と比較して自己集合系の単量体基礎単位を完全且つ正確に官能化する能力が非常に望まれる。本発明の1つの態様では、ペプチドを、その環境に応答して折畳まれるようにデザインすることができ、この性質の活用により、適時に形成される(または溶解する)高性能の材料を得ることができる。本発明の1つ以上の態様の実施で使用することができるペプチドの例には、以下が含まれるが、これらに限定されない。
【0037】
【化1】
上記で詳細に引用したアミノ酸に加えて、任意の上記ペプチドの任意の位置をXで示した(各Xは、独立して、当業者に公知の任意の天然もしくは非天然アミノ酸(LまたはD立体化学)またはアミノ酸の任意のアナログであり得る)。本出願では、D立体化学を、Dアミノ酸の前の上付き文字で示す(したがって、DPはD−プロリンである)。
【0038】
本発明のいくつかの実施形態では、ペプチドは、一般式VKVKVKVK(XXXX)aKVKVKV(XXXX)bKVKVKVKV(配列番号5)に適合し得る。本発明のこの実施形態の特定の例には、以下が含まれるが、これらに限定されない。
【0039】
【化2】
上記で詳細に引用したアミノ酸に加えて、任意の上記ペプチドの任意の位置をXで示した(各Xは、独立して、当業者に公知の任意の天然もしくは非天然アミノ酸(LまたはD立体化学)またはアミノ酸の任意のアナログであり得る)。好ましくは、各(XXXX)aおよび(XXXX)bは、ターン(例えば、β−ターン)を形成することができる配列を含み得る。
【0040】
本発明のいくつかの実施形態では、ペプチドは、以下の一般式に適合し得る。
【0041】
【化3】
mおよびnはそれぞれ独立して1〜100であってよく、mはnと等しくても等しくなくてもよい。
【0042】
ヒドロゲル調製の容易さおよびそれによって得られる材料特性(特に、処理可能性および形態学)が本発明で特に有利である。自己集合ストラテジーの使用により、化学的架橋が排除される。架橋を誘導する化学物質の使用は一般に選択性がなく、多くの架橋試薬は有毒であり、ヒドロゲル骨格から精製によって容易に除去されない。
【0043】
処理可能性に関して、物理的に架橋した自己集合網目構造から構築したヒドロゲルは、機械的剪断に応答することができる。この特徴により、剪断の適用および剪断停止後のゲル網目構造の完全な再形成(自己治癒)の間にフリーフロー懸濁液が得られる(Prudhomme,et al,Langmuir 1996,12,4651−4659およびNowak,et al,Nature 2002,417,424−428を参照のこと)。剪断薄膜化と自己治癒との組み合わせにより、空間分解様式で材料を形成可能である。例えば、本発明のいくつかの実施形態では、当業者は、所望の構成要素(増殖培地、増殖因子、生きた細胞など)の存在下にてex vivoで予め形成されたヒドロゲルを宿主に注射(剪断薄膜化)することができ、それにより、宿主が自己治癒して組織再生のための足場が得られる。さらに、自己集合を介して形成されたヒドロゲルは、上記のex vivo調製に制限されない。本発明は、塩、温度、およびpHなどの生物学的に関連する刺激に曝露した場合に自己集合するペプチド系を提供する(Pochan,et al.,Journal of the American Chemical Society 2003,125,11802−11803およびSchneider,et al,Journal of the American Chemical Society 2002,124,15030−15037を参照のこと)。これにより、ペプチド溶液の注射を介してin vivoで直接ヒドロゲルを形成することができる。ex vivoおよびin vivo調製が可能であるヒドロゲル系は、処理可能性に関して最も用途が広く、本発明の範囲内に含まれる。
【0044】
本明細書中に記載のβ−ヘアピンの自己集合により、最小固体材料から構成される強固な網目構造(2重量%以下のペプチドの剛性率(G’)が1000Pa超)を得ることができ、それにより、さらに処理する必要がない有意に希薄で多孔質な足場が得られる。これらのゲルは、ナノスケールおよびマイクロスケールの両方で多孔質である。この多孔質特性は、ヒドロゲル足場内の細胞移動を補助することができ、栄養の拡散が可能である。1つの実施形態では、本発明は、ヒドロゲル化が迅速に誘発して細胞適合性を示す強固で多孔質な材料を得ることができる正確にデザインされたペプチドを提供する。生理学的に関連する刺激によって自己集合を誘発し、それにより、剪断薄膜化されてこの系の処理の用途が広くなるヒドロゲルを得ることができる。
【0045】
提案したヒドロゲル化系は、ペプチドが自己集合に従う折り畳まれた高次構造を採用する(すなわち、ペプチドが正確に折畳まれない場合、ヒドロゲルに自己集合しない)能力に依存する。さらに、環境上の合図にのみ応答して折畳まれるペプチドをデザインすることが可能である。結果は、所望の刺激の存在下でもヒドロゲル化するペプチド系である。この環境依存性により、各ペプチドが折畳まれるまで系がゲル化しないという点で、本質的に「高性能な」ヒドロゲルが得られる。刺激(トリガー)の性質に依存して、この過程を介したヒドロゲル化を完全に可逆的であるようにデザインすることができ、刺激の簡潔な除去により、足場を含むペプチドが折畳まれなくなってヒドロゲルが分解する。環境トリガーのいくつかの例には、pH、イオン強度、特異的イオン結合(例えば、Ca2+)、および電磁放射(例えば、光)が含まれる。
【0046】
いくつかの態様では、本発明は、(a)β−ヘアピン高次構造への分子内ペプチド折り畳みを誘発する工程と、(b)ヘアピンのヒドロゲルに自己集合する工程とを含み得るヒドロゲルの生成方法を提供する。所望の材料特性(例えば、多孔性、強固/剛性率、生体機能性など、図1を参照のこと)を有するヒドロゲルを提供するようにペプチドをデザインすることができる。
【0047】
本発明のヒドロゲルの全ての所望の態様を、ペプチドデザインによって制御することができる。本発明で使用されるペプチドは、小ペプチド(例えば、約10〜約200残基、約10〜約100残基、約10〜約75残基、約10〜約50残基、約10〜約40残基、約10〜約30残基、約10〜約25残基、約10〜約20残基、約15〜約200残基、約15〜約100残基、約15〜約75残基、約15〜約50残基、約15〜約40残基、約15〜約30残基、約15〜約25残基、約15〜約20残基、約20〜約200残基、約20〜約100残基、約20〜約75残基、約20〜約50残基、約20〜約40残基、約20〜約30残基、または約20〜約25残基)であり得る。本発明のペプチドは、1つ以上の修飾アミノ酸残基(例えば、D−アミノ酸、天然に存在するアミノ酸のホモログ、修飾側鎖を有するアミノ酸など)を組み込むことができる。本発明のペプチドは、好ましくは、1つ以上のトリガーに応答して二次構造(例えば、β−ヘアピン二次構造)を採用する。トリガーは、1つ以上の環境条件の変化であり得る。特異的ペプチドを後に詳述するが、本発明の1つの態様では、断続的な4残基のターン配列に隣接する高β−シート性向残基から構成され得る。極性および無極性残基を、折り畳み状態で両親媒性表面が得られるように鎖領域中に連続的に配置することができる。
【0048】
本発明のいくつかの実施形態では、ペプチドが自己集合する能力は、その単分子折り畳み状態に依存する。例えば、折り畳み条件下で、ペプチドは所望の二次構造を採用することができる(例えば、ヘアピンのある表面が疎水性残基で裏打ちされており、他の表面が親水性残基で裏打ちされている両親媒性β−ヘアピン構造を採用することができる)。この例では、分子内折り畳みは、折り畳みの際の疎水性表面上の電荷密度の軽減、分子内疎水性ファンデルワールス相互作用の形成、ヘアピン内のβ鎖間の分子内水素結合の形成、およびターン配列のターン性向によって決定づけられる(図2)。分子内折り畳みを適用する要因の詳細な知識により、折り畳み事象の活性な誘導機構をデザインすることが可能である。分子内折り畳みの後、単量体ヘアピンのその後の自己集合について、折畳まれたヘアピンの疎水性表面の疎水性会合によって表面を容易にし、隣接ヘアピン間のH結合の形成および疎水性ファンデルワールス接触を介して側面を容易にする。これらのパラメーターの詳細な知識により、自己集合過程を制御し、それにより、最終材料の性質を制御することが可能である。これらの全デザインパラメーターを以下に概説し、図2に概略的に示す。当業者は、ペプチドの分子間会合およびゲル形成を好むペプチド残基の高次構造が得られる他の所望の二次構造を採用するようにペプチドをデザインすることができることを認識することができる。
【0049】
本発明のヒドロゲルで使用されるペプチドを、1つ以上の以下のパラメーターの変化によって任意の所望の特徴を有するように構築することができる:1)静電学的性質(例えば、ペプチド分子内折り畳み率の電荷によって変化させることができる);2)例えば、ペプチドの折り畳みおよび自己集合ならびにヒドロゲルの材料特性を変化させる、a)側面および表面の分子間疎水性相互作用、および/またはb)分子内疎水性相互作用が変化したペプチドを構築するファンデルワールス相互作用、3)水素結合(例えば、折り畳み、自己集合、および最終的な材料の性質を変化させるための、a)分子内および/またはb)分子間の水素結合の形成が変化したペプチドを構築することができる)、および4)ターン配列(例えば、折り畳みを制御し、それによって自己集合が誘発されるように本発明のペプチドのターン領域をデザインすることができる)。
【0050】
本発明により、当業者は、所望の分子内折り畳み、分子間自己集合、および材料特性を有するように所望の特徴(すなわち、適切な静電学的性質、分子内および分子間疎水性ファンデルワールス相互作用、およびターン配列)を有するペプチドをデザインすることが可能である。いくつかの実施形態では、静電学的および/または疎水性ファンデルワールス相互作用を使用して、活性な分子内折り畳みトリガーを有するペプチドをデザインすることができる。トリガーは、ペプチドを含む溶液の1つ以上の特徴(例えば、pH、塩濃度、特定のイオンの濃度、電磁放射、および/または温度)を変化させることができる。
【0051】
いくつかの実施形態では、本明細書中に記載のようにデザインしたペプチドを使用して、細胞の接着(例えば、哺乳動物細胞の接着)および増殖を支持する非細胞傷害性足場として使用することができるヒドロゲルを生成することが出来る。
【0052】
1つの特定の位実施形態では、MAX1(20残基のペプチド)を、ペプチドの分子内折り畳み、自己集合、および最終的なヒドロゲル材料の性質に対する静電学的性質の効果を探索するようにデザインした。配列は、II’型ターン構造を採用するようにデザインされた断続的テトラペプチド−VDPPT−に隣接する高β−シート性向を示すバリン残基およびリジン残基から構成される(図3A)。ヘアピン構造を安定化させるための要素の局所デザインの組み込みに加えて、自己集合状態でのβ−ヘアピン形成を好むような別の様式でのβ−ターンに隣接する極性残基および無極性残基の配置によって非局所効果も考慮した。さらに、i+l位でのトランスプロリルアミド結合の幾何学的性質の強いるために、β分岐残基はターンのi位(Val−9)に存在した。このデザイン要素により、折り畳み条件下で自己集合前に単量体ヘアピンの分子内折り畳みが確実に好まれる。再度デザインしたシスプロリル結合により、拡大した高次構造での各分子内の各β鎖を示すことができる。シスおよびトランス配座異性体の両方を採用することができるペプチドは、拡大して正確に折り畳まれた単量体を無差別に自己会合し、再度積極的にデザインすることができる。
【0053】
MAX1がヒドロゲル化する能力は、その単分子折り畳み状態に依存する。MAX1のいくつかのリジン側鎖が中性である塩基性水溶液条件下で(pH9.0、125mMホウ酸、10mM NaCl)、このペプチドは、両親媒性β−ヘアピンに分子内で折り畳まれる(pH7の生理学的条件下で、折り畳みを誘発することもできる)。ヘアピンのある表面が疎水性バリン残基で裏打ちされており、他の表面が親水性リジン残基で裏打ちされている(図2および3A)。分子内折り畳みの後、ヘアピンのその後の自己集合について、(a)個別のヘアピン間のH結合の形成および魅力的な疎水性相互作用を介して側面を容易にし、(b)ヒドロゲル化する折り畳まれたペプチドのバリンリッチ面の疎水性会合によって表面を容易にする(図3B)。リジン残基の荷電状態を、pHによって制御し、単分子折り畳みは可逆的である。リジン側鎖の固有のpKa未満へのpHの低下により、隣接リジン由来の鎖内電荷が反作用し、その後に各ヘアピンの非折り畳みが起こり、最終的に自己集合ヒドロゲル構造が溶解する。古典的な微小繊維ベースのデザインから調製したβ−シートリッチヒドロゲルと比較した場合、この可逆的挙動は固有である。これらの系では、自己集合過程は不可逆的である。これにより、両親媒性ヘアピンは、不可逆的に微小繊維が得られるペプチドと異なる自己集合機構を受けることが示唆される。実際、巨視的な材料の性質による分子レベル由来の本発明者らのゲル化過程の特徴付けを以下に示し、MAX1の分子内折り畳みとその自己集合する能力との関連によって反応性材料を調製することができる。
【0054】
時間依存性円偏光二色性(CD)研究は、分子内ヘアピン形成およびその後の自己集合と一致するヒドロゲル化機構を支持する。図4Aは、マイクロモル濃度でpHが5.5(非折り畳み条件)から9.0(折り畳み条件)に上昇した場合、MAX1の非撹拌溶液は、20℃で数時間かけてランダムコイルからβ−シートに移行することを示す。25℃を超える温度では、MAX1の折り畳みおよび自己集合は約数秒で起こる。406分でのCDスペクトルは、216nmで明らかに最小であり、MAX1がβ−シートが豊富な構造を採用することを示す54。図4Bは、認められたθ216は濃度依存性であり(両濃度についてのいずれか1つの時点でのθ216と比較して)、MAX1が自己集合し、濃度増加につれて集合速度が増加することを示す。この挙動は、MAX1を調査するために使用される全スペクトル法および顕微鏡法を通して一貫している。例えば、図4Aおよび4Bに示すCD研究のために使用したマイクロモル濃度では、20℃での自己集合は数時間かかる。ミリモル濃度では(下記の流動学研究で使用したミリモル濃度など)、ヒドロゲルを形成する自己集合は20℃で数分かかる。興味深いことに、自己集合速度は、濃度依存性だけでなく、混合速度にも依存し、激しく撹拌したサンプルは20℃で数秒以内にシート構造を採用する。
【0055】
折り畳みおよび自己集合過程の可逆性を、図4Cに示すように、pHおよび時間の関数としてのθ216の測定によって調査した。pH5.5でのMAX1の撹拌溶液のCDは、無作為なコイル高次構造を示す。NaOHの添加によるpH9.0へのpHの調整により、予想通り、β−シートが形成される。しかし、pH6.0へのその後の調整により、シートシグナルが完全に喪失し、ランダムコイルシグナルが全て回収される。この実験は、おそらく、単分子折り畳みおよび非折り畳みがそれぞれ起こるリジン側鎖の脱プロトン化および再プロトン化の結果として集合過程が可逆的であることを示す。また、この可逆的挙動は、アミロイド/プリオン様微小繊維から形成されたヒドロゲルでは認められない。
【0056】
ヒドロゲルマトリクス内のβ−シート構造の存在は、FTIR分光法によっても支持される(図5)。pH5.5の1重量%のMAX1のD2O溶液(ペプチドが可溶性の場合)は、1644cm−1にアミドIバンドを示し、ペプチドが折り畳まれていないことが示唆される。しかし、NaODの添加によってこの溶液をpH9.0に調整した場合、ゲル化が起こり、アミドIバンドが1615cm−1にシフトし、MAX1がβ−シートに富む構造を採用したことが強く示唆される。
【0057】
バルク(bulk)流動学試験は、単分子折り畳みおよびその後の弾性の開始および増殖によるゲル網目構造への自己集合を示す。図6Aは、一定の歪みおよび周波数での掃引時間実験の結果を示し、この実験中に2重量%MAX1溶液についてヒドロゲル化が開始された後に貯蔵剛性率(サンプル剛性)の増殖をモニタリングした。CDによって認められたβ−シート形成と類似して(図4B)、流動学は、20℃で30分後にMAX1の折り畳みおよび集合が有意に進行したことを示す(約600Paのゲル貯蔵剛性率)。ゲル形成は成熟まで持続し、2時間後にゲル剛性率は約1200Paに倍増した。ゲル形成の数時間後、約1600Paの平衡貯蔵剛性率に達した。この平衡挙動は、図6Bにおいて線形の周波数依存性剛性率測定によって明確に示される。比較のために、ストロベリーゼラチンの貯蔵剛性率は、約50Paである(データ示さず)。流動学は、主に液体様(G’’>G’)と主にゲル様(G’>G’’)応答とを分ける交差濃度が約1重量%であることを示す。文献のヒドロゲル剛性率のいくつかの例も比較のために図6Bに示す。
【0058】
自己集合ゲルの2つの顕著な特徴は、剪断終了後の剪断薄膜化およびその後の弾性の回復を示すことである。この粘度の低下(剪断薄膜化)は、歪みの適用による物理的架橋の破壊に起因する。図6Cは、歪み速度の増加につれてMAX1由来のゲルが剪断薄膜化し、粘度が低下することを明確に示す。重要なことには、MAX1の自己集合ゲルは、分子自己集合過程の迅速な緩和時間による剪断の停止後に迅速に再形成することができる。処理が容易な組織工学で使用される可能性があるβ−ヘアピンベースのヒドロゲルの1つの主な利点を本明細書中に記載する。ヒドロゲルを、高度に制御された条件下にてex vivoで予め形成し、シリンジ注射/剪断薄膜化を介して宿主に移入することができる。
【0059】
図6Dでは、元のゲル形成時と同一の掃引時間実験を、6Hzで180秒間の歪み1000%の適用直後に行った。再形成ゲルの初期剛性率は、650Paであり、これはたった30分後に回復した平衡剛性率の80%である。このことにより、迅速に回復し、且つ比較的強力なヒドロゲルが得られる。比較のために、2重量%のゼラチンヒドロゲルは、剪断薄膜化後に平衡剛性率の80%に回復するのに4時間かかる。
【0060】
図4Bでは、流動学データと濃度依存性CDデータとの間で興味深い比較を行うことができる。CDは、MAX1が分子内で折り畳まれて自己集合する速度がペプチド濃度に正に依存することを明確に示す。流動学的にモニタリングした2.0重量%ゲルの形成(濃度約6mM)を、より高いペプチド濃度での分子の折り畳みおよび自己集合と外観が類似すると見なすことができる(図6A)。折り畳みのあと始めてゲル足場にβ−ヘアピン自己集合することができ、粘弾性応答が得られる。実際、MAX1と配列が類似するがヘアピン形成を嫌うようにデザインしたコントロールペプチドは、同一の折り畳み条件に供した場合にヒドロゲル化されなかった(以下を参照のこと)。
【0061】
pH変化によってゲル化機構が可逆的であることを証明する流動学実験を、最終pHが約6.0になる2重量%ゲルへの少量の濃縮HClの添加によって試みた。系の流動学的応答は、本質的に純水の流動学的応答であり、これは装置の感度の閾値未満であった。これにより、自己集合足場およびゲル化の可逆性が明確に認められた。これは、CDによって積極的にモニタリングした酸性条件下でのMAX1の直接非折り畳みと完全に一致する(図4C)。したがって、MAX1の分子内折り畳みおよび分子間シート集合を積極的にモニタリングしたCD実験とペプチドのゲル足場への自己集合を積極的にモニタリングした流動学実験とを組み合わせて、どのようにして材料の性質が分子折り畳みおよびその後の集合機構に帰し得るのかということを示す明確な画像を形成する。
【0062】
レーザー走査型共焦点顕微鏡法(LSCM)により、水路が連続ゲルマトリクスを透過する異種ゲル微細構造が明らかとなる(図7A)。このマイクロスケールの多孔性を活用して、組織工学足場を生成することができる。多孔質微細構造を、伝統的な架橋親水性ポリマーから調整したヒドロゲルに処理しなければならない。対照的に、ヘアピンベースのゲルの微小孔性は、自己集合過程の結果であり、さらなる処理は必要ない。図7A中のゲルマトリクス領域は固体ペプチドではなく、むしろ、それ自体が水に透過するナノスケールの希薄なペプチド網目構造である(図7B)。
【0063】
1重量%ゲルを含むD2Oについての小さなおよび非常に小さな中性子散乱(SANS/USANS)データの組み合わせ(図8)は、ナノスケールのゲルマトリクスの微小繊維構造およびマイクロスケールの異種形態と一致する。低散乱ベクトルq(q=(4π/λ)sin(θ/2)、λ=中性子波長およびθ=散乱角)での強度は、勾配−4は2相間の鋭い界面からの散乱を明確に示す。このq体制(regime)では、2相は、図7AのLSCMで明確に認められたゲルマトリクスならびに1〜10μmのサイズの水孔および水路である。より高いqでのSANSデータでは、ほとんどの顕著な特徴は、qの生成物および粒子の回転半径(Rg)が1未満であるGunier領域中の約−1の勾配である。これはナノスケールにおけるロッド状の対象物からの散乱を示す。この説明は、cryoTEMデータ(図7B)中で認められたもの(ゲル足場が接合点を介して相互連結された微小繊維構造の短い領域からなる)と一致する。
【0064】
上記で考察された結果により、当業者は、ペプチドの静電特性を変化させ、それにより、本発明のヒドロゲルの形成および/または材料の性質を制御することが可能である。上記の特定の例では、自己集合および材料の性質により、ペプチドの折り畳みが一次配列のリジン電荷密度の減少によって部分的に支配されることを示す。これにより、全リジン残基が折り畳まれた両親媒性ヘアピンのある表面を占めることが可能である。当業者は、リジン以外のアミノ酸残基を使用することができることを認識する。例えば、環境条件の変化によって電荷を有するか電荷を有するようにできる任意の残基を使用することができる。さらに、複数の異なるアミノ酸残基を同じペプチド中で使用することができる。
【0065】
本発明のいくつかの実施形態では、溶液pHの変化の使用により、分子内折り畳み事象およびその後のヒドロゲル化を誘発することができ、これは可逆性を示し得る。したがって、化学的応答性を、分子内折り畳みとその後の分子間集合とを関連づけることによって実現することができる。得られたヒドロゲルは機械的に強固であり得るが、ナノスケールおよびマイクロスケールで多孔質であり、それにより、細胞適合材料として非常に良好な候補となる。さらに、ヒドロゲル足場の自己集合性によって機械的応答性材料(剪断薄膜化法によるヒドロゲル/細胞構築物の送達に活用することができる特質)が生成される。
【0066】
図2Aに関して、本発明は、ヘアピン内の残基間の分子内ファンデルワールス接触の形成によって折り畳み状態を安定化することができ、異なるヘアピン残基の側鎖間の分子間の側面および表面の接触によって自己集合状態を安定化することができることを示す。これにより、これらのパラメーターの調整によって本発明のヒドロゲルの特徴を変化させることが可能である。相対疎水性が変化するヘアピン折り畳みおよびペプチドの自己集合の温度依存性の研究によってこれを示す。水が低温で疎水基をより良好に溶媒和することができることが周知である。この現象を使用して、いくつかのタンパク質が低温で折り畳まれず、その疎水性の内部が水性溶媒に曝露されるタンパク質の低温変性を記載している。150μMのMAX1水溶液は、室温未満に冷却された場合に折り畳まれない。5℃のCDスペクトルは、ランダムコイル高次構造と一致する(図9A)。しかし、溶液の40℃への加温により、β−シート構造と一致するスペクトルが得られる。温度の関数としての218nmの平均残基楕円率のモニタリングにより、折り畳みおよびその後の自己集合が誘発される温度(Tgel)が約20℃であることが示される(図9B)。実際、MAX1のpH誘導性折り畳みおよび自己集合を含む以前の研究を20℃で行った。この温度では、折り畳みおよび自己集合は、都合良くモニタリングされるのに十分に遅い。より高い温度では、ヒドロゲル化する折り畳みおよび自己集合は迅速である(濃度に依存して一瞬から数秒まで)。疎水性相互作用の形成が折り畳みおよび自己集合過程に影響を与える場合、MAX1の疎水基含有量の変化によって折り畳みが誘発される温度に影響を与えるはずである。図9Bは、MAX1および2つのさらなるペプチド(MAX2およびMAX3)のCDデータを示す。MAX2は、1つのバリン残基が同種構造(isostructural)であるが、疎水性の低い残基(トレオニン)に置換されている以外はMAX1と同一である。MAX3では、2つのバリンがトレオニン残基と置換されている。得られたペプチドは、オクタノールから水への移動自由エネルギーの計算値の比較によって明らかなように、その疎水性が変化する(図9)。CDデータは、疎水性が減少するにつれて、折り畳みが誘導される温度が漸増することを示す。したがって、疎水性接触の形成は折り畳みおよび自己集合に影響を及ぼす。このデータはまた、pHに加えて、温度を使用して折り畳みを誘発することができることを示す。したがって、本発明のヒドロゲルの特徴を、使用するアミノ酸残基の疎水性を増減させるためのヒドロゲルを形成するために使用したペプチドのアミノ酸残基の調整によって変化させることができる。
【0067】
熱応答性の巨大ポリマーが報告されている。本発明は、自己集合を誘導するために温度誘発性折り畳みを使用する第1の系を提供する。熱誘導性ヒドロゲル化がMAX1およびMAX2で可逆性でないにもかかわらず、MAX3の折り畳み、自己集合、および最終的なヒドロゲル化は熱可逆性を示す。150μMのMAX3溶液のCDスペクトルは、このペプチドが5℃で折り畳まれないことを示す(図10A)。溶液の80℃への加熱により、スペクトルがβ−シート構造と一致する。その後の温度サイクルは、折り畳みおよび非折り畳みが可逆的であることを示す。図10Bでは、流動学は、2重量%のMAX3水溶液が数回の加熱/冷却サイクルによって熱可逆的ヒドロゲル化を受けることを示す。CDおよび流動学で使用した温度は、折り畳みおよびその後の自己集合が誘発される温度を括弧で囲んでいる(Tgel=60℃、図9)。CDおよび流動学データを合わせて、温度誘導性単分子折り畳みおよびその後の自己集合と一致するヒドロゲル化機構が示唆される。これらのペプチドによって示された温度応答性挙動は、処理の容易さのためにその用途が広くなる。例えば、フリーフローペプチド溶液を室温で調製し、in vivoで投与し、体温でゲル化を誘導することができる。いくつかのポリ−N−イソプロピルアクリルアミドポリマーを、このような移行が最終的に組織再生のための細胞外様足場を提供するように操作した。上記研究は、疎水性相互作用の形成が重要であるという考えを支持する。MAX1〜3は全て水素結合した位置でバリンおよびトレオニンを含む。これらの位置では、その側鎖はトランス回転異性体を採用することを好む。結果として、互いに反対に存在する疎水性残基は、ヘアピン点を横切ってその側鎖が互いに反対方向に向かう疎水性残基により、鎖を横切る分子内疎水性相互作用の形成を困難にしている(図11A)。しかし、これらの側鎖は、自己集合状態で隣接ヘアピンの疎水性側鎖と側面に沿って相互作用するように良好に位置づけられている(図11B)。これらの側面疎水性相互作用の形成は、自己集合を好む。MAX4(非水素結合位置でその全バリン残基が組み込まれたMAX1に匹敵する疎水性を有するペプチド)を使用して、これを示す。MAX4では、対向するバリン側鎖は、互いの方向を指すと予想される(図11B)。したがって、MAX4のバリン側鎖は、MAX1中のバリン側鎖よりも側面分子間疎水性相互作用を形成する可能性が低い。側面分子間疎水性相互作用の形成が分子内疎水性接触よりも自己集合に重要である場合、自己集合速度は、MAX4よりもMAX1の方が早いはずである。この速度の相違は、貯蔵剛性率(剛性)の増加から明らかなはずである。実際、図12は、流動学的応答を導く自己集合は、40℃でのMAX4と比較して1重量%のMAX1調製物でより迅速であることを示す(両ペプチドについて、Tgel=20℃)。したがって、静電学に加えて、自己集合時の側面疎水性接触の形成はヒドロゲル化にも寄与する。
【0068】
本発明のいくつかの実施形態では、種々の官能性(例えば、細胞接着エピトープ、受容体アゴニスト、受容体アンタゴニスト、リガンド、低分子など)を、ヘアピンヒドロゲル足場に組み込むことができる。これを達成するための1つの方法は、ペプチドの1つ以上のアミノ酸側鎖を官能化することであろう。疎水性表面上のトレオニンが置換されたMAX2およびMAX3は高温での折り畳みおよび自己集合が可能であり、それにより、類似のサイズの側鎖を有する中性残基を許容することができることを示す。いくつかの実施形態では、特定の側鎖に組み込まれるべき部分は、より大きく(例えば、RGDベースのモチーフ)、荷電残基を含み得る。MAX1の疎水性表面上の荷電残基を組み込む可能性を調査するために、MAX5(VKVKVKVKVDPPTKVKEKVKV−NH2)を調製した。配列は、MAX1が通常は折り畳まれて集合する塩基性pHで負に荷電した16位のグルタミン残基を含む。CD分光学により、MAX5の150μM溶液(pH9.0、125mMホウ酸、10mM NaCl)がランダムコイルとして存在することを確認し、疎水性表面では負電荷が許容されないことを示す(データ示さず)。しかし、MAX1の疎水性表面の変化は十分に許容される。1つ以上のリジン残基がシステイン、セリン、およびグルタミン酸などの他の残基に置換された配列は、折り畳まれて強固なヒドロゲルに自己集合することができる(データ示さず)。
【0069】
上記のペプチドは、同一のターン配列(すなわち、(−VDPPT−))を含む。この配列は、II’型ターンを形成する強い性向があり、それにより、分子内折り畳みの駆動を補助する。分子内折り畳みが自己集合前に起こらなければならないので、ターン構造形成を阻害するこのターン配列の変化を使用して、分子内折り畳みおよび自己集合を調整してヒドロゲル化させることができる。ターン領域(VKVKVKVKVLPPTKVKVKVKV−NH2)の重要性を探索するために、MAX9を調製した。MAX9は、10位のDProがLProに置換されていること以外はMAX1と同一である。II’型ターン形成を好むMAX1内に含まれるジペプチドDProLProと異なり、MAX9のLProLProモチーフは開いた高次構造を好む。開いたLProLPro高次構造から生じる2つの鎖が、反対方向に突出するであろう。したがって、β−ヘアピンを形成する分子内折り畳みはあまり好ましくないであろう。任意の観察可能な自己集合は、拡大したペプチド配座異性体の直接分子間会合に起因する可能性が高いであろう。折り畳み条件下のMAX9の150μM溶液のCDは、4時間後でさえもランダムコイルのみを示した(データ示さず)。また、2重量%のMAX9溶液はヒドロゲル化できず、低粘度溶液が数日間保持された。興味深いことに、1週間後、自己集合が起こったが、ヒドロゲル化は起こらなかった。代わりに、MAX9の自己集合した拡大配座異性体と大きさが一致した長い微小繊維が認められた(図13)。これは、β−ヘアピン高次構造への分子内折り畳みがヒドロゲルへの自己集合に必要であり、ヒドロゲルが得られる機構は拡大された微小繊維が得られる機構と異なることを強く支持する。いくつかの実施形態では、細胞接着部位として役立つターン配列を組み込むことができる。例えば、RGD結合エピトープは、一般に、細胞接着事象に重要なタンパク質のターン領域内で見出される。
【0070】
1つの態様では、本発明は、所望の環境刺激の存在下でのみ折畳まれて適時に誘発されるヒドロゲル化系が得られるデザインされた配列を有するペプチドを提供する。材料の情報を一過性および空間的に制御する能力により、材料の処理が完全に制御可能である。組織工学への適用のためのヒドロゲル−細胞構築物の調製に特異的に、誘発機構の性質に依存して、in vitroおよび/またはin vivoで制御可能なヒドロゲル形成を行うことができる。例えば、MAX1は、酸性溶液条件下(pH<9、10mM NaCl)では折畳まれないが、25℃を超える温度でpHを9に調整した場合(10mM NaCl)、数秒以内に折畳まれ、自己集合する。温度変化に応答して折畳まれるペプチドも存在した。例えば、MAX2は、温度が40℃未満の場合、pH9(10mM NaCl)で折畳まれないが、45℃を超える温度に加温した場合、折り畳み/自己集合する(図9B)。本発明者らはまた、折り畳みが起こる温度を予想通りに調製することができることを示す。折り畳みトリガーのデザインは、折り畳みおよび自己集合過程を支配する基本原理の理解によってのみ可能となることを指摘することが重要である。
【0071】
考察したペプチドのヒドロゲル化は、今までのところ、10mM NaClの存在下での塩基性溶液条件(pH=9)で起こっている。150mM NaClの存在下での生物学的関連する条件下で(すなわち、pH7)ヒドロゲル化を誘発することが望ましい。実際、これらの溶液条件での折り畳みおよび自己集合の開始は、MAX1を使用することで可能である。図14Aは、pH7(20mM Tris)でのMAX1の折り畳みは塩に依存することを示すCDデータを示す。20mM KFの存在下では、MAX1は折畳まれないが、150mM KFの存在下では、MAX1は折畳まれ、自己集合する。150mMのNaCl溶液が低波長で有意なシグナル散乱を起こすので、光学的に透明なKFをCD研究に使用する。折り畳み事象を誘発する塩を、FTIRによってさらに確認し、1重量%調製物についてpH7でKFを添加した際、1643cm−1でのアミドIバンド(ランダムコイル)が1616cm−1(β−シート)にシフトすることを示す(データ示さず)。NaClはまた、pH7でヒドロゲル化を誘発することができ、MAX1水溶液へのNaCl(最終濃度=150mM)の添加によって自己集合し、強固なヒドロゲルが得られる(2重量%で3000Pa、データ示さず)。ヒドロゲル−細胞構築物の形成のために、細胞増殖培地の添加によるヒドロゲル化の開始が理想的であろう。DMEM増殖培地がNaClおよび他の塩(総塩濃度は約400mM)を含むので、これをトリガーとして使用することができる。MAX1の2重量%水溶液は、無血清DMEM増殖培地の添加の際にヒドロゲル化する。図14Bは、得られた粘弾性の強固なゲルの周波数掃引データを示す。さらなる折り畳みトリガーを使用して、折り畳み(特異的イオン結合(例えば、カルシウム結合)が含まれるが、これに限定されない)および/または電磁放射(例えば、光)を開始させることができる。
【0072】
当業者は、材料基質の機械的性質は細胞−材料相互作用に影響を与えることを承知している。例えば、血管平滑筋細胞が架橋ポリアクリルアミド基質のあまり強固でない領域からより強固な領域に移動することが最近示された。また、後根神経節の軸索伸長速度とアガロースヒドロゲル剛性との間に直接的な相関関係が認められている。本発明により、ペプチド濃度およびペプチド折り畳み誘発条件(すなわち、塩濃度および温度)によってヒドロゲル(例えば、MAX1含有ヒドロゲル)の剛性を調整可能である。図15は、異なる温度で集合した一定の塩濃度およびpH7.4での2重量%のMAX1ヒドロゲルの剛性を示す。最終網目構造のバルク剛性率は、低温から高温まで30℃にわたり調整可能である。重要なことには、MAX1集合を温度を用いて誘発する場合、この過程は不可逆的であり、それにより、各誘発温度で達成されるゲルの剛性は、生理学的温度を用いた場合に維持される。ヒドロゲル剛性の調整可能性は、ヒドロゲル剛性と細胞挙動(接着および増殖など)との間に相関関係がある。
【0073】
本発明は、本明細書中に記載の折り畳みトリガーを使用して、線維芽細胞(NIH3T3、マウス)の接着および増殖を支持するヒドロゲルを形成することができることを示す。結合組織の発達におけるその重要性およびヒドロゲル足場に接着した場合に形態が容易に識別されるので、これらの細胞をモデル系として使用した。細胞をポリスチレンコントロールウェルまたはMAX1−ヒドロゲルの均一なスラブ(厚さ2mm)を含むウェルのいずれかに添加する定性研究を行った。光学顕微鏡によって初期細胞播種時間から4時間後に拡大した細胞を直接観察することによって細胞接着を定性的にモニタリングした(定量的細胞付着アッセイを以下に記載する。Akiyama,S.K.Functional analysis of cell adhesion:Quantitation of cell−matrix attachment,2002;Vol.69,pp281−296も参照のこと)。非接着線維芽細胞は円形であり、接着した線維芽細胞は広がった形態を有する。下記のように、光学顕微鏡によって定性的に増殖を測定し、3Hチミジンベースの細胞播種アッセイによって定量的に増殖を測定した。以下のプロトコールのいずれか1つによって、48ウェル組織培養プレート中にヒドロゲルを調製した。プロトコールI:MAX1を水に溶解し(酸性溶液(約pH5)が得られる)、同体積の緩衝液(pH9、250mMホウ酸、20mM NaCl)を室温で添加して折り畳み/自己集合を誘導し、2重量%ヒドロゲルを得た。得られたゲルを、10%ウシ血清および10mg/mLゲンタマイシンを含むDMEM中に浸漬した。これにより、培地がゲル全体に浸透し、pH7.3に調整された。プロトコール1によって調製されたゲルの貯蔵弾性率(剛性)は、約1600Paである。プロトコール2:MAX1を水に溶解し、同体積の10mg/mLゲンタマイシンを含む無血清DMEMを添加して折り畳み/自己集合を誘導し、2重量%ヒドロゲルを得た。実験によってはウシ血清を含むDMEMを添加して、血清タンパク質を導入することができる。プロトコール2によって調製されたゲルの貯蔵弾性率は、約1200Paである。図16Aは、拡大した形態によって示されるように、4時間後に(血清の存在下)、線維芽細胞がゲル表面に接着したことを示す。密集に到達するまで(104個の細胞を最初にプレートした場合、通常、72時間)、細胞は増殖する(図16B)。新鮮な培地を提供する限り、細胞は少なくとも1ヶ月生存したままである(本発明者らはこのアッセイを1ヶ月で停止させた)。ゲルを含まないコントロールウェルに添加した細胞は類似の挙動を示す(図16CおよびD)。図16A〜Dでは、10%ウシ血清を含むDMEMの存在下で細胞をプレートする。血清タンパク質は、足場への最初のコーティングによってヒドロゲルへの細胞の接着の媒介を補助することができる。ヒドロゲル足場のみが細胞接着を助長するかどうかを決定するために、細胞を、血清の非存在下で2重量%ヒドロゲルにプレートした。図16Eは、血清タンパク質の非存在下でさえも4時間後に線維芽細胞が付着および拡大し始めるが、血清含有培地と比較して範囲は狭いことを示す。4時間後の血清の添加により、細胞が増殖し、72時間後にほぼ密集に到達する(図16F)。ヒドロゲルを使用しないウェルで行った同一のコントロール実験では、細胞が同様に挙動することが示された。これらの実験は、ヒドロゲル足場によって提供されたペプチド表面が血清タンパク質の存在下で線維芽細胞を接着させ、血清が存在しない場合、その範囲はより狭いことを示す。ヒドロゲル形成で使用したペプチドへの細胞結合エピトープの組み込みを使用して、血清の非存在下での細胞接着を増強することができる。この光学顕微鏡データは、MAX1ヒドロゲルが線維芽細胞増殖を支持することを定性的に示す。
【0074】
細胞増殖速度を測定するために使用することができる定量的アッセイを開発した。これにより、異なる材料特性および構成要素のペプチド配列を有するゲルを、いかに良くこれらが増殖を支持するかに関して比較可能である。MTTおよびXTTなどの標準的な比色分析アッセイは、本発明のヒドロゲルと共に使用するのは不適切であることが証明されている。これらのアッセイは、細胞がテトラゾリウム誘導体をその対応する有色ホルマザンアナログに代謝する能力に依存する。アナログはヘアピンヒドロゲルに浸透して固定化し、それにより、UV分光法によるその後の定量が信頼できない。チミジンをDNA複製した増殖細胞に組み込む3Hチミジンベースのアッセイの使用によってこの問題に取り組んだ。この方法は、分析物を可溶化する必要がなく、非組み込みチミジンをヒドロゲルから容易に洗い流し、組み込まれたチミジンのみの定量が可能である。アッセイは2要素からなる。第1に、細胞が密集に到達しない所与の期間(ここでは、便宜上48時間を選択する)にわたって増殖を追跡することを望む場合、プレートすべき最適な細胞数を決定するための細胞播種実験を行うことができる。第2に、材料への細胞(最適密度)の播種により、および密集に到達した場合、48時間までの個別の測定点でのDNA複製された細胞数の決定によって、所与の材料についての増殖速度を決定することができる。本発明のヒドロゲルの最適細胞播種密度を決定することが可能な3Hチミジン細胞播種実験の結果を図17に示す。アッセイすべき細胞(例えば、線維芽細胞)を、MAX1ヒドロゲルまたはポリスチレンコントロール表面に異なる初期密度で播種することができる。48時間後、増殖培地を除去し、3Hチミジンを含む培地と交換し、3時間インキュベートすることができる。非組み込みチミジンを洗い流し(3Hチミジンがもはや検出されなくなるまで洗浄物をアッセイすることができる)、シンチレーションカウンティングのために細胞を死滅させることができる。データは、MAX1ヒドロゲルを含む2日間の増殖アッセイについて、最適な増殖速度を得るために40,000個の線維芽細胞を播種しなければならないことを示す。ポリスチレンの最適播種密度が異なる(20,000)ことに留意すべきであり、任意の所与の細胞型が異なる材料について異なる最適播種密度を示すこと(および異なる細胞型が1つの所与の材料について異なる最適播種密度を有すること)が公知である。したがって、各新規の材料のために、増殖速度を決定する前に類似の細胞播種実験を行うことができる。このデータにより、48時間後に、コントロール表面上よりもヒドロゲル上でより多くの細胞が増殖することも示唆される(各表面のそれぞれの最適播種密度での分解物(数)の比較)。ヒドロゲル足場の三次元多孔性により、細胞がヒドロゲル内で増殖可能であり、二次元ポリスチレン表面では不可能であると説明することができる。このデータは、定量的増殖が確立されたことを示す。当業者は、本明細書中に記載のアッセイを使用して、細胞増殖に対するペプチド配列およびヒドロゲル剛性の効果を研究することができる。
【0075】
図16および17に概説した実験は、ゲルの二次元表面上への細胞のプレーティングを必要とする。二次元ゲル表面上の接着および増殖を利用するアッセイにより、異なるヒドロゲル足場を容易に比較可能であり、これは材料−細胞相互作用を調査するための最も一般的な技術であるので、結果を文献に記載の材料と直接比較することができる。
【0076】
本発明はまた、細胞がゲル足場全体に三次元で組み込まれたヒドロゲル−細胞構築物を含む。線維芽細胞を含む無血清DMEMの添加によるMAX1水溶液のヒドロゲル化の開始によってこれを行うことができる。細胞の三次元in vivo環境を模倣する足場により、二次元研究で証明されなかった細胞−材料相互作用がさらに洞察される。図18は、この様式で調製された2重量%MAX1ヒドロゲルのLSCM画像を示す。側面からゲルの内部を見ることができるように画像を構築する。細胞トラッカーグリーン(CMFDA,Molecular Probes)で染色した細胞がゲル全体で認められ、ゲル化が完了する前に細胞が沈み始めるゲルの底付近がわずかに濃度がより高い。当業者は、目的の任意の細胞型を本発明と組み合わせて使用することができることを認識する。例えば、使用することができる細胞には、酵母細胞、植物細胞、および動物細胞が含まれるが、これらに限定されない。適切な細胞は、例えば、公知の培養物寄託機関(例えば、American Type Culture Collection(Manassas,VA))および当業者に公知の商業的供給元から市販されている。本発明の方法で使用される好ましい動物細胞には、昆虫細胞(最も好ましくは、ショウジョウバエ細胞、スポドプテラ細胞、およびトリコプルサ(Trichoplusa)細胞)、線形動細胞(最も好ましくは、線虫細胞)、および哺乳動物細胞(CHO細胞、COS細胞、VERO細胞、BHK細胞、AE−I細胞、SP2/0細胞、L5.1細胞、ハイブリドーマ細胞、最も好ましくは、293細胞、PER−C6細胞、およびHeLa細胞)などのヒト細胞が含まれるが、これらに限定されない。さらに、初代細胞培養物、組織ホモジネート、および/または組織ホモジネート由来の細胞を、本発明と組み合わせて使用することができる。
【0077】
図2に関して、静電学、水素結合、および疎水性相互作用は全て折り畳みおよび自己集合で役割を果たす。折り畳みおよび自己集合のための静電学および側面の分子内および分子間疎水性接触の変化を使用して、本発明のヒドロゲルの材料特性および/または形成条件を変化させることができる。さらに、ヘアピンのターン領域の変化を使用して、本発明のヒドロゲルの特徴を調整することもできる。理論に拘束されることを望まないが、自己集合は、以下の2つの相互作用に支配されると考えられる:1)ヘアピンの疎水性崩壊、および2)分子間水素結合形成。これら2つの要因の相互作用により、疎水的に会合した微小繊維接合が散在した微小繊維の短いセグメントから構成される足場が得られる(図1)。当業者は、本明細書中に記載の教示を前提として、これらの要因のいずれかまたは両方の寄与を調整するために使用したペプチドの配列(またはそれによる物理的性質)を変化させて、任意の所望の特徴を有するヒドロゲルを生成することができる。
【0078】
図2に概説した分子間相互作用を変化させて、自己集合過程および得られた材料の性質(細胞増殖に重要なゲルの多孔性など)を制御することができる。20残基から構成されるヘアピンの特定の例を本明細書中に提供した。本発明はまた、これよりも長いおよび短いペプチドを含み、得られたヒドロゲルの材料特性に影響を与えるために使用したペプチドの長さを変化させることができる。
【0079】
本発明はまた、特定の例で使用したペプチドのターンと比較して1つ以上のアミノ酸置換を有するペプチドを含む。当業者に公知の任意のターン配列を、本発明と組み合わせて使用することができる。
【0080】
本発明のいくつかの実施形態では、1つ以上の機能的部分は、ペプチド配列、有機分子、または他の分子であって良く、例えば、一方または両方のヘアピン鎖の一次配列内および/またはターン配列の一次配列内で、親水性表面から生じるアミノ酸側鎖、疎水性表面から生じるアミノ酸側鎖、ターン配列を含むアミノ酸側鎖のペプチドに組み込むことができる。このような機能的部分の特定の例には、ペプチド配列(例えば、細胞接着エピトープ、核局在化シグナルなど)、受容体アゴニストおよび/または受容体アンタゴニスト(例えば、コレステロール誘導体など)、ペプチド模倣物、環状ペプチド、金属キレート剤、蛍光およびスピン活性プローブ、ならびに低分子治療因子が含まれるが、これらに限定されない。1つ以上のこのような機能的部分を含むペプチドを、所望の折り畳み/自己集合特性および/または材料特性を有するヒドロゲルを生成するための本明細書中に記載の分光法、光および中性子散乱、顕微鏡法、および流動学技術によって分析することができる。
【0081】
一定の水素結合と比較して分子間表面および側面疎水性接触の形成への寄与を変化させるために、MAX1に基づいたペプチドを調製することができる。このようなペプチドは、同数の残基を有し得るが、折り畳まれたヘアピンの疎水性表面上に側鎖の疎水性が変化したアミノ酸を含む。したがって、折り畳みおよび自己集合の間に形成された可能な分子内および分子間水素結合の数は一定であるが、疎水性面の表面領域が変化している。調製することができるペプチドの例には、以下の一般的配列のペプチドが含まれるが、これらに限定されない:XKXKXKXKVDPPTKXKXKXKX−NH2(式中、X=バリン(MAX1);X=イソロイシン(MAX7);およびX=フェニルアラニン(MAX8)。分子モデリング(洞察/発見)は、これらのペプチドの表面領域はMAX1<MAX7<MAX8の順で増加することを示す。表面領域の変化は、ヒドロゲルの種々のパラメーターに影響を及ぼし、例えば、CDによってモニタリングしたところ、各ペプチドが折畳まれて自己集合する(Tgel)温度を減少させることができる(図9を参照のこと)。また、各ペプチドの流動学的性質は異なっていて良く、より疎水性の高いペプチドによりより迅速にヒドロゲルを得ることができる。さらに、ヒドロゲル化速度が影響を受ける可能性があり、ペプチドの疎水性が高いほどより迅速にヒドロゲルが形成される。ヒドロゲル化速度を、流動学によって決定することができる(図12を参照のこと)。Cryo−TEMを使用して、これらのデザインの変化がヒドロゲルのナノスケールの構造に影響を与え(例えば、疎水性表面領域が、自己集合状態の微小繊維の接合数を増加させる場合)、LSCMを使用してマイクロスケールの構造の変化(例えば、ゲル多孔性の変化)を評価することができるかどうかを決定することができる。折り畳まれたペプチドの疎水性面から生じたアミノ酸残基の性質および数の変化および/または折り畳まれたペプチドの疎水性面から生じた機能的部分の性質および数の変化を使用して、形成されたヒドロゲルの材料特性を変化させることができる。
【0082】
自己集合時の分子間水素結合形成への寄与を変化させるために、分子間水素結合の形成能力がより高いかより低いペプチドを調製することができる。例えば、Nαアルキル化リジン残基を含むMAX1の誘導体を調製することができる。MAX1中のリジン残基を、これらが自己集合時に隣接するヘアピンと分子間水素結合を形成することができるように連続して配置する(図3A)。各ヘアピン鎖内の1つ以上のリジン残基のアルキル化により、ヒドロゲル化を起こす自己集合を阻害することができる。このようなペプチドの例には、以下が含まれるが、これらに限定されない:
【0083】
【化A】
(太字の位置にNαブチル化リジン残基を含む)。当業者は、アミノ酸残基および/またはアミノ酸残基に付着した機能的部分の変化により、分子間水素結合の形成能力を調整された(増加または減少)他のペプチドを構築することができることを容易に認識する。ヒトアミリンの微小繊維への自己集合がN−ブチル残基を含むペプチドによって阻害されていることが公知である。CD分光法および流動学を使用して、これらのペプチドが種々の折り畳み条件下で折り畳まれてヒドロゲルに自己集合する能力を評価することができる。
【0084】
いくつかの実施形態では、分子間相互作用を活用して自己集合過程および得られる材料特性を制御するようにペプチドを構築することができる。疎水性崩壊と分子間水素結合との組み合わせを介して折り畳まれて自己集合するペプチドのために、温度を使用してこれらの各過程が自己集合機構に寄与する程度を制御することができる。上記で考察するように、各ペプチドは、折り畳まれて(Tgel)、自己集合し始める特徴的温度を有する。さらに、温度を使用して疎水性崩壊を駆動することができることが文献に十分に示されている。したがって、任意の所与のペプチドのために、折り畳まれるが疎水性崩壊をあまり増強しないように過不足なく加熱する場合、水素結合が自己集合機構を支配し、得られたヒドロゲル足場は、微小繊維構造のより長いセグメントから構成され得る。得られたヒドロゲルを、より巨大な孔サイズによって特徴づけることができる。逆に、水素結合よりも疎水性崩壊が速度的に好まれる極端な温度へのペプチドの曝露により、より短い微小繊維セグメントおよびより多数の微小繊維間架橋を有する足場を得ることができる。得られたゲルは、より小さな孔サイズを有し得る。したがって、温度制御により、ゲルの微視的およびナノスケールの特徴を制御する一般的方法が得られる。いくつかの実施形態では、2つの異なる温度(T1=TgelおよびT2=Tgel+30℃)でペプチドを集合させることができる。得られたヒドロゲルを、cryo−TEMおよびLSCMによって視覚化して、ナノスケールおよびマイクロスケールの性質を評価することができる。より多数の微小繊維間架橋を含むゲルはより強固であり得る。これを、流動学によって検証することができる。
【0085】
いくつかの実施形態では、例えば、MAX1の一般的構造に基づいて、配列の長さが異なるペプチド(すなわち、VK(VK)mVKVKVDPPTKVKV(KV)nKV−NH2(式中、m=1〜20およびn=1〜20、mは任意の所与のペプチドにおいてnと同一であっても異なっていても良い))を調製することができる。より長いヘアピンの疎水性表面が増加するので、より長い鎖領域から構成されるペプチドはより強固なゲルを形成することができる。より強固なゲルが得られるより長い鎖領域から構成されるペプチドを使用して、機能的部分を組み込むことができる。単量体ヘアピンは、その鎖が約7残基長である場合に最も安定しており、鎖の伸長によってヘアピンの安定性が増加しないことが示されている。より長い鎖領域を有するペプチドのヒドロゲルを調製し、例えば、同一の折り畳み条件下で流動学を介してその剛性を評価することができる。
【0086】
いくつかの実施形態では、ターン領域の1つ以上のアミノ酸を、MAX1のターン領域と比較して置換および/または修飾することができる。いくつかの実施形態では、構造的役割を果たすだけでなく、生物機能的役割も果たすターン配列を組み込むことができる。例えば、RGD結合エピトープは、通常、細胞接着事象で重要であることが公知のタンパク質のターン領域内に見出され、RGDに隣接する残基は結合事象にさらなる特異性を与える。これらのエピトープの自己集合ヘアピンのターン領域への組み込みにより、細胞接着特性が増強されたヒドロゲル足場を得ることができる。上記で考察されるように、ターン構造を形成することができない配列の組み込みにより、折り畳むことができずにヒドロゲルが形成されないペプチド(MAX9)が得られる。しかし、ターン構造を採用することができるが、MAX1の−VDPPT−ほど促進しない別のターン配列を含むペプチドは、依然として折り畳まれてヒドロゲル化し得る。MAX1の文脈では、弱いターン形成物(former)から強いターン形成物にターン構造を形成するためにテトラペプチド配列−VDPPT−をその元の性質を変化させる配列と置換したペプチドを調製することができる。各ペプチドが分子内で折り畳まれて自己集合する能力を、CDによって決定することができ、その対応するヒドロゲルの流動学的性質を研究して、所望の特徴(例えば、組織工学のための可能な足場としてのその適切性)を有するヒドロゲルを同定することができる。調製することができるペプチドの例には、以下が含まれるが、これらに限定されない:VKVKVKVK−XXXX−KVKVKVKV−NH2(配列番号12)(式中、−XXXX−は、−VDPGT−(非常に強いターン)、−ADPGT−(強いターン)、−VNGT−(中程度のターン)、−VGGT−(配列番号13)(弱いターン)である)。
【0087】
いくつかの実施形態では、機能低部分(例えば、細胞接着エピトープ)を、以下のうちの1つ以上に組み込むことができる:a)ペプチドのβ鎖部分中の1つ以上のアミノ酸側鎖上、b)β鎖の末端および中間部分の一次配列内、c)ターン領域に隣接する位置、d)ターン配列を形成する1つ以上のアミノ酸残基の側鎖上、および/またはe)ターン配列の一次配列内。使用することができる機能的部分の例は、簡潔なトリペプチドRGDである。配列を、ヒドロゲルの意図する使用によって変化させることができる。例えば、細胞の足場としてヒドロゲルを使用することを意図する場合、使用すべき特定の細胞型の接着を誘発することが公知の配列によって機能的部分を変化させることができる。種々の細胞型のための特異的接着配列の例を以下に示す。図19中のペプチドを調製し(十分に確立された直交保護ストラテジーを使用して、側鎖修飾ペプチドを合成する)、CDおよび流動学によって研究してその折り畳み特性および流動学的性質を確立することができる。この実施形態にしたがって調製されたヒドロゲルは、増強された細胞接着および増殖特性を有し得る。
【0088】
いくつかの実施形態では、所望の特徴を有するヒドロゲルを生成するために、自己集合前の折り畳み単量体の濃度を変化させることができる。動的光散乱(DLS)と組み合わせてCDを使用して、自己集合を引き起こすために存在しなければならない折り畳まれた単量体の濃度を決定することができる。DLSにより、自己集合過程によってナノ構造成長を直接モニタリングすることが可能である。5nmから1ミクロンまでの粒子サイズの分布を容易に観察することができる。例えば、MAX1を使用し、CDを使用して、20℃でのβ−シート形成速度をモニタリングすることができる。CDシグナルの大きさが分子内(折り畳みによる)β−シート形成対分子間(自己集合による)β−シート形成に帰し得る範囲を、同一溶液(μMペプチド濃度)についてのCDβ−シートシグナルの成長と自己集合粒子サイズの成長との実時間比較によって決定することができる。重要なことに、折り畳まれた単量体は、DLSにおける粒子サイズに成長しない。したがって、粒子サイズの成長が同時に起こることなくCDによって大量のβ−シート構造が認められる場合、自己集合を引き起こすために折り畳まれた単量体の集団が大量に存在しなければならないと結論づけることができる。逆に、CDβ−シートシグナル成長と同時に粒子が自己集合する場合、自己集合が分子内折り畳みの直後に起こると結論づけることができる。DLS中での粒子サイズの成長と流動学によってモニタリングされた貯蔵弾性率の成長との類似の実時間比較を行うことができる。DLSと流動学との間の直接比較を行うことができるようにミリモル粒子濃度を使用する。網目構造の流動学的性質が導入される粒子サイズの閾値を、直接観察する。
【0089】
いくつかの実施形態では、本発明のヒドロゲルは、増強された処理可能性を示し得る。ペプチド溶液が適時にヒドロゲル化可能な1つ以上の活性な分子内折り畳みトリガーを有するペプチドオンデザインの結果として、当該分野で公知のヒドロゲルと比較して、本発明のヒドロゲルの処理可能性を増強することができる。上記で示すように、折り畳みを誘発する能力により、自己集合およびその後のヒドロゲル形成を直接制御する手段が得られる。さらに、誘発エレメントを容易に組み込むことができる理想的な分子足場である適切なペプチド二次構造(すなわち、ヘアピンモチーフ)の特定の例を提供している。本発明のいくつかの実施形態では、pH、イオン強度、および温度などの生理学的に関連する刺激に応答してヒドロゲル化するようにヘアピンをデザインすることができる。処理可能性に関して、折り畳みの誘発により、in vitroおよびin vivoの両方でヒドロゲル化ストラテジーを実施することが可能である。本発明のさらなる実施形態では、さらなる手段(例えば、電磁放射(例えば、光)および特異的イオン結合(例えば、カルシウムイオン結合))を使用して、ヒドロゲル化を誘発することができる。ヒドロゲル化を誘発するための電磁放射の使用により、光ファイバーを介したin vivoでの空間分解材料の形成を誘導する都合の良い手段が得られる。例えば、低粘度のペプチド−細胞懸濁液を投与し、その後に照射によって局所的にゲル化することができる。組織中に天然に見出される刺激を介したex vivoでのヒドロゲル化を誘発する能力により、材料形成を制御する別の都合の良い手段が得られる。特異的イオン結合(例えば、カルシウムイオン結合)に応答してヒドロゲル化するペプチドを調製することができる。特異的イオンとしてカルシウムイオンを使用する場合、得られた材料は、細胞が増殖することができる足場として役立つだけでなく、ペプチド足場の生分解時に放出されるカルシウム供給源も提供する。
【0090】
本発明のヒドロゲルのデザインでは、折り畳みおよび自己集合過程を、当業者に公知の任意の適切な技術(例えば、CDおよびFTIR)によって特徴づけることができる。材料のナノスケールおよびマイクロスケールの多孔性を、cryo−TEM、LSCM、USANS、およびSANSによって決定することができ、材料の剛性を流動学(例えば、本明細書中に記載の方法を使用する)によって測定することができる。誘発過程の効率も試験することができる。いくつかの実施形態では、in vivoヒドロゲル化を意図するペプチドのための誘発系は、迅速にヒドロゲル化して強固な多孔質ゲルが得られる誘発系であり得る。希薄なペプチド溶液についてのトリガー誘導性シート形成速度をCDによって決定することができ、対応する1〜2重量%のヒドロゲルの剛性を振動流動学によって決定することができる。in vitro適用のために、ゲル化速度が迅速であることが望ましいが、必要ではない。特定の例では、MAX1を、以下で考察している各トリガーの有効性を調査するためのモデル系として使用することができる。しかし、一旦MAX1の状況で特定のトリガーの実行可能性が確立されると、トリガーを、MAX1と異なる所望の特質を示す他のペプチド配列に組み込むことができる。
【0091】
本発明のいくつかの実施形態では、電磁放射(例えば、光)を使用して、本発明のペプチドのヒドロゲル化を誘発することができる。適切な方法の例には、図20に示す方法が含まれる。図20に示す実施形態には、折り畳みを阻害するためのフォトケージ(photocage)の使用が含まれる。このようなフォトケージを、例えば、本発明のペプチドのヘアピンを含む残基の側鎖および/またはペプチド骨格内に組み込むことができる。折り畳みおよびそれによる自己集合を、光へのペプチド水溶液の曝露によって開始することができる。
【0092】
上記で考察するように、ヘアピンの疎水性表面(例えば、Glu)への荷電残基の組み込みにより、折り畳みおよびそれによる自己集合が阻害される。この阻害を使用して、疎水性表面への負電荷のフォトケージ(2−ニトロフェニル酢酸)の組み込みによって折り畳みを阻害することができる。1つの実施形態では、MAX1の16位のアミノ酸残基を、システイン(MAX6)と置換し、文献のプロトコールにしたがって2−ブロモ−2−(2−ニトロフェニル)酢酸でアルキル化してケージドペプチドを得ることができる(Pan & Bayley,Febs Letters 1997,405,81−85)。2−ニトロフェニル酢酸は一般的に使用されているフォトケージであり、ヘテロ原子由来のその光誘導性切断は十分に研究されている。ケージドペプチドの光(330〜360nm)への曝露により、負電荷のケージが放出され、疎水性表面上に中性Cysが生成される。光分解時に形成されたニトロソケトン副生成物が新規の形成されたCys側鎖をアルキル化する場合(潜在的に有害な副反応)、1mMジチオスレイトールを添加してニトロソケトン反応を停止させて還元環境を維持することができる。16位を最初に選択して修飾する。なぜならばこの位置でCysを含むコントロールペプチドが折り畳まれて自己集合するからである。当業者は、ペプチド中の1つ以上の他の位置を類似の様式で処理することができることを認識する。光分解効率は、配列位置に依存し得るので、フォトケージ化された(photocaged)残基の種々の位置および/または数を使用して、得られたヒドロゲルの性質を変化させることができる。いくつかの実施形態では、光分解速度は、マイクロ秒からミリ秒までの時間体制(time regime)であり得る。典型的には、照射時間は、約1マイクロ秒から数ミリ秒までであり得る(例えば、約0.01μs〜約1000ms、約0.01μs〜約100ms、約0.01μs〜約10ms、約0.01μs〜約1ms、約0.01μs〜約0.5ms、約0.1μs〜約1000ms、約0.1μs〜約100ms、約0.1μs〜約10ms、約0.1μs〜約1ms、約0.1μs〜約0.5ms、約1.0μs〜約1000ms、約1.0μs〜約100ms、約1.0μs〜約10ms、約1.0μs〜約1ms、約1.0μs〜約0.5ms、約10μs〜約1000ms、約10μs〜約100ms、約10μs〜約10ms、約10μs〜約1ms、または約10μs〜約0.5ms)。適切な照射供給源は、バルクおよび/または空間分解照射を行うことができ、水銀灯および空間分解照射が可能なUVレーザーを備えたNikon eclipse TE2000倒立蛍光顕微鏡(全て市販されている)が含まれるが、これらに限定されない。
【0093】
いくつかの実施形態では、ペプチドのN−アルキル化を使用して、水素結合駆動自己集合を阻害することができる。通常は分子内水素結合を形成する骨格のアミド窒素(例えば、MAX1の16位)にN−o−ニトロベンジルケージを組み込んだβ−ヘアピンを、標準的技術を使用して合成することができる。分子内水素結合が立体的に込み合っているので、この位置を占めるケージを使用して、分子内折り畳みを阻害することができる(図20)。o−ニトロベンジルケージは、その性質が十分に確立されている別のクラスの化合物である。光(330〜360nm)への曝露によってケージが放出され、水素結合の形成およびそれによる折り畳み/自己集合に関与し得るプロトン化アミドが再生される。光分解効率を配列の位置によって変化させることができ、これを使用して、ヒドロゲルの形成および/または特徴付けを変化させることができる。部位特異的N−アルキル化ペプチドを、文献のプロトコールによる固相ペプチド合成(SPPS)によって調製することができる(Reichwein,et al.,Tetrahedron Letters 1998,39,1243−1246およびTatsu,et al.,Febs Letters 2002,525,20−24を参照のこと)。上記の特定のケージを細胞で使用しているので、ケージの副生成物は細胞傷害性を示す可能性が低い。しかし、文献に記載の方法によってこの系の細胞適合性を測定することができる(Bryant,et al.,Journal of Biomaterials Science−Polymer Edition 2000,11,439−457を参照のこと)。上記のo−ニトロ誘導体は全て切断を誘導するための1つの光子励起を含むので、細胞の存在下でヒドロゲル化を誘発するための330〜360nmの光の使用によってUVベースの細胞の光損傷が起こり得るという別の可能性が懸念される。一般に、明らかな細胞損傷が認められない細胞ベースの実験を行う状況で1つの光子励起を使用するにもかかわらず、任意の特定の細胞型でUVベースの損傷が認められる場合、IRで吸収される2つの光子ケージ(例えば、クマリン誘導体)を使用することができる(Furuta,et al,Proceedings of the National Academy of Sciences of the United States of America 1999,96,1193−1200)。
【0094】
いくつかの実施形態では、特定のイオンの結合を使用して、ヒドロゲル化を誘発することができる。例えば、カルシウムイオン結合によってヒドロゲル化を誘発することができる。典型的に、カルシウムイオンの非存在下でのカルシウムイオンの結合を含む実施形態では、ペプチドが折り畳まれない一方で、カルシウムの添加は折り畳み/自己集合の核となる。上で示すように、ヘアピンのターン領域は、分子内折り畳みおよびそれによる自己集合の促進において重要であり得る。1つの特定の例では、4残基のターン(−VDPPT−)が金属イオンの非存在下で規則正しい構造を採用することができないターン/ループ配列または非天然キレートに置換されたMAX1の変異形を調製することができる。金属結合は、ターン形成に影響を与え、それにより、分子内折り畳みおよび自己集合を促進する。図20は、以下に詳述する金属トリガーを示す。
【0095】
1つの特定の実施形態では、4残基のターン(−VDPPT−)を、−DRKADGYIDFEE−と置換して、VKVKVKVKVDRKADGYIDFEEVKVKVKVKV(配列番号14)を得るMAX1の変異形がある。このペプチドは、金属の非存在下で規則正しくないはずであるが、Ca(II)の結合後に迅速な折り畳みおよび自己集合が起こる。配列DRKADGYIDFEEは、トロポニンCのEFハンドドメイン内に含まれるカルシウム結合ループに対応する。下線を引いた残基は、側鎖および主鎖カルボニルの両方を介してCa(II)に結合してエクアトリアル位の1つを占める水を有する五角錐の形状が得られる。重要なことには、この線形ペプチドが低mM KdでCa(II)と結合することができ、この結合事象により構造が形成されることが示されている。トロポニンCの結晶構造は、カルシウム結合ループのN末端およびC末端残基が約6Å以内であり、これらの末端残基がねじれ角を採用して逆平行シートの形成を促すことを示す。実際、このデザインしたペプチドを使用してエネルギー最小化および明示的溶媒(explicit solvent)を使用した力学的刺激を行い、この金属結合ループがMAX1の状況下でβ−ヘアピン構造の核となるように幾何学的に十分に適合していることが示された。
【0096】
別の特定の実施形態では、さらなる高親和性トリガーを使用することができる。このデザインにより、非天然Ca(II)キレート剤DOTAM(十分に研究されているオクダデンテートリガンドDOTAの誘導体である)の使用が求められる(図20)。DOTAMは、カルシウムと強固に結合して(Kd=29nM)、互いに5Å以内にリガンドのカルボニル含有アームが存在する四角反柱複合体を形成し、これら2つの位置がペプチド結合に理想的になる。したがって、図20のMAX1の4残基のターン(−VDPPT−)のDOTAM−誘導体との置換により、Ca(II)の非存在下で構築されない系が得られる。しかし、Ca(II)が結合する場合、2つのβ鎖が互いに5Å以内になり、折り畳みが開始される。このペプチド模倣物を、市販または容易に合成される出発材料を使用した手作業の固相合成を使用して調製することができる。
【0097】
いくつかの実施形態では、ペプチドの構造および材料の性質を使用して、細胞株(例えば、モデル線維芽細胞株および骨芽細胞株)の接着および増殖を調整することができる。上記のヒドロゲルは、組織工学への適用に有用であり得る(例えば、電解質濃度、温度、および光などの生体適合性刺激によって材料の形成を誘発し、自立に十分な剛性(10Pa超)を示す多孔質ゲルが得られる適用)。本開示により、当業者は、ペプチド構造と、材料の剛性と、細胞適合性とを相関させると同時に、最終的に組織工学に有用であり得るヒドロゲルを同定することが可能である。組織工学を実現するための3つの望ましい細胞レベルの生物学的特徴は、以下である:a)ヒドロゲルが所望の細胞に対して非細胞傷害性であること、b)ヒドロゲルが細胞接着(付着および拡大)を促進すること、およびc)ヒドロゲルによって細胞が増殖すること。本発明のヒドロゲルは、典型的には、1つ以上のこれらの特徴を有するが、かならずしもそうではない。第1に、細胞適合性を評価するための実験ストラテジー(パートa、b、およびc)を記載する。第2に、これらの研究に適切な細胞株の例を考察する。第3に、当業者が、ペプチド構造と、材料の剛性、細胞適合性との間の相互作用を描写し、適切なペプチドを選択し、特定の適用(例えば、特定の細胞株)を誘発することが可能な特定の例を記載する。いくつかの実施形態では、細胞結合エピトープをペプチドに組み込んで材料の細胞適合性を増強することができる。使用することができる細胞型の例には、哺乳動物細胞、NIH 3T3線維芽細胞、HEK293、HELA細胞株、およびラット頭蓋冠MC3T3−E1骨芽細胞、神経細胞、幹細胞、および組織サンプル(例えば、脳、肝臓、心臓など)由来の細胞が含まれるが、これらに限定されない。
【0098】
いくつかの実施形態では、細胞の接着および増殖を容易にするペプチドおよび誘発条件を選択することが望ましい。典型的には、線維芽細胞付着には4時間で十分であり、ほとんど全ての細胞が拡大するが増殖しない。文献のプロトコールには、増殖しない骨芽細胞の付着には12時間で十分であることが示されている。したがって、線維芽細胞の生存を、ヒドロゲルへの導入から4時間後に、たとえば生細胞対死細胞の優先的染色および蛍光ベースの画像化の実施によってアッセイすることができ、骨芽細胞を12時間後にアッセイすることができる。既知数の各細胞を、組織培養プレート中に予め形成したヒドロゲルに播種することができる。各細胞型についてそれぞれの時間をおいた後、カルセインAMまたはペンタフルオロベンゾイルアミノフルオレセイン二酢酸を使用して、生細胞の細胞質を染色することができる。ヨウ化プロピジウムを使用して、死細胞を染色することができる。ヒドロゲルを含まない組織培養プレートを使用した並行コントロールを実施することができる。ヒドロゲル中の死細胞数がコントロールプレートで認められた数よりも多い場合、材料は、特定の細胞型に不適切であると考えることができる。アポトーシスを受けた細胞が色素取り込みに抵抗し、ヒドロゲル細胞傷害性が不正確に評価される可能性がある。これらのゲルの細胞傷害性を、下記の接着および増殖研究でさらに検証することができる。予備実験の結果に基づいて、提案したゲルの大部分は、非細胞傷害性を示すようである。最後に、提案した色素に問題があると証明された場合、別の色素組(例えば、SYTO(SYTOX(死細胞の核酸指示薬)と組み合わせて使用される生細胞の核酸指示薬))を使用して細胞傷害性を評価することができる。
【0099】
一般に、合成材料を、細胞培養基質としてin vitroで使用する場合、細胞接着は、主に、非特異的に足場をコーティングする増殖培地中に存在する血清タンパク質(例えば、フィブロネクチンおよびビトロネクトリン(vitronectrin))によって媒介される。しかし、本明細書中に記載の機構の誘発によって最終的にin vivoで導入することができるヒドロゲルについて、ヒドロゲル足場は、血清タンパク質の非存在下で細胞接着を本質的に促進することができる。これにより、潜在的な宿主が足場に細胞接着タンパク質を導入する機構を提供しない場合、細胞付着が確実になる。本発明のヒドロゲルは、10%ウシ血清の存在下および非存在下の両方で細胞接着を促進することができる。穏やかな洗浄後の付着細胞画分対非付着細胞画分の決定によって細胞付着をX線で定量するアッセイを使用して、細胞付着に適切なヒドロゲルを同定することができる。詳細には、1)目的の細胞株(例えば、哺乳動物線維芽細胞または骨芽細胞)を培養し、3Hチミジンで24時間予め標識することができる。非組み込み標識を洗浄によって除去し、2時間追跡することができる。2)次いで、既知濃度の標識細胞を非処理組織培養ウェル(コントロール)および本発明のヒドロゲルを含むウェルの双方に導入することができる。ヒドロゲルを含むウェルでは、ウシ血清を含むか含まない細胞増殖培地を添加することができる。3)例えば、線維芽細胞については6時間にわたって細胞接着速度を最初にモニタリングし、その時間内の増加量を、日常的実験を使用して当業者が実験的に決定することができる。骨芽細胞接着速度を、24時間にわたって最初にモニタリングすることができる。これらの最終時点を実験的に精緻化することができ、付着および拡大するのに十分であるが、増殖しない時間を与えなければならない。また、終点が長すぎることに関する問題は、長時間インキュベートした場合、いくつかの細胞が、膜結合プロテアーゼまたは分泌されたマトリクスタンパク質によってヒドロゲル表面が修飾され、「非粘着(non−stick)」表面上でさえも付着し得ることである。このシナリオでは、細胞によって付着が促進され、必ずしも材料の表面によってではない。最適な終点を、熱変性BSAでコーティングした表面(典型的な非粘着表面)上の細胞のプレーティングによって決定することができる。2〜3%を超える細胞が付着した場合、最大終点に到達している。4)適切な時点で、非付着細胞を穏やかに洗い流し、液体シンチレーション分光法によって計数し、付着した細胞を、定量のための検量線を使用したシンチレーションカウンティングのために可溶化することができる。したがって、ポリスチレン組織培養物と血清タンパク質を含むか含まないヒドロゲル表面とを比較することができる。付着後の細胞拡大を、光学的に検証することができる。このようにして得た情報を使用して、任意の目的の細胞型の接着を促進するように適切なヒドロゲルをデザインすることができる。ペプチド構造の変化により、剛性および足場形態などの材料特性を、ヒドロゲルの細胞接着を助長する能力を至適化するように調整することができる。例えば、上記の簡潔な細胞拡大アッセイにより、線維芽細胞が血清タンパク質の存在下でMAX1ヒドロゲル(貯蔵剛性率=1200Pa)に効率良く付着したが、血清タンパク質の非存在下ではあまり効率が良くないことが示された。この特徴を有するヒドロゲルは、in vitro適用に有用であり得る。ペプチドへの構造修飾の導入および/または異なる剛性のヒドロゲルの調製により、血清タンパク質の非存在下で細胞接着を増強することができる。細胞接着が増強したヒドロゲルを、in vitroおよびin vivo適用のために使用することができ、細胞接着度がより低いヒドロゲルよりもin vivo適用により適切であり得る。
【0100】
いくつかの実施形態では、上記で概説の細胞接着を促進する非細胞傷害性ヒドロゲルを使用して、接着細胞の増殖を支持することができる。上記の3Hチミジンアッセイを使用して、増殖速度を評価することができる。所与のゲルについて、例えば、線維芽細胞についての48時間継続する速度実験の使用によって至適細胞播種密度を決定することができる。図17に関して、MAX1ヒドロゲルへの40,000個の線維芽細胞の最初の播種により、密集に到達するまで、2日間増殖が継続する。増殖を、任意の適切な期間(例えば、2日間またはより長い期間にわたり)継続させることができる。他の細胞型(例えば、骨芽細胞、神経細胞、幹細胞など)を含む速度実験の継続期間を、類似の様式で実験的に決定することができる。至適播種密度を決定した後、静止細胞をヒドロゲルに播種し、増殖細胞数を、3Hチミジン取り込みによる時間の関数として定量することができる。1つの実施形態では、光学密度での細胞を、10%ウシ血清を含む非標識培地の存在下で、24ウェル組織培養プレート中の予め形成されたヒドロゲルにプレートすることができる。三連で、細胞を、異なる期間(例えば、6、12、18時間など)増殖させることができる。各時間後、増殖培地を除去し、3Hチミジンを含む培地と置換することができる。細胞に3Hチミジンを3時間取り込ませることができる。非組み込みチミジンを洗い流し(3Hチミジンがもはや検出されなくなるまで洗浄物をアッセイすることができる)、シンチレーションカウンティングのために細胞を死滅させることができる。検量線(組織培養ポリスチレン上での所望の細胞の培養および血球計算板によって計数した細胞とシンチレーションカウントとの直接相関によって以前に決定することができる)との比較により、その時点でDNA複製を受けた細胞を定量することが可能である。いくつかの実施形態では、一定のヒドロゲル上での増殖細胞数は、ヒドロゲル表面の二次元領域内におそらく適合し得る数を超え得る。これらの場合、細胞を、ヒドロゲル足場の孔および流路に増殖させることができる。いくつかの実施形態では、本発明のヒドロゲルを、ヒドロゲル足場への細胞の増殖を促進するようにデザインすることができる。このヒドロゲル型を、例えば、時間の関数として三次元画像を収集することができるLSCMによる予め染色された細胞の増殖にしたがって容易に同定することができ、ヒドロゲル内部に移動した細胞が明らかに認められる。任意の適切な色素(例えば、Vybrant CFDA SE(Molecular Probes)(細胞の細胞質中に隔離され、娘細胞に伝えられる緑色蛍光プローブ(細胞生存率に影響を与えない)))を使用して、細胞を予め染色することができる。当業者は、細胞増殖を促進する所望の能力を有するヒドロゲルを同定するために、このアッセイ型を使用することができる。ペプチド構造およびそれによって得られるヒドロゲルの材料特性(剛性および足場形態など)の変化により、以下で考察しているように、細胞増殖を助長するヒドロゲルを生成することができる。
【0101】
上記アッセイは、二次元ヒドロゲル表面上接着および増殖の評価で有用であり、異なるヒドゲル足場が容易に比較可能であり、ほとんどの公開されている研究は二次元で実施されているので、結果を文献中の材料と直接比較することができる。図18に示すように、細胞の存在下での材料形成の誘発により、ヒドロゲル全体にこれらの細胞を三次元で組み込むことが可能である。細胞接着を促進する非細胞傷害性ヒドロゲル(例えば、上記で同定したヒドロゲル)を、三次元基質として使用することもできる。ヒドロゲル内部に隔離された非付着細胞を洗い流すことができないので、細胞付着は定量が困難であり得る。しかし、定性的細胞拡大アッセイを使用して、細胞拡大およびそれによる付着を測定することができる。細胞を、例えば、Vybrant CFDA SEで予め染色し、その後にヒドロゲルマトリクスに組み込むことができる。最初に組み込まれた細胞の細胞形態を、LSCM(非接着細胞は円形であり、接着した線維芽細胞は広がっている)によって三次元で視覚化することができる。さらに、色素が子孫(prodigy)に伝えられるので、細胞増殖を定性的に視覚化することができる。二次元研究のための上記と同一のアッセイを使用して、増殖速度を定量的に決定することができる。3Hチミジンはヒドロゲルマトリクスに容易に拡散し、DNA複製を受けた細胞でも利用可能なはずである。
【0102】
いくつかの実施形態では、本発明を使用して、細胞(例えば、細胞株)の接着および/または増殖を支持することができる。このような細胞または細胞株は、当業者に公知の任意の型であってよく、任意の目的のために(例えば、組織の生成または所望のタンパク質の発現のために)培養することができる。組織工学に関連する実施形態では、in vitro培養時のその高い能力に加えて、その結合組織および骨生成/治癒に関与するヒト細胞との関係に基づいて、モデル細胞株を選択することができる。この目的のための適切な細胞株の例には、NIH/3T3マウス線維芽細胞(ヒト結合組織の発達に関与する線維芽細胞モデルとして使用することができる不死化細胞株)およびラット頭蓋冠(MC3T3−E1サブクローン4)細胞(ヒト骨芽細胞モデルとして使用することができる)が含まれるが、これらに限定されない。両細胞株は十分に研究されており、重要な文献データベースにより、提案されたヒドロゲルの性能を以前に研究した材料を基準にして評価することが可能である。当業者は、MC3T3−E1細胞株の使用には限度がある可能性があることを承知している(その非分化線維芽細胞様表現型に戻す能力など)。いくつかの実施形態では、本発明のヒドロゲルを使用して、初代組織サンプル由来の細胞(例えば、頭蓋冠または骨髄由来の初代骨芽細胞)の接着および/または増殖を支持することができる。
【0103】
いくつかの実施形態では、本発明のヒドロゲルは、1つ以上の細胞および/または1つ以上の治療因子(例えば、医薬品)を含み得る。この目的のためのヒドロゲルを、動物モデル系で標準的技術を使用して同定することができる。動物への挿入前にヒドロゲルを予め形成することができるか、溶液の形態で挿入してin situでヒドロゲル化を誘発することができる。いくつかの実施形態では、生物学的に関連する刺激を介して材料形成を誘発するペプチドによってヒドロゲルを形成することができる。例えば、細胞および/または治療因子を含み得るペプチド溶液を、動物(例えば、ヒト)に挿入し、動物に対して内因的な1つ以上のトリガー(例えば、pH、イオン強度など)の結果としてヒドロゲル化することができる。他の実施形態では、外因性トリガー(例えば、電磁放射(例えば、光))を使用して、ヒドロゲル化を行うことができる。本発明の材料および方法を使用して、所望の剛性を有する足場を構築し、in vivoで使用することができる。このような足場は、細胞の接着および増殖ならびに/または治療因子の送達に最適な機械的特性を有する基質を提供することができる。
【0104】
いくつかの実施形態では、本発明は、ヒドロゲルの細胞適合性を予想する方法を提供する。例えば、上記の細胞適合性アッセイ由来の実験データを使用して、研究したヒドロゲル中に存在するペプチド構造と細胞適合性との間の相関関係を描写することができる。いくつかの実施形態では、細胞適合性と分子パラメーター(ペプチドの疎水性、β−ヘアピン鎖の長さ、および上記で考察した他のパラメーターなど)との間の相関関係を同定し、これを使用して、提案したヒドロゲルの細胞適合性を予想することができる。自己集合過程を理解および制御するために、分子パラメーターを変化させることができた。次いで、これらの相関関係により、反復デザイン過程における最適な材料特性および細胞適合性の両方を有する完全に新規のペプチドをデザインすることが可能である。
【0105】
特定の実施形態では、細胞接着エピトープをヘアピンベースのヒドロゲルに組み込んで、血清タンパク質の非存在下での細胞付着を増強することができる。また、増殖速度の増強を測定することが出来る。以前に記載の二次元の定量的細胞付着および増殖アッセイを使用することができる。1つの実施形態では、細胞接着エピトープを、ヒドロゲルに組み込むことができる。当業者は、組織工学材料内の細胞接着エピトープの添加によって細胞の付着および/または増殖が促進されるという明らかな実験的証拠が存在することを認識している(例えば、Urry,Angewandte Chemie−International Edition in English 1993,32,819−841,David,et al.,Bioconjugate Chemistry 2001,12,890−899,Rezania & Healy,Journal of Orthopaedic Research 1999,17,615−623,Cook,et al.,Journal of Biomedical Materials Research 1997,55,513−523,Burdick & Anseth,Biomaterials 2002,23,4315−4323,Houseman & Mrksich,Biomaterials 2001,22,943− 955,Kao,et al,Journal of Biomedical Materials Research 2001,55,79−88,およびSchmedlen,et al.,Biomaterials 2002,23,4325−4332を参照のこと)。1つ以上の細胞結合リガンドを、各ヘアピンに共有結合し、ヒドロゲルマトリクスに自己集合させることができる。1つ以上の配列エピトープを、ペプチドの任意の位置(例えば、1つ以上のリジン側鎖またはペプチド(例えば、MAX1)の一次配列内)に組み込むことができる。上記で考察した自己集合に悪影響を与えずにさらなるエピトープを許容することができる任意の位置を使用することができる。MAX1の構造内に、1つ以上のエピトープエピトープを組み込んで、示されたエピトープ濃度を正確に制御することができる。さらに、エピトープのコピーは同一であり得るか、同一性が変化し得る。例えば、相乗的リガンド結合部位は、α5β1中に存在することが公知であり、フィブロネクチンに対する変異誘発実験は、RGDがSDVおよびRNSエピトープに隣接したより有効なリガンドであることを示す(Ruoslahti,Annual Review of Cell and Developmental Biology 1996,12,697−715)。したがって、いくつかの実施形態では、多様なエピトープに富むヘアピンを調製し、これを使用して、細胞の接着および増殖を促進することができる。いくつかの実施形態では、1つ以上のインテグリン結合リガンドを組み込むことができる。このようなインテグリン結合リガンドは、線維芽細胞に特異的な細胞外マトリクスおよび骨芽細胞に特異的な骨シアロタンパク質由来のエピトープに基づき得る。ヒドロゲルと共に使用すべき細胞株と相補的であるように組み込むべきエピトープを選択することができる。
【0106】
1つの特定の実施形態では、3T3線維芽細胞との使用に適切なヒドロゲルを調製することができる。このようなヒドロゲルは、α5β1に結合することが公知の1つ以上のRGDエピトープを組み込むことができる。このインテグリンは、健康なヒト歯周結合組織から培養した線維芽細胞中に存在することが示されている(Hakkinen,et al.,Biochimica Et Biophysica Acta−Molecular Cell Research 1994,1224,33−42)。特定の実施形態では、1つ以上のペプチドエピトープ(TRGDSP(配列番号15)、RGDG(配列番号16)、RGDY(配列番号17)、およびRGDW(配列番号18))を、ペプチドの任意の位置に組み込み、これを使用して、本発明のヒドロゲルを調製することができる。各エピトープの1つ以上のコピーを単独で組み込むか、他のエピトープの1つ以上のコピーと組み合わせて組み込むことができる。
【0107】
別の特定の実施形態では、MC3T3骨芽細胞との使用に適切なヒドロゲルを調製することができる。このようなヒドロゲルは、エピトープFHRRIKA(配列番号19)(骨芽細胞表面上に示されたヘパラン硫酸プロテオグリカンに結合することが公知の骨シアロタンパク質由来のエピトープ)の1つ以上のコピーを組み込むことができる。他の実施形態では、本発明のヒドロゲルは、骨芽細胞接着に正の影響を及ぼすことが公知のさらなるエピトープ(GRGDSPY(配列番号20))を組み込むことができる。いずれかのエピトープの1つ以上のコピーまたは組み合わせた両エピトープの1つ以上のコピーを使用してヒドロゲルを調製することができる。Rezania & Healy(前出)は、RGD含有ペプチドおよびFHRRIKAを共に使用した場合、骨芽細胞増殖の増加を認めた。
【0108】
いくつかの実施形態では、細胞の結合および/または増殖の増強に適切なエピトープを、可溶性エピトープを使用した競合結合アッセイを使用して同定することができる。例えば、細胞の付着および/または増殖を、特異的リガンド結合事象によって媒介した場合、付着または増殖の増強は、添加した可溶性リガンドの濃度の増加によって競合的に阻害されるべきである。さらに、有意に増強させるRGD配列を含むペプチドについて、RGD配列中の1−アスパラギン酸残基をd−アスパラギン酸に置換したコントロール配列を調製する。このサンプルの立体化学の変化により、インテグリン結合が完全に悪影響を受けることが示された。
【0109】
本発明はまた、剛性が変化したヒドロゲルを含む。ゲルの剛性の変化を使用して、細胞−材料相互作用、詳細には、細胞の接着および増殖を調整することができる。剛性が変化する一方でペプチドの一次配列が一定に保持されたヒドロゲルを調製することができる。1つの特定の態様では、ペプチドMAX1を使用して、剛性が変化したヒドロゲルを調製することができる。MAX1ヒドロゲルの剛性を、ペプチド折り畳み誘発条件(すなわち、折り畳み温度)を変化させる一方でペプチド濃度を一定の保持することよって変化させることができる。予備データに基づいて、利用しやすい貯蔵弾性率(ゲル剛性)は、数Pa〜10,000Paの範囲である。異なる温度で形成されたMAX1ゲルは、37℃(例えば、細胞培養条件)での再平衡化後でさえもその剛性を保持する。剛性が変化したヒドロゲル上の二次元の細胞の接着および増殖をその後に観察することにより、任意の特定の細胞型(例えば、ニューロン材料、幹細胞、線維芽細胞−材料、および/または骨芽細胞−材料)の至適基質剛性を決定することができる。この目的のためにMAX1を使用することができるにもかかわらず、本明細書中に記載の任意のペプチドを使用してヒドロゲルの剛性を変化させることができる。ゲルの剛性の変化を使用して、ヒドロゲルの細胞適合性を至適化することもできる。
【0110】
本明細書中に記載の方法および適用の他の適切な修正および適合が明白であり、本発明またはその任意の実施形態の範囲を逸脱することなく修正および適合することができることが関連分野の当業者に容易に明らかである。ここに本発明を詳細に説明しているが、以下の実施例を参照することにより本発明がより明確に理解される。実施例は例示のみを目的として記載し、本発明を制限することを意図しない。
【実施例】
【0111】
(実施例1)
塩の存在によって誘発される分子内折り畳み事象は、生理学的条件下でのヒドロゲル網目構造へのβ−ヘアピンペプチドの集合を誘導する。pH7.4(低イオン強度溶液条件)では、希薄で均一なペプチド溶液(2重量%以下)は、純粋の粘度を示す。円偏光二色性分光法は、pH7.4且つ塩の非存在下ではペプチドは折り畳まれないことを示す。溶液のイオン強度の上昇により、ペプチド内の荷電アミノ酸間の静電相互作用をスクリーニングし、β−ヘアピン高次構造が採用される。折り畳まれたβ−ヘアピン分子は、疎水性崩壊および水素結合によって三次元ヒドロゲル網目構造に超分子的に集合する。FTIRおよびX線散乱データは、これらのヒドロゲルがβ−シートに富むことを示す。動的振動流動学測定は、得られた超分子構造が弾性材料を形成し、その構造(すなわち、剛性率)を、塩濃度および温度によって調整することができることを示す。ヒドロゲルの貯蔵弾性率は、塩濃度の増加と共に増加する。生理学的条件下での増殖培地においても強固なヒドロゲル化が認められる。透過型電子顕微鏡法により、ヒドロゲルの弾性は、半可動性微小繊維集合体からなる網目ナノ構造に起因することが明らかである。
【0112】
ヒドロゲルは、組織工学および薬物送達への適用で広く使用される集合的に重要な生体材料クラスである。自己集合ストラテジーは、環境条件(温度、pH、電場、イオン強度、または光など)に応答し得る所望のナノ構造およびミクロ構造を有する新規のヒドロゲル材料を構築するために正確に制御する。pHおよび温度トリガーを使用したβ−ヘアピンペプチドのヒドロゲル化が示されている(上記のPochan 2003およびSchneider 2002を参照のこと)。これらの分子は、pHおよび温度依存性分子内折り畳み事象を示し、可逆的に分子間で自己集合してヒドロゲル足場を形成する。一般に、ペプチド折り畳み事象の制御により、所望の刺激で自己集合を受ける応答性材料をデザインすることが可能である。さらに、自己集合過程を制御して、所望の形態的性質および機械的性質を操作することができる。下記の1つの特定の実施形態では、β−ヘアピン分子内折り畳み事象を誘導するためのトリガーとして塩濃度を使用し、その後に超分子弾性網目構造に自己集合する。折り畳み速度(すなわち、最終ゲルの流動学的性質)を、ペプチド溶液のイオン強度によって調整することができる。重要なことには、これらの物理的ヒドロゲルの形成は、いかなる化学的架橋も含まず、生理学的温度およびpHで実施することができる。
【0113】
刺激に応答する(例えば、物理的または化学的環境条件の変化に応答したゾル−ゲル転移または膨潤転移)高分子ゲルを作製するための異なるストラテジーが文献で報告されている。温度およびpHは最も一般的な刺激であるが、相転移、自己集合、またはポリマーの高次構造の変化によってゲル化を誘発するためにイオン強度も使用される。塩複合体の形成により、ブロックコポリマーの溶液および自己集合の性質を変化させ、球状または蠕虫様ミセルのような自己集合構造を形成することができる。例えば、塩の型および量により、凝集特性を変化させ、それにより、Pluronicベースのブロックコポリマー系によって形成された網目構造の弾性が変化することが示されている。LCST型相転移による熱応答性を示すことが周知のN−イソプロピルアクリルアミドなどの合成ポリマーの化学ヒドロゲルも塩誘導性体積相転移を示す。比較的高い塩濃度(またはLCSTより高い温度)で、疎水性相互作用によって化学的に架橋されたポリマー鎖が主に沈殿するか沈殿してゲルが崩壊する。イオン強度およびpHに起因するポリ(アクリル酸)ヒドロゲルおよびポリ(メタクリル酸)ヒドロゲルなどの疎水性網目構造の膨潤により、応答性生体材料が生成される可能性があるかどうかを調査した。
【0114】
生体高分子のゲル化挙動(ゼラチン、ポリサッカリド、およびβ−ラクトグロブリンなど)は、文献で広く研究されている。熱処理への適用により、これらの高分子が二次構造(高次構造)への相転移を受け、それにより、分子間網目構造を形成することが周知である。例えば、ゼラチンは、漸減温度での三重らせんの物理的連結点の形成によってヒドロゲルを形成する。ゼラチンゲルの流動学的研究により、粘弾性が処理条件(冷却速度、過冷却度、および濃度など)に非常に依存することが明らかとなった。しかし、生体高分子溶液のポリ多価電解性により、イオン強度も集合特性を制御するための重要なパラメーターである。生体高分子ゲル網目構造(ペクチン、β−ラクトグロブリン、ウェラン(welan)、およびゼラチン)に対するイオン強度の効果は、食品加工および薬学的適用におけるその重要性により、十分に研究されている。一般に、これらの溶液のイオン強度の増加により、分子間塩橋の物理的架橋の形成によって網目構造の弾性が増加する。
【0115】
応答性生体材料としてのその潜在的適用のために、オリゴペプチドおよびポリペプチドの十分に定義された配列を有する生体高分子の刺激応答性自己集合経路を模倣することは非常に興味深い。イオン強度およびpHによって誘発されたオリゴペプチドのβ−シートが豊富なβ−アミロイド様構造への自己集合は、生体模倣材料形成ストラテジーとして調査されている。例えば、Caplan et al.(Biomacromolecules 2000,1,627−631)は、オリゴペプチドの自己集合および得られたゲル(疎水性アミノ酸残基が正電荷または負電荷のアミノ酸に配列が変化している)の特性を、溶液のイオン強度によって変化させることができることを示した。さらに、エラスチン模倣ペプチド(McMillan and Conticello,Macromolecules 2000,33,4809−4821)、コイルドコイルタンパク質合成ポリマードメインを有するハイブリッド分子(Wang,et al.,Nature 1999,397,417−420)、およびさらなる多価電解質セグメントを有するロイシンジッパーポリペプチドドメイン(Petka et al.Science 1998,281,389−392)は、適切なイオン強度での温度またはpHを用いてゲル化される応答性ヒドロゲルを形成する。
【0116】
以下に詳述した特定の実施形態では、生理学的条件下での低ペプチド濃度(2重量%)の20アミノ酸β−ヘアピン分子(MAX1)の分子内折り畳みおよびその後の分子間自己集合を介した塩誘発性物理的ヒドロゲル形成を示す。分子は、II’型ターンとして公知の構造を採用するテトラペプチドターン配列(−VDPPT−)と連結したバリン(V)アミノ酸残基(イソプロピル水素側鎖)およびリジン(K)(第一級アミンキャッピング側鎖)アミノ酸残基の交互配列から構成される2つの鎖からなる。Vが無極性残基である一方で、Kは正電荷であり、ほぼ生理学的条件のpH値(約7.4)で親水性を示す。K残基の電荷および無極性V残基に起因して、静電気力および疎水性相互作用は、分子の折り畳みを制御するために使用することができるおもな分子内パラメーターである。折り畳み状態では、分子表面は親水性を示し、全V残基はヘアピンのある表面上に存在し、K残基は別の表面上に存在する。一旦分子が折り畳まれると、側面の分子内水素結合および表面上の疎水性相互作用の両方によって自己集合が駆動される。したがって、詳細には、これらの分子を、最初に折り畳まれ、その後に系のゲル特性が生じるβ−シートが豊富な四次構造に自己集合するようにデザインする。自己集合構造の最終的な弾性を、塩濃度によって調整することができる。さらに、表面上の疎水性会合により、ゲル化速度制御のパラメーターとして温度を使用することもできる。したがって、これらの分子を、溶液条件(イオン強度、温度、pH)が生理学的レベルに調整された場合にゾル−ゲル転移を示し、組織工学へ適用するために有意に使用することができるようにデザインする。
【0117】
サンプル調製:ABI 433Aペプチド合成機およびHBTU/HOBT活性化を使用した自動化Fmocベースの固相ペプチド合成によってアミド樹脂上にMAX1を調製した。ペプチド調製の詳細は、他で記載されている(上記のSchneider)。最初のDI水への凍結乾燥ペプチドの溶解およびその後の緩衝液および塩含有溶液の添加によって達成された所望の最終溶液条件によってヒドロゲルを調製する。TRIS緩衝液を使用したX線研究以外は、pH7.4での溶液の緩衝化のためにビス−トリスプロパンを使用する。
【0118】
円偏光二色性:Avivモデル215分光偏光計を使用して、CDスペクトルを収集した。20℃または37℃で測定した。0mMと150mMとの間のKF溶液を含む2重量%のMAX1(pH7.4、50mM BTP)についての190nmと260nmとの間の波長スキャンを、0.01mmの取り外し可能な石英セル中で行った。DI水および緩衝液へのペプチド溶解から2時間後にスペクトルを取った。時間依存性研究のために、平均残基楕円率(θ)を、218nmで測定した。式θ=θobs/l/c/r(式中、lはセルの光路長であり、cは濃度であり、rは残基数である)からθを計算した。
【0119】
赤外分光法:Nicolet Magna−IR860分光計にてIRスペクトルを取った。0.1M HClで1回およびD2Oで2回のペプチドのTFA塩の凍結乾燥によって、重水素化MAX1*nDClを調製した。サンプルを温度調節された水浴中に20℃で2時間保持し、その直後に測定物を室温で光路長30μmの亜鉛−セレニドフローセル中に入れた。装置を、1cm−1の分解能で操作し、記録したスペクトルは平均100スキャンであった。
【0120】
広角X線散乱:National Synchrotron Light Source,Brookhaven National Laboratory,beamline,X10AにてヒドロゲルのX線スペクトルを収集した。脱水を回避するために測定を行う直前にヒドロゲルをKaptonテープに塗布した。二次元Bruker CCDアレイ上に10分間データを収集した。ペプチド溶液を、125mM TRISで緩衝化した。室温で測定した。
【0121】
流動学:直径25.0mmの平行平板およびギャップ距離が0.5mmの歪み制御したRheometrics ARES流量計にて動的時間および周波数掃引実験を行った。凍結乾燥ペプチドを、10℃の脱イオン水および緩衝液または細胞増殖培地溶液に溶解して、サンプルを流量計にロードする前の折り畳みおよびゲル化を抑制した。サンプルをロードした後、温度を20℃または37℃のいずれかに上昇させた。標準的な低粘度鉱物油を使用して、プレート側面から隔離して蒸発を抑制した。コントロール実験は、鉱物油が流動学的測定に影響を与えないことを示した。歪み掃引実験を行って線形粘弾性体制を決定し、全流動学的実験を行った。6rad/sの周波数および1%の歪み用いて動的時間掃引実験を行った。0.1〜100rad/sの周波数および5%の歪みを使用して周波数掃引実験を行った。
【0122】
透過型電子顕微鏡法:非常に薄いヒドロゲル層を、カーボンコーティングした銅のグリッドに貼り付けた。2wt/v%の酢酸ウラニル溶液のグリッドへの滴下によって、サンプルを陰性染色した。過剰な溶液を濾紙で吸い取り、その後にサンプルを乾燥させた。塩および/または緩衝液の結晶の形成を回避し、各微小繊維を明確に画像化するために、2重量%のペプチド濃度のヒドロゲルを、DI水で約0.1重量%に希釈した。溶液中に微小繊維を均一に分散させるために、チップ超音波処理機を使用して穏やかに超音波処理を行った。Gatan CCDカメラおよびKodakネガフィルムを備えたJEOL 2000−FX透過型電子顕微鏡にて加速電圧200kVでヒドロゲルナノ構造の明視野画像を取った。
【0123】
図21Aは、pH7.4および20℃の2重量%のMAX1溶液の異なる塩濃度についてのCDスペクトルを示す。DI水および緩衝液でのMAX1溶解から2時間後に示した両スペクトルを取った。150mM KFを含むペプチド溶液が218nmで最小であることは、2重量%のゲル化濃度で、MAX1が折り畳まれてβ−シートに富む二次構造を採用することを示す。しかし、塩の非存在下での同一pHで2重量%のMAX1は2時間後でさえも有意に均一な二次構造を示さず、塩を添加しないペプチドは折り畳まれないままであることを示した。
【0124】
塩の存在下でのペプチド分子のβ−ヘアピン構造への折り畳みおよびその後のβ−シートの形成もFTIR分光法によって確認した。pH7.4および400mM NaCl、1580cm−1と1700cm−1との間のMAX1溶液のFTIRスペクトルを図21Bに示す。D2O中の不規則なペプチドについてのアミドIバンドは、1645cm−1付近に集中している。1644cm−1から1614cm−1へのアミドIバンドのシフトにより、400mM NaCl溶液中でペプチドがβ−シート高次構造であることが示唆される。1680cm−1のスペクトル中の弱いバンドは、逆平行β−シート構造を示し得る。NaClを含まないMAX1溶液については、プロット範囲で非常に広いスペクトルが得られる。1644cm−1の強いバンドにより、ペプチドが不規則な状態であることが示唆される。しかし、1614cm−1の弱いバンドは、溶液中の少量の折り畳まれたβ−ヘアピン分子の存在を示す。本発明者らは、いくつかのMAX1の折り畳みは、溶液中の過剰な緩衝液の塩および比較的高濃度のペプチドの両方に起因すると考える。ペプチド濃度がβ−シートの形成速度に影響を与えることが示された。したがって、ペプチド濃度が高く且つゲル化しない状況では、この少量のβ−シートアミドIシグナルは重要ではない。
【0125】
リジン残基状の第一級アミンによってMAX1はpH7.4で正味の電荷を有するので、データにより、鎖間の静電反発力によってMAX1は折り畳むことができないと示唆される。したがって、リジン残基の正電荷をCl−イオンによってスクリーニングした場合、分子内折り畳みが支持されてβ−ヘアピン構造が形成される。塩濃度の増加により、残基の疎水性会合を駆動することもでき、疎水性会合は折り畳みおよび自己集合にも寄与するはずである。CDデータおよびFTIRデータはどちらも、折り畳み機構が溶液のイオン強度によって誘発されることを示す。折り畳みをpHおよび温度によって誘発することができることも示されている(上記のPochan,Schneider)。塩の効果と類似して、刺激としてpHを使用した場合、高pHでリジン残基が脱プロトン化しMAX1が折り畳まれる。折り畳まれた場合、分子間水素結合および疎水性相互作用によってMAX1はより高次の構造に自己集合することができる。折り畳み速度、集合した凝集体の構造、および得られたヒドロゲル網目構造の性質を以下で考察する。
【0126】
X線散乱技術を使用して、最終ヒドロゲル中で結晶学的β−シート構造が認められる。図21Cは、3重量%のMAX1および150mMNaClからなるヒドロゲルについての広角曲線を示す。散乱ピークは、4.7Åに相当する。これはβ−シートに富む構造における鎖間距離の特徴的なサインである。ヒドロゲルを乾燥させずに測定したので、水構造に起因する高角度での散乱バックグラウンドの寄与が顕著である。
【0127】
動的振動技術によって観察したところ、分子内折り畳み機構によって形成されたヒドロゲルは、低濃度のMAX1ペプチドでさえも強固な粘弾性を示す。図22Aは、20、150、および400mMのNaCl濃度の2重量%のMAX1ペプチド溶液についての10−1〜102rad/sの周波数掃引測定を示す。周波数掃引測定の前に、ペプチド溶液を、流量計中で2.5時間ゲル化した(図22B)。400mMNaClを含むMAX1溶液について、平衡貯蔵弾性率(G’)は、約3000Paである。塩濃度の減少により、G’値および損失剛性率(G’’)が減少した。20mM NaClしか溶液中に存在しない場合、低剛性(約100PaのG’)のヒドロゲルが形成される。NaClを添加しないpH7.4のMAX1は、G’値が約1Paで3時間後でさえもいかなるゲル化も示さなかった(適用した歪みに対するサンプルの応答はごくわずかである。トルク値は装置の検出限界未満であり、測定値は重要ではない(示さず))。塩を含む全3サンプルについて、G’値は、G’’値よりも少なくとも1桁高い。また、全NaCl濃度について、研究された範囲では、G’値は本質的に周波数と無関係であり、低周波数ではG’とG’’との間に交差点は存在しない。これらの特徴は、架橋網目構造の明らかなサイン(signature)である。図22A中の周波数掃引データにより、これらの溶液が共有結合で架橋したポリマーゲルに類似の性質を有する強固な固体様ヒドロゲルを形成することが示唆される。
【0128】
図21中の分光法によるデータにより、塩を含まない溶液中で、リジン残基の静電反発力がペプチドの非折り畳みを維持し、それにより、網目構造への自己集合が阻止されることが示唆される。しかし、一旦分子が折り畳まれると、自己集合が起こり得る。全バリン残基が折り畳み状態の分子の片側に位置づけられるので、表面で疎水性二量体化が起こり得る。その後にさらなる分子間疎水性相互作用および水素結合が起こり、相互連結された網目構造が形成され得る。重要なことには、図22Aに示すように、異なる塩濃度のヒドロゲルの剛性の相違により、イオン強度によって自己集合を誘発することができるだけでなく、培地のイオン強度によって得られたナノ構造を予想通りに変化させることができることが明らかとなる。図22Bは、2重量%の同一のペプチド濃度についてのMAX1溶液の自己集合時のG’値の増加を示す。この実験は、異なる塩濃度では自己集合の速度および網目構造形成が異なることを明らかに示す。400mM NaClを含むMAX1溶液は、自己集合過程の最初の2分間でG’値が迅速に増加する一方で、20および150mM NaCl溶液で比較的低速で硬化することを示す。実際、20mM NaCl溶液のG’値は3000秒間で有意に変化せず、2〜3Paのままである。この遅延時間は、150mMではより短く(約1000秒)、400mMの塩溶液では示されない。図22Bにより、折り畳み速度が速いほどより高いイオン強度によって自己集合してより強固なゲルが得られることが明らかに示唆される。同一のペプチド濃度の溶液間の剛性の大きな相違は、おそらく、より速い折り畳みおよび集合速度によるより高度に架橋された網目構造に起因する。
【0129】
弾性が得られるヒドロゲルのナノ構造を、透過型電子顕微鏡法(TEM)によって研究した。図23Aは、400mM NaClを含むpH7.4で形成された2重量%のMAX1ヒドロゲル網目構造のTEM画像を示す。顕微鏡写真から、ナノスケールでの高度に相互連結した微小繊維網目構造が明らかである。TEM顕微鏡写真で直接認められないが、自己集合時に形成されたほとんどの接合点が微小繊維と交差し、長い非交差微小繊維の単純な縺れではないと考えられる(しかし、縺れは剛性率に確かに寄与している)。微小繊維間のこれらの架橋点は、元来は共有結合していないが、不変であり、水素結合および疎水性相互作用の両方によって形成される。図22Aに示す周波数掃引データもこの見解を支持している。G’は周波数に対して感受性がなく、G’’よりも非常に高く、G’’とG’との交差は認められない。画像化前の水の蒸発および比較的高い塩濃度のために、2重量%の構造は非常に密集しており、網目構造のいくつかの部分が沈殿した塩に埋め込まれている。したがって、ヒドロゲルを、DI水で約20倍(〜0.1重量%のペプチド)に希釈し、直ちにTEM画像化のためのサンプルを調製した。図23Bは、より希薄な微小繊維集合体を示す。微小繊維の輪郭の長さは、およそ数μmである。サンプル調製時の乾燥は微小繊維の軸に沿って高次構造を変化させ得るにもかかわらず、顕微鏡写真により、自己集合した微小繊維が半可動性を示すことが示唆される。重要なことには、微小繊維の幅が単分散サイズの約3nmである。提案された局所自己集合構造および寸法を、図24に示す。Insight IIモデリングは、折り畳まれたペプチド鎖の軸の長さは32Åであり、バリン面からリジン面までの距離が約10Åであることを示す。幅が約3nmの微小繊維は、これらの分子の寸法と非常に良好に一致する。CDデータと共に、TEMデータにより、自己集合過程時にMAX1が折り畳み状態にあることが強く示唆される。図24中の提案された構造では、簡潔にするために、β−ターンを微小繊維二重層の同一側面に存在するように示す。
【0130】
CDおよび流動学による結果は、β−ヘアピン分子の折り畳みおよびその後のゲル化をpH7.4での塩濃度の変化によって誘発することができることを明確に示す。この応答型により、生理学的条件下でこれらの材料を使用する機会が得られる。したがって、これらの分子の自己集合およびゲル化挙動を、生理学的温度(37℃)で研究した。MAX1の折り畳みおよびβ−シート形成が温度依存性であることが示された。CD研究により、MAX1について、不規則なランダムコイル状態から折り畳まれたβ−シート構造までの転移温度は、pH9および低濃度の塩(20mM)で約25℃であることが示された。この転移は、集合ペプチドの疎水性の変化によってより高い温度に調整することができることが示された。
【0131】
イオン強度によって誘発された自己集合に対する温度の効果を観察するために、20℃および37℃での動的掃引時間実験によって2.5時間にわたってゲル化をモニタリングした。図25Aでは、G’の変化を、pH7.4で150mMのNaCl溶液を含む2重量%のMAX1の時間の関数としてプロットする。両方の場合において、最初にMAX1溶液を10℃に保持して折り畳みを抑制し、その後に測定した。37℃でのG’の瞬間的な増加は、ペプチドが自己集合時に網目構造を直ちに形成したことを示す。対照的に、20℃では、ヒドロゲルの弾性応答の増加はより遅く、自己集合の最初の15分間についてはG’の増加は小さいことを示す。この挙動は、室温で粘性様挙動を示す一方で、体温および身体の塩濃度に曝露された場合に(例えば、in vivoでの注射)迅速なゲル化応答を示す溶液が望まれる潜在的用途で有利であり得る。図25Aは、生理学的に関連する条件下で(例えば、pH7.4、37℃)、有用な剛性を示す材料が得られるゲル化が約10分間で起こることを明確に示す。37℃での2.5時間のゲル化後に行った周波数掃引測定(図25B)により、G’値が約2000Paの強固なヒドロゲル(G’>G’’)が形成されることが示される。両温度でのゲル化時のG’’値(示さず)は一定であり、ゲル化過程を通してG’値よりも常に十分に低かった(1桁を超えて低い)。全サンプルでは、ゲル化の初期段階でさえも、G’>G’’であった。生体高分子のゲル化においても類似の挙動が認められた。イオン強度の効果に類似して、より高い温度で集合したより強固なゲルの形成は、ナノスケールでの構造の相違に起因し得る。より速い折り畳みおよびそれによる自己集合速度により、接合点の数においてより緻密であり、それによりG’値がより高い網目構造を形成することができる。このデータは、温度または塩濃度のいずれかによってMAX1ヒドロゲルの剛性を容易且つ予想通りに制御することができることを示す。
【0132】
MAX1ペプチドの折り畳みおよびβ−シート形成速度を、CDによって研究した。図25Cは、pH7.4で150mMのKF溶液を含む2重量%のMAX1についての20℃および37℃にて218nmで測定した楕円率の変化を示す。光学的にサイレントであり、且つCD測定を補助するので、電解質としてKFを使用する。CDデータにより、37℃での初期段階の自己集合β−シート形成が非常に迅速であることが明らかとなる。最初の5分間の折り畳み速度が有意に速く、15分後にプラトー領域に達する。しかし、20℃での折り畳み速度は遅い。β−シート形成およびそれによる自己集合の速度の相違は、図25に示す流動学的測定値と一致する。37℃では、自己集合の初期段階でG’値およびθ218値の変化は非常に迅速である一方で、20℃では、両値の変化速度は遥かに遅い。したがって、37℃で形成されたゲルのより高い貯蔵弾性率は、折り畳み速度の速さに起因し得る。自己集合の初期段階で、折り畳み速度が速いことによってβ−シートが豊富な微小繊維の成長のための核生成部位がより多くなり、その結果、より接合点の多い網目構造が形成される。この作用機構はこれまでに得た全データと一致する。
【0133】
生体材料への適用のために、生物学的に関連する条件に対するMAX1の応答を理解することが重要である。したがって、無血清DMEM細胞増殖培地においてMAX1のゲル化を研究した。図26Aは、37℃での細胞増殖培地中(哺乳動物細胞培養条件)でのMAX1のゲル化時のG’およびG’’を示す。2時間のゲル化の終了後、G’およびG’’値は、それぞれ2300Paおよび50Paであった。2時間後のG’値の増加速度が依然として有意であることが認められる。ペプチド濃度およびG’値を増加させる蒸発の影響を排除するために、2時間のゲル化後に動的掃引時間実験を中止した。(最終値を示さないG’値のこの継続的増加は、ゼラチンゲルでも認められた)。図26Bに示す周波数掃引データは、MAX1−細胞増殖培地溶液が約2500PaのG’値で強固なゲルを形成することを示す。培地誘導性ヒドロゲルの周波数掃引データの性質(G’>10×G’’、G’は周波数と無関係)は、図21Aに示す性質と類似している。有意な剪断に対するヒドロゲルの応答を、ゲル化溶液に対する大規模の歪み(6rad/sで1000%)の適用後のG’の回復をモニタリングすることによって研究した。適用した歪みが線形体制の十分に外側であるので、G’およびG’’値は示されない。図26Cは、剪断の停止およびその直後の6rad/sで5%の歪みの適用後、G’がその初期値のほぼ50%を即座に回復することを示す。回復データを、ゲル化データ(図26A)と比較した場合、G’の増加速度が初期ゲル化時の速度よりも早いことが認められる。これにより、大規模の歪みの適用時に物理的に架橋された自己集合網目構造が破壊され、接続性が減少し、それにより、材料の弾性が減少することが示唆される。剪断の中止後、有意な網目構造の剛性の即座な回復で明らかなように、網目構造が即座に再修復することができる。この実験は、これらのヒドロゲル処理可能であり、且つ機械的外力による破壊後に初期流動学的性質を回復することができることを示す。この処理および回復の容易さは、組織工学への適用(例えば、in vivo注射)に有利であり得る。
【0134】
β−ヘアピンペプチドの折り畳みおよび集合によって強固なヒドロゲルが形成される。MAX1は、生理学的pH(7.4)でランダムコイル高次構造で存在し、溶液への塩の添加により、β−シートに富む自己集合した構造を形成する。流動学的データは、ペプチド溶液が、G’’よりも少なくとも1桁高いG’でゲルを形成することを示す。さらに、G’が周波数と無関係であることは、網目構造が化学的に架橋したポリマーゲルの弾性と類似していることを示す。ヒドロゲルの自己集合速度および貯蔵弾性率を、ペプチド溶液のイオン強度によって調整することができる。網目構造は、おそらく表面の疎水性相互作用および水素結合に起因する物理的接合点によって互いに架橋している緻密な微小繊維集合体から構成される。微小繊維の幅は、約3nmであり、この寸法は、自己集合状態のヘアピンの折り畳み状態と良好に一致する。塩誘発性自己集合およびそれによるゲル化の性質も温度によって調整される。37℃で、β−シート形成およびゲル化の速度は、より強固なゲルが得られる20℃よりも速い。さらに、MAX1は、生理学的条件下での細胞増殖培地で強固且つ処理可能なヒドロゲルを形成する。したがって、適切にデザインされたペプチドの分子内折り畳みおよびそれによる超分子構造への分子間集合を塩によって誘発し、調製可能な剛性率を有するヒドロゲルを得ることができる。
【0135】
上記発明を、明示し、且つ理解する目的でいくらか詳細に説明しているが、この開示を読み取ることにより、当業者は、本発明の範囲および添付の特許請求の範囲を逸脱することなく形態および詳細の種々の変更を行うことができることを認識している。本明細書中で引用した全ての特許および刊行物は、その全体が本明細書中で参考として援用される。
【図面の簡単な説明】
【0136】
【図1】図1は、本発明のヒドロゲルを形成するためのペプチドの自己集合の略図を示す。非折り畳みペプチドは、他の折り畳み様ペプチドと自己集合される高次構造に個別に折り畳まれるように誘導される。折り畳み過程は、所望の刺激への曝露に起因し、自己集合過程により、相互連結した線維性のヒドロゲル網目構造が得られる。
【図2】図2は、本発明の所望の特徴を有するヒドロゲルの調製で使用されるペプチド中で変化し得るいくつかの要因の略図を示す。変化し得る特定の分子要因には、以下が含まれる。
【図2A】図2Aは、分子内折り畳みに影響を与える操作を記載し、以下が含まれる:1)各ペプチドのリジン残基の第1級アミン化学的官能基との間の静電相互作用、2)各ペプチドのペプチド骨格アーム間の疎水性のファンデルワールス相互作用、3)各ペプチド骨格のアーム間の水素結合、4)各ペプチドのターン性向(すなわち、特定の刺激と比較した折り畳みに対するペプチドの「強さ」)。
【図2B】図2Bは、分子間自己集合に影響を与える操作を記載し、以下が含まれる:5)隣接する折り畳みペプチドバリンリッチ表面間の疎水性のファンデルワールス相互作用、6)隣接ペプチドのペプチド骨格アーム間の疎水性のファンデルワールス相互作用、7)隣接ペプチドのペプチド骨格アーム間の水素結合。
【図3】図3Aは、本発明で使用されるペプチドの構造を示す。図3Bは、本発明のヒドロゲルの可逆的形成の略図を示す。詳細には、ペプチドMAX1は、酸性または中性pH溶液条件下では組織化されていない非折り畳み構造である。塩基性溶液条件に曝露された場合、MAX1ターン配列中のトランスプロピルアミド結合は、各ペプチドのアームを図2Aに列挙した相互作用によって安定化された並行高次構造に押し進める。したがって、自己集合は、図3Bに記載の相互作用によって起こる。得られたヒドロゲルを酸性pH条件に曝露した場合、ペプチドは折り畳まれずに分解されて希薄な低粘度の水溶液に戻る。
【図4】図4は、本発明にしたがって調製したヒドロゲルから得た実験の結果を示す。図4Aは、150μM MAX1溶液(pH9.0、125mMホウ酸、10mM NaCl)の時間依存性far UV−CDスペクトルを示す。図4Bは、同一条件下での150および250μM MAX1溶液についての時間関数としてモニタリングした[θ]216を示す。図4Cは、無作為なコイル−シート折り畳み/自己集合平衡が可逆性であることを示すMAX1の300μM溶液の時間およびpHの関数としてモニタリングした[θ]216を示す。全実験を、20℃で実施した。
【図5】図5は、FTIR(pH5.5(軌跡a)およびゲル化後のpH9.0(軌跡b)1重量% MAX1)を示す。(a)中の1664から(b)中の1615までのアミド1ストレッチにおける大きなシフトは、β−シート構造の成長を強く示す。
【図6】図6は、pH9での2重量%MAX1ヒドロゲルに関する流動学データを示す。図6Aは、貯蔵剛性率(G’、黒塗りの記号)および損失剛性率(G’’、白抜きの記号)の増加による時間の関数としての6Hzで1%染色でモニタリングした短期ヒドロゲル化を示す。図6Bは、周波数掃引データを示す。周波数に対する剛性率の非感受性(insensitivity)は、ペプチド折り畳みおよび自己集合による強く架橋した強固なヒドロゲルを示す。図6Cは、集合後過程(例えば、シリンジによる注射)が容易な剪断薄膜化(shear thinning)材料を示す掃引率データ(粘度、白抜きの記号;剪断応力、黒塗りの記号)を示す。図6Dは、歪み処理(6Hzで180秒の1000%歪み)の停止後の時間の関数としてのゲル剛性率の回復を示す。記号は、図6Aで定義の通りである。大歪みによるヒドロゲル破壊直後に、ゲルは即座にその前の剛性の約75%に戻って固化し、迅速に(約10〜20分)その歪み前の剛性を取り戻す。
【図7】図7は、レーザー走査型供焦点顕微鏡法(LSCM)および低温透過型電子顕微鏡法(cryoTEM)を使用して得た結果を示す。図7Aは、ヒドロゲル微小構造のLSCMを示す。緑色の領域は蛍光染色された自己集合ペプチドであり、黒の領域は水で満たされた孔およびチャネルである。スペースバーは20μmである。図7Bは、自己集合したナノ構造のcryoTEMを示す。暗い構造は、自己集合したペプチド足場であり、より明るい灰色の領域はガラス化水から構成される。スペースバーは200nmである。
【図8】1重量% MAX1ヒドロゲルの組み合わせたUSANS/SANSプロットを示す。(挿入図は、−1.1の非線形の最小二乗法での重ね合わせを用いた0.02<q<0.08のlog(強度)の拡大図である)。低qの勾配−4は、図7Aにて顕微鏡で認められる微孔構造を示す。挿入図中の勾配−1は、図7Bにて顕微鏡で認められる局所ロッド様構造を示す。
【図9】図9Aは、150μM MAX1溶液(125mMホウ酸、10mM NaCl、pH9.0)の温度依存性CDを示す。低温では、MAX1は折り畳まれない(その結果として、自己集合しない)。高温では、MAX1はβ−シートに折り畳まれる(その結果として、ヒドロゲルに自己集合する)。図9Bは、同一条件下(MAXl=VKVKVKVKVDPPTKVKVKVKV−NH2、MAX2=VKVKVKVKVDPTKVKTKVKV−NH2、およびMAX3=VKVKVKTKVDPPTKVKTKVKV−NH2)でのMAX1、2、および3の[θ]218の温度依存性を示す。相対疎水性は、下線を引いた位置でのバリンのトレオニンへの等構造(isostructural)であるが低疎水性の置換によって、MAX1>MAX2>MAX3である。最低温度で最高の疎水性で折り畳みが起こり、最高温度で最小の疎水性折り畳みが起こる。これは、図2に記載の分子パラメーター操作を端的に表す。25℃での水中のオクタノールからの全部で+8荷電状態のそれぞれの対応する非折り畳みペプチドの転移自由エネルギーの計算も示す。最も高い疎水性を示すMAX1は、7.60kcal/molと等しい最も高い自由エネルギーを有する。MAX2の転移自由エネルギーの計算値は6.88kcal/molであり、疎水性が最小のMAX3は、6.45kcal/molの最も低い転移エネルギーを有する。
【図10】図10Aは、150μM MAX3溶液(125mMホウ酸、10mMNaCl、pH9)の温度依存性CDを示す。白丸は、完全に折り畳まれていない特徴を示す5℃でのMAX3である。白四角は、80℃に加熱し、分子内折り畳みが起こったMAX3である。黒菱形は、完全に折り畳まれていない特徴を示す5℃に戻して冷却した後のMAX3である。黒三角は、80℃に再加熱し、分子内折り畳みが起こったMAX3である。図10Bは、同一条件下での2重量%のMAX3水性調製物の貯蔵弾性率(G’)の温度依存性を示す;20分間隔で表示の温度にてデータを収集し、おおよその装置/サンプル平衡が得られる時間を割り当てた。流動学は、可逆的自己集合および結果として起こる温度による固体ヒドロゲル剛性を明確に示す。
【図11】図11は、本発明の実施で使用することができる種々のペプチドの構造を示す。図11Aは、分子間側面疎水性接触(図2に記載)を可能にし、自己集合の駆動に役立つ、バリンが各ヘアピン突起内のH結合位に組み込まれ、且つ側鎖が外側に突出したMAX1の構造を示す。図11Bは、バリンが非H結合位に組み込まれ、且つその側鎖が内側に突出したMAX4の構造を示し、この構造はバリン駆動分子間相互作用の可能性が低く、自己集合があまり好まれない。
【図12】図12は、流動学研究の結果を示す。貯蔵弾性率は、40℃でのMAX1およびMAX4の1重量%調製物(pH9.0、125mMホウ酸、10mM NaCl)(周波数=6rad/秒)で増加する。MAX1は、分子間側面疎水性接触の増加のためにMAX4よりも明らかに迅速に集合する。
【図13】図13は、酢酸ウラニルで陰性に染色されたMAX9微小繊維のTEM顕微鏡写真を示す。この硬く不可逆な微小繊維は、MAX9の非折り畳み特性に起因し、ヒドロゲル化させない。
【図14】図14Aは、pH7(20mM Tris)での150μM MAX1の塩依存性CDを示す。20mM KF塩では、CDは、完全に折り畳まれていない高次構造を示す(黒丸)。150mM KFでは、CDは、強いβ−シート高次構造の形成を示す(黒四角)。図14Bは、無血清増殖培地を使用してpH7で形成されたMAX1ヒドロゲルの周波数掃引データを示す。剛性率(G’=黒丸、G’’=黒四角)の周波数非依存性は、重度に架橋された強固なヒドロゲル材料を明確に示す。
【図15】図15は、異なる温度での2重量%のMAX1の貯蔵弾性率対時間を示す(pH7.4、歪み1%、6rad/s)。黒丸=20℃、黒三角=37℃、および黒菱形=60℃。温度が高いほど、より迅速なペプチドの折り畳みおよび集合が起こり、得られたヒドロゲル網目構造がより強固になる。
【図16】図16は、光学顕微鏡法の結果を示す。図16Aは、2重量%Max1ゲルにプレートした10%ウシ血清を含むDMEM中の104個の線維芽細胞(t=4.5時間)を示す。図16Bは、t=72時間の16A由来の結果を示す。図16Cは、コントロール(ポリスチレンにプレートした10%ウシ血清を含むDMEM中の104個の線維芽細胞(t=4時間))を示す。図16Dは、t=72のコントロールを示す。図16Eは、2重量%Max1ゲルにプレートした血清を含まないDMEM中の104個の線維芽細胞(t=4.5時間)を示す。図16Fは、ウシ血清をサンプルに4時間添加し、72時間インキュベートした図16Eと同一の材料を示す。
【図17】図17は、種々の初期細胞播種密度でのMAX1ヒドロゲルに対するNIH3T3線維芽細胞の増殖アッセイ対細胞培養ポリスチレンの結果を示す。白抜きのバーは、組織培養ポリスチレンコントロール上での細胞増殖を示し、影をつけたバーは2重量%MAX1ヒドロゲル上での細胞増殖を示す。自己集合ヒドロゲル上での増殖は、全細胞播種密度でコントロールより高かった。
【図18】図18は、線維芽細胞がMAX1ゲル全体に充満していることを示すLSCM画像である。Z軸方向の重ね合わせ画像は、Z軸に対して垂直に見た。バー=100μm。したがって、ヒドロゲル形成の自己集合機構により、細胞を三次元に容易にカプセル化することができる。
【図19】図19は、修飾することができるペプチド中の位置を示す本発明で使用されるペプチドの略図を示す。細胞結合エピトープ(配列アルギニン、グリシン、アスパラギン酸(RGD)など)を、1)一方または両方のペプチド末端、2)一方または両方のペプチド鎖の中央、3)一方または両方のターン隣接位置、または4)共有結合を介した任意のリジン側鎖に組み込むことができる。
【図20】図20は、本発明と組み合わせて使用することができるトリガーの例を示す。330〜360nmの波長光を、図中に示すVal(16)(スキーム1および2)への図に示す化学物質の組み込みによる分子内折り畳み(それによる、ヒドロゲル化)トリガーとして使用することができる。カルシウムII結合を、ターン配列へのスキーム3および4に示す化学物質の組み込みによる細胞内折り畳み(それによる、ヒドロゲル化)トリガーとして使用することができる。
【図21】図21Aは、20℃で0mM(白丸)および150mM(黒丸)のKFを含む2重量%Max1(pH7.4)溶液のCDスペクトルを示し、これは、塩を使用した明確なβ−シート形成を示す。図21Bは、0および400mM NaClを含む2重量%Max1溶液(pH7.4)のFTIRスペクトルを示し、これも塩添加による明確なβ−シートの形成を示す。図21Cは、150mM NaClを含む3重量%Max1溶液(pH7)のWAXSスペクトルを示す。ピークは、4.7Åの間隔(β−シート高次構造中のペプチドの分子間間隔)を示す。
【図22】図22Aは、20℃で20mM(G’;黒丸、G’’;白丸)、150mM(G’;黒三角、G’’;白三角)、および400mM(G’;黒四角、G’’;白四角)のNaClを含む2重量%Max1(pH7.4)溶液の動的周波数掃引(歪み5%)を示す。より高い塩濃度の刺激ほど、ヒドロゲル網目構造がより強固になる。図22Bは、20℃で20mM(G’;黒丸)、150mM(G’;黒三角)、および400mM(G’;黒四角)のNaClを含む2重量%Max1溶液の動的掃引時間(歪み1%、6rad/s)を示す。塩濃度が高いほど、より迅速なペプチドの折り畳みおよび自己集合が起こり、最終的なヒドロゲル材料がより強固になる。
【図23】図23は、ヒドロゲルの自己集合構造の負に染色された(酢酸ウラニル)TEM画像を示す。図23Aは、2重量%Max1(pH7.4、400mM NaCl溶液)の密に相互連結した網目構造を示す。図23Bは、希釈ヒドロゲル(希釈後の最終濃度:約0.1重量%)の微小繊維集合を示す。ナノ構造は、図1および図24に概略的に示す網目構造と一致する。
【図24】図24は、微小繊維中の自己集合Max1β−ヘアピン分子の提案された構造を示す。分子の短軸(stand axis)は、32Åであり、横断面の厚さは20Åである。構造の長軸は、水素結合および微小繊維の成長方向を示す。
【図25】図25Aは、20℃(G’;白丸)および37℃(G’;白三角)で150mMのNaClを含む2重量%Max1溶液(pH7.4)の動的掃引時間データ(歪み1%、6rad/s)を示す。温度が高いほど、最終材料がより迅速に折畳まれ、より強固になる。図25Bは、2.5時間のゲル化後の(G’;黒丸、G’’;白丸)37℃でヒドロゲルの周波数掃引データ(歪み5%)を示す。図25Cは、20℃(白丸)および37℃(白三角)で150mM NaClを含む2重量%Max1溶液(pH7.4)の218nm([θ]218)での時間依存性平均モル楕円率を示す。
【図26】図26は、37℃での細胞増殖培地中での2重量%Max1溶液の流動学データ(G’;黒丸、G’’;白丸)を示す。図26Aは、ゲル形成の流動学データ(歪み1%、周波数6rad/s)を示す。図26Bは、周波数掃引データ(歪み5%)を示し、図26Cは、高歪み振幅(1000%、6rad/s)の停止後のヒドロゲルのG’およびG’’回復率(歪み1%、周波数6rad/s)を示す。pH折り畳みおよびアセンブリトリガーを比較した図6に示すように、細胞増殖培養形態へのペプチドの迅速な曝露によって生成されたゲルは有意に強固であり、剪断薄膜化することができ(例えば、シリンジ注射)、剪断停止後にその元の剛性に回復し得る。
【技術分野】
【0001】
本出願は、2004年7月28日出願の米国特許出願番号10/900,324号の優先権を主張する。本発明は、NIH助成金番号1P20RR17716−01によって一部支援されている。合衆国政府は、本発明に一定の権利を有し得る。
【背景技術】
【0002】
(発明の背景)
ヒドロゲルは、軟組織工学および骨工学での使用が非常に期待されているクラスの材料である。これらの用途において重要な材料となるヒドロゲルの一般的特徴は、その十分に水和した多孔質構造である。本発明は、既存のテクノロジーでは現在満たされない重要な材料要件を満たす新規のクラスの環境応答性を示すペプチドベースのヒドロゲルを提供する。本発明のヒドロゲルを、種々の細胞型(例えば、線維芽細胞および骨芽細胞)の接着および増殖に適合し、結合組織および骨の生成のための潜在的な組織工学足場となるようにデザインすることができる。組織再生用のヒドロゲルに必要な一連の生物学的性質および材料の性質が求められている。最終的な標的組織型と無関係に、ヒドロゲルは、一般的な一連の生物学的性質を示さなければならない。第1に、この材料は細胞適合性でなければならない。本明細書中で定義される、「細胞適合性」は、所望の細胞に対して細胞傷害性を示してはならないことを意味する。第2に、この材料は生体適合性を示さなければならない。本明細書中で定義される、「生体適合性」は、in vivoに置かれた場合、足場が組織再生に有意な免疫応答および炎症反応を引き起こさず、好ましくは、性分解によって無毒となる種であることを意味する。本発明は、新規の自己集合(self−assembly)法を使用した新規の材料の開発および得られた材料の細胞適合性の評価に関する。
【0003】
いくつかの所望の性質が一見したところ互いに矛盾するので、所望の材料の性質をいずれか1つの材料に包括的に組み込むことは困難である。例えば、理想的なヒドロゲルの形態は、細胞の運動性および栄養素/老廃物の拡散のために多孔性(ナノスケールからマイクロスケールの寸法にわたる)が高い。また、ヒドロゲルは、主に、細胞増殖および最終的に足場を容易に生分解するのに十分な体積を得るためにできるだけ固体材料が少ない水性溶媒から構成されるべきである。しかし、その希薄で多孔質であるにもかかわらず、これらの十分に水和した材料は、機械的に強固でもなければならない。ヒドロゲル化(hydrogelation)過程時に形成される構成分子架橋(化学的および/または物理的)のデザインによってこの明らかな矛盾(希薄な多孔質足場の剛性)に本質的に対処しなければならない。しかし、化学的架橋由来の副生成物が有毒である可能性があり、且つ足場からの除去が困難であるので、化学的架橋の導入は、生物学的に問題があり得る。理想的には、無害の生体適合性の化学的または物理的架橋法を、身体への最終的組み込みのためのin vitroゲル化または生理学的刺激(温度、イオン強度、pHなど)によって架橋の形成が誘発される直接的で迅速なin vivoゲル化のいずれかのために使用すべきである。自己集合を介した材料の形成を開始するための環境要因(trigger)を使用するという考え方が盛んに進められている。例えば、ペプチドの自己集合(およびそれによるゲル化)を、温度および光に感受性を示すリポソームからの塩の放出によって誘発することができることが示されている(Collier,et al.,Journal of the American Chemical Society 2001,123,9463−9464:非特許文献1)。一見したところヒドロゲルの剛性により、予め形成された足場におけるいかなる処理も実行不可能であることがデザインをさらに複雑にしている。例えば、強固な組織工学構築物をin vitroで形成することを望むことができるが、その後に組織再生のために宿主に注入する。恒久的に架橋した強固な網状組織を注入することは不可能である。
【0004】
現在のヒドロゲルテクノロジーは、天然由来の高分子および合成ポリマーの両方を使用する。一般に、天然ポリマーから調製されたヒドロゲルは、好ましい生物学的性質を示すが、所望の材料特性(例えば、低サンプル剛性)を欠き得る。対照的に、所望の材料特性を有する合成ポリマーを設計することができるが、細胞適合性が制限され得る。合成ポリマーの細胞適合性を増加させるための一般的アプローチは、ペプチドエピトープ(例えば、RGDモチーフ)を組み込むことである。しかし、位置特異的に制御された様式での予め形成したポリマーへのこれらのモチーフの組み込みは実現が非常に困難である。結果として、ポリマーの材料特性の制御は問題がある。例えば、細胞接着またはエピトープの接近可能性の制御のために表面上に提示されたエピトープの濃度の制御は困難である。さらに、これらの足場は、その基礎をなす分子網目構造によって微小規模で構造が均一(多孔質ではない)であり、細胞増殖が制限され得る。ゲル網目構造に微小規模で多孔質にするために、これらの系をさらに処理しなければならない(例えば、凍結融解サイクル、粒子浸出、ミクロスフェア焼結、および不織線維(non−woven fiver)形成)。要するに、現在、理想的な組織工学足場の必要な性質を全て首尾よく組み込まれた単一のヒドロゲル系が存在しない。
【0005】
新規のヒドロゲルストラテジーをデザインする機会および必要が存在する。in vitroまたはin vivoで容易に形成することができる強固、多孔質、且つ処理が容易で細胞適合性を示すヒドロゲルが必要である。
【非特許文献1】Collier,et al.,Journal of the American Chemical Society 2001,123,9463−9464
【発明の開示】
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、新規のヒドロゲルを提供する。さらに、本発明は、ヒドロゲル構築のための新規の過程を提供する。本明細書中で使用される、用語「ヒドロゲル」は、好ましくは、大量の水画分をカプセルされ、且つ機械的に自立した希薄な相互連結された足場を意味する。本明細書中で使用される、用語「ヒドロゲル」はまた、水性有機混合物および/または有機溶媒(例えば、DMF、DMSOなど)を含む希薄な相互連結された足場を含む。1つの態様では、本発明は、高性能のヒドロゲル化系を構築するための新規の過程と新規のペプチドとの組み合わせを提供する。1つ以上の環境シグナルまたは刺激(例えば、1つ以上の環境特徴の変化)に反応して二次構造が変化するように新規のペプチド(例えば、MAX1)をデザインした。1つの特定の態様では、本発明のペプチドは水溶液中に存在することができ、ペプチドの二次構造を変化させるために溶液の1つ以上のパラメーターを変化させることができる。特定の実施形態では、溶液のpH、イオン強度、特定のイオンの濃度、および/または温度のうち1つ以上を変化させてペプチドの二次構造を変化させることができる。典型的には、溶液の1つ以上のパラメーターの変化の結果としてペプチドの二次構造が変化した後、ペプチドをより高次の構造(例えば、ヒドロゲル)に集合させる。本発明の別の態様では、環境シグナルは、電磁放射(例えば、光)を含み得る。例えば、本発明のペプチドの構造を変化させることができ、この変化は、電磁放射に供した結果としての一次構造、二次構造、またはその両方の変化であり得る。典型的には、電子放射に曝露した後、本発明のこの態様を使用したペプチドは、所望の二次構造を有することが予想され、より高次の構造(例えば、ヒドロゲル)に自己集合する。
【0007】
1つの態様では、自己集合機構による低粘度の水溶液から強固なヒドロゲル材料(本質的に無限に粘度が変化する)への移行は、好ましくは、各ペプチドの所望の高次構造への折り畳みを前提とする。この分子内折り畳み過程を、所望の環境シグナルのみを用いて起こるように制御することができる。環境シグナルには、生理溶液の条件(37℃、pH7.4、および高塩濃度)が含まれるが、これらに限定されない。生理学的条件下で頑強且つ化学的に無害にゲル化するので、これらのゲルは、主に組織工学分野および創傷治癒分野で見込みがある。
【0008】
1つの特定の実施形態では、1つ以上の環境シグナルに応答してβ−ヘアピン二次構造を採用するようにペプチドをデザインすることができる。典型的には、β−ヘアピン構造の採用後、ペプチドは、より高次の構造(例えば、三次元網目構造、最終的にヒドロゲル)に自己集合する。1つの態様では、ペプチド分子上の側鎖がβ−ヘアピン高次構造中に固有に存在しない限り、自己集合は起こらない。
【0009】
したがって、1つの態様では、本発明は、1つ以上の環境刺激に応答して所望の二次構造を採用するようなペプチドをデザインする過程を提供する。さらに、本発明は、1つ以上の環境刺激に応答してより高次の構造(例えば、ヒドロゲル)を形成するペプチドをデザインする過程を提供する。本発明はまた、このようにして形成されたより高次の構造(例えば、ヒドロゲル)を含む。
【0010】
いくつかの態様では、二次構造の採用およびより高次の構造の形成は関連する。したがって、ペプチドの折り畳みおよび自己集合(例えば、ヒドロゲル)は関連する。本発明のこの態様により、ゲル化が制御される。特に、この態様により、ヒドロゲルの形成速度(すなわち、どれぐらい速くゲルが形成されるか)が制御される。また、自己集合過程の制御により、このようにして形成されたヒドロゲルの物理的特徴(例えば、得られたゲルの剛性)が制御される。1つ以上の環境刺激の適用後に、ヒドロゲルが、約1秒〜約5時間、約1秒〜約4時間、約1秒〜約3時間、約1秒〜約2時間、約1秒〜約1時間、約1秒〜約50分、約1秒〜約40分、約1秒〜約30分、約1秒〜約20分、約1秒〜約15分、約1秒〜約10分、約1秒〜約5分、約1秒〜約2分、約10秒〜約5時間、約10秒〜約4時間、約10秒〜約3時間、約10秒〜約2時間、約10秒〜約1時間、約10秒〜約50分、約10秒〜約40分、約10秒〜約30分、約10秒〜約20分、約10秒〜約15分、約10秒〜約10分、約10秒〜約5分、約10秒〜約2分、約30秒〜約5時間、約30秒〜約4時間、約30秒〜約3時間、約30秒〜約2時間、約30秒〜約1時間、約30秒〜約50分、約30秒〜約40分、約30秒〜約30分、約30秒〜約20分、約30秒〜約15分、約30秒〜約10分、約30秒〜約5分、約30秒〜約2分、約60秒〜約5時間、約60秒〜約4時間、約60秒〜約3時間、約60秒〜約2時間、約60秒〜約1時間、約60秒〜約50分、約60秒〜約40分、約60秒〜約30分、約60秒〜約20分、約60秒〜約15分、約60秒〜約10分、約60秒〜約5分、または約60秒〜約2分で形成することができるように、ペプチドをデザインし、そして/または環境刺激を選択することができる。
【0011】
本発明にしたがって形成されたヒドロゲルは、種々の量の固体材料を有し得る。例えば、約0.1重量%〜約10.0重量%、約0.1重量%〜約9.0重量%、約0.1重量%〜約8.0重量%、約0.1重量%〜約7.0重量%、約0.1重量%〜約6.0重量%、約0.1重量%〜約5.0重量%、約0.1重量%〜約4.0重量%、約0.1重量%〜約3.0重量%、約0.1重量%〜約2.0重量%、約0.1重量%〜約1.0重量%、約0.1重量%〜約0.75重量%、約0.1重量%〜約0.5重量%、約0.1重量%〜約0.25重量%、約0.25重量%〜約10.0重量%、約0.25重量%〜約9.0重量%、約0.25重量%〜約8.0重量%、約0.25重量%〜約7.0重量%、約0.25重量%〜約6.0重量%、約0.25重量%〜約5.0重量%、約0.25重量%〜約4.0重量%、約0.25重量%〜約3.0重量%、約0.25重量%〜約2.0重量%、約0.25重量%〜約1.0重量%、約0.25重量%〜約0.75重量%、約0.25重量%〜約0.5重量%、約0.5重量%〜約10.0重量%、約0.5重量%〜約9.0重量%、約0.5重量%〜約8.0重量%、約0.5重量%〜約7.0重量%、約0.5重量%〜約6.0重量%、約0.5重量%〜約5.0重量%、約0.5重量%〜約4.0重量%、約0.5重量%〜約3.0重量%、約0.5重量%〜約2.0重量%、約0.5重量%〜約1.0重量%、約0.5重量%〜約0.75重量%、約1.0重量%〜約10.0重量%、約1.0重量%〜約9.0重量%、約1.0重量%〜約8.0重量%、約1.0重量%〜約7.0重量%、約1.0重量%〜約6.0重量%、約1.0重量%〜約5.0重量%、約1.0重量%〜約4.0重量%、約1.0重量%〜約3.0重量%、約1.0重量%〜約2.0%、または約1.0重量%〜約1.5重量%のペプチドを含むヒドロゲルを形成することができる。
【0012】
1つの態様では、ペプチドの重量による量およびゲル化速度を変化させて、所望の剛性率(剛性)を有するヒドロゲルを生成することができる。本発明のヒドロゲルは、約1パスカル(Pa)〜約100,000Pa、約1Pa〜約50,000Pa、約1Pa〜約25,000Pa、約1Pa〜約10,000Pa、約1Pa〜約7,500Pa、約1Pa〜約5,000Pa、約1Pa〜約2,500Pa、約1Pa〜約2,000Pa、約1Pa〜約1,500Pa、約1Pa〜約1,000Pa、約1Pa〜約500Pa、約1Pa〜約250Pa、約1Pa〜約100Pa、約100Pa〜約100,000Pa、約100Pa〜約50,000Pa、約100Pa〜約25,000Pa、約100Pa〜約10,000Pa、約100Pa〜約7,500Pa、約100Pa〜約5,000Pa、約100Pa〜約2,500Pa、約100Pa〜約2,000Pa、約100Pa〜約1,500Pa、約100Pa〜約1,000Pa、約100Pa〜約500Pa、または約100Pa〜約250Paの剛性率を有し得る。
【0013】
本発明の1つの態様では、形成されたヒドロゲルを処理することができる。例えば、本発明のヒドロゲルを、動物(例えば、哺乳動物)に注入することができる。本発明のヒドロゲルが自己集合するので、強固なゲルを容易に処理することができる(例えば、シリンジによって注入することができる)一方で、処理の中止直後に再集合/硬化する。関連する態様では、ヒドロゲルを、低粘度溶液からの自己集合によって物理的に形成することができ、したがって、ヒドロゲルを、in vitroまたはin vivoで幾何学的に限定されて生成することができる。
【0014】
いくつかの好ましい実施形態では、本発明のペプチドベースのヒドロゲルは、完全に非細胞傷害性を示し、一般的な哺乳動物細胞(例えば、繊維芽細胞、骨芽細胞)の接着および増殖を促進することもできる。したがって、本発明のヒドロゲルを、細胞培養物中で使用することができる。細胞培養物を、自己集合機構によって三次元でカプセル化することができ、それにより、三次元での細胞付着および増殖が可能になる。いくつかの実施形態では、ヒドロゲルを三次元支持体として使用して、薬学的化合物、ペプチド、タンパク質、および抗体などの治療因子を生成するように操作された細胞株を増殖/維持することができる。ヒドロゲルおよび細胞を含むバイオリアクターによる培地の連続フローにより、化合物の迅速な単離および連続的細胞増殖手段が行われる。
【0015】
いくつかの態様では、本発明のペプチド(例えば、MAX−1および関連するヘアピンペプチド)から調製したヒドロゲルは、グラム陽性細菌およびグラム陰性細菌に対して抗菌挙動を示す。したがって、本発明のヒドロゲルは、臨床的状況(clinical setting)で抗菌性を示し得る。本発明のヒドロゲルのこの特徴により、細胞培養方法と同様に、ヒドロゲルが生きた動物(例えば、ヒトなどの哺乳動物)内におかれる状況でも有用となる。特定の実施形態では、本発明のヒドロゲルを、組織工学のために使用することができる。例えば、所望の量の1つ以上の細胞型を、1つ以上の本発明のペプチドを含む溶液中におくことができる。溶液を含む細胞をヒドロゲルに形成させることができ、この細胞をヒドロゲル全体に分散させることができる。次いで、ヒドロゲルを含む細胞を、例えば、損傷組織と置換するための組織として使用することができる。本発明のヒドロゲルの抗菌特性により、動物に移入された場合に感染防止に役立つ。いくつかの実施形態では、本発明のヒドロゲルを、可逆的にゲル化する(すなわち、1組の条件下でヒドロゲルを形成し、その後に他の条件下で溶液に戻る)ように構築することができる。細胞をヒドロゲル足場に移入し、最終的に溶解して細胞を適所に放出することができるように、これを組織工学への適用の際に使用することができる。抗菌ゲルは、創傷治癒への適用にも有用である。創傷治癒への適用で使用されるヒドロゲルは、ヒドロゲルに加えて治療因子を含み得る。例えば、生理学的条件下(すなわち、創傷との接触)におかれた場合にヒドロゲルが形成されるペプチド溶液は、凝固を促進する薬剤、鎮痛薬、および/または他の治療因子を含み得る。
【0016】
いくつかの態様では、本発明のヒドロゲルを、さらなる至適化のためのさらなる生化学(例えば、増殖因子または細胞接着ペプチドエピトープ)で容易に機能的にすることができる。したがって、本発明のペプチドは、本発明にヒドロゲルの特徴を変化させるペプチドであり得るさらなる成分を含み得る。
【0017】
本発明のヒドロゲルを、環境応答性遮断材料として微小流体(microfluidic)デバイスで使用することができる。例えば、微小流体デバイスのチャネルを、ペプチド水溶液に浸すことができる。所望の位置にチャネル障壁を取り付けるための空間分解能を有するヒドロゲル化を開始することができる。これらの障壁およびダムは、検出装置として作用することができる。溶液がこれらを通過してヒドロゲル溶解(ダム破壊)が起こる場合、初期のチャネルを開口して流動物を検出機に流し、分析物の分析手段が得られる。ヒドロゲル障壁の溶解を使用して、所望の化学転換および反応のための反応成分の混合を開始し、容易にすることもできる。
【0018】
本発明のヒドロゲルを、1つ以上の目的の分析物の検出のためのセンサとして使用することができる。例えば、本発明のペプチド溶液を、分析物を含み得るサンプルと接触させることができる。いくつかの例では、分析物の存在により、溶液のゲル化を誘導し得る。例えば、目的の分析物は金属イオンであってよく、ペプチド溶液の金属イオンとの接触により、溶液がゲル化し得る。他の例では、本発明のヒドロゲルを形成し、次いで、目的の分析物を含み得るサンプルと接触させることができる。分析物の存在により、ヒドロゲルを可溶化することができる。ヒドロゲルの形成または分解を、当業者に周知の標準的技術(例えば、光学的技術)を使用して検出することができる。
【0019】
いくつかの実施形態では、本発明のヒドロゲルを使用して、目的の分子(例えば、生体分子、タンパク質、DNA、RNAなど)の分離のためのマトリクスを調製することができる。ペプチドを、目的の分子の分離に有用な特徴を有するようにデザインすることができる。例えば、目的の分子と特異的に相互作用することができる部分を、ヒドロゲルを作製するために使用されるペプチドにデザインすることができる。特異的に相互作用することができる適切な部分には、エピトープ、リガンド、特異的低分子、および核酸配列などが含まれるが、これらに限定されない。このの型ヒドロゲルを、正の選択様式(すなわち、目的の分子の結合)または負の選択様式(すなわち、夾雑物の結合)のいずれかで使用することができる。この型の実施形態で使用するために、所望の分子を精製するためにヒドロゲルをナノ細孔形態および微小孔形態を制御することが望ましいかもしれない。例えば、本発明のヒドロゲルの孔サイズを、目的の分子の精製を至適化するための使用するペプチドの重量、ゲル化条件、および/またはペプチド構造の変化によって制御することができる。
【0020】
本発明のヒドロゲルを使用して、生きた生物中に存在する材料の耐性および/または接着を改良することができる。例えば、生きた生物中に移植すべき材料(例えば、人工器官、ペースメーカー、支持器(support)など)を、最初に本発明のヒドロゲルでコーティングすることができる。このようなヒドロゲルを、生物組織の移植デバイスへの接着を促進する1つ以上の部分を有するペプチドから作製することができる。このような部分には、接着エピトープなどが含まれ得る。ヒドロゲルはまた、低分子またはエピトープなどの免疫調整(例えば、抑制または刺激)部分を含み得る。
【0021】
別の実施形態では、本発明のヒドロゲルを、水溶液由来の有害金属イオンの改善のために使用することができる。有害金属イオンに結合する官能基を含むペプチドをデザインすることができる。ペプチドを、有害金属イオンを含む溶液に導入し、ペプチドのゲル化を誘導することができる。有害金属をヒドロゲル内に捕捉することができ、ヒドロゲルを、任意の適切な技術(例えば、濾過)によって残りの溶液から分離することができる。任意選択的に、ヒドロゲルを溶解し、有害金属イオンを単離することができる。
【0022】
いくつかの態様では、本発明は、ヒドロゲルの作製方法を提供する。このような方法は、ペプチドを含む溶液を提供し、この溶液の1つ以上の特徴を変化させ、ヒドロゲルが形成されることが必要であり得る。変化する特徴は、その変化の際にヒドロゲルが形成される任意の特徴であり得る。適切な例には、イオン強度、温度、特定のイオンの濃度、およびpHが含まれるが、これらに限定されない。いくつかの実施形態では、溶液の1つ以上の変化させる工程は、溶液を電磁放射と接触させることを含み得る。特定の実施形態では、変化する特徴は、溶液のpHであり得る。いくつかの態様では、溶液の1つ以上の特徴の変化により、塩濃度が約20mM〜約400mMになる。任意の塩(KCl、NaCl、MgCl2、KF、MgSO4)を使用することができる。1つの実施形態では、塩はNaClであり得る。いくつかの実施形態では、溶液は、所望のpH(例えば、pH9未満、約pH6.0〜約pH8.5、約pH7.0〜約pH8.0、または約pH7.4)を有することができ、ヒドロゲル形成の際に同一であっても変化してもよい。
【0023】
いくつかの態様では、本発明は、ヒドロゲルを提供する。このようなヒドロゲルは、ペプチドおよび約20mM〜約400mMの塩を含み得る。上記で考察しているように、任意の塩(例えば、NaCl)を使用することができる。ヒドロゲルを形成することができる任意のペプチド(例えば、MAX1)を使用することができる。
【0024】
1つの態様では、本発明は、ヒドロゲルの作製方法を提供する。このような方法は、ペプチドを含む溶液を動物に注入する工程と、前記溶液が前記動物内でヒドロゲルを形成することとを含み得る。任意の動物(例えば、哺乳動物(ヒトが含まれる))を使用することができる。本発明の態様で使用するための溶液は、いくつの成分を含んでもよい。いくつかの実施形態では、溶液は、1つ以上の治療因子を含み得る。当業者に公知の任意の治療因子を使用することができる。特定の実施形態では、溶液は、低分子、ペプチド、タンパク質、および細胞からなる群から選択される1つ以上の治療因子を含み得る。
【0025】
本発明の別の態様では、本発明は、必要とする動物に治療因子を送達する方法を提供する。このような方法は、治療因子および1つ以上のペプチドを含む溶液を動物に投与する工程と、前記溶液が前記動物内でヒドロゲルを形成することとを含み得る。このような方法を、任意の動物型(ヒトなどの哺乳動物が含まれる)に対して実施することができる。当業者に公知の任意の治療因子型(例えば、低分子、ペプチド、タンパク質、および細胞)を使用することができる。当業者は、治療因子が任意の望ましくない条件を防止および/または改善する任意の薬剤であることを認識している。
【0026】
別の態様では、本発明は、治療因子および1つ以上のペプチドを含むヒドロゲルを動物に投与する工程を含む、必要とする動物に治療因子を送達する方法を提供する。このような方法を、任意の動物型(ヒトなど哺乳動物が含まれる)に対して実施することができる。当業者に公知の任意の治療因子型(例えば、低分子、ペプチド、タンパク質、および細胞)を、使用することができる。
【0027】
別の態様では、本発明は、動物の創傷を処置する方法を提供する。このような方法は、創傷をペプチドを含む溶液と接触させる工程と、前記溶液がヒドロゲルを形成することとを含み得る。本発明のこの態様で使用する溶液は、1つ以上の治療因子をさらに含み得る。この型の方法を、任意の動物型(例えば、哺乳動物(ヒトが含まれる))に対して実施することができる。当業者に公知の任意の治療因子型(例えば、低分子、ペプチド、タンパク質、および細胞)を、使用することができる。
【0028】
別の態様では、本発明は、細胞を増殖させる方法を提供する。このような方法は、細胞を含むヒドロゲルを形成する工程と、細胞生存に適切な条件下で細胞を維持する工程とを含み得る。本発明のこの態様で使用するヒドロゲルは、典型的には、ペプチドを含む。ヒドロゲルを、例えば、ペプチドを含む溶液の1つ以上の特徴の調整によって形成することができる。調整される特徴は、1つ以上のpH、イオン強度、特定のイオンの濃度であり得る。1つの特定の実施形態では、調整される特徴は、イオン強度である。別の特定の実施形態では、調整される特徴はCa2+イオン濃度である。任意の型の細胞(哺乳動物細胞(ヒト細胞が含まれる)などの動物細胞)を、本発明の方法を使用して増殖させることができる。いくつかの特定の実施形態では、細胞は、骨芽細胞または線維芽細胞であり得る。本発明の方法を使用して増殖させるべき細胞は、組換え細胞(すなわち、1つ以上の外因性核酸分子を含み得る)であり得る。このような核酸分子を、細胞のゲノムに組み込み、そして/または染色体外に維持することができる。本発明の方法を使用して増殖させるべき細胞は、目的のタンパク質を発現し得る。目的のタンパク質の例には、抗体が含まれるが、これらに限定されない。
【0029】
別の態様では、本発明は、ヒドロゲルを含むセンサを提供する。本発明のセンサで使用するヒドロゲルは、ヒドロゲルが目的の分析物と接触した場合に変化する1つ以上の特徴を有し得る。目的の分析物は、検出されることが望ましい任意の材料である。いくつかの実施形態では、分析物に応答して変化したヒドロゲルの特徴は剛性である。他の実施形態では、変化した特徴は光学的性質である。光学的特徴には、吸光度、楕円率、および光散乱特性などが含まれるが、これらに限定されない。
【0030】
別の態様では、本発明は、環境条件を検出する方法を提供する。このような方法は、ヒドロゲルを含むセンサを環境条件を代表するサンプルと接触させる工程と、前記ヒドロゲルの1つ以上の特徴を決定する工程とを必要とし得る。典型的には、この型の方法では、ヒドロゲルが目的の分析物と接触した場合に、ヒドロゲルの1つ以上の特徴が変化する。ヒドロゲルの任意の特徴(例えば、剛性および/または光学的性質)を変化させることができる。この型の方法は、ヒドロゲルの特徴を異なる時間に決定した前記ヒドロゲルの同一の特徴と比較する工程も含み得る。
【0031】
別の態様では、本発明は、目的の分子を精製する方法を提供する。このような方法は、目的の分子および1つ以上の夾雑物を含む溶液を、目的の分子がヒドロゲルによって保持される条件下でヒドロゲルに接触させる工程と、ヒドロゲルから前記目的の分子を回収する工程とを含み得る。典型的には、この型の方法では、少なくとも1つの夾雑物がヒドロゲルによって保持されないか、目的の分子よりも低い程度で保持される。目的の分子は、当業者に公知の任意の分子であり得る。目的の分子の例には、タンパク質、核酸分子、および低分子などが含まれる。1つの特定の実施形態では、目的の分子は、抗体であり得る。目的の分子は、治療因子または治療因子の構成要素であり得る。治療因子の構成要素は、治療因子になるように修飾することができる材料である。治療因子の構成要素の1つの例は、治療因子になるように細胞傷害性化合物で共有結合によって修飾することができる抗体である。
【0032】
別の態様では、本発明は、目的の分子を精製する方法であって、目的の分子および1つ以上の夾雑物を含む溶液を、少なくとも1つの夾雑物がヒドロゲルによって保持される条件下でヒドロゲルに接触させる工程と、前記目的の分子を回収する工程とを含む方法を提供する。典型的には、この型の方法では、少なくとも1つの夾雑物がヒドロゲルによって保持されるか、目的の分子よりも高い程度で保持される。目的の分子は、当業者に公知の任意の分子であり得る。目的の分子の例には、タンパク質、核酸分子、および低分子などが含まれる。1つの特定の実施形態では、目的の分子は、抗体であり得る。目的の分子は、治療因子または治療因子の構成要素であり得る。治療因子の構成要素は、治療因子になるように修飾することができる材料である。治療因子の構成要素の1つの例は、治療因子になるように細胞傷害性化合物で共有結合によって修飾することができる抗体である。
【発明を実施するための最良の形態】
【0033】
(発明の詳細な説明)
1つの態様では、本発明は、ゲル足場基礎単位として小ペプチドを使用する自己集合ヒドロゲル化ストラテジーを提供する。ペプチドを、分子内で会合することができるβ−ヘアピン高次構造への分子内折り畳み後にのみ自己集合するようにデザインすることができる。折り畳み事象を、反応のよいヒドロゲル化系を与える環境刺激によって誘発することができる。ペプチドの使用は、化学合成が迅速であることならびにあつらえの材料および生物学的特性のための新規の残基および機能的エピトープの組み込みが可能なことにより有利である。ヒドロゲルの自己集合特性により、外因性架橋剤が必要ない。本明細書中に記載のペプチドベースのヒドロゲルは、強固であるが容易に処理される。このヒドロゲル化ストラテジーにより、潜在的なin vitroおよびin vivo組織工学構築物形成のために使用することができる細胞適合性ゲル化系が得られる。本発明は、ペプチド構造と最終的なヒドロゲルの形態学的、流動学的、および細胞レベルの生物学的性質との間の関係を確立する。以下によってこれを行った。1)ヒドロゲル形成させる折り畳みおよび自己集合および分子のデザインがどのように材料の性質に影響を与えるのかについての基本的理解を得ること。デザインしたペプチド構造と、折り畳み/自己集合過程と、最終的な材料の性質との間の関係の厳密な理解により、あつらえの組織工学特性を有するヒドロゲルを生成することができる。2)適時にペプチド溶液をヒドロゲル化することが可能な活性な分子内折り畳みトリガーのデザインによるヘアピンベースのヒドロゲルの処理可能性の増強。生物学的に適合する合図(例えば、pH、塩濃度、細胞培養培地、光、体温、または特定のイオン濃度(例えば、Ca2+)に応答して折畳まれるペプチドのデザインにより、当業者は、in vitroおよびin vivoの両方での組織工学への適用のためのヒドロゲル化過程をデザインすることができる。3)どのようにしてペプチド構造よび材料の性質がモデル細胞株(例えば、線維芽細胞株および骨芽細胞株)の接着および増殖に影響をあたえるのかを決定すること。β−ヘアピンヒドロゲルのペプチド性および多孔性により、これらの材料が、潜在的な骨および軟組織工学への適用のための細胞適合性の(非細胞傷害性を示し、細胞の接着および増殖を促進する)容易に官能化される基質としての材料の候補となる。ペプチド構造と、ヒドロゲル剛性と、細胞適合性との間に相関関係が確立されている。
【0034】
本発明は、1つの基本的分子デザイン内の1つ以上の上記材料特性を組み入れる。この別のストラテジーは、ヒドロゲルを生成するための以下の4つの基本的デザイン様相(facet)を使用する:1)小さなde novoでデザインされたペプチドを使用してヒドロゲルを調製することができる。2)外因性架橋剤を必要としない純粋な自己集合機構を介してヒドロゲルを構築することができる。3)ペプチドがターゲティングされた高次構造に正確に分子内で折畳まれない限り、自己集合せず、ヒドロゲル骨格形成されないようにペプチドをデザインすることができる。4)この分子内折り畳み事象、およびそれによるヒドロゲル化を、特異的環境刺激によって誘発することができる。
【0035】
図1は、この簡潔な材料構築デザインの根拠の1つの実施形態を示す。正確な溶液条件によってβ−ヘアピン高次構造への分子内折り畳みが決定づけられるまで水溶液中で構築されないように小ペプチドをデザインする。この表面が両親媒性のヘアピンが自己集合されて強固な多孔質のβ−シートリッチなヒドロゲルが得られる。本発明者らは、疎水性に会合した微小繊維内接合部が散在した微小繊維の短いセグメントと一致する自己集合状態のナノ構造(図1中の右側)を提案する(図7Bを参照のこと)。この形態は古典的な微小繊維ベースの集合物が得られる自己会合ペプチドから認められた形態と非常に異なることに留意すべきである。古典的な系において、ペプチドは、積層されたβ−シートリッチな微小繊維と会合する。自己集合過程は、通常は非常に遅く(数時間〜数ヵ月)、ミクロン単位の長さを有し得る微小組織が不可逆的に得られる。例は、十分に開発された成熟した微小繊維の絡みつきの際に形成されるヒドロゲルである。対照的に、本発明者らの提案したペプチド系は、新規の機構を提供し、この機構は、誘発された分子内折り畳みが所望の分子間自己集合前に起こらなければならない。したがって、ヒドロゲル化を、多様な一連の環境トリガーによって開始することができる。ヒドロゲル化事象は、非常に迅速であり(条件によって数秒以内)、必要に応じて、完全に可逆性であるようにデザインすることができる。得られたゲルは、ナノ細孔から微小孔の形態および98%を超える水から構成されているにもかかわらず有意な材料剛性を示すことによって特徴づけられる。
【0036】
複雑な材料を形成するための小ペプチドの実行可能性は、文献に十分に記載されている。ペプチドは、ヘリックスリボン、ナノチューブおよび小胞、表面集合構造、ならびにその他に自己集合することが認められている。小ペプチドからの材料の調製は、これらを迅速に化学合成することができ、新規のアミノ酸残基を組み込むことができるので、有利である。さらに、直交保護ストラテジーの使用により、化学的部分をアミノ酸側鎖と位置選択的に連結してあつらえの機能(例えば、細胞接着)を有する抱合体(conjugate)を得ることが可能である。位置選択性に関して、既存のポリマーの化学修飾に使用される比較的選択性のない方法と比較して自己集合系の単量体基礎単位を完全且つ正確に官能化する能力が非常に望まれる。本発明の1つの態様では、ペプチドを、その環境に応答して折畳まれるようにデザインすることができ、この性質の活用により、適時に形成される(または溶解する)高性能の材料を得ることができる。本発明の1つ以上の態様の実施で使用することができるペプチドの例には、以下が含まれるが、これらに限定されない。
【0037】
【化1】
上記で詳細に引用したアミノ酸に加えて、任意の上記ペプチドの任意の位置をXで示した(各Xは、独立して、当業者に公知の任意の天然もしくは非天然アミノ酸(LまたはD立体化学)またはアミノ酸の任意のアナログであり得る)。本出願では、D立体化学を、Dアミノ酸の前の上付き文字で示す(したがって、DPはD−プロリンである)。
【0038】
本発明のいくつかの実施形態では、ペプチドは、一般式VKVKVKVK(XXXX)aKVKVKV(XXXX)bKVKVKVKV(配列番号5)に適合し得る。本発明のこの実施形態の特定の例には、以下が含まれるが、これらに限定されない。
【0039】
【化2】
上記で詳細に引用したアミノ酸に加えて、任意の上記ペプチドの任意の位置をXで示した(各Xは、独立して、当業者に公知の任意の天然もしくは非天然アミノ酸(LまたはD立体化学)またはアミノ酸の任意のアナログであり得る)。好ましくは、各(XXXX)aおよび(XXXX)bは、ターン(例えば、β−ターン)を形成することができる配列を含み得る。
【0040】
本発明のいくつかの実施形態では、ペプチドは、以下の一般式に適合し得る。
【0041】
【化3】
mおよびnはそれぞれ独立して1〜100であってよく、mはnと等しくても等しくなくてもよい。
【0042】
ヒドロゲル調製の容易さおよびそれによって得られる材料特性(特に、処理可能性および形態学)が本発明で特に有利である。自己集合ストラテジーの使用により、化学的架橋が排除される。架橋を誘導する化学物質の使用は一般に選択性がなく、多くの架橋試薬は有毒であり、ヒドロゲル骨格から精製によって容易に除去されない。
【0043】
処理可能性に関して、物理的に架橋した自己集合網目構造から構築したヒドロゲルは、機械的剪断に応答することができる。この特徴により、剪断の適用および剪断停止後のゲル網目構造の完全な再形成(自己治癒)の間にフリーフロー懸濁液が得られる(Prudhomme,et al,Langmuir 1996,12,4651−4659およびNowak,et al,Nature 2002,417,424−428を参照のこと)。剪断薄膜化と自己治癒との組み合わせにより、空間分解様式で材料を形成可能である。例えば、本発明のいくつかの実施形態では、当業者は、所望の構成要素(増殖培地、増殖因子、生きた細胞など)の存在下にてex vivoで予め形成されたヒドロゲルを宿主に注射(剪断薄膜化)することができ、それにより、宿主が自己治癒して組織再生のための足場が得られる。さらに、自己集合を介して形成されたヒドロゲルは、上記のex vivo調製に制限されない。本発明は、塩、温度、およびpHなどの生物学的に関連する刺激に曝露した場合に自己集合するペプチド系を提供する(Pochan,et al.,Journal of the American Chemical Society 2003,125,11802−11803およびSchneider,et al,Journal of the American Chemical Society 2002,124,15030−15037を参照のこと)。これにより、ペプチド溶液の注射を介してin vivoで直接ヒドロゲルを形成することができる。ex vivoおよびin vivo調製が可能であるヒドロゲル系は、処理可能性に関して最も用途が広く、本発明の範囲内に含まれる。
【0044】
本明細書中に記載のβ−ヘアピンの自己集合により、最小固体材料から構成される強固な網目構造(2重量%以下のペプチドの剛性率(G’)が1000Pa超)を得ることができ、それにより、さらに処理する必要がない有意に希薄で多孔質な足場が得られる。これらのゲルは、ナノスケールおよびマイクロスケールの両方で多孔質である。この多孔質特性は、ヒドロゲル足場内の細胞移動を補助することができ、栄養の拡散が可能である。1つの実施形態では、本発明は、ヒドロゲル化が迅速に誘発して細胞適合性を示す強固で多孔質な材料を得ることができる正確にデザインされたペプチドを提供する。生理学的に関連する刺激によって自己集合を誘発し、それにより、剪断薄膜化されてこの系の処理の用途が広くなるヒドロゲルを得ることができる。
【0045】
提案したヒドロゲル化系は、ペプチドが自己集合に従う折り畳まれた高次構造を採用する(すなわち、ペプチドが正確に折畳まれない場合、ヒドロゲルに自己集合しない)能力に依存する。さらに、環境上の合図にのみ応答して折畳まれるペプチドをデザインすることが可能である。結果は、所望の刺激の存在下でもヒドロゲル化するペプチド系である。この環境依存性により、各ペプチドが折畳まれるまで系がゲル化しないという点で、本質的に「高性能な」ヒドロゲルが得られる。刺激(トリガー)の性質に依存して、この過程を介したヒドロゲル化を完全に可逆的であるようにデザインすることができ、刺激の簡潔な除去により、足場を含むペプチドが折畳まれなくなってヒドロゲルが分解する。環境トリガーのいくつかの例には、pH、イオン強度、特異的イオン結合(例えば、Ca2+)、および電磁放射(例えば、光)が含まれる。
【0046】
いくつかの態様では、本発明は、(a)β−ヘアピン高次構造への分子内ペプチド折り畳みを誘発する工程と、(b)ヘアピンのヒドロゲルに自己集合する工程とを含み得るヒドロゲルの生成方法を提供する。所望の材料特性(例えば、多孔性、強固/剛性率、生体機能性など、図1を参照のこと)を有するヒドロゲルを提供するようにペプチドをデザインすることができる。
【0047】
本発明のヒドロゲルの全ての所望の態様を、ペプチドデザインによって制御することができる。本発明で使用されるペプチドは、小ペプチド(例えば、約10〜約200残基、約10〜約100残基、約10〜約75残基、約10〜約50残基、約10〜約40残基、約10〜約30残基、約10〜約25残基、約10〜約20残基、約15〜約200残基、約15〜約100残基、約15〜約75残基、約15〜約50残基、約15〜約40残基、約15〜約30残基、約15〜約25残基、約15〜約20残基、約20〜約200残基、約20〜約100残基、約20〜約75残基、約20〜約50残基、約20〜約40残基、約20〜約30残基、または約20〜約25残基)であり得る。本発明のペプチドは、1つ以上の修飾アミノ酸残基(例えば、D−アミノ酸、天然に存在するアミノ酸のホモログ、修飾側鎖を有するアミノ酸など)を組み込むことができる。本発明のペプチドは、好ましくは、1つ以上のトリガーに応答して二次構造(例えば、β−ヘアピン二次構造)を採用する。トリガーは、1つ以上の環境条件の変化であり得る。特異的ペプチドを後に詳述するが、本発明の1つの態様では、断続的な4残基のターン配列に隣接する高β−シート性向残基から構成され得る。極性および無極性残基を、折り畳み状態で両親媒性表面が得られるように鎖領域中に連続的に配置することができる。
【0048】
本発明のいくつかの実施形態では、ペプチドが自己集合する能力は、その単分子折り畳み状態に依存する。例えば、折り畳み条件下で、ペプチドは所望の二次構造を採用することができる(例えば、ヘアピンのある表面が疎水性残基で裏打ちされており、他の表面が親水性残基で裏打ちされている両親媒性β−ヘアピン構造を採用することができる)。この例では、分子内折り畳みは、折り畳みの際の疎水性表面上の電荷密度の軽減、分子内疎水性ファンデルワールス相互作用の形成、ヘアピン内のβ鎖間の分子内水素結合の形成、およびターン配列のターン性向によって決定づけられる(図2)。分子内折り畳みを適用する要因の詳細な知識により、折り畳み事象の活性な誘導機構をデザインすることが可能である。分子内折り畳みの後、単量体ヘアピンのその後の自己集合について、折畳まれたヘアピンの疎水性表面の疎水性会合によって表面を容易にし、隣接ヘアピン間のH結合の形成および疎水性ファンデルワールス接触を介して側面を容易にする。これらのパラメーターの詳細な知識により、自己集合過程を制御し、それにより、最終材料の性質を制御することが可能である。これらの全デザインパラメーターを以下に概説し、図2に概略的に示す。当業者は、ペプチドの分子間会合およびゲル形成を好むペプチド残基の高次構造が得られる他の所望の二次構造を採用するようにペプチドをデザインすることができることを認識することができる。
【0049】
本発明のヒドロゲルで使用されるペプチドを、1つ以上の以下のパラメーターの変化によって任意の所望の特徴を有するように構築することができる:1)静電学的性質(例えば、ペプチド分子内折り畳み率の電荷によって変化させることができる);2)例えば、ペプチドの折り畳みおよび自己集合ならびにヒドロゲルの材料特性を変化させる、a)側面および表面の分子間疎水性相互作用、および/またはb)分子内疎水性相互作用が変化したペプチドを構築するファンデルワールス相互作用、3)水素結合(例えば、折り畳み、自己集合、および最終的な材料の性質を変化させるための、a)分子内および/またはb)分子間の水素結合の形成が変化したペプチドを構築することができる)、および4)ターン配列(例えば、折り畳みを制御し、それによって自己集合が誘発されるように本発明のペプチドのターン領域をデザインすることができる)。
【0050】
本発明により、当業者は、所望の分子内折り畳み、分子間自己集合、および材料特性を有するように所望の特徴(すなわち、適切な静電学的性質、分子内および分子間疎水性ファンデルワールス相互作用、およびターン配列)を有するペプチドをデザインすることが可能である。いくつかの実施形態では、静電学的および/または疎水性ファンデルワールス相互作用を使用して、活性な分子内折り畳みトリガーを有するペプチドをデザインすることができる。トリガーは、ペプチドを含む溶液の1つ以上の特徴(例えば、pH、塩濃度、特定のイオンの濃度、電磁放射、および/または温度)を変化させることができる。
【0051】
いくつかの実施形態では、本明細書中に記載のようにデザインしたペプチドを使用して、細胞の接着(例えば、哺乳動物細胞の接着)および増殖を支持する非細胞傷害性足場として使用することができるヒドロゲルを生成することが出来る。
【0052】
1つの特定の位実施形態では、MAX1(20残基のペプチド)を、ペプチドの分子内折り畳み、自己集合、および最終的なヒドロゲル材料の性質に対する静電学的性質の効果を探索するようにデザインした。配列は、II’型ターン構造を採用するようにデザインされた断続的テトラペプチド−VDPPT−に隣接する高β−シート性向を示すバリン残基およびリジン残基から構成される(図3A)。ヘアピン構造を安定化させるための要素の局所デザインの組み込みに加えて、自己集合状態でのβ−ヘアピン形成を好むような別の様式でのβ−ターンに隣接する極性残基および無極性残基の配置によって非局所効果も考慮した。さらに、i+l位でのトランスプロリルアミド結合の幾何学的性質の強いるために、β分岐残基はターンのi位(Val−9)に存在した。このデザイン要素により、折り畳み条件下で自己集合前に単量体ヘアピンの分子内折り畳みが確実に好まれる。再度デザインしたシスプロリル結合により、拡大した高次構造での各分子内の各β鎖を示すことができる。シスおよびトランス配座異性体の両方を採用することができるペプチドは、拡大して正確に折り畳まれた単量体を無差別に自己会合し、再度積極的にデザインすることができる。
【0053】
MAX1がヒドロゲル化する能力は、その単分子折り畳み状態に依存する。MAX1のいくつかのリジン側鎖が中性である塩基性水溶液条件下で(pH9.0、125mMホウ酸、10mM NaCl)、このペプチドは、両親媒性β−ヘアピンに分子内で折り畳まれる(pH7の生理学的条件下で、折り畳みを誘発することもできる)。ヘアピンのある表面が疎水性バリン残基で裏打ちされており、他の表面が親水性リジン残基で裏打ちされている(図2および3A)。分子内折り畳みの後、ヘアピンのその後の自己集合について、(a)個別のヘアピン間のH結合の形成および魅力的な疎水性相互作用を介して側面を容易にし、(b)ヒドロゲル化する折り畳まれたペプチドのバリンリッチ面の疎水性会合によって表面を容易にする(図3B)。リジン残基の荷電状態を、pHによって制御し、単分子折り畳みは可逆的である。リジン側鎖の固有のpKa未満へのpHの低下により、隣接リジン由来の鎖内電荷が反作用し、その後に各ヘアピンの非折り畳みが起こり、最終的に自己集合ヒドロゲル構造が溶解する。古典的な微小繊維ベースのデザインから調製したβ−シートリッチヒドロゲルと比較した場合、この可逆的挙動は固有である。これらの系では、自己集合過程は不可逆的である。これにより、両親媒性ヘアピンは、不可逆的に微小繊維が得られるペプチドと異なる自己集合機構を受けることが示唆される。実際、巨視的な材料の性質による分子レベル由来の本発明者らのゲル化過程の特徴付けを以下に示し、MAX1の分子内折り畳みとその自己集合する能力との関連によって反応性材料を調製することができる。
【0054】
時間依存性円偏光二色性(CD)研究は、分子内ヘアピン形成およびその後の自己集合と一致するヒドロゲル化機構を支持する。図4Aは、マイクロモル濃度でpHが5.5(非折り畳み条件)から9.0(折り畳み条件)に上昇した場合、MAX1の非撹拌溶液は、20℃で数時間かけてランダムコイルからβ−シートに移行することを示す。25℃を超える温度では、MAX1の折り畳みおよび自己集合は約数秒で起こる。406分でのCDスペクトルは、216nmで明らかに最小であり、MAX1がβ−シートが豊富な構造を採用することを示す54。図4Bは、認められたθ216は濃度依存性であり(両濃度についてのいずれか1つの時点でのθ216と比較して)、MAX1が自己集合し、濃度増加につれて集合速度が増加することを示す。この挙動は、MAX1を調査するために使用される全スペクトル法および顕微鏡法を通して一貫している。例えば、図4Aおよび4Bに示すCD研究のために使用したマイクロモル濃度では、20℃での自己集合は数時間かかる。ミリモル濃度では(下記の流動学研究で使用したミリモル濃度など)、ヒドロゲルを形成する自己集合は20℃で数分かかる。興味深いことに、自己集合速度は、濃度依存性だけでなく、混合速度にも依存し、激しく撹拌したサンプルは20℃で数秒以内にシート構造を採用する。
【0055】
折り畳みおよび自己集合過程の可逆性を、図4Cに示すように、pHおよび時間の関数としてのθ216の測定によって調査した。pH5.5でのMAX1の撹拌溶液のCDは、無作為なコイル高次構造を示す。NaOHの添加によるpH9.0へのpHの調整により、予想通り、β−シートが形成される。しかし、pH6.0へのその後の調整により、シートシグナルが完全に喪失し、ランダムコイルシグナルが全て回収される。この実験は、おそらく、単分子折り畳みおよび非折り畳みがそれぞれ起こるリジン側鎖の脱プロトン化および再プロトン化の結果として集合過程が可逆的であることを示す。また、この可逆的挙動は、アミロイド/プリオン様微小繊維から形成されたヒドロゲルでは認められない。
【0056】
ヒドロゲルマトリクス内のβ−シート構造の存在は、FTIR分光法によっても支持される(図5)。pH5.5の1重量%のMAX1のD2O溶液(ペプチドが可溶性の場合)は、1644cm−1にアミドIバンドを示し、ペプチドが折り畳まれていないことが示唆される。しかし、NaODの添加によってこの溶液をpH9.0に調整した場合、ゲル化が起こり、アミドIバンドが1615cm−1にシフトし、MAX1がβ−シートに富む構造を採用したことが強く示唆される。
【0057】
バルク(bulk)流動学試験は、単分子折り畳みおよびその後の弾性の開始および増殖によるゲル網目構造への自己集合を示す。図6Aは、一定の歪みおよび周波数での掃引時間実験の結果を示し、この実験中に2重量%MAX1溶液についてヒドロゲル化が開始された後に貯蔵剛性率(サンプル剛性)の増殖をモニタリングした。CDによって認められたβ−シート形成と類似して(図4B)、流動学は、20℃で30分後にMAX1の折り畳みおよび集合が有意に進行したことを示す(約600Paのゲル貯蔵剛性率)。ゲル形成は成熟まで持続し、2時間後にゲル剛性率は約1200Paに倍増した。ゲル形成の数時間後、約1600Paの平衡貯蔵剛性率に達した。この平衡挙動は、図6Bにおいて線形の周波数依存性剛性率測定によって明確に示される。比較のために、ストロベリーゼラチンの貯蔵剛性率は、約50Paである(データ示さず)。流動学は、主に液体様(G’’>G’)と主にゲル様(G’>G’’)応答とを分ける交差濃度が約1重量%であることを示す。文献のヒドロゲル剛性率のいくつかの例も比較のために図6Bに示す。
【0058】
自己集合ゲルの2つの顕著な特徴は、剪断終了後の剪断薄膜化およびその後の弾性の回復を示すことである。この粘度の低下(剪断薄膜化)は、歪みの適用による物理的架橋の破壊に起因する。図6Cは、歪み速度の増加につれてMAX1由来のゲルが剪断薄膜化し、粘度が低下することを明確に示す。重要なことには、MAX1の自己集合ゲルは、分子自己集合過程の迅速な緩和時間による剪断の停止後に迅速に再形成することができる。処理が容易な組織工学で使用される可能性があるβ−ヘアピンベースのヒドロゲルの1つの主な利点を本明細書中に記載する。ヒドロゲルを、高度に制御された条件下にてex vivoで予め形成し、シリンジ注射/剪断薄膜化を介して宿主に移入することができる。
【0059】
図6Dでは、元のゲル形成時と同一の掃引時間実験を、6Hzで180秒間の歪み1000%の適用直後に行った。再形成ゲルの初期剛性率は、650Paであり、これはたった30分後に回復した平衡剛性率の80%である。このことにより、迅速に回復し、且つ比較的強力なヒドロゲルが得られる。比較のために、2重量%のゼラチンヒドロゲルは、剪断薄膜化後に平衡剛性率の80%に回復するのに4時間かかる。
【0060】
図4Bでは、流動学データと濃度依存性CDデータとの間で興味深い比較を行うことができる。CDは、MAX1が分子内で折り畳まれて自己集合する速度がペプチド濃度に正に依存することを明確に示す。流動学的にモニタリングした2.0重量%ゲルの形成(濃度約6mM)を、より高いペプチド濃度での分子の折り畳みおよび自己集合と外観が類似すると見なすことができる(図6A)。折り畳みのあと始めてゲル足場にβ−ヘアピン自己集合することができ、粘弾性応答が得られる。実際、MAX1と配列が類似するがヘアピン形成を嫌うようにデザインしたコントロールペプチドは、同一の折り畳み条件に供した場合にヒドロゲル化されなかった(以下を参照のこと)。
【0061】
pH変化によってゲル化機構が可逆的であることを証明する流動学実験を、最終pHが約6.0になる2重量%ゲルへの少量の濃縮HClの添加によって試みた。系の流動学的応答は、本質的に純水の流動学的応答であり、これは装置の感度の閾値未満であった。これにより、自己集合足場およびゲル化の可逆性が明確に認められた。これは、CDによって積極的にモニタリングした酸性条件下でのMAX1の直接非折り畳みと完全に一致する(図4C)。したがって、MAX1の分子内折り畳みおよび分子間シート集合を積極的にモニタリングしたCD実験とペプチドのゲル足場への自己集合を積極的にモニタリングした流動学実験とを組み合わせて、どのようにして材料の性質が分子折り畳みおよびその後の集合機構に帰し得るのかということを示す明確な画像を形成する。
【0062】
レーザー走査型共焦点顕微鏡法(LSCM)により、水路が連続ゲルマトリクスを透過する異種ゲル微細構造が明らかとなる(図7A)。このマイクロスケールの多孔性を活用して、組織工学足場を生成することができる。多孔質微細構造を、伝統的な架橋親水性ポリマーから調整したヒドロゲルに処理しなければならない。対照的に、ヘアピンベースのゲルの微小孔性は、自己集合過程の結果であり、さらなる処理は必要ない。図7A中のゲルマトリクス領域は固体ペプチドではなく、むしろ、それ自体が水に透過するナノスケールの希薄なペプチド網目構造である(図7B)。
【0063】
1重量%ゲルを含むD2Oについての小さなおよび非常に小さな中性子散乱(SANS/USANS)データの組み合わせ(図8)は、ナノスケールのゲルマトリクスの微小繊維構造およびマイクロスケールの異種形態と一致する。低散乱ベクトルq(q=(4π/λ)sin(θ/2)、λ=中性子波長およびθ=散乱角)での強度は、勾配−4は2相間の鋭い界面からの散乱を明確に示す。このq体制(regime)では、2相は、図7AのLSCMで明確に認められたゲルマトリクスならびに1〜10μmのサイズの水孔および水路である。より高いqでのSANSデータでは、ほとんどの顕著な特徴は、qの生成物および粒子の回転半径(Rg)が1未満であるGunier領域中の約−1の勾配である。これはナノスケールにおけるロッド状の対象物からの散乱を示す。この説明は、cryoTEMデータ(図7B)中で認められたもの(ゲル足場が接合点を介して相互連結された微小繊維構造の短い領域からなる)と一致する。
【0064】
上記で考察された結果により、当業者は、ペプチドの静電特性を変化させ、それにより、本発明のヒドロゲルの形成および/または材料の性質を制御することが可能である。上記の特定の例では、自己集合および材料の性質により、ペプチドの折り畳みが一次配列のリジン電荷密度の減少によって部分的に支配されることを示す。これにより、全リジン残基が折り畳まれた両親媒性ヘアピンのある表面を占めることが可能である。当業者は、リジン以外のアミノ酸残基を使用することができることを認識する。例えば、環境条件の変化によって電荷を有するか電荷を有するようにできる任意の残基を使用することができる。さらに、複数の異なるアミノ酸残基を同じペプチド中で使用することができる。
【0065】
本発明のいくつかの実施形態では、溶液pHの変化の使用により、分子内折り畳み事象およびその後のヒドロゲル化を誘発することができ、これは可逆性を示し得る。したがって、化学的応答性を、分子内折り畳みとその後の分子間集合とを関連づけることによって実現することができる。得られたヒドロゲルは機械的に強固であり得るが、ナノスケールおよびマイクロスケールで多孔質であり、それにより、細胞適合材料として非常に良好な候補となる。さらに、ヒドロゲル足場の自己集合性によって機械的応答性材料(剪断薄膜化法によるヒドロゲル/細胞構築物の送達に活用することができる特質)が生成される。
【0066】
図2Aに関して、本発明は、ヘアピン内の残基間の分子内ファンデルワールス接触の形成によって折り畳み状態を安定化することができ、異なるヘアピン残基の側鎖間の分子間の側面および表面の接触によって自己集合状態を安定化することができることを示す。これにより、これらのパラメーターの調整によって本発明のヒドロゲルの特徴を変化させることが可能である。相対疎水性が変化するヘアピン折り畳みおよびペプチドの自己集合の温度依存性の研究によってこれを示す。水が低温で疎水基をより良好に溶媒和することができることが周知である。この現象を使用して、いくつかのタンパク質が低温で折り畳まれず、その疎水性の内部が水性溶媒に曝露されるタンパク質の低温変性を記載している。150μMのMAX1水溶液は、室温未満に冷却された場合に折り畳まれない。5℃のCDスペクトルは、ランダムコイル高次構造と一致する(図9A)。しかし、溶液の40℃への加温により、β−シート構造と一致するスペクトルが得られる。温度の関数としての218nmの平均残基楕円率のモニタリングにより、折り畳みおよびその後の自己集合が誘発される温度(Tgel)が約20℃であることが示される(図9B)。実際、MAX1のpH誘導性折り畳みおよび自己集合を含む以前の研究を20℃で行った。この温度では、折り畳みおよび自己集合は、都合良くモニタリングされるのに十分に遅い。より高い温度では、ヒドロゲル化する折り畳みおよび自己集合は迅速である(濃度に依存して一瞬から数秒まで)。疎水性相互作用の形成が折り畳みおよび自己集合過程に影響を与える場合、MAX1の疎水基含有量の変化によって折り畳みが誘発される温度に影響を与えるはずである。図9Bは、MAX1および2つのさらなるペプチド(MAX2およびMAX3)のCDデータを示す。MAX2は、1つのバリン残基が同種構造(isostructural)であるが、疎水性の低い残基(トレオニン)に置換されている以外はMAX1と同一である。MAX3では、2つのバリンがトレオニン残基と置換されている。得られたペプチドは、オクタノールから水への移動自由エネルギーの計算値の比較によって明らかなように、その疎水性が変化する(図9)。CDデータは、疎水性が減少するにつれて、折り畳みが誘導される温度が漸増することを示す。したがって、疎水性接触の形成は折り畳みおよび自己集合に影響を及ぼす。このデータはまた、pHに加えて、温度を使用して折り畳みを誘発することができることを示す。したがって、本発明のヒドロゲルの特徴を、使用するアミノ酸残基の疎水性を増減させるためのヒドロゲルを形成するために使用したペプチドのアミノ酸残基の調整によって変化させることができる。
【0067】
熱応答性の巨大ポリマーが報告されている。本発明は、自己集合を誘導するために温度誘発性折り畳みを使用する第1の系を提供する。熱誘導性ヒドロゲル化がMAX1およびMAX2で可逆性でないにもかかわらず、MAX3の折り畳み、自己集合、および最終的なヒドロゲル化は熱可逆性を示す。150μMのMAX3溶液のCDスペクトルは、このペプチドが5℃で折り畳まれないことを示す(図10A)。溶液の80℃への加熱により、スペクトルがβ−シート構造と一致する。その後の温度サイクルは、折り畳みおよび非折り畳みが可逆的であることを示す。図10Bでは、流動学は、2重量%のMAX3水溶液が数回の加熱/冷却サイクルによって熱可逆的ヒドロゲル化を受けることを示す。CDおよび流動学で使用した温度は、折り畳みおよびその後の自己集合が誘発される温度を括弧で囲んでいる(Tgel=60℃、図9)。CDおよび流動学データを合わせて、温度誘導性単分子折り畳みおよびその後の自己集合と一致するヒドロゲル化機構が示唆される。これらのペプチドによって示された温度応答性挙動は、処理の容易さのためにその用途が広くなる。例えば、フリーフローペプチド溶液を室温で調製し、in vivoで投与し、体温でゲル化を誘導することができる。いくつかのポリ−N−イソプロピルアクリルアミドポリマーを、このような移行が最終的に組織再生のための細胞外様足場を提供するように操作した。上記研究は、疎水性相互作用の形成が重要であるという考えを支持する。MAX1〜3は全て水素結合した位置でバリンおよびトレオニンを含む。これらの位置では、その側鎖はトランス回転異性体を採用することを好む。結果として、互いに反対に存在する疎水性残基は、ヘアピン点を横切ってその側鎖が互いに反対方向に向かう疎水性残基により、鎖を横切る分子内疎水性相互作用の形成を困難にしている(図11A)。しかし、これらの側鎖は、自己集合状態で隣接ヘアピンの疎水性側鎖と側面に沿って相互作用するように良好に位置づけられている(図11B)。これらの側面疎水性相互作用の形成は、自己集合を好む。MAX4(非水素結合位置でその全バリン残基が組み込まれたMAX1に匹敵する疎水性を有するペプチド)を使用して、これを示す。MAX4では、対向するバリン側鎖は、互いの方向を指すと予想される(図11B)。したがって、MAX4のバリン側鎖は、MAX1中のバリン側鎖よりも側面分子間疎水性相互作用を形成する可能性が低い。側面分子間疎水性相互作用の形成が分子内疎水性接触よりも自己集合に重要である場合、自己集合速度は、MAX4よりもMAX1の方が早いはずである。この速度の相違は、貯蔵剛性率(剛性)の増加から明らかなはずである。実際、図12は、流動学的応答を導く自己集合は、40℃でのMAX4と比較して1重量%のMAX1調製物でより迅速であることを示す(両ペプチドについて、Tgel=20℃)。したがって、静電学に加えて、自己集合時の側面疎水性接触の形成はヒドロゲル化にも寄与する。
【0068】
本発明のいくつかの実施形態では、種々の官能性(例えば、細胞接着エピトープ、受容体アゴニスト、受容体アンタゴニスト、リガンド、低分子など)を、ヘアピンヒドロゲル足場に組み込むことができる。これを達成するための1つの方法は、ペプチドの1つ以上のアミノ酸側鎖を官能化することであろう。疎水性表面上のトレオニンが置換されたMAX2およびMAX3は高温での折り畳みおよび自己集合が可能であり、それにより、類似のサイズの側鎖を有する中性残基を許容することができることを示す。いくつかの実施形態では、特定の側鎖に組み込まれるべき部分は、より大きく(例えば、RGDベースのモチーフ)、荷電残基を含み得る。MAX1の疎水性表面上の荷電残基を組み込む可能性を調査するために、MAX5(VKVKVKVKVDPPTKVKEKVKV−NH2)を調製した。配列は、MAX1が通常は折り畳まれて集合する塩基性pHで負に荷電した16位のグルタミン残基を含む。CD分光学により、MAX5の150μM溶液(pH9.0、125mMホウ酸、10mM NaCl)がランダムコイルとして存在することを確認し、疎水性表面では負電荷が許容されないことを示す(データ示さず)。しかし、MAX1の疎水性表面の変化は十分に許容される。1つ以上のリジン残基がシステイン、セリン、およびグルタミン酸などの他の残基に置換された配列は、折り畳まれて強固なヒドロゲルに自己集合することができる(データ示さず)。
【0069】
上記のペプチドは、同一のターン配列(すなわち、(−VDPPT−))を含む。この配列は、II’型ターンを形成する強い性向があり、それにより、分子内折り畳みの駆動を補助する。分子内折り畳みが自己集合前に起こらなければならないので、ターン構造形成を阻害するこのターン配列の変化を使用して、分子内折り畳みおよび自己集合を調整してヒドロゲル化させることができる。ターン領域(VKVKVKVKVLPPTKVKVKVKV−NH2)の重要性を探索するために、MAX9を調製した。MAX9は、10位のDProがLProに置換されていること以外はMAX1と同一である。II’型ターン形成を好むMAX1内に含まれるジペプチドDProLProと異なり、MAX9のLProLProモチーフは開いた高次構造を好む。開いたLProLPro高次構造から生じる2つの鎖が、反対方向に突出するであろう。したがって、β−ヘアピンを形成する分子内折り畳みはあまり好ましくないであろう。任意の観察可能な自己集合は、拡大したペプチド配座異性体の直接分子間会合に起因する可能性が高いであろう。折り畳み条件下のMAX9の150μM溶液のCDは、4時間後でさえもランダムコイルのみを示した(データ示さず)。また、2重量%のMAX9溶液はヒドロゲル化できず、低粘度溶液が数日間保持された。興味深いことに、1週間後、自己集合が起こったが、ヒドロゲル化は起こらなかった。代わりに、MAX9の自己集合した拡大配座異性体と大きさが一致した長い微小繊維が認められた(図13)。これは、β−ヘアピン高次構造への分子内折り畳みがヒドロゲルへの自己集合に必要であり、ヒドロゲルが得られる機構は拡大された微小繊維が得られる機構と異なることを強く支持する。いくつかの実施形態では、細胞接着部位として役立つターン配列を組み込むことができる。例えば、RGD結合エピトープは、一般に、細胞接着事象に重要なタンパク質のターン領域内で見出される。
【0070】
1つの態様では、本発明は、所望の環境刺激の存在下でのみ折畳まれて適時に誘発されるヒドロゲル化系が得られるデザインされた配列を有するペプチドを提供する。材料の情報を一過性および空間的に制御する能力により、材料の処理が完全に制御可能である。組織工学への適用のためのヒドロゲル−細胞構築物の調製に特異的に、誘発機構の性質に依存して、in vitroおよび/またはin vivoで制御可能なヒドロゲル形成を行うことができる。例えば、MAX1は、酸性溶液条件下(pH<9、10mM NaCl)では折畳まれないが、25℃を超える温度でpHを9に調整した場合(10mM NaCl)、数秒以内に折畳まれ、自己集合する。温度変化に応答して折畳まれるペプチドも存在した。例えば、MAX2は、温度が40℃未満の場合、pH9(10mM NaCl)で折畳まれないが、45℃を超える温度に加温した場合、折り畳み/自己集合する(図9B)。本発明者らはまた、折り畳みが起こる温度を予想通りに調製することができることを示す。折り畳みトリガーのデザインは、折り畳みおよび自己集合過程を支配する基本原理の理解によってのみ可能となることを指摘することが重要である。
【0071】
考察したペプチドのヒドロゲル化は、今までのところ、10mM NaClの存在下での塩基性溶液条件(pH=9)で起こっている。150mM NaClの存在下での生物学的関連する条件下で(すなわち、pH7)ヒドロゲル化を誘発することが望ましい。実際、これらの溶液条件での折り畳みおよび自己集合の開始は、MAX1を使用することで可能である。図14Aは、pH7(20mM Tris)でのMAX1の折り畳みは塩に依存することを示すCDデータを示す。20mM KFの存在下では、MAX1は折畳まれないが、150mM KFの存在下では、MAX1は折畳まれ、自己集合する。150mMのNaCl溶液が低波長で有意なシグナル散乱を起こすので、光学的に透明なKFをCD研究に使用する。折り畳み事象を誘発する塩を、FTIRによってさらに確認し、1重量%調製物についてpH7でKFを添加した際、1643cm−1でのアミドIバンド(ランダムコイル)が1616cm−1(β−シート)にシフトすることを示す(データ示さず)。NaClはまた、pH7でヒドロゲル化を誘発することができ、MAX1水溶液へのNaCl(最終濃度=150mM)の添加によって自己集合し、強固なヒドロゲルが得られる(2重量%で3000Pa、データ示さず)。ヒドロゲル−細胞構築物の形成のために、細胞増殖培地の添加によるヒドロゲル化の開始が理想的であろう。DMEM増殖培地がNaClおよび他の塩(総塩濃度は約400mM)を含むので、これをトリガーとして使用することができる。MAX1の2重量%水溶液は、無血清DMEM増殖培地の添加の際にヒドロゲル化する。図14Bは、得られた粘弾性の強固なゲルの周波数掃引データを示す。さらなる折り畳みトリガーを使用して、折り畳み(特異的イオン結合(例えば、カルシウム結合)が含まれるが、これに限定されない)および/または電磁放射(例えば、光)を開始させることができる。
【0072】
当業者は、材料基質の機械的性質は細胞−材料相互作用に影響を与えることを承知している。例えば、血管平滑筋細胞が架橋ポリアクリルアミド基質のあまり強固でない領域からより強固な領域に移動することが最近示された。また、後根神経節の軸索伸長速度とアガロースヒドロゲル剛性との間に直接的な相関関係が認められている。本発明により、ペプチド濃度およびペプチド折り畳み誘発条件(すなわち、塩濃度および温度)によってヒドロゲル(例えば、MAX1含有ヒドロゲル)の剛性を調整可能である。図15は、異なる温度で集合した一定の塩濃度およびpH7.4での2重量%のMAX1ヒドロゲルの剛性を示す。最終網目構造のバルク剛性率は、低温から高温まで30℃にわたり調整可能である。重要なことには、MAX1集合を温度を用いて誘発する場合、この過程は不可逆的であり、それにより、各誘発温度で達成されるゲルの剛性は、生理学的温度を用いた場合に維持される。ヒドロゲル剛性の調整可能性は、ヒドロゲル剛性と細胞挙動(接着および増殖など)との間に相関関係がある。
【0073】
本発明は、本明細書中に記載の折り畳みトリガーを使用して、線維芽細胞(NIH3T3、マウス)の接着および増殖を支持するヒドロゲルを形成することができることを示す。結合組織の発達におけるその重要性およびヒドロゲル足場に接着した場合に形態が容易に識別されるので、これらの細胞をモデル系として使用した。細胞をポリスチレンコントロールウェルまたはMAX1−ヒドロゲルの均一なスラブ(厚さ2mm)を含むウェルのいずれかに添加する定性研究を行った。光学顕微鏡によって初期細胞播種時間から4時間後に拡大した細胞を直接観察することによって細胞接着を定性的にモニタリングした(定量的細胞付着アッセイを以下に記載する。Akiyama,S.K.Functional analysis of cell adhesion:Quantitation of cell−matrix attachment,2002;Vol.69,pp281−296も参照のこと)。非接着線維芽細胞は円形であり、接着した線維芽細胞は広がった形態を有する。下記のように、光学顕微鏡によって定性的に増殖を測定し、3Hチミジンベースの細胞播種アッセイによって定量的に増殖を測定した。以下のプロトコールのいずれか1つによって、48ウェル組織培養プレート中にヒドロゲルを調製した。プロトコールI:MAX1を水に溶解し(酸性溶液(約pH5)が得られる)、同体積の緩衝液(pH9、250mMホウ酸、20mM NaCl)を室温で添加して折り畳み/自己集合を誘導し、2重量%ヒドロゲルを得た。得られたゲルを、10%ウシ血清および10mg/mLゲンタマイシンを含むDMEM中に浸漬した。これにより、培地がゲル全体に浸透し、pH7.3に調整された。プロトコール1によって調製されたゲルの貯蔵弾性率(剛性)は、約1600Paである。プロトコール2:MAX1を水に溶解し、同体積の10mg/mLゲンタマイシンを含む無血清DMEMを添加して折り畳み/自己集合を誘導し、2重量%ヒドロゲルを得た。実験によってはウシ血清を含むDMEMを添加して、血清タンパク質を導入することができる。プロトコール2によって調製されたゲルの貯蔵弾性率は、約1200Paである。図16Aは、拡大した形態によって示されるように、4時間後に(血清の存在下)、線維芽細胞がゲル表面に接着したことを示す。密集に到達するまで(104個の細胞を最初にプレートした場合、通常、72時間)、細胞は増殖する(図16B)。新鮮な培地を提供する限り、細胞は少なくとも1ヶ月生存したままである(本発明者らはこのアッセイを1ヶ月で停止させた)。ゲルを含まないコントロールウェルに添加した細胞は類似の挙動を示す(図16CおよびD)。図16A〜Dでは、10%ウシ血清を含むDMEMの存在下で細胞をプレートする。血清タンパク質は、足場への最初のコーティングによってヒドロゲルへの細胞の接着の媒介を補助することができる。ヒドロゲル足場のみが細胞接着を助長するかどうかを決定するために、細胞を、血清の非存在下で2重量%ヒドロゲルにプレートした。図16Eは、血清タンパク質の非存在下でさえも4時間後に線維芽細胞が付着および拡大し始めるが、血清含有培地と比較して範囲は狭いことを示す。4時間後の血清の添加により、細胞が増殖し、72時間後にほぼ密集に到達する(図16F)。ヒドロゲルを使用しないウェルで行った同一のコントロール実験では、細胞が同様に挙動することが示された。これらの実験は、ヒドロゲル足場によって提供されたペプチド表面が血清タンパク質の存在下で線維芽細胞を接着させ、血清が存在しない場合、その範囲はより狭いことを示す。ヒドロゲル形成で使用したペプチドへの細胞結合エピトープの組み込みを使用して、血清の非存在下での細胞接着を増強することができる。この光学顕微鏡データは、MAX1ヒドロゲルが線維芽細胞増殖を支持することを定性的に示す。
【0074】
細胞増殖速度を測定するために使用することができる定量的アッセイを開発した。これにより、異なる材料特性および構成要素のペプチド配列を有するゲルを、いかに良くこれらが増殖を支持するかに関して比較可能である。MTTおよびXTTなどの標準的な比色分析アッセイは、本発明のヒドロゲルと共に使用するのは不適切であることが証明されている。これらのアッセイは、細胞がテトラゾリウム誘導体をその対応する有色ホルマザンアナログに代謝する能力に依存する。アナログはヘアピンヒドロゲルに浸透して固定化し、それにより、UV分光法によるその後の定量が信頼できない。チミジンをDNA複製した増殖細胞に組み込む3Hチミジンベースのアッセイの使用によってこの問題に取り組んだ。この方法は、分析物を可溶化する必要がなく、非組み込みチミジンをヒドロゲルから容易に洗い流し、組み込まれたチミジンのみの定量が可能である。アッセイは2要素からなる。第1に、細胞が密集に到達しない所与の期間(ここでは、便宜上48時間を選択する)にわたって増殖を追跡することを望む場合、プレートすべき最適な細胞数を決定するための細胞播種実験を行うことができる。第2に、材料への細胞(最適密度)の播種により、および密集に到達した場合、48時間までの個別の測定点でのDNA複製された細胞数の決定によって、所与の材料についての増殖速度を決定することができる。本発明のヒドロゲルの最適細胞播種密度を決定することが可能な3Hチミジン細胞播種実験の結果を図17に示す。アッセイすべき細胞(例えば、線維芽細胞)を、MAX1ヒドロゲルまたはポリスチレンコントロール表面に異なる初期密度で播種することができる。48時間後、増殖培地を除去し、3Hチミジンを含む培地と交換し、3時間インキュベートすることができる。非組み込みチミジンを洗い流し(3Hチミジンがもはや検出されなくなるまで洗浄物をアッセイすることができる)、シンチレーションカウンティングのために細胞を死滅させることができる。データは、MAX1ヒドロゲルを含む2日間の増殖アッセイについて、最適な増殖速度を得るために40,000個の線維芽細胞を播種しなければならないことを示す。ポリスチレンの最適播種密度が異なる(20,000)ことに留意すべきであり、任意の所与の細胞型が異なる材料について異なる最適播種密度を示すこと(および異なる細胞型が1つの所与の材料について異なる最適播種密度を有すること)が公知である。したがって、各新規の材料のために、増殖速度を決定する前に類似の細胞播種実験を行うことができる。このデータにより、48時間後に、コントロール表面上よりもヒドロゲル上でより多くの細胞が増殖することも示唆される(各表面のそれぞれの最適播種密度での分解物(数)の比較)。ヒドロゲル足場の三次元多孔性により、細胞がヒドロゲル内で増殖可能であり、二次元ポリスチレン表面では不可能であると説明することができる。このデータは、定量的増殖が確立されたことを示す。当業者は、本明細書中に記載のアッセイを使用して、細胞増殖に対するペプチド配列およびヒドロゲル剛性の効果を研究することができる。
【0075】
図16および17に概説した実験は、ゲルの二次元表面上への細胞のプレーティングを必要とする。二次元ゲル表面上の接着および増殖を利用するアッセイにより、異なるヒドロゲル足場を容易に比較可能であり、これは材料−細胞相互作用を調査するための最も一般的な技術であるので、結果を文献に記載の材料と直接比較することができる。
【0076】
本発明はまた、細胞がゲル足場全体に三次元で組み込まれたヒドロゲル−細胞構築物を含む。線維芽細胞を含む無血清DMEMの添加によるMAX1水溶液のヒドロゲル化の開始によってこれを行うことができる。細胞の三次元in vivo環境を模倣する足場により、二次元研究で証明されなかった細胞−材料相互作用がさらに洞察される。図18は、この様式で調製された2重量%MAX1ヒドロゲルのLSCM画像を示す。側面からゲルの内部を見ることができるように画像を構築する。細胞トラッカーグリーン(CMFDA,Molecular Probes)で染色した細胞がゲル全体で認められ、ゲル化が完了する前に細胞が沈み始めるゲルの底付近がわずかに濃度がより高い。当業者は、目的の任意の細胞型を本発明と組み合わせて使用することができることを認識する。例えば、使用することができる細胞には、酵母細胞、植物細胞、および動物細胞が含まれるが、これらに限定されない。適切な細胞は、例えば、公知の培養物寄託機関(例えば、American Type Culture Collection(Manassas,VA))および当業者に公知の商業的供給元から市販されている。本発明の方法で使用される好ましい動物細胞には、昆虫細胞(最も好ましくは、ショウジョウバエ細胞、スポドプテラ細胞、およびトリコプルサ(Trichoplusa)細胞)、線形動細胞(最も好ましくは、線虫細胞)、および哺乳動物細胞(CHO細胞、COS細胞、VERO細胞、BHK細胞、AE−I細胞、SP2/0細胞、L5.1細胞、ハイブリドーマ細胞、最も好ましくは、293細胞、PER−C6細胞、およびHeLa細胞)などのヒト細胞が含まれるが、これらに限定されない。さらに、初代細胞培養物、組織ホモジネート、および/または組織ホモジネート由来の細胞を、本発明と組み合わせて使用することができる。
【0077】
図2に関して、静電学、水素結合、および疎水性相互作用は全て折り畳みおよび自己集合で役割を果たす。折り畳みおよび自己集合のための静電学および側面の分子内および分子間疎水性接触の変化を使用して、本発明のヒドロゲルの材料特性および/または形成条件を変化させることができる。さらに、ヘアピンのターン領域の変化を使用して、本発明のヒドロゲルの特徴を調整することもできる。理論に拘束されることを望まないが、自己集合は、以下の2つの相互作用に支配されると考えられる:1)ヘアピンの疎水性崩壊、および2)分子間水素結合形成。これら2つの要因の相互作用により、疎水的に会合した微小繊維接合が散在した微小繊維の短いセグメントから構成される足場が得られる(図1)。当業者は、本明細書中に記載の教示を前提として、これらの要因のいずれかまたは両方の寄与を調整するために使用したペプチドの配列(またはそれによる物理的性質)を変化させて、任意の所望の特徴を有するヒドロゲルを生成することができる。
【0078】
図2に概説した分子間相互作用を変化させて、自己集合過程および得られた材料の性質(細胞増殖に重要なゲルの多孔性など)を制御することができる。20残基から構成されるヘアピンの特定の例を本明細書中に提供した。本発明はまた、これよりも長いおよび短いペプチドを含み、得られたヒドロゲルの材料特性に影響を与えるために使用したペプチドの長さを変化させることができる。
【0079】
本発明はまた、特定の例で使用したペプチドのターンと比較して1つ以上のアミノ酸置換を有するペプチドを含む。当業者に公知の任意のターン配列を、本発明と組み合わせて使用することができる。
【0080】
本発明のいくつかの実施形態では、1つ以上の機能的部分は、ペプチド配列、有機分子、または他の分子であって良く、例えば、一方または両方のヘアピン鎖の一次配列内および/またはターン配列の一次配列内で、親水性表面から生じるアミノ酸側鎖、疎水性表面から生じるアミノ酸側鎖、ターン配列を含むアミノ酸側鎖のペプチドに組み込むことができる。このような機能的部分の特定の例には、ペプチド配列(例えば、細胞接着エピトープ、核局在化シグナルなど)、受容体アゴニストおよび/または受容体アンタゴニスト(例えば、コレステロール誘導体など)、ペプチド模倣物、環状ペプチド、金属キレート剤、蛍光およびスピン活性プローブ、ならびに低分子治療因子が含まれるが、これらに限定されない。1つ以上のこのような機能的部分を含むペプチドを、所望の折り畳み/自己集合特性および/または材料特性を有するヒドロゲルを生成するための本明細書中に記載の分光法、光および中性子散乱、顕微鏡法、および流動学技術によって分析することができる。
【0081】
一定の水素結合と比較して分子間表面および側面疎水性接触の形成への寄与を変化させるために、MAX1に基づいたペプチドを調製することができる。このようなペプチドは、同数の残基を有し得るが、折り畳まれたヘアピンの疎水性表面上に側鎖の疎水性が変化したアミノ酸を含む。したがって、折り畳みおよび自己集合の間に形成された可能な分子内および分子間水素結合の数は一定であるが、疎水性面の表面領域が変化している。調製することができるペプチドの例には、以下の一般的配列のペプチドが含まれるが、これらに限定されない:XKXKXKXKVDPPTKXKXKXKX−NH2(式中、X=バリン(MAX1);X=イソロイシン(MAX7);およびX=フェニルアラニン(MAX8)。分子モデリング(洞察/発見)は、これらのペプチドの表面領域はMAX1<MAX7<MAX8の順で増加することを示す。表面領域の変化は、ヒドロゲルの種々のパラメーターに影響を及ぼし、例えば、CDによってモニタリングしたところ、各ペプチドが折畳まれて自己集合する(Tgel)温度を減少させることができる(図9を参照のこと)。また、各ペプチドの流動学的性質は異なっていて良く、より疎水性の高いペプチドによりより迅速にヒドロゲルを得ることができる。さらに、ヒドロゲル化速度が影響を受ける可能性があり、ペプチドの疎水性が高いほどより迅速にヒドロゲルが形成される。ヒドロゲル化速度を、流動学によって決定することができる(図12を参照のこと)。Cryo−TEMを使用して、これらのデザインの変化がヒドロゲルのナノスケールの構造に影響を与え(例えば、疎水性表面領域が、自己集合状態の微小繊維の接合数を増加させる場合)、LSCMを使用してマイクロスケールの構造の変化(例えば、ゲル多孔性の変化)を評価することができるかどうかを決定することができる。折り畳まれたペプチドの疎水性面から生じたアミノ酸残基の性質および数の変化および/または折り畳まれたペプチドの疎水性面から生じた機能的部分の性質および数の変化を使用して、形成されたヒドロゲルの材料特性を変化させることができる。
【0082】
自己集合時の分子間水素結合形成への寄与を変化させるために、分子間水素結合の形成能力がより高いかより低いペプチドを調製することができる。例えば、Nαアルキル化リジン残基を含むMAX1の誘導体を調製することができる。MAX1中のリジン残基を、これらが自己集合時に隣接するヘアピンと分子間水素結合を形成することができるように連続して配置する(図3A)。各ヘアピン鎖内の1つ以上のリジン残基のアルキル化により、ヒドロゲル化を起こす自己集合を阻害することができる。このようなペプチドの例には、以下が含まれるが、これらに限定されない:
【0083】
【化A】
(太字の位置にNαブチル化リジン残基を含む)。当業者は、アミノ酸残基および/またはアミノ酸残基に付着した機能的部分の変化により、分子間水素結合の形成能力を調整された(増加または減少)他のペプチドを構築することができることを容易に認識する。ヒトアミリンの微小繊維への自己集合がN−ブチル残基を含むペプチドによって阻害されていることが公知である。CD分光法および流動学を使用して、これらのペプチドが種々の折り畳み条件下で折り畳まれてヒドロゲルに自己集合する能力を評価することができる。
【0084】
いくつかの実施形態では、分子間相互作用を活用して自己集合過程および得られる材料特性を制御するようにペプチドを構築することができる。疎水性崩壊と分子間水素結合との組み合わせを介して折り畳まれて自己集合するペプチドのために、温度を使用してこれらの各過程が自己集合機構に寄与する程度を制御することができる。上記で考察するように、各ペプチドは、折り畳まれて(Tgel)、自己集合し始める特徴的温度を有する。さらに、温度を使用して疎水性崩壊を駆動することができることが文献に十分に示されている。したがって、任意の所与のペプチドのために、折り畳まれるが疎水性崩壊をあまり増強しないように過不足なく加熱する場合、水素結合が自己集合機構を支配し、得られたヒドロゲル足場は、微小繊維構造のより長いセグメントから構成され得る。得られたヒドロゲルを、より巨大な孔サイズによって特徴づけることができる。逆に、水素結合よりも疎水性崩壊が速度的に好まれる極端な温度へのペプチドの曝露により、より短い微小繊維セグメントおよびより多数の微小繊維間架橋を有する足場を得ることができる。得られたゲルは、より小さな孔サイズを有し得る。したがって、温度制御により、ゲルの微視的およびナノスケールの特徴を制御する一般的方法が得られる。いくつかの実施形態では、2つの異なる温度(T1=TgelおよびT2=Tgel+30℃)でペプチドを集合させることができる。得られたヒドロゲルを、cryo−TEMおよびLSCMによって視覚化して、ナノスケールおよびマイクロスケールの性質を評価することができる。より多数の微小繊維間架橋を含むゲルはより強固であり得る。これを、流動学によって検証することができる。
【0085】
いくつかの実施形態では、例えば、MAX1の一般的構造に基づいて、配列の長さが異なるペプチド(すなわち、VK(VK)mVKVKVDPPTKVKV(KV)nKV−NH2(式中、m=1〜20およびn=1〜20、mは任意の所与のペプチドにおいてnと同一であっても異なっていても良い))を調製することができる。より長いヘアピンの疎水性表面が増加するので、より長い鎖領域から構成されるペプチドはより強固なゲルを形成することができる。より強固なゲルが得られるより長い鎖領域から構成されるペプチドを使用して、機能的部分を組み込むことができる。単量体ヘアピンは、その鎖が約7残基長である場合に最も安定しており、鎖の伸長によってヘアピンの安定性が増加しないことが示されている。より長い鎖領域を有するペプチドのヒドロゲルを調製し、例えば、同一の折り畳み条件下で流動学を介してその剛性を評価することができる。
【0086】
いくつかの実施形態では、ターン領域の1つ以上のアミノ酸を、MAX1のターン領域と比較して置換および/または修飾することができる。いくつかの実施形態では、構造的役割を果たすだけでなく、生物機能的役割も果たすターン配列を組み込むことができる。例えば、RGD結合エピトープは、通常、細胞接着事象で重要であることが公知のタンパク質のターン領域内に見出され、RGDに隣接する残基は結合事象にさらなる特異性を与える。これらのエピトープの自己集合ヘアピンのターン領域への組み込みにより、細胞接着特性が増強されたヒドロゲル足場を得ることができる。上記で考察されるように、ターン構造を形成することができない配列の組み込みにより、折り畳むことができずにヒドロゲルが形成されないペプチド(MAX9)が得られる。しかし、ターン構造を採用することができるが、MAX1の−VDPPT−ほど促進しない別のターン配列を含むペプチドは、依然として折り畳まれてヒドロゲル化し得る。MAX1の文脈では、弱いターン形成物(former)から強いターン形成物にターン構造を形成するためにテトラペプチド配列−VDPPT−をその元の性質を変化させる配列と置換したペプチドを調製することができる。各ペプチドが分子内で折り畳まれて自己集合する能力を、CDによって決定することができ、その対応するヒドロゲルの流動学的性質を研究して、所望の特徴(例えば、組織工学のための可能な足場としてのその適切性)を有するヒドロゲルを同定することができる。調製することができるペプチドの例には、以下が含まれるが、これらに限定されない:VKVKVKVK−XXXX−KVKVKVKV−NH2(配列番号12)(式中、−XXXX−は、−VDPGT−(非常に強いターン)、−ADPGT−(強いターン)、−VNGT−(中程度のターン)、−VGGT−(配列番号13)(弱いターン)である)。
【0087】
いくつかの実施形態では、機能低部分(例えば、細胞接着エピトープ)を、以下のうちの1つ以上に組み込むことができる:a)ペプチドのβ鎖部分中の1つ以上のアミノ酸側鎖上、b)β鎖の末端および中間部分の一次配列内、c)ターン領域に隣接する位置、d)ターン配列を形成する1つ以上のアミノ酸残基の側鎖上、および/またはe)ターン配列の一次配列内。使用することができる機能的部分の例は、簡潔なトリペプチドRGDである。配列を、ヒドロゲルの意図する使用によって変化させることができる。例えば、細胞の足場としてヒドロゲルを使用することを意図する場合、使用すべき特定の細胞型の接着を誘発することが公知の配列によって機能的部分を変化させることができる。種々の細胞型のための特異的接着配列の例を以下に示す。図19中のペプチドを調製し(十分に確立された直交保護ストラテジーを使用して、側鎖修飾ペプチドを合成する)、CDおよび流動学によって研究してその折り畳み特性および流動学的性質を確立することができる。この実施形態にしたがって調製されたヒドロゲルは、増強された細胞接着および増殖特性を有し得る。
【0088】
いくつかの実施形態では、所望の特徴を有するヒドロゲルを生成するために、自己集合前の折り畳み単量体の濃度を変化させることができる。動的光散乱(DLS)と組み合わせてCDを使用して、自己集合を引き起こすために存在しなければならない折り畳まれた単量体の濃度を決定することができる。DLSにより、自己集合過程によってナノ構造成長を直接モニタリングすることが可能である。5nmから1ミクロンまでの粒子サイズの分布を容易に観察することができる。例えば、MAX1を使用し、CDを使用して、20℃でのβ−シート形成速度をモニタリングすることができる。CDシグナルの大きさが分子内(折り畳みによる)β−シート形成対分子間(自己集合による)β−シート形成に帰し得る範囲を、同一溶液(μMペプチド濃度)についてのCDβ−シートシグナルの成長と自己集合粒子サイズの成長との実時間比較によって決定することができる。重要なことに、折り畳まれた単量体は、DLSにおける粒子サイズに成長しない。したがって、粒子サイズの成長が同時に起こることなくCDによって大量のβ−シート構造が認められる場合、自己集合を引き起こすために折り畳まれた単量体の集団が大量に存在しなければならないと結論づけることができる。逆に、CDβ−シートシグナル成長と同時に粒子が自己集合する場合、自己集合が分子内折り畳みの直後に起こると結論づけることができる。DLS中での粒子サイズの成長と流動学によってモニタリングされた貯蔵弾性率の成長との類似の実時間比較を行うことができる。DLSと流動学との間の直接比較を行うことができるようにミリモル粒子濃度を使用する。網目構造の流動学的性質が導入される粒子サイズの閾値を、直接観察する。
【0089】
いくつかの実施形態では、本発明のヒドロゲルは、増強された処理可能性を示し得る。ペプチド溶液が適時にヒドロゲル化可能な1つ以上の活性な分子内折り畳みトリガーを有するペプチドオンデザインの結果として、当該分野で公知のヒドロゲルと比較して、本発明のヒドロゲルの処理可能性を増強することができる。上記で示すように、折り畳みを誘発する能力により、自己集合およびその後のヒドロゲル形成を直接制御する手段が得られる。さらに、誘発エレメントを容易に組み込むことができる理想的な分子足場である適切なペプチド二次構造(すなわち、ヘアピンモチーフ)の特定の例を提供している。本発明のいくつかの実施形態では、pH、イオン強度、および温度などの生理学的に関連する刺激に応答してヒドロゲル化するようにヘアピンをデザインすることができる。処理可能性に関して、折り畳みの誘発により、in vitroおよびin vivoの両方でヒドロゲル化ストラテジーを実施することが可能である。本発明のさらなる実施形態では、さらなる手段(例えば、電磁放射(例えば、光)および特異的イオン結合(例えば、カルシウムイオン結合))を使用して、ヒドロゲル化を誘発することができる。ヒドロゲル化を誘発するための電磁放射の使用により、光ファイバーを介したin vivoでの空間分解材料の形成を誘導する都合の良い手段が得られる。例えば、低粘度のペプチド−細胞懸濁液を投与し、その後に照射によって局所的にゲル化することができる。組織中に天然に見出される刺激を介したex vivoでのヒドロゲル化を誘発する能力により、材料形成を制御する別の都合の良い手段が得られる。特異的イオン結合(例えば、カルシウムイオン結合)に応答してヒドロゲル化するペプチドを調製することができる。特異的イオンとしてカルシウムイオンを使用する場合、得られた材料は、細胞が増殖することができる足場として役立つだけでなく、ペプチド足場の生分解時に放出されるカルシウム供給源も提供する。
【0090】
本発明のヒドロゲルのデザインでは、折り畳みおよび自己集合過程を、当業者に公知の任意の適切な技術(例えば、CDおよびFTIR)によって特徴づけることができる。材料のナノスケールおよびマイクロスケールの多孔性を、cryo−TEM、LSCM、USANS、およびSANSによって決定することができ、材料の剛性を流動学(例えば、本明細書中に記載の方法を使用する)によって測定することができる。誘発過程の効率も試験することができる。いくつかの実施形態では、in vivoヒドロゲル化を意図するペプチドのための誘発系は、迅速にヒドロゲル化して強固な多孔質ゲルが得られる誘発系であり得る。希薄なペプチド溶液についてのトリガー誘導性シート形成速度をCDによって決定することができ、対応する1〜2重量%のヒドロゲルの剛性を振動流動学によって決定することができる。in vitro適用のために、ゲル化速度が迅速であることが望ましいが、必要ではない。特定の例では、MAX1を、以下で考察している各トリガーの有効性を調査するためのモデル系として使用することができる。しかし、一旦MAX1の状況で特定のトリガーの実行可能性が確立されると、トリガーを、MAX1と異なる所望の特質を示す他のペプチド配列に組み込むことができる。
【0091】
本発明のいくつかの実施形態では、電磁放射(例えば、光)を使用して、本発明のペプチドのヒドロゲル化を誘発することができる。適切な方法の例には、図20に示す方法が含まれる。図20に示す実施形態には、折り畳みを阻害するためのフォトケージ(photocage)の使用が含まれる。このようなフォトケージを、例えば、本発明のペプチドのヘアピンを含む残基の側鎖および/またはペプチド骨格内に組み込むことができる。折り畳みおよびそれによる自己集合を、光へのペプチド水溶液の曝露によって開始することができる。
【0092】
上記で考察するように、ヘアピンの疎水性表面(例えば、Glu)への荷電残基の組み込みにより、折り畳みおよびそれによる自己集合が阻害される。この阻害を使用して、疎水性表面への負電荷のフォトケージ(2−ニトロフェニル酢酸)の組み込みによって折り畳みを阻害することができる。1つの実施形態では、MAX1の16位のアミノ酸残基を、システイン(MAX6)と置換し、文献のプロトコールにしたがって2−ブロモ−2−(2−ニトロフェニル)酢酸でアルキル化してケージドペプチドを得ることができる(Pan & Bayley,Febs Letters 1997,405,81−85)。2−ニトロフェニル酢酸は一般的に使用されているフォトケージであり、ヘテロ原子由来のその光誘導性切断は十分に研究されている。ケージドペプチドの光(330〜360nm)への曝露により、負電荷のケージが放出され、疎水性表面上に中性Cysが生成される。光分解時に形成されたニトロソケトン副生成物が新規の形成されたCys側鎖をアルキル化する場合(潜在的に有害な副反応)、1mMジチオスレイトールを添加してニトロソケトン反応を停止させて還元環境を維持することができる。16位を最初に選択して修飾する。なぜならばこの位置でCysを含むコントロールペプチドが折り畳まれて自己集合するからである。当業者は、ペプチド中の1つ以上の他の位置を類似の様式で処理することができることを認識する。光分解効率は、配列位置に依存し得るので、フォトケージ化された(photocaged)残基の種々の位置および/または数を使用して、得られたヒドロゲルの性質を変化させることができる。いくつかの実施形態では、光分解速度は、マイクロ秒からミリ秒までの時間体制(time regime)であり得る。典型的には、照射時間は、約1マイクロ秒から数ミリ秒までであり得る(例えば、約0.01μs〜約1000ms、約0.01μs〜約100ms、約0.01μs〜約10ms、約0.01μs〜約1ms、約0.01μs〜約0.5ms、約0.1μs〜約1000ms、約0.1μs〜約100ms、約0.1μs〜約10ms、約0.1μs〜約1ms、約0.1μs〜約0.5ms、約1.0μs〜約1000ms、約1.0μs〜約100ms、約1.0μs〜約10ms、約1.0μs〜約1ms、約1.0μs〜約0.5ms、約10μs〜約1000ms、約10μs〜約100ms、約10μs〜約10ms、約10μs〜約1ms、または約10μs〜約0.5ms)。適切な照射供給源は、バルクおよび/または空間分解照射を行うことができ、水銀灯および空間分解照射が可能なUVレーザーを備えたNikon eclipse TE2000倒立蛍光顕微鏡(全て市販されている)が含まれるが、これらに限定されない。
【0093】
いくつかの実施形態では、ペプチドのN−アルキル化を使用して、水素結合駆動自己集合を阻害することができる。通常は分子内水素結合を形成する骨格のアミド窒素(例えば、MAX1の16位)にN−o−ニトロベンジルケージを組み込んだβ−ヘアピンを、標準的技術を使用して合成することができる。分子内水素結合が立体的に込み合っているので、この位置を占めるケージを使用して、分子内折り畳みを阻害することができる(図20)。o−ニトロベンジルケージは、その性質が十分に確立されている別のクラスの化合物である。光(330〜360nm)への曝露によってケージが放出され、水素結合の形成およびそれによる折り畳み/自己集合に関与し得るプロトン化アミドが再生される。光分解効率を配列の位置によって変化させることができ、これを使用して、ヒドロゲルの形成および/または特徴付けを変化させることができる。部位特異的N−アルキル化ペプチドを、文献のプロトコールによる固相ペプチド合成(SPPS)によって調製することができる(Reichwein,et al.,Tetrahedron Letters 1998,39,1243−1246およびTatsu,et al.,Febs Letters 2002,525,20−24を参照のこと)。上記の特定のケージを細胞で使用しているので、ケージの副生成物は細胞傷害性を示す可能性が低い。しかし、文献に記載の方法によってこの系の細胞適合性を測定することができる(Bryant,et al.,Journal of Biomaterials Science−Polymer Edition 2000,11,439−457を参照のこと)。上記のo−ニトロ誘導体は全て切断を誘導するための1つの光子励起を含むので、細胞の存在下でヒドロゲル化を誘発するための330〜360nmの光の使用によってUVベースの細胞の光損傷が起こり得るという別の可能性が懸念される。一般に、明らかな細胞損傷が認められない細胞ベースの実験を行う状況で1つの光子励起を使用するにもかかわらず、任意の特定の細胞型でUVベースの損傷が認められる場合、IRで吸収される2つの光子ケージ(例えば、クマリン誘導体)を使用することができる(Furuta,et al,Proceedings of the National Academy of Sciences of the United States of America 1999,96,1193−1200)。
【0094】
いくつかの実施形態では、特定のイオンの結合を使用して、ヒドロゲル化を誘発することができる。例えば、カルシウムイオン結合によってヒドロゲル化を誘発することができる。典型的に、カルシウムイオンの非存在下でのカルシウムイオンの結合を含む実施形態では、ペプチドが折り畳まれない一方で、カルシウムの添加は折り畳み/自己集合の核となる。上で示すように、ヘアピンのターン領域は、分子内折り畳みおよびそれによる自己集合の促進において重要であり得る。1つの特定の例では、4残基のターン(−VDPPT−)が金属イオンの非存在下で規則正しい構造を採用することができないターン/ループ配列または非天然キレートに置換されたMAX1の変異形を調製することができる。金属結合は、ターン形成に影響を与え、それにより、分子内折り畳みおよび自己集合を促進する。図20は、以下に詳述する金属トリガーを示す。
【0095】
1つの特定の実施形態では、4残基のターン(−VDPPT−)を、−DRKADGYIDFEE−と置換して、VKVKVKVKVDRKADGYIDFEEVKVKVKVKV(配列番号14)を得るMAX1の変異形がある。このペプチドは、金属の非存在下で規則正しくないはずであるが、Ca(II)の結合後に迅速な折り畳みおよび自己集合が起こる。配列DRKADGYIDFEEは、トロポニンCのEFハンドドメイン内に含まれるカルシウム結合ループに対応する。下線を引いた残基は、側鎖および主鎖カルボニルの両方を介してCa(II)に結合してエクアトリアル位の1つを占める水を有する五角錐の形状が得られる。重要なことには、この線形ペプチドが低mM KdでCa(II)と結合することができ、この結合事象により構造が形成されることが示されている。トロポニンCの結晶構造は、カルシウム結合ループのN末端およびC末端残基が約6Å以内であり、これらの末端残基がねじれ角を採用して逆平行シートの形成を促すことを示す。実際、このデザインしたペプチドを使用してエネルギー最小化および明示的溶媒(explicit solvent)を使用した力学的刺激を行い、この金属結合ループがMAX1の状況下でβ−ヘアピン構造の核となるように幾何学的に十分に適合していることが示された。
【0096】
別の特定の実施形態では、さらなる高親和性トリガーを使用することができる。このデザインにより、非天然Ca(II)キレート剤DOTAM(十分に研究されているオクダデンテートリガンドDOTAの誘導体である)の使用が求められる(図20)。DOTAMは、カルシウムと強固に結合して(Kd=29nM)、互いに5Å以内にリガンドのカルボニル含有アームが存在する四角反柱複合体を形成し、これら2つの位置がペプチド結合に理想的になる。したがって、図20のMAX1の4残基のターン(−VDPPT−)のDOTAM−誘導体との置換により、Ca(II)の非存在下で構築されない系が得られる。しかし、Ca(II)が結合する場合、2つのβ鎖が互いに5Å以内になり、折り畳みが開始される。このペプチド模倣物を、市販または容易に合成される出発材料を使用した手作業の固相合成を使用して調製することができる。
【0097】
いくつかの実施形態では、ペプチドの構造および材料の性質を使用して、細胞株(例えば、モデル線維芽細胞株および骨芽細胞株)の接着および増殖を調整することができる。上記のヒドロゲルは、組織工学への適用に有用であり得る(例えば、電解質濃度、温度、および光などの生体適合性刺激によって材料の形成を誘発し、自立に十分な剛性(10Pa超)を示す多孔質ゲルが得られる適用)。本開示により、当業者は、ペプチド構造と、材料の剛性と、細胞適合性とを相関させると同時に、最終的に組織工学に有用であり得るヒドロゲルを同定することが可能である。組織工学を実現するための3つの望ましい細胞レベルの生物学的特徴は、以下である:a)ヒドロゲルが所望の細胞に対して非細胞傷害性であること、b)ヒドロゲルが細胞接着(付着および拡大)を促進すること、およびc)ヒドロゲルによって細胞が増殖すること。本発明のヒドロゲルは、典型的には、1つ以上のこれらの特徴を有するが、かならずしもそうではない。第1に、細胞適合性を評価するための実験ストラテジー(パートa、b、およびc)を記載する。第2に、これらの研究に適切な細胞株の例を考察する。第3に、当業者が、ペプチド構造と、材料の剛性、細胞適合性との間の相互作用を描写し、適切なペプチドを選択し、特定の適用(例えば、特定の細胞株)を誘発することが可能な特定の例を記載する。いくつかの実施形態では、細胞結合エピトープをペプチドに組み込んで材料の細胞適合性を増強することができる。使用することができる細胞型の例には、哺乳動物細胞、NIH 3T3線維芽細胞、HEK293、HELA細胞株、およびラット頭蓋冠MC3T3−E1骨芽細胞、神経細胞、幹細胞、および組織サンプル(例えば、脳、肝臓、心臓など)由来の細胞が含まれるが、これらに限定されない。
【0098】
いくつかの実施形態では、細胞の接着および増殖を容易にするペプチドおよび誘発条件を選択することが望ましい。典型的には、線維芽細胞付着には4時間で十分であり、ほとんど全ての細胞が拡大するが増殖しない。文献のプロトコールには、増殖しない骨芽細胞の付着には12時間で十分であることが示されている。したがって、線維芽細胞の生存を、ヒドロゲルへの導入から4時間後に、たとえば生細胞対死細胞の優先的染色および蛍光ベースの画像化の実施によってアッセイすることができ、骨芽細胞を12時間後にアッセイすることができる。既知数の各細胞を、組織培養プレート中に予め形成したヒドロゲルに播種することができる。各細胞型についてそれぞれの時間をおいた後、カルセインAMまたはペンタフルオロベンゾイルアミノフルオレセイン二酢酸を使用して、生細胞の細胞質を染色することができる。ヨウ化プロピジウムを使用して、死細胞を染色することができる。ヒドロゲルを含まない組織培養プレートを使用した並行コントロールを実施することができる。ヒドロゲル中の死細胞数がコントロールプレートで認められた数よりも多い場合、材料は、特定の細胞型に不適切であると考えることができる。アポトーシスを受けた細胞が色素取り込みに抵抗し、ヒドロゲル細胞傷害性が不正確に評価される可能性がある。これらのゲルの細胞傷害性を、下記の接着および増殖研究でさらに検証することができる。予備実験の結果に基づいて、提案したゲルの大部分は、非細胞傷害性を示すようである。最後に、提案した色素に問題があると証明された場合、別の色素組(例えば、SYTO(SYTOX(死細胞の核酸指示薬)と組み合わせて使用される生細胞の核酸指示薬))を使用して細胞傷害性を評価することができる。
【0099】
一般に、合成材料を、細胞培養基質としてin vitroで使用する場合、細胞接着は、主に、非特異的に足場をコーティングする増殖培地中に存在する血清タンパク質(例えば、フィブロネクチンおよびビトロネクトリン(vitronectrin))によって媒介される。しかし、本明細書中に記載の機構の誘発によって最終的にin vivoで導入することができるヒドロゲルについて、ヒドロゲル足場は、血清タンパク質の非存在下で細胞接着を本質的に促進することができる。これにより、潜在的な宿主が足場に細胞接着タンパク質を導入する機構を提供しない場合、細胞付着が確実になる。本発明のヒドロゲルは、10%ウシ血清の存在下および非存在下の両方で細胞接着を促進することができる。穏やかな洗浄後の付着細胞画分対非付着細胞画分の決定によって細胞付着をX線で定量するアッセイを使用して、細胞付着に適切なヒドロゲルを同定することができる。詳細には、1)目的の細胞株(例えば、哺乳動物線維芽細胞または骨芽細胞)を培養し、3Hチミジンで24時間予め標識することができる。非組み込み標識を洗浄によって除去し、2時間追跡することができる。2)次いで、既知濃度の標識細胞を非処理組織培養ウェル(コントロール)および本発明のヒドロゲルを含むウェルの双方に導入することができる。ヒドロゲルを含むウェルでは、ウシ血清を含むか含まない細胞増殖培地を添加することができる。3)例えば、線維芽細胞については6時間にわたって細胞接着速度を最初にモニタリングし、その時間内の増加量を、日常的実験を使用して当業者が実験的に決定することができる。骨芽細胞接着速度を、24時間にわたって最初にモニタリングすることができる。これらの最終時点を実験的に精緻化することができ、付着および拡大するのに十分であるが、増殖しない時間を与えなければならない。また、終点が長すぎることに関する問題は、長時間インキュベートした場合、いくつかの細胞が、膜結合プロテアーゼまたは分泌されたマトリクスタンパク質によってヒドロゲル表面が修飾され、「非粘着(non−stick)」表面上でさえも付着し得ることである。このシナリオでは、細胞によって付着が促進され、必ずしも材料の表面によってではない。最適な終点を、熱変性BSAでコーティングした表面(典型的な非粘着表面)上の細胞のプレーティングによって決定することができる。2〜3%を超える細胞が付着した場合、最大終点に到達している。4)適切な時点で、非付着細胞を穏やかに洗い流し、液体シンチレーション分光法によって計数し、付着した細胞を、定量のための検量線を使用したシンチレーションカウンティングのために可溶化することができる。したがって、ポリスチレン組織培養物と血清タンパク質を含むか含まないヒドロゲル表面とを比較することができる。付着後の細胞拡大を、光学的に検証することができる。このようにして得た情報を使用して、任意の目的の細胞型の接着を促進するように適切なヒドロゲルをデザインすることができる。ペプチド構造の変化により、剛性および足場形態などの材料特性を、ヒドロゲルの細胞接着を助長する能力を至適化するように調整することができる。例えば、上記の簡潔な細胞拡大アッセイにより、線維芽細胞が血清タンパク質の存在下でMAX1ヒドロゲル(貯蔵剛性率=1200Pa)に効率良く付着したが、血清タンパク質の非存在下ではあまり効率が良くないことが示された。この特徴を有するヒドロゲルは、in vitro適用に有用であり得る。ペプチドへの構造修飾の導入および/または異なる剛性のヒドロゲルの調製により、血清タンパク質の非存在下で細胞接着を増強することができる。細胞接着が増強したヒドロゲルを、in vitroおよびin vivo適用のために使用することができ、細胞接着度がより低いヒドロゲルよりもin vivo適用により適切であり得る。
【0100】
いくつかの実施形態では、上記で概説の細胞接着を促進する非細胞傷害性ヒドロゲルを使用して、接着細胞の増殖を支持することができる。上記の3Hチミジンアッセイを使用して、増殖速度を評価することができる。所与のゲルについて、例えば、線維芽細胞についての48時間継続する速度実験の使用によって至適細胞播種密度を決定することができる。図17に関して、MAX1ヒドロゲルへの40,000個の線維芽細胞の最初の播種により、密集に到達するまで、2日間増殖が継続する。増殖を、任意の適切な期間(例えば、2日間またはより長い期間にわたり)継続させることができる。他の細胞型(例えば、骨芽細胞、神経細胞、幹細胞など)を含む速度実験の継続期間を、類似の様式で実験的に決定することができる。至適播種密度を決定した後、静止細胞をヒドロゲルに播種し、増殖細胞数を、3Hチミジン取り込みによる時間の関数として定量することができる。1つの実施形態では、光学密度での細胞を、10%ウシ血清を含む非標識培地の存在下で、24ウェル組織培養プレート中の予め形成されたヒドロゲルにプレートすることができる。三連で、細胞を、異なる期間(例えば、6、12、18時間など)増殖させることができる。各時間後、増殖培地を除去し、3Hチミジンを含む培地と置換することができる。細胞に3Hチミジンを3時間取り込ませることができる。非組み込みチミジンを洗い流し(3Hチミジンがもはや検出されなくなるまで洗浄物をアッセイすることができる)、シンチレーションカウンティングのために細胞を死滅させることができる。検量線(組織培養ポリスチレン上での所望の細胞の培養および血球計算板によって計数した細胞とシンチレーションカウントとの直接相関によって以前に決定することができる)との比較により、その時点でDNA複製を受けた細胞を定量することが可能である。いくつかの実施形態では、一定のヒドロゲル上での増殖細胞数は、ヒドロゲル表面の二次元領域内におそらく適合し得る数を超え得る。これらの場合、細胞を、ヒドロゲル足場の孔および流路に増殖させることができる。いくつかの実施形態では、本発明のヒドロゲルを、ヒドロゲル足場への細胞の増殖を促進するようにデザインすることができる。このヒドロゲル型を、例えば、時間の関数として三次元画像を収集することができるLSCMによる予め染色された細胞の増殖にしたがって容易に同定することができ、ヒドロゲル内部に移動した細胞が明らかに認められる。任意の適切な色素(例えば、Vybrant CFDA SE(Molecular Probes)(細胞の細胞質中に隔離され、娘細胞に伝えられる緑色蛍光プローブ(細胞生存率に影響を与えない)))を使用して、細胞を予め染色することができる。当業者は、細胞増殖を促進する所望の能力を有するヒドロゲルを同定するために、このアッセイ型を使用することができる。ペプチド構造およびそれによって得られるヒドロゲルの材料特性(剛性および足場形態など)の変化により、以下で考察しているように、細胞増殖を助長するヒドロゲルを生成することができる。
【0101】
上記アッセイは、二次元ヒドロゲル表面上接着および増殖の評価で有用であり、異なるヒドゲル足場が容易に比較可能であり、ほとんどの公開されている研究は二次元で実施されているので、結果を文献中の材料と直接比較することができる。図18に示すように、細胞の存在下での材料形成の誘発により、ヒドロゲル全体にこれらの細胞を三次元で組み込むことが可能である。細胞接着を促進する非細胞傷害性ヒドロゲル(例えば、上記で同定したヒドロゲル)を、三次元基質として使用することもできる。ヒドロゲル内部に隔離された非付着細胞を洗い流すことができないので、細胞付着は定量が困難であり得る。しかし、定性的細胞拡大アッセイを使用して、細胞拡大およびそれによる付着を測定することができる。細胞を、例えば、Vybrant CFDA SEで予め染色し、その後にヒドロゲルマトリクスに組み込むことができる。最初に組み込まれた細胞の細胞形態を、LSCM(非接着細胞は円形であり、接着した線維芽細胞は広がっている)によって三次元で視覚化することができる。さらに、色素が子孫(prodigy)に伝えられるので、細胞増殖を定性的に視覚化することができる。二次元研究のための上記と同一のアッセイを使用して、増殖速度を定量的に決定することができる。3Hチミジンはヒドロゲルマトリクスに容易に拡散し、DNA複製を受けた細胞でも利用可能なはずである。
【0102】
いくつかの実施形態では、本発明を使用して、細胞(例えば、細胞株)の接着および/または増殖を支持することができる。このような細胞または細胞株は、当業者に公知の任意の型であってよく、任意の目的のために(例えば、組織の生成または所望のタンパク質の発現のために)培養することができる。組織工学に関連する実施形態では、in vitro培養時のその高い能力に加えて、その結合組織および骨生成/治癒に関与するヒト細胞との関係に基づいて、モデル細胞株を選択することができる。この目的のための適切な細胞株の例には、NIH/3T3マウス線維芽細胞(ヒト結合組織の発達に関与する線維芽細胞モデルとして使用することができる不死化細胞株)およびラット頭蓋冠(MC3T3−E1サブクローン4)細胞(ヒト骨芽細胞モデルとして使用することができる)が含まれるが、これらに限定されない。両細胞株は十分に研究されており、重要な文献データベースにより、提案されたヒドロゲルの性能を以前に研究した材料を基準にして評価することが可能である。当業者は、MC3T3−E1細胞株の使用には限度がある可能性があることを承知している(その非分化線維芽細胞様表現型に戻す能力など)。いくつかの実施形態では、本発明のヒドロゲルを使用して、初代組織サンプル由来の細胞(例えば、頭蓋冠または骨髄由来の初代骨芽細胞)の接着および/または増殖を支持することができる。
【0103】
いくつかの実施形態では、本発明のヒドロゲルは、1つ以上の細胞および/または1つ以上の治療因子(例えば、医薬品)を含み得る。この目的のためのヒドロゲルを、動物モデル系で標準的技術を使用して同定することができる。動物への挿入前にヒドロゲルを予め形成することができるか、溶液の形態で挿入してin situでヒドロゲル化を誘発することができる。いくつかの実施形態では、生物学的に関連する刺激を介して材料形成を誘発するペプチドによってヒドロゲルを形成することができる。例えば、細胞および/または治療因子を含み得るペプチド溶液を、動物(例えば、ヒト)に挿入し、動物に対して内因的な1つ以上のトリガー(例えば、pH、イオン強度など)の結果としてヒドロゲル化することができる。他の実施形態では、外因性トリガー(例えば、電磁放射(例えば、光))を使用して、ヒドロゲル化を行うことができる。本発明の材料および方法を使用して、所望の剛性を有する足場を構築し、in vivoで使用することができる。このような足場は、細胞の接着および増殖ならびに/または治療因子の送達に最適な機械的特性を有する基質を提供することができる。
【0104】
いくつかの実施形態では、本発明は、ヒドロゲルの細胞適合性を予想する方法を提供する。例えば、上記の細胞適合性アッセイ由来の実験データを使用して、研究したヒドロゲル中に存在するペプチド構造と細胞適合性との間の相関関係を描写することができる。いくつかの実施形態では、細胞適合性と分子パラメーター(ペプチドの疎水性、β−ヘアピン鎖の長さ、および上記で考察した他のパラメーターなど)との間の相関関係を同定し、これを使用して、提案したヒドロゲルの細胞適合性を予想することができる。自己集合過程を理解および制御するために、分子パラメーターを変化させることができた。次いで、これらの相関関係により、反復デザイン過程における最適な材料特性および細胞適合性の両方を有する完全に新規のペプチドをデザインすることが可能である。
【0105】
特定の実施形態では、細胞接着エピトープをヘアピンベースのヒドロゲルに組み込んで、血清タンパク質の非存在下での細胞付着を増強することができる。また、増殖速度の増強を測定することが出来る。以前に記載の二次元の定量的細胞付着および増殖アッセイを使用することができる。1つの実施形態では、細胞接着エピトープを、ヒドロゲルに組み込むことができる。当業者は、組織工学材料内の細胞接着エピトープの添加によって細胞の付着および/または増殖が促進されるという明らかな実験的証拠が存在することを認識している(例えば、Urry,Angewandte Chemie−International Edition in English 1993,32,819−841,David,et al.,Bioconjugate Chemistry 2001,12,890−899,Rezania & Healy,Journal of Orthopaedic Research 1999,17,615−623,Cook,et al.,Journal of Biomedical Materials Research 1997,55,513−523,Burdick & Anseth,Biomaterials 2002,23,4315−4323,Houseman & Mrksich,Biomaterials 2001,22,943− 955,Kao,et al,Journal of Biomedical Materials Research 2001,55,79−88,およびSchmedlen,et al.,Biomaterials 2002,23,4325−4332を参照のこと)。1つ以上の細胞結合リガンドを、各ヘアピンに共有結合し、ヒドロゲルマトリクスに自己集合させることができる。1つ以上の配列エピトープを、ペプチドの任意の位置(例えば、1つ以上のリジン側鎖またはペプチド(例えば、MAX1)の一次配列内)に組み込むことができる。上記で考察した自己集合に悪影響を与えずにさらなるエピトープを許容することができる任意の位置を使用することができる。MAX1の構造内に、1つ以上のエピトープエピトープを組み込んで、示されたエピトープ濃度を正確に制御することができる。さらに、エピトープのコピーは同一であり得るか、同一性が変化し得る。例えば、相乗的リガンド結合部位は、α5β1中に存在することが公知であり、フィブロネクチンに対する変異誘発実験は、RGDがSDVおよびRNSエピトープに隣接したより有効なリガンドであることを示す(Ruoslahti,Annual Review of Cell and Developmental Biology 1996,12,697−715)。したがって、いくつかの実施形態では、多様なエピトープに富むヘアピンを調製し、これを使用して、細胞の接着および増殖を促進することができる。いくつかの実施形態では、1つ以上のインテグリン結合リガンドを組み込むことができる。このようなインテグリン結合リガンドは、線維芽細胞に特異的な細胞外マトリクスおよび骨芽細胞に特異的な骨シアロタンパク質由来のエピトープに基づき得る。ヒドロゲルと共に使用すべき細胞株と相補的であるように組み込むべきエピトープを選択することができる。
【0106】
1つの特定の実施形態では、3T3線維芽細胞との使用に適切なヒドロゲルを調製することができる。このようなヒドロゲルは、α5β1に結合することが公知の1つ以上のRGDエピトープを組み込むことができる。このインテグリンは、健康なヒト歯周結合組織から培養した線維芽細胞中に存在することが示されている(Hakkinen,et al.,Biochimica Et Biophysica Acta−Molecular Cell Research 1994,1224,33−42)。特定の実施形態では、1つ以上のペプチドエピトープ(TRGDSP(配列番号15)、RGDG(配列番号16)、RGDY(配列番号17)、およびRGDW(配列番号18))を、ペプチドの任意の位置に組み込み、これを使用して、本発明のヒドロゲルを調製することができる。各エピトープの1つ以上のコピーを単独で組み込むか、他のエピトープの1つ以上のコピーと組み合わせて組み込むことができる。
【0107】
別の特定の実施形態では、MC3T3骨芽細胞との使用に適切なヒドロゲルを調製することができる。このようなヒドロゲルは、エピトープFHRRIKA(配列番号19)(骨芽細胞表面上に示されたヘパラン硫酸プロテオグリカンに結合することが公知の骨シアロタンパク質由来のエピトープ)の1つ以上のコピーを組み込むことができる。他の実施形態では、本発明のヒドロゲルは、骨芽細胞接着に正の影響を及ぼすことが公知のさらなるエピトープ(GRGDSPY(配列番号20))を組み込むことができる。いずれかのエピトープの1つ以上のコピーまたは組み合わせた両エピトープの1つ以上のコピーを使用してヒドロゲルを調製することができる。Rezania & Healy(前出)は、RGD含有ペプチドおよびFHRRIKAを共に使用した場合、骨芽細胞増殖の増加を認めた。
【0108】
いくつかの実施形態では、細胞の結合および/または増殖の増強に適切なエピトープを、可溶性エピトープを使用した競合結合アッセイを使用して同定することができる。例えば、細胞の付着および/または増殖を、特異的リガンド結合事象によって媒介した場合、付着または増殖の増強は、添加した可溶性リガンドの濃度の増加によって競合的に阻害されるべきである。さらに、有意に増強させるRGD配列を含むペプチドについて、RGD配列中の1−アスパラギン酸残基をd−アスパラギン酸に置換したコントロール配列を調製する。このサンプルの立体化学の変化により、インテグリン結合が完全に悪影響を受けることが示された。
【0109】
本発明はまた、剛性が変化したヒドロゲルを含む。ゲルの剛性の変化を使用して、細胞−材料相互作用、詳細には、細胞の接着および増殖を調整することができる。剛性が変化する一方でペプチドの一次配列が一定に保持されたヒドロゲルを調製することができる。1つの特定の態様では、ペプチドMAX1を使用して、剛性が変化したヒドロゲルを調製することができる。MAX1ヒドロゲルの剛性を、ペプチド折り畳み誘発条件(すなわち、折り畳み温度)を変化させる一方でペプチド濃度を一定の保持することよって変化させることができる。予備データに基づいて、利用しやすい貯蔵弾性率(ゲル剛性)は、数Pa〜10,000Paの範囲である。異なる温度で形成されたMAX1ゲルは、37℃(例えば、細胞培養条件)での再平衡化後でさえもその剛性を保持する。剛性が変化したヒドロゲル上の二次元の細胞の接着および増殖をその後に観察することにより、任意の特定の細胞型(例えば、ニューロン材料、幹細胞、線維芽細胞−材料、および/または骨芽細胞−材料)の至適基質剛性を決定することができる。この目的のためにMAX1を使用することができるにもかかわらず、本明細書中に記載の任意のペプチドを使用してヒドロゲルの剛性を変化させることができる。ゲルの剛性の変化を使用して、ヒドロゲルの細胞適合性を至適化することもできる。
【0110】
本明細書中に記載の方法および適用の他の適切な修正および適合が明白であり、本発明またはその任意の実施形態の範囲を逸脱することなく修正および適合することができることが関連分野の当業者に容易に明らかである。ここに本発明を詳細に説明しているが、以下の実施例を参照することにより本発明がより明確に理解される。実施例は例示のみを目的として記載し、本発明を制限することを意図しない。
【実施例】
【0111】
(実施例1)
塩の存在によって誘発される分子内折り畳み事象は、生理学的条件下でのヒドロゲル網目構造へのβ−ヘアピンペプチドの集合を誘導する。pH7.4(低イオン強度溶液条件)では、希薄で均一なペプチド溶液(2重量%以下)は、純粋の粘度を示す。円偏光二色性分光法は、pH7.4且つ塩の非存在下ではペプチドは折り畳まれないことを示す。溶液のイオン強度の上昇により、ペプチド内の荷電アミノ酸間の静電相互作用をスクリーニングし、β−ヘアピン高次構造が採用される。折り畳まれたβ−ヘアピン分子は、疎水性崩壊および水素結合によって三次元ヒドロゲル網目構造に超分子的に集合する。FTIRおよびX線散乱データは、これらのヒドロゲルがβ−シートに富むことを示す。動的振動流動学測定は、得られた超分子構造が弾性材料を形成し、その構造(すなわち、剛性率)を、塩濃度および温度によって調整することができることを示す。ヒドロゲルの貯蔵弾性率は、塩濃度の増加と共に増加する。生理学的条件下での増殖培地においても強固なヒドロゲル化が認められる。透過型電子顕微鏡法により、ヒドロゲルの弾性は、半可動性微小繊維集合体からなる網目ナノ構造に起因することが明らかである。
【0112】
ヒドロゲルは、組織工学および薬物送達への適用で広く使用される集合的に重要な生体材料クラスである。自己集合ストラテジーは、環境条件(温度、pH、電場、イオン強度、または光など)に応答し得る所望のナノ構造およびミクロ構造を有する新規のヒドロゲル材料を構築するために正確に制御する。pHおよび温度トリガーを使用したβ−ヘアピンペプチドのヒドロゲル化が示されている(上記のPochan 2003およびSchneider 2002を参照のこと)。これらの分子は、pHおよび温度依存性分子内折り畳み事象を示し、可逆的に分子間で自己集合してヒドロゲル足場を形成する。一般に、ペプチド折り畳み事象の制御により、所望の刺激で自己集合を受ける応答性材料をデザインすることが可能である。さらに、自己集合過程を制御して、所望の形態的性質および機械的性質を操作することができる。下記の1つの特定の実施形態では、β−ヘアピン分子内折り畳み事象を誘導するためのトリガーとして塩濃度を使用し、その後に超分子弾性網目構造に自己集合する。折り畳み速度(すなわち、最終ゲルの流動学的性質)を、ペプチド溶液のイオン強度によって調整することができる。重要なことには、これらの物理的ヒドロゲルの形成は、いかなる化学的架橋も含まず、生理学的温度およびpHで実施することができる。
【0113】
刺激に応答する(例えば、物理的または化学的環境条件の変化に応答したゾル−ゲル転移または膨潤転移)高分子ゲルを作製するための異なるストラテジーが文献で報告されている。温度およびpHは最も一般的な刺激であるが、相転移、自己集合、またはポリマーの高次構造の変化によってゲル化を誘発するためにイオン強度も使用される。塩複合体の形成により、ブロックコポリマーの溶液および自己集合の性質を変化させ、球状または蠕虫様ミセルのような自己集合構造を形成することができる。例えば、塩の型および量により、凝集特性を変化させ、それにより、Pluronicベースのブロックコポリマー系によって形成された網目構造の弾性が変化することが示されている。LCST型相転移による熱応答性を示すことが周知のN−イソプロピルアクリルアミドなどの合成ポリマーの化学ヒドロゲルも塩誘導性体積相転移を示す。比較的高い塩濃度(またはLCSTより高い温度)で、疎水性相互作用によって化学的に架橋されたポリマー鎖が主に沈殿するか沈殿してゲルが崩壊する。イオン強度およびpHに起因するポリ(アクリル酸)ヒドロゲルおよびポリ(メタクリル酸)ヒドロゲルなどの疎水性網目構造の膨潤により、応答性生体材料が生成される可能性があるかどうかを調査した。
【0114】
生体高分子のゲル化挙動(ゼラチン、ポリサッカリド、およびβ−ラクトグロブリンなど)は、文献で広く研究されている。熱処理への適用により、これらの高分子が二次構造(高次構造)への相転移を受け、それにより、分子間網目構造を形成することが周知である。例えば、ゼラチンは、漸減温度での三重らせんの物理的連結点の形成によってヒドロゲルを形成する。ゼラチンゲルの流動学的研究により、粘弾性が処理条件(冷却速度、過冷却度、および濃度など)に非常に依存することが明らかとなった。しかし、生体高分子溶液のポリ多価電解性により、イオン強度も集合特性を制御するための重要なパラメーターである。生体高分子ゲル網目構造(ペクチン、β−ラクトグロブリン、ウェラン(welan)、およびゼラチン)に対するイオン強度の効果は、食品加工および薬学的適用におけるその重要性により、十分に研究されている。一般に、これらの溶液のイオン強度の増加により、分子間塩橋の物理的架橋の形成によって網目構造の弾性が増加する。
【0115】
応答性生体材料としてのその潜在的適用のために、オリゴペプチドおよびポリペプチドの十分に定義された配列を有する生体高分子の刺激応答性自己集合経路を模倣することは非常に興味深い。イオン強度およびpHによって誘発されたオリゴペプチドのβ−シートが豊富なβ−アミロイド様構造への自己集合は、生体模倣材料形成ストラテジーとして調査されている。例えば、Caplan et al.(Biomacromolecules 2000,1,627−631)は、オリゴペプチドの自己集合および得られたゲル(疎水性アミノ酸残基が正電荷または負電荷のアミノ酸に配列が変化している)の特性を、溶液のイオン強度によって変化させることができることを示した。さらに、エラスチン模倣ペプチド(McMillan and Conticello,Macromolecules 2000,33,4809−4821)、コイルドコイルタンパク質合成ポリマードメインを有するハイブリッド分子(Wang,et al.,Nature 1999,397,417−420)、およびさらなる多価電解質セグメントを有するロイシンジッパーポリペプチドドメイン(Petka et al.Science 1998,281,389−392)は、適切なイオン強度での温度またはpHを用いてゲル化される応答性ヒドロゲルを形成する。
【0116】
以下に詳述した特定の実施形態では、生理学的条件下での低ペプチド濃度(2重量%)の20アミノ酸β−ヘアピン分子(MAX1)の分子内折り畳みおよびその後の分子間自己集合を介した塩誘発性物理的ヒドロゲル形成を示す。分子は、II’型ターンとして公知の構造を採用するテトラペプチドターン配列(−VDPPT−)と連結したバリン(V)アミノ酸残基(イソプロピル水素側鎖)およびリジン(K)(第一級アミンキャッピング側鎖)アミノ酸残基の交互配列から構成される2つの鎖からなる。Vが無極性残基である一方で、Kは正電荷であり、ほぼ生理学的条件のpH値(約7.4)で親水性を示す。K残基の電荷および無極性V残基に起因して、静電気力および疎水性相互作用は、分子の折り畳みを制御するために使用することができるおもな分子内パラメーターである。折り畳み状態では、分子表面は親水性を示し、全V残基はヘアピンのある表面上に存在し、K残基は別の表面上に存在する。一旦分子が折り畳まれると、側面の分子内水素結合および表面上の疎水性相互作用の両方によって自己集合が駆動される。したがって、詳細には、これらの分子を、最初に折り畳まれ、その後に系のゲル特性が生じるβ−シートが豊富な四次構造に自己集合するようにデザインする。自己集合構造の最終的な弾性を、塩濃度によって調整することができる。さらに、表面上の疎水性会合により、ゲル化速度制御のパラメーターとして温度を使用することもできる。したがって、これらの分子を、溶液条件(イオン強度、温度、pH)が生理学的レベルに調整された場合にゾル−ゲル転移を示し、組織工学へ適用するために有意に使用することができるようにデザインする。
【0117】
サンプル調製:ABI 433Aペプチド合成機およびHBTU/HOBT活性化を使用した自動化Fmocベースの固相ペプチド合成によってアミド樹脂上にMAX1を調製した。ペプチド調製の詳細は、他で記載されている(上記のSchneider)。最初のDI水への凍結乾燥ペプチドの溶解およびその後の緩衝液および塩含有溶液の添加によって達成された所望の最終溶液条件によってヒドロゲルを調製する。TRIS緩衝液を使用したX線研究以外は、pH7.4での溶液の緩衝化のためにビス−トリスプロパンを使用する。
【0118】
円偏光二色性:Avivモデル215分光偏光計を使用して、CDスペクトルを収集した。20℃または37℃で測定した。0mMと150mMとの間のKF溶液を含む2重量%のMAX1(pH7.4、50mM BTP)についての190nmと260nmとの間の波長スキャンを、0.01mmの取り外し可能な石英セル中で行った。DI水および緩衝液へのペプチド溶解から2時間後にスペクトルを取った。時間依存性研究のために、平均残基楕円率(θ)を、218nmで測定した。式θ=θobs/l/c/r(式中、lはセルの光路長であり、cは濃度であり、rは残基数である)からθを計算した。
【0119】
赤外分光法:Nicolet Magna−IR860分光計にてIRスペクトルを取った。0.1M HClで1回およびD2Oで2回のペプチドのTFA塩の凍結乾燥によって、重水素化MAX1*nDClを調製した。サンプルを温度調節された水浴中に20℃で2時間保持し、その直後に測定物を室温で光路長30μmの亜鉛−セレニドフローセル中に入れた。装置を、1cm−1の分解能で操作し、記録したスペクトルは平均100スキャンであった。
【0120】
広角X線散乱:National Synchrotron Light Source,Brookhaven National Laboratory,beamline,X10AにてヒドロゲルのX線スペクトルを収集した。脱水を回避するために測定を行う直前にヒドロゲルをKaptonテープに塗布した。二次元Bruker CCDアレイ上に10分間データを収集した。ペプチド溶液を、125mM TRISで緩衝化した。室温で測定した。
【0121】
流動学:直径25.0mmの平行平板およびギャップ距離が0.5mmの歪み制御したRheometrics ARES流量計にて動的時間および周波数掃引実験を行った。凍結乾燥ペプチドを、10℃の脱イオン水および緩衝液または細胞増殖培地溶液に溶解して、サンプルを流量計にロードする前の折り畳みおよびゲル化を抑制した。サンプルをロードした後、温度を20℃または37℃のいずれかに上昇させた。標準的な低粘度鉱物油を使用して、プレート側面から隔離して蒸発を抑制した。コントロール実験は、鉱物油が流動学的測定に影響を与えないことを示した。歪み掃引実験を行って線形粘弾性体制を決定し、全流動学的実験を行った。6rad/sの周波数および1%の歪み用いて動的時間掃引実験を行った。0.1〜100rad/sの周波数および5%の歪みを使用して周波数掃引実験を行った。
【0122】
透過型電子顕微鏡法:非常に薄いヒドロゲル層を、カーボンコーティングした銅のグリッドに貼り付けた。2wt/v%の酢酸ウラニル溶液のグリッドへの滴下によって、サンプルを陰性染色した。過剰な溶液を濾紙で吸い取り、その後にサンプルを乾燥させた。塩および/または緩衝液の結晶の形成を回避し、各微小繊維を明確に画像化するために、2重量%のペプチド濃度のヒドロゲルを、DI水で約0.1重量%に希釈した。溶液中に微小繊維を均一に分散させるために、チップ超音波処理機を使用して穏やかに超音波処理を行った。Gatan CCDカメラおよびKodakネガフィルムを備えたJEOL 2000−FX透過型電子顕微鏡にて加速電圧200kVでヒドロゲルナノ構造の明視野画像を取った。
【0123】
図21Aは、pH7.4および20℃の2重量%のMAX1溶液の異なる塩濃度についてのCDスペクトルを示す。DI水および緩衝液でのMAX1溶解から2時間後に示した両スペクトルを取った。150mM KFを含むペプチド溶液が218nmで最小であることは、2重量%のゲル化濃度で、MAX1が折り畳まれてβ−シートに富む二次構造を採用することを示す。しかし、塩の非存在下での同一pHで2重量%のMAX1は2時間後でさえも有意に均一な二次構造を示さず、塩を添加しないペプチドは折り畳まれないままであることを示した。
【0124】
塩の存在下でのペプチド分子のβ−ヘアピン構造への折り畳みおよびその後のβ−シートの形成もFTIR分光法によって確認した。pH7.4および400mM NaCl、1580cm−1と1700cm−1との間のMAX1溶液のFTIRスペクトルを図21Bに示す。D2O中の不規則なペプチドについてのアミドIバンドは、1645cm−1付近に集中している。1644cm−1から1614cm−1へのアミドIバンドのシフトにより、400mM NaCl溶液中でペプチドがβ−シート高次構造であることが示唆される。1680cm−1のスペクトル中の弱いバンドは、逆平行β−シート構造を示し得る。NaClを含まないMAX1溶液については、プロット範囲で非常に広いスペクトルが得られる。1644cm−1の強いバンドにより、ペプチドが不規則な状態であることが示唆される。しかし、1614cm−1の弱いバンドは、溶液中の少量の折り畳まれたβ−ヘアピン分子の存在を示す。本発明者らは、いくつかのMAX1の折り畳みは、溶液中の過剰な緩衝液の塩および比較的高濃度のペプチドの両方に起因すると考える。ペプチド濃度がβ−シートの形成速度に影響を与えることが示された。したがって、ペプチド濃度が高く且つゲル化しない状況では、この少量のβ−シートアミドIシグナルは重要ではない。
【0125】
リジン残基状の第一級アミンによってMAX1はpH7.4で正味の電荷を有するので、データにより、鎖間の静電反発力によってMAX1は折り畳むことができないと示唆される。したがって、リジン残基の正電荷をCl−イオンによってスクリーニングした場合、分子内折り畳みが支持されてβ−ヘアピン構造が形成される。塩濃度の増加により、残基の疎水性会合を駆動することもでき、疎水性会合は折り畳みおよび自己集合にも寄与するはずである。CDデータおよびFTIRデータはどちらも、折り畳み機構が溶液のイオン強度によって誘発されることを示す。折り畳みをpHおよび温度によって誘発することができることも示されている(上記のPochan,Schneider)。塩の効果と類似して、刺激としてpHを使用した場合、高pHでリジン残基が脱プロトン化しMAX1が折り畳まれる。折り畳まれた場合、分子間水素結合および疎水性相互作用によってMAX1はより高次の構造に自己集合することができる。折り畳み速度、集合した凝集体の構造、および得られたヒドロゲル網目構造の性質を以下で考察する。
【0126】
X線散乱技術を使用して、最終ヒドロゲル中で結晶学的β−シート構造が認められる。図21Cは、3重量%のMAX1および150mMNaClからなるヒドロゲルについての広角曲線を示す。散乱ピークは、4.7Åに相当する。これはβ−シートに富む構造における鎖間距離の特徴的なサインである。ヒドロゲルを乾燥させずに測定したので、水構造に起因する高角度での散乱バックグラウンドの寄与が顕著である。
【0127】
動的振動技術によって観察したところ、分子内折り畳み機構によって形成されたヒドロゲルは、低濃度のMAX1ペプチドでさえも強固な粘弾性を示す。図22Aは、20、150、および400mMのNaCl濃度の2重量%のMAX1ペプチド溶液についての10−1〜102rad/sの周波数掃引測定を示す。周波数掃引測定の前に、ペプチド溶液を、流量計中で2.5時間ゲル化した(図22B)。400mMNaClを含むMAX1溶液について、平衡貯蔵弾性率(G’)は、約3000Paである。塩濃度の減少により、G’値および損失剛性率(G’’)が減少した。20mM NaClしか溶液中に存在しない場合、低剛性(約100PaのG’)のヒドロゲルが形成される。NaClを添加しないpH7.4のMAX1は、G’値が約1Paで3時間後でさえもいかなるゲル化も示さなかった(適用した歪みに対するサンプルの応答はごくわずかである。トルク値は装置の検出限界未満であり、測定値は重要ではない(示さず))。塩を含む全3サンプルについて、G’値は、G’’値よりも少なくとも1桁高い。また、全NaCl濃度について、研究された範囲では、G’値は本質的に周波数と無関係であり、低周波数ではG’とG’’との間に交差点は存在しない。これらの特徴は、架橋網目構造の明らかなサイン(signature)である。図22A中の周波数掃引データにより、これらの溶液が共有結合で架橋したポリマーゲルに類似の性質を有する強固な固体様ヒドロゲルを形成することが示唆される。
【0128】
図21中の分光法によるデータにより、塩を含まない溶液中で、リジン残基の静電反発力がペプチドの非折り畳みを維持し、それにより、網目構造への自己集合が阻止されることが示唆される。しかし、一旦分子が折り畳まれると、自己集合が起こり得る。全バリン残基が折り畳み状態の分子の片側に位置づけられるので、表面で疎水性二量体化が起こり得る。その後にさらなる分子間疎水性相互作用および水素結合が起こり、相互連結された網目構造が形成され得る。重要なことには、図22Aに示すように、異なる塩濃度のヒドロゲルの剛性の相違により、イオン強度によって自己集合を誘発することができるだけでなく、培地のイオン強度によって得られたナノ構造を予想通りに変化させることができることが明らかとなる。図22Bは、2重量%の同一のペプチド濃度についてのMAX1溶液の自己集合時のG’値の増加を示す。この実験は、異なる塩濃度では自己集合の速度および網目構造形成が異なることを明らかに示す。400mM NaClを含むMAX1溶液は、自己集合過程の最初の2分間でG’値が迅速に増加する一方で、20および150mM NaCl溶液で比較的低速で硬化することを示す。実際、20mM NaCl溶液のG’値は3000秒間で有意に変化せず、2〜3Paのままである。この遅延時間は、150mMではより短く(約1000秒)、400mMの塩溶液では示されない。図22Bにより、折り畳み速度が速いほどより高いイオン強度によって自己集合してより強固なゲルが得られることが明らかに示唆される。同一のペプチド濃度の溶液間の剛性の大きな相違は、おそらく、より速い折り畳みおよび集合速度によるより高度に架橋された網目構造に起因する。
【0129】
弾性が得られるヒドロゲルのナノ構造を、透過型電子顕微鏡法(TEM)によって研究した。図23Aは、400mM NaClを含むpH7.4で形成された2重量%のMAX1ヒドロゲル網目構造のTEM画像を示す。顕微鏡写真から、ナノスケールでの高度に相互連結した微小繊維網目構造が明らかである。TEM顕微鏡写真で直接認められないが、自己集合時に形成されたほとんどの接合点が微小繊維と交差し、長い非交差微小繊維の単純な縺れではないと考えられる(しかし、縺れは剛性率に確かに寄与している)。微小繊維間のこれらの架橋点は、元来は共有結合していないが、不変であり、水素結合および疎水性相互作用の両方によって形成される。図22Aに示す周波数掃引データもこの見解を支持している。G’は周波数に対して感受性がなく、G’’よりも非常に高く、G’’とG’との交差は認められない。画像化前の水の蒸発および比較的高い塩濃度のために、2重量%の構造は非常に密集しており、網目構造のいくつかの部分が沈殿した塩に埋め込まれている。したがって、ヒドロゲルを、DI水で約20倍(〜0.1重量%のペプチド)に希釈し、直ちにTEM画像化のためのサンプルを調製した。図23Bは、より希薄な微小繊維集合体を示す。微小繊維の輪郭の長さは、およそ数μmである。サンプル調製時の乾燥は微小繊維の軸に沿って高次構造を変化させ得るにもかかわらず、顕微鏡写真により、自己集合した微小繊維が半可動性を示すことが示唆される。重要なことには、微小繊維の幅が単分散サイズの約3nmである。提案された局所自己集合構造および寸法を、図24に示す。Insight IIモデリングは、折り畳まれたペプチド鎖の軸の長さは32Åであり、バリン面からリジン面までの距離が約10Åであることを示す。幅が約3nmの微小繊維は、これらの分子の寸法と非常に良好に一致する。CDデータと共に、TEMデータにより、自己集合過程時にMAX1が折り畳み状態にあることが強く示唆される。図24中の提案された構造では、簡潔にするために、β−ターンを微小繊維二重層の同一側面に存在するように示す。
【0130】
CDおよび流動学による結果は、β−ヘアピン分子の折り畳みおよびその後のゲル化をpH7.4での塩濃度の変化によって誘発することができることを明確に示す。この応答型により、生理学的条件下でこれらの材料を使用する機会が得られる。したがって、これらの分子の自己集合およびゲル化挙動を、生理学的温度(37℃)で研究した。MAX1の折り畳みおよびβ−シート形成が温度依存性であることが示された。CD研究により、MAX1について、不規則なランダムコイル状態から折り畳まれたβ−シート構造までの転移温度は、pH9および低濃度の塩(20mM)で約25℃であることが示された。この転移は、集合ペプチドの疎水性の変化によってより高い温度に調整することができることが示された。
【0131】
イオン強度によって誘発された自己集合に対する温度の効果を観察するために、20℃および37℃での動的掃引時間実験によって2.5時間にわたってゲル化をモニタリングした。図25Aでは、G’の変化を、pH7.4で150mMのNaCl溶液を含む2重量%のMAX1の時間の関数としてプロットする。両方の場合において、最初にMAX1溶液を10℃に保持して折り畳みを抑制し、その後に測定した。37℃でのG’の瞬間的な増加は、ペプチドが自己集合時に網目構造を直ちに形成したことを示す。対照的に、20℃では、ヒドロゲルの弾性応答の増加はより遅く、自己集合の最初の15分間についてはG’の増加は小さいことを示す。この挙動は、室温で粘性様挙動を示す一方で、体温および身体の塩濃度に曝露された場合に(例えば、in vivoでの注射)迅速なゲル化応答を示す溶液が望まれる潜在的用途で有利であり得る。図25Aは、生理学的に関連する条件下で(例えば、pH7.4、37℃)、有用な剛性を示す材料が得られるゲル化が約10分間で起こることを明確に示す。37℃での2.5時間のゲル化後に行った周波数掃引測定(図25B)により、G’値が約2000Paの強固なヒドロゲル(G’>G’’)が形成されることが示される。両温度でのゲル化時のG’’値(示さず)は一定であり、ゲル化過程を通してG’値よりも常に十分に低かった(1桁を超えて低い)。全サンプルでは、ゲル化の初期段階でさえも、G’>G’’であった。生体高分子のゲル化においても類似の挙動が認められた。イオン強度の効果に類似して、より高い温度で集合したより強固なゲルの形成は、ナノスケールでの構造の相違に起因し得る。より速い折り畳みおよびそれによる自己集合速度により、接合点の数においてより緻密であり、それによりG’値がより高い網目構造を形成することができる。このデータは、温度または塩濃度のいずれかによってMAX1ヒドロゲルの剛性を容易且つ予想通りに制御することができることを示す。
【0132】
MAX1ペプチドの折り畳みおよびβ−シート形成速度を、CDによって研究した。図25Cは、pH7.4で150mMのKF溶液を含む2重量%のMAX1についての20℃および37℃にて218nmで測定した楕円率の変化を示す。光学的にサイレントであり、且つCD測定を補助するので、電解質としてKFを使用する。CDデータにより、37℃での初期段階の自己集合β−シート形成が非常に迅速であることが明らかとなる。最初の5分間の折り畳み速度が有意に速く、15分後にプラトー領域に達する。しかし、20℃での折り畳み速度は遅い。β−シート形成およびそれによる自己集合の速度の相違は、図25に示す流動学的測定値と一致する。37℃では、自己集合の初期段階でG’値およびθ218値の変化は非常に迅速である一方で、20℃では、両値の変化速度は遥かに遅い。したがって、37℃で形成されたゲルのより高い貯蔵弾性率は、折り畳み速度の速さに起因し得る。自己集合の初期段階で、折り畳み速度が速いことによってβ−シートが豊富な微小繊維の成長のための核生成部位がより多くなり、その結果、より接合点の多い網目構造が形成される。この作用機構はこれまでに得た全データと一致する。
【0133】
生体材料への適用のために、生物学的に関連する条件に対するMAX1の応答を理解することが重要である。したがって、無血清DMEM細胞増殖培地においてMAX1のゲル化を研究した。図26Aは、37℃での細胞増殖培地中(哺乳動物細胞培養条件)でのMAX1のゲル化時のG’およびG’’を示す。2時間のゲル化の終了後、G’およびG’’値は、それぞれ2300Paおよび50Paであった。2時間後のG’値の増加速度が依然として有意であることが認められる。ペプチド濃度およびG’値を増加させる蒸発の影響を排除するために、2時間のゲル化後に動的掃引時間実験を中止した。(最終値を示さないG’値のこの継続的増加は、ゼラチンゲルでも認められた)。図26Bに示す周波数掃引データは、MAX1−細胞増殖培地溶液が約2500PaのG’値で強固なゲルを形成することを示す。培地誘導性ヒドロゲルの周波数掃引データの性質(G’>10×G’’、G’は周波数と無関係)は、図21Aに示す性質と類似している。有意な剪断に対するヒドロゲルの応答を、ゲル化溶液に対する大規模の歪み(6rad/sで1000%)の適用後のG’の回復をモニタリングすることによって研究した。適用した歪みが線形体制の十分に外側であるので、G’およびG’’値は示されない。図26Cは、剪断の停止およびその直後の6rad/sで5%の歪みの適用後、G’がその初期値のほぼ50%を即座に回復することを示す。回復データを、ゲル化データ(図26A)と比較した場合、G’の増加速度が初期ゲル化時の速度よりも早いことが認められる。これにより、大規模の歪みの適用時に物理的に架橋された自己集合網目構造が破壊され、接続性が減少し、それにより、材料の弾性が減少することが示唆される。剪断の中止後、有意な網目構造の剛性の即座な回復で明らかなように、網目構造が即座に再修復することができる。この実験は、これらのヒドロゲル処理可能であり、且つ機械的外力による破壊後に初期流動学的性質を回復することができることを示す。この処理および回復の容易さは、組織工学への適用(例えば、in vivo注射)に有利であり得る。
【0134】
β−ヘアピンペプチドの折り畳みおよび集合によって強固なヒドロゲルが形成される。MAX1は、生理学的pH(7.4)でランダムコイル高次構造で存在し、溶液への塩の添加により、β−シートに富む自己集合した構造を形成する。流動学的データは、ペプチド溶液が、G’’よりも少なくとも1桁高いG’でゲルを形成することを示す。さらに、G’が周波数と無関係であることは、網目構造が化学的に架橋したポリマーゲルの弾性と類似していることを示す。ヒドロゲルの自己集合速度および貯蔵弾性率を、ペプチド溶液のイオン強度によって調整することができる。網目構造は、おそらく表面の疎水性相互作用および水素結合に起因する物理的接合点によって互いに架橋している緻密な微小繊維集合体から構成される。微小繊維の幅は、約3nmであり、この寸法は、自己集合状態のヘアピンの折り畳み状態と良好に一致する。塩誘発性自己集合およびそれによるゲル化の性質も温度によって調整される。37℃で、β−シート形成およびゲル化の速度は、より強固なゲルが得られる20℃よりも速い。さらに、MAX1は、生理学的条件下での細胞増殖培地で強固且つ処理可能なヒドロゲルを形成する。したがって、適切にデザインされたペプチドの分子内折り畳みおよびそれによる超分子構造への分子間集合を塩によって誘発し、調製可能な剛性率を有するヒドロゲルを得ることができる。
【0135】
上記発明を、明示し、且つ理解する目的でいくらか詳細に説明しているが、この開示を読み取ることにより、当業者は、本発明の範囲および添付の特許請求の範囲を逸脱することなく形態および詳細の種々の変更を行うことができることを認識している。本明細書中で引用した全ての特許および刊行物は、その全体が本明細書中で参考として援用される。
【図面の簡単な説明】
【0136】
【図1】図1は、本発明のヒドロゲルを形成するためのペプチドの自己集合の略図を示す。非折り畳みペプチドは、他の折り畳み様ペプチドと自己集合される高次構造に個別に折り畳まれるように誘導される。折り畳み過程は、所望の刺激への曝露に起因し、自己集合過程により、相互連結した線維性のヒドロゲル網目構造が得られる。
【図2】図2は、本発明の所望の特徴を有するヒドロゲルの調製で使用されるペプチド中で変化し得るいくつかの要因の略図を示す。変化し得る特定の分子要因には、以下が含まれる。
【図2A】図2Aは、分子内折り畳みに影響を与える操作を記載し、以下が含まれる:1)各ペプチドのリジン残基の第1級アミン化学的官能基との間の静電相互作用、2)各ペプチドのペプチド骨格アーム間の疎水性のファンデルワールス相互作用、3)各ペプチド骨格のアーム間の水素結合、4)各ペプチドのターン性向(すなわち、特定の刺激と比較した折り畳みに対するペプチドの「強さ」)。
【図2B】図2Bは、分子間自己集合に影響を与える操作を記載し、以下が含まれる:5)隣接する折り畳みペプチドバリンリッチ表面間の疎水性のファンデルワールス相互作用、6)隣接ペプチドのペプチド骨格アーム間の疎水性のファンデルワールス相互作用、7)隣接ペプチドのペプチド骨格アーム間の水素結合。
【図3】図3Aは、本発明で使用されるペプチドの構造を示す。図3Bは、本発明のヒドロゲルの可逆的形成の略図を示す。詳細には、ペプチドMAX1は、酸性または中性pH溶液条件下では組織化されていない非折り畳み構造である。塩基性溶液条件に曝露された場合、MAX1ターン配列中のトランスプロピルアミド結合は、各ペプチドのアームを図2Aに列挙した相互作用によって安定化された並行高次構造に押し進める。したがって、自己集合は、図3Bに記載の相互作用によって起こる。得られたヒドロゲルを酸性pH条件に曝露した場合、ペプチドは折り畳まれずに分解されて希薄な低粘度の水溶液に戻る。
【図4】図4は、本発明にしたがって調製したヒドロゲルから得た実験の結果を示す。図4Aは、150μM MAX1溶液(pH9.0、125mMホウ酸、10mM NaCl)の時間依存性far UV−CDスペクトルを示す。図4Bは、同一条件下での150および250μM MAX1溶液についての時間関数としてモニタリングした[θ]216を示す。図4Cは、無作為なコイル−シート折り畳み/自己集合平衡が可逆性であることを示すMAX1の300μM溶液の時間およびpHの関数としてモニタリングした[θ]216を示す。全実験を、20℃で実施した。
【図5】図5は、FTIR(pH5.5(軌跡a)およびゲル化後のpH9.0(軌跡b)1重量% MAX1)を示す。(a)中の1664から(b)中の1615までのアミド1ストレッチにおける大きなシフトは、β−シート構造の成長を強く示す。
【図6】図6は、pH9での2重量%MAX1ヒドロゲルに関する流動学データを示す。図6Aは、貯蔵剛性率(G’、黒塗りの記号)および損失剛性率(G’’、白抜きの記号)の増加による時間の関数としての6Hzで1%染色でモニタリングした短期ヒドロゲル化を示す。図6Bは、周波数掃引データを示す。周波数に対する剛性率の非感受性(insensitivity)は、ペプチド折り畳みおよび自己集合による強く架橋した強固なヒドロゲルを示す。図6Cは、集合後過程(例えば、シリンジによる注射)が容易な剪断薄膜化(shear thinning)材料を示す掃引率データ(粘度、白抜きの記号;剪断応力、黒塗りの記号)を示す。図6Dは、歪み処理(6Hzで180秒の1000%歪み)の停止後の時間の関数としてのゲル剛性率の回復を示す。記号は、図6Aで定義の通りである。大歪みによるヒドロゲル破壊直後に、ゲルは即座にその前の剛性の約75%に戻って固化し、迅速に(約10〜20分)その歪み前の剛性を取り戻す。
【図7】図7は、レーザー走査型供焦点顕微鏡法(LSCM)および低温透過型電子顕微鏡法(cryoTEM)を使用して得た結果を示す。図7Aは、ヒドロゲル微小構造のLSCMを示す。緑色の領域は蛍光染色された自己集合ペプチドであり、黒の領域は水で満たされた孔およびチャネルである。スペースバーは20μmである。図7Bは、自己集合したナノ構造のcryoTEMを示す。暗い構造は、自己集合したペプチド足場であり、より明るい灰色の領域はガラス化水から構成される。スペースバーは200nmである。
【図8】1重量% MAX1ヒドロゲルの組み合わせたUSANS/SANSプロットを示す。(挿入図は、−1.1の非線形の最小二乗法での重ね合わせを用いた0.02<q<0.08のlog(強度)の拡大図である)。低qの勾配−4は、図7Aにて顕微鏡で認められる微孔構造を示す。挿入図中の勾配−1は、図7Bにて顕微鏡で認められる局所ロッド様構造を示す。
【図9】図9Aは、150μM MAX1溶液(125mMホウ酸、10mM NaCl、pH9.0)の温度依存性CDを示す。低温では、MAX1は折り畳まれない(その結果として、自己集合しない)。高温では、MAX1はβ−シートに折り畳まれる(その結果として、ヒドロゲルに自己集合する)。図9Bは、同一条件下(MAXl=VKVKVKVKVDPPTKVKVKVKV−NH2、MAX2=VKVKVKVKVDPTKVKTKVKV−NH2、およびMAX3=VKVKVKTKVDPPTKVKTKVKV−NH2)でのMAX1、2、および3の[θ]218の温度依存性を示す。相対疎水性は、下線を引いた位置でのバリンのトレオニンへの等構造(isostructural)であるが低疎水性の置換によって、MAX1>MAX2>MAX3である。最低温度で最高の疎水性で折り畳みが起こり、最高温度で最小の疎水性折り畳みが起こる。これは、図2に記載の分子パラメーター操作を端的に表す。25℃での水中のオクタノールからの全部で+8荷電状態のそれぞれの対応する非折り畳みペプチドの転移自由エネルギーの計算も示す。最も高い疎水性を示すMAX1は、7.60kcal/molと等しい最も高い自由エネルギーを有する。MAX2の転移自由エネルギーの計算値は6.88kcal/molであり、疎水性が最小のMAX3は、6.45kcal/molの最も低い転移エネルギーを有する。
【図10】図10Aは、150μM MAX3溶液(125mMホウ酸、10mMNaCl、pH9)の温度依存性CDを示す。白丸は、完全に折り畳まれていない特徴を示す5℃でのMAX3である。白四角は、80℃に加熱し、分子内折り畳みが起こったMAX3である。黒菱形は、完全に折り畳まれていない特徴を示す5℃に戻して冷却した後のMAX3である。黒三角は、80℃に再加熱し、分子内折り畳みが起こったMAX3である。図10Bは、同一条件下での2重量%のMAX3水性調製物の貯蔵弾性率(G’)の温度依存性を示す;20分間隔で表示の温度にてデータを収集し、おおよその装置/サンプル平衡が得られる時間を割り当てた。流動学は、可逆的自己集合および結果として起こる温度による固体ヒドロゲル剛性を明確に示す。
【図11】図11は、本発明の実施で使用することができる種々のペプチドの構造を示す。図11Aは、分子間側面疎水性接触(図2に記載)を可能にし、自己集合の駆動に役立つ、バリンが各ヘアピン突起内のH結合位に組み込まれ、且つ側鎖が外側に突出したMAX1の構造を示す。図11Bは、バリンが非H結合位に組み込まれ、且つその側鎖が内側に突出したMAX4の構造を示し、この構造はバリン駆動分子間相互作用の可能性が低く、自己集合があまり好まれない。
【図12】図12は、流動学研究の結果を示す。貯蔵弾性率は、40℃でのMAX1およびMAX4の1重量%調製物(pH9.0、125mMホウ酸、10mM NaCl)(周波数=6rad/秒)で増加する。MAX1は、分子間側面疎水性接触の増加のためにMAX4よりも明らかに迅速に集合する。
【図13】図13は、酢酸ウラニルで陰性に染色されたMAX9微小繊維のTEM顕微鏡写真を示す。この硬く不可逆な微小繊維は、MAX9の非折り畳み特性に起因し、ヒドロゲル化させない。
【図14】図14Aは、pH7(20mM Tris)での150μM MAX1の塩依存性CDを示す。20mM KF塩では、CDは、完全に折り畳まれていない高次構造を示す(黒丸)。150mM KFでは、CDは、強いβ−シート高次構造の形成を示す(黒四角)。図14Bは、無血清増殖培地を使用してpH7で形成されたMAX1ヒドロゲルの周波数掃引データを示す。剛性率(G’=黒丸、G’’=黒四角)の周波数非依存性は、重度に架橋された強固なヒドロゲル材料を明確に示す。
【図15】図15は、異なる温度での2重量%のMAX1の貯蔵弾性率対時間を示す(pH7.4、歪み1%、6rad/s)。黒丸=20℃、黒三角=37℃、および黒菱形=60℃。温度が高いほど、より迅速なペプチドの折り畳みおよび集合が起こり、得られたヒドロゲル網目構造がより強固になる。
【図16】図16は、光学顕微鏡法の結果を示す。図16Aは、2重量%Max1ゲルにプレートした10%ウシ血清を含むDMEM中の104個の線維芽細胞(t=4.5時間)を示す。図16Bは、t=72時間の16A由来の結果を示す。図16Cは、コントロール(ポリスチレンにプレートした10%ウシ血清を含むDMEM中の104個の線維芽細胞(t=4時間))を示す。図16Dは、t=72のコントロールを示す。図16Eは、2重量%Max1ゲルにプレートした血清を含まないDMEM中の104個の線維芽細胞(t=4.5時間)を示す。図16Fは、ウシ血清をサンプルに4時間添加し、72時間インキュベートした図16Eと同一の材料を示す。
【図17】図17は、種々の初期細胞播種密度でのMAX1ヒドロゲルに対するNIH3T3線維芽細胞の増殖アッセイ対細胞培養ポリスチレンの結果を示す。白抜きのバーは、組織培養ポリスチレンコントロール上での細胞増殖を示し、影をつけたバーは2重量%MAX1ヒドロゲル上での細胞増殖を示す。自己集合ヒドロゲル上での増殖は、全細胞播種密度でコントロールより高かった。
【図18】図18は、線維芽細胞がMAX1ゲル全体に充満していることを示すLSCM画像である。Z軸方向の重ね合わせ画像は、Z軸に対して垂直に見た。バー=100μm。したがって、ヒドロゲル形成の自己集合機構により、細胞を三次元に容易にカプセル化することができる。
【図19】図19は、修飾することができるペプチド中の位置を示す本発明で使用されるペプチドの略図を示す。細胞結合エピトープ(配列アルギニン、グリシン、アスパラギン酸(RGD)など)を、1)一方または両方のペプチド末端、2)一方または両方のペプチド鎖の中央、3)一方または両方のターン隣接位置、または4)共有結合を介した任意のリジン側鎖に組み込むことができる。
【図20】図20は、本発明と組み合わせて使用することができるトリガーの例を示す。330〜360nmの波長光を、図中に示すVal(16)(スキーム1および2)への図に示す化学物質の組み込みによる分子内折り畳み(それによる、ヒドロゲル化)トリガーとして使用することができる。カルシウムII結合を、ターン配列へのスキーム3および4に示す化学物質の組み込みによる細胞内折り畳み(それによる、ヒドロゲル化)トリガーとして使用することができる。
【図21】図21Aは、20℃で0mM(白丸)および150mM(黒丸)のKFを含む2重量%Max1(pH7.4)溶液のCDスペクトルを示し、これは、塩を使用した明確なβ−シート形成を示す。図21Bは、0および400mM NaClを含む2重量%Max1溶液(pH7.4)のFTIRスペクトルを示し、これも塩添加による明確なβ−シートの形成を示す。図21Cは、150mM NaClを含む3重量%Max1溶液(pH7)のWAXSスペクトルを示す。ピークは、4.7Åの間隔(β−シート高次構造中のペプチドの分子間間隔)を示す。
【図22】図22Aは、20℃で20mM(G’;黒丸、G’’;白丸)、150mM(G’;黒三角、G’’;白三角)、および400mM(G’;黒四角、G’’;白四角)のNaClを含む2重量%Max1(pH7.4)溶液の動的周波数掃引(歪み5%)を示す。より高い塩濃度の刺激ほど、ヒドロゲル網目構造がより強固になる。図22Bは、20℃で20mM(G’;黒丸)、150mM(G’;黒三角)、および400mM(G’;黒四角)のNaClを含む2重量%Max1溶液の動的掃引時間(歪み1%、6rad/s)を示す。塩濃度が高いほど、より迅速なペプチドの折り畳みおよび自己集合が起こり、最終的なヒドロゲル材料がより強固になる。
【図23】図23は、ヒドロゲルの自己集合構造の負に染色された(酢酸ウラニル)TEM画像を示す。図23Aは、2重量%Max1(pH7.4、400mM NaCl溶液)の密に相互連結した網目構造を示す。図23Bは、希釈ヒドロゲル(希釈後の最終濃度:約0.1重量%)の微小繊維集合を示す。ナノ構造は、図1および図24に概略的に示す網目構造と一致する。
【図24】図24は、微小繊維中の自己集合Max1β−ヘアピン分子の提案された構造を示す。分子の短軸(stand axis)は、32Åであり、横断面の厚さは20Åである。構造の長軸は、水素結合および微小繊維の成長方向を示す。
【図25】図25Aは、20℃(G’;白丸)および37℃(G’;白三角)で150mMのNaClを含む2重量%Max1溶液(pH7.4)の動的掃引時間データ(歪み1%、6rad/s)を示す。温度が高いほど、最終材料がより迅速に折畳まれ、より強固になる。図25Bは、2.5時間のゲル化後の(G’;黒丸、G’’;白丸)37℃でヒドロゲルの周波数掃引データ(歪み5%)を示す。図25Cは、20℃(白丸)および37℃(白三角)で150mM NaClを含む2重量%Max1溶液(pH7.4)の218nm([θ]218)での時間依存性平均モル楕円率を示す。
【図26】図26は、37℃での細胞増殖培地中での2重量%Max1溶液の流動学データ(G’;黒丸、G’’;白丸)を示す。図26Aは、ゲル形成の流動学データ(歪み1%、周波数6rad/s)を示す。図26Bは、周波数掃引データ(歪み5%)を示し、図26Cは、高歪み振幅(1000%、6rad/s)の停止後のヒドロゲルのG’およびG’’回復率(歪み1%、周波数6rad/s)を示す。pH折り畳みおよびアセンブリトリガーを比較した図6に示すように、細胞増殖培養形態へのペプチドの迅速な曝露によって生成されたゲルは有意に強固であり、剪断薄膜化することができ(例えば、シリンジ注射)、剪断停止後にその元の剛性に回復し得る。
【特許請求の範囲】
【請求項1】
ヒドロゲルの作製方法であって、
ペプチドを含む溶液を提供する工程と、
該溶液の1つ以上の特徴を変化させる工程とを包含する方法であって、ヒドロゲルが形成される、方法。
【請求項2】
前記特徴が、イオン強度、温度、特定のイオンの濃度、およびpHからなる群から選択される、請求項1に記載の方法。
【請求項3】
前記変化させる工程が、前記溶液を電磁放射と接触させることを含む、請求項1に記載の方法。
【請求項4】
前記変化させる工程が、前記溶液のpHを変化させることを含む、請求項1に記載の方法。
【請求項5】
前記溶液の1つ以上の特徴を変化させる工程により、塩濃度が約20mM〜約400mMになる、請求項1に記載の方法。
【請求項6】
前記塩がNaClである、請求項5に記載の方法。
【請求項7】
前記溶液のpHがpH9未満である、請求項1に記載の方法。
【請求項8】
前記溶液のpHが約pH6.0〜約pH8.5である、請求項1に記載の方法。
【請求項9】
前記溶液のpHが約pH7.0〜約pH8.0である、請求項1に記載の方法。
【請求項10】
前記溶液のpHが約pH7.4である、請求項1に記載の方法。
【請求項11】
ペプチドおよび約20mM〜約400mMの塩を含むヒドロゲル。
【請求項12】
前記塩がNaClである、請求項11に記載のヒドロゲル。
【請求項13】
MAX1を含む、請求項11に記載のヒドロゲル。
【請求項14】
ヒドロゲルの作製方法であって、
ペプチドを含む溶液を動物に注入する工程を包含し、該溶液が該動物内でヒドロゲルを形成する、方法。
【請求項15】
前記動物が哺乳動物である、請求項14に記載の方法。
【請求項16】
前記動物がヒトである、請求項14に記載の方法。
【請求項17】
前記溶液が、1つ以上の治療因子をさらに含む、請求項14に記載の方法。
【請求項18】
前記治療因子が、低分子、ペプチド、タンパク質、および細胞からなる群から選択される、請求項17に記載の方法。
【請求項19】
治療因子を必要とする動物に治療因子を送達する方法であって、
該治療因子および1つ以上のペプチドを含む溶液を該動物に投与する工程を包含し、該溶液が該動物内でヒドロゲルを形成する、方法。
【請求項20】
前記動物が哺乳動物である、請求項19に記載の方法。
【請求項21】
前記動物がヒトである、請求項19に記載の方法。
【請求項22】
前記治療因子が、低分子、ペプチド、タンパク質、および細胞からなる群から選択される、請求項19に記載の方法。
【請求項23】
治療因子を必要とする動物に治療因子を送達する方法であって、
該治療因子および1つ以上のペプチドを含むヒドロゲルを該動物に投与する工程を含む、方法。
【請求項24】
前記動物が哺乳動物である、請求項23に記載の方法。
【請求項25】
前記動物がヒトである、請求項23に記載の方法。
【請求項26】
前記治療因子が、低分子、ペプチド、タンパク質、および細胞からなる群から選択される、請求項23に記載の方法。
【請求項27】
動物の創傷を処置する方法であって、
該創傷と、ペプチドを含む溶液とを接触させる工程を包含し、該溶液がヒドロゲルを形成する、方法。
【請求項28】
前記溶液が治療因子を含む、請求項27に記載の方法。
【請求項29】
前記動物が哺乳動物である、請求項27に記載の方法。
【請求項30】
前記動物がヒトである、請求項27に記載の方法。
【請求項31】
前記治療因子が、低分子、ペプチド、タンパク質、および細胞からなる群から選択される、請求項28に記載の方法。
【請求項32】
細胞を増殖させる方法であって、
細胞を含むヒドロゲルを形成する工程と、
細胞生存に適切な条件下で該細胞を維持する工程と
を含む、方法。
【請求項33】
前記ヒドロゲルがペプチドを含む、請求項32に記載の方法。
【請求項34】
前記ヒドロゲルを形成する工程が、ペプチドを含む溶液の1つ以上の特徴を調整することを含む、請求項32に記載の方法。
【請求項35】
前記調整される特徴が、pH、イオン強度、特定のイオンの濃度からなる群から選択される、請求項34に記載の方法。
【請求項36】
前記調整される特徴がイオン強度である、請求項35に記載の方法。
【請求項37】
前記調整される特徴がCa2+イオン濃度である、請求項35に記載の方法。
【請求項38】
前記細胞が動物細胞である、請求項32に記載の方法。
【請求項39】
前記細胞が哺乳動物細胞である、請求項32に記載の方法。
【請求項40】
前記細胞がヒト細胞である、請求項32に記載の方法。
【請求項41】
前記細胞が骨芽細胞である、請求項32に記載の方法。
【請求項42】
前記細胞が線維芽細胞である、請求項32に記載の方法。
【請求項43】
前記細胞が組換え体である、請求項32に記載の方法。
【請求項44】
前記細胞が目的のタンパク質を発現する、請求項32に記載の方法。
【請求項45】
前記目的のタンパク質が抗体である、請求項44に記載の方法。
【請求項46】
ヒドロゲルを含むセンサ。
【請求項47】
前記ヒドロゲルの1つ以上の特徴が、該ヒドロゲルが目的の分析物と接触した場合に変化する、請求項46に記載の方法。
【請求項48】
前記変化した特徴が剛性である、請求項47に記載の方法。
【請求項49】
前記変化した特徴が光学的性質である、請求項47に記載の方法。
【請求項50】
環境条件を検出する方法であって、
ヒドロゲルを含むセンサを該環境条件を代表するサンプルと接触させる工程と、
該ヒドロゲルの1つ以上の特徴を決定する工程と
を包含する、方法。
【請求項51】
前記ヒドロゲルの1つ以上の特徴が、該ヒドロゲルが目的の分析物と接触した場合に変化する、請求項50に記載の方法。
【請求項52】
前記変化した特徴が剛性である、請求項51に記載の方法。
【請求項53】
前記変化した特徴が光学的性質である、請求項51に記載の方法。
【請求項54】
前記ヒドロゲルの特徴と、異なる時間に決定した該ヒドロゲルの同一の特徴とを比較する工程をさらに包含する、請求項50に記載の方法。
【請求項55】
目的の分子を精製する方法であって、
目的の分子および1つ以上の夾雑物を含む溶液を、目的の分子がヒドロゲルによって保持される条件下でヒドロゲルに接触させる工程と、
該ヒドロゲルから該目的の分子を回収する工程と
を包含する、方法。
【請求項56】
前記目的の分子がタンパク質である、請求項55に記載の方法。
【請求項57】
前記目的の分子が抗体である、請求項55に記載の方法。
【請求項58】
前記目的の分子が治療因子である、請求項55に記載の方法。
【請求項59】
目的の分子を精製する方法であって、
目的の分子および1つ以上の夾雑物を含む溶液を、少なくとも1つの夾雑物がヒドロゲルによって保持される条件下でヒドロゲルに接触させる工程と、
該目的の分子を回収する工程と
を包含する、方法。
【請求項60】
前記目的の分子がタンパク質である、請求項59に記載の方法。
【請求項61】
前記目的の分子が抗体である、請求項59に記載の方法。
【請求項62】
前記目的の分子が治療因子である、請求項59に記載の方法。
【請求項1】
ヒドロゲルの作製方法であって、
ペプチドを含む溶液を提供する工程と、
該溶液の1つ以上の特徴を変化させる工程とを包含する方法であって、ヒドロゲルが形成される、方法。
【請求項2】
前記特徴が、イオン強度、温度、特定のイオンの濃度、およびpHからなる群から選択される、請求項1に記載の方法。
【請求項3】
前記変化させる工程が、前記溶液を電磁放射と接触させることを含む、請求項1に記載の方法。
【請求項4】
前記変化させる工程が、前記溶液のpHを変化させることを含む、請求項1に記載の方法。
【請求項5】
前記溶液の1つ以上の特徴を変化させる工程により、塩濃度が約20mM〜約400mMになる、請求項1に記載の方法。
【請求項6】
前記塩がNaClである、請求項5に記載の方法。
【請求項7】
前記溶液のpHがpH9未満である、請求項1に記載の方法。
【請求項8】
前記溶液のpHが約pH6.0〜約pH8.5である、請求項1に記載の方法。
【請求項9】
前記溶液のpHが約pH7.0〜約pH8.0である、請求項1に記載の方法。
【請求項10】
前記溶液のpHが約pH7.4である、請求項1に記載の方法。
【請求項11】
ペプチドおよび約20mM〜約400mMの塩を含むヒドロゲル。
【請求項12】
前記塩がNaClである、請求項11に記載のヒドロゲル。
【請求項13】
MAX1を含む、請求項11に記載のヒドロゲル。
【請求項14】
ヒドロゲルの作製方法であって、
ペプチドを含む溶液を動物に注入する工程を包含し、該溶液が該動物内でヒドロゲルを形成する、方法。
【請求項15】
前記動物が哺乳動物である、請求項14に記載の方法。
【請求項16】
前記動物がヒトである、請求項14に記載の方法。
【請求項17】
前記溶液が、1つ以上の治療因子をさらに含む、請求項14に記載の方法。
【請求項18】
前記治療因子が、低分子、ペプチド、タンパク質、および細胞からなる群から選択される、請求項17に記載の方法。
【請求項19】
治療因子を必要とする動物に治療因子を送達する方法であって、
該治療因子および1つ以上のペプチドを含む溶液を該動物に投与する工程を包含し、該溶液が該動物内でヒドロゲルを形成する、方法。
【請求項20】
前記動物が哺乳動物である、請求項19に記載の方法。
【請求項21】
前記動物がヒトである、請求項19に記載の方法。
【請求項22】
前記治療因子が、低分子、ペプチド、タンパク質、および細胞からなる群から選択される、請求項19に記載の方法。
【請求項23】
治療因子を必要とする動物に治療因子を送達する方法であって、
該治療因子および1つ以上のペプチドを含むヒドロゲルを該動物に投与する工程を含む、方法。
【請求項24】
前記動物が哺乳動物である、請求項23に記載の方法。
【請求項25】
前記動物がヒトである、請求項23に記載の方法。
【請求項26】
前記治療因子が、低分子、ペプチド、タンパク質、および細胞からなる群から選択される、請求項23に記載の方法。
【請求項27】
動物の創傷を処置する方法であって、
該創傷と、ペプチドを含む溶液とを接触させる工程を包含し、該溶液がヒドロゲルを形成する、方法。
【請求項28】
前記溶液が治療因子を含む、請求項27に記載の方法。
【請求項29】
前記動物が哺乳動物である、請求項27に記載の方法。
【請求項30】
前記動物がヒトである、請求項27に記載の方法。
【請求項31】
前記治療因子が、低分子、ペプチド、タンパク質、および細胞からなる群から選択される、請求項28に記載の方法。
【請求項32】
細胞を増殖させる方法であって、
細胞を含むヒドロゲルを形成する工程と、
細胞生存に適切な条件下で該細胞を維持する工程と
を含む、方法。
【請求項33】
前記ヒドロゲルがペプチドを含む、請求項32に記載の方法。
【請求項34】
前記ヒドロゲルを形成する工程が、ペプチドを含む溶液の1つ以上の特徴を調整することを含む、請求項32に記載の方法。
【請求項35】
前記調整される特徴が、pH、イオン強度、特定のイオンの濃度からなる群から選択される、請求項34に記載の方法。
【請求項36】
前記調整される特徴がイオン強度である、請求項35に記載の方法。
【請求項37】
前記調整される特徴がCa2+イオン濃度である、請求項35に記載の方法。
【請求項38】
前記細胞が動物細胞である、請求項32に記載の方法。
【請求項39】
前記細胞が哺乳動物細胞である、請求項32に記載の方法。
【請求項40】
前記細胞がヒト細胞である、請求項32に記載の方法。
【請求項41】
前記細胞が骨芽細胞である、請求項32に記載の方法。
【請求項42】
前記細胞が線維芽細胞である、請求項32に記載の方法。
【請求項43】
前記細胞が組換え体である、請求項32に記載の方法。
【請求項44】
前記細胞が目的のタンパク質を発現する、請求項32に記載の方法。
【請求項45】
前記目的のタンパク質が抗体である、請求項44に記載の方法。
【請求項46】
ヒドロゲルを含むセンサ。
【請求項47】
前記ヒドロゲルの1つ以上の特徴が、該ヒドロゲルが目的の分析物と接触した場合に変化する、請求項46に記載の方法。
【請求項48】
前記変化した特徴が剛性である、請求項47に記載の方法。
【請求項49】
前記変化した特徴が光学的性質である、請求項47に記載の方法。
【請求項50】
環境条件を検出する方法であって、
ヒドロゲルを含むセンサを該環境条件を代表するサンプルと接触させる工程と、
該ヒドロゲルの1つ以上の特徴を決定する工程と
を包含する、方法。
【請求項51】
前記ヒドロゲルの1つ以上の特徴が、該ヒドロゲルが目的の分析物と接触した場合に変化する、請求項50に記載の方法。
【請求項52】
前記変化した特徴が剛性である、請求項51に記載の方法。
【請求項53】
前記変化した特徴が光学的性質である、請求項51に記載の方法。
【請求項54】
前記ヒドロゲルの特徴と、異なる時間に決定した該ヒドロゲルの同一の特徴とを比較する工程をさらに包含する、請求項50に記載の方法。
【請求項55】
目的の分子を精製する方法であって、
目的の分子および1つ以上の夾雑物を含む溶液を、目的の分子がヒドロゲルによって保持される条件下でヒドロゲルに接触させる工程と、
該ヒドロゲルから該目的の分子を回収する工程と
を包含する、方法。
【請求項56】
前記目的の分子がタンパク質である、請求項55に記載の方法。
【請求項57】
前記目的の分子が抗体である、請求項55に記載の方法。
【請求項58】
前記目的の分子が治療因子である、請求項55に記載の方法。
【請求項59】
目的の分子を精製する方法であって、
目的の分子および1つ以上の夾雑物を含む溶液を、少なくとも1つの夾雑物がヒドロゲルによって保持される条件下でヒドロゲルに接触させる工程と、
該目的の分子を回収する工程と
を包含する、方法。
【請求項60】
前記目的の分子がタンパク質である、請求項59に記載の方法。
【請求項61】
前記目的の分子が抗体である、請求項59に記載の方法。
【請求項62】
前記目的の分子が治療因子である、請求項59に記載の方法。
【図1】

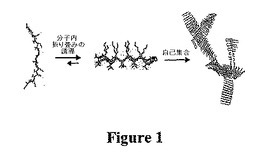
【図2A】
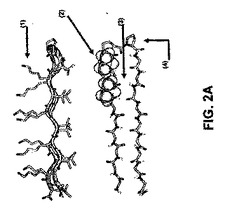
【図2B】
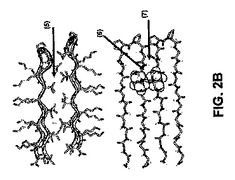
【図3】
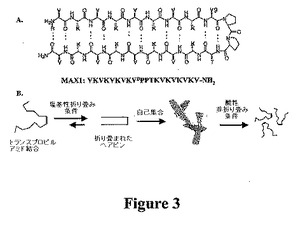
【図4】
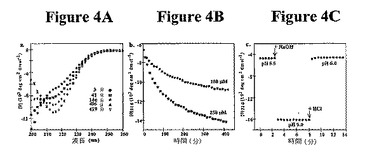
【図5】
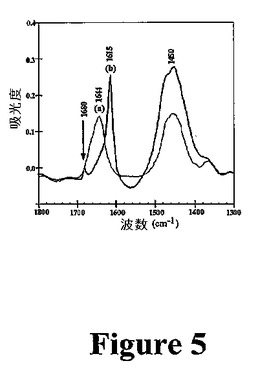
【図6】
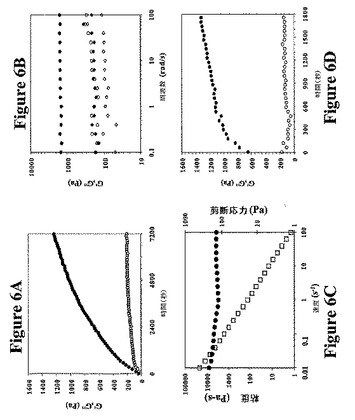
【図7A】
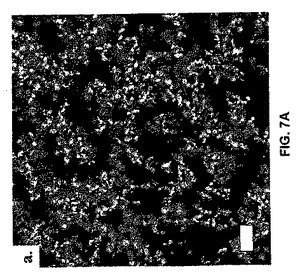
【図7B】

【図8】
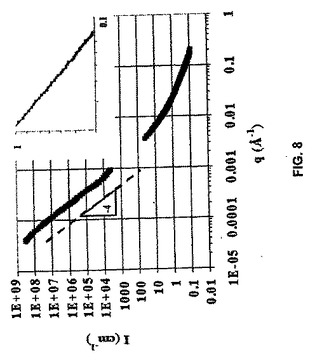
【図9】
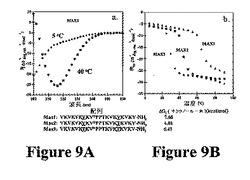
【図10】
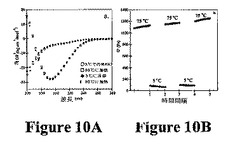
【図11】
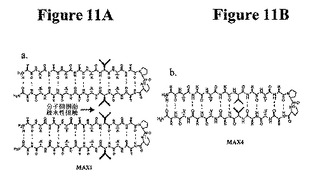
【図12】
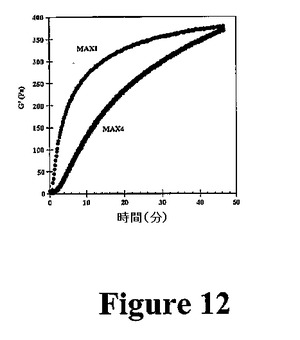
【図13】

【図14】
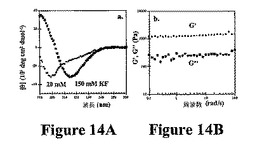
【図15】
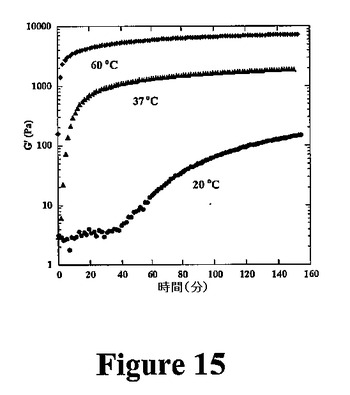
【図16A】

【図16B】

【図16C】

【図16D】

【図16E】

【図16F】

【図17】
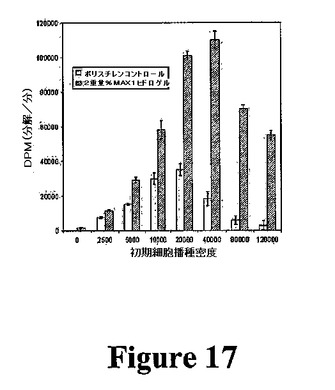
【図18】
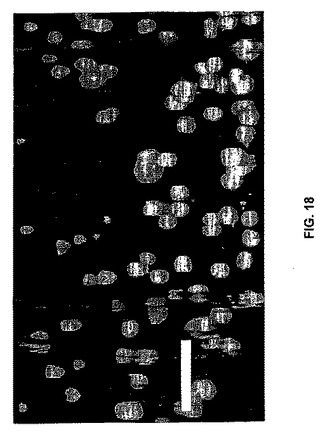
【図19】
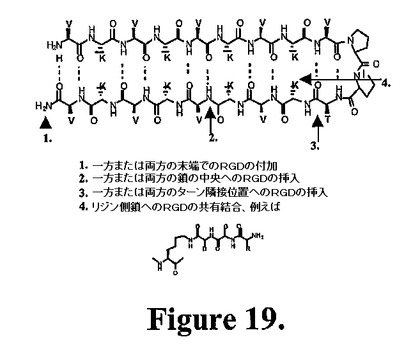
【図20】
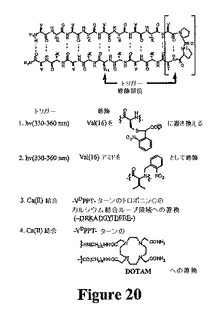
【図21】
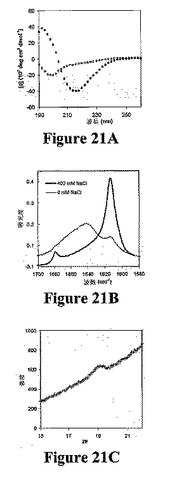
【図22】
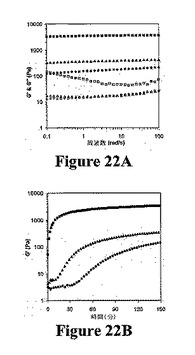
【図23A】

【図23B】

【図24】
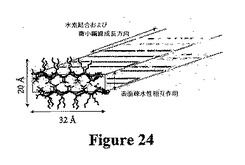
【図25】

【図26】
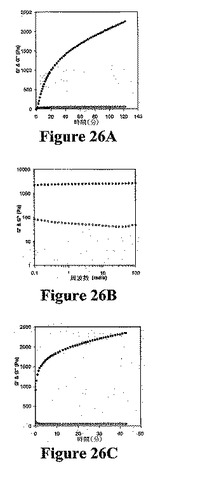
【図2A】
【図2B】
【図3】
【図4】
【図5】
【図6】
【図7A】
【図7B】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16A】
【図16B】
【図16C】
【図16D】
【図16E】
【図16F】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23A】
【図23B】
【図24】
【図25】
【図26】
【公表番号】特表2008−508292(P2008−508292A)
【公表日】平成20年3月21日(2008.3.21)
【国際特許分類】
【出願番号】特願2007−523740(P2007−523740)
【出願日】平成17年7月27日(2005.7.27)
【国際出願番号】PCT/US2005/026528
【国際公開番号】WO2006/076042
【国際公開日】平成18年7月20日(2006.7.20)
【出願人】(300077021)ユニバーシティ・オブ・デラウェア (3)
【氏名又は名称原語表記】UNIVERSITY OF DELAWARE
【出願人】(504131714)ユーディー テクノロジー コーポレーション (2)
【氏名又は名称原語表記】UD TECHNOLOGY CORPORATION
【Fターム(参考)】
【公表日】平成20年3月21日(2008.3.21)
【国際特許分類】
【出願日】平成17年7月27日(2005.7.27)
【国際出願番号】PCT/US2005/026528
【国際公開番号】WO2006/076042
【国際公開日】平成18年7月20日(2006.7.20)
【出願人】(300077021)ユニバーシティ・オブ・デラウェア (3)
【氏名又は名称原語表記】UNIVERSITY OF DELAWARE
【出願人】(504131714)ユーディー テクノロジー コーポレーション (2)
【氏名又は名称原語表記】UD TECHNOLOGY CORPORATION
【Fターム(参考)】
[ Back to top ]