
新規プロテインジスルフィドオキシドレダクターゼ
【課題】変性に強い新規なプロテインジスルフィドオキシドレダクターゼを提供すること、また、それらを含有する試薬・キットおよび製造法を提供すること、さらには、該新規プロテインジスルフィドオキシドレダクターゼを用いて効率的にタンパク質をリフォールディングもしくはフォールディングさせる方法、タンパク質の安定的保存方法を提供すること。
【解決手段】Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有することを特徴とする、成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【解決手段】Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有することを特徴とする、成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、タンパク質やペプチド等のシステインを介したジスルフィド結合の形成や、その逆反応、すなわちジスルフド結合の乖離に関わる反応を触媒する好熱菌由来のCPHCモチーフを有する新規プロテインジスルフィドオキシドレダクターゼ、およびその使用方法に関わる。更には、本プロテインジスルフィドオキシドレダクターゼを含有する試薬・キットにも関する。
【背景技術】
【0002】
大腸菌などの原核生物からヒトを始めとする真核生物まで、タンパク質中もしくはタンパク質間のジスルフィド結合はタンパク質に安定性を付与する手段として進化してきた。特に分泌タンパク質において広く採用されている。
【0003】
これらの結合は主に、原核生物ではペリプラズムにおいて、また真核生物においては小胞体において形成されることが知られており、合成されたタンパク質はそこに留まるものがあるが、より環境の厳しいリソソームのような他の細胞小器官へ移送されるか(真核生物生物の場合)、細胞外へ分泌される。グラム陰性菌の表層は、一般に細胞膜の外側に薄いペプチドグリカン層、更にその外側に外膜が存在するが、ペリプラズムとは細胞膜と外膜の間のことをいう。グラム陽性菌でも細胞膜と厚いペプチドグリカン層の間をペリプラズムと呼ぶ。この空間は、Archaeaにおいても同様に知られており、そこで働く酵素も存在する。
【0004】
ジスルフィド結合は、原核生物においてはDsbAもしくはDsbCと呼ばれるプロテインジスルフィドオキシドレダクターゼによって直接触媒されることが知られている。プロテインジスルフィドオキシドレダクターゼは、チオール:ジスルフィドインターチェンジプロテインとも呼ばれ、DsbAは主にジスルフィド結合の形成に、DsbCは主に形成されたジスルフィド結合を正しい組み合わせに組み換える異性化反応に関わっていることが調べられている。これらの酵素は、分子内にCXXCモチーフを有しており(DsbAは主にCPHC、まれにCPWCやCPYC、DsbCはCGYC)、これらが目的のタンパク質のシステインと相互作用することにより、反応を触媒する機構が提唱されている。またそれらのタンパク質のシステインの酸化還元を調整するためのタンパク質(DsbB、DsbD)も見出されている(たとえば非特許文献1を参照。)。
【非特許文献1】Cell, 99, 117−119 (1999)
【0005】
モチーフとは各種のタンパク質のアミノ酸配列中に認められる小さい構造部分を指す言葉である。それをコードするアミノ酸配列には特徴的なパターンがあり、その局所的に保存されたブロックにより定義されるものを配列モチーフと呼ぶ。多くの場合、ある機能を発現するために必要な最少の単位となるものである。
【0006】
一方、真核生物においては、プロテインジスルフィドイソメラーゼ(PDI)一分子でDsbAおよびDsbC双方の機能を担っていることが知られている。この酵素は、分子内に二つの保存されたCGHCモチーフを有している。異性化反応を触媒することが報告されているDsbCとPDIはDsbAと比較して、強いレダクターゼ活性を示すことが特徴である。DsbAにおけるCXXCモチーフの二つのシステインはジスルフィド結合による結合しており(酸化型)、DsbCのシステインはフリーのSH状態(還元型)であることが知られており、それが各酵素の性質に関わっていると考えられている。
【0007】
近年、様々なタンパク質の遺伝子が明らかとなり、大腸菌等の組換え細胞を用いたタンパク質生産が盛んに行われるようになっている。しかし、例えば真核生物由来のタンパク質を大腸菌で生産した場合、タンパク質が凝集した封入体(インクルージョンボディー)を形成するものが多く、問題となっている。その解決法として、封入体を一度尿素やグアニジン溶液などで完全に変性溶解し、徐々にそれらの変性剤成分を除去することにより、正しいフォールディングを形成させる方法(リフォールディング)が存在する。しかし、透析や希釈で単に変性剤の濃度を下げるだけでは、きちんとフォールディングされないタンパク質も多く、タンパク質のフォールディングを助ける機能を有するシャペロンと呼ばれるタンパク質を共存させることが有効であることが多く報告されている。さらに、ジスルフィド結合が必須なタンパク質のリフォールディングは困難な場合が多く、単に溶液を酸化状態に保っても、ジスルフィド結合の形成が不完全な場合や、正しい組み合わせのジスルフィド結合が形成されない場合が多いのが現状である。そこで、分離精製したプロテインジスルフィドオキシドレダクターゼをリフォールディング時に添加することが考案され、成果を上げている。また近年、プロテインジスルフィドオキシドレダクターゼには若干、シャペロンとしての機能が存在することも知られるようになっている。
【0008】
しかし、タンパク質のリフォールディングは先にも述べたように、まず凝集したタンパク質を尿素やグアニジン等の変性剤で処理し、それらを徐々に除去しながら(すなわちそれらが若干残存した状態)でリフォールディング反応を行うことが多い。そこで、変性に強いプロテインジスルフィドオキシドレダクターゼが求められていた。また、タンパク質の安定化剤や賦形剤等に使用する場合においても、安定性の高いプロテインジスルフィドオキシドレダクターゼが必要であり、同様に求められていた。
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明の目的は、変性に強い新規なプロテインジスルフィドオキシドレダクターゼを求めることにある。さらに、該新規プロテインジスルフィドオキシドレダクターゼを用いて効率的にタンパク質をリフォールディングもしくはフォールディングさせる方法を提供することにあり、さらに、タンパク質の安定的保存方法を提供することである。また、それらを含有する試薬・キットおよび製造法を提供することである。
【課題を解決するための手段】
【0010】
我々は、変性に強い好熱細菌に着目し、種々の好高熱菌について鋭意検討した結果、超好熱細菌であるArchaea(始原菌)から、ペリプラズムで機能していると推定される新規プロテインジスルフィドオキシドレダクターゼを分離し本発明を完成した。
【0011】
通常新規タンパク質の検索は、まず目的のタンパク質を分離してそのタンパク質の部分配列を指標としてその遺伝子を探索する方法や、遺伝子間のホモロジーを指標としたコロニーハイブリダイゼーションや、共通プライマーを用いるPCR法などを用いて行われるのが普通である。また、近年全ゲノムが明らかとなった生物種も多く、それらのデータベースを用いてホモロジーなどを指標として検索することによっても、目的タンパク質候補を絞り込むことができる。
【0012】
今回、本発明者らは、ゲノム配列が明らかとなり、インターネット上でその配列が入手可能な種々の微生物に着目し、それらの遺伝子配列中から新規プロテインジスルフィドオキシドレダクターゼの探索を実施した。まず、DsbA、DsbCおよびPDI等のすでに報告されているプロテインジスルフィドレダクトオキシダーゼの配列に着目し、それらのタンパク質とのホモロジーを指標として検索を実施した。しかし、ホモロジーのあるタンパク質はヒットしなかった。
【0013】
ところが、我々がさらに調査したところ、超好熱細菌であるArchaeaのPyrococcus furiosus(Pfu)から、PDIに類似したタンパク質が発見されていたことがわかった(J.Biol.Chem. 270,5748−5755(1995))。Archaeaは始原菌、もしくは古細菌と訳されるユニークな生物で、原核生物と真核生物の中間的な位置に分類されることもある。さらには、Pyrococcus horikoshiiからもホモログ(ORF ID:PH0178)が分離され、同様な検討が行なわれていた。
【0014】
これらのタンパク質をコードする遺伝子は、リーダー配列と思われる配列が見当たらないため、この分子は、目的のプロテインジスルフィドレダクトオキシダーゼではなく、細胞質内で機能しているタンパク質の一つ、グルタレドキシンの一種であると推定されていた。しかし我々は、これらのタンパク質とのホモロジーについても検討したところ、唯一、前述のPyrococcus furiosus由来の耐熱性プロテインジスルフィドオキシドレダクターゼとホモロジーを有するタンパク質(ORF ID:PH0178)のみがヒットした。このタンパク質には既に、アノテーション情報としてその情報が書き込まれていた。
【0015】
次に、本発明者らは、DsbA、DsbCおよびPDIに含まれる、コンセンサス配列(それぞれ CPHC(CPWC、CPFW)、CGYC、CGHC)に着目し検索を実施した。また、それらのモチーフと併せてリーダー配列(シグナル配列)を有するタンパク質の検索を行った。リーダー配列の有無を条件として挙げた理由としては、ペリプラズムへの移行には本配列が多くの場合必須であるからである。その結果、DsbAホモログと思われるタンパク質の存在が明らかとなった。このタンパク質はPyrococcus horikoshii遺伝子の1025340〜1025852(ORF IDはPH1130として登録)にコードされていることが分かった(配列番号3)。その配列から予想されるアミノ酸配列は配列番号1に示す通りであり(SWISS−PLOTアクセッション番号:D71054)、まだ機能は特定されていなかった。
【0016】
このタンパク質(以下PhDsbタンパク質)は、大腸菌DsbAとはほとんどホモロジーはないものの、CPHCコンセンサス配列、および5残基前のF(フェニルアラニン)も保存されていること、タンパク質のサイズ、およびモチーフが比較的N末端に近いところに存在することからみても、DsbAのホモログである可能性が高いと推測された。また、シグナルペプチド配列検索を実施したところ、本タンパク質はシグナルペプチダーゼによって切り出されると推定される配列をN末端に有していることが明らかとなった。
【0017】
本発明のタンパク質と大腸菌DsbAの比較を図1に示した。今まで報告されているDsbは大きく、酸性タンパク質に分類されるものと、塩基性タンパク質に分類されるものに分類されることが知られているが、本タンパク質は後者であることが、そのアミノ酸配列から推測された。アミノ酸配列(リーダー配列込み)から予想される等電点(pI)は大腸菌DsbAが5.93であるのに対し、PhDsbタンパク質は9.26と予測された。
【0018】
また、本PhDsbタンパク質のアミノ酸配列をBLAST検索(ホモロジー検索)を実施したところ、DDBJアクセッション番号AX041920に登録されているPyrococcus abyssi遺伝子の140922−140371(配列番号4)にコードされるタンパク質(配列番号6:SWISS−PLOTアクセッション番号F75086)に高いホモロジーを示すことが明らかとなった(図2)。この遺伝子にコードされるタンパク質の詳細な機能は明らかにされていないが(チオレドキシン様配列を有するとの記載はあり)、PhDsbタンパク質同様DsbAのホモログと予測される。このタンパク質を便宜上PaDsbと呼ぶこととする。
【0019】
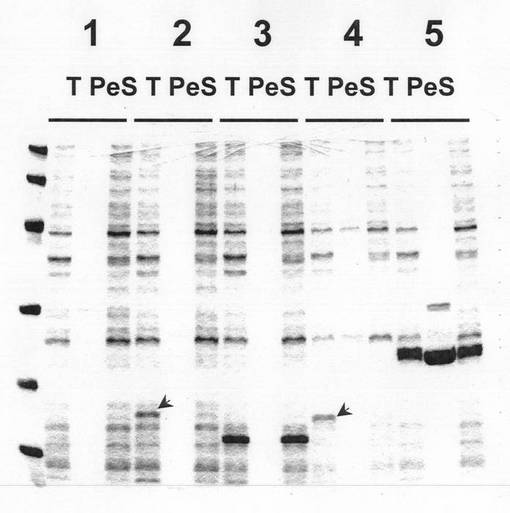
配列から本遺伝子のコードしているタンパク質が大腸菌DsbAのホモログであることはほぼ明らかであったが、実際にタンパク質を発現させて活性を調べる必要があるとの考えから、本発明者らは、次に、本遺伝子をクローニングし、大腸菌を用いた発現を試みた。しかし、最も効率的発現方法の一つとして知られているpET発現系を用いても、本遺伝子産物の発現量は大変低く、また、発現したものはペリプラズムへ移行せず不溶化してしまう傾向にあることが分かった(図3)。一方、コントロール実験として行った大腸菌DsbAにおいては、強い発現とペリプラズムへの移行が認められた。このことから、本タンパク質を機能を有する状態で発現させることに関しても何らかの工夫が必要であることが明らかとなった。
【0020】
次に、本遺伝子の発現効率が低調な理由として、リーダー配列(シグナル配列)が不適であることが予測された為、シグナルペプチダーゼで切断を受けると予測される領域に関して、大腸菌DsbAのリーダー配列と入れ替えて発現を試みたが、これに関してもほとんど改善が見られなかった(図3)。
【0021】
本発明者らは、これらの課題を解決するべく鋭意研究を進めた結果、予想されるリーダー配列を除去したものについて、ペリプラズムへの移行は見られないものの、細胞質内に強い発現を確認することができ、可溶化した状態で回収し得ることが明らかとなった(図3)。今回、シグナルペプチダーゼの切断予測はProtein Engineering, 10, 1−6 (1997)に従って実施した。具体的には、インターネットのSiganal Pのサイト(http://www.cbs.dtu.dk/services/SignalP/)の検索プログラム(Ver.1.1)を利用して予測を行なった。グラム陰性菌のデータを元に予測行った場合、27番目スレオニンと28番目のグルタミンの間(TQT−QT)、グラム陽性菌のデータを元にシグナルペプチダーゼ認識サイトの予測を行った場合は25番目のスレオニンと26番目のグルタミンの間(SQT−QT)が最も可能性の高い切断サイトであることが予測された。今回は、グラム陰性菌のデータを元に予測された切断部位を採用し、予測される切断末端に開始コドン(メチオニン)を付加した配列(配列番号2)を発現させ、実験に用いた。本検証実験から、本タンパク質の配列に起因する何らかの作用によって、本タンパク質の大腸菌細胞膜の透過性が極端に低くなったことの原因であると推察された。
【0022】
上にも記述したようにDsbAのリーダー配列を有するPhDsbによる試みにおける結果もこのことを示唆している。この実験で用いたDsbAリーダー配列を付加したPhDsbの配列は、あらかじめSignal P(Ver.1.1)を用いてシグナルペプチダーゼ認識サイトがあることを確認して用いている。
【0023】
また、不溶化にはシグナルペプチド配列が除去しきれなかったことが原因である可能性が示唆された。シグナルペプチドは大変疎水性に富むものであると予想される。また本実験は、グラム陽性菌のデータに基づいた予測サイトに基づいて調製したタンパク質を用いても、ほぼ同様の結果が得られたと予測される。さらに、Ver.2以降の本予測プログラムを用いた場合、25番目のスレオニンと26番目のグルタミンの間(SQT−QT)が最も可能性の高い切断サイトであると予測されるようであるが、実質は2アミノ酸の差であり、本質的には同様の結果が得られると予測される。
【0024】
一方、Pyrococcus abyssi由来のPaDsbにおいては、グラム陰性菌のデータを元にした予測では29番目のスレオニンと30番目のグルタミンの間(TTT−QT)、23番目のスレオニンと24セリンの間(SST−ST)で切断される可能性が高いことが予想され、この予想部位でリーダー配列を除去したタンパク質(当然メチオニンは付加する必要あり)は、PhDsb同様の活性を示すことが予想される。
【0025】
このようにして得られたプロテインジスルフィドオキシドレダクターゼは、大腸菌の細胞質内から超音波破砕することにより容易に回収することができ、また、塩基性タンパク質であることから、陽イオン交換(S−Sepharose)カラムクロマトグラフィーにより、容易に精製が可能であった。
【0026】
次に、この精製したタンパク質に関して、一般的にプロテインジスルフィドオキシドレダクターゼ活性を調べる際に用いられる、還元条件下でのインスリンを基質としたレダクターゼ活性測定(J. Biol. Chem. 254, 9627 (1979))、および酸化/還元型グルタチオン存在下における変性リボヌクレアーゼを基質としたオキシダーゼ活性測定(Biochem. 30, 613 (1991))を実施し、本酵素が目的の酵素であることを確認した。
【0027】
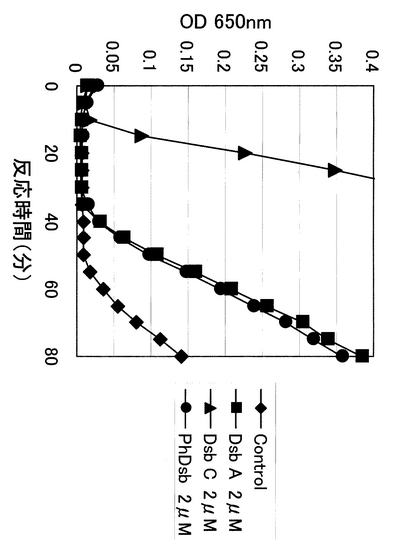
まず始めに、インスリンを基質としたジスルフィドレダクターゼ活性の測定を行なった。インスリンは2種類のペプチドがジスルフィド結合により結合したヘテロダイマーであるが、レダクターゼ活性によりジスルフィド結合が乖離すると、β鎖のペプチドが不溶化し濁度が上昇する。それを吸光度(650nm)測定することによりレダクターゼ活性を測定することが可能である。還元条件下でのインスリン乖離反応はDsbA、DsbC、およびPDIそれぞれに共通して見られる反応である。今回、対照とするため大腸菌DsbA、DsbCをそれぞれ大腸菌ゲノムDNAからクローニングし、文献に従って発現させペリプラズム画分より精製し使用した(J. Bacteriol. 179, 5333 (1997))。それぞれの酵素の最終濃度が2μMになるような一般的な測定条件を用い、反応時間と測定結果(OD 650nm)をプロットしたのが図4である。結果より、PhDsbタンパク質は明らかにDsbAに近いパターンを示すことが分かった。ちなみにDsbAおよびDsbCの反応パターンは報告させているものとほぼ同じ傾向を示していることを確認している。また、酵素が存在しない条件においても、還元剤の影響で徐々にインスリンのジスルフィド結合は乖離するためコントロールにおいても若干吸光度の上昇が見られたが、一般的な現象であることが知られている。
【0028】
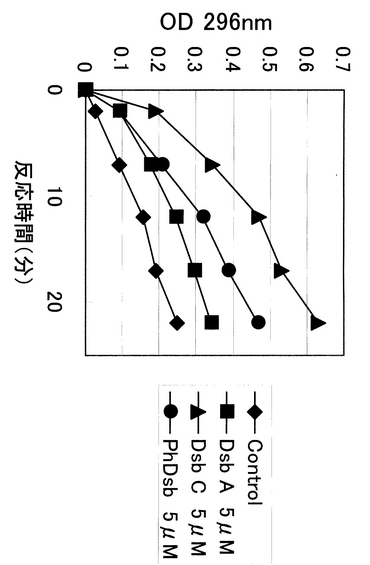
次に、変性リボヌクレアーゼを用いるジスルフィドオキシダーゼ活性の確認を行なった。酸化/還元型グルタチオン存在下における変性リボヌクレアーゼを基質としたオキシダーゼ活性測定はオキシダーゼ活性を測定する場合に頻繁に使用される方法である。リボヌクレアーゼAは活性にジスルフィド結合が必須であり、還元剤を含有する変性剤存在下で不活性化されることが知られている。このようにして変性させたリボヌクレアーゼAはゲル濾過や、希釈、透析などによって変性剤と還元剤濃度を低くすることによって徐々にリフォールディングにより活性が回復することが知られている。また、その回復速度はオキシダーゼ存在を共存させることで早まることが知られている。今回レダクターゼ活性同様に、本測定を用いてDsbAおよびDsbCをコントロールとしてPhDsbのオキシダーゼ活性を測定した。測定条件は文献に倣い最終濃度が5μMとなるように各酵素濃度を調整し、一定の比率にした酸化/還元型グルタチオンを含有する緩衝液添加して、変性還元リボヌクレアーゼAの活性回復をc−CMPを基質として用いOD 296nmの上昇を指標として測定を行なった。反応時間と測定結果(OD 296nm)をプロットしたのが図5である。結果より、PhDsbタンパク質はDsbCよりはむしろDsbAに近い中間的性質を示すことが明らかとなった。
【0029】
レダクターゼおよびオキシダーゼ活性の測定結果より、PhDsbはDsbAに近い性質を示すことが明らかとなり、本タンパク質は、アミノ酸配列のホモロジーには乏しいが、ほぼDsbAのホモログであることを確認することができた。アミノ酸配列が極端に異なっていることは、おそらく酵素を耐熱化させることと何らかの関連性があると推察される。
【0030】
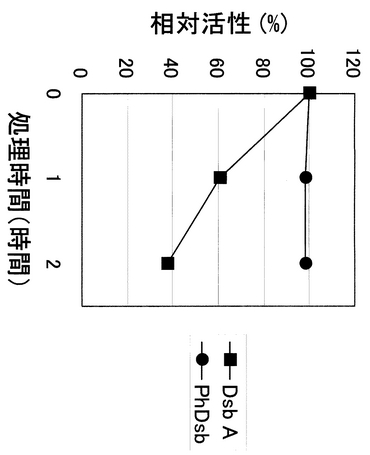
実際に、本発明のタンパク質の耐熱性を調べるために、100℃にて加熱処理し、残存プロテインジスルフィドレダクターゼ活性を指標として活性測定を行なったところ、2時間の加熱処理により大腸菌DsbAは約40%まで活性が低下したのに対し、PhDsbはほぼ100%活性を保っていることが明らかとなった。
【0031】
以上のような経緯を経て、ようやく、我々はArchaeaから、変性に強いプロテインジスルフィドオキシドレダクターゼを分離し、その酵素の性質を明らかにすることが出来た。
【0032】
本発明は以下の構成からなる。
(1)Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有することを特徴とする、成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(2)Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有し、下記理化学的特徴を有する、(1)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
熱安定性:pH7.0にて100℃、2時間の加熱で60%以上の残存活性を保持することができる。
(3)Pyrococcus属細菌由来であることを特徴とする(1)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(4)Pyrococcus horikoshii由来であることを特徴とする(3)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(5)該タンパク質の配列が、配列番号1に示される配列に由来することを特徴とする(4)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(6)該タンパク質の配列が、配列番号2に示される配列であることを特徴とする、(5)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(7)CPHCモチーフ配列が変更されたことを特徴とする、(1)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(8)(1)〜(7)に記載のタンパク質を発現するように、所望の遺伝子配列を挿入したベクター。
(9)(8)に記載のベクターで形質転換された形質転換体。
(10)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを生産する菌株を培養し、培養物より該タンパク質を採取することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
(11)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを細胞質内に生産する菌株を培養し、培養物より該タンパク質を採取することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
(12)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを、無細胞タンパク質合成法を用いて合成することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
(13)加熱処理工程を経ることを特徴とする、(10)〜(12)に記載の成熟型プロテインジスルフィドオキシドレダクターゼの製造方法。
(14)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質内もしくはタンパク質間のジスルフィド結合の切断を促進する方法。
(15)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質のリフォールディングを促進する方法。
(16)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質のフォールディングを促進する方法。
(17)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いて無細胞タンパク質合成時のフォールディングを促進する請求項13に記載の方法。
(18)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼをタンパク質安定化剤および/または賦形剤として用いることを特徴とするタンパク質の安定的保存方法。
(19)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを含有する試薬またはキット。
(20)生物機能を用いて所望のタンパク質を発現させる方法であって、(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを共発現させることにより、該タンパク質の活性を高める方法。
【発明の効果】
【0033】
本発明の、変性に強い新規なプロテインジスルフィドオキシドレダクターゼ、および、それらを含有する試薬・キットにより、効率的にタンパク質をリフォールディングもしくはフォールディングさせる方法、タンパク質の安定的保存方法が可能になった。
【発明を実施するための最良の形態】
【0034】
本発明は、Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有することを特徴とする、成熟型プロテインジスルフィドオキシドレダクターゼタンパク質である。Archaeaは別名始原菌や古細菌とも訳されることもある菌であり、原核生物と真核生物の中間的な位置に分類されることもある、大変ユニークな生物であり、Pyrococcus属以外にも、Thermococcus属等の種々の菌が報告されている。
【0035】
本発明のタンパク質は大腸菌等の好熱細菌以外の細菌で幅広く見出されているタンパク質であり、シグナルぺプチターゼで切り出されるリーダー配列をN末端に有し、それらの多くはN末端の近くにCHPC、もしくは稀にCPWCや、CPYC、CPFCなどのモチーフを有するのが特徴であるが、Archaeaでこの種の酵素が見出されたのは初めてである。
【0036】
本発明において用いられるタンパク質は、成熟型(Mature型)であることが重要である。本発明における成熟とは、リーダー配列(シグナル配列)部分、もしくはN末端側に存在する疎水アミノ酸等からなる膜貫通領域が切除されたタンパク質であることを指す。リーダー配列とは、タンパク質がペリプラズムや小胞体などへ膜を貫通して移行する際に必要な配列であり、移行後は速やかにシグナルペプチダーゼの働きで切断を受けることで、タンパク質は成熟化する。その場合、タンパク質はペリプラズム空間に成熟型酵素として、蓄積されることになる。実施例の、大腸菌DsbA、DsbCの調製はこの方法に準じている。シグナルペプチダーゼの作用する部位には法則性があり、現在、様々な予測用ソフトウエアを活用して切断サイトを予測することができるようになっている。
【0037】
一方、今回の場合のように他の生物種の機能を利用してタンパク質を生産する場合においては、リーダー配列の切除が困難な場合も予想される。例えば、今回のようにArchaeaのタンパク質を大腸菌で発現させるような場合は、タンパク質の膜透過、シグナルペプチダーゼによるリーダー配列の切断がうまく起こらないことがある。そのような場合、リーダー配列部分を除去したタンパク質をデザインし、細胞質内に発現させることで解決できることがある。その場合は、タンパク質合成開始のため、リーダー配列を除去した部分のN末端にメチオニンを付加する必要がある。
【0038】
その場合、リーダー配列の切断部位は、最も好ましくは天然のタンパク質を分離し、N末端解析を行って決定するが、それには多大なる労力と時間が必要な場合が多く、過去のデータを参考にして予測しても良い。
【0039】
シグナルペプチダーゼ切断サイトの予測には、種々開発されているソフトウエアを用いることができる。好ましくは、Signal P(http://www.cbs.dtu.dk/services/SignalP/)を用いると良い。この予測方法は、Protein Engineering, 10, 1−6 (1997)に基づいている。本ソフトウエアにも様々なバージョンがあり、予想配列も若干異なる場合があるが、それほど大差なく使用することが可能である。今回の予測にはVer.1.1を用いているが、基本的にはどのバージョンを用いても問題ない。また、グラム陰性菌、陽性菌、真核生物等で若干予測が異なるが、多くの場合は、グラム陰性菌のデータを用いることで問題なく予測可能である。特に今回は、宿主にグラム陰性菌である大腸菌を選択したため、グラム陰性菌のデータを用いることとした。
【0040】
酵素によっては、リーダー配列除去(成熟化)がタンパク質の溶解性や活性に大きな影響を有しているものも多く存在することが知られているが、本発明においては、その除去が大変重要であった。当然、Pyrococcus horikoshiiを培養して成熟型タンパク質を調製することも考えられるが、実際問題として現実的ではない。すなわち、本発明のタンパク質を得ようとすれば、大腸菌等の扱いの簡単な菌を宿主とすることがもっとも効率的であると考えられる。よって今回、本発明者らが用いた方法は、理にかなった方法であるといえる。
【0041】
今回実験に用いたタンパク質は、Signal P(Ver.1.1)で、グラム陰性菌のデータを元に予測を行い、PhDsbの27番目スレオニンと28番目のグルタミンの間(TQT−QT)で切断がおこると仮定して実験を進めた。すなわち、遺伝子をクローニングする段階で、27番目のスレオニンから上流の配列を除去し、28番目のグルタミンの上流に開始コドンとなるメチオニンの配列を導入した発現プラスミドを構築した。ちなみに、グラム陽性菌のデータを元に予測を行った場合は25番目のスレオニンと26番目のグルタミンの間(SQT−QT)が最も可能性の高い切断サイトであると予想されるが、本実験は、グラム陽性菌のデータに基づいた予測サイトに基づいて調製したタンパク質を用いても、ほぼ同様の結果が得られたと予測される。また、Ver.2以降の本予測プログラムを用いた場合、グラム陰性菌のデータを用いても、25番目のスレオニンと26番目のグルタミンの間(SQT−QT)が最も可能性の高い切断サイトであると予測されるようであるが、上記同様、実質は2アミノ酸の差であり、本質的には同様の結果が得られると予測される。
【0042】
一方、Pyrococcus abyssi由来のPaDsbにおいては、グラム陰性菌のデータを元にした予測では29番目のスレオニンと30番目のグルタミンの間(TTT−QT)、23番目のスレオニンと24セリンの間(SST−ST)で切断される可能性が高いことが予想され、このサイトで切断することで同様の活性型のプロテインジスルフィドオキシドレダクターゼを得ることが可能であることが容易に予測される。
【0043】
当然、これら予測以外の場所でリーダー配列切断部位を決定しても良い。すなわち、発現が向上し、タンパク質の溶解性や活性が向上する条件の場所で切断しさえすれば目的は達成されるのであって、当然、本発明の範囲に含めることができる。
【0044】
また、タンパク質を発現させる際に、種々のタグを付加して発現させても良い。実施例には示していないが、本発明においてはC末端側に6xHis−タグ配列を付加したものに関しても問題なく発現し、活性型タンパク質を得ることができることを確認している。当然、GST(グルタチオンSトランスフェラーゼ)やMBP(マルトースバインディングプロテイン)、GFP(緑色蛍光タンパク質)等との融合タンパク質と発現させても良い。タグの位置は、N末端でもC末端でも構わない。当然、機能を有する範囲において、本タンパク質の部分配列を用いてもよい。
【0045】
また、場合によってはプロテインジスルフィドオキシドレダクターゼ活性を変化させたり、活性を除去したりする場合が有効なことも考えられる。そのような場合は、活性中心であるCPHC配列もしくはその前後の配列に変異を導入することが有効である。特に、活性を除去する場合は、システイン残基を片方もしくは両方を別のアミノ酸に変化させることが有効である。また、活性を変化させる場合、CPHC配列の中のPHを変異させることが有効である。
【0046】
本発明のタンパク質の具体的な理化学的性質としては、好ましくは100℃、2時間の加熱で60%以上、更に好ましくは80%以上の活性を保持することを特徴とする。本活性の指標としては、上に挙げたインスリンを基質として測定するプロテインジスルフィドレダクターゼ活性を指標とするのが最も好ましい。条件としては、実施例10に示すように、十分酵素の初速を評価できる条件で測定する必要がある。また、好ましくは活性の測定は室温(25℃)前後で行う。
【0047】
実際に、レダクターゼ活性を指標として酵素の耐熱性の検討を行った例を図6に示した。方法は、100mMリン酸カリウム緩衝液(pH7.0)中、100℃で加熱した後、上記方法でレダクターゼ活性を測定する方法を用いた。100℃で加熱したのはDsbタンパク質自身が一種のシャペロンタンパク質であり、普通の酵素よりも耐熱性に優れているからである。その結果、大腸菌のDsbAでは100℃・2時間の加温で約40%まで活性が低下したのに対し、PhDsbタンパク質では98%以上の活性を保持したままであった。この結果より、PhDsbタンパク質が高度に耐熱化されていることが明らかとなった。
【0048】
本タンパク質(遺伝子)の由来としては、好ましくはArchaeaであり、さらに好ましくはPyrococcus属細菌由来である。また、最も好ましくはPyrococcus horikoshii由来である。同様に、Pyrococcus abyssi由来のものも好適に用いられる。
【0049】
該タンパク質の配列としては、配列番号1に示されるPyrococcus horikoshiiの配列に由来するものを用いるのがもっとも好ましい。一例としては、予測ソフトによって予測されたリーダー配列を除去し、N末端にメチオニンを付加した配列番号2に示される配列を用いる。ただ、配列はこれに限定されず、リーダー配列の切断サイトは幅を持って決めればよい。
【0050】
また同様に配列番号5に示されるPyrococcus abyssiのプロテインジスルフィドオキシドレダクターゼの配列に由来するものを用いることができる。これに関しても、任意の部位でリーダー配列を切断し、同時に成熟型タンパク質として使用することができる。
【0051】
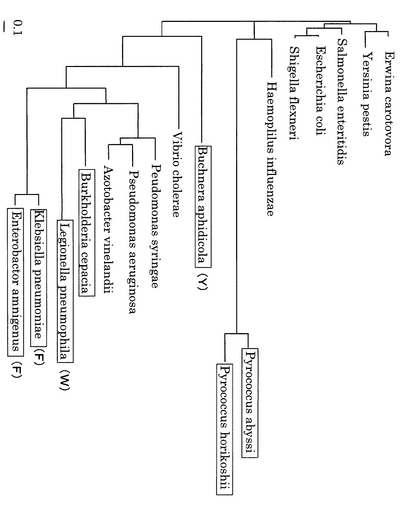
PhDsbとPaDsbのアミノ酸レベルでのホモロジーはGenetyx Ver.3.2を用いて調べたところ69.8%であった。一方、PhDsbと大腸菌のDsbAとのホモロジーは30.2%であり、明らかにArchaea内でのホモロジーは有意に高く、ほぼ同じ機能を有している可能性があるのではないかと推察される。図7には、Swiss−plotデータベースにDsbAとして登録されている由来の異なるタンパク質とPhDsb、PaDsbのアミノ酸配列を元にTreeVew(Ver.1.6.6)(http://taxonomy.zoology.gla.ac.uk/rod/treeview.htmlよりダウンロード)を用いて作成した系統図を示している。それぞれの、Swiss−plotにおけるアクセッション番号は表1に一覧した。その結果、図7から明らかにArchaea由来のDsbA(すなわち、PhDsbとPaDsbは進化的にも同じ傾向を有していることが分かる。
【0052】
【表1】

【0053】
表1は、DsbA類の、アクセッション番号、アミノ酸数、モチーフ、および等電点(pI)を示している。
【0054】
また、表1には、それぞれのアミノ酸数、モチーフ配列、およびGenetyx Ver.3.2を用いて予測した等電点(アミノ酸のpKaをアルギニン:12.5、ヒスチジン:6.0、リジン:10.5、アスパラギン酸:3.9、システイン:8.3、グルタミン酸:4.3、チロシン:10.1として計算)を示している。またそれを元に、図7においてpIが8以上の塩基性タンパク質は四角で囲み示している。また図7の( )にはモチーフCPXC(C:システイン、P:プロリン)のXにあたるところが多型を示していることから、主に用いられている残基であるH(ヒスチジン)以外のアミノ酸を採用しているもの関してそのアミノ酸を示している。これらの図から推察して、Pyrococcus属のDsbAは独自の進化を遂げた可能性が示唆される。
【0055】
本発明は、上記タンパク質の生産方法にも関し、該タンパク質を生産する菌株を適切な培地を用いて培養し、培養物より該タンパク質を採取することを特徴とする。菌としては、特に限定されないが、大腸菌が好ましく用いられ、K12株由来、B株由来等の大腸菌が好適に用いられる。今回の検討では、B株に由来するBL21株を用いて生産を行っているが、それに限定されるものではない。さらに詳細には、BL21−CodonPlus(DE3)−RIL(ストラタジーン社製)を用いているが、これは、ArchaeaのCodon usageが真核生物に近いためである。本菌株はCodon Usageが真核生物に近くなるように遺伝子工学的手法で改変が施されているものである。当然、大腸菌以外のバチルス属由来の微生物や、真核生物である酵母などを用いて生産しても良い。
【0056】
また、タンパク質生産時の誘導法としては、今回、pETベクター系(T7系)を用いたが、Lac系の誘導システムを用いて生産しても良く、特に限定されるものではない。また、合成法も特に問わない。
【0057】
また、本タンパク質は種々の無細胞タンパク質合成法によって合成しても良い。無細胞タンパク質合成法の種類については特に限定されないが、一般的に使われるコムギ胚芽抽出液や、大腸菌抽出液、網状赤血球抽出液、昆虫細胞抽出液、酵母抽出液などを好適に用いることができる。
【0058】
本タンパク質の調製方法に関しては特に限定されないが、大腸菌等を宿主として発現させる場合は、加熱処理することによりその精製効率を飛躍的に向上させることができる。今回は、実施例6で一度カラムクロマトしたフラクションに関して加熱処理を行った例を示しているが、微量混入している夾雑タンパク質を除去するのに大変有効であった。また、精製の初期段階での加熱処理も大変有効である。加熱することにより、立体構造が変化し、活性化されるArchaeaのタンパク質の例も報告されているが、今回調べた範囲で、本発明のタンパク質にそのような効果は確認されていない。しかし、今回試験した以外の性質においては、加熱処理が有効な場合も当然考えられる。
【0059】
該酵素の利用法の一つとして、そのレダクターゼ活性を利用したジスルフィド結合の乖離反応を挙げることができる。その用途は特に限定されないが、タンパク質の還元の促進反応が必要なすべての用途に用いることができる。また、産業としては、毛髪のパーマネントなど還元が必要なものへの応用が考えられ、本酵素は耐熱性であることからも、本用途への応用の可能性は高いといえる。また、タンパク質以外にもシスチン(システインがジスルフィド結合により結合したシステインの二量体)の還元促進等への応用にも持ちることが可能であることは容易に予測できる。また、本方法を様々なパターンのジスルフィド結合複合体に応用できることは容易に予想できる。
【0060】
また、該酵素の利用方法としては、タンパク質のリフォールディングを挙げることができる。リフォールディングとは一度変性させたタンパク質の立体構造を再度形成させ、機能を復活させることである。種々のタンパク質の合成においては、フォールディングがうまくいかずに、変性した凝集体として回収されることがあり、問題となっている。特に、微生物でタンパク質を合成する場合、インクルージョンボディー(封入体)の形成が問題となることが多い。また、無細胞タンパク質合成を行う場合も、タンパク質の凝集不溶化が問題になることがある。それらのタンパク質は一度、尿素やグアニジンなどのタンパク質変性剤で溶解した後、それらの成分を徐々に系から除去することにより、正しい立体構造をとらせることができる場合がある。また、ジスルフィド結合が必須な酵素の場合、変性時に還元剤を添加しておき、ジスルフィド結合も切断しておくと効果的である。その場合は、変性剤を除去する工程で、変性剤も除去し、ジスルフィド結合形成を促進する。ただ、変性剤や還元剤を系から除去するだけでは一般的にリフォールディングの効率は低いことが報告されている。そこで、種々のプロテインジスルフィドオキシドレダクターゼをその工程で働かせることが行なわれてきた。本酵素を用いても、同様に酵素をリフォールディングさせることが可能であることは実施例に示すリボヌクレアーゼを用いた実験例からも明らかである。また、本酵素は熱など過酷な条件に強いことから、種々の変性剤が存在するような条件での使用に関しても適しているといえる。また、本酵素を用いるリフォールディングは、ジスルフィド結合の形成を介さない場合にも有効であることが後述の実施例によっても明らかであり、様々なフォールディング一般への応用を考えることができる。
【0061】
本発明は、該酵素を用いてタンパク質のフォールディングを促進する方法を含む。フォールディングの促進としては、特に限定されないが、無細胞タンパク質合成等のタンパク質合成の場に本タンパク質を存在させることにより、フォールディング形成を促進させることができる。実際今回、高効率コムギ胚芽無細胞タンパク質合成反応液中でのヒト−S−アミラーゼの合成反応中に本酵素を存在させておく実験を実施したが、有意にフォールディングを促進する結果を得ている。アミラーゼは11のシステイン残基を有しており、それらがジスルフィド結合を形成することが活性発現に必須である。今回、酸化グルタチオンと還元グルタチオンを混合した、比較的ジスルフィド結合の形成されやすい条件を用いて無細胞タンパク質合成を実施している。
【0062】
実際のタンパク質合成には、重層法を用いた(FEBS Letters 514, 102−105(2002))。タンパク質合成には、ATPやアミノ酸などの低分子物質が必須であるが、通常のバッチ反応では2〜3時間でそれらの成分が枯渇してしまう。重層法は、反応溶液の上にバッファー層を重層することで、拡散反応によって低分子物質を連続的に供給する方法であり、バッチ法に比べ約10倍程度の合成量の向上を期待することができる。今回は、その下層すなわち、反応溶液にDsbタンパク質を混合し、実験を行った。バッファー層には一般的に、タンパク質成分を加えない方が良い結果が得られることが確かめられていることから、バッファー層にはタンパク質成分は添加していない。
【0063】
その結果、無添加(コントロール)でもある程度は活性を示したが、本酵素を添加することでコントロールに比べ約170%の活性を有するアミラーゼを合成することができた。また、この効果は大腸菌のDsbAよりも高く、むしろDsbCに近いものであった。これは、ジスルフィド結合形成に加え、シャペロンとしての作用が本酵素に高いことを示していると思われる。シャペロンとはヒットショックタンパク質に代表されるタンパク質のフォールディングを助ける作用を有するタンパク質を指す。近年、ジスルフィドオキシドレダクターゼとして報告されていたPDIやDsbタンパク質にも若干ではあるがシャペロン活性があることが知られてきており、PhDsbにはその活性が高いのではないかと予想される。
また、今回実験で証明したこれらの効果は、合成量の増大ではなく、質の向上であることはウェスタンブロッティング解析を用いて確認している。
【0064】
当然、本酵素は無細胞タンパク質合成によって目的タンパク質と同時合成しても良い。
【0065】
さらに本発明は、本酵素を賦形剤、安定化剤として用いることを特徴とするタンパク質の安定的保存方法も含有している。上記に記載したように、本酵素は、ジスルフィド結合形成の他にもシャペロンとしての働きも示唆されており、それらの用途には適していることが予想される。特に、本酵素は耐熱性であり、高温での酵素の安定性を向上させる用途とうへの応用も期待される。
【0066】
また、本発明は、生物機能を用いて所望のタンパク質を発現させる場合にも応用することができる。例えば、大腸菌等でタンパク質を発現させる際に、本発明のタンパク質を共発現させることにより、よりフォールディングのしっかりした質の高いタンパク質を取得することができる可能性が非常に高い。共発現の方法としては、同一ベクターまたは別のベクターでタンパク質を発現する遺伝子を供給する方法が考えられる。
【0067】
本発明は、本酵素を含有する試薬・キットである。
【0068】
本発明の実施の一態様としては、ジスルフィド結合乖離用試薬、および/またはキットである。
【0069】
本発明の実施の一態様としては、タンパク質リフォールディング用試薬、および/またはキットである。
【0070】
また、本発明の実施の一態様としては、タンパク質フォールディング用試薬、および/またはキットである。
【0071】
また、本発明の実施の一態様としては、タンパク質安定化剤、および/または賦形剤である。
【実施例】
【0072】
以下に本発明の実施例をあげることにより、本発明による効果をより一層明瞭なものとする。ただし、これらの実施例によって本発明の範囲は限定されるものではない。
【0073】
実施例1:PhDsbのクローニング
まず、P. HorikoshiiのゲノムDNAを鋳型として目的タンパク質のORFをPCR法にて増幅し、pET−11c(ストラタジーン社製)へクローニングした。具体的には、P. HorikoshiiのゲノムDNA(ATCCより購入)を鋳型として、配列番号6(NdeIサイトを有する)と配列番号7(BamHIサイトを有する)、また、リーダー配列を除去したタンパク質をクローニングする目的で配列番号8(NdeIサイトを有する)と配列番号7(BamHIサイトを有する)の配列を有するオリゴヌクレオチドをプライマーとして高正確性DNAポリメラーゼ(KOD −Plus− :東洋紡製)を用いて増幅を実施した(この酵素を用いて増幅したDNAは末端が平滑末端となる)。増幅はKOD −Plus−の取扱い説明書に従って実施した。増幅したDNAはMagExtractor−PCR&Gel Clean up−キット(東洋紡製)にて精製、末端を制限酵素BamHIで消化した後、更に同キットで精製した。また、一方で、pBluescript(pBS)ベクター(ストラタジーン製)をEcoRVとBamHIで消化し、同様に精製した。精製したDNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセルを形質転換した。37℃で一晩薬剤選択培地(LB/Ampプレート)にて培養した後、生じたコロニーを、LB/Amp培地を用いて液体培養し、それから分離精製したプラスミドを用いて配列確認を行った。
次に、配列を確認したプラスミド(それぞれpBS−PhDsb、pBS−PhDsb(L−)(L−:リーダー配列除去))をNdeIおよびBamHIで消化・精製し、目的遺伝子断片の調製を行った。同時に、pET−11c(ストラタジーン社製)も同様の制限酵素で消化・精製した。精製した目的DNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセル(東洋紡製)を形質転換した。最終的には、生じたコロニーからプラスミドDNA(pET−PhDsb、pET−PhDsb(L−))を抽出し、そのプラスミドDNAを用いて、BL21−CodonPlus(DE3)−RILコンピテントセル(ストラタジーン製)を形質転換し、下記実験に用いた。
【0074】
実施例2:大腸菌DsbA、DsbCのクローニング
まず、実施例1と同様に大腸菌ゲノムDNAを鋳型として目的タンパク質のORFをPCR法にて増幅し、pET−11c(ストラタジーン社製)へクローニングした。具体的には、大腸菌ゲノムDNA(JM109株より定法を用いて抽出)を鋳型として、配列番号9(NdeIサイトを有する)と配列番号10(BamHIサイトを有する)、および配列番号11(NdeIサイトを有する)と配列番号12(BamHIサイトを有する)に示すプライマーをそれぞれ用いて高正確性DNAポリメラーゼ(KOD −Plus− :東洋紡製)によって増幅を実施した(この酵素を用いて増幅したDNAは末端が平滑末端となる)。増幅はKOD −Plus−の取扱い説明書に従って実施した。増幅したDNAはMagExtractor−PCR&Gel Clean up−キット(東洋紡製)にて精製し、末端を制限酵素BamHIで消化した後、更に同キットで精製した。また一方で、pBluescriptベクター(ストラタジーン製)をEcoRVとBamHIで消化し、同様に精製した。精製した目的DNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセルを形質転換した。37℃で一晩薬剤選択培地(LB/Ampプレート)にて培養した後、生じたコロニーをLB/Amp培地を用いて液体培養し、それから分離精製したプラスミドを用いて配列確認を行った。
次に、配列確認を行ったプラスミド(pBS−DsbA、およびpBS−DsbC)をNdeIおよびBamHIで消化・精製し、目的遺伝子断片の調製を行った。同時に、pET−11c(ストラタジーン社製)も同様の制限酵素で消化・精製した。精製したDNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセル(東洋紡製)を形質転換した。最終的には、生じたコロニーからプラスミドDNAを抽出し、そのプラスミドDNA(pET−DsbA、pET−DsbC)を用いて、BL21−CodonPlus(DE3)−RILコンピテントセル(ストラタジーン製)を形質転換し、下記実験に用いた。
【0075】
実施例3:DsbAリーダー配列を有するPhDsbのクローニング
まず、上記実験によって調製されたpBS−DsbAを鋳型として、配列番号9と配列番号13(リンカー配列としてPhDsb配列を一部有する)に示すプライマーを用いて高正確性DNAポリメラーゼ(KOD −Plus−:東洋紡製)によってDsbAのシグナルペプチド配列部分の増幅を実施した(この酵素を用いて増幅したDNAは末端が平滑末端となる)。また一方、同じく上記実験によって調製されたpBS−PhDsbを鋳型として、配列番号14(リンカー配列としてDsbA配列の一部を有する)と、配列番号7をプライマーとして、同じく高正確性DNAポリメラーゼ(KOD −Plus−:東洋紡製)を用いてDsbAのシグナルペプチド配列部分の増幅を実施した。その後、それぞれの増幅産物を精製した後、10倍に希釈して混合し、プライマーを添加しない条件で、KOD −Plus−を用いたエクステンション反応を10サイクル実施した(この反応で、リンカー部分を解して、それぞれの増幅産物が連結される)。その後、反応液を鋳型に、配列番号9と配列番号7をプライマーとして、同じくKOD−Plus−を用いて融合遺伝子の全長を増幅した。増幅したDNAはMagExtractor−PCR&Gel Clean up−キット(東洋紡製)にて精製、末端を制限酵素BamHIで消化し、更に同キットで精製した。また一方で、pBluescriptベクター(ストラタジーン製)をEcoRVとBamHIで消化し、同様に精製した。精製したDNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセルを形質転換した。37℃で一晩薬剤選択培地(LB/Ampプレート)にて培養した後、生じたコロニーを用いて液体培養し、それから分離精製したプラスミドを用いて配列確認を行った。
次に、配列確認を行ったプラスミド(pBS−DsbAL/PhDsb)をNdeIおよびBamHIで消化・精製し、目的遺伝子の調製を行った。同時に、pET−11c(ストラタジーン社製)も同様の制限酵素で消化・精製した。精製した目的DNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセル(東洋紡製)を形質転換した。最終的には、生じたコロニーからプラスミドDNAを抽出し、そのプラスミドDNA(pET−DsbAL/PhDsb)を用いて、BL21−CodonPlus(DE3)−RILコンピテントセル(ストラタジーン製)を形質転換し、下記実験に供した。
【0076】
実施例4:PhDsb、大腸菌DsbA、およびDsbCタンパク質の発現条件の検討
それぞれの遺伝子を挿入したpET−11cで形質転換したBL21−CodonPlus(DE3)−RIL株(それぞれPhDsb、PhDsb(L−)、DsbAL/PhDsb、DsbA、およびDsbC遺伝子を保有)、を用いてタンパク質の発現を実施した。具体的には37℃でLB/Amp培地を用いて0.4ODになるまで培養した後、IPTGを1mMとなるように添加し、37℃で3時間培養し、目的タンパク質を発現させた。
培養の後、培養液を6,000r.p.m.で10分間遠心し、菌体を回収した。その後、10mM Tris−HCl(pH8.0)に菌体を懸濁し30分間放置し、ペリプラズムに発現されたタンパク質を菌外へ放出させた。その懸濁液を15,000r.p.m.で10分間遠心し、その上清を回収した(ペリプラズム画分)。このペリプラズムタンパク質の回収方法は、J. Bacteriol. 179(17) 5333−5339(1997)を参考にした。また、その時得られたペレットに10mM Tris−HCl(pH8.0)を適量添加、懸濁し、2分間超音波処理を行った後、15,000r.p.m.で10分間遠心しその上清を回収した(菌体内可溶性画分)。PhDsb、PhDsb(L−)、およびDsbAL/PhDsbに関して得られた画分をSDS−PAGE解析を行った結果を図3に示した。コントロールとして、DsbAの結果も示している。結果より、DsbAタンパク質はペリプラズムに回収されたのに対して、リーダー配列部分を含む完全長の遺伝子発現を行ったPhDsbタンパク質およびDsbAのリーダー配列を融合させたPhDsbタンパク質はペリプラズム画分ではまったく回収されなかった。また、その発現レベルも大変低いものであり、菌体内可溶性画分に回収されなかったことから不溶化しているものと推測された。一方、シグナルペプチド部分を除去したPhDsbL−は、当然ペリプラズム画分にはタンパク質は回収されなかったが、細胞質の可溶性画分に強い発現を認めることができ、可溶性タンパク質として回収できたことが明らかとなった。データは示していないが、DsbCもペリプラズム画分に強い発現を認めた。
【0077】
実施例5:DsbA、およびDsbCの精製
実施例3の方法に従って回収したDsbA、およびDsbCのペリプラズム画分を10mM Tris−HCl(pH7.5)にて平衡化させたDEAE−Sepharose(アマシャムバイオサイエンス社製)に供し、同緩衝液で5カラム容量分洗浄した後、0.1Mに続き0.2M NaClを含有する同緩衝液でタンパク質を溶出した。DsbAは0.1M NaCl画分に、DsbCは0.2M NaCl画分にそれぞれ溶出された。フラクションはそれぞれSDS−PAGE解析を行い、メインフラクションのみを集めて、20mM HEPES−KOH(pH7.6)に対して透析し、酵素溶液として種々の実験に使用した。文献等に従うと、この方法で得られるDsbタンパク質は、リーダー配列を切除されていると考えられ、以下の実験には、ここで得られたタンパク質(詳細に記述するとDsbA(L−)、DsbC(L−)となる)を用いて行った。
【0078】
実施例6:リーダー配列除去PhDsb(L−)の精製
実施例3の方法で発現誘導した菌体(BL21−CodonPlus(DE3)−RIL/pET−PhDsb(L−))を回収し、10mM Tris−HCl(pH7.5)にて懸濁後、超音波破砕の後、15,000r.p.m.にて遠心し、その上清を回収した。次に、その上清を10mM Tris−HCl(pH7.5)にて平衡化したS−Sepharose(アマシャムバイオサイエンス社製)に供し、同緩衝液にて5カラム容量分洗浄した後に、0.2M NaClを含有する同緩衝液にて溶出した。また、必要に応じて精製度を高める目的で、溶出された画分を80℃、20分間加熱処理した後、10mM Tris−HCl(pH7.5)にて10倍希釈し、同緩衝液にて平衡化したS−Sepharoseに供した。カラムを同様に同緩衝液にて洗浄した後に、0.2M NaClを含有する同緩衝液にて溶出した。溶出されたフラクションはSDS−PAGE解析を行い、メインフラクションのみを混合し、20mM HEPES−KOH(pH7.6)に対して透析し、酵素溶液として下記の種々の実験に用いた。
【0079】
実施例7:ヒト唾液(S)アミラーゼ遺伝子のクローニング
無細胞タンパク質合成実験に用いるヒト−S−アミラーゼ遺伝子は、ヒトcDNA(Clontech社製)からPCR法を用いてクローニングを行った。具体的には配列番号15および配列番号16(BamHI配列を含む)に記載のプライマーを使って、高正確性DNAポリメラーゼ(KOD −Plus−:東洋紡製)を用いて増幅を実施した(この酵素を用いて増幅したDNAは末端が平滑末端となる)。ヒトアミラーゼはリーダー配列を有するタンパク質であるが、無細胞タンパク質合成法においては除去されないことが分かっていることから、今回は最初から、既に報告されているリーダー配列部分を含まない形でクローニングできるよう、プライマー設計を行った。増幅はKOD −Plus−の取扱い説明書に従って実施した。増幅したDNAはMagExtractor−PCR&Gel Clean up−キット(東洋紡製)にて精製、末端を制限酵素BamHIで消化した後、更に同キットで精製した。また一方で、無細胞タンパク質合成用ベクター(pEU3−NIIベクター:東洋紡製)をEcoRVとBamHIで消化し、同様に精製した。精製した目的DNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセルを形質転換した。37℃で一晩薬剤選択培地(LB/Ampプレート)にて培養した後、生じたコロニーを用いて液体培養し、それから分離精製したプラスミドを用いて配列確認を行った。また、無細胞タンパク質合成用に、QIAFILTER MIDI KIT(QIAGEN社製)を用いてプラスミドを抽出し、さらにフェノール/クロロホルム抽出、エタノール沈殿法を用いてプラスミドを精製した。ここで構築したプラスミドを以下、pEU−S−AMYとする。
【0080】
実施例8:ジスルフィドレダクターゼ活性測定
各酵素のジスルフィドレダクターゼ活性は基本的には、J. Biol. Chem. 245(19)9627−9632(1979)に従って測定した。具体的には、アッセイ用緩衝液(1mg/ml ウシインスリン(ロシュダイアグノスティクス社製)、0.1M リン酸カリウム−酢酸緩衝液(pH7.0)、2mM EDTA)を96ウェルプレートに150μlづつ添加した後、酵素溶液を添加し180μlにした。酵素サンプルとしては、実施例5、および6にて調製した20mM HEPES−KOH(pH7.6)に溶解したDsbA(L−)、DsbC(L−)、PhDsb(L−)(以下(L−)略)を用い、測定時の終濃度が2μMとなるようにした。その後に、10mM ジチオスレイトール(DTT)を7.5μl添加、混合し(終濃度0.5mM)、反応を開始した。反応は、室温(25℃)にて行い、濁度(650nm)の変化をプレートリーダーにて5分毎に測定した。その結果を、図4に示す。図から明らかなように、PhDsbと大腸菌DsbAはほぼ同じ挙動を示し、PhDsbはDsbAのホモログであることが示唆された。コントロールも最終的には濁度の上昇が見られたが、これは還元状態における自発的な乖離現象であり、一般的な現象である。
【0081】
実施例9:ジスルフィドオキシダーゼ活性測定
各酵素のジスルフィドオキシダーゼ活性は基本的には、Biochemistry 30, 613−619 (1991)に従って実施した。すなわち、変性リボヌクレアーゼを酵素溶液で希釈し、リボヌクレアーゼ活性を指標としてその活性の復活を測定した。具体的には、まず、5mgのウシリボヌクレアーゼA(Sigama社製)を1mlの変性溶液(6M グアニジン塩酸塩、0.1M Tris−酢酸緩衝液(pH8.0)、2mM EDTA、0.14mM DTT)に溶解し、一晩放置し変性させた。その後、0.1%酢酸で平衡化したG−25スピンカラム(アマシャムバイオサイエンス社製)にてゲル濾過し、実験に用いた。
50μlの2倍濃度のアッセイ溶液(100mM Tris−酢酸(pH8.0)、9mMcCMP(Sigma社製)、0.4mM 酸化型グルタチオン(GSSG)、2mM 還元型グルタチオン(GSH))に50μlの10μMの各種酵素(DsbA(L−)、DsbC(L−)、およびPhDsb(L−)(以下(L−)略)(20mM HEPES−KOH(pH7.6))を混合した後、2.2μlのゲル濾過後の変性リボヌクレアーゼを添加、混合し、反応を開始した。反応は室温(25℃)で行い、ほぼ5分毎に296nmの変化を測定した。また、ブランクとして、変性リボヌクレアーゼAを添加しないものも測定し、コンタミ酵素によってバックグラウンドの吸光度が増加しないことを確認した。
その結果、図5に示すようにDsbC>PhDsb>DsbAの順番でリフォールディング活性を確認することができた。PhDsbはDsbAより若干活性が強く、DsbCとDsbAの中間的な性質を示した。また、コントロールにも若干吸光度の上昇が見られたが、これも一般的に知られている現象である。
【0082】
実施例10:耐熱性の評価
DsbAおよびPhDsbを100mMのリン酸カリウム緩衝液(pH7.0)にて60μMに調製し、サーマルサイクラーを用いて100℃で任意の時間加熱した後、基本的には実施例8の方法に従ってジスルフィドレダクターゼ活性を測定した。具体的には、酵素の終濃度は5μMとなるようにし、25分後のA650nmを測定した。25分後のコントロールの吸光度はブランクとほぼ同じであった。活性は、非加熱のサンプルの吸光度650nmを100%とし、それの相対値で示している。結果を図6に示すが、100℃、2時間の加熱で大腸菌由来のDsbAの活性は40%まで低下したのに対し、PhDsbはほぼ100%活性を保持していた。今回の測定においては、本条件で測定した実測地をそのまま活性値として評価しているが、今回の実験においては、酵素反応の初速をほぼ観察していると思われることから、妥当な測定方法であるといえる。
【0083】
実施例11:無細胞タンパク質合成への応用例
PhDsbタンパク質のフォールディングに及ぼす効果を、無細胞タンパク質合成法を用いて検証した。具体的には、ヒト−S−アミラーゼ(ジスルフィド結合の形成が活性発現に必須)を高効率コムギ胚芽無細胞タンパク質合成系(Proc.Natl.Acad.Sci.USA 96(2),559−564(2000))を用いて合成する際に、各種プロテインジスルフィドオキシドレダクターゼを共存させておき、合成後にアミラーゼ活性を測定することにより、そのフォールディングに及ぼす効果を測定した。ヒト−S−アミラーゼは11のシステイン残基を有し、それらが複雑なジスルフィド結合を形成して活性を発現すると考えられており、通常の還元条件を用いる無細胞タンパク質合成反応では活性型の酵素を合成することが困難である。よって、今回用いたタンパク質合成に用いる際の低分子成分組成は、参考文献(Nature Biotechnology, 15, 79−84 (1997))に報告されているように、還元剤(ジチオスレイトール:DTT)の代わりに、酸化グルタチオン(GSSG)と還元グルタチオン(GSH)を一定の組成になるように添加した組成を用いた。また、無細胞タンパク質合成に用いたバッファー以外の成分は、PROTEIOS Wheat germ cell−free protein synthesis kit(東洋紡製)のものを用い、キット添付の取扱い説明書に従い、重層法(FEBS Letters 514, 102−105(2002))を用いて実施した。詳細には、反応液(下層)として、容量の20%のWheat germ extract (キット添付;200OD)、800units/ml Ribonuclease inhibitor (東洋紡製)、3μM 評価用タンパク質(DsbA(L−)、DsbC(L−)、PhDsb(L−)、およびBSA(以下(L−)略))、バッファー成分(終濃度として;30mM HEPES−KOH (pH7.6)、95mM 酢酸カリウム、2.65mM 酢酸マグネシウム、0.25mM GTP、0.4mM 酸化型グルタチオン(GSSG)、1mM 還元型グルタチオン(GSH)、1.2mM ATP、16mM クレアチンリン酸、0.5mg/mlクレアチン(ホスホ)キナーゼ(Roche製)、0.38mM スペルミジン、20種類のL−アミノ酸(各0.3mM)、0.25mg/ml mRNA(PROTEIOSの取扱い説明書に従いpEU−S−AMYから転写し、精製したもの))、となるようにそれぞれ50μl調製した。使用したPhDsbは、フラクション後、加熱処理を行ない更に精製を行ったものを用いた(実施例6参照)。上層(バッファー層)としては、終濃度として;30mM HEPES−KOH (pH7.6)、95mM 酢酸カリウム、2.65mM 酢酸マグネシウム、0.25mM GTP、0.4mM 酸化型グルタチオン(GSSG)、1mM 還元型グルタチオン(GSH)、1.2mM ATP、16mM クレアチンリン酸、0.38mM スペルミジン、20種類のL−アミノ酸(各0.3mM)となるように調製した。反応は26℃で16時間行った。
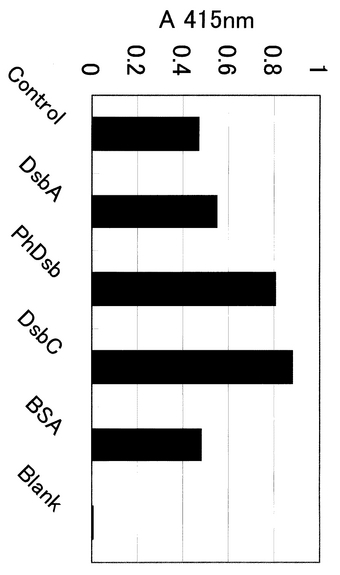
コムギ胚芽抽出液自体はアミラーゼ活性を有さず、また活性を阻害しないことを確認しているので、合成終了後、その液をそのまま用いてアミラーゼ活性の比較を行った。活性測定は、α−アミラーゼ測定キット・ダイヤカラー・リキッドAMY(東洋紡製)を用いて測定した。具体的には、酵素試薬(キット添付)300μlにサンプル5μlを添加した後に、基質試薬(キット添付)を150μl添加し、37℃にて11分間反応させ、415nmの吸光度をプレートリーダーで測定した。その結果を、図8に示した。図から明らかなように、コムギ胚芽抽出反応液のみを添加したBlankにはまったくアミラーゼ活性は認められなかった。一方、Dsbサンプルの代わりに20mM HEPES−KOD(pH7.6)のみを添加したControlについては活性の発現を認めることができた。これは、通常無細胞タンパク質合成反応で用いられるジチオスレイトール(DTT)を用いる代わりに、今回酸化/還元グルタチオン系を用いた効果であると思われた。また、BSAを添加したものの活性のレベルもControlとほぼ同じレベルであった。Dsb類を添加したものについては、DsbC>PhDsb>>DsbAの順番でアミラーゼ活性の向上を認めることができた。PhDsbはジスルフィドレダクターゼ活性レベルがDsbAとほとんど同レベルであることが確認されている(実施例8)ことから、DsbCで知られているようなジスルフィド結合を架けかえる(異性化)する活性(イソメラーゼ活性)は低いことが予想される。よって、この実験における高いリフォールディング活性は、ジスルフィドオキシダーゼ活性に加えて、シャペロンとしての効果が高かった可能性を示唆していると考えられる。
また、この実験における各条件でのアミラーゼの合成量は、別途実施した抗アミラーゼ抗体(Santa Cruz社製)を用いたウエスタンブロッティングの結果からほぼ同等であることを確認しており、合成量の違いによるものではなく、質の問題であることは確かである。また、ヒトS−アミラーゼ標品を用いた比較より、コントロールでタンパク質あたり約10%の活性であることが確認されている。よって、PhDsbの添加により、10%から17%程度(1.7倍)活性化されたことになる。
【産業上の利用可能性】
【0084】
本発明は、大腸菌等の組換え細胞を用いたタンパク質生産、あるいは、タンパク質研究の分野においてきわめて有用である。
【図面の簡単な説明】
【0085】
【図1】大腸菌DsbAとPhDsbのアミノ酸配列を比較した図。 四角で囲った部分がDsbAファミリーに共通するチオレドキシンモチーフを示す。また、*は共通のアミノ酸配列、−はギャップを示す。↓は、Signal Pプログラム(Ver.1.1)を用いて予測されたシグナルペプチダーゼ認識部位を示す。
【図2】PhDsbとPaDsbのアミノ酸配列を比較した図。 *は共通のアミノ酸配列、−はギャップを示す。
【図3】大腸菌(BL21−CodonPlus(DE3)−RIL)における種々のタンパク質の発現を示す図。10−20%グラジエントゲルを用いてSDS−PAGE解析を行った。1:ベクターのみ(コントロール)、2:PhDsb、3:PhDsb(L−)、4:DsbAL/PhDsb(DsbAのリーダー配列を有するPhDsb)、5:DsbA、T:大腸菌細胞ライゼート(Total)、Pe:ペリプラズム画分、S:菌体内可溶性画分。
【図4】種々の酵素のプロテインジスルフィドレダクターゼ活性の比較を示す図。縦軸に濁度(A650nm):レダクターゼ活性を、横軸に反応時間を示している。
【図5】種々の酵素のプロテインジスルフィドオキシダーゼ活性の比較を示す図。縦軸に吸光度(A296nm):活性を、横軸に反応時間を示している。
【図6】PhDsbの耐熱性を示す図。縦軸に相対プロテインジスルフィドレダクターゼ活性、横軸に100℃での処理時間を示している。
【図7】DsbA類の進化系統図を示す図。pI 8.0以上の塩基性タンパク質を四角で囲んで示している。また、括弧内のアルファベットはCPXCモチーフのXにH以外のアミノ酸を採用しているものについてそのアミノ酸を示している。
【図8】無細胞タンパク質合成でタンパク質を合成する際のフォールディングに及ぼす各酵素の効果を比較する図。縦軸にアミラーゼ活性(A415nm)を示している。
【技術分野】
【0001】
本発明は、タンパク質やペプチド等のシステインを介したジスルフィド結合の形成や、その逆反応、すなわちジスルフド結合の乖離に関わる反応を触媒する好熱菌由来のCPHCモチーフを有する新規プロテインジスルフィドオキシドレダクターゼ、およびその使用方法に関わる。更には、本プロテインジスルフィドオキシドレダクターゼを含有する試薬・キットにも関する。
【背景技術】
【0002】
大腸菌などの原核生物からヒトを始めとする真核生物まで、タンパク質中もしくはタンパク質間のジスルフィド結合はタンパク質に安定性を付与する手段として進化してきた。特に分泌タンパク質において広く採用されている。
【0003】
これらの結合は主に、原核生物ではペリプラズムにおいて、また真核生物においては小胞体において形成されることが知られており、合成されたタンパク質はそこに留まるものがあるが、より環境の厳しいリソソームのような他の細胞小器官へ移送されるか(真核生物生物の場合)、細胞外へ分泌される。グラム陰性菌の表層は、一般に細胞膜の外側に薄いペプチドグリカン層、更にその外側に外膜が存在するが、ペリプラズムとは細胞膜と外膜の間のことをいう。グラム陽性菌でも細胞膜と厚いペプチドグリカン層の間をペリプラズムと呼ぶ。この空間は、Archaeaにおいても同様に知られており、そこで働く酵素も存在する。
【0004】
ジスルフィド結合は、原核生物においてはDsbAもしくはDsbCと呼ばれるプロテインジスルフィドオキシドレダクターゼによって直接触媒されることが知られている。プロテインジスルフィドオキシドレダクターゼは、チオール:ジスルフィドインターチェンジプロテインとも呼ばれ、DsbAは主にジスルフィド結合の形成に、DsbCは主に形成されたジスルフィド結合を正しい組み合わせに組み換える異性化反応に関わっていることが調べられている。これらの酵素は、分子内にCXXCモチーフを有しており(DsbAは主にCPHC、まれにCPWCやCPYC、DsbCはCGYC)、これらが目的のタンパク質のシステインと相互作用することにより、反応を触媒する機構が提唱されている。またそれらのタンパク質のシステインの酸化還元を調整するためのタンパク質(DsbB、DsbD)も見出されている(たとえば非特許文献1を参照。)。
【非特許文献1】Cell, 99, 117−119 (1999)
【0005】
モチーフとは各種のタンパク質のアミノ酸配列中に認められる小さい構造部分を指す言葉である。それをコードするアミノ酸配列には特徴的なパターンがあり、その局所的に保存されたブロックにより定義されるものを配列モチーフと呼ぶ。多くの場合、ある機能を発現するために必要な最少の単位となるものである。
【0006】
一方、真核生物においては、プロテインジスルフィドイソメラーゼ(PDI)一分子でDsbAおよびDsbC双方の機能を担っていることが知られている。この酵素は、分子内に二つの保存されたCGHCモチーフを有している。異性化反応を触媒することが報告されているDsbCとPDIはDsbAと比較して、強いレダクターゼ活性を示すことが特徴である。DsbAにおけるCXXCモチーフの二つのシステインはジスルフィド結合による結合しており(酸化型)、DsbCのシステインはフリーのSH状態(還元型)であることが知られており、それが各酵素の性質に関わっていると考えられている。
【0007】
近年、様々なタンパク質の遺伝子が明らかとなり、大腸菌等の組換え細胞を用いたタンパク質生産が盛んに行われるようになっている。しかし、例えば真核生物由来のタンパク質を大腸菌で生産した場合、タンパク質が凝集した封入体(インクルージョンボディー)を形成するものが多く、問題となっている。その解決法として、封入体を一度尿素やグアニジン溶液などで完全に変性溶解し、徐々にそれらの変性剤成分を除去することにより、正しいフォールディングを形成させる方法(リフォールディング)が存在する。しかし、透析や希釈で単に変性剤の濃度を下げるだけでは、きちんとフォールディングされないタンパク質も多く、タンパク質のフォールディングを助ける機能を有するシャペロンと呼ばれるタンパク質を共存させることが有効であることが多く報告されている。さらに、ジスルフィド結合が必須なタンパク質のリフォールディングは困難な場合が多く、単に溶液を酸化状態に保っても、ジスルフィド結合の形成が不完全な場合や、正しい組み合わせのジスルフィド結合が形成されない場合が多いのが現状である。そこで、分離精製したプロテインジスルフィドオキシドレダクターゼをリフォールディング時に添加することが考案され、成果を上げている。また近年、プロテインジスルフィドオキシドレダクターゼには若干、シャペロンとしての機能が存在することも知られるようになっている。
【0008】
しかし、タンパク質のリフォールディングは先にも述べたように、まず凝集したタンパク質を尿素やグアニジン等の変性剤で処理し、それらを徐々に除去しながら(すなわちそれらが若干残存した状態)でリフォールディング反応を行うことが多い。そこで、変性に強いプロテインジスルフィドオキシドレダクターゼが求められていた。また、タンパク質の安定化剤や賦形剤等に使用する場合においても、安定性の高いプロテインジスルフィドオキシドレダクターゼが必要であり、同様に求められていた。
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明の目的は、変性に強い新規なプロテインジスルフィドオキシドレダクターゼを求めることにある。さらに、該新規プロテインジスルフィドオキシドレダクターゼを用いて効率的にタンパク質をリフォールディングもしくはフォールディングさせる方法を提供することにあり、さらに、タンパク質の安定的保存方法を提供することである。また、それらを含有する試薬・キットおよび製造法を提供することである。
【課題を解決するための手段】
【0010】
我々は、変性に強い好熱細菌に着目し、種々の好高熱菌について鋭意検討した結果、超好熱細菌であるArchaea(始原菌)から、ペリプラズムで機能していると推定される新規プロテインジスルフィドオキシドレダクターゼを分離し本発明を完成した。
【0011】
通常新規タンパク質の検索は、まず目的のタンパク質を分離してそのタンパク質の部分配列を指標としてその遺伝子を探索する方法や、遺伝子間のホモロジーを指標としたコロニーハイブリダイゼーションや、共通プライマーを用いるPCR法などを用いて行われるのが普通である。また、近年全ゲノムが明らかとなった生物種も多く、それらのデータベースを用いてホモロジーなどを指標として検索することによっても、目的タンパク質候補を絞り込むことができる。
【0012】
今回、本発明者らは、ゲノム配列が明らかとなり、インターネット上でその配列が入手可能な種々の微生物に着目し、それらの遺伝子配列中から新規プロテインジスルフィドオキシドレダクターゼの探索を実施した。まず、DsbA、DsbCおよびPDI等のすでに報告されているプロテインジスルフィドレダクトオキシダーゼの配列に着目し、それらのタンパク質とのホモロジーを指標として検索を実施した。しかし、ホモロジーのあるタンパク質はヒットしなかった。
【0013】
ところが、我々がさらに調査したところ、超好熱細菌であるArchaeaのPyrococcus furiosus(Pfu)から、PDIに類似したタンパク質が発見されていたことがわかった(J.Biol.Chem. 270,5748−5755(1995))。Archaeaは始原菌、もしくは古細菌と訳されるユニークな生物で、原核生物と真核生物の中間的な位置に分類されることもある。さらには、Pyrococcus horikoshiiからもホモログ(ORF ID:PH0178)が分離され、同様な検討が行なわれていた。
【0014】
これらのタンパク質をコードする遺伝子は、リーダー配列と思われる配列が見当たらないため、この分子は、目的のプロテインジスルフィドレダクトオキシダーゼではなく、細胞質内で機能しているタンパク質の一つ、グルタレドキシンの一種であると推定されていた。しかし我々は、これらのタンパク質とのホモロジーについても検討したところ、唯一、前述のPyrococcus furiosus由来の耐熱性プロテインジスルフィドオキシドレダクターゼとホモロジーを有するタンパク質(ORF ID:PH0178)のみがヒットした。このタンパク質には既に、アノテーション情報としてその情報が書き込まれていた。
【0015】
次に、本発明者らは、DsbA、DsbCおよびPDIに含まれる、コンセンサス配列(それぞれ CPHC(CPWC、CPFW)、CGYC、CGHC)に着目し検索を実施した。また、それらのモチーフと併せてリーダー配列(シグナル配列)を有するタンパク質の検索を行った。リーダー配列の有無を条件として挙げた理由としては、ペリプラズムへの移行には本配列が多くの場合必須であるからである。その結果、DsbAホモログと思われるタンパク質の存在が明らかとなった。このタンパク質はPyrococcus horikoshii遺伝子の1025340〜1025852(ORF IDはPH1130として登録)にコードされていることが分かった(配列番号3)。その配列から予想されるアミノ酸配列は配列番号1に示す通りであり(SWISS−PLOTアクセッション番号:D71054)、まだ機能は特定されていなかった。
【0016】
このタンパク質(以下PhDsbタンパク質)は、大腸菌DsbAとはほとんどホモロジーはないものの、CPHCコンセンサス配列、および5残基前のF(フェニルアラニン)も保存されていること、タンパク質のサイズ、およびモチーフが比較的N末端に近いところに存在することからみても、DsbAのホモログである可能性が高いと推測された。また、シグナルペプチド配列検索を実施したところ、本タンパク質はシグナルペプチダーゼによって切り出されると推定される配列をN末端に有していることが明らかとなった。
【0017】
本発明のタンパク質と大腸菌DsbAの比較を図1に示した。今まで報告されているDsbは大きく、酸性タンパク質に分類されるものと、塩基性タンパク質に分類されるものに分類されることが知られているが、本タンパク質は後者であることが、そのアミノ酸配列から推測された。アミノ酸配列(リーダー配列込み)から予想される等電点(pI)は大腸菌DsbAが5.93であるのに対し、PhDsbタンパク質は9.26と予測された。
【0018】
また、本PhDsbタンパク質のアミノ酸配列をBLAST検索(ホモロジー検索)を実施したところ、DDBJアクセッション番号AX041920に登録されているPyrococcus abyssi遺伝子の140922−140371(配列番号4)にコードされるタンパク質(配列番号6:SWISS−PLOTアクセッション番号F75086)に高いホモロジーを示すことが明らかとなった(図2)。この遺伝子にコードされるタンパク質の詳細な機能は明らかにされていないが(チオレドキシン様配列を有するとの記載はあり)、PhDsbタンパク質同様DsbAのホモログと予測される。このタンパク質を便宜上PaDsbと呼ぶこととする。
【0019】
配列から本遺伝子のコードしているタンパク質が大腸菌DsbAのホモログであることはほぼ明らかであったが、実際にタンパク質を発現させて活性を調べる必要があるとの考えから、本発明者らは、次に、本遺伝子をクローニングし、大腸菌を用いた発現を試みた。しかし、最も効率的発現方法の一つとして知られているpET発現系を用いても、本遺伝子産物の発現量は大変低く、また、発現したものはペリプラズムへ移行せず不溶化してしまう傾向にあることが分かった(図3)。一方、コントロール実験として行った大腸菌DsbAにおいては、強い発現とペリプラズムへの移行が認められた。このことから、本タンパク質を機能を有する状態で発現させることに関しても何らかの工夫が必要であることが明らかとなった。
【0020】
次に、本遺伝子の発現効率が低調な理由として、リーダー配列(シグナル配列)が不適であることが予測された為、シグナルペプチダーゼで切断を受けると予測される領域に関して、大腸菌DsbAのリーダー配列と入れ替えて発現を試みたが、これに関してもほとんど改善が見られなかった(図3)。
【0021】
本発明者らは、これらの課題を解決するべく鋭意研究を進めた結果、予想されるリーダー配列を除去したものについて、ペリプラズムへの移行は見られないものの、細胞質内に強い発現を確認することができ、可溶化した状態で回収し得ることが明らかとなった(図3)。今回、シグナルペプチダーゼの切断予測はProtein Engineering, 10, 1−6 (1997)に従って実施した。具体的には、インターネットのSiganal Pのサイト(http://www.cbs.dtu.dk/services/SignalP/)の検索プログラム(Ver.1.1)を利用して予測を行なった。グラム陰性菌のデータを元に予測行った場合、27番目スレオニンと28番目のグルタミンの間(TQT−QT)、グラム陽性菌のデータを元にシグナルペプチダーゼ認識サイトの予測を行った場合は25番目のスレオニンと26番目のグルタミンの間(SQT−QT)が最も可能性の高い切断サイトであることが予測された。今回は、グラム陰性菌のデータを元に予測された切断部位を採用し、予測される切断末端に開始コドン(メチオニン)を付加した配列(配列番号2)を発現させ、実験に用いた。本検証実験から、本タンパク質の配列に起因する何らかの作用によって、本タンパク質の大腸菌細胞膜の透過性が極端に低くなったことの原因であると推察された。
【0022】
上にも記述したようにDsbAのリーダー配列を有するPhDsbによる試みにおける結果もこのことを示唆している。この実験で用いたDsbAリーダー配列を付加したPhDsbの配列は、あらかじめSignal P(Ver.1.1)を用いてシグナルペプチダーゼ認識サイトがあることを確認して用いている。
【0023】
また、不溶化にはシグナルペプチド配列が除去しきれなかったことが原因である可能性が示唆された。シグナルペプチドは大変疎水性に富むものであると予想される。また本実験は、グラム陽性菌のデータに基づいた予測サイトに基づいて調製したタンパク質を用いても、ほぼ同様の結果が得られたと予測される。さらに、Ver.2以降の本予測プログラムを用いた場合、25番目のスレオニンと26番目のグルタミンの間(SQT−QT)が最も可能性の高い切断サイトであると予測されるようであるが、実質は2アミノ酸の差であり、本質的には同様の結果が得られると予測される。
【0024】
一方、Pyrococcus abyssi由来のPaDsbにおいては、グラム陰性菌のデータを元にした予測では29番目のスレオニンと30番目のグルタミンの間(TTT−QT)、23番目のスレオニンと24セリンの間(SST−ST)で切断される可能性が高いことが予想され、この予想部位でリーダー配列を除去したタンパク質(当然メチオニンは付加する必要あり)は、PhDsb同様の活性を示すことが予想される。
【0025】
このようにして得られたプロテインジスルフィドオキシドレダクターゼは、大腸菌の細胞質内から超音波破砕することにより容易に回収することができ、また、塩基性タンパク質であることから、陽イオン交換(S−Sepharose)カラムクロマトグラフィーにより、容易に精製が可能であった。
【0026】
次に、この精製したタンパク質に関して、一般的にプロテインジスルフィドオキシドレダクターゼ活性を調べる際に用いられる、還元条件下でのインスリンを基質としたレダクターゼ活性測定(J. Biol. Chem. 254, 9627 (1979))、および酸化/還元型グルタチオン存在下における変性リボヌクレアーゼを基質としたオキシダーゼ活性測定(Biochem. 30, 613 (1991))を実施し、本酵素が目的の酵素であることを確認した。
【0027】
まず始めに、インスリンを基質としたジスルフィドレダクターゼ活性の測定を行なった。インスリンは2種類のペプチドがジスルフィド結合により結合したヘテロダイマーであるが、レダクターゼ活性によりジスルフィド結合が乖離すると、β鎖のペプチドが不溶化し濁度が上昇する。それを吸光度(650nm)測定することによりレダクターゼ活性を測定することが可能である。還元条件下でのインスリン乖離反応はDsbA、DsbC、およびPDIそれぞれに共通して見られる反応である。今回、対照とするため大腸菌DsbA、DsbCをそれぞれ大腸菌ゲノムDNAからクローニングし、文献に従って発現させペリプラズム画分より精製し使用した(J. Bacteriol. 179, 5333 (1997))。それぞれの酵素の最終濃度が2μMになるような一般的な測定条件を用い、反応時間と測定結果(OD 650nm)をプロットしたのが図4である。結果より、PhDsbタンパク質は明らかにDsbAに近いパターンを示すことが分かった。ちなみにDsbAおよびDsbCの反応パターンは報告させているものとほぼ同じ傾向を示していることを確認している。また、酵素が存在しない条件においても、還元剤の影響で徐々にインスリンのジスルフィド結合は乖離するためコントロールにおいても若干吸光度の上昇が見られたが、一般的な現象であることが知られている。
【0028】
次に、変性リボヌクレアーゼを用いるジスルフィドオキシダーゼ活性の確認を行なった。酸化/還元型グルタチオン存在下における変性リボヌクレアーゼを基質としたオキシダーゼ活性測定はオキシダーゼ活性を測定する場合に頻繁に使用される方法である。リボヌクレアーゼAは活性にジスルフィド結合が必須であり、還元剤を含有する変性剤存在下で不活性化されることが知られている。このようにして変性させたリボヌクレアーゼAはゲル濾過や、希釈、透析などによって変性剤と還元剤濃度を低くすることによって徐々にリフォールディングにより活性が回復することが知られている。また、その回復速度はオキシダーゼ存在を共存させることで早まることが知られている。今回レダクターゼ活性同様に、本測定を用いてDsbAおよびDsbCをコントロールとしてPhDsbのオキシダーゼ活性を測定した。測定条件は文献に倣い最終濃度が5μMとなるように各酵素濃度を調整し、一定の比率にした酸化/還元型グルタチオンを含有する緩衝液添加して、変性還元リボヌクレアーゼAの活性回復をc−CMPを基質として用いOD 296nmの上昇を指標として測定を行なった。反応時間と測定結果(OD 296nm)をプロットしたのが図5である。結果より、PhDsbタンパク質はDsbCよりはむしろDsbAに近い中間的性質を示すことが明らかとなった。
【0029】
レダクターゼおよびオキシダーゼ活性の測定結果より、PhDsbはDsbAに近い性質を示すことが明らかとなり、本タンパク質は、アミノ酸配列のホモロジーには乏しいが、ほぼDsbAのホモログであることを確認することができた。アミノ酸配列が極端に異なっていることは、おそらく酵素を耐熱化させることと何らかの関連性があると推察される。
【0030】
実際に、本発明のタンパク質の耐熱性を調べるために、100℃にて加熱処理し、残存プロテインジスルフィドレダクターゼ活性を指標として活性測定を行なったところ、2時間の加熱処理により大腸菌DsbAは約40%まで活性が低下したのに対し、PhDsbはほぼ100%活性を保っていることが明らかとなった。
【0031】
以上のような経緯を経て、ようやく、我々はArchaeaから、変性に強いプロテインジスルフィドオキシドレダクターゼを分離し、その酵素の性質を明らかにすることが出来た。
【0032】
本発明は以下の構成からなる。
(1)Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有することを特徴とする、成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(2)Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有し、下記理化学的特徴を有する、(1)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
熱安定性:pH7.0にて100℃、2時間の加熱で60%以上の残存活性を保持することができる。
(3)Pyrococcus属細菌由来であることを特徴とする(1)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(4)Pyrococcus horikoshii由来であることを特徴とする(3)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(5)該タンパク質の配列が、配列番号1に示される配列に由来することを特徴とする(4)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(6)該タンパク質の配列が、配列番号2に示される配列であることを特徴とする、(5)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(7)CPHCモチーフ配列が変更されたことを特徴とする、(1)に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
(8)(1)〜(7)に記載のタンパク質を発現するように、所望の遺伝子配列を挿入したベクター。
(9)(8)に記載のベクターで形質転換された形質転換体。
(10)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを生産する菌株を培養し、培養物より該タンパク質を採取することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
(11)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを細胞質内に生産する菌株を培養し、培養物より該タンパク質を採取することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
(12)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを、無細胞タンパク質合成法を用いて合成することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
(13)加熱処理工程を経ることを特徴とする、(10)〜(12)に記載の成熟型プロテインジスルフィドオキシドレダクターゼの製造方法。
(14)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質内もしくはタンパク質間のジスルフィド結合の切断を促進する方法。
(15)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質のリフォールディングを促進する方法。
(16)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質のフォールディングを促進する方法。
(17)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いて無細胞タンパク質合成時のフォールディングを促進する請求項13に記載の方法。
(18)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼをタンパク質安定化剤および/または賦形剤として用いることを特徴とするタンパク質の安定的保存方法。
(19)(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを含有する試薬またはキット。
(20)生物機能を用いて所望のタンパク質を発現させる方法であって、(1)〜(7)に記載の成熟型プロテインジスルフィドオキシドレダクターゼを共発現させることにより、該タンパク質の活性を高める方法。
【発明の効果】
【0033】
本発明の、変性に強い新規なプロテインジスルフィドオキシドレダクターゼ、および、それらを含有する試薬・キットにより、効率的にタンパク質をリフォールディングもしくはフォールディングさせる方法、タンパク質の安定的保存方法が可能になった。
【発明を実施するための最良の形態】
【0034】
本発明は、Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有することを特徴とする、成熟型プロテインジスルフィドオキシドレダクターゼタンパク質である。Archaeaは別名始原菌や古細菌とも訳されることもある菌であり、原核生物と真核生物の中間的な位置に分類されることもある、大変ユニークな生物であり、Pyrococcus属以外にも、Thermococcus属等の種々の菌が報告されている。
【0035】
本発明のタンパク質は大腸菌等の好熱細菌以外の細菌で幅広く見出されているタンパク質であり、シグナルぺプチターゼで切り出されるリーダー配列をN末端に有し、それらの多くはN末端の近くにCHPC、もしくは稀にCPWCや、CPYC、CPFCなどのモチーフを有するのが特徴であるが、Archaeaでこの種の酵素が見出されたのは初めてである。
【0036】
本発明において用いられるタンパク質は、成熟型(Mature型)であることが重要である。本発明における成熟とは、リーダー配列(シグナル配列)部分、もしくはN末端側に存在する疎水アミノ酸等からなる膜貫通領域が切除されたタンパク質であることを指す。リーダー配列とは、タンパク質がペリプラズムや小胞体などへ膜を貫通して移行する際に必要な配列であり、移行後は速やかにシグナルペプチダーゼの働きで切断を受けることで、タンパク質は成熟化する。その場合、タンパク質はペリプラズム空間に成熟型酵素として、蓄積されることになる。実施例の、大腸菌DsbA、DsbCの調製はこの方法に準じている。シグナルペプチダーゼの作用する部位には法則性があり、現在、様々な予測用ソフトウエアを活用して切断サイトを予測することができるようになっている。
【0037】
一方、今回の場合のように他の生物種の機能を利用してタンパク質を生産する場合においては、リーダー配列の切除が困難な場合も予想される。例えば、今回のようにArchaeaのタンパク質を大腸菌で発現させるような場合は、タンパク質の膜透過、シグナルペプチダーゼによるリーダー配列の切断がうまく起こらないことがある。そのような場合、リーダー配列部分を除去したタンパク質をデザインし、細胞質内に発現させることで解決できることがある。その場合は、タンパク質合成開始のため、リーダー配列を除去した部分のN末端にメチオニンを付加する必要がある。
【0038】
その場合、リーダー配列の切断部位は、最も好ましくは天然のタンパク質を分離し、N末端解析を行って決定するが、それには多大なる労力と時間が必要な場合が多く、過去のデータを参考にして予測しても良い。
【0039】
シグナルペプチダーゼ切断サイトの予測には、種々開発されているソフトウエアを用いることができる。好ましくは、Signal P(http://www.cbs.dtu.dk/services/SignalP/)を用いると良い。この予測方法は、Protein Engineering, 10, 1−6 (1997)に基づいている。本ソフトウエアにも様々なバージョンがあり、予想配列も若干異なる場合があるが、それほど大差なく使用することが可能である。今回の予測にはVer.1.1を用いているが、基本的にはどのバージョンを用いても問題ない。また、グラム陰性菌、陽性菌、真核生物等で若干予測が異なるが、多くの場合は、グラム陰性菌のデータを用いることで問題なく予測可能である。特に今回は、宿主にグラム陰性菌である大腸菌を選択したため、グラム陰性菌のデータを用いることとした。
【0040】
酵素によっては、リーダー配列除去(成熟化)がタンパク質の溶解性や活性に大きな影響を有しているものも多く存在することが知られているが、本発明においては、その除去が大変重要であった。当然、Pyrococcus horikoshiiを培養して成熟型タンパク質を調製することも考えられるが、実際問題として現実的ではない。すなわち、本発明のタンパク質を得ようとすれば、大腸菌等の扱いの簡単な菌を宿主とすることがもっとも効率的であると考えられる。よって今回、本発明者らが用いた方法は、理にかなった方法であるといえる。
【0041】
今回実験に用いたタンパク質は、Signal P(Ver.1.1)で、グラム陰性菌のデータを元に予測を行い、PhDsbの27番目スレオニンと28番目のグルタミンの間(TQT−QT)で切断がおこると仮定して実験を進めた。すなわち、遺伝子をクローニングする段階で、27番目のスレオニンから上流の配列を除去し、28番目のグルタミンの上流に開始コドンとなるメチオニンの配列を導入した発現プラスミドを構築した。ちなみに、グラム陽性菌のデータを元に予測を行った場合は25番目のスレオニンと26番目のグルタミンの間(SQT−QT)が最も可能性の高い切断サイトであると予想されるが、本実験は、グラム陽性菌のデータに基づいた予測サイトに基づいて調製したタンパク質を用いても、ほぼ同様の結果が得られたと予測される。また、Ver.2以降の本予測プログラムを用いた場合、グラム陰性菌のデータを用いても、25番目のスレオニンと26番目のグルタミンの間(SQT−QT)が最も可能性の高い切断サイトであると予測されるようであるが、上記同様、実質は2アミノ酸の差であり、本質的には同様の結果が得られると予測される。
【0042】
一方、Pyrococcus abyssi由来のPaDsbにおいては、グラム陰性菌のデータを元にした予測では29番目のスレオニンと30番目のグルタミンの間(TTT−QT)、23番目のスレオニンと24セリンの間(SST−ST)で切断される可能性が高いことが予想され、このサイトで切断することで同様の活性型のプロテインジスルフィドオキシドレダクターゼを得ることが可能であることが容易に予測される。
【0043】
当然、これら予測以外の場所でリーダー配列切断部位を決定しても良い。すなわち、発現が向上し、タンパク質の溶解性や活性が向上する条件の場所で切断しさえすれば目的は達成されるのであって、当然、本発明の範囲に含めることができる。
【0044】
また、タンパク質を発現させる際に、種々のタグを付加して発現させても良い。実施例には示していないが、本発明においてはC末端側に6xHis−タグ配列を付加したものに関しても問題なく発現し、活性型タンパク質を得ることができることを確認している。当然、GST(グルタチオンSトランスフェラーゼ)やMBP(マルトースバインディングプロテイン)、GFP(緑色蛍光タンパク質)等との融合タンパク質と発現させても良い。タグの位置は、N末端でもC末端でも構わない。当然、機能を有する範囲において、本タンパク質の部分配列を用いてもよい。
【0045】
また、場合によってはプロテインジスルフィドオキシドレダクターゼ活性を変化させたり、活性を除去したりする場合が有効なことも考えられる。そのような場合は、活性中心であるCPHC配列もしくはその前後の配列に変異を導入することが有効である。特に、活性を除去する場合は、システイン残基を片方もしくは両方を別のアミノ酸に変化させることが有効である。また、活性を変化させる場合、CPHC配列の中のPHを変異させることが有効である。
【0046】
本発明のタンパク質の具体的な理化学的性質としては、好ましくは100℃、2時間の加熱で60%以上、更に好ましくは80%以上の活性を保持することを特徴とする。本活性の指標としては、上に挙げたインスリンを基質として測定するプロテインジスルフィドレダクターゼ活性を指標とするのが最も好ましい。条件としては、実施例10に示すように、十分酵素の初速を評価できる条件で測定する必要がある。また、好ましくは活性の測定は室温(25℃)前後で行う。
【0047】
実際に、レダクターゼ活性を指標として酵素の耐熱性の検討を行った例を図6に示した。方法は、100mMリン酸カリウム緩衝液(pH7.0)中、100℃で加熱した後、上記方法でレダクターゼ活性を測定する方法を用いた。100℃で加熱したのはDsbタンパク質自身が一種のシャペロンタンパク質であり、普通の酵素よりも耐熱性に優れているからである。その結果、大腸菌のDsbAでは100℃・2時間の加温で約40%まで活性が低下したのに対し、PhDsbタンパク質では98%以上の活性を保持したままであった。この結果より、PhDsbタンパク質が高度に耐熱化されていることが明らかとなった。
【0048】
本タンパク質(遺伝子)の由来としては、好ましくはArchaeaであり、さらに好ましくはPyrococcus属細菌由来である。また、最も好ましくはPyrococcus horikoshii由来である。同様に、Pyrococcus abyssi由来のものも好適に用いられる。
【0049】
該タンパク質の配列としては、配列番号1に示されるPyrococcus horikoshiiの配列に由来するものを用いるのがもっとも好ましい。一例としては、予測ソフトによって予測されたリーダー配列を除去し、N末端にメチオニンを付加した配列番号2に示される配列を用いる。ただ、配列はこれに限定されず、リーダー配列の切断サイトは幅を持って決めればよい。
【0050】
また同様に配列番号5に示されるPyrococcus abyssiのプロテインジスルフィドオキシドレダクターゼの配列に由来するものを用いることができる。これに関しても、任意の部位でリーダー配列を切断し、同時に成熟型タンパク質として使用することができる。
【0051】
PhDsbとPaDsbのアミノ酸レベルでのホモロジーはGenetyx Ver.3.2を用いて調べたところ69.8%であった。一方、PhDsbと大腸菌のDsbAとのホモロジーは30.2%であり、明らかにArchaea内でのホモロジーは有意に高く、ほぼ同じ機能を有している可能性があるのではないかと推察される。図7には、Swiss−plotデータベースにDsbAとして登録されている由来の異なるタンパク質とPhDsb、PaDsbのアミノ酸配列を元にTreeVew(Ver.1.6.6)(http://taxonomy.zoology.gla.ac.uk/rod/treeview.htmlよりダウンロード)を用いて作成した系統図を示している。それぞれの、Swiss−plotにおけるアクセッション番号は表1に一覧した。その結果、図7から明らかにArchaea由来のDsbA(すなわち、PhDsbとPaDsbは進化的にも同じ傾向を有していることが分かる。
【0052】
【表1】
【0053】
表1は、DsbA類の、アクセッション番号、アミノ酸数、モチーフ、および等電点(pI)を示している。
【0054】
また、表1には、それぞれのアミノ酸数、モチーフ配列、およびGenetyx Ver.3.2を用いて予測した等電点(アミノ酸のpKaをアルギニン:12.5、ヒスチジン:6.0、リジン:10.5、アスパラギン酸:3.9、システイン:8.3、グルタミン酸:4.3、チロシン:10.1として計算)を示している。またそれを元に、図7においてpIが8以上の塩基性タンパク質は四角で囲み示している。また図7の( )にはモチーフCPXC(C:システイン、P:プロリン)のXにあたるところが多型を示していることから、主に用いられている残基であるH(ヒスチジン)以外のアミノ酸を採用しているもの関してそのアミノ酸を示している。これらの図から推察して、Pyrococcus属のDsbAは独自の進化を遂げた可能性が示唆される。
【0055】
本発明は、上記タンパク質の生産方法にも関し、該タンパク質を生産する菌株を適切な培地を用いて培養し、培養物より該タンパク質を採取することを特徴とする。菌としては、特に限定されないが、大腸菌が好ましく用いられ、K12株由来、B株由来等の大腸菌が好適に用いられる。今回の検討では、B株に由来するBL21株を用いて生産を行っているが、それに限定されるものではない。さらに詳細には、BL21−CodonPlus(DE3)−RIL(ストラタジーン社製)を用いているが、これは、ArchaeaのCodon usageが真核生物に近いためである。本菌株はCodon Usageが真核生物に近くなるように遺伝子工学的手法で改変が施されているものである。当然、大腸菌以外のバチルス属由来の微生物や、真核生物である酵母などを用いて生産しても良い。
【0056】
また、タンパク質生産時の誘導法としては、今回、pETベクター系(T7系)を用いたが、Lac系の誘導システムを用いて生産しても良く、特に限定されるものではない。また、合成法も特に問わない。
【0057】
また、本タンパク質は種々の無細胞タンパク質合成法によって合成しても良い。無細胞タンパク質合成法の種類については特に限定されないが、一般的に使われるコムギ胚芽抽出液や、大腸菌抽出液、網状赤血球抽出液、昆虫細胞抽出液、酵母抽出液などを好適に用いることができる。
【0058】
本タンパク質の調製方法に関しては特に限定されないが、大腸菌等を宿主として発現させる場合は、加熱処理することによりその精製効率を飛躍的に向上させることができる。今回は、実施例6で一度カラムクロマトしたフラクションに関して加熱処理を行った例を示しているが、微量混入している夾雑タンパク質を除去するのに大変有効であった。また、精製の初期段階での加熱処理も大変有効である。加熱することにより、立体構造が変化し、活性化されるArchaeaのタンパク質の例も報告されているが、今回調べた範囲で、本発明のタンパク質にそのような効果は確認されていない。しかし、今回試験した以外の性質においては、加熱処理が有効な場合も当然考えられる。
【0059】
該酵素の利用法の一つとして、そのレダクターゼ活性を利用したジスルフィド結合の乖離反応を挙げることができる。その用途は特に限定されないが、タンパク質の還元の促進反応が必要なすべての用途に用いることができる。また、産業としては、毛髪のパーマネントなど還元が必要なものへの応用が考えられ、本酵素は耐熱性であることからも、本用途への応用の可能性は高いといえる。また、タンパク質以外にもシスチン(システインがジスルフィド結合により結合したシステインの二量体)の還元促進等への応用にも持ちることが可能であることは容易に予測できる。また、本方法を様々なパターンのジスルフィド結合複合体に応用できることは容易に予想できる。
【0060】
また、該酵素の利用方法としては、タンパク質のリフォールディングを挙げることができる。リフォールディングとは一度変性させたタンパク質の立体構造を再度形成させ、機能を復活させることである。種々のタンパク質の合成においては、フォールディングがうまくいかずに、変性した凝集体として回収されることがあり、問題となっている。特に、微生物でタンパク質を合成する場合、インクルージョンボディー(封入体)の形成が問題となることが多い。また、無細胞タンパク質合成を行う場合も、タンパク質の凝集不溶化が問題になることがある。それらのタンパク質は一度、尿素やグアニジンなどのタンパク質変性剤で溶解した後、それらの成分を徐々に系から除去することにより、正しい立体構造をとらせることができる場合がある。また、ジスルフィド結合が必須な酵素の場合、変性時に還元剤を添加しておき、ジスルフィド結合も切断しておくと効果的である。その場合は、変性剤を除去する工程で、変性剤も除去し、ジスルフィド結合形成を促進する。ただ、変性剤や還元剤を系から除去するだけでは一般的にリフォールディングの効率は低いことが報告されている。そこで、種々のプロテインジスルフィドオキシドレダクターゼをその工程で働かせることが行なわれてきた。本酵素を用いても、同様に酵素をリフォールディングさせることが可能であることは実施例に示すリボヌクレアーゼを用いた実験例からも明らかである。また、本酵素は熱など過酷な条件に強いことから、種々の変性剤が存在するような条件での使用に関しても適しているといえる。また、本酵素を用いるリフォールディングは、ジスルフィド結合の形成を介さない場合にも有効であることが後述の実施例によっても明らかであり、様々なフォールディング一般への応用を考えることができる。
【0061】
本発明は、該酵素を用いてタンパク質のフォールディングを促進する方法を含む。フォールディングの促進としては、特に限定されないが、無細胞タンパク質合成等のタンパク質合成の場に本タンパク質を存在させることにより、フォールディング形成を促進させることができる。実際今回、高効率コムギ胚芽無細胞タンパク質合成反応液中でのヒト−S−アミラーゼの合成反応中に本酵素を存在させておく実験を実施したが、有意にフォールディングを促進する結果を得ている。アミラーゼは11のシステイン残基を有しており、それらがジスルフィド結合を形成することが活性発現に必須である。今回、酸化グルタチオンと還元グルタチオンを混合した、比較的ジスルフィド結合の形成されやすい条件を用いて無細胞タンパク質合成を実施している。
【0062】
実際のタンパク質合成には、重層法を用いた(FEBS Letters 514, 102−105(2002))。タンパク質合成には、ATPやアミノ酸などの低分子物質が必須であるが、通常のバッチ反応では2〜3時間でそれらの成分が枯渇してしまう。重層法は、反応溶液の上にバッファー層を重層することで、拡散反応によって低分子物質を連続的に供給する方法であり、バッチ法に比べ約10倍程度の合成量の向上を期待することができる。今回は、その下層すなわち、反応溶液にDsbタンパク質を混合し、実験を行った。バッファー層には一般的に、タンパク質成分を加えない方が良い結果が得られることが確かめられていることから、バッファー層にはタンパク質成分は添加していない。
【0063】
その結果、無添加(コントロール)でもある程度は活性を示したが、本酵素を添加することでコントロールに比べ約170%の活性を有するアミラーゼを合成することができた。また、この効果は大腸菌のDsbAよりも高く、むしろDsbCに近いものであった。これは、ジスルフィド結合形成に加え、シャペロンとしての作用が本酵素に高いことを示していると思われる。シャペロンとはヒットショックタンパク質に代表されるタンパク質のフォールディングを助ける作用を有するタンパク質を指す。近年、ジスルフィドオキシドレダクターゼとして報告されていたPDIやDsbタンパク質にも若干ではあるがシャペロン活性があることが知られてきており、PhDsbにはその活性が高いのではないかと予想される。
また、今回実験で証明したこれらの効果は、合成量の増大ではなく、質の向上であることはウェスタンブロッティング解析を用いて確認している。
【0064】
当然、本酵素は無細胞タンパク質合成によって目的タンパク質と同時合成しても良い。
【0065】
さらに本発明は、本酵素を賦形剤、安定化剤として用いることを特徴とするタンパク質の安定的保存方法も含有している。上記に記載したように、本酵素は、ジスルフィド結合形成の他にもシャペロンとしての働きも示唆されており、それらの用途には適していることが予想される。特に、本酵素は耐熱性であり、高温での酵素の安定性を向上させる用途とうへの応用も期待される。
【0066】
また、本発明は、生物機能を用いて所望のタンパク質を発現させる場合にも応用することができる。例えば、大腸菌等でタンパク質を発現させる際に、本発明のタンパク質を共発現させることにより、よりフォールディングのしっかりした質の高いタンパク質を取得することができる可能性が非常に高い。共発現の方法としては、同一ベクターまたは別のベクターでタンパク質を発現する遺伝子を供給する方法が考えられる。
【0067】
本発明は、本酵素を含有する試薬・キットである。
【0068】
本発明の実施の一態様としては、ジスルフィド結合乖離用試薬、および/またはキットである。
【0069】
本発明の実施の一態様としては、タンパク質リフォールディング用試薬、および/またはキットである。
【0070】
また、本発明の実施の一態様としては、タンパク質フォールディング用試薬、および/またはキットである。
【0071】
また、本発明の実施の一態様としては、タンパク質安定化剤、および/または賦形剤である。
【実施例】
【0072】
以下に本発明の実施例をあげることにより、本発明による効果をより一層明瞭なものとする。ただし、これらの実施例によって本発明の範囲は限定されるものではない。
【0073】
実施例1:PhDsbのクローニング
まず、P. HorikoshiiのゲノムDNAを鋳型として目的タンパク質のORFをPCR法にて増幅し、pET−11c(ストラタジーン社製)へクローニングした。具体的には、P. HorikoshiiのゲノムDNA(ATCCより購入)を鋳型として、配列番号6(NdeIサイトを有する)と配列番号7(BamHIサイトを有する)、また、リーダー配列を除去したタンパク質をクローニングする目的で配列番号8(NdeIサイトを有する)と配列番号7(BamHIサイトを有する)の配列を有するオリゴヌクレオチドをプライマーとして高正確性DNAポリメラーゼ(KOD −Plus− :東洋紡製)を用いて増幅を実施した(この酵素を用いて増幅したDNAは末端が平滑末端となる)。増幅はKOD −Plus−の取扱い説明書に従って実施した。増幅したDNAはMagExtractor−PCR&Gel Clean up−キット(東洋紡製)にて精製、末端を制限酵素BamHIで消化した後、更に同キットで精製した。また、一方で、pBluescript(pBS)ベクター(ストラタジーン製)をEcoRVとBamHIで消化し、同様に精製した。精製したDNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセルを形質転換した。37℃で一晩薬剤選択培地(LB/Ampプレート)にて培養した後、生じたコロニーを、LB/Amp培地を用いて液体培養し、それから分離精製したプラスミドを用いて配列確認を行った。
次に、配列を確認したプラスミド(それぞれpBS−PhDsb、pBS−PhDsb(L−)(L−:リーダー配列除去))をNdeIおよびBamHIで消化・精製し、目的遺伝子断片の調製を行った。同時に、pET−11c(ストラタジーン社製)も同様の制限酵素で消化・精製した。精製した目的DNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセル(東洋紡製)を形質転換した。最終的には、生じたコロニーからプラスミドDNA(pET−PhDsb、pET−PhDsb(L−))を抽出し、そのプラスミドDNAを用いて、BL21−CodonPlus(DE3)−RILコンピテントセル(ストラタジーン製)を形質転換し、下記実験に用いた。
【0074】
実施例2:大腸菌DsbA、DsbCのクローニング
まず、実施例1と同様に大腸菌ゲノムDNAを鋳型として目的タンパク質のORFをPCR法にて増幅し、pET−11c(ストラタジーン社製)へクローニングした。具体的には、大腸菌ゲノムDNA(JM109株より定法を用いて抽出)を鋳型として、配列番号9(NdeIサイトを有する)と配列番号10(BamHIサイトを有する)、および配列番号11(NdeIサイトを有する)と配列番号12(BamHIサイトを有する)に示すプライマーをそれぞれ用いて高正確性DNAポリメラーゼ(KOD −Plus− :東洋紡製)によって増幅を実施した(この酵素を用いて増幅したDNAは末端が平滑末端となる)。増幅はKOD −Plus−の取扱い説明書に従って実施した。増幅したDNAはMagExtractor−PCR&Gel Clean up−キット(東洋紡製)にて精製し、末端を制限酵素BamHIで消化した後、更に同キットで精製した。また一方で、pBluescriptベクター(ストラタジーン製)をEcoRVとBamHIで消化し、同様に精製した。精製した目的DNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセルを形質転換した。37℃で一晩薬剤選択培地(LB/Ampプレート)にて培養した後、生じたコロニーをLB/Amp培地を用いて液体培養し、それから分離精製したプラスミドを用いて配列確認を行った。
次に、配列確認を行ったプラスミド(pBS−DsbA、およびpBS−DsbC)をNdeIおよびBamHIで消化・精製し、目的遺伝子断片の調製を行った。同時に、pET−11c(ストラタジーン社製)も同様の制限酵素で消化・精製した。精製したDNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセル(東洋紡製)を形質転換した。最終的には、生じたコロニーからプラスミドDNAを抽出し、そのプラスミドDNA(pET−DsbA、pET−DsbC)を用いて、BL21−CodonPlus(DE3)−RILコンピテントセル(ストラタジーン製)を形質転換し、下記実験に用いた。
【0075】
実施例3:DsbAリーダー配列を有するPhDsbのクローニング
まず、上記実験によって調製されたpBS−DsbAを鋳型として、配列番号9と配列番号13(リンカー配列としてPhDsb配列を一部有する)に示すプライマーを用いて高正確性DNAポリメラーゼ(KOD −Plus−:東洋紡製)によってDsbAのシグナルペプチド配列部分の増幅を実施した(この酵素を用いて増幅したDNAは末端が平滑末端となる)。また一方、同じく上記実験によって調製されたpBS−PhDsbを鋳型として、配列番号14(リンカー配列としてDsbA配列の一部を有する)と、配列番号7をプライマーとして、同じく高正確性DNAポリメラーゼ(KOD −Plus−:東洋紡製)を用いてDsbAのシグナルペプチド配列部分の増幅を実施した。その後、それぞれの増幅産物を精製した後、10倍に希釈して混合し、プライマーを添加しない条件で、KOD −Plus−を用いたエクステンション反応を10サイクル実施した(この反応で、リンカー部分を解して、それぞれの増幅産物が連結される)。その後、反応液を鋳型に、配列番号9と配列番号7をプライマーとして、同じくKOD−Plus−を用いて融合遺伝子の全長を増幅した。増幅したDNAはMagExtractor−PCR&Gel Clean up−キット(東洋紡製)にて精製、末端を制限酵素BamHIで消化し、更に同キットで精製した。また一方で、pBluescriptベクター(ストラタジーン製)をEcoRVとBamHIで消化し、同様に精製した。精製したDNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセルを形質転換した。37℃で一晩薬剤選択培地(LB/Ampプレート)にて培養した後、生じたコロニーを用いて液体培養し、それから分離精製したプラスミドを用いて配列確認を行った。
次に、配列確認を行ったプラスミド(pBS−DsbAL/PhDsb)をNdeIおよびBamHIで消化・精製し、目的遺伝子の調製を行った。同時に、pET−11c(ストラタジーン社製)も同様の制限酵素で消化・精製した。精製した目的DNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセル(東洋紡製)を形質転換した。最終的には、生じたコロニーからプラスミドDNAを抽出し、そのプラスミドDNA(pET−DsbAL/PhDsb)を用いて、BL21−CodonPlus(DE3)−RILコンピテントセル(ストラタジーン製)を形質転換し、下記実験に供した。
【0076】
実施例4:PhDsb、大腸菌DsbA、およびDsbCタンパク質の発現条件の検討
それぞれの遺伝子を挿入したpET−11cで形質転換したBL21−CodonPlus(DE3)−RIL株(それぞれPhDsb、PhDsb(L−)、DsbAL/PhDsb、DsbA、およびDsbC遺伝子を保有)、を用いてタンパク質の発現を実施した。具体的には37℃でLB/Amp培地を用いて0.4ODになるまで培養した後、IPTGを1mMとなるように添加し、37℃で3時間培養し、目的タンパク質を発現させた。
培養の後、培養液を6,000r.p.m.で10分間遠心し、菌体を回収した。その後、10mM Tris−HCl(pH8.0)に菌体を懸濁し30分間放置し、ペリプラズムに発現されたタンパク質を菌外へ放出させた。その懸濁液を15,000r.p.m.で10分間遠心し、その上清を回収した(ペリプラズム画分)。このペリプラズムタンパク質の回収方法は、J. Bacteriol. 179(17) 5333−5339(1997)を参考にした。また、その時得られたペレットに10mM Tris−HCl(pH8.0)を適量添加、懸濁し、2分間超音波処理を行った後、15,000r.p.m.で10分間遠心しその上清を回収した(菌体内可溶性画分)。PhDsb、PhDsb(L−)、およびDsbAL/PhDsbに関して得られた画分をSDS−PAGE解析を行った結果を図3に示した。コントロールとして、DsbAの結果も示している。結果より、DsbAタンパク質はペリプラズムに回収されたのに対して、リーダー配列部分を含む完全長の遺伝子発現を行ったPhDsbタンパク質およびDsbAのリーダー配列を融合させたPhDsbタンパク質はペリプラズム画分ではまったく回収されなかった。また、その発現レベルも大変低いものであり、菌体内可溶性画分に回収されなかったことから不溶化しているものと推測された。一方、シグナルペプチド部分を除去したPhDsbL−は、当然ペリプラズム画分にはタンパク質は回収されなかったが、細胞質の可溶性画分に強い発現を認めることができ、可溶性タンパク質として回収できたことが明らかとなった。データは示していないが、DsbCもペリプラズム画分に強い発現を認めた。
【0077】
実施例5:DsbA、およびDsbCの精製
実施例3の方法に従って回収したDsbA、およびDsbCのペリプラズム画分を10mM Tris−HCl(pH7.5)にて平衡化させたDEAE−Sepharose(アマシャムバイオサイエンス社製)に供し、同緩衝液で5カラム容量分洗浄した後、0.1Mに続き0.2M NaClを含有する同緩衝液でタンパク質を溶出した。DsbAは0.1M NaCl画分に、DsbCは0.2M NaCl画分にそれぞれ溶出された。フラクションはそれぞれSDS−PAGE解析を行い、メインフラクションのみを集めて、20mM HEPES−KOH(pH7.6)に対して透析し、酵素溶液として種々の実験に使用した。文献等に従うと、この方法で得られるDsbタンパク質は、リーダー配列を切除されていると考えられ、以下の実験には、ここで得られたタンパク質(詳細に記述するとDsbA(L−)、DsbC(L−)となる)を用いて行った。
【0078】
実施例6:リーダー配列除去PhDsb(L−)の精製
実施例3の方法で発現誘導した菌体(BL21−CodonPlus(DE3)−RIL/pET−PhDsb(L−))を回収し、10mM Tris−HCl(pH7.5)にて懸濁後、超音波破砕の後、15,000r.p.m.にて遠心し、その上清を回収した。次に、その上清を10mM Tris−HCl(pH7.5)にて平衡化したS−Sepharose(アマシャムバイオサイエンス社製)に供し、同緩衝液にて5カラム容量分洗浄した後に、0.2M NaClを含有する同緩衝液にて溶出した。また、必要に応じて精製度を高める目的で、溶出された画分を80℃、20分間加熱処理した後、10mM Tris−HCl(pH7.5)にて10倍希釈し、同緩衝液にて平衡化したS−Sepharoseに供した。カラムを同様に同緩衝液にて洗浄した後に、0.2M NaClを含有する同緩衝液にて溶出した。溶出されたフラクションはSDS−PAGE解析を行い、メインフラクションのみを混合し、20mM HEPES−KOH(pH7.6)に対して透析し、酵素溶液として下記の種々の実験に用いた。
【0079】
実施例7:ヒト唾液(S)アミラーゼ遺伝子のクローニング
無細胞タンパク質合成実験に用いるヒト−S−アミラーゼ遺伝子は、ヒトcDNA(Clontech社製)からPCR法を用いてクローニングを行った。具体的には配列番号15および配列番号16(BamHI配列を含む)に記載のプライマーを使って、高正確性DNAポリメラーゼ(KOD −Plus−:東洋紡製)を用いて増幅を実施した(この酵素を用いて増幅したDNAは末端が平滑末端となる)。ヒトアミラーゼはリーダー配列を有するタンパク質であるが、無細胞タンパク質合成法においては除去されないことが分かっていることから、今回は最初から、既に報告されているリーダー配列部分を含まない形でクローニングできるよう、プライマー設計を行った。増幅はKOD −Plus−の取扱い説明書に従って実施した。増幅したDNAはMagExtractor−PCR&Gel Clean up−キット(東洋紡製)にて精製、末端を制限酵素BamHIで消化した後、更に同キットで精製した。また一方で、無細胞タンパク質合成用ベクター(pEU3−NIIベクター:東洋紡製)をEcoRVとBamHIで消化し、同様に精製した。精製した目的DNA断片とベクターは2:1のモル比で混合し、それと等量のLigation試薬(Ligation high:東洋紡製)を添加し、16℃・1時間ライゲーション反応を行った後、大腸菌JM109コンピテントセルを形質転換した。37℃で一晩薬剤選択培地(LB/Ampプレート)にて培養した後、生じたコロニーを用いて液体培養し、それから分離精製したプラスミドを用いて配列確認を行った。また、無細胞タンパク質合成用に、QIAFILTER MIDI KIT(QIAGEN社製)を用いてプラスミドを抽出し、さらにフェノール/クロロホルム抽出、エタノール沈殿法を用いてプラスミドを精製した。ここで構築したプラスミドを以下、pEU−S−AMYとする。
【0080】
実施例8:ジスルフィドレダクターゼ活性測定
各酵素のジスルフィドレダクターゼ活性は基本的には、J. Biol. Chem. 245(19)9627−9632(1979)に従って測定した。具体的には、アッセイ用緩衝液(1mg/ml ウシインスリン(ロシュダイアグノスティクス社製)、0.1M リン酸カリウム−酢酸緩衝液(pH7.0)、2mM EDTA)を96ウェルプレートに150μlづつ添加した後、酵素溶液を添加し180μlにした。酵素サンプルとしては、実施例5、および6にて調製した20mM HEPES−KOH(pH7.6)に溶解したDsbA(L−)、DsbC(L−)、PhDsb(L−)(以下(L−)略)を用い、測定時の終濃度が2μMとなるようにした。その後に、10mM ジチオスレイトール(DTT)を7.5μl添加、混合し(終濃度0.5mM)、反応を開始した。反応は、室温(25℃)にて行い、濁度(650nm)の変化をプレートリーダーにて5分毎に測定した。その結果を、図4に示す。図から明らかなように、PhDsbと大腸菌DsbAはほぼ同じ挙動を示し、PhDsbはDsbAのホモログであることが示唆された。コントロールも最終的には濁度の上昇が見られたが、これは還元状態における自発的な乖離現象であり、一般的な現象である。
【0081】
実施例9:ジスルフィドオキシダーゼ活性測定
各酵素のジスルフィドオキシダーゼ活性は基本的には、Biochemistry 30, 613−619 (1991)に従って実施した。すなわち、変性リボヌクレアーゼを酵素溶液で希釈し、リボヌクレアーゼ活性を指標としてその活性の復活を測定した。具体的には、まず、5mgのウシリボヌクレアーゼA(Sigama社製)を1mlの変性溶液(6M グアニジン塩酸塩、0.1M Tris−酢酸緩衝液(pH8.0)、2mM EDTA、0.14mM DTT)に溶解し、一晩放置し変性させた。その後、0.1%酢酸で平衡化したG−25スピンカラム(アマシャムバイオサイエンス社製)にてゲル濾過し、実験に用いた。
50μlの2倍濃度のアッセイ溶液(100mM Tris−酢酸(pH8.0)、9mMcCMP(Sigma社製)、0.4mM 酸化型グルタチオン(GSSG)、2mM 還元型グルタチオン(GSH))に50μlの10μMの各種酵素(DsbA(L−)、DsbC(L−)、およびPhDsb(L−)(以下(L−)略)(20mM HEPES−KOH(pH7.6))を混合した後、2.2μlのゲル濾過後の変性リボヌクレアーゼを添加、混合し、反応を開始した。反応は室温(25℃)で行い、ほぼ5分毎に296nmの変化を測定した。また、ブランクとして、変性リボヌクレアーゼAを添加しないものも測定し、コンタミ酵素によってバックグラウンドの吸光度が増加しないことを確認した。
その結果、図5に示すようにDsbC>PhDsb>DsbAの順番でリフォールディング活性を確認することができた。PhDsbはDsbAより若干活性が強く、DsbCとDsbAの中間的な性質を示した。また、コントロールにも若干吸光度の上昇が見られたが、これも一般的に知られている現象である。
【0082】
実施例10:耐熱性の評価
DsbAおよびPhDsbを100mMのリン酸カリウム緩衝液(pH7.0)にて60μMに調製し、サーマルサイクラーを用いて100℃で任意の時間加熱した後、基本的には実施例8の方法に従ってジスルフィドレダクターゼ活性を測定した。具体的には、酵素の終濃度は5μMとなるようにし、25分後のA650nmを測定した。25分後のコントロールの吸光度はブランクとほぼ同じであった。活性は、非加熱のサンプルの吸光度650nmを100%とし、それの相対値で示している。結果を図6に示すが、100℃、2時間の加熱で大腸菌由来のDsbAの活性は40%まで低下したのに対し、PhDsbはほぼ100%活性を保持していた。今回の測定においては、本条件で測定した実測地をそのまま活性値として評価しているが、今回の実験においては、酵素反応の初速をほぼ観察していると思われることから、妥当な測定方法であるといえる。
【0083】
実施例11:無細胞タンパク質合成への応用例
PhDsbタンパク質のフォールディングに及ぼす効果を、無細胞タンパク質合成法を用いて検証した。具体的には、ヒト−S−アミラーゼ(ジスルフィド結合の形成が活性発現に必須)を高効率コムギ胚芽無細胞タンパク質合成系(Proc.Natl.Acad.Sci.USA 96(2),559−564(2000))を用いて合成する際に、各種プロテインジスルフィドオキシドレダクターゼを共存させておき、合成後にアミラーゼ活性を測定することにより、そのフォールディングに及ぼす効果を測定した。ヒト−S−アミラーゼは11のシステイン残基を有し、それらが複雑なジスルフィド結合を形成して活性を発現すると考えられており、通常の還元条件を用いる無細胞タンパク質合成反応では活性型の酵素を合成することが困難である。よって、今回用いたタンパク質合成に用いる際の低分子成分組成は、参考文献(Nature Biotechnology, 15, 79−84 (1997))に報告されているように、還元剤(ジチオスレイトール:DTT)の代わりに、酸化グルタチオン(GSSG)と還元グルタチオン(GSH)を一定の組成になるように添加した組成を用いた。また、無細胞タンパク質合成に用いたバッファー以外の成分は、PROTEIOS Wheat germ cell−free protein synthesis kit(東洋紡製)のものを用い、キット添付の取扱い説明書に従い、重層法(FEBS Letters 514, 102−105(2002))を用いて実施した。詳細には、反応液(下層)として、容量の20%のWheat germ extract (キット添付;200OD)、800units/ml Ribonuclease inhibitor (東洋紡製)、3μM 評価用タンパク質(DsbA(L−)、DsbC(L−)、PhDsb(L−)、およびBSA(以下(L−)略))、バッファー成分(終濃度として;30mM HEPES−KOH (pH7.6)、95mM 酢酸カリウム、2.65mM 酢酸マグネシウム、0.25mM GTP、0.4mM 酸化型グルタチオン(GSSG)、1mM 還元型グルタチオン(GSH)、1.2mM ATP、16mM クレアチンリン酸、0.5mg/mlクレアチン(ホスホ)キナーゼ(Roche製)、0.38mM スペルミジン、20種類のL−アミノ酸(各0.3mM)、0.25mg/ml mRNA(PROTEIOSの取扱い説明書に従いpEU−S−AMYから転写し、精製したもの))、となるようにそれぞれ50μl調製した。使用したPhDsbは、フラクション後、加熱処理を行ない更に精製を行ったものを用いた(実施例6参照)。上層(バッファー層)としては、終濃度として;30mM HEPES−KOH (pH7.6)、95mM 酢酸カリウム、2.65mM 酢酸マグネシウム、0.25mM GTP、0.4mM 酸化型グルタチオン(GSSG)、1mM 還元型グルタチオン(GSH)、1.2mM ATP、16mM クレアチンリン酸、0.38mM スペルミジン、20種類のL−アミノ酸(各0.3mM)となるように調製した。反応は26℃で16時間行った。
コムギ胚芽抽出液自体はアミラーゼ活性を有さず、また活性を阻害しないことを確認しているので、合成終了後、その液をそのまま用いてアミラーゼ活性の比較を行った。活性測定は、α−アミラーゼ測定キット・ダイヤカラー・リキッドAMY(東洋紡製)を用いて測定した。具体的には、酵素試薬(キット添付)300μlにサンプル5μlを添加した後に、基質試薬(キット添付)を150μl添加し、37℃にて11分間反応させ、415nmの吸光度をプレートリーダーで測定した。その結果を、図8に示した。図から明らかなように、コムギ胚芽抽出反応液のみを添加したBlankにはまったくアミラーゼ活性は認められなかった。一方、Dsbサンプルの代わりに20mM HEPES−KOD(pH7.6)のみを添加したControlについては活性の発現を認めることができた。これは、通常無細胞タンパク質合成反応で用いられるジチオスレイトール(DTT)を用いる代わりに、今回酸化/還元グルタチオン系を用いた効果であると思われた。また、BSAを添加したものの活性のレベルもControlとほぼ同じレベルであった。Dsb類を添加したものについては、DsbC>PhDsb>>DsbAの順番でアミラーゼ活性の向上を認めることができた。PhDsbはジスルフィドレダクターゼ活性レベルがDsbAとほとんど同レベルであることが確認されている(実施例8)ことから、DsbCで知られているようなジスルフィド結合を架けかえる(異性化)する活性(イソメラーゼ活性)は低いことが予想される。よって、この実験における高いリフォールディング活性は、ジスルフィドオキシダーゼ活性に加えて、シャペロンとしての効果が高かった可能性を示唆していると考えられる。
また、この実験における各条件でのアミラーゼの合成量は、別途実施した抗アミラーゼ抗体(Santa Cruz社製)を用いたウエスタンブロッティングの結果からほぼ同等であることを確認しており、合成量の違いによるものではなく、質の問題であることは確かである。また、ヒトS−アミラーゼ標品を用いた比較より、コントロールでタンパク質あたり約10%の活性であることが確認されている。よって、PhDsbの添加により、10%から17%程度(1.7倍)活性化されたことになる。
【産業上の利用可能性】
【0084】
本発明は、大腸菌等の組換え細胞を用いたタンパク質生産、あるいは、タンパク質研究の分野においてきわめて有用である。
【図面の簡単な説明】
【0085】
【図1】大腸菌DsbAとPhDsbのアミノ酸配列を比較した図。 四角で囲った部分がDsbAファミリーに共通するチオレドキシンモチーフを示す。また、*は共通のアミノ酸配列、−はギャップを示す。↓は、Signal Pプログラム(Ver.1.1)を用いて予測されたシグナルペプチダーゼ認識部位を示す。
【図2】PhDsbとPaDsbのアミノ酸配列を比較した図。 *は共通のアミノ酸配列、−はギャップを示す。
【図3】大腸菌(BL21−CodonPlus(DE3)−RIL)における種々のタンパク質の発現を示す図。10−20%グラジエントゲルを用いてSDS−PAGE解析を行った。1:ベクターのみ(コントロール)、2:PhDsb、3:PhDsb(L−)、4:DsbAL/PhDsb(DsbAのリーダー配列を有するPhDsb)、5:DsbA、T:大腸菌細胞ライゼート(Total)、Pe:ペリプラズム画分、S:菌体内可溶性画分。
【図4】種々の酵素のプロテインジスルフィドレダクターゼ活性の比較を示す図。縦軸に濁度(A650nm):レダクターゼ活性を、横軸に反応時間を示している。
【図5】種々の酵素のプロテインジスルフィドオキシダーゼ活性の比較を示す図。縦軸に吸光度(A296nm):活性を、横軸に反応時間を示している。
【図6】PhDsbの耐熱性を示す図。縦軸に相対プロテインジスルフィドレダクターゼ活性、横軸に100℃での処理時間を示している。
【図7】DsbA類の進化系統図を示す図。pI 8.0以上の塩基性タンパク質を四角で囲んで示している。また、括弧内のアルファベットはCPXCモチーフのXにH以外のアミノ酸を採用しているものについてそのアミノ酸を示している。
【図8】無細胞タンパク質合成でタンパク質を合成する際のフォールディングに及ぼす各酵素の効果を比較する図。縦軸にアミラーゼ活性(A415nm)を示している。
【特許請求の範囲】
【請求項1】
Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有することを特徴とする、成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項2】
Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有し、下記理化学的特徴を有する、請求項1に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
熱安定性:pH7.0にて100℃、2時間の加熱で60%以上の残存活性を保持することができる。
【請求項3】
Pyrococcus属細菌由来であることを特徴とする請求項1に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項4】
Pyrococcus horikoshii由来であることを特徴とする請求項3に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項5】
該タンパク質の配列が、配列番号1に示される配列に由来することを特徴とする請求項4に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項6】
該タンパク質の配列が、配列番号2に示される配列であることを特徴とする、請求項5に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項7】
CPHCモチーフ配列が変更されたことを特徴とする、請求項1に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項8】
請求項1〜7に記載のタンパク質を発現するように、所望の遺伝子配列を挿入したベクター。
【請求項9】
請求項8に記載のベクターで形質転換された形質転換体。
【請求項10】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを生産する菌株を培養し、培養物より該タンパク質を採取することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
【請求項11】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを細胞質内に生産する菌株を培養し、培養物より該タンパク質を採取することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
【請求項12】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを、無細胞タンパク質合成法を用いて合成することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
【請求項13】
加熱処理工程を経ることを特徴とする、請求項10〜12に記載の成熟型プロテインジスルフィドオキシドレダクターゼの製造方法。
【請求項14】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質内もしくはタンパク質間のジスルフィド結合の切断を促進する方法。
【請求項15】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質のリフォールディングを促進する方法。
【請求項16】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質のフォールディングを促進する方法。
【請求項17】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いて無細胞タンパク質合成時のフォールディングを促進する請求項13に記載の方法。
【請求項18】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼをタンパク質安定化剤および/または賦形剤として用いることを特徴とするタンパク質の安定的保存方法。
【請求項19】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを含有する試薬またはキット。
【請求項20】
生物機能を用いて所望のタンパク質を発現させる方法であって、請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを共発現させることにより、該タンパク質の活性を高める方法。
【請求項1】
Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有することを特徴とする、成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項2】
Archaea由来であって、CPHC(C:システイン、P:プロリン、H:ヒスチジン)モチーフを有し、下記理化学的特徴を有する、請求項1に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
熱安定性:pH7.0にて100℃、2時間の加熱で60%以上の残存活性を保持することができる。
【請求項3】
Pyrococcus属細菌由来であることを特徴とする請求項1に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項4】
Pyrococcus horikoshii由来であることを特徴とする請求項3に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項5】
該タンパク質の配列が、配列番号1に示される配列に由来することを特徴とする請求項4に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項6】
該タンパク質の配列が、配列番号2に示される配列であることを特徴とする、請求項5に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項7】
CPHCモチーフ配列が変更されたことを特徴とする、請求項1に記載の成熟型プロテインジスルフィドオキシドレダクターゼタンパク質。
【請求項8】
請求項1〜7に記載のタンパク質を発現するように、所望の遺伝子配列を挿入したベクター。
【請求項9】
請求項8に記載のベクターで形質転換された形質転換体。
【請求項10】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを生産する菌株を培養し、培養物より該タンパク質を採取することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
【請求項11】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを細胞質内に生産する菌株を培養し、培養物より該タンパク質を採取することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
【請求項12】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを、無細胞タンパク質合成法を用いて合成することを特徴とする成熟型プロテインジスルフィドオキシドレダクターゼタンパク質の製造方法。
【請求項13】
加熱処理工程を経ることを特徴とする、請求項10〜12に記載の成熟型プロテインジスルフィドオキシドレダクターゼの製造方法。
【請求項14】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質内もしくはタンパク質間のジスルフィド結合の切断を促進する方法。
【請求項15】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質のリフォールディングを促進する方法。
【請求項16】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いてタンパク質のフォールディングを促進する方法。
【請求項17】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを用いて無細胞タンパク質合成時のフォールディングを促進する請求項13に記載の方法。
【請求項18】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼをタンパク質安定化剤および/または賦形剤として用いることを特徴とするタンパク質の安定的保存方法。
【請求項19】
請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを含有する試薬またはキット。
【請求項20】
生物機能を用いて所望のタンパク質を発現させる方法であって、請求項1〜7に記載の成熟型プロテインジスルフィドオキシドレダクターゼを共発現させることにより、該タンパク質の活性を高める方法。
【図1】


【図2】

【図4】
【図5】
【図6】
【図7】
【図8】
【図3】
【図2】
【図4】
【図5】
【図6】
【図7】
【図8】
【図3】
【公開番号】特開2006−238734(P2006−238734A)
【公開日】平成18年9月14日(2006.9.14)
【国際特許分類】
【出願番号】特願2005−55971(P2005−55971)
【出願日】平成17年3月1日(2005.3.1)
【出願人】(000003160)東洋紡績株式会社 (3,622)
【Fターム(参考)】
【公開日】平成18年9月14日(2006.9.14)
【国際特許分類】
【出願日】平成17年3月1日(2005.3.1)
【出願人】(000003160)東洋紡績株式会社 (3,622)
【Fターム(参考)】
[ Back to top ]