
新規ペプチド化合物
【課題】癌免疫療法に有用な新規化合物を提供する。
【解決手段】式(1):

〔式中、Xはチロシン残基又はメチオニン残基を表し、Y及びZは単結合等を表し、R1は水素原子等を表し、R2は水酸基等を表し、R3は水素原子、アルキル基、アミノ基等を表し、R4は水素原子、アルキル基、カルボキシ基等を表し、mは1又は2を表し、nは0〜2の整数を表す。但し、nが0を表わす場合、R3は水素原子又はアルキル基を表す。〕で表される新規な化合物、又はその薬学上許容される塩を提供する。
【解決手段】式(1):
〔式中、Xはチロシン残基又はメチオニン残基を表し、Y及びZは単結合等を表し、R1は水素原子等を表し、R2は水酸基等を表し、R3は水素原子、アルキル基、アミノ基等を表し、R4は水素原子、アルキル基、カルボキシ基等を表し、mは1又は2を表し、nは0〜2の整数を表す。但し、nが0を表わす場合、R3は水素原子又はアルキル基を表す。〕で表される新規な化合物、又はその薬学上許容される塩を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、癌ワクチン療法の分野に属し、癌免疫療法剤として有用な新規ペプチド化合物に関する。詳しくは、イン・ビボでCTL誘導活性を有し、癌ワクチンとして有用な、WT1由来の癌抗原ペプチドの誘導体に関する。
【背景技術】
【0002】
生体による癌細胞やウイルス感染細胞等の排除には、細胞性免疫、とりわけ細胞傷害性T細胞(以下、CTLと称する)が重要な働きをしている。CTLは、癌細胞上の癌抗原タンパク質由来の抗原ペプチド(癌抗原ペプチド)とMHC(Major Histocompatibility Complex)クラスI抗原(ヒトの場合はHLA抗原と称する)により形成される複合体を認識し、癌細胞を攻撃・破壊する。
MHCクラスI分子に結合する癌抗原ペプチドは、細胞内でタンパク質が分解されることにより生成された8から12残基であることが一般に知られている。すなわち、一般に、癌抗原タンパク質の8から12残基の部分ペプチドが癌抗原ペプチドとして用いられる可能性がある。この場合、抗原ペプチド中にグルタミン残基やシステイン残基が含まれていると、通常、これらのアミノ酸残基が空気中で自然酸化される。これにより、ペプチド本来のMHCクラスI分子との結合性やT細胞受容体による認識が低くなることが報告されている(非特許文献1及び2を参照)。
【0003】
ところで、Wilms腫瘍の癌抑制遺伝子WT1(WT1遺伝子)は、Wilms癌、無紅彩、泌尿生殖異常、精神発達遅延などを合併するWAGR症候群の解析からWilms癌の原因遺伝子の1つとして染色体11p13から単離された遺伝子であり、そのアミノ酸配列は公知である(非特許文献3を参照)。WT1遺伝子はヒト白血病で高発現しており、白血病細胞をWT1アンチセンスオリゴマーで処理するとその細胞増殖が抑制されることなどから、WT1遺伝子は白血病細胞の増殖に促進的に働いていることが示唆されている。更に、WT1遺伝子は、胃癌、大腸癌、肺癌、乳癌、胚細胞癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌、卵巣癌等の固形癌においても高発現しており、白血病及び固形癌における新しい癌抗原タンパク質であることが判明した(非特許文献4及び5を参照)。更に、WT1タンパクの部分配列からなる癌抗原ペプチド、すなわち天然型の癌抗原ペプチドが同定された(特許文献1及び2を参照)。
【0004】
具体的には、癌抗原タンパク質WT1の第235位−第243位よりなるペプチドであるWT1235-243(Cys-Met-Thr-Trp-Asn-Gln-Met-Asn-Leu;配列番号:1)は、HLA-A24拘束性のCTL誘導活性を有する癌抗原ペプチドである(非特許文献6及び特許文献1を参照)。このWT1235-243の第2位のメチオニン残基をチロシン残基に改変した改変ペプチド(Cys-Tyr-Thr-Trp-Asn-Gln-Met-Asn-Leu;配列番号:2、以下当該改変ペプチドをWT1235-243(2M→Y)と称する場合もある)は、天然型ペプチドに比べてHLA-A24抗原への高い結合性を有している(特許文献3を参照)。これら天然型ペプチドWT1235-243及び改変型ペプチドWT1235-243(2M→Y)はいずれも免疫療法剤としての開発が期待されている。
更には、前記天然型ペプチド及び改変型ペプチドには、N末端にシステイン残基が存在し、空気中で酸化されてジスルフィド結合を介した二量体を形成し、当該二量体も又、癌抗原ペプチドとなり得ることがわかっている(特許文献4を参照)。
【特許文献1】国際公開第00/06602号パンフレット
【特許文献2】国際公開第00/18795号パンフレット
【特許文献3】国際公開第02/079253号パンフレット
【特許文献4】国際公開第2004/063217号パンフレット
【非特許文献1】Immunity., 6:273, 1997
【非特許文献2】J.Immunol., 160:2099, 1998
【非特許文献3】Cell, 60:509, 1990
【非特許文献4】J. Immunol., 164:1873-80, 2000
【非特許文献5】J. Clin. Immunol., 20, 195-202, 2000
【非特許文献6】Clin. Cancer. Res. 8: 2626, 2002
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明が解決しようとする課題は、イン・ビボでCTL誘導活性を示し、癌ワクチンとして癌免疫療法に用いられる、新規ペプチド化合物を提供することにある。
【課題を解決するための手段】
【0006】
発明者らは、WT1タンパク質由来の癌抗原ペプチド、WT1235-243又はWT1235-243(2M→Y)を改変し、物理化学的性質、安定性、生理活性の面で優れた癌抗原ペプチドを創製すべく鋭意検討を行った。すなわち、これらのペプチドを改変した化合物を製造し、免疫原性についてHLA−A2402/Kbトランスジェニックマウス(WO 02/47474号公報を参照、以下HLA−A24マウスとも称する)を用いて調べた。
【0007】
その結果、WT1235-243及びWT1235-243(2M→Y)のN末端システイン残基(Cys)を改変すること、詳しくは、N末端システイン残基のチオール基を修飾することによって、物理化学的性質及び安定性に優れたペプチド化合物を得ることに成功した。本発明のペプチド化合物は優れた免疫原性を有し、優れたCTL誘導活性を示す。また本発明のペプチド化合物により誘導されたぺプチド特異的T細胞は、癌細胞が本来有している天然型ペプチド(WT1235-243)と交叉反応性を示すため、癌免疫療法剤として有効である。
従来、WT1抗原ペプチド:WT1235-243及びWT1235-243(2M→Y)のシステイン残基を改変(修飾)したペプチド化合物が、癌抗原となり得る免疫原性を維持するか否かは全く知られていなかったが、本発明者らは、初めて、N末端システイン残基のチオール基と、システイン、グルタチオン、又はチオグリコール酸のチオール基とをジスルフィド結合で縮合させることにより得られる改変体が優れた癌抗原となり得ることを見出した。
本発明は、上記の知見を元に完成するに至ったものである。
【0008】
即ち本発明は、
〔1〕 式(1):
【化1】
〔式中、Xはチロシン残基又はメチオニン残基を表し、
Y及びZは、独立して、単結合又は1〜10残基のアミノ酸からなるペプチドの二価基を表し、
R1は水素原子又はアルキル基を表し、
R2は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3は水素原子、アルキル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4は水素原子、アルキル基、カルボキシ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、又は式(2):
【化2】
(式中、Wはアミノ酸残基を表す)
で表される基を表し、
mは1又は2を表し、
nは0〜2の整数を表す。但し、nが0を表わす場合、R3は水素原子又はアルキル基を表す。〕
で表される化合物、又はその薬学上許容される塩;
〔2〕 R3において、置換アルキルカルボニルアミノ基が、カルボキシ基、アミノ基、アルキルアミノ基及びジアルキルアミノ基から選択される1又は2の置換基で置換されたアルキルカルボニルアミノ基である、〔1〕に記載の化合物又はその薬学上許容される塩;
〔3〕 R3が水素原子、又は式(3):
【化3】
(式中、rは1〜3の整数を表す)
で表される基であり、
R4がカルボキシ基又は式(2’):
【化4】
(式中、W’はグリシン残基又はβ−アラニン残基を表す)
で表される基である、〔1〕又は〔2〕に記載の化合物、又はその薬学上許容される塩;
〔4〕 R3が式(3’):
【化5】
で表される基であり、
R4がカルボキシメチルカルバモイル基である、〔3〕に記載の化合物、又はその薬学上許容される塩;
〔5〕 R3が水素原子であり、R4がカルボキシ基である、〔3〕に記載の化合物、又はその薬学上許容される塩;
〔6〕 式(1’):
【化6】
〔式中、X'はチロシン残基又はメチオニン残基を表し、R1’は水素原子又はアルキル基を表し、R2’は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3’はアミノ基、アルキルアミノ基、ジアルキルアミノ基又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4’はカルボキシ基、カルバモイル基、アルキルカルバモイル基又はジアルキルカルバモイル基を表す。〕
で表される化合物、又はその薬学上許容される塩;
〔7〕 〔1〕〜〔6〕のいずれかで表される化合物又はその薬学上許容される塩に特異的に結合する抗体;
〔8〕 〔1〕〜〔6〕のいずれかで表される化合物又はその薬学上許容される塩とHLA−A24抗原との複合体が提示されている抗原提示細胞;
〔9〕 〔1〕〜〔6〕のいずれかで表される化合物又はその薬学上許容される塩により誘導されたCTL;
〔10〕 〔1〕〜〔6〕のいずれかで表される化合物又はその薬学上許容される塩とHLA−A24抗原との複合体を認識する、〔9〕に記載のCTL;
〔11〕 配列番号:1に記載のペプチドとHLA−A24抗原との複合体を認識する、〔9〕に記載のCTL;
〔12〕 〔1〕〜〔6〕のいずれかで表される化合物もしくはその薬学上許容される塩、〔8〕に記載の抗原提示細胞又は〔9〕〜〔11〕のいずれかに記載のCTL、及び薬学的に許容される担体を含有する医薬組成物;
〔13〕 癌ワクチンとして使用される、〔12〕に記載の医薬組成物;
〔14〕 〔1〕〜〔6〕のいずれかで表される化合物もしくはその薬学上許容される塩、〔8〕に記載の抗原提示細胞又は〔9〕〜〔11〕のいずれかに記載のCTLの、癌ワクチンを製造するための使用;
〔15〕 〔1〕〜〔6〕のいずれかで表される化合物もしくはその薬学上許容される塩、〔8〕に記載の抗原提示細胞又は〔9〕〜〔11〕のいずれかに記載のCTLを有効成分として含有する、癌免疫療法剤;
〔16〕 癌を治療又は予防するための方法であって、〔1〕〜〔6〕のいずれかで表される化合物もしくはその薬学上許容される塩、〔8〕に記載の抗原提示細胞又は〔9〕〜〔11〕のいずれかに記載のCTLの治療又は予防に有効な量を、それを必要としているHLA-A24陽性かつWT1陽性の癌患者に投与することからなる方法;
に関する。
【発明の効果】
【0009】
本発明により、癌免疫療法剤として有用な新規ペプチド化合物、詳しくは、イン・ビボでCTL誘導活性を有し、癌ワクチンとして有効な、WT1由来の癌抗原を提供することが可能となった。このペプチドは、WT1235-243及びWT1235-243(2M→Y)のN末端システインのメルカプト基が、その癌抗原ペプチドとしての活性を保持した形で修飾されており、物理化学的性質及び安定性に優れているので、治療や研究において広範に利用することができる。具体的には、本発明の新規ペプチドは、例えば、インビトロでの処理中に活性が低下する恐れがなく取り扱いが容易であり、安定した治療効果を発揮しうるという利点を有する。
【図面の簡単な説明】
【0010】
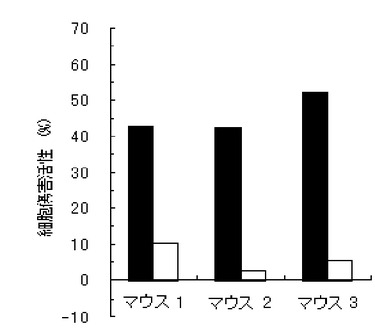
【図1】実施例1のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である(図中、黒棒)。図中、白棒はペプチド非パルスの結果を示す(以下同じ)。
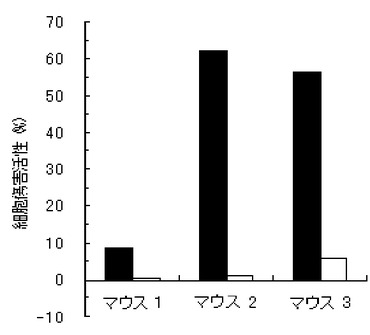
【図2】実施例2のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である。
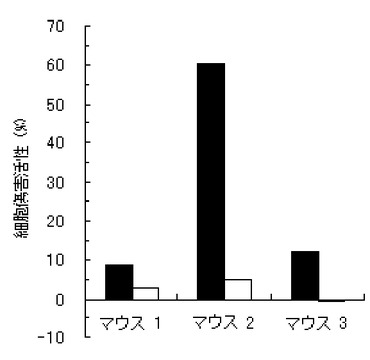
【図3】実施例3のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である。
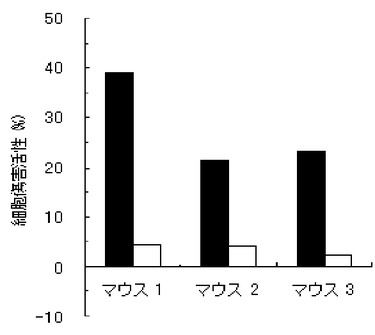
【図4】実施例4のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である。
【図5】実施例5のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である。
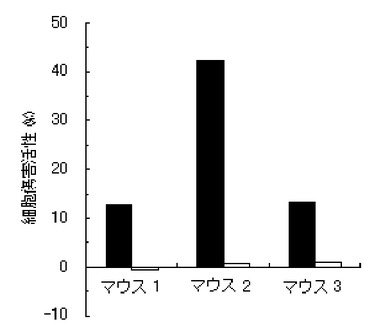
【図6】実施例6のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である。
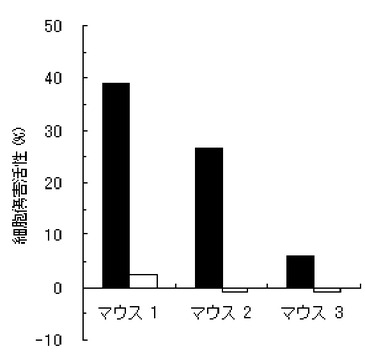
【図7】実施例1のペプチド化合物の用量依存的な細胞傷害活性(Specific Lysis)を示す図である。X軸には個体当たりの投与量(600μg、200μg、60μg、及び20μg)を示し、Y軸に細胞傷害活性(Specific Lysis)を示す。各用量においては、3匹のマウスを利用し、それぞれの細胞傷害活性平均値及び標準偏差(S.D.)を示す。
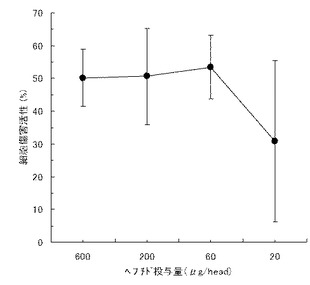
【図8】実施例1のペプチド化合物でマウスを免疫して得られたペプチド特異的T細胞の、各種ペプチドパルス細胞に対する反応性を示す図である。図中、斜線の棒は天然型ペプチド(WT1235-243)をパルスした細胞を用いた結果を、黒棒は実施例1のペプチド化合物をパルスした細胞を用いた結果を、白棒はインフルエンザ由来ペプチドをパルスした細胞を用いた結果を、それぞれ示す。また図の縦軸はスポットを示す。
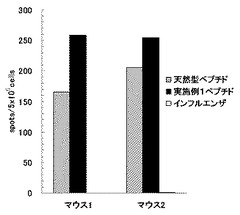
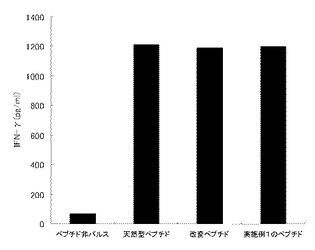
【図9】実施例1のペプチドの刺激によりヒトPBMCから誘導したペプチド特異的T細胞の、各種ペプチドパルス細胞に対する反応性を示す図である。図中「天然型ペプチド」は天然型ペプチド(WT1235-243)をパルスした細胞を用いた結果を、「改変ペプチド」は改変ペプチド(WT1235-243(2M→Y))をパルスした細胞を用いた結果を、「実施例1のペプチド」は実施例1のペプチド化合物をパルスした細胞を用いた結果を、「ペプチド非パルス」はペプチドをパルスしていない細胞を用いた結果を、それぞれ示す。また図の縦軸は、産生されたIFN-γの量を示す。
【発明を実施するための形態】
【0011】
本明細書及び図面において、アミノ酸残基を略号で表示する場合、次の略号で記述する。
Ala:アラニン残基
Arg:アルギニン残基
Asn:アスパラギン残基
Asp:アスパラギン酸残基
Cys:システイン残基
Gln:グルタミン残基
Glu:グルタミン酸残基
Gly:グリシン残基
His:ヒスチジン残基
Ile:イソロイシン残基
Leu:ロイシン残基
Lys:リジン残基
Met:メチオニン残基
Phe:フェニルアラニン残基
Pro:プロリン残基
Ser:セリン残基
Thr:トレオニン残基
Trp:トリプトファン残基
Tyr:チロシン残基
Val:バリン残基
【0012】
本明細書において、「アミノ酸残基」としては、天然もしくは非天然のα-アミノ酸残基、β−アミノ酸残基、γ-アミノ酸残基又はδ−アミノ酸残基が挙げられる。具体的には天然のα−アミノ酸(具体的には、Ala、Arg、Asn、Asp、Cys、Gln、Glu、Gly、His、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、Tyr又はVal)、オルニチン残基、ホモセリン残基、ホモシステイン残基、β−アラニン、γ−アミノブタン酸又はδ−アミノペンタン酸等が挙げられる。
前記アミノ酸残基に関し、光学異性体があり得る場合は、L体、D体のいずれであってもよいが、L体が好ましい。
本明細書において、ペプチド化合物のアミノ酸配列は常法に従って、そのN末端のアミノ酸残基が左側に位置し、C末端のアミノ酸残基が右側に位置するように記述する。
【0013】
(1)ペプチド化合物
本発明の第一の態様は、前記式(1)で表される化合物又はその薬学上許容される塩に関する。
式(1)中のXは、好ましくはチロシン残基(Tyr)を表す。
式(1)中のY及びZにおける「1〜10残基のアミノ酸残基からなるペプチドの二価基」としては、同一もしくは異なる、1〜10残基のアミノ酸残基からなるペプチドの二価基が挙げられ、そのアミノ酸配列は特に限定は無い。具体的には、ヒトWT1(Cell,60:509,1990、GenBank Acc.No.A38080)を構成するアミノ酸配列を挙げることができる。具体的には、Yとしては、ヒトWT1の第225位〜第234位からなる以下の10残基のペプチド:Asn-Leu-Tyr-Gln-Met-Thr-Ser-Gln-Leu-Glu(配列番号:3)、又は該配列番号:3で表されるペプチドからN末端の1〜9個のアミノ酸残基が欠損したペプチドの二価基を挙げることができる。また、Zとしては、ヒトWT1の第244位〜第253位からなる以下の10残基のペプチド:Gly-Ala-Thr-Leu-Lys-Gly-Val-Ala-Ala-Gly(配列番号:4)、又は該配列番号:4で表されるペプチドからC末端の1〜9個のアミノ酸残基が欠損したペプチドの二価基を挙げることができる。
Y及びZは好ましくは単結合を表す。
【0014】
本明細書において、アルキル基としては、炭素数1〜6の直鎖又は分枝のアルキル基が挙げられる。具体的には、メチル基、エチル基、プロピル基、1−メチルエチル基、ブチル基、1−メチルプロピル基、2-メチルプロピル基、1,1−ジメチルエチル基、ペンチル基等が挙げられる。
本明細書において、アルキルアミノ基としては、炭素数1〜6の直鎖又は分枝のアルキルアミノ基が挙げられる。具体的には、メチルアミノ基、エチルアミノ基、プロピルアミノ基、1−メチルエチルアミノ基、ブチルアミノ基、1−メチルプロピルアミノ基、2-メチルプロピルアミノ基、1,1−ジメチルエチルアミノ基、ペンチルアミノ基等が挙げられる。
本明細書において、ジアルキルアミノ基としては、2個の同一もしくは異なる炭素数1〜6の直鎖又は分枝のアルキルで置換されたアミノ基が挙げられる。具体的には、ジメチルアミノ基、エチルメチルアミノ基、ジエチルアミノ基、ジプロピルアミノ基、メチルプロピルアミノ基、ブチルメチルアミノ基、メチルペンチルアミノ基等が挙げられる。
本明細書において、アルキルカルボニルアミノ基における「アルキル」としては、前記アルキル基と同じものが挙げられる。アルキルカルバモイル基における「アルキル」としては、前記アルキルアミノ基におけるアルキルと同じものが挙げられる。ジアルキルカルバモイル基における「アルキル」としては、前記ジアルキルアミノ基におけるアルキルと同じものが挙げられ、2つのアルキルは同一もしくは異なっていてもよい。
【0015】
R1及びR2は好ましくは水素原子を表す。
R3が置換アルキルカルボニルアミノ基を表す場合の置換基としては、カルボキシ基、水酸基、アミノ基、アルキルアミノ基、又はジアルキルアミノ基が挙げられ同一もしくは異なる置換基が1〜4個、好ましくは1もしくは2個置換していてもよい。
式(2)中のWにおけるアミノ酸残基として、好ましくはグリシン残基(Gly)を挙げることができる。
【0016】
本発明のペプチド化合物として、具体的には、以下の式(4)〜(9):
【化7】
【化8】
【化9】
【化10】
【化11】
及び
【化12】
で表される化合物を挙げることができる。
【0017】
本発明のペプチド化合物は、本明細書実施例に記載された方法、又は通常のペプチド合成において用いられる方法に準じて製造することができる。製造方法としては、文献(ペプタイド・シンセシス(Peptide Synthesis), Interscience, New York, 1966; ザ・プロテインズ(The Proteins)、Vol.2,Academic Press Inc., New York, 1976; ペプチド合成、丸善(株)、1975;ペプチド合成の基礎と実験、丸善(株)、1985;医薬品の開発 続 第14巻・ペプチド合成、広川書店、1991)等に記載されている方法が挙げられる。例えば、Fmoc法もしくはBoc法を用いて固相合成機で製造する方法や、Boc−アミノ酸もしくはZ−アミノ酸を液相合成法で逐次縮合させて製造する方法が挙げられる(Fmocは9−フルオレニルメトキシカルボニル基、Bocはt−ブトキシカルボニル基、Zはベンジルオキシカルボニル基をそれぞれ表わす)。
本発明の化合物を製造するための中間体において、アミノ基、カルボキシ基、メルカプト基等の官能基は、必要に応じて保護、脱保護の技術を用い、適当な保護基で保護し、また脱保護することができる。好適な保護基、保護する方法、及び脱保護する方法としては、「Protective Groups in Organic Synthesis 2nd Edition (John Wiley & Sons, Inc.;1990)」等に詳細に記載されている。
【0018】
具体的には、以下の反応式で示される製造方法を例示することができる。
〔反応式1〕
【化13】
(式中、X、Y、Z、R1、R2、R3、R4、m及びnは前記と同義であり、R及びR’は独立して、水素原子もしくはメルカプト基の保護基を表わす。)
ここで、メルカプト基の保護基としてはアセトアミドメチル基又はトリチル基等が挙げられる。
【0019】
すなわち、式(1−1)の化合物と式(1−2)の化合物を、不活性溶媒中で酸化させることによって、式(1)の化合物を製造することができる。
酸化方法としては、通常のペプチド合成で、ジスルフィド結合を形成させる公知の方法を適宜選択すればよく、例えば、メルカプト基を持つ2つの中間体を適当な溶媒中に混合し酸化することにより形成できる。酸化法としては公知の酸化法、例えば空気酸化、ヨウ素酸化等の酸化法を用いることができる。溶媒としては水、酢酸、メタノール、クロロホルム、DMFもしくはDMSO等、又はこれらの混合液を用いることができる。酸化反応によりしばしば対称、非対称性ジスルフィド化合物の混合物を与える。目的の非対称性ジスルフィド化合物は種々のクロマトグラフィー、又は再結晶等で精製することによって得ることができる。あるいは活性化されたメルカプト基をもつ中間体とメルカプト基をもつ中間体を混合することにより選択的なジスルフィド結合を形成することができる。活性化されたメルカプト基をもつ中間体としては、Npys基(3−ニトロ−2−ピリジンスルフェニル基)が結合したメルカプト基等が挙げられる。あるいは、あらかじめ一方の中間体と例えば2,2’−ジチオビス(5−ニトロピリジン)を混合することによりメルカプト基を活性化した後、他方の中間体を加えることにより選択的なジスルフィド結合を形成することができる(Tetrahedron Letters. Vol. 37. No. 9, pp. 1347-1350)。
式(1−1)の化合物は、当業者に公知の液相もしくは固相のペプチド合成法に準じて調製することができる。
また、式(1−1)の化合物のN末端がアルキル化されている場合には、N末端のアミノ酸残基として、必要に応じて保護基で保護されたN−アルキルアミノ酸もしくはN,N−ジアルキルアミノ酸を用いて製造することができる。N−アルキルアミノ酸もしくはN,N−ジアルキルアミノ酸は、市販品を用いるか、アミノ酸又は保護アミノ酸を原料にして、塩基の存在下にアルキルハライドを反応させる等、当業者に周知の方法で調製することができる。例えば、下記〔反応式2〕に示されるとおり、t-ブトキシカルボニル基で保護されたアミノ酸に、水素化ナトリウム等の塩基の存在下に、アルキルハライドを反応させ、N末端のアミノ基を適宜アルキル化することができる。
【0020】
〔反応式2〕
(式中、X、Z、R1、R2、m及びRは前記と同義であり、Halは臭素原子若しくはヨウ素原子を表し、Protは保護基を表わす。)
また、C末端がアミド化又はアルキルアミド化されている場合には、原料として用いるC末端アミノ酸残基として、アミド化もしくはアルキルアミド化されたアミノ酸を原料に用いることができる。
【0021】
本発明の化合物、又はそれらを製造するための中間体は当業者に公知の方法で精製することができる。例えば、種々のクロマトグラフィー(例えば、シリカゲルカラムクロマトグラフィー、イオン交換カラムクロマトグラフィー、ゲルろ過、もしくは逆相クロマトグラフィー)、又は再結晶等で精製することができる。例えば、再結晶溶媒としては、メタノール、エタノールもしくは2−プロパノール等のアルコール系溶媒、ジエチルエーテル等のエーテル系溶媒、酢酸エチル等のエステル系溶媒、ベンゼンもしくはトルエン等の芳香族炭化水素系溶媒、アセトン等のケトン系溶媒、ヘキサン等の炭化水素系溶媒、ジメチルホルムアミドもしくはアセトニトリル等の非プロトン系溶媒、水、又はこれらの混合溶媒等を用いることができる。その他精製方法としては、実験化学講座(日本化学会編、丸善)1巻等に記載された方法等を用いることができる。
【0022】
本発明の化合物において、1つ以上の不斉点がある場合、通常の方法に従って、その不斉点を有する原料(アミノ酸)を用いることによって、製造することができる。また、本発明の化合物の光学純度を上げるために、製造工程の適当な段階で光学分割などを行ってもよい。光学分割法として例えば、本発明の化合物もしくはその中間体を不活性溶媒中(例えばメタノール、エタノール、もしくは2−プロパノール等のアルコール系溶媒、ジエチルエーテル等のエーテル系溶媒、酢酸エチル等のエステル系溶媒、トルエン等の炭化水素系溶媒、又はアセトニトリル等の非プロトン系溶媒、及びこれらの混合溶媒)、光学活性な酸(例えば、マンデル酸、N−ベンジルオキシアラニン、もしくは乳酸等のモノカルボン酸、酒石酸、o−ジイソプロピリデン酒石酸もしくはリンゴ酸等のジカルボン酸、又はカンファースルフォン酸もしくはブロモカンファースルフォン酸等のスルホン酸)と塩を形成させるジアステレオマー法により行うことができる。本発明の化合物もしくはその中間体がカルボキシ基等の酸性官能基を有する場合は、光学活性なアミン(例えばα−フェネチルアミン、キニン、キニジン、シンコニジン、シンコニン、ストリキニーネ等の有機アミン)と塩を形成させることにより光学分割を行うこともできる。
【0023】
塩を形成させる温度としては、室温から溶媒の沸点までの範囲から選択される。光学純度を向上させるためには、一旦、溶媒の沸点付近まで温度を上げることが望ましい。析出した塩を濾取する際、必要に応じて冷却し収率を向上させることができる。光学活性な酸、又はアミンの使用量は、基質に対し約0.5〜約2.0当量の範囲、好ましくは1当量前後の範囲が適当である。必要に応じ結晶を不活性溶媒中(例えばメタノール、エタノール、2−プロパノール等のアルコール系溶媒、ジエチルエーテル等のエーテル系溶媒、酢酸エチル等のエステル系溶媒、トルエン等の炭化水素系溶媒、アセトニトリル等の非プロトン系溶媒及びこれらの混合溶媒)で再結晶し、高純度の光学活性な塩を得ることもできる。また、必要に応じて光学分割した塩を通常の方法で酸又は塩基で処理しフリー体として得ることもできる。
【0024】
薬学上許容される塩としては、酸付加塩及び塩基付加塩が挙げられる。例えば、酸付加塩としては、塩酸塩、臭化水素酸塩、硫酸塩、ヨウ化水素酸塩、硝酸塩、リン酸塩等の無機酸塩、クエン酸塩、シュウ酸塩、酢酸塩、ギ酸塩、プロピオン酸塩、安息香酸塩、トリフルオロ酢酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、ベンゼンスルホン酸塩、パラトルエンスルホン酸塩等の有機酸塩が挙げられ、塩基付加塩としては、ナトリウム塩、カリウム塩、カルシウム塩、マグネシウム塩、アンモニウム塩等の無機塩基塩、トリエチルアンモニウム塩、トリエタノールアンモニウム塩、ピリジニウム塩、ジイソプロピルアンモニウム塩等の有機塩基塩等が挙げられ、さらにはアルギニン、アスパラギン酸、グルタミン酸などの塩基性あるいは酸性アミノ酸といったアミノ酸塩が挙げられる。
【0025】
また、本発明には、式(1)で示されるペプチド化合物又はその薬学上許容される塩の水和物、エタノール溶媒和物等の溶媒和物も含まれる。さらに、本発明には、式(1)で示されるペプチド化合物のあらゆるジアステレオマー、エナンチオマー等の存在するあらゆる立体異性体、及びあらゆる態様の結晶形のものも包含している。
一般にペプチド化合物の製造においては、光学活性なα−アミノ酸を縮合する工程、種々の保護基を除去する工程又はペプチドを樹脂から切り出す工程等で、アミノ酸が欠損したペプチド、加水分解や酸化等により分解したペプチド、アミノ酸がラセミ化したペプチド等種々の副生成物が生ずる。実験室スケールでは、種々のクロマトグラフィー(例えば、シリカゲルカラムクロマトグラフィー、イオン交換カラムクロマトグラフィー、ゲルろ過、もしくは逆相クロマトグラフィー)を組み合わせることによって、これらの不純物を除去し高純度のペプチド化合物を得ることができる。しかしながら、医薬品として提供するために工業的なスケールで高純度のペプチド化合物を得ることは容易ではない。
本発明のペプチド化合物は、その物理化学的性質においても、医薬品原薬として大量製造可能な性質を有する。具体的には、溶解性が高い、溶液中安定性に優れている、又は濃縮された際にゲル化しにくい等の性質を有し、逆相HPLC等のカラムクロマトグラフィーによる精製工程で、大量スケールにおいても容易に高純度のペプチド化合物を原薬として製造することができる。
【0026】
本発明のペプチド化合物は、癌免疫療法におけるCTL誘導剤の有効成分として、又癌ワクチンの有効成分として有用である。すなわち本発明のペプチド化合物は、本明細書実施例に示すとおり、優れた免疫原性を有し、優れたCTL誘導活性を示す。また、本発明のペプチド化合物によって誘導されるCTLは、驚くべきことに、癌細胞が本来保有するWT1の天然型ペプチドを認識することができる。従って、本発明のペプチド化合物は、WT1遺伝子が発現している癌、すなわち胃癌、大腸癌、肺癌、乳癌、胚細胞癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌又は卵巣癌等の治療薬又は予防薬(再発防止薬)として用いることができる。
【0027】
(2)抗体
本発明の第二の態様は、式(1)で表される本発明の化合物又はその薬学上許容される塩に特異的に結合する抗体(以下本発明の抗体と称する場合がある)に関する。本発明の抗体は、その形態に特に制限はなく、本発明の化合物を免疫原とするポリクローナル抗体であっても、またモノクローナル抗体であっても良い。
本発明の抗体は前記のように本発明の化合物に特異的に結合するものであれば特に制限されないが、具体的には、前述の式(4)〜式(9)のいずれかで表される化合物に特異的に結合する抗体を挙げることができる。
【0028】
これらの抗体の製造方法は、すでに周知であり、本発明の抗体もこれらの常法に従って製造することができる(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley and Sons. Section 11.12〜11.13、Antibodies; A Laboratory Manual, Lane, H, D.ら編, Cold Spring Harber Laboratory Press 出版 New York 1989)。
【0029】
具体的には、本発明の化合物(例えば式(4)〜式(9)のいずれかで表される化合物)を免疫原として用い、家兎等の非ヒト動物を免疫し、該免疫動物の血清から常法に従って得ることが可能である。一方、モノクローナル抗体の場合には、本発明の化合物(例えば式(4)〜式(9)のいずれかで表される化合物)をマウス等の非ヒト動物に免疫し、得られた脾臓細胞と骨髄腫細胞とを細胞融合させて調製したハイブリドーマ細胞の中から得ることができる(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley and Sons. Section 11.4〜11.11)。
【0030】
本発明の化合物に対する抗体の作製は、宿主に応じて種々のアジュバントを用いて免疫学的反応を高めることによって行うこともできる。そのようなアジュバントには、フロイントアジュバント、水酸化アルミニウムのようなミネラルゲル、並びにリゾレシチン、プルロニックポリオル、ポリアニオン、ペプチド、油乳剤、キーホールリンペットヘモシアニン及びジニトロフェノールのような表面活性物質、BCG(カルメット−ゲラン桿菌)やコリネバクテリウム-パルヴムなどのヒトアジュバントなどがある。
【0031】
以上のように本発明の化合物を用いて常法により適宜動物を免疫することにより、本発明の化合物を認識する抗体、さらにはその活性を中和する抗体が容易に作製できる。抗体の用途としては、アフィニティークロマトグラフィー、免疫学的診断等が挙げられる。免疫学的診断は、イムノブロット法、放射免疫測定法(RIA)、酵素免疫測定法(ELISA)、蛍光あるいは発光測定法等より適宜選択できる。このような免疫学的診断は、WT1遺伝子が発現している癌、すなわち胃癌、大腸癌、肺癌、乳癌、胚細胞癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌、卵巣癌等の診断において有効である。
【0032】
(3)抗原提示細胞
本発明の第三の態様は、本発明の化合物とHLA-A24抗原との複合体が提示された抗原提示細胞に関する。
後述の実施例において、本発明の化合物の投与によりCTL誘導活性が認められたが、これは、末梢血単核球中に、本発明の化合物とHLA-A24抗原との複合体の提示された抗原提示細胞が存在し、そして、この複合体の提示された細胞を特異的に認識するCTLが誘導されたことを示すものである。このような、HLA-A24抗原と本発明の化合物との複合体の提示された抗原提示細胞は、後述する細胞療法(DC療法)において有効に用いられる。
【0033】
本発明の抗原提示細胞は、本発明の化合物とHLA-A24抗原との複合体が提示された抗原提示細胞であれば良く、具体的には、例えば式(4)〜式(9)のいずれかに記載の化合物とHLA-A24抗原との複合体が樹状細胞の細胞表面に提示された抗原提示細胞を挙げることができる。
【0034】
細胞療法(DC療法)において用いられる抗原提示細胞は、癌患者から抗原提示能を有する細胞を単離し、この細胞に本発明の化合物を体外でパルスして、HLA-A24抗原と本発明の化合物との複合体を細胞表面に提示させることにより作製される。ここで「抗原提示能を有する細胞」とは、本発明の化合物を提示可能なHLA-A24抗原を細胞表面に発現している細胞であれば特に限定されないが、抗原提示能が高いとされている樹状細胞が好ましい。
【0035】
本発明の抗原提示細胞は、例えば癌患者から抗原提示能を有する細胞を単離し、該細胞に本発明の化合物(例えば式(4)〜式(9)のいずれかに記載の化合物)を体外でパルスし、HLA-A24抗原と本発明の化合物との複合体を作製することにより得られる(Cancer Immunol.Immunother.,46:82,1998、J.Immunol.,158:p1796,1997、Cancer Res., 59:p1184, 1999)。樹状細胞を用いる場合は、例えば、癌患者の末梢血からフィコール法によりリンパ球を分離し、その後非付着細胞を除き、付着細胞をGM-CSF及びIL-4存在下で培養して樹状細胞を誘導し、当該樹状細胞を本発明の化合物と共に培養してパルスすることなどにより、本発明の抗原提示細胞を調製することができる。
以上のようにして作製された本発明の抗原提示細胞は、後述するCTLの誘導剤、癌ワクチンの有効成分として、細胞療法(DC療法)において有効に用いられる。
【0036】
(4)CTL
本発明のペプチド化合物、は、ヒトWT1に由来し、HLA−A24拘束性のCTL誘導活性(免疫原性)を有する。すなわち、本発明の第四の態様は、本発明のペプチド化合物により誘導されるCTLに関する。
後述の実施例において、本発明の化合物の投与によりCTL誘導活性が認められた。これは、末梢血単核球中に、本発明の化合物とHLA-A24抗原との複合体の提示された抗原提示細胞が存在し、そして、この複合体の提示された細胞を認識するCTLが誘導されたことを示すものである。このような、本発明のペプチド化合物により誘導されたCTLは、後述する養子免疫療法において有効に用いられる。
【0037】
本発明のCTLは、本発明のペプチド化合物により誘導されたCTLであれば良いが、具体的には、例えば式(4)〜式(9)のいずれかで表される化合物とHLA-A24抗原との複合体を認識するCTL、天然型ペプチド(WT1235-243:配列番号:1)とHLA-A24抗原との複合体を認識するCTLを挙げることができる。
【0038】
養子免疫療法において用いられるCTLは、患者の末梢血リンパ球を単離し、これを本発明の化合物(例えば式(4)〜式(9)のいずれかで表される化合物)によってイン・ビトロで刺激する等により作製される(Journal of Experimental Medicine 1999, 190: 1669)。
以上のようにして作製された本発明のCTLは、癌ワクチンの有効成分として、養子免疫療法において有効に用いられる。
【0039】
(5)癌ワクチンとしての医薬組成物
上記(1)〜(4)に記載した本発明の化合物、本発明の抗原提示細胞、及び本発明のCTLは、それぞれの物質に応じた適切な形態とすることにより、CTLの誘導剤、すなわち癌ワクチンの有効成分とすることができる。以下、具体的に説明する。
【0040】
1) 本発明の化合物又はその薬学上許容される塩を有効成分とする癌ワクチン
本発明の化合物は、CTLの誘導能を有するものであり、誘導されたCTLは、細胞傷害作用やリンフォカインの産生を介して抗癌作用を発揮することができる。従って本発明の化合物は、癌の治療又は予防のための癌ワクチンの有効成分とすることができる。すなわち本発明は、本発明の化合物を有効成分として含有する癌ワクチン(癌ワクチンとしての医薬組成物)を提供する。本発明の癌ワクチンをHLA-A24陽性かつWT1陽性の患者に投与すると、抗原提示細胞のHLA-A24抗原に本発明化合物(例えば式(4)〜式(9)のいずれかで表される化合物)が提示され、提示されたHLA-A24抗原複合体特異的CTLが増殖して癌細胞を破壊することができ、従って、癌の治療又は予防が可能となる。本発明の癌ワクチンは、WT1遺伝子の発現レベルの上昇を伴う癌、例えば白血病、骨髄異形成症候群、多発性骨髄腫、悪性リンパ腫などの血液性の癌や、胃癌、大腸癌、肺癌、乳癌、胚細胞癌、肝癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌、卵巣癌等の固形癌の予防又は治療のために使用することができる。
よって、本発明は別の態様として、本発明の癌ワクチンの有効量をHLA-A24陽性かつWT1陽性の患者に投与することにより、癌を治療又は予防するための方法を提供する。
【0041】
本発明の化合物を有効成分とする癌ワクチンは、単一の癌抗原ペプチド、すなわちCTLエピトープ(例えば式(4)〜式(9)のいずれかで表される化合物)を有効成分とするものであっても、また他の癌抗原ペプチド(CTLエピトープ)やヘルパーエピトープを連結したエピトープペプチドを有効成分とするものであっても良い。すなわち近年、複数のCTLエピトープ(抗原ペプチド)を連結したエピトープペプチドが、イン・ビボで効率的にCTL誘導活性を有することが示されている。例えばJournal of Immunology 1998, 161: 3186-3194には、癌抗原タンパク質PSA由来のHLA-A2, -A3, -A11, B53拘束性CTLエピトープ(抗原ペプチド)を連結した約30merのエピトープペプチドが、イン・ビボでそれぞれのCTLエピトープに特異的なCTLを誘導したことが記載されている。またCTLエピトープとヘルパーエピトープとを連結させたエピトープペプチドにより、効率的にCTLが誘導されることも示されている。このようなエピトープペプチドの形態で投与した場合、抗原提示細胞内に取り込まれ、その後、細胞内分解を受けて生じた個々の抗原ペプチドがHLA抗原と結合して複合体を形成し、該複合体が抗原提示細胞表面に高密度に提示され、この複合体に特異的なCTLが体内で効率的に増殖し、癌細胞を破壊する。このようにして癌の治療又は予防が達成される。
【0042】
また本発明の化合物を有効成分とする癌ワクチンは、細胞性免疫が効果的に成立するように、医薬として許容されるキャリアー、例えば適当なアジュバントとともに投与したり、粒子状の剤型にして投与することができる。アジュバントとしては、文献(Clin. Microbiol.Rev., 7:277-289, 1994)に記載のものなどが応用可能であり、具体的には、菌体由来成分、GM-CSF、インターロイキン−2、インターロイキン−7もしくはインターロイキン−12等のサイトカイン、植物由来成分、海洋生物由来成分、水酸化アルミニウムの如き鉱物ゲル、リソレシチン、プルロニックポリオールの如き界面活性剤、ポリアニオン、ペプチド、又は油乳濁液(エマルジョン製剤)などを挙げることができる。菌体由来成分としては、リピドA(lipid A)、その誘導体であるモノホスホリルリピドA(monophosphoryl lipid A)、細菌(BCG菌等のMycobacterium属細菌が挙げられる)の死菌、細菌由来のタンパク質、ポリヌクレオチド)、フロイント不完全アジュバント(Freund's Incomplete Adjuvant)、フロイント完全アジュバント(Freund's Complete Adjuvant)、細胞壁骨格成分(例えば、BCG-CWS等が挙げられる)、トレハロースジミコレート(TDM)等が挙げられる。
また、リポソーム製剤、直径数μm のビーズに結合させた粒子状の製剤、リピッドを結合させた製剤なども考えられる。
【0043】
投与方法としては、皮内投与、皮下投与、筋肉内投与、静脈内投与などが挙げられる。製剤中の本発明の化合物の投与量は、治療目的の疾患、患者の年齢、体重等により適宜調整することができるが、通常0.0001mg〜1000mg、好ましくは 0.001mg〜1000mg、より好ましくは0.1mg〜10mgであり、これを数日ないし数月に1回投与するのが好ましい。
【0044】
2) 本発明の抗原提示細胞を有効成分とする癌ワクチン
本発明は、本発明の抗原提示細胞を有効成分とする癌ワクチンを提供する。
近年、癌患者の末梢血からリンパ球を分離し、その中から樹状細胞を誘導し、イン・ビトロで抗原ペプチド等をパルスして調製した抗原提示細胞を皮下投与などにより患者に戻す細胞療法(DC療法)が報告されている (Cancer Immunol.Immunother.,46:82,1998、J.Immunol.,158:p1796,1997、Cancer Res.,59:p1184,1999、Cancer Res.,56:p5672, 1996、J.Immunol.,161: p5607,1998、J.Exp.Med., 184: p465,1996)。従って前記本発明の抗原提示細胞を、細胞療法における癌ワクチンの有効成分として使用することができる。
【0045】
本発明の抗原提示細胞を有効成分とする癌ワクチンは、抗原提示細胞を安定に維持するために、生理食塩水、リン酸緩衝生理食塩水(PBS)、培地等を含むことが好ましい。投与方法としては、静脈内投与、皮下投与、皮内投与が挙げられる。また投与量は、前記文献記載の投与量が例示される。
前記癌ワクチンを患者の体内に戻すことにより、HLA-A24陽性かつWT1陽性の患者の体内で効率良く特異的なCTLが誘導され、癌を治療又は予防することができる。本発明の抗原提示細胞を有効成分とする癌ワクチンは、WT1遺伝子の発現レベルの上昇を伴う癌、例えば白血病、骨髄異形成症候群、多発性骨髄腫、悪性リンパ腫などの血液性の癌や、胃癌、大腸癌、肺癌、乳癌、胚細胞癌、肝癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌、卵巣癌等の固形癌の予防又は治療のために使用することができる。
【0046】
3) 本発明のCTLを有効成分とする癌ワクチン
本発明は、本発明のCTLを有効成分とする癌ワクチン(癌ワクチンとしての医薬組成物)を提供する。本発明のCTLは、以下の養子免疫療法において有効に用いられる。
メラノーマにおいて、患者本人の腫瘍内浸潤T細胞を体外で大量に培養し、これを患者に戻す養子免疫療法に治療効果が認められている(J. Natl. Cancer. Inst., 86: 1159, 1994)。またマウスのメラノーマでは、脾細胞をイン・ビトロで癌抗原ペプチドTRP−2で刺激し、癌抗原ペプチドに特異的なCTLを増殖させ、該CTLをメラノーマ移植マウスに投与することにより、転移抑制が認められている(J. Exp. Med., 185:453, 1997 )。これは、抗原提示細胞のHLA抗原と癌抗原ペプチドとの複合体を特異的に認識するCTLをイン・ビトロで増殖させた結果に基づくものである。従って、本発明の化合物を用いて、イン・ビトロで患者末梢血リンパ球を刺激して癌特異的CTLを増やした後、このCTLを患者に戻す治療法は有用であると考えられる。従って前記本発明のCTLを、養子免疫療法における癌ワクチンの有効成分として使用することができる。
【0047】
本発明のCTLを有効成分とする癌ワクチンは、CTLを安定に維持するために、生理食塩水、リン酸緩衝生理食塩水(PBS)、培地等を含むことが好ましい。投与方法としては、静脈内投与、皮下投与、皮内投与が挙げられる。また投与量としては、前記文献記載の投与量が例示される。
前記癌ワクチンを患者の体内に戻すことにより、HLA-A24陽性かつWT1陽性の患者の体内でCTLによる癌細胞の傷害作用が促進され、癌細胞を破壊することにより、癌を治療することができる。本発明のCTLを有効成分とする癌ワクチンは、WT1遺伝子の発現レベルの上昇を伴う癌、例えば白血病、骨髄異形成症候群、多発性骨髄腫、悪性リンパ腫などの血液性の癌や、胃癌、大腸癌、肺癌、乳癌、胚細胞癌、肝癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌、卵巣癌等の固形癌の予防又は治療のために使用することができる。
以下、実施例により本発明を具体的に説明するが、本発明の範囲はこれらに限定されるものではない。
また、以下の実施例において、実施例2の化合物、実施例4の化合物、及び実施例6の化合物はWT1235-243(配列番号:1)の修飾体に該当するものであり、それぞれシスチン修飾体、グルタチオン修飾体、及びチオグリコール酸修飾体である。一方、実施例1の化合物、実施例3の化合物、及び実施例5の化合物はWT1235-243(2M→Y)(配列番号:2)の修飾体に該当するものであり、それぞれシスチン修飾体、グルタチオン修飾体、及びチオグリコール酸修飾体である。
【実施例】
【0048】
実施例で用いられる略号は、以下のとおり:
Boc:t−ブトキシカルボニル
Npys:3-ニトロ-2-ピリジンスルフェニル
t-Bu:t−ブチル
Trt:トリフェニルメチル
Fmoc:9-フルオレニルメチルオキシカルボニル
DMF:ジメチルホルムアミド
HOBT:N-ヒドロキシベンゾトリアゾール
DIPCI:ジイソプロピルカルボジイミド。
【0049】
実施例1
式(4)で示されるペプチドの合成:
【化14】
下記の製造例1により得られたペプチド1.5g、Boc-Cys(Npys)-OH 600mgをジメチルスルホキシド20ml中に混合し20分間室温攪拌した。反応液にアセトニトリル600mlを加え氷冷下攪拌後、沈殿物を濾取した。濾上物はアセトニトリル、ジエチルエーテルで洗浄後減圧乾燥することによりBoc-Cys-OHがジスルフィド結合で結合したペプチド1.65gを得た。このペプチド0.53gをトリフルオロ酢酸5mlに溶解し10分間室温攪拌した。トリフルオロ酢酸を減圧留去後、残渣をアセトニトリル-酢酸-水(10/10/90)の混合液110mlに溶解しHPLC精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 3cmΦ×25cmL, 10μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:20ml/min
検出:UV220nm
2液濃度5%で平衡化させたカラムに粗ペプチド溶液をチャージした。その後2液濃度5%で10分間、15%で15分間送液しその後1分間あたり0.1%の割合で2液濃度を上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド300mgを得た。
・質量分析:LC-ESI/MS m/z =1292 [M+1]+ (理論値=1291.5)
【0050】
実施例2
式(5)で示されるペプチドの合成:
【化15】
実施例1と同様な方法で、下記の製造例2により得られたペプチド500mgとBoc-Cys(Npys)-OH 200mgを反応させた後、トリフルオロ酢酸中でBoc基を除去しHPLC精製することにより目的とするペプチド 104mgを得た。
・質量分析:LC-ESI/MS m/z =1260 [M+1]+ (理論値=1259.5)
【0051】
実施例3
式(8)で示されるペプチドの合成:
【化16】
製造例1により得られたペプチド240mgと2,2’-ジチオビス(5-ニトロピリジン)60mgをジメチルスルホキシド6ml中に混合し1時間室温攪拌した。反応液に還元型グルタチオン120mgを加え30℃にて1時間攪拌した。さらに還元型グルタチオン60mg、ジメチルスルホキシド4mlを追加し30分間攪拌後アセトニトリル200mlを加え生じた沈殿物を濾取、減圧乾燥することにより粗ペプチド440mgを得た。得られた粗ペプチドをアセトニトリル-酢酸-水(10/10/90)の混合液110mlに溶解しHPLC精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 3cmΦ×25cmL, 10μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:20ml/min
検出:UV220nm
2液濃度10%で平衡化させたカラムに粗ペプチド溶液をチャージした。その後2液濃度10%で10分間、17%で15分間送液しその後1分間あたり0.05%の割合で2液濃度を上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド107mgを得た。
・質量分析:LC-ESI/MS m/z =1477 [M+1]+ (理論値=1477.5)
【0052】
実施例4
式(9)で示されるペプチドの合成:
【化17】
製造例2により得られたペプチド120mgと2,2’-ジチオビス(5-ニトロピリジン)30mgをジメチルスルホキシド3ml中に混合し1時間室温攪拌した。反応液に還元型グルタチオン30mgを加え30分間室温攪拌後、水1mlを加え還元型グルタチオン30mgを追加しさらに20分間攪拌した。反応液にアセトニトリル100mlを加え生じた沈殿物を濾取、減圧乾燥することにより粗ペプチド160mgを得た。得られた粗ペプチドをアセトニトリル-酢酸-水(5/5/45)の混合液55mlに溶解しHPLC精製した。
ポンプ:Shimazu製;LC-6A型
カラム:YMC ODS-A 2cmΦ×25cmL, 10μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:10ml/min
検出:UV220nm
2液濃度10%で平衡化させたカラムに粗ペプチド溶液をチャージした。その後2液濃度10%で10分間、17%で15分間送液しその後1分間あたり0.05%の割合で2液濃度を上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド18mgを得た。
・質量分析:LC-ESI/MS m/z =1445 [M+1]+ (理論値=1445.5)
【0053】
実施例5
式(6)で示されるペプチドの合成:
【化18】
製造例1により得られたペプチド240mgと2,2’-ジチオビス(5-ニトロピリジン)60mgをジメチルスルホキシド6ml中に混合し1時間室温攪拌した。反応液にチオグリコール酸ナトリウム100mgを加え30℃にて30分間攪拌した。さらにチオグリコール酸ナトリウム50mg、ジメチルスルホキシド4ml、水2mlを追加し30分間攪拌後アセトニトリル200mlを加え生じた沈殿物を濾取、減圧乾燥することにより粗ペプチド305mgを得た。得られた粗ペプチドをアセトニトリル-酢酸-水(30/30/270)の混合液330mlに溶解し、濾過後濾液をHPLC精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 3cmΦ×25cmL, 10μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:20ml/min
検出:UV220nm
2液濃度10%で平衡化させたカラムに粗ペプチド溶液をチャージした。その後2液濃度10%で10分間、20%で15分間、23%で15分間送液しその後1分間あたり0.05%の割合で2液濃度を上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド15mgを得た。
・質量分析:LC-ESI/MS m/z =1263 [M+1]+ (理論値=1262.5)
【0054】
実施例6
式(7)で示されるペプチドの合成:
【化19】
製造例2により得られたペプチド240mgと2,2’-ジチオビス(5-ニトロピリジン)60mgをジメチルスルホキシド6ml中に混合し1時間室温攪拌した。反応液にチオグリコール酸ナトリウム50mgを加え30分間攪拌した。さらにチオグリコール酸ナトリウム50mgを追加し30℃にて1時間攪拌後アセトニトリル200mlを加え生じた沈殿物を濾取、減圧乾燥することにより粗ペプチド194mgを得た。得られた粗ペプチドをアセトニトリル-酢酸-水(10/20/90)の混合液120mlに溶解し、濾過後濾液をHPLC精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 3cmΦ×25cmL, 10μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:20ml/min
検出:UV220nm
2液濃度10%で平衡化させたカラムに粗ペプチド溶液をチャージした。その後2液濃度10%で10分間、18%で15分間送液しその後1分間あたり0.1%の割合で2液濃度を上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド30mgを得た。
・質量分析:LC-ESI/MS m/z =1230 [M+1]+ (理論値=1230.4)
【0055】
実施例7
式(4)で示されるペプチドの合成:
1. 保護ペプチド樹脂(Boc-Cys(Boc-Cys-OH)-Tyr(tBu)-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resinの合成
Fmoc-Leu-Alko-樹脂(ここで、Alkoはp−アルコキシベンジルアルコールを表す)10g(渡辺化学製;0.74mmol/g)をAdvanced ChemTech社製ACT90型固相合成機の500ml反応槽内に入れ、一旦この樹脂をDMF等で洗浄後(工程1)、25%ピペリジンで処理(3分×1回及び15分×1回)してFmoc基を切断後(工程2)、再びDMF等で樹脂を洗浄し(工程6)、ピペリジンを除去した。この反応槽内に、Fmoc-Asn(Trt)-OH 13.25gとHOBT(1−ヒドロキシベンゾトリアゾール)3.4gをNMP(N−メチルピロリジノン)200mlに溶解した溶液を加え、更にDIPCI(N,N’−ジイソプロピルカルボジイミド)3.42mlを加えて 60分間室温撹拌を行った(工程3)。NMPで樹脂を洗浄し(工程4)、Fmoc-Asn(Trt)-OH 13.25gとHOBT 3.4gとDIPCI 3.42mlを用いて再度カップリング反応を行った(工程3)。樹脂を洗浄後(工程6)25% Ac2O(無水酢酸)で3分x1回、15分×2回攪拌することにより未反応のアミノ基をキャッピングした(工程5)。樹脂を洗浄し(工程6)、脱保護(工程2)、洗浄操作(工程6)を行うことにより、H-Asn(Trt)-Leu-Alko-樹脂とした。同様にしてFmoc-Met-OH 8.25g、Fmoc-Gln(Trt)-OH 13.56g、Fmoc-Asn(Trt)-OH 13.25g、Fmoc-Trp(Boc)-OH 11.69g、Fmoc-Thr(tBu)-OH 8.82g、Fmoc-Tyr(tBu)-OH 10.2g、(Boc-Cys-OH)2 19.56gを用いてカップリング反応を行った。但しカップリングが困難な場合はカップリングを3回繰り返して行った。N末端の(Boc-Cys-OH)2(N,N'−t-ブトキシカルボニルシスチン)を縮合後、工程6の洗浄操作を実施し、ジエチルエーテル200mlで2回洗浄後減圧乾燥することによりBoc-Cys(Boc-Cys-OH)-Tyr(tBu)-Thr(tBu)-Trp(Boc) -Asn(Trt)- Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resin ((式(10)のペプチドレジン))22.87gを得た。上記合成工程の概要を表に示す。
<合成工程>
【0056】
【表1】
【0057】
2. 保護ペプチド樹脂の脱保護
上記操作によって得られた保護ペプチド樹脂(Boc-Cys(Boc-Cys-OH)-Tyr(tBu)-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resin 22.87gにトリフルオロ酢酸/エタンジチオール/水/トリイソプロピルシラン(94/2.5/2.5/1)混合液200mlを加え、室温で4時間攪拌した。反応液を濾過後濾液を氷冷下ジエチルエーテル 400mlに加えた。沈殿物をグラスフィルターで濾取しジエチルエーテルで洗浄後、減圧乾燥することにより粗ペプチド 8.27gを得た。
【0058】
3. 粗ペプチドの精製
得られた粗ペプチド 2.76gを20%酢酸水(1400ml)とアセトニトリル(35ml)の混合液に溶解後、不溶物を濾去し逆相液体クロマトグラフィーにより精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 5cmΦ×50cmL, 15-30μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:60ml/min
検出:UV280nm
カラムを2液濃度10%で平衡化させた後粗ペプチド溶液をチャージした。2液濃度10%で30分間送液した後120分間かけて2液濃度を34%に上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド H-Cys(H-Cys-OH)-Tyr-Thr-Trp-Asn-Gln-Met-Asn-Leu-OH(式(4)のペプチド) 0.91gを得た。
【0059】
試験例1
(マウスの免疫(1))
実施例1〜6で得た各抗原ペプチドの免疫原性について、HLA−A2402/Kbトランスジェニックマウス(WO 02/47474号公報、以下HLA−A24マウスとも称する)を利用することにより評価した。1ペプチドにつき3匹のトランスジェニックマウスに免疫することにより、それぞれのペプチドの免疫原性を評価した。
各合成ペプチドを用いた薬剤は以下にように調整した。即ち、実施例1〜3、5、6に記載の合成ペプチドをDMSOにて40mg/mlに調整した。その後、当該32.5μlを注射用水540μlと混合した。更に、当該550μlと700μlのフロイントの不完全アジュバント(Montanide ISA51)とをガラスシリンジを用いて混合することによりwater-in-oilエマルションを作製した。実施例4記載のペプチドについてはDMSOにて50mg/mlに調整し、一方ヘルパー合成ペプチド(FNNFTVSFWLRVPKVSASHLE、配列番号:5)をDMSOに20mg/mlに調整して、それぞれ30μlを注射用水540μlと混合した後、等量のフロイント不完全アジュバント(IFA)と混合することによりwater-in-oilエマルションを作製した。
その後、当該薬剤の200μlをHLA−A2402/Kbトランスジェニックマウスの尾根部の皮内に免疫した。実験開始7−8日後に脾臓を摘出し、スライドガラスのフロスト部分にて擦り破壊し、脾細胞を回収・調製した。ACKバッファー(0.15M NH4Cl、10mM KHCO3, 、0.1mM EDTA, pH7.2-7.4)にて溶血処理した脾細胞の一部に免疫した抗原ペプチドを100μg/mlで1時間パルスして7x106個/wellで24穴プレートに播種した。このとき、非ペプチドパルスの7x105個/wellの脾細胞を同時に加えて37℃下で5−6日間イン・ビトロ刺激培養した。ここでは、RPMI−1640培地に10%FCS、10mM HEPES、20mM L−グルタミン、1 mMピルビン酸ナトリウム、1mM MEM非必須アミノ酸、1%MEMビタミン、55μM 2-メルカプトエタノールを添加した培地にて培養した。
【0060】
次に、再刺激開始5−6日後に、常法に従って細胞傷害性試験を行った。標的細胞(T)として、EL−4細胞(大日本製薬株式会社、カタログNo. 06-39)にHLA−A2402/Kbをコードする遺伝子発現ベクターを導入して得られたEL4−A2402/Kb細胞、あるいは当該EL4−A2402/Kb細胞に抗原ペプチド:WT1235-243又はWT1235-243(2M→Y))をパルスした細胞を用いた。具体的には実施例1、実施例3、実施例5のペプチドの評価に関してはWT1235-243(2M→Y)をパルスした細胞を用い、実施例2、実施例4、実施例6のペプチドの評価に関してはWT1235-243をパルスした細胞を用いた。これらの細胞は1.85MBq/106個で51Crラベルし、ペプチドパルスは100μg/mlで1時間実施した(ラベル時間2時間、ラベル開始1時間後にペプチドを添加)。イン・ビトロ培養した脾細胞(E)による標的細胞(T)に対する傷害活性を51Crリリースアッセイ(J.Immunol., 159:4753, 1997)により測定した。このとき、E/T比80において作用させた。
【0061】
結果を、図1〜7に示す。図1〜6は、それぞれ、実施例1〜6のペプチド化合物に対応しており、図中、Y軸は細胞傷害活性(Specific Lysis)を示し、X軸は3匹のマウス個体毎の結果を示す。また、実施例1の化合物の用量依存性試験の結果を図7に示す。Y軸は細胞傷害活性(Specific Lysis)を示し、X軸は個体当たりの投与量(600μg、200μg、60μg、及び20μg)を示す。各用量においては、3匹のマウスを利用し、それぞれの細胞傷害活性平均値及び標準偏差(S.D.)を示す。図から明らかな通り、すべての合成ペプチドがCTL誘導活性を有すること、すなわち免疫原性を有することが判明した。
【0062】
試験例2
(マウスの免疫(2))
実施例1のペプチドをDMSOにて40mg/mlに調整した。その後、当該32.5μlを注射用水540μlと混合したのち、550μlを700μlのフロイントの不完全アジュバントMontanide ISA51(登録商標) (SEPPIC, Inc, Paris, France)とガラスシリンジを用いて混合することによりwater‐in-oilエマルションを作製した。
【0063】
次に、当該薬剤の200μlをHLA-A2402/Kbトランスジェニックマウスの尾根部の皮内に免疫した。実験開始7日後に、常法により脾臓の摘出および脾細胞を調製した(WO 02/47474号公報)。その後、mouse IFN gamma ELISPOT set (Enzyme - Linked Immunospot)(フジサワ、カタログ番号BD-551083)を用いて測定した。方法は添付書に従って行ない、1ウェルあたり5x105 cellsの脾細胞を注ぎ、更に、実施例1のペプチド、天然型ペプチド(WT1235-243)、またはインフルエンザウイルス由来ペプチド(ASNENMETM、HLA-A24非結合性のnegative controlペプチド)を含む細胞培地(最終濃度10-6 M)を注いだ。その後、約18時間37℃ CO2インキュベーターで培養した。
添付書に従ってプレートを洗浄し、KS Elispot Researchシステム(Carl Zeiss社製)にてスポット数を検出した。尚、Elispot法は細胞傷害性試験の代替法として知られている手法である(J.Immunological Methods, 1995, 181, 45-54)。結果を図8に示す。その結果、実施例1のペプチドは、天然型ペプチドに交叉反応性を示すHLA-A24特異的な細胞性免疫を誘導できることが判明した。
【0064】
試験例3
(ヒトPBMCを用いた試験)
HLA-A24陽性の健常人よりヘパリン入りの真空採血管にて末梢血50mlを採血した。2倍にPBS(-)で希釈した血液を半量のFicoll-Paque(Amersham Biosciences社)に重層した後、2000rpmで20分間遠心した。末梢血単核球(PBMC)を含む細胞層を回収し、3〜4倍量のPBS(-)を加え、1800rpmで10分間遠心した。細胞ペレットをPBS(-)で更に2回洗浄し、PBMCを回収した。
【0065】
PBMCをリンパ球用培養液(RPMI1640:AIM V=1:1、NEAA、10%FCS)に懸濁し、培養フラスコで2時間培養し、非付着細胞を回収した。非付着細胞からCD8+ T cell アイソレーションキットII(Miltenyi Biotec社)を用いてCD8陽性のT細胞を回収した。付着細胞は、1000U/mlのGM-CSFと1000U/mlのIL-4を含むリンパ球用培養液で7日間培養し、浮遊細胞を回収して樹状細胞(DC)を含む抗原提示細胞画分とした。回収した細胞は、実験に使用するまで凍結して保存した。
【0066】
上記で調製したDC細胞画分に実施例1のペプチドを50μg/mlのマイトマイシンを含むAIM-V培地に50μg/ml添加して1時間培養し、パルスした。その後、培地で3回洗浄後、更に実施例1のペプチドを50μg/mlの濃度で添加し、1時間パルスして抗原提示細胞とした。Day0にCD8陽性のT細胞にペプチドパルス抗原提示細胞を添加して1回目の刺激を行い、24穴プレートで10ng/mlのIL-7を含むリンパ球用培養液で培養を開始した。Day7にT細胞を回収し、洗浄後、1回目と同様にペプチドパルスした抗原提示細胞でペプチド刺激を行った。翌日(Day8)に50U/mlになるようにIL-2を添加した。Day14にT細胞を回収し、1回目、2回目と同様な方法で3回目の刺激を行った。翌日(Day15)に50U/mlになるようにIL-2を添加した。Day21にT細胞を回収して凍結保存した。
各種ペプチド(天然型ペプチド(WT1235-243)、改変ペプチド(WT1235-243(2M→Y))または実施例1のペプチド)をパルスした2×104個のVA13/A2402細胞(HLA-A2402発現)、またはペプチドをパルスしていない2×104個のVA13/A2402細胞に対して、1×105個のDay21のT細胞を添加し、18時間後に上清を回収して、ELISAでIFN-γ量を測定した。
【0067】
実施例1の合成ペプチドで刺激したT細胞のペプチド特異的反応性の検討結果を図9に示す。実施例1のペプチドで刺激したT細胞は、ペプチドをパルスしていない細胞に対してほとんど反応しないが、実施例1に記載のペプチドをパルスした細胞に良好に反応しており、ペプチド特異的なT細胞が誘導されていることが明らかになった。また誘導されたペプチド特異的なT細胞は、天然型ペプチド(WT1235-243)及び改変ペプチド(WT1235-243(2M→Y))をパルスした細胞に対しても同様に反応性を示した。
【0068】
製造例1
1. 保護ペプチド樹脂(H-Cys(Trt)-Tyr(tBu)-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resinの合成
Fmoc-Leu-Alko-樹脂(ここに、Alkoはp−アルコキシベンジルアルコール)10g(渡辺化学製;0.82mmol/g)をAdvanced ChemTech社製ACT90型固相合成機の500ml反応槽内に入れ、一旦この樹脂をDMF等で洗浄後(工程1)、25%Pip(ピペリジン)で処理(3分×1回及び15分×1回)してFmoc基を切断後(工程2)、再びDMF等で樹脂を洗浄し(工程1)、Pipを除去した。この反応槽内に、Fmoc-Asn(Trt)-OH 24.46gとHOBT(1−ヒドロキシベンゾトリアゾール)6.28gをNMP(N−メチルピロリジノン)200mlに溶解した溶液を加え、更にDIPCI(N,N’−ジイソプロピルカルボジイミド)6.3mlを加えて 30分間室温撹拌を行った(工程3)。30分後、NMPで樹脂を洗浄し(工程4)、Fmoc-Asn(Trt)-OH 24.46gとHOBT 6.28gとDIPCI 6.3mlを用いて再度カップリング反応を行い(工程5)、Fmoc-Asn(Trt)-Leu-Alko樹脂を合成した。その後、工程2の脱保護操作を行い、H-Asn(Trt)-Leu-Alko-樹脂とした。次いで、工程1の洗浄操作を行い、Fmoc-Met-OH 15.23g、Fmoc-Gln(Trt)-OH 25.04g、Fmoc-Asn(Trt)-OH 24.46g、Fmoc-Trp(Boc)-OH 21.59g、Fmoc-Thr(tBu)-OH 16.3g、Fmoc-Tyr(tBu)-OH 18.84g、Fmoc-Cys(Trt)-OH 24.01gを順次加え、工程3のカップリング反応を行った。但しFmoc-Thr(tBu)-OHについてはカップリングを3回繰り返して行った後、得られた樹脂をDMFで洗浄後、25% Ac2O(無水酢酸)で15分×2回で未反応のアミノ基をキャッピングした。N末端のFmoc-Cys(Trt)-OHを縮合後、工程2の脱保護操作を行い、工程6の洗浄操作を実施し、H-Cys(Trt)- Tyr(tBu)-Thr(tBu)-Trp(Boc) -Asn(Trt)- Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resinを得た。上記合成工程の概要を表に示す。
<合成工程>
【0069】
【表2】
【0070】
2. 保護ペプチド樹脂の脱保護
上記操作によって得られた保護ペプチド樹脂(H-Cys(Trt)-Tyr(tBu)-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resin 10.0gにトリフルオロ酢酸/エタンジチオール/水/トリイソプロピルシラン(94/2.5/2.5/1)混合液100mlを加え、室温で4時間攪拌した。反応液を氷冷下t−ブチルメチルエーテル 625mlに加え、15分間攪拌後不溶物をグラスフィルターで濾取した。濾上物を約100mlのt−ブチルメチルエーテルで5回洗浄し、洗浄後の濾上物を6M 塩酸グアニジン水溶液 1Lで抽出することにより粗ペプチド溶液を得た。
【0071】
3. 粗ペプチドの精製
得られた粗ペプチド溶液を逆相液体クロマトグラフィーにより精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 5cmΦ×50cmL, 15-30μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:60ml/min
検出:UV220nm
水浴で50℃に保温したカラムを2液濃度10%で平衡化させた後粗ペプチド溶液をチャージした。2液濃度10%で30分間送液した後40分間かけて2液濃度を20%に上昇させ更に360分間かけて2液濃度を40%に上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド(H-Cys-Tyr-Thr-Trp-Asn-Gln-Met-Asn-Leu-OH(配列番号:2) 1.50gを得た。
【0072】
・アミノ酸分析
加水分解:1%フェノール/6N塩酸水、110℃ 10時間
分析法:ニンヒドリン法
Asx:1.96(2) Thr:1.05(1) Glx:1.06(1) Met:1.05(1) * Leu:(1) Tyr:0.87(1)
*) Leu=基準アミノ酸 ( ) 内 理論値
・質量分析:LC-ESI/MS m/z =1173 [M+1]+ (理論値=1172.5)
・アミノ酸配列分析:N末2残基目 Tyrから、C末Leuまで順次確認した。
【0073】
製造例2
1. 保護ペプチド樹脂(H-Cys(Trt)-Met-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resinの合成
Fmoc-Leu-Alko-樹脂(ここに、Alkoはp−アルコキシベンジルアルコール)10g(渡辺化学製;0.81mmol/g)をAdvanced ChemTech社製ACT90型固相合成機の500ml反応槽内に入れ、一旦この樹脂をDMF等で洗浄後(工程1)、25%Pip(ピペリジン)で処理(3分×1回及び15分×1回)してFmoc基を切断後(工程2)、再びDMF等で樹脂を洗浄し(工程1)、Pipを除去した。この反応槽内に、Fmoc-Asn(Trt)-OH 24.17gとHOBT(1−ヒドロキシベンゾトリアゾール)6.2gをNMP(N−メチルピロリジノン)200mlに溶解した溶液を加え、更にDIPCI(N,N’−ジイソプロピルカルボジイミド)6.2mlを加えて 30分間室温撹拌を行った(工程3)。30分後、NMPで樹脂を洗浄し(工程4)、Fmoc-Asn(Trt)-OH 24.17g、HOBT 6.2g及びDIPCI 6.2mlを用いて再度カップリング反応を行い(工程5)、Fmoc-Asn(Trt)-Leu-Alko樹脂を合成した。その後、工程2の脱保護操作を行い、H-Asn(Trt)-Leu-Alko-樹脂とした。次いで、工程1の洗浄操作を行い、Fmoc-Met-OH 15.05g、Fmoc-Gln(Trt)-OH 24.73g、Fmoc-Asn(Trt)-OH 24.17g、Fmoc-Trp(Boc)-OH 21.33g、Fmoc-Thr(tBu)-OH 16.1g、Fmoc-Met-OH 15.05g、Fmoc-Cys(Trt)-OH 23.72gを順次加え、工程3のカップリング反応を行った。但しFmoc-Thr(tBu)-OHについてはカップリングを3回繰り返して行った後、得られた樹脂をDMFで洗浄後、25%Ac2O(無水酢酸)で15分×2回で未反応のアミノ基をキャッピングした。N末端のFmoc-Cys(Trt)-OHを縮合後、工程2の脱保護操作を行い、工程6の洗浄操作を実施し、H-Cys(Trt)-Met-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resinを得た。上記合成工程の概要は製造例1の表に記載された工程と同じである。
【0074】
2. 保護ペプチド樹脂の脱保護
上記操作によって得られた保護ペプチド樹脂(H-Cys(Trt)-Met-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resin 13.0gにトリフルオロ酢酸/エタンジチオール/水/トリイソプロピルシラン(94/2.5/2.5/1)混合液130mlを加え、室温で4時間攪拌した。反応液を氷冷下ジエチルエーテル800mlに加え、15分間攪拌後不溶物をグラスフィルターで濾取した。濾上物を約100mlのジエチルエーテルで5回洗浄し、洗浄後の濾上物を6M 塩酸グアニジン水溶液 1.3Lで抽出することにより粗ペプチド溶液を得た。
【0075】
3. 粗ペプチドの精製
得られた粗ペプチド溶液を逆相液体クロマトグラフィーにより精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 5cmΦ×50cmL, 15-30μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:60ml/min
検出:UV220nm
水浴で50℃に保温したカラムを2液濃度10%で平衡化させた後粗ペプチド溶液をチャージした。2液濃度10%で30分間送液した後40分間かけて2液濃度を20%に上昇させ更に360分間かけて2液濃度を40%に上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド(H-Cys-Met-Thr-Trp-Asn-Gln-Met-Asn-Leu-OH(配列番号:1) 2.32gを得た。
【0076】
・アミノ酸分析
加水分解:4N メタンスルホン酸、110℃ 17時間
分析法:ニンヒドリン法
Asx:1.87(2) Thr:0.93(1) Glx:0.95(1) Met:1.72(2) *Leu:(1) Trp:0.80(1)
*) Leu=基準アミノ酸 ( ) 内 理論値
・質量分析:LC-ESI/MS m/z =1141 [M+1]+ (理論値=1140.5)
・アミノ酸配列分析:N末2残基目 Metから、C末Leuまで順次確認した。
【産業上の利用可能性】
【0077】
本発明のペプチド化合物等は、癌免疫療法剤の有効成分として、有用である。
【0078】
配列表フリーテキスト
配列番号1:ペプチド誘導体
配列番号2:ペプチド誘導体
配列番号3:ペプチド誘導体
配列番号4:ペプチド誘導体
配列番号5:ヘルパー合成ペプチド
【技術分野】
【0001】
本発明は、癌ワクチン療法の分野に属し、癌免疫療法剤として有用な新規ペプチド化合物に関する。詳しくは、イン・ビボでCTL誘導活性を有し、癌ワクチンとして有用な、WT1由来の癌抗原ペプチドの誘導体に関する。
【背景技術】
【0002】
生体による癌細胞やウイルス感染細胞等の排除には、細胞性免疫、とりわけ細胞傷害性T細胞(以下、CTLと称する)が重要な働きをしている。CTLは、癌細胞上の癌抗原タンパク質由来の抗原ペプチド(癌抗原ペプチド)とMHC(Major Histocompatibility Complex)クラスI抗原(ヒトの場合はHLA抗原と称する)により形成される複合体を認識し、癌細胞を攻撃・破壊する。
MHCクラスI分子に結合する癌抗原ペプチドは、細胞内でタンパク質が分解されることにより生成された8から12残基であることが一般に知られている。すなわち、一般に、癌抗原タンパク質の8から12残基の部分ペプチドが癌抗原ペプチドとして用いられる可能性がある。この場合、抗原ペプチド中にグルタミン残基やシステイン残基が含まれていると、通常、これらのアミノ酸残基が空気中で自然酸化される。これにより、ペプチド本来のMHCクラスI分子との結合性やT細胞受容体による認識が低くなることが報告されている(非特許文献1及び2を参照)。
【0003】
ところで、Wilms腫瘍の癌抑制遺伝子WT1(WT1遺伝子)は、Wilms癌、無紅彩、泌尿生殖異常、精神発達遅延などを合併するWAGR症候群の解析からWilms癌の原因遺伝子の1つとして染色体11p13から単離された遺伝子であり、そのアミノ酸配列は公知である(非特許文献3を参照)。WT1遺伝子はヒト白血病で高発現しており、白血病細胞をWT1アンチセンスオリゴマーで処理するとその細胞増殖が抑制されることなどから、WT1遺伝子は白血病細胞の増殖に促進的に働いていることが示唆されている。更に、WT1遺伝子は、胃癌、大腸癌、肺癌、乳癌、胚細胞癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌、卵巣癌等の固形癌においても高発現しており、白血病及び固形癌における新しい癌抗原タンパク質であることが判明した(非特許文献4及び5を参照)。更に、WT1タンパクの部分配列からなる癌抗原ペプチド、すなわち天然型の癌抗原ペプチドが同定された(特許文献1及び2を参照)。
【0004】
具体的には、癌抗原タンパク質WT1の第235位−第243位よりなるペプチドであるWT1235-243(Cys-Met-Thr-Trp-Asn-Gln-Met-Asn-Leu;配列番号:1)は、HLA-A24拘束性のCTL誘導活性を有する癌抗原ペプチドである(非特許文献6及び特許文献1を参照)。このWT1235-243の第2位のメチオニン残基をチロシン残基に改変した改変ペプチド(Cys-Tyr-Thr-Trp-Asn-Gln-Met-Asn-Leu;配列番号:2、以下当該改変ペプチドをWT1235-243(2M→Y)と称する場合もある)は、天然型ペプチドに比べてHLA-A24抗原への高い結合性を有している(特許文献3を参照)。これら天然型ペプチドWT1235-243及び改変型ペプチドWT1235-243(2M→Y)はいずれも免疫療法剤としての開発が期待されている。
更には、前記天然型ペプチド及び改変型ペプチドには、N末端にシステイン残基が存在し、空気中で酸化されてジスルフィド結合を介した二量体を形成し、当該二量体も又、癌抗原ペプチドとなり得ることがわかっている(特許文献4を参照)。
【特許文献1】国際公開第00/06602号パンフレット
【特許文献2】国際公開第00/18795号パンフレット
【特許文献3】国際公開第02/079253号パンフレット
【特許文献4】国際公開第2004/063217号パンフレット
【非特許文献1】Immunity., 6:273, 1997
【非特許文献2】J.Immunol., 160:2099, 1998
【非特許文献3】Cell, 60:509, 1990
【非特許文献4】J. Immunol., 164:1873-80, 2000
【非特許文献5】J. Clin. Immunol., 20, 195-202, 2000
【非特許文献6】Clin. Cancer. Res. 8: 2626, 2002
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明が解決しようとする課題は、イン・ビボでCTL誘導活性を示し、癌ワクチンとして癌免疫療法に用いられる、新規ペプチド化合物を提供することにある。
【課題を解決するための手段】
【0006】
発明者らは、WT1タンパク質由来の癌抗原ペプチド、WT1235-243又はWT1235-243(2M→Y)を改変し、物理化学的性質、安定性、生理活性の面で優れた癌抗原ペプチドを創製すべく鋭意検討を行った。すなわち、これらのペプチドを改変した化合物を製造し、免疫原性についてHLA−A2402/Kbトランスジェニックマウス(WO 02/47474号公報を参照、以下HLA−A24マウスとも称する)を用いて調べた。
【0007】
その結果、WT1235-243及びWT1235-243(2M→Y)のN末端システイン残基(Cys)を改変すること、詳しくは、N末端システイン残基のチオール基を修飾することによって、物理化学的性質及び安定性に優れたペプチド化合物を得ることに成功した。本発明のペプチド化合物は優れた免疫原性を有し、優れたCTL誘導活性を示す。また本発明のペプチド化合物により誘導されたぺプチド特異的T細胞は、癌細胞が本来有している天然型ペプチド(WT1235-243)と交叉反応性を示すため、癌免疫療法剤として有効である。
従来、WT1抗原ペプチド:WT1235-243及びWT1235-243(2M→Y)のシステイン残基を改変(修飾)したペプチド化合物が、癌抗原となり得る免疫原性を維持するか否かは全く知られていなかったが、本発明者らは、初めて、N末端システイン残基のチオール基と、システイン、グルタチオン、又はチオグリコール酸のチオール基とをジスルフィド結合で縮合させることにより得られる改変体が優れた癌抗原となり得ることを見出した。
本発明は、上記の知見を元に完成するに至ったものである。
【0008】
即ち本発明は、
〔1〕 式(1):
【化1】
〔式中、Xはチロシン残基又はメチオニン残基を表し、
Y及びZは、独立して、単結合又は1〜10残基のアミノ酸からなるペプチドの二価基を表し、
R1は水素原子又はアルキル基を表し、
R2は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3は水素原子、アルキル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4は水素原子、アルキル基、カルボキシ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、又は式(2):
【化2】
(式中、Wはアミノ酸残基を表す)
で表される基を表し、
mは1又は2を表し、
nは0〜2の整数を表す。但し、nが0を表わす場合、R3は水素原子又はアルキル基を表す。〕
で表される化合物、又はその薬学上許容される塩;
〔2〕 R3において、置換アルキルカルボニルアミノ基が、カルボキシ基、アミノ基、アルキルアミノ基及びジアルキルアミノ基から選択される1又は2の置換基で置換されたアルキルカルボニルアミノ基である、〔1〕に記載の化合物又はその薬学上許容される塩;
〔3〕 R3が水素原子、又は式(3):
【化3】
(式中、rは1〜3の整数を表す)
で表される基であり、
R4がカルボキシ基又は式(2’):
【化4】
(式中、W’はグリシン残基又はβ−アラニン残基を表す)
で表される基である、〔1〕又は〔2〕に記載の化合物、又はその薬学上許容される塩;
〔4〕 R3が式(3’):
【化5】
で表される基であり、
R4がカルボキシメチルカルバモイル基である、〔3〕に記載の化合物、又はその薬学上許容される塩;
〔5〕 R3が水素原子であり、R4がカルボキシ基である、〔3〕に記載の化合物、又はその薬学上許容される塩;
〔6〕 式(1’):
【化6】
〔式中、X'はチロシン残基又はメチオニン残基を表し、R1’は水素原子又はアルキル基を表し、R2’は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3’はアミノ基、アルキルアミノ基、ジアルキルアミノ基又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4’はカルボキシ基、カルバモイル基、アルキルカルバモイル基又はジアルキルカルバモイル基を表す。〕
で表される化合物、又はその薬学上許容される塩;
〔7〕 〔1〕〜〔6〕のいずれかで表される化合物又はその薬学上許容される塩に特異的に結合する抗体;
〔8〕 〔1〕〜〔6〕のいずれかで表される化合物又はその薬学上許容される塩とHLA−A24抗原との複合体が提示されている抗原提示細胞;
〔9〕 〔1〕〜〔6〕のいずれかで表される化合物又はその薬学上許容される塩により誘導されたCTL;
〔10〕 〔1〕〜〔6〕のいずれかで表される化合物又はその薬学上許容される塩とHLA−A24抗原との複合体を認識する、〔9〕に記載のCTL;
〔11〕 配列番号:1に記載のペプチドとHLA−A24抗原との複合体を認識する、〔9〕に記載のCTL;
〔12〕 〔1〕〜〔6〕のいずれかで表される化合物もしくはその薬学上許容される塩、〔8〕に記載の抗原提示細胞又は〔9〕〜〔11〕のいずれかに記載のCTL、及び薬学的に許容される担体を含有する医薬組成物;
〔13〕 癌ワクチンとして使用される、〔12〕に記載の医薬組成物;
〔14〕 〔1〕〜〔6〕のいずれかで表される化合物もしくはその薬学上許容される塩、〔8〕に記載の抗原提示細胞又は〔9〕〜〔11〕のいずれかに記載のCTLの、癌ワクチンを製造するための使用;
〔15〕 〔1〕〜〔6〕のいずれかで表される化合物もしくはその薬学上許容される塩、〔8〕に記載の抗原提示細胞又は〔9〕〜〔11〕のいずれかに記載のCTLを有効成分として含有する、癌免疫療法剤;
〔16〕 癌を治療又は予防するための方法であって、〔1〕〜〔6〕のいずれかで表される化合物もしくはその薬学上許容される塩、〔8〕に記載の抗原提示細胞又は〔9〕〜〔11〕のいずれかに記載のCTLの治療又は予防に有効な量を、それを必要としているHLA-A24陽性かつWT1陽性の癌患者に投与することからなる方法;
に関する。
【発明の効果】
【0009】
本発明により、癌免疫療法剤として有用な新規ペプチド化合物、詳しくは、イン・ビボでCTL誘導活性を有し、癌ワクチンとして有効な、WT1由来の癌抗原を提供することが可能となった。このペプチドは、WT1235-243及びWT1235-243(2M→Y)のN末端システインのメルカプト基が、その癌抗原ペプチドとしての活性を保持した形で修飾されており、物理化学的性質及び安定性に優れているので、治療や研究において広範に利用することができる。具体的には、本発明の新規ペプチドは、例えば、インビトロでの処理中に活性が低下する恐れがなく取り扱いが容易であり、安定した治療効果を発揮しうるという利点を有する。
【図面の簡単な説明】
【0010】
【図1】実施例1のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である(図中、黒棒)。図中、白棒はペプチド非パルスの結果を示す(以下同じ)。
【図2】実施例2のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である。
【図3】実施例3のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である。
【図4】実施例4のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である。
【図5】実施例5のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である。
【図6】実施例6のペプチド化合物の、3匹のマウス個体毎の細胞傷害活性(Specific Lysis)を示す図である。
【図7】実施例1のペプチド化合物の用量依存的な細胞傷害活性(Specific Lysis)を示す図である。X軸には個体当たりの投与量(600μg、200μg、60μg、及び20μg)を示し、Y軸に細胞傷害活性(Specific Lysis)を示す。各用量においては、3匹のマウスを利用し、それぞれの細胞傷害活性平均値及び標準偏差(S.D.)を示す。
【図8】実施例1のペプチド化合物でマウスを免疫して得られたペプチド特異的T細胞の、各種ペプチドパルス細胞に対する反応性を示す図である。図中、斜線の棒は天然型ペプチド(WT1235-243)をパルスした細胞を用いた結果を、黒棒は実施例1のペプチド化合物をパルスした細胞を用いた結果を、白棒はインフルエンザ由来ペプチドをパルスした細胞を用いた結果を、それぞれ示す。また図の縦軸はスポットを示す。
【図9】実施例1のペプチドの刺激によりヒトPBMCから誘導したペプチド特異的T細胞の、各種ペプチドパルス細胞に対する反応性を示す図である。図中「天然型ペプチド」は天然型ペプチド(WT1235-243)をパルスした細胞を用いた結果を、「改変ペプチド」は改変ペプチド(WT1235-243(2M→Y))をパルスした細胞を用いた結果を、「実施例1のペプチド」は実施例1のペプチド化合物をパルスした細胞を用いた結果を、「ペプチド非パルス」はペプチドをパルスしていない細胞を用いた結果を、それぞれ示す。また図の縦軸は、産生されたIFN-γの量を示す。
【発明を実施するための形態】
【0011】
本明細書及び図面において、アミノ酸残基を略号で表示する場合、次の略号で記述する。
Ala:アラニン残基
Arg:アルギニン残基
Asn:アスパラギン残基
Asp:アスパラギン酸残基
Cys:システイン残基
Gln:グルタミン残基
Glu:グルタミン酸残基
Gly:グリシン残基
His:ヒスチジン残基
Ile:イソロイシン残基
Leu:ロイシン残基
Lys:リジン残基
Met:メチオニン残基
Phe:フェニルアラニン残基
Pro:プロリン残基
Ser:セリン残基
Thr:トレオニン残基
Trp:トリプトファン残基
Tyr:チロシン残基
Val:バリン残基
【0012】
本明細書において、「アミノ酸残基」としては、天然もしくは非天然のα-アミノ酸残基、β−アミノ酸残基、γ-アミノ酸残基又はδ−アミノ酸残基が挙げられる。具体的には天然のα−アミノ酸(具体的には、Ala、Arg、Asn、Asp、Cys、Gln、Glu、Gly、His、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、Tyr又はVal)、オルニチン残基、ホモセリン残基、ホモシステイン残基、β−アラニン、γ−アミノブタン酸又はδ−アミノペンタン酸等が挙げられる。
前記アミノ酸残基に関し、光学異性体があり得る場合は、L体、D体のいずれであってもよいが、L体が好ましい。
本明細書において、ペプチド化合物のアミノ酸配列は常法に従って、そのN末端のアミノ酸残基が左側に位置し、C末端のアミノ酸残基が右側に位置するように記述する。
【0013】
(1)ペプチド化合物
本発明の第一の態様は、前記式(1)で表される化合物又はその薬学上許容される塩に関する。
式(1)中のXは、好ましくはチロシン残基(Tyr)を表す。
式(1)中のY及びZにおける「1〜10残基のアミノ酸残基からなるペプチドの二価基」としては、同一もしくは異なる、1〜10残基のアミノ酸残基からなるペプチドの二価基が挙げられ、そのアミノ酸配列は特に限定は無い。具体的には、ヒトWT1(Cell,60:509,1990、GenBank Acc.No.A38080)を構成するアミノ酸配列を挙げることができる。具体的には、Yとしては、ヒトWT1の第225位〜第234位からなる以下の10残基のペプチド:Asn-Leu-Tyr-Gln-Met-Thr-Ser-Gln-Leu-Glu(配列番号:3)、又は該配列番号:3で表されるペプチドからN末端の1〜9個のアミノ酸残基が欠損したペプチドの二価基を挙げることができる。また、Zとしては、ヒトWT1の第244位〜第253位からなる以下の10残基のペプチド:Gly-Ala-Thr-Leu-Lys-Gly-Val-Ala-Ala-Gly(配列番号:4)、又は該配列番号:4で表されるペプチドからC末端の1〜9個のアミノ酸残基が欠損したペプチドの二価基を挙げることができる。
Y及びZは好ましくは単結合を表す。
【0014】
本明細書において、アルキル基としては、炭素数1〜6の直鎖又は分枝のアルキル基が挙げられる。具体的には、メチル基、エチル基、プロピル基、1−メチルエチル基、ブチル基、1−メチルプロピル基、2-メチルプロピル基、1,1−ジメチルエチル基、ペンチル基等が挙げられる。
本明細書において、アルキルアミノ基としては、炭素数1〜6の直鎖又は分枝のアルキルアミノ基が挙げられる。具体的には、メチルアミノ基、エチルアミノ基、プロピルアミノ基、1−メチルエチルアミノ基、ブチルアミノ基、1−メチルプロピルアミノ基、2-メチルプロピルアミノ基、1,1−ジメチルエチルアミノ基、ペンチルアミノ基等が挙げられる。
本明細書において、ジアルキルアミノ基としては、2個の同一もしくは異なる炭素数1〜6の直鎖又は分枝のアルキルで置換されたアミノ基が挙げられる。具体的には、ジメチルアミノ基、エチルメチルアミノ基、ジエチルアミノ基、ジプロピルアミノ基、メチルプロピルアミノ基、ブチルメチルアミノ基、メチルペンチルアミノ基等が挙げられる。
本明細書において、アルキルカルボニルアミノ基における「アルキル」としては、前記アルキル基と同じものが挙げられる。アルキルカルバモイル基における「アルキル」としては、前記アルキルアミノ基におけるアルキルと同じものが挙げられる。ジアルキルカルバモイル基における「アルキル」としては、前記ジアルキルアミノ基におけるアルキルと同じものが挙げられ、2つのアルキルは同一もしくは異なっていてもよい。
【0015】
R1及びR2は好ましくは水素原子を表す。
R3が置換アルキルカルボニルアミノ基を表す場合の置換基としては、カルボキシ基、水酸基、アミノ基、アルキルアミノ基、又はジアルキルアミノ基が挙げられ同一もしくは異なる置換基が1〜4個、好ましくは1もしくは2個置換していてもよい。
式(2)中のWにおけるアミノ酸残基として、好ましくはグリシン残基(Gly)を挙げることができる。
【0016】
本発明のペプチド化合物として、具体的には、以下の式(4)〜(9):
【化7】
【化8】
【化9】
【化10】
【化11】
及び
【化12】
で表される化合物を挙げることができる。
【0017】
本発明のペプチド化合物は、本明細書実施例に記載された方法、又は通常のペプチド合成において用いられる方法に準じて製造することができる。製造方法としては、文献(ペプタイド・シンセシス(Peptide Synthesis), Interscience, New York, 1966; ザ・プロテインズ(The Proteins)、Vol.2,Academic Press Inc., New York, 1976; ペプチド合成、丸善(株)、1975;ペプチド合成の基礎と実験、丸善(株)、1985;医薬品の開発 続 第14巻・ペプチド合成、広川書店、1991)等に記載されている方法が挙げられる。例えば、Fmoc法もしくはBoc法を用いて固相合成機で製造する方法や、Boc−アミノ酸もしくはZ−アミノ酸を液相合成法で逐次縮合させて製造する方法が挙げられる(Fmocは9−フルオレニルメトキシカルボニル基、Bocはt−ブトキシカルボニル基、Zはベンジルオキシカルボニル基をそれぞれ表わす)。
本発明の化合物を製造するための中間体において、アミノ基、カルボキシ基、メルカプト基等の官能基は、必要に応じて保護、脱保護の技術を用い、適当な保護基で保護し、また脱保護することができる。好適な保護基、保護する方法、及び脱保護する方法としては、「Protective Groups in Organic Synthesis 2nd Edition (John Wiley & Sons, Inc.;1990)」等に詳細に記載されている。
【0018】
具体的には、以下の反応式で示される製造方法を例示することができる。
〔反応式1〕
【化13】
(式中、X、Y、Z、R1、R2、R3、R4、m及びnは前記と同義であり、R及びR’は独立して、水素原子もしくはメルカプト基の保護基を表わす。)
ここで、メルカプト基の保護基としてはアセトアミドメチル基又はトリチル基等が挙げられる。
【0019】
すなわち、式(1−1)の化合物と式(1−2)の化合物を、不活性溶媒中で酸化させることによって、式(1)の化合物を製造することができる。
酸化方法としては、通常のペプチド合成で、ジスルフィド結合を形成させる公知の方法を適宜選択すればよく、例えば、メルカプト基を持つ2つの中間体を適当な溶媒中に混合し酸化することにより形成できる。酸化法としては公知の酸化法、例えば空気酸化、ヨウ素酸化等の酸化法を用いることができる。溶媒としては水、酢酸、メタノール、クロロホルム、DMFもしくはDMSO等、又はこれらの混合液を用いることができる。酸化反応によりしばしば対称、非対称性ジスルフィド化合物の混合物を与える。目的の非対称性ジスルフィド化合物は種々のクロマトグラフィー、又は再結晶等で精製することによって得ることができる。あるいは活性化されたメルカプト基をもつ中間体とメルカプト基をもつ中間体を混合することにより選択的なジスルフィド結合を形成することができる。活性化されたメルカプト基をもつ中間体としては、Npys基(3−ニトロ−2−ピリジンスルフェニル基)が結合したメルカプト基等が挙げられる。あるいは、あらかじめ一方の中間体と例えば2,2’−ジチオビス(5−ニトロピリジン)を混合することによりメルカプト基を活性化した後、他方の中間体を加えることにより選択的なジスルフィド結合を形成することができる(Tetrahedron Letters. Vol. 37. No. 9, pp. 1347-1350)。
式(1−1)の化合物は、当業者に公知の液相もしくは固相のペプチド合成法に準じて調製することができる。
また、式(1−1)の化合物のN末端がアルキル化されている場合には、N末端のアミノ酸残基として、必要に応じて保護基で保護されたN−アルキルアミノ酸もしくはN,N−ジアルキルアミノ酸を用いて製造することができる。N−アルキルアミノ酸もしくはN,N−ジアルキルアミノ酸は、市販品を用いるか、アミノ酸又は保護アミノ酸を原料にして、塩基の存在下にアルキルハライドを反応させる等、当業者に周知の方法で調製することができる。例えば、下記〔反応式2〕に示されるとおり、t-ブトキシカルボニル基で保護されたアミノ酸に、水素化ナトリウム等の塩基の存在下に、アルキルハライドを反応させ、N末端のアミノ基を適宜アルキル化することができる。
【0020】
〔反応式2〕
(式中、X、Z、R1、R2、m及びRは前記と同義であり、Halは臭素原子若しくはヨウ素原子を表し、Protは保護基を表わす。)
また、C末端がアミド化又はアルキルアミド化されている場合には、原料として用いるC末端アミノ酸残基として、アミド化もしくはアルキルアミド化されたアミノ酸を原料に用いることができる。
【0021】
本発明の化合物、又はそれらを製造するための中間体は当業者に公知の方法で精製することができる。例えば、種々のクロマトグラフィー(例えば、シリカゲルカラムクロマトグラフィー、イオン交換カラムクロマトグラフィー、ゲルろ過、もしくは逆相クロマトグラフィー)、又は再結晶等で精製することができる。例えば、再結晶溶媒としては、メタノール、エタノールもしくは2−プロパノール等のアルコール系溶媒、ジエチルエーテル等のエーテル系溶媒、酢酸エチル等のエステル系溶媒、ベンゼンもしくはトルエン等の芳香族炭化水素系溶媒、アセトン等のケトン系溶媒、ヘキサン等の炭化水素系溶媒、ジメチルホルムアミドもしくはアセトニトリル等の非プロトン系溶媒、水、又はこれらの混合溶媒等を用いることができる。その他精製方法としては、実験化学講座(日本化学会編、丸善)1巻等に記載された方法等を用いることができる。
【0022】
本発明の化合物において、1つ以上の不斉点がある場合、通常の方法に従って、その不斉点を有する原料(アミノ酸)を用いることによって、製造することができる。また、本発明の化合物の光学純度を上げるために、製造工程の適当な段階で光学分割などを行ってもよい。光学分割法として例えば、本発明の化合物もしくはその中間体を不活性溶媒中(例えばメタノール、エタノール、もしくは2−プロパノール等のアルコール系溶媒、ジエチルエーテル等のエーテル系溶媒、酢酸エチル等のエステル系溶媒、トルエン等の炭化水素系溶媒、又はアセトニトリル等の非プロトン系溶媒、及びこれらの混合溶媒)、光学活性な酸(例えば、マンデル酸、N−ベンジルオキシアラニン、もしくは乳酸等のモノカルボン酸、酒石酸、o−ジイソプロピリデン酒石酸もしくはリンゴ酸等のジカルボン酸、又はカンファースルフォン酸もしくはブロモカンファースルフォン酸等のスルホン酸)と塩を形成させるジアステレオマー法により行うことができる。本発明の化合物もしくはその中間体がカルボキシ基等の酸性官能基を有する場合は、光学活性なアミン(例えばα−フェネチルアミン、キニン、キニジン、シンコニジン、シンコニン、ストリキニーネ等の有機アミン)と塩を形成させることにより光学分割を行うこともできる。
【0023】
塩を形成させる温度としては、室温から溶媒の沸点までの範囲から選択される。光学純度を向上させるためには、一旦、溶媒の沸点付近まで温度を上げることが望ましい。析出した塩を濾取する際、必要に応じて冷却し収率を向上させることができる。光学活性な酸、又はアミンの使用量は、基質に対し約0.5〜約2.0当量の範囲、好ましくは1当量前後の範囲が適当である。必要に応じ結晶を不活性溶媒中(例えばメタノール、エタノール、2−プロパノール等のアルコール系溶媒、ジエチルエーテル等のエーテル系溶媒、酢酸エチル等のエステル系溶媒、トルエン等の炭化水素系溶媒、アセトニトリル等の非プロトン系溶媒及びこれらの混合溶媒)で再結晶し、高純度の光学活性な塩を得ることもできる。また、必要に応じて光学分割した塩を通常の方法で酸又は塩基で処理しフリー体として得ることもできる。
【0024】
薬学上許容される塩としては、酸付加塩及び塩基付加塩が挙げられる。例えば、酸付加塩としては、塩酸塩、臭化水素酸塩、硫酸塩、ヨウ化水素酸塩、硝酸塩、リン酸塩等の無機酸塩、クエン酸塩、シュウ酸塩、酢酸塩、ギ酸塩、プロピオン酸塩、安息香酸塩、トリフルオロ酢酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、ベンゼンスルホン酸塩、パラトルエンスルホン酸塩等の有機酸塩が挙げられ、塩基付加塩としては、ナトリウム塩、カリウム塩、カルシウム塩、マグネシウム塩、アンモニウム塩等の無機塩基塩、トリエチルアンモニウム塩、トリエタノールアンモニウム塩、ピリジニウム塩、ジイソプロピルアンモニウム塩等の有機塩基塩等が挙げられ、さらにはアルギニン、アスパラギン酸、グルタミン酸などの塩基性あるいは酸性アミノ酸といったアミノ酸塩が挙げられる。
【0025】
また、本発明には、式(1)で示されるペプチド化合物又はその薬学上許容される塩の水和物、エタノール溶媒和物等の溶媒和物も含まれる。さらに、本発明には、式(1)で示されるペプチド化合物のあらゆるジアステレオマー、エナンチオマー等の存在するあらゆる立体異性体、及びあらゆる態様の結晶形のものも包含している。
一般にペプチド化合物の製造においては、光学活性なα−アミノ酸を縮合する工程、種々の保護基を除去する工程又はペプチドを樹脂から切り出す工程等で、アミノ酸が欠損したペプチド、加水分解や酸化等により分解したペプチド、アミノ酸がラセミ化したペプチド等種々の副生成物が生ずる。実験室スケールでは、種々のクロマトグラフィー(例えば、シリカゲルカラムクロマトグラフィー、イオン交換カラムクロマトグラフィー、ゲルろ過、もしくは逆相クロマトグラフィー)を組み合わせることによって、これらの不純物を除去し高純度のペプチド化合物を得ることができる。しかしながら、医薬品として提供するために工業的なスケールで高純度のペプチド化合物を得ることは容易ではない。
本発明のペプチド化合物は、その物理化学的性質においても、医薬品原薬として大量製造可能な性質を有する。具体的には、溶解性が高い、溶液中安定性に優れている、又は濃縮された際にゲル化しにくい等の性質を有し、逆相HPLC等のカラムクロマトグラフィーによる精製工程で、大量スケールにおいても容易に高純度のペプチド化合物を原薬として製造することができる。
【0026】
本発明のペプチド化合物は、癌免疫療法におけるCTL誘導剤の有効成分として、又癌ワクチンの有効成分として有用である。すなわち本発明のペプチド化合物は、本明細書実施例に示すとおり、優れた免疫原性を有し、優れたCTL誘導活性を示す。また、本発明のペプチド化合物によって誘導されるCTLは、驚くべきことに、癌細胞が本来保有するWT1の天然型ペプチドを認識することができる。従って、本発明のペプチド化合物は、WT1遺伝子が発現している癌、すなわち胃癌、大腸癌、肺癌、乳癌、胚細胞癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌又は卵巣癌等の治療薬又は予防薬(再発防止薬)として用いることができる。
【0027】
(2)抗体
本発明の第二の態様は、式(1)で表される本発明の化合物又はその薬学上許容される塩に特異的に結合する抗体(以下本発明の抗体と称する場合がある)に関する。本発明の抗体は、その形態に特に制限はなく、本発明の化合物を免疫原とするポリクローナル抗体であっても、またモノクローナル抗体であっても良い。
本発明の抗体は前記のように本発明の化合物に特異的に結合するものであれば特に制限されないが、具体的には、前述の式(4)〜式(9)のいずれかで表される化合物に特異的に結合する抗体を挙げることができる。
【0028】
これらの抗体の製造方法は、すでに周知であり、本発明の抗体もこれらの常法に従って製造することができる(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley and Sons. Section 11.12〜11.13、Antibodies; A Laboratory Manual, Lane, H, D.ら編, Cold Spring Harber Laboratory Press 出版 New York 1989)。
【0029】
具体的には、本発明の化合物(例えば式(4)〜式(9)のいずれかで表される化合物)を免疫原として用い、家兎等の非ヒト動物を免疫し、該免疫動物の血清から常法に従って得ることが可能である。一方、モノクローナル抗体の場合には、本発明の化合物(例えば式(4)〜式(9)のいずれかで表される化合物)をマウス等の非ヒト動物に免疫し、得られた脾臓細胞と骨髄腫細胞とを細胞融合させて調製したハイブリドーマ細胞の中から得ることができる(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley and Sons. Section 11.4〜11.11)。
【0030】
本発明の化合物に対する抗体の作製は、宿主に応じて種々のアジュバントを用いて免疫学的反応を高めることによって行うこともできる。そのようなアジュバントには、フロイントアジュバント、水酸化アルミニウムのようなミネラルゲル、並びにリゾレシチン、プルロニックポリオル、ポリアニオン、ペプチド、油乳剤、キーホールリンペットヘモシアニン及びジニトロフェノールのような表面活性物質、BCG(カルメット−ゲラン桿菌)やコリネバクテリウム-パルヴムなどのヒトアジュバントなどがある。
【0031】
以上のように本発明の化合物を用いて常法により適宜動物を免疫することにより、本発明の化合物を認識する抗体、さらにはその活性を中和する抗体が容易に作製できる。抗体の用途としては、アフィニティークロマトグラフィー、免疫学的診断等が挙げられる。免疫学的診断は、イムノブロット法、放射免疫測定法(RIA)、酵素免疫測定法(ELISA)、蛍光あるいは発光測定法等より適宜選択できる。このような免疫学的診断は、WT1遺伝子が発現している癌、すなわち胃癌、大腸癌、肺癌、乳癌、胚細胞癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌、卵巣癌等の診断において有効である。
【0032】
(3)抗原提示細胞
本発明の第三の態様は、本発明の化合物とHLA-A24抗原との複合体が提示された抗原提示細胞に関する。
後述の実施例において、本発明の化合物の投与によりCTL誘導活性が認められたが、これは、末梢血単核球中に、本発明の化合物とHLA-A24抗原との複合体の提示された抗原提示細胞が存在し、そして、この複合体の提示された細胞を特異的に認識するCTLが誘導されたことを示すものである。このような、HLA-A24抗原と本発明の化合物との複合体の提示された抗原提示細胞は、後述する細胞療法(DC療法)において有効に用いられる。
【0033】
本発明の抗原提示細胞は、本発明の化合物とHLA-A24抗原との複合体が提示された抗原提示細胞であれば良く、具体的には、例えば式(4)〜式(9)のいずれかに記載の化合物とHLA-A24抗原との複合体が樹状細胞の細胞表面に提示された抗原提示細胞を挙げることができる。
【0034】
細胞療法(DC療法)において用いられる抗原提示細胞は、癌患者から抗原提示能を有する細胞を単離し、この細胞に本発明の化合物を体外でパルスして、HLA-A24抗原と本発明の化合物との複合体を細胞表面に提示させることにより作製される。ここで「抗原提示能を有する細胞」とは、本発明の化合物を提示可能なHLA-A24抗原を細胞表面に発現している細胞であれば特に限定されないが、抗原提示能が高いとされている樹状細胞が好ましい。
【0035】
本発明の抗原提示細胞は、例えば癌患者から抗原提示能を有する細胞を単離し、該細胞に本発明の化合物(例えば式(4)〜式(9)のいずれかに記載の化合物)を体外でパルスし、HLA-A24抗原と本発明の化合物との複合体を作製することにより得られる(Cancer Immunol.Immunother.,46:82,1998、J.Immunol.,158:p1796,1997、Cancer Res., 59:p1184, 1999)。樹状細胞を用いる場合は、例えば、癌患者の末梢血からフィコール法によりリンパ球を分離し、その後非付着細胞を除き、付着細胞をGM-CSF及びIL-4存在下で培養して樹状細胞を誘導し、当該樹状細胞を本発明の化合物と共に培養してパルスすることなどにより、本発明の抗原提示細胞を調製することができる。
以上のようにして作製された本発明の抗原提示細胞は、後述するCTLの誘導剤、癌ワクチンの有効成分として、細胞療法(DC療法)において有効に用いられる。
【0036】
(4)CTL
本発明のペプチド化合物、は、ヒトWT1に由来し、HLA−A24拘束性のCTL誘導活性(免疫原性)を有する。すなわち、本発明の第四の態様は、本発明のペプチド化合物により誘導されるCTLに関する。
後述の実施例において、本発明の化合物の投与によりCTL誘導活性が認められた。これは、末梢血単核球中に、本発明の化合物とHLA-A24抗原との複合体の提示された抗原提示細胞が存在し、そして、この複合体の提示された細胞を認識するCTLが誘導されたことを示すものである。このような、本発明のペプチド化合物により誘導されたCTLは、後述する養子免疫療法において有効に用いられる。
【0037】
本発明のCTLは、本発明のペプチド化合物により誘導されたCTLであれば良いが、具体的には、例えば式(4)〜式(9)のいずれかで表される化合物とHLA-A24抗原との複合体を認識するCTL、天然型ペプチド(WT1235-243:配列番号:1)とHLA-A24抗原との複合体を認識するCTLを挙げることができる。
【0038】
養子免疫療法において用いられるCTLは、患者の末梢血リンパ球を単離し、これを本発明の化合物(例えば式(4)〜式(9)のいずれかで表される化合物)によってイン・ビトロで刺激する等により作製される(Journal of Experimental Medicine 1999, 190: 1669)。
以上のようにして作製された本発明のCTLは、癌ワクチンの有効成分として、養子免疫療法において有効に用いられる。
【0039】
(5)癌ワクチンとしての医薬組成物
上記(1)〜(4)に記載した本発明の化合物、本発明の抗原提示細胞、及び本発明のCTLは、それぞれの物質に応じた適切な形態とすることにより、CTLの誘導剤、すなわち癌ワクチンの有効成分とすることができる。以下、具体的に説明する。
【0040】
1) 本発明の化合物又はその薬学上許容される塩を有効成分とする癌ワクチン
本発明の化合物は、CTLの誘導能を有するものであり、誘導されたCTLは、細胞傷害作用やリンフォカインの産生を介して抗癌作用を発揮することができる。従って本発明の化合物は、癌の治療又は予防のための癌ワクチンの有効成分とすることができる。すなわち本発明は、本発明の化合物を有効成分として含有する癌ワクチン(癌ワクチンとしての医薬組成物)を提供する。本発明の癌ワクチンをHLA-A24陽性かつWT1陽性の患者に投与すると、抗原提示細胞のHLA-A24抗原に本発明化合物(例えば式(4)〜式(9)のいずれかで表される化合物)が提示され、提示されたHLA-A24抗原複合体特異的CTLが増殖して癌細胞を破壊することができ、従って、癌の治療又は予防が可能となる。本発明の癌ワクチンは、WT1遺伝子の発現レベルの上昇を伴う癌、例えば白血病、骨髄異形成症候群、多発性骨髄腫、悪性リンパ腫などの血液性の癌や、胃癌、大腸癌、肺癌、乳癌、胚細胞癌、肝癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌、卵巣癌等の固形癌の予防又は治療のために使用することができる。
よって、本発明は別の態様として、本発明の癌ワクチンの有効量をHLA-A24陽性かつWT1陽性の患者に投与することにより、癌を治療又は予防するための方法を提供する。
【0041】
本発明の化合物を有効成分とする癌ワクチンは、単一の癌抗原ペプチド、すなわちCTLエピトープ(例えば式(4)〜式(9)のいずれかで表される化合物)を有効成分とするものであっても、また他の癌抗原ペプチド(CTLエピトープ)やヘルパーエピトープを連結したエピトープペプチドを有効成分とするものであっても良い。すなわち近年、複数のCTLエピトープ(抗原ペプチド)を連結したエピトープペプチドが、イン・ビボで効率的にCTL誘導活性を有することが示されている。例えばJournal of Immunology 1998, 161: 3186-3194には、癌抗原タンパク質PSA由来のHLA-A2, -A3, -A11, B53拘束性CTLエピトープ(抗原ペプチド)を連結した約30merのエピトープペプチドが、イン・ビボでそれぞれのCTLエピトープに特異的なCTLを誘導したことが記載されている。またCTLエピトープとヘルパーエピトープとを連結させたエピトープペプチドにより、効率的にCTLが誘導されることも示されている。このようなエピトープペプチドの形態で投与した場合、抗原提示細胞内に取り込まれ、その後、細胞内分解を受けて生じた個々の抗原ペプチドがHLA抗原と結合して複合体を形成し、該複合体が抗原提示細胞表面に高密度に提示され、この複合体に特異的なCTLが体内で効率的に増殖し、癌細胞を破壊する。このようにして癌の治療又は予防が達成される。
【0042】
また本発明の化合物を有効成分とする癌ワクチンは、細胞性免疫が効果的に成立するように、医薬として許容されるキャリアー、例えば適当なアジュバントとともに投与したり、粒子状の剤型にして投与することができる。アジュバントとしては、文献(Clin. Microbiol.Rev., 7:277-289, 1994)に記載のものなどが応用可能であり、具体的には、菌体由来成分、GM-CSF、インターロイキン−2、インターロイキン−7もしくはインターロイキン−12等のサイトカイン、植物由来成分、海洋生物由来成分、水酸化アルミニウムの如き鉱物ゲル、リソレシチン、プルロニックポリオールの如き界面活性剤、ポリアニオン、ペプチド、又は油乳濁液(エマルジョン製剤)などを挙げることができる。菌体由来成分としては、リピドA(lipid A)、その誘導体であるモノホスホリルリピドA(monophosphoryl lipid A)、細菌(BCG菌等のMycobacterium属細菌が挙げられる)の死菌、細菌由来のタンパク質、ポリヌクレオチド)、フロイント不完全アジュバント(Freund's Incomplete Adjuvant)、フロイント完全アジュバント(Freund's Complete Adjuvant)、細胞壁骨格成分(例えば、BCG-CWS等が挙げられる)、トレハロースジミコレート(TDM)等が挙げられる。
また、リポソーム製剤、直径数μm のビーズに結合させた粒子状の製剤、リピッドを結合させた製剤なども考えられる。
【0043】
投与方法としては、皮内投与、皮下投与、筋肉内投与、静脈内投与などが挙げられる。製剤中の本発明の化合物の投与量は、治療目的の疾患、患者の年齢、体重等により適宜調整することができるが、通常0.0001mg〜1000mg、好ましくは 0.001mg〜1000mg、より好ましくは0.1mg〜10mgであり、これを数日ないし数月に1回投与するのが好ましい。
【0044】
2) 本発明の抗原提示細胞を有効成分とする癌ワクチン
本発明は、本発明の抗原提示細胞を有効成分とする癌ワクチンを提供する。
近年、癌患者の末梢血からリンパ球を分離し、その中から樹状細胞を誘導し、イン・ビトロで抗原ペプチド等をパルスして調製した抗原提示細胞を皮下投与などにより患者に戻す細胞療法(DC療法)が報告されている (Cancer Immunol.Immunother.,46:82,1998、J.Immunol.,158:p1796,1997、Cancer Res.,59:p1184,1999、Cancer Res.,56:p5672, 1996、J.Immunol.,161: p5607,1998、J.Exp.Med., 184: p465,1996)。従って前記本発明の抗原提示細胞を、細胞療法における癌ワクチンの有効成分として使用することができる。
【0045】
本発明の抗原提示細胞を有効成分とする癌ワクチンは、抗原提示細胞を安定に維持するために、生理食塩水、リン酸緩衝生理食塩水(PBS)、培地等を含むことが好ましい。投与方法としては、静脈内投与、皮下投与、皮内投与が挙げられる。また投与量は、前記文献記載の投与量が例示される。
前記癌ワクチンを患者の体内に戻すことにより、HLA-A24陽性かつWT1陽性の患者の体内で効率良く特異的なCTLが誘導され、癌を治療又は予防することができる。本発明の抗原提示細胞を有効成分とする癌ワクチンは、WT1遺伝子の発現レベルの上昇を伴う癌、例えば白血病、骨髄異形成症候群、多発性骨髄腫、悪性リンパ腫などの血液性の癌や、胃癌、大腸癌、肺癌、乳癌、胚細胞癌、肝癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌、卵巣癌等の固形癌の予防又は治療のために使用することができる。
【0046】
3) 本発明のCTLを有効成分とする癌ワクチン
本発明は、本発明のCTLを有効成分とする癌ワクチン(癌ワクチンとしての医薬組成物)を提供する。本発明のCTLは、以下の養子免疫療法において有効に用いられる。
メラノーマにおいて、患者本人の腫瘍内浸潤T細胞を体外で大量に培養し、これを患者に戻す養子免疫療法に治療効果が認められている(J. Natl. Cancer. Inst., 86: 1159, 1994)。またマウスのメラノーマでは、脾細胞をイン・ビトロで癌抗原ペプチドTRP−2で刺激し、癌抗原ペプチドに特異的なCTLを増殖させ、該CTLをメラノーマ移植マウスに投与することにより、転移抑制が認められている(J. Exp. Med., 185:453, 1997 )。これは、抗原提示細胞のHLA抗原と癌抗原ペプチドとの複合体を特異的に認識するCTLをイン・ビトロで増殖させた結果に基づくものである。従って、本発明の化合物を用いて、イン・ビトロで患者末梢血リンパ球を刺激して癌特異的CTLを増やした後、このCTLを患者に戻す治療法は有用であると考えられる。従って前記本発明のCTLを、養子免疫療法における癌ワクチンの有効成分として使用することができる。
【0047】
本発明のCTLを有効成分とする癌ワクチンは、CTLを安定に維持するために、生理食塩水、リン酸緩衝生理食塩水(PBS)、培地等を含むことが好ましい。投与方法としては、静脈内投与、皮下投与、皮内投与が挙げられる。また投与量としては、前記文献記載の投与量が例示される。
前記癌ワクチンを患者の体内に戻すことにより、HLA-A24陽性かつWT1陽性の患者の体内でCTLによる癌細胞の傷害作用が促進され、癌細胞を破壊することにより、癌を治療することができる。本発明のCTLを有効成分とする癌ワクチンは、WT1遺伝子の発現レベルの上昇を伴う癌、例えば白血病、骨髄異形成症候群、多発性骨髄腫、悪性リンパ腫などの血液性の癌や、胃癌、大腸癌、肺癌、乳癌、胚細胞癌、肝癌、皮膚癌、膀胱癌、前立腺癌、子宮癌、子宮頸癌、卵巣癌等の固形癌の予防又は治療のために使用することができる。
以下、実施例により本発明を具体的に説明するが、本発明の範囲はこれらに限定されるものではない。
また、以下の実施例において、実施例2の化合物、実施例4の化合物、及び実施例6の化合物はWT1235-243(配列番号:1)の修飾体に該当するものであり、それぞれシスチン修飾体、グルタチオン修飾体、及びチオグリコール酸修飾体である。一方、実施例1の化合物、実施例3の化合物、及び実施例5の化合物はWT1235-243(2M→Y)(配列番号:2)の修飾体に該当するものであり、それぞれシスチン修飾体、グルタチオン修飾体、及びチオグリコール酸修飾体である。
【実施例】
【0048】
実施例で用いられる略号は、以下のとおり:
Boc:t−ブトキシカルボニル
Npys:3-ニトロ-2-ピリジンスルフェニル
t-Bu:t−ブチル
Trt:トリフェニルメチル
Fmoc:9-フルオレニルメチルオキシカルボニル
DMF:ジメチルホルムアミド
HOBT:N-ヒドロキシベンゾトリアゾール
DIPCI:ジイソプロピルカルボジイミド。
【0049】
実施例1
式(4)で示されるペプチドの合成:
【化14】
下記の製造例1により得られたペプチド1.5g、Boc-Cys(Npys)-OH 600mgをジメチルスルホキシド20ml中に混合し20分間室温攪拌した。反応液にアセトニトリル600mlを加え氷冷下攪拌後、沈殿物を濾取した。濾上物はアセトニトリル、ジエチルエーテルで洗浄後減圧乾燥することによりBoc-Cys-OHがジスルフィド結合で結合したペプチド1.65gを得た。このペプチド0.53gをトリフルオロ酢酸5mlに溶解し10分間室温攪拌した。トリフルオロ酢酸を減圧留去後、残渣をアセトニトリル-酢酸-水(10/10/90)の混合液110mlに溶解しHPLC精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 3cmΦ×25cmL, 10μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:20ml/min
検出:UV220nm
2液濃度5%で平衡化させたカラムに粗ペプチド溶液をチャージした。その後2液濃度5%で10分間、15%で15分間送液しその後1分間あたり0.1%の割合で2液濃度を上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド300mgを得た。
・質量分析:LC-ESI/MS m/z =1292 [M+1]+ (理論値=1291.5)
【0050】
実施例2
式(5)で示されるペプチドの合成:
【化15】
実施例1と同様な方法で、下記の製造例2により得られたペプチド500mgとBoc-Cys(Npys)-OH 200mgを反応させた後、トリフルオロ酢酸中でBoc基を除去しHPLC精製することにより目的とするペプチド 104mgを得た。
・質量分析:LC-ESI/MS m/z =1260 [M+1]+ (理論値=1259.5)
【0051】
実施例3
式(8)で示されるペプチドの合成:
【化16】
製造例1により得られたペプチド240mgと2,2’-ジチオビス(5-ニトロピリジン)60mgをジメチルスルホキシド6ml中に混合し1時間室温攪拌した。反応液に還元型グルタチオン120mgを加え30℃にて1時間攪拌した。さらに還元型グルタチオン60mg、ジメチルスルホキシド4mlを追加し30分間攪拌後アセトニトリル200mlを加え生じた沈殿物を濾取、減圧乾燥することにより粗ペプチド440mgを得た。得られた粗ペプチドをアセトニトリル-酢酸-水(10/10/90)の混合液110mlに溶解しHPLC精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 3cmΦ×25cmL, 10μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:20ml/min
検出:UV220nm
2液濃度10%で平衡化させたカラムに粗ペプチド溶液をチャージした。その後2液濃度10%で10分間、17%で15分間送液しその後1分間あたり0.05%の割合で2液濃度を上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド107mgを得た。
・質量分析:LC-ESI/MS m/z =1477 [M+1]+ (理論値=1477.5)
【0052】
実施例4
式(9)で示されるペプチドの合成:
【化17】
製造例2により得られたペプチド120mgと2,2’-ジチオビス(5-ニトロピリジン)30mgをジメチルスルホキシド3ml中に混合し1時間室温攪拌した。反応液に還元型グルタチオン30mgを加え30分間室温攪拌後、水1mlを加え還元型グルタチオン30mgを追加しさらに20分間攪拌した。反応液にアセトニトリル100mlを加え生じた沈殿物を濾取、減圧乾燥することにより粗ペプチド160mgを得た。得られた粗ペプチドをアセトニトリル-酢酸-水(5/5/45)の混合液55mlに溶解しHPLC精製した。
ポンプ:Shimazu製;LC-6A型
カラム:YMC ODS-A 2cmΦ×25cmL, 10μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:10ml/min
検出:UV220nm
2液濃度10%で平衡化させたカラムに粗ペプチド溶液をチャージした。その後2液濃度10%で10分間、17%で15分間送液しその後1分間あたり0.05%の割合で2液濃度を上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド18mgを得た。
・質量分析:LC-ESI/MS m/z =1445 [M+1]+ (理論値=1445.5)
【0053】
実施例5
式(6)で示されるペプチドの合成:
【化18】
製造例1により得られたペプチド240mgと2,2’-ジチオビス(5-ニトロピリジン)60mgをジメチルスルホキシド6ml中に混合し1時間室温攪拌した。反応液にチオグリコール酸ナトリウム100mgを加え30℃にて30分間攪拌した。さらにチオグリコール酸ナトリウム50mg、ジメチルスルホキシド4ml、水2mlを追加し30分間攪拌後アセトニトリル200mlを加え生じた沈殿物を濾取、減圧乾燥することにより粗ペプチド305mgを得た。得られた粗ペプチドをアセトニトリル-酢酸-水(30/30/270)の混合液330mlに溶解し、濾過後濾液をHPLC精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 3cmΦ×25cmL, 10μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:20ml/min
検出:UV220nm
2液濃度10%で平衡化させたカラムに粗ペプチド溶液をチャージした。その後2液濃度10%で10分間、20%で15分間、23%で15分間送液しその後1分間あたり0.05%の割合で2液濃度を上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド15mgを得た。
・質量分析:LC-ESI/MS m/z =1263 [M+1]+ (理論値=1262.5)
【0054】
実施例6
式(7)で示されるペプチドの合成:
【化19】
製造例2により得られたペプチド240mgと2,2’-ジチオビス(5-ニトロピリジン)60mgをジメチルスルホキシド6ml中に混合し1時間室温攪拌した。反応液にチオグリコール酸ナトリウム50mgを加え30分間攪拌した。さらにチオグリコール酸ナトリウム50mgを追加し30℃にて1時間攪拌後アセトニトリル200mlを加え生じた沈殿物を濾取、減圧乾燥することにより粗ペプチド194mgを得た。得られた粗ペプチドをアセトニトリル-酢酸-水(10/20/90)の混合液120mlに溶解し、濾過後濾液をHPLC精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 3cmΦ×25cmL, 10μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:20ml/min
検出:UV220nm
2液濃度10%で平衡化させたカラムに粗ペプチド溶液をチャージした。その後2液濃度10%で10分間、18%で15分間送液しその後1分間あたり0.1%の割合で2液濃度を上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド30mgを得た。
・質量分析:LC-ESI/MS m/z =1230 [M+1]+ (理論値=1230.4)
【0055】
実施例7
式(4)で示されるペプチドの合成:
1. 保護ペプチド樹脂(Boc-Cys(Boc-Cys-OH)-Tyr(tBu)-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resinの合成
Fmoc-Leu-Alko-樹脂(ここで、Alkoはp−アルコキシベンジルアルコールを表す)10g(渡辺化学製;0.74mmol/g)をAdvanced ChemTech社製ACT90型固相合成機の500ml反応槽内に入れ、一旦この樹脂をDMF等で洗浄後(工程1)、25%ピペリジンで処理(3分×1回及び15分×1回)してFmoc基を切断後(工程2)、再びDMF等で樹脂を洗浄し(工程6)、ピペリジンを除去した。この反応槽内に、Fmoc-Asn(Trt)-OH 13.25gとHOBT(1−ヒドロキシベンゾトリアゾール)3.4gをNMP(N−メチルピロリジノン)200mlに溶解した溶液を加え、更にDIPCI(N,N’−ジイソプロピルカルボジイミド)3.42mlを加えて 60分間室温撹拌を行った(工程3)。NMPで樹脂を洗浄し(工程4)、Fmoc-Asn(Trt)-OH 13.25gとHOBT 3.4gとDIPCI 3.42mlを用いて再度カップリング反応を行った(工程3)。樹脂を洗浄後(工程6)25% Ac2O(無水酢酸)で3分x1回、15分×2回攪拌することにより未反応のアミノ基をキャッピングした(工程5)。樹脂を洗浄し(工程6)、脱保護(工程2)、洗浄操作(工程6)を行うことにより、H-Asn(Trt)-Leu-Alko-樹脂とした。同様にしてFmoc-Met-OH 8.25g、Fmoc-Gln(Trt)-OH 13.56g、Fmoc-Asn(Trt)-OH 13.25g、Fmoc-Trp(Boc)-OH 11.69g、Fmoc-Thr(tBu)-OH 8.82g、Fmoc-Tyr(tBu)-OH 10.2g、(Boc-Cys-OH)2 19.56gを用いてカップリング反応を行った。但しカップリングが困難な場合はカップリングを3回繰り返して行った。N末端の(Boc-Cys-OH)2(N,N'−t-ブトキシカルボニルシスチン)を縮合後、工程6の洗浄操作を実施し、ジエチルエーテル200mlで2回洗浄後減圧乾燥することによりBoc-Cys(Boc-Cys-OH)-Tyr(tBu)-Thr(tBu)-Trp(Boc) -Asn(Trt)- Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resin ((式(10)のペプチドレジン))22.87gを得た。上記合成工程の概要を表に示す。
<合成工程>
【0056】
【表1】
【0057】
2. 保護ペプチド樹脂の脱保護
上記操作によって得られた保護ペプチド樹脂(Boc-Cys(Boc-Cys-OH)-Tyr(tBu)-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resin 22.87gにトリフルオロ酢酸/エタンジチオール/水/トリイソプロピルシラン(94/2.5/2.5/1)混合液200mlを加え、室温で4時間攪拌した。反応液を濾過後濾液を氷冷下ジエチルエーテル 400mlに加えた。沈殿物をグラスフィルターで濾取しジエチルエーテルで洗浄後、減圧乾燥することにより粗ペプチド 8.27gを得た。
【0058】
3. 粗ペプチドの精製
得られた粗ペプチド 2.76gを20%酢酸水(1400ml)とアセトニトリル(35ml)の混合液に溶解後、不溶物を濾去し逆相液体クロマトグラフィーにより精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 5cmΦ×50cmL, 15-30μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:60ml/min
検出:UV280nm
カラムを2液濃度10%で平衡化させた後粗ペプチド溶液をチャージした。2液濃度10%で30分間送液した後120分間かけて2液濃度を34%に上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド H-Cys(H-Cys-OH)-Tyr-Thr-Trp-Asn-Gln-Met-Asn-Leu-OH(式(4)のペプチド) 0.91gを得た。
【0059】
試験例1
(マウスの免疫(1))
実施例1〜6で得た各抗原ペプチドの免疫原性について、HLA−A2402/Kbトランスジェニックマウス(WO 02/47474号公報、以下HLA−A24マウスとも称する)を利用することにより評価した。1ペプチドにつき3匹のトランスジェニックマウスに免疫することにより、それぞれのペプチドの免疫原性を評価した。
各合成ペプチドを用いた薬剤は以下にように調整した。即ち、実施例1〜3、5、6に記載の合成ペプチドをDMSOにて40mg/mlに調整した。その後、当該32.5μlを注射用水540μlと混合した。更に、当該550μlと700μlのフロイントの不完全アジュバント(Montanide ISA51)とをガラスシリンジを用いて混合することによりwater-in-oilエマルションを作製した。実施例4記載のペプチドについてはDMSOにて50mg/mlに調整し、一方ヘルパー合成ペプチド(FNNFTVSFWLRVPKVSASHLE、配列番号:5)をDMSOに20mg/mlに調整して、それぞれ30μlを注射用水540μlと混合した後、等量のフロイント不完全アジュバント(IFA)と混合することによりwater-in-oilエマルションを作製した。
その後、当該薬剤の200μlをHLA−A2402/Kbトランスジェニックマウスの尾根部の皮内に免疫した。実験開始7−8日後に脾臓を摘出し、スライドガラスのフロスト部分にて擦り破壊し、脾細胞を回収・調製した。ACKバッファー(0.15M NH4Cl、10mM KHCO3, 、0.1mM EDTA, pH7.2-7.4)にて溶血処理した脾細胞の一部に免疫した抗原ペプチドを100μg/mlで1時間パルスして7x106個/wellで24穴プレートに播種した。このとき、非ペプチドパルスの7x105個/wellの脾細胞を同時に加えて37℃下で5−6日間イン・ビトロ刺激培養した。ここでは、RPMI−1640培地に10%FCS、10mM HEPES、20mM L−グルタミン、1 mMピルビン酸ナトリウム、1mM MEM非必須アミノ酸、1%MEMビタミン、55μM 2-メルカプトエタノールを添加した培地にて培養した。
【0060】
次に、再刺激開始5−6日後に、常法に従って細胞傷害性試験を行った。標的細胞(T)として、EL−4細胞(大日本製薬株式会社、カタログNo. 06-39)にHLA−A2402/Kbをコードする遺伝子発現ベクターを導入して得られたEL4−A2402/Kb細胞、あるいは当該EL4−A2402/Kb細胞に抗原ペプチド:WT1235-243又はWT1235-243(2M→Y))をパルスした細胞を用いた。具体的には実施例1、実施例3、実施例5のペプチドの評価に関してはWT1235-243(2M→Y)をパルスした細胞を用い、実施例2、実施例4、実施例6のペプチドの評価に関してはWT1235-243をパルスした細胞を用いた。これらの細胞は1.85MBq/106個で51Crラベルし、ペプチドパルスは100μg/mlで1時間実施した(ラベル時間2時間、ラベル開始1時間後にペプチドを添加)。イン・ビトロ培養した脾細胞(E)による標的細胞(T)に対する傷害活性を51Crリリースアッセイ(J.Immunol., 159:4753, 1997)により測定した。このとき、E/T比80において作用させた。
【0061】
結果を、図1〜7に示す。図1〜6は、それぞれ、実施例1〜6のペプチド化合物に対応しており、図中、Y軸は細胞傷害活性(Specific Lysis)を示し、X軸は3匹のマウス個体毎の結果を示す。また、実施例1の化合物の用量依存性試験の結果を図7に示す。Y軸は細胞傷害活性(Specific Lysis)を示し、X軸は個体当たりの投与量(600μg、200μg、60μg、及び20μg)を示す。各用量においては、3匹のマウスを利用し、それぞれの細胞傷害活性平均値及び標準偏差(S.D.)を示す。図から明らかな通り、すべての合成ペプチドがCTL誘導活性を有すること、すなわち免疫原性を有することが判明した。
【0062】
試験例2
(マウスの免疫(2))
実施例1のペプチドをDMSOにて40mg/mlに調整した。その後、当該32.5μlを注射用水540μlと混合したのち、550μlを700μlのフロイントの不完全アジュバントMontanide ISA51(登録商標) (SEPPIC, Inc, Paris, France)とガラスシリンジを用いて混合することによりwater‐in-oilエマルションを作製した。
【0063】
次に、当該薬剤の200μlをHLA-A2402/Kbトランスジェニックマウスの尾根部の皮内に免疫した。実験開始7日後に、常法により脾臓の摘出および脾細胞を調製した(WO 02/47474号公報)。その後、mouse IFN gamma ELISPOT set (Enzyme - Linked Immunospot)(フジサワ、カタログ番号BD-551083)を用いて測定した。方法は添付書に従って行ない、1ウェルあたり5x105 cellsの脾細胞を注ぎ、更に、実施例1のペプチド、天然型ペプチド(WT1235-243)、またはインフルエンザウイルス由来ペプチド(ASNENMETM、HLA-A24非結合性のnegative controlペプチド)を含む細胞培地(最終濃度10-6 M)を注いだ。その後、約18時間37℃ CO2インキュベーターで培養した。
添付書に従ってプレートを洗浄し、KS Elispot Researchシステム(Carl Zeiss社製)にてスポット数を検出した。尚、Elispot法は細胞傷害性試験の代替法として知られている手法である(J.Immunological Methods, 1995, 181, 45-54)。結果を図8に示す。その結果、実施例1のペプチドは、天然型ペプチドに交叉反応性を示すHLA-A24特異的な細胞性免疫を誘導できることが判明した。
【0064】
試験例3
(ヒトPBMCを用いた試験)
HLA-A24陽性の健常人よりヘパリン入りの真空採血管にて末梢血50mlを採血した。2倍にPBS(-)で希釈した血液を半量のFicoll-Paque(Amersham Biosciences社)に重層した後、2000rpmで20分間遠心した。末梢血単核球(PBMC)を含む細胞層を回収し、3〜4倍量のPBS(-)を加え、1800rpmで10分間遠心した。細胞ペレットをPBS(-)で更に2回洗浄し、PBMCを回収した。
【0065】
PBMCをリンパ球用培養液(RPMI1640:AIM V=1:1、NEAA、10%FCS)に懸濁し、培養フラスコで2時間培養し、非付着細胞を回収した。非付着細胞からCD8+ T cell アイソレーションキットII(Miltenyi Biotec社)を用いてCD8陽性のT細胞を回収した。付着細胞は、1000U/mlのGM-CSFと1000U/mlのIL-4を含むリンパ球用培養液で7日間培養し、浮遊細胞を回収して樹状細胞(DC)を含む抗原提示細胞画分とした。回収した細胞は、実験に使用するまで凍結して保存した。
【0066】
上記で調製したDC細胞画分に実施例1のペプチドを50μg/mlのマイトマイシンを含むAIM-V培地に50μg/ml添加して1時間培養し、パルスした。その後、培地で3回洗浄後、更に実施例1のペプチドを50μg/mlの濃度で添加し、1時間パルスして抗原提示細胞とした。Day0にCD8陽性のT細胞にペプチドパルス抗原提示細胞を添加して1回目の刺激を行い、24穴プレートで10ng/mlのIL-7を含むリンパ球用培養液で培養を開始した。Day7にT細胞を回収し、洗浄後、1回目と同様にペプチドパルスした抗原提示細胞でペプチド刺激を行った。翌日(Day8)に50U/mlになるようにIL-2を添加した。Day14にT細胞を回収し、1回目、2回目と同様な方法で3回目の刺激を行った。翌日(Day15)に50U/mlになるようにIL-2を添加した。Day21にT細胞を回収して凍結保存した。
各種ペプチド(天然型ペプチド(WT1235-243)、改変ペプチド(WT1235-243(2M→Y))または実施例1のペプチド)をパルスした2×104個のVA13/A2402細胞(HLA-A2402発現)、またはペプチドをパルスしていない2×104個のVA13/A2402細胞に対して、1×105個のDay21のT細胞を添加し、18時間後に上清を回収して、ELISAでIFN-γ量を測定した。
【0067】
実施例1の合成ペプチドで刺激したT細胞のペプチド特異的反応性の検討結果を図9に示す。実施例1のペプチドで刺激したT細胞は、ペプチドをパルスしていない細胞に対してほとんど反応しないが、実施例1に記載のペプチドをパルスした細胞に良好に反応しており、ペプチド特異的なT細胞が誘導されていることが明らかになった。また誘導されたペプチド特異的なT細胞は、天然型ペプチド(WT1235-243)及び改変ペプチド(WT1235-243(2M→Y))をパルスした細胞に対しても同様に反応性を示した。
【0068】
製造例1
1. 保護ペプチド樹脂(H-Cys(Trt)-Tyr(tBu)-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resinの合成
Fmoc-Leu-Alko-樹脂(ここに、Alkoはp−アルコキシベンジルアルコール)10g(渡辺化学製;0.82mmol/g)をAdvanced ChemTech社製ACT90型固相合成機の500ml反応槽内に入れ、一旦この樹脂をDMF等で洗浄後(工程1)、25%Pip(ピペリジン)で処理(3分×1回及び15分×1回)してFmoc基を切断後(工程2)、再びDMF等で樹脂を洗浄し(工程1)、Pipを除去した。この反応槽内に、Fmoc-Asn(Trt)-OH 24.46gとHOBT(1−ヒドロキシベンゾトリアゾール)6.28gをNMP(N−メチルピロリジノン)200mlに溶解した溶液を加え、更にDIPCI(N,N’−ジイソプロピルカルボジイミド)6.3mlを加えて 30分間室温撹拌を行った(工程3)。30分後、NMPで樹脂を洗浄し(工程4)、Fmoc-Asn(Trt)-OH 24.46gとHOBT 6.28gとDIPCI 6.3mlを用いて再度カップリング反応を行い(工程5)、Fmoc-Asn(Trt)-Leu-Alko樹脂を合成した。その後、工程2の脱保護操作を行い、H-Asn(Trt)-Leu-Alko-樹脂とした。次いで、工程1の洗浄操作を行い、Fmoc-Met-OH 15.23g、Fmoc-Gln(Trt)-OH 25.04g、Fmoc-Asn(Trt)-OH 24.46g、Fmoc-Trp(Boc)-OH 21.59g、Fmoc-Thr(tBu)-OH 16.3g、Fmoc-Tyr(tBu)-OH 18.84g、Fmoc-Cys(Trt)-OH 24.01gを順次加え、工程3のカップリング反応を行った。但しFmoc-Thr(tBu)-OHについてはカップリングを3回繰り返して行った後、得られた樹脂をDMFで洗浄後、25% Ac2O(無水酢酸)で15分×2回で未反応のアミノ基をキャッピングした。N末端のFmoc-Cys(Trt)-OHを縮合後、工程2の脱保護操作を行い、工程6の洗浄操作を実施し、H-Cys(Trt)- Tyr(tBu)-Thr(tBu)-Trp(Boc) -Asn(Trt)- Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resinを得た。上記合成工程の概要を表に示す。
<合成工程>
【0069】
【表2】
【0070】
2. 保護ペプチド樹脂の脱保護
上記操作によって得られた保護ペプチド樹脂(H-Cys(Trt)-Tyr(tBu)-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resin 10.0gにトリフルオロ酢酸/エタンジチオール/水/トリイソプロピルシラン(94/2.5/2.5/1)混合液100mlを加え、室温で4時間攪拌した。反応液を氷冷下t−ブチルメチルエーテル 625mlに加え、15分間攪拌後不溶物をグラスフィルターで濾取した。濾上物を約100mlのt−ブチルメチルエーテルで5回洗浄し、洗浄後の濾上物を6M 塩酸グアニジン水溶液 1Lで抽出することにより粗ペプチド溶液を得た。
【0071】
3. 粗ペプチドの精製
得られた粗ペプチド溶液を逆相液体クロマトグラフィーにより精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 5cmΦ×50cmL, 15-30μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:60ml/min
検出:UV220nm
水浴で50℃に保温したカラムを2液濃度10%で平衡化させた後粗ペプチド溶液をチャージした。2液濃度10%で30分間送液した後40分間かけて2液濃度を20%に上昇させ更に360分間かけて2液濃度を40%に上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド(H-Cys-Tyr-Thr-Trp-Asn-Gln-Met-Asn-Leu-OH(配列番号:2) 1.50gを得た。
【0072】
・アミノ酸分析
加水分解:1%フェノール/6N塩酸水、110℃ 10時間
分析法:ニンヒドリン法
Asx:1.96(2) Thr:1.05(1) Glx:1.06(1) Met:1.05(1) * Leu:(1) Tyr:0.87(1)
*) Leu=基準アミノ酸 ( ) 内 理論値
・質量分析:LC-ESI/MS m/z =1173 [M+1]+ (理論値=1172.5)
・アミノ酸配列分析:N末2残基目 Tyrから、C末Leuまで順次確認した。
【0073】
製造例2
1. 保護ペプチド樹脂(H-Cys(Trt)-Met-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resinの合成
Fmoc-Leu-Alko-樹脂(ここに、Alkoはp−アルコキシベンジルアルコール)10g(渡辺化学製;0.81mmol/g)をAdvanced ChemTech社製ACT90型固相合成機の500ml反応槽内に入れ、一旦この樹脂をDMF等で洗浄後(工程1)、25%Pip(ピペリジン)で処理(3分×1回及び15分×1回)してFmoc基を切断後(工程2)、再びDMF等で樹脂を洗浄し(工程1)、Pipを除去した。この反応槽内に、Fmoc-Asn(Trt)-OH 24.17gとHOBT(1−ヒドロキシベンゾトリアゾール)6.2gをNMP(N−メチルピロリジノン)200mlに溶解した溶液を加え、更にDIPCI(N,N’−ジイソプロピルカルボジイミド)6.2mlを加えて 30分間室温撹拌を行った(工程3)。30分後、NMPで樹脂を洗浄し(工程4)、Fmoc-Asn(Trt)-OH 24.17g、HOBT 6.2g及びDIPCI 6.2mlを用いて再度カップリング反応を行い(工程5)、Fmoc-Asn(Trt)-Leu-Alko樹脂を合成した。その後、工程2の脱保護操作を行い、H-Asn(Trt)-Leu-Alko-樹脂とした。次いで、工程1の洗浄操作を行い、Fmoc-Met-OH 15.05g、Fmoc-Gln(Trt)-OH 24.73g、Fmoc-Asn(Trt)-OH 24.17g、Fmoc-Trp(Boc)-OH 21.33g、Fmoc-Thr(tBu)-OH 16.1g、Fmoc-Met-OH 15.05g、Fmoc-Cys(Trt)-OH 23.72gを順次加え、工程3のカップリング反応を行った。但しFmoc-Thr(tBu)-OHについてはカップリングを3回繰り返して行った後、得られた樹脂をDMFで洗浄後、25%Ac2O(無水酢酸)で15分×2回で未反応のアミノ基をキャッピングした。N末端のFmoc-Cys(Trt)-OHを縮合後、工程2の脱保護操作を行い、工程6の洗浄操作を実施し、H-Cys(Trt)-Met-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resinを得た。上記合成工程の概要は製造例1の表に記載された工程と同じである。
【0074】
2. 保護ペプチド樹脂の脱保護
上記操作によって得られた保護ペプチド樹脂(H-Cys(Trt)-Met-Thr(tBu)-Trp(Boc)-Asn(Trt)-Gln(Trt)-Met-Asn(Trt)-Leu-Alko-Resin 13.0gにトリフルオロ酢酸/エタンジチオール/水/トリイソプロピルシラン(94/2.5/2.5/1)混合液130mlを加え、室温で4時間攪拌した。反応液を氷冷下ジエチルエーテル800mlに加え、15分間攪拌後不溶物をグラスフィルターで濾取した。濾上物を約100mlのジエチルエーテルで5回洗浄し、洗浄後の濾上物を6M 塩酸グアニジン水溶液 1.3Lで抽出することにより粗ペプチド溶液を得た。
【0075】
3. 粗ペプチドの精製
得られた粗ペプチド溶液を逆相液体クロマトグラフィーにより精製した。
ポンプ:Shimazu製;LC-8A型
カラム:YMC ODS-A 5cmΦ×50cmL, 15-30μm
溶出液1:H2O/0.1%TFA
溶出液2:CH3CN/0.1%TFA
流速:60ml/min
検出:UV220nm
水浴で50℃に保温したカラムを2液濃度10%で平衡化させた後粗ペプチド溶液をチャージした。2液濃度10%で30分間送液した後40分間かけて2液濃度を20%に上昇させ更に360分間かけて2液濃度を40%に上昇させた。目的物を含む分画を集め、アセトニトリルを減圧留去し凍結乾燥することにより目的とするペプチド(H-Cys-Met-Thr-Trp-Asn-Gln-Met-Asn-Leu-OH(配列番号:1) 2.32gを得た。
【0076】
・アミノ酸分析
加水分解:4N メタンスルホン酸、110℃ 17時間
分析法:ニンヒドリン法
Asx:1.87(2) Thr:0.93(1) Glx:0.95(1) Met:1.72(2) *Leu:(1) Trp:0.80(1)
*) Leu=基準アミノ酸 ( ) 内 理論値
・質量分析:LC-ESI/MS m/z =1141 [M+1]+ (理論値=1140.5)
・アミノ酸配列分析:N末2残基目 Metから、C末Leuまで順次確認した。
【産業上の利用可能性】
【0077】
本発明のペプチド化合物等は、癌免疫療法剤の有効成分として、有用である。
【0078】
配列表フリーテキスト
配列番号1:ペプチド誘導体
配列番号2:ペプチド誘導体
配列番号3:ペプチド誘導体
配列番号4:ペプチド誘導体
配列番号5:ヘルパー合成ペプチド
【特許請求の範囲】
【請求項1】
式(1):
【化1】
〔式中、Xはチロシン残基又はメチオニン残基を表し、
Y及びZは、独立して、単結合又は1〜10残基のアミノ酸からなるペプチドの二価基を表し、
R1は水素原子又はアルキル基を表し、
R2は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3は水素原子、アルキル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4は水素原子、アルキル基、カルボキシ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、又は式(2):
【化2】
(式中、Wはアミノ酸残基を表す)
で表される基を表し、
mは1又は2を表し、
nは0〜2の整数を表す。但し、nが0を表す場合、R3は水素原子又はアルキル基を表す。〕
で表される化合物、又はその薬学上許容される塩に特異的に結合する抗体。
【請求項2】
式(1):
【化3】
〔式中、Xはチロシン残基又はメチオニン残基を表し、
Y及びZは、独立して、単結合又は1〜10残基のアミノ酸からなるペプチドの二価基を表し、
R1は水素原子又はアルキル基を表し、
R2は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3は水素原子、アルキル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4は水素原子、アルキル基、カルボキシ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、又は式(2):
【化4】
(式中、Wはアミノ酸残基を表す)
で表される基を表し、
mは1又は2を表し、
nは0〜2の整数を表す。但し、nが0を表す場合、R3は水素原子又はアルキル基を表す。〕
で表される化合物、又はその薬学上許容される塩とHLA−A24抗原との複合体が提示されている抗原提示細胞。
【請求項3】
式(1):
【化5】
〔式中、Xはチロシン残基又はメチオニン残基を表し、
Y及びZは、独立して、単結合又は1〜10残基のアミノ酸からなるペプチドの二価基を表し、
R1は水素原子又はアルキル基を表し、
R2は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3は水素原子、アルキル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4は水素原子、アルキル基、カルボキシ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、又は式(2):
【化6】
(式中、Wはアミノ酸残基を表す)
で表される基を表し、
mは1又は2を表し、
nは0〜2の整数を表す。但し、nが0を表す場合、R3は水素原子又はアルキル基を表す。〕
で表される化合物、又はその薬学上許容される塩により誘導されたCTL。
【請求項4】
式(1):
【化7】
〔式中、Xはチロシン残基又はメチオニン残基を表し、
Y及びZは、独立して、単結合又は1〜10残基のアミノ酸からなるペプチドの二価基を表し、
R1は水素原子又はアルキル基を表し、
R2は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3は水素原子、アルキル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4は水素原子、アルキル基、カルボキシ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、又は式(2):
【化8】
(式中、Wはアミノ酸残基を表す)
で表される基を表し、
mは1又は2を表し、
nは0〜2の整数を表す。但し、nが0を表す場合、R3は水素原子又はアルキル基を表す。〕
で表される化合物、又はその薬学上許容される塩とHLA−A24抗原との複合体を認識する、請求項3に記載のCTL。
【請求項5】
配列番号:1に記載のペプチドとHLA−A24抗原との複合体を認識する、請求項3に記載のCTL。
【請求項6】
請求項2に記載の抗原提示細胞又は請求項3〜5のいずれかに記載のCTL、及び薬学的に許容される担体を含有する医薬組成物。
【請求項7】
癌ワクチンとして使用される、請求項6に記載の医薬組成物。
【請求項8】
請求項2に記載の抗原提示細胞又は請求項3〜5のいずれかに記載のCTLの、癌ワクチンを製造するための使用。
【請求項9】
請求項2に記載の抗原提示細胞又は請求項3〜5のいずれかに記載のCTLを有効成分として含有する、癌免疫療法剤。
【請求項1】
式(1):
【化1】
〔式中、Xはチロシン残基又はメチオニン残基を表し、
Y及びZは、独立して、単結合又は1〜10残基のアミノ酸からなるペプチドの二価基を表し、
R1は水素原子又はアルキル基を表し、
R2は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3は水素原子、アルキル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4は水素原子、アルキル基、カルボキシ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、又は式(2):
【化2】
(式中、Wはアミノ酸残基を表す)
で表される基を表し、
mは1又は2を表し、
nは0〜2の整数を表す。但し、nが0を表す場合、R3は水素原子又はアルキル基を表す。〕
で表される化合物、又はその薬学上許容される塩に特異的に結合する抗体。
【請求項2】
式(1):
【化3】
〔式中、Xはチロシン残基又はメチオニン残基を表し、
Y及びZは、独立して、単結合又は1〜10残基のアミノ酸からなるペプチドの二価基を表し、
R1は水素原子又はアルキル基を表し、
R2は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3は水素原子、アルキル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4は水素原子、アルキル基、カルボキシ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、又は式(2):
【化4】
(式中、Wはアミノ酸残基を表す)
で表される基を表し、
mは1又は2を表し、
nは0〜2の整数を表す。但し、nが0を表す場合、R3は水素原子又はアルキル基を表す。〕
で表される化合物、又はその薬学上許容される塩とHLA−A24抗原との複合体が提示されている抗原提示細胞。
【請求項3】
式(1):
【化5】
〔式中、Xはチロシン残基又はメチオニン残基を表し、
Y及びZは、独立して、単結合又は1〜10残基のアミノ酸からなるペプチドの二価基を表し、
R1は水素原子又はアルキル基を表し、
R2は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3は水素原子、アルキル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4は水素原子、アルキル基、カルボキシ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、又は式(2):
【化6】
(式中、Wはアミノ酸残基を表す)
で表される基を表し、
mは1又は2を表し、
nは0〜2の整数を表す。但し、nが0を表す場合、R3は水素原子又はアルキル基を表す。〕
で表される化合物、又はその薬学上許容される塩により誘導されたCTL。
【請求項4】
式(1):
【化7】
〔式中、Xはチロシン残基又はメチオニン残基を表し、
Y及びZは、独立して、単結合又は1〜10残基のアミノ酸からなるペプチドの二価基を表し、
R1は水素原子又はアルキル基を表し、
R2は水酸基、アミノ基、アルキルアミノ基又はジアルキルアミノ基を表し、
R3は水素原子、アルキル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、又は置換もしくは無置換のアルキルカルボニルアミノ基を表し、
R4は水素原子、アルキル基、カルボキシ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、又は式(2):
【化8】
(式中、Wはアミノ酸残基を表す)
で表される基を表し、
mは1又は2を表し、
nは0〜2の整数を表す。但し、nが0を表す場合、R3は水素原子又はアルキル基を表す。〕
で表される化合物、又はその薬学上許容される塩とHLA−A24抗原との複合体を認識する、請求項3に記載のCTL。
【請求項5】
配列番号:1に記載のペプチドとHLA−A24抗原との複合体を認識する、請求項3に記載のCTL。
【請求項6】
請求項2に記載の抗原提示細胞又は請求項3〜5のいずれかに記載のCTL、及び薬学的に許容される担体を含有する医薬組成物。
【請求項7】
癌ワクチンとして使用される、請求項6に記載の医薬組成物。
【請求項8】
請求項2に記載の抗原提示細胞又は請求項3〜5のいずれかに記載のCTLの、癌ワクチンを製造するための使用。
【請求項9】
請求項2に記載の抗原提示細胞又は請求項3〜5のいずれかに記載のCTLを有効成分として含有する、癌免疫療法剤。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2012−158597(P2012−158597A)
【公開日】平成24年8月23日(2012.8.23)
【国際特許分類】
【出願番号】特願2012−77513(P2012−77513)
【出願日】平成24年3月29日(2012.3.29)
【分割の表示】特願2009−200003(P2009−200003)の分割
【原出願日】平成18年11月29日(2006.11.29)
【出願人】(505443953)株式会社癌免疫研究所 (8)
【出願人】(000003311)中外製薬株式会社 (228)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【Fターム(参考)】
【公開日】平成24年8月23日(2012.8.23)
【国際特許分類】
【出願日】平成24年3月29日(2012.3.29)
【分割の表示】特願2009−200003(P2009−200003)の分割
【原出願日】平成18年11月29日(2006.11.29)
【出願人】(505443953)株式会社癌免疫研究所 (8)
【出願人】(000003311)中外製薬株式会社 (228)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【Fターム(参考)】
[ Back to top ]