
新規メイタンシノイド、および抗体とのコンジュゲートを調製するための前記メイタンシノイドの使用
本発明は、式(I)の化合物(式中、ALKは、(C1−C6)アルキレン基であり、X1およびX2は、それぞれ独立して、以下の基:−CH=CH−、−CO−、−CONR−、−NRCO−、−COO−、−OCO−、−OCONR−、−NRCOO−、−NRCONR’−、−NR−、−S(O)n(n=0、1または2)またはO−のうちの1つであり、RおよびR’は、独立して、Hまたは(C1−C6)アルキル基であり、Iは、1から40、好ましくは1から20、より好ましくは1から10の整数であり、jは、X2が−CH=CH−の場合、1に相当する整数であり、X2が−CH=CH−でない場合、2に相当する整数であり、Zbは、単一の結合、−O−または−NH−であり、RbはHまたは(C1−C6)アルキル、(C3−C7)シクロアルキル、アリール、ヘテロアリールまたは(C3−C7)ヘテロシクロアルキル基であり、またはZbは、一重結合であり、RbはHalである。)に関する。本発明は、腫瘍細胞に対して親和性を有する、抗体とのコンジュゲートを調製するための前記メイタンシノイドの使用に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規メイタンシノイド、および抗体とのコンジュゲートを調製するための前記メイタンシノイドの使用に関する。本発明はまた、前記メイタンシノイドおよび前記コンジュゲートを含む組成物に関する。
【背景技術】
【0002】
モノクローナル抗体−薬物コンジュゲートを用いて、腫瘍細胞を特異的に対象とする試みについて多くの論文が出現した(Selaら、Immunoconjugates、189−216頁(C.Vogel編.1987年);Ghoseら、Targeted Drugs、1−22頁(E.Goldberg編、1983年);Dienerら、Antibody mediated delivery systems、1−23頁(J.Rodwell編、1988年);Pieterszら、Antibody mediated delivery systems、25−53頁(J.Rodwell編、1988年);Bumolら、Antibody mediated delivery systems、55−79頁(J.Rodwell編、1988年)。また以下も参照されたい:Monneret Cら、Bulletin du Cancer、2000年、87(11)、829−38頁;Ricart A.D.ら、Nature Clinical Practice Oncology 2007年、4、245−255頁;Singh R.およびRickson H.K.、Therapeutic Antibodies:Methods and Protocols、2009年、525、445−467頁。タキサン誘導体(WO06061258)、レプトマイシン誘導体(WO07144709)、CC−1065および類似体(WO2007102069)など、またはメトトレキサート、ダウノルビシン、ドキソルビシン、ビンクリスチン、ビンブラスチン、メルファラン、マイトマイシンC、クロランブシルなどの異なるファミリーの細胞毒性薬が、抗体との結合に使用されてきた。
【0003】
腫瘍細胞に対する親和性を有する標的抗体の使用は、細胞毒性薬を腫瘍細胞の近くに直接または腫瘍細胞内に直接送達することを可能にし、したがって、細胞毒性薬に通常伴う副作用を最小限に抑えながら、細胞毒性薬の効率を増加させる。
【0004】
メイタンシノイドは、キャストアフリカンシュラブ、メイテナス・セラタ(Maytenus serrata)から単離した天然物であるメイタンシンから誘導される細胞毒性薬である(US3896111)。多くのメイタンシノイドが調製されてきた。以下を参照:US4151042;J.Med.Chem.1978年、21、31−37頁;Nature、1977年、270、721−722頁、Chem.Pharm.Bull.1984年、3441−3451頁;US4248870;US4137230;Chem.Bull.1984年、3441頁。
【0005】
従来技術
US5208020、US5416064、およびR.V.J.Chari、31 Advanced Drug Delivery Reviews、89−104頁(1998年)は、L−DM1(A)またはL−DM4(Α’):
【0006】
【化1】
などのメイタンシノイドのコンジュゲートについて記載している。
【0007】
メイタンシノイドのコンジュゲートは、EP0425235およびWO2004/103272で記載されている。EP0425235において、式(B1)、(B2)または(B3)の以下のメイタンシノイドが記載されている:
【0008】
【化2】
(式中、Z0、Z1またはZ2は、HまたはSRを表す。)。
【0009】
WO2004/103272では、式(C)のメイタンシノイドが記載されている:
【0010】
【化3】
(式中、Y’は、
(CR7CR8)i(CR9=CR10)pC≡CqAr(CR5CR6)mDu(CR11=CR12)r(C≡C)sBt(CR3CR4)uCR1R2SZを表し、A、BおよびDは、場合によって置換されているシクロアルキル、シクロアルケニル、ヘテロアリールまたはヘテロシクロアルキル基を表す。)。
【0011】
WO03/068144では、式(D)の化合物が記載されている。
【0012】
【化4】
(式中、Zは、細胞毒性薬であり、Qは、R2COO、R2R3NCOO、R2OCOO、R2O、R2CONR3、R2R3N、R2OCONR3またはSである。R2は、SCR4R5R6である。)。Zは、以下のメイタンシノイド誘導体の中から選択されるメイタンシノイド誘導体であってよい。
【0013】
【化5】
【0014】
さらに具体的には、以下の化合物(D)が開示されている:
【0015】
【化6】
したがって化合物(D)は、ペグ化リンカーの中に内部ジスルフィド結合を含有している。
【0016】
Immunogen Inc.はまた、2008年12月8日、GenevaのEuropean Antibody Congressにおいて、式(E)のコンジュゲートを開示し、
【0017】
【化7】
Genevaで2008年10月に開催された、第20回シンポジウムEORTC−NCI−AACRにおいて、式(Ε’)のコンジュゲートを開示した。
【0018】
【化8】
【先行技術文献】
【特許文献】
【0019】
【特許文献1】国際公開第06061258号
【特許文献2】国際公開第07144709号
【特許文献3】国際公開第2007102069号
【特許文献4】米国特許第4151042号明細書
【特許文献5】米国特許第4248870号明細書
【特許文献6】米国特許第4137230号明細書
【特許文献7】米国特許第5,208,020号明細書
【特許文献8】米国特許第5,416,064号明細書
【特許文献9】欧州特許第0425235号明細書
【特許文献10】国際公開第2004/103272号
【特許文献11】国際公開第03/068144号
【非特許文献】
【0020】
【非特許文献1】Selaら、Immunoconjugates、189−216頁(C.Vogel編.1987年)
【非特許文献2】Ghoseら、Targeted Drugs、1−22頁(E.Goldberg編、1983年)
【非特許文献3】Dienerら、Antibody mediated delivery systems、1−23頁(J.Rodwell編、1988年)
【非特許文献4】Pieterszら、Antibody mediated delivery systems、25−53頁(J.Rodwell編、1988年)
【非特許文献5】Bumolら、Antibody mediated delivery systems、55−79頁(J.Rodwell編、1988年)
【非特許文献6】Monneret Cら、Bulletin du Cancer、2000年、87(11)、829−38頁
【非特許文献7】Ricart A.D.ら、Nature Clinical Practice Oncology 2007年、4、245−255頁
【非特許文献8】Singh R.およびRickson H.K.、Therapeutic Antibodies:Methods and Protocols、2009年、525、445−467頁
【非特許文献9】J.Med.Chem.1978年、21、31−37頁
【非特許文献10】Nature、1977年、270、721−722頁
【非特許文献11】Chem.Pharm.Bull.1984年、3441−3451頁
【非特許文献12】Chem.Bull.1984年、3441頁
【非特許文献13】R.V.J.Chari、31 Advanced Drug Delivery Reviews、89−104頁(1998年)
【発明の概要】
【課題を解決するための手段】
【0021】
定義
「アルキル」は、1から20個の炭素原子を鎖内に有する直鎖もしくは分枝、または3から10個の炭素原子を有する環状であってよい脂肪族の炭化水素基を意味する。好ましいアルキル基は、鎖内に1から12個の炭素原子を有する。代表的アルキル基として、メチル、エチル、n−プロピル、i−プロピル、2,2−ジメチルプロピル、n−ブチル、t−ブチル、n−ペンチル、3−ペンチル、オクチル、ノニル、デシルが挙げられる。
「シクロアルキル」は、3から10個の炭素原子を有する環式の脂肪族炭化水素基を意味する。好ましいシクロアルキル基は、環状の鎖内に3から8個の炭素原子を有する。代表的なシクロアルキル基として、シクロプロピル、シクロブチル、シクロペンチルおよびシクロヘキシルが挙げられる。
「アリール」は、6から14個の炭素原子、好ましくは6から10個の炭素原子の芳香族単環式または多環式炭化水素環系を意味する。代表的なアリール基として、フェニルまたはナフチルが挙げられる。
「ヘテロアリール」は、不飽和の安定した3から14員、好ましくは5から10員の単環式、二環式または多環式の環を意味し、この環の少なくとも1つのメンバーはヘテロ原子である。通常、ヘテロ原子は、これらに限定されないが、酸素、窒素、硫黄、セレンまたはリン原子である。ヘテロ原子は、酸素、窒素または硫黄原子が好ましい。代表的なヘテロアリール基として、ピリジル、ピロリル、チエニル、フリル、ピリミジニルおよびトリアゾリルが挙げられる。
「ヘテロシクロアルキル」は、少なくとも1個のヘテロ原子を含有するシクロアルキル基を意味し、この環の少なくとも1つのメンバーはヘテロ原子である。
「アルコキシ」は、−O−アルキル基を意味し、アルキルは上記のように定義される。
「アルコキシルオキシ」は、−O−CO−アルキル基を意味し、アルキルは上記のように定義される。
「アルキレン」は、直鎖または分枝のアルカンから2個の水素原子を除去することにより形成される一般式−CmH2m−のアルキル基を意味する。代表的アルキレン基として、メチレン(−CH2−)、エチレン(−CH2CH2−)、プロピレン(−CH2CH2CH2−)、ブチレン(−CH2CH2CH2CH2−)、イソブチレン
【0022】
【化9】
、ヘキシレン(−CH2CH2CH2CH2CH2CH2−)が挙げられる。直鎖アルキレン基は、具体的には式−(CH2)m−(式中、mは、1から20の整数である。)で表すことができる。
「EphA2受容体」は、Eph受容体ファミリーに属するチロシンキナーゼ(Pasquale、E.B.ら、2005年、Nature Reviews Mol.Cell Biol.、6、462−475頁で概説されている)を指し、例えばGenbank受託番号NM_004431(ヒトEphA2)、NM_010139(マウスEphA2)またはNXM_345596(ラットEphA2)などのアミノ配列を含む。ヒトEphA2は、好ましいEph受容体である。「EphA2リガンド」という用語は、本明細書で使用する場合、EphA2受容体に結合し、場合によってはこれを活性化する(例えば、この自己リン酸化を刺激する)タンパク質を指す。本明細書中の好ましいEphA2リガンドは、「エフリンA1」であり、これは、EphA2受容体に結合し、例えばGenbank受託番号NM_004428(ヒトエフリンA1)などのようなアミノ配列を含む。
「ポリクローナル抗体」とは、1つ以上の他の、非同一の抗体の間でまたはそれらの存在下で産生された抗体である。一般的に、多クローン性抗体は、非同一の抗体を産生する他のいくつかのB−リンパ球の存在下でB−リンパ球から産生される。通常、多クローン性抗体は免疫された動物から直接得る。
「モノクローナル抗体」とは、実質的に同種の抗体の集団、すなわち微量で存在することがある、自然発生で起こり得る変異を除いて、基本的に同一であるこの集団を形成する抗体から得られる抗体である。これらの抗体は、単一のエピトープを対象とし、したがって極めて特異的である。
「裸の抗体」とは、メイタンシノイドと結合していない抗体である。
「エピトープ」とは、抗体が結合する抗原上の部位である。これは、抗原タンパク質のフォールディングにより近接近させられる隣接する残基または隣接しない残基で形成することができる。隣接するアミノ酸で形成されるエピトープは通常、非変性溶媒へ曝露されたまま保持されるが、隣接しないアミノ酸で形成されるエピトープは通常、前記曝露下で失われる。
【図面の簡単な説明】
【0023】
【図1】実施例1の脱グリコシル化コンジュゲートの逆畳み込み後のHRMSスペクトルを示す図である。
【図2】実施例2の脱グリコシル化コンジュゲートの逆畳み込み後のHRMSスペクトルを示す図である。
【図3】実施例3の脱グリコシル化コンジュゲートの逆畳み込み後のHRMSスペクトルを示す図である。
【図4】実施例4の脱グリコシル化コンジュゲートの逆畳み込み後のHRMSスペクトルを示す図である。
【図5】実施例5の脱グリコシル化コンジュゲートの逆畳み込み後のHRMSスペクトルを示す図である。
【図6A】配列を示す記号である。
【図6B】配列を示す記号である。
【図6C】配列を示す記号である。
【図7】様々なDARにおける、hu2H11R35R74コンジュゲートに対するPKパラメータ:HGS中のコンジュゲート20mg/kgをオスのCD−1マウス(n=4)に、単回投与の静脈内投与をした後、(AUC(O−inf);左)への曝露およびいくつかのコンジュゲートのクリアランス(CI;右)を、DARの関数として棒グラフにより説明するグラフである。グラフにおいて、2H11−DM4(底部)は、hu2H11R35R74−コンジュゲートを指す。
【発明を実施するための形態】
【0024】
これらの図は、各コンジュゲートについて、0から10のメイタンシノイド(D0:メイタンシノイドなし;Dx:x個のメイタンシノイド)を保持するコンジュゲートの分布が存在することを示している。
【0025】
新規メイタンシノイド
本発明は、式(I)の化合物
【0026】
【化10】
(式中、
ALKは、(C1−C6)アルキレン基であり、
X1およびX2は、それぞれ独立して、以下の基:−CH=CH−、−CO−、−CONR−、−NRCO−、−COO−、−OCO−、−OCONR−、−NRCOO−、−NRCONR’−、−NR−、−S(O)n(n=0、1または2)または−O−のうちの1つであり;
RおよびR’は、独立して、Hまたは(C1−C6)アルキル基であり;
iは、1から40、好ましくは1から20、より好ましくは1から10の整数であり、
jは、X2が−CH=CH−の場合、1に対応する整数であり、X2が−CH=CH−でない場合、2に対応する整数であり、
Zbは、単一結合、−O−または−NH−であり、Rbは、Hまたは(C1−C6)アルキル、(C3−C7)シクロアルキル、アリール、ヘテロアリールもしくは(C3−C7)ヘテロシクロアルキル基であり、またはZbは、一重結合であり、RbはHalである。)に関する。
【0027】
より具体的には、X2は、−CH=CH−または−CONR−であり、CO基は、−X1−ALK−基に連結し、Rは、Hまたは(C1−C6)アルキル基である。より具体的には、−X1−ALK−は−S−CH2−である。より具体的には、lは、3、4、5、6、7、8、9または10である。
【0028】
式(II)の化合物を区別することができる。
【0029】
【化11】
【0030】
式(II)は、以下の化合物を、さらに正確には網羅する。
【0031】
【化12】
【0032】
化合物は、塩基もしくは塩の形態で、または前記塩基もしくは前記塩の溶媒和物もしくは水和物の形態で存在することができる。
【0033】
本発明の化合物は、抗体上に存在する反応性化学基(GCR2)に対して反応性のある、反応性化学基−C(=O)ZbRb(GCR1)を含む。GCR1とGCR2の間の反応により、共有結合を介して抗体上に細胞毒性薬が結合できるようになる。したがって、この化合物は、抗体に結合する傾向がある。より具体的には、ZbはOである。このような場合、GCR1は、カルボン酸官能基(Rb=H)またはエステル官能基である。より具体的には、−C(=O)ZbRbは、−COOH、−COO(C1−C6)アルキル、例えば−COOCH3または−COOCH2CH=CH2である。エステル官能基の中でも、抗体(リジン基など)のアミノ基に対して優れた反応性を示す「活性化された」ものが好ましい。例えば活性化エステルは、以下のエステルであってよい:
【0034】
【化13】
または基
【0035】
【化14】
(式中、GIは、少なくとも1つの誘導性の基、例えば−NO2または−Hal、例えば−Fなどを表す。)。このような活性化エステルの例は、以下のエステルである。
【0036】
【化15】
別の−C(=0)ZbRbは、
【0037】
【化16】
である。
【0038】
GCR2は、例えば抗体の表面のリジン残基の側面上のリジン、ヒンジ領域のサッカライド基または鎖内S−S結合の還元後のシステインのチオール基により生じるε−アミノ基であってよい(Garnett M.C.ら、Advanced Drug Delivery Reviews、2001年、53、171−216頁)。さらに最近になって、新規な手法は、変異によりシステインを導入すること(Junutula J.R.ら、Nature Biotechnology、2008年、26、925−932頁;WO09026274)または新しいタイプのタンパク質の化学的性質の開発を可能とする非天然アミノ酸を導入すること(de Graaf A.J.ら、Bioconjugate Chem.2009年、2009年2月3日、(Review);DOI:10.1021/bc800294a;WO2006/069246およびChin J.W.ら、JACS、2002年、124、9026−9027頁(technology ReCode(登録商標)))を目標とした。
【0039】
本発明の化合物は、式:
【0040】
【化17】
の少なくとも1つのメイタンシノイドフラグメントに共有結合で付加しているコンジュゲートを調製するために使用することができる。
【0041】
したがって、式(I)の化合物を、メイタンシノイドフラグメントが抗体に共有結合で連結しているコンジュゲートを調製するために使用することができる。
【0042】
式(I)の化合物を調製するための一般的スキーム
式(I)の化合物をスキーム1に従い調製することができる:
【0043】
【化18】
【0044】
中間体P1は、中間体P2を含有するPEGに付加している反応性基RG2と反応することによって、X1を形成することができるRG1反応性基を含有する。例えば、X1=Sの形成は、DIEAなどの塩基の存在下、求核置換によるP1とRG1=−SHの反応ならびにP2とRG2=−Brの反応を介して行うことができる。この反応の例は、実施例1.2において示されている。
【0045】
RG1=−SHでのP1の例は、L−DM1およびL−DM4であり、またEP1313738の化合物11a、c、d、gでもある。
L−DM1:ALK−SH=−CH2CH2−SH;
L−DM4:ALK−SH=−CH2CH2CMe2−SH;
11a:ALK−SH=−CH2−SH;
11c:ALK−SH=−CH2CH2CH2−SH;
11d:ALK−SH=−CH2CH2CH2CH2−SH;
11g:ALK−SH=−CHMe−CH2−SH
【0046】
反応の他の例が、表Iに示されている。
【0047】
【表1】
【0048】
いくつかの式(I)の化合物については、P1とP2を反応させた後で、別の−ZbRb基の少なくとも1つを変換後、所望する最終の−ZbRb基を得てもよい。変換−ZbRb=−O−アリル→
【0049】
【化19】
と共に、スキーム1’で一例を示す。
【0050】
【化20】
【0051】
同様に、P1とP2との間の反応の前に、−ZbRb基を保持する中間体に対して少なくとも1つの変換を使用することもできる。
【0052】
別の例は、例えばSOCl2などのアシル化剤を必要とする、変換−ZbRb=−OH→ZbRb=Halである。
【0053】
P1の調製
メイタンシノールから開始して、スキーム2に従いP1を調製することができる。
【0054】
【化21】
【0055】
メイタンシノールを、反応性アシル基−COOZ(式中、Zは、Hまたはハロゲン原子である。)を含有する中間体P3と、エステル化反応により反応させる。この反応は、WO2004/103272の図3a−dに記載され、WO2007/021674にも記載されている。ZがHの場合、酸官能基の反応性を強化するカップリング剤の助けを借りてエステル化を行うことができる。
【0056】
P2の調製
P2を調製するための出発物質は、市販のPEG化合物または当業者に既知の少なくとも1つの化学反応を介して、前記市販のPEG化合物を用いて調製することができるPEG化合物である。PEG化合物は、例えばJenKem Technology USA Inc.、2033 W.McDermott Dr.Suite 320 #188、Allen、TX 75013−4675、USAから、市販されている。
【0057】
例えば、P2(式中、X2=−CONR−であり、RG2=Hal(P2=Hal−ALK−CONR−CH2CH2(OCH2CH2)i−COZbRb))の調製が、市販の化合物HOOCCH2CH2(OCH2CH2)i−OCH2CH2NH2を用いて以下に記載されている:
R=H
【0058】
【化22】
【0059】
ステップ(i):アミド結合の形成および酸性基の活性化;2つのステップを、DCMなどの極性の非プロトン性溶媒中で別々に行う:アミン基と、ハロゲノアルカン酸N−ヒドロキシスクシンイミジンエステル(例えば、ハロゲノ酢酸)とを反応させ、次いでDICなどのカップリング剤をその場で添加する。
R≠H
【0060】
【化23】
【0061】
ステップ(ii):エステルの形態でのカルボン酸の保護およびトリフルオロアセトアミドの形態でのアミンの保護;この反応は、DCMなどの極性の非プロトン性溶媒中、2つの別々のステップで行う:メタノールを用いて、トリメチルシリルジアゾメタンでの処理により酸を保護し、次いでTFAAおよびTEAの添加によりアミンを保護する。
【0062】
ステップ(iii):アミンのアルキル化およびエステルのアルカリ加水分解;この反応は、THFなどの極性の非プロトン性溶媒中、2つの別々のステップで行う:ハロゲン化アルキルRHalなどの脱離基を保持する反応体の存在下、NaHでの処理によりアミンをアルキル化し、水中でLiOHを添加する。
【0063】
ステップ(iv):ステップ(iii)に続いて、ステップ(i)の反応(式中、R=H)を行う。
【0064】
同様に、P2(式中、X2=−CH=CH−およびRG2=Hal(P2=Hal−CH2−CH=CH−CH2(OCH2CH2)i−COZbRb))の調製が、市販の化合物Hal−CH2CH=CHCH2(OCH2CH2)i−COOtBuを用いて以下に記載されている:
【0065】
【化24】
【0066】
コンジュゲートの調製方法
コンジュゲートは、以下のステップを含む方法により得ることができる:
(i)場合によって緩衝させた抗体水溶液を式(I)の化合物の溶液と接触させるステップ、
(ii)次いで、反応していない試薬および溶液中に存在し得るいかなる凝集体から(i)において形成されたコンジュゲートを場合によって分離するステップ。
【0067】
細胞結合剤の水溶液は、少なくとも1つの緩衝液、例えばリン酸カリウムまたはN−2−ヒドロキシエチルピペラジン−N’−2−エタンスルホン酸(Hepes緩衝液)など、または緩衝液混合物、例えば以下の実施例において開示されている緩衝液Aなどを用いて緩衝させることができる。緩衝液は、抗体の性質に依存する。式(I)の化合物は、有機極性溶媒(または極性溶媒の混合物)、例えばDMSOまたはDMA中で溶液である。
【0068】
反応の温度は通常、20から40℃の範囲で変動する。反応時間は、1から24時間の範囲で変動し得る。抗体と細胞毒性薬との間の反応は、屈折率検出器および/またはUV検出器を備えたサイズ排除クロマトグラフィー(SEC)でモニターすることができる。コンジュゲートの収率が低すぎる場合、反応時間を延長することができ、および/または式(I)の化合物を添加することができる。
【0069】
当業者であれば、多数の異なるクロマトグラフィー法を使用することによって、ステップ(ii)の分離を実施することができる:コンジュゲートは、例えばSEC、吸着クロマトグラフィー(例えばイオン交換クロマトグラフィー、IECなど)、疎水性相互作用クロマトグラフィー(HIC)、アフィニティクロマトグラフィー、混合担持クロマトグラフィー、例えばハイドロキシアパタイトクロマトグラフィーなど、または高速液体クロマトグラフィー(HPLC)により精製することができる。透析法または膜分離法による精製もまた使用することができる。
【0070】
使用することができる方法の例が、実施例1に記載されている。
【0071】
本明細書で使用する場合、「凝集体」という用語は、2つ以上の抗体の間に形成し得る連結を意味し、前記抗体は、改質されているまたは改質されていない。凝集体は、多数のパラメータ、例えば、溶液中の高濃度の抗体、溶液のpH、高い剪断力、結合したダイマーの数およびこれらの疎水性の性質、温度などの影響下で形成し得る(Wang & Gosh、2008年、J.Membrane Sci.、318:311−316頁および本明細書中に引用された参考文献を参照);これらパラメータのうちのいくつかの相対的影響は、明確に確立されていないことに注意されたい。タンパク質および抗体の場合、当業者であれば、Cromwellら(2006年、AAPS Journal、8(3):E572−E579)を参照することになる。凝集体中の含量は、当業者に周知の技法、例えばSEC(Walterら、1993年、Anal.Biochem.、212(2):469−480頁を参照)などを用いて求めることができる。
【0072】
ステップ(i)または(ii)の後、コンジュゲート含有溶液に、限外ろ過法および/または膜分離法の追加のステップ(iii)を施すことができる。
【0073】
コンジュゲートは、これらのステップの終わりに水溶液として回収される。
【0074】
抗体
「抗体」という用語は、もっとも広範な意味において本明細書中で使用され、任意のイソタイプ、例えばIgG、IgM、IgA、IgDおよびIgEなどのモノクローナル抗体(全長モノクローナル抗体を含む)、多クローン性抗体、多特異的抗体、キメラ抗体および抗体フラグメントを具体的に網羅する。特定の抗原に反応する抗体は、組換え型の方法、例えばファージまたは類似ベクターにおける組換え型抗体のライブラリーの選択などによって、または抗原もしくは抗原をコードする核酸で動物に免疫性を与えることによって生成することができる。
【0075】
典型的な抗体は、ジスルフィド結合で結合されている2つの同一の重鎖および2つの同一の軽鎖から構成される。各重鎖および軽鎖は、一定の領域および可変領域を含有する。本明細書で使用する場合、「VH」または「VH」は、抗体のイムノグロブリン重鎖の可変領域を指し、これには、Fv、scFv、dsFv、Fab、Fab’またはF(ab’)2フラグメントの重鎖が含まれる。「VL」または「VL」の言及は、抗体のイムノグロブリン軽鎖の可変領域を指し、これには、Fv、scFv、dsFv、Fab、Fab’またはF(ab’)2フラグメントの軽鎖が含まれる。各可変領域は、「相補性決定領域」(「CDR」)または「超可変領域」と呼ばれる3つのセグメントを含有し、これらの領域は、抗原のエピトープを結合することに主に関与している。これらは通常、N−末端から連番で、CDR1、CDR2およびCDR3と呼ばれる。可変領域のさらに高度に保護された部分は、「フレームワーク領域」(「FR」)と呼ばれる。ネイティブの重鎖および軽鎖の可変ドメインは、それぞれ4つのFR領域を含み、これらの領域は、βシート配置を広く採用し、3つのCDRで結合されており、これら3つのCDRは、βシート構造を結合しているループを形成し、場合によってβシート構造の一部を形成する。各鎖内のCDRは、FR領域により近接近で一緒にまとめて保持され、他の鎖からのCDRにより、抗体の抗原結合部位の形成に寄与する(Kabatら、Sequences of Proteins of Immunological Interest、第5版、National Institute of Health、Bethesda、MD、1991年を参照)。
【0076】
抗体(さらなる詳細については、Janewayら、「Immunobiology」、第5版、2001年、Garland Publishing、New Yorkを参照)は、例えばWO04043344、WO08010101、WO08047242またはWO05009369(抗−CA6)などに記述されている抗体の中から選択することができる。
【0077】
クラスAのEphレセプターファミリーメンバー、例えば好ましくはヒトのEphA2受容体などを認識し、前記受容体のアンタゴニストとして機能する抗体またはこのフラグメントもまた考慮することができる。この抗体には、いかなるアゴニスト活性もない。抗体またはこのエピトープ結合フラグメントは、請求項12から15に記載ものであってよい。
【0078】
ヒト化抗体またはこのエピトープ結合フラグメントは、EphA2受容体を発現する癌細胞の増殖を阻害する追加の能力を有することが好ましい。ヒト化抗体またはこのエピトープ結合フラグメントは、EphA2受容体を発現する転移性癌細胞の遊走を阻害する追加の能力を有することが好ましい。
【0079】
ヒト化抗体は、2H11R35R74抗体またはこのエピトープ結合フラグメントをヒト化することができる。ヒト化抗体は、hu53.2H11(WO2008/010101)をコードしているポリヌクレオチド配列の部位特異的突然変異誘発により得ることができる。好ましくは、再表面化またはヒト化した型の2H11R35R74抗体が提供され、この抗体またはこのフラグメントの表面曝露された残基が、軽鎖と重鎖の両方において置き換えられることによって、既知のヒト抗体の表面にさらによく類似する。ヒト化2H11R35R74抗体またはこのエピトープ結合フラグメントは、改善された特性を有する。例えば、ヒト化2H11R35R74抗体またはこのエピトープ結合フラグメントは、EphA2受容体を特異的に認識する。より好ましくは、ヒト化2H11R35R74抗体またはこのエピトープ結合フラグメントは、EphA2受容体発現細胞の増殖を阻害する追加の能力を有する。
【0080】
ヒト化型2H11R35R74抗体はまた、軽鎖と重鎖の両可変領域のこれらのそれぞれのアミノ酸配列、軽鎖と重鎖の可変領域に対する遺伝子のDNA配列、CDRの同定、これら表面アミノ酸の同定、ならびに組換え型の形態でのこれらの発現のための手段の開示に関して本明細書中で完全に特徴づけられている。しかし範囲はこれら配列を含む抗体およびフラグメントに限定されない。代わりに、EphA2受容体に特異的に結合するすべての抗体およびフラグメントも考慮される。EphA2受容体に特異的に結合する抗体およびフラグメントは、受容体の生物活性をアンタゴナイズすることが好ましい。このような抗体はさらに、アゴニスト活性が実質的にはないことがより好ましい。したがって、抗体およびエピトープ結合抗体フラグメントは、これらの骨格、CDR、ならびに/または軽鎖および重鎖のアミノ酸配列が2H11R35R74抗体またはこのヒト化誘導体とは異なってもよく、これらは依然として本発明の範囲内に含まれる。
【0081】
2H11R35R74抗体のCDRは、モデリングにより同定され、これらの分子構造が予測されてきた。ここでもまたCDRはエピトープ認知に対して重要であるが、これらは本発明の抗体およびフラグメントには必須ではない。したがって、例えば本発明の抗体の親和性の成熟により生じる改善された特性を有する抗体およびフラグメントが提供される。
【0082】
53.2H11が誘導される可能性のある、マウス軽鎖IgVkおよびJK生殖細胞系列遺伝子および重鎖IgVhおよびJh生殖細胞系列遺伝子が同定され、WO2008/010101で開示された。前記生殖細胞系列の配列の受託番号は、それぞれMMU231196およびAF303833である。このような生殖細胞系列遺伝子配列は、CDR内のものを含めて、抗体内の体細胞変異を同定するのに有用である。
【0083】
2H11R35R74抗体の重鎖および軽鎖の可変領域の配列ならびにこれらのCDRの配列は本出願の中に記載されている。このような情報は、ヒト化型の2H11R35R74抗体を産生するために使用することができる。本発明のヒト化2H11R35R74抗体をhu53.2H11の部位特異的突然変異誘発により得ることもできる。これらヒト化抗−EphA2抗体またはこれらの誘導体も本発明のコンジュゲートの細胞結合剤として使用することができる。
【0084】
したがって、一実施形態では、本発明は、SEQ ID NO:1、2、3、4、5、6からなる群から選択されるアミノ酸配列を有する1つ以上のCDRを含むヒト化抗体またはこのエピトープ結合フラグメントを提供する。好ましい実施形態において、本発明のヒト化抗体は、少なくとも1つの重鎖と少なくとも1つの軽鎖とを含み、前記重鎖は、SEQ ID NO:1、2および3で表されるアミノ酸配列を有する3つの連続したCDRを含み、前記軽鎖は、SEQ ID NO:4、5および6で表されるアミノ酸配列を有する3つの連続したCDRを含む。
【0085】
ヒト化2H11R35R74抗体またはこのフラグメントは、SEQ ID NO.12からなるアミノ酸配列を有するVHを含むことが好ましい。SEQ ID NO14からなるアミノ酸配列を有するVLを含むヒト化2H11R35R74抗体またはこのフラグメントもまた好ましい。ヒト化2H11R35R74抗体は、少なくとも1つの重鎖および少なくとも1つの軽鎖を含み、前記重鎖は、SEQ ID NO:1、2および3で表されるアミノ酸配列を有する3つの連続したCDRを含み、前記軽鎖は、SEQ ID NO:4、5および6で表されるアミノ酸配列を有する3つの連続したCDRを含み、前記重鎖は、SEQ ID NO.12からなるアミノ酸配列を有し、前記軽鎖は、SEQ ID NO.14からなるアミノ酸配列を有することが好ましい。
【0086】
コンジュゲート
コンジュゲートは、抗体に共有結合で付加しているメイタンシノイドの1から10個の分子を全般的に含む(いわゆる「薬物/抗体比」または「DAR」)。この数は、結合に対して使用される実験条件(例えば、メイタンシノイドと、抗体、反応時間、溶媒の性質および(存在する場合)共溶媒との比)と共に、抗体の性質および使用されるメイタンシノイドにより異なり得る。したがって、抗体とメイタンシノイドの接触により、異なる薬物/抗体比により互いに異なるものとなるいくつかのコンジュゲート、場合によって裸の抗体、場合によって凝集体を含む混合物が生成される。したがって求められるDARは、平均値である。
【0087】
したがって本発明はまた、請求項1から8の一項で定義された、抗体に共有結合で付加している1つ以上の化合物を含むコンジュゲートに関係している。この付加は、アミド結合を介しているのが好ましい。抗体は、請求項12から15のいずれか一項で定義された通りであることが好ましい。
【0088】
DARを求めるために本明細書中で使用される方法は、実質的に精製されたコンジュゲート溶液の252nmおよび280nmでの吸光度の比率を分光光度法により測定することからなる(これは、ステップ(ii)の後である)。特に、前記DARは、抗体に対してそれぞれ280および252nmで測定された吸光係数:εΑ280=224,000M−1cm−1およびεΑ252=82,880M−1cm−1を用いて、抗体に対して平均160,000分子量と仮定して、ならびにメイタンシノイドに対して、εD280=5,180M−1cm−1およびεD252=26,159M−1cm−1を用いて、分光光度法により求めることができる)。計算の方法は、Antony S.Dimitrov(編)、LLC、2009年、Therapeutic Antibodies and Protocols、vol525、445、Springer Scienceに基づき、以下でさらに詳細に記載されている:
252nm(A252)および280nm(A280)でのコンジュゲートに対する吸光度は、SEC分析の単量体ピーク(「DAR(SEC)」パラメータの計算を可能にする)に対して、または従来の分光光度計装置(「DAR(UV)」パラメータの計算を可能にする)を用いて測定される。吸光度は、以下の通り表現することができる:
A252=(cD×εD252)+(cA×εA252)
A280=(cD×εD280)+(cA×εA28O)
(式中、
cDおよびcAは、それぞれ、メイタンシノイドの溶液中および抗体の溶液中の濃度である。
εD252およびεD280は、それぞれ252nmおよび280nmでのメイタンシノイドのモル吸光係数である。
εA252およびεΑ280は、それぞれ、252nmおよび280nmでの抗体のモル吸光係数である。)。
2つの未知数があるこれら2つの方程式を解くことによって、以下の方程式が導かれる:
cD=[(εΑ280×A252)−(εA252×A280)]/[(εD252×εΑ280)−(εD252×εD280)]
cA=[A280−(cD×εD280)]/εΑ280ο
【0089】
次いで平均DARを、薬物濃度と抗体の濃度との比から計算する:
DAR=cD/cA
【0090】
UV分光光度計(DAR(UV))で測定した平均DARは、より具体的には4より上、より具体的には4と10の間、さらにより具体的には4と7の間である。
【0091】
コンジュゲートおよび式(I)の化合物も、抗癌剤として使用することができる。有利なことに、抗体は、腫瘍細胞に対して選択的な薬剤を有することを可能にし、したがってメイタンシノイドを前記細胞に極めて接近させ、またはメイタンシノイドが直接これら内部に狙いを定めるようにさせる(「Antibody−drug conjugates for cancer therapy」Carter P.J.ら、Cancer J.2008年、14、154−169頁;「Targeted cancer therapy:conferring specificity to cytotoxic drugs」Chari R.、Acc.Chem.Res.2008年、41、98−107頁を参照)。固体または液性腫瘍を治療することができる。
【0092】
コンジュゲートは、緩衝水溶液の形態で、好ましくは1と10mg/mlの間の濃度で製剤化することができる。溶液は、そのまま投与することができ、または溶液を希釈することによって、潅流用の溶液を形成することができる。
【実施例】
【0093】
方法A:高圧液体クロマトグラフィー−質量分析(LCMS)
スペクトルは、Waters UPLC−SQDシステム上で、正および/または負のエレクトロスプレーモード(ES+/−)で得た。クロマトグラフィーの条件は以下の通り:カラム:ACQUITY BEH C18、1.7μm−2.1×30mm;溶媒:A:H2O(0.1%ギ酸)B:CH3CN(0.1%ギ酸);カラム温度:45℃;流速:0.6ml/分;勾配(2分):5から50%のB、1分;1.3分:100%のB;1.45分:100%のB;1.75分:5%のB。
【0094】
方法B:高圧液体クロマトグラフィー−質量分析(LCMS)
スペクトルは、Waters ZQシステム上で、正および/または負のエレクトロスプレーモード(ES+/−)で得た。クロマトグラフィーの条件は以下の通り:カラム:XBridge C18 2.5μm 3×50mm;溶媒:A:H2O(0.1%ギ酸)B:CH3CN(0.1%ギ酸;カラム温度:70℃;流速:0.9ml/分;勾配(7分):5から100%のB、5.3分;5.5分:100%のB;6.3分:5%のB。
【0095】
方法C:質量分析(MS)
スペクトルは、Waters UPLC−SQDシステム上で、正および/または負のエレクトロスプレーモード(ES+/−)で得た。クロマトグラフィーの条件は以下の通り:カラム:ACQUITY BEH C18 1.7μm−2.1×50mm;溶媒:A:H2O(0.1%ギ酸)B:CH3CN(0.1%ギ酸);カラム温度:50℃;流速:1ml/分;勾配(2分):5から50%のB、0.8分;1.2分:100%のB;1.85分:100%のB;1.95:5%のB。
【0096】
方法D:高圧液体クロマトグラフィー−質量分析(LCMS)
スペクトルは、Waters UPLC−SQDシステム上で、正および/または負のエレクトロスプレーモード(ES+/−)で得た。クロマトグラフィーの条件は以下の通り:カラム:ACQUITY BEH C18 1.7μm−2.1×50mm;溶媒:A:H2O(0.1%ギ酸)B:CH3CN(0.1%ギ酸);カラム温度:50℃;流速:1ml/分;勾配(2分):5から50%のB、0.8分;1.2分:100%のB;1.85分:100%のB;1.95:5%のB。
【0097】
方法G:コンジュゲートの脱グリコシル化および高分解能質量分析(HRMS)
脱グリコシル化は、グリコシダーゼを用いた酵素消化の技法である。脱グリコシル化は、500μlのコンジュゲート+100μlのトリス緩衝液HCl 50mM+10μlのグリカナーゼ−F酵素(100単位の凍結乾燥した酵素/100μlの水)から生じる。培地をボルテックスし、一晩37℃で維持する。この脱グリコシル化した試料は、これでいつでもHRMSで分析を受けられる状態にある。Waters Q−Tof−2システム上で、エレクトロスプレー正モード(ES+)において質量スペクトルを得た。クロマトグラフィーの条件は、以下の通り:カラム:4μm BioSuite 250 URH SEC 4.6×300mm(Waters);溶媒:A:ギ酸アンモニウム25mM+1%ギ酸:B:CH3CN;カラム温度:30℃;流速0.4ml/分;定組成溶離70%A+30%B(15分)。
【0098】
方法H:分析用のサイズ排除クロマトグラフィー(SEC)
カラム:TSKgel G3000 SWXL 5μmカラム、7.8mm×30cm、TOSOH BIOSCIENCE、LLCパーツ#08541
移動相:KCl(0.2M)、KH2PO4(0.052M)、K2HPO4(0.107M)、iPrOH(量:20%)
分析条件:0.5ml/分で30分の定組成溶離
分光光度計検出器L2455DADを用いて、Lachrom Elite HPLCシステム(Merck)上で分析を実施。
【0099】
緩衝液の含量
緩衝液A(pH6.5):NaCl(50mM)、リン酸カリウム緩衝液(50mM)、EDTA(2mM)
緩衝液HGS(pH5.5):ヒスチジン(10mM)、グリシン(130mM)、ショ糖5%(w/v)、HCI(8mM)
【0100】
使用されている略語
AcOEt:酢酸エチル;ALK:(C1−C12)アルキレン基、特に(C1−C6)アルキレン;DAR:薬物抗体比;DBU:1,8−ジアザビシクロ[5.4.0]ウンデカ−7−エン;DCC:Ν,Ν’−ジシクロヘキシルカルボジイミド;DCM:ジクロロメタン;DEAD:アゾジカルボン酸ジエチル;DIC:Ν,Ν’−ジイソプロピルカルボジイミド;DIPEA:Ν,Ν−ジイソプロピルエチルアミン;DMA:ジメチルアセトアミド;DMAP:4−ジメチルアミノピリジン;DME:ジメトキシエタン;DMF:ジメチルホルムアミド;DMSO:ジメチルスルホキシド;ε:モル吸光係数;EEDQ:2−エトキシ−1−エトキシカルボニル−1,2−ジヒドロキノリン;EDCI:N−(3−ジメチルアミノプロピル)−N’−エチルカルボジイミド;EDTA:エチレンジアミン−テトラ酢酸;Fmoc:フルオレニルメトキシカルボニル;Hal:ハロゲン原子;HOBt:1−ヒドロキシベンゾトリアゾール;HEPES:4−(2−ヒドロキシエチル)−1−ピペラジン−エタンスルホン酸;HRMS:高分解能質量分析;NHS:N−ヒドロキシスクシンイミド;iPrOH:イソプロピルアルコール;NMP:N−メチルピロリジノン;Rf:保持因子;RP:減圧;RT:室温;SEC:サイズ排除クロマトグラフィー;TBDMS:tert−ブチルジメチルシリル;TEA:トリエチルアミン;TFA:トリフルオロ酢酸;TFAA:トリフルオロ酢酸無水物;TFF:タンジェンシャルフローフィルトレーション;THF:テトラヒドロフラン;TIPS:トリイソプロピルシリル;TLC:薄層クロマトグラフィー;tR:保持時間。
【0101】
実施例で使用されている抗体
以下の2つの抗体を使用してコンジュゲートを調製した。
hu2H11:(WO2008010101では、hu532H11としても参照されている):この抗体は、アメリカ培養細胞系統保存機関で、受託番号PTA−7662、ブダペスト条約下で預けられたハイブリドーマにより産生し、PCT出願WO2008/010101に記載されている。
hu2H11R35R74:このヒト化抗体は、EphA2受容体に結合し、配列SEQ ID NO:18の重鎖およびSEQ NO:16の軽鎖からなるhu532H11の部位特異的突然変異誘発により得られる。
【0102】
[実施例1]
【0103】
【化25】
1.1.コンジュゲートhu2H11R35R74−PEG4−NHAc−DM4の調製
磁気撹拌下、RTで、9mlのhu2H11R35R74(緩衝液A中14.36mg/ml)を添加し、次いで16.85mlの緩衝液A、3.23mlのHEPES 1M、1.59mlのDMA、続いてL−DM4−AcNH−PEG4−CONHS活性化エステルの、1.64mlの10mM DMA溶液を添加する。RTで1時間30分後、L−DM4−AcNH−PEG4−CONHS活性化エステルの追加の0.085mlの10mM DMA溶液を添加する。RTで1時間45分後、粗製の結合培地を60mlのHGS緩衝液で希釈し、Pellicon3カセット上のTFFで精製する。約10の試料容量のHGS緩衝液に対して試料を透析ろ過し、次いで収集する。TFFタンクおよびラインを追加の10mlのHGS緩衝液で洗浄する。2つの溶液を混合し、0.22μmのPVDFを介してろ過滅菌し、Amicon15上で濃縮し、0.22μmPVDFを介してろ過滅菌する。17mlのhu2H11R35R74−PEG4−NHAc−DM4コンジュゲート(c=5.76mg/ml)をこのようにして得た。次いで最終の薬剤含有量および単量体の純度についてこのコンジュゲートを分析する。SEC分析(方法H):DAR(SEC)=5.4;RT=16.757分;単量体の純度=99.5%;HRMSデータ:図1を参照。
【0104】
1.2.L−DM4−AcNH−PEG4−CONHS活性化エステルの調製
RTでの磁気撹拌下、154.3mgのL−DM4(WO04/103272に従い調製−化合物4bを参照)をガラスバイアル中に導入する。次いで、90mgの3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルの、0.94mlのDMA中溶液を添加し、続いて36μlのDIEAを添加する。RTで23時間後、この反応培地を5mlのAcOEtで希釈し、7mlの水で洗浄する。水相を5mlのAcOEtで抽出する。合わせた有機相をMgSO4で脱水し、RP下で濃縮乾燥させる。228mgの淡黄色の粘性油を得る。この生成物を最小量のDMAで希釈し、30gのC18−グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。RP下で画分2および3を濃縮後、無色の粘性の油を得る。この生成物を最小量のDMAで希釈し、30gのC18−グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。画分33から35をRP下で濃縮後、41mgのL−DM4−AcNH−PEG4−CONHS活性化エステルを白色メレンゲのような生成物の形態で得る。質量スペクトル(B):RT=4.06分;[M+H−H2O]+:m/z 1164;[M+H]+:m/z 1182;[M−H+HCO2H]−:m/z 1226;1H NMR(500MHz,δ:ppm,クロロホルム−d):0.80(s,3H);1.21(s,3H);1.22(s,3H);1.25(m,1H);1.29(d,J=6.7Hz,6H);1.46(m,1H);1.57(d,J=13.4Hz,1H);1.64(s,3H);1.76から1.83(m,1H);1.88から1.96(m,1H);2.18(dd,J=2.5および14.3Hz,1H);2.36(m,1H);2.53(m,1H);2.61(dd,J=12.5および14.3Hz,1H);2.82から2.92(m,10H;2.98(d,J=16.7Hz,1H);3.03(d,J=9.6Hz,1H);3.15(d,J=12.9Hz,1H);3.22(s,3H);3.32(s 広幅,1H);3.36(s,3H);3.42(m,2H);3v50(d,J=9.1Hz,1H);3.53(t,J=5.2Hz,2H);3.58から3v67(m,13H);3.84(t,J=6.4Hz,2H);3.99(s,3H);4.27(m,1H);4.77(dd,J=2.9および11.9Hz,1H);5.42(q,J=6.7Hz,1H);5.66(dd,J=9.1および15.4Hz,1H);6.23(s,1H);6.43(dd,J=11.3および15.4Hz,1H);6.64(d,J=1.1Hz,1H);6.74(d,J=11.3Hz,1H);6.85(d,J=1.1Hz,1H);7.08(t,J=5.2Hz,1H)
【0105】
1.3.3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルの調製
磁気撹拌下、RTで、671.4mgの3−(2−{2−[2−(2−アミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸(CA(PEG)4、Pierce)をガラスバイアル内に導入する。次いで、597.4mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルの、14mlのDCM中溶液を添加する。RTで15分後、0.396mlのDICを添加する。1時間30分後、粗製の反応培地を焼結ガラス上でろ過し、100gのCN−グラフトシリカゲル(勾配溶出、nヘプタン/iPrOH/AcOEt、iPrOH部分を増加)上で、フラッシュクロマトグラフィーにより濾液を精製する。画分30から45をRP下で濃縮後、761mgの3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルを無色の油の形態で得る。質量スペクトル(A):RT=0.74分;[M+H]+:m/z483/485(Brの2つの同位体による2つのピーク);[M−H+HCO2H]−:m/z527/529(Brの2つの同位体による2つのピーク)。
【0106】
ブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルは、公開プロトコル(Biochemistry、1974年、481頁)に従い調製することができた。
【0107】
[実施例2]
【0108】
【化26】
2.1.コンジュゲートhu2H11R35R74−PEG4−NMeAc−DM4の調製
RTでの磁気撹拌下、4mlのhu2H11R35R74(緩衝液A中14.36mg/ml)を添加し、次いで7.5mlの緩衝液A、1.45mlのHEPES 1M、1.05mlのDMA、続いてL−DM4−AcNMe−PEG4−CONHS活性化エステルの、0.39mlの10mM DMA溶液を添加する。RTで30分後、L−DM4−AcNMe−PEG4−CONHS活性化エステルの、追加の0.19mlの10mM DMA溶液を添加する。RTで3時間後、粗製の結合培地を65mlのHGS緩衝液で希釈し、Pellicon3カセット上でTFFにより精製する。約10の試料容量のHGS緩衝液に対して試料を透析ろ過し、次いで収集する。TFFタンクおよびラインを追加の10mlのHGS緩衝液で洗浄する。この2つの溶液を混合し、Amicon15上で濃縮し、0.22μmPVDFを介してろ過滅菌する。8.5mlのhu2H11R35R74−PEG4−NMeAc−DM4コンジュゲート(c=6.01mg/ml)をこのようにして得た。次いで最終の薬剤含有量および単量体の純度についてコンジュゲートを分析する。SEC分析(H):DAR(SEC)=5.5;RT=16.7分;単量体の純度=99.4%;HRMSデータ:図2を参照。
【0109】
2.2.L−DM4−AcNMe−PEG4−CONHS活性化エステルの調製
RTでの磁気撹拌下、133.4mgのL−DM4をガラスバイアル内に導入する。次いで、85mgの3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルの、0.2mlのDMA中溶液を添加し、続いて32.9μlのDIEAを添加する。RTで1時間後、この反応培地を、30gのC18−グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。所望の生成物を含有する画分をRP下で濃縮後、71.3mgのL−DM4−AcNMe−PEG4−CONHS活性化エステルを無色ガラスのような生成物の形態で得る。質量スペクトル(D):RT=0.98分;[M+H−H2O]+:m/z 1178(主要シグナル);[M+Na]+:m/z 1218;[M−H+HCO2H]−:m/z 1240;1H NMR(500MHz,δ:ppm,クロロホルム−d):0.81(s,3H);1.20から1.33(m,13H);1.42から1.52(m,1H);1.56から1.61(m,1H);1.65(s,3H);1.73から1.83(m,1H);1.96から2.04(m,1H);2.19(dd,J=2.8および14.4Hz,1H);2.29から2.41(m,1H);2.55から2.66(m,2H);2.83から2.93(m,12H);3.04(d,J=9.8Hz,1H);3.12(d,J=12.7Hz,1H);3.18から3.25(m,5H);3.37(s,3H);3.47から3.54(m,3H);3.57から3.68(m,15H);3.85(t,J=6.6Hz,2H);3.99(s,3H);4.29(m,1H);4.79(dd,J=2.8および12.2Hz,1H);5.41(q,J=6.7Hz,1H);5.68(dd,J=9.3および15.2Hz,1H);6.23(s,1H);6.43(dd,J=11.0および15.2Hz,1H);6.66(s,1H);6.74(d,J=11.0Hz,1H);6.83(s,1H)。
【0110】
2.3.3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2−ジオキソ−ピロリジン−1−イルエステルの調製
【0111】
【化27】
磁気撹拌下、RTで、115.1mgの3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸、1.5mlのDCM、97.3mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルを丸底フラスコ内に逐次的に導入する。2時間後、72μlのDIEAを添加し、RTでさらに1時間後、70.2μlのDICを添加する。粗製の反応培地をRTで4時間保持し、−20°Cで16時間保持し、次いで30gのシリカゲル(DCM:メタノール0:100から3:97(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。所望の生成物を含有する画分をRP下で濃縮後、85.8mgの3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルを白色固体の形態で得る。質量スペクトル(A):RT=0.84分;[M+H]+:m/z497/499
【0112】
2.4.3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸の調製
【0113】
【化28】
アルゴンの不活性雰囲気下、丸底フラスコ内で、磁気撹拌しながら、120.1mgの3−[2−(2−{2−[2−(2,2,2−トリフルオロ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステル、1mlの無水THFおよび59.8μlのCH3lを逐次的に導入する。この反応培地を氷/水槽を用いて約0℃で冷却し、16.1mgのNaH(油中、純度50%)を少しずつゆっくりと添加する。0°Cで15分およびRTで1時間後、粗製の反応培地をRP下で濃縮乾燥し、0.5mlのTHFおよび0.8mlの水で希釈する。次いでRTで、30.6mgのLiOHをこの反応物培地に添加する。粗製の反応培地をRTで2時間保持し、−20℃で16時間保持し、次いで30gのC18−グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。所望の生成物を含有する画分をRP下で濃縮後、115.3mgの3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸を黄色油の形態で得る。
【0114】
2.5.3−[2−(2−{2−[2−(2,2,2−トリフルオロ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルの調製
【0115】
【化29】
アルゴンの不活性雰囲気下、磁気撹拌しながら、230mgの3−(2−{2−[2−(2−アミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸(CA(PEG)4、Pierce)、2mlのDCMおよび1mlのメタノールを、丸底フラスコ内に、逐次的に導入する。RTで、1mlのトリメチルシリルジアゾメタン(ヘキサン中2M溶液)をこの反応物培地にゆっくりと添加する。RTで2時間後、過剰なトリメチルシリルジアゾメタンを、酢酸の添加により中和する。次いでRP下で粗製物を蒸発乾燥させる。得た残渣を2mlのDCMで希釈し、水−氷浴を用いて0℃に冷却し、次いで363μlのTEAおよび300μlのTFAAを逐次的に添加する。RTで2時間30分間および−20℃で19時間後、363μlのTEAおよび300μlのTFAAを逐次的に添加する。RTで4時間30分後、粗製物を−20℃でストックし、次いで30gのC18−グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。所望の生成物を含有する画分をRP下で濃縮後、123mgの3−[2−(2−{2−[2−(2,2,2−トリフルオロ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルを淡黄色の油の形態で得る。質量スペクトル(A):RT=0.90分;[M+H]+:m/z376;[M−H]−:m/z374。
【0116】
[実施例3]
【0117】
【化30】
3.1.hu2H11R35R74−PEG8−NHAc−DM4のコンジュゲートの調製
RTでの磁気撹拌下、4mlのhu2H11R35R74(緩衝液A中14.36mg/ml)を添加し、次いで7.5mlの緩衝液A、1.45mlのHEPES 1M、1.05mlのDMAを添加し、続いてL−DM4−AcNMe−PEG8−CONHS活性化エステルの0.405mlの10mM DMA溶液を添加する。RTで30分後、L−DM4−AcNMe−PEG8−CONHS活性化エステルの追加の0.1mlの10mMDMA溶液を添加する。RTで1時間45分後、粗製の結合培地を60mlのHGS緩衝液で希釈し、Pellicon3カセット上で、TFFで精製する。約10の試料容量のHGS緩衝液に対して試料を透析ろ過し、次いで収集する。TFFタンクおよびラインを追加の10mlのHGS緩衝液で洗浄する。2つの溶液を混合し、Amicon15上で濃縮し、0.22μmPVDFを介してろ過滅菌する。7.0mlのhu2H11R35R74−PEG8−AcNMe−DM4コンジュゲート(c=6.95mg/ml)をこのようにして得た。次いで最終の薬剤含有量および単量体の純度についてコンジュゲートを分析する。SEC分析(H):DAR(SEC)=5.0;RT=16.593分;単量体純度=99.5%:HRMSデータ:図3を参照。
【0118】
3.2.L−DM4−AcNH−PEG8−CONHS活性化エステルの調製
RTでの磁気撹拌下、65mgの3−{2−[2−(2−{2−[2−(2−{2−[2−(3−ブロモ−プロピオニルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルをガラスバイアル内に導入し、続いて67.7mgのL−DM4の、0.85mlのDMAおよび16.5μlのDIEA中溶液を導入する。RTで48時間後、10gのシリカゲル(DCM:MeOH 100:0から90:10(容量)の勾配溶出)上でフラッシュクロマトグラフィーによりこの反応培地を精製する。画分18から26をRP下で濃縮後、17mgのL−DM4−AcNH−PEG8−CONHS活性化エステルを無色のガラスの形態で得る。質量スペクトル(B)::RT=4.08分;[M+H−H2O]+:m/z 1340(主要シグナル);[M+Na]+:m/z 1380;[M−H+HCO2H]−:m/z 1402;1H NMR(400MHz,δ:ppm,クロロホルム−d):0.81(s,3H);1.22(s,3H);1.23(s,3H);1.26(m,1H);1.30(d,J=6.8Hz,6H);1.41から1.52(m,1H);1 .65(s,3H);1.80(m,1H);1.89から1.99(m,1H);2.19(m,1H);2.37(m,1H);2.47から2.67(m,2H);2.81から2.93(m,10H);2.99(d,J=16.6Hz,1H);3.04(d,J=9.8Hz,1H);3.16(d 広幅,J=13.7Hz,1H);3.23(s,3H);3.32(s 広幅,1H);3.37(s,3H);3.44(m,2H);3.51(d,J=9.1Hz,1H);3.54(t,J=5.4Hz,2H);3.59から3.73(m,29H);3.86(t,J=6.6Hz,2H);4.00(s,3H);4.22から4.33(m,1H);4.78(dd,J=2.9および12.2Hz,1H);5.43(q,J=6.8Hz,1H);5.67(dd,J=9.0および15.2Hz,1H);6.23(s,1H);6.44(dd,J=11.2および15.2Hz,1H);6.65(d,J=1.5Hz,1H);6.75(d,J=11.2Hz,1H);6.86(d,J=1.5Hz,1H);7.02から7.13(m,1H)。
【0119】
3.3.3−{2−[2−(2−{2−[2−(2−{2−[2−(3−ブロモ−プロピオニルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルの調製:
【0120】
【化31】
RTでの磁気撹拌下、100mgの3−(2−{2−[2−(2−アミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸(CA(PEG)4、Pierce)、2mlのDCMおよび53.5mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルをガラスバイアル内に逐次的に導入する。RTで1時間後、35.1μlのDICを添加する。1時間後、粗製の反応培地を焼結ガラス上でろ過し、RP下で濃縮乾燥させ、10mlのAcOEtで希釈し、焼結ガラス上でろ過し、RP下で濃縮乾燥する。76.5mgの3−{2−[2−(2−{2−[2−(2−{2−[2−(3−ブロモ−プロピオニルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルを無色の油の形態で得る。質量スペクトル(A):RT=0.80分;[M+H]+:m/z659/661;[M−H+HCO2H]−:m/z703/705
【0121】
[実施例4]
【0122】
【化32】
4.1.コンジュゲートhu2H11R35R74−PEG4−アリル−DM4の調製
RTでの磁気撹拌下、4mlのhu2H11R35R74(緩衝液A中14.36mg/ml)を添加し、次いで7.5mlの緩衝液A、1.45mlのHEPES 1M、1.14mlのDMAを添加し、続いてL−DM4−アリル−PEG4−CONHS活性化エステルの、0.3mlの10mM DMA溶液を添加する。RTで30分後、L−DM4−アリル−PEG4−CONHS活性化エステルの追加の0.125mlの10mM DMA溶液を添加する。RTで1時間25分後、粗製の結合培地を65mlのHGS緩衝液で希釈し、Pellicon3カセット上で、TFFで精製する。約10の試料容量のHGS緩衝液に対して試料を透析ろ過し、次いで収集する。TFFタンクおよびラインを追加の10mlのHGS緩衝液で洗浄する。2つの溶液剤を混合し、Amicon15上で濃縮し、0.22μmのPVDFを介してろ過滅菌する。8.0mlのhu2H11R35R74−PEG4−アリル−DM4コンジュゲート(c=5.22mg/ml)を得た。次いで、最終の薬剤含有量および単量体の純度についてコンジュゲートを分析する。SEC分析(H):DAR(SEC)=5.3;RT=16.767分;単量体の純度=99.4%;HRMSデータ:図4を参照。
【0123】
4.2.L−DM4−アリル−PEG4−CONHS活性化エステルの調製
RTでの磁気撹拌下、70mgのL−DM4、45mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステル(ブロモ−アリル−PEG4−CONHS)、0.5mlのDMAおよび23.5μlのDIEAをガラスバイアル内に逐次的に導入する。RTで2時間および−20℃で17時間後、50μlのDIEAを添加する。RTで24時間後、この反応培地を、30gのC−18グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。所期の生成物を含有する画分をRPで濃縮後、47.1mgのL−DM4−アリル−PEG4−CONHS活性化エステルを白色固体の形態で得る。質量スペクトル(D):RT=1.06分;[M+Na]+:m/z 1173;1H NMR(500MHz,δ:ppm,クロロホルム−d):0.81(s,3H);1.18から1.39(m,13H);1.42から1.52(m,1H);1.58(d,J=13.4Hz,1H);1.65(s,3H);1.73から1.82(m,1H);1.86から1.95(m,1H);2.19(d,J=14.3Hz,1H);2.40(m,1H);2.51から2.65(m,2H);2.82から2.95(m,9H);2.98から3.07(m,2H);3.12(d,J=12.6Hz,1H);3.18から3.27(m,1H);3.23(s,3H);3.36(s,3H);3.51(d,J=9.1Hz,1H);3.54から3.82(m,13H);3.86(t,J=6.4Hz,2H);3.91から3.95(m,2H);3.99(s,3H);4.28(t,J=11.0Hz,1H);4.78(dd,J=2.6および11.9Hz,1H);5.44(q,J=6.7Hz,1H);5.49から5.63(m,2H);5.68(dd,J=9.1および15.0Hz,1H);6.24(s,1H);6.43(dd,J=11.1および15.0Hz,1H);6.66(s,1H);6.77(d,J=11.1Hz,1H);6.83(s,1H)。
【0124】
4.3.3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルの調製
【0125】
【化33】
RTで、200mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸、4mlのDCMおよび232.3mgの担持されたDCC(2当量)をガラスバイアル内に逐次的に導入する。RTで1時間後、64.8mgのNHSを添加する。RTで5時間後、粗製物を焼結ガラスろ過し、固体をDCMで洗浄し、合わせた濾液をRP下で濃縮乾燥させる。15gのシリカゲル(MeOH:DCM 0:100から10:90(容量)の勾配溶出)上でのフラッシュクロマトグラフィーによる精製および所期の生成物を含有する画分のRP下での濃縮により、生成した46mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステル(ブロモ−アリル−PEG4−CONHS)を淡黄色の油の形態で得る。質量スペクトル(A):RT=1.02分;[M+H]+:m/z454/456;[M+Na]+:m/z476/478;[M−H+HCO2H]−:m/z498/500。
【0126】
4.4.3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸の調製
【0127】
【化34】
RTで、1gの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸tert−ブチルエステル(市販のもの)、6mlのTFAおよび3mlのDCMの溶液を3時間撹拌し、次いでRP下で濃縮乾燥させる。油性の残渣をトルエンで希釈し、RP下で濃縮乾燥させて、褐色油の形態で853mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸を生成する。
【0128】
[実施例5]
5.1.コンジュゲートhu2H11−PEG4−NHAc−DM4の調製
コンジュゲートhu2H11−PEG4−NHAc−DM4は、実施例1と類似の方法で調製することができた。撹拌下、RTで、1mlのhu2H11(緩衝液A中8.52mg/ml)を添加し、次いで0.7mlの緩衝液A、0.213mlのHEPES 1M、0.7mlのDMA、続いて0.085mlのL−DM4−AcNH−PEG4−CONHS活性化エステルの10mM DMA溶液を0.128mlのDMAで希釈する。RTで2時間後、粗製の培地をAmicon4上で、7000Gで濃縮し、Nap−10カラム上で緩衝液をHGS緩衝液と交換し、最後に5ml Zebaカラム上で精製する。1.15mlのhu2H11−PEG4−NHAc−DM4コンジュゲート(c=3.78mg/ml)をこのようにして得た。最終の薬剤含有量および単量体の純度についてコンジュゲートを分析する。SEC分析(方法H):DAR(UV)=6.6;DAR(SEC)=5.6;RT=15.387分;単量体の純度=99.7%;HRMSデータ:図5を参照。
【0129】
同様に、hu2H11の関与する他のコンジュゲートおよび実施例6から8に記載されたものを調製した(表IIaを参照)。
【0130】
[実施例9]
MDA−MB231腫瘍細胞の増殖の阻害(ECAAC ref.#92020424から)
指数増殖期の細胞をトリプシン処理し、適切な培地内に再懸濁させた(DMEM/F12 Gibco #21331;10%SVF Gibco #10500−056;2nMグルタミン Gibco #25030)。完全な血清含有培地の中、5000細胞/ウェルの密度で、細胞懸濁液を96−ウェルCytostar培養プレート(GE Healthcare Europe、#RPNQ0163)に分配した。4時間のコーティング後、三連のウェルに、10−7から10−12Mの間の範囲の濃度でコンジュゲートの段階希釈を添加した。コンジュゲートの存在下、37℃/5%CO2で3日間細胞を培養した。第4日目、10μlの14C−チミジン溶液(0.1μCi/ウェル(Perkin Elmer# NEC56825000))を各ウェルに添加した。microbeta放射性カウンター(Perkin Elmer)を用いて、実験が開始されてから96時間、14C−チミジンの取り込みを測定した。無細胞の試薬ブランクを試験ウェルの記録から引き算し、コンジュゲートで処理した細胞の記録を、ビヒクル処理した細胞の対照ウェルからの記録の平均値で割ることによって得た、残存する画分として、このデータをプロットした。これら実験において、実験開始時に、1μΜの濃縮で、裸の抗体(hu2H11またはhu2H11R35R74)を添加し、増殖の阻害を以前に述べた通り測定した。
【0131】
表IIaおよびIIbで報告された結果は、裸のhu2H11または/hu2H11R35R74の存在下で実施された競合実験によると、コンジュゲートは、MDA−MB231細胞に対して強力なインビトロの増殖阻害特性を示し、抗原への結合を介して作用することを示唆している。
【0132】
【表2】
【0133】
【表3】
【0134】
[実施例10]
L−DM4−AcNH−PEG4−COOMe
【0135】
【化35】
10.1.遊離薬物L−DM4−AcNH−PEG4−COOMeの調製
【0136】
【化36】
磁気撹拌下、RTで、Arの不活性雰囲気下、ガラスバイアル内に、57.4mgの3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステル、80mgのL−DM4の、0.44mlのDMA中溶液、最後に19.6μlのDIPEAを逐次的に導入する。RTで18時間後、粗製物を10mlの水で希釈し、3×7mlのAcOEtで抽出する。有機相を集め、MgSO4で脱水し、ろ過し、RP下で濃縮乾燥する。124mgの無色の油を得る。この生成物を最小量のDMAで希釈し、C18−グラフトシリカゲル(Merck、C18、5g、25−40μm、18ml/分、水:アセトニトリル100:0から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、12.8mgのメチルエステルL−DM4−AcNH−PEG4−COOMeを、無色の被膜の形態で得る。質量スペクトル(C):tR=0.97分;[M+H]+:m/z 1099;[M−H]−:m/z 1097;1H NMR(500MHz,ppm単位,クロロホルム−d):0.80(s,3H);1.21(s,3H);1.22(s,3H);1.24から1.35(m,7H);1.46(td,J=6.4および10.2Hz,1H);1.57(d,J=13.4Hz,1H);1.64(s,3H);1.80(ddd,J=4.9および11.5および14.6Hz,1H);1.92(m,1H);2.18(dd,J=2.3および14.7Hz,1H);2.36(ddd,J=4.8および11.4および16.2Hz,1H);2.52(ddd,J=5.1および11.3および16.3Hz,1H);2.59(t,J=6.4Hz,2H);2.63(m,1H);2.86(s,3H);2.88(m,1H);2.98(d,J=16.7Hz,1H);3.03(d,J=9.6Hz,1H);3.15(d,J=12.6Hz,1H);3.23(s,3H);3.36(s,3H);3.42(m,2H);3.50(d,J=9.1Hz,1H);3.54(m,2H);3.63(m,13H);3.68(s,3H);3.75(t,J=6.4Hz,2H);3.99(s,3H);4.27(t,J=11.3Hz,1H);4.78(dd,J=2.2および12.1Hz,1H);5.42(q,J=6.9Hz,1H);5.66(dd,J=9.1および15.4Hz,1H);6.21(s,1H);6.43(dd,J=11.0および15.4Hz,1H);6.64(s,1H);6.74(d,J=11.0Hz,1H);6.85(s,1H);7.05(t,J=5.5Hz,1H)。
【0137】
10.2.3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルの調製
【0138】
【化37】
磁気撹拌下、RTで、100mgの3−(2−{2−[2−(2−アミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸(CA(PEG)4、Pierce)、89mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルおよび2mlのDCMをガラスバイアル内に逐次的に導入する。RTで1時間後、0.7mlのMeOHおよび0.38mlの2Mトリメチルシリルジアゾメタンのヘキサン中溶液を添加する。RTで1時間後、粗製の反応混合物をRP下で濃縮乾燥し、次いで最小量のDMAで希釈し、C18−グラフトシリカゲル(Merck、C18、5g、25−40μm、18ml/分、水:アセトニトリル100:0から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、58mgの3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルを無色の油の形態で得る。質量スペクトル(A):tR=0.75分;[M+H]+:m/z400/402
ブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルは、公開プロトコル(Biochemistry、1974年、481頁)に従い調製することができた。
【0139】
[実施例11]
L−DM4−AcNMe−PEG4−COOMe
【0140】
【化38】
11.1.遊離薬物L−DM4−AcNMe−PEG4−COOMeの調製
【0141】
【化39】
磁気撹拌下、RTで、ガラスバイアル内に、30mgのL−DM4、20.8mgの3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルの、0.3mlのDMA中溶液、および7.4μlのDIPEAを逐次的に導入する。RTで18時間後、この反応培地を7mlのAcOEtで希釈し、5mlの水で2回洗浄する。有機相をブラインで洗浄し、MgSO4で脱水し、ろ過し、RP下で濃縮乾燥させる。39mgの無色のガラスを得る。この生成物を最小量のDMA/MeOH混合物で希釈し、C18−グラフトシリカゲル(XTerra(登録商標)C18、5μm、50×30mm、30ml/分、水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でクロマトグラフィーにより精製する。予想された化合物を含有する画分を減圧下で濃縮後、7.8mgのメチルエステルL−DM4−AcNMe−PEG4−COOMeを無色の固体の形態で得る。質量スペクトル(C):tR=1.00分;[M+H−H2O]+:m/z1095;[M+Na+]+:m/z 1135;[M−H+HCO2H]−:m/z 1157;[M−H]−:m/z 1111。1H NMR(500MHz,δ:ppm,DMSO−d6):0.78(s,3H);1.12(d,J=6.6Hz,3H);1.16(m,9H);1.26(m,1H);1.40から1.51(m,2H);1.59(s,3H);1.62(m,1H);1.87(m,1H);2.04(m,1H);2.28(m,1H);2.47から2.58(m:部分マスク,4H);2.73(s,3H);2.76(s,1H);2.80(d,J=9.6Hz,1H);2.95(s,2H);3.10(s,3H);3.16から3.48(m:部分マスク,21H);3.25(s,3H);3.59(s,3H);3.63(t,J=6.4Hz,2H);3.93(s,3H);4.08(m,1H);4.53(dd,J=2.9から11.9Hz,1H);5.32(q,J=6.6Hz,1H);5.58(dd,J=9.3から15.1Hz,1H);5.89(s,1H);6.49から6.58(m,2H);6.62(m,1H);6.84(s,1H);7.19(s,1H)。
【0142】
11.2.3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルの調製
【0143】
【化40】
磁気撹拌下、RTで、Arの不活性雰囲気下、127mgの3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステル、102.2mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルおよび1.2mlのDCMを逐次的に添加する。45分後、粗製物をRP下で濃縮し、5gのCN−グラフトシリカゲル(n.ヘプタン/iPrOH/AcOEtの勾配溶出、iPrOH部分を増加させる)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、122mgの3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルを無色の油の形態で得る。質量スペクトル(A):tR=0.84分;[M+H]+:m/z414/416。
【0144】
ブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルは、公開プロトコル(Biochemistry、1974年、481頁)に従い調製することができた。
【0145】
11.3.3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステルの調製
【0146】
【化41】
磁気撹拌下、RTで、Arの不活性雰囲気下で、271.7mgの3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルを、ジオキサン中の2mlの塩酸4N溶液の中で可溶化させる。18時間後、粗製の反応培地をRP下で濃縮し、4mlのMeOHの中で可溶化させ、3g SCX SPEカラム(10mlのMeOHで調整、10mlのMeOHで洗浄およびMeOH中アンモニア2Nで溶出)を通過させる。RP下で溶出画分の濃縮後、146mgの無色の油を得る。この油をAcOEt中に溶解し、MgSO4上で乾燥させ、ろ過し、RP下で蒸発させた。127mgの3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステルを無色の油として得る。質量スペクトル(A):tR=0.40分;[M+H]+:m/z294。
【0147】
11.4.3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルの調製
【0148】
【化42】
磁気撹拌下、RTで、Arの不活性雰囲気下、227mgの3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸の、0.644mlのDCMおよび0.644mlのMeOH中溶液を水−氷−塩化ナトリウムで約0℃まで冷却する。トリメチルシリルジアゾメタンの、0.449mlのヘキサン中2M溶液を添加し、RTで17時間後、酢酸のMeOH中0.5M溶液を添加することによって、5と6の間のpHを達成する。粗製物をAcOEt(30ml)で希釈し、水(2×15ml)で洗浄し、ブライン(15ml)で洗浄し、有機相をMgSO4上で脱水させ、ろ過し、RP下で蒸発させる。209.7mgの3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルを無色の油として得る。質量スペクトル(A):tR=1.18分;[M+H]+:m/z294、338、394。
【0149】
11.5.3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸の調製
【0150】
【化43】
磁気撹拌下、RTで、Arの不活性雰囲気下、330mgの市販の3−(2−{2−[2−(2−tert−ブトキシカルボニルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸の、4mlのTHF中溶液を、水−氷−塩化ナトリウムで、約0℃まで冷却する。72.2mgのNaH(油中70%)をゆっくりと添加し、25分後、0.174mlのMelを添加する。冷却槽を取り除き、RTで3時間後、120mgのNaHおよび0.2mlのMelを添加する。RTで1.5時間後、酢酸の希釈水溶液を添加することによって、5と6の間のpHを達成する。粗製の反応混合物をAcOEt(30ml)で希釈し、水(2×20ml)で洗浄し、ブライン(10ml)で洗浄し、有機相をMgSO4上で脱水させ、ろ過し、RP下で蒸発させる。227mgの3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸を無色の油として得る。質量スペクトル(A):tR=1.02分;[M+H]+:m/z280、380。
【0151】
[実施例12]
L−DM4−アリル−PEG4−COOMe
【0152】
【化44】
12.1遊離薬物L−DM4−アリル−PEG4−COOMeの調製
【0153】
【化45】
磁気撹拌下、RTで、ガラスバイアル内に、36.7mgのL−DM4、20.8mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステルの、0.37mlのDMA中溶液、最後に9.4μlのDIPEAを逐次的に導入する。RTで1.5時間後および−18℃で18時間後、この反応培地を、C18−グラフトシリカゲル(Merck、C18、5g、25−40μm、18ml/分、水:アセトニトリル100:0から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、18.2mgの白色固体を得て、これを、CN−グラフトシリカゲル(n.ヘプタン/iPrOH/AcOEtの勾配溶出、AcOEt部分を増加させる)上で、フラッシュクロマトグラフィーにより精製する。4.02mgのメチルエステルL−DM4−アリル−PEG4−COOMeを白色固体の形態で得る。質量スペクトル(C):tR=1.09分;[M−H + HCOOH]−:m/z 1112;[M+Na]+:m/z 1090。1H NMR(500MHz,ppm単位,クロロホルム−d):0.80(s,3H);1.18から1.26(m,7H);1.27から1.31(m,6H);1.40から1.50(m,1H);1.57(d,J=13.7Hz,3H);1.64(s,3H);1.77(ddd,J=4.8および11.7および14.5Hz,1H);1.91(ddd,J=4.8および11.7および14.5Hz,1H);2.18(dd,J=2.5および14.3Hz,1H);2.38(m,1H);2.57(m,4H);2.86(s,2H);2.89(m,1H);3.00(m,1H);3.04(d,J=9.9Hz,1H);3.11(d,J=12.3Hz,1H);3.23(s,3H);3.35(s,3H);3.50(d,J=9.1Hz,1H);3.55(m,2H);3.63(m,10H);3.69(s,3H);3.75(t,J=6.4Hz,2H);3.92(d,J=5.2Hz,2H);3.98(s,3H);4.27(ddd,J=1.6および10.4および12.3Hz,1H);4.78(dd,J=3.0および11.8Hz,1H);5.43(q,J=6.8Hz,1H);5.48から5.61(m,2H);5.67(dd,J=9.2および15.2Hz,1H);6.20(d,J=0.5Hz,1H);6.42(dd,J=11.3および15.4Hz,1H);6.65(d,J=1.9Hz,1H);6.77(d,J=11.3Hz,1H);6.82(d,J=1.1Hz,1H)。
【0154】
12.2.3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステルの調製
【0155】
【化46】
磁気撹拌下、50mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸の、1mlのDCMおよび0.25mlのMeOH中冷却溶液(水−氷浴)に、0.11mlの2Mトリメチルシリルジアゾメタンのヘキサン中溶液を添加する。水−氷浴を取り除いた後、粗製の反応混合物をRTで1.5hr撹拌し、2滴の酢酸で中和し、RP下で濃縮乾燥する。トルエンで共沸蒸発後、47mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステルをアンバー油の形態で得る。質量スペクトル(A):tR=1.12分;[M+H]+:m/z369/371。
【0156】
3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸の調製は実施例4で記載されている。
【0157】
[実施例13]
L−DM4−AcNH−PEG8−COOMe
【0158】
【化47】
13.1.遊離薬物L−DM4−AcNH−PEG8−COOMeの調製
【0159】
【化48】
磁気撹拌下、RTで、ガラスバイアル内に、60mgのL−DM4、11.4mgの炭酸カリウム、および75mgの3−{2−[2−(2−{2−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルの、0.7mlのDMA中溶液を逐次的に導入する。RTで22時間後、この反応培地を12mlの水で希釈し、2×10mlのAcOEtで抽出する。有機相を集め、MgSO4で脱水し、ろ過し、RP下で濃縮乾燥させる。50mgの無色の油を得る。この生成物を最小量のDCMで希釈し、5gのCN−グラフトシリカゲル(nヘプタン/iPrOH/AcOEtの勾配溶出、iPrOH部分を増加させる)上で、フラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分を減圧下で濃縮後、残渣を最小量のDMAで希釈し、C18−グラフトシリカゲル(Merck、C18、5g、25−40μm、12ml/分、水:アセトニトリル100:0から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、3.3mgのメチルエステルL−DM4−AcNH−PEG8−COOMeを無色の被膜の形態で得る。質量スペクトル(C):tR=0.97分;[M−H]− + HCOOH:m/z 1319。1H NMR(400MHz,ppm単位,クロロホルム−d):0.73(s,3H);1.10から1.19(m,7H);1.21(s,3H);1.23(s,3H);1.39(td,J=6.1および10.1Hz,1H);1.50(d,J=13.2Hz,1H);1.57(s,3H);1.71(m,1H);1.85(m,1H);2.11(dd,J=3.4および14.2Hz,1H);2.29(ddd,J=5.1および11.1および16.0Hz,1H);2.45(ddd,J=5.4および11.2および16.1Hz,1H);2.52(t,J=6.6Hz,3H);2.79(s,3H);2.82(m,1H);2.90(m,1H);2.96(d,J=9.3Hz,1H);3.07(d,J=13.2Hz,1H);3.15(s,3H);3.28(s,3H);3.35(m,2H);3.44(m,3H);3.56(s,29H);3.61(s,3H);3.68(t,J=6.6Hz,2H);3.91(s,3H);4.20(t,J=12.0Hz,1H);4.70(dd,J=3.2および12.0Hz,1H);5.35(q,J=7.0Hz,1H);5.59(dd,J=9.0および15.4Hz,1H);6.13(s,1H);6.35(dd,J=11.0および15.4Hz,1H);6.57(d,J=1 .5Hz,1H);6.67(d,J=11.2Hz,1H);6.78(d,J=1.5Hz,1H);6.96(t,J=5.6Hz,1H)。
【0160】
13.2.3−{2−[2−(2−{2−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルの調製
【0161】
【化49】
磁気撹拌下、Arの不活性雰囲気下、RTで、200mgの3−[2−(2−{2−[2−(2−{2−[2−(2−アミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸(CA(PEG)8、Pierce)、1.7mlのDCM、0.9mlのMeOH、および0.34mlの2Mトリメチルシリルジアゾメタンのヘキサン中溶液をガラスバイアル内に逐次的に導入する。RTで30分後、0.1mlの2Mトリメチルシリルジアゾメタンのヘキサン中溶液を添加する。RTで25分後、数滴の酢酸の添加により反応混合物を中和させ、RP下で濃縮乾燥させ、トルエンで共沸混合させる。こうして得た無色の油を、106.9mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルの、0.7mlのDCM中溶液で希釈する。RTで30分後および4℃で16時間後、粗製物を、20gのCN−グラフトシリカゲル(n.ヘプタン/iPrOH/AcOEtの勾配溶出、iPrOH部分を増加させる)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、175mgの3−{2−[2−(2−{2−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルを無色の油の形態で得る。質量スペクトル(B):tR=2.79分;[M+H]+:m/z576/578。
【0162】
ブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルは、公開プロトコル(Biochemistry、1974年、481頁)に従い調製することができた。
【0163】
[実施例14]
L−DM4−Mal−PEG4−COOMe
【0164】
【化50】
14.1遊離薬物L−DM4−Mal−PEG4−COOMeの調製
【0165】
【化51】
磁気撹拌下、RTで、160mgのL−DM4、115.8mgの3−{2−[2−(2−{2−[3−(2,5−ジオキソ−2,5−ジヒドロ−ピロール−1−イル)−プロピオニルアミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステル(市販のもの、SM(PEG)4、Pierce)、0.6mlのDMA、55.1mgの担持されたDIPEA(3.72mmol/g)、0.3mlの追加のDMAおよび6μlのDIPEAを逐次的に添加する。RTで1時間後および−20℃で16時間後、粗製の反応混合物をろ過し、DCMで洗浄し、20gのシリカゲル(勾配溶出、DCM:MeOH、MeOHの分担を増加)上でフラッシュクロマトグラフィーにより精製する。所期の生成物を含有する画分をRP下で濃縮後、110mgの無色の被膜を得て、これを10gのシリカゲル(勾配溶出、DCM:MeOH、MeOHの分担を増加)上で精製する。所期の生成物を含有する画分をRP下で濃縮後、19.6mgのメチルエステルL−DM4−Mal−PEG4−COOMeを無色のガラスの形態で得る。質量スペクトル(A):tR=1.31/1.32分(2つのジアステレオ異性体);[M−H]−:m/z 1208。1H NMR(500MHz,ppm単位,クロロホルム−d):0.73(s,3H);1.22(m,13H);1.39(m,1H);1.52(d,J=13.7Hz,1H);1.57(s,3H);1.78(m,1H);1.99(m,1H);2.11(ddd,J=1.8および1.9および14.1Hz,1H);2.24(ddd,J=4.7および11.5および15.9Hz,1H);2.38(m,3H);2.46(m,1H);2.53(m,3H);2.69(s,1H);2.82(s,3H);2.94(dd,J=4.7および9.6Hz,1H);3.08(m,3H);3.14(s,3H);3.29(s,3H);3.34(q,J=5.3Hz,2H);3.43(d,J=9.1Hz,1H);3.48(td,J=1.8および5.1Hz,2H);3.58(s,13H);3.67(m,7H);3.91(s,3H);4.22(t,J=11.3Hz,1H);4.70(m,1H);5.29(m,1H);5.59(m,1H);6.30(s 大,1H);6.35(dd,J=11.0および15.4Hz,1H);6.61(m,2H);6.76(s,1H)。
【0166】
[実施例15]
MDA−MB−231およびHCT116腫瘍細胞の増殖の阻害
指数増殖期の細胞をトリプシン処理し、これらのそれぞれの培地内に再懸濁させた(DMEM/F12 Gibco#21331;10%SVF Gibco#10500−056;MDA−MB231&MDA−A1細胞に対して2nMグルタミンGibco#25030;DMEM(Gibco#11960)10%SVF Gibco#10500−056;HCT116細胞に対して2nMグルタミンGibco#25030)。完全な血清含有培地の中で、5000細胞/ウェルの密度で(MDA−MB231、HCT116)、細胞懸濁液を96−ウェルCytostar培養物プレート(GE Healthcare Europe、#RPNQ0163)に分配した。4時間のコーティング後、3連ウェルに段階希釈を添加した。薬物の存在下、37°C/5%C02で3日間細胞を培養した。第4日目、10μlの14Cチミジン溶液(0.1μCi/ウェル(Perkin Elmer#NEC56825000)を各ウェルに添加した。microbeta放射性カウンター(Perkin Elmer)を用いて、実験が開始してから96時間、14Cチミジンの取り込みを測定した。無細胞の試薬ブランクを試験ウェル記録から引き算し、コンジュゲート処理した細胞の記録を、ビヒクル処理した細胞の対照ウェルからの記録の平均で割ることによって得た、残存する画分として、このデータをプロットした。
【0167】
【表4】
【0168】
これらから明白なように、エステル−COOMeの形態で試験された生成物は、2つの異なる細胞株に対して強力なインビトロの増殖阻害特性を示す。
【0169】
[実施例16]
hu2H11およびhu2H11R35R74のコンジュゲートのインビボの評価
コンジュゲートの制癌活性の評価のため、動物を毎日秤量し、週に2回キャリパーで腫瘍を測定した。質量(mg)=[長さ(mm)×幅(mm)2]/2の式を用いて腫瘍の重量を計算した。抗腫瘍活性の評価は、最大非毒性用量(HNTD)で行った。
【0170】
最下点(グループの平均)での20%の体重減(bwl)または10%以上の薬物死が生じる用量は、過剰に有毒な用量とみなされた。動物の体重には腫瘍の重量が含まれていた。主要評価項目は、ΔT/ΔC、メディアン退縮パーセント、部分退縮および完全退縮(PRおよびCR)および無腫瘍生存(TFS)である。
【0171】
特定された観察日の腫瘍量から最初の処理日(ステージング日)の腫瘍量を引くことによって、処理した腫瘍(T)および対照(C)のそれぞれの腫瘍量の変化を各腫瘍に対して計算する。メディアンΔΤを処理したグループに対して計算し、メディアンΔCを対照グループに対して計算する。次いで割合ΔΤ/ΔCを計算し、パーセンテージで表現する。
【0172】
【数1】
【0173】
用量は、ΔΤ/ΔCが40%より低い場合、治療的に活性があるとみなされ、ΔΤ/ΔCが10%よりも低い場合、非常に活性があるとみなされる。ΔΤ/ΔCが0より低い場合、この用量は、高活性とみなされ、この退縮パーセンテージの日付を記録する(ref1):
【0174】
%腫瘍退縮:これは、最初の処理の初日におけるこの量と比較した場合の、特定された観察日における処理したグループの腫瘍量の減少の%と定義される。特定の時間点および各動物に対して、%退縮を計算する。よって、グループに対するメディアン%退縮を計算する:%退縮(tにおいて)=
【0175】
【数2】
【0176】
部分退縮(PR):退縮は、処理開始時の腫瘍量の50%まで腫瘍量が低減した場合、部分的と定義される。
【0177】
完全退縮(CR):完全退縮は、腫瘍量=0mm3の場合達成される(腫瘍量が記録できない場合CRが考慮される)。
【0178】
TFS:無腫瘍とは、実験の終わりに、腫瘍が検出不能な動物と定義される。
【0179】
hu2H11−コンジュゲートとhu2H11R35R74−コンジュゲートとの比較
hu2H11−コンジュゲート=
【0180】
【化52】
hu2H11R35R74−コンジュゲート=
【0181】
【化53】
メスのSCIDマウスにS.C.移植した、強力に発現するターゲットである、測定可能な原発性結腸腫瘍、CR−LRB−004Pに対して、hu2h11−コンジュゲートおよびhu2h11R35R74−コンジュゲートの抗腫瘍活性を2つの用量レベルで評価した。対照グループは未処理のままにしておいた。用量は、1キログラム中のタンパク質をミリグラムで表現した。hu2h11R35R74−コンジュゲートを、40および10mg/kgの量で、静脈内(IV)大量瞬時投与により、第15日に投与した。等しい用量のDM4を与えるため、hu2h11−コンジュゲート、44および11mg/kgを投与した。結果を表IVに示す。
【0182】
CR−LRB−004P腫瘍の単回投与スケジュールを用いて、hu2h11R35R74−コンジュゲートは、40および10mg/kgにおいて活性があり、ΔΤ/ΔΟがそれぞれ28%および39%であり、hu2h11−コンジュゲートは、40mg/kgにおいてのみ活性があり、ΔΤ/ΔΟは26%であった。10mg/kgにおいて、hu2h11−コンジュゲートは、本モデルでは活性がなかった。これらの結果から、hu2h11R35R74−コンジュゲートは、低用量では、hu2h11−コンジュゲートよりも良い活性を示した。
【0183】
コンジュゲートの最適化、最適な薬物抗体比DARの選択−SCIDメスのマウスにおける前立腺の腺癌PC−3に対するhu2H11R35R74−コンジュゲートの制癌活性に対するDARの影響
メスのSCIDにS.C.移植した、前立腺のPC−3腫瘍について、6つの異なる薬物抗体比(DAR)での、2つの低い有効用量を比較して、hu2H11R35R74−コンジュゲートの抗腫瘍活性に対するDARの作用を評価した。対照グループは未処理のままにしておいた。用量は、1キログラム中のタンパク質をミリグラムで表現した。DARをUV法で求めた。hu2H11R35R74−コンジュゲートを、10および5mg/kgの量で、それぞれ3.4、4.4、5.9、6.2、7.4および8.4のDARで、静脈内(IV)大量瞬時投与により、第16日に投与した。結果を表Vに示す。
【0184】
【表5】
【0185】
【表6】
【0186】
単回投与スケジュールを用いて、hu2H11R35R74−コンジュゲートは、10mg/kgにおいて、DAR4.4から8.4で活性を示し、hu2H11R35R74−コンジュゲートは、5mg/kgにおいて、DAR5.9から4で活性を示した。結論として、DARは、hu2H11R35R74−コンジュゲートの腫瘍活性について効果を有する。これらの結果から、特定の腫瘍について、DAR(UV)は、4より上であるべきである。最適なDARは、少なくとも5.9に等しいことになる。
【0187】
[実施例17]
hu2H11R35R74−コンジュゲートのPKパラメータについてのDARの評価
異なる薬物−抗体比(DAR)でのhu2H11R35R74−コンジュゲートの薬物動態学的特性を、20mg/kgのコンジュゲートを単回の静脈内(IV)投与後、オスのCD−1マウスにおいて評価した。コンジュゲートの血漿レベルを測定することによって、標準的条件下での基本的な単回投与の薬物動態学的パラメータを確立した。PKパラメータを裸の親抗体のものと比較した。コンジュゲートおよびこれらの抗体成分の血漿中濃度(全抗体、結合した抗体とあらゆる非結合の抗体の和)を特定のELISA技法で測定した。結果を図7に示す。
【0188】
結果は、DAR値と全抗体成分への曝露との間の逆の相互関係を示し、AUC0−∞値は、0、3.4、4.3、5.9、6.6および7.4のDARに対して、それぞれ83,000,000、61,000,000、48,000,000、46,000,000、41,000,000および27,000,000ng・h/mLであった。
【0189】
同様に、DAR値とコンジュゲートへの曝露との間に逆の相互関係が存在し、AUC0−∞値は、3.4、4.3、5.9、6.6および7.4のDARに対して、それぞれ39,000,000、30,000,000、27,000,000、29,000,000および20,000,000ng・h/mLであった。
【0190】
DAR値と、抗体成分の排除との間に完璧な相互関係が存在し、CI値は、0、3.4、4.3、5.9、6.6および7.4のDARに対して、それぞれ0.00024、0.00033、0.00042、0.00043、0.00049および0.00074L/h/kgであった。
【0191】
同様に、DAR値とコンジュゲートの排除との間にほとんど完璧な相互関係が存在し、CI値は、3.4、4.3、5.9、6.6および7.4のDARに対してそれぞれ0.00051、0.00066、0.00075、0.00069、0.00099L/h/kgであった。
【0192】
結論として、DARは、PKパラメータに影響を及ぼし、DARが増加すると、曝露が減少し、除去が増加する。効力およびPKの評価結果によると、最適なDARは、5.9と7.4の間に含まれていることになる。
【0193】
[実施例18]
SCIDメスのマウスにおける前立腺の腺癌PC−3に対するhu2H11R35R74コンジュゲートの評価
メスのSCIDマウスにS.C.移植した、強力に発現するターゲットである、測定可能な前立腺PC−3腫瘍に対して、DAR=5.9を有する抗体薬物コンジュゲートhu2H11R35R74−コンジュゲートの抗腫瘍作用を、8つの用量レベルで評価した。対照グループは、未処理のままにしておいた。用量は、1キログラム中のタンパク質をミリグラムで表現した。これらを、160、120、80、40、20、10、5および2.5mg/kgの量で、静脈内(IV)大量瞬時投与により、第17日に投与した。結果を表VIに示す。
【0194】
単回投与スケジュールを用いて、体重減少および薬物に関連した死亡を誘発する有毒な量となる、試験したコンジュゲートの最大用量(160mg/kg)を発見した。HNTD(120mg/kg)および他の最小用量において、この化合物は高活性であった。2.5mg/kg以外のすべて用量に対して、hu2H11R35R74−コンジュゲートは、部分退縮を誘発し、120、80および20mg/kgに対して、完全退縮を誘発した。加えて、この腫瘍モデル悪液質であり、化合物の投与は、対照と比較して、最下点での体重減少を減少させた。結論として、hu2H11R35R74−コンジュゲートは、前立腺のPC−3腫瘍モデルにおいて、優れた用量−効果を有する高い活性を示した。
【0195】
【表7】
【技術分野】
【0001】
本発明は、新規メイタンシノイド、および抗体とのコンジュゲートを調製するための前記メイタンシノイドの使用に関する。本発明はまた、前記メイタンシノイドおよび前記コンジュゲートを含む組成物に関する。
【背景技術】
【0002】
モノクローナル抗体−薬物コンジュゲートを用いて、腫瘍細胞を特異的に対象とする試みについて多くの論文が出現した(Selaら、Immunoconjugates、189−216頁(C.Vogel編.1987年);Ghoseら、Targeted Drugs、1−22頁(E.Goldberg編、1983年);Dienerら、Antibody mediated delivery systems、1−23頁(J.Rodwell編、1988年);Pieterszら、Antibody mediated delivery systems、25−53頁(J.Rodwell編、1988年);Bumolら、Antibody mediated delivery systems、55−79頁(J.Rodwell編、1988年)。また以下も参照されたい:Monneret Cら、Bulletin du Cancer、2000年、87(11)、829−38頁;Ricart A.D.ら、Nature Clinical Practice Oncology 2007年、4、245−255頁;Singh R.およびRickson H.K.、Therapeutic Antibodies:Methods and Protocols、2009年、525、445−467頁。タキサン誘導体(WO06061258)、レプトマイシン誘導体(WO07144709)、CC−1065および類似体(WO2007102069)など、またはメトトレキサート、ダウノルビシン、ドキソルビシン、ビンクリスチン、ビンブラスチン、メルファラン、マイトマイシンC、クロランブシルなどの異なるファミリーの細胞毒性薬が、抗体との結合に使用されてきた。
【0003】
腫瘍細胞に対する親和性を有する標的抗体の使用は、細胞毒性薬を腫瘍細胞の近くに直接または腫瘍細胞内に直接送達することを可能にし、したがって、細胞毒性薬に通常伴う副作用を最小限に抑えながら、細胞毒性薬の効率を増加させる。
【0004】
メイタンシノイドは、キャストアフリカンシュラブ、メイテナス・セラタ(Maytenus serrata)から単離した天然物であるメイタンシンから誘導される細胞毒性薬である(US3896111)。多くのメイタンシノイドが調製されてきた。以下を参照:US4151042;J.Med.Chem.1978年、21、31−37頁;Nature、1977年、270、721−722頁、Chem.Pharm.Bull.1984年、3441−3451頁;US4248870;US4137230;Chem.Bull.1984年、3441頁。
【0005】
従来技術
US5208020、US5416064、およびR.V.J.Chari、31 Advanced Drug Delivery Reviews、89−104頁(1998年)は、L−DM1(A)またはL−DM4(Α’):
【0006】
【化1】
などのメイタンシノイドのコンジュゲートについて記載している。
【0007】
メイタンシノイドのコンジュゲートは、EP0425235およびWO2004/103272で記載されている。EP0425235において、式(B1)、(B2)または(B3)の以下のメイタンシノイドが記載されている:
【0008】
【化2】
(式中、Z0、Z1またはZ2は、HまたはSRを表す。)。
【0009】
WO2004/103272では、式(C)のメイタンシノイドが記載されている:
【0010】
【化3】
(式中、Y’は、
(CR7CR8)i(CR9=CR10)pC≡CqAr(CR5CR6)mDu(CR11=CR12)r(C≡C)sBt(CR3CR4)uCR1R2SZを表し、A、BおよびDは、場合によって置換されているシクロアルキル、シクロアルケニル、ヘテロアリールまたはヘテロシクロアルキル基を表す。)。
【0011】
WO03/068144では、式(D)の化合物が記載されている。
【0012】
【化4】
(式中、Zは、細胞毒性薬であり、Qは、R2COO、R2R3NCOO、R2OCOO、R2O、R2CONR3、R2R3N、R2OCONR3またはSである。R2は、SCR4R5R6である。)。Zは、以下のメイタンシノイド誘導体の中から選択されるメイタンシノイド誘導体であってよい。
【0013】
【化5】
【0014】
さらに具体的には、以下の化合物(D)が開示されている:
【0015】
【化6】
したがって化合物(D)は、ペグ化リンカーの中に内部ジスルフィド結合を含有している。
【0016】
Immunogen Inc.はまた、2008年12月8日、GenevaのEuropean Antibody Congressにおいて、式(E)のコンジュゲートを開示し、
【0017】
【化7】
Genevaで2008年10月に開催された、第20回シンポジウムEORTC−NCI−AACRにおいて、式(Ε’)のコンジュゲートを開示した。
【0018】
【化8】
【先行技術文献】
【特許文献】
【0019】
【特許文献1】国際公開第06061258号
【特許文献2】国際公開第07144709号
【特許文献3】国際公開第2007102069号
【特許文献4】米国特許第4151042号明細書
【特許文献5】米国特許第4248870号明細書
【特許文献6】米国特許第4137230号明細書
【特許文献7】米国特許第5,208,020号明細書
【特許文献8】米国特許第5,416,064号明細書
【特許文献9】欧州特許第0425235号明細書
【特許文献10】国際公開第2004/103272号
【特許文献11】国際公開第03/068144号
【非特許文献】
【0020】
【非特許文献1】Selaら、Immunoconjugates、189−216頁(C.Vogel編.1987年)
【非特許文献2】Ghoseら、Targeted Drugs、1−22頁(E.Goldberg編、1983年)
【非特許文献3】Dienerら、Antibody mediated delivery systems、1−23頁(J.Rodwell編、1988年)
【非特許文献4】Pieterszら、Antibody mediated delivery systems、25−53頁(J.Rodwell編、1988年)
【非特許文献5】Bumolら、Antibody mediated delivery systems、55−79頁(J.Rodwell編、1988年)
【非特許文献6】Monneret Cら、Bulletin du Cancer、2000年、87(11)、829−38頁
【非特許文献7】Ricart A.D.ら、Nature Clinical Practice Oncology 2007年、4、245−255頁
【非特許文献8】Singh R.およびRickson H.K.、Therapeutic Antibodies:Methods and Protocols、2009年、525、445−467頁
【非特許文献9】J.Med.Chem.1978年、21、31−37頁
【非特許文献10】Nature、1977年、270、721−722頁
【非特許文献11】Chem.Pharm.Bull.1984年、3441−3451頁
【非特許文献12】Chem.Bull.1984年、3441頁
【非特許文献13】R.V.J.Chari、31 Advanced Drug Delivery Reviews、89−104頁(1998年)
【発明の概要】
【課題を解決するための手段】
【0021】
定義
「アルキル」は、1から20個の炭素原子を鎖内に有する直鎖もしくは分枝、または3から10個の炭素原子を有する環状であってよい脂肪族の炭化水素基を意味する。好ましいアルキル基は、鎖内に1から12個の炭素原子を有する。代表的アルキル基として、メチル、エチル、n−プロピル、i−プロピル、2,2−ジメチルプロピル、n−ブチル、t−ブチル、n−ペンチル、3−ペンチル、オクチル、ノニル、デシルが挙げられる。
「シクロアルキル」は、3から10個の炭素原子を有する環式の脂肪族炭化水素基を意味する。好ましいシクロアルキル基は、環状の鎖内に3から8個の炭素原子を有する。代表的なシクロアルキル基として、シクロプロピル、シクロブチル、シクロペンチルおよびシクロヘキシルが挙げられる。
「アリール」は、6から14個の炭素原子、好ましくは6から10個の炭素原子の芳香族単環式または多環式炭化水素環系を意味する。代表的なアリール基として、フェニルまたはナフチルが挙げられる。
「ヘテロアリール」は、不飽和の安定した3から14員、好ましくは5から10員の単環式、二環式または多環式の環を意味し、この環の少なくとも1つのメンバーはヘテロ原子である。通常、ヘテロ原子は、これらに限定されないが、酸素、窒素、硫黄、セレンまたはリン原子である。ヘテロ原子は、酸素、窒素または硫黄原子が好ましい。代表的なヘテロアリール基として、ピリジル、ピロリル、チエニル、フリル、ピリミジニルおよびトリアゾリルが挙げられる。
「ヘテロシクロアルキル」は、少なくとも1個のヘテロ原子を含有するシクロアルキル基を意味し、この環の少なくとも1つのメンバーはヘテロ原子である。
「アルコキシ」は、−O−アルキル基を意味し、アルキルは上記のように定義される。
「アルコキシルオキシ」は、−O−CO−アルキル基を意味し、アルキルは上記のように定義される。
「アルキレン」は、直鎖または分枝のアルカンから2個の水素原子を除去することにより形成される一般式−CmH2m−のアルキル基を意味する。代表的アルキレン基として、メチレン(−CH2−)、エチレン(−CH2CH2−)、プロピレン(−CH2CH2CH2−)、ブチレン(−CH2CH2CH2CH2−)、イソブチレン
【0022】
【化9】
、ヘキシレン(−CH2CH2CH2CH2CH2CH2−)が挙げられる。直鎖アルキレン基は、具体的には式−(CH2)m−(式中、mは、1から20の整数である。)で表すことができる。
「EphA2受容体」は、Eph受容体ファミリーに属するチロシンキナーゼ(Pasquale、E.B.ら、2005年、Nature Reviews Mol.Cell Biol.、6、462−475頁で概説されている)を指し、例えばGenbank受託番号NM_004431(ヒトEphA2)、NM_010139(マウスEphA2)またはNXM_345596(ラットEphA2)などのアミノ配列を含む。ヒトEphA2は、好ましいEph受容体である。「EphA2リガンド」という用語は、本明細書で使用する場合、EphA2受容体に結合し、場合によってはこれを活性化する(例えば、この自己リン酸化を刺激する)タンパク質を指す。本明細書中の好ましいEphA2リガンドは、「エフリンA1」であり、これは、EphA2受容体に結合し、例えばGenbank受託番号NM_004428(ヒトエフリンA1)などのようなアミノ配列を含む。
「ポリクローナル抗体」とは、1つ以上の他の、非同一の抗体の間でまたはそれらの存在下で産生された抗体である。一般的に、多クローン性抗体は、非同一の抗体を産生する他のいくつかのB−リンパ球の存在下でB−リンパ球から産生される。通常、多クローン性抗体は免疫された動物から直接得る。
「モノクローナル抗体」とは、実質的に同種の抗体の集団、すなわち微量で存在することがある、自然発生で起こり得る変異を除いて、基本的に同一であるこの集団を形成する抗体から得られる抗体である。これらの抗体は、単一のエピトープを対象とし、したがって極めて特異的である。
「裸の抗体」とは、メイタンシノイドと結合していない抗体である。
「エピトープ」とは、抗体が結合する抗原上の部位である。これは、抗原タンパク質のフォールディングにより近接近させられる隣接する残基または隣接しない残基で形成することができる。隣接するアミノ酸で形成されるエピトープは通常、非変性溶媒へ曝露されたまま保持されるが、隣接しないアミノ酸で形成されるエピトープは通常、前記曝露下で失われる。
【図面の簡単な説明】
【0023】
【図1】実施例1の脱グリコシル化コンジュゲートの逆畳み込み後のHRMSスペクトルを示す図である。
【図2】実施例2の脱グリコシル化コンジュゲートの逆畳み込み後のHRMSスペクトルを示す図である。
【図3】実施例3の脱グリコシル化コンジュゲートの逆畳み込み後のHRMSスペクトルを示す図である。
【図4】実施例4の脱グリコシル化コンジュゲートの逆畳み込み後のHRMSスペクトルを示す図である。
【図5】実施例5の脱グリコシル化コンジュゲートの逆畳み込み後のHRMSスペクトルを示す図である。
【図6A】配列を示す記号である。
【図6B】配列を示す記号である。
【図6C】配列を示す記号である。
【図7】様々なDARにおける、hu2H11R35R74コンジュゲートに対するPKパラメータ:HGS中のコンジュゲート20mg/kgをオスのCD−1マウス(n=4)に、単回投与の静脈内投与をした後、(AUC(O−inf);左)への曝露およびいくつかのコンジュゲートのクリアランス(CI;右)を、DARの関数として棒グラフにより説明するグラフである。グラフにおいて、2H11−DM4(底部)は、hu2H11R35R74−コンジュゲートを指す。
【発明を実施するための形態】
【0024】
これらの図は、各コンジュゲートについて、0から10のメイタンシノイド(D0:メイタンシノイドなし;Dx:x個のメイタンシノイド)を保持するコンジュゲートの分布が存在することを示している。
【0025】
新規メイタンシノイド
本発明は、式(I)の化合物
【0026】
【化10】
(式中、
ALKは、(C1−C6)アルキレン基であり、
X1およびX2は、それぞれ独立して、以下の基:−CH=CH−、−CO−、−CONR−、−NRCO−、−COO−、−OCO−、−OCONR−、−NRCOO−、−NRCONR’−、−NR−、−S(O)n(n=0、1または2)または−O−のうちの1つであり;
RおよびR’は、独立して、Hまたは(C1−C6)アルキル基であり;
iは、1から40、好ましくは1から20、より好ましくは1から10の整数であり、
jは、X2が−CH=CH−の場合、1に対応する整数であり、X2が−CH=CH−でない場合、2に対応する整数であり、
Zbは、単一結合、−O−または−NH−であり、Rbは、Hまたは(C1−C6)アルキル、(C3−C7)シクロアルキル、アリール、ヘテロアリールもしくは(C3−C7)ヘテロシクロアルキル基であり、またはZbは、一重結合であり、RbはHalである。)に関する。
【0027】
より具体的には、X2は、−CH=CH−または−CONR−であり、CO基は、−X1−ALK−基に連結し、Rは、Hまたは(C1−C6)アルキル基である。より具体的には、−X1−ALK−は−S−CH2−である。より具体的には、lは、3、4、5、6、7、8、9または10である。
【0028】
式(II)の化合物を区別することができる。
【0029】
【化11】
【0030】
式(II)は、以下の化合物を、さらに正確には網羅する。
【0031】
【化12】
【0032】
化合物は、塩基もしくは塩の形態で、または前記塩基もしくは前記塩の溶媒和物もしくは水和物の形態で存在することができる。
【0033】
本発明の化合物は、抗体上に存在する反応性化学基(GCR2)に対して反応性のある、反応性化学基−C(=O)ZbRb(GCR1)を含む。GCR1とGCR2の間の反応により、共有結合を介して抗体上に細胞毒性薬が結合できるようになる。したがって、この化合物は、抗体に結合する傾向がある。より具体的には、ZbはOである。このような場合、GCR1は、カルボン酸官能基(Rb=H)またはエステル官能基である。より具体的には、−C(=O)ZbRbは、−COOH、−COO(C1−C6)アルキル、例えば−COOCH3または−COOCH2CH=CH2である。エステル官能基の中でも、抗体(リジン基など)のアミノ基に対して優れた反応性を示す「活性化された」ものが好ましい。例えば活性化エステルは、以下のエステルであってよい:
【0034】
【化13】
または基
【0035】
【化14】
(式中、GIは、少なくとも1つの誘導性の基、例えば−NO2または−Hal、例えば−Fなどを表す。)。このような活性化エステルの例は、以下のエステルである。
【0036】
【化15】
別の−C(=0)ZbRbは、
【0037】
【化16】
である。
【0038】
GCR2は、例えば抗体の表面のリジン残基の側面上のリジン、ヒンジ領域のサッカライド基または鎖内S−S結合の還元後のシステインのチオール基により生じるε−アミノ基であってよい(Garnett M.C.ら、Advanced Drug Delivery Reviews、2001年、53、171−216頁)。さらに最近になって、新規な手法は、変異によりシステインを導入すること(Junutula J.R.ら、Nature Biotechnology、2008年、26、925−932頁;WO09026274)または新しいタイプのタンパク質の化学的性質の開発を可能とする非天然アミノ酸を導入すること(de Graaf A.J.ら、Bioconjugate Chem.2009年、2009年2月3日、(Review);DOI:10.1021/bc800294a;WO2006/069246およびChin J.W.ら、JACS、2002年、124、9026−9027頁(technology ReCode(登録商標)))を目標とした。
【0039】
本発明の化合物は、式:
【0040】
【化17】
の少なくとも1つのメイタンシノイドフラグメントに共有結合で付加しているコンジュゲートを調製するために使用することができる。
【0041】
したがって、式(I)の化合物を、メイタンシノイドフラグメントが抗体に共有結合で連結しているコンジュゲートを調製するために使用することができる。
【0042】
式(I)の化合物を調製するための一般的スキーム
式(I)の化合物をスキーム1に従い調製することができる:
【0043】
【化18】
【0044】
中間体P1は、中間体P2を含有するPEGに付加している反応性基RG2と反応することによって、X1を形成することができるRG1反応性基を含有する。例えば、X1=Sの形成は、DIEAなどの塩基の存在下、求核置換によるP1とRG1=−SHの反応ならびにP2とRG2=−Brの反応を介して行うことができる。この反応の例は、実施例1.2において示されている。
【0045】
RG1=−SHでのP1の例は、L−DM1およびL−DM4であり、またEP1313738の化合物11a、c、d、gでもある。
L−DM1:ALK−SH=−CH2CH2−SH;
L−DM4:ALK−SH=−CH2CH2CMe2−SH;
11a:ALK−SH=−CH2−SH;
11c:ALK−SH=−CH2CH2CH2−SH;
11d:ALK−SH=−CH2CH2CH2CH2−SH;
11g:ALK−SH=−CHMe−CH2−SH
【0046】
反応の他の例が、表Iに示されている。
【0047】
【表1】
【0048】
いくつかの式(I)の化合物については、P1とP2を反応させた後で、別の−ZbRb基の少なくとも1つを変換後、所望する最終の−ZbRb基を得てもよい。変換−ZbRb=−O−アリル→
【0049】
【化19】
と共に、スキーム1’で一例を示す。
【0050】
【化20】
【0051】
同様に、P1とP2との間の反応の前に、−ZbRb基を保持する中間体に対して少なくとも1つの変換を使用することもできる。
【0052】
別の例は、例えばSOCl2などのアシル化剤を必要とする、変換−ZbRb=−OH→ZbRb=Halである。
【0053】
P1の調製
メイタンシノールから開始して、スキーム2に従いP1を調製することができる。
【0054】
【化21】
【0055】
メイタンシノールを、反応性アシル基−COOZ(式中、Zは、Hまたはハロゲン原子である。)を含有する中間体P3と、エステル化反応により反応させる。この反応は、WO2004/103272の図3a−dに記載され、WO2007/021674にも記載されている。ZがHの場合、酸官能基の反応性を強化するカップリング剤の助けを借りてエステル化を行うことができる。
【0056】
P2の調製
P2を調製するための出発物質は、市販のPEG化合物または当業者に既知の少なくとも1つの化学反応を介して、前記市販のPEG化合物を用いて調製することができるPEG化合物である。PEG化合物は、例えばJenKem Technology USA Inc.、2033 W.McDermott Dr.Suite 320 #188、Allen、TX 75013−4675、USAから、市販されている。
【0057】
例えば、P2(式中、X2=−CONR−であり、RG2=Hal(P2=Hal−ALK−CONR−CH2CH2(OCH2CH2)i−COZbRb))の調製が、市販の化合物HOOCCH2CH2(OCH2CH2)i−OCH2CH2NH2を用いて以下に記載されている:
R=H
【0058】
【化22】
【0059】
ステップ(i):アミド結合の形成および酸性基の活性化;2つのステップを、DCMなどの極性の非プロトン性溶媒中で別々に行う:アミン基と、ハロゲノアルカン酸N−ヒドロキシスクシンイミジンエステル(例えば、ハロゲノ酢酸)とを反応させ、次いでDICなどのカップリング剤をその場で添加する。
R≠H
【0060】
【化23】
【0061】
ステップ(ii):エステルの形態でのカルボン酸の保護およびトリフルオロアセトアミドの形態でのアミンの保護;この反応は、DCMなどの極性の非プロトン性溶媒中、2つの別々のステップで行う:メタノールを用いて、トリメチルシリルジアゾメタンでの処理により酸を保護し、次いでTFAAおよびTEAの添加によりアミンを保護する。
【0062】
ステップ(iii):アミンのアルキル化およびエステルのアルカリ加水分解;この反応は、THFなどの極性の非プロトン性溶媒中、2つの別々のステップで行う:ハロゲン化アルキルRHalなどの脱離基を保持する反応体の存在下、NaHでの処理によりアミンをアルキル化し、水中でLiOHを添加する。
【0063】
ステップ(iv):ステップ(iii)に続いて、ステップ(i)の反応(式中、R=H)を行う。
【0064】
同様に、P2(式中、X2=−CH=CH−およびRG2=Hal(P2=Hal−CH2−CH=CH−CH2(OCH2CH2)i−COZbRb))の調製が、市販の化合物Hal−CH2CH=CHCH2(OCH2CH2)i−COOtBuを用いて以下に記載されている:
【0065】
【化24】
【0066】
コンジュゲートの調製方法
コンジュゲートは、以下のステップを含む方法により得ることができる:
(i)場合によって緩衝させた抗体水溶液を式(I)の化合物の溶液と接触させるステップ、
(ii)次いで、反応していない試薬および溶液中に存在し得るいかなる凝集体から(i)において形成されたコンジュゲートを場合によって分離するステップ。
【0067】
細胞結合剤の水溶液は、少なくとも1つの緩衝液、例えばリン酸カリウムまたはN−2−ヒドロキシエチルピペラジン−N’−2−エタンスルホン酸(Hepes緩衝液)など、または緩衝液混合物、例えば以下の実施例において開示されている緩衝液Aなどを用いて緩衝させることができる。緩衝液は、抗体の性質に依存する。式(I)の化合物は、有機極性溶媒(または極性溶媒の混合物)、例えばDMSOまたはDMA中で溶液である。
【0068】
反応の温度は通常、20から40℃の範囲で変動する。反応時間は、1から24時間の範囲で変動し得る。抗体と細胞毒性薬との間の反応は、屈折率検出器および/またはUV検出器を備えたサイズ排除クロマトグラフィー(SEC)でモニターすることができる。コンジュゲートの収率が低すぎる場合、反応時間を延長することができ、および/または式(I)の化合物を添加することができる。
【0069】
当業者であれば、多数の異なるクロマトグラフィー法を使用することによって、ステップ(ii)の分離を実施することができる:コンジュゲートは、例えばSEC、吸着クロマトグラフィー(例えばイオン交換クロマトグラフィー、IECなど)、疎水性相互作用クロマトグラフィー(HIC)、アフィニティクロマトグラフィー、混合担持クロマトグラフィー、例えばハイドロキシアパタイトクロマトグラフィーなど、または高速液体クロマトグラフィー(HPLC)により精製することができる。透析法または膜分離法による精製もまた使用することができる。
【0070】
使用することができる方法の例が、実施例1に記載されている。
【0071】
本明細書で使用する場合、「凝集体」という用語は、2つ以上の抗体の間に形成し得る連結を意味し、前記抗体は、改質されているまたは改質されていない。凝集体は、多数のパラメータ、例えば、溶液中の高濃度の抗体、溶液のpH、高い剪断力、結合したダイマーの数およびこれらの疎水性の性質、温度などの影響下で形成し得る(Wang & Gosh、2008年、J.Membrane Sci.、318:311−316頁および本明細書中に引用された参考文献を参照);これらパラメータのうちのいくつかの相対的影響は、明確に確立されていないことに注意されたい。タンパク質および抗体の場合、当業者であれば、Cromwellら(2006年、AAPS Journal、8(3):E572−E579)を参照することになる。凝集体中の含量は、当業者に周知の技法、例えばSEC(Walterら、1993年、Anal.Biochem.、212(2):469−480頁を参照)などを用いて求めることができる。
【0072】
ステップ(i)または(ii)の後、コンジュゲート含有溶液に、限外ろ過法および/または膜分離法の追加のステップ(iii)を施すことができる。
【0073】
コンジュゲートは、これらのステップの終わりに水溶液として回収される。
【0074】
抗体
「抗体」という用語は、もっとも広範な意味において本明細書中で使用され、任意のイソタイプ、例えばIgG、IgM、IgA、IgDおよびIgEなどのモノクローナル抗体(全長モノクローナル抗体を含む)、多クローン性抗体、多特異的抗体、キメラ抗体および抗体フラグメントを具体的に網羅する。特定の抗原に反応する抗体は、組換え型の方法、例えばファージまたは類似ベクターにおける組換え型抗体のライブラリーの選択などによって、または抗原もしくは抗原をコードする核酸で動物に免疫性を与えることによって生成することができる。
【0075】
典型的な抗体は、ジスルフィド結合で結合されている2つの同一の重鎖および2つの同一の軽鎖から構成される。各重鎖および軽鎖は、一定の領域および可変領域を含有する。本明細書で使用する場合、「VH」または「VH」は、抗体のイムノグロブリン重鎖の可変領域を指し、これには、Fv、scFv、dsFv、Fab、Fab’またはF(ab’)2フラグメントの重鎖が含まれる。「VL」または「VL」の言及は、抗体のイムノグロブリン軽鎖の可変領域を指し、これには、Fv、scFv、dsFv、Fab、Fab’またはF(ab’)2フラグメントの軽鎖が含まれる。各可変領域は、「相補性決定領域」(「CDR」)または「超可変領域」と呼ばれる3つのセグメントを含有し、これらの領域は、抗原のエピトープを結合することに主に関与している。これらは通常、N−末端から連番で、CDR1、CDR2およびCDR3と呼ばれる。可変領域のさらに高度に保護された部分は、「フレームワーク領域」(「FR」)と呼ばれる。ネイティブの重鎖および軽鎖の可変ドメインは、それぞれ4つのFR領域を含み、これらの領域は、βシート配置を広く採用し、3つのCDRで結合されており、これら3つのCDRは、βシート構造を結合しているループを形成し、場合によってβシート構造の一部を形成する。各鎖内のCDRは、FR領域により近接近で一緒にまとめて保持され、他の鎖からのCDRにより、抗体の抗原結合部位の形成に寄与する(Kabatら、Sequences of Proteins of Immunological Interest、第5版、National Institute of Health、Bethesda、MD、1991年を参照)。
【0076】
抗体(さらなる詳細については、Janewayら、「Immunobiology」、第5版、2001年、Garland Publishing、New Yorkを参照)は、例えばWO04043344、WO08010101、WO08047242またはWO05009369(抗−CA6)などに記述されている抗体の中から選択することができる。
【0077】
クラスAのEphレセプターファミリーメンバー、例えば好ましくはヒトのEphA2受容体などを認識し、前記受容体のアンタゴニストとして機能する抗体またはこのフラグメントもまた考慮することができる。この抗体には、いかなるアゴニスト活性もない。抗体またはこのエピトープ結合フラグメントは、請求項12から15に記載ものであってよい。
【0078】
ヒト化抗体またはこのエピトープ結合フラグメントは、EphA2受容体を発現する癌細胞の増殖を阻害する追加の能力を有することが好ましい。ヒト化抗体またはこのエピトープ結合フラグメントは、EphA2受容体を発現する転移性癌細胞の遊走を阻害する追加の能力を有することが好ましい。
【0079】
ヒト化抗体は、2H11R35R74抗体またはこのエピトープ結合フラグメントをヒト化することができる。ヒト化抗体は、hu53.2H11(WO2008/010101)をコードしているポリヌクレオチド配列の部位特異的突然変異誘発により得ることができる。好ましくは、再表面化またはヒト化した型の2H11R35R74抗体が提供され、この抗体またはこのフラグメントの表面曝露された残基が、軽鎖と重鎖の両方において置き換えられることによって、既知のヒト抗体の表面にさらによく類似する。ヒト化2H11R35R74抗体またはこのエピトープ結合フラグメントは、改善された特性を有する。例えば、ヒト化2H11R35R74抗体またはこのエピトープ結合フラグメントは、EphA2受容体を特異的に認識する。より好ましくは、ヒト化2H11R35R74抗体またはこのエピトープ結合フラグメントは、EphA2受容体発現細胞の増殖を阻害する追加の能力を有する。
【0080】
ヒト化型2H11R35R74抗体はまた、軽鎖と重鎖の両可変領域のこれらのそれぞれのアミノ酸配列、軽鎖と重鎖の可変領域に対する遺伝子のDNA配列、CDRの同定、これら表面アミノ酸の同定、ならびに組換え型の形態でのこれらの発現のための手段の開示に関して本明細書中で完全に特徴づけられている。しかし範囲はこれら配列を含む抗体およびフラグメントに限定されない。代わりに、EphA2受容体に特異的に結合するすべての抗体およびフラグメントも考慮される。EphA2受容体に特異的に結合する抗体およびフラグメントは、受容体の生物活性をアンタゴナイズすることが好ましい。このような抗体はさらに、アゴニスト活性が実質的にはないことがより好ましい。したがって、抗体およびエピトープ結合抗体フラグメントは、これらの骨格、CDR、ならびに/または軽鎖および重鎖のアミノ酸配列が2H11R35R74抗体またはこのヒト化誘導体とは異なってもよく、これらは依然として本発明の範囲内に含まれる。
【0081】
2H11R35R74抗体のCDRは、モデリングにより同定され、これらの分子構造が予測されてきた。ここでもまたCDRはエピトープ認知に対して重要であるが、これらは本発明の抗体およびフラグメントには必須ではない。したがって、例えば本発明の抗体の親和性の成熟により生じる改善された特性を有する抗体およびフラグメントが提供される。
【0082】
53.2H11が誘導される可能性のある、マウス軽鎖IgVkおよびJK生殖細胞系列遺伝子および重鎖IgVhおよびJh生殖細胞系列遺伝子が同定され、WO2008/010101で開示された。前記生殖細胞系列の配列の受託番号は、それぞれMMU231196およびAF303833である。このような生殖細胞系列遺伝子配列は、CDR内のものを含めて、抗体内の体細胞変異を同定するのに有用である。
【0083】
2H11R35R74抗体の重鎖および軽鎖の可変領域の配列ならびにこれらのCDRの配列は本出願の中に記載されている。このような情報は、ヒト化型の2H11R35R74抗体を産生するために使用することができる。本発明のヒト化2H11R35R74抗体をhu53.2H11の部位特異的突然変異誘発により得ることもできる。これらヒト化抗−EphA2抗体またはこれらの誘導体も本発明のコンジュゲートの細胞結合剤として使用することができる。
【0084】
したがって、一実施形態では、本発明は、SEQ ID NO:1、2、3、4、5、6からなる群から選択されるアミノ酸配列を有する1つ以上のCDRを含むヒト化抗体またはこのエピトープ結合フラグメントを提供する。好ましい実施形態において、本発明のヒト化抗体は、少なくとも1つの重鎖と少なくとも1つの軽鎖とを含み、前記重鎖は、SEQ ID NO:1、2および3で表されるアミノ酸配列を有する3つの連続したCDRを含み、前記軽鎖は、SEQ ID NO:4、5および6で表されるアミノ酸配列を有する3つの連続したCDRを含む。
【0085】
ヒト化2H11R35R74抗体またはこのフラグメントは、SEQ ID NO.12からなるアミノ酸配列を有するVHを含むことが好ましい。SEQ ID NO14からなるアミノ酸配列を有するVLを含むヒト化2H11R35R74抗体またはこのフラグメントもまた好ましい。ヒト化2H11R35R74抗体は、少なくとも1つの重鎖および少なくとも1つの軽鎖を含み、前記重鎖は、SEQ ID NO:1、2および3で表されるアミノ酸配列を有する3つの連続したCDRを含み、前記軽鎖は、SEQ ID NO:4、5および6で表されるアミノ酸配列を有する3つの連続したCDRを含み、前記重鎖は、SEQ ID NO.12からなるアミノ酸配列を有し、前記軽鎖は、SEQ ID NO.14からなるアミノ酸配列を有することが好ましい。
【0086】
コンジュゲート
コンジュゲートは、抗体に共有結合で付加しているメイタンシノイドの1から10個の分子を全般的に含む(いわゆる「薬物/抗体比」または「DAR」)。この数は、結合に対して使用される実験条件(例えば、メイタンシノイドと、抗体、反応時間、溶媒の性質および(存在する場合)共溶媒との比)と共に、抗体の性質および使用されるメイタンシノイドにより異なり得る。したがって、抗体とメイタンシノイドの接触により、異なる薬物/抗体比により互いに異なるものとなるいくつかのコンジュゲート、場合によって裸の抗体、場合によって凝集体を含む混合物が生成される。したがって求められるDARは、平均値である。
【0087】
したがって本発明はまた、請求項1から8の一項で定義された、抗体に共有結合で付加している1つ以上の化合物を含むコンジュゲートに関係している。この付加は、アミド結合を介しているのが好ましい。抗体は、請求項12から15のいずれか一項で定義された通りであることが好ましい。
【0088】
DARを求めるために本明細書中で使用される方法は、実質的に精製されたコンジュゲート溶液の252nmおよび280nmでの吸光度の比率を分光光度法により測定することからなる(これは、ステップ(ii)の後である)。特に、前記DARは、抗体に対してそれぞれ280および252nmで測定された吸光係数:εΑ280=224,000M−1cm−1およびεΑ252=82,880M−1cm−1を用いて、抗体に対して平均160,000分子量と仮定して、ならびにメイタンシノイドに対して、εD280=5,180M−1cm−1およびεD252=26,159M−1cm−1を用いて、分光光度法により求めることができる)。計算の方法は、Antony S.Dimitrov(編)、LLC、2009年、Therapeutic Antibodies and Protocols、vol525、445、Springer Scienceに基づき、以下でさらに詳細に記載されている:
252nm(A252)および280nm(A280)でのコンジュゲートに対する吸光度は、SEC分析の単量体ピーク(「DAR(SEC)」パラメータの計算を可能にする)に対して、または従来の分光光度計装置(「DAR(UV)」パラメータの計算を可能にする)を用いて測定される。吸光度は、以下の通り表現することができる:
A252=(cD×εD252)+(cA×εA252)
A280=(cD×εD280)+(cA×εA28O)
(式中、
cDおよびcAは、それぞれ、メイタンシノイドの溶液中および抗体の溶液中の濃度である。
εD252およびεD280は、それぞれ252nmおよび280nmでのメイタンシノイドのモル吸光係数である。
εA252およびεΑ280は、それぞれ、252nmおよび280nmでの抗体のモル吸光係数である。)。
2つの未知数があるこれら2つの方程式を解くことによって、以下の方程式が導かれる:
cD=[(εΑ280×A252)−(εA252×A280)]/[(εD252×εΑ280)−(εD252×εD280)]
cA=[A280−(cD×εD280)]/εΑ280ο
【0089】
次いで平均DARを、薬物濃度と抗体の濃度との比から計算する:
DAR=cD/cA
【0090】
UV分光光度計(DAR(UV))で測定した平均DARは、より具体的には4より上、より具体的には4と10の間、さらにより具体的には4と7の間である。
【0091】
コンジュゲートおよび式(I)の化合物も、抗癌剤として使用することができる。有利なことに、抗体は、腫瘍細胞に対して選択的な薬剤を有することを可能にし、したがってメイタンシノイドを前記細胞に極めて接近させ、またはメイタンシノイドが直接これら内部に狙いを定めるようにさせる(「Antibody−drug conjugates for cancer therapy」Carter P.J.ら、Cancer J.2008年、14、154−169頁;「Targeted cancer therapy:conferring specificity to cytotoxic drugs」Chari R.、Acc.Chem.Res.2008年、41、98−107頁を参照)。固体または液性腫瘍を治療することができる。
【0092】
コンジュゲートは、緩衝水溶液の形態で、好ましくは1と10mg/mlの間の濃度で製剤化することができる。溶液は、そのまま投与することができ、または溶液を希釈することによって、潅流用の溶液を形成することができる。
【実施例】
【0093】
方法A:高圧液体クロマトグラフィー−質量分析(LCMS)
スペクトルは、Waters UPLC−SQDシステム上で、正および/または負のエレクトロスプレーモード(ES+/−)で得た。クロマトグラフィーの条件は以下の通り:カラム:ACQUITY BEH C18、1.7μm−2.1×30mm;溶媒:A:H2O(0.1%ギ酸)B:CH3CN(0.1%ギ酸);カラム温度:45℃;流速:0.6ml/分;勾配(2分):5から50%のB、1分;1.3分:100%のB;1.45分:100%のB;1.75分:5%のB。
【0094】
方法B:高圧液体クロマトグラフィー−質量分析(LCMS)
スペクトルは、Waters ZQシステム上で、正および/または負のエレクトロスプレーモード(ES+/−)で得た。クロマトグラフィーの条件は以下の通り:カラム:XBridge C18 2.5μm 3×50mm;溶媒:A:H2O(0.1%ギ酸)B:CH3CN(0.1%ギ酸;カラム温度:70℃;流速:0.9ml/分;勾配(7分):5から100%のB、5.3分;5.5分:100%のB;6.3分:5%のB。
【0095】
方法C:質量分析(MS)
スペクトルは、Waters UPLC−SQDシステム上で、正および/または負のエレクトロスプレーモード(ES+/−)で得た。クロマトグラフィーの条件は以下の通り:カラム:ACQUITY BEH C18 1.7μm−2.1×50mm;溶媒:A:H2O(0.1%ギ酸)B:CH3CN(0.1%ギ酸);カラム温度:50℃;流速:1ml/分;勾配(2分):5から50%のB、0.8分;1.2分:100%のB;1.85分:100%のB;1.95:5%のB。
【0096】
方法D:高圧液体クロマトグラフィー−質量分析(LCMS)
スペクトルは、Waters UPLC−SQDシステム上で、正および/または負のエレクトロスプレーモード(ES+/−)で得た。クロマトグラフィーの条件は以下の通り:カラム:ACQUITY BEH C18 1.7μm−2.1×50mm;溶媒:A:H2O(0.1%ギ酸)B:CH3CN(0.1%ギ酸);カラム温度:50℃;流速:1ml/分;勾配(2分):5から50%のB、0.8分;1.2分:100%のB;1.85分:100%のB;1.95:5%のB。
【0097】
方法G:コンジュゲートの脱グリコシル化および高分解能質量分析(HRMS)
脱グリコシル化は、グリコシダーゼを用いた酵素消化の技法である。脱グリコシル化は、500μlのコンジュゲート+100μlのトリス緩衝液HCl 50mM+10μlのグリカナーゼ−F酵素(100単位の凍結乾燥した酵素/100μlの水)から生じる。培地をボルテックスし、一晩37℃で維持する。この脱グリコシル化した試料は、これでいつでもHRMSで分析を受けられる状態にある。Waters Q−Tof−2システム上で、エレクトロスプレー正モード(ES+)において質量スペクトルを得た。クロマトグラフィーの条件は、以下の通り:カラム:4μm BioSuite 250 URH SEC 4.6×300mm(Waters);溶媒:A:ギ酸アンモニウム25mM+1%ギ酸:B:CH3CN;カラム温度:30℃;流速0.4ml/分;定組成溶離70%A+30%B(15分)。
【0098】
方法H:分析用のサイズ排除クロマトグラフィー(SEC)
カラム:TSKgel G3000 SWXL 5μmカラム、7.8mm×30cm、TOSOH BIOSCIENCE、LLCパーツ#08541
移動相:KCl(0.2M)、KH2PO4(0.052M)、K2HPO4(0.107M)、iPrOH(量:20%)
分析条件:0.5ml/分で30分の定組成溶離
分光光度計検出器L2455DADを用いて、Lachrom Elite HPLCシステム(Merck)上で分析を実施。
【0099】
緩衝液の含量
緩衝液A(pH6.5):NaCl(50mM)、リン酸カリウム緩衝液(50mM)、EDTA(2mM)
緩衝液HGS(pH5.5):ヒスチジン(10mM)、グリシン(130mM)、ショ糖5%(w/v)、HCI(8mM)
【0100】
使用されている略語
AcOEt:酢酸エチル;ALK:(C1−C12)アルキレン基、特に(C1−C6)アルキレン;DAR:薬物抗体比;DBU:1,8−ジアザビシクロ[5.4.0]ウンデカ−7−エン;DCC:Ν,Ν’−ジシクロヘキシルカルボジイミド;DCM:ジクロロメタン;DEAD:アゾジカルボン酸ジエチル;DIC:Ν,Ν’−ジイソプロピルカルボジイミド;DIPEA:Ν,Ν−ジイソプロピルエチルアミン;DMA:ジメチルアセトアミド;DMAP:4−ジメチルアミノピリジン;DME:ジメトキシエタン;DMF:ジメチルホルムアミド;DMSO:ジメチルスルホキシド;ε:モル吸光係数;EEDQ:2−エトキシ−1−エトキシカルボニル−1,2−ジヒドロキノリン;EDCI:N−(3−ジメチルアミノプロピル)−N’−エチルカルボジイミド;EDTA:エチレンジアミン−テトラ酢酸;Fmoc:フルオレニルメトキシカルボニル;Hal:ハロゲン原子;HOBt:1−ヒドロキシベンゾトリアゾール;HEPES:4−(2−ヒドロキシエチル)−1−ピペラジン−エタンスルホン酸;HRMS:高分解能質量分析;NHS:N−ヒドロキシスクシンイミド;iPrOH:イソプロピルアルコール;NMP:N−メチルピロリジノン;Rf:保持因子;RP:減圧;RT:室温;SEC:サイズ排除クロマトグラフィー;TBDMS:tert−ブチルジメチルシリル;TEA:トリエチルアミン;TFA:トリフルオロ酢酸;TFAA:トリフルオロ酢酸無水物;TFF:タンジェンシャルフローフィルトレーション;THF:テトラヒドロフラン;TIPS:トリイソプロピルシリル;TLC:薄層クロマトグラフィー;tR:保持時間。
【0101】
実施例で使用されている抗体
以下の2つの抗体を使用してコンジュゲートを調製した。
hu2H11:(WO2008010101では、hu532H11としても参照されている):この抗体は、アメリカ培養細胞系統保存機関で、受託番号PTA−7662、ブダペスト条約下で預けられたハイブリドーマにより産生し、PCT出願WO2008/010101に記載されている。
hu2H11R35R74:このヒト化抗体は、EphA2受容体に結合し、配列SEQ ID NO:18の重鎖およびSEQ NO:16の軽鎖からなるhu532H11の部位特異的突然変異誘発により得られる。
【0102】
[実施例1]
【0103】
【化25】
1.1.コンジュゲートhu2H11R35R74−PEG4−NHAc−DM4の調製
磁気撹拌下、RTで、9mlのhu2H11R35R74(緩衝液A中14.36mg/ml)を添加し、次いで16.85mlの緩衝液A、3.23mlのHEPES 1M、1.59mlのDMA、続いてL−DM4−AcNH−PEG4−CONHS活性化エステルの、1.64mlの10mM DMA溶液を添加する。RTで1時間30分後、L−DM4−AcNH−PEG4−CONHS活性化エステルの追加の0.085mlの10mM DMA溶液を添加する。RTで1時間45分後、粗製の結合培地を60mlのHGS緩衝液で希釈し、Pellicon3カセット上のTFFで精製する。約10の試料容量のHGS緩衝液に対して試料を透析ろ過し、次いで収集する。TFFタンクおよびラインを追加の10mlのHGS緩衝液で洗浄する。2つの溶液を混合し、0.22μmのPVDFを介してろ過滅菌し、Amicon15上で濃縮し、0.22μmPVDFを介してろ過滅菌する。17mlのhu2H11R35R74−PEG4−NHAc−DM4コンジュゲート(c=5.76mg/ml)をこのようにして得た。次いで最終の薬剤含有量および単量体の純度についてこのコンジュゲートを分析する。SEC分析(方法H):DAR(SEC)=5.4;RT=16.757分;単量体の純度=99.5%;HRMSデータ:図1を参照。
【0104】
1.2.L−DM4−AcNH−PEG4−CONHS活性化エステルの調製
RTでの磁気撹拌下、154.3mgのL−DM4(WO04/103272に従い調製−化合物4bを参照)をガラスバイアル中に導入する。次いで、90mgの3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルの、0.94mlのDMA中溶液を添加し、続いて36μlのDIEAを添加する。RTで23時間後、この反応培地を5mlのAcOEtで希釈し、7mlの水で洗浄する。水相を5mlのAcOEtで抽出する。合わせた有機相をMgSO4で脱水し、RP下で濃縮乾燥させる。228mgの淡黄色の粘性油を得る。この生成物を最小量のDMAで希釈し、30gのC18−グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。RP下で画分2および3を濃縮後、無色の粘性の油を得る。この生成物を最小量のDMAで希釈し、30gのC18−グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。画分33から35をRP下で濃縮後、41mgのL−DM4−AcNH−PEG4−CONHS活性化エステルを白色メレンゲのような生成物の形態で得る。質量スペクトル(B):RT=4.06分;[M+H−H2O]+:m/z 1164;[M+H]+:m/z 1182;[M−H+HCO2H]−:m/z 1226;1H NMR(500MHz,δ:ppm,クロロホルム−d):0.80(s,3H);1.21(s,3H);1.22(s,3H);1.25(m,1H);1.29(d,J=6.7Hz,6H);1.46(m,1H);1.57(d,J=13.4Hz,1H);1.64(s,3H);1.76から1.83(m,1H);1.88から1.96(m,1H);2.18(dd,J=2.5および14.3Hz,1H);2.36(m,1H);2.53(m,1H);2.61(dd,J=12.5および14.3Hz,1H);2.82から2.92(m,10H;2.98(d,J=16.7Hz,1H);3.03(d,J=9.6Hz,1H);3.15(d,J=12.9Hz,1H);3.22(s,3H);3.32(s 広幅,1H);3.36(s,3H);3.42(m,2H);3v50(d,J=9.1Hz,1H);3.53(t,J=5.2Hz,2H);3.58から3v67(m,13H);3.84(t,J=6.4Hz,2H);3.99(s,3H);4.27(m,1H);4.77(dd,J=2.9および11.9Hz,1H);5.42(q,J=6.7Hz,1H);5.66(dd,J=9.1および15.4Hz,1H);6.23(s,1H);6.43(dd,J=11.3および15.4Hz,1H);6.64(d,J=1.1Hz,1H);6.74(d,J=11.3Hz,1H);6.85(d,J=1.1Hz,1H);7.08(t,J=5.2Hz,1H)
【0105】
1.3.3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルの調製
磁気撹拌下、RTで、671.4mgの3−(2−{2−[2−(2−アミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸(CA(PEG)4、Pierce)をガラスバイアル内に導入する。次いで、597.4mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルの、14mlのDCM中溶液を添加する。RTで15分後、0.396mlのDICを添加する。1時間30分後、粗製の反応培地を焼結ガラス上でろ過し、100gのCN−グラフトシリカゲル(勾配溶出、nヘプタン/iPrOH/AcOEt、iPrOH部分を増加)上で、フラッシュクロマトグラフィーにより濾液を精製する。画分30から45をRP下で濃縮後、761mgの3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルを無色の油の形態で得る。質量スペクトル(A):RT=0.74分;[M+H]+:m/z483/485(Brの2つの同位体による2つのピーク);[M−H+HCO2H]−:m/z527/529(Brの2つの同位体による2つのピーク)。
【0106】
ブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルは、公開プロトコル(Biochemistry、1974年、481頁)に従い調製することができた。
【0107】
[実施例2]
【0108】
【化26】
2.1.コンジュゲートhu2H11R35R74−PEG4−NMeAc−DM4の調製
RTでの磁気撹拌下、4mlのhu2H11R35R74(緩衝液A中14.36mg/ml)を添加し、次いで7.5mlの緩衝液A、1.45mlのHEPES 1M、1.05mlのDMA、続いてL−DM4−AcNMe−PEG4−CONHS活性化エステルの、0.39mlの10mM DMA溶液を添加する。RTで30分後、L−DM4−AcNMe−PEG4−CONHS活性化エステルの、追加の0.19mlの10mM DMA溶液を添加する。RTで3時間後、粗製の結合培地を65mlのHGS緩衝液で希釈し、Pellicon3カセット上でTFFにより精製する。約10の試料容量のHGS緩衝液に対して試料を透析ろ過し、次いで収集する。TFFタンクおよびラインを追加の10mlのHGS緩衝液で洗浄する。この2つの溶液を混合し、Amicon15上で濃縮し、0.22μmPVDFを介してろ過滅菌する。8.5mlのhu2H11R35R74−PEG4−NMeAc−DM4コンジュゲート(c=6.01mg/ml)をこのようにして得た。次いで最終の薬剤含有量および単量体の純度についてコンジュゲートを分析する。SEC分析(H):DAR(SEC)=5.5;RT=16.7分;単量体の純度=99.4%;HRMSデータ:図2を参照。
【0109】
2.2.L−DM4−AcNMe−PEG4−CONHS活性化エステルの調製
RTでの磁気撹拌下、133.4mgのL−DM4をガラスバイアル内に導入する。次いで、85mgの3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルの、0.2mlのDMA中溶液を添加し、続いて32.9μlのDIEAを添加する。RTで1時間後、この反応培地を、30gのC18−グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。所望の生成物を含有する画分をRP下で濃縮後、71.3mgのL−DM4−AcNMe−PEG4−CONHS活性化エステルを無色ガラスのような生成物の形態で得る。質量スペクトル(D):RT=0.98分;[M+H−H2O]+:m/z 1178(主要シグナル);[M+Na]+:m/z 1218;[M−H+HCO2H]−:m/z 1240;1H NMR(500MHz,δ:ppm,クロロホルム−d):0.81(s,3H);1.20から1.33(m,13H);1.42から1.52(m,1H);1.56から1.61(m,1H);1.65(s,3H);1.73から1.83(m,1H);1.96から2.04(m,1H);2.19(dd,J=2.8および14.4Hz,1H);2.29から2.41(m,1H);2.55から2.66(m,2H);2.83から2.93(m,12H);3.04(d,J=9.8Hz,1H);3.12(d,J=12.7Hz,1H);3.18から3.25(m,5H);3.37(s,3H);3.47から3.54(m,3H);3.57から3.68(m,15H);3.85(t,J=6.6Hz,2H);3.99(s,3H);4.29(m,1H);4.79(dd,J=2.8および12.2Hz,1H);5.41(q,J=6.7Hz,1H);5.68(dd,J=9.3および15.2Hz,1H);6.23(s,1H);6.43(dd,J=11.0および15.2Hz,1H);6.66(s,1H);6.74(d,J=11.0Hz,1H);6.83(s,1H)。
【0110】
2.3.3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2−ジオキソ−ピロリジン−1−イルエステルの調製
【0111】
【化27】
磁気撹拌下、RTで、115.1mgの3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸、1.5mlのDCM、97.3mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルを丸底フラスコ内に逐次的に導入する。2時間後、72μlのDIEAを添加し、RTでさらに1時間後、70.2μlのDICを添加する。粗製の反応培地をRTで4時間保持し、−20°Cで16時間保持し、次いで30gのシリカゲル(DCM:メタノール0:100から3:97(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。所望の生成物を含有する画分をRP下で濃縮後、85.8mgの3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルを白色固体の形態で得る。質量スペクトル(A):RT=0.84分;[M+H]+:m/z497/499
【0112】
2.4.3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸の調製
【0113】
【化28】
アルゴンの不活性雰囲気下、丸底フラスコ内で、磁気撹拌しながら、120.1mgの3−[2−(2−{2−[2−(2,2,2−トリフルオロ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステル、1mlの無水THFおよび59.8μlのCH3lを逐次的に導入する。この反応培地を氷/水槽を用いて約0℃で冷却し、16.1mgのNaH(油中、純度50%)を少しずつゆっくりと添加する。0°Cで15分およびRTで1時間後、粗製の反応培地をRP下で濃縮乾燥し、0.5mlのTHFおよび0.8mlの水で希釈する。次いでRTで、30.6mgのLiOHをこの反応物培地に添加する。粗製の反応培地をRTで2時間保持し、−20℃で16時間保持し、次いで30gのC18−グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。所望の生成物を含有する画分をRP下で濃縮後、115.3mgの3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸を黄色油の形態で得る。
【0114】
2.5.3−[2−(2−{2−[2−(2,2,2−トリフルオロ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルの調製
【0115】
【化29】
アルゴンの不活性雰囲気下、磁気撹拌しながら、230mgの3−(2−{2−[2−(2−アミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸(CA(PEG)4、Pierce)、2mlのDCMおよび1mlのメタノールを、丸底フラスコ内に、逐次的に導入する。RTで、1mlのトリメチルシリルジアゾメタン(ヘキサン中2M溶液)をこの反応物培地にゆっくりと添加する。RTで2時間後、過剰なトリメチルシリルジアゾメタンを、酢酸の添加により中和する。次いでRP下で粗製物を蒸発乾燥させる。得た残渣を2mlのDCMで希釈し、水−氷浴を用いて0℃に冷却し、次いで363μlのTEAおよび300μlのTFAAを逐次的に添加する。RTで2時間30分間および−20℃で19時間後、363μlのTEAおよび300μlのTFAAを逐次的に添加する。RTで4時間30分後、粗製物を−20℃でストックし、次いで30gのC18−グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。所望の生成物を含有する画分をRP下で濃縮後、123mgの3−[2−(2−{2−[2−(2,2,2−トリフルオロ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルを淡黄色の油の形態で得る。質量スペクトル(A):RT=0.90分;[M+H]+:m/z376;[M−H]−:m/z374。
【0116】
[実施例3]
【0117】
【化30】
3.1.hu2H11R35R74−PEG8−NHAc−DM4のコンジュゲートの調製
RTでの磁気撹拌下、4mlのhu2H11R35R74(緩衝液A中14.36mg/ml)を添加し、次いで7.5mlの緩衝液A、1.45mlのHEPES 1M、1.05mlのDMAを添加し、続いてL−DM4−AcNMe−PEG8−CONHS活性化エステルの0.405mlの10mM DMA溶液を添加する。RTで30分後、L−DM4−AcNMe−PEG8−CONHS活性化エステルの追加の0.1mlの10mMDMA溶液を添加する。RTで1時間45分後、粗製の結合培地を60mlのHGS緩衝液で希釈し、Pellicon3カセット上で、TFFで精製する。約10の試料容量のHGS緩衝液に対して試料を透析ろ過し、次いで収集する。TFFタンクおよびラインを追加の10mlのHGS緩衝液で洗浄する。2つの溶液を混合し、Amicon15上で濃縮し、0.22μmPVDFを介してろ過滅菌する。7.0mlのhu2H11R35R74−PEG8−AcNMe−DM4コンジュゲート(c=6.95mg/ml)をこのようにして得た。次いで最終の薬剤含有量および単量体の純度についてコンジュゲートを分析する。SEC分析(H):DAR(SEC)=5.0;RT=16.593分;単量体純度=99.5%:HRMSデータ:図3を参照。
【0118】
3.2.L−DM4−AcNH−PEG8−CONHS活性化エステルの調製
RTでの磁気撹拌下、65mgの3−{2−[2−(2−{2−[2−(2−{2−[2−(3−ブロモ−プロピオニルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルをガラスバイアル内に導入し、続いて67.7mgのL−DM4の、0.85mlのDMAおよび16.5μlのDIEA中溶液を導入する。RTで48時間後、10gのシリカゲル(DCM:MeOH 100:0から90:10(容量)の勾配溶出)上でフラッシュクロマトグラフィーによりこの反応培地を精製する。画分18から26をRP下で濃縮後、17mgのL−DM4−AcNH−PEG8−CONHS活性化エステルを無色のガラスの形態で得る。質量スペクトル(B)::RT=4.08分;[M+H−H2O]+:m/z 1340(主要シグナル);[M+Na]+:m/z 1380;[M−H+HCO2H]−:m/z 1402;1H NMR(400MHz,δ:ppm,クロロホルム−d):0.81(s,3H);1.22(s,3H);1.23(s,3H);1.26(m,1H);1.30(d,J=6.8Hz,6H);1.41から1.52(m,1H);1 .65(s,3H);1.80(m,1H);1.89から1.99(m,1H);2.19(m,1H);2.37(m,1H);2.47から2.67(m,2H);2.81から2.93(m,10H);2.99(d,J=16.6Hz,1H);3.04(d,J=9.8Hz,1H);3.16(d 広幅,J=13.7Hz,1H);3.23(s,3H);3.32(s 広幅,1H);3.37(s,3H);3.44(m,2H);3.51(d,J=9.1Hz,1H);3.54(t,J=5.4Hz,2H);3.59から3.73(m,29H);3.86(t,J=6.6Hz,2H);4.00(s,3H);4.22から4.33(m,1H);4.78(dd,J=2.9および12.2Hz,1H);5.43(q,J=6.8Hz,1H);5.67(dd,J=9.0および15.2Hz,1H);6.23(s,1H);6.44(dd,J=11.2および15.2Hz,1H);6.65(d,J=1.5Hz,1H);6.75(d,J=11.2Hz,1H);6.86(d,J=1.5Hz,1H);7.02から7.13(m,1H)。
【0119】
3.3.3−{2−[2−(2−{2−[2−(2−{2−[2−(3−ブロモ−プロピオニルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルの調製:
【0120】
【化31】
RTでの磁気撹拌下、100mgの3−(2−{2−[2−(2−アミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸(CA(PEG)4、Pierce)、2mlのDCMおよび53.5mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルをガラスバイアル内に逐次的に導入する。RTで1時間後、35.1μlのDICを添加する。1時間後、粗製の反応培地を焼結ガラス上でろ過し、RP下で濃縮乾燥させ、10mlのAcOEtで希釈し、焼結ガラス上でろ過し、RP下で濃縮乾燥する。76.5mgの3−{2−[2−(2−{2−[2−(2−{2−[2−(3−ブロモ−プロピオニルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルを無色の油の形態で得る。質量スペクトル(A):RT=0.80分;[M+H]+:m/z659/661;[M−H+HCO2H]−:m/z703/705
【0121】
[実施例4]
【0122】
【化32】
4.1.コンジュゲートhu2H11R35R74−PEG4−アリル−DM4の調製
RTでの磁気撹拌下、4mlのhu2H11R35R74(緩衝液A中14.36mg/ml)を添加し、次いで7.5mlの緩衝液A、1.45mlのHEPES 1M、1.14mlのDMAを添加し、続いてL−DM4−アリル−PEG4−CONHS活性化エステルの、0.3mlの10mM DMA溶液を添加する。RTで30分後、L−DM4−アリル−PEG4−CONHS活性化エステルの追加の0.125mlの10mM DMA溶液を添加する。RTで1時間25分後、粗製の結合培地を65mlのHGS緩衝液で希釈し、Pellicon3カセット上で、TFFで精製する。約10の試料容量のHGS緩衝液に対して試料を透析ろ過し、次いで収集する。TFFタンクおよびラインを追加の10mlのHGS緩衝液で洗浄する。2つの溶液剤を混合し、Amicon15上で濃縮し、0.22μmのPVDFを介してろ過滅菌する。8.0mlのhu2H11R35R74−PEG4−アリル−DM4コンジュゲート(c=5.22mg/ml)を得た。次いで、最終の薬剤含有量および単量体の純度についてコンジュゲートを分析する。SEC分析(H):DAR(SEC)=5.3;RT=16.767分;単量体の純度=99.4%;HRMSデータ:図4を参照。
【0123】
4.2.L−DM4−アリル−PEG4−CONHS活性化エステルの調製
RTでの磁気撹拌下、70mgのL−DM4、45mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステル(ブロモ−アリル−PEG4−CONHS)、0.5mlのDMAおよび23.5μlのDIEAをガラスバイアル内に逐次的に導入する。RTで2時間および−20℃で17時間後、50μlのDIEAを添加する。RTで24時間後、この反応培地を、30gのC−18グラフトシリカゲル(水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。所期の生成物を含有する画分をRPで濃縮後、47.1mgのL−DM4−アリル−PEG4−CONHS活性化エステルを白色固体の形態で得る。質量スペクトル(D):RT=1.06分;[M+Na]+:m/z 1173;1H NMR(500MHz,δ:ppm,クロロホルム−d):0.81(s,3H);1.18から1.39(m,13H);1.42から1.52(m,1H);1.58(d,J=13.4Hz,1H);1.65(s,3H);1.73から1.82(m,1H);1.86から1.95(m,1H);2.19(d,J=14.3Hz,1H);2.40(m,1H);2.51から2.65(m,2H);2.82から2.95(m,9H);2.98から3.07(m,2H);3.12(d,J=12.6Hz,1H);3.18から3.27(m,1H);3.23(s,3H);3.36(s,3H);3.51(d,J=9.1Hz,1H);3.54から3.82(m,13H);3.86(t,J=6.4Hz,2H);3.91から3.95(m,2H);3.99(s,3H);4.28(t,J=11.0Hz,1H);4.78(dd,J=2.6および11.9Hz,1H);5.44(q,J=6.7Hz,1H);5.49から5.63(m,2H);5.68(dd,J=9.1および15.0Hz,1H);6.24(s,1H);6.43(dd,J=11.1および15.0Hz,1H);6.66(s,1H);6.77(d,J=11.1Hz,1H);6.83(s,1H)。
【0124】
4.3.3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステルの調製
【0125】
【化33】
RTで、200mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸、4mlのDCMおよび232.3mgの担持されたDCC(2当量)をガラスバイアル内に逐次的に導入する。RTで1時間後、64.8mgのNHSを添加する。RTで5時間後、粗製物を焼結ガラスろ過し、固体をDCMで洗浄し、合わせた濾液をRP下で濃縮乾燥させる。15gのシリカゲル(MeOH:DCM 0:100から10:90(容量)の勾配溶出)上でのフラッシュクロマトグラフィーによる精製および所期の生成物を含有する画分のRP下での濃縮により、生成した46mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステル(ブロモ−アリル−PEG4−CONHS)を淡黄色の油の形態で得る。質量スペクトル(A):RT=1.02分;[M+H]+:m/z454/456;[M+Na]+:m/z476/478;[M−H+HCO2H]−:m/z498/500。
【0126】
4.4.3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸の調製
【0127】
【化34】
RTで、1gの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸tert−ブチルエステル(市販のもの)、6mlのTFAおよび3mlのDCMの溶液を3時間撹拌し、次いでRP下で濃縮乾燥させる。油性の残渣をトルエンで希釈し、RP下で濃縮乾燥させて、褐色油の形態で853mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸を生成する。
【0128】
[実施例5]
5.1.コンジュゲートhu2H11−PEG4−NHAc−DM4の調製
コンジュゲートhu2H11−PEG4−NHAc−DM4は、実施例1と類似の方法で調製することができた。撹拌下、RTで、1mlのhu2H11(緩衝液A中8.52mg/ml)を添加し、次いで0.7mlの緩衝液A、0.213mlのHEPES 1M、0.7mlのDMA、続いて0.085mlのL−DM4−AcNH−PEG4−CONHS活性化エステルの10mM DMA溶液を0.128mlのDMAで希釈する。RTで2時間後、粗製の培地をAmicon4上で、7000Gで濃縮し、Nap−10カラム上で緩衝液をHGS緩衝液と交換し、最後に5ml Zebaカラム上で精製する。1.15mlのhu2H11−PEG4−NHAc−DM4コンジュゲート(c=3.78mg/ml)をこのようにして得た。最終の薬剤含有量および単量体の純度についてコンジュゲートを分析する。SEC分析(方法H):DAR(UV)=6.6;DAR(SEC)=5.6;RT=15.387分;単量体の純度=99.7%;HRMSデータ:図5を参照。
【0129】
同様に、hu2H11の関与する他のコンジュゲートおよび実施例6から8に記載されたものを調製した(表IIaを参照)。
【0130】
[実施例9]
MDA−MB231腫瘍細胞の増殖の阻害(ECAAC ref.#92020424から)
指数増殖期の細胞をトリプシン処理し、適切な培地内に再懸濁させた(DMEM/F12 Gibco #21331;10%SVF Gibco #10500−056;2nMグルタミン Gibco #25030)。完全な血清含有培地の中、5000細胞/ウェルの密度で、細胞懸濁液を96−ウェルCytostar培養プレート(GE Healthcare Europe、#RPNQ0163)に分配した。4時間のコーティング後、三連のウェルに、10−7から10−12Mの間の範囲の濃度でコンジュゲートの段階希釈を添加した。コンジュゲートの存在下、37℃/5%CO2で3日間細胞を培養した。第4日目、10μlの14C−チミジン溶液(0.1μCi/ウェル(Perkin Elmer# NEC56825000))を各ウェルに添加した。microbeta放射性カウンター(Perkin Elmer)を用いて、実験が開始されてから96時間、14C−チミジンの取り込みを測定した。無細胞の試薬ブランクを試験ウェルの記録から引き算し、コンジュゲートで処理した細胞の記録を、ビヒクル処理した細胞の対照ウェルからの記録の平均値で割ることによって得た、残存する画分として、このデータをプロットした。これら実験において、実験開始時に、1μΜの濃縮で、裸の抗体(hu2H11またはhu2H11R35R74)を添加し、増殖の阻害を以前に述べた通り測定した。
【0131】
表IIaおよびIIbで報告された結果は、裸のhu2H11または/hu2H11R35R74の存在下で実施された競合実験によると、コンジュゲートは、MDA−MB231細胞に対して強力なインビトロの増殖阻害特性を示し、抗原への結合を介して作用することを示唆している。
【0132】
【表2】
【0133】
【表3】
【0134】
[実施例10]
L−DM4−AcNH−PEG4−COOMe
【0135】
【化35】
10.1.遊離薬物L−DM4−AcNH−PEG4−COOMeの調製
【0136】
【化36】
磁気撹拌下、RTで、Arの不活性雰囲気下、ガラスバイアル内に、57.4mgの3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステル、80mgのL−DM4の、0.44mlのDMA中溶液、最後に19.6μlのDIPEAを逐次的に導入する。RTで18時間後、粗製物を10mlの水で希釈し、3×7mlのAcOEtで抽出する。有機相を集め、MgSO4で脱水し、ろ過し、RP下で濃縮乾燥する。124mgの無色の油を得る。この生成物を最小量のDMAで希釈し、C18−グラフトシリカゲル(Merck、C18、5g、25−40μm、18ml/分、水:アセトニトリル100:0から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、12.8mgのメチルエステルL−DM4−AcNH−PEG4−COOMeを、無色の被膜の形態で得る。質量スペクトル(C):tR=0.97分;[M+H]+:m/z 1099;[M−H]−:m/z 1097;1H NMR(500MHz,ppm単位,クロロホルム−d):0.80(s,3H);1.21(s,3H);1.22(s,3H);1.24から1.35(m,7H);1.46(td,J=6.4および10.2Hz,1H);1.57(d,J=13.4Hz,1H);1.64(s,3H);1.80(ddd,J=4.9および11.5および14.6Hz,1H);1.92(m,1H);2.18(dd,J=2.3および14.7Hz,1H);2.36(ddd,J=4.8および11.4および16.2Hz,1H);2.52(ddd,J=5.1および11.3および16.3Hz,1H);2.59(t,J=6.4Hz,2H);2.63(m,1H);2.86(s,3H);2.88(m,1H);2.98(d,J=16.7Hz,1H);3.03(d,J=9.6Hz,1H);3.15(d,J=12.6Hz,1H);3.23(s,3H);3.36(s,3H);3.42(m,2H);3.50(d,J=9.1Hz,1H);3.54(m,2H);3.63(m,13H);3.68(s,3H);3.75(t,J=6.4Hz,2H);3.99(s,3H);4.27(t,J=11.3Hz,1H);4.78(dd,J=2.2および12.1Hz,1H);5.42(q,J=6.9Hz,1H);5.66(dd,J=9.1および15.4Hz,1H);6.21(s,1H);6.43(dd,J=11.0および15.4Hz,1H);6.64(s,1H);6.74(d,J=11.0Hz,1H);6.85(s,1H);7.05(t,J=5.5Hz,1H)。
【0137】
10.2.3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルの調製
【0138】
【化37】
磁気撹拌下、RTで、100mgの3−(2−{2−[2−(2−アミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸(CA(PEG)4、Pierce)、89mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルおよび2mlのDCMをガラスバイアル内に逐次的に導入する。RTで1時間後、0.7mlのMeOHおよび0.38mlの2Mトリメチルシリルジアゾメタンのヘキサン中溶液を添加する。RTで1時間後、粗製の反応混合物をRP下で濃縮乾燥し、次いで最小量のDMAで希釈し、C18−グラフトシリカゲル(Merck、C18、5g、25−40μm、18ml/分、水:アセトニトリル100:0から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、58mgの3−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルを無色の油の形態で得る。質量スペクトル(A):tR=0.75分;[M+H]+:m/z400/402
ブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルは、公開プロトコル(Biochemistry、1974年、481頁)に従い調製することができた。
【0139】
[実施例11]
L−DM4−AcNMe−PEG4−COOMe
【0140】
【化38】
11.1.遊離薬物L−DM4−AcNMe−PEG4−COOMeの調製
【0141】
【化39】
磁気撹拌下、RTで、ガラスバイアル内に、30mgのL−DM4、20.8mgの3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルの、0.3mlのDMA中溶液、および7.4μlのDIPEAを逐次的に導入する。RTで18時間後、この反応培地を7mlのAcOEtで希釈し、5mlの水で2回洗浄する。有機相をブラインで洗浄し、MgSO4で脱水し、ろ過し、RP下で濃縮乾燥させる。39mgの無色のガラスを得る。この生成物を最小量のDMA/MeOH混合物で希釈し、C18−グラフトシリカゲル(XTerra(登録商標)C18、5μm、50×30mm、30ml/分、水:アセトニトリル95:5から5:95(容量)の勾配溶出)上でクロマトグラフィーにより精製する。予想された化合物を含有する画分を減圧下で濃縮後、7.8mgのメチルエステルL−DM4−AcNMe−PEG4−COOMeを無色の固体の形態で得る。質量スペクトル(C):tR=1.00分;[M+H−H2O]+:m/z1095;[M+Na+]+:m/z 1135;[M−H+HCO2H]−:m/z 1157;[M−H]−:m/z 1111。1H NMR(500MHz,δ:ppm,DMSO−d6):0.78(s,3H);1.12(d,J=6.6Hz,3H);1.16(m,9H);1.26(m,1H);1.40から1.51(m,2H);1.59(s,3H);1.62(m,1H);1.87(m,1H);2.04(m,1H);2.28(m,1H);2.47から2.58(m:部分マスク,4H);2.73(s,3H);2.76(s,1H);2.80(d,J=9.6Hz,1H);2.95(s,2H);3.10(s,3H);3.16から3.48(m:部分マスク,21H);3.25(s,3H);3.59(s,3H);3.63(t,J=6.4Hz,2H);3.93(s,3H);4.08(m,1H);4.53(dd,J=2.9から11.9Hz,1H);5.32(q,J=6.6Hz,1H);5.58(dd,J=9.3から15.1Hz,1H);5.89(s,1H);6.49から6.58(m,2H);6.62(m,1H);6.84(s,1H);7.19(s,1H)。
【0142】
11.2.3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルの調製
【0143】
【化40】
磁気撹拌下、RTで、Arの不活性雰囲気下、127mgの3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステル、102.2mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルおよび1.2mlのDCMを逐次的に添加する。45分後、粗製物をRP下で濃縮し、5gのCN−グラフトシリカゲル(n.ヘプタン/iPrOH/AcOEtの勾配溶出、iPrOH部分を増加させる)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、122mgの3−{2−[2−(2−{2−[(2−ブロモ−アセチル)−メチル−アミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルを無色の油の形態で得る。質量スペクトル(A):tR=0.84分;[M+H]+:m/z414/416。
【0144】
ブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルは、公開プロトコル(Biochemistry、1974年、481頁)に従い調製することができた。
【0145】
11.3.3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステルの調製
【0146】
【化41】
磁気撹拌下、RTで、Arの不活性雰囲気下で、271.7mgの3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルを、ジオキサン中の2mlの塩酸4N溶液の中で可溶化させる。18時間後、粗製の反応培地をRP下で濃縮し、4mlのMeOHの中で可溶化させ、3g SCX SPEカラム(10mlのMeOHで調整、10mlのMeOHで洗浄およびMeOH中アンモニア2Nで溶出)を通過させる。RP下で溶出画分の濃縮後、146mgの無色の油を得る。この油をAcOEt中に溶解し、MgSO4上で乾燥させ、ろ過し、RP下で蒸発させた。127mgの3−(2−{2−[2−(2−メチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステルを無色の油として得る。質量スペクトル(A):tR=0.40分;[M+H]+:m/z294。
【0147】
11.4.3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルの調製
【0148】
【化42】
磁気撹拌下、RTで、Arの不活性雰囲気下、227mgの3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸の、0.644mlのDCMおよび0.644mlのMeOH中溶液を水−氷−塩化ナトリウムで約0℃まで冷却する。トリメチルシリルジアゾメタンの、0.449mlのヘキサン中2M溶液を添加し、RTで17時間後、酢酸のMeOH中0.5M溶液を添加することによって、5と6の間のpHを達成する。粗製物をAcOEt(30ml)で希釈し、水(2×15ml)で洗浄し、ブライン(15ml)で洗浄し、有機相をMgSO4上で脱水させ、ろ過し、RP下で蒸発させる。209.7mgの3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸メチルエステルを無色の油として得る。質量スペクトル(A):tR=1.18分;[M+H]+:m/z294、338、394。
【0149】
11.5.3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸の調製
【0150】
【化43】
磁気撹拌下、RTで、Arの不活性雰囲気下、330mgの市販の3−(2−{2−[2−(2−tert−ブトキシカルボニルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸の、4mlのTHF中溶液を、水−氷−塩化ナトリウムで、約0℃まで冷却する。72.2mgのNaH(油中70%)をゆっくりと添加し、25分後、0.174mlのMelを添加する。冷却槽を取り除き、RTで3時間後、120mgのNaHおよび0.2mlのMelを添加する。RTで1.5時間後、酢酸の希釈水溶液を添加することによって、5と6の間のpHを達成する。粗製の反応混合物をAcOEt(30ml)で希釈し、水(2×20ml)で洗浄し、ブライン(10ml)で洗浄し、有機相をMgSO4上で脱水させ、ろ過し、RP下で蒸発させる。227mgの3−[2−(2−{2−[2−(tert−ブトキシカルボニル−メチル−アミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸を無色の油として得る。質量スペクトル(A):tR=1.02分;[M+H]+:m/z280、380。
【0151】
[実施例12]
L−DM4−アリル−PEG4−COOMe
【0152】
【化44】
12.1遊離薬物L−DM4−アリル−PEG4−COOMeの調製
【0153】
【化45】
磁気撹拌下、RTで、ガラスバイアル内に、36.7mgのL−DM4、20.8mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステルの、0.37mlのDMA中溶液、最後に9.4μlのDIPEAを逐次的に導入する。RTで1.5時間後および−18℃で18時間後、この反応培地を、C18−グラフトシリカゲル(Merck、C18、5g、25−40μm、18ml/分、水:アセトニトリル100:0から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、18.2mgの白色固体を得て、これを、CN−グラフトシリカゲル(n.ヘプタン/iPrOH/AcOEtの勾配溶出、AcOEt部分を増加させる)上で、フラッシュクロマトグラフィーにより精製する。4.02mgのメチルエステルL−DM4−アリル−PEG4−COOMeを白色固体の形態で得る。質量スペクトル(C):tR=1.09分;[M−H + HCOOH]−:m/z 1112;[M+Na]+:m/z 1090。1H NMR(500MHz,ppm単位,クロロホルム−d):0.80(s,3H);1.18から1.26(m,7H);1.27から1.31(m,6H);1.40から1.50(m,1H);1.57(d,J=13.7Hz,3H);1.64(s,3H);1.77(ddd,J=4.8および11.7および14.5Hz,1H);1.91(ddd,J=4.8および11.7および14.5Hz,1H);2.18(dd,J=2.5および14.3Hz,1H);2.38(m,1H);2.57(m,4H);2.86(s,2H);2.89(m,1H);3.00(m,1H);3.04(d,J=9.9Hz,1H);3.11(d,J=12.3Hz,1H);3.23(s,3H);3.35(s,3H);3.50(d,J=9.1Hz,1H);3.55(m,2H);3.63(m,10H);3.69(s,3H);3.75(t,J=6.4Hz,2H);3.92(d,J=5.2Hz,2H);3.98(s,3H);4.27(ddd,J=1.6および10.4および12.3Hz,1H);4.78(dd,J=3.0および11.8Hz,1H);5.43(q,J=6.8Hz,1H);5.48から5.61(m,2H);5.67(dd,J=9.2および15.2Hz,1H);6.20(d,J=0.5Hz,1H);6.42(dd,J=11.3および15.4Hz,1H);6.65(d,J=1.9Hz,1H);6.77(d,J=11.3Hz,1H);6.82(d,J=1.1Hz,1H)。
【0154】
12.2.3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステルの調製
【0155】
【化46】
磁気撹拌下、50mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸の、1mlのDCMおよび0.25mlのMeOH中冷却溶液(水−氷浴)に、0.11mlの2Mトリメチルシリルジアゾメタンのヘキサン中溶液を添加する。水−氷浴を取り除いた後、粗製の反応混合物をRTで1.5hr撹拌し、2滴の酢酸で中和し、RP下で濃縮乾燥する。トルエンで共沸蒸発後、47mgの3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸メチルエステルをアンバー油の形態で得る。質量スペクトル(A):tR=1.12分;[M+H]+:m/z369/371。
【0156】
3−(2−{2−[2−(4−ブロモ−ブタ−2−エニルオキシ)−エトキシ]−エトキシ}−エトキシ)−プロピオン酸の調製は実施例4で記載されている。
【0157】
[実施例13]
L−DM4−AcNH−PEG8−COOMe
【0158】
【化47】
13.1.遊離薬物L−DM4−AcNH−PEG8−COOMeの調製
【0159】
【化48】
磁気撹拌下、RTで、ガラスバイアル内に、60mgのL−DM4、11.4mgの炭酸カリウム、および75mgの3−{2−[2−(2−{2−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルの、0.7mlのDMA中溶液を逐次的に導入する。RTで22時間後、この反応培地を12mlの水で希釈し、2×10mlのAcOEtで抽出する。有機相を集め、MgSO4で脱水し、ろ過し、RP下で濃縮乾燥させる。50mgの無色の油を得る。この生成物を最小量のDCMで希釈し、5gのCN−グラフトシリカゲル(nヘプタン/iPrOH/AcOEtの勾配溶出、iPrOH部分を増加させる)上で、フラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分を減圧下で濃縮後、残渣を最小量のDMAで希釈し、C18−グラフトシリカゲル(Merck、C18、5g、25−40μm、12ml/分、水:アセトニトリル100:0から5:95(容量)の勾配溶出)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、3.3mgのメチルエステルL−DM4−AcNH−PEG8−COOMeを無色の被膜の形態で得る。質量スペクトル(C):tR=0.97分;[M−H]− + HCOOH:m/z 1319。1H NMR(400MHz,ppm単位,クロロホルム−d):0.73(s,3H);1.10から1.19(m,7H);1.21(s,3H);1.23(s,3H);1.39(td,J=6.1および10.1Hz,1H);1.50(d,J=13.2Hz,1H);1.57(s,3H);1.71(m,1H);1.85(m,1H);2.11(dd,J=3.4および14.2Hz,1H);2.29(ddd,J=5.1および11.1および16.0Hz,1H);2.45(ddd,J=5.4および11.2および16.1Hz,1H);2.52(t,J=6.6Hz,3H);2.79(s,3H);2.82(m,1H);2.90(m,1H);2.96(d,J=9.3Hz,1H);3.07(d,J=13.2Hz,1H);3.15(s,3H);3.28(s,3H);3.35(m,2H);3.44(m,3H);3.56(s,29H);3.61(s,3H);3.68(t,J=6.6Hz,2H);3.91(s,3H);4.20(t,J=12.0Hz,1H);4.70(dd,J=3.2および12.0Hz,1H);5.35(q,J=7.0Hz,1H);5.59(dd,J=9.0および15.4Hz,1H);6.13(s,1H);6.35(dd,J=11.0および15.4Hz,1H);6.57(d,J=1 .5Hz,1H);6.67(d,J=11.2Hz,1H);6.78(d,J=1.5Hz,1H);6.96(t,J=5.6Hz,1H)。
【0160】
13.2.3−{2−[2−(2−{2−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルの調製
【0161】
【化49】
磁気撹拌下、Arの不活性雰囲気下、RTで、200mgの3−[2−(2−{2−[2−(2−{2−[2−(2−アミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−プロピオン酸(CA(PEG)8、Pierce)、1.7mlのDCM、0.9mlのMeOH、および0.34mlの2Mトリメチルシリルジアゾメタンのヘキサン中溶液をガラスバイアル内に逐次的に導入する。RTで30分後、0.1mlの2Mトリメチルシリルジアゾメタンのヘキサン中溶液を添加する。RTで25分後、数滴の酢酸の添加により反応混合物を中和させ、RP下で濃縮乾燥させ、トルエンで共沸混合させる。こうして得た無色の油を、106.9mgのブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルの、0.7mlのDCM中溶液で希釈する。RTで30分後および4℃で16時間後、粗製物を、20gのCN−グラフトシリカゲル(n.ヘプタン/iPrOH/AcOEtの勾配溶出、iPrOH部分を増加させる)上でフラッシュクロマトグラフィーにより精製する。予想された化合物を含有する画分をRP下で濃縮後、175mgの3−{2−[2−(2−{2−[2−(2−{2−[2−(2−ブロモ−アセチルアミノ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸メチルエステルを無色の油の形態で得る。質量スペクトル(B):tR=2.79分;[M+H]+:m/z576/578。
【0162】
ブロモ−酢酸2,5−ジオキソ−ピロリジン−1−イルエステルは、公開プロトコル(Biochemistry、1974年、481頁)に従い調製することができた。
【0163】
[実施例14]
L−DM4−Mal−PEG4−COOMe
【0164】
【化50】
14.1遊離薬物L−DM4−Mal−PEG4−COOMeの調製
【0165】
【化51】
磁気撹拌下、RTで、160mgのL−DM4、115.8mgの3−{2−[2−(2−{2−[3−(2,5−ジオキソ−2,5−ジヒドロ−ピロール−1−イル)−プロピオニルアミノ]−エトキシ}−エトキシ)−エトキシ]−エトキシ}−プロピオン酸2,5−ジオキソ−ピロリジン−1−イルエステル(市販のもの、SM(PEG)4、Pierce)、0.6mlのDMA、55.1mgの担持されたDIPEA(3.72mmol/g)、0.3mlの追加のDMAおよび6μlのDIPEAを逐次的に添加する。RTで1時間後および−20℃で16時間後、粗製の反応混合物をろ過し、DCMで洗浄し、20gのシリカゲル(勾配溶出、DCM:MeOH、MeOHの分担を増加)上でフラッシュクロマトグラフィーにより精製する。所期の生成物を含有する画分をRP下で濃縮後、110mgの無色の被膜を得て、これを10gのシリカゲル(勾配溶出、DCM:MeOH、MeOHの分担を増加)上で精製する。所期の生成物を含有する画分をRP下で濃縮後、19.6mgのメチルエステルL−DM4−Mal−PEG4−COOMeを無色のガラスの形態で得る。質量スペクトル(A):tR=1.31/1.32分(2つのジアステレオ異性体);[M−H]−:m/z 1208。1H NMR(500MHz,ppm単位,クロロホルム−d):0.73(s,3H);1.22(m,13H);1.39(m,1H);1.52(d,J=13.7Hz,1H);1.57(s,3H);1.78(m,1H);1.99(m,1H);2.11(ddd,J=1.8および1.9および14.1Hz,1H);2.24(ddd,J=4.7および11.5および15.9Hz,1H);2.38(m,3H);2.46(m,1H);2.53(m,3H);2.69(s,1H);2.82(s,3H);2.94(dd,J=4.7および9.6Hz,1H);3.08(m,3H);3.14(s,3H);3.29(s,3H);3.34(q,J=5.3Hz,2H);3.43(d,J=9.1Hz,1H);3.48(td,J=1.8および5.1Hz,2H);3.58(s,13H);3.67(m,7H);3.91(s,3H);4.22(t,J=11.3Hz,1H);4.70(m,1H);5.29(m,1H);5.59(m,1H);6.30(s 大,1H);6.35(dd,J=11.0および15.4Hz,1H);6.61(m,2H);6.76(s,1H)。
【0166】
[実施例15]
MDA−MB−231およびHCT116腫瘍細胞の増殖の阻害
指数増殖期の細胞をトリプシン処理し、これらのそれぞれの培地内に再懸濁させた(DMEM/F12 Gibco#21331;10%SVF Gibco#10500−056;MDA−MB231&MDA−A1細胞に対して2nMグルタミンGibco#25030;DMEM(Gibco#11960)10%SVF Gibco#10500−056;HCT116細胞に対して2nMグルタミンGibco#25030)。完全な血清含有培地の中で、5000細胞/ウェルの密度で(MDA−MB231、HCT116)、細胞懸濁液を96−ウェルCytostar培養物プレート(GE Healthcare Europe、#RPNQ0163)に分配した。4時間のコーティング後、3連ウェルに段階希釈を添加した。薬物の存在下、37°C/5%C02で3日間細胞を培養した。第4日目、10μlの14Cチミジン溶液(0.1μCi/ウェル(Perkin Elmer#NEC56825000)を各ウェルに添加した。microbeta放射性カウンター(Perkin Elmer)を用いて、実験が開始してから96時間、14Cチミジンの取り込みを測定した。無細胞の試薬ブランクを試験ウェル記録から引き算し、コンジュゲート処理した細胞の記録を、ビヒクル処理した細胞の対照ウェルからの記録の平均で割ることによって得た、残存する画分として、このデータをプロットした。
【0167】
【表4】
【0168】
これらから明白なように、エステル−COOMeの形態で試験された生成物は、2つの異なる細胞株に対して強力なインビトロの増殖阻害特性を示す。
【0169】
[実施例16]
hu2H11およびhu2H11R35R74のコンジュゲートのインビボの評価
コンジュゲートの制癌活性の評価のため、動物を毎日秤量し、週に2回キャリパーで腫瘍を測定した。質量(mg)=[長さ(mm)×幅(mm)2]/2の式を用いて腫瘍の重量を計算した。抗腫瘍活性の評価は、最大非毒性用量(HNTD)で行った。
【0170】
最下点(グループの平均)での20%の体重減(bwl)または10%以上の薬物死が生じる用量は、過剰に有毒な用量とみなされた。動物の体重には腫瘍の重量が含まれていた。主要評価項目は、ΔT/ΔC、メディアン退縮パーセント、部分退縮および完全退縮(PRおよびCR)および無腫瘍生存(TFS)である。
【0171】
特定された観察日の腫瘍量から最初の処理日(ステージング日)の腫瘍量を引くことによって、処理した腫瘍(T)および対照(C)のそれぞれの腫瘍量の変化を各腫瘍に対して計算する。メディアンΔΤを処理したグループに対して計算し、メディアンΔCを対照グループに対して計算する。次いで割合ΔΤ/ΔCを計算し、パーセンテージで表現する。
【0172】
【数1】
【0173】
用量は、ΔΤ/ΔCが40%より低い場合、治療的に活性があるとみなされ、ΔΤ/ΔCが10%よりも低い場合、非常に活性があるとみなされる。ΔΤ/ΔCが0より低い場合、この用量は、高活性とみなされ、この退縮パーセンテージの日付を記録する(ref1):
【0174】
%腫瘍退縮:これは、最初の処理の初日におけるこの量と比較した場合の、特定された観察日における処理したグループの腫瘍量の減少の%と定義される。特定の時間点および各動物に対して、%退縮を計算する。よって、グループに対するメディアン%退縮を計算する:%退縮(tにおいて)=
【0175】
【数2】
【0176】
部分退縮(PR):退縮は、処理開始時の腫瘍量の50%まで腫瘍量が低減した場合、部分的と定義される。
【0177】
完全退縮(CR):完全退縮は、腫瘍量=0mm3の場合達成される(腫瘍量が記録できない場合CRが考慮される)。
【0178】
TFS:無腫瘍とは、実験の終わりに、腫瘍が検出不能な動物と定義される。
【0179】
hu2H11−コンジュゲートとhu2H11R35R74−コンジュゲートとの比較
hu2H11−コンジュゲート=
【0180】
【化52】
hu2H11R35R74−コンジュゲート=
【0181】
【化53】
メスのSCIDマウスにS.C.移植した、強力に発現するターゲットである、測定可能な原発性結腸腫瘍、CR−LRB−004Pに対して、hu2h11−コンジュゲートおよびhu2h11R35R74−コンジュゲートの抗腫瘍活性を2つの用量レベルで評価した。対照グループは未処理のままにしておいた。用量は、1キログラム中のタンパク質をミリグラムで表現した。hu2h11R35R74−コンジュゲートを、40および10mg/kgの量で、静脈内(IV)大量瞬時投与により、第15日に投与した。等しい用量のDM4を与えるため、hu2h11−コンジュゲート、44および11mg/kgを投与した。結果を表IVに示す。
【0182】
CR−LRB−004P腫瘍の単回投与スケジュールを用いて、hu2h11R35R74−コンジュゲートは、40および10mg/kgにおいて活性があり、ΔΤ/ΔΟがそれぞれ28%および39%であり、hu2h11−コンジュゲートは、40mg/kgにおいてのみ活性があり、ΔΤ/ΔΟは26%であった。10mg/kgにおいて、hu2h11−コンジュゲートは、本モデルでは活性がなかった。これらの結果から、hu2h11R35R74−コンジュゲートは、低用量では、hu2h11−コンジュゲートよりも良い活性を示した。
【0183】
コンジュゲートの最適化、最適な薬物抗体比DARの選択−SCIDメスのマウスにおける前立腺の腺癌PC−3に対するhu2H11R35R74−コンジュゲートの制癌活性に対するDARの影響
メスのSCIDにS.C.移植した、前立腺のPC−3腫瘍について、6つの異なる薬物抗体比(DAR)での、2つの低い有効用量を比較して、hu2H11R35R74−コンジュゲートの抗腫瘍活性に対するDARの作用を評価した。対照グループは未処理のままにしておいた。用量は、1キログラム中のタンパク質をミリグラムで表現した。DARをUV法で求めた。hu2H11R35R74−コンジュゲートを、10および5mg/kgの量で、それぞれ3.4、4.4、5.9、6.2、7.4および8.4のDARで、静脈内(IV)大量瞬時投与により、第16日に投与した。結果を表Vに示す。
【0184】
【表5】
【0185】
【表6】
【0186】
単回投与スケジュールを用いて、hu2H11R35R74−コンジュゲートは、10mg/kgにおいて、DAR4.4から8.4で活性を示し、hu2H11R35R74−コンジュゲートは、5mg/kgにおいて、DAR5.9から4で活性を示した。結論として、DARは、hu2H11R35R74−コンジュゲートの腫瘍活性について効果を有する。これらの結果から、特定の腫瘍について、DAR(UV)は、4より上であるべきである。最適なDARは、少なくとも5.9に等しいことになる。
【0187】
[実施例17]
hu2H11R35R74−コンジュゲートのPKパラメータについてのDARの評価
異なる薬物−抗体比(DAR)でのhu2H11R35R74−コンジュゲートの薬物動態学的特性を、20mg/kgのコンジュゲートを単回の静脈内(IV)投与後、オスのCD−1マウスにおいて評価した。コンジュゲートの血漿レベルを測定することによって、標準的条件下での基本的な単回投与の薬物動態学的パラメータを確立した。PKパラメータを裸の親抗体のものと比較した。コンジュゲートおよびこれらの抗体成分の血漿中濃度(全抗体、結合した抗体とあらゆる非結合の抗体の和)を特定のELISA技法で測定した。結果を図7に示す。
【0188】
結果は、DAR値と全抗体成分への曝露との間の逆の相互関係を示し、AUC0−∞値は、0、3.4、4.3、5.9、6.6および7.4のDARに対して、それぞれ83,000,000、61,000,000、48,000,000、46,000,000、41,000,000および27,000,000ng・h/mLであった。
【0189】
同様に、DAR値とコンジュゲートへの曝露との間に逆の相互関係が存在し、AUC0−∞値は、3.4、4.3、5.9、6.6および7.4のDARに対して、それぞれ39,000,000、30,000,000、27,000,000、29,000,000および20,000,000ng・h/mLであった。
【0190】
DAR値と、抗体成分の排除との間に完璧な相互関係が存在し、CI値は、0、3.4、4.3、5.9、6.6および7.4のDARに対して、それぞれ0.00024、0.00033、0.00042、0.00043、0.00049および0.00074L/h/kgであった。
【0191】
同様に、DAR値とコンジュゲートの排除との間にほとんど完璧な相互関係が存在し、CI値は、3.4、4.3、5.9、6.6および7.4のDARに対してそれぞれ0.00051、0.00066、0.00075、0.00069、0.00099L/h/kgであった。
【0192】
結論として、DARは、PKパラメータに影響を及ぼし、DARが増加すると、曝露が減少し、除去が増加する。効力およびPKの評価結果によると、最適なDARは、5.9と7.4の間に含まれていることになる。
【0193】
[実施例18]
SCIDメスのマウスにおける前立腺の腺癌PC−3に対するhu2H11R35R74コンジュゲートの評価
メスのSCIDマウスにS.C.移植した、強力に発現するターゲットである、測定可能な前立腺PC−3腫瘍に対して、DAR=5.9を有する抗体薬物コンジュゲートhu2H11R35R74−コンジュゲートの抗腫瘍作用を、8つの用量レベルで評価した。対照グループは、未処理のままにしておいた。用量は、1キログラム中のタンパク質をミリグラムで表現した。これらを、160、120、80、40、20、10、5および2.5mg/kgの量で、静脈内(IV)大量瞬時投与により、第17日に投与した。結果を表VIに示す。
【0194】
単回投与スケジュールを用いて、体重減少および薬物に関連した死亡を誘発する有毒な量となる、試験したコンジュゲートの最大用量(160mg/kg)を発見した。HNTD(120mg/kg)および他の最小用量において、この化合物は高活性であった。2.5mg/kg以外のすべて用量に対して、hu2H11R35R74−コンジュゲートは、部分退縮を誘発し、120、80および20mg/kgに対して、完全退縮を誘発した。加えて、この腫瘍モデル悪液質であり、化合物の投与は、対照と比較して、最下点での体重減少を減少させた。結論として、hu2H11R35R74−コンジュゲートは、前立腺のPC−3腫瘍モデルにおいて、優れた用量−効果を有する高い活性を示した。
【0195】
【表7】
【特許請求の範囲】
【請求項1】
式(I)の化合物:
【化1】
(式中、
ALKは、(C1−C6)アルキレン基であり、
X1およびX2は、それぞれ独立して、以下の基:−CH=CH−、−CO−、−CONR−、−NRCO−、−COO−、−OCO−、−OCONR−、−NRCOO−、−NRCONR’−、−NR−、−S(0)n(n=0、1または2)または−O−のうちの1つであり、
RおよびR’は、独立して、Hまたは(C1−C6)アルキル基であり、
iは、1から40、好ましくは1から20、より好ましくは1から10の整数であり、
jは、X2が−CH=CH−の場合、1に相当する整数であり、X2が−CH=CH−でない場合、2に相当する整数であり、
Zbは、単一結合、−O−または−NH−であり、Rbは、Hまたは(C1−C6)アルキル、(C3−C7)シクロアルキル、アリール、ヘテロアリールまたは(C3−C7)ヘテロシクロアルキル基であり、またはZbは一重結合であり、RbはHalである。)。
【請求項2】
X2が−CH=CH−または−CONR−であり、前記CO基が、−X1−ALK−基に連結されており、RがHまたは(C1−C6)アルキル基である、請求項1に記載の化合物。
【請求項3】
−X1−ALK−が−S−CH2−である、請求項1または2に記載の化合物。
【請求項4】
iが3、4、5、6、7、8、9または10である、請求項1から3に記載の化合物。
【請求項5】
式(II):
【化2】
である、請求項1から4に記載の化合物。
【請求項6】
以下の化合物:
【化3】
の中から選択される化合物。
【請求項7】
−COZbRbが、−COOH、−COO(C1−C6)アルキル、−COOMe、COOCH2CH=CH2、
【化4】
基
【化5】
(式中、Glは、少なくとも1つの誘導基−NO2、−Halまたは−Fを表す。)、または
【化6】
である、請求項1から6のいずれかに記載の化合物。
【請求項8】
塩基もしくは塩、または前記塩基もしくは前記塩の溶媒和物もしくは水和物の形態の、請求項1から7のいずれかに記載の化合物。
【請求項9】
(i)場合によって緩衝されている抗体水溶液と、請求項1から8に記載の化合物の溶液を接触させるステップ、ならびに
(ii)次いで、(i)で形成したコンジュゲートを、未反応の試薬および溶液中に存在し得るいかなる凝集体からも場合によって分離させるステップ
を含む、コンジュゲートの調製方法。
【請求項10】
反応温度が、通常20から40℃の範囲で変動し、および/または反応時間が1から24時間の範囲で変動する、請求項9に記載の方法。
【請求項11】
ステップ(i)または(ii)の後、コンジュゲート含有溶液に、追加のステップ(iii)である限外ろ過法および/または膜分離法を施す、請求項9から10に記載の方法。
【請求項12】
抗体が、EphA2受容体に特異的に結合し、少なくとも1つの重鎖および少なくとも1つの軽鎖を含む抗体またはこのエピトープ結合フラグメントであり、前記重鎖が、SEQ ID NO:1、2および3で表されるアミノ酸配列を有する3つの連続した相補性決定領域を含み、前記軽鎖が、SEQ ID NO:4、5および6で表されるアミノ酸配列を有する3つの連続した相補性決定領域を含む、請求項9から11に記載の方法。
【請求項13】
抗体またはエピトープ結合フラグメントが、ヒト化抗体もしくは再表面化抗体またはこのエピトープ結合フラグメントである、請求項12に記載の方法。
【請求項14】
前記重鎖が、SEQ ID NO:12からなるアミノ酸配列を含み、前記軽鎖が、SEQ ID NO:14からなるアミノ酸配列を含む、請求項12に記載の方法。
【請求項15】
前記重鎖が、アミノ酸配列SEQ ID NO:18にあり、前記軽鎖がアミノ酸配列SEQ ID NO:16にある、請求項12に記載の方法。
【請求項16】
請求項9から15に記載の方法を用いて入手可能なコンジュゲート。
【請求項17】
UV分光光度計で測定した平均DARが、4より上、より具体的には4と10の間、さらにより具体的には4と7の間にあり、前記DARが、
cD=[(εA280×A252)−(εA252×A280)]/[(εD252×εA280)]−(εA252×εD280)]
cA=[A280−(cD×εD280)]/εA280
εD252=26,159M−1cm−1
εD280=5,180M−1cm−1
εA280=224,000M−1cm−1
εA252=82,880M−1cm−1
A252およびA280は、それぞれ252および280nmにおいて、UV分光光度計で測定されたコンジュゲートの吸光度である
という条件で、方程式DAR=cD/cAから求められる、請求項16に記載のコンジュゲート。
【請求項18】
DARが、5と8の間、より具体的には5.5と8の間、さらにより具体的には5.9と7.5の間にある、請求項17に記載のコンジュゲート。
【請求項19】
請求項16から18に記載のコンジュゲートを含む水溶液。
【請求項20】
抗癌剤として使用するための、請求項1から8に記載の生成物。
【請求項21】
抗癌剤として使用するための、請求項16または18に記載のコンジュゲート。
【請求項22】
式:
【化7】
のメイタンシノイドフラグメントが抗体に共有結合で連結している、コンジュゲートを調製するための請求項1から8に記載の化合物の使用。
【請求項1】
式(I)の化合物:
【化1】
(式中、
ALKは、(C1−C6)アルキレン基であり、
X1およびX2は、それぞれ独立して、以下の基:−CH=CH−、−CO−、−CONR−、−NRCO−、−COO−、−OCO−、−OCONR−、−NRCOO−、−NRCONR’−、−NR−、−S(0)n(n=0、1または2)または−O−のうちの1つであり、
RおよびR’は、独立して、Hまたは(C1−C6)アルキル基であり、
iは、1から40、好ましくは1から20、より好ましくは1から10の整数であり、
jは、X2が−CH=CH−の場合、1に相当する整数であり、X2が−CH=CH−でない場合、2に相当する整数であり、
Zbは、単一結合、−O−または−NH−であり、Rbは、Hまたは(C1−C6)アルキル、(C3−C7)シクロアルキル、アリール、ヘテロアリールまたは(C3−C7)ヘテロシクロアルキル基であり、またはZbは一重結合であり、RbはHalである。)。
【請求項2】
X2が−CH=CH−または−CONR−であり、前記CO基が、−X1−ALK−基に連結されており、RがHまたは(C1−C6)アルキル基である、請求項1に記載の化合物。
【請求項3】
−X1−ALK−が−S−CH2−である、請求項1または2に記載の化合物。
【請求項4】
iが3、4、5、6、7、8、9または10である、請求項1から3に記載の化合物。
【請求項5】
式(II):
【化2】
である、請求項1から4に記載の化合物。
【請求項6】
以下の化合物:
【化3】
の中から選択される化合物。
【請求項7】
−COZbRbが、−COOH、−COO(C1−C6)アルキル、−COOMe、COOCH2CH=CH2、
【化4】
基
【化5】
(式中、Glは、少なくとも1つの誘導基−NO2、−Halまたは−Fを表す。)、または
【化6】
である、請求項1から6のいずれかに記載の化合物。
【請求項8】
塩基もしくは塩、または前記塩基もしくは前記塩の溶媒和物もしくは水和物の形態の、請求項1から7のいずれかに記載の化合物。
【請求項9】
(i)場合によって緩衝されている抗体水溶液と、請求項1から8に記載の化合物の溶液を接触させるステップ、ならびに
(ii)次いで、(i)で形成したコンジュゲートを、未反応の試薬および溶液中に存在し得るいかなる凝集体からも場合によって分離させるステップ
を含む、コンジュゲートの調製方法。
【請求項10】
反応温度が、通常20から40℃の範囲で変動し、および/または反応時間が1から24時間の範囲で変動する、請求項9に記載の方法。
【請求項11】
ステップ(i)または(ii)の後、コンジュゲート含有溶液に、追加のステップ(iii)である限外ろ過法および/または膜分離法を施す、請求項9から10に記載の方法。
【請求項12】
抗体が、EphA2受容体に特異的に結合し、少なくとも1つの重鎖および少なくとも1つの軽鎖を含む抗体またはこのエピトープ結合フラグメントであり、前記重鎖が、SEQ ID NO:1、2および3で表されるアミノ酸配列を有する3つの連続した相補性決定領域を含み、前記軽鎖が、SEQ ID NO:4、5および6で表されるアミノ酸配列を有する3つの連続した相補性決定領域を含む、請求項9から11に記載の方法。
【請求項13】
抗体またはエピトープ結合フラグメントが、ヒト化抗体もしくは再表面化抗体またはこのエピトープ結合フラグメントである、請求項12に記載の方法。
【請求項14】
前記重鎖が、SEQ ID NO:12からなるアミノ酸配列を含み、前記軽鎖が、SEQ ID NO:14からなるアミノ酸配列を含む、請求項12に記載の方法。
【請求項15】
前記重鎖が、アミノ酸配列SEQ ID NO:18にあり、前記軽鎖がアミノ酸配列SEQ ID NO:16にある、請求項12に記載の方法。
【請求項16】
請求項9から15に記載の方法を用いて入手可能なコンジュゲート。
【請求項17】
UV分光光度計で測定した平均DARが、4より上、より具体的には4と10の間、さらにより具体的には4と7の間にあり、前記DARが、
cD=[(εA280×A252)−(εA252×A280)]/[(εD252×εA280)]−(εA252×εD280)]
cA=[A280−(cD×εD280)]/εA280
εD252=26,159M−1cm−1
εD280=5,180M−1cm−1
εA280=224,000M−1cm−1
εA252=82,880M−1cm−1
A252およびA280は、それぞれ252および280nmにおいて、UV分光光度計で測定されたコンジュゲートの吸光度である
という条件で、方程式DAR=cD/cAから求められる、請求項16に記載のコンジュゲート。
【請求項18】
DARが、5と8の間、より具体的には5.5と8の間、さらにより具体的には5.9と7.5の間にある、請求項17に記載のコンジュゲート。
【請求項19】
請求項16から18に記載のコンジュゲートを含む水溶液。
【請求項20】
抗癌剤として使用するための、請求項1から8に記載の生成物。
【請求項21】
抗癌剤として使用するための、請求項16または18に記載のコンジュゲート。
【請求項22】
式:
【化7】
のメイタンシノイドフラグメントが抗体に共有結合で連結している、コンジュゲートを調製するための請求項1から8に記載の化合物の使用。
【図1】

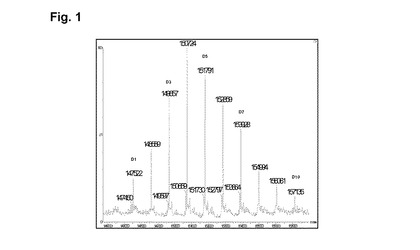
【図2】
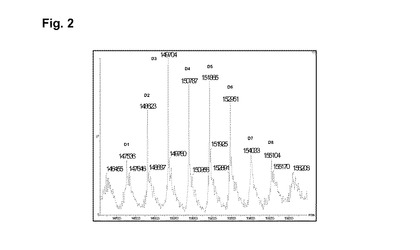
【図3】
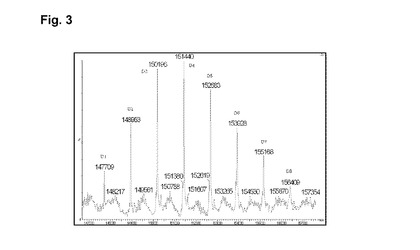
【図4】
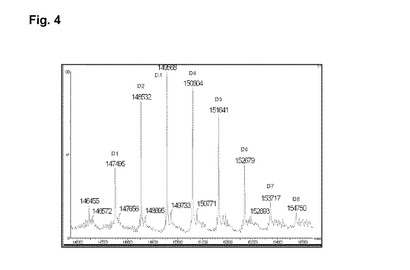
【図5】
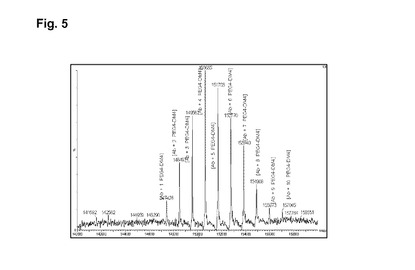
【図6A】

【図6B】

【図6C】

【図7】
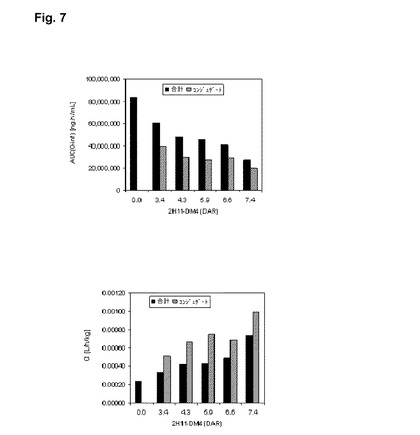
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図7】
【公表番号】特表2013−506653(P2013−506653A)
【公表日】平成25年2月28日(2013.2.28)
【国際特許分類】
【出願番号】特願2012−531540(P2012−531540)
【出願日】平成22年9月30日(2010.9.30)
【国際出願番号】PCT/IB2010/054417
【国際公開番号】WO2011/039721
【国際公開日】平成23年4月7日(2011.4.7)
【出願人】(504456798)サノフイ (433)
【Fターム(参考)】
【公表日】平成25年2月28日(2013.2.28)
【国際特許分類】
【出願日】平成22年9月30日(2010.9.30)
【国際出願番号】PCT/IB2010/054417
【国際公開番号】WO2011/039721
【国際公開日】平成23年4月7日(2011.4.7)
【出願人】(504456798)サノフイ (433)
【Fターム(参考)】
[ Back to top ]