
新規ルーメンバクテリア変異菌株及びそれを用いたコハク酸の製造方法
【課題】多様な有機酸を生産する従来の野生型菌株に比べて、他の有機酸をほとんど生成せず、高濃度でコハク酸を生成する特性を有している変異菌株を嫌気的な条件で培養することによるコハク酸の製造方法の提供。
【解決手段】ルーメンバクテリアで乳酸、蟻酸及び酢酸生成に関与する乳酸脱水素酵素遺伝子(ldhA)及びピルビン酸−蟻酸分解酵素遺伝子(pfl)が欠失された新規ルーメンバクテリア変異菌株(Mannheimia sp.LPK)。
【解決手段】ルーメンバクテリアで乳酸、蟻酸及び酢酸生成に関与する乳酸脱水素酵素遺伝子(ldhA)及びピルビン酸−蟻酸分解酵素遺伝子(pfl)が欠失された新規ルーメンバクテリア変異菌株(Mannheimia sp.LPK)。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、他の有機酸はほとんど生成せず、コハク酸を高濃度で生産するルーメンバクテリア変異菌株及び該変異菌株を嫌気的な条件で培養することを特徴とするコハク酸の製造方法に関する。
【背景技術】
【0002】
Succinivibrio dextrinosolvens、Fibrobacter succinogenes、及びRuminococcus flavefaciensなどを含む数種の嫌気性微生物が、ブドウ糖代謝を通じてコハク酸を最終的な産物として生成する(Zeikus,Annu.Rev.Microbiol.,34:423,1980)。CO2が過量存在するときにブドウ糖から高濃度かつ高収率でコハク酸を生産すると知られたAnaerobiospirillum succiniciproducensを除いては、産業的に有用な収率でコハク酸を生産する菌株は報告されていない(David et al.,Int.J.Syst.Bacteriol.,26:498,1976)。しかし、Anaerobiospirillum succiniciproducensは、絶対嫌気性の微生物であるので、これを用いてコハク酸を生産する発酵工程は、微量の酸素に露出しても工程自体が不安定になるという欠点を有する。
【0003】
このような欠点を改善するために、酸素に対して抵抗性を有し、かつ有機酸の生産性の高い菌株であるMannheimia succiniciproducens 55Eが開発された。しかし、前記菌株は、コハク酸以外にも蟻酸、酢酸及び乳酸を生成するため、収率が低く、かつコハク酸以外の他の有機酸を除去する精製工程で多くのコストがかかるという欠点を有する。
【0004】
コハク酸の生産のために、組換えられた大腸菌については多数の文献で報告されている。大腸菌の場合、乳酸脱水素酵素をコードする遺伝子(ldhA)及びピルビン酸−蟻酸の分解酵素をコードする遺伝子(pfl)が欠失されれば、嫌気的な条件ではほとんど生育できない。また、乳酸は、発酵産物として出ないが、他の代謝産物である酢酸及びエタノールがコハク酸の生産量の半分ほどをそれぞれ占めて、産業化するには収率が低すぎるという欠点がある。このような欠点を補完するために、好気条件で大腸菌の菌体を増殖させた後、再び嫌気条件に変えてコハク酸の発酵を誘導したが、依然として生産性が低いという欠点がある(Vemuri et al.,J.Ind.Microbiol.Biotechnol.,28:325,2002)。また、コハク酸の発酵の代謝経路でCO2を固定する、ピルビン酸カルボキシラーゼ(pyruvate carboxylase)、 ホスホエノールピルビン酸カルボキシラーゼ(phosphoenolpyruvate carboxylase)、 ピルビン酸カルボキシキナーゼ(pyruvate carboxykinase)、リンゴ酸酵素(malic enzyme)のような酵素の遺伝子を大腸菌に導入して、コハク酸の生産性を向上させた例が報告されている(Vemuri et al., Appl. Environ. Microbiol., 68:1715, 2002; Millard et al., Appl. Environ. Microbiol., 62:1808, 1996; Chao and Liao, Appl. Environ. Microbiol.,
59:4261, 1993; Stols and Donnelly, Appl. Environ. Microbiol., 63:2695, 1997)。
【0005】
一方、大腸菌でptsGの欠失が菌体産生及びコハク酸の生産性の向上に寄与すると報告されているが(Chatterjee et al.,Appl.Environ.Microbiol.,67:148,2001)、ほとんどのルーメンバクテリアはptsGを有していないため、大腸菌の場合と同様に、ptsGを除去する過程が不要であるという利点がある。最近、コハク酸の発酵の代謝経路において、CO2を固定する酵素の遺伝子を大腸菌以外にアクチノバチルス(Actinobacillus)属やアネロビオスピリルム(Anaerobiospirillum)属などのルーメンバクテリア(rumen bacteria)に導入する試みが行われているが、他の有機酸が過量生成されるか、または収率が低くて、産業的に適用し難い。
【発明の開示】
【発明が解決しようとする課題】
【0006】
したがって、本発明者らは、高収率でコハク酸を生産する菌株を開発するために鋭意努力した結果、ルーメンバクテリアの一種であるMannheimia succiniciproducens 55Eにおいて、ldhA及びpflを欠失させて、変異菌株であるMannheimia sp.LPK(KCTC 10558BP)を作製し、前記LPK菌株でホスホトランスアセチル化酵素遺伝子(pta)、酢酸キナーゼ遺伝子(ackA)及びホスホピルビン酸カルボキシラーゼ遺伝子(ppc)をそれぞれ欠失させて、変異菌株(Mannheimia sp.LPK7及びLPK4)を作製した後、これを嫌気的な条件で培養した結果、コハク酸が高収率で生産されるということを見出し、本発明を完成することに至った。
【0007】
従って、本発明の主な目的は、嫌気的な条件で他の有機酸は生成せず、コハク酸を高収率で生成するルーメンバクテリア変異菌株及びその製造方法を提供することにある。
【0008】
本発明の他の目的は、前記変異菌株を嫌気的な条件下で培養することを特徴とするコハク酸の製造方法を提供することにある。
【課題を解決するための手段】
【0009】
前記目的を達成するために、本発明は、ルーメンバクテリア変異菌株において、ldhA及びpflが欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株を提供する。
【0010】
また、本発明は、ルーメンバクテリア変異菌株において、ldhA、pfl、pta及びackAが欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株を提供する。
【0011】
また、本発明は、ルーメンバクテリア変異菌株において、ldhA、pfl及びppcが欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株を提供する。
【0012】
本発明において、前記ルーメンバクテリアは、マンヘミア(Mannheimia)属、アクチノバチルス(Actinobacillus)属及びアネロビオスピリルム(Anaerobiospirillum)属からなる群より選択され、他の有機酸はほとんど生成せず、コハク酸のみを生成する同型発酵菌株であることを特徴とすることができる。本発明の望ましい具現例において、前記ルーメンバクテリア変異菌株は、Mannheimia sp.LPK,LPK7またはLPK4である。
【0013】
また、本発明は、マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択されるルーメンバクテリアからldhA及びpflを欠失させることを特徴とする、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法を提供する。
【0014】
本発明に係るルーメンバクテリア変異菌株の製造方法において、前記ldhA及びpflの欠失は、相同組換えにより行われることを特徴とし、前記相同組換えは、欠失されたldhAを含む遺伝子交換ベクター及び欠失されたpflを含む遺伝子交換ベクターを使用して行われることを特徴とし、前記欠失されたldhAを含む遺伝子交換ベクターは、pMLKO−sacBであり、前記欠失されたpflを含む遺伝子交換ベクターは、pMPKO−sacBであることを特徴とすることができる。
【0015】
また、本発明は、マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択され、ldhA及びpflが欠失されているルーメンバクテリアからpta及びackAを追加で欠失させることを特徴とする、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法を提供する。
【0016】
前記pta及びackAの欠失は、相同組換えにより行われることを特徴とし、前記相同組換えは、欠失されたpta及びackAを含む遺伝子交換ベクターを使用して行われることを特徴とし、前記欠失されたpta及びackAを含む遺伝子交換ベクターは、pPTA−sacBであることを特徴とすることができる。
【0017】
また、本発明は、マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択され、ldhA及びpflが欠失されているルーメンバクテリアからppcを更に欠失させることを特徴とする、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法を提供する。
【0018】
前記ppcの欠失は、相同組換えにより行われることを特徴とし、前記相同組換えは、欠失されたppcを含む遺伝子交換ベクターを使用して行われることを特徴とし、前記欠失されたppcを含む遺伝子交換ベクターは、pPPC−sacBであることを特徴とすることができる。
【0019】
本発明において、前記ldhA及びpflが欠失されているルーメンバクテリアは、Mannheimia sp.LPK(KCTC 10558BP)であることを特徴とすることができる。
【0020】
また、本発明は、欠失されたldhAを含む遺伝子交換ベクターpMLKO−sacBと、欠失されたpflを含む遺伝子交換ベクターpMPKO−sacBと、欠失されたpta及びackAを含む遺伝子交換ベクターpPTA−sacBと、欠失されたppcを含む遺伝子交換ベクターpPPC−sacBと、を提供する。
【0021】
また、本発明は、前記ルーメンバクテリア変異菌株を嫌気的な条件で培養する段階と、前記培養液からコハク酸を回収する段階と、を含むコハク酸の製造方法を提供する。
【0022】
本発明において、‘欠失’という用語は、前記遺伝子によりコードされる酵素が生成されないように、該当遺伝子を変形させたものを全て含む。
【0023】
本発明では、まず、ルーメンバクテリアの一種であるMannheimia succiniciproducens 55Eの遺伝体情報からldhA及びpflをそれぞれ検出した後、遺伝子欠失ベクターを利用してMannheimia succiniciproducens 55Eの遺伝体から前記二つの遺伝子とも除去して、変異菌株(Mannheimia sp.LPK,KCTC 10558BP)を作製した。次いで、前記LPK(KCTC 10558BP)からpta−ackA及びppcをそれぞれ欠失させて変異菌株を作製した後、それらの変異菌株が他の有機酸はほとんど生成せず、コハク酸を高濃度で生産するということを確認した。
【0024】
本発明の変異菌株(Mannheimia sp.LPK、LPK4及びLPK7)は、通性嫌気性、グラム陰性、非運動性の桿菌(rod)または球桿菌(cocobacilli)菌であって、内生胞子(endospore)を生成しない特性を有し、嫌気条件でコハク酸が生産できる。
【図面の簡単な説明】
【0025】
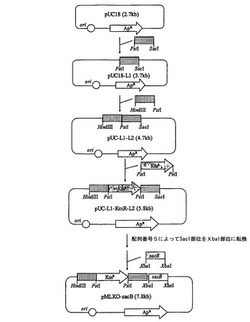
【図1】ldhA欠失ベクター(pMLKO−sacB)の作製過程を示す図である。
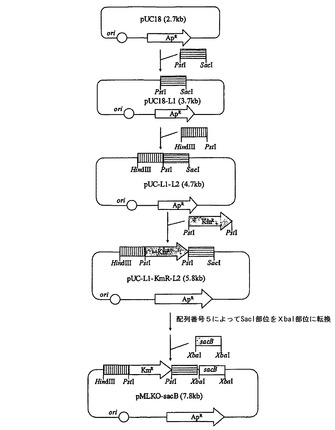
【図2】pfl欠失ベクター(pMPKO−sacB)の作製過程を示す図である。
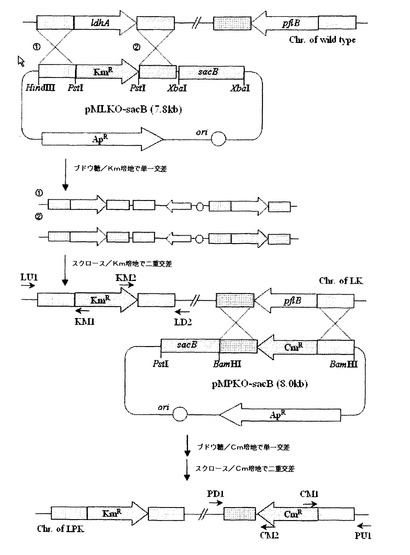
【図3】Mannheimia succiniciproducens 55EでldhA及びpflを欠失させて変異株(LPK)を作製する過程を示す図である。
【図4】Mannheimia sp.LPKでldhA及びpflの欠失を確認した電気泳動写真である[M:Lambda HindIIIサイズ・マーカー;レーン1〜3:PCR産物LU1及びKM1(1.5kb);レーン4〜6:PCR産物LD2及びKM2(1.7kb);レーン7〜9:PCR産物PU1及びCM1(2.2kb);及びレーン10〜12:PCR産物PD2及びCM2(1.6kb)]。
【図5】CO2で飽和された嫌気条件でMannheimia sp.LPKの培養特性を示すグラフである。
【図6】pta及びackAの欠失ベクター(pPTA−sacB)の作製過程を示す図である。
【図7】ppc欠失ベクター(pPPC−sacB)の作製過程を示す図である。
【図8】Mannheimia sp.LPKでpta及びackAを欠失させて変異株LPK7を作製する過程を示す図である。
【図9】Mannheimia sp.LPKでppcを欠失させて変異株LPK4を作製する過程を示す図である。
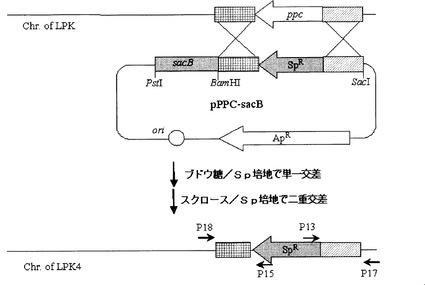
【図10】Mannheimia sp.LPK7でpta及びackAの欠失を確認した電気泳動写真である[M:1kbラダー・サイズ・マーカー;レーン1:PCR産物P13及びP14(1.1kb);及びレーン2:PCR産物P15及びP16(1.5kb)]。
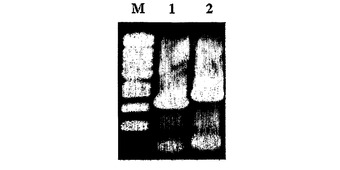
【図11】Mannheimia sp.LPK4でppcの欠失を確認した電気泳動写真である[M:1kbラダー・サイズ・マーカー;レーン1:PCR産物P13及びP17(1.1kb);及びレーン2:PCR産物P15及びP18(1.5kb)]。
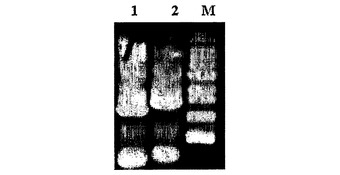
【図12】CO2で飽和された嫌気条件でMannheimia sp.LPK7の培養特性を示すグラフである。
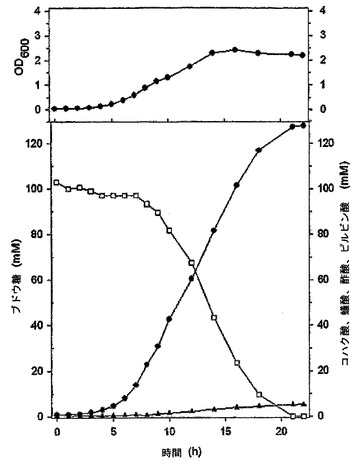
【図13】CO2で飽和された嫌気条件でMannheimia sp.LPK4の培養特性を示すグラフである。
【発明を実施するための最良の形態】
【0026】
以下、実施例を通じて本発明をさらに詳細に説明する。これらの実施例は、単に本発明をさらに具体的に説明するためのものであり、本発明の要旨によって、本発明の範囲がこれらの実施例により制限されないということは当業界に通常の知識を持つ者にとっては明らかである。
【0027】
特に、下記実施例では、マンヘミア属菌株から遺伝子を欠失させて変異菌株を得た後、これを利用してコハク酸を高濃度で生成することを含む方法のみを例示したが、アクチノバチルス属やアネロビオスピリルム属などの他のルーメンバクテリア菌株を用いて、前記遺伝子が欠失された変異菌株を得、また、これを用いてコハク酸を製造することも、当業界に通常の知識を持つ者にとっては明らかである。
【0028】
また、下記実施例では、特定の培地及び培養方法のみを例示したが、文献に報告されているように(Lee et al., Bioprocess Biosyst. Eng., 26:63, 2003; Lee et al., Appl. Microbiol. Biotechnol., 58:663, 2002; Lee et
al., Biotechnol. Lett., 25:111, 2003; Lee et al., Appl. Microbiol. Biotechnol.,
54:23, 2000; Lee et al., Biotechnol. Bioeng., 72:41, 2001)、乳清やCSL(corn steep liguor)などの糖化液と異なる培地を用いることや、流加培養(fed−batch culture)や連続培養などの多様な方法を使用することも、当業界に通常の知識を持つ者にとっては明らかである。
【0029】
実施例1:ldhA欠失ベクター(pMLKO−sacB)の作製
ldhAを相同組換え方法で破壊するために、遺伝子交換ベクターを次のように作製した。まず、Mannheimia succiniciproducens 55E(KCTC 0769BP)のゲノムDNAを鋳型とし、下記配列番号1及び2のプライマーを用いてPCRを行った後、得られたPCR断片をSacI及びPstIで切断し、これをpUC18(New England Biolabs, Inc., Beverly, Mass.)に導入してpUC18−L1を作製した。
配列番号1:5’−CAGTGAAGGAGCTCCGTAACGCATCCGCCG(LS1)
配列番号2:5’−CTTTATCGAATCTGCAGGCGGTTTCCAAAA(LP1)
【0030】
その後、Mannheimia succiniciproducens 55EのゲノムDNAを鋳型とし、下記配列番号3及び4のプライマーを用いてPCRを行った後、得られたPCR断片をPstI及びHindIIIで切断し、これを前記pUC18−L1に導入してpUC18−L1−L2を作製した。
配列番号3:5’−GTACTGTAAACTGCAGCTTTCATAGTTAGC(LP2)
配列番号4:5’−GCCGAAAGTCAAGCTTGCCGTCGTTTAGTG(LH2)
【0031】
pUC4K(Pharmacia,Freiburg,Germany)をPstIで切断して、得られたカナマイシン耐性遺伝子を、PstIで切断したpUC18−L1−L2と融合させてpUC18−L1−KmR−L2を作製した。SacIで切断したpUC18−L1−KmR−L2に下記配列番号5のリンカーを挿入して、新たなXbaI切断部位を作製した。
配列番号5:5’−TCTAGAAGCT
【0032】
pKmobsacB(Schafer et al.,Gene,145:69,1994)を鋳型とし、下記配列番号6及び7のプライマーを用いてPCRを行った後、得られた産物をXbaIで切断し、これを前記新たなXbaI制限酵素の位置に挿入してpMLKO−sacBを作製した(図1)。
配列番号6:5’−GCTCTAGACCTTCTATCGCCTTCTTGACG(SXF)
配列番号7:5’−GCTCTAGAGGCTACAAAATCACGGGCGTC(SXR)
【0033】
実施例2:pfl欠失ベクター(pMPKO−sacB)の作製
pflを相同組換え方法で破壊するために、遺伝子交換ベクターを次のように作製した。sacB遺伝子(Genbank 02730)を含有したpKmobsacBを鋳型とし、下記配列番号8及び9のプライマーを用いてPCRを行った後、得られたsacB産物をPstI及びBamHIで切断し、これをpUC19(Stratagene Cloning Systems.LaJolla,Calif.)に導入してpUC19−sacBを作製した。
配列番号8:5’−AGCGGATCCCCTTCTATCGCCTTCTTGACG(SBG)
配列番号9:5’−GTCCTGCAGGGCTACAAAATCACGGGCGTC(SPR)
【0034】
Mannheimia succiniciproducens 55EのゲノムDNAを鋳型とし、下記配列番号10と11のプライマーを用いてPCRを行った後、得られたPCR断片をBamHIで切断し、これをBamHIで切断したpUC19−sacBと融合してpUC19−sacB−pflを作製した。
配列番号10:5’−CATGGCGGATCCAGGTACGCTGATTTCGAT(PB1)
配列番号11:5’−CAAGGATCCAACGGATAAAGCTTTTATTAT(PB2)
【0035】
クロラムフェニコール耐性遺伝子を得るために、pACYC184(New England Biolabs,Inc.,Beverly,Mass.)を鋳型とし、下記配列番号12及び13のプライマーを用いてPCRを行った後、前記PCR産物をSmaIで切断し、これをBst1107Iで切断したpUC19−sacB−pflと融合してpMPKO−sacBを作製した(図2)。
配列番号12:5’−CTCGAGCCCGGGGTTTAAGGGCACCAATAA(CTR)
配列番号13:5’−CTCGAGCCCCGGGCTTTGCGCCGAATAAAT(CTF)
【0036】
実施例3:Mannheimia sp.LPK菌株の作製
図3は、Mannheimia succiniciproducens 55Eにおいて、ldhA及びpflを欠失させて変異株(LPK)を作製する過程を示す図である。Mannheimia succiniciproducens 55Eを10g/lのグルコースを含有するLB(Luria−Bertani)−グルコース培地に塗抹して、37℃で36時間培養した後、コロニーをLB−グルコース液体培地10mLに接種して12時間培養した。充分に成長した培養液をLB−グルコース液体培地100mLに1%接種して、200rpm、37℃の振盪培養器で培養した。
【0037】
約4〜5時間後にODが約0.2〜0.3になったとき、これを4℃、10分間、4000rpmの条件で遠心分離して細胞を得た後、4℃、10%のグリセロール溶液200mLで細胞を再懸濁した。また、4℃、10分間、4000rpmの条件で遠心分離して細胞を得た後、4℃、10%のグリセロール溶液200mLで細胞を再懸濁し、4℃、10分間、4000rpmの条件で遠心分離して細胞を得た。細胞とグリセロールとの体積比が1:1になるように再懸濁して細胞濃縮液を得た。
【0038】
このようにして得た細胞濃縮液と、実施例1及び実施例2で作製された遺伝子交換ベクターpMLKO−sacB及びpMPKO−sacBとを混合した後、1.8kV、25μF,200ohmsの条件でエレクトロポレーションを行った。電気衝撃後、LB−グルコース液体培地1mLを加えて、200rpm、37℃の振盪培養器で1時間培養した。培養液を適切な抗生剤[Km(最終濃度25μg/mL)またはCm(最終濃度6.8μg/mL)]を含有したLB−グルコース固体培地に塗抹して、37℃で48時間以上培養した。コロニーが形成されれば、二重交差(double crossover)のみ起こったものを選び出すために、これを、Km(25μg/mL)またはCm(6.8μg/mL)を含有したLB−スクロース(100g/lのスクロースを含有したLB培地)培地にストリークした後、24時間後に形成されたコロニーを同じプレートに再びストリークした。
【0039】
前記プレートで形成されたコロニー(変異株)を、抗生剤が含まれたLB−グルコース液体培地で培養し、培養された菌株から、ロッシェルなどの方法(Rochelle et al.,FEMS Microbiol.Lett.,100:59,1992)を応用して、ゲノムDNAを分離した。前記分離された変異株のゲノムDNAを鋳型としてPCRを行った後、得られたPCR産物を電気泳動することによってldhA及びpflの欠失の可否を確認した。
【0040】
ldhAの欠失の可否を確認するために、下記のように、PCRを2回行った。まず、前記変異株のゲノムDNAを鋳型とし、下記配列番号14及び15のプライマーを用いてPCRを行った。
配列番号14:5’−GACGTTTCCCGTTGAATATGGC(KM1)
配列番号15:5’−CATTGAGGCGTATTATCAGGAAAC(LU1)
【0041】
次いで、前記変異株のゲノムDNAを鋳型とし、下記配列番号16及び17のプライマーを用いてPCRを行った。前記2回のPCR反応で得られた反応物をゲル電気泳動して、そのサイズ(1.5kb)によってldhAの欠失の可否を確認した(図4)。
配列番号16:5’−GCAGTTTCATTTGATGCTCGATG(KM2)
配列番号17:5’−CCTCTTACGATGACGCATCTTTCC(LD2)
【0042】
pflの欠失の可否を確認するために、下記のようにPCRを2回行った。まず、前記変異株のゲノムDNAを鋳型とし、下記配列番号18及び19のプライマーを用いてPCRを行った。
配列番号18:5’−GGTGGTATATCCAGTGATTTTTTTCTCCAT(CM1)
配列番号19:5’−CTTTGCAACATTATGGTATGTATTGCCG(PU1)
【0043】
次いで、前記変異株のゲノムDNAを鋳型とし、下記配列番号20及び21のプライマーを用いてPCRを行った。前記2回のPCR反応で得られた反応物をゲル電気泳動して、そのサイズ(1.5kb)によってpflの欠失の可否を確認した(図4)。図4において、MはLambda HindIIIサイズ・マーカーを、レーン1〜3はPCR産物LU1 & KM1(1.5kb)を、レーン4〜6はPCR産物LD2 & KM2(1.7kb)を、レーン7〜9はPCR産物PU1 & CM1(2.2kb)を、レーン10〜12はPCR産物PD2 & CM2(1.6kb)をそれぞれ示す。
配列番号20:5’−TACTGCGATGAGTGGCAGGGCGGGGCGTAA(CM2)
配列番号21:5’−CCCCAGCATGTGCAAATCTTCGTCAC(PD2)
【0044】
ldhAの欠失は、配列番号14及び15のプライマー(LU1及びKM1)を用いたPCRの結果物が1.5kbであり、それと同時に、配列番号16及び17のプライマー(LD2及びKM2)を用いたPCRの結果物が1.7kbである事実によって確認された。そして、pflの欠失は、配列番号18及び19のプライマー(PU1及びCM1)を用いたPCRの結果物が2.2kbであり、それと同時に、配列番号20及び21のプライマー(PD2及びCM2)を用いたPCRの結果物が1.6kbで事実によってが確認された。それぞれのプライマーの位置は図3に示した。前記方法で作製された変異菌株、すなわち、ldhA及びpflが欠失された菌株を、Mannheimia sp.LPKと命名し、これを2003年11月26日付で国際寄託機関である韓国生命工学研究院(KRIBB)の遺伝子銀行(KCTC)に寄託番号‘KCTC 10558BP’として寄託した。
【0045】
実施例4:Mannheimia sp.LPKの発酵特性
前記実施例3で作製されたMannheimia sp.LPKの発酵特性を調べるために、CO2で飽和された嫌気的な条件で培養し、これから生産される反応産物を分析した。まず、20g/Lのブドウ糖、5g/Lのポリペプトン、5g/Lの酵母抽出物、3g/LのK2HPO4、1g/LのNaCl、1g/Lの(NH4)2SO4、0.2g/LのCaCl2・2H2O、0.2g/LのMgCl2・6H2O及び10g/LのMgCO3からなる前培養培地100mLに炭酸ガスを注入した後、Mannheimia sp.LPKを接種し、39℃で14時間前培養を行った。次いで、20g/Lのブドウ糖、5g/Lのポリペプトン、5g/Lの酵母抽出物、3g/LのK2HPO4、1g/LのNaCl、5g/Lの(NH4)2SO4、0.2g/LのCaCl2・2H2O、0.2g/LのMgCl2・6H2O及び5g/LのNa2CO3からなる本培養培地0.9Lを2.5Lの培養槽に入れ、100mLの前培養された微生物を接種した後、39℃でpH6.5の条件下で、炭酸ガスを0.25vvmの流速で供給しつつ回分培養を行った。
【0046】
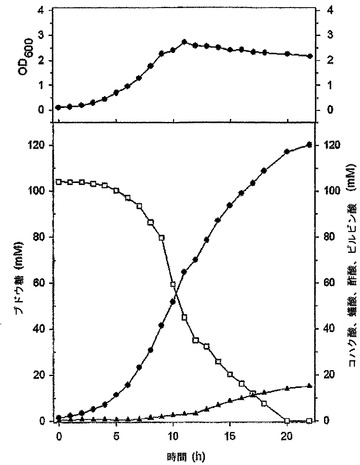
培養液内の細胞の濃度は、分光光度計(spectrophotometer,Ultraspec3000,Pharmacia Biotech.,Sweden)を利用して測定し、培地内に含まれたコハク酸、ブドウ糖、乳酸、酢酸及び蟻酸の量は、HPLC(Aminex HPX−87H column,Bio−Rad,USA)で測定した(図5)。図5において、シンボルは、培養時間による細胞の濃度(●)、コハク酸(○)、ブドウ糖(■)、蟻酸(◇)及び酢酸(△)の濃度の変化を表す。図5に示すように、14時間培養後に消耗されたブドウ糖の濃度は20g/Lであり、生成されたコハク酸の濃度は17.2g/Lであった。これによるコハク酸の収率(生成されたコハク酸の量/消費されたブドウ糖の量)は81%であり、コハク酸の体積生産性(生成されたコハク酸の濃度/所要時間)は1.23g/L/hであった。本発明のMannheimia sp.LPKを、CO2で飽和された嫌気的な条件で培養して得るコハク酸の生産方法は、親菌株であるMannheimia succiniciproducens 55Eを、CO2で飽和された嫌気的な条件で培養して得るコハク酸の生産方法より収率面において大きく向上し、コハク酸/酢酸の比率面では40.7:1であり、ほとんど副産物のないコハク酸が得られた。
【0047】
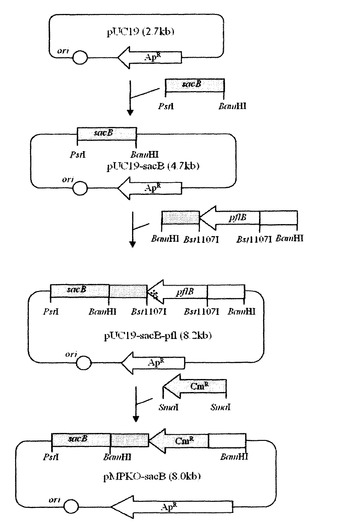
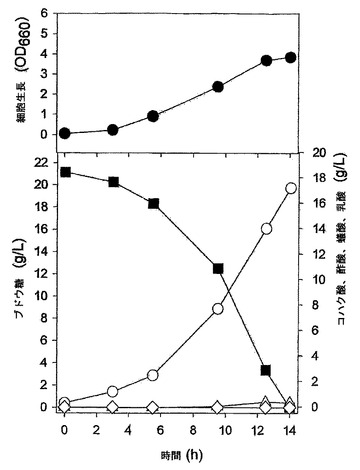
実施例5:pta及びackA欠失ベクター(pPTA−sacB)の作製
pta及びackAを相同組換え方法で破壊するために、遺伝子交換ベクターを次のように作製した。まず、Mannheimia sp.LPK(KCTC 10558BP)のゲノムDNAを鋳型とし、下記配列番号22及び23のプライマーを用いてPCRを行った後、得られたPCR断片をXbaI及びBamHIで切断し、これをpUC19に導入してpUC19−PTA1を作製した。
配列番号22:5’−GCTCTAGATATCCGCAGTATCACTTTCTGCGC
配列番号23:5’−TCCGCAGTCGGATCCGGGTTAACCGCACAG
【0048】
その後、Mannheimia sp.LPKのゲノムDNAを鋳型とし、下記配列番号24及び25のプライマーを用いてPCRを行った後、得られたPCR断片をXbaI及びSacIで切断し、これを前記pUC19−PTA1に導入してpUC19−PTA12を作製した。
配列番号24:5’−GGGGAGCTCGCTAACTTAGCTTCTAAAGGCCATGTTTCC
配列番号25:5’−GCTCTAGATATCCGGGTCAATATCGCCGCAAC
【0049】
スペクチノマイシン耐性遺伝子(GenBank X02588)を含有したプラスミドpIC156(Steinmetz et al.,Gene,142:79,1994)を鋳型とし、下記配列番号26及び27のプライマーを用いてPCRを行った後、得られたPCR断片(スペクチノマイシン耐性遺伝子)をEcoRVで切断し、これを前記pUC19−PTA12に導入してスペクチノマイシン耐性遺伝子を有するpUC19−PTA1S2を作製した。前記作製されたpUC19−PTA1S2をSacI及びBamHIで切断した後、pUC19−SacB(実施例2を参照)に導入してpPTA−sacBベクターを作製した(図6)。
配列番号26:5’−GAATTCGAGCTCGCCCGGGGATCGATCCTC
配列番号27:5’−CCCGGGCCGACAGGCTTTGAAGCATGCAAATGTCAC
【0050】
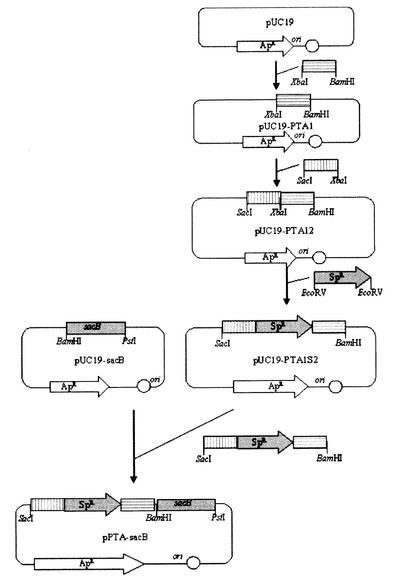
実施例6:ppc欠失ベクター(pPPC−sacB)の作製
ppcを相同組換え方法で破壊するために、遺伝子交換ベクターを次のように作製した。まず、Mannheimia sp.LPKのゲノムDNAを鋳型とし、下記配列番号28及び29のプライマーを用いてPCRを行った後、得られたPCR断片をXbaI及びBamHIで切断し、これをpUC19に導入してpUC19−PPC1を作製した。
配列番号28:5’−TACGGATCCCCAGAAAATCGCCCCCATGCCGA
配列番号29:5’−GCTCTAGATATCGTTTGATATTGTTCCGCCACATTTG
【0051】
その後、Mannheimia sp.LPKのゲノムDNAを鋳型とし、下記配列番号30及び31のプライマーを用いてPCRを行った後、得られたPCR断片をXbaI及びSacIで切断し、これを前記pUC19−PPC1に導入してpUC19−PPC12を作製した。
配列番号30:5’−GCTCTAGATATCCGTCAGGAAAGCACCCGCCATAGC
配列番号31:5’−GGGGAGCTCGTGTGGCGCTGCGGAAGTAAGGCAAAAATC
【0052】
EcoRVで切断したスペクチノマイシン耐性遺伝子(実施例5を参照)を前記pUC19−PPC12に導入してpUC19−PPC1S2を作製し、これをSacI及びBamHIで切断した後、前記pUC19−SacBに導入してpPPC−sacBベクターを作製した(図7)。
【0053】
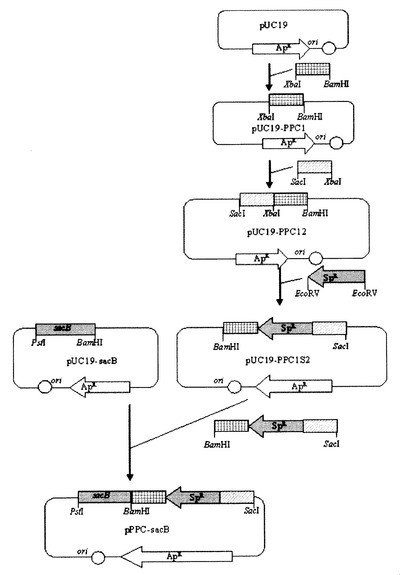
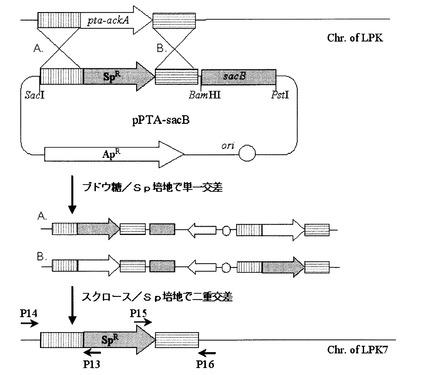
実施例7:Mannheimia sp.LPK7及びLPK4菌株の作製
図8及び図9は、Mannheimia sp.LPKでpta−ackA及びppcをそれぞれ欠失させて変異株、LPK7及びLPK4を作製する過程を示す図である。Mannheimia sp.LPKを、10g/lのグルコースを含有したLB−グルコース培地に塗抹して、37℃で36時間培養した後、コロニーをLB−グルコース液体培地10mLに接種して12時間培養した。充分に成長した培養液をLB−グルコース液体培地100mLに1%接種して、37℃の振盪培養器で培養した。
【0054】
前記培養液から、実施例3と同じ方法で細胞濃縮液を得た後、これを、実施例5及び実施例6で作製された遺伝子交換ベクターpPTA−sacB及びpPPC−sacBと混合した後、1.8kV、25μF,200ohmsの条件でエレクトロポレーションを行った。電気衝撃後、LB−グルコース液体培地1mLを加えて、200rpm、37℃の振盪培養器で1時間培養した。
【0055】
培養液を、スペクチノマイシン抗生剤(最終濃度50μg/mL)を含有したLB−グルコース固体培地に塗抹して、37℃で48時間以上培養した。コロニーが形成されれば、二重交差のみ起こったものを選び出すために、これを、スペクチノマイシン50μg/mLを含有したLB−スクロース培地(100g/lのスクロースを含有したLB培地)にストリークした。24時間後に形成されたコロニーをプレートに再びストリークした。前記プレートで形成されたコロニー(変異株)を、抗生剤が含まれたLB−グルコース液体培地で培養し、培養された菌株から、ロッシェルらの方法を応用してゲノムDNAを分離した。前記分離された変異株のゲノムDNAを鋳型としてPCRを行った後、PCR産物を電気泳動してpta−ackA及びppcの欠失の可否をそれぞれ確認した。
【0056】
pta−ackAの欠失の可否を確認するために、下記のようにPCRを2回行った。まず、前記変異株のゲノムDNAを鋳型とし、下記配列番号32及び33のプライマーを用いてPCRを行った。次に、前記変異株のゲノムDNAを鋳型とし、下記配列番号34及び35のプライマーを用いてPCRを行った。
配列番号32:5’−CCTGCAGGCATGCAAGCTTGGGCTGCAGGTCGACTC
配列番号33:5’−GCTGCCAAACAACCGAAAATACCGCAATAAACGGC
配列番号34:5’−GCATGTAACTTTACTGGATATAGCTAGAAAAGGCATCGGGGAG
配列番号35:5’−GCAACGCGAGGGTCAATACCGAAGGATTTCGCCG
【0057】
前記2回のPCR反応で得られた反応物をゲル電気泳動して、そのサイズによってpta−ackAの欠失の可否を確認した(図10)。図10において、Mは、1kbラダー・サイズ・マーカーを、レーン1は、PCR産物P13及びP14(1.1kb)を、レーン2は、PCR産物P15及びP16(1.5kb)を表す。pta−ackAの欠失は、配列番号32及び33のプライマー(P13及びP14)を用いたPCRの結果物が1.1kbであり、それと同時に、配列番号34及び35のプライマー(P15及びP16)を用いたPCRの結果物が1.5kbであるという事実によって確認した。プライマーの位置は図8に示した。前記方法で作製された変異菌株、すなわち、Mannheimia sp.LPKでpta−ackAが欠失された菌株を、Mannheimia sp.LPK7と命名し、これを国際寄託機関であるKCTCに寄託番号‘KCTC 10626BP’として寄託した。
【0058】
また、ppcの欠失の可否を確認するために、下記のように、PCRを2回行った。まず、前記変異株のゲノムDNAを鋳型とし、下記配列番号32及び36のプライマーを用いてPCRを行った。次いで、前記変異株のゲノムDNAを鋳型とし、下記配列番号34及び37のプライマーを用いてPCRを行った。
配列番号36:5’−GATCCAGGGAATGGCACGCAGGCTTTCAACGCCGCC
配列番号37:5’−GCAAAGCCAGAGGAATGGATGCCATTAACCAATAGCG
【0059】
前記2回のPCR反応で得られた反応物をゲル電気泳動して、そのサイズによってppcの欠失の可否を確認した(図11)。図11において、Mは、1kbラダー・サイズ・マーカーを、レーン1は、PCR産物P13及びP17(1.1kb)を、レーン2は、PCR産物P15及びP18(1.5kb)を表す。ppcの欠失は、配列番号32及び36のプライマー(P13及びP17)を用いたPCRの結果物が1.1kbであり、それと同時に、配列番号34及び37のプライマー(P15及びP18)を用いたPCRの結果物が1.5kbであるという事実によって確認した。プライマーの位置は図9に示した。前記方法で作製された変異菌株、すなわち、Mannheimia sp.LPKでppcが欠失された菌株を、Mannheimia sp.LPK4と命名した。
【0060】
実施例8:LPK7及びLPK4の発酵特性
前記実施例7で作製されたMannheimia sp.LPK7及びLPK4の発酵特性を調べるために、CO2で飽和された嫌気的な条件で培養し、これから生産される反応産物を分析した。まず、実施例4に記載の前培養培地200mLに炭酸ガスを注入した後、Mannheimia sp.LPK7及びLPK4をそれぞれ接種し、39℃で24時間前培養を行った。次いで、本培養培地(ブドウ糖濃度が18g/L(最終100mM)であることを除いては、実施例4と同じである)1.8Lを6.6Lの培養槽に入れ、100mLの前培養された微生物を接種した後、39℃でpH6.5の条件下で、炭酸ガスを0.25vvmの流速で供給しつつ回分式培養を行った。
【0061】
細胞濃度と、コハク酸、ブドウ糖、乳酸、酢酸及び蟻酸の濃度とは、実施例4と同じ方法で測定した(図12及び図13)。図12及び図13において、シンボルは、培養時間による細胞の濃度(上側の●)及び、コハク酸(下側の●)、ブドウ糖(□)、蟻酸(◆)及び酢酸(▲)の濃度の変化を表す。図12に示すように、Mannheimia sp.LPK7を22時間培養した後に消耗されたブドウ糖の濃度は100mMであり、生成されたコハク酸の濃度は124mMであった。これによるコハク酸の収率(生成されたコハク酸の量/消耗されたブドウ糖の量)は、モル比で124%であった。そして、酢酸の生成が著しく減った(表1)。また、図13に示すように、Mannheimia sp.LPK4を22時間培養した後に消耗されたブドウ糖の濃度は100mMであり、生成されたコハク酸の濃度は123.7mMであった。これによるコハク酸の収率(生成されたコハク酸の量/消費されたブドウ糖の量)は、モル比で123.7%であった。そして、酢酸の生成が野生型に比べて大きく減った(表1)。
【0062】
本発明のMannheimia sp.LPK7を、CO2で飽和された嫌気的な条件で培養して得るコハク酸の生産方法は、親菌株であるMannheimia succiniciproducens 55Eを、CO2で飽和された嫌気的な条件で培養して得るコハク酸の生産方法よりも、収率面において大きく向上し、コハク酸/酢酸の比率が9.8倍も向上することにより、ほとんど副産物のないコハク酸が得られた(表1)。
【0063】
【表1】

【0064】
Bulterらによると、これまで知られた微生物で酢酸生成遺伝子を全て破壊させても、まだ知られていないアミノ酸及び脂肪酸の物質代謝でも相当量生成されると知られている(Bulter et al.PNAS,101:2299,2004)。したがって、本発明は、これまで知られた酢酸の生成経路を全て遮断したものであって、高効率かつ高濃度のコハク酸の発酵を達成した。
【0065】
以上、本発明の内容の特定な部分を詳細に記述したが、当業界に通常の知識を持つ者にとってはこのような具体的な記述は単に好適な実施例に過ぎないものであり、本発明の範囲はこれによって限定されるものではない。したがって、本発明の実質的な範囲は、特許請求の範囲及びその等価物によって定義されると言えるであろう。
【産業上の利用可能性】
【0066】
以上詳細に説明及び立証したように、本発明に係るMannheimia sp.変異菌株(LPK、LPK7及びLPK4)は、CO2で飽和された嫌気的な条件ではコハク酸を生産し、酸素に対する抵抗性の高い通性嫌気性の菌株であるため、前記菌株を用いてコハク酸を製造する場合、絶対嫌気性の菌株を用いてコハク酸を生産する従来の方法に比べて、酸素露出などによる発酵工程の不安定性を画期的に除去できるだけでなく、他の有機酸の生成を除去することにより、精製工程及び生産収率の最適化及び極大化を達成することができる。
【技術分野】
【0001】
本発明は、他の有機酸はほとんど生成せず、コハク酸を高濃度で生産するルーメンバクテリア変異菌株及び該変異菌株を嫌気的な条件で培養することを特徴とするコハク酸の製造方法に関する。
【背景技術】
【0002】
Succinivibrio dextrinosolvens、Fibrobacter succinogenes、及びRuminococcus flavefaciensなどを含む数種の嫌気性微生物が、ブドウ糖代謝を通じてコハク酸を最終的な産物として生成する(Zeikus,Annu.Rev.Microbiol.,34:423,1980)。CO2が過量存在するときにブドウ糖から高濃度かつ高収率でコハク酸を生産すると知られたAnaerobiospirillum succiniciproducensを除いては、産業的に有用な収率でコハク酸を生産する菌株は報告されていない(David et al.,Int.J.Syst.Bacteriol.,26:498,1976)。しかし、Anaerobiospirillum succiniciproducensは、絶対嫌気性の微生物であるので、これを用いてコハク酸を生産する発酵工程は、微量の酸素に露出しても工程自体が不安定になるという欠点を有する。
【0003】
このような欠点を改善するために、酸素に対して抵抗性を有し、かつ有機酸の生産性の高い菌株であるMannheimia succiniciproducens 55Eが開発された。しかし、前記菌株は、コハク酸以外にも蟻酸、酢酸及び乳酸を生成するため、収率が低く、かつコハク酸以外の他の有機酸を除去する精製工程で多くのコストがかかるという欠点を有する。
【0004】
コハク酸の生産のために、組換えられた大腸菌については多数の文献で報告されている。大腸菌の場合、乳酸脱水素酵素をコードする遺伝子(ldhA)及びピルビン酸−蟻酸の分解酵素をコードする遺伝子(pfl)が欠失されれば、嫌気的な条件ではほとんど生育できない。また、乳酸は、発酵産物として出ないが、他の代謝産物である酢酸及びエタノールがコハク酸の生産量の半分ほどをそれぞれ占めて、産業化するには収率が低すぎるという欠点がある。このような欠点を補完するために、好気条件で大腸菌の菌体を増殖させた後、再び嫌気条件に変えてコハク酸の発酵を誘導したが、依然として生産性が低いという欠点がある(Vemuri et al.,J.Ind.Microbiol.Biotechnol.,28:325,2002)。また、コハク酸の発酵の代謝経路でCO2を固定する、ピルビン酸カルボキシラーゼ(pyruvate carboxylase)、 ホスホエノールピルビン酸カルボキシラーゼ(phosphoenolpyruvate carboxylase)、 ピルビン酸カルボキシキナーゼ(pyruvate carboxykinase)、リンゴ酸酵素(malic enzyme)のような酵素の遺伝子を大腸菌に導入して、コハク酸の生産性を向上させた例が報告されている(Vemuri et al., Appl. Environ. Microbiol., 68:1715, 2002; Millard et al., Appl. Environ. Microbiol., 62:1808, 1996; Chao and Liao, Appl. Environ. Microbiol.,
59:4261, 1993; Stols and Donnelly, Appl. Environ. Microbiol., 63:2695, 1997)。
【0005】
一方、大腸菌でptsGの欠失が菌体産生及びコハク酸の生産性の向上に寄与すると報告されているが(Chatterjee et al.,Appl.Environ.Microbiol.,67:148,2001)、ほとんどのルーメンバクテリアはptsGを有していないため、大腸菌の場合と同様に、ptsGを除去する過程が不要であるという利点がある。最近、コハク酸の発酵の代謝経路において、CO2を固定する酵素の遺伝子を大腸菌以外にアクチノバチルス(Actinobacillus)属やアネロビオスピリルム(Anaerobiospirillum)属などのルーメンバクテリア(rumen bacteria)に導入する試みが行われているが、他の有機酸が過量生成されるか、または収率が低くて、産業的に適用し難い。
【発明の開示】
【発明が解決しようとする課題】
【0006】
したがって、本発明者らは、高収率でコハク酸を生産する菌株を開発するために鋭意努力した結果、ルーメンバクテリアの一種であるMannheimia succiniciproducens 55Eにおいて、ldhA及びpflを欠失させて、変異菌株であるMannheimia sp.LPK(KCTC 10558BP)を作製し、前記LPK菌株でホスホトランスアセチル化酵素遺伝子(pta)、酢酸キナーゼ遺伝子(ackA)及びホスホピルビン酸カルボキシラーゼ遺伝子(ppc)をそれぞれ欠失させて、変異菌株(Mannheimia sp.LPK7及びLPK4)を作製した後、これを嫌気的な条件で培養した結果、コハク酸が高収率で生産されるということを見出し、本発明を完成することに至った。
【0007】
従って、本発明の主な目的は、嫌気的な条件で他の有機酸は生成せず、コハク酸を高収率で生成するルーメンバクテリア変異菌株及びその製造方法を提供することにある。
【0008】
本発明の他の目的は、前記変異菌株を嫌気的な条件下で培養することを特徴とするコハク酸の製造方法を提供することにある。
【課題を解決するための手段】
【0009】
前記目的を達成するために、本発明は、ルーメンバクテリア変異菌株において、ldhA及びpflが欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株を提供する。
【0010】
また、本発明は、ルーメンバクテリア変異菌株において、ldhA、pfl、pta及びackAが欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株を提供する。
【0011】
また、本発明は、ルーメンバクテリア変異菌株において、ldhA、pfl及びppcが欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株を提供する。
【0012】
本発明において、前記ルーメンバクテリアは、マンヘミア(Mannheimia)属、アクチノバチルス(Actinobacillus)属及びアネロビオスピリルム(Anaerobiospirillum)属からなる群より選択され、他の有機酸はほとんど生成せず、コハク酸のみを生成する同型発酵菌株であることを特徴とすることができる。本発明の望ましい具現例において、前記ルーメンバクテリア変異菌株は、Mannheimia sp.LPK,LPK7またはLPK4である。
【0013】
また、本発明は、マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択されるルーメンバクテリアからldhA及びpflを欠失させることを特徴とする、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法を提供する。
【0014】
本発明に係るルーメンバクテリア変異菌株の製造方法において、前記ldhA及びpflの欠失は、相同組換えにより行われることを特徴とし、前記相同組換えは、欠失されたldhAを含む遺伝子交換ベクター及び欠失されたpflを含む遺伝子交換ベクターを使用して行われることを特徴とし、前記欠失されたldhAを含む遺伝子交換ベクターは、pMLKO−sacBであり、前記欠失されたpflを含む遺伝子交換ベクターは、pMPKO−sacBであることを特徴とすることができる。
【0015】
また、本発明は、マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択され、ldhA及びpflが欠失されているルーメンバクテリアからpta及びackAを追加で欠失させることを特徴とする、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法を提供する。
【0016】
前記pta及びackAの欠失は、相同組換えにより行われることを特徴とし、前記相同組換えは、欠失されたpta及びackAを含む遺伝子交換ベクターを使用して行われることを特徴とし、前記欠失されたpta及びackAを含む遺伝子交換ベクターは、pPTA−sacBであることを特徴とすることができる。
【0017】
また、本発明は、マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択され、ldhA及びpflが欠失されているルーメンバクテリアからppcを更に欠失させることを特徴とする、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法を提供する。
【0018】
前記ppcの欠失は、相同組換えにより行われることを特徴とし、前記相同組換えは、欠失されたppcを含む遺伝子交換ベクターを使用して行われることを特徴とし、前記欠失されたppcを含む遺伝子交換ベクターは、pPPC−sacBであることを特徴とすることができる。
【0019】
本発明において、前記ldhA及びpflが欠失されているルーメンバクテリアは、Mannheimia sp.LPK(KCTC 10558BP)であることを特徴とすることができる。
【0020】
また、本発明は、欠失されたldhAを含む遺伝子交換ベクターpMLKO−sacBと、欠失されたpflを含む遺伝子交換ベクターpMPKO−sacBと、欠失されたpta及びackAを含む遺伝子交換ベクターpPTA−sacBと、欠失されたppcを含む遺伝子交換ベクターpPPC−sacBと、を提供する。
【0021】
また、本発明は、前記ルーメンバクテリア変異菌株を嫌気的な条件で培養する段階と、前記培養液からコハク酸を回収する段階と、を含むコハク酸の製造方法を提供する。
【0022】
本発明において、‘欠失’という用語は、前記遺伝子によりコードされる酵素が生成されないように、該当遺伝子を変形させたものを全て含む。
【0023】
本発明では、まず、ルーメンバクテリアの一種であるMannheimia succiniciproducens 55Eの遺伝体情報からldhA及びpflをそれぞれ検出した後、遺伝子欠失ベクターを利用してMannheimia succiniciproducens 55Eの遺伝体から前記二つの遺伝子とも除去して、変異菌株(Mannheimia sp.LPK,KCTC 10558BP)を作製した。次いで、前記LPK(KCTC 10558BP)からpta−ackA及びppcをそれぞれ欠失させて変異菌株を作製した後、それらの変異菌株が他の有機酸はほとんど生成せず、コハク酸を高濃度で生産するということを確認した。
【0024】
本発明の変異菌株(Mannheimia sp.LPK、LPK4及びLPK7)は、通性嫌気性、グラム陰性、非運動性の桿菌(rod)または球桿菌(cocobacilli)菌であって、内生胞子(endospore)を生成しない特性を有し、嫌気条件でコハク酸が生産できる。
【図面の簡単な説明】
【0025】
【図1】ldhA欠失ベクター(pMLKO−sacB)の作製過程を示す図である。
【図2】pfl欠失ベクター(pMPKO−sacB)の作製過程を示す図である。
【図3】Mannheimia succiniciproducens 55EでldhA及びpflを欠失させて変異株(LPK)を作製する過程を示す図である。
【図4】Mannheimia sp.LPKでldhA及びpflの欠失を確認した電気泳動写真である[M:Lambda HindIIIサイズ・マーカー;レーン1〜3:PCR産物LU1及びKM1(1.5kb);レーン4〜6:PCR産物LD2及びKM2(1.7kb);レーン7〜9:PCR産物PU1及びCM1(2.2kb);及びレーン10〜12:PCR産物PD2及びCM2(1.6kb)]。
【図5】CO2で飽和された嫌気条件でMannheimia sp.LPKの培養特性を示すグラフである。
【図6】pta及びackAの欠失ベクター(pPTA−sacB)の作製過程を示す図である。
【図7】ppc欠失ベクター(pPPC−sacB)の作製過程を示す図である。
【図8】Mannheimia sp.LPKでpta及びackAを欠失させて変異株LPK7を作製する過程を示す図である。
【図9】Mannheimia sp.LPKでppcを欠失させて変異株LPK4を作製する過程を示す図である。
【図10】Mannheimia sp.LPK7でpta及びackAの欠失を確認した電気泳動写真である[M:1kbラダー・サイズ・マーカー;レーン1:PCR産物P13及びP14(1.1kb);及びレーン2:PCR産物P15及びP16(1.5kb)]。
【図11】Mannheimia sp.LPK4でppcの欠失を確認した電気泳動写真である[M:1kbラダー・サイズ・マーカー;レーン1:PCR産物P13及びP17(1.1kb);及びレーン2:PCR産物P15及びP18(1.5kb)]。
【図12】CO2で飽和された嫌気条件でMannheimia sp.LPK7の培養特性を示すグラフである。
【図13】CO2で飽和された嫌気条件でMannheimia sp.LPK4の培養特性を示すグラフである。
【発明を実施するための最良の形態】
【0026】
以下、実施例を通じて本発明をさらに詳細に説明する。これらの実施例は、単に本発明をさらに具体的に説明するためのものであり、本発明の要旨によって、本発明の範囲がこれらの実施例により制限されないということは当業界に通常の知識を持つ者にとっては明らかである。
【0027】
特に、下記実施例では、マンヘミア属菌株から遺伝子を欠失させて変異菌株を得た後、これを利用してコハク酸を高濃度で生成することを含む方法のみを例示したが、アクチノバチルス属やアネロビオスピリルム属などの他のルーメンバクテリア菌株を用いて、前記遺伝子が欠失された変異菌株を得、また、これを用いてコハク酸を製造することも、当業界に通常の知識を持つ者にとっては明らかである。
【0028】
また、下記実施例では、特定の培地及び培養方法のみを例示したが、文献に報告されているように(Lee et al., Bioprocess Biosyst. Eng., 26:63, 2003; Lee et al., Appl. Microbiol. Biotechnol., 58:663, 2002; Lee et
al., Biotechnol. Lett., 25:111, 2003; Lee et al., Appl. Microbiol. Biotechnol.,
54:23, 2000; Lee et al., Biotechnol. Bioeng., 72:41, 2001)、乳清やCSL(corn steep liguor)などの糖化液と異なる培地を用いることや、流加培養(fed−batch culture)や連続培養などの多様な方法を使用することも、当業界に通常の知識を持つ者にとっては明らかである。
【0029】
実施例1:ldhA欠失ベクター(pMLKO−sacB)の作製
ldhAを相同組換え方法で破壊するために、遺伝子交換ベクターを次のように作製した。まず、Mannheimia succiniciproducens 55E(KCTC 0769BP)のゲノムDNAを鋳型とし、下記配列番号1及び2のプライマーを用いてPCRを行った後、得られたPCR断片をSacI及びPstIで切断し、これをpUC18(New England Biolabs, Inc., Beverly, Mass.)に導入してpUC18−L1を作製した。
配列番号1:5’−CAGTGAAGGAGCTCCGTAACGCATCCGCCG(LS1)
配列番号2:5’−CTTTATCGAATCTGCAGGCGGTTTCCAAAA(LP1)
【0030】
その後、Mannheimia succiniciproducens 55EのゲノムDNAを鋳型とし、下記配列番号3及び4のプライマーを用いてPCRを行った後、得られたPCR断片をPstI及びHindIIIで切断し、これを前記pUC18−L1に導入してpUC18−L1−L2を作製した。
配列番号3:5’−GTACTGTAAACTGCAGCTTTCATAGTTAGC(LP2)
配列番号4:5’−GCCGAAAGTCAAGCTTGCCGTCGTTTAGTG(LH2)
【0031】
pUC4K(Pharmacia,Freiburg,Germany)をPstIで切断して、得られたカナマイシン耐性遺伝子を、PstIで切断したpUC18−L1−L2と融合させてpUC18−L1−KmR−L2を作製した。SacIで切断したpUC18−L1−KmR−L2に下記配列番号5のリンカーを挿入して、新たなXbaI切断部位を作製した。
配列番号5:5’−TCTAGAAGCT
【0032】
pKmobsacB(Schafer et al.,Gene,145:69,1994)を鋳型とし、下記配列番号6及び7のプライマーを用いてPCRを行った後、得られた産物をXbaIで切断し、これを前記新たなXbaI制限酵素の位置に挿入してpMLKO−sacBを作製した(図1)。
配列番号6:5’−GCTCTAGACCTTCTATCGCCTTCTTGACG(SXF)
配列番号7:5’−GCTCTAGAGGCTACAAAATCACGGGCGTC(SXR)
【0033】
実施例2:pfl欠失ベクター(pMPKO−sacB)の作製
pflを相同組換え方法で破壊するために、遺伝子交換ベクターを次のように作製した。sacB遺伝子(Genbank 02730)を含有したpKmobsacBを鋳型とし、下記配列番号8及び9のプライマーを用いてPCRを行った後、得られたsacB産物をPstI及びBamHIで切断し、これをpUC19(Stratagene Cloning Systems.LaJolla,Calif.)に導入してpUC19−sacBを作製した。
配列番号8:5’−AGCGGATCCCCTTCTATCGCCTTCTTGACG(SBG)
配列番号9:5’−GTCCTGCAGGGCTACAAAATCACGGGCGTC(SPR)
【0034】
Mannheimia succiniciproducens 55EのゲノムDNAを鋳型とし、下記配列番号10と11のプライマーを用いてPCRを行った後、得られたPCR断片をBamHIで切断し、これをBamHIで切断したpUC19−sacBと融合してpUC19−sacB−pflを作製した。
配列番号10:5’−CATGGCGGATCCAGGTACGCTGATTTCGAT(PB1)
配列番号11:5’−CAAGGATCCAACGGATAAAGCTTTTATTAT(PB2)
【0035】
クロラムフェニコール耐性遺伝子を得るために、pACYC184(New England Biolabs,Inc.,Beverly,Mass.)を鋳型とし、下記配列番号12及び13のプライマーを用いてPCRを行った後、前記PCR産物をSmaIで切断し、これをBst1107Iで切断したpUC19−sacB−pflと融合してpMPKO−sacBを作製した(図2)。
配列番号12:5’−CTCGAGCCCGGGGTTTAAGGGCACCAATAA(CTR)
配列番号13:5’−CTCGAGCCCCGGGCTTTGCGCCGAATAAAT(CTF)
【0036】
実施例3:Mannheimia sp.LPK菌株の作製
図3は、Mannheimia succiniciproducens 55Eにおいて、ldhA及びpflを欠失させて変異株(LPK)を作製する過程を示す図である。Mannheimia succiniciproducens 55Eを10g/lのグルコースを含有するLB(Luria−Bertani)−グルコース培地に塗抹して、37℃で36時間培養した後、コロニーをLB−グルコース液体培地10mLに接種して12時間培養した。充分に成長した培養液をLB−グルコース液体培地100mLに1%接種して、200rpm、37℃の振盪培養器で培養した。
【0037】
約4〜5時間後にODが約0.2〜0.3になったとき、これを4℃、10分間、4000rpmの条件で遠心分離して細胞を得た後、4℃、10%のグリセロール溶液200mLで細胞を再懸濁した。また、4℃、10分間、4000rpmの条件で遠心分離して細胞を得た後、4℃、10%のグリセロール溶液200mLで細胞を再懸濁し、4℃、10分間、4000rpmの条件で遠心分離して細胞を得た。細胞とグリセロールとの体積比が1:1になるように再懸濁して細胞濃縮液を得た。
【0038】
このようにして得た細胞濃縮液と、実施例1及び実施例2で作製された遺伝子交換ベクターpMLKO−sacB及びpMPKO−sacBとを混合した後、1.8kV、25μF,200ohmsの条件でエレクトロポレーションを行った。電気衝撃後、LB−グルコース液体培地1mLを加えて、200rpm、37℃の振盪培養器で1時間培養した。培養液を適切な抗生剤[Km(最終濃度25μg/mL)またはCm(最終濃度6.8μg/mL)]を含有したLB−グルコース固体培地に塗抹して、37℃で48時間以上培養した。コロニーが形成されれば、二重交差(double crossover)のみ起こったものを選び出すために、これを、Km(25μg/mL)またはCm(6.8μg/mL)を含有したLB−スクロース(100g/lのスクロースを含有したLB培地)培地にストリークした後、24時間後に形成されたコロニーを同じプレートに再びストリークした。
【0039】
前記プレートで形成されたコロニー(変異株)を、抗生剤が含まれたLB−グルコース液体培地で培養し、培養された菌株から、ロッシェルなどの方法(Rochelle et al.,FEMS Microbiol.Lett.,100:59,1992)を応用して、ゲノムDNAを分離した。前記分離された変異株のゲノムDNAを鋳型としてPCRを行った後、得られたPCR産物を電気泳動することによってldhA及びpflの欠失の可否を確認した。
【0040】
ldhAの欠失の可否を確認するために、下記のように、PCRを2回行った。まず、前記変異株のゲノムDNAを鋳型とし、下記配列番号14及び15のプライマーを用いてPCRを行った。
配列番号14:5’−GACGTTTCCCGTTGAATATGGC(KM1)
配列番号15:5’−CATTGAGGCGTATTATCAGGAAAC(LU1)
【0041】
次いで、前記変異株のゲノムDNAを鋳型とし、下記配列番号16及び17のプライマーを用いてPCRを行った。前記2回のPCR反応で得られた反応物をゲル電気泳動して、そのサイズ(1.5kb)によってldhAの欠失の可否を確認した(図4)。
配列番号16:5’−GCAGTTTCATTTGATGCTCGATG(KM2)
配列番号17:5’−CCTCTTACGATGACGCATCTTTCC(LD2)
【0042】
pflの欠失の可否を確認するために、下記のようにPCRを2回行った。まず、前記変異株のゲノムDNAを鋳型とし、下記配列番号18及び19のプライマーを用いてPCRを行った。
配列番号18:5’−GGTGGTATATCCAGTGATTTTTTTCTCCAT(CM1)
配列番号19:5’−CTTTGCAACATTATGGTATGTATTGCCG(PU1)
【0043】
次いで、前記変異株のゲノムDNAを鋳型とし、下記配列番号20及び21のプライマーを用いてPCRを行った。前記2回のPCR反応で得られた反応物をゲル電気泳動して、そのサイズ(1.5kb)によってpflの欠失の可否を確認した(図4)。図4において、MはLambda HindIIIサイズ・マーカーを、レーン1〜3はPCR産物LU1 & KM1(1.5kb)を、レーン4〜6はPCR産物LD2 & KM2(1.7kb)を、レーン7〜9はPCR産物PU1 & CM1(2.2kb)を、レーン10〜12はPCR産物PD2 & CM2(1.6kb)をそれぞれ示す。
配列番号20:5’−TACTGCGATGAGTGGCAGGGCGGGGCGTAA(CM2)
配列番号21:5’−CCCCAGCATGTGCAAATCTTCGTCAC(PD2)
【0044】
ldhAの欠失は、配列番号14及び15のプライマー(LU1及びKM1)を用いたPCRの結果物が1.5kbであり、それと同時に、配列番号16及び17のプライマー(LD2及びKM2)を用いたPCRの結果物が1.7kbである事実によって確認された。そして、pflの欠失は、配列番号18及び19のプライマー(PU1及びCM1)を用いたPCRの結果物が2.2kbであり、それと同時に、配列番号20及び21のプライマー(PD2及びCM2)を用いたPCRの結果物が1.6kbで事実によってが確認された。それぞれのプライマーの位置は図3に示した。前記方法で作製された変異菌株、すなわち、ldhA及びpflが欠失された菌株を、Mannheimia sp.LPKと命名し、これを2003年11月26日付で国際寄託機関である韓国生命工学研究院(KRIBB)の遺伝子銀行(KCTC)に寄託番号‘KCTC 10558BP’として寄託した。
【0045】
実施例4:Mannheimia sp.LPKの発酵特性
前記実施例3で作製されたMannheimia sp.LPKの発酵特性を調べるために、CO2で飽和された嫌気的な条件で培養し、これから生産される反応産物を分析した。まず、20g/Lのブドウ糖、5g/Lのポリペプトン、5g/Lの酵母抽出物、3g/LのK2HPO4、1g/LのNaCl、1g/Lの(NH4)2SO4、0.2g/LのCaCl2・2H2O、0.2g/LのMgCl2・6H2O及び10g/LのMgCO3からなる前培養培地100mLに炭酸ガスを注入した後、Mannheimia sp.LPKを接種し、39℃で14時間前培養を行った。次いで、20g/Lのブドウ糖、5g/Lのポリペプトン、5g/Lの酵母抽出物、3g/LのK2HPO4、1g/LのNaCl、5g/Lの(NH4)2SO4、0.2g/LのCaCl2・2H2O、0.2g/LのMgCl2・6H2O及び5g/LのNa2CO3からなる本培養培地0.9Lを2.5Lの培養槽に入れ、100mLの前培養された微生物を接種した後、39℃でpH6.5の条件下で、炭酸ガスを0.25vvmの流速で供給しつつ回分培養を行った。
【0046】
培養液内の細胞の濃度は、分光光度計(spectrophotometer,Ultraspec3000,Pharmacia Biotech.,Sweden)を利用して測定し、培地内に含まれたコハク酸、ブドウ糖、乳酸、酢酸及び蟻酸の量は、HPLC(Aminex HPX−87H column,Bio−Rad,USA)で測定した(図5)。図5において、シンボルは、培養時間による細胞の濃度(●)、コハク酸(○)、ブドウ糖(■)、蟻酸(◇)及び酢酸(△)の濃度の変化を表す。図5に示すように、14時間培養後に消耗されたブドウ糖の濃度は20g/Lであり、生成されたコハク酸の濃度は17.2g/Lであった。これによるコハク酸の収率(生成されたコハク酸の量/消費されたブドウ糖の量)は81%であり、コハク酸の体積生産性(生成されたコハク酸の濃度/所要時間)は1.23g/L/hであった。本発明のMannheimia sp.LPKを、CO2で飽和された嫌気的な条件で培養して得るコハク酸の生産方法は、親菌株であるMannheimia succiniciproducens 55Eを、CO2で飽和された嫌気的な条件で培養して得るコハク酸の生産方法より収率面において大きく向上し、コハク酸/酢酸の比率面では40.7:1であり、ほとんど副産物のないコハク酸が得られた。
【0047】
実施例5:pta及びackA欠失ベクター(pPTA−sacB)の作製
pta及びackAを相同組換え方法で破壊するために、遺伝子交換ベクターを次のように作製した。まず、Mannheimia sp.LPK(KCTC 10558BP)のゲノムDNAを鋳型とし、下記配列番号22及び23のプライマーを用いてPCRを行った後、得られたPCR断片をXbaI及びBamHIで切断し、これをpUC19に導入してpUC19−PTA1を作製した。
配列番号22:5’−GCTCTAGATATCCGCAGTATCACTTTCTGCGC
配列番号23:5’−TCCGCAGTCGGATCCGGGTTAACCGCACAG
【0048】
その後、Mannheimia sp.LPKのゲノムDNAを鋳型とし、下記配列番号24及び25のプライマーを用いてPCRを行った後、得られたPCR断片をXbaI及びSacIで切断し、これを前記pUC19−PTA1に導入してpUC19−PTA12を作製した。
配列番号24:5’−GGGGAGCTCGCTAACTTAGCTTCTAAAGGCCATGTTTCC
配列番号25:5’−GCTCTAGATATCCGGGTCAATATCGCCGCAAC
【0049】
スペクチノマイシン耐性遺伝子(GenBank X02588)を含有したプラスミドpIC156(Steinmetz et al.,Gene,142:79,1994)を鋳型とし、下記配列番号26及び27のプライマーを用いてPCRを行った後、得られたPCR断片(スペクチノマイシン耐性遺伝子)をEcoRVで切断し、これを前記pUC19−PTA12に導入してスペクチノマイシン耐性遺伝子を有するpUC19−PTA1S2を作製した。前記作製されたpUC19−PTA1S2をSacI及びBamHIで切断した後、pUC19−SacB(実施例2を参照)に導入してpPTA−sacBベクターを作製した(図6)。
配列番号26:5’−GAATTCGAGCTCGCCCGGGGATCGATCCTC
配列番号27:5’−CCCGGGCCGACAGGCTTTGAAGCATGCAAATGTCAC
【0050】
実施例6:ppc欠失ベクター(pPPC−sacB)の作製
ppcを相同組換え方法で破壊するために、遺伝子交換ベクターを次のように作製した。まず、Mannheimia sp.LPKのゲノムDNAを鋳型とし、下記配列番号28及び29のプライマーを用いてPCRを行った後、得られたPCR断片をXbaI及びBamHIで切断し、これをpUC19に導入してpUC19−PPC1を作製した。
配列番号28:5’−TACGGATCCCCAGAAAATCGCCCCCATGCCGA
配列番号29:5’−GCTCTAGATATCGTTTGATATTGTTCCGCCACATTTG
【0051】
その後、Mannheimia sp.LPKのゲノムDNAを鋳型とし、下記配列番号30及び31のプライマーを用いてPCRを行った後、得られたPCR断片をXbaI及びSacIで切断し、これを前記pUC19−PPC1に導入してpUC19−PPC12を作製した。
配列番号30:5’−GCTCTAGATATCCGTCAGGAAAGCACCCGCCATAGC
配列番号31:5’−GGGGAGCTCGTGTGGCGCTGCGGAAGTAAGGCAAAAATC
【0052】
EcoRVで切断したスペクチノマイシン耐性遺伝子(実施例5を参照)を前記pUC19−PPC12に導入してpUC19−PPC1S2を作製し、これをSacI及びBamHIで切断した後、前記pUC19−SacBに導入してpPPC−sacBベクターを作製した(図7)。
【0053】
実施例7:Mannheimia sp.LPK7及びLPK4菌株の作製
図8及び図9は、Mannheimia sp.LPKでpta−ackA及びppcをそれぞれ欠失させて変異株、LPK7及びLPK4を作製する過程を示す図である。Mannheimia sp.LPKを、10g/lのグルコースを含有したLB−グルコース培地に塗抹して、37℃で36時間培養した後、コロニーをLB−グルコース液体培地10mLに接種して12時間培養した。充分に成長した培養液をLB−グルコース液体培地100mLに1%接種して、37℃の振盪培養器で培養した。
【0054】
前記培養液から、実施例3と同じ方法で細胞濃縮液を得た後、これを、実施例5及び実施例6で作製された遺伝子交換ベクターpPTA−sacB及びpPPC−sacBと混合した後、1.8kV、25μF,200ohmsの条件でエレクトロポレーションを行った。電気衝撃後、LB−グルコース液体培地1mLを加えて、200rpm、37℃の振盪培養器で1時間培養した。
【0055】
培養液を、スペクチノマイシン抗生剤(最終濃度50μg/mL)を含有したLB−グルコース固体培地に塗抹して、37℃で48時間以上培養した。コロニーが形成されれば、二重交差のみ起こったものを選び出すために、これを、スペクチノマイシン50μg/mLを含有したLB−スクロース培地(100g/lのスクロースを含有したLB培地)にストリークした。24時間後に形成されたコロニーをプレートに再びストリークした。前記プレートで形成されたコロニー(変異株)を、抗生剤が含まれたLB−グルコース液体培地で培養し、培養された菌株から、ロッシェルらの方法を応用してゲノムDNAを分離した。前記分離された変異株のゲノムDNAを鋳型としてPCRを行った後、PCR産物を電気泳動してpta−ackA及びppcの欠失の可否をそれぞれ確認した。
【0056】
pta−ackAの欠失の可否を確認するために、下記のようにPCRを2回行った。まず、前記変異株のゲノムDNAを鋳型とし、下記配列番号32及び33のプライマーを用いてPCRを行った。次に、前記変異株のゲノムDNAを鋳型とし、下記配列番号34及び35のプライマーを用いてPCRを行った。
配列番号32:5’−CCTGCAGGCATGCAAGCTTGGGCTGCAGGTCGACTC
配列番号33:5’−GCTGCCAAACAACCGAAAATACCGCAATAAACGGC
配列番号34:5’−GCATGTAACTTTACTGGATATAGCTAGAAAAGGCATCGGGGAG
配列番号35:5’−GCAACGCGAGGGTCAATACCGAAGGATTTCGCCG
【0057】
前記2回のPCR反応で得られた反応物をゲル電気泳動して、そのサイズによってpta−ackAの欠失の可否を確認した(図10)。図10において、Mは、1kbラダー・サイズ・マーカーを、レーン1は、PCR産物P13及びP14(1.1kb)を、レーン2は、PCR産物P15及びP16(1.5kb)を表す。pta−ackAの欠失は、配列番号32及び33のプライマー(P13及びP14)を用いたPCRの結果物が1.1kbであり、それと同時に、配列番号34及び35のプライマー(P15及びP16)を用いたPCRの結果物が1.5kbであるという事実によって確認した。プライマーの位置は図8に示した。前記方法で作製された変異菌株、すなわち、Mannheimia sp.LPKでpta−ackAが欠失された菌株を、Mannheimia sp.LPK7と命名し、これを国際寄託機関であるKCTCに寄託番号‘KCTC 10626BP’として寄託した。
【0058】
また、ppcの欠失の可否を確認するために、下記のように、PCRを2回行った。まず、前記変異株のゲノムDNAを鋳型とし、下記配列番号32及び36のプライマーを用いてPCRを行った。次いで、前記変異株のゲノムDNAを鋳型とし、下記配列番号34及び37のプライマーを用いてPCRを行った。
配列番号36:5’−GATCCAGGGAATGGCACGCAGGCTTTCAACGCCGCC
配列番号37:5’−GCAAAGCCAGAGGAATGGATGCCATTAACCAATAGCG
【0059】
前記2回のPCR反応で得られた反応物をゲル電気泳動して、そのサイズによってppcの欠失の可否を確認した(図11)。図11において、Mは、1kbラダー・サイズ・マーカーを、レーン1は、PCR産物P13及びP17(1.1kb)を、レーン2は、PCR産物P15及びP18(1.5kb)を表す。ppcの欠失は、配列番号32及び36のプライマー(P13及びP17)を用いたPCRの結果物が1.1kbであり、それと同時に、配列番号34及び37のプライマー(P15及びP18)を用いたPCRの結果物が1.5kbであるという事実によって確認した。プライマーの位置は図9に示した。前記方法で作製された変異菌株、すなわち、Mannheimia sp.LPKでppcが欠失された菌株を、Mannheimia sp.LPK4と命名した。
【0060】
実施例8:LPK7及びLPK4の発酵特性
前記実施例7で作製されたMannheimia sp.LPK7及びLPK4の発酵特性を調べるために、CO2で飽和された嫌気的な条件で培養し、これから生産される反応産物を分析した。まず、実施例4に記載の前培養培地200mLに炭酸ガスを注入した後、Mannheimia sp.LPK7及びLPK4をそれぞれ接種し、39℃で24時間前培養を行った。次いで、本培養培地(ブドウ糖濃度が18g/L(最終100mM)であることを除いては、実施例4と同じである)1.8Lを6.6Lの培養槽に入れ、100mLの前培養された微生物を接種した後、39℃でpH6.5の条件下で、炭酸ガスを0.25vvmの流速で供給しつつ回分式培養を行った。
【0061】
細胞濃度と、コハク酸、ブドウ糖、乳酸、酢酸及び蟻酸の濃度とは、実施例4と同じ方法で測定した(図12及び図13)。図12及び図13において、シンボルは、培養時間による細胞の濃度(上側の●)及び、コハク酸(下側の●)、ブドウ糖(□)、蟻酸(◆)及び酢酸(▲)の濃度の変化を表す。図12に示すように、Mannheimia sp.LPK7を22時間培養した後に消耗されたブドウ糖の濃度は100mMであり、生成されたコハク酸の濃度は124mMであった。これによるコハク酸の収率(生成されたコハク酸の量/消耗されたブドウ糖の量)は、モル比で124%であった。そして、酢酸の生成が著しく減った(表1)。また、図13に示すように、Mannheimia sp.LPK4を22時間培養した後に消耗されたブドウ糖の濃度は100mMであり、生成されたコハク酸の濃度は123.7mMであった。これによるコハク酸の収率(生成されたコハク酸の量/消費されたブドウ糖の量)は、モル比で123.7%であった。そして、酢酸の生成が野生型に比べて大きく減った(表1)。
【0062】
本発明のMannheimia sp.LPK7を、CO2で飽和された嫌気的な条件で培養して得るコハク酸の生産方法は、親菌株であるMannheimia succiniciproducens 55Eを、CO2で飽和された嫌気的な条件で培養して得るコハク酸の生産方法よりも、収率面において大きく向上し、コハク酸/酢酸の比率が9.8倍も向上することにより、ほとんど副産物のないコハク酸が得られた(表1)。
【0063】
【表1】
【0064】
Bulterらによると、これまで知られた微生物で酢酸生成遺伝子を全て破壊させても、まだ知られていないアミノ酸及び脂肪酸の物質代謝でも相当量生成されると知られている(Bulter et al.PNAS,101:2299,2004)。したがって、本発明は、これまで知られた酢酸の生成経路を全て遮断したものであって、高効率かつ高濃度のコハク酸の発酵を達成した。
【0065】
以上、本発明の内容の特定な部分を詳細に記述したが、当業界に通常の知識を持つ者にとってはこのような具体的な記述は単に好適な実施例に過ぎないものであり、本発明の範囲はこれによって限定されるものではない。したがって、本発明の実質的な範囲は、特許請求の範囲及びその等価物によって定義されると言えるであろう。
【産業上の利用可能性】
【0066】
以上詳細に説明及び立証したように、本発明に係るMannheimia sp.変異菌株(LPK、LPK7及びLPK4)は、CO2で飽和された嫌気的な条件ではコハク酸を生産し、酸素に対する抵抗性の高い通性嫌気性の菌株であるため、前記菌株を用いてコハク酸を製造する場合、絶対嫌気性の菌株を用いてコハク酸を生産する従来の方法に比べて、酸素露出などによる発酵工程の不安定性を画期的に除去できるだけでなく、他の有機酸の生成を除去することにより、精製工程及び生産収率の最適化及び極大化を達成することができる。
【特許請求の範囲】
【請求項1】
ルーメンバクテリア変異菌株において、乳酸脱水素酵素をコードする遺伝子(以下、「ldhA」という)及びピルビン酸−蟻酸の分解酵素をコードする遺伝子(以下、「pfl」という)が欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株。
【請求項2】
ルーメンバクテリア変異菌株において、ldhA、pfl、ホスホトランスアセチル化酵素遺伝子(以下、「pta」という)及び酢酸キナーゼ遺伝子(以下、「ackA」という)が欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株。
【請求項3】
ルーメンバクテリア変異菌株において、ldhA、pfl及びホスホピルビン酸カルボキシラーゼ遺伝子(以下、「ppc」という)が欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株。
【請求項4】
前記ルーメンバクテリアが、マンヘミア(Mannheimia)属、アクチノバチルス(Actinobacillus)属及びアネロビオスピリルム(Anaerobiospirillum)属からなる群より選択されることを特徴とする請求項1乃至請求項3の何れか一項に記載のルーメンバクテリア変異菌株。
【請求項5】
前記ルーメンバクテリアが他の有機酸はほとんど生成せず、コハク酸のみを生成する同型発酵菌株であることを特徴とする請求項1乃至請求項3の何れか一項に記載のルーメンバクテリア変異菌株。
【請求項6】
前記ルーメンバクテリア変異菌株が、Mannheimia sp.LPKであることを特徴とする請求項1記載の変異菌株。
【請求項7】
前記ルーメンバクテリア変異菌株が、KCTC10558BPであることを特徴とする請求項6記載の変異菌株。
【請求項8】
前記ルーメンバクテリア変異菌株が、Mannheimia sp.LPK7であることを特徴とする請求項2記載の変異菌株。
【請求項9】
前記ルーメンバクテリア変異菌株が、KCTC10626BPであることを特徴とする請求項8記載の変異菌株。
【請求項10】
前記ルーメンバクテリア変異菌株が、Mannheimia sp.LPK4であることを特徴とする請求項3記載の変異菌株。
【請求項11】
マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択されるルーメンバクテリアからldhA及びpflを欠失させることを含む、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法。
【請求項12】
マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択され、ldhA及びpflが欠失されているルーメンバクテリアからpta及びackAを更に欠失させることを含む、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法。
【請求項13】
マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択され、ldhA及びpflが欠失されているルーメンバクテリアからppcを追加で欠失させることを特徴とする、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法。
【請求項14】
前記ldhA及びpflが欠失されているルーメンバクテリアが、Mannheimia sp.LPK(KCTC 10558BP)であることを特徴とする請求項12または請求項13記載の方法。
【請求項15】
前記ldhA及びpflの欠失は、相同組換えにより行われることを特徴とする請求項11記載の方法。
【請求項16】
前記相同組換えは、欠失されたldhAを含む遺伝子交換ベクター及び欠失されたpflを含む遺伝子交換ベクターを使用して行われることを特徴とする請求項15記載の方法。
【請求項17】
前記欠失されたldhAを含む遺伝子交換ベクターが、pMLKO−sacBであり、前記欠失されたpflを含む遺伝子交換ベクターは、pMPKO−sacBであることを特徴とする請求項16記載の方法。
【請求項18】
前記pta及びackAの欠失は、相同組換えにより行われることを特徴とする請求項12記載の方法。
【請求項19】
前記相同組換えは、欠失されたpta及びackAを含む遺伝子交換ベクターを使用して行われることを特徴とする請求項18記載の方法。
【請求項20】
前記欠失されたpta及びackAを含む遺伝子交換ベクターは、pPTA−sacBであることを特徴とする請求項19記載の方法。
【請求項21】
前記ppcの欠失は、相同組換えにより行われることを特徴とする請求項13記載の方法。
【請求項22】
前記相同組換えは、欠失されたppcを含む遺伝子交換ベクターを使用して行われることを特徴とする請求項21記載の方法。
【請求項23】
前記欠失されたppcを含む遺伝子交換ベクターは、pPPC−sacBであることを特徴とする請求項22記載の方法。
【請求項24】
欠失されたldhAを含む遺伝子交換ベクターpMLKO−sacB。
【請求項25】
欠失されたpflを含む遺伝子交換ベクターpMPKO−sacB。
【請求項26】
欠失されたpta及びackAを含む遺伝子交換ベクターpPTA−sacB。
【請求項27】
欠失されたppcを含む遺伝子交換ベクターpPPC−sacB。
【請求項28】
請求項1乃至請求項3の何れか一項のルーメンバクテリア変異菌株を嫌気的な条件で培養する段階;及び前記培養液からコハク酸を回収する段階を含むコハク酸の製造方法。
【請求項29】
前記培養は他の有機酸はほとんど生成せず、コハク酸のみを生成する同型発酵であることを特徴とする請求項28記載のコハク酸の製造方法。
【請求項30】
前記ルーメンバクテリア変異菌株は Mannheimia sp.LPK、LPK7またはLPK4であることを特徴とする請求項28記載のコハク酸の製造方法。
【請求項1】
ルーメンバクテリア変異菌株において、乳酸脱水素酵素をコードする遺伝子(以下、「ldhA」という)及びピルビン酸−蟻酸の分解酵素をコードする遺伝子(以下、「pfl」という)が欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株。
【請求項2】
ルーメンバクテリア変異菌株において、ldhA、pfl、ホスホトランスアセチル化酵素遺伝子(以下、「pta」という)及び酢酸キナーゼ遺伝子(以下、「ackA」という)が欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株。
【請求項3】
ルーメンバクテリア変異菌株において、ldhA、pfl及びホスホピルビン酸カルボキシラーゼ遺伝子(以下、「ppc」という)が欠失されており、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株。
【請求項4】
前記ルーメンバクテリアが、マンヘミア(Mannheimia)属、アクチノバチルス(Actinobacillus)属及びアネロビオスピリルム(Anaerobiospirillum)属からなる群より選択されることを特徴とする請求項1乃至請求項3の何れか一項に記載のルーメンバクテリア変異菌株。
【請求項5】
前記ルーメンバクテリアが他の有機酸はほとんど生成せず、コハク酸のみを生成する同型発酵菌株であることを特徴とする請求項1乃至請求項3の何れか一項に記載のルーメンバクテリア変異菌株。
【請求項6】
前記ルーメンバクテリア変異菌株が、Mannheimia sp.LPKであることを特徴とする請求項1記載の変異菌株。
【請求項7】
前記ルーメンバクテリア変異菌株が、KCTC10558BPであることを特徴とする請求項6記載の変異菌株。
【請求項8】
前記ルーメンバクテリア変異菌株が、Mannheimia sp.LPK7であることを特徴とする請求項2記載の変異菌株。
【請求項9】
前記ルーメンバクテリア変異菌株が、KCTC10626BPであることを特徴とする請求項8記載の変異菌株。
【請求項10】
前記ルーメンバクテリア変異菌株が、Mannheimia sp.LPK4であることを特徴とする請求項3記載の変異菌株。
【請求項11】
マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択されるルーメンバクテリアからldhA及びpflを欠失させることを含む、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法。
【請求項12】
マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択され、ldhA及びpflが欠失されているルーメンバクテリアからpta及びackAを更に欠失させることを含む、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法。
【請求項13】
マンヘミア属、アクチノバチルス属及びアネロビオスピリルム属からなる群より選択され、ldhA及びpflが欠失されているルーメンバクテリアからppcを追加で欠失させることを特徴とする、嫌気的な条件で他の有機酸はほとんど生成せず、コハク酸を高濃度で生成する特性を有するルーメンバクテリア変異菌株の製造方法。
【請求項14】
前記ldhA及びpflが欠失されているルーメンバクテリアが、Mannheimia sp.LPK(KCTC 10558BP)であることを特徴とする請求項12または請求項13記載の方法。
【請求項15】
前記ldhA及びpflの欠失は、相同組換えにより行われることを特徴とする請求項11記載の方法。
【請求項16】
前記相同組換えは、欠失されたldhAを含む遺伝子交換ベクター及び欠失されたpflを含む遺伝子交換ベクターを使用して行われることを特徴とする請求項15記載の方法。
【請求項17】
前記欠失されたldhAを含む遺伝子交換ベクターが、pMLKO−sacBであり、前記欠失されたpflを含む遺伝子交換ベクターは、pMPKO−sacBであることを特徴とする請求項16記載の方法。
【請求項18】
前記pta及びackAの欠失は、相同組換えにより行われることを特徴とする請求項12記載の方法。
【請求項19】
前記相同組換えは、欠失されたpta及びackAを含む遺伝子交換ベクターを使用して行われることを特徴とする請求項18記載の方法。
【請求項20】
前記欠失されたpta及びackAを含む遺伝子交換ベクターは、pPTA−sacBであることを特徴とする請求項19記載の方法。
【請求項21】
前記ppcの欠失は、相同組換えにより行われることを特徴とする請求項13記載の方法。
【請求項22】
前記相同組換えは、欠失されたppcを含む遺伝子交換ベクターを使用して行われることを特徴とする請求項21記載の方法。
【請求項23】
前記欠失されたppcを含む遺伝子交換ベクターは、pPPC−sacBであることを特徴とする請求項22記載の方法。
【請求項24】
欠失されたldhAを含む遺伝子交換ベクターpMLKO−sacB。
【請求項25】
欠失されたpflを含む遺伝子交換ベクターpMPKO−sacB。
【請求項26】
欠失されたpta及びackAを含む遺伝子交換ベクターpPTA−sacB。
【請求項27】
欠失されたppcを含む遺伝子交換ベクターpPPC−sacB。
【請求項28】
請求項1乃至請求項3の何れか一項のルーメンバクテリア変異菌株を嫌気的な条件で培養する段階;及び前記培養液からコハク酸を回収する段階を含むコハク酸の製造方法。
【請求項29】
前記培養は他の有機酸はほとんど生成せず、コハク酸のみを生成する同型発酵であることを特徴とする請求項28記載のコハク酸の製造方法。
【請求項30】
前記ルーメンバクテリア変異菌株は Mannheimia sp.LPK、LPK7またはLPK4であることを特徴とする請求項28記載のコハク酸の製造方法。
【図1】

【図2】
【図3】
【図4】

【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公開番号】特開2010−263911(P2010−263911A)
【公開日】平成22年11月25日(2010.11.25)
【国際特許分類】
【出願番号】特願2010−165726(P2010−165726)
【出願日】平成22年7月23日(2010.7.23)
【分割の表示】特願2006−541014(P2006−541014)の分割
【原出願日】平成16年5月20日(2004.5.20)
【出願人】(502318478)コリア アドバンスド インスティチュート オブ サイエンス アンド テクノロジィ (27)
【Fターム(参考)】
【公開日】平成22年11月25日(2010.11.25)
【国際特許分類】
【出願日】平成22年7月23日(2010.7.23)
【分割の表示】特願2006−541014(P2006−541014)の分割
【原出願日】平成16年5月20日(2004.5.20)
【出願人】(502318478)コリア アドバンスド インスティチュート オブ サイエンス アンド テクノロジィ (27)
【Fターム(参考)】
[ Back to top ]