
新規ロンギシクレン誘導体および当該誘導体を含むチロシナーゼ活性阻害剤
【課題】本発明は、新規なロンギシクレン誘導体、好ましくはチロシナーゼ阻害活性を有する新規なロンギシクレン誘導体、及び当該誘導体を有効成分として含有するチロシナーゼ活性阻害剤を提供することを目的とする。
【解決手段】ロンギシクレンをアスペルギルス属に属する微生物またはその生体内酵素で処理することにより微生物変換して、得られたロンギシクレン誘導体を採取することによって、新規なロンギシクレン誘導体を得ることができる。
【解決手段】ロンギシクレンをアスペルギルス属に属する微生物またはその生体内酵素で処理することにより微生物変換して、得られたロンギシクレン誘導体を採取することによって、新規なロンギシクレン誘導体を得ることができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なロンギシクレン誘導体および当該ロンギシクレン誘導体を有効成分として含有するチロシナーゼ活性阻害剤に関する。
【背景技術】
【0002】
ロンギシクレンは、ロンギフォレン骨格を有する、セスキテルペン類に属するテルペノイド化合物の一種であり、抗真菌、抗寄生虫、及び強壮作用を有する。天然には、ロンギシクレンは日本国内に分布するCryptomeria Japonica D. DON(杉)やChamaecyparis obtusa (Sieb. et Zucc.) Endl.(檜)の根の精油中に含まれることが知られている(B.Shieh, Y.Iizuka and Y.Matsubara (1981), Agric. Biol. Chem., 6, 1493-1495、B.Shieh, YIizuka and Y.Matsubara (1981), Agric. Biol. Chem., 6, 1497-1499)。
【0003】
また、テルペノイド化合物は微生物変換によりテルペノイド誘導体とすることができることも知られている。微生物変換とは、目的の化合物を得るために、生物触媒として生体中の酵素類を使用した生物学的な合成プロセスであり、穏和な条件下で位置特異的に化合物を産生することを特徴とする。ゆえに微生物変換は、生物活性化合物の選択的製造のための有利な方法である。
【0004】
例えば、真菌の一種であるアスペルギルス ニガーは、(+)−フェンコン(M.Miyazawa, K.Yamamoto, Y.Noma and H.Kameoka, (1990), Chemistry Express, 5, 237-430)、1,4−シネオール(M.Miyazawa, Y.Noma, K.Yamamoto and H.Kameoka, (1992), Chemistry Express, 7, 305-308)、(−)−α−ビサボロール(M.Miyazawa, Y.Funatsu and H Kameoka, (1992), Chemistry Express, 7,573-576)、(−)−cis−ローズオキサイド(M.Miyazawa, K.Yokote and H.Kameoka (1995), Phytochemistry, 39, 85-89)、(+)−trans−ローズオキサイド(M.Miyazawa, K.Yokote and H.Kameoka (1995), Phytochemistry, 39, 85-89)等の種々の天然テルペノイド類を立体選択的に酸化し、テルペノイド誘導体に変換することが知られている。
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、新規なロンギシクレン誘導体、特にチロシナーゼ活性阻害作用を有するロンギシクレン誘導体を提供することを目的とする。また、本発明は、当該ロンギシクレン誘導体を有効成分とするチロシナーゼ活性阻害剤を提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明者らは、新規な生理活性物質の創製を目的として、種々の天然物化合物を様々な微生物により微生物変換し、その代謝産物を探索してきたところ、ロンギシクレンを基質としたアスペルギルス属に属する微生物の培養物中に、強いチロシナーゼ活性阻害作用を有する物質が産生されていることを見出した。このロンギシクレンを基質としたアスペルギルス属に属する微生物の培養物中に産生されるチロシナーゼ活性阻害物質を詳細に検討した結果、ロンギシクレン骨格を有する一連の化合物であることを確認すると共に、それらの単離、精製に成功した。
【0007】
したがって、第1の態様において本発明は、式
【化1】

〔一般名:
化合物〔II〕:(−)−(10R)−10−ヒドロキシ−ロンギシクレン酸
化合物〔III〕:(+)−(10S)−10−ヒドロキシ−ロンギシクレン酸
化合物〔IV〕:(+)−10−オキソ−ロンギシクレン酸〕
により表される遊離形または塩形のロンギシクレン誘導体を提供する。
【0008】
また、別の態様において、本発明は有効成分として上記ロンギシクレン誘導体を含有するチロシナーゼ活性阻害剤を提供する。
【0009】
さらに別の態様において、本発明は式
【化2】
のいずれかで表されるロンギシクレン誘導体の製造方法であって、
(1)ロンギシクレンを基質として、当該ロンギシクレン誘導体を生産する能力を有するアスペルギルス属に属する微生物によりロンギシクレンを処理する工程、そして
(2)当該誘導体を採取する工程
を含むことを特徴とする方法を提供する。
【本発明の効果】
【0010】
おどろくべきことに、本発明で得られるロンギシクレン誘導体は、チロシナーゼに対して顕著な活性抑制作用を示す。したがって、本発明のロンギシクレン誘導体は白斑や黒皮症などの色素疾患の治療薬や美白化粧品の原料、食品などの褐変抑制剤、さらには病害虫駆除薬として極めて有用である。
【0011】
チロシナーゼは、動物、植物、微生物界に広く分布する酵素であり、シミやソバカスの原因物質であるメラニン色素の合成を触媒する酵素である。従って、チロシナーゼ阻害剤は、白斑や黒皮症などの色素疾患の治療薬や美白化粧品の原料、食品などの褐変抑制剤、さらには病害虫駆除薬などに極めて有用である。従来、チロシナーゼ阻害活性を有する化合物としては、ハイドロキノン類、アスコルビン酸類、グルタチオン、コウジ酸、アルブチン等が知られている。しかし、これらの化合物には低い経時安定性、低い黒色防止作用、強い細胞毒性、水分存在下の酸化による活性低下等の欠点があったため(例えば、特開昭60−56912号公報、特開2001−316268号公報)、このような不都合を持たないチロシナーゼ活性阻害作用を有する化合物が求められていた。
【発明を実施するための最良の形態】
【0012】
用語の定義
本明細書中において使用される用語は、指示がない限り、当業者によって通常理解される通りの意味で用いられている。
【0013】
ロンギシクレン誘導体の製造方法
本発明の目的物質であるロンギシクレン誘導体を製造するには、ロンギシクレンを基質として、真菌の一種であるアスペルギルス属に属するロンギシクレン誘導体産生菌またはその生体内酵素によりロンギシクレンを処理し、当該ロンギシクレン誘導体を採取すればよい。ここで言う「処理」とは、ロンギシクレンと菌体との接触、ロンギシクレンを菌体の培養培地に含有させて行う培養、発酵等の常套の微生物変換手段を含む意味で用いられており、「採取」とは、常套の分離、抽出及び精製手段を含む工程を意味している。
【0014】
ロンギシクレン誘導体の製造に使用されるアスペルギルス属微生物の一例としては、アスペルギルス ニガー(Aspergillus niger)があり、これは独立行政法人製品評価技術基盤機構にNBRC4414として寄託されている。
【0015】
生体内酵素とは、菌が当該反応工程に利用している酵素として同定されるあらゆる酵素を意味し、適当な条件下、例えば菌体内の条件下におくことにより、菌そのものを利用している場合と同様に反応を進行させることが可能である。
【0016】
ロンギシクレン誘導体生産菌の培養に用いられる培地としては、使用する菌の育成あるいは所望の微生物変換に適した常套の培地を適宜選択することができ、当該菌が利用し得る栄養源を含むものであれば液体状でも固体状でもよいが、大量のロンギシクレン誘導体を得るためには液体培地を用いるのが好ましい。
【0017】
この培地には、当該菌を培養するために必要な物質、例えば当該菌が同化し得る炭素源、消化し得る窒素源および無機物等が適宜配合される。炭素源としては、ブドウ糖、ショ糖、麦芽糖、乳糖、デンプンなどが、窒素源としては、酵母エキス、肉エキス、ペプトン、ポリペプトン、マルトエキストラクト、硝酸ナトリウムなどが用いられる。また培地にはナトリウム、カリウム、カルシウム、マグネシウムなどの塩類が含まれ得る。
【0018】
培地のpH及び温度条件は使用する菌の育成に好適な範囲であればどのような条件でも使用することができ、このような条件は公知であるか、当業者であれば常套の手段により適宜設定することができる。例えば菌がアスペルギルス ニガーである場合、初発pHは約3〜4、好ましくは3.5の条件が、また、培養温度は約15〜約35℃、好ましくは25〜30℃の範囲が適当である。
【0019】
ロンギシクレン誘導体生産菌は静止した物体に付着して増殖する性質があるので静置培養が望ましく、また、当該菌が付着するような物体、例えばアルミ箔を液体培地中に入れておくと増殖が促進される。培養期間は一定しないが、生産されるべきロンギシクレン誘導体の濃度が最大となるまで培養するのが望ましい。これに要する日数は、液体培地を用いる静置培養の場合、通常10日間前後が適当である。
【0020】
得られた培養液は遠心分離または濾過などの手段により、菌体と培養液とに分離する。この両者は共にチロシナーゼ活性阻害作用を示すが、ロンギシクレン誘導体は培養液により多く含まれている。集められた培養液からロンギシクレン誘導体を抽出するには、塩化ナトリウム等の飽和溶液とした後、水と混和する有機溶媒、例えばメタノール、エタノールなどの低級アルコール類、アセトン、メチルエチルケトンなどを使用すればよい。また、水と混和しない有機溶媒、例えばクロロホルム、塩化メチレン、酢酸エチルなどを使用してもよい。
【0021】
このようにして得られた抽出液から減圧下に溶媒を留去すれば、ロンギシクレン誘導体を含む粗抽出物を得ることができる。
【0022】
この粗抽出物からロンギシクレン誘導体を単離、精製するには、通常の脂溶性低分子物質の単離、精製手段を適用することができる。すなわち、セファデックスLH−20(ファルマシア製、登録商標)などを用いるゲル濾過型クロマトグラフィー、シリカゲルなどの吸着剤を用いる吸着クロマトグラフィー、シリカゲルなどの順相系担体を用いる高速液体クロマトグラフィーなどを単独または組み合わせて実施すればよい。
【0023】
セファデックスLH−20を用いる場合は、一般に極性有機溶媒と非極性有機溶媒との組み合わせ、例えばメタノールとクロロホルムまたは塩化メチレンなどの混合溶媒により溶出される。
【0024】
シリカゲルを用いる吸着クロマトグラフィーを使用する場合は、ヘキサンとクロロホルム、酢酸エチルまたは塩化メチレンなどの混合溶媒を溶出溶媒とするのが適している。
【0025】
シリカゲルを担体とする高速液体クロマトグラフィーの場合には、塩化メチレンとベンゼンまたはメタノールの混合溶媒あるいは塩化メチレン単独を溶出溶媒として用いる。このような精製手段を適用することにより、前記に示されるロンギシクレン誘導体の1種またはそれ以上が単離される。
【0026】
チロシナーゼ活性阻害剤
本発明で得られるロンギシクレン誘導体、とりわけ上記化合物〔II〕〜〔IV〕は、チロシナーゼに対して顕著な活性抑制作用を示す。当該活性は適当なin vitroあるいはin vivo試験、例えば実施例2に記載の試験により容易に確認することができる。本発明のロンギシクレン誘導体は、好ましくは化合物0.5mMをチロシナーゼ(シグマ製)溶液(528ユニット/mL)に加え、37℃で60分間インキュベートしたとき、チロシナーゼの活性を10%以上、好ましくは20%以上、とりわけ好ましくは30%以上阻害することができる。
【0027】
したがって、本発明のロンギシクレン誘導体はチロシナーゼ活性阻害剤として使用することができる。好ましくは、本発明のロンギシクレン誘導体は、常套の製剤化手段により、賦形剤、pH調整剤、香料、界面活性剤、キレート剤、増粘剤、防腐剤等の常套の助剤を適宜使用して、液体、乳液、粉末、顆粒、錠剤、ローション、軟膏、注射溶液、クリームなど、任意の剤型に調製して用いることができる。例えば化粧料として本発明のロンギシクレン誘導体を製剤するとき、ローション、乳液、クリーム等の剤型が好ましい。
【0028】
また、本発明のロンギシクレン誘導体を含有するチロシナーゼ活性阻害剤は、動物に投与することを意図するとき、任意の形態で、例えば経口、経腸、非経腸、局所または経皮投与することができ、食品に配合することを意図するとき、常套の混合等の手段により食品中に配合することができる。
【実施例1】
【0029】
実験手法
NMRは500MHz(1H)、125MHz(13C)を用い、TMSを内部標準としCDCl3で測定した。
GCは水素炎イオン化型検出器、キャピラリーカラム(DB−5、島津製作所製:長さ30m×内径0.25mm)、及び20:1のスプリット注入ユニットを備えたHP 5890Aガスクロマトグラフ(ヒューレット・パッカード製)を使用した。移動相はヘリウムガスを1mL/分の流量で用いた。オーブン温度は4℃/分の昇温速度で90℃〜230℃にプログラムされた。注入口温度は270℃、検出器温度は280℃に設定した。ピーク面積はHP 3396 Series2検出器(ヒューレット・パッカード製)により計算した。
【0030】
GC/MSは、スプリット注入ユニット、キャピラリーカラム(HP−5MS、ヒューレット・パッカード製:長さ30m×内径0.25mm)を備えたガスクロマトグラフ(HP 5890A、ヒューレット・パッカード製)を質量分析計(HP 5972A、ヒューレット・パッカード製)に直結した。昇温プログラムはGCと同一である。移動相はヘリウムガスを1mL/分の流量で用いた。イオン源部温度は280℃、電子エネルギーは70電子ボルト(eV)であった。イオン化法は電子衝撃法(EI)を使用した。
【0031】
IRスペクトルはパーキン・エルマー製1760X型分光器により得た。
TLCは、CHCl3を展開溶媒としてシリカゲル60 F254を塗布したTLCプレート(メルク製:層厚0.25mm)を用いた。
シリカゲルカラムクロマトの展開溶媒は、ヘキサン−酢酸エチル系を用いた。
【0032】
アスペルギルス ニガーの前培養
低温で保存されたアスペルギルス ニガー(NBRC4414)の胞子を、滅菌した培養培地(ショ糖1.5%、グルコース1.5%、ポリペプトン0.5%、硫酸マグネシウム(七水和物)0.05%、塩化カリウム0.05%、リン酸二カリウム0.1%、硫酸第一鉄(七水和物)0.001%、及び蒸留水、pH7.2)を入れた振盪フラスコ中に植え付け、27℃で1日間培養した。
【0033】
経時変化
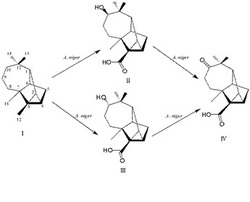
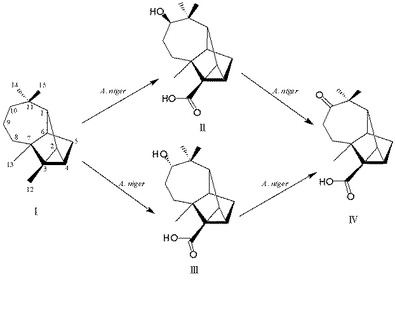
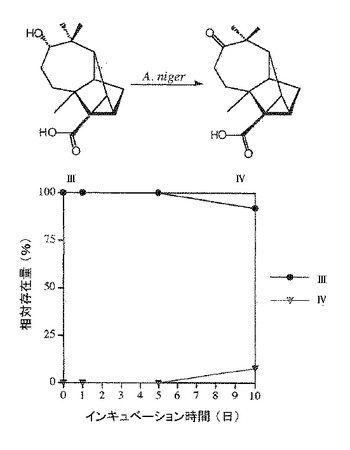
前培養したアスペルギルス ニガーを培養培地(50mLのペトリ皿中20mL)に移植し、1日間(菌糸体が培養培地の表面積の60〜80%を占めるまで)静置条件下で培養した。アスペルギルス ニガーが成長した後、(+)−ロンギシクレン〔I〕60mg(Fluka製)を培地に加えて27℃で10日間培養を続けたところ、化合物〔II〕〜〔IV〕に変換された。その後、塩化ナトリウムで飽和し、酢酸エチルで抽出した。抽出物を薄層クロマトグラフ(TLC)、ガスクロマトグラフィー(GC)及びガスクロマトグラフィー質量分析(GC/MS)で分析した。化合物〔II〕〜〔IV〕は、基質を全く添加しなかったアスペルギルス ニガー培養培地のTLC、GC及びGC−MS分析では検出されなかった。代謝産物の経時変化を図1に示す。〔I〕と化合物〔II〕、〔III〕、〔IV〕間の比率は、GCとGC/MSのピーク面積に基づいて決定した(図1)。10日後に(+)−ロンギシクレンの約80%が代謝された。10日間の変換後、化合物〔II〕は27%、化合物〔III〕は23%、化合物〔IV〕は30%であった。
【0034】
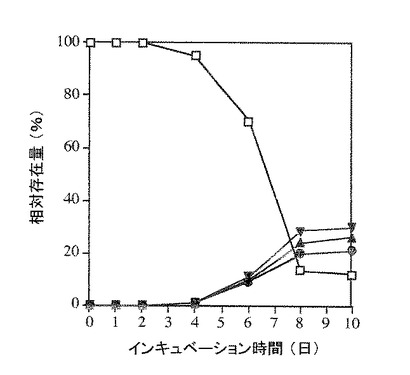
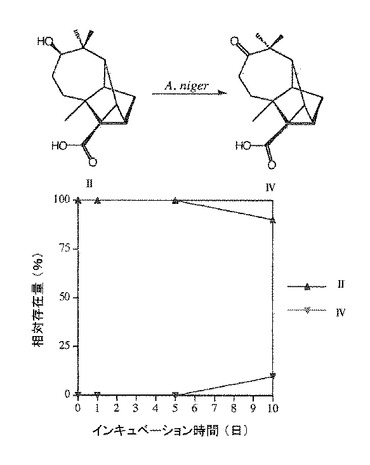
アスペルギルス ニガーによる微生物変換の代謝経路を明確にする目的で、少量の〔II〕、〔III〕及び〔IV〕を10日間アスペルギルス ニガーと共に27℃で培養した。TLC、GC 及びGC/MS法により検出したところ、〔II〕及び〔III〕はアスペルギルス ニガーにより〔IV〕に微生物変換された。そして、〔IV〕は代謝されなかった(図3及び4)。このようにして、アスペルギルス・ニガーによる〔I〕の微生物変換の代謝経路を経時変化(図2)及び代謝産物の構造より導いた(図1)。
図2中、□:化合物[I]、▲:化合物[II]、●:化合物[III]、▼:化合物[IV]である。
【0035】
(+)−ロンギシクレンの10日間の微生物変換
実施例1にて前培養したアスペルギルス ニガーを実施例1と同様の培地1800mLに移植し、27℃で曝気条件下にて1日間攪拌培養した。アスペルギルス ニガーが成長した後、(+)−ロンギシクレン600mgを培地に添加し、10日間培養を続けた。
【0036】
代謝産物の単離
10日間の微生物変換の後、濾過により培地と菌糸体を分離した。培地を塩化ナトリウムで飽和し、酢酸エチルで抽出した。さらに、菌糸体を酢酸エチルで抽出した。酢酸エチル抽出液を混ぜ、硫酸ナトリウムで脱水後、減圧下に溶媒を留去し、粗抽出物752mgを得た。抽出物をヘキサン−酢酸エチル混液と300メッシュのシリカゲルを用いたカラムクロマトグラフィーに供した。未反応の(+)−ロンギシクレン150mgが回収された。化合物〔II〕36mg、〔III〕28mg、及び〔IV〕83mgが単離された。
【0037】
化合物〔II〕
化合物〔II〕、(−)−(10R)−10−ヒドロキシ−ロンギシクレン酸の構造は以下のMS、IR、及びNMRデータから決定した。〔II〕の高分解能高速原子衝撃質量分析(HR−FABMS(POS.))では、m/z値251.1646、分子式C15H23O3であった。〔II〕のIRと13C−NMRスペクトルでは、水酸基(IR:3432cm−1、δC79.8ppm(CH))とカルボキシル基(IR:1683cm−1、δC 178.5ppm(C))の存在が示された。1H−及び13C−NMRシグナルでは、新規メチン基と非プロトン炭素の存在以外は基質と同様であった。また、メチレン基及びメチル基のシグナルは消失した。1H−NMRに関して、3個のメチル基の存在が示された。このことは、二次元NMR(COSY(correlation spectroscopy)、HMQC (heteronuclear multiple quantum correlation)、及びHMBC(heteronuclear multiple bond correlation))の帰属により確認された。特徴的なHMBCスペクトル相関では、2個のメチルプロトン(δH1.01, 1.08ppm;C−14, C−15)及び新規のメチン炭素(δC79.8ppm;C−10)を有するメチンプロトン(δH1.43;C−1)、新規の非プロトン炭素(δC178.5ppm;C−13)を有する2個のメチンプロトン(δH1.78, 2.11ppm;C−4, C−2)が観測された。このことより〔II〕は、〔I〕の10位の炭素がヒドロキシル化、12位の炭素がカルボキシル化されて生成したと考えられる。更に、〔II〕の絶対配置を新モッシャー法(I.Ohtani, T.Kusumi (1991), J. Am. Chem. Soc., 113, 4092-4096)により決定した。すなわち、〔II〕をジクロルメタン及びN,N−ジメチル−4−アミノピリジン(DMAP)中、(+)−(−)−塩化α−メトキシ−α−(トリフルオロメチル)フェニル酢酸(MTPA−Cl)で処理し、それぞれ(+)−MTPAエステル及び(−)−MTPAエステルを得た。Δδ値[δ(−)−δ(+)]は、10位の炭素の絶対配置がR体であることを示していた。比旋光度は(−)−体を示した。これらのデータから、〔II〕の構造は新規化合物である(−)−(10R)−10−ヒドロキシ−ロンギシクレン酸と決定された。
【0038】
化合物〔III〕
第二の代謝産物である化合物〔III〕、(+)−(10S)−10−ヒドロキシ−ロンギシクレン酸は、高分解能電子衝撃質量分析(HR−EIMS)により分子式C15H22O3、m/z値250.1580であった。〔III〕のIRと13C−NMRスペクトルでは、水酸基(IR:3430cm−1、δC76.7ppm(CH))とカルボキシル基(IR:1682cm−1、δC 178.7ppm(C))の存在が示された。1H−及び13C−NMRシグナルでは、新規メチン基と非プロトン炭素の存在以外は基質と同様であった。また、メチレン基及びメチル基のシグナルは消失した。1H−NMRに関して、3個のメチル基の存在が示された。このことは、二次元NMR(COSY、HMQC、及びHMBC)の帰属により確認された。特徴的なHMBCスペクトル相関では、2個のメチルプロトン(δH0.95, 1.08ppm;C−14, C−15)及び新規のメチン炭素(δC79.7ppm;C−10)を有するメチンプロトン(δH1.41−1.44;C−1)、新規の非プロトン炭素(δC178.7ppm;C−13)を有する2個のメチンプロトン(δH2.05, 1.74−1.77ppm;C−4, C−2)が観測された。このことより〔III〕は、〔I〕の10位の炭素がヒドロキシル化、12位の炭素がカルボキシル化されて生成したと考えられる。更に、〔III〕の絶対配置がS体であることをNOE分析により推測した。〔II〕の15位のα−メチル基の共鳴をデカップリングパルスで照射して飽和させたとき、10位の水素のシグナル強度の増強が観察された。しかしながら、14位のβ−メチル基と10位の水素間のNOE分析では、10位の炭素のヒドロキシル基はα位であることを示した。比旋光度は(+)−体を示した。これらのデータから、〔III〕の構造は新規化合物である(+)−(10S)−10−ヒドロキシ−ロンギシクレン酸と決定された。
【0039】
化合物〔IV〕
第三の代謝産物である化合物〔IV〕、(+)−10−オキソ−ロンギシクレンは、HR−EIMSにより分子式C15H20O3、m/z値248.1416であった。〔IV〕のIRと13C−NMRスペクトルでは、カルボニル基(IR:1674cm−1、δC215.9ppm(C))とカルボキシル基(IR:1698cm−1、δC 179.2ppm(C))の存在が示された。1H−及び13C−NMRシグナルでは、新規メチン基と非プロトン炭素の存在以外は基質と同様であった。また、メチレン基及びメチル基のシグナルは消失した。1H−NMRに関して、3個のメチル基の存在が示された。このことは、二次元NMR(COSY、HMQC、及びHMBC)の帰属により確認された。特徴的なHMBCスペクトル相関では、2個のメチルプロトン(δH1.15, 1.19ppm;C−14, C−15)及び新規の非プロトン炭素(δC215.9ppm;C−10)を有するメチンプロトン(δH1.57 −1.61;C−1)、新規の非プロトン炭素(δC179.2ppm;C−13)を有する2個のメチンプロトン(δH1.99, 2.20−2.21ppm;C−4, C−2)が観測された。このことより〔IV〕は、〔I〕の10位の炭素が酸化、12位の炭素がカルボキシル化されて生成したと考えられる。比旋光度は(+)−体を示した。これらのデータから、〔IV〕の構造は新規化合物である(+)−10−オキソ−ロンギシクレン酸と決定された。
【0040】
化合物〔II〕:無色結晶、[α]D21.5 -1.7(CHCl3;c=0.4)、HR-FABMS(POS.):m/z 251.1646 [M+H]+、分子式C15H23O3、EIMS m/z:[M]+ 250(5), 232(15), 204(12), 162(48), 108(59), 91(72), 69(100), 41(86)、IR(KBr法)νmaxcm-1:3407, 2962, 2930, 1677, 1434、1H及び13C−NMRを表1、2に示す。
【0041】
化合物〔III〕:無色結晶、[α]D21.5 +1.6(CHCl3;c=0.4)、HR-EIMS:m/z 250.1580 [M+H]+、分子式C15H22O3、EIMS m/z:[M]+ 250(4), 232(15), 204(14), 143(55), 105(68), 91(100), 69(94), 41(99)、IR(KBr法)νmaxcm-1:3420, 2960, 2923, 1682, 1456、1H及び13C−NMRを表1、2に示す。
【0042】
化合物〔IV〕:無色結晶、[α]D21.5 +22.3(CHCl3;c=0.5)、HR-EIMS:m/z 248.1416 [M+H]+、分子式C15H20O3、EIMS m/z:[M]+ 248(51), 230(34), 202(16), 123(69), 107(77), 93(100), 69(94), 41(90)、IR(KBr法)νmaxcm-1:2965, 2926, 1698, 1674、1H及び13C−NMRを表1、2に示す。
【0043】
【表1】
【表2】
【実施例2】
【0044】
チロシナーゼ阻害活性
L−チロシンおよびL−ドーパを基質としたチロシナーゼ活性を分光光度法により測定した。0.1Mリン酸緩衝液(pH6.8)680μL、20%ポリオキシエチレン−ノニル−フェニルエーテル80μL、0.03%L−チロシンまたはL−ドーパ100mL、および化合物〔II〕(DMSO溶液)40μLを試験管に加え、37℃で10分間インキュベートした。その後、チロシナーゼ(シグマ製)溶液(528units/mL、0.1Mリン酸緩衝液に溶解)を加え、37℃で60分間インキュベートした。酵素活性はチロシナーゼにより合成されるメラニンの吸光度変化(475nm)を測定することにより決定した。比較対照物質にはアルブチンを使用した。
【0045】
化合物〔III〕及び〔IV〕についても同様に試験した。チロシナーゼ活性は以下の式
チロシナーゼ活性(%)=[(A−B)/(Cp−Cn)]×100
〔この式において、Aは試験物質(0.1Mリン酸緩衝液、ポリオキシエチレン−ノニル−フェニルエーテル、L−チロシンまたはL−ドーパ、化合物溶液、およびチロシナーゼ)の吸光度、Bはブランク(0.1Mリン酸緩衝液、ポリオキシエチレン−ノニル−フェニルエーテル、蒸留水、化合物溶液、およびチロシナーゼ)の吸光度、Cpは陽性対照(0.1Mリン酸緩衝液、ポリオキシエチレン−ノニル−フェニルエーテル、L−チロシンまたはL−ドーパ、DMSO、およびチロシナーゼ)の吸光度、Cnは陰性対照(0.1Mリン酸緩衝液、ポリオキシエチレン−ノニル−フェニルエーテル、蒸留水、およびチロシナーゼ)の吸光度を示す。〕
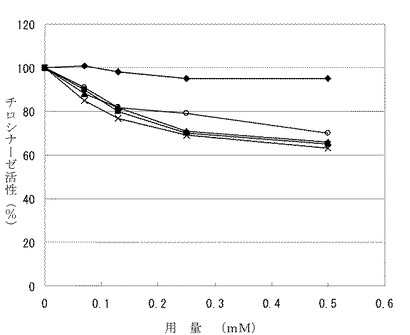
により得られる。結果を表3及び図5に示す。
【0046】
【表3】
図5中、◆:ロンギシクレン、■:化合物[II]、▲:化合物[III]、×:化合物[IV]、◇:アルブチンである。
【0047】
したがって、化合物〔II〕〜〔IV〕はロンギシクレン及びアルブチンよりも顕著にチロシナーゼ阻害活性を有する。
【図面の簡単な説明】
【0048】
【図1】(+)−ロンギシクレン〔I〕をアスペルギルス ニガーによって微生物変換したときの構造の変化を示す。
【図2】(+)−ロンギシクレン〔I〕をアスペルギルス ニガーによって微生物変換したときの、(+)−ロンギシクレン〔I〕および化合物〔II〕〜〔IV〕それぞれの相対存在量についての経時変化を示す。
【図3】化合物〔II〕の、アスペルギルス ニガーによる化合物〔IV〕への微生物変換とその経時変化を示す。
【図4】化合物〔III〕の、アスペルギルス ニガーによる化合物〔IV〕への微生物変換とその経時変化を示す。
【図5】(+)−ロンギシクレン〔I〕および化合物〔II〕〜〔IV〕、並びにアルブチン(対照)のチロシナーゼ阻害活性を示す。
【技術分野】
【0001】
本発明は、新規なロンギシクレン誘導体および当該ロンギシクレン誘導体を有効成分として含有するチロシナーゼ活性阻害剤に関する。
【背景技術】
【0002】
ロンギシクレンは、ロンギフォレン骨格を有する、セスキテルペン類に属するテルペノイド化合物の一種であり、抗真菌、抗寄生虫、及び強壮作用を有する。天然には、ロンギシクレンは日本国内に分布するCryptomeria Japonica D. DON(杉)やChamaecyparis obtusa (Sieb. et Zucc.) Endl.(檜)の根の精油中に含まれることが知られている(B.Shieh, Y.Iizuka and Y.Matsubara (1981), Agric. Biol. Chem., 6, 1493-1495、B.Shieh, YIizuka and Y.Matsubara (1981), Agric. Biol. Chem., 6, 1497-1499)。
【0003】
また、テルペノイド化合物は微生物変換によりテルペノイド誘導体とすることができることも知られている。微生物変換とは、目的の化合物を得るために、生物触媒として生体中の酵素類を使用した生物学的な合成プロセスであり、穏和な条件下で位置特異的に化合物を産生することを特徴とする。ゆえに微生物変換は、生物活性化合物の選択的製造のための有利な方法である。
【0004】
例えば、真菌の一種であるアスペルギルス ニガーは、(+)−フェンコン(M.Miyazawa, K.Yamamoto, Y.Noma and H.Kameoka, (1990), Chemistry Express, 5, 237-430)、1,4−シネオール(M.Miyazawa, Y.Noma, K.Yamamoto and H.Kameoka, (1992), Chemistry Express, 7, 305-308)、(−)−α−ビサボロール(M.Miyazawa, Y.Funatsu and H Kameoka, (1992), Chemistry Express, 7,573-576)、(−)−cis−ローズオキサイド(M.Miyazawa, K.Yokote and H.Kameoka (1995), Phytochemistry, 39, 85-89)、(+)−trans−ローズオキサイド(M.Miyazawa, K.Yokote and H.Kameoka (1995), Phytochemistry, 39, 85-89)等の種々の天然テルペノイド類を立体選択的に酸化し、テルペノイド誘導体に変換することが知られている。
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、新規なロンギシクレン誘導体、特にチロシナーゼ活性阻害作用を有するロンギシクレン誘導体を提供することを目的とする。また、本発明は、当該ロンギシクレン誘導体を有効成分とするチロシナーゼ活性阻害剤を提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明者らは、新規な生理活性物質の創製を目的として、種々の天然物化合物を様々な微生物により微生物変換し、その代謝産物を探索してきたところ、ロンギシクレンを基質としたアスペルギルス属に属する微生物の培養物中に、強いチロシナーゼ活性阻害作用を有する物質が産生されていることを見出した。このロンギシクレンを基質としたアスペルギルス属に属する微生物の培養物中に産生されるチロシナーゼ活性阻害物質を詳細に検討した結果、ロンギシクレン骨格を有する一連の化合物であることを確認すると共に、それらの単離、精製に成功した。
【0007】
したがって、第1の態様において本発明は、式
【化1】
〔一般名:
化合物〔II〕:(−)−(10R)−10−ヒドロキシ−ロンギシクレン酸
化合物〔III〕:(+)−(10S)−10−ヒドロキシ−ロンギシクレン酸
化合物〔IV〕:(+)−10−オキソ−ロンギシクレン酸〕
により表される遊離形または塩形のロンギシクレン誘導体を提供する。
【0008】
また、別の態様において、本発明は有効成分として上記ロンギシクレン誘導体を含有するチロシナーゼ活性阻害剤を提供する。
【0009】
さらに別の態様において、本発明は式
【化2】
のいずれかで表されるロンギシクレン誘導体の製造方法であって、
(1)ロンギシクレンを基質として、当該ロンギシクレン誘導体を生産する能力を有するアスペルギルス属に属する微生物によりロンギシクレンを処理する工程、そして
(2)当該誘導体を採取する工程
を含むことを特徴とする方法を提供する。
【本発明の効果】
【0010】
おどろくべきことに、本発明で得られるロンギシクレン誘導体は、チロシナーゼに対して顕著な活性抑制作用を示す。したがって、本発明のロンギシクレン誘導体は白斑や黒皮症などの色素疾患の治療薬や美白化粧品の原料、食品などの褐変抑制剤、さらには病害虫駆除薬として極めて有用である。
【0011】
チロシナーゼは、動物、植物、微生物界に広く分布する酵素であり、シミやソバカスの原因物質であるメラニン色素の合成を触媒する酵素である。従って、チロシナーゼ阻害剤は、白斑や黒皮症などの色素疾患の治療薬や美白化粧品の原料、食品などの褐変抑制剤、さらには病害虫駆除薬などに極めて有用である。従来、チロシナーゼ阻害活性を有する化合物としては、ハイドロキノン類、アスコルビン酸類、グルタチオン、コウジ酸、アルブチン等が知られている。しかし、これらの化合物には低い経時安定性、低い黒色防止作用、強い細胞毒性、水分存在下の酸化による活性低下等の欠点があったため(例えば、特開昭60−56912号公報、特開2001−316268号公報)、このような不都合を持たないチロシナーゼ活性阻害作用を有する化合物が求められていた。
【発明を実施するための最良の形態】
【0012】
用語の定義
本明細書中において使用される用語は、指示がない限り、当業者によって通常理解される通りの意味で用いられている。
【0013】
ロンギシクレン誘導体の製造方法
本発明の目的物質であるロンギシクレン誘導体を製造するには、ロンギシクレンを基質として、真菌の一種であるアスペルギルス属に属するロンギシクレン誘導体産生菌またはその生体内酵素によりロンギシクレンを処理し、当該ロンギシクレン誘導体を採取すればよい。ここで言う「処理」とは、ロンギシクレンと菌体との接触、ロンギシクレンを菌体の培養培地に含有させて行う培養、発酵等の常套の微生物変換手段を含む意味で用いられており、「採取」とは、常套の分離、抽出及び精製手段を含む工程を意味している。
【0014】
ロンギシクレン誘導体の製造に使用されるアスペルギルス属微生物の一例としては、アスペルギルス ニガー(Aspergillus niger)があり、これは独立行政法人製品評価技術基盤機構にNBRC4414として寄託されている。
【0015】
生体内酵素とは、菌が当該反応工程に利用している酵素として同定されるあらゆる酵素を意味し、適当な条件下、例えば菌体内の条件下におくことにより、菌そのものを利用している場合と同様に反応を進行させることが可能である。
【0016】
ロンギシクレン誘導体生産菌の培養に用いられる培地としては、使用する菌の育成あるいは所望の微生物変換に適した常套の培地を適宜選択することができ、当該菌が利用し得る栄養源を含むものであれば液体状でも固体状でもよいが、大量のロンギシクレン誘導体を得るためには液体培地を用いるのが好ましい。
【0017】
この培地には、当該菌を培養するために必要な物質、例えば当該菌が同化し得る炭素源、消化し得る窒素源および無機物等が適宜配合される。炭素源としては、ブドウ糖、ショ糖、麦芽糖、乳糖、デンプンなどが、窒素源としては、酵母エキス、肉エキス、ペプトン、ポリペプトン、マルトエキストラクト、硝酸ナトリウムなどが用いられる。また培地にはナトリウム、カリウム、カルシウム、マグネシウムなどの塩類が含まれ得る。
【0018】
培地のpH及び温度条件は使用する菌の育成に好適な範囲であればどのような条件でも使用することができ、このような条件は公知であるか、当業者であれば常套の手段により適宜設定することができる。例えば菌がアスペルギルス ニガーである場合、初発pHは約3〜4、好ましくは3.5の条件が、また、培養温度は約15〜約35℃、好ましくは25〜30℃の範囲が適当である。
【0019】
ロンギシクレン誘導体生産菌は静止した物体に付着して増殖する性質があるので静置培養が望ましく、また、当該菌が付着するような物体、例えばアルミ箔を液体培地中に入れておくと増殖が促進される。培養期間は一定しないが、生産されるべきロンギシクレン誘導体の濃度が最大となるまで培養するのが望ましい。これに要する日数は、液体培地を用いる静置培養の場合、通常10日間前後が適当である。
【0020】
得られた培養液は遠心分離または濾過などの手段により、菌体と培養液とに分離する。この両者は共にチロシナーゼ活性阻害作用を示すが、ロンギシクレン誘導体は培養液により多く含まれている。集められた培養液からロンギシクレン誘導体を抽出するには、塩化ナトリウム等の飽和溶液とした後、水と混和する有機溶媒、例えばメタノール、エタノールなどの低級アルコール類、アセトン、メチルエチルケトンなどを使用すればよい。また、水と混和しない有機溶媒、例えばクロロホルム、塩化メチレン、酢酸エチルなどを使用してもよい。
【0021】
このようにして得られた抽出液から減圧下に溶媒を留去すれば、ロンギシクレン誘導体を含む粗抽出物を得ることができる。
【0022】
この粗抽出物からロンギシクレン誘導体を単離、精製するには、通常の脂溶性低分子物質の単離、精製手段を適用することができる。すなわち、セファデックスLH−20(ファルマシア製、登録商標)などを用いるゲル濾過型クロマトグラフィー、シリカゲルなどの吸着剤を用いる吸着クロマトグラフィー、シリカゲルなどの順相系担体を用いる高速液体クロマトグラフィーなどを単独または組み合わせて実施すればよい。
【0023】
セファデックスLH−20を用いる場合は、一般に極性有機溶媒と非極性有機溶媒との組み合わせ、例えばメタノールとクロロホルムまたは塩化メチレンなどの混合溶媒により溶出される。
【0024】
シリカゲルを用いる吸着クロマトグラフィーを使用する場合は、ヘキサンとクロロホルム、酢酸エチルまたは塩化メチレンなどの混合溶媒を溶出溶媒とするのが適している。
【0025】
シリカゲルを担体とする高速液体クロマトグラフィーの場合には、塩化メチレンとベンゼンまたはメタノールの混合溶媒あるいは塩化メチレン単独を溶出溶媒として用いる。このような精製手段を適用することにより、前記に示されるロンギシクレン誘導体の1種またはそれ以上が単離される。
【0026】
チロシナーゼ活性阻害剤
本発明で得られるロンギシクレン誘導体、とりわけ上記化合物〔II〕〜〔IV〕は、チロシナーゼに対して顕著な活性抑制作用を示す。当該活性は適当なin vitroあるいはin vivo試験、例えば実施例2に記載の試験により容易に確認することができる。本発明のロンギシクレン誘導体は、好ましくは化合物0.5mMをチロシナーゼ(シグマ製)溶液(528ユニット/mL)に加え、37℃で60分間インキュベートしたとき、チロシナーゼの活性を10%以上、好ましくは20%以上、とりわけ好ましくは30%以上阻害することができる。
【0027】
したがって、本発明のロンギシクレン誘導体はチロシナーゼ活性阻害剤として使用することができる。好ましくは、本発明のロンギシクレン誘導体は、常套の製剤化手段により、賦形剤、pH調整剤、香料、界面活性剤、キレート剤、増粘剤、防腐剤等の常套の助剤を適宜使用して、液体、乳液、粉末、顆粒、錠剤、ローション、軟膏、注射溶液、クリームなど、任意の剤型に調製して用いることができる。例えば化粧料として本発明のロンギシクレン誘導体を製剤するとき、ローション、乳液、クリーム等の剤型が好ましい。
【0028】
また、本発明のロンギシクレン誘導体を含有するチロシナーゼ活性阻害剤は、動物に投与することを意図するとき、任意の形態で、例えば経口、経腸、非経腸、局所または経皮投与することができ、食品に配合することを意図するとき、常套の混合等の手段により食品中に配合することができる。
【実施例1】
【0029】
実験手法
NMRは500MHz(1H)、125MHz(13C)を用い、TMSを内部標準としCDCl3で測定した。
GCは水素炎イオン化型検出器、キャピラリーカラム(DB−5、島津製作所製:長さ30m×内径0.25mm)、及び20:1のスプリット注入ユニットを備えたHP 5890Aガスクロマトグラフ(ヒューレット・パッカード製)を使用した。移動相はヘリウムガスを1mL/分の流量で用いた。オーブン温度は4℃/分の昇温速度で90℃〜230℃にプログラムされた。注入口温度は270℃、検出器温度は280℃に設定した。ピーク面積はHP 3396 Series2検出器(ヒューレット・パッカード製)により計算した。
【0030】
GC/MSは、スプリット注入ユニット、キャピラリーカラム(HP−5MS、ヒューレット・パッカード製:長さ30m×内径0.25mm)を備えたガスクロマトグラフ(HP 5890A、ヒューレット・パッカード製)を質量分析計(HP 5972A、ヒューレット・パッカード製)に直結した。昇温プログラムはGCと同一である。移動相はヘリウムガスを1mL/分の流量で用いた。イオン源部温度は280℃、電子エネルギーは70電子ボルト(eV)であった。イオン化法は電子衝撃法(EI)を使用した。
【0031】
IRスペクトルはパーキン・エルマー製1760X型分光器により得た。
TLCは、CHCl3を展開溶媒としてシリカゲル60 F254を塗布したTLCプレート(メルク製:層厚0.25mm)を用いた。
シリカゲルカラムクロマトの展開溶媒は、ヘキサン−酢酸エチル系を用いた。
【0032】
アスペルギルス ニガーの前培養
低温で保存されたアスペルギルス ニガー(NBRC4414)の胞子を、滅菌した培養培地(ショ糖1.5%、グルコース1.5%、ポリペプトン0.5%、硫酸マグネシウム(七水和物)0.05%、塩化カリウム0.05%、リン酸二カリウム0.1%、硫酸第一鉄(七水和物)0.001%、及び蒸留水、pH7.2)を入れた振盪フラスコ中に植え付け、27℃で1日間培養した。
【0033】
経時変化
前培養したアスペルギルス ニガーを培養培地(50mLのペトリ皿中20mL)に移植し、1日間(菌糸体が培養培地の表面積の60〜80%を占めるまで)静置条件下で培養した。アスペルギルス ニガーが成長した後、(+)−ロンギシクレン〔I〕60mg(Fluka製)を培地に加えて27℃で10日間培養を続けたところ、化合物〔II〕〜〔IV〕に変換された。その後、塩化ナトリウムで飽和し、酢酸エチルで抽出した。抽出物を薄層クロマトグラフ(TLC)、ガスクロマトグラフィー(GC)及びガスクロマトグラフィー質量分析(GC/MS)で分析した。化合物〔II〕〜〔IV〕は、基質を全く添加しなかったアスペルギルス ニガー培養培地のTLC、GC及びGC−MS分析では検出されなかった。代謝産物の経時変化を図1に示す。〔I〕と化合物〔II〕、〔III〕、〔IV〕間の比率は、GCとGC/MSのピーク面積に基づいて決定した(図1)。10日後に(+)−ロンギシクレンの約80%が代謝された。10日間の変換後、化合物〔II〕は27%、化合物〔III〕は23%、化合物〔IV〕は30%であった。
【0034】
アスペルギルス ニガーによる微生物変換の代謝経路を明確にする目的で、少量の〔II〕、〔III〕及び〔IV〕を10日間アスペルギルス ニガーと共に27℃で培養した。TLC、GC 及びGC/MS法により検出したところ、〔II〕及び〔III〕はアスペルギルス ニガーにより〔IV〕に微生物変換された。そして、〔IV〕は代謝されなかった(図3及び4)。このようにして、アスペルギルス・ニガーによる〔I〕の微生物変換の代謝経路を経時変化(図2)及び代謝産物の構造より導いた(図1)。
図2中、□:化合物[I]、▲:化合物[II]、●:化合物[III]、▼:化合物[IV]である。
【0035】
(+)−ロンギシクレンの10日間の微生物変換
実施例1にて前培養したアスペルギルス ニガーを実施例1と同様の培地1800mLに移植し、27℃で曝気条件下にて1日間攪拌培養した。アスペルギルス ニガーが成長した後、(+)−ロンギシクレン600mgを培地に添加し、10日間培養を続けた。
【0036】
代謝産物の単離
10日間の微生物変換の後、濾過により培地と菌糸体を分離した。培地を塩化ナトリウムで飽和し、酢酸エチルで抽出した。さらに、菌糸体を酢酸エチルで抽出した。酢酸エチル抽出液を混ぜ、硫酸ナトリウムで脱水後、減圧下に溶媒を留去し、粗抽出物752mgを得た。抽出物をヘキサン−酢酸エチル混液と300メッシュのシリカゲルを用いたカラムクロマトグラフィーに供した。未反応の(+)−ロンギシクレン150mgが回収された。化合物〔II〕36mg、〔III〕28mg、及び〔IV〕83mgが単離された。
【0037】
化合物〔II〕
化合物〔II〕、(−)−(10R)−10−ヒドロキシ−ロンギシクレン酸の構造は以下のMS、IR、及びNMRデータから決定した。〔II〕の高分解能高速原子衝撃質量分析(HR−FABMS(POS.))では、m/z値251.1646、分子式C15H23O3であった。〔II〕のIRと13C−NMRスペクトルでは、水酸基(IR:3432cm−1、δC79.8ppm(CH))とカルボキシル基(IR:1683cm−1、δC 178.5ppm(C))の存在が示された。1H−及び13C−NMRシグナルでは、新規メチン基と非プロトン炭素の存在以外は基質と同様であった。また、メチレン基及びメチル基のシグナルは消失した。1H−NMRに関して、3個のメチル基の存在が示された。このことは、二次元NMR(COSY(correlation spectroscopy)、HMQC (heteronuclear multiple quantum correlation)、及びHMBC(heteronuclear multiple bond correlation))の帰属により確認された。特徴的なHMBCスペクトル相関では、2個のメチルプロトン(δH1.01, 1.08ppm;C−14, C−15)及び新規のメチン炭素(δC79.8ppm;C−10)を有するメチンプロトン(δH1.43;C−1)、新規の非プロトン炭素(δC178.5ppm;C−13)を有する2個のメチンプロトン(δH1.78, 2.11ppm;C−4, C−2)が観測された。このことより〔II〕は、〔I〕の10位の炭素がヒドロキシル化、12位の炭素がカルボキシル化されて生成したと考えられる。更に、〔II〕の絶対配置を新モッシャー法(I.Ohtani, T.Kusumi (1991), J. Am. Chem. Soc., 113, 4092-4096)により決定した。すなわち、〔II〕をジクロルメタン及びN,N−ジメチル−4−アミノピリジン(DMAP)中、(+)−(−)−塩化α−メトキシ−α−(トリフルオロメチル)フェニル酢酸(MTPA−Cl)で処理し、それぞれ(+)−MTPAエステル及び(−)−MTPAエステルを得た。Δδ値[δ(−)−δ(+)]は、10位の炭素の絶対配置がR体であることを示していた。比旋光度は(−)−体を示した。これらのデータから、〔II〕の構造は新規化合物である(−)−(10R)−10−ヒドロキシ−ロンギシクレン酸と決定された。
【0038】
化合物〔III〕
第二の代謝産物である化合物〔III〕、(+)−(10S)−10−ヒドロキシ−ロンギシクレン酸は、高分解能電子衝撃質量分析(HR−EIMS)により分子式C15H22O3、m/z値250.1580であった。〔III〕のIRと13C−NMRスペクトルでは、水酸基(IR:3430cm−1、δC76.7ppm(CH))とカルボキシル基(IR:1682cm−1、δC 178.7ppm(C))の存在が示された。1H−及び13C−NMRシグナルでは、新規メチン基と非プロトン炭素の存在以外は基質と同様であった。また、メチレン基及びメチル基のシグナルは消失した。1H−NMRに関して、3個のメチル基の存在が示された。このことは、二次元NMR(COSY、HMQC、及びHMBC)の帰属により確認された。特徴的なHMBCスペクトル相関では、2個のメチルプロトン(δH0.95, 1.08ppm;C−14, C−15)及び新規のメチン炭素(δC79.7ppm;C−10)を有するメチンプロトン(δH1.41−1.44;C−1)、新規の非プロトン炭素(δC178.7ppm;C−13)を有する2個のメチンプロトン(δH2.05, 1.74−1.77ppm;C−4, C−2)が観測された。このことより〔III〕は、〔I〕の10位の炭素がヒドロキシル化、12位の炭素がカルボキシル化されて生成したと考えられる。更に、〔III〕の絶対配置がS体であることをNOE分析により推測した。〔II〕の15位のα−メチル基の共鳴をデカップリングパルスで照射して飽和させたとき、10位の水素のシグナル強度の増強が観察された。しかしながら、14位のβ−メチル基と10位の水素間のNOE分析では、10位の炭素のヒドロキシル基はα位であることを示した。比旋光度は(+)−体を示した。これらのデータから、〔III〕の構造は新規化合物である(+)−(10S)−10−ヒドロキシ−ロンギシクレン酸と決定された。
【0039】
化合物〔IV〕
第三の代謝産物である化合物〔IV〕、(+)−10−オキソ−ロンギシクレンは、HR−EIMSにより分子式C15H20O3、m/z値248.1416であった。〔IV〕のIRと13C−NMRスペクトルでは、カルボニル基(IR:1674cm−1、δC215.9ppm(C))とカルボキシル基(IR:1698cm−1、δC 179.2ppm(C))の存在が示された。1H−及び13C−NMRシグナルでは、新規メチン基と非プロトン炭素の存在以外は基質と同様であった。また、メチレン基及びメチル基のシグナルは消失した。1H−NMRに関して、3個のメチル基の存在が示された。このことは、二次元NMR(COSY、HMQC、及びHMBC)の帰属により確認された。特徴的なHMBCスペクトル相関では、2個のメチルプロトン(δH1.15, 1.19ppm;C−14, C−15)及び新規の非プロトン炭素(δC215.9ppm;C−10)を有するメチンプロトン(δH1.57 −1.61;C−1)、新規の非プロトン炭素(δC179.2ppm;C−13)を有する2個のメチンプロトン(δH1.99, 2.20−2.21ppm;C−4, C−2)が観測された。このことより〔IV〕は、〔I〕の10位の炭素が酸化、12位の炭素がカルボキシル化されて生成したと考えられる。比旋光度は(+)−体を示した。これらのデータから、〔IV〕の構造は新規化合物である(+)−10−オキソ−ロンギシクレン酸と決定された。
【0040】
化合物〔II〕:無色結晶、[α]D21.5 -1.7(CHCl3;c=0.4)、HR-FABMS(POS.):m/z 251.1646 [M+H]+、分子式C15H23O3、EIMS m/z:[M]+ 250(5), 232(15), 204(12), 162(48), 108(59), 91(72), 69(100), 41(86)、IR(KBr法)νmaxcm-1:3407, 2962, 2930, 1677, 1434、1H及び13C−NMRを表1、2に示す。
【0041】
化合物〔III〕:無色結晶、[α]D21.5 +1.6(CHCl3;c=0.4)、HR-EIMS:m/z 250.1580 [M+H]+、分子式C15H22O3、EIMS m/z:[M]+ 250(4), 232(15), 204(14), 143(55), 105(68), 91(100), 69(94), 41(99)、IR(KBr法)νmaxcm-1:3420, 2960, 2923, 1682, 1456、1H及び13C−NMRを表1、2に示す。
【0042】
化合物〔IV〕:無色結晶、[α]D21.5 +22.3(CHCl3;c=0.5)、HR-EIMS:m/z 248.1416 [M+H]+、分子式C15H20O3、EIMS m/z:[M]+ 248(51), 230(34), 202(16), 123(69), 107(77), 93(100), 69(94), 41(90)、IR(KBr法)νmaxcm-1:2965, 2926, 1698, 1674、1H及び13C−NMRを表1、2に示す。
【0043】
【表1】
【表2】
【実施例2】
【0044】
チロシナーゼ阻害活性
L−チロシンおよびL−ドーパを基質としたチロシナーゼ活性を分光光度法により測定した。0.1Mリン酸緩衝液(pH6.8)680μL、20%ポリオキシエチレン−ノニル−フェニルエーテル80μL、0.03%L−チロシンまたはL−ドーパ100mL、および化合物〔II〕(DMSO溶液)40μLを試験管に加え、37℃で10分間インキュベートした。その後、チロシナーゼ(シグマ製)溶液(528units/mL、0.1Mリン酸緩衝液に溶解)を加え、37℃で60分間インキュベートした。酵素活性はチロシナーゼにより合成されるメラニンの吸光度変化(475nm)を測定することにより決定した。比較対照物質にはアルブチンを使用した。
【0045】
化合物〔III〕及び〔IV〕についても同様に試験した。チロシナーゼ活性は以下の式
チロシナーゼ活性(%)=[(A−B)/(Cp−Cn)]×100
〔この式において、Aは試験物質(0.1Mリン酸緩衝液、ポリオキシエチレン−ノニル−フェニルエーテル、L−チロシンまたはL−ドーパ、化合物溶液、およびチロシナーゼ)の吸光度、Bはブランク(0.1Mリン酸緩衝液、ポリオキシエチレン−ノニル−フェニルエーテル、蒸留水、化合物溶液、およびチロシナーゼ)の吸光度、Cpは陽性対照(0.1Mリン酸緩衝液、ポリオキシエチレン−ノニル−フェニルエーテル、L−チロシンまたはL−ドーパ、DMSO、およびチロシナーゼ)の吸光度、Cnは陰性対照(0.1Mリン酸緩衝液、ポリオキシエチレン−ノニル−フェニルエーテル、蒸留水、およびチロシナーゼ)の吸光度を示す。〕
により得られる。結果を表3及び図5に示す。
【0046】
【表3】
図5中、◆:ロンギシクレン、■:化合物[II]、▲:化合物[III]、×:化合物[IV]、◇:アルブチンである。
【0047】
したがって、化合物〔II〕〜〔IV〕はロンギシクレン及びアルブチンよりも顕著にチロシナーゼ阻害活性を有する。
【図面の簡単な説明】
【0048】
【図1】(+)−ロンギシクレン〔I〕をアスペルギルス ニガーによって微生物変換したときの構造の変化を示す。
【図2】(+)−ロンギシクレン〔I〕をアスペルギルス ニガーによって微生物変換したときの、(+)−ロンギシクレン〔I〕および化合物〔II〕〜〔IV〕それぞれの相対存在量についての経時変化を示す。
【図3】化合物〔II〕の、アスペルギルス ニガーによる化合物〔IV〕への微生物変換とその経時変化を示す。
【図4】化合物〔III〕の、アスペルギルス ニガーによる化合物〔IV〕への微生物変換とその経時変化を示す。
【図5】(+)−ロンギシクレン〔I〕および化合物〔II〕〜〔IV〕、並びにアルブチン(対照)のチロシナーゼ阻害活性を示す。
【特許請求の範囲】
【請求項1】
式
【化1】
である、遊離形または塩形のロンギシクレン誘導体。
【請求項2】
式
【化2】
である、遊離形または塩形のロンギシクレン誘導体。
【請求項3】
式
【化3】
である、遊離形または塩形のロンギシクレン誘導体。
【請求項4】
チロシナーゼ活性阻害作用を有する、請求項1〜3のいずれか1項に記載の誘導体。
【請求項5】
請求項1〜3のいずれか1項に記載のロンギシクレン誘導体を有効成分として含有するチロシナーゼ活性阻害剤。
【請求項6】
【化4】
のいずれかで表されるロンギシクレン誘導体の製造方法であって、
(1)ロンギシクレンを基質として、当該ロンギシクレン誘導体を生産する能力を有するアスペルギルス属に属する微生物によりロンギシクレンを処理する工程、そして
(2)当該誘導体を採取する工程
を含むことを特徴とする方法。
【請求項1】
式
【化1】
である、遊離形または塩形のロンギシクレン誘導体。
【請求項2】
式
【化2】
である、遊離形または塩形のロンギシクレン誘導体。
【請求項3】
式
【化3】
である、遊離形または塩形のロンギシクレン誘導体。
【請求項4】
チロシナーゼ活性阻害作用を有する、請求項1〜3のいずれか1項に記載の誘導体。
【請求項5】
請求項1〜3のいずれか1項に記載のロンギシクレン誘導体を有効成分として含有するチロシナーゼ活性阻害剤。
【請求項6】
【化4】
のいずれかで表されるロンギシクレン誘導体の製造方法であって、
(1)ロンギシクレンを基質として、当該ロンギシクレン誘導体を生産する能力を有するアスペルギルス属に属する微生物によりロンギシクレンを処理する工程、そして
(2)当該誘導体を採取する工程
を含むことを特徴とする方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2008−179546(P2008−179546A)
【公開日】平成20年8月7日(2008.8.7)
【国際特許分類】
【出願番号】特願2007−12602(P2007−12602)
【出願日】平成19年1月23日(2007.1.23)
【出願人】(000238201)扶桑薬品工業株式会社 (42)
【出願人】(000125347)学校法人近畿大学 (389)
【Fターム(参考)】
【公開日】平成20年8月7日(2008.8.7)
【国際特許分類】
【出願日】平成19年1月23日(2007.1.23)
【出願人】(000238201)扶桑薬品工業株式会社 (42)
【出願人】(000125347)学校法人近畿大学 (389)
【Fターム(参考)】
[ Back to top ]