
新規分解型リモノイド化合物
【課題】(+)-イソフラキシネロン又は(-)-フラキシネロン誘導体を生物変換して得られる化合物、該化合物を有効成分として含有する害虫摂食阻害剤を提供する。
【解決手段】一般式(A)又は(C):

[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]で表される化合物、並びに該化合物を有効成分として含む害虫摂食阻害剤に関する。
【解決手段】一般式(A)又は(C):
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]で表される化合物、並びに該化合物を有効成分として含む害虫摂食阻害剤に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規分解型リモノイド化合物、該化合物の製造方法、該化合物を有効成分として含有する害虫摂食阻害剤、該化合物を含有する組成物及び該組成物を害虫に適用する害虫防除方法に関する。
【背景技術】
【0002】
ミカン科の植物に含まれるリモノイドには、種々の分解型リモノイドが知られている。これらは、リモノイドと共通する部分構造を有しており、リモノイドよりも小さく単純な化合物である。このような分解型リモノイドに属し、Rutaceae(ミカン科)から得られる(-)-フラキシネロン((-)-fraxinellone))は、様々な生物活性を有することが報告されている(非特許文献1〜4)。
【0003】
【化1】
【0004】
本発明者らは、(+)-イソフラキシネロン((+)-isofraxinellone)の抗変異原活性(antimutagenic activites)について、既に報告している(非特許文献5)。また、Liuらは、(-)-フラキシネロンが、コクゾウムシ(Sitophilus zeamais)の成虫と同様、コクヌストモドキ(Tribolium castaneum)の成虫及び幼虫に対しても摂食阻害活性を有することを報告している(非特許文献6)。さらにMaiqueらは、下記一般式3
【0005】
【化2】
【0006】
で表される化合物(以下、化合物(3)ということがある)が、Trypaosoma cruziのトリポマスティゴート型原虫に対して、弱い摂食阻害活性を有することを報告している(非特許文献7)。
【0007】
生物変換は、生体中に存在する酵素を生体触媒として使用する簡易な手法であり、不活性な天然物から生理活性を有する医薬又は農薬化合物を製造するために用いられる。該手法の製造条件は温和であり、多くの場合、官能基の保護を必要としない。さらに、生物変換の特徴は、位置及び立体選択的反応であり、光学活性な生成物を与えることである。生物変換によって、天然物から生理活性物質を得る方法として、本発明者らは、フラボノイド(非特許文献8及び9)、テルペノイド(非特許文献10及び11)及びリグナン(非特許文献10及び11)の生物変換について報告している。
【0008】
現在、過剰な農薬使用が問題視されているが、通常、無農薬で食害を防ぐことは困難であり、環境負荷の少ない害虫防除法が求められている。害虫に対して摂食阻害活性を有する化合物は、低分子でありながら極めて低濃度で生物活性を示すため、安全かつ選択的な害虫防除剤としての利用が期待され、実用化に向けた研究が活発になされている。害虫摂食阻害剤の摂食阻害活性が強ければ害虫は餓死するので、その物質の使用濃度において人体に無害であれば、害虫摂食阻害剤は農薬として使用される可能性もある。
【非特許文献1】Okamura, H.; Yamauchi, K.; Miyawaki, K.; Iwagawa, T.; Nakatani, M. Tetrahedron Letters. 1997, 38, 263-266.
【非特許文献2】Nakatani, M.; Huang, RC.; Okamura, H.; Iwagawa, T.; Tadera, K. Phytochemistry. 1998, 49, 1773-1776.
【非特許文献3】Yu, S. M.; Ko, F. N.; Su, M. J.; Wu, T. S.; Wang, M. L.; Huang, T. F.; Teng, C. M. Naunyn-Schmiedeberg’s Arch Pharmacol. 1992, 345, 349-355.
【非特許文献4】Woo, W. S.; Lee, E. B.; Kang, S. S.; Shin, K. H.; Chi, H. J. Planta Med. 1987, 53, 399-401.
【非特許文献5】Miyazawa, M.; Shimamura, H.; Nakamura, S.; Kameoka, H. J. Agric, Food Chem. 1995, 4, 1428-1431.
【非特許文献6】Liu, Z. L.; Xu, Y. J.; Wu, J.; Goh, S. H.; Ho, S. H. J. Agric, Food Chem. 2002, 50, 1447-1450.
【非特許文献7】Maique, W. B.; Paulo, C. V. M.; Fatima, G. F. D. S.; Joao, B. F.; Sergio, A. Z. Naturforsch. 2001, 56c, 570-574.
【非特許文献8】Miyazawa, M.; Takahashi, K.; Araki, H. J Chem Technol Biotechnol. 2006, 81, 674-678.
【非特許文献9】Okuno, Y.; Miyazawa, M. J Chem Technol Biotechnol. 2006, 81, 29-33.
【非特許文献10】Miyazawa, M.; Miyamoto, Y. J Mol Catal B. 2004, 27, 83-89.
【非特許文献11】Miyazawa, M.; Nankai, H.; Kameoka, H. Phytochemistry. 1996, 43, 105-109.
【非特許文献12】Miyazawa, M.; Kasahara, H.; Kameoka, H. Phytochemistry. 1994, 35, 1191-1193.
【非特許文献13】Miyazawa, M.; Kasahara, H.; Kameoka, H. Phytochemistry. 1993, 34, 1501-1507.
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明は、新規分解型リモノイド化合物、該化合物の製造方法、該化合物を有効成分として含有する害虫摂食阻害剤、該化合物を含有する組成物及び該組成物を害虫に適用する害虫防除方法を提供することを目的とする。
【課題を解決するための手段】
【0010】
本発明者は、(+)-イソフラキシネロン((+)-isofraxinellone)又は(-)-フラキシネロン((-)-fraxinellone)誘導体をアスペルギルス・ニガー(Aspergillus niger)により生物変換したところ、(+)-イソフラキシネロン又は(-)-フラキシネロン誘導体がヒドロキシル化された新規化合物を得られることを見出した。さらに、これらの新規化合物は、害虫摂食抑制剤を有することを見出した。本発明は、更に検討を加えて完成されたものである。
【0011】
即ち、本発明は以下の化合物、及び該化合物を含む害虫摂食抑制剤を提供する。
項1. 一般式(A):
【0012】
【化3】
【0013】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する]
で表される化合物。
項2. 一般式(A)において、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合する項1に記載の化合物。
項3. 一般式(A−1):
【0014】
【化4】
【0015】
[式中、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合する]
で表される化合物。
項4. 項1〜3のいずれかに記載の化合物を有効成分として含む害虫摂食抑制剤。
項5. 害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である項4に記載の害虫摂食抑制剤。
項6. 一般式(A):
【0016】
【化5】
【0017】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する]
で表される化合物の製造方法であって、下記一般式(B)
【0018】
【化6】
【0019】
で表される化合物を生物変換することを特徴とする製造方法。
項7. 一般式(C):
【0020】
【化7】
【0021】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物。
項8. 一般式(C)において、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合する請求項7に記載の化合物。
項9. 一般式(C−1):
【0022】
【化8】
【0023】
[式中、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物。
項10. 項7〜9のいずれかに記載の化合物を有効成分として含む害虫摂食抑制剤。
項11. 害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である項8に記載の害虫摂食抑制剤。
項12. 一般式(C):
【0024】
【化9】
【0025】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物の製造方法であって、下記一般式(D)
【0026】
【化10】
【0027】
[式中、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物を生物変換することを特徴とする製造方法。
項13. 項1〜3及び項7〜9のいずれかに記載の化合物を含有する組成物。
項14. 項13に記載の組成物を害虫に適用することを特徴とする害虫防除方法。
項15. 一般式(B)
【0028】
【化11】
【0029】
及び一般式(D)
【0030】
【化12】
【0031】
[式中、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物からなる群から選ばれた少なくとも1種を有効成分として含む害虫摂食抑制剤であって、害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である害虫摂食抑制剤。
【発明の効果】
【0032】
本発明の一般式(A)及び(C)で表される化合物は新規であり、高い害虫摂食抑制活性を有しているため、害虫摂食抑制剤として有用である。また、本発明の一般式(B)及び(D)で表される化合物も高い害虫摂食抑制活性を有しているため、害虫摂食抑制剤として有用である。特に、本発明の化合物は、ヨトウムシに対して優れた害虫摂食抑制活性を有する。
【発明を実施するための最良の形態】
【0033】
本発明の化合物
本発明の第一の化合物は、下記一般式(A)で表される化合物(以下、化合物(A)ということがある)である。
【0034】
【化13】
【0035】
(式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する)
一般式(A)において、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合することが好ましく、4位又は6位のいずれか1つの炭素のみに1つ結合することがさらに好ましく、6位の炭素に1つ結合することが特に好ましい。
【0036】
化合物(A)の中でも、下記一般式(A−1)で表される化合物(以下、化合物(A−1)ということがある)が好ましい。
【0037】
【化14】
【0038】
(式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する。)
一般式(A−1)において、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合することが好ましく、4位又は6位のいずれか1つの炭素のみに1つ結合することがさらに好ましく、6位の炭素に1つ結合することが特に好ましい。
【0039】
また、化合物(A)の中でも、下記一般式(3-1)((+)-(6R)-6-ヒドロキシ-7,7a-エポキシ-フラキシネロン((+)-(6R)-6-hydroxy-7,7a-epoxy-fraxinellone))の絶対配置で表される化合物(以下、化合物(3-1)ということがある)が特に好ましい。
【0040】
【化15】
【0041】
本発明の第二の化合物は、下記一般式(C)で表される化合物(以下、化合物(C)ということがある)である。
【0042】
【化16】
【0043】
(式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。)
一般式(C)において、6位と7位との炭素間結合が一重結合である場合、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合することが好ましく、特に4位又は6位のいずれか1つの炭素のみに1つ結合することが好ましい。
【0044】
一般式(C)において、6位と7位との炭素間結合が二重結合である場合、OH基は4位又は5位のいずれか1つの炭素のみに1つ結合することが好ましく、特に4位の炭素に1つ結合することが好ましい。
【0045】
化合物(C)の中でも、下記一般式(C−1)で表される化合物(以下、化合物(C−1)ということがある)が好ましい。
【0046】
【化17】
【0047】
(式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。)
一般式(C−1)において、6位と7位との炭素間結合が一重結合である場合、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合することが好ましく、特に4位又は6位のいずれか1つの炭素のみに1つ結合することが好ましい。
【0048】
一般式(C−1)において、6位と7位との炭素間結合が二重結合である場合、OH基は4位又は5位のいずれか1つの炭素のみに1つ結合することが好ましく、特に4位の炭素に1つ結合することが好ましい。
【0049】
一般式(C−1)において、6位と7位との炭素間結合が二重結合である化合物の中でも、下記一般式(1-1)((+)-(4S)-4-ヒドロキシイソフラキシネロン)((+)-(4S)-4-hydroxyisofraxinellone))の絶対配置で表される化合物(以下、化合物(1-1)ということがある)が特に好ましい。
【0050】
【化18】
【0051】
一般式(C−1)において、6位と7位との炭素間結合が一重結合である化合物の中でも下記一般式(2-1)、(2-2)及び(2-3)の絶対配置で表される化合物(以下、それぞれ化合物(2-1)、(2-2)及び(2-3)ということがある)が好ましい。
【0052】
【化19】
【0053】
本発明の製造方法
本発明の一般式(A)で表される化合物は、下式に示すスキームにより得られる。
【0054】
【化20】
【0055】
(式(A)中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する。)
一般式(E)で表される化合物(以下、化合物(E)ということがある)は、非特許文献1〜4に記載の方法に準じた方法により得られる。また、化合物(E)の中でも、(-)-フラキシネロンは、非特許文献1〜4に記載の通り、和漢生薬白鮮皮より得られる。
【0056】
一般式(B)で表される化合物(以下、化合物(B)ということがある)は、例えば、従来公知の方法によって、化合物(E)をエポキシ化反応することにより得られる。化合物(B)の中でも、下記一般式(3)(化合物(3))で表される化合物は、例えば、(-)-フラキシネロンをエポキシ化反応することにより得られる(実施例2)。
【0057】
【化21】
【0058】
当該エポキシ化反応に使用されるエポキシ化反応の条件は、特に限定されず、従来公知の方法を採用すればよい。例えば、アルカリ条件下で、過酸化水素水、t-ブチルヒドロペルオキシド等を使用して分子内の二重結合をエポキシ化する。エポキシ化反応に使用される溶媒としては、例えば、メタノール、エタノール、テトラヒドロフラン、ジメチルホルムアミド等が挙げられる。反応温度は、通常、0℃〜室温程度とすればよく、反応時間は、通常、10分〜24時間程度である。必要に応じて、アルミナカラムクロマトグラフィー、シリカゲルクロマトグラフィー、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィー、疎水クロマトグラフィー、高速液体クロマトグラフィー等の適当な分離精製手段を1種若しくは2種以上組み合わせて精製することができる。
【0059】
本発明の化合物(A)は、化合物(B)を生物変換によりヒドロキシル化して得られる。例えば、本発明の化合物(3-1)は、化合物(3)を生物変換によりヒドロキシル化して得られる。
【0060】
また、 本発明の一般式(C)で表される化合物(以下、化合物(C)ということがある)は、下式に示すように、化合物(D)で表される化合物を生物変換によりヒドロキシル化して得られる。
【0061】
【化22】
【0062】
(式(C)中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。)
一般式(D)で表される化合物(以下、化合物(D)ということがある)において、6位と7位との炭素間結合が二重結合である化合物は、非特許文献5に記載の方法に準じて得られる。化合物(D)の中でも、化合物(1)((+)-イソフラキシネロン)は、ミカン科(Rutaceae)のハクセン(Dictamnus dasycarpus)の根皮を乾燥した和漢生薬白鮮皮より得られる分解型リモノイドであり、非特許文献5に記載の方法により抽出できる。
【0063】
一般式(D)で表される化合物において、6位と7位との炭素間結合が一重結合である化合物は、例えば、化合物(D)において6位と7位との炭素結合が二重結合である化合物を水素付加反応することによって得られる。また、上記一般式(E)で表される化合物の7位と7a位の二重結合を水素付加反応することによっても得られる。例えば、化合物(2)は、(-)-フラキシネロンを水素付加反応することにより得られる(実施例1)。
【0064】
水素付加反応は、従来公知の方法により行えばよい。水素付加反応に使用される試薬は、従来公知の試薬を使用すればよいが、例えば、水素化ホウ素ナトリウム、水素化ホウ素リチウム等を使用すればよい。必要に応じて、アルミナカラムクロマトグラフィー、シリカゲルクロマトグラフィー、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィー、疎水クロマトグラフィー、高速液体クロマトグラフィー等の適当な分離精製手段を1種若しくは2種以上組み合わせて精製することができる。
【0065】
本発明の化合物(C)の中でも、化合物(1-1)は、(+)-イソフラキシネロン(化合物(1))を生物変換することによりヒドロキシル化して得られる。
【0066】
また、本発明の化合物(C)の中でも、化合物(2-1)((-)-(4S)-4-ヒドロキシ-7,7a-ジヒドロフラキシネロン)、(2-2)((-)-(6R)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロン)及び(2-3)((-)-(6S)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロン)は、下記一般式(2)で表される化合物((-)-7,7a-ジヒドロフラキシネロン)(以下、化合物(2)ということがある)を生物変換によりヒドロキシル化して得られる。
【0067】
【化23】
【0068】
本発明の生物変換においては、黒麹菌であるアスペルギルス・ニガー(Aspergillus niger)を用いる。
【0069】
具体的には、培養されたアスペルギルス・ニガーを培地に移植し、基質(反応出発物質)を培地に加え、通常1〜11日間程度、好ましくは1〜15日間程度放置し、アスペルギルス・ニガー及び培地を抽出することにより、一般式(A)及び(C)で表される化合物(代謝物)が得られる。温度条件は、通常0〜25℃程度とすればよい。使用する培地は、特に限定されないが、例えば、万能培地(Czapek pepton培地)、寒天培地、LB培地等が挙げられ、これらの中でも、万能培地が好ましい。培養は、酸素の存在下で行うのが好ましい。必要に応じて、アルミナカラムクロマトグラフィー、シリカゲルクロマトグラフィー、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィー、疎水クロマトグラフィー、高速液体クロマトグラフィー等の適当な分離精製手段を1種若しくは2種以上組み合わせて精製することができる。具体的には実施例を参照。
【0070】
一般式(A)、(B)、(C)及び(D)で表される本発明の化合物は、害虫摂食阻害活性を有している。
【0071】
本発明の化合物が示す害虫摂食阻害活性とは、害虫の餌となる植物等に付着していると、害虫が餌を食べられなくなる活性をいう。
【0072】
本発明の化合物が有効に摂食阻害できる対象害虫としては、例えば、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類等に属する昆虫が挙げられる。本発明による摂食阻害は、特に鱗翅目に属する昆虫に対して有効であり、例えば、テンスジツトガ、ツトガ、シバツトガ、ロブノメイガ、ワモンノメイガ、アワノメイガ、アカフツヅリガ等のメイガ科; スゲドクガ等のドクガ科; オビヒトリ、キハラゴマダラヒトリ、シロヒトリ等のヒトリガ科; イネキンウワバ、ヨトウムシ(例えば、エゾチャイロヨトウ、シロシタヨトウ、フタオビキヨトウ、タンポキヨトウ、クサシロキヨトウ、イネヨトウ、スジギリヨトウ、ハスモンヨトウ、シロイチモジヨトウ、ヨトウガ、アワヨトウ等)等のヤガ科に属する昆虫に対して有効である。これらの中でもヨトウムシに対して特に有効である。対象害虫は、成虫及び幼虫のいずれでも良いが、特に幼虫が好ましい。
【0073】
本発明の化合物が有効に摂食阻害できる対象植物は、特に限定されないが、例えば、キャベツ、サツマイモ、サトイモ、ネギ、トマト、ピーマン、ナス、レタス、ハクサイ等が挙げられる。
【0074】
本発明の化合物(A)、(B)、(C)及び(D)は優れた害虫阻害活性を有している。これらの中でも、化合物(1)、(2)、(3)、(1-1)、(2-1)、(2-2)、(2-3)及び(3-1)は、特に優れた害虫阻害剤活性を有している。
【0075】
本発明の害虫摂食阻害剤
本発明の害虫摂食阻害剤は、本発明の化合物(A)、(B)、(C)及び(D)からなる群から選ばれた少なくとも1種を有効成分として含む。
【0076】
本発明の化合物は、直接植物の葉、実、茎等に散布、塗布等することにより、そのままでも害虫摂食阻害剤として作用するが、通常は、ローション、エアゾ−ル等の液剤や、クリーム剤等の各種形態に製剤化した組成物として使用される。液剤としては、ローション、エアゾール、油剤等を挙げることができ、これらの液剤に用いられる担体としては、例えば、水、メタノール、エタノール、イソプロピルアルコール、セチルアルコール等のアルコール類、石油ベンジン等の脂肪族炭化水素類、ミリスチン酸イソプロピル、酢酸セチル等のエステル類があげられる。液剤の種類により、適宜さらに乳化剤、分散剤、展着剤、湿潤剤、懸濁化剤、保存剤、噴射剤等の製剤用補助剤、塗膜形成剤などを加え、所望の製剤とすることができる。
【0077】
本発明の化合物は、通常用いられる製剤化方法により、害虫摂食阻害剤に製剤化することができる。すなわち、有効成分以外に必要な補助剤を含むことができる。製剤の形態としては、乳剤、水和剤、粉剤、エアロゾルなどを挙げることができる。好ましくは、水和剤である。本発明の害虫摂食阻害剤は、一般に散布によって害虫に適用する。
【0078】
乳化剤及び分散剤としては、例えば、石鹸類、ポリオキシエチレンオレイルエーテル等のポリオキシエチレン脂肪酸アルコールエーテル、ポリオキシエチレンノニルフェニルエーテル等のポリオキシエチレンアルキルアリールエーテル、ポリオキシエチレン脂肪酸エステル、脂肪酸グリセリド、ポリオキシエチレンソルビタンモノステアレート等のソルビタン脂肪酸エステル、高級アルコールの硫酸エステル、ドデシルベンゼンスルホン酸ソーダ等のアルキルアリールスルホン酸塩が挙げられ、展着剤または湿潤剤としては、例えば、グリセリン、プロピレングリコール、ポリエチレングリコールが挙げられる。
【0079】
また、懸濁化剤としては、例えば、カゼイン、ゼラチン、アルギン酸、カルボキシメチルセルロース、アラビアガム、ヒドロキシプロピルセルロース、ベントナイトが挙げられ、保存剤としては、例えば、サリチル酸、パラオキシ安息香酸エチル、パラオキシ安息香酸プロピル、パラオキシ安息香酸ブチルが挙げられる。
【0080】
噴射剤としては、例えば、ジメチルエーテル、クロロフルオロカーボン、炭酸ガス、LPGが挙げられ、塗膜形成剤としては、例えば、ニトロセルロース、アセチルセルロース、アセチルブチ ルセルロース、メチルセルロース等のセルロース誘導体、酢酸ビニル樹脂等のビニル系樹脂、ポリビニルアルコール、メチルポリシロキサン、オクチルメチルシクロテトラシロキサン、デカメチルシクロペンタシロキサン、ジメチルポリシロキサン、メチルフェニルポリシロキサンメチルポリシクロポリシロキサン、ジメチルシロキサン・メチル(ポリオキシエチレン)シロキサン共重合体、ジメチルシロキサン・メチル(ポリオキシエチレン・ポリオキシプロピレン)シロキサン共重合体、トリメチルシロキシケイ酸、オクタメチルシクロテトラシロキサンシリコーンポリエーテルポリマー等のシリコーン類が挙げられる。
【0081】
クリーム剤において用いられる担体としては、例えば、パラフィン、流動パラフィン、ワセリン等の炭化水素類、ジメチルシロキサン、コロイド状シリカ、ベントナイト等のケイ素化合物、エタノール、ステアリルアルコール、ラウリルアルコール、エチレングリコール、ポリエチレングリコール、グリセリン等のアルコール類、ラウリン酸、ステアリン酸等のカルボン酸類、蜜蝋、ラノリン等のエステル類等が挙げられる。さらに、液剤の製剤の際に用いられるのと同様の製剤用補助剤を適宜加えることにより目的の製剤とすることができる。
【0082】
これら製剤中の本発明の化合物の含有量は、製剤形態や適用方法等により異なるが、例えば、液剤として使用する場合、通常0.0001〜0.05重量%、より好ましくは0.0002〜0.002重量%の割合で含まれる。また、その処理量は、通常植物の葉表面積1cm2 当り本発明の化合物の量で0.5〜250 μg、好ましくは1〜100 μg相当量である。勿論、該処理量は、製剤形態や適用方法、対象とする害虫の種類や密度等により異なり、適宜上記の範囲にかかわることなく増加または減少させることもできる。
【0083】
本発明の害虫防除方法
本発明の害虫防除方法は、植物体内又は植物体の表面もしくは周辺に存在する害虫に対して本発明の化合物(A)、(B)、(C)及び(D)からなる群から選ばれた少なくとも1種を適用することにより、上記害虫による該植物体の摂食を阻害する方法である。本発明の害虫防除方法には、本発明の化合物をそのまま適用しても良いが、本発明の化合物を含む上記組成物又は害虫摂食抑制剤を使用する。
【0084】
次に、本発明を実施例を用いて具体的に説明するが、本発明がこれに限定されるものではない。
【0085】
化合物(1)は、Miyazawa, M.; Shimamura, H.; Nakamura, S.; Kameoka, H. J. Agric, Food Chem. 1995, 43, 1428-1431.に記載の方法と同様の方法により、ミカン科(Rutaceae)のハクセン(Dictamnus dasycarpus)の根皮から抽出した。
【0086】
実施例で使用した機器は、以下の通りである。
【0087】
ガスクロマトグラフィー(GC)
炎イオン化検出器(flame ionization detecotor(FID))を備えたヒューレットパッカード(Hewlett-Packard)社製5890A gas chromatographを使用した。カラムは、溶融石英(DB-5, 30m length, 0.25mm i.d.)を使用した。クロマトグラフィーのコンディションは以下の通りである。
オーブン温度は150℃から300℃に4℃/分で上昇するようにプログラムした。インジェクターと検出部の温度はそれぞれ270℃と280℃とした。スプリットインジェクションは19:1とした。ヘリウムガスのフローレートは、1.8 ml/minとした。
【0088】
EI−MS
EI−MS測定は、ガスクロマトグラフィー−質量分析(GC−MS)を使用した。GC−MSは、キャピラリーカラム(DB-5MS, 30 m length, 0.25 mm i.d.)を備えたヒューレットパッカード(Hewlett-Packard)社製5890A gas chromatograph−ヒューレットパッカード(Hewlett-Packard)社製5972A gas chromatographを使用した。クロマトグラフィーの条件は、上記のDB-5と同じとした。イオン源の温度は、230℃とし、電子エネルギーは70 eVとした。
【0089】
赤外線吸収スペクトル(IRスペクトル)
IRスペクトル測定は、日本分光株式会社製のJASCO FT/IR-470 plus fourier transform infrared spectrometerを使用した。
【0090】
NMRスペクトル
NMRスペクトル測定は、JEOL FX-500 (500.00 MHz, 1H; 125.65 MHz, 13C)spectrometerを使用した。CDCl3中のテトラメチルシラン(TMS)を標準物質として使用した。多重度は、DEPT pulse sequenceにより決定した。
【0091】
比旋光度
比旋光度の測定は、日本分光株式会社製のJASCO DIP-1000 digital polarimeterを使用した。
【0092】
実施例1(化合物(2)の合成)
化合物(2)は、(-)-フラキシネロンを以下の手順により還元して得た。(-)-フラキシネロン100mgを100%エタノール10ml中に溶かし、60mgのNaBH4を加えた。室温下に24時間攪拌下反応させた後、エバポレーターによりエタノールを留去し、得られた残渣に25mlの水を加えた。得られた混合物を30mlのCHCl3で4回で抽出した。得られたCHCl3溶液を中性になるまで水で洗浄し、エバポレーションを行い、明るい黄色のオイルを得た。得られたオイルをシリカゲルクロマトグラフィー(溶媒は、ヘキサン/酢酸エチルを9:1〜7:3に変化させた)で精製して、化合物(2)(52.4mg)を得た。化合物(2)が主生成物であり、収率は52%であった。化合物(2)の構造は、Weimin, Z.; Jean, L. W.; Kurt, H.; Rensheng, X.; Guowei, Q. Phytochemistry. 1998, 47, 7-11.に記載のスペクトルデータと比較して、(-)-7,7a-ジヒドロフラキシネロン((-)-7,7a-Dihydrofraxinellone)と決定された。
【0093】
実施例2(化合物(3)の合成)
化合物(3)は、(-)-フラキシネロンを以下の手順によりエポキシ化して得た。フラキシネロン50mgをメタノール1mlに溶解し、過酸化水素水(30%)60μl及び6N水酸化ナトリウム水溶液20μlを加えた。室温下に24時間攪拌下反応させた後、反応混合液に1N塩酸を加えてpH2に調整し、得られた混合液を10mlの水中に加えた。得られた混合液をジエチルエーテル10mlで4回抽出した。得られたエーテル溶液をエバポレーターで乾燥させると、無色の針状結晶が得られた。得られた結晶をシリカゲルクロマトグラフィー(溶媒は、ヘキサン/酢酸エチルを9:1〜7:3に変化させた)で精製して、化合物(3)(51mg)を得た。化合物(3)が主生成物であり、収率は95%(100%ee)であった。化合物(3)の構造は、Maique, W. B.; Paulo, C. V. M.; Fatima, G. F. D. S.; Joao, B. F.; Sergio, A. Z. Naturforsch. 2001, 56c, 570-574.に記載のスペクトルデータと比較して、(-)-7,7a-エポキシ-フラキシネロン((-)-7,7a-epoxy-fraxinellone)と決定された。
【0094】
実施例3(生物変換の手順)
アスペルギルス・ニガー(A.niger)による生物変換は、以下の手順により行った。万能培地(Czapek pepton培地)を用いて培養されたA. nigerを培養器中に2日間保持した(28℃攪拌下)。菌体(A.niger)を、万能培地(Czapek pepton培地)(50mlのペトリディッシュに20ml)に移植し、28℃静止下で培養した。1日後、成熟した生物体及び反応出発物質である各基質(0.4mg/ml)を培地に加え、生物体を4日〜12日培養した。ペトリディッシュ中の培地に1N塩酸を加えpH2に調整し、ジエチルエーテルで抽出し、エーテル層をエバポレーションした。得られた抽出物は、TLC、GC及びGC-MSで分析した。基質及び代謝生成物の比は、ガスクロマトグラフィーのピークエリアに基づき決定した。
【0095】
実施例4(化合物(1-1)の単離)
化合物(1)(90 mg)を実施例3の手順による生物変換の後、万能培地(Czapek pepton培地)と菌体をフィルターで分離した。培地に1N塩酸を加えてpH2に調整し、NaClで飽和し、ジエチルエーテルで抽出した。菌体もジエチルエーテルで抽出した。そして、両抽出物を混合した。この抽出物をジエチルエーテルと共に分液漏斗に入れて、水相とジエチルエーテル相とに分離した。ジエチルエーテル相を、Na2SO4で乾燥し、エバポレーターにかけ、シリカゲルクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=1:1)で溶出液を分割した。化合物(1)(48.6 mg)と代謝物である化合物(1-1)(31.4 mg)を分割したヘキサン/酢酸エチル溶出液から単離した。
【0096】
化合物(1-1)((+)-(4S)-4-ヒドロキシイソフラキシネロン): オイル; [α]25D -73.0o (c 0.19, CHCl3); EI-MS: m/z (rel int %): 248([M]+,6), 230(30), 203(5), 160(15), 145(27), 107(100), 95(41); IR(film, νmaxcm-1); 3442(OH), 1755(γ-lactone). 1H, 13C NMR (表1及び2)。
【0097】
実施例5(化合物(2-1)、(2-2)及び(2-3)の単離)
化合物(2)(100 mg)を実施例4と同様の操作により、化合物(2)(53.0 mg)と代謝物である化合物(2-1)(23.2mg)、化合物(2-2)(17.8mg)及び化合物(2-3)(6.0mg)を分割したヘキサン/酢酸エチル溶出液から単離した。
【0098】
化合物(2-1)((-)-(4S)-4-ヒドロキシ-7,7a-ジヒドロフラキシネロン): 黄色オイル; [α]25D-20.1o (c 0.40, CHCl3); EI-MS: m/z (rel int %): 250([M]+,25), 222(3), 204(3), 126(100), 108(91), 95(47), 84(64), 69(36); IR(film, νmaxcm-1); 3477(OH), 1765(γ-lactone). 1H, 13C NMR(表1及び2)。
【0099】
化合物(2-2)((-)-(6R)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロン):黄色オイル; [α]25D-45.5o (c 0.34, CHCl3); EI-MS: m/z (rel int %): 250([M]+,10), 204(4), 126(10), 108(100), 93(26), 82(73), 67(28); IR(film, νmaxcm-1); 3485(OH), 1766(γ-lactone). 1H, 13C NMR(表1及び2)。
【0100】
化合物(2-3)((-)-(6S)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロン): 黄色オイル; [α]25D-25.0o (c 0.22, CHCl3); EI-MS: m/z (rel int %): 250([M]+,11), 204(30), 154(10), 124(16), 109(67), 95(37), 82(100), 67(41); IR(film, νmaxcm-1); 3423(OH), 1766(γ-lactone). 1H, 13C NMR (表1及び2)。
【0101】
実施例6(化合物(3-1)の単離)
化合物(3)(90 mg)を同様の操作により、化合物(3)(31.5 mg)と代謝物である化合物(3-1)(18.5 mg)を分割したヘキサン/酢酸エチル溶出液から単離した。
【0102】
化合物(3-1)((+)-(6R)-6-ヒドロキシ-7,7a-エポキシ-フラキシネロン): 無色針状結晶; [α]25D+ 1.0o (c 1.00, CHCl3); EI-MS: m/z (rel int %): 264([M]+,13), 140(29), 124(31), 96(100), 77(15), 43(32); IR(film, νmaxcm-1); 3273(OH), 1780(γ-lactone). 1H, 13C NMR (表1及び2)。
【0103】
【表1】
【0104】
【表2】
【0105】
実施例7(害虫摂食阻害作用の評価方法)
日産化学工業(株)より入手したハスモンヨトウ(Spodoptera litura)を使用した。ハスモンヨトウの幼虫をナイロンメッシュスクリーンでカバーしたプラスチックケース(幅200 mm×300 mm, 高さ100 mm, 1容器当たり幼虫100個)内で飼育した。飼育環境は、25℃下、相対湿度70%、光周期:16時間を明、8時間を暗とした。市販食餌(Insecta LFS; 日本農産工業株式会社)を初齢の幼虫に与えた。実験方法は、Mallavadhani, U. V.; Mahapatra, A.; Raja, S. S.; Manjula, C. J. Agric, Food Chem. 2003, 51, 1952-1955、 Morimoto, M.; Kumeda, S.; Komai, K. J. Agric, Food Chem. 2000, 48, 1888-1891及びKumari, G. N. K.; Balachandran, J.; Aravind, S.; Ganesh, M. R. J. Agric, Food Chem. 2003, 51, 1555-1559に記載の方法に基づき行った。穿孔器を用いて直径2cmのリーフディスクを生のキャベツ(Brassica oleracea)の葉から準備した。2枚のディスクをアセトン溶液中の試験物質で処理し、他の2枚のディスクはコントロールとしてアセトンで処理した。4枚のディスクは同じペトリディッシュ内に交互に置いた。溶媒を完全に除去した後、10個の幼虫(三齢)を25℃、暗闇下、3〜5時間ペトリディッシュ内に置いた。モノトーンデータ変換のため、部分的に摂食されたリーフディスクをコピー用紙上にテープで貼り付けた。モノトーンデータをコピーし、エラーがないことを確認して、デジタルスキャナーを使用してデジタルデータに変換した。デジタルデータの分析は、public-domain NIH Image program (米国国立衛生研究所(U.S. National Institutes of Health)開発によってされ、インターネットを介しzippy, nimh, nih, govで匿名FTPで利用可能、又はthe National Technical Information Serice, Springfiled, Virginia, part number PB95-500195GEIによりフロッピー(登録商標)ディスクにより利用可能である)をマッキントッシュコンピューターを使用して行った。
【0106】
それぞれの実験は、摂食されていないディスクのデータファイルを測定し、処理されたディスクのものと比較して行った。摂食阻害活性パーセンテージ(%)の評価は、下式を用いて計算した。
【0107】
摂食阻害活性パーセンテージ(%)=100−(試験物質で処理されたディスクの摂食された部分(%)/コントロールディスクの摂食された部分(%))×10
試験物質として、化合物(1)、(2)、(3)、(1-1)、(2-1)、(2-2)、(2-3)、及び(3-1)を、葉面積当たり、それぞれ25、10及び5 μg/cm2使用して評価した。また、各試験物質について、ED50(μg/cm2)も測定した。
以上の結果を表3に示す。
【0108】
【表3】
【0109】
この結果から、化合物(1)、(2)、(3)、(1-1)、(2-1)、(2-2)、(2-3)、及び(3-1)はいずれも害虫摂食阻害活性が高いことがわかる。化合物(3-1)の害虫摂食阻害活性が最も高いと考えられる。
【0110】
実施例8(化合物(1)の生物変換)
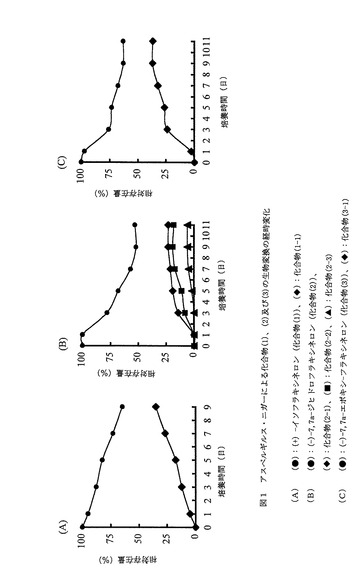
アスペルギルス・ニガーによる化合物(1)の生物変換の経時変化を調査するために、アスペルギルス・ニガーと共に少量の化合物(1)を9日間培養した。得られた代謝物は、TLC、GC及びGC−MSスペクトル分析で確認した。化合物(1)から(1-1)への変換の経時変化は、TLC及びGC(ガスクロマトグラフィー)による定量測定によってモニターした。結果を図1に示す。9日後、出発基質である化合物(1)がca35 %変換されたことがわかる。
【0111】
得られた代謝物を単離するために、アスペルギルス・ニガーを用いて大スケールで9日間培養した(実施例3の手順)。生物変換の後、実施例4に記載の通り、培地を抽出し、抽出液から代謝物である化合物(1-1)を得た。スペクトルデータから、化合物(1-1)の構造を確認した(実施例9)。
【0112】
実施例9(化合物(1-1)の構造決定)
高分解能質量分析及びNMRデータから、化合物(1-1)の分子式はC14H16O4と確認された。IRスペクトルより、3442 cm-1に水酸基由来のバンドが確認され、比旋光度は(−)を示した。化合物(1-1)のプロトン及びカーボンNMRスペクトルは、新規のメチン基の出現及びC−4位のメチレン基の消失以外は、化合物(1)のものと類似していた。プロトンNMRスペクトルに関しては、H-5’(7.46 ppm)が、H-4’(6.32 ppm)とH-2’(7.42 ppm)との特徴的なカップリング定数(J=1.6 Hz)を有していた。詳細には、二次元NMR(COSY、HMQC、HMBC及びNOE)によって確認された。
【0113】
COSYスペクトルは、H-4 (3.85 ppm)とδ2.37 及びδ2.12 (それぞれHα-5 及び Hβ-5,)並びにH-6 (5.48 ppm)とδ 2.37及びδ1.97 (それぞれHα-5及びH-8) に相関クロスピークが観測された。
【0114】
プロトンNMRスペクトルは、2つのメチル基がδ 1.97及びδ 0.88 (それぞれH-8 及び H-9)に観測され、2つのメチン基がδ5.56及びδ2.86 (それぞれH-3及びH-7a)に観測された。
【0115】
HMBCスペクトル相関は、新しいメチン炭素(68.1 ppm; C-4)と1つのメチル基(0.88 ppm; H-9)及び2つのメチン基(δ5.56及びδ2.86 (それぞれH-3及びH-7a))とに観測された。
【0116】
NOE差スペクトルは、H-4 (3.85 ppm)と1つのメチン基(δ 5.56 ppm (H-3))及びH-7a (2.86 ppm)と1つのメチル基(0.88 ppm (H-9))との間にNOEを観測した。
【0117】
従って、化合物(1-1)は、化合物(1)のC-4位をヒドロキシル化して得られるものである。C-4位の第2級アルコールの絶対配置を確認するため、Kusumi, T.; Fujita, Y.; Ohtani, I.; Kakisawa, H. Tetrahedron Letters. 1991, 32, 2923-2926に記載の手法に従い、化合物(1-1)を(S)-及び(R)-MTPAエステルに変換した。改良モッシャー法(Mosher's method)により、C-4位はS-配置を有することが確認された。以上の結果から、化合物(1-1)の構造が(-)-(4S)-4-ヒドロキシイソフラキシネロンであると確認された。
【0118】
実施例10(化合物(2)の生物変換)
アスペルギルス・ニガーによる化合物(2)の生物変換の経時変化を調査するために、アスペルギルス・ニガーと共に少量の化合物(2)を11日間培養した。得られた代謝物は、TLC、GC及びGC−MSスペクトル分析で確認した。化合物(2)から(2-1)、(2-2)及び(2-3)への変換の経時変化は、TLC及びGC(ガスクロマトグラフィー)による定量測定によってモニターした。結果を図1に示す。11日後、出発基質である化合物(2)がca53 %変換された。代謝物である化合物(2-1)、(2-2)及び(2-3)は、11日後にはそれぞれca23 % 、ca18 % 及びca6 %であった。
【0119】
これらの代謝物を単離するために、アスペルギルス・ニガーを用いて大スケールで11日間培養した(実施例3の手順)。生物変換の後、実施例5に記載の通り、培地を抽出し、抽出液から代謝物である化合物(2-1)、(2-2)及び(2-3)を得た。スペクトルデータから、化合物(2-1)、(2-2)及び(2-3)の構造を確認した(実施例11)。
【0120】
実施例11(化合物(2-1)の構造決定)
高分解能質量分析及びNMRデータから、化合物(2-1)の分子式はC14H18O4と確認された。IRスペクトルより、3477 cm-1に水酸基由来のバンドが確認され、比旋光度は(−)を示した。化合物(2-1)のプロトン及びカーボンNMRスペクトルは、新規のメチン基の出現及びC−4位のメチレン基の消失以外は、化合物(2)のものと類似していた。プロトンNMRスペクトルに関しては、H-5’(7.43 ppm)が、H-4’(6.30 ppm)とH-2’(7.39 ppm)との特徴的なカップリング定数(J=1.7 Hz)を有していた。詳細には、二次元NMR(COSY、HMQC、HMBC及びNOE)によって確認された。
【0121】
COSYスペクトルは、H-4 (3.68 ppm)とδ 1.81、δ 1.77、δ 1.64及びδ 1.54 (それぞれH-5β、H-7、H-6α及びH-5α)に相関クロスピークが観測された。
【0122】
プロトンNMRは、2つのメチル基がδ 1.32及びδ 0.92 (それぞれH-8及びH-9)に観測され、メチン基がδ 5.56、δ 2.50及びδ 1.77(それぞれH-3、H-7a及びH-7)に観測された。
【0123】
HMBCスペクトル相関は、新しいメチン炭素(71.9 ppm; C-4)と1つのメチル基(0.92 ppm; H-9)及び2つのメチン基(δ 5.40及びδ 2.50(それぞれH-3及びH-7a))とに観測された。従って、化合物(2-1)は、化合物(2)のC-4位をヒドロキシル化して得られるものである。
【0124】
NOE差スペクトルにおいて、H-4とH-3との間にNOEが観測されることから、OH基の立体化学はα配置と確認した。
【0125】
以上の結果から、化合物(2-1)の構造が(-)-(4S)-4-ヒドロキシ-7,7a-ジヒドロフラキシネロンであると確認された。
【0126】
実施例12(化合物(2-2)の構造決定)
高分解能質量分析及びNMRデータから、化合物(2-2)の分子式はC14H18O4と確認された。IRスペクトルより、3485 cm-1に水酸基由来のバンドが確認され、比旋光度は(−)を示した。化合物(2-2)のプロトン及びカーボンNMRスペクトルは、新規のメチン基の出現及びC−6位のメチレン基の消失以外は、化合物(2)のものと類似していた。プロトンNMRスペクトルに関しては、H-5’(7.44 ppm)が、H-4’(6.28 ppm)とH-2’(7.38 ppm)との特徴的なカップリング定数(J=1.6 Hz)を有していた。詳細には、二次元NMR(COSY、HMQC、HMBC及びNOE)によって確認された。
【0127】
COSYスペクトルは、H-6 (3.50 ppm)とδ 1.92、δ 1.74及びδ 1.51 (それぞれH-5β、H-7及びH-5α)に相関クロスピークが観測された。
【0128】
プロトンNMRは、2つのメチル基がδ 1.43及びδ 1.00(それぞれH-8及びH-9)に観測され、メチン基がδ 4.92、δ 2.52及びδ 1.74(それぞれH-3、H-7a及びH-7)に観測された。
【0129】
HMBCスペクトル相関は、新しいメチン炭素(72.0 ppm; C-6)と1つのメチル基(1.43 ppm; H-8)及び1つのメチン基(δ 2.52(H-7a))とに観測された。従って、化合物(2-2)は、化合物(2)のC-6位をヒドロキシル化して得られるものである。
【0130】
H-7aに照射すると、H-9の面積強度が強調されることから、C-7aの立体化学はNOE差によりα配置と決定した。
【0131】
C-7メチンプロトンのアキシアル(α)配置及びC-6メチンプロトンのアキシアル(β)配置は、1H NMRスペクトルにおけるH-7a、H-8及びH-6シグナルから観測されるH-7シグナルのスプリッティングパターン(H-7; ddq, J= 4.8, 7.0, 10.7 Hz)により判断した。
【0132】
以上の結果から、化合物(2-2)の構造が(-)-(6R)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロンであると確認された。
【0133】
実施例13(化合物(2-3)の構造決定)
高分解能質量分析及びNMRデータから、化合物(2-3)の分子式はC14H18O4と確認された。IRスペクトルより、3423 cm-1に水酸基由来のバンドが確認され、比旋光度は(−)を示した。化合物(2-3)のプロトン及びカーボンNMRスペクトルは、新規のメチン基の出現及びC−6位のメチレン基の消失以外は、化合物(2)のものと類似していた。プロトンNMRスペクトルに関しては、H-5’(7.43 ppm)が、H-4’(6.28 ppm)とH-2’(7.38 ppm)との特徴的なカップリング定数(J=1.6 Hz)を有していた。詳細には、二次元NMR(COSY、HMQC、HMBC及びNOE)によって確認された。
【0134】
COSYスペクトルは、H-6 (3.88 ppm)とδ 1.94、δ1.90及びδ1.61 (それぞれH-7、Hβ-5及びHα-5)に相関クロスピークが観測された。
【0135】
プロトンNMRは、2つのメチル基がδ 1.46及びδ 0.96(それぞれH-8及びH-9)に観測され、メチン基がδ 4.99、δ 2.31及びδ 1.94(それぞれH-3、H-7a及びH-7)に観測された。
【0136】
HMBCスペクトル相関は、新しいメチン炭素(69.6 ppm; C-6)と1つのメチル基(1.46 ppm; H-8)及び1つのメチン基(δ 2.31(H-7a))とに観測された。従って、化合物(2-3)は、化合物(2)のC-6位をヒドロキシル化して得られるものである。
【0137】
H-7aに照射すると、H-9の面積強度が強調されることから、C-7aの立体化学はNOE差によりα配置と決定した。
【0138】
C-7メチンプロトンのアキシアル(α)配置及びC-6メチンプロトンのエカトリアル(α)配置は、1H NMRスペクトルにおけるH-5α、H-6、H-7a及びH-8シグナルから観測されるH-7シグナルのスプリッティングパターン(H-7; dddq, J= 2.2, 2.2, 5.2, 7.4 Hz)により判断した。
【0139】
以上の結果から、化合物(2-3)の構造が(-)-(6S)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロンであると確認された。
【0140】
実施例14(化合物(3)の生物変換)
アスペルギルス・ニガーによる化合物(3)の生物変換の経時変化を調査するために、アスペルギルス・ニガーと共に少量の化合物(3)を11日間培養した。得られた代謝物は、TLC、GC及びGC−MSスペクトル分析で確認した。化合物(3)から(3-1)への変換の経時変化は、TLC及びGC(ガスクロマトグラフィー)による定量測定によってモニターした。結果を図1に示す。11日後、出発基質である化合物(3)がca37 %変換されたことがわかる。
【0141】
これらの代謝物を単離するために、アスペルギルス・ニガーを用いて大スケールで11日間培養した(実施例3の手順)。生物変換の後、実施例6に記載の通り、培地を抽出し、抽出液から代謝物である化合物(3-1)を得た。スペクトルデータから、化合物(3-1)の構造を確認した(実施例15)。
【0142】
実施例15(化合物(3-1)の構造決定)
高分解能質量分析及びNMRデータから、化合物(3-1)の分子式はC14H16O5と確認された。IRスペクトルより、3273 cm-1に水酸基由来のバンドが確認され、比旋光度は(+)を示した。化合物(3-1)のプロトン及びカーボンNMRスペクトルは、新規のメチン基の出現及びC−6位のメチレン基の消失以外は、化合物(3)のものと類似していた。プロトンNMRスペクトルに関しては、H-5’(7.47 ppm)が、H-4’(6.33 ppm)とH-2’(7.48 ppm)との特徴的なカップリング定数(J=1.7 Hz)を有していた。詳細には、二次元NMR(COSY、HMQC、HMBC及びNOE)によって確認された。
【0143】
COSYスペクトルは、H-6 (3.91 ppm)とδ 1.83、δ 1.59、δ 1.51及びδ 1.51 (それぞれHα-5、Hα-4、Hβ-5及びHβ-4)に相関クロスピークが観測された。
【0144】
プロトンNMRは、2つのメチル基がδ 1.68及びδ 0.93(それぞれH-8及びH-9)に観測され、メチン基がδ 5.19 (H-3)に観測された。
【0145】
HMBCスペクトル相関は、新しいメチン炭素(72.1 ppm; C-6)と1つのメチル基(1.68 ppm; H-8)及び2つのメチレン基(δ 1.83、δ 1.59、δ 1.51及びδ 1.51(それぞれHα-5、Hα-4、Hβ-5及びHβ-4))とに観測された。従って、化合物(3-1)は、化合物(3)のC-6位をヒドロキシル化して得られるものである。
【0146】
C-6位の第2級アルコールの絶対配置を確認するため、上述のKusumi, T.; Fujita, Y.; Ohtani, I.; Kakisawa, H. Tetrahedron Letters. 1991, 32, 2923-2926に記載の手法に従い、化合物(3-1)を(S)-及び(R)-MTPAエステル誘導体に変換した。
【0147】
化合物(3-1)の(S)-及び(R)-MTPAエステル誘導体は、Me-8に対してnegative Δδ(=δR-δS)values(- 0.15)及び Hα-5、Hβ-5及びH-4に対してそれぞれpositive values(H-5α(+ 0.11)、H-5β(+ 0.11)及びH-4(+ 0.03))を与えた。以上の改良モッシャー法(Mosher's method)により、C-6位はR-配置を有することが確認された。
【0148】
NOE差による化合物(3-1)の絶対配置の確認は、シグナルがオーバーラップしたことと、それぞれの水素に照射することの困難性により、行うことができなかった。
【0149】
以上の結果から、化合物(3-1)の構造が(+)-(6R)-6-ヒドロキシ-7,7a-エポキシ-フラキシネロンであると確認された。
【図面の簡単な説明】
【0150】
【図1】アスペルギルス・ニガーによる化合物(1)、(2)及び(3)の生物変換の経時変化を示す図である
【技術分野】
【0001】
本発明は、新規分解型リモノイド化合物、該化合物の製造方法、該化合物を有効成分として含有する害虫摂食阻害剤、該化合物を含有する組成物及び該組成物を害虫に適用する害虫防除方法に関する。
【背景技術】
【0002】
ミカン科の植物に含まれるリモノイドには、種々の分解型リモノイドが知られている。これらは、リモノイドと共通する部分構造を有しており、リモノイドよりも小さく単純な化合物である。このような分解型リモノイドに属し、Rutaceae(ミカン科)から得られる(-)-フラキシネロン((-)-fraxinellone))は、様々な生物活性を有することが報告されている(非特許文献1〜4)。
【0003】
【化1】
【0004】
本発明者らは、(+)-イソフラキシネロン((+)-isofraxinellone)の抗変異原活性(antimutagenic activites)について、既に報告している(非特許文献5)。また、Liuらは、(-)-フラキシネロンが、コクゾウムシ(Sitophilus zeamais)の成虫と同様、コクヌストモドキ(Tribolium castaneum)の成虫及び幼虫に対しても摂食阻害活性を有することを報告している(非特許文献6)。さらにMaiqueらは、下記一般式3
【0005】
【化2】
【0006】
で表される化合物(以下、化合物(3)ということがある)が、Trypaosoma cruziのトリポマスティゴート型原虫に対して、弱い摂食阻害活性を有することを報告している(非特許文献7)。
【0007】
生物変換は、生体中に存在する酵素を生体触媒として使用する簡易な手法であり、不活性な天然物から生理活性を有する医薬又は農薬化合物を製造するために用いられる。該手法の製造条件は温和であり、多くの場合、官能基の保護を必要としない。さらに、生物変換の特徴は、位置及び立体選択的反応であり、光学活性な生成物を与えることである。生物変換によって、天然物から生理活性物質を得る方法として、本発明者らは、フラボノイド(非特許文献8及び9)、テルペノイド(非特許文献10及び11)及びリグナン(非特許文献10及び11)の生物変換について報告している。
【0008】
現在、過剰な農薬使用が問題視されているが、通常、無農薬で食害を防ぐことは困難であり、環境負荷の少ない害虫防除法が求められている。害虫に対して摂食阻害活性を有する化合物は、低分子でありながら極めて低濃度で生物活性を示すため、安全かつ選択的な害虫防除剤としての利用が期待され、実用化に向けた研究が活発になされている。害虫摂食阻害剤の摂食阻害活性が強ければ害虫は餓死するので、その物質の使用濃度において人体に無害であれば、害虫摂食阻害剤は農薬として使用される可能性もある。
【非特許文献1】Okamura, H.; Yamauchi, K.; Miyawaki, K.; Iwagawa, T.; Nakatani, M. Tetrahedron Letters. 1997, 38, 263-266.
【非特許文献2】Nakatani, M.; Huang, RC.; Okamura, H.; Iwagawa, T.; Tadera, K. Phytochemistry. 1998, 49, 1773-1776.
【非特許文献3】Yu, S. M.; Ko, F. N.; Su, M. J.; Wu, T. S.; Wang, M. L.; Huang, T. F.; Teng, C. M. Naunyn-Schmiedeberg’s Arch Pharmacol. 1992, 345, 349-355.
【非特許文献4】Woo, W. S.; Lee, E. B.; Kang, S. S.; Shin, K. H.; Chi, H. J. Planta Med. 1987, 53, 399-401.
【非特許文献5】Miyazawa, M.; Shimamura, H.; Nakamura, S.; Kameoka, H. J. Agric, Food Chem. 1995, 4, 1428-1431.
【非特許文献6】Liu, Z. L.; Xu, Y. J.; Wu, J.; Goh, S. H.; Ho, S. H. J. Agric, Food Chem. 2002, 50, 1447-1450.
【非特許文献7】Maique, W. B.; Paulo, C. V. M.; Fatima, G. F. D. S.; Joao, B. F.; Sergio, A. Z. Naturforsch. 2001, 56c, 570-574.
【非特許文献8】Miyazawa, M.; Takahashi, K.; Araki, H. J Chem Technol Biotechnol. 2006, 81, 674-678.
【非特許文献9】Okuno, Y.; Miyazawa, M. J Chem Technol Biotechnol. 2006, 81, 29-33.
【非特許文献10】Miyazawa, M.; Miyamoto, Y. J Mol Catal B. 2004, 27, 83-89.
【非特許文献11】Miyazawa, M.; Nankai, H.; Kameoka, H. Phytochemistry. 1996, 43, 105-109.
【非特許文献12】Miyazawa, M.; Kasahara, H.; Kameoka, H. Phytochemistry. 1994, 35, 1191-1193.
【非特許文献13】Miyazawa, M.; Kasahara, H.; Kameoka, H. Phytochemistry. 1993, 34, 1501-1507.
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明は、新規分解型リモノイド化合物、該化合物の製造方法、該化合物を有効成分として含有する害虫摂食阻害剤、該化合物を含有する組成物及び該組成物を害虫に適用する害虫防除方法を提供することを目的とする。
【課題を解決するための手段】
【0010】
本発明者は、(+)-イソフラキシネロン((+)-isofraxinellone)又は(-)-フラキシネロン((-)-fraxinellone)誘導体をアスペルギルス・ニガー(Aspergillus niger)により生物変換したところ、(+)-イソフラキシネロン又は(-)-フラキシネロン誘導体がヒドロキシル化された新規化合物を得られることを見出した。さらに、これらの新規化合物は、害虫摂食抑制剤を有することを見出した。本発明は、更に検討を加えて完成されたものである。
【0011】
即ち、本発明は以下の化合物、及び該化合物を含む害虫摂食抑制剤を提供する。
項1. 一般式(A):
【0012】
【化3】
【0013】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する]
で表される化合物。
項2. 一般式(A)において、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合する項1に記載の化合物。
項3. 一般式(A−1):
【0014】
【化4】
【0015】
[式中、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合する]
で表される化合物。
項4. 項1〜3のいずれかに記載の化合物を有効成分として含む害虫摂食抑制剤。
項5. 害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である項4に記載の害虫摂食抑制剤。
項6. 一般式(A):
【0016】
【化5】
【0017】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する]
で表される化合物の製造方法であって、下記一般式(B)
【0018】
【化6】
【0019】
で表される化合物を生物変換することを特徴とする製造方法。
項7. 一般式(C):
【0020】
【化7】
【0021】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物。
項8. 一般式(C)において、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合する請求項7に記載の化合物。
項9. 一般式(C−1):
【0022】
【化8】
【0023】
[式中、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物。
項10. 項7〜9のいずれかに記載の化合物を有効成分として含む害虫摂食抑制剤。
項11. 害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である項8に記載の害虫摂食抑制剤。
項12. 一般式(C):
【0024】
【化9】
【0025】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物の製造方法であって、下記一般式(D)
【0026】
【化10】
【0027】
[式中、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物を生物変換することを特徴とする製造方法。
項13. 項1〜3及び項7〜9のいずれかに記載の化合物を含有する組成物。
項14. 項13に記載の組成物を害虫に適用することを特徴とする害虫防除方法。
項15. 一般式(B)
【0028】
【化11】
【0029】
及び一般式(D)
【0030】
【化12】
【0031】
[式中、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物からなる群から選ばれた少なくとも1種を有効成分として含む害虫摂食抑制剤であって、害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である害虫摂食抑制剤。
【発明の効果】
【0032】
本発明の一般式(A)及び(C)で表される化合物は新規であり、高い害虫摂食抑制活性を有しているため、害虫摂食抑制剤として有用である。また、本発明の一般式(B)及び(D)で表される化合物も高い害虫摂食抑制活性を有しているため、害虫摂食抑制剤として有用である。特に、本発明の化合物は、ヨトウムシに対して優れた害虫摂食抑制活性を有する。
【発明を実施するための最良の形態】
【0033】
本発明の化合物
本発明の第一の化合物は、下記一般式(A)で表される化合物(以下、化合物(A)ということがある)である。
【0034】
【化13】
【0035】
(式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する)
一般式(A)において、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合することが好ましく、4位又は6位のいずれか1つの炭素のみに1つ結合することがさらに好ましく、6位の炭素に1つ結合することが特に好ましい。
【0036】
化合物(A)の中でも、下記一般式(A−1)で表される化合物(以下、化合物(A−1)ということがある)が好ましい。
【0037】
【化14】
【0038】
(式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する。)
一般式(A−1)において、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合することが好ましく、4位又は6位のいずれか1つの炭素のみに1つ結合することがさらに好ましく、6位の炭素に1つ結合することが特に好ましい。
【0039】
また、化合物(A)の中でも、下記一般式(3-1)((+)-(6R)-6-ヒドロキシ-7,7a-エポキシ-フラキシネロン((+)-(6R)-6-hydroxy-7,7a-epoxy-fraxinellone))の絶対配置で表される化合物(以下、化合物(3-1)ということがある)が特に好ましい。
【0040】
【化15】
【0041】
本発明の第二の化合物は、下記一般式(C)で表される化合物(以下、化合物(C)ということがある)である。
【0042】
【化16】
【0043】
(式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。)
一般式(C)において、6位と7位との炭素間結合が一重結合である場合、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合することが好ましく、特に4位又は6位のいずれか1つの炭素のみに1つ結合することが好ましい。
【0044】
一般式(C)において、6位と7位との炭素間結合が二重結合である場合、OH基は4位又は5位のいずれか1つの炭素のみに1つ結合することが好ましく、特に4位の炭素に1つ結合することが好ましい。
【0045】
化合物(C)の中でも、下記一般式(C−1)で表される化合物(以下、化合物(C−1)ということがある)が好ましい。
【0046】
【化17】
【0047】
(式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。)
一般式(C−1)において、6位と7位との炭素間結合が一重結合である場合、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合することが好ましく、特に4位又は6位のいずれか1つの炭素のみに1つ結合することが好ましい。
【0048】
一般式(C−1)において、6位と7位との炭素間結合が二重結合である場合、OH基は4位又は5位のいずれか1つの炭素のみに1つ結合することが好ましく、特に4位の炭素に1つ結合することが好ましい。
【0049】
一般式(C−1)において、6位と7位との炭素間結合が二重結合である化合物の中でも、下記一般式(1-1)((+)-(4S)-4-ヒドロキシイソフラキシネロン)((+)-(4S)-4-hydroxyisofraxinellone))の絶対配置で表される化合物(以下、化合物(1-1)ということがある)が特に好ましい。
【0050】
【化18】
【0051】
一般式(C−1)において、6位と7位との炭素間結合が一重結合である化合物の中でも下記一般式(2-1)、(2-2)及び(2-3)の絶対配置で表される化合物(以下、それぞれ化合物(2-1)、(2-2)及び(2-3)ということがある)が好ましい。
【0052】
【化19】
【0053】
本発明の製造方法
本発明の一般式(A)で表される化合物は、下式に示すスキームにより得られる。
【0054】
【化20】
【0055】
(式(A)中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する。)
一般式(E)で表される化合物(以下、化合物(E)ということがある)は、非特許文献1〜4に記載の方法に準じた方法により得られる。また、化合物(E)の中でも、(-)-フラキシネロンは、非特許文献1〜4に記載の通り、和漢生薬白鮮皮より得られる。
【0056】
一般式(B)で表される化合物(以下、化合物(B)ということがある)は、例えば、従来公知の方法によって、化合物(E)をエポキシ化反応することにより得られる。化合物(B)の中でも、下記一般式(3)(化合物(3))で表される化合物は、例えば、(-)-フラキシネロンをエポキシ化反応することにより得られる(実施例2)。
【0057】
【化21】
【0058】
当該エポキシ化反応に使用されるエポキシ化反応の条件は、特に限定されず、従来公知の方法を採用すればよい。例えば、アルカリ条件下で、過酸化水素水、t-ブチルヒドロペルオキシド等を使用して分子内の二重結合をエポキシ化する。エポキシ化反応に使用される溶媒としては、例えば、メタノール、エタノール、テトラヒドロフラン、ジメチルホルムアミド等が挙げられる。反応温度は、通常、0℃〜室温程度とすればよく、反応時間は、通常、10分〜24時間程度である。必要に応じて、アルミナカラムクロマトグラフィー、シリカゲルクロマトグラフィー、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィー、疎水クロマトグラフィー、高速液体クロマトグラフィー等の適当な分離精製手段を1種若しくは2種以上組み合わせて精製することができる。
【0059】
本発明の化合物(A)は、化合物(B)を生物変換によりヒドロキシル化して得られる。例えば、本発明の化合物(3-1)は、化合物(3)を生物変換によりヒドロキシル化して得られる。
【0060】
また、 本発明の一般式(C)で表される化合物(以下、化合物(C)ということがある)は、下式に示すように、化合物(D)で表される化合物を生物変換によりヒドロキシル化して得られる。
【0061】
【化22】
【0062】
(式(C)中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。)
一般式(D)で表される化合物(以下、化合物(D)ということがある)において、6位と7位との炭素間結合が二重結合である化合物は、非特許文献5に記載の方法に準じて得られる。化合物(D)の中でも、化合物(1)((+)-イソフラキシネロン)は、ミカン科(Rutaceae)のハクセン(Dictamnus dasycarpus)の根皮を乾燥した和漢生薬白鮮皮より得られる分解型リモノイドであり、非特許文献5に記載の方法により抽出できる。
【0063】
一般式(D)で表される化合物において、6位と7位との炭素間結合が一重結合である化合物は、例えば、化合物(D)において6位と7位との炭素結合が二重結合である化合物を水素付加反応することによって得られる。また、上記一般式(E)で表される化合物の7位と7a位の二重結合を水素付加反応することによっても得られる。例えば、化合物(2)は、(-)-フラキシネロンを水素付加反応することにより得られる(実施例1)。
【0064】
水素付加反応は、従来公知の方法により行えばよい。水素付加反応に使用される試薬は、従来公知の試薬を使用すればよいが、例えば、水素化ホウ素ナトリウム、水素化ホウ素リチウム等を使用すればよい。必要に応じて、アルミナカラムクロマトグラフィー、シリカゲルクロマトグラフィー、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィー、疎水クロマトグラフィー、高速液体クロマトグラフィー等の適当な分離精製手段を1種若しくは2種以上組み合わせて精製することができる。
【0065】
本発明の化合物(C)の中でも、化合物(1-1)は、(+)-イソフラキシネロン(化合物(1))を生物変換することによりヒドロキシル化して得られる。
【0066】
また、本発明の化合物(C)の中でも、化合物(2-1)((-)-(4S)-4-ヒドロキシ-7,7a-ジヒドロフラキシネロン)、(2-2)((-)-(6R)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロン)及び(2-3)((-)-(6S)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロン)は、下記一般式(2)で表される化合物((-)-7,7a-ジヒドロフラキシネロン)(以下、化合物(2)ということがある)を生物変換によりヒドロキシル化して得られる。
【0067】
【化23】
【0068】
本発明の生物変換においては、黒麹菌であるアスペルギルス・ニガー(Aspergillus niger)を用いる。
【0069】
具体的には、培養されたアスペルギルス・ニガーを培地に移植し、基質(反応出発物質)を培地に加え、通常1〜11日間程度、好ましくは1〜15日間程度放置し、アスペルギルス・ニガー及び培地を抽出することにより、一般式(A)及び(C)で表される化合物(代謝物)が得られる。温度条件は、通常0〜25℃程度とすればよい。使用する培地は、特に限定されないが、例えば、万能培地(Czapek pepton培地)、寒天培地、LB培地等が挙げられ、これらの中でも、万能培地が好ましい。培養は、酸素の存在下で行うのが好ましい。必要に応じて、アルミナカラムクロマトグラフィー、シリカゲルクロマトグラフィー、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィー、疎水クロマトグラフィー、高速液体クロマトグラフィー等の適当な分離精製手段を1種若しくは2種以上組み合わせて精製することができる。具体的には実施例を参照。
【0070】
一般式(A)、(B)、(C)及び(D)で表される本発明の化合物は、害虫摂食阻害活性を有している。
【0071】
本発明の化合物が示す害虫摂食阻害活性とは、害虫の餌となる植物等に付着していると、害虫が餌を食べられなくなる活性をいう。
【0072】
本発明の化合物が有効に摂食阻害できる対象害虫としては、例えば、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類等に属する昆虫が挙げられる。本発明による摂食阻害は、特に鱗翅目に属する昆虫に対して有効であり、例えば、テンスジツトガ、ツトガ、シバツトガ、ロブノメイガ、ワモンノメイガ、アワノメイガ、アカフツヅリガ等のメイガ科; スゲドクガ等のドクガ科; オビヒトリ、キハラゴマダラヒトリ、シロヒトリ等のヒトリガ科; イネキンウワバ、ヨトウムシ(例えば、エゾチャイロヨトウ、シロシタヨトウ、フタオビキヨトウ、タンポキヨトウ、クサシロキヨトウ、イネヨトウ、スジギリヨトウ、ハスモンヨトウ、シロイチモジヨトウ、ヨトウガ、アワヨトウ等)等のヤガ科に属する昆虫に対して有効である。これらの中でもヨトウムシに対して特に有効である。対象害虫は、成虫及び幼虫のいずれでも良いが、特に幼虫が好ましい。
【0073】
本発明の化合物が有効に摂食阻害できる対象植物は、特に限定されないが、例えば、キャベツ、サツマイモ、サトイモ、ネギ、トマト、ピーマン、ナス、レタス、ハクサイ等が挙げられる。
【0074】
本発明の化合物(A)、(B)、(C)及び(D)は優れた害虫阻害活性を有している。これらの中でも、化合物(1)、(2)、(3)、(1-1)、(2-1)、(2-2)、(2-3)及び(3-1)は、特に優れた害虫阻害剤活性を有している。
【0075】
本発明の害虫摂食阻害剤
本発明の害虫摂食阻害剤は、本発明の化合物(A)、(B)、(C)及び(D)からなる群から選ばれた少なくとも1種を有効成分として含む。
【0076】
本発明の化合物は、直接植物の葉、実、茎等に散布、塗布等することにより、そのままでも害虫摂食阻害剤として作用するが、通常は、ローション、エアゾ−ル等の液剤や、クリーム剤等の各種形態に製剤化した組成物として使用される。液剤としては、ローション、エアゾール、油剤等を挙げることができ、これらの液剤に用いられる担体としては、例えば、水、メタノール、エタノール、イソプロピルアルコール、セチルアルコール等のアルコール類、石油ベンジン等の脂肪族炭化水素類、ミリスチン酸イソプロピル、酢酸セチル等のエステル類があげられる。液剤の種類により、適宜さらに乳化剤、分散剤、展着剤、湿潤剤、懸濁化剤、保存剤、噴射剤等の製剤用補助剤、塗膜形成剤などを加え、所望の製剤とすることができる。
【0077】
本発明の化合物は、通常用いられる製剤化方法により、害虫摂食阻害剤に製剤化することができる。すなわち、有効成分以外に必要な補助剤を含むことができる。製剤の形態としては、乳剤、水和剤、粉剤、エアロゾルなどを挙げることができる。好ましくは、水和剤である。本発明の害虫摂食阻害剤は、一般に散布によって害虫に適用する。
【0078】
乳化剤及び分散剤としては、例えば、石鹸類、ポリオキシエチレンオレイルエーテル等のポリオキシエチレン脂肪酸アルコールエーテル、ポリオキシエチレンノニルフェニルエーテル等のポリオキシエチレンアルキルアリールエーテル、ポリオキシエチレン脂肪酸エステル、脂肪酸グリセリド、ポリオキシエチレンソルビタンモノステアレート等のソルビタン脂肪酸エステル、高級アルコールの硫酸エステル、ドデシルベンゼンスルホン酸ソーダ等のアルキルアリールスルホン酸塩が挙げられ、展着剤または湿潤剤としては、例えば、グリセリン、プロピレングリコール、ポリエチレングリコールが挙げられる。
【0079】
また、懸濁化剤としては、例えば、カゼイン、ゼラチン、アルギン酸、カルボキシメチルセルロース、アラビアガム、ヒドロキシプロピルセルロース、ベントナイトが挙げられ、保存剤としては、例えば、サリチル酸、パラオキシ安息香酸エチル、パラオキシ安息香酸プロピル、パラオキシ安息香酸ブチルが挙げられる。
【0080】
噴射剤としては、例えば、ジメチルエーテル、クロロフルオロカーボン、炭酸ガス、LPGが挙げられ、塗膜形成剤としては、例えば、ニトロセルロース、アセチルセルロース、アセチルブチ ルセルロース、メチルセルロース等のセルロース誘導体、酢酸ビニル樹脂等のビニル系樹脂、ポリビニルアルコール、メチルポリシロキサン、オクチルメチルシクロテトラシロキサン、デカメチルシクロペンタシロキサン、ジメチルポリシロキサン、メチルフェニルポリシロキサンメチルポリシクロポリシロキサン、ジメチルシロキサン・メチル(ポリオキシエチレン)シロキサン共重合体、ジメチルシロキサン・メチル(ポリオキシエチレン・ポリオキシプロピレン)シロキサン共重合体、トリメチルシロキシケイ酸、オクタメチルシクロテトラシロキサンシリコーンポリエーテルポリマー等のシリコーン類が挙げられる。
【0081】
クリーム剤において用いられる担体としては、例えば、パラフィン、流動パラフィン、ワセリン等の炭化水素類、ジメチルシロキサン、コロイド状シリカ、ベントナイト等のケイ素化合物、エタノール、ステアリルアルコール、ラウリルアルコール、エチレングリコール、ポリエチレングリコール、グリセリン等のアルコール類、ラウリン酸、ステアリン酸等のカルボン酸類、蜜蝋、ラノリン等のエステル類等が挙げられる。さらに、液剤の製剤の際に用いられるのと同様の製剤用補助剤を適宜加えることにより目的の製剤とすることができる。
【0082】
これら製剤中の本発明の化合物の含有量は、製剤形態や適用方法等により異なるが、例えば、液剤として使用する場合、通常0.0001〜0.05重量%、より好ましくは0.0002〜0.002重量%の割合で含まれる。また、その処理量は、通常植物の葉表面積1cm2 当り本発明の化合物の量で0.5〜250 μg、好ましくは1〜100 μg相当量である。勿論、該処理量は、製剤形態や適用方法、対象とする害虫の種類や密度等により異なり、適宜上記の範囲にかかわることなく増加または減少させることもできる。
【0083】
本発明の害虫防除方法
本発明の害虫防除方法は、植物体内又は植物体の表面もしくは周辺に存在する害虫に対して本発明の化合物(A)、(B)、(C)及び(D)からなる群から選ばれた少なくとも1種を適用することにより、上記害虫による該植物体の摂食を阻害する方法である。本発明の害虫防除方法には、本発明の化合物をそのまま適用しても良いが、本発明の化合物を含む上記組成物又は害虫摂食抑制剤を使用する。
【0084】
次に、本発明を実施例を用いて具体的に説明するが、本発明がこれに限定されるものではない。
【0085】
化合物(1)は、Miyazawa, M.; Shimamura, H.; Nakamura, S.; Kameoka, H. J. Agric, Food Chem. 1995, 43, 1428-1431.に記載の方法と同様の方法により、ミカン科(Rutaceae)のハクセン(Dictamnus dasycarpus)の根皮から抽出した。
【0086】
実施例で使用した機器は、以下の通りである。
【0087】
ガスクロマトグラフィー(GC)
炎イオン化検出器(flame ionization detecotor(FID))を備えたヒューレットパッカード(Hewlett-Packard)社製5890A gas chromatographを使用した。カラムは、溶融石英(DB-5, 30m length, 0.25mm i.d.)を使用した。クロマトグラフィーのコンディションは以下の通りである。
オーブン温度は150℃から300℃に4℃/分で上昇するようにプログラムした。インジェクターと検出部の温度はそれぞれ270℃と280℃とした。スプリットインジェクションは19:1とした。ヘリウムガスのフローレートは、1.8 ml/minとした。
【0088】
EI−MS
EI−MS測定は、ガスクロマトグラフィー−質量分析(GC−MS)を使用した。GC−MSは、キャピラリーカラム(DB-5MS, 30 m length, 0.25 mm i.d.)を備えたヒューレットパッカード(Hewlett-Packard)社製5890A gas chromatograph−ヒューレットパッカード(Hewlett-Packard)社製5972A gas chromatographを使用した。クロマトグラフィーの条件は、上記のDB-5と同じとした。イオン源の温度は、230℃とし、電子エネルギーは70 eVとした。
【0089】
赤外線吸収スペクトル(IRスペクトル)
IRスペクトル測定は、日本分光株式会社製のJASCO FT/IR-470 plus fourier transform infrared spectrometerを使用した。
【0090】
NMRスペクトル
NMRスペクトル測定は、JEOL FX-500 (500.00 MHz, 1H; 125.65 MHz, 13C)spectrometerを使用した。CDCl3中のテトラメチルシラン(TMS)を標準物質として使用した。多重度は、DEPT pulse sequenceにより決定した。
【0091】
比旋光度
比旋光度の測定は、日本分光株式会社製のJASCO DIP-1000 digital polarimeterを使用した。
【0092】
実施例1(化合物(2)の合成)
化合物(2)は、(-)-フラキシネロンを以下の手順により還元して得た。(-)-フラキシネロン100mgを100%エタノール10ml中に溶かし、60mgのNaBH4を加えた。室温下に24時間攪拌下反応させた後、エバポレーターによりエタノールを留去し、得られた残渣に25mlの水を加えた。得られた混合物を30mlのCHCl3で4回で抽出した。得られたCHCl3溶液を中性になるまで水で洗浄し、エバポレーションを行い、明るい黄色のオイルを得た。得られたオイルをシリカゲルクロマトグラフィー(溶媒は、ヘキサン/酢酸エチルを9:1〜7:3に変化させた)で精製して、化合物(2)(52.4mg)を得た。化合物(2)が主生成物であり、収率は52%であった。化合物(2)の構造は、Weimin, Z.; Jean, L. W.; Kurt, H.; Rensheng, X.; Guowei, Q. Phytochemistry. 1998, 47, 7-11.に記載のスペクトルデータと比較して、(-)-7,7a-ジヒドロフラキシネロン((-)-7,7a-Dihydrofraxinellone)と決定された。
【0093】
実施例2(化合物(3)の合成)
化合物(3)は、(-)-フラキシネロンを以下の手順によりエポキシ化して得た。フラキシネロン50mgをメタノール1mlに溶解し、過酸化水素水(30%)60μl及び6N水酸化ナトリウム水溶液20μlを加えた。室温下に24時間攪拌下反応させた後、反応混合液に1N塩酸を加えてpH2に調整し、得られた混合液を10mlの水中に加えた。得られた混合液をジエチルエーテル10mlで4回抽出した。得られたエーテル溶液をエバポレーターで乾燥させると、無色の針状結晶が得られた。得られた結晶をシリカゲルクロマトグラフィー(溶媒は、ヘキサン/酢酸エチルを9:1〜7:3に変化させた)で精製して、化合物(3)(51mg)を得た。化合物(3)が主生成物であり、収率は95%(100%ee)であった。化合物(3)の構造は、Maique, W. B.; Paulo, C. V. M.; Fatima, G. F. D. S.; Joao, B. F.; Sergio, A. Z. Naturforsch. 2001, 56c, 570-574.に記載のスペクトルデータと比較して、(-)-7,7a-エポキシ-フラキシネロン((-)-7,7a-epoxy-fraxinellone)と決定された。
【0094】
実施例3(生物変換の手順)
アスペルギルス・ニガー(A.niger)による生物変換は、以下の手順により行った。万能培地(Czapek pepton培地)を用いて培養されたA. nigerを培養器中に2日間保持した(28℃攪拌下)。菌体(A.niger)を、万能培地(Czapek pepton培地)(50mlのペトリディッシュに20ml)に移植し、28℃静止下で培養した。1日後、成熟した生物体及び反応出発物質である各基質(0.4mg/ml)を培地に加え、生物体を4日〜12日培養した。ペトリディッシュ中の培地に1N塩酸を加えpH2に調整し、ジエチルエーテルで抽出し、エーテル層をエバポレーションした。得られた抽出物は、TLC、GC及びGC-MSで分析した。基質及び代謝生成物の比は、ガスクロマトグラフィーのピークエリアに基づき決定した。
【0095】
実施例4(化合物(1-1)の単離)
化合物(1)(90 mg)を実施例3の手順による生物変換の後、万能培地(Czapek pepton培地)と菌体をフィルターで分離した。培地に1N塩酸を加えてpH2に調整し、NaClで飽和し、ジエチルエーテルで抽出した。菌体もジエチルエーテルで抽出した。そして、両抽出物を混合した。この抽出物をジエチルエーテルと共に分液漏斗に入れて、水相とジエチルエーテル相とに分離した。ジエチルエーテル相を、Na2SO4で乾燥し、エバポレーターにかけ、シリカゲルクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=1:1)で溶出液を分割した。化合物(1)(48.6 mg)と代謝物である化合物(1-1)(31.4 mg)を分割したヘキサン/酢酸エチル溶出液から単離した。
【0096】
化合物(1-1)((+)-(4S)-4-ヒドロキシイソフラキシネロン): オイル; [α]25D -73.0o (c 0.19, CHCl3); EI-MS: m/z (rel int %): 248([M]+,6), 230(30), 203(5), 160(15), 145(27), 107(100), 95(41); IR(film, νmaxcm-1); 3442(OH), 1755(γ-lactone). 1H, 13C NMR (表1及び2)。
【0097】
実施例5(化合物(2-1)、(2-2)及び(2-3)の単離)
化合物(2)(100 mg)を実施例4と同様の操作により、化合物(2)(53.0 mg)と代謝物である化合物(2-1)(23.2mg)、化合物(2-2)(17.8mg)及び化合物(2-3)(6.0mg)を分割したヘキサン/酢酸エチル溶出液から単離した。
【0098】
化合物(2-1)((-)-(4S)-4-ヒドロキシ-7,7a-ジヒドロフラキシネロン): 黄色オイル; [α]25D-20.1o (c 0.40, CHCl3); EI-MS: m/z (rel int %): 250([M]+,25), 222(3), 204(3), 126(100), 108(91), 95(47), 84(64), 69(36); IR(film, νmaxcm-1); 3477(OH), 1765(γ-lactone). 1H, 13C NMR(表1及び2)。
【0099】
化合物(2-2)((-)-(6R)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロン):黄色オイル; [α]25D-45.5o (c 0.34, CHCl3); EI-MS: m/z (rel int %): 250([M]+,10), 204(4), 126(10), 108(100), 93(26), 82(73), 67(28); IR(film, νmaxcm-1); 3485(OH), 1766(γ-lactone). 1H, 13C NMR(表1及び2)。
【0100】
化合物(2-3)((-)-(6S)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロン): 黄色オイル; [α]25D-25.0o (c 0.22, CHCl3); EI-MS: m/z (rel int %): 250([M]+,11), 204(30), 154(10), 124(16), 109(67), 95(37), 82(100), 67(41); IR(film, νmaxcm-1); 3423(OH), 1766(γ-lactone). 1H, 13C NMR (表1及び2)。
【0101】
実施例6(化合物(3-1)の単離)
化合物(3)(90 mg)を同様の操作により、化合物(3)(31.5 mg)と代謝物である化合物(3-1)(18.5 mg)を分割したヘキサン/酢酸エチル溶出液から単離した。
【0102】
化合物(3-1)((+)-(6R)-6-ヒドロキシ-7,7a-エポキシ-フラキシネロン): 無色針状結晶; [α]25D+ 1.0o (c 1.00, CHCl3); EI-MS: m/z (rel int %): 264([M]+,13), 140(29), 124(31), 96(100), 77(15), 43(32); IR(film, νmaxcm-1); 3273(OH), 1780(γ-lactone). 1H, 13C NMR (表1及び2)。
【0103】
【表1】
【0104】
【表2】
【0105】
実施例7(害虫摂食阻害作用の評価方法)
日産化学工業(株)より入手したハスモンヨトウ(Spodoptera litura)を使用した。ハスモンヨトウの幼虫をナイロンメッシュスクリーンでカバーしたプラスチックケース(幅200 mm×300 mm, 高さ100 mm, 1容器当たり幼虫100個)内で飼育した。飼育環境は、25℃下、相対湿度70%、光周期:16時間を明、8時間を暗とした。市販食餌(Insecta LFS; 日本農産工業株式会社)を初齢の幼虫に与えた。実験方法は、Mallavadhani, U. V.; Mahapatra, A.; Raja, S. S.; Manjula, C. J. Agric, Food Chem. 2003, 51, 1952-1955、 Morimoto, M.; Kumeda, S.; Komai, K. J. Agric, Food Chem. 2000, 48, 1888-1891及びKumari, G. N. K.; Balachandran, J.; Aravind, S.; Ganesh, M. R. J. Agric, Food Chem. 2003, 51, 1555-1559に記載の方法に基づき行った。穿孔器を用いて直径2cmのリーフディスクを生のキャベツ(Brassica oleracea)の葉から準備した。2枚のディスクをアセトン溶液中の試験物質で処理し、他の2枚のディスクはコントロールとしてアセトンで処理した。4枚のディスクは同じペトリディッシュ内に交互に置いた。溶媒を完全に除去した後、10個の幼虫(三齢)を25℃、暗闇下、3〜5時間ペトリディッシュ内に置いた。モノトーンデータ変換のため、部分的に摂食されたリーフディスクをコピー用紙上にテープで貼り付けた。モノトーンデータをコピーし、エラーがないことを確認して、デジタルスキャナーを使用してデジタルデータに変換した。デジタルデータの分析は、public-domain NIH Image program (米国国立衛生研究所(U.S. National Institutes of Health)開発によってされ、インターネットを介しzippy, nimh, nih, govで匿名FTPで利用可能、又はthe National Technical Information Serice, Springfiled, Virginia, part number PB95-500195GEIによりフロッピー(登録商標)ディスクにより利用可能である)をマッキントッシュコンピューターを使用して行った。
【0106】
それぞれの実験は、摂食されていないディスクのデータファイルを測定し、処理されたディスクのものと比較して行った。摂食阻害活性パーセンテージ(%)の評価は、下式を用いて計算した。
【0107】
摂食阻害活性パーセンテージ(%)=100−(試験物質で処理されたディスクの摂食された部分(%)/コントロールディスクの摂食された部分(%))×10
試験物質として、化合物(1)、(2)、(3)、(1-1)、(2-1)、(2-2)、(2-3)、及び(3-1)を、葉面積当たり、それぞれ25、10及び5 μg/cm2使用して評価した。また、各試験物質について、ED50(μg/cm2)も測定した。
以上の結果を表3に示す。
【0108】
【表3】
【0109】
この結果から、化合物(1)、(2)、(3)、(1-1)、(2-1)、(2-2)、(2-3)、及び(3-1)はいずれも害虫摂食阻害活性が高いことがわかる。化合物(3-1)の害虫摂食阻害活性が最も高いと考えられる。
【0110】
実施例8(化合物(1)の生物変換)
アスペルギルス・ニガーによる化合物(1)の生物変換の経時変化を調査するために、アスペルギルス・ニガーと共に少量の化合物(1)を9日間培養した。得られた代謝物は、TLC、GC及びGC−MSスペクトル分析で確認した。化合物(1)から(1-1)への変換の経時変化は、TLC及びGC(ガスクロマトグラフィー)による定量測定によってモニターした。結果を図1に示す。9日後、出発基質である化合物(1)がca35 %変換されたことがわかる。
【0111】
得られた代謝物を単離するために、アスペルギルス・ニガーを用いて大スケールで9日間培養した(実施例3の手順)。生物変換の後、実施例4に記載の通り、培地を抽出し、抽出液から代謝物である化合物(1-1)を得た。スペクトルデータから、化合物(1-1)の構造を確認した(実施例9)。
【0112】
実施例9(化合物(1-1)の構造決定)
高分解能質量分析及びNMRデータから、化合物(1-1)の分子式はC14H16O4と確認された。IRスペクトルより、3442 cm-1に水酸基由来のバンドが確認され、比旋光度は(−)を示した。化合物(1-1)のプロトン及びカーボンNMRスペクトルは、新規のメチン基の出現及びC−4位のメチレン基の消失以外は、化合物(1)のものと類似していた。プロトンNMRスペクトルに関しては、H-5’(7.46 ppm)が、H-4’(6.32 ppm)とH-2’(7.42 ppm)との特徴的なカップリング定数(J=1.6 Hz)を有していた。詳細には、二次元NMR(COSY、HMQC、HMBC及びNOE)によって確認された。
【0113】
COSYスペクトルは、H-4 (3.85 ppm)とδ2.37 及びδ2.12 (それぞれHα-5 及び Hβ-5,)並びにH-6 (5.48 ppm)とδ 2.37及びδ1.97 (それぞれHα-5及びH-8) に相関クロスピークが観測された。
【0114】
プロトンNMRスペクトルは、2つのメチル基がδ 1.97及びδ 0.88 (それぞれH-8 及び H-9)に観測され、2つのメチン基がδ5.56及びδ2.86 (それぞれH-3及びH-7a)に観測された。
【0115】
HMBCスペクトル相関は、新しいメチン炭素(68.1 ppm; C-4)と1つのメチル基(0.88 ppm; H-9)及び2つのメチン基(δ5.56及びδ2.86 (それぞれH-3及びH-7a))とに観測された。
【0116】
NOE差スペクトルは、H-4 (3.85 ppm)と1つのメチン基(δ 5.56 ppm (H-3))及びH-7a (2.86 ppm)と1つのメチル基(0.88 ppm (H-9))との間にNOEを観測した。
【0117】
従って、化合物(1-1)は、化合物(1)のC-4位をヒドロキシル化して得られるものである。C-4位の第2級アルコールの絶対配置を確認するため、Kusumi, T.; Fujita, Y.; Ohtani, I.; Kakisawa, H. Tetrahedron Letters. 1991, 32, 2923-2926に記載の手法に従い、化合物(1-1)を(S)-及び(R)-MTPAエステルに変換した。改良モッシャー法(Mosher's method)により、C-4位はS-配置を有することが確認された。以上の結果から、化合物(1-1)の構造が(-)-(4S)-4-ヒドロキシイソフラキシネロンであると確認された。
【0118】
実施例10(化合物(2)の生物変換)
アスペルギルス・ニガーによる化合物(2)の生物変換の経時変化を調査するために、アスペルギルス・ニガーと共に少量の化合物(2)を11日間培養した。得られた代謝物は、TLC、GC及びGC−MSスペクトル分析で確認した。化合物(2)から(2-1)、(2-2)及び(2-3)への変換の経時変化は、TLC及びGC(ガスクロマトグラフィー)による定量測定によってモニターした。結果を図1に示す。11日後、出発基質である化合物(2)がca53 %変換された。代謝物である化合物(2-1)、(2-2)及び(2-3)は、11日後にはそれぞれca23 % 、ca18 % 及びca6 %であった。
【0119】
これらの代謝物を単離するために、アスペルギルス・ニガーを用いて大スケールで11日間培養した(実施例3の手順)。生物変換の後、実施例5に記載の通り、培地を抽出し、抽出液から代謝物である化合物(2-1)、(2-2)及び(2-3)を得た。スペクトルデータから、化合物(2-1)、(2-2)及び(2-3)の構造を確認した(実施例11)。
【0120】
実施例11(化合物(2-1)の構造決定)
高分解能質量分析及びNMRデータから、化合物(2-1)の分子式はC14H18O4と確認された。IRスペクトルより、3477 cm-1に水酸基由来のバンドが確認され、比旋光度は(−)を示した。化合物(2-1)のプロトン及びカーボンNMRスペクトルは、新規のメチン基の出現及びC−4位のメチレン基の消失以外は、化合物(2)のものと類似していた。プロトンNMRスペクトルに関しては、H-5’(7.43 ppm)が、H-4’(6.30 ppm)とH-2’(7.39 ppm)との特徴的なカップリング定数(J=1.7 Hz)を有していた。詳細には、二次元NMR(COSY、HMQC、HMBC及びNOE)によって確認された。
【0121】
COSYスペクトルは、H-4 (3.68 ppm)とδ 1.81、δ 1.77、δ 1.64及びδ 1.54 (それぞれH-5β、H-7、H-6α及びH-5α)に相関クロスピークが観測された。
【0122】
プロトンNMRは、2つのメチル基がδ 1.32及びδ 0.92 (それぞれH-8及びH-9)に観測され、メチン基がδ 5.56、δ 2.50及びδ 1.77(それぞれH-3、H-7a及びH-7)に観測された。
【0123】
HMBCスペクトル相関は、新しいメチン炭素(71.9 ppm; C-4)と1つのメチル基(0.92 ppm; H-9)及び2つのメチン基(δ 5.40及びδ 2.50(それぞれH-3及びH-7a))とに観測された。従って、化合物(2-1)は、化合物(2)のC-4位をヒドロキシル化して得られるものである。
【0124】
NOE差スペクトルにおいて、H-4とH-3との間にNOEが観測されることから、OH基の立体化学はα配置と確認した。
【0125】
以上の結果から、化合物(2-1)の構造が(-)-(4S)-4-ヒドロキシ-7,7a-ジヒドロフラキシネロンであると確認された。
【0126】
実施例12(化合物(2-2)の構造決定)
高分解能質量分析及びNMRデータから、化合物(2-2)の分子式はC14H18O4と確認された。IRスペクトルより、3485 cm-1に水酸基由来のバンドが確認され、比旋光度は(−)を示した。化合物(2-2)のプロトン及びカーボンNMRスペクトルは、新規のメチン基の出現及びC−6位のメチレン基の消失以外は、化合物(2)のものと類似していた。プロトンNMRスペクトルに関しては、H-5’(7.44 ppm)が、H-4’(6.28 ppm)とH-2’(7.38 ppm)との特徴的なカップリング定数(J=1.6 Hz)を有していた。詳細には、二次元NMR(COSY、HMQC、HMBC及びNOE)によって確認された。
【0127】
COSYスペクトルは、H-6 (3.50 ppm)とδ 1.92、δ 1.74及びδ 1.51 (それぞれH-5β、H-7及びH-5α)に相関クロスピークが観測された。
【0128】
プロトンNMRは、2つのメチル基がδ 1.43及びδ 1.00(それぞれH-8及びH-9)に観測され、メチン基がδ 4.92、δ 2.52及びδ 1.74(それぞれH-3、H-7a及びH-7)に観測された。
【0129】
HMBCスペクトル相関は、新しいメチン炭素(72.0 ppm; C-6)と1つのメチル基(1.43 ppm; H-8)及び1つのメチン基(δ 2.52(H-7a))とに観測された。従って、化合物(2-2)は、化合物(2)のC-6位をヒドロキシル化して得られるものである。
【0130】
H-7aに照射すると、H-9の面積強度が強調されることから、C-7aの立体化学はNOE差によりα配置と決定した。
【0131】
C-7メチンプロトンのアキシアル(α)配置及びC-6メチンプロトンのアキシアル(β)配置は、1H NMRスペクトルにおけるH-7a、H-8及びH-6シグナルから観測されるH-7シグナルのスプリッティングパターン(H-7; ddq, J= 4.8, 7.0, 10.7 Hz)により判断した。
【0132】
以上の結果から、化合物(2-2)の構造が(-)-(6R)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロンであると確認された。
【0133】
実施例13(化合物(2-3)の構造決定)
高分解能質量分析及びNMRデータから、化合物(2-3)の分子式はC14H18O4と確認された。IRスペクトルより、3423 cm-1に水酸基由来のバンドが確認され、比旋光度は(−)を示した。化合物(2-3)のプロトン及びカーボンNMRスペクトルは、新規のメチン基の出現及びC−6位のメチレン基の消失以外は、化合物(2)のものと類似していた。プロトンNMRスペクトルに関しては、H-5’(7.43 ppm)が、H-4’(6.28 ppm)とH-2’(7.38 ppm)との特徴的なカップリング定数(J=1.6 Hz)を有していた。詳細には、二次元NMR(COSY、HMQC、HMBC及びNOE)によって確認された。
【0134】
COSYスペクトルは、H-6 (3.88 ppm)とδ 1.94、δ1.90及びδ1.61 (それぞれH-7、Hβ-5及びHα-5)に相関クロスピークが観測された。
【0135】
プロトンNMRは、2つのメチル基がδ 1.46及びδ 0.96(それぞれH-8及びH-9)に観測され、メチン基がδ 4.99、δ 2.31及びδ 1.94(それぞれH-3、H-7a及びH-7)に観測された。
【0136】
HMBCスペクトル相関は、新しいメチン炭素(69.6 ppm; C-6)と1つのメチル基(1.46 ppm; H-8)及び1つのメチン基(δ 2.31(H-7a))とに観測された。従って、化合物(2-3)は、化合物(2)のC-6位をヒドロキシル化して得られるものである。
【0137】
H-7aに照射すると、H-9の面積強度が強調されることから、C-7aの立体化学はNOE差によりα配置と決定した。
【0138】
C-7メチンプロトンのアキシアル(α)配置及びC-6メチンプロトンのエカトリアル(α)配置は、1H NMRスペクトルにおけるH-5α、H-6、H-7a及びH-8シグナルから観測されるH-7シグナルのスプリッティングパターン(H-7; dddq, J= 2.2, 2.2, 5.2, 7.4 Hz)により判断した。
【0139】
以上の結果から、化合物(2-3)の構造が(-)-(6S)-6-ヒドロキシ-7,7a-ジヒドロフラキシネロンであると確認された。
【0140】
実施例14(化合物(3)の生物変換)
アスペルギルス・ニガーによる化合物(3)の生物変換の経時変化を調査するために、アスペルギルス・ニガーと共に少量の化合物(3)を11日間培養した。得られた代謝物は、TLC、GC及びGC−MSスペクトル分析で確認した。化合物(3)から(3-1)への変換の経時変化は、TLC及びGC(ガスクロマトグラフィー)による定量測定によってモニターした。結果を図1に示す。11日後、出発基質である化合物(3)がca37 %変換されたことがわかる。
【0141】
これらの代謝物を単離するために、アスペルギルス・ニガーを用いて大スケールで11日間培養した(実施例3の手順)。生物変換の後、実施例6に記載の通り、培地を抽出し、抽出液から代謝物である化合物(3-1)を得た。スペクトルデータから、化合物(3-1)の構造を確認した(実施例15)。
【0142】
実施例15(化合物(3-1)の構造決定)
高分解能質量分析及びNMRデータから、化合物(3-1)の分子式はC14H16O5と確認された。IRスペクトルより、3273 cm-1に水酸基由来のバンドが確認され、比旋光度は(+)を示した。化合物(3-1)のプロトン及びカーボンNMRスペクトルは、新規のメチン基の出現及びC−6位のメチレン基の消失以外は、化合物(3)のものと類似していた。プロトンNMRスペクトルに関しては、H-5’(7.47 ppm)が、H-4’(6.33 ppm)とH-2’(7.48 ppm)との特徴的なカップリング定数(J=1.7 Hz)を有していた。詳細には、二次元NMR(COSY、HMQC、HMBC及びNOE)によって確認された。
【0143】
COSYスペクトルは、H-6 (3.91 ppm)とδ 1.83、δ 1.59、δ 1.51及びδ 1.51 (それぞれHα-5、Hα-4、Hβ-5及びHβ-4)に相関クロスピークが観測された。
【0144】
プロトンNMRは、2つのメチル基がδ 1.68及びδ 0.93(それぞれH-8及びH-9)に観測され、メチン基がδ 5.19 (H-3)に観測された。
【0145】
HMBCスペクトル相関は、新しいメチン炭素(72.1 ppm; C-6)と1つのメチル基(1.68 ppm; H-8)及び2つのメチレン基(δ 1.83、δ 1.59、δ 1.51及びδ 1.51(それぞれHα-5、Hα-4、Hβ-5及びHβ-4))とに観測された。従って、化合物(3-1)は、化合物(3)のC-6位をヒドロキシル化して得られるものである。
【0146】
C-6位の第2級アルコールの絶対配置を確認するため、上述のKusumi, T.; Fujita, Y.; Ohtani, I.; Kakisawa, H. Tetrahedron Letters. 1991, 32, 2923-2926に記載の手法に従い、化合物(3-1)を(S)-及び(R)-MTPAエステル誘導体に変換した。
【0147】
化合物(3-1)の(S)-及び(R)-MTPAエステル誘導体は、Me-8に対してnegative Δδ(=δR-δS)values(- 0.15)及び Hα-5、Hβ-5及びH-4に対してそれぞれpositive values(H-5α(+ 0.11)、H-5β(+ 0.11)及びH-4(+ 0.03))を与えた。以上の改良モッシャー法(Mosher's method)により、C-6位はR-配置を有することが確認された。
【0148】
NOE差による化合物(3-1)の絶対配置の確認は、シグナルがオーバーラップしたことと、それぞれの水素に照射することの困難性により、行うことができなかった。
【0149】
以上の結果から、化合物(3-1)の構造が(+)-(6R)-6-ヒドロキシ-7,7a-エポキシ-フラキシネロンであると確認された。
【図面の簡単な説明】
【0150】
【図1】アスペルギルス・ニガーによる化合物(1)、(2)及び(3)の生物変換の経時変化を示す図である
【特許請求の範囲】
【請求項1】
一般式(A):
【化1】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する]
で表される化合物。
【請求項2】
一般式(A)において、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合する請求項1に記載の化合物。
【請求項3】
一般式(A−1):
【化2】
[式中、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合する]
で表される化合物。
【請求項4】
請求項1〜3のいずれかに記載の化合物を有効成分として含む害虫摂食抑制剤。
【請求項5】
害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である請求項4に記載の害虫摂食抑制剤。
【請求項6】
一般式(A):
【化3】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する]
で表される化合物の製造方法であって、下記一般式(B)
【化4】
で表される化合物を生物変換することを特徴とする製造方法。
【請求項7】
一般式(C):
【化5】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物。
【請求項8】
一般式(C)において、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合する請求項7に記載の化合物。
【請求項9】
一般式(C−1):
【化6】
[式中、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物。
【請求項10】
請求項7〜9のいずれかに記載の化合物を有効成分として含む害虫摂食抑制剤。
【請求項11】
害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である請求項8に記載の害虫摂食抑制剤。
【請求項12】
一般式(C):
【化7】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物の製造方法であって、下記一般式(D)
【化8】
[式中、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物を生物変換することを特徴とする製造方法。
【請求項13】
請求項1〜3及び請求項7〜9のいずれかに記載の化合物を含有する組成物。
【請求項14】
請求項13に記載の組成物を害虫に適用することを特徴とする害虫防除方法。
【請求項15】
一般式(B)
【化9】
及び一般式(D)
【化10】
[式中、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物からなる群から選ばれた少なくとも1種を有効成分として含む害虫摂食抑制剤であって、害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である害虫摂食抑制剤。
【請求項1】
一般式(A):
【化1】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する]
で表される化合物。
【請求項2】
一般式(A)において、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合する請求項1に記載の化合物。
【請求項3】
一般式(A−1):
【化2】
[式中、OH基は4位、5位又は6位のいずれか1つの炭素のみに1つ結合する]
で表される化合物。
【請求項4】
請求項1〜3のいずれかに記載の化合物を有効成分として含む害虫摂食抑制剤。
【請求項5】
害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である請求項4に記載の害虫摂食抑制剤。
【請求項6】
一般式(A):
【化3】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合する]
で表される化合物の製造方法であって、下記一般式(B)
【化4】
で表される化合物を生物変換することを特徴とする製造方法。
【請求項7】
一般式(C):
【化5】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物。
【請求項8】
一般式(C)において、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合する請求項7に記載の化合物。
【請求項9】
一般式(C−1):
【化6】
[式中、OH基が4位、5位又は6位のいずれか1つの炭素のみに1つ結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物。
【請求項10】
請求項7〜9のいずれかに記載の化合物を有効成分として含む害虫摂食抑制剤。
【請求項11】
害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である請求項8に記載の害虫摂食抑制剤。
【請求項12】
一般式(C):
【化7】
[式中、OH基は4位、5位及び6位からなる群から選ばれた少なくとも1種の炭素に結合し、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物の製造方法であって、下記一般式(D)
【化8】
[式中、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物を生物変換することを特徴とする製造方法。
【請求項13】
請求項1〜3及び請求項7〜9のいずれかに記載の化合物を含有する組成物。
【請求項14】
請求項13に記載の組成物を害虫に適用することを特徴とする害虫防除方法。
【請求項15】
一般式(B)
【化9】
及び一般式(D)
【化10】
[式中、6位と7位との炭素間結合は一重結合又は二重結合を示す。]
で表される化合物からなる群から選ばれた少なくとも1種を有効成分として含む害虫摂食抑制剤であって、害虫が、鱗翅目、アブラムシ類、ヨコバイ類、カメムシ類、コオロギ類、ハムシ類、ゾウムシ類、オサゾウムシ類、ゴミムシダマシ類、コガネムシ類、ガガンボ類、ウンカ類、バッタ類、イナゴ類からなる群から選ばれた少なくとも1種に属する昆虫である害虫摂食抑制剤。
【図1】

【公開番号】特開2009−179590(P2009−179590A)
【公開日】平成21年8月13日(2009.8.13)
【国際特許分類】
【出願番号】特願2008−19589(P2008−19589)
【出願日】平成20年1月30日(2008.1.30)
【出願人】(508031520)高砂薬業株式会社 (1)
【Fターム(参考)】
【公開日】平成21年8月13日(2009.8.13)
【国際特許分類】
【出願日】平成20年1月30日(2008.1.30)
【出願人】(508031520)高砂薬業株式会社 (1)
【Fターム(参考)】
[ Back to top ]