
新規化合物及びその製造方法、並びに有機半導体材料、及び有機半導体デバイス
【課題】電子移動度の良好な新規な化合物の提供。
【解決手段】ナフタレンの両ベンゼン環にそれぞれチオフェン環或いはセレノフェン環が結合した構造を有する化合物で、例えば、下式の反応で得られる化合物である。

該化合物は、電子移動度が良好であり、有機FETデバイスの有機半導体材料として使用される。
【解決手段】ナフタレンの両ベンゼン環にそれぞれチオフェン環或いはセレノフェン環が結合した構造を有する化合物で、例えば、下式の反応で得られる化合物である。
該化合物は、電子移動度が良好であり、有機FETデバイスの有機半導体材料として使用される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規化合物及びその製造方法、並びに有機半導体材料、及び有機半導体デバイスに関する。
【背景技術】
【0002】
近年、有機半導体材料を用いた有機ELデバイス、有機FET(電界効果トランジスタ)デバイス、有機薄膜光電変換デバイス等の薄膜デバイスが注目されており、実用化が始まっている。
【0003】
これらの薄膜デバイスに用いる有機半導体材料の基本的物性の中では、キャリアの移動度が重要である。例えば、有機ELデバイスにおいて、キャリアの移動度は、電荷の輸送効率に影響するため、高効率での発光或いは低電圧での駆動のために重要である。また、有機FETデバイスにおいて、キャリアの移動度は、スイッチング速度や駆動する装置の性能に直接影響するので、有機FETデバイスの実用化のためにも重要である。
【0004】
このような状況下、有機半導体材料として利用可能な種々の有機化合物の研究、開発が進められており、好適なキャリアの移動度を有するものとして、ベンゼン−チオフェン骨格を有する化合物について検討されている。非特許文献1には、種々のベンゼン−チオフェン骨格を有する化合物について例示されている。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Vibronic Coupling in Organic Semiconductors:The Case of Fused Polycyclic Benzene−Thiophene Structures ;Veaceslav Coropceanu,Ohyun Kwon,Brigitte Wex,Bilal R.Kaafarani,Nadine E.Gruhn,Jason C.Durivage,Douglas C.Neckers,and Jean−Luc Bredas ;Chem.Eur.J.2006,Vol.12,p2073−2080
【発明の概要】
【発明が解決しようとする課題】
【0006】
非特許文献1では、ナフタレン−チオフェン骨格を有する化合物の構造式が挙げられている。しかし、この化合物は現在のところ合成が成功していない化合物、いわば実在しない化合物の構造式を挙げているに過ぎない。これまでの有機合成化学の知見によると、ナフタレンにチオフェン環を導入することは極めて困難なためである。
【0007】
本発明の目的とするところは、ナフタレン−チオフェン骨格或いはナフタレン−セレノフェン骨格を有し、キャリアの移動度が良好な新規な化合物を提供すること、及び、この化合物の製造方法を提供すること、並びにこの化合物を含有する有機半導体材料及び有機半導体デバイスを提供することにある。
【課題を解決するための手段】
【0008】
本発明の第1の態様に係る化合物は、下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化1】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、又はフェニル基のいずれかを示す)で表されることを特徴とする。
【0009】
本発明の第2の態様に係る化合物は、下記一般式(5)、一般式(6)、一般式(7)、又は一般式(8)
【化2】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Xはハロゲン原子を示す)で表されることを特徴とする。
【0010】
本発明に係る化合物の製造方法は、
ジハロゲノジヒドロキシナフタレンと無水トリフルオロメタンスルホン酸とを反応させ、ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンを得る工程と、
前記ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンと末端アセチレン化合物とを反応させ、ジハロゲノ−ジエチニルナフタレン誘導体を得る工程と、
前記ジハロゲノ−ジエチニルナフタレン誘導体と硫化物塩或いはセレン化物塩とを反応させ、下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化3】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、或いはフェニル基のいずれかを示す)で表されるいずれかの化合物を得る工程と、
を含むことを特徴とする。
【0011】
更に、ジヒドロキシナフタレンとハロゲン化剤とを反応させ、前記ジハロゲノジヒドロキシナフタレンを得る工程を含んでいてもよい。
【0012】
また、前記ジヒドロキシナフタレンとして2,6−ジヒドロキシナフタレンを用い、前記一般式(1)或いは前記一般式(3)で表される化合物を得てもよい。
【0013】
また、前記ジヒドロキシナフタレンとして2,7−ジヒドロキシナフタレンを用い、前記一般式(2)で表される化合物を得てもよい。
【0014】
また、前記ジヒドロキシナフタレンとして1,5−ジヒドロキシナフタレンを用い、前記一般式(4)で表される化合物を得てもよい。
【0015】
また、前記ハロゲン化剤として臭素化剤或いは塩素化剤を用いることが望ましい。
【0016】
また、触媒を添加し、前記臭素化剤を複数回順次添加しながら反応させることが望ましい。
【0017】
また、前記末端アセチレン化合物としてトリメチルシリルアセチレン、フェニルアセチレン、或いは1−デシンのいずれかを用いることができる。
【0018】
また、前記ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンを極性溶媒に溶解して前記末端アセチレン化合物と反応させることが望ましい。
【0019】
また、前記極性溶媒として非プロトン性極性溶媒を用いることが望ましい。
【0020】
また、前記非プロトン性極性溶媒として、ジメチルホルムアミドを用いることが望ましい。
【0021】
更に、前記一般式(1)、前記一般式(2)、前記一般式(3)、又は、前記一般式(4)で表される化合物(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子を示す)にハロゲン化剤を添加して、下記一般式(5)、一般式(6)、一般式(7)、又は、一般式(8)
【化4】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Xはハロゲン原子を示す)で表される化合物を得る工程を含んでいてもよい。
【0022】
本発明に係る有機半導体材料は、下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化5】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、又はフェニル基のいずれかを示す)で表される化合物を少なくとも一種以上含むことを特徴とする。
【0023】
本発明に係る有機半導体デバイスは、前記有機半導体材料を含むことを特徴とする。
【発明の効果】
【0024】
本発明に係る化合物は、ナフタレン−チオフェン骨格或いはナフタレン−セレノフェン骨格を有する構造であり、π電子が存在すること、また、チオフェン環或いはセレノフェン環を有しており、硫黄原子或いはセレン原子を介した強い分子間相互作用を有することから、効果的なキャリアの移動が可能である。良好な電界移動度を有するので、有機半導体材料として利用でき、これを用いた有機半導体デバイスを構成することができる。
【0025】
また、本発明に係る化合物の製造方法によれば、ジハロゲノ−ジエチニルナフタレン誘導体を経ることで、ナフタレン−チオフェン骨格或いはナフタレン−セレノフェン骨格を有する化合物を製造することができる。
【0026】
また、ナフタレンの水素原子を選択的にハロゲン化できることから、ナフタレン−チオフェン骨格或いはナフタレン−セレノフェン骨格を有する化合物の収率を高めることができる。
【図面の簡単な説明】
【0027】
【図1】実施例にて作製したFET素子の概略構成を示す断面図(A)、及び平面図(B)である。
【図2】化合物Aを用いて作製したFET素子のVg−Id曲線(A)、及びVd−Id曲線(B)である。
【図3】化合物Bを用いて作製したFET素子のVg−Id曲線(A)、及びVd−Id曲線(B)である。
【図4】化合物Cを用いて作製したFET素子のVg−Id曲線(A)、及びVd−Id曲線(B)である。
【図5】化合物Dを用いて作製したFET素子のVg−Id曲線(A)、及びVd−Id曲線(B)である。
【発明を実施するための形態】
【0028】
以下に、本実施形態に係る新規化合物、その製造方法、有機半導体材料、及び有機半導体デバイスについて説明する。
【0029】
(新規化合物)
本実施形態の第1の態様に係る新規化合物は、一般式(1)、一般式(2)、一般式(3)、或いは、一般式(4)
【化6】
で表されるように、ナフタレンの両ベンゼン環にそれぞれチオフェン環或いはセレノフェン環が結合した化合物である。上記一般式(1)から一般式(4)中に示すZは、硫黄原子或いはセレン原子を表す。
【0030】
また、上記一般式(1)から一般式(4)中に示すRは、水素原子、アルキル基、或いはフェニル基のいずれかの置換基を表す。これらの置換基は同一の置換基が結合したものでも、異なる置換基が結合したものでもよいが、同一の置換基が結合していることが好ましい。
【0031】
アルキル基として、例えば、メチル基、エチル基、n−プロピル基、n−ペンチル基、n−ヘキシル基、n−ヘプチル基、n−オクチル基、n−ノニル基、n−デシル基、n−ウンデシル基、n−ドデシル基、n−トリデシル基、n−テトラデシル基、n−ペンタデシル基、n−ヘキサデシル基、n−ヘプタデシル基、n−オクタデシル基等の直鎖の飽和アルキル基、i−プロピル基、i−ブチル基、s−ブチル基、t−ブチル基等の分岐鎖の飽和アルキル基、シクロプロピル基、シクロブチル基等の環状の飽和アルキル基、1−プロペニル、2−プロペニル、1−ブチニル、2−ブチニル、3−ブチニル等の不飽和アルキル基が挙げられる。
【0032】
上記一般式(1)から一般式(4)に示す化合物は、π電子が存在すること、また、チオフェン環或いはセレノフェン環を有しており、硫黄原子或いはセレン原子を介した強い分子間相互作用により、効果的なキャリアの移動が可能であることから、良好な電界移動度を有し、有機半導体材料として利用可能である。
【0033】
本実施形態の第2の態様に係る新規化合物は、下記一般式(5)、一般式(6)、一般式(7)、又は、一般式(8)で表される。
【化7】
【0034】
一般式(5)から一般式(8)におけるZは、硫黄原子或いはセレン原子を表し、また、Xがハロゲン原子を示す。ハロゲン原子として、臭素或いはヨウ素が挙げられる。
【0035】
(化合物の製造方法)
続いて、上述した一般式(1)、一般式(2)、一般式(3)、及び一般式(4)に示される化合物の製造方法について段階的に説明する。
【0036】
まず、ジヒドロキシナフタレンとハロゲン化剤とを反応させ、ジハロゲノジヒドロキシナフタレンを合成する。
【0037】
ジヒドロキシナフタレンとして、ナフタレンの両ベンゼン環にそれぞれヒドロキシ基が結合したジヒドロキシナフタレンを用い、好ましくは、2,6−ジヒドロキシナフタレン、2,7−ジヒドロキシナフタレン、或いは1,5−ジヒドロキシナフタレンを用いるとよい。
【0038】
ハロゲン化剤として、臭素、N−ブロモスクシンイミド、過臭化ピリジニウムハイドロブロミド、又は、テトラアルキルアンモニウムトリブロミド等の臭素化剤、或いは、塩素、N−クロロスクシンイミド、テトラアルキルアンモニウムトリクロリド、塩化チオニル、塩化スルフリル等の塩素化剤を好適に使用することができる。
【0039】
得られたジハロゲノジヒドロキシナフタレンと無水トリフルオロメタンスルホン酸(CF3SO2−O−SO2CF3)とを反応させる。2つのヒドロキシ基がトリフルオロメタンスルフォニル基に置換され、ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンが得られる。
【0040】
得られたジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンと末端アセチレン化合物とを反応させ、ジハロゲノ−ジエチニルナフタレン誘導体を合成する。
【0041】
末端アセチレン化合物として、トリメチルシリルアセチレン(HC2Si(CH3)3)、フェニルアセチレン(C8H6)、1−デシン(C10H18)等を用いることができる。
【0042】
ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンは、極性溶媒に溶解して末端アセチレン化合物と反応させるとよい。極性溶媒により、トリフルオロメタンスルフォニル基が選択的に末端アセチレン化合物に置換されることとなる。これにより、得られるジハロゲノ−ジエチニルナフタレン誘導体の収率を高めることができる。ジハロゲノ−ジエチニルナフタレン誘導体の収率を高めることは、用いる試薬等の無駄がなくなるので、製造コストの低下につながる。
【0043】
極性溶媒として、非プロトン性極性溶媒を用いることが好ましく、例えば、ジメチルホルムアミド(DMF)、テトラヒドロフラン(THF)等が挙げられる。用いる非プロトン性極性溶媒の極性が高いほど、得られるジハロゲノ−ジエチニルナフタレン誘導体の収率が高くなるので、このなかで最も極性の高いジメチルホルムアミドを用いることがより好ましい。
【0044】
得られたジハロゲノ−ジエチニルナフタレン誘導体と硫化物塩或いはセレン化物塩とを反応させる。ハロゲン原子が硫黄原子或いはセレン原子に置換される。そして、この硫黄原子或いはセレン原子と、これらに隣接するアセチレン部とで環化して、チオフェン環或いはセレノフェン環が構成される。このようにして、上記した一般式(1)から一般式(4)に示した化合物を得ることができる。
【0045】
硫化物塩として、硫化物金属塩を用いることが好ましく、硫化物アルカリ金属塩を用いることがより好ましい。例えば、硫化ナトリウム・9水和物(Na2S・9H2O)、硫化ナトリウム・5水和物(Na2S・5H2O)、硫化ナトリウム無水物(Na2S)、水硫化ナトリウム水和物(NaSH・nH2O)等が挙げられる。
【0046】
反応に用いる硫化物塩は、ジハロゲノ−ジエチニルナフタレン誘導体に対して、通常1〜16モル使用すればよく、好ましくは2〜8モル、より好ましくは2〜5モルである。
【0047】
反応溶媒は使用しても使用しなくてもよいが、用いるジハロゲノ−ジエチニルナフタレン誘導体が固体である場合、溶媒を使用するとよい。また、沸点100℃以上の溶媒を反応混合物中に含有するのが好ましい。これにより、反応速度が向上する。
【0048】
沸点100℃以上の溶媒とは、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、及びN,N−ジメチルアセトアミド等のアミド類、エチレングリコール、プロピレングリコール、ポリエチレングリコール等のグリコール類、ジメチルスルホキシド等のスルホキシド類が挙げられる。
【0049】
上記の溶媒は、ジハロゲノ−ジエチニルナフタレン誘導体1モルに対して、0.01〜100モル、好ましくは0.1〜80モル、より好ましくは20〜50モル使用するとよい。
【0050】
反応温度は−50℃〜300℃で行うとよく、好ましくは−10℃〜250℃、より好ましくは40℃〜200℃である。
【0051】
また、触媒を添加することは必須ではないが、触媒を添加することにより反応が円滑に進行する場合は、触媒を添加するとよい。上記触媒として、銅原子、塩化銅(I)、塩化銅(II)、臭化銅(I)、臭化銅(II)、ヨウ化銅(I)、ヨウ化銅(II)等の金属ハロゲン化物が挙げられる。好ましくは銅原子、及び臭化銅(I)、臭化銅(II)等の銅ハロゲン化物である。
【0052】
また、必要に応じて公知の方法により、反応混合物から目的化合物を単離・精製してもよい。高純度の目的化合物を得るために、昇華精製、特に真空昇華精製を行うことも可能である。
【0053】
続いて、上記一般式(1)の製造方法について具体的に説明する。上記一般式(1)に示すトランス型の直線構造の化合物を合成する場合、2,6−ジヒドロキシナフタレンを用い、また、ハロゲン化剤として臭化剤を用いるとよい。一般式(1)のように直線構造の化合物を得るには、2,6−ジヒドロキシナフタレンの3位及び7位の炭素に結合している水素原子をハロゲン原子に置換する必要がある。例えば、塩素化剤を用いた場合、ジヒドロキシナフタレンの反応性の高い1位及び5位の炭素に結合する水素原子が塩素原子に置換されるが、3位及び7位の炭素に結合している水素原子は塩素原子に置換されにくい。
【0054】
臭素等の臭化剤を用いれば、後述のように1位及び5位の炭素に結合する水素原子が臭素原子に置換された後、3位及び7位の炭素に結合している水素原子が臭素原子に置換される。
【0055】
まず、臭化剤によって、反応性の高い1位及び5位の炭素に結合する水素原子が臭素原子に置換された後、更に、鉄等の触媒を添加し、臭化剤を複数回順次添加することにより、3位、及び7位の炭素に臭素原子が結合した1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンを得ることができる。
【0056】
上記手法によると、1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンを高い収率(50%以上)で得ることができる。これは、「Reaction of Tetrasulfur Tetranitride with Naphthalenols and Related Compouds」(Bull.Chem.Soc.Jpn,Vol.64,p68−73;Shuntaro Mataka,Kazufumi Takahashi,Youji Ikezaki,Taizo Hatta,Akiyoshi Torii,Masashi Tashiro)で報じられている1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンの合成の収率4%に比べて非常に高いことがわかる。
【0057】
この1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンを華状錫(フレーク状のスズ)等により還元して、1位及び5位に結合した臭素原子を水素原子に置換することにより、3,7−ジブロモ−2,6ジヒドロキシナフタレンを得ることができる。
【0058】
この3,7−ジブロモ−2,6ジヒドロキシナフタレンを用い、上述した無水トリフルオロメタンスルホン酸との反応、末端アセチレン化合物との反応、硫化物塩或いはセレン化物塩との反応の各工程を経ることで、一般式(1)に表されるトランス型の直線構造の化合物を選択的に得ることができる。また、上述のように高い収率で1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンを得ることができるので、試薬を効率的に利用することが可能であり、製造コストを安くすることも実現している。
【0059】
続いて、上記一般式(2)に示す化合物の製造方法について具体的に説明する。上記一般式(2)に示すシス型の直線構造の化合物を合成する場合、ジヒドロキシナフタレンとして、2,7−ジヒドロキシナフタレンを用い、また、ハロゲン化剤として臭化剤を用いるとよい。2,7−ジヒドロキシナフタレンと臭素等の臭化剤とを反応させると、8位の立体障害のため、三臭化物で反応が止まり、1,3,6−トリブロモ−2,7−ジヒドロキシナフタレンが得られる。これを、華状錫等で還元させることで、3,6−ジブロモ−2,7−ジヒドロキシナフタレンを得ることができる。この3,6−ジブロモ−2,7−ジヒドロキシナフタレンを用い、上述した無水トリフルオロメタンスルホン酸との反応、末端アセチレン化合物との反応、硫化物塩或いはセレン化物塩との反応の各工程を経ることで、一般式(2)に表されるシス型の直線構造の化合物を選択的に得ることができる。
【0060】
続いて、上記一般式(3)に示す化合物の製造方法について具体的に説明する。一般式(3)に示す化合物を合成する場合、ジヒドロキシナフタレンとして2,6−ジヒドロキシナフタレンを用い、塩素等の塩素化剤を用いるとよい。2,6−ジヒドロキシナフタレンと塩素化剤とを反応させることで、一段階で1,5−ジクロロ−2,6−ジヒドロキシナフタレンが得られる。この1,5−ジクロロ−2,6−ジヒドロキシナフタレンを用い、上述した無水トリフルオロメタンスルホン酸との反応、末端アセチレン化合物との反応、硫化物塩或いはセレン化物塩との反応の各工程を経ることで、一般式(3)に表される化合物を選択的に得ることができる。
【0061】
続いて、上記一般式(4)に示す化合物の製造方法について具体的に説明する。一般式(4)に示す化合物を合成する場合、ジヒドロキシナフタレンとして1,5−ジヒドロキシナフタレンを用い、臭素等の臭素化剤を用いるとよい。1,5−ジヒドロキシナフタレンと臭素化剤とを反応させることで、一段階で2,6−ジブロモ−1,5−ジヒドロキシナフタレンが得られる。この2,6−ジブロモ−1,5−ジヒドロキシナフタレンを用い、上述した無水トリフルオロメタンスルホン酸との反応、末端アセチレン化合物との反応、硫化物塩或いはセレン化物塩との反応の各工程を経ることで、一般式(4)に表される化合物を選択的に得ることができる。
【0062】
続いて、上記一般式(5)から一般式(8)で表される化合物の製造方法について説明する。
【0063】
上述のようにして得られた一般式(1)から一般式(4)で示される化合物に、ハロゲン化剤を添加する。具体的には、一般式(1)から一般式(4)においてRが水素原子で示される化合物をテトラヒドロフラン(THF)等の溶媒に溶解し、n−BuLiを添加後、これにジブロモテトラクロロエタン等のハロゲン化剤をTHFに溶解した溶液を滴下すればよい。
【0064】
n−BuLiを添加することで、硫黄或いはセレンの隣接炭素に結合している水素が引き抜かれてリチウム塩となり、これとハロゲン化剤が反応し、ハロゲン化反応が生じる。
【0065】
これにより、一般式(1)から一般式(4)においてRが水素原子で表される化合物の水素原子がハロゲン原子に置換され、析出した固体を濾取等により分離することで、一般式(5)から一般式(8)で表される化合物が得られる。
【0066】
ハロゲン化剤として、臭素化剤或いはヨウ素化剤を用いることができる。臭素化剤としては、ジブロモテトラクロロエタン、臭素、過臭化ピリジニウムハイドロブロミド、テトラアルキルアンモニウムトリブロミド等、ヨウ素化剤としては、ヨウ素、ジヨードエタン、ヨウ化パーフルオロヘキシル、テトラアルキルアンモニウムトリヨージド等を好適に使用することができる。
【0067】
n−BuLiを、一般式(1)から一般式(4)においてRが水素原子で表される化合物に対して、少なくとも2当量以上添加するとよい。一般式(1)から一般式(4)で表される化合物は、Rで示される置換基をそれぞれ2つ備えているためである。Rで示される置換基以外の部位との反応が遅い場合、また、一般式(1)から一般式(4)で表される化合物の溶解性が低い場合には、過剰のn−BuLiを加えてもよい。また、ハロゲン化剤は、添加したn−BuLi以上のモル比で加えるとよい。これらの配合比として、例えば、一般式(1)から一般式(4)で表される化合物1モルに対し、n−BuLiを3〜5モル程度、ハロゲン化剤を10モル程度添加すればよい。
【0068】
反応時間は、30分〜1時間程度でよく、30分未満であってもn−BuLiによる水素の引き抜き反応が完結する時間であればこれよりも短くて構わない。
【0069】
(有機半導体材料)
本実施形態に係る有機半導体材料は、上述した一般式(1)、一般式(2)、一般式(3)、或いは、一般式(4)に表される化合物を少なくとも1種以上含むものである。
【0070】
一般式(1)、一般式(2)、一般式(3)、及び一般式(4)に表される化合物は、いずれもナフタレン−チオフェン骨格或いはナフタレン−セレノフェン骨格を有しており、π電子が存在すること、また、チオフェン環或いはセレノフェン環を備えており、チオフェン環或いはセレノフェン環を構成する硫黄原子を介した強い分子間相互作用により、キャリアの移動が可能となる。従って、良好な電界移動度を有する有機半導体材料として利用することができる。
【0071】
有機半導体材料は、一般式(1)から一般式(4)で表される化合物一種のみ、或いはこれらの化合物を組み合わせた混合物から構成されていてもよいし、一般式(1)から一般式(4)で表される化合物の特性を阻害しない限り、他の物質を含んでいてもよい。また、既知の手法により不純物をドープして電界移動度を調整したものであってもよい。
【0072】
(有機半導体デバイス)
本実施形態に係る有機半導体デバイスは、上述した一般式(1)、一般式(2)、一般式(3)、或いは、一般式(4)で表される化合物を少なくとも1種以上含む有機半導体材料が用いられたデバイスである。この有機半導体デバイスとして、例えば、有機半導体層を有する薄膜トランジスタや、有機キャリア輸送層及び/又は発光層を有する発光デバイスが挙げられる。
【0073】
上述した本実施形態に係る有機半導体材料を使用する以外は、既知の材料及び構造を採用することができ、特に制限されない。
【0074】
有機半導体デバイスは、従来公知の種々の製造方法を用いて製造することができ、特に限定されるものではない。なお、有機半導体材料は溶解性がやや低いため、塗布法を用いることが困難な場合、真空蒸着法等によって製造することが好ましい。
【0075】
前述の有機半導体材料を用いることから、シリコンを用いた場合におけるコストを要する製造プロセスを必要とせず、有機半導体デバイスを製造することができる。
【0076】
また、有機半導体材料を用いることから、シリコンを用いたデバイスに比べ、機械的フレキシビリティに優れ、軽量である。これにより、軽量ディスプレイやスマートタグ等への応用も可能である。
【実施例1】
【0077】
一般式(1)に示した直線状の骨格を有する化合物の合成について説明する。なお、化合物の構造は、1H NMR(1H核磁気共鳴スペクトル)、EIMS(質量分析スペクトル)により決定した。使用した機器は以下の通りである。
1H NMR:JEOL Lambda 400 spectrometer
:JEOL EX−270 spectrometer
EIMS :Shimadzu QP−5050A
なお、これらの機器は後述の他の実施例においても同様に使用した。
【0078】
まず、ナフト[2,3−b:6,7−b’]ジチオフェンの合成について、段階的に説明する。
【0079】
(1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンの合成)
2,6−ジヒドロキシナフタレン(2g,12.5mol)を酢酸(60ml)に溶解し、そこへ臭素(2.6ml,50.7mol)を滴下し、還流温度下(120℃〜125℃)で反応させた。
【0080】
この段階では、2,6−ジヒドロキシナフタレンの反応性の高い1位及び5位にある水素が臭素に置換されるに留まり、1,5−ジブロモ−2,6−ジヒドロキシナフタレンが生成する。しかし、最終的に直線状の骨格を有するナフトジチオフェンを得るためには、3位及び7位の水素を臭素に置換させておく必要がある。
【0081】
このため、更に臭素(2.6ml)を5回ほど順次滴下すること、及び触媒として鉄粉(50mg,1.3mol)を加えることを行い、76時間反応させた。
【0082】
その後、室温まで冷却し、純水(50ml)を加え、析出した固体を濾取した。得られた反応混合物をアセトンで洗浄し、減圧下で乾燥させた。
【0083】
得られた粗精製物を1,4−ジオキサンから再結晶することで、無色針状結晶の1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレン(3.0g,収率51%)を得た。
【0084】
上記のように、臭素を順次滴下すること、及び、触媒として鉄粉を添加することにより、1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンを高い収率で合成することができた。
【0085】
上記反応の反応式を下記に示す。
【化8】
【0086】
得られた1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 6.18 (s, 2H, OH), 8.31 (s, 2H, ArH); EIMS (70 eV) m/z = 476 (M+).
という測定結果であった。
【0087】
(3,7−ジブロモ−2,6−ジヒドロキシナフタレンの合成)
1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレン(1.0g,2.1mmol)を酢酸(20ml)に溶解させ、華状錫(フレーク状のスズ)(499mg,4.2mmol)を加えた後、還流温度下で62時間攪拌した。
【0088】
室温まで冷却し、純水(20ml)を加え、析出した固体を濾取した。得られた反応混合物を減圧下で乾燥させることで、白色固体の3,7−ジブロモ−2,6−ジヒドロキシナフタレン(530mg,79%)を得た。
【0089】
上記反応の反応式を下記に示す。
【化9】
【0090】
得られた3,7−ジブロモ−2,6−ジヒドロキシナフタレンは、
1H-NMR (400 MHz, CDCl3) δ 5.58 (s, 2H, OH) , 7.25 (s, 2H, ArH), 7.89 (s, 2H, ArH); EIMS (70 eV) m/z = 318 (M+).
という測定結果であった。
【0091】
(3,7−ジブロモ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンの合成)
窒素雰囲気下、3,7−ジブロモ−2,6−ジヒドロキシナフタレン(636mg,2.0mmol)、ピリジン(1.0ml,12mmol)を塩化メチレン(20ml)に溶解した。
【0092】
更に、氷浴下で無水トリフルオロメタンスルホン酸(0.7ml,4.4mmol)をゆっくり加えた。室温で15時間半攪拌した後、純水(10ml)、1N塩酸(10ml)を加えた。
【0093】
溶液を塩化メチレン(20ml)で抽出した。なお、この抽出は同様の手法で3回行った。その後、有機相を飽和食塩水(20ml)で洗浄した。この洗浄は同様の手法で3回行った。
【0094】
無水硫酸マグネシウムを用いて有機相を乾燥し、ろ過した後、溶媒を減圧下で留去した。塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で得られた反応混合物を分離精製して、白色固体の3,7−ジブロモ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレン(970mg,収率84%)を得た。
【0095】
上記反応の反応式を下記に示す。
【化10】
【0096】
得られた3,7−ジブロモ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.14 (s, 2H, ArH), 8.25 (s, 2H, ArH); EIMS (70 eV) m/z = 582 (M+).
という測定結果であった。
【0097】
(2,6−ジブロモ−3,7−ビス(トリメチルシリルエチニル)ナフタレンの合成)
窒素雰囲気下で3,7−ジブロモ−2、6−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)をDMF(7ml)、ジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0098】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてトリメチルシリルアセチレン(0.28ml,2.0mmol)を加え、室温で11時間攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0099】
この反応溶液を、塩化メチレン(5ml)を用いて抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0100】
無水硫酸マグネシウムを用いて洗浄した有機相を乾燥し、ろ過した後、溶媒を減圧下で留去した。得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製し、白色固体の2,6−ジブロモ−3,7−ビス(トリメチルシリルエチニル)ナフタレン(162mg,収率34%)を得た。
【0101】
上記の反応式を以下に示す。
【化11】
【0102】
得られた2,6−ジブロモ−3,7−ビス(トリメチルシリルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.29 (s, 18H, TMS), 7.87 (s, 2H, ArH), 7.97 (s, 2H, ArH); EIMS (70 eV) m/z = 478 (M+).
という測定結果であった。
【0103】
(ナフト[2,3−b:6,7−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(101mg,0.42mmolmmol)をNMP(3ml)に懸濁させ、15分間攪拌した。
【0104】
そこに2,6−ジブロモ−3,7−ビス(トリメチルシリルエチニル)ナフタレン(50mg,0.1mmol)を加え、190℃で10時間攪拌した。
【0105】
これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取した。
【0106】
ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で得られた反応混合物を分離精製し、橙色固体のナフト[2,3−b:6,7−b’]ジチオフェン(26mg,収率100%)を得た。
【0107】
上記の反応式を下記に示す。
【化12】
【0108】
得られたナフト[2,3−b:6,7−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.43 (d, 2H, J = 5.8 Hz, ArH), 7.51 (d, 2H, J = 5.8 Hz, ArH), 8.41 (s, 2H, ArH), 8.52 (s, 2H, ArH); EIMS (70 eV) m/z = 240 (M+); mp >300 ℃
という測定結果であった。
【実施例2】
【0109】
続いて、2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェンの合成について、段階的に説明する。
【0110】
(2,6−ジブロモ−3、7−ビス(フェニルエチニル)ナフタレンの合成)
まず、前述のように合成した3,7−ジブロモ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、2,6−ジブロモ−3,7−ビス(フェニルエチニル)ナフタレンを合成する。
【0111】
窒素雰囲気下で、2,6−ジブロモ−3,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)、ジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0112】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてフェニルアセチレン(0.22ml,2.0mmol)を加え、室温で11時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0113】
この反応溶液を、塩化メチレン(5ml)を用いて抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0114】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0115】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.1)で分離精製することで、白色固体の2,6−ジブロモ−3,7−ビス(フェニルエチニル)ナフタレン(397mg,収率82%)を得た。
【0116】
上記の反応式を以下に示す。
【化13】
【0117】
得られた2,6−ジブロモ−3,7−ビス(フェニルエチニル)ナフタレンは、
1H-NMR (400 MHz, CDCl3) δ 7.39-7.41 (m, 6H, ArH), 7.62-7.64 (m, 4H, ArH) 7.97 (s, 2H, ArH), 8.07 (s, 2H, ArH); EIMS (70 eV) m/z = 486 (M+).
という測定結果であった。
【0118】
(2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(202mg,0.42mmol)をNMP(3ml)に懸濁させ、15分間攪拌した。
【0119】
そこに2,6−ジブロモ−3,7−ビス(フェニルエチニル)ナフタレン(100mg,0.2mmol)を加え、190℃で10時間攪拌した。
【0120】
これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した黄色固体(75mg,収率96%)を濾取した。
【0121】
得られた黄色固体を昇華精製し、2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェン(25mg,収率32%)を得た。
【0122】
上記の反応式を下記に示す。
【化14】
【0123】
得られた2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェンは、
EIMS (70 eV) m/z = 392 (M+)
という測定結果であった。なお、2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェンは難溶性であるため、1H NMR測定は行えなかった。
【実施例3】
【0124】
続いて、2,7−ジオクチルナフト[2,3−b:6,7−b’]ジチオフェンの合成について、段階的に説明する。
【0125】
(2,6−ジブロモ−3、7−ジ(デシン−1−イル)ナフタレンの合成)
まず、前述のように合成した2,6−ジブロモ−3,7−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、2,6−ジブロモ−3、7−ジ(デシン−1−イル)ナフタレンを合成する。
【0126】
窒素雰囲気下で、2,6−ジブロモ−3,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(493mg,1.0mmol)を、DMF(10ml)、ジイソプロピルアミン(0.42ml,3.0mmol)に溶解し、30分間脱気した。
【0127】
これに触媒としてPd(PPh3)2Cl2(70mg,0.1mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬として1−デシン(0.54ml,3.0mmol)を加え、室温で27時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0128】
この反応溶液から、塩化メチレン(10ml)を用い、有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(10ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0129】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0130】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.3)で分離精製することで、白色固体の2,6−ジブロモ−3,7−ジ(デシン−1−イル)ナフタレン(488mg,収率87%)を得た。
【0131】
上記の反応式を以下に示す。
【化15】
【0132】
得られた2,6−ジブロモ−3,7−ジ(デシン−1−イル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.89 (t, 6H, J = 7.02 Hz, CH2), 1.27-1.37 (m, 20H, CH2), 1.61-1.72 (m, 4H, CH2), 2.51 (t, 4H, J = 6.62 Hz, CH2) 7.79 (s, 2H, ArH), 7.95 (s, 2H, ArH); EIMS (70 eV) m/z = 558 (M+).
という測定結果であった。
【0133】
(2,7−ジオクチルナフト[2,3−b:6,7−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(346mg,1.44mmol)をNMP(12ml)に懸濁させ、15分間攪拌した。
【0134】
そこに2,6−ジブロモ−3,7−ジ(デシン−1−イル)ナフタレン(200mg,0.36mmol)を加え、190℃で9時間攪拌した。
【0135】
これを室温まで冷却した後、飽和塩化アンモニウム水溶液(30ml)に注ぎ、析出した固体を濾取した。
【0136】
得られた反応混合物を、塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)とクロロホルムからの再結晶により分離精製することで、黄色針状結晶の2,7−ジオクチルナフト[2,3−b:6,7−b’]ジチオフェン(130mg,収率78%)を得た。
【0137】
上記の反応式を下記に示す。
【化16】
【0138】
得られた2,7−ジオクチルナフト[2,3−b:6,7−b’]ジチオフェンは、
1H-NMR (400 MHz, CDCl3) δ 0.89 (t, 6H, J = 7.4 Hz, CH2), 1.28-1.50 (m, 20H, CH2), 1.75-1.83 (m, 4H, CH2), 2.92 (t, 4H, J = 7.4 Hz, CH2), 7.06 (s, 2H, ArH), 8.16 (s, 2H, ArH), 8.32 (s, 2H, ArH); EIMS (70 eV) m/z = 464 (M+) ; mp 269-271 °C.
という測定結果であった。
【実施例4】
【0139】
続いて、一般式(2)に表される化合物の合成例を具体的に記す。
【0140】
まず、2,7−ジフェニルナフト[2,3−b:7,6−b’]ジチオフェンの合成について、以下に段階的に説明する。
【0141】
(1,3,6−トリブロモ−2,7−ジヒドロキシナフタレンの合成)
窒素雰囲気下、2,7−ジヒドロキシナフタレン(5g,31mmol)を酢酸(150ml)に溶解した。なお、酢酸は溶媒として用いた。
【0142】
これに臭素(5.3ml,103mmol)を滴下し、還流温度下で反応させた。
【0143】
41時間後、室温まで冷却し、純水(50ml)を加えて、析出した固体を濾取した。この固体を純水で洗浄し、減圧下で乾燥し、白色固体の1,3,6−トリブロモ−2,7−ジヒドロキシナフタレン(10g,収率83%)を得た。
【0144】
上記反応式を下記に示す。
【化17】
【0145】
得られた1,3,6−トリブロモ−2,7−ジヒドロキシナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 5.88 (s, 1H, OH), 6.24 (s, 1H, OH), 7.60 (s, 1H, ArH), 7.88 (s, 1H, ArH), 7.89 (s, 1H, ArH); EIMS (70 eV) m/z = 396 (M+).
という測定結果であった。
【0146】
(3,6−ジブロモ−2,7−ジヒドロキシナフタレンの合成)
1,3,6−トリブロモ−2,7−ジヒドロキシナフタレン(5.0g,12.6mmol)を酢酸(20ml)に溶解させ、華状錫(フレーク状のスズ)(1.6g,12.6mmol)を加えた後、還流温度下で120時間攪拌した。
【0147】
室温まで冷却し、純水(100ml)を加え、析出した固体を濾取した。この固体を減圧下で乾燥し、白色固体の3,6−ジブロモ−2,7−ジヒドロキシナフタレン(3.4g,収率85%)を得た。
【0148】
上記反応の反応式を下記に示す。
【化18】
【0149】
得られた3,6−ジブロモ−2,7−ジヒドロキシナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 5.67 (s, 2H, OH), 7.24 (s, 2H, ArH), 7.87 (s, 2H, ArH); EIMS (70 eV) m/z = 318 (M+).
という測定結果であった。
【0150】
(3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレンの合成)
窒素雰囲気下、3,6−ジブロモ−2,7−ジヒドロキシナフタレン(3.0g,9.4mmol)、ピリジン(4.5ml,56mmol)を塩化メチレン(90ml)に溶解した。
【0151】
更に、氷浴下で無水トリフルオロメタンスルホン酸(3.3ml,21mmol)をゆっくり加えた。室温で4時間半攪拌した後、純水(10ml)、1N塩酸(10ml)を加えてクエンチした。
【0152】
この溶液から、塩化メチレン(20ml)を用いて有機相を抽出した。なお、この抽出は同様の手法で3回行った。その後、有機相を飽和食塩水(20ml)で洗浄した。この洗浄は同様の手法で3回行った。
【0153】
無水硫酸マグネシウムを用いて有機相を乾燥し、ろ過した後、溶媒を減圧下で留去した。塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で得られた反応混合物を分離精製して、白色固体の3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(3.3g,収率60%)を得た。
上記反応の反応式を下記に示す。
【化19】
【0154】
得られた3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレンは、
1H-NMR (400 MHz, CDCl3) δ 7.86 (s, 2H, ArH), 8.19 (s, 2H, ArH); EIMS (70 eV) m/z = 582 (M+).
という測定結果であった。
【0155】
(3,6−ジブロモ−2,7−ビス(フェニルエチニル)ナフタレンの合成)
窒素雰囲気下で、3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)、ジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0156】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてフェニルアセチレン(0.22ml,2.0mmol)を加え、室温で11時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0157】
この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0158】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0159】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.1)で分離精製することで、白色固体の3,6−ジブロモ−2,7−ビス(フェニルエチニル)ナフタレン(243mg,収率50%)を得た。
上記反応の反応式を下記に示す。
【化20】
【0160】
得られた3,6−ジブロモ−2,7−ビス(フェニルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.38-7.42 (m, 6H, ArH), 7.62-7.65 (m, 4H, ArH), 8.01 (s, 2H, ArH), 8.03 (s, 2H, ArH); EIMS (70 eV) m/z = 486 (M+).
という測定結果であった。
【0161】
(2,7−ジフェニルナフト[2,3−b:7,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(404mg,1.68mmol)をNMP(12ml)に懸濁させ、15分間攪拌した。
【0162】
そこに3,6−ジブロモ−2,7−ビス(フェニルエチニル)ナフタレン(200mg,0.4mmol)を加え、190℃で14時間攪拌した。
【0163】
これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取した。
【0164】
得られた固体を純水、エタノール、ヘキサン、塩化メチレン、熱クロロホルムで洗浄し、2,7−ジフェニルナフト[2,3−b:7,6−b’]ジチオフェン(73mg,収率45%)を得た。
上記反応の反応式を下記に示す。
【化21】
【0165】
得られた2,7−ジフェニルナフト[2,3−b:7,6−b’]ジチオフェンは、
EIMS (70 eV) m/z = 392 (M+)
という測定結果であった。なお、2,7−ジフェニルナフト[2,3−b:7,6−b’]ジチオフェンは難溶性のため、NMR測定はできなかった。
【実施例5】
【0166】
続いて、2,7−ジオクチルナフト[2,3−b:7,6−b’]ジチオフェンの合成について、以下に段階的に説明する。
(3,6−ジブロモ−2,7−ジ(デシン−1−イル)ナフタレンの合成)
【0167】
まず、前述のように合成した3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、3,6−ジブロモ−2,7−ジ(デシン−1−イル)ナフタレンを合成する。
【0168】
窒素雰囲気下で、3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)、ジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0169】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬として1−デシン(0.36ml,2.0mmol)を加え、室温で11時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0170】
この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0171】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0172】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.3)で分離精製し、白色固体の3,6−ジブロモ−2,7−ジ(デシン−1−イル)ナフタレン(444mg,収率80%)を得た。
【0173】
上記の反応式を以下に示す。
【化22】
【0174】
得られた3,6−ジブロモ−2,7−ジ(デシン−1−イル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.89 (t, 6H, J = 6.8 Hz, CH2), 1.27-1.72 (m, 24H, CH2), 2.50 (t, 4H, J = 6.9 Hz, CH2) 7.81 (s, 2H, ArH), 7.93 (s, 2H, ArH), EIMS (70 eV) m/z = 558 (M+).
という測定結果であった。
【0175】
(2,7−ジオクチルナフト[2,3−b:7,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(346mg,1.44mmol)をNMP(12ml)に懸濁させ、15分間攪拌した。
【0176】
これに3,6−ジブロモ−2,7−ジ(デシン−1−イル)ナフタレン(200mg,0.36mmol)を加え、190℃で12時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(30ml)に注ぎ、析出した固体を濾取した。
【0177】
濾取した固体を純水、エタノールで洗浄し、淡黄色固体の2,7−ジオクチルナフト[2,3−b:7,6−b’]ジチオフェン(168mg,収率100%)を得た。
【0178】
上記の反応式を下記に示す。
【化23】
【0179】
得られた2,7−ジオクチルナフト[2,3−b:7,6−b’]ジチオフェンは、
1H-NMR (400 MHz, CDCl3) δ 0.88 (t, 6H, J = 7.0 Hz, CH3), 1.28-1.81 (m, 24H, CH2), 2.92 (t, 4H, J = 7.3 Hz, CH2), 7.05 (s, 2H, ArH), 8.21 (s, 2H, ArH), 8.26 (s, 2H, ArH); EIMS (70 eV) m/z = 464 (M+)
という測定結果であった。
【実施例6】
【0180】
続いて、ナフト[2,3−b:7,6−b’]ジチオフェンの合成について、以下に段階的に説明する。
【0181】
(3,6−ジブロモ−2,7−ビス(トリメチルシリルエチニル)ナフタレンの合成)
【0182】
まず、前述のように合成した3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、3,6−ジブロモ−2,7−ビス(トリメチルシリルエチニル)ナフタレンを合成する。
【0183】
窒素雰囲気下で、3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)、ジイソプロピルアミンアミン(7ml)に溶解し、30分間脱気した。
【0184】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてトリメチルシリルアセチレン(0.22ml,2.0mmol)を加え、室温で11時間攪拌して反応させた。この反応溶液に純水(1ml)、ヘキサン(20ml)を加え、不溶性固体をスーパーハイフロセルを用いたろ過で取り除いた。
【0185】
この濾液から、ヘキサン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0186】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0187】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製し、白色固体の3,6−ジブロモ−2,7−ビス(トリメチルシリルエチニル)ナフタレン(92mg,収率19%)を得た。
【0188】
上記の反応式を以下に示す。
【化24】
【0189】
得られた3,6−ジブロモ−2,7−ビス(トリメチルシリルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.30 (s, 18H, TMS), 7.90 (s, 2H, ArH), 7.95 (s, 2H, ArH); EIMS (70 eV) m/z = 478 (M+).
という測定結果であった。
【0190】
(ナフト[2,3−b:7,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(101mg,0.42mmol)をNMP(3ml)に懸濁させ、15分間攪拌した。
【0191】
これに3,6−ジブロモ−2,7−ビス(トリメチルシリルエチニル)ナフタレン(50mg,0.10mmol)を加え、190℃で12時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取した。
【0192】
濾取した固体を純水、エタノール、ヘキサンで洗浄し、黄色固体のナフト[2,3−b:7,6−b’]ジチオフェン(73mg,収率45%)を得た。
【0193】
上記の反応式を下記に示す。
【化25】
【0194】
得られたナフト[2,3−b:7,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.43 (d, 2H, J = 5.5 Hz, ArH), 7.50 (d, 2H, J = 5.5 Hz ArH), 8.45 (s, 2H, ArH), 8.47 (s, 2H, ArH); EIMS (70 eV) m/z = 240 (M+).
という測定結果であった。
【実施例7】
【0195】
続いて、一般式(3)に表される化合物の合成例を具体的に記す。なお、化合物の構造は、1H NMR(1H核磁気共鳴スペクトル)、EIMS(質量分析スペクトル)により決定した。
【0196】
まず、ナフト[1,2−b:5,6−b’]ジチオフェンの合成について、以下に段階的に説明する。
【0197】
(1,5−ジクロロ−2,6−ジヒドロキシナフタレンの合成)
窒素雰囲気下、2,6−ジヒドロキシナフタレン(3.0g,18.7mmol)を、酢酸(90ml)に溶解した。なお、酢酸は溶媒として用いた。
【0198】
これに塩化スルフリル(3.0ml,37.5mmol)を滴下した。これを室温で5時間攪拌した後、純水(50ml)を加えて析出した固体を濾取し、減圧下で乾燥させて、白色固体の1,5−ジクロロ−2,6−ジヒドロキシナフタレン(3.3g,収率78%)を得た。
【0199】
上記反応式を下記に示す。
【化26】
【0200】
得られた1,5−ジクロロ−2,6−ジヒドロキシナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 5.79 (s, 2H, OH), 7.35 (d, 2H, J = 8.9 Hz, ArH), 7.96 (d, 2H, J = 8.9 Hz, ArH); EIMS (70 eV) m/z = 228 (M+).
という測定結果であった。
【0201】
(1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンの合成)
窒素雰囲気下、1,5−ジクロロ−2,6−ジヒドロキシナフタレン(2.3g,10mmol)及びピリジン(4.8ml,60mmol)を塩化メチレン(100ml)に溶解した。ピリジンは不要物を除去するための添加剤、また、塩化メチレンは溶媒として用いた。
【0202】
これに氷浴下で無水トリフルオロメタンスルホン酸(3.6ml,22mmol)をゆっくり加えた。これを室温で18時間攪拌した後、純水(10ml)及び1N塩酸(10ml)を加え、反応を停止した。
【0203】
この溶液を塩化メチレン(20ml)で抽出した。なお、この抽出は同様の手法で計3回行った。抽出後、有機相を飽和食塩水(20ml)で洗浄した。なお、この洗浄は同様に計3回行った。
【0204】
洗浄した有機相を、無水硫酸マグネシウムを用いて乾燥、ろ過した後、溶媒を減圧下で留去した。
【0205】
得られた反応混合物を、塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で分離精製し、白色固体の1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレン(4.9g,収率99%)を得た。
【0206】
上記の反応式を以下に示す。
【化27】
【0207】
得られた1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.68 (d, 2H, J = 9.3 Hz, ArH), 8.40 (d, 2H, J = 9.3 Hz, ArH); EIMS (70 eV) m/z = 492 (M+).
という測定結果であった。
【0208】
(1,5−ジクロロ−2,6−ビス(トリメチルシリルエチニル)ナフタレンの合成)
窒素雰囲気下で、1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレン(247mg,0.5mmol)及びトリエチルアミン(0.21ml,1.5mmol)をDMF(5ml)に溶解し、30分間脱気した。
【0209】
これに触媒としてPd(PPh3)2Cl2(35mg,0.05mmol,10mol%)及びCuI(19mg,0.1mmol,20mol%)、更に試薬としてトリメチルシリルアセチレン(0.21ml,15mmol)を加え、室温で17時間30分攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0210】
この反応溶液から、塩化メチレン(5ml)を用いて、有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0211】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0212】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製し、白色固体の1,5−ジクロロ−2,6−ビス(トリメチルシリルエチニル)ナフタレン(89mg,46%)を得た。
【0213】
上記の反応式を以下に示す。
【化28】
【0214】
得られた1,5−ジクロロ−2,6−ビス(トリメチルシリルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.31 (s, 18H, TMS), 7.61 (d, 2H, J = 8.8 Hz, ArH), 8.12 (d, 2H, J = 8.8 Hz, ArH); EIMS (70 eV) m/z = 388 (M+).
という測定結果であった。
【0215】
(ナフト[1,2−b:5,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(615mg,2.56mmol)をNMP(15ml)に懸濁させ、15分間攪拌した。
【0216】
これに、1,5−ジクロロ−2,6−ビス(トリメチルシリルエチニル)ナフタレン(250mg,0.64mmol)を加え、190℃で12時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(50ml)に注ぎ、析出した固体を濾取した。
【0217】
ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で得られた反応混合物を分離精製し、白色固体のナフト[1,2−b:5,6−b’]ジチオフェン(139mg,90%)を得た。
【0218】
上記の反応式を下記に示す。
【化29】
【0219】
得られたナフト[1,2−b:5,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.50 (d, 2H, J = 5.3 Hz, ArH), 7.54 (d, 2H, J = 5.3 Hz, ArH), 7.95 (d, 2H, J = 8.6 Hz, ArH), 8.07 (d, 2H, J = 8.6 Hz, ArH); EIMS (70 eV) m/z = 240 (M+); mp 150.4-150.8 ℃.
という測定結果であった。
【実施例8】
【0220】
続いて、2,7−ジフェニルナフト[1,2−b:5,6−b’]ジチオフェンの合成について段階的に説明する。
【0221】
(1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレンの合成)
まず、前述のように合成した1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレンを合成する。
【0222】
窒素雰囲気下で、1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレン(493mg,1.0mmol)、及びトリエチルアミン(0.42mg,3.0mmol)をDMF(10ml)に溶解し、30分間脱気した。
【0223】
これに触媒としてPd(PPh3)2Cl2(70mg,0.1mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてフェニルアセチレン(0.33ml,3.0mmol)を加え、室温で27時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0224】
この反応溶液から、塩化メチレン(10ml)を用い、有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(10ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0225】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0226】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製した後、ヘキサンで洗浄して、淡黄色固体の1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレン(180mg,収率45%)を得た。
【0227】
上記の反応式を以下に示す。
【化30】
【0228】
得られた1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.39-7.42 (m, 6H, ArH), 7.63-7.67 (m, 4H, ArH), 7.74 (d, 2H, J = 8.6 Hz, ArH), 8.25 (d, 2H, J = 8.6 Hz, ArH); EIMS (70 eV) m/z = 396 (M+).
という測定結果であった。
【0229】
(2,7−ジフェニルナフト[1,2−b:5,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(608mg,2.53mmol)をNMP(15ml)に懸濁させ、15分間攪拌した。
【0230】
これに、1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレン(250mg,0.63mmol)を加え、190℃で12時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(50ml)に注ぎ、析出した固体を濾取した。
【0231】
得られた固体を昇華精製し、2,7−ジフェニルナフト[1,2−b:5,6−b’]ジチオフェン(147mg,60%)を得た。
【0232】
上記の反応式を下記に示す。
【化31】
【0233】
得られた2,7−ジフェニルナフト[1,2−b:5,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.34-7.40 (m, 2H, ArH), 7.45-7.57 (m, 4H, ArH), 7.71 (s, 2H, ArH), 7.79-7.82 (m, 4H, ArH), 7.91 (d, 2H, J = 8.6 Hz, ArH), 8.05 (d, 2H, J = 8.6 Hz, ArH); EIMS (70 eV) m/z = 392 (M+); mp > 300 ℃.
という測定結果であった。
【実施例9】
【0234】
続いて、2,7−ジオクチルナフト[1,2−b:5,6−b’]ジチオフェンの合成について、段階的に説明する。
【0235】
(1,5−ジクロロ−2,6−ジ(デシン−1−イル)ナフタレンの合成)
まず、前述のように合成した1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、1,5−ジクロロ−2,6−ジ(デシン−1−イル)ナフタレンを合成する。
【0236】
窒素雰囲気下で、1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレン(493mg,1.0mmol)、及びトリエチルアミン(0.42mg,3.0mmol)をDMF(10ml)に溶解し、30分間脱気した。
【0237】
これに触媒としてPd(PPh3)2Cl2(70mg,0.1mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬として1−デシン(0.54ml,3.0mmol)を加え、室温で27時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0238】
この反応溶液を、塩化メチレン(10ml)を用いて抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(10ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0239】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0240】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.3)で分離精製し、白色固体の1,5−ジクロロ−2,6−ジ(デシン−1−イル)ナフタレン(408mg,収率87%)を得た。
【0241】
上記の反応式を以下に示す。
【化32】
【0242】
得られた1,5−ジクロロ−2,6−ジ(デシン−1−イル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.89 (t, 6H, J = 7.0 Hz, CH3), 1.23-1.71 (m, 24H, CH2), 2.53 (t, 4H, J = 7.0 Hz, CH2), 7.56 (d, 2H, J = 8.5 Hz, ArH), 8.13 (d, 2H, J = 8.5 Hz, ArH); EIMS (70 eV) m/z = 468 (M+).
という測定結果であった。
【0243】
(2,7−ジオクチルナフト[1,2−b:5,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(204mg,0.85mmol)をNMP(5ml)に懸濁させ、15分間攪拌した。
【0244】
これに1,5−ジクロロ−2,6−ジ(デシン−1−イル)ナフタレン(100mg,0.21mmol)を加え、190℃で13時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(30ml)に注ぎ、析出した固体を濾取した。
【0245】
ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.5)で得られた固体を分離精製し、白色固体の2,7−ジオクチルナフト[1,2−b:5,6−b’]ジチオフェン(147mg,60%)を得た。
【0246】
上記の反応式を下記に示す。
【化33】
【0247】
得られた2,7−ジオクチルナフト[1,2−b:5,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 0.88 (t, 6H, J = 6.8 Hz, CH3), 1.21-1.83 (m, 24H, CH2), 2.97 (t, 4H, J = 7.4 Hz, CH2), 7.14 (s, 2H, ArH), 7.77 (d, 2H, J = 8.6 Hz, ArH), 7.91 (d, 2H, J = 8.6 Hz, ArH); EIMS (70 eV) m/z = 464 (M+); mp 92-93 ℃.
という測定結果であった。
【実施例10】
【0248】
(ナフト[1,2−b:5,6−b’]ジセレノフェンの合成)
窒素雰囲気下、セレン(72mg,0.91mmol)をエタノール(3ml)に懸濁させ、氷浴温度下で水素化ホウ素ナトリウム(34mg,0.91mmol)を加え、40分攪拌した。
【0249】
そこにNMP(10ml)と、前述のように合成した1,5−ジクロロ−2,6−ビス(トリメチルシリルエチニル)ナフタレン(100mg,0.26mmol)を加え、190℃で12時間攪拌した。
【0250】
室温まで冷却した後、反応溶液を飽和塩化アンモニウム水溶液(50ml)に注ぎ、析出した固体を濾取した。ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で、得られた反応混合物を分離精製し、白色固体のナフト[1,2−b:5,6−b’]ジセレノフェン(70mg,収率81%)を得た。
【0251】
上記の反応式を下記に示す。
【化34】
【0252】
得られたナフト[1,2−b:5,6−b’]ジセレノフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.73 (d, 2H, J = 5.8 Hz, ArH), 7.92 (s, 4H, ArH), 8.08 (d, 2H, J = 5.9 Hz, ArH); 13C-NMR (100 MHz, CDCl3) δ 123.56, 124.40, 128.37, 128.39, 129.22, 139.95, 142.23; EIMS (70 eV) m/z = 336 (M+).
という測定結果であった。
【実施例11】
【0253】
(2,7−ジフェニルナフト[1,2−b:5,6−b’]ジセレノフェンの合成)
窒素雰囲気下、セレン(141mg,1.8mmol)をエタノール(4ml)に懸濁させ氷浴温度下で水素化ホウ素ナトリウム(68mg,1.8mmol)を加え、40分間攪拌した。
【0254】
そこにNMP(20ml)と、前述のように合成した1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレン(200mg,0.5mmol)を加え、190度で12時間攪拌した。室温まで冷却した後、反応溶液を飽和塩化アンモニウム水溶液(50mL)に注ぎ、析出した固体を濾取した。
【0255】
この固体を温度勾配熱昇華法により精製することで、淡黄色固体の2,7−ジフェニルナフト[1,2−b:5,6−b’]ジセレノフェン(66mg,収率27%)を得た。
【0256】
上記の反応式を下記に示す。
【化35】
【0257】
得られた2,7−ジフェニルナフト[1,2−b:5,6−b’]ジセレノフェンは、
EIMS (70 eV) m/z = 488 (M+).
という測定結果であった。
【実施例12】
【0258】
続いて、一般式(4)に表される化合物の合成例を具体的に記す。
【0259】
まず、ナフト[2,1−b:6,5−b’]ジチオフェンの合成について、以下に段階的に説明する。
【0260】
(2,6−ジブロモ−1,5−ジヒドロキシナフタレンの合成)
窒素雰囲気下、1,5−ジヒドロキシナフタレン(5.0g,31mmol)と少量のヨウ素を、酢酸(150ml)に溶解し、80℃まで加熱した。なお、酢酸は溶媒として用いた。
【0261】
これに臭素(3.2ml,62.4mmol)を滴下し、還流温度下で12時間反応させた。その後、室温まで冷却し、純水(50ml)を加えて析出した固体を濾取した。これを純水で洗浄し、減圧下で乾燥させて、白色固体の2,6−ジブロモ−1,5−ジヒドロキシナフタレン(8.2g,収率83%)を得た。
【0262】
上記反応式を下記に示す。
【化36】
【0263】
得られた2,6−ジブロモ−1,5−ジヒドロキシナフタレンは、
1H-NMR (400 MHz, CDCl3) δ 5.99 (s, 2H, OH), 7.39 (d, 2H, J = 9.4 Hz, ArH), 7.70 (d, 2H, J = 9.4 Hz, ArH); EIMS (70 eV) m/z = 318 (M+).
という測定結果であった。
【0264】
(2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレンの合成)
窒素雰囲気下、2,6−ジブロモ−1,5−ジヒドロキシナフタレン(3.0g,9.4mmol)及びピリジン(4.5ml,56mmol)を塩化メチレン(90ml)に溶解した。ピリジンは不要物を除去するための添加剤、また、塩化メチレンは溶媒として用いた。
【0265】
これに氷浴下で無水トリフルオロメタンスルホン酸(3.3ml,21mmol)をゆっくり加えた。これを室温で4時間30分攪拌した後、純水(10ml)及び1N塩酸(10ml)を加え、反応を停止した。
【0266】
この溶液から、塩化メチレン(20ml)を用いて有機相を抽出した。なお、この抽出は同様の手法で計3回行った。抽出後、有機相を飽和食塩水(20ml)で洗浄した。なお、この洗浄は同様に計3回行った。
【0267】
洗浄した有機相を、無水硫酸マグネシウムを用いて乾燥、ろ過した後、溶媒を減圧下で留去した。
【0268】
得られた反応混合物を、塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で分離精製し、白色固体の2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレン(3.2g,収率58%)を得た。
【0269】
上記の反応式を以下に示す。
【化37】
【0270】
得られた2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.89 (d, 2H, J = 9.2 Hz, ArH), 8.03 (d, 2H, J = 9.2 Hz, ArH); EIMS (70 eV) m/z = 582 (M+).
という測定結果であった。
【0271】
(2,6−ジブロモ−1,5−ビス(トリメチルシリルエチニル)ナフタレンの合成)
窒素雰囲気下で、2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)及びジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0272】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてトリメチルシリルアセチレン(0.28ml,2.0mmol)を加え、室温で11時間攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0273】
この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0274】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0275】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製し、白色固体の2,6−ジブロモ−1,5−ビス(トリメチルシリルエチニル)ナフタレンの粗生成物(234mg,収率49%)を得た。
【0276】
上記の反応式を以下に示す。
【化38】
【0277】
得られた2,6−ジブロモ−1,5−ビス(トリメチルシリルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.29 (s, 18H, TMS), 7.71 (d, 2H, J = 8.8 Hz, ArH), 8.14 (d, 2H, J = 8.8 Hz, ArH); EIMS (70 eV) m/z = 478 (M+).
という測定結果であった。
【0278】
(ナフト[2,1−b:6,5−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(202mg,0.84mmol)をNMP(6ml)に懸濁させ、15分間攪拌した。
【0279】
これに、2,6−ジブロモ−1,5−ビス(トリメチルシリルエチニル)ナフタレン(100mg,0.2mmol)を加え、190℃で14時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取し、ナフト[2,1−b:6,5−b’]ジチオフェンの粗生成物(62mg)を得た。
【0280】
上記の反応式を下記に示す。
【化39】
【0281】
得られたナフト[2,1−b:6,5−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.43 (d, 2H, J = 5.4 Hz, ArH), 8.05 (d, 2H, J = 5.5 Hz ArH), 8.05 (d, 2H, J = 8.9 Hz, ArH), 8.30 (d, 2H, J = 8.9 Hz, ArH); EIMS (70 eV) m/z = 240 (M+).
という測定結果であった。
【実施例13】
【0282】
続いて、2,7−ジフェニルナフト[2,1−b:6,5−b’]ジチオフェンの合成について、段階的に説明する。
【0283】
(2,6−ジブロモ−1,5−ビス(フェニルエチニル)ナフタレンの合成)
まず、前述のように合成した2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、2,6−ジブロモ−1,5−ビス(フェニルエチニル)ナフタレンを合成する。
【0284】
窒素雰囲気下で、2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)及びジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0285】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてフェニルアセチレン(0.22ml,2.0mmol)を加え、室温で11時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0286】
この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0287】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥し、ろ過した後、溶媒を減圧下で留去した。
【0288】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.1)で分離精製し、白色固体の2,6−ジブロモ−1,5−ビス(フェニルエチニル)ナフタレン(437mg,収率90%)を得た。
【0289】
上記の反応式を以下に示す。
【化40】
【0290】
得られた2,6−ジブロモ−1,5−ビス(フェニルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.42-7.44 (m, 6H, ArH), 7.69-7.72 (m, 4H, ArH), 7.79(d, 2H, J = 8.9 Hz, ArH), 8.27 (d, 2H, J = 8.9 Hz, ArH); EIMS (70 eV) m/z = 486 (M+).
という測定結果であった。
【0291】
(2,7−ジフェニルナフト[2,1−b:6,5−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(404mg,1.68mmol)をNMP(12ml)に懸濁させ、15分間攪拌した。
【0292】
これに、2,6−ジブロモ−1,5−ビス(フェニルエチニル)ナフタレン(200mg,0.4mmol)を加え、190℃で14時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取し、2,7−ジフェニルナフト[2,1−b:6,5−b’]ジチオフェンの粗生成物(192mg)を得た。
【0293】
上記の反応式を下記に示す。
【化41】
【0294】
得られた2,7−ジフェニルナフト[2,1−b:6,5−b’]ジチオフェンは、
1H-NMR (400 MHz, CDCl3) δ 7.39-7.40 (m, 2H, ArH), 7.47-7.51 (m, 4H, ArH), 7.82-7.84 (m, 4H, ArH), 8.01 (d, 2H, J = 8.6 Hz, ArH), 7.71 (s, 2H, ArH), 8.05 (d, 2H, J = 8.6 Hz, ArH); EIMS (70 eV) m/z = 392 (M+).
という測定結果であった。
【実施例14】
【0295】
続いて、2,7−ジオクチルナフト[2,1−b:6,5−b’]ジチオフェンの合成について、段階的に説明する。
【0296】
(2,6−ジブロモ−1,5−ジ(デシン−1−イル)ナフタレンの合成)
まず、前述のように合成した2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、2,6−ジブロモ−1,5−ジ(デシン−1−イル)ナフタレンを合成する。
【0297】
窒素雰囲気下で、2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)及びジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0298】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬として1−デシン(0.36ml,2.0mmol)を加え、室温で11時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0299】
この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0300】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥し、ろ過した後、溶媒を減圧下で留去した。
【0301】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製し、白色固体の2,6−ジブロモ−1,5−ジ(デシン−1−イル)ナフタレンの粗生成物(340mg,収率61%)を得た。
【0302】
上記の反応式を以下に示す。
【化42】
【0303】
得られた2,6−ジブロモ−1,5−ジ(デシン−1−イル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.89 (t, 6H, J = 7.0 Hz, CH3), 1.26-1.70 (m, 24H, CH2), 2.62 (t, 4H, J = 7.3 Hz, CH2), 7.68 (d, 2H, J = 9.4 Hz, ArH), 8.10 (d, 2H, J = 9.4 Hz, ArH); EIMS (70 eV) m/z = 558 (M+).
という測定結果であった。
【0304】
(2,7−ジオクチルナフト[2,1−b:6,5−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(404mg,1.68mmol)をNMP(12ml)に懸濁させ、15分間攪拌した。
【0305】
これに、2,6−ジブロモ−1,5−ジ(デシン−1−イル)ナフタレン(200mg,0.4mmol)を加え、190℃で14時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取し、2,7−ジオクチルナフト[2,1−b:6,5−b’]ジチオフェンの粗生成物(200mg,収率100%)を得た。
【0306】
上記の反応式を下記に示す。
【化43】
【0307】
得られた2,7−ジオクチルナフト[2,1−b:6,5−b’]ジチオフェンは、
1H-NMR (400 MHz, CDCl3) δ 0.88 (t, 6H, J = 7.0 Hz, CH3), 1.26-1.70 (m, 24H, CH2), 3.02 (t, 4H, J = 7.3 Hz, CH2), 7.68 (s, 2H, ArH), 7.89 (d, 2H, J = 8.8 Hz, ArH), 8.12 (d, 2H, J = 8.8 Hz, ArH); EIMS (70 eV) m/z = 464 (M+).
という測定結果であった。
【実施例15】
【0308】
続いて、一般式(5)で表される化合物の合成例を具体的に示す。
【0309】
(2,7−ジブロモナフト[2,3−b:6,7−b’]ジチオフェンの合成)
窒素雰囲気下、実施例1で合成したナフト[2,3−b:6,7−b’]ジチオフェン(50mg,0.21mmol)をTHF(10ml)に懸濁させた後、−78℃に冷却し、n−BuLi(0.4ml,0.63mmol,1.59M)を加えた。これを30分間攪拌した後、1,2−ジブロモ−1,1,2,2,−テトラクロロエタン(150mg,0.46mmol)のTHF(3mL)溶液を滴下した。
【0310】
これを室温まで昇温させて16時間攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。そして、析出した固体を濾取することで、2,7−ジブロモナフト[2,3−b:6,7−b’]ジチオフェン(15mg,収率18%)を得た。
【0311】
上記の反応式を下記に示す。
【化44】
【0312】
得られた2,7−ジブロモナフト[2,3−b:6,7−b’]ジチオフェンは、
1H-NMR (400 MHz, CDCl3) δ 7.43 (s, 2H, ArH), 8.22 (s, 2H, ArH), 8.31 (s, 2H, ArH);
EIMS (70 eV) m/z = 398 (M+)
という測定結果であった。
【実施例16】
【0313】
続いて、一般式(7)で表される化合物の合成例について、具体的に説明する。
(2,7−ジブロモナフト[1,2−b:5,6−b’]ジチオフェンの合成)
窒素雰囲気下、実施例7で合成したナフト[1,2−b:5,6−b’]ジチオフェン(50mg,0.21mmol)をTHF(5ml)に溶解した後、−78℃に冷却し、n−BuLi(0.4ml,0.63mmol,1.59M)を加えた。これを30分間攪拌した後、1,2−ジブロモ−1,1,2,2,−テトラクロロエタン(651mg,2mmol)のTHF(3mL)溶液を滴下した。
【0314】
これを室温まで昇温して16時間攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0315】
無水硫酸マグネシウムを用いて有機相を乾燥し、ろ過した後に、溶媒を減圧下で留去した。得られた反応混合物を塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で分離精製することで、白色固体の2,7−ジブロモナフト[1,2−b:5,6−b’]ジチオフェン(68mg,収率81%)を得た。
【0316】
上記の反応式を下記に示す。
【化45】
【0317】
得られた2,7−ジブロモナフト[1,2−b:5,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.48 (s, 2H, ArH), 7.80 (d, 2H, J = 8.5 Hz, ArH), 7.87 (d, 2H, J = 8.5 Hz, ArH); EIMS (70 eV) m/z = 398 (M+).
という測定結果であった。
【実施例17】
【0318】
(2,7−ジヨードナフト[1,2−b:5,6−b’]ジチオフェンの合成)
窒素雰囲気下、実施例7で合成したナフト[1,2−b:5,6−b’]ジチオフェン(50mg,0.21mmol)をTHF(5ml)に溶解した後、−78℃に冷却し、n−BuLi(0.4ml,0.63mmol,1.59M)を加えた。これを30分間攪拌した後、ヨウ素(117mg,0.46mmol)のTHF(3mL)溶液を滴下した。
【0319】
これを室温まで昇温させて10時間攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。そして、析出した固体を濾取することで、白色固体の2,7−ジヨードナフト[1,2−b:5,6−b’]ジチオフェン(82mg,収率80%)を得た。
【0320】
上記の反応式を下記に示す。
【化46】
【0321】
得られた2,7−ジヨードナフト[1,2−b:5,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.68 (s, 2H, ArH), 7.82 (d, 2H, J = 8.8 Hz, ArH), 7.86 (d, 2H, J = 8.8 Hz, ArH); EIMS (70 eV) m/z = 492 (M+)
という測定結果であった。
【実施例18】
【0322】
(FET特性)
実施例2で合成した2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェン(以下、化合物A)、実施例3で合成した2,7−ジオクチルナフト[2,3−b:6,7−b’]ジチオフェン(以下、化合物B)、実施例8で合成した2,7−ジフェニルナフト[1,2−b:5,6−b’]ジチオフェン(以下、化合物C)、実施例11で合成した2,7−ジフェニルナフト[1,2−b:5,6−b’]ジセレノフェン(以下、化合物D)を用いてそれぞれFET素子を作製し、FET特性を検証した。
【0323】
まず、SiO2基板を面積1cm×1cmの大きさに切り出し、裏面をフッ化水素酸で処理し、空気中で酸化されているシリカを取り除いた後、Auを真空蒸着してゲート電極を形成した。次に、SiO2基板表面上に、真空蒸着法で化合物Aの有機薄膜を形成した。なお、SiO2基板は、オクチルトリクロロシランで表面処理を施して用いた。
【0324】
形成した有機薄膜上にシャドウマスクを用いてAuを真空蒸着することでソース電極とドレイン電極とを形成した。
【0325】
作製したFET素子の概略構成を図1(図1(A)はFET素子の断面図、図1(B)はFET素子の平面図)に示す。作製したFET素子はトップコンタクト型で、チャネル長は50μm、チャネル幅は1.5mmである。
【0326】
上記と同様にして、化合物Cを用いたFET素子、及び、化合物Dを用いたFET素子をそれぞれ作成した。
【0327】
また、化合物Bを用いたFET素子は以下のようにして作成した。SiO2基板を面積1cm×1cmの大きさに切り出し、裏面をフッ化水素酸で処理し、空気中で酸化されているシリカを取り除いた後、Auを真空蒸着してゲート電極を形成した。次に、SiO2基板表面上に、0.4wt%に調製した化合物Bのクロロホルム溶液を用いてスピンコート法(有機薄膜作成条件:3000rpm,30sec)により有機薄膜を形成した。
【0328】
形成した有機薄膜上にシャドウマスクを用いてAuを真空蒸着することでソース電極とドレイン電極とを形成した。なお、FET素子の構造等については、上記他のFET素子と同様である。
【0329】
FET素子の性能は、ゲート電極に電位をかけた状態でソース電極とドレイン電極との間に電位をかけたときに流れた電流量に依存する。この電流値を測定することでFET素子の特性である電解移動度を決めることができる。電解移動度は、絶縁体としてのSiO2にゲート電圧を印加した結果、有機半導体層中に生じるキャリア種の電気的特性を表現する式(a)から求めることができる。
Id=WμCo(Vg−Vt)2/2L …(a)
【0330】
ここで、Idは飽和したソース−ドレイン電流値、Wはチャネル幅、Coはゲート電気容量、Vgはゲート電圧、Vtは閾値電圧、Lはチャネル長であり、μが決定する電解移動度(cm2/Vs)である。Coは用いたSiO2絶縁膜の誘電率、W及びLはFET素子の素子構造によって決まり、Id及びVgはFET素子の電流値の測定時に決まり、VtはId、Vgから求めることができる。式(a)に各値を代入することで、それぞれのゲート電位での電解移動度を算出することができる。なお、閾値電圧[Vt]は、Idの平方根をY軸に、VgをX軸に取り、Idの平方根が立ち上がるVg値として求めた。
【0331】
それぞれのFET素子について、p型FET特性を調べるために、負のゲート電圧をかけ、大気中にて駆動させて評価した。
【0332】
図2は、化合物Aを用いて作成したFET素子のFET特性結果である。また、図3は、化合物Bを用いて作成したFET素子のFET特性結果である。また、図4は、化合物Cを用いて作成したFET特性結果である。また、図5は、化合物Dを用いて作成したFET特性の結果である。
【0333】
ここで、図2(A)、図3(A)、図4(A)、及び図5(A)は、それぞれのFET素子のVg−Id曲線、また、図2(B)、図3(B)、図4(B)、及び図5(B)は、それぞれのFET素子のVd−Id曲線である。
【0334】
Vg−Id曲線は、アウトプット特性において、電流(Id)が飽和電流になる値になるようソース−ドレイン間の電圧(Vd)を固定した時の、ゲート電圧(Vg)と電流(Id)との関係を表す。すなわち、Vg−Id曲線は、当該FET素子のトランスファー特性(伝達特性)を示している。当該Vg−Id曲線において、off状態からon状態への立ち上がりが急なほど、スイッチング特性が良好であることを示しており、トランジスタ特性は優れていると言える。また、off電流が低ければ低いほど、on電流が高ければ高いほどon/off比が大きくなるので、良好なトランジスタであるといえる。
【0335】
一方、Vd−Id曲線は、ゲート電圧(Vg)を段階的に変化させた時の、ソース−ドレイン間の電圧(Vd)と電流(Id)との関係を表す。すなわち、Vd−Id曲線は、当該FET素子のアウトプット特性(出力特性)を示している。当該FET素子において、Vd−Id曲線が、いずれのVgにおいても、ソース−ドレイン間の高い電圧(Vd)領域で電流(Id)が飽和すること(飽和電流)、および、ソース−ドレイン間の低い電圧(Vd)領域で電流(Id)が直線的に立ち上がっていることを示せば、アウトプット特性が良く、そのFET素子は高性能であるといえる。
【0336】
図2(A)、図3(A)、図4(A)、及び図5(A)をみると、ゲート電圧(Vg)の印加により、いずれも電流(Id)が急峻に立ち上がっている。また、図2(B)図3(B)、図4(B)、及び図5(B)をみると、ソース−ドレイン間の電圧(Vd)の低い領域では、いずれもほぼ直線状に立ち上がっており、また、ソース−ドレイン間の電圧(Vd)の高い領域では、ドレイン電流が一定となっており、飽和電流が観測された。
【0337】
化合物Aを用いたFET素子では、電界移動度:0.7cm2/Vs、on/off比:106、化合物Cを用いたFET素子では、電界移動度:0.2cm2/Vs、on/off比:107、また、化合物Dを用いたFET素子では、電界移動度:0.2cm2/V、on/off比:107と良好な測定結果を得た。なお、on/off比は、それぞれのVg−Id曲線において、Vgが0〜−10V程度と小さいときをオフ状態とし、Vgが−60Vのときをオン状態として、オフ状態及びオン状態それぞれにおけるIdの値の比で表している。
【0338】
また、化合物Bを用い、塗布法にて製造したFET素子の電解移動度は10−3cm2/Vs台、また、on/off比は105と、化合物A、化合物C、及び化合物Dを用いたFET素子に比べてやや劣る結果となったものの、FET特性を備えていることから、塗布法も利用可能であることがわかる。
【0339】
このように、本実施例で合成した化合物A、化合物B、化合物C、及び化合物Dを用いたFET素子はp型トランジスタとして使用することができる。
【産業上の利用可能性】
【0340】
上記の化合物は、ナフタレンにチオフェン環或いはセレノフェン環が結合した構造であり、π電子が存在すること、また、チオフェン環或いはセレノフェン環を有しており、硫黄原子或いはセレン原子を介した強い分子間相互作用を有することから、効果的なキャリアの移動が可能である。良好な電界移動度を有するので、有機半導体材料として利用でき、これを用いた有機半導体デバイスを構成することができる。
【技術分野】
【0001】
本発明は、新規化合物及びその製造方法、並びに有機半導体材料、及び有機半導体デバイスに関する。
【背景技術】
【0002】
近年、有機半導体材料を用いた有機ELデバイス、有機FET(電界効果トランジスタ)デバイス、有機薄膜光電変換デバイス等の薄膜デバイスが注目されており、実用化が始まっている。
【0003】
これらの薄膜デバイスに用いる有機半導体材料の基本的物性の中では、キャリアの移動度が重要である。例えば、有機ELデバイスにおいて、キャリアの移動度は、電荷の輸送効率に影響するため、高効率での発光或いは低電圧での駆動のために重要である。また、有機FETデバイスにおいて、キャリアの移動度は、スイッチング速度や駆動する装置の性能に直接影響するので、有機FETデバイスの実用化のためにも重要である。
【0004】
このような状況下、有機半導体材料として利用可能な種々の有機化合物の研究、開発が進められており、好適なキャリアの移動度を有するものとして、ベンゼン−チオフェン骨格を有する化合物について検討されている。非特許文献1には、種々のベンゼン−チオフェン骨格を有する化合物について例示されている。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Vibronic Coupling in Organic Semiconductors:The Case of Fused Polycyclic Benzene−Thiophene Structures ;Veaceslav Coropceanu,Ohyun Kwon,Brigitte Wex,Bilal R.Kaafarani,Nadine E.Gruhn,Jason C.Durivage,Douglas C.Neckers,and Jean−Luc Bredas ;Chem.Eur.J.2006,Vol.12,p2073−2080
【発明の概要】
【発明が解決しようとする課題】
【0006】
非特許文献1では、ナフタレン−チオフェン骨格を有する化合物の構造式が挙げられている。しかし、この化合物は現在のところ合成が成功していない化合物、いわば実在しない化合物の構造式を挙げているに過ぎない。これまでの有機合成化学の知見によると、ナフタレンにチオフェン環を導入することは極めて困難なためである。
【0007】
本発明の目的とするところは、ナフタレン−チオフェン骨格或いはナフタレン−セレノフェン骨格を有し、キャリアの移動度が良好な新規な化合物を提供すること、及び、この化合物の製造方法を提供すること、並びにこの化合物を含有する有機半導体材料及び有機半導体デバイスを提供することにある。
【課題を解決するための手段】
【0008】
本発明の第1の態様に係る化合物は、下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化1】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、又はフェニル基のいずれかを示す)で表されることを特徴とする。
【0009】
本発明の第2の態様に係る化合物は、下記一般式(5)、一般式(6)、一般式(7)、又は一般式(8)
【化2】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Xはハロゲン原子を示す)で表されることを特徴とする。
【0010】
本発明に係る化合物の製造方法は、
ジハロゲノジヒドロキシナフタレンと無水トリフルオロメタンスルホン酸とを反応させ、ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンを得る工程と、
前記ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンと末端アセチレン化合物とを反応させ、ジハロゲノ−ジエチニルナフタレン誘導体を得る工程と、
前記ジハロゲノ−ジエチニルナフタレン誘導体と硫化物塩或いはセレン化物塩とを反応させ、下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化3】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、或いはフェニル基のいずれかを示す)で表されるいずれかの化合物を得る工程と、
を含むことを特徴とする。
【0011】
更に、ジヒドロキシナフタレンとハロゲン化剤とを反応させ、前記ジハロゲノジヒドロキシナフタレンを得る工程を含んでいてもよい。
【0012】
また、前記ジヒドロキシナフタレンとして2,6−ジヒドロキシナフタレンを用い、前記一般式(1)或いは前記一般式(3)で表される化合物を得てもよい。
【0013】
また、前記ジヒドロキシナフタレンとして2,7−ジヒドロキシナフタレンを用い、前記一般式(2)で表される化合物を得てもよい。
【0014】
また、前記ジヒドロキシナフタレンとして1,5−ジヒドロキシナフタレンを用い、前記一般式(4)で表される化合物を得てもよい。
【0015】
また、前記ハロゲン化剤として臭素化剤或いは塩素化剤を用いることが望ましい。
【0016】
また、触媒を添加し、前記臭素化剤を複数回順次添加しながら反応させることが望ましい。
【0017】
また、前記末端アセチレン化合物としてトリメチルシリルアセチレン、フェニルアセチレン、或いは1−デシンのいずれかを用いることができる。
【0018】
また、前記ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンを極性溶媒に溶解して前記末端アセチレン化合物と反応させることが望ましい。
【0019】
また、前記極性溶媒として非プロトン性極性溶媒を用いることが望ましい。
【0020】
また、前記非プロトン性極性溶媒として、ジメチルホルムアミドを用いることが望ましい。
【0021】
更に、前記一般式(1)、前記一般式(2)、前記一般式(3)、又は、前記一般式(4)で表される化合物(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子を示す)にハロゲン化剤を添加して、下記一般式(5)、一般式(6)、一般式(7)、又は、一般式(8)
【化4】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Xはハロゲン原子を示す)で表される化合物を得る工程を含んでいてもよい。
【0022】
本発明に係る有機半導体材料は、下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化5】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、又はフェニル基のいずれかを示す)で表される化合物を少なくとも一種以上含むことを特徴とする。
【0023】
本発明に係る有機半導体デバイスは、前記有機半導体材料を含むことを特徴とする。
【発明の効果】
【0024】
本発明に係る化合物は、ナフタレン−チオフェン骨格或いはナフタレン−セレノフェン骨格を有する構造であり、π電子が存在すること、また、チオフェン環或いはセレノフェン環を有しており、硫黄原子或いはセレン原子を介した強い分子間相互作用を有することから、効果的なキャリアの移動が可能である。良好な電界移動度を有するので、有機半導体材料として利用でき、これを用いた有機半導体デバイスを構成することができる。
【0025】
また、本発明に係る化合物の製造方法によれば、ジハロゲノ−ジエチニルナフタレン誘導体を経ることで、ナフタレン−チオフェン骨格或いはナフタレン−セレノフェン骨格を有する化合物を製造することができる。
【0026】
また、ナフタレンの水素原子を選択的にハロゲン化できることから、ナフタレン−チオフェン骨格或いはナフタレン−セレノフェン骨格を有する化合物の収率を高めることができる。
【図面の簡単な説明】
【0027】
【図1】実施例にて作製したFET素子の概略構成を示す断面図(A)、及び平面図(B)である。
【図2】化合物Aを用いて作製したFET素子のVg−Id曲線(A)、及びVd−Id曲線(B)である。
【図3】化合物Bを用いて作製したFET素子のVg−Id曲線(A)、及びVd−Id曲線(B)である。
【図4】化合物Cを用いて作製したFET素子のVg−Id曲線(A)、及びVd−Id曲線(B)である。
【図5】化合物Dを用いて作製したFET素子のVg−Id曲線(A)、及びVd−Id曲線(B)である。
【発明を実施するための形態】
【0028】
以下に、本実施形態に係る新規化合物、その製造方法、有機半導体材料、及び有機半導体デバイスについて説明する。
【0029】
(新規化合物)
本実施形態の第1の態様に係る新規化合物は、一般式(1)、一般式(2)、一般式(3)、或いは、一般式(4)
【化6】
で表されるように、ナフタレンの両ベンゼン環にそれぞれチオフェン環或いはセレノフェン環が結合した化合物である。上記一般式(1)から一般式(4)中に示すZは、硫黄原子或いはセレン原子を表す。
【0030】
また、上記一般式(1)から一般式(4)中に示すRは、水素原子、アルキル基、或いはフェニル基のいずれかの置換基を表す。これらの置換基は同一の置換基が結合したものでも、異なる置換基が結合したものでもよいが、同一の置換基が結合していることが好ましい。
【0031】
アルキル基として、例えば、メチル基、エチル基、n−プロピル基、n−ペンチル基、n−ヘキシル基、n−ヘプチル基、n−オクチル基、n−ノニル基、n−デシル基、n−ウンデシル基、n−ドデシル基、n−トリデシル基、n−テトラデシル基、n−ペンタデシル基、n−ヘキサデシル基、n−ヘプタデシル基、n−オクタデシル基等の直鎖の飽和アルキル基、i−プロピル基、i−ブチル基、s−ブチル基、t−ブチル基等の分岐鎖の飽和アルキル基、シクロプロピル基、シクロブチル基等の環状の飽和アルキル基、1−プロペニル、2−プロペニル、1−ブチニル、2−ブチニル、3−ブチニル等の不飽和アルキル基が挙げられる。
【0032】
上記一般式(1)から一般式(4)に示す化合物は、π電子が存在すること、また、チオフェン環或いはセレノフェン環を有しており、硫黄原子或いはセレン原子を介した強い分子間相互作用により、効果的なキャリアの移動が可能であることから、良好な電界移動度を有し、有機半導体材料として利用可能である。
【0033】
本実施形態の第2の態様に係る新規化合物は、下記一般式(5)、一般式(6)、一般式(7)、又は、一般式(8)で表される。
【化7】
【0034】
一般式(5)から一般式(8)におけるZは、硫黄原子或いはセレン原子を表し、また、Xがハロゲン原子を示す。ハロゲン原子として、臭素或いはヨウ素が挙げられる。
【0035】
(化合物の製造方法)
続いて、上述した一般式(1)、一般式(2)、一般式(3)、及び一般式(4)に示される化合物の製造方法について段階的に説明する。
【0036】
まず、ジヒドロキシナフタレンとハロゲン化剤とを反応させ、ジハロゲノジヒドロキシナフタレンを合成する。
【0037】
ジヒドロキシナフタレンとして、ナフタレンの両ベンゼン環にそれぞれヒドロキシ基が結合したジヒドロキシナフタレンを用い、好ましくは、2,6−ジヒドロキシナフタレン、2,7−ジヒドロキシナフタレン、或いは1,5−ジヒドロキシナフタレンを用いるとよい。
【0038】
ハロゲン化剤として、臭素、N−ブロモスクシンイミド、過臭化ピリジニウムハイドロブロミド、又は、テトラアルキルアンモニウムトリブロミド等の臭素化剤、或いは、塩素、N−クロロスクシンイミド、テトラアルキルアンモニウムトリクロリド、塩化チオニル、塩化スルフリル等の塩素化剤を好適に使用することができる。
【0039】
得られたジハロゲノジヒドロキシナフタレンと無水トリフルオロメタンスルホン酸(CF3SO2−O−SO2CF3)とを反応させる。2つのヒドロキシ基がトリフルオロメタンスルフォニル基に置換され、ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンが得られる。
【0040】
得られたジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンと末端アセチレン化合物とを反応させ、ジハロゲノ−ジエチニルナフタレン誘導体を合成する。
【0041】
末端アセチレン化合物として、トリメチルシリルアセチレン(HC2Si(CH3)3)、フェニルアセチレン(C8H6)、1−デシン(C10H18)等を用いることができる。
【0042】
ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンは、極性溶媒に溶解して末端アセチレン化合物と反応させるとよい。極性溶媒により、トリフルオロメタンスルフォニル基が選択的に末端アセチレン化合物に置換されることとなる。これにより、得られるジハロゲノ−ジエチニルナフタレン誘導体の収率を高めることができる。ジハロゲノ−ジエチニルナフタレン誘導体の収率を高めることは、用いる試薬等の無駄がなくなるので、製造コストの低下につながる。
【0043】
極性溶媒として、非プロトン性極性溶媒を用いることが好ましく、例えば、ジメチルホルムアミド(DMF)、テトラヒドロフラン(THF)等が挙げられる。用いる非プロトン性極性溶媒の極性が高いほど、得られるジハロゲノ−ジエチニルナフタレン誘導体の収率が高くなるので、このなかで最も極性の高いジメチルホルムアミドを用いることがより好ましい。
【0044】
得られたジハロゲノ−ジエチニルナフタレン誘導体と硫化物塩或いはセレン化物塩とを反応させる。ハロゲン原子が硫黄原子或いはセレン原子に置換される。そして、この硫黄原子或いはセレン原子と、これらに隣接するアセチレン部とで環化して、チオフェン環或いはセレノフェン環が構成される。このようにして、上記した一般式(1)から一般式(4)に示した化合物を得ることができる。
【0045】
硫化物塩として、硫化物金属塩を用いることが好ましく、硫化物アルカリ金属塩を用いることがより好ましい。例えば、硫化ナトリウム・9水和物(Na2S・9H2O)、硫化ナトリウム・5水和物(Na2S・5H2O)、硫化ナトリウム無水物(Na2S)、水硫化ナトリウム水和物(NaSH・nH2O)等が挙げられる。
【0046】
反応に用いる硫化物塩は、ジハロゲノ−ジエチニルナフタレン誘導体に対して、通常1〜16モル使用すればよく、好ましくは2〜8モル、より好ましくは2〜5モルである。
【0047】
反応溶媒は使用しても使用しなくてもよいが、用いるジハロゲノ−ジエチニルナフタレン誘導体が固体である場合、溶媒を使用するとよい。また、沸点100℃以上の溶媒を反応混合物中に含有するのが好ましい。これにより、反応速度が向上する。
【0048】
沸点100℃以上の溶媒とは、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、及びN,N−ジメチルアセトアミド等のアミド類、エチレングリコール、プロピレングリコール、ポリエチレングリコール等のグリコール類、ジメチルスルホキシド等のスルホキシド類が挙げられる。
【0049】
上記の溶媒は、ジハロゲノ−ジエチニルナフタレン誘導体1モルに対して、0.01〜100モル、好ましくは0.1〜80モル、より好ましくは20〜50モル使用するとよい。
【0050】
反応温度は−50℃〜300℃で行うとよく、好ましくは−10℃〜250℃、より好ましくは40℃〜200℃である。
【0051】
また、触媒を添加することは必須ではないが、触媒を添加することにより反応が円滑に進行する場合は、触媒を添加するとよい。上記触媒として、銅原子、塩化銅(I)、塩化銅(II)、臭化銅(I)、臭化銅(II)、ヨウ化銅(I)、ヨウ化銅(II)等の金属ハロゲン化物が挙げられる。好ましくは銅原子、及び臭化銅(I)、臭化銅(II)等の銅ハロゲン化物である。
【0052】
また、必要に応じて公知の方法により、反応混合物から目的化合物を単離・精製してもよい。高純度の目的化合物を得るために、昇華精製、特に真空昇華精製を行うことも可能である。
【0053】
続いて、上記一般式(1)の製造方法について具体的に説明する。上記一般式(1)に示すトランス型の直線構造の化合物を合成する場合、2,6−ジヒドロキシナフタレンを用い、また、ハロゲン化剤として臭化剤を用いるとよい。一般式(1)のように直線構造の化合物を得るには、2,6−ジヒドロキシナフタレンの3位及び7位の炭素に結合している水素原子をハロゲン原子に置換する必要がある。例えば、塩素化剤を用いた場合、ジヒドロキシナフタレンの反応性の高い1位及び5位の炭素に結合する水素原子が塩素原子に置換されるが、3位及び7位の炭素に結合している水素原子は塩素原子に置換されにくい。
【0054】
臭素等の臭化剤を用いれば、後述のように1位及び5位の炭素に結合する水素原子が臭素原子に置換された後、3位及び7位の炭素に結合している水素原子が臭素原子に置換される。
【0055】
まず、臭化剤によって、反応性の高い1位及び5位の炭素に結合する水素原子が臭素原子に置換された後、更に、鉄等の触媒を添加し、臭化剤を複数回順次添加することにより、3位、及び7位の炭素に臭素原子が結合した1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンを得ることができる。
【0056】
上記手法によると、1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンを高い収率(50%以上)で得ることができる。これは、「Reaction of Tetrasulfur Tetranitride with Naphthalenols and Related Compouds」(Bull.Chem.Soc.Jpn,Vol.64,p68−73;Shuntaro Mataka,Kazufumi Takahashi,Youji Ikezaki,Taizo Hatta,Akiyoshi Torii,Masashi Tashiro)で報じられている1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンの合成の収率4%に比べて非常に高いことがわかる。
【0057】
この1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンを華状錫(フレーク状のスズ)等により還元して、1位及び5位に結合した臭素原子を水素原子に置換することにより、3,7−ジブロモ−2,6ジヒドロキシナフタレンを得ることができる。
【0058】
この3,7−ジブロモ−2,6ジヒドロキシナフタレンを用い、上述した無水トリフルオロメタンスルホン酸との反応、末端アセチレン化合物との反応、硫化物塩或いはセレン化物塩との反応の各工程を経ることで、一般式(1)に表されるトランス型の直線構造の化合物を選択的に得ることができる。また、上述のように高い収率で1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンを得ることができるので、試薬を効率的に利用することが可能であり、製造コストを安くすることも実現している。
【0059】
続いて、上記一般式(2)に示す化合物の製造方法について具体的に説明する。上記一般式(2)に示すシス型の直線構造の化合物を合成する場合、ジヒドロキシナフタレンとして、2,7−ジヒドロキシナフタレンを用い、また、ハロゲン化剤として臭化剤を用いるとよい。2,7−ジヒドロキシナフタレンと臭素等の臭化剤とを反応させると、8位の立体障害のため、三臭化物で反応が止まり、1,3,6−トリブロモ−2,7−ジヒドロキシナフタレンが得られる。これを、華状錫等で還元させることで、3,6−ジブロモ−2,7−ジヒドロキシナフタレンを得ることができる。この3,6−ジブロモ−2,7−ジヒドロキシナフタレンを用い、上述した無水トリフルオロメタンスルホン酸との反応、末端アセチレン化合物との反応、硫化物塩或いはセレン化物塩との反応の各工程を経ることで、一般式(2)に表されるシス型の直線構造の化合物を選択的に得ることができる。
【0060】
続いて、上記一般式(3)に示す化合物の製造方法について具体的に説明する。一般式(3)に示す化合物を合成する場合、ジヒドロキシナフタレンとして2,6−ジヒドロキシナフタレンを用い、塩素等の塩素化剤を用いるとよい。2,6−ジヒドロキシナフタレンと塩素化剤とを反応させることで、一段階で1,5−ジクロロ−2,6−ジヒドロキシナフタレンが得られる。この1,5−ジクロロ−2,6−ジヒドロキシナフタレンを用い、上述した無水トリフルオロメタンスルホン酸との反応、末端アセチレン化合物との反応、硫化物塩或いはセレン化物塩との反応の各工程を経ることで、一般式(3)に表される化合物を選択的に得ることができる。
【0061】
続いて、上記一般式(4)に示す化合物の製造方法について具体的に説明する。一般式(4)に示す化合物を合成する場合、ジヒドロキシナフタレンとして1,5−ジヒドロキシナフタレンを用い、臭素等の臭素化剤を用いるとよい。1,5−ジヒドロキシナフタレンと臭素化剤とを反応させることで、一段階で2,6−ジブロモ−1,5−ジヒドロキシナフタレンが得られる。この2,6−ジブロモ−1,5−ジヒドロキシナフタレンを用い、上述した無水トリフルオロメタンスルホン酸との反応、末端アセチレン化合物との反応、硫化物塩或いはセレン化物塩との反応の各工程を経ることで、一般式(4)に表される化合物を選択的に得ることができる。
【0062】
続いて、上記一般式(5)から一般式(8)で表される化合物の製造方法について説明する。
【0063】
上述のようにして得られた一般式(1)から一般式(4)で示される化合物に、ハロゲン化剤を添加する。具体的には、一般式(1)から一般式(4)においてRが水素原子で示される化合物をテトラヒドロフラン(THF)等の溶媒に溶解し、n−BuLiを添加後、これにジブロモテトラクロロエタン等のハロゲン化剤をTHFに溶解した溶液を滴下すればよい。
【0064】
n−BuLiを添加することで、硫黄或いはセレンの隣接炭素に結合している水素が引き抜かれてリチウム塩となり、これとハロゲン化剤が反応し、ハロゲン化反応が生じる。
【0065】
これにより、一般式(1)から一般式(4)においてRが水素原子で表される化合物の水素原子がハロゲン原子に置換され、析出した固体を濾取等により分離することで、一般式(5)から一般式(8)で表される化合物が得られる。
【0066】
ハロゲン化剤として、臭素化剤或いはヨウ素化剤を用いることができる。臭素化剤としては、ジブロモテトラクロロエタン、臭素、過臭化ピリジニウムハイドロブロミド、テトラアルキルアンモニウムトリブロミド等、ヨウ素化剤としては、ヨウ素、ジヨードエタン、ヨウ化パーフルオロヘキシル、テトラアルキルアンモニウムトリヨージド等を好適に使用することができる。
【0067】
n−BuLiを、一般式(1)から一般式(4)においてRが水素原子で表される化合物に対して、少なくとも2当量以上添加するとよい。一般式(1)から一般式(4)で表される化合物は、Rで示される置換基をそれぞれ2つ備えているためである。Rで示される置換基以外の部位との反応が遅い場合、また、一般式(1)から一般式(4)で表される化合物の溶解性が低い場合には、過剰のn−BuLiを加えてもよい。また、ハロゲン化剤は、添加したn−BuLi以上のモル比で加えるとよい。これらの配合比として、例えば、一般式(1)から一般式(4)で表される化合物1モルに対し、n−BuLiを3〜5モル程度、ハロゲン化剤を10モル程度添加すればよい。
【0068】
反応時間は、30分〜1時間程度でよく、30分未満であってもn−BuLiによる水素の引き抜き反応が完結する時間であればこれよりも短くて構わない。
【0069】
(有機半導体材料)
本実施形態に係る有機半導体材料は、上述した一般式(1)、一般式(2)、一般式(3)、或いは、一般式(4)に表される化合物を少なくとも1種以上含むものである。
【0070】
一般式(1)、一般式(2)、一般式(3)、及び一般式(4)に表される化合物は、いずれもナフタレン−チオフェン骨格或いはナフタレン−セレノフェン骨格を有しており、π電子が存在すること、また、チオフェン環或いはセレノフェン環を備えており、チオフェン環或いはセレノフェン環を構成する硫黄原子を介した強い分子間相互作用により、キャリアの移動が可能となる。従って、良好な電界移動度を有する有機半導体材料として利用することができる。
【0071】
有機半導体材料は、一般式(1)から一般式(4)で表される化合物一種のみ、或いはこれらの化合物を組み合わせた混合物から構成されていてもよいし、一般式(1)から一般式(4)で表される化合物の特性を阻害しない限り、他の物質を含んでいてもよい。また、既知の手法により不純物をドープして電界移動度を調整したものであってもよい。
【0072】
(有機半導体デバイス)
本実施形態に係る有機半導体デバイスは、上述した一般式(1)、一般式(2)、一般式(3)、或いは、一般式(4)で表される化合物を少なくとも1種以上含む有機半導体材料が用いられたデバイスである。この有機半導体デバイスとして、例えば、有機半導体層を有する薄膜トランジスタや、有機キャリア輸送層及び/又は発光層を有する発光デバイスが挙げられる。
【0073】
上述した本実施形態に係る有機半導体材料を使用する以外は、既知の材料及び構造を採用することができ、特に制限されない。
【0074】
有機半導体デバイスは、従来公知の種々の製造方法を用いて製造することができ、特に限定されるものではない。なお、有機半導体材料は溶解性がやや低いため、塗布法を用いることが困難な場合、真空蒸着法等によって製造することが好ましい。
【0075】
前述の有機半導体材料を用いることから、シリコンを用いた場合におけるコストを要する製造プロセスを必要とせず、有機半導体デバイスを製造することができる。
【0076】
また、有機半導体材料を用いることから、シリコンを用いたデバイスに比べ、機械的フレキシビリティに優れ、軽量である。これにより、軽量ディスプレイやスマートタグ等への応用も可能である。
【実施例1】
【0077】
一般式(1)に示した直線状の骨格を有する化合物の合成について説明する。なお、化合物の構造は、1H NMR(1H核磁気共鳴スペクトル)、EIMS(質量分析スペクトル)により決定した。使用した機器は以下の通りである。
1H NMR:JEOL Lambda 400 spectrometer
:JEOL EX−270 spectrometer
EIMS :Shimadzu QP−5050A
なお、これらの機器は後述の他の実施例においても同様に使用した。
【0078】
まず、ナフト[2,3−b:6,7−b’]ジチオフェンの合成について、段階的に説明する。
【0079】
(1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンの合成)
2,6−ジヒドロキシナフタレン(2g,12.5mol)を酢酸(60ml)に溶解し、そこへ臭素(2.6ml,50.7mol)を滴下し、還流温度下(120℃〜125℃)で反応させた。
【0080】
この段階では、2,6−ジヒドロキシナフタレンの反応性の高い1位及び5位にある水素が臭素に置換されるに留まり、1,5−ジブロモ−2,6−ジヒドロキシナフタレンが生成する。しかし、最終的に直線状の骨格を有するナフトジチオフェンを得るためには、3位及び7位の水素を臭素に置換させておく必要がある。
【0081】
このため、更に臭素(2.6ml)を5回ほど順次滴下すること、及び触媒として鉄粉(50mg,1.3mol)を加えることを行い、76時間反応させた。
【0082】
その後、室温まで冷却し、純水(50ml)を加え、析出した固体を濾取した。得られた反応混合物をアセトンで洗浄し、減圧下で乾燥させた。
【0083】
得られた粗精製物を1,4−ジオキサンから再結晶することで、無色針状結晶の1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレン(3.0g,収率51%)を得た。
【0084】
上記のように、臭素を順次滴下すること、及び、触媒として鉄粉を添加することにより、1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンを高い収率で合成することができた。
【0085】
上記反応の反応式を下記に示す。
【化8】
【0086】
得られた1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 6.18 (s, 2H, OH), 8.31 (s, 2H, ArH); EIMS (70 eV) m/z = 476 (M+).
という測定結果であった。
【0087】
(3,7−ジブロモ−2,6−ジヒドロキシナフタレンの合成)
1,3,5,7−テトラブロモ−2,6−ジヒドロキシナフタレン(1.0g,2.1mmol)を酢酸(20ml)に溶解させ、華状錫(フレーク状のスズ)(499mg,4.2mmol)を加えた後、還流温度下で62時間攪拌した。
【0088】
室温まで冷却し、純水(20ml)を加え、析出した固体を濾取した。得られた反応混合物を減圧下で乾燥させることで、白色固体の3,7−ジブロモ−2,6−ジヒドロキシナフタレン(530mg,79%)を得た。
【0089】
上記反応の反応式を下記に示す。
【化9】
【0090】
得られた3,7−ジブロモ−2,6−ジヒドロキシナフタレンは、
1H-NMR (400 MHz, CDCl3) δ 5.58 (s, 2H, OH) , 7.25 (s, 2H, ArH), 7.89 (s, 2H, ArH); EIMS (70 eV) m/z = 318 (M+).
という測定結果であった。
【0091】
(3,7−ジブロモ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンの合成)
窒素雰囲気下、3,7−ジブロモ−2,6−ジヒドロキシナフタレン(636mg,2.0mmol)、ピリジン(1.0ml,12mmol)を塩化メチレン(20ml)に溶解した。
【0092】
更に、氷浴下で無水トリフルオロメタンスルホン酸(0.7ml,4.4mmol)をゆっくり加えた。室温で15時間半攪拌した後、純水(10ml)、1N塩酸(10ml)を加えた。
【0093】
溶液を塩化メチレン(20ml)で抽出した。なお、この抽出は同様の手法で3回行った。その後、有機相を飽和食塩水(20ml)で洗浄した。この洗浄は同様の手法で3回行った。
【0094】
無水硫酸マグネシウムを用いて有機相を乾燥し、ろ過した後、溶媒を減圧下で留去した。塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で得られた反応混合物を分離精製して、白色固体の3,7−ジブロモ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレン(970mg,収率84%)を得た。
【0095】
上記反応の反応式を下記に示す。
【化10】
【0096】
得られた3,7−ジブロモ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.14 (s, 2H, ArH), 8.25 (s, 2H, ArH); EIMS (70 eV) m/z = 582 (M+).
という測定結果であった。
【0097】
(2,6−ジブロモ−3,7−ビス(トリメチルシリルエチニル)ナフタレンの合成)
窒素雰囲気下で3,7−ジブロモ−2、6−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)をDMF(7ml)、ジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0098】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてトリメチルシリルアセチレン(0.28ml,2.0mmol)を加え、室温で11時間攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0099】
この反応溶液を、塩化メチレン(5ml)を用いて抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0100】
無水硫酸マグネシウムを用いて洗浄した有機相を乾燥し、ろ過した後、溶媒を減圧下で留去した。得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製し、白色固体の2,6−ジブロモ−3,7−ビス(トリメチルシリルエチニル)ナフタレン(162mg,収率34%)を得た。
【0101】
上記の反応式を以下に示す。
【化11】
【0102】
得られた2,6−ジブロモ−3,7−ビス(トリメチルシリルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.29 (s, 18H, TMS), 7.87 (s, 2H, ArH), 7.97 (s, 2H, ArH); EIMS (70 eV) m/z = 478 (M+).
という測定結果であった。
【0103】
(ナフト[2,3−b:6,7−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(101mg,0.42mmolmmol)をNMP(3ml)に懸濁させ、15分間攪拌した。
【0104】
そこに2,6−ジブロモ−3,7−ビス(トリメチルシリルエチニル)ナフタレン(50mg,0.1mmol)を加え、190℃で10時間攪拌した。
【0105】
これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取した。
【0106】
ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で得られた反応混合物を分離精製し、橙色固体のナフト[2,3−b:6,7−b’]ジチオフェン(26mg,収率100%)を得た。
【0107】
上記の反応式を下記に示す。
【化12】
【0108】
得られたナフト[2,3−b:6,7−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.43 (d, 2H, J = 5.8 Hz, ArH), 7.51 (d, 2H, J = 5.8 Hz, ArH), 8.41 (s, 2H, ArH), 8.52 (s, 2H, ArH); EIMS (70 eV) m/z = 240 (M+); mp >300 ℃
という測定結果であった。
【実施例2】
【0109】
続いて、2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェンの合成について、段階的に説明する。
【0110】
(2,6−ジブロモ−3、7−ビス(フェニルエチニル)ナフタレンの合成)
まず、前述のように合成した3,7−ジブロモ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、2,6−ジブロモ−3,7−ビス(フェニルエチニル)ナフタレンを合成する。
【0111】
窒素雰囲気下で、2,6−ジブロモ−3,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)、ジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0112】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてフェニルアセチレン(0.22ml,2.0mmol)を加え、室温で11時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0113】
この反応溶液を、塩化メチレン(5ml)を用いて抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0114】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0115】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.1)で分離精製することで、白色固体の2,6−ジブロモ−3,7−ビス(フェニルエチニル)ナフタレン(397mg,収率82%)を得た。
【0116】
上記の反応式を以下に示す。
【化13】
【0117】
得られた2,6−ジブロモ−3,7−ビス(フェニルエチニル)ナフタレンは、
1H-NMR (400 MHz, CDCl3) δ 7.39-7.41 (m, 6H, ArH), 7.62-7.64 (m, 4H, ArH) 7.97 (s, 2H, ArH), 8.07 (s, 2H, ArH); EIMS (70 eV) m/z = 486 (M+).
という測定結果であった。
【0118】
(2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(202mg,0.42mmol)をNMP(3ml)に懸濁させ、15分間攪拌した。
【0119】
そこに2,6−ジブロモ−3,7−ビス(フェニルエチニル)ナフタレン(100mg,0.2mmol)を加え、190℃で10時間攪拌した。
【0120】
これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した黄色固体(75mg,収率96%)を濾取した。
【0121】
得られた黄色固体を昇華精製し、2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェン(25mg,収率32%)を得た。
【0122】
上記の反応式を下記に示す。
【化14】
【0123】
得られた2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェンは、
EIMS (70 eV) m/z = 392 (M+)
という測定結果であった。なお、2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェンは難溶性であるため、1H NMR測定は行えなかった。
【実施例3】
【0124】
続いて、2,7−ジオクチルナフト[2,3−b:6,7−b’]ジチオフェンの合成について、段階的に説明する。
【0125】
(2,6−ジブロモ−3、7−ジ(デシン−1−イル)ナフタレンの合成)
まず、前述のように合成した2,6−ジブロモ−3,7−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、2,6−ジブロモ−3、7−ジ(デシン−1−イル)ナフタレンを合成する。
【0126】
窒素雰囲気下で、2,6−ジブロモ−3,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(493mg,1.0mmol)を、DMF(10ml)、ジイソプロピルアミン(0.42ml,3.0mmol)に溶解し、30分間脱気した。
【0127】
これに触媒としてPd(PPh3)2Cl2(70mg,0.1mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬として1−デシン(0.54ml,3.0mmol)を加え、室温で27時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0128】
この反応溶液から、塩化メチレン(10ml)を用い、有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(10ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0129】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0130】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.3)で分離精製することで、白色固体の2,6−ジブロモ−3,7−ジ(デシン−1−イル)ナフタレン(488mg,収率87%)を得た。
【0131】
上記の反応式を以下に示す。
【化15】
【0132】
得られた2,6−ジブロモ−3,7−ジ(デシン−1−イル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.89 (t, 6H, J = 7.02 Hz, CH2), 1.27-1.37 (m, 20H, CH2), 1.61-1.72 (m, 4H, CH2), 2.51 (t, 4H, J = 6.62 Hz, CH2) 7.79 (s, 2H, ArH), 7.95 (s, 2H, ArH); EIMS (70 eV) m/z = 558 (M+).
という測定結果であった。
【0133】
(2,7−ジオクチルナフト[2,3−b:6,7−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(346mg,1.44mmol)をNMP(12ml)に懸濁させ、15分間攪拌した。
【0134】
そこに2,6−ジブロモ−3,7−ジ(デシン−1−イル)ナフタレン(200mg,0.36mmol)を加え、190℃で9時間攪拌した。
【0135】
これを室温まで冷却した後、飽和塩化アンモニウム水溶液(30ml)に注ぎ、析出した固体を濾取した。
【0136】
得られた反応混合物を、塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)とクロロホルムからの再結晶により分離精製することで、黄色針状結晶の2,7−ジオクチルナフト[2,3−b:6,7−b’]ジチオフェン(130mg,収率78%)を得た。
【0137】
上記の反応式を下記に示す。
【化16】
【0138】
得られた2,7−ジオクチルナフト[2,3−b:6,7−b’]ジチオフェンは、
1H-NMR (400 MHz, CDCl3) δ 0.89 (t, 6H, J = 7.4 Hz, CH2), 1.28-1.50 (m, 20H, CH2), 1.75-1.83 (m, 4H, CH2), 2.92 (t, 4H, J = 7.4 Hz, CH2), 7.06 (s, 2H, ArH), 8.16 (s, 2H, ArH), 8.32 (s, 2H, ArH); EIMS (70 eV) m/z = 464 (M+) ; mp 269-271 °C.
という測定結果であった。
【実施例4】
【0139】
続いて、一般式(2)に表される化合物の合成例を具体的に記す。
【0140】
まず、2,7−ジフェニルナフト[2,3−b:7,6−b’]ジチオフェンの合成について、以下に段階的に説明する。
【0141】
(1,3,6−トリブロモ−2,7−ジヒドロキシナフタレンの合成)
窒素雰囲気下、2,7−ジヒドロキシナフタレン(5g,31mmol)を酢酸(150ml)に溶解した。なお、酢酸は溶媒として用いた。
【0142】
これに臭素(5.3ml,103mmol)を滴下し、還流温度下で反応させた。
【0143】
41時間後、室温まで冷却し、純水(50ml)を加えて、析出した固体を濾取した。この固体を純水で洗浄し、減圧下で乾燥し、白色固体の1,3,6−トリブロモ−2,7−ジヒドロキシナフタレン(10g,収率83%)を得た。
【0144】
上記反応式を下記に示す。
【化17】
【0145】
得られた1,3,6−トリブロモ−2,7−ジヒドロキシナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 5.88 (s, 1H, OH), 6.24 (s, 1H, OH), 7.60 (s, 1H, ArH), 7.88 (s, 1H, ArH), 7.89 (s, 1H, ArH); EIMS (70 eV) m/z = 396 (M+).
という測定結果であった。
【0146】
(3,6−ジブロモ−2,7−ジヒドロキシナフタレンの合成)
1,3,6−トリブロモ−2,7−ジヒドロキシナフタレン(5.0g,12.6mmol)を酢酸(20ml)に溶解させ、華状錫(フレーク状のスズ)(1.6g,12.6mmol)を加えた後、還流温度下で120時間攪拌した。
【0147】
室温まで冷却し、純水(100ml)を加え、析出した固体を濾取した。この固体を減圧下で乾燥し、白色固体の3,6−ジブロモ−2,7−ジヒドロキシナフタレン(3.4g,収率85%)を得た。
【0148】
上記反応の反応式を下記に示す。
【化18】
【0149】
得られた3,6−ジブロモ−2,7−ジヒドロキシナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 5.67 (s, 2H, OH), 7.24 (s, 2H, ArH), 7.87 (s, 2H, ArH); EIMS (70 eV) m/z = 318 (M+).
という測定結果であった。
【0150】
(3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレンの合成)
窒素雰囲気下、3,6−ジブロモ−2,7−ジヒドロキシナフタレン(3.0g,9.4mmol)、ピリジン(4.5ml,56mmol)を塩化メチレン(90ml)に溶解した。
【0151】
更に、氷浴下で無水トリフルオロメタンスルホン酸(3.3ml,21mmol)をゆっくり加えた。室温で4時間半攪拌した後、純水(10ml)、1N塩酸(10ml)を加えてクエンチした。
【0152】
この溶液から、塩化メチレン(20ml)を用いて有機相を抽出した。なお、この抽出は同様の手法で3回行った。その後、有機相を飽和食塩水(20ml)で洗浄した。この洗浄は同様の手法で3回行った。
【0153】
無水硫酸マグネシウムを用いて有機相を乾燥し、ろ過した後、溶媒を減圧下で留去した。塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で得られた反応混合物を分離精製して、白色固体の3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(3.3g,収率60%)を得た。
上記反応の反応式を下記に示す。
【化19】
【0154】
得られた3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレンは、
1H-NMR (400 MHz, CDCl3) δ 7.86 (s, 2H, ArH), 8.19 (s, 2H, ArH); EIMS (70 eV) m/z = 582 (M+).
という測定結果であった。
【0155】
(3,6−ジブロモ−2,7−ビス(フェニルエチニル)ナフタレンの合成)
窒素雰囲気下で、3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)、ジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0156】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてフェニルアセチレン(0.22ml,2.0mmol)を加え、室温で11時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0157】
この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0158】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0159】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.1)で分離精製することで、白色固体の3,6−ジブロモ−2,7−ビス(フェニルエチニル)ナフタレン(243mg,収率50%)を得た。
上記反応の反応式を下記に示す。
【化20】
【0160】
得られた3,6−ジブロモ−2,7−ビス(フェニルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.38-7.42 (m, 6H, ArH), 7.62-7.65 (m, 4H, ArH), 8.01 (s, 2H, ArH), 8.03 (s, 2H, ArH); EIMS (70 eV) m/z = 486 (M+).
という測定結果であった。
【0161】
(2,7−ジフェニルナフト[2,3−b:7,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(404mg,1.68mmol)をNMP(12ml)に懸濁させ、15分間攪拌した。
【0162】
そこに3,6−ジブロモ−2,7−ビス(フェニルエチニル)ナフタレン(200mg,0.4mmol)を加え、190℃で14時間攪拌した。
【0163】
これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取した。
【0164】
得られた固体を純水、エタノール、ヘキサン、塩化メチレン、熱クロロホルムで洗浄し、2,7−ジフェニルナフト[2,3−b:7,6−b’]ジチオフェン(73mg,収率45%)を得た。
上記反応の反応式を下記に示す。
【化21】
【0165】
得られた2,7−ジフェニルナフト[2,3−b:7,6−b’]ジチオフェンは、
EIMS (70 eV) m/z = 392 (M+)
という測定結果であった。なお、2,7−ジフェニルナフト[2,3−b:7,6−b’]ジチオフェンは難溶性のため、NMR測定はできなかった。
【実施例5】
【0166】
続いて、2,7−ジオクチルナフト[2,3−b:7,6−b’]ジチオフェンの合成について、以下に段階的に説明する。
(3,6−ジブロモ−2,7−ジ(デシン−1−イル)ナフタレンの合成)
【0167】
まず、前述のように合成した3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、3,6−ジブロモ−2,7−ジ(デシン−1−イル)ナフタレンを合成する。
【0168】
窒素雰囲気下で、3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)、ジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0169】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬として1−デシン(0.36ml,2.0mmol)を加え、室温で11時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0170】
この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0171】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0172】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.3)で分離精製し、白色固体の3,6−ジブロモ−2,7−ジ(デシン−1−イル)ナフタレン(444mg,収率80%)を得た。
【0173】
上記の反応式を以下に示す。
【化22】
【0174】
得られた3,6−ジブロモ−2,7−ジ(デシン−1−イル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.89 (t, 6H, J = 6.8 Hz, CH2), 1.27-1.72 (m, 24H, CH2), 2.50 (t, 4H, J = 6.9 Hz, CH2) 7.81 (s, 2H, ArH), 7.93 (s, 2H, ArH), EIMS (70 eV) m/z = 558 (M+).
という測定結果であった。
【0175】
(2,7−ジオクチルナフト[2,3−b:7,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(346mg,1.44mmol)をNMP(12ml)に懸濁させ、15分間攪拌した。
【0176】
これに3,6−ジブロモ−2,7−ジ(デシン−1−イル)ナフタレン(200mg,0.36mmol)を加え、190℃で12時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(30ml)に注ぎ、析出した固体を濾取した。
【0177】
濾取した固体を純水、エタノールで洗浄し、淡黄色固体の2,7−ジオクチルナフト[2,3−b:7,6−b’]ジチオフェン(168mg,収率100%)を得た。
【0178】
上記の反応式を下記に示す。
【化23】
【0179】
得られた2,7−ジオクチルナフト[2,3−b:7,6−b’]ジチオフェンは、
1H-NMR (400 MHz, CDCl3) δ 0.88 (t, 6H, J = 7.0 Hz, CH3), 1.28-1.81 (m, 24H, CH2), 2.92 (t, 4H, J = 7.3 Hz, CH2), 7.05 (s, 2H, ArH), 8.21 (s, 2H, ArH), 8.26 (s, 2H, ArH); EIMS (70 eV) m/z = 464 (M+)
という測定結果であった。
【実施例6】
【0180】
続いて、ナフト[2,3−b:7,6−b’]ジチオフェンの合成について、以下に段階的に説明する。
【0181】
(3,6−ジブロモ−2,7−ビス(トリメチルシリルエチニル)ナフタレンの合成)
【0182】
まず、前述のように合成した3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、3,6−ジブロモ−2,7−ビス(トリメチルシリルエチニル)ナフタレンを合成する。
【0183】
窒素雰囲気下で、3,6−ジブロモ−2,7−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)、ジイソプロピルアミンアミン(7ml)に溶解し、30分間脱気した。
【0184】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてトリメチルシリルアセチレン(0.22ml,2.0mmol)を加え、室温で11時間攪拌して反応させた。この反応溶液に純水(1ml)、ヘキサン(20ml)を加え、不溶性固体をスーパーハイフロセルを用いたろ過で取り除いた。
【0185】
この濾液から、ヘキサン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0186】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0187】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製し、白色固体の3,6−ジブロモ−2,7−ビス(トリメチルシリルエチニル)ナフタレン(92mg,収率19%)を得た。
【0188】
上記の反応式を以下に示す。
【化24】
【0189】
得られた3,6−ジブロモ−2,7−ビス(トリメチルシリルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.30 (s, 18H, TMS), 7.90 (s, 2H, ArH), 7.95 (s, 2H, ArH); EIMS (70 eV) m/z = 478 (M+).
という測定結果であった。
【0190】
(ナフト[2,3−b:7,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(101mg,0.42mmol)をNMP(3ml)に懸濁させ、15分間攪拌した。
【0191】
これに3,6−ジブロモ−2,7−ビス(トリメチルシリルエチニル)ナフタレン(50mg,0.10mmol)を加え、190℃で12時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取した。
【0192】
濾取した固体を純水、エタノール、ヘキサンで洗浄し、黄色固体のナフト[2,3−b:7,6−b’]ジチオフェン(73mg,収率45%)を得た。
【0193】
上記の反応式を下記に示す。
【化25】
【0194】
得られたナフト[2,3−b:7,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.43 (d, 2H, J = 5.5 Hz, ArH), 7.50 (d, 2H, J = 5.5 Hz ArH), 8.45 (s, 2H, ArH), 8.47 (s, 2H, ArH); EIMS (70 eV) m/z = 240 (M+).
という測定結果であった。
【実施例7】
【0195】
続いて、一般式(3)に表される化合物の合成例を具体的に記す。なお、化合物の構造は、1H NMR(1H核磁気共鳴スペクトル)、EIMS(質量分析スペクトル)により決定した。
【0196】
まず、ナフト[1,2−b:5,6−b’]ジチオフェンの合成について、以下に段階的に説明する。
【0197】
(1,5−ジクロロ−2,6−ジヒドロキシナフタレンの合成)
窒素雰囲気下、2,6−ジヒドロキシナフタレン(3.0g,18.7mmol)を、酢酸(90ml)に溶解した。なお、酢酸は溶媒として用いた。
【0198】
これに塩化スルフリル(3.0ml,37.5mmol)を滴下した。これを室温で5時間攪拌した後、純水(50ml)を加えて析出した固体を濾取し、減圧下で乾燥させて、白色固体の1,5−ジクロロ−2,6−ジヒドロキシナフタレン(3.3g,収率78%)を得た。
【0199】
上記反応式を下記に示す。
【化26】
【0200】
得られた1,5−ジクロロ−2,6−ジヒドロキシナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 5.79 (s, 2H, OH), 7.35 (d, 2H, J = 8.9 Hz, ArH), 7.96 (d, 2H, J = 8.9 Hz, ArH); EIMS (70 eV) m/z = 228 (M+).
という測定結果であった。
【0201】
(1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンの合成)
窒素雰囲気下、1,5−ジクロロ−2,6−ジヒドロキシナフタレン(2.3g,10mmol)及びピリジン(4.8ml,60mmol)を塩化メチレン(100ml)に溶解した。ピリジンは不要物を除去するための添加剤、また、塩化メチレンは溶媒として用いた。
【0202】
これに氷浴下で無水トリフルオロメタンスルホン酸(3.6ml,22mmol)をゆっくり加えた。これを室温で18時間攪拌した後、純水(10ml)及び1N塩酸(10ml)を加え、反応を停止した。
【0203】
この溶液を塩化メチレン(20ml)で抽出した。なお、この抽出は同様の手法で計3回行った。抽出後、有機相を飽和食塩水(20ml)で洗浄した。なお、この洗浄は同様に計3回行った。
【0204】
洗浄した有機相を、無水硫酸マグネシウムを用いて乾燥、ろ過した後、溶媒を減圧下で留去した。
【0205】
得られた反応混合物を、塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で分離精製し、白色固体の1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレン(4.9g,収率99%)を得た。
【0206】
上記の反応式を以下に示す。
【化27】
【0207】
得られた1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.68 (d, 2H, J = 9.3 Hz, ArH), 8.40 (d, 2H, J = 9.3 Hz, ArH); EIMS (70 eV) m/z = 492 (M+).
という測定結果であった。
【0208】
(1,5−ジクロロ−2,6−ビス(トリメチルシリルエチニル)ナフタレンの合成)
窒素雰囲気下で、1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレン(247mg,0.5mmol)及びトリエチルアミン(0.21ml,1.5mmol)をDMF(5ml)に溶解し、30分間脱気した。
【0209】
これに触媒としてPd(PPh3)2Cl2(35mg,0.05mmol,10mol%)及びCuI(19mg,0.1mmol,20mol%)、更に試薬としてトリメチルシリルアセチレン(0.21ml,15mmol)を加え、室温で17時間30分攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0210】
この反応溶液から、塩化メチレン(5ml)を用いて、有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0211】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0212】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製し、白色固体の1,5−ジクロロ−2,6−ビス(トリメチルシリルエチニル)ナフタレン(89mg,46%)を得た。
【0213】
上記の反応式を以下に示す。
【化28】
【0214】
得られた1,5−ジクロロ−2,6−ビス(トリメチルシリルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.31 (s, 18H, TMS), 7.61 (d, 2H, J = 8.8 Hz, ArH), 8.12 (d, 2H, J = 8.8 Hz, ArH); EIMS (70 eV) m/z = 388 (M+).
という測定結果であった。
【0215】
(ナフト[1,2−b:5,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(615mg,2.56mmol)をNMP(15ml)に懸濁させ、15分間攪拌した。
【0216】
これに、1,5−ジクロロ−2,6−ビス(トリメチルシリルエチニル)ナフタレン(250mg,0.64mmol)を加え、190℃で12時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(50ml)に注ぎ、析出した固体を濾取した。
【0217】
ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で得られた反応混合物を分離精製し、白色固体のナフト[1,2−b:5,6−b’]ジチオフェン(139mg,90%)を得た。
【0218】
上記の反応式を下記に示す。
【化29】
【0219】
得られたナフト[1,2−b:5,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.50 (d, 2H, J = 5.3 Hz, ArH), 7.54 (d, 2H, J = 5.3 Hz, ArH), 7.95 (d, 2H, J = 8.6 Hz, ArH), 8.07 (d, 2H, J = 8.6 Hz, ArH); EIMS (70 eV) m/z = 240 (M+); mp 150.4-150.8 ℃.
という測定結果であった。
【実施例8】
【0220】
続いて、2,7−ジフェニルナフト[1,2−b:5,6−b’]ジチオフェンの合成について段階的に説明する。
【0221】
(1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレンの合成)
まず、前述のように合成した1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレンを合成する。
【0222】
窒素雰囲気下で、1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレン(493mg,1.0mmol)、及びトリエチルアミン(0.42mg,3.0mmol)をDMF(10ml)に溶解し、30分間脱気した。
【0223】
これに触媒としてPd(PPh3)2Cl2(70mg,0.1mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてフェニルアセチレン(0.33ml,3.0mmol)を加え、室温で27時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0224】
この反応溶液から、塩化メチレン(10ml)を用い、有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(10ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0225】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0226】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製した後、ヘキサンで洗浄して、淡黄色固体の1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレン(180mg,収率45%)を得た。
【0227】
上記の反応式を以下に示す。
【化30】
【0228】
得られた1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.39-7.42 (m, 6H, ArH), 7.63-7.67 (m, 4H, ArH), 7.74 (d, 2H, J = 8.6 Hz, ArH), 8.25 (d, 2H, J = 8.6 Hz, ArH); EIMS (70 eV) m/z = 396 (M+).
という測定結果であった。
【0229】
(2,7−ジフェニルナフト[1,2−b:5,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(608mg,2.53mmol)をNMP(15ml)に懸濁させ、15分間攪拌した。
【0230】
これに、1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレン(250mg,0.63mmol)を加え、190℃で12時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(50ml)に注ぎ、析出した固体を濾取した。
【0231】
得られた固体を昇華精製し、2,7−ジフェニルナフト[1,2−b:5,6−b’]ジチオフェン(147mg,60%)を得た。
【0232】
上記の反応式を下記に示す。
【化31】
【0233】
得られた2,7−ジフェニルナフト[1,2−b:5,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.34-7.40 (m, 2H, ArH), 7.45-7.57 (m, 4H, ArH), 7.71 (s, 2H, ArH), 7.79-7.82 (m, 4H, ArH), 7.91 (d, 2H, J = 8.6 Hz, ArH), 8.05 (d, 2H, J = 8.6 Hz, ArH); EIMS (70 eV) m/z = 392 (M+); mp > 300 ℃.
という測定結果であった。
【実施例9】
【0234】
続いて、2,7−ジオクチルナフト[1,2−b:5,6−b’]ジチオフェンの合成について、段階的に説明する。
【0235】
(1,5−ジクロロ−2,6−ジ(デシン−1−イル)ナフタレンの合成)
まず、前述のように合成した1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、1,5−ジクロロ−2,6−ジ(デシン−1−イル)ナフタレンを合成する。
【0236】
窒素雰囲気下で、1,5−ジクロロ−2,6−ビス(トリフルオロメタンスルフォニル)ナフタレン(493mg,1.0mmol)、及びトリエチルアミン(0.42mg,3.0mmol)をDMF(10ml)に溶解し、30分間脱気した。
【0237】
これに触媒としてPd(PPh3)2Cl2(70mg,0.1mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬として1−デシン(0.54ml,3.0mmol)を加え、室温で27時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0238】
この反応溶液を、塩化メチレン(10ml)を用いて抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(10ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0239】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0240】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.3)で分離精製し、白色固体の1,5−ジクロロ−2,6−ジ(デシン−1−イル)ナフタレン(408mg,収率87%)を得た。
【0241】
上記の反応式を以下に示す。
【化32】
【0242】
得られた1,5−ジクロロ−2,6−ジ(デシン−1−イル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.89 (t, 6H, J = 7.0 Hz, CH3), 1.23-1.71 (m, 24H, CH2), 2.53 (t, 4H, J = 7.0 Hz, CH2), 7.56 (d, 2H, J = 8.5 Hz, ArH), 8.13 (d, 2H, J = 8.5 Hz, ArH); EIMS (70 eV) m/z = 468 (M+).
という測定結果であった。
【0243】
(2,7−ジオクチルナフト[1,2−b:5,6−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(204mg,0.85mmol)をNMP(5ml)に懸濁させ、15分間攪拌した。
【0244】
これに1,5−ジクロロ−2,6−ジ(デシン−1−イル)ナフタレン(100mg,0.21mmol)を加え、190℃で13時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(30ml)に注ぎ、析出した固体を濾取した。
【0245】
ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.5)で得られた固体を分離精製し、白色固体の2,7−ジオクチルナフト[1,2−b:5,6−b’]ジチオフェン(147mg,60%)を得た。
【0246】
上記の反応式を下記に示す。
【化33】
【0247】
得られた2,7−ジオクチルナフト[1,2−b:5,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 0.88 (t, 6H, J = 6.8 Hz, CH3), 1.21-1.83 (m, 24H, CH2), 2.97 (t, 4H, J = 7.4 Hz, CH2), 7.14 (s, 2H, ArH), 7.77 (d, 2H, J = 8.6 Hz, ArH), 7.91 (d, 2H, J = 8.6 Hz, ArH); EIMS (70 eV) m/z = 464 (M+); mp 92-93 ℃.
という測定結果であった。
【実施例10】
【0248】
(ナフト[1,2−b:5,6−b’]ジセレノフェンの合成)
窒素雰囲気下、セレン(72mg,0.91mmol)をエタノール(3ml)に懸濁させ、氷浴温度下で水素化ホウ素ナトリウム(34mg,0.91mmol)を加え、40分攪拌した。
【0249】
そこにNMP(10ml)と、前述のように合成した1,5−ジクロロ−2,6−ビス(トリメチルシリルエチニル)ナフタレン(100mg,0.26mmol)を加え、190℃で12時間攪拌した。
【0250】
室温まで冷却した後、反応溶液を飽和塩化アンモニウム水溶液(50ml)に注ぎ、析出した固体を濾取した。ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で、得られた反応混合物を分離精製し、白色固体のナフト[1,2−b:5,6−b’]ジセレノフェン(70mg,収率81%)を得た。
【0251】
上記の反応式を下記に示す。
【化34】
【0252】
得られたナフト[1,2−b:5,6−b’]ジセレノフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.73 (d, 2H, J = 5.8 Hz, ArH), 7.92 (s, 4H, ArH), 8.08 (d, 2H, J = 5.9 Hz, ArH); 13C-NMR (100 MHz, CDCl3) δ 123.56, 124.40, 128.37, 128.39, 129.22, 139.95, 142.23; EIMS (70 eV) m/z = 336 (M+).
という測定結果であった。
【実施例11】
【0253】
(2,7−ジフェニルナフト[1,2−b:5,6−b’]ジセレノフェンの合成)
窒素雰囲気下、セレン(141mg,1.8mmol)をエタノール(4ml)に懸濁させ氷浴温度下で水素化ホウ素ナトリウム(68mg,1.8mmol)を加え、40分間攪拌した。
【0254】
そこにNMP(20ml)と、前述のように合成した1,5−ジクロロ−2,6−ビス(フェニルエチニル)ナフタレン(200mg,0.5mmol)を加え、190度で12時間攪拌した。室温まで冷却した後、反応溶液を飽和塩化アンモニウム水溶液(50mL)に注ぎ、析出した固体を濾取した。
【0255】
この固体を温度勾配熱昇華法により精製することで、淡黄色固体の2,7−ジフェニルナフト[1,2−b:5,6−b’]ジセレノフェン(66mg,収率27%)を得た。
【0256】
上記の反応式を下記に示す。
【化35】
【0257】
得られた2,7−ジフェニルナフト[1,2−b:5,6−b’]ジセレノフェンは、
EIMS (70 eV) m/z = 488 (M+).
という測定結果であった。
【実施例12】
【0258】
続いて、一般式(4)に表される化合物の合成例を具体的に記す。
【0259】
まず、ナフト[2,1−b:6,5−b’]ジチオフェンの合成について、以下に段階的に説明する。
【0260】
(2,6−ジブロモ−1,5−ジヒドロキシナフタレンの合成)
窒素雰囲気下、1,5−ジヒドロキシナフタレン(5.0g,31mmol)と少量のヨウ素を、酢酸(150ml)に溶解し、80℃まで加熱した。なお、酢酸は溶媒として用いた。
【0261】
これに臭素(3.2ml,62.4mmol)を滴下し、還流温度下で12時間反応させた。その後、室温まで冷却し、純水(50ml)を加えて析出した固体を濾取した。これを純水で洗浄し、減圧下で乾燥させて、白色固体の2,6−ジブロモ−1,5−ジヒドロキシナフタレン(8.2g,収率83%)を得た。
【0262】
上記反応式を下記に示す。
【化36】
【0263】
得られた2,6−ジブロモ−1,5−ジヒドロキシナフタレンは、
1H-NMR (400 MHz, CDCl3) δ 5.99 (s, 2H, OH), 7.39 (d, 2H, J = 9.4 Hz, ArH), 7.70 (d, 2H, J = 9.4 Hz, ArH); EIMS (70 eV) m/z = 318 (M+).
という測定結果であった。
【0264】
(2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレンの合成)
窒素雰囲気下、2,6−ジブロモ−1,5−ジヒドロキシナフタレン(3.0g,9.4mmol)及びピリジン(4.5ml,56mmol)を塩化メチレン(90ml)に溶解した。ピリジンは不要物を除去するための添加剤、また、塩化メチレンは溶媒として用いた。
【0265】
これに氷浴下で無水トリフルオロメタンスルホン酸(3.3ml,21mmol)をゆっくり加えた。これを室温で4時間30分攪拌した後、純水(10ml)及び1N塩酸(10ml)を加え、反応を停止した。
【0266】
この溶液から、塩化メチレン(20ml)を用いて有機相を抽出した。なお、この抽出は同様の手法で計3回行った。抽出後、有機相を飽和食塩水(20ml)で洗浄した。なお、この洗浄は同様に計3回行った。
【0267】
洗浄した有機相を、無水硫酸マグネシウムを用いて乾燥、ろ過した後、溶媒を減圧下で留去した。
【0268】
得られた反応混合物を、塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で分離精製し、白色固体の2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレン(3.2g,収率58%)を得た。
【0269】
上記の反応式を以下に示す。
【化37】
【0270】
得られた2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.89 (d, 2H, J = 9.2 Hz, ArH), 8.03 (d, 2H, J = 9.2 Hz, ArH); EIMS (70 eV) m/z = 582 (M+).
という測定結果であった。
【0271】
(2,6−ジブロモ−1,5−ビス(トリメチルシリルエチニル)ナフタレンの合成)
窒素雰囲気下で、2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)及びジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0272】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてトリメチルシリルアセチレン(0.28ml,2.0mmol)を加え、室温で11時間攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0273】
この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0274】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥、ろ過した後、溶媒を減圧下で留去した。
【0275】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製し、白色固体の2,6−ジブロモ−1,5−ビス(トリメチルシリルエチニル)ナフタレンの粗生成物(234mg,収率49%)を得た。
【0276】
上記の反応式を以下に示す。
【化38】
【0277】
得られた2,6−ジブロモ−1,5−ビス(トリメチルシリルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.29 (s, 18H, TMS), 7.71 (d, 2H, J = 8.8 Hz, ArH), 8.14 (d, 2H, J = 8.8 Hz, ArH); EIMS (70 eV) m/z = 478 (M+).
という測定結果であった。
【0278】
(ナフト[2,1−b:6,5−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(202mg,0.84mmol)をNMP(6ml)に懸濁させ、15分間攪拌した。
【0279】
これに、2,6−ジブロモ−1,5−ビス(トリメチルシリルエチニル)ナフタレン(100mg,0.2mmol)を加え、190℃で14時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取し、ナフト[2,1−b:6,5−b’]ジチオフェンの粗生成物(62mg)を得た。
【0280】
上記の反応式を下記に示す。
【化39】
【0281】
得られたナフト[2,1−b:6,5−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.43 (d, 2H, J = 5.4 Hz, ArH), 8.05 (d, 2H, J = 5.5 Hz ArH), 8.05 (d, 2H, J = 8.9 Hz, ArH), 8.30 (d, 2H, J = 8.9 Hz, ArH); EIMS (70 eV) m/z = 240 (M+).
という測定結果であった。
【実施例13】
【0282】
続いて、2,7−ジフェニルナフト[2,1−b:6,5−b’]ジチオフェンの合成について、段階的に説明する。
【0283】
(2,6−ジブロモ−1,5−ビス(フェニルエチニル)ナフタレンの合成)
まず、前述のように合成した2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、2,6−ジブロモ−1,5−ビス(フェニルエチニル)ナフタレンを合成する。
【0284】
窒素雰囲気下で、2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)及びジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0285】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬としてフェニルアセチレン(0.22ml,2.0mmol)を加え、室温で11時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0286】
この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0287】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥し、ろ過した後、溶媒を減圧下で留去した。
【0288】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.1)で分離精製し、白色固体の2,6−ジブロモ−1,5−ビス(フェニルエチニル)ナフタレン(437mg,収率90%)を得た。
【0289】
上記の反応式を以下に示す。
【化40】
【0290】
得られた2,6−ジブロモ−1,5−ビス(フェニルエチニル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 7.42-7.44 (m, 6H, ArH), 7.69-7.72 (m, 4H, ArH), 7.79(d, 2H, J = 8.9 Hz, ArH), 8.27 (d, 2H, J = 8.9 Hz, ArH); EIMS (70 eV) m/z = 486 (M+).
という測定結果であった。
【0291】
(2,7−ジフェニルナフト[2,1−b:6,5−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(404mg,1.68mmol)をNMP(12ml)に懸濁させ、15分間攪拌した。
【0292】
これに、2,6−ジブロモ−1,5−ビス(フェニルエチニル)ナフタレン(200mg,0.4mmol)を加え、190℃で14時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取し、2,7−ジフェニルナフト[2,1−b:6,5−b’]ジチオフェンの粗生成物(192mg)を得た。
【0293】
上記の反応式を下記に示す。
【化41】
【0294】
得られた2,7−ジフェニルナフト[2,1−b:6,5−b’]ジチオフェンは、
1H-NMR (400 MHz, CDCl3) δ 7.39-7.40 (m, 2H, ArH), 7.47-7.51 (m, 4H, ArH), 7.82-7.84 (m, 4H, ArH), 8.01 (d, 2H, J = 8.6 Hz, ArH), 7.71 (s, 2H, ArH), 8.05 (d, 2H, J = 8.6 Hz, ArH); EIMS (70 eV) m/z = 392 (M+).
という測定結果であった。
【実施例14】
【0295】
続いて、2,7−ジオクチルナフト[2,1−b:6,5−b’]ジチオフェンの合成について、段階的に説明する。
【0296】
(2,6−ジブロモ−1,5−ジ(デシン−1−イル)ナフタレンの合成)
まず、前述のように合成した2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレンを用いて、2,6−ジブロモ−1,5−ジ(デシン−1−イル)ナフタレンを合成する。
【0297】
窒素雰囲気下で、2,6−ジブロモ−1,5−ビス(トリフルオロメタンスルフォニル)ナフタレン(582mg,1.0mmol)を、DMF(7ml)及びジイソプロピルアミン(7ml)に溶解し、30分間脱気した。
【0298】
これに触媒としてPd(PPh3)2Cl2(70mg,0.05mmol,10mol%)及びCuI(38mg,0.1mmol,20mol%)、更に試薬として1−デシン(0.36ml,2.0mmol)を加え、室温で11時間攪拌して反応させた後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。
【0299】
この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0300】
無水硫酸マグネシウムを用い、洗浄した有機相を乾燥し、ろ過した後、溶媒を減圧下で留去した。
【0301】
得られた反応物を、ヘキサンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.2)で分離精製し、白色固体の2,6−ジブロモ−1,5−ジ(デシン−1−イル)ナフタレンの粗生成物(340mg,収率61%)を得た。
【0302】
上記の反応式を以下に示す。
【化42】
【0303】
得られた2,6−ジブロモ−1,5−ジ(デシン−1−イル)ナフタレンは、
1H-NMR (270 MHz, CDCl3) δ 0.89 (t, 6H, J = 7.0 Hz, CH3), 1.26-1.70 (m, 24H, CH2), 2.62 (t, 4H, J = 7.3 Hz, CH2), 7.68 (d, 2H, J = 9.4 Hz, ArH), 8.10 (d, 2H, J = 9.4 Hz, ArH); EIMS (70 eV) m/z = 558 (M+).
という測定結果であった。
【0304】
(2,7−ジオクチルナフト[2,1−b:6,5−b’]ジチオフェンの合成)
窒素雰囲気下で、Na2S・9H2O(404mg,1.68mmol)をNMP(12ml)に懸濁させ、15分間攪拌した。
【0305】
これに、2,6−ジブロモ−1,5−ジ(デシン−1−イル)ナフタレン(200mg,0.4mmol)を加え、190℃で14時間攪拌した。これを室温まで冷却した後、飽和塩化アンモニウム水溶液(20ml)に注ぎ、析出した固体を濾取し、2,7−ジオクチルナフト[2,1−b:6,5−b’]ジチオフェンの粗生成物(200mg,収率100%)を得た。
【0306】
上記の反応式を下記に示す。
【化43】
【0307】
得られた2,7−ジオクチルナフト[2,1−b:6,5−b’]ジチオフェンは、
1H-NMR (400 MHz, CDCl3) δ 0.88 (t, 6H, J = 7.0 Hz, CH3), 1.26-1.70 (m, 24H, CH2), 3.02 (t, 4H, J = 7.3 Hz, CH2), 7.68 (s, 2H, ArH), 7.89 (d, 2H, J = 8.8 Hz, ArH), 8.12 (d, 2H, J = 8.8 Hz, ArH); EIMS (70 eV) m/z = 464 (M+).
という測定結果であった。
【実施例15】
【0308】
続いて、一般式(5)で表される化合物の合成例を具体的に示す。
【0309】
(2,7−ジブロモナフト[2,3−b:6,7−b’]ジチオフェンの合成)
窒素雰囲気下、実施例1で合成したナフト[2,3−b:6,7−b’]ジチオフェン(50mg,0.21mmol)をTHF(10ml)に懸濁させた後、−78℃に冷却し、n−BuLi(0.4ml,0.63mmol,1.59M)を加えた。これを30分間攪拌した後、1,2−ジブロモ−1,1,2,2,−テトラクロロエタン(150mg,0.46mmol)のTHF(3mL)溶液を滴下した。
【0310】
これを室温まで昇温させて16時間攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。そして、析出した固体を濾取することで、2,7−ジブロモナフト[2,3−b:6,7−b’]ジチオフェン(15mg,収率18%)を得た。
【0311】
上記の反応式を下記に示す。
【化44】
【0312】
得られた2,7−ジブロモナフト[2,3−b:6,7−b’]ジチオフェンは、
1H-NMR (400 MHz, CDCl3) δ 7.43 (s, 2H, ArH), 8.22 (s, 2H, ArH), 8.31 (s, 2H, ArH);
EIMS (70 eV) m/z = 398 (M+)
という測定結果であった。
【実施例16】
【0313】
続いて、一般式(7)で表される化合物の合成例について、具体的に説明する。
(2,7−ジブロモナフト[1,2−b:5,6−b’]ジチオフェンの合成)
窒素雰囲気下、実施例7で合成したナフト[1,2−b:5,6−b’]ジチオフェン(50mg,0.21mmol)をTHF(5ml)に溶解した後、−78℃に冷却し、n−BuLi(0.4ml,0.63mmol,1.59M)を加えた。これを30分間攪拌した後、1,2−ジブロモ−1,1,2,2,−テトラクロロエタン(651mg,2mmol)のTHF(3mL)溶液を滴下した。
【0314】
これを室温まで昇温して16時間攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。この反応溶液から、塩化メチレン(5ml)を用いて有機相を抽出した。この抽出は同様の手法で3回行った。抽出後、飽和食塩水(5ml)を用い、有機相を洗浄した。この洗浄は同様の手法で3回行った。
【0315】
無水硫酸マグネシウムを用いて有機相を乾燥し、ろ過した後に、溶媒を減圧下で留去した。得られた反応混合物を塩化メチレンを移動相とするシリカゲルカラムクロマトグラフィー(Rf=0.95)で分離精製することで、白色固体の2,7−ジブロモナフト[1,2−b:5,6−b’]ジチオフェン(68mg,収率81%)を得た。
【0316】
上記の反応式を下記に示す。
【化45】
【0317】
得られた2,7−ジブロモナフト[1,2−b:5,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.48 (s, 2H, ArH), 7.80 (d, 2H, J = 8.5 Hz, ArH), 7.87 (d, 2H, J = 8.5 Hz, ArH); EIMS (70 eV) m/z = 398 (M+).
という測定結果であった。
【実施例17】
【0318】
(2,7−ジヨードナフト[1,2−b:5,6−b’]ジチオフェンの合成)
窒素雰囲気下、実施例7で合成したナフト[1,2−b:5,6−b’]ジチオフェン(50mg,0.21mmol)をTHF(5ml)に溶解した後、−78℃に冷却し、n−BuLi(0.4ml,0.63mmol,1.59M)を加えた。これを30分間攪拌した後、ヨウ素(117mg,0.46mmol)のTHF(3mL)溶液を滴下した。
【0319】
これを室温まで昇温させて10時間攪拌した後、純水(1ml)、1N塩酸(1ml)を加えてクエンチした。そして、析出した固体を濾取することで、白色固体の2,7−ジヨードナフト[1,2−b:5,6−b’]ジチオフェン(82mg,収率80%)を得た。
【0320】
上記の反応式を下記に示す。
【化46】
【0321】
得られた2,7−ジヨードナフト[1,2−b:5,6−b’]ジチオフェンは、
1H-NMR (270 MHz, CDCl3) δ 7.68 (s, 2H, ArH), 7.82 (d, 2H, J = 8.8 Hz, ArH), 7.86 (d, 2H, J = 8.8 Hz, ArH); EIMS (70 eV) m/z = 492 (M+)
という測定結果であった。
【実施例18】
【0322】
(FET特性)
実施例2で合成した2,7−ジフェニルナフト[2,3−b:6,7−b’]ジチオフェン(以下、化合物A)、実施例3で合成した2,7−ジオクチルナフト[2,3−b:6,7−b’]ジチオフェン(以下、化合物B)、実施例8で合成した2,7−ジフェニルナフト[1,2−b:5,6−b’]ジチオフェン(以下、化合物C)、実施例11で合成した2,7−ジフェニルナフト[1,2−b:5,6−b’]ジセレノフェン(以下、化合物D)を用いてそれぞれFET素子を作製し、FET特性を検証した。
【0323】
まず、SiO2基板を面積1cm×1cmの大きさに切り出し、裏面をフッ化水素酸で処理し、空気中で酸化されているシリカを取り除いた後、Auを真空蒸着してゲート電極を形成した。次に、SiO2基板表面上に、真空蒸着法で化合物Aの有機薄膜を形成した。なお、SiO2基板は、オクチルトリクロロシランで表面処理を施して用いた。
【0324】
形成した有機薄膜上にシャドウマスクを用いてAuを真空蒸着することでソース電極とドレイン電極とを形成した。
【0325】
作製したFET素子の概略構成を図1(図1(A)はFET素子の断面図、図1(B)はFET素子の平面図)に示す。作製したFET素子はトップコンタクト型で、チャネル長は50μm、チャネル幅は1.5mmである。
【0326】
上記と同様にして、化合物Cを用いたFET素子、及び、化合物Dを用いたFET素子をそれぞれ作成した。
【0327】
また、化合物Bを用いたFET素子は以下のようにして作成した。SiO2基板を面積1cm×1cmの大きさに切り出し、裏面をフッ化水素酸で処理し、空気中で酸化されているシリカを取り除いた後、Auを真空蒸着してゲート電極を形成した。次に、SiO2基板表面上に、0.4wt%に調製した化合物Bのクロロホルム溶液を用いてスピンコート法(有機薄膜作成条件:3000rpm,30sec)により有機薄膜を形成した。
【0328】
形成した有機薄膜上にシャドウマスクを用いてAuを真空蒸着することでソース電極とドレイン電極とを形成した。なお、FET素子の構造等については、上記他のFET素子と同様である。
【0329】
FET素子の性能は、ゲート電極に電位をかけた状態でソース電極とドレイン電極との間に電位をかけたときに流れた電流量に依存する。この電流値を測定することでFET素子の特性である電解移動度を決めることができる。電解移動度は、絶縁体としてのSiO2にゲート電圧を印加した結果、有機半導体層中に生じるキャリア種の電気的特性を表現する式(a)から求めることができる。
Id=WμCo(Vg−Vt)2/2L …(a)
【0330】
ここで、Idは飽和したソース−ドレイン電流値、Wはチャネル幅、Coはゲート電気容量、Vgはゲート電圧、Vtは閾値電圧、Lはチャネル長であり、μが決定する電解移動度(cm2/Vs)である。Coは用いたSiO2絶縁膜の誘電率、W及びLはFET素子の素子構造によって決まり、Id及びVgはFET素子の電流値の測定時に決まり、VtはId、Vgから求めることができる。式(a)に各値を代入することで、それぞれのゲート電位での電解移動度を算出することができる。なお、閾値電圧[Vt]は、Idの平方根をY軸に、VgをX軸に取り、Idの平方根が立ち上がるVg値として求めた。
【0331】
それぞれのFET素子について、p型FET特性を調べるために、負のゲート電圧をかけ、大気中にて駆動させて評価した。
【0332】
図2は、化合物Aを用いて作成したFET素子のFET特性結果である。また、図3は、化合物Bを用いて作成したFET素子のFET特性結果である。また、図4は、化合物Cを用いて作成したFET特性結果である。また、図5は、化合物Dを用いて作成したFET特性の結果である。
【0333】
ここで、図2(A)、図3(A)、図4(A)、及び図5(A)は、それぞれのFET素子のVg−Id曲線、また、図2(B)、図3(B)、図4(B)、及び図5(B)は、それぞれのFET素子のVd−Id曲線である。
【0334】
Vg−Id曲線は、アウトプット特性において、電流(Id)が飽和電流になる値になるようソース−ドレイン間の電圧(Vd)を固定した時の、ゲート電圧(Vg)と電流(Id)との関係を表す。すなわち、Vg−Id曲線は、当該FET素子のトランスファー特性(伝達特性)を示している。当該Vg−Id曲線において、off状態からon状態への立ち上がりが急なほど、スイッチング特性が良好であることを示しており、トランジスタ特性は優れていると言える。また、off電流が低ければ低いほど、on電流が高ければ高いほどon/off比が大きくなるので、良好なトランジスタであるといえる。
【0335】
一方、Vd−Id曲線は、ゲート電圧(Vg)を段階的に変化させた時の、ソース−ドレイン間の電圧(Vd)と電流(Id)との関係を表す。すなわち、Vd−Id曲線は、当該FET素子のアウトプット特性(出力特性)を示している。当該FET素子において、Vd−Id曲線が、いずれのVgにおいても、ソース−ドレイン間の高い電圧(Vd)領域で電流(Id)が飽和すること(飽和電流)、および、ソース−ドレイン間の低い電圧(Vd)領域で電流(Id)が直線的に立ち上がっていることを示せば、アウトプット特性が良く、そのFET素子は高性能であるといえる。
【0336】
図2(A)、図3(A)、図4(A)、及び図5(A)をみると、ゲート電圧(Vg)の印加により、いずれも電流(Id)が急峻に立ち上がっている。また、図2(B)図3(B)、図4(B)、及び図5(B)をみると、ソース−ドレイン間の電圧(Vd)の低い領域では、いずれもほぼ直線状に立ち上がっており、また、ソース−ドレイン間の電圧(Vd)の高い領域では、ドレイン電流が一定となっており、飽和電流が観測された。
【0337】
化合物Aを用いたFET素子では、電界移動度:0.7cm2/Vs、on/off比:106、化合物Cを用いたFET素子では、電界移動度:0.2cm2/Vs、on/off比:107、また、化合物Dを用いたFET素子では、電界移動度:0.2cm2/V、on/off比:107と良好な測定結果を得た。なお、on/off比は、それぞれのVg−Id曲線において、Vgが0〜−10V程度と小さいときをオフ状態とし、Vgが−60Vのときをオン状態として、オフ状態及びオン状態それぞれにおけるIdの値の比で表している。
【0338】
また、化合物Bを用い、塗布法にて製造したFET素子の電解移動度は10−3cm2/Vs台、また、on/off比は105と、化合物A、化合物C、及び化合物Dを用いたFET素子に比べてやや劣る結果となったものの、FET特性を備えていることから、塗布法も利用可能であることがわかる。
【0339】
このように、本実施例で合成した化合物A、化合物B、化合物C、及び化合物Dを用いたFET素子はp型トランジスタとして使用することができる。
【産業上の利用可能性】
【0340】
上記の化合物は、ナフタレンにチオフェン環或いはセレノフェン環が結合した構造であり、π電子が存在すること、また、チオフェン環或いはセレノフェン環を有しており、硫黄原子或いはセレン原子を介した強い分子間相互作用を有することから、効果的なキャリアの移動が可能である。良好な電界移動度を有するので、有機半導体材料として利用でき、これを用いた有機半導体デバイスを構成することができる。
【特許請求の範囲】
【請求項1】
下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化1】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、又はフェニル基のいずれかを示す)で表されることを特徴とする化合物。
【請求項2】
下記一般式(5)、一般式(6)、一般式(7)、又は一般式(8)
【化2】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Xはハロゲン原子を示す)で表されることを特徴とする化合物。
【請求項3】
ジハロゲノジヒドロキシナフタレンと無水トリフルオロメタンスルホン酸とを反応させ、ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンを得る工程と、
前記ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンと末端アセチレン化合物とを反応させ、ジハロゲノ−ジエチニルナフタレン誘導体を得る工程と、
前記ジハロゲノ−ジエチニルナフタレン誘導体と硫化物塩或いはセレン化物塩とを反応させ、下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化3】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、或いはフェニル基のいずれかを示す)で表されるいずれかの化合物を得る工程と、
を含むことを特徴とする化合物の製造方法。
【請求項4】
更に、ジヒドロキシナフタレンとハロゲン化剤とを反応させ、前記ジハロゲノジヒドロキシナフタレンを得る工程を含むことを特徴とする請求項3に記載の化合物の製造方法。
【請求項5】
前記ジヒドロキシナフタレンとして2,6−ジヒドロキシナフタレンを用い、前記一般式(1)或いは前記一般式(3)で表される化合物を得ることを特徴とする請求項4に記載の化合物の製造方法。
【請求項6】
前記ジヒドロキシナフタレンとして2,7−ジヒドロキシナフタレンを用い、前記一般式(2)で表される化合物を得ることを特徴とする請求項4に記載の化合物の製造方法。
【請求項7】
前記ジヒドロキシナフタレンとして1,5−ジヒドロキシナフタレンを用い、前記一般式(4)で表される化合物を得ることを特徴とする請求項4に記載の化合物の製造方法。
【請求項8】
前記ハロゲン化剤として臭素化剤或いは塩素化剤を用いることを特徴とする請求項4に記載の化合物の製造方法。
【請求項9】
触媒を添加し、前記臭素化剤を複数回順次添加しながら反応させることを特徴とする請求項8に記載の化合物の製造方法。
【請求項10】
前記末端アセチレン化合物としてトリメチルシリルアセチレン、フェニルアセチレン、或いは1−デシンのいずれかを用いることを特徴とする請求項3に記載の化合物の製造方法。
【請求項11】
前記ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンを極性溶媒に溶解して前記末端アセチレン化合物と反応させることを特徴とする請求項3に記載の化合物の製造方法。
【請求項12】
前記極性溶媒として非プロトン性極性溶媒を用いることを特徴とする請求項11に記載の化合物の製造方法。
【請求項13】
前記非プロトン性極性溶媒として、ジメチルホルムアミドを用いることを特徴とする請求項12に記載の化合物の製造方法。
【請求項14】
更に、前記一般式(1)、前記一般式(2)、前記一般式(3)、又は、前記一般式(4)で表される化合物(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子を示す)にハロゲン化剤を添加して、下記一般式(5)、一般式(6)、一般式(7)、又は、一般式(8)
【化4】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Xはハロゲン原子を示す)で表される化合物を得る工程を含むことを特徴とする請求項3に記載の化合物の製造方法。
【請求項15】
下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化5】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、又はフェニル基のいずれかを示す)で表される化合物を少なくとも一種以上含むことを特徴とする有機半導体材料。
【請求項16】
請求項15に記載の前記有機半導体材料を含むことを特徴とする有機半導体デバイス。
【請求項1】
下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化1】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、又はフェニル基のいずれかを示す)で表されることを特徴とする化合物。
【請求項2】
下記一般式(5)、一般式(6)、一般式(7)、又は一般式(8)
【化2】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Xはハロゲン原子を示す)で表されることを特徴とする化合物。
【請求項3】
ジハロゲノジヒドロキシナフタレンと無水トリフルオロメタンスルホン酸とを反応させ、ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンを得る工程と、
前記ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンと末端アセチレン化合物とを反応させ、ジハロゲノ−ジエチニルナフタレン誘導体を得る工程と、
前記ジハロゲノ−ジエチニルナフタレン誘導体と硫化物塩或いはセレン化物塩とを反応させ、下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化3】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、或いはフェニル基のいずれかを示す)で表されるいずれかの化合物を得る工程と、
を含むことを特徴とする化合物の製造方法。
【請求項4】
更に、ジヒドロキシナフタレンとハロゲン化剤とを反応させ、前記ジハロゲノジヒドロキシナフタレンを得る工程を含むことを特徴とする請求項3に記載の化合物の製造方法。
【請求項5】
前記ジヒドロキシナフタレンとして2,6−ジヒドロキシナフタレンを用い、前記一般式(1)或いは前記一般式(3)で表される化合物を得ることを特徴とする請求項4に記載の化合物の製造方法。
【請求項6】
前記ジヒドロキシナフタレンとして2,7−ジヒドロキシナフタレンを用い、前記一般式(2)で表される化合物を得ることを特徴とする請求項4に記載の化合物の製造方法。
【請求項7】
前記ジヒドロキシナフタレンとして1,5−ジヒドロキシナフタレンを用い、前記一般式(4)で表される化合物を得ることを特徴とする請求項4に記載の化合物の製造方法。
【請求項8】
前記ハロゲン化剤として臭素化剤或いは塩素化剤を用いることを特徴とする請求項4に記載の化合物の製造方法。
【請求項9】
触媒を添加し、前記臭素化剤を複数回順次添加しながら反応させることを特徴とする請求項8に記載の化合物の製造方法。
【請求項10】
前記末端アセチレン化合物としてトリメチルシリルアセチレン、フェニルアセチレン、或いは1−デシンのいずれかを用いることを特徴とする請求項3に記載の化合物の製造方法。
【請求項11】
前記ジハロゲノ−ビス(トリフルオロメタンスルフォニル)ナフタレンを極性溶媒に溶解して前記末端アセチレン化合物と反応させることを特徴とする請求項3に記載の化合物の製造方法。
【請求項12】
前記極性溶媒として非プロトン性極性溶媒を用いることを特徴とする請求項11に記載の化合物の製造方法。
【請求項13】
前記非プロトン性極性溶媒として、ジメチルホルムアミドを用いることを特徴とする請求項12に記載の化合物の製造方法。
【請求項14】
更に、前記一般式(1)、前記一般式(2)、前記一般式(3)、又は、前記一般式(4)で表される化合物(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子を示す)にハロゲン化剤を添加して、下記一般式(5)、一般式(6)、一般式(7)、又は、一般式(8)
【化4】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Xはハロゲン原子を示す)で表される化合物を得る工程を含むことを特徴とする請求項3に記載の化合物の製造方法。
【請求項15】
下記一般式(1)、一般式(2)、一般式(3)、又は一般式(4)
【化5】
(式中、Zは硫黄原子又はセレン原子のいずれかを示し、Rは水素原子、アルキル基、又はフェニル基のいずれかを示す)で表される化合物を少なくとも一種以上含むことを特徴とする有機半導体材料。
【請求項16】
請求項15に記載の前記有機半導体材料を含むことを特徴とする有機半導体デバイス。
【図1】

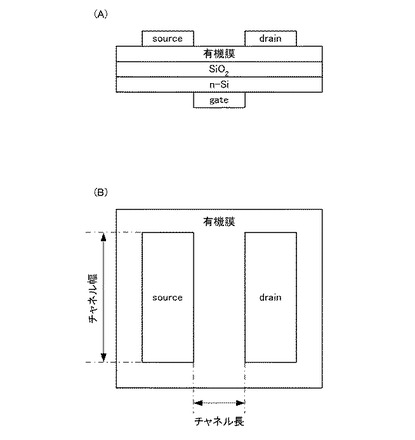
【図2】
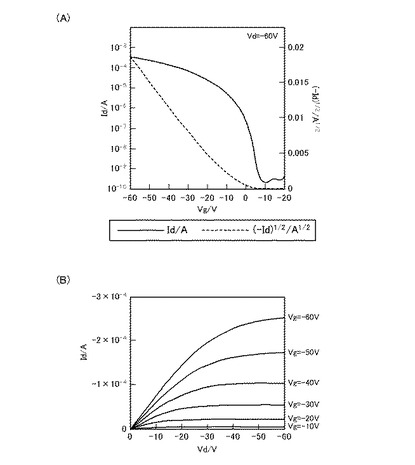
【図3】
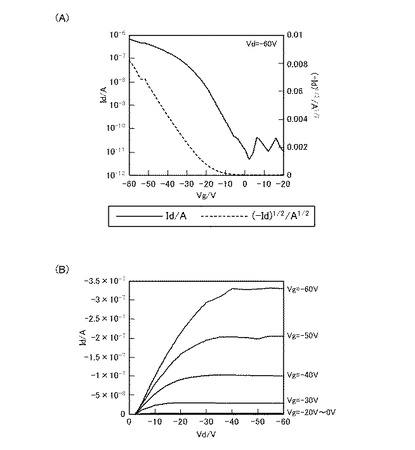
【図4】
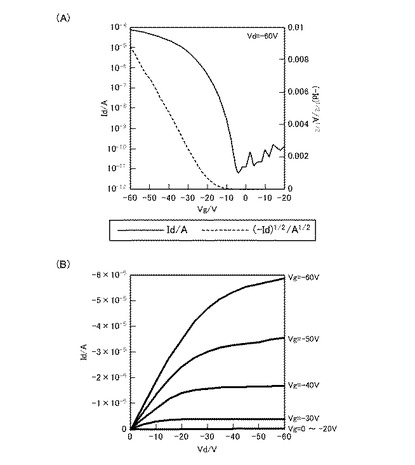
【図5】
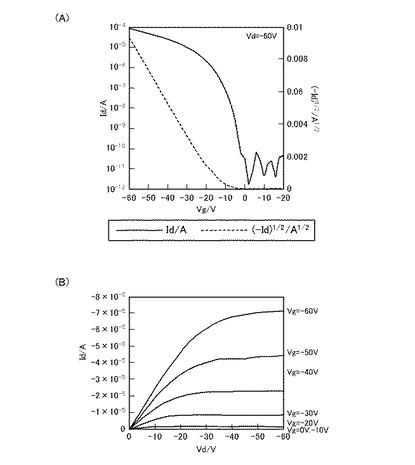
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2010−150229(P2010−150229A)
【公開日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願番号】特願2009−80527(P2009−80527)
【出願日】平成21年3月27日(2009.3.27)
【出願人】(504136568)国立大学法人広島大学 (924)
【Fターム(参考)】
【公開日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願日】平成21年3月27日(2009.3.27)
【出願人】(504136568)国立大学法人広島大学 (924)
【Fターム(参考)】
[ Back to top ]