
新規酵母およびそれを用いたΔ5,7−ステロール及びハイドロコルチゾンの製造方法
【課題】コレスタ−5,7,24−トリエン−3β−オールなどのΔ5,7−ステロール及びハイドロコルチゾンを効率よく製造する方法を提供すること。
【解決手段】アシルCoA:ステロールアシルトランスフェラーゼ遺伝子とステリルエステル加水分解酵素遺伝子の発現が増強されるように改変された酵母を用いてΔ5,7−ステロール及びハイドロコルチゾンを製造する。
【解決手段】アシルCoA:ステロールアシルトランスフェラーゼ遺伝子とステリルエステル加水分解酵素遺伝子の発現が増強されるように改変された酵母を用いてΔ5,7−ステロール及びハイドロコルチゾンを製造する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は医薬の中間体等として有用なΔ5,7−ステロール及びハイドロコルチゾンを製造する酵母およびそれを用いたΔ5,7−ステロール及びハイドロコルチゾンの製造法に関する。
【背景技術】
【0002】
ステロール類は動物、植物、真菌類に含まれている生命活動に必須の物質であり、細胞膜に遊離型ステロールの状態で含まれるほか、一部は脂肪酸とのエステル型として貯蔵される。このうち、C5,7−ジエン体ステロールはコレステロール骨格の5位と7位に炭素−炭素二重結合を有するステロールであるが、その中でもコレスタ−5,7,24−トリエン−3β−オール(別名、7−デヒドロデスモステロール、以下、TEOLと称する)は医薬品であるケノデオキシコール酸やウルソデオキシコール酸の合成原料として用いられるなど、医薬品の中間体などとして非常に有用な物質である。
【0003】
また、11β,17α,21-トリヒドロキシ-4-プレグネン-3,20-ジオン(ハイドロコルチゾン)は、その前駆物質であるプレグネノロン、プロゲステロン、7−デヒドロプレグネノロンとともに、医薬および医薬中間体として産業上有用な化合物である。プレグネノロンの既存の製造法として、コレステロールやジオスゲニン、スティグマステロールといった天然由来のステロール化合物を原料として、複数の有機合成反応により合成する方法が報告されている(非特許文献1)。
【0004】
従来、C5,7−ジエン体ステロールは動物組織の抽出物から精製されたものが使用されている。一方で、酵母等の真菌類においてエルゴステロールの生合成経路が明らかにされる(非特許文献2)につれて、近年ではその生合成経路を改変して特定のステロールを蓄積させるという研究が進められている。
【0005】
酵母、カビ等の真菌類において、エルゴステロール生合成酵素欠損株あるいはエルゴステロール生合成酵素遺伝子の過剰発現によりステロール組成を変えた研究例がいくつか報告されている。例えば、酵母サッカロミセス・セレビシエでステロールC−24メチルトランスフェラーゼをコードする遺伝子(ERG6)を欠損させることにより、側鎖24位にメチル基を持たないステロールのみが蓄積するようにした例(非特許文献3)や、酵母でステロールC−22デサチュラーゼをコードする遺伝子(ERG5)を欠損させアラビドプシスのΔ7−ステロールリダクターゼを高発現するように代謝工学的に改変し、エルゴスタ−5,24−ジエノール、エルゴスタ−5−エネノール、エルゴスタ−5,22−ジエノールが蓄積するようにした例が報告されている(非特許文献4)。
【0006】
また、特許文献1では、ステロールC−22デサチュラーゼ、および、ステロールC−24メチルトランスフェラーゼの両方の活性が低減化され、ステロールΔ7−リダクターゼ、および/またはステロールΔ5−デサチュラーゼが高発現化されるように代謝工学的に改変された真菌類(酵母、カビ)を培養し、培養物からステロール類を採取する方法が開示されている。
【0007】
一方、アシルCoA:ステロールアシルトランスフェラーゼはステロール化合物のエステル化を触媒する酵素であり、ステリルエステル加水分解酵素はステリルエステルの加水分解を触媒する酵素である。アシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質であるARE1の発現を強化した酵母は非特許文献5に開示されており、ス
テリルエステル加水分解酵素活性を有するYEH1のC末端にプロテインAを付加した融合タンパク質を導入した酵母が非特許文献6に開示されている。しかしながら、これらの文献ではARE1やYEH1の発現強化のC5,7−ジエン体ステロールやエルゴステロールなどのΔ5,7−ステロール産生への効果については記載されておらず、また、ARE1とYEH1の両方の発現を強化した酵母はこれまで知られていない。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2004-141125号公報
【非特許文献】
【0009】
【非特許文献1】J. Org. Chem. 44, 1582-1584 (1979)
【非特許文献2】Appl. Microbiol. Biotechnol. 63, 635-646(2004)
【非特許文献3】Biochem. Biophys. Res. Commun. 181, 509-517(1988)
【非特許文献4】J. Biol. Chem. 271(18), 10866-10873(1996)
【非特許文献5】Science 272, 1353-1356(1996)
【非特許文献6】Biochim. Biophys. Acta 1791, 118-124(2009)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、C5,7−ジエン体ステロールやエルゴステロールなどのΔ5,7−ステロール及びハイドロコルチゾンを効率よく産生する改変酵母を提供すること、およびこれを用いてC5,7−ジエン体ステロールやエルゴステロールなどのΔ5,7−ステロール及びハイドロコルチゾンを効率よく製造する方法を提供することを課題とする。
【課題を解決するための手段】
【0011】
本発明者らは上記課題を解決するために鋭意検討を行った。
上記のとおり、ステロール化合物はその大半がエステル体として細胞内に蓄積される。従って所望のステロール化合物を効率よく蓄積させるにはステロール化合物のエステル化を促進することが重要と一般的には考えられ、当業者であれば、Δ5,7−ステロールを効率よく産生するためにはステロールをエステル化するアシルCoA:ステロールアシルトランスフェラーゼ活性のみを増強し、逆にそのエステル体の加水分解を促進するステリルエステル加水分解酵素を低減化する、または改変しないと考えられる。しかしながら、本発明者らは、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子だけでなく、ステリルエステル加水分解酵素遺伝子の発現も同時に増強することを試みたところ、意外なことに、これらの両遺伝子の発現を増強することにより非常に効率よくΔ5,7−ステロールを産生させることができることを見出した。また、さらに酵母を改変することでハイドロコルチゾンも製造できることを見出し、本発明を完成させるに至った。
【0012】
すなわち、本発明は以下のとおりである。
[1]アシルCoA:ステロールアシルトランスフェラーゼ遺伝子とステリルエステル加水分解酵素遺伝子の発現が増強されるように改変された酵母。
[2]さらに、ステロールC−22デサチュラーゼ遺伝子(ERG5)及びステロールC−24メチルトランスフェラーゼ遺伝子(ERG6)の発現が低減するように改変された、[1]に記載の酵母。
[3]さらに、ステロールC−22デサチュラーゼ遺伝子(ERG5)の発現が低減するように改変された、[1]に記載の酵母。
[4]さらに、Δ7−還元活性、ステロール側鎖の20位及び22位との結合を切断する活性、ステロール3位酸化異性化活性、ステロイド17α水酸化活性、ステロイド21位水酸化活性、及びステロイド11β位水酸化活性から選択される少なくとも一つの活性を有
するように改変された[3]に記載の酵母。
[5]さらに、Δ7−還元活性、ステロール側鎖の20位及び22位との結合を切断する活性、ステロール3位酸化異性化活性、ステロイド17α水酸化活性、ステロイド21位水酸化活性、及びステロイド11β位水酸化活性を有するように改変された[3]に記載の酵母。
[6]サッカロミセス・セレビジエである、[1]〜[5]のいずれかに記載の酵母。
[7]親株がサッカロミセス・セレビジエYPH499株、サッカロミセス・セレビジエFY1679-06c株、サッカロミセス・セレビジエKA311A株またはサッカロミセス・セレビジエX2181-1B株である、[6]に記載の酵母。
[8] [1]〜[3]のいずれかに記載の酵母を培養してΔ5,7−ステロールを生成・蓄積させ、Δ5,7−ステロールを回収する、Δ5,7−ステロールの製造方法。
[9]培養する酵母が[1]又は[2]に記載の酵母であり、Δ5,7−ステロールがC5,7−ジエン体ステロールである、[8]に記載の方法。
[10]C5,7−ジエン体ステロールがコレスタ−5,7,24−トリエン−3β−オールまたは7−デヒドロコレステロールである、[9]に記載の方法。
[11]培養する酵母が[1]又は[3]に記載の酵母であり、Δ5,7−ステロールがエルゴスタ−5,7,24-トリエノールである、[8]に記載の方法。
[12]培養する酵母が[1]に記載の酵母であり、Δ5,7−ステロールがエルゴステロールである、[8]に記載の方法。
[13] [5]に記載の酵母を培養してハイドロコルチゾンを生成・蓄積させ、ハイドロコルチゾンを回収する、ハイドロコルチゾンの製造方法。
【発明の効果】
【0013】
本発明の酵母を用いることにより、Δ5,7−ステロール及びハイドロコルチゾンを産生させることができる。本発明の方法によってΔ5,7−ステロール及びハイドロコルチゾンを製造し、これらを用いて医薬を合成すれば、医薬の製造プロセスの効率化にも寄与すると考えられる。
【図面の簡単な説明】
【0014】
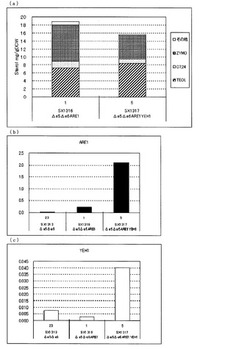
【図1】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。「C7,24」はコレスタ−7,24−ジエン−3β−オールを表し、「ZYMO」はザイモステロールを表し、「その他」はその他のステロール類を表す。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、Δe5Δe6は、ARE1とYEH1をいずれも増強していないerg5、erg6二重破壊株(SX1313)、Δe5Δe6ARE1は、ARE1のみを導入したerg5、erg6二重破壊株(SX1316)、Δe5Δe6ARE1YEH1は、ARE1とYEH1を導入したerg5、erg6破壊株(SX1317)を示す。いずれもYPH499を親株として作製された株である。
【図2】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、Δe5Δe6は、ARE1とYEH1をいずれも増強していないerg5、erg6二重破壊株(SX1312)、Δe5Δe6ARE1は、ARE1のみを導入したerg5、erg6二重破壊株(SX1319)、Δe5Δe6ARE1YEH1は、ARE1とYEH1を導入したerg5、erg6破壊株(SX1320)を示す。SX1319、SX1320については3クローンずつ評価した。いずれもFY1679−06cを親株として作製された株である。
【図3】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、Δe5Δe6は、ARE1とYEH1をいずれも増強していないerg5、erg6二重破壊株(SX1307)、Δe5Δe6ARE1は、ARE1のみを導入したerg5、erg6二重破壊株(SX1321)、Δe5Δe6ARE1YEH1は、ARE1とYEH1を導入したerg5、erg6破壊株(SX1322)を示す。いずれもKA311Aを親株として作製された株である。
【図4】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、Δe5Δe6は、ARE1とYEH1をいずれも増強していないerg5、erg6二重破壊株(SX1353)、Δe5Δe6ARE1は、ARE1のみを導入したerg5、erg6二重破壊株(SX1350)、Δe5Δe6ARE1YEH1は、ARE1とYEH1を導入したerg5、erg6破壊株(SX1351)を示す。いずれもX2181−1Bを親株として作製された株である。
【図5】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、−は、ARE1のみを導入したerg5、erg6二重破壊株(SX1319)、他はいずれもARE1とYEH1を導入したerg5、erg6二重破壊株であり、YEH1 locusは、YEH1をYEH1のlocusに導入した株(SX1320)、URA3 locusはYEH1をura3のlocusに導入した株(SX1332)、LEU2 locusは、YEH1をleu2のlocusに導入した株(SX1325)を示す。いずれもFY1679−06cを親株として作製された株である。
【図6】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、−は、ARE1とYEH1をいずれも導入していないerg5、erg6二重破壊株(SX1335)、他はいずれもARE1とYEH1を導入したerg5、erg6二重破壊株であり、ARE1 locusは、ARE1をARE1のlocusに導入した株(SX1320)、URA3 locusはARE1をura3のlocusに導入した株(SX1333)を示す。いずれもFY1679−06cを親株として作製された株である。
【図7】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE2とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE2とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、Δe5Δe6は、ARE2とYEH1をいずれも増強していないerg5、erg6二重破壊株(SX1335)、Δe5Δe6ARE2は、ARE2のみを導入したerg5、erg6二重破壊株(SX1347)、Δe5Δe6ARE2YEH1は、ARE2とYEH1を導入したerg5、erg6破壊株(SX1346)を示す。いずれもFY1679−06cを親株として作製された株である。
【図8】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。「ERGO」はエルゴステロールを表し、「ZYMO」はザイモステロールを表し、「その他」はその他のステロール類を表す。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、−は、ARE1とYEH1をいずれも増強していない株(SX1348)、ARE1は、ARE1のみを導入した株(SX1338)、ARE1YEH1は、ARE1とYEH1を導入した株(SX1337)を示す。いずれもFY1679−06cを親株として作製された株である。
【発明を実施するための形態】
【0015】
以下、本発明の実施の形態について詳細に説明する。
【0016】
(1)本発明の酵母
本発明の酵母は、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現とステリルエステル加水分解酵素遺伝子の発現が増強するように改変された酵母である。
【0017】
本発明において、Δ5,7−ステロールはステロール骨格の5位と7位に炭素−炭素二重結合を有するステロールを意味し、好ましくはエルゴステロールおよびC5,7−ジエン体ステロールである。また、C5,7−ジエン体ステロールはコレステロール骨格の5位と7位に炭素−炭素二重結合を有するステロールを意味するが、TEOL(コレスタ−5,7,24−トリエン−3β−オール)、7-デヒドロコレステロールが好ましく、TEOLが特に好ましい。また、ハイドロコルチゾンを目的生成物とする場合には、エルゴスタ−5,7,24−トリエノールが好ましい。Δ5,7−ステロールやC5,7−ジエン体ステロールには、それぞれΔ5,7−ステロールやC5,7−ジエン体ステロールのエステル体も含まれる。
【0018】
また、本発明において「アシルCoA:ステロールアシルトランスフェラーゼ遺伝子」とは、アシルCoA:ステロールアシルトランスフェラーゼ活性を有する蛋白質をコードする遺伝子を意味し、「ステリルエステル加水分解酵素遺伝子」とは、ステリルエステル加水分解酵素活性を有する蛋白質をコードする遺伝子を意味する。
【0019】
「アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現が増強した」とは、非改変株または親株と比較してアシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現が増強したことを意味する。アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現の増強は、非改変株または親株と比較して、単位菌体重量当たり5倍以上増強されていることが好ましく、10倍以上増強されていることがより好ましい。
なお、アシルCoA:ステロールアシルトランスフェラーゼは、ステロールのエステル化反応を触媒する酵素であり、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現は定量PCR法などによって測定することができる。
【0020】
「ステリルエステル加水分解酵素遺伝子の発現が増強した」とは、非改変株または親株と比較してステリルエステル加水分解酵素遺伝子の発現が増強したことを意味する。ステリルエステル加水分解酵素遺伝子発現の増強は、非改変株または親株と比較して、単位菌体重量当たり5倍以上増強されていることが好ましく、10倍以上増強されていることがより好ましい。
なお、ステリルエステル加水分解酵素遺伝子の発現は定量PCR法などによって測定することができる。
【0021】
本発明に用いる酵母は、以下に示すような酵母を親株として用い、該親株を改変することによって得ることができる。
親株として用いる酵母の種類は特に限定されないが、サッカロミセス属、デバリオマイセス属、シゾサッカロマイセス属、キャンディダ属、ヤロウィア属、ロドトルラ属、リポマイセス属、クルイベロマイセス属、ロドスポリジウム属、トリコデルマ属または、トルロプシス属、または、ピキア属に属する酵母であることが好ましく、例えば、サッカロミ
セス・セレビシエ(Saccharomyces cerevisiae)、サッカロミセス・バヤヌス(Saccharomyces bayanus)、デバリオマイセス・ニルソニ(Debaryomyces nilssonii)、デバリオマイセス・ハンセニ(Debaryomyces hansenii)、シゾサッカロマイセス・ポンベ(Schizosaccharomyces pombe)、キャンディダ・グラブラータ(Candida glabrata)、キャンディダ・トロピカリス(Candida tropicalis)、キャンディダ・ユティリス(Candida utilis)、キャンディダ・ボイディニィ(Candida boidinii)、ヤロウィア・リポリティカ(Yarrowia lipolytica)、ロドトルラ・グルティニス(Rhodotorula glutinis)、リポマイセス・リポフェラス(Lipomyces lipoferus)、クルイベロマイセス・ラクティス(Kluyveromyces lactis)、ロドスポリジウム・トルロイディス(Rhodosporidium toruloides)、トリコデルマ・リセイ(Trichoderma reesei)、トルロプシス・コリキュロサ(Torulopsis colliculosa)、ピキア・ファリノサ(Pichia farinosa)が挙げられる。
【0022】
これらの中で、代謝工学的手法が用いやすいことからサッカロミセス属が好適である。さらに、サッカロミセス属は、his3、leu2、trp1、ura3などの栄養要求性の遺伝型であることが望ましく、例えば、以下の株が例示される。
サッカロミセス・セレビジエYPH499株(MATa ura3 lys2 ade2 trp1 his3 leu2)(ATCC204679:American Type Culture Collection又はSTRATAGENE社より入手できる)、サッカロミセス・セレビジエFY1679-06c株(MATα ura3 leu2 trp1 his3)(Euroscarf社から入手できる)、サッカロミセス・セレビジエX2181-1B株(MATalpha his2 gal1 trp1 ade1
MAL SUC)(ATCC204822:American Type Culture Collectionより入手できる)。あるいは、サッカロミセス・セレビシエKA311A株(MATa his3 leu2 trp1 ura3)(Mol. Cell Biol. 13, 307-3083(1993))も用いることができ、KA311A株は、独立行政法人産業総合研究所特許生物寄託センターに受託番号:FERM P-19053として寄託されている。
【0023】
アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現およびステリルエステル加水分解酵素遺伝子の発現の増強は、遺伝子組換え法、例えば、該酵素をコードする遺伝子のコピー数を高めること、またはこの遺伝子のプロモーターを置換することによってこれらの遺伝子の発現量を増大させることによって行うことができる。
【0024】
アシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードする遺伝子として、ARE1遺伝子やARE2遺伝子が挙げられる。ARE1遺伝子およびARE2遺伝子は、多くの微生物で知られており、アシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードする限り特に限定されないが、例えば、それぞれ配列番号43または45の塩基配列を有するサッカロミセス・セレビジエ由来の遺伝子を挙げることができる。また、ARE1遺伝子およびARE2遺伝子は、アシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードするものである限り、配列番号43または45の塩基配列の相補配列を有するDNAとストリンジェントな条件でハイブリダイズするDNA、またはこれらの塩基配列と80%以上、好ましくは90%以上、より好ましくは95%以上、特に好ましくは99%以上の相同性を有するDNAのようなホモログであってもよい。ここで、ストリンジェントな条件としては、通常のサザンハイブリダイゼーションの洗いの条件である60℃、1×SSC,0.1%SDS、好ましくは、0.1×SSC、0.1%SDSに相当する塩濃度でハイブリダイズする条件が挙げられる。
【0025】
また、ARE1遺伝子およびARE2遺伝子は、それぞれ配列番号44または46のアミノ酸配列と80%以上、好ましくは90%以上、より好ましくは95%以上、特に好ましくは99%以上の同一性を有し、アシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードするDNAであってもよい。
【0026】
さらに、ARE1遺伝子およびARE2遺伝子は、配列番号44または46のアミノ酸配列において、1または数個のアミノ酸が置換、欠失、挿入または付加された配列を有し、アシル
CoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードするDNAであってもよい。ここで、1または数個とは、好ましくは、1〜20個、より好ましくは1〜10個、特に好ましくは1〜5個を意味する。
【0027】
また、サッカロミセス・セレビジエ以外の酵母、または他の微生物又は動植物由来のARE1遺伝子およびARE2遺伝子を使用することもできる。微生物または動植物由来のARE1遺伝子およびARE2遺伝子は、既にその塩基配列が決定されている遺伝子、またはホモロジー等に基づいてアシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードする遺伝子を微生物、動植物等の染色体より単離し、塩基配列を決定したものなどを使用することができる。また、塩基配列が決定された後には、その配列にしたがって合成した遺伝子を使用することもできる。これらはハイブリダイゼーション法やPCR法によりそのプロモーターおよびORF部分を含む領域を増幅することによって、取得することができる。
【0028】
ステリルエステル加水分解酵素活性を有するタンパク質をコードする遺伝子として、YEH1遺伝子が挙げられる。YEH1遺伝子は、多くの微生物で知られており、ステリルエステル加水分解酵素活性を有するタンパク質をコードする限り特に限定されないが、例えば、配列番号47の塩基配列を有するサッカロミセス・セレビジエ由来の遺伝子を挙げることができる。また、YEH1遺伝子は、ステリルエステル加水分解酵素活性を有するタンパク質をコードするものである限り、配列番号47の塩基配列の相補配列を有するDNAとストリンジェントな条件でハイブリダイズするDNA、またはこれらの塩基配列と80%以上、好ましくは90%以上、より好ましくは95%以上、特に好ましくは99%以上の相同性を有するDNAのようなホモログであってもよい。ここで、ストリンジェントな条件は、上記のとおりである。
【0029】
また、YEH1遺伝子は、それぞれ配列番号48のアミノ酸配列と80%以上、好ましくは90%以上、より好ましくは95%以上、特に好ましくは99%以上の同一性を有し、ステリルエステル加水分解酵素活性を有するタンパク質をコードするDNAであってもよい。
さらに、YEH1遺伝子は、配列番号48のアミノ酸配列において、1または数個のアミノ酸が置換、欠失、挿入または付加された配列を有し、ステリルエステル加水分解酵素活性を有するタンパク質をコードするDNAであってもよい。ここで、1または数個とは、好ましくは、1〜20個、より好ましくは1〜10個、特に好ましくは1〜5個を意味する。
【0030】
また、サッカロミセス・セレビジエ以外の酵母、または他の微生物又は動植物由来のYEH1遺伝子を使用することもできる。微生物または動植物由来のYEH1遺伝子は、既にその塩基配列が決定されている遺伝子、またはホモロジー等に基づいてステリルエステル加水分解酵素活性を有するタンパク質をコードする遺伝子を微生物、動植物等の染色体より単離し、塩基配列を決定したものなどを使用することができる。また、塩基配列が決定された後には、その配列にしたがって合成した遺伝子を使用することもできる。これらはハイブリダイゼーション法やPCR法によりそのプロモーターおよびORF部分を含む領域を増幅することによって、取得することができる。
【0031】
アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現、およびステリルエステル加水分解酵素遺伝子の発現を増強するためには、例えば、上記のようなDNAを宿主酵母で機能しうるプラスミドに発現可能に組込み、宿主酵母に導入すればよい。
【0032】
上記遺伝子を組込むことができるプラスミドベクターとしては、宿主酵母内での複製増殖機能を司る遺伝子を少なくとも含むものであれば特に制限されない。酵母に遺伝子を導入するために使用できるプラスミドの具体例としては、2μmプラスミド由来のpAURベクター(タカラバイオ社製)やpESCベクター(STRATAGENE社製)などが
挙げられる。これらのベクターは市販されており容易に入手可能である。
【0033】
なお、DNAの切断、連結、その他、染色体DNAの調製、PCR、プラスミドDNAの調製、形質転換、プライマーとして用いるオリゴヌクレオチドの設定等の方法は、当業者によく知られている通常の方法を採用することができる。これらの方法は、Sambrook, J., Fritsch, E. F., and Maniatis, T., "Molecular Cloning A Laboratory Manual, Second Edition", Cold Spring Harbor Laboratory Press, (1989)等に記載されている。
【0034】
ARE1(またはARE2遺伝子)およびYEH1遺伝子を、上記したような酵母内で複製可能なプラスミドベクターの適当な部位に挿入して得られる組換えベクターで、サッカロミセス・セレビジエなどの酵母を形質転換することにより、これらの遺伝子の発現が増加した酵母が得られる。
【0035】
形質転換は、例えば、エレクトロポレーション法(Fromm ME,et al.,(1986) Nature 319(6056):791-793)、スフェロプラスト法、酢酸リチウム法(Ito,H.et al, (1983) J. Bacteriol. 153(1), 163-168)等によって行うことができる。
また、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子およびステリルエステル加水分解酵素遺伝子の発現の増強は、公知の相同組換え法によって宿主のゲノム上でARE1(またはARE2遺伝子)およびYEH1遺伝子を多コピー化させることによって行うこともできる。例えば、ゲノム組込型プラスミドのうち多コピーがゲノムに組み込まれるベクターを用いる方法(Bio/Technol. 9, 1382-1385(1991))が挙げられる。ゲノム上の各遺伝子を含む発現単位の挿入箇所は、ステロール生合成関連遺伝子や生育関連遺伝子の発現が阻害されない箇所であれば何れのものでもよい。具体的には、ARE1遺伝子を適当なプロモーターの制御下となるように連結させたDNAを、宿主ゲノム上のARE1遺伝子座上などに挿入し、宿主ゲノム中のARE1遺伝子が2コピーとなるようにして、さらにYEH1遺伝子も同様のプロモーターの制御下となるように連結させたDNAを宿主ゲノム上のYEH1遺伝子座などに挿入して宿主ゲノム中のYEH1遺伝子が2コピーとなるようにゲノムを改変する方法などが好ましく用いられる。
【0036】
また、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子およびステリルエステル加水分解酵素遺伝子の発現の増強は、宿主ゲノム上でARE1(またはARE2遺伝子)およびYEH1遺伝子のプロモーターを置換または改変することによっても行うことができる。
上記組換えプラスミドによる導入または染色体上への相同組換えにおいて、ARE1(またはARE2遺伝子)およびYEH1遺伝子を発現させるためのプロモーター、または染色体上のARE1(またはARE2遺伝子)およびYEH1遺伝子のプロモーターを置換するために使用するプロモーターは、宿主酵母で機能しうるものであれば特に制限されないが、例えば、3−フォスフォグリセレートキナーゼ1遺伝子のプロモーター(以下、「PGK1プロモーター」と称することがある)あるいはアルコールデヒドロゲナーゼ遺伝子などの恒常的プロモーター、ないしは、GAL1やGAL10(Mol. Cell Biol. 4(8), 1440-1448(1984))などの誘導型プロモーター(例えば、STRATAGENE社製)などが挙げられる。
【0037】
なお、本発明の酵母は、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子、およびステリルエステル加水分解酵素遺伝子の発現の増強に加えて、他の酵素遺伝子の発現が非改変株と比べて増強または低下するように改変されたものでもよい。
他の酵素の改変は目的とするΔ5,7−ステロールの種類により適宜選択される。
【0038】
例えば、TEOLを生産する場合、ステロールC−22デサチュラーゼ遺伝子(ERG5)とステロールC−24メチルトランスフェラーゼ遺伝子(ERG6)の発現が低下するように改変することが好ましい。これらの改変は特開2004-141125に記載されている。
また、Δ5,7−ステロールとしてのエルゴスタ−5,7,24−トリエノールを中間体とし
てハイドロコルチゾンを目的生成物とする場合には、ステロールC−22デサチュラーゼ遺伝子(ERG5)のみの発現が低下するように改変することが好ましい。
【0039】
ステロールC−22デサチュラーゼは、各種ステロール類の側鎖の22位に二重結合を導入する能力を有する酵素であり、具体的には、例えばコレスタ−5,7,24−トリエノールをコレスタ−5,7,22,24−テトラエノールに、エルゴスタ−5,7−ジエノールをエルゴスタ−5,7,22−トリエノールに、エルゴスタ−5,24−ジエノールをエルゴスタ−5,22,24−トリエノールに、エルゴスタ−5,7,24−トリエノールをエルゴスタ−5,7,22,24−テトラエノールに変換する能力を有する酵素を指す。
【0040】
また、ステロールC−24メチルトランスフェラーゼは、S−アデノシルメチオニンから各種ステロール類の24位にメチル基を転移させる能力、さらには転移により付加されたメチル基にさらにメチル基を転移させる能力を有する酵素であり、具体的には、例えばザイモステロールをフェコステロールに変換する能力を有する酵素を指す。
これら酵素をコードする遺伝子は、既に単離され塩基配列が決定されているものもあり、例えば、ステロールC−22デサチュラーゼとしては、サッカロミセス・セレビシエ由来の遺伝子が報告されており(Biochem. Biophys. Acta 1299, 313-324(1996))、ステロールC−24メチルトランスフェラーゼとしては、サッカロミセス・セレビシエ由来の遺伝子が報告されている(Gene 169, 105-109(1996))。
【0041】
ステロールC−22デサチュラーゼ、および、ステロールC−24メチルトランスフェラーゼの両方の活性低減化方法としては、エチルメタンスルホン酸(EMS)、N−メチル−N'−ニトロ−N−ニトロソグアニジン(NTG)などの化学物質あるいはUVなどの変異源処理によって得られた両酵素遺伝子の欠損突然変異株を交配する方法、両遺伝子の転写をアンチセンス遺伝子の導入によりおさえる方法、両遺伝子の転写産物をRNAi技術によって抑制する方法、両酵素の特異的阻害剤で酵素活性を抑える方法などを用いることができる。
【0042】
酵母においては、ステロールC−22デサチュラーゼおよびステロールC−24メチルトランスフェラーゼをそれぞれコードしている遺伝子断片中に、HIS3、LEU2、TRP1、URA3などの栄養要求性遺伝子DNAないしはジェネティシンやオーレオバシジンAなどの抗生物質や抗真菌薬に対する耐性遺伝子が挿入された遺伝子断片、あるいはその遺伝子断片がさらにベクターに挿入されたプラスミドを作製し、相同組換えにより宿主染色体中の該遺伝子を破壊する方法が好適に用いられる。
【0043】
例えば、サッカロミセス・セレビシエにおいては、ステロールC−22デサチュラーゼをコードする遺伝子(ERG5)を含むDNA断片、ステロールC−24メチルトランスフェラーゼをコードする遺伝子(ERG6)を含むDNA断片を制限酵素切断ないしはポリメラーゼチェーンリアクション法(以下これを「PCR」と略称することがある)によって得てベクターに組み込んで中間ベクターを作製する。この中間ベクターをERG5遺伝子およびERG6遺伝子の遺伝子内部にある任意の制限酵素部位で2つに切断し、酵母のHIS3、LEU2、TRP1、URA3などの栄養要求性遺伝子cDNAのいずれか1つを2つの中間ベクター同士で重ならないように選択し挿入することにより遺伝子破壊用ベクター、ないしはそれをPCRで増幅して得られる遺伝子破壊用DNA断片を作製する。次いでこの遺伝子破壊用ベクターないしは遺伝子破壊用DNA断片を酵母に遺伝子導入することで相同組換えを誘発し、目的遺伝子を破壊することができる。
【0044】
かくして構築される遺伝子破壊用DNA断片若しくは遺伝子破壊用ベクターの具体例として、ERG5遺伝子の内部371bpを欠失させ、この欠失部分にTRP1遺伝子が挿
入されているDNA断片(erg5::TRP1)若しくは該DNA断片を有するプラスミドpSK−erg5::TRP1、ERG6遺伝子の内部476bpを欠失させ、この欠失部分にHIS3遺伝子が挿入されているDNA断片(erg6::HIS3)若しくは該DNA断片を有するプラスミドpUC−erg6::HIS3(特開2004-141125号公報を参照)等が挙げられる。
【0045】
酵母における遺伝子破壊の確認は、特定の栄養要求性遺伝子が挿入された遺伝子部分が相同組換えにより酵母ゲノム中に組み込まれるために、宿主株が持っていた複数の栄養要求性のうち該当する栄養要求性が消失することで知ることができる。さらにERG5およびERG6遺伝子の5'末端と3'末端にそれぞれ設定したPCRプライマーを用いて、遺伝子破壊操作を受けた酵母株から抽出した染色体DNAを鋳型としてPCRを行い、PCR断片の長さが挿入した栄養要求性遺伝子の長さだけ大きくなっていることを確認することにより遺伝子破壊の確認を行うことができる。
かくして得られる遺伝子破壊株の具体例としては、遺伝子破壊用ベクターpSK−erg5::TRP1のerg5::TRP1断片でERG5遺伝子を破壊した、サッカロミセス・セレビシエKAΔ5−5株、さらに該Δ5−5株のERG6遺伝子をpUC−erg6::HIS3のerg6::HIS3断片で破壊したKAΔ5Δ6−1株、KAΔ5Δ6−4株、KAΔ5Δ6−5株等が挙げられる。
さらに、本発明の酵母としては、以下の(3)に記載の酵母も含まれる。
【0046】
(2)Δ5,7−ステロールの製造方法
上記で説明した本発明の酵母を炭素源、窒素源、無機塩、アミノ酸、ビタミンなどを含有する培地中、好気条件下、温度、pHなどを調整しつつ培養を行えば、酵母菌体中にΔ5,7−ステロールが蓄積するのでこれを採取する。
【0047】
炭素源としては、例えばグルコース、グリセロール、フルクトース、シュークロース、マルトース、マンノース、澱粉、澱粉加水分解液、糖蜜、などの炭水化物、酢酸、ピルビン酸、メバロン酸などの各種有機酸が使用できる。さらに酵母の資化性によって、炭化水素、アルコールなども用いられる。特に廃糖蜜は好適にもちいられる。
窒素源としては、例えばアンモニア、または塩化アンモニウム、炭酸アンモニウムなどの各種無機塩および有機アンモニウム塩類あるいはペプトン、肉エキス、酵母エキス、カゼイン加水分解物など種々のものが使用可能である。
無機塩としては、例えばリン酸二水素カリウム、リン酸水素二カリウム、硫酸アンモニウム、塩化アンモニウム、硫酸マグネシウム、塩化ナトリウムなどを使用する。ビタミン、アミノ酸としては、使用する酵母の種類によってことなるが、必要に応じ添加する。また使用する菌株が、栄養要求性を示す場合は、その要求物質を添加する。
【0048】
培養は使用する菌株に応じて適切な条件で行うが、通常は、振盪培養または通気攪拌培養などの好気的条件下に行う。生産株として、サッカロミセス・セレビシエを用いる場合は、培養温度は20〜40℃が好適であり、培地のpHは弱酸性付近に維持する事が望ましい。培養期間は通常1〜15日間で菌体中にΔ5,7−ステロールが蓄積する。また、培養期間中に上記に記載した炭素源や窒素源、無機塩、ビタミン、アミノ酸を連続的、あるいは断続的に添加する流加培養や連続培養を行うことにより、菌体の生育やΔ5,7−ステロールの蓄積をさらに伸ばすことが可能である。
【0049】
培養終了後のΔ5,7−ステロールの抽出は一般に生物組織からステロール類を抽出する方法により行うことができる。具体的には、例えば、菌体を海砂とともにそのままアセトン中で破砕し酢酸エチルで抽出するか、又は、2MのNaOHを含む50%エタノール中で2時間煮沸し鹸化したのちヘプタンないしはクロロホルム/メタノール/水(体積比86:16:1)混液で抽出し、薄層クロマトグラフィーにかけステロール画分を掻き取
って、窒素気流下で乾燥させメタノール/クロロホルム混液に溶解することで総ステロールを菌体から抽出することができる。
【0050】
次いでステロールの種類の同定は、薄層クロマトグラフィーで分離抽出したステロール画分を、それ自体公知の適当な溶媒およびカラムの組み合わせでガスクロマトグラフィーないしは高速液体クロマトグラフィーにかけて、現れるピークの保持時間を標準試料の保持時間と比較することで行うことができる。また、同定された各種ステロールの総ステロール中にしめる割合は各ピークの面積から算出することができる。さらに、これらの主要なステロールのピークが目的化合物であることの確認は、マススペクトロメトリーを用いて、分子量および開裂パターンを調べることで行うことができる。また、GC−MSあるいはHPLC−MSを用いることによりさらに効率よく抽出されたステロール類を同定することができる。
【0051】
得られたΔ5,7−ステロールは各種医薬等の中間体となりうる。
例えば、TEOLはケノデオキシコール酸やウルソデオキシコール酸の合成原料として有用である。特開2007-210888号公報および特開2006-056877号公報に、TEOLからのウルソデオキシコール酸の合成方法が開示されている。
具体的には、TEOLを原料として、以下の4つの工程、すなわち
(I)3位水酸基の酸化と5位2重結合の4位への異性化を行う工程
(II)側鎖の酸化的切断により24位をカルボキシル基またはそのエステル誘導体とする工程
(III)7位に酸素官能基を導入する工程
(IV)4位2重結合の還元飽和化による5β立体構築を行う工程
を経て5β−3,7−ジオキソコラン酸を製造し、これを還元することによってウルソデオキシコール酸を製造することができる。
5β−3,7−ジオキソコラン酸のウルソデオキシコール酸への還元反応は、金属還元または接触水素化によって行うことができる。また、5β−3,7−ジオキソコラン酸からウルソデオキシコール酸への変換は、特開昭60-228500号公報及び特開平5-32692号公報の記載を参酌して行うことができる。
【0052】
(3)ハイドロコルチゾンの製造方法
上記酵母で、以下の活性を有するものを上記(2)と同様にして培養することによりハイドロコルチゾンが生成される。活性を有する酵母とは、該酵母自体がそれぞれの活性を有していてもよいし、該活性を有する物質を培養液中に添加してもよいし、該活性を有する酵母の菌体あるいはその処理物を培養液中に添加することでもよい。
【0053】
ハイドロコルチゾンの生成にさらに必要な活性とは、Δ7−還元活性、ステロール側鎖の20位及び22位との結合を切断する活性、ステロール3位酸化異性化活性、ステロイド17α水酸化活性、ステロイド21位水酸化活性、及びステロイド11β位水酸化活性である。本発明の酵母には、上記活性のうち少なくとも1つを有するものも含まれる。ハイドロコルチゾン生成を目的とする本発明の酵母としては、上記活性を有する蛋白質を生成するものが好ましく、また、発現するための遺伝子及びその活性に必要な補酵素などの遺伝子の全てを導入したものも好ましく挙げられる。上記活性を有する蛋白質を生成するためには、該蛋白質をコードするDNAを上記の発現ベクターに挿入して、上記酵母を形質転換した形質転換酵母を用いる方法が好ましく用いられる。
【0054】
ステロール側鎖の20位及び22位との結合を切断する活性を有する蛋白質を生成する活性を獲得する方法としては、 (i) 例えば、WO2010/079594公報に記載の酵素蛋白質(CYP204A1やCYPSS204A)をコードする遺伝子やその他の遺伝子がコードする同様の活性を有する酵素遺伝子と、該酵素蛋白質への電子伝達活性を有するフェレドキシン蛋白質、およ
び該フェレドキシン蛋白質への電子伝達活性を有するフェレドキシン還元酵素蛋白質のそれぞれをコードする遺伝子の混合物、あるいは(ii)上記酵素蛋白質と該酵素蛋白質への電子伝達活性を有するフェレドキシン蛋白質、および該フェレドキシン蛋白質への電子伝達活性を有するフェレドキシン還元酵素蛋白質の融合蛋白質をコードする遺伝子が発現するように該酵母に遺伝子導入する方法などが挙げられる。
【0055】
ここで、電子伝達活性を有するフェレドキシン蛋白質、および該フェレドキシン蛋白質への電子伝達活性を有するフェレドキシン還元酵素蛋白質は両者の活性を有する1種の蛋白質でもよい。例えば、Bacillus megateriumのP450-BM3遺伝子のP450還元酵素ドメインを酵素蛋白質(P450 2C11)に連結し、人工的に作製した融合蛋白質を利用できる(Helvig, C. and Capdevila, J. H., Biochemistry, (2000)39, 5196-5205)。
【0056】
フェレドキシン還元酵素は、例えば以下の中から選択される。
ホウレン草由来のフェレドキシンレダクターゼ、
シュードモナス プチダ(Pseudomonas putida)由来プチダレドキシンレダクターゼ、
動物由来のアドレノドキシンレダクターゼ、
ノボスフィンゴビウム サブテラニウム(Novosphingobium subterraneum)由来フェレドキシンレダクターゼ、
ノボスフィンゴビウム アロマティシボランス(Novosphingobium aromaticivorans)由来フェレドキシンレダクターゼ、
エシェリヒア コリ(Escherichia coli)由来フラボドキシンレダクターゼやフェレドキシンレダクターゼ、
サッカロミセス・セレビシエ(Saccharomyces cerevisiae)由来フェレドキシンレダクターゼ、
またはその他のフェレドキシンレダクターゼ。
【0057】
フェレドキシンは、例えば以下の中から選択される;
ホウレン草由来のフェレドキシン、
シュードモナス プチダ(Pseudomonas putida)由来プチダレドキシン、
動物由来のアドレノドキシン、
ノボスフィンゴビウム サブテラニウム(Novosphingobium subterraneum)由来フェレドキシン、
ノボスフィンゴビウム アロマティシボランス(Novosphingobium aromaticivorans)由来フェレドキシン、
エシェリヒア コリ(Escherichia coli)由来フラボドキシンやフェレドキシン、
サッカロミセス・セレビシエ(Saccharomyces cerevisiae)由来フェレドキシン、
またはその他のフェレドキシン型蛋白質。
【0058】
ステロイド17α位水酸化酵素蛋白質、ステロイド21位水酸化酵素蛋白質、ステロイド11β位水酸化酵素蛋白質、ステロール3位酸化異性化酵素蛋白質は、例えば、Molecular and Cellular Endocrinology 1990, 73, 73-80に記載のものが用いられる。
また、ステロールΔ7−還元酵素蛋白質は、Journal of Biological Chemistry 1996, 271, 10866-10873記載のシロイヌナズナ(Arabidopsis thaliana)由来のもの、あるいはApplied and Environmental Microbiology 2007, 73, 1736-1741記載のモルティエレラ(Mortierella)属カビ由来ものが用いられる。
具体的には、ステロールΔ7−還元酵素蛋白質はシロイヌナズナ由来NADPH−ステロールΔ7−還元酵素モルティエレラ・アルピナ(Mortierella alpina)由来ステロー
ルΔ7−還元酵素(MoDELTA7SR)が好ましい。
ステロイド17α位水酸化酵素蛋白質は、例えば、ウシ、マウス、ラット、あるいはヒト由来シトクロムP450c17が好ましい。
また、ステロイド21位水酸化酵素蛋白質は、ウシ、マウス、ラット、あるいはヒト由来シトクロムP450c21が好ましい。
ステロイド11β位水酸化酵素蛋白質としてはウシ、マウス、ラット、あるいはヒト由来シトクロムP450c11やクルブラリア・ルナタ(Curvularia lunata)由来P-450lun等が好ましい。
さらにステロール3位酸化異性化酵素蛋白質はウシ、マウス、ラット、あるいはヒト由来3β−ヒドロキシステロイド脱水素酵素(3βHSD)やストレプトミセス(Streptomyces)属細菌由来コレステロール酸化酵素が好ましい。
【0059】
ここで、好ましく用いられる酵母としては、サッカロミセス(Saccharomyces)属酵母、ピキア(Pichia)属酵母、シゾサッカロミセス(Schizosaccharomyces)属酵母、ヤロウィア(Yarrowia)属酵母、アスペルギルス(Aspergillus)属、モルティエレラ(Mortierella)属があげられる。より好ましくは、サッカロミセス属酵母があげられる。具体的にはサッカロミセス・セレビシエ(Saccharomyces cerevisiae)であり、より具体的には、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)FY1679-28c株(JP2004-528827 A1)、以下同様に、FY1679-18b株(JP2004-528827 A1)、KA311A株(FERM:P-19053)、YPH499株(ATCC#204679)、YPH500株(ATCC#204680)等が挙げられ、それぞれの酵素蛋白質をコードするDNAは、pESC-LEU(Stratagene社)等の発現ベクターや酵母菌の染色体に挿入されていることが好ましい。
【0060】
また、これらの酵母が、以下(1)−(5)の性質のいずれかまたはすべてを持つことが好ましい。
(1)ステロール−22デサチュラーゼ蛋白質をコードする遺伝子またはその発現を制御する領域のDNA配列を改変することで内在性のステロールの22位と23位の間が不飽和化されない、
(2)NADP−シトクロムP450還元酵素蛋白質の発現が本蛋白質をコードする遺伝子をその発現制御領域とともに1コピー以上宿主細胞に導入することで、強化されている、
(3)ステロールエステル加水分解酵素の発現が本蛋白質をコードする遺伝子をその発現制御領域とともに1コピー以上宿主細胞に導入することで、強化されている、
(4)ステロイド17α水酸化酵素、ステロイド21位水酸化酵素、ステロイド11β位水酸化酵素に電子を伝達するフェレドキシン型蛋白質および該蛋白質へ電子伝達活性を有するフェレドキシン還元酵素蛋白質のそれぞれをコードする遺伝子をその発現制御領域とともに宿主細胞に導入することで、該蛋白質の発現が強化されている、
(5)ステロール−24メチル基転移酵素蛋白質をコードする遺伝子またはその発現を制御する領域のDNA配列を改変することで内在性のステロールの24位にメチル基が存在しない。
【0061】
反応混合物からハイドロコルチゾンを分画する方法は特に限定されず、当業者に公知の分離又は精製のための手法を用いることができる。例えば、溶媒抽出、晶析、樹脂吸着、カラムクロマトグラフィー等により行うことができるが、これに限定されるものではない。
【0062】
また、上記で製造したハイドロコルチゾンからは、各種の誘導体を公知の方法で製造することができる。さらに、かくして製造されたハイドロコルチゾンおよびその誘導体は、それ自体既知の方法により試験管内、あるいは生体内における薬理学的または生理学的試験により、その医薬としての活性や安全性のスクリーニングを行なうことにより疾病治療薬とすることができる。この場合、上記工程により得た化合物を有効成分とする医薬組成物を製造する方法も本発明に含まれる。
【0063】
具体的には、ハイドロコルチゾンそれ自体を単独で用いることも可能であるが、薬学的に許容され得る担体と配合して医薬組成物として用いることもできる。かくして得られる医薬化合物はそれ自体を単独で用いることも可能であるが、薬学的に許容され得る担体と配合して医薬組成物として用いることもできる。この時の有効成分の担体に対する割合は、0.01〜90重量%の間で変動され得る。
【0064】
本発明で得られたΔ5,7−ステロールやハイドロコルチゾンを含む医薬組成物は、必要に応じて糖衣を施した錠剤、カプセル剤、軟カプセル剤、散剤、顆粒剤、細粒剤、シロップ剤、乳剤、懸濁剤、液剤等の剤形として経口的に投与してもよいし、あるいは薬学的に許容し得る液との無菌性溶液または懸濁剤形等の注射剤として、例えば静脈内投与、筋肉内投与、局所内投与、皮下投与してもよい。また坐薬や舌下錠、経鼻、経肺製剤等の粘膜投与、さらには軟膏や貼付剤等の外用剤としての投与も可能である。経口、非経口の組成物を調製する場合には、有機または無機の固体または液体の担体、希釈剤とともに通常用いられる単位容量形態で混和することによって行うことができる。これら製剤における有効成分量は、指示された範囲の適当な容量が得られるようにするものである。
【0065】
経口投与および粘膜投与のための固形製剤を製造する際に用いられる賦形剤としては、例えば乳糖、ショ糖、デンプン、タルク、セルロース、デキストリン、カオリン、炭酸カルシウム等が用いられる。経口投与のための液体製剤、すなわちシロップ剤、懸濁剤、液剤等は、一般的に用いられる不活性な希釈剤、例えば水、植物油等を含む。この製剤は、不活性な希釈剤以外に補助剤、例えば湿潤剤、懸濁補助剤、甘味剤、香味剤、着色剤、保存剤、安定剤等を含むこともできる。注射剤、粘膜投与剤等の製造に用いられる溶剤または懸濁化剤としては、例えば水、プロピレングリコール、ポリエチレングリコール、ベンジルアルコール、オレイン酸エチル、レシチン等が挙げられる。坐剤に用いられる基剤としては、例えばカカオ脂、乳化カカオ脂、ラウリン脂、ウィテップゾール等が挙げられる。外用剤の製造には、溶解剤または溶解補助剤としてアルコ−ル、脂肪酸エステル類、プロピレングリコール等が、粘着剤としてカルボキシビニルポリマ−、多糖類等が、乳化剤として界面活性剤等などが使用される。
【実施例】
【0066】
以下、実施例を挙げて本発明をさらに詳細に説明するが、本発明はこれら実施例により何ら限定されるものではない。
【0067】
(実施例1)
サッカロミセス・セレビジエ YPH499、FY1679−06c株、KA311A株、X2181−1B株のerg5およびerg6遺伝子の二重破壊及びARE1およびYEH1増強株の作製
(1)酵母染色体DNAの調製
サッカロミセス・セレビジエ S288C株(ATCCより入手 ATCC20458)の染色体DNAは、特開2004-141125と同様の方法で調製された。
【0068】
(2)遺伝子破壊用DNA断片erg5::TRP1の導入によるerg5遺伝子破壊株SX1311、SX1306、SX1334の作製
サッカロミセス・セレビジエ由来ステロールC−22デサチュラーゼをコードする遺伝子ERG5破壊用のプラスミドpSK−erg5::TRP1(特開2004−141125号公報を参照)を制限酵素Sac I(タカラバイオ社製、以下、特に記載のない限り、制限酵素はタカラバイオ社製である)とKpn Iで切断した。このDNA断片をサッカロミセス・セレビジエYPH499株(MATa ura3 lys2 ade2 trp1 his3 leu2)(STRATAGENE社より入手)とFY1679−06c株(MATα ura3 leu2 trp1 his3)(Euroscarf
社より入手)、X2181−1B(MATalpha his2 gal1 trp1 ade1 MAL SUC)(ATCCより入手)に酢酸リチウム法(Ito,H.et al, (1983) J. Bacteriol. 153(1), 163-168)、もしくはエレクトロポレーション法(Fromm ME,et al.,(1986) Nature 319(6056):791-793)で形質転換し、YPH499株は各20mg/lのアデニン、L−ヒスチジン、ウラシル、30mg/l L−リジン、100mg/l L−ロイシンを、FY1679−06c株は各20mg/lのL−ヒスチジン、ウラシル、100mg/l L−ロイシンを、X2181−1B株は20mg/l アデニンをそれぞれ含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0069】
寒天培地上に生育してきたトリプトファン非要求性のコロニーをYPD液体培地(1%
酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーTRP1−outF(配列番号1)とリバースプライマーERG5−D31(配列番号2)の組み合わせでPCRを行い、ERG5遺伝子座上での相同組換えの有無を調べた。PCRはTaKaRa ExTaq DNA ポリメラーゼ(タカラバイオ社製)を用いて(1)94℃、2分、(2)94℃、1分、(3)53℃、1分、(4)72℃、3分、(2)から(4)の反応を30回繰り返した後、4℃で冷却の条件で反応を行った。比較対照として、特開2004-141125に記載されたKAΔ5−5株、およびYPH499株、FY1679−06c株、X2181−1B株の染色体DNAを鋳型として同様にPCRを行った。
【0070】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1311(YPH499株由来)、SX1306(FY1679−06c株由来)、SX1334(X2181−1B株由来)と名付けた形質転換体は目的サイズと同じ約1.93kbpのDNA断片が検出された。従って、SX1311、SX1306、SX1334株は相同組換えによりERG5遺伝子が破壊されたことが確認された。
【0071】
(3)SX1311、SX1306株への遺伝子破壊用DNA断片erg6::HIS3の導入、およびSX1334株への遺伝子破壊用DNA断片erg6::HIS2の導入によるerg5、erg6二重遺伝子破壊株SX1313、SX1312、SX1340株の作製
(i)遺伝子破壊用DNA断片erg6::HIS3導入によるerg5、erg6二重破壊株SX1313、SX1312株の作製
サッカロミセス・セレビジエ由来ステロールC−24メチルトランスフェラーゼをコードする遺伝子ERG6破壊用のプラスミドpUC−erg6::HIS3(特開2004-141125を参照)を制限酵素ScaIとEcoRIで切断した。このDNA断片を上記(2)で作製したSX1311、SX1306株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SX1311株は各20mg/lのアデニン、ウラシル、30mg/l L−リジン、100mg/l L−ロイシンを、SX1306株は20mg/l ウラシル、100mg/l L−ロイシンをそれぞれ含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0072】
寒天培地上に生育してきたヒスチジンおよびトリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、 2%ポリペプトン、 2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーERG6−U52(配列
番号3)とリバースプライマーERG6−D34(配列番号4)の組み合わせでPCRを行い、ERG6遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。比較対照として、特開2004-141125に記載されたKAΔ5Δ6−1株、およびYPH499株、FY1679−06cの染色体DNAを鋳型として同様にPCRを行った。
【0073】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1313(YPH499株由来)、SX1312(FY1679−06c株由来)と名付けた形質転換体は目的サイズと同じ約1.78kbpのDNA断片が検出された。従って、SX1313,SX1312株は相同組換えによりERG6遺伝子が破壊されたことが確認された。
【0074】
(ii)破壊用ERG6、酵母発現用HIS2遺伝子断片の調製
サッカロミセス・セレビジエ由来ステロールC−24メチルトランスフェラーゼをコードする遺伝子(ERG6)を含むDNA断片、HIS2遺伝子を含むDNA断片は、上記(1)で取得したサッカロミセス・セレビジエ S288C株の染色体DNAを鋳型にしてPCRにて合成、単離した。以下にその方法の詳細を示す。
【0075】
ERG6遺伝子断片のPCRによる合成には、フォワードプライマーERG6−5L(配列番号5)とリバースプライマーERG6−3L(配列番号6)を用いた。このプライマーの組み合わせでERG6の蛋白質コード領域を含み、開始コドンの238塩基前から終始コドンの258塩基後までを増幅することができる。また、5’側には制限酵素PstIの認識配列を、3’側にはEcoRIの認識配列を含んだ形で合成することができる。
HIS2遺伝子断片のPCRによる合成には、フォワードプライマーHIS2−F(配列番号7)とリバースプライマーHIS2−R(配列番号8)を用いた。このプライマーの組み合わせでHIS2の蛋白質コード領域を含み、開始コドンの430塩基前から終始コドンの290塩基後までを増幅することができる。また、5’側には制限酵素SmaIの認識配列を、3’側にはEcoRIの認識配列を含んだ形で合成することができる。
PCRは、PrimeSTAR HS DNA ポリメラーゼ(タカラバイオ社製)を用いて、(1)98℃、2分、(2)98℃、10秒、(3)55℃、5秒、(4)72℃、2分、(2)から(4)の反応を30回繰り返し、(5)72℃、2分間反応させた後、4℃で冷却の条件で反応を行った。次にPCR産物をアガロースゲル電気泳動し、約1.65kbpのERG6遺伝子断片を、約1.73kbpのHIS2遺伝子断片を確認した。
【0076】
(iii)erg6破壊用プラスミドpUC−erg6::HIS2の作製
pUC19(タカラバイオ社製)を制限酵素PstIとEcoRIで切断したものと、上記(ii)でPCR合成したERG6遺伝子断片をPstIとEcoRIで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素HincIIによる切断で、約0.64kbpと3.66kbpの断片が確認できたものを目的プラスミドとして、pUC−ERG6Lと名付けた。
次に、pGEM−T Easy Vector Systems(プロメガ社製)を用いて、pGEM−T Easy(プロメガ社製)のクローニングサイトに上記(ii)でPCR合成したHIS2遺伝子断片をダイレクトクローニングにより連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素Sm
aIとEcoRIによる切断で、約3.0kbpと1.73kbpの断片が確認できたものを目的プラスミドとして、pGEM−T−HIS2と名付けた。
【0077】
さらに、上記pUC−ERG6Lを制限酵素BspT104Iで切断し、cloned
DNA Polymerase I(タカラバイオ社製)を用いてDNA末端平滑化処理を行った後に、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、プラスミドpGEM−T−HIS2を制限酵素SmaIとXbaIで切断し、DNA Polymerase I(タカラバイオ社製)を用いてDNA末端平滑化処理を行ったものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素ScaIによる切断で、約1.71kbpと1.67kbp、1.89kbpの断片が確認できたものを目的プラスミドとして、pUC−erg6::HIS2と名付けた。
【0078】
(iv)erg5、erg6二重遺伝子破壊株SX1340の作製
上記(iii)で作製したERG6破壊用のプラスミドpUC−erg6::HIS2を制限酵素EcoRIとPstIで切断した。このDNA断片を上記(2)で作製したSX1334株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、20mg/l アデニンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0079】
寒天培地上に生育してきたヒスチジンおよびトリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、 2%ポリペプトン、 2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーERG6−U52(配列番号3)とリバースプライマーERG6−D34(配列番号4)の組み合わせでPCRを行い、ERG6遺伝子座上での相同組換えの有無を調べた。PCRは上記(i)で述べた条件で行った。比較対照として、特開2004-141125に記載されたKAΔ5Δ6−1株、およびX2181−1B株の染色体DNAを鋳型として同様にPCRを行った。
PCR産物をアガロースゲル電気泳動で解析した結果、SX1340と名付けた形質転換体は目的サイズと同じ約2.76kbpのDNA断片が検出された。従って、SX1340株は相同組換えによりERG6遺伝子が破壊されたことが確認された。
【0080】
(4)erg5、erg6二重破壊株へのARE1遺伝子導入株の作製
(i)酵母発現用プラスミドpPG−LEUの作製
酵母細胞内で外来遺伝子を構成的に発現させるための3−ホスホグリセレートキナーゼ1遺伝子(PGK1)のプロモーター領域は、サッカロミセス・セレビジエKA311A株(受託番号:FERM P−19053)の染色体DNAを鋳型にし、フォワードプライマーPPGK1−5A(配列番号9)、リバースプライマーPPGK1−3A(配列番号10)を用いたPCRにて合成した。このプライマーの組み合わせで5'側に制限酵素BamH I、3'側に制限酵素EcoR Iの認識配列を含んだ形で合成することができる。PCRはTaKaRa ExTaq DNA ポリメラーゼ(タカラバイオ社製)を用いて(1)94℃、2分、(2)94℃、1分、(3)55℃、1分、(4)72℃、1分、(2)から(4)の反応を30回繰り返し、(5)72℃、4分間反応させた後、4℃で冷却の条件で反応を行った。次にPCR産物をアガロースゲル電気泳動し、約0.6kbpのPGK1プロモーターのDNA断片を確認した。
このPCR産物をpCR2.1(Invitrogen社製)に連結した。連結物を用
いてエッシェリシア・コリJM109に形質転換し、得られたコロニーからプラスミド抽出を行った。
得られたプラスミドをBamH I、EcoR Iで切断し、アガロースゲルから約0.6kbpのDNA断片(PGK1プロモーター)をGENE CLEAN II KIT(フナコシ社製)により抽出した。
【0081】
一方、酵母発現用ベクターpADNSΔE(特開2004-141125)も同じくBamH I、EcoR Iで切断し、アガロースゲルから約6.58kbpのDNA断片を抽出した。
上記2つのDNA断片をTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結し、エッシェリシア・コリJM109に形質転換し、得られたコロニーからプラスミドを抽出した。アガロースゲル電気泳動により約7.2kbpを確認したプラスミドを目的プラスミドとし、pPG−LEUと名付けた。pPG−LEUはプロモーターとしてPGK1プロモーター、ターミネーターとしてアルコールデヒドロゲナーゼ1遺伝子(ADH1)由来のADH1ターミネーターを持つ。
【0082】
(ii)酵母組換え用プラスミドpPG−LEU(−2μ)の作製
上記(i)で作製したプラスミドpPG−LEUを制限酵素Hpa IとEco47IIIで切断し、アガロースゲルから約5.4kbpのDNA断片を抽出することで2μm
DNAを除き、得られたDNA断片を自己連結し、エッシェリシア・コリJM109に形質転換し、得られたコロニーからプラスミドを抽出した。アガロースゲル電気泳動により約5.4kbpを確認したプラスミドを目的プラスミドとし、pPG−LEU(−2μ)と名付けた。
【0083】
(iii)酵母発現用のARE1、URA3、ADE1遺伝子断片の調製
サッカロミセス・セレビジエ由来アシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子(ARE1)を含むDNA断片、URA3、ADE1を含むDNA断片は、上記(1)で取得したサッカロミセス・セレビジエ S288C株の染色体DNAを鋳型にしてPCRにて合成、単離した。以下にその方法の詳細を示す。
ARE1遺伝子断片のPCRによる合成には、フォワードプライマーARE1−Sma
I(F)(配列番号11)とリバースプライマーARE1−EcoR I(R)(配列番号12)を用いた。このプライマーの組み合わせでARE1の蛋白質コード領域である開始コドンから終始コドンまでを増幅することができる。また、5'側には制限酵素Sma Iの認識配列を、3'側にはEcoR Iの認識配列を含んだ形で合成することができる。 URA3遺伝子断片のPCRによる合成には、フォワードプライマーURA3−2F(配列番号13)とリバースプライマーURA3−1R(配列番号14)を用いた。このプライマーの組み合わせでURA3の蛋白質コード領域を含み、開始コドンの254塩基前から終始コドンの178塩基後までを増幅することができる。また、合成された遺伝子断片中には開始コドンの223塩基前と終始コドンの136塩基後にHind IIIの認識配列が含まれた形で合成される。ADE1遺伝子断片のPCRによる合成には、フォワードプライマーADE1−F(配列番号15)とリバースプライマーADE1−R(配列番号16)を用いた。このプライマーの組み合わせでADE1の蛋白質コード領域を含み、開始コドンの346塩基前から終始コドンの312塩基後までを増幅することができる。また、合成された遺伝子断片中には終始コドンの106塩基後にEcoRVの認識配列が含まれた形で合成される。PCRは、PrimeSTAR HS DNA ポリメラーゼ(タカラバイオ社製)を用いて、(1)98℃、2分、(2)98℃、10秒、(3)55℃、5秒、(4)72℃、2分、(2)から(4)の反応を30回繰り返し、(5)72℃、7分間反応させた後、4℃で冷却の条件で反応を行った。次にPCR産物をアガロースゲル電気泳動し、約1.83kbpのARE1遺伝子断片を、約1.23kbpのURA3遺伝子断片を、約1.57kbpのADE1遺伝子断片を確認した。
【0084】
(5)ARE1発現用プラスミドpPGU−ARE1(−2μ)、pPGADE1−ARE1(−2μ)の作製
上記(2)で作製したプラスミドpPG−LEU(−2μ)を制限酵素SmaIとEcoRIで切断したものと、PCR合成したARE1遺伝子断片をSmaIとEcoRIで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素SmaIとEcoRIによる切断で、約5.4kbpと1.83kbpの断片が確認できたものを目的プラスミドとして、pPGL−ARE1(−2μ)と名付けた。
【0085】
次に、pUC18(タカラバイオ社製)をHindIIIで切断し、Alkaline
Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、PCR合成したURA3遺伝子断片をHindIIIで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素Hind IIIによる切断で、約2.68kbpと1.23kbpの断片が確認できたものを目的プラスミドとして、pUC−URA3と名付けた。
【0086】
また、pCR−Blunt(invitrogen社製)のクローニングサイトに上記(iii)でPCR合成したADE1遺伝子断片をダイレクトクローニングにより連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素SpeIとPstIによる切断で、約3.47kbpと1.62kbpの断片が確認できたものを目的プラスミドとして、pCR−B−ADE1と名付けた。
【0087】
続いて、pUC19(タカラバイオ社製)をXbaIとSmaIで切断し、cloned DNA Polymerase I(タカラバイオ社製)を用いてDNA末端平滑化処理を行った後に、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、上記pCR−B−ADE1をSpeIとEcoRVで切断し、cloned DNA Polymerase I(タカラバイオ社製)を用いてDNA末端平滑化処理を行ったものをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素EcoRIによる切断で、約1.01kbpと0.39kbp、2.68kbpの断片が確認できたものを目的プラスミドとして、pUC19−ADE1と名付けた。
【0088】
さらに、上記pPGL−ARE1(−2μ)をSspIで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、上記pUC−URA3をHindIIIで切断し、cloned DNA PolymeraseI(タカラバイオ社製)を用いてDNA末端平滑化処理を行ったもの、上記pUC19−ADE1をPstIとKpnIで切断し、clone
d DNA PolymeraseI(タカラバイオ社製)を用いてDNA末端平滑化処理を行ったものとをそれぞれTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素EcoR IとPst Iによる切断で、約3.56kbpと3.07kbpの断片が確認できたもの、制限酵素BspT104Iによる切断で、約3.74kbpと1.29kbp、1.85kbpの断片が確認できたものをそれぞれ目的プラスミドとして、pPGU−ARE1(−2μ)、pPGADE1−ARE1(−2μ)と名付けた。
【0089】
(6)erg5、erg6二重破壊株SX1313、SX1312、KAΔ5−4、SX1340へのARE1遺伝子導入株SX1314、SX1318、SX1315、SX1350株の作製
erg5、erg6二重破壊株SX1313、SX1312、特開2004-141125に記載されたKAΔ5−4(以後、SX1307と記載する)のゲノム上のARE1遺伝子座へさらにARE1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE1発現用のプラスミドpPGU−ARE1(−2μ)を制限酵素Xho I(ARE1遺伝子中)で切断した。このDNA断片を上記(3)で作製したSX1313、SX1312株、SX1307株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、100mg/l L−ロイシンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino
Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。なお、SX1313株の形質転換を行ったSD寒天培地にのみ20mg/l アデニン、30mg/l L−リジンを添加した。
【0090】
寒天培地上に生育してきたウラシルおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーARE1−check(配列番号18)の組み合わせでPCRを行い、ARE1遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。
【0091】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1314(YPH499株由来)、SX1318(FY1679−06c株由来)、SX1315(KA311A株由来)と名付けた形質転換体は目的サイズと同じ約2.6kbpのDNA断片が検出された。従って、SX1314、SX1318、SX1315株のゲノムのARE1遺伝子座に上記相同組換えによりARE1遺伝子が導入され、合計2コピー存在することが確認された。
【0092】
さらに、erg5、erg6二重破壊株SX1340のゲノム上のARE1遺伝子座へさらにARE1遺伝子を導入することを目的として、上記と同様に以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE1発現用のプラスミドpPGADE1−ARE1(−2μ)を制限酵素Xho I(ARE1遺伝子中)で切断した。また、ARE1を導入していない対照株を作製するため、上記(5)で作製したpUC19−ADE1を制限酵素HpaIで切断した。これらのDNA断片を上記(3)で作製したSX1340株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD
寒天培地(0.67% Yeast Nitrogen Base w/o Amino
Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0093】
寒天培地上に生育してきたアデニンおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーARE1−check(配列番号18)の組み合わせでPCRを行い、ARE1遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。
【0094】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1350(X2181−1B株由来)と名付けた形質転換体は目的サイズと同じ約2.6kbpのDNA断片が検出された。従って、SX1350株のゲノムのARE1遺伝子座に上記相同組換えによりARE1遺伝子が導入され、合計2コピー存在することが確認された。
また、pUC19−ADE1をHpaIで切断したDNA断片を用いて形質転換を行った株については、フォワードプライマーADE1−1F(配列番号19)とリバースプライマーM13 forward(配列番号20)の組み合わせでPCRを行い、ADE1遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。PCR産物をアガロースゲル電気泳動で解析した結果、SX1353と名付けた形質転換体は目的サイズと同じ約1.45kbpのDNA断片が検出された。従って、SX1353株は相同組換えによりADE1遺伝子が相補されたことが確認された。
【0095】
(7)ARE1遺伝子を導入したerg5、erg6二重破壊株へのYEH1遺伝子導入株の作製
(i)酵母発現用のYEH1、GAL1遺伝子断片の調製
サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子(YEH1)を含むDNA断片、GAL1を含むDNA断片は、上記(1)で取得したサッカロミセス・セレビジエS288C株の染色体DNAを鋳型にしてPCRにて合成、単離した。以下にその方法の詳細を示す。
YEH1遺伝子断片のPCRによる合成には、フォワードプライマーYLL−OF(配列番号21)とリバースプライマーYLL−OR(配列番号22)を用いた。このプライマーの組み合わせでYEH1の蛋白質コード領域である開始コドンから終始コドンまでを増幅することができる。また、5'側と3'側の両側に制限酵素EcoR Iの認識配列を含んだ形で合成することができる。
【0096】
GAL1遺伝子断片のPCRによる合成には、フォワードプライマーGAL1−51(配列番号23)とリバースプライマーGAL1−31(配列番号24)を用いた。このプライマーの組み合わせでGAL1の蛋白質コード領域を含み、開始コドンの499塩基前から終始コドンの230塩基後までを増幅することができる。また、5’側と3’側の両側に制限酵素SmaIの認識配列を含んだ形で合成することができる。
【0097】
PCRは、PrimeSTAR HS DNA ポリメラーゼ(タカラバイオ社製)を用いて、(1)98℃、2分、(2)98℃、10秒、(3)55℃、5秒、(4)72℃、2分、(2)から(4)の反応を30回繰り返し、(5)72℃、7分間反応させた後、4℃で冷却の条件で反応を行った。次にPCR産物をアガロースゲル電気泳動し、約1.72kbpのYEH1遺伝子断片を、約2.33kbpのGAL1遺伝子断片を確認した。
【0098】
(ii)YEH1発現用プラスミドpPGL−YEH1(−2μ)、pPGGAL−YEH1(−2μ)の作製
上記(4)(ii)で作製したプラスミドpPG−LEU(−2μ)を制限酵素EcoR Iで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、PCR合成したYEH1遺伝子断片をEcoR Iで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素EcoRIによる切断で、約5.4kbpと1.72kbpの断片が確認できたものを目的プラスミドとして、pPGL−YEH1(−2μ)と名付けた。
【0099】
次に、pUC19(タカラバイオ社製)をSmaIで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、PCR合成したGAL1遺伝子断片をSmaIで切断したものとをTAKARA DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素PvuIIによる切断で、約0.48kbpと0.72kbp、1.45kbp、2.36kbpの断片が確認できたものを目的プラスミドとして、pUC19−GAL1と名付けた。
続いて、上記pPGL−YEH1(−2μ)をSspIで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、上記pUC19−GAL1をSmaIで切断したものとをTAKARA DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素EcoRIによる切断で、約3.30kbpと2.66kbpの断片が確認でき、さらに制限酵素ScaIによる切断で、約1.28kbpと4.68kbpの断片が確認できたものを目的プラスミドとして、pPG−GAL1(−2μ)と名付けた。さらに、上記pPGL−YEH1(−2μ)をBamHIで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、上記pPG−GAL1(−2μ)をBamHIで切断したものとをTAKARA DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素HindIIIによる切断で、約0.37kbpと0.57kbpと0.19kbpと3.73kbpと2.82kbpの断片が確認できたものを目的プラスミドとして、pPGGAL1−YEH1(−2μ)と名付けた。
【0100】
(iii)ARE1遺伝子を導入したerg5、erg6二重破壊株SX1314、SX1318、SX1315、SX1350へのYEH1遺伝子導入株SX1317、SX1320、SX1322、SX1351株の作製
ARE1遺伝子を導入したerg5、erg6二重破壊株SX1314、SX1318、SX1315のゲノム上のYEH1遺伝子座へさらにYEH1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加
水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGL−YEH1(−2μ)を制限酵素Nde I(YEH1遺伝子中)で切断した。また、YEH1を導入していない対照株を作製するため、(4)(ii)で作製した酵母組換え用プラスミドpPG−LEU(−2μ)を制限酵素Bsp1407 Iで切断した。これらのDNA断片を上記(6)で作製したSX1314、SX1318、SX1315株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。なお、SX1314株に対して形質転換を行ったSD寒天培地にのみ20mg/lアデニン、30mg/l L−リジンを添加した。
【0101】
寒天培地上に生育してきたロイシンおよびウラシル、ヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーYEH1−RR(配列番号25)の組み合わせでPCRを行い、YEH1遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。
【0102】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1317(YPH499株由来)、SX1320(FY1679−06c株由来)、SX1322(KA311A株由来)と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1317,SX1320、SX1322株のゲノムには上記相同組換えによりYEH1遺伝子が導入され、2コピー存在していることが確認された。
【0103】
また、プラスミドpPG−LEU(−2μ)をBsp1407Iで切断したDNA断片を用いて形質転換を行った株については、フォワードプライマーPPGK1−R1(配列番号26)とリバースプライマーLEU2−R2(配列番号27)の組み合わせでPCRを行い、leu2遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。
【0104】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1316(YPH499株由来)、SX1319(FY1679−06c株由来)、SX1321(KA311A株由来)と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1316,SX1319、SX1321株は相同組換えによりLEU2遺伝子が相補されたことが確認された。
【0105】
さらに、ARE1遺伝子を導入したerg5、erg6二重破壊株SX1350のゲノム上のYEH1遺伝子座へさらにYEH1遺伝子を導入することを目的として、上記と同様に以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGGAL1−YEH1(−2μ)を制限酵素Bsp1407 I(YEH1遺伝子中)で切断した。このDNA断片を上記(6)で作製したSX1350株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SG寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%ガラクトース、2%寒天)に播き、30℃で3〜5日間培養した。
【0106】
ガラクトースを炭素源として寒天培地上に生育してきたアデニンおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養
液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーYEH1−RR(配列番号25)の組み合わせでPCRを行い、YEH1遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。
【0107】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1351(X2181−1B株由来)と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1351株のゲノムのARE1遺伝子座に上記相同組換えによりYEH1遺伝子が導入され、合計2コピー存在することが確認された。
【0108】
(実施例2)
erg5、erg6二重破壊株SX1312の発現locusの異なるARE1およびYEH1増強株の作製
(1)erg5、erg6二重破壊株SX1312へのARE1遺伝子導入株SX1326の作製
erg5、erg6二重破壊株SX1312のゲノム上のARE1遺伝子座へさらにARE1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE1発現用のプラスミドpPGL−ARE1(−2μ)を制限酵素Xho I(ARE1遺伝子中)で切断した。このDNA断片を上記実施例1(3)で作製したSX1312株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、20mg/l ウラシルを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0109】
寒天培地上に生育してきたロイシンおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーARE1−check(配列番号18)の組み合わせでPCRを行い、ARE1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
PCR産物をアガロースゲル電気泳動で解析した結果、SX1326と名付けた形質転換体は目的サイズと同じ約2.6kbpのDNA断片が検出された。従って、SX1326株のゲノムのARE1遺伝子座に上記相同組換えによりARE1遺伝子が導入され、合計2コピー存在することが確認された。
【0110】
(2)ARE1遺伝子を導入したSX1318、SX1326株への発現locusの異なるYEH1遺伝子導入株SX1325、SX1332の作製
(i)YEH1発現用プラスミドpPGU−YEH1(−2μ)の作製
上記実施例1(5)で作製したプラスミドpPGU−ARE1(−2μ)を制限酵素BamHIで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、pPGL−YEH1(−2μ)をBamH Iで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素SmaIによる切断で、約4.88kbpと1.64kbpの断片が確認できたものを目的プラスミドとして、pPGU−YEH1(−2μ)と名付けた。
【0111】
(ii)ARE1遺伝子を導入したerg5、erg6二重破壊株SX1318、1326への発現locusの異なるYEH1遺伝子導入株SX1325、SX1332株の作製
ARE1遺伝子を導入したerg5、erg6二重破壊株SX1318のleu2遺伝子座と、SX1326のゲノム上のura3遺伝子座へさらにYEH1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGL−YEH1(−2μ)を制限酵素BstEII(leu2遺伝子中)で切断し、pPGU−YEH1(−2μ)を制限酵素StuI(ura3遺伝子中)で切断した。これらのDNA断片をSX1318とSX1326株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0112】
寒天培地上に生育してきたロイシンおよびウラシル、ヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーLEU2−R2(配列番号27)とリバースプライマーYLL−Seq3R(配列番号28)、フォワードプライマーURA3−RR(配列番号29)とリバースプライマーYEH1−seqR1(配列番号30)の組み合わせでそれぞれPCRを行い、leu2遺伝子座上とura3遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0113】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1325(leu2遺伝子座)とSX1332(ura3遺伝子座)と名付けた形質転換体は目的サイズと同じ約2.8kbpと約2.6kbpのDNA断片がそれぞれ検出された。従って、SX1325,SX1332株のゲノムには上記相同組換えによりYEH1遺伝子が導入され、2コピー存在していることが確認された。
【0114】
(3)erg5、erg6二重破壊株SX1312への発現locusの異なるARE1遺伝子導入株SX1327の作製
erg5、erg6二重破壊株SX1312のゲノム上のura3遺伝子座へさらにARE1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE1発現用のプラスミドpPGU−ARE1(−2μ)を制限酵素StuI(ura3遺伝子中)で切断した。このDNA断片を上記実施例1(3)で作製したSX1312株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、100mg/l L−ロイシンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
寒天培地上に生育してきたウラシルおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーURA3−RR(配列番号29)とリバースプライマーARE1−RR(配列番号31)の組み合わせでPCRを行い、ARE1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0115】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1326と名付けた形質転
換体は目的サイズと同じ約2.5kbpのDNA断片が検出された。従って、SX1327株のゲノムのura3遺伝子座に上記相同組換えによりARE1遺伝子が導入され、合計2コピー存在することが確認された。
【0116】
(4)ARE1遺伝子を導入したSX1327株へのYEH1遺伝子導入株SX1333の作製
ARE1遺伝子を導入したerg5、erg6二重破壊株SX1327のゲノム上のYEH1遺伝子座へさらにYEH1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGL−YEH1(−2μ)を制限酵素NdeI(YEH1遺伝子中)で切断した。このDNA断片をSX1327株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
寒天培地上に生育してきたロイシンおよびウラシル、ヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーYEH1−RR(配列番号25)の組み合わせでそれぞれPCRを行い、YEH1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0117】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1333(YEH1遺伝子座)と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1333株のゲノムのYEH1遺伝子座には上記相同組換えによりYEH1遺伝子が導入され、2コピー存在していることが確認された。
【0118】
(5)ARE1、YEH1遺伝子を導入していないerg5、erg6二重破壊株SX1335株の作製
ARE1、YEH1を導入していない対照株を作製するため、上記実施例1(5)で作製した酵母組換え用プラスミドpUC−URA3を制限酵素StuIで切断した。これらのDNA断片を上記実施例1(3)で作製したerg5、erg6二重破壊株SX1312に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、100mg/l L−ロイシンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
寒天培地上に生育してきたウラシルおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーM13 forward(配列番号20)とリバースプライマーURA3−RR(配列番号29)の組み合わせでPCRを行い、ura3遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0119】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1329と名付けた形質転換体は目的サイズと同じ約1.2kbpのDNA断片が検出された。従って、SX1329株は相同組換えによりURA3遺伝子が相補されたことが確認された。
【0120】
次に、酵母組換え用プラスミドpPG−LEU(−2μ)を制限酵素Bsp1407 Iで切断した。このDNA断片を上記で作製したSX1329株に上記実施例1(2)と
同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0121】
寒天培地上に生育してきたロイシンおよびウラシル、ヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−R1(配列番号26)とリバースプライマーLEU2−R2(配列番号27)の組み合わせでPCRを行い、leu2遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0122】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1335と名付けた形質転換体は目的サイズと同じ約2.1kbpのDNA断片が検出された。従って、SX1335株は相同組換えによりLEU2遺伝子が相補されたことが確認された。
【0123】
(実施例3)
erg5、erg6二重破壊株SX1312のARE2およびYEH1増強株の作製
(1)erg5、erg6二重破壊株SX1312へのARE2遺伝子導入株SX1342の作製
(i)酵母発現用のARE2遺伝子断片の調製
サッカロミセス・セレビジエ由来アシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子(ARE2)を含むDNA断片は、上記実施例1(1)で取得したサッカロミセス・セレビジエ S288C株の染色体DNAを鋳型にしてPCRにて合成、単離した。以下にその方法の詳細を示す。
ARE2遺伝子断片のPCRによる合成には、フォワードプライマーARE2−SmaI(F)(配列番号32)とリバースプライマーARE2−EcoRI(R)(配列番号33)を用いた。このプライマーの組み合わせでARE2の蛋白質コード領域である開始コドンから終始コドンまでを増幅することができる。また、5’側には制限酵素SmaIの認識配列を、3’側にはEcoRIの認識配列を含んだ形で合成することができる。PCRは、PrimeSTAR HS DNA ポリメラーゼ(タカラバイオ社製)を用いて、(1)98℃、2分、(2)98℃、10秒、(3)55℃、5秒、(4)72℃、2分、(2)から(4)の反応を30回繰り返し、(5)72℃、7分間反応させた後、4℃で冷却の条件で反応を行った。次にPCR産物をアガロースゲル電気泳動し、約1.93kbpのARE2遺伝子断片を確認した。
【0124】
(ii)ARE2発現用プラスミドpPGL−ARE2(−2μ)の作製
上記実施例1(4)で作製したプラスミドpPG−LEU(−2μ)を制限酵素SmaIとEcoRIで切断したものと、PCR合成したARE2遺伝子断片をSmaIとEcoRIで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素SmaIとEcoRIによる切断で、約5.4kbpと1.93kbpの断片が確認できたものを目的プラスミドとして、pPGL−ARE2(−2μ)と名付けた。
【0125】
(iii)erg5、erg6二重破壊株SX1312へのARE2遺伝子導入株SX1342株の作製
erg5、erg6二重破壊株SX1312のゲノム上のARE2遺伝子座へさらにARE2遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE2発現用のプラスミドpPGL−ARE2(−2μ)を制限酵素SalI(ARE2遺伝子中)で切断した。このDNA断片を上記実施例1(3)で作製したSX1312株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、20mg/l ウラシルを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
寒天培地上に生育してきたロイシンおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーARE2−R1(配列番号34)の組み合わせでPCRを行い、ARE2遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0126】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1342と名付けた形質転換体は目的サイズと同じ約2.6kbpのDNA断片が検出された。従って、SX1342株のゲノムのARE2遺伝子座に上記相同組換えによりARE2遺伝子が導入され、合計2コピー存在することが確認された。
【0127】
(2)ARE2遺伝子を導入したSX1342株へのYEH1遺伝子導入株SX1346の作製
ARE2遺伝子を導入したerg5、erg6二重破壊株SX1342のゲノム上のYEH1遺伝子座へさらにYEH1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGU−YEH1(−2μ)を制限酵素Bsp1407I(YEH1遺伝子中)で切断した。また、YEH1を導入していない対照株を作製するため、上記実施例1(5)で作製した酵母組換え用プラスミドpUC−URA3を制限酵素StuIで切断した。これらのDNA断片を上記(1)で作製したSX1342株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0128】
寒天培地上に生育してきたロイシンおよびウラシル、ヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーYEH1−RR(配列番号25)の組み合わせでPCRを行い、YEH1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
PCR産物をアガロースゲル電気泳動で解析した結果、SX1346と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1346株のゲノムには上記相同組換えによりYEH1遺伝子が導入され、2コピー存在していることが確認された。
【0129】
また、プラスミドpUC−URA3をStuIで切断したDNA断片を用いて形質転換を行った株については、フォワードプライマーM13 forward(配列番号20)
とリバースプライマーURA3−RR(配列番号29)の組み合わせでPCRを行い、ura3遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0130】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1347と名付けた形質転換体は目的サイズと同じ約1.2kbpのDNA断片が検出された。従って、SX1347株は相同組換えによりURA3遺伝子が相補されたことが確認された。
【0131】
(実施例4)
サッカロミセス・セレビジエ FY1679−06c株のARE1およびYEH1増強株の作製
(1)FY1679−06c株へのARE1遺伝子増強株SX1330の作製
サッカロミセス・セレビジエ FY1679−06c株のゲノム上のARE1遺伝子座へさらにARE1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE1発現用のプラスミドpPGU−ARE1(−2μ)を制限酵素Xho I(ARE1遺伝子中)で切断した。このDNA断片をFY1679−06c株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、各20mg/l L−ヒスチジン、トリプトファン、100mg/l L−ロイシンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0132】
寒天培地上に生育してきたウラシル非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーARE1−check(配列番号18)の組み合わせでPCRを行い、ARE1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0133】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1330と名付けた形質転換体は目的サイズと同じ約2.6kbpのDNA断片が検出された。従って、SX1330株のゲノムのARE1遺伝子座に上記相同組換えによりARE1遺伝子が導入され、合計2コピー存在することが確認された。
【0134】
(2)ARE1遺伝子を導入したSX1330株へのYEH1遺伝子導入株SX1337の作製
ARE1遺伝子を導入したSX1330株のゲノム上のYEH1遺伝子座へさらにYEH1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGL−YEH1(−2μ)を制限酵素Nde I(YEH1遺伝子中)で切断した。また、YEH1を導入していない対照株を作製するため、上記実施例1(4)(ii)で作製した酵母組換え用プラスミドpPG−LEU(−2μ)を制限酵素Bsp1407 Iで切断した。これらのDNA断片を上記(1)で作製したSX1330株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、各20mg/l L−ヒスチジン、トリプトファンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0135】
寒天培地上に生育してきたロイシンおよびウラシル非要求性のコロニーをYPD液体培
地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーYEH1−RR(配列番号25)の組み合わせでPCRを行い、YEH1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0136】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1337と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1337株のゲノムには上記相同組換えによりYEH1遺伝子が導入され、2コピー存在していることが確認された。
【0137】
また、プラスミドpPG−LEU(−2μ)をBsp1407Iで切断したDNA断片を用いて形質転換を行った株については、フォワードプライマーPPGK1−R1(配列番号26)とリバースプライマーLEU2−R2(配列番号27)の組み合わせでPCRを行い、leu2遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0138】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1338と名付けた形質転換体は目的サイズと同じ約2.1kbpのDNA断片が検出された。従って、SX1338株は相同組換えによりLEU2遺伝子が相補されたことが確認された。
【0139】
(3)ARE1、YEH1遺伝子を導入していない対照株SX1348の作製
ARE1、YEH1を導入していない対照株を作製するため、上記実施例1(5)で作製した酵母組換え用プラスミドpUC−URA3を制限酵素StuIで切断した。このDNA断片をFY1679−06c株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、各20mg/l L−ヒスチジン、トリプトファン、100mg/l L−ロイシンを含むSD寒天培地(0.67% Yeast
Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0140】
寒天培地上に生育してきたウラシル非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーM13 forward(配列番号20)とリバースプライマーURA3−RR(配列番号29)の組み合わせでPCRを行い、ura3遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0141】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1343と名付けた形質転換体は目的サイズと同じ約1.2kbpのDNA断片が検出された。従って、SX1343株は相同組換えによりURA3遺伝子が相補されたことが確認された。
次に、酵母組換え用プラスミドpPG−LEU(−2μ)を制限酵素Bsp1407 Iで切断した。このDNA断片を上記で作製したSX1343株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、各20mg/l L−ヒスチジン、トリプトファンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0142】
寒天培地上に生育してきたロイシンおよびウラシル非要求性のコロニーをYPD液体培
地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−R1(配列番号26)とリバースプライマーLEU2−R2(配列番号27)の組み合わせでPCRを行い、leu2遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
PCR産物をアガロースゲル電気泳動で解析した結果、SX1348と名付けた形質転換体は目的サイズと同じ約2.1kbpのDNA断片が検出された。従って、SX1348株は相同組換えによりLEU2遺伝子が相補されたことが確認された。
【0143】
(実施例5)
ARE1、YEH1遺伝子を増強した各種株のステロール類の分析
(1)各種酵母株の培養、およびステロール類の分析
実施例1〜4で作製した各種株を0.5%グルコースを炭素源とした2mlのYPAD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース、40mg/lアデニン)に1白金耳植菌し、30℃、18時間培養し、種菌とした。この酵母培養液から初期OD660=0.2となる分量の培養液を回収し、4℃、10,000rpm、3分間遠心分離を行い、培養上清を除去した。菌体ペレットに0.1mlの0.85%生理食塩水を添加、懸濁し、2%グルコースを炭素源とした5mlのYPAD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース、40mg/lアデニン)に播種し、30℃、72時間培養を行った。酵母培養液0.5mlを4℃、14,000rpm、3分間遠心分離を行い、菌体を回収した。菌体ペレットにエタノール0.4mlと40%水酸化カリウム水溶液0.02mlを添加し、80℃、90分間振とうを行い、菌体内に蓄積したステロールエステルのケン化処理を行った。この液を35℃で減圧濃縮機にかけ、濃縮乾固させた。
得られた濃縮乾固物にエタノール0.1mlと滅菌水0.05mlを加え懸濁し、さらにヘプタン0.8mlを添加し激しく混和した後、室温で14,000rpm、1分間遠心分離することで水層と油層に分離した。上層のヘプタン層を別のチューブに取り、残った水層に再度、エタノール0.1mlとヘプタン0.8mlを添加し、抽出操作を繰り返した。抽出した約1.6mlのヘプタン溶液に滅菌水0.04mlを添加し、激しく混和した後、再度、室温で14,000rpm、1分間遠心分離することで水層と油層に分離した。約1.6mlのヘプタン層を回収し、35℃で減圧濃縮機にかけ、濃縮乾固させた。得られた濃縮乾固物は0.2mlトルエンに再溶解し、テストステロンを内部標準としてガスクロマトグラフィーによるステロール類の分析を行った。結果を図1〜8に示す。
【0144】
(2)ARE1、YEH1遺伝子を増強したerg5、erg6二重破壊株のステロール類の分析
図1(a)に示した通り、YPH499を親株として作製したARE1のみを増強したerg5、erg6二重破壊株(SX1316)に対して、ARE1とYEH1を増強したerg5、erg6二重破壊株(SX1317)では、ZYMOの蓄積量が減少し、TEOLの蓄積量が増加していることがわかった。このことからARE1増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることが明らかとなった。
一方、図2(a)に示した通り、FY1679−06cを親株として作製したARE1のみを増強したerg5、erg6二重破壊株(SX1319)に対して、さらにYEH1を増強したerg5、erg6二重破壊株(SX1320)でも、ZYMO蓄積量が減少し、TEOL蓄積量が増加した。これらのことからARE1増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることがFY1670−06cを親株とした場合でも明らかとなった。
【0145】
次に、図3(a)に示した通り、KA311Aを親株として作製したARE1のみを増強したerg5、erg6二重破壊株(SX1321)に対して、さらにYEH1を増強したerg5、erg6二重破壊株(SX1322)でも、ZYMO蓄積量が減少し、TEOL蓄積量が増加した。これらのことからARE1増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることがKA311Aを親株とした場合でも明らかとなった。
さらに、図4(a)に示した通り、X2181−1Bを親株として作製したARE1のみを増強したerg5、erg6二重破壊株(SX1350)に対して、さらにYEH1を増強したerg5、erg6二重破壊株(SX1351)でも、ZYMO蓄積量が減少し、TEOL蓄積量が増加した。これらのことからARE1増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることがX2181−1Bを親株とした場合でも明らかとなった。
【0146】
(3)発現locusの異なるARE1、YEH1を増強したerg5、erg6二重破壊株のステロール類の分析
図5(a)に示した通り、FY1679−06cを親株として作製した、発現locusの異なるYEH1を増強したerg5、erg6二重破壊、ARE1増強株において、YEH1、URA3、LEU2いずれのlocusでYEH1を発現させた株でも同等のTEOL蓄積を示した。このことからARE1増強下においてYEH1をどのようなlocusで発現増強させても、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることが明らかとなった。
一方、図6(a)に示した通り、FY1679−06cを親株として作製した、発現locusの異なるARE1を増強したerg5、erg6二重破壊、YEH1増強株において、ARE1、URA3いずれのlocusでARE1を発現させた株でも同等のTEOL蓄積を示した。このことからYEH1増強下においてARE1をどのようなlocusで発現増強させても、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることが明らかとなった。
【0147】
(4)ARE2、YEH1を増強したerg5、erg6二重破壊株のステロール類の分析
図7(a)に示した通り、FY1679−06cを親株として作製したARE2のみを増強したerg5、erg6二重破壊株(SX1347)に対して、さらにYEH1を増強したerg5、erg6二重破壊株(SX1346)でも、ZYMO蓄積量が減少し、TEOL蓄積量が増加した。これらのことからARE2増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることが、ARE2をステロールアシルCoA:ステロールアシルトランスフェラーゼとして用いた場合でも明らかとなった。
【0148】
(5)ARE1、YEH1を増強した株のステロール類の分析
図8(a)に示した通り、FY1679−06cを親株として作製したARE1のみを増強した株(SX1338)に対して、さらにYEH1を増強した株(SX1337)でも、ZYMO蓄積量が減少し、ERGO蓄積量が増加した。これらのことからARE1増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることが、erg5erg6二重破壊をしていない株を用いた場合でも明らかとなった。
【0149】
(実施例6)
ARE1、YEH1遺伝子を増強した各種株の転写量の分析
(1)酵母からの全RNAの調製
実施例5で調製した各種株について、RNeasy plus Mini Kitおよ
びRNase−Free DNase Set(いずれもQIAGEN社製)を用いて全RNAを以下のとおり調製した。
上記実施例5と同様に菌体を培養した後、回収した培養液を用いて、2×107個になるよう酵母菌体溶液を調製し(OD660=1が、約1.5×107個として細胞数を調製)、4℃、14,000rpm、1分間遠心分離して得られた菌体を0.85%生理食塩水に懸濁し、再度、4℃、10,000rpm、2分間遠心分離を行った。得られた菌体は、キット付属のBuffer RLT 0.6mlと2−メルカプトエタノール 6μlで懸濁し、この懸濁液をMagNA Lyser Green Beads(Roche社製)のチューブに移し、MagNA Lyser(Roche社製)を用いて室温で6,000rpm、1分間破砕を行った。この菌体破砕液をゲノムDNA除去ミニスピンカラムに入れ、RNeasy plus Mini Kit付属の使用説明書に従い、全RNAの調製を行った。オプションとして全RNAのカラム吸着後にDNase mix solution 0.08mlを添加し、室温で15分間静置し、染色体DNAの分解を行った。また、最後に50℃で保温したRNase−free waterを加え、室温で1分間静置し、遠心分離にて全RNAを溶出した。
【0150】
(2)逆転写反応
上記(1)で調製した全RNAを鋳型として、SuperScript III First−Strand Synthesis System for RT−PCR(invitrogen社製)を用いて、付属の使用説明書に従い、逆転写反応を行い、一本鎖cDNAを合成した。逆転写反応には、300ngの全RNA、プライマーとして50μM Oligo(dT)20を使用した。
【0151】
(3)定量PCR
定量PCRの反応は、SYBR premix Ex Taq(Perfect Real Time)(タカラバイオ社製)を用いて、付属の使用説明書に従って反応を行い、(1)95℃、10秒、(2)95℃、5秒、(3)60℃、20秒、(2)から(3)の反応を40回繰り返した。温度変化速度はいずれも20℃/秒とした。また、融解曲線分析を(1)95℃、0秒、(2)65℃、15秒、(3)95℃、0秒で行い、温度変化速度はいずれも20℃/秒とした。PCRプライマーとして、ARE1の定量PCRにはARE1−F(配列番号35)とARE1−R(配列番号36)、ARE2の定量PCRにはARE2−F(配列番号37)とARE2−R(配列番号38)、YEH1の定量PCRにはYEH1−F(配列番号39)とYEH1−R(配列番号40)を使用した。また、発現コントロールとしてアクチンをコードしているACT1を用い、ACT1−F(配列番号41)とACT1−R(配列番号42)をプライマーとして使用した。
定量分析には、Roche Diagnostics社製のLight−Cycler、およびLight−Cycler Software Ver.3.5を用い、Fit
Point法を用いて分析を行った。この結果を図1および図2の(b)、(c)に示す。
【0152】
(4)ARE1、YEH1遺伝子を増強したerg5、erg6二重破壊株の転写量の分析
図1(b)、(c)で示した通り、YPH499のerg5、erg6二重破壊株を親株とした株における転写解析を行った結果、ARE1のみを増強したSX1316ではARE1の転写量が、SX1313に比べ増加しており、発現増強されていることが確認できた。また、ARE1とYEH1をともに増強したSX1317のARE1、YEH1それぞれの転写量もSX1313に比べて増加していることから、両遺伝子とも発現増強されていることが確認できた。
同様に、FY1679−06cのerg5、erg6二重破壊株を親株とした株における転写解析の結果(図2(b)、(c))、SX1312に対して、ARE1のみを増強
したSX1319はARE1の転写量が増加し、ARE1とYEH1をともに増強したSX1320はARE1とYEH1それぞれの転写量が増加していることが確認できた。
【0153】
続いて、KA311Aのerg5、erg6二重破壊株を親株とした株における転写解析の結果(図3(b)、(c))、SX1307に対して、ARE1のみを増強したSX1321はARE1の転写量が増加し、ARE1とYEH1をともに増強したSX1322はARE1とYEH1それぞれの転写量が増加していることが確認できた。
さらに、X2181−1Bのerg5、erg6二重破壊株を親株とした株における転写解析の結果(図4(b)、(c))、SX1353に対して、ARE1のみを増強したSX1350はARE1の転写量が増加し、ARE1とYEH1をともに増強したSX1351はARE1とYEH1それぞれの転写量が増加していることが確認できた。
【0154】
(5)発現locusの異なるARE1、YEH1を増強したerg5、erg6二重破壊株の転写量の分析
FY1679−06cのerg5、erg6二重破壊、ARE1増強株を親株として、発現locusの異なるYEH1を強化した株における転写解析の結果(図5(b)、(c))、SX1319に対して、SX1320(YEH1 locus)、SX1332(URA3 locus)、SX1325(LEU2 locus)いずれの株も、YEH1の転写量が増加していることが確認できた。
【0155】
さらに、FY1679−06cのerg5、erg6二重破壊株を親株として、発現locusの異なるARE1をYEH1とともに強化した株における転写解析の結果(図6(b)、(c))、SX1335に対して、SX1320(ARE1 locus)、SX1333(URA3 locus)いずれの株も、ARE1とYEH1それぞれの転写量が増加していることが確認できた。
【0156】
(6)ARE2、YEH1を増強したerg5、erg6二重破壊株の転写量の分析
FY1679−06cのerg5、erg6二重破壊株を親株とした株における転写解析の結果(図7(b)、(c))、SX1335に対して、ARE2のみを増強したSX1347はARE2の転写量が増加し、ARE2とYEH1をともに増強したSX1346はARE2とYEH1それぞれの転写量が増加していることが確認できた。
【0157】
(7)ARE1、YEH1を増強した株の転写量の分析
FY1679−06cを親株とした株における転写解析の結果(図8(b)、(c))、SX1348に対して、ARE1のみを増強したSX1338はARE1の転写量が増加し、ARE1とYEH1をともに増強したSX1337はARE1とYEH1それぞれの転写量が増加していることが確認できた。
【0158】
なお、表1に、図1(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0159】
【表1】

【0160】
また、表2に、図2(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0161】
【表2】
【0162】
表3に、図3(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重
量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0163】
【表3】
【0164】
表4に、図4(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0165】
【表4】
【0166】
表5に、図5(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0167】
【表5】
【0168】
表6に、図6(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0169】
【表6】
【0170】
表7に、図7(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE2とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0171】
【表7】
【0172】
表8に、図8(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0173】
【表8】
【産業上の利用可能性】
【0174】
本発明によって製造されるΔ5,7−ステロールは、医薬分野、化粧品分野、食品分野等で有用である。
【技術分野】
【0001】
本発明は医薬の中間体等として有用なΔ5,7−ステロール及びハイドロコルチゾンを製造する酵母およびそれを用いたΔ5,7−ステロール及びハイドロコルチゾンの製造法に関する。
【背景技術】
【0002】
ステロール類は動物、植物、真菌類に含まれている生命活動に必須の物質であり、細胞膜に遊離型ステロールの状態で含まれるほか、一部は脂肪酸とのエステル型として貯蔵される。このうち、C5,7−ジエン体ステロールはコレステロール骨格の5位と7位に炭素−炭素二重結合を有するステロールであるが、その中でもコレスタ−5,7,24−トリエン−3β−オール(別名、7−デヒドロデスモステロール、以下、TEOLと称する)は医薬品であるケノデオキシコール酸やウルソデオキシコール酸の合成原料として用いられるなど、医薬品の中間体などとして非常に有用な物質である。
【0003】
また、11β,17α,21-トリヒドロキシ-4-プレグネン-3,20-ジオン(ハイドロコルチゾン)は、その前駆物質であるプレグネノロン、プロゲステロン、7−デヒドロプレグネノロンとともに、医薬および医薬中間体として産業上有用な化合物である。プレグネノロンの既存の製造法として、コレステロールやジオスゲニン、スティグマステロールといった天然由来のステロール化合物を原料として、複数の有機合成反応により合成する方法が報告されている(非特許文献1)。
【0004】
従来、C5,7−ジエン体ステロールは動物組織の抽出物から精製されたものが使用されている。一方で、酵母等の真菌類においてエルゴステロールの生合成経路が明らかにされる(非特許文献2)につれて、近年ではその生合成経路を改変して特定のステロールを蓄積させるという研究が進められている。
【0005】
酵母、カビ等の真菌類において、エルゴステロール生合成酵素欠損株あるいはエルゴステロール生合成酵素遺伝子の過剰発現によりステロール組成を変えた研究例がいくつか報告されている。例えば、酵母サッカロミセス・セレビシエでステロールC−24メチルトランスフェラーゼをコードする遺伝子(ERG6)を欠損させることにより、側鎖24位にメチル基を持たないステロールのみが蓄積するようにした例(非特許文献3)や、酵母でステロールC−22デサチュラーゼをコードする遺伝子(ERG5)を欠損させアラビドプシスのΔ7−ステロールリダクターゼを高発現するように代謝工学的に改変し、エルゴスタ−5,24−ジエノール、エルゴスタ−5−エネノール、エルゴスタ−5,22−ジエノールが蓄積するようにした例が報告されている(非特許文献4)。
【0006】
また、特許文献1では、ステロールC−22デサチュラーゼ、および、ステロールC−24メチルトランスフェラーゼの両方の活性が低減化され、ステロールΔ7−リダクターゼ、および/またはステロールΔ5−デサチュラーゼが高発現化されるように代謝工学的に改変された真菌類(酵母、カビ)を培養し、培養物からステロール類を採取する方法が開示されている。
【0007】
一方、アシルCoA:ステロールアシルトランスフェラーゼはステロール化合物のエステル化を触媒する酵素であり、ステリルエステル加水分解酵素はステリルエステルの加水分解を触媒する酵素である。アシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質であるARE1の発現を強化した酵母は非特許文献5に開示されており、ス
テリルエステル加水分解酵素活性を有するYEH1のC末端にプロテインAを付加した融合タンパク質を導入した酵母が非特許文献6に開示されている。しかしながら、これらの文献ではARE1やYEH1の発現強化のC5,7−ジエン体ステロールやエルゴステロールなどのΔ5,7−ステロール産生への効果については記載されておらず、また、ARE1とYEH1の両方の発現を強化した酵母はこれまで知られていない。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2004-141125号公報
【非特許文献】
【0009】
【非特許文献1】J. Org. Chem. 44, 1582-1584 (1979)
【非特許文献2】Appl. Microbiol. Biotechnol. 63, 635-646(2004)
【非特許文献3】Biochem. Biophys. Res. Commun. 181, 509-517(1988)
【非特許文献4】J. Biol. Chem. 271(18), 10866-10873(1996)
【非特許文献5】Science 272, 1353-1356(1996)
【非特許文献6】Biochim. Biophys. Acta 1791, 118-124(2009)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、C5,7−ジエン体ステロールやエルゴステロールなどのΔ5,7−ステロール及びハイドロコルチゾンを効率よく産生する改変酵母を提供すること、およびこれを用いてC5,7−ジエン体ステロールやエルゴステロールなどのΔ5,7−ステロール及びハイドロコルチゾンを効率よく製造する方法を提供することを課題とする。
【課題を解決するための手段】
【0011】
本発明者らは上記課題を解決するために鋭意検討を行った。
上記のとおり、ステロール化合物はその大半がエステル体として細胞内に蓄積される。従って所望のステロール化合物を効率よく蓄積させるにはステロール化合物のエステル化を促進することが重要と一般的には考えられ、当業者であれば、Δ5,7−ステロールを効率よく産生するためにはステロールをエステル化するアシルCoA:ステロールアシルトランスフェラーゼ活性のみを増強し、逆にそのエステル体の加水分解を促進するステリルエステル加水分解酵素を低減化する、または改変しないと考えられる。しかしながら、本発明者らは、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子だけでなく、ステリルエステル加水分解酵素遺伝子の発現も同時に増強することを試みたところ、意外なことに、これらの両遺伝子の発現を増強することにより非常に効率よくΔ5,7−ステロールを産生させることができることを見出した。また、さらに酵母を改変することでハイドロコルチゾンも製造できることを見出し、本発明を完成させるに至った。
【0012】
すなわち、本発明は以下のとおりである。
[1]アシルCoA:ステロールアシルトランスフェラーゼ遺伝子とステリルエステル加水分解酵素遺伝子の発現が増強されるように改変された酵母。
[2]さらに、ステロールC−22デサチュラーゼ遺伝子(ERG5)及びステロールC−24メチルトランスフェラーゼ遺伝子(ERG6)の発現が低減するように改変された、[1]に記載の酵母。
[3]さらに、ステロールC−22デサチュラーゼ遺伝子(ERG5)の発現が低減するように改変された、[1]に記載の酵母。
[4]さらに、Δ7−還元活性、ステロール側鎖の20位及び22位との結合を切断する活性、ステロール3位酸化異性化活性、ステロイド17α水酸化活性、ステロイド21位水酸化活性、及びステロイド11β位水酸化活性から選択される少なくとも一つの活性を有
するように改変された[3]に記載の酵母。
[5]さらに、Δ7−還元活性、ステロール側鎖の20位及び22位との結合を切断する活性、ステロール3位酸化異性化活性、ステロイド17α水酸化活性、ステロイド21位水酸化活性、及びステロイド11β位水酸化活性を有するように改変された[3]に記載の酵母。
[6]サッカロミセス・セレビジエである、[1]〜[5]のいずれかに記載の酵母。
[7]親株がサッカロミセス・セレビジエYPH499株、サッカロミセス・セレビジエFY1679-06c株、サッカロミセス・セレビジエKA311A株またはサッカロミセス・セレビジエX2181-1B株である、[6]に記載の酵母。
[8] [1]〜[3]のいずれかに記載の酵母を培養してΔ5,7−ステロールを生成・蓄積させ、Δ5,7−ステロールを回収する、Δ5,7−ステロールの製造方法。
[9]培養する酵母が[1]又は[2]に記載の酵母であり、Δ5,7−ステロールがC5,7−ジエン体ステロールである、[8]に記載の方法。
[10]C5,7−ジエン体ステロールがコレスタ−5,7,24−トリエン−3β−オールまたは7−デヒドロコレステロールである、[9]に記載の方法。
[11]培養する酵母が[1]又は[3]に記載の酵母であり、Δ5,7−ステロールがエルゴスタ−5,7,24-トリエノールである、[8]に記載の方法。
[12]培養する酵母が[1]に記載の酵母であり、Δ5,7−ステロールがエルゴステロールである、[8]に記載の方法。
[13] [5]に記載の酵母を培養してハイドロコルチゾンを生成・蓄積させ、ハイドロコルチゾンを回収する、ハイドロコルチゾンの製造方法。
【発明の効果】
【0013】
本発明の酵母を用いることにより、Δ5,7−ステロール及びハイドロコルチゾンを産生させることができる。本発明の方法によってΔ5,7−ステロール及びハイドロコルチゾンを製造し、これらを用いて医薬を合成すれば、医薬の製造プロセスの効率化にも寄与すると考えられる。
【図面の簡単な説明】
【0014】
【図1】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。「C7,24」はコレスタ−7,24−ジエン−3β−オールを表し、「ZYMO」はザイモステロールを表し、「その他」はその他のステロール類を表す。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、Δe5Δe6は、ARE1とYEH1をいずれも増強していないerg5、erg6二重破壊株(SX1313)、Δe5Δe6ARE1は、ARE1のみを導入したerg5、erg6二重破壊株(SX1316)、Δe5Δe6ARE1YEH1は、ARE1とYEH1を導入したerg5、erg6破壊株(SX1317)を示す。いずれもYPH499を親株として作製された株である。
【図2】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、Δe5Δe6は、ARE1とYEH1をいずれも増強していないerg5、erg6二重破壊株(SX1312)、Δe5Δe6ARE1は、ARE1のみを導入したerg5、erg6二重破壊株(SX1319)、Δe5Δe6ARE1YEH1は、ARE1とYEH1を導入したerg5、erg6破壊株(SX1320)を示す。SX1319、SX1320については3クローンずつ評価した。いずれもFY1679−06cを親株として作製された株である。
【図3】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、Δe5Δe6は、ARE1とYEH1をいずれも増強していないerg5、erg6二重破壊株(SX1307)、Δe5Δe6ARE1は、ARE1のみを導入したerg5、erg6二重破壊株(SX1321)、Δe5Δe6ARE1YEH1は、ARE1とYEH1を導入したerg5、erg6破壊株(SX1322)を示す。いずれもKA311Aを親株として作製された株である。
【図4】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、Δe5Δe6は、ARE1とYEH1をいずれも増強していないerg5、erg6二重破壊株(SX1353)、Δe5Δe6ARE1は、ARE1のみを導入したerg5、erg6二重破壊株(SX1350)、Δe5Δe6ARE1YEH1は、ARE1とYEH1を導入したerg5、erg6破壊株(SX1351)を示す。いずれもX2181−1Bを親株として作製された株である。
【図5】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、−は、ARE1のみを導入したerg5、erg6二重破壊株(SX1319)、他はいずれもARE1とYEH1を導入したerg5、erg6二重破壊株であり、YEH1 locusは、YEH1をYEH1のlocusに導入した株(SX1320)、URA3 locusはYEH1をura3のlocusに導入した株(SX1332)、LEU2 locusは、YEH1をleu2のlocusに導入した株(SX1325)を示す。いずれもFY1679−06cを親株として作製された株である。
【図6】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、−は、ARE1とYEH1をいずれも導入していないerg5、erg6二重破壊株(SX1335)、他はいずれもARE1とYEH1を導入したerg5、erg6二重破壊株であり、ARE1 locusは、ARE1をARE1のlocusに導入した株(SX1320)、URA3 locusはARE1をura3のlocusに導入した株(SX1333)を示す。いずれもFY1679−06cを親株として作製された株である。
【図7】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。(b)、(c)定量PCRによるARE2とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE2とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、Δe5Δe6は、ARE2とYEH1をいずれも増強していないerg5、erg6二重破壊株(SX1335)、Δe5Δe6ARE2は、ARE2のみを導入したerg5、erg6二重破壊株(SX1347)、Δe5Δe6ARE2YEH1は、ARE2とYEH1を導入したerg5、erg6破壊株(SX1346)を示す。いずれもFY1679−06cを親株として作製された株である。
【図8】(a)ガスクロマトグラフィーによるステロール類の分析結果を示す図である。「ERGO」はエルゴステロールを表し、「ZYMO」はザイモステロールを表し、「その他」はその他のステロール類を表す。(b)、(c)定量PCRによるARE1とYEH1の転写量を分析した結果を示す図である。転写量は、ACT1に対する比として示した。(b)は、ARE1とACT1の発現量比を示し、(c)は、YEH1とACT1の発現量比を示す。図中、−は、ARE1とYEH1をいずれも増強していない株(SX1348)、ARE1は、ARE1のみを導入した株(SX1338)、ARE1YEH1は、ARE1とYEH1を導入した株(SX1337)を示す。いずれもFY1679−06cを親株として作製された株である。
【発明を実施するための形態】
【0015】
以下、本発明の実施の形態について詳細に説明する。
【0016】
(1)本発明の酵母
本発明の酵母は、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現とステリルエステル加水分解酵素遺伝子の発現が増強するように改変された酵母である。
【0017】
本発明において、Δ5,7−ステロールはステロール骨格の5位と7位に炭素−炭素二重結合を有するステロールを意味し、好ましくはエルゴステロールおよびC5,7−ジエン体ステロールである。また、C5,7−ジエン体ステロールはコレステロール骨格の5位と7位に炭素−炭素二重結合を有するステロールを意味するが、TEOL(コレスタ−5,7,24−トリエン−3β−オール)、7-デヒドロコレステロールが好ましく、TEOLが特に好ましい。また、ハイドロコルチゾンを目的生成物とする場合には、エルゴスタ−5,7,24−トリエノールが好ましい。Δ5,7−ステロールやC5,7−ジエン体ステロールには、それぞれΔ5,7−ステロールやC5,7−ジエン体ステロールのエステル体も含まれる。
【0018】
また、本発明において「アシルCoA:ステロールアシルトランスフェラーゼ遺伝子」とは、アシルCoA:ステロールアシルトランスフェラーゼ活性を有する蛋白質をコードする遺伝子を意味し、「ステリルエステル加水分解酵素遺伝子」とは、ステリルエステル加水分解酵素活性を有する蛋白質をコードする遺伝子を意味する。
【0019】
「アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現が増強した」とは、非改変株または親株と比較してアシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現が増強したことを意味する。アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現の増強は、非改変株または親株と比較して、単位菌体重量当たり5倍以上増強されていることが好ましく、10倍以上増強されていることがより好ましい。
なお、アシルCoA:ステロールアシルトランスフェラーゼは、ステロールのエステル化反応を触媒する酵素であり、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現は定量PCR法などによって測定することができる。
【0020】
「ステリルエステル加水分解酵素遺伝子の発現が増強した」とは、非改変株または親株と比較してステリルエステル加水分解酵素遺伝子の発現が増強したことを意味する。ステリルエステル加水分解酵素遺伝子発現の増強は、非改変株または親株と比較して、単位菌体重量当たり5倍以上増強されていることが好ましく、10倍以上増強されていることがより好ましい。
なお、ステリルエステル加水分解酵素遺伝子の発現は定量PCR法などによって測定することができる。
【0021】
本発明に用いる酵母は、以下に示すような酵母を親株として用い、該親株を改変することによって得ることができる。
親株として用いる酵母の種類は特に限定されないが、サッカロミセス属、デバリオマイセス属、シゾサッカロマイセス属、キャンディダ属、ヤロウィア属、ロドトルラ属、リポマイセス属、クルイベロマイセス属、ロドスポリジウム属、トリコデルマ属または、トルロプシス属、または、ピキア属に属する酵母であることが好ましく、例えば、サッカロミ
セス・セレビシエ(Saccharomyces cerevisiae)、サッカロミセス・バヤヌス(Saccharomyces bayanus)、デバリオマイセス・ニルソニ(Debaryomyces nilssonii)、デバリオマイセス・ハンセニ(Debaryomyces hansenii)、シゾサッカロマイセス・ポンベ(Schizosaccharomyces pombe)、キャンディダ・グラブラータ(Candida glabrata)、キャンディダ・トロピカリス(Candida tropicalis)、キャンディダ・ユティリス(Candida utilis)、キャンディダ・ボイディニィ(Candida boidinii)、ヤロウィア・リポリティカ(Yarrowia lipolytica)、ロドトルラ・グルティニス(Rhodotorula glutinis)、リポマイセス・リポフェラス(Lipomyces lipoferus)、クルイベロマイセス・ラクティス(Kluyveromyces lactis)、ロドスポリジウム・トルロイディス(Rhodosporidium toruloides)、トリコデルマ・リセイ(Trichoderma reesei)、トルロプシス・コリキュロサ(Torulopsis colliculosa)、ピキア・ファリノサ(Pichia farinosa)が挙げられる。
【0022】
これらの中で、代謝工学的手法が用いやすいことからサッカロミセス属が好適である。さらに、サッカロミセス属は、his3、leu2、trp1、ura3などの栄養要求性の遺伝型であることが望ましく、例えば、以下の株が例示される。
サッカロミセス・セレビジエYPH499株(MATa ura3 lys2 ade2 trp1 his3 leu2)(ATCC204679:American Type Culture Collection又はSTRATAGENE社より入手できる)、サッカロミセス・セレビジエFY1679-06c株(MATα ura3 leu2 trp1 his3)(Euroscarf社から入手できる)、サッカロミセス・セレビジエX2181-1B株(MATalpha his2 gal1 trp1 ade1
MAL SUC)(ATCC204822:American Type Culture Collectionより入手できる)。あるいは、サッカロミセス・セレビシエKA311A株(MATa his3 leu2 trp1 ura3)(Mol. Cell Biol. 13, 307-3083(1993))も用いることができ、KA311A株は、独立行政法人産業総合研究所特許生物寄託センターに受託番号:FERM P-19053として寄託されている。
【0023】
アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現およびステリルエステル加水分解酵素遺伝子の発現の増強は、遺伝子組換え法、例えば、該酵素をコードする遺伝子のコピー数を高めること、またはこの遺伝子のプロモーターを置換することによってこれらの遺伝子の発現量を増大させることによって行うことができる。
【0024】
アシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードする遺伝子として、ARE1遺伝子やARE2遺伝子が挙げられる。ARE1遺伝子およびARE2遺伝子は、多くの微生物で知られており、アシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードする限り特に限定されないが、例えば、それぞれ配列番号43または45の塩基配列を有するサッカロミセス・セレビジエ由来の遺伝子を挙げることができる。また、ARE1遺伝子およびARE2遺伝子は、アシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードするものである限り、配列番号43または45の塩基配列の相補配列を有するDNAとストリンジェントな条件でハイブリダイズするDNA、またはこれらの塩基配列と80%以上、好ましくは90%以上、より好ましくは95%以上、特に好ましくは99%以上の相同性を有するDNAのようなホモログであってもよい。ここで、ストリンジェントな条件としては、通常のサザンハイブリダイゼーションの洗いの条件である60℃、1×SSC,0.1%SDS、好ましくは、0.1×SSC、0.1%SDSに相当する塩濃度でハイブリダイズする条件が挙げられる。
【0025】
また、ARE1遺伝子およびARE2遺伝子は、それぞれ配列番号44または46のアミノ酸配列と80%以上、好ましくは90%以上、より好ましくは95%以上、特に好ましくは99%以上の同一性を有し、アシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードするDNAであってもよい。
【0026】
さらに、ARE1遺伝子およびARE2遺伝子は、配列番号44または46のアミノ酸配列において、1または数個のアミノ酸が置換、欠失、挿入または付加された配列を有し、アシル
CoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードするDNAであってもよい。ここで、1または数個とは、好ましくは、1〜20個、より好ましくは1〜10個、特に好ましくは1〜5個を意味する。
【0027】
また、サッカロミセス・セレビジエ以外の酵母、または他の微生物又は動植物由来のARE1遺伝子およびARE2遺伝子を使用することもできる。微生物または動植物由来のARE1遺伝子およびARE2遺伝子は、既にその塩基配列が決定されている遺伝子、またはホモロジー等に基づいてアシルCoA:ステロールアシルトランスフェラーゼ活性を有するタンパク質をコードする遺伝子を微生物、動植物等の染色体より単離し、塩基配列を決定したものなどを使用することができる。また、塩基配列が決定された後には、その配列にしたがって合成した遺伝子を使用することもできる。これらはハイブリダイゼーション法やPCR法によりそのプロモーターおよびORF部分を含む領域を増幅することによって、取得することができる。
【0028】
ステリルエステル加水分解酵素活性を有するタンパク質をコードする遺伝子として、YEH1遺伝子が挙げられる。YEH1遺伝子は、多くの微生物で知られており、ステリルエステル加水分解酵素活性を有するタンパク質をコードする限り特に限定されないが、例えば、配列番号47の塩基配列を有するサッカロミセス・セレビジエ由来の遺伝子を挙げることができる。また、YEH1遺伝子は、ステリルエステル加水分解酵素活性を有するタンパク質をコードするものである限り、配列番号47の塩基配列の相補配列を有するDNAとストリンジェントな条件でハイブリダイズするDNA、またはこれらの塩基配列と80%以上、好ましくは90%以上、より好ましくは95%以上、特に好ましくは99%以上の相同性を有するDNAのようなホモログであってもよい。ここで、ストリンジェントな条件は、上記のとおりである。
【0029】
また、YEH1遺伝子は、それぞれ配列番号48のアミノ酸配列と80%以上、好ましくは90%以上、より好ましくは95%以上、特に好ましくは99%以上の同一性を有し、ステリルエステル加水分解酵素活性を有するタンパク質をコードするDNAであってもよい。
さらに、YEH1遺伝子は、配列番号48のアミノ酸配列において、1または数個のアミノ酸が置換、欠失、挿入または付加された配列を有し、ステリルエステル加水分解酵素活性を有するタンパク質をコードするDNAであってもよい。ここで、1または数個とは、好ましくは、1〜20個、より好ましくは1〜10個、特に好ましくは1〜5個を意味する。
【0030】
また、サッカロミセス・セレビジエ以外の酵母、または他の微生物又は動植物由来のYEH1遺伝子を使用することもできる。微生物または動植物由来のYEH1遺伝子は、既にその塩基配列が決定されている遺伝子、またはホモロジー等に基づいてステリルエステル加水分解酵素活性を有するタンパク質をコードする遺伝子を微生物、動植物等の染色体より単離し、塩基配列を決定したものなどを使用することができる。また、塩基配列が決定された後には、その配列にしたがって合成した遺伝子を使用することもできる。これらはハイブリダイゼーション法やPCR法によりそのプロモーターおよびORF部分を含む領域を増幅することによって、取得することができる。
【0031】
アシルCoA:ステロールアシルトランスフェラーゼ遺伝子の発現、およびステリルエステル加水分解酵素遺伝子の発現を増強するためには、例えば、上記のようなDNAを宿主酵母で機能しうるプラスミドに発現可能に組込み、宿主酵母に導入すればよい。
【0032】
上記遺伝子を組込むことができるプラスミドベクターとしては、宿主酵母内での複製増殖機能を司る遺伝子を少なくとも含むものであれば特に制限されない。酵母に遺伝子を導入するために使用できるプラスミドの具体例としては、2μmプラスミド由来のpAURベクター(タカラバイオ社製)やpESCベクター(STRATAGENE社製)などが
挙げられる。これらのベクターは市販されており容易に入手可能である。
【0033】
なお、DNAの切断、連結、その他、染色体DNAの調製、PCR、プラスミドDNAの調製、形質転換、プライマーとして用いるオリゴヌクレオチドの設定等の方法は、当業者によく知られている通常の方法を採用することができる。これらの方法は、Sambrook, J., Fritsch, E. F., and Maniatis, T., "Molecular Cloning A Laboratory Manual, Second Edition", Cold Spring Harbor Laboratory Press, (1989)等に記載されている。
【0034】
ARE1(またはARE2遺伝子)およびYEH1遺伝子を、上記したような酵母内で複製可能なプラスミドベクターの適当な部位に挿入して得られる組換えベクターで、サッカロミセス・セレビジエなどの酵母を形質転換することにより、これらの遺伝子の発現が増加した酵母が得られる。
【0035】
形質転換は、例えば、エレクトロポレーション法(Fromm ME,et al.,(1986) Nature 319(6056):791-793)、スフェロプラスト法、酢酸リチウム法(Ito,H.et al, (1983) J. Bacteriol. 153(1), 163-168)等によって行うことができる。
また、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子およびステリルエステル加水分解酵素遺伝子の発現の増強は、公知の相同組換え法によって宿主のゲノム上でARE1(またはARE2遺伝子)およびYEH1遺伝子を多コピー化させることによって行うこともできる。例えば、ゲノム組込型プラスミドのうち多コピーがゲノムに組み込まれるベクターを用いる方法(Bio/Technol. 9, 1382-1385(1991))が挙げられる。ゲノム上の各遺伝子を含む発現単位の挿入箇所は、ステロール生合成関連遺伝子や生育関連遺伝子の発現が阻害されない箇所であれば何れのものでもよい。具体的には、ARE1遺伝子を適当なプロモーターの制御下となるように連結させたDNAを、宿主ゲノム上のARE1遺伝子座上などに挿入し、宿主ゲノム中のARE1遺伝子が2コピーとなるようにして、さらにYEH1遺伝子も同様のプロモーターの制御下となるように連結させたDNAを宿主ゲノム上のYEH1遺伝子座などに挿入して宿主ゲノム中のYEH1遺伝子が2コピーとなるようにゲノムを改変する方法などが好ましく用いられる。
【0036】
また、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子およびステリルエステル加水分解酵素遺伝子の発現の増強は、宿主ゲノム上でARE1(またはARE2遺伝子)およびYEH1遺伝子のプロモーターを置換または改変することによっても行うことができる。
上記組換えプラスミドによる導入または染色体上への相同組換えにおいて、ARE1(またはARE2遺伝子)およびYEH1遺伝子を発現させるためのプロモーター、または染色体上のARE1(またはARE2遺伝子)およびYEH1遺伝子のプロモーターを置換するために使用するプロモーターは、宿主酵母で機能しうるものであれば特に制限されないが、例えば、3−フォスフォグリセレートキナーゼ1遺伝子のプロモーター(以下、「PGK1プロモーター」と称することがある)あるいはアルコールデヒドロゲナーゼ遺伝子などの恒常的プロモーター、ないしは、GAL1やGAL10(Mol. Cell Biol. 4(8), 1440-1448(1984))などの誘導型プロモーター(例えば、STRATAGENE社製)などが挙げられる。
【0037】
なお、本発明の酵母は、アシルCoA:ステロールアシルトランスフェラーゼ遺伝子、およびステリルエステル加水分解酵素遺伝子の発現の増強に加えて、他の酵素遺伝子の発現が非改変株と比べて増強または低下するように改変されたものでもよい。
他の酵素の改変は目的とするΔ5,7−ステロールの種類により適宜選択される。
【0038】
例えば、TEOLを生産する場合、ステロールC−22デサチュラーゼ遺伝子(ERG5)とステロールC−24メチルトランスフェラーゼ遺伝子(ERG6)の発現が低下するように改変することが好ましい。これらの改変は特開2004-141125に記載されている。
また、Δ5,7−ステロールとしてのエルゴスタ−5,7,24−トリエノールを中間体とし
てハイドロコルチゾンを目的生成物とする場合には、ステロールC−22デサチュラーゼ遺伝子(ERG5)のみの発現が低下するように改変することが好ましい。
【0039】
ステロールC−22デサチュラーゼは、各種ステロール類の側鎖の22位に二重結合を導入する能力を有する酵素であり、具体的には、例えばコレスタ−5,7,24−トリエノールをコレスタ−5,7,22,24−テトラエノールに、エルゴスタ−5,7−ジエノールをエルゴスタ−5,7,22−トリエノールに、エルゴスタ−5,24−ジエノールをエルゴスタ−5,22,24−トリエノールに、エルゴスタ−5,7,24−トリエノールをエルゴスタ−5,7,22,24−テトラエノールに変換する能力を有する酵素を指す。
【0040】
また、ステロールC−24メチルトランスフェラーゼは、S−アデノシルメチオニンから各種ステロール類の24位にメチル基を転移させる能力、さらには転移により付加されたメチル基にさらにメチル基を転移させる能力を有する酵素であり、具体的には、例えばザイモステロールをフェコステロールに変換する能力を有する酵素を指す。
これら酵素をコードする遺伝子は、既に単離され塩基配列が決定されているものもあり、例えば、ステロールC−22デサチュラーゼとしては、サッカロミセス・セレビシエ由来の遺伝子が報告されており(Biochem. Biophys. Acta 1299, 313-324(1996))、ステロールC−24メチルトランスフェラーゼとしては、サッカロミセス・セレビシエ由来の遺伝子が報告されている(Gene 169, 105-109(1996))。
【0041】
ステロールC−22デサチュラーゼ、および、ステロールC−24メチルトランスフェラーゼの両方の活性低減化方法としては、エチルメタンスルホン酸(EMS)、N−メチル−N'−ニトロ−N−ニトロソグアニジン(NTG)などの化学物質あるいはUVなどの変異源処理によって得られた両酵素遺伝子の欠損突然変異株を交配する方法、両遺伝子の転写をアンチセンス遺伝子の導入によりおさえる方法、両遺伝子の転写産物をRNAi技術によって抑制する方法、両酵素の特異的阻害剤で酵素活性を抑える方法などを用いることができる。
【0042】
酵母においては、ステロールC−22デサチュラーゼおよびステロールC−24メチルトランスフェラーゼをそれぞれコードしている遺伝子断片中に、HIS3、LEU2、TRP1、URA3などの栄養要求性遺伝子DNAないしはジェネティシンやオーレオバシジンAなどの抗生物質や抗真菌薬に対する耐性遺伝子が挿入された遺伝子断片、あるいはその遺伝子断片がさらにベクターに挿入されたプラスミドを作製し、相同組換えにより宿主染色体中の該遺伝子を破壊する方法が好適に用いられる。
【0043】
例えば、サッカロミセス・セレビシエにおいては、ステロールC−22デサチュラーゼをコードする遺伝子(ERG5)を含むDNA断片、ステロールC−24メチルトランスフェラーゼをコードする遺伝子(ERG6)を含むDNA断片を制限酵素切断ないしはポリメラーゼチェーンリアクション法(以下これを「PCR」と略称することがある)によって得てベクターに組み込んで中間ベクターを作製する。この中間ベクターをERG5遺伝子およびERG6遺伝子の遺伝子内部にある任意の制限酵素部位で2つに切断し、酵母のHIS3、LEU2、TRP1、URA3などの栄養要求性遺伝子cDNAのいずれか1つを2つの中間ベクター同士で重ならないように選択し挿入することにより遺伝子破壊用ベクター、ないしはそれをPCRで増幅して得られる遺伝子破壊用DNA断片を作製する。次いでこの遺伝子破壊用ベクターないしは遺伝子破壊用DNA断片を酵母に遺伝子導入することで相同組換えを誘発し、目的遺伝子を破壊することができる。
【0044】
かくして構築される遺伝子破壊用DNA断片若しくは遺伝子破壊用ベクターの具体例として、ERG5遺伝子の内部371bpを欠失させ、この欠失部分にTRP1遺伝子が挿
入されているDNA断片(erg5::TRP1)若しくは該DNA断片を有するプラスミドpSK−erg5::TRP1、ERG6遺伝子の内部476bpを欠失させ、この欠失部分にHIS3遺伝子が挿入されているDNA断片(erg6::HIS3)若しくは該DNA断片を有するプラスミドpUC−erg6::HIS3(特開2004-141125号公報を参照)等が挙げられる。
【0045】
酵母における遺伝子破壊の確認は、特定の栄養要求性遺伝子が挿入された遺伝子部分が相同組換えにより酵母ゲノム中に組み込まれるために、宿主株が持っていた複数の栄養要求性のうち該当する栄養要求性が消失することで知ることができる。さらにERG5およびERG6遺伝子の5'末端と3'末端にそれぞれ設定したPCRプライマーを用いて、遺伝子破壊操作を受けた酵母株から抽出した染色体DNAを鋳型としてPCRを行い、PCR断片の長さが挿入した栄養要求性遺伝子の長さだけ大きくなっていることを確認することにより遺伝子破壊の確認を行うことができる。
かくして得られる遺伝子破壊株の具体例としては、遺伝子破壊用ベクターpSK−erg5::TRP1のerg5::TRP1断片でERG5遺伝子を破壊した、サッカロミセス・セレビシエKAΔ5−5株、さらに該Δ5−5株のERG6遺伝子をpUC−erg6::HIS3のerg6::HIS3断片で破壊したKAΔ5Δ6−1株、KAΔ5Δ6−4株、KAΔ5Δ6−5株等が挙げられる。
さらに、本発明の酵母としては、以下の(3)に記載の酵母も含まれる。
【0046】
(2)Δ5,7−ステロールの製造方法
上記で説明した本発明の酵母を炭素源、窒素源、無機塩、アミノ酸、ビタミンなどを含有する培地中、好気条件下、温度、pHなどを調整しつつ培養を行えば、酵母菌体中にΔ5,7−ステロールが蓄積するのでこれを採取する。
【0047】
炭素源としては、例えばグルコース、グリセロール、フルクトース、シュークロース、マルトース、マンノース、澱粉、澱粉加水分解液、糖蜜、などの炭水化物、酢酸、ピルビン酸、メバロン酸などの各種有機酸が使用できる。さらに酵母の資化性によって、炭化水素、アルコールなども用いられる。特に廃糖蜜は好適にもちいられる。
窒素源としては、例えばアンモニア、または塩化アンモニウム、炭酸アンモニウムなどの各種無機塩および有機アンモニウム塩類あるいはペプトン、肉エキス、酵母エキス、カゼイン加水分解物など種々のものが使用可能である。
無機塩としては、例えばリン酸二水素カリウム、リン酸水素二カリウム、硫酸アンモニウム、塩化アンモニウム、硫酸マグネシウム、塩化ナトリウムなどを使用する。ビタミン、アミノ酸としては、使用する酵母の種類によってことなるが、必要に応じ添加する。また使用する菌株が、栄養要求性を示す場合は、その要求物質を添加する。
【0048】
培養は使用する菌株に応じて適切な条件で行うが、通常は、振盪培養または通気攪拌培養などの好気的条件下に行う。生産株として、サッカロミセス・セレビシエを用いる場合は、培養温度は20〜40℃が好適であり、培地のpHは弱酸性付近に維持する事が望ましい。培養期間は通常1〜15日間で菌体中にΔ5,7−ステロールが蓄積する。また、培養期間中に上記に記載した炭素源や窒素源、無機塩、ビタミン、アミノ酸を連続的、あるいは断続的に添加する流加培養や連続培養を行うことにより、菌体の生育やΔ5,7−ステロールの蓄積をさらに伸ばすことが可能である。
【0049】
培養終了後のΔ5,7−ステロールの抽出は一般に生物組織からステロール類を抽出する方法により行うことができる。具体的には、例えば、菌体を海砂とともにそのままアセトン中で破砕し酢酸エチルで抽出するか、又は、2MのNaOHを含む50%エタノール中で2時間煮沸し鹸化したのちヘプタンないしはクロロホルム/メタノール/水(体積比86:16:1)混液で抽出し、薄層クロマトグラフィーにかけステロール画分を掻き取
って、窒素気流下で乾燥させメタノール/クロロホルム混液に溶解することで総ステロールを菌体から抽出することができる。
【0050】
次いでステロールの種類の同定は、薄層クロマトグラフィーで分離抽出したステロール画分を、それ自体公知の適当な溶媒およびカラムの組み合わせでガスクロマトグラフィーないしは高速液体クロマトグラフィーにかけて、現れるピークの保持時間を標準試料の保持時間と比較することで行うことができる。また、同定された各種ステロールの総ステロール中にしめる割合は各ピークの面積から算出することができる。さらに、これらの主要なステロールのピークが目的化合物であることの確認は、マススペクトロメトリーを用いて、分子量および開裂パターンを調べることで行うことができる。また、GC−MSあるいはHPLC−MSを用いることによりさらに効率よく抽出されたステロール類を同定することができる。
【0051】
得られたΔ5,7−ステロールは各種医薬等の中間体となりうる。
例えば、TEOLはケノデオキシコール酸やウルソデオキシコール酸の合成原料として有用である。特開2007-210888号公報および特開2006-056877号公報に、TEOLからのウルソデオキシコール酸の合成方法が開示されている。
具体的には、TEOLを原料として、以下の4つの工程、すなわち
(I)3位水酸基の酸化と5位2重結合の4位への異性化を行う工程
(II)側鎖の酸化的切断により24位をカルボキシル基またはそのエステル誘導体とする工程
(III)7位に酸素官能基を導入する工程
(IV)4位2重結合の還元飽和化による5β立体構築を行う工程
を経て5β−3,7−ジオキソコラン酸を製造し、これを還元することによってウルソデオキシコール酸を製造することができる。
5β−3,7−ジオキソコラン酸のウルソデオキシコール酸への還元反応は、金属還元または接触水素化によって行うことができる。また、5β−3,7−ジオキソコラン酸からウルソデオキシコール酸への変換は、特開昭60-228500号公報及び特開平5-32692号公報の記載を参酌して行うことができる。
【0052】
(3)ハイドロコルチゾンの製造方法
上記酵母で、以下の活性を有するものを上記(2)と同様にして培養することによりハイドロコルチゾンが生成される。活性を有する酵母とは、該酵母自体がそれぞれの活性を有していてもよいし、該活性を有する物質を培養液中に添加してもよいし、該活性を有する酵母の菌体あるいはその処理物を培養液中に添加することでもよい。
【0053】
ハイドロコルチゾンの生成にさらに必要な活性とは、Δ7−還元活性、ステロール側鎖の20位及び22位との結合を切断する活性、ステロール3位酸化異性化活性、ステロイド17α水酸化活性、ステロイド21位水酸化活性、及びステロイド11β位水酸化活性である。本発明の酵母には、上記活性のうち少なくとも1つを有するものも含まれる。ハイドロコルチゾン生成を目的とする本発明の酵母としては、上記活性を有する蛋白質を生成するものが好ましく、また、発現するための遺伝子及びその活性に必要な補酵素などの遺伝子の全てを導入したものも好ましく挙げられる。上記活性を有する蛋白質を生成するためには、該蛋白質をコードするDNAを上記の発現ベクターに挿入して、上記酵母を形質転換した形質転換酵母を用いる方法が好ましく用いられる。
【0054】
ステロール側鎖の20位及び22位との結合を切断する活性を有する蛋白質を生成する活性を獲得する方法としては、 (i) 例えば、WO2010/079594公報に記載の酵素蛋白質(CYP204A1やCYPSS204A)をコードする遺伝子やその他の遺伝子がコードする同様の活性を有する酵素遺伝子と、該酵素蛋白質への電子伝達活性を有するフェレドキシン蛋白質、およ
び該フェレドキシン蛋白質への電子伝達活性を有するフェレドキシン還元酵素蛋白質のそれぞれをコードする遺伝子の混合物、あるいは(ii)上記酵素蛋白質と該酵素蛋白質への電子伝達活性を有するフェレドキシン蛋白質、および該フェレドキシン蛋白質への電子伝達活性を有するフェレドキシン還元酵素蛋白質の融合蛋白質をコードする遺伝子が発現するように該酵母に遺伝子導入する方法などが挙げられる。
【0055】
ここで、電子伝達活性を有するフェレドキシン蛋白質、および該フェレドキシン蛋白質への電子伝達活性を有するフェレドキシン還元酵素蛋白質は両者の活性を有する1種の蛋白質でもよい。例えば、Bacillus megateriumのP450-BM3遺伝子のP450還元酵素ドメインを酵素蛋白質(P450 2C11)に連結し、人工的に作製した融合蛋白質を利用できる(Helvig, C. and Capdevila, J. H., Biochemistry, (2000)39, 5196-5205)。
【0056】
フェレドキシン還元酵素は、例えば以下の中から選択される。
ホウレン草由来のフェレドキシンレダクターゼ、
シュードモナス プチダ(Pseudomonas putida)由来プチダレドキシンレダクターゼ、
動物由来のアドレノドキシンレダクターゼ、
ノボスフィンゴビウム サブテラニウム(Novosphingobium subterraneum)由来フェレドキシンレダクターゼ、
ノボスフィンゴビウム アロマティシボランス(Novosphingobium aromaticivorans)由来フェレドキシンレダクターゼ、
エシェリヒア コリ(Escherichia coli)由来フラボドキシンレダクターゼやフェレドキシンレダクターゼ、
サッカロミセス・セレビシエ(Saccharomyces cerevisiae)由来フェレドキシンレダクターゼ、
またはその他のフェレドキシンレダクターゼ。
【0057】
フェレドキシンは、例えば以下の中から選択される;
ホウレン草由来のフェレドキシン、
シュードモナス プチダ(Pseudomonas putida)由来プチダレドキシン、
動物由来のアドレノドキシン、
ノボスフィンゴビウム サブテラニウム(Novosphingobium subterraneum)由来フェレドキシン、
ノボスフィンゴビウム アロマティシボランス(Novosphingobium aromaticivorans)由来フェレドキシン、
エシェリヒア コリ(Escherichia coli)由来フラボドキシンやフェレドキシン、
サッカロミセス・セレビシエ(Saccharomyces cerevisiae)由来フェレドキシン、
またはその他のフェレドキシン型蛋白質。
【0058】
ステロイド17α位水酸化酵素蛋白質、ステロイド21位水酸化酵素蛋白質、ステロイド11β位水酸化酵素蛋白質、ステロール3位酸化異性化酵素蛋白質は、例えば、Molecular and Cellular Endocrinology 1990, 73, 73-80に記載のものが用いられる。
また、ステロールΔ7−還元酵素蛋白質は、Journal of Biological Chemistry 1996, 271, 10866-10873記載のシロイヌナズナ(Arabidopsis thaliana)由来のもの、あるいはApplied and Environmental Microbiology 2007, 73, 1736-1741記載のモルティエレラ(Mortierella)属カビ由来ものが用いられる。
具体的には、ステロールΔ7−還元酵素蛋白質はシロイヌナズナ由来NADPH−ステロールΔ7−還元酵素モルティエレラ・アルピナ(Mortierella alpina)由来ステロー
ルΔ7−還元酵素(MoDELTA7SR)が好ましい。
ステロイド17α位水酸化酵素蛋白質は、例えば、ウシ、マウス、ラット、あるいはヒト由来シトクロムP450c17が好ましい。
また、ステロイド21位水酸化酵素蛋白質は、ウシ、マウス、ラット、あるいはヒト由来シトクロムP450c21が好ましい。
ステロイド11β位水酸化酵素蛋白質としてはウシ、マウス、ラット、あるいはヒト由来シトクロムP450c11やクルブラリア・ルナタ(Curvularia lunata)由来P-450lun等が好ましい。
さらにステロール3位酸化異性化酵素蛋白質はウシ、マウス、ラット、あるいはヒト由来3β−ヒドロキシステロイド脱水素酵素(3βHSD)やストレプトミセス(Streptomyces)属細菌由来コレステロール酸化酵素が好ましい。
【0059】
ここで、好ましく用いられる酵母としては、サッカロミセス(Saccharomyces)属酵母、ピキア(Pichia)属酵母、シゾサッカロミセス(Schizosaccharomyces)属酵母、ヤロウィア(Yarrowia)属酵母、アスペルギルス(Aspergillus)属、モルティエレラ(Mortierella)属があげられる。より好ましくは、サッカロミセス属酵母があげられる。具体的にはサッカロミセス・セレビシエ(Saccharomyces cerevisiae)であり、より具体的には、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)FY1679-28c株(JP2004-528827 A1)、以下同様に、FY1679-18b株(JP2004-528827 A1)、KA311A株(FERM:P-19053)、YPH499株(ATCC#204679)、YPH500株(ATCC#204680)等が挙げられ、それぞれの酵素蛋白質をコードするDNAは、pESC-LEU(Stratagene社)等の発現ベクターや酵母菌の染色体に挿入されていることが好ましい。
【0060】
また、これらの酵母が、以下(1)−(5)の性質のいずれかまたはすべてを持つことが好ましい。
(1)ステロール−22デサチュラーゼ蛋白質をコードする遺伝子またはその発現を制御する領域のDNA配列を改変することで内在性のステロールの22位と23位の間が不飽和化されない、
(2)NADP−シトクロムP450還元酵素蛋白質の発現が本蛋白質をコードする遺伝子をその発現制御領域とともに1コピー以上宿主細胞に導入することで、強化されている、
(3)ステロールエステル加水分解酵素の発現が本蛋白質をコードする遺伝子をその発現制御領域とともに1コピー以上宿主細胞に導入することで、強化されている、
(4)ステロイド17α水酸化酵素、ステロイド21位水酸化酵素、ステロイド11β位水酸化酵素に電子を伝達するフェレドキシン型蛋白質および該蛋白質へ電子伝達活性を有するフェレドキシン還元酵素蛋白質のそれぞれをコードする遺伝子をその発現制御領域とともに宿主細胞に導入することで、該蛋白質の発現が強化されている、
(5)ステロール−24メチル基転移酵素蛋白質をコードする遺伝子またはその発現を制御する領域のDNA配列を改変することで内在性のステロールの24位にメチル基が存在しない。
【0061】
反応混合物からハイドロコルチゾンを分画する方法は特に限定されず、当業者に公知の分離又は精製のための手法を用いることができる。例えば、溶媒抽出、晶析、樹脂吸着、カラムクロマトグラフィー等により行うことができるが、これに限定されるものではない。
【0062】
また、上記で製造したハイドロコルチゾンからは、各種の誘導体を公知の方法で製造することができる。さらに、かくして製造されたハイドロコルチゾンおよびその誘導体は、それ自体既知の方法により試験管内、あるいは生体内における薬理学的または生理学的試験により、その医薬としての活性や安全性のスクリーニングを行なうことにより疾病治療薬とすることができる。この場合、上記工程により得た化合物を有効成分とする医薬組成物を製造する方法も本発明に含まれる。
【0063】
具体的には、ハイドロコルチゾンそれ自体を単独で用いることも可能であるが、薬学的に許容され得る担体と配合して医薬組成物として用いることもできる。かくして得られる医薬化合物はそれ自体を単独で用いることも可能であるが、薬学的に許容され得る担体と配合して医薬組成物として用いることもできる。この時の有効成分の担体に対する割合は、0.01〜90重量%の間で変動され得る。
【0064】
本発明で得られたΔ5,7−ステロールやハイドロコルチゾンを含む医薬組成物は、必要に応じて糖衣を施した錠剤、カプセル剤、軟カプセル剤、散剤、顆粒剤、細粒剤、シロップ剤、乳剤、懸濁剤、液剤等の剤形として経口的に投与してもよいし、あるいは薬学的に許容し得る液との無菌性溶液または懸濁剤形等の注射剤として、例えば静脈内投与、筋肉内投与、局所内投与、皮下投与してもよい。また坐薬や舌下錠、経鼻、経肺製剤等の粘膜投与、さらには軟膏や貼付剤等の外用剤としての投与も可能である。経口、非経口の組成物を調製する場合には、有機または無機の固体または液体の担体、希釈剤とともに通常用いられる単位容量形態で混和することによって行うことができる。これら製剤における有効成分量は、指示された範囲の適当な容量が得られるようにするものである。
【0065】
経口投与および粘膜投与のための固形製剤を製造する際に用いられる賦形剤としては、例えば乳糖、ショ糖、デンプン、タルク、セルロース、デキストリン、カオリン、炭酸カルシウム等が用いられる。経口投与のための液体製剤、すなわちシロップ剤、懸濁剤、液剤等は、一般的に用いられる不活性な希釈剤、例えば水、植物油等を含む。この製剤は、不活性な希釈剤以外に補助剤、例えば湿潤剤、懸濁補助剤、甘味剤、香味剤、着色剤、保存剤、安定剤等を含むこともできる。注射剤、粘膜投与剤等の製造に用いられる溶剤または懸濁化剤としては、例えば水、プロピレングリコール、ポリエチレングリコール、ベンジルアルコール、オレイン酸エチル、レシチン等が挙げられる。坐剤に用いられる基剤としては、例えばカカオ脂、乳化カカオ脂、ラウリン脂、ウィテップゾール等が挙げられる。外用剤の製造には、溶解剤または溶解補助剤としてアルコ−ル、脂肪酸エステル類、プロピレングリコール等が、粘着剤としてカルボキシビニルポリマ−、多糖類等が、乳化剤として界面活性剤等などが使用される。
【実施例】
【0066】
以下、実施例を挙げて本発明をさらに詳細に説明するが、本発明はこれら実施例により何ら限定されるものではない。
【0067】
(実施例1)
サッカロミセス・セレビジエ YPH499、FY1679−06c株、KA311A株、X2181−1B株のerg5およびerg6遺伝子の二重破壊及びARE1およびYEH1増強株の作製
(1)酵母染色体DNAの調製
サッカロミセス・セレビジエ S288C株(ATCCより入手 ATCC20458)の染色体DNAは、特開2004-141125と同様の方法で調製された。
【0068】
(2)遺伝子破壊用DNA断片erg5::TRP1の導入によるerg5遺伝子破壊株SX1311、SX1306、SX1334の作製
サッカロミセス・セレビジエ由来ステロールC−22デサチュラーゼをコードする遺伝子ERG5破壊用のプラスミドpSK−erg5::TRP1(特開2004−141125号公報を参照)を制限酵素Sac I(タカラバイオ社製、以下、特に記載のない限り、制限酵素はタカラバイオ社製である)とKpn Iで切断した。このDNA断片をサッカロミセス・セレビジエYPH499株(MATa ura3 lys2 ade2 trp1 his3 leu2)(STRATAGENE社より入手)とFY1679−06c株(MATα ura3 leu2 trp1 his3)(Euroscarf
社より入手)、X2181−1B(MATalpha his2 gal1 trp1 ade1 MAL SUC)(ATCCより入手)に酢酸リチウム法(Ito,H.et al, (1983) J. Bacteriol. 153(1), 163-168)、もしくはエレクトロポレーション法(Fromm ME,et al.,(1986) Nature 319(6056):791-793)で形質転換し、YPH499株は各20mg/lのアデニン、L−ヒスチジン、ウラシル、30mg/l L−リジン、100mg/l L−ロイシンを、FY1679−06c株は各20mg/lのL−ヒスチジン、ウラシル、100mg/l L−ロイシンを、X2181−1B株は20mg/l アデニンをそれぞれ含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0069】
寒天培地上に生育してきたトリプトファン非要求性のコロニーをYPD液体培地(1%
酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーTRP1−outF(配列番号1)とリバースプライマーERG5−D31(配列番号2)の組み合わせでPCRを行い、ERG5遺伝子座上での相同組換えの有無を調べた。PCRはTaKaRa ExTaq DNA ポリメラーゼ(タカラバイオ社製)を用いて(1)94℃、2分、(2)94℃、1分、(3)53℃、1分、(4)72℃、3分、(2)から(4)の反応を30回繰り返した後、4℃で冷却の条件で反応を行った。比較対照として、特開2004-141125に記載されたKAΔ5−5株、およびYPH499株、FY1679−06c株、X2181−1B株の染色体DNAを鋳型として同様にPCRを行った。
【0070】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1311(YPH499株由来)、SX1306(FY1679−06c株由来)、SX1334(X2181−1B株由来)と名付けた形質転換体は目的サイズと同じ約1.93kbpのDNA断片が検出された。従って、SX1311、SX1306、SX1334株は相同組換えによりERG5遺伝子が破壊されたことが確認された。
【0071】
(3)SX1311、SX1306株への遺伝子破壊用DNA断片erg6::HIS3の導入、およびSX1334株への遺伝子破壊用DNA断片erg6::HIS2の導入によるerg5、erg6二重遺伝子破壊株SX1313、SX1312、SX1340株の作製
(i)遺伝子破壊用DNA断片erg6::HIS3導入によるerg5、erg6二重破壊株SX1313、SX1312株の作製
サッカロミセス・セレビジエ由来ステロールC−24メチルトランスフェラーゼをコードする遺伝子ERG6破壊用のプラスミドpUC−erg6::HIS3(特開2004-141125を参照)を制限酵素ScaIとEcoRIで切断した。このDNA断片を上記(2)で作製したSX1311、SX1306株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SX1311株は各20mg/lのアデニン、ウラシル、30mg/l L−リジン、100mg/l L−ロイシンを、SX1306株は20mg/l ウラシル、100mg/l L−ロイシンをそれぞれ含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0072】
寒天培地上に生育してきたヒスチジンおよびトリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、 2%ポリペプトン、 2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーERG6−U52(配列
番号3)とリバースプライマーERG6−D34(配列番号4)の組み合わせでPCRを行い、ERG6遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。比較対照として、特開2004-141125に記載されたKAΔ5Δ6−1株、およびYPH499株、FY1679−06cの染色体DNAを鋳型として同様にPCRを行った。
【0073】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1313(YPH499株由来)、SX1312(FY1679−06c株由来)と名付けた形質転換体は目的サイズと同じ約1.78kbpのDNA断片が検出された。従って、SX1313,SX1312株は相同組換えによりERG6遺伝子が破壊されたことが確認された。
【0074】
(ii)破壊用ERG6、酵母発現用HIS2遺伝子断片の調製
サッカロミセス・セレビジエ由来ステロールC−24メチルトランスフェラーゼをコードする遺伝子(ERG6)を含むDNA断片、HIS2遺伝子を含むDNA断片は、上記(1)で取得したサッカロミセス・セレビジエ S288C株の染色体DNAを鋳型にしてPCRにて合成、単離した。以下にその方法の詳細を示す。
【0075】
ERG6遺伝子断片のPCRによる合成には、フォワードプライマーERG6−5L(配列番号5)とリバースプライマーERG6−3L(配列番号6)を用いた。このプライマーの組み合わせでERG6の蛋白質コード領域を含み、開始コドンの238塩基前から終始コドンの258塩基後までを増幅することができる。また、5’側には制限酵素PstIの認識配列を、3’側にはEcoRIの認識配列を含んだ形で合成することができる。
HIS2遺伝子断片のPCRによる合成には、フォワードプライマーHIS2−F(配列番号7)とリバースプライマーHIS2−R(配列番号8)を用いた。このプライマーの組み合わせでHIS2の蛋白質コード領域を含み、開始コドンの430塩基前から終始コドンの290塩基後までを増幅することができる。また、5’側には制限酵素SmaIの認識配列を、3’側にはEcoRIの認識配列を含んだ形で合成することができる。
PCRは、PrimeSTAR HS DNA ポリメラーゼ(タカラバイオ社製)を用いて、(1)98℃、2分、(2)98℃、10秒、(3)55℃、5秒、(4)72℃、2分、(2)から(4)の反応を30回繰り返し、(5)72℃、2分間反応させた後、4℃で冷却の条件で反応を行った。次にPCR産物をアガロースゲル電気泳動し、約1.65kbpのERG6遺伝子断片を、約1.73kbpのHIS2遺伝子断片を確認した。
【0076】
(iii)erg6破壊用プラスミドpUC−erg6::HIS2の作製
pUC19(タカラバイオ社製)を制限酵素PstIとEcoRIで切断したものと、上記(ii)でPCR合成したERG6遺伝子断片をPstIとEcoRIで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素HincIIによる切断で、約0.64kbpと3.66kbpの断片が確認できたものを目的プラスミドとして、pUC−ERG6Lと名付けた。
次に、pGEM−T Easy Vector Systems(プロメガ社製)を用いて、pGEM−T Easy(プロメガ社製)のクローニングサイトに上記(ii)でPCR合成したHIS2遺伝子断片をダイレクトクローニングにより連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素Sm
aIとEcoRIによる切断で、約3.0kbpと1.73kbpの断片が確認できたものを目的プラスミドとして、pGEM−T−HIS2と名付けた。
【0077】
さらに、上記pUC−ERG6Lを制限酵素BspT104Iで切断し、cloned
DNA Polymerase I(タカラバイオ社製)を用いてDNA末端平滑化処理を行った後に、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、プラスミドpGEM−T−HIS2を制限酵素SmaIとXbaIで切断し、DNA Polymerase I(タカラバイオ社製)を用いてDNA末端平滑化処理を行ったものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素ScaIによる切断で、約1.71kbpと1.67kbp、1.89kbpの断片が確認できたものを目的プラスミドとして、pUC−erg6::HIS2と名付けた。
【0078】
(iv)erg5、erg6二重遺伝子破壊株SX1340の作製
上記(iii)で作製したERG6破壊用のプラスミドpUC−erg6::HIS2を制限酵素EcoRIとPstIで切断した。このDNA断片を上記(2)で作製したSX1334株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、20mg/l アデニンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0079】
寒天培地上に生育してきたヒスチジンおよびトリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、 2%ポリペプトン、 2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーERG6−U52(配列番号3)とリバースプライマーERG6−D34(配列番号4)の組み合わせでPCRを行い、ERG6遺伝子座上での相同組換えの有無を調べた。PCRは上記(i)で述べた条件で行った。比較対照として、特開2004-141125に記載されたKAΔ5Δ6−1株、およびX2181−1B株の染色体DNAを鋳型として同様にPCRを行った。
PCR産物をアガロースゲル電気泳動で解析した結果、SX1340と名付けた形質転換体は目的サイズと同じ約2.76kbpのDNA断片が検出された。従って、SX1340株は相同組換えによりERG6遺伝子が破壊されたことが確認された。
【0080】
(4)erg5、erg6二重破壊株へのARE1遺伝子導入株の作製
(i)酵母発現用プラスミドpPG−LEUの作製
酵母細胞内で外来遺伝子を構成的に発現させるための3−ホスホグリセレートキナーゼ1遺伝子(PGK1)のプロモーター領域は、サッカロミセス・セレビジエKA311A株(受託番号:FERM P−19053)の染色体DNAを鋳型にし、フォワードプライマーPPGK1−5A(配列番号9)、リバースプライマーPPGK1−3A(配列番号10)を用いたPCRにて合成した。このプライマーの組み合わせで5'側に制限酵素BamH I、3'側に制限酵素EcoR Iの認識配列を含んだ形で合成することができる。PCRはTaKaRa ExTaq DNA ポリメラーゼ(タカラバイオ社製)を用いて(1)94℃、2分、(2)94℃、1分、(3)55℃、1分、(4)72℃、1分、(2)から(4)の反応を30回繰り返し、(5)72℃、4分間反応させた後、4℃で冷却の条件で反応を行った。次にPCR産物をアガロースゲル電気泳動し、約0.6kbpのPGK1プロモーターのDNA断片を確認した。
このPCR産物をpCR2.1(Invitrogen社製)に連結した。連結物を用
いてエッシェリシア・コリJM109に形質転換し、得られたコロニーからプラスミド抽出を行った。
得られたプラスミドをBamH I、EcoR Iで切断し、アガロースゲルから約0.6kbpのDNA断片(PGK1プロモーター)をGENE CLEAN II KIT(フナコシ社製)により抽出した。
【0081】
一方、酵母発現用ベクターpADNSΔE(特開2004-141125)も同じくBamH I、EcoR Iで切断し、アガロースゲルから約6.58kbpのDNA断片を抽出した。
上記2つのDNA断片をTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結し、エッシェリシア・コリJM109に形質転換し、得られたコロニーからプラスミドを抽出した。アガロースゲル電気泳動により約7.2kbpを確認したプラスミドを目的プラスミドとし、pPG−LEUと名付けた。pPG−LEUはプロモーターとしてPGK1プロモーター、ターミネーターとしてアルコールデヒドロゲナーゼ1遺伝子(ADH1)由来のADH1ターミネーターを持つ。
【0082】
(ii)酵母組換え用プラスミドpPG−LEU(−2μ)の作製
上記(i)で作製したプラスミドpPG−LEUを制限酵素Hpa IとEco47IIIで切断し、アガロースゲルから約5.4kbpのDNA断片を抽出することで2μm
DNAを除き、得られたDNA断片を自己連結し、エッシェリシア・コリJM109に形質転換し、得られたコロニーからプラスミドを抽出した。アガロースゲル電気泳動により約5.4kbpを確認したプラスミドを目的プラスミドとし、pPG−LEU(−2μ)と名付けた。
【0083】
(iii)酵母発現用のARE1、URA3、ADE1遺伝子断片の調製
サッカロミセス・セレビジエ由来アシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子(ARE1)を含むDNA断片、URA3、ADE1を含むDNA断片は、上記(1)で取得したサッカロミセス・セレビジエ S288C株の染色体DNAを鋳型にしてPCRにて合成、単離した。以下にその方法の詳細を示す。
ARE1遺伝子断片のPCRによる合成には、フォワードプライマーARE1−Sma
I(F)(配列番号11)とリバースプライマーARE1−EcoR I(R)(配列番号12)を用いた。このプライマーの組み合わせでARE1の蛋白質コード領域である開始コドンから終始コドンまでを増幅することができる。また、5'側には制限酵素Sma Iの認識配列を、3'側にはEcoR Iの認識配列を含んだ形で合成することができる。 URA3遺伝子断片のPCRによる合成には、フォワードプライマーURA3−2F(配列番号13)とリバースプライマーURA3−1R(配列番号14)を用いた。このプライマーの組み合わせでURA3の蛋白質コード領域を含み、開始コドンの254塩基前から終始コドンの178塩基後までを増幅することができる。また、合成された遺伝子断片中には開始コドンの223塩基前と終始コドンの136塩基後にHind IIIの認識配列が含まれた形で合成される。ADE1遺伝子断片のPCRによる合成には、フォワードプライマーADE1−F(配列番号15)とリバースプライマーADE1−R(配列番号16)を用いた。このプライマーの組み合わせでADE1の蛋白質コード領域を含み、開始コドンの346塩基前から終始コドンの312塩基後までを増幅することができる。また、合成された遺伝子断片中には終始コドンの106塩基後にEcoRVの認識配列が含まれた形で合成される。PCRは、PrimeSTAR HS DNA ポリメラーゼ(タカラバイオ社製)を用いて、(1)98℃、2分、(2)98℃、10秒、(3)55℃、5秒、(4)72℃、2分、(2)から(4)の反応を30回繰り返し、(5)72℃、7分間反応させた後、4℃で冷却の条件で反応を行った。次にPCR産物をアガロースゲル電気泳動し、約1.83kbpのARE1遺伝子断片を、約1.23kbpのURA3遺伝子断片を、約1.57kbpのADE1遺伝子断片を確認した。
【0084】
(5)ARE1発現用プラスミドpPGU−ARE1(−2μ)、pPGADE1−ARE1(−2μ)の作製
上記(2)で作製したプラスミドpPG−LEU(−2μ)を制限酵素SmaIとEcoRIで切断したものと、PCR合成したARE1遺伝子断片をSmaIとEcoRIで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素SmaIとEcoRIによる切断で、約5.4kbpと1.83kbpの断片が確認できたものを目的プラスミドとして、pPGL−ARE1(−2μ)と名付けた。
【0085】
次に、pUC18(タカラバイオ社製)をHindIIIで切断し、Alkaline
Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、PCR合成したURA3遺伝子断片をHindIIIで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素Hind IIIによる切断で、約2.68kbpと1.23kbpの断片が確認できたものを目的プラスミドとして、pUC−URA3と名付けた。
【0086】
また、pCR−Blunt(invitrogen社製)のクローニングサイトに上記(iii)でPCR合成したADE1遺伝子断片をダイレクトクローニングにより連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素SpeIとPstIによる切断で、約3.47kbpと1.62kbpの断片が確認できたものを目的プラスミドとして、pCR−B−ADE1と名付けた。
【0087】
続いて、pUC19(タカラバイオ社製)をXbaIとSmaIで切断し、cloned DNA Polymerase I(タカラバイオ社製)を用いてDNA末端平滑化処理を行った後に、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、上記pCR−B−ADE1をSpeIとEcoRVで切断し、cloned DNA Polymerase I(タカラバイオ社製)を用いてDNA末端平滑化処理を行ったものをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1% NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素EcoRIによる切断で、約1.01kbpと0.39kbp、2.68kbpの断片が確認できたものを目的プラスミドとして、pUC19−ADE1と名付けた。
【0088】
さらに、上記pPGL−ARE1(−2μ)をSspIで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、上記pUC−URA3をHindIIIで切断し、cloned DNA PolymeraseI(タカラバイオ社製)を用いてDNA末端平滑化処理を行ったもの、上記pUC19−ADE1をPstIとKpnIで切断し、clone
d DNA PolymeraseI(タカラバイオ社製)を用いてDNA末端平滑化処理を行ったものとをそれぞれTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素EcoR IとPst Iによる切断で、約3.56kbpと3.07kbpの断片が確認できたもの、制限酵素BspT104Iによる切断で、約3.74kbpと1.29kbp、1.85kbpの断片が確認できたものをそれぞれ目的プラスミドとして、pPGU−ARE1(−2μ)、pPGADE1−ARE1(−2μ)と名付けた。
【0089】
(6)erg5、erg6二重破壊株SX1313、SX1312、KAΔ5−4、SX1340へのARE1遺伝子導入株SX1314、SX1318、SX1315、SX1350株の作製
erg5、erg6二重破壊株SX1313、SX1312、特開2004-141125に記載されたKAΔ5−4(以後、SX1307と記載する)のゲノム上のARE1遺伝子座へさらにARE1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE1発現用のプラスミドpPGU−ARE1(−2μ)を制限酵素Xho I(ARE1遺伝子中)で切断した。このDNA断片を上記(3)で作製したSX1313、SX1312株、SX1307株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、100mg/l L−ロイシンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino
Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。なお、SX1313株の形質転換を行ったSD寒天培地にのみ20mg/l アデニン、30mg/l L−リジンを添加した。
【0090】
寒天培地上に生育してきたウラシルおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーARE1−check(配列番号18)の組み合わせでPCRを行い、ARE1遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。
【0091】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1314(YPH499株由来)、SX1318(FY1679−06c株由来)、SX1315(KA311A株由来)と名付けた形質転換体は目的サイズと同じ約2.6kbpのDNA断片が検出された。従って、SX1314、SX1318、SX1315株のゲノムのARE1遺伝子座に上記相同組換えによりARE1遺伝子が導入され、合計2コピー存在することが確認された。
【0092】
さらに、erg5、erg6二重破壊株SX1340のゲノム上のARE1遺伝子座へさらにARE1遺伝子を導入することを目的として、上記と同様に以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE1発現用のプラスミドpPGADE1−ARE1(−2μ)を制限酵素Xho I(ARE1遺伝子中)で切断した。また、ARE1を導入していない対照株を作製するため、上記(5)で作製したpUC19−ADE1を制限酵素HpaIで切断した。これらのDNA断片を上記(3)で作製したSX1340株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD
寒天培地(0.67% Yeast Nitrogen Base w/o Amino
Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0093】
寒天培地上に生育してきたアデニンおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーARE1−check(配列番号18)の組み合わせでPCRを行い、ARE1遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。
【0094】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1350(X2181−1B株由来)と名付けた形質転換体は目的サイズと同じ約2.6kbpのDNA断片が検出された。従って、SX1350株のゲノムのARE1遺伝子座に上記相同組換えによりARE1遺伝子が導入され、合計2コピー存在することが確認された。
また、pUC19−ADE1をHpaIで切断したDNA断片を用いて形質転換を行った株については、フォワードプライマーADE1−1F(配列番号19)とリバースプライマーM13 forward(配列番号20)の組み合わせでPCRを行い、ADE1遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。PCR産物をアガロースゲル電気泳動で解析した結果、SX1353と名付けた形質転換体は目的サイズと同じ約1.45kbpのDNA断片が検出された。従って、SX1353株は相同組換えによりADE1遺伝子が相補されたことが確認された。
【0095】
(7)ARE1遺伝子を導入したerg5、erg6二重破壊株へのYEH1遺伝子導入株の作製
(i)酵母発現用のYEH1、GAL1遺伝子断片の調製
サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子(YEH1)を含むDNA断片、GAL1を含むDNA断片は、上記(1)で取得したサッカロミセス・セレビジエS288C株の染色体DNAを鋳型にしてPCRにて合成、単離した。以下にその方法の詳細を示す。
YEH1遺伝子断片のPCRによる合成には、フォワードプライマーYLL−OF(配列番号21)とリバースプライマーYLL−OR(配列番号22)を用いた。このプライマーの組み合わせでYEH1の蛋白質コード領域である開始コドンから終始コドンまでを増幅することができる。また、5'側と3'側の両側に制限酵素EcoR Iの認識配列を含んだ形で合成することができる。
【0096】
GAL1遺伝子断片のPCRによる合成には、フォワードプライマーGAL1−51(配列番号23)とリバースプライマーGAL1−31(配列番号24)を用いた。このプライマーの組み合わせでGAL1の蛋白質コード領域を含み、開始コドンの499塩基前から終始コドンの230塩基後までを増幅することができる。また、5’側と3’側の両側に制限酵素SmaIの認識配列を含んだ形で合成することができる。
【0097】
PCRは、PrimeSTAR HS DNA ポリメラーゼ(タカラバイオ社製)を用いて、(1)98℃、2分、(2)98℃、10秒、(3)55℃、5秒、(4)72℃、2分、(2)から(4)の反応を30回繰り返し、(5)72℃、7分間反応させた後、4℃で冷却の条件で反応を行った。次にPCR産物をアガロースゲル電気泳動し、約1.72kbpのYEH1遺伝子断片を、約2.33kbpのGAL1遺伝子断片を確認した。
【0098】
(ii)YEH1発現用プラスミドpPGL−YEH1(−2μ)、pPGGAL−YEH1(−2μ)の作製
上記(4)(ii)で作製したプラスミドpPG−LEU(−2μ)を制限酵素EcoR Iで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、PCR合成したYEH1遺伝子断片をEcoR Iで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素EcoRIによる切断で、約5.4kbpと1.72kbpの断片が確認できたものを目的プラスミドとして、pPGL−YEH1(−2μ)と名付けた。
【0099】
次に、pUC19(タカラバイオ社製)をSmaIで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、PCR合成したGAL1遺伝子断片をSmaIで切断したものとをTAKARA DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素PvuIIによる切断で、約0.48kbpと0.72kbp、1.45kbp、2.36kbpの断片が確認できたものを目的プラスミドとして、pUC19−GAL1と名付けた。
続いて、上記pPGL−YEH1(−2μ)をSspIで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、上記pUC19−GAL1をSmaIで切断したものとをTAKARA DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素EcoRIによる切断で、約3.30kbpと2.66kbpの断片が確認でき、さらに制限酵素ScaIによる切断で、約1.28kbpと4.68kbpの断片が確認できたものを目的プラスミドとして、pPG−GAL1(−2μ)と名付けた。さらに、上記pPGL−YEH1(−2μ)をBamHIで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、上記pPG−GAL1(−2μ)をBamHIで切断したものとをTAKARA DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素HindIIIによる切断で、約0.37kbpと0.57kbpと0.19kbpと3.73kbpと2.82kbpの断片が確認できたものを目的プラスミドとして、pPGGAL1−YEH1(−2μ)と名付けた。
【0100】
(iii)ARE1遺伝子を導入したerg5、erg6二重破壊株SX1314、SX1318、SX1315、SX1350へのYEH1遺伝子導入株SX1317、SX1320、SX1322、SX1351株の作製
ARE1遺伝子を導入したerg5、erg6二重破壊株SX1314、SX1318、SX1315のゲノム上のYEH1遺伝子座へさらにYEH1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加
水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGL−YEH1(−2μ)を制限酵素Nde I(YEH1遺伝子中)で切断した。また、YEH1を導入していない対照株を作製するため、(4)(ii)で作製した酵母組換え用プラスミドpPG−LEU(−2μ)を制限酵素Bsp1407 Iで切断した。これらのDNA断片を上記(6)で作製したSX1314、SX1318、SX1315株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。なお、SX1314株に対して形質転換を行ったSD寒天培地にのみ20mg/lアデニン、30mg/l L−リジンを添加した。
【0101】
寒天培地上に生育してきたロイシンおよびウラシル、ヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーYEH1−RR(配列番号25)の組み合わせでPCRを行い、YEH1遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。
【0102】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1317(YPH499株由来)、SX1320(FY1679−06c株由来)、SX1322(KA311A株由来)と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1317,SX1320、SX1322株のゲノムには上記相同組換えによりYEH1遺伝子が導入され、2コピー存在していることが確認された。
【0103】
また、プラスミドpPG−LEU(−2μ)をBsp1407Iで切断したDNA断片を用いて形質転換を行った株については、フォワードプライマーPPGK1−R1(配列番号26)とリバースプライマーLEU2−R2(配列番号27)の組み合わせでPCRを行い、leu2遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。
【0104】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1316(YPH499株由来)、SX1319(FY1679−06c株由来)、SX1321(KA311A株由来)と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1316,SX1319、SX1321株は相同組換えによりLEU2遺伝子が相補されたことが確認された。
【0105】
さらに、ARE1遺伝子を導入したerg5、erg6二重破壊株SX1350のゲノム上のYEH1遺伝子座へさらにYEH1遺伝子を導入することを目的として、上記と同様に以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGGAL1−YEH1(−2μ)を制限酵素Bsp1407 I(YEH1遺伝子中)で切断した。このDNA断片を上記(6)で作製したSX1350株に上記(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SG寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%ガラクトース、2%寒天)に播き、30℃で3〜5日間培養した。
【0106】
ガラクトースを炭素源として寒天培地上に生育してきたアデニンおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養
液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーYEH1−RR(配列番号25)の組み合わせでPCRを行い、YEH1遺伝子座上での相同組換えの有無を調べた。PCRは上記(2)で述べた条件で行った。
【0107】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1351(X2181−1B株由来)と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1351株のゲノムのARE1遺伝子座に上記相同組換えによりYEH1遺伝子が導入され、合計2コピー存在することが確認された。
【0108】
(実施例2)
erg5、erg6二重破壊株SX1312の発現locusの異なるARE1およびYEH1増強株の作製
(1)erg5、erg6二重破壊株SX1312へのARE1遺伝子導入株SX1326の作製
erg5、erg6二重破壊株SX1312のゲノム上のARE1遺伝子座へさらにARE1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE1発現用のプラスミドpPGL−ARE1(−2μ)を制限酵素Xho I(ARE1遺伝子中)で切断した。このDNA断片を上記実施例1(3)で作製したSX1312株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、20mg/l ウラシルを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0109】
寒天培地上に生育してきたロイシンおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーARE1−check(配列番号18)の組み合わせでPCRを行い、ARE1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
PCR産物をアガロースゲル電気泳動で解析した結果、SX1326と名付けた形質転換体は目的サイズと同じ約2.6kbpのDNA断片が検出された。従って、SX1326株のゲノムのARE1遺伝子座に上記相同組換えによりARE1遺伝子が導入され、合計2コピー存在することが確認された。
【0110】
(2)ARE1遺伝子を導入したSX1318、SX1326株への発現locusの異なるYEH1遺伝子導入株SX1325、SX1332の作製
(i)YEH1発現用プラスミドpPGU−YEH1(−2μ)の作製
上記実施例1(5)で作製したプラスミドpPGU−ARE1(−2μ)を制限酵素BamHIで切断し、Alkaline Phosphatase(E.coli C75)(タカラバイオ社製)を用いて脱リン酸化処理を行ったものと、pPGL−YEH1(−2μ)をBamH Iで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素SmaIによる切断で、約4.88kbpと1.64kbpの断片が確認できたものを目的プラスミドとして、pPGU−YEH1(−2μ)と名付けた。
【0111】
(ii)ARE1遺伝子を導入したerg5、erg6二重破壊株SX1318、1326への発現locusの異なるYEH1遺伝子導入株SX1325、SX1332株の作製
ARE1遺伝子を導入したerg5、erg6二重破壊株SX1318のleu2遺伝子座と、SX1326のゲノム上のura3遺伝子座へさらにYEH1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGL−YEH1(−2μ)を制限酵素BstEII(leu2遺伝子中)で切断し、pPGU−YEH1(−2μ)を制限酵素StuI(ura3遺伝子中)で切断した。これらのDNA断片をSX1318とSX1326株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0112】
寒天培地上に生育してきたロイシンおよびウラシル、ヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーLEU2−R2(配列番号27)とリバースプライマーYLL−Seq3R(配列番号28)、フォワードプライマーURA3−RR(配列番号29)とリバースプライマーYEH1−seqR1(配列番号30)の組み合わせでそれぞれPCRを行い、leu2遺伝子座上とura3遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0113】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1325(leu2遺伝子座)とSX1332(ura3遺伝子座)と名付けた形質転換体は目的サイズと同じ約2.8kbpと約2.6kbpのDNA断片がそれぞれ検出された。従って、SX1325,SX1332株のゲノムには上記相同組換えによりYEH1遺伝子が導入され、2コピー存在していることが確認された。
【0114】
(3)erg5、erg6二重破壊株SX1312への発現locusの異なるARE1遺伝子導入株SX1327の作製
erg5、erg6二重破壊株SX1312のゲノム上のura3遺伝子座へさらにARE1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE1発現用のプラスミドpPGU−ARE1(−2μ)を制限酵素StuI(ura3遺伝子中)で切断した。このDNA断片を上記実施例1(3)で作製したSX1312株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、100mg/l L−ロイシンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
寒天培地上に生育してきたウラシルおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーURA3−RR(配列番号29)とリバースプライマーARE1−RR(配列番号31)の組み合わせでPCRを行い、ARE1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0115】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1326と名付けた形質転
換体は目的サイズと同じ約2.5kbpのDNA断片が検出された。従って、SX1327株のゲノムのura3遺伝子座に上記相同組換えによりARE1遺伝子が導入され、合計2コピー存在することが確認された。
【0116】
(4)ARE1遺伝子を導入したSX1327株へのYEH1遺伝子導入株SX1333の作製
ARE1遺伝子を導入したerg5、erg6二重破壊株SX1327のゲノム上のYEH1遺伝子座へさらにYEH1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGL−YEH1(−2μ)を制限酵素NdeI(YEH1遺伝子中)で切断した。このDNA断片をSX1327株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
寒天培地上に生育してきたロイシンおよびウラシル、ヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーYEH1−RR(配列番号25)の組み合わせでそれぞれPCRを行い、YEH1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0117】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1333(YEH1遺伝子座)と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1333株のゲノムのYEH1遺伝子座には上記相同組換えによりYEH1遺伝子が導入され、2コピー存在していることが確認された。
【0118】
(5)ARE1、YEH1遺伝子を導入していないerg5、erg6二重破壊株SX1335株の作製
ARE1、YEH1を導入していない対照株を作製するため、上記実施例1(5)で作製した酵母組換え用プラスミドpUC−URA3を制限酵素StuIで切断した。これらのDNA断片を上記実施例1(3)で作製したerg5、erg6二重破壊株SX1312に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、100mg/l L−ロイシンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
寒天培地上に生育してきたウラシルおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーM13 forward(配列番号20)とリバースプライマーURA3−RR(配列番号29)の組み合わせでPCRを行い、ura3遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0119】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1329と名付けた形質転換体は目的サイズと同じ約1.2kbpのDNA断片が検出された。従って、SX1329株は相同組換えによりURA3遺伝子が相補されたことが確認された。
【0120】
次に、酵母組換え用プラスミドpPG−LEU(−2μ)を制限酵素Bsp1407 Iで切断した。このDNA断片を上記で作製したSX1329株に上記実施例1(2)と
同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0121】
寒天培地上に生育してきたロイシンおよびウラシル、ヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−R1(配列番号26)とリバースプライマーLEU2−R2(配列番号27)の組み合わせでPCRを行い、leu2遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0122】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1335と名付けた形質転換体は目的サイズと同じ約2.1kbpのDNA断片が検出された。従って、SX1335株は相同組換えによりLEU2遺伝子が相補されたことが確認された。
【0123】
(実施例3)
erg5、erg6二重破壊株SX1312のARE2およびYEH1増強株の作製
(1)erg5、erg6二重破壊株SX1312へのARE2遺伝子導入株SX1342の作製
(i)酵母発現用のARE2遺伝子断片の調製
サッカロミセス・セレビジエ由来アシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子(ARE2)を含むDNA断片は、上記実施例1(1)で取得したサッカロミセス・セレビジエ S288C株の染色体DNAを鋳型にしてPCRにて合成、単離した。以下にその方法の詳細を示す。
ARE2遺伝子断片のPCRによる合成には、フォワードプライマーARE2−SmaI(F)(配列番号32)とリバースプライマーARE2−EcoRI(R)(配列番号33)を用いた。このプライマーの組み合わせでARE2の蛋白質コード領域である開始コドンから終始コドンまでを増幅することができる。また、5’側には制限酵素SmaIの認識配列を、3’側にはEcoRIの認識配列を含んだ形で合成することができる。PCRは、PrimeSTAR HS DNA ポリメラーゼ(タカラバイオ社製)を用いて、(1)98℃、2分、(2)98℃、10秒、(3)55℃、5秒、(4)72℃、2分、(2)から(4)の反応を30回繰り返し、(5)72℃、7分間反応させた後、4℃で冷却の条件で反応を行った。次にPCR産物をアガロースゲル電気泳動し、約1.93kbpのARE2遺伝子断片を確認した。
【0124】
(ii)ARE2発現用プラスミドpPGL−ARE2(−2μ)の作製
上記実施例1(4)で作製したプラスミドpPG−LEU(−2μ)を制限酵素SmaIとEcoRIで切断したものと、PCR合成したARE2遺伝子断片をSmaIとEcoRIで切断したものとをTaKaRa DNA Ligation Kit Ver.2(タカラバイオ社製)により連結した。これをエッシェリシア・コリJM109に形質転換し、100mg/lのアンピシリンを含むLB寒天培地(1%ポリペプトン、0.5%酵母エキス、1%NaCl、2%寒天)上に生育したコロニーからプラスミドを抽出した。アガロースゲル電気泳動により、制限酵素SmaIとEcoRIによる切断で、約5.4kbpと1.93kbpの断片が確認できたものを目的プラスミドとして、pPGL−ARE2(−2μ)と名付けた。
【0125】
(iii)erg5、erg6二重破壊株SX1312へのARE2遺伝子導入株SX1342株の作製
erg5、erg6二重破壊株SX1312のゲノム上のARE2遺伝子座へさらにARE2遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE2発現用のプラスミドpPGL−ARE2(−2μ)を制限酵素SalI(ARE2遺伝子中)で切断した。このDNA断片を上記実施例1(3)で作製したSX1312株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、20mg/l ウラシルを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
寒天培地上に生育してきたロイシンおよびヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーARE2−R1(配列番号34)の組み合わせでPCRを行い、ARE2遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0126】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1342と名付けた形質転換体は目的サイズと同じ約2.6kbpのDNA断片が検出された。従って、SX1342株のゲノムのARE2遺伝子座に上記相同組換えによりARE2遺伝子が導入され、合計2コピー存在することが確認された。
【0127】
(2)ARE2遺伝子を導入したSX1342株へのYEH1遺伝子導入株SX1346の作製
ARE2遺伝子を導入したerg5、erg6二重破壊株SX1342のゲノム上のYEH1遺伝子座へさらにYEH1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGU−YEH1(−2μ)を制限酵素Bsp1407I(YEH1遺伝子中)で切断した。また、YEH1を導入していない対照株を作製するため、上記実施例1(5)で作製した酵母組換え用プラスミドpUC−URA3を制限酵素StuIで切断した。これらのDNA断片を上記(1)で作製したSX1342株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、SD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0128】
寒天培地上に生育してきたロイシンおよびウラシル、ヒスチジン、トリプトファン非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーYEH1−RR(配列番号25)の組み合わせでPCRを行い、YEH1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
PCR産物をアガロースゲル電気泳動で解析した結果、SX1346と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1346株のゲノムには上記相同組換えによりYEH1遺伝子が導入され、2コピー存在していることが確認された。
【0129】
また、プラスミドpUC−URA3をStuIで切断したDNA断片を用いて形質転換を行った株については、フォワードプライマーM13 forward(配列番号20)
とリバースプライマーURA3−RR(配列番号29)の組み合わせでPCRを行い、ura3遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0130】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1347と名付けた形質転換体は目的サイズと同じ約1.2kbpのDNA断片が検出された。従って、SX1347株は相同組換えによりURA3遺伝子が相補されたことが確認された。
【0131】
(実施例4)
サッカロミセス・セレビジエ FY1679−06c株のARE1およびYEH1増強株の作製
(1)FY1679−06c株へのARE1遺伝子増強株SX1330の作製
サッカロミセス・セレビジエ FY1679−06c株のゲノム上のARE1遺伝子座へさらにARE1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステロールアシルCoA:ステロールアシルトランスフェラーゼをコードする遺伝子ARE1発現用のプラスミドpPGU−ARE1(−2μ)を制限酵素Xho I(ARE1遺伝子中)で切断した。このDNA断片をFY1679−06c株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、各20mg/l L−ヒスチジン、トリプトファン、100mg/l L−ロイシンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0132】
寒天培地上に生育してきたウラシル非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーARE1−check(配列番号18)の組み合わせでPCRを行い、ARE1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0133】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1330と名付けた形質転換体は目的サイズと同じ約2.6kbpのDNA断片が検出された。従って、SX1330株のゲノムのARE1遺伝子座に上記相同組換えによりARE1遺伝子が導入され、合計2コピー存在することが確認された。
【0134】
(2)ARE1遺伝子を導入したSX1330株へのYEH1遺伝子導入株SX1337の作製
ARE1遺伝子を導入したSX1330株のゲノム上のYEH1遺伝子座へさらにYEH1遺伝子を導入することを目的として、以下の工程を行った。サッカロミセス・セレビジエ由来ステリルエステル加水分解酵素をコードする遺伝子YEH1発現用のプラスミドpPGL−YEH1(−2μ)を制限酵素Nde I(YEH1遺伝子中)で切断した。また、YEH1を導入していない対照株を作製するため、上記実施例1(4)(ii)で作製した酵母組換え用プラスミドpPG−LEU(−2μ)を制限酵素Bsp1407 Iで切断した。これらのDNA断片を上記(1)で作製したSX1330株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、各20mg/l L−ヒスチジン、トリプトファンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0135】
寒天培地上に生育してきたロイシンおよびウラシル非要求性のコロニーをYPD液体培
地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−F(配列番号17)とリバースプライマーYEH1−RR(配列番号25)の組み合わせでPCRを行い、YEH1遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0136】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1337と名付けた形質転換体は目的サイズと同じ約2.0kbpのDNA断片が検出された。従って、SX1337株のゲノムには上記相同組換えによりYEH1遺伝子が導入され、2コピー存在していることが確認された。
【0137】
また、プラスミドpPG−LEU(−2μ)をBsp1407Iで切断したDNA断片を用いて形質転換を行った株については、フォワードプライマーPPGK1−R1(配列番号26)とリバースプライマーLEU2−R2(配列番号27)の組み合わせでPCRを行い、leu2遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0138】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1338と名付けた形質転換体は目的サイズと同じ約2.1kbpのDNA断片が検出された。従って、SX1338株は相同組換えによりLEU2遺伝子が相補されたことが確認された。
【0139】
(3)ARE1、YEH1遺伝子を導入していない対照株SX1348の作製
ARE1、YEH1を導入していない対照株を作製するため、上記実施例1(5)で作製した酵母組換え用プラスミドpUC−URA3を制限酵素StuIで切断した。このDNA断片をFY1679−06c株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、各20mg/l L−ヒスチジン、トリプトファン、100mg/l L−ロイシンを含むSD寒天培地(0.67% Yeast
Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0140】
寒天培地上に生育してきたウラシル非要求性のコロニーをYPD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーM13 forward(配列番号20)とリバースプライマーURA3−RR(配列番号29)の組み合わせでPCRを行い、ura3遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
【0141】
PCR産物をアガロースゲル電気泳動で解析した結果、SX1343と名付けた形質転換体は目的サイズと同じ約1.2kbpのDNA断片が検出された。従って、SX1343株は相同組換えによりURA3遺伝子が相補されたことが確認された。
次に、酵母組換え用プラスミドpPG−LEU(−2μ)を制限酵素Bsp1407 Iで切断した。このDNA断片を上記で作製したSX1343株に上記実施例1(2)と同様に酢酸リチウム法、もしくはエレクトロポレーション法で形質転換し、各20mg/l L−ヒスチジン、トリプトファンを含むSD寒天培地(0.67% Yeast Nitrogen Base w/o Amino Acids(Difco社製)、2%グルコース、2%寒天)に播き、30℃で3〜5日間培養した。
【0142】
寒天培地上に生育してきたロイシンおよびウラシル非要求性のコロニーをYPD液体培
地(1%酵母エキス、2%ポリペプトン、2%グルコース)2mlに植菌し、30℃で16時間、振とう培養を行った。この培養液から上記実施例1(1)で述べた方法で染色体DNAを抽出した。これを鋳型としてフォワードプライマーPPGK1−R1(配列番号26)とリバースプライマーLEU2−R2(配列番号27)の組み合わせでPCRを行い、leu2遺伝子座上での相同組換えの有無を調べた。PCRは上記実施例1(2)で述べた条件で行った。
PCR産物をアガロースゲル電気泳動で解析した結果、SX1348と名付けた形質転換体は目的サイズと同じ約2.1kbpのDNA断片が検出された。従って、SX1348株は相同組換えによりLEU2遺伝子が相補されたことが確認された。
【0143】
(実施例5)
ARE1、YEH1遺伝子を増強した各種株のステロール類の分析
(1)各種酵母株の培養、およびステロール類の分析
実施例1〜4で作製した各種株を0.5%グルコースを炭素源とした2mlのYPAD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース、40mg/lアデニン)に1白金耳植菌し、30℃、18時間培養し、種菌とした。この酵母培養液から初期OD660=0.2となる分量の培養液を回収し、4℃、10,000rpm、3分間遠心分離を行い、培養上清を除去した。菌体ペレットに0.1mlの0.85%生理食塩水を添加、懸濁し、2%グルコースを炭素源とした5mlのYPAD液体培地(1%酵母エキス、2%ポリペプトン、2%グルコース、40mg/lアデニン)に播種し、30℃、72時間培養を行った。酵母培養液0.5mlを4℃、14,000rpm、3分間遠心分離を行い、菌体を回収した。菌体ペレットにエタノール0.4mlと40%水酸化カリウム水溶液0.02mlを添加し、80℃、90分間振とうを行い、菌体内に蓄積したステロールエステルのケン化処理を行った。この液を35℃で減圧濃縮機にかけ、濃縮乾固させた。
得られた濃縮乾固物にエタノール0.1mlと滅菌水0.05mlを加え懸濁し、さらにヘプタン0.8mlを添加し激しく混和した後、室温で14,000rpm、1分間遠心分離することで水層と油層に分離した。上層のヘプタン層を別のチューブに取り、残った水層に再度、エタノール0.1mlとヘプタン0.8mlを添加し、抽出操作を繰り返した。抽出した約1.6mlのヘプタン溶液に滅菌水0.04mlを添加し、激しく混和した後、再度、室温で14,000rpm、1分間遠心分離することで水層と油層に分離した。約1.6mlのヘプタン層を回収し、35℃で減圧濃縮機にかけ、濃縮乾固させた。得られた濃縮乾固物は0.2mlトルエンに再溶解し、テストステロンを内部標準としてガスクロマトグラフィーによるステロール類の分析を行った。結果を図1〜8に示す。
【0144】
(2)ARE1、YEH1遺伝子を増強したerg5、erg6二重破壊株のステロール類の分析
図1(a)に示した通り、YPH499を親株として作製したARE1のみを増強したerg5、erg6二重破壊株(SX1316)に対して、ARE1とYEH1を増強したerg5、erg6二重破壊株(SX1317)では、ZYMOの蓄積量が減少し、TEOLの蓄積量が増加していることがわかった。このことからARE1増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることが明らかとなった。
一方、図2(a)に示した通り、FY1679−06cを親株として作製したARE1のみを増強したerg5、erg6二重破壊株(SX1319)に対して、さらにYEH1を増強したerg5、erg6二重破壊株(SX1320)でも、ZYMO蓄積量が減少し、TEOL蓄積量が増加した。これらのことからARE1増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることがFY1670−06cを親株とした場合でも明らかとなった。
【0145】
次に、図3(a)に示した通り、KA311Aを親株として作製したARE1のみを増強したerg5、erg6二重破壊株(SX1321)に対して、さらにYEH1を増強したerg5、erg6二重破壊株(SX1322)でも、ZYMO蓄積量が減少し、TEOL蓄積量が増加した。これらのことからARE1増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることがKA311Aを親株とした場合でも明らかとなった。
さらに、図4(a)に示した通り、X2181−1Bを親株として作製したARE1のみを増強したerg5、erg6二重破壊株(SX1350)に対して、さらにYEH1を増強したerg5、erg6二重破壊株(SX1351)でも、ZYMO蓄積量が減少し、TEOL蓄積量が増加した。これらのことからARE1増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることがX2181−1Bを親株とした場合でも明らかとなった。
【0146】
(3)発現locusの異なるARE1、YEH1を増強したerg5、erg6二重破壊株のステロール類の分析
図5(a)に示した通り、FY1679−06cを親株として作製した、発現locusの異なるYEH1を増強したerg5、erg6二重破壊、ARE1増強株において、YEH1、URA3、LEU2いずれのlocusでYEH1を発現させた株でも同等のTEOL蓄積を示した。このことからARE1増強下においてYEH1をどのようなlocusで発現増強させても、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることが明らかとなった。
一方、図6(a)に示した通り、FY1679−06cを親株として作製した、発現locusの異なるARE1を増強したerg5、erg6二重破壊、YEH1増強株において、ARE1、URA3いずれのlocusでARE1を発現させた株でも同等のTEOL蓄積を示した。このことからYEH1増強下においてARE1をどのようなlocusで発現増強させても、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることが明らかとなった。
【0147】
(4)ARE2、YEH1を増強したerg5、erg6二重破壊株のステロール類の分析
図7(a)に示した通り、FY1679−06cを親株として作製したARE2のみを増強したerg5、erg6二重破壊株(SX1347)に対して、さらにYEH1を増強したerg5、erg6二重破壊株(SX1346)でも、ZYMO蓄積量が減少し、TEOL蓄積量が増加した。これらのことからARE2増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることが、ARE2をステロールアシルCoA:ステロールアシルトランスフェラーゼとして用いた場合でも明らかとなった。
【0148】
(5)ARE1、YEH1を増強した株のステロール類の分析
図8(a)に示した通り、FY1679−06cを親株として作製したARE1のみを増強した株(SX1338)に対して、さらにYEH1を増強した株(SX1337)でも、ZYMO蓄積量が減少し、ERGO蓄積量が増加した。これらのことからARE1増強下においてYEH1を増強することにより、TEOLの前駆体であるZYMOを減らし、TEOLの蓄積を向上させることが、erg5erg6二重破壊をしていない株を用いた場合でも明らかとなった。
【0149】
(実施例6)
ARE1、YEH1遺伝子を増強した各種株の転写量の分析
(1)酵母からの全RNAの調製
実施例5で調製した各種株について、RNeasy plus Mini Kitおよ
びRNase−Free DNase Set(いずれもQIAGEN社製)を用いて全RNAを以下のとおり調製した。
上記実施例5と同様に菌体を培養した後、回収した培養液を用いて、2×107個になるよう酵母菌体溶液を調製し(OD660=1が、約1.5×107個として細胞数を調製)、4℃、14,000rpm、1分間遠心分離して得られた菌体を0.85%生理食塩水に懸濁し、再度、4℃、10,000rpm、2分間遠心分離を行った。得られた菌体は、キット付属のBuffer RLT 0.6mlと2−メルカプトエタノール 6μlで懸濁し、この懸濁液をMagNA Lyser Green Beads(Roche社製)のチューブに移し、MagNA Lyser(Roche社製)を用いて室温で6,000rpm、1分間破砕を行った。この菌体破砕液をゲノムDNA除去ミニスピンカラムに入れ、RNeasy plus Mini Kit付属の使用説明書に従い、全RNAの調製を行った。オプションとして全RNAのカラム吸着後にDNase mix solution 0.08mlを添加し、室温で15分間静置し、染色体DNAの分解を行った。また、最後に50℃で保温したRNase−free waterを加え、室温で1分間静置し、遠心分離にて全RNAを溶出した。
【0150】
(2)逆転写反応
上記(1)で調製した全RNAを鋳型として、SuperScript III First−Strand Synthesis System for RT−PCR(invitrogen社製)を用いて、付属の使用説明書に従い、逆転写反応を行い、一本鎖cDNAを合成した。逆転写反応には、300ngの全RNA、プライマーとして50μM Oligo(dT)20を使用した。
【0151】
(3)定量PCR
定量PCRの反応は、SYBR premix Ex Taq(Perfect Real Time)(タカラバイオ社製)を用いて、付属の使用説明書に従って反応を行い、(1)95℃、10秒、(2)95℃、5秒、(3)60℃、20秒、(2)から(3)の反応を40回繰り返した。温度変化速度はいずれも20℃/秒とした。また、融解曲線分析を(1)95℃、0秒、(2)65℃、15秒、(3)95℃、0秒で行い、温度変化速度はいずれも20℃/秒とした。PCRプライマーとして、ARE1の定量PCRにはARE1−F(配列番号35)とARE1−R(配列番号36)、ARE2の定量PCRにはARE2−F(配列番号37)とARE2−R(配列番号38)、YEH1の定量PCRにはYEH1−F(配列番号39)とYEH1−R(配列番号40)を使用した。また、発現コントロールとしてアクチンをコードしているACT1を用い、ACT1−F(配列番号41)とACT1−R(配列番号42)をプライマーとして使用した。
定量分析には、Roche Diagnostics社製のLight−Cycler、およびLight−Cycler Software Ver.3.5を用い、Fit
Point法を用いて分析を行った。この結果を図1および図2の(b)、(c)に示す。
【0152】
(4)ARE1、YEH1遺伝子を増強したerg5、erg6二重破壊株の転写量の分析
図1(b)、(c)で示した通り、YPH499のerg5、erg6二重破壊株を親株とした株における転写解析を行った結果、ARE1のみを増強したSX1316ではARE1の転写量が、SX1313に比べ増加しており、発現増強されていることが確認できた。また、ARE1とYEH1をともに増強したSX1317のARE1、YEH1それぞれの転写量もSX1313に比べて増加していることから、両遺伝子とも発現増強されていることが確認できた。
同様に、FY1679−06cのerg5、erg6二重破壊株を親株とした株における転写解析の結果(図2(b)、(c))、SX1312に対して、ARE1のみを増強
したSX1319はARE1の転写量が増加し、ARE1とYEH1をともに増強したSX1320はARE1とYEH1それぞれの転写量が増加していることが確認できた。
【0153】
続いて、KA311Aのerg5、erg6二重破壊株を親株とした株における転写解析の結果(図3(b)、(c))、SX1307に対して、ARE1のみを増強したSX1321はARE1の転写量が増加し、ARE1とYEH1をともに増強したSX1322はARE1とYEH1それぞれの転写量が増加していることが確認できた。
さらに、X2181−1Bのerg5、erg6二重破壊株を親株とした株における転写解析の結果(図4(b)、(c))、SX1353に対して、ARE1のみを増強したSX1350はARE1の転写量が増加し、ARE1とYEH1をともに増強したSX1351はARE1とYEH1それぞれの転写量が増加していることが確認できた。
【0154】
(5)発現locusの異なるARE1、YEH1を増強したerg5、erg6二重破壊株の転写量の分析
FY1679−06cのerg5、erg6二重破壊、ARE1増強株を親株として、発現locusの異なるYEH1を強化した株における転写解析の結果(図5(b)、(c))、SX1319に対して、SX1320(YEH1 locus)、SX1332(URA3 locus)、SX1325(LEU2 locus)いずれの株も、YEH1の転写量が増加していることが確認できた。
【0155】
さらに、FY1679−06cのerg5、erg6二重破壊株を親株として、発現locusの異なるARE1をYEH1とともに強化した株における転写解析の結果(図6(b)、(c))、SX1335に対して、SX1320(ARE1 locus)、SX1333(URA3 locus)いずれの株も、ARE1とYEH1それぞれの転写量が増加していることが確認できた。
【0156】
(6)ARE2、YEH1を増強したerg5、erg6二重破壊株の転写量の分析
FY1679−06cのerg5、erg6二重破壊株を親株とした株における転写解析の結果(図7(b)、(c))、SX1335に対して、ARE2のみを増強したSX1347はARE2の転写量が増加し、ARE2とYEH1をともに増強したSX1346はARE2とYEH1それぞれの転写量が増加していることが確認できた。
【0157】
(7)ARE1、YEH1を増強した株の転写量の分析
FY1679−06cを親株とした株における転写解析の結果(図8(b)、(c))、SX1348に対して、ARE1のみを増強したSX1338はARE1の転写量が増加し、ARE1とYEH1をともに増強したSX1337はARE1とYEH1それぞれの転写量が増加していることが確認できた。
【0158】
なお、表1に、図1(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0159】
【表1】
【0160】
また、表2に、図2(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0161】
【表2】
【0162】
表3に、図3(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重
量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0163】
【表3】
【0164】
表4に、図4(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0165】
【表4】
【0166】
表5に、図5(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0167】
【表5】
【0168】
表6に、図6(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0169】
【表6】
【0170】
表7に、図7(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE2とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0171】
【表7】
【0172】
表8に、図8(a)、(b)、(c)の数値データを示す。数値は、(A)乾燥菌体重量1gあたりのステロール蓄積量mgで示した。(B)ACT1に対する転写量比として、ARE1とACT1、YEH1とACT1の発現量比をそれぞれ示した。
【0173】
【表8】
【産業上の利用可能性】
【0174】
本発明によって製造されるΔ5,7−ステロールは、医薬分野、化粧品分野、食品分野等で有用である。
【特許請求の範囲】
【請求項1】
アシルCoA:ステロールアシルトランスフェラーゼ遺伝子とステリルエステル加水分解酵素遺伝子の発現が増強されるように改変された酵母。
【請求項2】
さらに、ステロールC−22デサチュラーゼ遺伝子(ERG5)及びステロールC−24メチルトランスフェラーゼ遺伝子(ERG6)の発現が低減するように改変された、請求項1に記載の酵母。
【請求項3】
さらに、ステロールC−22デサチュラーゼ遺伝子(ERG5)の発現が低減するように改変された、請求項1に記載の酵母。
【請求項4】
さらに、Δ7−還元活性、ステロール側鎖の20位及び22位との結合を切断する活性、ステロール3位酸化異性化活性、ステロイド17α水酸化活性、ステロイド21位水酸化活性、及びステロイド11β位水酸化活性から選択される少なくとも一つの活性を有するように改変された請求項3に記載の酵母。
【請求項5】
さらに、Δ7−還元活性、ステロール側鎖の20位及び22位との結合を切断する活性、ステロール3位酸化異性化活性、ステロイド17α水酸化活性、ステロイド21位水酸化活性、及びステロイド11β位水酸化活性を有するように改変された請求項3に記載の酵母。
【請求項6】
サッカロミセス・セレビジエである、請求項1〜5のいずれか一項に記載の酵母。
【請求項7】
親株がサッカロミセス・セレビジエYPH499株、サッカロミセス・セレビジエFY1679-06c株、サッカロミセス・セレビジエKA311A株またはサッカロミセス・セレビジエX2181-1B株である、請求項6に記載の酵母。
【請求項8】
請求項1〜3のいずれか一項に記載の酵母を培養してΔ5,7−ステロールを生成・蓄積させ、Δ5,7−ステロールを回収する、Δ5,7−ステロールの製造方法。
【請求項9】
培養する酵母が請求項1又は2に記載の酵母であり、Δ5,7−ステロールがC5,7−ジエン体ステロールである、請求項8に記載の方法。
【請求項10】
C5,7−ジエン体ステロールがコレスタ−5,7,24−トリエン−3β−オールまたは7−デヒドロコレステロールである、請求項9に記載の方法。
【請求項11】
培養する酵母が請求項1又は3に記載の酵母であり、Δ5,7−ステロールがエルゴスタ−5,7,24-トリエノールである、請求項8に記載の方法。
【請求項12】
培養する酵母が請求項1に記載の酵母であり、Δ5,7−ステロールがエルゴステロールである、請求項8に記載の方法。
【請求項13】
請求項5に記載の酵母を培養してハイドロコルチゾンを生成・蓄積させ、ハイドロコルチゾンを回収する、ハイドロコルチゾンの製造方法。
【請求項1】
アシルCoA:ステロールアシルトランスフェラーゼ遺伝子とステリルエステル加水分解酵素遺伝子の発現が増強されるように改変された酵母。
【請求項2】
さらに、ステロールC−22デサチュラーゼ遺伝子(ERG5)及びステロールC−24メチルトランスフェラーゼ遺伝子(ERG6)の発現が低減するように改変された、請求項1に記載の酵母。
【請求項3】
さらに、ステロールC−22デサチュラーゼ遺伝子(ERG5)の発現が低減するように改変された、請求項1に記載の酵母。
【請求項4】
さらに、Δ7−還元活性、ステロール側鎖の20位及び22位との結合を切断する活性、ステロール3位酸化異性化活性、ステロイド17α水酸化活性、ステロイド21位水酸化活性、及びステロイド11β位水酸化活性から選択される少なくとも一つの活性を有するように改変された請求項3に記載の酵母。
【請求項5】
さらに、Δ7−還元活性、ステロール側鎖の20位及び22位との結合を切断する活性、ステロール3位酸化異性化活性、ステロイド17α水酸化活性、ステロイド21位水酸化活性、及びステロイド11β位水酸化活性を有するように改変された請求項3に記載の酵母。
【請求項6】
サッカロミセス・セレビジエである、請求項1〜5のいずれか一項に記載の酵母。
【請求項7】
親株がサッカロミセス・セレビジエYPH499株、サッカロミセス・セレビジエFY1679-06c株、サッカロミセス・セレビジエKA311A株またはサッカロミセス・セレビジエX2181-1B株である、請求項6に記載の酵母。
【請求項8】
請求項1〜3のいずれか一項に記載の酵母を培養してΔ5,7−ステロールを生成・蓄積させ、Δ5,7−ステロールを回収する、Δ5,7−ステロールの製造方法。
【請求項9】
培養する酵母が請求項1又は2に記載の酵母であり、Δ5,7−ステロールがC5,7−ジエン体ステロールである、請求項8に記載の方法。
【請求項10】
C5,7−ジエン体ステロールがコレスタ−5,7,24−トリエン−3β−オールまたは7−デヒドロコレステロールである、請求項9に記載の方法。
【請求項11】
培養する酵母が請求項1又は3に記載の酵母であり、Δ5,7−ステロールがエルゴスタ−5,7,24-トリエノールである、請求項8に記載の方法。
【請求項12】
培養する酵母が請求項1に記載の酵母であり、Δ5,7−ステロールがエルゴステロールである、請求項8に記載の方法。
【請求項13】
請求項5に記載の酵母を培養してハイドロコルチゾンを生成・蓄積させ、ハイドロコルチゾンを回収する、ハイドロコルチゾンの製造方法。
【図1】

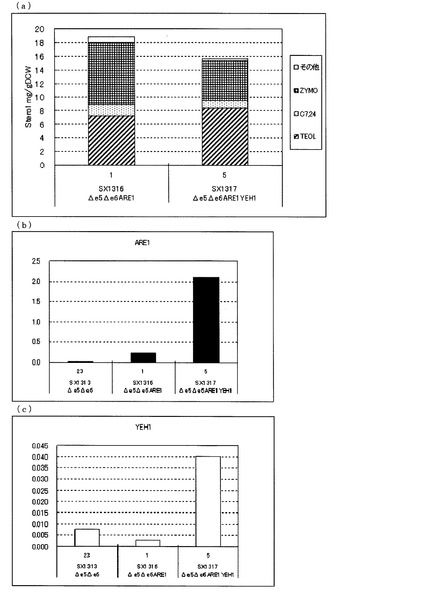
【図2】
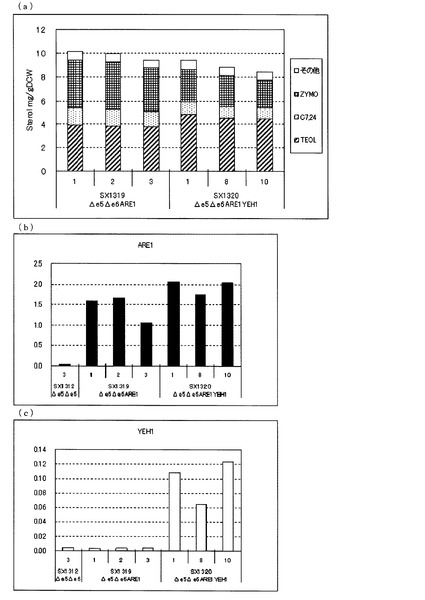
【図3】
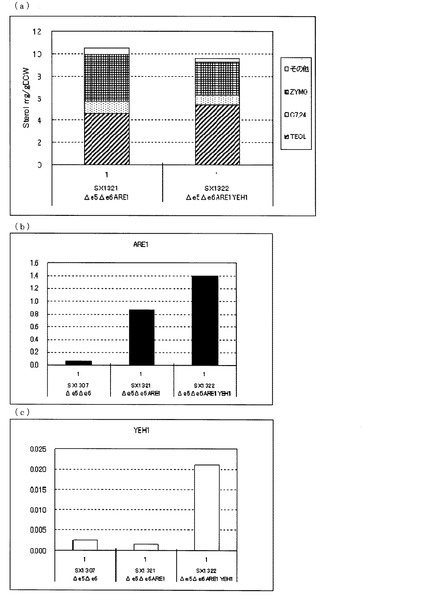
【図4】
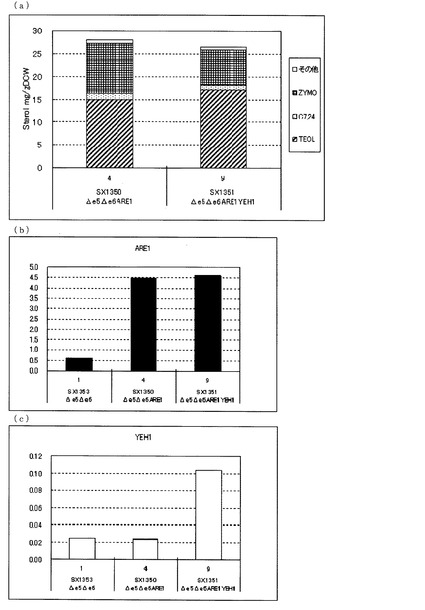
【図5】
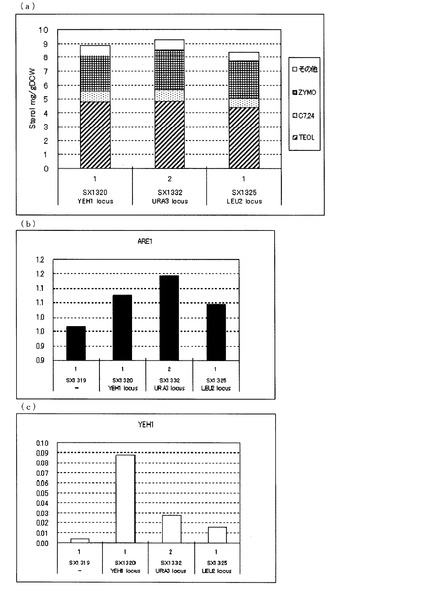
【図6】
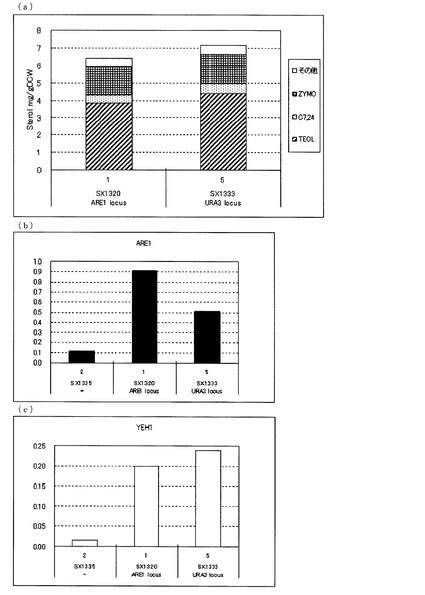
【図7】
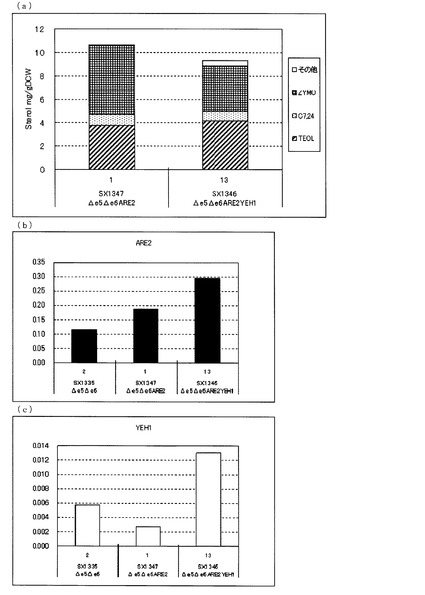
【図8】
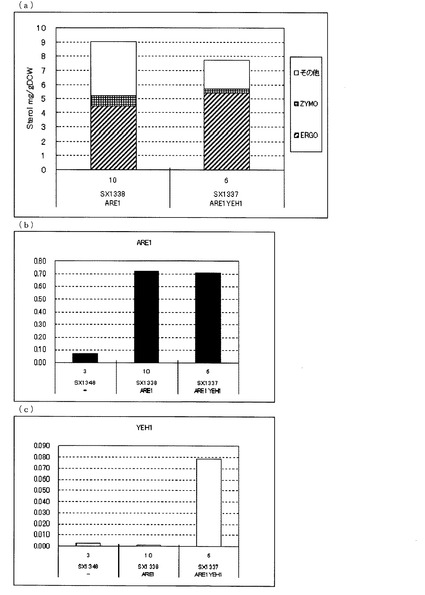
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2012−24084(P2012−24084A)
【公開日】平成24年2月9日(2012.2.9)
【国際特許分類】
【出願番号】特願2011−142282(P2011−142282)
【出願日】平成23年6月27日(2011.6.27)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
【公開日】平成24年2月9日(2012.2.9)
【国際特許分類】
【出願日】平成23年6月27日(2011.6.27)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
[ Back to top ]