
新規高等植物の作出方法、及び高等植物の生長促進方法
【課題】葉緑体のチラコイド内腔にシトクロムc6を有する新規な高等植物の作出方法を提供する。
【解決手段】葉緑体のチラコイド内腔にシトクロムc6を有する高等植物の作出方法であって、シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入することを特徴とする。さらに、高等植物の生長促進方法、ATP、NADPH、デンプン及びタンパク質からなる群から選ばれる少なくとも1つの合成促進方法並びに炭素固定促進方法。
【解決手段】葉緑体のチラコイド内腔にシトクロムc6を有する高等植物の作出方法であって、シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入することを特徴とする。さらに、高等植物の生長促進方法、ATP、NADPH、デンプン及びタンパク質からなる群から選ばれる少なくとも1つの合成促進方法並びに炭素固定促進方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、葉緑体のチラコイド内腔にシトクロムc6を有する新規な高等植物の作出方法、及び、上記チラコイド内腔にシトクロムc6を存在させ高等植物の生長を促進させる方法又は高等植物の炭素固定能を促進させる方法に関する。
【背景技術】
【0002】
従来、陸上植物等のいわゆる高等植物の生長促進に関する技術としては、リブロースビスリン酸カルボキシラーゼ等の酵素の活性を高めるなど、光合成暗反応(カルビン・ベンソンサイクル)に関する報告例、具体的には、関連酵素遺伝子の導入により葉を大きくした報告例(Shigeoka et al., Nature biotechnology, 19, 965-969(2001))等がある。しかしこのような技術は、様々な高等植物への適用の面で、汎用性に非常に乏しいものであった。
シトクロムc6は、光合成明反応における電子伝達タンパク質であって、本来、一部の藻類(ラン藻類等)にのみ存在するものであり、その電子伝達能力が極めて優れている(すなわち酸化還元電位が高電位である)ことが知られている(図1)。このため、様々な高等植物において、その葉緑体中に(詳しくはチラコイド内腔に)シトクロムc6を発現及び機能させ、光合成能を向上させる、汎用性に優れた技術の開発が強く望まれている。
ところで、シトクロムc6を細胞内で発現させるようにする技術としては、例えば、F. P. Molina-Heredia et al., Biochem. Biophys. Res. Commun., 243, 302-306(1998);T. Satoh et al., FEBS lett., 531, 543-547(2002);R. Gupta et al., Nature, 417, 567-571(2002);D. R. Hickey et al., Gene, 105, 73-81(1991) 等の文献に記載の技術が知られている。しかし、これら技術はいずれも、大腸菌、酵母あるいはラン藻といった高等植物ではない宿主細胞を用い、単に、上記シトクロムc6又はそれに類するタンパク質の大量生産や機能解析を目的としてなされたものであり、従来より当業者において通常行われている遺伝子発現法に含まれるものである。
そして、高等植物において光合成明反応の電子伝達体としてシトクロムc6を機能させること、つまり、高等植物細胞内の葉緑体中にあるチラコイドの内腔にシトクロムc6を存在させることに成功した報告例は全く無く、極めて困難であると考えられていた。
【発明の概要】
【0003】
そこで、本発明が解決しようとする課題は、葉緑体のチラコイド内腔にシトクロムc6を有する新規な高等植物の作出方法を提供すること、ひいては、高等植物の生長促進方法、ATP、NADPH、デンプン及びタンパク質からなる群から選ばれる少なくとも1つの合成促進方法並びに炭素固定促進方法を提供することにある。
本発明者は、上記課題を解決するべく鋭意検討を行った。その結果、特定のシグナルペプチドを付加したシトクロムc6タンパク質の遺伝子を、高等植物のゲノムDNAに導入し発現させるようにすれば、従来不可能であると考えられていた葉緑体包膜内及びチラコイド膜内へのシトクロムc6の輸送(通過)が可能となることを見出し、本発明を完成した。
すなわち、本発明は以下のとおりである。
(1) シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入することを特徴とする、葉緑体のチラコイド内腔にシトクロムc6を有する高等植物の作出方法。
(2) シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入して発現させ、葉緑体のチラコイド内腔にシトクロムc6を存在させることを特徴とする、高等植物の生長促進方法。
(3) シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入して発現させ、葉緑体のチラコイド内腔にシトクロムc6を存在させることを特徴とする、高等植物のATP、NADPH、デンプン及びタンパク質からなる群から選ばれる少なくとも1つの合成促進方法。
(4) シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入して発現させ、葉緑体のチラコイド内腔にシトクロムc6を存在させることを特徴とする、高等植物の炭素固定促進方法。
上記(1)〜(4)の方法においては、前記融合タンパク質が以下の(a)、(b)、(c)又は(d)のタンパク質であってもよい。
(a) 配列番号6に示されるアミノ酸配列を含むタンパク質
(b) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質
(c) 配列番号6に示されるアミノ酸配列のうちの前記シトクロムc6タンパク質に相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ電子伝達能を有するタンパク質
(d) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列及び前記シトクロムc6タンパク質に相当するアミノ酸配列においてそれぞれ1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質
また上記(1)〜(4)の方法においては、前記遺伝子が以下の(a)又は(b)のDNAを含む遺伝子であってもよい。
(a) 配列番号5に示される塩基配列を含むDNA
(b) 配列番号5に示される塩基配列を含むDNAと相補的な塩基配列を含むDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質をコードするDNA
(5) シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加されてなる融合タンパク質。
上記(5)の融合タンパク質は、以下の(a)、(b)、(c)又は(d)のタンパク質であってもよい。
(a) 配列番号6に示されるアミノ酸配列を含むタンパク質
(b) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質
(c) 配列番号6に示されるアミノ酸配列のうちの前記シトクロムc6タンパク質に相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ電子伝達能を有するタンパク質
(d) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列及び前記シトクロムc6タンパク質に相当するアミノ酸配列においてそれぞれ1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質
(6) 上記(5)の融合タンパク質をコードする遺伝子。
(7) 以下の(a)又は(b)のDNAを含む遺伝子。
(a) 配列番号5に示される塩基配列を含むDNA
(b) 配列番号5に示される塩基配列を含むDNAと相補的な塩基配列を含むDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質をコードするDNA
(8) 上記(6)又は(7)の遺伝子を含む組換えベクター。
(9) 上記(8)の組換えベクターを宿主に導入してなる形質転換体。
上記(9)の形質転換体は、宿主がアグロバクテリウム菌の形質転換体であってもよい。
(10) 上記(6)又は(7)の遺伝子が植物ゲノム中に導入されている、形質転換高等植物。
上記(10)の形質転換高等植物は、植物細胞中の葉緑体のチラコイド内腔にシトクロムc6を有するものであることが好ましい。
【図面の簡単な説明】
【0004】
【図1】図1は、葉緑体チラコイド膜の光合成電子伝達系を示す図である。
【図2】図2は、本発明におけるアグロバクテリウムを介したシトクロムc6遺伝子の導入と植物内におけるシトクロムc6の発現様式を示す模式図である。
【図3】図3は、シアノバクテリア及び藻類と高等植物の間における電子伝達系の相違を示す図である。
【図4】図4は、Porphyra yezoensisシトクロムc6のcDNAの塩基配列及び推定されるアミノ酸配列を示す図である。
【図5】図5は、Porphyra yezoensisシトクロムc6遺伝子の成熟タンパク質領域をPCRによって増幅した結果を示す図である。
【図6】図6は、植物導入用シトクロムc6遺伝子の構築に用いたpBluescript II SK+/- ベクターを示す図である。
【図7】図7は、Arabidopsis thaliana由来プラストシアニンのシグナルペプチドとPorphyra yezoensis由来シトクロムc6の成熟タンパク質領域を融合させた遺伝子の塩基配列と推定されるアミノ酸配列を示す図である。
【図8】図8は、トリペアレンタルメイティングによるアグロバクテリウム・チュメファシエンス(Agrobacterium tumefaciens)の形質転換法を示す模式図である。
【図9】図9は、植物に導入されたシトクロムc6遺伝子をPCRによって特定した電気泳動結果を示す図である。(A)は鋳型としてゲノムDNAを用いたときの結果であり、(B)は鋳型として全RNAを用いたときの結果である。
【図10】図10は、植物内で発現したシトクロムc6を電気泳動によって特定した結果を示す図である。(A)はCBB染色の結果であり、(B)はシトクロムc6抗体と反応させたウエスタンブロッティングの結果である。
【図11】図11は、生育日数の経過に伴う、野生型(○)及び形質転換A. thaliana(●)の背丈の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=20)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図12】図12は、生育日数の経過に伴う、野生型(○)及び形質転換A. thaliana(●)の根の長さの変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=20)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図13】図13は、生育日数の経過に伴う、野生型(○)及び形質転換A. thaliana(●)の葉の大きさの変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=20)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図14】図14は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後におけるクロロフィル含量の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=8)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図15】図15は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後におけるATP含量の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=8)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図16】図16は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後におけるNADPH含量の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=8)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図17】図17は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後における二酸化炭素同化能力の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=10)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図18】図18は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後におけるデンプン量の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=10)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図19】は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後におけるタンパク質量の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=8)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【発明を実施するための形態】
【0005】
以下、本発明について詳しく説明するが、本発明の範囲はこれらの説明に拘束されることはなく、以下の例示以外についても、本発明の趣旨を損なわない範囲で適宜変更し実施し得る。
なお、本明細書は、本願優先権主張の基礎となる特願2005-27012号明細書の全体を包含する。また、本明細書において引用された全ての先行技術文献、並びに公開公報、特許公報及びその他の特許文献は、参照として本明細書に組み入れられる。
1.本発明の概要
本発明は、シトクロムc6とシグナルペプチドとの融合タンパク質をコードする遺伝子を高等植物内に導入し、高等植物細胞内の葉緑体中のチラコイド内腔において、光合成明反応の電子伝達体としての役割を果たすシトクロムc6を発現させ且つ機能させることにより、高等植物の生長を促進させる方法に係るものである(図2)。本発明により、高等植物細胞内でのATP、NADPH、デンプン及びタンパク質の合成が促進され、またこれに伴い、二酸化炭素(CO2)を炭水化物に変換する光合成暗反応(炭素固定反応)も促進され得る。本発明は、このような、高等植物におけるATP等の合成や炭素固定を促進する方法を含み、また、生長促進、ATP等の合成促進、及び炭素固定の促進がされ得る高等植物の作出方法をも含むものである。
一般に、光合成明反応(光合成電子伝達反応)においては、クロロフィルの電子が太陽光のエネルギーを獲得し、チラコイド内腔(チラコイド膜中)の電子伝達鎖を次々と移動するが、その際、クロロフィルは水から電子を獲得し、酸素(O2)を遊離する一方、上記電子伝達反応に伴いチラコイド膜を介してH+のくみ出しが起こり、それで生じたプロトンの駆動力によって葉緑体のストロマ内でATPが合成される。そして、一連の反応の最後では、高エネルギー電子を(H+とともに)NADP+が受け取り、NADPHが合成される。
これに対し、光合成暗反応(炭素固定反応)では、光合成明反応で合成されたATPとNADPHがそれぞれエネルギー源と還元力として働くことで、葉緑体のストロマ内及び細胞質において、CO2が炭水化物に変換される。この炭素固定反応により、植物の葉等ではショ糖が合成される。ショ糖は、他の組織にも輸送され、様々な有機分子の合成材料として、ひいては植物自身の生長のためのエネルギー源として用いられる。
通常、高等植物における光合成明反応の電子伝達体(電子伝達タンパク質)は、プラストシアニン(PC)であるが、電子伝達能(酸化還元電位の高さ)に関しては、藻類における電子伝達体であるシトクロムc6の方が明らかに優れていることが分かっている(図3)。そのため、従来から、このシトクロムc6を高等植物内でも発現及び機能させ、光合成明反応及び暗反応を促進させようとする試みが種々なされてきた。しかしながら、このような試みは、実際のところ極めて困難であり、これまで成功例は無かった。
高等植物において、その細胞内のゲノム情報をもとに発現されたタンパク質が光合成明反応の電子伝達体としての機能を発揮するためには、チラコイド内腔に存在することが必須条件となる。そのためには、通常、「葉緑体包膜」及び「チラコイド膜」の2種の膜を通過する必要がある。この点が高等植物におけるシトクロムc6の機能発現を極めて困難にしている主要因であると考えられていた。そこで本発明者は、遺伝子組換え技術により、シトクロムc6遺伝子を高等植物のゲノム中に導入するにあたり、シトクロムc6タンパク質が、特定のシグナルペプチド(特定の長さ又は特定のアミノ酸配列のシグナルペプチド)が付加された融合タンパク質の状態で発現されるように、予めシグナル配列を付加した遺伝子を構築した上でその導入を行った。その結果、当該遺伝子導入により作出した植物体においては、チラコイド内腔にシトクロムc6タンパク質(シグナルペプチド無し)の存在が認められ、結果として、発現した融合タンパク質はそのシグナルペプチドの働きにより前述した2種の膜を通過し得るものであることが示され、本発明を完成した。
(1) PYC6遺伝子導入系の検討
これまでに、PYC6の酸化還元電位は高電位であることが知られており、また、そのPYC6遺伝子配列も明らかにされている。本発明においては、はじめにPYC6遺伝子をバイナリーベクターに連結し、形質転換用アグロバクテリウムを調製した。シロイヌナズナ(Arabidopsis thaliana)は、播種後低温処理を行ったのち長日条件で生育させ、抽台した段階で減圧浸潤により形質転換を行った。つづいて、得られた形質転換体において遺伝子の導入と発現を解析するため、植物体から抽出したゲノムDNAとRNAを鋳型とし、それぞれPCR、RT-PCRを行った。その結果、PYC6遺伝子の増幅が見られ、DNAシーケンサーによりその塩基配列はPYC6遺伝子と同様であることを確認した。次に、形質転換体中に確認されたPYC6遺伝子がタンパク質として発現しているかを解析するため、植物体から葉緑体タンパク質画分を抽出し、電気泳動後ウエスタンブロッティングを行った。その結果、PYC6タンパク質の発現を確認した。また、得られた形質転換体中のPYC6タンパク質のN末端アミノ酸配列を解析したところ、導入したPYC6のアミノ酸配列(PIR accession No. : JC5849)と一致した。この結果から、高等植物であるA. thaliana内で藻類由来cyt c6(PYC6)を発現させることに成功した。
(2) PYC6導入による植物体の影響
後述の実施例に示すように、PYC6導入植物の生育を観察したところ、葉の大きさは野生型(WT)に比べおよそ1.2〜1.3倍、背丈は約1.5倍増大していた。PYC6導入植物は生育初期段階においてWTと比較して成長速度が上昇していた。また、PYC6導入植物中のクロロフィル量を測定した結果、WTのおよそ1.1〜1.2倍であった。このPYC6導入により植物体にみられた現象は、明反応最終産物であるエネルギー物質を利用して光合成反応全体が活性化されたためではないかと考えられる。そこで、ルシフェリン・ルシフェラーゼ法によりPYC6導入植物のATP量を測定した結果、WTと比較して約1.7倍増大していた。これはA. thaliana において機能しているプラストシアニンに加え、さらに新たな電子伝達体として紅藻cyt c6が機能したことにより、明反応の電子伝達が活性化され、その最終生成物であるATP量が増大し、さらには植物全体の生育が活性化されたためと考えられた。
なお、当該融合タンパク質が前述した2種の膜を通過するに際しては、第1の膜(葉緑体包膜)の通過時にはシグナルペプチドの一部が、第2の膜(チラコイド膜)の通過時にはその残りの部分が使用され、それぞれ使用後は脱離した後、最終的にシグナルペプチドの無い状態でPYC6がチラコイド内腔に輸送される。そして輸送されたPYC6タンパク質は、このチラコイド内腔でヘム(ヘムc)と結合し且つフォールディングすることにより電子伝達体として機能し得るシトクロムc6になると考えられる。
2.新規高等植物の作出方法
本発明の作出方法は、前述の通り、葉緑体のチラコイド内腔にシトクロムc6を有する新規な高等植物の作出方法であり、シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入することを特徴とする方法である。
(1) 作出対象となる高等植物
本発明の作出方法に用い得る高等植物、すなわち葉緑体のチラコイド内腔にシトクロムc6を有するよう形質転換され得る高等植物としては、限定はされず、例えば以下の高等植物を挙げることができる。
本発明において対象となる植物は、植物体全体、植物器官(例えば葉、花弁、茎、根、種子等)、植物組織(例えば表皮、師部、柔組織、木部、維管束、柵状組織、海綿状組織等)又は植物培養細胞(カルスなどの組織培養を含む)のいずれをも意味するものである。形質転換に用いられる植物としては、C3、C4、CAM植物、及びこれらの中間植物等のすべてを含み、例えばアブラナ科、ナス科、イネ科、マメ科、アカザ科、バラ科、キク科、ユリ科、ナデシコ科、ウリ科、ヒルガオ科、ヒユ科、パイナップル科、サボテン科、及びアロエ科等に属する植物(果実、野菜、花卉等を含む)が挙げられるが(下記参照)、これらの植物に限定されるものではない。
〔C3植物〕
・アブラナ科:シロイヌナズナ(Arabidopsis thaliana) 、ダイコン(Raphanus) など
・ナス科:タバコ(Nicotiana tabacum)、ジャガイモ(Solanum)など
・イネ科:イネ(Oryza sativa)、トウモロコシ(Zea mays) 、コムギ(Triticun)など
・マメ科:ダイズ(Glycine max) 、エンドウ(Pisum) など
・アカザ科:ホウレンソウ(Spinacia)など
・バラ科:ヤマザクラ(Prunus)、バラ (Rosa) など
・キク科:ハルジオン(Erigeron)、タンポポ (Taraxacun) など
・ウリ科:カボチャ(Cucurdida)、キュウリ (Cucumis) など
・ヒルガオ科:サツマイモ(Ipomea)など
・ラン科:ウチョウラン(Poneorchis)など
〔C4植物〕
・イネ科:トウモロコシ(Zea mays)、サトウキビ(Saccharum officinarum)、アワ(Setaria itarica)など
・ヒユ科:アマランサス(Amaranthaceae)など
〔CAM植物〕
・パイナップル科:パイナップル(Ananas comosus)など
・サボテン科:デフューサ(Lophophora difusa)、オプンティア(Opuntia spp.)など
・アロエ科:キダチアロエ(Aloe arborescens)、アロエベラ(Aloe vera)など
〔C3-C4中間植物〕
・ツルナ科:クルマバザクロソウ(Mollugo verticillata)など
・イネ科:キビ(Panicum milioides)など
なかでも、本発明の作出方法の効果として、生長促進効果の高い植物が得られる点を考慮すると、例えば、より市場性の高い植物のほうが本発明の利用価値が高く、具体的には、葉物(ホウレンソウ、キャベツなど)、花物(バラ、コチョウランなど)、茎物(ジャガイモ、レンコンなど)、根物(ゴボウ、ダイコンなど)、穀物(イネ、コムギなど)、果実物(パイナップル、ブドウなど)、観賞用植物(マツ、カエデなど)、木材(スギ、ヒノキなど)などが好ましく挙げられる。
(2) 融合タンパク質
本発明の作出方法においては、高等植物のゲノム中に特定の融合タンパク質をコードする遺伝子を導入するが、当該融合タンパク質は、シトクロムc6タンパク質に特定の長さのシグナルペプチドが付加された融合タンパク質であることが重要である。
上記融合タンパク質は、シグナルペプチドの長さが50〜80アミノ酸残基であることが重要である。このようにシグナルペプチドが十分に長いことにより、シトクロムc6タンパク質を葉緑体包膜及びチラコイド膜のいずれにも通過させることができる。シグナルペプチドの配列は、高等植物から遺伝子クローニングすることにより決定することができる。
なお上記融合タンパク質におけるシグナルペプチドは、一般に、シトクロムc6タンパク質のN末端に付加されていることが好ましい。
本発明においては、上記融合タンパク質は、例えば、以下の(a)、(b)、(c)又は(d)のタンパク質であることが好ましい。
(a) 配列番号6に示されるアミノ酸配列を含むタンパク質
(b) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質
(c) 配列番号6に示されるアミノ酸配列のうちの前記シトクロムc6タンパク質に相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ電子伝達能を有するタンパク質
(d) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列及び前記シトクロムc6タンパク質に相当するアミノ酸配列においてそれぞれ1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質
上記(a)のタンパク質は、配列番号6に示すように、シトクロムc6タンパク質を構成する85個のアミノ酸(配列番号2)を含む配列のN末端側に、シグナルペプチドを構成する72個のアミノ酸(配列番号4)からなる配列が付加(連結)されてなる、合計157個のアミノ酸を含むものであるが、配列番号6で表されるアミノ酸配列からなる融合タンパク質であってもよい。但し、シグナルペプチドは、上記72個に限定されるものではない。ここで、シトクロムc6タンパク質の由来は特に限定されるものではなく、紅藻(Porphyra yezoensis)、ラン藻、褐藻、珪藻、および緑藻などを用いることができ、紅藻(P. yezoensis)由来のタンパク質であることが好ましい。P. yezoensis由来のシトクロムc6タンパク質のアミノ酸配列は公知であり(PIR accession No.: JC5849)、データベース検索することにより入手することができる。
上記(b)のタンパク質は、上記(a)のタンパク質を構成する全アミノ酸配列のうちの前記シグナルペプチドに相当する72個のアミノ酸配列(配列番号4)において、1個又は数個(例えば1個〜10個程度、好ましくは1個〜5個程度)のアミノ酸が欠失、置換又は付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質であればよく、限定はされない。
なお、上記(b)のタンパク質は、上記(a)のタンパク質と同様に高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質であることが重要であるため、例えば、第53番目〜第72番目のアミノ酸など、上記葉緑体包膜やチラコイド膜の通過に重要と考えられる部分のアミノ酸配列は、上記(a)のタンパク質のアミノ酸配列から変異置換されていないものが好ましい。
上記(c)のタンパク質は、上記(a)のタンパク質を構成する全アミノ酸配列のうちの前記シトクロムc6タンパク質に相当する85個のアミノ酸配列(配列番号2)において、1個又は数個(例えば1個〜10個程度、好ましくは1個〜5個程度)のアミノ酸が欠失、置換又は付加されたアミノ酸配列を含み、かつ(光合成明反応における)電子伝達能を有するタンパク質であればよく、限定はされない。
ここで、上記の「電子伝達能を有するタンパク質」とは、本発明においては、上記(c)のタンパク質を構成する全アミノ酸配列から前記シグナルペプチドに相当するアミノ酸配列が除かれたアミノ酸配列を含むタンパク質であって、ヘム(ヘムc)と結合した後において、電子伝達能を有するタンパク質を意味する。
なお、上記(c)のタンパク質は、シトクロムc6タンパク質と同様に電子伝達能を有するタンパク質であることが重要であるため、例えば、第14番目〜第18番目のアミノ酸、及び第47番目〜第60番目のアミノ酸など、高等植物の光合成明反応における電子伝達体として機能するために重要と考えられる部分のアミノ酸配列は、上記(a)のタンパク質のアミノ酸配列から変異置換されていないものが好ましい。
上記(d)のタンパク質は、上記(a)のタンパク質を構成する全アミノ酸配列のうちの、前記シグナルペプチドに相当する72個のアミノ酸配列(配列番号4)において1個又は数個(例えば1個〜10個程度、好ましくは1個〜5個程度)のアミノ酸が欠失、置換又は付加され、さらに前記シトクロムc6タンパク質に相当する85個のアミノ酸配列(配列番号2)においても1個又は数個(例えば1個〜10個程度、好ましくは1個〜5個程度)のアミノ酸が欠失、置換又は付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに(光合成明反応における)電子伝達能を有するタンパク質であればよく、限定はされない。
ここで、上記の「電子伝達能を有するタンパク質」とは、上記(c)のタンパク質の場合と同様の意味である。
なお、上記(d)のタンパク質は、上記(a)のタンパク質と同様に高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質であることに加え、シトクロムc6タンパク質と同様に電子伝達能を有するタンパク質であることも重要であるため、例えば、第53番目〜第72番目のアミノ酸などの上記葉緑体包膜やチラコイド膜の通過に重要と考えられる部分のアミノ酸配列や、第14番目〜第18番目のアミノ酸、及び第47番目〜第60番目のアミノ酸などの高等植物の光合成明反応における電子伝達体として機能するために重要と考えられる部分のアミノ酸配列は、上記(a)のタンパク質のアミノ酸配列から変異置換されていないものが好ましい。
上記(c)及び(d)のタンパク質の、電子伝達能の有無及び電子伝達能を有する場合、立体構造解析および酸化還元電位の測定を行うことにより確認・測定することができる。なお、シトクロムc6の酸化還元電位は、およそ350〜370mV程度である。
このような配列番号6で表されるアミノ酸配列において1若しくは複数個のアミノ酸が欠失、挿入または付加されたアミノ酸配列をコードするポリヌクレオチドは、「Molecular Cloning, A Laboratory Manual 2nd ed.」(Cold Spring Harbor Press (1989))、「Current Protocols in Molecular Biology」(John Wiley & Sons (1987-1997)、Kunkel (1985) Proc. Natl. Acad. Sci. USA 82: 488-92、Kunkel (1988) Method. Enzymol. 85: 2763-6)等に記載の部位特異的変異誘発法等の方法に従って調製することができる。このようなアミノ酸の欠失、置換又は付加等の変異型をコードする遺伝子の作製には、例えば、Kunkel法や Gapped duplex法等の公知手法により、部位特異的突然変異誘発法を利用した変異導入用キット、例えばQuikChangeTM Site-Directed Mutagenesis Kit(ストラタジーン社製)、GeneTailorTM Site-Directed Mutagenesis System(インビトロジェン社製)、TaKaRa Site-Directed Mutagenesis System(Mutan-K、Mutan-Super Express Km等:タカラバイオ社製)等を用いて行うことができる。
(3) 遺伝子
本発明の作出方法においては、高等植物のゲノム中に導入する遺伝子は、前述した融合タンパク質をコードする遺伝子であることが重要である。
本発明においては、上記遺伝子は、例えば、以下の(a)又は(b)のDNAを含む遺伝子であることが好ましい。なお、以下の(a)及び(b)のDNAは、いずれも前述した融合タンパク質の構造遺伝子(すなわち、シグナルペプチドをコードする遺伝子と、シトクロムc6又はこれと同様の電子伝達能を有するタンパク質の構造遺伝子とが連結した遺伝子)であるが、これらDNAを含む遺伝子としては、これらDNAのみからなるものであってもよいし、これらDNAを一部に含み、その他に当該融合タンパク質の構造遺伝子の発現に必要な公知の塩基配列(転写プロモーター、SD配列、Kozak配列、ターミネーター等)をも含むものであってもよく、限定はされない。なお、シトクロムc6をコードする塩基配列は、例えば日本DNAデータバンク(DDBJ)に公表されており公知である(accession number:AB40818)。
(a) 配列番号5に示される塩基配列を含むDNA
(b) 配列番号5に示される塩基配列を含むDNAと相補的な塩基配列を含むDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質をコードするDNA
上記(b)のDNAは、上記(a)のDNA若しくはそれと相補的な塩基配列を含むDNA、又はこれらの断片をプローブとして用い、コロニーハイブリダイゼーション、プラークハイブリダイゼーション、およびサザンブロット等の公知のハイブリダイゼーション法を実施し、cDNAライブラリーやゲノムライブラリーから得ることができる。ライブラリーは、公知の方法で作製されたものを利用してもよいし、市販のcDNAライブラリーやゲノムライブラリーを利用してもよく、限定はされない。
また、シグナルペプチド部分をコードする塩基配列(配列番号3)、シトクロムc6をコードする塩基配列(配列番号1)の両方又はいずれか一方の一部に変異が生じていてもよい。
ハイブリダイゼーション法の詳細な手順については、Molecular Cloning, A Laboratory Manual 2nd ed. (Cold Spring Harbor Laboratory Press (1989))等を適宜参照することができる。
ハイブリダイゼーション法を実施における「ストリンジェントな条件」とは、ハイブリダイゼーション後の洗浄時の条件であって、バッファーの塩濃度が15〜750mM、温度が25〜65℃、好ましくは塩濃度が15〜150mM、温度が45〜55℃の条件を意味する。具体的には、例えば、50mMで50℃等の条件を挙げることができる。さらに、このような塩濃度や温度等の条件に加えて、プローブ濃度、プローブの長さ、反応時間等の諸条件も考慮し、上記(b)のDNAを得るための条件を適宜設定することができる。
また、上記(b)のDNAは、当業者に公知の方法で適当な断片を用いてプローブを作製し、このプローブを用いてコロニーハイブリダイゼーション、プラークハイブリダイゼーション、サザンブロット等の公知のハイブリダイゼーション法により、cDNAライブラリーおよびゲノムライブラリーから得ることができる。上記ハイブリダイゼーションにおいてストリンジェントな条件としては、たとえば、1×SSC〜2×SSC、0.1%〜0.5%SDS及び30℃〜80℃の条件が挙げられ、より詳細には、60〜68℃で30分以上プレハイブリダイゼーションを行った後、プローブを添加して1時間以上68℃に保ってハイブリッド形成させ、その後、2×SSC、0.1%SDS中、室温で5〜15分の洗浄を1〜2回行う条件が挙げられる。
ハイブリダイズするDNAとしては、上記(a)のDNAの塩基配列に対して少なくとも70%以上の相同性を有する塩基配列であることが好ましく、より好ましくは80%以上、さらに好ましくは90%以上、さらに好ましくは95%以上である。
なお、上記(b)のDNAは、高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質をコードするDNAであることが重要であるため、翻訳後のアミノ酸配列を考慮したときに、例えば、第53番目〜第72番目のアミノ酸などの上記葉緑体包膜やチラコイド膜の通過に重要と考えられる部分のアミノ酸配列や、第14番目〜第18番目のアミノ酸、及び第47番目〜第60番目のアミノ酸などの高等植物の光合成明反応における電子伝達体として機能するために重要と考えられる部分のアミノ酸配列が、前述した(a)のタンパク質のアミノ酸配列から変異置換されないような、塩基配列を有するものが好ましい。
上記(b)のDNAとしては、例えば、上記(a)のDNAと完全に同一のものではないが、そのうちの第103番目の塩基を「T (チミン)」から「A (アデニン)」に置換し翻訳されるアミノ酸が「Ser (セリン)」から「Thr (トレオニン)」になるよう変異させたもの、第216番目の塩基を「C (シトシン)」から「T (チミン)」に置換するが翻訳されるアミノ酸には変化がないよう変異させたもの、及びこれらの塩基置換を組み合わせて変異させたもの、などが好ましく挙げられる。
本発明において、上記遺伝子は、高等植物において発現させるものであるため、アミノ酸に対応するコドンが、転写後、植物一般において通常用いられているコドン(好ましくは使用頻度の高いコドン)を示すDNAを含むものであることが必要である。
(4) 遺伝子の導入方法
高等植物の他の形質を変えずに、そのゲノム中に前述した融合タンパク質をコードする遺伝子(目的遺伝子)を導入して、新規な高等植物(形質転換高等植物)を作出する方法としては、限定はされないが、アグロバクテリウム法、エレクトロポレーション法、及びパーティクルガン法等の公知の遺伝子組換え技術を任意に採用することができる。例えば、限定はされないが、目的遺伝子を含むバイナリーベクターを導入したアグロバクテリウム属細菌(根頭がん種病菌)を用いる減圧浸潤法等が好適である。以下に、詳細に説明する。
(i) 組換えベクター
前述した融合タンパク質をコードする遺伝子を含む組換えベクターとしては、限定はされないが、上記バイナリーベクターとして用い得るものが好ましい。具体的には、Vir領域遺伝子の働きで切り出されるT-DNA領域を有するベクターの、当該T-DNA領域中に、植物ゲノムに組込みたい目的遺伝子を(GUS遺伝子の代わりに)挿入して構築される組換えベクターが好ましい。上記T-DNA領域を有するベクターとしては、例えば、pBI121ベクター(Clontech社製;カナマイシン耐性遺伝子を選択マーカーとし、35Sプロモーターの下流にGUS遺伝子を含む)、pBI101ベクター(Clontech社製;カナマイシン耐性遺伝子を選択マーカーとし、GUS遺伝子を含む)等が好ましく挙げられる。
また、減圧浸潤法に用い得るものではないが、宿主細胞に適した公知の発現ベクターに、目的遺伝子を挿入して構築される組換えベクターも、本発明の組換えベクターに含まれる。必要に応じ、当該遺伝子の上流に転写プロモーターやSD配列(宿主が原核細胞の場合)やKozak配列(宿主が真核細胞の場合)を、下流にターミネーターを、PCR等により付加しておく。上記転写プロモーター等、融合タンパク質の発現に必要な各要素は、目的遺伝子に含まれていてもよいし、もともと発現ベクターに含まれている場合はそれを利用してもよく、限定はされない。このような組換えベクターは、例えば、前述した融合タンパク質の大量生産等に用いることができる。
なお、これら各種組換えベクターの作製においては、制限酵素を用いる方法や、トポイソメラーゼを用いる方法等、公知の遺伝子組換え技術及び条件等を適宜採用して行うことができる。
(ii) 組換えベクターを導入してなる形質転換体
組換えベクターを導入する宿主としては、限定はされないが、最終的に減圧浸潤法を行う場合は、バイナリーベクターを導入する宿主としては、アグロバクテリウム属細菌(Agrobacterium tumefaciens等)を使用する。なお、アグロバクテリウム属細菌を用いる場合は、一般には、当該細菌が本来有しているTiプラスミド上のT-DNA領域中のがん遺伝子が、潰されているか又は取り除かれている等して、予め形質転換されている菌体(アグロバクテリウム属細菌の形質転換体)を用いる。その他、組換えベクターとして発現ベクターを導入する場合は、公知の宿主、例えば、大腸菌、枯草菌、酵母、カビ、ヒトやマウス等の各種動物細胞等を用いることができる。
宿主の形質転換の方法は、限定はされず、宿主と組換えベクターとの組み合わせを考慮して、公知の方法から適宜選択し実施することができる。例えば、電気穿孔法、リポフェクション法、ヒートショック法、PEG法、リン酸カルシウム法、DEAEデキストラン法等が好ましく実施できる。
得られる形質転換体においては、実際に用いた宿主と、組み換えベクターに含まれるEBNA1変異体遺伝子のコドン型の宿主とが、一致していてもよいし、異なっていてもよく、限定はされない。
(iii) 高等植物の形質転換、及び形質転換高等植物
高等植物のゲノム中に目的遺伝子を導入し、形質転換高等植物を作製する方法は、限定はされないが、前述した減圧浸潤法が好ましい。また、形質転換を行う植物体の状態は、成体またはカルスを用いることが好ましい。
減圧浸潤法による高等植物の形質転換方法では、具体的には、(a)前述した融合タンパク質をコードする遺伝子が挿入された組換えベクター(バイナリーベクター)を含む形質転換細菌(アグロバクテリウム属細菌の形質転換体)を高等植物の葉片に感染させ、(b)カナマイシン等の抗生物質を含む選択培地で培養し、(c)不定芽カルスを形成させ栽培して、形質転換高等植物を得るようにする。なお、減圧浸潤法を行うにあたり、各処理工程における手段および処理条件等は、すべて公知の範囲から適宜選択して行うようにすればよく、限定はされない。
上述のようにして得られた形質転換高等植物は、そのゲノム中に、前述した融合タンパク質をコードする遺伝子が導入されている。当該遺伝子を基にして発現された融合タンパク質は、前述したシグナルペプチドを有し植物細胞中の葉緑体包膜及びチラコイド膜の通過能を有するものである。よって、得られた形質転換高等植物は、チラコイド内腔にシトクロムc6(好ましくは光合成明反応の電子伝達体としてのシトクロムc6)を有するものである。
得られた形質転換高等植物においては、光合成明反応の電子伝達体として、プラストシアニン(PC)のみならずシトクロムc6も機能すると考えられ、効果的に生長促進が達成される。形質転換高等植物の成体の大きさ(背丈、根長、葉の大きさ等)は、限定はされないが、野生型のものに比べ、例えば、1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.5倍以上(例えば約1.9倍)となる。
また、形質転換高等植物においては、生長促進効果と同様に、各細胞中の各種分子数も増加する効果も得られる。例えば、クロロフィル分子の量は、限定はされないが、野生型のものに比べ、例えば、1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.5倍以上(例えば約1.6倍)となり、より一層光合成能が向上したものとなる。
さらに、得られた形質転換高等植物は、光合成効率が向上するため、その生産物であるATP合成量は、限定はされないが、野生型のものに比べ、例えば、1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.5倍以上(例えば約1.7倍)となる。同様に、NADPHの合成量は、限定はされないが、野生型のものに比べ、例えば、1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.5倍以上(例えば約1.7倍)となる。
さらに、得られた形質転換高等植物においては、ATP及びNADPHの合成量が増加することによって、これらがエネルギー源又は還元力となって働く、光合成暗反応の効率も向上したものとなる。例えば、形質転換高等植物は、二酸化炭素(CO2)を炭水化物に変換する炭素固定能が、野生型のものに比べ、例えば、1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.4倍以上となる。
さらに、得られた形質転換高等植物においては、光合成明反応・暗反応の効率が促進されたことによりタンパク質合成の効率も向上したものとなる。例えば、形質転換高等植物は、タンパク質の含量が、例えば1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.5倍以上となる。
また、硫酸および硝酸代謝や各種のアミノ酸合成、脂質合成量、色素ならびに花の増加なども期待できる。
なお、得られた形質転換高等植物においては、上述した各種作用効果を少なくとも1種有していることが好ましいが、より好ましくは2種以上、さらに好ましくは全て有していることである。
3.高等植物の生長促進方法、ATP、NADPH、デンプン及びタンパク質の合成促進方法、並びに炭素固定促進方法
前述したように、得られた形質転換高等植物においては、(i) 生長促進、(ii) ATP、NADPH、デンプン及びタンパク質の合成促進、並びに (iii) 炭素固定促進といった効果が認められる。よって、本発明は、高等植物の生長促進方法、高等植物のATP、NADPH、デンプン及びタンパク質からなる群から選ばれる少なくとも1つの合成促進方法、並びに高等植物の炭素固定促進方法をも含むものである。
これら方法は、具体的には、シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入して発現させ、葉緑体のチラコイド内腔にシトクロムc6を存在させることを特徴とするものである。これら方法において、融合タンパク質、当該タンパク質をコードする遺伝子、当該遺伝子の高等植物ゲノム中への導入方法、及び、得られる形質転換高等植物における種々の作用効果等については、前述した本発明の高等植物の作出方法における説明及び例示等が同様に適用できる。
以下に、実施例を挙げて本発明をより具体的に説明するが、本発明はこれらに限定されるものではない。
【実施例1】
【0006】
導入用シトクロムc 6遺伝子と植物発現用ベクターの構築
(1) Porphyra yezoensis由来シトクロムc6成熟タンパク領域遺伝子の調製
本実施例では、P. yezoensis由来シトクロムc6遺伝子をクローニングすることにより入手し、ベクターへと連結しプラスミドを作製した。このプラスミドを鋳型として、今回の目的DNAであるP. yezoensis 由来シトクロムc6成熟タンパク領域遺伝子を増幅させた。その後電気泳動を行い、ゲル抽出により目的遺伝子を抽出した後に制限酵素(Sac I, Pst I)で消化することで、遺伝子を調製した。
反応液は、以下の組成となるように0.5 ml アシストPCR用チューブに調製した。
【表1】

Primer(1):PYC6-C-term Sac I(10 pmol/μl)(30 mer, GC-Cont.:43.3%)
5’-GGA GCT CTT ACC AAC CTT TTT CAG ATT GAG - 3’(配列番号7)
Primer(2):Cyt-SD (10 pmol/μl)(29 mer, GC-Cont.:48.2%)
5’-CCG CGG AGA CGT TAA ATT GAA GAA GAA GC- 3’(配列番号8)
上記反応液にミネラルオイルを15μl重層した。その後、PTC-100TM Programmable Thermal Controllerを用いて反応を行った。反応は、94℃で48秒、62℃で48秒、72で1分24秒の反応を1サイクルとして、これを31サイクル行った。
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。
得られた PCR生成物20 μlにゲルローディング緩衝液4 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
2%(w/v)アガロースゲルから目的DNAと思われるバンドを切り出し、できるだけ細かく刻んだ。そして、そのゲルをAmicon Ultra free(登録商標) DAに入れ、7,300 rpm, 4℃, 10 min.遠心分離を行った。遠心後Ultra free MC(上側のカラム部分)を取りはずし、溶出してきた溶液に等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3 M NaOAc(pH 5.2)を1/20 vol.、99.5%エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75%エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、50 μlのTEに溶解した。
次に、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 120 min.インキュベートすることで制限酵素SacIによる消化を行った。
【表2】
インキュベート後のサンプル溶液に、等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5%エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75%エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
続いて、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 120 min.インキュベートすることで制限酵素 PstIによる消化を行った。
【表3】
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物20 μlにゲルローディング緩衝液4 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。その後、2%(w/v)アガロースゲルから目的DNAと思われるバンドを切り出し、できるだけ細かく刻んだ。そして、そのゲルをAmicon Ultra free(登録商標) DAに入れ、7,300 rpm, 4℃, 10 min.遠心分離を行った。遠心後Ultra free MC(上側のカラム部分)を取りはずし、溶出してきた溶液に等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75%エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
結果を以下に示す。
P. yezoensis由来シトクロムc6遺伝子(全長)が挿入されているプラスミドを鋳型とし、シトクロムc6成熟タンパク領域(PYC6)遺伝子をPCRにより増幅させた(図4)。その後PCR産物を2%(w/v)アガロースゲル(TBE)電気泳動を行ったところ、357 bp付近に単一なバンドを確認することができた(図5)。このバンドがPYC6遺伝子であると考えられた。そこでこのPYC6遺伝子を2つの制限酵素(Sac I, Pst I)で消化し、2%(w/v)アガロースゲル(TBE)電気泳動し、PYC6遺伝子(Sac I, Pst Iサイト保有)と思われる265 bp付近のバンドをゲルから抽出し、フェノール抽出、エタノール沈殿を行うことで、Sac I, Pst Iサイトを保有したPYC6遺伝子を得た。
(2) シトクロムc6への葉緑体包膜及びチラコイド膜通過ペプチド遺伝子の付加
本項では、BamH IとPst Iによる制限酵素処理と脱リン酸化(BAP処理)を行った葉緑体包膜およびチラコイド膜通過ペプチド遺伝子と、Sac IとPst Iによる制限酵素処理を行った P. yezoensis由来シトクロムc6成熟タンパク領域遺伝子をライゲーションさせた。その後クローニングベクター(pBluescript(登録商標) II SK+)にライゲーションさせ、サブクローニングした後、植物導入用シトクロムc6(sp+PYC6)遺伝子の塩基配列を確認した。なお、下記のユニバーサルプライマーをDNAシーケンスの際に使用した。
ユニバーサル FITC化 Forward primer(M13 Fw primer)(2 pmol/μl)
5’- CGC CAG GGT TTT CCC AGT CAC GAC - 3’(配列番号9)
ユニバーサル FITC化 Reverse primer(M13 Rv primer)(2 pmol/μl)
5’- GAG CGG ATA ACA ATT TCA CAC AGG - 3’(配列番号10)
(2-1) 植物導入用シトクロムc6遺伝子の調製
以下の組成になるように反応液を調製した。
【表4】
調製した反応液を低温恒温槽(16℃)で一晩インキュベートし、ライゲーション反応させた。次に、この溶液にアルカリフォスファターゼを加え、脱リン酸化を行った。以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 3 hrs.インキュベートすることでBacterial Alkaline Phosphataseによる脱リン酸化を行った。
【表5】
インキュベート後のサンプル溶液に、等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
(2-2) クローニングベクターpBluescript(登録商標)II SK+の調製
以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 5 hrs.インキュベートすることで制限酵素 SacIによる消化を行った。
【表6】
インキュベート後のサンプル溶液に、等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
続いて、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 5 hrs.インキュベートすることで制限酵素BamH Iによる消化を行った。
【表7】
反応終了後、1.5%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物20 μlにゲルローディング緩衝液4 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TAE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。その後、1.5%(w/v)アガロースゲルから目的DNAと思われるバンドを切り出し、ゲル100 μgを100 μlに換算し、それに対しNaI solutionを3 vol.加えよく撹拌し、55℃で5分インキュベートしゲルを溶解させた。これにGLASSMILKを10 μl加え、室温で15分間激しく撹拌し、14,000 rpm, 4℃, 5 sec.遠心分離を行い、沈殿を回収した。この沈殿にNEW WASH 300 μlを加え、よく撹拌後、14,000 rpm, 4℃, 5 sec.遠心分離を行い、沈殿を回収した。この操作を3度繰り返して得られた沈殿を乾燥させ、20 μlの滅菌水に溶解させた。その後、溶液を0.5 ml アシストPCR用チューブへと移し、14,000 rpm, 4℃, 30 sec.遠心分離を行った。得られた上清を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
(2-3) 植物導入用シトクロムc6遺伝子のサブクローニング
シトクロムc6遺伝子のサブクローニングは、公知方法に従って行った(Hanahan, D., J. Mol. Biol., 166(4), 557-580 (1983).; 中山 広樹等 バイオイラストレイテッドII 遺伝子解析の基礎, 秀潤社, 83-88 (1996).)。
植物導入用遺伝子と、クローニングベクターを以下の組成になるように調製した。
【表8】
調製した反応液を低温恒温槽(16℃)で一晩インキュベートし、ライゲーション反応させた。大腸菌への形質転換は、以下の通りに行った。まず、150 μlのコンピテントセル(DH5a)を氷上である程度溶かし、そこにライゲーションさせた反応液4 μlを穏やかに加え、チップの先で軽くかき混ぜ、氷上で30分間放置した。放置終了後、42℃で20秒間ヒートショックを行った。その後氷上で3分間静置し、LB- Ampicillin-X-Gal-IPTG プレートにVector : Insert = 1:1、1:3それぞれ、菌体液100 μl、50μlをコンラージ棒を用いて、プレート全体に塗り広げ、37℃で一晩培養した。
(2-4) アルカリSDS法によるプラスミド調製
アルカリSDS法は、公知手法に従って行った(Weiss. B, et al., J. Biol. Chem., 243, 4543-4555 (1968); Birnboim, H. C., Methods Enzymol., 100, 243 (1983))。すなわち、3ml LB-Ampicillin 液体培地で200 rpm, 37℃, 16 hrs.震盪培養した菌体液を1.5 mlずつ1.5 ml micro centrifuged tubeに分注し、6,000 rpm, 4℃, 10 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。沈殿(菌体)に100 μlのSolution I(50 mM グルコース-25 mM Tris-HCl(pH 8.0)-10 mM EDTA)を加え、ボルテックスでよく撹拌し、200 μlのSolution IIを加え、静かに転倒させ撹拌した。5分間室温で放置後、さらに150 μlのSolution III(0.2N NaOH-1% SDS)を加え、氷上で30分間放置後、100 μlのクロロホルムを加え、14,000 rpm, 4℃, 10 min.遠心分離した。上清を新しいチューブに回収し、それに2倍量の99.5% エタノールを加え、静かに転倒させ撹拌し、15分間室温で放置した。その後、500 μlの70% エタノールを加え14,000 rpm, 4℃, 10 min.遠心分離し、得られた沈殿を3〜5 分間TOMY MICRO Vacで乾燥させ、20 μlのTEに溶かした。
得られたプラスミドに目的DNA が組み込まれていることを確認するために、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 120 min.インキュベートすることで制限酵素SacI, BamH Iによる消化を行った。
【表9】
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。この結果、目的DNAが組み込まれているサンプルに関してのみ、3 ml LB-Ampicillin液体培地での培養の残りの菌体液(1.5 ml)から、100 μlを5 ml LB-Ampicillin液体培地に植菌し、200 rpm, 37℃, 16 hrs.震盪培養した。
(2-5) QIAGEN Spin Miniprep Kitによるプラスミドの調製
5 ml LB-Ampicillin液体培地で培養した菌体液を6,000 rpm, 4℃, 10 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。沈殿(菌体)にキット(QIAGEN Spin Miniprep Kit(250)(QIAGEN))に含まれているBuffer P1を250 μl加え、十分に懸濁させた。
なお、QIAGEN Spin Miniprep Kit(250)(QIAGEN)にはCollection tube、QIA prep Spin Column、Buffer P1、Buffer P2、Buffer N3、Buffer PB、Buffer PE、Buffer EBが含まれている。
上記懸濁液に、Buffer P2を250 μl加え、静かに転倒させ溶液が均一になるまで撹拌した。さらにBuffer N3を350 μl加え静かに転倒させ撹拌し、13,000 rpm, 4℃, 10 min.遠心分離した。ここで得られた上清をあらかじめCollection tubeにセットしておいたQIA prep spin columnに移し、13,000 rpm, 4℃, 1 min.遠心分離した。Collection tubeにたまった濾液を捨て、QIA prep spin columnに、500 μlのBuffer PBを加え、13,000 rpm, 4℃, 1 min.遠心分離し、再び濾液を捨てた。さらにQIA prep spin columnにBuffer PE を750 μl加え、13,000 rpm, 4℃, 1 min.遠心分離し濾液を捨て、再び13,000 rpm, 4℃, 1 min.遠心分離し、QIA prep spin columnのみを新しい1.5 ml micro centrifuged tubeに移した。QIA prep spin columnの中央に50 μlのBuffer EBを加え、室温で1分間放置し、13,000 rpm, 4℃, 1 min.遠心分離し、この濾液をプラスミドサンプルとした。
得られたプラスミドに目的DNAが組み込まれていることを確認するために、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃、120 min. インキュベートすることで制限酵素 SacI, BamH Iによる消化を行った。
【表10】
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。ここで目的DNAが組み込まれており、単一性が確認されたものをシークエンスサンプルとした。
(2-6) ダイデオキシ法による塩基配列の決定
塩基配列の決定は、オートシークエンサーを用いて公知方法(Sanger, F., Sanger, F., Determination of nucleotide sequence in DNA., Science, 214, 1205-1210 (1981))により決定した。塩基配列決定後は、解析ソフトであるGENETYX-MACを用いてデータの分析を行った。
(2-7) 結果
2つの制限酵素(BamH I, Pst I)消化と脱リン酸化(BAP 処理)を行ったsp遺伝子を、2つの制限酵素(Sac I, Pst I)消化を行ったPYC6遺伝子とライゲーションさせ、さらに再度BAP処理を行い、フェノール抽出、エタノール沈殿により植物導入用シトクロムc6(sp+PYC6)遺伝子を作製することができた。
このsp+PYC6遺伝子の塩基配列を確認するため、クローニングベクター(pBluescript II SK+(図6)を2つの制限酵素(BamH I, Sac I)で消化したものにライゲーションし、サブクローニング後、ダイデオキシ法によるシークエンスを行い、sp+PYC6遺伝子の全塩基配列474bpを決定した(図7)。
そのシークエンス結果をGENETYX-MACを用いて既知のsp遺伝子とPYC6遺伝子と比較したところ、シグナルペプチド配列中の103番目のT(チミン塩基)がA(アデニン塩基)に変異している以外は完全に一致した。またPrimer配列中に付加した制限酵素サイトも確認した(図7)。今回確認された塩基の変異は、コードするアミノ酸をSerからThrに変えるものであったが、ともに性質と分子量も類似しており、それによる二次構造(aヘリックスなど)の変化はなく、膜通過というシグナルペプチドとしての機能には影響がないと考えられたため、このサンプル(sp+PYC6)を以後の実験に用いた
(3) 植物導入用シトクロムc6発現ベクターの作製
本実施例では、塩基配列を確認した植物導入用シトクロムc6(sp+PYC6)遺伝子を発現ベクターpBI121に挿入し、植物へ導入用シトクロムc6発現ベクター(pBI cyt c6)を構築した。
(3-1) 植物発現用ベクターpBI121の調製
以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、 37℃, 5 hrs.インキュベートすることで制限酵素SacIによる消化を行った。
【表11】
インキュベート後のサンプル溶液に、等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
続いて、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tube に調製し、 37℃, 5 hrs. インキュベートすることで制限酵素BamH Iによる消化を行った。
【表12】
反応終了後、1.5%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物20 μlにゲルローディング緩衝液4 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TAE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。その後、1.5%(w/v)アガロースゲルから目的DNAと思われるバンドを切り出し、ゲル100 μgを100 μlに換算し、それに対しNaI solutionを3 vol.加えよく撹拌し、55℃で5分インキュベートしゲルを溶解させた。これにGLASSMILKを10 μl加え、室温で15分間激しく撹拌し、14,000 rpm, 4℃, 5 sec.遠心分離を行い、沈殿を回収した。この沈殿にNEW WASH 300 μlを加え、よく撹拌後、14,000 rpm, 4℃, 5 sec.遠心分離を行い、沈殿を回収した。この操作を3度繰り返して得られた沈殿を乾燥させ、20 μlの滅菌水に溶解させた。その後、溶液を0.5 ml アシストPCR用チューブへと移し、14,000 rpm, 4℃, 30 sec.遠心分離を行った。得られた上清を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
(3-2) 植物発現用シトクロムc6発現ベクター(pBI cyt c6)の調製
前記の通り作製した導入用シトクロムc6(sp+PYC6)遺伝子は、以下の組成になるように反応液を調製した。
【表13】
調製した反応液を低温恒温槽(16℃)で一晩インキュベートし、ライゲーション反応させた。
結果を以下に示す。
塩基配列を確認したsp+PYC6遺伝子(BamH I, Sac I消化、BAP処理済み)を、植物発現用ベクター(pBI121)を2つの制限酵素(BamH I, Sac I)で消化したものにライゲーションし、植物導入用シトクロムc6発現ベクター(pBI cyt c6)を調製することができた。
【実施例2】
【0007】
形質転換植物(シトクロムc 6導入Arabidopsis thaliana)の作製
(1) 植物感染用Agrobacterium tumefaciensの調製
(1-1) シトクロムc6発現ベクター(pBI cyt c6)導入大腸菌の調製
本実施例では、植物導入用シトクロムc6遺伝子を挿入した植物発現用ベクター(pBI cyt c6)を宿主大腸菌 HB101に形質転換した。その後、数個のコロニーを選び3 mlのLB-Kanamycin 液体培地で培養し、アルカリSDS法によるプラスミド調製を行うことでベクターの構築と宿主大腸菌HB101への形質転換の有無を確認した。
大腸菌への形質転換は、以下の通りに行った。まず、150 μlのコンピテントセル(HB101)を氷上である程度溶かし、そこにライゲーションさせた反応液4 μlを穏やかに加え、チップの先で軽くかき混ぜ、氷上で30分間放置した。放置終了後、42℃で20秒間ヒートショックを行った。その後氷上で3分間静置し、LB-Kanamycinにそれぞれ菌体液 100 μl、50μlをコンラージ棒を用いて、プレート全体に塗り広げ、37℃で一晩培養した。
培養後、形質転換体(コロニー)をランダムに20クローンピックアップし、そのコロニーを3 ml LB-Kanamycin培地に植菌し、200 rpm, 37℃, 16 hrs.震盪培養した。培養した菌体液を1.5 mlずつ1.5 ml micro centrifuged tubeに分注し、6,000 rpm, 4℃, 10 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。沈殿(菌体)に100 μlのSolution I(前出)を加え、ボルテックスでよく撹拌し、200 μlのSolution II(0.2 N NaOH-1% SDS)を加え、静かに転倒させ撹拌した。5分間室温で放置後、さらに150 μl の Solution III(前出)を加え、氷上で30分間放置後、100 μlのクロロホルムを加え、14,000 rpm, 4℃, 10 min.遠心分離した。上清を新しい回収し、それに 2 倍量の99.5% エタノールを加え、静かに転倒させ撹拌し、15分間室温で放置した。その後、500 μlの70% エタノールを加え14,000 rpm, 4℃, 10 min.遠心分離し、得られた沈殿を3〜5分間 TOMY MICRO Vacで乾燥させ、20 μlのTEに溶かした。
得られたプラスミドに目的DNAが組み込まれていることを確認するために、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 120 min. インキュベートすることで制限酵素BamHIと SacIによる消化を行った。
【表14】
反応終了後、1.5%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TAE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
結果を以下に示す。
植物導入用シトクロムc6発現ベクター(pBI cyt c6)を、宿主大腸菌HB101へと形質転換させた(図8)。その後、形質転換体(コロニー)をランダムに20クローンピックアップした。その結果、20サンプル中 3クローンにsp+PYC6遺伝子と思われるサイズ(474 bp)付近に単一なバンドを確認することができた。よってこのプラスミド内にはsp+PYC6遺伝子が挿入されていることが示された。このプラスミドを保有する大腸菌HB101を以後の実験に用いた。
(1-2) 形質転換Agrobacterium tumefaciensの調製と選抜
本項では、高等植物 A. thalianaへと目的遺伝子を導入するために必要な宿主アグロバクテリウム(Agrobacterium tumefaciens)を調製するため、三者混合培養(トリペアレンタルメイティング)を行い、得られた形質転換体のうち、数個のコロニーを選び3 mlのLB-Kanamycin液体培地で培養し、アルカリSDS法によるプラスミド調製を行うことで宿主アグロバクテリウムへの形質転換の有無を確認した。
(1-2-1) 各種菌体の培養
(a) Agrobacterium tumefaciens(LBA4404)*pAL4404保有
菌体のグリセロールストック溶液をYEP-Streptomycin寒天培地(プレート)(30 μg/ml Streptomycin)に白金耳を用いて、プレート全体に線画状に塗り、30℃でおよそ 40 hrs.培養した。培養後、コロニーをピックアップし、そのコロニーを5 ml YEP-Streptomycin(30 μg/ml Streptomycin)培地で200 rpm, 30℃, 30 hrs.震盪培養した。
(b) Escherichia coli(HB101)*pRK2013保有
菌体のグリセロールストック溶液をLB-Kanamycin 寒天培地(プレート)(40 μg/ml Kanamycin)に白金耳を用いて、プレート全体に線画状に塗り、37℃で16 hrs.培養した。培養後、コロニーをピックアップし、そのコロニーを5 ml LB-Kanamycin(40 μg/ml Kanamycin)培地で200 rpm, 37℃, 16 hrs.震盪培養した。
(c) Escherichia coli(HB101)*pBI cyt c6 保有
菌体のグリセロールストック溶液をLB-Kanamycin 寒天培地(プレート)(40 μg/ml Kanamycin)に白金耳を用いて、プレート全体に線画状に塗り、37℃で16 hrs.培養した。培養後、コロニーをピックアップし、そのコロニーを5 ml LB-Kanamycin(40 μg/ml Kanamycin)培地で200 rpm, 37℃, 16 hrs.震盪培養した。
(1-2-2) トリペアレンタルメイティング
トリペアレンタルメイティングは、公知方法により行った(駒嶺 穆, 野村 港二, 「生物化学実験法 41」植物細胞工学入門, 学会出版センター, 298-302 (1998))。すなわち、5 mlのYEP-Sm.液体培地あるいはLB-Km.液体培地で震盪培養した三種の菌体液(A. tumefaciens(LBA4404)*pAL4404保有、E. coli(HB101)*pBI cyt c6 保有、E. coli(HB101)*pRK2013保有)を 5,000 rpm, 4℃, 5 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。洗浄のため培養時に使用した液体培地(YEP 培地あるいは LB 培地)を沈殿(菌体)に加えよく撹拌し、もう一度 5,000 rpm, 4℃, 5 min.遠心分離した。遠心分離後、菌体を培養時に使用した液体培地(YEP培地あるいはLB培地)3 mlに溶解させた。次にそれぞれの菌体液を30 μlずつMin A-Kanamycin寒天培地(プレート)に重ねるように滴下し、そのまま静置した状態で30℃, 3 days培養した。培養後、最少培地である Min A-Kanamycin 寒天培地(プレート)に生えてきた菌体を白金耳を用いてすべてかき取り、その菌体を10 mM MgSO4 200 μlに溶解させた。その後、その菌体溶液を再び Min A-Kanamycin 寒天培地(プレート)に白金耳を用いて、プレート全体に線画状に塗り、30℃でおよそ3 days.培養した。培養後、コロニーをピックアップし、そのコロニーを5 ml YEP-Kanamycin(40 μg/ml Kanamycin)培地で 200 rpm, 30℃, 40 hrs.震盪培養した。
(1-2-3) アルカリSDS法によるTiプラスミド調製
5 ml YEP-Kanamycin(40 μg/ml Kanamycin)液体培地で200 rpm, 30℃, 40 hrs.震盪培養した菌体液を1.5 mlずつ1.5 ml micro centrifuged tubeに分注し、5,000 rpm, 4℃, 10 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。1 mlのS-bufferを沈殿(菌体)に加え、ボルテックスでよく撹拌し、8,000 rpm, 4℃, 10 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。
なお、S-bufferは、C6H12O6(グルコース(M.W. : 180.16))を13.512 g秤取り、1M Tris-HCl(pH 8.0)12.5 ml、0.5M EDTA(pH 8.0)10 ml加え、超純水で500 mlに定容し、オートクレーブ処理(121℃、10分)を行うことにより調製した。
沈殿に再び100 μlのS-bufferと10 mg/ml リゾチームを5 μl加え、10分間室温で放置した。そこに200 μl Solution II、TE-saturated Phenolを30 μl加え、静かに転倒させ撹拌した。5分間室温で放置後、さらに150 μlのSolution IIIを加え、-20℃で15分放置した後、12,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに 14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
得られたプラスミドに1 mlのRNase Aを加え、37℃, 90 min.インキュベートすることでRNase処理を行った。反応終了後、1.5%(w/v)アガロースゲル電気泳動にて確認を行った。得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TAE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
結果を以下に示す。A. tumefaciens(LBA4404)はYEP-Sm.培地で培養し、E. coli(HB101)はLB-Km.培地で培養した。その後、トリペアレンタルメイティング(三者混合培養)によりA. tumefaciens(LBA4404)へpBI cyt c6を導入し、形質転換させた。次にトリペアレンタルメイティングによりTiプラスミド(pBI cyt c6)が導入された形質転換体A. tumefaciensをランダムに10クローンピックアップした。そのコロニーを3 ml YEP-Km.培地にて培養し、その後、アルカリSDS法によってプラスミドを調製し、これを1%(w/v)アガロースゲル(TAE)電気泳動した。その結果、1クローンにおいて、pBI cyt c6と思われるサイズ(13.5 kbp)にバンドを確認することができた。よって形質転換体A. tumefaciensにはTiプラスミド(pBI cyt c6)が導入されていることが示された。
(2) 形質転換植物(シトクロムc6導入Arabidopsis thaliana)の作製
(2-1) Arabidopsis thalianaの栽培
よく水洗いした7.5×7.5丸型ポットにバーミキュライトを九分目位まで入れた。次に、HYPONeX原液を2,000倍希釈した溶液を500 ml用意し、カッターで厚さ1 cmほどにスライスしておいたロックウールを入れた。十分に水分を含んで湿ったロックウールをピンセットでつまみ、7.5×7.5丸ポットの上に3つずつのせた。台所用三角コーナーのメッシュをポットの口を覆える位の大きさに切り、よく水洗いし、ポットの口にかぶせて輪ゴムでとめた。このポットに、種子を1粒ずつ播種した。種子を播種した後、4℃, 3 days低温処理を行った。低温処理終了後、植物育成用光照射インキュベーター内に移し、22℃・長日条件下で栽培した。
発芽後は2日に1度程度水を与え、週に1度程1,000倍希釈したHYPONeXを液体肥料として与えた。適当に間引きを行い、およそ3週間後植物体が抽台し始めたときに摘心し、竹串(支柱)を利用して成長した植物体を支え、種子が完全に熟したら採種し、乾燥状態で保存しておいた。
(2-2) 形質転換Agrobacterium tumefaciensの植物体への感染
次に、高等植物A. thaliana(col-0)に植物導入用シトクロムc6(sp+PYC6遺伝子)を導入するためアグロバクテリウム法の一つである減圧浸潤法により、形質転換植物を作製した(Bechtold, N., Pelletier, G., Methods in Molecular Biology, 82 (1998))。具体的には以下の通りである。
(2-2-1) Agrobacterium tumefaciens の培養
菌体のグリセロールストック溶液をMin A-Km.寒天培地(プレート)(400 μg/ml Kanamycin)に白金耳を用いて、プレート全体に線画状に塗り、30℃でおよそ40 hrs.培養した。培養後、コロニーをピックアップし、そのコロニーを10 ml YEP-Km.(40 μg/ml Kanamycin)培地で200 rpm, 30℃, 30 hrs.震盪培養(前培養)し、その培養液を新たに100 ml YEP-Km.(40 μg/ml Kanamycin)培地に全量加え、さらに200 rpm, 30℃, 30 hrs.震盪培養(本培養)した。
(2-2-2) 植物体 Arabidopsis thaliana(col-0)の栽培
形質転換させる植物として用いるA. thaliana(col-0)の播種から種子の採種までの栽培条件は以下のとおりである。
低温処理:4℃(暗所)で3日間静置
植物育成温度:22℃
照射光:長日条件(100 μE m-2 s-1)
液体肥料:HYPONeX(1/1,000 倍希釈)
植物個体によって多少差はあるが、Wild type株の場合、播種後7日(低温処理3日間を含める)で発芽し、発芽後20日前後で植物が抽台した。その後、摘心処理し5〜6日で側枝が伸長し始め、同時に最初の花の開花・結実が始まった。採種は緑色のさやが薄茶色になり縦裂し始めたものから回収した。一つのさやからおよそ50粒の種子が得られた。回収した種子は、播種まで乾燥させた状態で保存しておいた。
(2-2-3) 植物体への感染
感染用植物体Arabidopsis thaliana(col-0)を上記(2-2-2)に記載の方法に従って栽培し、抽台を始めたころまで生育させた。その後、茎から茎葉が誘導され始めたときに摘心し、すでに開花・結実している花を切除した。なお、実験は、この植物体と上記(2-2-1)の菌体培養液の準備が同時に整うよう調整した。
まず、浸潤操作を行う実験台にキムタオル敷いておいた。
次に、A. tumefaciens 培養液を浸潤用懸濁培地でおよそ2/3倍希釈し、1L容ビーカーに入れた。そのビーカーを吸引鐘内にセットし、そこへ培養懸濁液を吸引しすぎないよう直前に十分に吸水させた植物体A. thalianaを植えたポットごとビーカーの上に逆さにして固定し、植物体を培養懸濁液に浸けた。
減圧浸潤法で用いたA. tumefaciens培養液は、OD600=1.2〜1.5のものをOD600=0.8となるように浸潤用懸濁培地で約2/3倍希釈したものを使用した。また減圧浸潤時間は30秒、1分、5分、10分と検討した結果、5分が最も適していた。それは、減圧浸潤時間が長くなるほどその後の植物体の生育は悪くなったが、5分間の処理の場合、それほど生育の悪化は見られず、花数及び結実したさや(種子)数も1分間処理したものと大差がなかったためである。
次に、培養懸濁液に浸した植物体を吸引鐘に入れ、アスピレーターで減圧した。減圧終了後、吸引鐘から植物体を取り出し、キムタオルで余分な培養懸濁液を落とした。その植物体をプラスチックトレイに横倒しにして、端に水を滴下し、ふたをして20℃で2〜3日放置した(その間は水を与えなかった)。その後、ポットをおこし、植物育成用光照射インキュベーター内で22℃、長日条件で通常どおり栽培を行った。2日に1度程度水やりを行い、週に1度程1,000 倍希釈したHYPONeXを液体肥料として与えた。適当に間引きを行い、およそ3週間後植物体が抽台し始めたら摘心し、竹串(支柱)を利用して成長した植物体を支え、種子が完全に熟したときは採種し、乾燥状態で保存しておいた。
【実施例3】
【0008】
形質転換植物におけるシトクロムc 6遺伝子導入と発現の解析
(1) PCR法を用いたゲノムDNA中の導入遺伝子の解析
本実施例では、形質転換体Arabidopsis thalianaよりゲノムDNAを抽出し、そのゲノムDNAを鋳型としてPCRにより導入遺伝子の増幅の有無をもって植物体内への導入を確認した。
(1-1) 形質転換体からゲノムDNAの抽出
葉2〜3枚程度の形質転換体Arabidopsis thalianaをあらかじめ冷却した乳鉢の中に入れ、素早く液体窒素を加え、植物体がパウダー状になるまでホモジナイズした。その後、パウダー状になった試料を1.5 ml micro centrifuged tubeに入れ秤量した。Wash bufferに終濃度0.5%ととなるように2-Mercaptoethanolを加えた溶液を、上記秤量後の試料に1 ml加えよく混ぜた。12,500 rpm, 4℃, 10 min.遠心分離後、上清を捨てた。そこへSolution Iに終濃度1%となるように2-Mercaptoethanolを加えた溶液を300 μl加えボルテックスでよく混ぜた。次に、1% NaBH4を30 μl、Solution IIを150 μl加え、数秒ボルテックスにかけた。50℃で10分インキュベートし、その溶液にISOPLANT II(NIPPON GENE)に含まれる100 μlのSolution III-Aと120 μlのSolution III-Bを入れ、氷上で10分間放置した。
12,500 rpm, 4℃, 10 min.遠心分離し、水相を回収した。そこへ2倍量の99.5% エタノールを加え、直ちに8,000 rpm, 室温, 1 min遠心分離し、上清を捨てた。さらに70% エタノールを1 ml加え8,000 rpm, 室温, 1 min遠心分離し、上清を捨てた。沈殿物をある程度まで風乾させ、50 μlのTEに溶解させた。この溶液に1 mg/ml RNase Aを1 μl加え、37℃で30分間インキュベートした。その後、ゲノムDNAの抽出を確認するために、得られたゲノムDNA5 μlにゲルローディング緩衝液1 μlを加え、マーカーにλHindIIIを用いて1.5%(w/v)アガロースゲルにて電気泳動を行った。泳動は泳動緩衝液(1×TAE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
電気泳動によるゲノムDNA 存在の確認後、その濃度測定を行うためゲノムDNA溶液を100倍希釈し、分光光度計でUV測定した。算出された濃度をもとに10 ng/μlとなるようにゲノムDNA溶液を調製し、今後の実験に用いた。
(1-2) PCR
PCRは、公知方法に従って行った(Sambrook, J. D., W. Russell, Molecular Cloning : a Laboratory Manual, 3rd edn. Corld Spring Harbor Laboratory Press, Corld Spring Harbor, New York)。
以下の組成となるように反応液を0.5 ml アシストPCR用チューブに調製した。
【表15】
この反応液にミネラルオイルを15 μl重層した。その後、PTC-100TM Programmable Thermal Controllerを用いて反応を行った。
プライマーとして以下のものを用いた。
Primer(3) ATP-NA1 Bam(10 pmol/μl)(31mer, GC-Cont.:54.8%)
5’-GGA TCC ATG GCC GCA ATT ACA TCA GCT ACC G-3’(配列番号11)
Primer(1) PYC6-C-term SacI(10 pmol/μl)(30mer, GC-Cont.:43.3%)
5’- GGA GCT CTT ACC AAC CTT TTT CAG ATT GAG - 3’(配列番号7)
ユニバーサル FITC化 Forward primer(M13 Fw primer)(2 pmol/μl)
5’- CGC CAG GGT TTT CCC AGT CAC GAC - 3’(配列番号9)
ユニバーサル FITC化 Reverse primer(M13 Rv primer)(2 pmol/μl)
5’- GAG CGG ATA ACA ATT TCA CAC AGG - 3’(配列番号10)
反応は、(94℃で48秒、64℃で48秒、72℃で1分24秒)×31サイクルの条件で行った。
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
(1-3) PCR産物のゲル抽出
2%(w/v)アガロースゲルから、目的DNAと思われるバンドを切り出し、できるだけ細かく刻んだ。そして、そのゲルをAmicon Ultra free (登録商標) DAに入れた。7,300 rpm, 4℃, 10 min.遠心分離を行った。遠心後Ultra free MC(上側のカラム部分)を取りはずし、溶出してきた溶液に等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを 2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解させた。この溶液に目的のDNAが単一に抽出・精製されているかを確認するため、2%(w/v)アガロースゲル電気泳動を行った。得られたサンプル10 μlにゲルローディング緩衝液2 μlを加え、泳動には泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
(1-4) PCR 産物のサブクローニング
PCR産物のサブクローニングは、公知手法に従って行った(Hanahan, D., J. Mol. Biol., 166(4), 557-580 (1983).; 中山 広樹等 バイオイラストレイテッドII 遺伝子解析の基礎, 秀潤社, 83-88 (1996).)。
PCR 生成物に以下の組成になるように調製した。
【表16】
調製した反応液を低温恒温槽(16℃)で一晩インキュベートし、ライゲーション反応させた。大腸菌への形質転換は、以下の通りに行った。まず、150 μlのコンピテントセル(DH5a)を氷上である程度溶かし、そこにライゲーションさせた反応液 4 μlを穏やかに加え、チップの先で軽くかき混ぜ、氷上で30分間放置した。放置終了後、42℃で20秒間ヒートショックを行った。その後氷上で3分間静置し、LB-Ampicillin-X-Gal-IPTGプレートにVector : Insert = 1:1、1:3それぞれ、菌体液100 μl、50μlをコンラージ棒を用いて、プレート全体に塗り広げ、37℃で一晩培養した。
(1-5) アルカリSDS法によるプラスミド調製
本項におけるプラスミド調製は、実施例1(2-4)に記載した内容と同様にして行った。
(1-6) QIAGEN Spin Miniprep Kitによるプラスミドの調製
本項におけるプラスミド調製は、実施例1(2-5)に記載した内容と同様にして行った。
(1-7) ダイデオキシ法による塩基配列の決定
本項における塩基配列の決定は、実施例1(2-6)に記載の方法と同様にして行った。結果を以下に示す。
形質転換体A. thalianaの葉2〜3枚ほどから、ISOPLANT II を用いて純度の高いゲノムDNAを抽出した。そのゲノムDNA溶液を鋳型とし、植物導入用シトクロムc6遺伝子の導入の有無を確認するためPCRにより目的遺伝子を増幅させた。その後PCR産物を2%(w/v)アガロースゲル(TBE)電気泳動を行ったところ、474 bp付近に単一なバンドを確認した(図9)。この増幅した遺伝子の塩基配列を確認するため、ゲル抽出後、サブクローニングを行い、ダイデオキシ法によるシークエンスにより増幅した遺伝子の全塩基配列474 bpを決定した。そのシークエンス結果をGENETYX-MACを用いて既知のsp遺伝子配列とPYC6遺伝子配列と比較したところ、導入遺伝子同様シグナルペプチド遺伝子配列中の103番目のT(チミン塩基)がA(アデニン塩基)に変異している以外は完全に一致した。以上の結果より植物体(A. thaliana)内にsp+PYC6遺伝子が導入されたことが示された。
(2) RT-PCR法を用いた全RNA中の発現遺伝子の解析
本項では、形質転換体Arabidopsis thalianaより全RNAを抽出し、その全RNAを鋳型としてRT-PCRにより導入遺伝子の増幅の有無をもって植物体内での遺伝子の発現を確認した。
Primer(3) ATP-NA1 Bam (10 pmol/μl)(31 mer, GC-Cont.:54.8%)
5’- GGA TCC ATG GCC GCA ATT ACA TCA GCT ACC G-3’(配列番号11)
Primer(1) PYC6-C-term SacI (10 pmol/μl)(30 mer, GC-Cont.:43.3%)
5’- GGA GCT CTT ACC AAC CTT TTT CAG ATT GAG - 3’(配列番号7)
ユニバーサル FITC化 Forward primer(M13 Fw primer)(2 pmol/μl)
5’- CGC CAG GGT TTT CCC AGT CAC GAC - 3’(配列番号9)
ユニバーサル FITC化 Reverse primer(M13 Rv primer)(2 pmol/μl)
5’- GAG CGG ATA ACA ATT TCA CAC AGG - 3’(配列番号10)
(2-1) 形質転換体から 全RNAの抽出
形質転換体から全RNAの抽出は、市販のキット(RNeasy Plant Mini Kit(QIAGEN))を用い、説明書に従って行った。
(2-2) PCR
PCRは、公知手法に従って行った(Sambrook, J. D., W. Russell, Molecular Cloning : a Laboratory Manual, 3rd edn. Corld Spring Harbor Laboratory Press, Corld Spring Harbor, New York)。
以下の組成となるように反応液を0.5 ml アシストPCR用チューブに調製した。
【表17】
この反応液にミネラルオイルを15 μl重層した。その後、PTC-100TM Programmable Thermal Controllerを用いて反応を行った。
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
(2-3) PCR産物のゲル抽出
本項におけるPCR産物のゲル抽出は、実施例3(1-3)と同様にして行った。
(2-4) PCR 産物のサブクローニング
本項におけるPCR産物のサブクローニングは、実施例3(1-4)に記載の方法と同様にして行った。
(2-5) アルカリSDS法によるプラスミド調製
本項におけるアルカリSDS法は、公知手法に従って、実施例1(2-4)に記載の方法と同様に行った(Weiss. B, et al., J. Biol. Chem., 243, 4543-4555 (1968); Birnboim, H. C., Methods Enzymol., 100, 243 (1983))。
(2-6) QIAGEN Spin Miniprep Kitによるプラスミドの調製
本項におけるQIAGEN Spin Miniprep Kitによるプラスミドの調製は、実施例1(2-5)に記載の方法と同様に行った。
(2-7) ダイデオキシ法による塩基配列の決定
本項における塩基配列の決定は、実施例1(2-6)に記載の方法と同様にして行った(Sanger, F., Sanger, F., Determination of nucleotide sequence in DNA., Science, 214, 1205-1210 (1981))。
(2-8) 結果
遺伝子の導入が確認された形質転換植物(PYC6導入植物)の葉2〜3枚ほどから、RNeasy Plant Mini Kitを用いて発現している全RNAを抽出した。その全RNA溶液を鋳型とし、sp+PYC6遺伝子の発現の有無を確認するためRT-PCRにより目的遺伝子を増幅させた。その後PCR産物を2%(w/v)アガロースゲル(TBE)電気泳動を行ったところ474 bp付近に単一なバンドを確認した(図9)。この増幅した遺伝子の塩基配列を確認するため、ゲル抽出後、サブクローニングを行い、ダイデオキシ法によるシークエンスにより増幅した遺伝子の全塩基配列474 bpを決定した。そのシークエンス結果をGENETYX-MACを用いて既知のsp遺伝子配列とPYC6遺伝子配列と比較したところ、導入遺伝子同様シグナルペプチド遺伝子配列中の103番目のT(チミン塩基)がA(アデニン塩基)に変異している以外は完全に一致した。以上の結果より植物体(A. thaliana)内にsp+PYC6遺伝子が発現していると考えられた。
(3) ウエスタンブロッティング法を用いた発現タンパク質の解析
本項では、形質転換体Arabidopsis thalianaより葉緑体を抽出し、そこから得られた葉緑体タンパク質画分に導入したシトクロムc6が発現していることを確認した。
(3-1) 葉緑体画分の調製
葉緑体画分は公知手法に従って調製した(中村 研三等(監修),植物のタンパク質実験プロトコール, 細胞工学シリーズ第9巻, 秀潤社, 114-117 (1998))。
5〜6 g程度の形質転換体Arabidopsis thalianaをあらかじめ冷却した乳鉢の中に入れ、素早く液体窒素を加え、植物体がパウダー状になるまでホモジナイズした。その後、パウダー状になった試料に氷冷しておいた破砕Bufferを20 ml加えよく撹拌し、4重にしたミラクロスで濾過した。得られた濾液を15 ml容コーニングチューブに移し、7,000 rpm, 4℃, 15 sec.遠心分離し上清を丁寧に取り除いた。沈殿にその5 vol.の破砕Bufferを加え均一になるまで混ぜ、あらかじめ10 mlの30%パーコールをいれておいたガラス遠沈管に静かに重層し、2,000 rpm, 4℃, 3 min.遠心分離した。終了後上清のパーコールを取り除き、沈殿を5 mlの破砕Bufferで均一になるまで混ぜ、再び2,000 rpm, 4℃, 3 min.遠心分離した(この作業を2度繰り返す)。最後に沈殿を1 mlの破砕Bufferに溶解させ、その溶液を葉緑体画分とした。
(3-2) ウエスタンブロッティング
上記(3-1)で得られた葉緑体画分溶液にCelLytic P Plant Cell Lysis/Extraction reagentを1 ml加え、10,000 rpm, 4℃, 10 min.遠心分離し、得られた上清を葉緑体タンパク抽出溶液とした。1.5 ml microcentrifuged tubeに1 μg/μl分子量標準マーカー5 μlと超純水を5 μl、葉緑体タンパク抽出溶液を加え、それぞれのチューブに等量の試料用緩衝液を加え、よく撹拌した。調製したチューブを40℃、30 min.湯浴中でインキュベートし、泳動用試料とした。
泳動槽に陽極および陰極電極液、SDS-PAGEゲルをセットし、ウェルに泳動用試料20 μlを慎重にアプライし、30 Vの定電圧で泳動用試料を分離ゲルの上端まで泳動し、100 Vの定電圧でマーカーがゲルの下端から約5 mm程度まで泳動した。泳動中にあらかじめ、濾紙をブロッティング緩衝液A、B、Cに各2枚ずつ浸し、PVDF膜は少量のメタノールに5 sec.浸して、ブロッティングC液に浸した。泳動が終了したら、ゲルをガラス板から外し、ブロッティングC液に5 min.ほど浸した。セミドライブロッティング装置に下からAの濾紙2枚、Bの濾紙2枚、PVDF膜、ゲル、Cの濾紙2枚の順で重ね、セット後に60 mAの定電流で90 min.転写した。ブロッティング終了後、直ちに以下に示す順に操作し、抗原抗体反応の操作を行った。
(i) PVDF膜をTBSに浸し5分間震盪する(2回)。
(ii) 5% BSA in TBSに浸し1時間から一晩震盪する。
(iii) TBSに浸し5分間震盪する。
(iv) st-antibodyに浸し2時間震盪する。
(v) TTBSに浸し5分間震盪する(2回)。
(vi) 2nd-antibodyに浸し1時間30分震盪する。
(vii) TTBSに浸し5分間震盪する(2回)。
(viii) TBSに浸し5分間震盪する(2回)。
(ix) 2nd-antibody development reagentに浸し20分震盪する(遮光)。
(x) 超純水で洗浄する。
(3-3) 結果
PYC6導入植物よりパーコールの密度勾配遠心により葉緑体画分を分離し、得られた葉緑体タンパク質をSDS-PAGEにより電気泳動的に分子量でふるい分けた。次に、分離した全てのタンパク質をPVDF膜へと転写した後、P. yezoensis 由来シトクロムc6を認識する一次抗体(ウサギ抗体)と抗ウサギ抗体(二次抗体)を用いて抗原抗体反応を行ったところ、PYC6導入植物のみに抗原抗体反応を検出することができた(図10)。以上の結果から、PYC6導入植物において導入したPYC6は葉緑体中で発現していることが明らかとなった。
(4) 発現したシトクロムc6タンパク質のN末端アミノ酸配列解析
本項では、上記(3)で抗原抗体反応を示したPYC6タンパクのN末端アミノ酸配列を解析し、シグナルペプチド切断の有無を確認した。
上記(3-1)で得られた葉緑体画分溶液にCelLytic P Plant Cell Lysis/Extraction reagent を1 ml加え、10,000 rpm, 4℃, 10 min.遠心分離し、得られた上清を葉緑体タンパク抽出溶液とした。1.5 ml microcentrifuged tubeに1 μg/μl分子量標準マーカー5 μlと超純水を5 μl、葉緑体タンパク抽出溶液を加え、それぞれのチューブに等量の試料用緩衝液を加え、よく撹拌した。調製したチューブを40℃、30 min.湯浴中でインキュベートし、泳動用試料とした。
泳動槽に陽極および陰極電極液、SDS-PAGEゲルをセットし、ウェルに泳動用試料20 μlを慎重にアプライし、30 Vの定電圧で泳動用試料を分離ゲルの上端まで泳動し、100 Vの定電圧でマーカーがゲルの下端から約5 mm程度まで泳動した。泳動中にあらかじめ、濾紙をブロッティング緩衝液A、B、Cに各2枚ずつ浸し、PVDF膜は少量のメタノールに5 sec.浸して、ブロッティングC液に浸した。泳動が終了したら、ゲルをガラス板から外し、ブロッティングC液に5 min.ほど浸した。セミドライブロッティング装置に下からAの濾紙2枚、Bの濾紙2枚、PVDF膜、ゲル、Cの濾紙2枚の順で重ね、セット後に60 mAの定電流で90 min.転写した。ブロッティング終了後、CBB染色、脱色操作を行い、目的タンパク質部分を1 mm x 5 mmの大きさに切り1.5 ml micro centrifuged tubeに移した。これをシーケンスサンプルとしてApplied Biosystems Protein Sequencer 492型でアミノ酸配列の解析を行った。
形質転換植物より得られたPYC6抗体と抗原抗体反応を示す分子量およそ9.1 kDaのタンパク質のN末端アミノ酸配列をエドマン法により解析した結果、その配列はN末端1残基目から順に ADLDNGEKVF(配列番号12)であった(下記表を参照)。
【表18】
これはPYC6の成熟タンパク領域のN末端アミノ酸配列と完全に一致しており、植物体内でシグナルペプチドの切断が行われ、正確に葉緑体チラコイド膜を通過しルーメン側で発現していることが明らかになった。すなわち、得られた形質転換体中のPYC6タンパク質のN末端アミノ酸配列の解析結果より、葉緑体輸送シグナルが切断された成熟領域アミノ酸配列と一致したことから、sp+PYC6遺伝子は転写・翻訳後葉緑体へと輸送されていることが確認できた。
【実施例4】
【0009】
形質転換植物の生育測定と表現型の観察
(1) 生育観察(背丈、根長、葉の大きさ)
本項では、形質転換体Arabidopsis thalianaの播種後の生育を観察した。
形質転換体を播種した後、植物体の「背丈」、「根長」及び「葉の大きさ」を10日おきに観察測定した。植物試料は野生株と形質転換体共に20個体行い、その平均を算出し、統計処理により有意差を求めた。
PYC6導入植物の生育(表現型)の観察を行った結果、PYC6導入植物の背丈は、最大で1.9倍(播種後40日)野生型(WT)に比べて大きくなり、約60日目で1.3倍増大していた(図11)。根長は、最大で1.35倍(播種後40日)WTに比べ大きくなり、約60日目で1.17倍増大していた(図12)。葉の大きさは、最大で1.9倍(播種後40日)の差が見られ、約60日目で1.3倍増大していた(図13)。これらPYC6導入植物にみられた現象は、植物体内でのPYC6の発現により何らかの形で植物の生育に非常に重要な光合成反応が活性化されたためと考えられ、その影響により植物体の生育速度が上昇したと思われた。
(2) 総クロロフィル量の測定
本項では、形質転換体Arabidopsis thalianaより全クロロフィルをアセトンにより抽出し、定量した。
形質転換体Arabidopsis thalianaの葉をリーフパンチで採取し、秤量した。あらかじめ冷却した乳鉢の中に試料を入れ、素早く液体窒素を加え、植物体がパウダー状になるまでホモジナイズした。その後、パウダー状になった試料に80%アセトンを加え4 mlに定容した。その溶液の663 nmと645 nmの吸光度を分光光度計UV-VISIBLE SPECTROPHOTOMETERで測定した。
得られた663 nmと645 nmの吸光値を以下に示した式に代入し、クロロフィル量を算出した。
総クロロフィル量(μg/ml)
= 8.02×ABS(663) + 20.21×ABS(645)
PYC6導入植物中のクロロフィル量を測定した結果、播種後40日から70日の間においてはWTよりも1.1〜1.2倍増大していた(図14)。これは、PYC6の導入により明反応が活性化され、それにより増大した明反応最終産物であるエネルギー物質(ATP)がクロロフィルの合成に利用された影響ではないかと考えられた。
【実施例5】
【0010】
形質転換植物の光合成活性測定
(1) 方法及び結果
光合成活性の指標として、アデニンヌクレオチド(ATP)含量、NADPH量、二酸化炭素同化能力、デンプン量、及びタンパク質量を、それぞれ、以下に示す方法で測定した。
(1-1) 総ATP含量の測定
本実施例では、形質転換体Arabidopsis thalianaより全ATPを抽出し、ルシフェリン・ルシフェラーゼ法を用いて定量した。
形質転換体Arabidopsis thalianaの葉を予め秤量した。秤量後、冷却した乳鉢の中に試料を入れ、素早く液体窒素を加えて、植物体がパウダー状になるまで乳棒でホモジナイズした。その後、パウダー状になった試料に0.25M 過塩素酸を2 ml加え、その溶液を15 ml容コーニングチューブに回収した。10,000 rpm, 4℃, 10 min.遠心分離し、得られた上清を別の15 ml容コーニングチューブに移し、1N KOHでpH7.0に調整した。調製後、0.2M リン酸緩衝液(pH 7.2)で5 mlに定容し、これをATP抽出液とした。
ATP抽出液100 μlを1.5 ml microcentrifuged tubeに準備し、ENLITEN Luciferase/Luciferin Reagentを20 μl添加してから90 sec.後ルミノメーター(GENE LIGHT 55)で10 sec.反応液の発光強度を積算し、得られたRLU値から溶液のATP濃度を算出し、そこから植物重量当たりのATP量を換算した。
その結果、PYC6導入植物の総ATP量は、最大で1.93倍(播種後60日)増大していた(図15)。これは、PYC6導入植物において機能しているプラストシアニンに加え、さらに新たな電子伝達体として紅藻cyt c6(PYC6)が機能したことにより、明反応の電子伝達が活性化され、その最終生成物であるATP量が増大したものと考えられた。
(1-2) 総NADPH含量の測定
本実施例では、形質転換体Arabidopsis thalianaより全NADPHを抽出し、定量した。
形質転換体Arabidopsis thalianaを予め秤量した。その後、70℃程度のホットバスで温めた0.1 N NaOHを入れ、ポリトロンホモジェナイザーを用いて破砕した。その後、アイスバスに移し、約2 mlの0.1 N HClを加え、pH試験紙でpH 7.5に調整した。次に0.1 mlのグリシルグリシン緩衝液(pH 7.5)を加え、よく撹拌し、メスシリンダーに移し、液量を測定する。測定後、15 ml容コーニングチューブに移し、10,000 rpm、20 min、4℃で遠心分離した。遠心分離後、上清を15 ml容コーニングチューブに回収し、-80℃で一晩冷却し、溶解後、10,000 rpm、20 min、4℃で遠心分離し、定量に用いた。
NADPH抽出液100.0 μl、0.1 M グリシルグリシン緩衝液(pH 7.4)346.0 μl、0.2 M ニコチンアミド200.0 μl、3.2 mg/ml フェナジンメトサルフェート(PMS)82.0 μl、5 mg/mlチアゾールブルー67.0 μl、グルコース-6-リン酸2ナトリウム水和物200 μlを混ぜ、分光光度計U3310にセットした。を用いて570 nm、25℃で測定する。U3310に温度制御ユニットを取り付け、25℃にセットする。その後、570 nmにおける吸光度の変化を30秒おきに2分間測定し、得られた吸光値の差からNADPH量を算出し、そこから植物重量あたりのNADPH量を算出した。
その結果、PYC6導入植物の総NADPH量は播種後40〜70日で1.2〜1.4倍増大していた(図16)。これは、PYC6導入の高等植物への導入によって、野生型よりも多く電子が伝達され、その最終生成物であるNADPH量が増大したものと考えられた。
(1-3) 二酸化炭素同化能力の測定
本実施例では、形質転換体Arabidopsis thalianaの葉を用いて二酸化炭素の同化能力を測定した。
二酸化炭素の同化能力は、CO2ガス分析機CIRAS-1(小糸工業製)を用いて行った。また、二酸化炭素の供給量は350 ppmに設定し、飽和光下で測定した。
その結果、PYC6導入植物の二酸化炭素の同化能力は最大で2.2倍増大していた(播種後50日)(図17)。これは、PYC6導入の高等植物への導入によって、ATPやNADPH量が増大したことにより、そのエネルギーを利用する暗反応(カルビン・ベンソンサイクル)が活性化され、二酸化炭素の同化能力が増大したものと考えられた。
(1-4) デンプン量の測定
本実施例では、形質転換体Arabidopsis thalianaよりデンプンを抽出し、定量した。
植物葉を採取後、乾燥させる(70度、2日)。約5 mgを量り取り乳鉢に入れ、3 mlの32%過塩素酸を加えて、乳棒でよく破砕する。その後、全て試験管に移し、20℃で20分静置した。次に、6.0 cmのWhatman GF/A grass fibre diskで濾過し、濾液に5 mlのヨウ素溶液を加え、よく混ぜ、約4℃で30分間静置する。その後、1.5 cmのWhatman GF/A grass fibre discで濾過し、フィルターを乾燥させる。
乾燥したフィルターを試験管に入れ、4 mlの0.75M硫酸を加え、沸騰湯浴中で30分間インキュベートする。その後、上清を回収し、フェノール硫酸法により発色させ、分光光度計を用いて485 nmの吸収を測定した。
その結果、PYC6導入植物のデンプン量は播種後40〜70日で1.15〜1.25倍増大していた(図18)。これは、PYC6導入の高等植物への導入によって、CO2の同化能が増大し、それに伴いデンプンの合成も促進されたと考えられた。
(1-5) タンパク質量の測定
本実施例では、形質転換体Arabidopsis thalianaよりタンパク質を抽出し、Lowry法を用いて定量した。
植物体の重量を測定し、記録する。乳鉢に適量の植物体を入れ、液体窒素下、乳棒で充分に破砕する。常温まで戻した後、植物体1 gあたり2 mlのCelLytic P(Sigma製)を入れ、乳棒でよく磨砕し、試験管に移す。15分室温で転倒混和後、遠心分離する。この上清を抽出タンパク質とする。
次に、Lowry A液 : 0.1 Nの水酸化ナトリウム溶液に2%になるように炭酸ナトリウムを加えたもの、Lowry B液 : 0.5% 硫酸銅五水和物と1% クエン酸ナトリウムになるように調製したもの、Lowry C液 : 1 N フェノール溶液、Lowry D液 : Lowry A液 + Lowry B液を作製した。その後、抽出したタンパク質溶液、0.1 mlにLowry D液 1.0 mlを加え、ボルテックスミキサーで撹拌した後、30°C、15 minインキュベートした。次にLowry C液 0.1 mlを加えて、速やかにボルテックスミキサーで撹拌した後、30°C、30 minインキュベートし、770 nmの吸光値を測定した。
その結果、その結果、PYC6導入植物のタンパク質量は、最大1.3倍(播種後50日)増大していた(図19)。これは、PYC6導入の高等植物への導入によって、増産されたATPやNADPHがタンパク質の合成を活性化したのではないかと考えられた。
(2) 考察
本発明で得られた知見として、PYC6導入植物はWTと比較して成長速度が上昇していた。これはPYC6導入植物中のATP及びNADPHが増大していたことから、A. thalianaにおいて機能しているプラストシアニンに加え、さらに新たに導入した電子伝達体である紅藻cyt c6 が機能したことにより、明反応の電子伝達が2回路となったためにその最終生成物であるATP及びNADPH量が増大し、さらに植物全体の生育が活性化される結果となったと推測された。これまで陸上植物への暗反応(カルビン・ベンソンサイクル)の関連酵素の補強による成分量の増進についての研究は行われているが、本発明のように明反応を活性化させた例はない。
また、これまでcyt c6 を機能させることを目的として植物へと導入した例はなく、本発明の内容は今後他種の植物にも十分応用可能であり、有用な植物の生長促進法や植物の生産技術(果実物、葉物、生花など)などの基盤となる汎用性の高い発明であるといえる。さらに、植物体内で増産されたATP及びNADPHを利用することで他の物質代謝も活性化され、汚染物質(CO2など)の除去法の分野にも極めて有効と考えられた。
【産業上の利用可能性】
【0011】
本発明によれば、葉緑体のチラコイド内腔にシトクロムc6を有する新規な高等植物の作出方法を提供することができ、ひいては、高等植物の生長促進方法、ATP、NADPH、デンプン及びタンパク質の合成促進方法及び炭素固定促進方法を提供することができる。
また、葉緑体のチラコイド内腔にまで輸送され得るシグナルペプチド付加シトクロムc6タンパク質、当該タンパク質をコードする遺伝子、当該遺伝子を含む組換えベクター、当該組換えベクターを含む形質転換体、及び当該遺伝子が植物ゲノム中に導入されている形質転換高等植物を提供することもできる。
【配列表フリーテキスト】
【0012】
配列番号7:プライマー
配列番号8:プライマー
配列番号9:プライマー
配列番号10:プライマー
【技術分野】
【0001】
本発明は、葉緑体のチラコイド内腔にシトクロムc6を有する新規な高等植物の作出方法、及び、上記チラコイド内腔にシトクロムc6を存在させ高等植物の生長を促進させる方法又は高等植物の炭素固定能を促進させる方法に関する。
【背景技術】
【0002】
従来、陸上植物等のいわゆる高等植物の生長促進に関する技術としては、リブロースビスリン酸カルボキシラーゼ等の酵素の活性を高めるなど、光合成暗反応(カルビン・ベンソンサイクル)に関する報告例、具体的には、関連酵素遺伝子の導入により葉を大きくした報告例(Shigeoka et al., Nature biotechnology, 19, 965-969(2001))等がある。しかしこのような技術は、様々な高等植物への適用の面で、汎用性に非常に乏しいものであった。
シトクロムc6は、光合成明反応における電子伝達タンパク質であって、本来、一部の藻類(ラン藻類等)にのみ存在するものであり、その電子伝達能力が極めて優れている(すなわち酸化還元電位が高電位である)ことが知られている(図1)。このため、様々な高等植物において、その葉緑体中に(詳しくはチラコイド内腔に)シトクロムc6を発現及び機能させ、光合成能を向上させる、汎用性に優れた技術の開発が強く望まれている。
ところで、シトクロムc6を細胞内で発現させるようにする技術としては、例えば、F. P. Molina-Heredia et al., Biochem. Biophys. Res. Commun., 243, 302-306(1998);T. Satoh et al., FEBS lett., 531, 543-547(2002);R. Gupta et al., Nature, 417, 567-571(2002);D. R. Hickey et al., Gene, 105, 73-81(1991) 等の文献に記載の技術が知られている。しかし、これら技術はいずれも、大腸菌、酵母あるいはラン藻といった高等植物ではない宿主細胞を用い、単に、上記シトクロムc6又はそれに類するタンパク質の大量生産や機能解析を目的としてなされたものであり、従来より当業者において通常行われている遺伝子発現法に含まれるものである。
そして、高等植物において光合成明反応の電子伝達体としてシトクロムc6を機能させること、つまり、高等植物細胞内の葉緑体中にあるチラコイドの内腔にシトクロムc6を存在させることに成功した報告例は全く無く、極めて困難であると考えられていた。
【発明の概要】
【0003】
そこで、本発明が解決しようとする課題は、葉緑体のチラコイド内腔にシトクロムc6を有する新規な高等植物の作出方法を提供すること、ひいては、高等植物の生長促進方法、ATP、NADPH、デンプン及びタンパク質からなる群から選ばれる少なくとも1つの合成促進方法並びに炭素固定促進方法を提供することにある。
本発明者は、上記課題を解決するべく鋭意検討を行った。その結果、特定のシグナルペプチドを付加したシトクロムc6タンパク質の遺伝子を、高等植物のゲノムDNAに導入し発現させるようにすれば、従来不可能であると考えられていた葉緑体包膜内及びチラコイド膜内へのシトクロムc6の輸送(通過)が可能となることを見出し、本発明を完成した。
すなわち、本発明は以下のとおりである。
(1) シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入することを特徴とする、葉緑体のチラコイド内腔にシトクロムc6を有する高等植物の作出方法。
(2) シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入して発現させ、葉緑体のチラコイド内腔にシトクロムc6を存在させることを特徴とする、高等植物の生長促進方法。
(3) シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入して発現させ、葉緑体のチラコイド内腔にシトクロムc6を存在させることを特徴とする、高等植物のATP、NADPH、デンプン及びタンパク質からなる群から選ばれる少なくとも1つの合成促進方法。
(4) シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入して発現させ、葉緑体のチラコイド内腔にシトクロムc6を存在させることを特徴とする、高等植物の炭素固定促進方法。
上記(1)〜(4)の方法においては、前記融合タンパク質が以下の(a)、(b)、(c)又は(d)のタンパク質であってもよい。
(a) 配列番号6に示されるアミノ酸配列を含むタンパク質
(b) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質
(c) 配列番号6に示されるアミノ酸配列のうちの前記シトクロムc6タンパク質に相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ電子伝達能を有するタンパク質
(d) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列及び前記シトクロムc6タンパク質に相当するアミノ酸配列においてそれぞれ1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質
また上記(1)〜(4)の方法においては、前記遺伝子が以下の(a)又は(b)のDNAを含む遺伝子であってもよい。
(a) 配列番号5に示される塩基配列を含むDNA
(b) 配列番号5に示される塩基配列を含むDNAと相補的な塩基配列を含むDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質をコードするDNA
(5) シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加されてなる融合タンパク質。
上記(5)の融合タンパク質は、以下の(a)、(b)、(c)又は(d)のタンパク質であってもよい。
(a) 配列番号6に示されるアミノ酸配列を含むタンパク質
(b) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質
(c) 配列番号6に示されるアミノ酸配列のうちの前記シトクロムc6タンパク質に相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ電子伝達能を有するタンパク質
(d) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列及び前記シトクロムc6タンパク質に相当するアミノ酸配列においてそれぞれ1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質
(6) 上記(5)の融合タンパク質をコードする遺伝子。
(7) 以下の(a)又は(b)のDNAを含む遺伝子。
(a) 配列番号5に示される塩基配列を含むDNA
(b) 配列番号5に示される塩基配列を含むDNAと相補的な塩基配列を含むDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質をコードするDNA
(8) 上記(6)又は(7)の遺伝子を含む組換えベクター。
(9) 上記(8)の組換えベクターを宿主に導入してなる形質転換体。
上記(9)の形質転換体は、宿主がアグロバクテリウム菌の形質転換体であってもよい。
(10) 上記(6)又は(7)の遺伝子が植物ゲノム中に導入されている、形質転換高等植物。
上記(10)の形質転換高等植物は、植物細胞中の葉緑体のチラコイド内腔にシトクロムc6を有するものであることが好ましい。
【図面の簡単な説明】
【0004】
【図1】図1は、葉緑体チラコイド膜の光合成電子伝達系を示す図である。
【図2】図2は、本発明におけるアグロバクテリウムを介したシトクロムc6遺伝子の導入と植物内におけるシトクロムc6の発現様式を示す模式図である。
【図3】図3は、シアノバクテリア及び藻類と高等植物の間における電子伝達系の相違を示す図である。
【図4】図4は、Porphyra yezoensisシトクロムc6のcDNAの塩基配列及び推定されるアミノ酸配列を示す図である。
【図5】図5は、Porphyra yezoensisシトクロムc6遺伝子の成熟タンパク質領域をPCRによって増幅した結果を示す図である。
【図6】図6は、植物導入用シトクロムc6遺伝子の構築に用いたpBluescript II SK+/- ベクターを示す図である。
【図7】図7は、Arabidopsis thaliana由来プラストシアニンのシグナルペプチドとPorphyra yezoensis由来シトクロムc6の成熟タンパク質領域を融合させた遺伝子の塩基配列と推定されるアミノ酸配列を示す図である。
【図8】図8は、トリペアレンタルメイティングによるアグロバクテリウム・チュメファシエンス(Agrobacterium tumefaciens)の形質転換法を示す模式図である。
【図9】図9は、植物に導入されたシトクロムc6遺伝子をPCRによって特定した電気泳動結果を示す図である。(A)は鋳型としてゲノムDNAを用いたときの結果であり、(B)は鋳型として全RNAを用いたときの結果である。
【図10】図10は、植物内で発現したシトクロムc6を電気泳動によって特定した結果を示す図である。(A)はCBB染色の結果であり、(B)はシトクロムc6抗体と反応させたウエスタンブロッティングの結果である。
【図11】図11は、生育日数の経過に伴う、野生型(○)及び形質転換A. thaliana(●)の背丈の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=20)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図12】図12は、生育日数の経過に伴う、野生型(○)及び形質転換A. thaliana(●)の根の長さの変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=20)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図13】図13は、生育日数の経過に伴う、野生型(○)及び形質転換A. thaliana(●)の葉の大きさの変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=20)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図14】図14は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後におけるクロロフィル含量の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=8)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図15】図15は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後におけるATP含量の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=8)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図16】図16は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後におけるNADPH含量の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=8)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図17】図17は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後における二酸化炭素同化能力の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=10)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図18】図18は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後におけるデンプン量の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=10)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【図19】は、生育日数の経過に伴う、野生型及び形質転換A. thalianaの光照射3時間後におけるタンパク質量の変化を示す図である。グラフの各値は、平均±S.D.(標準偏差) (n=8)を表す。アスタリスク(*)は、5%の有意水準で有意に差が見られた値であることを示す。
【発明を実施するための形態】
【0005】
以下、本発明について詳しく説明するが、本発明の範囲はこれらの説明に拘束されることはなく、以下の例示以外についても、本発明の趣旨を損なわない範囲で適宜変更し実施し得る。
なお、本明細書は、本願優先権主張の基礎となる特願2005-27012号明細書の全体を包含する。また、本明細書において引用された全ての先行技術文献、並びに公開公報、特許公報及びその他の特許文献は、参照として本明細書に組み入れられる。
1.本発明の概要
本発明は、シトクロムc6とシグナルペプチドとの融合タンパク質をコードする遺伝子を高等植物内に導入し、高等植物細胞内の葉緑体中のチラコイド内腔において、光合成明反応の電子伝達体としての役割を果たすシトクロムc6を発現させ且つ機能させることにより、高等植物の生長を促進させる方法に係るものである(図2)。本発明により、高等植物細胞内でのATP、NADPH、デンプン及びタンパク質の合成が促進され、またこれに伴い、二酸化炭素(CO2)を炭水化物に変換する光合成暗反応(炭素固定反応)も促進され得る。本発明は、このような、高等植物におけるATP等の合成や炭素固定を促進する方法を含み、また、生長促進、ATP等の合成促進、及び炭素固定の促進がされ得る高等植物の作出方法をも含むものである。
一般に、光合成明反応(光合成電子伝達反応)においては、クロロフィルの電子が太陽光のエネルギーを獲得し、チラコイド内腔(チラコイド膜中)の電子伝達鎖を次々と移動するが、その際、クロロフィルは水から電子を獲得し、酸素(O2)を遊離する一方、上記電子伝達反応に伴いチラコイド膜を介してH+のくみ出しが起こり、それで生じたプロトンの駆動力によって葉緑体のストロマ内でATPが合成される。そして、一連の反応の最後では、高エネルギー電子を(H+とともに)NADP+が受け取り、NADPHが合成される。
これに対し、光合成暗反応(炭素固定反応)では、光合成明反応で合成されたATPとNADPHがそれぞれエネルギー源と還元力として働くことで、葉緑体のストロマ内及び細胞質において、CO2が炭水化物に変換される。この炭素固定反応により、植物の葉等ではショ糖が合成される。ショ糖は、他の組織にも輸送され、様々な有機分子の合成材料として、ひいては植物自身の生長のためのエネルギー源として用いられる。
通常、高等植物における光合成明反応の電子伝達体(電子伝達タンパク質)は、プラストシアニン(PC)であるが、電子伝達能(酸化還元電位の高さ)に関しては、藻類における電子伝達体であるシトクロムc6の方が明らかに優れていることが分かっている(図3)。そのため、従来から、このシトクロムc6を高等植物内でも発現及び機能させ、光合成明反応及び暗反応を促進させようとする試みが種々なされてきた。しかしながら、このような試みは、実際のところ極めて困難であり、これまで成功例は無かった。
高等植物において、その細胞内のゲノム情報をもとに発現されたタンパク質が光合成明反応の電子伝達体としての機能を発揮するためには、チラコイド内腔に存在することが必須条件となる。そのためには、通常、「葉緑体包膜」及び「チラコイド膜」の2種の膜を通過する必要がある。この点が高等植物におけるシトクロムc6の機能発現を極めて困難にしている主要因であると考えられていた。そこで本発明者は、遺伝子組換え技術により、シトクロムc6遺伝子を高等植物のゲノム中に導入するにあたり、シトクロムc6タンパク質が、特定のシグナルペプチド(特定の長さ又は特定のアミノ酸配列のシグナルペプチド)が付加された融合タンパク質の状態で発現されるように、予めシグナル配列を付加した遺伝子を構築した上でその導入を行った。その結果、当該遺伝子導入により作出した植物体においては、チラコイド内腔にシトクロムc6タンパク質(シグナルペプチド無し)の存在が認められ、結果として、発現した融合タンパク質はそのシグナルペプチドの働きにより前述した2種の膜を通過し得るものであることが示され、本発明を完成した。
(1) PYC6遺伝子導入系の検討
これまでに、PYC6の酸化還元電位は高電位であることが知られており、また、そのPYC6遺伝子配列も明らかにされている。本発明においては、はじめにPYC6遺伝子をバイナリーベクターに連結し、形質転換用アグロバクテリウムを調製した。シロイヌナズナ(Arabidopsis thaliana)は、播種後低温処理を行ったのち長日条件で生育させ、抽台した段階で減圧浸潤により形質転換を行った。つづいて、得られた形質転換体において遺伝子の導入と発現を解析するため、植物体から抽出したゲノムDNAとRNAを鋳型とし、それぞれPCR、RT-PCRを行った。その結果、PYC6遺伝子の増幅が見られ、DNAシーケンサーによりその塩基配列はPYC6遺伝子と同様であることを確認した。次に、形質転換体中に確認されたPYC6遺伝子がタンパク質として発現しているかを解析するため、植物体から葉緑体タンパク質画分を抽出し、電気泳動後ウエスタンブロッティングを行った。その結果、PYC6タンパク質の発現を確認した。また、得られた形質転換体中のPYC6タンパク質のN末端アミノ酸配列を解析したところ、導入したPYC6のアミノ酸配列(PIR accession No. : JC5849)と一致した。この結果から、高等植物であるA. thaliana内で藻類由来cyt c6(PYC6)を発現させることに成功した。
(2) PYC6導入による植物体の影響
後述の実施例に示すように、PYC6導入植物の生育を観察したところ、葉の大きさは野生型(WT)に比べおよそ1.2〜1.3倍、背丈は約1.5倍増大していた。PYC6導入植物は生育初期段階においてWTと比較して成長速度が上昇していた。また、PYC6導入植物中のクロロフィル量を測定した結果、WTのおよそ1.1〜1.2倍であった。このPYC6導入により植物体にみられた現象は、明反応最終産物であるエネルギー物質を利用して光合成反応全体が活性化されたためではないかと考えられる。そこで、ルシフェリン・ルシフェラーゼ法によりPYC6導入植物のATP量を測定した結果、WTと比較して約1.7倍増大していた。これはA. thaliana において機能しているプラストシアニンに加え、さらに新たな電子伝達体として紅藻cyt c6が機能したことにより、明反応の電子伝達が活性化され、その最終生成物であるATP量が増大し、さらには植物全体の生育が活性化されたためと考えられた。
なお、当該融合タンパク質が前述した2種の膜を通過するに際しては、第1の膜(葉緑体包膜)の通過時にはシグナルペプチドの一部が、第2の膜(チラコイド膜)の通過時にはその残りの部分が使用され、それぞれ使用後は脱離した後、最終的にシグナルペプチドの無い状態でPYC6がチラコイド内腔に輸送される。そして輸送されたPYC6タンパク質は、このチラコイド内腔でヘム(ヘムc)と結合し且つフォールディングすることにより電子伝達体として機能し得るシトクロムc6になると考えられる。
2.新規高等植物の作出方法
本発明の作出方法は、前述の通り、葉緑体のチラコイド内腔にシトクロムc6を有する新規な高等植物の作出方法であり、シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入することを特徴とする方法である。
(1) 作出対象となる高等植物
本発明の作出方法に用い得る高等植物、すなわち葉緑体のチラコイド内腔にシトクロムc6を有するよう形質転換され得る高等植物としては、限定はされず、例えば以下の高等植物を挙げることができる。
本発明において対象となる植物は、植物体全体、植物器官(例えば葉、花弁、茎、根、種子等)、植物組織(例えば表皮、師部、柔組織、木部、維管束、柵状組織、海綿状組織等)又は植物培養細胞(カルスなどの組織培養を含む)のいずれをも意味するものである。形質転換に用いられる植物としては、C3、C4、CAM植物、及びこれらの中間植物等のすべてを含み、例えばアブラナ科、ナス科、イネ科、マメ科、アカザ科、バラ科、キク科、ユリ科、ナデシコ科、ウリ科、ヒルガオ科、ヒユ科、パイナップル科、サボテン科、及びアロエ科等に属する植物(果実、野菜、花卉等を含む)が挙げられるが(下記参照)、これらの植物に限定されるものではない。
〔C3植物〕
・アブラナ科:シロイヌナズナ(Arabidopsis thaliana) 、ダイコン(Raphanus) など
・ナス科:タバコ(Nicotiana tabacum)、ジャガイモ(Solanum)など
・イネ科:イネ(Oryza sativa)、トウモロコシ(Zea mays) 、コムギ(Triticun)など
・マメ科:ダイズ(Glycine max) 、エンドウ(Pisum) など
・アカザ科:ホウレンソウ(Spinacia)など
・バラ科:ヤマザクラ(Prunus)、バラ (Rosa) など
・キク科:ハルジオン(Erigeron)、タンポポ (Taraxacun) など
・ウリ科:カボチャ(Cucurdida)、キュウリ (Cucumis) など
・ヒルガオ科:サツマイモ(Ipomea)など
・ラン科:ウチョウラン(Poneorchis)など
〔C4植物〕
・イネ科:トウモロコシ(Zea mays)、サトウキビ(Saccharum officinarum)、アワ(Setaria itarica)など
・ヒユ科:アマランサス(Amaranthaceae)など
〔CAM植物〕
・パイナップル科:パイナップル(Ananas comosus)など
・サボテン科:デフューサ(Lophophora difusa)、オプンティア(Opuntia spp.)など
・アロエ科:キダチアロエ(Aloe arborescens)、アロエベラ(Aloe vera)など
〔C3-C4中間植物〕
・ツルナ科:クルマバザクロソウ(Mollugo verticillata)など
・イネ科:キビ(Panicum milioides)など
なかでも、本発明の作出方法の効果として、生長促進効果の高い植物が得られる点を考慮すると、例えば、より市場性の高い植物のほうが本発明の利用価値が高く、具体的には、葉物(ホウレンソウ、キャベツなど)、花物(バラ、コチョウランなど)、茎物(ジャガイモ、レンコンなど)、根物(ゴボウ、ダイコンなど)、穀物(イネ、コムギなど)、果実物(パイナップル、ブドウなど)、観賞用植物(マツ、カエデなど)、木材(スギ、ヒノキなど)などが好ましく挙げられる。
(2) 融合タンパク質
本発明の作出方法においては、高等植物のゲノム中に特定の融合タンパク質をコードする遺伝子を導入するが、当該融合タンパク質は、シトクロムc6タンパク質に特定の長さのシグナルペプチドが付加された融合タンパク質であることが重要である。
上記融合タンパク質は、シグナルペプチドの長さが50〜80アミノ酸残基であることが重要である。このようにシグナルペプチドが十分に長いことにより、シトクロムc6タンパク質を葉緑体包膜及びチラコイド膜のいずれにも通過させることができる。シグナルペプチドの配列は、高等植物から遺伝子クローニングすることにより決定することができる。
なお上記融合タンパク質におけるシグナルペプチドは、一般に、シトクロムc6タンパク質のN末端に付加されていることが好ましい。
本発明においては、上記融合タンパク質は、例えば、以下の(a)、(b)、(c)又は(d)のタンパク質であることが好ましい。
(a) 配列番号6に示されるアミノ酸配列を含むタンパク質
(b) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質
(c) 配列番号6に示されるアミノ酸配列のうちの前記シトクロムc6タンパク質に相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ電子伝達能を有するタンパク質
(d) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列及び前記シトクロムc6タンパク質に相当するアミノ酸配列においてそれぞれ1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質
上記(a)のタンパク質は、配列番号6に示すように、シトクロムc6タンパク質を構成する85個のアミノ酸(配列番号2)を含む配列のN末端側に、シグナルペプチドを構成する72個のアミノ酸(配列番号4)からなる配列が付加(連結)されてなる、合計157個のアミノ酸を含むものであるが、配列番号6で表されるアミノ酸配列からなる融合タンパク質であってもよい。但し、シグナルペプチドは、上記72個に限定されるものではない。ここで、シトクロムc6タンパク質の由来は特に限定されるものではなく、紅藻(Porphyra yezoensis)、ラン藻、褐藻、珪藻、および緑藻などを用いることができ、紅藻(P. yezoensis)由来のタンパク質であることが好ましい。P. yezoensis由来のシトクロムc6タンパク質のアミノ酸配列は公知であり(PIR accession No.: JC5849)、データベース検索することにより入手することができる。
上記(b)のタンパク質は、上記(a)のタンパク質を構成する全アミノ酸配列のうちの前記シグナルペプチドに相当する72個のアミノ酸配列(配列番号4)において、1個又は数個(例えば1個〜10個程度、好ましくは1個〜5個程度)のアミノ酸が欠失、置換又は付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質であればよく、限定はされない。
なお、上記(b)のタンパク質は、上記(a)のタンパク質と同様に高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質であることが重要であるため、例えば、第53番目〜第72番目のアミノ酸など、上記葉緑体包膜やチラコイド膜の通過に重要と考えられる部分のアミノ酸配列は、上記(a)のタンパク質のアミノ酸配列から変異置換されていないものが好ましい。
上記(c)のタンパク質は、上記(a)のタンパク質を構成する全アミノ酸配列のうちの前記シトクロムc6タンパク質に相当する85個のアミノ酸配列(配列番号2)において、1個又は数個(例えば1個〜10個程度、好ましくは1個〜5個程度)のアミノ酸が欠失、置換又は付加されたアミノ酸配列を含み、かつ(光合成明反応における)電子伝達能を有するタンパク質であればよく、限定はされない。
ここで、上記の「電子伝達能を有するタンパク質」とは、本発明においては、上記(c)のタンパク質を構成する全アミノ酸配列から前記シグナルペプチドに相当するアミノ酸配列が除かれたアミノ酸配列を含むタンパク質であって、ヘム(ヘムc)と結合した後において、電子伝達能を有するタンパク質を意味する。
なお、上記(c)のタンパク質は、シトクロムc6タンパク質と同様に電子伝達能を有するタンパク質であることが重要であるため、例えば、第14番目〜第18番目のアミノ酸、及び第47番目〜第60番目のアミノ酸など、高等植物の光合成明反応における電子伝達体として機能するために重要と考えられる部分のアミノ酸配列は、上記(a)のタンパク質のアミノ酸配列から変異置換されていないものが好ましい。
上記(d)のタンパク質は、上記(a)のタンパク質を構成する全アミノ酸配列のうちの、前記シグナルペプチドに相当する72個のアミノ酸配列(配列番号4)において1個又は数個(例えば1個〜10個程度、好ましくは1個〜5個程度)のアミノ酸が欠失、置換又は付加され、さらに前記シトクロムc6タンパク質に相当する85個のアミノ酸配列(配列番号2)においても1個又は数個(例えば1個〜10個程度、好ましくは1個〜5個程度)のアミノ酸が欠失、置換又は付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに(光合成明反応における)電子伝達能を有するタンパク質であればよく、限定はされない。
ここで、上記の「電子伝達能を有するタンパク質」とは、上記(c)のタンパク質の場合と同様の意味である。
なお、上記(d)のタンパク質は、上記(a)のタンパク質と同様に高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質であることに加え、シトクロムc6タンパク質と同様に電子伝達能を有するタンパク質であることも重要であるため、例えば、第53番目〜第72番目のアミノ酸などの上記葉緑体包膜やチラコイド膜の通過に重要と考えられる部分のアミノ酸配列や、第14番目〜第18番目のアミノ酸、及び第47番目〜第60番目のアミノ酸などの高等植物の光合成明反応における電子伝達体として機能するために重要と考えられる部分のアミノ酸配列は、上記(a)のタンパク質のアミノ酸配列から変異置換されていないものが好ましい。
上記(c)及び(d)のタンパク質の、電子伝達能の有無及び電子伝達能を有する場合、立体構造解析および酸化還元電位の測定を行うことにより確認・測定することができる。なお、シトクロムc6の酸化還元電位は、およそ350〜370mV程度である。
このような配列番号6で表されるアミノ酸配列において1若しくは複数個のアミノ酸が欠失、挿入または付加されたアミノ酸配列をコードするポリヌクレオチドは、「Molecular Cloning, A Laboratory Manual 2nd ed.」(Cold Spring Harbor Press (1989))、「Current Protocols in Molecular Biology」(John Wiley & Sons (1987-1997)、Kunkel (1985) Proc. Natl. Acad. Sci. USA 82: 488-92、Kunkel (1988) Method. Enzymol. 85: 2763-6)等に記載の部位特異的変異誘発法等の方法に従って調製することができる。このようなアミノ酸の欠失、置換又は付加等の変異型をコードする遺伝子の作製には、例えば、Kunkel法や Gapped duplex法等の公知手法により、部位特異的突然変異誘発法を利用した変異導入用キット、例えばQuikChangeTM Site-Directed Mutagenesis Kit(ストラタジーン社製)、GeneTailorTM Site-Directed Mutagenesis System(インビトロジェン社製)、TaKaRa Site-Directed Mutagenesis System(Mutan-K、Mutan-Super Express Km等:タカラバイオ社製)等を用いて行うことができる。
(3) 遺伝子
本発明の作出方法においては、高等植物のゲノム中に導入する遺伝子は、前述した融合タンパク質をコードする遺伝子であることが重要である。
本発明においては、上記遺伝子は、例えば、以下の(a)又は(b)のDNAを含む遺伝子であることが好ましい。なお、以下の(a)及び(b)のDNAは、いずれも前述した融合タンパク質の構造遺伝子(すなわち、シグナルペプチドをコードする遺伝子と、シトクロムc6又はこれと同様の電子伝達能を有するタンパク質の構造遺伝子とが連結した遺伝子)であるが、これらDNAを含む遺伝子としては、これらDNAのみからなるものであってもよいし、これらDNAを一部に含み、その他に当該融合タンパク質の構造遺伝子の発現に必要な公知の塩基配列(転写プロモーター、SD配列、Kozak配列、ターミネーター等)をも含むものであってもよく、限定はされない。なお、シトクロムc6をコードする塩基配列は、例えば日本DNAデータバンク(DDBJ)に公表されており公知である(accession number:AB40818)。
(a) 配列番号5に示される塩基配列を含むDNA
(b) 配列番号5に示される塩基配列を含むDNAと相補的な塩基配列を含むDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質をコードするDNA
上記(b)のDNAは、上記(a)のDNA若しくはそれと相補的な塩基配列を含むDNA、又はこれらの断片をプローブとして用い、コロニーハイブリダイゼーション、プラークハイブリダイゼーション、およびサザンブロット等の公知のハイブリダイゼーション法を実施し、cDNAライブラリーやゲノムライブラリーから得ることができる。ライブラリーは、公知の方法で作製されたものを利用してもよいし、市販のcDNAライブラリーやゲノムライブラリーを利用してもよく、限定はされない。
また、シグナルペプチド部分をコードする塩基配列(配列番号3)、シトクロムc6をコードする塩基配列(配列番号1)の両方又はいずれか一方の一部に変異が生じていてもよい。
ハイブリダイゼーション法の詳細な手順については、Molecular Cloning, A Laboratory Manual 2nd ed. (Cold Spring Harbor Laboratory Press (1989))等を適宜参照することができる。
ハイブリダイゼーション法を実施における「ストリンジェントな条件」とは、ハイブリダイゼーション後の洗浄時の条件であって、バッファーの塩濃度が15〜750mM、温度が25〜65℃、好ましくは塩濃度が15〜150mM、温度が45〜55℃の条件を意味する。具体的には、例えば、50mMで50℃等の条件を挙げることができる。さらに、このような塩濃度や温度等の条件に加えて、プローブ濃度、プローブの長さ、反応時間等の諸条件も考慮し、上記(b)のDNAを得るための条件を適宜設定することができる。
また、上記(b)のDNAは、当業者に公知の方法で適当な断片を用いてプローブを作製し、このプローブを用いてコロニーハイブリダイゼーション、プラークハイブリダイゼーション、サザンブロット等の公知のハイブリダイゼーション法により、cDNAライブラリーおよびゲノムライブラリーから得ることができる。上記ハイブリダイゼーションにおいてストリンジェントな条件としては、たとえば、1×SSC〜2×SSC、0.1%〜0.5%SDS及び30℃〜80℃の条件が挙げられ、より詳細には、60〜68℃で30分以上プレハイブリダイゼーションを行った後、プローブを添加して1時間以上68℃に保ってハイブリッド形成させ、その後、2×SSC、0.1%SDS中、室温で5〜15分の洗浄を1〜2回行う条件が挙げられる。
ハイブリダイズするDNAとしては、上記(a)のDNAの塩基配列に対して少なくとも70%以上の相同性を有する塩基配列であることが好ましく、より好ましくは80%以上、さらに好ましくは90%以上、さらに好ましくは95%以上である。
なお、上記(b)のDNAは、高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質をコードするDNAであることが重要であるため、翻訳後のアミノ酸配列を考慮したときに、例えば、第53番目〜第72番目のアミノ酸などの上記葉緑体包膜やチラコイド膜の通過に重要と考えられる部分のアミノ酸配列や、第14番目〜第18番目のアミノ酸、及び第47番目〜第60番目のアミノ酸などの高等植物の光合成明反応における電子伝達体として機能するために重要と考えられる部分のアミノ酸配列が、前述した(a)のタンパク質のアミノ酸配列から変異置換されないような、塩基配列を有するものが好ましい。
上記(b)のDNAとしては、例えば、上記(a)のDNAと完全に同一のものではないが、そのうちの第103番目の塩基を「T (チミン)」から「A (アデニン)」に置換し翻訳されるアミノ酸が「Ser (セリン)」から「Thr (トレオニン)」になるよう変異させたもの、第216番目の塩基を「C (シトシン)」から「T (チミン)」に置換するが翻訳されるアミノ酸には変化がないよう変異させたもの、及びこれらの塩基置換を組み合わせて変異させたもの、などが好ましく挙げられる。
本発明において、上記遺伝子は、高等植物において発現させるものであるため、アミノ酸に対応するコドンが、転写後、植物一般において通常用いられているコドン(好ましくは使用頻度の高いコドン)を示すDNAを含むものであることが必要である。
(4) 遺伝子の導入方法
高等植物の他の形質を変えずに、そのゲノム中に前述した融合タンパク質をコードする遺伝子(目的遺伝子)を導入して、新規な高等植物(形質転換高等植物)を作出する方法としては、限定はされないが、アグロバクテリウム法、エレクトロポレーション法、及びパーティクルガン法等の公知の遺伝子組換え技術を任意に採用することができる。例えば、限定はされないが、目的遺伝子を含むバイナリーベクターを導入したアグロバクテリウム属細菌(根頭がん種病菌)を用いる減圧浸潤法等が好適である。以下に、詳細に説明する。
(i) 組換えベクター
前述した融合タンパク質をコードする遺伝子を含む組換えベクターとしては、限定はされないが、上記バイナリーベクターとして用い得るものが好ましい。具体的には、Vir領域遺伝子の働きで切り出されるT-DNA領域を有するベクターの、当該T-DNA領域中に、植物ゲノムに組込みたい目的遺伝子を(GUS遺伝子の代わりに)挿入して構築される組換えベクターが好ましい。上記T-DNA領域を有するベクターとしては、例えば、pBI121ベクター(Clontech社製;カナマイシン耐性遺伝子を選択マーカーとし、35Sプロモーターの下流にGUS遺伝子を含む)、pBI101ベクター(Clontech社製;カナマイシン耐性遺伝子を選択マーカーとし、GUS遺伝子を含む)等が好ましく挙げられる。
また、減圧浸潤法に用い得るものではないが、宿主細胞に適した公知の発現ベクターに、目的遺伝子を挿入して構築される組換えベクターも、本発明の組換えベクターに含まれる。必要に応じ、当該遺伝子の上流に転写プロモーターやSD配列(宿主が原核細胞の場合)やKozak配列(宿主が真核細胞の場合)を、下流にターミネーターを、PCR等により付加しておく。上記転写プロモーター等、融合タンパク質の発現に必要な各要素は、目的遺伝子に含まれていてもよいし、もともと発現ベクターに含まれている場合はそれを利用してもよく、限定はされない。このような組換えベクターは、例えば、前述した融合タンパク質の大量生産等に用いることができる。
なお、これら各種組換えベクターの作製においては、制限酵素を用いる方法や、トポイソメラーゼを用いる方法等、公知の遺伝子組換え技術及び条件等を適宜採用して行うことができる。
(ii) 組換えベクターを導入してなる形質転換体
組換えベクターを導入する宿主としては、限定はされないが、最終的に減圧浸潤法を行う場合は、バイナリーベクターを導入する宿主としては、アグロバクテリウム属細菌(Agrobacterium tumefaciens等)を使用する。なお、アグロバクテリウム属細菌を用いる場合は、一般には、当該細菌が本来有しているTiプラスミド上のT-DNA領域中のがん遺伝子が、潰されているか又は取り除かれている等して、予め形質転換されている菌体(アグロバクテリウム属細菌の形質転換体)を用いる。その他、組換えベクターとして発現ベクターを導入する場合は、公知の宿主、例えば、大腸菌、枯草菌、酵母、カビ、ヒトやマウス等の各種動物細胞等を用いることができる。
宿主の形質転換の方法は、限定はされず、宿主と組換えベクターとの組み合わせを考慮して、公知の方法から適宜選択し実施することができる。例えば、電気穿孔法、リポフェクション法、ヒートショック法、PEG法、リン酸カルシウム法、DEAEデキストラン法等が好ましく実施できる。
得られる形質転換体においては、実際に用いた宿主と、組み換えベクターに含まれるEBNA1変異体遺伝子のコドン型の宿主とが、一致していてもよいし、異なっていてもよく、限定はされない。
(iii) 高等植物の形質転換、及び形質転換高等植物
高等植物のゲノム中に目的遺伝子を導入し、形質転換高等植物を作製する方法は、限定はされないが、前述した減圧浸潤法が好ましい。また、形質転換を行う植物体の状態は、成体またはカルスを用いることが好ましい。
減圧浸潤法による高等植物の形質転換方法では、具体的には、(a)前述した融合タンパク質をコードする遺伝子が挿入された組換えベクター(バイナリーベクター)を含む形質転換細菌(アグロバクテリウム属細菌の形質転換体)を高等植物の葉片に感染させ、(b)カナマイシン等の抗生物質を含む選択培地で培養し、(c)不定芽カルスを形成させ栽培して、形質転換高等植物を得るようにする。なお、減圧浸潤法を行うにあたり、各処理工程における手段および処理条件等は、すべて公知の範囲から適宜選択して行うようにすればよく、限定はされない。
上述のようにして得られた形質転換高等植物は、そのゲノム中に、前述した融合タンパク質をコードする遺伝子が導入されている。当該遺伝子を基にして発現された融合タンパク質は、前述したシグナルペプチドを有し植物細胞中の葉緑体包膜及びチラコイド膜の通過能を有するものである。よって、得られた形質転換高等植物は、チラコイド内腔にシトクロムc6(好ましくは光合成明反応の電子伝達体としてのシトクロムc6)を有するものである。
得られた形質転換高等植物においては、光合成明反応の電子伝達体として、プラストシアニン(PC)のみならずシトクロムc6も機能すると考えられ、効果的に生長促進が達成される。形質転換高等植物の成体の大きさ(背丈、根長、葉の大きさ等)は、限定はされないが、野生型のものに比べ、例えば、1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.5倍以上(例えば約1.9倍)となる。
また、形質転換高等植物においては、生長促進効果と同様に、各細胞中の各種分子数も増加する効果も得られる。例えば、クロロフィル分子の量は、限定はされないが、野生型のものに比べ、例えば、1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.5倍以上(例えば約1.6倍)となり、より一層光合成能が向上したものとなる。
さらに、得られた形質転換高等植物は、光合成効率が向上するため、その生産物であるATP合成量は、限定はされないが、野生型のものに比べ、例えば、1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.5倍以上(例えば約1.7倍)となる。同様に、NADPHの合成量は、限定はされないが、野生型のものに比べ、例えば、1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.5倍以上(例えば約1.7倍)となる。
さらに、得られた形質転換高等植物においては、ATP及びNADPHの合成量が増加することによって、これらがエネルギー源又は還元力となって働く、光合成暗反応の効率も向上したものとなる。例えば、形質転換高等植物は、二酸化炭素(CO2)を炭水化物に変換する炭素固定能が、野生型のものに比べ、例えば、1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.4倍以上となる。
さらに、得られた形質転換高等植物においては、光合成明反応・暗反応の効率が促進されたことによりタンパク質合成の効率も向上したものとなる。例えば、形質転換高等植物は、タンパク質の含量が、例えば1.1倍以上、好ましくは1.2倍以上、さらに好ましくは1.5倍以上となる。
また、硫酸および硝酸代謝や各種のアミノ酸合成、脂質合成量、色素ならびに花の増加なども期待できる。
なお、得られた形質転換高等植物においては、上述した各種作用効果を少なくとも1種有していることが好ましいが、より好ましくは2種以上、さらに好ましくは全て有していることである。
3.高等植物の生長促進方法、ATP、NADPH、デンプン及びタンパク質の合成促進方法、並びに炭素固定促進方法
前述したように、得られた形質転換高等植物においては、(i) 生長促進、(ii) ATP、NADPH、デンプン及びタンパク質の合成促進、並びに (iii) 炭素固定促進といった効果が認められる。よって、本発明は、高等植物の生長促進方法、高等植物のATP、NADPH、デンプン及びタンパク質からなる群から選ばれる少なくとも1つの合成促進方法、並びに高等植物の炭素固定促進方法をも含むものである。
これら方法は、具体的には、シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加された融合タンパク質をコードする遺伝子を高等植物のゲノム中に導入して発現させ、葉緑体のチラコイド内腔にシトクロムc6を存在させることを特徴とするものである。これら方法において、融合タンパク質、当該タンパク質をコードする遺伝子、当該遺伝子の高等植物ゲノム中への導入方法、及び、得られる形質転換高等植物における種々の作用効果等については、前述した本発明の高等植物の作出方法における説明及び例示等が同様に適用できる。
以下に、実施例を挙げて本発明をより具体的に説明するが、本発明はこれらに限定されるものではない。
【実施例1】
【0006】
導入用シトクロムc 6遺伝子と植物発現用ベクターの構築
(1) Porphyra yezoensis由来シトクロムc6成熟タンパク領域遺伝子の調製
本実施例では、P. yezoensis由来シトクロムc6遺伝子をクローニングすることにより入手し、ベクターへと連結しプラスミドを作製した。このプラスミドを鋳型として、今回の目的DNAであるP. yezoensis 由来シトクロムc6成熟タンパク領域遺伝子を増幅させた。その後電気泳動を行い、ゲル抽出により目的遺伝子を抽出した後に制限酵素(Sac I, Pst I)で消化することで、遺伝子を調製した。
反応液は、以下の組成となるように0.5 ml アシストPCR用チューブに調製した。
【表1】
Primer(1):PYC6-C-term Sac I(10 pmol/μl)(30 mer, GC-Cont.:43.3%)
5’-GGA GCT CTT ACC AAC CTT TTT CAG ATT GAG - 3’(配列番号7)
Primer(2):Cyt-SD (10 pmol/μl)(29 mer, GC-Cont.:48.2%)
5’-CCG CGG AGA CGT TAA ATT GAA GAA GAA GC- 3’(配列番号8)
上記反応液にミネラルオイルを15μl重層した。その後、PTC-100TM Programmable Thermal Controllerを用いて反応を行った。反応は、94℃で48秒、62℃で48秒、72で1分24秒の反応を1サイクルとして、これを31サイクル行った。
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。
得られた PCR生成物20 μlにゲルローディング緩衝液4 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
2%(w/v)アガロースゲルから目的DNAと思われるバンドを切り出し、できるだけ細かく刻んだ。そして、そのゲルをAmicon Ultra free(登録商標) DAに入れ、7,300 rpm, 4℃, 10 min.遠心分離を行った。遠心後Ultra free MC(上側のカラム部分)を取りはずし、溶出してきた溶液に等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3 M NaOAc(pH 5.2)を1/20 vol.、99.5%エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75%エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、50 μlのTEに溶解した。
次に、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 120 min.インキュベートすることで制限酵素SacIによる消化を行った。
【表2】
インキュベート後のサンプル溶液に、等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5%エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75%エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
続いて、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 120 min.インキュベートすることで制限酵素 PstIによる消化を行った。
【表3】
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物20 μlにゲルローディング緩衝液4 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。その後、2%(w/v)アガロースゲルから目的DNAと思われるバンドを切り出し、できるだけ細かく刻んだ。そして、そのゲルをAmicon Ultra free(登録商標) DAに入れ、7,300 rpm, 4℃, 10 min.遠心分離を行った。遠心後Ultra free MC(上側のカラム部分)を取りはずし、溶出してきた溶液に等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75%エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
結果を以下に示す。
P. yezoensis由来シトクロムc6遺伝子(全長)が挿入されているプラスミドを鋳型とし、シトクロムc6成熟タンパク領域(PYC6)遺伝子をPCRにより増幅させた(図4)。その後PCR産物を2%(w/v)アガロースゲル(TBE)電気泳動を行ったところ、357 bp付近に単一なバンドを確認することができた(図5)。このバンドがPYC6遺伝子であると考えられた。そこでこのPYC6遺伝子を2つの制限酵素(Sac I, Pst I)で消化し、2%(w/v)アガロースゲル(TBE)電気泳動し、PYC6遺伝子(Sac I, Pst Iサイト保有)と思われる265 bp付近のバンドをゲルから抽出し、フェノール抽出、エタノール沈殿を行うことで、Sac I, Pst Iサイトを保有したPYC6遺伝子を得た。
(2) シトクロムc6への葉緑体包膜及びチラコイド膜通過ペプチド遺伝子の付加
本項では、BamH IとPst Iによる制限酵素処理と脱リン酸化(BAP処理)を行った葉緑体包膜およびチラコイド膜通過ペプチド遺伝子と、Sac IとPst Iによる制限酵素処理を行った P. yezoensis由来シトクロムc6成熟タンパク領域遺伝子をライゲーションさせた。その後クローニングベクター(pBluescript(登録商標) II SK+)にライゲーションさせ、サブクローニングした後、植物導入用シトクロムc6(sp+PYC6)遺伝子の塩基配列を確認した。なお、下記のユニバーサルプライマーをDNAシーケンスの際に使用した。
ユニバーサル FITC化 Forward primer(M13 Fw primer)(2 pmol/μl)
5’- CGC CAG GGT TTT CCC AGT CAC GAC - 3’(配列番号9)
ユニバーサル FITC化 Reverse primer(M13 Rv primer)(2 pmol/μl)
5’- GAG CGG ATA ACA ATT TCA CAC AGG - 3’(配列番号10)
(2-1) 植物導入用シトクロムc6遺伝子の調製
以下の組成になるように反応液を調製した。
【表4】
調製した反応液を低温恒温槽(16℃)で一晩インキュベートし、ライゲーション反応させた。次に、この溶液にアルカリフォスファターゼを加え、脱リン酸化を行った。以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 3 hrs.インキュベートすることでBacterial Alkaline Phosphataseによる脱リン酸化を行った。
【表5】
インキュベート後のサンプル溶液に、等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
(2-2) クローニングベクターpBluescript(登録商標)II SK+の調製
以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 5 hrs.インキュベートすることで制限酵素 SacIによる消化を行った。
【表6】
インキュベート後のサンプル溶液に、等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
続いて、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 5 hrs.インキュベートすることで制限酵素BamH Iによる消化を行った。
【表7】
反応終了後、1.5%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物20 μlにゲルローディング緩衝液4 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TAE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。その後、1.5%(w/v)アガロースゲルから目的DNAと思われるバンドを切り出し、ゲル100 μgを100 μlに換算し、それに対しNaI solutionを3 vol.加えよく撹拌し、55℃で5分インキュベートしゲルを溶解させた。これにGLASSMILKを10 μl加え、室温で15分間激しく撹拌し、14,000 rpm, 4℃, 5 sec.遠心分離を行い、沈殿を回収した。この沈殿にNEW WASH 300 μlを加え、よく撹拌後、14,000 rpm, 4℃, 5 sec.遠心分離を行い、沈殿を回収した。この操作を3度繰り返して得られた沈殿を乾燥させ、20 μlの滅菌水に溶解させた。その後、溶液を0.5 ml アシストPCR用チューブへと移し、14,000 rpm, 4℃, 30 sec.遠心分離を行った。得られた上清を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
(2-3) 植物導入用シトクロムc6遺伝子のサブクローニング
シトクロムc6遺伝子のサブクローニングは、公知方法に従って行った(Hanahan, D., J. Mol. Biol., 166(4), 557-580 (1983).; 中山 広樹等 バイオイラストレイテッドII 遺伝子解析の基礎, 秀潤社, 83-88 (1996).)。
植物導入用遺伝子と、クローニングベクターを以下の組成になるように調製した。
【表8】
調製した反応液を低温恒温槽(16℃)で一晩インキュベートし、ライゲーション反応させた。大腸菌への形質転換は、以下の通りに行った。まず、150 μlのコンピテントセル(DH5a)を氷上である程度溶かし、そこにライゲーションさせた反応液4 μlを穏やかに加え、チップの先で軽くかき混ぜ、氷上で30分間放置した。放置終了後、42℃で20秒間ヒートショックを行った。その後氷上で3分間静置し、LB- Ampicillin-X-Gal-IPTG プレートにVector : Insert = 1:1、1:3それぞれ、菌体液100 μl、50μlをコンラージ棒を用いて、プレート全体に塗り広げ、37℃で一晩培養した。
(2-4) アルカリSDS法によるプラスミド調製
アルカリSDS法は、公知手法に従って行った(Weiss. B, et al., J. Biol. Chem., 243, 4543-4555 (1968); Birnboim, H. C., Methods Enzymol., 100, 243 (1983))。すなわち、3ml LB-Ampicillin 液体培地で200 rpm, 37℃, 16 hrs.震盪培養した菌体液を1.5 mlずつ1.5 ml micro centrifuged tubeに分注し、6,000 rpm, 4℃, 10 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。沈殿(菌体)に100 μlのSolution I(50 mM グルコース-25 mM Tris-HCl(pH 8.0)-10 mM EDTA)を加え、ボルテックスでよく撹拌し、200 μlのSolution IIを加え、静かに転倒させ撹拌した。5分間室温で放置後、さらに150 μlのSolution III(0.2N NaOH-1% SDS)を加え、氷上で30分間放置後、100 μlのクロロホルムを加え、14,000 rpm, 4℃, 10 min.遠心分離した。上清を新しいチューブに回収し、それに2倍量の99.5% エタノールを加え、静かに転倒させ撹拌し、15分間室温で放置した。その後、500 μlの70% エタノールを加え14,000 rpm, 4℃, 10 min.遠心分離し、得られた沈殿を3〜5 分間TOMY MICRO Vacで乾燥させ、20 μlのTEに溶かした。
得られたプラスミドに目的DNA が組み込まれていることを確認するために、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 120 min.インキュベートすることで制限酵素SacI, BamH Iによる消化を行った。
【表9】
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。この結果、目的DNAが組み込まれているサンプルに関してのみ、3 ml LB-Ampicillin液体培地での培養の残りの菌体液(1.5 ml)から、100 μlを5 ml LB-Ampicillin液体培地に植菌し、200 rpm, 37℃, 16 hrs.震盪培養した。
(2-5) QIAGEN Spin Miniprep Kitによるプラスミドの調製
5 ml LB-Ampicillin液体培地で培養した菌体液を6,000 rpm, 4℃, 10 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。沈殿(菌体)にキット(QIAGEN Spin Miniprep Kit(250)(QIAGEN))に含まれているBuffer P1を250 μl加え、十分に懸濁させた。
なお、QIAGEN Spin Miniprep Kit(250)(QIAGEN)にはCollection tube、QIA prep Spin Column、Buffer P1、Buffer P2、Buffer N3、Buffer PB、Buffer PE、Buffer EBが含まれている。
上記懸濁液に、Buffer P2を250 μl加え、静かに転倒させ溶液が均一になるまで撹拌した。さらにBuffer N3を350 μl加え静かに転倒させ撹拌し、13,000 rpm, 4℃, 10 min.遠心分離した。ここで得られた上清をあらかじめCollection tubeにセットしておいたQIA prep spin columnに移し、13,000 rpm, 4℃, 1 min.遠心分離した。Collection tubeにたまった濾液を捨て、QIA prep spin columnに、500 μlのBuffer PBを加え、13,000 rpm, 4℃, 1 min.遠心分離し、再び濾液を捨てた。さらにQIA prep spin columnにBuffer PE を750 μl加え、13,000 rpm, 4℃, 1 min.遠心分離し濾液を捨て、再び13,000 rpm, 4℃, 1 min.遠心分離し、QIA prep spin columnのみを新しい1.5 ml micro centrifuged tubeに移した。QIA prep spin columnの中央に50 μlのBuffer EBを加え、室温で1分間放置し、13,000 rpm, 4℃, 1 min.遠心分離し、この濾液をプラスミドサンプルとした。
得られたプラスミドに目的DNAが組み込まれていることを確認するために、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃、120 min. インキュベートすることで制限酵素 SacI, BamH Iによる消化を行った。
【表10】
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。ここで目的DNAが組み込まれており、単一性が確認されたものをシークエンスサンプルとした。
(2-6) ダイデオキシ法による塩基配列の決定
塩基配列の決定は、オートシークエンサーを用いて公知方法(Sanger, F., Sanger, F., Determination of nucleotide sequence in DNA., Science, 214, 1205-1210 (1981))により決定した。塩基配列決定後は、解析ソフトであるGENETYX-MACを用いてデータの分析を行った。
(2-7) 結果
2つの制限酵素(BamH I, Pst I)消化と脱リン酸化(BAP 処理)を行ったsp遺伝子を、2つの制限酵素(Sac I, Pst I)消化を行ったPYC6遺伝子とライゲーションさせ、さらに再度BAP処理を行い、フェノール抽出、エタノール沈殿により植物導入用シトクロムc6(sp+PYC6)遺伝子を作製することができた。
このsp+PYC6遺伝子の塩基配列を確認するため、クローニングベクター(pBluescript II SK+(図6)を2つの制限酵素(BamH I, Sac I)で消化したものにライゲーションし、サブクローニング後、ダイデオキシ法によるシークエンスを行い、sp+PYC6遺伝子の全塩基配列474bpを決定した(図7)。
そのシークエンス結果をGENETYX-MACを用いて既知のsp遺伝子とPYC6遺伝子と比較したところ、シグナルペプチド配列中の103番目のT(チミン塩基)がA(アデニン塩基)に変異している以外は完全に一致した。またPrimer配列中に付加した制限酵素サイトも確認した(図7)。今回確認された塩基の変異は、コードするアミノ酸をSerからThrに変えるものであったが、ともに性質と分子量も類似しており、それによる二次構造(aヘリックスなど)の変化はなく、膜通過というシグナルペプチドとしての機能には影響がないと考えられたため、このサンプル(sp+PYC6)を以後の実験に用いた
(3) 植物導入用シトクロムc6発現ベクターの作製
本実施例では、塩基配列を確認した植物導入用シトクロムc6(sp+PYC6)遺伝子を発現ベクターpBI121に挿入し、植物へ導入用シトクロムc6発現ベクター(pBI cyt c6)を構築した。
(3-1) 植物発現用ベクターpBI121の調製
以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、 37℃, 5 hrs.インキュベートすることで制限酵素SacIによる消化を行った。
【表11】
インキュベート後のサンプル溶液に、等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
続いて、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tube に調製し、 37℃, 5 hrs. インキュベートすることで制限酵素BamH Iによる消化を行った。
【表12】
反応終了後、1.5%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物20 μlにゲルローディング緩衝液4 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TAE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。その後、1.5%(w/v)アガロースゲルから目的DNAと思われるバンドを切り出し、ゲル100 μgを100 μlに換算し、それに対しNaI solutionを3 vol.加えよく撹拌し、55℃で5分インキュベートしゲルを溶解させた。これにGLASSMILKを10 μl加え、室温で15分間激しく撹拌し、14,000 rpm, 4℃, 5 sec.遠心分離を行い、沈殿を回収した。この沈殿にNEW WASH 300 μlを加え、よく撹拌後、14,000 rpm, 4℃, 5 sec.遠心分離を行い、沈殿を回収した。この操作を3度繰り返して得られた沈殿を乾燥させ、20 μlの滅菌水に溶解させた。その後、溶液を0.5 ml アシストPCR用チューブへと移し、14,000 rpm, 4℃, 30 sec.遠心分離を行った。得られた上清を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
(3-2) 植物発現用シトクロムc6発現ベクター(pBI cyt c6)の調製
前記の通り作製した導入用シトクロムc6(sp+PYC6)遺伝子は、以下の組成になるように反応液を調製した。
【表13】
調製した反応液を低温恒温槽(16℃)で一晩インキュベートし、ライゲーション反応させた。
結果を以下に示す。
塩基配列を確認したsp+PYC6遺伝子(BamH I, Sac I消化、BAP処理済み)を、植物発現用ベクター(pBI121)を2つの制限酵素(BamH I, Sac I)で消化したものにライゲーションし、植物導入用シトクロムc6発現ベクター(pBI cyt c6)を調製することができた。
【実施例2】
【0007】
形質転換植物(シトクロムc 6導入Arabidopsis thaliana)の作製
(1) 植物感染用Agrobacterium tumefaciensの調製
(1-1) シトクロムc6発現ベクター(pBI cyt c6)導入大腸菌の調製
本実施例では、植物導入用シトクロムc6遺伝子を挿入した植物発現用ベクター(pBI cyt c6)を宿主大腸菌 HB101に形質転換した。その後、数個のコロニーを選び3 mlのLB-Kanamycin 液体培地で培養し、アルカリSDS法によるプラスミド調製を行うことでベクターの構築と宿主大腸菌HB101への形質転換の有無を確認した。
大腸菌への形質転換は、以下の通りに行った。まず、150 μlのコンピテントセル(HB101)を氷上である程度溶かし、そこにライゲーションさせた反応液4 μlを穏やかに加え、チップの先で軽くかき混ぜ、氷上で30分間放置した。放置終了後、42℃で20秒間ヒートショックを行った。その後氷上で3分間静置し、LB-Kanamycinにそれぞれ菌体液 100 μl、50μlをコンラージ棒を用いて、プレート全体に塗り広げ、37℃で一晩培養した。
培養後、形質転換体(コロニー)をランダムに20クローンピックアップし、そのコロニーを3 ml LB-Kanamycin培地に植菌し、200 rpm, 37℃, 16 hrs.震盪培養した。培養した菌体液を1.5 mlずつ1.5 ml micro centrifuged tubeに分注し、6,000 rpm, 4℃, 10 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。沈殿(菌体)に100 μlのSolution I(前出)を加え、ボルテックスでよく撹拌し、200 μlのSolution II(0.2 N NaOH-1% SDS)を加え、静かに転倒させ撹拌した。5分間室温で放置後、さらに150 μl の Solution III(前出)を加え、氷上で30分間放置後、100 μlのクロロホルムを加え、14,000 rpm, 4℃, 10 min.遠心分離した。上清を新しい回収し、それに 2 倍量の99.5% エタノールを加え、静かに転倒させ撹拌し、15分間室温で放置した。その後、500 μlの70% エタノールを加え14,000 rpm, 4℃, 10 min.遠心分離し、得られた沈殿を3〜5分間 TOMY MICRO Vacで乾燥させ、20 μlのTEに溶かした。
得られたプラスミドに目的DNAが組み込まれていることを確認するために、以下に示す組成となるようにサンプルを1.5 ml micro centrifuged tubeに調製し、37℃, 120 min. インキュベートすることで制限酵素BamHIと SacIによる消化を行った。
【表14】
反応終了後、1.5%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TAE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
結果を以下に示す。
植物導入用シトクロムc6発現ベクター(pBI cyt c6)を、宿主大腸菌HB101へと形質転換させた(図8)。その後、形質転換体(コロニー)をランダムに20クローンピックアップした。その結果、20サンプル中 3クローンにsp+PYC6遺伝子と思われるサイズ(474 bp)付近に単一なバンドを確認することができた。よってこのプラスミド内にはsp+PYC6遺伝子が挿入されていることが示された。このプラスミドを保有する大腸菌HB101を以後の実験に用いた。
(1-2) 形質転換Agrobacterium tumefaciensの調製と選抜
本項では、高等植物 A. thalianaへと目的遺伝子を導入するために必要な宿主アグロバクテリウム(Agrobacterium tumefaciens)を調製するため、三者混合培養(トリペアレンタルメイティング)を行い、得られた形質転換体のうち、数個のコロニーを選び3 mlのLB-Kanamycin液体培地で培養し、アルカリSDS法によるプラスミド調製を行うことで宿主アグロバクテリウムへの形質転換の有無を確認した。
(1-2-1) 各種菌体の培養
(a) Agrobacterium tumefaciens(LBA4404)*pAL4404保有
菌体のグリセロールストック溶液をYEP-Streptomycin寒天培地(プレート)(30 μg/ml Streptomycin)に白金耳を用いて、プレート全体に線画状に塗り、30℃でおよそ 40 hrs.培養した。培養後、コロニーをピックアップし、そのコロニーを5 ml YEP-Streptomycin(30 μg/ml Streptomycin)培地で200 rpm, 30℃, 30 hrs.震盪培養した。
(b) Escherichia coli(HB101)*pRK2013保有
菌体のグリセロールストック溶液をLB-Kanamycin 寒天培地(プレート)(40 μg/ml Kanamycin)に白金耳を用いて、プレート全体に線画状に塗り、37℃で16 hrs.培養した。培養後、コロニーをピックアップし、そのコロニーを5 ml LB-Kanamycin(40 μg/ml Kanamycin)培地で200 rpm, 37℃, 16 hrs.震盪培養した。
(c) Escherichia coli(HB101)*pBI cyt c6 保有
菌体のグリセロールストック溶液をLB-Kanamycin 寒天培地(プレート)(40 μg/ml Kanamycin)に白金耳を用いて、プレート全体に線画状に塗り、37℃で16 hrs.培養した。培養後、コロニーをピックアップし、そのコロニーを5 ml LB-Kanamycin(40 μg/ml Kanamycin)培地で200 rpm, 37℃, 16 hrs.震盪培養した。
(1-2-2) トリペアレンタルメイティング
トリペアレンタルメイティングは、公知方法により行った(駒嶺 穆, 野村 港二, 「生物化学実験法 41」植物細胞工学入門, 学会出版センター, 298-302 (1998))。すなわち、5 mlのYEP-Sm.液体培地あるいはLB-Km.液体培地で震盪培養した三種の菌体液(A. tumefaciens(LBA4404)*pAL4404保有、E. coli(HB101)*pBI cyt c6 保有、E. coli(HB101)*pRK2013保有)を 5,000 rpm, 4℃, 5 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。洗浄のため培養時に使用した液体培地(YEP 培地あるいは LB 培地)を沈殿(菌体)に加えよく撹拌し、もう一度 5,000 rpm, 4℃, 5 min.遠心分離した。遠心分離後、菌体を培養時に使用した液体培地(YEP培地あるいはLB培地)3 mlに溶解させた。次にそれぞれの菌体液を30 μlずつMin A-Kanamycin寒天培地(プレート)に重ねるように滴下し、そのまま静置した状態で30℃, 3 days培養した。培養後、最少培地である Min A-Kanamycin 寒天培地(プレート)に生えてきた菌体を白金耳を用いてすべてかき取り、その菌体を10 mM MgSO4 200 μlに溶解させた。その後、その菌体溶液を再び Min A-Kanamycin 寒天培地(プレート)に白金耳を用いて、プレート全体に線画状に塗り、30℃でおよそ3 days.培養した。培養後、コロニーをピックアップし、そのコロニーを5 ml YEP-Kanamycin(40 μg/ml Kanamycin)培地で 200 rpm, 30℃, 40 hrs.震盪培養した。
(1-2-3) アルカリSDS法によるTiプラスミド調製
5 ml YEP-Kanamycin(40 μg/ml Kanamycin)液体培地で200 rpm, 30℃, 40 hrs.震盪培養した菌体液を1.5 mlずつ1.5 ml micro centrifuged tubeに分注し、5,000 rpm, 4℃, 10 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。1 mlのS-bufferを沈殿(菌体)に加え、ボルテックスでよく撹拌し、8,000 rpm, 4℃, 10 min.遠心分離し、アスピレーターで上清を吸引して取り除いた。
なお、S-bufferは、C6H12O6(グルコース(M.W. : 180.16))を13.512 g秤取り、1M Tris-HCl(pH 8.0)12.5 ml、0.5M EDTA(pH 8.0)10 ml加え、超純水で500 mlに定容し、オートクレーブ処理(121℃、10分)を行うことにより調製した。
沈殿に再び100 μlのS-bufferと10 mg/ml リゾチームを5 μl加え、10分間室温で放置した。そこに200 μl Solution II、TE-saturated Phenolを30 μl加え、静かに転倒させ撹拌した。5分間室温で放置後、さらに150 μlのSolution IIIを加え、-20℃で15分放置した後、12,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに 14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解した。
得られたプラスミドに1 mlのRNase Aを加え、37℃, 90 min.インキュベートすることでRNase処理を行った。反応終了後、1.5%(w/v)アガロースゲル電気泳動にて確認を行った。得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TAE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
結果を以下に示す。A. tumefaciens(LBA4404)はYEP-Sm.培地で培養し、E. coli(HB101)はLB-Km.培地で培養した。その後、トリペアレンタルメイティング(三者混合培養)によりA. tumefaciens(LBA4404)へpBI cyt c6を導入し、形質転換させた。次にトリペアレンタルメイティングによりTiプラスミド(pBI cyt c6)が導入された形質転換体A. tumefaciensをランダムに10クローンピックアップした。そのコロニーを3 ml YEP-Km.培地にて培養し、その後、アルカリSDS法によってプラスミドを調製し、これを1%(w/v)アガロースゲル(TAE)電気泳動した。その結果、1クローンにおいて、pBI cyt c6と思われるサイズ(13.5 kbp)にバンドを確認することができた。よって形質転換体A. tumefaciensにはTiプラスミド(pBI cyt c6)が導入されていることが示された。
(2) 形質転換植物(シトクロムc6導入Arabidopsis thaliana)の作製
(2-1) Arabidopsis thalianaの栽培
よく水洗いした7.5×7.5丸型ポットにバーミキュライトを九分目位まで入れた。次に、HYPONeX原液を2,000倍希釈した溶液を500 ml用意し、カッターで厚さ1 cmほどにスライスしておいたロックウールを入れた。十分に水分を含んで湿ったロックウールをピンセットでつまみ、7.5×7.5丸ポットの上に3つずつのせた。台所用三角コーナーのメッシュをポットの口を覆える位の大きさに切り、よく水洗いし、ポットの口にかぶせて輪ゴムでとめた。このポットに、種子を1粒ずつ播種した。種子を播種した後、4℃, 3 days低温処理を行った。低温処理終了後、植物育成用光照射インキュベーター内に移し、22℃・長日条件下で栽培した。
発芽後は2日に1度程度水を与え、週に1度程1,000倍希釈したHYPONeXを液体肥料として与えた。適当に間引きを行い、およそ3週間後植物体が抽台し始めたときに摘心し、竹串(支柱)を利用して成長した植物体を支え、種子が完全に熟したら採種し、乾燥状態で保存しておいた。
(2-2) 形質転換Agrobacterium tumefaciensの植物体への感染
次に、高等植物A. thaliana(col-0)に植物導入用シトクロムc6(sp+PYC6遺伝子)を導入するためアグロバクテリウム法の一つである減圧浸潤法により、形質転換植物を作製した(Bechtold, N., Pelletier, G., Methods in Molecular Biology, 82 (1998))。具体的には以下の通りである。
(2-2-1) Agrobacterium tumefaciens の培養
菌体のグリセロールストック溶液をMin A-Km.寒天培地(プレート)(400 μg/ml Kanamycin)に白金耳を用いて、プレート全体に線画状に塗り、30℃でおよそ40 hrs.培養した。培養後、コロニーをピックアップし、そのコロニーを10 ml YEP-Km.(40 μg/ml Kanamycin)培地で200 rpm, 30℃, 30 hrs.震盪培養(前培養)し、その培養液を新たに100 ml YEP-Km.(40 μg/ml Kanamycin)培地に全量加え、さらに200 rpm, 30℃, 30 hrs.震盪培養(本培養)した。
(2-2-2) 植物体 Arabidopsis thaliana(col-0)の栽培
形質転換させる植物として用いるA. thaliana(col-0)の播種から種子の採種までの栽培条件は以下のとおりである。
低温処理:4℃(暗所)で3日間静置
植物育成温度:22℃
照射光:長日条件(100 μE m-2 s-1)
液体肥料:HYPONeX(1/1,000 倍希釈)
植物個体によって多少差はあるが、Wild type株の場合、播種後7日(低温処理3日間を含める)で発芽し、発芽後20日前後で植物が抽台した。その後、摘心処理し5〜6日で側枝が伸長し始め、同時に最初の花の開花・結実が始まった。採種は緑色のさやが薄茶色になり縦裂し始めたものから回収した。一つのさやからおよそ50粒の種子が得られた。回収した種子は、播種まで乾燥させた状態で保存しておいた。
(2-2-3) 植物体への感染
感染用植物体Arabidopsis thaliana(col-0)を上記(2-2-2)に記載の方法に従って栽培し、抽台を始めたころまで生育させた。その後、茎から茎葉が誘導され始めたときに摘心し、すでに開花・結実している花を切除した。なお、実験は、この植物体と上記(2-2-1)の菌体培養液の準備が同時に整うよう調整した。
まず、浸潤操作を行う実験台にキムタオル敷いておいた。
次に、A. tumefaciens 培養液を浸潤用懸濁培地でおよそ2/3倍希釈し、1L容ビーカーに入れた。そのビーカーを吸引鐘内にセットし、そこへ培養懸濁液を吸引しすぎないよう直前に十分に吸水させた植物体A. thalianaを植えたポットごとビーカーの上に逆さにして固定し、植物体を培養懸濁液に浸けた。
減圧浸潤法で用いたA. tumefaciens培養液は、OD600=1.2〜1.5のものをOD600=0.8となるように浸潤用懸濁培地で約2/3倍希釈したものを使用した。また減圧浸潤時間は30秒、1分、5分、10分と検討した結果、5分が最も適していた。それは、減圧浸潤時間が長くなるほどその後の植物体の生育は悪くなったが、5分間の処理の場合、それほど生育の悪化は見られず、花数及び結実したさや(種子)数も1分間処理したものと大差がなかったためである。
次に、培養懸濁液に浸した植物体を吸引鐘に入れ、アスピレーターで減圧した。減圧終了後、吸引鐘から植物体を取り出し、キムタオルで余分な培養懸濁液を落とした。その植物体をプラスチックトレイに横倒しにして、端に水を滴下し、ふたをして20℃で2〜3日放置した(その間は水を与えなかった)。その後、ポットをおこし、植物育成用光照射インキュベーター内で22℃、長日条件で通常どおり栽培を行った。2日に1度程度水やりを行い、週に1度程1,000 倍希釈したHYPONeXを液体肥料として与えた。適当に間引きを行い、およそ3週間後植物体が抽台し始めたら摘心し、竹串(支柱)を利用して成長した植物体を支え、種子が完全に熟したときは採種し、乾燥状態で保存しておいた。
【実施例3】
【0008】
形質転換植物におけるシトクロムc 6遺伝子導入と発現の解析
(1) PCR法を用いたゲノムDNA中の導入遺伝子の解析
本実施例では、形質転換体Arabidopsis thalianaよりゲノムDNAを抽出し、そのゲノムDNAを鋳型としてPCRにより導入遺伝子の増幅の有無をもって植物体内への導入を確認した。
(1-1) 形質転換体からゲノムDNAの抽出
葉2〜3枚程度の形質転換体Arabidopsis thalianaをあらかじめ冷却した乳鉢の中に入れ、素早く液体窒素を加え、植物体がパウダー状になるまでホモジナイズした。その後、パウダー状になった試料を1.5 ml micro centrifuged tubeに入れ秤量した。Wash bufferに終濃度0.5%ととなるように2-Mercaptoethanolを加えた溶液を、上記秤量後の試料に1 ml加えよく混ぜた。12,500 rpm, 4℃, 10 min.遠心分離後、上清を捨てた。そこへSolution Iに終濃度1%となるように2-Mercaptoethanolを加えた溶液を300 μl加えボルテックスでよく混ぜた。次に、1% NaBH4を30 μl、Solution IIを150 μl加え、数秒ボルテックスにかけた。50℃で10分インキュベートし、その溶液にISOPLANT II(NIPPON GENE)に含まれる100 μlのSolution III-Aと120 μlのSolution III-Bを入れ、氷上で10分間放置した。
12,500 rpm, 4℃, 10 min.遠心分離し、水相を回収した。そこへ2倍量の99.5% エタノールを加え、直ちに8,000 rpm, 室温, 1 min遠心分離し、上清を捨てた。さらに70% エタノールを1 ml加え8,000 rpm, 室温, 1 min遠心分離し、上清を捨てた。沈殿物をある程度まで風乾させ、50 μlのTEに溶解させた。この溶液に1 mg/ml RNase Aを1 μl加え、37℃で30分間インキュベートした。その後、ゲノムDNAの抽出を確認するために、得られたゲノムDNA5 μlにゲルローディング緩衝液1 μlを加え、マーカーにλHindIIIを用いて1.5%(w/v)アガロースゲルにて電気泳動を行った。泳動は泳動緩衝液(1×TAE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
電気泳動によるゲノムDNA 存在の確認後、その濃度測定を行うためゲノムDNA溶液を100倍希釈し、分光光度計でUV測定した。算出された濃度をもとに10 ng/μlとなるようにゲノムDNA溶液を調製し、今後の実験に用いた。
(1-2) PCR
PCRは、公知方法に従って行った(Sambrook, J. D., W. Russell, Molecular Cloning : a Laboratory Manual, 3rd edn. Corld Spring Harbor Laboratory Press, Corld Spring Harbor, New York)。
以下の組成となるように反応液を0.5 ml アシストPCR用チューブに調製した。
【表15】
この反応液にミネラルオイルを15 μl重層した。その後、PTC-100TM Programmable Thermal Controllerを用いて反応を行った。
プライマーとして以下のものを用いた。
Primer(3) ATP-NA1 Bam(10 pmol/μl)(31mer, GC-Cont.:54.8%)
5’-GGA TCC ATG GCC GCA ATT ACA TCA GCT ACC G-3’(配列番号11)
Primer(1) PYC6-C-term SacI(10 pmol/μl)(30mer, GC-Cont.:43.3%)
5’- GGA GCT CTT ACC AAC CTT TTT CAG ATT GAG - 3’(配列番号7)
ユニバーサル FITC化 Forward primer(M13 Fw primer)(2 pmol/μl)
5’- CGC CAG GGT TTT CCC AGT CAC GAC - 3’(配列番号9)
ユニバーサル FITC化 Reverse primer(M13 Rv primer)(2 pmol/μl)
5’- GAG CGG ATA ACA ATT TCA CAC AGG - 3’(配列番号10)
反応は、(94℃で48秒、64℃で48秒、72℃で1分24秒)×31サイクルの条件で行った。
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。
得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
(1-3) PCR産物のゲル抽出
2%(w/v)アガロースゲルから、目的DNAと思われるバンドを切り出し、できるだけ細かく刻んだ。そして、そのゲルをAmicon Ultra free (登録商標) DAに入れた。7,300 rpm, 4℃, 10 min.遠心分離を行った。遠心後Ultra free MC(上側のカラム部分)を取りはずし、溶出してきた溶液に等量のTE-saturated Phenolを加えボルテックスを用いてよく撹拌した。そして、14,000 rpm, 4℃, 10 min.遠心分離を行った。水層を新しい1.5 ml micro centrifuged tubeに移し、3M NaOAc(pH 5.2)を1/20 vol.、99.5% エタノールを 2 vol.加えた。よく撹拌した後、-20℃で30分放置した。その後14,000 rpm, 4℃, 10 min.遠心分離を行い、上清を捨てた。沈殿に75% エタノールを500 μl加え沈殿をよく洗浄した。さらに14,000 rpm, 4℃, 5 min.遠心分離を行い、上清を捨てた。沈殿をある程度風乾させた後、20 μlのTEに溶解させた。この溶液に目的のDNAが単一に抽出・精製されているかを確認するため、2%(w/v)アガロースゲル電気泳動を行った。得られたサンプル10 μlにゲルローディング緩衝液2 μlを加え、泳動には泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
(1-4) PCR 産物のサブクローニング
PCR産物のサブクローニングは、公知手法に従って行った(Hanahan, D., J. Mol. Biol., 166(4), 557-580 (1983).; 中山 広樹等 バイオイラストレイテッドII 遺伝子解析の基礎, 秀潤社, 83-88 (1996).)。
PCR 生成物に以下の組成になるように調製した。
【表16】
調製した反応液を低温恒温槽(16℃)で一晩インキュベートし、ライゲーション反応させた。大腸菌への形質転換は、以下の通りに行った。まず、150 μlのコンピテントセル(DH5a)を氷上である程度溶かし、そこにライゲーションさせた反応液 4 μlを穏やかに加え、チップの先で軽くかき混ぜ、氷上で30分間放置した。放置終了後、42℃で20秒間ヒートショックを行った。その後氷上で3分間静置し、LB-Ampicillin-X-Gal-IPTGプレートにVector : Insert = 1:1、1:3それぞれ、菌体液100 μl、50μlをコンラージ棒を用いて、プレート全体に塗り広げ、37℃で一晩培養した。
(1-5) アルカリSDS法によるプラスミド調製
本項におけるプラスミド調製は、実施例1(2-4)に記載した内容と同様にして行った。
(1-6) QIAGEN Spin Miniprep Kitによるプラスミドの調製
本項におけるプラスミド調製は、実施例1(2-5)に記載した内容と同様にして行った。
(1-7) ダイデオキシ法による塩基配列の決定
本項における塩基配列の決定は、実施例1(2-6)に記載の方法と同様にして行った。結果を以下に示す。
形質転換体A. thalianaの葉2〜3枚ほどから、ISOPLANT II を用いて純度の高いゲノムDNAを抽出した。そのゲノムDNA溶液を鋳型とし、植物導入用シトクロムc6遺伝子の導入の有無を確認するためPCRにより目的遺伝子を増幅させた。その後PCR産物を2%(w/v)アガロースゲル(TBE)電気泳動を行ったところ、474 bp付近に単一なバンドを確認した(図9)。この増幅した遺伝子の塩基配列を確認するため、ゲル抽出後、サブクローニングを行い、ダイデオキシ法によるシークエンスにより増幅した遺伝子の全塩基配列474 bpを決定した。そのシークエンス結果をGENETYX-MACを用いて既知のsp遺伝子配列とPYC6遺伝子配列と比較したところ、導入遺伝子同様シグナルペプチド遺伝子配列中の103番目のT(チミン塩基)がA(アデニン塩基)に変異している以外は完全に一致した。以上の結果より植物体(A. thaliana)内にsp+PYC6遺伝子が導入されたことが示された。
(2) RT-PCR法を用いた全RNA中の発現遺伝子の解析
本項では、形質転換体Arabidopsis thalianaより全RNAを抽出し、その全RNAを鋳型としてRT-PCRにより導入遺伝子の増幅の有無をもって植物体内での遺伝子の発現を確認した。
Primer(3) ATP-NA1 Bam (10 pmol/μl)(31 mer, GC-Cont.:54.8%)
5’- GGA TCC ATG GCC GCA ATT ACA TCA GCT ACC G-3’(配列番号11)
Primer(1) PYC6-C-term SacI (10 pmol/μl)(30 mer, GC-Cont.:43.3%)
5’- GGA GCT CTT ACC AAC CTT TTT CAG ATT GAG - 3’(配列番号7)
ユニバーサル FITC化 Forward primer(M13 Fw primer)(2 pmol/μl)
5’- CGC CAG GGT TTT CCC AGT CAC GAC - 3’(配列番号9)
ユニバーサル FITC化 Reverse primer(M13 Rv primer)(2 pmol/μl)
5’- GAG CGG ATA ACA ATT TCA CAC AGG - 3’(配列番号10)
(2-1) 形質転換体から 全RNAの抽出
形質転換体から全RNAの抽出は、市販のキット(RNeasy Plant Mini Kit(QIAGEN))を用い、説明書に従って行った。
(2-2) PCR
PCRは、公知手法に従って行った(Sambrook, J. D., W. Russell, Molecular Cloning : a Laboratory Manual, 3rd edn. Corld Spring Harbor Laboratory Press, Corld Spring Harbor, New York)。
以下の組成となるように反応液を0.5 ml アシストPCR用チューブに調製した。
【表17】
この反応液にミネラルオイルを15 μl重層した。その後、PTC-100TM Programmable Thermal Controllerを用いて反応を行った。
反応終了後、2%(w/v)アガロースゲル電気泳動にて確認を行った。得られたPCR生成物10 μlにゲルローディング緩衝液2 μlを加え、泳動用サンプルとした。泳動は泳動緩衝液(1×TBE)を用い、100 Vで30分間行った。泳動終了後、0.1 mg/ml エチジウムブロマイドで7分間染色を行い、10分間超純水で脱色を行った。脱色を行ったゲルをUVトランスイルミネーター上にのせ、手動式フード付きポラロイド(登録商標)カメラで写真に記録した。
(2-3) PCR産物のゲル抽出
本項におけるPCR産物のゲル抽出は、実施例3(1-3)と同様にして行った。
(2-4) PCR 産物のサブクローニング
本項におけるPCR産物のサブクローニングは、実施例3(1-4)に記載の方法と同様にして行った。
(2-5) アルカリSDS法によるプラスミド調製
本項におけるアルカリSDS法は、公知手法に従って、実施例1(2-4)に記載の方法と同様に行った(Weiss. B, et al., J. Biol. Chem., 243, 4543-4555 (1968); Birnboim, H. C., Methods Enzymol., 100, 243 (1983))。
(2-6) QIAGEN Spin Miniprep Kitによるプラスミドの調製
本項におけるQIAGEN Spin Miniprep Kitによるプラスミドの調製は、実施例1(2-5)に記載の方法と同様に行った。
(2-7) ダイデオキシ法による塩基配列の決定
本項における塩基配列の決定は、実施例1(2-6)に記載の方法と同様にして行った(Sanger, F., Sanger, F., Determination of nucleotide sequence in DNA., Science, 214, 1205-1210 (1981))。
(2-8) 結果
遺伝子の導入が確認された形質転換植物(PYC6導入植物)の葉2〜3枚ほどから、RNeasy Plant Mini Kitを用いて発現している全RNAを抽出した。その全RNA溶液を鋳型とし、sp+PYC6遺伝子の発現の有無を確認するためRT-PCRにより目的遺伝子を増幅させた。その後PCR産物を2%(w/v)アガロースゲル(TBE)電気泳動を行ったところ474 bp付近に単一なバンドを確認した(図9)。この増幅した遺伝子の塩基配列を確認するため、ゲル抽出後、サブクローニングを行い、ダイデオキシ法によるシークエンスにより増幅した遺伝子の全塩基配列474 bpを決定した。そのシークエンス結果をGENETYX-MACを用いて既知のsp遺伝子配列とPYC6遺伝子配列と比較したところ、導入遺伝子同様シグナルペプチド遺伝子配列中の103番目のT(チミン塩基)がA(アデニン塩基)に変異している以外は完全に一致した。以上の結果より植物体(A. thaliana)内にsp+PYC6遺伝子が発現していると考えられた。
(3) ウエスタンブロッティング法を用いた発現タンパク質の解析
本項では、形質転換体Arabidopsis thalianaより葉緑体を抽出し、そこから得られた葉緑体タンパク質画分に導入したシトクロムc6が発現していることを確認した。
(3-1) 葉緑体画分の調製
葉緑体画分は公知手法に従って調製した(中村 研三等(監修),植物のタンパク質実験プロトコール, 細胞工学シリーズ第9巻, 秀潤社, 114-117 (1998))。
5〜6 g程度の形質転換体Arabidopsis thalianaをあらかじめ冷却した乳鉢の中に入れ、素早く液体窒素を加え、植物体がパウダー状になるまでホモジナイズした。その後、パウダー状になった試料に氷冷しておいた破砕Bufferを20 ml加えよく撹拌し、4重にしたミラクロスで濾過した。得られた濾液を15 ml容コーニングチューブに移し、7,000 rpm, 4℃, 15 sec.遠心分離し上清を丁寧に取り除いた。沈殿にその5 vol.の破砕Bufferを加え均一になるまで混ぜ、あらかじめ10 mlの30%パーコールをいれておいたガラス遠沈管に静かに重層し、2,000 rpm, 4℃, 3 min.遠心分離した。終了後上清のパーコールを取り除き、沈殿を5 mlの破砕Bufferで均一になるまで混ぜ、再び2,000 rpm, 4℃, 3 min.遠心分離した(この作業を2度繰り返す)。最後に沈殿を1 mlの破砕Bufferに溶解させ、その溶液を葉緑体画分とした。
(3-2) ウエスタンブロッティング
上記(3-1)で得られた葉緑体画分溶液にCelLytic P Plant Cell Lysis/Extraction reagentを1 ml加え、10,000 rpm, 4℃, 10 min.遠心分離し、得られた上清を葉緑体タンパク抽出溶液とした。1.5 ml microcentrifuged tubeに1 μg/μl分子量標準マーカー5 μlと超純水を5 μl、葉緑体タンパク抽出溶液を加え、それぞれのチューブに等量の試料用緩衝液を加え、よく撹拌した。調製したチューブを40℃、30 min.湯浴中でインキュベートし、泳動用試料とした。
泳動槽に陽極および陰極電極液、SDS-PAGEゲルをセットし、ウェルに泳動用試料20 μlを慎重にアプライし、30 Vの定電圧で泳動用試料を分離ゲルの上端まで泳動し、100 Vの定電圧でマーカーがゲルの下端から約5 mm程度まで泳動した。泳動中にあらかじめ、濾紙をブロッティング緩衝液A、B、Cに各2枚ずつ浸し、PVDF膜は少量のメタノールに5 sec.浸して、ブロッティングC液に浸した。泳動が終了したら、ゲルをガラス板から外し、ブロッティングC液に5 min.ほど浸した。セミドライブロッティング装置に下からAの濾紙2枚、Bの濾紙2枚、PVDF膜、ゲル、Cの濾紙2枚の順で重ね、セット後に60 mAの定電流で90 min.転写した。ブロッティング終了後、直ちに以下に示す順に操作し、抗原抗体反応の操作を行った。
(i) PVDF膜をTBSに浸し5分間震盪する(2回)。
(ii) 5% BSA in TBSに浸し1時間から一晩震盪する。
(iii) TBSに浸し5分間震盪する。
(iv) st-antibodyに浸し2時間震盪する。
(v) TTBSに浸し5分間震盪する(2回)。
(vi) 2nd-antibodyに浸し1時間30分震盪する。
(vii) TTBSに浸し5分間震盪する(2回)。
(viii) TBSに浸し5分間震盪する(2回)。
(ix) 2nd-antibody development reagentに浸し20分震盪する(遮光)。
(x) 超純水で洗浄する。
(3-3) 結果
PYC6導入植物よりパーコールの密度勾配遠心により葉緑体画分を分離し、得られた葉緑体タンパク質をSDS-PAGEにより電気泳動的に分子量でふるい分けた。次に、分離した全てのタンパク質をPVDF膜へと転写した後、P. yezoensis 由来シトクロムc6を認識する一次抗体(ウサギ抗体)と抗ウサギ抗体(二次抗体)を用いて抗原抗体反応を行ったところ、PYC6導入植物のみに抗原抗体反応を検出することができた(図10)。以上の結果から、PYC6導入植物において導入したPYC6は葉緑体中で発現していることが明らかとなった。
(4) 発現したシトクロムc6タンパク質のN末端アミノ酸配列解析
本項では、上記(3)で抗原抗体反応を示したPYC6タンパクのN末端アミノ酸配列を解析し、シグナルペプチド切断の有無を確認した。
上記(3-1)で得られた葉緑体画分溶液にCelLytic P Plant Cell Lysis/Extraction reagent を1 ml加え、10,000 rpm, 4℃, 10 min.遠心分離し、得られた上清を葉緑体タンパク抽出溶液とした。1.5 ml microcentrifuged tubeに1 μg/μl分子量標準マーカー5 μlと超純水を5 μl、葉緑体タンパク抽出溶液を加え、それぞれのチューブに等量の試料用緩衝液を加え、よく撹拌した。調製したチューブを40℃、30 min.湯浴中でインキュベートし、泳動用試料とした。
泳動槽に陽極および陰極電極液、SDS-PAGEゲルをセットし、ウェルに泳動用試料20 μlを慎重にアプライし、30 Vの定電圧で泳動用試料を分離ゲルの上端まで泳動し、100 Vの定電圧でマーカーがゲルの下端から約5 mm程度まで泳動した。泳動中にあらかじめ、濾紙をブロッティング緩衝液A、B、Cに各2枚ずつ浸し、PVDF膜は少量のメタノールに5 sec.浸して、ブロッティングC液に浸した。泳動が終了したら、ゲルをガラス板から外し、ブロッティングC液に5 min.ほど浸した。セミドライブロッティング装置に下からAの濾紙2枚、Bの濾紙2枚、PVDF膜、ゲル、Cの濾紙2枚の順で重ね、セット後に60 mAの定電流で90 min.転写した。ブロッティング終了後、CBB染色、脱色操作を行い、目的タンパク質部分を1 mm x 5 mmの大きさに切り1.5 ml micro centrifuged tubeに移した。これをシーケンスサンプルとしてApplied Biosystems Protein Sequencer 492型でアミノ酸配列の解析を行った。
形質転換植物より得られたPYC6抗体と抗原抗体反応を示す分子量およそ9.1 kDaのタンパク質のN末端アミノ酸配列をエドマン法により解析した結果、その配列はN末端1残基目から順に ADLDNGEKVF(配列番号12)であった(下記表を参照)。
【表18】
これはPYC6の成熟タンパク領域のN末端アミノ酸配列と完全に一致しており、植物体内でシグナルペプチドの切断が行われ、正確に葉緑体チラコイド膜を通過しルーメン側で発現していることが明らかになった。すなわち、得られた形質転換体中のPYC6タンパク質のN末端アミノ酸配列の解析結果より、葉緑体輸送シグナルが切断された成熟領域アミノ酸配列と一致したことから、sp+PYC6遺伝子は転写・翻訳後葉緑体へと輸送されていることが確認できた。
【実施例4】
【0009】
形質転換植物の生育測定と表現型の観察
(1) 生育観察(背丈、根長、葉の大きさ)
本項では、形質転換体Arabidopsis thalianaの播種後の生育を観察した。
形質転換体を播種した後、植物体の「背丈」、「根長」及び「葉の大きさ」を10日おきに観察測定した。植物試料は野生株と形質転換体共に20個体行い、その平均を算出し、統計処理により有意差を求めた。
PYC6導入植物の生育(表現型)の観察を行った結果、PYC6導入植物の背丈は、最大で1.9倍(播種後40日)野生型(WT)に比べて大きくなり、約60日目で1.3倍増大していた(図11)。根長は、最大で1.35倍(播種後40日)WTに比べ大きくなり、約60日目で1.17倍増大していた(図12)。葉の大きさは、最大で1.9倍(播種後40日)の差が見られ、約60日目で1.3倍増大していた(図13)。これらPYC6導入植物にみられた現象は、植物体内でのPYC6の発現により何らかの形で植物の生育に非常に重要な光合成反応が活性化されたためと考えられ、その影響により植物体の生育速度が上昇したと思われた。
(2) 総クロロフィル量の測定
本項では、形質転換体Arabidopsis thalianaより全クロロフィルをアセトンにより抽出し、定量した。
形質転換体Arabidopsis thalianaの葉をリーフパンチで採取し、秤量した。あらかじめ冷却した乳鉢の中に試料を入れ、素早く液体窒素を加え、植物体がパウダー状になるまでホモジナイズした。その後、パウダー状になった試料に80%アセトンを加え4 mlに定容した。その溶液の663 nmと645 nmの吸光度を分光光度計UV-VISIBLE SPECTROPHOTOMETERで測定した。
得られた663 nmと645 nmの吸光値を以下に示した式に代入し、クロロフィル量を算出した。
総クロロフィル量(μg/ml)
= 8.02×ABS(663) + 20.21×ABS(645)
PYC6導入植物中のクロロフィル量を測定した結果、播種後40日から70日の間においてはWTよりも1.1〜1.2倍増大していた(図14)。これは、PYC6の導入により明反応が活性化され、それにより増大した明反応最終産物であるエネルギー物質(ATP)がクロロフィルの合成に利用された影響ではないかと考えられた。
【実施例5】
【0010】
形質転換植物の光合成活性測定
(1) 方法及び結果
光合成活性の指標として、アデニンヌクレオチド(ATP)含量、NADPH量、二酸化炭素同化能力、デンプン量、及びタンパク質量を、それぞれ、以下に示す方法で測定した。
(1-1) 総ATP含量の測定
本実施例では、形質転換体Arabidopsis thalianaより全ATPを抽出し、ルシフェリン・ルシフェラーゼ法を用いて定量した。
形質転換体Arabidopsis thalianaの葉を予め秤量した。秤量後、冷却した乳鉢の中に試料を入れ、素早く液体窒素を加えて、植物体がパウダー状になるまで乳棒でホモジナイズした。その後、パウダー状になった試料に0.25M 過塩素酸を2 ml加え、その溶液を15 ml容コーニングチューブに回収した。10,000 rpm, 4℃, 10 min.遠心分離し、得られた上清を別の15 ml容コーニングチューブに移し、1N KOHでpH7.0に調整した。調製後、0.2M リン酸緩衝液(pH 7.2)で5 mlに定容し、これをATP抽出液とした。
ATP抽出液100 μlを1.5 ml microcentrifuged tubeに準備し、ENLITEN Luciferase/Luciferin Reagentを20 μl添加してから90 sec.後ルミノメーター(GENE LIGHT 55)で10 sec.反応液の発光強度を積算し、得られたRLU値から溶液のATP濃度を算出し、そこから植物重量当たりのATP量を換算した。
その結果、PYC6導入植物の総ATP量は、最大で1.93倍(播種後60日)増大していた(図15)。これは、PYC6導入植物において機能しているプラストシアニンに加え、さらに新たな電子伝達体として紅藻cyt c6(PYC6)が機能したことにより、明反応の電子伝達が活性化され、その最終生成物であるATP量が増大したものと考えられた。
(1-2) 総NADPH含量の測定
本実施例では、形質転換体Arabidopsis thalianaより全NADPHを抽出し、定量した。
形質転換体Arabidopsis thalianaを予め秤量した。その後、70℃程度のホットバスで温めた0.1 N NaOHを入れ、ポリトロンホモジェナイザーを用いて破砕した。その後、アイスバスに移し、約2 mlの0.1 N HClを加え、pH試験紙でpH 7.5に調整した。次に0.1 mlのグリシルグリシン緩衝液(pH 7.5)を加え、よく撹拌し、メスシリンダーに移し、液量を測定する。測定後、15 ml容コーニングチューブに移し、10,000 rpm、20 min、4℃で遠心分離した。遠心分離後、上清を15 ml容コーニングチューブに回収し、-80℃で一晩冷却し、溶解後、10,000 rpm、20 min、4℃で遠心分離し、定量に用いた。
NADPH抽出液100.0 μl、0.1 M グリシルグリシン緩衝液(pH 7.4)346.0 μl、0.2 M ニコチンアミド200.0 μl、3.2 mg/ml フェナジンメトサルフェート(PMS)82.0 μl、5 mg/mlチアゾールブルー67.0 μl、グルコース-6-リン酸2ナトリウム水和物200 μlを混ぜ、分光光度計U3310にセットした。を用いて570 nm、25℃で測定する。U3310に温度制御ユニットを取り付け、25℃にセットする。その後、570 nmにおける吸光度の変化を30秒おきに2分間測定し、得られた吸光値の差からNADPH量を算出し、そこから植物重量あたりのNADPH量を算出した。
その結果、PYC6導入植物の総NADPH量は播種後40〜70日で1.2〜1.4倍増大していた(図16)。これは、PYC6導入の高等植物への導入によって、野生型よりも多く電子が伝達され、その最終生成物であるNADPH量が増大したものと考えられた。
(1-3) 二酸化炭素同化能力の測定
本実施例では、形質転換体Arabidopsis thalianaの葉を用いて二酸化炭素の同化能力を測定した。
二酸化炭素の同化能力は、CO2ガス分析機CIRAS-1(小糸工業製)を用いて行った。また、二酸化炭素の供給量は350 ppmに設定し、飽和光下で測定した。
その結果、PYC6導入植物の二酸化炭素の同化能力は最大で2.2倍増大していた(播種後50日)(図17)。これは、PYC6導入の高等植物への導入によって、ATPやNADPH量が増大したことにより、そのエネルギーを利用する暗反応(カルビン・ベンソンサイクル)が活性化され、二酸化炭素の同化能力が増大したものと考えられた。
(1-4) デンプン量の測定
本実施例では、形質転換体Arabidopsis thalianaよりデンプンを抽出し、定量した。
植物葉を採取後、乾燥させる(70度、2日)。約5 mgを量り取り乳鉢に入れ、3 mlの32%過塩素酸を加えて、乳棒でよく破砕する。その後、全て試験管に移し、20℃で20分静置した。次に、6.0 cmのWhatman GF/A grass fibre diskで濾過し、濾液に5 mlのヨウ素溶液を加え、よく混ぜ、約4℃で30分間静置する。その後、1.5 cmのWhatman GF/A grass fibre discで濾過し、フィルターを乾燥させる。
乾燥したフィルターを試験管に入れ、4 mlの0.75M硫酸を加え、沸騰湯浴中で30分間インキュベートする。その後、上清を回収し、フェノール硫酸法により発色させ、分光光度計を用いて485 nmの吸収を測定した。
その結果、PYC6導入植物のデンプン量は播種後40〜70日で1.15〜1.25倍増大していた(図18)。これは、PYC6導入の高等植物への導入によって、CO2の同化能が増大し、それに伴いデンプンの合成も促進されたと考えられた。
(1-5) タンパク質量の測定
本実施例では、形質転換体Arabidopsis thalianaよりタンパク質を抽出し、Lowry法を用いて定量した。
植物体の重量を測定し、記録する。乳鉢に適量の植物体を入れ、液体窒素下、乳棒で充分に破砕する。常温まで戻した後、植物体1 gあたり2 mlのCelLytic P(Sigma製)を入れ、乳棒でよく磨砕し、試験管に移す。15分室温で転倒混和後、遠心分離する。この上清を抽出タンパク質とする。
次に、Lowry A液 : 0.1 Nの水酸化ナトリウム溶液に2%になるように炭酸ナトリウムを加えたもの、Lowry B液 : 0.5% 硫酸銅五水和物と1% クエン酸ナトリウムになるように調製したもの、Lowry C液 : 1 N フェノール溶液、Lowry D液 : Lowry A液 + Lowry B液を作製した。その後、抽出したタンパク質溶液、0.1 mlにLowry D液 1.0 mlを加え、ボルテックスミキサーで撹拌した後、30°C、15 minインキュベートした。次にLowry C液 0.1 mlを加えて、速やかにボルテックスミキサーで撹拌した後、30°C、30 minインキュベートし、770 nmの吸光値を測定した。
その結果、その結果、PYC6導入植物のタンパク質量は、最大1.3倍(播種後50日)増大していた(図19)。これは、PYC6導入の高等植物への導入によって、増産されたATPやNADPHがタンパク質の合成を活性化したのではないかと考えられた。
(2) 考察
本発明で得られた知見として、PYC6導入植物はWTと比較して成長速度が上昇していた。これはPYC6導入植物中のATP及びNADPHが増大していたことから、A. thalianaにおいて機能しているプラストシアニンに加え、さらに新たに導入した電子伝達体である紅藻cyt c6 が機能したことにより、明反応の電子伝達が2回路となったためにその最終生成物であるATP及びNADPH量が増大し、さらに植物全体の生育が活性化される結果となったと推測された。これまで陸上植物への暗反応(カルビン・ベンソンサイクル)の関連酵素の補強による成分量の増進についての研究は行われているが、本発明のように明反応を活性化させた例はない。
また、これまでcyt c6 を機能させることを目的として植物へと導入した例はなく、本発明の内容は今後他種の植物にも十分応用可能であり、有用な植物の生長促進法や植物の生産技術(果実物、葉物、生花など)などの基盤となる汎用性の高い発明であるといえる。さらに、植物体内で増産されたATP及びNADPHを利用することで他の物質代謝も活性化され、汚染物質(CO2など)の除去法の分野にも極めて有効と考えられた。
【産業上の利用可能性】
【0011】
本発明によれば、葉緑体のチラコイド内腔にシトクロムc6を有する新規な高等植物の作出方法を提供することができ、ひいては、高等植物の生長促進方法、ATP、NADPH、デンプン及びタンパク質の合成促進方法及び炭素固定促進方法を提供することができる。
また、葉緑体のチラコイド内腔にまで輸送され得るシグナルペプチド付加シトクロムc6タンパク質、当該タンパク質をコードする遺伝子、当該遺伝子を含む組換えベクター、当該組換えベクターを含む形質転換体、及び当該遺伝子が植物ゲノム中に導入されている形質転換高等植物を提供することもできる。
【配列表フリーテキスト】
【0012】
配列番号7:プライマー
配列番号8:プライマー
配列番号9:プライマー
配列番号10:プライマー
【特許請求の範囲】
【請求項1】
シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加されてなる融合タンパク質又はその変異型タンパク質であって、以下の(a)、(b)、(c)若しくは(d)のタンパク質であることを特徴とする、前記融合タンパク質又はその変異型タンパク質。
(a) 配列番号6に示されるアミノ酸配列を含むタンパク質
(b) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質
(c) 配列番号6に示されるアミノ酸配列のうちの前記シトクロムc6タンパク質に相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ電子伝達能を有するタンパク質
(d) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列及び前記シトクロムc6タンパク質に相当するアミノ酸配列においてそれぞれ1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質
【請求項2】
請求項1に記載の融合タンパク質又はその変異型タンパク質をコードする遺伝子であって、以下の(a)若しくは(b)のDNAを含むことを特徴とする、前記遺伝子。
(a) 配列番号5に示される塩基配列を含むDNA
(b) 配列番号5に示される塩基配列を含むDNAと相補的な塩基配列を含むDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質をコードするDNA
【請求項3】
請求項2に記載の遺伝子を含む組換えベクター。
【請求項4】
請求項3に記載の組換えベクターを宿主に導入してなる形質転換体。
【請求項5】
宿主がアグロバクテリウム属に属する微生物である、請求項4に記載の形質転換体。
【請求項6】
請求項2に記載の遺伝子が植物ゲノム中に導入されている、形質転換高等植物。
【請求項7】
植物細胞中の葉緑体のチラコイド内腔にシトクロムc6を有するものである、請求項6に記載の形質転換高等植物。
【請求項1】
シトクロムc6タンパク質に50〜80アミノ酸残基のシグナルペプチドが付加されてなる融合タンパク質又はその変異型タンパク質であって、以下の(a)、(b)、(c)若しくは(d)のタンパク質であることを特徴とする、前記融合タンパク質又はその変異型タンパク質。
(a) 配列番号6に示されるアミノ酸配列を含むタンパク質
(b) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能を有するタンパク質
(c) 配列番号6に示されるアミノ酸配列のうちの前記シトクロムc6タンパク質に相当するアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ電子伝達能を有するタンパク質
(d) 配列番号6に示されるアミノ酸配列のうちの前記シグナルペプチドに相当するアミノ酸配列及び前記シトクロムc6タンパク質に相当するアミノ酸配列においてそれぞれ1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を含み、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質
【請求項2】
請求項1に記載の融合タンパク質又はその変異型タンパク質をコードする遺伝子であって、以下の(a)若しくは(b)のDNAを含むことを特徴とする、前記遺伝子。
(a) 配列番号5に示される塩基配列を含むDNA
(b) 配列番号5に示される塩基配列を含むDNAと相補的な塩基配列を含むDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ高等植物の葉緑体包膜及びチラコイド膜の通過能、並びに電子伝達能を有するタンパク質をコードするDNA
【請求項3】
請求項2に記載の遺伝子を含む組換えベクター。
【請求項4】
請求項3に記載の組換えベクターを宿主に導入してなる形質転換体。
【請求項5】
宿主がアグロバクテリウム属に属する微生物である、請求項4に記載の形質転換体。
【請求項6】
請求項2に記載の遺伝子が植物ゲノム中に導入されている、形質転換高等植物。
【請求項7】
植物細胞中の葉緑体のチラコイド内腔にシトクロムc6を有するものである、請求項6に記載の形質転換高等植物。
【図1】

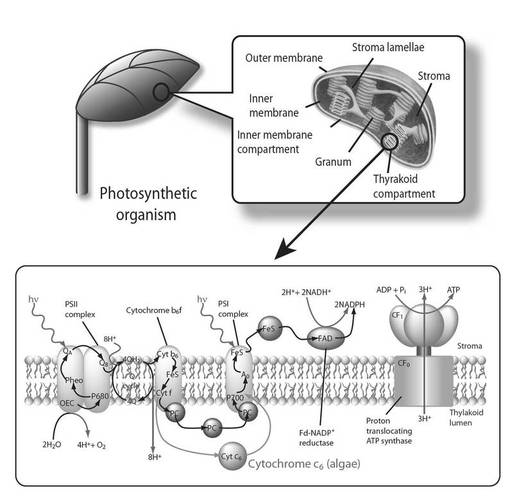
【図2】
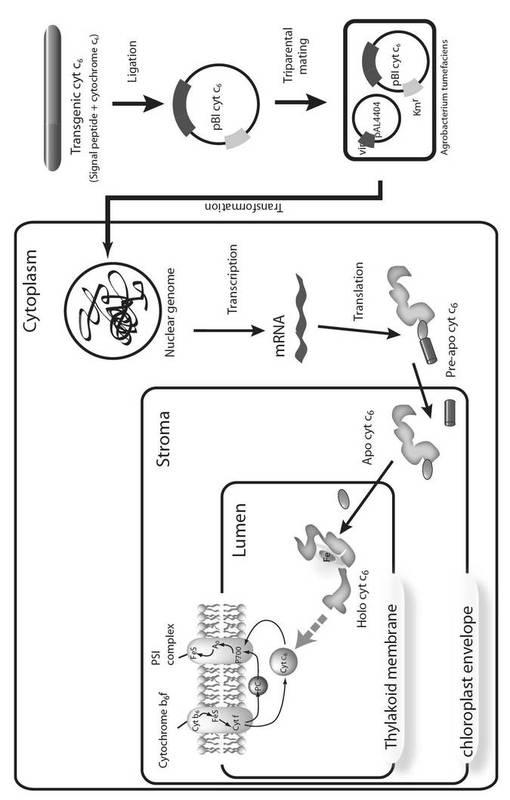
【図3】
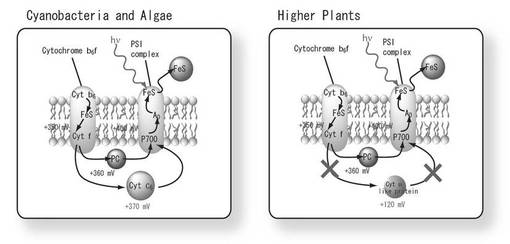
【図4】
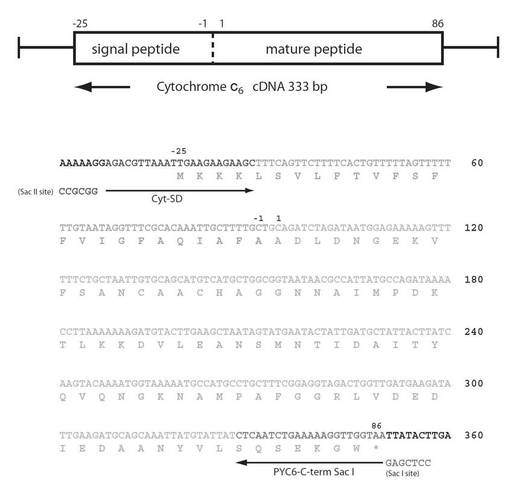
【図5】
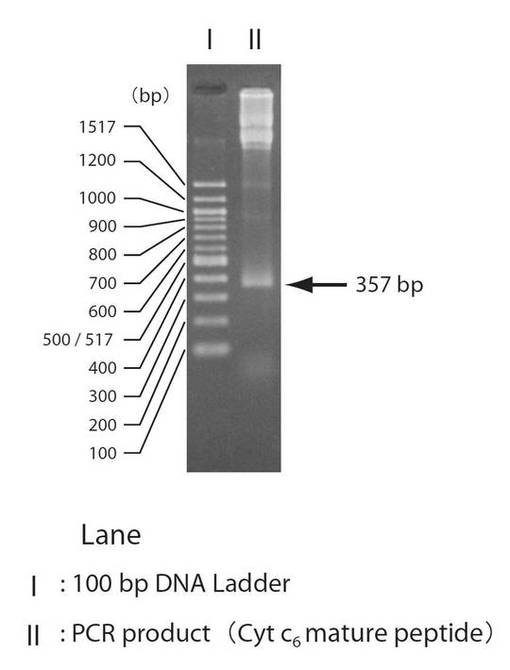
【図6】
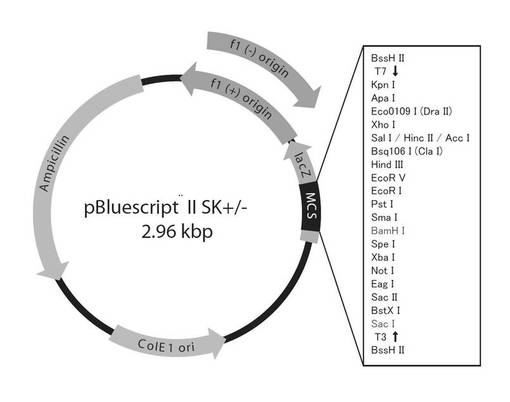
【図7】

【図8】
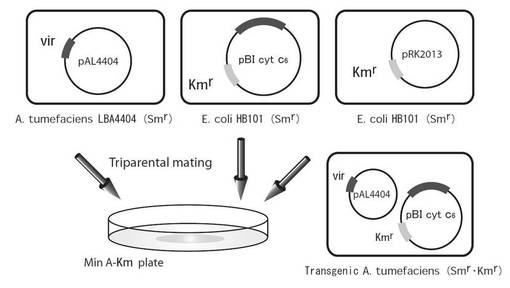
【図9】
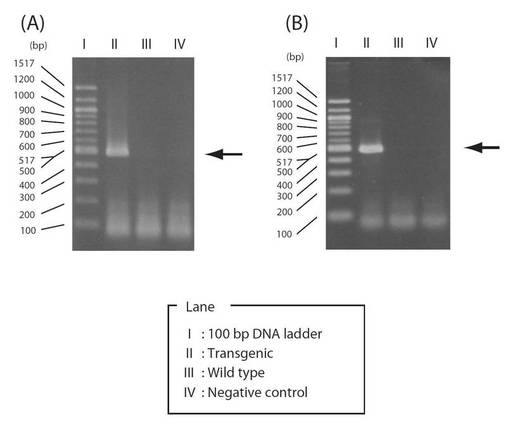
【図10】
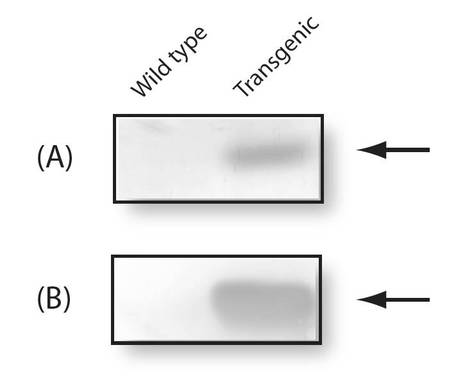
【図11】
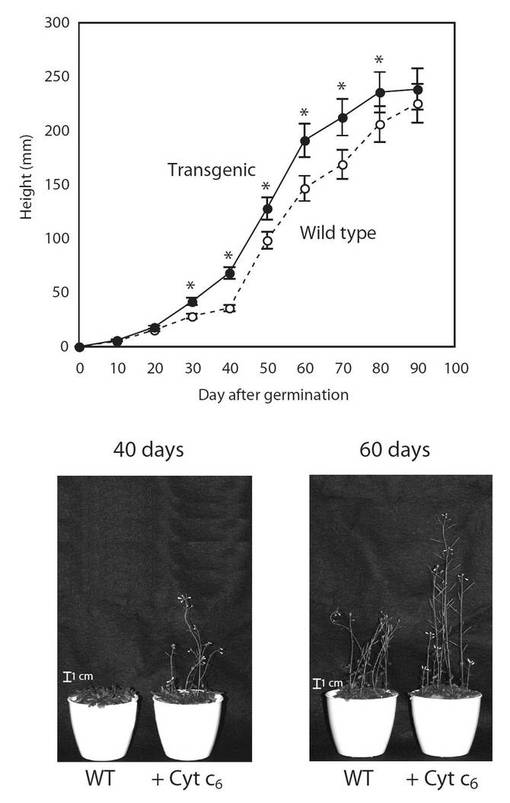
【図12】
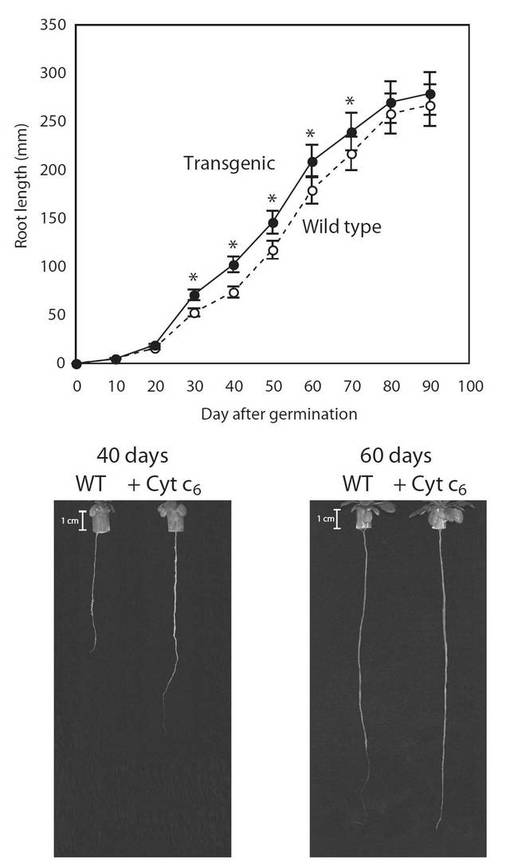
【図13】
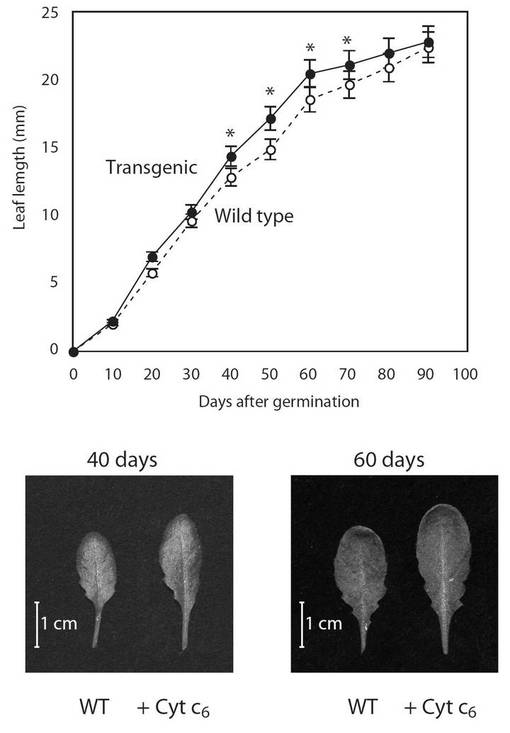
【図14】
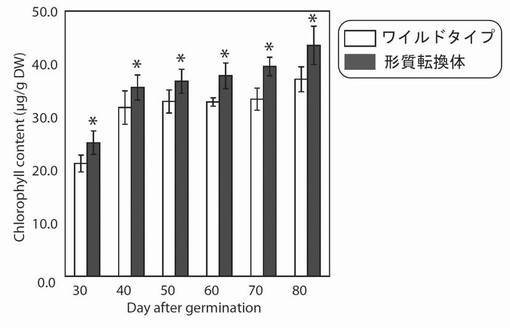
【図15】
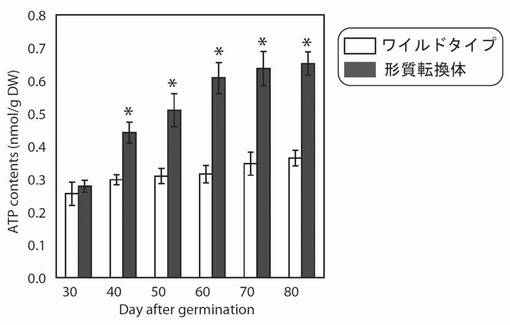
【図16】
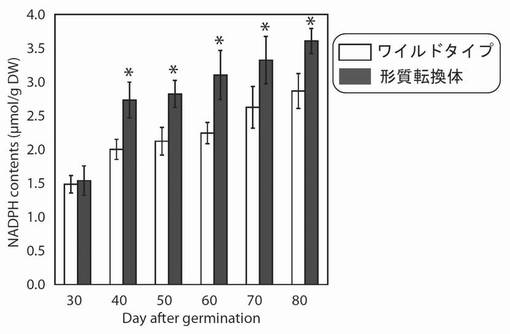
【図17】
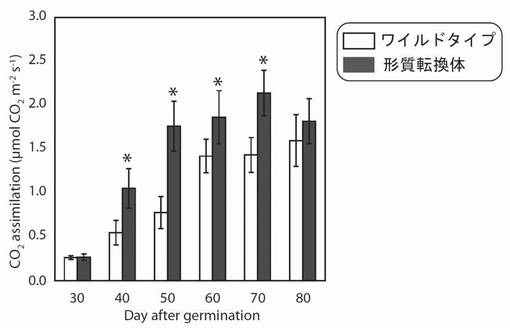
【図18】
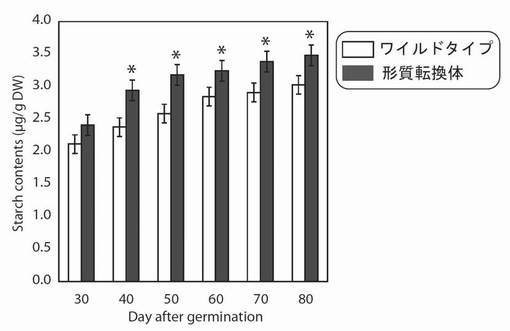
【図19】
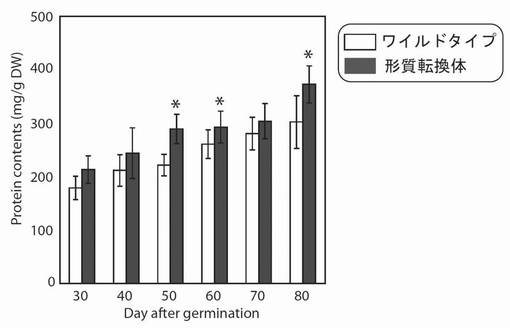
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公開番号】特開2012−110330(P2012−110330A)
【公開日】平成24年6月14日(2012.6.14)
【国際特許分類】
【出願番号】特願2011−284942(P2011−284942)
【出願日】平成23年12月27日(2011.12.27)
【分割の表示】特願2007−501678(P2007−501678)の分割
【原出願日】平成18年2月1日(2006.2.1)
【出願人】(899000057)学校法人日本大学 (650)
【Fターム(参考)】
【公開日】平成24年6月14日(2012.6.14)
【国際特許分類】
【出願日】平成23年12月27日(2011.12.27)
【分割の表示】特願2007−501678(P2007−501678)の分割
【原出願日】平成18年2月1日(2006.2.1)
【出願人】(899000057)学校法人日本大学 (650)
【Fターム(参考)】
[ Back to top ]