
新規N−アセチルグルコサミン−2−エピメラーゼ及びCMP−N−アセチルノイラミン酸の製造方法
本発明は新規N−アセチルグルコサミン−2−エピメラーゼ及びCMP−N−アセチルノイラミン酸の製造方法に係り、さらに詳しくは、バクテロイデスフラギリスNCTC9343(Bacteroides fragilis NCTC 9343)由来のN−アセチルグルコサミン−2−エピメラーゼ及び前記N−アセチルグルコサミン−2−エピメラーゼを用いてCMP−N−アセチルノイラミン酸を製造する方法に関する。本発明によれば、安価な基質であるシチジンモノホスフェートとN−アセチル−D−グルコサミンを用いて一回の反応により経済的にCMP−N−アセチルノイラミン酸を量産することができる。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は新規N−アセチルグルコサミン−2−エピメラーゼ及びCMP−N−アセチルノイラミン酸の製造方法に係り、さらに詳しくは、バクテロイデスフラギリスNCTC9343(Bacteroides fragilisNCTC 9343)由来のN−アセチルグルコサミン−2−エピメラーゼ及び前記N−アセチルグルコサミン−2−エピメラーゼを用いてCMP−N−アセチルノイラミン酸を製造する方法に関する。
【背景技術】
【0002】
最近、糖鎖に関する構造及び機能に関する研究が盛んになされるに伴い、生理活性を有するオリゴ糖、糖脂質、糖タンパク質などの医薬品または機能性素材としての用途開発が注目を浴びている。中でも、末端にN−アセチルノイラミン酸(NeuAc)を含有するシアル酸含有糖鎖は、細胞接着やウィルス感染時に受容体となるなど重要な機能を有する糖鎖である。
【0003】
シアル酸含有糖鎖は、一般的に、シアル酸転移酵素の触媒作用により合成される。シアル酸転移酵素は、CMP−N−アセチルノイラミン酸(CMP−NeuAc)を糖供給体として糖鎖などの受容体にシアル酸を転移させる酵素である。しかしながら、糖供給体として使用するCMP−NeuAcは極めて高価であり、しかも、量的にも試薬レベルの少量でしか供給されていないのが現状である。
【0004】
CMP−NeuAcの製造方法としては、シチジン5’−トリリン酸(CTP)とN−アセチルノイラミン酸(NeuAc)を基質としてCMP−NeuAcシンテターゼの触媒により合成する方法が知られているが、その原料となるCTP及びNeuAcは高価であるため、直接原料として合成されたCMP−NeuAcも高価にならざるを得ない。
【0005】
近年、オロチン酸からウリジン5’−トリリン酸(UTP)への変換を司るブレビバクテリウムアンモニアゲネス(Brevibacterium ammoniagenes)菌体、UTPからCTPへの変換反応を触媒するCTP合成酵素を生産する組換え大腸菌及びCMP−NeuAc合成酵素を生産する組換え大腸菌を組み合わせ、オロチン酸とNeuAcを原料としてCMP−NeuAcを合成する方法が開発されている。上記の方法は、高価なCTPを使用しない方法ではあるが、複数種の菌体を調製しなければならないなど工程が煩雑であると共に、それを行うための大型の設備を準備することを余儀なくされ、しかも、高価なNeuAcを原料とするという点において、実用的な方法であるとはいえない。
【0006】
一方、NeuAcの製造方法に関しては、シアル酸の多量体であるコロミン酸を微生物から回収し、これを化学分解して回収する方法が知られているが、最近、酵素を用いた方法が開発されている。
【0007】
CMP−N−アセチルノイラミン酸(CMP-neuraminic acid;CMP−NeuAc)の生産方法としては、(1)N−アセチルノイラミン酸リアーゼまたはN−アセチルノイラミン酸シンテターゼを用いてN−アセチルマンノサミン(N-acetyl-D-mannosamine;ManNAc)から製造する方法(J. Am. Chem. Soc., 110:6481, 1988; J. Am. Chem. Soc., 110:7159, 1988;特開平10−4961号公報)、(2)アルカリの条件下においてN−アセチルグルコサミン(GlcNAc)をN−アセチルマンノサミン(ManNAc)に変換し、ここにN−アセチルノイラミン酸リアーゼまたはN−アセチルノイラミン酸シンテターゼを添加してN−アセチルノイラミン酸(NeuAc)を製造する方法(特開平05−211884号公報;Biotechnol. Bioeng., 66:2, 1999; Enzyme Microb. Technol., 20, 1997)、(3)GlcNAcからMamNAcへの変換を触媒するN−アセチルグルコサミン(GlcNAc)2−エピメラーゼとNeuAcリアーゼまたはNeuAcシンテターゼを用いてGlcNAcからCMP−NeuAcを製造する方法(WO95/26399;特開平03−180190号公報;特開2001−136982号公報)、(4)大腸菌と酵母菌体を用いてCMP−N−アセチルノイラミン酸を合成する方法(WO2004/009830)などが報告されている。
【0008】
しかしながら、(1)の方法は原料となるN−アセチルマンノサミン(ManNAc)が高価であり、(2)の方法は安価なN−アセチルグルコサミン(GlcNAc)を原料とする方法ではあるが、GlcNAcとN−アセチルマンノサミン(ManNAc)との混合物においてManNAcを精製する工程が複雑であるという問題がある。さらに、(3)の方法において使用するGlcNAc2−エピメラーゼは、ATPを必要とするため、高価なATPを添加したり、微生物を用いてATPの前駆体であるアデニンからATPを生成しなければならないという欠点があり、(4)の方法は大腸菌と酵母菌体を利用する方法がその工程からみて複雑であるという問題がある。
【0009】
N−アセチルグルコサミン−2−エピメラーゼに関しては、豚やラットの腎臓、肝、脾臓、脳、腸内粘膜、甲状腺、膵臓及び唾液腺に存在することが知られており、豚由来の酵素に関してその特性が調べられており、主として動物由来の遺伝子などから抽出されて研究されている(Biochemistry, 17:3363, 1970; Biochem. J., 210:21, 1983; Proc. Natl. Acad. Sci. U.S.A., 52:371, 1964)。また、N−アセチルグルコサミン−2−エピメラーゼの遺伝子については豚由来の遺伝子が知られている(J. Biol. Chem., 271:16294, 1996)。微生物の中では、藍藻の一種であるシネコシスティス属PCC6803(Synechocystis sp. PCC 6803)はそのゲノムの全体配列が決定されており、ゲノムからN−アセチルグルコサミン−2−エピメラーゼを確認した(DNA Research, 3:109, 1996; Nucleic Acids Research, 26:63, 1998)。
【0010】
一方、大韓民国公開特許(第10−2001−0102019号)は、N−アセチルグルコサミン−2−エピメラーゼ及び前記酵素をコードするDNAに関するものであり、N−アセチルグルコサミン−2−エピメラーゼをコードするDNAを藍藻(Cyanobacteria)のうちシネコシスティス(Synechocystis)から発見して使用した。
【0011】
また、大韓民国公開特許(第10−2006−0010706号)は、CMP−N−アセチルノイラミン酸の製造方法に関するものであり、N−アセチルグルコサミン−2−エピメラーゼをコードする遺伝子及びN−アセチルノイラミン酸アルドラーゼをコードする遺伝子を含む共発現ベクター導入の形質転換体にシチジンモノホスフェート(CMP)、N−アセチルグルコサミン、ピルビン酸及び酵母を添加してノイラミン酸を合成した後、CMP−N−アセチルノイラミン酸合成酵素を添加したり、N−アセチルノイラミン酸アルドラーゼをコードする遺伝子及びCMP−N−アセチルノイラミン酸合成酵素をコードする遺伝子を含む共発現ベクター導入の形質転換体にシチジンモノホスフェート(CMP)、N−アセチルグルコサミン、ピルビン酸及び酵母を添加してCMP−N−アセチルノイラミン酸を製造する方法に関するものであるが、CMP−N−アセチルノイラミン酸を製造するために多段階を経なければならず、且つ、基質として用いられるシチジンモノホスフェート(CMP)がシチジントリホスフェート(CTP)に転換される収率が低いという問題点がある。
【0012】
そこで、本発明者らは、CMP−N−アセチルノイラミン酸を経済的で且つ簡単な方法により製造する方法を開発するために鋭意努力した結果、バクテロイデスフラギリスNCTC9343(Bacteroides fragilis NCTC 9343)において新規N−アセチルグルコサミン−2−エピメラーゼ酵素を確認し、前記酵素の遺伝子を組換えベクターで形質転換して得られたN−アセチルグルコサミン−2−エピメラーゼ、安価な基質であるシチジンモノホスフェート(CMP)及び極少量のNTPを用いて多数の基質と酵素を一気に混ぜてワンポット連鎖反応により高収率にてCMP−N−アセチルノイラミン酸を製造することができるということを確認し、本発明を完成するに至った。
【発明の詳細な説明】
【0013】
本発明の主たる目的は、新規N−アセチルグルコサミン−2−エピメラーゼ及び前記酵素をコードする遺伝子を含有する組換えベクターで形質転換された組換え微生物を培養することを含む、N−アセチルグルコサミン−2−エピメラーゼの製造方法を提供することにある。
【0014】
本発明の他の目的は、前記N−アセチルグルコサミン−2−エピメラーゼを用いて多数の基質と酵素を一気に混ぜてワンポット連鎖反応によりCMP−N−アセチルノイラミン酸を製造する方法を提供することにある。
【0015】
上記の目的を達成するために、本発明は、配列番号2のアミノ酸配列を有するN−アセチルグルコサミン−2−エピメラーゼを提供する。
【0016】
また、本発明は、前記N−アセチルグルコサミン−2−エピメラーゼをコードする遺伝子、前記遺伝子を含有する組換えベクター、前記組換えベクターで形質転換された組換え微生物及び前記組換え微生物を培養すること、及びN−アセチルグルコサミン−2−エピメラーゼを回収することを含む、N−アセチルグルコサミン−2−エピメラーゼの製造方法を提供する。
【0017】
さらに、本発明は、N−アセチル−D−グルコサミンを含む反応混合物を前記N−アセチルグルコサミン−2−エピメラーゼの存在下で反応させること、及びN−アセチル−D−マンノサミンを回収することを含む、N−アセチル−D−マンノサミンの製造方法を提供する。
【0018】
さらに、本発明は、ピルビン酸及び前記N−アセチル−D−マンノサミンを含む反応混合物をN−アセチルノイラミン酸アルドラーゼの存在下で反応させること、及びノイラミン酸を回収することを含む、ノイラミン酸製造方法を提供する。
【0019】
さらに、本発明は、(i)シチジン5’モノホスフェート(cytidine 5' monophosphate;CMP)、(ii)ヌクレオチドトリ−ホスフェート(nucleotide tri-phosphate;NTP)またはATP、(iii)アセチルホスフェート、(iv)N−アセチル−D−グルコサミン及び(v)ピルビン酸を含む反応混合物を(a)N−アセチルグルコサミン−2−エピメラーゼ、(b)N−アセチルノイラミン酸アルドラーゼ、(c)シチジン5’モノホスフェートキナーゼ、(d)アセテートキナーゼ及び(e)CMP−N−アセチルノイラミン酸合成酵素の存在下で反応させること、及びCMP−N−アセチルノイラミン酸を回収することを含む、CMP−N−アセチルノイラミン酸の製造方法を提供する。
【0020】
本発明の他の特徴及び実施態様は、下記の詳細な説明及び特許請求の範囲から一層明らかになる。
【図面の簡単な説明】
【0021】
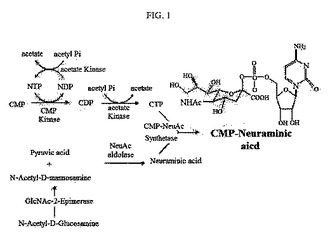
【図1】CMP−N−アセチルノイラミン酸の合成過程を示す模式図である。
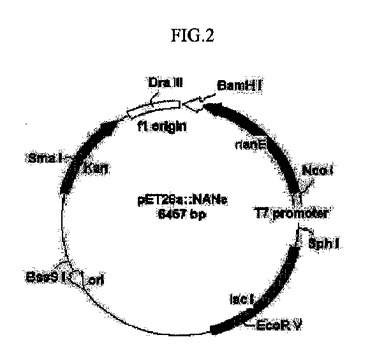
【図2】N−アセチルグルコサミン−2−エピメラーゼをコードする遺伝子(nanE)を含有する組換えベクターを示すものである。
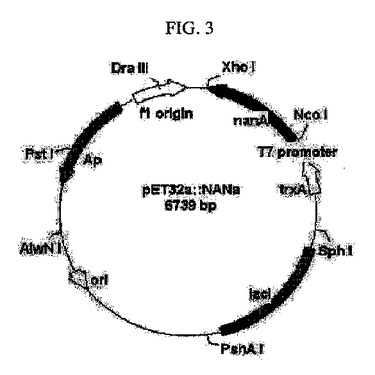
【図3】N−アセチルノイラミン酸アルドラーゼをコードする遺伝子(nan)を含有する組換えベクターを示すものである。
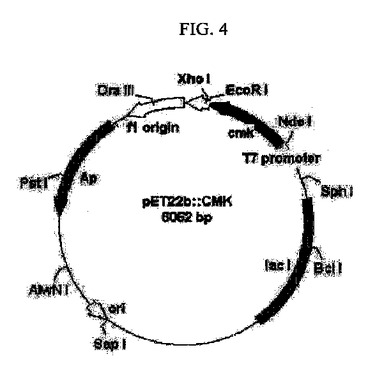
【図4】シチジン5’モノホスフェートキナーゼをコードする遺伝子(cmk)を含有する組換えベクターを示すものである。
【図5】アセテートキナーゼをコードする遺伝子(ack)を含有する組換えベクターを示すものである。
【図6】CMP−N−アセチルノイラミン酸合成酵素をコードする遺伝子(neu)を含有する組換えベクターを示すものである。
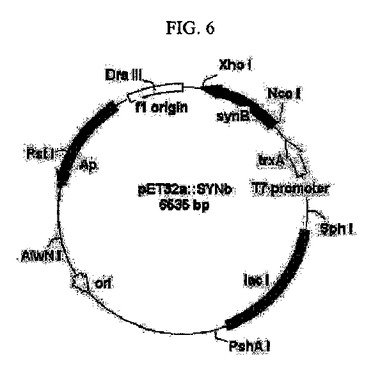
【図7】各酵素の発現の有無をSDS−PAGEゲルを通じて確認した結果を示すものである(レーン1:マーカー、レーン2:シチジン5’モノホスフェートキナーゼ、レーン3:アセテートキナーゼ、レーン4:CMP−N−アセチルノイラミン酸合成酵素、レーン5:N−アセチルノイラミン酸アルドラーゼ、レーン6:アシルグルコサミン−2−エピメラーゼ)。
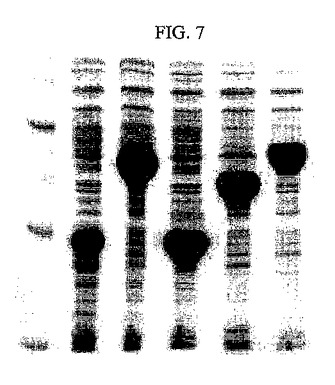
【図8】精製された各酵素の発現の有無をSDS−PAGEゲルを通じて確認した結果を示すものである(レーン1:マーカー、レーン2:精製されたシチジン5’モノホスフェートキナーゼ、レーン3:精製されたアセテートキナーゼ、レーン4:精製されたCMP−N−アセチルノイラミン酸合成酵素、レーン5:精製されたN−アセチルノイラミン酸アルドラーゼ、レーン6:精製されたアシルグルコサミン−2−エピメラーゼ)。
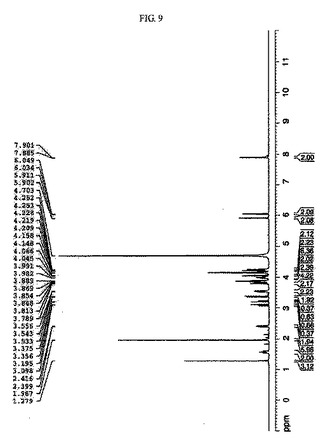
【図9】CMP−N−アセチルノイラミン酸をH1NMRにより分析した結果を示すものである。
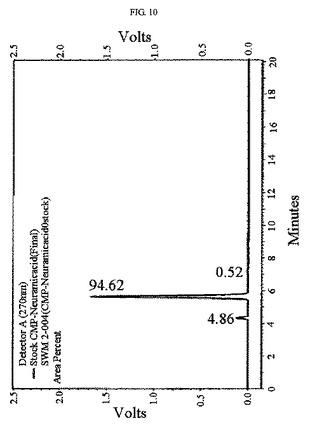
【図10】CMP−N−アセチルノイラミン酸をHPLCにより分析した結果を示すものである。
【発明を実施するための最良の形態】
【0022】
本発明は、一観点において、CMP−N−アセチルノイラミン酸の合成に関与するN−アセチルグルコサミン−2−エピメラーゼ酵素、前記N−アセチルグルコサミン−2−エピメラーゼをコードする遺伝子、前記遺伝子を含有する組換えベクター、前記組換えベクターで形質転換された組換え微生物及び前記組換え微生物を培養することを特徴とするN−アセチルグルコサミン−2−エピメラーゼの製造方法に関するものである。
【0023】
本発明において、前記遺伝子は、配列番号1の塩基配列を有することを特徴とする。
【0024】
前記N−アセチルグルコサミン−2−エピメラーゼは、バクテロイデスフラギリスNCTC9343由来の酵素であり、シネコシスティス属とバクテロイデスセタイオタオミクロンの該当遺伝子と35.06〜32.9%の相同性を示している。
【0025】
本発明は、他の観点において、前記N−アセチルグルコサミン−2−エピメラーゼを用いることを特徴とするN−アセチル−D−マンノサミン及びノイラミン酸を製造する方法に関する。
【0026】
本発明は、さらに他の観点において、前記N−アセチルグルコサミン−2−エピメラーゼを用いてワンポット連鎖反応によりCMP−N−アセチルノイラミン酸を製造する方法に関する。
【0027】
本発明によるCMP−N−アセチルノイラミン酸は、(i)シチジン5’モノホスフェート、(ii)ヌクレオチドトリホスフェートまたはATP、(iii)アセチルホスフェート、(iv)N−アセチル−D−グルコサミン及び(v)ピルビン酸を含む反応混合物を(a)N−アセチルグルコサミン−2−エピメラーゼ、(b)N−アセチルノイラミン酸アルドラーゼ、(c)シチジン5’モノホスフェートキナーゼ、(d)アセテートキナーゼ及び(e)CMP−N−アセチルノイラミン酸合成酵素の存在下で反応させることにより製造することができる。
【0028】
本発明によれば、安価な基質であるシチジンモノホスフェートは、CMP−キナーゼ及びアセテートキナーゼによりシチジンダイホスフェート(cytidine diphosphate;CDP)を経てシチジントリホスフェートに97%以上の高収率にて合成される。前記CDPに用いられるホスフェートの供与体としては極少量のNTPを使用し、NDPは過量のアセチルホスフェートとアセテートキナーゼにより再利用されて経済的である。特に、本発明においては、CDPに供されるホスフェート供与体として用いられるATPの代わりにCTPを使用することにより、反応終了後に不純物として存在するADPを除去しなければならないという煩雑さを解消することができる。
【0029】
また、N−アセチル−D−グルコサミン(N-acetyl-D-glucosamine;GlacNAc)は、本発明によるN−アセチルグルコサミン−2−エピメラーゼによりN−アセチル−D−マンノサミン(N-acetyl-D-mannosamine;ManNAc)に転換され、ManNAcとピルビン酸はN−アセチルノイラミン酸アルドラーゼによりノイラミン酸に合成される。以上において合成されたCTPとノイラミン酸は、CMP−N−アセチルノイラミン酸合成酵素(CMP-NeuAc synthetase)によりCMP−N−アセチルノイラミン酸に合成される(図1)。
【0030】
本発明において、前記ヌクレオチドトリホスフェート(NTP)またはATPはアセチルホスフェート及びアセテートキナーゼにより再生されることを特徴とし、前記ヌクレオチドトリホスフェート(NTP)はシチジントリホスフェート(cytidinetri-phosphate;CTP)であることを特徴とする。
【0031】
本発明において、前記N−アセチルグルコサミン−2−エピメラーゼ、N−アセチルノイラミン酸アルドラーゼ、シチジン5’モノホスフェートキナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素は担体に固定化されていることを特徴とし、前記N−アセチルグルコサミン−2−エピメラーゼ、N−アセチルノイラミン酸アルドラーゼ、シチジン5’モノホスフェートキナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素はそれぞれ配列番号1、配列番号5、配列番号8、配列番号11及び配列番号14の塩基配列によりコードされることを特徴とする。
【0032】
本発明においては、高発現された各酵素を精製した後、キトサンなどの高分子ビーズを用いて固定化することにより反復的な合成段階を経ることなく1回で合成することができ、合成コストを大幅に下げることができる。酵素の固定化方法としては、通常の酵素の固定化手段(共有付着法、多機能試薬を用いた架橋、吸着法、微細カプセル化方法など)を使用することができる。酵素の精製方法としては、通常の酵素の精製手段(塩析処理、等電点沈殿処理、透析処理、各種のクロマトグラフィ処理など)を使用することができ、前記精製方法により得られる助酵素または精製酵素を利用することができ、前記酵素を高分子ビーズに固定化する方法としては、通常の酵素固定化方法を使用することができる。上記の方法により精製された酵素は高分子ビーズに固定化して20回以上使用することができるため、酵素を回収するコストと工数を減らすことができる。
【0033】
本発明によれば、前記多数の基質と酵素を一回に混ぜてワンポット連鎖反応を行うことにより各酵素の反応性を極大化させ、少量の酵素及び基質を使用しても、CMP−N−アセチルノイラミン酸を90%以上の高収率にて合成することができる。すなわち、N−アセチルグルコサミン−2−エピメラーゼの場合、エピメラーゼ単独反応を行うと、CMP−N−アセチルノイラミン酸の合成収率は50%にもならないが、ワンポット連鎖反応を行う場合に90%以上の高収率にてCMP−N−アセチルノイラミン酸を合成することができる。
【0034】
本発明において用いられるN−アセチルノイラミン酸アルドラーゼ、CMP−キナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素は公知の酵素であり、通常の方法により調製することができるが、前記遺伝子のクローニング、クローニングしたDNA断片を含む組換えベクターの製作、組換えベクターの導入された形質転換体の製作、形質転換体の培養による目的酵素タンパク質の生産などの組換えDNA法を使用することが好ましい。形質転換に用いられる宿主は特に限定されないが、操作し易さの観点から、大腸菌を使用することが好ましい。
【0035】
本発明において用いられるN−アセチルグルコサミン−2−エピメラーゼは、バクテロイデスフラギリスNCTC9343からN−アセチルグルコサミン−2−エピメラーゼをコードする新規遺伝子を分離したものである。バクテロイデスフラギリスのゲノムDNAの全体配列は決定されたが(US7,090,973)、N−アセチルグルコサミン−2−エピメラーゼ遺伝子に対する機能は全く知られていない。
【0036】
本発明において、「ベクター」とは、適合する宿主内においてDNAを発現可能な適合する調節配列に作動可能に連結されたDNA配列を含有するDNA製造物のことを言う。ベクターは、プラスミド、ファージ粒子または簡単に潜在的ゲノム挿入物であってもよい。適当な宿主で形質転換されれば、ベクターは宿主ゲノムとは無関係に複製して機能できるか、あるいは、場合によってはゲノムそのものに統合可能である。現在、プラスミドがベクターの最も汎用される形態であるため、本発明の明細書における「プラスミド」及び「ベクター」は時々相互交換的に使用される。本発明の目的からみて、プラスミドベクターを用いることが好ましい。このような目的に使用可能な典型的なプラスミドベクターは、(a)1つの宿主細胞当たりに数百個のプラスミドベクターを含むように複製が効率よく行われるようにする複製開始点、(b)プラスミドベクターで形質転換された宿主細胞を選抜できるようにする抗生剤耐性遺伝子及び(c)外来DNA切片が挿入可能な制限酵素切断部位を含む構造を有している。たとえ適切な制限酵素切断部位が存在しないとしても、通常の方法による合成オリゴヌクレオチドアダプターまたはリンカーを使用すれば、ベクターと外来DNAを容易にライゲート(ligation)することができる。
【0037】
ライゲーション後に、ベクターは適切な宿主細胞で形質転換される必要がある。形質転換はカルシウムクロリド方法を用いて容易に行うことができる。選択的に、電気穿孔法(Neumann, et al., EMBO J., 1:841, 1982)もこれらの細胞の形質転換に使用可能である。
【0038】
本発明による遺伝子の過発現のために用いられるベクターとしては、当業界における公知の発現ベクターが使用可能である。
【0039】
核酸は他の核酸配列と機能的関係を持つように配置されるときに「作動可能に連結」される。これは、適切な分子(例えば、転写活性化タンパク質)が調節配列に結合されるときに遺伝子発現を可能にする方式により連結された遺伝子及び調節配列であってもよい。例えば、前配列または分泌リーダーに対するDNAはポリペプチドの分泌に関与するプレタンパク質として発現される場合、ポリペプチドに対するDNAに作動可能に連結され、プロモーターまたはエンハンサーは配列の転写に影響する場合にコード配列に作動可能に連結されるか、あるいは、リボソーム結合部位は配列の転写に影響する場合にコード配列に作動可能に連結されるか、あるいは、リボソーム結合部位は翻訳を容易ならしめるために配置される場合にコード配列に作動可能に連結される。一般的に、「作動可能に連結された」とは、連結されたDNA配列が接触し、また、分泌リーダーの場合、接触してリーディングフレーム内に存在することを意味する。しかしながら、エンハンサーは接触する必要がない。これら配列の連結は便利な制限酵素部位においてライゲーションにより行われる。このような部位が存在しない場合、通常の方法による合成オリゴヌクレオチドアダプターまたはリンカーを使用する。
【0040】
当業界において周知されたように、宿主細胞における形質感染遺伝子の発現レベルを高めるためには、当該遺伝子が選択された発現宿主内において機能を発揮する転写及び解読発現調節配列に作動可能に連結されなければならない。好ましくは、発現調節配列及び当該遺伝子は細菌選択マーカー及び複製開始点を一緒に含んでいる一つの組換えベクター内に含まれることになる。宿主細胞が真核細胞である場合には、組換えベクターは真核発現宿主内において有用な発現マーカーをさらに含む必要がある。
【0041】
上述した組換えベクターにより形質転換された宿主細胞は本発明の他の側面を構成する。本願明細書における使用用語「形質転換」とは、DNAを宿主に導入してDNAが染色体外因子としてまたは染色体統合完成により複製可能になることを意味する。
【0042】
もちろん、全てのベクターが本発明のDNA配列を発現する上で一概に同等に機能を発揮することはないということを理解する必要がある。同様に、全ての宿主が同じ発現システムに対して一概に同様な機能を発揮することはない。しかしながら、当業者であれば、過度な実験的負担なしに本発明の範囲を逸脱しないままで種々のベクター、発現調節配列及び宿主から適切な選択をすることができる。例えば、ベクターを選択するに当たっては宿主を考慮しなければならないが、これは、ベクターがその内部において複製される必要があるためである。ベクターの複製数、複製数を調節可能な能力及び当該ベクターによりコードされる他のタンパク質、例えば、抗生剤マーカーの発現も考慮さるべきである。
【実施例】
【0043】
以下、本発明を実施例により詳述する。これらの実施例は単に本発明をより具体的に説明するためのものであり、本発明の範囲がこれらの実施例に制限されないことは当業界において通常の知識を持った者にとって自明である。
【0044】
《実施例1:遺伝子のクローニング及び形質転換》
実施例1−1.N−アセチルグルコサミン−2−エピメラーゼをコードするnanE遺伝子(配列番号1)のクローニング及び形質転換
CMP−N−アセチルノイラミン酸を製造するに当たって収率を高め、しかも、コストを下げるための最も大きな問題点として指摘されたN−アセチル−D−グルコサミンからN−アセチル−D−マンノサミンへの異性質化段階を解決するために、N−アセチルグルコサミン−2−エピメラーゼを種々のソースから発見した。
【0045】
嫌気性微生物であるバクテロイデスフラギリスNCTC9343(Bacteroides fragilis NCTC 9343, NC_003228)から3種のN−アセチルグルコサミン−2−エピメラーゼ類似遺伝子を発見した。
【0046】
N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子1番は947856〜949034区域にあり、1179bpでコードされており、リゾビウムメリローティ(Rhizobium meliloti)とホモサピエンス(Homo sapiens)の当該酵素遺伝子と23.68%〜24.44%の相同性を示している。
【0047】
N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子2番は1996625〜1997809区域にあり、1185bpでコードされており、シネコシスティス属(Synechocystis sp.)とバクテロイデスセタイオタオミクロン(Bacteroides thetaiotaomicron)の当該酵素遺伝子と35.06%〜32.9%の相同性を示している。
【0048】
N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子3番は4765592〜4766353区域にあり、762bpでコードされており、ロドピレルーラ・バルティカ(Rhodopirellula baltica)とシネコシスティス属(Synechocystis sp.)の当該酵素遺伝子と48.18%〜34.97%の相同性を示している。
【0049】
各遺伝子をpET発現ベクター(Novagen)に挿入し、これをBL21(DE3)、BL21(DE3)pLysS発現菌株に導入して形質転換した。その結果、N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子1番及び2番は可溶性タンパク質として高発現したが、候補遺伝子3番は可溶性タンパク質として発現されず、非可溶性として発現されることを確認した。
【0050】
高発現された候補遺伝子酵素1番及び2番の活性を調べるために、基質としてN−アセチル−D−グルコサミン及びピルビン酸を使用し、N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子1番、2番及び活性が確認されたN−アセチルノイラミン酸アルドラーゼを用いて2段階を1回に反応させて生成されたノイラミン酸を糖分析装置(Metrohm)を用いて確認した。
【0051】
その結果、N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子1番は活性がなく、N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子2番の方が活性に優れていることを確認することができた。また、ヒトN−アセチルグルコサミン−2−エピメラーゼ(organism="Homo sapiens,ene="RENBP", coded_by="NM_002910.4:203..1456")を同じ条件下(50mM N−アセチルD−グルコサミン、100〜200mM ピルビン酸、10mM MgCl2、50mM〜100mM Tris HCl、30℃〜40℃)で活性を比較したとき、N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子2番の反応速度及び収率が極めて高いことが分かり、前記候補遺伝子2番をN−アセチルグルコサミン−2−エピメラーゼとして確認した。
【0052】
前記N−アセチルグルコサミン−2−エピメラーゼ(GlcNAc 2−epimerase;配列番号2)をコードするnanE遺伝子をクローニングするために、バクテロイデスフラギリスNCTC9343(Bacteroides fragilis NCTC 9343)菌株の染色体DNAを鋳型とし、配列番号3及び配列番号4のプライマーを用いてPCR法によりnanE遺伝子を増幅した。
【0053】
配列番号3:5'-ct gcc atg gtt atg aat act aca g
配列番号4:5'-aat gga tcc tta ttt ttc tga cag
【0054】
PCR条件は鋳型DNA(10pmol)1μL、プライマーそれぞれ1μL、Primix(Genotech)4μL、蒸留水14μLをMastercycler gradient PCR(Ependorf)を用いて変性(96℃において1分)、アニーリング(40.2℃において1分)、伸長(72℃において2分)段階を合計で30回実施した。
【0055】
前記増幅されたPCR産物をプラスミドミニプレップキット(Plasmid mini-prep kit(Solgent))で精製し、70%エタノールを添加してDNAを添加した。前記沈殿されたDNAをアガロスゲル電気泳動により確認し、1.2kb相当のDNA断片を精製した。前記精製されたDNAを制限酵素NcoI及びBamHIで切断し、上記と同じ制限酵素NcoI及びBamHIで切断されたプラスミドpET28a(+)(Novagen)T4DNA(Takara)にリガーゼを用いてライゲートした(図2)。
【0056】
前記組換えベクターをE. coliXL-Blueに導入して得られたカナマイシン耐性形質転換体からプラスミドpNANEeを分離した。前記pNANEeプラスミドは、pET28a(+)のT7プロモーター下流のNcoI-BamHI切断部位にバクテロイデスフラギリスのnanE遺伝子の構造遺伝子を含有するDNA断片が挿入されたものである。
【0057】
前記組換え遺伝子を塩化カルシウムで処理して菌体内にプラスミドを導入する方法(J. Mol. Biol., 53:159, 1970)により高発現菌株E. coli BL21(DE3)(Invitrogen)に導入し、これをE. coli/pNANeと命名した。
【0058】
実施例1−2.N−アセチルノイラミン酸アルドラーゼをコードするnanA遺伝子のクローニング及び形質転換
N−アセチルノイラミン酸アルドラーゼ(NeuNAc aldolase)をコードするnanA遺伝子(配列番号5)をクローニングするために、大腸菌K−12(E. coliK-12 C600)菌株の染色体DNAを鋳型とし、配列番号6及び配列番号7のプライマーを用いてPCR法によりnanA遺伝子を増幅した。
【0059】
配列番号6:5'-ggtatccatggcaacgaatttacg
配列番号7:5'-ggtaggctcgagcgaggggaaac
【0060】
PCR条件は鋳型DNA(10pmol)1μL、プライマーそれぞれ1μL、Primix(Genotech)4μL、蒸留水14μLをMastercycler gradient PCR(Ependorf)を用いて変性(96℃において1分)、アニーリング(50.2℃において1分)、伸長(72℃において2分)段階を合計で30回実施した。
【0061】
前記増幅されたPCR産物を遺伝子増幅後、反応液をプラスミドミニプレップキット(Plasmid mini-prep kit (Solgent))で精製し、70%エタノールを添加してDNAを沈殿させた。沈殿回収したDNAをアガロスゲル電気泳動により確認し、0.9kb相当のDNA断片を精製した。前記DNAを制限酵素NcoI及びXhoIで切断し、上記と同じ制限酵素NcoI及びXhoIで切断されたプラスミドpET32a(+)(Novagen)T4DNA(Takara)にリガーゼを用いて連結した(図3)。前記反応遺伝体をE. coliXL-Blueに導入して得られたアンピシリン耐性形質転換体からプラスミドpNANaを単離した。前記pNANaは、pET32a(+)のT7プロモーター下流のNcoI-XhoI切断部位にE. coli K-12 C600のnanA遺伝子の構造遺伝子を含有するDNA断片が挿入されたものである。
【0062】
前記組換え遺伝子を塩化カルシウムで処理して菌体内にプラスミドを導入する方法(J. Mol. Biol., 53:159, 1970)により高発現菌株E. coli BL21(DE3)pLysS(Invitrogen)に導入し、これをE. coli/pNANaと命名した。
【0063】
実施例1−3.シチジン5’モノホスフェートキナーゼをコードするcmk遺伝子のクローニング及び形質転換
シチジン5’モノホスフェートキナーゼをコードするcmk遺伝子(配列番号8)をクローニングするために、大腸菌K−12(E. coli K-12)菌株の染色体DNAを鋳型とし、配列番号9及び配列番号10のプライマーを用いてPCR法によりcmk遺伝子を増幅した。
【0064】
配列番号9:5'-cat atg acggca att gcc ccg gtt att ac
配列番号10:5'-gaa ttc ggt cgc tta tgc gag agc c
【0065】
PCR条件は鋳型DNA(10pmol)1μL、プライマーそれぞれ1μL、Primix(Genotech)4μLl、蒸留水14μLをMastercycler gradient PCR(Ependorf)を用いて変性(96℃において1分)、アニーリング(55.7℃において1分)、伸長(72℃において2分)段階を合計で30回実施した。
【0066】
前記増幅されたPCR産物の反応液をプラスミドミニプレップキット(Plasmid mini-prep kit (Solgent))で精製し、70%エタノールを添加してDNAを沈殿させた。前記沈殿回収したDNAをアガロスゲル電気泳動により確認し、0.7kb相当のDNA断片を精製した。前記精製されたDNAを制限酵素NdeI及びEcoRIで切断し、上記と同じ制限酵素NdeI及びEcoRIで切断したプラスミドpET22b(+)(Novagen)T4DNA(Takara)にリガーゼを用いて連結した(図4)。前記反応遺伝体をE. coliXL-Blueに導入して得られたアンピシリン耐性形質転換体からプラスミドpCMKを単離した。前記pCMKは、pET22b(+)のT7プロモーター下流のNdeI及びEcoRI切断部位にE.coli K-12C600のcmk遺伝子の構造遺伝子を含有するDNA断片が挿入されたものである。
【0067】
前記組換え遺伝子を塩化カルシウムで処理して菌体内にプラスミドを導入する方法(J. Mol. Biol., 53:159, 1970)により高発現菌株E.coli BL21(DE3)pLysS(Invitrogen)に導入し、これをE. coli/pCMKと命名した。
【0068】
実施例1−4.アセテートキナーゼをコードするack遺伝子のクローニング及び形質転換
アセテートキナーゼをコードするack遺伝子(配列番号11)をクローニングするために、大腸菌K−12(E. coliK-12)菌株の染色体DNAを鋳型とし、配列番号12及び配列番号13のプライマーを用いて、PCR法によりack遺伝子を増幅した。
【0069】
配列番号12:5'-catatgtcgagtaagttagtttctg
配列番号13:5'-gaatcctcaggcagtcaggcggctcgcgtc
【0070】
PCR条件は鋳型DNA(10pmol)1μL、プライマーそれぞれ1μL、Primix(Genotech)4μL、蒸留水14μLをMastercycler gradient PCR(Ependorf)を用いて変性(96℃において1分)、アニーリング(58.7℃において1分)、伸長(72℃において2分)段階を合計で30回実施した。
【0071】
前記増幅されたPCR産物の反応液をプラスミドミニプレップキット(Plasmid mini-prep kit (Solgent))で精製し、70%エタノールを添加してDNAを沈殿させた。前記沈殿回収したDNAをアガロスゲル電気泳動により確認し、1.2kb相当のDNA断片を精製した。前記精製されたDNAを制限酵素NdeI及びEcoRIで切断し、上記と同じ制限酵素NdeI及びEcoRIで切断したプラスミドpET24ma(+)(Novagen)T4DNA(Takara)にリガーゼを用いて連結した(図5)。前記反応遺伝体をE.coli XL-Blueに導入して得られたカナマイシン耐性形質転換体からプラスミドpACKaを単離した。
【0072】
前記pACKaは、pET24ma(+)のT7プロモーター下流のNdeI及びEcoRI切断部位にE. coliK-12 C600のackA遺伝子の構造遺伝子を含有するDNA断片が挿入されたものである。
【0073】
前記組換え遺伝子を塩化カルシウムで処理して菌体内にプラスミドを導入する方法(J. Mol. Biol., 53:159, 1970)により高発現菌株E. coli BL21(DE3)pLysS(Invitrogen)に導入し、これをE. coli/pACKaと命名した。
【0074】
実施例1−5.CMP−N−アセチルノイラミン酸合成酵素をコードするneu遺伝子のクローニング及び形質転換
CMP−N−アセチルノイラミン酸合成酵素をコードするneu遺伝子(配列番号14)をクローニングするために、ナイセリア・メニンジャイティディス(Neisseria meningitidis, Koram Biotech)菌株の染色体DNAを鋳型とし、配列番号15及び配列番号16のプライマーDNAを用いてPCR法によりneu遺伝子を増幅した。
【0075】
配列番号15:5'-aagcatatggaaaaacaaaatattgcg
配列番号16:5'-gtggaattcttagctttccttgtg
【0076】
PCR条件は鋳型DNA(10pmol)1μL、プライマーそれぞれ1μL、Primix(Genotech)4μL、蒸留水14μLをMastercycler gradient PCR(Ependorf)を用いて変性(96℃において1分)、アニーリング(57.7℃において1分)、伸長(72℃において2分)段階を合計で30回実施した。
【0077】
前記増幅されたPCR産物反応液をプラスミドミニプレップキット(Plasmid mini-prep kit (Solgent))で精製し、70%エタノールを添加してDNAを沈殿させた。前記沈殿回収したDNAをアガロスゲル電気泳動により確認し、0.7kb相当のDNA断片を精製した。前記精製されたDNAを制限酵素NdeI及びEcoRIで切断し、上記と同じ制限酵素NdeI及びEcoRIで切断したプラスミドpET32a(+)(Novagen)T4DNA(Takara)にリガーゼを用いて連結した(図6)。前記反応遺伝体をE. coliXL-Blueに導入して得られたアンピシリン耐性形質転換体からプラスミドpSYNbを単離した。前記pSYNbは、pET24ma(+)のT7プロモーター下流のNdeI及びEcoRI切断部位にナイセリア・メニンジャイティディスのsynB遺伝子の構造遺伝子を含有するDNA断片が挿入されたものである。
【0078】
前記組換え遺伝子を塩化カルシウムで処理して菌体内にプラスミドを導入する方法(J. Mol. Biol., 53:159, 1970)により高発現菌株E. coli BL21(DE3)(Invitrogen)に導入し、これをE. coli/pSYNbと命名した。
【0079】
《実施例2:酵素発現確認》
前記形質転換体E. coli/pNANe、E. coli/pNANa、E. coli/pCMK、E. coli/pACKa及びE. coli/pSYNbをLB培地において培養した種菌500mLを本培養LB培地5Lに接種し、細胞密度(OD600)が約3〜5であるときに細胞を得た。各酵素の形質転換体の培養条件を表1に示す。前記得られた細胞を超音波破砕器またはフレンチプレスで破砕した後、SDS−PAGEゲルを通じてそれぞれの酵素発現の度合いを確認した(図7)。
【0080】
その結果、それぞれの形質転換体において挿入された遺伝子によりコードされる酵素が過発現されることを確認することができた。
【0081】
【表1】
【0082】
《実施例3:酵素の精製》
前記N−アセチルグルコサミン−2−エピメラーゼ、N−アセチルノイラミン酸アルドラーゼ、シチジン5’モノホスフェートキナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素を硫酸アンモニウムを用いた沈殿及びイオン交換樹脂カラムにより精製した(Protein purification techniqes第2版、Oxford University Press, 2001)。
【0083】
各酵素沈殿に用いられる硫酸アンモニウムの濃度は、30〜80%の範囲において適切に使用することができる。前記それぞれの酵素を硫酸アンモニウムが含まれた状態で10℃以下、好ましくは、3〜5℃の冷蔵庫において1〜5時間かけて攪拌しながら沈殿した。
【0084】
前記酵素沈殿物を最少量の緩衝溶液50mM Tris HCl(pH7.5)に溶かした後、透析用膜を用いて脱塩した。精製及び濃縮された酵素液は強イオン交換樹脂UNO sphere Q(Bio−RAD)が充填されたカラムに溶出液A(20mM Tris HCl(pH7.5))、溶出液B(20mM Tris HCl/1M NaCl(pH7.5))を用いたA液−B液のグラディアントにより精製した。上記において精製された酵素をSDS−PAGEゲルで確認した(図8)。
【0085】
《実施例4:酵素の固定化》
高分子ビーズ1kg(約1L)に2.5%グルタアルデヒド3Lを添加した後、常温において2時間かけて攪拌して高分子ビーズを活性化させた。前記活性化されたビーズをろ過した後に洗浄し、前記実施例3において精製されたそれぞれの酵素を高分子ビーズ1g当たりに3mg混合した後、常温において6時間かけて攪拌して酵素を固定化した。酵素の固定化の度合い及び定量はブラッドフォード方法(Bradford method, Anal. Biochem., 72:248, 1976)を用いて確認した。
【0086】
《実施例5:CMP−N−アセチルノイラミン酸の合成》
反応混合液(20〜80mMシチジン5’モノホスフェート(CMP、Shanghai QZU Bioscience & Biotechnology)20〜100g、20〜80mMN−アセチル−D−グルコサミン(Shanghai Jiubang Chemical)34.15〜136.6g、40〜120mMピルビン酸33.9〜101.7g、20mMMgCl2・H2O(Duksan)30.9g、1mMCTP(Sigma)3.72g、80〜500mMアセチルホスフェート(Sigma)84〜525g、50mM Tris HClバッファー(pH7.0)7L、37℃2MNaOHを用いてpH6.5〜8.0維持)を前記実施例4において製造したシチジンモノホスフェートキナーゼ(cytidine monophosphate kinase;CMK)、アセテートキナーゼ(acetate kinase;ACK)、N−アセチルノイラミンアルドラーゼ(NeuAc aldolase、NAN)、CMP−N−アセチルノイラミン合成酵素(CMP-NeuAc synthetase、NEU)及びN−アセチルグルコサミン−2−エピメラーゼ(GlcNAc-2-epimerase)がそれぞれ固定された高分子ビーズと混合した後、反応器において攪拌して5〜12時間反応させた。
【0087】
前記反応上澄み液を分離、検出するためにThermo 42カラム(Thermo electron corporation)を使用し、溶出液はA液(100mMKH2PO4/K2HPO4)とB液(メタノール)を用いて、A液(90%):B液(10%)によりHPLCを実施した。その結果、約90%の収率にてCMP−N−アセチルノイラミン酸が合成されていることを確認することができた。
【0088】
《実施例6:CMP−N−アセチルノイラミン酸の精製》
前記実施例5において合成したCMP−N−アセチルノイラミン酸反応液を陰イオン交換樹脂ダウエックスヤギ型(Dowex-1×2, Cl--form)カラムの上部に注入し、塩の濃度勾配を用いて分離した。前記目的物が含まれている溶出液を再濃縮してセファデックスG−10ゲル透過法を用いて塩を除去することにより高純度の最終生成物を得、これを凍結乾燥して濃縮した。各精製段階を経て最終的に81%のCMP−N−アセチルノイラミン酸を得、NMR及びHPLCを用いて最終収得物を確認した(図9及び図10)。
【産業上の利用可能性】
【0089】
以上詳述したように、本発明は、新規N−アセチルグルコサミン−2−エピメラーゼ及びCMP−N−アセチルノイラミン酸を経済的に量産可能な方法を提供するという効果がある。本発明によれば、安価な基質であるシチジンモノホスフェートとN−アセチル−D−グルコサミンを用いて1回の反応によりCMP−N−アセチルノイラミン酸を量産することができ、ヌクレオチドトリホスフェート(NTP)またはATPはアセチルホスフェート及びアセテートキナーゼにより再生されるため、CMP−N−アセチルノイラミン酸を低廉に生産することができる。
【0090】
以上、本発明の内容の特定の部分を詳述したが、当業界における通常の知識を持った者にとって、このような具体的な記述は単なる好適な実施態様に過ぎず、これにより本発明の範囲が制限されることはないという点は明らかである。よって、本発明の実質的な範囲は特許請求の範囲とこれらの等価物により定義されると言える。
【技術分野】
【0001】
本発明は新規N−アセチルグルコサミン−2−エピメラーゼ及びCMP−N−アセチルノイラミン酸の製造方法に係り、さらに詳しくは、バクテロイデスフラギリスNCTC9343(Bacteroides fragilisNCTC 9343)由来のN−アセチルグルコサミン−2−エピメラーゼ及び前記N−アセチルグルコサミン−2−エピメラーゼを用いてCMP−N−アセチルノイラミン酸を製造する方法に関する。
【背景技術】
【0002】
最近、糖鎖に関する構造及び機能に関する研究が盛んになされるに伴い、生理活性を有するオリゴ糖、糖脂質、糖タンパク質などの医薬品または機能性素材としての用途開発が注目を浴びている。中でも、末端にN−アセチルノイラミン酸(NeuAc)を含有するシアル酸含有糖鎖は、細胞接着やウィルス感染時に受容体となるなど重要な機能を有する糖鎖である。
【0003】
シアル酸含有糖鎖は、一般的に、シアル酸転移酵素の触媒作用により合成される。シアル酸転移酵素は、CMP−N−アセチルノイラミン酸(CMP−NeuAc)を糖供給体として糖鎖などの受容体にシアル酸を転移させる酵素である。しかしながら、糖供給体として使用するCMP−NeuAcは極めて高価であり、しかも、量的にも試薬レベルの少量でしか供給されていないのが現状である。
【0004】
CMP−NeuAcの製造方法としては、シチジン5’−トリリン酸(CTP)とN−アセチルノイラミン酸(NeuAc)を基質としてCMP−NeuAcシンテターゼの触媒により合成する方法が知られているが、その原料となるCTP及びNeuAcは高価であるため、直接原料として合成されたCMP−NeuAcも高価にならざるを得ない。
【0005】
近年、オロチン酸からウリジン5’−トリリン酸(UTP)への変換を司るブレビバクテリウムアンモニアゲネス(Brevibacterium ammoniagenes)菌体、UTPからCTPへの変換反応を触媒するCTP合成酵素を生産する組換え大腸菌及びCMP−NeuAc合成酵素を生産する組換え大腸菌を組み合わせ、オロチン酸とNeuAcを原料としてCMP−NeuAcを合成する方法が開発されている。上記の方法は、高価なCTPを使用しない方法ではあるが、複数種の菌体を調製しなければならないなど工程が煩雑であると共に、それを行うための大型の設備を準備することを余儀なくされ、しかも、高価なNeuAcを原料とするという点において、実用的な方法であるとはいえない。
【0006】
一方、NeuAcの製造方法に関しては、シアル酸の多量体であるコロミン酸を微生物から回収し、これを化学分解して回収する方法が知られているが、最近、酵素を用いた方法が開発されている。
【0007】
CMP−N−アセチルノイラミン酸(CMP-neuraminic acid;CMP−NeuAc)の生産方法としては、(1)N−アセチルノイラミン酸リアーゼまたはN−アセチルノイラミン酸シンテターゼを用いてN−アセチルマンノサミン(N-acetyl-D-mannosamine;ManNAc)から製造する方法(J. Am. Chem. Soc., 110:6481, 1988; J. Am. Chem. Soc., 110:7159, 1988;特開平10−4961号公報)、(2)アルカリの条件下においてN−アセチルグルコサミン(GlcNAc)をN−アセチルマンノサミン(ManNAc)に変換し、ここにN−アセチルノイラミン酸リアーゼまたはN−アセチルノイラミン酸シンテターゼを添加してN−アセチルノイラミン酸(NeuAc)を製造する方法(特開平05−211884号公報;Biotechnol. Bioeng., 66:2, 1999; Enzyme Microb. Technol., 20, 1997)、(3)GlcNAcからMamNAcへの変換を触媒するN−アセチルグルコサミン(GlcNAc)2−エピメラーゼとNeuAcリアーゼまたはNeuAcシンテターゼを用いてGlcNAcからCMP−NeuAcを製造する方法(WO95/26399;特開平03−180190号公報;特開2001−136982号公報)、(4)大腸菌と酵母菌体を用いてCMP−N−アセチルノイラミン酸を合成する方法(WO2004/009830)などが報告されている。
【0008】
しかしながら、(1)の方法は原料となるN−アセチルマンノサミン(ManNAc)が高価であり、(2)の方法は安価なN−アセチルグルコサミン(GlcNAc)を原料とする方法ではあるが、GlcNAcとN−アセチルマンノサミン(ManNAc)との混合物においてManNAcを精製する工程が複雑であるという問題がある。さらに、(3)の方法において使用するGlcNAc2−エピメラーゼは、ATPを必要とするため、高価なATPを添加したり、微生物を用いてATPの前駆体であるアデニンからATPを生成しなければならないという欠点があり、(4)の方法は大腸菌と酵母菌体を利用する方法がその工程からみて複雑であるという問題がある。
【0009】
N−アセチルグルコサミン−2−エピメラーゼに関しては、豚やラットの腎臓、肝、脾臓、脳、腸内粘膜、甲状腺、膵臓及び唾液腺に存在することが知られており、豚由来の酵素に関してその特性が調べられており、主として動物由来の遺伝子などから抽出されて研究されている(Biochemistry, 17:3363, 1970; Biochem. J., 210:21, 1983; Proc. Natl. Acad. Sci. U.S.A., 52:371, 1964)。また、N−アセチルグルコサミン−2−エピメラーゼの遺伝子については豚由来の遺伝子が知られている(J. Biol. Chem., 271:16294, 1996)。微生物の中では、藍藻の一種であるシネコシスティス属PCC6803(Synechocystis sp. PCC 6803)はそのゲノムの全体配列が決定されており、ゲノムからN−アセチルグルコサミン−2−エピメラーゼを確認した(DNA Research, 3:109, 1996; Nucleic Acids Research, 26:63, 1998)。
【0010】
一方、大韓民国公開特許(第10−2001−0102019号)は、N−アセチルグルコサミン−2−エピメラーゼ及び前記酵素をコードするDNAに関するものであり、N−アセチルグルコサミン−2−エピメラーゼをコードするDNAを藍藻(Cyanobacteria)のうちシネコシスティス(Synechocystis)から発見して使用した。
【0011】
また、大韓民国公開特許(第10−2006−0010706号)は、CMP−N−アセチルノイラミン酸の製造方法に関するものであり、N−アセチルグルコサミン−2−エピメラーゼをコードする遺伝子及びN−アセチルノイラミン酸アルドラーゼをコードする遺伝子を含む共発現ベクター導入の形質転換体にシチジンモノホスフェート(CMP)、N−アセチルグルコサミン、ピルビン酸及び酵母を添加してノイラミン酸を合成した後、CMP−N−アセチルノイラミン酸合成酵素を添加したり、N−アセチルノイラミン酸アルドラーゼをコードする遺伝子及びCMP−N−アセチルノイラミン酸合成酵素をコードする遺伝子を含む共発現ベクター導入の形質転換体にシチジンモノホスフェート(CMP)、N−アセチルグルコサミン、ピルビン酸及び酵母を添加してCMP−N−アセチルノイラミン酸を製造する方法に関するものであるが、CMP−N−アセチルノイラミン酸を製造するために多段階を経なければならず、且つ、基質として用いられるシチジンモノホスフェート(CMP)がシチジントリホスフェート(CTP)に転換される収率が低いという問題点がある。
【0012】
そこで、本発明者らは、CMP−N−アセチルノイラミン酸を経済的で且つ簡単な方法により製造する方法を開発するために鋭意努力した結果、バクテロイデスフラギリスNCTC9343(Bacteroides fragilis NCTC 9343)において新規N−アセチルグルコサミン−2−エピメラーゼ酵素を確認し、前記酵素の遺伝子を組換えベクターで形質転換して得られたN−アセチルグルコサミン−2−エピメラーゼ、安価な基質であるシチジンモノホスフェート(CMP)及び極少量のNTPを用いて多数の基質と酵素を一気に混ぜてワンポット連鎖反応により高収率にてCMP−N−アセチルノイラミン酸を製造することができるということを確認し、本発明を完成するに至った。
【発明の詳細な説明】
【0013】
本発明の主たる目的は、新規N−アセチルグルコサミン−2−エピメラーゼ及び前記酵素をコードする遺伝子を含有する組換えベクターで形質転換された組換え微生物を培養することを含む、N−アセチルグルコサミン−2−エピメラーゼの製造方法を提供することにある。
【0014】
本発明の他の目的は、前記N−アセチルグルコサミン−2−エピメラーゼを用いて多数の基質と酵素を一気に混ぜてワンポット連鎖反応によりCMP−N−アセチルノイラミン酸を製造する方法を提供することにある。
【0015】
上記の目的を達成するために、本発明は、配列番号2のアミノ酸配列を有するN−アセチルグルコサミン−2−エピメラーゼを提供する。
【0016】
また、本発明は、前記N−アセチルグルコサミン−2−エピメラーゼをコードする遺伝子、前記遺伝子を含有する組換えベクター、前記組換えベクターで形質転換された組換え微生物及び前記組換え微生物を培養すること、及びN−アセチルグルコサミン−2−エピメラーゼを回収することを含む、N−アセチルグルコサミン−2−エピメラーゼの製造方法を提供する。
【0017】
さらに、本発明は、N−アセチル−D−グルコサミンを含む反応混合物を前記N−アセチルグルコサミン−2−エピメラーゼの存在下で反応させること、及びN−アセチル−D−マンノサミンを回収することを含む、N−アセチル−D−マンノサミンの製造方法を提供する。
【0018】
さらに、本発明は、ピルビン酸及び前記N−アセチル−D−マンノサミンを含む反応混合物をN−アセチルノイラミン酸アルドラーゼの存在下で反応させること、及びノイラミン酸を回収することを含む、ノイラミン酸製造方法を提供する。
【0019】
さらに、本発明は、(i)シチジン5’モノホスフェート(cytidine 5' monophosphate;CMP)、(ii)ヌクレオチドトリ−ホスフェート(nucleotide tri-phosphate;NTP)またはATP、(iii)アセチルホスフェート、(iv)N−アセチル−D−グルコサミン及び(v)ピルビン酸を含む反応混合物を(a)N−アセチルグルコサミン−2−エピメラーゼ、(b)N−アセチルノイラミン酸アルドラーゼ、(c)シチジン5’モノホスフェートキナーゼ、(d)アセテートキナーゼ及び(e)CMP−N−アセチルノイラミン酸合成酵素の存在下で反応させること、及びCMP−N−アセチルノイラミン酸を回収することを含む、CMP−N−アセチルノイラミン酸の製造方法を提供する。
【0020】
本発明の他の特徴及び実施態様は、下記の詳細な説明及び特許請求の範囲から一層明らかになる。
【図面の簡単な説明】
【0021】
【図1】CMP−N−アセチルノイラミン酸の合成過程を示す模式図である。
【図2】N−アセチルグルコサミン−2−エピメラーゼをコードする遺伝子(nanE)を含有する組換えベクターを示すものである。
【図3】N−アセチルノイラミン酸アルドラーゼをコードする遺伝子(nan)を含有する組換えベクターを示すものである。
【図4】シチジン5’モノホスフェートキナーゼをコードする遺伝子(cmk)を含有する組換えベクターを示すものである。
【図5】アセテートキナーゼをコードする遺伝子(ack)を含有する組換えベクターを示すものである。
【図6】CMP−N−アセチルノイラミン酸合成酵素をコードする遺伝子(neu)を含有する組換えベクターを示すものである。
【図7】各酵素の発現の有無をSDS−PAGEゲルを通じて確認した結果を示すものである(レーン1:マーカー、レーン2:シチジン5’モノホスフェートキナーゼ、レーン3:アセテートキナーゼ、レーン4:CMP−N−アセチルノイラミン酸合成酵素、レーン5:N−アセチルノイラミン酸アルドラーゼ、レーン6:アシルグルコサミン−2−エピメラーゼ)。
【図8】精製された各酵素の発現の有無をSDS−PAGEゲルを通じて確認した結果を示すものである(レーン1:マーカー、レーン2:精製されたシチジン5’モノホスフェートキナーゼ、レーン3:精製されたアセテートキナーゼ、レーン4:精製されたCMP−N−アセチルノイラミン酸合成酵素、レーン5:精製されたN−アセチルノイラミン酸アルドラーゼ、レーン6:精製されたアシルグルコサミン−2−エピメラーゼ)。
【図9】CMP−N−アセチルノイラミン酸をH1NMRにより分析した結果を示すものである。
【図10】CMP−N−アセチルノイラミン酸をHPLCにより分析した結果を示すものである。
【発明を実施するための最良の形態】
【0022】
本発明は、一観点において、CMP−N−アセチルノイラミン酸の合成に関与するN−アセチルグルコサミン−2−エピメラーゼ酵素、前記N−アセチルグルコサミン−2−エピメラーゼをコードする遺伝子、前記遺伝子を含有する組換えベクター、前記組換えベクターで形質転換された組換え微生物及び前記組換え微生物を培養することを特徴とするN−アセチルグルコサミン−2−エピメラーゼの製造方法に関するものである。
【0023】
本発明において、前記遺伝子は、配列番号1の塩基配列を有することを特徴とする。
【0024】
前記N−アセチルグルコサミン−2−エピメラーゼは、バクテロイデスフラギリスNCTC9343由来の酵素であり、シネコシスティス属とバクテロイデスセタイオタオミクロンの該当遺伝子と35.06〜32.9%の相同性を示している。
【0025】
本発明は、他の観点において、前記N−アセチルグルコサミン−2−エピメラーゼを用いることを特徴とするN−アセチル−D−マンノサミン及びノイラミン酸を製造する方法に関する。
【0026】
本発明は、さらに他の観点において、前記N−アセチルグルコサミン−2−エピメラーゼを用いてワンポット連鎖反応によりCMP−N−アセチルノイラミン酸を製造する方法に関する。
【0027】
本発明によるCMP−N−アセチルノイラミン酸は、(i)シチジン5’モノホスフェート、(ii)ヌクレオチドトリホスフェートまたはATP、(iii)アセチルホスフェート、(iv)N−アセチル−D−グルコサミン及び(v)ピルビン酸を含む反応混合物を(a)N−アセチルグルコサミン−2−エピメラーゼ、(b)N−アセチルノイラミン酸アルドラーゼ、(c)シチジン5’モノホスフェートキナーゼ、(d)アセテートキナーゼ及び(e)CMP−N−アセチルノイラミン酸合成酵素の存在下で反応させることにより製造することができる。
【0028】
本発明によれば、安価な基質であるシチジンモノホスフェートは、CMP−キナーゼ及びアセテートキナーゼによりシチジンダイホスフェート(cytidine diphosphate;CDP)を経てシチジントリホスフェートに97%以上の高収率にて合成される。前記CDPに用いられるホスフェートの供与体としては極少量のNTPを使用し、NDPは過量のアセチルホスフェートとアセテートキナーゼにより再利用されて経済的である。特に、本発明においては、CDPに供されるホスフェート供与体として用いられるATPの代わりにCTPを使用することにより、反応終了後に不純物として存在するADPを除去しなければならないという煩雑さを解消することができる。
【0029】
また、N−アセチル−D−グルコサミン(N-acetyl-D-glucosamine;GlacNAc)は、本発明によるN−アセチルグルコサミン−2−エピメラーゼによりN−アセチル−D−マンノサミン(N-acetyl-D-mannosamine;ManNAc)に転換され、ManNAcとピルビン酸はN−アセチルノイラミン酸アルドラーゼによりノイラミン酸に合成される。以上において合成されたCTPとノイラミン酸は、CMP−N−アセチルノイラミン酸合成酵素(CMP-NeuAc synthetase)によりCMP−N−アセチルノイラミン酸に合成される(図1)。
【0030】
本発明において、前記ヌクレオチドトリホスフェート(NTP)またはATPはアセチルホスフェート及びアセテートキナーゼにより再生されることを特徴とし、前記ヌクレオチドトリホスフェート(NTP)はシチジントリホスフェート(cytidinetri-phosphate;CTP)であることを特徴とする。
【0031】
本発明において、前記N−アセチルグルコサミン−2−エピメラーゼ、N−アセチルノイラミン酸アルドラーゼ、シチジン5’モノホスフェートキナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素は担体に固定化されていることを特徴とし、前記N−アセチルグルコサミン−2−エピメラーゼ、N−アセチルノイラミン酸アルドラーゼ、シチジン5’モノホスフェートキナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素はそれぞれ配列番号1、配列番号5、配列番号8、配列番号11及び配列番号14の塩基配列によりコードされることを特徴とする。
【0032】
本発明においては、高発現された各酵素を精製した後、キトサンなどの高分子ビーズを用いて固定化することにより反復的な合成段階を経ることなく1回で合成することができ、合成コストを大幅に下げることができる。酵素の固定化方法としては、通常の酵素の固定化手段(共有付着法、多機能試薬を用いた架橋、吸着法、微細カプセル化方法など)を使用することができる。酵素の精製方法としては、通常の酵素の精製手段(塩析処理、等電点沈殿処理、透析処理、各種のクロマトグラフィ処理など)を使用することができ、前記精製方法により得られる助酵素または精製酵素を利用することができ、前記酵素を高分子ビーズに固定化する方法としては、通常の酵素固定化方法を使用することができる。上記の方法により精製された酵素は高分子ビーズに固定化して20回以上使用することができるため、酵素を回収するコストと工数を減らすことができる。
【0033】
本発明によれば、前記多数の基質と酵素を一回に混ぜてワンポット連鎖反応を行うことにより各酵素の反応性を極大化させ、少量の酵素及び基質を使用しても、CMP−N−アセチルノイラミン酸を90%以上の高収率にて合成することができる。すなわち、N−アセチルグルコサミン−2−エピメラーゼの場合、エピメラーゼ単独反応を行うと、CMP−N−アセチルノイラミン酸の合成収率は50%にもならないが、ワンポット連鎖反応を行う場合に90%以上の高収率にてCMP−N−アセチルノイラミン酸を合成することができる。
【0034】
本発明において用いられるN−アセチルノイラミン酸アルドラーゼ、CMP−キナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素は公知の酵素であり、通常の方法により調製することができるが、前記遺伝子のクローニング、クローニングしたDNA断片を含む組換えベクターの製作、組換えベクターの導入された形質転換体の製作、形質転換体の培養による目的酵素タンパク質の生産などの組換えDNA法を使用することが好ましい。形質転換に用いられる宿主は特に限定されないが、操作し易さの観点から、大腸菌を使用することが好ましい。
【0035】
本発明において用いられるN−アセチルグルコサミン−2−エピメラーゼは、バクテロイデスフラギリスNCTC9343からN−アセチルグルコサミン−2−エピメラーゼをコードする新規遺伝子を分離したものである。バクテロイデスフラギリスのゲノムDNAの全体配列は決定されたが(US7,090,973)、N−アセチルグルコサミン−2−エピメラーゼ遺伝子に対する機能は全く知られていない。
【0036】
本発明において、「ベクター」とは、適合する宿主内においてDNAを発現可能な適合する調節配列に作動可能に連結されたDNA配列を含有するDNA製造物のことを言う。ベクターは、プラスミド、ファージ粒子または簡単に潜在的ゲノム挿入物であってもよい。適当な宿主で形質転換されれば、ベクターは宿主ゲノムとは無関係に複製して機能できるか、あるいは、場合によってはゲノムそのものに統合可能である。現在、プラスミドがベクターの最も汎用される形態であるため、本発明の明細書における「プラスミド」及び「ベクター」は時々相互交換的に使用される。本発明の目的からみて、プラスミドベクターを用いることが好ましい。このような目的に使用可能な典型的なプラスミドベクターは、(a)1つの宿主細胞当たりに数百個のプラスミドベクターを含むように複製が効率よく行われるようにする複製開始点、(b)プラスミドベクターで形質転換された宿主細胞を選抜できるようにする抗生剤耐性遺伝子及び(c)外来DNA切片が挿入可能な制限酵素切断部位を含む構造を有している。たとえ適切な制限酵素切断部位が存在しないとしても、通常の方法による合成オリゴヌクレオチドアダプターまたはリンカーを使用すれば、ベクターと外来DNAを容易にライゲート(ligation)することができる。
【0037】
ライゲーション後に、ベクターは適切な宿主細胞で形質転換される必要がある。形質転換はカルシウムクロリド方法を用いて容易に行うことができる。選択的に、電気穿孔法(Neumann, et al., EMBO J., 1:841, 1982)もこれらの細胞の形質転換に使用可能である。
【0038】
本発明による遺伝子の過発現のために用いられるベクターとしては、当業界における公知の発現ベクターが使用可能である。
【0039】
核酸は他の核酸配列と機能的関係を持つように配置されるときに「作動可能に連結」される。これは、適切な分子(例えば、転写活性化タンパク質)が調節配列に結合されるときに遺伝子発現を可能にする方式により連結された遺伝子及び調節配列であってもよい。例えば、前配列または分泌リーダーに対するDNAはポリペプチドの分泌に関与するプレタンパク質として発現される場合、ポリペプチドに対するDNAに作動可能に連結され、プロモーターまたはエンハンサーは配列の転写に影響する場合にコード配列に作動可能に連結されるか、あるいは、リボソーム結合部位は配列の転写に影響する場合にコード配列に作動可能に連結されるか、あるいは、リボソーム結合部位は翻訳を容易ならしめるために配置される場合にコード配列に作動可能に連結される。一般的に、「作動可能に連結された」とは、連結されたDNA配列が接触し、また、分泌リーダーの場合、接触してリーディングフレーム内に存在することを意味する。しかしながら、エンハンサーは接触する必要がない。これら配列の連結は便利な制限酵素部位においてライゲーションにより行われる。このような部位が存在しない場合、通常の方法による合成オリゴヌクレオチドアダプターまたはリンカーを使用する。
【0040】
当業界において周知されたように、宿主細胞における形質感染遺伝子の発現レベルを高めるためには、当該遺伝子が選択された発現宿主内において機能を発揮する転写及び解読発現調節配列に作動可能に連結されなければならない。好ましくは、発現調節配列及び当該遺伝子は細菌選択マーカー及び複製開始点を一緒に含んでいる一つの組換えベクター内に含まれることになる。宿主細胞が真核細胞である場合には、組換えベクターは真核発現宿主内において有用な発現マーカーをさらに含む必要がある。
【0041】
上述した組換えベクターにより形質転換された宿主細胞は本発明の他の側面を構成する。本願明細書における使用用語「形質転換」とは、DNAを宿主に導入してDNAが染色体外因子としてまたは染色体統合完成により複製可能になることを意味する。
【0042】
もちろん、全てのベクターが本発明のDNA配列を発現する上で一概に同等に機能を発揮することはないということを理解する必要がある。同様に、全ての宿主が同じ発現システムに対して一概に同様な機能を発揮することはない。しかしながら、当業者であれば、過度な実験的負担なしに本発明の範囲を逸脱しないままで種々のベクター、発現調節配列及び宿主から適切な選択をすることができる。例えば、ベクターを選択するに当たっては宿主を考慮しなければならないが、これは、ベクターがその内部において複製される必要があるためである。ベクターの複製数、複製数を調節可能な能力及び当該ベクターによりコードされる他のタンパク質、例えば、抗生剤マーカーの発現も考慮さるべきである。
【実施例】
【0043】
以下、本発明を実施例により詳述する。これらの実施例は単に本発明をより具体的に説明するためのものであり、本発明の範囲がこれらの実施例に制限されないことは当業界において通常の知識を持った者にとって自明である。
【0044】
《実施例1:遺伝子のクローニング及び形質転換》
実施例1−1.N−アセチルグルコサミン−2−エピメラーゼをコードするnanE遺伝子(配列番号1)のクローニング及び形質転換
CMP−N−アセチルノイラミン酸を製造するに当たって収率を高め、しかも、コストを下げるための最も大きな問題点として指摘されたN−アセチル−D−グルコサミンからN−アセチル−D−マンノサミンへの異性質化段階を解決するために、N−アセチルグルコサミン−2−エピメラーゼを種々のソースから発見した。
【0045】
嫌気性微生物であるバクテロイデスフラギリスNCTC9343(Bacteroides fragilis NCTC 9343, NC_003228)から3種のN−アセチルグルコサミン−2−エピメラーゼ類似遺伝子を発見した。
【0046】
N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子1番は947856〜949034区域にあり、1179bpでコードされており、リゾビウムメリローティ(Rhizobium meliloti)とホモサピエンス(Homo sapiens)の当該酵素遺伝子と23.68%〜24.44%の相同性を示している。
【0047】
N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子2番は1996625〜1997809区域にあり、1185bpでコードされており、シネコシスティス属(Synechocystis sp.)とバクテロイデスセタイオタオミクロン(Bacteroides thetaiotaomicron)の当該酵素遺伝子と35.06%〜32.9%の相同性を示している。
【0048】
N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子3番は4765592〜4766353区域にあり、762bpでコードされており、ロドピレルーラ・バルティカ(Rhodopirellula baltica)とシネコシスティス属(Synechocystis sp.)の当該酵素遺伝子と48.18%〜34.97%の相同性を示している。
【0049】
各遺伝子をpET発現ベクター(Novagen)に挿入し、これをBL21(DE3)、BL21(DE3)pLysS発現菌株に導入して形質転換した。その結果、N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子1番及び2番は可溶性タンパク質として高発現したが、候補遺伝子3番は可溶性タンパク質として発現されず、非可溶性として発現されることを確認した。
【0050】
高発現された候補遺伝子酵素1番及び2番の活性を調べるために、基質としてN−アセチル−D−グルコサミン及びピルビン酸を使用し、N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子1番、2番及び活性が確認されたN−アセチルノイラミン酸アルドラーゼを用いて2段階を1回に反応させて生成されたノイラミン酸を糖分析装置(Metrohm)を用いて確認した。
【0051】
その結果、N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子1番は活性がなく、N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子2番の方が活性に優れていることを確認することができた。また、ヒトN−アセチルグルコサミン−2−エピメラーゼ(organism="Homo sapiens,ene="RENBP", coded_by="NM_002910.4:203..1456")を同じ条件下(50mM N−アセチルD−グルコサミン、100〜200mM ピルビン酸、10mM MgCl2、50mM〜100mM Tris HCl、30℃〜40℃)で活性を比較したとき、N−アセチルグルコサミン−2−エピメラーゼ候補遺伝子2番の反応速度及び収率が極めて高いことが分かり、前記候補遺伝子2番をN−アセチルグルコサミン−2−エピメラーゼとして確認した。
【0052】
前記N−アセチルグルコサミン−2−エピメラーゼ(GlcNAc 2−epimerase;配列番号2)をコードするnanE遺伝子をクローニングするために、バクテロイデスフラギリスNCTC9343(Bacteroides fragilis NCTC 9343)菌株の染色体DNAを鋳型とし、配列番号3及び配列番号4のプライマーを用いてPCR法によりnanE遺伝子を増幅した。
【0053】
配列番号3:5'-ct gcc atg gtt atg aat act aca g
配列番号4:5'-aat gga tcc tta ttt ttc tga cag
【0054】
PCR条件は鋳型DNA(10pmol)1μL、プライマーそれぞれ1μL、Primix(Genotech)4μL、蒸留水14μLをMastercycler gradient PCR(Ependorf)を用いて変性(96℃において1分)、アニーリング(40.2℃において1分)、伸長(72℃において2分)段階を合計で30回実施した。
【0055】
前記増幅されたPCR産物をプラスミドミニプレップキット(Plasmid mini-prep kit(Solgent))で精製し、70%エタノールを添加してDNAを添加した。前記沈殿されたDNAをアガロスゲル電気泳動により確認し、1.2kb相当のDNA断片を精製した。前記精製されたDNAを制限酵素NcoI及びBamHIで切断し、上記と同じ制限酵素NcoI及びBamHIで切断されたプラスミドpET28a(+)(Novagen)T4DNA(Takara)にリガーゼを用いてライゲートした(図2)。
【0056】
前記組換えベクターをE. coliXL-Blueに導入して得られたカナマイシン耐性形質転換体からプラスミドpNANEeを分離した。前記pNANEeプラスミドは、pET28a(+)のT7プロモーター下流のNcoI-BamHI切断部位にバクテロイデスフラギリスのnanE遺伝子の構造遺伝子を含有するDNA断片が挿入されたものである。
【0057】
前記組換え遺伝子を塩化カルシウムで処理して菌体内にプラスミドを導入する方法(J. Mol. Biol., 53:159, 1970)により高発現菌株E. coli BL21(DE3)(Invitrogen)に導入し、これをE. coli/pNANeと命名した。
【0058】
実施例1−2.N−アセチルノイラミン酸アルドラーゼをコードするnanA遺伝子のクローニング及び形質転換
N−アセチルノイラミン酸アルドラーゼ(NeuNAc aldolase)をコードするnanA遺伝子(配列番号5)をクローニングするために、大腸菌K−12(E. coliK-12 C600)菌株の染色体DNAを鋳型とし、配列番号6及び配列番号7のプライマーを用いてPCR法によりnanA遺伝子を増幅した。
【0059】
配列番号6:5'-ggtatccatggcaacgaatttacg
配列番号7:5'-ggtaggctcgagcgaggggaaac
【0060】
PCR条件は鋳型DNA(10pmol)1μL、プライマーそれぞれ1μL、Primix(Genotech)4μL、蒸留水14μLをMastercycler gradient PCR(Ependorf)を用いて変性(96℃において1分)、アニーリング(50.2℃において1分)、伸長(72℃において2分)段階を合計で30回実施した。
【0061】
前記増幅されたPCR産物を遺伝子増幅後、反応液をプラスミドミニプレップキット(Plasmid mini-prep kit (Solgent))で精製し、70%エタノールを添加してDNAを沈殿させた。沈殿回収したDNAをアガロスゲル電気泳動により確認し、0.9kb相当のDNA断片を精製した。前記DNAを制限酵素NcoI及びXhoIで切断し、上記と同じ制限酵素NcoI及びXhoIで切断されたプラスミドpET32a(+)(Novagen)T4DNA(Takara)にリガーゼを用いて連結した(図3)。前記反応遺伝体をE. coliXL-Blueに導入して得られたアンピシリン耐性形質転換体からプラスミドpNANaを単離した。前記pNANaは、pET32a(+)のT7プロモーター下流のNcoI-XhoI切断部位にE. coli K-12 C600のnanA遺伝子の構造遺伝子を含有するDNA断片が挿入されたものである。
【0062】
前記組換え遺伝子を塩化カルシウムで処理して菌体内にプラスミドを導入する方法(J. Mol. Biol., 53:159, 1970)により高発現菌株E. coli BL21(DE3)pLysS(Invitrogen)に導入し、これをE. coli/pNANaと命名した。
【0063】
実施例1−3.シチジン5’モノホスフェートキナーゼをコードするcmk遺伝子のクローニング及び形質転換
シチジン5’モノホスフェートキナーゼをコードするcmk遺伝子(配列番号8)をクローニングするために、大腸菌K−12(E. coli K-12)菌株の染色体DNAを鋳型とし、配列番号9及び配列番号10のプライマーを用いてPCR法によりcmk遺伝子を増幅した。
【0064】
配列番号9:5'-cat atg acggca att gcc ccg gtt att ac
配列番号10:5'-gaa ttc ggt cgc tta tgc gag agc c
【0065】
PCR条件は鋳型DNA(10pmol)1μL、プライマーそれぞれ1μL、Primix(Genotech)4μLl、蒸留水14μLをMastercycler gradient PCR(Ependorf)を用いて変性(96℃において1分)、アニーリング(55.7℃において1分)、伸長(72℃において2分)段階を合計で30回実施した。
【0066】
前記増幅されたPCR産物の反応液をプラスミドミニプレップキット(Plasmid mini-prep kit (Solgent))で精製し、70%エタノールを添加してDNAを沈殿させた。前記沈殿回収したDNAをアガロスゲル電気泳動により確認し、0.7kb相当のDNA断片を精製した。前記精製されたDNAを制限酵素NdeI及びEcoRIで切断し、上記と同じ制限酵素NdeI及びEcoRIで切断したプラスミドpET22b(+)(Novagen)T4DNA(Takara)にリガーゼを用いて連結した(図4)。前記反応遺伝体をE. coliXL-Blueに導入して得られたアンピシリン耐性形質転換体からプラスミドpCMKを単離した。前記pCMKは、pET22b(+)のT7プロモーター下流のNdeI及びEcoRI切断部位にE.coli K-12C600のcmk遺伝子の構造遺伝子を含有するDNA断片が挿入されたものである。
【0067】
前記組換え遺伝子を塩化カルシウムで処理して菌体内にプラスミドを導入する方法(J. Mol. Biol., 53:159, 1970)により高発現菌株E.coli BL21(DE3)pLysS(Invitrogen)に導入し、これをE. coli/pCMKと命名した。
【0068】
実施例1−4.アセテートキナーゼをコードするack遺伝子のクローニング及び形質転換
アセテートキナーゼをコードするack遺伝子(配列番号11)をクローニングするために、大腸菌K−12(E. coliK-12)菌株の染色体DNAを鋳型とし、配列番号12及び配列番号13のプライマーを用いて、PCR法によりack遺伝子を増幅した。
【0069】
配列番号12:5'-catatgtcgagtaagttagtttctg
配列番号13:5'-gaatcctcaggcagtcaggcggctcgcgtc
【0070】
PCR条件は鋳型DNA(10pmol)1μL、プライマーそれぞれ1μL、Primix(Genotech)4μL、蒸留水14μLをMastercycler gradient PCR(Ependorf)を用いて変性(96℃において1分)、アニーリング(58.7℃において1分)、伸長(72℃において2分)段階を合計で30回実施した。
【0071】
前記増幅されたPCR産物の反応液をプラスミドミニプレップキット(Plasmid mini-prep kit (Solgent))で精製し、70%エタノールを添加してDNAを沈殿させた。前記沈殿回収したDNAをアガロスゲル電気泳動により確認し、1.2kb相当のDNA断片を精製した。前記精製されたDNAを制限酵素NdeI及びEcoRIで切断し、上記と同じ制限酵素NdeI及びEcoRIで切断したプラスミドpET24ma(+)(Novagen)T4DNA(Takara)にリガーゼを用いて連結した(図5)。前記反応遺伝体をE.coli XL-Blueに導入して得られたカナマイシン耐性形質転換体からプラスミドpACKaを単離した。
【0072】
前記pACKaは、pET24ma(+)のT7プロモーター下流のNdeI及びEcoRI切断部位にE. coliK-12 C600のackA遺伝子の構造遺伝子を含有するDNA断片が挿入されたものである。
【0073】
前記組換え遺伝子を塩化カルシウムで処理して菌体内にプラスミドを導入する方法(J. Mol. Biol., 53:159, 1970)により高発現菌株E. coli BL21(DE3)pLysS(Invitrogen)に導入し、これをE. coli/pACKaと命名した。
【0074】
実施例1−5.CMP−N−アセチルノイラミン酸合成酵素をコードするneu遺伝子のクローニング及び形質転換
CMP−N−アセチルノイラミン酸合成酵素をコードするneu遺伝子(配列番号14)をクローニングするために、ナイセリア・メニンジャイティディス(Neisseria meningitidis, Koram Biotech)菌株の染色体DNAを鋳型とし、配列番号15及び配列番号16のプライマーDNAを用いてPCR法によりneu遺伝子を増幅した。
【0075】
配列番号15:5'-aagcatatggaaaaacaaaatattgcg
配列番号16:5'-gtggaattcttagctttccttgtg
【0076】
PCR条件は鋳型DNA(10pmol)1μL、プライマーそれぞれ1μL、Primix(Genotech)4μL、蒸留水14μLをMastercycler gradient PCR(Ependorf)を用いて変性(96℃において1分)、アニーリング(57.7℃において1分)、伸長(72℃において2分)段階を合計で30回実施した。
【0077】
前記増幅されたPCR産物反応液をプラスミドミニプレップキット(Plasmid mini-prep kit (Solgent))で精製し、70%エタノールを添加してDNAを沈殿させた。前記沈殿回収したDNAをアガロスゲル電気泳動により確認し、0.7kb相当のDNA断片を精製した。前記精製されたDNAを制限酵素NdeI及びEcoRIで切断し、上記と同じ制限酵素NdeI及びEcoRIで切断したプラスミドpET32a(+)(Novagen)T4DNA(Takara)にリガーゼを用いて連結した(図6)。前記反応遺伝体をE. coliXL-Blueに導入して得られたアンピシリン耐性形質転換体からプラスミドpSYNbを単離した。前記pSYNbは、pET24ma(+)のT7プロモーター下流のNdeI及びEcoRI切断部位にナイセリア・メニンジャイティディスのsynB遺伝子の構造遺伝子を含有するDNA断片が挿入されたものである。
【0078】
前記組換え遺伝子を塩化カルシウムで処理して菌体内にプラスミドを導入する方法(J. Mol. Biol., 53:159, 1970)により高発現菌株E. coli BL21(DE3)(Invitrogen)に導入し、これをE. coli/pSYNbと命名した。
【0079】
《実施例2:酵素発現確認》
前記形質転換体E. coli/pNANe、E. coli/pNANa、E. coli/pCMK、E. coli/pACKa及びE. coli/pSYNbをLB培地において培養した種菌500mLを本培養LB培地5Lに接種し、細胞密度(OD600)が約3〜5であるときに細胞を得た。各酵素の形質転換体の培養条件を表1に示す。前記得られた細胞を超音波破砕器またはフレンチプレスで破砕した後、SDS−PAGEゲルを通じてそれぞれの酵素発現の度合いを確認した(図7)。
【0080】
その結果、それぞれの形質転換体において挿入された遺伝子によりコードされる酵素が過発現されることを確認することができた。
【0081】
【表1】
【0082】
《実施例3:酵素の精製》
前記N−アセチルグルコサミン−2−エピメラーゼ、N−アセチルノイラミン酸アルドラーゼ、シチジン5’モノホスフェートキナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素を硫酸アンモニウムを用いた沈殿及びイオン交換樹脂カラムにより精製した(Protein purification techniqes第2版、Oxford University Press, 2001)。
【0083】
各酵素沈殿に用いられる硫酸アンモニウムの濃度は、30〜80%の範囲において適切に使用することができる。前記それぞれの酵素を硫酸アンモニウムが含まれた状態で10℃以下、好ましくは、3〜5℃の冷蔵庫において1〜5時間かけて攪拌しながら沈殿した。
【0084】
前記酵素沈殿物を最少量の緩衝溶液50mM Tris HCl(pH7.5)に溶かした後、透析用膜を用いて脱塩した。精製及び濃縮された酵素液は強イオン交換樹脂UNO sphere Q(Bio−RAD)が充填されたカラムに溶出液A(20mM Tris HCl(pH7.5))、溶出液B(20mM Tris HCl/1M NaCl(pH7.5))を用いたA液−B液のグラディアントにより精製した。上記において精製された酵素をSDS−PAGEゲルで確認した(図8)。
【0085】
《実施例4:酵素の固定化》
高分子ビーズ1kg(約1L)に2.5%グルタアルデヒド3Lを添加した後、常温において2時間かけて攪拌して高分子ビーズを活性化させた。前記活性化されたビーズをろ過した後に洗浄し、前記実施例3において精製されたそれぞれの酵素を高分子ビーズ1g当たりに3mg混合した後、常温において6時間かけて攪拌して酵素を固定化した。酵素の固定化の度合い及び定量はブラッドフォード方法(Bradford method, Anal. Biochem., 72:248, 1976)を用いて確認した。
【0086】
《実施例5:CMP−N−アセチルノイラミン酸の合成》
反応混合液(20〜80mMシチジン5’モノホスフェート(CMP、Shanghai QZU Bioscience & Biotechnology)20〜100g、20〜80mMN−アセチル−D−グルコサミン(Shanghai Jiubang Chemical)34.15〜136.6g、40〜120mMピルビン酸33.9〜101.7g、20mMMgCl2・H2O(Duksan)30.9g、1mMCTP(Sigma)3.72g、80〜500mMアセチルホスフェート(Sigma)84〜525g、50mM Tris HClバッファー(pH7.0)7L、37℃2MNaOHを用いてpH6.5〜8.0維持)を前記実施例4において製造したシチジンモノホスフェートキナーゼ(cytidine monophosphate kinase;CMK)、アセテートキナーゼ(acetate kinase;ACK)、N−アセチルノイラミンアルドラーゼ(NeuAc aldolase、NAN)、CMP−N−アセチルノイラミン合成酵素(CMP-NeuAc synthetase、NEU)及びN−アセチルグルコサミン−2−エピメラーゼ(GlcNAc-2-epimerase)がそれぞれ固定された高分子ビーズと混合した後、反応器において攪拌して5〜12時間反応させた。
【0087】
前記反応上澄み液を分離、検出するためにThermo 42カラム(Thermo electron corporation)を使用し、溶出液はA液(100mMKH2PO4/K2HPO4)とB液(メタノール)を用いて、A液(90%):B液(10%)によりHPLCを実施した。その結果、約90%の収率にてCMP−N−アセチルノイラミン酸が合成されていることを確認することができた。
【0088】
《実施例6:CMP−N−アセチルノイラミン酸の精製》
前記実施例5において合成したCMP−N−アセチルノイラミン酸反応液を陰イオン交換樹脂ダウエックスヤギ型(Dowex-1×2, Cl--form)カラムの上部に注入し、塩の濃度勾配を用いて分離した。前記目的物が含まれている溶出液を再濃縮してセファデックスG−10ゲル透過法を用いて塩を除去することにより高純度の最終生成物を得、これを凍結乾燥して濃縮した。各精製段階を経て最終的に81%のCMP−N−アセチルノイラミン酸を得、NMR及びHPLCを用いて最終収得物を確認した(図9及び図10)。
【産業上の利用可能性】
【0089】
以上詳述したように、本発明は、新規N−アセチルグルコサミン−2−エピメラーゼ及びCMP−N−アセチルノイラミン酸を経済的に量産可能な方法を提供するという効果がある。本発明によれば、安価な基質であるシチジンモノホスフェートとN−アセチル−D−グルコサミンを用いて1回の反応によりCMP−N−アセチルノイラミン酸を量産することができ、ヌクレオチドトリホスフェート(NTP)またはATPはアセチルホスフェート及びアセテートキナーゼにより再生されるため、CMP−N−アセチルノイラミン酸を低廉に生産することができる。
【0090】
以上、本発明の内容の特定の部分を詳述したが、当業界における通常の知識を持った者にとって、このような具体的な記述は単なる好適な実施態様に過ぎず、これにより本発明の範囲が制限されることはないという点は明らかである。よって、本発明の実質的な範囲は特許請求の範囲とこれらの等価物により定義されると言える。
【特許請求の範囲】
【請求項1】
配列番号2のアミノ酸配列を有するN−アセチルグルコサミン−2−エピメラーゼ。
【請求項2】
請求項1に記載のN−アセチルグルコサミン−2−エピメラーゼをコードする遺伝子。
【請求項3】
配列番号1の塩基配列を有する請求項2に記載の遺伝子。
【請求項4】
請求項2に記載の遺伝子を含有する組換えベクター。
【請求項5】
請求項4に記載の組換えベクターで形質転換された組換え微生物。
【請求項6】
請求項5に記載の組換え微生物を培養すること、及びN−アセチルグルコサミン−2−エピメラーゼを回収することを含む、N−アセチルグルコサミン−2−エピメラーゼの製造方法。
【請求項7】
N−アセチル−D−グルコサミンを含む反応混合物を請求項1に記載のN−アセチルグルコサミン−2−エピメラーゼの存在下で反応させること、及びN−アセチル−D−マンノサミンを回収することを含む、N−アセチル−D−マンノサミンの製造方法。
【請求項8】
ピルビン酸及び請求項7に記載の方法により製造されたN−アセチル−D−マンノサミンを含む反応混合物をN−アセチルノイラミン酸アルドラーゼの存在下で反応させること、及びノイラミン酸を回収することを含む、ノイラミン酸の製造方法。
【請求項9】
(i)シチジン5’モノホスフェート(CMP)、(ii)ヌクレオチドトリホスフェート(NTP)またはATP、(iii)アセチルホスフェート、(iv)N−アセチル−D−グルコサミン及び(v)ピルビン酸を含む反応混合物を(a)N−アセチルグルコサミン−2−エピメラーゼ、(b)N−アセチルノイラミン酸アルドラーゼ、(c)シチジン5’モノホスフェートキナーゼ、(d)アセテートキナーゼ及び(e)CMP−N−アセチルノイラミン酸合成酵素の存在下で反応させること、及びCMP−N−アセチルノイラミン酸を回収することを含む、CMP−N−アセチルノイラミン酸の製造方法。
【請求項10】
前記ヌクレオチドトリホスフェート(NTP)またはATPはアセチルホスフェート及びアセテートキナーゼにより再生されることを特徴とする請求項9に記載の方法。
【請求項11】
前記ヌクレオチドトリホスフェート(NTP)はシチジントリホスフェート(CTP)であることを特徴とする請求項9または請求項10に記載の方法。
【請求項12】
前記N−アセチルグルコサミン−2−エピメラーゼは配列番号2のアミノ酸配列を有することを特徴とする請求項9に記載の方法。
【請求項13】
前記N−アセチルグルコサミン−2−エピメラーゼ、N−アセチルノイラミン酸アルドラーゼ、シチジン5’モノホスフェートキナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素は担体に固定化されていることを特徴とする請求項9に記載の方法。
【請求項14】
前記N−アセチルグルコサミン−2−エピメラーゼ、N−アセチルノイラミン酸アルドラーゼ、シチジン5’モノホスフェートキナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素はそれぞれ配列番号1、配列番号5、配列番号8、配列番号11及び配列番号14の塩基配列によりコードされることを特徴とする請求項9に記載の方法。
【請求項1】
配列番号2のアミノ酸配列を有するN−アセチルグルコサミン−2−エピメラーゼ。
【請求項2】
請求項1に記載のN−アセチルグルコサミン−2−エピメラーゼをコードする遺伝子。
【請求項3】
配列番号1の塩基配列を有する請求項2に記載の遺伝子。
【請求項4】
請求項2に記載の遺伝子を含有する組換えベクター。
【請求項5】
請求項4に記載の組換えベクターで形質転換された組換え微生物。
【請求項6】
請求項5に記載の組換え微生物を培養すること、及びN−アセチルグルコサミン−2−エピメラーゼを回収することを含む、N−アセチルグルコサミン−2−エピメラーゼの製造方法。
【請求項7】
N−アセチル−D−グルコサミンを含む反応混合物を請求項1に記載のN−アセチルグルコサミン−2−エピメラーゼの存在下で反応させること、及びN−アセチル−D−マンノサミンを回収することを含む、N−アセチル−D−マンノサミンの製造方法。
【請求項8】
ピルビン酸及び請求項7に記載の方法により製造されたN−アセチル−D−マンノサミンを含む反応混合物をN−アセチルノイラミン酸アルドラーゼの存在下で反応させること、及びノイラミン酸を回収することを含む、ノイラミン酸の製造方法。
【請求項9】
(i)シチジン5’モノホスフェート(CMP)、(ii)ヌクレオチドトリホスフェート(NTP)またはATP、(iii)アセチルホスフェート、(iv)N−アセチル−D−グルコサミン及び(v)ピルビン酸を含む反応混合物を(a)N−アセチルグルコサミン−2−エピメラーゼ、(b)N−アセチルノイラミン酸アルドラーゼ、(c)シチジン5’モノホスフェートキナーゼ、(d)アセテートキナーゼ及び(e)CMP−N−アセチルノイラミン酸合成酵素の存在下で反応させること、及びCMP−N−アセチルノイラミン酸を回収することを含む、CMP−N−アセチルノイラミン酸の製造方法。
【請求項10】
前記ヌクレオチドトリホスフェート(NTP)またはATPはアセチルホスフェート及びアセテートキナーゼにより再生されることを特徴とする請求項9に記載の方法。
【請求項11】
前記ヌクレオチドトリホスフェート(NTP)はシチジントリホスフェート(CTP)であることを特徴とする請求項9または請求項10に記載の方法。
【請求項12】
前記N−アセチルグルコサミン−2−エピメラーゼは配列番号2のアミノ酸配列を有することを特徴とする請求項9に記載の方法。
【請求項13】
前記N−アセチルグルコサミン−2−エピメラーゼ、N−アセチルノイラミン酸アルドラーゼ、シチジン5’モノホスフェートキナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素は担体に固定化されていることを特徴とする請求項9に記載の方法。
【請求項14】
前記N−アセチルグルコサミン−2−エピメラーゼ、N−アセチルノイラミン酸アルドラーゼ、シチジン5’モノホスフェートキナーゼ、アセテートキナーゼ及びCMP−N−アセチルノイラミン酸合成酵素はそれぞれ配列番号1、配列番号5、配列番号8、配列番号11及び配列番号14の塩基配列によりコードされることを特徴とする請求項9に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2010−512741(P2010−512741A)
【公表日】平成22年4月30日(2010.4.30)
【国際特許分類】
【出願番号】特願2009−541215(P2009−541215)
【出願日】平成19年12月7日(2007.12.7)
【国際出願番号】PCT/KR2007/006367
【国際公開番号】WO2008/072861
【国際公開日】平成20年6月19日(2008.6.19)
【出願人】(509168900)ジーン ケム インコーポレーテッド (1)
【Fターム(参考)】
【公表日】平成22年4月30日(2010.4.30)
【国際特許分類】
【出願日】平成19年12月7日(2007.12.7)
【国際出願番号】PCT/KR2007/006367
【国際公開番号】WO2008/072861
【国際公開日】平成20年6月19日(2008.6.19)
【出願人】(509168900)ジーン ケム インコーポレーテッド (1)
【Fターム(参考)】
[ Back to top ]