
新規PLC様タンパク質およびその利用
【課題】生体内で重要な機能を担っている新規なPLC様タンパク質およびそれをコードする遺伝子の提供、および該タンパク質または遺伝子の、医薬としての用途の提供、および該タンパク質および該遺伝子の発現を制御する化合物のスクリーニング方法の提供。
【解決手段】(a)特定のアミノ酸配列からなるタンパク質をコードするポリヌクレオチド、(b)特定の塩基配列のコード領域を含むポリヌクレオチド、(c)特定のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなり、PLC活性を有するタンパク質をコードする塩基配列を含むポリヌクレオチド、(d)特定の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチドであって、PLC活性を有するタンパク質をコードするポリヌクレオチド、のいずれかに記載のポリヌクレオチド。
【解決手段】(a)特定のアミノ酸配列からなるタンパク質をコードするポリヌクレオチド、(b)特定の塩基配列のコード領域を含むポリヌクレオチド、(c)特定のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなり、PLC活性を有するタンパク質をコードする塩基配列を含むポリヌクレオチド、(d)特定の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチドであって、PLC活性を有するタンパク質をコードするポリヌクレオチド、のいずれかに記載のポリヌクレオチド。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なホスホリパーゼC(Phospholipase
C:以下PLCという。)様タンパク質および該タンパク質をコードする遺伝子、並びに該タンパク質または該遺伝子を組成物として含む医薬品、さらには該タンパク質または該遺伝子が関与する疾患の治療のための化合物のスクリーニング方法に関する。
【背景技術】
【0002】
PLCとは
細胞膜のリン脂質は様々な酵素によって分解され、多くの活性物質に合成される。例えば、ホスホリパーゼA2が作用すると、プロスタグランジンやロイコトリエンが合成される。また、ホスファチジルイノシトール-4,5-二リン酸(PI(4,5)P2)にPLCが作用すると、セカンドメッセンジャーであるイノシトール3リン酸(IP3)やジアシルグリセロール(DAG)ができる。
【0003】
PLCは受容体にカップルしたホスホリパーゼで、細胞外からのホルモン、神経伝達物質、成長因子などの刺激に応答し、細胞膜に存在するリン脂質のうちPI(4,5)P2のみを分解する酵素である。IP3は小胞体のIP3受容体に作用し、細胞内のカルシウム濃度を上昇させ、DAGはプロテインキナーゼC(PKC)を活性化する。PI(4,5)P2は細胞骨格の再構成や、エキソサイトーシス、イオンチャンネル等を直接的に制御していることから、PLC等によるPI(4,5)P2の正確な量的制御は、細胞の恒常性の維持において、非常に重要な役割を担っていると考えられている。それらは、その構造的な特徴や活性制御のメカニズムにより現在では、β(1,2,3,4)、γ(1,2)、δ(1,3,4)、ε(1)、ζ(1)の5つのサブタイプに分けられている。サブタイプによって発現分布が異なり、生体内において、それぞれ異なった機能を担っている(例えば非特許文献1または2参照)。
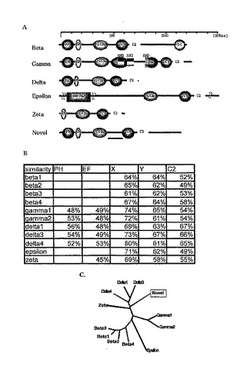
【0004】
サブタイプについて
βタイプのPLCは主にGタンパクによって制御を受けている。線虫のゲノム配列において、2種のPLCβが存在していることから、βタイプのPLCは線虫からヒトまで、広く保存されているPLCである事が分かる。PLCβ2は造血系の細胞、PLCβ4は網膜と小脳に限定されて発現しているが、PLCβ1とβ3はより広範囲に発現が見られ、PLCβ1は大脳皮質や海馬に特に多く、β3は肝臓、耳下腺、脳などに多い。
【0005】
γタイプのPLCはXドメインとYドメインの間に2つのSH2ドメインとSH3ドメインを持っている事が特徴である。SH2ドメインはリン酸化されたチロシン残基を認識し、SH3ドメインはプロリンに富んだ配列と結合することが知られているドメインである。PLCγ1のSH2ドメインはチロシンキナーゼ型レセプターの自己リン酸化されたチロシン残基を認識して結合する。そして、PLCγ1のチロシン残基がリン酸化されることで活性化される事が分かっている。PLCγ1の発現分布はユビキタスであるが、特に脳や肺に多い。脳においてはニューロンに最も多く、次いでオリゴデンドロサイト、アストロサイトに多く発現している事が知られている。PLCγ2は造血系の細胞において多く発現しており、B細胞の分化にPLCγ2が必要であるという報告を裏付けている。
【0006】
δタイプのPLCは真核細胞において進化的に最もよく保存されているPLCである。酵母、粘菌、植物はδタイプのPLCしか持っておらず、βタイプやγタイプのPLCはより高等な生物のみが有するタンパク質である。PLCδの分子量は小さく、さらに構造は単純で、PH、EF-Hand、PLCX、PLCY、C2ドメインから構成されている。PLCδ1が膜へと結合するにはまず、PHドメインが細胞膜のPI 4,5)P2と結合するが、この状態ではPLCδ1は不活性型である。PLCが活性化される為にはさらにC2ドメインが細胞膜に結合する事が必要であり、このとき活性中心が露出される。この活性化型のPLCδ1は効率的にPI(4,5)P2を加水分解し、IP3の産生に付随して細胞膜から遊離する。PLC活性にはカルシウムイオンが必要なことも分かっているが、中でもδタイプのPLCはカルシウムに対する感受性が最も強いことから、生体内に於いてPLCδはカルシウムイオンによって厳密に制御されていることが示唆されている。PLCδタイプの発現分布はδ1が最も多く、そして広範囲に分布している。PLCδ1は脳、心臓、肺、骨格筋、精巣に多く分布しており、ラット脳においてはアストログリアに多く、ニューロンにはあまり発現していない事が分かっている。また、アルツハイマー病患者の脳において、PLCδ1の蓄積が見られることも報告されている。PLCδ3は脳、骨格筋、心臓などに多く、PLCδ4は精巣、脳、骨格筋に分布している。
【0007】
PLCεは近年Rasのエフェクターとして発見された新しいPLCで、Rasと結合するRAドメインを持っている。Rasは単量体Gタンパク質であり、細胞の成長、分化やガン化など、様々な局面で作用している因子である。ヒトのガンのうち、約15%の症例においてras遺伝子に変異が起こっている事などからも、Rasによるシグナル伝達を解明することは非常に重要であると言える。PLCεはRasやそのファミリータンパクであるRap1とGTP依存的に結合することが示された 。さらに、PLCεは進化上PLCβに近いことから、三量体Gタンパクによる制御が検討され、Gα12、Gβ1γ2によって活性化されることも示された。また、PLCεはRas-GEFドメインを持つことから、Ras、Rap1、Rap2A、Rhoに対してGTP-exchange活性を示すかどうかが検討され、Rap1特異的にGEF活性を持つ事が示された。PLCεのmRNAレベルでの発現パターンはいくつか報告されているが、心臓に最も多く、次いで肺、腎臓、脾臓、膵臓に発現しているという報告と、肝臓に最も多く、次いで肺、骨格筋、心臓、小腸に多いという報告がある。
【0008】
PLCζは近年同定された最も新しいPLCである。PLCζの制御メカニズムについてはまだあまりよく分かっていないが、カルシウムに対する感受性が非常に強いことが報告されている。また、PLCζの局在は精子に限定されており、受精の際に起こる卵のカルシウムオシレーションを引き起こす作用があることが分かっている(例えば非特許文献3参照)。
【0009】
本発明者らは、2004年度日本脂質生化学会、および2004年度日本生化学会において、本願発明に係る新規PLC様タンパク質の概要に関して発表を行った(非特許文献4および5参照)。本願発明に係る新規PLC様タンパク質が、PLCに特徴的な構造を有していること、脳に発現していること、カルシウム感受性であること等について公表したものの、新規PLC様タンパク質のアミノ酸および遺伝子配列、発現部位の詳細な特定、機能等については全く明らかにしていない。
【0010】
新規PLC様タンパクの有用性
既知のPLCが上記のように生体内において重要な機能を担っていること、各サブタイプ毎に特徴的な機能を有していること等を鑑みると、新規PLCを見つけることができれば、そのサブタイプに特有の重要な機能を担っている可能性も高く、これまで治療が不可能であった疾患との関わりなどが存在する可能性も非常に高く、産業上有効に利用することができると思われる。
【非特許文献1】K. Fukami, Structure, regulation, and function ofphospholipase C isozymes, J. Biochem. 131 (2002) 293-299.
【非特許文献2】M. J. Rebecchi, and S. N. Pentyala, Structure,function, and control of phosphoinositide-specific phospholipase C, Physiol.Rev. 80 (2000) 1291-1335.
【非特許文献3】M. Katan, Families of phosphoinositide-specificphospholipase C: structure and function, Biochim. Biophys. Acta 1436 (1998)5-17.
【非特許文献4】2004年度 日本脂質生化学会要旨集、2004年5月20日発行
【非特許文献5】2004年度 日本生化学会要旨集、2004年8月25日発行
【発明の開示】
【発明が解決しようとする課題】
【0011】
本発明は、このような状況に鑑みてなされたものであり、その目的は新規なPLC様タンパク質遺伝子を提供することにある。また、本発明は、このようにして同定された新規なPLC様タンパク質または遺伝子の医薬としての用途、特に神経再生医療に有効である薬剤を提供すること、さらには該タンパク質または該遺伝子が関与する疾患の治療のための化合物のスクリーニング方法を提供することも目的とする。
【0012】
マウスゲノム解析が完了したことから、そのゲノムシークエンスを検索することにより、PLCに特徴的な構造(例えばPH, EF-hand, PLCX, PLCY, C2など)を手がかりとすれば、これまで知られていないPLC様構造を持つ配列の存在は、比較的容易に見つけ出すことができるようになった。しかし、PLC様構造を持つ配列は複数存在し、それらのうちどれが生体内で実際にタンパクとして発現し、PI(4,5)P2をIP3とDAGに加水分解するPLC活性を有するかということは全く明らかではなかった。また、データベースに示されている配列は、PLC様タンパクの全長配列ではないこと、cDNAクローン情報には多くのミスがあること、オープンリーディングフレームが示されていないこと、メチオニンも複数存在し、タンパクをコードしている最初の塩基がどこであるのかも不明であったことなどから、真の新規PLC様タンパク質を見出すことは決して容易なことではなく、実際マウスゲノム解析が完了してから2年が過ぎようとしている現在でも、本願発明者以外で新規PLCタンパク質の同定に成功した者はいないのが現状である。
【課題を解決するための手段】
【0013】
本願発明者らは、上記課題を解決すべく鋭意研究を行った結果、新規PLC様タンパク質の全長cDNAのクローニングおよびアミノ酸配列の決定に成功し、生体内における発現、PLC活性の確認に成功した。さらに、本タンパク質が神経突起伸展活性を有するという予想だにしなかった効果をも見出し、神経再生医療への有用性を示すことができた。
【0014】
具体的には、セレラ社(MD, USA) のデータベースを検索し、既知PLCが有する共通した特徴的な構造(PH, EF-hand, PLCX, PLCY, C2)を手がかりに、マウスの4番染色体にPLCと思われるドメイン構造を持つ予想アミノ酸配列を見いだすことが出来た。この予想タンパク質mCP21687は約1600アミノ酸から構成されていたが、開始コドンが含まれて
おらず、完全長のものではなかった。そこで、Genbankより類似の配列を検索したところ、RIKEN cDNAクローンA930027K05がmCP21687をコードしている遺伝子である事と考えられた。しかし、A930027K05クローンは開始メチオニンを 含むタンパク質をコードしていたが、予想転写産物にはPLCYドメインとC2ドメインは含まれていなかった。これはクローニングの際に起こったフレーム
シフト変異又はシークエンシングミスと思われたため、真偽を確かめるためにA930027K05クローンの配列を基にマウス脳cDNAよりPCR反応を 行ったところ、A930027K05クローンには2箇所に1塩基の挿入がある事が判明し、さらにはC2ドメインをコードする配列の約600塩基後にストップコドンが存在し、C末端側のアミノ酸配列は当初の予想よりも400アミノ酸短い事が判明した。
【0015】
単離したクローンの全長をシークエンスし、アミノ酸配列に変換した。その結果、この遺伝子の転写産物は全長1164アミノ酸からなるタンパク質である事が分かった。推定分子量は約125kDaであった。また既知のPLCに特徴的なPHドメイン、EF-handドメイン、PLCXドメイン、C2ドメインを有していることも明らかとなった。新規PLC様タンパク質は、PLCXドメインとPLCYドメインの間の配列が他のPLCよりも長いこと、C末端側に200アミノ酸ほどの付加的な配列を持つことが、既知PLCタンパクと大きく違う点であった。また、各ドメイン毎の類似性を比較したところ、PLCδタイプに最も類似性が高いことが示された(図1)ものの、全配列間の相同性は約30%程度であった。またインターネット上に公開されているプログラムCLUSTALWを用いて系統樹を作成したところ、新規PLC様タンパク質はPLCδに近いことが示されたが、δタイプのPLCが進化的に高く保存されているのに対して、新規PLC様タンパク質は線虫においてホモログが見当たらないことや、PLCδファミリーは構造が比較的単純であり約750アミノ酸からなるタンパク質であるのに対し、新規PLC様タンパク質は1164アミノ酸から構成されていることなどを考え合わせると、本願発明にかかるタンパク質は、新しいサブタイプのPLCタンパク質であると考えられた。
【0016】
また、Sf-9/バキュロウイルス発現系を用いて新規PLC様タンパク質を発現させたところ、実際にPLC活性を有していることが確認できた。さらに、このタンパクに特徴的な配列部分のペプチドに対するポリクローナル抗体を作成し、それを利用して、生体内に存在する新規PLC様タンパク質を精製したところ、Sf-9/バキュロウイルス発現系を用いて発現させたものと同様のPLC活性を有していることを確認した。また、この新規PLC様タンパク質は0.1〜1uMという生体内に近い低カルシウム濃度で最大の活性を示すことも明らかになった。
【0017】
また生体内での発現分布を調べたところ、脳に特異的に発現しており、その中でも特に嗅球、海馬、大脳皮質に多く発現していたことから、新規PLC様タンパク質は脳における記憶のメカニズムに関与していると考えられた。さらに詳細な解析により、嗅球のうち僧帽細胞に発現が限局していることが判明し、神経細胞のシナプス形成への関与も強く示唆された。海馬神経初代培養細胞に新規PLC様タンパク質を強制発現させると、神経突起数が顕著に増大することも明らかになり、新規PLC様タンパク質は、脳梗塞等の脳疾患およびパーキンソン病等における神経再生医療等にも極めて有用なタンパク質であることが示唆された。
【0018】
本願発明の内容は、新規なPLC様タンパク質およびそれをコードする遺伝子を提供すること、及びこのようにして同定された新規なPLC様タンパク質または該遺伝子の用途を提供することである。即ち、本願発明は以下の通りである。
(1)下記(a)から(d)のいずれかに記載のポリヌクレオチド。
(a)配列番号:2に記載のアミノ酸配列からなるタンパク質をコードするポリヌクレオチド。
(b)配列番号:1に記載の塩基配列のコード領域を含むポリヌクレオチド。
(c)配列番号:2に記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなり、PLC活性を有するタンパク質をコードする塩基配列を含むポリヌクレオチド。
(d)配列番号:1に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチドであって、PLC活性を有するタンパク質をコードするポリヌクレオチド。
(2)(1)に記載のポリヌクレオチドによりコードされるポリペプチド。
(3)(1)に記載のポリヌクレオチドが挿入された組み換えベクター。
(4)(1)に記載のポリヌクレオチドまたは(3)に記載のベクターを保持する形質転換体。
(5)(1)に記載のポリヌクレオチドとストリンジェントな条件下で特異的にハイブリダイズするポリヌクレオチドであって、少なくとも15ヌクレオチドの鎖長を持つポリヌクレオチド。
(6)(1)に記載のポリヌクレオチドもしくはその一部に対するアンチセンスポリヌクレオチド。
(7)(2)に記載のポリペプチドに結合する抗体。
(8)配列番号;2で表されるアミノ酸配列中、第497番から第624番目の部分アミノ酸配列部と反応性を示すことを特徴とする(7)に記載の抗体。
(9)(2)に記載のポリペプチドまたは(3)に記載の組み換えベクター及び薬学的に許容されうる担体を含んでなる医薬組成物
(10)該医薬組成物が、神経再生剤である請求項9に記載の医薬組成物
(11)以下の(a)〜(c)の工程を含む、請求項2に記載のポリペプチドまたはその部分ペプチドに結合する候補化合物のスクリーニング方法。
(a)(2)に記載のポリペプチドまたはその部分ペプチドと被検化合物を接触させる工程、
(b)該ポリペプチドまたはその部分ペプチドと被検化合物との結合活性を検出する工程、
(c)被検化合物を接触させない場合と比較して、該ポリペプチドまたはその部分ペプチドに結合する活性を有する化合物を選択する工程
(12)以下の(a)〜(c)の工程を含む、神経再生剤として有効である候補化合物のスクリーニング方法。
(a)(1)に記載のポリヌクレオチドまたは(2)に記載のポリペプチドを発現する細胞と被検化合物を接触させる工程
(b)上記細胞の(1)に記載のポリヌクレオチドまたは(2)に記載のポリペプチドの発現量を測定する工程
(c)被検化合物を接触させない場合と比較して、上記発現量を変化させる化合物を選択する工程
(13)(1)に記載のポリヌクレオチドの転写を制御する領域の下流にレポーター遺伝子の連結されたDNAを含むプラスミドで形質転換された形質転換体と被験化合物とを接触させ、被験化合物より(1)に記載のポリヌクレオチドの発現を制御する化合物を選択することを特徴とする、(1)に記載のポリヌクレオチドの発現を制御する化合物のスクリーニング方法
。
【発明の効果】
【0019】
本発明に係るタンパク質は、既知PLCとは、構造的に大きく異なるものであり、Sf-9/バキュロウイルス系を用いて発現させたもの、内在性のもの、両方にPLC活性があることが確認できた。また、本タンパク質は0.1〜1uMという生体内に近い低カルシウム濃度で最大の活性を示し、既知PLCのサブタイプのうち最もカルシウム感受性の高いδタイプの約10倍の感受性を示すものであった。
【0020】
さらに本タンパク質の生体内における発現は、脳に特異的であり、その中でも特に嗅球、海馬、大脳皮質に多く発現していたことから、脳における記憶のメカニズムに関与していると考えられた。さらに詳細な解析により、嗅球のうち僧帽細胞に発現が限局していることが判明し、神経細胞のシナプス形成への関与も強く示唆された。海馬神経初代培養細胞に新規PLC様タンパク質を強制発現させると、神経突起数が増大するという予想だにしない効果を見出した。このことは、新規PLC様タンパク質が、脳梗塞等の脳疾患およびパーキンソン病等における神経再生医療等に極めて有用なタンパク質であることを示すものである。
【発明を実施するための最良の形態】
【0021】
本発明は、新規なPLC様タンパク質、及びそれをコードするポリヌクレオチドを提供する。以下、本発明のために、好ましい実施形態に関して詳述する。
【0022】
(1)ポリヌクレオチド
本発明は、配列番号2で示されるアミノ酸配列をコードする核酸、及び配列番号2で示されるアミノ酸配列のうち1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなり、PLC活性を有するポリヌクレオチド)のアミノ酸配列をコードするポリヌクレオチドも提供する。
本発明のポリヌクレオチドは、一本鎖および二本鎖型両方のDNA、およびそのRNA相補体も含む。DNAには、例えば、天然由来のDNA、組換えDNA、化学合成したDNA、PCRによって増幅されたDNA、およびそれらの組み合わせが含まれる。本発明のポリヌクレオチドとしてはDNAが好ましい。なお、周知の通り、コドンには縮重があり、1つのアミノ酸をコードする塩基配列が複数存在するアミノ酸もあるが、上記アミノ酸配列をコードする塩基配列であれば、いずれの塩基配列を有するものも本願発明の範囲に含まれる。なお、下記の実施例1において実際にクローニングされたcDNAの塩基配列が配列番号1に示されている。よって、本発明のポリヌクレオチドは、好ましくは配列番号:1の塩基1−3497を有する。
【0023】
本明細書において、「ストリンジェントな条件下」とは、中程度又は高程度なストリンジェントな条件においてハイブリダイズすることを意味する。具体的には、中程度のストリンジェントな条件は、例えば、DNAの長さに基づき、一般の技術を有する当業者によって、容易に決定することが可能である。基本的な条件は、Sambrook,J.ら、Molecular
Cloning,A Laboratory Manual(3rd edition),Cold Spring
Harbor Laboratory,7.42−7.45(2001)に示され、そしてニトロセルロースフィルターに関し、5×SSC、0.5% SDS、1.0mM EDTA(pH8.0)の前洗浄溶液、約40−50℃での、約50%ホルムアミド、2×SSC−6×SSC(又は約42℃での約50%ホルムアミド中の、スターク溶液(Stark’s solution)などの他の同様のハイブリダイゼーション溶液)のハイブリダイゼーション条件、および約60℃、0.5×SSC、0.1% SDSの洗浄条件の使用が含まれる。高ストリンジェントな条件もまた、例えばDNAの長さに基づき、当業者によって、容易に決定することが可能である。一般的に、こうした条件は、中程度にストリンジェントな条件よりも高い温度及び/又は低い塩濃度でのハイブリダイゼーション及び/又は洗浄を含み、例えば上記のようなハイブリダイゼーション条件、及びおよそ68℃、0.2×SSC、0.1% SDSの洗浄を伴うと定義される。当業者は、温度および洗浄溶液塩濃度は、プローブの長さ等の要因に従って、必要に応じて調整可能であることを認識するであろう。
【0024】
本発明のポリヌクレオチドは、例えば以下の方法により調製することが可能である。
公知のPLCに特徴的な塩基配列(PH, EF-hand, PLCX, PLCY, C2)をクエリーとして塩基配列の検索を行い、マウスゲノムよりPLC様タンパク質候補を見出す。見出したゲノム配列は又はその一部を利用して、ハイブリダイゼーションや核酸増幅反応等の遺伝子工学の基本的手法を用いて、cDNAライブラリーなどから本発明ポリヌクレオチド(例えば本発明DNA)を調製することができる。
【0025】
ここで、核酸増幅反応は、例えば、ポリメラーゼ連鎖反応(PCR)[Saiki
R.K.et al.,Science,230,1350−1354(1985)],ライゲース連鎖反応(LCR)[Wu
D.Y.et al.,Genomics,4,560−569(1989);
Barringer K.J.et al.,Gene,89,117−122(1990);
Barany F.,Proc.Natl.Acad.Sci.USA,88,189−193(1991)]、及び転写に基づく増幅[Kwoh
D.Y.et al.,Proc.Natl.Acad.Sci.USA,86,1173−1177(1989)]等の温度循環を必要とする反応、並びに鎖置換反応(SDA)[Walker
G.T.et al.,Proc.Natl.Acad.Sci.USA,89,392−396(1992);
Walker G.T.et
al.,Nuc.Acids Res.,20,1691−1696(1992)]、自己保持配列複製(3SR)[Guatelli J.C.,Proc.Natl.Acad.Sci.USA,87,1874−1878(1990)]、及びQβレプリカーゼシステム[リザイルディら、BioTechnology,6,1197−1202(1988)]等の恒温反応を含む。また、欧州特許第0525882号に記載されている標的核酸と変異配列の競合増幅による核酸配列に基づく増幅(Nucleic
Acid Sequence Based
Amplification: NASABA)反応等も利用可能である。好ましくはPCR法である。
【0026】
実施例1に示すように、配列番号3及び4のプライマーを使用した場合、PCR産物として約1.5kbpのDNA断片が得られるので、これを例えばアガロースゲル電気泳動等の分子量によりDNA断片を篩い分ける方法で分離し、特定のバンドを切り出す方法等の常法に従って単離して本発明の核酸を得ることができる。
【0027】
上記のようなハイブリダイゼーション、核酸増幅反応等を使用してクローニングされる相同な核酸は、配列表の配列番号1に記載の塩基配列に対して少なくとも20%以上、好ましくは30%以上、より好ましくは50%以上、さらにより好ましくは70%以上、最も好ましくは90%以上の同一性を有する。
【0028】
同一性パーセントは、視覚的検査および数学的計算によって決定することが可能である。あるいは、2つの核酸配列の同一性パーセントは、Devereuxら,Nucl.Acids
Res.,12,387(1984)に記載され、そしてウィスコンシン大学遺伝学コンピューターグループ(UWGCG)より入手可能なGAPコンピュータープログラム、バージョン6.0を用いて、配列情報を比較することによって、決定可能である。GAPプログラムの好ましいデフォルトパラメーターには:(1)ヌクレオチドに関する単一(unary)比較マトリックス(同一に対し1および非同一に対し0の値を含む)、並びにSchwartz及びDayhoff監修,Atlas
of Protein
Sequence and Structure,pp.353−358,National
Biomedical Research Foundation(1979)に記載されるような、Gribskov及びBurgess,Nucl.Acids
Res.,14,6745(1986)の加重比較マトリックス;(2)各ギャップに対する3.0のペナルティおよび各ギャップ中の各記号に対しさらに0.10のペナルティ;及び(3)末端ギャップに対するペナルティなし、が含まれる。当業者に用いられる、配列比較の他のプログラムもまた、使用可能である。
【0029】
本発明は、本発明者らにより同定されたポリヌクレオチド(配列番号:1に記載の塩基配列からなるポリヌクレオチドまたはその相補鎖)に相補的な、少なくとも15ヌクレオチドの鎖長を有するヌクレオチドを提供する。ここで「相補鎖」とは、A:T(ただしRNAの場合は U)、G:Cの塩基対からなる2本鎖核酸の一方の鎖に対する他方の鎖を指す。また、「相補的」とは、少なくとも15個の連続したヌクレオチド領域で完全に相補配列である場合に限られず、少なくとも70%、好ましくは少なくとも80%、より好ましくは90%、さらに好ましくは95%以上の塩基配列上の相同性を有すればよい。相同性を決定するためのアルゴリズムは本明細書に記載したものを使用すればよい。このようなヌクレオチドは、本発明のポリヌクレオチドを検出、単離するためのプローブとして、また、本発明のヌクレオチドを増幅するためのプライマーとして利用することが可能である。プライマーとして用いる場合には、通常、15〜100ヌクレオチド、好ましくは15〜35ヌクレオチドの鎖長を有する。また、プローブとして用いる場合には、本発明のDNAの少なくとも一部若しくは全部の配列を含む少なくとも15ヌクレオチド、好ましくは少なくとも30ヌクレオチドの鎖長のポリヌクレオチドが用いられる。このようなポリヌクレオチドは、好ましくは本発明のポリペプチドをコードするポリヌクレオチドに特異的にハイブリダイズするものである。「特異的にハイブリダイズする」とは、通常のハイブリダイゼーション条件下、好ましくはストリンジェントな条件下で、本発明者らにより同定されたポリヌクレオチド(配列番号:1または3)とハイブリダイズし、他のポリペプチドをコードするDNAとはハイブリダイズしないことを意味する。
【0030】
また、このポリヌクレオチドには、本発明のポリペプチドをコードする遺伝子の発現を抑制するポリヌクレオチドが含まれる。このようなポリヌクレオチドには、アンチセンスポリヌクレオチド(アンチセンスDNA/RNA;本発明のポリペプチドをコードする遺伝子の転写産物と相補的なアンチセンスRNA、および該RNAをコードするDNA)やリボザイム(本発明のポリペプチドをコードする遺伝子の転写産物を特異的に開裂するリボザイム活性を有するRNAをコードするDNA)が含まれる。
【0031】
アンチセンスポリヌクレオチドが標的遺伝子の発現を抑制する作用としては、以下のような複数の要因が存在する。すなわち、三重鎖形成による転写開始阻害、RNAポリメラーゼによって局部的に開状ループ構造がつくられた部位とのハイブリッド形成による転写抑制、合成の進みつつあるRNAとのハイブリッド形成による転写阻害、イントロンとエキソンとの接合点でのハイブリッド形成によるスプライシング抑制、スプライソソーム形成部位とのハイブリッド形成によるスプライシング抑制、mRNAとのハイブリッド形成による核から細胞質への移行抑制、キャッピング部位やポリ(A)付加部位とのハイブリッド形成によるスプライシング抑制、翻訳開始因子結合部位とのハイブリッド形成による翻訳開始抑制、開始コドン近傍のリボソーム結合部位とのハイブリッド形成による翻訳抑制、mRNAの翻訳領域やポリソーム結合部位とのハイブリッド形成によるペプチド鎖の伸長阻止、および核酸とタンパク質との相互作用部位とのハイブリッド形成による遺伝子発現抑制などである。これらは、転写、スプライシング、または翻訳の過程を阻害して、標的遺伝子の発現を抑制する(平島および井上「新生化学実験講座2 核酸IV 遺伝子の複製と発現」,日本生化学会編,東京化学同人,pp.319-347,1993)。
【0032】
本発明で用いられるアンチセンスポリヌクレオチドは、上記のいずれの作用で標的遺伝子の発現を抑制してもよい。一つの態様としては、遺伝子のmRNAの5'端近傍の非翻訳領域に相補的なアンチセンス配列を設計すれば、遺伝子の翻訳阻害に効果的と考えられる。しかし、コード領域もしくは3'側の非翻訳領域に相補的な配列も使用し得る。このように、遺伝子の翻訳領域だけでなく非翻訳領域の配列のアンチセンス配列を含むポリヌクレオチドも、本発明で利用されるアンチセンスポリヌクレオチドに含まれる。使用されるアンチセンスポリヌクレオチドは、適当なプロモーターの下流に連結され、好ましくは3'側に転写終結シグナルを含む配列が連結される。アンチセンスポリヌクレオチド配列は、標的遺伝子またはその一部と相補的な配列であることが好ましいが、遺伝子の発現を有効に阻害できる限り、完全に相補的でなくてもよい。転写されたRNAは、標的とする遺伝子の転写産物に対して好ましくは90%以上、最も好ましくは95%以上の相補性を有する。アンチセンス配列を用いて、効果的に標的遺伝子の発現を阻害するには、アンチセンスポリヌクレオチドは、アンチセンス効果を引き起こすために、少なくとも15ヌクレオチド以上、好ましくは100ヌクレオチド、さらに好ましくは500ヌクレオチド以上の鎖長を有し、通常、3000ヌクレオチド以内、好ましくは2000ヌクレオチド以内の鎖長を有する。
【0033】
該アンチセンスポリヌクレオチドは、例えば、本発明のポリペプチドをコードするポリヌクレオチド(例えば、配列番号:1)の配列情報を基にホスホロチオネート法(Stein, 1988 Physicochemical properties of phosphorothioate
oligodeoxynucleotides. Nucleic Acids Res 16, 3209-21 (1988))などにより調製することが可能である。
【0034】
内在性遺伝子の発現の抑制は、また、リボザイムをコードするポリヌクレオチドを利用して行うことも可能である。リボザイムとは触媒活性を有するRNA分子のことをいう。リボザイムには種々の活性を有するものがあるが、中でもRNAを切断する酵素としてのリボザイムの研究により、RNAの部位特異的な切断を目的とするリボザイムの設計が可能となった。リボザイムには、グループIイントロン型や、RNasePに含まれるM1RNAのように400ヌクレオチド以上の大きさのものもあるが、ハンマーヘッド型やヘアピン型と呼ばれる40ヌクレオチド程度の活性ドメインを有するものもある(小泉誠および大塚栄子, (1990) 蛋白質核酸酵素,35:2191)。
【0035】
例えば、ハンマーヘッド型リボザイムの自己切断ドメインは、G13U14C15のC15の3'側を切断するが、活性にはU14が9位のAと塩基対を形成することが重要とされ、15位の塩基はCの他にAまたはUでも切断されることが示されている(M.Koizumiら,(1988) FEBS Lett.228:225)。リボザイムの基質結合部を標的部位近傍のRNA配列と相補的になるように設計すれば、標的RNA中のUC、UUまたはUAという配列を認識する制限酵素的なRNA切断リボザイムを作出することが可能である(M.Koizumiら,(1988) FEBS Lett. 239:285、小泉誠および大塚栄子,(1990) 蛋白質核酸酵素,35:2191、 M.Koizumiら, (1989) Nucleic Acids
Res. 17:7059)。
【0036】
また、ヘアピン型リボザイムも、本発明の目的のために有用である。ヘアピン型リボザイムは、例えばタバコリングスポットウイルスのサテライトRNAのマイナス鎖に見出される(J.M.Buzayan Nature 323:349,1986)。このリボザイムも、標的特異的なRNA切断を起こすように設計できることが示されている(Y.Kikuchi
およびN.Sasaki
(1992) Nucleic Acids Res. 19:6751、
菊池洋, (1992) 化学と生物 30:112)。
【0037】
本発明のポリペプチドをコードする遺伝子の発現を抑制するポリヌクレオチドは、遺伝子治療に用いる場合には、例えば、レトロウイルスベクター、アデノウイルスベクター、アデノ随伴ウイルスベクターなどのウイルスベクターやリポソームなどの非ウイルスベクターなどを利用して、ex vivo法やin vivo法などにより患者へ投与を行うことが考えられる。
【0038】
(2)ポリペプチド
下記実施例1において詳述する方法によりクローニングされた、本発明者らにより同定されたマウス新規PLC様タンパク質をコードするポリヌクレオチドの塩基配列を配列番号:1に、該ポリヌクレオチドによってコードされるポリペプチドのアミノ酸配列は配列番号:2に示す。
【0039】
本発明はまた、本発明者らにより同定されたポリぺプチドと機能的に同等なポリペプチド、および該ポリペプチドをコードするポリヌクレオチドに関する。ここで「機能的に同等」とは、対象となるポリペプチドが本発明者らにより同定されたポリぺプチドと同等の生物学的特性を有していることを意味する。本発明ポリペプチドが持つ生物学的特性としては、PLC活性(PIP2をIP3及びDAGに分解する活性)、低カルシウム感受性(本発明ポリペプチドは、低カルシウム濃度0.1〜1uMにおいて、最大のPLC活性を示すという特性を有する。)、および発現(本発明ポリペプチドは、動物の組織、好ましくは哺乳動物の組織で発現される。また発現部位としては、脳で高い発現を示す。好ましくは、嗅球、海馬、大脳皮質で高い発現を示す。さらに好ましくは、僧帽細胞において高い発現を示す。)等が挙げられる。
【0040】
本発明ポリペプチドは、典型的には、配列番号2に記載したアミノ酸残基1164個からなるアミノ酸配列を有する。なお一般的に、酵素のような生理活性を有するポリペプチドにおいて、そのアミノ酸配列のうち、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加された場合であっても、該生理活性が維持されることがあることは周知である。また天然産のポリペプチドの中には、それを生産する生物種の品種の違いや生態型の違いによる遺伝子の変異、あるいはよく似たアイソザイムの存在等に起因して、1から複数個のアミノ酸変異を有する変異タンパク質が存在することは知られている。従って、配列番号2に示されるアミノ酸配列において1若しくは複数個のアミノ酸が置換し若しくは欠失し、若しくは該アミノ酸配列に1若しくは複数個のアミノ酸が挿入され若しくは付加されたアミノ酸配列を有し、PLC活性を有するポリペプチド(以下、便宜的に「修飾ポリペプチド」)も本発明の範囲に含まれる。
【0041】
ここで、「複数個」とは、好ましくは1−200個、より好ましくは1−100個、さらにより好ましくは1−50個、最も好ましくは1−20個である。一般的には、部位特異的な変異によってアミノ酸が置換された場合に、元々のポリペプチドが有する活性は保持される程度に置換が可能なアミノ酸の個数は、好ましくは1−10個である。
【0042】
本発明のポリペプチドは、クローニングされた核酸の塩基配列からの推定に基づいて、配列番号2のアミノ酸配列を有するが、その配列を有するポリペプチドのみに限定されるわけではなく、本願明細書に記載した特性を有する限り全ての相同ポリペプチドを含むことが意図される。
【0043】
このような修飾ポリペプチドのアミノ酸配列は、配列番号2に示されるアミノ酸配列と少なくとも20%以上、好ましくは30%以上、より好ましくは50%、さらにより好ましくは70%以上、最も好ましくは90%以上の同一性を有することが好ましい。
【0044】
本発明のポリペプチドについて、NCBIの提供するblast 2 sequence (http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html)
を利用して相同性の確認を行うと、最も似ているマウスPLCδとは約30%程度の同一性であった。したがって、本発明のポリペプチドは新規なポリペプチドであると考えられた。また、特に、本発明のポリペプチドにおいて相同性の高い領域は、配列番号2に示されるアミノ酸配列のうち48−157、173―238、326−471、633−757及び776−874である。
【0045】
一般的に、同様の性質を有するアミノ酸同士の置換(例えば、ある疎水性アミノ酸から別の疎水性アミノ酸への置換、ある親水性アミノ酸から別の親水性アミノ酸への置換、ある酸性アミノ酸から別の酸性アミノ酸への置換、あるいはある塩基性アミノ酸から別の塩基性アミノ酸への置換)を導入した場合、得られる修飾ポリペプチドはもとのポリペプチドと同様の性質を有することが多い。遺伝子組換え技術を使用して、このような所望の変異を有する組換えポリペプチドを作製する手法は当業者に周知であり、このような修飾ポリペプチドも本発明の範囲に含まれる。
【0046】
本願明細書において、同一性のパーセントは、例えば、Altschulら(Nucl.Acids.Res.,25.3389−3402(1997))に記載されているBLASTプログラム、あるいはPearsonら(Proc.Natl.Acad.Sci.USA,2444−2448(1988))に記載されているFASTAを用いて配列情報と比較し決定することが可能である。当該プログラムは、インターネット上でNational
Center for
Biotechnology Information(NCBI)、あるいはDNA Data
Bank of
Japan(DDBJ)のウェブサイトから利用することが可能である。各プログラムによる同一性検索の各種条件(パラメーター)は同サイトに詳しく記載されており、一部の設定を適宜変更することが可能であるが、検索は通常デフォルト値を用いて行う。なお、当業者に用いられる、配列比較の他のプログラムもまた使用可能である。
【0047】
本発明のポリペプチドは、例えば、後述の実施例1に従って、本発明の核酸による配列番号1に記載のDNA配列を大腸菌、酵母、昆虫、または動物細胞に、それぞれの宿主で増幅可能な発現ベクターを用いて導入および発現させることにより、当該ポリペプチドを大量に得ることができる。
【0048】
本発明によって、このポリペプチドのアミノ酸配列およびそれをコードするDNA配列が開示されれば、当該配列またはその一部を利用して、ハイブリダイゼーション、PCR等の核酸増幅反応等の遺伝子工学的手法を用いて、他の生物種から同様の生理活性を有するポリペプチドをコードする遺伝子を容易に単離することができる。このような場合、それらの遺伝子がコードする新規ポリペプチドも本発明の範囲に含まれる。
【0049】
なお、本発明のポリペプチドは、そのアミノ酸配列が上述した通りのものであり、前記酵素活性を有するものであれば、ポリペプチドに糖鎖が結合していてもよい。
【0050】
(3)組み換えベクターと形質転換体
本発明によれば、単離した本発明のポリヌクレオチドを含む組換えベクターが提供される。
【0051】
プラスミド等のベクターに本発明核酸のDNA断片を組込む方法としては、例えば、Sambrook,J.ら,Molecular
Cloning,A Laboratory Manual(3rd edition),Cold Spring
Harbor Laboratory,1.1(2001)に記載の方法などが挙げられる。簡便には、市販のライゲーションキット(例えば、宝酒造製等)を用いることもできる。このようにして得られる組換えベクター(例えば、組換えプラスミド)は、宿主細胞(例えば、大腸菌DH5α、TB1、LE392、XL−392、又はXL−1Blue等)に導入される。
【0052】
プラスミドを宿主細胞に導入する方法としては、Sambrook,J.ら,Molecular
Cloning,A Laboratory Manual(3rd edition),Cold Spring
Harbor Laboratory,16.1(2001)に記載の塩化カルシウム法または塩化カルシウム/塩化ルビジウム法、リン酸カルシウム法、リポフェクション法、エレクトロポレーション法、エレクトロインジェクション法、PEGなどの化学的な処理による方法、遺伝子銃などを用いる方法などが挙げられる。
【0053】
ベクターは、簡単には当業界において入手可能な組換え用ベクター(例えば、プラスミドDNA等)に所望の遺伝子を常法により連結することによって調製することができる。用いられるベクターの具体例としては、大腸菌由来のプラスミドとして、例えば、pFLAG−CMV1、pDONR201、pBluescript、pUC18、pUC19、pBR322等が例示されるが、これらに限定されない。
【0054】
当業者であれば制限末端は発現ベクターに適合するように適宜選択することが可能である。発現ベクターは、「本発明タンパク質を発現させたい宿主細胞」に適したものを当業者であれば適宜選択することができる。このように本発明発現ベクターは上記の本発明核酸が目的の宿主細胞中で発現し得るように遺伝子発現に関与する領域(プロモーター領域、エンハンサー領域、オペレーター領域等)が適切に配列されており、さらに本発明核酸が適切に発現するように構築されていることが好ましい。
【0055】
また、発現ベクターの構築は、制限処理及び連結作業を必要としないGatewayシステム(インビトロジェン社)を用いることもできる。Gatewayシステムとは、PCR産物の方向性を維持したままクローニングができ、また、DNA断片を適切に改変した発現ベクターにサブクローニングを可能にした部位特異的な組換えを利用したシステムである。具体的には、PCR産物とドナーベクターとから部位特異的な組換え酵素であるBPクロナーゼによってエントリークローンを作成し、その後、このクローンと別の組換え酵素であるLBクロナーゼによって組換え可能なデスティネーションベクターにPCR産物を移入することにより、発現系に対応した発現クローンを調製するものである。最初にエントリークローンを作成すれば、制限酵素やリガーゼで作業する手間の係るサブクローニングステップが不要となる点を特徴の一つとする。
【0056】
発現ベクターの種類は、原核細胞および/または真核細胞の各種の宿主細胞中で所望の遺伝子を発現し、所望のタンパク質を生産する機能を有するものであれば特に限定されないが、例えば、哺乳類用発現ベクターとしてpFLAG−CMV1、pcDNA3.1、pGreenLanternなどが好ましく、大腸菌用発現ベクターとしてpQE−30、pQE−60、pMAL−C2、pMAL−p2、pSE420、pET又はpCAL若しくはそれらの人工的修飾物(pQE30、pET又はpCALを適当な制限酵素で処理して得られるDNAフラグメント)などが好ましく、酵母用発現べクターとしてpYES2(サッカロマイセス属)、pPIC3.5K、pPIC9K、pA0815(以上ピキア属)、昆虫用発現ベクターとしてpFastBacp、pBacPAK8/9、pBK283、pVL1392、pBlueBac4.5などが好ましい。
【0057】
上記「本発明発現ベクター」を宿主細胞に組み込み、形質転換体を得ることができる。上記「宿主細胞」として真核細胞(哺乳類細胞、酵母、昆虫細胞等)であっても原核細胞(大腸菌、枯草菌等)であっても使用することができる。本発明の形質転換体を得るための宿主細胞は、特に限定されず、さらに、または、ヒト(例えば、HeLa、293T、SH−SY5Y)、マウス(例えば、Neuro2a、NIH3T3)、サル(例えば、COS−1)由来の培養細胞でもよい。これらはいずれも公知であり、市販されているか(例えば、大日本製薬社)、あるいは公共の研究機関(例えば、理研セルバンク)より入手可能である。あるいは、胚、器官、組織若しくは非ヒト個体も使用可能である。
【0058】
ところで、「本発明のポリヌクレオチド」は、マウスゲノムライブラリーから発見されたポリヌクレオチドであるため、本発明においては真核細胞を本発明の形質転換体の宿主細胞として用いるとより天然物に近い性質を有した「本発明酵素」が得られる(例えば糖鎖が付加された態様など)と考えられる。従って、「宿主細胞」としては真核細胞、特に哺乳類細胞を選択することが好ましい。動物細胞としてはマウス由来、アフリカツメガエル由来、ラット由来、ハムスタ−由来、サル由来またはヒト由来の細胞若しくはそれらの細胞から樹立した培養細胞株などが例示される。
【0059】
宿主細胞として細菌、特に大腸菌を用いる場合、一般に発現べクターは少なくとも、プロモーター/オペレーター領域、開始コドン、所望のタンパク質をコードする遺伝子、終止コドン、ターミネーターおよび複製可能単位から構成される。宿主細胞として酵母、植物細胞、動物細胞または昆虫細胞を用いる場合には、一般に発現べクターは少なくとも、プロモーター、関始コドン、所望のタンパク質をコードする遺伝子、終止コドン、ターミネーターを含んでいることが好ましい。またシグナルペブチドをコードするDNA、エンハンサー配列、所望の遺伝子の5’側および3’側の非翻訳領域、選択マーカー領域または複製可能単位などを適宜含んでいてもよい。
【0060】
本発明のべクタ−において、好適な開始コドンとしては、メチオニンコドン(ATG)が例示される。また、終止コドンとしては、常用の終止コドン(例えば、TAG、TGA、TAAなど)が例示される。
【0061】
複製可能単位とは、宿主細胞中でその全DNA配列を複製することができる能力をもつDNAを意味し、天然のプラスミド、人工的に修飾されたプラスミド(天然のプラスミドから調製されたプラスミド)および合成プラスミド等が含まれる。
【0062】
エンハンサー配列、ターミネーター配列については、例えば、それぞれSV40に由来するもの等、当業者において通常使用されるものを用いることができる。
【0063】
選択マーカーとしては、通常使用されるものを常法により用いることができる。例えばテトラサイクリン、アンピシリン、またはカナマイシンもしくはネオマイシン、ハイグロマイシンまたはスペクチノマイシン等の抗生物質耐性遺伝子などが例示される。
【0064】
発現べクターは、少なくとも、上述のプロモータ−、開始コドン、所望のタンパク質をコードする遺伝子、終止コドン、およびターミネーター領域を連続的かつ環状に適当な複製可能単位に連結することによって調製することができる。またこの際、所望により制限酵素での消化やT4
DNAリガーゼを用いるライゲーション等の常法により適当なDNAフラグメント(例えば、リンカー、他の制限酵素部位など)を用いることができる。
【0065】
本発明の発現べクターの宿主細胞への導入[形質転換(形質移入)]は従来公知の方法を用いて行うことができる。
例えば、細菌(E.coli,Bacillus subtilis s
等)の場合は、例えばCohenらの方法[Proc.Natl.Acad.Sci.USA,69,2110(1972)]、プロトプラスト法[Mol.Gen.Genet.,168,111(1979)]やコンピテント法[J.Mol.Biol.,56,209(1971)]によって、Saccharomyces
cervisiaeの場合は、例えばHinnenらの方法[Proc.Natl.Acad.Sci.USA,75,1927(1978)]やリチウム法[J.B.Bacteriol.,153,163(1983)]によって、植物細胞の場合は、例えばリーフディスク法[Science,227,129(1985)]、エレクトロポレ−ション法[Nature,319,791(1986)]によって、動物細胞の場合は、例えばGrahamの方法[Virology,52,456(1973)]、昆虫細胞の場合は、例えばSummerらの方法[Mol.Cell
Biol.,3,2156−2165(1983)]によってそれぞれ形質転換することができる。
【0066】
なお、組換えベクターの構築及びそれを用いて本発明の核酸を宿主細胞に導入する方法の具体例が下記実施例4に詳述されている。
【0067】
形質転換体において発現したポリペプチドを小胞体の内腔に、細胞周辺腔に、または細胞外の環境に分泌させるために、適当な分泌シグナルを目的のポリペプチドに組み込むことができる。これらのシグナルは目的のポリペプチドに対して内因性であっても、異種シグナルであってもよい。
【0068】
本発明のポリペプチドの回収は、本発明のポリペプチドが培地に分泌される場合は、培地を回収する。本発明のポリペプチドが細胞内に産生される場合は、その細胞をまず溶解し、その後にポリペプチドを回収する。
【0069】
組換え細胞培養物から本発明のポリペプチドを回収し精製するには、硫酸アンモニウムまたはエタノール沈殿、酸抽出、アニオンまたはカチオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティークロマトグラフィー、ヒドロキシルアパタイトクロマトグラフィーおよびレクチンクロマトグラフィーを含めた公知の方法を用いることができる。
【0070】
(4)抗体
本発明は、本発明のポリペプチドに結合する抗体を提供する。ここで「抗体」には、ポリクローナルおよびモノクローナル抗体、キメラ抗体、一本鎖抗体、ヒト化抗体、さらにFabまたは他の免疫グロブリン発現ライブラリーの産物を含むFabフラグメントが含まれる。
【0071】
本発明のポリペプチドまたはその断片もしくは類似体、またはそれらを発現する細胞は、本発明のポリペプチドに結合する抗体を産生するための免疫原としても使用することができる。抗体は、好ましくは、本発明のポリペプチドに免疫特異的である。「免疫特異的」とは、その抗体が他のポリペプチドに対するその親和性よりも本発明のポリペプチドに対して実質的に高い親和性を有することを意味する。
【0072】
本発明のポリペプチドに結合する抗体は、当業者に公知の方法により調製することが可能である。ポリクローナル抗体であれば、例えば、次のようにして得ることができる。本発明のポリペプチドあるいはそのGSTとの融合タンパク質をウサギ等の小動物に免疫し血清を得る。これを、例えば、硫安沈殿、プロテインA、プロテインGカラム、DEAEイオン交換クロマトグラフィー、本発明のポリペプチドをカップリングしたアフィニティーカラム等により精製することにより調製する。具体例については実施例7に詳述されている。また、モノクローナル抗体であれば、例えば、本発明のポリペプチドをマウスなどの小動物に免疫を行い、同マウスより脾臓を摘出し、これをすりつぶして細胞を分離し、マウスミエローマ細胞とポリエチレングリコールなどの試薬により融合させ、これによりできた融合細胞(ハイブリドーマ)の中から、本発明のポリペプチドに結合する抗体を産生するクローンを選択する。次いで、得られたハイブリドーマをマウス腹腔内に移植し、同マウスより腹水を回収し、得られたモノクローナル抗体を、例えば、硫安沈殿、プロテインA、プロテインGカラム、DEAEイオン交換クロマトグラフィー、本発明のポリペプチドをカップリングしたアフィニティーカラム等により精製することで、調製することが可能である。
【0073】
本発明の抗体は、本発明のポリペプチドやこれを発現する細胞の単離、同定、および精製に利用することができる。
【0074】
(5)医薬組成物
本発明の「医薬組成物 」とは、前記で定義される本発明の「ポリペプチド」、「ポリヌクレオチド」または「抗体」のいずれかと、薬学的に許容され得る担体とからなる医薬組成物
である。
【0075】
ここで「薬学的に許容され得る担体」とは、賦形剤、希釈剤、増量剤、崩壊剤、安定剤、保存剤、緩衝剤、乳化剤、芳香剤、着色剤、甘味剤、粘稠剤、矯味剤、溶解補助剤あるいはその他の添加剤等が挙げられる。そのような担体の一つ以上を用いることにより、錠剤、丸剤、散剤、顆粒剤、注射剤、液剤、カプセル剤、トロー剤、エリキシル剤、懸濁剤、乳剤あるいはシロップ剤等の形態の医薬
組成物 を調製することができる。これらの医薬
組成物 は、経口あるいは非経口的に投与することができる。非経口投与のためのその他の形態としては、一つまたはそれ以上の活性物質を含み、常法により処方される外用液剤、腸溶内投与のための坐剤およびペッサリーなどが含まれる。
【0076】
投与量は、患者の年齢、性別、体重及び症状、治療効果、投与方法、処理時間、あるいは該医薬
組成物 に含有される活性成分(前記ポリペプチドや抗体など)の種類などにより異なるが、通常成人一人当たり、一回につき10μgから1000mg(あるいは10μgから500mg)の範囲で投与することができる。しかしながら、投与量は種々の条件により変動するため、上記投与量より少ない量で十分な場合もあり、また上記の範囲を越える投与量が必要な場合もある。
【0077】
とりわけ注射剤の場合には、例えば生理食塩水あるいは市販の注射用蒸留水等の非毒性の薬学的に許容され得る担体中に0.1μg抗体/ml担体〜10mg抗体/ml担体の濃度となるように溶解または懸濁することにより製造することができる。このようにして製造された注射剤は、処置を必要とするヒト患者に対し、1回の投与において1kg体重あたり、1μg〜100mgの割合で、好ましくは50μg〜50mgの割合で、1日あたり1回〜数回投与することができる。投与の形態としては、静脈内注射、皮下注射、皮内注射、筋肉内注射あるいは腹腔内注射のような医療上適当な投与形態が例示できる。好ましくは静脈内注射である。
【0078】
また、注射剤は、場合により、非水性の希釈剤(例えばプロピレングリコール、ポリエチレングリコール、オリーブ油のような植物油、エタノールのようなアルコール類など)、懸濁剤あるいは乳濁剤として調製することもできる。
【0079】
そのような注射剤の無菌化は、バクテリア保留フィルターを通す濾過滅菌、殺菌剤の配合または照射により行うことができる。注射剤は、用時調製の形態として製造することができる。即ち、凍結乾燥法などによって無菌の固体組成物
とし、使用前に無菌の注射用蒸留水または他の溶媒に溶解して使用することができる。
【0080】
本発明の医薬組成物は、脳疾患、特に神経再生を必要とする疾患の治療及び予防に適用が可能である。ここで、脳疾患とは、脳腫瘍、脳出血、脳梗塞、クモ膜下出血、脳動脈瘤、脳静脈瘤、脳血管の奇形や増殖、先天性奇形、水頭症、脳性マヒ、パーキンソン、ウィルス感染や予防ワクチン注射後の脳脊髄炎や脳炎などを意味する。また本願明細書において神経再生医療とは、神経再生を必要とする疾患の治療及び予防などを意味する。
【0081】
また、本発明の医薬組成物の種々疾患症状の治療効果については、常法に従って、既知の疾患モデル動物に投与することにより試験、検討することができる。
【0082】
(7)スクリーニング方法
本発明は、本発明のポリペプチドに結合する化合物のスクリーニング方法を提供する。この方法は、本発明のポリペプチドとこれに結合する化合物を含むと予想される被検試料とを接触せしめ、該ポリペプチドと被検試料との結合活性を検出し、そして本発明のポリペプチドに結合する活性を有する化合物を選択する、ことを含む。
【0083】
スクリーニングに用いられる本発明のポリペプチドは組換えポリペプチドであっても、天然由来のポリペプチドであってもよい。また部分ペプチドであってもよい。また細胞表面に発現させた形態、または膜画分としての形態であってもよい。被検試料としては特に制限はなく、例えば、細胞抽出物、細胞培養上清、発酵微生物産生物、海洋生物抽出物、植物抽出物、精製若しくは粗精製ポリペプチド、非ペプチド性化合物、合成低分子化合物、天然化合物が挙げられる。被検試料を接触させる本発明のポリペプチドは、例えば、精製したポリペプチドとして、可溶型ポリペプチドとして、担体に結合させた形態として、他のポリペプチドとの融合ポリペプチドとして、細胞膜上に発現させた形態として、膜画分として被検試料に接触させることができる。
【0084】
本発明のポリペプチドを用いて、これに結合するポリペプチドをスクリーニングする方法としては、当業者に公知の多くの方法を用いることが可能である。このようなスクリーニングは、例えば、免疫沈降法により行うことができる。具体的には、以下のように行うことができる。本発明のポリペプチドをコードする遺伝子を、pSV2neo,
pcDNA I,
pCD8 などの外来遺伝子発現用のベクターに挿入することで動物細胞などで当該遺伝子を発現させる。発現に用いるプロモーターとしては
SV40 early
promoter (Rigby In Williamson (ed.),
Genetic Engineering,
Vol.3. Academic
Press, London,
p.83−141(1982)), EF−1 α promoter (Kimら
Gene 91, p.217−223
(1990)), CAG promoter (Niwa et
al. Gene
108, p.193−200 (1991)), RSV
LTR promoter
(Cullen Methods
in Enzymology
152, p.684−704 (1987), SR α promoter (Takebe et
al. Mol.
Cell. Biol.
8,
p.466 (1988)),
CMV immediate
early promoter
(Seed and
Aruffo Proc.
Natl. Acad.
Sci. USA
84, p.3365−3369 (1987)), SV40
late promoter
(Gheysen and Fiers J. Mol.
Appl. Genet.
1,
p.385−394 (1982)), Adenovirus late promoter
(Kaufman et al. Mol. Cell.
Biol. 9, p. 946
(1989)), HSV TK promoter 等の一般的に使用できるプロモーターであれば何を用いてもよい。
【0085】
動物細胞に遺伝子を導入することで外来遺伝子を発現させるためには、エレクトロポレーション法
(Chu, G.
et al.
Nucl. Acid
Res. 15, 1311−1326
(1987))、リン酸カルシウム法 (Chen, C
and Okayama,
H. Mol.
Cell. Biol.
7,
2745−2752 (1987))、DEAEデキストラン法 (Lopata,
M. A.
et al.
Nucl. Acids
Res. 12, 5707−5717
(1984); Sussman,
D. J.
and Milman,
G. Mol.
Cell. Biol.
4,
1642−1643 (1985))、リポフェクチン法 (Derijard,
B. Cell
7,
1025−1037 (1994); Lamb, B. T.
et al.
Nature Genetics
5,
22−30 (1993);
Rabindran, S. K. et al.
Science 259, 230−234 (1993))等の方法があるが、いずれの方法によってもよい。
【0086】
特異性の明らかとなっているモノクローナル抗体の認識部位(エピトープ)を本発明のポリペプチドのN末またはC末に導入することにより、モノクローナル抗体の認識部位を有する融合ポリペプチドとして本発明のポリペプチドを発現させることができる。用いるエピトープ−抗体系としては市販されているものを利用することができる(実験医学
13, 85−90 (1995))。マルチクローニングサイトを介して、β−ガラクトシダーゼ、マルトース結合蛋白質、グルタチオンS−トランスフェラーゼ、緑色蛍光蛋白質(GFP)などとの融合ポリペプチドを発現することができるベクターが市販されている。
【0087】
融合ポリペプチドにすることにより本発明のポリペプチドの性質をできるだけ変化させないようにするために数個から十数個のアミノ酸からなる小さなエピトープ部分のみを導入して、融合ポリペプチドを調製する方法も報告されている。例えば、ポリヒスチジン(His−tag)、インフルエンザ凝集素
HA、ヒトc−myc、FLAG、Vesicular stomatitis ウイルス糖蛋白質(VSV−GP)、T7
gene10 蛋白質(T7−tag)、ヒト単純ヘルペスウイルス糖蛋白質(HSV−tag)、E−tag(モノクローナルファージ上のエピトープ)などのエピトープとそれを認識するモノクローナル抗体を、本発明のポリペプチドに結合するポリペプチドのスクリーニングのためのエピトープ−抗体系として利用できる(実験医学
13, 85−90 (1995))。
【0088】
免疫沈降においては、これらの抗体を、適当な界面活性剤を利用して調製した細胞溶解液に添加することにより免疫複合体を形成させる。この免疫複合体は本発明のポリペプチド、それと結合能を有するポリペプチド、および抗体からなる。上記エピトープに対する抗体を用いる以外に、本発明のポリペプチドに対する抗体を利用して免疫沈降を行うことも可能である。本発明のポリペプチドに対する抗体は、例えば、本発明のポリペプチドをコードする遺伝子を適当な大腸菌発現ベクターに導入して大腸菌内で発現させ、発現させたポリペプチドを精製し、これをウサギやマウス、ラット、ヤギ、ニワトリなどに免疫することで調製することができる。また、合成した本発明のポリペプチドの部分ペプチドを上記の動物に免疫することによって調製することもできる。
【0089】
免疫複合体は、例えば、抗体がマウスIgG 抗体であれば、Protein A SepharoseやProtein G Sepharoseを用いて沈降させることができる。また、本発明のポリペプチドを、例えば、GSTなどのエピトープとの融合ポリペプチドとして調製した場合には、グルタチオン−Sepharose
4Bなどのこれらエピトープに特異的に結合する物質を利用して、本発明のポリペプチドの抗体を利用した場合と同様に、免疫複合体を形成させることができる。
【0090】
免疫沈降の一般的な方法については、例えば、文献(Harlow,E.
and Lane,
D.: Antibodies,
pp.511−552, Cold Spring Harbor Laboratory
publications, New York (1988) )記載の方法に従って、または準じて行えばよい。
【0091】
免疫沈降されたポリペプチドの解析にはSDS−PAGEが一般的であり、適当な濃度のゲルを用いることでポリペプチドの分子量により結合していたポリペプチドを解析することができる。また、この際、一般的には本発明のポリペプチドに結合したポリペプチドは、クマシー染色や銀染色といったポリペプチドの通常の染色法では検出することは困難であるので、放射性同位元素である35S−メチオニンや35S−システインを含んだ培養液で細胞を培養し、該細胞内のポリペプチドを標識して、これを検出することで検出感度を向上させることができる。ポリペプチドの分子量が判明すれば直接SDS−ポリアクリルアミドゲルから目的のポリペプチドを精製し、その配列を決定することもできる。
【0092】
また、本発明のポリペプチドを用いて、該ポリペプチドに結合するポリペプチドを単離する方法としては、例えば、ウエストウエスタンブロッティング法(Skolnik,
E. Y.
et al.,Cell
(1991) 65,
83−90)を用いて行うことができる。すなわち、本発明のポリペプチドと結合するポリペプチドを発現していることが予想される細胞、組織、臓器(例えば、精巣)よりファージベクター(λgt11, ZAPなど)を用いたcDNAライブラリーを作製し、これをLB−アガロース上で発現させフィルターに発現させたポリペプチドを固定し、精製して標識した本発明のポリペプチドと上記フィルターとを反応させ、本発明のポリペプチドと結合したポリペプチドを発現するプラークを標識により検出すればよい。本発明のポリペプチドを標識する方法としては、ビオチンとアビジンの結合性を利用する方法、本発明のポリペプチド又は本発明のポリペプチドに融合したポリペプチド(例えばGSTなど)に特異的に結合する抗体を利用する方法、ラジオアイソトープを利用する方法又は蛍光を利用する方法等が挙げられる。
【0093】
また、本発明のスクリーニング方法の他の態様としては、細胞を用いた2−ハイブリッドシステム(Fields,
S., and
Sternglanz, R.,Trends. Genet. (1994) 10,
286−292、Dalton S, and Treisman R
(1992) Characterization
of SAP−1,
a protein
recruited by serum response factor
to the
c−fos serum
response element. Cell 68, 597−612、「MATCHMARKER
Two−Hybrid System」,「Mammalian MATCHMAKER
Two−Hybrid Assay Kit」,「MATCHMAKER
One−Hybrid System」(いずれもクロンテック社製)、「HybriZAP Two−Hybrid
Vector System」(ストラタジーン社製))を用いて行う方法が挙げられる。
【0094】
2−ハイブリッドシステムにおいては、本発明のポリペプチドまたはその部分ペプチドをSRF
DNA結合領域またはGAL4 DNA結合領域と融合させて酵母細胞の中で発現させ、本発明のポリペプチドと結合するポリペプチドを発現していることが予想される細胞より、VP16またはGAL4転写活性化領域と融合する形で発現するようなcDNAライブラリーを作製し、これを上記酵母細胞に導入し、検出された陽性クローンからライブラリー由来cDNAを単離する(酵母細胞内で本発明のポリペプチドと結合するポリペプチドが発現すると、両者の結合によりレポーター遺伝子が活性化され、陽性のクローンが確認できる)。単離したcDNAを大腸菌に導入して発現させることにより、該cDNAがコードするポリペプチドを得ることができる。これにより本発明のポリペプチドに結合するポリペプチドまたはその遺伝子を調製することが可能である。
【0095】
2−ハイブリッドシステムにおいて用いられるレポーター遺伝子としては、例えば、HIS3遺伝子の他、Ade2遺伝子、LacZ遺伝子、CAT遺伝子、ルシフェラーゼ遺伝子、PAI−1(Plasminogen
activator inhibitor type1)遺伝子等が挙げられるが、これらに制限されない。2ハイブリッド法によるスクリーニングは、酵母の他、哺乳動物細胞などを使って行うこともできる。
【0096】
本発明のポリペプチドと結合する化合物のスクリーニングは、アフィニティクロマトグラフィーを用いて行うこともできる。例えば、本発明のポリペプチドをアフィニティーカラムの担体に固定し、ここに本発明のポリペプチドと結合するポリペプチドを発現していることが予想される被検試料を適用する。この場合の被検試料としては、例えば細胞抽出物、細胞溶解物等が挙げられる。被検試料を適用した後、カラムを洗浄し、本発明のポリペプチドに結合したポリペプチドを調製することができる。
【0097】
得られたポリペプチドは、そのアミノ酸配列を分析し、それを基にオリゴDNAを合成し、該DNAをプローブとしてcDNAライブラリーをスクリーニングすることにより、該ポリペプチドをコードするDNAを得ることができる。
【0098】
また、ポリペプチドに限らず、本発明のポリペプチドに結合する化合物(アゴニストおよびアンタゴニストを含む)を単離する方法としては、例えば、固定した本発明のポリペプチドに、合成化合物、天然物バンク、もしくはランダムファージペプチドディスプレイライブラリーを作用させ、本発明のポリペプチドに結合する分子をスクリーニングする方法や、コンビナトリアルケミストリー技術によるハイスループットを用いたスクリーニング方法(Wrighton
NC; Farrell
FX; Chang
R; Kashyap
AK; Barbone
FP; Mulcahy
LS;Johnson DL; Barrett RW; Jolliffe
LK; Dower
WJ., Small
peptides as potent mimetics of
the protein
hormone erythropoietin,
Science (UNITED
STATES) Jul
26 1996,
273 p458−64、Verdine
GL., The
combinatorial chemistry of nature. Nature
(ENGLAND) Nov 7 1996, 384
p11−13、Hogan JC Jr.,Directed combinatorial chemistry.
Nature (ENGLAND)
Nov 7
1996, 384
p17−9)が当業者に公知である。
【0099】
本発明において、結合した化合物を検出又は測定する手段として表面プラズモン共鳴現象を利用したバイオセンサーを使用することもできる。表面プラズモン共鳴現象を利用したバイオセンサーは、本発明のポリペプチドと被検化合物との間の相互作用を微量のポリペプチドを用いてかつ標識することなく、表面プラズモン共鳴シグナルとしてリアルタイムに観察することが可能である(例えばBIAcore、Pharmacia製)。したがって、BIAcore等のバイオセンサーを用いることにより本発明のポリペプチドと被検化合物との結合を評価することが可能である。
【0100】
また、本発明は、細胞における本発明のポリヌクレオチド及びポリペプチドの発現を制御する化合物のスクリーニング方法を提供する。
【0101】
細胞における本発明のポリヌクレオチドの発現は、当業者に公知の方法で検出することができる。このような方法としては、例えば、ノーザンブロッティング法やRT−PCR法などを例示することができるがこれらに制限されない。ノーザンブロッティング法においては、検査対象となる細胞のRNAを精製し、アガロースゲル中にて電気泳動し、メンブレンにブロットしたものに、本発明の遺伝子を放射能などで標識したプローブをハイブリダイズさせ、特異的に出現するバンドの有無、濃淡で本発明のポリヌクレオチドの発現を測定する。また、RT−PCR法では、検査対象となる細胞のRNAを精製し、逆転写酵素によりcDNAとし、これを鋳型として、本発明のポリヌクレオチドに特徴的な配列をプライマーとして、本発明の遺伝子の転写産物由来のcDNAをPCRにより増幅する。これにより増幅されたcDNAの量は鋳型となったcDNAの量、ひいては本発明のポリヌクレオチドの転写産物の量に比例すると考えられるので、このPCRにより増幅されたDNA断片の量を電気泳動法などを用いて測定することで、本発明のポリヌクレオチドの発現量を測定できる。
【0102】
細胞における本発明のポリペプチドの発現は、当業者に公知の方法で検出することができる。例えば、本発明の抗体を用い、抗原抗体反応を行わせることにより、本発明のポリペプチドまたは該ポリペプチドを含む細胞を免疫学的に検出することができる。該検出方法は、本発明のポリペプチドをコードする遺伝子(ゲノムDNA等)の変異または発現異常が原因となり得る疾患の診断にも利用することができる。また、本発明のポリペプチドの定量方法としては、免疫学的に検出する方法として、マイクロタイタープレートを用いるELISA法・蛍光抗体法、ウェスタンブロット法、免疫組織染色法等があげられる。また、液相中で本発明のポリペプチドと反応する抗体のうちエピトープが異なる2種類のモノクローナル抗体を用いたサンドイッチELISA法、放射性ヨード等の放射性同位体で標識した本発明のポリペプチドと本発明のポリペプチドを認識する抗体とを用いるラジオイムノアッセイ法等があげられる。
【0103】
また、本発明は、本発明のポリヌクレオチドの転写を制御する領域の下流にレポーター遺伝子の連結されたDNAを含むプラスミドで形質転換された形質転換体と被験化合物とを接触させ、本発明のポリヌクレオチドの発現を制御する化合物を選択することを特徴とする、本発明のポリヌクレオチドの発現を制御する化合物のスクリーニング方法を提供する。
【0104】
本発明のスクリーニング方法の別の態様においては、まず、本発明のポリヌクレオチドのプロモータ―領域の下流にレポーター遺伝子が機能的に結合したDNAを有する細胞または細胞抽出液を提供する。
【0105】
ここで、「機能的に結合した」とは、本発明のポリヌクレオチドのプロモータ―領域に転写因子が結合することにより、レポーター遺伝子の発現が誘導されるように、本発明のポリヌクレオチドのプロモータ―領域とレポーター遺伝子とが結合していることをいう。従って、レポーター遺伝子が他の遺伝子と結合しており、他の遺伝子産物との融合タンパク質を形成する場合であっても、本発明のポリヌクレオチドのプロモータ―領域に転写因子が結合することによって、該融合タンパク質の発現が誘導されるものであれば、上記「機能的に結合した」の意に含まれる。
【0106】
上記レポーター遺伝子としては、その発現が検出可能なものであれば特に制限されず、例えば、当業者において一般的に使用されるCAT遺伝子、lacZ遺伝子、ルシフェラーゼ遺伝子、β−グルクロニダーゼ遺伝子(GUS)およびGFP遺伝子等を挙げることができる。また、上記レポーター遺伝子には、本発明のポリヌクレオチドをコードするDNAもまた含まれる。
【0107】
本発明の評価方法の別の態様では、次いで、上記細胞または上記細胞抽出液に被験試料を接触させる。次いで、該細胞または該細胞抽出液における上記レポーター遺伝子の発現レベルを測定する。
【0108】
レポーター遺伝子の発現レベルは、使用するレポーター遺伝子の種類に応じて、当業者に公知の方法により測定することができる。例えば、レポーター遺伝子がCAT遺伝子である場合には、該遺伝子産物によるクロラムフェニコールのアセチル化を検出することによって、レポーター遺伝子の発現レベルを測定することができる。レポーター遺伝子がlacZ遺伝子である場合には、該遺伝子発現産物の触媒作用による色素化合物の発色を検出することにより、また、ルシフェラーゼ遺伝子である場合には、該遺伝子発現産物の触媒作用による蛍光化合物の蛍光を検出することにより、また、β−グルクロニダーゼ遺伝子(GUS)である場合には、該遺伝子発現産物の触媒作用によるGlucuron(ICN社)の発光や5−ブロモ−4−クロロ−3−インドリル−β−グルクロニド(X−Gluc)の発色を検出することにより、さらに、GFP遺伝子である場合には、GFPタンパク質による蛍光を検出することにより、レポーター遺伝子の発現レベルを測定することができる。
【0109】
また、本発明のポリヌクレオチドをレポーターとする場合、該遺伝子の発現レベルの測定は、当業者に公知の方法によって行うことができる。例えば、該遺伝子のmRNAを定法に従って抽出し、このmRNAを鋳型としたノーザンハイブリダイゼーション法、またはRT−PCR法を実施することによって該遺伝子の転写レベルの測定を行うことができる。さらに、DNAアレイ技術を用いて、該遺伝子の発現レベルを測定することも可能である。
【0110】
また、該遺伝子からコードされる本発明のポリヌクレオチドを含む画分を定法に従って回収し、該ポリヌクレオチドの発現をSDS−PAGE等の電気泳動法で検出することにより、遺伝子の翻訳レベルの測定を行うこともできる。また、本発明のポリペプチドに対する抗体を用いて、ウェスタンブロッティング法などを実施し、該ポリペプチドの発現を検出することにより、遺伝子の翻訳レベルの測定を行うことも可能である。
【0111】
本発明のスクリーニングにより単離しうる化合物は、本発明の蛋白質の活性を調節するための薬剤の候補となり、本発明の蛋白質の発現異常や機能異常などに起因する疾患や本発明の蛋白質の活性を制御することにより治療可能な疾患の治療への応用が考えられる。治療や予防の対象となる疾患としては、例えば、脳疾患、特に神経再生を必要とする疾患が考えられる。本発明のスクリーニング方法を用いて単離しうる化合物の構造の一部を、付加、欠失及び/又は置換により変換される物質も、本発明の蛋白質に結合する化合物に含まれる。
以下、本発明を実施例によりさらに詳細に説明するが、本発明はこれら実施例に制限されるものではない。
【実施例1】
【0112】
マウスゲノムデータベースによる新規PLC様タンパク質の検索:
セレラ社(MD, USA) のデータベースを検索した。その結果、マウスの4番染色体にPLCと思われるドメイン構造
(PH, EF-hand, PLCX, PLCY, C2) を持つ予想アミノ酸配列を見いだすことが出来た。この予想タンパク質mCP21687は約1600アミノ酸から構成されていたが、開始コドンが含まれておらず、完全長のものでは無いと考えられた。そこで、Genbankより類似の配列を検索したところ、RIKEN
cDNAクローンA930027K05がmCP21687をコードしている遺伝子である事が分かった。A930027K05クローンは開始メチオニンを含むタンパク質をコードしていたが、予想転写産物にはPLCYドメインとC2ドメインは含まれていなかった。これはクローニングの際に起こったフレーム シフト変異又はシークエンシングミスと思われたため、真偽を確かめるためにA930027K05クローンの配列を基にマウス脳cDNAよりPCR反応を 行ったところ、A930027K05クローンには2箇所に1塩基の挿入がある事が判明、さらにはC2ドメインをコードする配列の約600塩基後にストッ プコドンが存在し、C末端側のアミノ酸配列は当初の予想よりも400アミノ酸短い事が判明した。以上の事より、この遺伝子の転写産物は全長1164アミノ酸(配列番号:2)からなるタンパク質である事が分かった。
【実施例2】
【0113】
新規PLC様タンパク質のcDNAクローニング:
マウス脳よりRNeasy miniキット (QIAGEN, Hilden, Germany) を用いてマウス脳 total RNAを精製した。ついで、SuperScript III First-Strand
Synthesis System (Invitrogen, CA, USA) を用いて逆転写反応を行い、マウス脳cDNAを得た。得られたcDNAをテンプレートとして、以下に示すオリゴヌクレオチドプライマーと反応条件にてKOD plus polymerase (TOYOBO, Tokyo, Japan)を用いてPCR反応を行った。プライマー配列中の赤字はそれぞれ開始コドンと終止コドンの位置を示しており、下線部はクローニング用に付加した制限酵素サイト(Bgl IIサイトおよびSal Iサイト)を示している。
N末端側プライマー5' agatctatgcctggtccccagccgtc 3'(配列番号:3に示した。)
C末端側プライマー5' gtcgacgtctcaatccctaacggtgg 3'(配列番号:4に示した。)
PCRのサイクルは94℃ 2分、[94℃ 20秒、55℃ 20秒、68℃ 4分]を35回繰り返し、68℃ 5分の条件で行った。得られた約3500 bpのPCR産物をpBluescript
II SK (+) のEco RVサイトにクローニングして、ABI
PRISM 3100 Genetic Analyzer (Applied Biosystems, CA, USA) を用いて全長のシークエンスを確認した。全長配列は配列番号:1に示したとおりである。
【実施例3】
【0114】
既知PLCとの比較:
ドメイン領域の割り出しにはSMART (http://smart.embl-heidelberg.de/) を利用した。また、既知のPLCとのドメイン構造の比較には、NCBIの提供するblast 2 sequence (http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html)を利用した。その結果を図1に示す。
【0115】
このPLCの構造上の特徴としてはPLCXドメインとPLCYドメインの間の配列が他のPLCよりも長いことと、C末端に200アミノ酸ほどの長い付加的な配列を持つことが挙げられる。また、各ドメインごとの類似性を比較したところ、PLCδタイプに最も類似性が高いことが示された(図1B)。さらに、インターネット上に公開されているプログラム CLUSTALW (http://clustalw.genome.jp/) を用いて系統樹を作成したところ、新規PLC様タンパク質はPLCδタイプに近いという結果を示した(図1C) 。ドメイン間の比較と系統樹から、新規PLC様タンパク質はPLCδに近い事が示されたが、δタイプのPLCが進化的に高く保存されているのに対し、新規PLCは線虫においてホモログが見あたらないことや、PLCδファミリーは構造が比較的単純であり、約750アミノ酸からなるタンパク質であるのに対し、新規PLCは1164アミノ酸から構成される事などから、この新規PLC様タンパク質が新しいサブタイプの物であると考えられた。
【実施例4】
【0116】
新規PLC様タンパク質遺伝子の発現ベクターへの組み込みおよび形質転換体の作成:
新規PLC様タンパク質の全長cDNAを、Sf-9/バキュロウィルス発現用pFastBac-GSTベクター、哺乳類発現用ベクターpCDNA3.1
(Invitrogen, CA, USA) にサブクローニングし、in vitroや細胞を用いた実験に使用した。また、一部のトランケート変異体を用いた実験では遺伝子をpFlag-CMV2ベクター (SIGMA,MO,USA)にサブクローニングして実験に用いた。
【実施例5】
【0117】
PLC活性の測定:
新規PLC様タンパク質が実際にPLCとしての活性、つまりはPI (4,5) P2を加水分解してIP3とDAGを産生する酵素活性を有するのかどうかを検討した。まず、マウス脳を摘出し、その抽出液より得られた免疫沈降物の酵素活性を測定することで、内在性の新規PLC様タンパク質がPLC活性を持つかどうかを検討した。測定は[3H]-PI (4,5) P2を基質として行い、反応後に得られた[3H]-IP3を分離し、その放射線の強度を液体シンチレーションカウンターで計測することで数値化した。具体的には以下の方法により行った。
【0118】
酵素の調製1(内在性タンパク質の調製)
マウス脳を20
mM Hepes-KOH pH 7.0, 120 mM KCl , protease inhibitor cocktail EDTA free, 0.1%
sodium deoxycholateを含むバッファーに懸濁し、超音波処理を行った。次いで15000 rpmにて10分間遠心分離を行い、その上清を42000 rpmにて30分間遠心した。得られた上清に新規PLC抗体とprotein A-sepharose (Amersham Bioscience, NJ, USA) を加えて4℃にて2時間ゆっくりと撹拌しながら反応させ、免疫沈降を行った。sepharoseビーズを20 mM Hepes-KOH pH 7.0, 120 mM
KCl , protease inhibitor cocktail EDTA free, 0.1% sodium deoxycholateを含むバッファーで5回洗浄したものを活性測定に用いた。
【0119】
酵素の調製2(バキュロウィルス発現系タンパクの調整)
Sf-9/バキュロウィルス発現系にて、GST-PLC融合タンパクを発現させた。回収した細胞を 20 mM Hepes-KOH
pH 7.0, 120 mM KCl , protease inhibitor cocktail EDTA free, 0.1% sodium
deoxycholateを含むバッファーに懸濁し、超音波処理を行った。次いで15000 rpmにて10分間遠心分離を行い、その上清にglutathione sepharose 4B
(Amersham Bioscience, NJ, USA)を加えて4℃にて1時間ゆっくりと撹拌しながら反応させた。sepharoseビーズを20 mM Hepes-KOH pH 7.0, 120 mM KCl , protease inhibitor cocktail
EDTA free, 0.1% sodium deoxycholateを含むバッファーで3回洗浄したものを活性測定に用いた。精製したタンパク量はSDS-PAGEにてBSAスタンダードと共に泳動し、CBB染色を行うことで定量した。
【0120】
基質の調製
PI(4,5)P2
(SIGMA, MO, USA) 、 PE (Doosan Serdary Research Lab.,
Kyungki Korea) 、[3H]-PI(4,5)P2 (PerkinElmer, MA, USA) を混合し、窒素ガスで完全に乾燥させた。これに滅菌水を加え、超音波処理を行って均質化したものを反応用の基質溶液とした。[最終濃度 : 200 μg/ml
PI(4,5)P2 , 100μg/ml PE, 2x106 DPM/ml
[3H]-PI(4,5)P2 ]
【0121】
PLC酵素反応
5 x PLC assay buffer(250
mM Hepes-KOH pH 7.0, 380 mM KCl, 2.5 mg/ml BSA) 10 μl、5
x Ca2+ EGTA溶液(CaCl2溶液と100 mM EGTA溶液、滅菌水を混合して調製)10 μl、酵素溶液 20 μl、反応基質溶液 10 μlを混ぜ合わせ、37℃にて5分間ゆっくりと振盪しながら反応させた。反応はクロロホルム : メタノール=2 : 1溶液 1 mlを加えることで停止させた。この溶液に1N HClを250 μl加えて14,000
rpmにて2分間遠心分離を行うことで、PLC活性によって生成したIP3を反応液上層に単離した。得られた反応液の上層450 μlに対してクリアゾルII (nacalai tesque, Kyoto, Japan) を2
ml加えて良く混ぜ合わせた後、液体シンチレーションカウンター (Aloka, Tokyo, Japan) にてトリチウムの壊変数を測定した。なお、特にカルシウム濃度について明記していない実験では、カルシウム濃度は10 μMで行った。
【0122】
上記方法に従って、新規PLC様タンパク質のPLC活性を測定した。ポジティブコントロールには脳において多く発現が見られ、かつ非常に強いPLC活性を持つことが知られているPLCδ1とPLCδ4を用いた。ネガティブコントロールにはNormal rabbit IgG (Santa Cruz, CA, USA) を用いた。測定の結果、PLCδ1の8,500 dpmのカウントに対して、新規PLCの場合は5,500 dpmという若干低い値ではあったが、新規PLC様タンパク質にはPLCとしての活性がある事が分かった (図5)。
【実施例6】
【0123】
新規PLC様タンパク質のカルシウム感受性、pH感受性、及び変異体の酵素活性の測定:
新規PLCがカルシウムによって制御を受けるのか否かを検討した。sf-9/バキュロウィルス発現系にてN末端にGSTタグを融合させた新規PLCとPLCδ1を発現させ、精製してタンパク量を定量した。これを用いてPLC活性を測定し、1分間で1mgのPLCが生成したIP3量
(nmol/min/mgPLC) を算出した。その結果、カルシウムに対して非常に強い感受性を持つことが知られているPLCδ1が10 μMのカルシウム濃度で最大の活性を示したのに対し、新規PLCは1 μMという、より生体内に近いカルシウム濃度で最大の活性を示すことが判明した (図6A) 。さらに、新規PLCの活性がpHに依存するかどうかを検討した。反応溶液のpHが6.5、7.0、7.4、8.0の4点で同様の活性測定を行った所、新規PLCは既知のPLCと同じくpH 7.0付近で最大の活性を示すことが分かった (図6B) 。さらに、活性ドメインに変異を導入した変異体による活性測定を行った。PLCファミリーのXドメインには非常に高く保存されているHis残基があり、このHis残基に変異を導入することでPLC活性が消失することが分かっている (Ellis et al., 1998) 。そこで、新規PLCにおけるこのHis残基である341番目のHis残基をAla残基に置換した変異体
(H341A) を作製して活性測定を行ったところ、PLC活性は完全に消失した (図6C) 。以上のことより、今回単離した新規タンパク質がPLCとしての活性を持っている、すなわち新規PLCである事が示された。
【実施例7】
【0124】
新規PLC様タンパク質に対するポリクローナル抗体の作成:
新規PLCのXドメインとYドメインの間の配列(配列番号:5)をpQE30ベクター (QIAGEN, Hilden, Germany) にクローニングし、大腸菌にて6 x Hisタグ融合タンパク質を発現、変性条件下にてNi-NTA Agarose ビーズ (QIAGEN, Hilden, Germany) を用いて精製した。作成したペプチドのアミノ酸配列を配列番号:6に示した。これをウサギ (日本白色種) に2週間おきに300〜400μgずつ免疫し、十分な抗体価が得られたところで全採血を行った。得られた血清より、以下のように抗体を精製した。まず、抗原部位と同じ遺伝子配列をpGEX-6p2ベクター (Amersham Bioscience, NJ, USA) にクローニングし、大腸菌にてGST融合タンパク質を発現させ、非変性条件下にてglutathione
sepharose 4B(Amersham Bioscience, NJ, USA)を用いて精製した。精製したGST融合抗原タンパク質をマニュアルに従いHiTrap NHS-activated HPカラム (Amersham
Bioscience, NJ, USA) に吸着させて抗原カラムを作製し、アフィニティー精製により精製抗体を得た。
【実施例8】
【0125】
新規PLC様タンパク質の組織分布と発現時期の検討:
〈動物組織抽出液の調製〉
マウスより臓器を摘出後、1 x PBS, 1% Nonitet P-40, 0.1% SDS, protease inhibitor cocktail EDTA
free (Roche, Mannheim, Germany) に懸濁し、超音波処理を行った。次いで15,000
rpmにて10分間遠心分離を行い、その上清をさらに42,000
rpmにて30分間遠心分離を行った。得られた上清に1/2量の3 x SDSサンプルバッファーを加え、100℃で2分間加熱した物をサンプルとした。成長段階における新規PLC様タンパク質の発現パターンを解析する際には、生後0日目、1日目、3日目、5日目、7日目、9日目、14日目、21日目のマウス脳の抽出液を、上述した方法により調整した。コントロールには生体マウスの脳を用いた。
【0126】
〈ウェスタンブロットによる新規PLCの検出〉
サンプルを7.5%
SDS-PAGEにて展開後、Immobilon-Pメンブレン
(Millipore, MA, USA) にセミドライ型トランスファー装置 (Bio-Rad, CA,
USA) を用いて2.1 mA/cm2で転写した。タンパクが転写されたメンブレンを0.1%アミドブラックを含む50%メタノール/10%酢酸溶液を用いて1分間固定処理し、水ですすいだ。次いで10%スキムミルクを含むTBSを用いて37℃にて1時間ブロッキング反応を行った。適量のブロッキング溶液に1/1000量の新規PLC抗体を加え、1時間抗体反応を行った後、0.1% Tween-20、1%スキムミルクを含むTBSで5分間 x 3回洗浄した。適量の1次抗体洗浄液に1/2000量のHRP-conjugated anti-rabbit
immunoglobulins抗体 (DAKO, Kyoto, Japan) を加え、室温にて30分間抗体反応を行った。反応後、メンブレンを0.1% Tween-20を含むTBSで5分間 x 5回洗浄し、ECL detection kit (Amersham Bioscience, NJ, USA) を用いて発光させ、X線フィルムNewA (コニカ,
Tokyo, Japan) を用いて可視化した。
【0127】
上記方法に従って、マウス各組織における新規PLC様タンパク質の分布を調べた。マウス脳、心臓、骨格筋、腎臓、肝臓、小腸、肺、精巣、膵臓を摘出し、その抽出液を用いてウェスタンブロットを行った (図2A) 。その結果、推定分子量約125 kDaの新規PLC様タンパク質は脳においてのみ発現を確認することができた。このことより、新規PLC様タンパク質が脳において何らかの機能を担っていると予想できる。
【0128】
脳におけるさらに細かい分布を調べる為に、マウス脳を嗅球、大脳皮質、海馬、終脳 (線条体付近) 、間脳、中脳、小脳、橋/延髄に分類し、これに加えて脊髄も摘出した。これらの臓器より得られた抽出液を用いてウェスタンブロットを行い、各部位における発現量を比較したものが図2Bである。新規PLC様タンパク質は嗅球、大脳皮質、海馬に強く発現し、小脳、間脳、中脳、終脳にもバンドが確認されたが、延髄、脊髄には全く発現が見られなかった。
【0129】
また、東京大学大学院 新領域創成科学研究科分子認識化学分野 東原和成助教授のご好意によりマウス鋤鼻器官、嗅上皮、副嗅球、主嗅球を単離したサンプルをいただき、ウェスタンブロットによりそれぞれにおける発現を確認したところ、鋤鼻器官や嗅上皮には発現が全く見られなかったのに対し、副嗅球と主嗅球には発現を確認することが出来た(図3C)。このことより、新規PLC様タンパク質が嗅球全体に分布しており、末梢の受容器である鋤鼻器官や嗅上皮には全く発現がみられない事が分かった。嗅球は臭いやフェロモンの情報を記憶し、識別する部分であり、海馬は様々な記憶を司る部位であること、大脳皮質は長期記憶に関与すると言われていることなどから、新規PLCは脳における記憶のメカニズムと何らかの関係を持っていることが強く示唆された。
【0130】
マウスの成長段階における新規PLC様タンパク質の発現パターンを解析した。サンプルには生後0日目、1日目、3日目、5日目、7日目、9日目、14日目、21日目のマウスの脳の抽出液を用い、コントロールには成体マウスの脳抽出液を用いた (図3A) 。その結果、新規PLC様タンパク質は1週齢もしくは2週齢付近から徐々に発現量が上昇するタンパク質である事が判明した。このことは、マウスが成長し、外部からの様々な刺激をうけて脳が発達すると共に新規PLCの発現量が増加する事を示唆している。
【実施例9】
【0131】
内在性新規PLCと強制発現新規PLCの比較:
マウス脳からの免疫沈降物と、HeLa細胞にて過剰発現させた新規PLC様タンパク質の移動度をウェスタンブロットにより比較した。サンプルの調製は、上記ウエスタンブロットの際の動物組織抽出液の調整法に従って行った。その結果、図3Bに示すとおり、両タンパクの移動度が一致したことから、今回単離した遺伝子は完全長であることが確認できた。
【実施例10】
【0132】
In situ
hybridizationによるマウス脳における新規PLC様タンパク質の局在の検討:
プローブ配列として、新規PLCに特徴的である抗体作成にも利用したXドメインとYドメインの間の配列(配列番号5)を用いた。PCR反応により増幅させたあと、Multiple Cloning siteの両端にT7プロモーター配列とT3プロモーター配列を持つpBluescript II SK (+)にクローニングした。染色する組織切片はジェノスタッフ有限会社に委託して作成した8週齢のマウス脳のパラフィン切片を用いた。プローブ作成と染色はアロカ株式会社に委託した。
【0133】
脳細胞には神経細胞を始め、アストログリア、ミクログリア、オリゴデンドログリアなど、様々な細胞が存在することが知られているため、脳細胞のうちどの細胞に新規PLC様タンパク質の発現が見られるのかどうかを検証することはタンパク質の機能を類推する上で非常に有効な手がかりになる。そこで、マウス脳における新規PLC様タンパク質のさらに細かい局在を調べるために、上記方法に従って、in situ
hybridizationによる検証を行った。その結果、図4に示すように、嗅球の先端部分に強いシグナルを確認することが出来た。これを拡大して確認したところ、この部分は僧帽細胞と思われる神経細胞であった。
【実施例11】
【0134】
新規PLC様タンパク質の細胞内局在:
カバーガラスを入れたφ35 mmディッシュに1 x 105個のHeLa細胞を蒔き、37℃、5% CO2存在下にて24時間培養した。次にDMRIE-C transfection reagent (Invitrogen, CA, USA) のプロトコールに従ってpCDNA3.1ベクターに組み込んだ新規PLCまたはpFlag-CMV2ベクターに組み込んだ新規PLCの欠損変異体をトランスフェクションした。さらに24時間培養した後、PBS (-) 2 mlで細胞を洗浄し、3.7%ホルマリン/PBSで10分間固定した。2 mlのPBS (-) で細胞を2回洗浄した後、0.2% TritonX-100/PBSにて2分間処理した。2 mlのPBS (-) で細胞を4回洗浄した後、10% スキムミルクを含むPBSで30分間ブロッキング反応を行った。一時抗体反応は1/100量の抗新規PLC抗体または抗Flag抗体 (SIGMA, MO, USA) を含むブロッキング溶液80 μlをカバーガラスにのせ、室温にて30分間行った。次いで、2 mlのPBS (-) を加えて5分間静置することを4回繰り返して余分な一時抗体を洗い流し、二次抗体反応を行った。二次抗体には1/100量のAlexa 488 conjugated anti-rabbit IgG抗体 (Molecular Probes, OR, USA) を含むブロッキング溶液を100
μl用い、室温にて30分間反応させた。また、二次抗体と同時に1/100量のAlexa 594 conjugated Phalloidin
(Molecular Probes, OR, USA) を用いてアクチンを染色した。2 mlのPBS (-) で5分間 x 4回細胞を洗浄した後、Slow-Fade Light Antifade Kit (Molecular Probes, OR, USA) を用いて細胞をスライドグラス上に封入し、共焦点顕微鏡 (Leica, Wetzlar, Germany) にて観察した。
【0135】
その結果、新規PLCはアクチンと共局在を示し、形質膜付近に多く存在していたが (図7A) 、Delta N変異体は細胞質に局在を変化し、アクチンとの共局在は全く観察されなかった(図7B)。次に、新規PLCのPHドメイン (PH; 48〜157
a.a.) と、開始メチオニンからPHドメインまでを含む領域
(1-PH; 1〜157 a.a.) をFlagタグ融合タンパク質としてHeLa細胞に強制発現させた。これを同様に蛍光顕微鏡下にて観察したところ、PHドメインは細胞質全体に広がっているのに対し (図7C) 、1-PHはアクチンと同様の局在を示し、膜付近に多く存在している事が分かった (図7D) 。これらの事は、N末端の45アミノ酸には新規PLCを細胞膜付近へと移行させるなんらかの機能があることを示唆している。
【実施例12】
【0136】
新規PLC様タンパク質の海馬神経初代培養細胞における局在:
新規PLCの組織における局在や発現時期の解析から、新規PLCが神経突起の伸長や、記憶など、脳の成長や成熟に伴う現象に関わっている可能性が強く示唆されたので、次に神経の伸長における役割を明らかにするため、海馬神経初代培養細胞における局在を検討した。
【0137】
生後1―2日目のマウスから海馬を単離し、トリプシンで細胞をバラバラにした後、神経に誘導する培地で5日間培養した。これを神経細胞のマーカーであるMAP2抗体と新規PLC抗体で染色した所、樹状突起とシナプス様部位に新規PLC抗体が点在するのが観察された(図8A)。更に、神経誘導培地で培養2日目に新規PLCをアデノウイルスにより強制発現させ、培養5日目にその形態を観察したところ、新規PLCを強制発現させることにより神経突起数が増大することがわかった(図8B)。
【産業上の利用可能性】
【0138】
本発明は、新規なホスホリパーゼC様タンパク質および該タンパク質をコードする遺伝子、並びに該タンパク質または該遺伝子を組成物として含む医薬品に関する。さらには該タンパク質または該遺伝子が関与する疾患の治療のための化合物のスクリーニング方法に関する。該タンパク質は、脳の記憶に関与する部位において重要な機能を果たしている分子であり、海馬初代培養細胞において神経突起伸展活性を示したことから、該タンパク質、該タンパク質および該遺伝子の発現を制御する化合物は、脳疾患を代表例とするような、神経再生を必要とする疾患の治療及び予防に適用が可能である。
【図面の簡単な説明】
【0139】
【図1】既知PLCとの比較を行った結果を示した図である。A.既知PLCと新規PLC様タンパク質のドメイン構造の比較を示したものである。B.ドメイン毎の既知PLCとの類似性を示したものである。C.ソフトウェアを用いて系統樹を作成したものである。
【図2】新規PLC様タンパク質の発現パターンの解析を行った結果を示した図である。A. マウスの脳、心臓、骨格筋、腎臓、肝臓、小腸、肺、精巣、膵臓における新規PLCの発現パターンを示している。B. マウス嗅球、大脳皮質、海馬、終脳、間脳、中脳、小脳、延髄、脊髄を摘出し、Aと同様にウェスタンブロットを行い新規PLCの発現パターンを解析した結果である。
【図3】新規PLC様タンパク質の発現パターンの解析を行った結果を示した図である。A. マウスの成長段階における新規PLC様タンパク質の発現パターンを解析した結果である。B. マウス脳から免疫沈降物と、HeLa細胞にて過剰発現させた新規PLC様タンパク質の移動度をウェスタンブロットにより比較した結果である。C. マウス主嗅球、副嗅球、嗅上皮、鋤鼻器官の抽出液を用いてウェスタンブロットにより新規PLC様タンパク質の発現量を比較したものである。
【図4】In situ hybridizationによる新規PLC mRNAの局在を示した図である。A. アンチセンスプローブを用いて検出した場合のマウス嗅球先端部の拡大図である。B. Aのスライドにエオジン染色を施した時の染色像である。C. センスプローブを用いた場合の染色像である。D. Cのスライドにエオジン染色を施した時の染色像である。E. 図Aの拡大図である。
【図5】新規PLC様タンパク質の酵素活性を測定した結果のうち、内在性のタンパクによるPLC活性を示したものである。マウス脳抽出液より新規PLC抗体、PLCδ1抗体、PLCδ4抗体、Control IgG抗体を用いて免疫沈降を行い、得られた免疫沈降物のPLC活性をそれぞれ示した。
【図6】新規PLC様タンパク質の酵素活性を測定した結果である。A.新規PLCのカルシウム感受性の結果を示している。B.新規PLCの活性に対するpHの影響を示している。C.PLCのXドメインに保存されている341番目のHis残基をAla残基に置換した変異体の活性測定を示している。
【図7】新規PLCの細胞内における局在を示した図である。A.新規PLCの局在を示している。B.N末端を欠く変異体の局在を示している。C、D : A、Bと同様の方法でpFlag-CMVベクターに組み込んだ新規PLCのPHドメインまたは開始メチオニンからPHドメインまでのコンストラクト(1-PH)をトランスフェクションし、一時抗体に抗FLAG抗体を用いて染色したものである。
【図8】海馬primarycultureにおける新規PLCの局在を示した図である。A,B:内在性の新規PLCの局在を示している。C.新規PLCをアデノウイルスで過剰発現させたものの局在を示している。
【技術分野】
【0001】
本発明は、新規なホスホリパーゼC(Phospholipase
C:以下PLCという。)様タンパク質および該タンパク質をコードする遺伝子、並びに該タンパク質または該遺伝子を組成物として含む医薬品、さらには該タンパク質または該遺伝子が関与する疾患の治療のための化合物のスクリーニング方法に関する。
【背景技術】
【0002】
PLCとは
細胞膜のリン脂質は様々な酵素によって分解され、多くの活性物質に合成される。例えば、ホスホリパーゼA2が作用すると、プロスタグランジンやロイコトリエンが合成される。また、ホスファチジルイノシトール-4,5-二リン酸(PI(4,5)P2)にPLCが作用すると、セカンドメッセンジャーであるイノシトール3リン酸(IP3)やジアシルグリセロール(DAG)ができる。
【0003】
PLCは受容体にカップルしたホスホリパーゼで、細胞外からのホルモン、神経伝達物質、成長因子などの刺激に応答し、細胞膜に存在するリン脂質のうちPI(4,5)P2のみを分解する酵素である。IP3は小胞体のIP3受容体に作用し、細胞内のカルシウム濃度を上昇させ、DAGはプロテインキナーゼC(PKC)を活性化する。PI(4,5)P2は細胞骨格の再構成や、エキソサイトーシス、イオンチャンネル等を直接的に制御していることから、PLC等によるPI(4,5)P2の正確な量的制御は、細胞の恒常性の維持において、非常に重要な役割を担っていると考えられている。それらは、その構造的な特徴や活性制御のメカニズムにより現在では、β(1,2,3,4)、γ(1,2)、δ(1,3,4)、ε(1)、ζ(1)の5つのサブタイプに分けられている。サブタイプによって発現分布が異なり、生体内において、それぞれ異なった機能を担っている(例えば非特許文献1または2参照)。
【0004】
サブタイプについて
βタイプのPLCは主にGタンパクによって制御を受けている。線虫のゲノム配列において、2種のPLCβが存在していることから、βタイプのPLCは線虫からヒトまで、広く保存されているPLCである事が分かる。PLCβ2は造血系の細胞、PLCβ4は網膜と小脳に限定されて発現しているが、PLCβ1とβ3はより広範囲に発現が見られ、PLCβ1は大脳皮質や海馬に特に多く、β3は肝臓、耳下腺、脳などに多い。
【0005】
γタイプのPLCはXドメインとYドメインの間に2つのSH2ドメインとSH3ドメインを持っている事が特徴である。SH2ドメインはリン酸化されたチロシン残基を認識し、SH3ドメインはプロリンに富んだ配列と結合することが知られているドメインである。PLCγ1のSH2ドメインはチロシンキナーゼ型レセプターの自己リン酸化されたチロシン残基を認識して結合する。そして、PLCγ1のチロシン残基がリン酸化されることで活性化される事が分かっている。PLCγ1の発現分布はユビキタスであるが、特に脳や肺に多い。脳においてはニューロンに最も多く、次いでオリゴデンドロサイト、アストロサイトに多く発現している事が知られている。PLCγ2は造血系の細胞において多く発現しており、B細胞の分化にPLCγ2が必要であるという報告を裏付けている。
【0006】
δタイプのPLCは真核細胞において進化的に最もよく保存されているPLCである。酵母、粘菌、植物はδタイプのPLCしか持っておらず、βタイプやγタイプのPLCはより高等な生物のみが有するタンパク質である。PLCδの分子量は小さく、さらに構造は単純で、PH、EF-Hand、PLCX、PLCY、C2ドメインから構成されている。PLCδ1が膜へと結合するにはまず、PHドメインが細胞膜のPI 4,5)P2と結合するが、この状態ではPLCδ1は不活性型である。PLCが活性化される為にはさらにC2ドメインが細胞膜に結合する事が必要であり、このとき活性中心が露出される。この活性化型のPLCδ1は効率的にPI(4,5)P2を加水分解し、IP3の産生に付随して細胞膜から遊離する。PLC活性にはカルシウムイオンが必要なことも分かっているが、中でもδタイプのPLCはカルシウムに対する感受性が最も強いことから、生体内に於いてPLCδはカルシウムイオンによって厳密に制御されていることが示唆されている。PLCδタイプの発現分布はδ1が最も多く、そして広範囲に分布している。PLCδ1は脳、心臓、肺、骨格筋、精巣に多く分布しており、ラット脳においてはアストログリアに多く、ニューロンにはあまり発現していない事が分かっている。また、アルツハイマー病患者の脳において、PLCδ1の蓄積が見られることも報告されている。PLCδ3は脳、骨格筋、心臓などに多く、PLCδ4は精巣、脳、骨格筋に分布している。
【0007】
PLCεは近年Rasのエフェクターとして発見された新しいPLCで、Rasと結合するRAドメインを持っている。Rasは単量体Gタンパク質であり、細胞の成長、分化やガン化など、様々な局面で作用している因子である。ヒトのガンのうち、約15%の症例においてras遺伝子に変異が起こっている事などからも、Rasによるシグナル伝達を解明することは非常に重要であると言える。PLCεはRasやそのファミリータンパクであるRap1とGTP依存的に結合することが示された 。さらに、PLCεは進化上PLCβに近いことから、三量体Gタンパクによる制御が検討され、Gα12、Gβ1γ2によって活性化されることも示された。また、PLCεはRas-GEFドメインを持つことから、Ras、Rap1、Rap2A、Rhoに対してGTP-exchange活性を示すかどうかが検討され、Rap1特異的にGEF活性を持つ事が示された。PLCεのmRNAレベルでの発現パターンはいくつか報告されているが、心臓に最も多く、次いで肺、腎臓、脾臓、膵臓に発現しているという報告と、肝臓に最も多く、次いで肺、骨格筋、心臓、小腸に多いという報告がある。
【0008】
PLCζは近年同定された最も新しいPLCである。PLCζの制御メカニズムについてはまだあまりよく分かっていないが、カルシウムに対する感受性が非常に強いことが報告されている。また、PLCζの局在は精子に限定されており、受精の際に起こる卵のカルシウムオシレーションを引き起こす作用があることが分かっている(例えば非特許文献3参照)。
【0009】
本発明者らは、2004年度日本脂質生化学会、および2004年度日本生化学会において、本願発明に係る新規PLC様タンパク質の概要に関して発表を行った(非特許文献4および5参照)。本願発明に係る新規PLC様タンパク質が、PLCに特徴的な構造を有していること、脳に発現していること、カルシウム感受性であること等について公表したものの、新規PLC様タンパク質のアミノ酸および遺伝子配列、発現部位の詳細な特定、機能等については全く明らかにしていない。
【0010】
新規PLC様タンパクの有用性
既知のPLCが上記のように生体内において重要な機能を担っていること、各サブタイプ毎に特徴的な機能を有していること等を鑑みると、新規PLCを見つけることができれば、そのサブタイプに特有の重要な機能を担っている可能性も高く、これまで治療が不可能であった疾患との関わりなどが存在する可能性も非常に高く、産業上有効に利用することができると思われる。
【非特許文献1】K. Fukami, Structure, regulation, and function ofphospholipase C isozymes, J. Biochem. 131 (2002) 293-299.
【非特許文献2】M. J. Rebecchi, and S. N. Pentyala, Structure,function, and control of phosphoinositide-specific phospholipase C, Physiol.Rev. 80 (2000) 1291-1335.
【非特許文献3】M. Katan, Families of phosphoinositide-specificphospholipase C: structure and function, Biochim. Biophys. Acta 1436 (1998)5-17.
【非特許文献4】2004年度 日本脂質生化学会要旨集、2004年5月20日発行
【非特許文献5】2004年度 日本生化学会要旨集、2004年8月25日発行
【発明の開示】
【発明が解決しようとする課題】
【0011】
本発明は、このような状況に鑑みてなされたものであり、その目的は新規なPLC様タンパク質遺伝子を提供することにある。また、本発明は、このようにして同定された新規なPLC様タンパク質または遺伝子の医薬としての用途、特に神経再生医療に有効である薬剤を提供すること、さらには該タンパク質または該遺伝子が関与する疾患の治療のための化合物のスクリーニング方法を提供することも目的とする。
【0012】
マウスゲノム解析が完了したことから、そのゲノムシークエンスを検索することにより、PLCに特徴的な構造(例えばPH, EF-hand, PLCX, PLCY, C2など)を手がかりとすれば、これまで知られていないPLC様構造を持つ配列の存在は、比較的容易に見つけ出すことができるようになった。しかし、PLC様構造を持つ配列は複数存在し、それらのうちどれが生体内で実際にタンパクとして発現し、PI(4,5)P2をIP3とDAGに加水分解するPLC活性を有するかということは全く明らかではなかった。また、データベースに示されている配列は、PLC様タンパクの全長配列ではないこと、cDNAクローン情報には多くのミスがあること、オープンリーディングフレームが示されていないこと、メチオニンも複数存在し、タンパクをコードしている最初の塩基がどこであるのかも不明であったことなどから、真の新規PLC様タンパク質を見出すことは決して容易なことではなく、実際マウスゲノム解析が完了してから2年が過ぎようとしている現在でも、本願発明者以外で新規PLCタンパク質の同定に成功した者はいないのが現状である。
【課題を解決するための手段】
【0013】
本願発明者らは、上記課題を解決すべく鋭意研究を行った結果、新規PLC様タンパク質の全長cDNAのクローニングおよびアミノ酸配列の決定に成功し、生体内における発現、PLC活性の確認に成功した。さらに、本タンパク質が神経突起伸展活性を有するという予想だにしなかった効果をも見出し、神経再生医療への有用性を示すことができた。
【0014】
具体的には、セレラ社(MD, USA) のデータベースを検索し、既知PLCが有する共通した特徴的な構造(PH, EF-hand, PLCX, PLCY, C2)を手がかりに、マウスの4番染色体にPLCと思われるドメイン構造を持つ予想アミノ酸配列を見いだすことが出来た。この予想タンパク質mCP21687は約1600アミノ酸から構成されていたが、開始コドンが含まれて
おらず、完全長のものではなかった。そこで、Genbankより類似の配列を検索したところ、RIKEN cDNAクローンA930027K05がmCP21687をコードしている遺伝子である事と考えられた。しかし、A930027K05クローンは開始メチオニンを 含むタンパク質をコードしていたが、予想転写産物にはPLCYドメインとC2ドメインは含まれていなかった。これはクローニングの際に起こったフレーム
シフト変異又はシークエンシングミスと思われたため、真偽を確かめるためにA930027K05クローンの配列を基にマウス脳cDNAよりPCR反応を 行ったところ、A930027K05クローンには2箇所に1塩基の挿入がある事が判明し、さらにはC2ドメインをコードする配列の約600塩基後にストップコドンが存在し、C末端側のアミノ酸配列は当初の予想よりも400アミノ酸短い事が判明した。
【0015】
単離したクローンの全長をシークエンスし、アミノ酸配列に変換した。その結果、この遺伝子の転写産物は全長1164アミノ酸からなるタンパク質である事が分かった。推定分子量は約125kDaであった。また既知のPLCに特徴的なPHドメイン、EF-handドメイン、PLCXドメイン、C2ドメインを有していることも明らかとなった。新規PLC様タンパク質は、PLCXドメインとPLCYドメインの間の配列が他のPLCよりも長いこと、C末端側に200アミノ酸ほどの付加的な配列を持つことが、既知PLCタンパクと大きく違う点であった。また、各ドメイン毎の類似性を比較したところ、PLCδタイプに最も類似性が高いことが示された(図1)ものの、全配列間の相同性は約30%程度であった。またインターネット上に公開されているプログラムCLUSTALWを用いて系統樹を作成したところ、新規PLC様タンパク質はPLCδに近いことが示されたが、δタイプのPLCが進化的に高く保存されているのに対して、新規PLC様タンパク質は線虫においてホモログが見当たらないことや、PLCδファミリーは構造が比較的単純であり約750アミノ酸からなるタンパク質であるのに対し、新規PLC様タンパク質は1164アミノ酸から構成されていることなどを考え合わせると、本願発明にかかるタンパク質は、新しいサブタイプのPLCタンパク質であると考えられた。
【0016】
また、Sf-9/バキュロウイルス発現系を用いて新規PLC様タンパク質を発現させたところ、実際にPLC活性を有していることが確認できた。さらに、このタンパクに特徴的な配列部分のペプチドに対するポリクローナル抗体を作成し、それを利用して、生体内に存在する新規PLC様タンパク質を精製したところ、Sf-9/バキュロウイルス発現系を用いて発現させたものと同様のPLC活性を有していることを確認した。また、この新規PLC様タンパク質は0.1〜1uMという生体内に近い低カルシウム濃度で最大の活性を示すことも明らかになった。
【0017】
また生体内での発現分布を調べたところ、脳に特異的に発現しており、その中でも特に嗅球、海馬、大脳皮質に多く発現していたことから、新規PLC様タンパク質は脳における記憶のメカニズムに関与していると考えられた。さらに詳細な解析により、嗅球のうち僧帽細胞に発現が限局していることが判明し、神経細胞のシナプス形成への関与も強く示唆された。海馬神経初代培養細胞に新規PLC様タンパク質を強制発現させると、神経突起数が顕著に増大することも明らかになり、新規PLC様タンパク質は、脳梗塞等の脳疾患およびパーキンソン病等における神経再生医療等にも極めて有用なタンパク質であることが示唆された。
【0018】
本願発明の内容は、新規なPLC様タンパク質およびそれをコードする遺伝子を提供すること、及びこのようにして同定された新規なPLC様タンパク質または該遺伝子の用途を提供することである。即ち、本願発明は以下の通りである。
(1)下記(a)から(d)のいずれかに記載のポリヌクレオチド。
(a)配列番号:2に記載のアミノ酸配列からなるタンパク質をコードするポリヌクレオチド。
(b)配列番号:1に記載の塩基配列のコード領域を含むポリヌクレオチド。
(c)配列番号:2に記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなり、PLC活性を有するタンパク質をコードする塩基配列を含むポリヌクレオチド。
(d)配列番号:1に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチドであって、PLC活性を有するタンパク質をコードするポリヌクレオチド。
(2)(1)に記載のポリヌクレオチドによりコードされるポリペプチド。
(3)(1)に記載のポリヌクレオチドが挿入された組み換えベクター。
(4)(1)に記載のポリヌクレオチドまたは(3)に記載のベクターを保持する形質転換体。
(5)(1)に記載のポリヌクレオチドとストリンジェントな条件下で特異的にハイブリダイズするポリヌクレオチドであって、少なくとも15ヌクレオチドの鎖長を持つポリヌクレオチド。
(6)(1)に記載のポリヌクレオチドもしくはその一部に対するアンチセンスポリヌクレオチド。
(7)(2)に記載のポリペプチドに結合する抗体。
(8)配列番号;2で表されるアミノ酸配列中、第497番から第624番目の部分アミノ酸配列部と反応性を示すことを特徴とする(7)に記載の抗体。
(9)(2)に記載のポリペプチドまたは(3)に記載の組み換えベクター及び薬学的に許容されうる担体を含んでなる医薬組成物
(10)該医薬組成物が、神経再生剤である請求項9に記載の医薬組成物
(11)以下の(a)〜(c)の工程を含む、請求項2に記載のポリペプチドまたはその部分ペプチドに結合する候補化合物のスクリーニング方法。
(a)(2)に記載のポリペプチドまたはその部分ペプチドと被検化合物を接触させる工程、
(b)該ポリペプチドまたはその部分ペプチドと被検化合物との結合活性を検出する工程、
(c)被検化合物を接触させない場合と比較して、該ポリペプチドまたはその部分ペプチドに結合する活性を有する化合物を選択する工程
(12)以下の(a)〜(c)の工程を含む、神経再生剤として有効である候補化合物のスクリーニング方法。
(a)(1)に記載のポリヌクレオチドまたは(2)に記載のポリペプチドを発現する細胞と被検化合物を接触させる工程
(b)上記細胞の(1)に記載のポリヌクレオチドまたは(2)に記載のポリペプチドの発現量を測定する工程
(c)被検化合物を接触させない場合と比較して、上記発現量を変化させる化合物を選択する工程
(13)(1)に記載のポリヌクレオチドの転写を制御する領域の下流にレポーター遺伝子の連結されたDNAを含むプラスミドで形質転換された形質転換体と被験化合物とを接触させ、被験化合物より(1)に記載のポリヌクレオチドの発現を制御する化合物を選択することを特徴とする、(1)に記載のポリヌクレオチドの発現を制御する化合物のスクリーニング方法
。
【発明の効果】
【0019】
本発明に係るタンパク質は、既知PLCとは、構造的に大きく異なるものであり、Sf-9/バキュロウイルス系を用いて発現させたもの、内在性のもの、両方にPLC活性があることが確認できた。また、本タンパク質は0.1〜1uMという生体内に近い低カルシウム濃度で最大の活性を示し、既知PLCのサブタイプのうち最もカルシウム感受性の高いδタイプの約10倍の感受性を示すものであった。
【0020】
さらに本タンパク質の生体内における発現は、脳に特異的であり、その中でも特に嗅球、海馬、大脳皮質に多く発現していたことから、脳における記憶のメカニズムに関与していると考えられた。さらに詳細な解析により、嗅球のうち僧帽細胞に発現が限局していることが判明し、神経細胞のシナプス形成への関与も強く示唆された。海馬神経初代培養細胞に新規PLC様タンパク質を強制発現させると、神経突起数が増大するという予想だにしない効果を見出した。このことは、新規PLC様タンパク質が、脳梗塞等の脳疾患およびパーキンソン病等における神経再生医療等に極めて有用なタンパク質であることを示すものである。
【発明を実施するための最良の形態】
【0021】
本発明は、新規なPLC様タンパク質、及びそれをコードするポリヌクレオチドを提供する。以下、本発明のために、好ましい実施形態に関して詳述する。
【0022】
(1)ポリヌクレオチド
本発明は、配列番号2で示されるアミノ酸配列をコードする核酸、及び配列番号2で示されるアミノ酸配列のうち1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなり、PLC活性を有するポリヌクレオチド)のアミノ酸配列をコードするポリヌクレオチドも提供する。
本発明のポリヌクレオチドは、一本鎖および二本鎖型両方のDNA、およびそのRNA相補体も含む。DNAには、例えば、天然由来のDNA、組換えDNA、化学合成したDNA、PCRによって増幅されたDNA、およびそれらの組み合わせが含まれる。本発明のポリヌクレオチドとしてはDNAが好ましい。なお、周知の通り、コドンには縮重があり、1つのアミノ酸をコードする塩基配列が複数存在するアミノ酸もあるが、上記アミノ酸配列をコードする塩基配列であれば、いずれの塩基配列を有するものも本願発明の範囲に含まれる。なお、下記の実施例1において実際にクローニングされたcDNAの塩基配列が配列番号1に示されている。よって、本発明のポリヌクレオチドは、好ましくは配列番号:1の塩基1−3497を有する。
【0023】
本明細書において、「ストリンジェントな条件下」とは、中程度又は高程度なストリンジェントな条件においてハイブリダイズすることを意味する。具体的には、中程度のストリンジェントな条件は、例えば、DNAの長さに基づき、一般の技術を有する当業者によって、容易に決定することが可能である。基本的な条件は、Sambrook,J.ら、Molecular
Cloning,A Laboratory Manual(3rd edition),Cold Spring
Harbor Laboratory,7.42−7.45(2001)に示され、そしてニトロセルロースフィルターに関し、5×SSC、0.5% SDS、1.0mM EDTA(pH8.0)の前洗浄溶液、約40−50℃での、約50%ホルムアミド、2×SSC−6×SSC(又は約42℃での約50%ホルムアミド中の、スターク溶液(Stark’s solution)などの他の同様のハイブリダイゼーション溶液)のハイブリダイゼーション条件、および約60℃、0.5×SSC、0.1% SDSの洗浄条件の使用が含まれる。高ストリンジェントな条件もまた、例えばDNAの長さに基づき、当業者によって、容易に決定することが可能である。一般的に、こうした条件は、中程度にストリンジェントな条件よりも高い温度及び/又は低い塩濃度でのハイブリダイゼーション及び/又は洗浄を含み、例えば上記のようなハイブリダイゼーション条件、及びおよそ68℃、0.2×SSC、0.1% SDSの洗浄を伴うと定義される。当業者は、温度および洗浄溶液塩濃度は、プローブの長さ等の要因に従って、必要に応じて調整可能であることを認識するであろう。
【0024】
本発明のポリヌクレオチドは、例えば以下の方法により調製することが可能である。
公知のPLCに特徴的な塩基配列(PH, EF-hand, PLCX, PLCY, C2)をクエリーとして塩基配列の検索を行い、マウスゲノムよりPLC様タンパク質候補を見出す。見出したゲノム配列は又はその一部を利用して、ハイブリダイゼーションや核酸増幅反応等の遺伝子工学の基本的手法を用いて、cDNAライブラリーなどから本発明ポリヌクレオチド(例えば本発明DNA)を調製することができる。
【0025】
ここで、核酸増幅反応は、例えば、ポリメラーゼ連鎖反応(PCR)[Saiki
R.K.et al.,Science,230,1350−1354(1985)],ライゲース連鎖反応(LCR)[Wu
D.Y.et al.,Genomics,4,560−569(1989);
Barringer K.J.et al.,Gene,89,117−122(1990);
Barany F.,Proc.Natl.Acad.Sci.USA,88,189−193(1991)]、及び転写に基づく増幅[Kwoh
D.Y.et al.,Proc.Natl.Acad.Sci.USA,86,1173−1177(1989)]等の温度循環を必要とする反応、並びに鎖置換反応(SDA)[Walker
G.T.et al.,Proc.Natl.Acad.Sci.USA,89,392−396(1992);
Walker G.T.et
al.,Nuc.Acids Res.,20,1691−1696(1992)]、自己保持配列複製(3SR)[Guatelli J.C.,Proc.Natl.Acad.Sci.USA,87,1874−1878(1990)]、及びQβレプリカーゼシステム[リザイルディら、BioTechnology,6,1197−1202(1988)]等の恒温反応を含む。また、欧州特許第0525882号に記載されている標的核酸と変異配列の競合増幅による核酸配列に基づく増幅(Nucleic
Acid Sequence Based
Amplification: NASABA)反応等も利用可能である。好ましくはPCR法である。
【0026】
実施例1に示すように、配列番号3及び4のプライマーを使用した場合、PCR産物として約1.5kbpのDNA断片が得られるので、これを例えばアガロースゲル電気泳動等の分子量によりDNA断片を篩い分ける方法で分離し、特定のバンドを切り出す方法等の常法に従って単離して本発明の核酸を得ることができる。
【0027】
上記のようなハイブリダイゼーション、核酸増幅反応等を使用してクローニングされる相同な核酸は、配列表の配列番号1に記載の塩基配列に対して少なくとも20%以上、好ましくは30%以上、より好ましくは50%以上、さらにより好ましくは70%以上、最も好ましくは90%以上の同一性を有する。
【0028】
同一性パーセントは、視覚的検査および数学的計算によって決定することが可能である。あるいは、2つの核酸配列の同一性パーセントは、Devereuxら,Nucl.Acids
Res.,12,387(1984)に記載され、そしてウィスコンシン大学遺伝学コンピューターグループ(UWGCG)より入手可能なGAPコンピュータープログラム、バージョン6.0を用いて、配列情報を比較することによって、決定可能である。GAPプログラムの好ましいデフォルトパラメーターには:(1)ヌクレオチドに関する単一(unary)比較マトリックス(同一に対し1および非同一に対し0の値を含む)、並びにSchwartz及びDayhoff監修,Atlas
of Protein
Sequence and Structure,pp.353−358,National
Biomedical Research Foundation(1979)に記載されるような、Gribskov及びBurgess,Nucl.Acids
Res.,14,6745(1986)の加重比較マトリックス;(2)各ギャップに対する3.0のペナルティおよび各ギャップ中の各記号に対しさらに0.10のペナルティ;及び(3)末端ギャップに対するペナルティなし、が含まれる。当業者に用いられる、配列比較の他のプログラムもまた、使用可能である。
【0029】
本発明は、本発明者らにより同定されたポリヌクレオチド(配列番号:1に記載の塩基配列からなるポリヌクレオチドまたはその相補鎖)に相補的な、少なくとも15ヌクレオチドの鎖長を有するヌクレオチドを提供する。ここで「相補鎖」とは、A:T(ただしRNAの場合は U)、G:Cの塩基対からなる2本鎖核酸の一方の鎖に対する他方の鎖を指す。また、「相補的」とは、少なくとも15個の連続したヌクレオチド領域で完全に相補配列である場合に限られず、少なくとも70%、好ましくは少なくとも80%、より好ましくは90%、さらに好ましくは95%以上の塩基配列上の相同性を有すればよい。相同性を決定するためのアルゴリズムは本明細書に記載したものを使用すればよい。このようなヌクレオチドは、本発明のポリヌクレオチドを検出、単離するためのプローブとして、また、本発明のヌクレオチドを増幅するためのプライマーとして利用することが可能である。プライマーとして用いる場合には、通常、15〜100ヌクレオチド、好ましくは15〜35ヌクレオチドの鎖長を有する。また、プローブとして用いる場合には、本発明のDNAの少なくとも一部若しくは全部の配列を含む少なくとも15ヌクレオチド、好ましくは少なくとも30ヌクレオチドの鎖長のポリヌクレオチドが用いられる。このようなポリヌクレオチドは、好ましくは本発明のポリペプチドをコードするポリヌクレオチドに特異的にハイブリダイズするものである。「特異的にハイブリダイズする」とは、通常のハイブリダイゼーション条件下、好ましくはストリンジェントな条件下で、本発明者らにより同定されたポリヌクレオチド(配列番号:1または3)とハイブリダイズし、他のポリペプチドをコードするDNAとはハイブリダイズしないことを意味する。
【0030】
また、このポリヌクレオチドには、本発明のポリペプチドをコードする遺伝子の発現を抑制するポリヌクレオチドが含まれる。このようなポリヌクレオチドには、アンチセンスポリヌクレオチド(アンチセンスDNA/RNA;本発明のポリペプチドをコードする遺伝子の転写産物と相補的なアンチセンスRNA、および該RNAをコードするDNA)やリボザイム(本発明のポリペプチドをコードする遺伝子の転写産物を特異的に開裂するリボザイム活性を有するRNAをコードするDNA)が含まれる。
【0031】
アンチセンスポリヌクレオチドが標的遺伝子の発現を抑制する作用としては、以下のような複数の要因が存在する。すなわち、三重鎖形成による転写開始阻害、RNAポリメラーゼによって局部的に開状ループ構造がつくられた部位とのハイブリッド形成による転写抑制、合成の進みつつあるRNAとのハイブリッド形成による転写阻害、イントロンとエキソンとの接合点でのハイブリッド形成によるスプライシング抑制、スプライソソーム形成部位とのハイブリッド形成によるスプライシング抑制、mRNAとのハイブリッド形成による核から細胞質への移行抑制、キャッピング部位やポリ(A)付加部位とのハイブリッド形成によるスプライシング抑制、翻訳開始因子結合部位とのハイブリッド形成による翻訳開始抑制、開始コドン近傍のリボソーム結合部位とのハイブリッド形成による翻訳抑制、mRNAの翻訳領域やポリソーム結合部位とのハイブリッド形成によるペプチド鎖の伸長阻止、および核酸とタンパク質との相互作用部位とのハイブリッド形成による遺伝子発現抑制などである。これらは、転写、スプライシング、または翻訳の過程を阻害して、標的遺伝子の発現を抑制する(平島および井上「新生化学実験講座2 核酸IV 遺伝子の複製と発現」,日本生化学会編,東京化学同人,pp.319-347,1993)。
【0032】
本発明で用いられるアンチセンスポリヌクレオチドは、上記のいずれの作用で標的遺伝子の発現を抑制してもよい。一つの態様としては、遺伝子のmRNAの5'端近傍の非翻訳領域に相補的なアンチセンス配列を設計すれば、遺伝子の翻訳阻害に効果的と考えられる。しかし、コード領域もしくは3'側の非翻訳領域に相補的な配列も使用し得る。このように、遺伝子の翻訳領域だけでなく非翻訳領域の配列のアンチセンス配列を含むポリヌクレオチドも、本発明で利用されるアンチセンスポリヌクレオチドに含まれる。使用されるアンチセンスポリヌクレオチドは、適当なプロモーターの下流に連結され、好ましくは3'側に転写終結シグナルを含む配列が連結される。アンチセンスポリヌクレオチド配列は、標的遺伝子またはその一部と相補的な配列であることが好ましいが、遺伝子の発現を有効に阻害できる限り、完全に相補的でなくてもよい。転写されたRNAは、標的とする遺伝子の転写産物に対して好ましくは90%以上、最も好ましくは95%以上の相補性を有する。アンチセンス配列を用いて、効果的に標的遺伝子の発現を阻害するには、アンチセンスポリヌクレオチドは、アンチセンス効果を引き起こすために、少なくとも15ヌクレオチド以上、好ましくは100ヌクレオチド、さらに好ましくは500ヌクレオチド以上の鎖長を有し、通常、3000ヌクレオチド以内、好ましくは2000ヌクレオチド以内の鎖長を有する。
【0033】
該アンチセンスポリヌクレオチドは、例えば、本発明のポリペプチドをコードするポリヌクレオチド(例えば、配列番号:1)の配列情報を基にホスホロチオネート法(Stein, 1988 Physicochemical properties of phosphorothioate
oligodeoxynucleotides. Nucleic Acids Res 16, 3209-21 (1988))などにより調製することが可能である。
【0034】
内在性遺伝子の発現の抑制は、また、リボザイムをコードするポリヌクレオチドを利用して行うことも可能である。リボザイムとは触媒活性を有するRNA分子のことをいう。リボザイムには種々の活性を有するものがあるが、中でもRNAを切断する酵素としてのリボザイムの研究により、RNAの部位特異的な切断を目的とするリボザイムの設計が可能となった。リボザイムには、グループIイントロン型や、RNasePに含まれるM1RNAのように400ヌクレオチド以上の大きさのものもあるが、ハンマーヘッド型やヘアピン型と呼ばれる40ヌクレオチド程度の活性ドメインを有するものもある(小泉誠および大塚栄子, (1990) 蛋白質核酸酵素,35:2191)。
【0035】
例えば、ハンマーヘッド型リボザイムの自己切断ドメインは、G13U14C15のC15の3'側を切断するが、活性にはU14が9位のAと塩基対を形成することが重要とされ、15位の塩基はCの他にAまたはUでも切断されることが示されている(M.Koizumiら,(1988) FEBS Lett.228:225)。リボザイムの基質結合部を標的部位近傍のRNA配列と相補的になるように設計すれば、標的RNA中のUC、UUまたはUAという配列を認識する制限酵素的なRNA切断リボザイムを作出することが可能である(M.Koizumiら,(1988) FEBS Lett. 239:285、小泉誠および大塚栄子,(1990) 蛋白質核酸酵素,35:2191、 M.Koizumiら, (1989) Nucleic Acids
Res. 17:7059)。
【0036】
また、ヘアピン型リボザイムも、本発明の目的のために有用である。ヘアピン型リボザイムは、例えばタバコリングスポットウイルスのサテライトRNAのマイナス鎖に見出される(J.M.Buzayan Nature 323:349,1986)。このリボザイムも、標的特異的なRNA切断を起こすように設計できることが示されている(Y.Kikuchi
およびN.Sasaki
(1992) Nucleic Acids Res. 19:6751、
菊池洋, (1992) 化学と生物 30:112)。
【0037】
本発明のポリペプチドをコードする遺伝子の発現を抑制するポリヌクレオチドは、遺伝子治療に用いる場合には、例えば、レトロウイルスベクター、アデノウイルスベクター、アデノ随伴ウイルスベクターなどのウイルスベクターやリポソームなどの非ウイルスベクターなどを利用して、ex vivo法やin vivo法などにより患者へ投与を行うことが考えられる。
【0038】
(2)ポリペプチド
下記実施例1において詳述する方法によりクローニングされた、本発明者らにより同定されたマウス新規PLC様タンパク質をコードするポリヌクレオチドの塩基配列を配列番号:1に、該ポリヌクレオチドによってコードされるポリペプチドのアミノ酸配列は配列番号:2に示す。
【0039】
本発明はまた、本発明者らにより同定されたポリぺプチドと機能的に同等なポリペプチド、および該ポリペプチドをコードするポリヌクレオチドに関する。ここで「機能的に同等」とは、対象となるポリペプチドが本発明者らにより同定されたポリぺプチドと同等の生物学的特性を有していることを意味する。本発明ポリペプチドが持つ生物学的特性としては、PLC活性(PIP2をIP3及びDAGに分解する活性)、低カルシウム感受性(本発明ポリペプチドは、低カルシウム濃度0.1〜1uMにおいて、最大のPLC活性を示すという特性を有する。)、および発現(本発明ポリペプチドは、動物の組織、好ましくは哺乳動物の組織で発現される。また発現部位としては、脳で高い発現を示す。好ましくは、嗅球、海馬、大脳皮質で高い発現を示す。さらに好ましくは、僧帽細胞において高い発現を示す。)等が挙げられる。
【0040】
本発明ポリペプチドは、典型的には、配列番号2に記載したアミノ酸残基1164個からなるアミノ酸配列を有する。なお一般的に、酵素のような生理活性を有するポリペプチドにおいて、そのアミノ酸配列のうち、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加された場合であっても、該生理活性が維持されることがあることは周知である。また天然産のポリペプチドの中には、それを生産する生物種の品種の違いや生態型の違いによる遺伝子の変異、あるいはよく似たアイソザイムの存在等に起因して、1から複数個のアミノ酸変異を有する変異タンパク質が存在することは知られている。従って、配列番号2に示されるアミノ酸配列において1若しくは複数個のアミノ酸が置換し若しくは欠失し、若しくは該アミノ酸配列に1若しくは複数個のアミノ酸が挿入され若しくは付加されたアミノ酸配列を有し、PLC活性を有するポリペプチド(以下、便宜的に「修飾ポリペプチド」)も本発明の範囲に含まれる。
【0041】
ここで、「複数個」とは、好ましくは1−200個、より好ましくは1−100個、さらにより好ましくは1−50個、最も好ましくは1−20個である。一般的には、部位特異的な変異によってアミノ酸が置換された場合に、元々のポリペプチドが有する活性は保持される程度に置換が可能なアミノ酸の個数は、好ましくは1−10個である。
【0042】
本発明のポリペプチドは、クローニングされた核酸の塩基配列からの推定に基づいて、配列番号2のアミノ酸配列を有するが、その配列を有するポリペプチドのみに限定されるわけではなく、本願明細書に記載した特性を有する限り全ての相同ポリペプチドを含むことが意図される。
【0043】
このような修飾ポリペプチドのアミノ酸配列は、配列番号2に示されるアミノ酸配列と少なくとも20%以上、好ましくは30%以上、より好ましくは50%、さらにより好ましくは70%以上、最も好ましくは90%以上の同一性を有することが好ましい。
【0044】
本発明のポリペプチドについて、NCBIの提供するblast 2 sequence (http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html)
を利用して相同性の確認を行うと、最も似ているマウスPLCδとは約30%程度の同一性であった。したがって、本発明のポリペプチドは新規なポリペプチドであると考えられた。また、特に、本発明のポリペプチドにおいて相同性の高い領域は、配列番号2に示されるアミノ酸配列のうち48−157、173―238、326−471、633−757及び776−874である。
【0045】
一般的に、同様の性質を有するアミノ酸同士の置換(例えば、ある疎水性アミノ酸から別の疎水性アミノ酸への置換、ある親水性アミノ酸から別の親水性アミノ酸への置換、ある酸性アミノ酸から別の酸性アミノ酸への置換、あるいはある塩基性アミノ酸から別の塩基性アミノ酸への置換)を導入した場合、得られる修飾ポリペプチドはもとのポリペプチドと同様の性質を有することが多い。遺伝子組換え技術を使用して、このような所望の変異を有する組換えポリペプチドを作製する手法は当業者に周知であり、このような修飾ポリペプチドも本発明の範囲に含まれる。
【0046】
本願明細書において、同一性のパーセントは、例えば、Altschulら(Nucl.Acids.Res.,25.3389−3402(1997))に記載されているBLASTプログラム、あるいはPearsonら(Proc.Natl.Acad.Sci.USA,2444−2448(1988))に記載されているFASTAを用いて配列情報と比較し決定することが可能である。当該プログラムは、インターネット上でNational
Center for
Biotechnology Information(NCBI)、あるいはDNA Data
Bank of
Japan(DDBJ)のウェブサイトから利用することが可能である。各プログラムによる同一性検索の各種条件(パラメーター)は同サイトに詳しく記載されており、一部の設定を適宜変更することが可能であるが、検索は通常デフォルト値を用いて行う。なお、当業者に用いられる、配列比較の他のプログラムもまた使用可能である。
【0047】
本発明のポリペプチドは、例えば、後述の実施例1に従って、本発明の核酸による配列番号1に記載のDNA配列を大腸菌、酵母、昆虫、または動物細胞に、それぞれの宿主で増幅可能な発現ベクターを用いて導入および発現させることにより、当該ポリペプチドを大量に得ることができる。
【0048】
本発明によって、このポリペプチドのアミノ酸配列およびそれをコードするDNA配列が開示されれば、当該配列またはその一部を利用して、ハイブリダイゼーション、PCR等の核酸増幅反応等の遺伝子工学的手法を用いて、他の生物種から同様の生理活性を有するポリペプチドをコードする遺伝子を容易に単離することができる。このような場合、それらの遺伝子がコードする新規ポリペプチドも本発明の範囲に含まれる。
【0049】
なお、本発明のポリペプチドは、そのアミノ酸配列が上述した通りのものであり、前記酵素活性を有するものであれば、ポリペプチドに糖鎖が結合していてもよい。
【0050】
(3)組み換えベクターと形質転換体
本発明によれば、単離した本発明のポリヌクレオチドを含む組換えベクターが提供される。
【0051】
プラスミド等のベクターに本発明核酸のDNA断片を組込む方法としては、例えば、Sambrook,J.ら,Molecular
Cloning,A Laboratory Manual(3rd edition),Cold Spring
Harbor Laboratory,1.1(2001)に記載の方法などが挙げられる。簡便には、市販のライゲーションキット(例えば、宝酒造製等)を用いることもできる。このようにして得られる組換えベクター(例えば、組換えプラスミド)は、宿主細胞(例えば、大腸菌DH5α、TB1、LE392、XL−392、又はXL−1Blue等)に導入される。
【0052】
プラスミドを宿主細胞に導入する方法としては、Sambrook,J.ら,Molecular
Cloning,A Laboratory Manual(3rd edition),Cold Spring
Harbor Laboratory,16.1(2001)に記載の塩化カルシウム法または塩化カルシウム/塩化ルビジウム法、リン酸カルシウム法、リポフェクション法、エレクトロポレーション法、エレクトロインジェクション法、PEGなどの化学的な処理による方法、遺伝子銃などを用いる方法などが挙げられる。
【0053】
ベクターは、簡単には当業界において入手可能な組換え用ベクター(例えば、プラスミドDNA等)に所望の遺伝子を常法により連結することによって調製することができる。用いられるベクターの具体例としては、大腸菌由来のプラスミドとして、例えば、pFLAG−CMV1、pDONR201、pBluescript、pUC18、pUC19、pBR322等が例示されるが、これらに限定されない。
【0054】
当業者であれば制限末端は発現ベクターに適合するように適宜選択することが可能である。発現ベクターは、「本発明タンパク質を発現させたい宿主細胞」に適したものを当業者であれば適宜選択することができる。このように本発明発現ベクターは上記の本発明核酸が目的の宿主細胞中で発現し得るように遺伝子発現に関与する領域(プロモーター領域、エンハンサー領域、オペレーター領域等)が適切に配列されており、さらに本発明核酸が適切に発現するように構築されていることが好ましい。
【0055】
また、発現ベクターの構築は、制限処理及び連結作業を必要としないGatewayシステム(インビトロジェン社)を用いることもできる。Gatewayシステムとは、PCR産物の方向性を維持したままクローニングができ、また、DNA断片を適切に改変した発現ベクターにサブクローニングを可能にした部位特異的な組換えを利用したシステムである。具体的には、PCR産物とドナーベクターとから部位特異的な組換え酵素であるBPクロナーゼによってエントリークローンを作成し、その後、このクローンと別の組換え酵素であるLBクロナーゼによって組換え可能なデスティネーションベクターにPCR産物を移入することにより、発現系に対応した発現クローンを調製するものである。最初にエントリークローンを作成すれば、制限酵素やリガーゼで作業する手間の係るサブクローニングステップが不要となる点を特徴の一つとする。
【0056】
発現ベクターの種類は、原核細胞および/または真核細胞の各種の宿主細胞中で所望の遺伝子を発現し、所望のタンパク質を生産する機能を有するものであれば特に限定されないが、例えば、哺乳類用発現ベクターとしてpFLAG−CMV1、pcDNA3.1、pGreenLanternなどが好ましく、大腸菌用発現ベクターとしてpQE−30、pQE−60、pMAL−C2、pMAL−p2、pSE420、pET又はpCAL若しくはそれらの人工的修飾物(pQE30、pET又はpCALを適当な制限酵素で処理して得られるDNAフラグメント)などが好ましく、酵母用発現べクターとしてpYES2(サッカロマイセス属)、pPIC3.5K、pPIC9K、pA0815(以上ピキア属)、昆虫用発現ベクターとしてpFastBacp、pBacPAK8/9、pBK283、pVL1392、pBlueBac4.5などが好ましい。
【0057】
上記「本発明発現ベクター」を宿主細胞に組み込み、形質転換体を得ることができる。上記「宿主細胞」として真核細胞(哺乳類細胞、酵母、昆虫細胞等)であっても原核細胞(大腸菌、枯草菌等)であっても使用することができる。本発明の形質転換体を得るための宿主細胞は、特に限定されず、さらに、または、ヒト(例えば、HeLa、293T、SH−SY5Y)、マウス(例えば、Neuro2a、NIH3T3)、サル(例えば、COS−1)由来の培養細胞でもよい。これらはいずれも公知であり、市販されているか(例えば、大日本製薬社)、あるいは公共の研究機関(例えば、理研セルバンク)より入手可能である。あるいは、胚、器官、組織若しくは非ヒト個体も使用可能である。
【0058】
ところで、「本発明のポリヌクレオチド」は、マウスゲノムライブラリーから発見されたポリヌクレオチドであるため、本発明においては真核細胞を本発明の形質転換体の宿主細胞として用いるとより天然物に近い性質を有した「本発明酵素」が得られる(例えば糖鎖が付加された態様など)と考えられる。従って、「宿主細胞」としては真核細胞、特に哺乳類細胞を選択することが好ましい。動物細胞としてはマウス由来、アフリカツメガエル由来、ラット由来、ハムスタ−由来、サル由来またはヒト由来の細胞若しくはそれらの細胞から樹立した培養細胞株などが例示される。
【0059】
宿主細胞として細菌、特に大腸菌を用いる場合、一般に発現べクターは少なくとも、プロモーター/オペレーター領域、開始コドン、所望のタンパク質をコードする遺伝子、終止コドン、ターミネーターおよび複製可能単位から構成される。宿主細胞として酵母、植物細胞、動物細胞または昆虫細胞を用いる場合には、一般に発現べクターは少なくとも、プロモーター、関始コドン、所望のタンパク質をコードする遺伝子、終止コドン、ターミネーターを含んでいることが好ましい。またシグナルペブチドをコードするDNA、エンハンサー配列、所望の遺伝子の5’側および3’側の非翻訳領域、選択マーカー領域または複製可能単位などを適宜含んでいてもよい。
【0060】
本発明のべクタ−において、好適な開始コドンとしては、メチオニンコドン(ATG)が例示される。また、終止コドンとしては、常用の終止コドン(例えば、TAG、TGA、TAAなど)が例示される。
【0061】
複製可能単位とは、宿主細胞中でその全DNA配列を複製することができる能力をもつDNAを意味し、天然のプラスミド、人工的に修飾されたプラスミド(天然のプラスミドから調製されたプラスミド)および合成プラスミド等が含まれる。
【0062】
エンハンサー配列、ターミネーター配列については、例えば、それぞれSV40に由来するもの等、当業者において通常使用されるものを用いることができる。
【0063】
選択マーカーとしては、通常使用されるものを常法により用いることができる。例えばテトラサイクリン、アンピシリン、またはカナマイシンもしくはネオマイシン、ハイグロマイシンまたはスペクチノマイシン等の抗生物質耐性遺伝子などが例示される。
【0064】
発現べクターは、少なくとも、上述のプロモータ−、開始コドン、所望のタンパク質をコードする遺伝子、終止コドン、およびターミネーター領域を連続的かつ環状に適当な複製可能単位に連結することによって調製することができる。またこの際、所望により制限酵素での消化やT4
DNAリガーゼを用いるライゲーション等の常法により適当なDNAフラグメント(例えば、リンカー、他の制限酵素部位など)を用いることができる。
【0065】
本発明の発現べクターの宿主細胞への導入[形質転換(形質移入)]は従来公知の方法を用いて行うことができる。
例えば、細菌(E.coli,Bacillus subtilis s
等)の場合は、例えばCohenらの方法[Proc.Natl.Acad.Sci.USA,69,2110(1972)]、プロトプラスト法[Mol.Gen.Genet.,168,111(1979)]やコンピテント法[J.Mol.Biol.,56,209(1971)]によって、Saccharomyces
cervisiaeの場合は、例えばHinnenらの方法[Proc.Natl.Acad.Sci.USA,75,1927(1978)]やリチウム法[J.B.Bacteriol.,153,163(1983)]によって、植物細胞の場合は、例えばリーフディスク法[Science,227,129(1985)]、エレクトロポレ−ション法[Nature,319,791(1986)]によって、動物細胞の場合は、例えばGrahamの方法[Virology,52,456(1973)]、昆虫細胞の場合は、例えばSummerらの方法[Mol.Cell
Biol.,3,2156−2165(1983)]によってそれぞれ形質転換することができる。
【0066】
なお、組換えベクターの構築及びそれを用いて本発明の核酸を宿主細胞に導入する方法の具体例が下記実施例4に詳述されている。
【0067】
形質転換体において発現したポリペプチドを小胞体の内腔に、細胞周辺腔に、または細胞外の環境に分泌させるために、適当な分泌シグナルを目的のポリペプチドに組み込むことができる。これらのシグナルは目的のポリペプチドに対して内因性であっても、異種シグナルであってもよい。
【0068】
本発明のポリペプチドの回収は、本発明のポリペプチドが培地に分泌される場合は、培地を回収する。本発明のポリペプチドが細胞内に産生される場合は、その細胞をまず溶解し、その後にポリペプチドを回収する。
【0069】
組換え細胞培養物から本発明のポリペプチドを回収し精製するには、硫酸アンモニウムまたはエタノール沈殿、酸抽出、アニオンまたはカチオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティークロマトグラフィー、ヒドロキシルアパタイトクロマトグラフィーおよびレクチンクロマトグラフィーを含めた公知の方法を用いることができる。
【0070】
(4)抗体
本発明は、本発明のポリペプチドに結合する抗体を提供する。ここで「抗体」には、ポリクローナルおよびモノクローナル抗体、キメラ抗体、一本鎖抗体、ヒト化抗体、さらにFabまたは他の免疫グロブリン発現ライブラリーの産物を含むFabフラグメントが含まれる。
【0071】
本発明のポリペプチドまたはその断片もしくは類似体、またはそれらを発現する細胞は、本発明のポリペプチドに結合する抗体を産生するための免疫原としても使用することができる。抗体は、好ましくは、本発明のポリペプチドに免疫特異的である。「免疫特異的」とは、その抗体が他のポリペプチドに対するその親和性よりも本発明のポリペプチドに対して実質的に高い親和性を有することを意味する。
【0072】
本発明のポリペプチドに結合する抗体は、当業者に公知の方法により調製することが可能である。ポリクローナル抗体であれば、例えば、次のようにして得ることができる。本発明のポリペプチドあるいはそのGSTとの融合タンパク質をウサギ等の小動物に免疫し血清を得る。これを、例えば、硫安沈殿、プロテインA、プロテインGカラム、DEAEイオン交換クロマトグラフィー、本発明のポリペプチドをカップリングしたアフィニティーカラム等により精製することにより調製する。具体例については実施例7に詳述されている。また、モノクローナル抗体であれば、例えば、本発明のポリペプチドをマウスなどの小動物に免疫を行い、同マウスより脾臓を摘出し、これをすりつぶして細胞を分離し、マウスミエローマ細胞とポリエチレングリコールなどの試薬により融合させ、これによりできた融合細胞(ハイブリドーマ)の中から、本発明のポリペプチドに結合する抗体を産生するクローンを選択する。次いで、得られたハイブリドーマをマウス腹腔内に移植し、同マウスより腹水を回収し、得られたモノクローナル抗体を、例えば、硫安沈殿、プロテインA、プロテインGカラム、DEAEイオン交換クロマトグラフィー、本発明のポリペプチドをカップリングしたアフィニティーカラム等により精製することで、調製することが可能である。
【0073】
本発明の抗体は、本発明のポリペプチドやこれを発現する細胞の単離、同定、および精製に利用することができる。
【0074】
(5)医薬組成物
本発明の「医薬組成物 」とは、前記で定義される本発明の「ポリペプチド」、「ポリヌクレオチド」または「抗体」のいずれかと、薬学的に許容され得る担体とからなる医薬組成物
である。
【0075】
ここで「薬学的に許容され得る担体」とは、賦形剤、希釈剤、増量剤、崩壊剤、安定剤、保存剤、緩衝剤、乳化剤、芳香剤、着色剤、甘味剤、粘稠剤、矯味剤、溶解補助剤あるいはその他の添加剤等が挙げられる。そのような担体の一つ以上を用いることにより、錠剤、丸剤、散剤、顆粒剤、注射剤、液剤、カプセル剤、トロー剤、エリキシル剤、懸濁剤、乳剤あるいはシロップ剤等の形態の医薬
組成物 を調製することができる。これらの医薬
組成物 は、経口あるいは非経口的に投与することができる。非経口投与のためのその他の形態としては、一つまたはそれ以上の活性物質を含み、常法により処方される外用液剤、腸溶内投与のための坐剤およびペッサリーなどが含まれる。
【0076】
投与量は、患者の年齢、性別、体重及び症状、治療効果、投与方法、処理時間、あるいは該医薬
組成物 に含有される活性成分(前記ポリペプチドや抗体など)の種類などにより異なるが、通常成人一人当たり、一回につき10μgから1000mg(あるいは10μgから500mg)の範囲で投与することができる。しかしながら、投与量は種々の条件により変動するため、上記投与量より少ない量で十分な場合もあり、また上記の範囲を越える投与量が必要な場合もある。
【0077】
とりわけ注射剤の場合には、例えば生理食塩水あるいは市販の注射用蒸留水等の非毒性の薬学的に許容され得る担体中に0.1μg抗体/ml担体〜10mg抗体/ml担体の濃度となるように溶解または懸濁することにより製造することができる。このようにして製造された注射剤は、処置を必要とするヒト患者に対し、1回の投与において1kg体重あたり、1μg〜100mgの割合で、好ましくは50μg〜50mgの割合で、1日あたり1回〜数回投与することができる。投与の形態としては、静脈内注射、皮下注射、皮内注射、筋肉内注射あるいは腹腔内注射のような医療上適当な投与形態が例示できる。好ましくは静脈内注射である。
【0078】
また、注射剤は、場合により、非水性の希釈剤(例えばプロピレングリコール、ポリエチレングリコール、オリーブ油のような植物油、エタノールのようなアルコール類など)、懸濁剤あるいは乳濁剤として調製することもできる。
【0079】
そのような注射剤の無菌化は、バクテリア保留フィルターを通す濾過滅菌、殺菌剤の配合または照射により行うことができる。注射剤は、用時調製の形態として製造することができる。即ち、凍結乾燥法などによって無菌の固体組成物
とし、使用前に無菌の注射用蒸留水または他の溶媒に溶解して使用することができる。
【0080】
本発明の医薬組成物は、脳疾患、特に神経再生を必要とする疾患の治療及び予防に適用が可能である。ここで、脳疾患とは、脳腫瘍、脳出血、脳梗塞、クモ膜下出血、脳動脈瘤、脳静脈瘤、脳血管の奇形や増殖、先天性奇形、水頭症、脳性マヒ、パーキンソン、ウィルス感染や予防ワクチン注射後の脳脊髄炎や脳炎などを意味する。また本願明細書において神経再生医療とは、神経再生を必要とする疾患の治療及び予防などを意味する。
【0081】
また、本発明の医薬組成物の種々疾患症状の治療効果については、常法に従って、既知の疾患モデル動物に投与することにより試験、検討することができる。
【0082】
(7)スクリーニング方法
本発明は、本発明のポリペプチドに結合する化合物のスクリーニング方法を提供する。この方法は、本発明のポリペプチドとこれに結合する化合物を含むと予想される被検試料とを接触せしめ、該ポリペプチドと被検試料との結合活性を検出し、そして本発明のポリペプチドに結合する活性を有する化合物を選択する、ことを含む。
【0083】
スクリーニングに用いられる本発明のポリペプチドは組換えポリペプチドであっても、天然由来のポリペプチドであってもよい。また部分ペプチドであってもよい。また細胞表面に発現させた形態、または膜画分としての形態であってもよい。被検試料としては特に制限はなく、例えば、細胞抽出物、細胞培養上清、発酵微生物産生物、海洋生物抽出物、植物抽出物、精製若しくは粗精製ポリペプチド、非ペプチド性化合物、合成低分子化合物、天然化合物が挙げられる。被検試料を接触させる本発明のポリペプチドは、例えば、精製したポリペプチドとして、可溶型ポリペプチドとして、担体に結合させた形態として、他のポリペプチドとの融合ポリペプチドとして、細胞膜上に発現させた形態として、膜画分として被検試料に接触させることができる。
【0084】
本発明のポリペプチドを用いて、これに結合するポリペプチドをスクリーニングする方法としては、当業者に公知の多くの方法を用いることが可能である。このようなスクリーニングは、例えば、免疫沈降法により行うことができる。具体的には、以下のように行うことができる。本発明のポリペプチドをコードする遺伝子を、pSV2neo,
pcDNA I,
pCD8 などの外来遺伝子発現用のベクターに挿入することで動物細胞などで当該遺伝子を発現させる。発現に用いるプロモーターとしては
SV40 early
promoter (Rigby In Williamson (ed.),
Genetic Engineering,
Vol.3. Academic
Press, London,
p.83−141(1982)), EF−1 α promoter (Kimら
Gene 91, p.217−223
(1990)), CAG promoter (Niwa et
al. Gene
108, p.193−200 (1991)), RSV
LTR promoter
(Cullen Methods
in Enzymology
152, p.684−704 (1987), SR α promoter (Takebe et
al. Mol.
Cell. Biol.
8,
p.466 (1988)),
CMV immediate
early promoter
(Seed and
Aruffo Proc.
Natl. Acad.
Sci. USA
84, p.3365−3369 (1987)), SV40
late promoter
(Gheysen and Fiers J. Mol.
Appl. Genet.
1,
p.385−394 (1982)), Adenovirus late promoter
(Kaufman et al. Mol. Cell.
Biol. 9, p. 946
(1989)), HSV TK promoter 等の一般的に使用できるプロモーターであれば何を用いてもよい。
【0085】
動物細胞に遺伝子を導入することで外来遺伝子を発現させるためには、エレクトロポレーション法
(Chu, G.
et al.
Nucl. Acid
Res. 15, 1311−1326
(1987))、リン酸カルシウム法 (Chen, C
and Okayama,
H. Mol.
Cell. Biol.
7,
2745−2752 (1987))、DEAEデキストラン法 (Lopata,
M. A.
et al.
Nucl. Acids
Res. 12, 5707−5717
(1984); Sussman,
D. J.
and Milman,
G. Mol.
Cell. Biol.
4,
1642−1643 (1985))、リポフェクチン法 (Derijard,
B. Cell
7,
1025−1037 (1994); Lamb, B. T.
et al.
Nature Genetics
5,
22−30 (1993);
Rabindran, S. K. et al.
Science 259, 230−234 (1993))等の方法があるが、いずれの方法によってもよい。
【0086】
特異性の明らかとなっているモノクローナル抗体の認識部位(エピトープ)を本発明のポリペプチドのN末またはC末に導入することにより、モノクローナル抗体の認識部位を有する融合ポリペプチドとして本発明のポリペプチドを発現させることができる。用いるエピトープ−抗体系としては市販されているものを利用することができる(実験医学
13, 85−90 (1995))。マルチクローニングサイトを介して、β−ガラクトシダーゼ、マルトース結合蛋白質、グルタチオンS−トランスフェラーゼ、緑色蛍光蛋白質(GFP)などとの融合ポリペプチドを発現することができるベクターが市販されている。
【0087】
融合ポリペプチドにすることにより本発明のポリペプチドの性質をできるだけ変化させないようにするために数個から十数個のアミノ酸からなる小さなエピトープ部分のみを導入して、融合ポリペプチドを調製する方法も報告されている。例えば、ポリヒスチジン(His−tag)、インフルエンザ凝集素
HA、ヒトc−myc、FLAG、Vesicular stomatitis ウイルス糖蛋白質(VSV−GP)、T7
gene10 蛋白質(T7−tag)、ヒト単純ヘルペスウイルス糖蛋白質(HSV−tag)、E−tag(モノクローナルファージ上のエピトープ)などのエピトープとそれを認識するモノクローナル抗体を、本発明のポリペプチドに結合するポリペプチドのスクリーニングのためのエピトープ−抗体系として利用できる(実験医学
13, 85−90 (1995))。
【0088】
免疫沈降においては、これらの抗体を、適当な界面活性剤を利用して調製した細胞溶解液に添加することにより免疫複合体を形成させる。この免疫複合体は本発明のポリペプチド、それと結合能を有するポリペプチド、および抗体からなる。上記エピトープに対する抗体を用いる以外に、本発明のポリペプチドに対する抗体を利用して免疫沈降を行うことも可能である。本発明のポリペプチドに対する抗体は、例えば、本発明のポリペプチドをコードする遺伝子を適当な大腸菌発現ベクターに導入して大腸菌内で発現させ、発現させたポリペプチドを精製し、これをウサギやマウス、ラット、ヤギ、ニワトリなどに免疫することで調製することができる。また、合成した本発明のポリペプチドの部分ペプチドを上記の動物に免疫することによって調製することもできる。
【0089】
免疫複合体は、例えば、抗体がマウスIgG 抗体であれば、Protein A SepharoseやProtein G Sepharoseを用いて沈降させることができる。また、本発明のポリペプチドを、例えば、GSTなどのエピトープとの融合ポリペプチドとして調製した場合には、グルタチオン−Sepharose
4Bなどのこれらエピトープに特異的に結合する物質を利用して、本発明のポリペプチドの抗体を利用した場合と同様に、免疫複合体を形成させることができる。
【0090】
免疫沈降の一般的な方法については、例えば、文献(Harlow,E.
and Lane,
D.: Antibodies,
pp.511−552, Cold Spring Harbor Laboratory
publications, New York (1988) )記載の方法に従って、または準じて行えばよい。
【0091】
免疫沈降されたポリペプチドの解析にはSDS−PAGEが一般的であり、適当な濃度のゲルを用いることでポリペプチドの分子量により結合していたポリペプチドを解析することができる。また、この際、一般的には本発明のポリペプチドに結合したポリペプチドは、クマシー染色や銀染色といったポリペプチドの通常の染色法では検出することは困難であるので、放射性同位元素である35S−メチオニンや35S−システインを含んだ培養液で細胞を培養し、該細胞内のポリペプチドを標識して、これを検出することで検出感度を向上させることができる。ポリペプチドの分子量が判明すれば直接SDS−ポリアクリルアミドゲルから目的のポリペプチドを精製し、その配列を決定することもできる。
【0092】
また、本発明のポリペプチドを用いて、該ポリペプチドに結合するポリペプチドを単離する方法としては、例えば、ウエストウエスタンブロッティング法(Skolnik,
E. Y.
et al.,Cell
(1991) 65,
83−90)を用いて行うことができる。すなわち、本発明のポリペプチドと結合するポリペプチドを発現していることが予想される細胞、組織、臓器(例えば、精巣)よりファージベクター(λgt11, ZAPなど)を用いたcDNAライブラリーを作製し、これをLB−アガロース上で発現させフィルターに発現させたポリペプチドを固定し、精製して標識した本発明のポリペプチドと上記フィルターとを反応させ、本発明のポリペプチドと結合したポリペプチドを発現するプラークを標識により検出すればよい。本発明のポリペプチドを標識する方法としては、ビオチンとアビジンの結合性を利用する方法、本発明のポリペプチド又は本発明のポリペプチドに融合したポリペプチド(例えばGSTなど)に特異的に結合する抗体を利用する方法、ラジオアイソトープを利用する方法又は蛍光を利用する方法等が挙げられる。
【0093】
また、本発明のスクリーニング方法の他の態様としては、細胞を用いた2−ハイブリッドシステム(Fields,
S., and
Sternglanz, R.,Trends. Genet. (1994) 10,
286−292、Dalton S, and Treisman R
(1992) Characterization
of SAP−1,
a protein
recruited by serum response factor
to the
c−fos serum
response element. Cell 68, 597−612、「MATCHMARKER
Two−Hybrid System」,「Mammalian MATCHMAKER
Two−Hybrid Assay Kit」,「MATCHMAKER
One−Hybrid System」(いずれもクロンテック社製)、「HybriZAP Two−Hybrid
Vector System」(ストラタジーン社製))を用いて行う方法が挙げられる。
【0094】
2−ハイブリッドシステムにおいては、本発明のポリペプチドまたはその部分ペプチドをSRF
DNA結合領域またはGAL4 DNA結合領域と融合させて酵母細胞の中で発現させ、本発明のポリペプチドと結合するポリペプチドを発現していることが予想される細胞より、VP16またはGAL4転写活性化領域と融合する形で発現するようなcDNAライブラリーを作製し、これを上記酵母細胞に導入し、検出された陽性クローンからライブラリー由来cDNAを単離する(酵母細胞内で本発明のポリペプチドと結合するポリペプチドが発現すると、両者の結合によりレポーター遺伝子が活性化され、陽性のクローンが確認できる)。単離したcDNAを大腸菌に導入して発現させることにより、該cDNAがコードするポリペプチドを得ることができる。これにより本発明のポリペプチドに結合するポリペプチドまたはその遺伝子を調製することが可能である。
【0095】
2−ハイブリッドシステムにおいて用いられるレポーター遺伝子としては、例えば、HIS3遺伝子の他、Ade2遺伝子、LacZ遺伝子、CAT遺伝子、ルシフェラーゼ遺伝子、PAI−1(Plasminogen
activator inhibitor type1)遺伝子等が挙げられるが、これらに制限されない。2ハイブリッド法によるスクリーニングは、酵母の他、哺乳動物細胞などを使って行うこともできる。
【0096】
本発明のポリペプチドと結合する化合物のスクリーニングは、アフィニティクロマトグラフィーを用いて行うこともできる。例えば、本発明のポリペプチドをアフィニティーカラムの担体に固定し、ここに本発明のポリペプチドと結合するポリペプチドを発現していることが予想される被検試料を適用する。この場合の被検試料としては、例えば細胞抽出物、細胞溶解物等が挙げられる。被検試料を適用した後、カラムを洗浄し、本発明のポリペプチドに結合したポリペプチドを調製することができる。
【0097】
得られたポリペプチドは、そのアミノ酸配列を分析し、それを基にオリゴDNAを合成し、該DNAをプローブとしてcDNAライブラリーをスクリーニングすることにより、該ポリペプチドをコードするDNAを得ることができる。
【0098】
また、ポリペプチドに限らず、本発明のポリペプチドに結合する化合物(アゴニストおよびアンタゴニストを含む)を単離する方法としては、例えば、固定した本発明のポリペプチドに、合成化合物、天然物バンク、もしくはランダムファージペプチドディスプレイライブラリーを作用させ、本発明のポリペプチドに結合する分子をスクリーニングする方法や、コンビナトリアルケミストリー技術によるハイスループットを用いたスクリーニング方法(Wrighton
NC; Farrell
FX; Chang
R; Kashyap
AK; Barbone
FP; Mulcahy
LS;Johnson DL; Barrett RW; Jolliffe
LK; Dower
WJ., Small
peptides as potent mimetics of
the protein
hormone erythropoietin,
Science (UNITED
STATES) Jul
26 1996,
273 p458−64、Verdine
GL., The
combinatorial chemistry of nature. Nature
(ENGLAND) Nov 7 1996, 384
p11−13、Hogan JC Jr.,Directed combinatorial chemistry.
Nature (ENGLAND)
Nov 7
1996, 384
p17−9)が当業者に公知である。
【0099】
本発明において、結合した化合物を検出又は測定する手段として表面プラズモン共鳴現象を利用したバイオセンサーを使用することもできる。表面プラズモン共鳴現象を利用したバイオセンサーは、本発明のポリペプチドと被検化合物との間の相互作用を微量のポリペプチドを用いてかつ標識することなく、表面プラズモン共鳴シグナルとしてリアルタイムに観察することが可能である(例えばBIAcore、Pharmacia製)。したがって、BIAcore等のバイオセンサーを用いることにより本発明のポリペプチドと被検化合物との結合を評価することが可能である。
【0100】
また、本発明は、細胞における本発明のポリヌクレオチド及びポリペプチドの発現を制御する化合物のスクリーニング方法を提供する。
【0101】
細胞における本発明のポリヌクレオチドの発現は、当業者に公知の方法で検出することができる。このような方法としては、例えば、ノーザンブロッティング法やRT−PCR法などを例示することができるがこれらに制限されない。ノーザンブロッティング法においては、検査対象となる細胞のRNAを精製し、アガロースゲル中にて電気泳動し、メンブレンにブロットしたものに、本発明の遺伝子を放射能などで標識したプローブをハイブリダイズさせ、特異的に出現するバンドの有無、濃淡で本発明のポリヌクレオチドの発現を測定する。また、RT−PCR法では、検査対象となる細胞のRNAを精製し、逆転写酵素によりcDNAとし、これを鋳型として、本発明のポリヌクレオチドに特徴的な配列をプライマーとして、本発明の遺伝子の転写産物由来のcDNAをPCRにより増幅する。これにより増幅されたcDNAの量は鋳型となったcDNAの量、ひいては本発明のポリヌクレオチドの転写産物の量に比例すると考えられるので、このPCRにより増幅されたDNA断片の量を電気泳動法などを用いて測定することで、本発明のポリヌクレオチドの発現量を測定できる。
【0102】
細胞における本発明のポリペプチドの発現は、当業者に公知の方法で検出することができる。例えば、本発明の抗体を用い、抗原抗体反応を行わせることにより、本発明のポリペプチドまたは該ポリペプチドを含む細胞を免疫学的に検出することができる。該検出方法は、本発明のポリペプチドをコードする遺伝子(ゲノムDNA等)の変異または発現異常が原因となり得る疾患の診断にも利用することができる。また、本発明のポリペプチドの定量方法としては、免疫学的に検出する方法として、マイクロタイタープレートを用いるELISA法・蛍光抗体法、ウェスタンブロット法、免疫組織染色法等があげられる。また、液相中で本発明のポリペプチドと反応する抗体のうちエピトープが異なる2種類のモノクローナル抗体を用いたサンドイッチELISA法、放射性ヨード等の放射性同位体で標識した本発明のポリペプチドと本発明のポリペプチドを認識する抗体とを用いるラジオイムノアッセイ法等があげられる。
【0103】
また、本発明は、本発明のポリヌクレオチドの転写を制御する領域の下流にレポーター遺伝子の連結されたDNAを含むプラスミドで形質転換された形質転換体と被験化合物とを接触させ、本発明のポリヌクレオチドの発現を制御する化合物を選択することを特徴とする、本発明のポリヌクレオチドの発現を制御する化合物のスクリーニング方法を提供する。
【0104】
本発明のスクリーニング方法の別の態様においては、まず、本発明のポリヌクレオチドのプロモータ―領域の下流にレポーター遺伝子が機能的に結合したDNAを有する細胞または細胞抽出液を提供する。
【0105】
ここで、「機能的に結合した」とは、本発明のポリヌクレオチドのプロモータ―領域に転写因子が結合することにより、レポーター遺伝子の発現が誘導されるように、本発明のポリヌクレオチドのプロモータ―領域とレポーター遺伝子とが結合していることをいう。従って、レポーター遺伝子が他の遺伝子と結合しており、他の遺伝子産物との融合タンパク質を形成する場合であっても、本発明のポリヌクレオチドのプロモータ―領域に転写因子が結合することによって、該融合タンパク質の発現が誘導されるものであれば、上記「機能的に結合した」の意に含まれる。
【0106】
上記レポーター遺伝子としては、その発現が検出可能なものであれば特に制限されず、例えば、当業者において一般的に使用されるCAT遺伝子、lacZ遺伝子、ルシフェラーゼ遺伝子、β−グルクロニダーゼ遺伝子(GUS)およびGFP遺伝子等を挙げることができる。また、上記レポーター遺伝子には、本発明のポリヌクレオチドをコードするDNAもまた含まれる。
【0107】
本発明の評価方法の別の態様では、次いで、上記細胞または上記細胞抽出液に被験試料を接触させる。次いで、該細胞または該細胞抽出液における上記レポーター遺伝子の発現レベルを測定する。
【0108】
レポーター遺伝子の発現レベルは、使用するレポーター遺伝子の種類に応じて、当業者に公知の方法により測定することができる。例えば、レポーター遺伝子がCAT遺伝子である場合には、該遺伝子産物によるクロラムフェニコールのアセチル化を検出することによって、レポーター遺伝子の発現レベルを測定することができる。レポーター遺伝子がlacZ遺伝子である場合には、該遺伝子発現産物の触媒作用による色素化合物の発色を検出することにより、また、ルシフェラーゼ遺伝子である場合には、該遺伝子発現産物の触媒作用による蛍光化合物の蛍光を検出することにより、また、β−グルクロニダーゼ遺伝子(GUS)である場合には、該遺伝子発現産物の触媒作用によるGlucuron(ICN社)の発光や5−ブロモ−4−クロロ−3−インドリル−β−グルクロニド(X−Gluc)の発色を検出することにより、さらに、GFP遺伝子である場合には、GFPタンパク質による蛍光を検出することにより、レポーター遺伝子の発現レベルを測定することができる。
【0109】
また、本発明のポリヌクレオチドをレポーターとする場合、該遺伝子の発現レベルの測定は、当業者に公知の方法によって行うことができる。例えば、該遺伝子のmRNAを定法に従って抽出し、このmRNAを鋳型としたノーザンハイブリダイゼーション法、またはRT−PCR法を実施することによって該遺伝子の転写レベルの測定を行うことができる。さらに、DNAアレイ技術を用いて、該遺伝子の発現レベルを測定することも可能である。
【0110】
また、該遺伝子からコードされる本発明のポリヌクレオチドを含む画分を定法に従って回収し、該ポリヌクレオチドの発現をSDS−PAGE等の電気泳動法で検出することにより、遺伝子の翻訳レベルの測定を行うこともできる。また、本発明のポリペプチドに対する抗体を用いて、ウェスタンブロッティング法などを実施し、該ポリペプチドの発現を検出することにより、遺伝子の翻訳レベルの測定を行うことも可能である。
【0111】
本発明のスクリーニングにより単離しうる化合物は、本発明の蛋白質の活性を調節するための薬剤の候補となり、本発明の蛋白質の発現異常や機能異常などに起因する疾患や本発明の蛋白質の活性を制御することにより治療可能な疾患の治療への応用が考えられる。治療や予防の対象となる疾患としては、例えば、脳疾患、特に神経再生を必要とする疾患が考えられる。本発明のスクリーニング方法を用いて単離しうる化合物の構造の一部を、付加、欠失及び/又は置換により変換される物質も、本発明の蛋白質に結合する化合物に含まれる。
以下、本発明を実施例によりさらに詳細に説明するが、本発明はこれら実施例に制限されるものではない。
【実施例1】
【0112】
マウスゲノムデータベースによる新規PLC様タンパク質の検索:
セレラ社(MD, USA) のデータベースを検索した。その結果、マウスの4番染色体にPLCと思われるドメイン構造
(PH, EF-hand, PLCX, PLCY, C2) を持つ予想アミノ酸配列を見いだすことが出来た。この予想タンパク質mCP21687は約1600アミノ酸から構成されていたが、開始コドンが含まれておらず、完全長のものでは無いと考えられた。そこで、Genbankより類似の配列を検索したところ、RIKEN
cDNAクローンA930027K05がmCP21687をコードしている遺伝子である事が分かった。A930027K05クローンは開始メチオニンを含むタンパク質をコードしていたが、予想転写産物にはPLCYドメインとC2ドメインは含まれていなかった。これはクローニングの際に起こったフレーム シフト変異又はシークエンシングミスと思われたため、真偽を確かめるためにA930027K05クローンの配列を基にマウス脳cDNAよりPCR反応を 行ったところ、A930027K05クローンには2箇所に1塩基の挿入がある事が判明、さらにはC2ドメインをコードする配列の約600塩基後にストッ プコドンが存在し、C末端側のアミノ酸配列は当初の予想よりも400アミノ酸短い事が判明した。以上の事より、この遺伝子の転写産物は全長1164アミノ酸(配列番号:2)からなるタンパク質である事が分かった。
【実施例2】
【0113】
新規PLC様タンパク質のcDNAクローニング:
マウス脳よりRNeasy miniキット (QIAGEN, Hilden, Germany) を用いてマウス脳 total RNAを精製した。ついで、SuperScript III First-Strand
Synthesis System (Invitrogen, CA, USA) を用いて逆転写反応を行い、マウス脳cDNAを得た。得られたcDNAをテンプレートとして、以下に示すオリゴヌクレオチドプライマーと反応条件にてKOD plus polymerase (TOYOBO, Tokyo, Japan)を用いてPCR反応を行った。プライマー配列中の赤字はそれぞれ開始コドンと終止コドンの位置を示しており、下線部はクローニング用に付加した制限酵素サイト(Bgl IIサイトおよびSal Iサイト)を示している。
N末端側プライマー5' agatctatgcctggtccccagccgtc 3'(配列番号:3に示した。)
C末端側プライマー5' gtcgacgtctcaatccctaacggtgg 3'(配列番号:4に示した。)
PCRのサイクルは94℃ 2分、[94℃ 20秒、55℃ 20秒、68℃ 4分]を35回繰り返し、68℃ 5分の条件で行った。得られた約3500 bpのPCR産物をpBluescript
II SK (+) のEco RVサイトにクローニングして、ABI
PRISM 3100 Genetic Analyzer (Applied Biosystems, CA, USA) を用いて全長のシークエンスを確認した。全長配列は配列番号:1に示したとおりである。
【実施例3】
【0114】
既知PLCとの比較:
ドメイン領域の割り出しにはSMART (http://smart.embl-heidelberg.de/) を利用した。また、既知のPLCとのドメイン構造の比較には、NCBIの提供するblast 2 sequence (http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html)を利用した。その結果を図1に示す。
【0115】
このPLCの構造上の特徴としてはPLCXドメインとPLCYドメインの間の配列が他のPLCよりも長いことと、C末端に200アミノ酸ほどの長い付加的な配列を持つことが挙げられる。また、各ドメインごとの類似性を比較したところ、PLCδタイプに最も類似性が高いことが示された(図1B)。さらに、インターネット上に公開されているプログラム CLUSTALW (http://clustalw.genome.jp/) を用いて系統樹を作成したところ、新規PLC様タンパク質はPLCδタイプに近いという結果を示した(図1C) 。ドメイン間の比較と系統樹から、新規PLC様タンパク質はPLCδに近い事が示されたが、δタイプのPLCが進化的に高く保存されているのに対し、新規PLCは線虫においてホモログが見あたらないことや、PLCδファミリーは構造が比較的単純であり、約750アミノ酸からなるタンパク質であるのに対し、新規PLCは1164アミノ酸から構成される事などから、この新規PLC様タンパク質が新しいサブタイプの物であると考えられた。
【実施例4】
【0116】
新規PLC様タンパク質遺伝子の発現ベクターへの組み込みおよび形質転換体の作成:
新規PLC様タンパク質の全長cDNAを、Sf-9/バキュロウィルス発現用pFastBac-GSTベクター、哺乳類発現用ベクターpCDNA3.1
(Invitrogen, CA, USA) にサブクローニングし、in vitroや細胞を用いた実験に使用した。また、一部のトランケート変異体を用いた実験では遺伝子をpFlag-CMV2ベクター (SIGMA,MO,USA)にサブクローニングして実験に用いた。
【実施例5】
【0117】
PLC活性の測定:
新規PLC様タンパク質が実際にPLCとしての活性、つまりはPI (4,5) P2を加水分解してIP3とDAGを産生する酵素活性を有するのかどうかを検討した。まず、マウス脳を摘出し、その抽出液より得られた免疫沈降物の酵素活性を測定することで、内在性の新規PLC様タンパク質がPLC活性を持つかどうかを検討した。測定は[3H]-PI (4,5) P2を基質として行い、反応後に得られた[3H]-IP3を分離し、その放射線の強度を液体シンチレーションカウンターで計測することで数値化した。具体的には以下の方法により行った。
【0118】
酵素の調製1(内在性タンパク質の調製)
マウス脳を20
mM Hepes-KOH pH 7.0, 120 mM KCl , protease inhibitor cocktail EDTA free, 0.1%
sodium deoxycholateを含むバッファーに懸濁し、超音波処理を行った。次いで15000 rpmにて10分間遠心分離を行い、その上清を42000 rpmにて30分間遠心した。得られた上清に新規PLC抗体とprotein A-sepharose (Amersham Bioscience, NJ, USA) を加えて4℃にて2時間ゆっくりと撹拌しながら反応させ、免疫沈降を行った。sepharoseビーズを20 mM Hepes-KOH pH 7.0, 120 mM
KCl , protease inhibitor cocktail EDTA free, 0.1% sodium deoxycholateを含むバッファーで5回洗浄したものを活性測定に用いた。
【0119】
酵素の調製2(バキュロウィルス発現系タンパクの調整)
Sf-9/バキュロウィルス発現系にて、GST-PLC融合タンパクを発現させた。回収した細胞を 20 mM Hepes-KOH
pH 7.0, 120 mM KCl , protease inhibitor cocktail EDTA free, 0.1% sodium
deoxycholateを含むバッファーに懸濁し、超音波処理を行った。次いで15000 rpmにて10分間遠心分離を行い、その上清にglutathione sepharose 4B
(Amersham Bioscience, NJ, USA)を加えて4℃にて1時間ゆっくりと撹拌しながら反応させた。sepharoseビーズを20 mM Hepes-KOH pH 7.0, 120 mM KCl , protease inhibitor cocktail
EDTA free, 0.1% sodium deoxycholateを含むバッファーで3回洗浄したものを活性測定に用いた。精製したタンパク量はSDS-PAGEにてBSAスタンダードと共に泳動し、CBB染色を行うことで定量した。
【0120】
基質の調製
PI(4,5)P2
(SIGMA, MO, USA) 、 PE (Doosan Serdary Research Lab.,
Kyungki Korea) 、[3H]-PI(4,5)P2 (PerkinElmer, MA, USA) を混合し、窒素ガスで完全に乾燥させた。これに滅菌水を加え、超音波処理を行って均質化したものを反応用の基質溶液とした。[最終濃度 : 200 μg/ml
PI(4,5)P2 , 100μg/ml PE, 2x106 DPM/ml
[3H]-PI(4,5)P2 ]
【0121】
PLC酵素反応
5 x PLC assay buffer(250
mM Hepes-KOH pH 7.0, 380 mM KCl, 2.5 mg/ml BSA) 10 μl、5
x Ca2+ EGTA溶液(CaCl2溶液と100 mM EGTA溶液、滅菌水を混合して調製)10 μl、酵素溶液 20 μl、反応基質溶液 10 μlを混ぜ合わせ、37℃にて5分間ゆっくりと振盪しながら反応させた。反応はクロロホルム : メタノール=2 : 1溶液 1 mlを加えることで停止させた。この溶液に1N HClを250 μl加えて14,000
rpmにて2分間遠心分離を行うことで、PLC活性によって生成したIP3を反応液上層に単離した。得られた反応液の上層450 μlに対してクリアゾルII (nacalai tesque, Kyoto, Japan) を2
ml加えて良く混ぜ合わせた後、液体シンチレーションカウンター (Aloka, Tokyo, Japan) にてトリチウムの壊変数を測定した。なお、特にカルシウム濃度について明記していない実験では、カルシウム濃度は10 μMで行った。
【0122】
上記方法に従って、新規PLC様タンパク質のPLC活性を測定した。ポジティブコントロールには脳において多く発現が見られ、かつ非常に強いPLC活性を持つことが知られているPLCδ1とPLCδ4を用いた。ネガティブコントロールにはNormal rabbit IgG (Santa Cruz, CA, USA) を用いた。測定の結果、PLCδ1の8,500 dpmのカウントに対して、新規PLCの場合は5,500 dpmという若干低い値ではあったが、新規PLC様タンパク質にはPLCとしての活性がある事が分かった (図5)。
【実施例6】
【0123】
新規PLC様タンパク質のカルシウム感受性、pH感受性、及び変異体の酵素活性の測定:
新規PLCがカルシウムによって制御を受けるのか否かを検討した。sf-9/バキュロウィルス発現系にてN末端にGSTタグを融合させた新規PLCとPLCδ1を発現させ、精製してタンパク量を定量した。これを用いてPLC活性を測定し、1分間で1mgのPLCが生成したIP3量
(nmol/min/mgPLC) を算出した。その結果、カルシウムに対して非常に強い感受性を持つことが知られているPLCδ1が10 μMのカルシウム濃度で最大の活性を示したのに対し、新規PLCは1 μMという、より生体内に近いカルシウム濃度で最大の活性を示すことが判明した (図6A) 。さらに、新規PLCの活性がpHに依存するかどうかを検討した。反応溶液のpHが6.5、7.0、7.4、8.0の4点で同様の活性測定を行った所、新規PLCは既知のPLCと同じくpH 7.0付近で最大の活性を示すことが分かった (図6B) 。さらに、活性ドメインに変異を導入した変異体による活性測定を行った。PLCファミリーのXドメインには非常に高く保存されているHis残基があり、このHis残基に変異を導入することでPLC活性が消失することが分かっている (Ellis et al., 1998) 。そこで、新規PLCにおけるこのHis残基である341番目のHis残基をAla残基に置換した変異体
(H341A) を作製して活性測定を行ったところ、PLC活性は完全に消失した (図6C) 。以上のことより、今回単離した新規タンパク質がPLCとしての活性を持っている、すなわち新規PLCである事が示された。
【実施例7】
【0124】
新規PLC様タンパク質に対するポリクローナル抗体の作成:
新規PLCのXドメインとYドメインの間の配列(配列番号:5)をpQE30ベクター (QIAGEN, Hilden, Germany) にクローニングし、大腸菌にて6 x Hisタグ融合タンパク質を発現、変性条件下にてNi-NTA Agarose ビーズ (QIAGEN, Hilden, Germany) を用いて精製した。作成したペプチドのアミノ酸配列を配列番号:6に示した。これをウサギ (日本白色種) に2週間おきに300〜400μgずつ免疫し、十分な抗体価が得られたところで全採血を行った。得られた血清より、以下のように抗体を精製した。まず、抗原部位と同じ遺伝子配列をpGEX-6p2ベクター (Amersham Bioscience, NJ, USA) にクローニングし、大腸菌にてGST融合タンパク質を発現させ、非変性条件下にてglutathione
sepharose 4B(Amersham Bioscience, NJ, USA)を用いて精製した。精製したGST融合抗原タンパク質をマニュアルに従いHiTrap NHS-activated HPカラム (Amersham
Bioscience, NJ, USA) に吸着させて抗原カラムを作製し、アフィニティー精製により精製抗体を得た。
【実施例8】
【0125】
新規PLC様タンパク質の組織分布と発現時期の検討:
〈動物組織抽出液の調製〉
マウスより臓器を摘出後、1 x PBS, 1% Nonitet P-40, 0.1% SDS, protease inhibitor cocktail EDTA
free (Roche, Mannheim, Germany) に懸濁し、超音波処理を行った。次いで15,000
rpmにて10分間遠心分離を行い、その上清をさらに42,000
rpmにて30分間遠心分離を行った。得られた上清に1/2量の3 x SDSサンプルバッファーを加え、100℃で2分間加熱した物をサンプルとした。成長段階における新規PLC様タンパク質の発現パターンを解析する際には、生後0日目、1日目、3日目、5日目、7日目、9日目、14日目、21日目のマウス脳の抽出液を、上述した方法により調整した。コントロールには生体マウスの脳を用いた。
【0126】
〈ウェスタンブロットによる新規PLCの検出〉
サンプルを7.5%
SDS-PAGEにて展開後、Immobilon-Pメンブレン
(Millipore, MA, USA) にセミドライ型トランスファー装置 (Bio-Rad, CA,
USA) を用いて2.1 mA/cm2で転写した。タンパクが転写されたメンブレンを0.1%アミドブラックを含む50%メタノール/10%酢酸溶液を用いて1分間固定処理し、水ですすいだ。次いで10%スキムミルクを含むTBSを用いて37℃にて1時間ブロッキング反応を行った。適量のブロッキング溶液に1/1000量の新規PLC抗体を加え、1時間抗体反応を行った後、0.1% Tween-20、1%スキムミルクを含むTBSで5分間 x 3回洗浄した。適量の1次抗体洗浄液に1/2000量のHRP-conjugated anti-rabbit
immunoglobulins抗体 (DAKO, Kyoto, Japan) を加え、室温にて30分間抗体反応を行った。反応後、メンブレンを0.1% Tween-20を含むTBSで5分間 x 5回洗浄し、ECL detection kit (Amersham Bioscience, NJ, USA) を用いて発光させ、X線フィルムNewA (コニカ,
Tokyo, Japan) を用いて可視化した。
【0127】
上記方法に従って、マウス各組織における新規PLC様タンパク質の分布を調べた。マウス脳、心臓、骨格筋、腎臓、肝臓、小腸、肺、精巣、膵臓を摘出し、その抽出液を用いてウェスタンブロットを行った (図2A) 。その結果、推定分子量約125 kDaの新規PLC様タンパク質は脳においてのみ発現を確認することができた。このことより、新規PLC様タンパク質が脳において何らかの機能を担っていると予想できる。
【0128】
脳におけるさらに細かい分布を調べる為に、マウス脳を嗅球、大脳皮質、海馬、終脳 (線条体付近) 、間脳、中脳、小脳、橋/延髄に分類し、これに加えて脊髄も摘出した。これらの臓器より得られた抽出液を用いてウェスタンブロットを行い、各部位における発現量を比較したものが図2Bである。新規PLC様タンパク質は嗅球、大脳皮質、海馬に強く発現し、小脳、間脳、中脳、終脳にもバンドが確認されたが、延髄、脊髄には全く発現が見られなかった。
【0129】
また、東京大学大学院 新領域創成科学研究科分子認識化学分野 東原和成助教授のご好意によりマウス鋤鼻器官、嗅上皮、副嗅球、主嗅球を単離したサンプルをいただき、ウェスタンブロットによりそれぞれにおける発現を確認したところ、鋤鼻器官や嗅上皮には発現が全く見られなかったのに対し、副嗅球と主嗅球には発現を確認することが出来た(図3C)。このことより、新規PLC様タンパク質が嗅球全体に分布しており、末梢の受容器である鋤鼻器官や嗅上皮には全く発現がみられない事が分かった。嗅球は臭いやフェロモンの情報を記憶し、識別する部分であり、海馬は様々な記憶を司る部位であること、大脳皮質は長期記憶に関与すると言われていることなどから、新規PLCは脳における記憶のメカニズムと何らかの関係を持っていることが強く示唆された。
【0130】
マウスの成長段階における新規PLC様タンパク質の発現パターンを解析した。サンプルには生後0日目、1日目、3日目、5日目、7日目、9日目、14日目、21日目のマウスの脳の抽出液を用い、コントロールには成体マウスの脳抽出液を用いた (図3A) 。その結果、新規PLC様タンパク質は1週齢もしくは2週齢付近から徐々に発現量が上昇するタンパク質である事が判明した。このことは、マウスが成長し、外部からの様々な刺激をうけて脳が発達すると共に新規PLCの発現量が増加する事を示唆している。
【実施例9】
【0131】
内在性新規PLCと強制発現新規PLCの比較:
マウス脳からの免疫沈降物と、HeLa細胞にて過剰発現させた新規PLC様タンパク質の移動度をウェスタンブロットにより比較した。サンプルの調製は、上記ウエスタンブロットの際の動物組織抽出液の調整法に従って行った。その結果、図3Bに示すとおり、両タンパクの移動度が一致したことから、今回単離した遺伝子は完全長であることが確認できた。
【実施例10】
【0132】
In situ
hybridizationによるマウス脳における新規PLC様タンパク質の局在の検討:
プローブ配列として、新規PLCに特徴的である抗体作成にも利用したXドメインとYドメインの間の配列(配列番号5)を用いた。PCR反応により増幅させたあと、Multiple Cloning siteの両端にT7プロモーター配列とT3プロモーター配列を持つpBluescript II SK (+)にクローニングした。染色する組織切片はジェノスタッフ有限会社に委託して作成した8週齢のマウス脳のパラフィン切片を用いた。プローブ作成と染色はアロカ株式会社に委託した。
【0133】
脳細胞には神経細胞を始め、アストログリア、ミクログリア、オリゴデンドログリアなど、様々な細胞が存在することが知られているため、脳細胞のうちどの細胞に新規PLC様タンパク質の発現が見られるのかどうかを検証することはタンパク質の機能を類推する上で非常に有効な手がかりになる。そこで、マウス脳における新規PLC様タンパク質のさらに細かい局在を調べるために、上記方法に従って、in situ
hybridizationによる検証を行った。その結果、図4に示すように、嗅球の先端部分に強いシグナルを確認することが出来た。これを拡大して確認したところ、この部分は僧帽細胞と思われる神経細胞であった。
【実施例11】
【0134】
新規PLC様タンパク質の細胞内局在:
カバーガラスを入れたφ35 mmディッシュに1 x 105個のHeLa細胞を蒔き、37℃、5% CO2存在下にて24時間培養した。次にDMRIE-C transfection reagent (Invitrogen, CA, USA) のプロトコールに従ってpCDNA3.1ベクターに組み込んだ新規PLCまたはpFlag-CMV2ベクターに組み込んだ新規PLCの欠損変異体をトランスフェクションした。さらに24時間培養した後、PBS (-) 2 mlで細胞を洗浄し、3.7%ホルマリン/PBSで10分間固定した。2 mlのPBS (-) で細胞を2回洗浄した後、0.2% TritonX-100/PBSにて2分間処理した。2 mlのPBS (-) で細胞を4回洗浄した後、10% スキムミルクを含むPBSで30分間ブロッキング反応を行った。一時抗体反応は1/100量の抗新規PLC抗体または抗Flag抗体 (SIGMA, MO, USA) を含むブロッキング溶液80 μlをカバーガラスにのせ、室温にて30分間行った。次いで、2 mlのPBS (-) を加えて5分間静置することを4回繰り返して余分な一時抗体を洗い流し、二次抗体反応を行った。二次抗体には1/100量のAlexa 488 conjugated anti-rabbit IgG抗体 (Molecular Probes, OR, USA) を含むブロッキング溶液を100
μl用い、室温にて30分間反応させた。また、二次抗体と同時に1/100量のAlexa 594 conjugated Phalloidin
(Molecular Probes, OR, USA) を用いてアクチンを染色した。2 mlのPBS (-) で5分間 x 4回細胞を洗浄した後、Slow-Fade Light Antifade Kit (Molecular Probes, OR, USA) を用いて細胞をスライドグラス上に封入し、共焦点顕微鏡 (Leica, Wetzlar, Germany) にて観察した。
【0135】
その結果、新規PLCはアクチンと共局在を示し、形質膜付近に多く存在していたが (図7A) 、Delta N変異体は細胞質に局在を変化し、アクチンとの共局在は全く観察されなかった(図7B)。次に、新規PLCのPHドメイン (PH; 48〜157
a.a.) と、開始メチオニンからPHドメインまでを含む領域
(1-PH; 1〜157 a.a.) をFlagタグ融合タンパク質としてHeLa細胞に強制発現させた。これを同様に蛍光顕微鏡下にて観察したところ、PHドメインは細胞質全体に広がっているのに対し (図7C) 、1-PHはアクチンと同様の局在を示し、膜付近に多く存在している事が分かった (図7D) 。これらの事は、N末端の45アミノ酸には新規PLCを細胞膜付近へと移行させるなんらかの機能があることを示唆している。
【実施例12】
【0136】
新規PLC様タンパク質の海馬神経初代培養細胞における局在:
新規PLCの組織における局在や発現時期の解析から、新規PLCが神経突起の伸長や、記憶など、脳の成長や成熟に伴う現象に関わっている可能性が強く示唆されたので、次に神経の伸長における役割を明らかにするため、海馬神経初代培養細胞における局在を検討した。
【0137】
生後1―2日目のマウスから海馬を単離し、トリプシンで細胞をバラバラにした後、神経に誘導する培地で5日間培養した。これを神経細胞のマーカーであるMAP2抗体と新規PLC抗体で染色した所、樹状突起とシナプス様部位に新規PLC抗体が点在するのが観察された(図8A)。更に、神経誘導培地で培養2日目に新規PLCをアデノウイルスにより強制発現させ、培養5日目にその形態を観察したところ、新規PLCを強制発現させることにより神経突起数が増大することがわかった(図8B)。
【産業上の利用可能性】
【0138】
本発明は、新規なホスホリパーゼC様タンパク質および該タンパク質をコードする遺伝子、並びに該タンパク質または該遺伝子を組成物として含む医薬品に関する。さらには該タンパク質または該遺伝子が関与する疾患の治療のための化合物のスクリーニング方法に関する。該タンパク質は、脳の記憶に関与する部位において重要な機能を果たしている分子であり、海馬初代培養細胞において神経突起伸展活性を示したことから、該タンパク質、該タンパク質および該遺伝子の発現を制御する化合物は、脳疾患を代表例とするような、神経再生を必要とする疾患の治療及び予防に適用が可能である。
【図面の簡単な説明】
【0139】
【図1】既知PLCとの比較を行った結果を示した図である。A.既知PLCと新規PLC様タンパク質のドメイン構造の比較を示したものである。B.ドメイン毎の既知PLCとの類似性を示したものである。C.ソフトウェアを用いて系統樹を作成したものである。
【図2】新規PLC様タンパク質の発現パターンの解析を行った結果を示した図である。A. マウスの脳、心臓、骨格筋、腎臓、肝臓、小腸、肺、精巣、膵臓における新規PLCの発現パターンを示している。B. マウス嗅球、大脳皮質、海馬、終脳、間脳、中脳、小脳、延髄、脊髄を摘出し、Aと同様にウェスタンブロットを行い新規PLCの発現パターンを解析した結果である。
【図3】新規PLC様タンパク質の発現パターンの解析を行った結果を示した図である。A. マウスの成長段階における新規PLC様タンパク質の発現パターンを解析した結果である。B. マウス脳から免疫沈降物と、HeLa細胞にて過剰発現させた新規PLC様タンパク質の移動度をウェスタンブロットにより比較した結果である。C. マウス主嗅球、副嗅球、嗅上皮、鋤鼻器官の抽出液を用いてウェスタンブロットにより新規PLC様タンパク質の発現量を比較したものである。
【図4】In situ hybridizationによる新規PLC mRNAの局在を示した図である。A. アンチセンスプローブを用いて検出した場合のマウス嗅球先端部の拡大図である。B. Aのスライドにエオジン染色を施した時の染色像である。C. センスプローブを用いた場合の染色像である。D. Cのスライドにエオジン染色を施した時の染色像である。E. 図Aの拡大図である。
【図5】新規PLC様タンパク質の酵素活性を測定した結果のうち、内在性のタンパクによるPLC活性を示したものである。マウス脳抽出液より新規PLC抗体、PLCδ1抗体、PLCδ4抗体、Control IgG抗体を用いて免疫沈降を行い、得られた免疫沈降物のPLC活性をそれぞれ示した。
【図6】新規PLC様タンパク質の酵素活性を測定した結果である。A.新規PLCのカルシウム感受性の結果を示している。B.新規PLCの活性に対するpHの影響を示している。C.PLCのXドメインに保存されている341番目のHis残基をAla残基に置換した変異体の活性測定を示している。
【図7】新規PLCの細胞内における局在を示した図である。A.新規PLCの局在を示している。B.N末端を欠く変異体の局在を示している。C、D : A、Bと同様の方法でpFlag-CMVベクターに組み込んだ新規PLCのPHドメインまたは開始メチオニンからPHドメインまでのコンストラクト(1-PH)をトランスフェクションし、一時抗体に抗FLAG抗体を用いて染色したものである。
【図8】海馬primarycultureにおける新規PLCの局在を示した図である。A,B:内在性の新規PLCの局在を示している。C.新規PLCをアデノウイルスで過剰発現させたものの局在を示している。
【特許請求の範囲】
【請求項1】
下記(a)から(d)のいずれかに記載のポリヌクレオチド。
(a)配列番号:2に記載のアミノ酸配列からなるタンパク質をコードするポリヌクレオチド。
(b)配列番号:1に記載の塩基配列のコード領域を含むポリヌクレオチド。
(c)配列番号:2に記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなり、PLC活性を有するタンパク質をコードする塩基配列を含むポリヌクレオチド。
(d)配列番号:1に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチドであって、PLC活性を有するタンパク質をコードするポリヌクレオチド。
【請求項2】
請求項1に記載のポリヌクレオチドによりコードされるポリペプチド。
【請求項3】
請求項1に記載のポリヌクレオチドが挿入された組み換えベクター。
【請求項4】
請求項1に記載のポリヌクレオチドまたは請求項3に記載のベクターを保持する形質転換体。
【請求項5】
請求項1に記載のポリヌクレオチドとストリンジェントな条件下で特異的にハイブリダイズするポリヌクレオチドであって、少なくとも15ヌクレオチドの鎖長を持つポリヌクレオチド。
【請求項6】
請求項1に記載のポリヌクレオチドもしくはその一部に対するアンチセンスポリヌクレオチド。
【請求項7】
請求項2に記載のポリペプチドに結合する抗体。
【請求項8】
配列番号;2で表されるアミノ酸配列中、第497番から第624番目の部分アミノ酸配列部と反応性を示すことを特徴とする請求項7に記載の抗体。
【請求項9】
請求項2に記載のポリペプチドまたは請求項3に記載の組み換えベクター及び薬学的に許容されうる担体を含んでなる医薬組成物
【請求項10】
該医薬組成物が、神経再生剤である請求項9に記載の医薬組成物
【請求項11】
以下の(a)〜(c)の工程を含む、請求項2に記載のポリペプチドまたはその部分ペプチドに結合する候補化合物のスクリーニング方法。
(a)請求項2に記載のポリペプチドまたはその部分ペプチドと被検化合物を接触させる工程、
(b)該ポリペプチドまたはその部分ペプチドと被検化合物との結合活性を検出する工程、
(c)被検化合物を接触させない場合と比較して、該ポリペプチドまたはその部分ペプチドに結合する活性を有する化合物を選択する工程
【請求項12】
以下の(a)〜(c)の工程を含む、神経再生剤として有効である候補化合物のスクリーニング方法。
(a)請求項1に記載のポリヌクレオチドまたは請求項2に記載のポリペプチドを発現する細胞と被検化合物を接触させる工程
(b)上記細胞の請求項1に記載のポリヌクレオチドまたは請求項2に記載のポリペプチドの発現量を測定する工程
(c)被検化合物を接触させない場合と比較して、上記発現量を変化させる化合物を選択する工程
【請求項13】
請求項1に記載のポリヌクレオチドの転写を制御する領域の下流にレポーター遺伝子の連結されたDNAを含むプラスミドで形質転換された形質転換体と被験化合物とを接触させ、被験化合物より請求項1に記載のポリヌクレオチドの発現を制御する化合物を選択することを特徴とする、請求項1に記載のポリヌクレオチドの発現を制御する化合物のスクリーニング方法 。
【請求項1】
下記(a)から(d)のいずれかに記載のポリヌクレオチド。
(a)配列番号:2に記載のアミノ酸配列からなるタンパク質をコードするポリヌクレオチド。
(b)配列番号:1に記載の塩基配列のコード領域を含むポリヌクレオチド。
(c)配列番号:2に記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなり、PLC活性を有するタンパク質をコードする塩基配列を含むポリヌクレオチド。
(d)配列番号:1に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチドであって、PLC活性を有するタンパク質をコードするポリヌクレオチド。
【請求項2】
請求項1に記載のポリヌクレオチドによりコードされるポリペプチド。
【請求項3】
請求項1に記載のポリヌクレオチドが挿入された組み換えベクター。
【請求項4】
請求項1に記載のポリヌクレオチドまたは請求項3に記載のベクターを保持する形質転換体。
【請求項5】
請求項1に記載のポリヌクレオチドとストリンジェントな条件下で特異的にハイブリダイズするポリヌクレオチドであって、少なくとも15ヌクレオチドの鎖長を持つポリヌクレオチド。
【請求項6】
請求項1に記載のポリヌクレオチドもしくはその一部に対するアンチセンスポリヌクレオチド。
【請求項7】
請求項2に記載のポリペプチドに結合する抗体。
【請求項8】
配列番号;2で表されるアミノ酸配列中、第497番から第624番目の部分アミノ酸配列部と反応性を示すことを特徴とする請求項7に記載の抗体。
【請求項9】
請求項2に記載のポリペプチドまたは請求項3に記載の組み換えベクター及び薬学的に許容されうる担体を含んでなる医薬組成物
【請求項10】
該医薬組成物が、神経再生剤である請求項9に記載の医薬組成物
【請求項11】
以下の(a)〜(c)の工程を含む、請求項2に記載のポリペプチドまたはその部分ペプチドに結合する候補化合物のスクリーニング方法。
(a)請求項2に記載のポリペプチドまたはその部分ペプチドと被検化合物を接触させる工程、
(b)該ポリペプチドまたはその部分ペプチドと被検化合物との結合活性を検出する工程、
(c)被検化合物を接触させない場合と比較して、該ポリペプチドまたはその部分ペプチドに結合する活性を有する化合物を選択する工程
【請求項12】
以下の(a)〜(c)の工程を含む、神経再生剤として有効である候補化合物のスクリーニング方法。
(a)請求項1に記載のポリヌクレオチドまたは請求項2に記載のポリペプチドを発現する細胞と被検化合物を接触させる工程
(b)上記細胞の請求項1に記載のポリヌクレオチドまたは請求項2に記載のポリペプチドの発現量を測定する工程
(c)被検化合物を接触させない場合と比較して、上記発現量を変化させる化合物を選択する工程
【請求項13】
請求項1に記載のポリヌクレオチドの転写を制御する領域の下流にレポーター遺伝子の連結されたDNAを含むプラスミドで形質転換された形質転換体と被験化合物とを接触させ、被験化合物より請求項1に記載のポリヌクレオチドの発現を制御する化合物を選択することを特徴とする、請求項1に記載のポリヌクレオチドの発現を制御する化合物のスクリーニング方法 。
【図1】


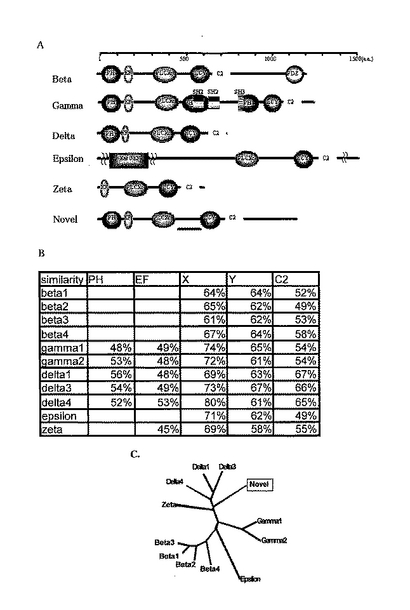
【図2】
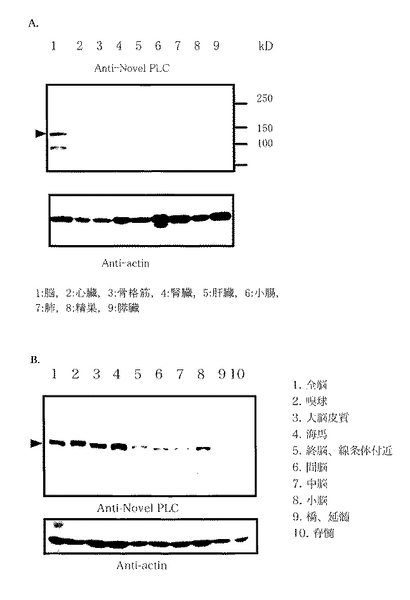
【図3】
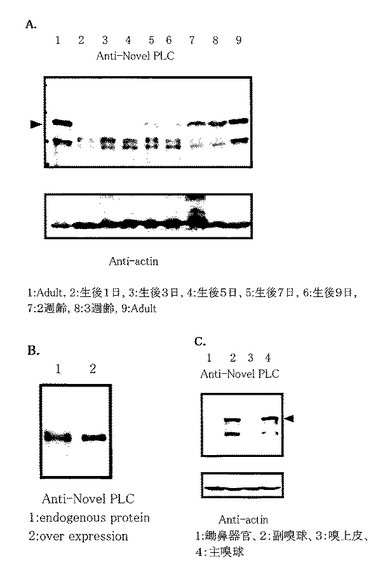
【図4】
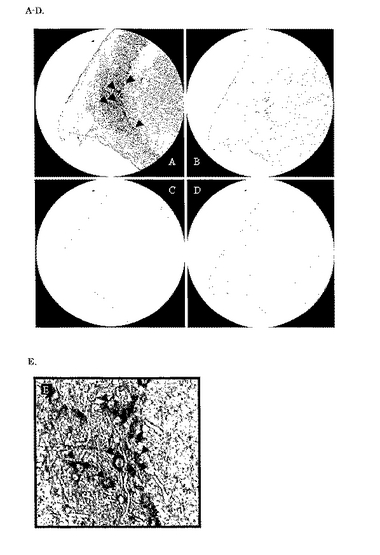
【図5】
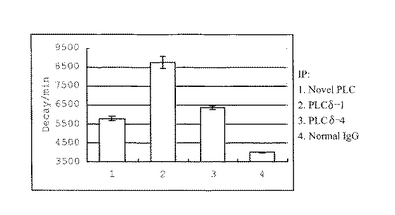
【図6】
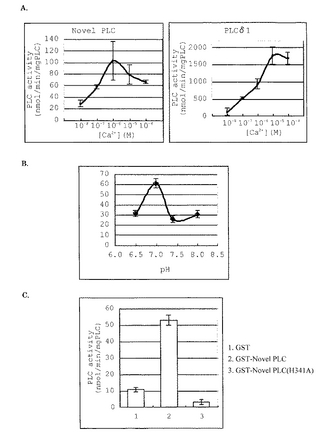
【図7】
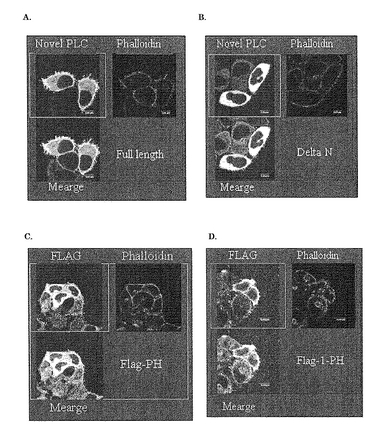
【図8】
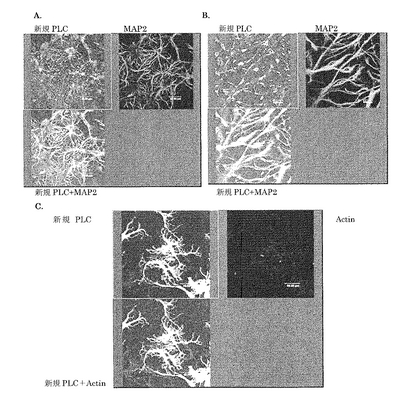
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2006−180738(P2006−180738A)
【公開日】平成18年7月13日(2006.7.13)
【国際特許分類】
【出願番号】特願2004−375846(P2004−375846)
【出願日】平成16年12月27日(2004.12.27)
【出願人】(592068200)学校法人東京薬科大学 (32)
【Fターム(参考)】
【公開日】平成18年7月13日(2006.7.13)
【国際特許分類】
【出願日】平成16年12月27日(2004.12.27)
【出願人】(592068200)学校法人東京薬科大学 (32)
【Fターム(参考)】
[ Back to top ]