
方法および生成物
本発明は共結晶を生成する方法を付与し、この方法は第一物質および第二物質を付与して、第一および第二の物質は共結晶を形成するのに適合する工程と、前記第一および第二の物質を共に混合する工程と、前記第一および第二の物質の混合物を、圧力およびせん断の長期で持続した条件にさらして、前記第一および第二物質の共結晶を十分に形成する工程とを含有する。圧力およびせん断の長期で持続した条件を、押出成形工程に好ましくは適応する。関連した組成物およびその使用を、同様に提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、製薬関係において役立つ生成物を形成するために役立つ方法、およびこのような方法によって形成される生成物に関する。本発明は、特に押出成形機を使用して共結晶生成物を形成する方法、およびそのような方法によって得られたか、または得ることができる生成物に関するが、これらに限らない。
【背景技術】
【0002】
結晶工学は、最近、活性薬剤の生理化学的性質を調整する方法として研究されている。医薬品へのそれの適用は、活性医薬品成分(API)を含有する幅広い範囲の複数の成分構造の組織的発見に関して、結晶性複合体と医薬品を形成するために使用することができる分子および分子間相互作用の種類を再考することによって、新規な経路を提供する。結晶工学は、薬物の溶解度、分解度および生体利用効率を向上させるために利用できる興味深い潜在的な代替方法を提供する。
【0003】
共結晶は、二つ以上の成分で構成される結晶性物質であり、通常化学両論的比率において、それぞれの成分が原子、イオン性化合物または非共有結合によって共に保持される分子であると考えることができる。共結晶は、二元化合物または分子複合体と以前呼ばれた。APIの生理化学的性質および共結晶を形成する物質の性質は改良することができるとともに、薬物分子の内因性活性を保持することができる。
【0004】
薬学的な共結晶は、製剤設計の際のAPIの改良において、顆粒球、塩および溶媒和物の魅力的な代替として現れる(Blagden N.,de Matas M.,Gavan P.T.,York P.;2007Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates.Advanced Drug Delivery Reviews.59,617−630)。ゲストとともに活性剤を共結晶化することによって、一つは、その活性薬剤の既存の固体相に関する性質を改善することができる新規固体相を生成することができる。例えば、医薬品成分(APIs)と薬学的に許容されるゲストの共結晶を含有する新規な薬学製剤は、既存の製剤に関して優れた性質を有することができる。製薬分野において、活性薬剤はAPIであることができ、共結晶(ゲスト)の他の成分は、薬学的に許容される化合物である必要がある(また必要に応じてAPIであることができる)。活性薬剤およびゲストは、機能性食品、農薬、色素、染料、爆薬、高分子添加物、潤滑剤添加物、光学化学製品、並びに構造的および電子的な物質を含有することができる。活性薬剤またはそれらの塩の物理的性質は、共結晶を形成することによって修飾することができる。前記性質は融点、密度、吸湿性、結晶形態学、容積負荷、圧縮性および製剤の性能に改良を加えることとなる貯蔵寿命を含有する。さらに他の性質、例えば生物学的利用能、毒性、味覚、物理的安定性、化学的安定性、生産コストおよび製造方法を、活性薬剤単独または塩としてよりむしろ共結晶を使用することによって改良することができる(Scott L.C,2007. Co−crystallization methods.WO2007/038524)。
【0005】
共結晶形成の成功率は通常極めて低く、相当するホモマー結晶における分子間より強い場合に、前記ヘテロマー系は、二つ(またはそれ以上)の分子間の非共有結合を形成するのみであるためである。様々な結晶工学技術は、現在共結晶設計で考慮中である。
【0006】
溶液の結晶化は、最も好ましい方法である(Sudhakar P.,Srivijaya R.,Sreekanth B.R.,Jayanthi P.K.,Peddy Vishweshwar.,Moses J.Babu.,Vyas K.,Javed Iqbal.,2007.Carboxylic acid−pyridine supramolecular heterocatemer in a co−crystal. Journal of Molecular Structure.In press;Renata Dreos.,Lassaad Mechi.,Giorgio Nardin.,Lucio Randaccio.,Pathzia Siega.,2005.Alternative co−crystallization of “almost” enantiomers and true enantiomers in some cis−b−organocobalt salen−type complexes with a−amino acids.Journal of Organometallic Chemistry.690,3815−382;Hickey B. Magali.,Matthew L.,Peterson A.,Lisa A., et al.,2007.Performance comparison of a co−crystal of carbamazepine with marketed product.European Journal of Pharmaceutics and Biopharmaceutics.67,112−119;Scott L.C,Kenneth I.H.,2007.4.Co−crystals of Piroxicam with Carboxylic Acids. Crystal Growth and Design.1−14)。この方法は、特に構造分析で単結晶を得るための使用である――化学的な化合物または要素の多数のサンプルを種々の異なる凝結状態にて溶液から凝固された。温度、溶媒または非溶媒含量、種晶、濃度、振動、純度および他の要因の一つ以上を変更することによって、一つは溶液から固形形態を凝固させるのに必要な状態を作り出すことができる。この方法の限定は、二つの成分(活性/ゲスト)が、極めて同程度の溶解度を有する必要がある;一方、ほとんど溶解度のない一成分は沈殿するであろう。同様に、多くの場合、顆粒球形成および本技術の明らかな失敗が、観測された。
【0007】
固体技術、例えばグラインディングまたはミリングにおいて、共結晶は、少なくとも二つの結晶性化合物を含有する混合物をグラインドすることによって生成される(Xyrofin O.Y.1996.Composition comprising co−crystals methods for its manufacture and its use.WO96/07331;Scott L.C,Kenneth I.H.,2007.2.Co−crystals ofPiroxicam with Carboxylic Acids. Crystal Growth and Design.1−14)。
【0008】
溶液に基づく実験は、グラインディングと比較して、それぞれのゲストに関して幅広い様々な形状を生成する。しかしながら、スクリーニング技術として、グラインディング実験は、それらが溶液から容易に得られない形状を特定することができるため、従来の溶液に基づく実験に対して良好な補足となる。重要さを、共結晶に対してスクリーニングする多数の実験技術の使用で与えた。しかしながら、固体技術、例えばグラインディングまたはミリングは、労力が集約し、しばしば小さい容器、例えばμLのウェルプレートにて実施することが困難である。しかし、これらの後者の経験的な方法の開発は、良く理解されてなく、それらの成功は共結晶形成に不可解な雰囲気を与えた(Chiarella R.A.,Davey R.J.,Peterson M.L.,2007.Making Co−Crystals−The Utility of Ternary Phase Diagrams. Crystal Growth and Design. Vol.0,1−4)。しかし、極性に基づいてグラインディング時に適切な溶媒の選択によって、共結晶技術の著しい欠点である顆粒球の形成を制御することができる(Andrew V. Trask., W. D. Samuel Motherwellb and William Jones.,2004.Solvent−drop grinding: green polymorph control of cocrystallisation. Chem. Commun.890−891)。
【0009】
超音波結晶化および超音波を使用して共結晶化による固体形態に関するスクリーニング技術を調査した(Scott L.C,2005.Screening for solid forms by ultrasound crystallization and cocrystallization using ultrasound.WO/2005/089375)。溶液の超音波処理を実施して共結晶を得た−超音波によって発生するキャビテーションエネルギーは、共結晶の成長に実用的であると考えられる。過飽和を核形成および結晶成長を制御する際の超音波の重要性を同様に研究した(Hong Li., Hairong Li., Zhichao Guo., Yu Liu., 2006. The application of power ultrasound to reaction crystallization. Ultrasonics Sonochemistry 13, 359−363; 2. Ruecroft G., Hipkiss D., Tuan Ly., Maxted N., Peter W. Cains., 2005. Sonocrystallization: The Use of Ultrasound for Improved Industrial Crystallization. Organic Process Research and Development. 9, 923−932; Castro et al., 2007)。Bucar et al.は(Dejan−Kresimir Bucar.,Leonard R.MacGillivray.,2007.Preparation and Reactivity of Nanocrystalline Cocrystals Formed via Sonocrystallization.J.Am.Chem.Soc.129(1))、ナノサイズの大きさの有機共結晶の調製をし、本方法が他の共結晶系に適応して、生理活性物質の物理的性質(例えば、溶解度)に影響することができることを予測する方法として超音波結晶化を導入した。しかしながら、酸化に作用を受けやすいAPIsまたはゲストは、本技術によって処理することができない。溶媒および貧溶媒の対の選択がまた重要である。
【0010】
米国特許第5,158,789号明細書には、二つの多価アルコール、ソルビトールおよびキシリトールの溶解物の押出成形品であり、菓子類に関連の性質を改善した製品を凝固で産生することを報告した。
【0011】
広範囲にわたる工業用の押出成形機の使用は、従来プラスチック、ゴムおよび食品業界においてであった。最近において、押出成形機の可能性が薬物応用において理解され始め、主に多くの機能が単一の連続動作において実行することができるためである。従って、多くの分離バッチ作業によって従来実施される工程を組み合わせることができ、製造効率を上昇して、潜在的に生成物の一貫性を改善する。しかしながら、押出成形に基づく製薬方法設計は、専門的な供給および下流の取り扱い技術と共に従来のプラスチック処理操作から開発した−それは様々な形態で重合体マトリックス内にAPIの分散剤を含む。ほとんどの従来の高分子処理機は、医薬品及び医薬部外品の製造管理及び品質管理の基準(GMP)の環境での使用に適応することができる。押出成形処理操作は、実験室から製造規模までに容易に拡大することができ、それら自身が、工程分析技術(PAT)として製薬業界において知られている製造過程の観測技術で十分に役立てることができる。製薬の押出成形品の応用の例を、以下に簡潔に記載する:
【0012】
固体分散剤:
固体分散剤は、活性薬剤物質(溶質)および希釈剤またはキャリア(溶媒または連続相)の完全混和として規定する。従来の技術において、薬剤の固体分散剤は、溶解または溶媒蒸発方法によって通常生成し、その後生成した物質を、カプセル化または錠剤に圧縮する前に、粉末状にし、ふるいにかけ、賦形剤と混合する。溶融押出成形は、これらの系の製造において改善を提供し、微粒子および分子分散剤で使用されてきた。
【0013】
放出制御ドラッグデリバリー:
放出制御ドラッグデリバリーシステムは、従来の服用形態にわたって数々の利点を提供する。放出制御錠剤の製造で最も一般的な工程は、湿式造粒法および直接的な圧縮技術を含み、それらは共に容量の均一化および分離の問題の対象となる。溶融押出成形技術は、放出制御経口投与形態の設計および開発を、水または溶媒を用いずに容易にする。下流のマイクロ造粒または球形化の性能を有する単軸または二軸押出成形機を使用して、顆粒剤または押出成形による錠剤を生成する。親水性および疎水性の物質を処理することができ、一成分のみを溶解または軟化して、物質流動を促進する必要がある。
【0014】
経皮フィルム:
押出成形したシートおよびフィルムを、製薬業界において、生成物包装および経皮薬剤のデリバリーシステムで使用する。後者の適用の場合、活性成分をキャリアと密接に混合し、基体に適用する。従来の押出成形は、創傷を水冷ローラー、30μm未満の厚さで直接巻回した連続性の薄いフィルムを製造するために幅広く、薄い鋳型と連結した。巻き取り速度およびローラー温度の制御は、フィルム結晶化度および分子方位性のある程度の制御を許容する。多層フィルムを、共押出成形、薄層化またはカプセル化によって生成することができる。
【0015】
湿式造粒:
造粒とは、製薬用語において、小粒子の凝集による粒子形態の過程とする。造粒は、直接圧縮、湿式造粒または乾燥した造粒から達成することができる。湿式造粒を、物理的な振動、流動化またはその両方で粉体層の混合物のせん断によるバッチモードで従来通りに実施した。二軸押出成形機は、湿式造粒の他の方法を超える利点を、滞留時間ならびに配分および分散混合のレベルを許容するスクリュー構造の柔軟性により提供する。同様に、これは、凝集および均質化の度合いの制御が比較的短い工程滞留時間、1分の度合いで可能とする。その結果、二軸スクリュー押出成形機は、改善した品質、空間利用および減少した開発時間を、従来の湿式造粒方法を超えて提供する。
【0016】
押出成形球状化:
これは均一な大きさの球状粒子を製造する複数の段階工程であり、即時放出および制御放出の両方の適応において使用することができる。前記粒子を使用して、ゼラチンカプセルを補充、または錠剤に圧縮する。押出成形球形化の主な利点は、活性な医薬品原料の高水準を比較的小さい分子の範囲内において組み込む能力である。押出成形球形化工程は、湿式造粒と、球状粒子の形成、乾燥、サイズ分布でスクリーニングおよび場合によりコーティングを組み合わせたものである。球形化工程は、固定壁を有するボウルと、溝面を有する急速回転基体の作用で通常の球状粒子に押出成形することによって、棒状小粒生成物に変形したものである。
【0017】
上述するように、製薬における応用で使用する既存の押出成形に基づく方法は、ポリマーキャリア組成物とAPIの組み合わせに基づくものである。
【発明の概要】
【発明が解決しようとする課題】
【0018】
製薬分野および他の分野において、共結晶の形成を改善した方法の必要性がある。
【課題を解決するための手段】
【0019】
第一の態様によれば、共結晶を生成する方法において、
第一および第二の物質を付与し、第一および第二の物質は共結晶を形成するのに適合する工程と、
前記第一および第二の物質を共に混合する工程と、
前記第一および第二の物質の混合物を、圧力およびせん断の長期の持続した条件にさらして、前記第一および第二の物質の共結晶を十分に形成する工程と
を含有する方法。
【0020】
共結晶という用語は、二つ以上の成分から生成される結晶性物質であると考えることができる組成物とする;特に二つ以上の成分は、二つの成分の結晶成分がそれぞれ異なる結晶構造に関与する。同様に、共結晶を、多成分分子結晶であると考えることができ、それでしばしば称する。実用的な定義は:共結晶が、非共有結合によって共に保持した二つ以上の分子種(電気的に中性)からなる結晶性物質と規定し、両方の成分が室温にて固形である(Akeroy, Crystal engineering: strategies and architectures, Acta Cryst. B53 (1997) 569−586; and S. L. Mohssette, O. Almarsson, M. L. Peterson, J. F. Remenar, M.J. Read, A.V. Lemmo, S. Ellis, M.J. Cima, CR. Gardner, High Throughput crystallisation: polymorphs, salts, co−crystals and solvates of pharmaceutical solids, Adv. Drug DeNv. Rev. 56 (2004) 275−300.参照)。
【0021】
一般的なレベルにおいて、共結晶は固体分散剤と異なる点に留意すべきである。“固体分散剤”は、不活性キャリアで化合物の分子および分子分散に近いものとして記載して使用される総称語である。これは、単純な共融混合物および固体溶液を含有する。単純な共融混合物は、二つの化合物からなり、液相に完全に混合するが、固体相において非常に限られた範囲で混合する。2つの溶融物質の混合物を冷却したとき、それら両方は非常に微細な(しかし分離した)結晶の物理的混合物を同時に生成して結晶化する。固溶体は、成分の数に関わりなく、丁度1つの相からなる液体溶液と同等である。典型的な固溶体は結晶構造を有し、溶質分子は結晶格子で溶媒分子と置換する。しかしながら、新規な結晶構造の形成が、共結晶の場合には全くない−溶質原子は、単に溶媒の分子を構造変化なしに置換、または溶媒分子の間の間隔の範囲内に位置する。侵入型固溶体において、溶質分子の体積は、通常溶媒の20%未満のレベルにて存在する。これは共結晶から結晶性固体溶液を区別し、規定した規則正しいパッキングモチーフ、対応するゲスト、および非共有結合によって結合したホスト分子を、新規な結晶構造を形成するための格子全体に化学量論的比率で示す。非結晶性固溶体において、溶質分子は、分子的であるが、不規則に非結晶性溶媒に分散している。固体分散剤を、また非結晶性または半結晶性マトリックスに分散した超微細な結晶質の薬剤粒子を含むことができる(Improving drug solubility for oral delivery using solid dispersion,C.Leuner and J.Dressman,Eur. J. Pharm and Biopharm50(2000)47−60参照)。
【0022】
さらに、既存のポリマーおよびAPI押出成形技術に関して、共結晶が固溶体および固体分散剤の双方と異なる構造を有し、その双方が、上記に簡潔に記載するように、ポリマーに基づく押出成形物において一般的な固体形状であることを明白にすべきである。ポリマーに基づく押出成形物の関連において、前者はAPIおよびポリマーの非結晶性混合物であり、後者は半結晶性ポリマーマトリックスの範囲内においてAPIの分散剤である。
【0023】
共結晶の存在は、当業者に知られる数々の分析手法によって特定することができる。これらの最も厳密なものは、おそらくX線結晶解析であり、それは結晶構造の詳細な観測および単一の原子規模での潜在的なそれの解像度を含む。しかしながら、X線結晶解析は、時間がかかり、複雑な工程である。共結晶形成によく適している結晶構造を調査して、時間的に効率的且つ単純な他の方法は、粉末X線回折測定(PXRD)特徴の使用である。一般的に、共結晶性構造の存在は、1つ以上の新規PXRDピークの出現によって識別することができる。非結晶性形状は、拡散した粉末X線回折パターンを示す一方、共結晶は単一成分またはそれらの物理的混合物のPXRDパターンで示さない、付加的な特有の(単一の)ピーク/(複数の)ピークを示す。第一および第二の物質(いくつかの場合、前記物質の様々な高分子の形態)のPXRDパターンの知見によれば、第一および第二の物質と比較して1つ以上の新規PXRDピークの出現によって共結晶構造の存在を同定することが可能である。従って本発明の実施形態は、上記に示したような方法の産出生成物のPXRDパターンと、第一および第二の物質の単独または混合物のPXRDパターン、または関連する共結晶の既知のPXRDパターンを比較することによって、共結晶の存在を同定する工程を含むことができる。
【0024】
本発明の方法が、連続フロー法であることが好ましい。連続工程における合成方法を実行する能力は、従来のバッチ方法と比較して、十分な利点がある。バッチ工程を超える利点は、改善した効率、簡単なスケールアップ、一貫した生成物の性質および減少した洗浄の必要性を含有する。
【0025】
好ましい実施形態において、第一物質は医薬品原料(API)である。上記のように、製薬分野において、共結晶を製造する改善した方法への特有の必要性がある。本分野において、既存の技術は、労働力の集中、遅延、一貫性がなくおよび/または信頼性がなく、規模の拡大の影響を受けないまたはこれらの問題の組み合わせであることを含む不利な点を被る。しかしながら、本発明は製薬分野を超えて、例えば他にも農薬、爆薬、機能性食品、色素、染料、潤滑添加剤、光学的な化学製品、構造物質および電子物質を含む適用がある点に留意すべきである。
【0026】
第一および第二の物質を、少なくとも1分間、好ましくは2分間以上、特に2〜40分間、特に2〜30分間圧力およびせん断の持続状態にさらすことが好ましい。共結晶を形成するために必要とされる時間の長さは、第一および第二の物質がさらされる圧力およびせん断の状態の厳正性に通常依存することはいうまでもないが、長期に渡る、持続してさらすことは通常、改善した共結晶形成に至ることを見出した。しかしながら、多くの場合、共結晶形成に必要とされる時間量に対して、せん断および圧力の条件下で費やされる過度な時間による可能なAPI分解の間のバランスがある。このようなバランスは、使用される物質および前記物質に課される状態に依存する最適化の事項である:このような最適化は通常当業者にとって一連の方法である。
【0027】
前記方法が、少なくとも20w/w%共結晶体、40w/w%共結晶体、より好ましくは少なくとも60w/w%共結晶体、特に共結晶の80w/w%共結晶体を含有する産出生成物を得るために適切であることが好ましい。本発明に記載の方法において、90w/w%共結晶体以上の共結晶の産出生成物を達成することができることができ、著しく高い割合の精製を共結晶の生成において示すことを見出した。
【0028】
従って、本発明の非常に好ましい実施形態において、前記方法は、90w/w%共結晶体以上、好ましくは95w/w%共結晶体以上、特に99w/w%共結晶体以上の産出生成物の形成に適切である。一般的に、それは比較的高い割合の収率を、透過時間および/または圧力およびせん断状態の強度を最適化することによって得ることができるようである。
【0029】
特定の実施形態において、圧力およびせん断を押出成形方法に適応した。驚くべきことに、押出成形の工程を使用して共結晶を得ることができ、それは上述のように、現在得ることが困難なものである。さらに押出成形は、共結晶の高収率且つ大量の生成方法を提供する。これは既存の共結晶化技術を超える非常に有意な利点を提供する。
【0030】
押出成形とは、圧力およびせん断を適応されている間に、細長いルーメンを通る物質の搬送を意味する;通常圧力およびせん断は、少なくとも部分的に、ルーメンを通る物質を搬送する手段によって適応される。押出成形は、物質が鋳型を通過して成形、または同様に押出成形工程の生成物を処理する工程を含むが、これは一般的に共結晶形成では必要でない。
【0031】
押出成形は、スクリューに基づく押出成形方法であることが一般的に好ましい。単一スクリューの押出成形はいくつかの実施形態において適用することができるが、この方法は、二つ以上のスクリューが前記第一および第二の物質の混合物と押出成形工程中に作用する場合に、スクリューに基づく押出成形方法であることが一般的に好ましい。前記方法は、混合や混合物を処理して、所望の共結晶化を得る多く過程で提供する。
【0032】
好ましい実施形態において、スクリューに基づく押出成形方法は、2軸押出成形方法である。2軸方法は、押出成形装置の複雑さを最小にする有効なバランスを提供するとともに、所望のように押出成形工程を処理する能力を提供する。当然、3つ以上のスクリューが相互に影響する押出成形方法を用いることは可能である−前記システムは、ポリマーの押出成形としてよく知られている。
【0033】
2軸押出成形方法が使用されるとき、それは共回転方法であることが、一般的に好ましい。しかしながら、いくつかの実施形態において、逆回転方法がいくつかの利点を提供することを見出すことができる。
【0034】
逆回転スクリューを、非常に高いせん断を必要とするとき使用し、それらは2つの逆回転スクリューの間の高い圧力およびせん断力を発生するためである。従って、逆回転スクリューは、非常に高いレベルのせん断および圧力が、共結晶を形成するのに好ましい場合、実用的である。しかしながら、逆回転スクリュー系が、空気の閉じ込め、低い最大スクリュー速度および産出物に関する課題を受けるおそれがある;これらは特定の応用において不利になるかもしれない。
【0035】
共回転システムは、良好なレベルの混合および物質の搬送を達成することができ、また高速で操作することができ、従って高出力速度を達成することができる。それらは、逆回転系より摩耗する傾向が低い。
【0036】
一つ以上のスクリューが存在するとき、スクリューが少なくとも実質的に噛合、好ましくは完全に噛合することが好ましい。一対のスクリューは、それぞれのスクリューの螺旋形のねじ部位のフライトチップが、実質的に他のスクリュー底に達したとき、完全に噛合であると考えることができる;通常機械的なクリアランスを提供する小さい隙間があるが、一般的に間隙を最小限に保つだろう。本用語を定量化するために、一対のスクリューは、一つのスクリューのフライトチップと他方の底までの間隙が、スクリューの底の全体の深さの10%以下、より好ましくは5%以下であるとき、実質的に噛合であることを示唆することができる。噛合系は、それらが自己ワイピングであり、系内における物質の局部的過熱を防ぐという利点を有する。
【0037】
当然、本発明の特定の実施形態において、非噛合システムを使用することが好ましいことを留意すべきである。非噛合システムは、大量の揮発性物質をシステムから取り除くのが望ましい場合や、または非常に高い粘着性物質がシステムに作用して容認できないほど高いレベルのトルクを生じる場合に使用することができる。
【0038】
本発明の方法において用いる、他の潜在的な種類の押出成形機は、再循環押出成形機である。リサイクル押出成形機は、通常2軸システムであり、バッチの原料をシステムから排出されるまでの所定の時間処理することができる。前記押出成形機、例えばHaake Minilabは、様々な用途に実用的であることができるが、比較的従来の再循環でない押出成形機ほど幅広く使用されていない。それらは一般的に従来の押出成形機のように一定比率増加に適切ではない、バッチシステムである。しかしながら、少量、即ち5gより低い量を処理するそれらの性能は、それらがいくつかの製薬研究に適切であるとする。
【0039】
前記方法は共結晶、特にAPIおよび共結晶形成剤または“ゲスト”物質(それ自身がAPIであることができる)形成することができる第一および第二の物質で単に実施することが一般的に好ましい。2つを超える物質が、共結晶を形成するために共に働くことができ、従ってさらなる共結晶を形成する物質が存在することができることが可能な点に留意する必要がある。従って本発明の方法を、あらゆる結晶形成しない物質、例えば溶媒または潤滑剤の非存在下にて実質的に実施することが好ましい。共結晶を溶媒または他の付加的な物質がない場合で形成することができることが、本発明の方法の有意な利点である。製造後の前記付加的な物質の除去は、困難または不可能であり、前記物質の存在は、低レベルでさえ、十分に安全関連または少なくとも調整のハードルであることができる。当然、例えば、潤滑剤または溶媒の存在が望ましいかもしれない状況であることができることを理解され、本発明の方法が前述の添加剤の包含物と互換性があるが、それらが前記方法から省略することができることは、有意な利点である。
【0040】
誤解を避けるために、共結晶を形成に関与している物質は、結晶構造を形成できる必要があることを言及すべきである。従って、非結晶性または“半結晶性”構造を形成するポリマーは、本発明の工程における共結晶形成で適切ではない。
【0041】
前記第一および第二の物質の混合物を、付加的な熱にさらすことが望ましい。“付加的な熱”とは、混合物がそれに適用され、周囲温度を越え、押出成形工程の間の摩擦によって発生される熱を越える熱であることを意味する。
【0042】
特定の実施例において、前記工程は、工程の少なく一部の期間で、最も低い融点を有する共結晶を形成する物質の融点付近の温度で実施することが好ましい。一般的に、前記温度は、最も低い融点を有する共結晶を形成する物質の融点より若干低いが、融点または融点より若干高くすることが好ましい。好ましい実施形態において、前記温度は融点の20℃の範囲内、好ましくは融点の10℃の範囲であることができる。前記成分のうちの1つが共融混合物である場合、関連する融点はその混合の融点である。前述の温度が使われる場合、共結晶形成に関して利点があることが分かっていた。
【0043】
所望の性質および収率の共結晶を得るため、押出成形工程時に必要とされるせん断、圧力のおよび滞留時間の条件に依存して、(単数の)スクリューまたは(複数の)スクリューの形状を変更することができる。一般的に2軸または他の多数のスクリューの配置が、形状の変更に比較的従うが、それは狭い範囲にて単軸押出成形機が可能である。押出成形の装置または工程の以下の態様に変更することが可能である:特にバレルの長さ、長さの比:バレルの直径(L/Dの比率)、スクリュー成分の組成物(例えば、分散または分布の混合成分、前進または後進の供給成分、スクリューの谷底の深さ(即ち、ネジ山の深さ)スクリュー回転速度、供給方法(スターベーション供給対フラッド供給)、押出成形機を通過する数である。これらの態様は、押出成形工程にわたる高度な調節、および結果として生じる共結晶生成物を可能とする。
【0044】
押出成形時に、L/Dの比率が15/1以上(即ち、長さはスクリューの直径の15×以上)であることが好ましいことが分かった。好ましくは、L/D比率は20/1以上、いくつかの実施形態において30/1以上の比率が好ましい。40/1のL/D比率が、共結晶の形成に適切であることを見出した。前記比率は、特に2軸システムに適用するが、同様に他の押出成形システムにも適用することができる。
【0045】
押出成形最中に混合物を、分布または分散混合に少なくとも一定期間さらすことが好ましい。混合物を分散混合物に少なくとも一定期間さらすことが一般的に好ましい;分散混合物は、せん断、圧力および熱発生に関して比較的強力であり、従って共結晶の形成を促進する際、しばしば実用的である。一般的に混合物を、それぞれ分布および分散の混合物のそれぞれに少なくとも一定の期間さらすことが最も好ましい。
【0046】
押出成形機のスクリュー、特に2軸または他の多数のスクリュー押出成形機は、多くの異なる部材を含み、物質が押出成形時に受ける条件を決めることができる。前記部材は、それらが連続らせん形のねじ山を含むことができない点で必ずしも最も厳密な意味で“スクリュー”であるのではないが、スクリューという用語を、組成物を問わず全体として組立物に関して用いる。一般的に、スクリューの長さの重要な部分は、らせん形のねじ山を含み、通常その長さの半分以上はらせん形のねじ山を含むであろう。
【0047】
スクリューを形成する部材を、通常、シャフト上に組み立て、完全なスクリューを形成する。シャフトは、通常、シャフトに関連する部材の回転を防ぐ横断面、例えば多角形、多くの場合六角形である。それぞれの部材は、通常スクリューの全長に関して非常に短い。押出成形機のスクリューの直径の比率に関して、部材の長さを述べることが最も適切である。
【0048】
らせん形のスクリュー部材を使用して、押出成形機による物質を搬送し、それらは比較的低レベルの混合、並びに圧力およびせん断の適応を与える。前記らせん形部材によって適用される圧力およびせん断のレベルを、例えば前記らせん形部材を多数のスクリュー押出成形機で変えたり、前記部材の深さおよび/またはピッチを変えたりすることによって変更することができる。異なったらせん形のスクリューの種類、例えば前進搬送部材、排出部材または反対のスクリュー部材が存在することができる。
【0049】
比較的激しい混合、せん断および圧力の適応が必要である場合、これは部材の組み合わせ、特に混合パドルを使用して達成することができる。混合パドルは通常、丸い突出部材(lobed element)、例えば楕円または類似した形状の部材を含み、らせん型のねじ山は含まない。パドルは曲線且つ平坦な混合表面を設ける。2軸押出成形機において、一つ以上の対応する対の丸い突出部材を、それぞれのスクリューに設けることができる。1本のスクリューでの丸い突出部材を、それが他方のスクリューでの丸い突出部材に対して回転方向にオフセット、通常二つの丸い突出部に90°(即ち、一般的に楕円)のパドルであり、例えば部材を回転して、突出部材の混合表面を狭い溝に分離して、パドル対に相応する形状による回転時に、実質的に一定のままであることができるか、または回転時に多少程度変化する可能性があるように配置した。オフセットの異なった程度は、三つの丸い突出部材、または混合部材の他の形状で適切に使用することができる。前記混合パドルの効果は、混合物をパドル対の間に塗布し、従って比較的強度の混合で、高いせん断および圧力を受ける。さらに、混合表面の平坦な性質は、前進搬送が、強く促進されずに、それ自体、混合物がそのような部材に滞留する傾向があることを意味する;混合物の前進搬送が、上流搬送部材によって強制される上流混合物によって動作した圧力で主に促進するが、下記のように、混合部材の特定の構成は、前進搬送の度合いを提供することができる。
【0050】
混合、並びにせん断および圧力の適応の度合いは、混合部材の数および形状によって定めることができる。分布混合は、押出成形の技術において知られている用語であり、“分布混合は、優れた空間分布を達成するために、微量成分をマトリックスにわたって分布する工程である”として規定することができる。分布混合は、混合部材(例えば、突出部材)の一連の対を、混合部材のそれぞれの対は、前記対に対して回転方向にオフセット、即ちスタガー角である場合に達成することができる。一般的に、次の混合部材は、前進搬送を提供するらせん部の方向と同じ方向でオフセットする。通常、それぞれの混合部材(例えば、突出部材)の長さは、スクリューの直径の0.25×まで、好ましくはスクリューの直径の少なくとも0.125×までである;例えば、スクリューの直径16mmに関しては、それぞれの部材は4mmの長さであることができる。分布混合は、物質の混合物の流路を再配置することによって主に混合することを考慮することができる;本質的に、それぞれの混合物部材の相関的に短い長さは、混合物が混合部材の間にて撹拌され、高く強制的に塗布されるもののレベルは比較的低いことを意味する。回転方向のオフセット量は、分布混合配列を提供するように搬送量、およびある程度混合の重要性を決定する。一対が供給スクリューでのらせんと同じ方向に約10°から45°(通常30°)までで、前記対からオフセットする場合、前進搬送の十分な度合いを提供し;約46°から65°(通常60°)のオフセットは、いくらか少ない搬送を提供し;約75°から約90°のオフセットは、著しく少ない搬送を提供する−90°のオフセットは本来混合物の搬送を全く提供しない。
【0051】
分散混合は、混合の強度の態様であり、せん断および圧力の高いレベルを混合物に与える。分散混合は、押出成形の技術において知られる用語であり、“分散混合は結合力のある微量成分、例えば固体粒子の集合体または液体の液滴の大きさにおける減少を含む”として規定することができる。分散混合を、混合物が圧縮されて、混合部材の混合表面の間を塗付する細長い混合部位を通過して圧入されたときに、達成することができる。分散混合は一つ以上の混合部材、例えば二つの突出部材で提供して、混合表面の細長い部位をあらゆる回転方向にオフセットなしに提供することができる;即ち、混合表面の細長い部位を、比較的長い混合部材(2軸システムのそれぞれのスクリューの一つ)の対と、実質的に回転方向に全くオフセットすることなく提供することができるか、または次の部材の間で実質的に回転方向にオフセットが全くない、複数の一連の比較的短い混合部材であることができる。例えば、回転方向にオフセットを全く有さない突出部混合部材を含むスクリューの直径の0.5×またはそれ以上の長さの部位を、分散混合物に提供するであろう。都合よく、分散混合ゾーンは、互いにはオフセットしてない二つ以上の突出部材を含むことができ、即ちそれらは実質的に継続した混合表面を提供する。本質的には、分散混合の有意な態様は、混合物の少なくとも一部が、混合物を塗付して、圧力およびせん断の高い度合いを適応する混合部材の通過を制限する−これは上述のような混合部材を使用して達成することができる。
【0052】
しかしながら、上記分布および分散混合システムは、本発明の使用に対して好ましいシステムを示す点に留意する必要がある。分布または分散の混合を達成する他の方法を、当業者は想到することができる。分散および分布の混合の提案を、Rheology Bulletin Vol.66,No.1(January1997)−Analysis of Mixing in Polymer Processing Equipment by lea Manas−Zloczowerで提供する。
【0053】
本方法にて使用される押出成形装置が、分散混合領域(即ち、混合部材を含有する部位)を、スクリューの全長の少なくとも1/40、好ましくは少なくとも1/30、より好ましくはスクリューの全長の少なくとも1/20を有することが好ましい。好ましくは、分散混合の少なくとも一部があり、その部位は少なくとも0.5倍の直径の長さである。より好ましくは、分散混合の少なくとも一部位があり、分散混合の全ての全長は、少なくとも直径の1.5倍以上、好ましくは直径の2倍以上である。
【0054】
本発明の実施形態において、混合および分散混合の部の両方、即ち混合部材を含有する部を有することが好ましい。好ましい実施形態において、構造は少なくとも一部を分布混合した後、少なくとも一部を分散混合することを含む。本発明の好ましい実施形態において、分布混合の少なくとも二つの領域、および分布混合の少なくとも二つの領域を提供する。分布混合部のそれぞれが少なくとも直径の長さの1倍、より好ましくは直径の長さの1.5倍、そしてそれらは直径の長さの2倍以上であることが好ましい。それぞれの分散混合の部位が直径の長さの少なくとも0.5倍であり、それらが直径の長さ1倍以上、それらが直径の長さの1.5倍以上にすることができる。一般的に混合部の合計5倍以上の長さ、より好ましくは混合部の直径の長さの10倍以上であることが好ましい。
【0055】
一般的にスクリューの全長の半分以下の押出成形システムが、混合部材を含有することが好ましい。一般的にスクリューの全長の2/5以下または1/4以下が、混合部材を含有する。当然、実際の割合は、ネジの全長に依存して変更することができ、全長の半分を超える場合は混合部材含む状況を想定することができる。
【0056】
上記のように、第一および第二の物質は共結晶を形成する必要がある。共結晶の形成を予測するために利用できる確かな規則は全くない。しかしながら、一般的に第一および第二の物質が、水素結合形成で顕著な可能性を伴う優遇の群を有するべきである。一般的に所望の部類に分類される物質は、カルボン酸、アミン、アミド、スルホンアミド、ヒドロキシルアルコール、ケトン、アミノ酸、糖類および複素環式塩基を含み、従ってこのような物質、すなわち適切な活性基を含有する物質は本方法における使用に好ましい。一般的に、特定のAPIの共結晶を形成することが好ましく、適切なゲストまたは共結晶形成のためコフォーマー(co−former)を選択することが必要であるだろう;即ち、ゲストを変更する自由度があるが、通常APIはない。適切なゲストを同定することは、一般的にある程度の試行錯誤を伴うが、APIの化学作用の知識によってしばしば導かれる。適切なゲストを同定することは、一般的にある程度の試行錯誤を伴うが、APIの化学作用についての知識によってしばしば導かれる。しかしながら、一般的に、カルボン酸、アミドおよび複素環式塩基を含むAPIおよびゲストが、本発明の使用に好ましい。
【0057】
適切なAPIの例を、下記の表1に記載する。
【0058】
【表1】

【0059】
適切なゲストの例を、下記の表2に記載する。
【0060】
【表2】
【0061】
本発明の実施形態において、第一および第二の物質が化学量論的比率で形成されることが好ましい。その比率は1:1であることができ、またはそれは他の整数比率、例えば1:2、2:1等、であることができる。
【0062】
本発明の実施形態において、第一物質はフェニルアルカン酸である。好ましくは、第一物質はイブプロフェンであり、第二物質はニコチンアミドである。適切な、イブプロフェンとニコチンアミドの重量比は、約41.2:26、即ち1:1モル比である。
【0063】
本発明の他の実施形態において、第一物質はカルバマゼピン、第二物質はサッカリンである。カルバマゼピンとサッカリンの適切な重量比は、約47:37、即ち1:1モル比率である。
【0064】
本発明の他の実施形態において、第一物質はカルバマゼピン、第二物質はニコチンアミドである。カルバマゼピンとニコチンアミドの適切な重量比は、約118:61、即ち1:1モル比である。
【0065】
本発明の他の実施形態において、第一物質はカフェイン、第二物質はマレイン酸である。カフェインとマレイン酸の適切な重量比は、約194:116、即ち1:1モル比、または97:29、即ち2:1モル比である。
【0066】
本発明の他の実施形態において、第一物質はテオフィリン、第二物質はマレイン酸である。テオフィリンとマレイン酸の適切な重量比は、45:29、即ち1:1モル比である。
【0067】
本発明の他の実施形態において、第一物質はサリチル酸、第二物質はニコチンアミドである。サリチル酸とニコチンアミドの適切な重量比は、69:61、即ち1:1モル比である。
【0068】
押出成形工程の生成物が一般的に共結晶の凝集物を含む粒子であることが、本発明の注目に値する態様である。前記粒子は、一般的にそれらの最大の大きさで2から3000μmの大きさであり、錠剤または他の単位用量形状に直接圧縮されることに非常に適切である。さらに、凝集物は、水および胃の生体外モデルの両方に容易に溶解し、薬物送達において極めて実用的であることを示した。
【0069】
従って、更なる態様において、本発明は、上で述べられる方法を含む、凝集された共結晶を含有する前記粒子の形成方法を提供する。
【0070】
別の態様において、本発明は、凝集された共結晶の粒子、好ましくは直径2から3000μmを形成するものを提供する。好ましくは、粒子は、少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含有し、それらは90w/w%共結晶体以上を含むことができる。
【0071】
任意に、前記方法は改質剤化合物を押出成形工程に導入する段階を含むことができる。適切な改質剤は、密度改質剤(ラクトース、微結晶性セルロース等)、結合剤(澱粉、セルロース、ポリビニルピロリドン等)、崩壊剤(ナトリウム澱粉グリコール酸塩、架橋ポリビニルピロリドン)および湿潤剤を包含する。前記改質剤を、共結晶化を実質的に完了した後、押出成形工程に、適切に導入することができる。このように、改質剤を、工程の産出生成物内に取り込むが、共結晶化工程を妨害しない。例えば、改質剤を、分散混合の全ての領域の下流に導入することができる。
【0072】
更なる実施形態において、上述のような方法を実施して凝集された共結晶の粒子を形成し、任意に適切なモールドで前記粒子を圧縮して、単位用量形状を形成する工程を含む活性薬剤の単位用量形状を形成する方法を提供する。適切には、単位用量は錠剤または同様のものである。任意に、前記方法は、製薬学的に許容される賦形剤を提供することを含むことができる。適切な賦形剤は、改質剤として上で述べられた化合物を包含することができる。
【0073】
他の態様において、本発明は、共結晶を含む組成物を付与し、前記共結晶は第一および第二の物質を含み、第一および第二の物質を上記のような工程に露出した。
【0074】
一実施形態において、本発明は、フェニルアルカン酸およびニコチンアミド、好ましくはイブプロフェンおよびニコチンアミドの共結晶を含む組成物を提供する。共結晶の存在は、3.2°2−θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は、少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体、特に少なくとも90w/w%共結晶体を含む。
【0075】
一実施形態において、本発明は、カルバマゼピンおよびサッカリンの共結晶を含む生成物を提供する。共結晶の存在は、7°2−θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は、少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0076】
一実施形態において、本発明は、カルバマゼピンおよびニコチンアミドの共結晶を含む生成物を提供する。共結晶の存在は、20.4 2θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0077】
一実施形態において、本発明は、カフェインとマレイン酸を1:1のモル比にて含有する生成物を提供する。共結晶の存在は、9、11.1、13.2、14.2、15.5および13.2 2θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0078】
一実施形態において、本発明は、カフェインとマレイン酸を2:1のモル比にて含有する生成物を提供する。共結晶の存在は、8.8、10.1、13.5および16 2θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0079】
一実施形態において、本発明は、テオフィリンおよびマレイン酸の共結晶を含む生成物を提供する。共結晶の存在は、9、11.5、13.6および16.8 2θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は、少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0080】
一実施形態において、本発明は、サリチル酸およびニコチンアミドの共結晶を含む生成物を提供する。共結晶の存在は、7.8、8.4、および9.1 2θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0081】
さらなる態様において、本発明は、上記の工程から得られるか、または得ることができる共結晶を含む組成物を提供する。
【0082】
さらなる態様において、本発明は、上記の共結晶を含有する医薬品、任意に製薬学的に許容される賦形剤と組み合わせたものを提供する。医薬品は、投与のためのあらゆる適切な形状、例えば、顆粒剤、錠剤、カプセル等であることができる。
【0083】
さらなる態様において、本発明は、痛みの緩和または炎症状態の治療のためのイブプロフェンおよびニコチンアミドの共結晶を含む組成物を提供する。炎症状態の例は、外傷によって誘導された炎症および自己免疫状態であり、例えば慢性関節リウマチ、紅斑性狼瘡、重症筋無力症、天疱瘡、シェーグレン症候群、強直性脊椎炎、炎症性腸疾患等である。
適切な共結晶のさらなる詳細を、上に記載する。
【0084】
さらなる態様において、本発明は、カルバマゼピンおよびサッカリンまたはカルバマゼピンおよびニコチンアミドの共結晶を含有する組成物を、心因性障害、例えば癲癇、躁うつ病注意欠陥多動性障害(ADD)または注意欠陥多動性障害(ADHD)、精神分裂症、幻肢症候群および三叉神経痛の治療のために提供する。適切な共結晶のさらなる詳細を、上に記載する。
【0085】
さらなる態様において、本発明は、カフェインおよびマレイン酸の共結晶を含有する組成物を、呼吸器系の中枢神経系の疾患の治療のために提供する。
【0086】
さらなる態様において、本発明は、テオフィリンおよびマレイン酸の共結晶を含有する組成物を、気管の慢性閉塞性肺疾患(例えば、COPD)、喘息(特に気管支)または無呼吸発作(特に小児無呼吸発作)の治療のために提供する。
【0087】
さらなる態様において、本発明は、サリチル酸およびニコチンアミドの共結晶を含有する組成物を、痛みの減少、熱の緩和、またはざ瘡、乾癬、皮膚硬結、うおのめ、毛孔性角化症および疣(例えば局所投与による)の治療の使用で提供する。
【0088】
さらなる態様において、本発明は上で述べられる方法に従って得ることができるか、または得られた凝集した共結晶を含む粒子を提供する。前記粒子は、溶解および凝集形態に関して好ましい性質を有する。
【0089】
さらなる態様において、本発明は、治療において使用するために上記の方法のような共結晶を含有する組成物を提供する。本発明は、同様に上記のように共結晶を含有する組成物の使用を、薬剤の製造において、一つ以上の上記病状の治療にて使用するために提供する。
【0090】
本発明の実施形態を、例としてのみに、添付の図を参照にしてここで記載する:
【図面の簡単な説明】
【0091】
【図1】2軸押出成形機のスクリュー部材の例を示す図である。
【図2】イブプロフェンおよびニコチンアミドを含有する物理的混合のPXRDパターンを示す図である。
【図3】選択的な供給および分布の混合ゾーンで15:1スクリュー構成を使用して押出成形後に、得られたイブプロフェンおよびニコチンアミドの共結晶のPXRDパターンを示す図である。
【図4】選択的な供給および分布の混合ゾーンで40:1スクリュー構成を使用して押出成形後に、得られたイブプロフェンおよびニコチンアミドの共結晶のPXRDパターンを示す図である。
【図5】供給、分布および分散の混合ゾーンで40:1スクリュー構成を使用して押出成形後に、得られたイブプロフェンおよびニコチンアミドの共結晶のPXRDパターンを示す図である。
【図6】イブプロフェンおよびニコチンアミドの単一の共結晶を使用して、得られたPXRDパターンを示す図である。図6a〜dは、凝集されたイブプロフェンおよびニコチンアミド共結晶のSEMイメージを、それぞれ350×、1100×、1800×および3500×の倍率にて示す図である。
【図7】供給、分布および分散のゾーンで40:1スクリュー構造を使用して押出成形後に、得られたカルバマゼピンおよびサッカリンの共結晶のPXRDパターンを示す図である。
【図8】図8a〜dは、構成A、BおよびCにおいて使用したような2軸部材の4種類の写真を示す図である:a)搬送構造b)交互(即ち、分布)混合構造c)分散混合構造d)放出構造。
【図9】図9a〜9dは、カルバマゼピンおよびサッカリンのそれぞれ構成A、BまたはCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す図である(全てのPXRD結果をグラフで示し、X軸は2−θおよびY軸はカウントを示す)。図9e〜9hは、構成Cを使用して生成した凝集したカルバマゼピンおよびサッカリン共結晶の600×、600×、1000×および1800×にそれぞれ拡大したSEMイメージを示す図である。
【図10】図10a〜10dは、カルバマゼピンおよびニコチンアミドのそれぞれ構成A、BまたはCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す図である。図10eは、構成Cを1000×倍率を使用して製造した凝集カルバマゼピンおよびニコチンアミド共結晶のSEMイメージを示す図である。
【図11】図11a〜11cは、カフェインおよびマレイン酸(モル比1:1)のそれぞれ構成A、BおよびCを使用して、押出成型後のPXRD結果のグラフを示す図である。図11d〜11gは、構成Cをそれぞれ137×、550×、600×および820×倍率を使用して生成した凝集カフェインとマレイン酸の共結晶(モル比1:1)のSEMイメージを示す図である。
【図12】図12a〜12cは、カフェインとマレイン酸(モル比2:1)のそれぞれの構成A、BまたはCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す図である。図12dおよび12eは、構成Cのそれぞれ800および2000×倍率を使用して生成した凝集したカフェインおよびマレイン酸共結晶(モル比2:1)のSEMイメージを示す図である。
【図13】図13a〜13dは、テオフィリンおよびマレイン酸のそれぞれ構成A、BおよびCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す図である。図13eは、構成Cの倍率500×を使用して製造した凝集テオフィリンおよびマレイン酸共結晶のSEMイメージを示す図である。
【図14】図14a〜14cは、サリチル酸およびニコチンアミドのそれぞれ構成A、BまたはCを使用して、押出成形後のPXRD結果のグラフを示す図である。図14dおよび14eは、構成Cを使用して生成した凝集サリチル酸−ニコチンアミド共結晶のそれぞれ200×および800×倍率におけるSEMイメージを示す図である。
【図15a】図15aは、押出成形装置自体の表示に対するスクリュー構成A、BおよびCの概略図を示す図である。
【図15b】図15bは、押出成形装置自体の表示に対するスクリュー構成A、BおよびCの概略図を示す図である。
【発明を実施するための形態】
【0092】
押出成形−背景
押出成形は、開口部または鋳型によって物質を圧入することによって生成物を形成する方法として規定することができる。本方法は、溶解物の押出成形の場合、加熱バレルにて回転しているアルキメデスのねじの作用によって、継続的な方式で通常実施される。ポリマーに関して、溶解は、バレル壁およびポリマーの粘性のせん断により、伝導加温の二重作用によって達成される。押出成形機の最も単純で広く使用される形態は、単スクリューを使用するものであり、それは一般的に、溶融物質の溶解および測定することを達成するために、単純な単一のフライトデザインである。
【0093】
二軸押出成形機(TSE)は、単軸押出成形機の低い混合性能を克服するために、二軸を使用して開発され、通常並べて、同じ(共回転)または逆(異方向)方向に回転するように配置された。スクリューは、通常、物理的な間隔を除いて、密接または完全に噛み合う、即ち、それぞれのスクリューのフライトの先端は、対向するスクリューの底に達するように構成されている。これは、高度な混合を、二つのスクリューの間で“噛合”領域において可能とする。TSEは、粘性抵抗流体に依存することよりむしろ強制的搬送によって作動し、スクリューの自己ワイピング動作は、押出成形機をより衛生的にして、単スクリュー構造より少ない沈滞である。TSEスクリューは、通常、交換可能なスクリュー部材が配置される六角形のシャフトからなる。これはスクリュー構造で高度な柔軟性を可能とし、応用では、搬送、混練、混合および放出の混合を提供するように容易に構成することができる。TSEは通常、少量供給(starve−fed)され、不完全に満たされたチャネルを実施した。
【0094】
逆回転押出成形機は、押出成形機内の物質移動のため、低い度合いの混合であるが、高い物質供給および搬送の特性を有する。それぞれのスクリューのフライトが他のスクリューのチャネルに適合し、完全に満たされる場合、物質をスクリューで回転するのを完全に妨げられ、従って軸方向に積極的に移動する。前記移動は、物質の粘度およびバレルおよびスクリューの金属面への粘着に独立している。逆回転TSEの滞留時間および溶解温度は、非常に一定である。スクリューの間の物質には、高いせん断力を受け、高圧の進展が生じ、従って逆回転TSEは、スクリューの間で生じる高圧による共回転より、低いスクリュー速度にて作動する。逆回転TSEの典型的重合例は、熱分解に感度が高く、低い滞留時間、例えばPVCおよび木材複合体ポリマーを要求する物質を包含する。
【0095】
共回転押出成形機は、TSEの中で最も産業的に重要な種類であり、密接または完全に噛合するスクリュー構造を有する傾向がある。スクリュー部材は自己ワイピングであり、高いスクリューの速度および処理能力が、本構造によって可能である。共回転TSEは、物質を長手方向並びに横断方向に混合する能力を有するため、物質がスクリューの1つのチャンバから他のチャンバまで輸送され、そして優れた混合および混合物内に高いエネルギー入力を生じる。共回転スクリューは、半回転システムと比較して、高度な柔軟性を提供する。典型的な構成は、搬送、混練および混合部材を含有する。バリア部材を使用して、高低の圧力の溶融封止および領域を設け、液体の注射または揮発性物質の除去を可能にする。共回転TSEの一般的な適用は、重合樹脂が広範囲にわたる補強用充填剤および添加剤と混合する場合に、ほとんどの樹脂混合の実施を含む。混合および反応性の押出成形品は、同様に応用で幅広く使われている。共回転TSEからの押出成形品は、一般的に次の形成工程で使用して小球形にされる;TSEのみでは、製造にて、発生する低いヘッド圧力および固有の変動による生成物の製造で特に適切でない。
【実施例】
【0096】
二軸押出成形による共結晶形成−実験方法
器材
2つの共回転二軸押出成形機を、共結晶の形成に使用し、双方ともスクリュー直径16mmである。第一は、スクリューの長さと直径(LD)の比率15:1(Thermo Prism TSE16TC)を有し、3つの温度制御バレルゾーンおよび1つの鋳型ゾーンを組み込む短い押出成形機である。LDの比率40:1(Thermo Prism Eurolab)を有する長い押出成形機を同様に使用して、合計10の温度制御バレルおよび鋳型ゾーンを組み込む。スクリュー構造と組み合わせた押出成形機の長さは、押出成形時の可能な残余時間および混合程度を決定する。
【0097】
一つのスクリュー構成を、搬送部材および一つの分布混合ゾーンからなる単純なスクリュー構造を有して、15:1LD押出成形機に使用した。二つのスクリュー構成を、40:1LD押出成形機に使用し、一つは搬送部材および三つは主に分布混合部位である。第2の構成は、分布および分散混合ゾーンおよび逆搬送部材の比較的複雑な組合せを使用した。これは高い滞在時間および厳格な混合環境を提供した。表3a〜3cは、使用される3つのスクリュー構成を集約し、主要な種類のスクリュー部材の例を示す写真を図1に示す。
【0098】
実験手順
洗浄した押出成形機を、選択された処理温度まで予熱した。バレル温度プロフィールの範囲を使用し、通常は冷却された供給ゾーンから最大の中央位置点までバレルに沿って増加し、鋳型の端部に向かって減少する。前記試験のために、押出成形機を鋳型なしに実施した。押出成形機のスクリュー回転速度を設定した;広範囲の速度を達成することができ、押出成形機とともに最高200回転/分(rpm)までを本明細書で使用した。一般的なスクリュー回転速度を、20から50rpmの間に設定した。その後活性薬剤およびコフォーマーの予備混合した混合物を、押出成形機のフィードホッパに導入した。手動投与することは、小さいバッチ容積(通常10〜30gの間)にて便利であることを証明することができた。大きいバッチ容積に関しては、重量系供給システムを比較的便利に使用することができる。その後薬物およびコフォーマーの押出混合物を、スクリューの出口にて、構成部材および設定された作動状態によって、粉末、粘着性の塊または溶解形態で収集した。収集した物質を、その後共結晶形成で分析した。
【0099】
実験の方針の間、以下のパラメータを調整することができる:
設定温度
スクリュー回転速度
処理量
スクリュー構造(即ち、分布および分散的な混合の度合い)
押出成形機による通過数
【0100】
表3a−スクリュー部材の概略図、構成1。
【表3a】
【0101】
表3b−スクリュー部材の概略図、構成2。
【表3b】
【0102】
表3c−スクリュー部材の概略図、構成3。
【表3c】
【0103】
実施例1
イブプロフェンおよびニコチンアミドの物理的な混合物を、41.2gのイブプロフェンおよび26gのニコチンアミド(モル比1:1)を撹拌混合機で30分間混合することによって調製した。LD比率15:1およびスクリュー直径16mm(Thermo Prism TSE 16TC)を有する押出成形機を用いた。これはスクリュー構成1を組み込み、主に前進供給部材および小さい分布混合ゾーンからなる。詳細なスクリュー構成を、上に示した。バレル温度を80℃に設定した。一旦温度が15分間安定したとき、物理的混合物を押出成形機にゆっくりと供給して、スクリューを20rpmにて回転した。微細な凝集生成物を、押出成形機の出口にて収集した。押出成形機による物質の残在時間は、約3分であった。粉末を室温まで冷却し、その後粉末X線回折測定(PXRD)評価の対象とした。
【0104】
図2は、イブプロフェン−ニコチンアミドを含有する物理的混合のPXRDパターンを示す。イブプロフェンの特徴的なピークは、6°2θにて観測することができる。
【0105】
図3は供給および単一の分布混合のゾーンで15:1スクリュー構成を使用して、押出成形後に得られた共結晶のPXRDパターンを示す。3.2°2θで共結晶の特徴的ピークが、共結晶が押出成形工程時に形成されることを明らかに示すものを観測した。PXRDパターンは、同様にイブプロフェン結晶の特徴的なピークの出現を6°2θにて示した。これはイブプロフェンの部分が共結晶形状に変換されなかったことを示す。
【0106】
実施例2
イブプロフェンおよびニコチンアミドの物理的混合物を、41.2gイブプロフェンおよび26gニコチンアミド(モル比1:1)を撹拌混合機で30分間混合することによって調製した。LD比率40:1およびスクリュー直径16mmを有する押出成形機を使用した(Thermo Prism Eurolab)。これはスクリュー構成2を組み込み、供給および分布混合のゾーンからなる。詳細なスクリュー構成を、上に示した。バレル温度を80℃に設定した。一旦温度が15分間安定したとき、物理的混合物を押出成形機にゆっくりと供給して、スクリューを20rpmにて回転した。押出成形機による物質の残在時間は、約20分であった。微細な凝集生成物を、押出成形機の出口にて収集した。粉末を室温まで冷却し、その後粉末X線回折測定(PXRD)評価の対象とした。
【0107】
上記のように、図2はイブプロフェン−ニコチンアミドを含有する物理的混合物のPXRDパターンを示す。イブプロフェンの特徴的なピークは、6°2θにて観測することができる。
【0108】
図4は、上記のように、代替供給および分布混合のゾーンを有する40:1スクリュー構成を使用して押出成形後に得られた共結晶のPXRDパターンを示す。それは共結晶の特徴的なピークの出現を、3.2°2θにて示した。これは共結晶が押出成形工程時に形成されることを明らかに示す。PXRDパターンは、同様にイブプロフェン結晶の小さい特徴的なピークを、6°2θにて示した。PXRDパターンを適切な算出方法を使用して分析し、混合物が約72%共結晶を含有することを示した。
【0109】
実施例3
イブプロフェンおよびニコチンアミドの物理的混合物を、41.2gイブプロフェンおよび26gニコチンアミド(モル比1:1)を、撹拌混合機で30分間混合することによって調整した。LD比率40:1(Thermo Prism Eurolab)、ならびに最後の供給ゾーン(構成3)より前に逆供給スクリュー領域を有する交互供給、分布および分散混合のスクリューゾーンを有する押出成形機を、80℃に設定した。一旦温度が15分間安定したとき、物理的混合物を押出成形機にゆっくりと供給して、スクリューを20rpmにて回転した。押出成形機による物質の残在時間は、約33分であった。微細な凝集生成物を、エンドゾーンにて収集した。粉末を室温まで冷却し、その後粉末X線回折測定(PXRD)評価の対象とした。
【0110】
上記のように、図2は、イブプロフェン−ニコチンアミドを含む物理的混合のPXRDパターンを示す。イブプロフェンの特徴的なピークは、6°2θにて観測することができる。
【0111】
図5は、40:1スクリュー構成3を使用して、押出成形後得られた共結晶のPXRDパターンを示す。それは共結晶の特徴的なピークの出現を、3.2°2θにて示す。これは共結晶が押出成形工程時に形成されたことを明らかに示した。PXRDは、イブプロフェン結晶の特徴的なピークを6°2θにて示さなかった。適切な算出方法によって分析したPXRDパターンは、約94%共結晶含量を示した。
【0112】
図6は、イブプロフェン−ニコチンアミドの単一共結晶を使用して得られたPXRDパターンを示す(これを比較として溶媒技術によって生成した)。
【0113】
図6aから6dは、スクリュー構成3を使用して生成したイブプロフェン−ニコチンアミドから形成した共結晶の凝集物の走査型電子顕微鏡写真(SEM)を示す(350×、1100×、1800×および3500×倍率)。
【0114】
実施例4
カルバマゼピンおよびサッカリンの物理的混合物を、47gカルバマゼピンおよび37gサッカリン(モル比1:1)を、撹拌混合機で30分間混合して調製した。押出成形はスクリュー構成3を組み込んだTSEにて実施し、供給、分布および分散混合のゾーンからなる。詳細なスクリュー構成を上に示した。バレル温度を140℃に設定した。一旦温度が15分間安定したとき、物理的混合物を押出成形機にゆっくりと供給し、スクリューを20rpmにて回転した。押出成形機による物質の残在時間は、約33分であった。微細な凝集生成物をエンドゾーンにて収集した。粉末を室温まで冷却し、その後粉末X線回折測定(PXRD)評価の対象とした。
【0115】
図7は、40:1スクリュー構成3を使用して、押出成形後に得られた共結晶のPXRDパターンを示す。それは、カルバマゼピン−サッカリン共結晶の特徴的なピークの出現を7°2θにて示した。これは共結晶が押出成形工程時に形成されたことを明らかに示す。
【0116】
二軸スクリュー押出成形機による共結晶の形成−更なる実験方法
更なる実験を、共結晶収率に関するせん断、混合および滞留時間の効果を評価するために実施した。
【0117】
実施例5〜10の全ての実験を、スクリュー直径16mm、長さと直径(L:D)の比率40:1を伴うThermo Pharmalab HME16共回転二軸押出成形機で実施した。
【0118】
三軸構成を(概略的に図15aおよび15bに示す)使用して、共結晶収率に関するせん断、混合および滞留時間の効果を評価する。スクリュー構成を以後A、BおよびCとして、それぞれ混合のレベルを低、中および高として示す。構成を以下にさらに詳細に記載する:
【0119】
構成A
これは混合強度の最小レベルを提供し、スクリュー先端にて計測部材を有する単なる前進搬送部材からなる。この構成は、下記の表に示すように要約することができる:
【0120】
表4:スクリュー構成A、供給から排出までを整理した。
【表4】
【0121】
構成B
これは、分布混合の中レベルを提供し、従来のポリマー合成(混合)実施において使用されるスクリュー構成の一般的な種類である。分布混合(流路を再配置することによって混合)を、前記部材から特定の角度−30、60または90°で配置で、長さD/4の二つの突出物の一連の混合パドルを使用して、本明細書にて達成した。前記パドルを、前進搬送方向に配置し、即ち30°混合パドルは最も前進する搬送、60°はより少なく、90°は0搬送活動および単なる混合を提供する。この構成は、下記の表に示すように要約することができる:
【0122】
表5:スクリュー構成B、供給から排出までを整理した。
【表5】
【0123】
構成C
これは高い度合いの分布および分散の混合を提供する。分散混合(凝集物を分解するための高いせん断作用)を、混合パドルの対を同じ方向に共に配置、即ちスタガー角によって達成した。これは、多くの物質を圧入して、パドルの高いせん断先端を通過する広いパドルを効果的に製造した。この構成は、下記の表に示すように、簡単に要約することができる:
【0124】
表6:スクリュー構成C、供給から排出までを整理した。
【表6】
【0125】
図8a〜8dは、構成A、BおよびCにて使用した二軸スクリュー部材の、以下の種類の写真を示す。
a)搬送構成または供給スクリュー
b)交互(即ち、分布)の混合構成
c)分散混合構成
d)排出構成
【0126】
図15aおよび15bにおいて、押出成形装置のバレルの概略図を左側に示す。装置の長さを10個のゾーンに分割する(図において“ブロック”として分類)−温度ゾーンに対応する前記ゾーンは、下記の表に記載する。この長さをさらに40単位の長さ、スクリュー直径に対応するそれぞれの単位に分割する−それゆえに直径を示す注釈“D”とする。右側に、3つのスクリュー構成A、BおよびCを概略的に記載する。また直径測定を使用して、構成の中のそれぞれの部材の長さを示す。FSは“供給スクリュー”、即ち最小限の混合を提供する搬送スクリューを表す。混合パドルを設ける場合には、回転方向のオフセットの度合いである度数を数値的に示し、部材の数とする−それぞれの混合パドルは長さ0.25Dである。30、60または90度のオフセットは、分布混合に一致し、0度のオフセットは、分散混合領域に対応する。混合部材のオフセット角に対する注釈“f”および“a”は、重要でない。全ての3つの構成を排出スクリューとして仕上げる。
【0127】
実施例5
カルバマゼピン:サッカリン(1:1)
方法:
236gのカルバマゼピンを、183gサッカリンと(即ち、化学両論的に1:1の関係にて)、撹拌混合機で30分間混合した。構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1(Pharmalab HME16,Thermo)を有する押出成形機を、鋳型なしで、表8に記載の温度設定に設定した。一旦、温度が安定したとき、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、室温まで冷却し、分析の対象とした。
【0128】
【表7】
【0129】
【表8】
【0130】
図9a〜9dは、それぞれ構成A、BまたはCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す(全てのPXRD結果のグラフに関するN.B.、X軸は2シータ(θ)を示し、Y軸はカウントを示す)。
【0131】
図9aは、実質的に共結晶が、2−θ=7のピークの欠如による証明することにより、混合物のみから得られないことを示した。
【0132】
図9bに示すように、共結晶は構成Aを使用して全く得られなかった:
2−θ=7のピーク、共結晶の特徴は観測しなかった。
2−θ=8.9のピークにおいて、脱水カルバマゼピンの特徴を顕著に観測した。
【0133】
図9cに示すように、低率純度共結晶を構成Bの使用によって得た:
2−θ=7のピークにおいて、共結晶の特徴であるが、低強度を有する。
2−θ=8.9のピークにおいて、カルバマゼピンの脱水の特徴を観測した。
【0134】
図9dに示すように、高い度合で共結晶を、構成Cを使用して得た:
2−θ=7で特徴的なピーク。
2−θ=8.9で脱水カルバマゼピンの特徴的なピークを全く観測しなかった。
【0135】
図9e〜9hは、構成Cを使用して得られたカルバマゼピン−サッカリン共結晶(1:1)の凝集物のSEMイメージを示す(それぞれ600×、600×、1000×および1800×倍率)。
【0136】
実施例6
カルバマゼピン:ニコチンアミド(1:1)
方法:
236gのカルバマゼピンを、122gのニコチンアミドと(即ち、1:1の化学両論的な関係)、撹拌混合機で30分間混合した。構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1(Pharmalab HME16、Thermo)を有する押出成形機を、鋳型なしで、表10に記載の温度に設定した。一旦、温度が安定すると、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、室温まで冷却し、分析の対象とした。
【0137】
【表9】
【0138】
【表10】
【0139】
図10a〜10dは、それぞれ構成A、BまたはCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す(全てのPXRD結果のグラフに関して、X軸は2シータ(θ)を示し、Y軸はカウントを示す)。
【0140】
図10aに示すように、実質的に共結晶は、2−θ=20.4でピークの欠如により証明したように、混合物のみからは全く得られなかった。
【0141】
図10bに示すように、低率純度は、構成Aを使用して得られた共結晶であった。
2−θ=20.4のピーク、観測された共結晶の特徴であるが、構成Cより非常に低い強度である。
【0142】
図10cに示すように、低率純度の共結晶を、構成Bを用いて得た。
2−θ=20.4のピーク、観測された共結晶の特徴であるが、構成Cよりも低強度である。
2−θ=6.6、8.9、10.1、13.3、15.5、17.8および26.5のピークを観測した;前記ピークは、同様に共結晶の代表的であるように考えられるが、20.4より優位でない。
【0143】
図10dに示すように、良好な純度の共結晶を、構成Cを使用して得た。
2−θ=20.4のピーク、共結晶の特徴は観測した。
2−θ=6.6、8.9、10.1、13.3、15.5、17.8および26.5のピークが、同様に観測された。
【0144】
図10eは、構成Cを使用して得られたカルバマゼピンおよびニコチンアミドの共結晶(1:1)の凝集物のSEMを示す(倍率1000×)。
【0145】
実施例7
カフェイン:マレイン酸(1:1)
方法:
194gカフェインを、116gマレイン酸と(即ち、1:1の化学両論関係)、30分間撹拌混合機で混合した。鋳型を有さない構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1を有する押出成形機(Pharmalab HME16,Thermo)を、表12に記載した温度に設定した。一旦、温度が安定すると、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、室温まで冷却し、分析の対象とした。
【0146】
【表11】
【0147】
【表12】
【0148】
図11a〜11cは、それぞれ構成A、BまたはCを使用して、押出成形後のPXRD結果のグラフを示す(全てのPXRD結果のグラフに関して、X軸は2シータ(θ)を示し、Y軸はカウントを示す)。
【0149】
図11aに示すように、非常に低率純度の共結晶を、構成Aを使用して得て、PXRDパターンが、高いせん断のバッチと比較して、2−θ=9、11.1、13.2、14.2および15.5にて非常に低強度の特徴的なピークを示す一方、2−θ=12のピーク、無水β−カフェインの特徴は非常に高かった。
【0150】
図11bに示すように、低率純度の共結晶を、構成Bを使用して得て、PXRDパターンが、2−θ=9、11.1、13.2、14.2および15.5にて特徴的なピークを、高いせん断のバッチと比較して低い強度で示す一方、2−θ=12の小ピーク、無水β−カフェインの特徴を観測した。
【0151】
図11cに示すように、良好な純度の共結晶を、構成Cを使用して得て、PXRDパターンが、2−θ=13.2のピークと、2−θ=9、11.1、13.2、14.2および15.5にて特徴的なピークを、最も高い強度を有して示した。
【0152】
図11d〜11gは、構成Cを使用して生成されたカフェインおよびマレイン酸の共結晶の凝集物のSEMイメージを、それぞれ倍率137×、550×、600×および820×にて示す。
【0153】
実施例8
カフェイン:マレイン酸(2:1)
方法:
388gカフェインを、116gマレイン酸と(即ち、化学両論的な関係2:1)、撹拌混合機で30分混合した。鋳型を有さない構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1を有する押出成形機(Pharmalab HME16,Thermo)を、表14に記載した温度に設定した。一旦、温度が安定したとき、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、分析の対象とした。
【0154】
【表13】
【0155】
【表14】
【0156】
図12a〜12cは、それぞれ構成A、BまたはCを使用して、押出成形後のPXRDの結果のグラフを示す(全てのPXRD結果のグラフに関して、X軸は2シータ(θ)を示し、Y軸はカウントを示す)。
【0157】
図12aに示すように、非常に低率純度の共結晶を、構成Aを使用して得た。
2−θ=8.8、10.1、13.5および16のピーク、高いせん断でのバッチと比較して、非常に低い強度を有する。
2−θ=12のピークの強度、無水β−カフェインの特徴は非常に高い。
【0158】
図12bに示すように、低率の純度の共結晶を、構成Bを使用して得た。
2:1共結晶の2−θ=8.8、10.1、13.5および16でピークを観測した。
無水β−カフェインの2−θ=12の特徴でピークの強度は、比較的高かった。
【0159】
図12cに示すように、良好な純度の共結晶を、構成Cを使用して得た。
2−θ=8.8、10.1、13.5および16で2:1共結晶の特徴的なピークを観測した。
無水β−カフェインの2−θ=12のピークの特徴は、重要でない。
【0160】
図12dおよび12eは、構成Cを使用して得たカフェインおよびマレイン酸の共結晶(2:1)の凝集物のSEMイメージを示す(倍率880×および2000×)。
【0161】
実施例9
テオフィリン:マレイン酸(1:1)
方法:
180gテオフィリンを、116gマレイン酸と(即ち、化学両論関係1:1)、撹拌混合機で30分間混合した。鋳型を有さない構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1である押出成形機(Pharmalab HME16、Thermo)を、表16に記載した温度に設定した。一旦、温度が安定すると、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、分析の対象とした。
【0162】
【表15】
【0163】
【表16】
【0164】
図13a〜13dは、それぞれ構成A、BまたはCを使用して、粉砕または押出成形後におけるPXRDの結果のグラフを示す(全てのPXRDの結果のグラフに関して、X軸は2シータ(θ)を示し、Y軸はカウントを示す)。
【0165】
図13aに示すように、実質的に共結晶は、共結晶のピークの特徴の欠如によって証明されるように、混合物のみからは全く得られなかった。2θ=12.5のピークは、テオフィリンの特徴である。
【0166】
図13bに示すように、非常に低率純度の共結晶を、構成Aを使用して得た。
2θ=9、11.5、13.6および16.8で共結晶の特徴的なピークを観測したが、非常に低い、微々たるピーク強度を有する。
2θ=12.5でテオフィリンの特徴的なピークを、顕著に観測した。
【0167】
図13cに示すように、低率の純度の共結晶を、構成Bを使用して得た。
2θ=9、11.5、12、13.6および16.8で共結晶の特徴的なピークを観測したが、バッチと比較すると低強度を有する。
2θ=12.5でテオフィリンの特徴的なピークは微々たるものである。
【0168】
図13dに示すように、良好な純度の共結晶を、構成Cを使用して得た。
2θ=9、11.5、12、13.6および16.8で共結晶の特徴的なピークを観測した。
2θ=12.5でテオフィリンの特徴的なピークは微々たるものである。
【0169】
図13eは、構成Cを使用して得られたテオフィリンおよびマレイン酸の共結晶(モル比1:1)の凝集物のSEMイメージを示す。
【0170】
実施例10
サリチル酸:ニコチンアミド(1:1)
方法:
138gサリチル酸を、122gマレイン酸と(即ち、化学両論関係1:1)、撹拌混合機で30分混合した。鋳型を有さない構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1を有する押出成形機(Pharmalab HME16,Thermo)を、表18に記載した温度に設定した。一旦、温度が安定すると、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、分析の対象とした。
【0171】
【表17】
【0172】
【表18】
【0173】
図14a〜14cは、それぞれ構成A、BまたはCを使用して、押出成形後のPXRDの結果のグラフを示す(全てのPXRD結果のグラフに関して、X軸は2シータ(θ)を示すが、Y軸はカウントを示す)。
【0174】
図14aに示すように、低純度の共結晶を、構成Aを使用して得た。
2−θ=7.8、8.4および9.1で共結晶に特徴的なピークを観測したが、高いせん断の押出成形と比較して、低い強度を有する。
【0175】
図14bに示すように、良好な純度の共結晶を得た。
2−θ=7.8、8.4および9.1で共結晶に特徴的なピークを観測した。
【0176】
図14cに示すように、良好な純度の共結晶を、構成Cを使用して得た。
2θ=7.8、8.4および9.1のピーク、共結晶の特徴は観測した。
【0177】
図14dおよび14eは、構成Cを使用して形成したサリチル酸およびニコチンアミドの共結晶の凝集物を示す(倍率それぞれ200×および800×)。
【0178】
実施例5〜10から、構成Cが比較的高い純度の共結晶を一貫して提供することが分かる。従って、以下を決定することができる:
押出成形は、多種多様なAPIsおよびゲスト物質から共結晶を生成するための確実な方法を提供する。
混合の厳格さは、共結晶を達成する際に重要である。
分散混合の領域の供給は、高レベルな共結晶形成の達成において重要である。
【0179】
溶解データ(生体外薬剤放出):
以下の実験を、水中および人口胃液中の純カルバマゼピンと実施例5に従って形成された共結晶の飽和溶解度および生体外における分解と比較して実施した。
【0180】
カルバマゼピン:サッカリン
純カルバマゼピンおよびカルバマゼピン−サッカリンの共結晶の飽和溶解度測定を、20mLの水に既知の過剰量のサンプルを添加することによって実施した。サンプルを、水浴(37±0.5℃)にて20rpm、24時間撹拌した。その後サンプルを0.45μmの膜濾過で濾過し、水と希釈し、その後分光光度計(Jasco V−630,Jasco Corporation, Japan)によって320nmにて分析した。カルバマゼピン−サッカリンの共結晶(0.78mg/mL)は、カルバマゼピンと(0.29mg/mL)比較して著しい改善を飽和溶解度にて示した。
【0181】
【表19】
【0182】
カルバマゼピン:サッカリン−生体外溶出試験
純カルバマゼピン、および上記の実施例5に従って構成Cを使用して生成したカルバマゼピン−サッカリン共結晶の凝集物(カルバマゼピン200mgに等しい)の溶解研究を、人口胃液(SGF)900mLにてUSP24タイプII溶出試験装置を使用して実施した。SGFを、2g/LNaClおよび1g/LTritonX−100と調製し、pH2までHClで酸性化した。浴槽温度を37±0.5℃に維持し、100rpmで撹拌した。サンプルを、15、30および60分にて収集され、新鮮な溶出媒体と交換した。遠心分離後、0.45μm膜濾過器を通して濾過し、適切な希釈度の、カルバマゼピンの濃度を分光光度法にて320nmで測定した。
【0183】
【表20】
【0184】
結論
純カルバマゼピン粉末は、69.11%のみの薬剤放出を60分間のSGFにて示した。一方、カルバマゼピン−サッカリン共結晶は、86.79%薬剤放出を示した。共結晶凝集物からの初期薬剤放出は、純薬と比較して急速であった。共結晶は15分にて薬剤放出74.74%、30分にて薬剤放出82.89%を示す一方、純カルバマゼピンは15分にて薬剤放出43.48%、30分にて薬剤放出59.29%を示すのみであった。従って、この結果は、本方法の生成物が、共結晶構造に含有した活性薬剤の生物学的利用能を増加させることに極めて適切であることを示す。
【0185】
圧縮研究(錠剤形成)
構成Cを使用して押出成形したバッチの産出生成物を、それらの圧縮化および錠剤形成の性質に関して分析した。
【0186】
カルバマゼピン:サッカリンの共結晶(1:1)
圧縮性の研究を、10mm直径の平板対面パンチを取り付ける圧縮研究のプレス(Caleva Process Solutions Ltd., England)を使用して実施した。鋳型壁を、それぞれの圧縮前に、アセトンで清浄し、前もってステアリン酸マグネシウムで滑らかにした。カルバマゼピン−サッカリン共結晶の凝集物300mgを、鋳型内に手動充填した。圧縮および減圧は、100mm/min、残留負荷度合い10,000N、残留時間0.1秒にて実施し、圧縮力に対する体積変化を記録した。錠剤の厚さは、シックネスゲージ(Mitutoyo,Japan)を使用して測定し、硬度は硬度計(Schleuniger−4M,Copley)を使用して試験した。
【0187】
【表21】
【0188】
イブプロフェン−ニコチンアミドの共結晶(1:1):
圧縮性の研究を、10mm直径の平板対面パンチを取り付ける圧縮研究のプレス(Caleva Process Solutions Ltd., England)を使用して実施した。鋳型壁を、それぞれの圧縮前に、アセトンで清浄し、ステアリン酸マグネシウムで前もって滑らかにした。イブプロフェン−ニコチンアミド共結晶の凝集物400mgを、鋳型内に手動充填した。圧縮および減圧は、100mm/min、残留負荷度合い5,000N、残留時間0.1秒にて実施し、圧縮力に対する体積変化を記録した。錠剤の厚さは、シックネスゲージ(Mitutoyo,Japan)を使用して測定し、硬度は硬度計(Schleuniger−4M,Copley)を使用して試験した。
【0189】
【表22】
【0190】
結論
この技術によって生成した共結晶凝集は、直接圧縮可能である一方、成分の物理的混合は、同様の条件に基づいて圧縮形成できなかった。圧縮物の破壊強度に関して本発明によって得られる粉末物質の圧縮能力は、共結晶の凝集物を使用して得られた圧縮物が、物理的混合物を使用して得たものと比較して、高度な破壊強度を有することを示した。
【0191】
前記実験結果は、上記に示したように押出成形工程の生成物を、凝固によって錠剤に直接形成するのに適していることを示す。これは錠剤製造を簡単にするために、極めて実用的である。
【技術分野】
【0001】
本発明は、製薬関係において役立つ生成物を形成するために役立つ方法、およびこのような方法によって形成される生成物に関する。本発明は、特に押出成形機を使用して共結晶生成物を形成する方法、およびそのような方法によって得られたか、または得ることができる生成物に関するが、これらに限らない。
【背景技術】
【0002】
結晶工学は、最近、活性薬剤の生理化学的性質を調整する方法として研究されている。医薬品へのそれの適用は、活性医薬品成分(API)を含有する幅広い範囲の複数の成分構造の組織的発見に関して、結晶性複合体と医薬品を形成するために使用することができる分子および分子間相互作用の種類を再考することによって、新規な経路を提供する。結晶工学は、薬物の溶解度、分解度および生体利用効率を向上させるために利用できる興味深い潜在的な代替方法を提供する。
【0003】
共結晶は、二つ以上の成分で構成される結晶性物質であり、通常化学両論的比率において、それぞれの成分が原子、イオン性化合物または非共有結合によって共に保持される分子であると考えることができる。共結晶は、二元化合物または分子複合体と以前呼ばれた。APIの生理化学的性質および共結晶を形成する物質の性質は改良することができるとともに、薬物分子の内因性活性を保持することができる。
【0004】
薬学的な共結晶は、製剤設計の際のAPIの改良において、顆粒球、塩および溶媒和物の魅力的な代替として現れる(Blagden N.,de Matas M.,Gavan P.T.,York P.;2007Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates.Advanced Drug Delivery Reviews.59,617−630)。ゲストとともに活性剤を共結晶化することによって、一つは、その活性薬剤の既存の固体相に関する性質を改善することができる新規固体相を生成することができる。例えば、医薬品成分(APIs)と薬学的に許容されるゲストの共結晶を含有する新規な薬学製剤は、既存の製剤に関して優れた性質を有することができる。製薬分野において、活性薬剤はAPIであることができ、共結晶(ゲスト)の他の成分は、薬学的に許容される化合物である必要がある(また必要に応じてAPIであることができる)。活性薬剤およびゲストは、機能性食品、農薬、色素、染料、爆薬、高分子添加物、潤滑剤添加物、光学化学製品、並びに構造的および電子的な物質を含有することができる。活性薬剤またはそれらの塩の物理的性質は、共結晶を形成することによって修飾することができる。前記性質は融点、密度、吸湿性、結晶形態学、容積負荷、圧縮性および製剤の性能に改良を加えることとなる貯蔵寿命を含有する。さらに他の性質、例えば生物学的利用能、毒性、味覚、物理的安定性、化学的安定性、生産コストおよび製造方法を、活性薬剤単独または塩としてよりむしろ共結晶を使用することによって改良することができる(Scott L.C,2007. Co−crystallization methods.WO2007/038524)。
【0005】
共結晶形成の成功率は通常極めて低く、相当するホモマー結晶における分子間より強い場合に、前記ヘテロマー系は、二つ(またはそれ以上)の分子間の非共有結合を形成するのみであるためである。様々な結晶工学技術は、現在共結晶設計で考慮中である。
【0006】
溶液の結晶化は、最も好ましい方法である(Sudhakar P.,Srivijaya R.,Sreekanth B.R.,Jayanthi P.K.,Peddy Vishweshwar.,Moses J.Babu.,Vyas K.,Javed Iqbal.,2007.Carboxylic acid−pyridine supramolecular heterocatemer in a co−crystal. Journal of Molecular Structure.In press;Renata Dreos.,Lassaad Mechi.,Giorgio Nardin.,Lucio Randaccio.,Pathzia Siega.,2005.Alternative co−crystallization of “almost” enantiomers and true enantiomers in some cis−b−organocobalt salen−type complexes with a−amino acids.Journal of Organometallic Chemistry.690,3815−382;Hickey B. Magali.,Matthew L.,Peterson A.,Lisa A., et al.,2007.Performance comparison of a co−crystal of carbamazepine with marketed product.European Journal of Pharmaceutics and Biopharmaceutics.67,112−119;Scott L.C,Kenneth I.H.,2007.4.Co−crystals of Piroxicam with Carboxylic Acids. Crystal Growth and Design.1−14)。この方法は、特に構造分析で単結晶を得るための使用である――化学的な化合物または要素の多数のサンプルを種々の異なる凝結状態にて溶液から凝固された。温度、溶媒または非溶媒含量、種晶、濃度、振動、純度および他の要因の一つ以上を変更することによって、一つは溶液から固形形態を凝固させるのに必要な状態を作り出すことができる。この方法の限定は、二つの成分(活性/ゲスト)が、極めて同程度の溶解度を有する必要がある;一方、ほとんど溶解度のない一成分は沈殿するであろう。同様に、多くの場合、顆粒球形成および本技術の明らかな失敗が、観測された。
【0007】
固体技術、例えばグラインディングまたはミリングにおいて、共結晶は、少なくとも二つの結晶性化合物を含有する混合物をグラインドすることによって生成される(Xyrofin O.Y.1996.Composition comprising co−crystals methods for its manufacture and its use.WO96/07331;Scott L.C,Kenneth I.H.,2007.2.Co−crystals ofPiroxicam with Carboxylic Acids. Crystal Growth and Design.1−14)。
【0008】
溶液に基づく実験は、グラインディングと比較して、それぞれのゲストに関して幅広い様々な形状を生成する。しかしながら、スクリーニング技術として、グラインディング実験は、それらが溶液から容易に得られない形状を特定することができるため、従来の溶液に基づく実験に対して良好な補足となる。重要さを、共結晶に対してスクリーニングする多数の実験技術の使用で与えた。しかしながら、固体技術、例えばグラインディングまたはミリングは、労力が集約し、しばしば小さい容器、例えばμLのウェルプレートにて実施することが困難である。しかし、これらの後者の経験的な方法の開発は、良く理解されてなく、それらの成功は共結晶形成に不可解な雰囲気を与えた(Chiarella R.A.,Davey R.J.,Peterson M.L.,2007.Making Co−Crystals−The Utility of Ternary Phase Diagrams. Crystal Growth and Design. Vol.0,1−4)。しかし、極性に基づいてグラインディング時に適切な溶媒の選択によって、共結晶技術の著しい欠点である顆粒球の形成を制御することができる(Andrew V. Trask., W. D. Samuel Motherwellb and William Jones.,2004.Solvent−drop grinding: green polymorph control of cocrystallisation. Chem. Commun.890−891)。
【0009】
超音波結晶化および超音波を使用して共結晶化による固体形態に関するスクリーニング技術を調査した(Scott L.C,2005.Screening for solid forms by ultrasound crystallization and cocrystallization using ultrasound.WO/2005/089375)。溶液の超音波処理を実施して共結晶を得た−超音波によって発生するキャビテーションエネルギーは、共結晶の成長に実用的であると考えられる。過飽和を核形成および結晶成長を制御する際の超音波の重要性を同様に研究した(Hong Li., Hairong Li., Zhichao Guo., Yu Liu., 2006. The application of power ultrasound to reaction crystallization. Ultrasonics Sonochemistry 13, 359−363; 2. Ruecroft G., Hipkiss D., Tuan Ly., Maxted N., Peter W. Cains., 2005. Sonocrystallization: The Use of Ultrasound for Improved Industrial Crystallization. Organic Process Research and Development. 9, 923−932; Castro et al., 2007)。Bucar et al.は(Dejan−Kresimir Bucar.,Leonard R.MacGillivray.,2007.Preparation and Reactivity of Nanocrystalline Cocrystals Formed via Sonocrystallization.J.Am.Chem.Soc.129(1))、ナノサイズの大きさの有機共結晶の調製をし、本方法が他の共結晶系に適応して、生理活性物質の物理的性質(例えば、溶解度)に影響することができることを予測する方法として超音波結晶化を導入した。しかしながら、酸化に作用を受けやすいAPIsまたはゲストは、本技術によって処理することができない。溶媒および貧溶媒の対の選択がまた重要である。
【0010】
米国特許第5,158,789号明細書には、二つの多価アルコール、ソルビトールおよびキシリトールの溶解物の押出成形品であり、菓子類に関連の性質を改善した製品を凝固で産生することを報告した。
【0011】
広範囲にわたる工業用の押出成形機の使用は、従来プラスチック、ゴムおよび食品業界においてであった。最近において、押出成形機の可能性が薬物応用において理解され始め、主に多くの機能が単一の連続動作において実行することができるためである。従って、多くの分離バッチ作業によって従来実施される工程を組み合わせることができ、製造効率を上昇して、潜在的に生成物の一貫性を改善する。しかしながら、押出成形に基づく製薬方法設計は、専門的な供給および下流の取り扱い技術と共に従来のプラスチック処理操作から開発した−それは様々な形態で重合体マトリックス内にAPIの分散剤を含む。ほとんどの従来の高分子処理機は、医薬品及び医薬部外品の製造管理及び品質管理の基準(GMP)の環境での使用に適応することができる。押出成形処理操作は、実験室から製造規模までに容易に拡大することができ、それら自身が、工程分析技術(PAT)として製薬業界において知られている製造過程の観測技術で十分に役立てることができる。製薬の押出成形品の応用の例を、以下に簡潔に記載する:
【0012】
固体分散剤:
固体分散剤は、活性薬剤物質(溶質)および希釈剤またはキャリア(溶媒または連続相)の完全混和として規定する。従来の技術において、薬剤の固体分散剤は、溶解または溶媒蒸発方法によって通常生成し、その後生成した物質を、カプセル化または錠剤に圧縮する前に、粉末状にし、ふるいにかけ、賦形剤と混合する。溶融押出成形は、これらの系の製造において改善を提供し、微粒子および分子分散剤で使用されてきた。
【0013】
放出制御ドラッグデリバリー:
放出制御ドラッグデリバリーシステムは、従来の服用形態にわたって数々の利点を提供する。放出制御錠剤の製造で最も一般的な工程は、湿式造粒法および直接的な圧縮技術を含み、それらは共に容量の均一化および分離の問題の対象となる。溶融押出成形技術は、放出制御経口投与形態の設計および開発を、水または溶媒を用いずに容易にする。下流のマイクロ造粒または球形化の性能を有する単軸または二軸押出成形機を使用して、顆粒剤または押出成形による錠剤を生成する。親水性および疎水性の物質を処理することができ、一成分のみを溶解または軟化して、物質流動を促進する必要がある。
【0014】
経皮フィルム:
押出成形したシートおよびフィルムを、製薬業界において、生成物包装および経皮薬剤のデリバリーシステムで使用する。後者の適用の場合、活性成分をキャリアと密接に混合し、基体に適用する。従来の押出成形は、創傷を水冷ローラー、30μm未満の厚さで直接巻回した連続性の薄いフィルムを製造するために幅広く、薄い鋳型と連結した。巻き取り速度およびローラー温度の制御は、フィルム結晶化度および分子方位性のある程度の制御を許容する。多層フィルムを、共押出成形、薄層化またはカプセル化によって生成することができる。
【0015】
湿式造粒:
造粒とは、製薬用語において、小粒子の凝集による粒子形態の過程とする。造粒は、直接圧縮、湿式造粒または乾燥した造粒から達成することができる。湿式造粒を、物理的な振動、流動化またはその両方で粉体層の混合物のせん断によるバッチモードで従来通りに実施した。二軸押出成形機は、湿式造粒の他の方法を超える利点を、滞留時間ならびに配分および分散混合のレベルを許容するスクリュー構造の柔軟性により提供する。同様に、これは、凝集および均質化の度合いの制御が比較的短い工程滞留時間、1分の度合いで可能とする。その結果、二軸スクリュー押出成形機は、改善した品質、空間利用および減少した開発時間を、従来の湿式造粒方法を超えて提供する。
【0016】
押出成形球状化:
これは均一な大きさの球状粒子を製造する複数の段階工程であり、即時放出および制御放出の両方の適応において使用することができる。前記粒子を使用して、ゼラチンカプセルを補充、または錠剤に圧縮する。押出成形球形化の主な利点は、活性な医薬品原料の高水準を比較的小さい分子の範囲内において組み込む能力である。押出成形球形化工程は、湿式造粒と、球状粒子の形成、乾燥、サイズ分布でスクリーニングおよび場合によりコーティングを組み合わせたものである。球形化工程は、固定壁を有するボウルと、溝面を有する急速回転基体の作用で通常の球状粒子に押出成形することによって、棒状小粒生成物に変形したものである。
【0017】
上述するように、製薬における応用で使用する既存の押出成形に基づく方法は、ポリマーキャリア組成物とAPIの組み合わせに基づくものである。
【発明の概要】
【発明が解決しようとする課題】
【0018】
製薬分野および他の分野において、共結晶の形成を改善した方法の必要性がある。
【課題を解決するための手段】
【0019】
第一の態様によれば、共結晶を生成する方法において、
第一および第二の物質を付与し、第一および第二の物質は共結晶を形成するのに適合する工程と、
前記第一および第二の物質を共に混合する工程と、
前記第一および第二の物質の混合物を、圧力およびせん断の長期の持続した条件にさらして、前記第一および第二の物質の共結晶を十分に形成する工程と
を含有する方法。
【0020】
共結晶という用語は、二つ以上の成分から生成される結晶性物質であると考えることができる組成物とする;特に二つ以上の成分は、二つの成分の結晶成分がそれぞれ異なる結晶構造に関与する。同様に、共結晶を、多成分分子結晶であると考えることができ、それでしばしば称する。実用的な定義は:共結晶が、非共有結合によって共に保持した二つ以上の分子種(電気的に中性)からなる結晶性物質と規定し、両方の成分が室温にて固形である(Akeroy, Crystal engineering: strategies and architectures, Acta Cryst. B53 (1997) 569−586; and S. L. Mohssette, O. Almarsson, M. L. Peterson, J. F. Remenar, M.J. Read, A.V. Lemmo, S. Ellis, M.J. Cima, CR. Gardner, High Throughput crystallisation: polymorphs, salts, co−crystals and solvates of pharmaceutical solids, Adv. Drug DeNv. Rev. 56 (2004) 275−300.参照)。
【0021】
一般的なレベルにおいて、共結晶は固体分散剤と異なる点に留意すべきである。“固体分散剤”は、不活性キャリアで化合物の分子および分子分散に近いものとして記載して使用される総称語である。これは、単純な共融混合物および固体溶液を含有する。単純な共融混合物は、二つの化合物からなり、液相に完全に混合するが、固体相において非常に限られた範囲で混合する。2つの溶融物質の混合物を冷却したとき、それら両方は非常に微細な(しかし分離した)結晶の物理的混合物を同時に生成して結晶化する。固溶体は、成分の数に関わりなく、丁度1つの相からなる液体溶液と同等である。典型的な固溶体は結晶構造を有し、溶質分子は結晶格子で溶媒分子と置換する。しかしながら、新規な結晶構造の形成が、共結晶の場合には全くない−溶質原子は、単に溶媒の分子を構造変化なしに置換、または溶媒分子の間の間隔の範囲内に位置する。侵入型固溶体において、溶質分子の体積は、通常溶媒の20%未満のレベルにて存在する。これは共結晶から結晶性固体溶液を区別し、規定した規則正しいパッキングモチーフ、対応するゲスト、および非共有結合によって結合したホスト分子を、新規な結晶構造を形成するための格子全体に化学量論的比率で示す。非結晶性固溶体において、溶質分子は、分子的であるが、不規則に非結晶性溶媒に分散している。固体分散剤を、また非結晶性または半結晶性マトリックスに分散した超微細な結晶質の薬剤粒子を含むことができる(Improving drug solubility for oral delivery using solid dispersion,C.Leuner and J.Dressman,Eur. J. Pharm and Biopharm50(2000)47−60参照)。
【0022】
さらに、既存のポリマーおよびAPI押出成形技術に関して、共結晶が固溶体および固体分散剤の双方と異なる構造を有し、その双方が、上記に簡潔に記載するように、ポリマーに基づく押出成形物において一般的な固体形状であることを明白にすべきである。ポリマーに基づく押出成形物の関連において、前者はAPIおよびポリマーの非結晶性混合物であり、後者は半結晶性ポリマーマトリックスの範囲内においてAPIの分散剤である。
【0023】
共結晶の存在は、当業者に知られる数々の分析手法によって特定することができる。これらの最も厳密なものは、おそらくX線結晶解析であり、それは結晶構造の詳細な観測および単一の原子規模での潜在的なそれの解像度を含む。しかしながら、X線結晶解析は、時間がかかり、複雑な工程である。共結晶形成によく適している結晶構造を調査して、時間的に効率的且つ単純な他の方法は、粉末X線回折測定(PXRD)特徴の使用である。一般的に、共結晶性構造の存在は、1つ以上の新規PXRDピークの出現によって識別することができる。非結晶性形状は、拡散した粉末X線回折パターンを示す一方、共結晶は単一成分またはそれらの物理的混合物のPXRDパターンで示さない、付加的な特有の(単一の)ピーク/(複数の)ピークを示す。第一および第二の物質(いくつかの場合、前記物質の様々な高分子の形態)のPXRDパターンの知見によれば、第一および第二の物質と比較して1つ以上の新規PXRDピークの出現によって共結晶構造の存在を同定することが可能である。従って本発明の実施形態は、上記に示したような方法の産出生成物のPXRDパターンと、第一および第二の物質の単独または混合物のPXRDパターン、または関連する共結晶の既知のPXRDパターンを比較することによって、共結晶の存在を同定する工程を含むことができる。
【0024】
本発明の方法が、連続フロー法であることが好ましい。連続工程における合成方法を実行する能力は、従来のバッチ方法と比較して、十分な利点がある。バッチ工程を超える利点は、改善した効率、簡単なスケールアップ、一貫した生成物の性質および減少した洗浄の必要性を含有する。
【0025】
好ましい実施形態において、第一物質は医薬品原料(API)である。上記のように、製薬分野において、共結晶を製造する改善した方法への特有の必要性がある。本分野において、既存の技術は、労働力の集中、遅延、一貫性がなくおよび/または信頼性がなく、規模の拡大の影響を受けないまたはこれらの問題の組み合わせであることを含む不利な点を被る。しかしながら、本発明は製薬分野を超えて、例えば他にも農薬、爆薬、機能性食品、色素、染料、潤滑添加剤、光学的な化学製品、構造物質および電子物質を含む適用がある点に留意すべきである。
【0026】
第一および第二の物質を、少なくとも1分間、好ましくは2分間以上、特に2〜40分間、特に2〜30分間圧力およびせん断の持続状態にさらすことが好ましい。共結晶を形成するために必要とされる時間の長さは、第一および第二の物質がさらされる圧力およびせん断の状態の厳正性に通常依存することはいうまでもないが、長期に渡る、持続してさらすことは通常、改善した共結晶形成に至ることを見出した。しかしながら、多くの場合、共結晶形成に必要とされる時間量に対して、せん断および圧力の条件下で費やされる過度な時間による可能なAPI分解の間のバランスがある。このようなバランスは、使用される物質および前記物質に課される状態に依存する最適化の事項である:このような最適化は通常当業者にとって一連の方法である。
【0027】
前記方法が、少なくとも20w/w%共結晶体、40w/w%共結晶体、より好ましくは少なくとも60w/w%共結晶体、特に共結晶の80w/w%共結晶体を含有する産出生成物を得るために適切であることが好ましい。本発明に記載の方法において、90w/w%共結晶体以上の共結晶の産出生成物を達成することができることができ、著しく高い割合の精製を共結晶の生成において示すことを見出した。
【0028】
従って、本発明の非常に好ましい実施形態において、前記方法は、90w/w%共結晶体以上、好ましくは95w/w%共結晶体以上、特に99w/w%共結晶体以上の産出生成物の形成に適切である。一般的に、それは比較的高い割合の収率を、透過時間および/または圧力およびせん断状態の強度を最適化することによって得ることができるようである。
【0029】
特定の実施形態において、圧力およびせん断を押出成形方法に適応した。驚くべきことに、押出成形の工程を使用して共結晶を得ることができ、それは上述のように、現在得ることが困難なものである。さらに押出成形は、共結晶の高収率且つ大量の生成方法を提供する。これは既存の共結晶化技術を超える非常に有意な利点を提供する。
【0030】
押出成形とは、圧力およびせん断を適応されている間に、細長いルーメンを通る物質の搬送を意味する;通常圧力およびせん断は、少なくとも部分的に、ルーメンを通る物質を搬送する手段によって適応される。押出成形は、物質が鋳型を通過して成形、または同様に押出成形工程の生成物を処理する工程を含むが、これは一般的に共結晶形成では必要でない。
【0031】
押出成形は、スクリューに基づく押出成形方法であることが一般的に好ましい。単一スクリューの押出成形はいくつかの実施形態において適用することができるが、この方法は、二つ以上のスクリューが前記第一および第二の物質の混合物と押出成形工程中に作用する場合に、スクリューに基づく押出成形方法であることが一般的に好ましい。前記方法は、混合や混合物を処理して、所望の共結晶化を得る多く過程で提供する。
【0032】
好ましい実施形態において、スクリューに基づく押出成形方法は、2軸押出成形方法である。2軸方法は、押出成形装置の複雑さを最小にする有効なバランスを提供するとともに、所望のように押出成形工程を処理する能力を提供する。当然、3つ以上のスクリューが相互に影響する押出成形方法を用いることは可能である−前記システムは、ポリマーの押出成形としてよく知られている。
【0033】
2軸押出成形方法が使用されるとき、それは共回転方法であることが、一般的に好ましい。しかしながら、いくつかの実施形態において、逆回転方法がいくつかの利点を提供することを見出すことができる。
【0034】
逆回転スクリューを、非常に高いせん断を必要とするとき使用し、それらは2つの逆回転スクリューの間の高い圧力およびせん断力を発生するためである。従って、逆回転スクリューは、非常に高いレベルのせん断および圧力が、共結晶を形成するのに好ましい場合、実用的である。しかしながら、逆回転スクリュー系が、空気の閉じ込め、低い最大スクリュー速度および産出物に関する課題を受けるおそれがある;これらは特定の応用において不利になるかもしれない。
【0035】
共回転システムは、良好なレベルの混合および物質の搬送を達成することができ、また高速で操作することができ、従って高出力速度を達成することができる。それらは、逆回転系より摩耗する傾向が低い。
【0036】
一つ以上のスクリューが存在するとき、スクリューが少なくとも実質的に噛合、好ましくは完全に噛合することが好ましい。一対のスクリューは、それぞれのスクリューの螺旋形のねじ部位のフライトチップが、実質的に他のスクリュー底に達したとき、完全に噛合であると考えることができる;通常機械的なクリアランスを提供する小さい隙間があるが、一般的に間隙を最小限に保つだろう。本用語を定量化するために、一対のスクリューは、一つのスクリューのフライトチップと他方の底までの間隙が、スクリューの底の全体の深さの10%以下、より好ましくは5%以下であるとき、実質的に噛合であることを示唆することができる。噛合系は、それらが自己ワイピングであり、系内における物質の局部的過熱を防ぐという利点を有する。
【0037】
当然、本発明の特定の実施形態において、非噛合システムを使用することが好ましいことを留意すべきである。非噛合システムは、大量の揮発性物質をシステムから取り除くのが望ましい場合や、または非常に高い粘着性物質がシステムに作用して容認できないほど高いレベルのトルクを生じる場合に使用することができる。
【0038】
本発明の方法において用いる、他の潜在的な種類の押出成形機は、再循環押出成形機である。リサイクル押出成形機は、通常2軸システムであり、バッチの原料をシステムから排出されるまでの所定の時間処理することができる。前記押出成形機、例えばHaake Minilabは、様々な用途に実用的であることができるが、比較的従来の再循環でない押出成形機ほど幅広く使用されていない。それらは一般的に従来の押出成形機のように一定比率増加に適切ではない、バッチシステムである。しかしながら、少量、即ち5gより低い量を処理するそれらの性能は、それらがいくつかの製薬研究に適切であるとする。
【0039】
前記方法は共結晶、特にAPIおよび共結晶形成剤または“ゲスト”物質(それ自身がAPIであることができる)形成することができる第一および第二の物質で単に実施することが一般的に好ましい。2つを超える物質が、共結晶を形成するために共に働くことができ、従ってさらなる共結晶を形成する物質が存在することができることが可能な点に留意する必要がある。従って本発明の方法を、あらゆる結晶形成しない物質、例えば溶媒または潤滑剤の非存在下にて実質的に実施することが好ましい。共結晶を溶媒または他の付加的な物質がない場合で形成することができることが、本発明の方法の有意な利点である。製造後の前記付加的な物質の除去は、困難または不可能であり、前記物質の存在は、低レベルでさえ、十分に安全関連または少なくとも調整のハードルであることができる。当然、例えば、潤滑剤または溶媒の存在が望ましいかもしれない状況であることができることを理解され、本発明の方法が前述の添加剤の包含物と互換性があるが、それらが前記方法から省略することができることは、有意な利点である。
【0040】
誤解を避けるために、共結晶を形成に関与している物質は、結晶構造を形成できる必要があることを言及すべきである。従って、非結晶性または“半結晶性”構造を形成するポリマーは、本発明の工程における共結晶形成で適切ではない。
【0041】
前記第一および第二の物質の混合物を、付加的な熱にさらすことが望ましい。“付加的な熱”とは、混合物がそれに適用され、周囲温度を越え、押出成形工程の間の摩擦によって発生される熱を越える熱であることを意味する。
【0042】
特定の実施例において、前記工程は、工程の少なく一部の期間で、最も低い融点を有する共結晶を形成する物質の融点付近の温度で実施することが好ましい。一般的に、前記温度は、最も低い融点を有する共結晶を形成する物質の融点より若干低いが、融点または融点より若干高くすることが好ましい。好ましい実施形態において、前記温度は融点の20℃の範囲内、好ましくは融点の10℃の範囲であることができる。前記成分のうちの1つが共融混合物である場合、関連する融点はその混合の融点である。前述の温度が使われる場合、共結晶形成に関して利点があることが分かっていた。
【0043】
所望の性質および収率の共結晶を得るため、押出成形工程時に必要とされるせん断、圧力のおよび滞留時間の条件に依存して、(単数の)スクリューまたは(複数の)スクリューの形状を変更することができる。一般的に2軸または他の多数のスクリューの配置が、形状の変更に比較的従うが、それは狭い範囲にて単軸押出成形機が可能である。押出成形の装置または工程の以下の態様に変更することが可能である:特にバレルの長さ、長さの比:バレルの直径(L/Dの比率)、スクリュー成分の組成物(例えば、分散または分布の混合成分、前進または後進の供給成分、スクリューの谷底の深さ(即ち、ネジ山の深さ)スクリュー回転速度、供給方法(スターベーション供給対フラッド供給)、押出成形機を通過する数である。これらの態様は、押出成形工程にわたる高度な調節、および結果として生じる共結晶生成物を可能とする。
【0044】
押出成形時に、L/Dの比率が15/1以上(即ち、長さはスクリューの直径の15×以上)であることが好ましいことが分かった。好ましくは、L/D比率は20/1以上、いくつかの実施形態において30/1以上の比率が好ましい。40/1のL/D比率が、共結晶の形成に適切であることを見出した。前記比率は、特に2軸システムに適用するが、同様に他の押出成形システムにも適用することができる。
【0045】
押出成形最中に混合物を、分布または分散混合に少なくとも一定期間さらすことが好ましい。混合物を分散混合物に少なくとも一定期間さらすことが一般的に好ましい;分散混合物は、せん断、圧力および熱発生に関して比較的強力であり、従って共結晶の形成を促進する際、しばしば実用的である。一般的に混合物を、それぞれ分布および分散の混合物のそれぞれに少なくとも一定の期間さらすことが最も好ましい。
【0046】
押出成形機のスクリュー、特に2軸または他の多数のスクリュー押出成形機は、多くの異なる部材を含み、物質が押出成形時に受ける条件を決めることができる。前記部材は、それらが連続らせん形のねじ山を含むことができない点で必ずしも最も厳密な意味で“スクリュー”であるのではないが、スクリューという用語を、組成物を問わず全体として組立物に関して用いる。一般的に、スクリューの長さの重要な部分は、らせん形のねじ山を含み、通常その長さの半分以上はらせん形のねじ山を含むであろう。
【0047】
スクリューを形成する部材を、通常、シャフト上に組み立て、完全なスクリューを形成する。シャフトは、通常、シャフトに関連する部材の回転を防ぐ横断面、例えば多角形、多くの場合六角形である。それぞれの部材は、通常スクリューの全長に関して非常に短い。押出成形機のスクリューの直径の比率に関して、部材の長さを述べることが最も適切である。
【0048】
らせん形のスクリュー部材を使用して、押出成形機による物質を搬送し、それらは比較的低レベルの混合、並びに圧力およびせん断の適応を与える。前記らせん形部材によって適用される圧力およびせん断のレベルを、例えば前記らせん形部材を多数のスクリュー押出成形機で変えたり、前記部材の深さおよび/またはピッチを変えたりすることによって変更することができる。異なったらせん形のスクリューの種類、例えば前進搬送部材、排出部材または反対のスクリュー部材が存在することができる。
【0049】
比較的激しい混合、せん断および圧力の適応が必要である場合、これは部材の組み合わせ、特に混合パドルを使用して達成することができる。混合パドルは通常、丸い突出部材(lobed element)、例えば楕円または類似した形状の部材を含み、らせん型のねじ山は含まない。パドルは曲線且つ平坦な混合表面を設ける。2軸押出成形機において、一つ以上の対応する対の丸い突出部材を、それぞれのスクリューに設けることができる。1本のスクリューでの丸い突出部材を、それが他方のスクリューでの丸い突出部材に対して回転方向にオフセット、通常二つの丸い突出部に90°(即ち、一般的に楕円)のパドルであり、例えば部材を回転して、突出部材の混合表面を狭い溝に分離して、パドル対に相応する形状による回転時に、実質的に一定のままであることができるか、または回転時に多少程度変化する可能性があるように配置した。オフセットの異なった程度は、三つの丸い突出部材、または混合部材の他の形状で適切に使用することができる。前記混合パドルの効果は、混合物をパドル対の間に塗布し、従って比較的強度の混合で、高いせん断および圧力を受ける。さらに、混合表面の平坦な性質は、前進搬送が、強く促進されずに、それ自体、混合物がそのような部材に滞留する傾向があることを意味する;混合物の前進搬送が、上流搬送部材によって強制される上流混合物によって動作した圧力で主に促進するが、下記のように、混合部材の特定の構成は、前進搬送の度合いを提供することができる。
【0050】
混合、並びにせん断および圧力の適応の度合いは、混合部材の数および形状によって定めることができる。分布混合は、押出成形の技術において知られている用語であり、“分布混合は、優れた空間分布を達成するために、微量成分をマトリックスにわたって分布する工程である”として規定することができる。分布混合は、混合部材(例えば、突出部材)の一連の対を、混合部材のそれぞれの対は、前記対に対して回転方向にオフセット、即ちスタガー角である場合に達成することができる。一般的に、次の混合部材は、前進搬送を提供するらせん部の方向と同じ方向でオフセットする。通常、それぞれの混合部材(例えば、突出部材)の長さは、スクリューの直径の0.25×まで、好ましくはスクリューの直径の少なくとも0.125×までである;例えば、スクリューの直径16mmに関しては、それぞれの部材は4mmの長さであることができる。分布混合は、物質の混合物の流路を再配置することによって主に混合することを考慮することができる;本質的に、それぞれの混合物部材の相関的に短い長さは、混合物が混合部材の間にて撹拌され、高く強制的に塗布されるもののレベルは比較的低いことを意味する。回転方向のオフセット量は、分布混合配列を提供するように搬送量、およびある程度混合の重要性を決定する。一対が供給スクリューでのらせんと同じ方向に約10°から45°(通常30°)までで、前記対からオフセットする場合、前進搬送の十分な度合いを提供し;約46°から65°(通常60°)のオフセットは、いくらか少ない搬送を提供し;約75°から約90°のオフセットは、著しく少ない搬送を提供する−90°のオフセットは本来混合物の搬送を全く提供しない。
【0051】
分散混合は、混合の強度の態様であり、せん断および圧力の高いレベルを混合物に与える。分散混合は、押出成形の技術において知られる用語であり、“分散混合は結合力のある微量成分、例えば固体粒子の集合体または液体の液滴の大きさにおける減少を含む”として規定することができる。分散混合を、混合物が圧縮されて、混合部材の混合表面の間を塗付する細長い混合部位を通過して圧入されたときに、達成することができる。分散混合は一つ以上の混合部材、例えば二つの突出部材で提供して、混合表面の細長い部位をあらゆる回転方向にオフセットなしに提供することができる;即ち、混合表面の細長い部位を、比較的長い混合部材(2軸システムのそれぞれのスクリューの一つ)の対と、実質的に回転方向に全くオフセットすることなく提供することができるか、または次の部材の間で実質的に回転方向にオフセットが全くない、複数の一連の比較的短い混合部材であることができる。例えば、回転方向にオフセットを全く有さない突出部混合部材を含むスクリューの直径の0.5×またはそれ以上の長さの部位を、分散混合物に提供するであろう。都合よく、分散混合ゾーンは、互いにはオフセットしてない二つ以上の突出部材を含むことができ、即ちそれらは実質的に継続した混合表面を提供する。本質的には、分散混合の有意な態様は、混合物の少なくとも一部が、混合物を塗付して、圧力およびせん断の高い度合いを適応する混合部材の通過を制限する−これは上述のような混合部材を使用して達成することができる。
【0052】
しかしながら、上記分布および分散混合システムは、本発明の使用に対して好ましいシステムを示す点に留意する必要がある。分布または分散の混合を達成する他の方法を、当業者は想到することができる。分散および分布の混合の提案を、Rheology Bulletin Vol.66,No.1(January1997)−Analysis of Mixing in Polymer Processing Equipment by lea Manas−Zloczowerで提供する。
【0053】
本方法にて使用される押出成形装置が、分散混合領域(即ち、混合部材を含有する部位)を、スクリューの全長の少なくとも1/40、好ましくは少なくとも1/30、より好ましくはスクリューの全長の少なくとも1/20を有することが好ましい。好ましくは、分散混合の少なくとも一部があり、その部位は少なくとも0.5倍の直径の長さである。より好ましくは、分散混合の少なくとも一部位があり、分散混合の全ての全長は、少なくとも直径の1.5倍以上、好ましくは直径の2倍以上である。
【0054】
本発明の実施形態において、混合および分散混合の部の両方、即ち混合部材を含有する部を有することが好ましい。好ましい実施形態において、構造は少なくとも一部を分布混合した後、少なくとも一部を分散混合することを含む。本発明の好ましい実施形態において、分布混合の少なくとも二つの領域、および分布混合の少なくとも二つの領域を提供する。分布混合部のそれぞれが少なくとも直径の長さの1倍、より好ましくは直径の長さの1.5倍、そしてそれらは直径の長さの2倍以上であることが好ましい。それぞれの分散混合の部位が直径の長さの少なくとも0.5倍であり、それらが直径の長さ1倍以上、それらが直径の長さの1.5倍以上にすることができる。一般的に混合部の合計5倍以上の長さ、より好ましくは混合部の直径の長さの10倍以上であることが好ましい。
【0055】
一般的にスクリューの全長の半分以下の押出成形システムが、混合部材を含有することが好ましい。一般的にスクリューの全長の2/5以下または1/4以下が、混合部材を含有する。当然、実際の割合は、ネジの全長に依存して変更することができ、全長の半分を超える場合は混合部材含む状況を想定することができる。
【0056】
上記のように、第一および第二の物質は共結晶を形成する必要がある。共結晶の形成を予測するために利用できる確かな規則は全くない。しかしながら、一般的に第一および第二の物質が、水素結合形成で顕著な可能性を伴う優遇の群を有するべきである。一般的に所望の部類に分類される物質は、カルボン酸、アミン、アミド、スルホンアミド、ヒドロキシルアルコール、ケトン、アミノ酸、糖類および複素環式塩基を含み、従ってこのような物質、すなわち適切な活性基を含有する物質は本方法における使用に好ましい。一般的に、特定のAPIの共結晶を形成することが好ましく、適切なゲストまたは共結晶形成のためコフォーマー(co−former)を選択することが必要であるだろう;即ち、ゲストを変更する自由度があるが、通常APIはない。適切なゲストを同定することは、一般的にある程度の試行錯誤を伴うが、APIの化学作用の知識によってしばしば導かれる。適切なゲストを同定することは、一般的にある程度の試行錯誤を伴うが、APIの化学作用についての知識によってしばしば導かれる。しかしながら、一般的に、カルボン酸、アミドおよび複素環式塩基を含むAPIおよびゲストが、本発明の使用に好ましい。
【0057】
適切なAPIの例を、下記の表1に記載する。
【0058】
【表1】
【0059】
適切なゲストの例を、下記の表2に記載する。
【0060】
【表2】
【0061】
本発明の実施形態において、第一および第二の物質が化学量論的比率で形成されることが好ましい。その比率は1:1であることができ、またはそれは他の整数比率、例えば1:2、2:1等、であることができる。
【0062】
本発明の実施形態において、第一物質はフェニルアルカン酸である。好ましくは、第一物質はイブプロフェンであり、第二物質はニコチンアミドである。適切な、イブプロフェンとニコチンアミドの重量比は、約41.2:26、即ち1:1モル比である。
【0063】
本発明の他の実施形態において、第一物質はカルバマゼピン、第二物質はサッカリンである。カルバマゼピンとサッカリンの適切な重量比は、約47:37、即ち1:1モル比率である。
【0064】
本発明の他の実施形態において、第一物質はカルバマゼピン、第二物質はニコチンアミドである。カルバマゼピンとニコチンアミドの適切な重量比は、約118:61、即ち1:1モル比である。
【0065】
本発明の他の実施形態において、第一物質はカフェイン、第二物質はマレイン酸である。カフェインとマレイン酸の適切な重量比は、約194:116、即ち1:1モル比、または97:29、即ち2:1モル比である。
【0066】
本発明の他の実施形態において、第一物質はテオフィリン、第二物質はマレイン酸である。テオフィリンとマレイン酸の適切な重量比は、45:29、即ち1:1モル比である。
【0067】
本発明の他の実施形態において、第一物質はサリチル酸、第二物質はニコチンアミドである。サリチル酸とニコチンアミドの適切な重量比は、69:61、即ち1:1モル比である。
【0068】
押出成形工程の生成物が一般的に共結晶の凝集物を含む粒子であることが、本発明の注目に値する態様である。前記粒子は、一般的にそれらの最大の大きさで2から3000μmの大きさであり、錠剤または他の単位用量形状に直接圧縮されることに非常に適切である。さらに、凝集物は、水および胃の生体外モデルの両方に容易に溶解し、薬物送達において極めて実用的であることを示した。
【0069】
従って、更なる態様において、本発明は、上で述べられる方法を含む、凝集された共結晶を含有する前記粒子の形成方法を提供する。
【0070】
別の態様において、本発明は、凝集された共結晶の粒子、好ましくは直径2から3000μmを形成するものを提供する。好ましくは、粒子は、少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含有し、それらは90w/w%共結晶体以上を含むことができる。
【0071】
任意に、前記方法は改質剤化合物を押出成形工程に導入する段階を含むことができる。適切な改質剤は、密度改質剤(ラクトース、微結晶性セルロース等)、結合剤(澱粉、セルロース、ポリビニルピロリドン等)、崩壊剤(ナトリウム澱粉グリコール酸塩、架橋ポリビニルピロリドン)および湿潤剤を包含する。前記改質剤を、共結晶化を実質的に完了した後、押出成形工程に、適切に導入することができる。このように、改質剤を、工程の産出生成物内に取り込むが、共結晶化工程を妨害しない。例えば、改質剤を、分散混合の全ての領域の下流に導入することができる。
【0072】
更なる実施形態において、上述のような方法を実施して凝集された共結晶の粒子を形成し、任意に適切なモールドで前記粒子を圧縮して、単位用量形状を形成する工程を含む活性薬剤の単位用量形状を形成する方法を提供する。適切には、単位用量は錠剤または同様のものである。任意に、前記方法は、製薬学的に許容される賦形剤を提供することを含むことができる。適切な賦形剤は、改質剤として上で述べられた化合物を包含することができる。
【0073】
他の態様において、本発明は、共結晶を含む組成物を付与し、前記共結晶は第一および第二の物質を含み、第一および第二の物質を上記のような工程に露出した。
【0074】
一実施形態において、本発明は、フェニルアルカン酸およびニコチンアミド、好ましくはイブプロフェンおよびニコチンアミドの共結晶を含む組成物を提供する。共結晶の存在は、3.2°2−θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は、少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体、特に少なくとも90w/w%共結晶体を含む。
【0075】
一実施形態において、本発明は、カルバマゼピンおよびサッカリンの共結晶を含む生成物を提供する。共結晶の存在は、7°2−θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は、少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0076】
一実施形態において、本発明は、カルバマゼピンおよびニコチンアミドの共結晶を含む生成物を提供する。共結晶の存在は、20.4 2θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0077】
一実施形態において、本発明は、カフェインとマレイン酸を1:1のモル比にて含有する生成物を提供する。共結晶の存在は、9、11.1、13.2、14.2、15.5および13.2 2θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0078】
一実施形態において、本発明は、カフェインとマレイン酸を2:1のモル比にて含有する生成物を提供する。共結晶の存在は、8.8、10.1、13.5および16 2θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0079】
一実施形態において、本発明は、テオフィリンおよびマレイン酸の共結晶を含む生成物を提供する。共結晶の存在は、9、11.5、13.6および16.8 2θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は、少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0080】
一実施形態において、本発明は、サリチル酸およびニコチンアミドの共結晶を含む生成物を提供する。共結晶の存在は、7.8、8.4、および9.1 2θにて、共結晶の特徴的なPXRDピークの存在によって同定することができる。好ましくは、生成物は少なくとも50w/w%共結晶体、より好ましくは少なくとも75w/w%共結晶体を含む。
【0081】
さらなる態様において、本発明は、上記の工程から得られるか、または得ることができる共結晶を含む組成物を提供する。
【0082】
さらなる態様において、本発明は、上記の共結晶を含有する医薬品、任意に製薬学的に許容される賦形剤と組み合わせたものを提供する。医薬品は、投与のためのあらゆる適切な形状、例えば、顆粒剤、錠剤、カプセル等であることができる。
【0083】
さらなる態様において、本発明は、痛みの緩和または炎症状態の治療のためのイブプロフェンおよびニコチンアミドの共結晶を含む組成物を提供する。炎症状態の例は、外傷によって誘導された炎症および自己免疫状態であり、例えば慢性関節リウマチ、紅斑性狼瘡、重症筋無力症、天疱瘡、シェーグレン症候群、強直性脊椎炎、炎症性腸疾患等である。
適切な共結晶のさらなる詳細を、上に記載する。
【0084】
さらなる態様において、本発明は、カルバマゼピンおよびサッカリンまたはカルバマゼピンおよびニコチンアミドの共結晶を含有する組成物を、心因性障害、例えば癲癇、躁うつ病注意欠陥多動性障害(ADD)または注意欠陥多動性障害(ADHD)、精神分裂症、幻肢症候群および三叉神経痛の治療のために提供する。適切な共結晶のさらなる詳細を、上に記載する。
【0085】
さらなる態様において、本発明は、カフェインおよびマレイン酸の共結晶を含有する組成物を、呼吸器系の中枢神経系の疾患の治療のために提供する。
【0086】
さらなる態様において、本発明は、テオフィリンおよびマレイン酸の共結晶を含有する組成物を、気管の慢性閉塞性肺疾患(例えば、COPD)、喘息(特に気管支)または無呼吸発作(特に小児無呼吸発作)の治療のために提供する。
【0087】
さらなる態様において、本発明は、サリチル酸およびニコチンアミドの共結晶を含有する組成物を、痛みの減少、熱の緩和、またはざ瘡、乾癬、皮膚硬結、うおのめ、毛孔性角化症および疣(例えば局所投与による)の治療の使用で提供する。
【0088】
さらなる態様において、本発明は上で述べられる方法に従って得ることができるか、または得られた凝集した共結晶を含む粒子を提供する。前記粒子は、溶解および凝集形態に関して好ましい性質を有する。
【0089】
さらなる態様において、本発明は、治療において使用するために上記の方法のような共結晶を含有する組成物を提供する。本発明は、同様に上記のように共結晶を含有する組成物の使用を、薬剤の製造において、一つ以上の上記病状の治療にて使用するために提供する。
【0090】
本発明の実施形態を、例としてのみに、添付の図を参照にしてここで記載する:
【図面の簡単な説明】
【0091】
【図1】2軸押出成形機のスクリュー部材の例を示す図である。
【図2】イブプロフェンおよびニコチンアミドを含有する物理的混合のPXRDパターンを示す図である。
【図3】選択的な供給および分布の混合ゾーンで15:1スクリュー構成を使用して押出成形後に、得られたイブプロフェンおよびニコチンアミドの共結晶のPXRDパターンを示す図である。
【図4】選択的な供給および分布の混合ゾーンで40:1スクリュー構成を使用して押出成形後に、得られたイブプロフェンおよびニコチンアミドの共結晶のPXRDパターンを示す図である。
【図5】供給、分布および分散の混合ゾーンで40:1スクリュー構成を使用して押出成形後に、得られたイブプロフェンおよびニコチンアミドの共結晶のPXRDパターンを示す図である。
【図6】イブプロフェンおよびニコチンアミドの単一の共結晶を使用して、得られたPXRDパターンを示す図である。図6a〜dは、凝集されたイブプロフェンおよびニコチンアミド共結晶のSEMイメージを、それぞれ350×、1100×、1800×および3500×の倍率にて示す図である。
【図7】供給、分布および分散のゾーンで40:1スクリュー構造を使用して押出成形後に、得られたカルバマゼピンおよびサッカリンの共結晶のPXRDパターンを示す図である。
【図8】図8a〜dは、構成A、BおよびCにおいて使用したような2軸部材の4種類の写真を示す図である:a)搬送構造b)交互(即ち、分布)混合構造c)分散混合構造d)放出構造。
【図9】図9a〜9dは、カルバマゼピンおよびサッカリンのそれぞれ構成A、BまたはCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す図である(全てのPXRD結果をグラフで示し、X軸は2−θおよびY軸はカウントを示す)。図9e〜9hは、構成Cを使用して生成した凝集したカルバマゼピンおよびサッカリン共結晶の600×、600×、1000×および1800×にそれぞれ拡大したSEMイメージを示す図である。
【図10】図10a〜10dは、カルバマゼピンおよびニコチンアミドのそれぞれ構成A、BまたはCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す図である。図10eは、構成Cを1000×倍率を使用して製造した凝集カルバマゼピンおよびニコチンアミド共結晶のSEMイメージを示す図である。
【図11】図11a〜11cは、カフェインおよびマレイン酸(モル比1:1)のそれぞれ構成A、BおよびCを使用して、押出成型後のPXRD結果のグラフを示す図である。図11d〜11gは、構成Cをそれぞれ137×、550×、600×および820×倍率を使用して生成した凝集カフェインとマレイン酸の共結晶(モル比1:1)のSEMイメージを示す図である。
【図12】図12a〜12cは、カフェインとマレイン酸(モル比2:1)のそれぞれの構成A、BまたはCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す図である。図12dおよび12eは、構成Cのそれぞれ800および2000×倍率を使用して生成した凝集したカフェインおよびマレイン酸共結晶(モル比2:1)のSEMイメージを示す図である。
【図13】図13a〜13dは、テオフィリンおよびマレイン酸のそれぞれ構成A、BおよびCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す図である。図13eは、構成Cの倍率500×を使用して製造した凝集テオフィリンおよびマレイン酸共結晶のSEMイメージを示す図である。
【図14】図14a〜14cは、サリチル酸およびニコチンアミドのそれぞれ構成A、BまたはCを使用して、押出成形後のPXRD結果のグラフを示す図である。図14dおよび14eは、構成Cを使用して生成した凝集サリチル酸−ニコチンアミド共結晶のそれぞれ200×および800×倍率におけるSEMイメージを示す図である。
【図15a】図15aは、押出成形装置自体の表示に対するスクリュー構成A、BおよびCの概略図を示す図である。
【図15b】図15bは、押出成形装置自体の表示に対するスクリュー構成A、BおよびCの概略図を示す図である。
【発明を実施するための形態】
【0092】
押出成形−背景
押出成形は、開口部または鋳型によって物質を圧入することによって生成物を形成する方法として規定することができる。本方法は、溶解物の押出成形の場合、加熱バレルにて回転しているアルキメデスのねじの作用によって、継続的な方式で通常実施される。ポリマーに関して、溶解は、バレル壁およびポリマーの粘性のせん断により、伝導加温の二重作用によって達成される。押出成形機の最も単純で広く使用される形態は、単スクリューを使用するものであり、それは一般的に、溶融物質の溶解および測定することを達成するために、単純な単一のフライトデザインである。
【0093】
二軸押出成形機(TSE)は、単軸押出成形機の低い混合性能を克服するために、二軸を使用して開発され、通常並べて、同じ(共回転)または逆(異方向)方向に回転するように配置された。スクリューは、通常、物理的な間隔を除いて、密接または完全に噛み合う、即ち、それぞれのスクリューのフライトの先端は、対向するスクリューの底に達するように構成されている。これは、高度な混合を、二つのスクリューの間で“噛合”領域において可能とする。TSEは、粘性抵抗流体に依存することよりむしろ強制的搬送によって作動し、スクリューの自己ワイピング動作は、押出成形機をより衛生的にして、単スクリュー構造より少ない沈滞である。TSEスクリューは、通常、交換可能なスクリュー部材が配置される六角形のシャフトからなる。これはスクリュー構造で高度な柔軟性を可能とし、応用では、搬送、混練、混合および放出の混合を提供するように容易に構成することができる。TSEは通常、少量供給(starve−fed)され、不完全に満たされたチャネルを実施した。
【0094】
逆回転押出成形機は、押出成形機内の物質移動のため、低い度合いの混合であるが、高い物質供給および搬送の特性を有する。それぞれのスクリューのフライトが他のスクリューのチャネルに適合し、完全に満たされる場合、物質をスクリューで回転するのを完全に妨げられ、従って軸方向に積極的に移動する。前記移動は、物質の粘度およびバレルおよびスクリューの金属面への粘着に独立している。逆回転TSEの滞留時間および溶解温度は、非常に一定である。スクリューの間の物質には、高いせん断力を受け、高圧の進展が生じ、従って逆回転TSEは、スクリューの間で生じる高圧による共回転より、低いスクリュー速度にて作動する。逆回転TSEの典型的重合例は、熱分解に感度が高く、低い滞留時間、例えばPVCおよび木材複合体ポリマーを要求する物質を包含する。
【0095】
共回転押出成形機は、TSEの中で最も産業的に重要な種類であり、密接または完全に噛合するスクリュー構造を有する傾向がある。スクリュー部材は自己ワイピングであり、高いスクリューの速度および処理能力が、本構造によって可能である。共回転TSEは、物質を長手方向並びに横断方向に混合する能力を有するため、物質がスクリューの1つのチャンバから他のチャンバまで輸送され、そして優れた混合および混合物内に高いエネルギー入力を生じる。共回転スクリューは、半回転システムと比較して、高度な柔軟性を提供する。典型的な構成は、搬送、混練および混合部材を含有する。バリア部材を使用して、高低の圧力の溶融封止および領域を設け、液体の注射または揮発性物質の除去を可能にする。共回転TSEの一般的な適用は、重合樹脂が広範囲にわたる補強用充填剤および添加剤と混合する場合に、ほとんどの樹脂混合の実施を含む。混合および反応性の押出成形品は、同様に応用で幅広く使われている。共回転TSEからの押出成形品は、一般的に次の形成工程で使用して小球形にされる;TSEのみでは、製造にて、発生する低いヘッド圧力および固有の変動による生成物の製造で特に適切でない。
【実施例】
【0096】
二軸押出成形による共結晶形成−実験方法
器材
2つの共回転二軸押出成形機を、共結晶の形成に使用し、双方ともスクリュー直径16mmである。第一は、スクリューの長さと直径(LD)の比率15:1(Thermo Prism TSE16TC)を有し、3つの温度制御バレルゾーンおよび1つの鋳型ゾーンを組み込む短い押出成形機である。LDの比率40:1(Thermo Prism Eurolab)を有する長い押出成形機を同様に使用して、合計10の温度制御バレルおよび鋳型ゾーンを組み込む。スクリュー構造と組み合わせた押出成形機の長さは、押出成形時の可能な残余時間および混合程度を決定する。
【0097】
一つのスクリュー構成を、搬送部材および一つの分布混合ゾーンからなる単純なスクリュー構造を有して、15:1LD押出成形機に使用した。二つのスクリュー構成を、40:1LD押出成形機に使用し、一つは搬送部材および三つは主に分布混合部位である。第2の構成は、分布および分散混合ゾーンおよび逆搬送部材の比較的複雑な組合せを使用した。これは高い滞在時間および厳格な混合環境を提供した。表3a〜3cは、使用される3つのスクリュー構成を集約し、主要な種類のスクリュー部材の例を示す写真を図1に示す。
【0098】
実験手順
洗浄した押出成形機を、選択された処理温度まで予熱した。バレル温度プロフィールの範囲を使用し、通常は冷却された供給ゾーンから最大の中央位置点までバレルに沿って増加し、鋳型の端部に向かって減少する。前記試験のために、押出成形機を鋳型なしに実施した。押出成形機のスクリュー回転速度を設定した;広範囲の速度を達成することができ、押出成形機とともに最高200回転/分(rpm)までを本明細書で使用した。一般的なスクリュー回転速度を、20から50rpmの間に設定した。その後活性薬剤およびコフォーマーの予備混合した混合物を、押出成形機のフィードホッパに導入した。手動投与することは、小さいバッチ容積(通常10〜30gの間)にて便利であることを証明することができた。大きいバッチ容積に関しては、重量系供給システムを比較的便利に使用することができる。その後薬物およびコフォーマーの押出混合物を、スクリューの出口にて、構成部材および設定された作動状態によって、粉末、粘着性の塊または溶解形態で収集した。収集した物質を、その後共結晶形成で分析した。
【0099】
実験の方針の間、以下のパラメータを調整することができる:
設定温度
スクリュー回転速度
処理量
スクリュー構造(即ち、分布および分散的な混合の度合い)
押出成形機による通過数
【0100】
表3a−スクリュー部材の概略図、構成1。
【表3a】
【0101】
表3b−スクリュー部材の概略図、構成2。
【表3b】
【0102】
表3c−スクリュー部材の概略図、構成3。
【表3c】
【0103】
実施例1
イブプロフェンおよびニコチンアミドの物理的な混合物を、41.2gのイブプロフェンおよび26gのニコチンアミド(モル比1:1)を撹拌混合機で30分間混合することによって調製した。LD比率15:1およびスクリュー直径16mm(Thermo Prism TSE 16TC)を有する押出成形機を用いた。これはスクリュー構成1を組み込み、主に前進供給部材および小さい分布混合ゾーンからなる。詳細なスクリュー構成を、上に示した。バレル温度を80℃に設定した。一旦温度が15分間安定したとき、物理的混合物を押出成形機にゆっくりと供給して、スクリューを20rpmにて回転した。微細な凝集生成物を、押出成形機の出口にて収集した。押出成形機による物質の残在時間は、約3分であった。粉末を室温まで冷却し、その後粉末X線回折測定(PXRD)評価の対象とした。
【0104】
図2は、イブプロフェン−ニコチンアミドを含有する物理的混合のPXRDパターンを示す。イブプロフェンの特徴的なピークは、6°2θにて観測することができる。
【0105】
図3は供給および単一の分布混合のゾーンで15:1スクリュー構成を使用して、押出成形後に得られた共結晶のPXRDパターンを示す。3.2°2θで共結晶の特徴的ピークが、共結晶が押出成形工程時に形成されることを明らかに示すものを観測した。PXRDパターンは、同様にイブプロフェン結晶の特徴的なピークの出現を6°2θにて示した。これはイブプロフェンの部分が共結晶形状に変換されなかったことを示す。
【0106】
実施例2
イブプロフェンおよびニコチンアミドの物理的混合物を、41.2gイブプロフェンおよび26gニコチンアミド(モル比1:1)を撹拌混合機で30分間混合することによって調製した。LD比率40:1およびスクリュー直径16mmを有する押出成形機を使用した(Thermo Prism Eurolab)。これはスクリュー構成2を組み込み、供給および分布混合のゾーンからなる。詳細なスクリュー構成を、上に示した。バレル温度を80℃に設定した。一旦温度が15分間安定したとき、物理的混合物を押出成形機にゆっくりと供給して、スクリューを20rpmにて回転した。押出成形機による物質の残在時間は、約20分であった。微細な凝集生成物を、押出成形機の出口にて収集した。粉末を室温まで冷却し、その後粉末X線回折測定(PXRD)評価の対象とした。
【0107】
上記のように、図2はイブプロフェン−ニコチンアミドを含有する物理的混合物のPXRDパターンを示す。イブプロフェンの特徴的なピークは、6°2θにて観測することができる。
【0108】
図4は、上記のように、代替供給および分布混合のゾーンを有する40:1スクリュー構成を使用して押出成形後に得られた共結晶のPXRDパターンを示す。それは共結晶の特徴的なピークの出現を、3.2°2θにて示した。これは共結晶が押出成形工程時に形成されることを明らかに示す。PXRDパターンは、同様にイブプロフェン結晶の小さい特徴的なピークを、6°2θにて示した。PXRDパターンを適切な算出方法を使用して分析し、混合物が約72%共結晶を含有することを示した。
【0109】
実施例3
イブプロフェンおよびニコチンアミドの物理的混合物を、41.2gイブプロフェンおよび26gニコチンアミド(モル比1:1)を、撹拌混合機で30分間混合することによって調整した。LD比率40:1(Thermo Prism Eurolab)、ならびに最後の供給ゾーン(構成3)より前に逆供給スクリュー領域を有する交互供給、分布および分散混合のスクリューゾーンを有する押出成形機を、80℃に設定した。一旦温度が15分間安定したとき、物理的混合物を押出成形機にゆっくりと供給して、スクリューを20rpmにて回転した。押出成形機による物質の残在時間は、約33分であった。微細な凝集生成物を、エンドゾーンにて収集した。粉末を室温まで冷却し、その後粉末X線回折測定(PXRD)評価の対象とした。
【0110】
上記のように、図2は、イブプロフェン−ニコチンアミドを含む物理的混合のPXRDパターンを示す。イブプロフェンの特徴的なピークは、6°2θにて観測することができる。
【0111】
図5は、40:1スクリュー構成3を使用して、押出成形後得られた共結晶のPXRDパターンを示す。それは共結晶の特徴的なピークの出現を、3.2°2θにて示す。これは共結晶が押出成形工程時に形成されたことを明らかに示した。PXRDは、イブプロフェン結晶の特徴的なピークを6°2θにて示さなかった。適切な算出方法によって分析したPXRDパターンは、約94%共結晶含量を示した。
【0112】
図6は、イブプロフェン−ニコチンアミドの単一共結晶を使用して得られたPXRDパターンを示す(これを比較として溶媒技術によって生成した)。
【0113】
図6aから6dは、スクリュー構成3を使用して生成したイブプロフェン−ニコチンアミドから形成した共結晶の凝集物の走査型電子顕微鏡写真(SEM)を示す(350×、1100×、1800×および3500×倍率)。
【0114】
実施例4
カルバマゼピンおよびサッカリンの物理的混合物を、47gカルバマゼピンおよび37gサッカリン(モル比1:1)を、撹拌混合機で30分間混合して調製した。押出成形はスクリュー構成3を組み込んだTSEにて実施し、供給、分布および分散混合のゾーンからなる。詳細なスクリュー構成を上に示した。バレル温度を140℃に設定した。一旦温度が15分間安定したとき、物理的混合物を押出成形機にゆっくりと供給し、スクリューを20rpmにて回転した。押出成形機による物質の残在時間は、約33分であった。微細な凝集生成物をエンドゾーンにて収集した。粉末を室温まで冷却し、その後粉末X線回折測定(PXRD)評価の対象とした。
【0115】
図7は、40:1スクリュー構成3を使用して、押出成形後に得られた共結晶のPXRDパターンを示す。それは、カルバマゼピン−サッカリン共結晶の特徴的なピークの出現を7°2θにて示した。これは共結晶が押出成形工程時に形成されたことを明らかに示す。
【0116】
二軸スクリュー押出成形機による共結晶の形成−更なる実験方法
更なる実験を、共結晶収率に関するせん断、混合および滞留時間の効果を評価するために実施した。
【0117】
実施例5〜10の全ての実験を、スクリュー直径16mm、長さと直径(L:D)の比率40:1を伴うThermo Pharmalab HME16共回転二軸押出成形機で実施した。
【0118】
三軸構成を(概略的に図15aおよび15bに示す)使用して、共結晶収率に関するせん断、混合および滞留時間の効果を評価する。スクリュー構成を以後A、BおよびCとして、それぞれ混合のレベルを低、中および高として示す。構成を以下にさらに詳細に記載する:
【0119】
構成A
これは混合強度の最小レベルを提供し、スクリュー先端にて計測部材を有する単なる前進搬送部材からなる。この構成は、下記の表に示すように要約することができる:
【0120】
表4:スクリュー構成A、供給から排出までを整理した。
【表4】
【0121】
構成B
これは、分布混合の中レベルを提供し、従来のポリマー合成(混合)実施において使用されるスクリュー構成の一般的な種類である。分布混合(流路を再配置することによって混合)を、前記部材から特定の角度−30、60または90°で配置で、長さD/4の二つの突出物の一連の混合パドルを使用して、本明細書にて達成した。前記パドルを、前進搬送方向に配置し、即ち30°混合パドルは最も前進する搬送、60°はより少なく、90°は0搬送活動および単なる混合を提供する。この構成は、下記の表に示すように要約することができる:
【0122】
表5:スクリュー構成B、供給から排出までを整理した。
【表5】
【0123】
構成C
これは高い度合いの分布および分散の混合を提供する。分散混合(凝集物を分解するための高いせん断作用)を、混合パドルの対を同じ方向に共に配置、即ちスタガー角によって達成した。これは、多くの物質を圧入して、パドルの高いせん断先端を通過する広いパドルを効果的に製造した。この構成は、下記の表に示すように、簡単に要約することができる:
【0124】
表6:スクリュー構成C、供給から排出までを整理した。
【表6】
【0125】
図8a〜8dは、構成A、BおよびCにて使用した二軸スクリュー部材の、以下の種類の写真を示す。
a)搬送構成または供給スクリュー
b)交互(即ち、分布)の混合構成
c)分散混合構成
d)排出構成
【0126】
図15aおよび15bにおいて、押出成形装置のバレルの概略図を左側に示す。装置の長さを10個のゾーンに分割する(図において“ブロック”として分類)−温度ゾーンに対応する前記ゾーンは、下記の表に記載する。この長さをさらに40単位の長さ、スクリュー直径に対応するそれぞれの単位に分割する−それゆえに直径を示す注釈“D”とする。右側に、3つのスクリュー構成A、BおよびCを概略的に記載する。また直径測定を使用して、構成の中のそれぞれの部材の長さを示す。FSは“供給スクリュー”、即ち最小限の混合を提供する搬送スクリューを表す。混合パドルを設ける場合には、回転方向のオフセットの度合いである度数を数値的に示し、部材の数とする−それぞれの混合パドルは長さ0.25Dである。30、60または90度のオフセットは、分布混合に一致し、0度のオフセットは、分散混合領域に対応する。混合部材のオフセット角に対する注釈“f”および“a”は、重要でない。全ての3つの構成を排出スクリューとして仕上げる。
【0127】
実施例5
カルバマゼピン:サッカリン(1:1)
方法:
236gのカルバマゼピンを、183gサッカリンと(即ち、化学両論的に1:1の関係にて)、撹拌混合機で30分間混合した。構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1(Pharmalab HME16,Thermo)を有する押出成形機を、鋳型なしで、表8に記載の温度設定に設定した。一旦、温度が安定したとき、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、室温まで冷却し、分析の対象とした。
【0128】
【表7】
【0129】
【表8】
【0130】
図9a〜9dは、それぞれ構成A、BまたはCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す(全てのPXRD結果のグラフに関するN.B.、X軸は2シータ(θ)を示し、Y軸はカウントを示す)。
【0131】
図9aは、実質的に共結晶が、2−θ=7のピークの欠如による証明することにより、混合物のみから得られないことを示した。
【0132】
図9bに示すように、共結晶は構成Aを使用して全く得られなかった:
2−θ=7のピーク、共結晶の特徴は観測しなかった。
2−θ=8.9のピークにおいて、脱水カルバマゼピンの特徴を顕著に観測した。
【0133】
図9cに示すように、低率純度共結晶を構成Bの使用によって得た:
2−θ=7のピークにおいて、共結晶の特徴であるが、低強度を有する。
2−θ=8.9のピークにおいて、カルバマゼピンの脱水の特徴を観測した。
【0134】
図9dに示すように、高い度合で共結晶を、構成Cを使用して得た:
2−θ=7で特徴的なピーク。
2−θ=8.9で脱水カルバマゼピンの特徴的なピークを全く観測しなかった。
【0135】
図9e〜9hは、構成Cを使用して得られたカルバマゼピン−サッカリン共結晶(1:1)の凝集物のSEMイメージを示す(それぞれ600×、600×、1000×および1800×倍率)。
【0136】
実施例6
カルバマゼピン:ニコチンアミド(1:1)
方法:
236gのカルバマゼピンを、122gのニコチンアミドと(即ち、1:1の化学両論的な関係)、撹拌混合機で30分間混合した。構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1(Pharmalab HME16、Thermo)を有する押出成形機を、鋳型なしで、表10に記載の温度に設定した。一旦、温度が安定すると、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、室温まで冷却し、分析の対象とした。
【0137】
【表9】
【0138】
【表10】
【0139】
図10a〜10dは、それぞれ構成A、BまたはCを使用して、粉砕または押出成形後のPXRD結果のグラフを示す(全てのPXRD結果のグラフに関して、X軸は2シータ(θ)を示し、Y軸はカウントを示す)。
【0140】
図10aに示すように、実質的に共結晶は、2−θ=20.4でピークの欠如により証明したように、混合物のみからは全く得られなかった。
【0141】
図10bに示すように、低率純度は、構成Aを使用して得られた共結晶であった。
2−θ=20.4のピーク、観測された共結晶の特徴であるが、構成Cより非常に低い強度である。
【0142】
図10cに示すように、低率純度の共結晶を、構成Bを用いて得た。
2−θ=20.4のピーク、観測された共結晶の特徴であるが、構成Cよりも低強度である。
2−θ=6.6、8.9、10.1、13.3、15.5、17.8および26.5のピークを観測した;前記ピークは、同様に共結晶の代表的であるように考えられるが、20.4より優位でない。
【0143】
図10dに示すように、良好な純度の共結晶を、構成Cを使用して得た。
2−θ=20.4のピーク、共結晶の特徴は観測した。
2−θ=6.6、8.9、10.1、13.3、15.5、17.8および26.5のピークが、同様に観測された。
【0144】
図10eは、構成Cを使用して得られたカルバマゼピンおよびニコチンアミドの共結晶(1:1)の凝集物のSEMを示す(倍率1000×)。
【0145】
実施例7
カフェイン:マレイン酸(1:1)
方法:
194gカフェインを、116gマレイン酸と(即ち、1:1の化学両論関係)、30分間撹拌混合機で混合した。鋳型を有さない構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1を有する押出成形機(Pharmalab HME16,Thermo)を、表12に記載した温度に設定した。一旦、温度が安定すると、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、室温まで冷却し、分析の対象とした。
【0146】
【表11】
【0147】
【表12】
【0148】
図11a〜11cは、それぞれ構成A、BまたはCを使用して、押出成形後のPXRD結果のグラフを示す(全てのPXRD結果のグラフに関して、X軸は2シータ(θ)を示し、Y軸はカウントを示す)。
【0149】
図11aに示すように、非常に低率純度の共結晶を、構成Aを使用して得て、PXRDパターンが、高いせん断のバッチと比較して、2−θ=9、11.1、13.2、14.2および15.5にて非常に低強度の特徴的なピークを示す一方、2−θ=12のピーク、無水β−カフェインの特徴は非常に高かった。
【0150】
図11bに示すように、低率純度の共結晶を、構成Bを使用して得て、PXRDパターンが、2−θ=9、11.1、13.2、14.2および15.5にて特徴的なピークを、高いせん断のバッチと比較して低い強度で示す一方、2−θ=12の小ピーク、無水β−カフェインの特徴を観測した。
【0151】
図11cに示すように、良好な純度の共結晶を、構成Cを使用して得て、PXRDパターンが、2−θ=13.2のピークと、2−θ=9、11.1、13.2、14.2および15.5にて特徴的なピークを、最も高い強度を有して示した。
【0152】
図11d〜11gは、構成Cを使用して生成されたカフェインおよびマレイン酸の共結晶の凝集物のSEMイメージを、それぞれ倍率137×、550×、600×および820×にて示す。
【0153】
実施例8
カフェイン:マレイン酸(2:1)
方法:
388gカフェインを、116gマレイン酸と(即ち、化学両論的な関係2:1)、撹拌混合機で30分混合した。鋳型を有さない構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1を有する押出成形機(Pharmalab HME16,Thermo)を、表14に記載した温度に設定した。一旦、温度が安定したとき、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、分析の対象とした。
【0154】
【表13】
【0155】
【表14】
【0156】
図12a〜12cは、それぞれ構成A、BまたはCを使用して、押出成形後のPXRDの結果のグラフを示す(全てのPXRD結果のグラフに関して、X軸は2シータ(θ)を示し、Y軸はカウントを示す)。
【0157】
図12aに示すように、非常に低率純度の共結晶を、構成Aを使用して得た。
2−θ=8.8、10.1、13.5および16のピーク、高いせん断でのバッチと比較して、非常に低い強度を有する。
2−θ=12のピークの強度、無水β−カフェインの特徴は非常に高い。
【0158】
図12bに示すように、低率の純度の共結晶を、構成Bを使用して得た。
2:1共結晶の2−θ=8.8、10.1、13.5および16でピークを観測した。
無水β−カフェインの2−θ=12の特徴でピークの強度は、比較的高かった。
【0159】
図12cに示すように、良好な純度の共結晶を、構成Cを使用して得た。
2−θ=8.8、10.1、13.5および16で2:1共結晶の特徴的なピークを観測した。
無水β−カフェインの2−θ=12のピークの特徴は、重要でない。
【0160】
図12dおよび12eは、構成Cを使用して得たカフェインおよびマレイン酸の共結晶(2:1)の凝集物のSEMイメージを示す(倍率880×および2000×)。
【0161】
実施例9
テオフィリン:マレイン酸(1:1)
方法:
180gテオフィリンを、116gマレイン酸と(即ち、化学両論関係1:1)、撹拌混合機で30分間混合した。鋳型を有さない構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1である押出成形機(Pharmalab HME16、Thermo)を、表16に記載した温度に設定した。一旦、温度が安定すると、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、分析の対象とした。
【0162】
【表15】
【0163】
【表16】
【0164】
図13a〜13dは、それぞれ構成A、BまたはCを使用して、粉砕または押出成形後におけるPXRDの結果のグラフを示す(全てのPXRDの結果のグラフに関して、X軸は2シータ(θ)を示し、Y軸はカウントを示す)。
【0165】
図13aに示すように、実質的に共結晶は、共結晶のピークの特徴の欠如によって証明されるように、混合物のみからは全く得られなかった。2θ=12.5のピークは、テオフィリンの特徴である。
【0166】
図13bに示すように、非常に低率純度の共結晶を、構成Aを使用して得た。
2θ=9、11.5、13.6および16.8で共結晶の特徴的なピークを観測したが、非常に低い、微々たるピーク強度を有する。
2θ=12.5でテオフィリンの特徴的なピークを、顕著に観測した。
【0167】
図13cに示すように、低率の純度の共結晶を、構成Bを使用して得た。
2θ=9、11.5、12、13.6および16.8で共結晶の特徴的なピークを観測したが、バッチと比較すると低強度を有する。
2θ=12.5でテオフィリンの特徴的なピークは微々たるものである。
【0168】
図13dに示すように、良好な純度の共結晶を、構成Cを使用して得た。
2θ=9、11.5、12、13.6および16.8で共結晶の特徴的なピークを観測した。
2θ=12.5でテオフィリンの特徴的なピークは微々たるものである。
【0169】
図13eは、構成Cを使用して得られたテオフィリンおよびマレイン酸の共結晶(モル比1:1)の凝集物のSEMイメージを示す。
【0170】
実施例10
サリチル酸:ニコチンアミド(1:1)
方法:
138gサリチル酸を、122gマレイン酸と(即ち、化学両論関係1:1)、撹拌混合機で30分混合した。鋳型を有さない構成A、BまたはCで記載した異なったスクリュー幾何学的形状で、LD比率40:1を有する押出成形機(Pharmalab HME16,Thermo)を、表18に記載した温度に設定した。一旦、温度が安定すると、混合物を押出成形機に供給し、スクリューを20rpmにて回転した。微細な凝集生成物をエンドゾーン10にて収集し、分析の対象とした。
【0171】
【表17】
【0172】
【表18】
【0173】
図14a〜14cは、それぞれ構成A、BまたはCを使用して、押出成形後のPXRDの結果のグラフを示す(全てのPXRD結果のグラフに関して、X軸は2シータ(θ)を示すが、Y軸はカウントを示す)。
【0174】
図14aに示すように、低純度の共結晶を、構成Aを使用して得た。
2−θ=7.8、8.4および9.1で共結晶に特徴的なピークを観測したが、高いせん断の押出成形と比較して、低い強度を有する。
【0175】
図14bに示すように、良好な純度の共結晶を得た。
2−θ=7.8、8.4および9.1で共結晶に特徴的なピークを観測した。
【0176】
図14cに示すように、良好な純度の共結晶を、構成Cを使用して得た。
2θ=7.8、8.4および9.1のピーク、共結晶の特徴は観測した。
【0177】
図14dおよび14eは、構成Cを使用して形成したサリチル酸およびニコチンアミドの共結晶の凝集物を示す(倍率それぞれ200×および800×)。
【0178】
実施例5〜10から、構成Cが比較的高い純度の共結晶を一貫して提供することが分かる。従って、以下を決定することができる:
押出成形は、多種多様なAPIsおよびゲスト物質から共結晶を生成するための確実な方法を提供する。
混合の厳格さは、共結晶を達成する際に重要である。
分散混合の領域の供給は、高レベルな共結晶形成の達成において重要である。
【0179】
溶解データ(生体外薬剤放出):
以下の実験を、水中および人口胃液中の純カルバマゼピンと実施例5に従って形成された共結晶の飽和溶解度および生体外における分解と比較して実施した。
【0180】
カルバマゼピン:サッカリン
純カルバマゼピンおよびカルバマゼピン−サッカリンの共結晶の飽和溶解度測定を、20mLの水に既知の過剰量のサンプルを添加することによって実施した。サンプルを、水浴(37±0.5℃)にて20rpm、24時間撹拌した。その後サンプルを0.45μmの膜濾過で濾過し、水と希釈し、その後分光光度計(Jasco V−630,Jasco Corporation, Japan)によって320nmにて分析した。カルバマゼピン−サッカリンの共結晶(0.78mg/mL)は、カルバマゼピンと(0.29mg/mL)比較して著しい改善を飽和溶解度にて示した。
【0181】
【表19】
【0182】
カルバマゼピン:サッカリン−生体外溶出試験
純カルバマゼピン、および上記の実施例5に従って構成Cを使用して生成したカルバマゼピン−サッカリン共結晶の凝集物(カルバマゼピン200mgに等しい)の溶解研究を、人口胃液(SGF)900mLにてUSP24タイプII溶出試験装置を使用して実施した。SGFを、2g/LNaClおよび1g/LTritonX−100と調製し、pH2までHClで酸性化した。浴槽温度を37±0.5℃に維持し、100rpmで撹拌した。サンプルを、15、30および60分にて収集され、新鮮な溶出媒体と交換した。遠心分離後、0.45μm膜濾過器を通して濾過し、適切な希釈度の、カルバマゼピンの濃度を分光光度法にて320nmで測定した。
【0183】
【表20】
【0184】
結論
純カルバマゼピン粉末は、69.11%のみの薬剤放出を60分間のSGFにて示した。一方、カルバマゼピン−サッカリン共結晶は、86.79%薬剤放出を示した。共結晶凝集物からの初期薬剤放出は、純薬と比較して急速であった。共結晶は15分にて薬剤放出74.74%、30分にて薬剤放出82.89%を示す一方、純カルバマゼピンは15分にて薬剤放出43.48%、30分にて薬剤放出59.29%を示すのみであった。従って、この結果は、本方法の生成物が、共結晶構造に含有した活性薬剤の生物学的利用能を増加させることに極めて適切であることを示す。
【0185】
圧縮研究(錠剤形成)
構成Cを使用して押出成形したバッチの産出生成物を、それらの圧縮化および錠剤形成の性質に関して分析した。
【0186】
カルバマゼピン:サッカリンの共結晶(1:1)
圧縮性の研究を、10mm直径の平板対面パンチを取り付ける圧縮研究のプレス(Caleva Process Solutions Ltd., England)を使用して実施した。鋳型壁を、それぞれの圧縮前に、アセトンで清浄し、前もってステアリン酸マグネシウムで滑らかにした。カルバマゼピン−サッカリン共結晶の凝集物300mgを、鋳型内に手動充填した。圧縮および減圧は、100mm/min、残留負荷度合い10,000N、残留時間0.1秒にて実施し、圧縮力に対する体積変化を記録した。錠剤の厚さは、シックネスゲージ(Mitutoyo,Japan)を使用して測定し、硬度は硬度計(Schleuniger−4M,Copley)を使用して試験した。
【0187】
【表21】
【0188】
イブプロフェン−ニコチンアミドの共結晶(1:1):
圧縮性の研究を、10mm直径の平板対面パンチを取り付ける圧縮研究のプレス(Caleva Process Solutions Ltd., England)を使用して実施した。鋳型壁を、それぞれの圧縮前に、アセトンで清浄し、ステアリン酸マグネシウムで前もって滑らかにした。イブプロフェン−ニコチンアミド共結晶の凝集物400mgを、鋳型内に手動充填した。圧縮および減圧は、100mm/min、残留負荷度合い5,000N、残留時間0.1秒にて実施し、圧縮力に対する体積変化を記録した。錠剤の厚さは、シックネスゲージ(Mitutoyo,Japan)を使用して測定し、硬度は硬度計(Schleuniger−4M,Copley)を使用して試験した。
【0189】
【表22】
【0190】
結論
この技術によって生成した共結晶凝集は、直接圧縮可能である一方、成分の物理的混合は、同様の条件に基づいて圧縮形成できなかった。圧縮物の破壊強度に関して本発明によって得られる粉末物質の圧縮能力は、共結晶の凝集物を使用して得られた圧縮物が、物理的混合物を使用して得たものと比較して、高度な破壊強度を有することを示した。
【0191】
前記実験結果は、上記に示したように押出成形工程の生成物を、凝固によって錠剤に直接形成するのに適していることを示す。これは錠剤製造を簡単にするために、極めて実用的である。
【特許請求の範囲】
【請求項1】
共結晶を生成する方法において、
第一および第二の物質を付与し、第一および第二の物質は共結晶を形成するのに適合する工程と、
前記第一および第二の物質を共に混合する工程と、
前記第一および第二の物質の混合物を、圧力およびせん断の長期の持続した条件にさらして、前記第一および第二の物質の共結晶を十分に形成する工程と
を含有する方法。
【請求項2】
上述の方法の産出生成物のPXRDパターンを、第一および第二の物質のみもしくは混合物のPXRDパターン、または関連のある共結晶の既知のPXRDパターンと比較することによって、共結晶の存在を同定する工程を含有する請求項1に記載の方法。
【請求項3】
連続流動方法である、請求項1または2に記載の方法。
【請求項4】
第一物質は、活性医薬品成分(API)である、前記いずれか一項に記載の方法。
【請求項5】
第一および第二の物質を、圧力およびせん断の持続した条件に少なくとも1分、好ましくは2分以上、特に2〜40分、とりわけ2〜30分さらす、前記いずれか一項に記載の方法。
【請求項6】
前記方法は、少なくとも20w/w%共結晶体、40w/w%共結晶体、より好ましくは少なくとも60w/w%共結晶体、特に少なくとも共結晶の80w/w%共結晶体を含有する産出生成物を得るのに適切である、前記いずれか一項に記載の方法。
【請求項7】
前記方法は、少なくとも90w/w%共結晶体以上の純度を達成することができるものを含有する産出生成物を得るのに適切である、前記いずれか一項に記載の方法。
【請求項8】
圧力およびせん断を押出方法にて適応する、前記いずれか一項に記載の方法。
【請求項9】
圧力およびせん断をスクリューに基づく押出方法に適応する、前記いずれか一項に記載の方法。
【請求項10】
スクリューに基づく押出成形方法は、複数のスクリューに基づく押出成形方法である、請求項9に記載の方法。
【請求項11】
スクリューに基づく押出方法は、二軸押出方法である、請求項10に記載の方法。
【請求項12】
二軸押出方法は、共回転方法である、請求項11に記載の方法。
【請求項13】
スクリューは少なくとも実質的に噛合する、請求項10〜12のいずれか一項に記載の方法。
【請求項14】
前記方法は、共結晶を形成することができる第一および第二の物質で単に実行する、前記いずれか一項に記載の方法。
【請求項15】
前記第一および第二の物質を、付加的な熱にさらす、前記いずれか一項に記載の方法。
【請求項16】
処理を、処理の持続期間の少なくとも一部で、最も低い融点を有する共結晶を形成する物質の融点付近の温度にて実行する、前記いずれか一項に記載の工程方法。
【請求項17】
押出機の以下一つ以上の性質を選択する工程:スクリューまたはバレルの長さ、長さの比:スクリューの直径(L/D比)、スクリュー部材の組成物(例えば、分布または分散混合部材、前進または逆供給部材)、スクリュー底の深さ(即ち、ネジ山の深さ)、スクリュー回転速度、供給方法(例えば、スターベーション供給対フラッド供給)、および押出成形機を通過する数、を含有する前記いずれか一項に記載の方法。
【請求項18】
スクリューの長さとスクリューの直径の割合は、15/1以上、好ましくは20/1以上、好ましくは30/1以上、任意に約40/1である、前記いずれか一項に記載の方法。
【請求項19】
混合物を、、分布または分散の混合の少なくとも一定期間にさらす、前記いずれか一項に記載の方法。
【請求項20】
混合物を、分散混合の少なくとも一定期間にさらす、請求項19に記載の方法。
【請求項21】
前記方法にて使用した押出装置は、スクリューの全長の少なくとも1/40、好ましくは少なくとも1/30、より好ましくはスクリューの全長の少なくとも1/20で、分散混合領域を含有する、請求項20に記載の方法。
【請求項22】
少なくとも一つの分散散混合領域があり、分散混合の全ての領域の全長は、少なくともスクリュー直径の1.5倍以上、好ましくはスクリュー直径の2倍以上である、請求項20または21に記載の方法。
【請求項23】
第一および第二の物質のそれぞれは、以下の一つ:カルボン酸、アミン、アミド、スルホンアミド、ヒドロキシアルコール、ケトン、アミノ酸、糖類、複素環式塩基である、前記いずれか一項に記載の方法。
【請求項24】
第一物質は、ナプロキセン、イブプロフェン、ケトプロフェン、トルメチン、フェノプロフェン、インドメタシン、サリチル酸、ナブメトン、ピロキシカム、ピオグリタゾン、グリピジド、グリメピリド、トルブタミド、ワルファリン、アトルバスタチン(スタチン)、プラゾシン、カプトプリル、ニフェジピン、フェロジピン、アムロジピン、リドカイン、ラモトリギン、アンフェタミン、メトホルミン、フルオキセチン、プリマキン、カフェイン、テオフィリン、カルバマゼピン、セレコキシブ、バルデコキシブ、アテノロールおよびプロプラノロールの一つである、前記いずれか一項に記載の方法。
【請求項25】
第二物質は、グルタル酸、クエン酸、フマル酸、マロン酸、シュウ酸、安息香酸、リンゴ酸、マレイン酸、酒石酸、コハク酸、アジピン酸、サリチル酸、桂皮酸、アントラニル酸、馬尿酸、チロシン、リジン、アルギニン、イソロイシン、トリプトファン、ヒスチジン、システイン、サッカリン、フルクトース、マンニトール、ブドウ糖、アスパルテームおよびニコチンアミドの一つである、前記いずれか一項に記載の方法。
【請求項26】
第一および第二の物質を化学両論的比率で付与する、前記いずれか一項に記載の方法。
【請求項27】
第一物質はフェニルアルカン酸である、前記いずれか一項に記載の方法。
【請求項28】
第一物質はイブプロフェン、第二物質はニコチンアミドである、請求項27に記載の方法。
【請求項29】
第一物質はカルバマゼピン、第二物質はサッカリンである、請求項1〜26のいずれか一項に記載の方法。
【請求項30】
第一物質はカルバマゼピン、第二物質はニコチンアミドである、請求項1〜26のいずれか一項に記載の方法。
【請求項31】
第一物質はカフェイン、第二物質はマレイン酸である、請求項1〜26のいずれか一項に記載の方法。
【請求項32】
第一物質はテオフィリン、第二物質はマレイン酸である、請求項1〜26のいずれか一項に記載の方法。
【請求項33】
第一物質はサリチル酸、第二物質はニコチンアミドである、請求項1〜26のいずれか一項に記載の方法。
【請求項34】
共結晶を実質的に完了したに後に、押出成形処理で修飾化合物を導入する工程を含有する、前記いずれか一項に記載の方法。
【請求項35】
請求項1〜34のいずれか一項に記載の方法を含む、凝集した共結晶の粒子を含有する粒子形成の方法。
【請求項36】
請求項1〜35のいずれか一項に記載の方法を実施する工程と、前記粒子を、任意に適切な鋳型において圧縮して、単位用量形状を形成する工程とを含有する、活性薬剤の単位用量形状を形成する方法。
【請求項37】
凝集された共結晶の粒子を圧縮して単位用量形状を形成する工程を含有する、請求項36に記載の方法。
【請求項38】
共結晶を含有する組成物であって、共結晶は第一および第二の物質を含有し、第一および第二物質を請求項1〜37のいずれか一項に記載の処理にさらした組成物。
【請求項39】
フェニルアルカン酸およびニコチンアミド、好ましくはイブプロフェンおよびニコチンアミド;
カルバマゼピンおよびサッカリン;
カルバマゼピンおよびニコチンアミド;
カフェインおよびマレイン酸;
テオフィリンおよびマレイン酸;
サリチル酸およびニコチンアミド
の一つ以上の共結晶を含有する生成物。
【請求項40】
少なくとも50w/w%共結晶体、より好ましくは共結晶の75w/w%共結晶体を含有する、請求項39に記載の生成物。
【請求項41】
請求項1〜37のいずれか一項に記載の処理によって得たか、または得ることができる共結晶を含有する組成物。
【請求項42】
凝集された共結晶の粒子で、好ましくは直径2〜3000μmであるものを含有する、請求項41に記載の組成物。
【請求項43】
請求項38〜42のいずれか一項に記載の共結晶を、任意に製薬学的に許容される賦形剤と組み合わせて含む組成物を含有する医薬品。
【請求項44】
薬物治療で使用する、請求項38〜43のいずれか一項に記載の共結晶。
【請求項45】
病状の治療のための薬剤の製造における、請求項38〜42のいずれか一項に記載の共結晶の使用。
【請求項1】
共結晶を生成する方法において、
第一および第二の物質を付与し、第一および第二の物質は共結晶を形成するのに適合する工程と、
前記第一および第二の物質を共に混合する工程と、
前記第一および第二の物質の混合物を、圧力およびせん断の長期の持続した条件にさらして、前記第一および第二の物質の共結晶を十分に形成する工程と
を含有する方法。
【請求項2】
上述の方法の産出生成物のPXRDパターンを、第一および第二の物質のみもしくは混合物のPXRDパターン、または関連のある共結晶の既知のPXRDパターンと比較することによって、共結晶の存在を同定する工程を含有する請求項1に記載の方法。
【請求項3】
連続流動方法である、請求項1または2に記載の方法。
【請求項4】
第一物質は、活性医薬品成分(API)である、前記いずれか一項に記載の方法。
【請求項5】
第一および第二の物質を、圧力およびせん断の持続した条件に少なくとも1分、好ましくは2分以上、特に2〜40分、とりわけ2〜30分さらす、前記いずれか一項に記載の方法。
【請求項6】
前記方法は、少なくとも20w/w%共結晶体、40w/w%共結晶体、より好ましくは少なくとも60w/w%共結晶体、特に少なくとも共結晶の80w/w%共結晶体を含有する産出生成物を得るのに適切である、前記いずれか一項に記載の方法。
【請求項7】
前記方法は、少なくとも90w/w%共結晶体以上の純度を達成することができるものを含有する産出生成物を得るのに適切である、前記いずれか一項に記載の方法。
【請求項8】
圧力およびせん断を押出方法にて適応する、前記いずれか一項に記載の方法。
【請求項9】
圧力およびせん断をスクリューに基づく押出方法に適応する、前記いずれか一項に記載の方法。
【請求項10】
スクリューに基づく押出成形方法は、複数のスクリューに基づく押出成形方法である、請求項9に記載の方法。
【請求項11】
スクリューに基づく押出方法は、二軸押出方法である、請求項10に記載の方法。
【請求項12】
二軸押出方法は、共回転方法である、請求項11に記載の方法。
【請求項13】
スクリューは少なくとも実質的に噛合する、請求項10〜12のいずれか一項に記載の方法。
【請求項14】
前記方法は、共結晶を形成することができる第一および第二の物質で単に実行する、前記いずれか一項に記載の方法。
【請求項15】
前記第一および第二の物質を、付加的な熱にさらす、前記いずれか一項に記載の方法。
【請求項16】
処理を、処理の持続期間の少なくとも一部で、最も低い融点を有する共結晶を形成する物質の融点付近の温度にて実行する、前記いずれか一項に記載の工程方法。
【請求項17】
押出機の以下一つ以上の性質を選択する工程:スクリューまたはバレルの長さ、長さの比:スクリューの直径(L/D比)、スクリュー部材の組成物(例えば、分布または分散混合部材、前進または逆供給部材)、スクリュー底の深さ(即ち、ネジ山の深さ)、スクリュー回転速度、供給方法(例えば、スターベーション供給対フラッド供給)、および押出成形機を通過する数、を含有する前記いずれか一項に記載の方法。
【請求項18】
スクリューの長さとスクリューの直径の割合は、15/1以上、好ましくは20/1以上、好ましくは30/1以上、任意に約40/1である、前記いずれか一項に記載の方法。
【請求項19】
混合物を、、分布または分散の混合の少なくとも一定期間にさらす、前記いずれか一項に記載の方法。
【請求項20】
混合物を、分散混合の少なくとも一定期間にさらす、請求項19に記載の方法。
【請求項21】
前記方法にて使用した押出装置は、スクリューの全長の少なくとも1/40、好ましくは少なくとも1/30、より好ましくはスクリューの全長の少なくとも1/20で、分散混合領域を含有する、請求項20に記載の方法。
【請求項22】
少なくとも一つの分散散混合領域があり、分散混合の全ての領域の全長は、少なくともスクリュー直径の1.5倍以上、好ましくはスクリュー直径の2倍以上である、請求項20または21に記載の方法。
【請求項23】
第一および第二の物質のそれぞれは、以下の一つ:カルボン酸、アミン、アミド、スルホンアミド、ヒドロキシアルコール、ケトン、アミノ酸、糖類、複素環式塩基である、前記いずれか一項に記載の方法。
【請求項24】
第一物質は、ナプロキセン、イブプロフェン、ケトプロフェン、トルメチン、フェノプロフェン、インドメタシン、サリチル酸、ナブメトン、ピロキシカム、ピオグリタゾン、グリピジド、グリメピリド、トルブタミド、ワルファリン、アトルバスタチン(スタチン)、プラゾシン、カプトプリル、ニフェジピン、フェロジピン、アムロジピン、リドカイン、ラモトリギン、アンフェタミン、メトホルミン、フルオキセチン、プリマキン、カフェイン、テオフィリン、カルバマゼピン、セレコキシブ、バルデコキシブ、アテノロールおよびプロプラノロールの一つである、前記いずれか一項に記載の方法。
【請求項25】
第二物質は、グルタル酸、クエン酸、フマル酸、マロン酸、シュウ酸、安息香酸、リンゴ酸、マレイン酸、酒石酸、コハク酸、アジピン酸、サリチル酸、桂皮酸、アントラニル酸、馬尿酸、チロシン、リジン、アルギニン、イソロイシン、トリプトファン、ヒスチジン、システイン、サッカリン、フルクトース、マンニトール、ブドウ糖、アスパルテームおよびニコチンアミドの一つである、前記いずれか一項に記載の方法。
【請求項26】
第一および第二の物質を化学両論的比率で付与する、前記いずれか一項に記載の方法。
【請求項27】
第一物質はフェニルアルカン酸である、前記いずれか一項に記載の方法。
【請求項28】
第一物質はイブプロフェン、第二物質はニコチンアミドである、請求項27に記載の方法。
【請求項29】
第一物質はカルバマゼピン、第二物質はサッカリンである、請求項1〜26のいずれか一項に記載の方法。
【請求項30】
第一物質はカルバマゼピン、第二物質はニコチンアミドである、請求項1〜26のいずれか一項に記載の方法。
【請求項31】
第一物質はカフェイン、第二物質はマレイン酸である、請求項1〜26のいずれか一項に記載の方法。
【請求項32】
第一物質はテオフィリン、第二物質はマレイン酸である、請求項1〜26のいずれか一項に記載の方法。
【請求項33】
第一物質はサリチル酸、第二物質はニコチンアミドである、請求項1〜26のいずれか一項に記載の方法。
【請求項34】
共結晶を実質的に完了したに後に、押出成形処理で修飾化合物を導入する工程を含有する、前記いずれか一項に記載の方法。
【請求項35】
請求項1〜34のいずれか一項に記載の方法を含む、凝集した共結晶の粒子を含有する粒子形成の方法。
【請求項36】
請求項1〜35のいずれか一項に記載の方法を実施する工程と、前記粒子を、任意に適切な鋳型において圧縮して、単位用量形状を形成する工程とを含有する、活性薬剤の単位用量形状を形成する方法。
【請求項37】
凝集された共結晶の粒子を圧縮して単位用量形状を形成する工程を含有する、請求項36に記載の方法。
【請求項38】
共結晶を含有する組成物であって、共結晶は第一および第二の物質を含有し、第一および第二物質を請求項1〜37のいずれか一項に記載の処理にさらした組成物。
【請求項39】
フェニルアルカン酸およびニコチンアミド、好ましくはイブプロフェンおよびニコチンアミド;
カルバマゼピンおよびサッカリン;
カルバマゼピンおよびニコチンアミド;
カフェインおよびマレイン酸;
テオフィリンおよびマレイン酸;
サリチル酸およびニコチンアミド
の一つ以上の共結晶を含有する生成物。
【請求項40】
少なくとも50w/w%共結晶体、より好ましくは共結晶の75w/w%共結晶体を含有する、請求項39に記載の生成物。
【請求項41】
請求項1〜37のいずれか一項に記載の処理によって得たか、または得ることができる共結晶を含有する組成物。
【請求項42】
凝集された共結晶の粒子で、好ましくは直径2〜3000μmであるものを含有する、請求項41に記載の組成物。
【請求項43】
請求項38〜42のいずれか一項に記載の共結晶を、任意に製薬学的に許容される賦形剤と組み合わせて含む組成物を含有する医薬品。
【請求項44】
薬物治療で使用する、請求項38〜43のいずれか一項に記載の共結晶。
【請求項45】
病状の治療のための薬剤の製造における、請求項38〜42のいずれか一項に記載の共結晶の使用。
【図1】


【図2】
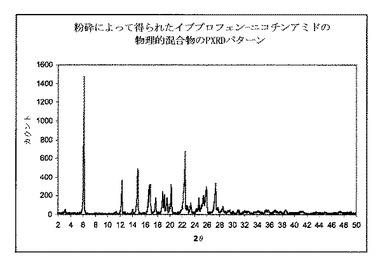
【図3】
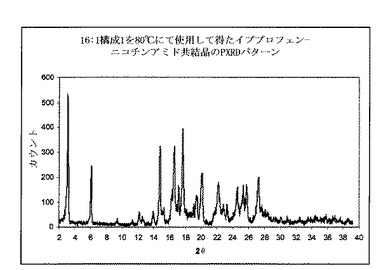
【図4】
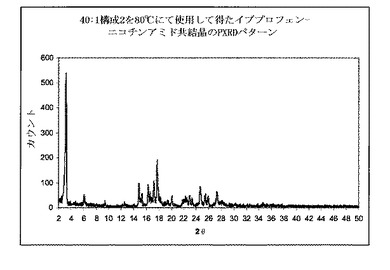
【図5】
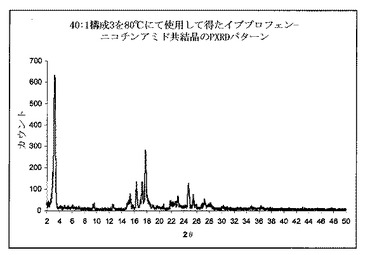
【図6】
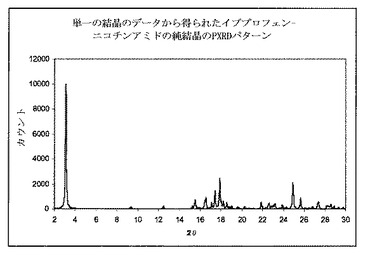
【図6a】

【図6b】

【図6c】

【図6d】

【図7】
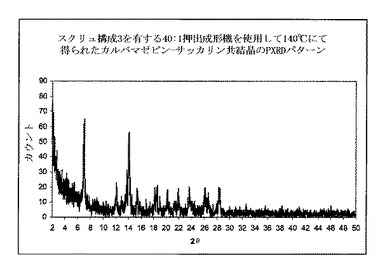
【図8a】
【図8b】
【図8c】
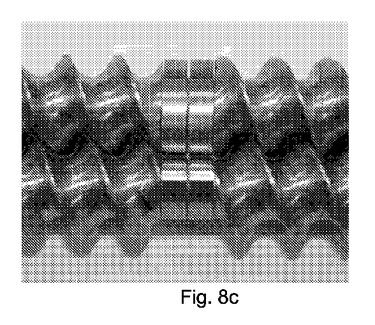
【図8d】
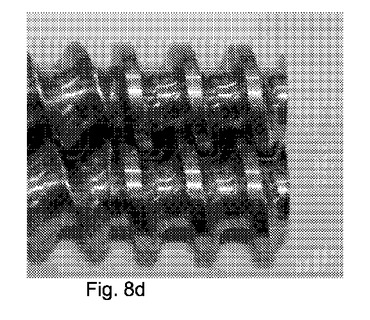
【図9a】
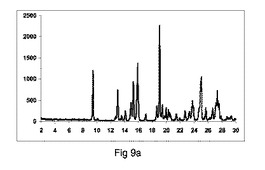
【図9b】
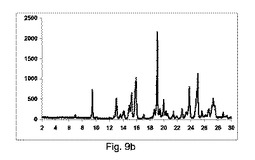
【図9c】
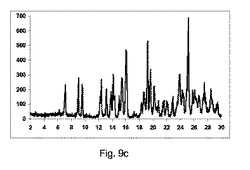
【図9d】
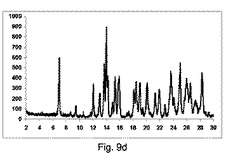
【図9e】

【図9f】
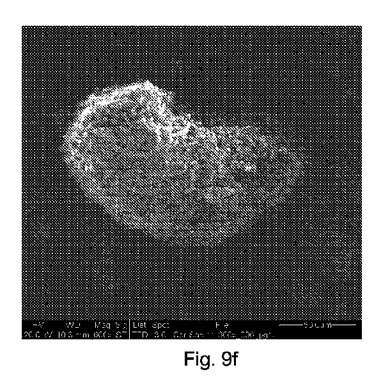
【図9g】

【図9h】

【図10a】
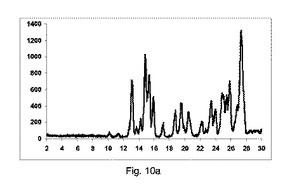
【図10b】
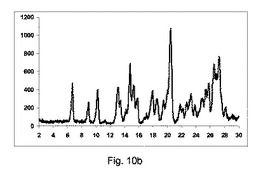
【図10c】
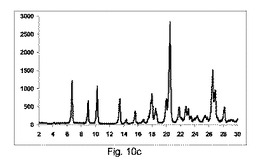
【図10d】
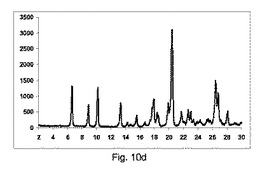
【図10e】

【図11a】
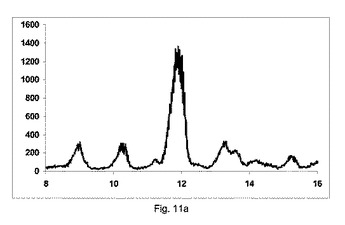
【図11b】
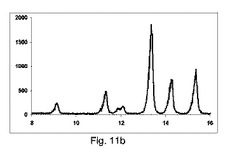
【図11c】
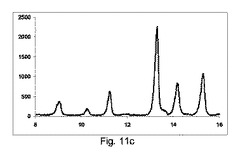
【図11d】

【図11e】

【図11f】

【図11g】

【図12a】
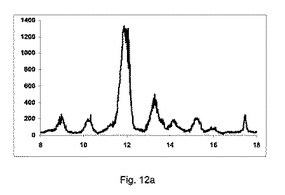
【図12b】
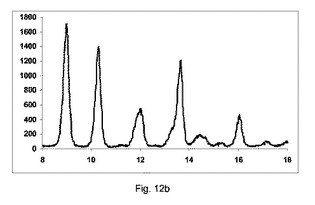
【図12c】
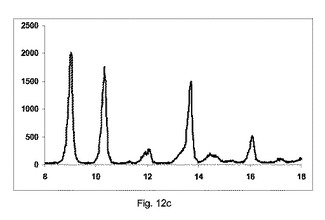
【図12d】

【図12e】

【図13a】
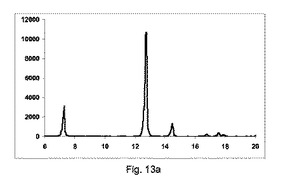
【図13b】
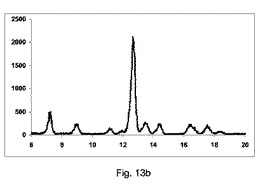
【図13c】
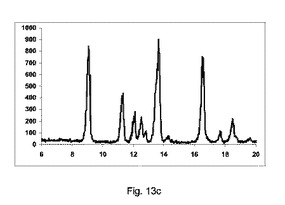
【図13d】
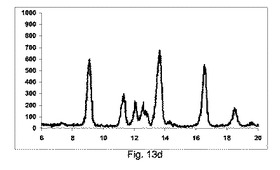
【図13e】
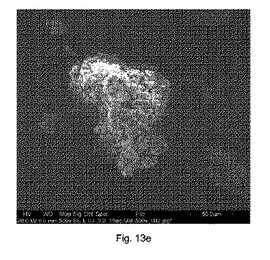
【図14a】
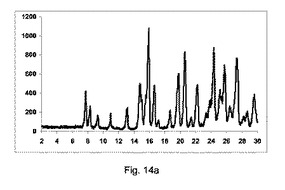
【図14b】
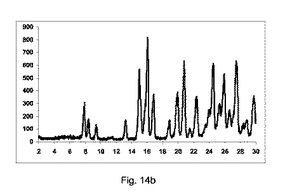
【図14c】
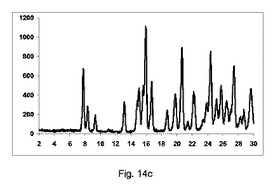
【図14d】

【図14e】

【図15a】

【図15b】

【図2】
【図3】
【図4】
【図5】
【図6】
【図6a】
【図6b】
【図6c】
【図6d】
【図7】
【図8a】
【図8b】
【図8c】
【図8d】
【図9a】
【図9b】
【図9c】
【図9d】
【図9e】
【図9f】
【図9g】
【図9h】
【図10a】
【図10b】
【図10c】
【図10d】
【図10e】
【図11a】
【図11b】
【図11c】
【図11d】
【図11e】
【図11f】
【図11g】
【図12a】
【図12b】
【図12c】
【図12d】
【図12e】
【図13a】
【図13b】
【図13c】
【図13d】
【図13e】
【図14a】
【図14b】
【図14c】
【図14d】
【図14e】
【図15a】
【図15b】
【公表番号】特表2011−529101(P2011−529101A)
【公表日】平成23年12月1日(2011.12.1)
【国際特許分類】
【出願番号】特願2011−520593(P2011−520593)
【出願日】平成21年7月27日(2009.7.27)
【国際出願番号】PCT/GB2009/050924
【国際公開番号】WO2010/013035
【国際公開日】平成22年2月4日(2010.2.4)
【出願人】(511060076)ユニバーシティー オブ ブラッドフォード (2)
【氏名又は名称原語表記】UNIVERSITY OF BRADFORD
【Fターム(参考)】
【公表日】平成23年12月1日(2011.12.1)
【国際特許分類】
【出願日】平成21年7月27日(2009.7.27)
【国際出願番号】PCT/GB2009/050924
【国際公開番号】WO2010/013035
【国際公開日】平成22年2月4日(2010.2.4)
【出願人】(511060076)ユニバーシティー オブ ブラッドフォード (2)
【氏名又は名称原語表記】UNIVERSITY OF BRADFORD
【Fターム(参考)】
[ Back to top ]