
有機分子の結晶多形の定量方法
【課題】内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができる方法を提供する。
【解決手段】有機分子の結晶多形を2相以上含有する試料を、リートベルト法による多相解析に付すことにより前記結晶多形を定量する方法であって、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いて多相解析を行う。
【解決手段】有機分子の結晶多形を2相以上含有する試料を、リートベルト法による多相解析に付すことにより前記結晶多形を定量する方法であって、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いて多相解析を行う。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、有機分子の結晶多形の定量方法に関するものである。
【背景技術】
【0002】
有機分子の結晶多形は重要である。例えば医薬分野においては、ICH ガイドラインQ6A (International Conference on Harmonization Harmonized Guideline. Q6A Specifications, Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products, Chemical Substances, 1999)において、結晶多形の存在形(どの結晶形を含んでいるか)を規定すべきであることが記載されており、結晶多形が医薬品の品質や機能に影響を及ぼす場合には、その含量を精密に定量することが重要になっている。
【0003】
有機分子の結晶多形の定量方法として、例えば非特許文献1には、有機分子の結晶多形が混合した混合粉末試料を、フッ化リチウムを内部標準試料として用いたリートベルト (Rietveld) 法を用いて定量測定する方法が記載されている。そして、上記非特許文献1には、4相の有機分子の結晶多形が混合した粉末試料であっても微量まで定量することができること、特に、2相の有機分子の結晶多形が混合した粉末試料では複相を1重量%以下まで定量することができることが記載されている。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Sampath S. Iyengar et al. (Powder Diffraction 16, 1, 20. (2001))
【非特許文献2】Zeltan Nemet et al. (Journal of Pharmaceutical and Biomedical Analysis, 51, 572. (2010))
【発明の概要】
【発明が解決しようとする課題】
【0005】
ところが、非特許文献1に記載の方法では、フッ化リチウムを内部標準試料として用いている。このため、測定対象の試料とフッ化リチウムとが反応し、これによって結晶多形が変化しているおそれを排除することができない。また、内部標準試料としてフッ化リチウム以外の化合物を用いたとしても、当該化合物と測定対象の試料との反応性や親和性を予め調査する必要がある。
【0006】
内部標準試料を用いない方法として、非特許文献2には、Famotidineの結晶多形の混合試料を用いて、透過測定法を用いたリートベルト法による多相解析法が記載されている。当該非特許文献2においては、X線照射体積が一定になり定量測定に好適な方法を採用することにより、即ち、一組のフィルムに挟んで円盤状(foil)に成形した試料を用いることで選択配向を軽減して定量ができることが記載されている。そして、非特許文献2においては、ブラッグの条件で計算される面間隔値dが最低角反射に相当する角度を大きく上回る範囲におけるX線回折データを用いることにより、測定下限が3重量%で測定した例が記載されている。
【0007】
本発明は、上記状況に鑑みてなされたものであり、その主たる目的は、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中の前記結晶多形を、従来の方法よりも低い検出下限で定量することができる方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明者は上記状況の下、照射体積一定条件で収集され、最低角反射に相当する角度を含む範囲における粉末X線回折データを用い、有機分子の結晶多形の定量を、リートベルト法による多相解析法により実施することにより、内部標準試料を用いることなく、従来の方法よりも低い検出下限で定量することができることを見出した。
【0009】
即ち、本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料を、リートベルト法による多相解析に付すことにより前記結晶多形を定量する方法であって、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いて多相解析を行うことを特徴としている。
【0010】
上記照射体積一定条件とは、デバイ・シェラー (Debye-Scherrer) 法による透過測定法であることがより好ましい。また、粉砕工程を経ていない粉末試料を用いることがより好ましい。さらに、上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよい。
【0011】
上記方法によれば、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いるので、精度が向上し、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができる。
【0012】
また、本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料中の前記結晶多形を、粉末X線回折強度比を用いて作成した検量線を用いて定量する方法であって、照射体積一定条件であるデバイ・シェラー法による透過測定法を用い、かつ、粉砕工程を経ていない粉末試料を用いることを特徴としている。
【0013】
ブラッグの条件で計算される面間隔値dが測定対象の粉末試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いることがより好ましい。また、上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよい。さらに、上記検量線が、結晶多形に含まれる全ての単一結晶の粉末X線回折強度を含んで作成されていることがより好ましい。
【0014】
上記方法によれば、照射体積一定条件であるデバイ・シェラー法による透過測定法を用い、かつ、粉砕工程を経ていない粉末試料を用い、粉末X線回折強度比を用いて作成した検量線を用いて定量するので、精度が向上し、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができる。
【発明の効果】
【0015】
本発明に係る有機分子の結晶多形の定量方法によれば、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができるという効果を奏する。
【図面の簡単な説明】
【0016】
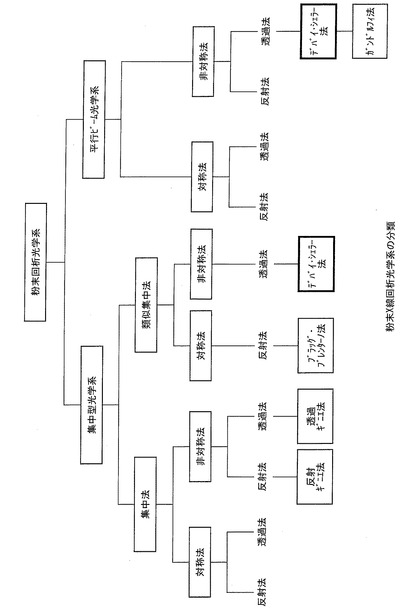
【図1】粉末X線回折光学系の分類を示すチャートである。
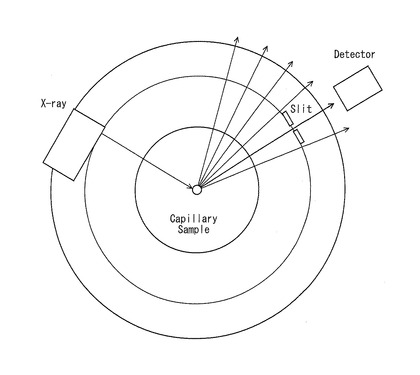
【図2】透過測定法に用いる装置の概略の構成を示す平面図である。
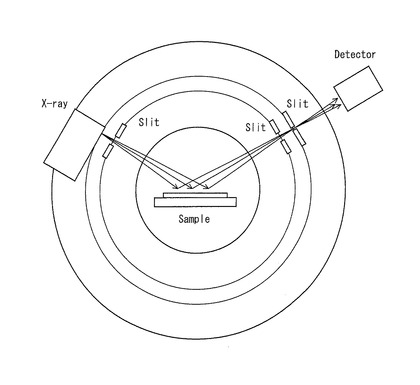
【図3】反射測定法に用いる装置の概略の構成を示す平面図である。
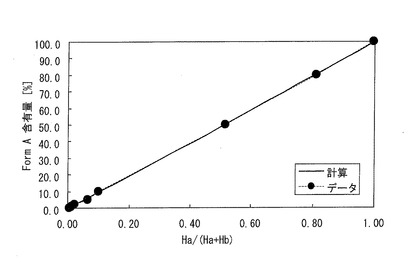
【図4】検量線を示すグラフである。
【図5】実施例で調製した3−(5−メチル−1,2,4−オキサジアゾール−3−イル)−5−(3−メトキシフェニル)−1,6−ナフチリジン−2(1H)オンのForm Aの粉末結晶の粉末X線回折データ(XRDデータ)を示すチャートである。
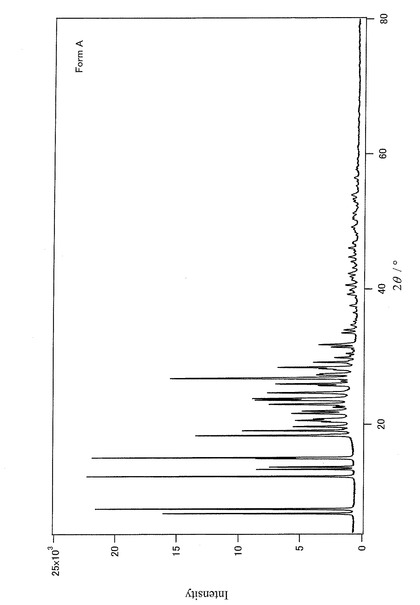
【図6】実施例で調製した3−(5−メチル−1,2,4−オキサジアゾール−3−イル)−5−(3−メトキシフェニル)−1,6−ナフチリジン−2(1H)オンのForm Bの粉末結晶の粉末X線回折データを示すチャートである。
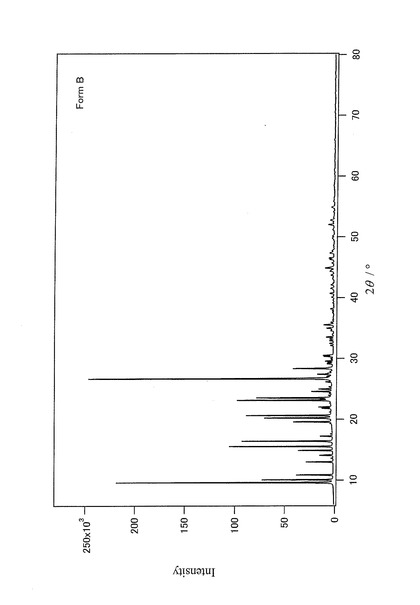
【図7】上記Form Aの粉末結晶を50重量%、上記Form Bの粉末結晶を50重量%含む混合試料の粉末X線回折データを示すチャートである。
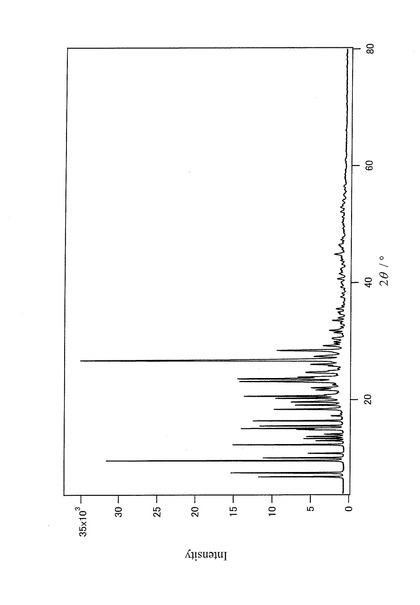
【図8】上記Form Aの粉末結晶を0.5重量%、上記Form Bの粉末結晶を99.5重量%含む混合試料の粉末X線回折データを示すチャートである。
【発明を実施するための形態】
【0017】
本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料を、リートベルト法による多相解析に付すことにより前記結晶多形を定量する方法であって、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いて多相解析を行う方法である。また、本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料中の前記結晶多形を、粉末X線回折強度比を用いて作成した検量線を用いて定量する方法であって、照射体積一定条件であるデバイ・シェラー法による透過測定法を用い、かつ、粉砕工程を経ていない粉末試料を用いる方法である。尚、本発明における「有機分子」には、白金、金等の金属原子に有機配位子が配位してなる錯体も含まれる。
【0018】
一言で粉末X線回折測定といっても、時代や光源、施設等により種々の測定法が利用されている。例えば、中井,泉らによる粉末X線解析の実際第2版(株式会社朝倉書店,2009)には、代表的な測定法(粉末X線回折光学系の分類)が紹介されている。市販の粉末X線回折装置においては、さらに拡張した疑似集中法非対称測定透過法の一つがデバイ・シェラー法として認識されている。本発明においてデバイ・シェラー法とは、図1に示す分類において、疑似集中法非対称測定透過法の一つ、または、平行ビームを用いた非対称測定透過法の一つである。また、本明細書で「反射(測定)法」・「透過(測定)法」と記載があるときは、図1に示す分類とする。尚、図1に示す「平行ビーム」とは、Ge,Siからなる完全性の高い結晶モノクロメーターを用いてX線を平行化したビームを示す。シンクロトロン放射光施設では一般的であり、実験室装置でも実用化されている。
【0019】
本発明において「照射体積一定条件」とは、「定量測定」を意味する。粉末X線回折法は、定性測定および定量測定が可能であり、目的や用途によって使い分けられている。但し、定性測定が簡便に測定できるのに対して、定量測定は、測定対象の試料に応じて個別に測定条件を設定しなければならない。粉末X線回折を使用する主目的は、種々の結晶・非結晶成分からなる粉末試料に含まれる結晶成分の検出、および、物質の帰属判定である。現在、他の分析法では代替することができず、また、非破壊分析で検出・帰属の精度が高く有用であるため、広く利用されるに至る。一方、例えば1990年以降、利用者の増えたリートベルト解析法に用いることが必須の定量測定は、定性測定と比較して使用頻度が少ない。そのため、定量測定は粉末X線回折の専門家以外は、あまり馴染みがない。本発明においては、粉末X線回折法による定量測定を行う。
【0020】
「ブラッグの条件」は、nλ=2d sinθで表される。ここで、nは整数を表し、粉末X線回折においては通常「1」と置いて差し支えないので、本発明では「1」とする。また、λはX線の波長、dは結晶格子の面間隔値、θは測定角度を示す。このため、本発明における「ブラッグの条件で計算される面間隔値dが測定対象の粉末試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データ」とは、以下のように表現することができる。即ち、「最低角反射を示す角度を2θ1、sinθ=λ/(2×1.55)を満たす角度を2θ2とするとき、少なくとも2θ1以上、2θ2以下の測定角度範囲を含む粉末X線回折データ」である。
【0021】
照射体積一定条件で収集した粉末X線回折データは定量性が高く、種々の測定装置に利用することができる。本発明においては、特に、疑似集中法を用いたデバイ・シェラー光学系を備えた装置が好適である。もちろん、SPring-8,Photon Factory等で利用されている平行ビームを用いたデバイ・シェラー光学系を備えた装置も好適である。但し、利用コストや汎用性の面から鑑みて、市販の装置を用いることが望ましい。透過測定法に用いる装置の概略の構成を示す平面図を図2に示す。図2に示すように、透過測定法に用いる装置においては、X線源(X-ray) から照射されたX線が回転する試料、即ち、ガラスキャピラリーに入れられた試料(Capillary Sample)で回折され、回折されたX線がスリット(Slit)を介して検出器(Detector)で検出されるようになっている。従って、試料に照射されるX線は、一定の照射幅で照射されることになるので、照射体積一定条件となる。
【0022】
また、照射体積一定条件とすることができるのであれば、最も汎用的なブラッグ・ブレンターノ光学系装置を利用することもできる。但し、有機分子では吸収係数が小さすぎるために、一般に用いられている試料ホルダーでは、汎用のCuKα線等の長波長X線を用いても、多くの入射X線が試料を透過してしまって定量することができない。それゆえ、照射体積一定条件とするには、特殊な平板状の深型試料ホルダーが必要になるだけでなく、大量の試料を必要とするので、現実的には試料(粉末試料)に重金属が含まれている(有機分子が錯体である)場合に限られる。反射測定法に用いる装置の概略の構成を示す平面図を図3に示す。図3に示すように、反射測定法に用いる装置においては、X線源(X-ray) からスリット(Slit)を介して照射されたX線が平板状の深型試料ホルダーに入れられた試料(Sample)で反射され、反射されたX線が複数のスリット(Slit)を介して検出器(Detector)で検出されるようになっている。従って、試料に照射されるX線は、深型試料ホルダーに入れられた試料の深さ(厚さ)が充分であれば、試料を透過することなく反射されることになるので、照射体積一定条件となる。
【0023】
X線は、汎用のCuKα線等の長波長X線が、利用コストやデータ解像度の面から鑑みてより好ましい。但し、試料(粉末試料)に重金属が含まれている(有機分子が錯体である)場合であって特に透過測定法を用いる場合には、試料吸収による回折強度の低下という問題も生じるため、CuKα線よりも短い波長のX線が好ましい。
【0024】
X線源は、市販の装置で利用することができる管球が好ましく、より高輝度を実現することができる回転対陰極の光源がより好ましく、ビームの平行性および輝度の観点から、大型放射光施設を用いるX線源がさらに好ましいものの、特に限定されるものではない。
【0025】
X線は、モノクロメーターを用いて単色化されていることが、リートベルト法による計算時間を短くするために望ましいものの、特に、単色化されている必要はない。
【0026】
光学系としては、疑似集中法を用いたデバイ・シェラー光学系が、利用コストや汎用性の面から鑑みて好ましい。もちろん、X線として平行ビームを用いたデバイ・シェラー光学系も好適であり、さらに、格子面間隔傾斜型放物面人工格子を用いた装置や、大型放射光施設がより好ましいものの、特に限定されるものではない。格子面間隔傾斜型放物面人工格子は、一般にパラボリック・ミラーや多相膜ミラーとも呼ばれており、実験室X線源のような典型的な発散特性X線源においても高効率でX線を平行化することができる、優れた人工格子である。
【0027】
粉末試料を入れる試料ホルダーとしては、通常、デバイ・シェラー光学系を用いた測定装置で用いられるガラスキャピラリーが最も好ましい。ガラスキャピラリーは、粉末X線回折法で最大の問題となる選択配向の緩和には、最も効果が高い。測定装置には、測定中にガラスキャピラリーを一定速度で回転させるスピナー機構が装備されていることが必要である。ガラスキャピラリーの材質は、各種化学薬品との親和性が低いホウ珪酸ガラスが最も好ましい。また、粉末試料と反応しないのであれば、ホウ珪酸ガラスよりも吸収係数が小さいリンデマンガラス等の種々のガラス材料も用いることができる。
【0028】
また、ガラスキャピラリーを用いる代りに、粉末試料を、X線の回折が無く、吸収係数の小さいフィルム、例えば、膜透過測定法に用いられるマイラーフィルムやカプトンフィルム等のフィルムに挟んで測定することもできる。選択配向が少ない場合には、粉末試料を、反射法に用いられる汎用の平板のホルダーに挟んで測定しても構わない。ホルダーは、平滑性に優れるガラス、アルミニウム板、Si無反射基板、PMMA(ポリメチルメタクリレート)板等の、測定領域においてX線回折の生じない材質で形成されていることが望ましい。フィルムやホルダーを用いる場合、測定装置には、平面回転機構が装備されていることが必要である。
【0029】
測定装置の検出器としては、多チャンネル型検出器が好ましい。特にガラスキャピラリーを用いるデバイ・シェラー光学系の汎用装置(通常、実験室で用いる装置)では、回折強度が著しく弱いため、従来用いられているシンチレーションカウンター等の検出器では充分なS/N比のデータを得たい場合に測定時間がかかる不都合がある。上記多チャンネル型検出器は、その不都合を解決することができるため好適である。検出方式は、特に限定されるものではなく、位置敏感型および半導体型の両方が適している。
【0030】
上記デバイ・シェラー光学系を用いた測定装置としては、具体的には、例えば、Bruker AXS社製D8 ADVANCE Vαrio1、PANalytical 社製X' Pert PRO MPD、リガク社製 Smart Labを挙げることができる。
【0031】
「リートベルト法」は、例えば、中井,泉らによる粉末X線解析の実際第2版(株式会社朝倉書店,2009)に詳述されている。本発明においては、当該書籍に記載されているリートベルト解析の手法を採用することとするが、具体的な手法は、上記手法にのみ限定されるものではない。
【0032】
リートベルト解析では、粉末X線回折装置によって、一定の2θ間隔で一連の回折強度yi (i=1,2,3,…)を測定する場合に、全粉末X線回折パターンに含まれている情報を最大限に抽出するために、近似構造モデルに基づいて計算した回折パターンを実測回折パターンに当てはめる。即ち、i番目の回折点2θi に対する計算強度をf(2θi ;x1 ,x2 ,x3 ,…)≡fi (x)、統計的重みをwi (=1/yi )としたとき、重み付き残差二乗和
【0033】
【数1】

【0034】
を最小とする一組の可変パラメーターxを最小二乗法により精密化する。fi (x)はxについて非線形なので、xの初期値を与え、非線形最小二乗法によりxを反復改良せねばならない。波長一定で2θ可変の粉末X線回折法の場合には、実質的にyi に寄与するブラッグ反射の強度を合計し、バックグラウンド強度yb (2θi )を加えるとi番目の測定点でのfi (x)が下式から得られる。
【0035】
【数2】
【0036】
ここで、sは回折装置、測定条件、試料(粉末試料)に依存する種々の定数を全て吸収させた尺度因子、SR (θi )はブラッグ・ブレンターノ光学系における平板試料の表面粗さの補正因子、A(θi )は吸収因子、D(θi )はブラッグ・ブレンターノ光学系において照射幅が一定となるように発散角を自動的に可変する発散スリットを用いるときの補正因子、Kは2θi におけるブラッグ反射強度に実質的に寄与する反射の番号、mKはブラッグ反射の多重度、F(hk )は結晶構造因子、hk は回折指数hklを表すベクトル、Pk は試料の選択配向による回折強度の変動を補正するための選択配向関数、L(θi )はローレンツ・偏光因子、θiKはブラッグ角、G(Δ2θiK)≡G(2θi −2θK )は回折プロファイル形を近似するためのプロファイル関数を示す。
【0037】
このうち、結晶構造因子F(hk )は、jを単位胞内の原子の番号、gi を占有率、f0iを原子散乱因子、fi'をX線分散補正の実数部、fj'' をX線分散補正の虚数部、Tj をデバイーワラー因子(温度因子と呼ぶことが多い)、xi ,yi ,zi を分率座標とすると、
【0038】
【数3】
【0039】
となる。
【0040】
【数4】
【0041】
は単位胞内の全原子についての和を表す。Tj は原子の熱振動に起因する回折強度の減少を表す因子であり、等方性調和熱振動で近似する場合、
【0042】
【数5】
【0043】
で与えられる。ここで、Bj とUj は等方性原子変位パラメーター(等方性温度因子や等方性熱振動パラメーターとも呼ばれる)である。
【0044】
リートベルト法プログラムとしては、例えば、RIETAN-FP ,GSAS,FullProf,TOPAS 等を好適に用いることができるものの、これら特定のプログラムに限定されるものではなく、種々のプログラムを用いることができる。積算時間は、測定対象の試料に応じて、適宜設定すればよい。
【0045】
本発明における粉末X線回折データの測定範囲として、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å(0.155nm)以下(λ=1.5405Åの場合、2θ>61.797°)の範囲における粉末X線回折データを用いる。即ち、実質的に全ての粉末X線回折データをリートベルト法による多相解析法に用いるので、精度が向上し、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で含まれる結晶多形の重量分率を精密に定量することができる。
【0046】
面間隔値dを1.55Å以下(λ=1.5405Åの場合、2θ>61.797°)、より好ましくは1.2Å以下にすることで、バックグラウンドが複雑になりやすいデバイ・シェラー光学系においても、等方性原子変位パラメーターと強く相関するものの、最小二乗法を比較的安定して収束させることができる。
【0047】
また、測定対象の試料の最低角反射に相当する面間隔を含むことで、分子組成比の関係等で席占有率が1.0に満たない成分を含む粉末試料であっても、定量性を損なうことなくリートベルト法による解析を実施することができる。尚、有機分子においては、最低角反射に相当する面間隔は必ず1.55Å以上(通常は4.43Å以上)に存在する。
【0048】
リートベルト法による精密化の目安として、Rwp値が一つの指標になる。Rwp値は10%以下まで精密化されることが好ましく、7%以下まで精密化されることがより好ましい。それと共に、Rp 値も10%以下となることが好ましい。これらRwp値およびRp 値は、R.A.Young ら (The Rietveld Method, Oxford, 1993)に従って以下のように定義される。
【0049】
【数6】
【0050】
ここで、wi は統計的重み、yi は観測強度、fi (x)は理論回折強度を示す。
【0051】
リートベルト法による有機分子の結晶多形の定量方法としては、多相解析における各々の結晶相の尺度因子により算出する計算方法が好ましい。例えば、中井,泉らによる粉末X線解析の実際第2版(株式会社朝倉書店,2009)によると、リートベルト解析プログラムは通常、二つ以上の結晶相からなる混合物も取り扱うことができる。この機能を活用して、結晶相iの質量分率wi (上記統計的重みwi とは異なる)は、リートベルト法により精密化した尺度因子sj (j=1,2,3,…)から、
【0052】
【数7】
【0053】
という式で求められる。ここで、Zは単位胞中の化学式単位の数、Mは化学式単位の質量、Vは化学式単位の体積、
【0054】
【数8】
【0055】
は全相についての和を表す。
【0056】
リートベルト法における有機分子の結晶多形の多相解析において、粒子径および吸収係数から生じるmicroabsorption の問題は、汎用のCuKα線のような長波長X線においても無視して構わない。結晶多形のため、即ち同一分子・同一組成のため、基本的な吸収係数は同じであり、粒子径による効果だけが異なるからである。有機分子を構成するC,H,N,O,S等では吸収係数も小さく問題になることはない。本発明においてもこの考え方に従う。もちろん、粒子径が著しく異なる場合、または、重元素が含まれる場合は、補正することによってより好適になるため、この内容に限定されるものではない。
【0057】
以上のように、本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料を、リートベルト法による多相解析に付すことにより前記結晶多形を定量する方法であって、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いて多相解析を行う方法である。そして、上記照射体積一定条件とは、デバイ・シェラー法による透過測定法であることが好ましい。これにより、実質的に全ての粉末X線回折データをリートベルト法による多相解析に付すので精度が向上し、分子量が大きく複雑な化学構造を有し互いの有機分子のピーク重畳が激しい試料であっても測定することができ、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができる。
【0058】
従って、本発明に係る有機分子の結晶多形の定量方法の更なる特徴は、粉砕工程を経ていない粉末試料を用いること、さらには、上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよいことにある。Zeltan Nemetら (Journal of Pharmaceutical and Biomedical Analysis, 49, 338. (2009)) の報告では、Famotidineの結晶相の定量においては、測定の前処理として行う粉砕工程による影響は無いと記載されている。しかしながら、多くの有機分子の結晶は、粉砕における応力により、結晶形に影響を受けているおそれがある。本発明に係る定量方法によれば、乳鉢を用いてすり潰す(摩砕)等の粉砕工程を経ていない粉末試料を用いることで、粉砕による影響を受けずに測定することができるので、選択配向を生じ易い結晶の有機分子からなる結晶多形であっても、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、精密に定量することができる。
【0059】
また、以上のように、本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料中の前記結晶多形を、粉末X線回折強度比を用いて作成した検量線を用いて定量する方法であって、照射体積一定条件であるデバイ・シェラー法による透過測定法を用い、かつ、粉砕工程を経ていない粉末試料を用いる方法である。そして、ブラッグの条件で計算される面間隔値dが測定対象の粉末試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いることが好ましい。これにより、分子量が大きく複雑な化学構造を有し互いの有機分子のピーク重畳が激しい試料であっても測定することができ、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少なくとも1重量%以上、99重量%以下の範囲で含まれる結晶多形を、定量することができる。上記検量線は、有機分子の含有量が既知でそれぞれ含有量が異なる5点以上の試料(混合試料)を用いて測定した粉末X線回折強度を用いて、例えば最小二乗法で求められていることがより好ましい。
【0060】
また、本発明に係る有機分子の結晶多形の定量方法は、上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよいことにある。そして、上記検量線が、結晶多形に含まれる全ての単一結晶の粉末X線回折強度を含んで作成されていることが好ましい。
【0061】
即ち、本発明に係る有機分子の結晶多形の定量方法の更なる特徴は、各結晶相の分率座標を固定しながら精密化することで、粉末試料が目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいても、当該粗大粒子による回折強度の致命的な変化を防止し、かつ、実質的な解析精度を下げることなく定量することができることを見出したことにある。リートベルト法において、粗大粒子による強い回折は、最小二乗残差として目立つことが多く、弊害が大きい。そのため、リートベルト法プログラムでは、選択配向因子や原子の分率座標を調整して精密化してしまう。このとき、分子間の結合距離、結合角、または、二面角を或る程度の許容範囲で拘束条件をかけながら解析することは、熟練測定者の勘所となっている(熟練を要する作業となっている)。また、分率座標は、有機分子からなる結晶ではパラメーター数が多く、実質的に精密化が不可能な水素原子も多数有しているため、煩雑な解析になることが多い。そのため、最小二乗残差の誤差の掃き溜めとなり易いという問題がある。これに対して、本発明においては、任意性が生じ難いように結晶構造の分率座標だけを全て固定する方法を実施したところ、定量性は実質的に維持されることを見出した。本発明に係る定量方法によれば、粉砕による影響を受けずに測定することができるので、選択配向を生じ易い結晶の有機分子からなる結晶多形であっても、有機分子の結晶多形を2相以上含有する試料中に、少なくとも1重量%以上、99重量%以下の範囲で含まれる結晶多形を、定量することができる。
【実施例】
【0062】
以下、実施例および比較例によって本発明をより詳細に説明する。
〔試料の調製方法〕
下記実施例に用いる試料を、以下の方法で調製した。
【0063】
公知の製造方法である特許第3390453号公報の第23頁(実施例86)に記載されている方法によって、3−(5−メチル−1,2,4−オキサジアゾール−3−イル)−5−(3−メトキシフェニル)−1,6−ナフチリジン−2(1H)オンの無色粉末を製造した。そして、上記無色粉末を、ジメチルホルムアミドおよび2−プロパノールの混合溶媒(ジメチルホルムアミド:2−プロパノール=4:7(容量比))に溶解させた後、再結晶させた。その後、得られた固形分を濾取して乾燥させることにより、3−(5−メチル−1,2,4−オキサジアゾール−3−イル)−5−(3−メトキシフェニル)−1,6−ナフチリジン−2(1H)オンの粉末結晶(以下、単にForm Aと記す)を得た。
【0064】
一方、上記混合溶媒の代りに水およびエタノールの混合溶媒(水:エタノール=1:4(容量比))を用いた以外は、上記操作と同様にして無色粉末を溶解させて再結晶させ、得られた固形分を濾取して乾燥させることにより、3−(5−メチル−1,2,4−オキサジアゾール−3−イル)−5−(3−メトキシフェニル)−1,6−ナフチリジン−2(1H)オンの粉末結晶(以下、単にForm Bと記す)を得た。
【0065】
上記Form Aの粉末X線回折データ(XRDデータ)を図5に示すと共に、上記Form Bの粉末X線回折データを図6に示す。
〔実施例1〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量(仕込み量)が80重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢にForm A1.00499gとForm B1.00040gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm A1.00401gを加えて同様に30分間手動で混合した後、さらにForm A1.99976gを加えて30分間手動で混合して、Form Aの含有量が80重量%の混合試料(以下、80%混合試料と記す)を作成した。得られた80%混合試料を目開き150μmの篩を用いて篩別した後、Hilgenberg社製1.0mmφほう珪酸ガラスキャピラリーに封管した。
【0066】
次に、ブルカーAXS社製高分解能粉末回折装置 D8 ADVANCE with Vαrio1(CuKα,40kV,40mA)に1次元高速PSD検出器(VÅNTEC-1) を組み合わせた装置を用いて、疑似デバイ・シェラー光学系で測定対象の試料の最低角反射に相当する面間隔から1.19Å(0.109nm)以下(2θ>80.676°)の範囲における粉末X線回折データ(XRDデータ)を得た。尚、粉末X線回折法の弱点である選択配向の寄与を低減するために、ほう珪酸ガラスキャピラリーを毎分60回転で回転させながら室温で測定を行った。
【0067】
そして、透過法測定であることから、試料のX線透過率の実測値を用いて線吸収係数を次式より求めた。
【0068】
Ix =I0 exp(−μt)
ここで、μは線吸収係数、tは試料厚さ、Ix は試料透過X線強度、I0 はX線強度である。
【0069】
得られたXRDパターンに対し、RIETAN-FP プログラムパッケージ (Fujio Izumi and Koichi Momma, Solid. State Phenom., 130 15 (2007))を用いてリートベルト2相解析を実施した。ほう珪酸ガラスキャピラリーによる非晶質からの散乱は、こぶ状のバックグラウンドとして測定データの精密化の妨げとなる。そのため、11次のルジャンドル直交多項式を重畳した複合バックグラウンド関数で、全体のバックグラウンドを見積もった。そのとき、初期のバックグラウンドデータは プログラムPowderX (Cheng Dong, J. Appl. Crystallogr. 32 838 (1999)) にて近似した。プロファイル関数には虎谷の拡張分割Pseude-Voigt関数を使用した。しかしながら、粉末法の貧弱なデータを用い、水素原子まで付加された2相の結晶多形を含む全てのパラメーターを精密化することは、困難であると予測された。そこで、以下のような解析条件を前提に、2相解析を実施した。
(1)二つの結晶構造の分率座標は固定する。
(2)結晶構造に関するパラメーターでは、格子定数、等方性原子変位パラメーターのみを精密化の対象とする。但し、最小二乗法が発散するときには固定する。
(3)その他、尺度因子、プロファイル関数、バックグラウンド関数は精密化する。
【0070】
リートベルト解析による解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。ここでRwp値、Rp 値およびS値は、R.A.Young ら (The Rietveld Method, Oxford, 1993)の定義に従った。
〔実施例2〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量が50重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢にForm A1.00074gとForm B1.00031gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm A0.50103gとForm B0.50076gとを加えて同様に30分間手動で混合した後、さらにForm A1.00406gとForm B1.000008gとを加えて30分間手動で混合して、Form Aの含有量が50重量%の混合試料(以下、50%混合試料と記す)を作成した。上記50%混合試料の粉末X線回折データを図7に示す。得られた50%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
〔実施例3〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量が10重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢にForm A0.50025gとForm B0.50133gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm B2.00203gを加えて同様に30分間手動で混合した後、さらにForm B2.00169gを加えて30分間手動で混合して、Form Aの含有量が10重量%の混合試料(以下、10%混合試料と記す)を作成した。得られた10%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
〔実施例4〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量が5重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢にForm A0.25017gとForm B0.25039gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm B0.50213gを加えて同様に30分間手動で混合した後、さらにForm B2.00094gを加えて同様に30分間手動で混合し、次いでForm B1.99975gを加えて同様に30分間手動で混合して、Form Aの含有量が5重量%の混合試料(以下、5%混合試料と記す)を作成した。得られた5%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
〔実施例5〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量が2重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢にForm A0.18138gとForm B0.320839gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm B1.00049gを加えて同様に30分間手動で混合した後、さらにForm B1.49992gを加えて同様に30分間手動で混合した。次いで、この混合試料にForm B3.001975gを加えて同様に30分間手動で混合した後、さらにForm B3.00796gを加えて同様に30分間手動で混合して、Form Aの含有量が2重量%の混合試料(以下、2%混合試料と記す)を作成した。得られた2%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
〔実施例6〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量が1重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢に実施例5で作成した2%混合試料1.00201gとForm B1.00097gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料に2%混合試料1.00103gとForm B1.00735gとを加えて同様に30分間手動で混合した後、さらに2%混合試料0.50408gとForm B0.50602gとを加えて同様に30分間手動で混合した。次いで、この混合試料に2%混合試料2.50712gを加えて同様に30分間手動で混合して、Form Aの含有量が1重量%の混合試料(以下、1%混合試料と記す)を作成した。得られた1%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
〔実施例7〕
粉末結晶であるForm AおよびForm Bを、を、Form Aの含有量が0.5重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢に実施例5で作成した2%混合試料1.25136gとForm B1.25156gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm B1.00294gを加えて同様に30分間手動で混合した後、さらにForm B1.50119gを加えて同様に30分間手動で混合して、Form Aの含有量が0.5重量%の混合試料(以下、0.5%混合試料と記す)を作成した。上記0.5%混合試料の粉末X線回折データを図8に示す。得られた0.5%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
【0071】
【表1】
【0072】
【表2】
【0073】
〔実施例8〕
実施例1〜7で作成した80%混合試料,50%混合試料,10%混合試料,5%混合試料,2%混合試料,1%混合試料,0.5%混合試料の粉末X線回折データ、並びに、実施例1と同じ方法で測定した粉末結晶であるForm A(以下、Form A100%試料と記す)およびForm B(以下、Form B100%試料と記す)の粉末X線回折データを用いて、ピーク重畳が無く、Form Aに特徴的な2θ=6.9°の回折ピークの強度とForm Bに特徴的な2θ=10.1°の回折ピークの強度との比(ピーク強度比)から、以下に示す式により検量線を作成して、各混合試料におけるForm AおよびForm Bの定量を行った。
【0074】
y=100×(Ha/(Ha+Hb))x
ここで、yはForm Aの量(重量%)、Haは2θ=6.9°の回折ピーク強度、Hbは2θ=10.1°の回折ピーク強度、xは最小二乗法を用い、理論値と観測値の差が最小になるようにして求めた値(1.0375)である。
【0075】
80%混合試料,50%混合試料,10%混合試料,5%混合試料,2%混合試料,1%混合試料,0.5%混合試料の粉末X線回折データから得られた2θ=6.9°および2θ=10.1°のピーク強度の一覧を表3に示す。また、Form A100%試料およびForm B100%試料の粉末X線回折データから得られた2θ=6.9°および2θ=10.1°のピーク強度(counts)の一覧を表4に示す。そして、検量線法による解析結果、即ち、定量分析結果(検出量)を表5に示す。また、検量線のグラフを図4に示す。
【0076】
【表3】
【0077】
【表4】
【0078】
【表5】
【0079】
〔比較例1〕
実施例6で作成した1%混合試料を、目開き150μmの篩を用いて篩別した後、直径25mm,深さ0.2mmのガラス製試料台に充填した。次に、株式会社リガク製粉末回折装置RINT-2100V(CuKα,40kV,40mA)を用いてブラッグ・ブレンターノ光学系で測定対象の試料の最低角反射に相当する面間隔から1.176Å以下(2θ>80.0°)の範囲における粉末X線回折データ(XRDデータ)を得た。尚、粉末X線回折法の弱点である選択配向の寄与を低減するために、ガラス製試料台を毎分30回転で回転させながら室温で測定を行った。さらに、発散スリット1/2°、散乱スリット1/2°、受光スリット0.3mmとし、0.03°/2θステップ、積算時間4.0sec で、Fixed time法を用いた。
【0080】
得られたXRDパターンに対し、RIETAN-FP プログラムパッケージを用いてリートベルト2相解析を実施した。11次のルジャンドル直交多項式を重畳した複合バックグラウンド関数で、全体のバックグラウンドを見積もった。そのとき、初期のバックグラウンドデータは プログラムPowderX (Cheng Dong, J. Appl. Crystallogr. 32 838 (1999)) にて近似した。プロファイル関数には虎谷の拡張分割Pseude-Voigt関数を使用した。また、実施例1と同じ結晶構造を初期モデルとした。しかしながら、粉末法の貧弱なデータを用い、水素原子まで付加された2相の結晶多形を含む全てのパラメーターを精密化することは、困難であると予測された。そこで、実施例1に記載した(1)〜(3)の解析条件を前提に、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表6に示す。また、最終の信頼度因子を表7に示す。
〔比較例2〕
実施例7で作成した0.5%混合試料を用いた以外は、比較例1と同じ方法で2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表6に示す。また、最終の信頼度因子を表7に示す。
〔比較例3〕
実施例7で得られた粉末X線回折データ(XRDデータ)を用い、リートベルト解析に用いる計算範囲を9〜35°/2θに制限した以外は、実施例7と同じ方法で2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表8に示す。また、最終の信頼度因子を表9に示す。
【0081】
【表6】
【0082】
【表7】
【0083】
【表8】
【0084】
【表9】
【0085】
〔まとめ〕
上記実施例1〜8の結果と比較例1〜3の結果との対比から明らかなように、本発明に係る定量方法によれば、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で含まれる結晶多形を、精密に定量することができることが判る。
【0086】
尚、本発明は、上述した実施形態に限定されるものではなく、記述した範囲内で種々の変形を加えた態様で実施することができ、従って、請求項に示した範囲で種々の変更が可能である。
【産業上の利用可能性】
【0087】
本発明に係る有機分子の結晶多形の定量方法によれば、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができる方法を提供することができる。
【0088】
それゆえ、本発明に係る有機分子の結晶多形の定量方法は、例えば、医薬品の原体の定量性に好適に利用することができる。
【技術分野】
【0001】
本発明は、有機分子の結晶多形の定量方法に関するものである。
【背景技術】
【0002】
有機分子の結晶多形は重要である。例えば医薬分野においては、ICH ガイドラインQ6A (International Conference on Harmonization Harmonized Guideline. Q6A Specifications, Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products, Chemical Substances, 1999)において、結晶多形の存在形(どの結晶形を含んでいるか)を規定すべきであることが記載されており、結晶多形が医薬品の品質や機能に影響を及ぼす場合には、その含量を精密に定量することが重要になっている。
【0003】
有機分子の結晶多形の定量方法として、例えば非特許文献1には、有機分子の結晶多形が混合した混合粉末試料を、フッ化リチウムを内部標準試料として用いたリートベルト (Rietveld) 法を用いて定量測定する方法が記載されている。そして、上記非特許文献1には、4相の有機分子の結晶多形が混合した粉末試料であっても微量まで定量することができること、特に、2相の有機分子の結晶多形が混合した粉末試料では複相を1重量%以下まで定量することができることが記載されている。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Sampath S. Iyengar et al. (Powder Diffraction 16, 1, 20. (2001))
【非特許文献2】Zeltan Nemet et al. (Journal of Pharmaceutical and Biomedical Analysis, 51, 572. (2010))
【発明の概要】
【発明が解決しようとする課題】
【0005】
ところが、非特許文献1に記載の方法では、フッ化リチウムを内部標準試料として用いている。このため、測定対象の試料とフッ化リチウムとが反応し、これによって結晶多形が変化しているおそれを排除することができない。また、内部標準試料としてフッ化リチウム以外の化合物を用いたとしても、当該化合物と測定対象の試料との反応性や親和性を予め調査する必要がある。
【0006】
内部標準試料を用いない方法として、非特許文献2には、Famotidineの結晶多形の混合試料を用いて、透過測定法を用いたリートベルト法による多相解析法が記載されている。当該非特許文献2においては、X線照射体積が一定になり定量測定に好適な方法を採用することにより、即ち、一組のフィルムに挟んで円盤状(foil)に成形した試料を用いることで選択配向を軽減して定量ができることが記載されている。そして、非特許文献2においては、ブラッグの条件で計算される面間隔値dが最低角反射に相当する角度を大きく上回る範囲におけるX線回折データを用いることにより、測定下限が3重量%で測定した例が記載されている。
【0007】
本発明は、上記状況に鑑みてなされたものであり、その主たる目的は、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中の前記結晶多形を、従来の方法よりも低い検出下限で定量することができる方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明者は上記状況の下、照射体積一定条件で収集され、最低角反射に相当する角度を含む範囲における粉末X線回折データを用い、有機分子の結晶多形の定量を、リートベルト法による多相解析法により実施することにより、内部標準試料を用いることなく、従来の方法よりも低い検出下限で定量することができることを見出した。
【0009】
即ち、本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料を、リートベルト法による多相解析に付すことにより前記結晶多形を定量する方法であって、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いて多相解析を行うことを特徴としている。
【0010】
上記照射体積一定条件とは、デバイ・シェラー (Debye-Scherrer) 法による透過測定法であることがより好ましい。また、粉砕工程を経ていない粉末試料を用いることがより好ましい。さらに、上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよい。
【0011】
上記方法によれば、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いるので、精度が向上し、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができる。
【0012】
また、本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料中の前記結晶多形を、粉末X線回折強度比を用いて作成した検量線を用いて定量する方法であって、照射体積一定条件であるデバイ・シェラー法による透過測定法を用い、かつ、粉砕工程を経ていない粉末試料を用いることを特徴としている。
【0013】
ブラッグの条件で計算される面間隔値dが測定対象の粉末試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いることがより好ましい。また、上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよい。さらに、上記検量線が、結晶多形に含まれる全ての単一結晶の粉末X線回折強度を含んで作成されていることがより好ましい。
【0014】
上記方法によれば、照射体積一定条件であるデバイ・シェラー法による透過測定法を用い、かつ、粉砕工程を経ていない粉末試料を用い、粉末X線回折強度比を用いて作成した検量線を用いて定量するので、精度が向上し、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができる。
【発明の効果】
【0015】
本発明に係る有機分子の結晶多形の定量方法によれば、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができるという効果を奏する。
【図面の簡単な説明】
【0016】
【図1】粉末X線回折光学系の分類を示すチャートである。
【図2】透過測定法に用いる装置の概略の構成を示す平面図である。
【図3】反射測定法に用いる装置の概略の構成を示す平面図である。
【図4】検量線を示すグラフである。
【図5】実施例で調製した3−(5−メチル−1,2,4−オキサジアゾール−3−イル)−5−(3−メトキシフェニル)−1,6−ナフチリジン−2(1H)オンのForm Aの粉末結晶の粉末X線回折データ(XRDデータ)を示すチャートである。
【図6】実施例で調製した3−(5−メチル−1,2,4−オキサジアゾール−3−イル)−5−(3−メトキシフェニル)−1,6−ナフチリジン−2(1H)オンのForm Bの粉末結晶の粉末X線回折データを示すチャートである。
【図7】上記Form Aの粉末結晶を50重量%、上記Form Bの粉末結晶を50重量%含む混合試料の粉末X線回折データを示すチャートである。
【図8】上記Form Aの粉末結晶を0.5重量%、上記Form Bの粉末結晶を99.5重量%含む混合試料の粉末X線回折データを示すチャートである。
【発明を実施するための形態】
【0017】
本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料を、リートベルト法による多相解析に付すことにより前記結晶多形を定量する方法であって、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いて多相解析を行う方法である。また、本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料中の前記結晶多形を、粉末X線回折強度比を用いて作成した検量線を用いて定量する方法であって、照射体積一定条件であるデバイ・シェラー法による透過測定法を用い、かつ、粉砕工程を経ていない粉末試料を用いる方法である。尚、本発明における「有機分子」には、白金、金等の金属原子に有機配位子が配位してなる錯体も含まれる。
【0018】
一言で粉末X線回折測定といっても、時代や光源、施設等により種々の測定法が利用されている。例えば、中井,泉らによる粉末X線解析の実際第2版(株式会社朝倉書店,2009)には、代表的な測定法(粉末X線回折光学系の分類)が紹介されている。市販の粉末X線回折装置においては、さらに拡張した疑似集中法非対称測定透過法の一つがデバイ・シェラー法として認識されている。本発明においてデバイ・シェラー法とは、図1に示す分類において、疑似集中法非対称測定透過法の一つ、または、平行ビームを用いた非対称測定透過法の一つである。また、本明細書で「反射(測定)法」・「透過(測定)法」と記載があるときは、図1に示す分類とする。尚、図1に示す「平行ビーム」とは、Ge,Siからなる完全性の高い結晶モノクロメーターを用いてX線を平行化したビームを示す。シンクロトロン放射光施設では一般的であり、実験室装置でも実用化されている。
【0019】
本発明において「照射体積一定条件」とは、「定量測定」を意味する。粉末X線回折法は、定性測定および定量測定が可能であり、目的や用途によって使い分けられている。但し、定性測定が簡便に測定できるのに対して、定量測定は、測定対象の試料に応じて個別に測定条件を設定しなければならない。粉末X線回折を使用する主目的は、種々の結晶・非結晶成分からなる粉末試料に含まれる結晶成分の検出、および、物質の帰属判定である。現在、他の分析法では代替することができず、また、非破壊分析で検出・帰属の精度が高く有用であるため、広く利用されるに至る。一方、例えば1990年以降、利用者の増えたリートベルト解析法に用いることが必須の定量測定は、定性測定と比較して使用頻度が少ない。そのため、定量測定は粉末X線回折の専門家以外は、あまり馴染みがない。本発明においては、粉末X線回折法による定量測定を行う。
【0020】
「ブラッグの条件」は、nλ=2d sinθで表される。ここで、nは整数を表し、粉末X線回折においては通常「1」と置いて差し支えないので、本発明では「1」とする。また、λはX線の波長、dは結晶格子の面間隔値、θは測定角度を示す。このため、本発明における「ブラッグの条件で計算される面間隔値dが測定対象の粉末試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データ」とは、以下のように表現することができる。即ち、「最低角反射を示す角度を2θ1、sinθ=λ/(2×1.55)を満たす角度を2θ2とするとき、少なくとも2θ1以上、2θ2以下の測定角度範囲を含む粉末X線回折データ」である。
【0021】
照射体積一定条件で収集した粉末X線回折データは定量性が高く、種々の測定装置に利用することができる。本発明においては、特に、疑似集中法を用いたデバイ・シェラー光学系を備えた装置が好適である。もちろん、SPring-8,Photon Factory等で利用されている平行ビームを用いたデバイ・シェラー光学系を備えた装置も好適である。但し、利用コストや汎用性の面から鑑みて、市販の装置を用いることが望ましい。透過測定法に用いる装置の概略の構成を示す平面図を図2に示す。図2に示すように、透過測定法に用いる装置においては、X線源(X-ray) から照射されたX線が回転する試料、即ち、ガラスキャピラリーに入れられた試料(Capillary Sample)で回折され、回折されたX線がスリット(Slit)を介して検出器(Detector)で検出されるようになっている。従って、試料に照射されるX線は、一定の照射幅で照射されることになるので、照射体積一定条件となる。
【0022】
また、照射体積一定条件とすることができるのであれば、最も汎用的なブラッグ・ブレンターノ光学系装置を利用することもできる。但し、有機分子では吸収係数が小さすぎるために、一般に用いられている試料ホルダーでは、汎用のCuKα線等の長波長X線を用いても、多くの入射X線が試料を透過してしまって定量することができない。それゆえ、照射体積一定条件とするには、特殊な平板状の深型試料ホルダーが必要になるだけでなく、大量の試料を必要とするので、現実的には試料(粉末試料)に重金属が含まれている(有機分子が錯体である)場合に限られる。反射測定法に用いる装置の概略の構成を示す平面図を図3に示す。図3に示すように、反射測定法に用いる装置においては、X線源(X-ray) からスリット(Slit)を介して照射されたX線が平板状の深型試料ホルダーに入れられた試料(Sample)で反射され、反射されたX線が複数のスリット(Slit)を介して検出器(Detector)で検出されるようになっている。従って、試料に照射されるX線は、深型試料ホルダーに入れられた試料の深さ(厚さ)が充分であれば、試料を透過することなく反射されることになるので、照射体積一定条件となる。
【0023】
X線は、汎用のCuKα線等の長波長X線が、利用コストやデータ解像度の面から鑑みてより好ましい。但し、試料(粉末試料)に重金属が含まれている(有機分子が錯体である)場合であって特に透過測定法を用いる場合には、試料吸収による回折強度の低下という問題も生じるため、CuKα線よりも短い波長のX線が好ましい。
【0024】
X線源は、市販の装置で利用することができる管球が好ましく、より高輝度を実現することができる回転対陰極の光源がより好ましく、ビームの平行性および輝度の観点から、大型放射光施設を用いるX線源がさらに好ましいものの、特に限定されるものではない。
【0025】
X線は、モノクロメーターを用いて単色化されていることが、リートベルト法による計算時間を短くするために望ましいものの、特に、単色化されている必要はない。
【0026】
光学系としては、疑似集中法を用いたデバイ・シェラー光学系が、利用コストや汎用性の面から鑑みて好ましい。もちろん、X線として平行ビームを用いたデバイ・シェラー光学系も好適であり、さらに、格子面間隔傾斜型放物面人工格子を用いた装置や、大型放射光施設がより好ましいものの、特に限定されるものではない。格子面間隔傾斜型放物面人工格子は、一般にパラボリック・ミラーや多相膜ミラーとも呼ばれており、実験室X線源のような典型的な発散特性X線源においても高効率でX線を平行化することができる、優れた人工格子である。
【0027】
粉末試料を入れる試料ホルダーとしては、通常、デバイ・シェラー光学系を用いた測定装置で用いられるガラスキャピラリーが最も好ましい。ガラスキャピラリーは、粉末X線回折法で最大の問題となる選択配向の緩和には、最も効果が高い。測定装置には、測定中にガラスキャピラリーを一定速度で回転させるスピナー機構が装備されていることが必要である。ガラスキャピラリーの材質は、各種化学薬品との親和性が低いホウ珪酸ガラスが最も好ましい。また、粉末試料と反応しないのであれば、ホウ珪酸ガラスよりも吸収係数が小さいリンデマンガラス等の種々のガラス材料も用いることができる。
【0028】
また、ガラスキャピラリーを用いる代りに、粉末試料を、X線の回折が無く、吸収係数の小さいフィルム、例えば、膜透過測定法に用いられるマイラーフィルムやカプトンフィルム等のフィルムに挟んで測定することもできる。選択配向が少ない場合には、粉末試料を、反射法に用いられる汎用の平板のホルダーに挟んで測定しても構わない。ホルダーは、平滑性に優れるガラス、アルミニウム板、Si無反射基板、PMMA(ポリメチルメタクリレート)板等の、測定領域においてX線回折の生じない材質で形成されていることが望ましい。フィルムやホルダーを用いる場合、測定装置には、平面回転機構が装備されていることが必要である。
【0029】
測定装置の検出器としては、多チャンネル型検出器が好ましい。特にガラスキャピラリーを用いるデバイ・シェラー光学系の汎用装置(通常、実験室で用いる装置)では、回折強度が著しく弱いため、従来用いられているシンチレーションカウンター等の検出器では充分なS/N比のデータを得たい場合に測定時間がかかる不都合がある。上記多チャンネル型検出器は、その不都合を解決することができるため好適である。検出方式は、特に限定されるものではなく、位置敏感型および半導体型の両方が適している。
【0030】
上記デバイ・シェラー光学系を用いた測定装置としては、具体的には、例えば、Bruker AXS社製D8 ADVANCE Vαrio1、PANalytical 社製X' Pert PRO MPD、リガク社製 Smart Labを挙げることができる。
【0031】
「リートベルト法」は、例えば、中井,泉らによる粉末X線解析の実際第2版(株式会社朝倉書店,2009)に詳述されている。本発明においては、当該書籍に記載されているリートベルト解析の手法を採用することとするが、具体的な手法は、上記手法にのみ限定されるものではない。
【0032】
リートベルト解析では、粉末X線回折装置によって、一定の2θ間隔で一連の回折強度yi (i=1,2,3,…)を測定する場合に、全粉末X線回折パターンに含まれている情報を最大限に抽出するために、近似構造モデルに基づいて計算した回折パターンを実測回折パターンに当てはめる。即ち、i番目の回折点2θi に対する計算強度をf(2θi ;x1 ,x2 ,x3 ,…)≡fi (x)、統計的重みをwi (=1/yi )としたとき、重み付き残差二乗和
【0033】
【数1】
【0034】
を最小とする一組の可変パラメーターxを最小二乗法により精密化する。fi (x)はxについて非線形なので、xの初期値を与え、非線形最小二乗法によりxを反復改良せねばならない。波長一定で2θ可変の粉末X線回折法の場合には、実質的にyi に寄与するブラッグ反射の強度を合計し、バックグラウンド強度yb (2θi )を加えるとi番目の測定点でのfi (x)が下式から得られる。
【0035】
【数2】
【0036】
ここで、sは回折装置、測定条件、試料(粉末試料)に依存する種々の定数を全て吸収させた尺度因子、SR (θi )はブラッグ・ブレンターノ光学系における平板試料の表面粗さの補正因子、A(θi )は吸収因子、D(θi )はブラッグ・ブレンターノ光学系において照射幅が一定となるように発散角を自動的に可変する発散スリットを用いるときの補正因子、Kは2θi におけるブラッグ反射強度に実質的に寄与する反射の番号、mKはブラッグ反射の多重度、F(hk )は結晶構造因子、hk は回折指数hklを表すベクトル、Pk は試料の選択配向による回折強度の変動を補正するための選択配向関数、L(θi )はローレンツ・偏光因子、θiKはブラッグ角、G(Δ2θiK)≡G(2θi −2θK )は回折プロファイル形を近似するためのプロファイル関数を示す。
【0037】
このうち、結晶構造因子F(hk )は、jを単位胞内の原子の番号、gi を占有率、f0iを原子散乱因子、fi'をX線分散補正の実数部、fj'' をX線分散補正の虚数部、Tj をデバイーワラー因子(温度因子と呼ぶことが多い)、xi ,yi ,zi を分率座標とすると、
【0038】
【数3】
【0039】
となる。
【0040】
【数4】
【0041】
は単位胞内の全原子についての和を表す。Tj は原子の熱振動に起因する回折強度の減少を表す因子であり、等方性調和熱振動で近似する場合、
【0042】
【数5】
【0043】
で与えられる。ここで、Bj とUj は等方性原子変位パラメーター(等方性温度因子や等方性熱振動パラメーターとも呼ばれる)である。
【0044】
リートベルト法プログラムとしては、例えば、RIETAN-FP ,GSAS,FullProf,TOPAS 等を好適に用いることができるものの、これら特定のプログラムに限定されるものではなく、種々のプログラムを用いることができる。積算時間は、測定対象の試料に応じて、適宜設定すればよい。
【0045】
本発明における粉末X線回折データの測定範囲として、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å(0.155nm)以下(λ=1.5405Åの場合、2θ>61.797°)の範囲における粉末X線回折データを用いる。即ち、実質的に全ての粉末X線回折データをリートベルト法による多相解析法に用いるので、精度が向上し、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で含まれる結晶多形の重量分率を精密に定量することができる。
【0046】
面間隔値dを1.55Å以下(λ=1.5405Åの場合、2θ>61.797°)、より好ましくは1.2Å以下にすることで、バックグラウンドが複雑になりやすいデバイ・シェラー光学系においても、等方性原子変位パラメーターと強く相関するものの、最小二乗法を比較的安定して収束させることができる。
【0047】
また、測定対象の試料の最低角反射に相当する面間隔を含むことで、分子組成比の関係等で席占有率が1.0に満たない成分を含む粉末試料であっても、定量性を損なうことなくリートベルト法による解析を実施することができる。尚、有機分子においては、最低角反射に相当する面間隔は必ず1.55Å以上(通常は4.43Å以上)に存在する。
【0048】
リートベルト法による精密化の目安として、Rwp値が一つの指標になる。Rwp値は10%以下まで精密化されることが好ましく、7%以下まで精密化されることがより好ましい。それと共に、Rp 値も10%以下となることが好ましい。これらRwp値およびRp 値は、R.A.Young ら (The Rietveld Method, Oxford, 1993)に従って以下のように定義される。
【0049】
【数6】
【0050】
ここで、wi は統計的重み、yi は観測強度、fi (x)は理論回折強度を示す。
【0051】
リートベルト法による有機分子の結晶多形の定量方法としては、多相解析における各々の結晶相の尺度因子により算出する計算方法が好ましい。例えば、中井,泉らによる粉末X線解析の実際第2版(株式会社朝倉書店,2009)によると、リートベルト解析プログラムは通常、二つ以上の結晶相からなる混合物も取り扱うことができる。この機能を活用して、結晶相iの質量分率wi (上記統計的重みwi とは異なる)は、リートベルト法により精密化した尺度因子sj (j=1,2,3,…)から、
【0052】
【数7】
【0053】
という式で求められる。ここで、Zは単位胞中の化学式単位の数、Mは化学式単位の質量、Vは化学式単位の体積、
【0054】
【数8】
【0055】
は全相についての和を表す。
【0056】
リートベルト法における有機分子の結晶多形の多相解析において、粒子径および吸収係数から生じるmicroabsorption の問題は、汎用のCuKα線のような長波長X線においても無視して構わない。結晶多形のため、即ち同一分子・同一組成のため、基本的な吸収係数は同じであり、粒子径による効果だけが異なるからである。有機分子を構成するC,H,N,O,S等では吸収係数も小さく問題になることはない。本発明においてもこの考え方に従う。もちろん、粒子径が著しく異なる場合、または、重元素が含まれる場合は、補正することによってより好適になるため、この内容に限定されるものではない。
【0057】
以上のように、本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料を、リートベルト法による多相解析に付すことにより前記結晶多形を定量する方法であって、照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いて多相解析を行う方法である。そして、上記照射体積一定条件とは、デバイ・シェラー法による透過測定法であることが好ましい。これにより、実質的に全ての粉末X線回折データをリートベルト法による多相解析に付すので精度が向上し、分子量が大きく複雑な化学構造を有し互いの有機分子のピーク重畳が激しい試料であっても測定することができ、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができる。
【0058】
従って、本発明に係る有機分子の結晶多形の定量方法の更なる特徴は、粉砕工程を経ていない粉末試料を用いること、さらには、上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよいことにある。Zeltan Nemetら (Journal of Pharmaceutical and Biomedical Analysis, 49, 338. (2009)) の報告では、Famotidineの結晶相の定量においては、測定の前処理として行う粉砕工程による影響は無いと記載されている。しかしながら、多くの有機分子の結晶は、粉砕における応力により、結晶形に影響を受けているおそれがある。本発明に係る定量方法によれば、乳鉢を用いてすり潰す(摩砕)等の粉砕工程を経ていない粉末試料を用いることで、粉砕による影響を受けずに測定することができるので、選択配向を生じ易い結晶の有機分子からなる結晶多形であっても、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、精密に定量することができる。
【0059】
また、以上のように、本発明に係る有機分子の結晶多形の定量方法は、有機分子の結晶多形を2相以上含有する試料中の前記結晶多形を、粉末X線回折強度比を用いて作成した検量線を用いて定量する方法であって、照射体積一定条件であるデバイ・シェラー法による透過測定法を用い、かつ、粉砕工程を経ていない粉末試料を用いる方法である。そして、ブラッグの条件で計算される面間隔値dが測定対象の粉末試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いることが好ましい。これにより、分子量が大きく複雑な化学構造を有し互いの有機分子のピーク重畳が激しい試料であっても測定することができ、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少なくとも1重量%以上、99重量%以下の範囲で含まれる結晶多形を、定量することができる。上記検量線は、有機分子の含有量が既知でそれぞれ含有量が異なる5点以上の試料(混合試料)を用いて測定した粉末X線回折強度を用いて、例えば最小二乗法で求められていることがより好ましい。
【0060】
また、本発明に係る有機分子の結晶多形の定量方法は、上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよいことにある。そして、上記検量線が、結晶多形に含まれる全ての単一結晶の粉末X線回折強度を含んで作成されていることが好ましい。
【0061】
即ち、本発明に係る有機分子の結晶多形の定量方法の更なる特徴は、各結晶相の分率座標を固定しながら精密化することで、粉末試料が目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいても、当該粗大粒子による回折強度の致命的な変化を防止し、かつ、実質的な解析精度を下げることなく定量することができることを見出したことにある。リートベルト法において、粗大粒子による強い回折は、最小二乗残差として目立つことが多く、弊害が大きい。そのため、リートベルト法プログラムでは、選択配向因子や原子の分率座標を調整して精密化してしまう。このとき、分子間の結合距離、結合角、または、二面角を或る程度の許容範囲で拘束条件をかけながら解析することは、熟練測定者の勘所となっている(熟練を要する作業となっている)。また、分率座標は、有機分子からなる結晶ではパラメーター数が多く、実質的に精密化が不可能な水素原子も多数有しているため、煩雑な解析になることが多い。そのため、最小二乗残差の誤差の掃き溜めとなり易いという問題がある。これに対して、本発明においては、任意性が生じ難いように結晶構造の分率座標だけを全て固定する方法を実施したところ、定量性は実質的に維持されることを見出した。本発明に係る定量方法によれば、粉砕による影響を受けずに測定することができるので、選択配向を生じ易い結晶の有機分子からなる結晶多形であっても、有機分子の結晶多形を2相以上含有する試料中に、少なくとも1重量%以上、99重量%以下の範囲で含まれる結晶多形を、定量することができる。
【実施例】
【0062】
以下、実施例および比較例によって本発明をより詳細に説明する。
〔試料の調製方法〕
下記実施例に用いる試料を、以下の方法で調製した。
【0063】
公知の製造方法である特許第3390453号公報の第23頁(実施例86)に記載されている方法によって、3−(5−メチル−1,2,4−オキサジアゾール−3−イル)−5−(3−メトキシフェニル)−1,6−ナフチリジン−2(1H)オンの無色粉末を製造した。そして、上記無色粉末を、ジメチルホルムアミドおよび2−プロパノールの混合溶媒(ジメチルホルムアミド:2−プロパノール=4:7(容量比))に溶解させた後、再結晶させた。その後、得られた固形分を濾取して乾燥させることにより、3−(5−メチル−1,2,4−オキサジアゾール−3−イル)−5−(3−メトキシフェニル)−1,6−ナフチリジン−2(1H)オンの粉末結晶(以下、単にForm Aと記す)を得た。
【0064】
一方、上記混合溶媒の代りに水およびエタノールの混合溶媒(水:エタノール=1:4(容量比))を用いた以外は、上記操作と同様にして無色粉末を溶解させて再結晶させ、得られた固形分を濾取して乾燥させることにより、3−(5−メチル−1,2,4−オキサジアゾール−3−イル)−5−(3−メトキシフェニル)−1,6−ナフチリジン−2(1H)オンの粉末結晶(以下、単にForm Bと記す)を得た。
【0065】
上記Form Aの粉末X線回折データ(XRDデータ)を図5に示すと共に、上記Form Bの粉末X線回折データを図6に示す。
〔実施例1〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量(仕込み量)が80重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢にForm A1.00499gとForm B1.00040gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm A1.00401gを加えて同様に30分間手動で混合した後、さらにForm A1.99976gを加えて30分間手動で混合して、Form Aの含有量が80重量%の混合試料(以下、80%混合試料と記す)を作成した。得られた80%混合試料を目開き150μmの篩を用いて篩別した後、Hilgenberg社製1.0mmφほう珪酸ガラスキャピラリーに封管した。
【0066】
次に、ブルカーAXS社製高分解能粉末回折装置 D8 ADVANCE with Vαrio1(CuKα,40kV,40mA)に1次元高速PSD検出器(VÅNTEC-1) を組み合わせた装置を用いて、疑似デバイ・シェラー光学系で測定対象の試料の最低角反射に相当する面間隔から1.19Å(0.109nm)以下(2θ>80.676°)の範囲における粉末X線回折データ(XRDデータ)を得た。尚、粉末X線回折法の弱点である選択配向の寄与を低減するために、ほう珪酸ガラスキャピラリーを毎分60回転で回転させながら室温で測定を行った。
【0067】
そして、透過法測定であることから、試料のX線透過率の実測値を用いて線吸収係数を次式より求めた。
【0068】
Ix =I0 exp(−μt)
ここで、μは線吸収係数、tは試料厚さ、Ix は試料透過X線強度、I0 はX線強度である。
【0069】
得られたXRDパターンに対し、RIETAN-FP プログラムパッケージ (Fujio Izumi and Koichi Momma, Solid. State Phenom., 130 15 (2007))を用いてリートベルト2相解析を実施した。ほう珪酸ガラスキャピラリーによる非晶質からの散乱は、こぶ状のバックグラウンドとして測定データの精密化の妨げとなる。そのため、11次のルジャンドル直交多項式を重畳した複合バックグラウンド関数で、全体のバックグラウンドを見積もった。そのとき、初期のバックグラウンドデータは プログラムPowderX (Cheng Dong, J. Appl. Crystallogr. 32 838 (1999)) にて近似した。プロファイル関数には虎谷の拡張分割Pseude-Voigt関数を使用した。しかしながら、粉末法の貧弱なデータを用い、水素原子まで付加された2相の結晶多形を含む全てのパラメーターを精密化することは、困難であると予測された。そこで、以下のような解析条件を前提に、2相解析を実施した。
(1)二つの結晶構造の分率座標は固定する。
(2)結晶構造に関するパラメーターでは、格子定数、等方性原子変位パラメーターのみを精密化の対象とする。但し、最小二乗法が発散するときには固定する。
(3)その他、尺度因子、プロファイル関数、バックグラウンド関数は精密化する。
【0070】
リートベルト解析による解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。ここでRwp値、Rp 値およびS値は、R.A.Young ら (The Rietveld Method, Oxford, 1993)の定義に従った。
〔実施例2〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量が50重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢にForm A1.00074gとForm B1.00031gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm A0.50103gとForm B0.50076gとを加えて同様に30分間手動で混合した後、さらにForm A1.00406gとForm B1.000008gとを加えて30分間手動で混合して、Form Aの含有量が50重量%の混合試料(以下、50%混合試料と記す)を作成した。上記50%混合試料の粉末X線回折データを図7に示す。得られた50%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
〔実施例3〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量が10重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢にForm A0.50025gとForm B0.50133gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm B2.00203gを加えて同様に30分間手動で混合した後、さらにForm B2.00169gを加えて30分間手動で混合して、Form Aの含有量が10重量%の混合試料(以下、10%混合試料と記す)を作成した。得られた10%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
〔実施例4〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量が5重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢にForm A0.25017gとForm B0.25039gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm B0.50213gを加えて同様に30分間手動で混合した後、さらにForm B2.00094gを加えて同様に30分間手動で混合し、次いでForm B1.99975gを加えて同様に30分間手動で混合して、Form Aの含有量が5重量%の混合試料(以下、5%混合試料と記す)を作成した。得られた5%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
〔実施例5〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量が2重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢にForm A0.18138gとForm B0.320839gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm B1.00049gを加えて同様に30分間手動で混合した後、さらにForm B1.49992gを加えて同様に30分間手動で混合した。次いで、この混合試料にForm B3.001975gを加えて同様に30分間手動で混合した後、さらにForm B3.00796gを加えて同様に30分間手動で混合して、Form Aの含有量が2重量%の混合試料(以下、2%混合試料と記す)を作成した。得られた2%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
〔実施例6〕
粉末結晶であるForm AおよびForm Bを、Form Aの含有量が1重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢に実施例5で作成した2%混合試料1.00201gとForm B1.00097gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料に2%混合試料1.00103gとForm B1.00735gとを加えて同様に30分間手動で混合した後、さらに2%混合試料0.50408gとForm B0.50602gとを加えて同様に30分間手動で混合した。次いで、この混合試料に2%混合試料2.50712gを加えて同様に30分間手動で混合して、Form Aの含有量が1重量%の混合試料(以下、1%混合試料と記す)を作成した。得られた1%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
〔実施例7〕
粉末結晶であるForm AおよびForm Bを、を、Form Aの含有量が0.5重量%になるように、以下の手順で均一に混合した。試料の汚染を避けるために、メノウ製の乳鉢を容器として使用した。乳鉢に実施例5で作成した2%混合試料1.25136gとForm B1.25156gとを入れて、ステンレス製スパーテルを用いて30分間手動で混合した。次に、この混合試料にForm B1.00294gを加えて同様に30分間手動で混合した後、さらにForm B1.50119gを加えて同様に30分間手動で混合して、Form Aの含有量が0.5重量%の混合試料(以下、0.5%混合試料と記す)を作成した。上記0.5%混合試料の粉末X線回折データを図8に示す。得られた0.5%混合試料を用いて、実施例1と同じ方法で、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表1に示す。また、最終の信頼度因子を表2に示す。
【0071】
【表1】
【0072】
【表2】
【0073】
〔実施例8〕
実施例1〜7で作成した80%混合試料,50%混合試料,10%混合試料,5%混合試料,2%混合試料,1%混合試料,0.5%混合試料の粉末X線回折データ、並びに、実施例1と同じ方法で測定した粉末結晶であるForm A(以下、Form A100%試料と記す)およびForm B(以下、Form B100%試料と記す)の粉末X線回折データを用いて、ピーク重畳が無く、Form Aに特徴的な2θ=6.9°の回折ピークの強度とForm Bに特徴的な2θ=10.1°の回折ピークの強度との比(ピーク強度比)から、以下に示す式により検量線を作成して、各混合試料におけるForm AおよびForm Bの定量を行った。
【0074】
y=100×(Ha/(Ha+Hb))x
ここで、yはForm Aの量(重量%)、Haは2θ=6.9°の回折ピーク強度、Hbは2θ=10.1°の回折ピーク強度、xは最小二乗法を用い、理論値と観測値の差が最小になるようにして求めた値(1.0375)である。
【0075】
80%混合試料,50%混合試料,10%混合試料,5%混合試料,2%混合試料,1%混合試料,0.5%混合試料の粉末X線回折データから得られた2θ=6.9°および2θ=10.1°のピーク強度の一覧を表3に示す。また、Form A100%試料およびForm B100%試料の粉末X線回折データから得られた2θ=6.9°および2θ=10.1°のピーク強度(counts)の一覧を表4に示す。そして、検量線法による解析結果、即ち、定量分析結果(検出量)を表5に示す。また、検量線のグラフを図4に示す。
【0076】
【表3】
【0077】
【表4】
【0078】
【表5】
【0079】
〔比較例1〕
実施例6で作成した1%混合試料を、目開き150μmの篩を用いて篩別した後、直径25mm,深さ0.2mmのガラス製試料台に充填した。次に、株式会社リガク製粉末回折装置RINT-2100V(CuKα,40kV,40mA)を用いてブラッグ・ブレンターノ光学系で測定対象の試料の最低角反射に相当する面間隔から1.176Å以下(2θ>80.0°)の範囲における粉末X線回折データ(XRDデータ)を得た。尚、粉末X線回折法の弱点である選択配向の寄与を低減するために、ガラス製試料台を毎分30回転で回転させながら室温で測定を行った。さらに、発散スリット1/2°、散乱スリット1/2°、受光スリット0.3mmとし、0.03°/2θステップ、積算時間4.0sec で、Fixed time法を用いた。
【0080】
得られたXRDパターンに対し、RIETAN-FP プログラムパッケージを用いてリートベルト2相解析を実施した。11次のルジャンドル直交多項式を重畳した複合バックグラウンド関数で、全体のバックグラウンドを見積もった。そのとき、初期のバックグラウンドデータは プログラムPowderX (Cheng Dong, J. Appl. Crystallogr. 32 838 (1999)) にて近似した。プロファイル関数には虎谷の拡張分割Pseude-Voigt関数を使用した。また、実施例1と同じ結晶構造を初期モデルとした。しかしながら、粉末法の貧弱なデータを用い、水素原子まで付加された2相の結晶多形を含む全てのパラメーターを精密化することは、困難であると予測された。そこで、実施例1に記載した(1)〜(3)の解析条件を前提に、2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表6に示す。また、最終の信頼度因子を表7に示す。
〔比較例2〕
実施例7で作成した0.5%混合試料を用いた以外は、比較例1と同じ方法で2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表6に示す。また、最終の信頼度因子を表7に示す。
〔比較例3〕
実施例7で得られた粉末X線回折データ(XRDデータ)を用い、リートベルト解析に用いる計算範囲を9〜35°/2θに制限した以外は、実施例7と同じ方法で2相解析を実施した。解析結果、即ち、定量分析結果(検出量)を表8に示す。また、最終の信頼度因子を表9に示す。
【0081】
【表6】
【0082】
【表7】
【0083】
【表8】
【0084】
【表9】
【0085】
〔まとめ〕
上記実施例1〜8の結果と比較例1〜3の結果との対比から明らかなように、本発明に係る定量方法によれば、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で含まれる結晶多形を、精密に定量することができることが判る。
【0086】
尚、本発明は、上述した実施形態に限定されるものではなく、記述した範囲内で種々の変形を加えた態様で実施することができ、従って、請求項に示した範囲で種々の変更が可能である。
【産業上の利用可能性】
【0087】
本発明に係る有機分子の結晶多形の定量方法によれば、内部標準試料を用いることなく、有機分子の結晶多形を2相以上含有する試料中に、少ない含有量で、具体的には、1重量%で含まれる結晶多形を、定量することができる方法を提供することができる。
【0088】
それゆえ、本発明に係る有機分子の結晶多形の定量方法は、例えば、医薬品の原体の定量性に好適に利用することができる。
【特許請求の範囲】
【請求項1】
有機分子の結晶多形を2相以上含有する試料を、リートベルト法による多相解析に付すことにより前記結晶多形を定量する方法であって、
照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いて多相解析を行うことを特徴とする有機分子の結晶多形の定量方法。
【請求項2】
上記照射体積一定条件とは、デバイ・シェラー法による透過測定法であることを特徴とする請求項1に記載の有機分子の結晶多形の定量方法。
【請求項3】
粉砕工程を経ていない粉末試料を用いることを特徴とする請求項1または2に記載の有機分子の結晶多形の定量方法。
【請求項4】
上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよいことを特徴とする請求項3に記載の有機分子の結晶多形の定量方法。
【請求項5】
有機分子の結晶多形を2相以上含有する試料中の前記結晶多形を、粉末X線回折強度比を用いて作成した検量線を用いて定量する方法であって、
照射体積一定条件であるデバイ・シェラー法による透過測定法を用い、かつ、粉砕工程を経ていない粉末試料を用いることを特徴とする有機分子の結晶多形の定量方法。
【請求項6】
ブラッグの条件で計算される面間隔値dが測定対象の粉末試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いることを特徴とする請求項5に記載の有機分子の結晶多形の定量方法。
【請求項7】
上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよいことを特徴とする請求項5または6に記載の有機分子の結晶多形の定量方法。
【請求項8】
上記検量線が、結晶多形に含まれる全ての単一結晶の粉末X線回折強度を含んで作成されていることを特徴とする請求項5,6または7に記載の有機分子の結晶多形の定量方法。
【請求項1】
有機分子の結晶多形を2相以上含有する試料を、リートベルト法による多相解析に付すことにより前記結晶多形を定量する方法であって、
照射体積一定条件で収集され、ブラッグの条件で計算される面間隔値dが前記試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いて多相解析を行うことを特徴とする有機分子の結晶多形の定量方法。
【請求項2】
上記照射体積一定条件とは、デバイ・シェラー法による透過測定法であることを特徴とする請求項1に記載の有機分子の結晶多形の定量方法。
【請求項3】
粉砕工程を経ていない粉末試料を用いることを特徴とする請求項1または2に記載の有機分子の結晶多形の定量方法。
【請求項4】
上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよいことを特徴とする請求項3に記載の有機分子の結晶多形の定量方法。
【請求項5】
有機分子の結晶多形を2相以上含有する試料中の前記結晶多形を、粉末X線回折強度比を用いて作成した検量線を用いて定量する方法であって、
照射体積一定条件であるデバイ・シェラー法による透過測定法を用い、かつ、粉砕工程を経ていない粉末試料を用いることを特徴とする有機分子の結晶多形の定量方法。
【請求項6】
ブラッグの条件で計算される面間隔値dが測定対象の粉末試料の最低角反射に相当する面間隔以上、1.55Å以下の範囲における粉末X線回折データを用いることを特徴とする請求項5に記載の有機分子の結晶多形の定量方法。
【請求項7】
上記粉末試料が、目開き150μmの篩にて篩別した10μm以上の粗大粒子を含んでいてもよいことを特徴とする請求項5または6に記載の有機分子の結晶多形の定量方法。
【請求項8】
上記検量線が、結晶多形に含まれる全ての単一結晶の粉末X線回折強度を含んで作成されていることを特徴とする請求項5,6または7に記載の有機分子の結晶多形の定量方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2012−117941(P2012−117941A)
【公開日】平成24年6月21日(2012.6.21)
【国際特許分類】
【出願番号】特願2010−268700(P2010−268700)
【出願日】平成22年12月1日(2010.12.1)
【出願人】(000002093)住友化学株式会社 (8,981)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【Fターム(参考)】
【公開日】平成24年6月21日(2012.6.21)
【国際特許分類】
【出願日】平成22年12月1日(2010.12.1)
【出願人】(000002093)住友化学株式会社 (8,981)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【Fターム(参考)】
[ Back to top ]