
有機無機複合材料
【課題】 熱膨張が小さい、脆性破壊を防止した有機無機複合材料を提供する。
【解決手段】 少なくとも(A)熱可塑性アクリル樹脂と、平均一次粒子径が1nm以上20nm以下で、表面が親水性である(B)シリカ微粒子と、(C)熱可塑性エラストマーを含有してなり、前記(B)シリカ微粒子の体積分率が、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して24体積%以上63体積%以下である有機無機複合材料。前記有機無機複合材料に含有される(C)熱可塑性エラストマーの体積分率が(A)熱可塑性アクリル樹脂の体積分率以下である。
【解決手段】 少なくとも(A)熱可塑性アクリル樹脂と、平均一次粒子径が1nm以上20nm以下で、表面が親水性である(B)シリカ微粒子と、(C)熱可塑性エラストマーを含有してなり、前記(B)シリカ微粒子の体積分率が、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して24体積%以上63体積%以下である有機無機複合材料。前記有機無機複合材料に含有される(C)熱可塑性エラストマーの体積分率が(A)熱可塑性アクリル樹脂の体積分率以下である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は有機無機複合材料に関し、特に熱膨張が小さい、脆性破壊を防止した有機無機複合材料に関するものである。
【背景技術】
【0002】
有機材料である樹脂材料は無機材料と比べて成形加工しやすいことが特徴である。特に熱可塑性樹脂は成形時間が短く生産性が高いことから、射出成型、圧縮成形、押出成形などを介して幅広い部材に利用されている。しかし樹脂材料は無機材料と比較して熱膨張が大きく、金属部材に対する位置精度のずれや熱応力による密着性の低下、また温度環境による物性の変化が大きい等の問題があり、用途が限定される問題がある。
【0003】
熱可塑性樹脂の熱膨張を低減するため、樹脂材料と無機材料を混合し複合材料とする技術が広く知られている。例えば特許文献1では熱可塑性アクリル樹脂にシリカ粒子を混合した熱膨張の小さい材料が開示されている。
【0004】
ところが、無機粒子を添加すればするほど熱膨張を低減することができる反面、材料の靱性が低下し、過荷重の負荷がかかった際に脆性破壊を起こしやすくなる。特に薄肉の部材において高濃度に無機微粒子が添加された樹脂材料を用いると、わずかな外力で部材が粉砕されることもある。無機微粒子が多く添加された樹脂材料が脆性破壊すると破片や粉じんが大量に発生し、周囲の汚染や他の部材への付着の原因となる。一般的に樹脂材料の靱性を改善するためにはゴム状材料を添加することが有効である。特許文献2では、ゴム状重合体を用いて無機フィラー含有ポリフェニレンエーテル系樹脂の靱性を改善する方法が開示されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開平11−343349号公報
【特許文献2】特開2005−281448号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、特許文献2の方法では、ゴム状重合体は樹脂材料と比較して線膨張係数が高いため、樹脂材料に添加すると線膨張係数が悪化して、靱性の改善と線膨張係数の低減を両立することは難しかった。
【0007】
本発明は、この様な背景技術に鑑みてなされたものであり、熱膨張が小さい、脆性破壊を防止した有機無機複合材料を提供するものである。
【課題を解決するための手段】
【0008】
上記の課題を解決する有機無機複合材料は、少なくとも(A)熱可塑性アクリル樹脂と、平均一次粒子径が1nm以上20nm以下で、表面が親水性である(B)シリカ微粒子と、(C)熱可塑性エラストマーを含有してなり、前記(B)シリカ微粒子の体積分率が、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して24体積%以上63体積%以下であり、且つ前記(C)熱可塑性エラストマーの体積分率が(A)熱可塑性アクリル樹脂の体積分率以下であることを特徴とする。
【発明の効果】
【0009】
本発明によれば、熱膨張が小さい、脆性破壊を防止した有機無機複合材料を提供することができる。
【図面の簡単な説明】
【0010】
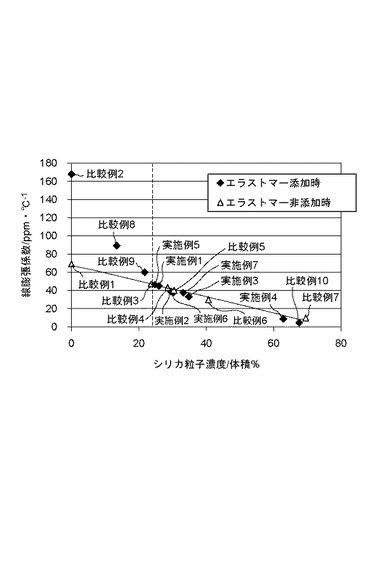
【図1】本発明の有機無機複合材料の線膨張係数を示すグラフである。
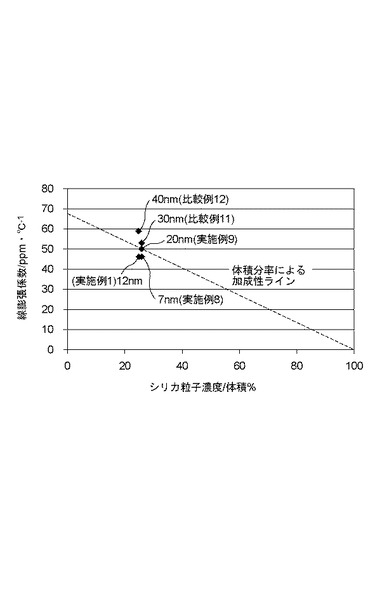
【図2】本発明の有機無機複合材料に含有されるシリカ微粒子の粒子径の影響を示すグラフである。
【発明を実施するための形態】
【0011】
以下、本発明の実施の形態について詳細に説明する。
【0012】
本発明に係る有機無機複合材料は、少なくとも(A)熱可塑性アクリル樹脂と、平均一次粒子径が1nm以上20nm以下で、表面が親水性である(B)シリカ微粒子と、(C)熱可塑性エラストマーを含有してなり、前記(B)シリカ微粒子の体積分率が、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して24体積%以上63体積%以下であり、且つ前記(C)熱可塑性エラストマーの体積分率が(A)熱可塑性アクリル樹脂の体積分率以下であることを特徴とする。
【0013】
本発明に係る有機無機複合材料は、少なくとも(A)熱可塑性アクリル樹脂、(B)シリカ微粒子、(C)熱可塑性エラストマーを含有する組成物から成る。以降の説明において、(A)(B)(C)の各成分を混合した後の組成物からなる有機無機複合材料を「複合材料」と呼ぶ。(A)(B)を含み(C)を含まない系を「二成分系」、(A)(B)(C)を含む系を「三成分系」と呼ぶ。
【0014】
<複合材料の熱膨張の低減について>
(C)成分の熱可塑性エラストマーはゴム状重合体であり、熱膨張が非常に大きいため、樹脂材料の熱膨張を悪化させると考えられる。しかし、本発明者は複合材料を三成分系にした場合、無機微粒子の添加濃度や表面官能基、粒子径の変化による複合材料の熱膨張変化に特徴があることを見出した。また、鋭意検討の結果、無機微粒子の添加濃度や表面官能基、粒子径を最適に選択することにより、三成分系の熱膨張を二成分系と同等まで低減することができた。熱膨張の大きい熱可塑性エラストマーを添加した三成分系でも熱膨張が二成分系と同等まで熱膨張を低減できる理由は、例えば文献1:No.920−17 日本機械学会第69期通常総会講演会講演論文集(Vol.A)、および文献2:No.930−63 日本機械学会第71期全国大会講演論文集(Vol.B)に記載されている。
【0015】
上記の文献1および2では弾性率の高い材質(以下強化材)、マトリクス樹脂、ボイドを混合することで熱膨張を低減している。温度上昇時に強化材とマトリクス樹脂の近傍に発生する圧縮応力がボイドを潰し、マクロ的な熱膨張を低減している。ここで文献1によれば、ボイドの代替として弾性率の小さい材料を用いることも可能であることが記載されている。本発明で用いる熱可塑性エラストマーは弾性率が非常に小さいため、ボイドと同様のメカニズムで熱膨張を低減することが可能と推察される。
【0016】
しかしボイドの代替として熱可塑性エラストマーを用いると、強化材はマトリクス樹脂だけではなく熱可塑性エラストマー内にも混入するため、マトリクス樹脂が熱可塑性エラストマーを潰そうとする圧縮応力が小さくなると考えられる。またマトリクス樹脂との相溶性によって複合材料中の熱可塑性エラストマーの分布状態も変化する。熱可塑性エラストマーが複合材料の表層に多く分布する場合、熱可塑性エラストマーの膨張が直接マクロ物性として発現しやすくなるため、複合材料の熱膨張が大きくなることが推測される。さらに熱可塑性エラストマーは材質としての弾性率を有するため、ボイドよりも潰れにくい。以上の観点から、熱可塑性エラストマーをボイドの代替として用いた場合、ボイドを混合した系に比べて熱膨張の低減が非効率的になる。
【0017】
上記のメカニズムを整理すると、複合材料は、(I)圧縮応力に起因する熱可塑性エラストマーの潰れによる熱膨張の低減、および(II)熱可塑性エラストマーの大きい熱膨張による熱膨張の悪化、の2つの影響を受けると考えられる。熱膨張を低減するためには(I)の効果を大きくする必要がある。
【0018】
(I)の効果を大きくするためには、強化材とマトリクス樹脂の近傍に発生する圧縮応力をより大きく発生させればよいため、強化材の表面積を増やすことが有効な手段となる。強化材の表面積を増やすためには、強化材の添加量を増やすか、強化材のサイズを小さくすればよい。また、複合材料中の各成分の分布状態も(I)と(II)のそれぞれに影響すると考えられる。各成分の分布状態に影響を与える要因として、強化材の表面官能基が挙げられる。従って熱膨張を低減するためには上述のパラメーターを最適に選択する必要がある。
【0019】
以下、本発明の複合材料に含まれる(A)熱可塑性アクリル樹脂、(B)シリカ微粒子、(C)熱可塑性エラストマーについて説明する。
【0020】
(A)熱可塑性アクリル樹脂
熱可塑性アクリル樹脂は本発明の母材となる成分であり、本発明で用いる熱可塑性アクリル樹脂とは、下記一般式(1)に示されるポリメタクリル酸メチル(以下PMMA)構造を主として有するポリマーを示す。熱可塑性アクリル樹脂は、物性値が大幅に悪化する、または最終的に作られる複合材料の混合状態を大きく変化させることがない限り、主骨格中の官能基が置換されていても構わない。例えばポリアクリル酸メチル、ポリアクリル酸エチル、ポリメタクリル酸エチルなどのアクリル酸誘導体のポリマーも主骨格として用いることができる。同様に熱可塑性アクリル樹脂は、物性値が大幅に悪化する、または最終的に作られる複合材料の混合状態を大きく変化させることがない限り、別のモノマー成分が含まれる共重合体を用いてもよい。
【0021】
【化1】

【0022】
本発明の複合材料に含有される(A)熱可塑性アクリル樹脂の体積分率は、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して34体積%以上75体積%以下、好ましくは40体積%以上60体積%以下であることが望ましい。
【0023】
(B)シリカ微粒子
本発明で用いられるシリカ微粒子は、表面が親水性であることを特徴とする。ここで表面が親水性であるとは、シリカ微粒子の表面に存在する官能基として水酸基またはアミノ基を含む官能基がもっとも強く検出される状態を表す。樹脂材料に無機微粒子を添加して熱膨張を低減する方法はよく知られているが、本発明者はシリカ微粒子の表面修飾基により熱膨張が変化し、この傾向が二成分系よりも三成分系で顕著であることを見出した。特に三成分系で表面がトリメチルシリル基で修飾されたシリカ微粒子を用いた場合、マトリクス樹脂そのものよりも熱膨張が悪化する現象が確認できている。これは先述の複合材料中の各成分の分布状態が変化することに起因していると考えられる。
【0024】
温度上昇時に圧縮応力を十分に発生させるため、シリカ微粒子の表面積が大きいこと、すなわち粒子径が十分に小さいことが重要である。三成分系において平均一次粒子径が20nmを超えると、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子の二成分系の体積分率から求められる熱膨張の値(加成性値)よりも、複合材料の熱膨張が悪化する。従って無機微粒子の平均一次粒子径は1nm以上20nm以下、好ましくは7nm以上12nm以下であることが望ましい。ここで平均一次粒子径は同体積の球に換算した場合の直径を示す。
【0025】
さらに、圧縮応力を十分に発生させるためにはシリカ微粒子の添加量が十分である必要がある。図1に示すように、表面が親水性で平均一次粒子径が十分に小さい場合、シリカ微粒子が24体積%以上になると二成分系と三成分系の熱膨張が同程度まで低下する。シリカ微粒子が多量に添加されるほど熱膨張を低減できるが、複合材料中に含まれる高分子成分の相対的な含有量が少なくなり、靱性や破壊強度が低下する。本発明の複合材料に含有される(B)シリカ微粒子の体積分率は、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して24体積%以上63体積%以下、好ましくは24体積%以上35体積%以下であることが望ましい。シリカ微粒子が24体積%未満では、(C)熱可塑性エラストマーの有する大きい熱膨張が顕在化し、線膨張係数は二成分系のときよりも悪化する。シリカ微粒子が63体積%を超えると複合材料の脆性破壊の防止改善効果がほとんど得られなくなり、また破壊強度の低下に伴い成形が困難になる。
【0026】
混合するシリカ微粒子は粉体であることが望ましいが、液体混合法を用いる場合には分散剤などの影響がない範囲でスラリーを使用しても構わない。
【0027】
(C)熱可塑性エラストマー
熱可塑性エラストマーは複合材料に脆性破壊の防止を与えるために添加されるゴム状重合体であり、弾性率は100MPa以下のものを用いる。熱可塑性エラストマーを添加するほど複合材料の脆性破壊の防止を改善できるが、破壊強度が弱くなる。また添加しすぎると熱膨張の悪化要因にもなる。
【0028】
本発明の複合材料に含有される(C)熱可塑性エラストマーの体積分率は、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して1体積%以上34体積%以下、好ましくは12体積%以上24体積%以下であることが望ましい。また、複合材料に含有される(C)熱可塑性エラストマーの体積分率は、(A)熱可塑性アクリル樹脂の体積分率以下であることが好ましい。
【0029】
熱可塑性エラストマーは硬度が低いものを使用する必要がある。熱可塑性エラストマーの硬度が高いと弾性率が高くなるため、温度上昇で発生した圧縮応力による熱可塑性エラストマーの潰れが不十分となり、熱膨張が低減できない。従って熱可塑性エラストマーは硬度(デュロメーターA)が90度以下のものを用いることが好ましい。しかし熱可塑性エラストマーは硬度が低くなると耐熱性が低下するため、複合材料として熱膨張と耐熱性のバランスがよくなるものを適宜選択する必要がある。
【0030】
熱可塑性エラストマーは分子量が小さいと流動性が良好になるが、流動性が高すぎると熱膨張悪化の原因となる。成形加工時の温度において熱可塑性エラストマーの流動性は(A)熱可塑性アクリル樹脂よりも低いことが好ましい。
【0031】
本発明では硬度の低いスチレン系またはオレフィン系の熱可塑性エラストマーを用いる。スチレン系とはスチレンブロック共重合体であり、スチレンブロックの他にブタジエン、エチレン、イソプレン、プロピレン等のオレフィンブロック(水添されていてもよい)を有していることが特徴である。またオレフィン系とはオレフィンブロック共重合体であり、オレフィンブロック以外にスチレンブロックが含まれていてもよい。
【0032】
<複合材料の製造方法>
複合材料を製造する方法は、(A)熱可塑性アクリル樹脂、(B)シリカ微粒子、(C)熱可塑性エラストマーの各成分を均一に混合できる方法であれば特に限定されない。例えば、(A)を加熱し溶融させた状態で混合する溶融混練法や、(A)と(C)を溶媒中に溶解させて(B)とともによく撹拌し脱溶媒することで複合材料を得る溶媒混合法が挙げられる。
【0033】
熱膨張や脆性破壊の防止を極端に悪化しない限り、適宜添加剤を加えてもよい。添加剤とは、例えば耐熱安定剤、耐光安定剤、難燃剤、酸化防止剤、潤滑剤、離型剤、着色剤、帯電防止剤、光増感剤、抗菌剤などが挙げられる。また熱膨張や脆性破壊の防止が極端に悪化しない限り、複合材料中にボイドが含まれても構わない。
【0034】
本発明の複合材料に含有される添加剤の含有量は、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して0体積%以上5体積%以下が好ましい。
【0035】
本発明の複合材料は、例えば精密機器用外装材、光学用ミラーなどの用途に用いることができる。
【実施例】
【0036】
以下に、本発明の実施例について説明するが、本発明はこれらの例によって何ら限定されるものではない。
【0037】
<実施例1>
PMMA(デルペット70NH;旭化成ケミカルズ社商品名)をテトラヒドロフラン(以下THF)溶媒に対して5重量%になるように混合し、常温にてPMMAを溶解させ、PMMA/THF溶液を作製した。
【0038】
スチレン−ブタジエン系熱可塑性エラストマー(タフテックH1221;旭化成ケミカルズ社商品名)をTHF溶媒に対して5重量%になるように混合し、常温にて熱可塑性エラストマーを溶解させ、熱可塑性エラストマー/THF溶液を作製した。
【0039】
シリカ粒子(アエロジル200;アエロジル社商品名、平均一次粒子径12nm、表面は水酸基)0.56g、PMMA/THF溶液12g、熱可塑性エラストマー/THF溶液4.8gをよく混合する。シリカ粒子は非常にかさ高いため、溶液中に浸漬するようにTHFを任意量添加し、シリカ粒子を十分に浸漬させた状態で混合する。マグネチックスターラーで撹拌しながら溶媒を除去した後、真空乾燥炉を用い200℃で3から4時間の乾燥を行い、三成分系複合材料1を得た。
【0040】
<実施例2から4:シリカ粒子含有量の増加>
(実施例2)
実施例1において、シリカ粒子の添加量を0.72gに変えた以外は同様にして、三成分系複合材料2を得た。
【0041】
(実施例3)
実施例1において、シリカ粒子の添加量を0.84gに変えた以外は同様にして、三成分系複合材料3を得た。
【0042】
(実施例4)
実施例1において、シリカ粒子を0.84g、PMMA/THF溶液を2.4g、熱可塑性エラストマー/THF溶液を1.8gと変更して混合した以外は同様にして、三成分系複合材料4を得た。
【0043】
<実施例5から6:熱可塑性エラストマー含有量の減少>
(実施例5)
実施例1において、PMMA/THF溶液を14.4g、熱可塑性エラストマー/THF溶液を2.4gと変更して混合した以外は同様にして、三成分系複合材料5を得た。
【0044】
(実施例6)
実施例1において、シリカ粒子を0.72g、PMMA/THF溶液を14.4g、熱可塑性エラストマー/THF溶液を2.4gと変更して混合した以外は同様にして、三成分系複合材料6を得た。
【0045】
<実施例7:熱可塑性エラストマー含有量の増加>
(実施例7)
実施例1において、シリカ粒子を0.72g、PMMA/THF溶液を9.6g、熱可塑性エラストマー/THF溶液を7.2gと変更して混合した以外は同様にして、三成分系複合材料7を得た。
【0046】
<実施例8から9:シリカ粒子の平均一次粒子径の変更>
(実施例8)
実施例1において、シリカ粒子をアエロジル300(日本アエロジル社商品名、平均一次粒子径7nm、表面は水酸基)に変えた以外は同様にして、三成分系複合材料8を得た。
【0047】
(実施例9)
実施例1において、シリカ粒子をアエロジル90G(日本アエロジル社商品名、平均一次粒子径20nm、表面は水酸基)に変えた以外は同様にして、三成分系複合材料9を得た。
【0048】
次に、比較例を示す。なお比較例中、シリカ粒子を含む複合材料の番号は3ケタで表記している。
【0049】
<比較例1から2:シリカ非添加系>
(比較例1)
熱可塑性樹脂のPMMA(デルペット70NH;旭化成ケミカルズ社商品名)だけを用いた。
【0050】
(比較例2)
実施例1において、シリカ粒子を添加しない場合をPMMA/エラストマー混合体とし、比較例2とする。
【0051】
<比較例3から7:PMMA二成分系>
実施例1において、熱可塑性エラストマーを添加せず、シリカ粒子とPMMA/THF溶液の添加量の割合を下記のように変えて、二成分系材料103から107を得た。
【0052】
(比較例3)
二成分系材料番号103:シリカ粒子量0.36g:PMMA/THF溶液量12.8g
(比較例4)
二成分系材料番号104:シリカ粒子量0.40g:PMMA/THF溶液量12.0g
(比較例5)
二成分系材料番号105:シリカ粒子量0.44g:PMMA/THF溶液量11.2g
(比較例6)
二成分系材料番号106:シリカ粒子量0.50g:PMMA/THF溶液量10.0g
(比較例7)
二成分系材料番号107:シリカ粒子量0.80g:PMMA/THF溶液量4.0g
【0053】
<比較例8から9:三成分系、シリカ粒子含有量不足>
(比較例8)
実施例1においてシリカ粒子の添加量を0.26gに変えた以外は同様にして、三成分系複合材料108を得た。
【0054】
(比較例9)
実施例1においてシリカ粒子の添加量を0.40gに変えた以外は同様にして、三成分系複合材料109を得た。
【0055】
<比較例10:三成分系、シリカ粒子含有量が多い>
(比較例10)
実施例1において、シリカ粒子を0.96g、PMMA/THF溶液を2.4g、熱可塑性エラストマー/THF溶液を1.8gと変更して混合した以外は同様にして、三成分系複合材料110を得た。
【0056】
<比較例11から12:シリカ粒子径が大きい>
(比較例11)
実施例1において、シリカ粒子をアエロジル50(アエロジル社商品名、平均一次粒子径30nm、表面は水酸基)に変えた以外は同様にして、三成分系複合材料111を得た。
【0057】
(比較例12)
実施例1において、シリカ粒子をアエロジルOX50(アエロジル社商品名、平均一次粒子径40nm、表面は水酸基)に変えた以外は同様にして、三成分系複合材料112を得た。
【0058】
<比較例13から14:疎水性シリカ表面>
(比較例13)
実施例1において、シリカ粒子をアエロジルRX200(アエロジル社商品名、平均一次粒子径12nm、表面はトリメチルシリル基修飾)に変えた以外は同様にして、三成分系複合材料113を得た。
【0059】
<比較例14:熱可塑性エラストマー添加量が多い>
(比較例14)
実施例1において、シリカ粒子を0.72g、PMMA/THF溶液を7.2g、熱可塑性エラストマー/THF溶液を9.6gと変更して混合した以外は同様にして、三成分系複合材料114を得た。
【0060】
次に、上記の実施例、比較例の材料の評価を示す。
【0061】
<試料の作製>
上記の実施例、比較例で作製した材料を評価するため、試験片を作製した。6mm角のプレス成形用金型の面に離型剤としてノベック−1720(住友スリーエム社商品名)を滴下してよく拭き取る。上記の実施例、比較例で作製した材料をプレス成形用金型に充填し、小型熱プレス機(アズワン社製)にセットしながら250℃まで加熱した。加熱小型熱プレス機の上面と下面の温度が250℃に達した後に110MPaの荷重を付与し、100℃まで風冷しながら荷重を自然開放させた。100℃で完全に荷重を除き、金型から成形体を離型することで直方体の試験片(6mm×6mm、厚さおよそ3mm)を得た。
【0062】
<線膨張係数の評価>
試験片をTMA(TMA Q400;TAインスツルメント社商品名)にて−10から65℃で3サイクル温度負荷を与え、厚み方向に対する20から40℃の線膨張係数を算出した。変位の測定には膨張プローブを使用した。
【0063】
<体積分率の評価>
シリカ微粒子の含有量の測定はTGA(TGA Q500;TAインスツルメント社商品名)を用いて行った。シリカ微粒子の含有量を体積%へ換算するために用いた各材料の密度値は以下である。
PMMA 1.19、シリカ 2.00、スチレン−ブタジエン系熱可塑性エラストマー 0.89。
複合材料中の各成分の体積分率は、添加した熱可塑性アクリル樹脂と熱可塑性エラストマーの重量比が一定であると仮定して算出した。
【0064】
<破壊モード、破壊強度の評価>
得られた6mm角の試験片をプレス装置で圧縮破壊させ、破壊モードの確認と破壊時の荷重(破壊強度)の測定を行った。
破壊モードについては、破壊後の試験片が粉々に粉砕されていた場合を脆性破壊、破壊後の試験片が餅状に圧潰した場合を延性破壊とした。破壊のタイミングが明確でないものは、破壊荷重を「検出できず」とした。
【0065】
上記の実施例と比較例の評価結果を表1に示す(表1中の体積分率の表記は実際の値を四捨五入して整数値に揃えたため、合計が100体積%になっていない例もある)。
【0066】
表中、粒子径12nmかつ水酸基表面を有するシリカ粒子を用いたもの、および比較例1から7の結果を抽出し、シリカ粒子体積分率と線膨張係数でプロットしたものを図1に示す。また水酸基表面を有するシリカ粒子でシリカ粒子の体積分率が25体積%に近いものを抽出し、プロットしたものを図2に示す。図2中のnmの数値は、シリカ粒子の平均一次粒子径を示す。
【0067】
【表1】
【0068】
図1により、シリカ粒子が24体積%以上において、二成分系と三成分系の線膨張係数が同レベルとなることが明らかである。
【0069】
表1および図2より、粒子径が大きくなるほど線膨張係数が悪化することがわかる。シリカ粒子の線膨張係数を石英と同等(0.5ppm/℃)と仮定すると、粒子径20nm以下において体積分率から線形的に計算される線膨張係数の値(加成性値)を下回る。
【0070】
表1より、粒子表面が疎水性基で修飾されているほど線膨張係数が悪化することがわかる。
【0071】
表1より、複合材料は熱可塑性エラストマーが添加されていない場合は必ず脆性破壊を起こすが、熱可塑性エラストマーが添加されている場合は複合材料が延性破壊しやすく、材料の脆性破壊の防止が改善されていることがわかる。
【0072】
表1より、熱可塑性エラストマーの添加量が大きすぎると線膨張係数が悪化することがわかる。
【産業上の利用可能性】
【0073】
本発明の有機無機複合材料は、熱膨張が小さく、脆性破壊を防止できるので、精密機器用外装材や光学用ミラーに利用することができる。
【技術分野】
【0001】
本発明は有機無機複合材料に関し、特に熱膨張が小さい、脆性破壊を防止した有機無機複合材料に関するものである。
【背景技術】
【0002】
有機材料である樹脂材料は無機材料と比べて成形加工しやすいことが特徴である。特に熱可塑性樹脂は成形時間が短く生産性が高いことから、射出成型、圧縮成形、押出成形などを介して幅広い部材に利用されている。しかし樹脂材料は無機材料と比較して熱膨張が大きく、金属部材に対する位置精度のずれや熱応力による密着性の低下、また温度環境による物性の変化が大きい等の問題があり、用途が限定される問題がある。
【0003】
熱可塑性樹脂の熱膨張を低減するため、樹脂材料と無機材料を混合し複合材料とする技術が広く知られている。例えば特許文献1では熱可塑性アクリル樹脂にシリカ粒子を混合した熱膨張の小さい材料が開示されている。
【0004】
ところが、無機粒子を添加すればするほど熱膨張を低減することができる反面、材料の靱性が低下し、過荷重の負荷がかかった際に脆性破壊を起こしやすくなる。特に薄肉の部材において高濃度に無機微粒子が添加された樹脂材料を用いると、わずかな外力で部材が粉砕されることもある。無機微粒子が多く添加された樹脂材料が脆性破壊すると破片や粉じんが大量に発生し、周囲の汚染や他の部材への付着の原因となる。一般的に樹脂材料の靱性を改善するためにはゴム状材料を添加することが有効である。特許文献2では、ゴム状重合体を用いて無機フィラー含有ポリフェニレンエーテル系樹脂の靱性を改善する方法が開示されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開平11−343349号公報
【特許文献2】特開2005−281448号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、特許文献2の方法では、ゴム状重合体は樹脂材料と比較して線膨張係数が高いため、樹脂材料に添加すると線膨張係数が悪化して、靱性の改善と線膨張係数の低減を両立することは難しかった。
【0007】
本発明は、この様な背景技術に鑑みてなされたものであり、熱膨張が小さい、脆性破壊を防止した有機無機複合材料を提供するものである。
【課題を解決するための手段】
【0008】
上記の課題を解決する有機無機複合材料は、少なくとも(A)熱可塑性アクリル樹脂と、平均一次粒子径が1nm以上20nm以下で、表面が親水性である(B)シリカ微粒子と、(C)熱可塑性エラストマーを含有してなり、前記(B)シリカ微粒子の体積分率が、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して24体積%以上63体積%以下であり、且つ前記(C)熱可塑性エラストマーの体積分率が(A)熱可塑性アクリル樹脂の体積分率以下であることを特徴とする。
【発明の効果】
【0009】
本発明によれば、熱膨張が小さい、脆性破壊を防止した有機無機複合材料を提供することができる。
【図面の簡単な説明】
【0010】
【図1】本発明の有機無機複合材料の線膨張係数を示すグラフである。
【図2】本発明の有機無機複合材料に含有されるシリカ微粒子の粒子径の影響を示すグラフである。
【発明を実施するための形態】
【0011】
以下、本発明の実施の形態について詳細に説明する。
【0012】
本発明に係る有機無機複合材料は、少なくとも(A)熱可塑性アクリル樹脂と、平均一次粒子径が1nm以上20nm以下で、表面が親水性である(B)シリカ微粒子と、(C)熱可塑性エラストマーを含有してなり、前記(B)シリカ微粒子の体積分率が、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して24体積%以上63体積%以下であり、且つ前記(C)熱可塑性エラストマーの体積分率が(A)熱可塑性アクリル樹脂の体積分率以下であることを特徴とする。
【0013】
本発明に係る有機無機複合材料は、少なくとも(A)熱可塑性アクリル樹脂、(B)シリカ微粒子、(C)熱可塑性エラストマーを含有する組成物から成る。以降の説明において、(A)(B)(C)の各成分を混合した後の組成物からなる有機無機複合材料を「複合材料」と呼ぶ。(A)(B)を含み(C)を含まない系を「二成分系」、(A)(B)(C)を含む系を「三成分系」と呼ぶ。
【0014】
<複合材料の熱膨張の低減について>
(C)成分の熱可塑性エラストマーはゴム状重合体であり、熱膨張が非常に大きいため、樹脂材料の熱膨張を悪化させると考えられる。しかし、本発明者は複合材料を三成分系にした場合、無機微粒子の添加濃度や表面官能基、粒子径の変化による複合材料の熱膨張変化に特徴があることを見出した。また、鋭意検討の結果、無機微粒子の添加濃度や表面官能基、粒子径を最適に選択することにより、三成分系の熱膨張を二成分系と同等まで低減することができた。熱膨張の大きい熱可塑性エラストマーを添加した三成分系でも熱膨張が二成分系と同等まで熱膨張を低減できる理由は、例えば文献1:No.920−17 日本機械学会第69期通常総会講演会講演論文集(Vol.A)、および文献2:No.930−63 日本機械学会第71期全国大会講演論文集(Vol.B)に記載されている。
【0015】
上記の文献1および2では弾性率の高い材質(以下強化材)、マトリクス樹脂、ボイドを混合することで熱膨張を低減している。温度上昇時に強化材とマトリクス樹脂の近傍に発生する圧縮応力がボイドを潰し、マクロ的な熱膨張を低減している。ここで文献1によれば、ボイドの代替として弾性率の小さい材料を用いることも可能であることが記載されている。本発明で用いる熱可塑性エラストマーは弾性率が非常に小さいため、ボイドと同様のメカニズムで熱膨張を低減することが可能と推察される。
【0016】
しかしボイドの代替として熱可塑性エラストマーを用いると、強化材はマトリクス樹脂だけではなく熱可塑性エラストマー内にも混入するため、マトリクス樹脂が熱可塑性エラストマーを潰そうとする圧縮応力が小さくなると考えられる。またマトリクス樹脂との相溶性によって複合材料中の熱可塑性エラストマーの分布状態も変化する。熱可塑性エラストマーが複合材料の表層に多く分布する場合、熱可塑性エラストマーの膨張が直接マクロ物性として発現しやすくなるため、複合材料の熱膨張が大きくなることが推測される。さらに熱可塑性エラストマーは材質としての弾性率を有するため、ボイドよりも潰れにくい。以上の観点から、熱可塑性エラストマーをボイドの代替として用いた場合、ボイドを混合した系に比べて熱膨張の低減が非効率的になる。
【0017】
上記のメカニズムを整理すると、複合材料は、(I)圧縮応力に起因する熱可塑性エラストマーの潰れによる熱膨張の低減、および(II)熱可塑性エラストマーの大きい熱膨張による熱膨張の悪化、の2つの影響を受けると考えられる。熱膨張を低減するためには(I)の効果を大きくする必要がある。
【0018】
(I)の効果を大きくするためには、強化材とマトリクス樹脂の近傍に発生する圧縮応力をより大きく発生させればよいため、強化材の表面積を増やすことが有効な手段となる。強化材の表面積を増やすためには、強化材の添加量を増やすか、強化材のサイズを小さくすればよい。また、複合材料中の各成分の分布状態も(I)と(II)のそれぞれに影響すると考えられる。各成分の分布状態に影響を与える要因として、強化材の表面官能基が挙げられる。従って熱膨張を低減するためには上述のパラメーターを最適に選択する必要がある。
【0019】
以下、本発明の複合材料に含まれる(A)熱可塑性アクリル樹脂、(B)シリカ微粒子、(C)熱可塑性エラストマーについて説明する。
【0020】
(A)熱可塑性アクリル樹脂
熱可塑性アクリル樹脂は本発明の母材となる成分であり、本発明で用いる熱可塑性アクリル樹脂とは、下記一般式(1)に示されるポリメタクリル酸メチル(以下PMMA)構造を主として有するポリマーを示す。熱可塑性アクリル樹脂は、物性値が大幅に悪化する、または最終的に作られる複合材料の混合状態を大きく変化させることがない限り、主骨格中の官能基が置換されていても構わない。例えばポリアクリル酸メチル、ポリアクリル酸エチル、ポリメタクリル酸エチルなどのアクリル酸誘導体のポリマーも主骨格として用いることができる。同様に熱可塑性アクリル樹脂は、物性値が大幅に悪化する、または最終的に作られる複合材料の混合状態を大きく変化させることがない限り、別のモノマー成分が含まれる共重合体を用いてもよい。
【0021】
【化1】
【0022】
本発明の複合材料に含有される(A)熱可塑性アクリル樹脂の体積分率は、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して34体積%以上75体積%以下、好ましくは40体積%以上60体積%以下であることが望ましい。
【0023】
(B)シリカ微粒子
本発明で用いられるシリカ微粒子は、表面が親水性であることを特徴とする。ここで表面が親水性であるとは、シリカ微粒子の表面に存在する官能基として水酸基またはアミノ基を含む官能基がもっとも強く検出される状態を表す。樹脂材料に無機微粒子を添加して熱膨張を低減する方法はよく知られているが、本発明者はシリカ微粒子の表面修飾基により熱膨張が変化し、この傾向が二成分系よりも三成分系で顕著であることを見出した。特に三成分系で表面がトリメチルシリル基で修飾されたシリカ微粒子を用いた場合、マトリクス樹脂そのものよりも熱膨張が悪化する現象が確認できている。これは先述の複合材料中の各成分の分布状態が変化することに起因していると考えられる。
【0024】
温度上昇時に圧縮応力を十分に発生させるため、シリカ微粒子の表面積が大きいこと、すなわち粒子径が十分に小さいことが重要である。三成分系において平均一次粒子径が20nmを超えると、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子の二成分系の体積分率から求められる熱膨張の値(加成性値)よりも、複合材料の熱膨張が悪化する。従って無機微粒子の平均一次粒子径は1nm以上20nm以下、好ましくは7nm以上12nm以下であることが望ましい。ここで平均一次粒子径は同体積の球に換算した場合の直径を示す。
【0025】
さらに、圧縮応力を十分に発生させるためにはシリカ微粒子の添加量が十分である必要がある。図1に示すように、表面が親水性で平均一次粒子径が十分に小さい場合、シリカ微粒子が24体積%以上になると二成分系と三成分系の熱膨張が同程度まで低下する。シリカ微粒子が多量に添加されるほど熱膨張を低減できるが、複合材料中に含まれる高分子成分の相対的な含有量が少なくなり、靱性や破壊強度が低下する。本発明の複合材料に含有される(B)シリカ微粒子の体積分率は、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して24体積%以上63体積%以下、好ましくは24体積%以上35体積%以下であることが望ましい。シリカ微粒子が24体積%未満では、(C)熱可塑性エラストマーの有する大きい熱膨張が顕在化し、線膨張係数は二成分系のときよりも悪化する。シリカ微粒子が63体積%を超えると複合材料の脆性破壊の防止改善効果がほとんど得られなくなり、また破壊強度の低下に伴い成形が困難になる。
【0026】
混合するシリカ微粒子は粉体であることが望ましいが、液体混合法を用いる場合には分散剤などの影響がない範囲でスラリーを使用しても構わない。
【0027】
(C)熱可塑性エラストマー
熱可塑性エラストマーは複合材料に脆性破壊の防止を与えるために添加されるゴム状重合体であり、弾性率は100MPa以下のものを用いる。熱可塑性エラストマーを添加するほど複合材料の脆性破壊の防止を改善できるが、破壊強度が弱くなる。また添加しすぎると熱膨張の悪化要因にもなる。
【0028】
本発明の複合材料に含有される(C)熱可塑性エラストマーの体積分率は、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して1体積%以上34体積%以下、好ましくは12体積%以上24体積%以下であることが望ましい。また、複合材料に含有される(C)熱可塑性エラストマーの体積分率は、(A)熱可塑性アクリル樹脂の体積分率以下であることが好ましい。
【0029】
熱可塑性エラストマーは硬度が低いものを使用する必要がある。熱可塑性エラストマーの硬度が高いと弾性率が高くなるため、温度上昇で発生した圧縮応力による熱可塑性エラストマーの潰れが不十分となり、熱膨張が低減できない。従って熱可塑性エラストマーは硬度(デュロメーターA)が90度以下のものを用いることが好ましい。しかし熱可塑性エラストマーは硬度が低くなると耐熱性が低下するため、複合材料として熱膨張と耐熱性のバランスがよくなるものを適宜選択する必要がある。
【0030】
熱可塑性エラストマーは分子量が小さいと流動性が良好になるが、流動性が高すぎると熱膨張悪化の原因となる。成形加工時の温度において熱可塑性エラストマーの流動性は(A)熱可塑性アクリル樹脂よりも低いことが好ましい。
【0031】
本発明では硬度の低いスチレン系またはオレフィン系の熱可塑性エラストマーを用いる。スチレン系とはスチレンブロック共重合体であり、スチレンブロックの他にブタジエン、エチレン、イソプレン、プロピレン等のオレフィンブロック(水添されていてもよい)を有していることが特徴である。またオレフィン系とはオレフィンブロック共重合体であり、オレフィンブロック以外にスチレンブロックが含まれていてもよい。
【0032】
<複合材料の製造方法>
複合材料を製造する方法は、(A)熱可塑性アクリル樹脂、(B)シリカ微粒子、(C)熱可塑性エラストマーの各成分を均一に混合できる方法であれば特に限定されない。例えば、(A)を加熱し溶融させた状態で混合する溶融混練法や、(A)と(C)を溶媒中に溶解させて(B)とともによく撹拌し脱溶媒することで複合材料を得る溶媒混合法が挙げられる。
【0033】
熱膨張や脆性破壊の防止を極端に悪化しない限り、適宜添加剤を加えてもよい。添加剤とは、例えば耐熱安定剤、耐光安定剤、難燃剤、酸化防止剤、潤滑剤、離型剤、着色剤、帯電防止剤、光増感剤、抗菌剤などが挙げられる。また熱膨張や脆性破壊の防止が極端に悪化しない限り、複合材料中にボイドが含まれても構わない。
【0034】
本発明の複合材料に含有される添加剤の含有量は、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して0体積%以上5体積%以下が好ましい。
【0035】
本発明の複合材料は、例えば精密機器用外装材、光学用ミラーなどの用途に用いることができる。
【実施例】
【0036】
以下に、本発明の実施例について説明するが、本発明はこれらの例によって何ら限定されるものではない。
【0037】
<実施例1>
PMMA(デルペット70NH;旭化成ケミカルズ社商品名)をテトラヒドロフラン(以下THF)溶媒に対して5重量%になるように混合し、常温にてPMMAを溶解させ、PMMA/THF溶液を作製した。
【0038】
スチレン−ブタジエン系熱可塑性エラストマー(タフテックH1221;旭化成ケミカルズ社商品名)をTHF溶媒に対して5重量%になるように混合し、常温にて熱可塑性エラストマーを溶解させ、熱可塑性エラストマー/THF溶液を作製した。
【0039】
シリカ粒子(アエロジル200;アエロジル社商品名、平均一次粒子径12nm、表面は水酸基)0.56g、PMMA/THF溶液12g、熱可塑性エラストマー/THF溶液4.8gをよく混合する。シリカ粒子は非常にかさ高いため、溶液中に浸漬するようにTHFを任意量添加し、シリカ粒子を十分に浸漬させた状態で混合する。マグネチックスターラーで撹拌しながら溶媒を除去した後、真空乾燥炉を用い200℃で3から4時間の乾燥を行い、三成分系複合材料1を得た。
【0040】
<実施例2から4:シリカ粒子含有量の増加>
(実施例2)
実施例1において、シリカ粒子の添加量を0.72gに変えた以外は同様にして、三成分系複合材料2を得た。
【0041】
(実施例3)
実施例1において、シリカ粒子の添加量を0.84gに変えた以外は同様にして、三成分系複合材料3を得た。
【0042】
(実施例4)
実施例1において、シリカ粒子を0.84g、PMMA/THF溶液を2.4g、熱可塑性エラストマー/THF溶液を1.8gと変更して混合した以外は同様にして、三成分系複合材料4を得た。
【0043】
<実施例5から6:熱可塑性エラストマー含有量の減少>
(実施例5)
実施例1において、PMMA/THF溶液を14.4g、熱可塑性エラストマー/THF溶液を2.4gと変更して混合した以外は同様にして、三成分系複合材料5を得た。
【0044】
(実施例6)
実施例1において、シリカ粒子を0.72g、PMMA/THF溶液を14.4g、熱可塑性エラストマー/THF溶液を2.4gと変更して混合した以外は同様にして、三成分系複合材料6を得た。
【0045】
<実施例7:熱可塑性エラストマー含有量の増加>
(実施例7)
実施例1において、シリカ粒子を0.72g、PMMA/THF溶液を9.6g、熱可塑性エラストマー/THF溶液を7.2gと変更して混合した以外は同様にして、三成分系複合材料7を得た。
【0046】
<実施例8から9:シリカ粒子の平均一次粒子径の変更>
(実施例8)
実施例1において、シリカ粒子をアエロジル300(日本アエロジル社商品名、平均一次粒子径7nm、表面は水酸基)に変えた以外は同様にして、三成分系複合材料8を得た。
【0047】
(実施例9)
実施例1において、シリカ粒子をアエロジル90G(日本アエロジル社商品名、平均一次粒子径20nm、表面は水酸基)に変えた以外は同様にして、三成分系複合材料9を得た。
【0048】
次に、比較例を示す。なお比較例中、シリカ粒子を含む複合材料の番号は3ケタで表記している。
【0049】
<比較例1から2:シリカ非添加系>
(比較例1)
熱可塑性樹脂のPMMA(デルペット70NH;旭化成ケミカルズ社商品名)だけを用いた。
【0050】
(比較例2)
実施例1において、シリカ粒子を添加しない場合をPMMA/エラストマー混合体とし、比較例2とする。
【0051】
<比較例3から7:PMMA二成分系>
実施例1において、熱可塑性エラストマーを添加せず、シリカ粒子とPMMA/THF溶液の添加量の割合を下記のように変えて、二成分系材料103から107を得た。
【0052】
(比較例3)
二成分系材料番号103:シリカ粒子量0.36g:PMMA/THF溶液量12.8g
(比較例4)
二成分系材料番号104:シリカ粒子量0.40g:PMMA/THF溶液量12.0g
(比較例5)
二成分系材料番号105:シリカ粒子量0.44g:PMMA/THF溶液量11.2g
(比較例6)
二成分系材料番号106:シリカ粒子量0.50g:PMMA/THF溶液量10.0g
(比較例7)
二成分系材料番号107:シリカ粒子量0.80g:PMMA/THF溶液量4.0g
【0053】
<比較例8から9:三成分系、シリカ粒子含有量不足>
(比較例8)
実施例1においてシリカ粒子の添加量を0.26gに変えた以外は同様にして、三成分系複合材料108を得た。
【0054】
(比較例9)
実施例1においてシリカ粒子の添加量を0.40gに変えた以外は同様にして、三成分系複合材料109を得た。
【0055】
<比較例10:三成分系、シリカ粒子含有量が多い>
(比較例10)
実施例1において、シリカ粒子を0.96g、PMMA/THF溶液を2.4g、熱可塑性エラストマー/THF溶液を1.8gと変更して混合した以外は同様にして、三成分系複合材料110を得た。
【0056】
<比較例11から12:シリカ粒子径が大きい>
(比較例11)
実施例1において、シリカ粒子をアエロジル50(アエロジル社商品名、平均一次粒子径30nm、表面は水酸基)に変えた以外は同様にして、三成分系複合材料111を得た。
【0057】
(比較例12)
実施例1において、シリカ粒子をアエロジルOX50(アエロジル社商品名、平均一次粒子径40nm、表面は水酸基)に変えた以外は同様にして、三成分系複合材料112を得た。
【0058】
<比較例13から14:疎水性シリカ表面>
(比較例13)
実施例1において、シリカ粒子をアエロジルRX200(アエロジル社商品名、平均一次粒子径12nm、表面はトリメチルシリル基修飾)に変えた以外は同様にして、三成分系複合材料113を得た。
【0059】
<比較例14:熱可塑性エラストマー添加量が多い>
(比較例14)
実施例1において、シリカ粒子を0.72g、PMMA/THF溶液を7.2g、熱可塑性エラストマー/THF溶液を9.6gと変更して混合した以外は同様にして、三成分系複合材料114を得た。
【0060】
次に、上記の実施例、比較例の材料の評価を示す。
【0061】
<試料の作製>
上記の実施例、比較例で作製した材料を評価するため、試験片を作製した。6mm角のプレス成形用金型の面に離型剤としてノベック−1720(住友スリーエム社商品名)を滴下してよく拭き取る。上記の実施例、比較例で作製した材料をプレス成形用金型に充填し、小型熱プレス機(アズワン社製)にセットしながら250℃まで加熱した。加熱小型熱プレス機の上面と下面の温度が250℃に達した後に110MPaの荷重を付与し、100℃まで風冷しながら荷重を自然開放させた。100℃で完全に荷重を除き、金型から成形体を離型することで直方体の試験片(6mm×6mm、厚さおよそ3mm)を得た。
【0062】
<線膨張係数の評価>
試験片をTMA(TMA Q400;TAインスツルメント社商品名)にて−10から65℃で3サイクル温度負荷を与え、厚み方向に対する20から40℃の線膨張係数を算出した。変位の測定には膨張プローブを使用した。
【0063】
<体積分率の評価>
シリカ微粒子の含有量の測定はTGA(TGA Q500;TAインスツルメント社商品名)を用いて行った。シリカ微粒子の含有量を体積%へ換算するために用いた各材料の密度値は以下である。
PMMA 1.19、シリカ 2.00、スチレン−ブタジエン系熱可塑性エラストマー 0.89。
複合材料中の各成分の体積分率は、添加した熱可塑性アクリル樹脂と熱可塑性エラストマーの重量比が一定であると仮定して算出した。
【0064】
<破壊モード、破壊強度の評価>
得られた6mm角の試験片をプレス装置で圧縮破壊させ、破壊モードの確認と破壊時の荷重(破壊強度)の測定を行った。
破壊モードについては、破壊後の試験片が粉々に粉砕されていた場合を脆性破壊、破壊後の試験片が餅状に圧潰した場合を延性破壊とした。破壊のタイミングが明確でないものは、破壊荷重を「検出できず」とした。
【0065】
上記の実施例と比較例の評価結果を表1に示す(表1中の体積分率の表記は実際の値を四捨五入して整数値に揃えたため、合計が100体積%になっていない例もある)。
【0066】
表中、粒子径12nmかつ水酸基表面を有するシリカ粒子を用いたもの、および比較例1から7の結果を抽出し、シリカ粒子体積分率と線膨張係数でプロットしたものを図1に示す。また水酸基表面を有するシリカ粒子でシリカ粒子の体積分率が25体積%に近いものを抽出し、プロットしたものを図2に示す。図2中のnmの数値は、シリカ粒子の平均一次粒子径を示す。
【0067】
【表1】
【0068】
図1により、シリカ粒子が24体積%以上において、二成分系と三成分系の線膨張係数が同レベルとなることが明らかである。
【0069】
表1および図2より、粒子径が大きくなるほど線膨張係数が悪化することがわかる。シリカ粒子の線膨張係数を石英と同等(0.5ppm/℃)と仮定すると、粒子径20nm以下において体積分率から線形的に計算される線膨張係数の値(加成性値)を下回る。
【0070】
表1より、粒子表面が疎水性基で修飾されているほど線膨張係数が悪化することがわかる。
【0071】
表1より、複合材料は熱可塑性エラストマーが添加されていない場合は必ず脆性破壊を起こすが、熱可塑性エラストマーが添加されている場合は複合材料が延性破壊しやすく、材料の脆性破壊の防止が改善されていることがわかる。
【0072】
表1より、熱可塑性エラストマーの添加量が大きすぎると線膨張係数が悪化することがわかる。
【産業上の利用可能性】
【0073】
本発明の有機無機複合材料は、熱膨張が小さく、脆性破壊を防止できるので、精密機器用外装材や光学用ミラーに利用することができる。
【特許請求の範囲】
【請求項1】
少なくとも(A)熱可塑性アクリル樹脂と、平均一次粒子径が1nm以上20nm以下で、表面が親水性である(B)シリカ微粒子と、(C)熱可塑性エラストマーを含有してなり、前記(B)シリカ微粒子の体積分率が、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して24体積%以上63体積%以下であり、且つ前記(C)熱可塑性エラストマーの体積分率が(A)熱可塑性アクリル樹脂の体積分率以下であることを特徴とする有機無機複合材料。
【請求項2】
前記(C)熱可塑性エラストマーがスチレン系、オレフィン系の熱可塑性エラストマーから選ばれることを特徴とする請求項1に記載の有機無機複合材料。
【請求項1】
少なくとも(A)熱可塑性アクリル樹脂と、平均一次粒子径が1nm以上20nm以下で、表面が親水性である(B)シリカ微粒子と、(C)熱可塑性エラストマーを含有してなり、前記(B)シリカ微粒子の体積分率が、(A)熱可塑性アクリル樹脂と(B)シリカ微粒子と(C)熱可塑性エラストマーの全体に対して24体積%以上63体積%以下であり、且つ前記(C)熱可塑性エラストマーの体積分率が(A)熱可塑性アクリル樹脂の体積分率以下であることを特徴とする有機無機複合材料。
【請求項2】
前記(C)熱可塑性エラストマーがスチレン系、オレフィン系の熱可塑性エラストマーから選ばれることを特徴とする請求項1に記載の有機無機複合材料。
【図1】

【図2】
【図2】
【公開番号】特開2013−53202(P2013−53202A)
【公開日】平成25年3月21日(2013.3.21)
【国際特許分類】
【出願番号】特願2011−191429(P2011−191429)
【出願日】平成23年9月2日(2011.9.2)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
【公開日】平成25年3月21日(2013.3.21)
【国際特許分類】
【出願日】平成23年9月2日(2011.9.2)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
[ Back to top ]