
未分化細胞の製造方法
【課題】未分化細胞の製造方法の提供。
【解決手段】拘束された細胞を含む細胞群におけるCD34+細胞の相対数を増加させる方法であって、(i)該細胞群を、該拘束された細胞に操作可能に密着する薬剤と接触させること、ここで、該薬剤は、環状アデノシン一リン酸(cAMP)、CD4分子、CD8分子、T−細胞受容体の一部もしくは全部、リガンド(固定化または遊離)、ペプチド、T−細胞受容体(TCR)、抗体、交差反応性抗体、モノクローナル抗体、およびポリクローナル抗体からなる群から選択される;および(ii)該薬剤と密着した拘束された細胞をインキュベートして、該密着の結果としてCD34+細胞の相対数を増加させることを含む方法。
【解決手段】拘束された細胞を含む細胞群におけるCD34+細胞の相対数を増加させる方法であって、(i)該細胞群を、該拘束された細胞に操作可能に密着する薬剤と接触させること、ここで、該薬剤は、環状アデノシン一リン酸(cAMP)、CD4分子、CD8分子、T−細胞受容体の一部もしくは全部、リガンド(固定化または遊離)、ペプチド、T−細胞受容体(TCR)、抗体、交差反応性抗体、モノクローナル抗体、およびポリクローナル抗体からなる群から選択される;および(ii)該薬剤と密着した拘束された細胞をインキュベートして、該密着の結果としてCD34+細胞の相対数を増加させることを含む方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、未分化細胞の製造方法に関する。
【0002】
特に、本発明は、より拘束された細胞(more committed cell)からの未分化細胞の製造方法に関する。
【0003】
さらに本発明は、新しいより拘束された細胞、すなわち、再拘束細胞(recommitted cell)の製造のための本発明の未分化細胞の使用に関する。
【0004】
本発明は、また、免疫学的症状または疾患に関連する症状の緩和または免疫学的症状または疾患からの部分的もしくは完全な治療などの免疫系に対して(直接的に、または、それから得られる生成物の使用を介して間接的に)効果を有するための本発明の未分化細胞または本発明の再拘束細胞の使用にも関する。
【背景技術】
【0005】
概論として、分化は、細胞の構造および機能が段々に拘束されて、例えばT細胞またはB細胞の形成のようにより特殊化された細胞を生じるプロセスである。したがって、細胞がより拘束されると、該細胞は、さらに分化される。
【0006】
対照的に、逆分化(retrodifferentiation)は、細胞の構造および機能があまり特殊化されていない細胞を生じるように段々に変えられるプロセスである。
【0007】
未分化細胞は、多系譜(multilineage)分化能を有する。すなわち、該未分化細胞は、2つ以上のタイプの特殊化された細胞に分化する能力を有する。未分化細胞の典型的な例は、幹細胞である。
【0008】
対照的に、分化細胞は、多系譜分化能を有しない。分化細胞の典型的な例は、T細胞である。
【0009】
多くの未分化細胞および分化細胞がイン・ビボで見られ、一般的な技術は、それらについての一般的な教示を充分にもっている。
【0010】
例証として、とりわけ、LevittおよびMertelsman 1995(Marcel Dekker Incにより発行されたHaematopoietic Stem Cells, 特に第45−59頁)(非特許文献1)およびRoittら(Immunology, 4th Edition, Roitt, Brostoff および Male 編 1996, Publ. Mosby, 特に第10章)(非特許文献2)が引用される。
【0011】
しかしながら、要するに、未分化細胞の例としては、リンパ造血前駆細胞(lymphohaematopoietic progenitor cell)(LPC)が挙げられる。LPCとしては、多能性幹細胞(PSC)、リンパ系幹細胞(LSC)および骨髄性幹細胞(MSC)が挙げられる。LSCおよびMSCは、各々、PSCの分化により形成される。したがって、LSCおよびMSCは、PSCよりも拘束されている。
【0012】
分化細胞の例としては、T細胞、B細胞、好酸球、好塩基球、好中球、骨髄巨核球、単球、赤血球、顆粒球、マスト細胞およびリンパ球が挙げられる。
【0013】
T細胞およびB細胞は、LSCの分化により形成される。したがって、T細胞およびB細胞は、LSCよりも拘束されている。
【0014】
好酸球、好塩基球、好中球、骨髄巨核球、単球、赤血球、顆粒球、マスト細胞、NKおよびリンパ球は、MSCの分化により形成される。したがって、これらの細胞の各々は、MSCよりも拘束されている。
【0015】
抗原は、未分化細胞および分化細胞と関連している。ここでは、「関連する」なる用語は、各々の抗原を発現するかもしくは発現する能力を有する細胞、または各々の抗原を提供するかもしくは提供させる能力を有する細胞、または各々の抗原からなる細胞を意味する。
【0016】
ほとんどの未分化細胞および分化細胞は、主要組織適合遺伝子複合体(MHC)クラスI抗原および/またはクラスII抗原からなる。これらの抗原がこれらの細胞と関連している場合、それらは、クラスI+および/またはクラスII+細胞と呼ばれる。
【0017】
未分化細胞または分化細胞と関連する各々の特異的抗原は、マーカーとして作用することができる。したがって、種々のタイプの細胞を、それらの関連する特定の抗原に基づいて、または、関連抗原の特定の組合せに基づいて、お互いに区別することができる。
【0018】
これらのマーカー抗原の例としては、抗原CD34、CD19およびCD3が挙げられる。これらの抗原が存在する場合、これらの個々の細胞は、各々、CD34+、CD19+およびCD3+細胞と呼ばれる。これらの抗原が存在しない場合、これらの細胞は、各々、CD34−、CD19−およびCD3−細胞と呼ばれる。
【0019】
より詳しくは、PSCは、CD34+細胞である。LSCは、DR+、CD34+およびTdT+細胞である。MSCは、CD34+、DR+、CD13+、CD33+、CD7+およびTdT+細胞である。B細胞は、CD19+、CD21+、CD22+およびDR+細胞である。T細胞は、CD2+、CD3+、およびCD4+またはCD8+細胞である。未熟リンパ球は、CD4+およびCD8+細胞である。活性化T細胞は、DR+細胞である。ナチュラルキラー細胞(NK)は、CD56+およびCD16+細胞である。Tリンパ球は、CD7+細胞である。白血球は、CD45+細胞である。顆粒球は、CD13+およびCD33+細胞である。単球マクロファージ細胞は、CD14+およびDR+細胞である。
【0020】
したがって、前記抗原マーカーの存在について検索することにより、ある種の細胞タイプ(例えば、細胞が未分化細胞であるか分化細胞であるか)およびその細胞タイプの特殊化(例えば、細胞がT細胞であるかB細胞であるか)を同定することが可能である。
【0021】
逆分化の一般的概念は、新しくない。実際、1976年に、Jose Uriel(Cancer Research 36, 4269−4275, 1976年11月)(非特許文献3)は、このトピックについての評論を開示している;すなわち:
「逆分化は、変化した病因(物理的、化学的およびウイルス性)の有害因子に対する細胞完全性の維持について一般的な適応プロセスとして明白である。そのゲノム上でコードされている情報全体を保存すると同時に、逆分化を受けている細胞は、細胞質構造の自己遺伝子欠損のプロセスおよび遺伝子発現のより幼若なパターンへの移行により形態学的および機能的複雑さを失う。これにより、初めから異なる細胞表現型の漸進性均一化、および成人細胞において操作的な調節シグナルに対する応答性の減少が生じる。逆分化は、通常、復帰突然変異(reversion)が始まった、終末表現型を回復させようとする再個体発生のプロセスにより相殺される。これは、逆分化が常に細胞再生および組織修復に関連している理由を説明している。」
【0022】
Uriel(同書)(非特許文献3)は、次いで、報告された逆分化のケース、例えば、ツメガエル(Xenopus)のオタマジャクシの消化管内皮細胞からの核に関連するGurdonの研究(Advances in Morphogenesis [1966] 第4巻,第1−43頁. New York Academic Press, Abercrombie および Bracher 編)(非特許文献4)、および肝臓の再生に関連するBresnickの研究(Methods in Cancer Research [1971] 第6巻, 第347−391頁)(非特許文献5)を続けて検討した。
【0023】
Uriel(同書)(非特許文献3)は、また、イン・ビトロ培養のための単離された肝実質細胞に関係する研究についても報告した。Urielによると、以下のとおりである:
「胎児もしくは新生児肝実質細胞についての結果、または再生している肝臓もしくは樹立した肝細胞癌からの肝実質細胞についての結果と対照的に、静止している成人肝実質細胞から永久クラス系を得るのは困難であった。」
【0024】
Uriel(同書)(非特許文献3)は、また、癌における明らかな逆分化についても報告した;すなわち:
「多くの腫瘍の生化学的表現型は、肝発癌現象の前新生物期の間、・・・未熟に向かう復帰突然変異の類似変化を示し、細胞は、逆分化する。」
【0025】
逆分化についてのより最近の発見としては、Minoru Fukudaの研究(Cancer Research [1981] 第41巻, 第4621−4628頁)(非特許文献6)が挙げられる。Fukudaは、腫瘍促進性ホルボールエステル、12−O−テトラデカノイル−ホルボール−13−アセテート(TPA)の使用により、K562ヒト白血病細胞の細胞表面糖タンパク質プロフィルの特異的な変化をひき起こした。Fukudaによると、TPAは、K562ヒト白血病細胞を逆分化段階に誘導すると思われた。
【0026】
また、Hassら(Cell Growth & Differentiation [1991] 第2巻, 第541−548頁)(非特許文献7)は、ホルボールエステルの不在下でのTPA分化したU−937白血病細胞の32〜36日間の長期培養により逆分化のプロセスが生じること、および逆分化した細胞が基質から分離され、増殖を再開したことを報告した。
【0027】
逆分化の別のケースは、CurtinおよびSnellの研究(Br. J. Cancer [1983] 第48巻, 第495−505頁)(非特許文献8)である。これらの研究者は、ジエチルニトロサミン誘発肝発癌現象の間に生じる酵素的変化および部分肝切除後の肝臓再生を正常な肝臓分化と比較した。これらの研究者は、分化の段階を追っての逆転と似ていた発癌現象の間の酵素活性の変化を見いだした。これらの研究者によると、それらの結果は、基礎をなす逆分化プロセスが肝発癌現象および肝臓再生の両方のプロセスに共通していることを示唆している。
【0028】
より最近では、Chastreら(FEBS Letters [1985] 第188巻, 第2号, 第2810−2811頁)(非特許文献9)は、ヒト大腸癌性サブクローンHT29−18の逆分化について報告した。
【0029】
さらに最近では、Kobayashiら(Leukaemia Research [1994] 第18巻, 第12号, 第929−933頁)(非特許文献10)は、リポ多糖類(LPS)による処置によりマクロファージ様細胞に分化した単一のラット骨髄単球性白血病細胞由来の逆分化細胞系(RD−1)の樹立について報告した。
【0030】
現在の解釈によると、Molecular Biology of the Cell(Garland Publishers Inc. 発行. 1983)の第911頁(非特許文献11)の第911頁およびさらに最近ではLevittおよびMertelsman(非特許文献1)に見られる教示により立証されているように、PSCのような幹細胞は、以下の4つの特徴を有する:
i.未分化細胞である。すなわち、最終的に分化されない;
ii.制限なく分割する能力を有する;
iii.前記分化細胞などの分化した子孫を生じる能力を有する;
iv.分割すると、各々の娘は、選択の自由を有する:その親と同様のPSCとして維持することができるか、または、終末分化に不可逆的に誘導する経路に入ることができる。
最後の資格、すなわち、当該技術分野における一般的な教示によると、未分化細胞は、より拘束された細胞に分化すると、逆分化することができないということを理解すべきである。Uriel(同書)(非特許文献3)、Fukunda(同書)(非特許文献6)、Hassら(同書)(非特許文献7)、CurtinおよびSnell(同書)(非特許文献8)、Chastreら(同書)(非特許文献9)、ならびにKobayashiら(同書)(非特許文献10)は、あるタイプの分化細胞を逆分化したが、これらの細胞が同一系譜に拘束されたままであった場合には、それらは、未分化細胞に逆分化しなかったので、この解釈は、これらの研究者の教示によってさえ支持された。
【先行技術文献】
【非特許文献】
【0031】
【非特許文献1】LevittおよびMertelsman 1995(Marcel Dekker Incにより発行されたHaematopoietic Stem Cells, 特に第45−59頁)
【非特許文献2】Roittら(Immunology, 4th Edition, Roitt, Brostoff および Male 編 1996, Publ. Mosby, 特に第10章)
【非特許文献3】Jose Uriel(Cancer Research 36, 4269−4275, 1976年11月)
【非特許文献4】Advances in Morphogenesis [1966] 第4巻,第1−43頁. New York Academic Press, Abercrombie および Bracher 編
【非特許文献5】Methods in Cancer Research [1971] 第6巻, 第347−391頁
【非特許文献6】Cancer Research [1981] 第41巻, 第4621−4628頁
【非特許文献7】Hassら(Cell Growth & Differentiation [1991] 第2巻, 第541−548頁)
【非特許文献8】Br. J. Cancer [1983] 第48巻, 第495−505頁
【非特許文献9】Chastreら(FEBS Letters [1985] 第188巻, 第2号, 第2810−2811頁)
【非特許文献10】Kobayashiら(Leukaemia Research [1994] 第18巻, 第12号, 第929−933頁)
【非特許文献11】Molecular Biology of the Cell(Garland Publishers Inc. 発行. 1983)の第911頁
【発明の概要】
【発明が解決しようとする課題】
【0032】
したがって、本発明以前の技術の状態によると、より拘束された細胞から、幹細胞などの未分化細胞を形成することはできなかったと思われた。しかしながら、本発明は、この確信が誤っており、より拘束された細胞から未分化細胞を形成することができることを示す。
【課題を解決するための手段】
【0033】
かくして、第1の態様によると、本発明は、より拘束された細胞を、このより拘束された細胞を未分化細胞に逆分化させる薬剤と接触せさることからなる、未分化細胞の製造方法を提供するものである。
【0034】
第2の態様によると、本発明は、より拘束された細胞を、このより拘束された細胞を未分化細胞に逆分化させる薬剤と接触させ、次いで、該未分化細胞を再拘束細胞に拘束することからなる方法を提供するものである。
【0035】
「再拘束細胞」なる用語は、未分化細胞から誘導された細胞、すなわち新しいより拘束された細胞を意味する。
【0036】
第3の態様によると、本発明は、本発明の方法により生産された未分化細胞を提供するものである。
【0037】
第4の態様によると、本発明は、医薬としてのまたは医薬の調製における本発明の方法により生産された未分化細胞を提供するものである。
【0038】
第5の態様によると、本発明は、免疫学的障害または疾患の治療薬の製造における本発明の方法により生産された未分化細胞の使用を提供するものである。
【0039】
第6の態様によると、本発明は、本発明の方法により生産された再拘束細胞を提供するものである。
【0040】
第7の態様によると、本発明は、医薬としてのまたは医薬の調製における本発明の方法により生産された再拘束細胞を提供するものである。
【0041】
第8の態様によると、本発明は、免疫学的障害または疾患の治療薬の製造における本発明の方法により生産された再拘束細胞の使用を提供するものである。
【0042】
第9の態様によると、本発明は、より拘束された細胞を未分化細胞に逆分化させることができる薬物を付着している、より拘束された細胞を提供するものである。
【0043】
第10の態様によると、本発明は、CD19+およびCD3+細胞を提供するものである。
【0044】
かくして、最も広い意味では、本発明は、より拘束された細胞から未分化細胞を形成する能力を有するという非常に驚くべき発見に基づいている。
【発明を実施するための形態】
【0045】
本発明は、より拘束された細胞から未分化細胞を製造し、次いで、これらの未分化細胞を、障害の治療のためのイン・ビトロもしくはイン・ビボまたはその組合せで薬物として、または、該薬物を調製するために使用することが可能であるので、非常に好都合である。
【0046】
本発明は、また、初期のより拘束された細胞を修正または除去するために、または、その生成物を修正または除去するために、逆分化により製造された未分化細胞を新しい分化細胞のような再拘束細胞に拘束することができるので、好都合である。
【0047】
好ましくは、より拘束された細胞は、MHCクラスI+および/またはMHCクラスII+未分化細胞に逆分化する能力を有する。
【0048】
好ましくは、より拘束された細胞は、幹細胞抗原からなる未分化細胞に逆分化する能力を有する。
【0049】
好ましくは、より拘束された細胞は、CD34+未分化細胞に逆分化する能力を有する。
【0050】
好ましくは、より拘束された細胞は、リンパ造血前駆細胞に逆分化する能力を有する。
【0051】
好ましくは、より拘束された細胞は、多能性幹細胞に逆分化する能力を有する。
【0052】
未分化細胞は、抗原発現、捕捉または認識に関係がある成分からなってもよい。好ましくは、未分化細胞は、MHCクラスI+および/またはMHCクラスII+細胞である。
【0053】
好ましくは、未分化細胞は、幹細胞抗原からなる。
【0054】
好ましくは、未分化細胞は、CD34+未分化細胞である。
【0055】
好ましくは、未分化細胞は、リンパ造血前駆細胞である。
【0056】
好ましくは、未分化細胞は、多能性幹細胞である。
【0057】
より拘束された細胞は、抗原発現、捕捉または認識に関係がある成分からなってもよい。好ましくは、より拘束された細胞は、MHCクラスI+および/またはMHCクラスII+細胞である。
【0058】
好ましくは、該薬剤は、より拘束された細胞の細胞外に作用する。
【0059】
好ましくは、より拘束された細胞は、薬剤により操作可能に密着可能な(engageable)受容体からなる(該薬剤は、該受容体に操作可能に密着する(engage))。
【0060】
好ましくは、受容体は、細胞表面受容体である。
【0061】
好ましくは、受容体は、α−成分および/またはβ−成分からなる。
【0062】
好ましくは、受容体は、相同領域を有するβ−鎖からなる。
【0063】
好ましくは、受容体は、少なくともHLA−DRのβ−鎖の相同領域からなる。
【0064】
好ましくは、受容体は、相同領域を有するα−鎖からなる。
【0065】
好ましくは、受容体は、少なくともHLA−DRのα−鎖の相同領域からなる。
【0066】
好ましくは、薬剤は、受容体に対する抗体である。
【0067】
好ましくは、薬剤は、受容体に対するモノクローナル抗体である。
【0068】
好ましくは、薬剤は、HLA−DRのβ−鎖の相同領域に対する抗体、好ましくは、モノクローナル抗体である。
【0069】
好ましくは、薬剤は、HLA−DRのα−鎖の相同領域に対する抗体、好ましくは、モノクローナル抗体である。
【0070】
好ましくは、薬剤は、生物学的応答調節剤と共に用いられる。
【0071】
好ましくは、生物学的応答調節剤は、アルキル化剤である。
【0072】
好ましくは、アルキル化剤は、シクロホスファミドであるか、または、シクロホスファミドからなる。
【0073】
1つの好ましい具体例では、より拘束された細胞は、分化細胞である。
【0074】
好ましくは、より拘束された細胞は、B細胞またはT細胞のいずれか1つである。
【0075】
別の好ましい具体例では、より拘束された細胞は、より成熟した未分化細胞である。
【0076】
1つの好ましい具体例では、未分化細胞が再拘束細胞に拘束された場合、再拘束細胞は、逆分化前のより拘束された細胞と同一の系譜のものである。
【0077】
別の好ましい具体例では、未分化細胞が再拘束細胞に拘束された場合、再拘束細胞は、逆分化前のより拘束された細胞と異なる系譜のものである。
【0078】
好ましくは、再拘束細胞は、B細胞、T細胞または顆粒球のいずれか1つである。
【0079】
好ましくは、該方法は、イン・ビトロ方法である。
【0080】
好ましくは、薬剤は、MHC遺伝子発現を調節し、好ましくは、この場合、該薬剤は、MHC I+および/またはMHC II+発現を調節する。
【0081】
該薬剤は、より拘束された細胞を未分化細胞に逆分化させるために、このより拘束された細胞に操作可能に密着する。これに関して、より拘束された細胞の未分化細胞への逆分化のための薬剤は、より拘束された細胞との直接的な密着(engagement)または間接的な密着において作用する。
【0082】
直接的密着の例は、より拘束された細胞が、B細胞上で見いだされるもののような相同領域(一般的に、同一または類似の配列を有することが判明している領域)を有するβ−鎖のような、その細胞表面上に少なくとも1つの細胞表面受容体を有する場合および薬剤が該細胞表面受容体に直接的に密着する場合である。別の例は、より拘束された細胞が、T細胞上で見られるもののような相同領域を有するα−鎖のような、その細胞表面上に細胞表面受容体を有する場合および該薬剤が細胞表面受容体に直接的に密着する場合である。
【0083】
間接的な密着の例は、より拘束された細胞がその細胞表面上に少なくとも2つの細胞表面受容体を有する場合であり、薬剤の、受容体のうちの1つとの密着は、他の受容体に影響を及ぼし、次いで、より拘束された細胞の逆分化を誘発する。
【0084】
より拘束された細胞の未分化細胞への逆分化のための薬剤は、化学化合物または組成物である。しかしながら、好ましくは、該薬剤は、より拘束された細胞の表面上で細胞表面受容体を密着させる能力を有する。例えば、好ましい薬剤としては、環状アデノシン一リン酸(cAMP)、CD4分子、CD8分子、T−細胞受容体の一部もしくは全部、リガンド(固定化または遊離)、ペプチド、T−細胞受容体(TCR)、抗体、交差反応性抗体、モノクローナル抗体、またはポリクローナル抗体の1つ以上が挙げられる。
【0085】
薬剤が抗体、交差反応性抗体、モノクローナル抗体、またはポリクローナル抗体である場合、好ましくは、該薬剤は、MHCクラスII抗原のβ鎖、MHC HLA−DR抗原のβ鎖、MHCクラスIもしくはクラスII抗原のα鎖、HLA−DR抗原のα鎖、MHCクラスII抗原もしくはMHCクラスI抗原のαおよびβ鎖のうちのいずれか1つ以上に対する抗体、交差反応性抗体、モノクローナル抗体、またはポリクローナル抗体のうちのいずれか1つ以上である。適切な抗体の例は、CR3/43(ダコ(Dako)より供給された)である。
【0086】
より拘束された細胞は、未分化細胞から誘導されたかまたは誘導可能な細胞である。
【0087】
かくして、1つの具体例では、より拘束された細胞は、未分化細胞でもある。したがって、例証として、未分化細胞は、リンパ系幹細胞または骨髄幹細胞であることができ、未分化細胞は、多能性幹細胞である。
【0088】
別の好ましい具体例では、より拘束された細胞は、CFC−T細胞、CFC−B細胞、CFC−エオシン細胞、CFC−バス(Bas)細胞、CFC−バス細胞、CFC−GM細胞、CFC−MEG細胞、BFC−E細胞、CFC−E細胞、T細胞、B細胞、好酸球、好塩基球、好中球、単球、骨髄巨核細胞または赤血球などの分化細胞であり;未分化細胞は、骨髄幹細胞、リンパ系幹細胞または多能性幹細胞である。
【0089】
より拘束された細胞が分化細胞である場合、好ましくは、該分化細胞は、Bリンパ球(活性化または非活性化)、Tリンパ球(活性化または非活性化)、マクロファージ単球系譜からの細胞、クラスIまたはクラスII抗原を発現させる能力を有する有核細胞、クラスIまたはクラスII抗原を発現させることができる細胞、または除核細胞(すなわち、赤血球のような核を含有しない細胞)である。
【0090】
別の好ましい具体例は、分化細胞は、各々、CD56および/またはCD16細胞表面受容体を発現する、大きな顆粒状リンパ球、ヌルリンパ球、およびナチュラルキラー細胞からなる細胞のグループのいずれか1つから選択される。
【0091】
分化細胞は、除核細胞の核形成により形成されてもよい。
【0092】
薬剤は、より拘束された細胞内で細胞内に作用してもよい。しかしながら、好ましくは、薬剤は、より拘束された細胞の細胞外に作用する。
【0093】
好ましい具体例では、薬剤は、より拘束された細胞の表面上に存在する受容体に操作可能に密着する。該受容体は、より拘束された細胞により発現させられる能力を有する受容体のように、より拘束された細胞により発現される。
【0094】
好ましくは、受容体は、主要組織適合遺伝子複合体(MHC)のクラスIまたはクラスII抗原である。好ましい具体例では、細胞表面受容体は、HLA−DR受容体、DM受容体、DP受容体、DQ受容体、HLA−A受容体、HLA−B受容体、HLA−C受容体、HLA−E受容体、HLA−F受容体、またはHLA−G受容体のいずれか1つである。
【0095】
さらに好ましい具体例では、細胞表面受容体は、HLA−DR受容体である。
【0096】
好ましくは、接触工程は、薬剤を以下のものの1つ以上と密着させることからなる:クラスI抗原のα−鎖の相同領域、クラスII抗原のα−鎖相同領域、CD4細胞表面受容体、CD8細胞表面受容体、リンパ球の存在下でのクラスII抗原のβ−鎖の相同領域、リンパ球の存在下でのクラスI抗原のα−鎖の相同領域、またはリンパ球の存在下でのクラスII抗原のα−鎖の相同領域。
【0097】
好ましくは、接触工程は、生物学的応答調節剤の存在下で生じる。
【0098】
好ましくは、生物学的応答調節剤は、免疫調節剤、成長因子、サイトカイン、細胞表面受容体、ホルモン、核酸、ヌクレオチド配列、抗原またはペプチドなどの調節剤の1つ以上である。
【0099】
本発明の好ましい具体例では、未分化細胞は、次いで、分化細胞のような再拘束細胞に拘束される。
【0100】
再拘束細胞は、未分化細胞が誘導された、より拘束された細胞と同一の系譜のものである。
【0101】
別法としては、再拘束細胞は、未分化細胞が誘導された、より拘束された細胞と異なる系譜のものである。
【0102】
さらに、本発明は、未分化細胞を再拘束細胞に拘束し、次いで、再拘束細胞を骨髄腫に縮合させることを含む、本発明の未分化細胞の製造方法を包含する。これは、抗体または抗原またはホルモンなどの多量の所望の生産物をイン・ビトロで発現させる。
【0103】
本発明の他の態様は、以下のものを含む:
【0104】
より拘束された細胞から未分化細胞を製造するための本発明の薬物のいずれか1つの使用。
【0105】
B−リンパ球またはT−リンパ球からモノクローナル抗体またはポリクローナル抗体または特異的抗体のいずれか1つを製造するための本発明の方法により生産される未分化細胞;マクロファージ単球系譜からの細胞;クラスIまたはクラスII抗原を発現する能力を有する核形成細胞;クラスIまたはクラスII抗原を発現させる能力を有する細胞;除核細胞;断片化細胞;またはアポプティック(apoptic)細胞の使用。
【0106】
B−リンパ球からエフェクターT−リンパ球を生産するための(逆もまた同様)本発明の方法により生産された未分化細胞の使用。
【0107】
B−リンパ球、T−リンパ球からなるかもしくはこれから調製される医薬、マクロファージ単球系譜からの細胞、クラスIもしくはクラスII抗原を発現する能力を有する核形成細胞、クラスIもしくはクラスII抗原を発現させる能力を有する細胞、または除核細胞のいずれか1つ以上を生産するための本発明の方法により生産される未分化細胞の使用。
【0108】
本発明は、前記使用を利用するプロセス、およびかかるプロセスから製造された生成物または組成物も包含する。
【0109】
本発明は、適切な希釈剤、担体または賦形剤と混合した、本発明の未分化細胞またはそれから得られた生成物からなる医薬を包含する。
【0110】
1つの好ましい具体例では、医薬は、適切な希釈剤、担体または賦形剤と混合した、本発明の未分化細胞から得られた抗体または抗原からなる。
【0111】
好ましくは、医薬は、癌、自己免疫疾患、血液障害、細胞もしくは組織再生、臓器再生、臓器もしくは組織移植の処置、または先天性代謝障害のいずれか1つの処置のためのである。
【0112】
好ましい具体例では、本発明は、遺伝子をより拘束された細胞に導入し、次いで、本発明の方法により未分化細胞を製造し、これにより、遺伝子が未分化細胞中に存在することからなる、遺伝子を未分化細胞のゲノムに導入するプロセスに関する。
【0113】
より好ましい具体例では、本発明は、遺伝子をより拘束された細胞のゲノムに挿入し、次いで、本発明の方法により未分化細胞を製造し、これにより、遺伝子が未分化細胞中に存在することからなる、遺伝子を未分化細胞のゲノムに導入するプロセスに関する。
【0114】
さらに好ましい具体例では、本発明は、遺伝子をより拘束された細胞のゲノムに挿入し、次いで、本発明の方法により未分化細胞を製造し、これにより、遺伝子が未分化細胞のゲノム中に存在することからなる、遺伝子のゲノムを未分化細胞に導入するプロセスに関する。
【0115】
本発明は、本発明のこれらのプロセスのいずれか1つにより製造される未分化細胞を包含する。
【0116】
既に記載したとおり、本発明は、適切な希釈剤、担体または賦形剤と混合した、これらのプロセスのいずれか1つにより製造された未分化細胞からなる医薬を包含する。正しくないゲノム構造を有するより拘束された細胞により生じるかまたは該細胞と関連する症状または病状を緩和するために、かかる医薬について、未分化細胞を用いて、正しいゲノム構造を有するものなど有益なより拘束された細胞を生産することができる。
【0117】
かくして、本発明は、また、本発明の方法により未分化細胞を形成し、未分化細胞を再拘束細胞に拘束し、これにより、細胞のゲノムおよび/または核の整列または再整列により突然変異が除去されることからなる、より拘束された細胞から後天性突然変異を除去するプロセスを提供するものでもある。
【0118】
好ましくは、遺伝子は、ゲノムの免疫グロブリン部位またはTCR部位に挿入される。
【0119】
別法としては、未分化細胞を用いて、正しくないゲノム構造を有するより拘束された細胞により生じるかまたは該細胞と関連する症状または病状を治療する実体を生産するより拘束された細胞を生産することができる。
【0120】
例えば、本発明を用いて、未分化細胞に逆分化したより拘束された細胞により発現される抗原に対する抗体またはT細胞受容体を製造することができる。これに関して、抗原は、胎児特異的抗原または交差反応胎児特異的抗原であってよい。
【0121】
本発明は、また、未分化細胞およびより拘束された細胞のレベルを制御するプロセスをも含む。例えば、本発明は、本発明の方法により未分化細胞を形成し、次いで、アポプトーシス遺伝子を活性化して、その死をもたらすなど未分化細胞に影響を及ぼすことからなる方法を含む。
【0122】
本発明の1つの好ましい具体例では、より拘束された細胞は、癌細胞ではない。本発明の別の好ましい具体例では、薬剤は、発癌性でなく、癌増殖を促進させる能力も有しない。
【0123】
本発明は、また、より拘束された細胞を、このより拘束された細胞を未分化細胞に逆分化させる薬剤と接触させることにより未分化細胞を製造し、次いで、所望により、該未分化細胞を再拘束細胞に拘束することからなる欠損細胞または不必要な細胞により生じる疾患または障害に罹っている患者の治療方法であって、該未分化細胞または再拘束細胞が欠損細胞または不必要な細胞に影響を及ぼして、該疾患または障害の症状を緩和するかまたは疾患または病状の患者を治療する治療方法をも包含している。
【0124】
要約すると、本発明は、より拘束された細胞からの未分化細胞の製造に関する。
【0125】
本発明は、以下の図面を参照して、例証として説明される。
【図面の簡単な説明】
【0126】
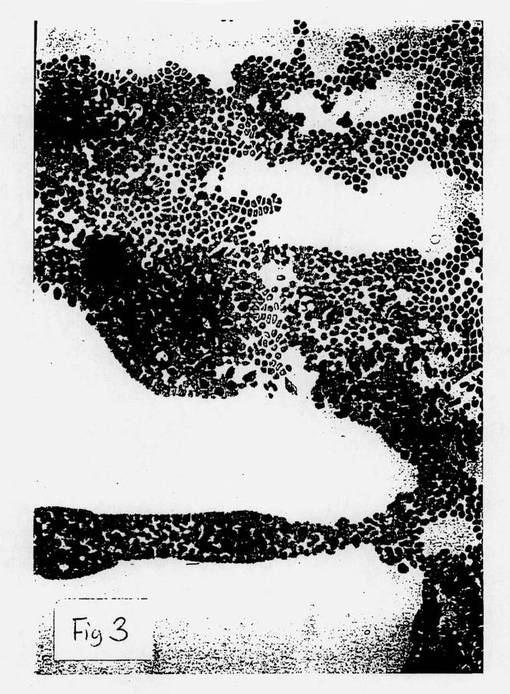
【図1】図1は、本発明方法以前の細胞の顕微鏡写真である。
【図2】図2は、本発明方法により製造された細胞の顕微鏡写真である。
【図3】図3は、低倍率であるが、本発明の方法により製造された顕微鏡写真である。
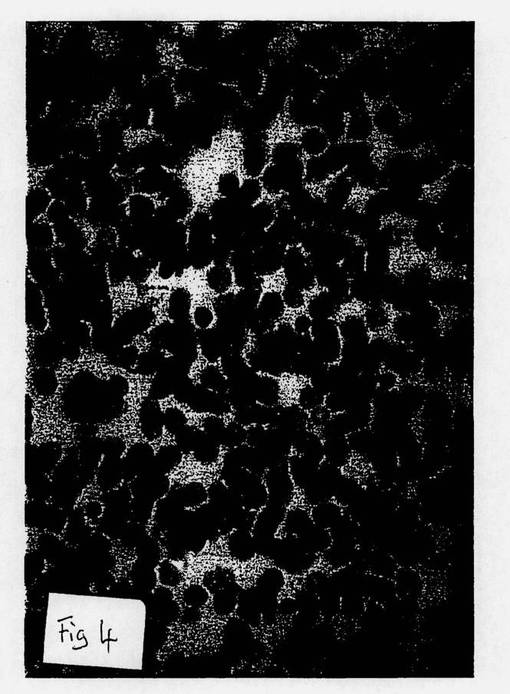
【図4】図4は、本発明方法以前の細胞の顕微鏡写真である。
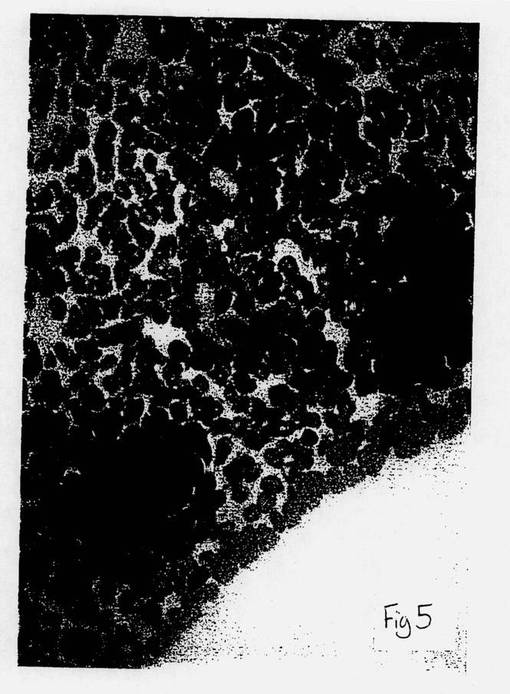
【図5】図5は、本発明方法により製造された細胞の顕微鏡写真である。
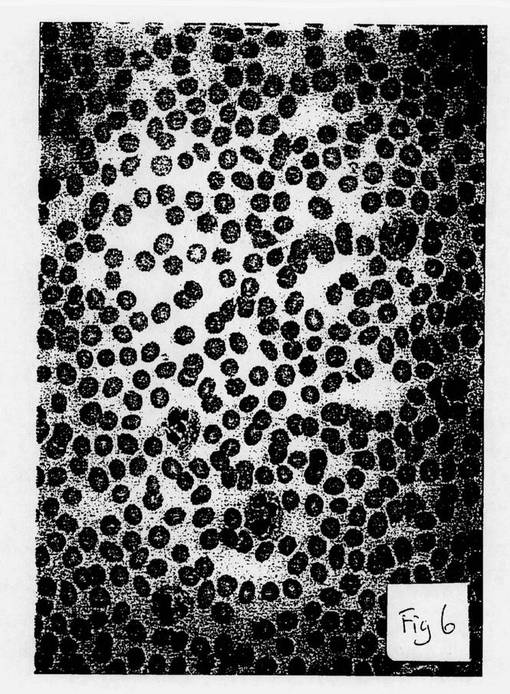
【図6】図6は、本発明方法により製造された細胞の顕微鏡写真である。
【実施例】
【0127】
A.物質および方法
患者
血液試料は、B−細胞慢性リンパ球性白血病患者、抗体欠損(IgA欠損およびX染色体性小児性低ガンマグロブリン血症を含む)患者、HIV感染およびAIDS症候群患者、CMV感染患者、ホジキンリンパ腫患者、急性T−細胞白血病患者、ホジキンリンパ腫の6日齢乳児、急性T−細胞白血病患者、胚芽細胞増殖症の6日齢乳児、種々の感染および慢性症状の種々の患者、臍帯血、骨髄、および健康な供血者からの富化B−リンパ球調製物から、EDTAを含有するラベンダートップ管中に得た。
【0128】
臨床条件および実験条件
血液試料に適用された種々のタイプの処置を含む患者の臨床および実験処置条件を表1に記載する。コールターカウンターを用いて白血球(WBC)分画を得た。これらは、同表に含まれる。
【0129】
血液の処置
得られた血液試料をすぐに、HLA−DR抗原(DAKO)のβ−鎖の相同領域に対する純粋な抗体で処理し、最長24時間、室温で、頭−頭ローラー(head to head roller)で混合した。まず、いくつかの試料を頭−頭ローラーで15分間混合した後、それらを22℃でインキュベーター中でインキュベートした。血液試料に添加したモノクローナル抗体の濃度は、血液ml当たり10〜50μlを変化した。
【0130】
さらに、同一の濃度で他の処置を適用し、これらは、HLA−DR抗原のα−鎖の相同領域に対するモノクローナル抗体、クラスI抗原の相同領域に対するモノクローナル抗体、CD4に対するモノクローナル抗体、CD8に対するモノクローナル抗体、およびHLA−DR抗原のβ−鎖の相同領域に対するPEコンジュゲートモノクローナル抗体の添加を含んだ。
【0131】
他の処置は、血液試料への、HLA−DR抗原のαおよびβ−鎖の相同領域に対するモノクローナル抗体の同時添加を含んだ。
【0132】
さらにまた、シクロホスファミドなどのアルキル化剤を、HLA−DR抗原のβ−鎖の相同領域に対する純粋なモノクローナル抗体と合わせて血液試料に添加した。
【0133】
これらの処置に続いて、製造者の指示により指示されるように標識モノクローナル抗体のパネルを用いて血液試料を染色し、次いで、フローサイトメトリーを用いて分析した。
【0134】
モノクローナル抗体によりインキュベーション期間は、2時間、4時間、6時間、12時間〜24時間の範囲であった。
【0135】
標識抗体
以下のモノクローナル抗体を用いて、フローサイトメトリーにより細胞上の以下のマーカーを検出した:CD19およびCD3、CD4およびCD8、DRおよびCD3、CD56&16およびCD3、CD45およびCD14、CD8およびCD3、CD8およびCD28、サイマルテスト(simultest)対照(IgG1 FITC+IgG2a PE)、CD34およびCD2、CD7およびCD13&13、CD10およびCD25、CD5およびCD10、CD5およびCD21、CD7およびCD5、CD13およびCD20、CD23およびCD57、ならびにCD25およびCD45 RA(ベクトン・アンド・デッケンソン(Becton & dickenson)およびDAKO)。
【0136】
種々の処置に付随した免疫表現型変化について説明するために、前記パネルの大部分を用いて、処置および未処置の各々の患者の血液試料を分析し、これらは、同一血液試料の異なるアリコットについて別々に行った。未処置試料および他の対照処置を染色し、同時に分析した。
【0137】
フローサイトメトリー
白血球を染色し、製造者の指示に従って溶解させた。負の対照バックトラッキングを含んだサイマルテスト(simultest)またはPAINT A GATEソフトウエアー(BDSI)によりFACScan@でフローサイトメトリー分析を行った。10,000〜20,000個の事象が得られ、リストモードファイルに保存された。
【0138】
形態学
形態学は、顕微鏡およびライト染色を用いて分析した。
【0139】
B. 結果
CD19およびCD3パネル
血液試料をHLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体で処理すると、CD19+細胞の相対数が常に減少した。このマーカーは、panB−細胞抗原である(表参照)。この抗原は、成熟の全段階で、全てのヒトBリンパ球に存在するが、終末に分化した形質細胞には存在しない。従って、これは、B細胞が未分化細胞へ逆分化していたことを示す。
【0140】
同様の処理により、CD3+細胞の相対数が、劇的なことに、特に、B−CLL患者の血液中で増加し、常に、CD3−CD19−細胞における相対数の増加を伴った。CD3は、全ての成熟Tリンパ球および65%−85%の胸腺細胞に存在する。このマーカーは、常にα/βまたはガンマ/デルタT−細胞受容体(TCR)と関連して知られ、一緒にこれらの複合体は、細胞内部に対するシグナル変換において重要である。従って、これは、B細胞が未分化細胞に逆分化し、それから、T細胞と称する新しい分化細胞に拘束されていたことを示す。
【0141】
細胞の新しいクローンは、CD19およびCD3マーカー、すなわち、CD19+およびCD3+細胞を共同発現しているB−CLL患者の処理血液において、現れた(チャート1.処理開始後2時間、6時間および24時間の患者2、3および4参照)。他の病状の患者は、細胞のこれらのクローンの相対数の増加を示した。これらの細胞は、非常に大きく、非常に顆粒状であり、極度に高いレベルのCD19が、それらの細胞膜に発現した。CD3マーカーは、正常な成熟リンパ球に発現するのと同様なレベルにて、これらの細胞で発現されるようである。
【0142】
表2において、患者番号2、3および4は、実際には、同じ患者を表す番号であり、それらの説明は、単に、経時的な血液処理の効果を示すものであった(この患者の実験条件および臨床条件については表1を参照)。
【0143】
処理試料におけるCD19+CD3+クローンは、経時的に減少し、初期レベルが2時間目、6時間目および24時間目の未処理試料において測定されたレベルに達する。
【0144】
同じサイズおよび粒状の別のタイプの細胞が処理試料において検出され、これらの細胞は、それらの表面で発現した高レベルのCD19を有するが、CD3マーカーに対して陰性であり、FC受容体において豊富であった。しかしながら、これらの細胞の相対数は、時間に伴って減少することが明らかになった。興味深いことに、血液試料(2、3および4)の処理の24時間目に、初めに血液試料の処理の2および6時間後に増加することが観察された細胞群において、CD19−CD3−細胞の相対数が減少した。しかしながら、WBC群のコールターカウントは、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体による血液の処理により減少した。この発見は、このタイプの処理により、コールターによって検出できないが(表1)、表面マーカーに基づいて細胞をカウントするフローサイトメトリーによって測定した場合にサイズおよび粒状について説明され得る非定形細胞が生じることを示す。さらに、これらの非定形細胞は、顕微鏡下でライト染色を用いて形態を分析することにより説明される。これらの現象のフローサイトメトリーチャートは、チャート(1、2、3および4)に示され、血液試料の処理により観察された免疫発現型変化は、CD19+およびCD3+リンパ球が相互に連絡された細胞群であるが、幹細胞と比較したCD19およびCD3相対発現に基づいて別個のままであることを示唆すると思われる。
【0145】
表2において、患者番号5および6は、同じ患者を示すが、処理および未処理の血液試料の分析は、同時に経時的にモニターされた(表1参照)。
【0146】
B−細胞悪性腫瘍を有さない患者血液は、B−CLL患者の血液と比較すると、類似した傾向の免疫発現型変化を示したが、その変化は、同程度ではなかった。しかしながら、これらの患者の血液中のB−リンパ球およびMHCクラスII陽性細胞の相対的および絶対的な数は、B−CLL患者の血液中でみられた数と比較して、極度に低い。
【0147】
どちらもB細胞欠損であるX−結合乳児低ガンマグロブリン血症である2人の兄弟は、彼らの血液の処理によりCD3+細胞の相対数において異なる免疫発現型変化を示した。2カ月齢であり、病状が重くなかった弟は、血液の処理によりCD3+の相対数のわずかな増加を示し、それはCD3−CD19−細胞の相対数の減少を伴った。他方、2才で重症であり、比較的高い数のDR抗原を発現している活性T細胞を有する兄は、血液の処理によりCD3+細胞の数の減少を示した。これら二人の患者から得られた血液試料は、極度に少量だったので、他のマーカーを使用して引き起される可能性のある他の表現型変化を測定するようなことはしなかった(表2.ID43/BDおよび04/BD)。
【0148】
表2において患者91は、血液の処理後のCD3+細胞の相対数の減少を示し、それは、CD3−CD19−細胞の相対数の増加を伴った。しかしながら、CD4およびCD8のような他の表面マーカーの分析において(表3参照)、患者は、血液中にCD4+CD8+細胞の高い相対数を有することが観察され、これは、DR抗原のβ−鎖に対するモノクローナル抗体で血液試料を処理する前に示され、これらの二重陽性細胞は、血液処理後、かなり減少した。さらに、さらなるマーカーを分析すると、CD3+細胞の相対数が上昇したことが示された(表4参照)。
【0149】
健康な供血者から得られたB−リンパ球の豊富な他のは、DR抗原のβ−鎖に対するモノクローナル抗体で処理すると、CD3+細胞の相対数の劇的な増加を示し、それは、常にCD19+細胞の相対数の減少およびCD19−CD3−細胞の相対数の増加を伴った。CD4およびCD8のようなマーカーを使用するさらなる分析は、これらのマーカーの相対数の付随した増加を示す。しかしながら、同一供血者のTリンパ球の豊富な調製物は、同一のモノクローナル抗体で処理した場合、同様の変化を示さなかった。
【0150】
CD4およびCD8パネル
CD4抗原は、ヒト免疫不全ウイルスに対する受容体である。CD4分子は、クラスI抗原上のCD8結合部位と同様な領域であるB2ドメインにおいてMHCクラスII抗原を結合する。CD4のクラスII抗原への結合は、抗原へのT細胞応答を増強し、それゆえCD8のクラスI抗原への結合を行う。CD8抗原は、ヒトサプレッサー/細胞障害性Tリンパ球サブセット上ならびにナチュラルキラー(NK)リンパ球のサブセットおよび大部分の正常胸腺細胞上に存在する。CD4およびCD8抗原は、胸腺細胞で共同発現し、これらの細胞は、それらがT−リンパ球へと成熟するにつれて、どちらのマーカーも失う。
【0151】
CD4およびCD8マーカーの分析(下記参照)にて、および表2に示された血液試料の大部分から、B−リンパ球の未分化細胞への逆分化プロセスの存在およびT−リンパ球へのその後の分化を支持する染色パターンが明らかになる。
【0152】
二重陽性細胞であるCD4+CD8+細胞は、常に、β−鎖の相同領域に対するモノクローナル抗体での血液試料の処理に従うことが明白であり、これらのタイプの細胞は、B−CLL患者の処理試料の血液において著しく増加し、それは、未処理試料において全く見られなかった(表3およびチャート1、2、3および4参照)。同様の試料において、CD8+およびCD4+細胞のような単陽性細胞の相対数もまた、同時に増加することが示された。さらに、少なくともB−CLLの場合においてB細胞に相当するCD4−CD8−細胞の相対数の減少は、経時的に測定すると、同一レベルのままであった未処理試料と比較して、処理試料では劇的に減少することを示した。しかしながら、処理試料中におけるCD4+CD8+細胞の相対数の経時的な測定は、二重陽性細胞の相対数の減少に付随して単陽性細胞数の増加が起こることを示した。このタイプの免疫発現型変化は、胸腺におけるT−リンパ球系譜の前駆細胞の胸腺発達を特徴とする(患者番号2、3、および4)。CD4抗原は、ヘルパー/インデューサーT−リンパ球サブセット(CD4+CD3+)および正常な胸腺細胞の大部分に存在する。しかしながら、この抗原は、単球の細胞表面上ならびに単球およびマクロファージの細胞質に低密度で存在する(CD3−CD4−)。
【0153】
CD4+低細胞の相対数は、処理後の種々の血液試料中で、異なって影響された。このタイプの細胞の相対数は、未処理試料と比較すると、処理後のB−CLL患者の血液試料中で影響されなかったようである。そのような低レベルのCD4発現は、単球および非常に初期の胸腺細胞で見られる。
【0154】
処理後のHIV+25患者は、CD4およびCD8を同時に発現している二重陽性細胞の数の実質的な増加を示した。一方、処理後の患者91は、細胞のこのサブタイプの減少を示し、そのような現象の観察は、時間に依存する。CD8+細胞の相対数は、経時的に測定すると、B−CLL患者の未処理血液試料において増加することが観察されるが、CD4+およびCD4+低細胞の相対数は、同時に減少することが観察された(表3 患者2、3および4)。
【0155】
DRおよびCD3パネル
DRマーカーは、単球、樹状細胞、B−細胞および活性T−リンパ球に存在する。
【0156】
このパネルで分析された処理および未処理試料は、血液試料をCD19およびCD3マーカーで分析した場合に得られたものと同様な免疫発現型変化を示し(表2参照)、これらの抗原は、前記したように、各々、panBおよびT−細胞マーカーである。
【0157】
血液のモノクローナル抗体での処理は、DR+B−リンパ球の相対数に影響し、その結果、DR+細胞のレベルが減少するようである。対照的に、CD3+(T−細胞)細胞の相対数は、有意に増加する(表4およびチャート参照)。さらに、活性T細胞の相対数は、B−CLL患者の処理血液試料の大部分において増加し、これらのタイプの細胞は、他の病状の患者の処理試料において不定に影響された。さらに、DR高陽性細胞の相対数は、B−CLL患者および、血液中のDR+CD34+芽球が増加した6日齢の乳児の処理血液試料における有意な数において明白となった。しかしながら、この患者の血液中に存在した芽球は、処理前および処理後にTおよびB−細胞マーカーに対して陰性であったが、処理後の骨髄系譜抗原に対してより陽性になったことに注意すべきである。CD3−DR−細胞の相対数は、処理血液試料の大部分において増加し、CD3+細胞(T−細胞)の相対数の増加に比例し、DR+細胞(B−細胞)の相対数の減少に反比例した。
【0158】
CD56&16およびCD3パネル
CD56&CD16マーカーは、細胞の不均一群、一般に大きな粒状リンパ球として知られるリンパ球のサブセット、およびナチュラルキラー(NK)リンパ球に見られる。CD16抗原は、実質的に全ての休止NKリンパ球に発現し、ある個体由来のいくつかのCD3+Tリンパ球に若干発現する。この抗原は、顆粒球に少量見られ、大きなアズール親和性顆粒を含有するリンパ球と結合する。CD16抗原は、IgG FC受容体IIIである。
【0159】
可変数のCD16+リンパ球は、CD57抗原もしくは低密度CD8抗原のいずれかか、または両方を共同発現する。ほとんどの個体では、実質的に、CD5、CD4またはCD3抗原のような他のT−リンパ球抗原とオーバーラップしない。CD56抗原は、本質的に全ての休止および活性CD16+NKリンパ球に存在し、細胞のこれらのサブセットは、非主要組織適合遺伝子複合体制限細胞障害性を行う。
【0160】
B−CLLおよびその他の病状の患者の処理および未処理血液試料の免疫発現は、非常に顆粒状の中くらいのサイズのCD56&CD16を共同発現している細胞の相対数の増加を示した(表5およびチャート1、2、3および4を参照)。これらの観察はまた、CD3抗原のみを発現する(CD56およびCD16マーカーの発現は無い)細胞、およびCD56&CD16およびCD3マーカーを一緒に共同発現する細胞の相対数の著しい増加を伴った。
【0161】
表5において、患者番号2、3および4は、同一の血液試料を表すが、それぞれ2時間、6時間および24時間(処理前および処理後)で分析している。この試料は、DR抗原のβ−鎖の相同領域に対するモノクローナル抗体による血液の処理により、CD56+およびCD16+細胞、CD3+細胞およびCD56+およびCD16+CD3+細胞の自発的生成を引き起こすようであり、これらの観察は、常にB−細胞マーカー(CD19、DR、CD56、CD16−CD3−)の消失を伴った。
【0162】
処理前および処理後のこの血液試料の前進的な分析は、CD56+およびCD16+細胞のレベルが経時的に減少すること、CD3+細胞のレベルが経時的に増加することを示した。
【0163】
B−CLL患者7の血液試料は、処理および未処理試料において観察された免疫発現型変化と比較して、CD56、CD16およびCD3抗原を発現する細胞数のいかなる変化も示さず、これは、モノクローナル抗体の添加量がBリンパ球の数に比べて極度に少なかったためである。しかしながら、適量のモノクローナル抗体による別の場合のこの患者の血液試料の処理は、CD3+、CD56+&CD16+およびCD56+およびCD16+CD3+細胞の相対数の有意な増加を示した。
【0164】
他の病状の他の患者の血液試料は、これらの細胞のレベルの不定な変化を示し、これは、処理前、処理期間の血液中に存在するB−リンパ球の数およびおそらく患者の臨床条件に依存すると思われる。
【0165】
CD45およびCD14パネル
CD45抗原は、末梢血、胸腺、脾臓および扁桃中のリンパ球、単球、多形核細胞、好酸球および好塩基球、ならびに骨髄中の白血球前駆体を含む全てのヒト白血球に存在する。
【0166】
CD14は、70%ないし93%の正常な末梢血単球、77%ないし90%の胸膜液または腹膜液食細胞に存在する。この抗原は、顆粒球に若干発現し、非刺激リンパ球、マイトジェン−活性Tリンパ球、赤血球、または血小板には存在しない。
【0167】
CD45抗原は、タンパク質チロシンホスファターゼのファミリーを表し、この分子は、外部刺激(抗原)と相互作用し、細胞増殖および分化の調節へ導くScr−ファミリーメンバーを経てシグナル変換に影響する。
【0168】
特にB−CLL患者から得られた処理血液試料におけるDR抗原のβ−鎖の密着は、そのような処理がB−リンパ球でのCD45抗原のレベルに影響することを示唆する。DR抗原のβ−鎖の刺激により生じた全免疫発現型変化は、CD45およびCD14発現レベルならびに前方分散および側面分散によって測定されたような形態(それぞれサイズおよび粒状)に基づいて分離され得る種々のタイプの細胞を生じさせるようであり、これらの結果は、表6およびチャート(1、2、3、4および5)に示す。
【0169】
処理によりCD45低細胞の相対数は(未処理試料と比較して)、有意に増加し、それゆえCD45およびCD14抗原を共同発現する細胞の相対数は増加した。このタイプの免疫発現型変化は、CD45高細胞の相対数(未処理試料と比較して)の減少と同時に起こった。しかしながら、この後者の細胞群は、形態およびCD45発現度に基づいて、さらに分割することができる。一のタイプは、極度に大きく、チャートにある残りの細胞と比較して極度に高いレベルのCD45抗原を有した(チャート1、2、3および4参照)。経時的な処理後のこのパネル分析(表 患者2、3および4ならびにチャート参照)により、CD45+細胞の相対数は、始めに経時的に急激に減少し、CD45低細胞を生じた。しかしながら、24時間後の血液の分析は、反対の状態を示した。
【0170】
試料5および7は、他のB−CLL患者から得られた他の試料で得られた変化とは反対の免疫発現型変化を表し、これは、この試料がモノクローナル抗体との非常に短いインキュベーション時間で分析したからである。実際、処理後の血液試料の一連の分析は、Bリンパ球によって調節された免疫発現型変化が発達の段階を表すので時間依存性であること、時間Xで測定された免疫発現型変化は、時間Xプラスでは同じでないだろうということ(それは一度引き起こされると固定されない)を示唆すると思われる。しかしながら、これらのタイプの変化は、体内でのより厳しい条件で起こっているにちがいなく、さもなければ免疫病理学が、結果として起こるであろう。B−細胞悪性腫瘍を有しない他の患者由来の血液試料の処理の影響は、細胞の免疫発現型の不定な変化を示し、これは、B−リンパ球がより少量で存在するからである。しかしながら、健康な供血者から得られたB−リンパ球の豊富なフラクションの処理は、高いB−リンパ球数を有するB−CLLで得られたのと同様な免疫発現型変化を示す。
【0171】
CD8およびCD3パネル
CD8抗原決定因子は、クラスIMHC分子と相互作用し、その結果、CD8+Tリンパ球とその標的細胞との間の付着を増加させる。このタイプの相互作用は、休止リンパ球の活性化を増強する。CD8抗原は、タンパク質チロシンキナーゼ(p56ick)と結合し、次に、CD8/p56ick複合体がT−リンパ球活性化における役割を果す。
【0172】
B−CLL患者から得られた血液試料の、B鎖に対するモノクローナル抗体での処理は、CD3CD8およびCD3(CD4CD3に非常によく似ている)陽性細胞の相対数の有意な増加を引き起こし、したがって、より明らかに、始めに生じた二重陽性細胞が成熟T−リンパ球に発達していることを示す。これは、直接的にCD19によっておよびDRによって、間接的にCD8−CD3−抗原によって測定することができるプロセスである。同一患者の処理血液試料の経時的な連続的な評価は、胸腺細胞発達と同一であるプロセスと一致すると思われる(表7、患者2、3および4、ならびにチャート1)。
【0173】
CD8+細胞の相対数は、処理および未処理試料において経時的に増加したが、程度がより大きかったのは、未処理試料においてであった。一方、CD8+CD3+細胞の相対数は、未処理試料において経時的に減少した。しかしながら、CD3+細胞の相対数は、経時的に測定すると、処理血液試料において増加し、これらのタイプの細胞は、CD4+CD3+単陽性細胞:胸腺細胞の成熟型に大いに一致した。さらに、これらの試料は、他のパネルでも免疫発現されたので(表3、4、5および6中に前記した)、その全変化は、極度にTリンパ球前駆体および後継体の生成においてB細胞に原因があることを示す。
【0174】
別々のアリコットでB−CLL患者由来の血液試料(番号2、3および4、表1、2、3、4、5、6、7)は、処理しないか、DR抗原のβ−鎖の相同領域に対するPEコンジュゲートモノクローナル抗体および同モノクローナル抗体の非コンジュゲート形態で処理した。PEコンジュゲートの比較によると、処理は、明らかに、CD3陽性細胞、および同血液試料を抗体の非コンジュゲート形態で処理した場合に有意なレベルで観察されたCD4のような関連マーカーの相対数の変化を示す。しかしながら、表面で発現されているDR抗原を有しないCD45陽性細胞数の増加は、経時的に測定した場合に示された(表8を参照)。その発見は、経時的に免疫発現した場合に未処理試料において示されたのと同様であった(表6)。さらに、CD45低発現細胞の相対数は、経時的に減少し、その現象は、同一患者の未処理試料においても示された(経時的に測定した場合)(チャート1Aを参照)。
【0175】
C. 異なる特性を有する他のモノクローナル抗体のT−リンパ球生成に対する影響の比較
CD19およびCD3パネル
DR抗原のα−鎖の相同領域およびMHCクラスI抗原の相同領域に対するモノクローナル抗体による血液試料の処理により、CD3+細胞の数が減少し、CD19+細胞の数が増加した。DR抗原のβ−鎖の相同領域に対するモノクローナル抗体による血液試料の処理により、CD19+細胞の数が減少し、CD3+細胞の数が増加した。シクロホスファミドとともに後者のモノローナル抗体で処理すると、同様な効果が現れた(表14 B−CLL患者5/6、2時間処理)。
【0176】
同じ試料におけるCD19+およびCD3+細胞の前進的な分析により、DR抗原のβ−鎖の相同領域に対するモノクローナル抗体で処理した血液中においてのみ、CD3+の相対数の増加がさらに明らかになった(表14 患者5/6、処理後24時間)。しかしながら、シクロホスファミドおよびDR抗原のβ−鎖に対するモノクローナル抗体で処理した血液試料の前進的な分析(24時間後の患者5/6 表14)は、正確に同じ条件下で、2時間のインキュベーション時間で観察されたものと比較して、CD19+およびCD3+細胞の相対数において逆転を示す。
【0177】
一般に、同一患者の血液試料の、DR抗原のα−鎖の相同領域に対するモノクローナル抗体またはクラスI抗原のα−鎖の相同領域に対するモノクローナル抗体での処理は、未処理試料と比較して、CD19−細胞(panBマーカー)の相対数の増加を示す。CD19−CD3−細胞の相対数は、DR抗原のα−鎖に対するモノクローナル抗体で処理したか、またはクラスI抗原に対するモノクローナル抗体で処理した血液試料中で、わずかに減少した(表14およびチャート2、3、および4を参照)。クラスI抗原に対するモノクローナル抗体での、患者09の血液試料の処理により、CD3+細胞の相対数が増加し、CD19+およびCD19−CD3−細胞の相対数がわずかに減少した。しかしながら、健康な供血者から得られたB−リンパ球の豊富な調製物の、DR抗原のβ−鎖またはα−鎖に対するモノクローナル抗体での処理により、B−CLL患者で得られたのと同様な免疫発現型変化が示された。
【0178】
HIV+およびIgA欠損患者の、DR抗原のβ−鎖に対するモノクローナル抗体での処理により、CD3+細胞の相対数が増加し、CD19+細胞の相対数が減少した。しかしながら、同一血液試料の、クラスI抗原の相同領域に対するモノクローナル抗体による処理は、同様の効果を生じなかった。B−細胞欠損患者(34/BDおよび04/BD)から得られた血液試料の処理により、DR抗原のβ−鎖、クラスI抗原およびCD4抗原に対するモノクローナル抗体で処理した場合、不定な免疫発現型変化が示された。
【0179】
CD4およびCD8パネル
CD19およびCD3パネル(表14)を用いて分析された血液試料も、また、CD4およびCD8パネル(表15)で免疫発現した。どちらのパネルも、相互に一致し、確定すると思われる。B−CLL患者(表15、患者5/6および10、チャート2、3および4)の血液試料をDR抗原のβ−鎖の相同領域に対するモノクローナル抗体と一緒に、またはこのモノクローナル抗体およびシクロホスファミドと一緒に2時間インキュベートすると、CD8+およびCD4+細胞ならびに両方のマーカーを共同発現している細胞の相対数が増加した。一方、同一試料の、DR抗原のα−鎖の相同領域またはクラスI抗原のα−鎖の相同領域に対するモノクローナル抗体による処理は、同様の効果を生じなかった。
【0180】
DR抗原のβ−鎖に対するモノクローナル抗体およびシクロホスファミドで2時間および24時間インキュベートして得られた免疫発現型の傾向を比較すると、CD4およびCD8陽性細胞の相対数における逆の変化が明らかになり(表15、2時間および24時間のB−CLL患者5/6)、かかる変化は、同一血液試料がCD19およびCD3パネル(表14同患者)で分析された場合に得られたのと一致した。後者の発見は、未分化細胞がT−リンパ球またはB−リンパ球に分化することができる場合、その後の分化が可逆的であることを示す。
【0181】
DRおよびCD3パネル
DRおよびCD3(表16)パネルで得られた免疫発現型変化は、2時間分析で、DR抗原のベータ−もしくはアルファ−側の相同領域に対するモノクローナル抗体、またはクラスI抗原に対するモノクローナル抗体、またはDR抗原のβ−鎖に対するモノクローナル抗体およびシクロホスファミドによる同一血液試料の処理の後の、CD19およびCD3パネル、ならびにCD4およびCD8パネル(表14および15、ならびにチャート2、3および4)で得られた発見を確認する。
【0182】
結果から、DR抗原のβ−鎖の相同領域に対するモノクローナル抗体は、非常にDR+細胞からのCD3陽性細胞の生産を促す能力を有することが明らかであろう。
【0183】
さらに、DR抗原のα−鎖の密着またはシクロホスファミドと共にその分子のβ−側の密着(インキュベーション時間を長くした)を含むような処理は、CD19+細胞またはDR+細胞の相対数の増加を促進した。
【0184】
CD56&16およびCD3パネル
血液試料、特に高いB−リンパ球数を有するB−CLL患者の血液試料の、DR抗原のβ−鎖の相同領域に対するモノクローナル抗体での処理により、CD56&16陽性細胞の相対数が増加した。
【0185】
これらの患者において、CD3+およびCD56+ならびにCD16+CD3+細胞の相対数もまた、β−鎖に対するモノクローナル抗体での血液試料の処理の後に増加し、これは、同一血液試料をCD3およびCD19ならびにDRおよびCD3パネルで分析した場合に同一試料で示された先の観察結果を確かなものとした。
【0186】
CD45およびCD14パネル
DR抗原のβ−またはアルファ−鎖に対するモノクローナル抗体または、β−鎖に対するモノクローナル抗体およびシクロホスファミド、またはクラスI抗原に対するモノクローナル抗体で処理した血液試料もまた、CD45およびCD14パネルで分析した(表18)。CD45低、CD45高およびCD45中の描写は、恣意的である。血液試料5/6の、DR抗原のβ−鎖に対するモノクローナル抗体、またはこのモノクローナル抗体およびシクロホスファミドでの処理(2時間)は、CD45+低細胞を生じ、CD45+中細胞の相対数を増加させた。しかしながら、前者の処理は、CD45+高細胞の相対数を増加させ、後者の処理は、CD45+中細胞の相対数を減少させ、これらの変化は、時間に依存することが明らかになった。
【0187】
クラスI抗原に対するモノクローナル抗体で処理した、患者5/6および10(B−CLL)の血液試料は、CD45+中細胞の相対数の減少を示し、未処理試料と比較して、同一処理後の血液試料09およびHIV+において同様の観察結果が示された。HIV+およびIgA/D患者の血液試料をクラスI抗原に対するモノクローナル抗体で処理すると、未処理試料またはDR抗原のβ−鎖に対するモノクローナル抗体で処理した試料と比較して、CD45+低細胞の相対数が増加した。しかしながら、これらの患者の血液試料は、DR抗原のβ−鎖の相同領域に対するモノクローナル抗体での処理によるCD45+中細胞の相対数の減少を示した。中CD45+細胞は、クラスI抗原に対するモノクローナル抗体処理後のIgA/D患者の血液試料中で増加した。極度に大きく、非常顆粒状であり、CD45抗原を高レベルで発現している細胞は、MHCクラスII抗原のDR抗原のβ−鎖の相同領域に対するモノクローナル抗体で処理した血液試料において示された(チャート1、2、3、4および5参照)。
【0188】
D8およびCD28パネル
CD28抗原は、およそ60%ないし80%の末梢血T(CD3+)リンパ球、50%のCD8+Tリンパ球および5%の未熟CD3−胸腺細胞に存在する。胸腺細胞成熟の間、CD28抗原発現は、ほとんどのCD4+CD8+未熟胸腺細胞での低密度から事実上全ての成熟CD3+、CD4+またはCD8+胸腺細胞での高密度へ増加する。細胞活性化は、さらにCD28抗原密度を増加させる。CD28の発現もまた、CD8+リンパ球を二つの機能群に分割する。CD8+CD28+リンパ球は、同種抗原−特異的細胞毒性を媒介し、それは、主要組織適合遺伝子複合体(MHC)クラスIに制限されている。細胞増殖の抑制は、CD8+CD28+サブセットによって媒介される。CD28抗原は、細胞付着分子であり、活性Bリンパ球に存在するB7/BB−1抗原のリガンドとして機能する。
【0189】
B−CLL患者(表19、患者5/6および8)の血液試料をDR抗原のβ−鎖の相同領域に対するモノクローナル抗体で処理すると、CD8+、CD28+およびCD8+CD28+細胞の相対数が増加し、全てのその他のタイプの処理では、増加しなかった。
【0190】
CD34およびCD2パネル
CD34抗原は、未熟造血前駆細胞および骨髄中の全ての造血コロニー形成細胞に存在し、それは、単分化能の(CFU−GM,BFU−E)および多能性の前駆体(CFU−GEMM,CFU−MixおよびCFU−blast)を含む。CD34もまた、間質細胞前駆体で発現する。正常な骨における末端デオキシヌクレオチジルトランスフェラーゼ(TdT)+B−およびT−リンパ系前駆体は、CD34−である。CD34抗原は、CD33抗原を発現するがCD14およびCD15抗原を欠損する初期の骨髄細胞、およびCD71抗原を発現し、かすかにCD45抗原を発現する初期の赤血球細胞に存在する。CD34抗原もまた、毛細管内皮細胞およびおよそ1%のヒト胸腺細胞で見られる。正常な末梢血リンパ球、単球、顆粒球および血小板は、CD34抗原を発現しない。CD34抗原密度は、初期の造血前駆細胞で最も高く、該細胞の成熟につれて減少する。該抗原は、完全に分化した造血細胞には存在しない。
【0191】
拘束されていないCD34+前駆細胞は、CD38−、DR−であり、CD71、CD33、CD10およびCD5のような系譜−特異的抗原を欠損しており、一方、系譜−拘束されたCD34+細胞は、高密度でCD38抗原を発現する。
【0192】
ほとんどのCD34−細胞は、相互に、CD45ROまたはCD45RA抗原のいずれかを発現する。およそ60%の急性B−リンパ性白血病および急性骨髄性白血病は、CD34抗原を発現する。該抗原は、慢性リンパ性白血病(BまたはT系譜)またはリンパ腫では発現されない。CD2抗原は、Tリンパ球およびナチュラルキラーリンパ球(NK)のサブセットに存在する。
【0193】
結果は、チャート2、3および5に示す。
【0194】
DR抗原のβ−鎖または同抗原のα−鎖に対するモノクローナル抗体での処理後のB−CLL患者の血液試料の分析(表20、患者5/6、2時間)により、前者の抗体での処理後、CD34+およびCD34−CD2+細胞の相対数の著しい増加が明らかになった。同血液試料は、他のマーカーに対して、前記したパネル(表14ないし19を参照)で免疫発現されたので、ここで観察されたCD34+およびCD34+CD2−細胞の相対数の増加は、CD4+CD8+、CD8+CD3+およびCD4+CD3+単陽性(SP)細胞の相対数の増加と同時に起こると思われる。さらに、唯一、HLA−DR抗原のβ−鎖の密着であろうと思われるこれらの発見は、該プロセスがBリンパ球退行を経てT−リンパ球を生じていることを直接支持する。
【0195】
24時間後の同様の処理の分析により、CD34+細胞は、レベルが減少して、Tリンパ球の相対数がさらに増加すると思われた。始めにT−リンパ球生成を引き起こした逆分化のプロセスは、逆転して、B−リンパ球生成を引き起こすことができる。前者の現象は、HLA−DR抗原のβ−鎖に対するモノクローナル抗体およびシクロホスファミドと一緒の2時間インキュベーション時間で観察され、一方、後者のプロセスは、同一試料を同一処理で24時間インキュベーション時間で示された(チャート2)。
【0196】
HIV+患者(表20患者HIV+)の血液試料をHLA−DR抗原のβ−鎖に対するモノクローナル抗体で処理すると、CD34+およびCD2+CD34+細胞の相対数が著しく増加し、同様に、同一血液試料をHLA−DR抗原のβ−鎖に対するモノクローナル抗体および同抗原のα−鎖に対するモノクローナル抗体で処理した。しかしながら、この血液試料の、HLA−DR抗原のα−鎖に対するモノクローナル抗体での処理は、CD34+細胞のレベルに影響しなかった。そのとき白血病について調べられ、血液中に非常に高い不定形細胞(芽球)数を有した6日齢の乳児から得られた血液試料(BB/ST表20)を、HLA−DR抗原のβ−鎖に対するモノクローナル抗体、または同抗原のα−鎖に対するモノクローナル抗体、または一緒に添加した両モノクローナル抗体で処理すると、その結果、以下の免疫発現型変化が起こった。
【0197】
未処理血液試料の分析により、CD34+およびDR+細胞の相対数は、著しく増加し、β−鎖に対するモノクローナル抗体での処理により、CD34+細胞の相対数は、さらに増加したが、HLA−DR抗原のα−鎖に対するモノクローナル抗体での処理、または一緒に添加した場合の該分子のαおよびβ−鎖に対するモノクローナル抗体での処理により、減少が示された。しかしながら、後者の処理により、CD34+CD2+細胞の相対数が増加し、同一血液試料をHLA−DR抗原のβ−鎖に対するモノクローナル抗体のみで処理した場合にその反対のことが起こった。同一患者の24時間後の処理および未処理血液アリコットの分析で、CD34+の相対数は、HLA−DR抗原のβ−鎖に対するモノクローナル抗体での処理で非常に高いレベルで維持されたことを除いて、前記した全ての処理で減少した。後者の処理で、24時間後、CD34+CD2+細胞の相対数が減少し続けた。
【0198】
これらの結果は、β−鎖を経たHLA−DR抗原の密着が、CD2+CD34+プールから、またはB−CLL患者のB−リンパ球のようなより成熟したタイプの細胞からのCD34+細胞の生産を促進することを示し、これらの結果は、このタイプの処理が逆分化を促進することを示す。しかしながら、24時間後の血液試料の免疫発現は、これらのタイプの細胞が別の系譜全体において存在すると思われ、この場合、細胞がCD7およびCD13&33パネルで処理された血液試料の分析で観察された骨髄系譜に存在するか、またはむしろ、それら自身を該骨髄系譜に拘束すると思われることを示唆する。
【0199】
形態は、MHCクラスII抗原のβ−鎖の相同領域に対するモノクローナル抗体での処理で、B−CLLのB−リンパ球および健康な個体(CD19ビーズを用いる)の豊富なフラクションの免疫発現型特徴を変化させる。これらは、B−リンパ球の形態の変化を伴った。B−リンパ球は、未処理血液スミア中で、スライドガラスでのコロニー形成が、顆粒球、単球、多数の原始様細胞および有核赤血球によって代わられることが観察された。有糸分裂像または有意な細胞死は、処理または非処理血液スミア中で観察されなかった。
【0200】
表20の結果もまた、本発明方法によると、より成熟した未分化細胞の逆分化によって未分化細胞を調製することが可能であるというさらに重要な発見を証明する。
【0201】
D. 顕微鏡写真
前記したような抗原試験に加えて、本発明の方法を、顕微鏡を用いて視覚的に行った。
【0202】
この点において、図1は、本発明の方法以前の分化したB細胞の顕微鏡写真である。図2は、本発明の方法(ここでは、薬剤は、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体であった)に従って、B細胞の逆分化によって形成された未分化細胞の顕微鏡写真である。該未分化細胞は、暗く染色された細胞凝集群である。図3は、同未分化細胞の顕微鏡写真であるが、より低い倍率で拡大した。
【0203】
図1ないし3は、したがって、本発明の方法によるB細胞の未分化幹細胞への逆分化を視覚的に示す。
【0204】
図4は、本発明の方法以前の分化したB細胞の顕微鏡写真である。図5は、本発明に従って(ここで使用した薬剤は、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体であった)、B細胞の逆分化によって形成された未分化細胞の顕微鏡写真である。また、未分化細胞は、暗く染色された細胞凝集群である。図6は、図5の同未分化細胞から分化した顆粒球の形成の顕微鏡写真である。
【0205】
従って、図4ないし6は、本発明の方法によって、B細胞が未分化幹細胞へ逆分化し、次いで、その未分化細胞が、最初の分化細胞とは異なる系譜である新たな分化細胞に拘束されることを視覚的に証明する。
【0206】
本発明の方法によってT細胞が未分化幹細胞へ逆分化し、次いで、その未分化細胞が、最初の分化細胞とは異なる系譜である新たな分化細胞に拘束されることもまた、顕微鏡によって追跡された。
【0207】
E. 概要
要するに、該実施例は、幹細胞の発生において利用され得るTおよびBリンパ球の発生および発達に関係する非常に興味深い発見を明らかにし、およそ数時間で末梢血試料中でのリンパ造血に影響する、in vitroでの実験を記述する。
【0208】
高Bリンパ球数を伴うB−細胞慢性リンパ性白血病(B−CLL)患者から得られた末梢血試料の、クラス−II抗原のβ−鎖の相同領域に対するモノクローナル抗体での処理は、単陽性(SP)T−リンパ球および、同時に共同発現される胸腺細胞マーカーCD4およびCD8抗原に対して二重陽性であるその前駆体の相対数の著しい増加を引き起こした。しかしながら、これらの現象は、常に、B−リンパ球の相対数の有意な減少を伴った。これらの観察は、同一血液試料をクラス−II抗原のα−鎖の相同領域またはクラス−I抗原の相同領域に対するモノクローナル抗体で処理した場合には示されなかった。
【0209】
B−細胞慢性リンパ性白血病(CLL)患者から得られた全血の、HLA−DR抗原のB鎖の相同領域に対するモノクローナル抗体での処理は、T−リンパ球生成を引き起こすと思われた。この事象は、CD4およびCD8マーカーを共同発現している二重陽性細胞の出現、CD34を発現している細胞の出現およびそれに付随する単陽性CD4+CD3+およびCD8+CD3+リンパ球の相対数の増加によってマークされた。さらに、かかる細胞の発生において生じた免疫発現型変化は、特に経時的に測定した場合に、胸腺細胞発達について例証されたものと一致した。
【0210】
全血をDR抗原のβ−鎖の相同領域に対するモノクローナル抗体と一緒に2時間インキュベートして生じた二重陽性細胞(DP)のパーセントは、経時的に減少し、これらの事象は、同時に、および遅れても、単陽性CD4+CD3+およびCD8+CD3細胞のパーセントの増加を伴った。TCRαおよびβ鎖もまた、これらのタイプの細胞で発現された。
【0211】
B−リンパ球は、常に、CD19、CD21、CD23、IgMおよびDRのようなマーカーを失うことが観察され、これは、CD34+およびCD34+CD2+細胞の出現、CD7+細胞の増加、CD8+CD28+およびCD28+細胞の増加、CD25+細胞の増加、CD10+およびCD34+細胞ならびにCD34+およびCD19+細胞の出現、CD5+細胞およびCD45抗原を低レベルで発現している細胞の増加と同時に起こった。これらの変化は、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体での血液の処理が原因であった。
【0212】
処理前のB−CLL患者の血液における白血球の大部分はBリンパ球であったので、かかる処理と関連した免疫発現型変化は、Bリンパ球の逆分化およびその後の拘束(すなわち、再拘束)と一致する。さらに、シクロホスファミドおよびHLA−DR抗原のβ−鎖に対するモノクローナル抗体での処理後にT−リンパ球になることを促されたB−CLL患者のB−リンパ球は、インキュベーションを延長してこの処理をした後、B−リンパ球に逆戻りすることができた。
【0213】
HLA−DR抗原のβ−鎖に対するモノクローナル抗体で処理した試料の、CD16&56およびCD3およびCD8およびCD3パネルでの分析で、これらのマーカーを発現している細胞の相対数は、CD19およびCD3およびDRおよびCD3のようなパネルで測定したものと一致した増加において、着々と増加する。比濁分析および免疫電気泳動を用いたHIV感染患者の処理および未処理試料の上清の研究により、IgGレベルの増加が明らかになり、それは、B−細胞が形質細胞段階を経たにちがいないことを示す。前記した全ての細胞の相対数の増加もまた、CD56&16抗原を非常に多数発現している中サイズの非常に顆粒状の細胞の出現を伴った。極度に大きく、非常に顆粒状の他の細胞が一時的に観察され、これらは、CD34に対して陽性であり、CD4CD8マーカーに対して二重陽性であった。他の一過性の細胞もまた観察され、これらは、大きくかつ顆粒状であり、CD3およびCD19受容体に対して陽性であった。B−リンパ球の大部分に存在するCD25は、失われ、常に数の増加が観察された新たに形成されたT−リンパ球によって発現されるようになった。
【0214】
CD28+CD8+およびCD28+細胞は、B−CLL患者の全血の、DR抗原のB鎖の相同領域に対するモノクローナル抗体での処理後に現れた。これらの発見は、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体での血液の処理に起因した。
【0215】
この方法で生じたT−リンパ球生成は、健康な供血者の末梢血、臍帯血、骨髄、HIV+患者およびAIDS患者を包含する様々な感染患者、健康な供血者の血液試料から得られたBリンパ球の豊富なフラクション、IgA欠損患者および様々な他の病状の他の患者においても観察された。さらに、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体で処理したB−CLL患者二人の試料における骨髄マーカーの分析は、CD13およびCD33のような骨髄マーカーを発現している細胞の相対数の有意な増加を示した。これらのマーカーは、CD56&16またはCD7抗原と共同発現された。しかしながら、T−リンパ球マーカーを有しており、骨髄抗原を有していないCD7+細胞の相対数は、細胞の別の群で観察された。これらの個々の観察は、未処理試料において、またはクラスI抗原もしくはHLA−DR抗原のα−鎖の相同領域に対するモノクローナル抗体で処理した試料において、見られなかった(チャート2&3を参照)。これらの最終結果は、HLA−DR抗原のβ−鎖を経て一度引き起こされたB−リンパ球は、T−リンパ球前駆細胞に退行することができるだけでなく、骨髄系譜および赤血球系譜にも存在し得ることを示唆する。
【0216】
本発明の方法によって生産される幹細胞は、いずれの組織の幹細胞でもあってよく、必ずしもリンパ造血前駆細胞に限定されないことに注意すべきである。
【0217】
本発明の他の変形例は、当事者には明白であろう。
【0218】
【表1−1】

【表1−2】
【表1−3】
【表1−4】
【表2】
【表3】
【表4】
【表5】
【表6】
【表7】
【表8】
【表9】
【表10】
【表11】
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【表18】
【表19】
【表20】
【表21】
【表22】
【表23−1】
【表23−2】
【表24】
【表25】
【技術分野】
【0001】
本発明は、未分化細胞の製造方法に関する。
【0002】
特に、本発明は、より拘束された細胞(more committed cell)からの未分化細胞の製造方法に関する。
【0003】
さらに本発明は、新しいより拘束された細胞、すなわち、再拘束細胞(recommitted cell)の製造のための本発明の未分化細胞の使用に関する。
【0004】
本発明は、また、免疫学的症状または疾患に関連する症状の緩和または免疫学的症状または疾患からの部分的もしくは完全な治療などの免疫系に対して(直接的に、または、それから得られる生成物の使用を介して間接的に)効果を有するための本発明の未分化細胞または本発明の再拘束細胞の使用にも関する。
【背景技術】
【0005】
概論として、分化は、細胞の構造および機能が段々に拘束されて、例えばT細胞またはB細胞の形成のようにより特殊化された細胞を生じるプロセスである。したがって、細胞がより拘束されると、該細胞は、さらに分化される。
【0006】
対照的に、逆分化(retrodifferentiation)は、細胞の構造および機能があまり特殊化されていない細胞を生じるように段々に変えられるプロセスである。
【0007】
未分化細胞は、多系譜(multilineage)分化能を有する。すなわち、該未分化細胞は、2つ以上のタイプの特殊化された細胞に分化する能力を有する。未分化細胞の典型的な例は、幹細胞である。
【0008】
対照的に、分化細胞は、多系譜分化能を有しない。分化細胞の典型的な例は、T細胞である。
【0009】
多くの未分化細胞および分化細胞がイン・ビボで見られ、一般的な技術は、それらについての一般的な教示を充分にもっている。
【0010】
例証として、とりわけ、LevittおよびMertelsman 1995(Marcel Dekker Incにより発行されたHaematopoietic Stem Cells, 特に第45−59頁)(非特許文献1)およびRoittら(Immunology, 4th Edition, Roitt, Brostoff および Male 編 1996, Publ. Mosby, 特に第10章)(非特許文献2)が引用される。
【0011】
しかしながら、要するに、未分化細胞の例としては、リンパ造血前駆細胞(lymphohaematopoietic progenitor cell)(LPC)が挙げられる。LPCとしては、多能性幹細胞(PSC)、リンパ系幹細胞(LSC)および骨髄性幹細胞(MSC)が挙げられる。LSCおよびMSCは、各々、PSCの分化により形成される。したがって、LSCおよびMSCは、PSCよりも拘束されている。
【0012】
分化細胞の例としては、T細胞、B細胞、好酸球、好塩基球、好中球、骨髄巨核球、単球、赤血球、顆粒球、マスト細胞およびリンパ球が挙げられる。
【0013】
T細胞およびB細胞は、LSCの分化により形成される。したがって、T細胞およびB細胞は、LSCよりも拘束されている。
【0014】
好酸球、好塩基球、好中球、骨髄巨核球、単球、赤血球、顆粒球、マスト細胞、NKおよびリンパ球は、MSCの分化により形成される。したがって、これらの細胞の各々は、MSCよりも拘束されている。
【0015】
抗原は、未分化細胞および分化細胞と関連している。ここでは、「関連する」なる用語は、各々の抗原を発現するかもしくは発現する能力を有する細胞、または各々の抗原を提供するかもしくは提供させる能力を有する細胞、または各々の抗原からなる細胞を意味する。
【0016】
ほとんどの未分化細胞および分化細胞は、主要組織適合遺伝子複合体(MHC)クラスI抗原および/またはクラスII抗原からなる。これらの抗原がこれらの細胞と関連している場合、それらは、クラスI+および/またはクラスII+細胞と呼ばれる。
【0017】
未分化細胞または分化細胞と関連する各々の特異的抗原は、マーカーとして作用することができる。したがって、種々のタイプの細胞を、それらの関連する特定の抗原に基づいて、または、関連抗原の特定の組合せに基づいて、お互いに区別することができる。
【0018】
これらのマーカー抗原の例としては、抗原CD34、CD19およびCD3が挙げられる。これらの抗原が存在する場合、これらの個々の細胞は、各々、CD34+、CD19+およびCD3+細胞と呼ばれる。これらの抗原が存在しない場合、これらの細胞は、各々、CD34−、CD19−およびCD3−細胞と呼ばれる。
【0019】
より詳しくは、PSCは、CD34+細胞である。LSCは、DR+、CD34+およびTdT+細胞である。MSCは、CD34+、DR+、CD13+、CD33+、CD7+およびTdT+細胞である。B細胞は、CD19+、CD21+、CD22+およびDR+細胞である。T細胞は、CD2+、CD3+、およびCD4+またはCD8+細胞である。未熟リンパ球は、CD4+およびCD8+細胞である。活性化T細胞は、DR+細胞である。ナチュラルキラー細胞(NK)は、CD56+およびCD16+細胞である。Tリンパ球は、CD7+細胞である。白血球は、CD45+細胞である。顆粒球は、CD13+およびCD33+細胞である。単球マクロファージ細胞は、CD14+およびDR+細胞である。
【0020】
したがって、前記抗原マーカーの存在について検索することにより、ある種の細胞タイプ(例えば、細胞が未分化細胞であるか分化細胞であるか)およびその細胞タイプの特殊化(例えば、細胞がT細胞であるかB細胞であるか)を同定することが可能である。
【0021】
逆分化の一般的概念は、新しくない。実際、1976年に、Jose Uriel(Cancer Research 36, 4269−4275, 1976年11月)(非特許文献3)は、このトピックについての評論を開示している;すなわち:
「逆分化は、変化した病因(物理的、化学的およびウイルス性)の有害因子に対する細胞完全性の維持について一般的な適応プロセスとして明白である。そのゲノム上でコードされている情報全体を保存すると同時に、逆分化を受けている細胞は、細胞質構造の自己遺伝子欠損のプロセスおよび遺伝子発現のより幼若なパターンへの移行により形態学的および機能的複雑さを失う。これにより、初めから異なる細胞表現型の漸進性均一化、および成人細胞において操作的な調節シグナルに対する応答性の減少が生じる。逆分化は、通常、復帰突然変異(reversion)が始まった、終末表現型を回復させようとする再個体発生のプロセスにより相殺される。これは、逆分化が常に細胞再生および組織修復に関連している理由を説明している。」
【0022】
Uriel(同書)(非特許文献3)は、次いで、報告された逆分化のケース、例えば、ツメガエル(Xenopus)のオタマジャクシの消化管内皮細胞からの核に関連するGurdonの研究(Advances in Morphogenesis [1966] 第4巻,第1−43頁. New York Academic Press, Abercrombie および Bracher 編)(非特許文献4)、および肝臓の再生に関連するBresnickの研究(Methods in Cancer Research [1971] 第6巻, 第347−391頁)(非特許文献5)を続けて検討した。
【0023】
Uriel(同書)(非特許文献3)は、また、イン・ビトロ培養のための単離された肝実質細胞に関係する研究についても報告した。Urielによると、以下のとおりである:
「胎児もしくは新生児肝実質細胞についての結果、または再生している肝臓もしくは樹立した肝細胞癌からの肝実質細胞についての結果と対照的に、静止している成人肝実質細胞から永久クラス系を得るのは困難であった。」
【0024】
Uriel(同書)(非特許文献3)は、また、癌における明らかな逆分化についても報告した;すなわち:
「多くの腫瘍の生化学的表現型は、肝発癌現象の前新生物期の間、・・・未熟に向かう復帰突然変異の類似変化を示し、細胞は、逆分化する。」
【0025】
逆分化についてのより最近の発見としては、Minoru Fukudaの研究(Cancer Research [1981] 第41巻, 第4621−4628頁)(非特許文献6)が挙げられる。Fukudaは、腫瘍促進性ホルボールエステル、12−O−テトラデカノイル−ホルボール−13−アセテート(TPA)の使用により、K562ヒト白血病細胞の細胞表面糖タンパク質プロフィルの特異的な変化をひき起こした。Fukudaによると、TPAは、K562ヒト白血病細胞を逆分化段階に誘導すると思われた。
【0026】
また、Hassら(Cell Growth & Differentiation [1991] 第2巻, 第541−548頁)(非特許文献7)は、ホルボールエステルの不在下でのTPA分化したU−937白血病細胞の32〜36日間の長期培養により逆分化のプロセスが生じること、および逆分化した細胞が基質から分離され、増殖を再開したことを報告した。
【0027】
逆分化の別のケースは、CurtinおよびSnellの研究(Br. J. Cancer [1983] 第48巻, 第495−505頁)(非特許文献8)である。これらの研究者は、ジエチルニトロサミン誘発肝発癌現象の間に生じる酵素的変化および部分肝切除後の肝臓再生を正常な肝臓分化と比較した。これらの研究者は、分化の段階を追っての逆転と似ていた発癌現象の間の酵素活性の変化を見いだした。これらの研究者によると、それらの結果は、基礎をなす逆分化プロセスが肝発癌現象および肝臓再生の両方のプロセスに共通していることを示唆している。
【0028】
より最近では、Chastreら(FEBS Letters [1985] 第188巻, 第2号, 第2810−2811頁)(非特許文献9)は、ヒト大腸癌性サブクローンHT29−18の逆分化について報告した。
【0029】
さらに最近では、Kobayashiら(Leukaemia Research [1994] 第18巻, 第12号, 第929−933頁)(非特許文献10)は、リポ多糖類(LPS)による処置によりマクロファージ様細胞に分化した単一のラット骨髄単球性白血病細胞由来の逆分化細胞系(RD−1)の樹立について報告した。
【0030】
現在の解釈によると、Molecular Biology of the Cell(Garland Publishers Inc. 発行. 1983)の第911頁(非特許文献11)の第911頁およびさらに最近ではLevittおよびMertelsman(非特許文献1)に見られる教示により立証されているように、PSCのような幹細胞は、以下の4つの特徴を有する:
i.未分化細胞である。すなわち、最終的に分化されない;
ii.制限なく分割する能力を有する;
iii.前記分化細胞などの分化した子孫を生じる能力を有する;
iv.分割すると、各々の娘は、選択の自由を有する:その親と同様のPSCとして維持することができるか、または、終末分化に不可逆的に誘導する経路に入ることができる。
最後の資格、すなわち、当該技術分野における一般的な教示によると、未分化細胞は、より拘束された細胞に分化すると、逆分化することができないということを理解すべきである。Uriel(同書)(非特許文献3)、Fukunda(同書)(非特許文献6)、Hassら(同書)(非特許文献7)、CurtinおよびSnell(同書)(非特許文献8)、Chastreら(同書)(非特許文献9)、ならびにKobayashiら(同書)(非特許文献10)は、あるタイプの分化細胞を逆分化したが、これらの細胞が同一系譜に拘束されたままであった場合には、それらは、未分化細胞に逆分化しなかったので、この解釈は、これらの研究者の教示によってさえ支持された。
【先行技術文献】
【非特許文献】
【0031】
【非特許文献1】LevittおよびMertelsman 1995(Marcel Dekker Incにより発行されたHaematopoietic Stem Cells, 特に第45−59頁)
【非特許文献2】Roittら(Immunology, 4th Edition, Roitt, Brostoff および Male 編 1996, Publ. Mosby, 特に第10章)
【非特許文献3】Jose Uriel(Cancer Research 36, 4269−4275, 1976年11月)
【非特許文献4】Advances in Morphogenesis [1966] 第4巻,第1−43頁. New York Academic Press, Abercrombie および Bracher 編
【非特許文献5】Methods in Cancer Research [1971] 第6巻, 第347−391頁
【非特許文献6】Cancer Research [1981] 第41巻, 第4621−4628頁
【非特許文献7】Hassら(Cell Growth & Differentiation [1991] 第2巻, 第541−548頁)
【非特許文献8】Br. J. Cancer [1983] 第48巻, 第495−505頁
【非特許文献9】Chastreら(FEBS Letters [1985] 第188巻, 第2号, 第2810−2811頁)
【非特許文献10】Kobayashiら(Leukaemia Research [1994] 第18巻, 第12号, 第929−933頁)
【非特許文献11】Molecular Biology of the Cell(Garland Publishers Inc. 発行. 1983)の第911頁
【発明の概要】
【発明が解決しようとする課題】
【0032】
したがって、本発明以前の技術の状態によると、より拘束された細胞から、幹細胞などの未分化細胞を形成することはできなかったと思われた。しかしながら、本発明は、この確信が誤っており、より拘束された細胞から未分化細胞を形成することができることを示す。
【課題を解決するための手段】
【0033】
かくして、第1の態様によると、本発明は、より拘束された細胞を、このより拘束された細胞を未分化細胞に逆分化させる薬剤と接触せさることからなる、未分化細胞の製造方法を提供するものである。
【0034】
第2の態様によると、本発明は、より拘束された細胞を、このより拘束された細胞を未分化細胞に逆分化させる薬剤と接触させ、次いで、該未分化細胞を再拘束細胞に拘束することからなる方法を提供するものである。
【0035】
「再拘束細胞」なる用語は、未分化細胞から誘導された細胞、すなわち新しいより拘束された細胞を意味する。
【0036】
第3の態様によると、本発明は、本発明の方法により生産された未分化細胞を提供するものである。
【0037】
第4の態様によると、本発明は、医薬としてのまたは医薬の調製における本発明の方法により生産された未分化細胞を提供するものである。
【0038】
第5の態様によると、本発明は、免疫学的障害または疾患の治療薬の製造における本発明の方法により生産された未分化細胞の使用を提供するものである。
【0039】
第6の態様によると、本発明は、本発明の方法により生産された再拘束細胞を提供するものである。
【0040】
第7の態様によると、本発明は、医薬としてのまたは医薬の調製における本発明の方法により生産された再拘束細胞を提供するものである。
【0041】
第8の態様によると、本発明は、免疫学的障害または疾患の治療薬の製造における本発明の方法により生産された再拘束細胞の使用を提供するものである。
【0042】
第9の態様によると、本発明は、より拘束された細胞を未分化細胞に逆分化させることができる薬物を付着している、より拘束された細胞を提供するものである。
【0043】
第10の態様によると、本発明は、CD19+およびCD3+細胞を提供するものである。
【0044】
かくして、最も広い意味では、本発明は、より拘束された細胞から未分化細胞を形成する能力を有するという非常に驚くべき発見に基づいている。
【発明を実施するための形態】
【0045】
本発明は、より拘束された細胞から未分化細胞を製造し、次いで、これらの未分化細胞を、障害の治療のためのイン・ビトロもしくはイン・ビボまたはその組合せで薬物として、または、該薬物を調製するために使用することが可能であるので、非常に好都合である。
【0046】
本発明は、また、初期のより拘束された細胞を修正または除去するために、または、その生成物を修正または除去するために、逆分化により製造された未分化細胞を新しい分化細胞のような再拘束細胞に拘束することができるので、好都合である。
【0047】
好ましくは、より拘束された細胞は、MHCクラスI+および/またはMHCクラスII+未分化細胞に逆分化する能力を有する。
【0048】
好ましくは、より拘束された細胞は、幹細胞抗原からなる未分化細胞に逆分化する能力を有する。
【0049】
好ましくは、より拘束された細胞は、CD34+未分化細胞に逆分化する能力を有する。
【0050】
好ましくは、より拘束された細胞は、リンパ造血前駆細胞に逆分化する能力を有する。
【0051】
好ましくは、より拘束された細胞は、多能性幹細胞に逆分化する能力を有する。
【0052】
未分化細胞は、抗原発現、捕捉または認識に関係がある成分からなってもよい。好ましくは、未分化細胞は、MHCクラスI+および/またはMHCクラスII+細胞である。
【0053】
好ましくは、未分化細胞は、幹細胞抗原からなる。
【0054】
好ましくは、未分化細胞は、CD34+未分化細胞である。
【0055】
好ましくは、未分化細胞は、リンパ造血前駆細胞である。
【0056】
好ましくは、未分化細胞は、多能性幹細胞である。
【0057】
より拘束された細胞は、抗原発現、捕捉または認識に関係がある成分からなってもよい。好ましくは、より拘束された細胞は、MHCクラスI+および/またはMHCクラスII+細胞である。
【0058】
好ましくは、該薬剤は、より拘束された細胞の細胞外に作用する。
【0059】
好ましくは、より拘束された細胞は、薬剤により操作可能に密着可能な(engageable)受容体からなる(該薬剤は、該受容体に操作可能に密着する(engage))。
【0060】
好ましくは、受容体は、細胞表面受容体である。
【0061】
好ましくは、受容体は、α−成分および/またはβ−成分からなる。
【0062】
好ましくは、受容体は、相同領域を有するβ−鎖からなる。
【0063】
好ましくは、受容体は、少なくともHLA−DRのβ−鎖の相同領域からなる。
【0064】
好ましくは、受容体は、相同領域を有するα−鎖からなる。
【0065】
好ましくは、受容体は、少なくともHLA−DRのα−鎖の相同領域からなる。
【0066】
好ましくは、薬剤は、受容体に対する抗体である。
【0067】
好ましくは、薬剤は、受容体に対するモノクローナル抗体である。
【0068】
好ましくは、薬剤は、HLA−DRのβ−鎖の相同領域に対する抗体、好ましくは、モノクローナル抗体である。
【0069】
好ましくは、薬剤は、HLA−DRのα−鎖の相同領域に対する抗体、好ましくは、モノクローナル抗体である。
【0070】
好ましくは、薬剤は、生物学的応答調節剤と共に用いられる。
【0071】
好ましくは、生物学的応答調節剤は、アルキル化剤である。
【0072】
好ましくは、アルキル化剤は、シクロホスファミドであるか、または、シクロホスファミドからなる。
【0073】
1つの好ましい具体例では、より拘束された細胞は、分化細胞である。
【0074】
好ましくは、より拘束された細胞は、B細胞またはT細胞のいずれか1つである。
【0075】
別の好ましい具体例では、より拘束された細胞は、より成熟した未分化細胞である。
【0076】
1つの好ましい具体例では、未分化細胞が再拘束細胞に拘束された場合、再拘束細胞は、逆分化前のより拘束された細胞と同一の系譜のものである。
【0077】
別の好ましい具体例では、未分化細胞が再拘束細胞に拘束された場合、再拘束細胞は、逆分化前のより拘束された細胞と異なる系譜のものである。
【0078】
好ましくは、再拘束細胞は、B細胞、T細胞または顆粒球のいずれか1つである。
【0079】
好ましくは、該方法は、イン・ビトロ方法である。
【0080】
好ましくは、薬剤は、MHC遺伝子発現を調節し、好ましくは、この場合、該薬剤は、MHC I+および/またはMHC II+発現を調節する。
【0081】
該薬剤は、より拘束された細胞を未分化細胞に逆分化させるために、このより拘束された細胞に操作可能に密着する。これに関して、より拘束された細胞の未分化細胞への逆分化のための薬剤は、より拘束された細胞との直接的な密着(engagement)または間接的な密着において作用する。
【0082】
直接的密着の例は、より拘束された細胞が、B細胞上で見いだされるもののような相同領域(一般的に、同一または類似の配列を有することが判明している領域)を有するβ−鎖のような、その細胞表面上に少なくとも1つの細胞表面受容体を有する場合および薬剤が該細胞表面受容体に直接的に密着する場合である。別の例は、より拘束された細胞が、T細胞上で見られるもののような相同領域を有するα−鎖のような、その細胞表面上に細胞表面受容体を有する場合および該薬剤が細胞表面受容体に直接的に密着する場合である。
【0083】
間接的な密着の例は、より拘束された細胞がその細胞表面上に少なくとも2つの細胞表面受容体を有する場合であり、薬剤の、受容体のうちの1つとの密着は、他の受容体に影響を及ぼし、次いで、より拘束された細胞の逆分化を誘発する。
【0084】
より拘束された細胞の未分化細胞への逆分化のための薬剤は、化学化合物または組成物である。しかしながら、好ましくは、該薬剤は、より拘束された細胞の表面上で細胞表面受容体を密着させる能力を有する。例えば、好ましい薬剤としては、環状アデノシン一リン酸(cAMP)、CD4分子、CD8分子、T−細胞受容体の一部もしくは全部、リガンド(固定化または遊離)、ペプチド、T−細胞受容体(TCR)、抗体、交差反応性抗体、モノクローナル抗体、またはポリクローナル抗体の1つ以上が挙げられる。
【0085】
薬剤が抗体、交差反応性抗体、モノクローナル抗体、またはポリクローナル抗体である場合、好ましくは、該薬剤は、MHCクラスII抗原のβ鎖、MHC HLA−DR抗原のβ鎖、MHCクラスIもしくはクラスII抗原のα鎖、HLA−DR抗原のα鎖、MHCクラスII抗原もしくはMHCクラスI抗原のαおよびβ鎖のうちのいずれか1つ以上に対する抗体、交差反応性抗体、モノクローナル抗体、またはポリクローナル抗体のうちのいずれか1つ以上である。適切な抗体の例は、CR3/43(ダコ(Dako)より供給された)である。
【0086】
より拘束された細胞は、未分化細胞から誘導されたかまたは誘導可能な細胞である。
【0087】
かくして、1つの具体例では、より拘束された細胞は、未分化細胞でもある。したがって、例証として、未分化細胞は、リンパ系幹細胞または骨髄幹細胞であることができ、未分化細胞は、多能性幹細胞である。
【0088】
別の好ましい具体例では、より拘束された細胞は、CFC−T細胞、CFC−B細胞、CFC−エオシン細胞、CFC−バス(Bas)細胞、CFC−バス細胞、CFC−GM細胞、CFC−MEG細胞、BFC−E細胞、CFC−E細胞、T細胞、B細胞、好酸球、好塩基球、好中球、単球、骨髄巨核細胞または赤血球などの分化細胞であり;未分化細胞は、骨髄幹細胞、リンパ系幹細胞または多能性幹細胞である。
【0089】
より拘束された細胞が分化細胞である場合、好ましくは、該分化細胞は、Bリンパ球(活性化または非活性化)、Tリンパ球(活性化または非活性化)、マクロファージ単球系譜からの細胞、クラスIまたはクラスII抗原を発現させる能力を有する有核細胞、クラスIまたはクラスII抗原を発現させることができる細胞、または除核細胞(すなわち、赤血球のような核を含有しない細胞)である。
【0090】
別の好ましい具体例は、分化細胞は、各々、CD56および/またはCD16細胞表面受容体を発現する、大きな顆粒状リンパ球、ヌルリンパ球、およびナチュラルキラー細胞からなる細胞のグループのいずれか1つから選択される。
【0091】
分化細胞は、除核細胞の核形成により形成されてもよい。
【0092】
薬剤は、より拘束された細胞内で細胞内に作用してもよい。しかしながら、好ましくは、薬剤は、より拘束された細胞の細胞外に作用する。
【0093】
好ましい具体例では、薬剤は、より拘束された細胞の表面上に存在する受容体に操作可能に密着する。該受容体は、より拘束された細胞により発現させられる能力を有する受容体のように、より拘束された細胞により発現される。
【0094】
好ましくは、受容体は、主要組織適合遺伝子複合体(MHC)のクラスIまたはクラスII抗原である。好ましい具体例では、細胞表面受容体は、HLA−DR受容体、DM受容体、DP受容体、DQ受容体、HLA−A受容体、HLA−B受容体、HLA−C受容体、HLA−E受容体、HLA−F受容体、またはHLA−G受容体のいずれか1つである。
【0095】
さらに好ましい具体例では、細胞表面受容体は、HLA−DR受容体である。
【0096】
好ましくは、接触工程は、薬剤を以下のものの1つ以上と密着させることからなる:クラスI抗原のα−鎖の相同領域、クラスII抗原のα−鎖相同領域、CD4細胞表面受容体、CD8細胞表面受容体、リンパ球の存在下でのクラスII抗原のβ−鎖の相同領域、リンパ球の存在下でのクラスI抗原のα−鎖の相同領域、またはリンパ球の存在下でのクラスII抗原のα−鎖の相同領域。
【0097】
好ましくは、接触工程は、生物学的応答調節剤の存在下で生じる。
【0098】
好ましくは、生物学的応答調節剤は、免疫調節剤、成長因子、サイトカイン、細胞表面受容体、ホルモン、核酸、ヌクレオチド配列、抗原またはペプチドなどの調節剤の1つ以上である。
【0099】
本発明の好ましい具体例では、未分化細胞は、次いで、分化細胞のような再拘束細胞に拘束される。
【0100】
再拘束細胞は、未分化細胞が誘導された、より拘束された細胞と同一の系譜のものである。
【0101】
別法としては、再拘束細胞は、未分化細胞が誘導された、より拘束された細胞と異なる系譜のものである。
【0102】
さらに、本発明は、未分化細胞を再拘束細胞に拘束し、次いで、再拘束細胞を骨髄腫に縮合させることを含む、本発明の未分化細胞の製造方法を包含する。これは、抗体または抗原またはホルモンなどの多量の所望の生産物をイン・ビトロで発現させる。
【0103】
本発明の他の態様は、以下のものを含む:
【0104】
より拘束された細胞から未分化細胞を製造するための本発明の薬物のいずれか1つの使用。
【0105】
B−リンパ球またはT−リンパ球からモノクローナル抗体またはポリクローナル抗体または特異的抗体のいずれか1つを製造するための本発明の方法により生産される未分化細胞;マクロファージ単球系譜からの細胞;クラスIまたはクラスII抗原を発現する能力を有する核形成細胞;クラスIまたはクラスII抗原を発現させる能力を有する細胞;除核細胞;断片化細胞;またはアポプティック(apoptic)細胞の使用。
【0106】
B−リンパ球からエフェクターT−リンパ球を生産するための(逆もまた同様)本発明の方法により生産された未分化細胞の使用。
【0107】
B−リンパ球、T−リンパ球からなるかもしくはこれから調製される医薬、マクロファージ単球系譜からの細胞、クラスIもしくはクラスII抗原を発現する能力を有する核形成細胞、クラスIもしくはクラスII抗原を発現させる能力を有する細胞、または除核細胞のいずれか1つ以上を生産するための本発明の方法により生産される未分化細胞の使用。
【0108】
本発明は、前記使用を利用するプロセス、およびかかるプロセスから製造された生成物または組成物も包含する。
【0109】
本発明は、適切な希釈剤、担体または賦形剤と混合した、本発明の未分化細胞またはそれから得られた生成物からなる医薬を包含する。
【0110】
1つの好ましい具体例では、医薬は、適切な希釈剤、担体または賦形剤と混合した、本発明の未分化細胞から得られた抗体または抗原からなる。
【0111】
好ましくは、医薬は、癌、自己免疫疾患、血液障害、細胞もしくは組織再生、臓器再生、臓器もしくは組織移植の処置、または先天性代謝障害のいずれか1つの処置のためのである。
【0112】
好ましい具体例では、本発明は、遺伝子をより拘束された細胞に導入し、次いで、本発明の方法により未分化細胞を製造し、これにより、遺伝子が未分化細胞中に存在することからなる、遺伝子を未分化細胞のゲノムに導入するプロセスに関する。
【0113】
より好ましい具体例では、本発明は、遺伝子をより拘束された細胞のゲノムに挿入し、次いで、本発明の方法により未分化細胞を製造し、これにより、遺伝子が未分化細胞中に存在することからなる、遺伝子を未分化細胞のゲノムに導入するプロセスに関する。
【0114】
さらに好ましい具体例では、本発明は、遺伝子をより拘束された細胞のゲノムに挿入し、次いで、本発明の方法により未分化細胞を製造し、これにより、遺伝子が未分化細胞のゲノム中に存在することからなる、遺伝子のゲノムを未分化細胞に導入するプロセスに関する。
【0115】
本発明は、本発明のこれらのプロセスのいずれか1つにより製造される未分化細胞を包含する。
【0116】
既に記載したとおり、本発明は、適切な希釈剤、担体または賦形剤と混合した、これらのプロセスのいずれか1つにより製造された未分化細胞からなる医薬を包含する。正しくないゲノム構造を有するより拘束された細胞により生じるかまたは該細胞と関連する症状または病状を緩和するために、かかる医薬について、未分化細胞を用いて、正しいゲノム構造を有するものなど有益なより拘束された細胞を生産することができる。
【0117】
かくして、本発明は、また、本発明の方法により未分化細胞を形成し、未分化細胞を再拘束細胞に拘束し、これにより、細胞のゲノムおよび/または核の整列または再整列により突然変異が除去されることからなる、より拘束された細胞から後天性突然変異を除去するプロセスを提供するものでもある。
【0118】
好ましくは、遺伝子は、ゲノムの免疫グロブリン部位またはTCR部位に挿入される。
【0119】
別法としては、未分化細胞を用いて、正しくないゲノム構造を有するより拘束された細胞により生じるかまたは該細胞と関連する症状または病状を治療する実体を生産するより拘束された細胞を生産することができる。
【0120】
例えば、本発明を用いて、未分化細胞に逆分化したより拘束された細胞により発現される抗原に対する抗体またはT細胞受容体を製造することができる。これに関して、抗原は、胎児特異的抗原または交差反応胎児特異的抗原であってよい。
【0121】
本発明は、また、未分化細胞およびより拘束された細胞のレベルを制御するプロセスをも含む。例えば、本発明は、本発明の方法により未分化細胞を形成し、次いで、アポプトーシス遺伝子を活性化して、その死をもたらすなど未分化細胞に影響を及ぼすことからなる方法を含む。
【0122】
本発明の1つの好ましい具体例では、より拘束された細胞は、癌細胞ではない。本発明の別の好ましい具体例では、薬剤は、発癌性でなく、癌増殖を促進させる能力も有しない。
【0123】
本発明は、また、より拘束された細胞を、このより拘束された細胞を未分化細胞に逆分化させる薬剤と接触させることにより未分化細胞を製造し、次いで、所望により、該未分化細胞を再拘束細胞に拘束することからなる欠損細胞または不必要な細胞により生じる疾患または障害に罹っている患者の治療方法であって、該未分化細胞または再拘束細胞が欠損細胞または不必要な細胞に影響を及ぼして、該疾患または障害の症状を緩和するかまたは疾患または病状の患者を治療する治療方法をも包含している。
【0124】
要約すると、本発明は、より拘束された細胞からの未分化細胞の製造に関する。
【0125】
本発明は、以下の図面を参照して、例証として説明される。
【図面の簡単な説明】
【0126】
【図1】図1は、本発明方法以前の細胞の顕微鏡写真である。
【図2】図2は、本発明方法により製造された細胞の顕微鏡写真である。
【図3】図3は、低倍率であるが、本発明の方法により製造された顕微鏡写真である。
【図4】図4は、本発明方法以前の細胞の顕微鏡写真である。
【図5】図5は、本発明方法により製造された細胞の顕微鏡写真である。
【図6】図6は、本発明方法により製造された細胞の顕微鏡写真である。
【実施例】
【0127】
A.物質および方法
患者
血液試料は、B−細胞慢性リンパ球性白血病患者、抗体欠損(IgA欠損およびX染色体性小児性低ガンマグロブリン血症を含む)患者、HIV感染およびAIDS症候群患者、CMV感染患者、ホジキンリンパ腫患者、急性T−細胞白血病患者、ホジキンリンパ腫の6日齢乳児、急性T−細胞白血病患者、胚芽細胞増殖症の6日齢乳児、種々の感染および慢性症状の種々の患者、臍帯血、骨髄、および健康な供血者からの富化B−リンパ球調製物から、EDTAを含有するラベンダートップ管中に得た。
【0128】
臨床条件および実験条件
血液試料に適用された種々のタイプの処置を含む患者の臨床および実験処置条件を表1に記載する。コールターカウンターを用いて白血球(WBC)分画を得た。これらは、同表に含まれる。
【0129】
血液の処置
得られた血液試料をすぐに、HLA−DR抗原(DAKO)のβ−鎖の相同領域に対する純粋な抗体で処理し、最長24時間、室温で、頭−頭ローラー(head to head roller)で混合した。まず、いくつかの試料を頭−頭ローラーで15分間混合した後、それらを22℃でインキュベーター中でインキュベートした。血液試料に添加したモノクローナル抗体の濃度は、血液ml当たり10〜50μlを変化した。
【0130】
さらに、同一の濃度で他の処置を適用し、これらは、HLA−DR抗原のα−鎖の相同領域に対するモノクローナル抗体、クラスI抗原の相同領域に対するモノクローナル抗体、CD4に対するモノクローナル抗体、CD8に対するモノクローナル抗体、およびHLA−DR抗原のβ−鎖の相同領域に対するPEコンジュゲートモノクローナル抗体の添加を含んだ。
【0131】
他の処置は、血液試料への、HLA−DR抗原のαおよびβ−鎖の相同領域に対するモノクローナル抗体の同時添加を含んだ。
【0132】
さらにまた、シクロホスファミドなどのアルキル化剤を、HLA−DR抗原のβ−鎖の相同領域に対する純粋なモノクローナル抗体と合わせて血液試料に添加した。
【0133】
これらの処置に続いて、製造者の指示により指示されるように標識モノクローナル抗体のパネルを用いて血液試料を染色し、次いで、フローサイトメトリーを用いて分析した。
【0134】
モノクローナル抗体によりインキュベーション期間は、2時間、4時間、6時間、12時間〜24時間の範囲であった。
【0135】
標識抗体
以下のモノクローナル抗体を用いて、フローサイトメトリーにより細胞上の以下のマーカーを検出した:CD19およびCD3、CD4およびCD8、DRおよびCD3、CD56&16およびCD3、CD45およびCD14、CD8およびCD3、CD8およびCD28、サイマルテスト(simultest)対照(IgG1 FITC+IgG2a PE)、CD34およびCD2、CD7およびCD13&13、CD10およびCD25、CD5およびCD10、CD5およびCD21、CD7およびCD5、CD13およびCD20、CD23およびCD57、ならびにCD25およびCD45 RA(ベクトン・アンド・デッケンソン(Becton & dickenson)およびDAKO)。
【0136】
種々の処置に付随した免疫表現型変化について説明するために、前記パネルの大部分を用いて、処置および未処置の各々の患者の血液試料を分析し、これらは、同一血液試料の異なるアリコットについて別々に行った。未処置試料および他の対照処置を染色し、同時に分析した。
【0137】
フローサイトメトリー
白血球を染色し、製造者の指示に従って溶解させた。負の対照バックトラッキングを含んだサイマルテスト(simultest)またはPAINT A GATEソフトウエアー(BDSI)によりFACScan@でフローサイトメトリー分析を行った。10,000〜20,000個の事象が得られ、リストモードファイルに保存された。
【0138】
形態学
形態学は、顕微鏡およびライト染色を用いて分析した。
【0139】
B. 結果
CD19およびCD3パネル
血液試料をHLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体で処理すると、CD19+細胞の相対数が常に減少した。このマーカーは、panB−細胞抗原である(表参照)。この抗原は、成熟の全段階で、全てのヒトBリンパ球に存在するが、終末に分化した形質細胞には存在しない。従って、これは、B細胞が未分化細胞へ逆分化していたことを示す。
【0140】
同様の処理により、CD3+細胞の相対数が、劇的なことに、特に、B−CLL患者の血液中で増加し、常に、CD3−CD19−細胞における相対数の増加を伴った。CD3は、全ての成熟Tリンパ球および65%−85%の胸腺細胞に存在する。このマーカーは、常にα/βまたはガンマ/デルタT−細胞受容体(TCR)と関連して知られ、一緒にこれらの複合体は、細胞内部に対するシグナル変換において重要である。従って、これは、B細胞が未分化細胞に逆分化し、それから、T細胞と称する新しい分化細胞に拘束されていたことを示す。
【0141】
細胞の新しいクローンは、CD19およびCD3マーカー、すなわち、CD19+およびCD3+細胞を共同発現しているB−CLL患者の処理血液において、現れた(チャート1.処理開始後2時間、6時間および24時間の患者2、3および4参照)。他の病状の患者は、細胞のこれらのクローンの相対数の増加を示した。これらの細胞は、非常に大きく、非常に顆粒状であり、極度に高いレベルのCD19が、それらの細胞膜に発現した。CD3マーカーは、正常な成熟リンパ球に発現するのと同様なレベルにて、これらの細胞で発現されるようである。
【0142】
表2において、患者番号2、3および4は、実際には、同じ患者を表す番号であり、それらの説明は、単に、経時的な血液処理の効果を示すものであった(この患者の実験条件および臨床条件については表1を参照)。
【0143】
処理試料におけるCD19+CD3+クローンは、経時的に減少し、初期レベルが2時間目、6時間目および24時間目の未処理試料において測定されたレベルに達する。
【0144】
同じサイズおよび粒状の別のタイプの細胞が処理試料において検出され、これらの細胞は、それらの表面で発現した高レベルのCD19を有するが、CD3マーカーに対して陰性であり、FC受容体において豊富であった。しかしながら、これらの細胞の相対数は、時間に伴って減少することが明らかになった。興味深いことに、血液試料(2、3および4)の処理の24時間目に、初めに血液試料の処理の2および6時間後に増加することが観察された細胞群において、CD19−CD3−細胞の相対数が減少した。しかしながら、WBC群のコールターカウントは、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体による血液の処理により減少した。この発見は、このタイプの処理により、コールターによって検出できないが(表1)、表面マーカーに基づいて細胞をカウントするフローサイトメトリーによって測定した場合にサイズおよび粒状について説明され得る非定形細胞が生じることを示す。さらに、これらの非定形細胞は、顕微鏡下でライト染色を用いて形態を分析することにより説明される。これらの現象のフローサイトメトリーチャートは、チャート(1、2、3および4)に示され、血液試料の処理により観察された免疫発現型変化は、CD19+およびCD3+リンパ球が相互に連絡された細胞群であるが、幹細胞と比較したCD19およびCD3相対発現に基づいて別個のままであることを示唆すると思われる。
【0145】
表2において、患者番号5および6は、同じ患者を示すが、処理および未処理の血液試料の分析は、同時に経時的にモニターされた(表1参照)。
【0146】
B−細胞悪性腫瘍を有さない患者血液は、B−CLL患者の血液と比較すると、類似した傾向の免疫発現型変化を示したが、その変化は、同程度ではなかった。しかしながら、これらの患者の血液中のB−リンパ球およびMHCクラスII陽性細胞の相対的および絶対的な数は、B−CLL患者の血液中でみられた数と比較して、極度に低い。
【0147】
どちらもB細胞欠損であるX−結合乳児低ガンマグロブリン血症である2人の兄弟は、彼らの血液の処理によりCD3+細胞の相対数において異なる免疫発現型変化を示した。2カ月齢であり、病状が重くなかった弟は、血液の処理によりCD3+の相対数のわずかな増加を示し、それはCD3−CD19−細胞の相対数の減少を伴った。他方、2才で重症であり、比較的高い数のDR抗原を発現している活性T細胞を有する兄は、血液の処理によりCD3+細胞の数の減少を示した。これら二人の患者から得られた血液試料は、極度に少量だったので、他のマーカーを使用して引き起される可能性のある他の表現型変化を測定するようなことはしなかった(表2.ID43/BDおよび04/BD)。
【0148】
表2において患者91は、血液の処理後のCD3+細胞の相対数の減少を示し、それは、CD3−CD19−細胞の相対数の増加を伴った。しかしながら、CD4およびCD8のような他の表面マーカーの分析において(表3参照)、患者は、血液中にCD4+CD8+細胞の高い相対数を有することが観察され、これは、DR抗原のβ−鎖に対するモノクローナル抗体で血液試料を処理する前に示され、これらの二重陽性細胞は、血液処理後、かなり減少した。さらに、さらなるマーカーを分析すると、CD3+細胞の相対数が上昇したことが示された(表4参照)。
【0149】
健康な供血者から得られたB−リンパ球の豊富な他のは、DR抗原のβ−鎖に対するモノクローナル抗体で処理すると、CD3+細胞の相対数の劇的な増加を示し、それは、常にCD19+細胞の相対数の減少およびCD19−CD3−細胞の相対数の増加を伴った。CD4およびCD8のようなマーカーを使用するさらなる分析は、これらのマーカーの相対数の付随した増加を示す。しかしながら、同一供血者のTリンパ球の豊富な調製物は、同一のモノクローナル抗体で処理した場合、同様の変化を示さなかった。
【0150】
CD4およびCD8パネル
CD4抗原は、ヒト免疫不全ウイルスに対する受容体である。CD4分子は、クラスI抗原上のCD8結合部位と同様な領域であるB2ドメインにおいてMHCクラスII抗原を結合する。CD4のクラスII抗原への結合は、抗原へのT細胞応答を増強し、それゆえCD8のクラスI抗原への結合を行う。CD8抗原は、ヒトサプレッサー/細胞障害性Tリンパ球サブセット上ならびにナチュラルキラー(NK)リンパ球のサブセットおよび大部分の正常胸腺細胞上に存在する。CD4およびCD8抗原は、胸腺細胞で共同発現し、これらの細胞は、それらがT−リンパ球へと成熟するにつれて、どちらのマーカーも失う。
【0151】
CD4およびCD8マーカーの分析(下記参照)にて、および表2に示された血液試料の大部分から、B−リンパ球の未分化細胞への逆分化プロセスの存在およびT−リンパ球へのその後の分化を支持する染色パターンが明らかになる。
【0152】
二重陽性細胞であるCD4+CD8+細胞は、常に、β−鎖の相同領域に対するモノクローナル抗体での血液試料の処理に従うことが明白であり、これらのタイプの細胞は、B−CLL患者の処理試料の血液において著しく増加し、それは、未処理試料において全く見られなかった(表3およびチャート1、2、3および4参照)。同様の試料において、CD8+およびCD4+細胞のような単陽性細胞の相対数もまた、同時に増加することが示された。さらに、少なくともB−CLLの場合においてB細胞に相当するCD4−CD8−細胞の相対数の減少は、経時的に測定すると、同一レベルのままであった未処理試料と比較して、処理試料では劇的に減少することを示した。しかしながら、処理試料中におけるCD4+CD8+細胞の相対数の経時的な測定は、二重陽性細胞の相対数の減少に付随して単陽性細胞数の増加が起こることを示した。このタイプの免疫発現型変化は、胸腺におけるT−リンパ球系譜の前駆細胞の胸腺発達を特徴とする(患者番号2、3、および4)。CD4抗原は、ヘルパー/インデューサーT−リンパ球サブセット(CD4+CD3+)および正常な胸腺細胞の大部分に存在する。しかしながら、この抗原は、単球の細胞表面上ならびに単球およびマクロファージの細胞質に低密度で存在する(CD3−CD4−)。
【0153】
CD4+低細胞の相対数は、処理後の種々の血液試料中で、異なって影響された。このタイプの細胞の相対数は、未処理試料と比較すると、処理後のB−CLL患者の血液試料中で影響されなかったようである。そのような低レベルのCD4発現は、単球および非常に初期の胸腺細胞で見られる。
【0154】
処理後のHIV+25患者は、CD4およびCD8を同時に発現している二重陽性細胞の数の実質的な増加を示した。一方、処理後の患者91は、細胞のこのサブタイプの減少を示し、そのような現象の観察は、時間に依存する。CD8+細胞の相対数は、経時的に測定すると、B−CLL患者の未処理血液試料において増加することが観察されるが、CD4+およびCD4+低細胞の相対数は、同時に減少することが観察された(表3 患者2、3および4)。
【0155】
DRおよびCD3パネル
DRマーカーは、単球、樹状細胞、B−細胞および活性T−リンパ球に存在する。
【0156】
このパネルで分析された処理および未処理試料は、血液試料をCD19およびCD3マーカーで分析した場合に得られたものと同様な免疫発現型変化を示し(表2参照)、これらの抗原は、前記したように、各々、panBおよびT−細胞マーカーである。
【0157】
血液のモノクローナル抗体での処理は、DR+B−リンパ球の相対数に影響し、その結果、DR+細胞のレベルが減少するようである。対照的に、CD3+(T−細胞)細胞の相対数は、有意に増加する(表4およびチャート参照)。さらに、活性T細胞の相対数は、B−CLL患者の処理血液試料の大部分において増加し、これらのタイプの細胞は、他の病状の患者の処理試料において不定に影響された。さらに、DR高陽性細胞の相対数は、B−CLL患者および、血液中のDR+CD34+芽球が増加した6日齢の乳児の処理血液試料における有意な数において明白となった。しかしながら、この患者の血液中に存在した芽球は、処理前および処理後にTおよびB−細胞マーカーに対して陰性であったが、処理後の骨髄系譜抗原に対してより陽性になったことに注意すべきである。CD3−DR−細胞の相対数は、処理血液試料の大部分において増加し、CD3+細胞(T−細胞)の相対数の増加に比例し、DR+細胞(B−細胞)の相対数の減少に反比例した。
【0158】
CD56&16およびCD3パネル
CD56&CD16マーカーは、細胞の不均一群、一般に大きな粒状リンパ球として知られるリンパ球のサブセット、およびナチュラルキラー(NK)リンパ球に見られる。CD16抗原は、実質的に全ての休止NKリンパ球に発現し、ある個体由来のいくつかのCD3+Tリンパ球に若干発現する。この抗原は、顆粒球に少量見られ、大きなアズール親和性顆粒を含有するリンパ球と結合する。CD16抗原は、IgG FC受容体IIIである。
【0159】
可変数のCD16+リンパ球は、CD57抗原もしくは低密度CD8抗原のいずれかか、または両方を共同発現する。ほとんどの個体では、実質的に、CD5、CD4またはCD3抗原のような他のT−リンパ球抗原とオーバーラップしない。CD56抗原は、本質的に全ての休止および活性CD16+NKリンパ球に存在し、細胞のこれらのサブセットは、非主要組織適合遺伝子複合体制限細胞障害性を行う。
【0160】
B−CLLおよびその他の病状の患者の処理および未処理血液試料の免疫発現は、非常に顆粒状の中くらいのサイズのCD56&CD16を共同発現している細胞の相対数の増加を示した(表5およびチャート1、2、3および4を参照)。これらの観察はまた、CD3抗原のみを発現する(CD56およびCD16マーカーの発現は無い)細胞、およびCD56&CD16およびCD3マーカーを一緒に共同発現する細胞の相対数の著しい増加を伴った。
【0161】
表5において、患者番号2、3および4は、同一の血液試料を表すが、それぞれ2時間、6時間および24時間(処理前および処理後)で分析している。この試料は、DR抗原のβ−鎖の相同領域に対するモノクローナル抗体による血液の処理により、CD56+およびCD16+細胞、CD3+細胞およびCD56+およびCD16+CD3+細胞の自発的生成を引き起こすようであり、これらの観察は、常にB−細胞マーカー(CD19、DR、CD56、CD16−CD3−)の消失を伴った。
【0162】
処理前および処理後のこの血液試料の前進的な分析は、CD56+およびCD16+細胞のレベルが経時的に減少すること、CD3+細胞のレベルが経時的に増加することを示した。
【0163】
B−CLL患者7の血液試料は、処理および未処理試料において観察された免疫発現型変化と比較して、CD56、CD16およびCD3抗原を発現する細胞数のいかなる変化も示さず、これは、モノクローナル抗体の添加量がBリンパ球の数に比べて極度に少なかったためである。しかしながら、適量のモノクローナル抗体による別の場合のこの患者の血液試料の処理は、CD3+、CD56+&CD16+およびCD56+およびCD16+CD3+細胞の相対数の有意な増加を示した。
【0164】
他の病状の他の患者の血液試料は、これらの細胞のレベルの不定な変化を示し、これは、処理前、処理期間の血液中に存在するB−リンパ球の数およびおそらく患者の臨床条件に依存すると思われる。
【0165】
CD45およびCD14パネル
CD45抗原は、末梢血、胸腺、脾臓および扁桃中のリンパ球、単球、多形核細胞、好酸球および好塩基球、ならびに骨髄中の白血球前駆体を含む全てのヒト白血球に存在する。
【0166】
CD14は、70%ないし93%の正常な末梢血単球、77%ないし90%の胸膜液または腹膜液食細胞に存在する。この抗原は、顆粒球に若干発現し、非刺激リンパ球、マイトジェン−活性Tリンパ球、赤血球、または血小板には存在しない。
【0167】
CD45抗原は、タンパク質チロシンホスファターゼのファミリーを表し、この分子は、外部刺激(抗原)と相互作用し、細胞増殖および分化の調節へ導くScr−ファミリーメンバーを経てシグナル変換に影響する。
【0168】
特にB−CLL患者から得られた処理血液試料におけるDR抗原のβ−鎖の密着は、そのような処理がB−リンパ球でのCD45抗原のレベルに影響することを示唆する。DR抗原のβ−鎖の刺激により生じた全免疫発現型変化は、CD45およびCD14発現レベルならびに前方分散および側面分散によって測定されたような形態(それぞれサイズおよび粒状)に基づいて分離され得る種々のタイプの細胞を生じさせるようであり、これらの結果は、表6およびチャート(1、2、3、4および5)に示す。
【0169】
処理によりCD45低細胞の相対数は(未処理試料と比較して)、有意に増加し、それゆえCD45およびCD14抗原を共同発現する細胞の相対数は増加した。このタイプの免疫発現型変化は、CD45高細胞の相対数(未処理試料と比較して)の減少と同時に起こった。しかしながら、この後者の細胞群は、形態およびCD45発現度に基づいて、さらに分割することができる。一のタイプは、極度に大きく、チャートにある残りの細胞と比較して極度に高いレベルのCD45抗原を有した(チャート1、2、3および4参照)。経時的な処理後のこのパネル分析(表 患者2、3および4ならびにチャート参照)により、CD45+細胞の相対数は、始めに経時的に急激に減少し、CD45低細胞を生じた。しかしながら、24時間後の血液の分析は、反対の状態を示した。
【0170】
試料5および7は、他のB−CLL患者から得られた他の試料で得られた変化とは反対の免疫発現型変化を表し、これは、この試料がモノクローナル抗体との非常に短いインキュベーション時間で分析したからである。実際、処理後の血液試料の一連の分析は、Bリンパ球によって調節された免疫発現型変化が発達の段階を表すので時間依存性であること、時間Xで測定された免疫発現型変化は、時間Xプラスでは同じでないだろうということ(それは一度引き起こされると固定されない)を示唆すると思われる。しかしながら、これらのタイプの変化は、体内でのより厳しい条件で起こっているにちがいなく、さもなければ免疫病理学が、結果として起こるであろう。B−細胞悪性腫瘍を有しない他の患者由来の血液試料の処理の影響は、細胞の免疫発現型の不定な変化を示し、これは、B−リンパ球がより少量で存在するからである。しかしながら、健康な供血者から得られたB−リンパ球の豊富なフラクションの処理は、高いB−リンパ球数を有するB−CLLで得られたのと同様な免疫発現型変化を示す。
【0171】
CD8およびCD3パネル
CD8抗原決定因子は、クラスIMHC分子と相互作用し、その結果、CD8+Tリンパ球とその標的細胞との間の付着を増加させる。このタイプの相互作用は、休止リンパ球の活性化を増強する。CD8抗原は、タンパク質チロシンキナーゼ(p56ick)と結合し、次に、CD8/p56ick複合体がT−リンパ球活性化における役割を果す。
【0172】
B−CLL患者から得られた血液試料の、B鎖に対するモノクローナル抗体での処理は、CD3CD8およびCD3(CD4CD3に非常によく似ている)陽性細胞の相対数の有意な増加を引き起こし、したがって、より明らかに、始めに生じた二重陽性細胞が成熟T−リンパ球に発達していることを示す。これは、直接的にCD19によっておよびDRによって、間接的にCD8−CD3−抗原によって測定することができるプロセスである。同一患者の処理血液試料の経時的な連続的な評価は、胸腺細胞発達と同一であるプロセスと一致すると思われる(表7、患者2、3および4、ならびにチャート1)。
【0173】
CD8+細胞の相対数は、処理および未処理試料において経時的に増加したが、程度がより大きかったのは、未処理試料においてであった。一方、CD8+CD3+細胞の相対数は、未処理試料において経時的に減少した。しかしながら、CD3+細胞の相対数は、経時的に測定すると、処理血液試料において増加し、これらのタイプの細胞は、CD4+CD3+単陽性細胞:胸腺細胞の成熟型に大いに一致した。さらに、これらの試料は、他のパネルでも免疫発現されたので(表3、4、5および6中に前記した)、その全変化は、極度にTリンパ球前駆体および後継体の生成においてB細胞に原因があることを示す。
【0174】
別々のアリコットでB−CLL患者由来の血液試料(番号2、3および4、表1、2、3、4、5、6、7)は、処理しないか、DR抗原のβ−鎖の相同領域に対するPEコンジュゲートモノクローナル抗体および同モノクローナル抗体の非コンジュゲート形態で処理した。PEコンジュゲートの比較によると、処理は、明らかに、CD3陽性細胞、および同血液試料を抗体の非コンジュゲート形態で処理した場合に有意なレベルで観察されたCD4のような関連マーカーの相対数の変化を示す。しかしながら、表面で発現されているDR抗原を有しないCD45陽性細胞数の増加は、経時的に測定した場合に示された(表8を参照)。その発見は、経時的に免疫発現した場合に未処理試料において示されたのと同様であった(表6)。さらに、CD45低発現細胞の相対数は、経時的に減少し、その現象は、同一患者の未処理試料においても示された(経時的に測定した場合)(チャート1Aを参照)。
【0175】
C. 異なる特性を有する他のモノクローナル抗体のT−リンパ球生成に対する影響の比較
CD19およびCD3パネル
DR抗原のα−鎖の相同領域およびMHCクラスI抗原の相同領域に対するモノクローナル抗体による血液試料の処理により、CD3+細胞の数が減少し、CD19+細胞の数が増加した。DR抗原のβ−鎖の相同領域に対するモノクローナル抗体による血液試料の処理により、CD19+細胞の数が減少し、CD3+細胞の数が増加した。シクロホスファミドとともに後者のモノローナル抗体で処理すると、同様な効果が現れた(表14 B−CLL患者5/6、2時間処理)。
【0176】
同じ試料におけるCD19+およびCD3+細胞の前進的な分析により、DR抗原のβ−鎖の相同領域に対するモノクローナル抗体で処理した血液中においてのみ、CD3+の相対数の増加がさらに明らかになった(表14 患者5/6、処理後24時間)。しかしながら、シクロホスファミドおよびDR抗原のβ−鎖に対するモノクローナル抗体で処理した血液試料の前進的な分析(24時間後の患者5/6 表14)は、正確に同じ条件下で、2時間のインキュベーション時間で観察されたものと比較して、CD19+およびCD3+細胞の相対数において逆転を示す。
【0177】
一般に、同一患者の血液試料の、DR抗原のα−鎖の相同領域に対するモノクローナル抗体またはクラスI抗原のα−鎖の相同領域に対するモノクローナル抗体での処理は、未処理試料と比較して、CD19−細胞(panBマーカー)の相対数の増加を示す。CD19−CD3−細胞の相対数は、DR抗原のα−鎖に対するモノクローナル抗体で処理したか、またはクラスI抗原に対するモノクローナル抗体で処理した血液試料中で、わずかに減少した(表14およびチャート2、3、および4を参照)。クラスI抗原に対するモノクローナル抗体での、患者09の血液試料の処理により、CD3+細胞の相対数が増加し、CD19+およびCD19−CD3−細胞の相対数がわずかに減少した。しかしながら、健康な供血者から得られたB−リンパ球の豊富な調製物の、DR抗原のβ−鎖またはα−鎖に対するモノクローナル抗体での処理により、B−CLL患者で得られたのと同様な免疫発現型変化が示された。
【0178】
HIV+およびIgA欠損患者の、DR抗原のβ−鎖に対するモノクローナル抗体での処理により、CD3+細胞の相対数が増加し、CD19+細胞の相対数が減少した。しかしながら、同一血液試料の、クラスI抗原の相同領域に対するモノクローナル抗体による処理は、同様の効果を生じなかった。B−細胞欠損患者(34/BDおよび04/BD)から得られた血液試料の処理により、DR抗原のβ−鎖、クラスI抗原およびCD4抗原に対するモノクローナル抗体で処理した場合、不定な免疫発現型変化が示された。
【0179】
CD4およびCD8パネル
CD19およびCD3パネル(表14)を用いて分析された血液試料も、また、CD4およびCD8パネル(表15)で免疫発現した。どちらのパネルも、相互に一致し、確定すると思われる。B−CLL患者(表15、患者5/6および10、チャート2、3および4)の血液試料をDR抗原のβ−鎖の相同領域に対するモノクローナル抗体と一緒に、またはこのモノクローナル抗体およびシクロホスファミドと一緒に2時間インキュベートすると、CD8+およびCD4+細胞ならびに両方のマーカーを共同発現している細胞の相対数が増加した。一方、同一試料の、DR抗原のα−鎖の相同領域またはクラスI抗原のα−鎖の相同領域に対するモノクローナル抗体による処理は、同様の効果を生じなかった。
【0180】
DR抗原のβ−鎖に対するモノクローナル抗体およびシクロホスファミドで2時間および24時間インキュベートして得られた免疫発現型の傾向を比較すると、CD4およびCD8陽性細胞の相対数における逆の変化が明らかになり(表15、2時間および24時間のB−CLL患者5/6)、かかる変化は、同一血液試料がCD19およびCD3パネル(表14同患者)で分析された場合に得られたのと一致した。後者の発見は、未分化細胞がT−リンパ球またはB−リンパ球に分化することができる場合、その後の分化が可逆的であることを示す。
【0181】
DRおよびCD3パネル
DRおよびCD3(表16)パネルで得られた免疫発現型変化は、2時間分析で、DR抗原のベータ−もしくはアルファ−側の相同領域に対するモノクローナル抗体、またはクラスI抗原に対するモノクローナル抗体、またはDR抗原のβ−鎖に対するモノクローナル抗体およびシクロホスファミドによる同一血液試料の処理の後の、CD19およびCD3パネル、ならびにCD4およびCD8パネル(表14および15、ならびにチャート2、3および4)で得られた発見を確認する。
【0182】
結果から、DR抗原のβ−鎖の相同領域に対するモノクローナル抗体は、非常にDR+細胞からのCD3陽性細胞の生産を促す能力を有することが明らかであろう。
【0183】
さらに、DR抗原のα−鎖の密着またはシクロホスファミドと共にその分子のβ−側の密着(インキュベーション時間を長くした)を含むような処理は、CD19+細胞またはDR+細胞の相対数の増加を促進した。
【0184】
CD56&16およびCD3パネル
血液試料、特に高いB−リンパ球数を有するB−CLL患者の血液試料の、DR抗原のβ−鎖の相同領域に対するモノクローナル抗体での処理により、CD56&16陽性細胞の相対数が増加した。
【0185】
これらの患者において、CD3+およびCD56+ならびにCD16+CD3+細胞の相対数もまた、β−鎖に対するモノクローナル抗体での血液試料の処理の後に増加し、これは、同一血液試料をCD3およびCD19ならびにDRおよびCD3パネルで分析した場合に同一試料で示された先の観察結果を確かなものとした。
【0186】
CD45およびCD14パネル
DR抗原のβ−またはアルファ−鎖に対するモノクローナル抗体または、β−鎖に対するモノクローナル抗体およびシクロホスファミド、またはクラスI抗原に対するモノクローナル抗体で処理した血液試料もまた、CD45およびCD14パネルで分析した(表18)。CD45低、CD45高およびCD45中の描写は、恣意的である。血液試料5/6の、DR抗原のβ−鎖に対するモノクローナル抗体、またはこのモノクローナル抗体およびシクロホスファミドでの処理(2時間)は、CD45+低細胞を生じ、CD45+中細胞の相対数を増加させた。しかしながら、前者の処理は、CD45+高細胞の相対数を増加させ、後者の処理は、CD45+中細胞の相対数を減少させ、これらの変化は、時間に依存することが明らかになった。
【0187】
クラスI抗原に対するモノクローナル抗体で処理した、患者5/6および10(B−CLL)の血液試料は、CD45+中細胞の相対数の減少を示し、未処理試料と比較して、同一処理後の血液試料09およびHIV+において同様の観察結果が示された。HIV+およびIgA/D患者の血液試料をクラスI抗原に対するモノクローナル抗体で処理すると、未処理試料またはDR抗原のβ−鎖に対するモノクローナル抗体で処理した試料と比較して、CD45+低細胞の相対数が増加した。しかしながら、これらの患者の血液試料は、DR抗原のβ−鎖の相同領域に対するモノクローナル抗体での処理によるCD45+中細胞の相対数の減少を示した。中CD45+細胞は、クラスI抗原に対するモノクローナル抗体処理後のIgA/D患者の血液試料中で増加した。極度に大きく、非常顆粒状であり、CD45抗原を高レベルで発現している細胞は、MHCクラスII抗原のDR抗原のβ−鎖の相同領域に対するモノクローナル抗体で処理した血液試料において示された(チャート1、2、3、4および5参照)。
【0188】
D8およびCD28パネル
CD28抗原は、およそ60%ないし80%の末梢血T(CD3+)リンパ球、50%のCD8+Tリンパ球および5%の未熟CD3−胸腺細胞に存在する。胸腺細胞成熟の間、CD28抗原発現は、ほとんどのCD4+CD8+未熟胸腺細胞での低密度から事実上全ての成熟CD3+、CD4+またはCD8+胸腺細胞での高密度へ増加する。細胞活性化は、さらにCD28抗原密度を増加させる。CD28の発現もまた、CD8+リンパ球を二つの機能群に分割する。CD8+CD28+リンパ球は、同種抗原−特異的細胞毒性を媒介し、それは、主要組織適合遺伝子複合体(MHC)クラスIに制限されている。細胞増殖の抑制は、CD8+CD28+サブセットによって媒介される。CD28抗原は、細胞付着分子であり、活性Bリンパ球に存在するB7/BB−1抗原のリガンドとして機能する。
【0189】
B−CLL患者(表19、患者5/6および8)の血液試料をDR抗原のβ−鎖の相同領域に対するモノクローナル抗体で処理すると、CD8+、CD28+およびCD8+CD28+細胞の相対数が増加し、全てのその他のタイプの処理では、増加しなかった。
【0190】
CD34およびCD2パネル
CD34抗原は、未熟造血前駆細胞および骨髄中の全ての造血コロニー形成細胞に存在し、それは、単分化能の(CFU−GM,BFU−E)および多能性の前駆体(CFU−GEMM,CFU−MixおよびCFU−blast)を含む。CD34もまた、間質細胞前駆体で発現する。正常な骨における末端デオキシヌクレオチジルトランスフェラーゼ(TdT)+B−およびT−リンパ系前駆体は、CD34−である。CD34抗原は、CD33抗原を発現するがCD14およびCD15抗原を欠損する初期の骨髄細胞、およびCD71抗原を発現し、かすかにCD45抗原を発現する初期の赤血球細胞に存在する。CD34抗原もまた、毛細管内皮細胞およびおよそ1%のヒト胸腺細胞で見られる。正常な末梢血リンパ球、単球、顆粒球および血小板は、CD34抗原を発現しない。CD34抗原密度は、初期の造血前駆細胞で最も高く、該細胞の成熟につれて減少する。該抗原は、完全に分化した造血細胞には存在しない。
【0191】
拘束されていないCD34+前駆細胞は、CD38−、DR−であり、CD71、CD33、CD10およびCD5のような系譜−特異的抗原を欠損しており、一方、系譜−拘束されたCD34+細胞は、高密度でCD38抗原を発現する。
【0192】
ほとんどのCD34−細胞は、相互に、CD45ROまたはCD45RA抗原のいずれかを発現する。およそ60%の急性B−リンパ性白血病および急性骨髄性白血病は、CD34抗原を発現する。該抗原は、慢性リンパ性白血病(BまたはT系譜)またはリンパ腫では発現されない。CD2抗原は、Tリンパ球およびナチュラルキラーリンパ球(NK)のサブセットに存在する。
【0193】
結果は、チャート2、3および5に示す。
【0194】
DR抗原のβ−鎖または同抗原のα−鎖に対するモノクローナル抗体での処理後のB−CLL患者の血液試料の分析(表20、患者5/6、2時間)により、前者の抗体での処理後、CD34+およびCD34−CD2+細胞の相対数の著しい増加が明らかになった。同血液試料は、他のマーカーに対して、前記したパネル(表14ないし19を参照)で免疫発現されたので、ここで観察されたCD34+およびCD34+CD2−細胞の相対数の増加は、CD4+CD8+、CD8+CD3+およびCD4+CD3+単陽性(SP)細胞の相対数の増加と同時に起こると思われる。さらに、唯一、HLA−DR抗原のβ−鎖の密着であろうと思われるこれらの発見は、該プロセスがBリンパ球退行を経てT−リンパ球を生じていることを直接支持する。
【0195】
24時間後の同様の処理の分析により、CD34+細胞は、レベルが減少して、Tリンパ球の相対数がさらに増加すると思われた。始めにT−リンパ球生成を引き起こした逆分化のプロセスは、逆転して、B−リンパ球生成を引き起こすことができる。前者の現象は、HLA−DR抗原のβ−鎖に対するモノクローナル抗体およびシクロホスファミドと一緒の2時間インキュベーション時間で観察され、一方、後者のプロセスは、同一試料を同一処理で24時間インキュベーション時間で示された(チャート2)。
【0196】
HIV+患者(表20患者HIV+)の血液試料をHLA−DR抗原のβ−鎖に対するモノクローナル抗体で処理すると、CD34+およびCD2+CD34+細胞の相対数が著しく増加し、同様に、同一血液試料をHLA−DR抗原のβ−鎖に対するモノクローナル抗体および同抗原のα−鎖に対するモノクローナル抗体で処理した。しかしながら、この血液試料の、HLA−DR抗原のα−鎖に対するモノクローナル抗体での処理は、CD34+細胞のレベルに影響しなかった。そのとき白血病について調べられ、血液中に非常に高い不定形細胞(芽球)数を有した6日齢の乳児から得られた血液試料(BB/ST表20)を、HLA−DR抗原のβ−鎖に対するモノクローナル抗体、または同抗原のα−鎖に対するモノクローナル抗体、または一緒に添加した両モノクローナル抗体で処理すると、その結果、以下の免疫発現型変化が起こった。
【0197】
未処理血液試料の分析により、CD34+およびDR+細胞の相対数は、著しく増加し、β−鎖に対するモノクローナル抗体での処理により、CD34+細胞の相対数は、さらに増加したが、HLA−DR抗原のα−鎖に対するモノクローナル抗体での処理、または一緒に添加した場合の該分子のαおよびβ−鎖に対するモノクローナル抗体での処理により、減少が示された。しかしながら、後者の処理により、CD34+CD2+細胞の相対数が増加し、同一血液試料をHLA−DR抗原のβ−鎖に対するモノクローナル抗体のみで処理した場合にその反対のことが起こった。同一患者の24時間後の処理および未処理血液アリコットの分析で、CD34+の相対数は、HLA−DR抗原のβ−鎖に対するモノクローナル抗体での処理で非常に高いレベルで維持されたことを除いて、前記した全ての処理で減少した。後者の処理で、24時間後、CD34+CD2+細胞の相対数が減少し続けた。
【0198】
これらの結果は、β−鎖を経たHLA−DR抗原の密着が、CD2+CD34+プールから、またはB−CLL患者のB−リンパ球のようなより成熟したタイプの細胞からのCD34+細胞の生産を促進することを示し、これらの結果は、このタイプの処理が逆分化を促進することを示す。しかしながら、24時間後の血液試料の免疫発現は、これらのタイプの細胞が別の系譜全体において存在すると思われ、この場合、細胞がCD7およびCD13&33パネルで処理された血液試料の分析で観察された骨髄系譜に存在するか、またはむしろ、それら自身を該骨髄系譜に拘束すると思われることを示唆する。
【0199】
形態は、MHCクラスII抗原のβ−鎖の相同領域に対するモノクローナル抗体での処理で、B−CLLのB−リンパ球および健康な個体(CD19ビーズを用いる)の豊富なフラクションの免疫発現型特徴を変化させる。これらは、B−リンパ球の形態の変化を伴った。B−リンパ球は、未処理血液スミア中で、スライドガラスでのコロニー形成が、顆粒球、単球、多数の原始様細胞および有核赤血球によって代わられることが観察された。有糸分裂像または有意な細胞死は、処理または非処理血液スミア中で観察されなかった。
【0200】
表20の結果もまた、本発明方法によると、より成熟した未分化細胞の逆分化によって未分化細胞を調製することが可能であるというさらに重要な発見を証明する。
【0201】
D. 顕微鏡写真
前記したような抗原試験に加えて、本発明の方法を、顕微鏡を用いて視覚的に行った。
【0202】
この点において、図1は、本発明の方法以前の分化したB細胞の顕微鏡写真である。図2は、本発明の方法(ここでは、薬剤は、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体であった)に従って、B細胞の逆分化によって形成された未分化細胞の顕微鏡写真である。該未分化細胞は、暗く染色された細胞凝集群である。図3は、同未分化細胞の顕微鏡写真であるが、より低い倍率で拡大した。
【0203】
図1ないし3は、したがって、本発明の方法によるB細胞の未分化幹細胞への逆分化を視覚的に示す。
【0204】
図4は、本発明の方法以前の分化したB細胞の顕微鏡写真である。図5は、本発明に従って(ここで使用した薬剤は、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体であった)、B細胞の逆分化によって形成された未分化細胞の顕微鏡写真である。また、未分化細胞は、暗く染色された細胞凝集群である。図6は、図5の同未分化細胞から分化した顆粒球の形成の顕微鏡写真である。
【0205】
従って、図4ないし6は、本発明の方法によって、B細胞が未分化幹細胞へ逆分化し、次いで、その未分化細胞が、最初の分化細胞とは異なる系譜である新たな分化細胞に拘束されることを視覚的に証明する。
【0206】
本発明の方法によってT細胞が未分化幹細胞へ逆分化し、次いで、その未分化細胞が、最初の分化細胞とは異なる系譜である新たな分化細胞に拘束されることもまた、顕微鏡によって追跡された。
【0207】
E. 概要
要するに、該実施例は、幹細胞の発生において利用され得るTおよびBリンパ球の発生および発達に関係する非常に興味深い発見を明らかにし、およそ数時間で末梢血試料中でのリンパ造血に影響する、in vitroでの実験を記述する。
【0208】
高Bリンパ球数を伴うB−細胞慢性リンパ性白血病(B−CLL)患者から得られた末梢血試料の、クラス−II抗原のβ−鎖の相同領域に対するモノクローナル抗体での処理は、単陽性(SP)T−リンパ球および、同時に共同発現される胸腺細胞マーカーCD4およびCD8抗原に対して二重陽性であるその前駆体の相対数の著しい増加を引き起こした。しかしながら、これらの現象は、常に、B−リンパ球の相対数の有意な減少を伴った。これらの観察は、同一血液試料をクラス−II抗原のα−鎖の相同領域またはクラス−I抗原の相同領域に対するモノクローナル抗体で処理した場合には示されなかった。
【0209】
B−細胞慢性リンパ性白血病(CLL)患者から得られた全血の、HLA−DR抗原のB鎖の相同領域に対するモノクローナル抗体での処理は、T−リンパ球生成を引き起こすと思われた。この事象は、CD4およびCD8マーカーを共同発現している二重陽性細胞の出現、CD34を発現している細胞の出現およびそれに付随する単陽性CD4+CD3+およびCD8+CD3+リンパ球の相対数の増加によってマークされた。さらに、かかる細胞の発生において生じた免疫発現型変化は、特に経時的に測定した場合に、胸腺細胞発達について例証されたものと一致した。
【0210】
全血をDR抗原のβ−鎖の相同領域に対するモノクローナル抗体と一緒に2時間インキュベートして生じた二重陽性細胞(DP)のパーセントは、経時的に減少し、これらの事象は、同時に、および遅れても、単陽性CD4+CD3+およびCD8+CD3細胞のパーセントの増加を伴った。TCRαおよびβ鎖もまた、これらのタイプの細胞で発現された。
【0211】
B−リンパ球は、常に、CD19、CD21、CD23、IgMおよびDRのようなマーカーを失うことが観察され、これは、CD34+およびCD34+CD2+細胞の出現、CD7+細胞の増加、CD8+CD28+およびCD28+細胞の増加、CD25+細胞の増加、CD10+およびCD34+細胞ならびにCD34+およびCD19+細胞の出現、CD5+細胞およびCD45抗原を低レベルで発現している細胞の増加と同時に起こった。これらの変化は、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体での血液の処理が原因であった。
【0212】
処理前のB−CLL患者の血液における白血球の大部分はBリンパ球であったので、かかる処理と関連した免疫発現型変化は、Bリンパ球の逆分化およびその後の拘束(すなわち、再拘束)と一致する。さらに、シクロホスファミドおよびHLA−DR抗原のβ−鎖に対するモノクローナル抗体での処理後にT−リンパ球になることを促されたB−CLL患者のB−リンパ球は、インキュベーションを延長してこの処理をした後、B−リンパ球に逆戻りすることができた。
【0213】
HLA−DR抗原のβ−鎖に対するモノクローナル抗体で処理した試料の、CD16&56およびCD3およびCD8およびCD3パネルでの分析で、これらのマーカーを発現している細胞の相対数は、CD19およびCD3およびDRおよびCD3のようなパネルで測定したものと一致した増加において、着々と増加する。比濁分析および免疫電気泳動を用いたHIV感染患者の処理および未処理試料の上清の研究により、IgGレベルの増加が明らかになり、それは、B−細胞が形質細胞段階を経たにちがいないことを示す。前記した全ての細胞の相対数の増加もまた、CD56&16抗原を非常に多数発現している中サイズの非常に顆粒状の細胞の出現を伴った。極度に大きく、非常に顆粒状の他の細胞が一時的に観察され、これらは、CD34に対して陽性であり、CD4CD8マーカーに対して二重陽性であった。他の一過性の細胞もまた観察され、これらは、大きくかつ顆粒状であり、CD3およびCD19受容体に対して陽性であった。B−リンパ球の大部分に存在するCD25は、失われ、常に数の増加が観察された新たに形成されたT−リンパ球によって発現されるようになった。
【0214】
CD28+CD8+およびCD28+細胞は、B−CLL患者の全血の、DR抗原のB鎖の相同領域に対するモノクローナル抗体での処理後に現れた。これらの発見は、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体での血液の処理に起因した。
【0215】
この方法で生じたT−リンパ球生成は、健康な供血者の末梢血、臍帯血、骨髄、HIV+患者およびAIDS患者を包含する様々な感染患者、健康な供血者の血液試料から得られたBリンパ球の豊富なフラクション、IgA欠損患者および様々な他の病状の他の患者においても観察された。さらに、HLA−DR抗原のβ−鎖の相同領域に対するモノクローナル抗体で処理したB−CLL患者二人の試料における骨髄マーカーの分析は、CD13およびCD33のような骨髄マーカーを発現している細胞の相対数の有意な増加を示した。これらのマーカーは、CD56&16またはCD7抗原と共同発現された。しかしながら、T−リンパ球マーカーを有しており、骨髄抗原を有していないCD7+細胞の相対数は、細胞の別の群で観察された。これらの個々の観察は、未処理試料において、またはクラスI抗原もしくはHLA−DR抗原のα−鎖の相同領域に対するモノクローナル抗体で処理した試料において、見られなかった(チャート2&3を参照)。これらの最終結果は、HLA−DR抗原のβ−鎖を経て一度引き起こされたB−リンパ球は、T−リンパ球前駆細胞に退行することができるだけでなく、骨髄系譜および赤血球系譜にも存在し得ることを示唆する。
【0216】
本発明の方法によって生産される幹細胞は、いずれの組織の幹細胞でもあってよく、必ずしもリンパ造血前駆細胞に限定されないことに注意すべきである。
【0217】
本発明の他の変形例は、当事者には明白であろう。
【0218】
【表1−1】
【表1−2】
【表1−3】
【表1−4】
【表2】
【表3】
【表4】
【表5】
【表6】
【表7】
【表8】
【表9】
【表10】
【表11】
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【表18】
【表19】
【表20】
【表21】
【表22】
【表23−1】
【表23−2】
【表24】
【表25】
【特許請求の範囲】
【請求項1】
拘束された細胞を含む細胞群におけるCD34+細胞の相対数をイン・ビトロで増加させる方法であって、
(i)該細胞群を、該拘束された細胞に操作可能に密着する薬剤と接触させるステップであって、該密着は、前記拘束された細胞の表面上で、抗原の捕捉、認識または提供を媒介する受容体を密着することを含み、該受容体はMHCクラスI抗原またはMHCクラスII抗原であるステップと、
(ii)該薬剤と密着した拘束された細胞をインキュベートして、該密着の結果としてCD34+細胞の相対数を増加させるステップと、
を含む方法。
【請求項1】
拘束された細胞を含む細胞群におけるCD34+細胞の相対数をイン・ビトロで増加させる方法であって、
(i)該細胞群を、該拘束された細胞に操作可能に密着する薬剤と接触させるステップであって、該密着は、前記拘束された細胞の表面上で、抗原の捕捉、認識または提供を媒介する受容体を密着することを含み、該受容体はMHCクラスI抗原またはMHCクラスII抗原であるステップと、
(ii)該薬剤と密着した拘束された細胞をインキュベートして、該密着の結果としてCD34+細胞の相対数を増加させるステップと、
を含む方法。
【図1】


【図2】

【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2012−40011(P2012−40011A)
【公開日】平成24年3月1日(2012.3.1)
【国際特許分類】
【出願番号】特願2011−220926(P2011−220926)
【出願日】平成23年10月5日(2011.10.5)
【分割の表示】特願2008−330347(P2008−330347)の分割
【原出願日】平成8年1月31日(1996.1.31)
【出願人】(507237819)トライステム・トレイディング・(キプロス)・リミテッド (5)
【氏名又は名称原語表記】TriStem Trading (Cyprus) Limited
【Fターム(参考)】
【公開日】平成24年3月1日(2012.3.1)
【国際特許分類】
【出願日】平成23年10月5日(2011.10.5)
【分割の表示】特願2008−330347(P2008−330347)の分割
【原出願日】平成8年1月31日(1996.1.31)
【出願人】(507237819)トライステム・トレイディング・(キプロス)・リミテッド (5)
【氏名又は名称原語表記】TriStem Trading (Cyprus) Limited
【Fターム(参考)】
[ Back to top ]