
末梢動脈閉塞症治療剤
【課題】 式(I)で示されるデプシペプチド化合物、又はその製薬学的に許容される塩の具体的な疾患に対する治療の可能性の解明と新規治療薬の創製
【解決手段】 G蛋白質であるGq/11に対する阻害作用を有する式(I)で示される化合物、又はその製薬学的に許容される塩を有効成分として含有する末梢動脈閉塞症予防及び/又は治療剤、特に、安静時疼痛を軽減、潰瘍・壊死の進展を阻止、救肢率を向上させる重症末梢動脈閉塞症予防及び/又は治療剤
【解決手段】 G蛋白質であるGq/11に対する阻害作用を有する式(I)で示される化合物、又はその製薬学的に許容される塩を有効成分として含有する末梢動脈閉塞症予防及び/又は治療剤、特に、安静時疼痛を軽減、潰瘍・壊死の進展を阻止、救肢率を向上させる重症末梢動脈閉塞症予防及び/又は治療剤
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬、殊に末梢動脈閉塞症の予防及び/又は治療剤に関するものであり、特に、重症末梢動脈閉塞症の予防及び/又は治療剤、すなわち安静時疼痛、潰瘍・壊死を改善する予防及び/又は治療剤に係るものである。
【背景技術】
【0002】
末梢動脈閉塞症(peripheral arterial disease: PAD、以下PADと記す。)とは、四肢への主幹動脈に生じた動脈硬化性閉塞性病変により、主に下肢を中心として虚血症状を呈する疾患である(非特許文献1)。PADには、閉塞性動脈硬化症やBuerger病が知られている。
【0003】
PADの臨床症状・徴候についての進行度の目安としてFontain分類が知られており、症状の程度により、[I度:冷感・しびれ感、II度:間歇性跂行、III度:安静時疼痛、IV度:潰瘍・壊死]に分類されている。PADの中でも、III度又はIV度の症状を呈した場合には重症PADと呼ぶ。
PADは、50歳以上の喫煙男性に多く、近年、高齢化社会の到来に加えて、急速な食生活の欧米化や車社会化などによる生活様式の変化により疾病の傾向が大きく変化し、他の動脈硬化性血管疾患の急速な増加と同様にPADが増加している。
【0004】
PADは高血圧や糖尿病、高脂血症などを合併することもある。前述の通り、PADは、全身的な動脈硬化性変化の一分症でもあり、多くの例で虚血性心疾患や脳血管障害、腎機能障害などを生じる他臓器の閉塞性血管病変を有している。特に最近においては、動脈硬化の危険因子としてインスリン抵抗性の概念が導入され注目されているが、PADはこうした危険因子を有する代表的疾患でもある。特に日本における糖尿病患者の急速な増加は直接的に本症の増加につながっていると考えられており、例えば、糖尿病性腎症による透析例が増加することにより、重症虚血肢が増加し、本症の糖尿病併存頻度は35%以上と考えられている。
【0005】
PADの診断と治療について、『下肢閉塞性動脈硬化症の診断・治療指針』(Trans Atlantic Inter-Society Consensus(TASC))がガイドラインを示している(非特許文献2)。安静時疼痛や潰瘍・壊死に分類される重症患者は、PADの総患者数の約25%を占める。重症虚血肢において、肢虚血の重症度、閉塞性病変の存在部位や範囲、側副路様式や末梢血流の良否、併存疾患の重症度、肢機能の治療目標、生命予後などを考慮して治療方針が選択されるが、薬剤あるいは物理的方法によって血行の再開を図る治療が行なわれている。
【0006】
重症PADの治療剤としては、日本ではプロスタグランジンE1製剤(以下、PGE1製剤)やアルガトロバンが用いられている。PGE1製剤は主に血管拡張作用を期待して処方されているが血小板凝集阻害作用は弱い。一方、アルガトロバンは、トロンビンによる血小板活性化・凝集ばかりか血液凝固も強く阻害するが血管拡張作用はない。
【0007】
一方、物理的な血流改善の手法としては、外科的血行再建術のほかに、バルーンやレーザー、アテレクトミーカテーテルなどによる経皮的血管形成術(PTA)に加えてステント留置術など、種々の血管内治療器具が開発されている。しかしながら、PTAやステント留置術においては内皮細胞を含む血管組織を傷害し、急性再閉塞、さらに慢性期に起こる再狭窄が問題となっている。このような血行再建療法後の種々の血栓性弊害には血小板が重要な役割を果たしており、クロピドグレルやチクロピジンをはじめとする血小板凝集阻害剤が汎用されている(非特許文献3)。
従って、重症PADの治療において、強力な血小板凝集阻害作用と血管拡張作用を併せ持つ薬剤は有用であると考えられる。
【0008】
東京都奥多摩町で採取したクロモバクテリウム(Chromobacterium)属の土壌細菌QS3666株の培養上清から単離、構造決定された化合物(I)が、ADP惹起ヒト血小板凝集を強く阻害する化合物 YM−254890として報告されている(特許文献1、非特許文献4)。また、化合物(I)の絶対立体構造は、マーフィー法およびキラルHPLC分析法によって決定されている(非特許文献5)。
【化2】

化合物(I)は特異的なGq/11阻害剤である(非特許文献6)。Gq/11とは、G蛋白質の一種である。G蛋白質はα、β、γサブユニットから成る三量体蛋白質であり、アミノ酸配列の相同性と標的とする効果器の違いからGs,Gi,Gq,G12のサブファミリーに分類される。さらにサブファミリーにはサブタイプが存在し、Gqには、Gq, G11, G14, G15, G16のサブタイプの存在が報告されている。G蛋白質を直接修飾してその機能を阻害する細菌毒素としてコレラ毒素および百日咳毒素が知られているが低分子阻害剤は報告されていない。化合物(I)はGqおよびG11を強く阻害するがG14, G15およびG16は阻害しないGq/11特異的低分子阻害薬である。
また、化合物(I)は高ずり応力条件下における血小板凝集やコラーゲン表層上での血小板壁血栓形成を阻害するばかりか、種々のin vivo血栓症モデルにおいても強力な抗血栓作用を発現し、血栓溶解剤との併用効果も確認されている(非特許文献7,8)。さらに、化合物(I)はマウス血管内膜肥厚モデルにおいて、血管傷害3週間後の新生内膜肥厚形成を抑制した(非特許文献7)。しかしながら、PAD治療効果に関する具体的開示はない。
【特許文献1】特開2003−210190号公報
【非特許文献1】Lancet, 358, 1257, 2001
【非特許文献2】Journal of Vascular Surgery, 31, S9, 2000
【非特許文献3】New England Journal of Medicine, 344, 1608, 2001
【非特許文献4】Journal of Antibiotics, 56(4), 358, 2003
【非特許文献5】Tetrahedron, 59, 4533-4538, 2003
【非特許文献6】Journal of Biological Chemistry, 279, 47438, 2004
【非特許文献7】Thrombosis and Haemostasis, 94, 184, 2005
【非特許文献8】Thrombosis and Haemostasis, 90, 406, 2003
【非特許文献9】European Journal of Clinical Investigation, 18, 399, 1988
【非特許文献10】Drugs, 36 (suppl.1), 87, 1988
【非特許文献11】Journal of Cardiovascular Pharmacology, 36 (5 suppl.1), S93, 2000
【非特許文献12】Thrombosis Research, 18, 55, 1980
【非特許文献13】Prostaglandins, 49, 175, 1995
【非特許文献14】Journal of Cardiovascular Pharmacology, 35, 323, 2000
【非特許文献15】Arzneimittelforschung, 39, 856, 1989
【非特許文献16】European Journal of Pharmacology, 536, 154, 2006
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明者は、強力な血小板凝集阻害作用と血管拡張作用を併せ持ち、かつ安全性の高い重症PAD治療薬を提供すること、さらには、薬理効果が高く、薬理効果と安全性とのバランスに優れた重症PAD治療薬を提供すること、およびGq/11阻害剤の有用な用途を提供することを目的として研究を行った。
本発明者は特異的Gq/11阻害剤の病変局所への限局的な投与が、閉塞性動脈硬化症やBuerger病などのPAD治療に有効であることを見出し、発明を完成させた。
【課題を解決するための手段】
【0010】
即ち、本発明は化合物(I)又はその製薬学的に許容される塩を有効成分として含有するPAD用治療剤に関する。特に、本発明は化合物(I)又はその製薬学的に許容される塩を有効成分として含有する重症PADの予防及び/又は治療剤、すなわち安静時疼痛の軽減、潰瘍・壊死の進展阻止、救肢率向上の予防及び/又は治療剤に関する。
【発明の効果】
【0011】
本発明者は、化合物(I)又はその塩が各種血管収縮物質で惹起した血管収縮反応に対して有効であることを見出し、本発明を完成させた。
化合物(I)は、直接型α1作動薬フェニレフリン、脳内伝達物質セロトニン(以下、5−HTと記す。)、血管内皮細胞から分泌されるエンドセリン(以下、ET−1と記す。)、又は、塩化カリウムによる血管収縮に対する作用を検討した結果、塩化カリウムで血管を収縮させた場合を除いて、濃度依存的な阻害を示した。フェニレフリン、5−HT、又はET−1で惹起した血管収縮に対する50 %阻害濃度は、8.6−18 nMであった。また、化合物(I)は、内皮の有無に関わらず、大動脈中フェニレフリン誘発の血管収縮に影響した。フェニレフリン(1 μM)で惹起した血管収縮において血管内皮を除去した血管を用いた場合の50 %阻害濃度は11 nMであり、血管内皮のある血管と同様であった。即ち、化合物(I)が直接動脈の平滑筋細胞に影響を与えた。5−HTは、PAD患者の血漿中に高濃度存在すること(非特許文献9)、また、5−HTに対する血小板の反応性は、若齢の健常人よりも心血管疾患の罹患歴のある老齢者の方が高いこと(非特許文献10)、さらに、PAD患者から摘出された大腿動脈片においてはET−1またはその受容体の発現が高いこと(非特許文献11)が報告されている。これらより、化合物(I)はPAD患者に対して有効な血管拡張剤であると言える。
【0012】
さらに、本発明者は化合物(I)又はその塩が大腿動脈内投与後の血小板凝集の抑制に有効であることを見出した。
化合物(I)の大腿動脈内投与後の血小板凝集阻害活性は、以前報告した静脈内投与後の場合(非特許文献8)とほぼ同様であった。化合物(I)のADP惹起血小板凝集阻害活性は、Gq共役型のADP受容体であるP2Y1の阻害に基づくものと考えられた。また、化合物(I)の遊離血漿中濃度の消失は極めて速い(T1/2=3.7分)が、血小板凝集阻害活性は持続的であることより、血小板凝集阻害活性は必ずしも遊離血漿中濃度には依存せず、血小板内のGq阻害作用に基づくことにより発現することが示唆される。
【0013】
さらに、本発明者は化合物(I)又はその塩がラットラウリン酸惹起下肢壊死モデルに対する壊死進展抑制作用を示すことを見出した。
ラットラウリン酸惹起下肢壊死モデルは重症PADモデルとして汎用され、ラウリン酸を動脈内に注入することで内皮傷害が惹起され、引き続き血小板凝集や血小板粘着が起こり末梢循環が障害される。本モデルを用いてチクロピジン(非特許文献12)、PGE1(非特許文献13)、5−HT拮抗薬(非特許文献14)などの薬剤の薬効評価が報告されている。これらの報告において薬剤はラウリン酸注入前に投与されており、また、ラウリン酸注入後投与では薬効の減弱が報告されている(非特許文献14、15)。すなわち、ラウリン酸注入時の内皮傷害の重症度が高く、従来薬では充分な薬効が発現しにくいモデルである。しかしながら、化合物(I)は、ラウリン酸注入15分後に動脈内に投与しても用量依存的かつ強力な下肢壊死進展予防効果が確認された。因みに、化合物(I)をラウリン酸注入前に投与した場合においても充分な薬効が確認されている(非特許文献16)。
化合物(I)は、ラウリン酸注入前だけではなく注入後投与でも有効であったことから、化合物(I)が極めて強力な血管拡張作用と抗血小板作用を併せ持つ治療薬であると言える。
【0014】
さらに、本発明者は化合物(I)又はその塩がラウリン酸注入後の下肢皮膚血流量低下に対して有効であることを見出した。
一方、PGE1はラウリン酸注入により惹起される皮膚血流量の低下を抑制できなかった。また、クロピドグレルにおいては、ラウリン酸注入により惹起される皮膚血流量の低下を軽度抑制した。試験例3において、両剤とも(クロピドグレルについては前投与のみ)ラウリン酸注入3日後における下肢壊死進展を有意に抑制した。PGE1は、血管拡張作用があるものの血小板凝集阻害作用は弱い。また、クロピドグレルは、血管拡張作用はないものの血小板凝集阻害作用は強い。
従来薬が明確な薬効を検出できない本試験系においても、化合物(I)はすぐれた薬効を発現したことから、化合物(I)が極めて強力な血管拡張作用と抗血小板作用を併せ持つ薬剤であると言える。
【0015】
さらに、本発明者は化合物(I)又はその塩がラット大腿動脈内に累積投与したところ、本発明化合物は用量依存的に全身平均血圧を低下させることを見出した。化合物(I)をラット静脈内に累積投与してもほぼ同様の降圧作用が見られる(非特許文献8)。
化合物(I)の降圧作用は、Gq/11共役型血管シグナルの阻害によるものであると考えられた。試験例3における最小有効用量は3 μg/kgであり、試験例5における最小影響用量は30 μg/kgであることから用量に10 倍の乖離があることを見出した。
化合物(I)は、静脈内投与や経口投与においては薬効用量と降圧用量との間乖離は1〜3倍だったが(非特許文献7、8)、動脈内への局所投与により、より効果的な薬効が発現され、降圧用量との乖離も確保できる薬剤であると言える。
【発明を実施するための最良の形態】
【0016】
以下に実施例及び試験例に基づいて本発明をより詳細に説明するが、本発明はこれらの実施例等に限定されるものではない。尚、以下の実施例等において用いる化合物は化合物(I)の遊離体を意味する。
【0017】
本発明の医薬の有効成分は、化合物(I)又は、その製薬学的に許容される塩である。化合物(I)は、酸付加塩を形成する場合もある。かかる塩としては、製薬学的に許容される塩であり、具体的には、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸などの無機酸、ギ酸、酢酸、プロピオン酸、シュウ酸、マロン酸、コハク酸、フマル酸、マレイン酸、乳酸、リンゴ酸、酒石酸、クエン酸、メタンスルホン酸、エタンスルホン酸、アスパラギン酸、グルタミン酸などの有機酸との付加酸塩、等が挙げられる。
また、本発明の有効成分は不斉炭素原子を有するため、これに基づく光学活性体が存在し、ジアステレオマー、エナンチオマーの混合物及びその単離されたものを包含する。また、本発明の有効成分には水和物、エタノール等の溶媒和物や結晶多形の物質も全て包含する。本発明において特に好ましいのは、化合物(I)の遊離体である。
【0018】
これらの化合物は前記の特開2003−210190号公報に記載された製法により、或いはそれに準じて入手可能である。菌株はクロモバクテリウム エスピー QS3666として独立行政法人産業技術総合研究所 特許生物寄託センター(日本国茨城県つくば市東1丁目1番地1中央第6(郵便番号305−8566))に受託番号FERM BP−10786号として国際寄託されている。この菌株の菌学的性状は特開2003−210190号公報に記載された通りである。また、微生物は人工的に又は自然に変異を起こしやすいので、本発明において用いられるクロモバクテリウム エスピー QS3666株は、天然から分離された微生物の他に、これに紫外線、放射線、化学薬剤などで人工的に変異させたもの及びそれらの天然変異体についても包含する。 本発明の薬剤は局所投与に適した有機又は無機の担体、賦形剤、その他の添加剤を用いて、常法に従って、注射剤またはステント溶出剤として調製し提供することができる。
【0019】
注射剤としては、無菌の水性又は非水性の溶液剤、懸濁剤、乳濁剤を包含する。水性の溶液剤、懸濁剤の希釈剤としては、例えば、プロピレングリコール、ポリエチレングリコール、オリーブ油のような植物油、エタノールのようなアルコール類、ポリソルベート80等がある。このような組成物は、さらに防腐剤、湿潤剤、乳化剤、分散剤、安定化剤(例えばラクトース)、溶解補助剤(例えば、グルタミン酸、アスパラギン酸)のような補助剤を含んでいても良い。これらは例えば、バクテリア保管フィルターを通す濾過、殺菌剤の配合又は照射によって無菌化される。これらはまた無菌の固体組成物を製造し、使用前に無菌水又は無菌の注射用溶媒に溶解して使用することもできる。
【0020】
本発明の有効成分化合物の投与量は、投与ルート、疾患の症状、投与対象の年齢、性別等を考慮して個々の場合に応じて適宜決定されるが、通常動脈注射投与の場合、成人1人当たり有効成分は0.001 mg乃至100 mgであり、好ましくは、0.005 mg乃至20 mgであり、更に好ましくは、0.05 mg乃至 5 mgであり、これを一日に1〜複数回に分けて投与される。
【実施例】
【0021】
実験例1 動脈注射用サンプルの調整
化合物(I)を秤量後、100 %エタノールにて20 mg/mlを調製した。次に生理食塩水で200倍希釈して0.1 mg/ml (0.5%エタノール)を調製し、0.5 %エタノール生理食塩水により必要に応じて希釈して実験に供した。
【0022】
試験例1 ラット大動脈リング標本における各種血管収縮物質で惹起した血管収縮反応に対する化合物(I)の阻害作用
(方法)
実験には、一群あたり4匹の雄性SDラット(日本クレア)を用いた。ラットから大動脈を摘出し、輪状方向の血管標本(血管幅2-3 mM)を作製した。血管内腔を綿棒で擦り内皮を剥離した血管標本もあわせて作製した。各標本を37℃のKrebs-Henseleit溶液を満たしたオルガンバスに懸垂し、95 % O2-5% CO2ガスを通気した。静止張力は1gとし、張力変化は等尺性に測定した。なお、ET−1の吸着を防止するためオルガンバスにはあらかじめシリコン処理を施した。60分間の平衡時間をおいた後、本実験を開始した。
内皮を剥離した血管標本あるいは内皮を剥離していない血管標本に対し、1 μMフェニレフリンを添加して血管収縮反応を惹起した。次に化合物(I)を累積的に投与し、濃度反応曲線を得た。化合物(I)の阻害率は、化合物(I)の添加前の収縮を基に算出した。次に、内皮を剥離していない血管標本に対し、10 μMの5−HT、30 nMのET−1あるいは60 mMの塩化カリウムで惹起した血管収縮に対する化合物(I)の弛緩作用を検討した。10 μMの5−HTあるいは60 mMの塩化カリウムの場合は、化合物(I)を収縮惹起剤投与30分前に投与し、血管標本で化合物(I)の濃度依存性を検討した。化合物(I)の阻害率は溶媒投与後の惹起剤の収縮率を基に算出した。エンドセリンの場合は、一旦血管収縮を惹起すると脱感作を起こすため、一濃度の阻害率評価は一標本を用いて行った。
(結果)
図1に示すように、フェニレフリン惹起血管収縮反応に対して化合物(I)は濃度依存的な阻害を示し、内皮存在下および非存在下における50%阻害濃度はそれぞれ16±2.3 nMおよび11±2.9 nM(n=4、平均値±標準誤差)であった。また、化合物(I)は5−HTあるいはET−1惹起血管収縮反応を濃度依存的に阻害し50 %阻害濃度はそれぞれ8.6±1.0 nM(n=4、平均値±標準誤差)および18 nM(n=4、平均値)であった。一方、塩化カリウム惹起血管収縮に対し、化合物(I)は1 μMでも阻害作用を示さなかった。
【0023】
試験例2 化合物(I)のラット大腿動脈内単回投与後のex vivo血小板凝集阻害作用
(方法)
実験には、一群あたり4匹の雄性SDラット(日本クレア)を用いた。一晩絶食したラットをペントバルビタール(60 mg/kg)の腹腔内投与により麻酔した。薬剤投与用のカテーテルを左大腿動脈内に留置した。溶媒(0.5 %濃度のエタノールを含む生理食塩水)あるいは化合物(I)(1−10 μg/kg、0.5 ml/kg)を上記カテーテルを介して大腿動脈内に単回投与した。投与5分後、動物を開腹し後大静脈より3.8 %クエン酸ナトリウムを抗凝固剤として6 mlの採血を行い、室温下で200g、5分の遠心操作により多血小板血漿を、2000g、10分の遠心操作により乏血小板血漿を得て血小板数を血球計数器(日本光電、MEK−6258)にてカウントして30万/μlの多血小板血漿を調製した。血小板凝集能は凝集計(MCメディカル、MCM Hema Tracer 212)を用いて測定した。90 μlの多血小板血漿を37 ℃で1分間インキュベート後、50 μM のADPを10 μl添加し、5分間の血小板凝集をモニターした。化合物(I)の血小板凝集阻害率は溶媒投与群の血小板凝集能を基に算出した。
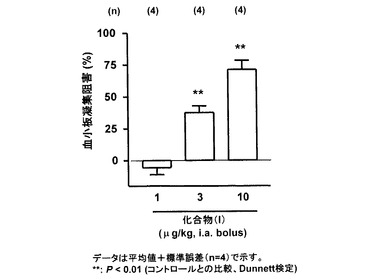
(結果)
図2に示すように、化合物(I)は用量依存的にADP惹起血小板凝集を阻害した。1、3および10 μg/kgにおける血小板凝集阻害活性はそれぞれ-5.6±5.4 %、38±5.1 %および71±7.0 %であった(n=4、平均値±標準誤差)。
【0024】
試験例3 ラットラウリン酸惹起下肢壊死モデルに対する化合物(I)の壊死進展抑制作用
(方法)
ラットラウリン酸下肢壊死モデルはアシダらの方法を用いて作成した(非特許文献12)。実験には、一群あたり6−24匹の雄性SDラット(日本クレア)を用いた。一晩絶食したラットをペントバルビタール(60 mg/kg)の腹腔内投与により麻酔し、薬剤およびラウリン酸注入用のカテーテルを左大腿動脈内に留置した。また、PGE1の静脈内投与用に右頸静脈にカニューレを留置した。薬剤としては化合物(I)の他、PGE1(小野薬品)およびクロピドグレル(APIN Chemicals)を使用した。コントロール群には大腿動脈カニューレよりラウリン酸(7.5 mg/mlを0.33 ml/kg)の注入のみを行なった。偽手術群は大腿動脈カニューレより同容量の生理食塩水の投与を行なった。化合物(I)(0.5%濃度のエタノールを含む生理食塩水溶液)はラウリン酸注入後15分後に同カテーテルより単回投与した。PGE1は、ラウリン酸投与5分前から30分間、頸静脈カニューレを介して1 μg/kg/分で持続静脈内投与するかあるいはラウリン酸注入後5分後に同カテーテルより0.2 μg/kg/分の速度で15分間の持続動脈内投与を行なった。クロピドグレルはラウリン酸投与4時間前あるいは2時間後に30 mg/kg経口投与した。また、クロピドグレルはラウリン酸投与後1日1回3日間ゾンデを用いて胃内に強制投与した。ラウリン酸投与3日後、以下の病変重症度スコアを用いて5指の総計を算出し、薬効判定を行なった。0:変化なし、1:爪の黒変、2:指の黒変(第一関節まで)、3:指の黒変(第一関節以上)、4:指の脱落、脱落が足蹠に及ぶ場合にはさらに5ポイントを追加した。
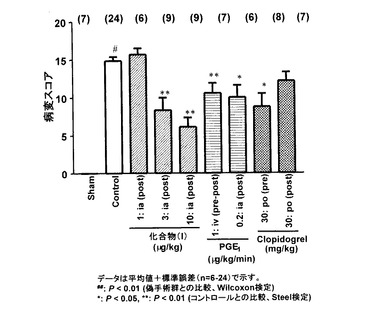
(結果)
図3に示すように、偽手術群ではまったく下肢壊死症状は起こらなかった。一方、コントロール群ではラウリン酸投与3日後には明確な下肢壊死や脱落病変が認められた。ラウリン酸注入後15分後に化合物(I)を投与すると病変スコアの用量依存的な改善が認められ、3 μg/kg以上で有意な改善を示した。PGE1では、ラウリン酸投与前から1 μg/kg/分の持続静脈内投与およびラウリン酸投与5分後からの0.2 μg/kg/分の持続動脈内投与において有意な病変スコアの改善が認められた。クロピドグレルでは、ラウリン酸投与4時間前から投与した場合には有効であったが、ラウリン酸投与後に投与開始した場合には薬効は検出できなかった。
【0025】
試験例4 ラット大腿動脈へのラウリン酸注入後の下肢皮膚血流量低下に対する化合物(I)の影響
(方法)
実験には、一群あたり3匹の雄性SDラット(日本クレア)を用いた。レーザードップラー血流計(Laser Doppler Perfusion Imager Systemlisca)を用いて左(患肢)および右(正常肢)下肢における皮膚表面の血流量をカラーイメージで記録した。一晩絶食したラットをペントバルビタール(60 mg/kg)の腹腔内投与により麻酔し、薬剤およびラウリン酸注入用のカテーテルを左大腿動脈内に留置した。また、PGE1の静脈内投与用に右頸静脈にカニューレを留置した。薬剤としては化合物(I)の他、PGE1およびベラプロストを使用した。ラウリン酸の注入は既述の条件(7.5 mg/mlを0.33 ml/kg)で行なった。溶媒(i.a.)群は、大腿動脈カニューレよりラウリン酸を注入し15分後に同カニューレより0.5 %エタノールを含む生理食塩水溶液を0.5 ml/kg投与した。化合物(I)群(溶媒は0.5%エタノールを含む生理食塩水、投与量:10 μg/kg)は、大腿動脈カニューレよりラウリン酸を注入し15分後に同カニューレより化合物(I)を単回投与した。血流量観察は、ラウリン酸注入前、ラウリン酸注入10分後、および溶媒あるいは化合物(I)投与10分後(ラウリン酸投与25分後)に行なった。PGE1は、ラウリン酸投与5分前から30分間、頸静脈カニューレを介して1 μg/kg/分の静脈内投与を行なった。血流量観察は、ラウリン酸注入前、ラウリン酸注入直前(PGE1投与開始5分後)、およびPGE1投与開始30分後(ラウリン酸投与25分後)に行なった。溶媒(0.5 %メチルセルロース)およびクロピドグレル(30 mg/kg)は無麻酔下でラウリン酸投与4時間前に強制経口投与した。
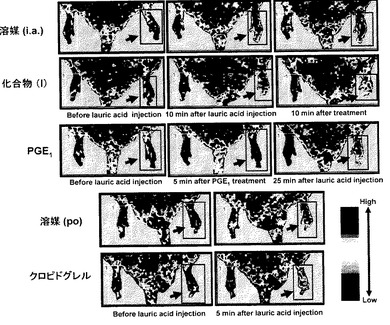
(結果)
図4には、ラウリン酸および各薬剤投与後の皮膚血流量に及ぼす影響をドップラーイメージで示した。図4右下に示すように血流量が少ないと黒色〜青色として表され、血流量が多いと赤色で示される。患肢を図中矢印で明示したが、溶媒(i.a.)投与群では、ラウリン酸注入後皮膚血流量の低下が観察されたが、化合物(I)の投与後、皮膚血流量は顕著に回復した。PGE1の投与開始5分後、両側性に下肢皮膚血流量の増加が見られたが(図4、PGE1中列)、ラウリン酸注入後、皮膚血流量は顕著に低下した(図4、PGE1右列)。溶媒(p.o.)投与群では、ラウリン酸注入後皮膚血流量の低下が観察された。クロピドグレルの前投与によりラウリン酸による皮膚血流量低下は軽度抑制された(図4、クロピドグレル右列)。
【0026】
試験例5 化合物(I)のラット大腿動脈内単回投与後の全身血圧および心拍数に及ぼす影響
(方法)
実験には、一群あたり5匹の雄性SDラット(日本クレア)を用いた。一晩絶食したラットをウレタン(1.4 g/kg)の腹腔内投与により麻酔した。血圧・心拍数測定用のカニューレを左総頸動脈に挿入した。また、薬剤投与用のカテーテルを左大腿動脈内に留置した。全身血圧は圧トランスデューサー(AP−200T、日本光電)により、心拍数はタコメーター(AT−600G、日本光電)により測定し、感熱式レコーダー(WT−685G、日本光電)を用いて持続的にモニターした。溶媒(0.5%濃度のエタノールを含む生理食塩水)あるいは化合物(I)(10−100 μg/kg、0.5 ml/kg)を上記カテーテルを介して10−20分間隔で大腿動脈内に累積投与した。溶媒投与時の全身平均血圧および心拍数を100%として各用量投与後の最大変化率を相対変化率%として表した。
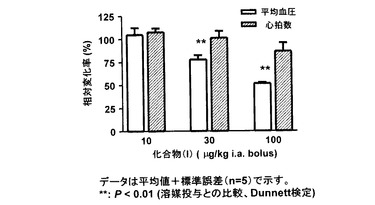
(結果)
図5に示すように、化合物(I)は用量依存的に全身平均血圧を低下した。10 μg/kgでは有意な変動は見られなかったが、30および100 μg/kgでは全身血圧をそれぞれ25±5.2 %、50±2.1 %有意に低下した。また、100 μg/kgまで心拍数には有意な変動は認められなかった。
【産業上の利用可能性】
【0027】
本発明の有効成分である化合物(I)又はその塩は、強力な血小板凝集阻害作用と血管拡張作用を有しており、化合物(I)の使用を局所に限定することで、安全性に優れ、効果も期待できるPAD治療剤/重症PAD治療剤を提供できる。本発明により、重症PADの症状である安静時疼痛の軽減、潰瘍・壊死の進展阻止、救肢率の向上が期待できる。具体的には、PADのうち閉塞性動脈硬化症やBuerger病にも有効であることが期待できる。
【図面の簡単な説明】
【0028】
【図1】は、各濃度における塩酸フェニレフリン(内皮あり、内皮なしの両条件)、5−HT、ET−1、又は、塩化カリウムによるラット大動脈標本の収縮度に対する化合物(I)の阻害作用を示すグラフである。
【図2】は、ラット大腿動脈内に化合物(I)の各用量を単回投与5分後のADP惹起血小板凝集阻害活性を示すグラフである。
【図3】は、ラット大腿動脈内にラウリン酸を投与後3日における各薬剤投与群における病変スコアを示すグラフである。
【図4】は、ラット大腿動脈内にラウリン酸を投与した前後における後肢皮膚血流量の変動をレーザードップラー皮膚血流計で観察した際のカラー画像である。
【図5】は、化合物(I)をラット大腿動脈内に各用量、単回投与した5分後での平均血圧および心拍数を示すグラフである。
【技術分野】
【0001】
本発明は、医薬、殊に末梢動脈閉塞症の予防及び/又は治療剤に関するものであり、特に、重症末梢動脈閉塞症の予防及び/又は治療剤、すなわち安静時疼痛、潰瘍・壊死を改善する予防及び/又は治療剤に係るものである。
【背景技術】
【0002】
末梢動脈閉塞症(peripheral arterial disease: PAD、以下PADと記す。)とは、四肢への主幹動脈に生じた動脈硬化性閉塞性病変により、主に下肢を中心として虚血症状を呈する疾患である(非特許文献1)。PADには、閉塞性動脈硬化症やBuerger病が知られている。
【0003】
PADの臨床症状・徴候についての進行度の目安としてFontain分類が知られており、症状の程度により、[I度:冷感・しびれ感、II度:間歇性跂行、III度:安静時疼痛、IV度:潰瘍・壊死]に分類されている。PADの中でも、III度又はIV度の症状を呈した場合には重症PADと呼ぶ。
PADは、50歳以上の喫煙男性に多く、近年、高齢化社会の到来に加えて、急速な食生活の欧米化や車社会化などによる生活様式の変化により疾病の傾向が大きく変化し、他の動脈硬化性血管疾患の急速な増加と同様にPADが増加している。
【0004】
PADは高血圧や糖尿病、高脂血症などを合併することもある。前述の通り、PADは、全身的な動脈硬化性変化の一分症でもあり、多くの例で虚血性心疾患や脳血管障害、腎機能障害などを生じる他臓器の閉塞性血管病変を有している。特に最近においては、動脈硬化の危険因子としてインスリン抵抗性の概念が導入され注目されているが、PADはこうした危険因子を有する代表的疾患でもある。特に日本における糖尿病患者の急速な増加は直接的に本症の増加につながっていると考えられており、例えば、糖尿病性腎症による透析例が増加することにより、重症虚血肢が増加し、本症の糖尿病併存頻度は35%以上と考えられている。
【0005】
PADの診断と治療について、『下肢閉塞性動脈硬化症の診断・治療指針』(Trans Atlantic Inter-Society Consensus(TASC))がガイドラインを示している(非特許文献2)。安静時疼痛や潰瘍・壊死に分類される重症患者は、PADの総患者数の約25%を占める。重症虚血肢において、肢虚血の重症度、閉塞性病変の存在部位や範囲、側副路様式や末梢血流の良否、併存疾患の重症度、肢機能の治療目標、生命予後などを考慮して治療方針が選択されるが、薬剤あるいは物理的方法によって血行の再開を図る治療が行なわれている。
【0006】
重症PADの治療剤としては、日本ではプロスタグランジンE1製剤(以下、PGE1製剤)やアルガトロバンが用いられている。PGE1製剤は主に血管拡張作用を期待して処方されているが血小板凝集阻害作用は弱い。一方、アルガトロバンは、トロンビンによる血小板活性化・凝集ばかりか血液凝固も強く阻害するが血管拡張作用はない。
【0007】
一方、物理的な血流改善の手法としては、外科的血行再建術のほかに、バルーンやレーザー、アテレクトミーカテーテルなどによる経皮的血管形成術(PTA)に加えてステント留置術など、種々の血管内治療器具が開発されている。しかしながら、PTAやステント留置術においては内皮細胞を含む血管組織を傷害し、急性再閉塞、さらに慢性期に起こる再狭窄が問題となっている。このような血行再建療法後の種々の血栓性弊害には血小板が重要な役割を果たしており、クロピドグレルやチクロピジンをはじめとする血小板凝集阻害剤が汎用されている(非特許文献3)。
従って、重症PADの治療において、強力な血小板凝集阻害作用と血管拡張作用を併せ持つ薬剤は有用であると考えられる。
【0008】
東京都奥多摩町で採取したクロモバクテリウム(Chromobacterium)属の土壌細菌QS3666株の培養上清から単離、構造決定された化合物(I)が、ADP惹起ヒト血小板凝集を強く阻害する化合物 YM−254890として報告されている(特許文献1、非特許文献4)。また、化合物(I)の絶対立体構造は、マーフィー法およびキラルHPLC分析法によって決定されている(非特許文献5)。
【化2】
化合物(I)は特異的なGq/11阻害剤である(非特許文献6)。Gq/11とは、G蛋白質の一種である。G蛋白質はα、β、γサブユニットから成る三量体蛋白質であり、アミノ酸配列の相同性と標的とする効果器の違いからGs,Gi,Gq,G12のサブファミリーに分類される。さらにサブファミリーにはサブタイプが存在し、Gqには、Gq, G11, G14, G15, G16のサブタイプの存在が報告されている。G蛋白質を直接修飾してその機能を阻害する細菌毒素としてコレラ毒素および百日咳毒素が知られているが低分子阻害剤は報告されていない。化合物(I)はGqおよびG11を強く阻害するがG14, G15およびG16は阻害しないGq/11特異的低分子阻害薬である。
また、化合物(I)は高ずり応力条件下における血小板凝集やコラーゲン表層上での血小板壁血栓形成を阻害するばかりか、種々のin vivo血栓症モデルにおいても強力な抗血栓作用を発現し、血栓溶解剤との併用効果も確認されている(非特許文献7,8)。さらに、化合物(I)はマウス血管内膜肥厚モデルにおいて、血管傷害3週間後の新生内膜肥厚形成を抑制した(非特許文献7)。しかしながら、PAD治療効果に関する具体的開示はない。
【特許文献1】特開2003−210190号公報
【非特許文献1】Lancet, 358, 1257, 2001
【非特許文献2】Journal of Vascular Surgery, 31, S9, 2000
【非特許文献3】New England Journal of Medicine, 344, 1608, 2001
【非特許文献4】Journal of Antibiotics, 56(4), 358, 2003
【非特許文献5】Tetrahedron, 59, 4533-4538, 2003
【非特許文献6】Journal of Biological Chemistry, 279, 47438, 2004
【非特許文献7】Thrombosis and Haemostasis, 94, 184, 2005
【非特許文献8】Thrombosis and Haemostasis, 90, 406, 2003
【非特許文献9】European Journal of Clinical Investigation, 18, 399, 1988
【非特許文献10】Drugs, 36 (suppl.1), 87, 1988
【非特許文献11】Journal of Cardiovascular Pharmacology, 36 (5 suppl.1), S93, 2000
【非特許文献12】Thrombosis Research, 18, 55, 1980
【非特許文献13】Prostaglandins, 49, 175, 1995
【非特許文献14】Journal of Cardiovascular Pharmacology, 35, 323, 2000
【非特許文献15】Arzneimittelforschung, 39, 856, 1989
【非特許文献16】European Journal of Pharmacology, 536, 154, 2006
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明者は、強力な血小板凝集阻害作用と血管拡張作用を併せ持ち、かつ安全性の高い重症PAD治療薬を提供すること、さらには、薬理効果が高く、薬理効果と安全性とのバランスに優れた重症PAD治療薬を提供すること、およびGq/11阻害剤の有用な用途を提供することを目的として研究を行った。
本発明者は特異的Gq/11阻害剤の病変局所への限局的な投与が、閉塞性動脈硬化症やBuerger病などのPAD治療に有効であることを見出し、発明を完成させた。
【課題を解決するための手段】
【0010】
即ち、本発明は化合物(I)又はその製薬学的に許容される塩を有効成分として含有するPAD用治療剤に関する。特に、本発明は化合物(I)又はその製薬学的に許容される塩を有効成分として含有する重症PADの予防及び/又は治療剤、すなわち安静時疼痛の軽減、潰瘍・壊死の進展阻止、救肢率向上の予防及び/又は治療剤に関する。
【発明の効果】
【0011】
本発明者は、化合物(I)又はその塩が各種血管収縮物質で惹起した血管収縮反応に対して有効であることを見出し、本発明を完成させた。
化合物(I)は、直接型α1作動薬フェニレフリン、脳内伝達物質セロトニン(以下、5−HTと記す。)、血管内皮細胞から分泌されるエンドセリン(以下、ET−1と記す。)、又は、塩化カリウムによる血管収縮に対する作用を検討した結果、塩化カリウムで血管を収縮させた場合を除いて、濃度依存的な阻害を示した。フェニレフリン、5−HT、又はET−1で惹起した血管収縮に対する50 %阻害濃度は、8.6−18 nMであった。また、化合物(I)は、内皮の有無に関わらず、大動脈中フェニレフリン誘発の血管収縮に影響した。フェニレフリン(1 μM)で惹起した血管収縮において血管内皮を除去した血管を用いた場合の50 %阻害濃度は11 nMであり、血管内皮のある血管と同様であった。即ち、化合物(I)が直接動脈の平滑筋細胞に影響を与えた。5−HTは、PAD患者の血漿中に高濃度存在すること(非特許文献9)、また、5−HTに対する血小板の反応性は、若齢の健常人よりも心血管疾患の罹患歴のある老齢者の方が高いこと(非特許文献10)、さらに、PAD患者から摘出された大腿動脈片においてはET−1またはその受容体の発現が高いこと(非特許文献11)が報告されている。これらより、化合物(I)はPAD患者に対して有効な血管拡張剤であると言える。
【0012】
さらに、本発明者は化合物(I)又はその塩が大腿動脈内投与後の血小板凝集の抑制に有効であることを見出した。
化合物(I)の大腿動脈内投与後の血小板凝集阻害活性は、以前報告した静脈内投与後の場合(非特許文献8)とほぼ同様であった。化合物(I)のADP惹起血小板凝集阻害活性は、Gq共役型のADP受容体であるP2Y1の阻害に基づくものと考えられた。また、化合物(I)の遊離血漿中濃度の消失は極めて速い(T1/2=3.7分)が、血小板凝集阻害活性は持続的であることより、血小板凝集阻害活性は必ずしも遊離血漿中濃度には依存せず、血小板内のGq阻害作用に基づくことにより発現することが示唆される。
【0013】
さらに、本発明者は化合物(I)又はその塩がラットラウリン酸惹起下肢壊死モデルに対する壊死進展抑制作用を示すことを見出した。
ラットラウリン酸惹起下肢壊死モデルは重症PADモデルとして汎用され、ラウリン酸を動脈内に注入することで内皮傷害が惹起され、引き続き血小板凝集や血小板粘着が起こり末梢循環が障害される。本モデルを用いてチクロピジン(非特許文献12)、PGE1(非特許文献13)、5−HT拮抗薬(非特許文献14)などの薬剤の薬効評価が報告されている。これらの報告において薬剤はラウリン酸注入前に投与されており、また、ラウリン酸注入後投与では薬効の減弱が報告されている(非特許文献14、15)。すなわち、ラウリン酸注入時の内皮傷害の重症度が高く、従来薬では充分な薬効が発現しにくいモデルである。しかしながら、化合物(I)は、ラウリン酸注入15分後に動脈内に投与しても用量依存的かつ強力な下肢壊死進展予防効果が確認された。因みに、化合物(I)をラウリン酸注入前に投与した場合においても充分な薬効が確認されている(非特許文献16)。
化合物(I)は、ラウリン酸注入前だけではなく注入後投与でも有効であったことから、化合物(I)が極めて強力な血管拡張作用と抗血小板作用を併せ持つ治療薬であると言える。
【0014】
さらに、本発明者は化合物(I)又はその塩がラウリン酸注入後の下肢皮膚血流量低下に対して有効であることを見出した。
一方、PGE1はラウリン酸注入により惹起される皮膚血流量の低下を抑制できなかった。また、クロピドグレルにおいては、ラウリン酸注入により惹起される皮膚血流量の低下を軽度抑制した。試験例3において、両剤とも(クロピドグレルについては前投与のみ)ラウリン酸注入3日後における下肢壊死進展を有意に抑制した。PGE1は、血管拡張作用があるものの血小板凝集阻害作用は弱い。また、クロピドグレルは、血管拡張作用はないものの血小板凝集阻害作用は強い。
従来薬が明確な薬効を検出できない本試験系においても、化合物(I)はすぐれた薬効を発現したことから、化合物(I)が極めて強力な血管拡張作用と抗血小板作用を併せ持つ薬剤であると言える。
【0015】
さらに、本発明者は化合物(I)又はその塩がラット大腿動脈内に累積投与したところ、本発明化合物は用量依存的に全身平均血圧を低下させることを見出した。化合物(I)をラット静脈内に累積投与してもほぼ同様の降圧作用が見られる(非特許文献8)。
化合物(I)の降圧作用は、Gq/11共役型血管シグナルの阻害によるものであると考えられた。試験例3における最小有効用量は3 μg/kgであり、試験例5における最小影響用量は30 μg/kgであることから用量に10 倍の乖離があることを見出した。
化合物(I)は、静脈内投与や経口投与においては薬効用量と降圧用量との間乖離は1〜3倍だったが(非特許文献7、8)、動脈内への局所投与により、より効果的な薬効が発現され、降圧用量との乖離も確保できる薬剤であると言える。
【発明を実施するための最良の形態】
【0016】
以下に実施例及び試験例に基づいて本発明をより詳細に説明するが、本発明はこれらの実施例等に限定されるものではない。尚、以下の実施例等において用いる化合物は化合物(I)の遊離体を意味する。
【0017】
本発明の医薬の有効成分は、化合物(I)又は、その製薬学的に許容される塩である。化合物(I)は、酸付加塩を形成する場合もある。かかる塩としては、製薬学的に許容される塩であり、具体的には、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸などの無機酸、ギ酸、酢酸、プロピオン酸、シュウ酸、マロン酸、コハク酸、フマル酸、マレイン酸、乳酸、リンゴ酸、酒石酸、クエン酸、メタンスルホン酸、エタンスルホン酸、アスパラギン酸、グルタミン酸などの有機酸との付加酸塩、等が挙げられる。
また、本発明の有効成分は不斉炭素原子を有するため、これに基づく光学活性体が存在し、ジアステレオマー、エナンチオマーの混合物及びその単離されたものを包含する。また、本発明の有効成分には水和物、エタノール等の溶媒和物や結晶多形の物質も全て包含する。本発明において特に好ましいのは、化合物(I)の遊離体である。
【0018】
これらの化合物は前記の特開2003−210190号公報に記載された製法により、或いはそれに準じて入手可能である。菌株はクロモバクテリウム エスピー QS3666として独立行政法人産業技術総合研究所 特許生物寄託センター(日本国茨城県つくば市東1丁目1番地1中央第6(郵便番号305−8566))に受託番号FERM BP−10786号として国際寄託されている。この菌株の菌学的性状は特開2003−210190号公報に記載された通りである。また、微生物は人工的に又は自然に変異を起こしやすいので、本発明において用いられるクロモバクテリウム エスピー QS3666株は、天然から分離された微生物の他に、これに紫外線、放射線、化学薬剤などで人工的に変異させたもの及びそれらの天然変異体についても包含する。 本発明の薬剤は局所投与に適した有機又は無機の担体、賦形剤、その他の添加剤を用いて、常法に従って、注射剤またはステント溶出剤として調製し提供することができる。
【0019】
注射剤としては、無菌の水性又は非水性の溶液剤、懸濁剤、乳濁剤を包含する。水性の溶液剤、懸濁剤の希釈剤としては、例えば、プロピレングリコール、ポリエチレングリコール、オリーブ油のような植物油、エタノールのようなアルコール類、ポリソルベート80等がある。このような組成物は、さらに防腐剤、湿潤剤、乳化剤、分散剤、安定化剤(例えばラクトース)、溶解補助剤(例えば、グルタミン酸、アスパラギン酸)のような補助剤を含んでいても良い。これらは例えば、バクテリア保管フィルターを通す濾過、殺菌剤の配合又は照射によって無菌化される。これらはまた無菌の固体組成物を製造し、使用前に無菌水又は無菌の注射用溶媒に溶解して使用することもできる。
【0020】
本発明の有効成分化合物の投与量は、投与ルート、疾患の症状、投与対象の年齢、性別等を考慮して個々の場合に応じて適宜決定されるが、通常動脈注射投与の場合、成人1人当たり有効成分は0.001 mg乃至100 mgであり、好ましくは、0.005 mg乃至20 mgであり、更に好ましくは、0.05 mg乃至 5 mgであり、これを一日に1〜複数回に分けて投与される。
【実施例】
【0021】
実験例1 動脈注射用サンプルの調整
化合物(I)を秤量後、100 %エタノールにて20 mg/mlを調製した。次に生理食塩水で200倍希釈して0.1 mg/ml (0.5%エタノール)を調製し、0.5 %エタノール生理食塩水により必要に応じて希釈して実験に供した。
【0022】
試験例1 ラット大動脈リング標本における各種血管収縮物質で惹起した血管収縮反応に対する化合物(I)の阻害作用
(方法)
実験には、一群あたり4匹の雄性SDラット(日本クレア)を用いた。ラットから大動脈を摘出し、輪状方向の血管標本(血管幅2-3 mM)を作製した。血管内腔を綿棒で擦り内皮を剥離した血管標本もあわせて作製した。各標本を37℃のKrebs-Henseleit溶液を満たしたオルガンバスに懸垂し、95 % O2-5% CO2ガスを通気した。静止張力は1gとし、張力変化は等尺性に測定した。なお、ET−1の吸着を防止するためオルガンバスにはあらかじめシリコン処理を施した。60分間の平衡時間をおいた後、本実験を開始した。
内皮を剥離した血管標本あるいは内皮を剥離していない血管標本に対し、1 μMフェニレフリンを添加して血管収縮反応を惹起した。次に化合物(I)を累積的に投与し、濃度反応曲線を得た。化合物(I)の阻害率は、化合物(I)の添加前の収縮を基に算出した。次に、内皮を剥離していない血管標本に対し、10 μMの5−HT、30 nMのET−1あるいは60 mMの塩化カリウムで惹起した血管収縮に対する化合物(I)の弛緩作用を検討した。10 μMの5−HTあるいは60 mMの塩化カリウムの場合は、化合物(I)を収縮惹起剤投与30分前に投与し、血管標本で化合物(I)の濃度依存性を検討した。化合物(I)の阻害率は溶媒投与後の惹起剤の収縮率を基に算出した。エンドセリンの場合は、一旦血管収縮を惹起すると脱感作を起こすため、一濃度の阻害率評価は一標本を用いて行った。
(結果)
図1に示すように、フェニレフリン惹起血管収縮反応に対して化合物(I)は濃度依存的な阻害を示し、内皮存在下および非存在下における50%阻害濃度はそれぞれ16±2.3 nMおよび11±2.9 nM(n=4、平均値±標準誤差)であった。また、化合物(I)は5−HTあるいはET−1惹起血管収縮反応を濃度依存的に阻害し50 %阻害濃度はそれぞれ8.6±1.0 nM(n=4、平均値±標準誤差)および18 nM(n=4、平均値)であった。一方、塩化カリウム惹起血管収縮に対し、化合物(I)は1 μMでも阻害作用を示さなかった。
【0023】
試験例2 化合物(I)のラット大腿動脈内単回投与後のex vivo血小板凝集阻害作用
(方法)
実験には、一群あたり4匹の雄性SDラット(日本クレア)を用いた。一晩絶食したラットをペントバルビタール(60 mg/kg)の腹腔内投与により麻酔した。薬剤投与用のカテーテルを左大腿動脈内に留置した。溶媒(0.5 %濃度のエタノールを含む生理食塩水)あるいは化合物(I)(1−10 μg/kg、0.5 ml/kg)を上記カテーテルを介して大腿動脈内に単回投与した。投与5分後、動物を開腹し後大静脈より3.8 %クエン酸ナトリウムを抗凝固剤として6 mlの採血を行い、室温下で200g、5分の遠心操作により多血小板血漿を、2000g、10分の遠心操作により乏血小板血漿を得て血小板数を血球計数器(日本光電、MEK−6258)にてカウントして30万/μlの多血小板血漿を調製した。血小板凝集能は凝集計(MCメディカル、MCM Hema Tracer 212)を用いて測定した。90 μlの多血小板血漿を37 ℃で1分間インキュベート後、50 μM のADPを10 μl添加し、5分間の血小板凝集をモニターした。化合物(I)の血小板凝集阻害率は溶媒投与群の血小板凝集能を基に算出した。
(結果)
図2に示すように、化合物(I)は用量依存的にADP惹起血小板凝集を阻害した。1、3および10 μg/kgにおける血小板凝集阻害活性はそれぞれ-5.6±5.4 %、38±5.1 %および71±7.0 %であった(n=4、平均値±標準誤差)。
【0024】
試験例3 ラットラウリン酸惹起下肢壊死モデルに対する化合物(I)の壊死進展抑制作用
(方法)
ラットラウリン酸下肢壊死モデルはアシダらの方法を用いて作成した(非特許文献12)。実験には、一群あたり6−24匹の雄性SDラット(日本クレア)を用いた。一晩絶食したラットをペントバルビタール(60 mg/kg)の腹腔内投与により麻酔し、薬剤およびラウリン酸注入用のカテーテルを左大腿動脈内に留置した。また、PGE1の静脈内投与用に右頸静脈にカニューレを留置した。薬剤としては化合物(I)の他、PGE1(小野薬品)およびクロピドグレル(APIN Chemicals)を使用した。コントロール群には大腿動脈カニューレよりラウリン酸(7.5 mg/mlを0.33 ml/kg)の注入のみを行なった。偽手術群は大腿動脈カニューレより同容量の生理食塩水の投与を行なった。化合物(I)(0.5%濃度のエタノールを含む生理食塩水溶液)はラウリン酸注入後15分後に同カテーテルより単回投与した。PGE1は、ラウリン酸投与5分前から30分間、頸静脈カニューレを介して1 μg/kg/分で持続静脈内投与するかあるいはラウリン酸注入後5分後に同カテーテルより0.2 μg/kg/分の速度で15分間の持続動脈内投与を行なった。クロピドグレルはラウリン酸投与4時間前あるいは2時間後に30 mg/kg経口投与した。また、クロピドグレルはラウリン酸投与後1日1回3日間ゾンデを用いて胃内に強制投与した。ラウリン酸投与3日後、以下の病変重症度スコアを用いて5指の総計を算出し、薬効判定を行なった。0:変化なし、1:爪の黒変、2:指の黒変(第一関節まで)、3:指の黒変(第一関節以上)、4:指の脱落、脱落が足蹠に及ぶ場合にはさらに5ポイントを追加した。
(結果)
図3に示すように、偽手術群ではまったく下肢壊死症状は起こらなかった。一方、コントロール群ではラウリン酸投与3日後には明確な下肢壊死や脱落病変が認められた。ラウリン酸注入後15分後に化合物(I)を投与すると病変スコアの用量依存的な改善が認められ、3 μg/kg以上で有意な改善を示した。PGE1では、ラウリン酸投与前から1 μg/kg/分の持続静脈内投与およびラウリン酸投与5分後からの0.2 μg/kg/分の持続動脈内投与において有意な病変スコアの改善が認められた。クロピドグレルでは、ラウリン酸投与4時間前から投与した場合には有効であったが、ラウリン酸投与後に投与開始した場合には薬効は検出できなかった。
【0025】
試験例4 ラット大腿動脈へのラウリン酸注入後の下肢皮膚血流量低下に対する化合物(I)の影響
(方法)
実験には、一群あたり3匹の雄性SDラット(日本クレア)を用いた。レーザードップラー血流計(Laser Doppler Perfusion Imager Systemlisca)を用いて左(患肢)および右(正常肢)下肢における皮膚表面の血流量をカラーイメージで記録した。一晩絶食したラットをペントバルビタール(60 mg/kg)の腹腔内投与により麻酔し、薬剤およびラウリン酸注入用のカテーテルを左大腿動脈内に留置した。また、PGE1の静脈内投与用に右頸静脈にカニューレを留置した。薬剤としては化合物(I)の他、PGE1およびベラプロストを使用した。ラウリン酸の注入は既述の条件(7.5 mg/mlを0.33 ml/kg)で行なった。溶媒(i.a.)群は、大腿動脈カニューレよりラウリン酸を注入し15分後に同カニューレより0.5 %エタノールを含む生理食塩水溶液を0.5 ml/kg投与した。化合物(I)群(溶媒は0.5%エタノールを含む生理食塩水、投与量:10 μg/kg)は、大腿動脈カニューレよりラウリン酸を注入し15分後に同カニューレより化合物(I)を単回投与した。血流量観察は、ラウリン酸注入前、ラウリン酸注入10分後、および溶媒あるいは化合物(I)投与10分後(ラウリン酸投与25分後)に行なった。PGE1は、ラウリン酸投与5分前から30分間、頸静脈カニューレを介して1 μg/kg/分の静脈内投与を行なった。血流量観察は、ラウリン酸注入前、ラウリン酸注入直前(PGE1投与開始5分後)、およびPGE1投与開始30分後(ラウリン酸投与25分後)に行なった。溶媒(0.5 %メチルセルロース)およびクロピドグレル(30 mg/kg)は無麻酔下でラウリン酸投与4時間前に強制経口投与した。
(結果)
図4には、ラウリン酸および各薬剤投与後の皮膚血流量に及ぼす影響をドップラーイメージで示した。図4右下に示すように血流量が少ないと黒色〜青色として表され、血流量が多いと赤色で示される。患肢を図中矢印で明示したが、溶媒(i.a.)投与群では、ラウリン酸注入後皮膚血流量の低下が観察されたが、化合物(I)の投与後、皮膚血流量は顕著に回復した。PGE1の投与開始5分後、両側性に下肢皮膚血流量の増加が見られたが(図4、PGE1中列)、ラウリン酸注入後、皮膚血流量は顕著に低下した(図4、PGE1右列)。溶媒(p.o.)投与群では、ラウリン酸注入後皮膚血流量の低下が観察された。クロピドグレルの前投与によりラウリン酸による皮膚血流量低下は軽度抑制された(図4、クロピドグレル右列)。
【0026】
試験例5 化合物(I)のラット大腿動脈内単回投与後の全身血圧および心拍数に及ぼす影響
(方法)
実験には、一群あたり5匹の雄性SDラット(日本クレア)を用いた。一晩絶食したラットをウレタン(1.4 g/kg)の腹腔内投与により麻酔した。血圧・心拍数測定用のカニューレを左総頸動脈に挿入した。また、薬剤投与用のカテーテルを左大腿動脈内に留置した。全身血圧は圧トランスデューサー(AP−200T、日本光電)により、心拍数はタコメーター(AT−600G、日本光電)により測定し、感熱式レコーダー(WT−685G、日本光電)を用いて持続的にモニターした。溶媒(0.5%濃度のエタノールを含む生理食塩水)あるいは化合物(I)(10−100 μg/kg、0.5 ml/kg)を上記カテーテルを介して10−20分間隔で大腿動脈内に累積投与した。溶媒投与時の全身平均血圧および心拍数を100%として各用量投与後の最大変化率を相対変化率%として表した。
(結果)
図5に示すように、化合物(I)は用量依存的に全身平均血圧を低下した。10 μg/kgでは有意な変動は見られなかったが、30および100 μg/kgでは全身血圧をそれぞれ25±5.2 %、50±2.1 %有意に低下した。また、100 μg/kgまで心拍数には有意な変動は認められなかった。
【産業上の利用可能性】
【0027】
本発明の有効成分である化合物(I)又はその塩は、強力な血小板凝集阻害作用と血管拡張作用を有しており、化合物(I)の使用を局所に限定することで、安全性に優れ、効果も期待できるPAD治療剤/重症PAD治療剤を提供できる。本発明により、重症PADの症状である安静時疼痛の軽減、潰瘍・壊死の進展阻止、救肢率の向上が期待できる。具体的には、PADのうち閉塞性動脈硬化症やBuerger病にも有効であることが期待できる。
【図面の簡単な説明】
【0028】
【図1】は、各濃度における塩酸フェニレフリン(内皮あり、内皮なしの両条件)、5−HT、ET−1、又は、塩化カリウムによるラット大動脈標本の収縮度に対する化合物(I)の阻害作用を示すグラフである。
【図2】は、ラット大腿動脈内に化合物(I)の各用量を単回投与5分後のADP惹起血小板凝集阻害活性を示すグラフである。
【図3】は、ラット大腿動脈内にラウリン酸を投与後3日における各薬剤投与群における病変スコアを示すグラフである。
【図4】は、ラット大腿動脈内にラウリン酸を投与した前後における後肢皮膚血流量の変動をレーザードップラー皮膚血流計で観察した際のカラー画像である。
【図5】は、化合物(I)をラット大腿動脈内に各用量、単回投与した5分後での平均血圧および心拍数を示すグラフである。
【特許請求の範囲】
【請求項1】
G蛋白質Gq/11阻害作用を有する式(I)で示される化合物を有効成分として含有する末梢動脈閉塞症予防及び/又は治療剤。
【化1】
【請求項2】
注射剤あるいはステント溶出剤である、請求項1に記載の末梢動脈閉塞症予防及び/又は治療剤。
【請求項3】
末梢動脈閉塞症予防及び/又は治療剤製造のための、化合物(I)又はその製薬学的に許容される塩の使用。
【請求項4】
化合物(I)又はその製薬学的に許容される塩の治療有効量を患者に投与することを含む末梢動脈閉塞症予防及び/又は治療の方法。
【請求項1】
G蛋白質Gq/11阻害作用を有する式(I)で示される化合物を有効成分として含有する末梢動脈閉塞症予防及び/又は治療剤。
【化1】
【請求項2】
注射剤あるいはステント溶出剤である、請求項1に記載の末梢動脈閉塞症予防及び/又は治療剤。
【請求項3】
末梢動脈閉塞症予防及び/又は治療剤製造のための、化合物(I)又はその製薬学的に許容される塩の使用。
【請求項4】
化合物(I)又はその製薬学的に許容される塩の治療有効量を患者に投与することを含む末梢動脈閉塞症予防及び/又は治療の方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2008−56657(P2008−56657A)
【公開日】平成20年3月13日(2008.3.13)
【国際特許分類】
【出願番号】特願2007−116394(P2007−116394)
【出願日】平成19年4月26日(2007.4.26)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 1)日本薬理学会会誌128,23−31,2006(平成18年7月1日) 2)European Journal of Pharmacology,Volume 536,Issues 1−2,24 April 2006,Pages154−161(平成18年3月3日電子ジャーナル公開) URL:http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6T1J−4JD0FCR−2−C&_cdi=4892&_user=126699&_orig=search&_coverDate=04%2F24%2F2006&_sk=994639998&view=c&wchp=dGLbVtz−zSkWz&md5=54445c876ff3d5a0a9c32ca98a4a21ad&ie=/sdarticle.pdf
【出願人】(000006677)アステラス製薬株式会社 (274)
【Fターム(参考)】
【公開日】平成20年3月13日(2008.3.13)
【国際特許分類】
【出願日】平成19年4月26日(2007.4.26)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 1)日本薬理学会会誌128,23−31,2006(平成18年7月1日) 2)European Journal of Pharmacology,Volume 536,Issues 1−2,24 April 2006,Pages154−161(平成18年3月3日電子ジャーナル公開) URL:http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6T1J−4JD0FCR−2−C&_cdi=4892&_user=126699&_orig=search&_coverDate=04%2F24%2F2006&_sk=994639998&view=c&wchp=dGLbVtz−zSkWz&md5=54445c876ff3d5a0a9c32ca98a4a21ad&ie=/sdarticle.pdf
【出願人】(000006677)アステラス製薬株式会社 (274)
【Fターム(参考)】
[ Back to top ]