
架橋グリコペプチド−セファロスポリン抗生物質
【課題】独自の化学構造を有する架橋グリコペプチド−セファロスポリン新規抗生物質を提供する。
【解決手段】抗生物質として有用な架橋グリコペプチド−セファロスポリン化合物およびその薬学的に受容可能な塩。また、このような化合物を含有する薬学的組成物;このような化合物を用いて哺乳動物における細菌感染を処置するための方法;ならびにこのような化合物を調製するために有用なプロセスおよび中間体。特性のなかでもとりわけ、化合物は、メチシリン耐性Staphylococci aureus(MRSA)を含むグラム陽性細菌に対して、驚くべき予測されなかった能力を有することが見出されている。
【解決手段】抗生物質として有用な架橋グリコペプチド−セファロスポリン化合物およびその薬学的に受容可能な塩。また、このような化合物を含有する薬学的組成物;このような化合物を用いて哺乳動物における細菌感染を処置するための方法;ならびにこのような化合物を調製するために有用なプロセスおよび中間体。特性のなかでもとりわけ、化合物は、メチシリン耐性Staphylococci aureus(MRSA)を含むグラム陽性細菌に対して、驚くべき予測されなかった能力を有することが見出されている。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の背景)
(発明の分野)
本発明は、抗生物質として有用な新規架橋バンコマイシン−セファロスポリン化合物に関する。本発明はまた、このような化合物を含有する薬学的組成物;このような化合物を抗細菌剤として使用する方法;ならびにこのような化合物を調製するために有用なプロセスおよび中間体に関する。
【背景技術】
【0002】
(当該分野の現状)
種々のクラスの抗生物質化合物は、当該分野において公知であり、これらの化合物としては、例えば、β−ラクタム抗生物質(例えば、セファロスポリン)およびグリコペプチド抗生物質(例えば、バンコマイシン)が挙げられる。架橋抗生物質化合物はまた、当該分野において公知である。例えば、W.L.Truettに対して発行され、そして「Antibiotics and Process for Preparation」と題された特許文献1;1999年12月16日に発行され、そして「Novel Antibacterial Agents」と題された特許文献2を参照のこと。さらに、2003年4月17日に発行され、そして「Cross−Linked Glycopeptide−Cephalosporin Antibiotics」と題された特許文献3は、セファロスポリン基のオキシム部分に共有結合したグリコペプチド基を有する化合物を開示している。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】米国特許第5,693,791号明細書
【特許文献2】国際公開第99/64049号パンフレット
【特許文献3】国際公開第03/031449号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0004】
しかし、細菌が抗生物質に対する耐性を発達する能力に起因して、独自の化学構造を有する新規抗生物質に対する必要性が存在している。さらに、改善された抗細菌特性を有する新規抗生物質に対する必要性が存在し、これらの抗細菌特性としては、例えば、グラム陽性細菌に対する増強した能力が挙げられる。特に、細菌の抗生物質耐性株(例えば、メチシリン耐性Staphylococci aureus(MRSA))に対して高度に効果的な新規抗生物質に対する必要性が存在している。
【課題を解決するための手段】
【0005】
(発明の要旨)
本発明は、抗生物質として有用な架橋グリコペプチド−セファロスポリン化合物を提供する。本発明の化合物は、独自の化学構造を有し、この化学構造において、グリコペプチド基は、セファロスポリン基のピリジニウム部分に共有結合している。特性のなかでもとりわけ、本発明の化合物は、メチシリン耐性Staphylococci aureus(MRSA)を含むグラム陽性細菌に対して、驚くべき予測されなかった能力を有することが見出されている。
【0006】
従って、本発明の1つの局面において、本発明は、式I:
【0007】
【化8】

の化合物またはその薬学的に受容可能な塩を提供し、ここで、
X1およびX2の各々は、独立して、水素またはクロロであり;
Wは、NおよびCClからなる群より選択され;
R2は、水素またはC1〜6アルキルであり;
R4およびR5のうちの一方は、ヒドロキシであり、そして他方は、水素であり;
R6およびR7の各々は、独立して、水素またはメチルであり;
R8は、水素または式:
【0008】
【化9】
の基であり;
R9は、水素、C1〜6アルキルおよびC3〜6シクロアルキルから選択され、ここで、アルキルおよびシクロアルキルは、−COOHまたは1個〜3個のフッ素原子で置換され得;
各R3は、独立して、C1〜6アルキル、−OR、ハロ、−SR、−S(O)R、−S(O)2Rおよび−S(O)2ORから選択され、ここで、各Rは、独立して、C1〜6アルキルであり、このC1〜6アルキルは、COOHまたは1個〜3個のフッ素原子で置換され得;
nは、0、1、2または3であり;
xは、0、1または2であり;
yは、0、1または2であり;
Raは、−Y−R”−であり、ここで、
R”は、C1〜12アルキレン、C2〜12アルケニレン、C2〜12アルキニレン、C3〜6シクロアルキレン、C6〜10アリーレン、C2〜9ヘテロアリーレン、C3〜6複素環およびその組み合わせから選択され、そして必要に応じて、Zから選択される1個〜2個の基で置換され、ここで、Zは、−OR’、−SR’、−F、−Cl、−N(R’)2、−OC(O)R’、−C(O)OR’、−NHC(O)R’、−C(O)N(R’)2、−CF3および−OCF3、ならびに天然に存在するアミノ酸の側鎖からなり、ここで、各R’は、独立して、水素またはC1〜4アルキルであり;そしてR”は、最も多くて20個の非水素原子を含み;
Yは、メタ位またはパラ位でR”をピリジニウム環に結合し、直接結合、NR’、O(エーテル)、S(スルフィド)、C(O)(カルボニル)、NR’(CO)およびC(O)NR’から選択されて、Yにおけるヘテロ原子とR”におけるヘテロ原子とも間の直接結合を除外し;
各RbおよびRdは、独立して、水素、C1〜6アルキル、C2〜6アルケニルおよびC2〜6アルキニルからなる群より選択され;
各Rcは、独立して、−Y’−R”−Y’−であり、ここで、各Y’は、独立して、直接結合、O(エーテル)およびNR’からなる群よりされ、Y’におけるヘテロ原子とR”におけるヘテロ原子との間の直接結合を除外し;そして
各Reは、独立して、上記R”により規定される基から選択される。
【0009】
別の局面において、本発明は、式II:
【0010】
【化10】
の化合物またはその薬学的に受容可能な塩を提供し;ここで、
Wは、NおよびCClからなる群より選択され;
R9は、水素、C1〜6アルキルおよびC3〜6シクロアルキルから選択され、ここで、アルキルおよびシクロアルキルは、−COOHまたは1個〜3個のフッ素原子で置換され得;
ピリジニウム環は、メタ置換またはパラ置換を有し
R10は、水素またはC1〜6アルキルであり;そして
R11は、C1〜12アルキレンである。
【0011】
別のその組成物の局面において、本発明は、薬学的に受容可能なキャリアおよび治療有効量の式Iもしくは式IIの化合物またはその薬学的に受容可能な塩を含有する薬学的組成物を提供し、本明細書中で議論されるあらゆる特定の実施形態を含む。
【0012】
細菌と、増殖阻害量の式Iもしくは式IIの化合物またはその薬学的に受容可能な塩とを接触させることによって、細菌の増殖を阻害する方法および/または細菌の細胞壁生合成を阻害する方法がまた、提供され、本明細書中で議論されるあらゆる特定の実施形態を含む。特に、上記方法は、上記化合物が、本明細書中でIa、Ib、Ic、Id、IeおよびIfとして示される化合物からなる群より選択される実施形態を含む。
【0013】
関連する局面において、本発明は、哺乳動物における細菌感染を処置する方法を提供し、この方法は、薬学的に受容可能なキャリアおよび治療有効量の式Iもしくは式IIの化合物、またはその薬学的に受容可能な塩を含有する薬学的組成物を、哺乳動物に投与する工程を包含し、本明細書中で議論されるあらゆる特定の実施形態を含む。
【0014】
本発明はまた、式Iもしくは式IIの化合物、またはその塩を調製するためのプロセスに関する。従って、別のその方法の局面において、本発明は、式Iの化合物またはその塩を調製するためのプロセスを提供し;このプロセスは、式1の化合物またはその塩、その活性化誘導体もしくはその保護された誘導体と、式3もしくは式4の化合物またはその塩、その活性化誘導体もしくはその保護された誘導体とを反応させて;式Iの化合物またはその塩を提供する工程を包含し;ここで、上記式1、式3および式4の化合物は、本明細書中で規定されるとおりである。
【0015】
さらに、別のその方法の局面において、本発明は、式Iの化合物またはその塩を調製するためのプロセスを提供し;このプロセスは、式2の化合物またはその塩、その活性化誘導体もしくはその保護された誘導体と;式5の化合物またはその塩、その活性化誘導体もしくはその保護された誘導体とを反応させて;式Iの化合物またはその塩を提供する工程を包含し;ここで、上記式2および式5の化合物は、本明細書中に規定されるとおりである。
【0016】
1つの実施形態において、これらのプロセスは、式Iの化合物の薬学的に受容可能な塩を形成する工程をさらに包含する。本発明はまた、あらゆるこれらのプロセスによって調製される生成物に関する。
【0017】
本発明はまた、治療における使用のための、式Iもしくは式IIの化合物、またはその薬学的に受容可能な塩に関する。さらに、本発明は、医薬の製造のための、式Iもしくは式IIの化合物、またはその薬学的に受容可能な塩の使用に関し、これらの医薬としては、哺乳動物における細菌感染を処置するための医薬が挙げられる。
本発明は、例えば以下の項目を提供する。
(項目1)
式I:
【化1】
の化合物またはその薬学的に受容可能な塩であって、ここで:
X1およびX2の各々は、独立して、水素またはクロロであり;
Wは、NおよびCClから選択され;
R2は、水素またはC1〜6アルキルであり;
R4およびR5のうちの一方は、ヒドロキシであり、そして他方は、水素であり;
R6およびR7の各々は、独立して、水素またはメチルであり;
R8は、水素または式:
【化2】
の基であり;
R9は、水素、C1〜6アルキルおよびC3〜6シクロアルキルから選択され、ここで、アルキルおよびシクロアルキルは、−COOHまたは1個〜3個のフッ素原子で置換され得;
各R3は、独立して、C1〜6アルキル、−OR、ハロ、−SR、−S(O)R、−S(O)2Rおよび−S(O)2ORから選択され、ここで、各Rは、独立して、C1〜6アルキルであり、このC1〜6アルキルは、COOHまたは1個〜3個のフッ素原子で置換され得;
nは、0、1、2または3であり;
xは、0、1または2であり;
yは、0、1または2であり;
Raは、−Y−R”−であり、ここで、
R”は、C1〜12アルキレン、C2〜12アルケニレン、C2〜12アルキニレン、C3〜6シクロアルキレン、C6〜10アリーレン、C2〜9ヘテロアリーレン、C3〜6複素環およびその組み合わせから選択され、そして必要に応じて、Zから選択される1個〜2個の基で置換され、ここで、Zは、−OR’、−SR’、−F、−Cl、−N(R’)2、−OC(O)R’、−C(O)OR’、−NHC(O)R’、−C(O)N(R’)2、−CF3および−OCF3、ならびに天然に存在するアミノ酸の側鎖からなり、ここで、各R’は、独立して、水素またはC1〜4アルキルであり;そしてR”は、最も多くて20個の非水素原子を含み;
Yは、メタ位またはパラ位でR”をピリジニウム環に結合し、直接結合、NR’、O、S、C(O)、NR’C(O)およびC(O)NR’から選択されて、Yにおけるヘテロ原子とR”におけるヘテロ原子との間の直接結合除外し;
各RbおよびRdは、独立して、水素、C1〜6アルキル、C2〜6アルケニルおよびC2〜6アルキニルからなる群より選択され;
各Rcは、独立して、−Y’−R”−Y’−であり、ここで、各Y’は、独立して、直接結合、OおよびNR’から選択され、Y’におけるヘテロ原子とR”におけるヘテロ原子との間の直接結合を除外し;そして
各Reは、独立して、R”から選択される、化合物またはその薬学的に受容可能な塩。
(項目2)
項目1に記載の化合物であって、ここで、R9は、水素またはC1〜4アルキルである、化合物。
(項目3)
項目2に記載の化合物であって、ここで、R9は、水素またはメチルである、化合物。
(項目4)
項目1〜3のいずれか1項に記載の化合物であって、ここで、Wは、CClである、化合物。
(項目5)
項目1〜3のいずれか1項に記載の化合物であって、ここで、Wは、Nである、化合物。
(項目6)
項目1〜5のいずれか1項に記載の化合物であって、ここで、各R3は、独立して、非置換C1〜4アルキル、非置換C1〜4アルコキシ、フルオロおよびクロロから選択される、化合物。
(項目7)
項目1〜6のいずれか1項に記載の化合物であって、ここで、nは、0である、化合物。
(項目8)
項目1〜7のいずれか1項に記載の化合物であって、ここで、xは、1であり、そしてyは、0である、化合物。
(項目9)
項目8に記載の化合物であって、ここで、Raは、−Y−R”−であり;Yは、直接結合であり;そしてR”は、C1〜6アルキレンである、化合物。
(項目10)
項目8または9に記載の化合物であって、ここで、Rcは、−Y’−R”−Y’−であり、ここで、各Y’は、直接結合であり;そしてR”は、必要に応じて−COOH基で置換されたC1〜12アルキレンである、化合物。
(項目11)
項目8〜10のいずれか1項に記載の化合物であって、ここで、Rbは、水素またはC1〜4アルキルである、化合物。
(項目12)
項目1〜7のいずれか1項に記載の化合物であって、ここで、xは、1であり、そしてyは、1である化合物。
(項目13)
項目12に記載の化合物であって、ここで、Raは、−Y−R”−であり;Yは、直接結合であり;そしてR”は、C1〜6アルキレンである、化合物。
(項目14)
項目12または13に記載の化合物であって、ここで、Rcは、−Y’−R”−Y’−であり、ここで、各Y’は、直接結合であり;そしてR”は、必要に応じて−COOH基で置換されるC1〜12アルキレンである、化合物。
(項目15)
項目12〜14のいずれか1項に記載の化合物であって、ここで、Reは、C1〜12アルキレンである、化合物。
(項目16)
項目12〜15のいずれか1項に記載の化合物であって、ここで、RbおよびRdは、独立して、水素またはC1〜4アルキルである、化合物。
(項目17)
項目1〜7のいずれか1項に記載の化合物であって、ここで、xは、0であり、そしてyは、0である、化合物。
(項目18)
項目17に記載の化合物であって、ここで、Raは、−Y−R”−であり;Yは、直接結合であり;そしてR”は、C1〜6アルキレンである、化合物。
(項目19)
項目1〜18のいずれか1項に記載の化合物であって、ここで、R2は、水素である、化合物。
(項目20)
式II:
【化3】
の化合物またはその薬学的に受容可能な塩であって、ここで、
Wは、NおよびCClから選択され;
R9は、水素、C1〜6アルキルおよびC3〜6シクロアルキルから選択され、ここで、アルキルおよびシクロアルキルは、−COOHまたは1個〜3個のフッ素原子で置換され得;
ピリジニウム環は、メタ置換またはパラ置換を有し;
R10は、水素またはC1〜6アルキルであり;そして
R11は、C1〜12アルキレンである、化合物またはその薬学的に受容可能な塩。
(項目21)
項目20に記載の化合物であって、ここで、Wは、CClである、化合物。
(項目22)
項目20または21に記載の化合物であって、ここで、ここで、R9は、水素またはC1〜4アルキルである、化合物。
(項目23)
項目22に記載の化合物であって、ここで、R9は、水素である、化合物。
(項目24)
項目22に記載の化合物であって、ここで、R9は、メチルである、化合物。
(項目25)
項目20〜24のいずれか1項に記載の化合物であって、ここで、前記ピリジニウム環は、パラ置換されている、化合物。
(項目26)
項目20〜25のいずれか1項に記載の化合物であって、ここで、R10は、水素またはC1〜4アルキルである、化合物。
(項目27)
項目26に記載の化合物であって、ここで、R10は、水素、メチルまたはエチルである、化合物。
(項目28)
項目20〜27のいずれか1項に記載の化合物であって、ここで、R11は、C1〜10アルキレンである、化合物。
(項目29)
項目28に記載の化合物であって、ここで、R11は、−(CH2)2−または−(CH2)5−である、化合物。
(項目30)
項目20に記載の化合物であって、ここで、Wは、CClであり;R9は、メチル
であり;R10は、エチルであり;R11は、−(CH2)2−であり;そして前記ピリジニウム環は、パラ置換されている、化合物。
(項目31)
薬学的に受容可能なキャリアおよび治療有効量の項目1〜30のいずれか1項に記載の化合物を含有する、薬学的組成物。
(項目32)
細菌の増殖を阻害する方法であって、該方法は、細菌と、増殖阻害量の項目1〜30のいずれか1項に記載の化合物とを接触させる工程を包含する、方法。
(項目33)
哺乳動物における細菌感染を処置する方法であって、該方法は、哺乳動物に、薬学的に受容可能なキャリアおよび治療有効量の項目1〜30のいずれか1項に記載の化合物を含有する薬学的組成物を投与する工程を包含する、方法。
(項目34)
項目1〜30のいずれか1項に記載の化合物を調製するためのプロセスであって;該プロセスは、式1:
【化4】
の化合物またはその塩もしくはその活性化誘導体もしくはその保護された誘導体と、式3または4:
【化5】
の化合物またはその塩もしくはその活性化誘導体もしくはその保護された誘導体を反応させて、式Iの化合物またはその塩を得る工程を包含する、プロセス。
(項目35)
項目1〜30のいずれか1項に記載の化合物を調製するためのプロセスであって;該プロセスは、式5:
【化6】
の化合物またはその塩もしくはその活性化誘導体もしくは保護された誘導体を;式2:
【化7】
の化合物またはその塩もしくはその活性化誘導体もしくは保護された誘導体と反応させて、式Iの化合物またはその塩を得る工程を包含する、プロセス。
(項目36)
項目34または35に記載のプロセスによって調製される生成物。
【図面の簡単な説明】
【0018】
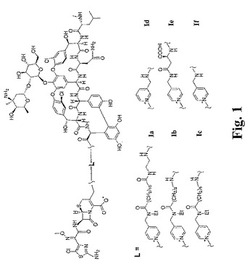
【図1】図1は、本発明の選択された実施形態に従う架橋グリコペプチド−セファロスポリン抗生物質の例を示す。
【図2】図2は、架橋グリコペプチド−セファロスポリン抗生物質を調製するために有用なセファロスポリン中間体を調製するための合成スキームを示す。
【図3】図3〜図6は、本明細書中で、それぞれ、Ia、Ib、IdおよびIeとして示される代表的な架橋グリコペプチド−セファロスポリン抗生物質を調製するための合成スキームを示す。
【図4】図3〜図6は、本明細書中で、それぞれ、Ia、Ib、IdおよびIeとして示される代表的な架橋グリコペプチド−セファロスポリン抗生物質を調製するための合成スキームを示す。
【図5】図3〜図6は、本明細書中で、それぞれ、Ia、Ib、IdおよびIeとして示される代表的な架橋グリコペプチド−セファロスポリン抗生物質を調製するための合成スキームを示す。
【図6】図3〜図6は、本明細書中で、それぞれ、Ia、Ib、IdおよびIeとして示される代表的な架橋グリコペプチド−セファロスポリン抗生物質を調製するための合成スキームを示す。
【図7】図7および図8は、比較のための架橋グリコペプチド−セファロスポリン抗生物質のデス−クロロアナログを調製するための合成スキームを示す。
【図8】図7および図8は、比較のための架橋グリコペプチド−セファロスポリン抗生物質のデス−クロロアナログを調製するための合成スキームを示す。
【図9】図9および図10は、代表的な架橋グリコペプチド−セファロスポリン抗生物質をさらに調製するための合成スキームを示す。
【図10】図9および図10は、代表的な架橋グリコペプチド−セファロスポリン抗生物質をさらに調製するための合成スキームを示す。
【発明を実施するための形態】
【0019】
(発明の詳細な説明)
本発明は、式Iもしくは式IIの新規グリコペプチド−セファロスポリン化合物、またはその薬学的に受容可能な塩を提供する。これらの化合物は、複数のキラル中心を有し、そして、この点において、上記化合物は、示される立体化学を有することが意図される。特に、上記化合物のグリコペプチド部分は、対応する天然に存在するグリコペプチド(すなわち、バンコマイシン、クロロオリエンチシンAなど)の立体化学を有することが意図される。上記分子のセファロスポリン部分は、公知のセファロスポリン化合物の立体化学を有することが意図される。しかし、示されるものとは異なる立体化学を有する少量の異性体が、本発明の組成物中に存在し得、ただし、全体としての上記組成物の有用性は、このような異性体の存在によってそれほど減少されないことを、当業者は理解する。
【0020】
さらに、本発明の化合物の結合部分は、1つ以上のキラル中心を含み得る。代表的に、上記分子のこの部分は、ラセミ混合物として調製される。しかし、所望される場合、純粋な立体異性体(すなわち、個々のエナンチオマーまたはジアステレオマー)が使用され得るか、または立体異性体エンリッチ混合物が用いられ得る。全てのこのような立体異性体およびエンリッチ混合物は、本発明の範囲内に含まれる。
【0021】
さらに、本発明の化合物は、いくつかの酸性基(すなわち、カルボン酸基)およびいくつかの塩基性基(すなわち、一級アミン基および二級アミン基)を含み、そして、そのため、上記式Iの化合物は、種々の塩形態で存在し得る。全てのこのような塩形態は、本発明の範囲内に含まれる。また、上記式Iの化合物は、ピリジニウム環を含むので、上記ピリジニウム基のための陰イオン性対イオンが、必要に応じて存在し、これらの陰イオン性対イオンとしては、ハライド(例えば、クロライド);カルボキシレート(例えば、アセテート)などが挙げられるが、これらに限定されない。
(定義)
以下の用語は、本明細書中で使用される場合、別に示されなければ、以下の意味を有する:
用語「アルキル」とは、一価飽和炭化水素基をいい、直鎖状でも分枝していてもよい。別に規定されなければ、このようなアルキル基は、代表的に、1個〜10個の炭素原子を含む。代表的なアルキル基としては、例として、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、sec−ブチル、イソブチル、tert−ブチル、n−ペンチル、n−ヘキシル、n−ヘプチル、n−オクチル、n−ノニル、n−デシルなどが挙げられる。
【0022】
用語「アルキレン」とは、二価飽和炭化水素基をいい、直鎖状でも分枝していてもよい。別に規定されなければ、このようなアルキレン基は、代表的に、1個〜10個の炭素原子を含む。代表的なアルキレン基としては、例として、メチレン、エタン−1,2−ジイル(「エチレン」)、プロパン−1,2−ジイル、プロパン−1,3−ジイル、ブタン−1,4−ジイル、ペンタン−1,5−ジイルなどが挙げられる。
【0023】
用語「アルケニル」とは、一価不飽和炭化水素基をいい、直鎖状でも分枝していてもよく、少なくとも1つ、そして代表的には、1つ、2つまたは3つの炭素−炭素二重結合を有する。別に規定されなければ、このようなアルケニル基は、代表的に、2個〜10個の炭素原子を含む。代表的なアルケニル基としては、例として、エテニル、n−プロペニル、イソプロペニル、n−ブタ−2−エニル、n−ヘキサ−3−エニルなどが挙げられる。
【0024】
用語「アルキニル」とは、一価不飽和炭化水素基をいい、直鎖状でも分枝していてもよく、少なくとも1つ、そして代表的には、1つ、2つまたは3つの炭素−炭素三重結合を有する。別に規定されなければ、このようなアルキニル基は、代表的に、2個〜10個の炭素原子を含む。代表的なアルキニル基としては、例として、エチニル、n−プロピニル、n−ブタ−2−イニル、n−ヘキサ−3−イニルなどが挙げられる。
【0025】
用語「アリール」とは、単一の環(すなわち、フェニル)または縮合環(すなわち、ナフタレン)を有する一価芳香族炭化水素をいう。別に規定されなければ、このようなアリール基は、代表的に、6個〜10個の炭素環原子を含む。代表的なアリール基としては、例として、フェニルおよびナフタレン−1−イル、ナフタレン−2−イルなどが挙げられる。
【0026】
用語「アリーレン(arylene)」とは、単一の環(すなわち、フェニレン)または縮合環(すなわち、ナフタレンジイル)を有する二価芳香族炭化水素をいう。別に規定されなければ、このようなアリーレン基は、代表的に、6個〜10個の炭素環原子を含む。代表的なアリーレン基としては、例として、1,2−フェニレン、1,3−フェニレン、1,4−フェニレン、ナフタレン−1,5−ジイル、ナフタレン−2,7−ジイルなどが挙げられる。
【0027】
用語「シクロアルキル」とは、一価飽和炭素環式炭化水素基をいう。別に規定されなければ、このようなシクロアルキル基は、代表的に、3個〜10個の炭素原子を含む。代表的なシクロアルキル基としては、例として、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルなどが挙げられる。
【0028】
用語「シクロアルキレン」とは、二価飽和炭素環式炭化水素基をいう。別に規定されなければ、このようなシクロアルキル基は、代表的に、3個〜10個の炭素原子を含む。代表的なシクロアルキレン基としては、例として、シクロプロパン−1,2−ジイル、シクロブチル−1,2−ジイル、シクロブチル−1,3−ジイル、シクロペンチル−1,2−ジイル、シクロペンチル−1,3−ジイル、シクロヘキシル−1,2−ジイル、シクロヘキシル−1,3−ジイル、シクロヘキシル−1,4−ジイルなどが挙げられる。
【0029】
用語「ハロ」とは、フルオロ、クロロ、ブロモおよびヨードをいう。
【0030】
用語「ヘテロアリール」とは、単一の環または2つの縮合環を有し、そしてその環に、窒素、酸素または硫黄から選択される少なくとも1個のヘテロ原子(代表的には、1個〜3個のヘテロ原子)を含む一価芳香族基をいう。別に規定されなければ、このようなヘテロアリール基は、代表的に、全体で5個〜10個の環原子を含む。代表的なヘテロアリール基としては、例として、ピロール、イミダゾール、チアゾール、オキサゾール、フラン、チオフェン、トリアゾール、ピラゾール、イソキサゾール、イソチアゾール、ピリジン、ピラジン、ピリダジン、ピリミジン、トリアジン、インドール、ベンゾフラン、ベンゾチオフェン、ベンズイミダゾール、ベンズチアゾール、キノリン、イソキノリン、キナゾリン、キノキサリンなどの一価の種が挙げられ、ここで、その結合点は、あらゆる利用可能な炭素環原子または窒素環原子にある。
【0031】
用語「ヘテロアリーレン」とは、単一の環または2つの縮合環を有し、そしてその環に、窒素、酸素または硫黄から選択される少なくとも1個のヘテロ原子(代表的には、1個〜3個のヘテロ原子)を含む二価芳香族基をいう。別に規定されなければ、このようなヘテロアリーレン基は、代表的に、全体で5個〜10個の環原子を含む。代表的なヘテロアリーレン基としては、例として、ピロール、イミダゾール、チアゾール、オキサゾール、フラン、チオフェン、トリアゾール、ピラゾール、イソキサゾール、イソチアゾール、ピリジン、ピラジン、ピリダジン、ピリミジン、トリアジン、インドール、ベンゾフラン、ベンゾチオフェン、ベンズイミダゾール、ベンズチアゾール、キノリン、イソキノリン、キナゾリン、キノキサリンなどの二価の種が挙げられ、ここで、その結合点は、あらゆる利用可能な炭素環原子または窒素環原子にある。
【0032】
用語「ヘテロサイクリル」または「ヘテロサイクリック」とは、単一の環または複数の縮合環を有し、そしてその環に、窒素、酸素または硫黄から選択される少なくとも1個のヘテロ原子(代表的には、1個〜3個のヘテロ原子)を含む一価または二価の飽和基または不飽和(非芳香族)基をいう。別に規定されなければ、このようなヘテロサイクリック基は、代表的に、全体で2個〜9個の環原子を含む。代表的なヘテロサイクリック基としては、例として、ピロリジニン、イミダゾリジン、ピラゾリジン、ピペリジン、1,4−ジオキサン、モルホリン、チオモルホリン、ピペラジン、3−ピロリンなどの一価の種が挙げられ、ここで、その結合点は、あらゆる利用可能な炭素環原子または窒素環原子にある。
【0033】
用語「セファロスポリン」は、本明細書中でその当該分野で認識される様式で使用されて、以下の一般式および番号システム:
【0034】
【化11】
を有するβ−ラクタム環系をいい、ここで、RxおよびRyは、上記セファロスポリンの残りの部分を示す。
【0035】
用語「グリコペプチド抗生物質」または「グリコペプチド」は、本明細書中でその当該分野で認識される様式で使用されて、グリコペプチドまたはダルバー(dalbah)ペプチドとして公知の抗生物質のクラスをいう。例えば、R.Nagarajan,「Glycopepitide Antibiotics」,Marcel Dekker,Inc.(1994)およびそこに引用される参考文献を参照のこと。代表的なグリコペプチドとしては、バンコマイシン、A82846A(エレモマイシン)、A82846B(クロロオリエンチシンA)、A82846C、PA−42867−A(オリエンチシンA)、PA−42867−C、PA−42867−Dなどが挙げられる。
【0036】
用語「バンコマイシン」は、本明細書中でその当該分野で認識される様式で使用されて、バンコマイシンとして公知のグリコペプチド抗生物質をいう。本発明の化合物において、上記結合部分のための結合点は、バンコマイシンの「C末端」にある。
【0037】
用語「架橋グリコペプチド−セファロスポリン抗生物質」とは、グリコペプチド成分とセファロスポリン成分との共有結合体をいう。
【0038】
用語「薬学的に受容可能な塩」とは、患者(例えば、哺乳動物)への投与のために受容可能な塩(例えば、所与の投薬レジメンのために受容可能な哺乳動物安全性を有する塩)をいう。このような塩は、薬学的に受容可能な無機塩基または有機塩基および薬学的に受容可能な無機酸または有機酸に由来し得る。薬学的に受容可能な無機塩基由来の塩としては、アルミニウム、アンモニウム、カルシウム、銅、第二鉄、第一鉄、リチウム、マグネシウム、第二マンガン、第一マンガン、カリウム、ナトリウム、亜鉛などが挙げられる。アンモニウム塩、カルシウム塩、マグネシウム塩、カリウム塩およびナトリウム塩が、特に好ましい。薬学的に受容可能な有機塩基由来の塩としては、置換アミン、環状アミン、天然に存在するアミンなど(例えば、アルギニン、ベタイン、カフェイン、コリン、N,N’−ジベンジルエチレンジアミン、ジエチルアミン、2−ジエチルアミノエタノール、2−ジメチルアミノエタノール、エタノールアミン、エチレンジアミン、N−エチルモルホリン、N−エチルピペリジン、グルカミン、グルコサミン、ヒスチジン、ヒドラバミン、イソプロピルアミン、リジン、メチルグルカミン、モルホリン、ピペラジン、ピペリジン、ポリアミン樹脂、プロカイン、プリン、テオブロミン、トリエチルアミン、トリメチルアミン、トリプロピルアミン、トロメタミンなど)を含む、一級アミン、二級アミンおよび三級アミンの塩が挙げられる。薬学的に受容可能な酸由来の塩としては、酢酸、アスコルビン酸、ベンゼンスルホン酸、安息香酸、樟脳スルホン酸、クエン酸、エタンスルホン酸、フマル酸、グルコン酸、グルコロン酸、グルタミン酸、馬尿酸、臭化水素酸、塩酸、イセチオン酸、乳酸、ラクトビオン酸、マレイン酸、リンゴ酸、マンデル酸、メタンスルホン酸、ムチン酸、ナフタレンスルホン酸、ニコチン酸、硝酸、パモ酸、パントテン酸、リン酸、コハク酸、硫酸、酒石酸、p−トルエンスルホン酸など由来の塩が挙げられる。特に好ましいのは、クエン酸、臭化水素酸、塩酸、マレイン酸、リン酸、硫酸および酒石酸である。
【0039】
用語「その塩」とは、酸の水素が、陽イオン(例えば、金属陽イオンまたは有機陽イオンなど(例えば、NH4+陽イオンなど))によって置換される場合に形成される化合物をいう。好ましくは、上記塩は、薬学的に受容可能な塩であるが、これは、患者への投与が意図されない中間体化合物の塩については必要とされない。
【0040】
用語「治療有効量」とは、処置を必要としている患者に投与される場合、有効な処置のために充分な量をいう。
【0041】
用語「処置する」または「処置」とは、本明細書中で使用される場合、患者(例えば、哺乳動物(特に、ヒトまたは伴生種動物(companion animal)))における疾患もしくは医学的状態(例えば、細菌感染)を処置することまたはその処置をいい:
(a)上記疾患または医学的状態の発症を防止する工程(すなわち、患者の予防処置);
(b)上記疾患または医学的状態を改善する工程(すなわち、患者における上記疾患もしくは医学的状態を除去する工程またはその後退を引き起こす工程);
(c)上記疾患または医学的状態を抑制する工程(すなわち、患者における上記疾患もしくは医学的状態の進行を遅延させる工程または妨げる工程);あるいは
(d)患者における上記疾患または医学的状態の症状を軽減する工程
を包含する。
【0042】
用語「増殖阻害量」とは、微生物の増殖もしくは生殖を阻害するために充分な量または上記微生物(グラム陽性細菌を含む)の死もしくは溶解を引き起こすために充分な量をいう。
【0043】
用語「細胞壁生合成阻害量」とは、微生物(グラム陽性細菌を含む)における細胞壁生合成を阻害するために充分な量をいう。
【0044】
用語「脱離基」とは、置換反応(例えば、求核置換反応)において別の官能基または原子によって置換され得る官能基または原子をいう。例として、代表的な脱離基としては、クロロ基、ブロモ基およびヨード基;ならびにスルホン酸エステル基(例えば、メシレート、トシレート、ブロシレート、ノシレートなど);活性化エステル基(例えば、7−アザベンゾトリアゾール−1−オキシなど);アシルオキシ基(例えば、アセトキシ、トリフルオロアセトキシなど)が挙げられる。
【0045】
用語「その保護された誘導体」とは、保護基またはブロック基により、上記化合物の1つ以上の官能基を、所望されない反応から保護する特定の化合物の誘導体をいう。保護され得る官能基としては、例として、カルボン酸基、アミノ基、ヒドロキシ基、チオール基、カルボニル基などが挙げられる。カルボン酸のための代表的な保護基としては、エステル(例えば、p−メトキシベンジルエステル)、アミドおよびヒドラジド;アミノ基のためには、カルバメート(例えば、tert−ブトキシカルボニル)およびアミド;ヒドロキシ基のためには、エーテルおよびエステル;チオール基のためには、チオエーテルおよびチオエステル;カルボニル基のためには、アセタールおよびケタール;などが挙げられる。このような保護基は、当業者に周知であり、そして例えば、T.W.GreeneおよびG.M.Wuts,Protecting Groups in Organic Synthesis,第3版,Wiley,New York,1999ならびにそこに引用される参考文献に記載される。
【0046】
用語「アミノ保護基」とは、アミノ基における所望されない反応を防止するために適切な保護基をいう。代表的なアミノ保護基としては、tert−ブトキシカルボニル(BOC)、トリチル(Tr)、ベンジルオキシカルボニル(Cbz)、9−フルオレニルメトキシカルボニル(Fmoc)、ホルミル、トリメチルシリル(TMS)、tert−ブチルジメチルシリル(TBS)などが挙げられるが、これらに限定されない。
【0047】
用語「カルボキシ保護基」とは、カルボキシ基における所望されない反応を防止するために適切な保護基をいう。代表的なカルボキシ保護基としては、エステル(例えば、メチル、エチル、tert−ブチル、ベンジル(Bn)、p−メトキシベンジル(PMB)、9−フルオレニルメチル(Fm)、トリメチルシリル(TMS)、tert−ブチルジメチルシリル(TBS)、ジフェニルメチル(ベンズヒドリル、DPM)のエステル)などが挙げられるが、これらに限定されない。
【0048】
カルボン酸またはその保護された誘導体に関する「活性化誘導体」とは、上記カルボン酸または誘導体と、活性化(カップリング)剤(例えば、1−ヒドロキシベンゾトリアゾール(HOBT)、1−ヒドロキシ−7−アザベンゾトリアゾール(HOAT)または以下に記載されるか、もしくはそうでなければ当該分野で公知の他のもの)との反応によりもたらされる生成物(代表的には、反応性エステル)をいう。
【0049】
「天然に存在するアミノ酸の側鎖」とは、式HOOC−CHR−NH2における基であるRをいい、この式は、アラニン、アルギニン、アスパラギン、アスパラギン酸、システイン、グルタミン、グルタミン酸、グリシン、ヒスチジン、イソロイシン、ロイシン、リジン、メチオニン、フェニルアラニン、プロリン、セリン、スレオニン、トリプトファン、チロシンおよびバリンから選択されるアミノ酸、そして好ましくは、アラニン、アルギニン、アスパラギン、アスパラギン酸、システイン、グルタミン、グルタミン酸、グリシン、イソロイシン、ロイシン、リジン、メチオニン、セリン、スレオニンおよびバリンから選択されるアミノ酸を示す。
【0050】
(本発明の化合物の代表的な実施形態)
以下の置換基および値は、本発明の種々の局面の代表的な例および実施形態を提供することを意図する。これらの代表的な値は、このような局面および実施形態をさらに規定することを意図し、そして他の実施形態を除外することも本発明の範囲を限定することも意図しない。この点において、特定の値または置換基が好ましいという説明は、特に示されない限り、どのような方法においても、他の値または置換基を本発明から除外することを意図しない。
【0051】
式Iの化合物において、R”に存在するあらゆるヘテロアリーレン基またはヘテロサイクリック基は、好ましくは、全体で5個または6個、代表的には、6個の環原子を有し、各アリール基は、全体で6個の環原子を有する。上記R”基は、好ましくは、アルキレン鎖であり、そして好ましくは、直鎖状である。
【0052】
各R3基は、存在する場合、好ましくは、独立して、不飽和C1〜4アルキル、不飽和C1〜4アルコキシ、フルオロおよびクロロから選択される。1つの実施形態において、nは、1または2であり、各R3は、独立して、メチル、メトキシ、フルオロおよびクロロから選択される。別の実施形態において、nは、0であり、そのため、R3基は、存在しない。
【0053】
1つの好ましい実施形態において、R8は、水素である。別の好ましい実施形態において、R8は、式:
【0054】
【化12】
の基である。
【0055】
選択される実施形態において、R9は、水素またはC1〜4アルキルであり、例えば、水素またはメチルが挙げられる。1つの実施形態において、R9は、水素である。別の実施形態において、R9は、メチルである。
【0056】
1つの実施形態において、Wは、CClである。別の実施形態において、Wは、Nである。
【0057】
式Iの他の変更の選択される実施形態としては、互いに独立して、以下が挙げられる;X1およびX2については、クロロ;R2については、水素またはC1〜4アルキル;R4およびR5については、それぞれ、ヒドロキシおよび水素;R6およびR7については、それぞれ、水素およびメチル;そしてR8については、水素。
【0058】
他の選択された実施形態において、Raは、−Y−R”−であって、ここで、R”は、C1〜6アルキレン、C2〜6アルケニレンまはたC2〜6アルキニレンであり、そして、Yは、直接結合、NR’、O、S、C(O)、NR’(CO)および(CO)NR’から選択され、ここで、R’は、水素またはメチルである。一実施形態において、Yは、直接結合である。基Raのさらなる実施形態において、R”は、C1〜6アルキレン、またより好ましくは、C1〜4アルキレン(例えば、メチレン、エチレン、プロピレンまたはブチレン)である。
【0059】
好ましくは、基Raにおいて、Yは直接結合であり、そして、R”は、C1〜6アルキレンであるか、またはより好ましくは、C1〜4アルキレン(例えば、メチレン)である。
【0060】
式Iのピリジニウム環は、代表的には、メタ置換もしくはパラ置換されており、より一般的には、パラ置換されている。
【0061】
好ましくは、変数xは0または1である。xが0でない場合、基Rbは好ましくは、水素、C1〜4アルキルまたはC2〜4アルケニルからなる群より選択される。一実施形態において、Rbは、水素またはC1〜4アルキルである。
【0062】
xが0でない場合、基Rcは、好ましくは、−Y’−R”−Y’−であり、ここで、各Y’は、独立して、直接結合、OおよびNR’から選択され、ここで、R’は水素またはメチルであり、そして、R”は、C1〜12アルキレン、C2〜12アルケニレンおよびC2〜12アルキニレンから選択され、ここで、アルキレン基、アルケニレン基またはアルキニレン基は、必要に応じて、1〜2つの基で置換され、この基は、Zまたは天然に存在するアミノ酸の側鎖より選択される。好ましくは、基Rcにおいて、各Y’は直接結合であり、そしてR”はZおよび天然に存在するアミノ酸の側鎖より選択される1〜2つの基で置換され得るC1〜12アルキニレンまたはC2〜12アルケニレンである。基Rcの別の実施形態において、R”はC1〜12アルキレン(C1〜6アルキレンを含む)であり、この基は、非置換であるか、または−COOH基で置換されている。
【0063】
好ましくは、変数yは0または1である。yが0でない場合、基Rdは、好ましくは、水素およびC1〜4アルキルからなる群より選択され、選択された実施形態においては、水素およびメチルである。一実施形態において、RdはHである。yが0でない場合、基Reは、好ましくは、C1〜12アルキレン、C2〜12アルケニレンおよびC2〜12アルキニレンより選択される。一実施形態において、Reは、C1〜12アルキレンである。より好ましくは、Reは、C1〜6アルキレン、C2〜6アルケニレンおよびC2〜6アルキニレンから選択され、最も好ましくは、C2〜4アルケニレンおよびC1〜4アルキレンより選択される。
【0064】
選択された実施形態において、基R2は、水素またはC1〜4アルキルである。さらなる実施形態において、R2は、水素またはメチルであり;一実施形態において、R2は水素である。
【0065】
選択された実施形態において、xおよびyは独立して0および1より選択される。従って、好ましい実施形態としては、x+y=0の化合物、x+y=1の化合物およびx+y=2の化合物が挙げられる。あるいは、好ましい実施形態としては、図1において「L」で表される「リンカー」構造が、ピリジニウム環を除いてわずか約30個の炭素原子を含む化合物が挙げられる。
【0066】
x=y=1である本発明の化合物の例は、本明細書においてIaと命名される化合物である。x=1かつy=0である本発明の化合物の例としては、本明細書においてIb、IcおよびIeと命名される化合物が挙げられる。x=y=0である本発明の化合物の例としては、本明細書においてIdおよびIfとして命名される化合物が挙げられる(図1を参照のこと)。
【0067】
構造Iの化合物の一つの例示的な分類は、xが0または1であり;yが0または1であり;Raがメチレン;Rb(xが1の場合)が水素、メチルまたはエチルであり;Rc(xが1の場合)が−COOHで置換され得るC1〜12アルキレン、好ましくは、C1〜6アルキレン(例えば、エチレンまたはn−ペンチレン(−(CH2)5−))であり;Rd(yが1の場合)が水素であり;そしてRe(yが1の場合)がエチレン(−CH2CH2−)である化合物である。構造Iのこの分類の特定の実施形態としては、本明細書においてIa、Ib、Ic、Id、IeおよびIfと命名される化合物が挙げられる。この分類の一実施形態において、xは1であり;yは0であり;Raはメチレンであり;Rbは水素、メチレンまたはエチルであり;そしてRcはC2〜6アルキレン(例えば、エチレンまたはn−ペンチレン(−(CH2)5−))である。構造Iのこの分類の特定の実施形態としては、本明細書においてIbおよびIcと命名される化合物が挙げられる。
【0068】
式Iの本発明の化合物のサブセットはまた、式IIにより規定され得る。式IIの選択される実施形態において、WはCClである。他の選択される実施形態において、R9は、水素またはC1〜4アルキル(水素またはメチルを含む)である。
【0069】
R10は、水素またはC1〜6アルキルであり、好ましくは、水素またはC1〜4アルキル(水素、メチルまたはエチルを含む)である。別の実施形態において、R10はエチルである。
【0070】
さらなる選択される実施形態において、R11はC1〜10アルキレンであり、好ましくはR11はC2〜10アルキレンであり、より好ましくは、C2〜6アルキレン(例えば、−(CH2)2または−(CH2)5−)である。
【0071】
上記式Iと同様に、式IIのピリジニウム環は、代表的には、メタ置換もしくはパラ置換されており、より一般的には、パラ置換されている。
【0072】
式IIに従う本発明の化合物の例は、本明細書においてIbおよびIcと命名される化合物が挙げられる(図1を参照のこと)。
【0073】
理論に限定されることは意図しないが、式IおよびIIの化合物は、細菌の細胞壁生合成を阻害し、それにより、細菌の増殖を阻害するか、または細菌の溶解を引き起こすと考えられる。従って、これらは、抗生物質として有用である。
【0074】
他の特性の中でもとりわけ、本発明の化合物は、以下にさらに説明するように、グラム陽性細菌(例えば、メチシリン耐性Staphylococci aureus(MRSA)およびメチシリン耐性Staphylococci epidermitis(MRSE))に対して驚くべき予期せぬ効力を有することが見出された。
【0075】
(一般的な合成手順)
本発明の架橋グリコペプチド−セファロスポリン化合物は、容易に入手可能な出発物質から、好ましくは、本明細書中に記載される中間体化合物1〜5を介して、調製され得る。代表的または好ましいプロセス条件(すなわち、反応の温度、時間、反応物質のモル比、溶媒、圧力など)が与えられるが、特に言及されない限り、当業者に決定されるような他のプロセス条件もまた使用され得ることが理解される。最適な反応条件は、使用される特定の反応物質または溶媒によって変化し得るが、このような条件は、慣用的な最適化手順によって、当業者により容易に決定され得る。
【0076】
さらに、当業者に理解されるように、生じる所望されない反応から特定の官能基を保護するために、慣用的な保護基が必須であるか、または所望され得る。特定の官能基についての適切な保護基、ならびにこのような官能基の保護および脱保護に適切な条件は、当該分野で周知である。所望される場合、本明細書において記載される手順に例示されるもの以外の保護基が使用され得る。例えば、多数の保護基、ならびにその導入および除去の手段は、T.W.GreeneおよびG.M.Wuts,Protecting Groups in Organic Synthesis,第3版,Wiley,New York,1999ならびにこの文献で引用される参考文献に記載される。
【0077】
合成の好ましい方法において、yが0である式Iの化合物、または選択される実施形態においては式IIの化合物は、式1:
【0078】
【化13】
のグリコペプチド、またはその塩、もしくはC末端のカルボキシルが活性化された誘導体および/もしくはアミン保護された誘導体であって、R4〜R8、X1およびX2が本明細書において定義されるようなものである、化合物を、式3または4:
【0079】
【化14】
の化合物またはその塩もしくはカルボキシル基が保護された誘導体であって、W、R2、R3、R9、Ra〜e、n、xおよびyが、本明細書において定義されるようなものである、化合物と反応させることによって調製され;式IもしくはIIの化合物、またはその塩もしくは保護された誘導体を提供する。1、3および4の好ましい実施形態は、上に記載されるようなものである。
【0080】
式IIの化合物を調製する際に、構造1、3および/または4の変数は、以下のように定義される:nは0である;xは1である;yは0である;RaはCH2である;Rbは水素またはC1〜6アルキル(R10について上で定義された通り)である;RcはC1〜12アルキレン(R11について上で定義された通り)である;R2、R5およびR6は水素である;R7はCH3である;R9は本明細書において定義される通りである;R4はOHである;そして、X1およびX2はClである。
【0081】
代表的には、この反応は、以下にさらに議論するように、不活性な希釈剤(例えば、DMF)において、慣用的なカルボン酸−アミン(ペプチド)カップリング試薬を使用して、グリコペプチド1またはその塩を、約0.5当量〜約1.5当量、好ましくは、約0.9当量〜約1.1当量の式3または4の化合物とカップリングすることによって実施される。この反応において、グリコペプチド1またはその塩は、代表的には、まず、過剰(好ましくは、約1.8当量〜約2.2当量)のアミン(例えば、ジイソプロピルエチルアミン)の存在下、約−20℃〜約25℃の範囲の温度(好ましくは周囲温度)にて、約0.25時間〜約3時間、カップリング試薬に接触される。好ましくは、次いで、過剰のトリ
フルオロ酢酸(代表的には約2当量)が添加され、任意の過剰のジイソプロピルエチルアミンのTFA塩を形成する。次いで、この反応は、一般に、約−20℃〜約10℃の温度(好ましくは、約0℃)まで冷却され、そして、中間体3または4が添加され、その後、過剰の2,4,6−コリジンが添加される。この反応は、代表的には、約0℃において、約1時間〜約6時間、または反応が実質的に完了するまで維持される。
【0082】
あるいは、式I(yが0でない場合)の化合物の調製については、式2のグリコペプチド誘導体(R2、R4〜R8、Rd〜e、X1およびX2は本明細書において定義される通り)またはその塩もしくは保護された誘導体は、式5(W、R3、R9、Ra〜c、nおよびxは本明細書において定義される通り)の中間体あるいはその塩または活性化された誘導体もしくは保護された誘導体と反応させられ得る。2および5の好ましい実施形態は、上で記載された通りである。
【0083】
【化15】
代表的に、このようなカップリング反応は、コリジンのような塩基の存在下で、中間体2の塩(例えば、トリフルオロ酢酸塩)を、約0.5当量〜1.5当量の中間体5の活性化されたエステルと反応させることによって実施される。好ましくは、Rdは、この場合水素であり、その結果、反応は、C末端の第一級アミンにおいて優先的に生じる。例えば、図3に例示され、実施例4においてさらに説明されるIaの調製を参照のこと。
【0084】
式I(y>1の場合)の化合物の調製について、類似のストラテジーに従い得、このストラテジーにおいて、1つ以上のアミノ酸(例えば、構造NHRd−Re−COOH)の、中間体2のC末端−NHRd基への付加が、中間体5との反応を進める。
【0085】
これらの反応において使用するための、好ましいカップリング試薬または活性化試薬としては、ベンゾトリアゾール−1−イルオキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBOP)が挙げられ、これらは、好ましくは、約0.5当量〜1.5当量の量、好ましくは、約0.9当量〜1.1当量の量で、約0.5当量〜1.5当量、好ましくは約0.9当量〜1.1当量の1−ヒドロキシベンゾトリアゾール(HOBT)または1−ヒドロキシ−7−アザベンゾトリアゾール(HOAT)と組み合せて使用される。他の適切なカップリング試薬としては、O−(7−アザベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HATU);ビス(2−オキソ−3−オキサゾリジニル)ホスフィン酸クロリド(BOP−Cl);ジフェニルホスホリルアジド(DPPA);ジフェニルホスフィン酸クロリド;ジフェニルクロロホスフェート(DPCP)およびHOAT;1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(EDAC);ペンタフルオロフェニルジフェニルホスフィネートなどが挙げられる。
【0086】
カップリング反応が完了した後、次いで、生成物中に存在する任意の保護基が、従来の手順および試薬を使用して除去される。例えば、N−トリチル、N−BOC(N−tert−ブトキシカルボニル)および/またはCOO−PMB(パラ−メトキシベンジルエステル)の脱保護は、周囲温度において、不活性な希釈剤(例えば、ジクロロメタンまたはヘプタン)中で、過剰のトリフルオロ酢酸および過剰のアニソールまたはトリエチルシランを用いて、約1〜約12時間、または反応が完了するまで処理するより達成され得る。脱保護された生成物は、カラムクロマトグラフィー、HPLC、再結晶化などのような慣用的な手順を使用して精製され得る。
【0087】
上記の手順における使用に適切な式Iのグリコペプチドは、市販されているか、または、適切なグリコペプチド産生生物の発酵、その後に、当該分野で認識される手順および機器を使用して、生じた発酵ブロスからグリコペプチドを単離することによって調製され得る。誘導体2は、例えば、実施例3に記載されるように、1のジアミンとの反応により容易に調製される。
【0088】
上記の手順において使用されるセファロスポリン中間体3および4は、慣用的な手順を使用して、市販の出発物質および試薬から容易に調製される。一例として、式4の中間体は、図2に示され、実施例1および2に記載されるように調製され得る。簡単には、2−アミノ−5−クロロ−α−メトキシイミノ−4−チアゾール酢酸(6)を、EDACで触媒して、アミノセファロスポリン酸エステル7と反応させて、アミド結合を形成した。この生成物(8)を、次いで、TFAおよびアニソールで処理して、PMBエステルを解離させ、その後、第一級塩素をピリジン誘導体で置き換えた。このピリジン誘導体は、上で構造4に示されるように、置換基−Ra−NHR2、そして必要に応じて、置換基R3を含む。図2に示される調製において、化合物4−(N−t−BOC−N−エチル)アミノメチルピリジン(9)が使用され、その結果、Raはメチレンであり、R2はエチルである。この反応は、保護された形態の中間体(10)を生じ;TFAを用いた脱保護により、中間体4a(WがCClであり、R9がMeであり、nが0であり、RaがCH2であり、そしてR2がEtである、中間体4)を生じる。
【0089】
代替的なプロセスにおいて、生成物8を、アセトン中でヨウ化ナトリウムと反応させ、その後、9と反応させた。次いで、TFA/アニソールを用いる反応を使用して、Boc保護基およびPMB保護基の両方を除去した。
【0090】
上記の反応において使用するための種々の置換ピリジンは、市販されているか、または、慣用的な手順を使用して、市販の出発物質および試薬から調製され得る。例えば、種々のアミノアルキル置換ピリジン(例えば、Raがメチレンであるアミノメチルピリジン、およびRaがエチレンであるアミノエチルピリジン)は、市販されているか、または、標準的な有機合成手順を使用して調製され得る。この反応において使用するための代表的な置換ピリジン誘導体としては、R3が、メチル、メトキシ、チオメトキシ、カルボキシチオメトキシ、フルオロ、クロロ、フェニル、シクロプロピル、カルボン酸、カルボキサミドおよびその組み合わせから選択されるものが挙げられる。R”をピリジニウム環に結合するYが、NR’、O(エーテル)、S(スルフィド)、C(O)(カルボニル)、NR’(CO)、および(CO)NR’より選択される化合物の調製について、出発ピリジン化合物は、市販されているか、または、周知の手順により調製され得る。例えば、3−ヒドロキシピリジン、4−ヒドロキシピリジン、3−アミノピリジン、4−アミノピリジン、4−メルカプトピリジン、ニコチン酸およびイソニコチン酸は、Aldrich Chemical Co,Milwaukee,WIから市販されている。
【0091】
中間体4は、さらに、式3の中間体を形成するように置換され得る。例えば、図4は、脱保護後の、式3の中間体(xが1の場合)を形成するための、式4の中間体のN−(t−BOC)−β−アラニン(実施例5、Ibの調製を参照のこと)との反応、アスパラギン酸(実施例8、Ieの調製を参照のこと)との反応を示す。β−アラニンまたは類似の化合物(例えば、他のアミノ酸)のさらなる付加を採用すると、x>1の場合の式3の中間体を形成し得る。
【0092】
中間体4と、二酸(例えば、HOOC−Rc−COOHの構造のもの)との反応を採用すると、式5の中間体(xが1の場合)を形成し得る(図3;実施例4;Iaの調製を参照のこと)。式5の中間体(x>1の場合)を形成するために、中間体4は、まず、1モル以上のアミノ酸(例えば、構造HOOC−Rc−NHRbのもの)と反応させる。
【0093】
基Rcにおいて、Y’がO(エーテル)およびNR’(直接結合以外)から選択される化合物の調製において、Rcを含む結合部分は、アミド結合ではなく、1つ以上のカルバメート結合または尿素結合を含む。このような結合は、従来の方法により形成され得る。例えば、アミン(例えば、中間体3または4における−NHR2)は、イソシアネートまたはクロロホルメートと反応して、それぞれ、尿素結合またはカルバメート結合を形成し得る。
【0094】
本発明の代表的な化合物またはそれに対する中間体を調製するための特定の反応条件および手順に関するさらなる詳細は、以下に示す実施例に記載されている。
【0095】
(薬学的組成物)
本発明の架橋グリコペプチド−セファロスポリン化合物は、代表的に、薬学的組成物の形態で患者に投与される。従って、その組成物局面の1つでは、本発明は、薬学的に受容可能なキャリアまたは賦形剤と、式Iもしくは式IIの化合物またはそれらの薬学的に受容可能な塩の治療有効量とを含有する薬学的組成物に関する。
【0096】
本発明の薬学的組成物では、任意の通常のキャリアまたは賦形剤が使用され得る。特定のキャリアまたは賦形剤、またはキャリアまたは賦形剤の組合せの選択は、特定の患者または病気または疾患状態を治療するのに使用される投与様式に依存している。このことに関して、特定の投与様式(例えば、経口投与、局所投与、吸入による投与、または非経口投与)に適当な薬学的組成物の調製は、十分に、医薬分野の当業者の範囲内である。さらに、このような組成物の成分は、例えば、Sigma,P.O.Box 14508,St.Louis,MO 63178から市販されている。さらなる例として、通常の処方技術は、RemingtonのPharmaceutical Sciences,Mace Publishing Co.,Philadelphia,PA 第17版(1985)および「Modern Pharmaceutics」、Marcel Dekker,Inc.第3版(G.S.Banker & C.T.Rhodes編)で記述されている。
【0097】
本発明の薬学的組成物は、代表的には、式IまたはIIの化合物またはそれらの薬学的に受容可能な塩の治療有効量を含有する。代表的には、このような薬学的組成物は、約0.1〜約90重量%の活性因子、より一般的には、約10〜約30%の活性因子を含有する。
【0098】
本発明の好ましい薬学的組成物は、非経口投与、特に静脈内投与に適切な組成物である。このような薬学的組成物は、代表的には、式Iもしくは式IIの化合物またはそれらの薬学的に受容可能な塩の治療有効量を含有する生理学的に受容可能な滅菌水溶液を含む。
【0099】
活性成分の静脈内投与に適切な生理学的に受容可能なキャリア水溶液は、当該分野で周知である。このような水溶液としては、例として、5%デキストロース、Ringer溶液(乳酸化Ringer注入物、乳酸化Ringer5%デキストロース添加注入物、アクリル化Ringer注入物)、ノルモソール−M(Normosol−M)、イソライトE(Isolyte E)などが挙げられる。
【0100】
必要に応じて、このような水溶液は、共溶媒(例えば、ポリエチレングリコール;キレート試薬、例えばエチレンジアミン4酢酸;可溶化剤、例えば、シクロデキストリン;抗酸化剤、例えば、メタ重亜硫酸ナトリウム)など)を含有し得る。
【0101】
所望の場合、本発明の水性薬学的組成物は、投与前に適切なキャリアにより、脂質化され、その後再形成され得る。好ましい実施形態において、薬学的組成物は、薬学的に受容可能なキャリアおよび式Iまたは式IIの化合物あるいはそれらの薬学的に受容可能な塩の治療有効量を含有する脂質化組成物である。好ましくは、この組成物中のキャリアは、スクロース、マンニトール、デキストロース、デキストラン、ラクトースまたはこれらの組み合わせを含有する。より好ましくは、このキャリアは、スクロース、マンニトール、またはこれらの組み合せを含有する。
【0102】
1つの実施形態において、本発明の薬学的組成物は、シクロデキストリンを含有する。本発明の薬学的組成物中で使用される場合、このシクロデキストリンは、好ましくは、ヒドロキシプロピル−β−シクロデキストリンまたはスルホブチルエーテルβ−シクロデキストリンである。このような処方物中で、シクロデキストリンは、処方物の約1〜25重量%;好ましくは、約2〜10重量%を含む。さらに、活性成分に対するシクロデキストリンの重量比は、代表的には、約1:1〜約10:1の範囲である。
【0103】
本発明の薬学的組成物は、好ましくは、単位投薬形態中にパッケージングされる。用語「ユニット投薬形態」は、患者に投薬するのに適切な物理的に分離したユニット(すなわち、単独でか、または一つ以上のさらなるユニットと組み合わせるかのどちらかで所望の治療効果を生じるように計算された所定量の活性成分を含む各ユニット)を指す。例えば、このようなユニット投薬形態は、滅菌した、気密されたアンプルなどにパッケージングされ得る。
【0104】
以下の処方物は、本発明の代表的な薬学的組成物を示す。
【0105】
(処方実施例A)
注射可能溶液の調製に適した凍結溶液を以下のとおりに調製する。
【0106】
成分 量
活性化合物 10〜1000mg
賦形剤(例えば、デキストロース) 0〜50g
注射用水 10〜1000mL
(代表的手順)
賦形剤(もしあるならば)を、約80%の注射用水に溶解し、活性化合物を添加し、溶解する。pHを1M水酸化ナトリウムで3〜4.5に調整し、次いで容量を、注射用水を用いて最終容量の95%に調節する。pHを確認し、必要な場合調節し、そして容量を、注射用水を用いて最終容量に調節する。次いで、処方物を0.22ミクロンフィルタを通して滅菌濾過し、無菌条件下で、滅菌バイアル中に入れる。このバイアルをキャップし、ラベルし、そして凍結貯蔵する。
【0107】
(処方実施例B)
注射可能溶液の調製に適切な脂質化粉末を、以下のとおりに調製する。
【0108】
成分 量
活性化合物 10〜1000mg
賦形剤(例えば、マンニトールおよび/またはスクロース) 0〜50g
緩衝材(例えば、クエン酸塩) 0〜500mg
注射用水 10〜100mL
(代表的な手順)
賦形剤および/または緩衝剤(もし、あるならば)を、約60%の注射用水に溶解する。活性化合物を添加して溶解し、そのpHを、1M水酸化ナトリウムを用いて3〜4.5に調節して、その容量を注射用水を用いて最終容量の95%に調節する。pHを確認し、必要な場合調節し、そして容量を注射用水を用いて最終容量に調節する。次いで、この処方物を0.22ミクロンのフィルタを通して滅菌濾過し、無菌条件下にて滅菌バイアルに入れる。次いで、この処方物を適切な凍結乾燥サイクルを使用して凍結乾燥する。このバイアルをキャップし(必要に応じて、部分的減圧下、または乾燥窒素下)、ラベルし、そして凍結下で貯蔵する。
【0109】
(処方実施例C)
患者への静脈内投与のための注射可能溶液を、上記処方実施例Bより、以下のとおりに調製する。
【0110】
(代表的な手順)
処方実施例Bの凍結乾燥粉末(例えば、10〜1000mgの活性化合物を含有する)を、20mLの滅菌水で再形成し、得られた溶液を、100mL中の注入バッグ内で80mLの滅菌生理食塩水でさらに希釈する。次いで、この希釈した溶液を、30〜120分間に渡って患者に静脈内投与する。
【0111】
(利用)
本発明の架橋グリコペプチド−セファロスポリン化合物は、抗生物質として有用である。例えば、本発明の化合物は、細菌感染、および哺乳動物(ヒトおよびペット(すなわち、イヌ、ネコなど)を含む)において本発明の化合物に感受性の微生物により引き起こされる他の細菌関連の医学的状態の処置または予防に有用である。
【0112】
従って、その方法の1つの局面において、本発明は、哺乳動物における細菌感染症の処置方法、薬学的に受容可能なキャリアおよび式Iまたは式IIの化合物もしくはこれらの薬学的に受容可能な塩の治療有効量を含む薬学的組成物を、このような処置を必要とする哺乳動物に投与する工程を包含する方法を提供する。
【0113】
例示目的で、本発明の化合物は、グラム陽性菌および関連する微生物により引き起こされる感染症の処置または予防に特に有用である。例えば、本発明の化合物は、特定のEnterococcus spp.;Staphylococcus spp.(コアグラーゼ陰性staphylococci(CNS)を含む);Streptococcus
spp.;Listeria spp.;Clostridium spp.;Bacillus spp.などにより引き起こされる感染症を処置または予防するのに有効である。本発明の化合物を使用して有効に処置される細菌種の例としては、メチシリン抵抗性Staphylococcus aureus(MRSA);メチシリン感受性Staphylococcus aureus(MSSA);グリコペプチド中間体感受性Staphylococcus aureus(GISA);メチシリン抵抗性Staphylococcus epidermitis(MRSE);メチシリン感受性Staph
ylococcus epidermitis(MSSE);バンコマイシン感受性Enterococcus faecalis(EFSVS);バンコマイシン感受性Enterococcus faecium(EFMVS);ペニシリン抵抗性Streptococcus pneumoniae(PRSP);Streprococcus pyogenesなどが挙げられるが、これらに限定されない。
【0114】
以下の実施例16の表2に示すように、本発明のいくつかの化合物Ia〜f、Im、およびInは、メチシリン感受性Staphylococcus aureusおよびメチシリン抵抗性Staphylococcus aureusに対して、10倍以上のバンコマイシンよりも有効であった。これらの化合物はまた、これらのデス−クロロアナログIg、IjおよびIkよりも有意に活性であるが、これらの化合物もまたバンコマイシンよりもより活性であった。「時間殺傷(time−kill)」アッセイにおいて、実施例17に記載されるように、式Iの化合物(すなわち化合物Ib)は、4時間で、1.0μg/mL以下の濃度にて、MRSAに対し殺菌性であった。比較すると、バンコマイシンは、24時間で、4μg/mLの濃度にて、MRSAに対し殺菌性であった。神経栄養性(neutropenic)マウスにおけるインビボアッセイにおいて、実施例18に記載されるように、式Iの化合物(すなわち、化合物Ib)は、バンコマイシンについてのivの9mg/kgのED50と比較して、ivの0.1mg/kg以下のED50を有した。
【0115】
一般に、本発明の化合物は、グリコペプチドまたはセファロスポリンのどちらかに感受性である細菌株により引き起こされる感染症の処置または予防に好ましい。
【0116】
本発明の化合物を使用して処置または予防され得る感染症または細菌関連の医学状態の代表的な型としては、皮膚感染症および皮膚構造感染症、尿路感染症、肺炎、心内膜炎、カテーテル関連血流感染症、骨髄炎などが挙げられるが、これらに限定されない。このような状態の処置において、患者は、処置されるべき微生物にすでに感染しているか、または感染症に感受性であると疑わしいだけかもしれず、このような場合において、活性成分が予防的に投与される。
【0117】
本発明の化合物は、代表的に、任意の受容可能な投与経路により治療有効量を投与される。好ましくは、この化合物は、非経口投与される。この化合物は、一日一回の投薬、または一日あたり複数回の投薬において、投与され得る。この処置レジメンは、延長された期間(例えば、数日間または1〜6週間以上にわたって)にわたる投与を必要とし得る。投薬あたりに投与される活性剤の量または投与される総量は、代表的には、患者の担当医により決定され、感染症の性質および重篤度、患者の年齢および健康状態、患者の活性成分に対する耐性、感染症を起こす細菌、投与経路などのような因子に依存する。
【0118】
一般に、適切な投薬は、活性成分の約0.25〜約10.0mg/kg/日、好ましくは、約0.5〜約2mg/kg/日の範囲である。平均的な70kgのヒトについて、これは、一日あたり約15〜約700mg、または好ましくは、一日あたり約35〜約150mgの量の活性成分である。
【0119】
さらに、本発明の化合物は、細菌の増殖を阻害するのに有効である。この実施形態において、細菌は、インビトロまたはインビボのどちらかで、式Iまたは式IIの化合物あるいはこれらの薬学的に受容可能な塩の増殖阻害量に接触される。代表的には、増殖阻害量は、約0.008μg/mL〜約50μg/mL;好ましくは、約0.008μg/mL〜約25μg/mL;そしてより好ましくは、約0.008μg/mL〜約10μg/mLの範囲である。細菌増殖の阻害は、代表的には、細菌繁殖の減少もしくは欠損、および/または細菌崩壊の溶菌(すなわち、未処置の細菌と比較した所与の処置期間(すなわち、1時間あたり)にわたる所与の容量(すなわち、mLあたり)中のコロニー形成単位の減少)により、証明される。
【0120】
本発明の化合物はまた、細菌の細胞壁生合成を阻害するのに有効である。この実施形態において、細菌は、インビトロまたはインビボのどちらかで、式Iまたは式IIの化合物あるいはこれらの薬学的に受容可能な塩の細胞壁生合成の阻害量に接触される。代表的には、細胞壁生合成阻害量は、約0.04μg/mL〜約50μg/mL;好ましくは約0.04μg/mL〜約25μg/mL;そして、より好ましくは、約0.04μg/mL〜約10μg/mLの範囲である。細菌における細胞壁生合成の阻害は、代表的には、細菌の溶菌を含む、細菌の増殖の阻害または欠損により証明される。
【0121】
さらに、本発明の化合物は、特定の細菌(メチシリン抵抗性Staphylococci aureus(MRSA)およびメチシリン抵抗性Staphylococci epidermitis(MRSE)を含む)に対して、驚きべき、かつ予想外の迅速な死滅率(cidelite)を有することが見出されている。本発明の化合物のこれらの特性、および抗生物質的な利用性は、当業者に周知の種々のインビトロアッセイおよびインビボアッセイを使用して実施され得る。例えば、代表的なアッセイは、以下の実施例においてさらに詳述される。
【実施例】
【0122】
(実施例)
以下の合成実施例および生物学的実施例は、本発明を例示するため提供されるが、いかなる場合においても本発明の範囲を制限するようには解釈されるべきではない。
【0123】
以下の実施例において、他に指定されない限り、以下の略語は、下の意味を持つ。以下に規定されない略語は、その一般に認められた意味を有する。
【0124】
BOC = tert−ブトキシカルボニル
CFU = コロニー形成ユニット
DCM = ジクロロメタン
DIPEA = ジイソプロピルエチルアミン
DMF = N,N−ジメチルホルムアミド
DMSO = ジメチルスルホキシド
EtOAc = 酢酸エチル
HOBT = 1−ヒドロキシベンゾトリアゾール
HOAT = 1−ヒドロキシ−7アザベンゾトリアゾール
HPLC = 高速液体クロマトグラフィー
MIC = 最少阻害濃度
MS = 質量分析
PMB = p−メトキシベンジル
PyBOP = ベンゾトリアゾール−1−イルオキシトリピロリジノホスホニウムヘキサフルオロリン酸
THF = テトラヒドロフラン
TLC = 薄層クロマトグラフィー
TFA = トリフルオロ酢酸
以下の実施例において、記録された全ての温度は、他に指示されない限り、セ氏温度(℃)である。また、他に注記されない限り、試薬、出発物質および溶媒を、市販業者(例えば、Aldrich,Fluka,Sigmaなど)から購入し、さらに精製することなく使用した。バンコマイシン塩化物の半水和物をAlpharma,Inc.,Fort Lee,NJ07024(Alpharma AS、Oslo、Norway)から購入した。
【0125】
逆相HPLCを、他に規定されない限り、代表的にC18カラムおよび(A)(98%水、2%アセトニトリル、0.1%TFA)と、(B)(10%水、90%アセトニトリル、0.1%TFA増加勾配)の増加勾配(例えば、0〜約70%)を使用して行った。
【0126】
以下に記載される合成において、中間体4a〜4gを、以下のとおりに規定する。これらの化合物の各々において、nは0であり、Raは、−CH2−である。
【0127】
【化16】
(実施例1)
(4a:(7R)−7−[(Z)−2−(2−アミノ−5−クロロチアゾール−4−イル)−2−(メトキシイミノ)アセトアミド]−3−[((4−エチルアミノ)メチル)−1−ピリジノ)メチル]−3−セフェン−4−カルボキシレート ビス−トリフルオロ酢酸塩の調製)(図2および図3を参照のこと)
(A.2−アミノ−5−クロロ−α−(メトキシイミノ)−4−チアゾール酢酸(6)の調製)
500mLの酢酸に、40.0g(0.20mol)の2−アミノ−α−(メトキシイミノ)−4−チアゾール酢酸(Ardrich Chemical Company,Milwaukee,WI)および27.9g(0.21mol)のN−クロロスクシンイミドを添加した。この混合物を、70℃の浴中で加熱した。その固形物を30分以内に溶解し、そして45分後にこの反応をMSにより完了した。この黒色溶液を室温まで冷却し、減圧下で濃縮し、黒色の固形物を得て、この固形物を精製することなく使用した。
【0128】
(B.セファロスポリン誘導体(8)の調製)
700mLのDMF中の47.5g(0.20mol)の(6)に、81.1g(0.20mol)のアミノセファロスポリンエステル(7)(Otsuka,Osaka,JP)および27.0g(0.20mol)のHOATを添加した。この混合物を0℃まで冷却し、そして26.7g(0.22mol)の2,4,6−コリジンを添加した。この溶液に、42.2g(0.22mol)の1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミドヒドロクロリド(EDAC)を添加した。5時間後、この溶液を、1:1のEtOAc/エーテルと水とに分配し、この相を分離した。この水溶液をEtOAcで2回逆抽出した。合わせた有機相を0.5Mのクエン酸、水、50%飽和NaHCO3、水、およびブラインで各々1回洗浄した。この有機相をMgSO4で乾燥し、濾過し、減圧下で濃縮した。この残渣を最少容量の塩化メチレンに溶解し、この生成物を、その溶液をエーテルに注ぐことにより沈澱させた。この固形物を濾過により単離して、48gの化合物(8)を得た。
【0129】
分析データ:MS m/z 計算値 586.04、実測値 586.2(M+1);1H NMR(DMSO−d6):d9:60(d,1H)、7.35(m,3H)、6.91(d,2H)、5.82(m,1H)、5.17(m,3H)、4.56(m,2H),3.84(s,3H)、3.76(s,3H)、3.62(m,2H)。
【0130】
(C.ピリジニウム部分への結合)
400mLの1,2−ジクロロエタン中の48.5g(82.7mol)のクロロメチルセファロスポリンエステル8および40mLのアニソールに、300mLのTFAを添加した。40分後、この溶液を減圧下で1/2の容量に濃縮し、生成物を、この濃縮物を1Lのエーテルに注ぐことにより沈澱させた。この固形物を、濾過により単離し、エーテルでリンスし、乾燥して42.5gの粗製遊離酸を得た。この物質(25.0g、53.6mol)を200mLの1:1のアセトニトリルDMFに溶解した。この溶液に、10.1g(83.3mmol)の2,4,6−コリジン、および15.2g(64.5mmol)の4−(N−t−ブトキシカルボニル)−エチルアミノメチルピリジン(9)(従来の手順により、4−エチルアミノメチルピリジンより調製、これは、Aldrich Chemical Company,Milwaukee,WIより市販されている)を添加した。3.5時間後、生成物を、この溶液を1Lのエーテルに注ぐことにより沈澱させた。この固形物を、濾過により単離し、エーテルで洗浄し、減圧下で乾燥して26gの粗製物質を得た。この生成物を、分取逆相HPLCにより精製し、化合物10をオフホワイトの粉末として得た。
【0131】
分析データ:MS m/z 計算値 666.12、実測値 666.2(M+1);1H NMR(DMSO−d6):d 9.36(d,1H)、8.91(d,2H)、7.95(d,2H)、7.24(b,1H)、5.83(m,1H)、5.50(m、2H)、5.17(d,1H)、4.66(s,2H)、3.83(s,3H)、3.52(d,1H)、3.30(m,3H)、1.37(s,9H)、1.08(t,3H)。
【0132】
(D.10の脱保護)
610mg(0.78mmol)の化合物10および1mLのアニソールに、8mLのトリフルオロ酢酸を添加した。20分後、この反応物を100mLのエーテルに注ぐことにより、その生成物を沈殿させた。この固体を濾過により単離し、エーテルで洗浄し、そして真空下で乾燥し、淡黄色固体として4aを得た。
【0133】
分析データ:1H NMR(DMSO−d6)d 9.56(d,1H),9.44(br,1H),9.10(d,2H),8.21(d,2H),7.38(b,1H),5.86(m,1H),5.54(m,2H),5.15(d,1H),4.51(s,2H),3.80(s,3H),3.40(m,2H),3.30(m,2H),1.22(t,3H)。
【0134】
(実施例2)
(4bの調製:(7R)−7−[(Z)−2−(2−アミノ−5−クロロチアゾール−4−イル)−2−(メトキシイミノ)アセトアミド]−3−[(4−(アミノエチル)−1−ピリジニオ)メチル]−3−セフェム−4−カルボキシレート ビス−トリフルオロ酢酸塩)
(A.2−アミノ−5−クロロ−α−(メトキシイミノ)−4−チアゾール酢酸(6)の調製)
500mLのDMFに、50.0g(250mmol)の2−アミノ−α−(メトキシイミノ)−4−チアゾール酢酸および35g(260mmol)のN−クロロスクシンイミドを添加した。この混合物を室温で一晩攪拌し、その後質量スペクトル分析がもはや出発物質が存在しないことを示した。この淡褐色の溶液を、さらなる操作なしに使用した。
【0135】
(B.セファロスポリン誘導体(8)の調製)
工程(a)からのDMF(101.5g、250mmol)中の酸6の溶液に、アミノセファロスポロン酸エステル7(34g、250mmol)を添加した。この混合物を0℃に冷却し、そして33.5mL(250mmol)の2,4,6−コリジンを添加した。この溶液に、53g(275mmol)の1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩を添加した。2時間後、その溶液を3Lの水に沈殿させ、濾過した。この固体を水(2×1L)、飽和炭酸水素ナトリウム(500mL)および水(4×500mL)で洗浄し、真空下で乾燥した。乾燥した固体を、室温で500mLの塩化メチレンに溶解させ、その溶液をゆっくりと攪拌し、沈殿を形成させた。結晶を濾過により回収し、洗浄液がもはや褐色でなくなるまで塩化メチレンで洗浄し、そして真空下で乾燥し、アミド8(74g)を得た。
【0136】
(C.ピリジニウム部分へのカップリング)
アセトン(250mL)を、窒素下、暗所で50g(85mmol)のクロロメチルセファロスポリンエステル8および13g(85mmol)のヨウ化ナトリウムの混合物に添加した。30分間の攪拌後、27g(130mmol)の4−(N−tert−ブトキシカルボニル)アミノメチルピリジンおよび30mLのアセトンを添加した。さらなる2時間の攪拌後、1.4Lの0.1N HClを添加し、粘着性の沈殿を生成した。溶媒部分を傾斜し、そして粘着性の残渣を800mLの水で処理し、固体を得た。その水を傾斜し、そしてその固体を1Lの4:1 酢酸エチル/エタノールに溶解した。その溶液を500mLの飽和ブラインで洗浄し、硫酸マグネシウムで乾燥し、そして乾燥状態までエバポレートし、70g(79mmol、93%)の生成物(10に類似、NEtBOCの代わりに−NHBOCを有する)を得、その生成物は、HPLC(254nm)で決定した場合、78%の純度を有した。
【0137】
この物質を、さらなる精製なしに、以下のようにして脱保護化した。粗生成物(70g、79mmol)を、窒素下で、550mLの塩化メチレンに溶解させ、そして35mL(320mmol)のアニソールを添加し、続いて150mLのトリフルオロ酢酸を添加した。2時間後、この混合物を真空下で濃縮した。1Lのジエチルエーテルの添加の際に、生成物は沈殿した。この固体を濾過により単離し、エーテルで洗浄し、200mLの水中で攪拌し、そして濾過した。この濾液を乾燥状態まで凍結乾燥し、逆相HPLCにより精製し、30g(約50%)の4bを得た。
【0138】
(実施例3)
(2aの調製:C−アミノエチルアミドバンコマイシン(R2、R5、R6、R8がHであり;R4がOHであり;R7がMeであり;X1、X2がClであり;RdがHであり;Reが−CH2CH2−である構造2))
75mLのDMSO中の塩酸バンコマイシン一水和物(7.3g、4.7mmol)の溶液に、室温で、4.1mL(23.5mmol)のDIPEAを添加し、続いて1.8g(5.6mmol)の9−フルオレニルメチル N−(2−アミノエチル)カルバメート塩酸塩(Fmoc−NH−CH2−CH2−NH3Clを添加した(図3を参照のこと)。75mLのN,N’−ジメチルプロピレン尿素(DMPU)中のPyBOP(2.7g、5.2mmol)およびHOBT(0.63g、4.7mmol)の溶液を、次いで手早く滴下した。この生成した溶液を、室温で2時間攪拌し、次いで800mLのジエチルエーテルに注ぎ、ゴム状物を得た。このエーテルを傾斜し、このゴム状物をさらなるエーテルで洗浄し、粗[C]−(2−Fmoc−アミノエチル)バンコマイシンを得た。
【0139】
この生成物(Fmoc−2a)を40mLのDMF中に溶解させ、10mLのピペリジンを添加し、そしてこの溶液を室温で20分間静置し、次いで450mLのアセトニトリルに滴下し、沈殿を形成させた。この混合物を遠心分離し、そのアセトニトリルを傾斜し、そしてこの残渣を450mLのアセトニトリルで2回、450mLのジエチルエーテルで1回洗浄し、次いで風乾した。この残渣を、次いで水に溶解させ、3N HClでpH<5まで酸性化し、そして0.1%のトリフルオロ酢酸を含む水中の2〜30%アセトニトリルの勾配を用いて逆相HPLCで精製した。このことにより、生成物2aをトリ(TFA)塩として得た。
【0140】
(実施例4)
(化合物Iaの調製)
上の実施例1に記載したように調製したピリジニウムラクタムビストリフルオロアセテート4a(2.4g)を、窒素下でN,N−ジメチルホルムアミド(DMF、40mL)に溶解し、0℃まで冷却した。ドデカン二酸ビス−1−ヒドロキシ−7−アザトリアゾールエステル(7.0g)を添加し(11)、続いて2,4,6−コリジン(1.2mL)を添加し、そしてこの混合物を65分間攪拌し、次いで酢酸エチル(50mL)に添加し、そしてこの混合物をジエチルエーテル(400mL)中に沈殿させた。この活性化エステルラクタム5aを濾過により回収し、そして真空下で乾燥し、そして一部(14mg)をDMF(122μL)中に溶解し、0℃で、DMF(500μL)中の、実施例3に記載するように調製したC−アミノエチルアミドバンコマイシントリフルオロアセテート2a(50mg)および2,4,6−コリジン(5.4μL)の混合物に添加した。この混合物を20分間0℃に保ち、次いでトリフルオロ酢酸(7.3μL)を添加した。この混合物を−20℃に一晩保ち、次いで逆相HPLCにより精製し、生成物Iaを得た。
【0141】
分析データ:MS m/z 実測値2252.8、計算値2252.7。
【0142】
(実施例5)
(化合物Ibの調製)
窒素下、室温で1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(740mg)、1−ヒドロキシ−7−アザトリアゾール(520mg)およびN−(tert−ブトキシカルボニル)−β−アラニン(660mg)をDMF(10mL)中で7時間攪拌し、次いで0℃まで冷却した。ピリジニウムラクタムビストリフルオロアセテート4a(2g)(実施例1を参照のこと)を添加し、それが完全に溶解するまで攪拌し、次いで2,4,6−コリジン(700μL)を添加し、そしてこの混合物を0℃で100分間攪拌した。トリフルオロ酢酸(700μL)および水(30mL)を添加し、この生成物を逆相HPLCで精製した。凍結乾燥後、この生成物を20mLの50%トリフルオロ酢酸/ジクロロメタンで40分間処理した。このラクタムアミン3a(図4を参照のこと)を、ジエチルエーテル中への沈殿によって、そのビストリフルオロアセテート塩として回収し、真空下で乾燥した。
【0143】
窒素下、室温で塩酸バンコマイシン一水和物1a(900mg)(R5、R6、R8がHであり;R4がOHであり;R7がMeであり;X1、X2がClである構造1;図において「バンコマイシン」とも呼ばれる)を、ジメチルスルホキシド(5mL)に溶解し、1−ヒドロキシ−7−アザトリアゾールを添加し(80mg)、続いてDMF(5mL)中のPyBOP(300mg)およびN,N−ジイソプロピルエチルアミン(100μL)を添加した。この混合物を20分間攪拌し、トリフルオロ酢酸(45μL)を添加し、そしてこの混合物を0℃に冷却した。ピリジニウムラクタムアミン3a(250mg)を添加し、続いて2,4,6−コリジン(270μL)を添加し、そしてこの混合物を0℃で30分間攪拌し、次いでアセトニトリル中に沈殿させた。粗生成物を遠心分離により回収し、逆相HPLCにより精製し、Ibを得た。
【0144】
分析データ:MS m/z 実測値2069.7、計算値2069.4。
【0145】
(実施例6)
(化合物Icの調製)
この化合物を、N−BOC−β−アラニンの代わりに6−(N−BOC)アミノヘキサン酸に変えて、実施例5に記載した手順に従って調製した。
【0146】
分析データ:MS m/z 実測値2110.5、計算値2111.5。
【0147】
(実施例7)
(化合物Idの調製)
窒素下、室温で塩酸バンコマイシン一水和物1a(900mg)を、ジメチルスルホキシド(5mL)に溶解し、1−ヒドロキシ−7−アザトリアゾール(80mg)を添加し、続いてDMF(5mL)中のPyBOP(300mg)およびN,N−ジイソプロピルエチルアミン(100μL)を添加した。この混合物を20分間攪拌し、トリフルオロ酢酸(45μL)を添加し、そしてこの混合物を0℃に冷却した。ピリジニウムラクタムビストリフルオロアセテート4c(250mg)(ピリジン上でのメタ置換を除いて4bと同一;図5を参照のこと)を添加し、続いて2,4,6−コリジン(270μL)を添加し、そしてこの混合物を0℃で30分間攪拌し、次いでアセトニトリル中に沈殿させた。粗生成物を遠心分離により回収し、逆相HPLCにより精製し、生成物Idを得た。
【0148】
分析データ:MS m/z 実測値1970.9、計算値1970.3。
【0149】
(実施例8)
(化合物Ieの調製)
(A.3bの調製(WがClであり、R9がMeであり、nが0であり、RaがCH2であり、Rbが水素であり、Rcが−CH2CH(COOH)−であり、R2が水素であり、そしてxが1である構造3))
HOAT(56mg、0.2mmol)、PyBOP(208mg、0.2mmol)およびN−BOCアスパラギン酸α−tert−ブチルエステルをDMF(500μL)中に溶解した。DIPEA(70μL、0.2mmol)を次いで添加し、そしてその反応物を室温で20分間攪拌した。この反応物を次いで−10℃まで冷却し、そしてTFA(32μL、0.2mmol)を添加した。C3−ピリジニウムセファロスポリン4b(306mg、0.2mmol)をこの反応物にDMF(500μL)中の溶液として添加した。コリジン(160μL、0.6mmol)を添加し、この反応物を−10℃で1時間攪拌した。この反応混合物を真空中で濃縮し、次いで逆相HPLC(60分間にわたる10〜90%勾配)を用いて精製し、凍結乾燥の後で、保護されたセファロスポリンアスパラギン酸誘導体のTFA塩を、白色の非晶質粉末(193mg)として得た。この生成物(162mg)をTFA(10mL)中に溶解し、室温で60分間攪拌した。この反応物を真空中で濃縮し、そしてこの残渣をCH3CN/H2O(1:1;0.1%TFA)に溶解し、凍結乾燥し、所望の脱保護されたセファロスポリンアスパラギン酸誘導体(3b)のTFA塩を白色固体(92mg)として得た。
【0150】
分析データ:MS m/z 652.9(MH+)。
【0151】
(B.3bとバンコマイシンとの反応)
塩酸バンコマイシン1a(0.32g、0.21mmol)を3mLのDMSO中に溶解した。PyBOP(0.11g、0.21mmol)およびHOAT(0.03g、0.21mmol)の0.5mL DMF溶液を添加し、続いてDIPEA(0.04mL、0.21mmol)を添加した。この反応物を30分間攪拌し、次いでTFA(0.02mL、0.21mmol)を添加し、そしてこの混合物を0℃まで冷却した。DMF(1mL)中の3b(0.15g、0.17mmol)(図6を参照のこと)の溶液を次いで添加し、続いてコリジン(0.08mL、0.63mmol)を添加した。この反応物を4℃で一晩攪拌し、次いでEtOAcから沈殿させ、遠心分離し、そして生成した固体をMeCNで洗浄した。所望の化合物をHPLC(2〜30% MeCN)により精製し、0.3gのIeを白色固体として得た。
【0152】
分析データ:MS m/z 1042.6[(M+2H)/2)]2+。
【0153】
(実施例9)
(化合物Ifの調製)
塩酸バンコマイシン1a(134.0mg、0.09mmol)およびHOAT(12.3mg、0.09mmol)を含む0.6mLのDMSOの溶液に、0.6mLのDMF中のPyBOP(46.9mg、0.09mmol)の溶液を添加し、続いてジイソプロピルエチルアミン(31.4μL、0.2mmol)を添加した。周囲温度での20分間の攪拌の後、0℃で冷却しつつ、この反応混合物をTFA(13.9μL、0.2mmol)で処理した。バンコマイシンのこの活性化エステルに、69mg(0.09mmol)のピリジニウムラクタム4aおよびコリジン(51.3μL、0.4mmol)を添加した。この最終混合物を室温で3時間攪拌し、その後TFA(33.4μL、0.40mmol)でクエンチした。この粗生成物を、酢酸エチルを添加することにより、反応混合物から沈殿させた。この固体を遠心沈降させることにより回収し、乾燥し、そして分取HPLCにより精製した。この所望の生成物Ifを、白色の綿毛状固体(89mg)として得た。
【0154】
分析データ:保持時間(分析HPLC:6分にわたる10〜70%MeCN/H2O)=2.00分。MS m/z 計算値1970.27(C86H94Cl3N16O28S2);実測値985.7[(M+2H)/2]]2+。
【0155】
(実施例10)
(化合物Ikの調製(化合物Ifのdes−クロロアナログ))
ヨウ化ナトリウム(0.271g、1.81mmol)を、光から保護してアセトン25mLに溶解したセファロスポリン−β−ラクタム12(0.999g、1.81mmol)(図7を参照のこと)の溶液に添加し、窒素下で攪拌した。25分後、4−(N−BOC−アミノメチル)ピリジン(0.390g、1.87mmol)を添加し、そしてその反応物を2時間攪拌した。この生成物を大過剰の酢酸エチルで沈殿させ、濾過により回収した。
【0156】
この生成物(0.353g、0.488mmol)をDCM(2.5mL)に溶解し、アニソール(0.21mL、1.93mmol)およびTFA(1.5mL、19.47mmol)を添加した。この反応混合物を、次いで、窒素下で45分間室温で攪拌した。この生成物を、ジエチルエーテルを用いて沈殿させ、遠心分離し、そしてそのペレットをジエチルエーテルで2回洗浄し、そして乾燥した。この生成物を、水に溶解し、不溶物を濾別した。水性物質を凍結乾燥し、0.166gの4dのTFA塩を橙色固体として得た(WがCHであることを除いて4bと同一である)。
【0157】
分析データ:MS m/z 504.1(M+1);HPLC(2〜30 アセトニトリル/水、254nm)保持時間=0.270分。
【0158】
HOAt(0.0626g、0.460mmol)を、DMSO(3.5mL)中の塩酸バンコマイシン(0.7078g、0.455mmol)の溶液に添加した。DMF(3.5mL)に溶解したPyBOP(0.2338g、0.449mmol)およびDIPEA(79.0μL、0.453mmol)を添加し、そしてこの反応物を、室温、窒素下でで20分間攪拌し、続いてTFA(35.0μL、0.454mmol)を加えた。この反応物を、次いで氷浴で冷却した。ラクタムアミド4d(0.166g、0.227mmol)を添加し、溶解の際に、コリジン(180.0μL、1.365mmol)を添加した。50分の攪拌の後、TFA(125.0μL、1.62mmol)を添加し、そして生成物をアセトニトリルで沈殿させ、遠心分離した。このペレットを最小量のDMFに再溶解し、アセトニトリルで3回再沈殿させ、次いで真空下で乾燥し、0.8gの粗生成物を得た。この粗生成物を分取HPLC(2〜35アセトニトリル/水)で精製し、化合物Ikを含む画分を集め、そして凍結乾燥した。
【0159】
分析データ:MS m/z 1539.6(断片)、HPLC:(2〜30 アセトニトリル/水、254nm)保持時間=2.92分。
【0160】
(実施例11)
(化合物Igの調製(化合物Ibのdes−クロロアナログ))
(A.N−エチル−N−(4−ピリジルメチル)−3−(N−BOC)−アミノプロパンアミド(13)の調製)
DCM(50mL)中のN−t−BOC−β−アラニン(4.678g、24.7mmol)の溶液に、ジイソプロピルカルボジイミド(3.87g、24.7mmol)を添加した。この混合物を氷浴で冷却し、そして4−(エチルアミノメチル)ピリジン(3.37g)を添加した。この反応混合物を、次いで3.5時間攪拌した。この固体を濾過により除去し、そして濾液を水で洗浄し、真空下で乾燥し、5.34gの標題中間体を得た。
【0161】
分析データ:MS m/z 308.1(M+1)。
【0162】
(B.化合物Igの調製)
ヨウ化ナトリウム(0.485g、3.22mmol)を、光から保護しかつ窒素下でアセトン(50mL)に溶解したセファロスポリン−β−ラクタム12(2.031g、3.22mmol)(図8を参照のこと)の溶液に添加した。20分後、上に記載したように調製した、アセトン(10mL)中の13(1.29g、3.40mmol)を添加し、そしてこの反応物を6時間攪拌した。この生成物を大過剰の酢酸エチルを用いて沈殿させ、濾過により回収した。この生成物(1.70g、1.90mmol)をDCM(5mL)に溶解し、そしてアニソール(0.825mL、7.6mmol)およびTFA(5mL、64.9mmol)を添加した。この反応混合物を、次いで窒素下、室温で1時間攪拌した。この生成物をジエチルエーテルを用いて沈殿させ、遠心分離し、そしてこのペレットをジエチルエーテルで2回洗浄して乾燥し、1.7gの3cのTFA塩(WがCHであることを除いて3aと同一である)を得た。
【0163】
分析データ:MS m/z 603.3(M+1);HPLC:(2〜90 アセトニトリル/水、254nm)保持時間=3.42分。
【0164】
HOAt(0.560g、4.11mmol)を、30mLのDMSO中の塩酸バンコマイシン(6.350g、4.08mmol)の溶液に添加した。DMF(30mL)に溶解したPyBOP(2.127g、4.09mmol)およびDIPEA(0.711mL、4.08mmol)を添加し、そしてこの反応物を、窒素下、室温で20分間攪拌し、続いてTFA(0.314mL、4.08mmol)を添加した。この反応物を、次いで氷浴で冷却した。DMSO(25mL)およびDMF(10mL)に溶解したラクタムアミン3c(1.7g、2.04mmol)を添加し、そして溶解の際にコリジン(1.89mL、14.3mmol)を添加した。この反応物を50分間攪拌し、TFA(1.26mL、16.3mmol)を添加し、そしてこの生成物をアセトニトリルで沈殿させ、遠心沈降させた。このペレットを、最小量のDMFに再溶解し、アセトニトリルで3回再沈殿させ、次いで真空下で乾燥し、7.4gの粗生成物を得た。この粗生成物を、分取HPLC(2〜35 アセトニトリル/水)で精製し、この生成物Igを含有する画分を集めて凍結乾燥した。
【0165】
分析データ:MS m/z 1639.5(断片);HPLC:(2〜30 アセトニトリル/水、254nm)保持時間=3.76分。
【0166】
(実施例12)
(化合物Ijの調製(化合物Idのdes−クロロアナログ))
ヨウ化ナトリウム(1.4g、9.1mmol)を、35mLのアセトン中の(7R)−7−[(Z)−2−(2−アミノ−チアゾール−4−イル)−2−(メトキシイミノ)アセトアミド]−3−(クロロメチル)−3−セフェム−4−カルボキシレート(5g、9.1mmol)に添加した。この反応容器を箔中に包み、この反応物を1時間攪拌した。5mLのアセトン中の3−アミノメチルピリジン(2.8g、13.8mmol)の溶液を添加し、この反応物を70分間攪拌し、次いでエーテル中に沈殿させ、5.4gの粗収量の4e(WがCHであることを除いて4cと同一である)を得た。この固体(4.4g)を20mLのDCMに懸濁し、20mLのTFAを添加した。この反応混合物を室温で90分間攪拌し、次いで濃縮した。得られた油状物をエーテルから沈殿させ、分取HPLC(2〜10% MeCN)により精製し、0.6gの4eのTFA塩を白色の綿毛状固体として得た。
【0167】
分析データ:Rt=0.8分(10〜70% MeCN)。1H NMR(DMSO−d6)δ 3.25(dd,2H),3.8(s,3H),4.2(bs,2H),5.05(d,1H),5.45(dd,2H),5.8(dd,1H),7.05(s,1H),8.2(m,1H),8.45(bs,2H),8.6(d,1H),9.0(d,1H),9.05(s,1H),9.4(dd,1H)。
【0168】
塩酸バンコマイシン(0.73g、0.49mmol)を、2.5mLのDMSOに溶解した。2mLのDMF中のPyBOP(0.23g、0.45mmol)およびHOAT(0.06g、0.45mmol)の溶液を添加し、続いてDIPEA(0.16mL、0.82mmol)を添加した。この反応物を、25分間攪拌し、次いでTFA(0.07mL、8.2mmol)を添加し、そしてこの反応物を0℃まで冷却した。コリジンを添加し(0.22mL、1.6mmol)、次いで2mLのDMF中の上記ラクタム4eの溶液を添加した。この反応物を2時間攪拌し、次いでエーテルから沈殿させ、遠心分離した。この生成物をHPLCにより精製し、次いで凍結乾燥し、0.23gの生成物Ijを得た。
【0169】
(実施例13:化合物Imの調製)
J.Antibiotics 53(10):1061(2000)に従って調製した2−アミノ−α−(メトキシイミノ)−4−チアジアゾール酢酸(15)(4.0g、19.8mmol)を、50mL DMF中の、Bull.Chem.Soc.Jpn.43:2925−33(1970)に従って調製したセファロスポリン誘導体14(8.42g、20.79mmol)(図9を参照のこと)およびHOAT(3.33g、21.78mmol)と混合した。その水溶液を窒素でパージし、0℃に冷却して、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド(3.99g、20.79mmol)、続いて1,3,5−コリジン(2.75mL、20.79mmol)を添加した。その水溶液を、0℃で2時間攪拌し、次いで、その反応混合物を、300mLの水の中に注いだ。その結果として生じた固体を、濾過し、飽和NaHCO3水溶液で粉砕し、再び濾過し、水で2回洗浄し、そして一晩、空気乾燥し、灰色がかった白色の粉末としてセファロスポリン中間体16を得た。この化合物を、さらに精製することなく使用した。
【0170】
分析データ:MS m/z 計算値.553.01;実測値.553.0(M+、35Cl)、555.0(M+、37Cl).
クロロ化合物16(4.0g、7.23mmol)を、4−t−BOC−アミノメチルピリジン(2.26g、10.85mmol)およびヨウ化ナトリウム(1.08g、7.23mmol)と一緒にフラスコの中に入れ、窒素でパージした。アセトン(60mL)を添加し、その反応混合物を、室温で2時間攪拌し、次いで、300mLの0.2M HClの中に注ぎ、結果としてフラスコの側面に赤いゴムの形成物が生じた。アセトンをデカントし、そのゴムを減圧下で乾燥させた。乾燥させるとすぐに、その残渣を20mLのDCMに溶解して、TFA(20mL)を添加した。その反応液を1時間攪拌し、その後、LC/MS解析によって脱保護が完了したことが示された。この溶液を次いで、200mLのEt2Oに注ぎ、生じた沈殿物を濾過し、Et2Oで洗浄し、そして減圧下で乾燥させた。乾燥させるとすぐに、粗製固体を、100mLの水で3時間、粉砕した。この水溶液を濾過し、そして生成物を凍結乾燥して、bis−TFA塩として粗製化合物4f(WがNであることを除いて4bと同一)を得た。その化合物を、さらに精製することなく使用した。
【0171】
分析データ:MS m/z 計算値.491.0;実測値.491.5
塩酸バンコマイシン(0.2g、0.135mmol)およびHOAt(20.6mg、0.135mmol)を、2mLのDMSO中に溶解した。この溶液に2mLのDMF中のPyBOP(70.2mg、0.0135mmol)の溶液を添加した。DIPEA(23.5μL、0.135mmol)を添加し、溶液を室温で20分間攪拌した。この時間の後、1mLのDMF中の4fの溶液(49mg、0.0675mmol)を添加し、その溶液を0℃に冷却した。次いで、1,3,5−コリジン(62.5μL、0.473mmol)を添加し、その溶液を0℃で45分間攪拌した。次いで、TFA(150μL)を添加し、その溶液を70mLのEt2Oの中に注いだ。生じた沈殿物を濾過し、Et2Oで洗浄し、そして減圧下で乾燥させた。その生成物を、逆相HPLCによって精製し、凍結乾燥によって単離して、トリ−TFA塩としてImを得た。
【0172】
分析データ:MS m/z 計算値.1937.8;実測値.1937.6.
(実施例14:化合物Inの調製)
DMF(100mL)中の2−アミノ−α−(トリチロキシイミノ)−4−チアゾール酢酸(17)(15.60g、36.3mmol)(2−アミノ−α−(ヒドロキシイミノ)−4−チアゾール酢酸エチルのトリチル化、続いて、加水分解によって調製した)の攪拌した溶液に、4.85g(36.3mmol)のN−クロロスクシンイミドを添加した(図10を参照のこと)。その反応混合物を、室温で15時間、攪拌し、次いで、水(500mL)の中に注ぎ、沈殿した固体を濾過して乾燥させ、灰色がかった白色の固体として5−クロロ化合物(15.11g、90%)を得た。
【0173】
分析データ:1H NMR(DMSO−d、300MHz):δ7.18−7.33(m、15H);MS m/z 実測値.486.4[M+Na]+.
250mLのDMF中のこの化合物(28.0g、60mmol)の攪拌した溶液に、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド(12.7g、66mmol)、HOBT(9.0g、66mmol)および2,4,6−コリジン(8.8mL、66mmol)を添加した。その反応混合物を0℃に冷却し、5分後、14(実施例13;図9を参照のこと)(24.4g、60mmol)を添加した。3.5時間後、その反応混合物を、酢酸エチル(1200mL)で希釈し、1.0M HCL(250mL)、飽和炭酸水素ナトリウム水溶液(250mL)およびブライン(250mL)で洗浄した。有機層を、乾燥(MgSO4)し、濾過し、真空化で濃縮して、灰色がかった白色の固体としてセファロスポリン中間体18(43.2g、88%)を得、それをさらに精製することなく使用した。
【0174】
分析データ:1H NMR(DMSO−d6、300MHz):δ3.65(q、2H)、3.76g(s、3H)、4.54(q、2H)、5.24(s、2H)、5.27(d、1H)、6.03(q、1H)、6.95(d、2H)、7.18−7.41(m、19H)、9.90(s、1H).
ヨウ化ナトリウム(920mg、61mmol)およびN−t−BOC−4−(アミノメチル)ピリジン(1.8g、86mmol)を、暗所(アルミホイルに包まれたフラスコ)でアセトン(20mL)中の18(5g)の攪拌した溶液に添加した。その反応混合物を、室温で2.5時間、攪拌し、次いで、ジエチルエーテル(200mL)に一部分ずつ添加し、生じた固体を濾過によって回収した。TFA/DCM(40ml、1:1)中の粗製固体(5.9g)の溶液を、室温で1時間攪拌し、次いで、ジエチルエーテル(300mL)に一部分ずつ添加した。生じた個体を、固体残渣を得るために濾過によって回収し、それを、分取用HPLCによって精製し、各々の白い固体として、4g(R9が水素であることを除いて4bと同一)の2個のヒドロキシイミノ立体異性体(それぞれ、240mgおよび120mg)のTFA塩を得た。
【0175】
分析データ:
4gの主要な立体異性体:1H NMR(DMSO−d6、300MHz):δ3.40(q、2H)、4.45(s、2H)、5.18(d、1H)、5.54(q、2H)、5.87(q、1H)、7.31(bs、2H)、8.18(d、2H)、8.71(bs、3H)、9.09(d、2H)、9.39(d、1H)、11.73(s、1H);MS m/z 実測値.524.2[M+H]+.
4gの少ない立体異性体:1H NMR(DMSO−d6、300MHz):δ3.50(q、2H)、4.45(s、2H)、5.19(d、1H)、5.58(q、2H)、5.83(q、1H)、7.27(bs、2H)、8.20(d、2H)、8.77(bs、3H)、8.81(d、1H)、9.10(d、2H)、12.51(s、1H);MS m/z 実測値.524.2[M+H]+.
DMF(1mL)中のPyBOP(170mg、0.33mmol)およびHOAt(44mg、0.33mmol)の溶液を、DMSO/DMF(6mL、3:1)中の塩酸バンコマイシン(485mg、0.33mmol)の攪拌した溶液に添加した。続いて、DIPEA(0.057mL、0.33mmol)を添加し、その反応混合物を、室温で30分間、攪拌した。TFA(0.026mL、0.33mmol)を、その反応混合物に添加し、次いで、それを0℃に冷却し、次いで、コリジン(0.097mL、0.73mmol)およびDMF(1mL)中の4g(123mg、0.16mmol)の溶液を添加した。その反応混合物を、4時間にわたって、室温で暖め、次いで、酢酸エチル(50mL)に一部分ずつ添加し、生じた固体を、濾過によって回収し、分取用HPLCによって精製し、白い固体として生成物In(174mg、46%)のTFA塩を得た。
【0176】
分析データ:MS m/z 実測値.979.4[(M−ピリジン)/2]+.
(実施例15:水溶性の決定)
本発明の化合物の水溶性を、以下の手順を使用して決定した。pH2.2での5重量%のデキストロース緩衝液を、99mLの5重量%デキストロース水溶液(Baxter)に1mlの1N 塩酸(Aldrich)を添加して調製した。次いで、較正標準物のための1mg/mLのストック溶液を、1mLのDMSO中に1mgの試験化合物を溶解することによって調製した。この溶液を、30秒間、ボルテックスし、次いで、10分間、超音波処理した。次いで、このストック溶液を、以下の濃度:50μg/mL、125μg/mL、250μg/mL、375μg/mLおよび500μg/mLを有する較正標準物を調製するために水で希釈した。
【0177】
各試験化合物(30mg)を、Millipore non−sterile,Ultrafree−MC 0.1μmフィルターユニット(Millipore UFC30VVOO)に秤量して入れ、磁気攪拌子を、各ユニットに添加した。次いで、5重量%のデキストロース緩衝液(750μL)を、各ユニットに添加し、これらの混合物を5分間、ボルテックスした。次いで、フィルターユニットを、エッペンドルフチューブラックに入れ、そのチューブラックを、磁気攪拌機の上に置いた。次いで、各ユニットを、1N NaOH(VWR)を使用してpH3に滴定し、得られた溶液を5分間、7000rpmで遠心分離した。次いで、各ユニットを、5%デキストロース緩衝液で200倍希釈し、希釈したサンプルを、分析のための自動サンプラーバイアルに移した。
【0178】
較正標準物および試験サンプルを、以下の条件を使用する逆相HPLCによって分析した:
カラム:Luna 150×4.6mm;C18;5μ
移動相:A=5/95、B=95/5、両方=MeCN/H2O;0.1%TFA
方法:10m Libo 100(6分で、0〜100%のB)
注入容量:20μL
波長:214nm
各試験サンプルの溶解性を、較正曲線と試験サンプルのピーク領域とを比較し、そして希釈係数を乗じることによって算出した。
【0179】
【表1】
(実施例16:最小発育阻止濃度(MIC)の決定)
最小発育阻止濃度(MIC)アッセイを、NCCLSガイドラインに記載の微量液体希釈法を用いて行った(broth microdilution method)(NCCLS.2000.Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically;Approved Standard−第5編、第20巻、No.2を参照のこと)。細菌株を、American Type Tissue Culture Collection(ATCC)、Stanford University Hospital(SU)、Kaiser Permanente
Regional Laboratory in Berkeley(KPB)、Massachusetts General Hospital(MGH)、the Centers for Disease Control(CDC)、the San Francisco Veterans’Administration Hospital(SFVA)またはthe University of California San Francisco Hospital(UCSF)から入手した。バンコマイシン耐性腸球菌は、それらのテイコプラニンに対する感受性に基づいて、VanAまたは
Van Bとして表現型決定した。Van A、Van B、Van C1またはVan
C2として遺伝子型決定した、いくつかのバンコマイシン耐性腸球菌をまた、Mayo
Clinicから入手した。
【0180】
このアッセイにおいて、低温保存した参照の細菌培養物および臨床株を、適切な寒天培地(すなわち、トリプティケースソイ(Trypticase Soy)寒天、脱繊維素化ヒツジ赤血球含有トリプティケースソイ寒天、ブレインハートインフュージョン(Brain Heart Infusion)寒天、チョコレート(Chocolate)寒天)での単離のために線画した。コロニーの形成を可能にするためのインキュベーション後、これらのプレートをパラフィルム(登録商標)で密閉し、そして2週間まで冷凍保存した。アッセイの接種材料を調製し、低い可変性を確実にするために、寒天プレートに培養した細菌単離物からのいくつかのコロニーを、接種ループを用いてつつき、そして無菌的にMueller−Hinton Broth(製造業者の認証に基づいて、必要とされるレベルの二価のカチオンを補充した)に移した。ブロス培養物を、35℃で一晩増殖させ、新鮮な、予め温めたブロスに希釈し、そして対数期まで増殖させた;これは、0.5 MacFarland standardまたは1mlあたり1×108コロニー形成単位(CFU/mL)に等しい。懸度がMacFarland標準に等しい場合、種可変性に起因して、全ての細胞懸濁物で1×108CFU/mLを含むとは限らない。従って、受容可能な調節(NCCLSガイドラインに基づく)を、異なる細菌株の希釈物において作製した。96ウェルマイクロタイタープレートにおいて、2倍連続希釈した抗生物質濃度シリーズ(やはり、対応する培地(100μL)中)に希釈した場合に、Mueller−Hinton Broth、補充したMueller−Hinton Broth、またはHaemophilus試験培地中でこの培養物(100μL)が最初の細菌濃度(5×105CFU/mL)を得るように、接種材料を希釈した。次いで、プレートを、35℃で18〜24時間インキュベートした。MICを、細菌の増殖を伴わない最低濃度のウェルとして目視で読み取った。細菌の増殖を、3つより多くのピンポイントコロニー、直径が2mmより大きな沈殿した細胞のボタン(button)、または明確な懸度として規定する。
【0181】
最初のスクリーニングにおいて慣用的に試験される株としては、メチシリン感受性Staphylococcus aureus(MSSA)、メチシリン耐性Staphylococcus aureus(MRSA)、ペニシリナーゼを生成するStaphylococcus aureus、メチシリン感受性Staphylococcus epidermidis(MSSE)、メチシリン耐性Staphylococcus epidermidis(MRSE)、バンコマイシン感受性Enterococcus faecium(EFMVS)、バンコマイシン感受性Enterococcus faecalis(EFSVS)、テイコプラニンにも耐性のバンコマイシン耐性Enterococcus faecium(EFMVR VanA)、テイコプラニンに感受性のバンコマイシン耐性Enterococcus faecium(EFMVR VanB
)、テイコプラニンにも耐性のバンコマイシン耐性Enterococcus faecalis(EFSVR VanA)、テイコプラニンに感受性のバンコマイシン耐性Enterococcus faecalis(EFSVR VanB)、ペニシリン感受
性Streptococcus pneumoniae(PSSP)およびペニシリン耐性Streptococcus pneumoniae(PSRP)が、挙げられる。Mueller−Hinton brothにおける十分な増殖についての、PSSPおよびPSRPの無能性に起因して、これらの株についてのMICを、脱線維素血液を補充したTSブロスまたはHaemophilus試験培地を補充したTSブロスのいずれかを用いて決定した。
【0182】
次いで、上記の株に対して十分な活性を有する試験化合物を、臨床単離物(上に列挙した種、ならびに種分化していない(non−speciated)コアグラーゼ陰性の、メチシリンに対して感受性のStaphylococcus(MS−CNS)および耐性のStaphylococcus(MR−CNS)の両方を含む)のより多くのパネルにおけるMIC値について試験した。さらに、これらの試験化合物をまた、グラム陰性微生物(例えば、Escherichia coli、Pseudomonas aeruginosa、Klebsiella pneumoniae、Enterobacter
cloacae、Acinetobacter baumannii、Haemophilius influenzaeおよびMoraxella catarrhalis)に対してMICについてアッセイした。
【0183】
表2は、既知の抗生物質であるバンコマイシンと比較した、メチシリン耐性S.aureus(MRSA)およびメチシリン感受性S.aureus(MSSA)に対する本発明の化合物についてのMIC90データを示す。
【0184】
【表2】
表2のデータは、本発明の化合物(すなわち、Ia、Ib、Ic、Id、Ie、If、ImおよびIn)が、des−クロロアナログIg、IjおよびIkまたはバンコマイシンのいずれかと比較して、MRSA33591に対して驚くべき予期しない抗菌活性を有すること;ならびに本発明の化合物が、バンコマイシンと比較して、MSSA 13709に対して驚くべき予期しない抗菌活性を有することを示す。
【0185】
(実施例17:時間−殺傷アッセイ)
この時間−殺傷アッセイは、試験化合物の細菌活性の速度を測定するための方法である。これらの手順は、V.Lorian、「Antibiotics in Laboratory Medicine」、第4版、WilliamsおよびWilkins(1996)、104〜105頁に記載されるのと類似の手順である。細菌コロニー形成を迅速に防護し、そして宿主組織損傷を減少させるために、迅速な時間−殺傷が所望される。
【0186】
細菌性接種材料を、MICの決定のために実施例16に記載されるように調製した。細菌を、振盪フラスコ中の予め温めた培地に希釈し、そして振盪しながらインキュベートした(200rpm、35℃)。0時間、1時間、4時間、および24時間で、サンプルをフラスコから取り出し、細菌を、プレート計数によって数えた。最初のサンプリングに続いて、アッセイされるべき化合物を振盪フラスコ培養物に添加した。化合物の添加前および添加後の、これらの間隔でのプレート計数を、時間−殺傷曲線に図式的に表した。細菌活性は、24時間まで、細菌細胞数において3対数以上の減少(同程度〜99.9%より多くの減少)として規定される。
【0187】
このアッセイにおいて、式Iの化合物(すなわち、化合物Ib)は、4時間において1.0μg/mL以下の濃度で、MRSA 33591に対して殺菌性であった。比較すると、バンコマイシンは、24時間において4μg/mLの濃度で、MRSA 33591に対して殺菌性であった。
【0188】
(実施例18:好中球減少マウスにおけるインビボでの有効性研究)
動物(雄性CD−1マウス、20−30g)を、Charles Rivers Laboratories(Gilroy、CA)から入手し、そして餌および水を自由に摂取させた。好中球減少症を、細菌接種の4日前および2日前に与えられたシクロホスファミドの腹腔内(IP)注射(200mg/kg)を介して誘導した。
【0189】
使用した生物は、診療的に関連のあるグラム陽性病原体の感受性株または耐性株(例えば、メチシリン感受性Staphylococcus aureus(MSSA13709)およびメチシリン耐性Staphylococcus aureus(MRSA3
3591))のいずれかであった。細菌接種材料の濃度は約106CFU/mLであった。動物を、イソフルランで軽く麻酔し、そして50mLの細菌接種材料を前大腿に注射した。接種の1時間後、動物に、ビヒクルまたは試験化合物の適切な用量を静脈内投与した。処置の0時間後および24時間後に、これらの動物を安楽死させ(CO2窒息)、そして前大腿および後大腿を無菌的に収集した。この大腿を、10mLの滅菌生理食塩水に入れ、ホモジナイズした。ホモジネートの希釈物を、トリプティクソイ(tripticsoy)寒天プレート上にプレートし、これを一晩インキュベートした。所定のプレート上の細菌コロニーの数は、希釈係数を乗じ、大腿重量(グラム)で除算し、対数CFU/gとして表した。ED50(大腿力価において最大減少の50%を生成するために必要とした用量)を、各試験化合物について評価した。
【0190】
このアッセイにおいて、式Iの化合物(すなわち、化合物Ib)は、バンコマイシンについてのivで9mg/kgのED50と比べて、ivで0.10mg/kg未満のED50を有した。
【0191】
本発明は、その特定の実施形態を参照して記載されているが、種々の変更がなされ得、本発明の真の精神および範囲から逸脱することなしに等価物が置換され得ることは、当業者によって理解されるべきである。さらに、多くの改変が、特定の状況、物質、成分の組成物、プロセス、プロセス工程または工程を、本発明の目的、精神および範囲に適応させるためになされ得る。全てのこのような改変は、本明細書に添付される特許請求の範囲内であることが意図される。さらに、本明細書中で引用された全ての刊行物、特許および特許書類は、それらが参考として個々に援用されるかのような同じ程度で、本明細書中にその全体が参考として援用される。
【技術分野】
【0001】
(発明の背景)
(発明の分野)
本発明は、抗生物質として有用な新規架橋バンコマイシン−セファロスポリン化合物に関する。本発明はまた、このような化合物を含有する薬学的組成物;このような化合物を抗細菌剤として使用する方法;ならびにこのような化合物を調製するために有用なプロセスおよび中間体に関する。
【背景技術】
【0002】
(当該分野の現状)
種々のクラスの抗生物質化合物は、当該分野において公知であり、これらの化合物としては、例えば、β−ラクタム抗生物質(例えば、セファロスポリン)およびグリコペプチド抗生物質(例えば、バンコマイシン)が挙げられる。架橋抗生物質化合物はまた、当該分野において公知である。例えば、W.L.Truettに対して発行され、そして「Antibiotics and Process for Preparation」と題された特許文献1;1999年12月16日に発行され、そして「Novel Antibacterial Agents」と題された特許文献2を参照のこと。さらに、2003年4月17日に発行され、そして「Cross−Linked Glycopeptide−Cephalosporin Antibiotics」と題された特許文献3は、セファロスポリン基のオキシム部分に共有結合したグリコペプチド基を有する化合物を開示している。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】米国特許第5,693,791号明細書
【特許文献2】国際公開第99/64049号パンフレット
【特許文献3】国際公開第03/031449号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0004】
しかし、細菌が抗生物質に対する耐性を発達する能力に起因して、独自の化学構造を有する新規抗生物質に対する必要性が存在している。さらに、改善された抗細菌特性を有する新規抗生物質に対する必要性が存在し、これらの抗細菌特性としては、例えば、グラム陽性細菌に対する増強した能力が挙げられる。特に、細菌の抗生物質耐性株(例えば、メチシリン耐性Staphylococci aureus(MRSA))に対して高度に効果的な新規抗生物質に対する必要性が存在している。
【課題を解決するための手段】
【0005】
(発明の要旨)
本発明は、抗生物質として有用な架橋グリコペプチド−セファロスポリン化合物を提供する。本発明の化合物は、独自の化学構造を有し、この化学構造において、グリコペプチド基は、セファロスポリン基のピリジニウム部分に共有結合している。特性のなかでもとりわけ、本発明の化合物は、メチシリン耐性Staphylococci aureus(MRSA)を含むグラム陽性細菌に対して、驚くべき予測されなかった能力を有することが見出されている。
【0006】
従って、本発明の1つの局面において、本発明は、式I:
【0007】
【化8】
の化合物またはその薬学的に受容可能な塩を提供し、ここで、
X1およびX2の各々は、独立して、水素またはクロロであり;
Wは、NおよびCClからなる群より選択され;
R2は、水素またはC1〜6アルキルであり;
R4およびR5のうちの一方は、ヒドロキシであり、そして他方は、水素であり;
R6およびR7の各々は、独立して、水素またはメチルであり;
R8は、水素または式:
【0008】
【化9】
の基であり;
R9は、水素、C1〜6アルキルおよびC3〜6シクロアルキルから選択され、ここで、アルキルおよびシクロアルキルは、−COOHまたは1個〜3個のフッ素原子で置換され得;
各R3は、独立して、C1〜6アルキル、−OR、ハロ、−SR、−S(O)R、−S(O)2Rおよび−S(O)2ORから選択され、ここで、各Rは、独立して、C1〜6アルキルであり、このC1〜6アルキルは、COOHまたは1個〜3個のフッ素原子で置換され得;
nは、0、1、2または3であり;
xは、0、1または2であり;
yは、0、1または2であり;
Raは、−Y−R”−であり、ここで、
R”は、C1〜12アルキレン、C2〜12アルケニレン、C2〜12アルキニレン、C3〜6シクロアルキレン、C6〜10アリーレン、C2〜9ヘテロアリーレン、C3〜6複素環およびその組み合わせから選択され、そして必要に応じて、Zから選択される1個〜2個の基で置換され、ここで、Zは、−OR’、−SR’、−F、−Cl、−N(R’)2、−OC(O)R’、−C(O)OR’、−NHC(O)R’、−C(O)N(R’)2、−CF3および−OCF3、ならびに天然に存在するアミノ酸の側鎖からなり、ここで、各R’は、独立して、水素またはC1〜4アルキルであり;そしてR”は、最も多くて20個の非水素原子を含み;
Yは、メタ位またはパラ位でR”をピリジニウム環に結合し、直接結合、NR’、O(エーテル)、S(スルフィド)、C(O)(カルボニル)、NR’(CO)およびC(O)NR’から選択されて、Yにおけるヘテロ原子とR”におけるヘテロ原子とも間の直接結合を除外し;
各RbおよびRdは、独立して、水素、C1〜6アルキル、C2〜6アルケニルおよびC2〜6アルキニルからなる群より選択され;
各Rcは、独立して、−Y’−R”−Y’−であり、ここで、各Y’は、独立して、直接結合、O(エーテル)およびNR’からなる群よりされ、Y’におけるヘテロ原子とR”におけるヘテロ原子との間の直接結合を除外し;そして
各Reは、独立して、上記R”により規定される基から選択される。
【0009】
別の局面において、本発明は、式II:
【0010】
【化10】
の化合物またはその薬学的に受容可能な塩を提供し;ここで、
Wは、NおよびCClからなる群より選択され;
R9は、水素、C1〜6アルキルおよびC3〜6シクロアルキルから選択され、ここで、アルキルおよびシクロアルキルは、−COOHまたは1個〜3個のフッ素原子で置換され得;
ピリジニウム環は、メタ置換またはパラ置換を有し
R10は、水素またはC1〜6アルキルであり;そして
R11は、C1〜12アルキレンである。
【0011】
別のその組成物の局面において、本発明は、薬学的に受容可能なキャリアおよび治療有効量の式Iもしくは式IIの化合物またはその薬学的に受容可能な塩を含有する薬学的組成物を提供し、本明細書中で議論されるあらゆる特定の実施形態を含む。
【0012】
細菌と、増殖阻害量の式Iもしくは式IIの化合物またはその薬学的に受容可能な塩とを接触させることによって、細菌の増殖を阻害する方法および/または細菌の細胞壁生合成を阻害する方法がまた、提供され、本明細書中で議論されるあらゆる特定の実施形態を含む。特に、上記方法は、上記化合物が、本明細書中でIa、Ib、Ic、Id、IeおよびIfとして示される化合物からなる群より選択される実施形態を含む。
【0013】
関連する局面において、本発明は、哺乳動物における細菌感染を処置する方法を提供し、この方法は、薬学的に受容可能なキャリアおよび治療有効量の式Iもしくは式IIの化合物、またはその薬学的に受容可能な塩を含有する薬学的組成物を、哺乳動物に投与する工程を包含し、本明細書中で議論されるあらゆる特定の実施形態を含む。
【0014】
本発明はまた、式Iもしくは式IIの化合物、またはその塩を調製するためのプロセスに関する。従って、別のその方法の局面において、本発明は、式Iの化合物またはその塩を調製するためのプロセスを提供し;このプロセスは、式1の化合物またはその塩、その活性化誘導体もしくはその保護された誘導体と、式3もしくは式4の化合物またはその塩、その活性化誘導体もしくはその保護された誘導体とを反応させて;式Iの化合物またはその塩を提供する工程を包含し;ここで、上記式1、式3および式4の化合物は、本明細書中で規定されるとおりである。
【0015】
さらに、別のその方法の局面において、本発明は、式Iの化合物またはその塩を調製するためのプロセスを提供し;このプロセスは、式2の化合物またはその塩、その活性化誘導体もしくはその保護された誘導体と;式5の化合物またはその塩、その活性化誘導体もしくはその保護された誘導体とを反応させて;式Iの化合物またはその塩を提供する工程を包含し;ここで、上記式2および式5の化合物は、本明細書中に規定されるとおりである。
【0016】
1つの実施形態において、これらのプロセスは、式Iの化合物の薬学的に受容可能な塩を形成する工程をさらに包含する。本発明はまた、あらゆるこれらのプロセスによって調製される生成物に関する。
【0017】
本発明はまた、治療における使用のための、式Iもしくは式IIの化合物、またはその薬学的に受容可能な塩に関する。さらに、本発明は、医薬の製造のための、式Iもしくは式IIの化合物、またはその薬学的に受容可能な塩の使用に関し、これらの医薬としては、哺乳動物における細菌感染を処置するための医薬が挙げられる。
本発明は、例えば以下の項目を提供する。
(項目1)
式I:
【化1】
の化合物またはその薬学的に受容可能な塩であって、ここで:
X1およびX2の各々は、独立して、水素またはクロロであり;
Wは、NおよびCClから選択され;
R2は、水素またはC1〜6アルキルであり;
R4およびR5のうちの一方は、ヒドロキシであり、そして他方は、水素であり;
R6およびR7の各々は、独立して、水素またはメチルであり;
R8は、水素または式:
【化2】
の基であり;
R9は、水素、C1〜6アルキルおよびC3〜6シクロアルキルから選択され、ここで、アルキルおよびシクロアルキルは、−COOHまたは1個〜3個のフッ素原子で置換され得;
各R3は、独立して、C1〜6アルキル、−OR、ハロ、−SR、−S(O)R、−S(O)2Rおよび−S(O)2ORから選択され、ここで、各Rは、独立して、C1〜6アルキルであり、このC1〜6アルキルは、COOHまたは1個〜3個のフッ素原子で置換され得;
nは、0、1、2または3であり;
xは、0、1または2であり;
yは、0、1または2であり;
Raは、−Y−R”−であり、ここで、
R”は、C1〜12アルキレン、C2〜12アルケニレン、C2〜12アルキニレン、C3〜6シクロアルキレン、C6〜10アリーレン、C2〜9ヘテロアリーレン、C3〜6複素環およびその組み合わせから選択され、そして必要に応じて、Zから選択される1個〜2個の基で置換され、ここで、Zは、−OR’、−SR’、−F、−Cl、−N(R’)2、−OC(O)R’、−C(O)OR’、−NHC(O)R’、−C(O)N(R’)2、−CF3および−OCF3、ならびに天然に存在するアミノ酸の側鎖からなり、ここで、各R’は、独立して、水素またはC1〜4アルキルであり;そしてR”は、最も多くて20個の非水素原子を含み;
Yは、メタ位またはパラ位でR”をピリジニウム環に結合し、直接結合、NR’、O、S、C(O)、NR’C(O)およびC(O)NR’から選択されて、Yにおけるヘテロ原子とR”におけるヘテロ原子との間の直接結合除外し;
各RbおよびRdは、独立して、水素、C1〜6アルキル、C2〜6アルケニルおよびC2〜6アルキニルからなる群より選択され;
各Rcは、独立して、−Y’−R”−Y’−であり、ここで、各Y’は、独立して、直接結合、OおよびNR’から選択され、Y’におけるヘテロ原子とR”におけるヘテロ原子との間の直接結合を除外し;そして
各Reは、独立して、R”から選択される、化合物またはその薬学的に受容可能な塩。
(項目2)
項目1に記載の化合物であって、ここで、R9は、水素またはC1〜4アルキルである、化合物。
(項目3)
項目2に記載の化合物であって、ここで、R9は、水素またはメチルである、化合物。
(項目4)
項目1〜3のいずれか1項に記載の化合物であって、ここで、Wは、CClである、化合物。
(項目5)
項目1〜3のいずれか1項に記載の化合物であって、ここで、Wは、Nである、化合物。
(項目6)
項目1〜5のいずれか1項に記載の化合物であって、ここで、各R3は、独立して、非置換C1〜4アルキル、非置換C1〜4アルコキシ、フルオロおよびクロロから選択される、化合物。
(項目7)
項目1〜6のいずれか1項に記載の化合物であって、ここで、nは、0である、化合物。
(項目8)
項目1〜7のいずれか1項に記載の化合物であって、ここで、xは、1であり、そしてyは、0である、化合物。
(項目9)
項目8に記載の化合物であって、ここで、Raは、−Y−R”−であり;Yは、直接結合であり;そしてR”は、C1〜6アルキレンである、化合物。
(項目10)
項目8または9に記載の化合物であって、ここで、Rcは、−Y’−R”−Y’−であり、ここで、各Y’は、直接結合であり;そしてR”は、必要に応じて−COOH基で置換されたC1〜12アルキレンである、化合物。
(項目11)
項目8〜10のいずれか1項に記載の化合物であって、ここで、Rbは、水素またはC1〜4アルキルである、化合物。
(項目12)
項目1〜7のいずれか1項に記載の化合物であって、ここで、xは、1であり、そしてyは、1である化合物。
(項目13)
項目12に記載の化合物であって、ここで、Raは、−Y−R”−であり;Yは、直接結合であり;そしてR”は、C1〜6アルキレンである、化合物。
(項目14)
項目12または13に記載の化合物であって、ここで、Rcは、−Y’−R”−Y’−であり、ここで、各Y’は、直接結合であり;そしてR”は、必要に応じて−COOH基で置換されるC1〜12アルキレンである、化合物。
(項目15)
項目12〜14のいずれか1項に記載の化合物であって、ここで、Reは、C1〜12アルキレンである、化合物。
(項目16)
項目12〜15のいずれか1項に記載の化合物であって、ここで、RbおよびRdは、独立して、水素またはC1〜4アルキルである、化合物。
(項目17)
項目1〜7のいずれか1項に記載の化合物であって、ここで、xは、0であり、そしてyは、0である、化合物。
(項目18)
項目17に記載の化合物であって、ここで、Raは、−Y−R”−であり;Yは、直接結合であり;そしてR”は、C1〜6アルキレンである、化合物。
(項目19)
項目1〜18のいずれか1項に記載の化合物であって、ここで、R2は、水素である、化合物。
(項目20)
式II:
【化3】
の化合物またはその薬学的に受容可能な塩であって、ここで、
Wは、NおよびCClから選択され;
R9は、水素、C1〜6アルキルおよびC3〜6シクロアルキルから選択され、ここで、アルキルおよびシクロアルキルは、−COOHまたは1個〜3個のフッ素原子で置換され得;
ピリジニウム環は、メタ置換またはパラ置換を有し;
R10は、水素またはC1〜6アルキルであり;そして
R11は、C1〜12アルキレンである、化合物またはその薬学的に受容可能な塩。
(項目21)
項目20に記載の化合物であって、ここで、Wは、CClである、化合物。
(項目22)
項目20または21に記載の化合物であって、ここで、ここで、R9は、水素またはC1〜4アルキルである、化合物。
(項目23)
項目22に記載の化合物であって、ここで、R9は、水素である、化合物。
(項目24)
項目22に記載の化合物であって、ここで、R9は、メチルである、化合物。
(項目25)
項目20〜24のいずれか1項に記載の化合物であって、ここで、前記ピリジニウム環は、パラ置換されている、化合物。
(項目26)
項目20〜25のいずれか1項に記載の化合物であって、ここで、R10は、水素またはC1〜4アルキルである、化合物。
(項目27)
項目26に記載の化合物であって、ここで、R10は、水素、メチルまたはエチルである、化合物。
(項目28)
項目20〜27のいずれか1項に記載の化合物であって、ここで、R11は、C1〜10アルキレンである、化合物。
(項目29)
項目28に記載の化合物であって、ここで、R11は、−(CH2)2−または−(CH2)5−である、化合物。
(項目30)
項目20に記載の化合物であって、ここで、Wは、CClであり;R9は、メチル
であり;R10は、エチルであり;R11は、−(CH2)2−であり;そして前記ピリジニウム環は、パラ置換されている、化合物。
(項目31)
薬学的に受容可能なキャリアおよび治療有効量の項目1〜30のいずれか1項に記載の化合物を含有する、薬学的組成物。
(項目32)
細菌の増殖を阻害する方法であって、該方法は、細菌と、増殖阻害量の項目1〜30のいずれか1項に記載の化合物とを接触させる工程を包含する、方法。
(項目33)
哺乳動物における細菌感染を処置する方法であって、該方法は、哺乳動物に、薬学的に受容可能なキャリアおよび治療有効量の項目1〜30のいずれか1項に記載の化合物を含有する薬学的組成物を投与する工程を包含する、方法。
(項目34)
項目1〜30のいずれか1項に記載の化合物を調製するためのプロセスであって;該プロセスは、式1:
【化4】
の化合物またはその塩もしくはその活性化誘導体もしくはその保護された誘導体と、式3または4:
【化5】
の化合物またはその塩もしくはその活性化誘導体もしくはその保護された誘導体を反応させて、式Iの化合物またはその塩を得る工程を包含する、プロセス。
(項目35)
項目1〜30のいずれか1項に記載の化合物を調製するためのプロセスであって;該プロセスは、式5:
【化6】
の化合物またはその塩もしくはその活性化誘導体もしくは保護された誘導体を;式2:
【化7】
の化合物またはその塩もしくはその活性化誘導体もしくは保護された誘導体と反応させて、式Iの化合物またはその塩を得る工程を包含する、プロセス。
(項目36)
項目34または35に記載のプロセスによって調製される生成物。
【図面の簡単な説明】
【0018】
【図1】図1は、本発明の選択された実施形態に従う架橋グリコペプチド−セファロスポリン抗生物質の例を示す。
【図2】図2は、架橋グリコペプチド−セファロスポリン抗生物質を調製するために有用なセファロスポリン中間体を調製するための合成スキームを示す。
【図3】図3〜図6は、本明細書中で、それぞれ、Ia、Ib、IdおよびIeとして示される代表的な架橋グリコペプチド−セファロスポリン抗生物質を調製するための合成スキームを示す。
【図4】図3〜図6は、本明細書中で、それぞれ、Ia、Ib、IdおよびIeとして示される代表的な架橋グリコペプチド−セファロスポリン抗生物質を調製するための合成スキームを示す。
【図5】図3〜図6は、本明細書中で、それぞれ、Ia、Ib、IdおよびIeとして示される代表的な架橋グリコペプチド−セファロスポリン抗生物質を調製するための合成スキームを示す。
【図6】図3〜図6は、本明細書中で、それぞれ、Ia、Ib、IdおよびIeとして示される代表的な架橋グリコペプチド−セファロスポリン抗生物質を調製するための合成スキームを示す。
【図7】図7および図8は、比較のための架橋グリコペプチド−セファロスポリン抗生物質のデス−クロロアナログを調製するための合成スキームを示す。
【図8】図7および図8は、比較のための架橋グリコペプチド−セファロスポリン抗生物質のデス−クロロアナログを調製するための合成スキームを示す。
【図9】図9および図10は、代表的な架橋グリコペプチド−セファロスポリン抗生物質をさらに調製するための合成スキームを示す。
【図10】図9および図10は、代表的な架橋グリコペプチド−セファロスポリン抗生物質をさらに調製するための合成スキームを示す。
【発明を実施するための形態】
【0019】
(発明の詳細な説明)
本発明は、式Iもしくは式IIの新規グリコペプチド−セファロスポリン化合物、またはその薬学的に受容可能な塩を提供する。これらの化合物は、複数のキラル中心を有し、そして、この点において、上記化合物は、示される立体化学を有することが意図される。特に、上記化合物のグリコペプチド部分は、対応する天然に存在するグリコペプチド(すなわち、バンコマイシン、クロロオリエンチシンAなど)の立体化学を有することが意図される。上記分子のセファロスポリン部分は、公知のセファロスポリン化合物の立体化学を有することが意図される。しかし、示されるものとは異なる立体化学を有する少量の異性体が、本発明の組成物中に存在し得、ただし、全体としての上記組成物の有用性は、このような異性体の存在によってそれほど減少されないことを、当業者は理解する。
【0020】
さらに、本発明の化合物の結合部分は、1つ以上のキラル中心を含み得る。代表的に、上記分子のこの部分は、ラセミ混合物として調製される。しかし、所望される場合、純粋な立体異性体(すなわち、個々のエナンチオマーまたはジアステレオマー)が使用され得るか、または立体異性体エンリッチ混合物が用いられ得る。全てのこのような立体異性体およびエンリッチ混合物は、本発明の範囲内に含まれる。
【0021】
さらに、本発明の化合物は、いくつかの酸性基(すなわち、カルボン酸基)およびいくつかの塩基性基(すなわち、一級アミン基および二級アミン基)を含み、そして、そのため、上記式Iの化合物は、種々の塩形態で存在し得る。全てのこのような塩形態は、本発明の範囲内に含まれる。また、上記式Iの化合物は、ピリジニウム環を含むので、上記ピリジニウム基のための陰イオン性対イオンが、必要に応じて存在し、これらの陰イオン性対イオンとしては、ハライド(例えば、クロライド);カルボキシレート(例えば、アセテート)などが挙げられるが、これらに限定されない。
(定義)
以下の用語は、本明細書中で使用される場合、別に示されなければ、以下の意味を有する:
用語「アルキル」とは、一価飽和炭化水素基をいい、直鎖状でも分枝していてもよい。別に規定されなければ、このようなアルキル基は、代表的に、1個〜10個の炭素原子を含む。代表的なアルキル基としては、例として、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、sec−ブチル、イソブチル、tert−ブチル、n−ペンチル、n−ヘキシル、n−ヘプチル、n−オクチル、n−ノニル、n−デシルなどが挙げられる。
【0022】
用語「アルキレン」とは、二価飽和炭化水素基をいい、直鎖状でも分枝していてもよい。別に規定されなければ、このようなアルキレン基は、代表的に、1個〜10個の炭素原子を含む。代表的なアルキレン基としては、例として、メチレン、エタン−1,2−ジイル(「エチレン」)、プロパン−1,2−ジイル、プロパン−1,3−ジイル、ブタン−1,4−ジイル、ペンタン−1,5−ジイルなどが挙げられる。
【0023】
用語「アルケニル」とは、一価不飽和炭化水素基をいい、直鎖状でも分枝していてもよく、少なくとも1つ、そして代表的には、1つ、2つまたは3つの炭素−炭素二重結合を有する。別に規定されなければ、このようなアルケニル基は、代表的に、2個〜10個の炭素原子を含む。代表的なアルケニル基としては、例として、エテニル、n−プロペニル、イソプロペニル、n−ブタ−2−エニル、n−ヘキサ−3−エニルなどが挙げられる。
【0024】
用語「アルキニル」とは、一価不飽和炭化水素基をいい、直鎖状でも分枝していてもよく、少なくとも1つ、そして代表的には、1つ、2つまたは3つの炭素−炭素三重結合を有する。別に規定されなければ、このようなアルキニル基は、代表的に、2個〜10個の炭素原子を含む。代表的なアルキニル基としては、例として、エチニル、n−プロピニル、n−ブタ−2−イニル、n−ヘキサ−3−イニルなどが挙げられる。
【0025】
用語「アリール」とは、単一の環(すなわち、フェニル)または縮合環(すなわち、ナフタレン)を有する一価芳香族炭化水素をいう。別に規定されなければ、このようなアリール基は、代表的に、6個〜10個の炭素環原子を含む。代表的なアリール基としては、例として、フェニルおよびナフタレン−1−イル、ナフタレン−2−イルなどが挙げられる。
【0026】
用語「アリーレン(arylene)」とは、単一の環(すなわち、フェニレン)または縮合環(すなわち、ナフタレンジイル)を有する二価芳香族炭化水素をいう。別に規定されなければ、このようなアリーレン基は、代表的に、6個〜10個の炭素環原子を含む。代表的なアリーレン基としては、例として、1,2−フェニレン、1,3−フェニレン、1,4−フェニレン、ナフタレン−1,5−ジイル、ナフタレン−2,7−ジイルなどが挙げられる。
【0027】
用語「シクロアルキル」とは、一価飽和炭素環式炭化水素基をいう。別に規定されなければ、このようなシクロアルキル基は、代表的に、3個〜10個の炭素原子を含む。代表的なシクロアルキル基としては、例として、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルなどが挙げられる。
【0028】
用語「シクロアルキレン」とは、二価飽和炭素環式炭化水素基をいう。別に規定されなければ、このようなシクロアルキル基は、代表的に、3個〜10個の炭素原子を含む。代表的なシクロアルキレン基としては、例として、シクロプロパン−1,2−ジイル、シクロブチル−1,2−ジイル、シクロブチル−1,3−ジイル、シクロペンチル−1,2−ジイル、シクロペンチル−1,3−ジイル、シクロヘキシル−1,2−ジイル、シクロヘキシル−1,3−ジイル、シクロヘキシル−1,4−ジイルなどが挙げられる。
【0029】
用語「ハロ」とは、フルオロ、クロロ、ブロモおよびヨードをいう。
【0030】
用語「ヘテロアリール」とは、単一の環または2つの縮合環を有し、そしてその環に、窒素、酸素または硫黄から選択される少なくとも1個のヘテロ原子(代表的には、1個〜3個のヘテロ原子)を含む一価芳香族基をいう。別に規定されなければ、このようなヘテロアリール基は、代表的に、全体で5個〜10個の環原子を含む。代表的なヘテロアリール基としては、例として、ピロール、イミダゾール、チアゾール、オキサゾール、フラン、チオフェン、トリアゾール、ピラゾール、イソキサゾール、イソチアゾール、ピリジン、ピラジン、ピリダジン、ピリミジン、トリアジン、インドール、ベンゾフラン、ベンゾチオフェン、ベンズイミダゾール、ベンズチアゾール、キノリン、イソキノリン、キナゾリン、キノキサリンなどの一価の種が挙げられ、ここで、その結合点は、あらゆる利用可能な炭素環原子または窒素環原子にある。
【0031】
用語「ヘテロアリーレン」とは、単一の環または2つの縮合環を有し、そしてその環に、窒素、酸素または硫黄から選択される少なくとも1個のヘテロ原子(代表的には、1個〜3個のヘテロ原子)を含む二価芳香族基をいう。別に規定されなければ、このようなヘテロアリーレン基は、代表的に、全体で5個〜10個の環原子を含む。代表的なヘテロアリーレン基としては、例として、ピロール、イミダゾール、チアゾール、オキサゾール、フラン、チオフェン、トリアゾール、ピラゾール、イソキサゾール、イソチアゾール、ピリジン、ピラジン、ピリダジン、ピリミジン、トリアジン、インドール、ベンゾフラン、ベンゾチオフェン、ベンズイミダゾール、ベンズチアゾール、キノリン、イソキノリン、キナゾリン、キノキサリンなどの二価の種が挙げられ、ここで、その結合点は、あらゆる利用可能な炭素環原子または窒素環原子にある。
【0032】
用語「ヘテロサイクリル」または「ヘテロサイクリック」とは、単一の環または複数の縮合環を有し、そしてその環に、窒素、酸素または硫黄から選択される少なくとも1個のヘテロ原子(代表的には、1個〜3個のヘテロ原子)を含む一価または二価の飽和基または不飽和(非芳香族)基をいう。別に規定されなければ、このようなヘテロサイクリック基は、代表的に、全体で2個〜9個の環原子を含む。代表的なヘテロサイクリック基としては、例として、ピロリジニン、イミダゾリジン、ピラゾリジン、ピペリジン、1,4−ジオキサン、モルホリン、チオモルホリン、ピペラジン、3−ピロリンなどの一価の種が挙げられ、ここで、その結合点は、あらゆる利用可能な炭素環原子または窒素環原子にある。
【0033】
用語「セファロスポリン」は、本明細書中でその当該分野で認識される様式で使用されて、以下の一般式および番号システム:
【0034】
【化11】
を有するβ−ラクタム環系をいい、ここで、RxおよびRyは、上記セファロスポリンの残りの部分を示す。
【0035】
用語「グリコペプチド抗生物質」または「グリコペプチド」は、本明細書中でその当該分野で認識される様式で使用されて、グリコペプチドまたはダルバー(dalbah)ペプチドとして公知の抗生物質のクラスをいう。例えば、R.Nagarajan,「Glycopepitide Antibiotics」,Marcel Dekker,Inc.(1994)およびそこに引用される参考文献を参照のこと。代表的なグリコペプチドとしては、バンコマイシン、A82846A(エレモマイシン)、A82846B(クロロオリエンチシンA)、A82846C、PA−42867−A(オリエンチシンA)、PA−42867−C、PA−42867−Dなどが挙げられる。
【0036】
用語「バンコマイシン」は、本明細書中でその当該分野で認識される様式で使用されて、バンコマイシンとして公知のグリコペプチド抗生物質をいう。本発明の化合物において、上記結合部分のための結合点は、バンコマイシンの「C末端」にある。
【0037】
用語「架橋グリコペプチド−セファロスポリン抗生物質」とは、グリコペプチド成分とセファロスポリン成分との共有結合体をいう。
【0038】
用語「薬学的に受容可能な塩」とは、患者(例えば、哺乳動物)への投与のために受容可能な塩(例えば、所与の投薬レジメンのために受容可能な哺乳動物安全性を有する塩)をいう。このような塩は、薬学的に受容可能な無機塩基または有機塩基および薬学的に受容可能な無機酸または有機酸に由来し得る。薬学的に受容可能な無機塩基由来の塩としては、アルミニウム、アンモニウム、カルシウム、銅、第二鉄、第一鉄、リチウム、マグネシウム、第二マンガン、第一マンガン、カリウム、ナトリウム、亜鉛などが挙げられる。アンモニウム塩、カルシウム塩、マグネシウム塩、カリウム塩およびナトリウム塩が、特に好ましい。薬学的に受容可能な有機塩基由来の塩としては、置換アミン、環状アミン、天然に存在するアミンなど(例えば、アルギニン、ベタイン、カフェイン、コリン、N,N’−ジベンジルエチレンジアミン、ジエチルアミン、2−ジエチルアミノエタノール、2−ジメチルアミノエタノール、エタノールアミン、エチレンジアミン、N−エチルモルホリン、N−エチルピペリジン、グルカミン、グルコサミン、ヒスチジン、ヒドラバミン、イソプロピルアミン、リジン、メチルグルカミン、モルホリン、ピペラジン、ピペリジン、ポリアミン樹脂、プロカイン、プリン、テオブロミン、トリエチルアミン、トリメチルアミン、トリプロピルアミン、トロメタミンなど)を含む、一級アミン、二級アミンおよび三級アミンの塩が挙げられる。薬学的に受容可能な酸由来の塩としては、酢酸、アスコルビン酸、ベンゼンスルホン酸、安息香酸、樟脳スルホン酸、クエン酸、エタンスルホン酸、フマル酸、グルコン酸、グルコロン酸、グルタミン酸、馬尿酸、臭化水素酸、塩酸、イセチオン酸、乳酸、ラクトビオン酸、マレイン酸、リンゴ酸、マンデル酸、メタンスルホン酸、ムチン酸、ナフタレンスルホン酸、ニコチン酸、硝酸、パモ酸、パントテン酸、リン酸、コハク酸、硫酸、酒石酸、p−トルエンスルホン酸など由来の塩が挙げられる。特に好ましいのは、クエン酸、臭化水素酸、塩酸、マレイン酸、リン酸、硫酸および酒石酸である。
【0039】
用語「その塩」とは、酸の水素が、陽イオン(例えば、金属陽イオンまたは有機陽イオンなど(例えば、NH4+陽イオンなど))によって置換される場合に形成される化合物をいう。好ましくは、上記塩は、薬学的に受容可能な塩であるが、これは、患者への投与が意図されない中間体化合物の塩については必要とされない。
【0040】
用語「治療有効量」とは、処置を必要としている患者に投与される場合、有効な処置のために充分な量をいう。
【0041】
用語「処置する」または「処置」とは、本明細書中で使用される場合、患者(例えば、哺乳動物(特に、ヒトまたは伴生種動物(companion animal)))における疾患もしくは医学的状態(例えば、細菌感染)を処置することまたはその処置をいい:
(a)上記疾患または医学的状態の発症を防止する工程(すなわち、患者の予防処置);
(b)上記疾患または医学的状態を改善する工程(すなわち、患者における上記疾患もしくは医学的状態を除去する工程またはその後退を引き起こす工程);
(c)上記疾患または医学的状態を抑制する工程(すなわち、患者における上記疾患もしくは医学的状態の進行を遅延させる工程または妨げる工程);あるいは
(d)患者における上記疾患または医学的状態の症状を軽減する工程
を包含する。
【0042】
用語「増殖阻害量」とは、微生物の増殖もしくは生殖を阻害するために充分な量または上記微生物(グラム陽性細菌を含む)の死もしくは溶解を引き起こすために充分な量をいう。
【0043】
用語「細胞壁生合成阻害量」とは、微生物(グラム陽性細菌を含む)における細胞壁生合成を阻害するために充分な量をいう。
【0044】
用語「脱離基」とは、置換反応(例えば、求核置換反応)において別の官能基または原子によって置換され得る官能基または原子をいう。例として、代表的な脱離基としては、クロロ基、ブロモ基およびヨード基;ならびにスルホン酸エステル基(例えば、メシレート、トシレート、ブロシレート、ノシレートなど);活性化エステル基(例えば、7−アザベンゾトリアゾール−1−オキシなど);アシルオキシ基(例えば、アセトキシ、トリフルオロアセトキシなど)が挙げられる。
【0045】
用語「その保護された誘導体」とは、保護基またはブロック基により、上記化合物の1つ以上の官能基を、所望されない反応から保護する特定の化合物の誘導体をいう。保護され得る官能基としては、例として、カルボン酸基、アミノ基、ヒドロキシ基、チオール基、カルボニル基などが挙げられる。カルボン酸のための代表的な保護基としては、エステル(例えば、p−メトキシベンジルエステル)、アミドおよびヒドラジド;アミノ基のためには、カルバメート(例えば、tert−ブトキシカルボニル)およびアミド;ヒドロキシ基のためには、エーテルおよびエステル;チオール基のためには、チオエーテルおよびチオエステル;カルボニル基のためには、アセタールおよびケタール;などが挙げられる。このような保護基は、当業者に周知であり、そして例えば、T.W.GreeneおよびG.M.Wuts,Protecting Groups in Organic Synthesis,第3版,Wiley,New York,1999ならびにそこに引用される参考文献に記載される。
【0046】
用語「アミノ保護基」とは、アミノ基における所望されない反応を防止するために適切な保護基をいう。代表的なアミノ保護基としては、tert−ブトキシカルボニル(BOC)、トリチル(Tr)、ベンジルオキシカルボニル(Cbz)、9−フルオレニルメトキシカルボニル(Fmoc)、ホルミル、トリメチルシリル(TMS)、tert−ブチルジメチルシリル(TBS)などが挙げられるが、これらに限定されない。
【0047】
用語「カルボキシ保護基」とは、カルボキシ基における所望されない反応を防止するために適切な保護基をいう。代表的なカルボキシ保護基としては、エステル(例えば、メチル、エチル、tert−ブチル、ベンジル(Bn)、p−メトキシベンジル(PMB)、9−フルオレニルメチル(Fm)、トリメチルシリル(TMS)、tert−ブチルジメチルシリル(TBS)、ジフェニルメチル(ベンズヒドリル、DPM)のエステル)などが挙げられるが、これらに限定されない。
【0048】
カルボン酸またはその保護された誘導体に関する「活性化誘導体」とは、上記カルボン酸または誘導体と、活性化(カップリング)剤(例えば、1−ヒドロキシベンゾトリアゾール(HOBT)、1−ヒドロキシ−7−アザベンゾトリアゾール(HOAT)または以下に記載されるか、もしくはそうでなければ当該分野で公知の他のもの)との反応によりもたらされる生成物(代表的には、反応性エステル)をいう。
【0049】
「天然に存在するアミノ酸の側鎖」とは、式HOOC−CHR−NH2における基であるRをいい、この式は、アラニン、アルギニン、アスパラギン、アスパラギン酸、システイン、グルタミン、グルタミン酸、グリシン、ヒスチジン、イソロイシン、ロイシン、リジン、メチオニン、フェニルアラニン、プロリン、セリン、スレオニン、トリプトファン、チロシンおよびバリンから選択されるアミノ酸、そして好ましくは、アラニン、アルギニン、アスパラギン、アスパラギン酸、システイン、グルタミン、グルタミン酸、グリシン、イソロイシン、ロイシン、リジン、メチオニン、セリン、スレオニンおよびバリンから選択されるアミノ酸を示す。
【0050】
(本発明の化合物の代表的な実施形態)
以下の置換基および値は、本発明の種々の局面の代表的な例および実施形態を提供することを意図する。これらの代表的な値は、このような局面および実施形態をさらに規定することを意図し、そして他の実施形態を除外することも本発明の範囲を限定することも意図しない。この点において、特定の値または置換基が好ましいという説明は、特に示されない限り、どのような方法においても、他の値または置換基を本発明から除外することを意図しない。
【0051】
式Iの化合物において、R”に存在するあらゆるヘテロアリーレン基またはヘテロサイクリック基は、好ましくは、全体で5個または6個、代表的には、6個の環原子を有し、各アリール基は、全体で6個の環原子を有する。上記R”基は、好ましくは、アルキレン鎖であり、そして好ましくは、直鎖状である。
【0052】
各R3基は、存在する場合、好ましくは、独立して、不飽和C1〜4アルキル、不飽和C1〜4アルコキシ、フルオロおよびクロロから選択される。1つの実施形態において、nは、1または2であり、各R3は、独立して、メチル、メトキシ、フルオロおよびクロロから選択される。別の実施形態において、nは、0であり、そのため、R3基は、存在しない。
【0053】
1つの好ましい実施形態において、R8は、水素である。別の好ましい実施形態において、R8は、式:
【0054】
【化12】
の基である。
【0055】
選択される実施形態において、R9は、水素またはC1〜4アルキルであり、例えば、水素またはメチルが挙げられる。1つの実施形態において、R9は、水素である。別の実施形態において、R9は、メチルである。
【0056】
1つの実施形態において、Wは、CClである。別の実施形態において、Wは、Nである。
【0057】
式Iの他の変更の選択される実施形態としては、互いに独立して、以下が挙げられる;X1およびX2については、クロロ;R2については、水素またはC1〜4アルキル;R4およびR5については、それぞれ、ヒドロキシおよび水素;R6およびR7については、それぞれ、水素およびメチル;そしてR8については、水素。
【0058】
他の選択された実施形態において、Raは、−Y−R”−であって、ここで、R”は、C1〜6アルキレン、C2〜6アルケニレンまはたC2〜6アルキニレンであり、そして、Yは、直接結合、NR’、O、S、C(O)、NR’(CO)および(CO)NR’から選択され、ここで、R’は、水素またはメチルである。一実施形態において、Yは、直接結合である。基Raのさらなる実施形態において、R”は、C1〜6アルキレン、またより好ましくは、C1〜4アルキレン(例えば、メチレン、エチレン、プロピレンまたはブチレン)である。
【0059】
好ましくは、基Raにおいて、Yは直接結合であり、そして、R”は、C1〜6アルキレンであるか、またはより好ましくは、C1〜4アルキレン(例えば、メチレン)である。
【0060】
式Iのピリジニウム環は、代表的には、メタ置換もしくはパラ置換されており、より一般的には、パラ置換されている。
【0061】
好ましくは、変数xは0または1である。xが0でない場合、基Rbは好ましくは、水素、C1〜4アルキルまたはC2〜4アルケニルからなる群より選択される。一実施形態において、Rbは、水素またはC1〜4アルキルである。
【0062】
xが0でない場合、基Rcは、好ましくは、−Y’−R”−Y’−であり、ここで、各Y’は、独立して、直接結合、OおよびNR’から選択され、ここで、R’は水素またはメチルであり、そして、R”は、C1〜12アルキレン、C2〜12アルケニレンおよびC2〜12アルキニレンから選択され、ここで、アルキレン基、アルケニレン基またはアルキニレン基は、必要に応じて、1〜2つの基で置換され、この基は、Zまたは天然に存在するアミノ酸の側鎖より選択される。好ましくは、基Rcにおいて、各Y’は直接結合であり、そしてR”はZおよび天然に存在するアミノ酸の側鎖より選択される1〜2つの基で置換され得るC1〜12アルキニレンまたはC2〜12アルケニレンである。基Rcの別の実施形態において、R”はC1〜12アルキレン(C1〜6アルキレンを含む)であり、この基は、非置換であるか、または−COOH基で置換されている。
【0063】
好ましくは、変数yは0または1である。yが0でない場合、基Rdは、好ましくは、水素およびC1〜4アルキルからなる群より選択され、選択された実施形態においては、水素およびメチルである。一実施形態において、RdはHである。yが0でない場合、基Reは、好ましくは、C1〜12アルキレン、C2〜12アルケニレンおよびC2〜12アルキニレンより選択される。一実施形態において、Reは、C1〜12アルキレンである。より好ましくは、Reは、C1〜6アルキレン、C2〜6アルケニレンおよびC2〜6アルキニレンから選択され、最も好ましくは、C2〜4アルケニレンおよびC1〜4アルキレンより選択される。
【0064】
選択された実施形態において、基R2は、水素またはC1〜4アルキルである。さらなる実施形態において、R2は、水素またはメチルであり;一実施形態において、R2は水素である。
【0065】
選択された実施形態において、xおよびyは独立して0および1より選択される。従って、好ましい実施形態としては、x+y=0の化合物、x+y=1の化合物およびx+y=2の化合物が挙げられる。あるいは、好ましい実施形態としては、図1において「L」で表される「リンカー」構造が、ピリジニウム環を除いてわずか約30個の炭素原子を含む化合物が挙げられる。
【0066】
x=y=1である本発明の化合物の例は、本明細書においてIaと命名される化合物である。x=1かつy=0である本発明の化合物の例としては、本明細書においてIb、IcおよびIeと命名される化合物が挙げられる。x=y=0である本発明の化合物の例としては、本明細書においてIdおよびIfとして命名される化合物が挙げられる(図1を参照のこと)。
【0067】
構造Iの化合物の一つの例示的な分類は、xが0または1であり;yが0または1であり;Raがメチレン;Rb(xが1の場合)が水素、メチルまたはエチルであり;Rc(xが1の場合)が−COOHで置換され得るC1〜12アルキレン、好ましくは、C1〜6アルキレン(例えば、エチレンまたはn−ペンチレン(−(CH2)5−))であり;Rd(yが1の場合)が水素であり;そしてRe(yが1の場合)がエチレン(−CH2CH2−)である化合物である。構造Iのこの分類の特定の実施形態としては、本明細書においてIa、Ib、Ic、Id、IeおよびIfと命名される化合物が挙げられる。この分類の一実施形態において、xは1であり;yは0であり;Raはメチレンであり;Rbは水素、メチレンまたはエチルであり;そしてRcはC2〜6アルキレン(例えば、エチレンまたはn−ペンチレン(−(CH2)5−))である。構造Iのこの分類の特定の実施形態としては、本明細書においてIbおよびIcと命名される化合物が挙げられる。
【0068】
式Iの本発明の化合物のサブセットはまた、式IIにより規定され得る。式IIの選択される実施形態において、WはCClである。他の選択される実施形態において、R9は、水素またはC1〜4アルキル(水素またはメチルを含む)である。
【0069】
R10は、水素またはC1〜6アルキルであり、好ましくは、水素またはC1〜4アルキル(水素、メチルまたはエチルを含む)である。別の実施形態において、R10はエチルである。
【0070】
さらなる選択される実施形態において、R11はC1〜10アルキレンであり、好ましくはR11はC2〜10アルキレンであり、より好ましくは、C2〜6アルキレン(例えば、−(CH2)2または−(CH2)5−)である。
【0071】
上記式Iと同様に、式IIのピリジニウム環は、代表的には、メタ置換もしくはパラ置換されており、より一般的には、パラ置換されている。
【0072】
式IIに従う本発明の化合物の例は、本明細書においてIbおよびIcと命名される化合物が挙げられる(図1を参照のこと)。
【0073】
理論に限定されることは意図しないが、式IおよびIIの化合物は、細菌の細胞壁生合成を阻害し、それにより、細菌の増殖を阻害するか、または細菌の溶解を引き起こすと考えられる。従って、これらは、抗生物質として有用である。
【0074】
他の特性の中でもとりわけ、本発明の化合物は、以下にさらに説明するように、グラム陽性細菌(例えば、メチシリン耐性Staphylococci aureus(MRSA)およびメチシリン耐性Staphylococci epidermitis(MRSE))に対して驚くべき予期せぬ効力を有することが見出された。
【0075】
(一般的な合成手順)
本発明の架橋グリコペプチド−セファロスポリン化合物は、容易に入手可能な出発物質から、好ましくは、本明細書中に記載される中間体化合物1〜5を介して、調製され得る。代表的または好ましいプロセス条件(すなわち、反応の温度、時間、反応物質のモル比、溶媒、圧力など)が与えられるが、特に言及されない限り、当業者に決定されるような他のプロセス条件もまた使用され得ることが理解される。最適な反応条件は、使用される特定の反応物質または溶媒によって変化し得るが、このような条件は、慣用的な最適化手順によって、当業者により容易に決定され得る。
【0076】
さらに、当業者に理解されるように、生じる所望されない反応から特定の官能基を保護するために、慣用的な保護基が必須であるか、または所望され得る。特定の官能基についての適切な保護基、ならびにこのような官能基の保護および脱保護に適切な条件は、当該分野で周知である。所望される場合、本明細書において記載される手順に例示されるもの以外の保護基が使用され得る。例えば、多数の保護基、ならびにその導入および除去の手段は、T.W.GreeneおよびG.M.Wuts,Protecting Groups in Organic Synthesis,第3版,Wiley,New York,1999ならびにこの文献で引用される参考文献に記載される。
【0077】
合成の好ましい方法において、yが0である式Iの化合物、または選択される実施形態においては式IIの化合物は、式1:
【0078】
【化13】
のグリコペプチド、またはその塩、もしくはC末端のカルボキシルが活性化された誘導体および/もしくはアミン保護された誘導体であって、R4〜R8、X1およびX2が本明細書において定義されるようなものである、化合物を、式3または4:
【0079】
【化14】
の化合物またはその塩もしくはカルボキシル基が保護された誘導体であって、W、R2、R3、R9、Ra〜e、n、xおよびyが、本明細書において定義されるようなものである、化合物と反応させることによって調製され;式IもしくはIIの化合物、またはその塩もしくは保護された誘導体を提供する。1、3および4の好ましい実施形態は、上に記載されるようなものである。
【0080】
式IIの化合物を調製する際に、構造1、3および/または4の変数は、以下のように定義される:nは0である;xは1である;yは0である;RaはCH2である;Rbは水素またはC1〜6アルキル(R10について上で定義された通り)である;RcはC1〜12アルキレン(R11について上で定義された通り)である;R2、R5およびR6は水素である;R7はCH3である;R9は本明細書において定義される通りである;R4はOHである;そして、X1およびX2はClである。
【0081】
代表的には、この反応は、以下にさらに議論するように、不活性な希釈剤(例えば、DMF)において、慣用的なカルボン酸−アミン(ペプチド)カップリング試薬を使用して、グリコペプチド1またはその塩を、約0.5当量〜約1.5当量、好ましくは、約0.9当量〜約1.1当量の式3または4の化合物とカップリングすることによって実施される。この反応において、グリコペプチド1またはその塩は、代表的には、まず、過剰(好ましくは、約1.8当量〜約2.2当量)のアミン(例えば、ジイソプロピルエチルアミン)の存在下、約−20℃〜約25℃の範囲の温度(好ましくは周囲温度)にて、約0.25時間〜約3時間、カップリング試薬に接触される。好ましくは、次いで、過剰のトリ
フルオロ酢酸(代表的には約2当量)が添加され、任意の過剰のジイソプロピルエチルアミンのTFA塩を形成する。次いで、この反応は、一般に、約−20℃〜約10℃の温度(好ましくは、約0℃)まで冷却され、そして、中間体3または4が添加され、その後、過剰の2,4,6−コリジンが添加される。この反応は、代表的には、約0℃において、約1時間〜約6時間、または反応が実質的に完了するまで維持される。
【0082】
あるいは、式I(yが0でない場合)の化合物の調製については、式2のグリコペプチド誘導体(R2、R4〜R8、Rd〜e、X1およびX2は本明細書において定義される通り)またはその塩もしくは保護された誘導体は、式5(W、R3、R9、Ra〜c、nおよびxは本明細書において定義される通り)の中間体あるいはその塩または活性化された誘導体もしくは保護された誘導体と反応させられ得る。2および5の好ましい実施形態は、上で記載された通りである。
【0083】
【化15】
代表的に、このようなカップリング反応は、コリジンのような塩基の存在下で、中間体2の塩(例えば、トリフルオロ酢酸塩)を、約0.5当量〜1.5当量の中間体5の活性化されたエステルと反応させることによって実施される。好ましくは、Rdは、この場合水素であり、その結果、反応は、C末端の第一級アミンにおいて優先的に生じる。例えば、図3に例示され、実施例4においてさらに説明されるIaの調製を参照のこと。
【0084】
式I(y>1の場合)の化合物の調製について、類似のストラテジーに従い得、このストラテジーにおいて、1つ以上のアミノ酸(例えば、構造NHRd−Re−COOH)の、中間体2のC末端−NHRd基への付加が、中間体5との反応を進める。
【0085】
これらの反応において使用するための、好ましいカップリング試薬または活性化試薬としては、ベンゾトリアゾール−1−イルオキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBOP)が挙げられ、これらは、好ましくは、約0.5当量〜1.5当量の量、好ましくは、約0.9当量〜1.1当量の量で、約0.5当量〜1.5当量、好ましくは約0.9当量〜1.1当量の1−ヒドロキシベンゾトリアゾール(HOBT)または1−ヒドロキシ−7−アザベンゾトリアゾール(HOAT)と組み合せて使用される。他の適切なカップリング試薬としては、O−(7−アザベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HATU);ビス(2−オキソ−3−オキサゾリジニル)ホスフィン酸クロリド(BOP−Cl);ジフェニルホスホリルアジド(DPPA);ジフェニルホスフィン酸クロリド;ジフェニルクロロホスフェート(DPCP)およびHOAT;1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(EDAC);ペンタフルオロフェニルジフェニルホスフィネートなどが挙げられる。
【0086】
カップリング反応が完了した後、次いで、生成物中に存在する任意の保護基が、従来の手順および試薬を使用して除去される。例えば、N−トリチル、N−BOC(N−tert−ブトキシカルボニル)および/またはCOO−PMB(パラ−メトキシベンジルエステル)の脱保護は、周囲温度において、不活性な希釈剤(例えば、ジクロロメタンまたはヘプタン)中で、過剰のトリフルオロ酢酸および過剰のアニソールまたはトリエチルシランを用いて、約1〜約12時間、または反応が完了するまで処理するより達成され得る。脱保護された生成物は、カラムクロマトグラフィー、HPLC、再結晶化などのような慣用的な手順を使用して精製され得る。
【0087】
上記の手順における使用に適切な式Iのグリコペプチドは、市販されているか、または、適切なグリコペプチド産生生物の発酵、その後に、当該分野で認識される手順および機器を使用して、生じた発酵ブロスからグリコペプチドを単離することによって調製され得る。誘導体2は、例えば、実施例3に記載されるように、1のジアミンとの反応により容易に調製される。
【0088】
上記の手順において使用されるセファロスポリン中間体3および4は、慣用的な手順を使用して、市販の出発物質および試薬から容易に調製される。一例として、式4の中間体は、図2に示され、実施例1および2に記載されるように調製され得る。簡単には、2−アミノ−5−クロロ−α−メトキシイミノ−4−チアゾール酢酸(6)を、EDACで触媒して、アミノセファロスポリン酸エステル7と反応させて、アミド結合を形成した。この生成物(8)を、次いで、TFAおよびアニソールで処理して、PMBエステルを解離させ、その後、第一級塩素をピリジン誘導体で置き換えた。このピリジン誘導体は、上で構造4に示されるように、置換基−Ra−NHR2、そして必要に応じて、置換基R3を含む。図2に示される調製において、化合物4−(N−t−BOC−N−エチル)アミノメチルピリジン(9)が使用され、その結果、Raはメチレンであり、R2はエチルである。この反応は、保護された形態の中間体(10)を生じ;TFAを用いた脱保護により、中間体4a(WがCClであり、R9がMeであり、nが0であり、RaがCH2であり、そしてR2がEtである、中間体4)を生じる。
【0089】
代替的なプロセスにおいて、生成物8を、アセトン中でヨウ化ナトリウムと反応させ、その後、9と反応させた。次いで、TFA/アニソールを用いる反応を使用して、Boc保護基およびPMB保護基の両方を除去した。
【0090】
上記の反応において使用するための種々の置換ピリジンは、市販されているか、または、慣用的な手順を使用して、市販の出発物質および試薬から調製され得る。例えば、種々のアミノアルキル置換ピリジン(例えば、Raがメチレンであるアミノメチルピリジン、およびRaがエチレンであるアミノエチルピリジン)は、市販されているか、または、標準的な有機合成手順を使用して調製され得る。この反応において使用するための代表的な置換ピリジン誘導体としては、R3が、メチル、メトキシ、チオメトキシ、カルボキシチオメトキシ、フルオロ、クロロ、フェニル、シクロプロピル、カルボン酸、カルボキサミドおよびその組み合わせから選択されるものが挙げられる。R”をピリジニウム環に結合するYが、NR’、O(エーテル)、S(スルフィド)、C(O)(カルボニル)、NR’(CO)、および(CO)NR’より選択される化合物の調製について、出発ピリジン化合物は、市販されているか、または、周知の手順により調製され得る。例えば、3−ヒドロキシピリジン、4−ヒドロキシピリジン、3−アミノピリジン、4−アミノピリジン、4−メルカプトピリジン、ニコチン酸およびイソニコチン酸は、Aldrich Chemical Co,Milwaukee,WIから市販されている。
【0091】
中間体4は、さらに、式3の中間体を形成するように置換され得る。例えば、図4は、脱保護後の、式3の中間体(xが1の場合)を形成するための、式4の中間体のN−(t−BOC)−β−アラニン(実施例5、Ibの調製を参照のこと)との反応、アスパラギン酸(実施例8、Ieの調製を参照のこと)との反応を示す。β−アラニンまたは類似の化合物(例えば、他のアミノ酸)のさらなる付加を採用すると、x>1の場合の式3の中間体を形成し得る。
【0092】
中間体4と、二酸(例えば、HOOC−Rc−COOHの構造のもの)との反応を採用すると、式5の中間体(xが1の場合)を形成し得る(図3;実施例4;Iaの調製を参照のこと)。式5の中間体(x>1の場合)を形成するために、中間体4は、まず、1モル以上のアミノ酸(例えば、構造HOOC−Rc−NHRbのもの)と反応させる。
【0093】
基Rcにおいて、Y’がO(エーテル)およびNR’(直接結合以外)から選択される化合物の調製において、Rcを含む結合部分は、アミド結合ではなく、1つ以上のカルバメート結合または尿素結合を含む。このような結合は、従来の方法により形成され得る。例えば、アミン(例えば、中間体3または4における−NHR2)は、イソシアネートまたはクロロホルメートと反応して、それぞれ、尿素結合またはカルバメート結合を形成し得る。
【0094】
本発明の代表的な化合物またはそれに対する中間体を調製するための特定の反応条件および手順に関するさらなる詳細は、以下に示す実施例に記載されている。
【0095】
(薬学的組成物)
本発明の架橋グリコペプチド−セファロスポリン化合物は、代表的に、薬学的組成物の形態で患者に投与される。従って、その組成物局面の1つでは、本発明は、薬学的に受容可能なキャリアまたは賦形剤と、式Iもしくは式IIの化合物またはそれらの薬学的に受容可能な塩の治療有効量とを含有する薬学的組成物に関する。
【0096】
本発明の薬学的組成物では、任意の通常のキャリアまたは賦形剤が使用され得る。特定のキャリアまたは賦形剤、またはキャリアまたは賦形剤の組合せの選択は、特定の患者または病気または疾患状態を治療するのに使用される投与様式に依存している。このことに関して、特定の投与様式(例えば、経口投与、局所投与、吸入による投与、または非経口投与)に適当な薬学的組成物の調製は、十分に、医薬分野の当業者の範囲内である。さらに、このような組成物の成分は、例えば、Sigma,P.O.Box 14508,St.Louis,MO 63178から市販されている。さらなる例として、通常の処方技術は、RemingtonのPharmaceutical Sciences,Mace Publishing Co.,Philadelphia,PA 第17版(1985)および「Modern Pharmaceutics」、Marcel Dekker,Inc.第3版(G.S.Banker & C.T.Rhodes編)で記述されている。
【0097】
本発明の薬学的組成物は、代表的には、式IまたはIIの化合物またはそれらの薬学的に受容可能な塩の治療有効量を含有する。代表的には、このような薬学的組成物は、約0.1〜約90重量%の活性因子、より一般的には、約10〜約30%の活性因子を含有する。
【0098】
本発明の好ましい薬学的組成物は、非経口投与、特に静脈内投与に適切な組成物である。このような薬学的組成物は、代表的には、式Iもしくは式IIの化合物またはそれらの薬学的に受容可能な塩の治療有効量を含有する生理学的に受容可能な滅菌水溶液を含む。
【0099】
活性成分の静脈内投与に適切な生理学的に受容可能なキャリア水溶液は、当該分野で周知である。このような水溶液としては、例として、5%デキストロース、Ringer溶液(乳酸化Ringer注入物、乳酸化Ringer5%デキストロース添加注入物、アクリル化Ringer注入物)、ノルモソール−M(Normosol−M)、イソライトE(Isolyte E)などが挙げられる。
【0100】
必要に応じて、このような水溶液は、共溶媒(例えば、ポリエチレングリコール;キレート試薬、例えばエチレンジアミン4酢酸;可溶化剤、例えば、シクロデキストリン;抗酸化剤、例えば、メタ重亜硫酸ナトリウム)など)を含有し得る。
【0101】
所望の場合、本発明の水性薬学的組成物は、投与前に適切なキャリアにより、脂質化され、その後再形成され得る。好ましい実施形態において、薬学的組成物は、薬学的に受容可能なキャリアおよび式Iまたは式IIの化合物あるいはそれらの薬学的に受容可能な塩の治療有効量を含有する脂質化組成物である。好ましくは、この組成物中のキャリアは、スクロース、マンニトール、デキストロース、デキストラン、ラクトースまたはこれらの組み合わせを含有する。より好ましくは、このキャリアは、スクロース、マンニトール、またはこれらの組み合せを含有する。
【0102】
1つの実施形態において、本発明の薬学的組成物は、シクロデキストリンを含有する。本発明の薬学的組成物中で使用される場合、このシクロデキストリンは、好ましくは、ヒドロキシプロピル−β−シクロデキストリンまたはスルホブチルエーテルβ−シクロデキストリンである。このような処方物中で、シクロデキストリンは、処方物の約1〜25重量%;好ましくは、約2〜10重量%を含む。さらに、活性成分に対するシクロデキストリンの重量比は、代表的には、約1:1〜約10:1の範囲である。
【0103】
本発明の薬学的組成物は、好ましくは、単位投薬形態中にパッケージングされる。用語「ユニット投薬形態」は、患者に投薬するのに適切な物理的に分離したユニット(すなわち、単独でか、または一つ以上のさらなるユニットと組み合わせるかのどちらかで所望の治療効果を生じるように計算された所定量の活性成分を含む各ユニット)を指す。例えば、このようなユニット投薬形態は、滅菌した、気密されたアンプルなどにパッケージングされ得る。
【0104】
以下の処方物は、本発明の代表的な薬学的組成物を示す。
【0105】
(処方実施例A)
注射可能溶液の調製に適した凍結溶液を以下のとおりに調製する。
【0106】
成分 量
活性化合物 10〜1000mg
賦形剤(例えば、デキストロース) 0〜50g
注射用水 10〜1000mL
(代表的手順)
賦形剤(もしあるならば)を、約80%の注射用水に溶解し、活性化合物を添加し、溶解する。pHを1M水酸化ナトリウムで3〜4.5に調整し、次いで容量を、注射用水を用いて最終容量の95%に調節する。pHを確認し、必要な場合調節し、そして容量を、注射用水を用いて最終容量に調節する。次いで、処方物を0.22ミクロンフィルタを通して滅菌濾過し、無菌条件下で、滅菌バイアル中に入れる。このバイアルをキャップし、ラベルし、そして凍結貯蔵する。
【0107】
(処方実施例B)
注射可能溶液の調製に適切な脂質化粉末を、以下のとおりに調製する。
【0108】
成分 量
活性化合物 10〜1000mg
賦形剤(例えば、マンニトールおよび/またはスクロース) 0〜50g
緩衝材(例えば、クエン酸塩) 0〜500mg
注射用水 10〜100mL
(代表的な手順)
賦形剤および/または緩衝剤(もし、あるならば)を、約60%の注射用水に溶解する。活性化合物を添加して溶解し、そのpHを、1M水酸化ナトリウムを用いて3〜4.5に調節して、その容量を注射用水を用いて最終容量の95%に調節する。pHを確認し、必要な場合調節し、そして容量を注射用水を用いて最終容量に調節する。次いで、この処方物を0.22ミクロンのフィルタを通して滅菌濾過し、無菌条件下にて滅菌バイアルに入れる。次いで、この処方物を適切な凍結乾燥サイクルを使用して凍結乾燥する。このバイアルをキャップし(必要に応じて、部分的減圧下、または乾燥窒素下)、ラベルし、そして凍結下で貯蔵する。
【0109】
(処方実施例C)
患者への静脈内投与のための注射可能溶液を、上記処方実施例Bより、以下のとおりに調製する。
【0110】
(代表的な手順)
処方実施例Bの凍結乾燥粉末(例えば、10〜1000mgの活性化合物を含有する)を、20mLの滅菌水で再形成し、得られた溶液を、100mL中の注入バッグ内で80mLの滅菌生理食塩水でさらに希釈する。次いで、この希釈した溶液を、30〜120分間に渡って患者に静脈内投与する。
【0111】
(利用)
本発明の架橋グリコペプチド−セファロスポリン化合物は、抗生物質として有用である。例えば、本発明の化合物は、細菌感染、および哺乳動物(ヒトおよびペット(すなわち、イヌ、ネコなど)を含む)において本発明の化合物に感受性の微生物により引き起こされる他の細菌関連の医学的状態の処置または予防に有用である。
【0112】
従って、その方法の1つの局面において、本発明は、哺乳動物における細菌感染症の処置方法、薬学的に受容可能なキャリアおよび式Iまたは式IIの化合物もしくはこれらの薬学的に受容可能な塩の治療有効量を含む薬学的組成物を、このような処置を必要とする哺乳動物に投与する工程を包含する方法を提供する。
【0113】
例示目的で、本発明の化合物は、グラム陽性菌および関連する微生物により引き起こされる感染症の処置または予防に特に有用である。例えば、本発明の化合物は、特定のEnterococcus spp.;Staphylococcus spp.(コアグラーゼ陰性staphylococci(CNS)を含む);Streptococcus
spp.;Listeria spp.;Clostridium spp.;Bacillus spp.などにより引き起こされる感染症を処置または予防するのに有効である。本発明の化合物を使用して有効に処置される細菌種の例としては、メチシリン抵抗性Staphylococcus aureus(MRSA);メチシリン感受性Staphylococcus aureus(MSSA);グリコペプチド中間体感受性Staphylococcus aureus(GISA);メチシリン抵抗性Staphylococcus epidermitis(MRSE);メチシリン感受性Staph
ylococcus epidermitis(MSSE);バンコマイシン感受性Enterococcus faecalis(EFSVS);バンコマイシン感受性Enterococcus faecium(EFMVS);ペニシリン抵抗性Streptococcus pneumoniae(PRSP);Streprococcus pyogenesなどが挙げられるが、これらに限定されない。
【0114】
以下の実施例16の表2に示すように、本発明のいくつかの化合物Ia〜f、Im、およびInは、メチシリン感受性Staphylococcus aureusおよびメチシリン抵抗性Staphylococcus aureusに対して、10倍以上のバンコマイシンよりも有効であった。これらの化合物はまた、これらのデス−クロロアナログIg、IjおよびIkよりも有意に活性であるが、これらの化合物もまたバンコマイシンよりもより活性であった。「時間殺傷(time−kill)」アッセイにおいて、実施例17に記載されるように、式Iの化合物(すなわち化合物Ib)は、4時間で、1.0μg/mL以下の濃度にて、MRSAに対し殺菌性であった。比較すると、バンコマイシンは、24時間で、4μg/mLの濃度にて、MRSAに対し殺菌性であった。神経栄養性(neutropenic)マウスにおけるインビボアッセイにおいて、実施例18に記載されるように、式Iの化合物(すなわち、化合物Ib)は、バンコマイシンについてのivの9mg/kgのED50と比較して、ivの0.1mg/kg以下のED50を有した。
【0115】
一般に、本発明の化合物は、グリコペプチドまたはセファロスポリンのどちらかに感受性である細菌株により引き起こされる感染症の処置または予防に好ましい。
【0116】
本発明の化合物を使用して処置または予防され得る感染症または細菌関連の医学状態の代表的な型としては、皮膚感染症および皮膚構造感染症、尿路感染症、肺炎、心内膜炎、カテーテル関連血流感染症、骨髄炎などが挙げられるが、これらに限定されない。このような状態の処置において、患者は、処置されるべき微生物にすでに感染しているか、または感染症に感受性であると疑わしいだけかもしれず、このような場合において、活性成分が予防的に投与される。
【0117】
本発明の化合物は、代表的に、任意の受容可能な投与経路により治療有効量を投与される。好ましくは、この化合物は、非経口投与される。この化合物は、一日一回の投薬、または一日あたり複数回の投薬において、投与され得る。この処置レジメンは、延長された期間(例えば、数日間または1〜6週間以上にわたって)にわたる投与を必要とし得る。投薬あたりに投与される活性剤の量または投与される総量は、代表的には、患者の担当医により決定され、感染症の性質および重篤度、患者の年齢および健康状態、患者の活性成分に対する耐性、感染症を起こす細菌、投与経路などのような因子に依存する。
【0118】
一般に、適切な投薬は、活性成分の約0.25〜約10.0mg/kg/日、好ましくは、約0.5〜約2mg/kg/日の範囲である。平均的な70kgのヒトについて、これは、一日あたり約15〜約700mg、または好ましくは、一日あたり約35〜約150mgの量の活性成分である。
【0119】
さらに、本発明の化合物は、細菌の増殖を阻害するのに有効である。この実施形態において、細菌は、インビトロまたはインビボのどちらかで、式Iまたは式IIの化合物あるいはこれらの薬学的に受容可能な塩の増殖阻害量に接触される。代表的には、増殖阻害量は、約0.008μg/mL〜約50μg/mL;好ましくは、約0.008μg/mL〜約25μg/mL;そしてより好ましくは、約0.008μg/mL〜約10μg/mLの範囲である。細菌増殖の阻害は、代表的には、細菌繁殖の減少もしくは欠損、および/または細菌崩壊の溶菌(すなわち、未処置の細菌と比較した所与の処置期間(すなわち、1時間あたり)にわたる所与の容量(すなわち、mLあたり)中のコロニー形成単位の減少)により、証明される。
【0120】
本発明の化合物はまた、細菌の細胞壁生合成を阻害するのに有効である。この実施形態において、細菌は、インビトロまたはインビボのどちらかで、式Iまたは式IIの化合物あるいはこれらの薬学的に受容可能な塩の細胞壁生合成の阻害量に接触される。代表的には、細胞壁生合成阻害量は、約0.04μg/mL〜約50μg/mL;好ましくは約0.04μg/mL〜約25μg/mL;そして、より好ましくは、約0.04μg/mL〜約10μg/mLの範囲である。細菌における細胞壁生合成の阻害は、代表的には、細菌の溶菌を含む、細菌の増殖の阻害または欠損により証明される。
【0121】
さらに、本発明の化合物は、特定の細菌(メチシリン抵抗性Staphylococci aureus(MRSA)およびメチシリン抵抗性Staphylococci epidermitis(MRSE)を含む)に対して、驚きべき、かつ予想外の迅速な死滅率(cidelite)を有することが見出されている。本発明の化合物のこれらの特性、および抗生物質的な利用性は、当業者に周知の種々のインビトロアッセイおよびインビボアッセイを使用して実施され得る。例えば、代表的なアッセイは、以下の実施例においてさらに詳述される。
【実施例】
【0122】
(実施例)
以下の合成実施例および生物学的実施例は、本発明を例示するため提供されるが、いかなる場合においても本発明の範囲を制限するようには解釈されるべきではない。
【0123】
以下の実施例において、他に指定されない限り、以下の略語は、下の意味を持つ。以下に規定されない略語は、その一般に認められた意味を有する。
【0124】
BOC = tert−ブトキシカルボニル
CFU = コロニー形成ユニット
DCM = ジクロロメタン
DIPEA = ジイソプロピルエチルアミン
DMF = N,N−ジメチルホルムアミド
DMSO = ジメチルスルホキシド
EtOAc = 酢酸エチル
HOBT = 1−ヒドロキシベンゾトリアゾール
HOAT = 1−ヒドロキシ−7アザベンゾトリアゾール
HPLC = 高速液体クロマトグラフィー
MIC = 最少阻害濃度
MS = 質量分析
PMB = p−メトキシベンジル
PyBOP = ベンゾトリアゾール−1−イルオキシトリピロリジノホスホニウムヘキサフルオロリン酸
THF = テトラヒドロフラン
TLC = 薄層クロマトグラフィー
TFA = トリフルオロ酢酸
以下の実施例において、記録された全ての温度は、他に指示されない限り、セ氏温度(℃)である。また、他に注記されない限り、試薬、出発物質および溶媒を、市販業者(例えば、Aldrich,Fluka,Sigmaなど)から購入し、さらに精製することなく使用した。バンコマイシン塩化物の半水和物をAlpharma,Inc.,Fort Lee,NJ07024(Alpharma AS、Oslo、Norway)から購入した。
【0125】
逆相HPLCを、他に規定されない限り、代表的にC18カラムおよび(A)(98%水、2%アセトニトリル、0.1%TFA)と、(B)(10%水、90%アセトニトリル、0.1%TFA増加勾配)の増加勾配(例えば、0〜約70%)を使用して行った。
【0126】
以下に記載される合成において、中間体4a〜4gを、以下のとおりに規定する。これらの化合物の各々において、nは0であり、Raは、−CH2−である。
【0127】
【化16】
(実施例1)
(4a:(7R)−7−[(Z)−2−(2−アミノ−5−クロロチアゾール−4−イル)−2−(メトキシイミノ)アセトアミド]−3−[((4−エチルアミノ)メチル)−1−ピリジノ)メチル]−3−セフェン−4−カルボキシレート ビス−トリフルオロ酢酸塩の調製)(図2および図3を参照のこと)
(A.2−アミノ−5−クロロ−α−(メトキシイミノ)−4−チアゾール酢酸(6)の調製)
500mLの酢酸に、40.0g(0.20mol)の2−アミノ−α−(メトキシイミノ)−4−チアゾール酢酸(Ardrich Chemical Company,Milwaukee,WI)および27.9g(0.21mol)のN−クロロスクシンイミドを添加した。この混合物を、70℃の浴中で加熱した。その固形物を30分以内に溶解し、そして45分後にこの反応をMSにより完了した。この黒色溶液を室温まで冷却し、減圧下で濃縮し、黒色の固形物を得て、この固形物を精製することなく使用した。
【0128】
(B.セファロスポリン誘導体(8)の調製)
700mLのDMF中の47.5g(0.20mol)の(6)に、81.1g(0.20mol)のアミノセファロスポリンエステル(7)(Otsuka,Osaka,JP)および27.0g(0.20mol)のHOATを添加した。この混合物を0℃まで冷却し、そして26.7g(0.22mol)の2,4,6−コリジンを添加した。この溶液に、42.2g(0.22mol)の1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミドヒドロクロリド(EDAC)を添加した。5時間後、この溶液を、1:1のEtOAc/エーテルと水とに分配し、この相を分離した。この水溶液をEtOAcで2回逆抽出した。合わせた有機相を0.5Mのクエン酸、水、50%飽和NaHCO3、水、およびブラインで各々1回洗浄した。この有機相をMgSO4で乾燥し、濾過し、減圧下で濃縮した。この残渣を最少容量の塩化メチレンに溶解し、この生成物を、その溶液をエーテルに注ぐことにより沈澱させた。この固形物を濾過により単離して、48gの化合物(8)を得た。
【0129】
分析データ:MS m/z 計算値 586.04、実測値 586.2(M+1);1H NMR(DMSO−d6):d9:60(d,1H)、7.35(m,3H)、6.91(d,2H)、5.82(m,1H)、5.17(m,3H)、4.56(m,2H),3.84(s,3H)、3.76(s,3H)、3.62(m,2H)。
【0130】
(C.ピリジニウム部分への結合)
400mLの1,2−ジクロロエタン中の48.5g(82.7mol)のクロロメチルセファロスポリンエステル8および40mLのアニソールに、300mLのTFAを添加した。40分後、この溶液を減圧下で1/2の容量に濃縮し、生成物を、この濃縮物を1Lのエーテルに注ぐことにより沈澱させた。この固形物を、濾過により単離し、エーテルでリンスし、乾燥して42.5gの粗製遊離酸を得た。この物質(25.0g、53.6mol)を200mLの1:1のアセトニトリルDMFに溶解した。この溶液に、10.1g(83.3mmol)の2,4,6−コリジン、および15.2g(64.5mmol)の4−(N−t−ブトキシカルボニル)−エチルアミノメチルピリジン(9)(従来の手順により、4−エチルアミノメチルピリジンより調製、これは、Aldrich Chemical Company,Milwaukee,WIより市販されている)を添加した。3.5時間後、生成物を、この溶液を1Lのエーテルに注ぐことにより沈澱させた。この固形物を、濾過により単離し、エーテルで洗浄し、減圧下で乾燥して26gの粗製物質を得た。この生成物を、分取逆相HPLCにより精製し、化合物10をオフホワイトの粉末として得た。
【0131】
分析データ:MS m/z 計算値 666.12、実測値 666.2(M+1);1H NMR(DMSO−d6):d 9.36(d,1H)、8.91(d,2H)、7.95(d,2H)、7.24(b,1H)、5.83(m,1H)、5.50(m、2H)、5.17(d,1H)、4.66(s,2H)、3.83(s,3H)、3.52(d,1H)、3.30(m,3H)、1.37(s,9H)、1.08(t,3H)。
【0132】
(D.10の脱保護)
610mg(0.78mmol)の化合物10および1mLのアニソールに、8mLのトリフルオロ酢酸を添加した。20分後、この反応物を100mLのエーテルに注ぐことにより、その生成物を沈殿させた。この固体を濾過により単離し、エーテルで洗浄し、そして真空下で乾燥し、淡黄色固体として4aを得た。
【0133】
分析データ:1H NMR(DMSO−d6)d 9.56(d,1H),9.44(br,1H),9.10(d,2H),8.21(d,2H),7.38(b,1H),5.86(m,1H),5.54(m,2H),5.15(d,1H),4.51(s,2H),3.80(s,3H),3.40(m,2H),3.30(m,2H),1.22(t,3H)。
【0134】
(実施例2)
(4bの調製:(7R)−7−[(Z)−2−(2−アミノ−5−クロロチアゾール−4−イル)−2−(メトキシイミノ)アセトアミド]−3−[(4−(アミノエチル)−1−ピリジニオ)メチル]−3−セフェム−4−カルボキシレート ビス−トリフルオロ酢酸塩)
(A.2−アミノ−5−クロロ−α−(メトキシイミノ)−4−チアゾール酢酸(6)の調製)
500mLのDMFに、50.0g(250mmol)の2−アミノ−α−(メトキシイミノ)−4−チアゾール酢酸および35g(260mmol)のN−クロロスクシンイミドを添加した。この混合物を室温で一晩攪拌し、その後質量スペクトル分析がもはや出発物質が存在しないことを示した。この淡褐色の溶液を、さらなる操作なしに使用した。
【0135】
(B.セファロスポリン誘導体(8)の調製)
工程(a)からのDMF(101.5g、250mmol)中の酸6の溶液に、アミノセファロスポロン酸エステル7(34g、250mmol)を添加した。この混合物を0℃に冷却し、そして33.5mL(250mmol)の2,4,6−コリジンを添加した。この溶液に、53g(275mmol)の1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩を添加した。2時間後、その溶液を3Lの水に沈殿させ、濾過した。この固体を水(2×1L)、飽和炭酸水素ナトリウム(500mL)および水(4×500mL)で洗浄し、真空下で乾燥した。乾燥した固体を、室温で500mLの塩化メチレンに溶解させ、その溶液をゆっくりと攪拌し、沈殿を形成させた。結晶を濾過により回収し、洗浄液がもはや褐色でなくなるまで塩化メチレンで洗浄し、そして真空下で乾燥し、アミド8(74g)を得た。
【0136】
(C.ピリジニウム部分へのカップリング)
アセトン(250mL)を、窒素下、暗所で50g(85mmol)のクロロメチルセファロスポリンエステル8および13g(85mmol)のヨウ化ナトリウムの混合物に添加した。30分間の攪拌後、27g(130mmol)の4−(N−tert−ブトキシカルボニル)アミノメチルピリジンおよび30mLのアセトンを添加した。さらなる2時間の攪拌後、1.4Lの0.1N HClを添加し、粘着性の沈殿を生成した。溶媒部分を傾斜し、そして粘着性の残渣を800mLの水で処理し、固体を得た。その水を傾斜し、そしてその固体を1Lの4:1 酢酸エチル/エタノールに溶解した。その溶液を500mLの飽和ブラインで洗浄し、硫酸マグネシウムで乾燥し、そして乾燥状態までエバポレートし、70g(79mmol、93%)の生成物(10に類似、NEtBOCの代わりに−NHBOCを有する)を得、その生成物は、HPLC(254nm)で決定した場合、78%の純度を有した。
【0137】
この物質を、さらなる精製なしに、以下のようにして脱保護化した。粗生成物(70g、79mmol)を、窒素下で、550mLの塩化メチレンに溶解させ、そして35mL(320mmol)のアニソールを添加し、続いて150mLのトリフルオロ酢酸を添加した。2時間後、この混合物を真空下で濃縮した。1Lのジエチルエーテルの添加の際に、生成物は沈殿した。この固体を濾過により単離し、エーテルで洗浄し、200mLの水中で攪拌し、そして濾過した。この濾液を乾燥状態まで凍結乾燥し、逆相HPLCにより精製し、30g(約50%)の4bを得た。
【0138】
(実施例3)
(2aの調製:C−アミノエチルアミドバンコマイシン(R2、R5、R6、R8がHであり;R4がOHであり;R7がMeであり;X1、X2がClであり;RdがHであり;Reが−CH2CH2−である構造2))
75mLのDMSO中の塩酸バンコマイシン一水和物(7.3g、4.7mmol)の溶液に、室温で、4.1mL(23.5mmol)のDIPEAを添加し、続いて1.8g(5.6mmol)の9−フルオレニルメチル N−(2−アミノエチル)カルバメート塩酸塩(Fmoc−NH−CH2−CH2−NH3Clを添加した(図3を参照のこと)。75mLのN,N’−ジメチルプロピレン尿素(DMPU)中のPyBOP(2.7g、5.2mmol)およびHOBT(0.63g、4.7mmol)の溶液を、次いで手早く滴下した。この生成した溶液を、室温で2時間攪拌し、次いで800mLのジエチルエーテルに注ぎ、ゴム状物を得た。このエーテルを傾斜し、このゴム状物をさらなるエーテルで洗浄し、粗[C]−(2−Fmoc−アミノエチル)バンコマイシンを得た。
【0139】
この生成物(Fmoc−2a)を40mLのDMF中に溶解させ、10mLのピペリジンを添加し、そしてこの溶液を室温で20分間静置し、次いで450mLのアセトニトリルに滴下し、沈殿を形成させた。この混合物を遠心分離し、そのアセトニトリルを傾斜し、そしてこの残渣を450mLのアセトニトリルで2回、450mLのジエチルエーテルで1回洗浄し、次いで風乾した。この残渣を、次いで水に溶解させ、3N HClでpH<5まで酸性化し、そして0.1%のトリフルオロ酢酸を含む水中の2〜30%アセトニトリルの勾配を用いて逆相HPLCで精製した。このことにより、生成物2aをトリ(TFA)塩として得た。
【0140】
(実施例4)
(化合物Iaの調製)
上の実施例1に記載したように調製したピリジニウムラクタムビストリフルオロアセテート4a(2.4g)を、窒素下でN,N−ジメチルホルムアミド(DMF、40mL)に溶解し、0℃まで冷却した。ドデカン二酸ビス−1−ヒドロキシ−7−アザトリアゾールエステル(7.0g)を添加し(11)、続いて2,4,6−コリジン(1.2mL)を添加し、そしてこの混合物を65分間攪拌し、次いで酢酸エチル(50mL)に添加し、そしてこの混合物をジエチルエーテル(400mL)中に沈殿させた。この活性化エステルラクタム5aを濾過により回収し、そして真空下で乾燥し、そして一部(14mg)をDMF(122μL)中に溶解し、0℃で、DMF(500μL)中の、実施例3に記載するように調製したC−アミノエチルアミドバンコマイシントリフルオロアセテート2a(50mg)および2,4,6−コリジン(5.4μL)の混合物に添加した。この混合物を20分間0℃に保ち、次いでトリフルオロ酢酸(7.3μL)を添加した。この混合物を−20℃に一晩保ち、次いで逆相HPLCにより精製し、生成物Iaを得た。
【0141】
分析データ:MS m/z 実測値2252.8、計算値2252.7。
【0142】
(実施例5)
(化合物Ibの調製)
窒素下、室温で1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(740mg)、1−ヒドロキシ−7−アザトリアゾール(520mg)およびN−(tert−ブトキシカルボニル)−β−アラニン(660mg)をDMF(10mL)中で7時間攪拌し、次いで0℃まで冷却した。ピリジニウムラクタムビストリフルオロアセテート4a(2g)(実施例1を参照のこと)を添加し、それが完全に溶解するまで攪拌し、次いで2,4,6−コリジン(700μL)を添加し、そしてこの混合物を0℃で100分間攪拌した。トリフルオロ酢酸(700μL)および水(30mL)を添加し、この生成物を逆相HPLCで精製した。凍結乾燥後、この生成物を20mLの50%トリフルオロ酢酸/ジクロロメタンで40分間処理した。このラクタムアミン3a(図4を参照のこと)を、ジエチルエーテル中への沈殿によって、そのビストリフルオロアセテート塩として回収し、真空下で乾燥した。
【0143】
窒素下、室温で塩酸バンコマイシン一水和物1a(900mg)(R5、R6、R8がHであり;R4がOHであり;R7がMeであり;X1、X2がClである構造1;図において「バンコマイシン」とも呼ばれる)を、ジメチルスルホキシド(5mL)に溶解し、1−ヒドロキシ−7−アザトリアゾールを添加し(80mg)、続いてDMF(5mL)中のPyBOP(300mg)およびN,N−ジイソプロピルエチルアミン(100μL)を添加した。この混合物を20分間攪拌し、トリフルオロ酢酸(45μL)を添加し、そしてこの混合物を0℃に冷却した。ピリジニウムラクタムアミン3a(250mg)を添加し、続いて2,4,6−コリジン(270μL)を添加し、そしてこの混合物を0℃で30分間攪拌し、次いでアセトニトリル中に沈殿させた。粗生成物を遠心分離により回収し、逆相HPLCにより精製し、Ibを得た。
【0144】
分析データ:MS m/z 実測値2069.7、計算値2069.4。
【0145】
(実施例6)
(化合物Icの調製)
この化合物を、N−BOC−β−アラニンの代わりに6−(N−BOC)アミノヘキサン酸に変えて、実施例5に記載した手順に従って調製した。
【0146】
分析データ:MS m/z 実測値2110.5、計算値2111.5。
【0147】
(実施例7)
(化合物Idの調製)
窒素下、室温で塩酸バンコマイシン一水和物1a(900mg)を、ジメチルスルホキシド(5mL)に溶解し、1−ヒドロキシ−7−アザトリアゾール(80mg)を添加し、続いてDMF(5mL)中のPyBOP(300mg)およびN,N−ジイソプロピルエチルアミン(100μL)を添加した。この混合物を20分間攪拌し、トリフルオロ酢酸(45μL)を添加し、そしてこの混合物を0℃に冷却した。ピリジニウムラクタムビストリフルオロアセテート4c(250mg)(ピリジン上でのメタ置換を除いて4bと同一;図5を参照のこと)を添加し、続いて2,4,6−コリジン(270μL)を添加し、そしてこの混合物を0℃で30分間攪拌し、次いでアセトニトリル中に沈殿させた。粗生成物を遠心分離により回収し、逆相HPLCにより精製し、生成物Idを得た。
【0148】
分析データ:MS m/z 実測値1970.9、計算値1970.3。
【0149】
(実施例8)
(化合物Ieの調製)
(A.3bの調製(WがClであり、R9がMeであり、nが0であり、RaがCH2であり、Rbが水素であり、Rcが−CH2CH(COOH)−であり、R2が水素であり、そしてxが1である構造3))
HOAT(56mg、0.2mmol)、PyBOP(208mg、0.2mmol)およびN−BOCアスパラギン酸α−tert−ブチルエステルをDMF(500μL)中に溶解した。DIPEA(70μL、0.2mmol)を次いで添加し、そしてその反応物を室温で20分間攪拌した。この反応物を次いで−10℃まで冷却し、そしてTFA(32μL、0.2mmol)を添加した。C3−ピリジニウムセファロスポリン4b(306mg、0.2mmol)をこの反応物にDMF(500μL)中の溶液として添加した。コリジン(160μL、0.6mmol)を添加し、この反応物を−10℃で1時間攪拌した。この反応混合物を真空中で濃縮し、次いで逆相HPLC(60分間にわたる10〜90%勾配)を用いて精製し、凍結乾燥の後で、保護されたセファロスポリンアスパラギン酸誘導体のTFA塩を、白色の非晶質粉末(193mg)として得た。この生成物(162mg)をTFA(10mL)中に溶解し、室温で60分間攪拌した。この反応物を真空中で濃縮し、そしてこの残渣をCH3CN/H2O(1:1;0.1%TFA)に溶解し、凍結乾燥し、所望の脱保護されたセファロスポリンアスパラギン酸誘導体(3b)のTFA塩を白色固体(92mg)として得た。
【0150】
分析データ:MS m/z 652.9(MH+)。
【0151】
(B.3bとバンコマイシンとの反応)
塩酸バンコマイシン1a(0.32g、0.21mmol)を3mLのDMSO中に溶解した。PyBOP(0.11g、0.21mmol)およびHOAT(0.03g、0.21mmol)の0.5mL DMF溶液を添加し、続いてDIPEA(0.04mL、0.21mmol)を添加した。この反応物を30分間攪拌し、次いでTFA(0.02mL、0.21mmol)を添加し、そしてこの混合物を0℃まで冷却した。DMF(1mL)中の3b(0.15g、0.17mmol)(図6を参照のこと)の溶液を次いで添加し、続いてコリジン(0.08mL、0.63mmol)を添加した。この反応物を4℃で一晩攪拌し、次いでEtOAcから沈殿させ、遠心分離し、そして生成した固体をMeCNで洗浄した。所望の化合物をHPLC(2〜30% MeCN)により精製し、0.3gのIeを白色固体として得た。
【0152】
分析データ:MS m/z 1042.6[(M+2H)/2)]2+。
【0153】
(実施例9)
(化合物Ifの調製)
塩酸バンコマイシン1a(134.0mg、0.09mmol)およびHOAT(12.3mg、0.09mmol)を含む0.6mLのDMSOの溶液に、0.6mLのDMF中のPyBOP(46.9mg、0.09mmol)の溶液を添加し、続いてジイソプロピルエチルアミン(31.4μL、0.2mmol)を添加した。周囲温度での20分間の攪拌の後、0℃で冷却しつつ、この反応混合物をTFA(13.9μL、0.2mmol)で処理した。バンコマイシンのこの活性化エステルに、69mg(0.09mmol)のピリジニウムラクタム4aおよびコリジン(51.3μL、0.4mmol)を添加した。この最終混合物を室温で3時間攪拌し、その後TFA(33.4μL、0.40mmol)でクエンチした。この粗生成物を、酢酸エチルを添加することにより、反応混合物から沈殿させた。この固体を遠心沈降させることにより回収し、乾燥し、そして分取HPLCにより精製した。この所望の生成物Ifを、白色の綿毛状固体(89mg)として得た。
【0154】
分析データ:保持時間(分析HPLC:6分にわたる10〜70%MeCN/H2O)=2.00分。MS m/z 計算値1970.27(C86H94Cl3N16O28S2);実測値985.7[(M+2H)/2]]2+。
【0155】
(実施例10)
(化合物Ikの調製(化合物Ifのdes−クロロアナログ))
ヨウ化ナトリウム(0.271g、1.81mmol)を、光から保護してアセトン25mLに溶解したセファロスポリン−β−ラクタム12(0.999g、1.81mmol)(図7を参照のこと)の溶液に添加し、窒素下で攪拌した。25分後、4−(N−BOC−アミノメチル)ピリジン(0.390g、1.87mmol)を添加し、そしてその反応物を2時間攪拌した。この生成物を大過剰の酢酸エチルで沈殿させ、濾過により回収した。
【0156】
この生成物(0.353g、0.488mmol)をDCM(2.5mL)に溶解し、アニソール(0.21mL、1.93mmol)およびTFA(1.5mL、19.47mmol)を添加した。この反応混合物を、次いで、窒素下で45分間室温で攪拌した。この生成物を、ジエチルエーテルを用いて沈殿させ、遠心分離し、そしてそのペレットをジエチルエーテルで2回洗浄し、そして乾燥した。この生成物を、水に溶解し、不溶物を濾別した。水性物質を凍結乾燥し、0.166gの4dのTFA塩を橙色固体として得た(WがCHであることを除いて4bと同一である)。
【0157】
分析データ:MS m/z 504.1(M+1);HPLC(2〜30 アセトニトリル/水、254nm)保持時間=0.270分。
【0158】
HOAt(0.0626g、0.460mmol)を、DMSO(3.5mL)中の塩酸バンコマイシン(0.7078g、0.455mmol)の溶液に添加した。DMF(3.5mL)に溶解したPyBOP(0.2338g、0.449mmol)およびDIPEA(79.0μL、0.453mmol)を添加し、そしてこの反応物を、室温、窒素下でで20分間攪拌し、続いてTFA(35.0μL、0.454mmol)を加えた。この反応物を、次いで氷浴で冷却した。ラクタムアミド4d(0.166g、0.227mmol)を添加し、溶解の際に、コリジン(180.0μL、1.365mmol)を添加した。50分の攪拌の後、TFA(125.0μL、1.62mmol)を添加し、そして生成物をアセトニトリルで沈殿させ、遠心分離した。このペレットを最小量のDMFに再溶解し、アセトニトリルで3回再沈殿させ、次いで真空下で乾燥し、0.8gの粗生成物を得た。この粗生成物を分取HPLC(2〜35アセトニトリル/水)で精製し、化合物Ikを含む画分を集め、そして凍結乾燥した。
【0159】
分析データ:MS m/z 1539.6(断片)、HPLC:(2〜30 アセトニトリル/水、254nm)保持時間=2.92分。
【0160】
(実施例11)
(化合物Igの調製(化合物Ibのdes−クロロアナログ))
(A.N−エチル−N−(4−ピリジルメチル)−3−(N−BOC)−アミノプロパンアミド(13)の調製)
DCM(50mL)中のN−t−BOC−β−アラニン(4.678g、24.7mmol)の溶液に、ジイソプロピルカルボジイミド(3.87g、24.7mmol)を添加した。この混合物を氷浴で冷却し、そして4−(エチルアミノメチル)ピリジン(3.37g)を添加した。この反応混合物を、次いで3.5時間攪拌した。この固体を濾過により除去し、そして濾液を水で洗浄し、真空下で乾燥し、5.34gの標題中間体を得た。
【0161】
分析データ:MS m/z 308.1(M+1)。
【0162】
(B.化合物Igの調製)
ヨウ化ナトリウム(0.485g、3.22mmol)を、光から保護しかつ窒素下でアセトン(50mL)に溶解したセファロスポリン−β−ラクタム12(2.031g、3.22mmol)(図8を参照のこと)の溶液に添加した。20分後、上に記載したように調製した、アセトン(10mL)中の13(1.29g、3.40mmol)を添加し、そしてこの反応物を6時間攪拌した。この生成物を大過剰の酢酸エチルを用いて沈殿させ、濾過により回収した。この生成物(1.70g、1.90mmol)をDCM(5mL)に溶解し、そしてアニソール(0.825mL、7.6mmol)およびTFA(5mL、64.9mmol)を添加した。この反応混合物を、次いで窒素下、室温で1時間攪拌した。この生成物をジエチルエーテルを用いて沈殿させ、遠心分離し、そしてこのペレットをジエチルエーテルで2回洗浄して乾燥し、1.7gの3cのTFA塩(WがCHであることを除いて3aと同一である)を得た。
【0163】
分析データ:MS m/z 603.3(M+1);HPLC:(2〜90 アセトニトリル/水、254nm)保持時間=3.42分。
【0164】
HOAt(0.560g、4.11mmol)を、30mLのDMSO中の塩酸バンコマイシン(6.350g、4.08mmol)の溶液に添加した。DMF(30mL)に溶解したPyBOP(2.127g、4.09mmol)およびDIPEA(0.711mL、4.08mmol)を添加し、そしてこの反応物を、窒素下、室温で20分間攪拌し、続いてTFA(0.314mL、4.08mmol)を添加した。この反応物を、次いで氷浴で冷却した。DMSO(25mL)およびDMF(10mL)に溶解したラクタムアミン3c(1.7g、2.04mmol)を添加し、そして溶解の際にコリジン(1.89mL、14.3mmol)を添加した。この反応物を50分間攪拌し、TFA(1.26mL、16.3mmol)を添加し、そしてこの生成物をアセトニトリルで沈殿させ、遠心沈降させた。このペレットを、最小量のDMFに再溶解し、アセトニトリルで3回再沈殿させ、次いで真空下で乾燥し、7.4gの粗生成物を得た。この粗生成物を、分取HPLC(2〜35 アセトニトリル/水)で精製し、この生成物Igを含有する画分を集めて凍結乾燥した。
【0165】
分析データ:MS m/z 1639.5(断片);HPLC:(2〜30 アセトニトリル/水、254nm)保持時間=3.76分。
【0166】
(実施例12)
(化合物Ijの調製(化合物Idのdes−クロロアナログ))
ヨウ化ナトリウム(1.4g、9.1mmol)を、35mLのアセトン中の(7R)−7−[(Z)−2−(2−アミノ−チアゾール−4−イル)−2−(メトキシイミノ)アセトアミド]−3−(クロロメチル)−3−セフェム−4−カルボキシレート(5g、9.1mmol)に添加した。この反応容器を箔中に包み、この反応物を1時間攪拌した。5mLのアセトン中の3−アミノメチルピリジン(2.8g、13.8mmol)の溶液を添加し、この反応物を70分間攪拌し、次いでエーテル中に沈殿させ、5.4gの粗収量の4e(WがCHであることを除いて4cと同一である)を得た。この固体(4.4g)を20mLのDCMに懸濁し、20mLのTFAを添加した。この反応混合物を室温で90分間攪拌し、次いで濃縮した。得られた油状物をエーテルから沈殿させ、分取HPLC(2〜10% MeCN)により精製し、0.6gの4eのTFA塩を白色の綿毛状固体として得た。
【0167】
分析データ:Rt=0.8分(10〜70% MeCN)。1H NMR(DMSO−d6)δ 3.25(dd,2H),3.8(s,3H),4.2(bs,2H),5.05(d,1H),5.45(dd,2H),5.8(dd,1H),7.05(s,1H),8.2(m,1H),8.45(bs,2H),8.6(d,1H),9.0(d,1H),9.05(s,1H),9.4(dd,1H)。
【0168】
塩酸バンコマイシン(0.73g、0.49mmol)を、2.5mLのDMSOに溶解した。2mLのDMF中のPyBOP(0.23g、0.45mmol)およびHOAT(0.06g、0.45mmol)の溶液を添加し、続いてDIPEA(0.16mL、0.82mmol)を添加した。この反応物を、25分間攪拌し、次いでTFA(0.07mL、8.2mmol)を添加し、そしてこの反応物を0℃まで冷却した。コリジンを添加し(0.22mL、1.6mmol)、次いで2mLのDMF中の上記ラクタム4eの溶液を添加した。この反応物を2時間攪拌し、次いでエーテルから沈殿させ、遠心分離した。この生成物をHPLCにより精製し、次いで凍結乾燥し、0.23gの生成物Ijを得た。
【0169】
(実施例13:化合物Imの調製)
J.Antibiotics 53(10):1061(2000)に従って調製した2−アミノ−α−(メトキシイミノ)−4−チアジアゾール酢酸(15)(4.0g、19.8mmol)を、50mL DMF中の、Bull.Chem.Soc.Jpn.43:2925−33(1970)に従って調製したセファロスポリン誘導体14(8.42g、20.79mmol)(図9を参照のこと)およびHOAT(3.33g、21.78mmol)と混合した。その水溶液を窒素でパージし、0℃に冷却して、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド(3.99g、20.79mmol)、続いて1,3,5−コリジン(2.75mL、20.79mmol)を添加した。その水溶液を、0℃で2時間攪拌し、次いで、その反応混合物を、300mLの水の中に注いだ。その結果として生じた固体を、濾過し、飽和NaHCO3水溶液で粉砕し、再び濾過し、水で2回洗浄し、そして一晩、空気乾燥し、灰色がかった白色の粉末としてセファロスポリン中間体16を得た。この化合物を、さらに精製することなく使用した。
【0170】
分析データ:MS m/z 計算値.553.01;実測値.553.0(M+、35Cl)、555.0(M+、37Cl).
クロロ化合物16(4.0g、7.23mmol)を、4−t−BOC−アミノメチルピリジン(2.26g、10.85mmol)およびヨウ化ナトリウム(1.08g、7.23mmol)と一緒にフラスコの中に入れ、窒素でパージした。アセトン(60mL)を添加し、その反応混合物を、室温で2時間攪拌し、次いで、300mLの0.2M HClの中に注ぎ、結果としてフラスコの側面に赤いゴムの形成物が生じた。アセトンをデカントし、そのゴムを減圧下で乾燥させた。乾燥させるとすぐに、その残渣を20mLのDCMに溶解して、TFA(20mL)を添加した。その反応液を1時間攪拌し、その後、LC/MS解析によって脱保護が完了したことが示された。この溶液を次いで、200mLのEt2Oに注ぎ、生じた沈殿物を濾過し、Et2Oで洗浄し、そして減圧下で乾燥させた。乾燥させるとすぐに、粗製固体を、100mLの水で3時間、粉砕した。この水溶液を濾過し、そして生成物を凍結乾燥して、bis−TFA塩として粗製化合物4f(WがNであることを除いて4bと同一)を得た。その化合物を、さらに精製することなく使用した。
【0171】
分析データ:MS m/z 計算値.491.0;実測値.491.5
塩酸バンコマイシン(0.2g、0.135mmol)およびHOAt(20.6mg、0.135mmol)を、2mLのDMSO中に溶解した。この溶液に2mLのDMF中のPyBOP(70.2mg、0.0135mmol)の溶液を添加した。DIPEA(23.5μL、0.135mmol)を添加し、溶液を室温で20分間攪拌した。この時間の後、1mLのDMF中の4fの溶液(49mg、0.0675mmol)を添加し、その溶液を0℃に冷却した。次いで、1,3,5−コリジン(62.5μL、0.473mmol)を添加し、その溶液を0℃で45分間攪拌した。次いで、TFA(150μL)を添加し、その溶液を70mLのEt2Oの中に注いだ。生じた沈殿物を濾過し、Et2Oで洗浄し、そして減圧下で乾燥させた。その生成物を、逆相HPLCによって精製し、凍結乾燥によって単離して、トリ−TFA塩としてImを得た。
【0172】
分析データ:MS m/z 計算値.1937.8;実測値.1937.6.
(実施例14:化合物Inの調製)
DMF(100mL)中の2−アミノ−α−(トリチロキシイミノ)−4−チアゾール酢酸(17)(15.60g、36.3mmol)(2−アミノ−α−(ヒドロキシイミノ)−4−チアゾール酢酸エチルのトリチル化、続いて、加水分解によって調製した)の攪拌した溶液に、4.85g(36.3mmol)のN−クロロスクシンイミドを添加した(図10を参照のこと)。その反応混合物を、室温で15時間、攪拌し、次いで、水(500mL)の中に注ぎ、沈殿した固体を濾過して乾燥させ、灰色がかった白色の固体として5−クロロ化合物(15.11g、90%)を得た。
【0173】
分析データ:1H NMR(DMSO−d、300MHz):δ7.18−7.33(m、15H);MS m/z 実測値.486.4[M+Na]+.
250mLのDMF中のこの化合物(28.0g、60mmol)の攪拌した溶液に、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド(12.7g、66mmol)、HOBT(9.0g、66mmol)および2,4,6−コリジン(8.8mL、66mmol)を添加した。その反応混合物を0℃に冷却し、5分後、14(実施例13;図9を参照のこと)(24.4g、60mmol)を添加した。3.5時間後、その反応混合物を、酢酸エチル(1200mL)で希釈し、1.0M HCL(250mL)、飽和炭酸水素ナトリウム水溶液(250mL)およびブライン(250mL)で洗浄した。有機層を、乾燥(MgSO4)し、濾過し、真空化で濃縮して、灰色がかった白色の固体としてセファロスポリン中間体18(43.2g、88%)を得、それをさらに精製することなく使用した。
【0174】
分析データ:1H NMR(DMSO−d6、300MHz):δ3.65(q、2H)、3.76g(s、3H)、4.54(q、2H)、5.24(s、2H)、5.27(d、1H)、6.03(q、1H)、6.95(d、2H)、7.18−7.41(m、19H)、9.90(s、1H).
ヨウ化ナトリウム(920mg、61mmol)およびN−t−BOC−4−(アミノメチル)ピリジン(1.8g、86mmol)を、暗所(アルミホイルに包まれたフラスコ)でアセトン(20mL)中の18(5g)の攪拌した溶液に添加した。その反応混合物を、室温で2.5時間、攪拌し、次いで、ジエチルエーテル(200mL)に一部分ずつ添加し、生じた固体を濾過によって回収した。TFA/DCM(40ml、1:1)中の粗製固体(5.9g)の溶液を、室温で1時間攪拌し、次いで、ジエチルエーテル(300mL)に一部分ずつ添加した。生じた個体を、固体残渣を得るために濾過によって回収し、それを、分取用HPLCによって精製し、各々の白い固体として、4g(R9が水素であることを除いて4bと同一)の2個のヒドロキシイミノ立体異性体(それぞれ、240mgおよび120mg)のTFA塩を得た。
【0175】
分析データ:
4gの主要な立体異性体:1H NMR(DMSO−d6、300MHz):δ3.40(q、2H)、4.45(s、2H)、5.18(d、1H)、5.54(q、2H)、5.87(q、1H)、7.31(bs、2H)、8.18(d、2H)、8.71(bs、3H)、9.09(d、2H)、9.39(d、1H)、11.73(s、1H);MS m/z 実測値.524.2[M+H]+.
4gの少ない立体異性体:1H NMR(DMSO−d6、300MHz):δ3.50(q、2H)、4.45(s、2H)、5.19(d、1H)、5.58(q、2H)、5.83(q、1H)、7.27(bs、2H)、8.20(d、2H)、8.77(bs、3H)、8.81(d、1H)、9.10(d、2H)、12.51(s、1H);MS m/z 実測値.524.2[M+H]+.
DMF(1mL)中のPyBOP(170mg、0.33mmol)およびHOAt(44mg、0.33mmol)の溶液を、DMSO/DMF(6mL、3:1)中の塩酸バンコマイシン(485mg、0.33mmol)の攪拌した溶液に添加した。続いて、DIPEA(0.057mL、0.33mmol)を添加し、その反応混合物を、室温で30分間、攪拌した。TFA(0.026mL、0.33mmol)を、その反応混合物に添加し、次いで、それを0℃に冷却し、次いで、コリジン(0.097mL、0.73mmol)およびDMF(1mL)中の4g(123mg、0.16mmol)の溶液を添加した。その反応混合物を、4時間にわたって、室温で暖め、次いで、酢酸エチル(50mL)に一部分ずつ添加し、生じた固体を、濾過によって回収し、分取用HPLCによって精製し、白い固体として生成物In(174mg、46%)のTFA塩を得た。
【0176】
分析データ:MS m/z 実測値.979.4[(M−ピリジン)/2]+.
(実施例15:水溶性の決定)
本発明の化合物の水溶性を、以下の手順を使用して決定した。pH2.2での5重量%のデキストロース緩衝液を、99mLの5重量%デキストロース水溶液(Baxter)に1mlの1N 塩酸(Aldrich)を添加して調製した。次いで、較正標準物のための1mg/mLのストック溶液を、1mLのDMSO中に1mgの試験化合物を溶解することによって調製した。この溶液を、30秒間、ボルテックスし、次いで、10分間、超音波処理した。次いで、このストック溶液を、以下の濃度:50μg/mL、125μg/mL、250μg/mL、375μg/mLおよび500μg/mLを有する較正標準物を調製するために水で希釈した。
【0177】
各試験化合物(30mg)を、Millipore non−sterile,Ultrafree−MC 0.1μmフィルターユニット(Millipore UFC30VVOO)に秤量して入れ、磁気攪拌子を、各ユニットに添加した。次いで、5重量%のデキストロース緩衝液(750μL)を、各ユニットに添加し、これらの混合物を5分間、ボルテックスした。次いで、フィルターユニットを、エッペンドルフチューブラックに入れ、そのチューブラックを、磁気攪拌機の上に置いた。次いで、各ユニットを、1N NaOH(VWR)を使用してpH3に滴定し、得られた溶液を5分間、7000rpmで遠心分離した。次いで、各ユニットを、5%デキストロース緩衝液で200倍希釈し、希釈したサンプルを、分析のための自動サンプラーバイアルに移した。
【0178】
較正標準物および試験サンプルを、以下の条件を使用する逆相HPLCによって分析した:
カラム:Luna 150×4.6mm;C18;5μ
移動相:A=5/95、B=95/5、両方=MeCN/H2O;0.1%TFA
方法:10m Libo 100(6分で、0〜100%のB)
注入容量:20μL
波長:214nm
各試験サンプルの溶解性を、較正曲線と試験サンプルのピーク領域とを比較し、そして希釈係数を乗じることによって算出した。
【0179】
【表1】
(実施例16:最小発育阻止濃度(MIC)の決定)
最小発育阻止濃度(MIC)アッセイを、NCCLSガイドラインに記載の微量液体希釈法を用いて行った(broth microdilution method)(NCCLS.2000.Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically;Approved Standard−第5編、第20巻、No.2を参照のこと)。細菌株を、American Type Tissue Culture Collection(ATCC)、Stanford University Hospital(SU)、Kaiser Permanente
Regional Laboratory in Berkeley(KPB)、Massachusetts General Hospital(MGH)、the Centers for Disease Control(CDC)、the San Francisco Veterans’Administration Hospital(SFVA)またはthe University of California San Francisco Hospital(UCSF)から入手した。バンコマイシン耐性腸球菌は、それらのテイコプラニンに対する感受性に基づいて、VanAまたは
Van Bとして表現型決定した。Van A、Van B、Van C1またはVan
C2として遺伝子型決定した、いくつかのバンコマイシン耐性腸球菌をまた、Mayo
Clinicから入手した。
【0180】
このアッセイにおいて、低温保存した参照の細菌培養物および臨床株を、適切な寒天培地(すなわち、トリプティケースソイ(Trypticase Soy)寒天、脱繊維素化ヒツジ赤血球含有トリプティケースソイ寒天、ブレインハートインフュージョン(Brain Heart Infusion)寒天、チョコレート(Chocolate)寒天)での単離のために線画した。コロニーの形成を可能にするためのインキュベーション後、これらのプレートをパラフィルム(登録商標)で密閉し、そして2週間まで冷凍保存した。アッセイの接種材料を調製し、低い可変性を確実にするために、寒天プレートに培養した細菌単離物からのいくつかのコロニーを、接種ループを用いてつつき、そして無菌的にMueller−Hinton Broth(製造業者の認証に基づいて、必要とされるレベルの二価のカチオンを補充した)に移した。ブロス培養物を、35℃で一晩増殖させ、新鮮な、予め温めたブロスに希釈し、そして対数期まで増殖させた;これは、0.5 MacFarland standardまたは1mlあたり1×108コロニー形成単位(CFU/mL)に等しい。懸度がMacFarland標準に等しい場合、種可変性に起因して、全ての細胞懸濁物で1×108CFU/mLを含むとは限らない。従って、受容可能な調節(NCCLSガイドラインに基づく)を、異なる細菌株の希釈物において作製した。96ウェルマイクロタイタープレートにおいて、2倍連続希釈した抗生物質濃度シリーズ(やはり、対応する培地(100μL)中)に希釈した場合に、Mueller−Hinton Broth、補充したMueller−Hinton Broth、またはHaemophilus試験培地中でこの培養物(100μL)が最初の細菌濃度(5×105CFU/mL)を得るように、接種材料を希釈した。次いで、プレートを、35℃で18〜24時間インキュベートした。MICを、細菌の増殖を伴わない最低濃度のウェルとして目視で読み取った。細菌の増殖を、3つより多くのピンポイントコロニー、直径が2mmより大きな沈殿した細胞のボタン(button)、または明確な懸度として規定する。
【0181】
最初のスクリーニングにおいて慣用的に試験される株としては、メチシリン感受性Staphylococcus aureus(MSSA)、メチシリン耐性Staphylococcus aureus(MRSA)、ペニシリナーゼを生成するStaphylococcus aureus、メチシリン感受性Staphylococcus epidermidis(MSSE)、メチシリン耐性Staphylococcus epidermidis(MRSE)、バンコマイシン感受性Enterococcus faecium(EFMVS)、バンコマイシン感受性Enterococcus faecalis(EFSVS)、テイコプラニンにも耐性のバンコマイシン耐性Enterococcus faecium(EFMVR VanA)、テイコプラニンに感受性のバンコマイシン耐性Enterococcus faecium(EFMVR VanB
)、テイコプラニンにも耐性のバンコマイシン耐性Enterococcus faecalis(EFSVR VanA)、テイコプラニンに感受性のバンコマイシン耐性Enterococcus faecalis(EFSVR VanB)、ペニシリン感受
性Streptococcus pneumoniae(PSSP)およびペニシリン耐性Streptococcus pneumoniae(PSRP)が、挙げられる。Mueller−Hinton brothにおける十分な増殖についての、PSSPおよびPSRPの無能性に起因して、これらの株についてのMICを、脱線維素血液を補充したTSブロスまたはHaemophilus試験培地を補充したTSブロスのいずれかを用いて決定した。
【0182】
次いで、上記の株に対して十分な活性を有する試験化合物を、臨床単離物(上に列挙した種、ならびに種分化していない(non−speciated)コアグラーゼ陰性の、メチシリンに対して感受性のStaphylococcus(MS−CNS)および耐性のStaphylococcus(MR−CNS)の両方を含む)のより多くのパネルにおけるMIC値について試験した。さらに、これらの試験化合物をまた、グラム陰性微生物(例えば、Escherichia coli、Pseudomonas aeruginosa、Klebsiella pneumoniae、Enterobacter
cloacae、Acinetobacter baumannii、Haemophilius influenzaeおよびMoraxella catarrhalis)に対してMICについてアッセイした。
【0183】
表2は、既知の抗生物質であるバンコマイシンと比較した、メチシリン耐性S.aureus(MRSA)およびメチシリン感受性S.aureus(MSSA)に対する本発明の化合物についてのMIC90データを示す。
【0184】
【表2】
表2のデータは、本発明の化合物(すなわち、Ia、Ib、Ic、Id、Ie、If、ImおよびIn)が、des−クロロアナログIg、IjおよびIkまたはバンコマイシンのいずれかと比較して、MRSA33591に対して驚くべき予期しない抗菌活性を有すること;ならびに本発明の化合物が、バンコマイシンと比較して、MSSA 13709に対して驚くべき予期しない抗菌活性を有することを示す。
【0185】
(実施例17:時間−殺傷アッセイ)
この時間−殺傷アッセイは、試験化合物の細菌活性の速度を測定するための方法である。これらの手順は、V.Lorian、「Antibiotics in Laboratory Medicine」、第4版、WilliamsおよびWilkins(1996)、104〜105頁に記載されるのと類似の手順である。細菌コロニー形成を迅速に防護し、そして宿主組織損傷を減少させるために、迅速な時間−殺傷が所望される。
【0186】
細菌性接種材料を、MICの決定のために実施例16に記載されるように調製した。細菌を、振盪フラスコ中の予め温めた培地に希釈し、そして振盪しながらインキュベートした(200rpm、35℃)。0時間、1時間、4時間、および24時間で、サンプルをフラスコから取り出し、細菌を、プレート計数によって数えた。最初のサンプリングに続いて、アッセイされるべき化合物を振盪フラスコ培養物に添加した。化合物の添加前および添加後の、これらの間隔でのプレート計数を、時間−殺傷曲線に図式的に表した。細菌活性は、24時間まで、細菌細胞数において3対数以上の減少(同程度〜99.9%より多くの減少)として規定される。
【0187】
このアッセイにおいて、式Iの化合物(すなわち、化合物Ib)は、4時間において1.0μg/mL以下の濃度で、MRSA 33591に対して殺菌性であった。比較すると、バンコマイシンは、24時間において4μg/mLの濃度で、MRSA 33591に対して殺菌性であった。
【0188】
(実施例18:好中球減少マウスにおけるインビボでの有効性研究)
動物(雄性CD−1マウス、20−30g)を、Charles Rivers Laboratories(Gilroy、CA)から入手し、そして餌および水を自由に摂取させた。好中球減少症を、細菌接種の4日前および2日前に与えられたシクロホスファミドの腹腔内(IP)注射(200mg/kg)を介して誘導した。
【0189】
使用した生物は、診療的に関連のあるグラム陽性病原体の感受性株または耐性株(例えば、メチシリン感受性Staphylococcus aureus(MSSA13709)およびメチシリン耐性Staphylococcus aureus(MRSA3
3591))のいずれかであった。細菌接種材料の濃度は約106CFU/mLであった。動物を、イソフルランで軽く麻酔し、そして50mLの細菌接種材料を前大腿に注射した。接種の1時間後、動物に、ビヒクルまたは試験化合物の適切な用量を静脈内投与した。処置の0時間後および24時間後に、これらの動物を安楽死させ(CO2窒息)、そして前大腿および後大腿を無菌的に収集した。この大腿を、10mLの滅菌生理食塩水に入れ、ホモジナイズした。ホモジネートの希釈物を、トリプティクソイ(tripticsoy)寒天プレート上にプレートし、これを一晩インキュベートした。所定のプレート上の細菌コロニーの数は、希釈係数を乗じ、大腿重量(グラム)で除算し、対数CFU/gとして表した。ED50(大腿力価において最大減少の50%を生成するために必要とした用量)を、各試験化合物について評価した。
【0190】
このアッセイにおいて、式Iの化合物(すなわち、化合物Ib)は、バンコマイシンについてのivで9mg/kgのED50と比べて、ivで0.10mg/kg未満のED50を有した。
【0191】
本発明は、その特定の実施形態を参照して記載されているが、種々の変更がなされ得、本発明の真の精神および範囲から逸脱することなしに等価物が置換され得ることは、当業者によって理解されるべきである。さらに、多くの改変が、特定の状況、物質、成分の組成物、プロセス、プロセス工程または工程を、本発明の目的、精神および範囲に適応させるためになされ得る。全てのこのような改変は、本明細書に添付される特許請求の範囲内であることが意図される。さらに、本明細書中で引用された全ての刊行物、特許および特許書類は、それらが参考として個々に援用されるかのような同じ程度で、本明細書中にその全体が参考として援用される。
【特許請求の範囲】
【請求項1】
明細書に記載の発明。
【請求項1】
明細書に記載の発明。
【図1】

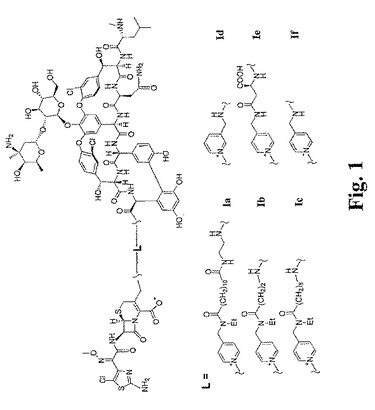
【図2】
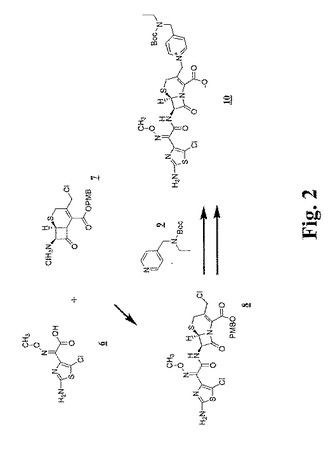
【図3】
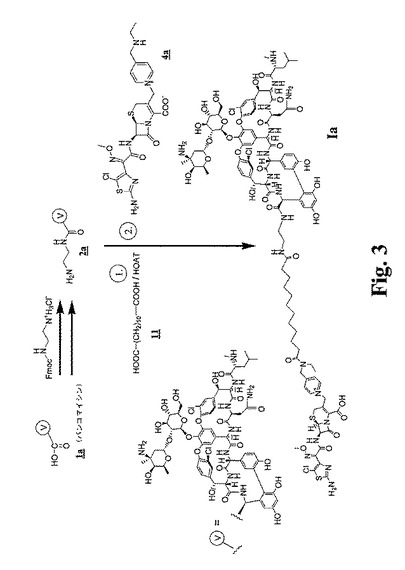
【図4】
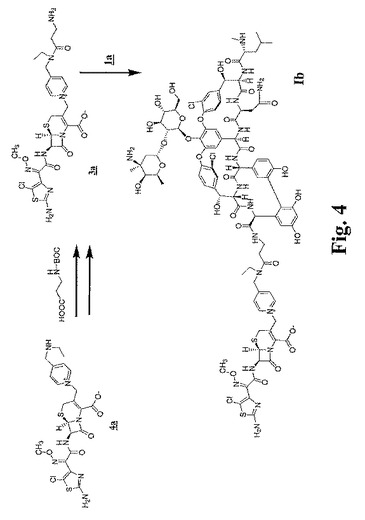
【図5】
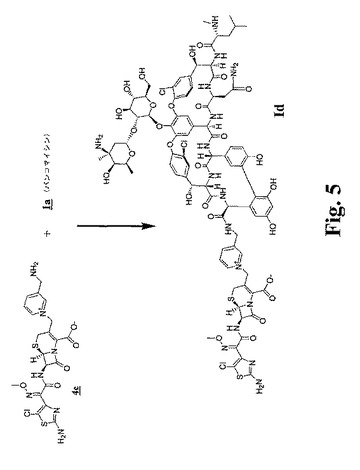
【図6】
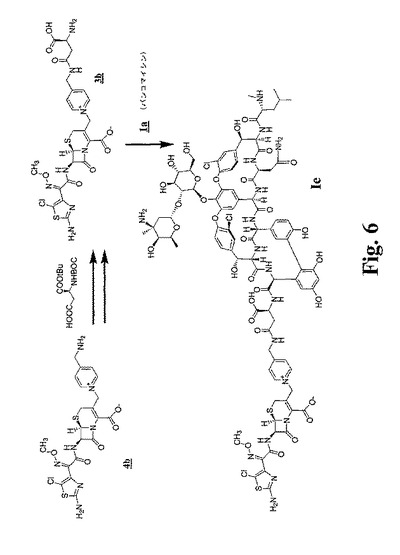
【図7】
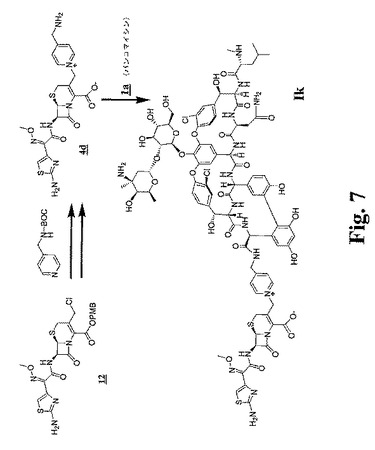
【図8】
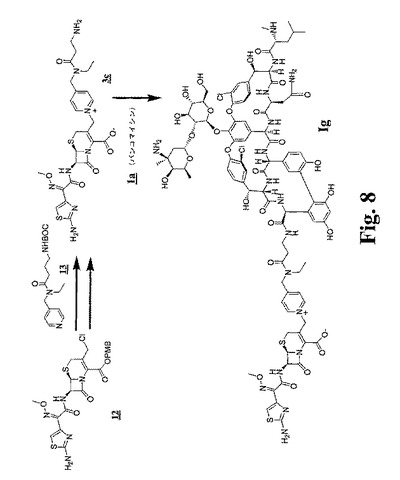
【図9】
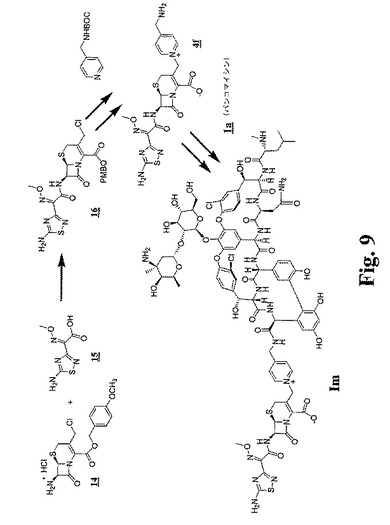
【図10】
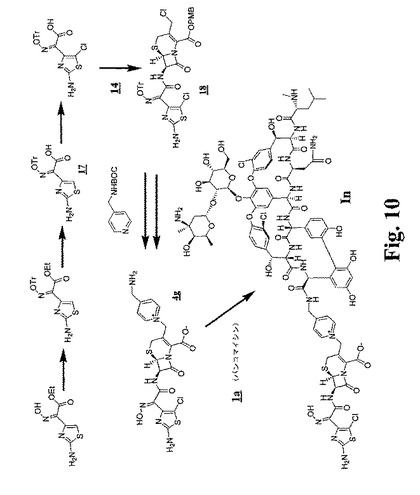
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2010−254701(P2010−254701A)
【公開日】平成22年11月11日(2010.11.11)
【国際特許分類】
【出願番号】特願2010−130496(P2010−130496)
【出願日】平成22年6月7日(2010.6.7)
【分割の表示】特願2006−533289(P2006−533289)の分割
【原出願日】平成16年5月21日(2004.5.21)
【出願人】(500154711)セラヴァンス, インコーポレーテッド (129)
【Fターム(参考)】
【公開日】平成22年11月11日(2010.11.11)
【国際特許分類】
【出願日】平成22年6月7日(2010.6.7)
【分割の表示】特願2006−533289(P2006−533289)の分割
【原出願日】平成16年5月21日(2004.5.21)
【出願人】(500154711)セラヴァンス, インコーポレーテッド (129)
【Fターム(参考)】
[ Back to top ]