
核酸の精製方法
本発明は、8〜13のpKaを有する不足量の核酸結合性のA基を備えているか、またはA基と、結合中および好ましくは溶出中、中性電荷を有する結合阻害性のN基とを有する、核酸結合相を用いて核酸を精製する方法であって、(a)核酸結合性のA基のpKよりも小さいpH(結合pH)で、核酸を核酸結合相に結合させる工程;(b)結合pHよりも大きいpH(溶出pH)で、該核酸を溶出させる工程を含む方法に関する。さらに、核酸の精製に使用され得る対応するキットおよび核酸結合相も開示する。本発明による手法により、低塩濃度の使用による核酸の精製、特に溶出が可能になり、そのため、精製された核酸の処理過程を直接行なうことができる、例えば、PCRに使用することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、核酸結合支持材を用いて核酸を精製するための方法およびキットに関する。また、適切な支持材を精製する好適な方法を記載する。
【背景技術】
【0002】
核酸を精製および単離する種々の方法、例えば、フェノールクロロホルムの使用、塩析法、イオン交換体およびシリカ粒子の使用が先行技術において開示されている。
【0003】
既知の核酸精製方法の一例は、「電荷スイッチ切り替え(charge−switch)法」である。これは、主に弱塩基性のポリマー、例えば、ポリBis−Tris、ポリTris、ポリヒスチジン、ポリヒドロキシル化アミン、キトサンまたはトリエタノールアミンなどを含む核酸結合相を、第1のpHで核酸含有試料と接触させることを伴い、この接触中、核酸結合相は正電荷を有する。これにより、負電荷を有する核酸の前記相に対する結合が促進される。核酸を放出/溶出させるため、正電荷を逆転または中和するために、電荷スイッチ切り替え原理に従って、核酸結合相のpKaよりも大きい第2のpHが設定される。このpHを該固相の核酸結合基のpKaよりも大きく設定することにより、核酸結合相からの結合核酸の分離が促進される。
【0004】
先行技術には、可溶性相(例えば特許文献1参照)と固相(例えば特許文献2参照)の両方が開示されている。種々の溶液、例えば、非常に高いpHを有する溶液あるいは生物学的バッファー、特に、例えばTrisバッファーなどの低塩バッファーが溶出に使用されている。
【0005】
精製核酸は、通常、このほかにも処理される。前記の処理は、例えば、増幅(ポリメラーゼ連鎖反応(PCR)など)、酵素反応(制限、ライゲーション、リン酸化、脱リン酸化またはRNA転写など)、ハイブリッド捕捉アッセイおよび電気泳動を含む。このような「下流」反応は、多くの場合、比較的高い塩濃度に対する耐性がほとんどなく、したがって、核酸のさらなる処理過程を行なう前に、しばしば脱塩工程を行なわれなければならない。
【0006】
したがって、含まれる塩があまり多過ぎず、さらに、あまり塩基性が高過ぎない溶出バッファーは、さらなるバッファー交換または脱塩工程なしでの、精製核酸の直接後続する使用に望ましい。
【0007】
しかしながら、アニオン交換体の溶出能は、溶出バッファーのpHと塩濃度の増大の関数として増大する。したがって、クロマトグラフィーの要件は、DNAの処理過程の実施に関する要件と相反する。このため、先行技術において、適当な核酸結合基とpH条件を選択することにより、核酸の溶出を特異的に促進させることが試みられている。
【0008】
このような核酸精製方法が知られているにもかかわらず、既存の方法改善する必要性、より詳しくは、直後に試料の処理過程も行なうことができるような様式で核酸を精製する必要性がある。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】欧州特許第0707077号明細書
【特許文献2】国際公開第99/29703号
【発明の概要】
【課題を解決するための手段】
【0010】
したがって、本発明の目的は、直後に試料の処理過程を行なうことが可能な核酸の精製方法を提供することである。より詳しくは、本発明は、低塩濃度で溶出が可能な方法を利用可能にすることを意図するものである。
【0011】
この目的は、本発明により、特別な核酸結合相を用いる核酸精製方法によって達成される。
【0012】
第1の実施形態によれば、核酸結合相は、8〜13のpKaを有する核酸結合性のA基と、結合を減弱させ、かつ使用される結合pHにおいて中性電荷を有するN基も有する。
【0013】
この核酸結合相を用いた精製方法は、以下の工程:
(a)核酸結合性のA基のpKよりも小さいpH(結合pH)で、核酸を核酸結合相に結合させる工程;
(b)結合pHよりも大きいpH(溶出pH)で、該核酸を溶出させる工程
を有する。
【図面の簡単な説明】
【0014】
【図1】図1は、核酸結合性のA基と結合減弱性のN基を備えており、前記の基が別々のリガンドに存在している支持材の模式的概略図を示す(請求項2(i)も参照のこと)。
【図2】図2は、核酸結合性のA基と結合阻害性のN基を備えており、前記の基が単一のリガンド内に混合物として存在している支持材の模式的概略図を示す(請求項2(ii)も参照のこと)。(A)は、最初にA基を備え、次いでN基を備えた支持材を示す;(B)は、単一のリガンド内のAとNの交互配列を示す。
【図3】図3は、核酸結合性のA基と立体遮蔽性のN基を備えており、前記の基を、図1および2に示したスキームによる核酸結合性のA基と組み合わせることが可能な支持材の模式的概略図を示す。請求項2(iii)参照。
【図4】図4は、一例として核酸結合性のA基と、結合阻害性および/または立体遮蔽性のN基とを備えており、前記の基が、図1〜3に示したスキームの組合せによって形成されている支持材の模式的概略図を示す。請求項2(iv)参照。
【図5】図5は、準化学量論量の核酸結合性のA基を備えた支持材の模式的概略図を示す。請求項3(iv)参照。
【図6】図6は、異なるpH値での実施例A3a)による溶出プロフィールを示す。
【図7】図7は、異なるpH値での実施例A3b)による溶出プロフィールを示す。
【図8】図8は、実施例C.1)に従って調製したシリカゲルの溶出図を示す。
【発明を実施するための形態】
【0015】
本発明は、核酸結合性のA基に加えて結合減弱性のN基も有する核酸結合相によって核酸の精製に関する。本発明によれば、核酸結合性のA基は8〜13のpKaを有する。核酸は、前記A基の少なくとも1つのpKaよりも下のpHで結合させる。したがって、A基はプロトンを取り込み、その結果、正電荷を有する状態になり、核酸結合相が、負電荷を有する核酸に結合することが可能になる。溶出は結合pHより大きいpHで行ない、それにより、核酸結合相の正電荷を減少させる。
【0016】
さらなる基のN基は、結合pHで、好ましくは溶出pHでも電荷中性である。したがって、N基は、いくつかの態様において、A基の結合強度に影響を及ぼす:(1.)A基の配置に割り込み、したがって、A基の密度が低下するため、核酸結合相に対する核酸の結合強度に影響を及ぼす。(2.)弱い相互作用に参与し(例えば、ファンデルワールス相互作用によって)、中間強度の相互作用まで参与し得る(例えば、水素結合によって)官能基によって、結合強度を特異的にモジュレートし得る。(3.)N基は、大きさおよび/または数が増大すると核酸結合性のA基を立体遮蔽することがあり得、それにより結合強度を低下させる。したがって、N基は、核酸結合強度の一様でpH非依存性の低下を引き起こす。この結果として、結合される核酸が少なくなり、そのうえ、より容易に核酸結合相から分離され得る。N基の機能は、特異的かつ制御された様式で、A基の結合強度および/または数を減少させ、それにより、本発明による核酸結合相において所望の頻度を設定することである。N基の比率が高いほど、A基の比率が低く、電荷密度が低く、核酸結合相に対する核酸の結合が弱い。N基に対するA基の比率は、対応する核酸結合相の調製方法(例えば、重合、(重)縮合によって、特に、支持材のコーティングによって)において、または該方法によって、所望の強度の核酸結合が事前に確立されることを可能にするための調製プロセスによって特異的に調整され得る。
【0017】
このことの大きな利点は、溶出させた核酸を、精製直後に、さらに処理することが可能な条件下で溶出を実施できることである。多くの生物生体工学的方法、例えば、核酸の増幅(特に、PCRによるもの)、配列決定、逆転写、制限解析、およびその他(上記参照)などは、溶出液中の夾雑物に対して、特に、高い塩濃度に対して敏感である。本発明による精製方法では、好都合には、低塩濃度または低イオン強度での溶出が可能となり、それにより、後続の生物生体工学的方法、特にPCR反応において、さらなる処理過程を直接行なうことが可能になる。
【0018】
N基を用いることにより核酸の結合を減弱させるという概念により、驚くべき良好な結果がもたらされる。先行技術では、方法の最適化のために核酸の最適な結合を想定し、次いで、結合条件、特に溶出の最適化に注力する。対照的に、結合強度の減弱は、例えば、核酸が、結合させる工程において不充分にしか核酸結合相に結合しないか、または洗浄中に溶出されることがあり得るため、予測される収率に関して不都合であると考えられていた。このことに鑑みると、N基の存在により結合強度が低下するにもかかわらず成功裡で緩徐な核酸精製という本発明による概念は、極めて驚くべきこととみなされるはずである。
【0019】
N基に対するA基の比率は、1%〜99%、1〜50%、好ましくは1%〜25%であり得る。
【0020】
好ましい一実施形態によれば、核酸結合相は、A基とN基を導入するために修飾される。本発明によれば、これは、以下により詳細に説明する種々の実施形態を包含する。
【0021】
一実施形態によれば、核酸結合相は異なるリガンド(リガンドIとリガンドII)を備えており、リガンドIは少なくとも1つのA基を有し、リガンドIIは少なくとも1つのN基を有する。この実施形態を図1Aに図解する。このとき、A基とN基は直接近接しており、また、ポリマーコーティングを形成させてもよい。N基担持リガンドIIの存在のため、A基の数は減少し、それにより、特に支持材が使用されている場合、核酸の結合の強固さが低下する。したがって、N基のため、該支持体表面上のA基の密度は低下し、結果として、核酸に対する結合親和性が低下する。その結果、核酸は、より容易に溶出され得る。用語「リガンド(1つまたは複数)」は、特に、好ましくは、少なくとも1つのA基および/または1つのN基を有する支持材が、本発明による核酸結合相を提供するために修飾される表面の官能基をいう。リガンドIとIIは、好ましくは支持材に結合させており、例えば、反応性の個々の分子のモノマー、ダイマー、オリゴマーまたはポリマーであり得る。変形例の一例において、支持材と反応する化合物の一部としてのA基またはN基は、直接またはリンカーもしくはスペーサーを介して支持材に結合させる。好ましい一実施形態によれば、この修飾は、支持材を、異なるリガンドIとIIを有する混合物と接触させることによりなされる。リガンドIに対するリガンドIIの比率が高いほど、核酸に対する結合強度が低い。この比は、単に、コーティングプロセスにおけるリガンドI反応体とリガンドII反応体の対応する比によって制御され得る。詳細を、以下に、対応する支持材の合成または調製方法に関して説明する。また、リガンドは、ポリマーコーティングを形成していてもよい。また、支持材は、A基と開始剤分子T(Initiatormolekuelen)を備えたものであってもよく、1つ以上のN基を前記開始剤分子に結合させる。この実施形態を図1B)に示す。N基は、この場合、支持材の表面に対してA基の「上部」に存在する。また、A基も開始剤分子を介して結合させることも可能である。
【0022】
本発明のさらなる一実施形態によれば、支持材は、1つ以上のA基と1つ以上のN基とが単一のリガンド内に存在する核酸結合リガンドで修飾される。
この実施形態を図2により詳細に示す。この概念の実現には種々の可能性がある。
【0023】
変形例の一例によれば、支持材は、N基を含む1つ以上の化合物を担持しているA基(直接または例えば開始剤分子Tを介してのいずれかにより結合)を備えている(図2Aも参照のこと)。複数のN基が存在する場合、これらはまた、非分枝鎖の形態であってもよく、分枝型樹状構造としての形態であってもよく、対応する混合物としての形態であってもよい。これには、リガンド1つあたり例えば、1〜100個のN基、1〜20、好ましくは1〜10、特に好ましくは1〜5個のN基が含まれ得る。また、所望により、なおさらなるA基とN基を導入してもよい。この連結ストラテジーでは、実質的に純粋にA基で占有された比較的均一な結合面と、突出部と隣接し、N基による占有のためA基の結合強度が低下した1つ以上の面とがもたらされる。
【0024】
この実施形態の変形例のさらなる一例によれば、支持材は、N基とA基の混合物を担持しているA基(直接または例えば開始剤分子Tを介してのいずれかにより結合)を備えている(図2Bも参照のこと)。これは、最初に1つ以上のA基を支持材に結合させることを伴い得る。この後、このようなA含有主リガンドに、少なくとも1つ以上のN基を導入する化合物を結合させる。反応条件の制御により、各場合において、リガンド内にA基とN基を1つだけ存在させることが可能になる;しかしながら、リガンドはまた、オリゴマーまたはポリマーの形態であってもよい。オリゴマーおよびポリマーとして設計されるリガンドは、N基がランダムな分布(例えば図2B、右側の図参照)、交互配列(例えば図2B、左側の図参照)、または他の配置のブロックコポリマーを有するものであり得る。また、組合せも可能である。適切なA含有およびN含有官能基/リガンドの好適な設計方法を以下に詳細に説明する。
【0025】
さらなる実施形態によれば、支持材は、A基が、少なくとも1つ、好ましくは多数のN基を有する化合物によって立体遮蔽されたA基とN基で修飾されている。この実施形態にも、種々の変形例が存在する。
【0026】
変形例の一例によれば、立体遮蔽は、好ましくは、N基を有するオリゴマーまたはポリマーによって行なう。前記オリゴマーまたはポリマーは、例えば、A基(1つまたは複数)を有するリガンドに結合させてもよい(図3Bも参照のこと)。しかしながら、立体遮蔽が確保される限り、前記オリゴマーまたはポリマーはA基に隣接して配置するのがよい(図3Aも参照のこと)。N基を有するオリゴマー/ポリマーはA基を、多かれ少なかれ規則的な二次構造(例えば、「ランダムコイル」またはらせん)を形成することにより、周囲環境から、いわば蓋をすることによって遮蔽し、したがって、核酸の結合を減弱させることが可能である。また、該オリゴマー/ポリマーは、N基から調製したものであってもよく、また、ブロックコポリマーによって導入されたものであってもよい。
【0027】
この変形例は、例えば、第1の工程において開始剤基Tを支持材に、例えばシラン処理によって適用することにより調製され得る。次いで、第2の工程において、A基を含むモノマーが、例えばATRP(原子移動ラジカル重合)によってグラフト化され得る。このモノマーを充分反応させた後、中性のN基を有する第2のモノマーが導入され得る。これにより、アニオン交換体基を担持している第1のホモポリマーと、第1のホモポリマーに連結され、中性のN基を担持している第2のホモポリマーからなるコポリマーが、該支持体上に得られる。発生期ポリマー鎖の長さ、したがって、立体遮蔽の強度は、N基を担持している第2のモノマーの量によって制御され得る。
【0028】
したがって、立体遮蔽の程度は、鎖長およびモノマー置換によって制御され得る。N基を有するオリゴマー/ポリマーの好ましい鎖長は、n=10〜1000、n=10〜500、特に好ましくは、n=10〜100である。N基を担持しているオリゴマー/ポリマーは、本出願書類に記載のモノマーを用いて設計してもよく、これを詳細に説明する。ポリアクリレートが特に好適である。
【0029】
使用される好ましいポリマーは、下記式:
【0030】
【化1】

によるポリアクリレートである。
【0031】
立体遮蔽がなされるのに特に好適な試薬は、加水分解後に中性のジオールN基が生成されるメタクリル酸グリシジルである。この目的のため、開始剤分子Tは、例えばシラン処理によって適用され得る。この後、第1のモノマー、例えば、メタクリル酸N,N−ジメチルアミノプロピルを支持体上で、例えばATRPによって重合させる。第2の重合工程では、次いで、この基を立体遮蔽する。これは、例えば、モノマーを充分反応させた後、第2のモノマー、例えば、加水分解後に中性のジオールN基が生成されるメタクリル酸グリシジルを導入することによりなされ得る。該立体効果により、核酸とA基の密接な接触が抑制され、その結果、後者はあまり強く結合されず、低イオン強度で、したがって低塩濃度で核酸結合相から溶出され得る。
【0032】
また、N基の立体遮蔽は、「バルキー」な置換基によってもなされ得、挙げられる得る該置換基の一例は、分枝アルキル原子団、例えば、イソプロピル、ジイソプロピル、第3級ブチル、脂肪族または芳香族環(炭素環として、もしくは複素環としてのいずれも)である。また、このような立体阻害性のN基も、オリゴマー/ポリマーの形態で使用され得る。
【0033】
したがって、立体遮蔽は、特に、(i)さらに導入されるN基の比率;(ii)N基を担持する化合物の合成;(iii)N基の選択および構造、ならびに(iv)N基を有するオリゴマー/ポリマーの鎖長によって制御され得る。
【0034】
したがって、A基(1つまたは複数)は、同じリガンド内に存在しているか、または近接して配置されたN基によって特に良好に遮蔽され得る。また、A基に結合させることも可能である。立体遮蔽は、N基(1つまたは複数)を有する化合物の大きさの増大、また、N基の大きさの増大の関数として増大し、鎖長の増大および炭素鎖の高い分枝性または環状基の環の大きさの増大は、ともに立体遮蔽を増大させる。
【0035】
特定の一実施形態では、支持材を、上記のすべての連結ストラテジーの組合せを用いて修飾する。一例を図4に示す。したがって、支持材は、A基、立体遮蔽性のN基および開始剤分子Tが互いに直接連結されたものであり得、前記開始剤分子は、さらなる立体遮蔽性のN基を備えている(例えば、図4Aを参照のこと)。この型の連結は、例えば、通常のN基を用いて行なわれ得る(例えば、図4Bを参照のこと)。さらに、支持材は、例えば、A基を備えており(直接または開始剤分子Tを介して結合)、N基とA基が連結された混合物を担持しており、N基の全部または一部が立体阻害性基であるものであってもよい(図4Dおよび4Eを参照のこと)。
【0036】
本出願書類のさらなる一態様によれば、該方法は、支持材を有し、かつA基で修飾された核酸結合相を用いて行なわれ、該支持材は、一部のみがA基で占有されており、これは、提供される核酸結合基が通常よりも相当少ないことを意味する。したがって、支持材の単位面積あたり、および/または支持材の1グラムあたりに存在する核酸結合性のA基が少なくなることにより、核酸の結合強度が先の連結ストラテジーと同程度に低減される。図5は、この概念の一実施形態を示す。
したがって、この実施形態でも、特に、核酸結合相からの核酸の溶出に低塩濃度が使用され得るため、前記核酸のさらなる処理過程を直接行なうことが可能な条件下で核酸を溶出させることが可能である。
【0037】
支持材上のA基の本発明による準化学量論量は、支持材に対して準化学量論量のA基導入化合物を用いることにより得られ得る。また、支持材が有するA基を結合させ得る潜在的結合部位の量を少なくすることも考えられ得る。さらなる一実施形態としては、結合部位の減少または前記結合部位の反応性の低減を引き起こす支持材の化学的または物理的前処理が挙げられる。可能な択一的ストラテジーは、A基担持化合物を、同様にその支持材に結合する別の化合物との混合物にて適用することである。この競合性結合により、支持材の表面上のA基の比率が低減される。
【0038】
準化学量論量のA基に基づく実施形態を、さらに、A基とN基での支持材修飾のために上記の1つ以上の実施形態と組み合わせてもよい。
【0039】
準化学量論量のA基を有する支持体を提供することは、シリカ支持材が使用される場合、後者がシラノール基を有するため特に好都合である。A基は、好ましくはシランまたはシラン混合物によって導入する。支持体の完全な被覆に必要とされるシランは最小量である。この最小量は、支持材の具体的な表面によって規定される。シラン(1種類または複数種)の量が支持体表面の占有に不充分な場合、電荷密度を下げるとよい。したがって、A基の量、結果として核酸結合相の結合強度は、適用されるシランの量によって調整され得る。
【0040】
一実施形態によれば、支持材は、A基を有するシランでコートされたシリカ表面を有し、シランの量は0.1〜50μmol(マイクロモル、本明細書ではumolとも称する)、好ましくは0.1〜10umolである。シランの量を、溶出が比較的低いイオン強度または比較的低い(所望の)塩濃度で可能であるほど大きく低減させることは重要である。これは、例えば、実験によって試験し、クロマトグラムによって確認することができる。したがって、シランの量は、好ましくは、所望の結合特性/溶出特性を得るために、使用される支持材に応じて最適化され得る。
【0041】
核酸結合相は、好ましくは固相である。これは、例えば、A基、結合減弱性のN基、および開始剤分子T(存在する場合)を固相支持材に結合させることにより調製され得る。詳細を以下に説明する。固相を用いると、結合された核酸を試料から除去することが容易になる。したがって、一実施形態によれば、核酸の結合の後に、固相または未結合浮遊物を除去する。
【0042】
該核酸結合基に適した支持体の例は、酸化物材料である。これは、特に、Al2O3、TiO2、ZrO2、Ta2O5、SiO2およびポリケイ酸などの酸化物であり、好ましい支持材はSiO2またはポリケイ酸である。また、ポリスチレンおよびその誘導体、ポリアクリレートおよびポリメタクリレート、ならびにその誘導体、またはポリウレタン、ナイロン、ポリエチレン、ポリプロピレン、ポリブチリデン、ならびにこれらの材料のコポリマーなどの有機ポリマーも好適な支持体である。また、このような核酸結合基を、多糖類、特に、ヒドロゲル、例えば、アガロース、セルロース、デキストラン、セファデックス、セファクリルおよびキトサンに連結させてもよい。さらに、核酸結合基を、無機系支持体(例えば、ガラスなど)または金属表面(例えば、金など)に結合させてもよい。磁性粒子の使用は特に好都合である。核酸結合性のA基および/またはN基は、このような支持体に、直接結合させてもよく、あるいは他の化学分子(例えば、開始剤分子もしくはリンカー)を介して結合させてもよい。また、大きな分子の一部分であってもよく、支持材が結合部位としての適当な官能基を有していない場合、それ自体は既知の様式にて導入され得る。
【0043】
支持材のさらなる実施形態は、非磁性および磁性の粒子、カラム材料、膜、ならびに表面コーティングを含む。また、官能性付与したチューブ、膜、不織布、紙、反応槽(PCR槽など)、「エッペンドルフチューブ」、マルチプレート、チップおよびマイクロアレイなどの支持体も挙げられよう。
【0044】
一実施形態によれば、該核酸結合相は、結合中および溶出中、どちらも正電荷を有する。
【0045】
本発明のさらなる一実施形態は、核酸に、本発明による原理に従って可逆的に結合する可溶性核酸結合相に関する。可溶性ポリマーは、A基とN基を、例えば、交互またはランダムに有するものであり得る(例えば図2B参照)。また、A基の側鎖をN基、特にアルキル原子団(挙げられ得る一例は、窒素上のジイソプロピル原子団である)によって遮蔽することも可能である。支持材に関する記載内容は、相応して可溶性核酸結合相に適用される。
【0046】
一実施形態によれば、核酸結合性のA基はイオン交換体、好ましくはアニオン交換体である。核酸の結合において実証された好ましいA基はアミノ基であり、第1級、第2級および第3級アミノ基が好ましい。これらは、置換されていても非置換であってもよい。また、環状アミン、芳香族アミンまたはアミノ官能性付与された複素環も使用され得る。該アミンは、置換基、例えば、アルキル、アルケニル、アルキニルまたは芳香族置換基を有するものであってもよく、また、該炭化水素鎖は、環状になった閉鎖型であってもよい。また、該炭化水素鎖は、ヘテロ原子(酸素、窒素、イオウもしくはケイ素など)、または分枝構造を有するものであってもよい。記載のように、A基としてのアミノ基は、8〜13、好ましくは9〜13、特に好ましくは10〜12のpKaを有する。
【0047】
A基は、重合または縮合によってオリゴマーまたはポリマーを形成し、したがって、リガンドの形成に特に好適な化合物の一部分であってもよい。かかる鎖形成性化合物の例は、アミノ基を含むアクリレート、例えば、
N−(3−アミノメチル)メタクリルアミド、N−(3−アミノエチル)メタクリルアミド、
N−(3−アミノプロピル)メタクリルアミド、N−(3−アミノイソプロピル)メタクリルアミド、
N,N−ジメチルアクリルアミド、N,N−ジエチルアクリルアミド、N,N−ジイソプロピルアクリルアミド、
N,N−(ジメチルアミノ)エチルアクリルアミド、N,N−(ジメチルアミノ)エチルアクリレート、
N,N−(ジメチルアミノ)エチルメタクリルアミド、N,N−(ジメチルアミノ)エチルメタクリレート、
N,N−(ジメチルアミノ)プロピルアクリルアミド、N,N−(ジメチルアミノ)プロピルアクリレート、
N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート、
N,N−(ジエチルアミノ)エチルアクリルアミド、N,N−(ジエチルアミノ)エチルアクリレート、
N,N−(ジエチルアミノ)エチルメタクリルアミド、N,N−(ジエチルアミノ)エチルメタクリレート、
N,N−(ジエチルアミノ)プロピルアクリルアミド、N,N−(ジエチルアミノ)プロピルアクリレート、
N,N−(ジエチルアミノ)プロピルメタクリルアミド、N,N−(ジエチルアミノ)プロピルメタクリレート、
N,N−(ジイソプロピルアミノ)エチルアクリルアミド、N,N−(ジイソプロピルアミノ)エチルアクリレート、
N,N−(ジイソプロピルアミノ)エチルメタクリルアミド、N,N−(ジイソプロピルアミノ)エチルメタクリレート、
N,N−(ジイソプロピルアミノ)プロピルアクリルアミド、N,N−(ジイソプロピルアミノ)プロピルアクリレート、
N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート、2−(ジメチルアミノ)エチルメタクリレート(DMAEMA)および2−(ジイソプロピルアミノ)エチルメタクリレート
などである。
【0048】
これらのうち、N,N−(ジメチルアミノ)プロピルメタクリレートが特に好ましい。
【0049】
A基は、シラン内、好ましくは反応性シラン内に存在するものであり得る。反応性シランは、加水分解不安定性のSi結合、例えば、Si−NまたはSi−O結合を有する化合物をいう。少なくとも1つのA基を含む反応性シランの例は:
【0050】
【化2】
(式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基であり、
*は、アミノ、アミノメチル、アミノエチル、アミノプロピル、ジメチルアミノ、ジエチルアミノ、ジイソプロピルアミノ、ジプロピルアミノ、ジエタノールアミノ、ジプロパノールアミノ、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、エーテルアミン、ポリエーテルアミン、4−ジイソブチルアミノ−1−ブタン、6−ジプロピルアミノ−1−ヘキサンである)
である。
【0051】
対応する反応性シランが、A基の導入に使用され得る。特に好ましいものは、ジエチルアミノプロピルトリメトキシシラン(DEAPS)、ジメチルアミノプロピルトリメトキシシランおよびN,N−ジイソプロピルアミノプロピルトリメトキシシランである。
【0052】
本発明の一実施形態によれば、核酸は、2〜8、好ましくは4〜7.5のpHで結合させる。これは、結合中、したがって試料中のpHをいう。したがって、核酸結合相の設計によるが、本発明による方法はまた、非常に穏やかな条件下で、実質的に中性の範囲で行なわれ得る。核酸結合相のプロトン化性基が8〜13、好ましくは9〜13、特に好ましくは10〜12のpKaを有することにより、該相は、比較的中性のpHであっても、有効な核酸結合を可能にするための充分な正電荷を有する。その結果、結合は、所望により非常に穏やかな条件下で行なわれ得る。
【0053】
出発材料によるが、核酸を放出させるための少なくとも1回の通常の溶解工程が結合させる工程の前に行なわれ得る。
【0054】
核酸の溶出は、本発明の方法の別の重要な工程である。記載のように、核酸は、結合pHより高いpHで放出させる。この結果として、溶出中、A基が有する正電荷が少なくなり、これは核酸の放出に有利である。慣用的なアニオン交換体と比べ、A基の低比率および/または結合減弱性のN基の存在により、支持材に対する核酸の結合の低減がもたらされる。この結果として、上記のように、溶出も低塩濃度で行なうことが可能である。
【0055】
溶出pHは、好ましくはA基のpKより低い。使用される核酸結合性のA基または核酸結合相によるが、溶出は、好ましくは8〜11、8〜10のpHで、好ましくは8.0〜9、特に好ましくは8.5〜9のpHで行なわれる。しかしながら、原理的には、より高いpH値を使用することも可能である。しかしながら、低塩濃度にもかかわらず核酸が放出され得、さらに条件が穏やかであるため、好ましい低pH値により特に好都合な結果が得られる。
【0056】
単離された核酸の処理過程を、引き続いて直接、溶出バッファー中で行なうことを可能にするため、後者は、記載のように、好ましくは低塩濃度を有するものである。これは、本発明による核酸結合相の設計によって可能になる。したがって、一実施形態によれば、塩濃度は1mM〜1000mM、特に好ましくは1mM〜200mM、1mM〜250mM、または1mM〜100mMである。好適な塩は、アルカリ金属およびアルカリ土類金属またはアンモニアの塩化物、無機酸の他の塩、酢酸塩、ホウ酸塩、ならびにTris、Bis−Trisおよび有機バッファー(例えば、MIS、CHAPS、HEPESなど)などの化合物などであり得る。同じものが結合バッファーに適用される。さらに、溶出のための好適な物質は先行技術において知られている。塩濃度は結合させる工程と溶出させる工程で変更しないか、または溶出中でわずかに上昇させる。しかしながら、好ましくは、該濃度は、後続する反応を障害するような様式では増大させない。さらに、結合中と溶出中の温度は同じであってもよく、あるいは溶出中で上昇させる。
【0057】
精製を助長するため、好ましくは、核酸の結合後、溶出の前に少なくとも1回の洗浄工程を行なう。低塩濃度を有する水溶液での洗浄が好ましいが、水での洗浄も好ましい。洗浄バッファー中に存在させる塩は、好ましくは1mM〜1000mM、特に好ましくは1mM〜200mM、1mM〜250mM、または1mM〜100mMの濃度である。バッファーは、有機化合物(糖質など)、好ましくは、有機溶媒(例えば、アルコール、ポリオール、ポリエチレングリコール、エーテル、ポリエーテル、ジメチルスルホキシド、アセトンまたはアセトニトリルなど)を含むものであり得る。しかしながら、下流での適用が障害されないようにするため、洗浄バッファーは、干渉量の対応有機成分を有しないものであるのがよい。
【0058】
使用され得る結合減弱性のN基は、電荷中性基、例えば、ヒドロキシル基、ジオール基、トリオール基、糖類、エポキシド基、C1〜C6アルキル、アルケンもしくはアルキン基、ポリオール基、エーテル、ポリエーテル、ハライドまたはイミドなどである。親水性のN基の使用により、水性バッファーでのイオン交換体の継続的に良好なぬれ性が確保される。N基は、重合または縮合によってオリゴマーまたはポリマーを形成する化合物の一部分であってもよい。N基を導入し得る化合物の例は、アクリレート、例えば、アクリル酸ブチル、アクリル酸プロピル、アクリル酸エチル、アクリル酸メチル、メタクリル酸グリシジル、メタクリル酸ヒドロキシエチル(HEMA)、メタクリル酸グリシドキシプロピル、グリセロールモノメタクリレート(異性体混合物)、グリコールモノメタクリレートおよびN−アクリルオキシスクシンイミドなどである。
【0059】
また、N基はシラン内、好ましくは反応性シラン内に存在するものであってもよい。N基を含む反応性シランの例は:
【0060】
【化3】
(式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基であり;
*は、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、エタンジオール、プロパンジオール、プロパントリオール、ブタントリオール、3−グリシドオキシプロピル、エチルグリシジルエーテル、アルキル原子団、特に、C1〜C4アルキル原子団、ハライドまたは水素である)
である。
【0061】
一実施形態によれば、支持材は、少なくとも一部が、A基および/またはN基を有する化合物で官能性付与された開始剤基Tを有する。好適な例は、本明細書において以下に詳細に説明する。
【0062】
さらに、本発明は、対応する核酸結合支持材の合成またはその調製方法に関する。この目的のためには、種々の実施形態がある。
【0063】
代替法(A):この方法は、少なくとも2種類の異なるリガンドIとIIの混合物を用いて支持材に官能性付与することを含み、リガンドIは少なくとも1つのA基を有し、リガンドIIは、少なくとも1つのN基を有するか、または少なくとも1つのN基である。この実施形態に関する詳細は、上記において既に説明した。上記の開示内容を参照されたい。
【0064】
代替法(B):この方法は、第1の工程において、開始剤分子Tと、少なくとも1つのA基を有するリガンドとの混合物を用いて支持材に官能性付与することを含む。ここで、A基を有する化合物は、さらなる分子は結合され得ないような様式で選択されたもの、または修飾されたものである。第2の工程では、次いで、少なくとも1つのN基を有し、開始剤分子Tに結合する化合物が添加される。
【0065】
代替法(C):核酸結合リガンドでの支持材の官能性付与。前記のリガンドのそれぞれは、1つ以上のA基と1つ以上のN基とを有する。詳細は、本発明による方法に関して上記において説明している。第1の工程において、開始剤分子Tを支持材に結合させ、該分子を、少なくとも1つのA基および/または少なくとも1つのN基を有する化合物を添加し、前記化合物を開始剤分子Tに結合させてコポリマー鎖を伸長させるための開始点として使用することが好ましい。該ポリマー鎖は、少なくとも1つのA基および/または少なくとも1つのN基を有する化合物をさらに添加することにより伸長される。
【0066】
これに関連して、該重合工程はまた、A基とN基の両方をオリゴマーによって組み込み、したがってブロックコポリマーが得られるような様式で制御してもよい。本発明に従って使用され得る重合反応の制御の一例は、例えば、「原子移動ラジカル重合」(ATRP)である。ATRPは、遷移金属錯体を添加すること、およびオルガノハライドを伴う原子転移プロセスとの組合せにより、フリーラジカルの濃度を、不均衡または組換えなどの連鎖停止反応が大幅に抑制されるような程度まで低減させることを特徴とする。
【0067】
代替法(D):A基が、少なくとも1つのN基を有する化合物によって立体遮蔽されるA基とN基での支持材の官能性付与。この変形例の一実施形態によれば、支持材は、第1の工程において、開始剤基Tで官能性付与される。この後、少なくとも1つのA基を有する化合物を添加し、これを開始剤分子Tに結合させる。さらなる工程において、少なくとも1つのN基を有する少なくとも1種類の化合物が結合することにより、A基が遮蔽される。N基は、その3次元構造によってA基を立体遮蔽するオリゴマー/ポリマー(上記参照)であることが好ましい。詳細は、本発明による方法に関して上記において説明している。
【0068】
代替法(E):この実施形態によれば、支持材は、準化学量論量のA基で官能性付与される。この変形例は、上記において詳細に説明した。上記の開示内容を参照されたい。
【0069】
また、代替法A〜Eは任意の組合せで使用され得る。核酸結合性のA基、N基および開始剤分子Tに関する詳細は、上記において詳細に説明しており、また、本明細書に示した支持体の修飾方法に関しても適用され、その場合に使用される成分の特徴を示す。上記の開示内容を参照されたい。
【0070】
A基とN基で支持材に官能性付与する方法の一実施形態によれば、前記の基は、単官能性、二官能性もしくは三官能性の反応性シランによって、または異なる官能基を有する少なくとも2種類の反応性シランの混合物によって導入される。使用され得る反応性シランは、例えば、アミノシラン、ジシラザン、クロロシランまたはアルコキシシランである。N基に対するA基の比率は1%〜99%、1〜50%、好ましくは1%〜25%である。
【0071】
三官能性および二官能性の反応性シランは、支持体上で、重縮合によって架橋された厚い層を形成する傾向にある。対照的に、単官能性の反応性シランは、例えば、支持材のシラノール基と反応してシロキサン(Si−O−Si)結合を形成し、支持体上に、むしろ単分子層をもたらす。支持材を反応性シランと、気相中または溶媒中での懸濁状態で反応させてもよく、後者においては、反応性シランの化学的性質に応じて有機溶媒、好ましくは水性溶媒を使用することが可能である。
【0072】
好適で好ましい支持材は、上記において詳細に説明した。該方法の一実施形態によれば、支持材を、まず開始剤分子Tで修飾した後、次の工程で、A基および/またはN基をモノマーの形態で導入する。開始剤分子Tは、例えば、(重)縮合または重合、特に、フリーラジカル重合(特に、ATRP(原子移動ラジカル重合)など)によって導入されるさらなる化合物の結合部位として使用してもよい。開始剤分子Tの例は:
【0073】
【化4】
【0074】
【化5】
(式中、Xは、ハロゲン、特にCl、Br、Iであり、
Y1、Y2、Y3またはY4は、互いに独立して、R、OR、OHまたはHであり、
Rは、C1〜C3アルキルである)
である。
【0075】
2−(クロロメチル)アリルトリメトキシシランまたは[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシラン(BPDS)を開始剤分子として使用することが好ましい。
【0076】
本発明による方法に従ってA基および/またはN基を導入し得る反応性シランの例を以下に示す:
【0077】
【化6】
式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基、特に、メチル、エチル、プロピル、イソプロピルであり;
*は、アミノ、アミノメチル、アミノエチル、アミノプロピル、ジメチルアミノ、ジエチルアミノ、ジイソプロピルアミノ、ジプロピルアミノ、ジエタノールアミノ、ジプロパノールアミノ、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、エーテルアミン、ポリエーテルアミン、4−ジイソブチルアミノ−1−ブタン、6−ジプロピルアミノ−1−ヘキサン、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、エタンジオール、プロパンジオール、プロパントリオール、ブタントリオール、3−グリシドオキシプロピル、エチルグリシジルエーテル、アルキル原子団、特に、C1〜C4アルキル原子団、特に、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、ハライドまたは水素である。
【0078】
詳細に説明したシラン処理と準化学量論でのシラン処理の混合型の方法に加え、支持材上のA基の密度を規定の様式で調整できる可能性が他にもある。記載のように、例えば、支持材上で「表面開始グラフト重合(grafting−from)」プロセスを行なうことが可能である。これは、例えば、上記に記載した原子移動ラジカル重合(ATRP)法によって行なわれ得る。この方法は、まず、開始剤基T、N基および/またはA基を支持体に、シラン処理によって適用することを含む。開始剤基Tは、この基によって表面開始グラフト重合プロセスが開始されるため(連鎖開始)、このプロセスには重要である。
【0079】
例えば、ハライド含有シランを使用し、ATRP(原子移動ラジカル重合)によって、ホモポリマー、コポリマーおよびブロックコポリマーを伸長させることができる。ATRPは、リビング/制御フリーラジカル重合(LFRP)の特別な形態であり、これは、遷移金属錯体を添加すること、およびオルガノハライドを伴う原子転移プロセスとの組合せにより、フリーラジカルの濃度を、不均衡または組換えなどの連鎖停止反応が大幅に抑制されるような程度まで低減させることを含む。この方法により、支持体表面の電荷密度を、モノマーおよびモノマー混合物の使用によって特異的に調整し、それにより、核酸の結合強度に影響を及ぼすことが可能になる。A基および/またはN基は、モノマーの適切な選択によって特異的に適用され得る。
【0080】
このようにして作製される核酸結合支持材は、特に、本発明による精製方法に使用され得る。
【0081】
さらに、本発明は、核酸を精製するための本発明による核酸結合相の使用に関する。本発明によれば、核酸は、特に、DNAおよびRNA、特にゲノムDNA、プラスミドDNA、ならびにPCR断片、cDNA、miRNA、siRNA、また、オリゴヌクレオチドおよび修飾核酸(例えば、PMAまたはLMAなど)を含む。また、ウイルスもしくは細菌のRNAおよびDNA、またはヒト、動物もしくは植物源由来の核酸を精製することも可能である。さらに、DNA/RNAハイブリッドおよび修飾核酸も本発明による精製に適している。
【0082】
また、本発明は、少なくとも1つのプロトン化性基を有し、かつ8〜13、好ましくは9〜13、特に好ましくは10〜12のpKaを有する核酸結合性のA基を有する本発明による核酸結合支持材を有することを特徴とする、核酸を精製するためのキットを提供する。本発明による支持材およびその官能基に関する詳細は上記に記載している;上記の記載内容を参照されたい。
【0083】
また、キットは、例えば、精製方法に関して上記に記載の結合バッファー、洗浄バッファーおよび/または溶出バッファーを有するものであってもよい。また、溶解バッファーおよび中和バッファーを有するものであってもよい。
【0084】
一実施形態によれば、キットは、好ましくは以下の特徴:
(a)1〜13のpH;および/または
(b)1mM〜1000mM、特に好ましくは1mM〜200mM、1mM〜250mM、または1mM〜100mMの塩濃度
の少なくとも1つを有する結合バッファーを有する。
【0085】
対応する特徴の利点は、方法に関して上記に説明しており、上記の開示内容を参照されたい。
【0086】
また、一実施形態によれば、キットは、好ましくは以下の特徴:
(a)2〜7、好ましくは4〜7のpH;
および/または
(b)1mM〜1000mM、特に好ましくは1mM〜800mM、1mM〜600mMの塩濃度;および/または
(c)水、生物学的バッファー、有機バッファー、特に、Tris、Tris−Bis、MIS、MOPS、CHAPSおよびHEPESからなる群より選択されること
の少なくとも1つを有する洗浄バッファーを有する。
【0087】
また、さらなる実施形態によれば、キットは、好ましくは以下の特徴:
(a)8〜10、好ましくは8〜9のpH
および/または
(b)1mM〜1000mM、特に好ましくは1mM〜200mM、1mM〜250mM、または1mM〜100mMの塩濃度;および/または
(c)水、生物学的バッファー、有機バッファー、特に、Tris、Tris−Bis、MIS、MOPS、CHAPSおよびHEPESからなる群より選択されること
の少なくとも1つを有する溶出バッファーを有する。
【0088】
核酸結合相および溶出条件に関する詳細は上記に記載しており、本発明によるキットに関しても適用され、その場合に使用される成分/バッファーの特徴を示す。上記の開示内容を参照されたい。
【0089】
本発明によるキットは、特に、本発明による方法の枠内で適用され得る。本発明の方法、キットおよび核酸結合固相は、特に、分子生物学、分子診断、法医学、食品分析、および応用試験の分野において使用され得る。本発明によるキットの適用により、精製核酸を、「下流」適用、特にPCR反応において、さらに直接処理することが可能になる。
【0090】
記載のパラメータ、特に、溶出促進性結合減弱性のN基と核酸結合基の低減を選択/組み合わせることにより、核酸結合相のpHが溶出条件に関して最適化され得る。相応して、核酸結合相の溶出プロフィール、特に、塩濃度と溶出pHが制御または調整され得る。
【0091】
本発明による系によって精製され得る核酸は、体液中(血液、尿、大便、唾液など)、生体源中、例えば、組織、細胞(特に、動物細胞、ヒト細胞、植物細胞、細菌細胞など)、器官(肝臓、腎臓または肺など)に存在するものであり得、また、核酸を、支持材(綿棒など)、PapSmears、および安定化媒体(PreServCytもしくはSurepathなど)、または他の液状物(例えば、搾汁、水性試料もしくは食品一般など)に由来する他のものから入手することも可能である。また、核酸は、植物材料、細菌溶解物、パラフィン包埋組織、水溶液またはゲルから得られるものであってもよい。
【0092】
溶出された核酸は、好ましくは、さらなる処理過程が直接行なわれるものであり得、したがって、例えば、PCR、RT−PCR、制限消化または転写に使用され得るものである。溶出バッファーが上記のように設計されている限り、好ましくは低塩濃度を有する限り、さらなる精製は必要とされない。
【実施例】
【0093】
本発明を、いくつかの実施例に基づいて以下に説明する。前記実施例は、本発明の限定ではなく、好ましい実施形態である。また、参考文献はすべて、本開示の主題に関して本明細書に挙げたものである。
【0094】
実施例
本実験では、プラスミドDNAを核酸モデル系として使用した。
【0095】
A) 隣接するA基とN基での支持材の修飾
A.1) 1:1のモル比2−(クロロメチル)アリルトリメトキシシランおよびDEAPSでのシリカゲルのシラン処理(AAK01−10)
材料
支持材:ほぼ150nmの孔径を有するシリカ/シリカゲル。比表面積は、ほぼ25m2/gである。
【0096】
コーティング試薬:2−(クロロメチル)アリルトリメトキシシラン、DEAPS;QSP1バッファー(酸性酢酸バッファー)。
【0097】
調製プロトコル
三つ口フラスコに、70mlの水、2.5mlのQSP1バッファー(QIAGEN)、322μlの2−(クロロメチル)アリルトリメトキシシランおよび413μlのDEAPSを仕込み(pHは5.3となった)、次いで、18gのシリカゲルを添加した。混合物を攪拌しながら95℃まで20分間以内で加熱し、この温度でさらに4時間攪拌し、次いで、攪拌しながら1時間冷却した。シリカゲルをP3ガラスフリットによって除去し、逐次、32.3gのTris/NaClバッファー、30mlの脱イオン水で2回、35mlのメタノールで2回、最後に30mlのメタノールで洗浄した。最終支持材AAK01−10を125℃で一晩乾燥させた。
【0098】
比較のため、同じ支持材を、A基を有する化合物DEAPSのみで修飾した。三つ口フラスコに、70mlの水、2.5mlのQSP1バッファー、825μlのDEAPSを仕込み(pHは5.5となった)、次いで、18gのシリカゲルを添加した。混合物を攪拌しながら95℃まで20分間以内で加熱し、この温度でさらに4時間攪拌し、次いで、攪拌しながら1時間冷却した。シリカゲルをP3ガラスフリットによって除去し、逐次、32.3gのTris/NaClバッファー、30mlの脱イオン水で2回、35mlのメタノールで2回、最後に30mlのメタノールでで洗浄した。最終支持材AAK01−30を125℃で一晩乾燥させた。
【0099】
溶出点の測定を伴う修飾支持材AAK01−10を用いたプラスミドDNAの精製
結合は、50mM Tris−HCl(pH7.0)、15%エタノールのバッファー中で行なった。溶出は、バッファーB(50mM Tris−HCl(pH7.0),15%エタノール,2M NaCl)の0%〜100%の段階的勾配の23分間にわたる流動、およびUV分光法による連続的な溶出DNAの量の測定を伴った。有意な量のDNAが最初に溶出されたNaCl濃度を溶出点として記録した。ここでは、N基の導入により、溶出点が有意に低下することが示された。
【0100】
プロトン化性のA基(DEAPS)のみでコートした支持材(AAK01−30)は、pH7.0で1600mM NaClに溶出点を示す。したがって、核酸の溶出には、かなり高い塩濃度が必要とされる。混合型A/N修飾シリカゲルAAK01−10の溶出点は、約700mM NaClである。したがって、溶出に必要とされるイオン強度は50%より大きく低下した。
【0101】
A.2)メタクリル酸ヒドロキシエチル(HEMA)でのA/N修飾シリカゲルシラン処理(AAK01−11)
材料
出発材料:修飾シリカゲルAAK01−10(プロトコルA.1を参照のこと)
コーティング試薬:メタクリル酸ヒドロキシエチル(HEMA)
調製プロトコル
HEMAオリゴマーを、Cu(I)触媒型原子移動ラジカル重合(ATRP)の補助により、事前に結合させたクロロシラン上で合成する。
【0102】
反応フラスコ内で、先にAl2O3によって活性化させておいた20mlのHEMAと、20mlの脱イオン水を、アルゴンで30分間リンス処理し、次いで、68mgのCuCl2、46mgのCuBr2および313mgのビスピリジンと混合し、混合後、10gのAAK01−20を添加する。混合物を室温で4時間攪拌し、その直後、P3ガラスフリットによって吸引しながら濾別する。ガラスフリット上の物質を、まず、100mM NaCl/100mM EDTAバッファーで数回洗浄し、次いで、VE水で数回洗浄し、8mlのメタノールで1回、7mlのメタノールで2回洗浄する。最終支持材AAK01−11を60℃で14時間乾燥させた。
【0103】
比較のため、DEAPS修飾支持材AAK01−30をHEMAと反応させ、同様に処理した。この反応の最終生成物をAAK01−31と命名した。官能基としてDEAPSは第3級アミンのみを有し、重合性アリル基は有しないため、HEMAカップリングはないはずである。
【0104】
溶出点の測定を伴う修飾支持材AAK01−11を用いたプラスミドDNAの精製
実験は、A.1に記載のようにして行なった。プロトン化性基(DEAPS)のみでコートした支持材AAK01−31は、pH7.0で1500mM NaClに溶出点を示す。したがって、核酸の溶出には、かなり高い塩濃度が必要とされる。HEMA修飾シリカゲルAK01−11の溶出点は約400mM NaClである。したがって、溶出に必要とされるイオン強度は、ほぼ75%低下した。
【0105】
A.3)種々のモル比のシランでのシリカビーズのシラン処理
a)FFシリカビーズ
以下の実験設定を選択した。使用した支持材はFFシリカビーズであった。
【0106】
【表1】
*:溶出バッファーの組成:20mM 塩化カリウム;50mM Tris水溶液,pHXに調整;
**:QNバッファーの組成:1600mM 塩化ナトリウム;50mM Tris15%エタノール水溶液,pH7に調整
結果を図6に示す。
b)MagAttract Beads G(QIAGEN)
以下の実験設定を選択した。使用した支持材は、磁性シリカビーズであるMagAttract Beads G(QIAGEN製)であった。
【0107】
【表2】
*:溶出バッファーの組成:20mM 塩化カリウム;水中50mM Tris,pHXに調整;
**:QNバッファーの組成:1600mM 塩化ナトリウム;50mM Tris15%エタノール水溶液,pH7に調整.
結果を図7に示す。
【0108】
B) 開始剤分子での支持材の修飾
本発明によるランダムおよび/またはブロック(コ)ポリマーは、まず、開始剤基Tを適切な支持材に、本明細書において以下に一例として示すようにして適用することにより調製する。次いで、前記開始剤基を、後続の工程においてA基および/またはN基のリガンド/ポリマー鎖を適用する開始点として使用する。
【0109】
B.1)シリカゲル上での[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシランのシラン処理
材料
支持材:シリカゲル(Fuji MB 1500−40/75/ロット:HT70594)
コーティング試薬:[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシラン(BPDS)
シランの量:800μmol/gの支持材
調製プロトコル
還流冷却器および排水器を備えた250ml容KPG攪拌器(乾燥)に、まず、10.0gの支持材、200mlの乾燥シクロヘキサンおよび2490mgの[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシランを仕込んだ。
【0110】
混合物を、攪拌しながら(100l/分)85℃(油浴温度)で一晩(16時間)反応させた。次いで油浴を除き、懸濁液を、攪拌しながら1/2時間以内で冷却した。修飾支持材/シリカゲル開始剤(KI04)をP3フリットによって除去し、その上面を7×20mlのヘキサンで洗浄した。次いで、修飾支持材を、40℃の真空乾燥キャビネット内で、一晩(ほぼ12時間)乾燥させた。
【0111】
5つのロットのシリカゲル開始剤(KI 04 a〜e)を、上記の調製プロトコルに従って調製し、後続の工程で本発明の弱いイオン交換体を調製するための開始点として使用した。
【0112】
C)ATRPによる弱いイオン交換体の調製
本発明の弱いイオン交換体は、A基および/またはN基のランダムポリマー鎖を、出発材料である開始剤基T(KI04)担持修飾支持材上で伸長させることにより調製した。
【0113】
C.1)B.1に記載の修飾支持材/シリカゲル開始剤上へのA基/Nのランダムポリマーのグラフト化
この実験の組の出発材料は、開始剤としてBPDSを有するシリカゲル開始剤(KI04d)とした。
【0114】
このために、モノマーDMAEMA(2−ジメチルアミノ)エチルメタクリレート/A基)およびHEMA(メタクリル酸ヒドロキシエチル/N基)を、互いに以下の比で適用した:
DMAEMA:HEMA
100:0 T 16.0mmol:0.0mmol(KI04d−01)
50:50 T 8.0mmol:8.0mmol(KI04d−03)
30:70 T 4.8mmol:11.2mmol(KI04d−04)
20:80 T 3.2mmol:12.8mmol(KI04d−05)
10:90 T 1.6mmol:14.4mmol(KI04d−07)
材料
シリカゲル開始剤:KI04d
モノマー:DMAEMAおよびHEMA
リガンド:0.96mmolの2,2’−ビピリジル(bdy)
銅塩:0.48mmolの臭化銅I(精製済)
溶媒:36mlのジメチルホルムアミド(DMF)
開始剤:Cu(I):リガンド比=1:4:8
開始剤:モノマー(1種類または複数種)比=1:133
調製プロトコル
KPG攪拌器(ガラス製ガイドスリーブ)、アルゴン接続口および計泡器(またはダイヤフラムポンプへの接続口)を備えた100ml容三つ口フラスコに、まず、150mgのbdyと68.8mgの臭化銅Iを仕込んだ。
【0115】
装置内を、ダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、空気中の酸素が入るのを抑制するために、緩徐なアルゴン流を装置内に永続的に通した。
【0116】
並行して、36mlのDMF(リガンド)およびモノマーを脱気した。
【0117】
次いで、これらの液体を、調製物に応じて以下の初期重量比を有するモノマーと共に、排水器を備えた二つ口フラスコ内に導入した。
【0118】
KI04d−01=2700μlのDMAEMA:0.0μlのHEMA
KI04d−03=1350μlのDMAEMA:972μlのHEMA
KI04d−04=810μlのDMAEMA:1360μlのHEMA
KI04d−05=540μlのDMAEMA:1555μlのHEMA
KI04d−07=270μlのDMAEMA:1750μlのHEMA
リガンドとモノマー(1種類または複数種)の混合物を、まず、超音波浴内で5分間処理した後、ガス空間を高真空下で5分間真空にした。
【0119】
リガンド−モノマー混合物をピペット使用により、先で導入したbdy/臭化銅Iに添加した。装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。この後、装置内に永続的に緩徐なアルゴン流を通した。
【0120】
銅リガンド錯体を、攪拌しながら(200l/分)15分間以内で溶解させた。この後、1.2gのKI 04dシリカゲル開始剤を添加した。
【0121】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、装置内に緩徐なアルゴン流を永続的に通した。
【0122】
混合物を40℃(油浴温度)で4時間攪拌した(200l/分)。
【0123】
上面に該基が伸長した支持材(以下、本明細書において単にシリカゲルと記載する)を、P3フリットによって直ちに取り出し、続いて、逐次、
5×8mlのDMF
5×8mlのTHF
5×8mlの0.1M EDTA/酢酸バッファー(pH3.6)(氷酢酸を含む0.2M酢酸ナトリウム)
2×8mlのTris/NaClバッファー(pH7.0)
3×8mlの水および
3×8mlのメタノール
で洗浄した。
【0124】
次いで、シリカゲルを、重量が一定になるまで、真空乾燥キャビネット内で40℃にて乾燥させた。
【0125】
収率:
KI04d−01=1.31g
KI04d−03=1.20g
KI04d−04=1.23g
KI04d−05=1.29g
KI04d−07=1.21g
得られたアニオン交換体(修飾支持材、すなわちシリカゲル)および出発材料(シリカゲル開始剤)を、そのpDNA結合能に関して試験し(これは、本明細書において以下により詳細に説明する)、対応する溶出プロフィールを記録した。
【0126】
また、アニオン交換体をCHNの含有量について試験し、このポリマー鎖長から、Nに対するAの比を求めた。使用したモノマーの(モル)質量比は、得られるシリカゲルに反映されることを示すことができた。
【0127】
溶出点の測定を伴う、修飾支持材KI04d−01〜KI04d−07を用いたプラスミドDNAの精製
【0128】
【表3】
手順
単独の測定を、シリカゲル毎、および2種類の溶出バッファーのいずれかについて行なった。
【0129】
このために、各場合において、50.00mg(±5mg)のシリカゲルを、エッペンドルフの2ml容Safe Lock反応チューブ(以下、本明細書においてエッペンドルフチューブという)内に計り入れ、DNA溶液のため、計量した各シリカゲルに対して100μgのpcmvbを結合バッファーと混合して1mlにした。
【0130】
続いて、1mlのDNA溶液を、計量したシリカゲルの入った各エッペンドルフチューブに添加し、エッペンドルフチューブを密封し、短時間ボルテックスした。
【0131】
次いで、エッペンドルフチューブを、回転式(end−over−end)振盪機内に中速度で5分間載置した。振盪後、エッペンドルフチューブを短時間遠心分離した。
【0132】
次いで、シリカゲル−DNA懸濁液を、ピペットにより、切断先端を用いて、ボトムフリットを有する調製Tip 20ブロック(以下、本明細書において、Tipという)内に移した。Tipの下方には、上清み収集のための2ml容エッペンドルフチューブを準備した。
【0133】
エッペンドルフチューブ内に生じ得る残留物質を捕捉するため、各場合において、該チューブを0.9mlの結合バッファーでリンス処理し、該バッファーは、対応するTipにも同様に適用した。
【0134】
液体内部からの滴下が停止したら、残留液状物をすべて内部で押圧し、新たなエッペンドルフ収集チューブをTipカラムの下方に配置した。
【0135】
続いて、1mlの洗浄バッファーを、洗浄のためにピペットで各Tip内に移した。液状物が内部から滴下しなくなったら、残留液状物をすべて内部で押圧し、エッペンドルフ収集チューブ(洗浄液が入っている)を脇に置き、新たなエッペンドルフ収集チューブをTipカラムの下方に配置した。
【0136】
続いて、1mlの適切な溶出バッファーをピペットで各Tip内に移した。この場合も、液状物が内部から滴下しなくなったら、残留液状物をすべて内部で押圧し、エッペンドルフ収集チューブ(溶出液が入っている)を脇に置き、新たなエッペンドルフ収集チューブをTipカラムの下方に配置した。
【0137】
残留結合DNAを分離させるため、1mlのTris−NaClバッファーをピペットで各Tip内に移した。液状物が内部から滴下しなくなったら、残留液状物をすべて内部で押圧し、エッペンドルフ収集チューブ(Tris−NaCl溶液が入っている)を脇に置き、Tipカラムを処分した。
【0138】
DNA濃度をSpectraMaxによって測定するため、各場合において、採取した100μlの溶液をUVプレートに移した。
【0139】
上清みのブランクを、10μlのEBバッファーと180μlの結合バッファーを混合することにより調製した。100μlのこの溶液をUVプレート内に導入した。使用したDNAの量の参照として、100μlのDNA溶液を90μlの結合バッファーと混合し、100μlのこの溶液をUVプレート内に導入した。
【0140】
溶出液に対して使用したブランクは、100μlの対応する溶出バッファーとした。
【0141】
100μlのTris−NaClバッファーを、Tris−NaCl溶液ブランクとして使用した。
【0142】
有意な量のDNAが最初に溶出されたNaCl濃度を溶出点として記録した。
【0143】
【表4】
この場合も、結果(上記の表に示し、図8に示したもの)により、N基の導入により溶出点が有意に低下することが示された。
【0144】
pH8.5では、A基(DMAEMA)のみでコートした支持材/シリカゲル(KI04d−01)での溶出点は、1032mM NaClであった。したがって、核酸を溶出させるためには、かなり高い塩濃度が必要であった。混合型A/N修飾シリカゲルKI04d−05の溶出点は、約580mM NaClであった。したがって、溶出に必要とされるイオン強度はほぼ50%低下した。
【0145】
C.2)B.1に記載の修飾支持材/シリカゲル開始剤上へのA基/N基のブロックポリマーのグラフティング
この実験の組の出発材料は、BPDSを開始剤とするシリカゲル開始剤(KI04e)とした。
【0146】
この後、A基の少なくとも1種類のモノマーのアミノポリマー鎖をグラフティングする第1の工程を行なった。そして、この後、このようにして調製したシリカゲルに、N基の少なくとも1種類のモノマー(好ましくは、HEMA)のポリマー鎖を適用する第2の工程を行なった。
【0147】
C.2.1)工程1
材料
シリカゲル開始剤:KI04e
モノマー:DMAEMA
リガンド:7.2mmolの2,2’−ビピリジル(bdy)
銅塩:3.6mmolの臭化銅I(精製済)
溶媒:270mlのジメチルホルムアミド(DMF)
開始剤:Cu(I):リガンド比=1:4:8
開始剤:モノマー比=1:133
調製プロトコル
KPG攪拌器(ガラス製ガイドスリーブ)、アルゴン接続口および計泡器(またはダイヤフラムポンプへの接続口)を備えた500ml容三つ口フラスコに、まず、1125mgのbdyと516mgの臭化銅Iを仕込んだ。
【0148】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、装置内に緩徐なアルゴン流を永続的に通した。
【0149】
並行して、270mlのDMFと20.25mlのDMAEMAを脱気した。この目的のため、この2種類の液体を、排水器を備えた二つ口フラスコ内に導入した。混合物を、まず、超音波浴内で5分間処理し、この後、ガス空間を高真空下で5分間真空にした。
【0150】
DMF−DMAEMA混合物を、ピペット使用によりbdy/臭化銅Iに添加した。
【0151】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。この後、装置内に永続的に緩徐なアルゴン流を通した。
【0152】
銅リガンド錯体を、攪拌しながら(200l/分)15分間以内で溶解させた。この後、9gのKI04eシリカゲル開始剤を添加した。
【0153】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、装置内に緩徐なアルゴン流を永続的に通した。
【0154】
混合物を40℃(油浴温度)で4時間攪拌した(200l/分)。
【0155】
上面にA基が伸長したKI04e−01シリカゲルを、P3フリットによって直ちに取り出し、続いて、逐次、
5×60mlのDMF
5×60mlのTHF
5×60mlの0.1 M EDTA/酢酸バッファー(pH3.6)(氷酢酸を含む0.2M酢酸ナトリウム)
2×60mlのTris/NaClバッファー(pH7.0)および
3×60mlの水
で洗浄した。
【0156】
次いで、KI04e−01シリカゲルを、重量が一定になるまで、真空乾燥キャビネット内で40℃にて乾燥させた。
【0157】
C.2.2)工程2
材料
シリカゲル開始剤:KI04e−01
モノマー:HEMA
リガンド:0.96mmolの2,2’−ビピリジル(bdy)
銅塩:0.48mmolの臭化銅I(精製済)
溶媒:36mlのジメチルホルムアミド(DMF)
開始剤:Cu(I):リガンド比=1:4:8
開始剤:モノマー比=1:200
調製プロトコル
KPG攪拌器(ガラス製ガイドスリーブ)、アルゴン接続口および計泡器(またはダイヤフラムポンプへの接続口)を備えた100ml容三つ口フラスコに、まず、150mgのbdyと68.8mgの臭化銅Iを仕込んだ。
【0158】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、空気中の酸素が入るのを抑制するために、装置内に緩徐なアルゴン流を永続的に通した。
【0159】
並行して、36mlのDMFと2916μlのHEMAを脱気した。この目的のため、この2種類の液体を、排水器を備えた二つ口フラスコ内に導入した。混合物を、まず、超音波浴内で5分間処理し、この後、ガス空間を高真空下で5分間真空にした。
【0160】
DMF−HEMA混合物を、ピペット使用によりbdy/臭化銅Iに添加した。
【0161】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。この後、装置内に永続的に緩徐なアルゴン流を通した。
【0162】
銅リガンド錯体を、攪拌しながら(200l/分)15分間以内で溶解させた。この後、1.2gのKI04e−01シリカゲル開始剤を添加した。
【0163】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、装置内に緩徐なアルゴン流を永続的に通した。
【0164】
混合物を、40℃(油浴温度)で6時間攪拌した(200l/分)。
【0165】
上面にA基が伸長したKI04e−01−01シリカゲルを、P3フリットによって直ちに取り出し、続いて、逐次、
5×8mlのDMF
5×8mlのTHF
5×8mlの0.1 M EDTA/酢酸バッファー(pH3.6)(氷酢酸を含む0.2M酢酸ナトリウム)
2×8mlのTris/NaClバッファー(pH7.0)
3×8mlの水および
3×8mlのメタノール
で洗浄した。
【0166】
次いで、KI04e−01−01シリカゲルを、重量が一定になるまで、真空乾燥キャビネット内で40℃にて乾燥させた。
【0167】
このシリカゲルで行なった溶出実験により、N基モノマーのグラフトポリマー鎖もまた、核酸(1つまたは複数)の立体遮蔽に有意に寄与し得、それにより、前記核酸(1つまたは複数)の結合強度が有意に低下し得ることが実証され得る。
【技術分野】
【0001】
本発明は、核酸結合支持材を用いて核酸を精製するための方法およびキットに関する。また、適切な支持材を精製する好適な方法を記載する。
【背景技術】
【0002】
核酸を精製および単離する種々の方法、例えば、フェノールクロロホルムの使用、塩析法、イオン交換体およびシリカ粒子の使用が先行技術において開示されている。
【0003】
既知の核酸精製方法の一例は、「電荷スイッチ切り替え(charge−switch)法」である。これは、主に弱塩基性のポリマー、例えば、ポリBis−Tris、ポリTris、ポリヒスチジン、ポリヒドロキシル化アミン、キトサンまたはトリエタノールアミンなどを含む核酸結合相を、第1のpHで核酸含有試料と接触させることを伴い、この接触中、核酸結合相は正電荷を有する。これにより、負電荷を有する核酸の前記相に対する結合が促進される。核酸を放出/溶出させるため、正電荷を逆転または中和するために、電荷スイッチ切り替え原理に従って、核酸結合相のpKaよりも大きい第2のpHが設定される。このpHを該固相の核酸結合基のpKaよりも大きく設定することにより、核酸結合相からの結合核酸の分離が促進される。
【0004】
先行技術には、可溶性相(例えば特許文献1参照)と固相(例えば特許文献2参照)の両方が開示されている。種々の溶液、例えば、非常に高いpHを有する溶液あるいは生物学的バッファー、特に、例えばTrisバッファーなどの低塩バッファーが溶出に使用されている。
【0005】
精製核酸は、通常、このほかにも処理される。前記の処理は、例えば、増幅(ポリメラーゼ連鎖反応(PCR)など)、酵素反応(制限、ライゲーション、リン酸化、脱リン酸化またはRNA転写など)、ハイブリッド捕捉アッセイおよび電気泳動を含む。このような「下流」反応は、多くの場合、比較的高い塩濃度に対する耐性がほとんどなく、したがって、核酸のさらなる処理過程を行なう前に、しばしば脱塩工程を行なわれなければならない。
【0006】
したがって、含まれる塩があまり多過ぎず、さらに、あまり塩基性が高過ぎない溶出バッファーは、さらなるバッファー交換または脱塩工程なしでの、精製核酸の直接後続する使用に望ましい。
【0007】
しかしながら、アニオン交換体の溶出能は、溶出バッファーのpHと塩濃度の増大の関数として増大する。したがって、クロマトグラフィーの要件は、DNAの処理過程の実施に関する要件と相反する。このため、先行技術において、適当な核酸結合基とpH条件を選択することにより、核酸の溶出を特異的に促進させることが試みられている。
【0008】
このような核酸精製方法が知られているにもかかわらず、既存の方法改善する必要性、より詳しくは、直後に試料の処理過程も行なうことができるような様式で核酸を精製する必要性がある。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】欧州特許第0707077号明細書
【特許文献2】国際公開第99/29703号
【発明の概要】
【課題を解決するための手段】
【0010】
したがって、本発明の目的は、直後に試料の処理過程を行なうことが可能な核酸の精製方法を提供することである。より詳しくは、本発明は、低塩濃度で溶出が可能な方法を利用可能にすることを意図するものである。
【0011】
この目的は、本発明により、特別な核酸結合相を用いる核酸精製方法によって達成される。
【0012】
第1の実施形態によれば、核酸結合相は、8〜13のpKaを有する核酸結合性のA基と、結合を減弱させ、かつ使用される結合pHにおいて中性電荷を有するN基も有する。
【0013】
この核酸結合相を用いた精製方法は、以下の工程:
(a)核酸結合性のA基のpKよりも小さいpH(結合pH)で、核酸を核酸結合相に結合させる工程;
(b)結合pHよりも大きいpH(溶出pH)で、該核酸を溶出させる工程
を有する。
【図面の簡単な説明】
【0014】
【図1】図1は、核酸結合性のA基と結合減弱性のN基を備えており、前記の基が別々のリガンドに存在している支持材の模式的概略図を示す(請求項2(i)も参照のこと)。
【図2】図2は、核酸結合性のA基と結合阻害性のN基を備えており、前記の基が単一のリガンド内に混合物として存在している支持材の模式的概略図を示す(請求項2(ii)も参照のこと)。(A)は、最初にA基を備え、次いでN基を備えた支持材を示す;(B)は、単一のリガンド内のAとNの交互配列を示す。
【図3】図3は、核酸結合性のA基と立体遮蔽性のN基を備えており、前記の基を、図1および2に示したスキームによる核酸結合性のA基と組み合わせることが可能な支持材の模式的概略図を示す。請求項2(iii)参照。
【図4】図4は、一例として核酸結合性のA基と、結合阻害性および/または立体遮蔽性のN基とを備えており、前記の基が、図1〜3に示したスキームの組合せによって形成されている支持材の模式的概略図を示す。請求項2(iv)参照。
【図5】図5は、準化学量論量の核酸結合性のA基を備えた支持材の模式的概略図を示す。請求項3(iv)参照。
【図6】図6は、異なるpH値での実施例A3a)による溶出プロフィールを示す。
【図7】図7は、異なるpH値での実施例A3b)による溶出プロフィールを示す。
【図8】図8は、実施例C.1)に従って調製したシリカゲルの溶出図を示す。
【発明を実施するための形態】
【0015】
本発明は、核酸結合性のA基に加えて結合減弱性のN基も有する核酸結合相によって核酸の精製に関する。本発明によれば、核酸結合性のA基は8〜13のpKaを有する。核酸は、前記A基の少なくとも1つのpKaよりも下のpHで結合させる。したがって、A基はプロトンを取り込み、その結果、正電荷を有する状態になり、核酸結合相が、負電荷を有する核酸に結合することが可能になる。溶出は結合pHより大きいpHで行ない、それにより、核酸結合相の正電荷を減少させる。
【0016】
さらなる基のN基は、結合pHで、好ましくは溶出pHでも電荷中性である。したがって、N基は、いくつかの態様において、A基の結合強度に影響を及ぼす:(1.)A基の配置に割り込み、したがって、A基の密度が低下するため、核酸結合相に対する核酸の結合強度に影響を及ぼす。(2.)弱い相互作用に参与し(例えば、ファンデルワールス相互作用によって)、中間強度の相互作用まで参与し得る(例えば、水素結合によって)官能基によって、結合強度を特異的にモジュレートし得る。(3.)N基は、大きさおよび/または数が増大すると核酸結合性のA基を立体遮蔽することがあり得、それにより結合強度を低下させる。したがって、N基は、核酸結合強度の一様でpH非依存性の低下を引き起こす。この結果として、結合される核酸が少なくなり、そのうえ、より容易に核酸結合相から分離され得る。N基の機能は、特異的かつ制御された様式で、A基の結合強度および/または数を減少させ、それにより、本発明による核酸結合相において所望の頻度を設定することである。N基の比率が高いほど、A基の比率が低く、電荷密度が低く、核酸結合相に対する核酸の結合が弱い。N基に対するA基の比率は、対応する核酸結合相の調製方法(例えば、重合、(重)縮合によって、特に、支持材のコーティングによって)において、または該方法によって、所望の強度の核酸結合が事前に確立されることを可能にするための調製プロセスによって特異的に調整され得る。
【0017】
このことの大きな利点は、溶出させた核酸を、精製直後に、さらに処理することが可能な条件下で溶出を実施できることである。多くの生物生体工学的方法、例えば、核酸の増幅(特に、PCRによるもの)、配列決定、逆転写、制限解析、およびその他(上記参照)などは、溶出液中の夾雑物に対して、特に、高い塩濃度に対して敏感である。本発明による精製方法では、好都合には、低塩濃度または低イオン強度での溶出が可能となり、それにより、後続の生物生体工学的方法、特にPCR反応において、さらなる処理過程を直接行なうことが可能になる。
【0018】
N基を用いることにより核酸の結合を減弱させるという概念により、驚くべき良好な結果がもたらされる。先行技術では、方法の最適化のために核酸の最適な結合を想定し、次いで、結合条件、特に溶出の最適化に注力する。対照的に、結合強度の減弱は、例えば、核酸が、結合させる工程において不充分にしか核酸結合相に結合しないか、または洗浄中に溶出されることがあり得るため、予測される収率に関して不都合であると考えられていた。このことに鑑みると、N基の存在により結合強度が低下するにもかかわらず成功裡で緩徐な核酸精製という本発明による概念は、極めて驚くべきこととみなされるはずである。
【0019】
N基に対するA基の比率は、1%〜99%、1〜50%、好ましくは1%〜25%であり得る。
【0020】
好ましい一実施形態によれば、核酸結合相は、A基とN基を導入するために修飾される。本発明によれば、これは、以下により詳細に説明する種々の実施形態を包含する。
【0021】
一実施形態によれば、核酸結合相は異なるリガンド(リガンドIとリガンドII)を備えており、リガンドIは少なくとも1つのA基を有し、リガンドIIは少なくとも1つのN基を有する。この実施形態を図1Aに図解する。このとき、A基とN基は直接近接しており、また、ポリマーコーティングを形成させてもよい。N基担持リガンドIIの存在のため、A基の数は減少し、それにより、特に支持材が使用されている場合、核酸の結合の強固さが低下する。したがって、N基のため、該支持体表面上のA基の密度は低下し、結果として、核酸に対する結合親和性が低下する。その結果、核酸は、より容易に溶出され得る。用語「リガンド(1つまたは複数)」は、特に、好ましくは、少なくとも1つのA基および/または1つのN基を有する支持材が、本発明による核酸結合相を提供するために修飾される表面の官能基をいう。リガンドIとIIは、好ましくは支持材に結合させており、例えば、反応性の個々の分子のモノマー、ダイマー、オリゴマーまたはポリマーであり得る。変形例の一例において、支持材と反応する化合物の一部としてのA基またはN基は、直接またはリンカーもしくはスペーサーを介して支持材に結合させる。好ましい一実施形態によれば、この修飾は、支持材を、異なるリガンドIとIIを有する混合物と接触させることによりなされる。リガンドIに対するリガンドIIの比率が高いほど、核酸に対する結合強度が低い。この比は、単に、コーティングプロセスにおけるリガンドI反応体とリガンドII反応体の対応する比によって制御され得る。詳細を、以下に、対応する支持材の合成または調製方法に関して説明する。また、リガンドは、ポリマーコーティングを形成していてもよい。また、支持材は、A基と開始剤分子T(Initiatormolekuelen)を備えたものであってもよく、1つ以上のN基を前記開始剤分子に結合させる。この実施形態を図1B)に示す。N基は、この場合、支持材の表面に対してA基の「上部」に存在する。また、A基も開始剤分子を介して結合させることも可能である。
【0022】
本発明のさらなる一実施形態によれば、支持材は、1つ以上のA基と1つ以上のN基とが単一のリガンド内に存在する核酸結合リガンドで修飾される。
この実施形態を図2により詳細に示す。この概念の実現には種々の可能性がある。
【0023】
変形例の一例によれば、支持材は、N基を含む1つ以上の化合物を担持しているA基(直接または例えば開始剤分子Tを介してのいずれかにより結合)を備えている(図2Aも参照のこと)。複数のN基が存在する場合、これらはまた、非分枝鎖の形態であってもよく、分枝型樹状構造としての形態であってもよく、対応する混合物としての形態であってもよい。これには、リガンド1つあたり例えば、1〜100個のN基、1〜20、好ましくは1〜10、特に好ましくは1〜5個のN基が含まれ得る。また、所望により、なおさらなるA基とN基を導入してもよい。この連結ストラテジーでは、実質的に純粋にA基で占有された比較的均一な結合面と、突出部と隣接し、N基による占有のためA基の結合強度が低下した1つ以上の面とがもたらされる。
【0024】
この実施形態の変形例のさらなる一例によれば、支持材は、N基とA基の混合物を担持しているA基(直接または例えば開始剤分子Tを介してのいずれかにより結合)を備えている(図2Bも参照のこと)。これは、最初に1つ以上のA基を支持材に結合させることを伴い得る。この後、このようなA含有主リガンドに、少なくとも1つ以上のN基を導入する化合物を結合させる。反応条件の制御により、各場合において、リガンド内にA基とN基を1つだけ存在させることが可能になる;しかしながら、リガンドはまた、オリゴマーまたはポリマーの形態であってもよい。オリゴマーおよびポリマーとして設計されるリガンドは、N基がランダムな分布(例えば図2B、右側の図参照)、交互配列(例えば図2B、左側の図参照)、または他の配置のブロックコポリマーを有するものであり得る。また、組合せも可能である。適切なA含有およびN含有官能基/リガンドの好適な設計方法を以下に詳細に説明する。
【0025】
さらなる実施形態によれば、支持材は、A基が、少なくとも1つ、好ましくは多数のN基を有する化合物によって立体遮蔽されたA基とN基で修飾されている。この実施形態にも、種々の変形例が存在する。
【0026】
変形例の一例によれば、立体遮蔽は、好ましくは、N基を有するオリゴマーまたはポリマーによって行なう。前記オリゴマーまたはポリマーは、例えば、A基(1つまたは複数)を有するリガンドに結合させてもよい(図3Bも参照のこと)。しかしながら、立体遮蔽が確保される限り、前記オリゴマーまたはポリマーはA基に隣接して配置するのがよい(図3Aも参照のこと)。N基を有するオリゴマー/ポリマーはA基を、多かれ少なかれ規則的な二次構造(例えば、「ランダムコイル」またはらせん)を形成することにより、周囲環境から、いわば蓋をすることによって遮蔽し、したがって、核酸の結合を減弱させることが可能である。また、該オリゴマー/ポリマーは、N基から調製したものであってもよく、また、ブロックコポリマーによって導入されたものであってもよい。
【0027】
この変形例は、例えば、第1の工程において開始剤基Tを支持材に、例えばシラン処理によって適用することにより調製され得る。次いで、第2の工程において、A基を含むモノマーが、例えばATRP(原子移動ラジカル重合)によってグラフト化され得る。このモノマーを充分反応させた後、中性のN基を有する第2のモノマーが導入され得る。これにより、アニオン交換体基を担持している第1のホモポリマーと、第1のホモポリマーに連結され、中性のN基を担持している第2のホモポリマーからなるコポリマーが、該支持体上に得られる。発生期ポリマー鎖の長さ、したがって、立体遮蔽の強度は、N基を担持している第2のモノマーの量によって制御され得る。
【0028】
したがって、立体遮蔽の程度は、鎖長およびモノマー置換によって制御され得る。N基を有するオリゴマー/ポリマーの好ましい鎖長は、n=10〜1000、n=10〜500、特に好ましくは、n=10〜100である。N基を担持しているオリゴマー/ポリマーは、本出願書類に記載のモノマーを用いて設計してもよく、これを詳細に説明する。ポリアクリレートが特に好適である。
【0029】
使用される好ましいポリマーは、下記式:
【0030】
【化1】
によるポリアクリレートである。
【0031】
立体遮蔽がなされるのに特に好適な試薬は、加水分解後に中性のジオールN基が生成されるメタクリル酸グリシジルである。この目的のため、開始剤分子Tは、例えばシラン処理によって適用され得る。この後、第1のモノマー、例えば、メタクリル酸N,N−ジメチルアミノプロピルを支持体上で、例えばATRPによって重合させる。第2の重合工程では、次いで、この基を立体遮蔽する。これは、例えば、モノマーを充分反応させた後、第2のモノマー、例えば、加水分解後に中性のジオールN基が生成されるメタクリル酸グリシジルを導入することによりなされ得る。該立体効果により、核酸とA基の密接な接触が抑制され、その結果、後者はあまり強く結合されず、低イオン強度で、したがって低塩濃度で核酸結合相から溶出され得る。
【0032】
また、N基の立体遮蔽は、「バルキー」な置換基によってもなされ得、挙げられる得る該置換基の一例は、分枝アルキル原子団、例えば、イソプロピル、ジイソプロピル、第3級ブチル、脂肪族または芳香族環(炭素環として、もしくは複素環としてのいずれも)である。また、このような立体阻害性のN基も、オリゴマー/ポリマーの形態で使用され得る。
【0033】
したがって、立体遮蔽は、特に、(i)さらに導入されるN基の比率;(ii)N基を担持する化合物の合成;(iii)N基の選択および構造、ならびに(iv)N基を有するオリゴマー/ポリマーの鎖長によって制御され得る。
【0034】
したがって、A基(1つまたは複数)は、同じリガンド内に存在しているか、または近接して配置されたN基によって特に良好に遮蔽され得る。また、A基に結合させることも可能である。立体遮蔽は、N基(1つまたは複数)を有する化合物の大きさの増大、また、N基の大きさの増大の関数として増大し、鎖長の増大および炭素鎖の高い分枝性または環状基の環の大きさの増大は、ともに立体遮蔽を増大させる。
【0035】
特定の一実施形態では、支持材を、上記のすべての連結ストラテジーの組合せを用いて修飾する。一例を図4に示す。したがって、支持材は、A基、立体遮蔽性のN基および開始剤分子Tが互いに直接連結されたものであり得、前記開始剤分子は、さらなる立体遮蔽性のN基を備えている(例えば、図4Aを参照のこと)。この型の連結は、例えば、通常のN基を用いて行なわれ得る(例えば、図4Bを参照のこと)。さらに、支持材は、例えば、A基を備えており(直接または開始剤分子Tを介して結合)、N基とA基が連結された混合物を担持しており、N基の全部または一部が立体阻害性基であるものであってもよい(図4Dおよび4Eを参照のこと)。
【0036】
本出願書類のさらなる一態様によれば、該方法は、支持材を有し、かつA基で修飾された核酸結合相を用いて行なわれ、該支持材は、一部のみがA基で占有されており、これは、提供される核酸結合基が通常よりも相当少ないことを意味する。したがって、支持材の単位面積あたり、および/または支持材の1グラムあたりに存在する核酸結合性のA基が少なくなることにより、核酸の結合強度が先の連結ストラテジーと同程度に低減される。図5は、この概念の一実施形態を示す。
したがって、この実施形態でも、特に、核酸結合相からの核酸の溶出に低塩濃度が使用され得るため、前記核酸のさらなる処理過程を直接行なうことが可能な条件下で核酸を溶出させることが可能である。
【0037】
支持材上のA基の本発明による準化学量論量は、支持材に対して準化学量論量のA基導入化合物を用いることにより得られ得る。また、支持材が有するA基を結合させ得る潜在的結合部位の量を少なくすることも考えられ得る。さらなる一実施形態としては、結合部位の減少または前記結合部位の反応性の低減を引き起こす支持材の化学的または物理的前処理が挙げられる。可能な択一的ストラテジーは、A基担持化合物を、同様にその支持材に結合する別の化合物との混合物にて適用することである。この競合性結合により、支持材の表面上のA基の比率が低減される。
【0038】
準化学量論量のA基に基づく実施形態を、さらに、A基とN基での支持材修飾のために上記の1つ以上の実施形態と組み合わせてもよい。
【0039】
準化学量論量のA基を有する支持体を提供することは、シリカ支持材が使用される場合、後者がシラノール基を有するため特に好都合である。A基は、好ましくはシランまたはシラン混合物によって導入する。支持体の完全な被覆に必要とされるシランは最小量である。この最小量は、支持材の具体的な表面によって規定される。シラン(1種類または複数種)の量が支持体表面の占有に不充分な場合、電荷密度を下げるとよい。したがって、A基の量、結果として核酸結合相の結合強度は、適用されるシランの量によって調整され得る。
【0040】
一実施形態によれば、支持材は、A基を有するシランでコートされたシリカ表面を有し、シランの量は0.1〜50μmol(マイクロモル、本明細書ではumolとも称する)、好ましくは0.1〜10umolである。シランの量を、溶出が比較的低いイオン強度または比較的低い(所望の)塩濃度で可能であるほど大きく低減させることは重要である。これは、例えば、実験によって試験し、クロマトグラムによって確認することができる。したがって、シランの量は、好ましくは、所望の結合特性/溶出特性を得るために、使用される支持材に応じて最適化され得る。
【0041】
核酸結合相は、好ましくは固相である。これは、例えば、A基、結合減弱性のN基、および開始剤分子T(存在する場合)を固相支持材に結合させることにより調製され得る。詳細を以下に説明する。固相を用いると、結合された核酸を試料から除去することが容易になる。したがって、一実施形態によれば、核酸の結合の後に、固相または未結合浮遊物を除去する。
【0042】
該核酸結合基に適した支持体の例は、酸化物材料である。これは、特に、Al2O3、TiO2、ZrO2、Ta2O5、SiO2およびポリケイ酸などの酸化物であり、好ましい支持材はSiO2またはポリケイ酸である。また、ポリスチレンおよびその誘導体、ポリアクリレートおよびポリメタクリレート、ならびにその誘導体、またはポリウレタン、ナイロン、ポリエチレン、ポリプロピレン、ポリブチリデン、ならびにこれらの材料のコポリマーなどの有機ポリマーも好適な支持体である。また、このような核酸結合基を、多糖類、特に、ヒドロゲル、例えば、アガロース、セルロース、デキストラン、セファデックス、セファクリルおよびキトサンに連結させてもよい。さらに、核酸結合基を、無機系支持体(例えば、ガラスなど)または金属表面(例えば、金など)に結合させてもよい。磁性粒子の使用は特に好都合である。核酸結合性のA基および/またはN基は、このような支持体に、直接結合させてもよく、あるいは他の化学分子(例えば、開始剤分子もしくはリンカー)を介して結合させてもよい。また、大きな分子の一部分であってもよく、支持材が結合部位としての適当な官能基を有していない場合、それ自体は既知の様式にて導入され得る。
【0043】
支持材のさらなる実施形態は、非磁性および磁性の粒子、カラム材料、膜、ならびに表面コーティングを含む。また、官能性付与したチューブ、膜、不織布、紙、反応槽(PCR槽など)、「エッペンドルフチューブ」、マルチプレート、チップおよびマイクロアレイなどの支持体も挙げられよう。
【0044】
一実施形態によれば、該核酸結合相は、結合中および溶出中、どちらも正電荷を有する。
【0045】
本発明のさらなる一実施形態は、核酸に、本発明による原理に従って可逆的に結合する可溶性核酸結合相に関する。可溶性ポリマーは、A基とN基を、例えば、交互またはランダムに有するものであり得る(例えば図2B参照)。また、A基の側鎖をN基、特にアルキル原子団(挙げられ得る一例は、窒素上のジイソプロピル原子団である)によって遮蔽することも可能である。支持材に関する記載内容は、相応して可溶性核酸結合相に適用される。
【0046】
一実施形態によれば、核酸結合性のA基はイオン交換体、好ましくはアニオン交換体である。核酸の結合において実証された好ましいA基はアミノ基であり、第1級、第2級および第3級アミノ基が好ましい。これらは、置換されていても非置換であってもよい。また、環状アミン、芳香族アミンまたはアミノ官能性付与された複素環も使用され得る。該アミンは、置換基、例えば、アルキル、アルケニル、アルキニルまたは芳香族置換基を有するものであってもよく、また、該炭化水素鎖は、環状になった閉鎖型であってもよい。また、該炭化水素鎖は、ヘテロ原子(酸素、窒素、イオウもしくはケイ素など)、または分枝構造を有するものであってもよい。記載のように、A基としてのアミノ基は、8〜13、好ましくは9〜13、特に好ましくは10〜12のpKaを有する。
【0047】
A基は、重合または縮合によってオリゴマーまたはポリマーを形成し、したがって、リガンドの形成に特に好適な化合物の一部分であってもよい。かかる鎖形成性化合物の例は、アミノ基を含むアクリレート、例えば、
N−(3−アミノメチル)メタクリルアミド、N−(3−アミノエチル)メタクリルアミド、
N−(3−アミノプロピル)メタクリルアミド、N−(3−アミノイソプロピル)メタクリルアミド、
N,N−ジメチルアクリルアミド、N,N−ジエチルアクリルアミド、N,N−ジイソプロピルアクリルアミド、
N,N−(ジメチルアミノ)エチルアクリルアミド、N,N−(ジメチルアミノ)エチルアクリレート、
N,N−(ジメチルアミノ)エチルメタクリルアミド、N,N−(ジメチルアミノ)エチルメタクリレート、
N,N−(ジメチルアミノ)プロピルアクリルアミド、N,N−(ジメチルアミノ)プロピルアクリレート、
N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート、
N,N−(ジエチルアミノ)エチルアクリルアミド、N,N−(ジエチルアミノ)エチルアクリレート、
N,N−(ジエチルアミノ)エチルメタクリルアミド、N,N−(ジエチルアミノ)エチルメタクリレート、
N,N−(ジエチルアミノ)プロピルアクリルアミド、N,N−(ジエチルアミノ)プロピルアクリレート、
N,N−(ジエチルアミノ)プロピルメタクリルアミド、N,N−(ジエチルアミノ)プロピルメタクリレート、
N,N−(ジイソプロピルアミノ)エチルアクリルアミド、N,N−(ジイソプロピルアミノ)エチルアクリレート、
N,N−(ジイソプロピルアミノ)エチルメタクリルアミド、N,N−(ジイソプロピルアミノ)エチルメタクリレート、
N,N−(ジイソプロピルアミノ)プロピルアクリルアミド、N,N−(ジイソプロピルアミノ)プロピルアクリレート、
N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート、2−(ジメチルアミノ)エチルメタクリレート(DMAEMA)および2−(ジイソプロピルアミノ)エチルメタクリレート
などである。
【0048】
これらのうち、N,N−(ジメチルアミノ)プロピルメタクリレートが特に好ましい。
【0049】
A基は、シラン内、好ましくは反応性シラン内に存在するものであり得る。反応性シランは、加水分解不安定性のSi結合、例えば、Si−NまたはSi−O結合を有する化合物をいう。少なくとも1つのA基を含む反応性シランの例は:
【0050】
【化2】
(式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基であり、
*は、アミノ、アミノメチル、アミノエチル、アミノプロピル、ジメチルアミノ、ジエチルアミノ、ジイソプロピルアミノ、ジプロピルアミノ、ジエタノールアミノ、ジプロパノールアミノ、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、エーテルアミン、ポリエーテルアミン、4−ジイソブチルアミノ−1−ブタン、6−ジプロピルアミノ−1−ヘキサンである)
である。
【0051】
対応する反応性シランが、A基の導入に使用され得る。特に好ましいものは、ジエチルアミノプロピルトリメトキシシラン(DEAPS)、ジメチルアミノプロピルトリメトキシシランおよびN,N−ジイソプロピルアミノプロピルトリメトキシシランである。
【0052】
本発明の一実施形態によれば、核酸は、2〜8、好ましくは4〜7.5のpHで結合させる。これは、結合中、したがって試料中のpHをいう。したがって、核酸結合相の設計によるが、本発明による方法はまた、非常に穏やかな条件下で、実質的に中性の範囲で行なわれ得る。核酸結合相のプロトン化性基が8〜13、好ましくは9〜13、特に好ましくは10〜12のpKaを有することにより、該相は、比較的中性のpHであっても、有効な核酸結合を可能にするための充分な正電荷を有する。その結果、結合は、所望により非常に穏やかな条件下で行なわれ得る。
【0053】
出発材料によるが、核酸を放出させるための少なくとも1回の通常の溶解工程が結合させる工程の前に行なわれ得る。
【0054】
核酸の溶出は、本発明の方法の別の重要な工程である。記載のように、核酸は、結合pHより高いpHで放出させる。この結果として、溶出中、A基が有する正電荷が少なくなり、これは核酸の放出に有利である。慣用的なアニオン交換体と比べ、A基の低比率および/または結合減弱性のN基の存在により、支持材に対する核酸の結合の低減がもたらされる。この結果として、上記のように、溶出も低塩濃度で行なうことが可能である。
【0055】
溶出pHは、好ましくはA基のpKより低い。使用される核酸結合性のA基または核酸結合相によるが、溶出は、好ましくは8〜11、8〜10のpHで、好ましくは8.0〜9、特に好ましくは8.5〜9のpHで行なわれる。しかしながら、原理的には、より高いpH値を使用することも可能である。しかしながら、低塩濃度にもかかわらず核酸が放出され得、さらに条件が穏やかであるため、好ましい低pH値により特に好都合な結果が得られる。
【0056】
単離された核酸の処理過程を、引き続いて直接、溶出バッファー中で行なうことを可能にするため、後者は、記載のように、好ましくは低塩濃度を有するものである。これは、本発明による核酸結合相の設計によって可能になる。したがって、一実施形態によれば、塩濃度は1mM〜1000mM、特に好ましくは1mM〜200mM、1mM〜250mM、または1mM〜100mMである。好適な塩は、アルカリ金属およびアルカリ土類金属またはアンモニアの塩化物、無機酸の他の塩、酢酸塩、ホウ酸塩、ならびにTris、Bis−Trisおよび有機バッファー(例えば、MIS、CHAPS、HEPESなど)などの化合物などであり得る。同じものが結合バッファーに適用される。さらに、溶出のための好適な物質は先行技術において知られている。塩濃度は結合させる工程と溶出させる工程で変更しないか、または溶出中でわずかに上昇させる。しかしながら、好ましくは、該濃度は、後続する反応を障害するような様式では増大させない。さらに、結合中と溶出中の温度は同じであってもよく、あるいは溶出中で上昇させる。
【0057】
精製を助長するため、好ましくは、核酸の結合後、溶出の前に少なくとも1回の洗浄工程を行なう。低塩濃度を有する水溶液での洗浄が好ましいが、水での洗浄も好ましい。洗浄バッファー中に存在させる塩は、好ましくは1mM〜1000mM、特に好ましくは1mM〜200mM、1mM〜250mM、または1mM〜100mMの濃度である。バッファーは、有機化合物(糖質など)、好ましくは、有機溶媒(例えば、アルコール、ポリオール、ポリエチレングリコール、エーテル、ポリエーテル、ジメチルスルホキシド、アセトンまたはアセトニトリルなど)を含むものであり得る。しかしながら、下流での適用が障害されないようにするため、洗浄バッファーは、干渉量の対応有機成分を有しないものであるのがよい。
【0058】
使用され得る結合減弱性のN基は、電荷中性基、例えば、ヒドロキシル基、ジオール基、トリオール基、糖類、エポキシド基、C1〜C6アルキル、アルケンもしくはアルキン基、ポリオール基、エーテル、ポリエーテル、ハライドまたはイミドなどである。親水性のN基の使用により、水性バッファーでのイオン交換体の継続的に良好なぬれ性が確保される。N基は、重合または縮合によってオリゴマーまたはポリマーを形成する化合物の一部分であってもよい。N基を導入し得る化合物の例は、アクリレート、例えば、アクリル酸ブチル、アクリル酸プロピル、アクリル酸エチル、アクリル酸メチル、メタクリル酸グリシジル、メタクリル酸ヒドロキシエチル(HEMA)、メタクリル酸グリシドキシプロピル、グリセロールモノメタクリレート(異性体混合物)、グリコールモノメタクリレートおよびN−アクリルオキシスクシンイミドなどである。
【0059】
また、N基はシラン内、好ましくは反応性シラン内に存在するものであってもよい。N基を含む反応性シランの例は:
【0060】
【化3】
(式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基であり;
*は、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、エタンジオール、プロパンジオール、プロパントリオール、ブタントリオール、3−グリシドオキシプロピル、エチルグリシジルエーテル、アルキル原子団、特に、C1〜C4アルキル原子団、ハライドまたは水素である)
である。
【0061】
一実施形態によれば、支持材は、少なくとも一部が、A基および/またはN基を有する化合物で官能性付与された開始剤基Tを有する。好適な例は、本明細書において以下に詳細に説明する。
【0062】
さらに、本発明は、対応する核酸結合支持材の合成またはその調製方法に関する。この目的のためには、種々の実施形態がある。
【0063】
代替法(A):この方法は、少なくとも2種類の異なるリガンドIとIIの混合物を用いて支持材に官能性付与することを含み、リガンドIは少なくとも1つのA基を有し、リガンドIIは、少なくとも1つのN基を有するか、または少なくとも1つのN基である。この実施形態に関する詳細は、上記において既に説明した。上記の開示内容を参照されたい。
【0064】
代替法(B):この方法は、第1の工程において、開始剤分子Tと、少なくとも1つのA基を有するリガンドとの混合物を用いて支持材に官能性付与することを含む。ここで、A基を有する化合物は、さらなる分子は結合され得ないような様式で選択されたもの、または修飾されたものである。第2の工程では、次いで、少なくとも1つのN基を有し、開始剤分子Tに結合する化合物が添加される。
【0065】
代替法(C):核酸結合リガンドでの支持材の官能性付与。前記のリガンドのそれぞれは、1つ以上のA基と1つ以上のN基とを有する。詳細は、本発明による方法に関して上記において説明している。第1の工程において、開始剤分子Tを支持材に結合させ、該分子を、少なくとも1つのA基および/または少なくとも1つのN基を有する化合物を添加し、前記化合物を開始剤分子Tに結合させてコポリマー鎖を伸長させるための開始点として使用することが好ましい。該ポリマー鎖は、少なくとも1つのA基および/または少なくとも1つのN基を有する化合物をさらに添加することにより伸長される。
【0066】
これに関連して、該重合工程はまた、A基とN基の両方をオリゴマーによって組み込み、したがってブロックコポリマーが得られるような様式で制御してもよい。本発明に従って使用され得る重合反応の制御の一例は、例えば、「原子移動ラジカル重合」(ATRP)である。ATRPは、遷移金属錯体を添加すること、およびオルガノハライドを伴う原子転移プロセスとの組合せにより、フリーラジカルの濃度を、不均衡または組換えなどの連鎖停止反応が大幅に抑制されるような程度まで低減させることを特徴とする。
【0067】
代替法(D):A基が、少なくとも1つのN基を有する化合物によって立体遮蔽されるA基とN基での支持材の官能性付与。この変形例の一実施形態によれば、支持材は、第1の工程において、開始剤基Tで官能性付与される。この後、少なくとも1つのA基を有する化合物を添加し、これを開始剤分子Tに結合させる。さらなる工程において、少なくとも1つのN基を有する少なくとも1種類の化合物が結合することにより、A基が遮蔽される。N基は、その3次元構造によってA基を立体遮蔽するオリゴマー/ポリマー(上記参照)であることが好ましい。詳細は、本発明による方法に関して上記において説明している。
【0068】
代替法(E):この実施形態によれば、支持材は、準化学量論量のA基で官能性付与される。この変形例は、上記において詳細に説明した。上記の開示内容を参照されたい。
【0069】
また、代替法A〜Eは任意の組合せで使用され得る。核酸結合性のA基、N基および開始剤分子Tに関する詳細は、上記において詳細に説明しており、また、本明細書に示した支持体の修飾方法に関しても適用され、その場合に使用される成分の特徴を示す。上記の開示内容を参照されたい。
【0070】
A基とN基で支持材に官能性付与する方法の一実施形態によれば、前記の基は、単官能性、二官能性もしくは三官能性の反応性シランによって、または異なる官能基を有する少なくとも2種類の反応性シランの混合物によって導入される。使用され得る反応性シランは、例えば、アミノシラン、ジシラザン、クロロシランまたはアルコキシシランである。N基に対するA基の比率は1%〜99%、1〜50%、好ましくは1%〜25%である。
【0071】
三官能性および二官能性の反応性シランは、支持体上で、重縮合によって架橋された厚い層を形成する傾向にある。対照的に、単官能性の反応性シランは、例えば、支持材のシラノール基と反応してシロキサン(Si−O−Si)結合を形成し、支持体上に、むしろ単分子層をもたらす。支持材を反応性シランと、気相中または溶媒中での懸濁状態で反応させてもよく、後者においては、反応性シランの化学的性質に応じて有機溶媒、好ましくは水性溶媒を使用することが可能である。
【0072】
好適で好ましい支持材は、上記において詳細に説明した。該方法の一実施形態によれば、支持材を、まず開始剤分子Tで修飾した後、次の工程で、A基および/またはN基をモノマーの形態で導入する。開始剤分子Tは、例えば、(重)縮合または重合、特に、フリーラジカル重合(特に、ATRP(原子移動ラジカル重合)など)によって導入されるさらなる化合物の結合部位として使用してもよい。開始剤分子Tの例は:
【0073】
【化4】
【0074】
【化5】
(式中、Xは、ハロゲン、特にCl、Br、Iであり、
Y1、Y2、Y3またはY4は、互いに独立して、R、OR、OHまたはHであり、
Rは、C1〜C3アルキルである)
である。
【0075】
2−(クロロメチル)アリルトリメトキシシランまたは[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシラン(BPDS)を開始剤分子として使用することが好ましい。
【0076】
本発明による方法に従ってA基および/またはN基を導入し得る反応性シランの例を以下に示す:
【0077】
【化6】
式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基、特に、メチル、エチル、プロピル、イソプロピルであり;
*は、アミノ、アミノメチル、アミノエチル、アミノプロピル、ジメチルアミノ、ジエチルアミノ、ジイソプロピルアミノ、ジプロピルアミノ、ジエタノールアミノ、ジプロパノールアミノ、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、エーテルアミン、ポリエーテルアミン、4−ジイソブチルアミノ−1−ブタン、6−ジプロピルアミノ−1−ヘキサン、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、エタンジオール、プロパンジオール、プロパントリオール、ブタントリオール、3−グリシドオキシプロピル、エチルグリシジルエーテル、アルキル原子団、特に、C1〜C4アルキル原子団、特に、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、ハライドまたは水素である。
【0078】
詳細に説明したシラン処理と準化学量論でのシラン処理の混合型の方法に加え、支持材上のA基の密度を規定の様式で調整できる可能性が他にもある。記載のように、例えば、支持材上で「表面開始グラフト重合(grafting−from)」プロセスを行なうことが可能である。これは、例えば、上記に記載した原子移動ラジカル重合(ATRP)法によって行なわれ得る。この方法は、まず、開始剤基T、N基および/またはA基を支持体に、シラン処理によって適用することを含む。開始剤基Tは、この基によって表面開始グラフト重合プロセスが開始されるため(連鎖開始)、このプロセスには重要である。
【0079】
例えば、ハライド含有シランを使用し、ATRP(原子移動ラジカル重合)によって、ホモポリマー、コポリマーおよびブロックコポリマーを伸長させることができる。ATRPは、リビング/制御フリーラジカル重合(LFRP)の特別な形態であり、これは、遷移金属錯体を添加すること、およびオルガノハライドを伴う原子転移プロセスとの組合せにより、フリーラジカルの濃度を、不均衡または組換えなどの連鎖停止反応が大幅に抑制されるような程度まで低減させることを含む。この方法により、支持体表面の電荷密度を、モノマーおよびモノマー混合物の使用によって特異的に調整し、それにより、核酸の結合強度に影響を及ぼすことが可能になる。A基および/またはN基は、モノマーの適切な選択によって特異的に適用され得る。
【0080】
このようにして作製される核酸結合支持材は、特に、本発明による精製方法に使用され得る。
【0081】
さらに、本発明は、核酸を精製するための本発明による核酸結合相の使用に関する。本発明によれば、核酸は、特に、DNAおよびRNA、特にゲノムDNA、プラスミドDNA、ならびにPCR断片、cDNA、miRNA、siRNA、また、オリゴヌクレオチドおよび修飾核酸(例えば、PMAまたはLMAなど)を含む。また、ウイルスもしくは細菌のRNAおよびDNA、またはヒト、動物もしくは植物源由来の核酸を精製することも可能である。さらに、DNA/RNAハイブリッドおよび修飾核酸も本発明による精製に適している。
【0082】
また、本発明は、少なくとも1つのプロトン化性基を有し、かつ8〜13、好ましくは9〜13、特に好ましくは10〜12のpKaを有する核酸結合性のA基を有する本発明による核酸結合支持材を有することを特徴とする、核酸を精製するためのキットを提供する。本発明による支持材およびその官能基に関する詳細は上記に記載している;上記の記載内容を参照されたい。
【0083】
また、キットは、例えば、精製方法に関して上記に記載の結合バッファー、洗浄バッファーおよび/または溶出バッファーを有するものであってもよい。また、溶解バッファーおよび中和バッファーを有するものであってもよい。
【0084】
一実施形態によれば、キットは、好ましくは以下の特徴:
(a)1〜13のpH;および/または
(b)1mM〜1000mM、特に好ましくは1mM〜200mM、1mM〜250mM、または1mM〜100mMの塩濃度
の少なくとも1つを有する結合バッファーを有する。
【0085】
対応する特徴の利点は、方法に関して上記に説明しており、上記の開示内容を参照されたい。
【0086】
また、一実施形態によれば、キットは、好ましくは以下の特徴:
(a)2〜7、好ましくは4〜7のpH;
および/または
(b)1mM〜1000mM、特に好ましくは1mM〜800mM、1mM〜600mMの塩濃度;および/または
(c)水、生物学的バッファー、有機バッファー、特に、Tris、Tris−Bis、MIS、MOPS、CHAPSおよびHEPESからなる群より選択されること
の少なくとも1つを有する洗浄バッファーを有する。
【0087】
また、さらなる実施形態によれば、キットは、好ましくは以下の特徴:
(a)8〜10、好ましくは8〜9のpH
および/または
(b)1mM〜1000mM、特に好ましくは1mM〜200mM、1mM〜250mM、または1mM〜100mMの塩濃度;および/または
(c)水、生物学的バッファー、有機バッファー、特に、Tris、Tris−Bis、MIS、MOPS、CHAPSおよびHEPESからなる群より選択されること
の少なくとも1つを有する溶出バッファーを有する。
【0088】
核酸結合相および溶出条件に関する詳細は上記に記載しており、本発明によるキットに関しても適用され、その場合に使用される成分/バッファーの特徴を示す。上記の開示内容を参照されたい。
【0089】
本発明によるキットは、特に、本発明による方法の枠内で適用され得る。本発明の方法、キットおよび核酸結合固相は、特に、分子生物学、分子診断、法医学、食品分析、および応用試験の分野において使用され得る。本発明によるキットの適用により、精製核酸を、「下流」適用、特にPCR反応において、さらに直接処理することが可能になる。
【0090】
記載のパラメータ、特に、溶出促進性結合減弱性のN基と核酸結合基の低減を選択/組み合わせることにより、核酸結合相のpHが溶出条件に関して最適化され得る。相応して、核酸結合相の溶出プロフィール、特に、塩濃度と溶出pHが制御または調整され得る。
【0091】
本発明による系によって精製され得る核酸は、体液中(血液、尿、大便、唾液など)、生体源中、例えば、組織、細胞(特に、動物細胞、ヒト細胞、植物細胞、細菌細胞など)、器官(肝臓、腎臓または肺など)に存在するものであり得、また、核酸を、支持材(綿棒など)、PapSmears、および安定化媒体(PreServCytもしくはSurepathなど)、または他の液状物(例えば、搾汁、水性試料もしくは食品一般など)に由来する他のものから入手することも可能である。また、核酸は、植物材料、細菌溶解物、パラフィン包埋組織、水溶液またはゲルから得られるものであってもよい。
【0092】
溶出された核酸は、好ましくは、さらなる処理過程が直接行なわれるものであり得、したがって、例えば、PCR、RT−PCR、制限消化または転写に使用され得るものである。溶出バッファーが上記のように設計されている限り、好ましくは低塩濃度を有する限り、さらなる精製は必要とされない。
【実施例】
【0093】
本発明を、いくつかの実施例に基づいて以下に説明する。前記実施例は、本発明の限定ではなく、好ましい実施形態である。また、参考文献はすべて、本開示の主題に関して本明細書に挙げたものである。
【0094】
実施例
本実験では、プラスミドDNAを核酸モデル系として使用した。
【0095】
A) 隣接するA基とN基での支持材の修飾
A.1) 1:1のモル比2−(クロロメチル)アリルトリメトキシシランおよびDEAPSでのシリカゲルのシラン処理(AAK01−10)
材料
支持材:ほぼ150nmの孔径を有するシリカ/シリカゲル。比表面積は、ほぼ25m2/gである。
【0096】
コーティング試薬:2−(クロロメチル)アリルトリメトキシシラン、DEAPS;QSP1バッファー(酸性酢酸バッファー)。
【0097】
調製プロトコル
三つ口フラスコに、70mlの水、2.5mlのQSP1バッファー(QIAGEN)、322μlの2−(クロロメチル)アリルトリメトキシシランおよび413μlのDEAPSを仕込み(pHは5.3となった)、次いで、18gのシリカゲルを添加した。混合物を攪拌しながら95℃まで20分間以内で加熱し、この温度でさらに4時間攪拌し、次いで、攪拌しながら1時間冷却した。シリカゲルをP3ガラスフリットによって除去し、逐次、32.3gのTris/NaClバッファー、30mlの脱イオン水で2回、35mlのメタノールで2回、最後に30mlのメタノールで洗浄した。最終支持材AAK01−10を125℃で一晩乾燥させた。
【0098】
比較のため、同じ支持材を、A基を有する化合物DEAPSのみで修飾した。三つ口フラスコに、70mlの水、2.5mlのQSP1バッファー、825μlのDEAPSを仕込み(pHは5.5となった)、次いで、18gのシリカゲルを添加した。混合物を攪拌しながら95℃まで20分間以内で加熱し、この温度でさらに4時間攪拌し、次いで、攪拌しながら1時間冷却した。シリカゲルをP3ガラスフリットによって除去し、逐次、32.3gのTris/NaClバッファー、30mlの脱イオン水で2回、35mlのメタノールで2回、最後に30mlのメタノールでで洗浄した。最終支持材AAK01−30を125℃で一晩乾燥させた。
【0099】
溶出点の測定を伴う修飾支持材AAK01−10を用いたプラスミドDNAの精製
結合は、50mM Tris−HCl(pH7.0)、15%エタノールのバッファー中で行なった。溶出は、バッファーB(50mM Tris−HCl(pH7.0),15%エタノール,2M NaCl)の0%〜100%の段階的勾配の23分間にわたる流動、およびUV分光法による連続的な溶出DNAの量の測定を伴った。有意な量のDNAが最初に溶出されたNaCl濃度を溶出点として記録した。ここでは、N基の導入により、溶出点が有意に低下することが示された。
【0100】
プロトン化性のA基(DEAPS)のみでコートした支持材(AAK01−30)は、pH7.0で1600mM NaClに溶出点を示す。したがって、核酸の溶出には、かなり高い塩濃度が必要とされる。混合型A/N修飾シリカゲルAAK01−10の溶出点は、約700mM NaClである。したがって、溶出に必要とされるイオン強度は50%より大きく低下した。
【0101】
A.2)メタクリル酸ヒドロキシエチル(HEMA)でのA/N修飾シリカゲルシラン処理(AAK01−11)
材料
出発材料:修飾シリカゲルAAK01−10(プロトコルA.1を参照のこと)
コーティング試薬:メタクリル酸ヒドロキシエチル(HEMA)
調製プロトコル
HEMAオリゴマーを、Cu(I)触媒型原子移動ラジカル重合(ATRP)の補助により、事前に結合させたクロロシラン上で合成する。
【0102】
反応フラスコ内で、先にAl2O3によって活性化させておいた20mlのHEMAと、20mlの脱イオン水を、アルゴンで30分間リンス処理し、次いで、68mgのCuCl2、46mgのCuBr2および313mgのビスピリジンと混合し、混合後、10gのAAK01−20を添加する。混合物を室温で4時間攪拌し、その直後、P3ガラスフリットによって吸引しながら濾別する。ガラスフリット上の物質を、まず、100mM NaCl/100mM EDTAバッファーで数回洗浄し、次いで、VE水で数回洗浄し、8mlのメタノールで1回、7mlのメタノールで2回洗浄する。最終支持材AAK01−11を60℃で14時間乾燥させた。
【0103】
比較のため、DEAPS修飾支持材AAK01−30をHEMAと反応させ、同様に処理した。この反応の最終生成物をAAK01−31と命名した。官能基としてDEAPSは第3級アミンのみを有し、重合性アリル基は有しないため、HEMAカップリングはないはずである。
【0104】
溶出点の測定を伴う修飾支持材AAK01−11を用いたプラスミドDNAの精製
実験は、A.1に記載のようにして行なった。プロトン化性基(DEAPS)のみでコートした支持材AAK01−31は、pH7.0で1500mM NaClに溶出点を示す。したがって、核酸の溶出には、かなり高い塩濃度が必要とされる。HEMA修飾シリカゲルAK01−11の溶出点は約400mM NaClである。したがって、溶出に必要とされるイオン強度は、ほぼ75%低下した。
【0105】
A.3)種々のモル比のシランでのシリカビーズのシラン処理
a)FFシリカビーズ
以下の実験設定を選択した。使用した支持材はFFシリカビーズであった。
【0106】
【表1】
*:溶出バッファーの組成:20mM 塩化カリウム;50mM Tris水溶液,pHXに調整;
**:QNバッファーの組成:1600mM 塩化ナトリウム;50mM Tris15%エタノール水溶液,pH7に調整
結果を図6に示す。
b)MagAttract Beads G(QIAGEN)
以下の実験設定を選択した。使用した支持材は、磁性シリカビーズであるMagAttract Beads G(QIAGEN製)であった。
【0107】
【表2】
*:溶出バッファーの組成:20mM 塩化カリウム;水中50mM Tris,pHXに調整;
**:QNバッファーの組成:1600mM 塩化ナトリウム;50mM Tris15%エタノール水溶液,pH7に調整.
結果を図7に示す。
【0108】
B) 開始剤分子での支持材の修飾
本発明によるランダムおよび/またはブロック(コ)ポリマーは、まず、開始剤基Tを適切な支持材に、本明細書において以下に一例として示すようにして適用することにより調製する。次いで、前記開始剤基を、後続の工程においてA基および/またはN基のリガンド/ポリマー鎖を適用する開始点として使用する。
【0109】
B.1)シリカゲル上での[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシランのシラン処理
材料
支持材:シリカゲル(Fuji MB 1500−40/75/ロット:HT70594)
コーティング試薬:[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシラン(BPDS)
シランの量:800μmol/gの支持材
調製プロトコル
還流冷却器および排水器を備えた250ml容KPG攪拌器(乾燥)に、まず、10.0gの支持材、200mlの乾燥シクロヘキサンおよび2490mgの[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシランを仕込んだ。
【0110】
混合物を、攪拌しながら(100l/分)85℃(油浴温度)で一晩(16時間)反応させた。次いで油浴を除き、懸濁液を、攪拌しながら1/2時間以内で冷却した。修飾支持材/シリカゲル開始剤(KI04)をP3フリットによって除去し、その上面を7×20mlのヘキサンで洗浄した。次いで、修飾支持材を、40℃の真空乾燥キャビネット内で、一晩(ほぼ12時間)乾燥させた。
【0111】
5つのロットのシリカゲル開始剤(KI 04 a〜e)を、上記の調製プロトコルに従って調製し、後続の工程で本発明の弱いイオン交換体を調製するための開始点として使用した。
【0112】
C)ATRPによる弱いイオン交換体の調製
本発明の弱いイオン交換体は、A基および/またはN基のランダムポリマー鎖を、出発材料である開始剤基T(KI04)担持修飾支持材上で伸長させることにより調製した。
【0113】
C.1)B.1に記載の修飾支持材/シリカゲル開始剤上へのA基/Nのランダムポリマーのグラフト化
この実験の組の出発材料は、開始剤としてBPDSを有するシリカゲル開始剤(KI04d)とした。
【0114】
このために、モノマーDMAEMA(2−ジメチルアミノ)エチルメタクリレート/A基)およびHEMA(メタクリル酸ヒドロキシエチル/N基)を、互いに以下の比で適用した:
DMAEMA:HEMA
100:0 T 16.0mmol:0.0mmol(KI04d−01)
50:50 T 8.0mmol:8.0mmol(KI04d−03)
30:70 T 4.8mmol:11.2mmol(KI04d−04)
20:80 T 3.2mmol:12.8mmol(KI04d−05)
10:90 T 1.6mmol:14.4mmol(KI04d−07)
材料
シリカゲル開始剤:KI04d
モノマー:DMAEMAおよびHEMA
リガンド:0.96mmolの2,2’−ビピリジル(bdy)
銅塩:0.48mmolの臭化銅I(精製済)
溶媒:36mlのジメチルホルムアミド(DMF)
開始剤:Cu(I):リガンド比=1:4:8
開始剤:モノマー(1種類または複数種)比=1:133
調製プロトコル
KPG攪拌器(ガラス製ガイドスリーブ)、アルゴン接続口および計泡器(またはダイヤフラムポンプへの接続口)を備えた100ml容三つ口フラスコに、まず、150mgのbdyと68.8mgの臭化銅Iを仕込んだ。
【0115】
装置内を、ダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、空気中の酸素が入るのを抑制するために、緩徐なアルゴン流を装置内に永続的に通した。
【0116】
並行して、36mlのDMF(リガンド)およびモノマーを脱気した。
【0117】
次いで、これらの液体を、調製物に応じて以下の初期重量比を有するモノマーと共に、排水器を備えた二つ口フラスコ内に導入した。
【0118】
KI04d−01=2700μlのDMAEMA:0.0μlのHEMA
KI04d−03=1350μlのDMAEMA:972μlのHEMA
KI04d−04=810μlのDMAEMA:1360μlのHEMA
KI04d−05=540μlのDMAEMA:1555μlのHEMA
KI04d−07=270μlのDMAEMA:1750μlのHEMA
リガンドとモノマー(1種類または複数種)の混合物を、まず、超音波浴内で5分間処理した後、ガス空間を高真空下で5分間真空にした。
【0119】
リガンド−モノマー混合物をピペット使用により、先で導入したbdy/臭化銅Iに添加した。装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。この後、装置内に永続的に緩徐なアルゴン流を通した。
【0120】
銅リガンド錯体を、攪拌しながら(200l/分)15分間以内で溶解させた。この後、1.2gのKI 04dシリカゲル開始剤を添加した。
【0121】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、装置内に緩徐なアルゴン流を永続的に通した。
【0122】
混合物を40℃(油浴温度)で4時間攪拌した(200l/分)。
【0123】
上面に該基が伸長した支持材(以下、本明細書において単にシリカゲルと記載する)を、P3フリットによって直ちに取り出し、続いて、逐次、
5×8mlのDMF
5×8mlのTHF
5×8mlの0.1M EDTA/酢酸バッファー(pH3.6)(氷酢酸を含む0.2M酢酸ナトリウム)
2×8mlのTris/NaClバッファー(pH7.0)
3×8mlの水および
3×8mlのメタノール
で洗浄した。
【0124】
次いで、シリカゲルを、重量が一定になるまで、真空乾燥キャビネット内で40℃にて乾燥させた。
【0125】
収率:
KI04d−01=1.31g
KI04d−03=1.20g
KI04d−04=1.23g
KI04d−05=1.29g
KI04d−07=1.21g
得られたアニオン交換体(修飾支持材、すなわちシリカゲル)および出発材料(シリカゲル開始剤)を、そのpDNA結合能に関して試験し(これは、本明細書において以下により詳細に説明する)、対応する溶出プロフィールを記録した。
【0126】
また、アニオン交換体をCHNの含有量について試験し、このポリマー鎖長から、Nに対するAの比を求めた。使用したモノマーの(モル)質量比は、得られるシリカゲルに反映されることを示すことができた。
【0127】
溶出点の測定を伴う、修飾支持材KI04d−01〜KI04d−07を用いたプラスミドDNAの精製
【0128】
【表3】
手順
単独の測定を、シリカゲル毎、および2種類の溶出バッファーのいずれかについて行なった。
【0129】
このために、各場合において、50.00mg(±5mg)のシリカゲルを、エッペンドルフの2ml容Safe Lock反応チューブ(以下、本明細書においてエッペンドルフチューブという)内に計り入れ、DNA溶液のため、計量した各シリカゲルに対して100μgのpcmvbを結合バッファーと混合して1mlにした。
【0130】
続いて、1mlのDNA溶液を、計量したシリカゲルの入った各エッペンドルフチューブに添加し、エッペンドルフチューブを密封し、短時間ボルテックスした。
【0131】
次いで、エッペンドルフチューブを、回転式(end−over−end)振盪機内に中速度で5分間載置した。振盪後、エッペンドルフチューブを短時間遠心分離した。
【0132】
次いで、シリカゲル−DNA懸濁液を、ピペットにより、切断先端を用いて、ボトムフリットを有する調製Tip 20ブロック(以下、本明細書において、Tipという)内に移した。Tipの下方には、上清み収集のための2ml容エッペンドルフチューブを準備した。
【0133】
エッペンドルフチューブ内に生じ得る残留物質を捕捉するため、各場合において、該チューブを0.9mlの結合バッファーでリンス処理し、該バッファーは、対応するTipにも同様に適用した。
【0134】
液体内部からの滴下が停止したら、残留液状物をすべて内部で押圧し、新たなエッペンドルフ収集チューブをTipカラムの下方に配置した。
【0135】
続いて、1mlの洗浄バッファーを、洗浄のためにピペットで各Tip内に移した。液状物が内部から滴下しなくなったら、残留液状物をすべて内部で押圧し、エッペンドルフ収集チューブ(洗浄液が入っている)を脇に置き、新たなエッペンドルフ収集チューブをTipカラムの下方に配置した。
【0136】
続いて、1mlの適切な溶出バッファーをピペットで各Tip内に移した。この場合も、液状物が内部から滴下しなくなったら、残留液状物をすべて内部で押圧し、エッペンドルフ収集チューブ(溶出液が入っている)を脇に置き、新たなエッペンドルフ収集チューブをTipカラムの下方に配置した。
【0137】
残留結合DNAを分離させるため、1mlのTris−NaClバッファーをピペットで各Tip内に移した。液状物が内部から滴下しなくなったら、残留液状物をすべて内部で押圧し、エッペンドルフ収集チューブ(Tris−NaCl溶液が入っている)を脇に置き、Tipカラムを処分した。
【0138】
DNA濃度をSpectraMaxによって測定するため、各場合において、採取した100μlの溶液をUVプレートに移した。
【0139】
上清みのブランクを、10μlのEBバッファーと180μlの結合バッファーを混合することにより調製した。100μlのこの溶液をUVプレート内に導入した。使用したDNAの量の参照として、100μlのDNA溶液を90μlの結合バッファーと混合し、100μlのこの溶液をUVプレート内に導入した。
【0140】
溶出液に対して使用したブランクは、100μlの対応する溶出バッファーとした。
【0141】
100μlのTris−NaClバッファーを、Tris−NaCl溶液ブランクとして使用した。
【0142】
有意な量のDNAが最初に溶出されたNaCl濃度を溶出点として記録した。
【0143】
【表4】
この場合も、結果(上記の表に示し、図8に示したもの)により、N基の導入により溶出点が有意に低下することが示された。
【0144】
pH8.5では、A基(DMAEMA)のみでコートした支持材/シリカゲル(KI04d−01)での溶出点は、1032mM NaClであった。したがって、核酸を溶出させるためには、かなり高い塩濃度が必要であった。混合型A/N修飾シリカゲルKI04d−05の溶出点は、約580mM NaClであった。したがって、溶出に必要とされるイオン強度はほぼ50%低下した。
【0145】
C.2)B.1に記載の修飾支持材/シリカゲル開始剤上へのA基/N基のブロックポリマーのグラフティング
この実験の組の出発材料は、BPDSを開始剤とするシリカゲル開始剤(KI04e)とした。
【0146】
この後、A基の少なくとも1種類のモノマーのアミノポリマー鎖をグラフティングする第1の工程を行なった。そして、この後、このようにして調製したシリカゲルに、N基の少なくとも1種類のモノマー(好ましくは、HEMA)のポリマー鎖を適用する第2の工程を行なった。
【0147】
C.2.1)工程1
材料
シリカゲル開始剤:KI04e
モノマー:DMAEMA
リガンド:7.2mmolの2,2’−ビピリジル(bdy)
銅塩:3.6mmolの臭化銅I(精製済)
溶媒:270mlのジメチルホルムアミド(DMF)
開始剤:Cu(I):リガンド比=1:4:8
開始剤:モノマー比=1:133
調製プロトコル
KPG攪拌器(ガラス製ガイドスリーブ)、アルゴン接続口および計泡器(またはダイヤフラムポンプへの接続口)を備えた500ml容三つ口フラスコに、まず、1125mgのbdyと516mgの臭化銅Iを仕込んだ。
【0148】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、装置内に緩徐なアルゴン流を永続的に通した。
【0149】
並行して、270mlのDMFと20.25mlのDMAEMAを脱気した。この目的のため、この2種類の液体を、排水器を備えた二つ口フラスコ内に導入した。混合物を、まず、超音波浴内で5分間処理し、この後、ガス空間を高真空下で5分間真空にした。
【0150】
DMF−DMAEMA混合物を、ピペット使用によりbdy/臭化銅Iに添加した。
【0151】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。この後、装置内に永続的に緩徐なアルゴン流を通した。
【0152】
銅リガンド錯体を、攪拌しながら(200l/分)15分間以内で溶解させた。この後、9gのKI04eシリカゲル開始剤を添加した。
【0153】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、装置内に緩徐なアルゴン流を永続的に通した。
【0154】
混合物を40℃(油浴温度)で4時間攪拌した(200l/分)。
【0155】
上面にA基が伸長したKI04e−01シリカゲルを、P3フリットによって直ちに取り出し、続いて、逐次、
5×60mlのDMF
5×60mlのTHF
5×60mlの0.1 M EDTA/酢酸バッファー(pH3.6)(氷酢酸を含む0.2M酢酸ナトリウム)
2×60mlのTris/NaClバッファー(pH7.0)および
3×60mlの水
で洗浄した。
【0156】
次いで、KI04e−01シリカゲルを、重量が一定になるまで、真空乾燥キャビネット内で40℃にて乾燥させた。
【0157】
C.2.2)工程2
材料
シリカゲル開始剤:KI04e−01
モノマー:HEMA
リガンド:0.96mmolの2,2’−ビピリジル(bdy)
銅塩:0.48mmolの臭化銅I(精製済)
溶媒:36mlのジメチルホルムアミド(DMF)
開始剤:Cu(I):リガンド比=1:4:8
開始剤:モノマー比=1:200
調製プロトコル
KPG攪拌器(ガラス製ガイドスリーブ)、アルゴン接続口および計泡器(またはダイヤフラムポンプへの接続口)を備えた100ml容三つ口フラスコに、まず、150mgのbdyと68.8mgの臭化銅Iを仕込んだ。
【0158】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、空気中の酸素が入るのを抑制するために、装置内に緩徐なアルゴン流を永続的に通した。
【0159】
並行して、36mlのDMFと2916μlのHEMAを脱気した。この目的のため、この2種類の液体を、排水器を備えた二つ口フラスコ内に導入した。混合物を、まず、超音波浴内で5分間処理し、この後、ガス空間を高真空下で5分間真空にした。
【0160】
DMF−HEMA混合物を、ピペット使用によりbdy/臭化銅Iに添加した。
【0161】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。この後、装置内に永続的に緩徐なアルゴン流を通した。
【0162】
銅リガンド錯体を、攪拌しながら(200l/分)15分間以内で溶解させた。この後、1.2gのKI04e−01シリカゲル開始剤を添加した。
【0163】
装置内をダイヤフラムポンプによって1分間真空にし、次いでアルゴンで満たした。この手順を3回行なった。続いて、装置内に緩徐なアルゴン流を永続的に通した。
【0164】
混合物を、40℃(油浴温度)で6時間攪拌した(200l/分)。
【0165】
上面にA基が伸長したKI04e−01−01シリカゲルを、P3フリットによって直ちに取り出し、続いて、逐次、
5×8mlのDMF
5×8mlのTHF
5×8mlの0.1 M EDTA/酢酸バッファー(pH3.6)(氷酢酸を含む0.2M酢酸ナトリウム)
2×8mlのTris/NaClバッファー(pH7.0)
3×8mlの水および
3×8mlのメタノール
で洗浄した。
【0166】
次いで、KI04e−01−01シリカゲルを、重量が一定になるまで、真空乾燥キャビネット内で40℃にて乾燥させた。
【0167】
このシリカゲルで行なった溶出実験により、N基モノマーのグラフトポリマー鎖もまた、核酸(1つまたは複数)の立体遮蔽に有意に寄与し得、それにより、前記核酸(1つまたは複数)の結合強度が有意に低下し得ることが実証され得る。
【特許請求の範囲】
【請求項1】
核酸結合相を用いて結合と溶出によって核酸を精製する方法であって、該核酸結合相が:
(i)8〜13のpKを有する核酸結合性のA基と、
(ii)使用される結合pHにおいて中性電荷を有する電荷中性のN基と
を有すること;および
該方法が、少なくとも以下の工程:
(a)該核酸結合性のA基のpKよりも小さいpH(結合pH)で、該核酸を該核酸結合相に結合させる工程と、
(b)該結合pHよりも大きいpH(溶出pH)で、該核酸を溶出させる工程と
を有すること
を特徴とする方法。
【請求項2】
前記核酸結合相が修飾支持材であり、前記修飾が:
(i)リガンドIが少なくとも1つのA基を有し、リガンドIIが少なくとも1つのN基を有する、少なくとも該リガンドIと該リガンドIIとを有する混合物での該支持材の修飾;
(ii)核酸結合リガンドでの該支持材の修飾であって、該リガンドのそれぞれが該リガンド内に1つ以上のA基と1つ以上のN基とを有する、修飾;
(iii)該A基が、少なくとも1つのN基を有する化合物によって立体遮蔽される、該A基および該N基での該支持材の修飾;および
(iv)該実施形態(i)〜(iii)の少なくとも2つの組合せによる該支持材の修飾
からなる群より選択されることを特徴とする、請求項1に記載の方法。
【請求項3】
前記支持材が核酸結合性のA基で修飾されており、該支持材の一部だけがA基で占有されていることを特徴とする、核酸結合支持材を用いて結合と溶出によって核酸を精製する方法。
【請求項4】
前記支持材が、以下の特徴:
(i)該支持材が、A基を有するシランでコートされたシリカ表面を有し、シランの量が0.1〜50μmol、好ましくは0.1〜10μmolであること;および/または
(ii)該支持材が、さらに、請求項1によるN基を有すること;および/または
(iii)該支持材が、請求項2において規定される1つ以上の修飾を有すること
の1つ以上を有することを特徴とする、請求項3に記載の方法。
【請求項5】
前記核酸結合相が、以下の特徴:
(i)前記支持材が酸化物材料であること;および/または
(ii)該支持材が、Al2O3、TiO2、ZrO2、Ta2O5、SiO2およびポリケイ酸からなる群より選択されること;および/または
(iii)該支持材がSiO2もしくはポリケイ酸であること;および/または
(iv)該支持材が、特に、ポリスチレンおよびその誘導体、ポリアクリレートおよびポリメタクリレート、ならびにその誘導体、ポリウレタン、ナイロン、ポリエチレン、ポリプロピレン、ポリブチリデン、ならびにこれらの材料のコポリマーからなる群より選択される有機ポリマーであること;および/または
(v)該支持材が多糖、特に、ヒドロゲル、例えば、アガロース、セルロース、デキストラン、セファデックス、セファクリルもしくはキトサンであること;および/または
(vi)該支持材がガラスもしくは金属であること;および/または
(vii)該支持材が磁性であること
の1つ以上を満たす支持材を有することを特徴とする、請求項1〜4の1つ以上に記載の方法。
【請求項6】
前記核酸結合相が、以下の特徴:
(i)前記A基が、9〜13、好ましくは10〜12、特に好ましくは10〜11のpKを有すること;および/または
(ii)該A基が、第3級、第2級もしくは第1級アミノ基であること;および/または
(iii)該A基が、重合性モノマーの一部であること;および/または
(iv)該A基が、事前に反応性シランとして適用しておいた開始剤分子に結合されていること;および/または
(v)該A基が、反応性シラン基の一部として導入したものであること;および/または
(vi)該A基が:
【化7】
(式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基であり、
*は、アミノ、アミノメチル、アミノエチル、アミノプロピル、ジメチルアミノ、ジエチルアミノ、ジイソプロピルアミノ、ジプロピルアミノ、ジエタノールアミノ、ジプロパノールアミノ、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、エーテルアミン、ポリエーテルアミン、4−ジイソブチルアミノ−1−ブタン、6−ジプロピルアミノ−1−ヘキサンである)
からなる群より選択される化合物の一部として導入したものであること;および/または
(vi)該A基が、ジエチルアミノプロピルトリメトキシシラン、ジメチルアミノプロピルトリメトキシシランおよびN,N−ジイソプロピルアミノプロピルトリメトキシシランからなる群より選択される化合物の一部として導入したものであること
の1つ以上を満たすA基を有することを特徴とする、請求項1〜5の1つ以上に記載の方法。
【請求項7】
前記結合させる工程(a)および/または前記溶出させる工程(b)が、以下の特徴:
(i)該溶出させる工程(b)において、前記溶出pHはA基の前記pKよりも小さいこと;および/または
(ii)結合バッファーおよび/または溶出バッファーの塩濃度が1mM〜1000mM、1mM〜500mM、1mM〜250mMまたは1mM〜100mMであること;および/または
(iii)該塩濃度を、該結合させる工程(a)および/または該溶出させる工程(b)で変更しないか、あるいは溶出中で上昇させること;および/または
(iv)該結合バッファーのpHがpH2〜pH8、pH2〜pH7.5、pH4〜pH8またはpH4〜pH7.5であること、および/または
(v)該溶出バッファーのpHがpH2〜pH10、pH4〜pH10、pH7〜pH10またはpH8〜pH9であること;および/または
(vi)温度を、該結合させる工程(a)および/または該溶出させる工程(b)で変更しないか、あるいは溶出中で上昇させること;および/または
(vii)該溶出させる工程の温度が2℃〜95℃または21℃〜60℃であること
の1つ以上を満たす条件下で行なわれることを特徴とする、請求項1〜6の1つ以上に記載の方法。
【請求項8】
前記N基が、以下の特徴:
(i)該N基が、ヒドロキシル基、ジオール基、トリオール基、ポリオール基、糖類、エポキシド基、ハライド、アルキル基、好ましくは、C1〜C6アルキル基、アルケン基、アルキレン基、イミド基、エーテル基、もしくはポリエーテル基からなる群より選択されること、および/または
(ii)該N基が、2<pH<12のpH範囲の電荷中性であること;および/または
(iii)該N基が、使用される前記結合pHにおいて中性電荷を有すること;および/または
(iv)該N基が、重合性モノマーの一部として導入したものであること;および/または
(v)該N基が、事前に反応性シランとして適用しておいた開始剤分子に結合されていること;および/または
(vi)該N基が、反応性シラン基の一部として導入したものであること;および/または
(vii)該N基が、
【化8】
(式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基、特に、メチル、エチル、プロピル、イソプロピルであり;
*は、アミノ、アミノメチル、アミノエチル、アミノプロピル、ジメチルアミノ、ジエチルアミノ、ジイソプロピルアミノ、ジプロピルアミノ、ジエタノールアミノ、ジプロパノールアミノ、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、エーテルアミン、ポリエーテルアミン、4−ジイソブチルアミノ−1−ブタン、6−ジプロピルアミノ−1−ヘキサン、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、エタンジオール、プロパンジオール、プロパントリオール、ブタントリオール、3−グリシドオキシプロピル、エチルグリシジルエーテル、アルキル原子団、特に、C1〜C4アルキル原子団、特に、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、ハライドまたは水素である)
からなる群より選択される化合物の一部として導入したものであること
の1つ以上を満たすことを特徴とする、請求項1〜7の1つ以上に記載の方法。
【請求項9】
以下の特徴:
(i)前記支持材が、シラン処理によって前記A基と前記N基とを備えたものであること;および/または
(ii)該A基および/または該N基および/またはT基が、反応性シランによって導入されたものであること;および/または
(iii)該A基および/または該N基が、単官能性、二官能性もしくは三官能性の反応性シランまたは異なる官能基を有する少なくとも2つの反応性シランの混合物によって導入されたものであること;および/または
(iv)該A基および/または該N基が、アミノシラン、ジシラザン、クロロシランもしくはアルコキシシランからなる群より選択される反応性シランによって導入されたものであること;および/または
(v)該N基に対する該A基の比率が1%〜99%、1〜50%、好ましくは1%〜25%であること
の1つ以上を満たすことを特徴とする、請求項1〜8の1つ以上に記載の方法。
【請求項10】
以下の特徴:
(i)前記支持材が、開始剤分子Tで修飾し、続いてモノマーの形態で導入された前記A基および/または前記N基で修飾したものであること;および/または
(ii)該支持材が、以下の特徴:
(aa)該開始剤分子が反応性シランであること;および/または
(bb)該開始剤分子が、
【化9】
(式中、Xは、ハロゲン、特にCl、Br、Iであり、
Y1、Y2、Y3またはY4は、互いに独立して、R、OR、OHまたはHであり、
Rは、C1−C3アルキルである)
からなる群より選択されること
の1つ以上を有する該開始剤分子Tで修飾したものであること、
(iii)該開始剤分子が2−(クロロメチル)アリルトリメトキシシランであること;
(iv)該開始剤分子が[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシランであること、
(v)該A基が、
N−(3−アミノメチル)メタクリルアミド、N−(3−アミノエチル)メタクリルアミド、N−(3−アミノプロピル)メタクリルアミド、N−(3−アミノイソプロピル)メタクリルアミド、N,N−ジメチルアクリルアミド、N,N−ジエチルアクリルアミド、N,N−ジイソプロピルアクリルアミド、N,N−(ジメチルアミノ)エチルアクリルアミド、N,N−(ジメチルアミノ)エチルアクリレート、N,N−(ジメチルアミノ)エチルメタクリルアミド、N,N−(ジメチルアミノ)エチルメタクリレート、N,N−(ジメチルアミノ)プロピルアクリルアミド、N,N−(ジメチルアミノ)プロピルアクリレート、N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート、N,N−(ジエチルアミノ)エチルアクリルアミド、N,N−(ジエチルアミノ)エチルアクリレート、N,N−(ジエチルアミノ)エチルメタクリルアミド、N,N−(ジエチルアミノ)エチルメタクリレート、N,N−(ジエチルアミノ)プロピルアクリルアミド、N,N−(ジエチルアミノ)プロピルアクリレート、N,N−(ジエチルアミノ)プロピルメタクリルアミド、N,N−(ジエチルアミノ)プロピルメタクリレート、N,N−(ジイソプロピルアミノ)エチルアクリルアミド、N,N−(ジイソプロピルアミノ)エチルアクリレート、N,N−(ジイソプロピルアミノ)エチルメタクリルアミド、N,N−(ジイソプロピルアミノ)エチルメタクリレート、N,N−(ジイソプロピルアミノ)プロピルアクリルアミド、N,N−(ジイソプロピルアミノ)プロピルアクリレート、N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート、2−(ジイソプロピルアミノ)エチルメタクリレート
からなる群より選択されるモノマーによって導入されたものであること、および/または
(vi)該N基が、
アクリル酸ブチル、アクリル酸プロピル、アクリル酸エチル、アクリル酸メチル、メタクリル酸グリシジル、メタクリル酸ヒドロキシエチル(HEMA)、メタクリル酸グリシドオキシプロピル、グリセロールモノメタクリレート(異性体混合物)、グリコールモノメタクリレート、アクリル酸グリシジル、アクリル酸ヒドロキシエチル、アクリル酸グリシドオキシプロピル、グリセロールモノアクリレート(異性体混合物)、グリコールモノアクリレートおよびN−アクリルオキシスクシンイミド
からなる群より選択されるモノマーによって導入されたものであること
の1つ以上を特徴とする、請求項2〜9の1つ以上に記載の方法。
【請求項11】
前記支持材が、A基を有する化合物で少なくとも一部が官能性付与されている開始剤基Tを有し、該A基がN基によって立体遮蔽されており、該立体遮蔽が、以下の修飾:
(i)該N基が、該A基を立体遮蔽する分枝鎖もしくは非分枝鎖のオリゴマーもしくはポリマーの形態であること;および/または
(ii)該N基が、n=10〜1000、好ましくはn=10〜100の鎖長を有するモノマーで構成されたオリゴマーもしくはポリマーの形態であること;および/または
(iii)該N基が、好ましくは、下記式:
【化10】
によるポリアクリレートとして存在していること、
(iv)該N基が、例えば、イソプロピル、イソブチル、tert−ブチルもしくはn−ヘキシル原子団などの空間的に広がった基を有すること、あるいは該基であること
の1つ以上によってもたらされることを特徴とする、請求項1〜10の1つ以上に記載の方法。
【請求項12】
以下の代替法(A)〜(F):
代替法(A):前記リガンドIが少なくとも1つのA基を有し、前記リガンドIIが少なくとも1つのN基を有する、少なくとも該リガンドIと該リガンドIIとを有する混合物での前記支持材の官能性付与;および/または
代替法(B):
(a)開始剤分子Tと少なくとも1つのA基を有するリガンドとの混合物での該支持材の官能性付与ならびに、(b)少なくとも1つのN基を有する化合物の添加ならびに該開始剤分子Tへの該化合物の結合;および/または
代替法(C):核酸結合リガンドでの該支持材の官能性付与であって、該リガンドのそれぞれが該リガンド内に1つ以上のA基と1つ以上のN基とを有する官能性付与;および/または
代替法(D):該A基が、少なくとも1つのN基を有する化合物によって立体遮蔽される、該A基および該N基での該支持材の官能性付与;および/または
代替法(E):準化学量論量のA基での該支持材の官能性付与;および/または
代替法(F):代替法(A)〜(E)の少なくとも2つの組合せによる該支持材の官能性付与
の少なくとも1つに従って行なわれる、請求項1〜6および8〜11の1つ以上において規定される核酸結合支持材の調製方法。
【請求項13】
以下の特徴:
(i)前記A基が、第3級、第2級もしくは第1級アミノ基であること;および/または
(ii)前記N基が、ヒドロキシル基、ジオール基、トリオール基、ポリオール基、糖類、エポキシド基、ハライド、C1〜C6アルキル基、アルケン基、アルキレン基、イミド基、エーテル基、もしくはポリエーテル基からなる群より選択されること、および/または
(iii)該N基が、前記溶出pHにおいて、好ましくは2<pH<12の特定のpH範囲において電荷中性であること;および/または
(iv)該A基および/または該N基がモノマーによって導入されること;および/または
(v)該A基および/または該N基がシラン処理によって導入されること;および/または
(vi)該A基および/または該N基が反応性シランによって導入されること;および/または
(vii)該A基および/または該N基が、単官能性または二官能性および/または三官能性の反応性シランの使用によって導入されること;および/または
(viii)該A基および/または該N基が、アミノシラン、ジシラザン、クロロシランもしくはアルコキシシランからなる群より選択される反応性シランの使用によって導入されること;および/または
(ix)該A基および/または該N基が:
【化11】
(式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基、特に、メチル、エチル、プロピル、イソプロピルであり;
*は、アミノ、アミノメチル、アミノエチル、アミノプロピル、ジメチルアミノ、ジエチルアミノ、ジイソプロピルアミノ、ジプロピルアミノ、ジエタノールアミノ、ジプロパノールアミノ、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、エーテルアミン、ポリエーテルアミン、4−ジイソブチルアミノ−1−ブタン、6−ジプロピルアミノ−1−ヘキサン、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、エタンジオール、プロパンジオール、プロパントリオール、ブタントリオール、3−グリシドオキシプロピル、エチルグリシジルエーテル、アルキル原子団、特に、C1〜C4アルキル原子団、特に、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、ハライドまたは水素である)
からなる群より選択される反応性シランによって導入されること、
(x)該A基および該N基が、該A基の比率が1%〜99%、1〜50%、好ましくは1%〜25%となるような比で導入されること;および/または
(xi)該支持材が、以下の特徴:
(aa)該開始剤分子がシラン処理によって適用されること;および/または
(bb)該開始剤分子がシランであること;および/または
(cc)該開始剤分子が、
【化12】
【化13】
(式中、Xは、ハロゲン、特にCl、Br、Iであり、
Y1、Y2、Y3またはY4は、互いに独立して、R、OR、OHまたはHであり、
Rは、C1〜C3アルキルである)
からなる群より選択されること
の1つ以上を有する開始剤分子Tで修飾されること、
(xii)使用される該開始剤分子が、2−(クロロメチル)アリルトリメトキシシランであること;
(xiii)使用される該開始剤分子が、[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシランであること;および/または
(xiv)該A基が、
2−(ジイソプロピルアミノ)エチルメタクリレート、
N−(3−アミノメチル)メタクリルアミド、N−(3−アミノエチル)メタクリルアミド、
N−(3−アミノプロピル)メタクリルアミド、N−(3−アミノイソプロピル)メタクリルアミド、
N,N−ジメチルアクリルアミド、N,N−ジエチルアクリルアミド、N,N−ジイソプロピルアクリルアミド、
N,N−(ジメチルアミノ)エチルアクリルアミド、N,N−(ジメチルアミノ)エチルアクリレート、
N,N−(ジメチルアミノ)エチルメタクリルアミド、N,N−(ジメチルアミノ)エチルメタクリレート、
N,N−(ジメチルアミノ)プロピルアクリルアミド、N,N−(ジメチルアミノ)プロピルアクリレート、
N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート、
N,N−(ジエチルアミノ)エチルアクリルアミド、N,N−(ジエチルアミノ)エチルアクリレート、
N,N−(ジエチルアミノ)エチルメタクリルアミド、N,N−(ジエチルアミノ)エチルメタクリレート、
N,N−(ジエチルアミノ)プロピルアクリルアミド、N,N−(ジエチルアミノ)プロピルアクリレート、
N,N−(ジエチルアミノ)プロピルメタクリルアミド、N,N−(ジエチルアミノ)プロピルメタクリレート、
N,N−(ジイソプロピルアミノ)エチルアクリルアミド、N,N−(ジイソプロピルアミノ)エチルアクリレート、
N,N−(ジイソプロピルアミノ)エチルメタクリルアミド、N,N−(ジイソプロピルアミノ)エチルメタクリレート、
N,N−(ジイソプロピルアミノ)プロピルアクリルアミド、N,N−(ジイソプロピルアミノ)プロピルアクリレート、
N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート
からなる群より選択されるモノマーによって導入されること、および/または
(xiv)N基が、アクリル酸ブチル、アクリル酸プロピル、アクリル酸エチル、アクリル酸メチル、メタクリル酸ヒドロキシエチル(HEMA)、メタクリル酸グリシジル、メタクリル酸グリシドオキシプロピル、グリセロールモノメタクリレート(異性体混合物)、N−アクリルオキシスクシンイミドおよびグリコールモノメタクリレート
からなる群より選択されるモノマーによって導入されること
の少なくとも1つを満たすことを特徴とする、請求項12に記載の方法。
【請求項14】
核酸を精製するための、請求項1〜6および8〜11の1つ以上において規定される、または請求項12と13のいずれかにより得られ得る、核酸結合支持材の使用。
【請求項15】
請求項1〜6および8〜11の1つ以上による支持材、または請求項12もしくは13により得られ得る支持材を有することを特徴とする、核酸を精製するためのキット。
【請求項1】
核酸結合相を用いて結合と溶出によって核酸を精製する方法であって、該核酸結合相が:
(i)8〜13のpKを有する核酸結合性のA基と、
(ii)使用される結合pHにおいて中性電荷を有する電荷中性のN基と
を有すること;および
該方法が、少なくとも以下の工程:
(a)該核酸結合性のA基のpKよりも小さいpH(結合pH)で、該核酸を該核酸結合相に結合させる工程と、
(b)該結合pHよりも大きいpH(溶出pH)で、該核酸を溶出させる工程と
を有すること
を特徴とする方法。
【請求項2】
前記核酸結合相が修飾支持材であり、前記修飾が:
(i)リガンドIが少なくとも1つのA基を有し、リガンドIIが少なくとも1つのN基を有する、少なくとも該リガンドIと該リガンドIIとを有する混合物での該支持材の修飾;
(ii)核酸結合リガンドでの該支持材の修飾であって、該リガンドのそれぞれが該リガンド内に1つ以上のA基と1つ以上のN基とを有する、修飾;
(iii)該A基が、少なくとも1つのN基を有する化合物によって立体遮蔽される、該A基および該N基での該支持材の修飾;および
(iv)該実施形態(i)〜(iii)の少なくとも2つの組合せによる該支持材の修飾
からなる群より選択されることを特徴とする、請求項1に記載の方法。
【請求項3】
前記支持材が核酸結合性のA基で修飾されており、該支持材の一部だけがA基で占有されていることを特徴とする、核酸結合支持材を用いて結合と溶出によって核酸を精製する方法。
【請求項4】
前記支持材が、以下の特徴:
(i)該支持材が、A基を有するシランでコートされたシリカ表面を有し、シランの量が0.1〜50μmol、好ましくは0.1〜10μmolであること;および/または
(ii)該支持材が、さらに、請求項1によるN基を有すること;および/または
(iii)該支持材が、請求項2において規定される1つ以上の修飾を有すること
の1つ以上を有することを特徴とする、請求項3に記載の方法。
【請求項5】
前記核酸結合相が、以下の特徴:
(i)前記支持材が酸化物材料であること;および/または
(ii)該支持材が、Al2O3、TiO2、ZrO2、Ta2O5、SiO2およびポリケイ酸からなる群より選択されること;および/または
(iii)該支持材がSiO2もしくはポリケイ酸であること;および/または
(iv)該支持材が、特に、ポリスチレンおよびその誘導体、ポリアクリレートおよびポリメタクリレート、ならびにその誘導体、ポリウレタン、ナイロン、ポリエチレン、ポリプロピレン、ポリブチリデン、ならびにこれらの材料のコポリマーからなる群より選択される有機ポリマーであること;および/または
(v)該支持材が多糖、特に、ヒドロゲル、例えば、アガロース、セルロース、デキストラン、セファデックス、セファクリルもしくはキトサンであること;および/または
(vi)該支持材がガラスもしくは金属であること;および/または
(vii)該支持材が磁性であること
の1つ以上を満たす支持材を有することを特徴とする、請求項1〜4の1つ以上に記載の方法。
【請求項6】
前記核酸結合相が、以下の特徴:
(i)前記A基が、9〜13、好ましくは10〜12、特に好ましくは10〜11のpKを有すること;および/または
(ii)該A基が、第3級、第2級もしくは第1級アミノ基であること;および/または
(iii)該A基が、重合性モノマーの一部であること;および/または
(iv)該A基が、事前に反応性シランとして適用しておいた開始剤分子に結合されていること;および/または
(v)該A基が、反応性シラン基の一部として導入したものであること;および/または
(vi)該A基が:
【化7】
(式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基であり、
*は、アミノ、アミノメチル、アミノエチル、アミノプロピル、ジメチルアミノ、ジエチルアミノ、ジイソプロピルアミノ、ジプロピルアミノ、ジエタノールアミノ、ジプロパノールアミノ、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、エーテルアミン、ポリエーテルアミン、4−ジイソブチルアミノ−1−ブタン、6−ジプロピルアミノ−1−ヘキサンである)
からなる群より選択される化合物の一部として導入したものであること;および/または
(vi)該A基が、ジエチルアミノプロピルトリメトキシシラン、ジメチルアミノプロピルトリメトキシシランおよびN,N−ジイソプロピルアミノプロピルトリメトキシシランからなる群より選択される化合物の一部として導入したものであること
の1つ以上を満たすA基を有することを特徴とする、請求項1〜5の1つ以上に記載の方法。
【請求項7】
前記結合させる工程(a)および/または前記溶出させる工程(b)が、以下の特徴:
(i)該溶出させる工程(b)において、前記溶出pHはA基の前記pKよりも小さいこと;および/または
(ii)結合バッファーおよび/または溶出バッファーの塩濃度が1mM〜1000mM、1mM〜500mM、1mM〜250mMまたは1mM〜100mMであること;および/または
(iii)該塩濃度を、該結合させる工程(a)および/または該溶出させる工程(b)で変更しないか、あるいは溶出中で上昇させること;および/または
(iv)該結合バッファーのpHがpH2〜pH8、pH2〜pH7.5、pH4〜pH8またはpH4〜pH7.5であること、および/または
(v)該溶出バッファーのpHがpH2〜pH10、pH4〜pH10、pH7〜pH10またはpH8〜pH9であること;および/または
(vi)温度を、該結合させる工程(a)および/または該溶出させる工程(b)で変更しないか、あるいは溶出中で上昇させること;および/または
(vii)該溶出させる工程の温度が2℃〜95℃または21℃〜60℃であること
の1つ以上を満たす条件下で行なわれることを特徴とする、請求項1〜6の1つ以上に記載の方法。
【請求項8】
前記N基が、以下の特徴:
(i)該N基が、ヒドロキシル基、ジオール基、トリオール基、ポリオール基、糖類、エポキシド基、ハライド、アルキル基、好ましくは、C1〜C6アルキル基、アルケン基、アルキレン基、イミド基、エーテル基、もしくはポリエーテル基からなる群より選択されること、および/または
(ii)該N基が、2<pH<12のpH範囲の電荷中性であること;および/または
(iii)該N基が、使用される前記結合pHにおいて中性電荷を有すること;および/または
(iv)該N基が、重合性モノマーの一部として導入したものであること;および/または
(v)該N基が、事前に反応性シランとして適用しておいた開始剤分子に結合されていること;および/または
(vi)該N基が、反応性シラン基の一部として導入したものであること;および/または
(vii)該N基が、
【化8】
(式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基、特に、メチル、エチル、プロピル、イソプロピルであり;
*は、アミノ、アミノメチル、アミノエチル、アミノプロピル、ジメチルアミノ、ジエチルアミノ、ジイソプロピルアミノ、ジプロピルアミノ、ジエタノールアミノ、ジプロパノールアミノ、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、エーテルアミン、ポリエーテルアミン、4−ジイソブチルアミノ−1−ブタン、6−ジプロピルアミノ−1−ヘキサン、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、エタンジオール、プロパンジオール、プロパントリオール、ブタントリオール、3−グリシドオキシプロピル、エチルグリシジルエーテル、アルキル原子団、特に、C1〜C4アルキル原子団、特に、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、ハライドまたは水素である)
からなる群より選択される化合物の一部として導入したものであること
の1つ以上を満たすことを特徴とする、請求項1〜7の1つ以上に記載の方法。
【請求項9】
以下の特徴:
(i)前記支持材が、シラン処理によって前記A基と前記N基とを備えたものであること;および/または
(ii)該A基および/または該N基および/またはT基が、反応性シランによって導入されたものであること;および/または
(iii)該A基および/または該N基が、単官能性、二官能性もしくは三官能性の反応性シランまたは異なる官能基を有する少なくとも2つの反応性シランの混合物によって導入されたものであること;および/または
(iv)該A基および/または該N基が、アミノシラン、ジシラザン、クロロシランもしくはアルコキシシランからなる群より選択される反応性シランによって導入されたものであること;および/または
(v)該N基に対する該A基の比率が1%〜99%、1〜50%、好ましくは1%〜25%であること
の1つ以上を満たすことを特徴とする、請求項1〜8の1つ以上に記載の方法。
【請求項10】
以下の特徴:
(i)前記支持材が、開始剤分子Tで修飾し、続いてモノマーの形態で導入された前記A基および/または前記N基で修飾したものであること;および/または
(ii)該支持材が、以下の特徴:
(aa)該開始剤分子が反応性シランであること;および/または
(bb)該開始剤分子が、
【化9】
(式中、Xは、ハロゲン、特にCl、Br、Iであり、
Y1、Y2、Y3またはY4は、互いに独立して、R、OR、OHまたはHであり、
Rは、C1−C3アルキルである)
からなる群より選択されること
の1つ以上を有する該開始剤分子Tで修飾したものであること、
(iii)該開始剤分子が2−(クロロメチル)アリルトリメトキシシランであること;
(iv)該開始剤分子が[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシランであること、
(v)該A基が、
N−(3−アミノメチル)メタクリルアミド、N−(3−アミノエチル)メタクリルアミド、N−(3−アミノプロピル)メタクリルアミド、N−(3−アミノイソプロピル)メタクリルアミド、N,N−ジメチルアクリルアミド、N,N−ジエチルアクリルアミド、N,N−ジイソプロピルアクリルアミド、N,N−(ジメチルアミノ)エチルアクリルアミド、N,N−(ジメチルアミノ)エチルアクリレート、N,N−(ジメチルアミノ)エチルメタクリルアミド、N,N−(ジメチルアミノ)エチルメタクリレート、N,N−(ジメチルアミノ)プロピルアクリルアミド、N,N−(ジメチルアミノ)プロピルアクリレート、N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート、N,N−(ジエチルアミノ)エチルアクリルアミド、N,N−(ジエチルアミノ)エチルアクリレート、N,N−(ジエチルアミノ)エチルメタクリルアミド、N,N−(ジエチルアミノ)エチルメタクリレート、N,N−(ジエチルアミノ)プロピルアクリルアミド、N,N−(ジエチルアミノ)プロピルアクリレート、N,N−(ジエチルアミノ)プロピルメタクリルアミド、N,N−(ジエチルアミノ)プロピルメタクリレート、N,N−(ジイソプロピルアミノ)エチルアクリルアミド、N,N−(ジイソプロピルアミノ)エチルアクリレート、N,N−(ジイソプロピルアミノ)エチルメタクリルアミド、N,N−(ジイソプロピルアミノ)エチルメタクリレート、N,N−(ジイソプロピルアミノ)プロピルアクリルアミド、N,N−(ジイソプロピルアミノ)プロピルアクリレート、N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート、2−(ジイソプロピルアミノ)エチルメタクリレート
からなる群より選択されるモノマーによって導入されたものであること、および/または
(vi)該N基が、
アクリル酸ブチル、アクリル酸プロピル、アクリル酸エチル、アクリル酸メチル、メタクリル酸グリシジル、メタクリル酸ヒドロキシエチル(HEMA)、メタクリル酸グリシドオキシプロピル、グリセロールモノメタクリレート(異性体混合物)、グリコールモノメタクリレート、アクリル酸グリシジル、アクリル酸ヒドロキシエチル、アクリル酸グリシドオキシプロピル、グリセロールモノアクリレート(異性体混合物)、グリコールモノアクリレートおよびN−アクリルオキシスクシンイミド
からなる群より選択されるモノマーによって導入されたものであること
の1つ以上を特徴とする、請求項2〜9の1つ以上に記載の方法。
【請求項11】
前記支持材が、A基を有する化合物で少なくとも一部が官能性付与されている開始剤基Tを有し、該A基がN基によって立体遮蔽されており、該立体遮蔽が、以下の修飾:
(i)該N基が、該A基を立体遮蔽する分枝鎖もしくは非分枝鎖のオリゴマーもしくはポリマーの形態であること;および/または
(ii)該N基が、n=10〜1000、好ましくはn=10〜100の鎖長を有するモノマーで構成されたオリゴマーもしくはポリマーの形態であること;および/または
(iii)該N基が、好ましくは、下記式:
【化10】
によるポリアクリレートとして存在していること、
(iv)該N基が、例えば、イソプロピル、イソブチル、tert−ブチルもしくはn−ヘキシル原子団などの空間的に広がった基を有すること、あるいは該基であること
の1つ以上によってもたらされることを特徴とする、請求項1〜10の1つ以上に記載の方法。
【請求項12】
以下の代替法(A)〜(F):
代替法(A):前記リガンドIが少なくとも1つのA基を有し、前記リガンドIIが少なくとも1つのN基を有する、少なくとも該リガンドIと該リガンドIIとを有する混合物での前記支持材の官能性付与;および/または
代替法(B):
(a)開始剤分子Tと少なくとも1つのA基を有するリガンドとの混合物での該支持材の官能性付与ならびに、(b)少なくとも1つのN基を有する化合物の添加ならびに該開始剤分子Tへの該化合物の結合;および/または
代替法(C):核酸結合リガンドでの該支持材の官能性付与であって、該リガンドのそれぞれが該リガンド内に1つ以上のA基と1つ以上のN基とを有する官能性付与;および/または
代替法(D):該A基が、少なくとも1つのN基を有する化合物によって立体遮蔽される、該A基および該N基での該支持材の官能性付与;および/または
代替法(E):準化学量論量のA基での該支持材の官能性付与;および/または
代替法(F):代替法(A)〜(E)の少なくとも2つの組合せによる該支持材の官能性付与
の少なくとも1つに従って行なわれる、請求項1〜6および8〜11の1つ以上において規定される核酸結合支持材の調製方法。
【請求項13】
以下の特徴:
(i)前記A基が、第3級、第2級もしくは第1級アミノ基であること;および/または
(ii)前記N基が、ヒドロキシル基、ジオール基、トリオール基、ポリオール基、糖類、エポキシド基、ハライド、C1〜C6アルキル基、アルケン基、アルキレン基、イミド基、エーテル基、もしくはポリエーテル基からなる群より選択されること、および/または
(iii)該N基が、前記溶出pHにおいて、好ましくは2<pH<12の特定のpH範囲において電荷中性であること;および/または
(iv)該A基および/または該N基がモノマーによって導入されること;および/または
(v)該A基および/または該N基がシラン処理によって導入されること;および/または
(vi)該A基および/または該N基が反応性シランによって導入されること;および/または
(vii)該A基および/または該N基が、単官能性または二官能性および/または三官能性の反応性シランの使用によって導入されること;および/または
(viii)該A基および/または該N基が、アミノシラン、ジシラザン、クロロシランもしくはアルコキシシランからなる群より選択される反応性シランの使用によって導入されること;および/または
(ix)該A基および/または該N基が:
【化11】
(式中
nは、1〜5であり、
Rは、C1〜C6、好ましくはC1〜C3アルキル基、特に、メチル、エチル、プロピル、イソプロピルであり;
*は、アミノ、アミノメチル、アミノエチル、アミノプロピル、ジメチルアミノ、ジエチルアミノ、ジイソプロピルアミノ、ジプロピルアミノ、ジエタノールアミノ、ジプロパノールアミノ、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、エーテルアミン、ポリエーテルアミン、4−ジイソブチルアミノ−1−ブタン、6−ジプロピルアミノ−1−ヘキサン、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル、エタンジオール、プロパンジオール、プロパントリオール、ブタントリオール、3−グリシドオキシプロピル、エチルグリシジルエーテル、アルキル原子団、特に、C1〜C4アルキル原子団、特に、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、ハライドまたは水素である)
からなる群より選択される反応性シランによって導入されること、
(x)該A基および該N基が、該A基の比率が1%〜99%、1〜50%、好ましくは1%〜25%となるような比で導入されること;および/または
(xi)該支持材が、以下の特徴:
(aa)該開始剤分子がシラン処理によって適用されること;および/または
(bb)該開始剤分子がシランであること;および/または
(cc)該開始剤分子が、
【化12】
【化13】
(式中、Xは、ハロゲン、特にCl、Br、Iであり、
Y1、Y2、Y3またはY4は、互いに独立して、R、OR、OHまたはHであり、
Rは、C1〜C3アルキルである)
からなる群より選択されること
の1つ以上を有する開始剤分子Tで修飾されること、
(xii)使用される該開始剤分子が、2−(クロロメチル)アリルトリメトキシシランであること;
(xiii)使用される該開始剤分子が、[3−(2−ブロモイソブチリル)プロピル]エトキシジメチルシランであること;および/または
(xiv)該A基が、
2−(ジイソプロピルアミノ)エチルメタクリレート、
N−(3−アミノメチル)メタクリルアミド、N−(3−アミノエチル)メタクリルアミド、
N−(3−アミノプロピル)メタクリルアミド、N−(3−アミノイソプロピル)メタクリルアミド、
N,N−ジメチルアクリルアミド、N,N−ジエチルアクリルアミド、N,N−ジイソプロピルアクリルアミド、
N,N−(ジメチルアミノ)エチルアクリルアミド、N,N−(ジメチルアミノ)エチルアクリレート、
N,N−(ジメチルアミノ)エチルメタクリルアミド、N,N−(ジメチルアミノ)エチルメタクリレート、
N,N−(ジメチルアミノ)プロピルアクリルアミド、N,N−(ジメチルアミノ)プロピルアクリレート、
N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート、
N,N−(ジエチルアミノ)エチルアクリルアミド、N,N−(ジエチルアミノ)エチルアクリレート、
N,N−(ジエチルアミノ)エチルメタクリルアミド、N,N−(ジエチルアミノ)エチルメタクリレート、
N,N−(ジエチルアミノ)プロピルアクリルアミド、N,N−(ジエチルアミノ)プロピルアクリレート、
N,N−(ジエチルアミノ)プロピルメタクリルアミド、N,N−(ジエチルアミノ)プロピルメタクリレート、
N,N−(ジイソプロピルアミノ)エチルアクリルアミド、N,N−(ジイソプロピルアミノ)エチルアクリレート、
N,N−(ジイソプロピルアミノ)エチルメタクリルアミド、N,N−(ジイソプロピルアミノ)エチルメタクリレート、
N,N−(ジイソプロピルアミノ)プロピルアクリルアミド、N,N−(ジイソプロピルアミノ)プロピルアクリレート、
N,N−(ジメチルアミノ)プロピルメタクリルアミド、N,N−(ジメチルアミノ)プロピルメタクリレート
からなる群より選択されるモノマーによって導入されること、および/または
(xiv)N基が、アクリル酸ブチル、アクリル酸プロピル、アクリル酸エチル、アクリル酸メチル、メタクリル酸ヒドロキシエチル(HEMA)、メタクリル酸グリシジル、メタクリル酸グリシドオキシプロピル、グリセロールモノメタクリレート(異性体混合物)、N−アクリルオキシスクシンイミドおよびグリコールモノメタクリレート
からなる群より選択されるモノマーによって導入されること
の少なくとも1つを満たすことを特徴とする、請求項12に記載の方法。
【請求項14】
核酸を精製するための、請求項1〜6および8〜11の1つ以上において規定される、または請求項12と13のいずれかにより得られ得る、核酸結合支持材の使用。
【請求項15】
請求項1〜6および8〜11の1つ以上による支持材、または請求項12もしくは13により得られ得る支持材を有することを特徴とする、核酸を精製するためのキット。
【図1】


【図2】
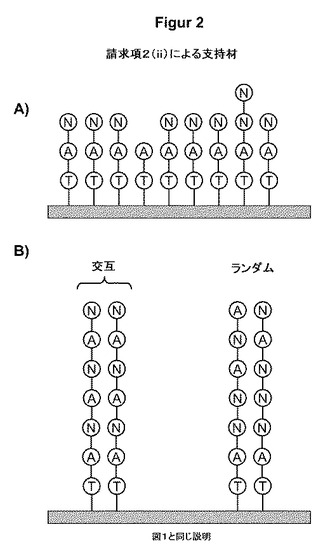
【図3】

【図4】

【図5】
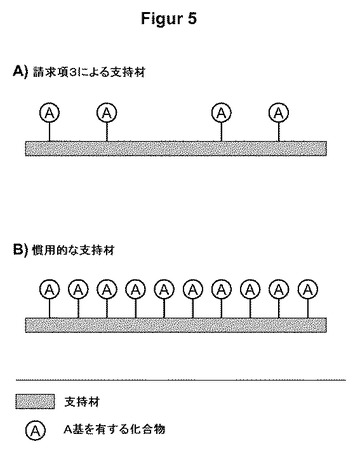
【図6】

【図7】
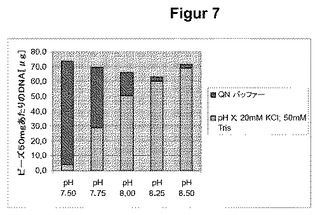
【図8】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2012−513386(P2012−513386A)
【公表日】平成24年6月14日(2012.6.14)
【国際特許分類】
【出願番号】特願2011−541520(P2011−541520)
【出願日】平成21年12月23日(2009.12.23)
【国際出願番号】PCT/EP2009/067909
【国際公開番号】WO2010/072834
【国際公開日】平成22年7月1日(2010.7.1)
【出願人】(507214038)キアゲン ゲーエムベーハー (16)
【Fターム(参考)】
【公表日】平成24年6月14日(2012.6.14)
【国際特許分類】
【出願日】平成21年12月23日(2009.12.23)
【国際出願番号】PCT/EP2009/067909
【国際公開番号】WO2010/072834
【国際公開日】平成22年7月1日(2010.7.1)
【出願人】(507214038)キアゲン ゲーエムベーハー (16)
【Fターム(参考)】
[ Back to top ]