
核酸の迅速な検出方法
【課題】従来の標的核酸とのハイブリダイゼーション時に蛍光が消光する核酸プローブまたは核酸プライマーを用いた方法では、核酸プローブが核酸増幅工程、特に伸長工程を阻害するため、短時間化の際に感度が低下する問題があり、核酸プライマーの非特異増幅によって標的核酸特異的検出が妨げられる問題があった。本発明では、これらの問題点を克服し、標的核酸の検出を特異性および高感度を維持しながら迅速に行うことを課題とした。
【解決手段】第一プライマー及び第二プライマーのTm値と標識プローブのTm値の関係、さらにプライマーの濃度と標識プローブの濃度の関係を調節し、最適化することによって特異性および高感度を維持しながら迅速に標的核酸を検出する。
【解決手段】第一プライマー及び第二プライマーのTm値と標識プローブのTm値の関係、さらにプライマーの濃度と標識プローブの濃度の関係を調節し、最適化することによって特異性および高感度を維持しながら迅速に標的核酸を検出する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、試料中に存在する特定の標的核酸分子を迅速に検出する方法及びこれらの方法により利用するプローブとキットに関する。
【背景技術】
【0002】
一般に生物の保持する核酸は極めて少量であり、それらを検出する際には核酸増幅工程を伴うことがほとんどである。既に知られている核酸増幅方法としてPCR(特許文献1、特許文献2、特許文献3)、NASBA(非特許文献1)、LCR(特許文献4、特許文献5)、SDA(非特許文献2)、RCR(特許文献6)、TMA(非特許文献3)LAMP(非特許文献4)、ICAN(非特許文献5)などが挙げられる。
【0003】
なかでもPCR法は、標的核酸、4種類のデオキシヌクレオシド三リン酸、一対のオリゴヌクレオチドプライマー及び耐熱性DNAポリメラーゼの存在下で、温度の上昇、下降を繰り返すことにより、上記一対のオリゴヌクレオチドプライマーで挟まれる標的核酸の特定領域を指数関数的に増幅させることができる。
【0004】
通常PCR法は特許文献3に示されているように、以下のa)〜c)からなる増幅サイクルを用いる。
a)90〜105℃の範囲内の温度で(好ましくは90〜100℃)0.5〜5分間(好ましくは0.5〜3分間)、変性させる工程、b)35〜65℃の範囲内の温度で(好ましくは37〜60℃)0.5〜5分間(好ましくは1〜3分間)、プライマーと鋳型のハイブリッドを形成させる(アニーリング)工程、及びc)40〜80℃の範囲内の温度で(好ましくは50〜75℃)0.5〜40分間(好ましくは1〜3分間)、プライマー伸長生成物を形成させる(伸長)工程。
【0005】
PCR法のサイクルを繰り返し行うことで増幅させた標的核酸の複製は、各種の検出方法を用いて検出できるようになる。現在利用されている代表的な検出方法として、アガロースゲル電気泳動法がある。しかしながら、電気泳動による検出では指数関数的に増幅された標的核酸の複製を取り扱う必要がありコンタミネーションの起こる危険性が高かった。また、操作も煩雑であり検出終了までの時間も1時間以上を要した。
【0006】
電気泳動以外の検出方法として、二重鎖核酸に結合した時に増強された蛍光を示すDNA挿入色素(インターカレーター)を利用する方法が一般的に用いられている。増幅中のDNA濃度の上昇による蛍光増強は、反応の進行を測定するのに、および標的分子のコピー数を決定するのに利用できる。さらに、制御された温度変化に伴う蛍光をモニターすることにより、例えばPCRサイクル反応の終了時点で、DNA融解曲線が作成できる。
【0007】
一般的な核酸検出法が核酸濃度の上昇をモニターするのに利用される場合、前述のインターカレーターを利用する方法は比較的短時間で核酸を検出することができる。各反応の同じ時点で、単一の蛍光シグナル値が取得される。最終時点での融解曲線解析は、二重鎖核酸の性質をある程度識別できる。融解曲線解析での融解温度が極端に低い時はプライマーダイマーと考えられるし、主なピークが一本でない時は目的のPCR産物ではない非特異増幅産物が存在することを示している。
【0008】
しかし、一般的なインターカレーター法は二重鎖核酸特異的に蛍光が生じる。それゆえ目的の核酸配列特異的な検出が必要とされる場合は目的の核酸配列に非特異的な二重鎖核酸をも検出する可能性があるため、それほど有効ではない。
【0009】
核酸配列特異的プローブ法は、増幅反応の進行をモニターするのにさらなる核酸反応成分を利用する。これらの方法は、検出の基本として、蛍光エネルギー転移(FET)を利用することが多い。一つあるいはそれ以上の核酸プローブを蛍光分子で標識するが、そのうちの一つはエネルギー供与体として働くことができ、もう一方はエネルギー受容体分子である。これらは時として、それぞれレポーター分子および消光分子として知られる。供与体分子は励起スペクトラム範囲の光の特異的波長で励起され、引き続いて蛍光放出波長の範囲で光を放出する。受容体分子も、様々な距離依存性エネルギー転移機構により、供与体分子からエネルギーを受け取ることにより、この波長で励起される。起こり得る蛍光エネルギー移動の特別の例には、蛍光共鳴エネルギー転移(FRET)がある。一般に、受容体分子と供与体分子が近接している時に(例えば、同じまたは隣接する分子上)、受容体分子は供与体分子の放出エネルギーを受け取る。FET検出の基本は、供与体と受容体放出波長における変化をモニターすることである。
【0010】
FETまたはFRETプローブには通常用いられる二つの種類があり、受容体から供与体を分離するために核酸プローブの加水分解を用いるものと、供与体と受容体分子の空間的関係を変化させるためにハイブリダイゼーションを利用するものである。
【0011】
加水分解プローブは、TaqMan(登録商標)プローブとして市販されている。これらは、供与体および受容体分子で標識されたDNAオリゴヌクレオチドからなる。このプローブは、PCR増幅産物の一方の鎖の特異的領域に結合するよう設計される。PCRプライマーがこの鎖にアニールした後、Taq酵素が5’から3’へのポリメラーゼ活性によりDNAを伸長する。TaqMan(登録商標)プローブは、Taq伸長の開始を防ぐために、3’末端がリン酸化で保護されている。もしTaqMan(登録商標)プローブが産物の鎖にハイブリダイズしているのならば、伸長するTaq分子がプローブを加水分解し、検出の基本として受容体から供与体を遊離する。この場合のシグナルは累積的であり、遊離の供与体と受容体分子の濃度は増幅反応の各サイクルで上昇する。
【0012】
蛍光シグナルの生成がプローブの加水分解反応の発生に依存するという事実は、この方法に伴う時間的不利益が存在することを意味する。さらに、プローブの存在がPCR過程のプライマーの伸長過程を妨げる可能性もあった。また、50サイクル以上というような多数回の増幅サイクルが必要な場合、加水分解が非特異的になり得ることも見出されている。
【0013】
ハイブリダイゼーションプローブは、数多くの型式のものが利用可能である。分子ビーコンは、ヘアピンループを形成するような相補的な5’および3’配列を有するオリゴヌクレオチドである。末端の蛍光標識は、ヘアピン構造が形成されるためにFRETの近接にある。分子ビーコンの相補的配列へのハイブリダイゼーションに続き、蛍光標識は分離され、そのためFRETは生じず、これが検出の基本となる。
【0014】
標識プローブの対合も利用可能である。供与体分子を標識したプローブと受容体分子を標識したプローブがPCR産物の鎖上で近接してハイブリダイゼーションすることによって、FRETが生じ得る。この時のFRETによって生じた波長の蛍光が検出の基本となる。このタイプの変種には、標識増幅プライマーと単一の近接する標識プローブの利用が含まれる。
【0015】
二つのプローブ、または二つの標識分子を含む分子ビーコンタイプの使用は、その過程に伴うコストを増やす。さらに近接して特異的に結合するに十分な長さの二つのプローブを知るため、この方法では合理的な長さの既知配列の存在が必要となる。これはある診断応用で問題となり、例えばHIVウイルスのように、保存されている配列の長さが比較的短い場合に、有効なプローブを設計することが難しいことがある。
【0016】
特許文献7では、ハイブリダイゼーション時に蛍光が消光する核酸プローブを用いた方法が提供されている。蛍光色素で標識された核酸プローブを標的核酸にハイブリダイゼーションさせ、ハイブリダイゼーション前後における蛍光色素の発光の減少量を測定する方法または蛍光色素で標識された核酸プライマーを標的核酸にハイブリダイゼーションさせた後伸長させ、核酸変性時の蛍光量に対してアニーリング反応及び伸長反応時の蛍光量の減少を測定する方法が示されている。さらに該方法を用いたリアルタイム定量的PCR法、及び該PCR法で得られるデータの解析の際、アニーリング反応時の蛍光強度値を、変性反応時のもので補正する過程を有するデータ解析法が記載されている。しかしながら、特許文献7の記載のみでは特異性および高感度を維持しながら迅速に核酸を検出する方法を容易に想像し得るものではない。つまり、ハイブリダイゼーション時に蛍光が消光する核酸プローブを使用した場合は核酸プローブが核酸増幅工程、特に伸長工程を阻害するため、短時間化しようとすると感度が低下する問題点、またハイブリダイゼーション時に蛍光が消光する核酸プライマーを使用した場合は核酸プライマーの非特異ハイブリダイゼーションから伸長が開始され、非特異増幅産物となって蛍光の減少を検出してしまい特異的な検出が難しいという問題点があり、特許文献7に開示された技術を用いて特異性および高感度を維持しながら迅速に核酸を検出することは困難であった。
【特許文献1】米国特許第4,683,195号
【特許文献2】米国特許第4,683,202号
【特許文献3】米国特許第4,965,188号
【特許文献4】国際公開89/12696号
【特許文献5】特開平2−2934号
【特許文献6】国際公開90/1069号公報
【特許文献7】特許第3437816号
【非特許文献1】Nucleic acid sequence−basedamplification method;Nature 第350巻、第91頁(1991)
【非特許文献2】Strand Displacement Amplification:Nucleic acid research 第20巻、第1691頁(1992)
【非特許文献3】Transcription mediated amplification method;J.Clin.Microbiol. 第31巻、第3270頁(1993)
【非特許文献4】loop−mediated isothermal amplification method :J Clin Microbiol. 2004 第42巻:第1,956頁
【非特許文献5】isothermal and chimeric primer−initiated amplification of nucleic acids: Kekkaku. 2003 第78巻、第533頁
【発明の開示】
【発明が解決しようとする課題】
【0017】
前述のように、標的核酸とのハイブリダイゼーション時に蛍光が消光する核酸プローブまたは核酸プライマーを用いた方法では、核酸プローブが核酸増幅工程、特に伸長工程を阻害するため、短時間化の際に感度が低下する問題があり、核酸プライマーの非特異増幅によって標的核酸特異的検出が妨げられる問題があった。本発明では、これらの問題点を克服し、標的核酸の検出を特異性および高感度を維持しながら迅速に行うことを課題とした。
【課題を解決するための手段】
【0018】
本発明者らは、上記の点に鑑み、鋭意検討を重ねた結果、第一プライマー及び第二プライマーのTm値と標識プローブのTm値の関係、さらにプライマーの濃度と標識プローブの濃度の関係を調節し、最適化することによって標的核酸の検出をより迅速に、より高感度に行うことができることを見出し、本発明に至った。すなわち、本発明は以下のような構成からなる。
【0019】
1.標的核酸の存在または量を確認したい試料と少なくとも下記(a)と(b)の存在下、連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該標識物質の蛍光を測定しながら、高速PCRを行い、該標的核酸の存在または量を調べることを特徴とする検出方法。
(a)次の(i)〜(iv)の性質を有する標識プローブ
(i) 標的核酸に相補的な配列と二重鎖核酸構造を形成でき、かつ14塩基以上30塩基以下の長さのオリゴヌクレオチドを有する
(ii) 該オリゴヌクレオチドの末端部が該二重鎖核酸構造形成時に消光する性質を有する蛍光物質で修飾されている
(iii) 該二重鎖核酸構造形成時に、蛍光物質で修飾されている該末端部にG(グアニン)とC(シトシン)の対が少なくとも1つ以上存在する
(iv)該標識プローブの濃度が第二プライマーの濃度に対して10倍以下である
(b)次の(i)〜(iii)の性質を有する第一プライマーおよび第二プライマー
(i) 第一プライマーの伸長産物が標的核酸と相補的な配列を有しかつ第二プライマーの鋳型となる性質を有し、該第二プライマーの伸長産物が標的核酸と相同な配列を有しかつ該第一プライマーの鋳型となる性質を有する
(ii) 該第一プライマーおよび該第二プライマーによる増幅産物の長さが50bp以上500bp以下である
(iii) 該第一プライマー及び該第二プライマーのTm値が70℃以上90℃以下であり、かつ該第二プライマーのTm値は前記標識プローブのTm値より大きい
2.(b)の第一プライマーおよび第二プライマーが、さらに
(iv) 該第一プライマーの伸長産物において、該伸長産物の5’末端から前記標識プローブが二重鎖核酸構造を形成する領域までの距離を(A)、該標識プローブが二重鎖核酸構造を形成する領域から該伸長産物の3’末端までの距離を(B)とした場合に(A/B)の値が0.1以上3.0以下であること、
を特徴とする1の検出方法。
3.(a)の標識プローブに、さらに
(v) 標的部位と3個以下の非相補的塩基を導入すること、
を特徴とする1または2の検出方法。
4.標識プローブの中央部に、非相補的塩基を導入することを特徴とする3載の検出方法。
5.(a)の標識プローブに修飾されている蛍光物質が該標識プローブの末端に修飾され、該末端の塩基がGもしくはCであることを特徴とする1〜4のいずれかの検出方法。
6.第一プライマーの濃度が第二プライマーの濃度に対して4倍以上であることを特徴とする1〜5のいずれかの検出方法。
7.高速PCRが、10℃/秒以上の速度で温度の上昇及び下降を繰り返すことを特徴とする1〜6のいずれかの検出方法。
8.さらに、
(c)100塩基/秒以上のDNA合成速度を有するDNAポリメラーゼ
の存在下、
高速PCRを行うことを特徴とする1〜7のいずれかの検出方法。
9.DNAポリメラーゼが、さらに5’エキソヌクレアーゼ活性を持たないことを特徴とする8の検出方法。
10.DNAポリメラーゼが、KOD DNAポリメラーゼ(Thermococcus kodakaraensis KOD1由来)またはその変異体であることを特徴とする8または9の検出方法。
11.標的核酸がピロリ菌の23S rRNA遺伝子のセンス鎖であり、
(a)の標識プローブのオリゴヌクレオチドの配列が配列番号2より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べることを特徴とする1〜10のいずれか載の検出方法。
12.標的核酸がピロリ菌の23S rRNA遺伝子のアンチセンス鎖であり、
(a)の標識プローブのオリゴヌクレオチドの配列が配列番号1より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べることを特徴とする1〜10のいずれかの検出方法。
13.1〜12のいずれかの方法に引き続いて、密封状態のまま、標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、0.1℃/秒以上の連続的な温度上昇又は下降を伴う融解曲線解析を行い、標的核酸中の変異の存在を調べることを特徴とする検出方法。
14.変異がピロリ菌の23S rRNA遺伝子の第2142位または第2143位であり、抗生物質耐性を判別する変異の存在を調べることを特徴とする13の検出方法。
15.標識プローブのオリゴヌクレオチドの配列の中央部に変異の部位が位置することを特徴とする13または14の検出方法。
16.少なくとも次の(a)及び(b)を含むことを特徴とするピロリ菌の存在および/またはピロリ菌の抗生物質耐性の存在を検出するためのキット。
(a)次の(i)〜(ii)の性質を有する標識プローブ
(i)配列番号1または配列番号2より選ばれる14塩基以上30塩基以下の連続した塩基配列のオリゴヌクレオチドを有する
(ii)該オリゴヌクレオチドが、該オリゴヌクレオチドと相補的な配列と二重鎖核酸構造を形成する時に消光する性質を有する蛍光物質が、該オリゴヌクレオチドの末端部に標識されている
(b)Tm値が70℃以上90℃以下であり配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列を含むプライマーと、Tm値が70℃以上90℃以下であり配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列を含むプライマー
17.(a)の標識プローブと二重鎖核酸構造を形成しうる伸長産物を生成せしめる側のプライマーの濃度が、他方のプライマーの濃度に対して4倍以上であることを特徴とする16のキット。
18.さらに、
(c)100塩基/秒以上のDNA合成速度を有するDNAポリメラーゼ
を含むことを特徴とする16または17のキット。
20.DNAポリメラーゼが、さらに5’エキソヌクレアーゼ活性を持たないことを特徴とする18のキット。
20.DNAポリメラーゼが、KOD DNAポリメラーゼ(Thermococcus kodakaraensis KOD1由来)またはその変異体であることを特徴とする18または19のキット。
21.標識プローブの中央部に、ピロリ菌の23S rRNA遺伝子の第2142位または第2143位に相当する部位が位置することを特徴とする16〜20のキット。
【発明の効果】
【0020】
本発明によれば、特異性および高感度を維持しながら迅速な核酸検出が可能となる。具体的には、標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少が生じることと、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、PCRの同一サイクル中に達成することが可能になった。さらに伸長速度が速いDNAポリメラーゼを使用することにより増幅反応時間を短縮し、これまでになく迅速かつ高感度な核酸検出方法を実現できた。
さらに、本発明を利用し、ピロリ菌の23S rRNA遺伝子の存在または量を調べること、ピロリ菌の23S rRNA遺伝子の変異の存在を調べること、23S rRNA遺伝子の第2142位または第2143位における変異の存在を調べピロリ菌の抗生物質耐性を判別することも迅速かつ高感度に行えるようになった。
【発明を実施するための最良の形態】
【0021】
本発明では、高速PCRは核酸増幅工程、すなわち後述するような「変性」、「アニーリング」、「伸長」からなる一連の工程、または本工程のうちアニーリングおよび伸長を同時に行い「変性」および「アニーリングと伸長」とした一連の工程、に要する時間を短縮することにより、迅速性が高い利点を有している。また、核酸増幅を行うためのプライマーとは別にハイブリダイゼーション時に蛍光を消光させる標識プローブを使用することにより、標的核酸の存在または量を反映した特異的な増幅核酸を、標的核酸に依存しない非特異的な増幅核酸と明確に区別し得るため、感度および正確性が高い利点を有している。しかし、これらを単純に組み合わせても、特異性および高感度を維持しながらの迅速な核酸検出はできない。なぜならば、高速PCRにおいては標的核酸を増幅する工程として、標的核酸へのプライマーのアニーリングおよび標的核酸を鋳型とするプライマーの伸長(伸長反応)と、標的核酸およびプライマー伸長産物の解離(変性反応)が短時間のうちに繰り返し行われており、蛍光が消光する標識プローブは標的核酸へのハイブリダイゼーションによって初めて蛍光シグナルの減少が生じるため、標的核酸へのプライマーのアニーリングを起点とする核酸増幅と、標的核酸への標識プローブのハイブリダイゼーションに起因する蛍光シグナルの減少を、PCRの同一サイクル中に起こすことが困難だからである。つまり、高速PCR溶液中に単純に標的核酸とのハイブリダイゼーションによって蛍光の減少を生じる標識プローブを加えても、核酸増幅のためのプライマーのアニーリングと、蛍光シグナルの減少を生じさせるための標識プローブのハイブリダイゼーションとの間で、標的核酸に対する競合が起こり、標的核酸は増幅するが蛍光シグナルの減少が生じない、または標識プローブのハイブリダイゼーションで蛍光シグナルの減少は生じるが標的核酸が増幅しないことになり、どちらも標的核酸の存在または量、並びに標的核酸中の変異の存在を、特異性および高感度を維持しながら迅速に調べることはできない。すなわち、本発明では、様々な条件を検討し、高速PCRの迅速性が高い利点と、ハイブリダイゼーションによって蛍光の減少を生じる標識プローブの感度および正確性が高い利点を両立し、標的核酸の存在または量、並びに標的核酸中の変異の存在を、特異性および高感度を維持しながら迅速に調べる核酸検出方法を確立したものである。
【0022】
本発明の標的核酸とは生物(細菌も含む)の保持する核酸の塩基配列またはその相補的な塩基配列のいずれかを指す。
【0023】
本発明における高速PCR を以下に示す。PCR法を高速化する方法は例えば特許公開2005−328709に示されている。「変性」、「アニーリング」、「伸長」の各温度へ達する温度変化に要する時間の短縮、各工程の温度を保持する時間の短縮、サイクル数の減少などが挙げられている。特に重要となるのは温度変化に要する時間の短縮及び各工程の温度保持時間の短縮である。温度変化に要する時間の短縮は、具体的に温度の上昇及び下降を10℃/秒以上の速度で行うことを示す。10℃/秒以上の温度上昇・下降を達成する手段には特に制限がないが、一例としてロシュ・ダイアグノスティック社製のLight Cycler(登録商標)システムが挙げられる。このシステムでは温度上昇・下降に空気を使って温度制御する方式を採用し、温風と冷風で反応液の入ったガラスキャピラリーの温度を直接変化させている。それにより約20℃/秒の温度上昇・下降速度を達成している。
【0024】
また高速核酸増幅を達成するための別の手段として伸長工程に要する時間の短縮が考えられる。伸長工程時間を短縮する方策は、いくつか考えられる。最も有力な方策は、PCRによる増幅領域を極限まで少なくすることである。そうすれば当然、伸長に要する時間は短縮可能になる。このような場合でも、増幅領域は第一プライマー及び第二プライマーを含めて60塩基対程度が最低限度である。検出の目的で標識プローブを設定すれば、更にその部分を確保する必要があり、100塩基対程度の増幅が必要となる。増幅産物の長さは、高速増幅かつ標識プローブによる検出を行うために50bpから500bpの短い産物が好ましく、100bpから300bpが好ましい。
【0025】
さらにこの増幅産物の長さはDNAポリメラーゼの伸長速度により制限される。現在知られているDNAポリメラーゼ(単独あるいは組みあわせ)としてのTaqポリメラーゼや,EX−Taq,LA−Taq,Expandシリーズ,Plutinumシリーズ,Tbr,Tfl,Tru,Tth,T1i,Tac,Tne,Tma,Tih、Tfi(以上はPolI型酵素),Pfu,Pfutubo,Pyrobest(登録商標),Pwo,KOD,Bst,Sac,Sso,Poc,Pab,Mth,Pho,ES4,VENT,DEEPVENT(以上はα型酵素)などが挙げられる。天然型のポリメラーゼのアミノ酸配列を公知の手段により、1もしくは数個が欠失、置換若しくは付加させたもの(変異体)であっても良い。あるいは、上記の酵素(天然型、変異体)に化学修飾などの手段によりさらに改変を加えたものであっても良い。例えば、KOD DNAポリメラーゼは、最も一般的であるTaqポリメラーゼの倍にあたる100〜140塩基/秒以上のDNA合成速度を有する。また、プロセッシビティーなども優れていることが、伸長速度の速い要因のひとつであると考えられる。KOD DNAポリメラーゼは、東洋紡績製のもの(製品コードKOD−101など)を容易に入手することができる。
【0026】
近年、上記のDNAポリメラーゼをさらに変異、改変などの改良を加えて100塩基/秒以上のデオキシリボ核酸合成速度を達成させたもの、あるいは、組み合わせにより当該性能を達成させたもの(組合せにはKOD DNAポリメラーゼを含んでいても良い)も、本願発明のDNAポリメラーゼとして用いることができる。例えば、上記KOD DNAポリメラーゼ以外に100塩基/秒以上のデオキシリボ核酸合成速度を有するDNAポリメラーゼとして、KOD Dash DNAポリメラーゼ(東洋紡績製)、PrimeSTAR MAX DNAポリメラーゼ(TAKARA製)、Platinum Pfx DNAポリメラーゼ(インビトロジェン製)なども市販されている。
【0027】
DNAポリメラーゼはさらに、5’エキソヌクレアーゼ活性を持たないことが好ましい。本発明では標識プローブからの蛍光の減少量を標的核酸の検出に利用している。そのため、高速PCR工程中で第一のプライマーによる伸長産物に対して標識プローブ及び第二のプライマーがハイブリダイゼーションしている場合に、5’エキソヌクレアーゼ活性を持つDNAポリメラーゼ、例えばTaqポリメラーゼでは標識プローブを分解し、非特異シグナルの増加を伴う。本発明では標的核酸への標識プローブのハイブリダイゼーション時に起こる蛍光の減少量を検出に利用していることから、非特異シグナルの増加はバックグランドの上昇となり正確な定量もしくは検出ができなくなる。5’エキソヌクレアーゼ活性を持たないDNAポリメラーゼは特に限定はされないが、例えばKODポリメラーゼが本発明に適合する。
【0028】
二重鎖核酸構造は、標的核酸中の塩基とその相補的な核酸配列中の塩基の間で起こる塩基対生成によって形成される。塩基対生成はシトシンとグアニン、アデニンとチミンまたはアデニンとウラシルの間で起こる水素結合により生じる。標的核酸と標識プローブがハイブリダイゼーションすることにより二重鎖核酸構造が形成され、標識プローブの蛍光の減少が生じるようになる。
【0029】
本発明における「末端」とは標識プローブの塩基配列5‘側の最も端に存在する塩基、もしくは標識プローブの塩基配列で3’側の最も端に存在する塩基を指す。「末端部」とは5‘末端の塩基から3’末端に向かって5塩基以内または3‘末端の塩基から5’末端に向かって5塩基以内の塩基を示している。
【0030】
標識プローブに標識する蛍光物質は、核酸増幅工程中分解もしくは蛍光が減衰してなくならなければよく、ハイブリダイゼーション時に消光を生じる蛍光物質であればよい。特に標識プローブの末端においてグアニンとシトシンの塩基対形成時に蛍光の消光を生じる蛍光物質が好ましい。具体的には、フルオロセインまたはその誘導体(例えばフルオロセインイソチオシアネート(FITC))、BODIPY(商標) シリーズ、ローダミンまたはその誘導体(例えば5−カルボキシローダミン6G(CR6G)やテトラメチルローダミン(TAMRA))などを使用できるが、BODIPYシリーズやCR6Gの使用が特に好ましい。蛍光色素のオリゴヌクレオチドへの結合方法は、通常の方法に従って行うことができる。蛍光色素の消光を利用すれば、インターカレーターなどの他の二重鎖核酸構造への挿入色素を用いることなく、またFRET現象を起こす二種類のプローブを用いることなく、一種類の蛍光物質に標識されたプローブを用いて単純かつ特異的に標的核酸配列を検出することができる。標識プローブの塩基配列中の蛍光物質の標識位置は、特に限定されないが末端部に標識されていることが好ましく、末端に標識されていることがより好ましい。
【0031】
標識プローブの塩基配列とそれに対し相補的な標的核酸の塩基配列の選択はハイブリダイゼーション時に蛍光の消光を生じるために重要である。標識プローブの塩基配列は標的核酸とのハイブリダイゼーション時に消光を生じる塩基配列であればよく、二重鎖核酸構造形成時に、蛍光物質が修飾されている標識プローブ末端部にG(グアニン)とC(シトシン)の対が少なくとも1つ以上存在するように設計すればよい。標識プローブの末端が標識されている場合は、その末端塩基がGもしくはCであること、または標識プローブの末端と塩基対を形成する標的核酸の塩基から1もしくは3塩基離れてGが存在するように標識プローブの塩基配列が設計されていることがより好ましい。標識プローブの末端が標識されており、かつ末端塩基がCであるように設計することがさらに好ましい。
【0032】
標的核酸の塩基配列から、どうしても末端部がGまたはCに設計できない場合、第一プライマーまたは第二プライマーの塩基配列中にGまたはCを故意に導入することで標識プローブをハイブリダイゼーションさせ、かつ標識プローブの蛍光を消光することができる相補的な塩基配列を作り出すことが可能である。この場合、蛍光シグナルの減少が生じることと、第二プライマーの伸長による核酸の増幅とを、PCRの同一サイクル中に達成するためにも第一プライマーにGまたはCを導入することが好ましい。また標識プローブの第一プライマーに対する非特異のハイブリダイゼーションを避けるために故意のGまたはCの導入位置は第一プライマーの3‘末端から末端塩基を含めて5’末端側へ4塩基以上8塩基以下離れて設計することが好ましい。3塩基以下の位置に設計するとプライマーが伸長できなくなり、9塩基以上離れて設計すると増幅によらない標識プローブとプライマーとの非特異ハイブリダイゼーションが起こりやすくなる。
【0033】
本発明を達成する手段として各プライマー及び標識プローブのTm値が規定される。Tm値とはプライマーが相補塩基とアニーリングし、50%が解離する時の温度を表す。Tm値は計算により算出することも可能である。Tm値の計算方法は、ハイブリダイゼーションにおける塩濃度等をパラメーターにして各種の計算方法が考案されている。一般的に良く用いられる計算方法としては、最近接塩基対法(Schildkraut C., Lifson S. (1965) Dependence of the melting temperature of DNA on salt concentration − Biopolymers 3, 195−208 、Breslauer K.J., Frank R., Blocker H., Markey L.A. (1986) Predicting DNA duplex stability from the base sequence − Proc. Natl. Acad. Sci. USA 83, 3746−3750)やWallance法(Wallace, R.B.; Shaffer, J.; Murphy R.F.; Bonner, J.; Hirose, T.; Itakura, K.; (1979) Nucelic Acid Res. 6, 3543)、GC%法(Dependence of the Melting Temperature of DNA on Salt Concentration、Schildkraut C.,Lifson S. (1965) BIOPOLYMERS 3.195−208、Optimization of the annealing temperature for DNA amplification in vitro、W.Rychlik、 Nucl.Acids Res.(1990) 18(21)6409−6412)などを用いることができ、標的核酸の配列または検出の条件などによっては計算方法によるTm値の差異が影響するが、当業者にとって容易に想到し得る検討により実験的に当該Tm値の差異を修正し、本発明の効果を有するプライマーおよび標識プローブを設定することができる。
【0034】
第一プライマーおよび第二プライマーのTm値と標識プローブのTm値の関係が、本発明に記載の効果を得るために重要な要素の一つとなる。そこで、発明をより具体的に説明するための一例として、本発明では、Breslauer K.J., Frank R., Blocker H., Markey L.A. (1986) Predicting DNA duplex stability from the base sequence − Proc. Natl. Acad. Sci. USA 83, 3746−3750に記載の方法にて計算したTm値を用いる。
【0035】
具体的には上記計算方法によれば、配列番号3より選ばれる24塩基の連続した塩基配列(5’−GAGATGGGAGCTGTCTCAACCAGA−3’:配列番号5)のTm値は70.52、配列番号4より選ばれる25塩基の連続した塩基配列(5’−TGCGCATGATATTCCCATTAGCAGT−3’:配列番号10)のTm値は73.39となる。
【0036】
プライマーのTm値は70℃以上が好ましい。より好ましくは70℃以上90℃以下である。本プライマーの使用で65℃以上の温度で安定したアニーリングが可能となる。91℃以上のプライマーを使用する場合、非特異的なハイブリダイゼーションが起こりやすくなり結果的に目的とする増幅産物の増幅効率を下げてしまう。
【0037】
本発明では標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とのバランスが重要となる。このバランスは第二プライマー及び標識プローブのTm値、さらに第二プライマー及び標識プローブの濃度によって最適化される。上記バランスを最適化するにはTm値は標識プローブよりも第二プライマーが大きくなるように設計する必要がある。本検出方法は標識プローブの発する蛍光がハイブリダイゼーションによって減少することを利用しているため、標識プローブ濃度が濃い場合はバックグランドの蛍光シグナルが高くなる。標識プローブ濃度はハイブリダイゼーション時の蛍光の減少した蛍光シグナルに対してバックグランドの蛍光シグナルが少なくなる濃度が好ましい。具体的には第二プライマーの10倍以下であることが好ましい。より好ましくは5倍以下である。10倍を超える場合はバックグランド蛍光シグナルの影響により、蛍光が減少したときの蛍光強度の差を検出できなくなる。標識プローブ濃度の下限は特に限定されないが標識プローブと標的核酸または第一プライマーの伸長産物とのハイブリダイゼーション効率を極端に下げない濃度がよい。上記の蛍光シグナルの減少と核酸の増幅のバランスをとるためには標識プローブの濃度は第二プライマーの濃度よりも高いほうが好ましい。標識プローブの濃度は具体的には10nM以上が好ましい。10nM未満の場合は蛍光シグナルを感度良く検出することが難しい。
【0038】
上記Tm値及び濃度の関係を満たすことにより、従来法では達成できなかった、標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、高速PCRの同一サイクル中に達成することが可能になる。
【0039】
本発明の重要な開示について、図1を用いて、より詳しく説明する。従来の方法では第二プライマーのアニーリングおよび伸長により標的核酸は増幅するが、標識プローブが標的核酸または第一プライマーの伸長産物にハイブリダイゼーションできないために蛍光シグナルの減少が生じない場合(図1(1)−A及び図1(2)−A)、または標識プローブの標的核酸または第一プライマーの伸長産物へのハイブリダイゼーションにより蛍光シグナルの減少が生じるが、標識プローブのハイブリダイゼーションがプライマーの伸長を阻害し、標的核酸が増幅しない場合(図1(1)−B及び図1(2)−B)のどちらか一方が起こる割合が大きく、どちらの場合も標的核酸の存在または量、並びに標的核酸中の変異の存在を、迅速かつ高感度に調べることはできなかった。一方、本発明の方法によれば、第一プライマー及び第二プライマーのTm値と標識プローブのTm値の関係、さらにプライマーの濃度と標識プローブの濃度の関係を上記のように規定することで高速PCR工程における変性温度からアニーリング・伸長温度までの温度下降中に標識プローブが標的核酸または第一プライマーの伸長産物にハイブリダイゼーションしつつ、その二重鎖核酸構造を形成する領域まで第二プライマーが伸長する。続いて、アニーリング・伸長温度から次サイクルとなる変性温度までの温度上昇中には標識プローブは標的核酸または第一プライマーの伸長産物から解離することにより、二重鎖核酸構造を形成した領域まで伸長した第二プライマーのさらなる伸長を阻害しないため、第一プライマーの鋳型となるような第二プライマーの伸長産物が形成される(図1(1)−C及び図1(2)−C)。このように、標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、高速PCRの同一サイクル中に達成することで、高速PCRの迅速性が高い利点と、標識プローブの感度および正確性が高い利点を両立し、標的核酸の存在または量、並びに標的核酸中の変異の存在を、迅速かつ高感度に調べる核酸検出方法を確立することができた。
【0040】
さらには、前記に記載の伸長速度の速いDNAポリメラーゼを使用することにより第二プライマーの伸長が第一プライマーと相補的な塩基配列まで到達することが可能となり、より効率的に第一プライマーの鋳型となるような第二プライマーの伸長産物が形成され得る。
【0041】
また、本発明の別の重要な開示として、上記のように蛍光シグナルの減少と核酸の増幅とを効率よく行うためには、第一プライマーの伸長産物において標識プローブが二重鎖核酸構造を形成する位置も重要である。前述の通り、本発明の方法では、標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、PCRの同一サイクル中に達成することが重要であり、さらには高速PCR工程において変性温度からアニーリング・伸長温度までの温度下降中に、標識プローブの標的核酸または第一プライマーの伸長産物へのハイブリダイゼーションより二重鎖核酸構造を形成する領域まで第二プライマーが伸長し、続いて、アニーリング・伸長温度から次サイクルとなる変性温度までの温度上昇中に、当該二重鎖核酸構造を形成した領域まで伸長した第二プライマーがさらなる伸長を行うことにより、第一プライマーの鋳型となるような第二プライマーの伸長産物が形成されることが重要である。すなわち、第一プライマーの伸長産物の5’末端から標識プローブが二重鎖核酸構造を形成する領域までの距離を(A)、該標識プローブが二重鎖核酸構造を形成する領域から第一プライマーの伸長産物の3’末端までの距離を(B)とした場合(図2)に、(A)と(B)のバランスが重要である。より詳細には(A)は第一プライマーの伸長産物の5’末端から二重鎖核酸構造を形成している標識プローブの3‘末端まで、(B)は二重鎖核酸構造を形成している標識プローブの5‘末端から第一プライマーの伸長産物の3’末端までの距離である。特に(A)が(B)より大きすぎると、第二プライマーの伸長が第一プライマーまで届かず、第一プライマーの鋳型となるような第二プライマーの伸長産物を形成することができないため、標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、PCRの同一サイクル中に達成することができない。蛍光シグナルの減少と核酸の増幅とを効率よく行うための(A)と(B)のバランスとして、具体的には、(A/B)の値が0.1以上3.0以下であることが好ましく、0.3以上3.0以下かつ第一プライマーと第二プライマーの伸長産物が80bp〜300bpであることがより好ましい。さらに好ましくは(A/B)の値が0.4以上2.0以下かつ第一プライマーと第二プライマーの伸長産物が100bp〜300bpである。
【0042】
核酸増幅工程で使用する第一プライマーと第二プライマーの濃度に差をつけることによって標的核酸の特定領域の一本鎖核酸を優先的に増幅する方法も、本発明に適用できる。例えば図3に示すような第一プライマーと第二プライマー、標識プローブの設計において、第一プライマーの濃度を第二プライマーの濃度より高く設定することにより、第一プライマーによる伸長産物を優先的に生成させることもできる。第二プライマーによる伸長産物および標識プローブは第一プライマーの伸長産物に対して競合的に作用するため、このように標的核酸を含む一本鎖核酸を意図的に増幅させることにより、蛍光検出工程において標識プローブのハイブリダイゼーション効率を上げ、より多くの蛍光シグナルを発生させることができる。第二プライマーに対する第一プライマーの濃度比は、特に限定されないが好ましくは4倍以上、より好ましくは5倍以上、さらに好ましくは10倍以上がよい。また、50倍以下がよく、より好ましくは30倍以下がよい。好ましくは4倍以上50倍以下であり、より好ましくは10倍以上30倍以下である。本発明の第二プライマーの濃度は特に限定されないが、25nM〜500nMが好ましい。より好ましくは50nM〜250nMである。
【0043】
蛍光検出は例えば標識プローブと、標的核酸または第一プライマーの伸長産物をハイブリダイゼーションさせ二重鎖核酸構造を形成した後、温度上昇により標識プローブと標的核酸が解離することで生じる蛍光の増加量を検出してもよい。温度上昇速度は好ましくは0.1℃/秒以上であり、0.1℃/秒以上5℃/秒以下であり、より好ましくは0.5℃/秒以上2℃/秒以下である。5℃/秒を超える場合は核酸プローブと標的核酸の解離温度を正確に捉えることができない。
【0044】
非相補的塩基の定義は標識プローブがハイブリダイゼーションする時に標的核酸中の塩基と塩基対を形成しない標識プローブ中の塩基のことである。野生型の塩基配列と一致する標識プローブと野生型の標識核酸がハイブリダイゼーションする場合は完全に一致するため非相補的塩基はない。しかし、野生型の塩基配列と一致する標識プローブと野生型に対して一塩基変異をもつ標的核酸をハイブリダイゼーションする場合には、標識プローブに非相補的塩基がある。本発明の変異は野生型に対して異なる塩基を有することであり、標的核酸中での事象である。標識プローブ塩基配列中の標的部位との非相補的塩基の存在は、前記の標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、PCRの同一サイクル中に達成するために有利に働く。その理由としては、非相補的塩基を含んだ標識プローブは温度上昇時に解離しやすくなるため、第二プライマーの伸長がより円滑に達成されることが挙げられる。この場合、4個以上の非相補的塩基が一つの標識プローブ中に存在すると、前記の標識プローブ14塩基〜30塩基の範囲内ではハイブリダイゼーションが起こりにくくなる。非相補的塩基の数は好ましくは3個以下である。標識プローブ中の非相補的塩基の導入位置は特に限定されないが、標的核酸において検出対象となる変異塩基を除く位置に導入することが好ましく、標識プローブの末端から末端を含めて2塩基以上離れていることが好ましい。また、標識プローブ中に塩基対結合力の比較的強いグアニンやシトシンが3塩基以上連続して存在する場合に、グアニンやシトシンの連続した領域に導入することが好ましい。
【0045】
標識プローブの塩基配列と変異における位置関係は、例えば核酸プローブと標的核酸または第一プライマーの伸長産物をハイブリダイゼーションさせ、温度上昇により核酸プローブと標的核酸が解離することで生じる蛍光の減少を検出する方法を用いる場合に、完全一致配列と1塩基変異が存在する配列の解離温度の違いがわかれば特に限定されない。好ましくは当該標識プローブの塩基配列の両末端から3塩基以上離れて存在すればよく、より好ましくは両末端から5塩基以上離れて存在すればよい。特に好ましくは標識プローブの中央部に変異の部位が位置すればよい。中央部は当該オリゴヌクレオチド配列の中心から両末端に向かって3塩基以内のことをいう。当該オリゴヌクレオチド配列が偶数個の場合の中心とは当該オリゴヌクレオチド配列数を2で割った数を、両末端を含んで数える数え方で両末端塩基より内側に向かって数えた塩基をいい、当該オリゴヌクレオチド配列が奇数個の場合の中心とは当該オリゴヌクレオチド配列数に1を加えて2で割った数を、両末端を含んで数える数え方で両末端塩基より内側に向かって数えた塩基をいう。
【0046】
3’末端を蛍光色素で標識するか、3’末端にリン酸化などの処置を加えるなどして高速PCR中に伸長して標識プローブのTm値が上昇しないような工夫をすることが好ましい。ただし、蛍光検出方法として、例えば標識プローブと標的核酸または第一プライマーの伸長産物をハイブリダイゼーションさせ、温度上昇により標識プローブと標的核酸が解離することで生じる蛍光の増加を検出する方法を用いる場合は、高速PCR中に標識プローブが伸長すると標識プローブと標的核酸の解離温度を正確に測定できなくなるため、前記のような工夫が特に必要である。
【0047】
また、本発明は下記も含む。
標的核酸の存在または量を確認したい試料と少なくとも下記(a)、(b)の存在下、連続的または間欠的に標識プローブに標識されている蛍光物質の励起波長の光を与えかつ標識プローブからの蛍光を測定しながら、高速PCRを行い、該標的核酸の存在または量を調べ、引き続いて、密封状態のまま、標識プローブに標識されている蛍光物質の励起波長の光を与えかつ標識プローブからの蛍光を測定しながら、0.1℃/秒以上の連続的な温度上昇又は下降を伴う融解曲線解析を行い、標的核酸中の変異の存在を調べることを特徴とする検出方法。この場合において、
(a)次の(i)〜(iv)の性質を有する標識プローブ
(i) 標的核酸に相補的な配列と二重鎖核酸構造を形成でき、かつ14塩基以上30塩基以下の長さのオリゴヌクレオチドを有する
(ii) 該オリゴヌクレオチドの末端部が該二重鎖核酸構造形成時に消光する性質を有する蛍光物質で修飾されている
(iii) 該二重鎖核酸構造形成時に、蛍光物質が修飾されている該末端部のヌクレオチドから3塩基以内にG(グアニン)とC(シトシン)の対が少なくとも1つ以上存在する
(iv)該標識プローブの濃度が該第二プライマーの濃度に対して10倍以下である
(b)次の(i)〜(iii)の性質を有する第一プライマーおよび第二プライマー
(i) 第一プライマーの伸長産物が標的核酸と相補的な配列を有しかつ第二プライマーの鋳型となる性質を有し、該第二プライマーの伸長産物が標的核酸と相同な配列を有しかつ該第一プライマーの鋳型となる性質を有する
(ii) 該第一プライマーおよび該第二プライマーによる増幅産物の長さが50bp以上500bp以下である
(iii) 該第一プライマー及び該第二プライマーのTm値が70℃以上90℃以下であり、かつ該第二プライマーのTm値は前記標識プローブのTm値より大きい
を示すものである。
【0048】
標的核酸の例として、感染症の原因となる細菌の保持している特定の核酸が考えられる。例えばピロリ菌(Helicobacter pylori)の23SrRNA遺伝子の検出が挙げられる。ピロリ菌は、胃・十二指腸潰瘍の起因菌とされ、近年胃癌との関連性も指摘されてきている。潰瘍の疑いがある場合、呼気試験、血中または便中ピロリ菌抗体検査等、を行った結果、ピロリ菌感染の可能性がある場合、さらに内視鏡検査において胃生検を行い、ピロリ菌の存在を迅速ウレアーゼ試験あるいは培養法によって確かめられる。内視鏡検査においてピロリ菌の存在が確認された場合、除菌療法等の処置が行なわれる。ピロリ菌の除菌療法としては、プロトンポンプ阻害剤とアモキシシリンとクラリスロマイシンの3剤併用療法が有効とされ、本邦においては、西暦2000年より保険診療として広く実施されている。本発明によりピロリ菌を検出するために、第一プライマー及び第二プライマー、標識プローブをピロリ菌の23SrRNA遺伝子配列をターゲットとして設計した。第一プライマー及び第二プライマーにより23SrRNA遺伝子配列の特定領域を増幅する工程において、標識プローブを用い、「伸長」工程または「アニーリング・伸長」工程での蛍光シグナルの検出を行うことで、より高感度なピロリ菌検出が可能となる。
【0049】
また、近年クラリスロマイシン耐性ピロリ菌(以下CAM耐性菌と称する)の出現が報告され、ピロリ菌(Helicobacter pylori)のCAM耐性遺伝子の検出が注目を集めている。CAM耐性ピロリ菌はピロリ菌除菌成功率の低下の大きな要因の一つとして挙げられるようになってきている。CAMは、ピロリ菌の23SrRNAのペプチジルトランスフェラーゼループに結合し、そのタンパク質合成を阻害することによって作用を発揮する。しかしながら、23SrRNA遺伝子の点突然変異によってピロリ菌はCAM耐性能を取得することが明らかにされている(James versalovic et. al. Antimicrobial Agents and Chemotherapy p477−480 1996)。この点について、CAM耐性菌の93%は、前記の領域にある2142位あるいは2143位のアデニンがグアニンに変異しており(A2142GまたはA2143G)、CAM耐性菌の7%は、2142位がシトシンに変異している(A2142C)ことが報告されている(Stone,G.G. et. al. Antimicrobial Agents & Chemotherapy. 41(3):712−4, 1997)。
【0050】
ピロリ菌のCAM耐性を判定するには、例えば以下記述の方法が考えられる。野生型ピロリ菌とCAM耐性ピロリ菌に共通の配列を用いて設計した第一及び第二プライマーによりピロリ菌の23SrRNA遺伝子の特定領域を増幅する。この工程において増幅される産物(野生型のものとCAM耐性の一塩基変異が存在する増幅産物の二種類)を標的核酸とした野生型ピロリ菌の23SrRNA遺伝子と完全に一致する標識プローブまたは前記二種類のCAM耐性ピロリ菌の23SrRNA遺伝子と完全に一致する標識プローブを同時に使用する。PCR工程中の「伸長」工程または「アニーリング・伸長」工程での蛍光シグナルの検出を行う。この検出では野生型ピロリ菌の23SrRNA遺伝子と完全に一致する標識プローブを用いた場合は、野生型ピロリ菌の特異的な検出が可能であり、前記二種類のCAM耐性ピロリ菌の23SrRNA遺伝子と完全に一致する標識プローブを用いた場合はCAM耐性ピロリ菌の特異的な検出が可能である。さらに引き続き融解曲線解析を行うことにより、一つの標識プローブで野生型ピロリ菌とCAM耐性ピロリ菌の検出が同時に行えることも本発明の範疇に含まれる。CAM耐性ピロリ菌に関して、一塩基変異は前記のとおり2142位あるいは2143位のアデニンがグアニンに変異(A2142GまたはA2143G)、または2142位がシトシンに変異している(A2142C)ピロリ菌が大半を占めているが、この限りではない。ピロリ菌における標的核酸とは野生型の保有する核酸を含み、一塩基変異が存在しているCAM耐性菌の保有する核酸を含んでいる。この場合の非相補的塩基の数は、野生型に完全一致の塩基配列を持つ標識プローブではCAM耐性ピロリ菌に対して1塩基である。つまり3塩基以下の非相補的塩基とは標識プローブが同じで標的核酸が異なる場合に標的核酸中の1塩基変異を含んで3塩基以下ということを示している。
【0051】
すなわち、本発明を利用し、下記の検出方法を実施しても良い。
連続的または間欠的に標識プローブに標識されている蛍光物質の励起波長の光を与えかつ(a)の標識プローブからの蛍光を測定しながら、(b)の第一プライマーと第二プライマーを用いて高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べることを特徴とする検出方法。
【0052】
さらに、本発明を利用し、下記の検出方法を実施しても良い。
連続的または間欠的に標識プローブに標識されている蛍光物質の励起波長の光を与えかつ(a)の標識プローブからの蛍光を測定しながら、(b)の第一プライマーと第二プライマーを用いて高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べ、引き続いて、密封状態のまま、標識プローブに標識されている蛍光物質の励起波長の光を与えかつ標識プローブからの蛍光を測定しながら、0.1℃/秒以上の連続的な温度上昇又は下降を伴う融解曲線解析を行い、ピロリ菌の23S rRNA遺伝子の変異の存在を調べることを特徴とする検出方法。
【0053】
さらに、本発明を利用し、下記の検出方法を実施しても良い。
連続的または間欠的に標識プローブに標識されている蛍光物質の励起波長の光を与えかつ(a)の標識プローブからの蛍光を測定しながら、(b)の第一プライマーと第二プライマーを用いて高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べ、引き続いて、密封状態のまま、標識プローブに標識されている蛍光物質の励起波長の光を与えかつ(a)の標識プローブからの蛍光を測定しながら、0.1℃/秒以上の連続的な温度上昇又は下降を伴う融解曲線解析を行い、23S rRNA遺伝子の第2142位または第2143位における変異の存在を調べピロリ菌の抗生物質耐性を判別することを特徴とする検出方法。
【0054】
上記のピロリ菌の23S rRNA遺伝子の存在または量を調べる方法、ピロリ菌の23S rRNA遺伝子の変異の存在を調べる方法、23S rRNA遺伝子の第2142位または第2143位における変異の存在を調べピロリ菌の抗生物質耐性を判別する方法においては、標的核酸がピロリ菌の23S rRNA遺伝子のセンス鎖の場合は、
(a)の標識プローブのオリゴヌクレオチドの配列が、配列番号2より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
標的核酸がピロリ菌の23S rRNA遺伝子のアンチセンス鎖の場合は、
(a)の標識プローブのオリゴヌクレオチドの配列が配列番号1より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列
であればよい。
【0055】
また、限定されるわけではないが本発明は抗酸菌の検出にも利用できる。抗酸菌の同定には培養法が用いられているが、抗酸菌は増殖速度が遅い。小川培地などによる培養では同定に約1ヶ月を要することもある。近年抗酸菌の検査に遺伝子検査が導入され、短時間化されたといっても1日から2日必要となる。本発明の方法を適用すれば30分以内に検査を完了することができ、極めて短時間で抗酸菌を検出することが可能となる。
【0056】
その他にも、より迅速に検出を行う必要がある感染症項目にも適用できる。例えば性感染症のクラミジアや淋菌、C型肝炎ウイルス、B型肝炎ウイルス、多剤耐性黄色ブドウ球菌、多剤耐性緑膿菌、敗血症菌などの検出に利用できる。さらに、ヒトの遺伝子検査にも適用できる。例えば癌遺伝子検査では30分以内に検出ができることから術中診断も可能になる。
【0057】
1つの容器中とは検体を含む溶液がプラスチック、あるいはガラスなどから成る容器中に充填され、増幅から検出に至るまでに別の容器に移し替えるもしくは取り出す操作がないことを示す。密封状態は外部からの不純物の進入がなく、内部からの溶液の蒸発がないことを示す。不純物などのコンタミネーションの防止、ならびに溶液組成の変化による結果の相違を防ぐことは重要である。
【実施例】
【0058】
以下に実施例を示して本発明を具体的に説明するが、本発明は実施例に限定されるものではない。
【0059】
以下にピロリ菌の23S rRNA遺伝子上のクラリスロマシン耐性を判定する方法を示す。
【0060】
(実施例1)
(検体の採取)
胃生検材料より遺伝子をQIAamp(登録商標) DNA micro Kit(Qiagen社製)を用いて抽出した。抽出されたDNAは−20℃で保存した。
(テンプレートプラスミドの調製)
1μlの抽出されたDNA溶液を鋳型として用い、PCRを実施した。ここで使用したプライマーペアは、ピロリ菌23SrRNA遺伝子を全長増幅できるプライマーを使用した。いずれもピロリ菌の23SrRNA遺伝子 (GenBank accession number U27270)の公知の配列から設計した。
【0061】
PCRは、サーマルサイクラーとしてGeneAmp(登録商標)9700(Applied Biosystems社製.)を使用し、全量を26μlとして実施した。1μlのDNA溶液と試薬混合物(KOD−plus 0.5U、10XPCR緩衝液2.5μl、200μMのdNTP、各々5μMのプライマー)を混合した。標的DNAの初期変性を94℃で2分間行った後、94℃・15秒の変性工程、60℃・30秒のアニーリング工程、68℃・60秒の伸長工程のサイクルを35回繰り返した。なお、その他のPCR条件は常法通りである(例えば、遺伝子 操作技術マニュアル、医学書院、1995年発行を参照)。得られた核酸断片を制限酵素によって平滑末端切断されたpUC18へライゲーションし、コンピテントセルJM109を用いて形質転換体の取得を行なった。目的核酸断片が挿入された形質転換体を液体培養しプラスミドを抽出した。
【0062】
得られたプラスミドの核酸配列をオートシークエンサーにより解析し、野生型の塩基配列を有するプラスミド、ピロリ菌の23S rRNA遺伝子の第2142位または第2143位のアデニンがグアニンに変異した塩基配列を持つプラスミドの計3種類を得た。以下、野生型はWT、23S rRNA遺伝子の2142位変異型は2142G、同じく23S rRNA遺伝子の2143位変異型は2143Gと示す。
【0063】
(実施例2)
(プライマー及び標識プローブの合成)
検出に使用するプライマー及び標識プローブは核酸合成受託会社((株)日本バイオサービス、(株)サワディー、GENSET KK、シグマアルドリッチジャパン(株)等)に依頼した。
【0064】
使用した標識プローブ及びプライマーの塩基配列を表1に示す。標識プローブは配列番号1より選ばれる12塩基から22塩基の塩基配列を使用した。またプライマーについてもプライマーF1〜F4はそれぞれ配列番号3より選ばれる24塩基、29塩基、29塩基、22塩基からなる塩基配列である。プライマーR1〜R4はそれぞれ配列番号4より選ばれる25塩基、31塩基、24塩基、26塩基からなる塩基配列である。プライマーF5はピロリ菌23SrRNA遺伝子の塩基配列中、配列番号1で示される塩基配列までの距離が配列番号3で示される塩基配列より長くなる領域に設計した24塩基からなる塩基配列である。プライマーR5はピロリ菌23SrRNA遺伝子の塩基配列中、配列番号2で示される塩基配列までの距離が配列番号4で示される塩基配列より長くなる領域に設計した22塩基からなる塩基配列である。
【0065】
【表1】

【0066】
(実施例3)
(野生型ピロリ菌の検出)
WT、2142G、2143Gの各プラスミドDNAは表2に示す反応液にテンプレートとして混合した。以下、表2の反応組成を基準とし、反応液を構成するいずれかの溶液の最終濃度を変更した場合はミリQ水で調節し全量20μLを維持した。また、表2の組成は一例であり反応組成の最適化は同業者であれば容易に行えるものである。プラスミド量は104コピー及び102コピーを使用した。プライマーは表1に記載のプライマーF4とプライマーR4を使用した。標識プローブは表1記載の標識プローブAを使用した。標識プローブAは野生型に対して完全に一致する塩基配列の標識プローブに故意の非相補的塩基1塩基を導入したものである。例えば標識プローブAにおいて、故意の非相補的塩基とは、変異部位に相当する標識プローブ中の非相補的塩基、すなわち2142位および2143位を除いた標識プローブの塩基配列に非相補的塩基を導入することをいう。標識プローブAはCAM耐性ピロリ菌(変異型)2142G、2143Gに対して、いずれも非相補的塩基2塩基が存在している標識プローブである。PCR増幅時の反応条件は表3で示される(核酸増幅サイクル)を用いた。増幅及び蛍光の測定にはロシュ・ダイアグノスティック社製ライトサイクラー(登録商標)を使用した。測定モードは530nmを利用した。以下特に記載がない場合はすべてライトサイクラー(登録商標)での測定とする。図4〜6は、グラフの横軸をサイクル数n、縦軸を蛍光シグナル値として解析結果を表した図である。ライトサイクラーソフトウェアの解析により、バックグランドとして現れる標識プローブAの発する蛍光シグナル値を0として計算されているため、縦軸の数値は小さくなるほど標識プローブに対する標的核酸が多いことを示している。
【0067】
【表2】
【0068】
【表3】
【0069】
解析の結果、標識プローブAのプラスミド量104コピー及び102コピーではそれぞれ図4、図5に示す結果が得られた。なお、図4,図5中、2143Gはピロリ菌23S rRNA遺伝子の2143位変異型、2142Gはピロリ菌23S rRNA遺伝子の2142位変異型、NCはテンプレートにミリQ水を使用したネガティブコントロール、WTはピロリ菌23S rRNA遺伝子の野生型を示し、図6〜9においても同様である。野生型のピロリ菌23S rRNA遺伝子を導入したプラスミドをテンプレートにした場合、104コピーでは30サイクルを越えた付近から、102コピーでは38サイクルを越えた付近から標識プローブが標的核酸の増幅産物にハイブリダイゼーションした時の蛍光シグナル値の減少を確認した。一塩基変異が存在している変異型のプラスミドを用いた場合は標識プローブAに2個の非相補的塩基が存在し、増幅反応中の標的核酸に対する標識プローブAのハイブリダイゼーション能力が低下するため、蛍光シグナル値の減少は見られなかった。このように標識プローブ中の非相補的塩基の数を調節することによりWTを特異的に検出することができた。標識プローブへの故意の非相補的塩基の導入は標識プローブの標的核酸に対するハイブリダイゼーション能力及び解離のしやすさを調整するための手法として有効である。プラスミド量102コピーでも標識プローブ特異的かつ短時間での検出が可能であることがわかった。
【0070】
(実施例4)
(ピロリ菌CAM耐性遺伝子変異の検出)
表1に記載の標識プローブBを用いてCAM耐性ピロリ菌(変異型)の検出を行った。標識プローブBは2142G変異型に対して完全一致する塩基配列中に故意の非相補的塩基1塩基を導入したものである。野生型に対しては変異部位に存在する非相補的塩基1塩基と故意の非相補的塩基1塩基で合計非相補的塩基が2塩基存在していることになる。2143G変異型に対しては非相補的塩基3塩基が存在する標識プローブである。標識プローブ以外は実施例3の条件を用いた。
【0071】
標識プローブBでは図4、図5とは異なり2142G変異型のプラスミドをテンプレートとした場合にのみ蛍光シグナル値の減少が確認できた。NC、WT、2143Gのテンプレートを用いた時は蛍光シグナル値の減少が生じなかった。本発明によれば標的核酸の塩基配列特異的に蛍光シグナルの減少が生じることで、標的核酸を検出できることがわかる。
【0072】
(実施例5)
プライマーF1〜F5とプライマーR1〜R5で組み合わせを変えた検討を行った。プライマー以外の条件は実施例3に従った。テンプレートはWTの104コピープラスミドを使用した。その結果を表4に示す。表4の数値は、ライトサイクラーリアルタイムPCRにおける測定時、標識プローブAに標識されたBODIPY−FL(商標)の発する蛍光シグナル値がある一定値(−0.1)を下回った時のサイクル数(Ct値)を示している。蛍光シグナルの減少が見られない組み合わせは数値の記載の代わりに(−)を示した。(A/B)の数値は(標的核酸におけるプライマーRの5‘末端相同的な塩基から標識プローブの3’末端と相補的な塩基までの距離/標的核酸におけるプライマーFの5‘末端と相補的な塩基から標識プローブの5’末端と相補的な塩基までの距離)を示している。
なお、使用した条件は、標識プローブが14塩基以上30塩基以下、標識プローブの濃度が第二プライマーの濃度に対して10倍以下、第一プライマー及び第二プライマーのTm値が70℃以上90℃以下でありかつ第二プライマーのTm値は標識プローブのTm値より大きいと言う全ての条件を満たしている。
【0073】
表4より、(A/B)の数値で0.5〜1.5かつ増幅産物の大きさが300bpまでのプライマーの組み合わせである場合、プライマーF4及びR4の組み合わせと同等の結果を得ることが可能であった。Aの値が大きなR5を使用した場合は、各プライマーFによる伸長反応がプライマーR5まで完了せず増幅がみられなかった。またBが極端に短い場合(A/Bの数値で0.3以下)でも蛍光シグナル値が得られなかった。この結果より増幅産物長が300bp以内で、A/Bの数値が1に近いほうが有利であることがわかる。
また、表4の結果は全て高速PCRの条件で行っているが、例えばプライマーF5とプライマーR1の組み合わせでは検出に至っていない。プライマーF5及びプライマーR1を用いた反応液をアガロースゲル電気泳動により確認したところ、泳動バンドは確認できなかった。従来法の高速PCR条件を用いるだけでは標的核酸の迅速な検出はできないことがわかる。
【0074】
【表4】
【0075】
蛍光シグナルが(−0.1)を下回った時のサイクル数は増幅検出効率の一つの指標となる。表4で該サイクル数を34以下、34を超え35.5以下、35.5を超え38以下、38を超え42以下に分けると、増幅検出効率は34以下>34を超え35.5以下>35.5を超え38以下>38を超え42以下となる。34以下の結果を示したのは(A/B)の数値で0.50〜1.5かつ増幅産物の大きさが100〜300bpに含まれる条件であった。34以下ではプライマーとプローブの位置及び増幅産物長のバランスが最も良好であった。34を超え35.5以下の結果を示したのは(A/B)の数値で0.40〜2.0かつ増幅産物の大きさが100〜300bpに含まれる条件であった。次に35.5を超え38以下の結果を示したのは(A/B)の数値で0.30〜3.0かつ増幅産物の大きさが100〜300bpに含まれる条件であった。38を超え42以下の結果は実施例5の条件において検出限界付近となり、(A/B)の数値で0.30〜3.0かつ増幅産物の大きさが〜400bpに含まれる条件であった。
結果を例示しないが、他の遺伝子配列をターゲットとした時に、(A/B)の数値で0.10〜3.0かつ増幅産物の大きさが50bp〜500bpに含まれる条件であれば、本発明におけるプライマー・プローブの位置及び増幅産物長以外の条件を最適化すると迅速な検出は可能である。本発明に開示されている情報を用いれば条件の最適化は困難ではない。
【0076】
(実施例6)
(第一プライマー濃度及び第二プライマー濃度比の検討)
第二プライマーに対する第一プライマーの濃度比が1倍と10倍の検討を行った。1倍ではプライマーF4 500nM、プライマーR4 500nMを使用した。また、10倍ではプライマーF4 150nM、プライマーR4 1500nMを使用した。プライマー濃度以外の条件は実施例3と同じ条件を使用した。テンプレートはWT、2142G、2143Gの各プラスミド(プラスミド量102コピー)を使用した。濃度比1倍の結果を図6(比較例)、10倍の結果を図5に示す。
【0077】
濃度比が10倍の時は標識プローブに対しての一本鎖標的核酸がより多く増幅することにより、プライマーの伸長及び標識プローブのハイブリダイゼーションの関係が良好に維持されていた。そのため完全一致テンプレートの時のみ特異的に蛍光シグナルの減少が生じた。濃度比1倍の時はテンプレートと塩基配列が完全一致する標識プローブを用いた場合にも蛍光シグナルの減少による検出はできなかった。非対称PCRを行うと標的核酸がより多く増幅するため、対称PCRより特異的検出を感度良く行えることがわかる。
【0078】
(実施例7)
(高速増幅用DNAポリメラーゼの選択)
核酸増幅工程の高速化に適用し得るDNAポリメラーゼを検討した。DNAポリメラーゼとしてはTaqポリメラーゼ(Thermus aquaticus由来)とKODポリメラーゼ(Thermococcus kodakaraensis KOD1由来)の2種類を検討した。Taqポリメラーゼは東洋紡績社製Blend Taq−Plus−(登録商標)、KODポリメラーゼは東洋紡績社製KOD−Plus− ver.2(登録商標)を使用した。Taqポリメラーゼに関しては表1に示す標識プローブA、プライマーF4,R4を使用し、その他の試薬組成はプロトコール記載の試薬組成を参考にした。反応条件は表3に示す(核酸増幅サイクル)を利用した。KODポリメラーゼに関してはTaqポリメラーゼと同じく標識プローブA、プライマーF4,R4を使用し、基本反応条件は実施例3と同様の条件を使用した。テンプレートはWT、2142G、2143Gの各プラスミド(プラスミド量104コピー)を使用した。KODポリメラーゼを用いた場合の結果は図4に示すとおりである。また、それぞれの増幅産物を3%アガロースゲルで電気泳動し、エチジウムブロマイドで染色して増幅産物量を確認した結果を表5に示す。
【0079】
【表5】
【0080】
Taqポリメラーゼを使用した場合、反応条件を固定し上記試薬組成の他にも様々な試薬組成検討を行ったが特異的なピロリ菌の検出はできなかった。これに対しKODポリメラーゼを使用すると高速増幅に対応して特異的にピロリ菌の検出が可能であった。電気泳動の結果から、Taqポリメラーゼによる増幅は高速増幅の場合WTでわずかにバンドが見える程度の増幅であり、WT のプラスミド(102コピー)をテンプレートにした時には電気泳動バンドの検出もできなかった。KODポリメラーゼは高速増幅に適応できるが、Taqポリメラーゼは高速増幅に適応できず、目的の核酸増幅が見られないためピロリ菌の検出ができなかった。伸長速度が速くかつ5‘エキソヌクレアーゼ活性を持たないKODポリメラーゼのほうが本発明のDNAポリメラーゼとして適していることがわかる。
【0081】
(実施例8)
核酸増幅工程に引き続き融解曲線解析を行うことで1塩基変異が存在する場合のテンプレートと存在しない場合のテンプレートを1つの標識プローブで検出できるかどうかを検討した。テンプレートはピロリ菌WT、2142G、2143Gの三種類のプラスミドを使用し、プラスミド量は104コピー及び102コピーを使用した。実施例3と同じ試薬組成ならびに反応条件は表3の(核酸増幅サイクル)・(融解曲線解析)・(クールダウン)に示すものを使用した。プライマー及び標識プローブはそれぞれ表1に示すプライマーF4,R4、核酸プローブAを使用した。得られた蛍光シグナル値の補正は行わず、ライトサイクラー(登録商標)における融解曲線解析の結果をそのまま利用した。測定レンジは530nmを使用した。
【0082】
104コピー及び102コピーのプラスミドをテンプレートとして用いた時の蛍光測定の結果をそれぞれ図7、図8に示す。ライトサイクラー(登録商標)を使用した融解曲線解析においては標識プローブが標的核酸から解離する時の蛍光量の増加を検出している。図の縦軸は蛍光強度の一次導関数の逆符号の値(−dF/dt)、横軸は温度(℃)である。図7、図8からはそれぞれ2つのピークが得られた。61℃に見られるピークはWTのピークであり、50℃に見られるピークはWTに対して1塩基変異を持つ二種類の変異型のピロリ菌のピークである。この結果から、1つの標識プローブを使用し融解曲線解析を行うことでWTと変異型を別々のピークで検出できることがわかる。
【0083】
(実施例9)
(菌体を直接テンプレートとした融解曲線解析による検出)
ピロリ菌のATCC株2種(野生型26695株及び2143G変異型UA1182株)を入手した。そのうちそれぞれ109菌体を分取し、95℃5分の熱処理を行った。その後それぞれ105熱処理後菌体溶液を表2のテンプレートとした。その他は融解曲線解析を行った実施例8と同様の方法にてピロリ菌の検出を行った。その結果を図9に示す。
【0084】
融解曲線解析によりピロリ菌野生型及びCAM耐性ピロリ菌の検出を行ったところ、いずれも融解曲線解析によって単独ピークとして検出することができた。菌体をサンプルとして検出(CAM耐性判定)まで30分以内で行うことが可能である。
【0085】
(比較例1)
(ピロリ菌の検出までの時間)
一般的に使用されるPCRサイクル94℃15秒、65℃30秒、68℃30秒でPCRを行い、融解曲線解析で検出を行った場合、サーマルサイクラーの中でも温度の上昇・下降速度の早いとされているライトサイクラー(登録商標)でも増幅・検出あわせて1時間以上を要した。
【0086】
実施例3の反応条件において、ライトサイクラー(登録商標)を使用した標的核酸の検出までに要する時間は15分以内、さらに融解曲線解析を用いてもピロリ菌の検出及びピロリ菌CAM耐性遺伝子の存在を検出するのに要する時間は20分以内であった。本発明の検出方法を用いると極めて短時間で、かつ高感度なピロリ菌の検出が可能である。
【0087】
(実施例10)
(第二プライマーと標識プローブ濃度比の検討)
表1に記載の標識プローブAとプライマーF4、プライマーR4を用いてプライマーとプローブの濃度比の検討を行った。プライマー、プローブ濃度以外の条件に関しては実施例3の条件を用いた。第二プライマー(プライマーF4)とプローブ濃度に関しては以下の4条件を用いた。
第二プライマー150nM、プローブ150nM(プローブ/プライマー比=1):図4
第二プライマー100nM、プローブ500nM(プローブ/プライマー比=5):図10
第二プライマー100nM、プローブ1000nM(プローブ/プライマー比=10):図11
第二プライマー100nM、プローブ1100nM(プローブ/プライマー比=11)
テンプレートはピロリ菌WTのプラスミド104を用いた。
【0088】
図4、図10、11よりプローブ濃度/プライマー濃度が1の場合は問題なくMTを検出できている。濃度比5の時はプローブが多いことによる増幅制限を受けているため、Ct値は濃度比1の時と比較してやや遅くなっているがMTを検出できていた。濃度比10では、プローブ過多により増幅制限が強く現れるとともに、標的核酸にハイブリダイゼーションして消光しているプローブに対してハイブリダイゼーションせずに蛍光を発しているプローブが多くなりバックグラウンドシグナルが増えたことで、濃度比1や5よりもシグナルが弱くなっているが、MTを検出できていた。データは示していないが、さらに濃度比が11と高くなった場合は、増幅制限を受けていることとシグナル検出温度においてハイブリダイゼーションしていないプローブが極端に多くなり、そのバックグランドシグナルによって蛍光シグナルの差が見られなくなったことの2つの理由により検出できなかった。
【0089】
(実施例11)
(プローブ塩基数の検討)
表1に記載の標識プローブA、C〜Eを用いてプローブ塩基数の検討を行った。標識プローブ以外の条件に関しては実施例3の条件を用いた。標識プローブAは19塩基(Tm=62.20)、標識プローブCは12塩基(Tm=33.47)、標識プローブDは16塩基(Tm=54.34)、標識プローブEは32塩基(Tm=80.65)である。標識プローブCについてはアニール温度を50℃に設定変更して検出を行った。テンプレートはピロリ菌WTのプラスミド104を用いた。標識プローブA、標識プローブDを用いて検出した結果をそれぞれ図4、図12に示す。
【0090】
図4、図12より、標識プローブ塩基数が14〜30塩基に含まれる標識プローブAおよび標識プローブDではWTの検出が確認できた。12塩基である標識プローブCおよび32塩基である標識プローブEを用いた場合は蛍光シグナルの減少が見られずWTの検出が確認できなかった。特に32塩基の標識プローブEについてはTm値が80.65であり、第二プライマーのTm値71.66よりも高くなっており、標識プローブEの標的核酸へのハイブリダイゼーションによりプライマーの伸長反応が阻害されたと考えられる。またこの結果より、従来の高速PCR法に蛍光標識された核酸プローブを添加するだけでは標的核酸の迅速な検出が実現できないことがわかる。
【0091】
(実施例12)
(プローブ塩基数の検討:融解曲線解析)
表1に記載の標識プローブA、C〜Eを用いてプローブ塩基数の検討で融解曲線解析による検出を行った。標識プローブ以外の条件に関しては実施例8の条件を用いた。テンプレートはピロリ菌WT、2142G、2143Gの三種類のプラスミドを使用し、プラスミド量は104コピーを使用した。
【0092】
図7、図13より標識プローブ塩基数が14〜30塩基に含まれる標識プローブAおよび標識プローブDではWT、2142G、2143Gのそれぞれの検出ピークが確認できた。12塩基である標識プローブCおよび32塩基である標識プローブEを用いた場合はWT、2142G、2143Gのそれぞれの検出ピークが確認できなかった。標識プローブEについては増幅産物をアガロースゲル電気泳動にて確認したところ、目的の位置に泳動バンドが見られず、増幅阻害が起きていた。
【0093】
以下に16S rRNA遺伝子を標的とした抗酸菌(マイコバクテリウム属菌)を検出する方法を示す。
【0094】
(実施例13)
(テンプレートの調製)
ヒト型結核菌(Mycobacterium tuberculosis)、トリ型結核菌(Mycobacterium avium)、マイコバクテリウムカンサシイ(Mycobacterium kansasii)をそれぞれ小川培地(日水社製)にて37℃約2週間培養した。培養した各菌体をリン酸緩衝液に懸濁した液を用いて、常法に従いフェノール・クロロホルム抽出を行い、ゲノムDNAを抽出した。抽出されたゲノムDNAは−20℃で保存した。ヒト型結核菌はMT、トリ型結核菌はMA、マイコバクテリウムカンサシイはMKと示す。
【0095】
(実施例14)
(プライマー及び標識プローブの合成)
検出に使用するプライマー及び標識プローブは核酸合成受託会社((株)日本バイオサービス、(株)サワディー、GENSET KK、シグマアルドリッチジャパン(株)等)に依頼した。
【0096】
使用した標識プローブ及びプライマーの塩基配列を表6に示す。配列番号34〜38は標識プローブで、配列番号34、37,38はMTを、配列番号35はMAを、配列番号36はMKを検出するためのプローブである。配列番号20〜33は抗酸菌16SrRNA遺伝子の一部の領域を増幅できる共通のプライマーである。
【0097】
【表6】
【0098】
(実施例15)
(MT、MA、MKの検出)
表7または表2に示す反応液にMT、MA、MKの各ゲノムDNA20ngをテンプレートとして混合した。以下、表7または表2の反応組成を基準とし、反応液を構成するいずれかの溶液の最終濃度を変更した場合はミリQ水で調節し全量20μLを維持した。また、表7または表2の組成は一例であり反応組成の最適化は同業者であれば容易に行えるものである。プライマーは表6に記載のプライマーF6とプライマーR6を使用した。標識プローブは表6記載の標識プローブF〜Hを使用した。標識プローブFはMTの塩基配列に対して完全に一致する塩基配列を持つ標識プローブである。標識プローブGはMAの塩基配列に対して完全に一致する塩基配列を持つ標識プローブである。標識プローブHはMKの塩基配列に対して完全に一致する塩基配列を持つ標識プローブである。標識プローブFを使用する場合は表7の組成、標識プローブG、Hを使用する場合は表2の組成を利用した。増幅及び蛍光の測定にはロシュ・ダイアグノスティック社製ライトサイクラー(登録商標)を使用した。PCRの条件は表8に示す条件を使用した。測定モードは530nmを利用した。以下、特に記載がない場合はすべてライトサイクラー(登録商標)での測定とする。図14〜18は、グラフの横軸をサイクル数n、縦軸を蛍光シグナル値として解析結果を表した図である。ライトサイクラーソフトウェアの解析により、バックグランドとして現れる標識プローブの発する蛍光シグナル値を0として計算されているため、縦軸の数値は小さくなるほど標識プローブに対する標的核酸が多いことを示している。
【0099】
【表7】
【0100】
【表8】
【0101】
解析の結果、標識プローブFを使用してMT、標識プローブGを利用してMA、標識プローブHを利用してMKを検出した結果をそれぞれ図14〜16に示す。なお、図14〜16中、NCはテンプレートにミリQ水を使用したネガティブコントロールを示す。標識プローブFを用いた図14からはMTに特異的な検出シグナルが、標識プローブGを用いた図15からはMAに特異的な検出シグナルが、標識プローブHを用いた図16からはMKに特異的な検出シグナルが、それぞれ確認できた。
【0102】
(実施例16)
(第二プライマーとプローブ濃度比の検討)
表6に記載の標識プローブFとプライマーF6、プライマーR6を用いてプライマーとプローブの濃度比の検討を行った。プライマー、プローブ濃度以外の条件に関しては実施例12の条件を用いた。テンプレートはMTゲノムDNA20ngを用いた。第二プライマー(プライマーR6)とプローブ濃度に関しては以下の3条件を用いた。
第二プライマー150nM、プローブ150nM(プローブ/プライマー比=1):図14
第二プライマー150nM、プローブ450nM(プローブ/プライマー比=5):図17
第二プライマー150nM、プローブ1650nM(プローブ/プライマー比=11):図18
【0103】
図14、17,18より、プローブ濃度/プライマー濃度が1の場合は問題なくMTを検出できているのに対して、濃度比5の時はプローブが多いことによる増幅制限を受けてCt値が遅くなっている。さらに濃度比が11と高くなった図18では、増幅制限とともに、シグナル検出温度においてハイブリダイゼーションしていないプローブが極端に多くなり、そのバックグランドシグナルによって蛍光シグナルの差が見られなくなった。標識プローブFとプライマーF6、プライマーR6を用いた検討ではプローブ濃度/プライマー濃度=11での検出はできなかったが、標識プローブの蛍光強度が低く、第二プライマーのTmがプローブ濃度のTmよりも極端に大きい場合に、プローブ濃度/プライマー濃度=10では検出が確認できる組み合わせも存在する。
【0104】
(実施例17)
(プライマーのTmとプローブのTmの検討)
表6に記載の標識プローブF、I、JとプライマーF6〜8、プライマーR6〜8を用いてプライマーとプローブのTmによってMT検出に差が現れるかどうかの検討を行った。プライマーF6とR6、プライマーF7とR7、プライマーF8とR8をプライマーセットとして固定し、それぞれTmの異なるプライマーセットを得た。標識プローブ3種類とプライマーセット3種類を組み合わせて検討した。標識プローブとプライマーセット以外の条件に関しては基本的に実施例15の条件を用いた。標識プローブIの場合のみ、表8におけるPCR条件のアニール・伸長温度を60℃から57℃に変更して実施した。結果を表9に示す。表9の「○」はCt値=40未満、「△」はCt値=40以上、「×」は検出されないことを示している。例えば、標識プローブFとプライマーF6、R6の組み合わせの結果は、図14に示されるが、この場合Ct値=31.35で「○」となる。その他の組み合わせについても図14様の図が得られているが、ここでは結果のみ示す。
【0105】
【表9】
【0106】
表9より、プライマーとプローブのTmによって検出されるCt値に差が見られた。例えばプライマーF6とR6、標識プローブFの組み合わせでは、プローブTm<プライマーTmのため問題なく検出が可能であった。一方、プライマーF8とR8、標識プローブJの組み合わせではプローブTm>プライマーTmのため、検出ができなかった。高速検出においてプローブTm>プライマーTmの場合は、プローブのハイブリダイゼーションによりプライマーの伸長が阻害され、増幅が阻害されたと考えられる。
【0107】
(実施例18)
(プライマーのTmの検討)
表6に記載の標識プローブF及びプライマーF6、F8〜12、プライマーR6、R8〜12を用いてプライマーTm値によってMT検出に差が現れるかどうかの検討を行った。プライマーF6種類とプライマーR6種類を組み合わせて検出を実施した。プライマー以外の条件は基本的に実施例15の条件を用いた。ただし、PCR条件におけるアニール・伸長温度は各プライマーセットのTmに合わせて50℃〜60℃の範囲で設定した。結果を表10に示す。判定基準は実施例17と同様とした。
【0108】
【表10】
【0109】
表10より、プライマーのTmが70以上であれば問題なく検出されているが、プライマーF,RのどちらかのプライマーTmが70未満となると高速検出の条件バランスがくずれてCt値が遅延しているのがわかる。本願発明ではプライマー及び標識プローブの濃度やTmなどの条件バランスが重要となってくるため、安定して検出を行うにはプライマーのTmは70℃以上が適している。
【0110】
(実施例19)
(MTとMAの融解曲線解析による検出)
核酸増幅工程に引き続き融解曲線解析を行うことで1塩基変異が存在する場合のテンプレートと存在しない場合のテンプレートを1つの標識プローブで検出できるかどうかを検討した。テンプレートはMT、MAのゲノム20ngを使用した。実施例15と同じ試薬組成ならびに反応条件は表8の(核酸増幅サイクル)・(融解曲線解析)・(クールダウン)に示すものを使用した。プライマー及び標識プローブはそれぞれ表6に示すプライマーF6,R6、核酸プローブFを使用した。得られた蛍光シグナル値の補正は行わず、ライトサイクラー(登録商標)における融解曲線解析の結果をそのまま利用した。測定レンジは530nmを使用した。
【0111】
MT及びMAゲノムをテンプレートとして用いた時の蛍光測定の結果を図19に示す。ライトサイクラー(登録商標)を使用した融解曲線解析においては標識プローブが標的核酸から解離する時の蛍光量の増加を検出している。図の縦軸は蛍光強度の一次導関数の逆符号の値(−dF/dt)、横軸は温度(℃)である。図19では2つのピークが確認できた。60℃に見られるピークは標識プローブFに対して完全に一致する塩基配列を持つMTの検出ピークである。55℃に見られるピークは標識プローブFに対して1塩基変異が存在する塩基配列を持つMAの検出ピークである。この結果から、1つの標識プローブを使用し融解曲線解析を行うことで抗酸菌であるMTとMA種を別々のピークとして検出できることがわかった。
【0112】
(比較例2)
(MTの検出までの時間)
一般的にMTの遺伝子検査に使用されるロシュダイアグノスティック社のアンプリコアマイコバクテリウムツベルクローシスキットを用いて、陽性コントロールを検出するのに要する時間はPCR及び検出できるまで最短で約6時間かかる。
【0113】
実施例15の反応条件において、ライトサイクラー(登録商標)を使用した標的核酸の検出までに要する時間は約20分、さらに融解曲線解析を用いてもMTを検出するのに要する時間は30分以内であった。本発明の検出方法を用いれば極めて短時間でのMT検出が可能となる。
【産業上の利用可能性】
【0114】
本発明によれば、迅速かつ高感度な核酸検出が可能となる。具体的には、第二プライマーの伸長反応を阻害しない標識プローブの性質及び濃度により、増幅効率を保持したまま不要なバックグランド蛍光シグナル値を極力減らし、必要な蛍光シグナルの減少量を測定することが可能になる。さらに伸長速度が速いDNAポリメラーゼを使用することにより増幅反応時間を短縮し、これまでになく迅速かつ高感度な核酸検出方法を実現でき、さらに、ピロリ菌の23S rRNA遺伝子の存在または量を調べること、ピロリ菌の23S rRNA遺伝子の変異の存在を調べること、23S rRNA遺伝子の第2142位または第2143位における変異の存在を調べピロリ菌の抗生物質耐性を判別することなども迅速かつ高感度に行えるようになり、産業界に大きく貢献する。
【図面の簡単な説明】
【0115】
【図1】プライマー及びプローブの濃度とそれぞれのTm値の関係により可能となる迅速かつ高感度な検出方法の原理を示す。
【図2】標的核酸または第一プライマーの伸長産物と標識プローブの位置関係A、Bを模式的に示す。
【図3】本発明における非対称PCRの方法について、第一プライマー、第二プライマー及び標識プローブの位置関係を示す。
【図4】プラスミド104コピーを鋳型としてピロリ菌のリアルタイム検出結果を示す。
【図5】プラスミド102コピーを鋳型としてピロリ菌のリアルタイム検出結果を示す。
【図6】第一プライマーと第二プライマーの濃度比が1:1である時のピロリ菌のリアルタイム検出結果を示す。
【図7】プラスミド104コピーを鋳型として融解曲線解析によるピロリ菌の野生型と変異型を同時検出した結果を示す。
【図8】プラスミド102コピーを鋳型として融解曲線解析によるピロリ菌の野生型と変異型を同時検出した結果を示す。
【図9】菌体を簡易処理したものを鋳型としてピロリ菌の検出を行った結果を示す。
【図10】プローブ/プライマー比=5の条件におけるピロリ菌のリアルタイム検出結果を示す。
【図11】プローブ/プライマー比=10の条件におけるピロリ菌のリアルタイム検出結果を示す。
【図12】標識プローブDを用いた場合のピロリ菌のリアルタイム検出を示す。
【図13】標識プローブDを用いた場合の融解曲線解析によるピロリ菌の野生型と変異型同時検出結果を示す。
【図14】16SrRNA遺伝子をターゲットとした時のMTのリアルタイム検出結果を示す。
【図15】16SrRNA遺伝子をターゲットとした時のMAのリアルタイム検出結果を示す。
【図16】16SrRNA遺伝子をターゲットとした時のMKのリアルタイム検出結果を示す。
【図17】プローブ/プライマー比=5の条件におけるMTのリアルタイム検出結果を示す。
【図18】プローブ/プライマー比=11の条件におけるMTのリアルタイム検出結果を示す。
【図19】MT及びMAゲノムを鋳型として融解曲線解析によるMTとMAをピークのTmで判別した結果を示す。
【技術分野】
【0001】
本発明は、試料中に存在する特定の標的核酸分子を迅速に検出する方法及びこれらの方法により利用するプローブとキットに関する。
【背景技術】
【0002】
一般に生物の保持する核酸は極めて少量であり、それらを検出する際には核酸増幅工程を伴うことがほとんどである。既に知られている核酸増幅方法としてPCR(特許文献1、特許文献2、特許文献3)、NASBA(非特許文献1)、LCR(特許文献4、特許文献5)、SDA(非特許文献2)、RCR(特許文献6)、TMA(非特許文献3)LAMP(非特許文献4)、ICAN(非特許文献5)などが挙げられる。
【0003】
なかでもPCR法は、標的核酸、4種類のデオキシヌクレオシド三リン酸、一対のオリゴヌクレオチドプライマー及び耐熱性DNAポリメラーゼの存在下で、温度の上昇、下降を繰り返すことにより、上記一対のオリゴヌクレオチドプライマーで挟まれる標的核酸の特定領域を指数関数的に増幅させることができる。
【0004】
通常PCR法は特許文献3に示されているように、以下のa)〜c)からなる増幅サイクルを用いる。
a)90〜105℃の範囲内の温度で(好ましくは90〜100℃)0.5〜5分間(好ましくは0.5〜3分間)、変性させる工程、b)35〜65℃の範囲内の温度で(好ましくは37〜60℃)0.5〜5分間(好ましくは1〜3分間)、プライマーと鋳型のハイブリッドを形成させる(アニーリング)工程、及びc)40〜80℃の範囲内の温度で(好ましくは50〜75℃)0.5〜40分間(好ましくは1〜3分間)、プライマー伸長生成物を形成させる(伸長)工程。
【0005】
PCR法のサイクルを繰り返し行うことで増幅させた標的核酸の複製は、各種の検出方法を用いて検出できるようになる。現在利用されている代表的な検出方法として、アガロースゲル電気泳動法がある。しかしながら、電気泳動による検出では指数関数的に増幅された標的核酸の複製を取り扱う必要がありコンタミネーションの起こる危険性が高かった。また、操作も煩雑であり検出終了までの時間も1時間以上を要した。
【0006】
電気泳動以外の検出方法として、二重鎖核酸に結合した時に増強された蛍光を示すDNA挿入色素(インターカレーター)を利用する方法が一般的に用いられている。増幅中のDNA濃度の上昇による蛍光増強は、反応の進行を測定するのに、および標的分子のコピー数を決定するのに利用できる。さらに、制御された温度変化に伴う蛍光をモニターすることにより、例えばPCRサイクル反応の終了時点で、DNA融解曲線が作成できる。
【0007】
一般的な核酸検出法が核酸濃度の上昇をモニターするのに利用される場合、前述のインターカレーターを利用する方法は比較的短時間で核酸を検出することができる。各反応の同じ時点で、単一の蛍光シグナル値が取得される。最終時点での融解曲線解析は、二重鎖核酸の性質をある程度識別できる。融解曲線解析での融解温度が極端に低い時はプライマーダイマーと考えられるし、主なピークが一本でない時は目的のPCR産物ではない非特異増幅産物が存在することを示している。
【0008】
しかし、一般的なインターカレーター法は二重鎖核酸特異的に蛍光が生じる。それゆえ目的の核酸配列特異的な検出が必要とされる場合は目的の核酸配列に非特異的な二重鎖核酸をも検出する可能性があるため、それほど有効ではない。
【0009】
核酸配列特異的プローブ法は、増幅反応の進行をモニターするのにさらなる核酸反応成分を利用する。これらの方法は、検出の基本として、蛍光エネルギー転移(FET)を利用することが多い。一つあるいはそれ以上の核酸プローブを蛍光分子で標識するが、そのうちの一つはエネルギー供与体として働くことができ、もう一方はエネルギー受容体分子である。これらは時として、それぞれレポーター分子および消光分子として知られる。供与体分子は励起スペクトラム範囲の光の特異的波長で励起され、引き続いて蛍光放出波長の範囲で光を放出する。受容体分子も、様々な距離依存性エネルギー転移機構により、供与体分子からエネルギーを受け取ることにより、この波長で励起される。起こり得る蛍光エネルギー移動の特別の例には、蛍光共鳴エネルギー転移(FRET)がある。一般に、受容体分子と供与体分子が近接している時に(例えば、同じまたは隣接する分子上)、受容体分子は供与体分子の放出エネルギーを受け取る。FET検出の基本は、供与体と受容体放出波長における変化をモニターすることである。
【0010】
FETまたはFRETプローブには通常用いられる二つの種類があり、受容体から供与体を分離するために核酸プローブの加水分解を用いるものと、供与体と受容体分子の空間的関係を変化させるためにハイブリダイゼーションを利用するものである。
【0011】
加水分解プローブは、TaqMan(登録商標)プローブとして市販されている。これらは、供与体および受容体分子で標識されたDNAオリゴヌクレオチドからなる。このプローブは、PCR増幅産物の一方の鎖の特異的領域に結合するよう設計される。PCRプライマーがこの鎖にアニールした後、Taq酵素が5’から3’へのポリメラーゼ活性によりDNAを伸長する。TaqMan(登録商標)プローブは、Taq伸長の開始を防ぐために、3’末端がリン酸化で保護されている。もしTaqMan(登録商標)プローブが産物の鎖にハイブリダイズしているのならば、伸長するTaq分子がプローブを加水分解し、検出の基本として受容体から供与体を遊離する。この場合のシグナルは累積的であり、遊離の供与体と受容体分子の濃度は増幅反応の各サイクルで上昇する。
【0012】
蛍光シグナルの生成がプローブの加水分解反応の発生に依存するという事実は、この方法に伴う時間的不利益が存在することを意味する。さらに、プローブの存在がPCR過程のプライマーの伸長過程を妨げる可能性もあった。また、50サイクル以上というような多数回の増幅サイクルが必要な場合、加水分解が非特異的になり得ることも見出されている。
【0013】
ハイブリダイゼーションプローブは、数多くの型式のものが利用可能である。分子ビーコンは、ヘアピンループを形成するような相補的な5’および3’配列を有するオリゴヌクレオチドである。末端の蛍光標識は、ヘアピン構造が形成されるためにFRETの近接にある。分子ビーコンの相補的配列へのハイブリダイゼーションに続き、蛍光標識は分離され、そのためFRETは生じず、これが検出の基本となる。
【0014】
標識プローブの対合も利用可能である。供与体分子を標識したプローブと受容体分子を標識したプローブがPCR産物の鎖上で近接してハイブリダイゼーションすることによって、FRETが生じ得る。この時のFRETによって生じた波長の蛍光が検出の基本となる。このタイプの変種には、標識増幅プライマーと単一の近接する標識プローブの利用が含まれる。
【0015】
二つのプローブ、または二つの標識分子を含む分子ビーコンタイプの使用は、その過程に伴うコストを増やす。さらに近接して特異的に結合するに十分な長さの二つのプローブを知るため、この方法では合理的な長さの既知配列の存在が必要となる。これはある診断応用で問題となり、例えばHIVウイルスのように、保存されている配列の長さが比較的短い場合に、有効なプローブを設計することが難しいことがある。
【0016】
特許文献7では、ハイブリダイゼーション時に蛍光が消光する核酸プローブを用いた方法が提供されている。蛍光色素で標識された核酸プローブを標的核酸にハイブリダイゼーションさせ、ハイブリダイゼーション前後における蛍光色素の発光の減少量を測定する方法または蛍光色素で標識された核酸プライマーを標的核酸にハイブリダイゼーションさせた後伸長させ、核酸変性時の蛍光量に対してアニーリング反応及び伸長反応時の蛍光量の減少を測定する方法が示されている。さらに該方法を用いたリアルタイム定量的PCR法、及び該PCR法で得られるデータの解析の際、アニーリング反応時の蛍光強度値を、変性反応時のもので補正する過程を有するデータ解析法が記載されている。しかしながら、特許文献7の記載のみでは特異性および高感度を維持しながら迅速に核酸を検出する方法を容易に想像し得るものではない。つまり、ハイブリダイゼーション時に蛍光が消光する核酸プローブを使用した場合は核酸プローブが核酸増幅工程、特に伸長工程を阻害するため、短時間化しようとすると感度が低下する問題点、またハイブリダイゼーション時に蛍光が消光する核酸プライマーを使用した場合は核酸プライマーの非特異ハイブリダイゼーションから伸長が開始され、非特異増幅産物となって蛍光の減少を検出してしまい特異的な検出が難しいという問題点があり、特許文献7に開示された技術を用いて特異性および高感度を維持しながら迅速に核酸を検出することは困難であった。
【特許文献1】米国特許第4,683,195号
【特許文献2】米国特許第4,683,202号
【特許文献3】米国特許第4,965,188号
【特許文献4】国際公開89/12696号
【特許文献5】特開平2−2934号
【特許文献6】国際公開90/1069号公報
【特許文献7】特許第3437816号
【非特許文献1】Nucleic acid sequence−basedamplification method;Nature 第350巻、第91頁(1991)
【非特許文献2】Strand Displacement Amplification:Nucleic acid research 第20巻、第1691頁(1992)
【非特許文献3】Transcription mediated amplification method;J.Clin.Microbiol. 第31巻、第3270頁(1993)
【非特許文献4】loop−mediated isothermal amplification method :J Clin Microbiol. 2004 第42巻:第1,956頁
【非特許文献5】isothermal and chimeric primer−initiated amplification of nucleic acids: Kekkaku. 2003 第78巻、第533頁
【発明の開示】
【発明が解決しようとする課題】
【0017】
前述のように、標的核酸とのハイブリダイゼーション時に蛍光が消光する核酸プローブまたは核酸プライマーを用いた方法では、核酸プローブが核酸増幅工程、特に伸長工程を阻害するため、短時間化の際に感度が低下する問題があり、核酸プライマーの非特異増幅によって標的核酸特異的検出が妨げられる問題があった。本発明では、これらの問題点を克服し、標的核酸の検出を特異性および高感度を維持しながら迅速に行うことを課題とした。
【課題を解決するための手段】
【0018】
本発明者らは、上記の点に鑑み、鋭意検討を重ねた結果、第一プライマー及び第二プライマーのTm値と標識プローブのTm値の関係、さらにプライマーの濃度と標識プローブの濃度の関係を調節し、最適化することによって標的核酸の検出をより迅速に、より高感度に行うことができることを見出し、本発明に至った。すなわち、本発明は以下のような構成からなる。
【0019】
1.標的核酸の存在または量を確認したい試料と少なくとも下記(a)と(b)の存在下、連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該標識物質の蛍光を測定しながら、高速PCRを行い、該標的核酸の存在または量を調べることを特徴とする検出方法。
(a)次の(i)〜(iv)の性質を有する標識プローブ
(i) 標的核酸に相補的な配列と二重鎖核酸構造を形成でき、かつ14塩基以上30塩基以下の長さのオリゴヌクレオチドを有する
(ii) 該オリゴヌクレオチドの末端部が該二重鎖核酸構造形成時に消光する性質を有する蛍光物質で修飾されている
(iii) 該二重鎖核酸構造形成時に、蛍光物質で修飾されている該末端部にG(グアニン)とC(シトシン)の対が少なくとも1つ以上存在する
(iv)該標識プローブの濃度が第二プライマーの濃度に対して10倍以下である
(b)次の(i)〜(iii)の性質を有する第一プライマーおよび第二プライマー
(i) 第一プライマーの伸長産物が標的核酸と相補的な配列を有しかつ第二プライマーの鋳型となる性質を有し、該第二プライマーの伸長産物が標的核酸と相同な配列を有しかつ該第一プライマーの鋳型となる性質を有する
(ii) 該第一プライマーおよび該第二プライマーによる増幅産物の長さが50bp以上500bp以下である
(iii) 該第一プライマー及び該第二プライマーのTm値が70℃以上90℃以下であり、かつ該第二プライマーのTm値は前記標識プローブのTm値より大きい
2.(b)の第一プライマーおよび第二プライマーが、さらに
(iv) 該第一プライマーの伸長産物において、該伸長産物の5’末端から前記標識プローブが二重鎖核酸構造を形成する領域までの距離を(A)、該標識プローブが二重鎖核酸構造を形成する領域から該伸長産物の3’末端までの距離を(B)とした場合に(A/B)の値が0.1以上3.0以下であること、
を特徴とする1の検出方法。
3.(a)の標識プローブに、さらに
(v) 標的部位と3個以下の非相補的塩基を導入すること、
を特徴とする1または2の検出方法。
4.標識プローブの中央部に、非相補的塩基を導入することを特徴とする3載の検出方法。
5.(a)の標識プローブに修飾されている蛍光物質が該標識プローブの末端に修飾され、該末端の塩基がGもしくはCであることを特徴とする1〜4のいずれかの検出方法。
6.第一プライマーの濃度が第二プライマーの濃度に対して4倍以上であることを特徴とする1〜5のいずれかの検出方法。
7.高速PCRが、10℃/秒以上の速度で温度の上昇及び下降を繰り返すことを特徴とする1〜6のいずれかの検出方法。
8.さらに、
(c)100塩基/秒以上のDNA合成速度を有するDNAポリメラーゼ
の存在下、
高速PCRを行うことを特徴とする1〜7のいずれかの検出方法。
9.DNAポリメラーゼが、さらに5’エキソヌクレアーゼ活性を持たないことを特徴とする8の検出方法。
10.DNAポリメラーゼが、KOD DNAポリメラーゼ(Thermococcus kodakaraensis KOD1由来)またはその変異体であることを特徴とする8または9の検出方法。
11.標的核酸がピロリ菌の23S rRNA遺伝子のセンス鎖であり、
(a)の標識プローブのオリゴヌクレオチドの配列が配列番号2より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べることを特徴とする1〜10のいずれか載の検出方法。
12.標的核酸がピロリ菌の23S rRNA遺伝子のアンチセンス鎖であり、
(a)の標識プローブのオリゴヌクレオチドの配列が配列番号1より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べることを特徴とする1〜10のいずれかの検出方法。
13.1〜12のいずれかの方法に引き続いて、密封状態のまま、標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、0.1℃/秒以上の連続的な温度上昇又は下降を伴う融解曲線解析を行い、標的核酸中の変異の存在を調べることを特徴とする検出方法。
14.変異がピロリ菌の23S rRNA遺伝子の第2142位または第2143位であり、抗生物質耐性を判別する変異の存在を調べることを特徴とする13の検出方法。
15.標識プローブのオリゴヌクレオチドの配列の中央部に変異の部位が位置することを特徴とする13または14の検出方法。
16.少なくとも次の(a)及び(b)を含むことを特徴とするピロリ菌の存在および/またはピロリ菌の抗生物質耐性の存在を検出するためのキット。
(a)次の(i)〜(ii)の性質を有する標識プローブ
(i)配列番号1または配列番号2より選ばれる14塩基以上30塩基以下の連続した塩基配列のオリゴヌクレオチドを有する
(ii)該オリゴヌクレオチドが、該オリゴヌクレオチドと相補的な配列と二重鎖核酸構造を形成する時に消光する性質を有する蛍光物質が、該オリゴヌクレオチドの末端部に標識されている
(b)Tm値が70℃以上90℃以下であり配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列を含むプライマーと、Tm値が70℃以上90℃以下であり配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列を含むプライマー
17.(a)の標識プローブと二重鎖核酸構造を形成しうる伸長産物を生成せしめる側のプライマーの濃度が、他方のプライマーの濃度に対して4倍以上であることを特徴とする16のキット。
18.さらに、
(c)100塩基/秒以上のDNA合成速度を有するDNAポリメラーゼ
を含むことを特徴とする16または17のキット。
20.DNAポリメラーゼが、さらに5’エキソヌクレアーゼ活性を持たないことを特徴とする18のキット。
20.DNAポリメラーゼが、KOD DNAポリメラーゼ(Thermococcus kodakaraensis KOD1由来)またはその変異体であることを特徴とする18または19のキット。
21.標識プローブの中央部に、ピロリ菌の23S rRNA遺伝子の第2142位または第2143位に相当する部位が位置することを特徴とする16〜20のキット。
【発明の効果】
【0020】
本発明によれば、特異性および高感度を維持しながら迅速な核酸検出が可能となる。具体的には、標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少が生じることと、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、PCRの同一サイクル中に達成することが可能になった。さらに伸長速度が速いDNAポリメラーゼを使用することにより増幅反応時間を短縮し、これまでになく迅速かつ高感度な核酸検出方法を実現できた。
さらに、本発明を利用し、ピロリ菌の23S rRNA遺伝子の存在または量を調べること、ピロリ菌の23S rRNA遺伝子の変異の存在を調べること、23S rRNA遺伝子の第2142位または第2143位における変異の存在を調べピロリ菌の抗生物質耐性を判別することも迅速かつ高感度に行えるようになった。
【発明を実施するための最良の形態】
【0021】
本発明では、高速PCRは核酸増幅工程、すなわち後述するような「変性」、「アニーリング」、「伸長」からなる一連の工程、または本工程のうちアニーリングおよび伸長を同時に行い「変性」および「アニーリングと伸長」とした一連の工程、に要する時間を短縮することにより、迅速性が高い利点を有している。また、核酸増幅を行うためのプライマーとは別にハイブリダイゼーション時に蛍光を消光させる標識プローブを使用することにより、標的核酸の存在または量を反映した特異的な増幅核酸を、標的核酸に依存しない非特異的な増幅核酸と明確に区別し得るため、感度および正確性が高い利点を有している。しかし、これらを単純に組み合わせても、特異性および高感度を維持しながらの迅速な核酸検出はできない。なぜならば、高速PCRにおいては標的核酸を増幅する工程として、標的核酸へのプライマーのアニーリングおよび標的核酸を鋳型とするプライマーの伸長(伸長反応)と、標的核酸およびプライマー伸長産物の解離(変性反応)が短時間のうちに繰り返し行われており、蛍光が消光する標識プローブは標的核酸へのハイブリダイゼーションによって初めて蛍光シグナルの減少が生じるため、標的核酸へのプライマーのアニーリングを起点とする核酸増幅と、標的核酸への標識プローブのハイブリダイゼーションに起因する蛍光シグナルの減少を、PCRの同一サイクル中に起こすことが困難だからである。つまり、高速PCR溶液中に単純に標的核酸とのハイブリダイゼーションによって蛍光の減少を生じる標識プローブを加えても、核酸増幅のためのプライマーのアニーリングと、蛍光シグナルの減少を生じさせるための標識プローブのハイブリダイゼーションとの間で、標的核酸に対する競合が起こり、標的核酸は増幅するが蛍光シグナルの減少が生じない、または標識プローブのハイブリダイゼーションで蛍光シグナルの減少は生じるが標的核酸が増幅しないことになり、どちらも標的核酸の存在または量、並びに標的核酸中の変異の存在を、特異性および高感度を維持しながら迅速に調べることはできない。すなわち、本発明では、様々な条件を検討し、高速PCRの迅速性が高い利点と、ハイブリダイゼーションによって蛍光の減少を生じる標識プローブの感度および正確性が高い利点を両立し、標的核酸の存在または量、並びに標的核酸中の変異の存在を、特異性および高感度を維持しながら迅速に調べる核酸検出方法を確立したものである。
【0022】
本発明の標的核酸とは生物(細菌も含む)の保持する核酸の塩基配列またはその相補的な塩基配列のいずれかを指す。
【0023】
本発明における高速PCR を以下に示す。PCR法を高速化する方法は例えば特許公開2005−328709に示されている。「変性」、「アニーリング」、「伸長」の各温度へ達する温度変化に要する時間の短縮、各工程の温度を保持する時間の短縮、サイクル数の減少などが挙げられている。特に重要となるのは温度変化に要する時間の短縮及び各工程の温度保持時間の短縮である。温度変化に要する時間の短縮は、具体的に温度の上昇及び下降を10℃/秒以上の速度で行うことを示す。10℃/秒以上の温度上昇・下降を達成する手段には特に制限がないが、一例としてロシュ・ダイアグノスティック社製のLight Cycler(登録商標)システムが挙げられる。このシステムでは温度上昇・下降に空気を使って温度制御する方式を採用し、温風と冷風で反応液の入ったガラスキャピラリーの温度を直接変化させている。それにより約20℃/秒の温度上昇・下降速度を達成している。
【0024】
また高速核酸増幅を達成するための別の手段として伸長工程に要する時間の短縮が考えられる。伸長工程時間を短縮する方策は、いくつか考えられる。最も有力な方策は、PCRによる増幅領域を極限まで少なくすることである。そうすれば当然、伸長に要する時間は短縮可能になる。このような場合でも、増幅領域は第一プライマー及び第二プライマーを含めて60塩基対程度が最低限度である。検出の目的で標識プローブを設定すれば、更にその部分を確保する必要があり、100塩基対程度の増幅が必要となる。増幅産物の長さは、高速増幅かつ標識プローブによる検出を行うために50bpから500bpの短い産物が好ましく、100bpから300bpが好ましい。
【0025】
さらにこの増幅産物の長さはDNAポリメラーゼの伸長速度により制限される。現在知られているDNAポリメラーゼ(単独あるいは組みあわせ)としてのTaqポリメラーゼや,EX−Taq,LA−Taq,Expandシリーズ,Plutinumシリーズ,Tbr,Tfl,Tru,Tth,T1i,Tac,Tne,Tma,Tih、Tfi(以上はPolI型酵素),Pfu,Pfutubo,Pyrobest(登録商標),Pwo,KOD,Bst,Sac,Sso,Poc,Pab,Mth,Pho,ES4,VENT,DEEPVENT(以上はα型酵素)などが挙げられる。天然型のポリメラーゼのアミノ酸配列を公知の手段により、1もしくは数個が欠失、置換若しくは付加させたもの(変異体)であっても良い。あるいは、上記の酵素(天然型、変異体)に化学修飾などの手段によりさらに改変を加えたものであっても良い。例えば、KOD DNAポリメラーゼは、最も一般的であるTaqポリメラーゼの倍にあたる100〜140塩基/秒以上のDNA合成速度を有する。また、プロセッシビティーなども優れていることが、伸長速度の速い要因のひとつであると考えられる。KOD DNAポリメラーゼは、東洋紡績製のもの(製品コードKOD−101など)を容易に入手することができる。
【0026】
近年、上記のDNAポリメラーゼをさらに変異、改変などの改良を加えて100塩基/秒以上のデオキシリボ核酸合成速度を達成させたもの、あるいは、組み合わせにより当該性能を達成させたもの(組合せにはKOD DNAポリメラーゼを含んでいても良い)も、本願発明のDNAポリメラーゼとして用いることができる。例えば、上記KOD DNAポリメラーゼ以外に100塩基/秒以上のデオキシリボ核酸合成速度を有するDNAポリメラーゼとして、KOD Dash DNAポリメラーゼ(東洋紡績製)、PrimeSTAR MAX DNAポリメラーゼ(TAKARA製)、Platinum Pfx DNAポリメラーゼ(インビトロジェン製)なども市販されている。
【0027】
DNAポリメラーゼはさらに、5’エキソヌクレアーゼ活性を持たないことが好ましい。本発明では標識プローブからの蛍光の減少量を標的核酸の検出に利用している。そのため、高速PCR工程中で第一のプライマーによる伸長産物に対して標識プローブ及び第二のプライマーがハイブリダイゼーションしている場合に、5’エキソヌクレアーゼ活性を持つDNAポリメラーゼ、例えばTaqポリメラーゼでは標識プローブを分解し、非特異シグナルの増加を伴う。本発明では標的核酸への標識プローブのハイブリダイゼーション時に起こる蛍光の減少量を検出に利用していることから、非特異シグナルの増加はバックグランドの上昇となり正確な定量もしくは検出ができなくなる。5’エキソヌクレアーゼ活性を持たないDNAポリメラーゼは特に限定はされないが、例えばKODポリメラーゼが本発明に適合する。
【0028】
二重鎖核酸構造は、標的核酸中の塩基とその相補的な核酸配列中の塩基の間で起こる塩基対生成によって形成される。塩基対生成はシトシンとグアニン、アデニンとチミンまたはアデニンとウラシルの間で起こる水素結合により生じる。標的核酸と標識プローブがハイブリダイゼーションすることにより二重鎖核酸構造が形成され、標識プローブの蛍光の減少が生じるようになる。
【0029】
本発明における「末端」とは標識プローブの塩基配列5‘側の最も端に存在する塩基、もしくは標識プローブの塩基配列で3’側の最も端に存在する塩基を指す。「末端部」とは5‘末端の塩基から3’末端に向かって5塩基以内または3‘末端の塩基から5’末端に向かって5塩基以内の塩基を示している。
【0030】
標識プローブに標識する蛍光物質は、核酸増幅工程中分解もしくは蛍光が減衰してなくならなければよく、ハイブリダイゼーション時に消光を生じる蛍光物質であればよい。特に標識プローブの末端においてグアニンとシトシンの塩基対形成時に蛍光の消光を生じる蛍光物質が好ましい。具体的には、フルオロセインまたはその誘導体(例えばフルオロセインイソチオシアネート(FITC))、BODIPY(商標) シリーズ、ローダミンまたはその誘導体(例えば5−カルボキシローダミン6G(CR6G)やテトラメチルローダミン(TAMRA))などを使用できるが、BODIPYシリーズやCR6Gの使用が特に好ましい。蛍光色素のオリゴヌクレオチドへの結合方法は、通常の方法に従って行うことができる。蛍光色素の消光を利用すれば、インターカレーターなどの他の二重鎖核酸構造への挿入色素を用いることなく、またFRET現象を起こす二種類のプローブを用いることなく、一種類の蛍光物質に標識されたプローブを用いて単純かつ特異的に標的核酸配列を検出することができる。標識プローブの塩基配列中の蛍光物質の標識位置は、特に限定されないが末端部に標識されていることが好ましく、末端に標識されていることがより好ましい。
【0031】
標識プローブの塩基配列とそれに対し相補的な標的核酸の塩基配列の選択はハイブリダイゼーション時に蛍光の消光を生じるために重要である。標識プローブの塩基配列は標的核酸とのハイブリダイゼーション時に消光を生じる塩基配列であればよく、二重鎖核酸構造形成時に、蛍光物質が修飾されている標識プローブ末端部にG(グアニン)とC(シトシン)の対が少なくとも1つ以上存在するように設計すればよい。標識プローブの末端が標識されている場合は、その末端塩基がGもしくはCであること、または標識プローブの末端と塩基対を形成する標的核酸の塩基から1もしくは3塩基離れてGが存在するように標識プローブの塩基配列が設計されていることがより好ましい。標識プローブの末端が標識されており、かつ末端塩基がCであるように設計することがさらに好ましい。
【0032】
標的核酸の塩基配列から、どうしても末端部がGまたはCに設計できない場合、第一プライマーまたは第二プライマーの塩基配列中にGまたはCを故意に導入することで標識プローブをハイブリダイゼーションさせ、かつ標識プローブの蛍光を消光することができる相補的な塩基配列を作り出すことが可能である。この場合、蛍光シグナルの減少が生じることと、第二プライマーの伸長による核酸の増幅とを、PCRの同一サイクル中に達成するためにも第一プライマーにGまたはCを導入することが好ましい。また標識プローブの第一プライマーに対する非特異のハイブリダイゼーションを避けるために故意のGまたはCの導入位置は第一プライマーの3‘末端から末端塩基を含めて5’末端側へ4塩基以上8塩基以下離れて設計することが好ましい。3塩基以下の位置に設計するとプライマーが伸長できなくなり、9塩基以上離れて設計すると増幅によらない標識プローブとプライマーとの非特異ハイブリダイゼーションが起こりやすくなる。
【0033】
本発明を達成する手段として各プライマー及び標識プローブのTm値が規定される。Tm値とはプライマーが相補塩基とアニーリングし、50%が解離する時の温度を表す。Tm値は計算により算出することも可能である。Tm値の計算方法は、ハイブリダイゼーションにおける塩濃度等をパラメーターにして各種の計算方法が考案されている。一般的に良く用いられる計算方法としては、最近接塩基対法(Schildkraut C., Lifson S. (1965) Dependence of the melting temperature of DNA on salt concentration − Biopolymers 3, 195−208 、Breslauer K.J., Frank R., Blocker H., Markey L.A. (1986) Predicting DNA duplex stability from the base sequence − Proc. Natl. Acad. Sci. USA 83, 3746−3750)やWallance法(Wallace, R.B.; Shaffer, J.; Murphy R.F.; Bonner, J.; Hirose, T.; Itakura, K.; (1979) Nucelic Acid Res. 6, 3543)、GC%法(Dependence of the Melting Temperature of DNA on Salt Concentration、Schildkraut C.,Lifson S. (1965) BIOPOLYMERS 3.195−208、Optimization of the annealing temperature for DNA amplification in vitro、W.Rychlik、 Nucl.Acids Res.(1990) 18(21)6409−6412)などを用いることができ、標的核酸の配列または検出の条件などによっては計算方法によるTm値の差異が影響するが、当業者にとって容易に想到し得る検討により実験的に当該Tm値の差異を修正し、本発明の効果を有するプライマーおよび標識プローブを設定することができる。
【0034】
第一プライマーおよび第二プライマーのTm値と標識プローブのTm値の関係が、本発明に記載の効果を得るために重要な要素の一つとなる。そこで、発明をより具体的に説明するための一例として、本発明では、Breslauer K.J., Frank R., Blocker H., Markey L.A. (1986) Predicting DNA duplex stability from the base sequence − Proc. Natl. Acad. Sci. USA 83, 3746−3750に記載の方法にて計算したTm値を用いる。
【0035】
具体的には上記計算方法によれば、配列番号3より選ばれる24塩基の連続した塩基配列(5’−GAGATGGGAGCTGTCTCAACCAGA−3’:配列番号5)のTm値は70.52、配列番号4より選ばれる25塩基の連続した塩基配列(5’−TGCGCATGATATTCCCATTAGCAGT−3’:配列番号10)のTm値は73.39となる。
【0036】
プライマーのTm値は70℃以上が好ましい。より好ましくは70℃以上90℃以下である。本プライマーの使用で65℃以上の温度で安定したアニーリングが可能となる。91℃以上のプライマーを使用する場合、非特異的なハイブリダイゼーションが起こりやすくなり結果的に目的とする増幅産物の増幅効率を下げてしまう。
【0037】
本発明では標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とのバランスが重要となる。このバランスは第二プライマー及び標識プローブのTm値、さらに第二プライマー及び標識プローブの濃度によって最適化される。上記バランスを最適化するにはTm値は標識プローブよりも第二プライマーが大きくなるように設計する必要がある。本検出方法は標識プローブの発する蛍光がハイブリダイゼーションによって減少することを利用しているため、標識プローブ濃度が濃い場合はバックグランドの蛍光シグナルが高くなる。標識プローブ濃度はハイブリダイゼーション時の蛍光の減少した蛍光シグナルに対してバックグランドの蛍光シグナルが少なくなる濃度が好ましい。具体的には第二プライマーの10倍以下であることが好ましい。より好ましくは5倍以下である。10倍を超える場合はバックグランド蛍光シグナルの影響により、蛍光が減少したときの蛍光強度の差を検出できなくなる。標識プローブ濃度の下限は特に限定されないが標識プローブと標的核酸または第一プライマーの伸長産物とのハイブリダイゼーション効率を極端に下げない濃度がよい。上記の蛍光シグナルの減少と核酸の増幅のバランスをとるためには標識プローブの濃度は第二プライマーの濃度よりも高いほうが好ましい。標識プローブの濃度は具体的には10nM以上が好ましい。10nM未満の場合は蛍光シグナルを感度良く検出することが難しい。
【0038】
上記Tm値及び濃度の関係を満たすことにより、従来法では達成できなかった、標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、高速PCRの同一サイクル中に達成することが可能になる。
【0039】
本発明の重要な開示について、図1を用いて、より詳しく説明する。従来の方法では第二プライマーのアニーリングおよび伸長により標的核酸は増幅するが、標識プローブが標的核酸または第一プライマーの伸長産物にハイブリダイゼーションできないために蛍光シグナルの減少が生じない場合(図1(1)−A及び図1(2)−A)、または標識プローブの標的核酸または第一プライマーの伸長産物へのハイブリダイゼーションにより蛍光シグナルの減少が生じるが、標識プローブのハイブリダイゼーションがプライマーの伸長を阻害し、標的核酸が増幅しない場合(図1(1)−B及び図1(2)−B)のどちらか一方が起こる割合が大きく、どちらの場合も標的核酸の存在または量、並びに標的核酸中の変異の存在を、迅速かつ高感度に調べることはできなかった。一方、本発明の方法によれば、第一プライマー及び第二プライマーのTm値と標識プローブのTm値の関係、さらにプライマーの濃度と標識プローブの濃度の関係を上記のように規定することで高速PCR工程における変性温度からアニーリング・伸長温度までの温度下降中に標識プローブが標的核酸または第一プライマーの伸長産物にハイブリダイゼーションしつつ、その二重鎖核酸構造を形成する領域まで第二プライマーが伸長する。続いて、アニーリング・伸長温度から次サイクルとなる変性温度までの温度上昇中には標識プローブは標的核酸または第一プライマーの伸長産物から解離することにより、二重鎖核酸構造を形成した領域まで伸長した第二プライマーのさらなる伸長を阻害しないため、第一プライマーの鋳型となるような第二プライマーの伸長産物が形成される(図1(1)−C及び図1(2)−C)。このように、標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、高速PCRの同一サイクル中に達成することで、高速PCRの迅速性が高い利点と、標識プローブの感度および正確性が高い利点を両立し、標的核酸の存在または量、並びに標的核酸中の変異の存在を、迅速かつ高感度に調べる核酸検出方法を確立することができた。
【0040】
さらには、前記に記載の伸長速度の速いDNAポリメラーゼを使用することにより第二プライマーの伸長が第一プライマーと相補的な塩基配列まで到達することが可能となり、より効率的に第一プライマーの鋳型となるような第二プライマーの伸長産物が形成され得る。
【0041】
また、本発明の別の重要な開示として、上記のように蛍光シグナルの減少と核酸の増幅とを効率よく行うためには、第一プライマーの伸長産物において標識プローブが二重鎖核酸構造を形成する位置も重要である。前述の通り、本発明の方法では、標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、PCRの同一サイクル中に達成することが重要であり、さらには高速PCR工程において変性温度からアニーリング・伸長温度までの温度下降中に、標識プローブの標的核酸または第一プライマーの伸長産物へのハイブリダイゼーションより二重鎖核酸構造を形成する領域まで第二プライマーが伸長し、続いて、アニーリング・伸長温度から次サイクルとなる変性温度までの温度上昇中に、当該二重鎖核酸構造を形成した領域まで伸長した第二プライマーがさらなる伸長を行うことにより、第一プライマーの鋳型となるような第二プライマーの伸長産物が形成されることが重要である。すなわち、第一プライマーの伸長産物の5’末端から標識プローブが二重鎖核酸構造を形成する領域までの距離を(A)、該標識プローブが二重鎖核酸構造を形成する領域から第一プライマーの伸長産物の3’末端までの距離を(B)とした場合(図2)に、(A)と(B)のバランスが重要である。より詳細には(A)は第一プライマーの伸長産物の5’末端から二重鎖核酸構造を形成している標識プローブの3‘末端まで、(B)は二重鎖核酸構造を形成している標識プローブの5‘末端から第一プライマーの伸長産物の3’末端までの距離である。特に(A)が(B)より大きすぎると、第二プライマーの伸長が第一プライマーまで届かず、第一プライマーの鋳型となるような第二プライマーの伸長産物を形成することができないため、標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、PCRの同一サイクル中に達成することができない。蛍光シグナルの減少と核酸の増幅とを効率よく行うための(A)と(B)のバランスとして、具体的には、(A/B)の値が0.1以上3.0以下であることが好ましく、0.3以上3.0以下かつ第一プライマーと第二プライマーの伸長産物が80bp〜300bpであることがより好ましい。さらに好ましくは(A/B)の値が0.4以上2.0以下かつ第一プライマーと第二プライマーの伸長産物が100bp〜300bpである。
【0042】
核酸増幅工程で使用する第一プライマーと第二プライマーの濃度に差をつけることによって標的核酸の特定領域の一本鎖核酸を優先的に増幅する方法も、本発明に適用できる。例えば図3に示すような第一プライマーと第二プライマー、標識プローブの設計において、第一プライマーの濃度を第二プライマーの濃度より高く設定することにより、第一プライマーによる伸長産物を優先的に生成させることもできる。第二プライマーによる伸長産物および標識プローブは第一プライマーの伸長産物に対して競合的に作用するため、このように標的核酸を含む一本鎖核酸を意図的に増幅させることにより、蛍光検出工程において標識プローブのハイブリダイゼーション効率を上げ、より多くの蛍光シグナルを発生させることができる。第二プライマーに対する第一プライマーの濃度比は、特に限定されないが好ましくは4倍以上、より好ましくは5倍以上、さらに好ましくは10倍以上がよい。また、50倍以下がよく、より好ましくは30倍以下がよい。好ましくは4倍以上50倍以下であり、より好ましくは10倍以上30倍以下である。本発明の第二プライマーの濃度は特に限定されないが、25nM〜500nMが好ましい。より好ましくは50nM〜250nMである。
【0043】
蛍光検出は例えば標識プローブと、標的核酸または第一プライマーの伸長産物をハイブリダイゼーションさせ二重鎖核酸構造を形成した後、温度上昇により標識プローブと標的核酸が解離することで生じる蛍光の増加量を検出してもよい。温度上昇速度は好ましくは0.1℃/秒以上であり、0.1℃/秒以上5℃/秒以下であり、より好ましくは0.5℃/秒以上2℃/秒以下である。5℃/秒を超える場合は核酸プローブと標的核酸の解離温度を正確に捉えることができない。
【0044】
非相補的塩基の定義は標識プローブがハイブリダイゼーションする時に標的核酸中の塩基と塩基対を形成しない標識プローブ中の塩基のことである。野生型の塩基配列と一致する標識プローブと野生型の標識核酸がハイブリダイゼーションする場合は完全に一致するため非相補的塩基はない。しかし、野生型の塩基配列と一致する標識プローブと野生型に対して一塩基変異をもつ標的核酸をハイブリダイゼーションする場合には、標識プローブに非相補的塩基がある。本発明の変異は野生型に対して異なる塩基を有することであり、標的核酸中での事象である。標識プローブ塩基配列中の標的部位との非相補的塩基の存在は、前記の標的核酸または第一プライマーの伸長産物への標識プローブのハイブリダイゼーションによる蛍光シグナルの減少と、標的核酸または第一プライマーの伸長産物を鋳型とする第二プライマーの伸長による核酸の増幅とを、PCRの同一サイクル中に達成するために有利に働く。その理由としては、非相補的塩基を含んだ標識プローブは温度上昇時に解離しやすくなるため、第二プライマーの伸長がより円滑に達成されることが挙げられる。この場合、4個以上の非相補的塩基が一つの標識プローブ中に存在すると、前記の標識プローブ14塩基〜30塩基の範囲内ではハイブリダイゼーションが起こりにくくなる。非相補的塩基の数は好ましくは3個以下である。標識プローブ中の非相補的塩基の導入位置は特に限定されないが、標的核酸において検出対象となる変異塩基を除く位置に導入することが好ましく、標識プローブの末端から末端を含めて2塩基以上離れていることが好ましい。また、標識プローブ中に塩基対結合力の比較的強いグアニンやシトシンが3塩基以上連続して存在する場合に、グアニンやシトシンの連続した領域に導入することが好ましい。
【0045】
標識プローブの塩基配列と変異における位置関係は、例えば核酸プローブと標的核酸または第一プライマーの伸長産物をハイブリダイゼーションさせ、温度上昇により核酸プローブと標的核酸が解離することで生じる蛍光の減少を検出する方法を用いる場合に、完全一致配列と1塩基変異が存在する配列の解離温度の違いがわかれば特に限定されない。好ましくは当該標識プローブの塩基配列の両末端から3塩基以上離れて存在すればよく、より好ましくは両末端から5塩基以上離れて存在すればよい。特に好ましくは標識プローブの中央部に変異の部位が位置すればよい。中央部は当該オリゴヌクレオチド配列の中心から両末端に向かって3塩基以内のことをいう。当該オリゴヌクレオチド配列が偶数個の場合の中心とは当該オリゴヌクレオチド配列数を2で割った数を、両末端を含んで数える数え方で両末端塩基より内側に向かって数えた塩基をいい、当該オリゴヌクレオチド配列が奇数個の場合の中心とは当該オリゴヌクレオチド配列数に1を加えて2で割った数を、両末端を含んで数える数え方で両末端塩基より内側に向かって数えた塩基をいう。
【0046】
3’末端を蛍光色素で標識するか、3’末端にリン酸化などの処置を加えるなどして高速PCR中に伸長して標識プローブのTm値が上昇しないような工夫をすることが好ましい。ただし、蛍光検出方法として、例えば標識プローブと標的核酸または第一プライマーの伸長産物をハイブリダイゼーションさせ、温度上昇により標識プローブと標的核酸が解離することで生じる蛍光の増加を検出する方法を用いる場合は、高速PCR中に標識プローブが伸長すると標識プローブと標的核酸の解離温度を正確に測定できなくなるため、前記のような工夫が特に必要である。
【0047】
また、本発明は下記も含む。
標的核酸の存在または量を確認したい試料と少なくとも下記(a)、(b)の存在下、連続的または間欠的に標識プローブに標識されている蛍光物質の励起波長の光を与えかつ標識プローブからの蛍光を測定しながら、高速PCRを行い、該標的核酸の存在または量を調べ、引き続いて、密封状態のまま、標識プローブに標識されている蛍光物質の励起波長の光を与えかつ標識プローブからの蛍光を測定しながら、0.1℃/秒以上の連続的な温度上昇又は下降を伴う融解曲線解析を行い、標的核酸中の変異の存在を調べることを特徴とする検出方法。この場合において、
(a)次の(i)〜(iv)の性質を有する標識プローブ
(i) 標的核酸に相補的な配列と二重鎖核酸構造を形成でき、かつ14塩基以上30塩基以下の長さのオリゴヌクレオチドを有する
(ii) 該オリゴヌクレオチドの末端部が該二重鎖核酸構造形成時に消光する性質を有する蛍光物質で修飾されている
(iii) 該二重鎖核酸構造形成時に、蛍光物質が修飾されている該末端部のヌクレオチドから3塩基以内にG(グアニン)とC(シトシン)の対が少なくとも1つ以上存在する
(iv)該標識プローブの濃度が該第二プライマーの濃度に対して10倍以下である
(b)次の(i)〜(iii)の性質を有する第一プライマーおよび第二プライマー
(i) 第一プライマーの伸長産物が標的核酸と相補的な配列を有しかつ第二プライマーの鋳型となる性質を有し、該第二プライマーの伸長産物が標的核酸と相同な配列を有しかつ該第一プライマーの鋳型となる性質を有する
(ii) 該第一プライマーおよび該第二プライマーによる増幅産物の長さが50bp以上500bp以下である
(iii) 該第一プライマー及び該第二プライマーのTm値が70℃以上90℃以下であり、かつ該第二プライマーのTm値は前記標識プローブのTm値より大きい
を示すものである。
【0048】
標的核酸の例として、感染症の原因となる細菌の保持している特定の核酸が考えられる。例えばピロリ菌(Helicobacter pylori)の23SrRNA遺伝子の検出が挙げられる。ピロリ菌は、胃・十二指腸潰瘍の起因菌とされ、近年胃癌との関連性も指摘されてきている。潰瘍の疑いがある場合、呼気試験、血中または便中ピロリ菌抗体検査等、を行った結果、ピロリ菌感染の可能性がある場合、さらに内視鏡検査において胃生検を行い、ピロリ菌の存在を迅速ウレアーゼ試験あるいは培養法によって確かめられる。内視鏡検査においてピロリ菌の存在が確認された場合、除菌療法等の処置が行なわれる。ピロリ菌の除菌療法としては、プロトンポンプ阻害剤とアモキシシリンとクラリスロマイシンの3剤併用療法が有効とされ、本邦においては、西暦2000年より保険診療として広く実施されている。本発明によりピロリ菌を検出するために、第一プライマー及び第二プライマー、標識プローブをピロリ菌の23SrRNA遺伝子配列をターゲットとして設計した。第一プライマー及び第二プライマーにより23SrRNA遺伝子配列の特定領域を増幅する工程において、標識プローブを用い、「伸長」工程または「アニーリング・伸長」工程での蛍光シグナルの検出を行うことで、より高感度なピロリ菌検出が可能となる。
【0049】
また、近年クラリスロマイシン耐性ピロリ菌(以下CAM耐性菌と称する)の出現が報告され、ピロリ菌(Helicobacter pylori)のCAM耐性遺伝子の検出が注目を集めている。CAM耐性ピロリ菌はピロリ菌除菌成功率の低下の大きな要因の一つとして挙げられるようになってきている。CAMは、ピロリ菌の23SrRNAのペプチジルトランスフェラーゼループに結合し、そのタンパク質合成を阻害することによって作用を発揮する。しかしながら、23SrRNA遺伝子の点突然変異によってピロリ菌はCAM耐性能を取得することが明らかにされている(James versalovic et. al. Antimicrobial Agents and Chemotherapy p477−480 1996)。この点について、CAM耐性菌の93%は、前記の領域にある2142位あるいは2143位のアデニンがグアニンに変異しており(A2142GまたはA2143G)、CAM耐性菌の7%は、2142位がシトシンに変異している(A2142C)ことが報告されている(Stone,G.G. et. al. Antimicrobial Agents & Chemotherapy. 41(3):712−4, 1997)。
【0050】
ピロリ菌のCAM耐性を判定するには、例えば以下記述の方法が考えられる。野生型ピロリ菌とCAM耐性ピロリ菌に共通の配列を用いて設計した第一及び第二プライマーによりピロリ菌の23SrRNA遺伝子の特定領域を増幅する。この工程において増幅される産物(野生型のものとCAM耐性の一塩基変異が存在する増幅産物の二種類)を標的核酸とした野生型ピロリ菌の23SrRNA遺伝子と完全に一致する標識プローブまたは前記二種類のCAM耐性ピロリ菌の23SrRNA遺伝子と完全に一致する標識プローブを同時に使用する。PCR工程中の「伸長」工程または「アニーリング・伸長」工程での蛍光シグナルの検出を行う。この検出では野生型ピロリ菌の23SrRNA遺伝子と完全に一致する標識プローブを用いた場合は、野生型ピロリ菌の特異的な検出が可能であり、前記二種類のCAM耐性ピロリ菌の23SrRNA遺伝子と完全に一致する標識プローブを用いた場合はCAM耐性ピロリ菌の特異的な検出が可能である。さらに引き続き融解曲線解析を行うことにより、一つの標識プローブで野生型ピロリ菌とCAM耐性ピロリ菌の検出が同時に行えることも本発明の範疇に含まれる。CAM耐性ピロリ菌に関して、一塩基変異は前記のとおり2142位あるいは2143位のアデニンがグアニンに変異(A2142GまたはA2143G)、または2142位がシトシンに変異している(A2142C)ピロリ菌が大半を占めているが、この限りではない。ピロリ菌における標的核酸とは野生型の保有する核酸を含み、一塩基変異が存在しているCAM耐性菌の保有する核酸を含んでいる。この場合の非相補的塩基の数は、野生型に完全一致の塩基配列を持つ標識プローブではCAM耐性ピロリ菌に対して1塩基である。つまり3塩基以下の非相補的塩基とは標識プローブが同じで標的核酸が異なる場合に標的核酸中の1塩基変異を含んで3塩基以下ということを示している。
【0051】
すなわち、本発明を利用し、下記の検出方法を実施しても良い。
連続的または間欠的に標識プローブに標識されている蛍光物質の励起波長の光を与えかつ(a)の標識プローブからの蛍光を測定しながら、(b)の第一プライマーと第二プライマーを用いて高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べることを特徴とする検出方法。
【0052】
さらに、本発明を利用し、下記の検出方法を実施しても良い。
連続的または間欠的に標識プローブに標識されている蛍光物質の励起波長の光を与えかつ(a)の標識プローブからの蛍光を測定しながら、(b)の第一プライマーと第二プライマーを用いて高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べ、引き続いて、密封状態のまま、標識プローブに標識されている蛍光物質の励起波長の光を与えかつ標識プローブからの蛍光を測定しながら、0.1℃/秒以上の連続的な温度上昇又は下降を伴う融解曲線解析を行い、ピロリ菌の23S rRNA遺伝子の変異の存在を調べることを特徴とする検出方法。
【0053】
さらに、本発明を利用し、下記の検出方法を実施しても良い。
連続的または間欠的に標識プローブに標識されている蛍光物質の励起波長の光を与えかつ(a)の標識プローブからの蛍光を測定しながら、(b)の第一プライマーと第二プライマーを用いて高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べ、引き続いて、密封状態のまま、標識プローブに標識されている蛍光物質の励起波長の光を与えかつ(a)の標識プローブからの蛍光を測定しながら、0.1℃/秒以上の連続的な温度上昇又は下降を伴う融解曲線解析を行い、23S rRNA遺伝子の第2142位または第2143位における変異の存在を調べピロリ菌の抗生物質耐性を判別することを特徴とする検出方法。
【0054】
上記のピロリ菌の23S rRNA遺伝子の存在または量を調べる方法、ピロリ菌の23S rRNA遺伝子の変異の存在を調べる方法、23S rRNA遺伝子の第2142位または第2143位における変異の存在を調べピロリ菌の抗生物質耐性を判別する方法においては、標的核酸がピロリ菌の23S rRNA遺伝子のセンス鎖の場合は、
(a)の標識プローブのオリゴヌクレオチドの配列が、配列番号2より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
標的核酸がピロリ菌の23S rRNA遺伝子のアンチセンス鎖の場合は、
(a)の標識プローブのオリゴヌクレオチドの配列が配列番号1より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列
であればよい。
【0055】
また、限定されるわけではないが本発明は抗酸菌の検出にも利用できる。抗酸菌の同定には培養法が用いられているが、抗酸菌は増殖速度が遅い。小川培地などによる培養では同定に約1ヶ月を要することもある。近年抗酸菌の検査に遺伝子検査が導入され、短時間化されたといっても1日から2日必要となる。本発明の方法を適用すれば30分以内に検査を完了することができ、極めて短時間で抗酸菌を検出することが可能となる。
【0056】
その他にも、より迅速に検出を行う必要がある感染症項目にも適用できる。例えば性感染症のクラミジアや淋菌、C型肝炎ウイルス、B型肝炎ウイルス、多剤耐性黄色ブドウ球菌、多剤耐性緑膿菌、敗血症菌などの検出に利用できる。さらに、ヒトの遺伝子検査にも適用できる。例えば癌遺伝子検査では30分以内に検出ができることから術中診断も可能になる。
【0057】
1つの容器中とは検体を含む溶液がプラスチック、あるいはガラスなどから成る容器中に充填され、増幅から検出に至るまでに別の容器に移し替えるもしくは取り出す操作がないことを示す。密封状態は外部からの不純物の進入がなく、内部からの溶液の蒸発がないことを示す。不純物などのコンタミネーションの防止、ならびに溶液組成の変化による結果の相違を防ぐことは重要である。
【実施例】
【0058】
以下に実施例を示して本発明を具体的に説明するが、本発明は実施例に限定されるものではない。
【0059】
以下にピロリ菌の23S rRNA遺伝子上のクラリスロマシン耐性を判定する方法を示す。
【0060】
(実施例1)
(検体の採取)
胃生検材料より遺伝子をQIAamp(登録商標) DNA micro Kit(Qiagen社製)を用いて抽出した。抽出されたDNAは−20℃で保存した。
(テンプレートプラスミドの調製)
1μlの抽出されたDNA溶液を鋳型として用い、PCRを実施した。ここで使用したプライマーペアは、ピロリ菌23SrRNA遺伝子を全長増幅できるプライマーを使用した。いずれもピロリ菌の23SrRNA遺伝子 (GenBank accession number U27270)の公知の配列から設計した。
【0061】
PCRは、サーマルサイクラーとしてGeneAmp(登録商標)9700(Applied Biosystems社製.)を使用し、全量を26μlとして実施した。1μlのDNA溶液と試薬混合物(KOD−plus 0.5U、10XPCR緩衝液2.5μl、200μMのdNTP、各々5μMのプライマー)を混合した。標的DNAの初期変性を94℃で2分間行った後、94℃・15秒の変性工程、60℃・30秒のアニーリング工程、68℃・60秒の伸長工程のサイクルを35回繰り返した。なお、その他のPCR条件は常法通りである(例えば、遺伝子 操作技術マニュアル、医学書院、1995年発行を参照)。得られた核酸断片を制限酵素によって平滑末端切断されたpUC18へライゲーションし、コンピテントセルJM109を用いて形質転換体の取得を行なった。目的核酸断片が挿入された形質転換体を液体培養しプラスミドを抽出した。
【0062】
得られたプラスミドの核酸配列をオートシークエンサーにより解析し、野生型の塩基配列を有するプラスミド、ピロリ菌の23S rRNA遺伝子の第2142位または第2143位のアデニンがグアニンに変異した塩基配列を持つプラスミドの計3種類を得た。以下、野生型はWT、23S rRNA遺伝子の2142位変異型は2142G、同じく23S rRNA遺伝子の2143位変異型は2143Gと示す。
【0063】
(実施例2)
(プライマー及び標識プローブの合成)
検出に使用するプライマー及び標識プローブは核酸合成受託会社((株)日本バイオサービス、(株)サワディー、GENSET KK、シグマアルドリッチジャパン(株)等)に依頼した。
【0064】
使用した標識プローブ及びプライマーの塩基配列を表1に示す。標識プローブは配列番号1より選ばれる12塩基から22塩基の塩基配列を使用した。またプライマーについてもプライマーF1〜F4はそれぞれ配列番号3より選ばれる24塩基、29塩基、29塩基、22塩基からなる塩基配列である。プライマーR1〜R4はそれぞれ配列番号4より選ばれる25塩基、31塩基、24塩基、26塩基からなる塩基配列である。プライマーF5はピロリ菌23SrRNA遺伝子の塩基配列中、配列番号1で示される塩基配列までの距離が配列番号3で示される塩基配列より長くなる領域に設計した24塩基からなる塩基配列である。プライマーR5はピロリ菌23SrRNA遺伝子の塩基配列中、配列番号2で示される塩基配列までの距離が配列番号4で示される塩基配列より長くなる領域に設計した22塩基からなる塩基配列である。
【0065】
【表1】
【0066】
(実施例3)
(野生型ピロリ菌の検出)
WT、2142G、2143Gの各プラスミドDNAは表2に示す反応液にテンプレートとして混合した。以下、表2の反応組成を基準とし、反応液を構成するいずれかの溶液の最終濃度を変更した場合はミリQ水で調節し全量20μLを維持した。また、表2の組成は一例であり反応組成の最適化は同業者であれば容易に行えるものである。プラスミド量は104コピー及び102コピーを使用した。プライマーは表1に記載のプライマーF4とプライマーR4を使用した。標識プローブは表1記載の標識プローブAを使用した。標識プローブAは野生型に対して完全に一致する塩基配列の標識プローブに故意の非相補的塩基1塩基を導入したものである。例えば標識プローブAにおいて、故意の非相補的塩基とは、変異部位に相当する標識プローブ中の非相補的塩基、すなわち2142位および2143位を除いた標識プローブの塩基配列に非相補的塩基を導入することをいう。標識プローブAはCAM耐性ピロリ菌(変異型)2142G、2143Gに対して、いずれも非相補的塩基2塩基が存在している標識プローブである。PCR増幅時の反応条件は表3で示される(核酸増幅サイクル)を用いた。増幅及び蛍光の測定にはロシュ・ダイアグノスティック社製ライトサイクラー(登録商標)を使用した。測定モードは530nmを利用した。以下特に記載がない場合はすべてライトサイクラー(登録商標)での測定とする。図4〜6は、グラフの横軸をサイクル数n、縦軸を蛍光シグナル値として解析結果を表した図である。ライトサイクラーソフトウェアの解析により、バックグランドとして現れる標識プローブAの発する蛍光シグナル値を0として計算されているため、縦軸の数値は小さくなるほど標識プローブに対する標的核酸が多いことを示している。
【0067】
【表2】
【0068】
【表3】
【0069】
解析の結果、標識プローブAのプラスミド量104コピー及び102コピーではそれぞれ図4、図5に示す結果が得られた。なお、図4,図5中、2143Gはピロリ菌23S rRNA遺伝子の2143位変異型、2142Gはピロリ菌23S rRNA遺伝子の2142位変異型、NCはテンプレートにミリQ水を使用したネガティブコントロール、WTはピロリ菌23S rRNA遺伝子の野生型を示し、図6〜9においても同様である。野生型のピロリ菌23S rRNA遺伝子を導入したプラスミドをテンプレートにした場合、104コピーでは30サイクルを越えた付近から、102コピーでは38サイクルを越えた付近から標識プローブが標的核酸の増幅産物にハイブリダイゼーションした時の蛍光シグナル値の減少を確認した。一塩基変異が存在している変異型のプラスミドを用いた場合は標識プローブAに2個の非相補的塩基が存在し、増幅反応中の標的核酸に対する標識プローブAのハイブリダイゼーション能力が低下するため、蛍光シグナル値の減少は見られなかった。このように標識プローブ中の非相補的塩基の数を調節することによりWTを特異的に検出することができた。標識プローブへの故意の非相補的塩基の導入は標識プローブの標的核酸に対するハイブリダイゼーション能力及び解離のしやすさを調整するための手法として有効である。プラスミド量102コピーでも標識プローブ特異的かつ短時間での検出が可能であることがわかった。
【0070】
(実施例4)
(ピロリ菌CAM耐性遺伝子変異の検出)
表1に記載の標識プローブBを用いてCAM耐性ピロリ菌(変異型)の検出を行った。標識プローブBは2142G変異型に対して完全一致する塩基配列中に故意の非相補的塩基1塩基を導入したものである。野生型に対しては変異部位に存在する非相補的塩基1塩基と故意の非相補的塩基1塩基で合計非相補的塩基が2塩基存在していることになる。2143G変異型に対しては非相補的塩基3塩基が存在する標識プローブである。標識プローブ以外は実施例3の条件を用いた。
【0071】
標識プローブBでは図4、図5とは異なり2142G変異型のプラスミドをテンプレートとした場合にのみ蛍光シグナル値の減少が確認できた。NC、WT、2143Gのテンプレートを用いた時は蛍光シグナル値の減少が生じなかった。本発明によれば標的核酸の塩基配列特異的に蛍光シグナルの減少が生じることで、標的核酸を検出できることがわかる。
【0072】
(実施例5)
プライマーF1〜F5とプライマーR1〜R5で組み合わせを変えた検討を行った。プライマー以外の条件は実施例3に従った。テンプレートはWTの104コピープラスミドを使用した。その結果を表4に示す。表4の数値は、ライトサイクラーリアルタイムPCRにおける測定時、標識プローブAに標識されたBODIPY−FL(商標)の発する蛍光シグナル値がある一定値(−0.1)を下回った時のサイクル数(Ct値)を示している。蛍光シグナルの減少が見られない組み合わせは数値の記載の代わりに(−)を示した。(A/B)の数値は(標的核酸におけるプライマーRの5‘末端相同的な塩基から標識プローブの3’末端と相補的な塩基までの距離/標的核酸におけるプライマーFの5‘末端と相補的な塩基から標識プローブの5’末端と相補的な塩基までの距離)を示している。
なお、使用した条件は、標識プローブが14塩基以上30塩基以下、標識プローブの濃度が第二プライマーの濃度に対して10倍以下、第一プライマー及び第二プライマーのTm値が70℃以上90℃以下でありかつ第二プライマーのTm値は標識プローブのTm値より大きいと言う全ての条件を満たしている。
【0073】
表4より、(A/B)の数値で0.5〜1.5かつ増幅産物の大きさが300bpまでのプライマーの組み合わせである場合、プライマーF4及びR4の組み合わせと同等の結果を得ることが可能であった。Aの値が大きなR5を使用した場合は、各プライマーFによる伸長反応がプライマーR5まで完了せず増幅がみられなかった。またBが極端に短い場合(A/Bの数値で0.3以下)でも蛍光シグナル値が得られなかった。この結果より増幅産物長が300bp以内で、A/Bの数値が1に近いほうが有利であることがわかる。
また、表4の結果は全て高速PCRの条件で行っているが、例えばプライマーF5とプライマーR1の組み合わせでは検出に至っていない。プライマーF5及びプライマーR1を用いた反応液をアガロースゲル電気泳動により確認したところ、泳動バンドは確認できなかった。従来法の高速PCR条件を用いるだけでは標的核酸の迅速な検出はできないことがわかる。
【0074】
【表4】
【0075】
蛍光シグナルが(−0.1)を下回った時のサイクル数は増幅検出効率の一つの指標となる。表4で該サイクル数を34以下、34を超え35.5以下、35.5を超え38以下、38を超え42以下に分けると、増幅検出効率は34以下>34を超え35.5以下>35.5を超え38以下>38を超え42以下となる。34以下の結果を示したのは(A/B)の数値で0.50〜1.5かつ増幅産物の大きさが100〜300bpに含まれる条件であった。34以下ではプライマーとプローブの位置及び増幅産物長のバランスが最も良好であった。34を超え35.5以下の結果を示したのは(A/B)の数値で0.40〜2.0かつ増幅産物の大きさが100〜300bpに含まれる条件であった。次に35.5を超え38以下の結果を示したのは(A/B)の数値で0.30〜3.0かつ増幅産物の大きさが100〜300bpに含まれる条件であった。38を超え42以下の結果は実施例5の条件において検出限界付近となり、(A/B)の数値で0.30〜3.0かつ増幅産物の大きさが〜400bpに含まれる条件であった。
結果を例示しないが、他の遺伝子配列をターゲットとした時に、(A/B)の数値で0.10〜3.0かつ増幅産物の大きさが50bp〜500bpに含まれる条件であれば、本発明におけるプライマー・プローブの位置及び増幅産物長以外の条件を最適化すると迅速な検出は可能である。本発明に開示されている情報を用いれば条件の最適化は困難ではない。
【0076】
(実施例6)
(第一プライマー濃度及び第二プライマー濃度比の検討)
第二プライマーに対する第一プライマーの濃度比が1倍と10倍の検討を行った。1倍ではプライマーF4 500nM、プライマーR4 500nMを使用した。また、10倍ではプライマーF4 150nM、プライマーR4 1500nMを使用した。プライマー濃度以外の条件は実施例3と同じ条件を使用した。テンプレートはWT、2142G、2143Gの各プラスミド(プラスミド量102コピー)を使用した。濃度比1倍の結果を図6(比較例)、10倍の結果を図5に示す。
【0077】
濃度比が10倍の時は標識プローブに対しての一本鎖標的核酸がより多く増幅することにより、プライマーの伸長及び標識プローブのハイブリダイゼーションの関係が良好に維持されていた。そのため完全一致テンプレートの時のみ特異的に蛍光シグナルの減少が生じた。濃度比1倍の時はテンプレートと塩基配列が完全一致する標識プローブを用いた場合にも蛍光シグナルの減少による検出はできなかった。非対称PCRを行うと標的核酸がより多く増幅するため、対称PCRより特異的検出を感度良く行えることがわかる。
【0078】
(実施例7)
(高速増幅用DNAポリメラーゼの選択)
核酸増幅工程の高速化に適用し得るDNAポリメラーゼを検討した。DNAポリメラーゼとしてはTaqポリメラーゼ(Thermus aquaticus由来)とKODポリメラーゼ(Thermococcus kodakaraensis KOD1由来)の2種類を検討した。Taqポリメラーゼは東洋紡績社製Blend Taq−Plus−(登録商標)、KODポリメラーゼは東洋紡績社製KOD−Plus− ver.2(登録商標)を使用した。Taqポリメラーゼに関しては表1に示す標識プローブA、プライマーF4,R4を使用し、その他の試薬組成はプロトコール記載の試薬組成を参考にした。反応条件は表3に示す(核酸増幅サイクル)を利用した。KODポリメラーゼに関してはTaqポリメラーゼと同じく標識プローブA、プライマーF4,R4を使用し、基本反応条件は実施例3と同様の条件を使用した。テンプレートはWT、2142G、2143Gの各プラスミド(プラスミド量104コピー)を使用した。KODポリメラーゼを用いた場合の結果は図4に示すとおりである。また、それぞれの増幅産物を3%アガロースゲルで電気泳動し、エチジウムブロマイドで染色して増幅産物量を確認した結果を表5に示す。
【0079】
【表5】
【0080】
Taqポリメラーゼを使用した場合、反応条件を固定し上記試薬組成の他にも様々な試薬組成検討を行ったが特異的なピロリ菌の検出はできなかった。これに対しKODポリメラーゼを使用すると高速増幅に対応して特異的にピロリ菌の検出が可能であった。電気泳動の結果から、Taqポリメラーゼによる増幅は高速増幅の場合WTでわずかにバンドが見える程度の増幅であり、WT のプラスミド(102コピー)をテンプレートにした時には電気泳動バンドの検出もできなかった。KODポリメラーゼは高速増幅に適応できるが、Taqポリメラーゼは高速増幅に適応できず、目的の核酸増幅が見られないためピロリ菌の検出ができなかった。伸長速度が速くかつ5‘エキソヌクレアーゼ活性を持たないKODポリメラーゼのほうが本発明のDNAポリメラーゼとして適していることがわかる。
【0081】
(実施例8)
核酸増幅工程に引き続き融解曲線解析を行うことで1塩基変異が存在する場合のテンプレートと存在しない場合のテンプレートを1つの標識プローブで検出できるかどうかを検討した。テンプレートはピロリ菌WT、2142G、2143Gの三種類のプラスミドを使用し、プラスミド量は104コピー及び102コピーを使用した。実施例3と同じ試薬組成ならびに反応条件は表3の(核酸増幅サイクル)・(融解曲線解析)・(クールダウン)に示すものを使用した。プライマー及び標識プローブはそれぞれ表1に示すプライマーF4,R4、核酸プローブAを使用した。得られた蛍光シグナル値の補正は行わず、ライトサイクラー(登録商標)における融解曲線解析の結果をそのまま利用した。測定レンジは530nmを使用した。
【0082】
104コピー及び102コピーのプラスミドをテンプレートとして用いた時の蛍光測定の結果をそれぞれ図7、図8に示す。ライトサイクラー(登録商標)を使用した融解曲線解析においては標識プローブが標的核酸から解離する時の蛍光量の増加を検出している。図の縦軸は蛍光強度の一次導関数の逆符号の値(−dF/dt)、横軸は温度(℃)である。図7、図8からはそれぞれ2つのピークが得られた。61℃に見られるピークはWTのピークであり、50℃に見られるピークはWTに対して1塩基変異を持つ二種類の変異型のピロリ菌のピークである。この結果から、1つの標識プローブを使用し融解曲線解析を行うことでWTと変異型を別々のピークで検出できることがわかる。
【0083】
(実施例9)
(菌体を直接テンプレートとした融解曲線解析による検出)
ピロリ菌のATCC株2種(野生型26695株及び2143G変異型UA1182株)を入手した。そのうちそれぞれ109菌体を分取し、95℃5分の熱処理を行った。その後それぞれ105熱処理後菌体溶液を表2のテンプレートとした。その他は融解曲線解析を行った実施例8と同様の方法にてピロリ菌の検出を行った。その結果を図9に示す。
【0084】
融解曲線解析によりピロリ菌野生型及びCAM耐性ピロリ菌の検出を行ったところ、いずれも融解曲線解析によって単独ピークとして検出することができた。菌体をサンプルとして検出(CAM耐性判定)まで30分以内で行うことが可能である。
【0085】
(比較例1)
(ピロリ菌の検出までの時間)
一般的に使用されるPCRサイクル94℃15秒、65℃30秒、68℃30秒でPCRを行い、融解曲線解析で検出を行った場合、サーマルサイクラーの中でも温度の上昇・下降速度の早いとされているライトサイクラー(登録商標)でも増幅・検出あわせて1時間以上を要した。
【0086】
実施例3の反応条件において、ライトサイクラー(登録商標)を使用した標的核酸の検出までに要する時間は15分以内、さらに融解曲線解析を用いてもピロリ菌の検出及びピロリ菌CAM耐性遺伝子の存在を検出するのに要する時間は20分以内であった。本発明の検出方法を用いると極めて短時間で、かつ高感度なピロリ菌の検出が可能である。
【0087】
(実施例10)
(第二プライマーと標識プローブ濃度比の検討)
表1に記載の標識プローブAとプライマーF4、プライマーR4を用いてプライマーとプローブの濃度比の検討を行った。プライマー、プローブ濃度以外の条件に関しては実施例3の条件を用いた。第二プライマー(プライマーF4)とプローブ濃度に関しては以下の4条件を用いた。
第二プライマー150nM、プローブ150nM(プローブ/プライマー比=1):図4
第二プライマー100nM、プローブ500nM(プローブ/プライマー比=5):図10
第二プライマー100nM、プローブ1000nM(プローブ/プライマー比=10):図11
第二プライマー100nM、プローブ1100nM(プローブ/プライマー比=11)
テンプレートはピロリ菌WTのプラスミド104を用いた。
【0088】
図4、図10、11よりプローブ濃度/プライマー濃度が1の場合は問題なくMTを検出できている。濃度比5の時はプローブが多いことによる増幅制限を受けているため、Ct値は濃度比1の時と比較してやや遅くなっているがMTを検出できていた。濃度比10では、プローブ過多により増幅制限が強く現れるとともに、標的核酸にハイブリダイゼーションして消光しているプローブに対してハイブリダイゼーションせずに蛍光を発しているプローブが多くなりバックグラウンドシグナルが増えたことで、濃度比1や5よりもシグナルが弱くなっているが、MTを検出できていた。データは示していないが、さらに濃度比が11と高くなった場合は、増幅制限を受けていることとシグナル検出温度においてハイブリダイゼーションしていないプローブが極端に多くなり、そのバックグランドシグナルによって蛍光シグナルの差が見られなくなったことの2つの理由により検出できなかった。
【0089】
(実施例11)
(プローブ塩基数の検討)
表1に記載の標識プローブA、C〜Eを用いてプローブ塩基数の検討を行った。標識プローブ以外の条件に関しては実施例3の条件を用いた。標識プローブAは19塩基(Tm=62.20)、標識プローブCは12塩基(Tm=33.47)、標識プローブDは16塩基(Tm=54.34)、標識プローブEは32塩基(Tm=80.65)である。標識プローブCについてはアニール温度を50℃に設定変更して検出を行った。テンプレートはピロリ菌WTのプラスミド104を用いた。標識プローブA、標識プローブDを用いて検出した結果をそれぞれ図4、図12に示す。
【0090】
図4、図12より、標識プローブ塩基数が14〜30塩基に含まれる標識プローブAおよび標識プローブDではWTの検出が確認できた。12塩基である標識プローブCおよび32塩基である標識プローブEを用いた場合は蛍光シグナルの減少が見られずWTの検出が確認できなかった。特に32塩基の標識プローブEについてはTm値が80.65であり、第二プライマーのTm値71.66よりも高くなっており、標識プローブEの標的核酸へのハイブリダイゼーションによりプライマーの伸長反応が阻害されたと考えられる。またこの結果より、従来の高速PCR法に蛍光標識された核酸プローブを添加するだけでは標的核酸の迅速な検出が実現できないことがわかる。
【0091】
(実施例12)
(プローブ塩基数の検討:融解曲線解析)
表1に記載の標識プローブA、C〜Eを用いてプローブ塩基数の検討で融解曲線解析による検出を行った。標識プローブ以外の条件に関しては実施例8の条件を用いた。テンプレートはピロリ菌WT、2142G、2143Gの三種類のプラスミドを使用し、プラスミド量は104コピーを使用した。
【0092】
図7、図13より標識プローブ塩基数が14〜30塩基に含まれる標識プローブAおよび標識プローブDではWT、2142G、2143Gのそれぞれの検出ピークが確認できた。12塩基である標識プローブCおよび32塩基である標識プローブEを用いた場合はWT、2142G、2143Gのそれぞれの検出ピークが確認できなかった。標識プローブEについては増幅産物をアガロースゲル電気泳動にて確認したところ、目的の位置に泳動バンドが見られず、増幅阻害が起きていた。
【0093】
以下に16S rRNA遺伝子を標的とした抗酸菌(マイコバクテリウム属菌)を検出する方法を示す。
【0094】
(実施例13)
(テンプレートの調製)
ヒト型結核菌(Mycobacterium tuberculosis)、トリ型結核菌(Mycobacterium avium)、マイコバクテリウムカンサシイ(Mycobacterium kansasii)をそれぞれ小川培地(日水社製)にて37℃約2週間培養した。培養した各菌体をリン酸緩衝液に懸濁した液を用いて、常法に従いフェノール・クロロホルム抽出を行い、ゲノムDNAを抽出した。抽出されたゲノムDNAは−20℃で保存した。ヒト型結核菌はMT、トリ型結核菌はMA、マイコバクテリウムカンサシイはMKと示す。
【0095】
(実施例14)
(プライマー及び標識プローブの合成)
検出に使用するプライマー及び標識プローブは核酸合成受託会社((株)日本バイオサービス、(株)サワディー、GENSET KK、シグマアルドリッチジャパン(株)等)に依頼した。
【0096】
使用した標識プローブ及びプライマーの塩基配列を表6に示す。配列番号34〜38は標識プローブで、配列番号34、37,38はMTを、配列番号35はMAを、配列番号36はMKを検出するためのプローブである。配列番号20〜33は抗酸菌16SrRNA遺伝子の一部の領域を増幅できる共通のプライマーである。
【0097】
【表6】
【0098】
(実施例15)
(MT、MA、MKの検出)
表7または表2に示す反応液にMT、MA、MKの各ゲノムDNA20ngをテンプレートとして混合した。以下、表7または表2の反応組成を基準とし、反応液を構成するいずれかの溶液の最終濃度を変更した場合はミリQ水で調節し全量20μLを維持した。また、表7または表2の組成は一例であり反応組成の最適化は同業者であれば容易に行えるものである。プライマーは表6に記載のプライマーF6とプライマーR6を使用した。標識プローブは表6記載の標識プローブF〜Hを使用した。標識プローブFはMTの塩基配列に対して完全に一致する塩基配列を持つ標識プローブである。標識プローブGはMAの塩基配列に対して完全に一致する塩基配列を持つ標識プローブである。標識プローブHはMKの塩基配列に対して完全に一致する塩基配列を持つ標識プローブである。標識プローブFを使用する場合は表7の組成、標識プローブG、Hを使用する場合は表2の組成を利用した。増幅及び蛍光の測定にはロシュ・ダイアグノスティック社製ライトサイクラー(登録商標)を使用した。PCRの条件は表8に示す条件を使用した。測定モードは530nmを利用した。以下、特に記載がない場合はすべてライトサイクラー(登録商標)での測定とする。図14〜18は、グラフの横軸をサイクル数n、縦軸を蛍光シグナル値として解析結果を表した図である。ライトサイクラーソフトウェアの解析により、バックグランドとして現れる標識プローブの発する蛍光シグナル値を0として計算されているため、縦軸の数値は小さくなるほど標識プローブに対する標的核酸が多いことを示している。
【0099】
【表7】
【0100】
【表8】
【0101】
解析の結果、標識プローブFを使用してMT、標識プローブGを利用してMA、標識プローブHを利用してMKを検出した結果をそれぞれ図14〜16に示す。なお、図14〜16中、NCはテンプレートにミリQ水を使用したネガティブコントロールを示す。標識プローブFを用いた図14からはMTに特異的な検出シグナルが、標識プローブGを用いた図15からはMAに特異的な検出シグナルが、標識プローブHを用いた図16からはMKに特異的な検出シグナルが、それぞれ確認できた。
【0102】
(実施例16)
(第二プライマーとプローブ濃度比の検討)
表6に記載の標識プローブFとプライマーF6、プライマーR6を用いてプライマーとプローブの濃度比の検討を行った。プライマー、プローブ濃度以外の条件に関しては実施例12の条件を用いた。テンプレートはMTゲノムDNA20ngを用いた。第二プライマー(プライマーR6)とプローブ濃度に関しては以下の3条件を用いた。
第二プライマー150nM、プローブ150nM(プローブ/プライマー比=1):図14
第二プライマー150nM、プローブ450nM(プローブ/プライマー比=5):図17
第二プライマー150nM、プローブ1650nM(プローブ/プライマー比=11):図18
【0103】
図14、17,18より、プローブ濃度/プライマー濃度が1の場合は問題なくMTを検出できているのに対して、濃度比5の時はプローブが多いことによる増幅制限を受けてCt値が遅くなっている。さらに濃度比が11と高くなった図18では、増幅制限とともに、シグナル検出温度においてハイブリダイゼーションしていないプローブが極端に多くなり、そのバックグランドシグナルによって蛍光シグナルの差が見られなくなった。標識プローブFとプライマーF6、プライマーR6を用いた検討ではプローブ濃度/プライマー濃度=11での検出はできなかったが、標識プローブの蛍光強度が低く、第二プライマーのTmがプローブ濃度のTmよりも極端に大きい場合に、プローブ濃度/プライマー濃度=10では検出が確認できる組み合わせも存在する。
【0104】
(実施例17)
(プライマーのTmとプローブのTmの検討)
表6に記載の標識プローブF、I、JとプライマーF6〜8、プライマーR6〜8を用いてプライマーとプローブのTmによってMT検出に差が現れるかどうかの検討を行った。プライマーF6とR6、プライマーF7とR7、プライマーF8とR8をプライマーセットとして固定し、それぞれTmの異なるプライマーセットを得た。標識プローブ3種類とプライマーセット3種類を組み合わせて検討した。標識プローブとプライマーセット以外の条件に関しては基本的に実施例15の条件を用いた。標識プローブIの場合のみ、表8におけるPCR条件のアニール・伸長温度を60℃から57℃に変更して実施した。結果を表9に示す。表9の「○」はCt値=40未満、「△」はCt値=40以上、「×」は検出されないことを示している。例えば、標識プローブFとプライマーF6、R6の組み合わせの結果は、図14に示されるが、この場合Ct値=31.35で「○」となる。その他の組み合わせについても図14様の図が得られているが、ここでは結果のみ示す。
【0105】
【表9】
【0106】
表9より、プライマーとプローブのTmによって検出されるCt値に差が見られた。例えばプライマーF6とR6、標識プローブFの組み合わせでは、プローブTm<プライマーTmのため問題なく検出が可能であった。一方、プライマーF8とR8、標識プローブJの組み合わせではプローブTm>プライマーTmのため、検出ができなかった。高速検出においてプローブTm>プライマーTmの場合は、プローブのハイブリダイゼーションによりプライマーの伸長が阻害され、増幅が阻害されたと考えられる。
【0107】
(実施例18)
(プライマーのTmの検討)
表6に記載の標識プローブF及びプライマーF6、F8〜12、プライマーR6、R8〜12を用いてプライマーTm値によってMT検出に差が現れるかどうかの検討を行った。プライマーF6種類とプライマーR6種類を組み合わせて検出を実施した。プライマー以外の条件は基本的に実施例15の条件を用いた。ただし、PCR条件におけるアニール・伸長温度は各プライマーセットのTmに合わせて50℃〜60℃の範囲で設定した。結果を表10に示す。判定基準は実施例17と同様とした。
【0108】
【表10】
【0109】
表10より、プライマーのTmが70以上であれば問題なく検出されているが、プライマーF,RのどちらかのプライマーTmが70未満となると高速検出の条件バランスがくずれてCt値が遅延しているのがわかる。本願発明ではプライマー及び標識プローブの濃度やTmなどの条件バランスが重要となってくるため、安定して検出を行うにはプライマーのTmは70℃以上が適している。
【0110】
(実施例19)
(MTとMAの融解曲線解析による検出)
核酸増幅工程に引き続き融解曲線解析を行うことで1塩基変異が存在する場合のテンプレートと存在しない場合のテンプレートを1つの標識プローブで検出できるかどうかを検討した。テンプレートはMT、MAのゲノム20ngを使用した。実施例15と同じ試薬組成ならびに反応条件は表8の(核酸増幅サイクル)・(融解曲線解析)・(クールダウン)に示すものを使用した。プライマー及び標識プローブはそれぞれ表6に示すプライマーF6,R6、核酸プローブFを使用した。得られた蛍光シグナル値の補正は行わず、ライトサイクラー(登録商標)における融解曲線解析の結果をそのまま利用した。測定レンジは530nmを使用した。
【0111】
MT及びMAゲノムをテンプレートとして用いた時の蛍光測定の結果を図19に示す。ライトサイクラー(登録商標)を使用した融解曲線解析においては標識プローブが標的核酸から解離する時の蛍光量の増加を検出している。図の縦軸は蛍光強度の一次導関数の逆符号の値(−dF/dt)、横軸は温度(℃)である。図19では2つのピークが確認できた。60℃に見られるピークは標識プローブFに対して完全に一致する塩基配列を持つMTの検出ピークである。55℃に見られるピークは標識プローブFに対して1塩基変異が存在する塩基配列を持つMAの検出ピークである。この結果から、1つの標識プローブを使用し融解曲線解析を行うことで抗酸菌であるMTとMA種を別々のピークとして検出できることがわかった。
【0112】
(比較例2)
(MTの検出までの時間)
一般的にMTの遺伝子検査に使用されるロシュダイアグノスティック社のアンプリコアマイコバクテリウムツベルクローシスキットを用いて、陽性コントロールを検出するのに要する時間はPCR及び検出できるまで最短で約6時間かかる。
【0113】
実施例15の反応条件において、ライトサイクラー(登録商標)を使用した標的核酸の検出までに要する時間は約20分、さらに融解曲線解析を用いてもMTを検出するのに要する時間は30分以内であった。本発明の検出方法を用いれば極めて短時間でのMT検出が可能となる。
【産業上の利用可能性】
【0114】
本発明によれば、迅速かつ高感度な核酸検出が可能となる。具体的には、第二プライマーの伸長反応を阻害しない標識プローブの性質及び濃度により、増幅効率を保持したまま不要なバックグランド蛍光シグナル値を極力減らし、必要な蛍光シグナルの減少量を測定することが可能になる。さらに伸長速度が速いDNAポリメラーゼを使用することにより増幅反応時間を短縮し、これまでになく迅速かつ高感度な核酸検出方法を実現でき、さらに、ピロリ菌の23S rRNA遺伝子の存在または量を調べること、ピロリ菌の23S rRNA遺伝子の変異の存在を調べること、23S rRNA遺伝子の第2142位または第2143位における変異の存在を調べピロリ菌の抗生物質耐性を判別することなども迅速かつ高感度に行えるようになり、産業界に大きく貢献する。
【図面の簡単な説明】
【0115】
【図1】プライマー及びプローブの濃度とそれぞれのTm値の関係により可能となる迅速かつ高感度な検出方法の原理を示す。
【図2】標的核酸または第一プライマーの伸長産物と標識プローブの位置関係A、Bを模式的に示す。
【図3】本発明における非対称PCRの方法について、第一プライマー、第二プライマー及び標識プローブの位置関係を示す。
【図4】プラスミド104コピーを鋳型としてピロリ菌のリアルタイム検出結果を示す。
【図5】プラスミド102コピーを鋳型としてピロリ菌のリアルタイム検出結果を示す。
【図6】第一プライマーと第二プライマーの濃度比が1:1である時のピロリ菌のリアルタイム検出結果を示す。
【図7】プラスミド104コピーを鋳型として融解曲線解析によるピロリ菌の野生型と変異型を同時検出した結果を示す。
【図8】プラスミド102コピーを鋳型として融解曲線解析によるピロリ菌の野生型と変異型を同時検出した結果を示す。
【図9】菌体を簡易処理したものを鋳型としてピロリ菌の検出を行った結果を示す。
【図10】プローブ/プライマー比=5の条件におけるピロリ菌のリアルタイム検出結果を示す。
【図11】プローブ/プライマー比=10の条件におけるピロリ菌のリアルタイム検出結果を示す。
【図12】標識プローブDを用いた場合のピロリ菌のリアルタイム検出を示す。
【図13】標識プローブDを用いた場合の融解曲線解析によるピロリ菌の野生型と変異型同時検出結果を示す。
【図14】16SrRNA遺伝子をターゲットとした時のMTのリアルタイム検出結果を示す。
【図15】16SrRNA遺伝子をターゲットとした時のMAのリアルタイム検出結果を示す。
【図16】16SrRNA遺伝子をターゲットとした時のMKのリアルタイム検出結果を示す。
【図17】プローブ/プライマー比=5の条件におけるMTのリアルタイム検出結果を示す。
【図18】プローブ/プライマー比=11の条件におけるMTのリアルタイム検出結果を示す。
【図19】MT及びMAゲノムを鋳型として融解曲線解析によるMTとMAをピークのTmで判別した結果を示す。
【特許請求の範囲】
【請求項1】
標的核酸の存在または量を確認したい試料と少なくとも下記(a)と(b)の存在下、連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該標識物質の蛍光を測定しながら、高速PCRを行い、該標的核酸の存在または量を調べることを特徴とする検出方法。
(a)次の(i)〜(iv)の性質を有する標識プローブ
(i) 標的核酸に相補的な配列と二重鎖核酸構造を形成でき、かつ14塩基以上30塩基以下の長さのオリゴヌクレオチドを有する
(ii) 該オリゴヌクレオチドの末端部が該二重鎖核酸構造形成時に消光する性質を有する蛍光物質で修飾されている
(iii) 該二重鎖核酸構造形成時に、蛍光物質で修飾されている該末端部にG(グアニン)とC(シトシン)の対が少なくとも1つ以上存在する
(iv)該標識プローブの濃度が第二プライマーの濃度に対して10倍以下である
(b)次の(i)〜(iii)の性質を有する第一プライマーおよび第二プライマー
(i) 第一プライマーの伸長産物が標的核酸と相補的な配列を有しかつ第二プライマーの鋳型となる性質を有し、該第二プライマーの伸長産物が標的核酸と相同な配列を有しかつ該第一プライマーの鋳型となる性質を有する
(ii) 該第一プライマーおよび該第二プライマーによる増幅産物の長さが50bp以上500bp以下である
(iii) 該第一プライマー及び該第二プライマーのTm値が70℃以上90℃以下であり、かつ該第二プライマーのTm値は前記標識プローブのTm値より大きい
【請求項2】
(b)の第一プライマーおよび第二プライマーが、さらに
(iv) 該第一プライマーの伸長産物において、該伸長産物の5’末端から前記標識プローブが二重鎖核酸構造を形成する領域までの距離を(A)、該標識プローブが二重鎖核酸構造を形成する領域から該伸長産物の3’末端までの距離を(B)とした場合に(A/B)の値が0.1以上3.0以下であること、
を特徴とする請求項1に記載の検出方法。
【請求項3】
(a)の標識プローブに、さらに
(v) 標的部位と3個以下の非相補的塩基を導入すること、
を特徴とする請求項1または2に記載の検出方法。
【請求項4】
標識プローブの中央部に、非相補的塩基を導入することを特徴とする請求項3に記載の検出方法。
【請求項5】
(a)の標識プローブに修飾されている蛍光物質が該標識プローブの末端に修飾され、該末端の塩基がGもしくはCであることを特徴とする請求項1〜4のいずれかに記載の検出方法。
【請求項6】
第一プライマーの濃度が第二プライマーの濃度に対して4倍以上であることを特徴とする請求項1〜5のいずれかに記載の検出方法。
【請求項7】
高速PCRが、10℃/秒以上の速度で温度の上昇及び下降を繰り返すことを特徴とする請求項1〜6のいずれかに記載の検出方法。
【請求項8】
さらに、
(c)100塩基/秒以上のDNA合成速度を有するDNAポリメラーゼ
の存在下、
高速PCRを行うことを特徴とする請求項1〜7のいずれかに記載の検出方法。
【請求項9】
DNAポリメラーゼが、さらに5’エキソヌクレアーゼ活性を持たないことを特徴とする請求項8に記載の検出方法。
【請求項10】
DNAポリメラーゼが、KOD DNAポリメラーゼ(Thermococcus kodakaraensis KOD1由来)またはその変異体であることを特徴とする請求項8または9に記載の検出方法。
【請求項11】
標的核酸がピロリ菌の23S rRNA遺伝子のセンス鎖であり、
(a)の標識プローブのオリゴヌクレオチドの配列が配列番号2より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べることを特徴とする請求項1〜10のいずれかに記載の検出方法。
【請求項12】
標的核酸がピロリ菌の23S rRNA遺伝子のアンチセンス鎖であり、
(a)の標識プローブのオリゴヌクレオチドの配列が配列番号1より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べることを特徴とする請求項1〜10のいずれかに記載の検出方法。
【請求項13】
請求項1〜12のいずれかに記載の方法に引き続いて、密封状態のまま、標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、0.1℃/秒以上の連続的な温度上昇又は下降を伴う融解曲線解析を行い、標的核酸中の変異の存在を調べることを特徴とする検出方法。
【請求項14】
変異がピロリ菌の23S rRNA遺伝子の第2142位または第2143位であり、抗生物質耐性を判別する変異の存在を調べることを特徴とする請求項13に記載の検出方法。
【請求項15】
標識プローブのオリゴヌクレオチドの配列の中央部に変異の部位が位置することを特徴とする請求項13または14に記載の検出方法。
【請求項16】
少なくとも次の(a)及び(b)を含むことを特徴とするピロリ菌の存在および/またはピロリ菌の抗生物質耐性の存在を検出するためのキット。
(a)次の(i)〜(ii)の性質を有する標識プローブ
(i)配列番号1または配列番号2より選ばれる14塩基以上30塩基以下の連続した塩基配列のオリゴヌクレオチドを有する
(ii) 該オリゴヌクレオチドが、該オリゴヌクレオチドと相補的な配列と二重鎖核酸構造を形成する時に消光する性質を有する蛍光物質が、該オリゴヌクレオチドの末端部に標識されている
(b)Tm値が70℃以上90℃以下であり配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列を含むプライマーと、Tm値が70℃以上90℃以下であり配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列を含むプライマー
【請求項17】
(a)の標識プローブと二重鎖核酸構造を形成しうる伸長産物を生成せしめる側のプライマーの濃度が、他方のプライマーの濃度に対して4倍以上であることを特徴とする請求項16に記載のキット。
【請求項18】
さらに、
(c)100塩基/秒以上のDNA合成速度を有するDNAポリメラーゼ
を含むことを特徴とする請求項16または17に記載のキット。
【請求項19】
DNAポリメラーゼが、さらに5’エキソヌクレアーゼ活性を持たないことを特徴とする請求項18に記載のキット。
【請求項20】
DNAポリメラーゼが、KOD DNAポリメラーゼ(Thermococcus kodakaraensis KOD1由来)またはその変異体であることを特徴とする請求項18または19に記載のキット。
【請求項21】
標識プローブの中央部に、ピロリ菌の23S rRNA遺伝子の第2142位または第2143位に相当する部位が位置することを特徴とする請求項16〜20に記載のキット。
【請求項1】
標的核酸の存在または量を確認したい試料と少なくとも下記(a)と(b)の存在下、連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該標識物質の蛍光を測定しながら、高速PCRを行い、該標的核酸の存在または量を調べることを特徴とする検出方法。
(a)次の(i)〜(iv)の性質を有する標識プローブ
(i) 標的核酸に相補的な配列と二重鎖核酸構造を形成でき、かつ14塩基以上30塩基以下の長さのオリゴヌクレオチドを有する
(ii) 該オリゴヌクレオチドの末端部が該二重鎖核酸構造形成時に消光する性質を有する蛍光物質で修飾されている
(iii) 該二重鎖核酸構造形成時に、蛍光物質で修飾されている該末端部にG(グアニン)とC(シトシン)の対が少なくとも1つ以上存在する
(iv)該標識プローブの濃度が第二プライマーの濃度に対して10倍以下である
(b)次の(i)〜(iii)の性質を有する第一プライマーおよび第二プライマー
(i) 第一プライマーの伸長産物が標的核酸と相補的な配列を有しかつ第二プライマーの鋳型となる性質を有し、該第二プライマーの伸長産物が標的核酸と相同な配列を有しかつ該第一プライマーの鋳型となる性質を有する
(ii) 該第一プライマーおよび該第二プライマーによる増幅産物の長さが50bp以上500bp以下である
(iii) 該第一プライマー及び該第二プライマーのTm値が70℃以上90℃以下であり、かつ該第二プライマーのTm値は前記標識プローブのTm値より大きい
【請求項2】
(b)の第一プライマーおよび第二プライマーが、さらに
(iv) 該第一プライマーの伸長産物において、該伸長産物の5’末端から前記標識プローブが二重鎖核酸構造を形成する領域までの距離を(A)、該標識プローブが二重鎖核酸構造を形成する領域から該伸長産物の3’末端までの距離を(B)とした場合に(A/B)の値が0.1以上3.0以下であること、
を特徴とする請求項1に記載の検出方法。
【請求項3】
(a)の標識プローブに、さらに
(v) 標的部位と3個以下の非相補的塩基を導入すること、
を特徴とする請求項1または2に記載の検出方法。
【請求項4】
標識プローブの中央部に、非相補的塩基を導入することを特徴とする請求項3に記載の検出方法。
【請求項5】
(a)の標識プローブに修飾されている蛍光物質が該標識プローブの末端に修飾され、該末端の塩基がGもしくはCであることを特徴とする請求項1〜4のいずれかに記載の検出方法。
【請求項6】
第一プライマーの濃度が第二プライマーの濃度に対して4倍以上であることを特徴とする請求項1〜5のいずれかに記載の検出方法。
【請求項7】
高速PCRが、10℃/秒以上の速度で温度の上昇及び下降を繰り返すことを特徴とする請求項1〜6のいずれかに記載の検出方法。
【請求項8】
さらに、
(c)100塩基/秒以上のDNA合成速度を有するDNAポリメラーゼ
の存在下、
高速PCRを行うことを特徴とする請求項1〜7のいずれかに記載の検出方法。
【請求項9】
DNAポリメラーゼが、さらに5’エキソヌクレアーゼ活性を持たないことを特徴とする請求項8に記載の検出方法。
【請求項10】
DNAポリメラーゼが、KOD DNAポリメラーゼ(Thermococcus kodakaraensis KOD1由来)またはその変異体であることを特徴とする請求項8または9に記載の検出方法。
【請求項11】
標的核酸がピロリ菌の23S rRNA遺伝子のセンス鎖であり、
(a)の標識プローブのオリゴヌクレオチドの配列が配列番号2より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べることを特徴とする請求項1〜10のいずれかに記載の検出方法。
【請求項12】
標的核酸がピロリ菌の23S rRNA遺伝子のアンチセンス鎖であり、
(a)の標識プローブのオリゴヌクレオチドの配列が配列番号1より選ばれる14塩基以上30塩基以下の連続した塩基配列であり、
(b)の第一プライマーが、配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
(b)の第二プライマーが、配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列であり、
連続的または間欠的に標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、高速PCRを行い、ピロリ菌の23S rRNA遺伝子の存在または量を調べることを特徴とする請求項1〜10のいずれかに記載の検出方法。
【請求項13】
請求項1〜12のいずれかに記載の方法に引き続いて、密封状態のまま、標識プローブの蛍光物質の励起波長の光を与えかつ該蛍光物質の蛍光を測定しながら、0.1℃/秒以上の連続的な温度上昇又は下降を伴う融解曲線解析を行い、標的核酸中の変異の存在を調べることを特徴とする検出方法。
【請求項14】
変異がピロリ菌の23S rRNA遺伝子の第2142位または第2143位であり、抗生物質耐性を判別する変異の存在を調べることを特徴とする請求項13に記載の検出方法。
【請求項15】
標識プローブのオリゴヌクレオチドの配列の中央部に変異の部位が位置することを特徴とする請求項13または14に記載の検出方法。
【請求項16】
少なくとも次の(a)及び(b)を含むことを特徴とするピロリ菌の存在および/またはピロリ菌の抗生物質耐性の存在を検出するためのキット。
(a)次の(i)〜(ii)の性質を有する標識プローブ
(i)配列番号1または配列番号2より選ばれる14塩基以上30塩基以下の連続した塩基配列のオリゴヌクレオチドを有する
(ii) 該オリゴヌクレオチドが、該オリゴヌクレオチドと相補的な配列と二重鎖核酸構造を形成する時に消光する性質を有する蛍光物質が、該オリゴヌクレオチドの末端部に標識されている
(b)Tm値が70℃以上90℃以下であり配列番号3より選ばれる20塩基以上35塩基以下の連続した塩基配列を含むプライマーと、Tm値が70℃以上90℃以下であり配列番号4より選ばれる20塩基以上35塩基以下の連続した塩基配列を含むプライマー
【請求項17】
(a)の標識プローブと二重鎖核酸構造を形成しうる伸長産物を生成せしめる側のプライマーの濃度が、他方のプライマーの濃度に対して4倍以上であることを特徴とする請求項16に記載のキット。
【請求項18】
さらに、
(c)100塩基/秒以上のDNA合成速度を有するDNAポリメラーゼ
を含むことを特徴とする請求項16または17に記載のキット。
【請求項19】
DNAポリメラーゼが、さらに5’エキソヌクレアーゼ活性を持たないことを特徴とする請求項18に記載のキット。
【請求項20】
DNAポリメラーゼが、KOD DNAポリメラーゼ(Thermococcus kodakaraensis KOD1由来)またはその変異体であることを特徴とする請求項18または19に記載のキット。
【請求項21】
標識プローブの中央部に、ピロリ菌の23S rRNA遺伝子の第2142位または第2143位に相当する部位が位置することを特徴とする請求項16〜20に記載のキット。
【図1】

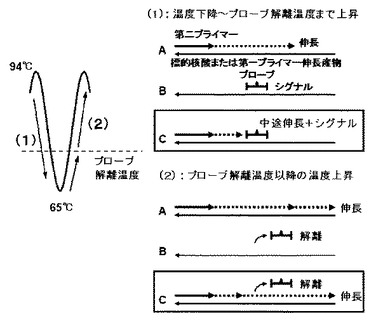
【図2】
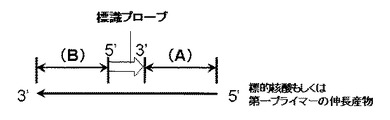
【図3】
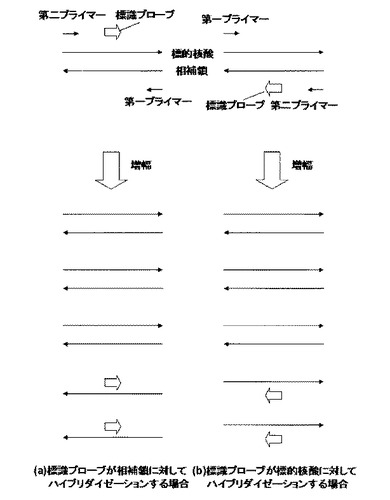
【図4】
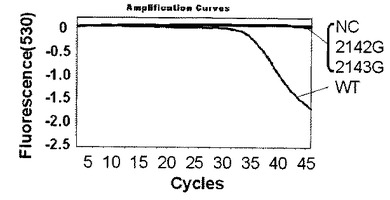
【図5】
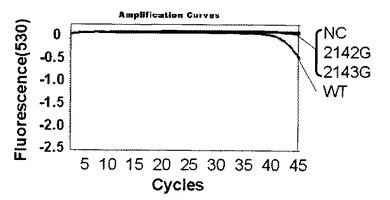
【図6】
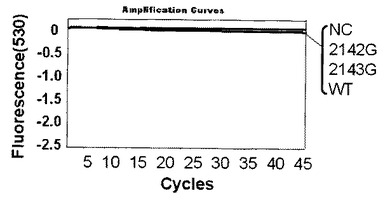
【図7】
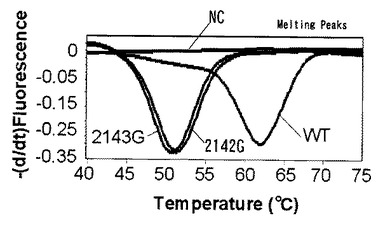
【図8】
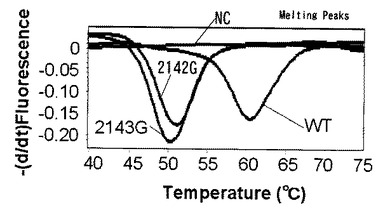
【図9】
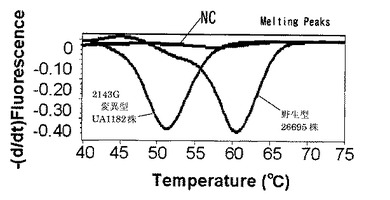
【図10】
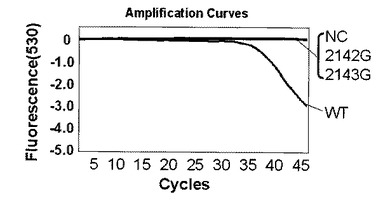
【図11】
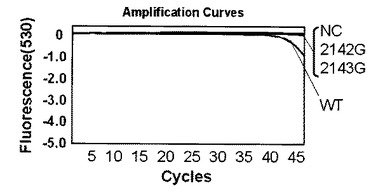
【図12】
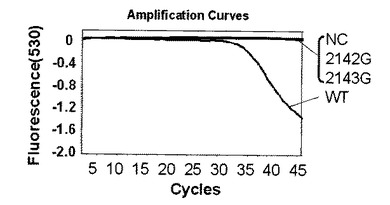
【図13】
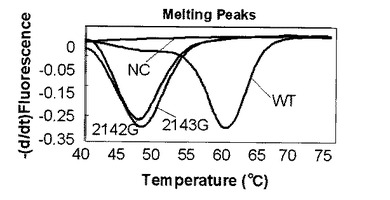
【図14】
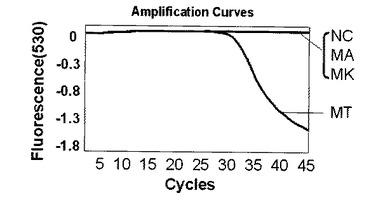
【図15】
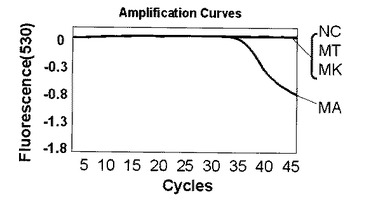
【図16】
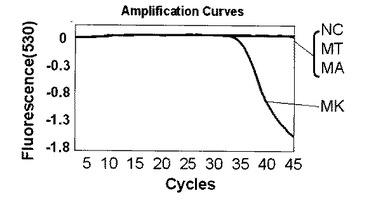
【図17】
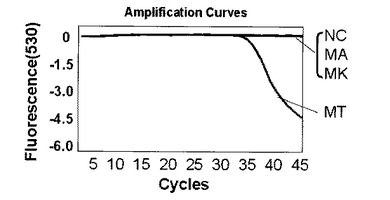
【図18】
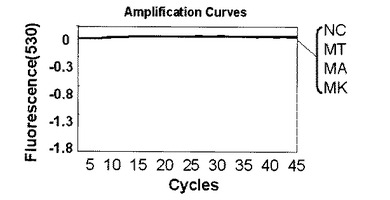
【図19】
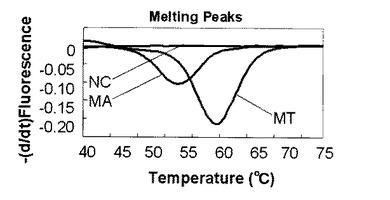
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公開番号】特開2009−148245(P2009−148245A)
【公開日】平成21年7月9日(2009.7.9)
【国際特許分類】
【出願番号】特願2008−153069(P2008−153069)
【出願日】平成20年6月11日(2008.6.11)
【出願人】(000003160)東洋紡績株式会社 (3,622)
【Fターム(参考)】
【公開日】平成21年7月9日(2009.7.9)
【国際特許分類】
【出願日】平成20年6月11日(2008.6.11)
【出願人】(000003160)東洋紡績株式会社 (3,622)
【Fターム(参考)】
[ Back to top ]