
核酸パッケージングシステム
本発明は、ファージパッケージング機構を使用した、標的核酸のクローニングに関する。パッケージング開始部位を、標的DNAの中に導入することができる。ファージパッケージングシステムの構成要素を標的DNAと組み合わせて、ファージカプシドの中にDNAをパッケージすることができる。パッケージDNAを使用して、標的核酸のライブラリーを作り出すこともでき、またはそのDNAを配列決定することもできる。例えば、本発明の一実施形態において、核酸を配列決定するための方法が提供され、この方法は、核酸を含むカプシドを提供するステップと、核酸を単離するステップと、核酸を配列決定するステップとを含み得る。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、一般に、分子生物学の分野に関する。より具体的には、本発明は、核酸分子のクローニングおよび核酸ライブラリーの産生に関する。
【背景技術】
【0002】
ゲノム学は、生物医学研究の中心的で統合的な学問になった。ゲノム配列決定プロジェクトは、事実上すべての生命過程の研究を変えた、安定した一連のさらに大きく、より複雑なゲノムデータセットを生成する。遺伝学および比較ゲノム学の進歩は、疾患が、分子の細部についての、前例がないレベルで分析され、かつ理解されることを可能にする。
【0003】
ゲノム研究は、高品質で、大きな挿入断片のゲノムライブラリーの有用性を必要とする。高品質ゲノムライブラリーは、狭いサイズ範囲のDNA断片を必要とする。DNA断片のサイズ選択は、パルスフィールドゲル電気泳動(PFGE)を使用して、典型的に行なわれるが、これは、いくつかの制限を有する。PFGEは、非常に時間がかかり、再現不可能である。さらに、PFGEからの断片DNAの収量は、非常に乏しい。
【0004】
高品質ゲノムライブラリーはまた、全ゲノムが、最小限のバイアスを伴って表わされることをも必要とする。ゲノムライブラリーのDNA断片は、ゲノムを部分的に消化するための制限酵素を使用して、産生されることが多いが、これは、いくつかの制限を有する。ゲノムの部分的な消化は、制限部位の間の距離が、適用されるサイズ選択限界未満であり、またはそれを超えるゲノムの領域に対してバイアスをもたらす。部分的な消化と関連するクローニングバイアスに対する代案として、DNA断片は、ゲノムをランダムに剪断することによって産生することができる。しかしながら、クローニング効率は、ランダムに剪断されたDNAを使用すると、著しく低下する。
【発明の概要】
【発明が解決しようとする課題】
【0005】
当技術分野が必要とするのは、全ゲノムを正確に表わすことができる高品質ゲノムライブラリーである。ゲノムライブラリーは、効率的に、かつ高収量を伴って産生されるべきである。ゲノムライブラリーはまた、容易に増殖され、かつ分析されるべきでもある。本発明は、これらの必要性を満たす。
【課題を解決するための手段】
【0006】
本発明は、核酸をクローニングし配列決定するため、および複数の核酸をクローニングし配列決定するためのパッケージングシステムの使用に関する。パッケージングシステムは、たとえば、ファージベースのパッケージングメカニズムであってもよい。核酸をカプシドの中にパッケージすることができ、このカプシドはバクテリオファージカプシドであってもよい。クローニングされた核酸は、たとえば、核酸ライブラリーの産生において使用することができる。
【0007】
核酸を配列決定するための方法であって、核酸を含むカプシドを提供するステップと、核酸を単離するステップと、核酸を配列決定するステップとを含み得る方法が本明細書において提供される。複数の核酸を配列決定するための方法であって、それぞれが核酸を含む複数のカプシドを提供するステップと、核酸を単離するステップと、核酸を配列決定するステップとを含み得る方法もまた本明細書において提供される。
【0008】
配列決定は、(a)水性マイクロリアクターが、核酸の単一コピー、核酸に結合することができる単一ビーズ、および核酸増幅を行なうために必要な試薬を含有する増幅反応溶液を含むように、油中水型エマルション中で水性マイクロリアクターの中に核酸を送達するステップと、(b)マイクロリアクター中で核酸を増幅して核酸の増幅コピーを形成し、マイクロリアクター中で増幅コピーをビーズに結合させるステップと、(c)平面表面上にある少なくとも10,000個の反応チャンバーのアレイにビーズを送達するステップであって、複数の反応チャンバーが単一核酸結合ビーズしか含まないステップと、(d)複数の反応チャンバー上で配列決定反応を同時に行なうステップとを含んでもよい。
【0009】
配列決定はまた、(a)複数の水性マイクロリアクターが、核酸の単一コピー、核酸に結合することができる単一ビーズ、および核酸増幅を行なうために必要な試薬を含有する増幅反応溶液を含むように、油中水型エマルション中で水性マイクロリアクターの中に複数の核酸を送達するステップと、(b)マイクロリアクター中で核酸を増幅して核酸の増幅コピーを形成し、マイクロリアクター中で増幅コピーをビーズに結合させるステップと、(c)平面表面上にある少なくとも10,000個の反応チャンバーのアレイにビーズを送達するステップであって、複数の反応チャンバーが単一核酸結合ビーズしか含まないステップと、
(d)複数の反応チャンバー上で配列決定反応を同時に行なうステップとを含んでもよい。
【0010】
ビーズは、少なくとも10,000個の増幅コピーに結合してもよい。核酸の増幅は、ポリメラーゼ連鎖反応を使用することによって成し遂げることができる。配列決定反応は、ピロリン酸ベースの配列決定反応であってもよい。配列決定反応は、(a)有効量の配列決定プライマーを核酸の増幅コピーにアニールし、ポリメラーゼおよび所定のヌクレオチド三リン酸を用いて配列決定プライマーを伸長させて、配列決定産物、および所定のヌクレオチド三リン酸が配列決定プライマーの3’末端上に取り込まれる場合には配列決定反応副産物を得るステップと、(b)配列決定反応副産物を同定し、それによって、反応チャンバー中で核酸の配列を決定するステップとを含んでもよい。
【0011】
核酸または複数の核酸の単離は、(a)第1のアダプター末端および第2のアダプター末端を少なくとも1つの核酸にライゲーションするステップであって、第1のアダプター末端が、オリゴヌクレオチド配列Yを含み、少なくとも1つの核酸の5’末端にライゲーションし、第2のアダプター末端が、オリゴヌクレオチド配列Zを含み、少なくとも1つの核酸の3’末端にライゲーションし、第1のアダプターが、5’末端で固体支持体に少なくとも1つの核酸を固定するための手段を有するステップと、(b)ステップ(a)の少なくとも1つの核酸を、固体支持体の存在下において、1つまたは複数のコロニープライマーXと混合するステップであって、それぞれのコロニープライマーXが、オリゴヌクレオチド配列Zにハイブリダイズすることができ、5’末端で固体支持体にコロニープライマーを固定するための手段を有し、それによって、少なくとも1つの核酸およびコロニープライマーの両方の5’末端が、固体支持体に固定され、少なくとも1つの核酸およびコロニープライマーの両方の上述の5’末端が、DNA変性条件下での水または水性緩衝液を用いる洗浄によって、それらを除去することができないように、上述の固体支持体に固定されるステップと、(c)核酸コロニーが生成されるように、固定された少なくとも1つの核酸上で1つまたは複数の核酸増幅反応を行なうステップとを含んでもよい。コロニープライマーは、縮重プライマーであってもよい。
【0012】
オリゴヌクレオチド配列Zは、オリゴヌクレオチド配列Yに相補的であってもよく、コロニープライマーXは、オリゴヌクレオチド配列Yと同じ配列であってもよい。核酸にアダプター末端をライゲーションする場合、2つの異なるコロニープライマーXは、少なくとも1つの核酸と混合してもよく、オリゴヌクレオチド配列Zは、コロニープライマーXのうち1つにハイブリダイズしてもよく、オリゴヌクレオチドYは、コロニープライマーXのうち1つの配列と同じであってもよい。
【0013】
少なくとも1つの核酸は、1つまたは複数の核酸コロニーにおいて配列決定することができる。配列決定は、標識ヌクレオチドの取り込みおよび検出を含んでもよく、さらに、核酸コロニーを視覚化することを含んでもよい。視覚化は、標識核酸プローブまたは非標識核酸プローブの使用を含んでもよい。
【0014】
少なくとも1つの核酸およびコロニープライマーを固体支持体に固定するための手段は、少なくとも1つの核酸およびコロニープライマーを支持体に共有結合で固定するための手段を含んでもよい。少なくとも1つの核酸およびコロニープライマーを固体支持体に共有結合で固定するための手段は、化学的に修飾された基であってもよい。少なくとも1つの核酸およびコロニープライマーを固体支持体に固定するための手段は、少なくとも1つの核酸およびコロニープライマーを支持体に共有結合で固定するための手段を含んでもよい。化学的に修飾可能な官能基は、アミノ基であってもよい。
【0015】
少なくとも1つの核酸およびコロニープライマーの両方の上述の5’末端が固定される固体支持体は、ラテックスビーズ、デキストランビーズ、ポリスチレンおよびポリプロピレン表面、ポリアクリルアミドゲル、金表面、ガラス表面、ならびにシリコンウェーハからなる群から選択することができる。固体支持体上の核酸コロニーの密度は、10,000/mm2〜100,000/mm2であってもよい。固体支持体に付着したコロニープライマーXの密度は、少なくとも1fmol/mm2であってもよい。少なくとも1つの核酸の密度は、10,000/mm2〜100,000/mm2であってもよい。少なくとも1つの核酸およびコロニープライマーの両方の5’末端は、共有結合を介して上述の固体支持体に固定することができる。
【0016】
核酸をクローニングするための方法もまた本明細書において提供される。パッケージング開始部位(PIS)を標的核酸の中に導入することができ、これは、標的核酸からクローニングされる核酸の上流末端を確立する。PISは、λ、P1、P7、またはT4を含むが、これらに限定されないファージパッケージング開始部位であってもよい。標的核酸は、ベクター、染色体DNA、または全ゲノムを含むが、これらに限定されない、核酸の任意の形態であってもよい。標的核酸としての全ゲノムの使用により、ゲノムライブラリーの産生が可能となる可能性がある。
【0017】
PISは、標的核酸にランダムに導入することができ、これは、ゲノムの範囲を増加させる可能性がある。PISは、転位およびライゲーションを含むが、これらに限定されない任意の方式において標的核酸の中に導入することができる。PISは、第1の核酸の転位によって導入することができる。第1の核酸は、Tn7、Tn5、Tn10、Mu、およびMarinerを含むが、これらに限定されない転位性末端を含んでもよい。
【0018】
次いで、パッケージングは、PISで開始することができ、これは、標的核酸に及ぶ、PISから下流の核酸のクローニングをもたらす。パッケージングは、in vitroにおいてまたはin vivoにおいて生じてもよい。クローニングされた核酸は、カプシド中にパッケージすることができ、これは、クローンを実質的に一定の長さにする可能性がある。
【0019】
第1の核酸はまた、ベクターエレメントを含んでもよい。ベクターエレメントは、PISの下流にあってもよく、クローニングされた核酸中に存在してもよい。ベクターエレメントは、低コピー複製開始点および高コピー複製開始点を含むが、これらに限定されない複製開始点であってもよい。ベクターエレメントはまた、高コピー複製開始点および低コピー複製開始点であってもよい。高コピー複製開始点は、複製誘発剤に応答性であってもよい。第1の核酸は、複製誘発剤をコードする核酸を含んでもよい。複製誘発剤は、TrfAであってもよい。
【図面の簡単な説明】
【0020】
【図1】λdoc粒子を使用する、トランスポゾン媒介クローニングの手法を示す図である。ステップ1:in vitro転位反応は、転位性末端(外向きの矢印)を介して、ゲノムDNA(長い直線)の全体にわたる多数の部位に、トランスポゾンを含有するPAC/oriVをランダムに挿入する。ステップ2:DNAは、in vitroにおいてパッケージされ、cos部位で始まり、頭部がいっぱいになるまで続く。次いで、はみ出たDNAは、Sau3Aを用いて消化されるが、頭部内のDNAは、保護される。ステップ3:次いで、ファージ頭部は、精製され、溶解され、Sau3A突出を有するゲノムDNAに融合された、一定の長さの、一般的な組成のcos−PAC/oriV(太い矢印)の断片がもたらされる。次いで、Sau3A突出およびcos突出を有するオリゴは、断片にライゲーションされる。ステップ4:断片は、再度パッケージされ、入ってくるDNAがcos末端を通して環状になる、DH10Bなどの宿主を感染させるために使用される。クローンは、CamRコロニーとして単離される。
【図2】in vitro転位およびファージパッケージングを介してクローニングするためのさらなる模式図を示す図である。
【図3】Sau3A突出を有するDNA断片にcos部位を追加するための、2つの核酸(配列番号1および2)を含むリンカーを示す図である。
【図4】トランスポゾン媒介クローニングの使用に基づくin vivo組換えのための手順を示す図である。ステップ1:図1におけるように、in vitro転位反応は、転位性末端(外向きの矢印)を介して、ゲノムDNA(長い直線)の全体にわたる多数の部位に、トランスポゾンをランダムに挿入する。しかしながら、この場合、トランスポゾンは、複製開始点を有してないcos−attB−KanRを含有する。ステップ2:DNAは、in vitroにおいてパッケージされ、cos部位で始まり、頭部がいっぱいになるまで続く。次いで、はみ出たDNAは、Sau3Aを用いて消化されるが、頭部内のDNAは、保護される。ステップ3:次いで、ファージ頭部は、精製され、溶解され、Sau3A突出を有するゲノムDNAに融合された、一定の長さの、一般的な組成のcos−PAC/oriV(太い矢印)の断片がもたらされる。次いで、Sau3A突出およびcos突出を有するオリゴが、断片にライゲーションされる。ステップ4:断片は、再度パッケージされ、新しいpPAC/oriV/attPベクターを含有し、Intを発現するように誘発された、DH10Bなどの宿主を感染させるために使用される。cos末端を介しての次に続く環化は、CamR付与プラスミドをもたらす。
【図5】転位性の/パッケージすることができるベクターを示す図である。ベクターのPvuII切断は、in vitroにおいて転位を可能にするTn5モザイク末端(ME)が側面に位置する線状の断片をもたらす。次いで、pac部位およびcos部位は、転位産物がin vitroにおいてパッケージされることを可能にする。
【図6】ファージカプシドのアフィニティー精製およびパッケージDNAの単離が後に続くλファージベースのパッケージングシステムを使用して、ゲノムDNAライブラリーを生成するための戦略を示す図である。
【図7】P22HT「ヘッドフル(headful)」パッケージングシステムを使用して、ゲノムDNAライブラリーを生成するための戦略を示す図である。
【図8】ウイルスカプシドパッケージDNAの末端の配列をクローニングし、かつ特徴づけるための戦略を示す図である。
【図9】ファージパッケージDNAの中へのクラスIII制限酵素部位およびリーチングプライマー対結合部位のクローニングが、DNAの末端の配列を特徴づけるためにどのように使用することができるかを示す図である。
【図10】DNAの中にリーチングプライマー対挿入断片をクローニングした後に、ファージパッケージDNAの末端をどのようにしてPCR増幅することができるかを示す図である。
【図11】DNAの中にリーチングプライマー対挿入断片をクローニングした後に、ファージパッケージDNAの末端をどのように配列決定することができるかを示す図である。
【発明を実施するための形態】
【0021】
発明者らは、より正確に表わす、標的DNAを単離し、パッケージするために使用することができるファージパッケージングシステムを開発した。使用されるファージパッケージングシステムに依存して、システムは、特定の、より一定のサイズのDNA断片を生成する。さらに、標的DNAは、標的DNAを、扱いやすく、クローニングすることができるサイズに低下させるために剪断ステップまたは制限消化ステップを必要とする方法と比較して、よりライゲーション可能である。したがって、標的DNAを得るための効率は、他の方法と比較して高い。標的DNAのパッケージングに続いて、DNAは、ライブラリーの生成などの多数の目的のために使用することができ、それを抽出することができ、それを増幅することができ、またはパッケージDNA断片の末端のクローニングおよび配列決定などによってDNAを配列決定することができる。
【0022】
DNA断片は、標的DNAの中にパッケージング開始部位(PIS)を導入することによって生成することができる。PISは、標的核酸からクローニングされる核酸の上流末端を確立する。パッケージングシステムによるPISの認識は、PISでのパッケージングの開始をもたらしてもよく、これは、標的核酸に及ぶ、PISから下流の核酸のクローニングをもたらす。核酸のライブラリーは、標的核酸から複数の核酸をクローニングすることによって産生することができる。ゲノムライブラリーは、ゲノムから複数の核酸をクローニングすることによって産生することができる。
【0023】
1.パッケージングシステム
核酸は、パッケージング開始部位(PIS)でのパッケージングを開始することによって、標的核酸からクローニングすることができる。PISは、ターミナーゼによって認識されてもよく、これは、部位特異的にまたはランダムに標的核酸に切れ目を入れてもよく、これは、カプシドの中に核酸を挿入してもよい。したがって、クローニングされた核酸は、カプシド中にパッケージすることができる。ターミナーゼは、2つのタンパク質を含んでもよく、これらは、パッケージングシステムが機能することを可能にするATPアーゼ活性を有していてもよい。
【0024】
カプシドは、5〜160kbのサイズであってもよい核酸をパッケージすることができてもよい。複数のカプシドは、それぞれ、核酸をパッケージすることができてもよく、カプシドによってパッケージされる核酸は、ほぼ同じサイズであってもよい。サイズ選択のそのような形態は、PFGE電気泳動と関連する非効率に対する主な改善である。さらに、カプシドの感染特性は、後の操作において、エレクトロポレーションよりも高い率で、パッケージされた核酸の導入を可能にすることができる。
【0025】
使用されるパッケージングシステムは、真核生物または原核生物のものであってもよい。たとえば、システムは、表1に挙げられるバクテリオファージから選択することができる。
【0026】
【表1】

パッケージングシステムはまた、ポックスウイルス、ヘルペスウイルス、アデノウイルス、レンチウイルス、またはエプスタイン−バーウイルスから選択することができる。
【0027】
a.パッケージング開始部位
PISは、パッケージング開始部位であってもよい。パッケージング開始部位の代表的な例は、バクテリオファージλ、P1、P7、T4、T7、Φ29、またはP22パッケージング開始部位を含むが、これらに限定されない。PISは、pac部位またはcos部位であってもよい。PISはまた、長末端反復などの真核生物ウイルスPISであってもよい。
【0028】
PISは、標的核酸の中に、PISを含む第1の核酸を導入することによって、標的核酸の中に導入することができる。第1の核酸はまた、ベクターエレメントを含んでもよい。第1の核酸はまた、転位性エレメントを含んでもよい。転位性エレメントは、PISを含む核酸セグメントの側面に位置する1対の転位性末端を含んでもよい。転位性エレメントは、Tn7、Tn5、Tn10、Minos、Mu、およびMarinerを含むが、これらに限定されない、システムからの転位性末端を含んでもよい。核酸セグメントはまた、ベクターエレメントを含んでもよい。
【0029】
第1の核酸は、ランダム方式または非ランダム方式のいずれかにおいて標的核酸の中に導入することができる。PISを含む第1の核酸はまた、突然変異誘発を含むが、これに限定されない多くの標準的な分子生物学手法のいずれかによって、標的核酸の中に導入することができる。PISを含む第1の核酸は、in vitroにおいてまたはin vivoにおいて、転位によって標的核酸の中に導入することができる。第1の核酸は、標的DNAの中への転位性エレメントの転位を可能にする任意の形態をしていてもよい。第1の核酸は、線状DNAまたは環状DNAの形態をしていてもよく、これは、スーパーコイル核酸またはリラックス核酸であってもよい。第1の核酸は、プラスミドの一部であってもよい。第1の核酸はまた、バクテリオファージを含むが、これに限定されない複製可能な遺伝子パッケージであってもよい。
【0030】
トランスポサーゼは、標的核酸の中への第1の核酸の転位を触媒するために、in vitroにおいてまたはin vivoにおいて使用することができる。転位は、ランダムに生じてもよく、その程度は、特定のトランスポゾンシステムに依存して異なっていてもよい。
【0031】
転位は、たとえば、市販で入手可能な転位システムを使用して、in vitroにおいて生じてもよい。転位はまた、米国特許第5,965,443号、第5,948,622号、または第5,925,545号において開示される方法を使用して、in vitroにおいて生じてもよく、これらの内容は、参照によって本明細書において組み込まれる。in vitro転位については、標的核酸は、染色体DNA、消化DNA、剪断DNA、サイズ特異的DNA、および全ゲノムを含むが、これらに限定されない、核酸の任意の形態であってもよい。
【0032】
転位はまた、トランスポサーゼを発現する宿主細胞の中に第1の核酸を導入することによってin vivoにおいて生じてもよい。第1の核酸は、エレクトロポレーションおよび感染を含むが、これらに限定されない方法によって宿主細胞の中に導入することができる。in vivo転位については、標的核酸は、レプリコンを含む、核酸の任意の形態であってもよい。レプリコンは、ベクターまたは染色体であってもよい。
【0033】
PISを含む第1の核酸はまた、標的核酸に第1の核酸をライゲーションすることによって、標的核酸の中に導入することができる。ライゲーションされた産物は、線状または環状であってもよい。標的核酸は、in vitro転位について上記に説明される核酸の形態を含むが、これらに限定されない、核酸の任意の形態であってもよい。標的核酸は、ランダム剪断および制限消化を含むが、これらに限定されない方法によって生成することができる。第1の核酸および標的核酸は、適合性の末端を有していてもよい。その代わりに、リンカーが使用することができる。
【0034】
標的核酸は、HindIIIなどの第1の制限酵素を用いて消化されてもよく、これは、部分的であってもよい。標的核酸はまた、第2の制限酵素を用いて消化することができる。第1の核酸は、消化された標的核酸と適合性の末端を有していてもよい。第1の核酸および標的核酸をライゲーションした後に、PISを含む、結果として生じる標的核酸は、ファージパッケージングのための基質としてもよい。
【0035】
PISは、標的核酸に固有のものであってもよく、または標的DNA中に先在していてもよい。PISはまた、DNA切断によって標的核酸の中に導入することができる。切断は、ターミナーゼと標的核酸を接触させることによって行なわれてもよく、これは、ランダムな位置で標的核酸を切断してもよい。ターミナーゼは、ファージP22からのなどのように、ファージターミナーゼであってもよく、これは、高形質導入P22であってもよい。ターミナーゼは、ファージP22のポリペプチドGp3およびGp2を含んでもよい。
【0036】
b.ベクターエレメント
PISは、ベクターエレメントと共に標的核酸の中に導入することができる。ベクターエレメントは、PISの下流または上流にあってもよい。下流PISは、クローニングされた核酸中に含まれていてもよい。クローニングされた核酸中のベクターエレメントは、クローニングDNAの、後の操作および増殖のために使用することができる。
【0037】
(1)複製開始点
ベクターエレメントは、複製開始点であってもよい。複製開始点は、低コピー複製開始点であってもよい。低コピー複製開始点はまた、再編成を最小限にしながら、宿主細胞中のクローニングDNAを維持するために使用することができる。低コピー複製開始点の代表的な例は、oriSである。
【0038】
複製開始点はまた、高コピー複製開始点であってもよい。高コピー複製開始点は、より多量のクローニングDNAを産生するために使用することができる。低コピー複製開始点の代表的な例は、oriVである。
【0039】
ベクターエレメントはまた、高コピー複製開始点および低コピー複製開始点であってもよい。高コピー複製開始点は、誘発性プロモーターのコントロール下としてもよい。クローニングされた核酸は、低コピー複製開始点を使用して維持できるが、高コピー複製開始点の誘発によって多量に産生することができる。そのようなシステムは、Hradecnaら1998年およびWildら1998年によって記載され、これらの内容は、参照によって本明細書において組み込まれる。
【0040】
(2)マーカー
ベクターエレメントはまた、選択可能マーカーまたは可視マーカーであってもよい。選択可能マーカーまたは可視マーカーは、クローニングDNAを維持し、かつ操作するために使用することができる。マーカーの代表的な例は、AmpR、CamR、KanR、lacZ、およびGFPを含むが、これらに限定されない。
【0041】
(3)組み込み部位
ベクターエレメントはまた、組み込み部位であってもよい。組み込み部位は、相補的な組み込み部位を含む第2の核酸の中にクローニングされた核酸を組み込むことを可能にすることができる。第2の核酸はまた、ベクターエレメントを含んでもよい。総合システムの使用は、いくつかの重要な利点を有する。クローニングされた核酸の、後の操作に必要とされる多くの配列が、第1の核酸の代わりに複製プラスミドベクター上に配置することができる。このことから、クローニングされた核酸をパッケージするためにカプシドを使用する場合に、より多くの標的核酸をクローニングすることが可能となる可能性がある。
【0042】
組み込み部位および相補的な組み込み部位の例は、λシステムおよびCre−loxシステムを含むが、これらに限定されない。λファージが、E.coliに感染する場合、それは、細胞を溶解して発達する可能性があり、または細菌染色体中の特異的なポイント、attBにおいて組み込むことによって、溶原菌を確立する可能性がある。組み込みは、λ上のattPおよびattBの間の部位特異的組換えによって媒介され、連続した染色体の中へのλの線状の挿入をもたらす。attPは、約250bp長であるが、attBは、わずか約20bpである。ファージint遺伝子の産物は、宿主因子IHFと共に、attPおよびattBの間のクロスオーバー反応を触媒し、それによって組み込みを引き起こす。
【0043】
第1の核酸は、PISの下流にattBを含んでもよく、第2の核酸は、複製可能であり、attPを含む。パッケージングを開始した後に、クローニングされたものは、複製可能な第2の核酸の中に組み込むことができる。同様の手法を用いて、DNA断片は、Janusベクターの中に挿入された(Burlandら1993年)、これは、参照によって本明細書において組み込まれる。
【0044】
c.λパッケージング
パッケージングシステムは、パッケージングを開始し終了するための特異的な部位を使用してもよい。正常なファージの発達において、ビリオン当たりのDNAの長さは、頭部の容量によって直接決定されないので、λは「ヘッドフル」タイプのファージではない。代わりに、λは、パッケージングを開始し、かつ終了するために高度に特異的なcos部位を使用する。これらの部位は、ターミナーゼによって切断されて、パッケージDNAの末端に12塩基突出(cosLおよびcosR)を残す。λの自然のローリングサークルパッケージング基質中で、cos部位は、頭部サイズによって決定されるフルパッケージングリミットよりも、共に近くに、間隔をおいて配置される。その結果として、頭部容量ではなくcos部位間隔が、通常、ビリオンDNAの長さを決定する。
【0045】
d.ヘッドフルパッケージング
「ヘッドフル」パッケージングシステムもまた使用することができる。ヘッドフルパッケージングシステムは、線状の連続移動方式において、ファージプロヘッドの空洞の中にクローニングされた核酸を供給し、それが、内部のDNAが、進行を停止するのに十分な、内壁に対する圧力をかける限界に達するまで、頭部を拡張させてもよい。これは、頭部における立体構造変化を誘発してもよく、これは、入ってくるDNAのエンドヌクレアーゼによる切断を活性化し、感染粒子を作製するためのファージ尾部の付着を可能にする。いっぱいの頭部は、狭いサイズ範囲内のDNA分子を含有してもよい。カプシドの容量は、パッケージDNAに対する最大サイズの制限を設定してもよい。代表的なヘッドフルパッケージングシステムは、P1、P7、T4、KVP40、P22、φ29、およびT7を含むが、これらに限定されない。
【0046】
(1)P1
バクテリオファージP1およびP7は、110〜115kbのヘッドフル容量を有する。パッケージング開始は、pac部位で生じ、頭部がいっぱいになり、非特異的末端の切断が成されるまで続く。市販で入手可能な段階1 P1パッケージング抽出物またはpacase抽出物は、λのcos部位に類似のpac部位でパッケージされるようにDNAを切断してもよい。それは、am10.1突然変異、P1遺伝子10中のナンセンス突然変異を含有してもよい。それは、頭部および尾部のタンパク質を含むすべての後期ファージタンパク質合成を欠く。段階2抽出物は、頭部の中にDNAをカプシド形成するファージパッケージングタンパク質を含有し、尾部を追加してもよい。それは、pac−切断活性を欠く、遺伝子9のam9.16ナンセンス突然変異を含有してもよい。
【0047】
パッケージング株はまた、2段階パッケージング反応のために構築することができる。パッケージング株構築のために、ナンセンス突然変異は、主な尾管タンパク質をコードするTubまたは主な尾鞘タンパク質をコードする遺伝子22を含むが、これに限定されない主な尾部遺伝子のうち1つにおいて作り出すことができる。これにより、pacase−頭部成熟/尾部欠乏段階1パッケージング抽出物をもたらしてもよい。段階2pacase−頭部欠乏/尾部成熟株は、P1の主な頭部タンパク質をコードする遺伝子23中にナンセンス突然変異を導入することによって、構築することができる。P1ゲノム内にCamrマーカーを置換することもまた望ましい可能性がある。
【0048】
cre−lox部位特異的組換えシステムは、P1パッケージクローニングされた核酸を環状にするために使用することができる。第1の核酸は、pacパッケージング開始配列の下流にloxP部位を含んでもよい。クローニングされた核酸の転位およびパッケージングの後に、P1頭部からはみ出たDNAは、切り取られてもよく、第2のloxP組換え部位を含有する、λについて提唱されるようなリンカーは、この時、追加することができる。段階2パッケージング反応の間の尾部の追加に続いて、パッケージDNAはCre+宿主の中に注入することができる。宿主細胞の中への注入に続いて、DNAは、Creリコンビナーゼによる2つのファージloxP部位の間の組換えによって環状にすることができる。これは、環状のクローニングされた核酸が複製され、安定して維持されることを可能にすることができる。
【0049】
(2)T4
バクテリオファージT4は、169kbのゲノムサイズを有する、最大の、十分に特徴づけられたファージのうち1つである。P1およびP7のように、T4は、ヘッドフル方式においてそのDNAをパッケージし、パッケージングメカニズムの構成要素は、徹底的に研究されてきた。λ、P1、およびP7と異なり、T4 DNAのパッケージングは、特異的な部位で開始しない。T4のためのin vitroパッケージングシステムが開発されてきた。T4 in vitroシステムは、外来DNAを、効率的にパッケージすることができ、160kbの長さまでの大きなDNA断片をクローニングするのに適応させることができる。
【0050】
P1およびP7 in vitroパッケージングについて上記に記載される同様の方法は、T4パッケージクローンを環状にするために使用されるcre−lox部位特異的組換えシステムを使用して、利用することができる。T4システムの1つの利点は、より大きな変異体およびより小さな変異体の両方の多くの頭部長変異体の同定であり、パッケージされるDNAの量は、適宜、変えられる。特定の頭部長突然変異と組み合わせてパッケージング株を使用することは、クローニングされた核酸の可能なサイズ範囲を拡張するのに有益であると分かる可能性がある。
【0051】
(3)KVP40
KVP40は、T4関連のものであると比較ゲノム学によって示された広い宿主範囲のビブリオファージである。それは、245kbのゲノムサイズを有する、大きく、尾がある、二本鎖DNAファージである。このファージの遺伝的性質および生化学的活性に関して現在、ほとんど知られていないが、KVP40のゲノム配列および構成は、T4との広範囲な保存の領域を示す。最大の保存領域のうち1つは、ビリオン構造遺伝子を構成する遺伝子クラスター中にあり、それらのDNAパッケージングメカニズムにおける類似性を同様に示唆する。これは、KVP40を、in vitroパッケージングを使用するクローニングシステムの候補にする。
【0052】
KVP40 in vitroパッケージングシステムの開発はこの拡張のために必要とされるだろう。主な構成要素が十分に特徴づけられているT4 DNAパッケージングについての知識の増加と共に、類似の株は、KVP40のin vitroパッケージング抽出物のために開発される可能性がある。推定上のターミナーゼ、頭部、および尾部に関連する遺伝子は既に同定されており、株構築のための可能な突然変異誘発標的を確実に提供するであろう。
【0053】
KVP40の宿主範囲は、少なくとも8つのビブリオ種および1つのフォトバクテリウム種を含むが、それは、より望ましい宿主であるE.coliに感染しない。V.parahaemolyticusのompK遺伝子は、KVP40の宿主受容体として同定された外膜タンパク質(OMP)をコードする。JM109などのE.coli株におけるompK遺伝子の発現は、KVP40による感染を可能にする。
【0054】
(4)P22
バクテリオファージP22は、Salmonella typhimuriumの株に感染する可能性があるポドウイルスである。P22は、大サブユニット(Gp2)および小サブユニット(Gp3)を含むターミナーゼを用いるヘッドフルメカニズムを使用して、約42kbと同じくらいのDNAをパッケージする。ターミナーゼは、pac部位またはpac様部位でDNAに結合し、切断してもよく、次いで、ウイルスプロカプシドの中へのDNAのATP加水分解ベースの転座に関与してもよい。プロカプシドがいっぱいになるまで、DNAは、プロカプシドの中にパッケージすることができる。パッケージングの間に、カプシドシェルは広がり、立体構造変化を受けてもよい。パッケージングに続いて、ターミナーゼは、再び、パッケージDNAの末端を切断してもよく、次いで、放出され、なお、切断された、非パッケージDNAに付着してもよい。次いで、ターミナーゼは、新しいプロカプシドの中に挿入され、連続サイクルにおいて隣接標的核酸断片をパッケージするプロセスを続けてもよい。
【0055】
(a)P22突然変異体
P22は、高形質導入P22(P22HT)などのP22突然変異体であってもよい。P22HT突然変異は、遺伝子3において生じてもよく、これは、突然変異体Gp3タンパク質をもたらしてもよい。P22HTは、HT105/1突然変異を持っていてもよい。P22HTターミナーゼによるパッケージングの開始は、明らかな配列特異性をほとんどまたはまったく有していなくてもよく、したがって、より低い特異性でDNAを認識してもよい。ターミナーゼは、ランダムな位置でDNAに結合し、切断してもよく、次いで、ウイルスプロカプシドの中へのDNAのATP加水分解ベースの転座に関与してもよい(図7)。P22HTによってパッケージされる標的核酸は、46kbであってもよい。
【0056】
P22突然変異体は、また所与の標的核酸に沿っての処理能力が増加していてもよい。突然変異体は、標的核酸から10までのカプシドをパッケージすることができる可能性がある。P22突然変異体もまた、E.coliに感染することができる可能性がある。
【0057】
(5)φ29
φ29は、Bacillus subtilisの株に感染できるバクテリオファージである。ファージは、扁長20面体カプシドの内部に約30kbと同じくらいのDNAを詰めてもよい。DNAは、頭部−尾部コネクター(Gp10)、パッケージングATPアーゼ(Gp16)、およびRNA分子の環(pRNA)を含むATP依存性モーターによってプロカプシドの中に転座することができる。
【0058】
(6)λパッケージングおよびDoc粒子
パッケージされる核酸は、まれに、間隔をおいて配置されるcos部位を含有してもよい。パッケージングは、cos部位、cosLで始まってもよく、これは、切断されて、頭部の中に挿入される12塩基突出を残す。cos部位は、標的核酸の末端にライゲーションされた第1の核酸によって、標的核酸の末端に存在していてもよい。パッケージングは、頭部がいっぱいになるまで、一方向に進んでもよい。ターミナーゼが、切断するための第2のcos部位を見つけない場合、プロセスは、DNAが頭部から外にはみ出すと共に止まってもよい。はみ出たDNAは、DNアーゼIなどのヌクレアーゼまたはSau3Aなどの頻繁に切断する制限酵素によって非特異的に除去することができる。ヌクレアーゼがはみ出たDNAを切断する位置は、ランダムであってもよい。
【0059】
次いで、ファージ尾部を付着させて、λdocLと呼ばれる、たった1つのcos部位を有する粒子を産生してもよい。これらの粒子は、E.coliの中にそれらのDNAを効率的に注入する可能性があるが、それらは、λ分子の右の末端に通常、見つけられる付着末端であるcosRを欠くので、それは、細胞に入る際、効率的に環化しなくてもよい。λdocL粒子は、組み込みによって宿主細胞の中に核酸を導入するために使用することができる。尾部追加の代案として、第2のパッケージングが行なわれてもよい。ヌクレアーゼ消化およびDNA抽出の後に、リンカーをライゲーションし、後の産物を再度パッケージし、形質移入し、クローンを選択することができる。
【0060】
loxPシステムは、尾部追加の代案としてクローンのin vitro環状化のために使用することができる。loxP部位は、第1の核酸上のPISの下流に追加することができる。クローニングされた核酸は、in vitroパッケージングによってサイズ選択され、抽出され、loxP部位を含有するリンカーは、ライゲーションすることができる。クローンは、in vitroCreリコンビナーゼによって環状にされ、プラスミドとして安定して維持されるように、宿主細胞の中にエレクトロポレーションすることができる。
【0061】
2.パッケージDNAの単離
カプシド形成後に、パッケージDNAは、カプシドを単離することによって回収することができる。カプシドは、等密度遠心分離もしくは速度遠心分離または差次的沈降を含むが、これらに限定されない方法によって単離することができる。カプシドはまた、アフィニティー精製によって単離することができ、これは、拡張されたカプシドの表面上に含有されるタンパク質のエピトープに特異的に結合することができる抗体を使用することによって行なわれてもよい。たとえば、カプシドタンパク質は、λファージのDタンパク質であってもよい。
【0062】
抗カプシドタンパク質抗体は、固体支持体に付着することができる。固体支持体は、ビーズ、微粒子、カラム、試験ストリップ、カートリッジ、マイクロタイタープレート、顕微鏡スライドガラス、またはナイロン、ニトロセルロース、もしくは他の適した材料などの膜であってもよい。微粒子は、0.2μm〜7.0μmのサイズであってもよい。微粒子はまた、ハプテン化することができる。微粒子はまた、少なくとも1つまたは2つの蛍光標識によって含浸させてもよい。微粒子はまた、約0.1μm未満のサイズの強磁性流体または磁性粒子を形成することができてもよい。微粒子はまた、ろ過によって収集可能、または除去可能である可能性がある。
【0063】
パッケージDNAは、フェノール抽出などによってカプシドから単離することができる。抽出に先立って、DNアーゼIなどのヌクレアーゼは、非パッケージDNAまたは非標的DNAを消化するために使用することができる。フェノールは、カプシドを含む溶液に追加することができる。この混合物を遠心分離した後に、水相は、有機相から分離されてもよく、クロロホルムが追加することができる。この混合物を遠心分離した後に、水相は、有機相から分離することができる。単離DNAは、水相に冷エタノールを追加することによって、水相から濃縮され、その後、遠心分離が続けられてもよい。DNAは、1回または複数回、エタノールを用いて洗浄してもよく、水または緩衝液中に再懸濁することができる。フェノール−クロロホルム抽出は、少なくとも1回、繰り返することができる。
【0064】
パッケージDNAはまた、DNA結合カラムを使用して、カプシドから単離することができる。たとえば、QIAGENラムダ手順(QIAGEN、Valencia、Ca)が使用することができる。非パッケージDNAまたは非標的DNAは、ヌクレアーゼを用いて消化することができる。カプシドは、沈澱され、次いで、溶解することができる。単離DNAは、カラムに結合させ、洗浄してもよい。DNAは、カラムから溶出され、イソプロパノールなどのアルコールを用いて溶液から沈澱することができる。DNAは、水または緩衝液中に再懸濁することができる。
【0065】
カプシドが、カプシドタンパク質アフィニティーカラムを使用して単離される場合、カプシドは、溶出され、溶解することができる。パッケージDNAは、上記に記載されるように、カラムベースのシステムを使用して、フェノール抽出することができ、または単離することができる。
【0066】
単離DNAの末端は、フィルインされてもよく、これは、DNAポリメラーゼを使用することによって行なわれてもよい。アダプター挿入断片は、DNAの末端にライゲーションされてもよく、これは、環状DNAをもたらしてもよい。ライゲーション反応は、アダプター挿入断片のそれぞれの末端が、単離DNA分子の末端にライゲーションするのに十分な濃度のアダプター挿入断片を含んでもよい。アダプター挿入断片の末端は、リン酸化されなくてもよく、これは、アダプター挿入断片がモノマー環を形成するのを予防してもよい。ライゲーション反応は、油中水型エマルション中で行なわれてもよい。エマルションの水性液滴は、環状にされる約1つの単離DNA分子を含有してもよい。
【0067】
アダプター挿入断片は、2つのアダプタープライマー結合部位を含んでもよく、これは、tail−to−tailなどのように、反対の向きを有していてもよい。結合部位は、10〜50または16〜25塩基対長であってもよい。アダプタープライマー挿入断片はまた、増幅およびヌクレオチド配列決定の両方を支援することができるプライミング領域を含んでもよい。それらの部位に結合してもよいプライマーは、国際公開第2007/145612 A1号または米国特許第7,115,400号もしくは第7,323,305号において記載されるような配列を有していてもよく、これらの内容は、参照によって本明細書において組み込まれる。たとえば、リーチングプライマー(reaching primer)は、プライマーAおよびプライマーBであってもよい。アダプター挿入断片はまた、454配列決定システム(454 Life Systems、Branford、CT)上でうまく見つけるために使用することができる、短い(たとえば4ヌクレオチド)「配列決定キー」配列を含んでもよい。アダプター挿入断片はまた、セパレーターエレメントを含んでもよく、これは、知られている配列を有していてもよい。配列は、ローリングサークル増幅のためのプライミング部位を含んでもよい。配列はまた、単離DNAの2つの末端を同定するために使用することができる。単離DNAの、後の配列決定の間に、セパレーターエレメントの配列は、全単離DNAが配列決定されたことを示してもよい。アダプター挿入断片は、複製開始点を含んでもよく、かつさらにマーカーを含んでもよい。
【0068】
アダプター挿入断片はまた、平滑末端切断制限酵素などの制限酵素のための認識配列を含んでもよく、この配列は、2つのアダプタープライマー結合部位の間に位置してもよい。単離DNAにアダプター挿入断片をライゲーションする際に、結果として生じるDNAは、アダプタープライマー結合部位の間でDNAを切断することができる制限酵素を用いて消化することができる。次いで、アダプター末端は、結果として生じる線状DNAの末端にライゲーションすることができる。
【0069】
アダプター挿入断片はまた、「リーチング」制限酵素認識部位を含んでもよい。認識部位は、プライマー結合部位に隣接していてもよい。アダプター挿入断片はまた、2つのアダプタープライマー結合部位の側面に位置するリーチング制限酵素認識部位を含んでもよく、認識部位は、制限酵素認識部位から離れて、逆方向に向けられてもよい。リーチング制限酵素は、クラスIIS酵素であってもよく、これは、Fok I、Alw26 I、Bbv I、Bsr I、Ear I、Hph I、Mme I、Mbo II、SfaN I、またはTth111 Iであってもよい。リーチング制限酵素はまた、クラスIII制限酵素であってもよく、これは、EcoP15 I、EcoP I、Hinf III、またはStyLT Iであってもよい。単離DNAにアダプター挿入断片をライゲーションする際に、リーチング制限酵素は、単離DNA内の位置でなどのように、認識部位から離れた位置でDNAを切断してもよい。アダプター挿入断片はまた、特異的な結合メンバーを含んでもよく、これは、ビオチンであってもよい。
【0070】
単離DNAに挿入断片をライゲーションした後に、結果として生じる環状DNAは、リーチング制限酵素を用いて消化されてもよく、これは、線状DNA断片をもたらしてもよく、これは、それぞれの末端上に、単離DNAの末端に対応し得る配列を含んでもよい。DNA断片は、環状にされてもよく、これは、DNA断片の末端をフィルインし、次いで、ライゲーションを行なうことによって行なわれてもよい。
【0071】
アダプタープライマー結合部位を含むアダプター末端はまた、単離DNAの末端にライゲーションすることができる。アダプター断片は、増幅およびヌクレオチド配列決定の両方を支援することができるプライミング領域を含んでもよく、454配列決定システム(454 Life Systems、Branford、CT)上でうまく見つけるために使用することができる、短い(たとえば4ヌクレオチド)「配列決定キー」配列を含んでもよい。部位に結合することができるプライマーは、プライマーAまたはプライマーBであってもよい。アダプター末端は、オリゴヌクレオチド配列Yを有する第1のアダプター末端であってもよい。アダプター末端はまた、オリゴヌクレオチド配列Zを有する第2のアダプター末端であってもよい。アダプター末端は、固体支持体に核酸を固定するための手段を含んでもよい。
【0072】
アダプター末端は、縮重2塩基一本鎖3’突出を有していてもよい。縮重は、2つの突出塩基が、ランダムであってもよいこと(つまり、それらは、それぞれ、G、A、T、またはCであってもよいこと)を意味し得る。アダプター末端は、単離DNAのリーチング制限酵素切断末端へのアダプターの、方向性をもつライゲーションを強く促進するために設計することができる。アダプター末端は、多量のモル過剰のアダプター末端(15:1アダプター末端:単離DNA比)を含有してもよいライゲーション反応において単離DNA末端と組み合わせられてもよく、これは、単離DNA末端の利用を最大限にしてもよく、単離DNAコンカテマーを形成する可能性を最小限にしてもよい。アダプター末端は、リン酸化されなくてもよく、これはアダプター末端ダイマーの形成を最小限にしてもよい。ライゲーションに続いて、ライゲーション産物は、鎖置換DNAポリメラーゼの使用などによって、フィルイン反応の使用によって修復することができる。DNAポリメラーゼは、Bst DNAポリメラーゼ(Large Fragment)、φ29DNAポリメラーゼ、DNAポリメラーゼI(Klenow Fragment)、またはVent(登録商標)DNAポリメラーゼであってもよい。
【0073】
アダプター末端は、米国特許第7,323,305号において記載されるように設計されてもよく、これらの内容は、参照によって本明細書において組み込まれる。アダプター末端は、ホスホジエステル結合の代わりにホスホロチオネート結合を含んでもよい。アダプター末端はまた、特異的な結合メンバーを含んでもよく、これは、ビオチンであってもよい。特異的な結合メンバーは、第1のアダプター末端に追加されてもよく、これは、5’末端であってもよく、特異的な結合メンバーは、第2のアダプター末端に追加されなくてもよい。第1および第2のアダプター末端の単離DNAへのライゲーションに続いて、単離DNAは、2つの第1のアダプター末端、1つの第1のアダプター末端および1つの第2のアダプター末端、ならびに2つの第2のアダプター末端を含んでもよい。アダプター末端ライゲーション単離DNAは、ストレプトアビジンなどの第1のアダプター末端の特異的な結合メンバーのための特異的な結合パートナーに結合された固体基質と接触させてもよい。固体基質は磁性ビーズであってもよい。
【0074】
固体基質とアダプター末端ライゲーション単離DNAを接触させる際に、少なくとも1つの第1のアダプター末端を含む単離DNAのみが結合するであろう。1つの第1のアダプター末端のみを含む単離DNAは、単離DNAの一方の末端で結合するであろうが、2つの第1のアダプター末端を含む単離DNAは、2つの末端で結合するであろう。非結合単離DNAは、固体基質から洗い流することができる。固体基質は、低塩類(「融解」または変性)溶液にさらされてもよく、これは、1つの第1のアダプター末端のみを含む単離DNAのみを放出してもよい。2つの第1のアダプター末端を含む単離DNAは、固体基質に結合したままであろう。固体基質から放出される単離一本鎖DNAは、PCR増幅および配列決定においてなどのように、さらなる使用のために収集することができる。
【0075】
3.パッケージDNAの増殖
カプシド形成後に、クローニングされた核酸はまた、適切な宿主細胞に感染させるために使用することができる。宿主細胞において、クローニングされた核酸は、環状にされ、増殖することができる。次いで、クローニングされた核酸は、DNAの単離のために標準的な手法を使用して単離することができる。クローニングされた核酸のIS混入を予防するために、宿主株は、ISエレメントを含んでいなくてもよい。
【0076】
クローニングされた核酸が環状ではない場合、それは、カプシドから単離され、in vitroにおいて環状にすることができる。環状化は、線状DNAの末端と適合性の1つもしくは複数のリンカーのライゲーションまたは組換えを含むが、これらに限定されない方法によって行なわれてもよい。
【0077】
環状核酸は、oriおよび選択可能マーカーまたは可視マーカーを含んでもよい。環状核酸は、細菌などの宿主細胞中で増殖させることができる核酸であってもよく、細菌人工染色体(BAC)であってもよい。環状核酸は、宿主細胞の中に形質転換することができる。
【0078】
4.単離DNAの増幅
単離DNAは、PCRなどによって増幅することができる。単離DNAが環状にされている場合、それは、アダプタープライマー結合部位の間に位置する部位で、DNAを切断することができる制限酵素を用いてそれを消化することによって線状にすることができる。
【0079】
a.エマルションPCR
アダプタープライマー結合部位を含む単離DNAは、ビーズエマルションPCR増幅(emPCR)の使用などによってPCR増幅することができる。emPCRは、米国特許第7,323,305号および国際公開第2007/145612号において記載されるように行なわれてもよく、これらの内容は、参照によって本明細書において組み込まれる。単離DNAは、一本鎖であってもよく、アダプタープライマーにアニールすることができる。アダプタープライマーは、固体支持体に付着させてもよく、これは、球状ビーズであってもよい。固体支持体は、複数の結合アダプタープライマーを含んでもよい。固体支持体は、水性反応混合物中に懸濁され、次いで、油中水型エマルション中にカプセル化することができる。エマルションは、分散した水相微小滴を含んでもよく、これは、60〜100μmの直径であってもよく、耐熱性の油相によって包まれてもよい。それぞれの微小滴は、増幅反応溶液(つまり核酸増幅のために必要な試薬)を含有してもよい。増幅は、PCR反応ミックス(ポリメラーゼ、塩類、dNTPs)ならびに1対のアダプタープライマー(プライマーAおよびプライマーB)を含んでもよい。微小滴集団のサブセットはまた、DNA鋳型を含むDNAビーズを含有してもよい。微小滴のこのサブセットは、増幅のための基本となってもよい。このサブセット内にないマイクロカプセルは、鋳型DNAを有していなくてもよく、増幅に参加しなくてもよい。増幅技術は、PCRであってもよく、PCRプライマーは、非対称PCRを行なうために8:1または16:1の比で存在する(つまり、1つの第2のプライマーに対して8または16の第1のプライマー)。
【0080】
単離DNAは、ビーズに固定することができるオリゴヌクレオチド(プライマーB)にアニールすることができる。サーモサイクリングの間に、一本鎖DNA鋳型およびビーズ上の固定Bプライマーの間の結合は切断される可能性があり、これは、周囲のマイクロカプセル化溶液の中に鋳型を放出する可能性がある。増幅溶液は、追加の溶液相プライマーAおよびプライマーBを含有してもよい。結合反応速度が、固定プライマーよりも溶液相プライマーについてより速いので、溶液相Bプライマーは、鋳型の相補的b’領域に容易に結合してもよい。初期PCRにおいて、A鎖およびB鎖の両方は、等しく十分に増幅することができる。
【0081】
中期増幅(つまり、サイクル10および30の間)によって、Bプライマーは、消耗されてもよく、これは、指数関数的増幅を停止させてもよい。次いで、反応は、非対称増幅に入ってもよく、単位複製配列集団は、A鎖が優位を占めるようになってもよい。後期相増幅において、30〜40のサイクル後に、非対称増幅は、溶液中でA鎖の濃度を増加させてもよい。過剰A鎖は、ビーズ固定Bプライマーにアニールし始める。次いで、耐熱性ポリメラーゼは、単位複製配列の固定ビーズ結合B鎖を合成するために、鋳型としてA鎖を用いる。
【0082】
終期増幅において、熱サイクリングを続けると、ビーズ結合プライマーへのさらなるアニーリングが強いられる可能性がある。溶液相増幅はこの段階で最小限となる可能性があるが、固定B鎖の濃度は増加する可能性がある。次いで、エマルションは、破壊されてもよく、固定産物は、変性によって(熱、pHなどによって)一本鎖にされてもよく、これは、相補的A鎖を取り出してもよい。Aプライマーは、固定鎖のA’領域にアニールされてもよく、固定鎖に、配列決定酵素および任意の必要な補助タンパク質が載せられてもよい。次いで、ビーズは、米国特許第6,274,320号、第6258,568号、および第6,210,891号において記載されるものなどのように、認識されるピロリン酸技術を使用して配列決定されてもよく、これらの内容は、参照によって本明細書において組み込まれる。
【0083】
(1)捕捉ビーズへのアダプタープライマーの結合
アダプタープライマーは、捕捉ビーズに付着することができる。プライマーは、当技術分野において知られている任意の方式において固体支持体捕捉ビーズに付着することができる。多数の方法は、微視的ビーズなどの固体支持体にDNAを付着させるために当技術分野において存在する。ビーズへのDNAの共有化学結合は、ホスホアミデート結合を通してDNA上の5’−リン酸をアミノコート捕捉ビーズに結合するために、水溶性カルボジイミドなどの標準的なカップリング剤の使用によって成し遂げることができる。他の代案は、同様の化学作用を使用して、第1に特異的なオリゴヌクレオチドリンカーをビーズにカップルし、次いで、DNAをビーズ上のリンカーに結合するためにDNAリガーゼを使用することである。ビーズにオリゴヌクレオチドをつなぐための他の結合化学作用は、N−ヒドロキシスクシンアミド(NHS)およびその誘導体の使用を含む。そのような方法において、オリゴヌクレオチドの一方の末端は、固体支持体と共有結合を形成する反応基(アミド基など)を含有してもよく、リンカーの他方の末端は、固定されるオリゴヌクレオチドと結合することができる第2の反応基を含有する。オリゴヌクレオチドは、共有結合によってDNA捕捉ビーズに結合することができる。しかしながら、キレート化または抗原抗体複合体などの非共有結合もまた、ビーズにオリゴヌクレオチドをつなぐために使用することができる。
【0084】
制限酵素部位からのオーバーラップ末端またはバクテリオファージラムダベースのクローニングベクターの「付着末端」などのように、DNA断片の末端でユニークな配列に特異的にハイブリダイズするオリゴヌクレオチドリンカーが、利用することができるが、平滑末端ライゲーションもまた有益に使用することができる。これらの方法は、米国特許第5,674,743号において詳細に記載される。ビーズを固定するために使用される方法は、本発明の方法におけるステップの全体にわたって、固定オリゴヌクレオチドを結合させるために続けてもよい。
【0085】
それぞれの捕捉ビーズは、単離DNAの一部を認識する(つまり、相補的である)複数のプライマーを有するように設計されてもよく、単離DNAは、したがって、捕捉ビーズにハイブリダイズされる。単離DNAのクローン増幅を成し遂げるために、1つのユニークな単離DNAのみがいずれか1つの捕捉ビーズに付着してもよい。
【0086】
本明細書において使用されるビーズは、任意の好都合なサイズであってもよく、任意の数の知られている材料から製造することができる。そのような材料の例は、無機物、天然ポリマー、および合成ポリマーを含む。これらの材料の特定の例は、セルロース、セルロース誘導体、アクリル樹脂、グラス、シリカゲル、ポリスチレン、ゼラチン、ポリビニルピロリドン、ビニールおよびアクリルアミドのコポリマー、ジビニルベンゼンと架橋されたポリスチレン、またはその他同種のもの(たとえばMerrifield、Biochemistry 1964年、3巻、1385〜1390頁において記載される)、ポリアクリルアミド、ラテックスゲル、ポリスチレン、デキストラン、ゴム、シリコン、プラスチック、ニトロセルロース、天然スポンジ、シリカゲル、制御孔ガラス、金属、架橋デキストラン(たとえばSephadex(商標))アガロースゲル(Sepharose(商標))、および当業者らに知られている固相支持体を含む。捕捉ビーズは、約25〜40μmの直径のSepharoseビーズであってもよい。
【0087】
(2)乳化
付着した一本鎖鋳型核酸を有する捕捉ビーズは、熱安定性の油中水型エマルションとして乳化することができる。エマルションは、当技術分野において知られている任意の適した方法によって形成することができる。エマルションを作り出すための1つの方法が下記に記載されるが、エマルションを作製するための任意の方法が使用することができる。これらの方法は、当技術分野において知られており、補助剤法、向流法、逆流法、回転ドラム法、および膜法を含む。そのうえ、マイクロカプセルのサイズは、構成要素の流量および速度を変動させることによって調整することができる。たとえば、液滴追加において、滴のサイズおよび送達の総時間は変動させてもよい。エマルションは、マイクロリットル当たり約3,000ビーズの密度で、ある程度の密度のビーズ「マイクロリアクター」を含有してもよい。
【0088】
エマルションは、増幅溶液中で鋳型付着ビーズを懸濁することによって生成することができる。本明細書において使用されるように、「増幅溶液」という用語は、鋳型DNAの増幅を行なうのに必要な、試薬の十分な混合物を意味し得る。
【0089】
ビーズ/増幅溶液混合物は、液滴に追加して、生物学的適合油(たとえば軽鉱物油、Sigma)の回転混合物にし、乳化させてもよい。使用される油は、1つまたは複数の生物学的適合乳化安定剤で補足することができる。これらの乳化安定剤はAtlox 4912、Span 80、および他の認識され、市販で入手可能な適した安定剤を含んでもよい。形成される液滴は、5ミクロン〜500ミクロン、約50〜300ミクロン、または100〜150ミクロンのサイズに及んでもよい。
【0090】
マイクロリアクターのサイズの制限はない。マイクロリアクターは、必要とされる増幅の程度にとって十分な増幅試薬を包含するのに十分に大きいものであってもよい。マイクロリアクターはまた、それぞれが、DNAライブラリーのメンバーを含有するマイクロリアクターの集団が、従来の実験装置(たとえばPCRサーモサイクリング装置、試験管、インキュベーター、およびその他同種のもの)によって増幅することができるように、十分に小さいものであってもよい。
【0091】
マイクロリアクターの最適サイズは、100〜200ミクロンの直径としてもよい。このサイズのマイクロリアクターは、10ml未満の容量のマイクロリアクターの懸濁液中で、約600,000メンバーを含むDNAライブラリーの増幅を可能にすることができる。たとえば、PCRが選ばれた増幅方法である場合、10mlは、96本の管の容量を有する一般的なサーモサイクラーの96本の管に一致するであろう。600,000マイクロリアクターの懸濁液は、1ml未満の容量を有していてもよい。1ml未満の懸濁液は、従来のPCRサーモサイクラーの約10本の管において増幅することができる。600,000マイクロリアクターの懸濁液はまた、0.5ml未満の容量を有していてもよい。
【0092】
(3)増幅
カプセル化後に、鋳型核酸は、転写ベースの増幅システム(Kwoh D.ら、Proc.Natl. Acad. Sci. (U.S.A.)86巻:1173頁(1989年);Gingeras T. R.ら、PCT特許出願国際公開第88/10315号;Davey,C.ら、欧州特許出願公開第329,822号;Miller, H. I.ら、PCT特許出願国際公開第89/06700号および「race」(Frohman, M.A.、In: PCR Protocols: A Guide to Methods and Applications、Academic Press、NY(1990年))および「片側PCR」(Ohara,O.ら、Proc. Natl. Acad. Sci. (U.S.A.)86巻、5673〜5677頁(1989年))を含むDNA増幅の任意の適した方法によって増幅することができる。なお、「ジオリゴヌクレオチド」増幅、等温増幅(Walker,G. T.ら、Proc. Natl. Acad. Sci. (U.S.A.)89巻:392〜396頁(1992年))、およびローリングサークル増幅(米国特許第5,714,320号において考察される)などのそれほど一般的ではない方法が、本発明において使用することができる。
【0093】
DNA増幅は、PCRによって行なわれてもよい。PCRは、PCRに必要な試薬をすべて含むPCR溶液を用いて、ビーズに結合された単離DNAをカプセル化することによって行なわれてもよい。次いで、PCRは、当技術分野において知られている任意の適したサーモサイクリングレジメンにエマルションをさらすことによって成し遂げることができる。30および50サイクルの間のまたは約40サイクルの増幅が行なわれてもよい。増幅手順に続いて、1つまたは複数のハイブリダイゼーションおよび伸長のサイクルが増幅サイクルに続いてあってもよい。10および30サイクルの間のまたは約25サイクルのハイブリダイゼーションおよび伸長が行なわれてもよい。鋳型DNAは、典型的に、鋳型DNAの少なくとも200万〜5000万のコピーまたは約1000万〜3000万のコピーがビーズ当たりに固定されるまで増幅することができる。
【0094】
(4)エマルションの破壊およびビーズ回収
単離DNAの増幅に続いて、エマルションは、「破壊」することができる(「解乳化」とも呼ばれる)。米国特許第5,989,892号においてなどのように、エマルションを破壊するための多くの方法があり、これらの内容は、参照によって本明細書において組み込まれる。エマルションは、さらなる油を追加して、エマルションを2つの相に分離することによって破壊することができる。次いで、油相は、除去されてもよく、適した有機溶媒(たとえばヘキサン)が追加することができる。混合後に、油/有機溶媒相は除去される。このステップは数回繰り返することができる。最後に、ビーズ上の水層は除去することができる。次いで、ビーズは、有機溶媒/アニーリング緩衝混合液を用いて洗浄してもよく、次いで、アニーリング緩衝液中で再び洗浄してもよい。適した有機溶媒は、メタノール、エタノール、およびその他同種のものなどのアルコールを含む。
【0095】
次いで、増幅された単離DNA含有ビーズは、たとえば知られている技術による配列決定反応における使用のために水溶液中に再懸濁することができる(Sanger, F.ら、Proc. Natl. Acad. Sci. U.S.A.75巻、5463〜5467頁(1977年);Maxam,A. M. & Gilbert、W. Proc Natl Acad Sci USA74巻、560〜564頁(1977年);Ronaghi, M.ら、Science281巻、363巻、365巻(1998年);Lysov,I.ら、Dokl Akad Nauk SSSR303巻、1508〜1511頁(1988年);Bains W. & Smith G. C. J.TheorBiol135巻、303〜307頁(1988年);Drnanac, R.ら、Genomics4巻、114〜128頁(1989年);Khrapko,K. R.ら、FEBS Lett256巻、118〜122頁(1989年);Pevzner P. A. J Biomol Struct Dyn7巻、63〜73頁(1989年);Southern,E. M.ら、Genomics13巻、1008〜1017頁(1992年)を参照されたい)。ビーズは、ピロリン酸ベースの配列決定反応において使用されてもよく(たとえば米国特許第6,274,320号、第6258,568号、および第6,210,891号において記載される、これらの内容は、参照によって本明細書において組み込まれる)、PCR産物の第2の鎖は除去されてもよく、配列決定プライマーは、ビーズに結合された一本鎖単離DNAにアニールすることができる。
【0096】
第2の鎖は、NaOH、低イオン(たとえば塩)強度、または熱処理などの一般的に知られている任意の数の方法を使用して融解することができる。この融解ステップに続いて、ビーズはペレットにされてもよく、上清は捨てられてもよい。ビーズは、アニーリング緩衝液中に再懸濁されてもよく、配列決定プライマーは、追加されてもよく、標準的なアニーリングサイクルを使用して、ビーズ付着一本鎖鋳型にアニールすることができる。
【0097】
(5)ビーズの精製
ビーズ上の増幅DNAはまた、直接ビーズ上でまたは異なる反応槽中で配列決定することができる。DNAは、反応槽にビーズを移し、DNAを配列決定反応にかけることによって、直接ビーズ上で配列決定することができる(たとえばピロリン酸配列決定またはSanger配列決定)。その代わりに、ビーズは、単離することができ、DNAは、それぞれの各ビーズから取り出され、配列決定することができる。いずれの場合においても、配列決定ステップは、それぞれの個々のビーズについて行なわれてもよい。ビーズはまた、米国特許第7,323,305号において記載される方法によって精製されてもよく、これらの内容は、参照によって本明細書において組み込まれる。
【0098】
b.ブリッジングPCR
環状であってもよく、アダプター挿入断片を含んでもよい単離DNAは、「ブリッジング」PCRを使用してPCR増幅することができる。ブリッジングPCRは、米国特許第7,115,400号において記載される方法によって行なわれてもよく、これらの内容は、参照によって本明細書において組み込まれる。増幅に先立って、単離DNAは、2つのプライマー結合部位の間の部位を切断する制限酵素を用いてそれを消化することによって線状にすることができる。単離DNAの一方の末端(5’末端)の配列は、オリゴヌクレオチド配列Yを有していてもよく、「コロニー」プライマーXの配列と同一の配列を含んでもよい。オリゴヌクレオチド配列Yは、知られている配列であってもよく、可変長であってもよい。オリゴヌクレオチド配列Yは、少なくとも5ヌクレオチド長、5〜100ヌクレオチド長、または約20ヌクレオチド長であってもよい。自然発生ヌクレオチドまたは非自然発生ヌクレオチドはオリゴヌクレオチド配列Y中に存在してもよい。
【0099】
配列Yの反対側の単離DNAの末端(3’末端)に含有されるオリゴヌクレオチド配列は、オリゴヌクレオチド配列Zを有していてもよい。オリゴヌクレオチド配列Zは、知られている配列であってもよく、可変長であってもよい。オリゴヌクレオチド配列Zは、少なくとも5ヌクレオチド長、5〜100ヌクレオチド長、または約20ヌクレオチド長であってもよい。自然発生ヌクレオチドまたは非自然発生ヌクレオチドはオリゴヌクレオチド配列Z中に存在してもよい。オリゴヌクレオチド配列Zは、それがコロニープライマーXとハイブリダイズするように設計されてもよく、それが、本明細書においてY’と呼ばれるオリゴヌクレオチド配列Yに相補的となるように設計することができる。核酸鋳型のそれぞれ5’および3’末端に含有されるオリゴヌクレオチド配列YおよびZは、鋳型の末端の先端に位置する必要はない。たとえば、オリゴヌクレオチド配列YおよびZは、核酸鋳型のそれぞれ5’および3’末端(もしくは末端)にまたはその近くに位置してもよいが(たとえば、5’および3’末端の0〜100ヌクレオチド以内)、それらは、核酸鋳型の5’または3’末端からさらに離れて(たとえば100ヌクレオチドを超えて)位置してもよい。したがって、配列YおよびZが、どちらかの側にある、つまり、増幅される核酸配列の側面に位置するという条件で、オリゴヌクレオチド配列YおよびZは核酸鋳型内の任意の位置に位置してもよい。
【0100】
本明細書において使用されるような「核酸鋳型」はまた、二本鎖形態の、増幅され、配列決定される核酸を含む実体を含む。核酸鋳型が二本鎖形態をしている場合、オリゴヌクレオチド配列YおよびZは、鎖のうち1つのそれぞれ5’および3’末端に含有される。他の鎖は、DNAの塩基対合則により、オリゴヌクレオチド配列YおよびZを含有する鎖に相補的であり、したがって、5’末端にオリゴヌクレオチド配列Z’および3’末端にオリゴヌクレオチド配列Y’を含有する。
【0101】
本明細書において使用されるような「コロニープライマー」は、相補的配列にハイブリダイズし、特異的なポリメラーゼ反応を開始することができるオリゴヌクレオチド配列を含む実体を指し得る。コロニープライマーを含む配列は、それが、その相補的配列との最大のハイブリダイズ活性およびあらゆる他の配列への非常に低い非特異的ハイブリダイズ活性を有するように選ばれる。コロニープライマーとして使用される配列は、任意の配列を含むことができるが、5'-AGAAGGAGAAGGAAAGGGAAAGGG(配列番号1)または5'-CACCAACCCAAACCAACCCAAACC(配列番号2)を含んでもよい。コロニープライマーは、5〜100塩基長または15〜25塩基長とすることができる。自然発生ヌクレオチドまたは非自然発生ヌクレオチドがプライマー中に存在してもよい。1つまたは2つの異なるコロニープライマーは、本発明の方法において核酸コロニーを生成するために使用することができる。
【0102】
本明細書において使用されるような「縮重プライマー配列」は、上述の核酸断片の配列と無関係の任意の核酸断片にハイブリダイズすることができる、短いオリゴヌクレオチド配列を指し得る。したがって、そのような縮重プライマーは、鋳型へのハイブリダイゼーションが生じるのに(1つまたは複数の)核酸鋳型におけるオリゴヌクレオチド配列YまたはZの存在を必要としないが、オリゴヌクレオチド配列XまたはYを含有する鋳型にハイブリダイズするための縮重プライマーの使用は除外されない。しかしながら、明白に、本発明の増幅方法における使用のために、縮重プライマーは、どちらかの側のまたは増幅される核酸配列の側面に位置する部位で鋳型中の核酸配列にハイブリダイズしなければならない。
【0103】
本明細書において使用されるような「固体支持体」は、たとえばラテックスビーズ、デキストランビーズ、ポリスチレン、ポリプロピレン表面、ポリアクリルアミドゲル、金表面、ガラス表面、およびシリコンウェーハなどの、核酸が共有結合することができる任意の固体表面を指し得る。
【0104】
本明細書において使用されるような「固体支持体に核酸を付着させるための手段」は、化学的に修飾可能な官能基を含む任意の化学的または非化学的な付着方法を指し得る。「付着」は、共有結合によるまたは不可逆的受動的吸着を介してのまたは分子の間のアフィニティーを介しての、固体支持体上の核酸の固定に関する(たとえばビオチン化分子によるアビジンコート表面上の固定)。付着は、それが、DNA変性条件下で水または水性緩衝液を用いて洗浄することによって除去され得ない十分な強度のものでなければならない。
【0105】
本明細書において使用されるような「化学的に修飾可能な官能基」は、たとえばリン酸基、カルボキシ部分もしくはアルデヒド部分、チオール基、またはアミノ基などの基を指し得る。
【0106】
本明細書において使用されるような「核酸コロニー」は、核酸鎖の多数のコピーを含む分散したエリアを指し得る。核酸鎖に対する相補鎖の多数のコピーもまた、同じコロニー中に存在してもよい。コロニーを構成する核酸鎖の多数のコピーは、固体支持体上に一般に固定され、一本鎖または二本鎖の形態をしていてもよい。本発明の核酸コロニーは、使用される条件に依存して異なるサイズおよび密度で生成することができる。コロニーのサイズは、0.2μm〜6μmまたは0.3μm〜4μmであってもよい。本発明の方法における使用のための核酸コロニーの密度は、10,000/mm2〜100,000/mm2であってもよい。より高い密度、たとえば、100,000/mm2〜1,000,000/mm2および1,000,000/mm2〜10,000,000/mm2が達成することができると考えられる。
【0107】
核酸コロニーは、単離DNAから生成することができる。それぞれが複数の異なる単離DNAのうち1つを表わす複数のコロニーもまた生成することができる。
【0108】
増幅される核酸を含む複数の単離DNAが生成することができ、核酸はそれらの5’末端にオリゴヌクレオチド配列Yおよび3’末端にオリゴヌクレオチド配列Zを含有し、さらに、(1つまたは複数の)核酸は、5’末端に、固体支持体に(1つまたは複数の)核酸を付着させるための手段を有する。複数の単離DNAは、オリゴヌクレオチド配列Zにハイブリダイズし、5’末端に、固体支持体にコロニープライマーを付着させるための手段を持っていてもよい複数のコロニープライマーXと混合される。複数の単離DNAおよびコロニープライマーは、固体支持体に共有結合してもよい。
【0109】
多数の2つの異なるコロニープライマーXは、複数の核酸鋳型と混合することができる。コロニープライマーXの配列は、オリゴヌクレオチド配列Zが、コロニープライマーXのうち1つにハイブリダイズすることができ、オリゴヌクレオチド配列Yが、コロニープライマーXのうち1つの配列と同じであるようなものであってもよい。
【0110】
オリゴヌクレオチド配列Zはまた、オリゴヌクレオチド配列Y(Y’)に相補的であってもよく、複数のコロニープライマーXは、オリゴヌクレオチド配列Yと同じ配列であってもよい。
【0111】
複数のコロニープライマーXは、縮重プライマー配列を含んでもよく、複数の単離DNAは、増幅される核酸を含んでもよく、それぞれ5’および3’末端にオリゴヌクレオチド配列YまたはZを含有していなくてもよい。
【0112】
単離核酸の5’末端に含有されるオリゴヌクレオチド配列は、任意の配列および任意の長さであってもよく、配列Yとして本明細書において示される。オリゴヌクレオチドの適した長さおよび配列は、十分に知られている方法を使用して、選択することができ、当技術分野をにおいて立証されている。たとえば、増幅される核酸のそれぞれの末端に付着されるオリゴヌクレオチド配列は、5および100ヌクレオチドの間の長さの通常、比較的短いヌクレオチド配列である。核酸の3’末端に含有されるオリゴヌクレオチド配列は、任意の配列および任意の長さとすることができ、配列Zとして本明細書において示される。オリゴヌクレオチドの適した長さおよび配列は、十分に知られており、当技術分野において立証されている方法を使用して、選択することができる。たとえば、増幅される核酸のそれぞれの末端に含有されるオリゴヌクレオチド配列は、5および100ヌクレオチドの間の長さの通常、比較的短いヌクレオチド配列である。
【0113】
オリゴヌクレオチド配列Zの配列は、それがコロニープライマーXのうち1つにハイブリダイズすることができるようなものである。オリゴヌクレオチド配列Yの配列は、それがコロニープライマーXのうち1つと同じであるようなものとしてもよい。オリゴヌクレオチド配列Zは、オリゴヌクレオチド配列Y(Y’)に相補的であってもよく、コロニープライマーXは、オリゴヌクレオチド配列Yと同じ配列である。
【0114】
オリゴヌクレオチド配列YおよびZは、当技術分野において標準的もしくは従来のものである技術を使用して調製されてもよく、または商業的供給源から購入することができる。
【0115】
本発明の単離DNAを産生する場合、さらなる望ましい配列は、十分に知られており、当技術分野において立証されている方法によって導入することができる。そのようなさらなる配列は、たとえば、制限酵素部位またはある核酸タグを含み、所与の核酸鋳型配列の増幅産物を同定することを可能にする。他の望ましい配列は、フォールドバックDNA配列(一本鎖にされた場合にヘアピンループまたは他の第2の構造を形成する)、たとえば、核酸ポリメラーゼによって認識されるプロモーターDNA配列などの、タンパク質/DNA相互作用を指示する「コントロール」DNA配列、またはDNA結合タンパク質によって認識されるオペレーターDNA配列を含む。
【0116】
増幅される複数の核酸配列がある場合、オリゴヌクレオチドYおよびZの付着は、同じまたは異なる反応において実行することができる。
【0117】
単離DNAが一度調製されたら、それは、本明細書において記載される方法において使用される前に増幅することができる。そのような増幅は、たとえば、発現ベクターの中に鋳型核酸を挿入し、かつ適した生物学的宿主においてそれを増幅するまたはPCRによってそれを増幅することによって、十分に知られており、当技術分野をにおいて立証されている方法を使用して実行することができる。しかしながら、本発明の方法は、核酸鋳型の多数のコピーが、核酸鋳型の単一コピーから生成される核酸コロニーにおいて産生されるのを可能にするので、この増幅ステップは、必須ではない。
【0118】
単離DNAの5’末端は、固体支持体に核酸鋳型を共有結合させるための手段を有するように修飾することができる。そのような手段は、たとえば、たとえばリン酸基、カルボキシ部分もしくはアルデヒド部分、チオール基、またはアミノ基などの化学的に修飾可能な官能基とすることができる。チオール基、リン酸基、またはアミノ基は、核酸の5’−修飾のために使用することができる。
【0119】
コロニープライマーは、当技術分野において標準的なまたは従来のものである技術を使用して調製することができる。一般に、本発明のコロニープライマーは、十分に知られており、当技術分野において立証されている方法によって生成される合成オリゴヌクレオチドであり、または商業的供給源から購入することができる。
【0120】
1つまたは2つの異なるコロニープライマーXは、任意の核酸配列を増幅するために使用することができる。コロニープライマーXの5’末端は、固体支持体にコロニープライマーを共有結合させるための手段を有するように修飾することができる。共有結合は、上記に記載されるように、化学的に修飾可能な官能基であってもよい。コロニープライマーは、たとえば制限エンドヌクレアーゼ部位またはそれぞれリボザイム切断部位のような他のタイプの切断部位などのさらなる所望の配列を含むように設計することができる。さらなる配列は、フォールドバックDNA配列(一本鎖にされた場合にヘアピンループまたは他の第2の構造を形成する)、たとえば、核酸ポリメラーゼによって認識されるプロモーターDNA配列などの、タンパク質/DNA相互作用を指示する「コントロール」DNA配列、またはDNA結合タンパク質によって認識されるオペレーターDNA配列を含む。
【0121】
5’末端による支持体へのコロニープライマーXの固定は、単離DNAの3’末端に含有される相補的なオリゴヌクレオチド配列とのハイブリダイゼーションが一度起こったら、コロニープライマーがポリメラーゼによる鎖伸長のために利用可能となるように、その3’末端を支持体から遠くにしてもよい。
【0122】
一度、本発明の単離DNAおよびコロニープライマーの両方が合成されたら、それらは、それらが、固体支持体に付着している場合に、付着された単離DNAおよびコロニープライマーの適切な密度が得られるように、適切な割合で共に混合される。混合物中のコロニープライマーの割合は、単離DNAの割合よりも高くてもよい。単離DNAに対するコロニープライマーの比は、コロニープライマーおよび単離DNAが固体支持体に固定される場合に、固体支持体の全体または明示されるエリアにほぼ一定の密度で位置する複数のコロニープライマーを含むコロニープライマーの「ローン(lawn)」が、コロニープライマーのローン内に間隔を置いて個々に固定される1つまたは複数の単離DNAと共に、形成されるようなものであってもよい。
【0123】
単離DNAは、一本鎖形態で提供することができる。しかしながら、それはまた、全体としてまたは部分的に、支持体への付着を可能にするように一方の5’末端または両方の5’末端が修飾された二本鎖形態で提供することができる。その場合、付着プロセスが完了した後に、たとえば放出される鎖を洗浄する前に94℃まで加熱することによって、当技術分野において知られている手段によって鎖を分離することが望まれる。二本鎖分子の両方の鎖が表面と反応し、両方とも付着する場合、結果は、一方の鎖のみが付着し、1回の増幅ステップが行なわれる場合と同じである可能性がある。言いかえれば、二本鎖単離DNAの両方の鎖が付着した場合において、両方の鎖は、必然的に、互いに近くに付着し、一方の鎖のみを付着させ、1回の増幅ステップを行なう結果と区別不能である。したがって、一本鎖および二本鎖単離DNAは、表面に付着され、コロニー生成のために適した鋳型核酸を提供するために使用することができる。
【0124】
個々のコロニープライマーおよび個々の単離DNAの間の距離(したがって、コロニープライマーおよび単離DNAの密度)は、コロニープライマーおよび支持体に固定される単離DNAの濃度を変えることによってコントロールすることができる。コロニープライマーの密度は、少なくとも1fmol/mm2、少なくとも10fmol/mm2、30〜60fmol/mm2としてもよい。単離DNAの密度は、10,000/mm2〜100,000/mm2であってもよい。100,000/mm2〜1,000,000/mm2および1,000,000/mm2〜10,000,000/mm2のより高い密度が達成することができる。
【0125】
付着した単離DNAおよびコロニープライマーの密度のコントロールは、支持体の表面上の核酸コロニーの最終密度をコントロールすることを可能にすることができる。これは、本発明の方法によれば、1つの核酸コロニーが、コロニープライマーが、固体支持体上の適した位置に存在するという条件で、1つの単離DNAの付着の結果として生じ得るといった事実によるものである。単一コロニー内の単離DNAの密度もまた、付着したコロニープライマーの密度をコントロールすることによってコントロールすることができる。
【0126】
コロニープライマーおよび単離DNAが一度、適切な密度で固体支持体上に固定されたら、核酸コロニーは、それぞれのコロニーが、もとの固定された単離DNAおよびその相補的配列の多数のコピーを含むように、共有結合した単離DNA上で適切な数の増幅のサイクルを実行することによって生成することができる。増幅の1回のサイクルは、ハイブリダイゼーション、伸長、および変性のステップからなり、これらのステップは、PCRについて当技術分野において十分に知られている試薬および条件を使用して、一般に行なわれる。
【0127】
典型的な増幅反応は、固体支持体、付着した単離DNA、およびコロニープライマーをプライマーハイブリダイゼーションを誘発する条件にかけること、たとえば、それらを約65℃の温度にかけることを含む。これらの条件下で、単離DNAの3’末端のオリゴヌクレオチド配列Zは、固定されたコロニープライマーXにハイブリダイズするであろう、またヌクレオシド三リン酸分子または任意の他のヌクレオチド前駆体、たとえば修飾ヌクレオシド三リン酸分子の供給を加えての、プライマー伸長を支援するための条件および試薬、たとえば、約72℃の温度、核酸ポリメラーゼ、たとえばDNA依存性DNAポリメラーゼ分子もしくは逆転写酵素分子(つまりRNA依存性DNAポリメラーゼ)またはRNAポリメラーゼの存在下において、コロニープライマーは、単離DNAに相補的なヌクレオチドの追加によって伸長するであろう。
【0128】
核酸ポリメラーゼは、DNAポリメラーゼ(Klenow fragment、T4 DNAポリメラーゼ)、耐熱性細菌からの熱安定性DNAポリメラーゼ(Taq、VENT、Pfu、Tfl DNAポリメラーゼなど)、またはその遺伝子改変誘導体(TaqGold、VENTexo、Pfu exo)であってもよい。RNAポリメラーゼおよび逆転写酵素の組み合わせを使用して、DNAコロニーの増幅を生じさせることもできる。コロニープライマー伸長のために使用される核酸ポリメラーゼは、PCR反応条件、つまり、加熱および冷却のサイクルの繰り返し下で安定性である可能性があり、使用される変性温度、普通、約94℃で安定性である可能性がある。
【0129】
ヌクレオシド三リン酸分子は、デオキシリボヌクレオチド三リン酸、たとえばDATP、dTTP、dCTP、dGTPであってもよく、またはリボヌクレオシド三リン酸、たとえばdATP、dUTP、dCTP、dGTPであってもよい。ヌクレオシド三リン酸分子は、自然発生または非自然発生のものであってもよい。
【0130】
ハイブリダイゼーションおよび伸長のステップの後に、支持体および付着した核酸を変性条件にさらす上で、2つの固定された核酸が存在するであろう、第1は、最初の固定された単離DNAであり、第2は、それに相補的な核酸であり、固定されたコロニープライマーXのうち1つから伸長する。もとの固定された単離DNAおよび形成された固定された伸長コロニープライマーの両方は、次いで、ハイブリダイゼーション、伸長、および変性のさらなるサイクルに支持体をかける上で、さらなる回の増幅を開始することができる。そのようなさらなる回の増幅は、単離DNAの多数の固定されたコピーおよびその相補的配列を含む核酸コロニーをもたらすであろう。
【0131】
単離DNAの最初の固定は、鋳型核酸が、全長の単離DNA内で離れて位置するコロニープライマーとハイブリダイズすることができるのみであることを意味する。したがって、形成される核酸コロニーの境界は、最初の単離DNAが固定されるエリアに対して、比較的局所的なエリアに制限されている。明らかに、単離DNAおよびその相補体のより多くのコピーが、増幅のさらなる回、つまりハイブリダイゼーション、伸長、および変性のさらなる回を実行することによって、一度、合成されたら、次いで、形成されるコロニーの境界が、最初の単離DNAが固定されるエリアに対して、なお、比較的局所的なエリアに制限される可能性があるが、生成されている核酸コロニーの境界は、さらに拡張することができるであろう。
【0132】
単一の固定された単離DNAからの核酸コロニーを生成することができ、これらのコロニーのサイズは、単離DNAがかけられる増幅の回数を変えることによってコントロールすることができる。したがって、それぞれの固定された単離DNAの場所内に十分な数の固定されたコロニープライマーがあるという条件で、固体支持体の表面上に形成される核酸コロニーの数は、支持体に最初に固定される単離DNAの数に依存する。コロニープライマーおよび単離DNAが固定された固体支持体が、プライマーのローン内に間隔を置いて固定された単離DNAと共に適切な密度で、固定されたコロニープライマーのローンを含んでいる可能性があるのは、この理由のためである。
【0133】
核酸コロニーのそのようないわゆる「オートパターニング」は、単離DNAがもともと固定される密度を調節することによって密度をコントロールすることができるという事実によりより高い密度の核酸コロニーが得ることができるという点において、他の方法に対して利点を有している可能性がある。そのような方法は、したがって、たとえば、支持体の特定の局所的なエリア上に特異的なプライマーを特異的に整列させ、次いで、プライマーの同じ局所的なエリア上に単離DNAを含有する特定のサンプルを配置することによってコロニー形成を開始することによって制限されない。先行技術方法を使用して整列することができるコロニーの数、たとえば国際公開第96/04404号(Mosaic Technologies,Inc.)において開示されるものは、したがって、特異的なプライマーエリアが最初のステップにおいて整列され得る密度/間隔によって制限される。
【0134】
単離DNAの最初の密度、したがって、単離DNAから結果として生じる核酸コロニーの密度をコントロールすることができることによって、形成される核酸コロニーのサイズ、さらに個々のコロニー内の単離DNAの密度をコントロールすることができることと共に、高密度の個々の核酸コロニーを十分に大きなサイズの、十分に多くの増幅配列を含有する固体支持体上で産生することができ、核酸コロニーについて行なわれる後の分析またはモニタリングを可能にする、最適の状況に達することができる。
【0135】
一度、核酸コロニーが生成されたら、たとえばコロニー視覚化または配列決定などの、さらなるステップを行なうことが望ましい可能性がある。たとえば、特定の核酸断片の一部または全部の存在または不在について、生成されるコロニーをスクリーニングすることが必要である場合、たとえば、コロニー視覚化は、必要とされる可能性がある。この場合、特定の核酸断片を含有する1つまたは複数のコロニーは、対象とする核酸断片に特異的にハイブリダイズする核酸プローブを設計することによって検出することができる。
【0136】
そのような核酸プローブは、蛍光基、ビオチンを含有する実体(たとえば、蛍光基を用いて標識されたストレプトアビジンとのインキュベーションによって検出することができる)、放射標識(当技術分野において十分に知られており、立証されている方法によって核酸プローブの中に取り込むことができ、たとえばシンチレーション液とのインキュベーションによって放射活性を検出することによって検出することができる)、もしくは染料または他の染色剤などの検出可能な実体を用いて標識してもよい。
【0137】
核酸プローブはまた、非標識であってもよく、核酸ポリメラーゼを用いる多くの標識ヌクレオチドの取り込みのためのプライマーとして作用するように設計することができる。次いで、取り込まれた標識、したがって核酸コロニーの検出を実行することができる。
【0138】
次いで、核酸コロニーは、ハイブリダイゼーションのために調製することができる。そのような調製は、コロニーを構成する核酸鋳型のすべてまたは一部分が、一本鎖形態で存在するように、コロニーの処理を含んでもよい。これは、たとえば、コロニー中の任意の二本鎖DNAの熱変性によって達成することができる。その代わりに、コロニーは、鋳型核酸中の配列の二本鎖形態に特異的な制限エンドヌクレアーゼを用いて処理することができる。したがって、エンドヌクレアーゼは、オリゴヌクレオチド配列YまたはZ中に含有される配列または単離DNA中に存在する他の配列に特異的であってもよい。消化後に、コロニーは、二本鎖DNA分子が分離されるように、加熱され、コロニーは、固定されていない鎖を除去するために洗浄されて、したがって、コロニー中に付着した一本鎖DNAを残す。
【0139】
ハイブリダイゼーションのためのコロニーの調製の後に、標識プローブまたは非標識プローブは、次いで、その特異的なDNA配列とのプローブのハイブリダイゼーションのための適切な条件下でコロニーに追加してもよい。
【0140】
次いで、プローブは、熱変性によって除去されてもよく、所望の場合、第2の核酸のための特異的なプローブが、ハイブリダイズされ、検出することができる。これらのステップは、所望されるだけ繰り返することができる。
【0141】
次いで、核酸コロニーにハイブリダイズされる標識プローブは、適切な検出デバイスを含む機器を使用して検出することができる。蛍光標識のための検出システムは、電荷結合素子(CCD)カメラであってもよく、これは、拡大デバイス、たとえば顕微鏡に任意選択で連結することができる。そのような技術を使用して、並行して、多くのコロニーを同時にモニターすることが可能である。たとえばCCDカメラおよび10×または20×対物レンズを有する顕微鏡を使用すると、1mm2および4mm2の間の表面に対してコロニーを観察することが可能であり、これは、10000および200000コロニーの間のコロニーを並行してモニターすることに対応する。
【0142】
生成されるコロニーをモニターするための代替方法は、コロニーで覆われる表面をスキャンすることである。たとえば、100000000個までのコロニーを同時に整列することができ、全表面に対してCCDカメラを用いて写真を撮ることによってモニターすることができるシステムを使用することができる。このように、100000000個までのコロニーを短時間でモニターすることができることを理解することができる。
【0143】
表面上の蛍光の検出および定量化を可能にする任意の他のデバイスもまた、本発明の核酸コロニーをモニターするために使用することができる。たとえば、蛍光画像装置または共焦点顕微鏡を使用することができる。標識が放射性である場合、放射活性検出システムが必要とすることができる。
【0144】
単離DNAの配列は、任意の適切な固相配列決定技術を使用することによって決定することができる。たとえば、本発明において使用することができる配列決定の1つの技術は、「配列決定プライマー」と本明細書において時に呼ばれる適切なプライマーを、配列決定される核酸鋳型とハイブリダイズすること、プライマーを伸長させること、およびプライマーを伸長させるために使用されるヌクレオチドを検出することを含む。プライマーを伸長させるために使用される核酸は、さらなるヌクレオチドが、成長している核酸鎖に追加される前に、検出されてもよく、したがって、1塩基ずつのin situ核酸配列決定を可能にする。
【0145】
取り込まれるヌクレオチドの検出は、プライマー伸長反応において1つまたは複数の標識ヌクレオチドを含むことによって容易にすることができる。任意の適切な検出可能な標識、たとえば蛍光体、放射標識などが使用することができる。蛍光標識が使用することができる。同じまたは異なる標識が、それぞれの異なるタイプのヌクレオチドについて使用することができる。標識は蛍光体であり、同じ標識が、それぞれの異なるタイプのヌクレオチドについて使用される場合、それぞれのヌクレオチド取り込みは、特定の波長で検出されるシグナルの累積的な増加を提供してもよい。異なる標識が使用される場合、これらのシグナルは、異なる適切な波長で検出することができる。所望の場合、標識ヌクレオチドおよび非標識ヌクレオチドの混合物が提供される。
【0146】
配列決定される単離DNAへの適切な配列決定プライマーのハイブリダイゼーションを可能にするために、核酸鋳型は、一本鎖形態をしていてもよい。核酸コロニーを構成する核酸鋳型が二本鎖形態で存在する場合、これらは、当技術分野において十分に知られている方法を使用して、たとえば変性、切断などによって、一本鎖単離DNAを提供するために処理することができる。
【0147】
単離DNAにハイブリダイズされ、プライマー伸長に使用される配列決定プライマーは、たとえば15〜25ヌクレオチド長の短いオリゴヌクレオチドであってもよい。プライマーの配列は、それらが、配列決定される単離DNAの一部にハイブリダイズするように設計されてもよく、ストリンジェントな条件下であってもよい。配列決定のために使用されるプライマーの配列は、核酸コロニーを生成するために使用されるコロニープライマーの配列と同じまたは同様の配列を有していてもよい。配列決定プライマーは、溶液または固定された形態で提供することができる。
【0148】
一度、配列決定プライマーが、単離DNAおよび配列決定プライマーを、当技術分野において十分に知られている方法によって決定される適切な条件にかけることによって、配列決定される単離DNAにアニールされたら、プライマー伸長は、たとえば、核酸ポリメラーゼおよび少なくともいくつかが、標識形態で提供されるヌクレオチドの供給および適したヌクレオチドが提供される場合に、プライマー伸長に適した条件を使用して、行なわれてもよい。
【0149】
それぞれのプライマー伸長ステップ後に、後のステップに干渉する可能性がある、取り込まれていないヌクレオチドを除去するために、洗浄ステップが含まれていてもよい。一度、プライマー伸長ステップが行なわれたら、核酸コロニーは、標識ヌクレオチドが伸長プライマーの中に取り込まれたかどうかを決定するために、モニターすることができる。次いで、プライマー伸長ステップは、伸長プライマーの中に取り込まれる次のおよび後のヌクレオチドを決定するために、繰り返することができる。
【0150】
適切な標識、たとえば蛍光または放射活性の検出および定量化を可能にする任意のデバイスが配列決定に使用することができる。標識が蛍光である場合、拡大デバイスに任意選択で付着されたCCDカメラが使用することができる。実際に、配列決定のために使用されるデバイスは、増幅核酸コロニーをモニターするための、上記に記載されるデバイスと同じであってもよい。
【0151】
検出システムは、プライマー伸長のそれぞれのステップの後にそれぞれのコロニーに取り込まれるヌクレオチドの数および性質を決定するために、分析システムと組み合わせて使用することができる。この分析は、それぞれのプライマー伸長ステップ直後に実行されてもよくまたは記録されたデータを使用して後に実行されてもよく、所与のコロニー内の核酸鋳型の配列を決定することを可能にする。
【0152】
決定されている配列が未知の場合、所与のコロニーに適用されるヌクレオチドは、次いで、分析の全体にわたって繰り返される選ばれた順、たとえばdATP、dTTP、dCTP、dGTPで適用することができる。しかしながら、決定されている配列が知られており、たとえば、知られている配列からの配列中に小さな差異が存在するかどうかを分析するために、再度配列決定されている場合、配列決定プロセスは、知られている配列に従って選ばれる適切な順で、それぞれのステップで、ヌクレオチドを追加することによって、より速く成することができる。所与の配列との差異は、したがって、プライマー伸長の特定の段階での、あるヌクレオチドの取り込みの欠如によって検出される。
【0153】
固体支持体へのコロニープライマーおよび核酸鋳型の付着は、支持体が、核酸増幅反応の間にかけられてもよい温度、たとえば、約100℃までの温度、たとえば約94℃で耐熱性であってもよい。付着は、共有結合であってもよく、米国特許第7,115,400号において記載されるように成し遂げることができ、これらの内容は、参照によって本明細書において組み込まれる。
【0154】
5.単離DNAの配列決定
配列決定は、国際公開第2007/145612号もしくは国際公開第05003375または米国特許第7,323,305号もしくは第7,115,400号において記載される方法によって行なわれてもよく、これらの内容は、参照によって本明細書において組み込まれる。機械は、Roche(454)GS FLX(454 Life Systems、Branford、CT)、Illuminaゲノム分析機器配列決定プライマー(Illumina Inc.、San Diego、CA)であってもよい。
【0155】
たとえば、ピロリン酸配列決定が使用することができる。この技術は、Hyman、1988年Anew method of sequencing DNA. Anal Biochem.174巻:423〜36頁およびRonaghi、2001年Pyrosequencingsheds light on DNA sequencing. Genome Res11巻:3〜11頁において記載されるように、DNA合成の間に放出されるピロリン酸(Ppi)の検出に基づく。
【0156】
酵素反応のカスケードにおいて、可視光は、取り込まれるヌクレオチドの数に比例して生成し得る。カスケードは、無機Ppiが、ポリメラーゼによるヌクレオチド取り込みと共に放出することができる核酸重合反応を用いて始まってもよい。放出されるPpiは、ATPスルフリラーゼによってATPに変換されてもよく、これは、ルシフェラーゼにエネルギーを提供して、ルシフェリンを酸化してもよく、光を生成してもよい。追加されるヌクレオチドが知られているので、鋳型の配列は決定される可能性がある。固相ピロリン酸配列決定は、3つの酵素システムにおいて固定されたDNAを用いる。シグナル対ノイズ比を増加させるために、自然dATPは、dATPαSと交換することができる。dATPαSは、2つの異性体(SpおよびRp)の混合物であってもよい。ピロリン酸中の純粋2’−デオキシアデノシン−5’−O’−(1−チオ三リン酸)Sp−異性体は、読み取り長の2倍まで、実質的により長い読み取りを可能にするために、配列決定において使用することができる。
【0157】
ピロリン酸ベースの配列決定は、ヌクレオチド三リン酸の存在下において、単離DNAおよび伸長プライマーをポリメラーゼ反応にかけることによって行なわれてもよく、それによって、ヌクレオチド三リン酸は、それが、標的位置における塩基に相補的である場合、取り込まれるようになり、ピロリン酸(PPi)を放出するにすぎないであろう。ヌクレオチド三リン酸は、サンプル−プライマー混合物の別々の一定分量にまたは同じサンプル−プライマー混合物に引き続いて追加される。PPiの放出は、次いで、どのヌクレオチドが取り込まれるかを示すために検出される。
【0158】
配列産物の領域は、単離DNAの領域に配列決定プライマーをアニールすることおよびDNAポリメラーゼおよび知られているヌクレオチド三リン酸、つまり、dATP、dCTP、dGTP、dTTP、またはこれらのヌクレオチドのうち1つの類似体と配列決定プライマーを接触させることによって、決定することができる。配列は、下記に記載されるように、配列反応副産物を検出することによって決定することができる。
【0159】
配列プライマーは、それが、増幅核酸鋳型の領域に特異的にアニールすることができる限り、任意の長さまたは塩基組成とすることができる。配列決定プライマーのための特定の構造は、それが、増幅鋳型核酸上の領域に特異的にプライムすることができる限り、必要とされない。配列決定プライマーは、特徴づけられる配列およびアンカープライマーにハイブリダイズすることができる配列の間にある鋳型の領域に相補的であってもよい。配列決定プライマーは、配列産物を形成するためにDNAポリメラーゼを用いて伸長される。伸長は、1つまたは複数のタイプのヌクレオチド三リン酸および所望の場合、補助結合タンパク質の存在下において行なわれる。
【0160】
dNTPの取り込みは、配列決定副産物の存在についてアッセイすることによって決定することができる。配列決定産物のヌクレオチド配列はまた、dNMPが、伸長配列プライマーの中に取り込まれる時に、ヌクレオチド三リン酸(dNTP)から放たれる無機ピロリン酸(PPi)を測定することによって決定することができる。Pyrosequencing(商標)技術(Pyrosequencing AB、Stockholm、Sweden)と称される配列決定のこの方法は、溶液(液相)中でまたは固相技術として行なわれてもよい。PPiベースの配列決定方法は、たとえば国際公開第WO9813523A1号,Ronaghiら、1996年Anal. Biochem.242巻:84〜89頁、Ronaghiら、1998年Science281巻:363〜365頁(1998年)、および米国特許出願第2001/0024790号において一般に記載され、これらの内容は、参照によって本明細書において組み込まれる。たとえば米国特許第6,210,891号および第6,258,568号もまた参照されたい。これらの内容は、参照によって本明細書において組み込まれる。
【0161】
これらの条件下で放出されるピロリン酸は、酵素で(たとえばルシフェラーゼ−ルシフェリン反応における光の生成によって)検出することができる。そのような方法は、電気泳動の必要性および可能性として危険な放射標識の使用を回避しながら、ヌクレオチドを所与の標的位置において同定することおよびDNAを簡単にかつ急速に配列決定することを可能にする。
【0162】
PPiは、多くの異なる手法によって検出されてもよく、様々な酵素法は、以前に記載されている(たとえばReevesら、1969年Anal. Biochem.28巻:282〜287頁;Guilloryら、1971年Anal. Biochem.39巻:170〜180頁;Johnsonら、1968年Anal.Biochem. 15:273頁;Cookら、1978年Anal. Biochem. 91:557〜565頁、およびDrakeら、1979年Anal.Biochem. 94:117〜120頁を参照されたい)。
【0163】
ポリメラーゼによるdNTPの取り込みの結果として放たれるPPiは、たとえばATPスルフリラーゼを使用して、ATPに変換することができる。この酵素は、硫黄代謝に関与するとして同定された。硫黄は、還元形態および酸化形態の両方において、植物および動物の成長のための、必須の無機栄養素である(たとえば、SchmidtおよびJager、1992年Ann. Rev. Plant Physiol. Plant Mol. Biol.43巻:325〜349頁を参照されたい)。植物および微生物の両方において、硫酸の活発な取り込みの後に硫化物への還元が続く。硫酸は、利用可能な細胞の還元剤に比べて非常に低い酸化/還元電位を有するので、同化における最初のステップは、ATP依存性反応を介してのその活性化を必要とする(たとえばLeyh、1993年Crit.Rev. Biochem. Mol. Biol. 28:515〜542頁を参照されたい)。ATPスルフリラーゼ(ATP:硫酸アデニリルトランスフェラーゼ;EC 2.7.7.4)は、無機硫酸(SO4−2)の代謝における最初の反応を触媒する。たとえば、RobbinsおよびLipmann、1958年J.Biol. Chem.233巻:686〜690頁;HawesおよびNicholas、1973年Biochem. J.133巻:541〜550頁を参照されたい)。この反応において、SO4−2は、アデノシン5’−ホスホ硫酸(APS)に活性化される。
【0164】
ATPスルフリラーゼは、Saccharomyces cerevisiae(たとえばHawesおよびNicholas、1973年Biochem.J.133巻:541〜550頁を参照されたい)、Penicillium chrysogenum(たとえばRenostoら、1990年J. Biol.Chem.265巻:10300〜10308頁を参照されたい)、ラット肝臓(たとえばYuら、1989年Arch. Biochem. Biophys.269巻:165〜174頁を参照されたい)、および植物(たとえばShawおよびAnderson、1972年Biochem.J.127巻:237〜247頁;Osslundら、1982年Plant Physiol.70巻:39〜45頁を参照されたい)などのいくつかの供給源から高度に精製されてきた。そのうえ、ATPスルフリラーゼ遺伝子は、原核生物(たとえばLeyhら、1992年J.Biol. Chem.267巻:10405〜10410頁;SchwedockおよびLong、1989年Mol. Plant Microbe Interaction2巻:181〜194頁;LaueおよびNelson、1994年J.Bacteriol.176巻:3723〜3729頁を参照されたい)、真核生物(たとえばCherestら、1987年Mol. Gen. Genet.210巻:307〜313頁;MountainおよびKorch、1991年Yeast7巻:873〜880頁;Fosterら、1994年J.Biol. Chem.269巻:19777〜19786頁を参照されたい)、植物(たとえばLeustekら、1994年Plant Physiol.105巻:897〜90216頁を参照されたい)、および動物(たとえばLiら、1995年J.Biol. Chem.270巻:29453〜29459頁を参照されたい)からクローニングされてきた。酵素は、特定の供給源に依存して、ホモオリゴマーまたはヘテロダイマーである(たとえばLeyhおよびSuo、1992年J.Biol. Chem.267巻:542〜545頁を参照されたい)。
【0165】
耐熱性スルフリラーゼを使用することができる。耐熱性スルフリラーゼは、たとえばArchaeoglobus種またはPyrococcus種から得られてもよく、耐熱性スルフリラーゼの配列は、データベース受入番号028606、受入番号Q9YCR4、および受入番号P56863で入手可能である。
【0166】
ATPスルフリラーゼは、多くの異なる適用、たとえば高濃度のATPでのADPのバイオルミノメトリック検出(たとえばSchultzら、1993年Anal. Biochem.215巻:302〜304頁を参照されたい)、DNAポリメラーゼ活性の継続的なモニタリング(たとえばNyrbn、1987年Anal.Biochem.167巻:235〜238頁を参照されたい)、およびDNA配列決定(たとえばRonaghiら、1996年Anal. Biochem.242巻:84〜89頁;Ronaghiら、1998年Science281巻:363〜365頁;Ronaghiら、1998年Anal.Biochem.267巻:65〜71頁を参照されたい)に使用されてきた。
【0167】
いくつかのアッセイが、前のATPスルフリラーゼ反応の検出のために開発されてきた。比色モリブドリシスアッセイは、リン酸検出に基づくものであってもよいのに対して(たとえばWilsonおよびBandurski、1958年J. Biol. Chem.233巻:975〜981頁を参照されたい)、継続的な分光光度モリブドリシスアッセイは、NADH酸化の検出に基づくものであってもよい(たとえばSeubertら、1983年Arch.Biochem. Biophys.225巻:679〜691頁;Seubertら、1985年Arch. Biochem. Biophys.240巻:509〜523頁を参照されたい)。後のアッセイは、いくつかの検出酵素の存在を必要としてもよい。さらに、いくつかの放射性アッセイもまた文献において記載されている(たとえばDaleyら、1986年Anal.Biochem.157巻:385〜395頁を参照されたい)。たとえば、あるアッセイは、32P標識ATPから放出される32PPiの検出に基づくものであり(たとえばSeubertら、1985年Arch.Biochem. Biophys.240巻:509〜523頁を参照されたい)、他のアッセイは、[35S]標識APSへの35Sの取り込みに基づき(このアッセイはまた、カップリング酵素として、精製APSキナーゼをも必要とする;たとえばSeubertら、1983年Arch.Biochem. Biophys.225巻:679〜691頁を参照されたい)、第3の反応は、35標識APSからの35SO4−2の放出に依存する(たとえばDaleyら、1986年Anal.Biochem.157巻:385〜395頁を参照されたい)。
【0168】
逆ATPスルフリラーゼ反応の検出については、継続的な分光光度アッセイ(たとえばSegelら、1987年MethodsEnzymol.143巻:334〜349頁を参照されたい)、バイオルミノメトリックアッセイ(たとえばBalharryおよびNicholas、1971年Anal.Biochem.40巻:1〜17頁を参照されたい)、35SO4−2放出アッセイ(たとえばSeubertら、1985年Arch.Biochem. Biophys.240巻:509〜523頁を参照されたい)、または32PPi取り込みアッセイ(たとえばOsslundら、1982年PlantPhysiol.70巻:39〜45頁を参照されたい)が使用することができる。
【0169】
ATPスルフリラーゼによって産生されるATPは、光を生成するために、酵素反応を使用して加水分解することができる。光放出化学反応(つまり化学発光)および生物学的反応(つまり生物発光)は、様々な代謝物質の高感度測定のために分析生化学において広く使用される。生物発光反応において、光の放出をもたらす化学反応は、酵素触媒することができる。たとえば、ルシフェリン−ルシフェラーゼシステムは、ATPの特異的なアッセイを可能にし、細菌ルシフェラーゼ−オキシドリダクターゼシステムは、NAD(P)Hをモニタリングするために使用することができる。両システムは、ATPまたはNAD(P)Hの産生または利用を含むカップル反応の手段によって、多数の物質の分析まで拡張されてきた(たとえばKricka、1991年Chemiluminescent and bioluminescent techniques. Clin.Chem.37巻:1472〜1281頁を参照されたい)。
【0170】
新しい試薬の開発は、ATP(たとえばLundin、1982年Applicationsof firefly luciferase In; Luminescent Assays(Raven Press、New Yorkを参照されたい)またはNAD(P)H(たとえばLovgrenら、Continuousmonitoring of NADH-converting reactions by bacterial luminescence. J. Appl.Biochem.4巻:103〜111頁を参照されたい)の濃度に比例した、安定性の光放出を得ることを可能にしてきた。そのような安定性の光放出試薬を用いると、知られている量のATPまたはNAD(P)Hの追加によって終点アッセイをし、個々のアッセイを較正することが可能である。さらに、安定性の光放出システムはまたATPまたはNAD(P)H変換システムの継続的なモニタリングを可能にする。
【0171】
ATPを光に変換するための適した酵素は、ルシフェラーゼ、たとえば昆虫ルシフェラーゼを含む。ルシフェラーゼは、触媒作用の最終産物として光を産生する。最もよく知られている光放出酵素は、ホタル、Photinus pyralis(甲虫目)の酵素である。対応する遺伝子は、クローニングされ、細菌(たとえばde Wetら、1985年Proc.Natl. Acad. Sci. USA80巻:7870〜7873頁を参照されたい)および植物において(たとえばOwら、1986年Science234巻:856〜859頁を参照されたい)ならびに昆虫(たとえばJhaら、1990年FEBSLett.274巻:24〜26頁)および哺乳動物細胞において(たとえばde Wetら、1987年Mol. Cell. Biol.7巻:725〜7373頁;Kellerら、1987年Proc.Natl. Acad. Sci. USA82巻:3264〜3268頁を参照されたい)発現されてきた。さらに、ジャマイカコメツキムシ、Pyroplorusplagiophihalamus(甲虫目)からの多くのルシフェラーゼ遺伝子は、最近、クローニングされ、部分的に特徴づけられてきた(たとえばWoodら、1989年J.Biolumin. Chemilumin.4巻:289〜301頁;Woodら、1989年Science244巻:700〜702頁を参照されたい)。別のルシフェラーゼは、時に、異なる波長の光を産生することができ、これにより、異なる波長での光放出の同時のモニタリングが可能となる可能性がある。したがって、これらの前述の特徴は、ユニークであり、現在のレポーターシステムの利用に関して新しい特質を追加する。
【0172】
ホタルルシフェラーゼは、ルシフェリン、アデノシン5’−3リン酸(ATP)、マグネシウムイオン、および酸素の存在下において生物発光を触媒し、0.88の量子収量をもたらしてもよい(たとえばMcElroyおよびSelinger、1960年Arch. Biochem. Biophys.88巻:136〜145頁を参照されたい)。ホタルルシフェラーゼ生物発光反応は、約1×10−13Mの検出限界で、ATPの検出のためのアッセイとして用いることができる(たとえばLeach、1981年J.Appl. Biochem.3巻:473〜517頁を参照されたい)。さらに、ルシフェラーゼ媒介検出システムの感度の全体的な程度および利便性は、ホタルルシフェラーゼベースのバイオセンサーの開発への相当な関心を引き起こした(たとえばGreenおよびKricka、1984年Talanta31巻:173〜176頁;Blumら、1989年J.Biolumin. Chemilumin.4巻:543〜550頁を参照されたい)。
【0173】
上記の酵素を使用して、配列プライマーを、ポリメラーゼおよび知られているdNTPにさらすことができる。dNTPが、プライマー配列の3’末端上に取り込まれる場合、dNTPが切断されてもよく、PPi分子が放たれてもよい。次いで、PPiは、ATPスルフリラーゼを用いてATPに変換することができる。ATPスルフリラーゼは、PPiの変換がPPiに関しての一次速度式で進む十分に高い濃度で存在してもよい。ルシフェラーゼの存在下において、ATPは、加水分解されて、光子を生成する。反応は、反応、ATP−−>DP+PO43−+光子(光)が、ATPに関しての一次速度式で進むように、反応混合物内に存在する十分な濃度のルシフェラーゼを有していてもよい。光子は、下記に記載される方法および器具を使用して測定することができる。PPiおよびカップルスルフリラーゼ/ルシフェラーゼ反応は、検出のための光を生成するために使用することができる。スルフリラーゼおよびルシフェラーゼのどちらかまたは両方は、それぞれの反応部位に配置される1つまたは複数の可動性固体支持体上に固定することができる。
【0174】
PPiは、リアルタイムシグナルを発するポリメラーゼ反応の間に検出されるように放出することができる。配列決定反応は、リアルタイムで継続的にモニターすることができる。反応は、2秒未満で起こってもよい(NyrenおよびLundin、前掲)。律速段階は、ATPスルフリラーゼによるATPへのPPiの変換であってもよいが、ルシフェラーゼ反応は速く、0.2秒未満を要すると推測された。ポリメラーゼの取り込み速度もまた、様々な方法によって推測され、たとえば、Klenowポリメラーゼの場合において、1つの塩基の完全な取り込みは、0.5秒未満を要する可能性があることが分かった。したがって、1つの塩基の取り込みおよびこの酵素アッセイによる検出のための推測される総時間は、約3秒である。そのため、非常に速い反応時間が可能であり、リアルタイム検出を可能にすることが分かるであろう。反応時間は、より耐熱性のルシフェラーゼを使用することによってさらに減少し得る。
【0175】
ほとんどの適用については、ATPおよびPPiのような混入物がない試薬が使用することができる。これらの混入物は、樹脂に結合されたアピラーゼおよび/またはピロホスファターゼを含有するプレカラムに試薬を流すことによって除去することができる。その代わりに、アピラーゼまたはピロホスファターゼは、磁性ビーズに結合されてもよく、試薬中に存在する混入しているATPおよびPPiを除去するために使用することができる。さらに、拡散性の配列決定試薬、たとえば取り込まれていないdNTPsは、洗浄緩衝液を用いて洗い流することができる。ピロリン酸配列決定において使用される任意の洗浄緩衝液が使用することができる。
【0176】
配列決定反応における反応物の濃度は、0.2ml緩衝液中に1pmol DNA、3pmolポリメラーゼ、40pmol dNTPを含んでもよい。Ronaghiら、Anal. Biochem.242巻:84〜89頁(1996年)を参照されたい。
【0177】
所望の場合、配列決定反応は、それぞれの4つの所定のヌクレオチドを用いて行なわれてもよい。「完全」サイクルは、所定の順において、ヌクレオチドDATP、dGTP、dCTP、およびdTTP(またはdUTP)のそれぞれについての配列決定試薬を順次、与えることを含んでもよい。取り込まれていないdNTPsは、それぞれのヌクレオチド追加の間に洗い流することができる。その代わりに、取り込まれていないdNTPsは、アピラーゼによって分解することができる。サイクルは、配列産物の所望の量の配列が得られるまで、所望されるように繰り返することができる。約10〜1000、10〜100、10〜75、20〜50、または約30ヌクレオチドの配列情報が、1つのアニールされた配列決定プライマーの伸長から得られてもよい。
【0178】
ヌクレオチドは、ビオチンなどのハプテンのジスルフィド誘導体を含有するように修飾することができる。アンカー基質にアニールした新生プライマーへの修飾ヌクレオチドの追加は、i)修飾がビオチンである例において、酵素分子に結合されたアビジンコンジュゲート部分またはストレプトアビジンコンジュゲート部分の順次結合、ii)過剰なアビジン結合酵素またはストレプトアビジン結合酵素の洗い流し、iii)酵素活性に適用可能な条件下での適した酵素基質の流入、およびiv)1つまたは複数の酵素基質反応産物の検出を含む重合後ステップによって分析することができる。ハプテンは還元剤の追加を通じて除去することができる。そのような方法は、電気泳動の必要性および可能性として危険な放射標識の使用を回避しながら、ヌクレオチドを所与の標的位置において同定することおよびDNAを簡単にかつ急速に配列決定することを可能にする。
【0179】
ハプテンを検出するための酵素は、ホースラディッシュペルオキシダーゼであってもよい。洗浄緩衝液は、本明細書において、様々な反応物の追加の間に使用することができる。アピラーゼは、配列決定プライマーを伸長させるために使用される、未反応dNTPを除去するために使用することができる。洗浄緩衝液は、アピラーゼを含んでもよい。
【0180】
例としてのハプテン、たとえばビオチン、ジゴキシゲニン、蛍光染料分子cy3およびcy5、ならびにフルオレスセインは、伸長DNA分子の中に様々な効率で取り込まれてもよい。ハプテンの付着は、糖、塩基を介しての、およびヌクレオチド上のリン酸部分を介しての結合を通して生じてもよい。シグナル増幅のための例としての手段は、蛍光手段、電気化学手段、および酵素手段を含む。酵素増幅が使用される場合、酵素、たとえばアルカリホスファターゼ(AP)、ホースラディッシュペルオキシダーゼ(HRP)、ベータ−ガラクトシダーゼ、ルシフェラーゼは、光生成基質が知られているものを含んでもよく、光生成(化学発光)基質の検出のための手段はCCDカメラを含んでもよい。
【0181】
修飾塩基は、追加されてもよく、検出が生じてもよく、ハプテンコンジュゲート部分は、除去されてもよく、または切断剤もしくは不活性化剤の使用によって不活性化することができる。たとえば、切断可能なリンカーがジスルフィドである場合、切断剤は、還元剤、たとえばジチオスレイトール(DTT)、ベータ−メルカプトエタノールなどであってもよい。不活性化はまた、熱、冷却、化学的変性剤、界面活性剤、疎水性試薬、または自殺型阻害剤によって成し遂げることができる。
【0182】
ルシフェラーゼは、光子の同時に起こる放出を伴ってdATPを直接、加水分解する可能性がある。加水分解は、伸長配列決定プライマーの中へのdATPの取り込みと無関係に生じるので、これは、偽陽性シグナルをもたらす可能性がある。この問題を回避するために、DNAの中に取り込まれるdATP類似体が使用することができる、つまり、それは、DNAポリメラーゼの基質であるが、ルシフェラーゼの基質ではない。1つのそのような類似体は、α−チオ−dATPである。したがって、α−チオ−dATPの使用により、成長している核酸鎖の中に取り込まれることなく、dATPが加水分解される場合に生じ得る偽性の光子生成を回避してもよい。
【0183】
PPiベースの検出は、配列決定プライマーの追加直後の、配列決定反応混合物へのコントロールヌクレオチドの追加に続いて放出される光の測定によって較正することができる。これにより、反応条件の標準化を可能にすることができる。2つ以上の連続した同一のヌクレオチドの取り込みは、放出される光の量における対応する増加によって明らかにすることができる。したがって、コントロールヌクレオチドと比較した、放出される光の2倍の増加により、伸長プライマーの中への2つの連続のdNTPsの取り込みを明らかにしてもよい。
【0184】
アピラーゼは、配列決定反応混合物内で、あらゆる残りの取り込まれなかったdNTPsの分解を容易にするために、固体支持体の表面に「薄く塗られてもよい」または「流することができる」。アピラーゼはまた、生成されるATPをも分解し、したがって、反応から生成される光を「消す」。アピラーゼを用いる処理の際に、あらゆる残りの反応物は、次に続くdNTPインキュベーションおよび光子検出のステップの調製中に洗い流することができる。その代わりに、アピラーゼは、固体支持体または可動性固体支持体に結合することができる。
【0185】
a.配列決定反応の検出
固体支持体は、画像システム230に光学的に結合されてもよく、これは、従来の光学機械または光ファイバー束と連携してCCDシステムを含んでもよい。灌流チャンバー基質は、水性インターフェースの近くで生成される光が、基質またはチャンバーの外部に光ファイバーを通して直接伝達することができるように、光ファイバーアレイウェーハを含んでもよい。CCDシステムが光ファイバーコネクターを含む場合、画像化は、灌流チャンバー基質をコネクターと直接接触させて配置することによって成し遂げることができる。その代わりに、従来の光学機械は、たとえば、光ファイバー基質の外部から直接、CCDセンサー上への1−1倍率高開口数レンズシステムを使用することによって、光を画像化するために使用することができる。基質が、光ファイバーカップリングを提供しない場合、レンズシステムはまた、上記に記載されるように使用されてもよく、この場合において、基質または灌流チャンバーカバーは光学的に透明である。
【0186】
画像システム230は、基質表面上で反応器から光を収集するために使用することができる。光は、たとえば、当技術分野において知られている高感度低ノイズ器具を使用してCCD上に画像化することができる。光ファイバーベースの画像化については、光ファイバーは、カバースリップの中に直接またはFORAについては、同様に、検出器に光を伝える光ファイバーであるマイクロウェルを形成する光ファイバーを有するように取り込まれてもよい。
【0187】
画像システムは、コンピューター制御およびデータ収集システム240に結合することができる。任意の一般的に入手可能なハードウェアおよびソフトウェアパッケージが使用することができる。コンピューター制御およびデータ収集システムはまた、試薬送達をコントロールするためにコンジット200に結合することができる。
【0188】
ピロリン酸配列決定反応によって生成される光子は、それらが焦点調整デバイス(たとえば光学用レンズまたは光ファイバー)を通過し、CCDエレメント上に集中する場合のみ、CCDによって捕捉することができる。しかしながら、放出される光子は、全方向に等しく流出する可能性がある。平面状のアレイ(たとえばDNAチップ)を用いる場合に、それらの、後の「捕捉」および定量化を最大限にするために、光子は、それらが生成されるポイントにできるだけ近くに、たとえば、平面状の固体支持体の直近に収集することができる。これは、(i)カバースリップおよび従来の光学用レンズもしくは光ファイバー束の間に光学用液浸油を用いることまたは(ii)カバースリップ自体の中に光ファイバーを直接、取り込むことによって成し遂げることができる。同様に、薄く、光学的に透明な平面状の表面が使用される場合、光ファイバー束はまた、その背面に対して配置され、全反応/灌流チャンバーの奥行きを通して「画像化する」必要性を排除してもよい。
【0189】
反応イベント、たとえば、ルシフェラーゼによって生成される光子は、多種多様の検出器具、たとえば、光電子増倍管、CCD、CMOS、吸光度光度計、ルミノメーター、電荷注入デバイス(CID)、または他の固体検出器および本明細書において記載される器具を使用して検出され、定量化することができる。放出される光子の定量化は、溶融光ファイバー束が取り付けられたCCDカメラの使用によって成し遂げることができる。放出される光子の定量化はまた、マイクロチャネルプレート増幅装置が取り付けられたCCDカメラの使用によって成し遂げることができる。背面薄化CCDは、感度を増加させるために使用することができる。CCD検出器は、たとえばBronksら、1995年Anal. Chem.65巻:2750〜2757頁において記載される。
【0190】
CCDシステムは、Lockheed−Martin LM485 CCDチップおよび6−8μmの個々のファイバー直径を有する1−1光ファイバーコネクター(束)を有するSpectral Instruments,Inc.(Tucson、Ariz.)シリーズ600 4ポートカメラであってもよい。このシステムは、4096×4096または1600万超のピクセルを有していてもよく、10%〜>40%に及ぶ量子効率を有する。したがって、波長に依存して、CCDセンサー上に画像化された40%もの光子は、検出可能な電子に変換される可能性がある。
【0191】
蛍光部分は、標識として使用されてもよく、反応イベントの検出は、レーザーを用いてアレイの表面をスキャンするために、共焦点走査型顕微鏡を使用して実行されてもよく、またはより小さな光学解像度が可能な走査型近接場光学顕微鏡(SNOM)などの他の技術が入手可能であり、それによって、「より密な」アレイの使用を可能にする。たとえば、SNOMを使用して、個々のポリヌクレオチドは、100nm、たとえば10nm×10nm未満の距離、離れている場合、識別される可能性がある。さらに、走査トンネル顕微鏡(Binningら、Helvetica Physica Acta、55巻:726〜735頁、1982年)および原子間力顕微鏡(Hanswaら、AnnuRev Biophys Biomol Struct、23巻:115〜139頁、1994年)が使用することができる。
【0192】
本発明は、次に続く非限定的な実施例によって示される、多数の態様を有する。
【実施例】
【0193】
(実施例1)
PACクローニングベクターの中にDNAをクローニングするためのλdoc粒子の使用
Tn7ドナープラスミドpGPS3(New England Biolabs)を使用して、pPAC/oriVベクターの完全なコピーを加えた、λcos部位を含有する転位性カセットを構築する(図1−1)。次いで、転位性カセットは、標的DNAの中に転位する(図1−1)。転位は、サザンブロッティングによって確認することができる。
【0194】
標的DNAの中への転位性エレメントの転位後に、それは、λ抽出物を用いてin vitroにおいてパッケージする(図1−2)。プロヘッドは満たされるであろうが、パッケージングは、第2のcos部位の欠如により完了しなくてもよい。欠けているcos部位を提供するために、頭部からはみ出たあらゆるDNAを除去するために、調製物は、Sau3Aを用いて消化する。次に、ファージ尾部を追加して、ヘッドフルDNAを含有するλdocLビリオンを産生する。ビリオンDNAの一方の末端は、cosLで終了し、他方は、Sau3A突出で終了する。
【0195】
パッケージDNA分子を環状にし、安定性のプラスミドを形成するために、第2のcos部位を、図3において示されるように、リンカーをアニールすることによってSau3Aに追加する。リンカーによって追加したcos部位は、DNAが環状になるまたはコンカテマーを形成することを可能にする。次いで、第2回のin vitroパッケージングは、それぞれの末端にcos突出を有するDNA分子を産生する。DNA分子は、環状になり、両端にλcos部位を有するカプシド形成DNAを有する完全に感染性の形態で再度パッケージされる。これらの調製物は、次いで、高効率でかつ好都合に、細胞の中にPACを導入するために使用される。
【0196】
(実施例2)
PACクローニングベクターの中にDNAをクローニングするためのλdoc粒子のさらなる使用
誘発性複製開始点を含むベクター構築物と一致する、ファージベースのサイズ選択およびランダムクローニングのためのバクテリオファージλ in vitroパッケージングを使用するクローニングシステムを設計した。このクローニングシステムは、バクテリオファージP1またはP7 in vitroパッケージングおよびより大きな(ヘッドフル容量)バクテリオファージを取り込むように拡張することができる。バクテリオファージin vitroパッケージングを使用するためのそのような手法を図2において示す。
【0197】
ファージパッケージング開始認識部位は、in vitro転位によって標的DNAの中に導入する。トランスポゾンは、ファージ特異的パッケージング開始部位、転位のための19bp Tn5モザイク末端、クローニングDNAの増幅のためのoriV、およびプラスミド選択のための抗生物質抵抗性を付与する遺伝子を含む。転位に続いて、バクテリオファージin vitroパッケージングシステムは、クローニングDNAをパッケージするために使用される。パッケージングは、ファージ特異的パッケージング部位で開始され、ファージカプシドのヘッドフル容量に達するまで続く。この容量、およびしたがってクローン挿入断片サイズは、使用する特異的なファージに基づくものであってもよい。
【0198】
ファージ頭部からはみ出たDNAおよびあらゆる非パッケージDNAは、DNアーゼIまたは4塩基認識制限エンドヌクレアーゼによって消化する。パッケージDNAは、ヌクレアーゼ消化から保護される。次いで、クローンの、後の環状化を可能にするDNAリンカーを、パッケージDNAの末端の末端にライゲーションする。その代わりに、パッケージDNAは、ファージ頭部から抽出し、その後、リンカーライゲーションおよび再度パッケージングを続けてもよい。次いで、ファージ尾部を追加し、クローニングDNAを含有するビリオンは、E.coliを形質移入し、その後、抗生物質抵抗性の選択を続けるために使用する。
【0199】
(実施例3)
PACクローニングベクターの中にDNAをクローニングするためのλdoc粒子の代替の使用
実施例1の手順を、プロヘッドがDNAで満たされるポイントまで行ない、次いで、Sau3Aを用いて切断する。ファージ尾部を直接、追加する代わりに、図3のリンカーを使用して、DNAがカプシドからはみ出ている一方で、cosR部位をλdoc粒子にライゲーションする。リンカーをライゲーションした後に、実施例1におけるように、尾部を追加する。正常なcosRおよびcosL末端を有するファージ粒子は、カプシドを破壊して開け、DNAを環状にし、再度パッケージすることが必要ではなく、細胞の中にPACを導入するために直接、使用されるので、これは、全体的なプロセスを単純化する。
【0200】
(実施例4)
in vivoトランスポゾン組換えクローニング
実施例1および3において、かなりの量のクローニング容量がベクターによって取られる。これは、PACベクターが、相当な長さを有し、したがって、そうでなければクローニングDNAに当てることができる、カプシドにおける空間を占めるという事実によって引き起こされる。この制限について検討するために、cos部位の側面に位置する修飾Tn5転位性末端、組換えのためのattP部位、および選択可能マーカーとしてのKanRを含む転位カセットを調製する。トランスポゾンは、ゲノムDNAの中にランダムにin vitroにおいて導入し、cos部位からパッケージし、Sau3A消化し、cosRリンカーを追加し、結果として生じた断片を精製し、再度パッケージする。次いで、挿入断片は、attP部位を含有する修飾PACベクターを含む株の中に導入する。これは、attBに対する同起源の組換え部位である。株は、IPTG誘発性Ptacプロモーター下でpHS3−1プラスミドからIntタンパク質を発現する(Leeら1990年)。宿主はまた、組換えがプラスミドとのみ生じることを確実にするためにIS−ともする。一度、細胞において、挿入断片がcos突出を介して環化したら、それらは、Int媒介性の組換えを通してPACの中に組換えられる。組換え体は、ベクター(CamR)および挿入断片(KanR)の両方に対する抗生物質を含有する培地上で平板培養することによって選択する。
【0201】
(実施例5)
転位性クローニングベクター
一連の転位性クローニングベクターは、バクテリオファージin vitroパッケージングを使用するクローニングのための形質を含むように構築した。これらの形質は、1)バクテリオファージλのファージ特異的パッケージング開始部位(cos)および162bp P1パッケージング開始部位(pac)、2)誘発性複製開始点、oriV、ならびに3)転位のための19bpモザイク末端配列が側面に位置するカナマイシン抵抗性遺伝子を含む。この構築物におけるモザイク末端は、高活性Tn5 in vitro転位システムに基づくものであり、トランスポゾンの発生を可能にするように設計し、パッケージ部位、CAT(クロラムフェニコールアセチルトランスフェラーゼ)遺伝子、oriV、およびPvuII制限酵素を用いる消化後のモザイク末端を含む。標的DNAの中へのトランスポゾンの組み込みは、クロラムフェニコール(Camr)についての選択によって確認することができ、カナマイシン(Kans)に対する感度についてさらにスクリーニングすることができる。cosファージパッケージング開始部位およびpacファージパッケージング開始部位の位置は、組み込みに続いて、パッケージングが、これらの部位で始まり、最終的に隣接標的DNAまで、ベクターを通して時計回りに続く(図4を参照)ように設計する。これは、クローンが、安定して選択され、パッケージング、形質移入、および環状化に続いてプラスミドとして維持されることを可能にする。
【0202】
cos、pac、ならびにcosおよびpacの両方を含有する一連の3つのプラスミドを構築した。最後に、Kanr遺伝子の側面に位置する19bpモザイク末端配列を、最終産物の3つのベクターに追加した。ベクターシリーズのそれぞれの構成要素を個々に試験し、予想通りに機能することを示した。3つのベクターはすべて、in vivoにおいて効率的に転位し、DH10Bゲノムの中に組み込まれることが示された。転位に続いて分析した形質転換体の約98%が、Camr Kansであり、組み込みが確認された。候補からのゲノムDNAのサザンブロッティングにより、ゲノムの中へのランダムな組み込みを確認した。
【0203】
cos機能性を2つの実験によって確認した。精製λターミナーゼ(Epicentre)を用いる単純な消化により、ベクターのcos切断を確認した。さらに、プラスミドマルチマーから構成されるコンカテマーが、λプロファージによってin vivoにおいてパッケージされ得るかどうかを決定するために、in vivoコスミドパッケージングアッセイを行なった。cosを含有する2つのベクターから産生されるコンカテマーは、Camr形質導入粒子の数によって測定されるように、効率的にパッケージされたが、pacのみを有するベクターは、in vivoにおいてλによってパッケージすることができなかった。
【0204】
(実施例6)
ファージパッケージング部位のライゲーション
パッケージ部位は、線状転位性クローニングベクターを、部分的に消化したゲノムDNAに直接、ライゲーションすることによって、標的DNAの中に導入する。EcoRI、BamHI、およびHindIII部位(または全多重クローニング部位)を、tL3ターミネーターおよびモザイク末端の間に位置するユニークなPmeI部位で、転位性クローニングベクターの中に導入する。EcoRI部分的消化物を使用する場合、転位性クローニングベクターは、EcoRIおよびPvuIIによって消化し、精製し、一方の末端に平滑末端化PvuIIおよびpac/cos部位ならびに反対の末端にEcoRI付着末端を有する線状転位性クローニングベクターがもたらされる。EcoRI/PvuII二重消化物は、ベクターエレメントを含有するクローンを用いる一方向のパッケージングをもたらす。
【0205】
線状転位性クローニングベクターを、部分的に消化したゲノムDNAにライゲーションする。次いで、in vitroパッケージング反応を行ない、転位性クローニングベクターの構成要素を通して、転位性クローニングベクター上のパッケージ部位でパッケージングを開始し、ファージ頭部のヘッドフル容量に達するまで、ライゲーションゲノムDNAまで続ける。転位によるパッケージング開始部位の導入と同様に、はみ出たDNAは消化し、適切なリンカーは、DNAの環状化のためにライゲーションし、ファージ尾部を追加し、使用したクローニングDNAを含有するビリオンをE.coliに形質移入する。
【0206】
(実施例7)
λまたはP1を用いるin vitroパッケージング
市販で入手可能なλ in vitroパッケージング抽出物は、シングルチューブシステムとして、段階1および段階2のパッケージング抽出物を組み合わせたものである。クローニングシステムは、順次、2つのパッケージ段階を順次使用してもよい。2つの従来の相補的な溶原性E.coli株BHB2690(段階1)およびBHB2688(段階2)からの抽出物を生成し、試験した。2段階パッケージングシステムの効率は、市販で入手可能なシングルチューブシステムに匹敵した。さらに、発明者らは、段階1抽出物を使用して、パッケージベクターのcos切断を示し、実施例5において記載されるベクターの機能性を確認することができた。バクテリオファージP1のin vitroパッケージングシステムについては、発明者らは、それぞれ、株NS3208およびNS3210から、P1溶原菌の段階1および段階2の抽出物を生成した(Corenら、J Mol Biol、249巻:176〜84頁、1995)。発明者らはまた、段階1(pacase)抽出物を使用して、パッケージベクターのpac部位の切断をも示した。
【0207】
(実施例8)
λファージパッケージングを使用するゲノムDNAのクローニングおよびファージカプシドのアフィニティー精製
本実施例は、λファージパッケージングを使用するDNAのクローニングおよびファージカプシドのアフィニティー精製について記載する。cos部位、複製開始点、および薬剤抵抗性マーカーを含有する核酸は、たとえばin vitro転位によって、ゲノムDNAの中にランダムに挿入する。λターミナーゼ(Nu1およびAの遺伝子の産物を含有する)は、cos部位におよびλプロヘッドに結合し、次いで、cos部位でDNAを切断する(図6)。次いで、ATP駆動モーターは、プロヘッドの中にDNAを詰め込む。ウイルスカプシドは、カプシドへのDタンパク質の追加によって、DNAがその中に挿入されるとともに拡張する。約50kbのDNAがカプシドを満たすまで、このプロセスは続く。カプシドからぶら下がったままのDNAは、頻繁に切断する制限部位を用いて切断することができる。カプシドの内部にあるDNAは保護されるので、カプシドの外部にあるDNAのみを制限酵素によって切断することができる。
【0208】
DNAの制限に続いて、ファージカプシドは、カラムに結合した抗Dタンパク質抗体を使用してアフィニティー精製する。精製カプシドからのDNAは、フェノール抽出によって単離することができる。次いで、DNAは環状にし、細菌の中に形質転換する。
【0209】
(実施例9)
P22HTファージパッケージングを使用するゲノムDNAのクローニング
本実施例は、P22HTファージパッケージングを使用するゲノムDNAのクローニングについて記載する。Gp2およびGp3のタンパク質を含有するP22HTターミナーゼは、ランダムな部位でゲノムDNAに結合し、それを切断する(図7)。ターミナーゼは、DNAに結合したままであり、プロヘッドがいっぱいになるまで、P22プロヘッドの中にそれを詰め込む。カプシドが拡張するとともに、それは、広がり、破壊されたDNAになお付着しているターミナーゼを放出する立体構造変化を受ける。次いで、ターミナーゼは、新しいプロヘッドの中にDNAを挿入し、DNAパッケージングの新しいサイクルを始めることができる。ターミナーゼは、隣接ゲノムDNAセグメントの連続パッケージングの多くのサイクルを続けることができる。
【0210】
パッケージDNAをクローニングするために、ファージカプシドは、拡張されたカプシドの外部に存在するエピトープに結合することができる抗体を含有するカラムを使用してアフィニティー精製することができる。カプシドはまた、差次的沈降または等密度遠心分離によって単離することもできる。これらの方法は、DNアーゼ消化が伴い得る。DNAは、上記に記載されるように、さらにそれを抽出し、環状にし、細菌の中に形質転換することによってクローニングすることができる。
【0211】
(実施例10)
ファージパッケージングによってクローニングされたゲノムDNAの末端の配列の特徴づけ
本実施例は、ファージベースのシステムを使用してパッケージされたゲノムDNAの末端の配列をどのように特徴づけることができるかについて記載する。ゲノムDNAは、ファージカプシドから最初に単離される。次に、EcoP15I部位が側面に位置する2つの外向きリーチングプライマー配列を含有する核酸を、単離DNA断片の末端にライゲーションする(図8)。図9Bは、リーチングプライマー対核酸の構造を示す。リーチングプライマー対断片は、2つの異なるゲノムDNA分子へのリーチングプライマー対断片のライゲーションを回避するのに十分に低い稀釈度で単離DNAにライゲーションする。リーチングプライマー対断片の末端は、それとモノマー環を形成することを回避するためにリン酸化されなくてもよい。
【0212】
ライゲーションに続いて、DNAをEcoP15Iを用いて消化し、これにより、EcoP15I認識部位から27bp離れたところでDNAを切断する。この制限酵素を用いるEcoR消化は、2つのリーチングプライマー配列、側面に位置するEcoP15I部位、およびそれぞれの末端に27bpのゲノムDNAを含有するDNA断片を放出する。放出断片におけるゲノムDNAは、パッケージゲノムDNAのそれぞれの末端を表わす。放出断片は、フィルインすることができる。図9Aは、平滑末端を作り出すために、放出断片のEcoP15I切断末端をどのようにフィルインすることができるかを示す。次いで、放出平滑断片は、ライゲーションして、対になった末端環DNAを作り出すことができる。次いで、このDNAは、2つのリーチングプライマーを使用して、PCR増幅することができ(図10)、次いで、増幅産物は、配列決定することができる。リーチングプライマー対部位の間に制限部位がある場合、対になった末端環は、PCR増幅の前に適切な制限酵素を用いて切断することによって線状にしてもよい。DNAはまた、固相核酸増幅を使用して増幅を行なうことによって、PCR増幅し、直接配列決定してもよい。上記に記載されるアプローチはまた、ビーズエマルションPCR増幅および配列決定における使用のために、磁性ビーズに結合させたリーチングプライマーのうち1つに適応することができる(図11)。
【技術分野】
【0001】
本発明は、一般に、分子生物学の分野に関する。より具体的には、本発明は、核酸分子のクローニングおよび核酸ライブラリーの産生に関する。
【背景技術】
【0002】
ゲノム学は、生物医学研究の中心的で統合的な学問になった。ゲノム配列決定プロジェクトは、事実上すべての生命過程の研究を変えた、安定した一連のさらに大きく、より複雑なゲノムデータセットを生成する。遺伝学および比較ゲノム学の進歩は、疾患が、分子の細部についての、前例がないレベルで分析され、かつ理解されることを可能にする。
【0003】
ゲノム研究は、高品質で、大きな挿入断片のゲノムライブラリーの有用性を必要とする。高品質ゲノムライブラリーは、狭いサイズ範囲のDNA断片を必要とする。DNA断片のサイズ選択は、パルスフィールドゲル電気泳動(PFGE)を使用して、典型的に行なわれるが、これは、いくつかの制限を有する。PFGEは、非常に時間がかかり、再現不可能である。さらに、PFGEからの断片DNAの収量は、非常に乏しい。
【0004】
高品質ゲノムライブラリーはまた、全ゲノムが、最小限のバイアスを伴って表わされることをも必要とする。ゲノムライブラリーのDNA断片は、ゲノムを部分的に消化するための制限酵素を使用して、産生されることが多いが、これは、いくつかの制限を有する。ゲノムの部分的な消化は、制限部位の間の距離が、適用されるサイズ選択限界未満であり、またはそれを超えるゲノムの領域に対してバイアスをもたらす。部分的な消化と関連するクローニングバイアスに対する代案として、DNA断片は、ゲノムをランダムに剪断することによって産生することができる。しかしながら、クローニング効率は、ランダムに剪断されたDNAを使用すると、著しく低下する。
【発明の概要】
【発明が解決しようとする課題】
【0005】
当技術分野が必要とするのは、全ゲノムを正確に表わすことができる高品質ゲノムライブラリーである。ゲノムライブラリーは、効率的に、かつ高収量を伴って産生されるべきである。ゲノムライブラリーはまた、容易に増殖され、かつ分析されるべきでもある。本発明は、これらの必要性を満たす。
【課題を解決するための手段】
【0006】
本発明は、核酸をクローニングし配列決定するため、および複数の核酸をクローニングし配列決定するためのパッケージングシステムの使用に関する。パッケージングシステムは、たとえば、ファージベースのパッケージングメカニズムであってもよい。核酸をカプシドの中にパッケージすることができ、このカプシドはバクテリオファージカプシドであってもよい。クローニングされた核酸は、たとえば、核酸ライブラリーの産生において使用することができる。
【0007】
核酸を配列決定するための方法であって、核酸を含むカプシドを提供するステップと、核酸を単離するステップと、核酸を配列決定するステップとを含み得る方法が本明細書において提供される。複数の核酸を配列決定するための方法であって、それぞれが核酸を含む複数のカプシドを提供するステップと、核酸を単離するステップと、核酸を配列決定するステップとを含み得る方法もまた本明細書において提供される。
【0008】
配列決定は、(a)水性マイクロリアクターが、核酸の単一コピー、核酸に結合することができる単一ビーズ、および核酸増幅を行なうために必要な試薬を含有する増幅反応溶液を含むように、油中水型エマルション中で水性マイクロリアクターの中に核酸を送達するステップと、(b)マイクロリアクター中で核酸を増幅して核酸の増幅コピーを形成し、マイクロリアクター中で増幅コピーをビーズに結合させるステップと、(c)平面表面上にある少なくとも10,000個の反応チャンバーのアレイにビーズを送達するステップであって、複数の反応チャンバーが単一核酸結合ビーズしか含まないステップと、(d)複数の反応チャンバー上で配列決定反応を同時に行なうステップとを含んでもよい。
【0009】
配列決定はまた、(a)複数の水性マイクロリアクターが、核酸の単一コピー、核酸に結合することができる単一ビーズ、および核酸増幅を行なうために必要な試薬を含有する増幅反応溶液を含むように、油中水型エマルション中で水性マイクロリアクターの中に複数の核酸を送達するステップと、(b)マイクロリアクター中で核酸を増幅して核酸の増幅コピーを形成し、マイクロリアクター中で増幅コピーをビーズに結合させるステップと、(c)平面表面上にある少なくとも10,000個の反応チャンバーのアレイにビーズを送達するステップであって、複数の反応チャンバーが単一核酸結合ビーズしか含まないステップと、
(d)複数の反応チャンバー上で配列決定反応を同時に行なうステップとを含んでもよい。
【0010】
ビーズは、少なくとも10,000個の増幅コピーに結合してもよい。核酸の増幅は、ポリメラーゼ連鎖反応を使用することによって成し遂げることができる。配列決定反応は、ピロリン酸ベースの配列決定反応であってもよい。配列決定反応は、(a)有効量の配列決定プライマーを核酸の増幅コピーにアニールし、ポリメラーゼおよび所定のヌクレオチド三リン酸を用いて配列決定プライマーを伸長させて、配列決定産物、および所定のヌクレオチド三リン酸が配列決定プライマーの3’末端上に取り込まれる場合には配列決定反応副産物を得るステップと、(b)配列決定反応副産物を同定し、それによって、反応チャンバー中で核酸の配列を決定するステップとを含んでもよい。
【0011】
核酸または複数の核酸の単離は、(a)第1のアダプター末端および第2のアダプター末端を少なくとも1つの核酸にライゲーションするステップであって、第1のアダプター末端が、オリゴヌクレオチド配列Yを含み、少なくとも1つの核酸の5’末端にライゲーションし、第2のアダプター末端が、オリゴヌクレオチド配列Zを含み、少なくとも1つの核酸の3’末端にライゲーションし、第1のアダプターが、5’末端で固体支持体に少なくとも1つの核酸を固定するための手段を有するステップと、(b)ステップ(a)の少なくとも1つの核酸を、固体支持体の存在下において、1つまたは複数のコロニープライマーXと混合するステップであって、それぞれのコロニープライマーXが、オリゴヌクレオチド配列Zにハイブリダイズすることができ、5’末端で固体支持体にコロニープライマーを固定するための手段を有し、それによって、少なくとも1つの核酸およびコロニープライマーの両方の5’末端が、固体支持体に固定され、少なくとも1つの核酸およびコロニープライマーの両方の上述の5’末端が、DNA変性条件下での水または水性緩衝液を用いる洗浄によって、それらを除去することができないように、上述の固体支持体に固定されるステップと、(c)核酸コロニーが生成されるように、固定された少なくとも1つの核酸上で1つまたは複数の核酸増幅反応を行なうステップとを含んでもよい。コロニープライマーは、縮重プライマーであってもよい。
【0012】
オリゴヌクレオチド配列Zは、オリゴヌクレオチド配列Yに相補的であってもよく、コロニープライマーXは、オリゴヌクレオチド配列Yと同じ配列であってもよい。核酸にアダプター末端をライゲーションする場合、2つの異なるコロニープライマーXは、少なくとも1つの核酸と混合してもよく、オリゴヌクレオチド配列Zは、コロニープライマーXのうち1つにハイブリダイズしてもよく、オリゴヌクレオチドYは、コロニープライマーXのうち1つの配列と同じであってもよい。
【0013】
少なくとも1つの核酸は、1つまたは複数の核酸コロニーにおいて配列決定することができる。配列決定は、標識ヌクレオチドの取り込みおよび検出を含んでもよく、さらに、核酸コロニーを視覚化することを含んでもよい。視覚化は、標識核酸プローブまたは非標識核酸プローブの使用を含んでもよい。
【0014】
少なくとも1つの核酸およびコロニープライマーを固体支持体に固定するための手段は、少なくとも1つの核酸およびコロニープライマーを支持体に共有結合で固定するための手段を含んでもよい。少なくとも1つの核酸およびコロニープライマーを固体支持体に共有結合で固定するための手段は、化学的に修飾された基であってもよい。少なくとも1つの核酸およびコロニープライマーを固体支持体に固定するための手段は、少なくとも1つの核酸およびコロニープライマーを支持体に共有結合で固定するための手段を含んでもよい。化学的に修飾可能な官能基は、アミノ基であってもよい。
【0015】
少なくとも1つの核酸およびコロニープライマーの両方の上述の5’末端が固定される固体支持体は、ラテックスビーズ、デキストランビーズ、ポリスチレンおよびポリプロピレン表面、ポリアクリルアミドゲル、金表面、ガラス表面、ならびにシリコンウェーハからなる群から選択することができる。固体支持体上の核酸コロニーの密度は、10,000/mm2〜100,000/mm2であってもよい。固体支持体に付着したコロニープライマーXの密度は、少なくとも1fmol/mm2であってもよい。少なくとも1つの核酸の密度は、10,000/mm2〜100,000/mm2であってもよい。少なくとも1つの核酸およびコロニープライマーの両方の5’末端は、共有結合を介して上述の固体支持体に固定することができる。
【0016】
核酸をクローニングするための方法もまた本明細書において提供される。パッケージング開始部位(PIS)を標的核酸の中に導入することができ、これは、標的核酸からクローニングされる核酸の上流末端を確立する。PISは、λ、P1、P7、またはT4を含むが、これらに限定されないファージパッケージング開始部位であってもよい。標的核酸は、ベクター、染色体DNA、または全ゲノムを含むが、これらに限定されない、核酸の任意の形態であってもよい。標的核酸としての全ゲノムの使用により、ゲノムライブラリーの産生が可能となる可能性がある。
【0017】
PISは、標的核酸にランダムに導入することができ、これは、ゲノムの範囲を増加させる可能性がある。PISは、転位およびライゲーションを含むが、これらに限定されない任意の方式において標的核酸の中に導入することができる。PISは、第1の核酸の転位によって導入することができる。第1の核酸は、Tn7、Tn5、Tn10、Mu、およびMarinerを含むが、これらに限定されない転位性末端を含んでもよい。
【0018】
次いで、パッケージングは、PISで開始することができ、これは、標的核酸に及ぶ、PISから下流の核酸のクローニングをもたらす。パッケージングは、in vitroにおいてまたはin vivoにおいて生じてもよい。クローニングされた核酸は、カプシド中にパッケージすることができ、これは、クローンを実質的に一定の長さにする可能性がある。
【0019】
第1の核酸はまた、ベクターエレメントを含んでもよい。ベクターエレメントは、PISの下流にあってもよく、クローニングされた核酸中に存在してもよい。ベクターエレメントは、低コピー複製開始点および高コピー複製開始点を含むが、これらに限定されない複製開始点であってもよい。ベクターエレメントはまた、高コピー複製開始点および低コピー複製開始点であってもよい。高コピー複製開始点は、複製誘発剤に応答性であってもよい。第1の核酸は、複製誘発剤をコードする核酸を含んでもよい。複製誘発剤は、TrfAであってもよい。
【図面の簡単な説明】
【0020】
【図1】λdoc粒子を使用する、トランスポゾン媒介クローニングの手法を示す図である。ステップ1:in vitro転位反応は、転位性末端(外向きの矢印)を介して、ゲノムDNA(長い直線)の全体にわたる多数の部位に、トランスポゾンを含有するPAC/oriVをランダムに挿入する。ステップ2:DNAは、in vitroにおいてパッケージされ、cos部位で始まり、頭部がいっぱいになるまで続く。次いで、はみ出たDNAは、Sau3Aを用いて消化されるが、頭部内のDNAは、保護される。ステップ3:次いで、ファージ頭部は、精製され、溶解され、Sau3A突出を有するゲノムDNAに融合された、一定の長さの、一般的な組成のcos−PAC/oriV(太い矢印)の断片がもたらされる。次いで、Sau3A突出およびcos突出を有するオリゴは、断片にライゲーションされる。ステップ4:断片は、再度パッケージされ、入ってくるDNAがcos末端を通して環状になる、DH10Bなどの宿主を感染させるために使用される。クローンは、CamRコロニーとして単離される。
【図2】in vitro転位およびファージパッケージングを介してクローニングするためのさらなる模式図を示す図である。
【図3】Sau3A突出を有するDNA断片にcos部位を追加するための、2つの核酸(配列番号1および2)を含むリンカーを示す図である。
【図4】トランスポゾン媒介クローニングの使用に基づくin vivo組換えのための手順を示す図である。ステップ1:図1におけるように、in vitro転位反応は、転位性末端(外向きの矢印)を介して、ゲノムDNA(長い直線)の全体にわたる多数の部位に、トランスポゾンをランダムに挿入する。しかしながら、この場合、トランスポゾンは、複製開始点を有してないcos−attB−KanRを含有する。ステップ2:DNAは、in vitroにおいてパッケージされ、cos部位で始まり、頭部がいっぱいになるまで続く。次いで、はみ出たDNAは、Sau3Aを用いて消化されるが、頭部内のDNAは、保護される。ステップ3:次いで、ファージ頭部は、精製され、溶解され、Sau3A突出を有するゲノムDNAに融合された、一定の長さの、一般的な組成のcos−PAC/oriV(太い矢印)の断片がもたらされる。次いで、Sau3A突出およびcos突出を有するオリゴが、断片にライゲーションされる。ステップ4:断片は、再度パッケージされ、新しいpPAC/oriV/attPベクターを含有し、Intを発現するように誘発された、DH10Bなどの宿主を感染させるために使用される。cos末端を介しての次に続く環化は、CamR付与プラスミドをもたらす。
【図5】転位性の/パッケージすることができるベクターを示す図である。ベクターのPvuII切断は、in vitroにおいて転位を可能にするTn5モザイク末端(ME)が側面に位置する線状の断片をもたらす。次いで、pac部位およびcos部位は、転位産物がin vitroにおいてパッケージされることを可能にする。
【図6】ファージカプシドのアフィニティー精製およびパッケージDNAの単離が後に続くλファージベースのパッケージングシステムを使用して、ゲノムDNAライブラリーを生成するための戦略を示す図である。
【図7】P22HT「ヘッドフル(headful)」パッケージングシステムを使用して、ゲノムDNAライブラリーを生成するための戦略を示す図である。
【図8】ウイルスカプシドパッケージDNAの末端の配列をクローニングし、かつ特徴づけるための戦略を示す図である。
【図9】ファージパッケージDNAの中へのクラスIII制限酵素部位およびリーチングプライマー対結合部位のクローニングが、DNAの末端の配列を特徴づけるためにどのように使用することができるかを示す図である。
【図10】DNAの中にリーチングプライマー対挿入断片をクローニングした後に、ファージパッケージDNAの末端をどのようにしてPCR増幅することができるかを示す図である。
【図11】DNAの中にリーチングプライマー対挿入断片をクローニングした後に、ファージパッケージDNAの末端をどのように配列決定することができるかを示す図である。
【発明を実施するための形態】
【0021】
発明者らは、より正確に表わす、標的DNAを単離し、パッケージするために使用することができるファージパッケージングシステムを開発した。使用されるファージパッケージングシステムに依存して、システムは、特定の、より一定のサイズのDNA断片を生成する。さらに、標的DNAは、標的DNAを、扱いやすく、クローニングすることができるサイズに低下させるために剪断ステップまたは制限消化ステップを必要とする方法と比較して、よりライゲーション可能である。したがって、標的DNAを得るための効率は、他の方法と比較して高い。標的DNAのパッケージングに続いて、DNAは、ライブラリーの生成などの多数の目的のために使用することができ、それを抽出することができ、それを増幅することができ、またはパッケージDNA断片の末端のクローニングおよび配列決定などによってDNAを配列決定することができる。
【0022】
DNA断片は、標的DNAの中にパッケージング開始部位(PIS)を導入することによって生成することができる。PISは、標的核酸からクローニングされる核酸の上流末端を確立する。パッケージングシステムによるPISの認識は、PISでのパッケージングの開始をもたらしてもよく、これは、標的核酸に及ぶ、PISから下流の核酸のクローニングをもたらす。核酸のライブラリーは、標的核酸から複数の核酸をクローニングすることによって産生することができる。ゲノムライブラリーは、ゲノムから複数の核酸をクローニングすることによって産生することができる。
【0023】
1.パッケージングシステム
核酸は、パッケージング開始部位(PIS)でのパッケージングを開始することによって、標的核酸からクローニングすることができる。PISは、ターミナーゼによって認識されてもよく、これは、部位特異的にまたはランダムに標的核酸に切れ目を入れてもよく、これは、カプシドの中に核酸を挿入してもよい。したがって、クローニングされた核酸は、カプシド中にパッケージすることができる。ターミナーゼは、2つのタンパク質を含んでもよく、これらは、パッケージングシステムが機能することを可能にするATPアーゼ活性を有していてもよい。
【0024】
カプシドは、5〜160kbのサイズであってもよい核酸をパッケージすることができてもよい。複数のカプシドは、それぞれ、核酸をパッケージすることができてもよく、カプシドによってパッケージされる核酸は、ほぼ同じサイズであってもよい。サイズ選択のそのような形態は、PFGE電気泳動と関連する非効率に対する主な改善である。さらに、カプシドの感染特性は、後の操作において、エレクトロポレーションよりも高い率で、パッケージされた核酸の導入を可能にすることができる。
【0025】
使用されるパッケージングシステムは、真核生物または原核生物のものであってもよい。たとえば、システムは、表1に挙げられるバクテリオファージから選択することができる。
【0026】
【表1】
パッケージングシステムはまた、ポックスウイルス、ヘルペスウイルス、アデノウイルス、レンチウイルス、またはエプスタイン−バーウイルスから選択することができる。
【0027】
a.パッケージング開始部位
PISは、パッケージング開始部位であってもよい。パッケージング開始部位の代表的な例は、バクテリオファージλ、P1、P7、T4、T7、Φ29、またはP22パッケージング開始部位を含むが、これらに限定されない。PISは、pac部位またはcos部位であってもよい。PISはまた、長末端反復などの真核生物ウイルスPISであってもよい。
【0028】
PISは、標的核酸の中に、PISを含む第1の核酸を導入することによって、標的核酸の中に導入することができる。第1の核酸はまた、ベクターエレメントを含んでもよい。第1の核酸はまた、転位性エレメントを含んでもよい。転位性エレメントは、PISを含む核酸セグメントの側面に位置する1対の転位性末端を含んでもよい。転位性エレメントは、Tn7、Tn5、Tn10、Minos、Mu、およびMarinerを含むが、これらに限定されない、システムからの転位性末端を含んでもよい。核酸セグメントはまた、ベクターエレメントを含んでもよい。
【0029】
第1の核酸は、ランダム方式または非ランダム方式のいずれかにおいて標的核酸の中に導入することができる。PISを含む第1の核酸はまた、突然変異誘発を含むが、これに限定されない多くの標準的な分子生物学手法のいずれかによって、標的核酸の中に導入することができる。PISを含む第1の核酸は、in vitroにおいてまたはin vivoにおいて、転位によって標的核酸の中に導入することができる。第1の核酸は、標的DNAの中への転位性エレメントの転位を可能にする任意の形態をしていてもよい。第1の核酸は、線状DNAまたは環状DNAの形態をしていてもよく、これは、スーパーコイル核酸またはリラックス核酸であってもよい。第1の核酸は、プラスミドの一部であってもよい。第1の核酸はまた、バクテリオファージを含むが、これに限定されない複製可能な遺伝子パッケージであってもよい。
【0030】
トランスポサーゼは、標的核酸の中への第1の核酸の転位を触媒するために、in vitroにおいてまたはin vivoにおいて使用することができる。転位は、ランダムに生じてもよく、その程度は、特定のトランスポゾンシステムに依存して異なっていてもよい。
【0031】
転位は、たとえば、市販で入手可能な転位システムを使用して、in vitroにおいて生じてもよい。転位はまた、米国特許第5,965,443号、第5,948,622号、または第5,925,545号において開示される方法を使用して、in vitroにおいて生じてもよく、これらの内容は、参照によって本明細書において組み込まれる。in vitro転位については、標的核酸は、染色体DNA、消化DNA、剪断DNA、サイズ特異的DNA、および全ゲノムを含むが、これらに限定されない、核酸の任意の形態であってもよい。
【0032】
転位はまた、トランスポサーゼを発現する宿主細胞の中に第1の核酸を導入することによってin vivoにおいて生じてもよい。第1の核酸は、エレクトロポレーションおよび感染を含むが、これらに限定されない方法によって宿主細胞の中に導入することができる。in vivo転位については、標的核酸は、レプリコンを含む、核酸の任意の形態であってもよい。レプリコンは、ベクターまたは染色体であってもよい。
【0033】
PISを含む第1の核酸はまた、標的核酸に第1の核酸をライゲーションすることによって、標的核酸の中に導入することができる。ライゲーションされた産物は、線状または環状であってもよい。標的核酸は、in vitro転位について上記に説明される核酸の形態を含むが、これらに限定されない、核酸の任意の形態であってもよい。標的核酸は、ランダム剪断および制限消化を含むが、これらに限定されない方法によって生成することができる。第1の核酸および標的核酸は、適合性の末端を有していてもよい。その代わりに、リンカーが使用することができる。
【0034】
標的核酸は、HindIIIなどの第1の制限酵素を用いて消化されてもよく、これは、部分的であってもよい。標的核酸はまた、第2の制限酵素を用いて消化することができる。第1の核酸は、消化された標的核酸と適合性の末端を有していてもよい。第1の核酸および標的核酸をライゲーションした後に、PISを含む、結果として生じる標的核酸は、ファージパッケージングのための基質としてもよい。
【0035】
PISは、標的核酸に固有のものであってもよく、または標的DNA中に先在していてもよい。PISはまた、DNA切断によって標的核酸の中に導入することができる。切断は、ターミナーゼと標的核酸を接触させることによって行なわれてもよく、これは、ランダムな位置で標的核酸を切断してもよい。ターミナーゼは、ファージP22からのなどのように、ファージターミナーゼであってもよく、これは、高形質導入P22であってもよい。ターミナーゼは、ファージP22のポリペプチドGp3およびGp2を含んでもよい。
【0036】
b.ベクターエレメント
PISは、ベクターエレメントと共に標的核酸の中に導入することができる。ベクターエレメントは、PISの下流または上流にあってもよい。下流PISは、クローニングされた核酸中に含まれていてもよい。クローニングされた核酸中のベクターエレメントは、クローニングDNAの、後の操作および増殖のために使用することができる。
【0037】
(1)複製開始点
ベクターエレメントは、複製開始点であってもよい。複製開始点は、低コピー複製開始点であってもよい。低コピー複製開始点はまた、再編成を最小限にしながら、宿主細胞中のクローニングDNAを維持するために使用することができる。低コピー複製開始点の代表的な例は、oriSである。
【0038】
複製開始点はまた、高コピー複製開始点であってもよい。高コピー複製開始点は、より多量のクローニングDNAを産生するために使用することができる。低コピー複製開始点の代表的な例は、oriVである。
【0039】
ベクターエレメントはまた、高コピー複製開始点および低コピー複製開始点であってもよい。高コピー複製開始点は、誘発性プロモーターのコントロール下としてもよい。クローニングされた核酸は、低コピー複製開始点を使用して維持できるが、高コピー複製開始点の誘発によって多量に産生することができる。そのようなシステムは、Hradecnaら1998年およびWildら1998年によって記載され、これらの内容は、参照によって本明細書において組み込まれる。
【0040】
(2)マーカー
ベクターエレメントはまた、選択可能マーカーまたは可視マーカーであってもよい。選択可能マーカーまたは可視マーカーは、クローニングDNAを維持し、かつ操作するために使用することができる。マーカーの代表的な例は、AmpR、CamR、KanR、lacZ、およびGFPを含むが、これらに限定されない。
【0041】
(3)組み込み部位
ベクターエレメントはまた、組み込み部位であってもよい。組み込み部位は、相補的な組み込み部位を含む第2の核酸の中にクローニングされた核酸を組み込むことを可能にすることができる。第2の核酸はまた、ベクターエレメントを含んでもよい。総合システムの使用は、いくつかの重要な利点を有する。クローニングされた核酸の、後の操作に必要とされる多くの配列が、第1の核酸の代わりに複製プラスミドベクター上に配置することができる。このことから、クローニングされた核酸をパッケージするためにカプシドを使用する場合に、より多くの標的核酸をクローニングすることが可能となる可能性がある。
【0042】
組み込み部位および相補的な組み込み部位の例は、λシステムおよびCre−loxシステムを含むが、これらに限定されない。λファージが、E.coliに感染する場合、それは、細胞を溶解して発達する可能性があり、または細菌染色体中の特異的なポイント、attBにおいて組み込むことによって、溶原菌を確立する可能性がある。組み込みは、λ上のattPおよびattBの間の部位特異的組換えによって媒介され、連続した染色体の中へのλの線状の挿入をもたらす。attPは、約250bp長であるが、attBは、わずか約20bpである。ファージint遺伝子の産物は、宿主因子IHFと共に、attPおよびattBの間のクロスオーバー反応を触媒し、それによって組み込みを引き起こす。
【0043】
第1の核酸は、PISの下流にattBを含んでもよく、第2の核酸は、複製可能であり、attPを含む。パッケージングを開始した後に、クローニングされたものは、複製可能な第2の核酸の中に組み込むことができる。同様の手法を用いて、DNA断片は、Janusベクターの中に挿入された(Burlandら1993年)、これは、参照によって本明細書において組み込まれる。
【0044】
c.λパッケージング
パッケージングシステムは、パッケージングを開始し終了するための特異的な部位を使用してもよい。正常なファージの発達において、ビリオン当たりのDNAの長さは、頭部の容量によって直接決定されないので、λは「ヘッドフル」タイプのファージではない。代わりに、λは、パッケージングを開始し、かつ終了するために高度に特異的なcos部位を使用する。これらの部位は、ターミナーゼによって切断されて、パッケージDNAの末端に12塩基突出(cosLおよびcosR)を残す。λの自然のローリングサークルパッケージング基質中で、cos部位は、頭部サイズによって決定されるフルパッケージングリミットよりも、共に近くに、間隔をおいて配置される。その結果として、頭部容量ではなくcos部位間隔が、通常、ビリオンDNAの長さを決定する。
【0045】
d.ヘッドフルパッケージング
「ヘッドフル」パッケージングシステムもまた使用することができる。ヘッドフルパッケージングシステムは、線状の連続移動方式において、ファージプロヘッドの空洞の中にクローニングされた核酸を供給し、それが、内部のDNAが、進行を停止するのに十分な、内壁に対する圧力をかける限界に達するまで、頭部を拡張させてもよい。これは、頭部における立体構造変化を誘発してもよく、これは、入ってくるDNAのエンドヌクレアーゼによる切断を活性化し、感染粒子を作製するためのファージ尾部の付着を可能にする。いっぱいの頭部は、狭いサイズ範囲内のDNA分子を含有してもよい。カプシドの容量は、パッケージDNAに対する最大サイズの制限を設定してもよい。代表的なヘッドフルパッケージングシステムは、P1、P7、T4、KVP40、P22、φ29、およびT7を含むが、これらに限定されない。
【0046】
(1)P1
バクテリオファージP1およびP7は、110〜115kbのヘッドフル容量を有する。パッケージング開始は、pac部位で生じ、頭部がいっぱいになり、非特異的末端の切断が成されるまで続く。市販で入手可能な段階1 P1パッケージング抽出物またはpacase抽出物は、λのcos部位に類似のpac部位でパッケージされるようにDNAを切断してもよい。それは、am10.1突然変異、P1遺伝子10中のナンセンス突然変異を含有してもよい。それは、頭部および尾部のタンパク質を含むすべての後期ファージタンパク質合成を欠く。段階2抽出物は、頭部の中にDNAをカプシド形成するファージパッケージングタンパク質を含有し、尾部を追加してもよい。それは、pac−切断活性を欠く、遺伝子9のam9.16ナンセンス突然変異を含有してもよい。
【0047】
パッケージング株はまた、2段階パッケージング反応のために構築することができる。パッケージング株構築のために、ナンセンス突然変異は、主な尾管タンパク質をコードするTubまたは主な尾鞘タンパク質をコードする遺伝子22を含むが、これに限定されない主な尾部遺伝子のうち1つにおいて作り出すことができる。これにより、pacase−頭部成熟/尾部欠乏段階1パッケージング抽出物をもたらしてもよい。段階2pacase−頭部欠乏/尾部成熟株は、P1の主な頭部タンパク質をコードする遺伝子23中にナンセンス突然変異を導入することによって、構築することができる。P1ゲノム内にCamrマーカーを置換することもまた望ましい可能性がある。
【0048】
cre−lox部位特異的組換えシステムは、P1パッケージクローニングされた核酸を環状にするために使用することができる。第1の核酸は、pacパッケージング開始配列の下流にloxP部位を含んでもよい。クローニングされた核酸の転位およびパッケージングの後に、P1頭部からはみ出たDNAは、切り取られてもよく、第2のloxP組換え部位を含有する、λについて提唱されるようなリンカーは、この時、追加することができる。段階2パッケージング反応の間の尾部の追加に続いて、パッケージDNAはCre+宿主の中に注入することができる。宿主細胞の中への注入に続いて、DNAは、Creリコンビナーゼによる2つのファージloxP部位の間の組換えによって環状にすることができる。これは、環状のクローニングされた核酸が複製され、安定して維持されることを可能にすることができる。
【0049】
(2)T4
バクテリオファージT4は、169kbのゲノムサイズを有する、最大の、十分に特徴づけられたファージのうち1つである。P1およびP7のように、T4は、ヘッドフル方式においてそのDNAをパッケージし、パッケージングメカニズムの構成要素は、徹底的に研究されてきた。λ、P1、およびP7と異なり、T4 DNAのパッケージングは、特異的な部位で開始しない。T4のためのin vitroパッケージングシステムが開発されてきた。T4 in vitroシステムは、外来DNAを、効率的にパッケージすることができ、160kbの長さまでの大きなDNA断片をクローニングするのに適応させることができる。
【0050】
P1およびP7 in vitroパッケージングについて上記に記載される同様の方法は、T4パッケージクローンを環状にするために使用されるcre−lox部位特異的組換えシステムを使用して、利用することができる。T4システムの1つの利点は、より大きな変異体およびより小さな変異体の両方の多くの頭部長変異体の同定であり、パッケージされるDNAの量は、適宜、変えられる。特定の頭部長突然変異と組み合わせてパッケージング株を使用することは、クローニングされた核酸の可能なサイズ範囲を拡張するのに有益であると分かる可能性がある。
【0051】
(3)KVP40
KVP40は、T4関連のものであると比較ゲノム学によって示された広い宿主範囲のビブリオファージである。それは、245kbのゲノムサイズを有する、大きく、尾がある、二本鎖DNAファージである。このファージの遺伝的性質および生化学的活性に関して現在、ほとんど知られていないが、KVP40のゲノム配列および構成は、T4との広範囲な保存の領域を示す。最大の保存領域のうち1つは、ビリオン構造遺伝子を構成する遺伝子クラスター中にあり、それらのDNAパッケージングメカニズムにおける類似性を同様に示唆する。これは、KVP40を、in vitroパッケージングを使用するクローニングシステムの候補にする。
【0052】
KVP40 in vitroパッケージングシステムの開発はこの拡張のために必要とされるだろう。主な構成要素が十分に特徴づけられているT4 DNAパッケージングについての知識の増加と共に、類似の株は、KVP40のin vitroパッケージング抽出物のために開発される可能性がある。推定上のターミナーゼ、頭部、および尾部に関連する遺伝子は既に同定されており、株構築のための可能な突然変異誘発標的を確実に提供するであろう。
【0053】
KVP40の宿主範囲は、少なくとも8つのビブリオ種および1つのフォトバクテリウム種を含むが、それは、より望ましい宿主であるE.coliに感染しない。V.parahaemolyticusのompK遺伝子は、KVP40の宿主受容体として同定された外膜タンパク質(OMP)をコードする。JM109などのE.coli株におけるompK遺伝子の発現は、KVP40による感染を可能にする。
【0054】
(4)P22
バクテリオファージP22は、Salmonella typhimuriumの株に感染する可能性があるポドウイルスである。P22は、大サブユニット(Gp2)および小サブユニット(Gp3)を含むターミナーゼを用いるヘッドフルメカニズムを使用して、約42kbと同じくらいのDNAをパッケージする。ターミナーゼは、pac部位またはpac様部位でDNAに結合し、切断してもよく、次いで、ウイルスプロカプシドの中へのDNAのATP加水分解ベースの転座に関与してもよい。プロカプシドがいっぱいになるまで、DNAは、プロカプシドの中にパッケージすることができる。パッケージングの間に、カプシドシェルは広がり、立体構造変化を受けてもよい。パッケージングに続いて、ターミナーゼは、再び、パッケージDNAの末端を切断してもよく、次いで、放出され、なお、切断された、非パッケージDNAに付着してもよい。次いで、ターミナーゼは、新しいプロカプシドの中に挿入され、連続サイクルにおいて隣接標的核酸断片をパッケージするプロセスを続けてもよい。
【0055】
(a)P22突然変異体
P22は、高形質導入P22(P22HT)などのP22突然変異体であってもよい。P22HT突然変異は、遺伝子3において生じてもよく、これは、突然変異体Gp3タンパク質をもたらしてもよい。P22HTは、HT105/1突然変異を持っていてもよい。P22HTターミナーゼによるパッケージングの開始は、明らかな配列特異性をほとんどまたはまったく有していなくてもよく、したがって、より低い特異性でDNAを認識してもよい。ターミナーゼは、ランダムな位置でDNAに結合し、切断してもよく、次いで、ウイルスプロカプシドの中へのDNAのATP加水分解ベースの転座に関与してもよい(図7)。P22HTによってパッケージされる標的核酸は、46kbであってもよい。
【0056】
P22突然変異体は、また所与の標的核酸に沿っての処理能力が増加していてもよい。突然変異体は、標的核酸から10までのカプシドをパッケージすることができる可能性がある。P22突然変異体もまた、E.coliに感染することができる可能性がある。
【0057】
(5)φ29
φ29は、Bacillus subtilisの株に感染できるバクテリオファージである。ファージは、扁長20面体カプシドの内部に約30kbと同じくらいのDNAを詰めてもよい。DNAは、頭部−尾部コネクター(Gp10)、パッケージングATPアーゼ(Gp16)、およびRNA分子の環(pRNA)を含むATP依存性モーターによってプロカプシドの中に転座することができる。
【0058】
(6)λパッケージングおよびDoc粒子
パッケージされる核酸は、まれに、間隔をおいて配置されるcos部位を含有してもよい。パッケージングは、cos部位、cosLで始まってもよく、これは、切断されて、頭部の中に挿入される12塩基突出を残す。cos部位は、標的核酸の末端にライゲーションされた第1の核酸によって、標的核酸の末端に存在していてもよい。パッケージングは、頭部がいっぱいになるまで、一方向に進んでもよい。ターミナーゼが、切断するための第2のcos部位を見つけない場合、プロセスは、DNAが頭部から外にはみ出すと共に止まってもよい。はみ出たDNAは、DNアーゼIなどのヌクレアーゼまたはSau3Aなどの頻繁に切断する制限酵素によって非特異的に除去することができる。ヌクレアーゼがはみ出たDNAを切断する位置は、ランダムであってもよい。
【0059】
次いで、ファージ尾部を付着させて、λdocLと呼ばれる、たった1つのcos部位を有する粒子を産生してもよい。これらの粒子は、E.coliの中にそれらのDNAを効率的に注入する可能性があるが、それらは、λ分子の右の末端に通常、見つけられる付着末端であるcosRを欠くので、それは、細胞に入る際、効率的に環化しなくてもよい。λdocL粒子は、組み込みによって宿主細胞の中に核酸を導入するために使用することができる。尾部追加の代案として、第2のパッケージングが行なわれてもよい。ヌクレアーゼ消化およびDNA抽出の後に、リンカーをライゲーションし、後の産物を再度パッケージし、形質移入し、クローンを選択することができる。
【0060】
loxPシステムは、尾部追加の代案としてクローンのin vitro環状化のために使用することができる。loxP部位は、第1の核酸上のPISの下流に追加することができる。クローニングされた核酸は、in vitroパッケージングによってサイズ選択され、抽出され、loxP部位を含有するリンカーは、ライゲーションすることができる。クローンは、in vitroCreリコンビナーゼによって環状にされ、プラスミドとして安定して維持されるように、宿主細胞の中にエレクトロポレーションすることができる。
【0061】
2.パッケージDNAの単離
カプシド形成後に、パッケージDNAは、カプシドを単離することによって回収することができる。カプシドは、等密度遠心分離もしくは速度遠心分離または差次的沈降を含むが、これらに限定されない方法によって単離することができる。カプシドはまた、アフィニティー精製によって単離することができ、これは、拡張されたカプシドの表面上に含有されるタンパク質のエピトープに特異的に結合することができる抗体を使用することによって行なわれてもよい。たとえば、カプシドタンパク質は、λファージのDタンパク質であってもよい。
【0062】
抗カプシドタンパク質抗体は、固体支持体に付着することができる。固体支持体は、ビーズ、微粒子、カラム、試験ストリップ、カートリッジ、マイクロタイタープレート、顕微鏡スライドガラス、またはナイロン、ニトロセルロース、もしくは他の適した材料などの膜であってもよい。微粒子は、0.2μm〜7.0μmのサイズであってもよい。微粒子はまた、ハプテン化することができる。微粒子はまた、少なくとも1つまたは2つの蛍光標識によって含浸させてもよい。微粒子はまた、約0.1μm未満のサイズの強磁性流体または磁性粒子を形成することができてもよい。微粒子はまた、ろ過によって収集可能、または除去可能である可能性がある。
【0063】
パッケージDNAは、フェノール抽出などによってカプシドから単離することができる。抽出に先立って、DNアーゼIなどのヌクレアーゼは、非パッケージDNAまたは非標的DNAを消化するために使用することができる。フェノールは、カプシドを含む溶液に追加することができる。この混合物を遠心分離した後に、水相は、有機相から分離されてもよく、クロロホルムが追加することができる。この混合物を遠心分離した後に、水相は、有機相から分離することができる。単離DNAは、水相に冷エタノールを追加することによって、水相から濃縮され、その後、遠心分離が続けられてもよい。DNAは、1回または複数回、エタノールを用いて洗浄してもよく、水または緩衝液中に再懸濁することができる。フェノール−クロロホルム抽出は、少なくとも1回、繰り返することができる。
【0064】
パッケージDNAはまた、DNA結合カラムを使用して、カプシドから単離することができる。たとえば、QIAGENラムダ手順(QIAGEN、Valencia、Ca)が使用することができる。非パッケージDNAまたは非標的DNAは、ヌクレアーゼを用いて消化することができる。カプシドは、沈澱され、次いで、溶解することができる。単離DNAは、カラムに結合させ、洗浄してもよい。DNAは、カラムから溶出され、イソプロパノールなどのアルコールを用いて溶液から沈澱することができる。DNAは、水または緩衝液中に再懸濁することができる。
【0065】
カプシドが、カプシドタンパク質アフィニティーカラムを使用して単離される場合、カプシドは、溶出され、溶解することができる。パッケージDNAは、上記に記載されるように、カラムベースのシステムを使用して、フェノール抽出することができ、または単離することができる。
【0066】
単離DNAの末端は、フィルインされてもよく、これは、DNAポリメラーゼを使用することによって行なわれてもよい。アダプター挿入断片は、DNAの末端にライゲーションされてもよく、これは、環状DNAをもたらしてもよい。ライゲーション反応は、アダプター挿入断片のそれぞれの末端が、単離DNA分子の末端にライゲーションするのに十分な濃度のアダプター挿入断片を含んでもよい。アダプター挿入断片の末端は、リン酸化されなくてもよく、これは、アダプター挿入断片がモノマー環を形成するのを予防してもよい。ライゲーション反応は、油中水型エマルション中で行なわれてもよい。エマルションの水性液滴は、環状にされる約1つの単離DNA分子を含有してもよい。
【0067】
アダプター挿入断片は、2つのアダプタープライマー結合部位を含んでもよく、これは、tail−to−tailなどのように、反対の向きを有していてもよい。結合部位は、10〜50または16〜25塩基対長であってもよい。アダプタープライマー挿入断片はまた、増幅およびヌクレオチド配列決定の両方を支援することができるプライミング領域を含んでもよい。それらの部位に結合してもよいプライマーは、国際公開第2007/145612 A1号または米国特許第7,115,400号もしくは第7,323,305号において記載されるような配列を有していてもよく、これらの内容は、参照によって本明細書において組み込まれる。たとえば、リーチングプライマー(reaching primer)は、プライマーAおよびプライマーBであってもよい。アダプター挿入断片はまた、454配列決定システム(454 Life Systems、Branford、CT)上でうまく見つけるために使用することができる、短い(たとえば4ヌクレオチド)「配列決定キー」配列を含んでもよい。アダプター挿入断片はまた、セパレーターエレメントを含んでもよく、これは、知られている配列を有していてもよい。配列は、ローリングサークル増幅のためのプライミング部位を含んでもよい。配列はまた、単離DNAの2つの末端を同定するために使用することができる。単離DNAの、後の配列決定の間に、セパレーターエレメントの配列は、全単離DNAが配列決定されたことを示してもよい。アダプター挿入断片は、複製開始点を含んでもよく、かつさらにマーカーを含んでもよい。
【0068】
アダプター挿入断片はまた、平滑末端切断制限酵素などの制限酵素のための認識配列を含んでもよく、この配列は、2つのアダプタープライマー結合部位の間に位置してもよい。単離DNAにアダプター挿入断片をライゲーションする際に、結果として生じるDNAは、アダプタープライマー結合部位の間でDNAを切断することができる制限酵素を用いて消化することができる。次いで、アダプター末端は、結果として生じる線状DNAの末端にライゲーションすることができる。
【0069】
アダプター挿入断片はまた、「リーチング」制限酵素認識部位を含んでもよい。認識部位は、プライマー結合部位に隣接していてもよい。アダプター挿入断片はまた、2つのアダプタープライマー結合部位の側面に位置するリーチング制限酵素認識部位を含んでもよく、認識部位は、制限酵素認識部位から離れて、逆方向に向けられてもよい。リーチング制限酵素は、クラスIIS酵素であってもよく、これは、Fok I、Alw26 I、Bbv I、Bsr I、Ear I、Hph I、Mme I、Mbo II、SfaN I、またはTth111 Iであってもよい。リーチング制限酵素はまた、クラスIII制限酵素であってもよく、これは、EcoP15 I、EcoP I、Hinf III、またはStyLT Iであってもよい。単離DNAにアダプター挿入断片をライゲーションする際に、リーチング制限酵素は、単離DNA内の位置でなどのように、認識部位から離れた位置でDNAを切断してもよい。アダプター挿入断片はまた、特異的な結合メンバーを含んでもよく、これは、ビオチンであってもよい。
【0070】
単離DNAに挿入断片をライゲーションした後に、結果として生じる環状DNAは、リーチング制限酵素を用いて消化されてもよく、これは、線状DNA断片をもたらしてもよく、これは、それぞれの末端上に、単離DNAの末端に対応し得る配列を含んでもよい。DNA断片は、環状にされてもよく、これは、DNA断片の末端をフィルインし、次いで、ライゲーションを行なうことによって行なわれてもよい。
【0071】
アダプタープライマー結合部位を含むアダプター末端はまた、単離DNAの末端にライゲーションすることができる。アダプター断片は、増幅およびヌクレオチド配列決定の両方を支援することができるプライミング領域を含んでもよく、454配列決定システム(454 Life Systems、Branford、CT)上でうまく見つけるために使用することができる、短い(たとえば4ヌクレオチド)「配列決定キー」配列を含んでもよい。部位に結合することができるプライマーは、プライマーAまたはプライマーBであってもよい。アダプター末端は、オリゴヌクレオチド配列Yを有する第1のアダプター末端であってもよい。アダプター末端はまた、オリゴヌクレオチド配列Zを有する第2のアダプター末端であってもよい。アダプター末端は、固体支持体に核酸を固定するための手段を含んでもよい。
【0072】
アダプター末端は、縮重2塩基一本鎖3’突出を有していてもよい。縮重は、2つの突出塩基が、ランダムであってもよいこと(つまり、それらは、それぞれ、G、A、T、またはCであってもよいこと)を意味し得る。アダプター末端は、単離DNAのリーチング制限酵素切断末端へのアダプターの、方向性をもつライゲーションを強く促進するために設計することができる。アダプター末端は、多量のモル過剰のアダプター末端(15:1アダプター末端:単離DNA比)を含有してもよいライゲーション反応において単離DNA末端と組み合わせられてもよく、これは、単離DNA末端の利用を最大限にしてもよく、単離DNAコンカテマーを形成する可能性を最小限にしてもよい。アダプター末端は、リン酸化されなくてもよく、これはアダプター末端ダイマーの形成を最小限にしてもよい。ライゲーションに続いて、ライゲーション産物は、鎖置換DNAポリメラーゼの使用などによって、フィルイン反応の使用によって修復することができる。DNAポリメラーゼは、Bst DNAポリメラーゼ(Large Fragment)、φ29DNAポリメラーゼ、DNAポリメラーゼI(Klenow Fragment)、またはVent(登録商標)DNAポリメラーゼであってもよい。
【0073】
アダプター末端は、米国特許第7,323,305号において記載されるように設計されてもよく、これらの内容は、参照によって本明細書において組み込まれる。アダプター末端は、ホスホジエステル結合の代わりにホスホロチオネート結合を含んでもよい。アダプター末端はまた、特異的な結合メンバーを含んでもよく、これは、ビオチンであってもよい。特異的な結合メンバーは、第1のアダプター末端に追加されてもよく、これは、5’末端であってもよく、特異的な結合メンバーは、第2のアダプター末端に追加されなくてもよい。第1および第2のアダプター末端の単離DNAへのライゲーションに続いて、単離DNAは、2つの第1のアダプター末端、1つの第1のアダプター末端および1つの第2のアダプター末端、ならびに2つの第2のアダプター末端を含んでもよい。アダプター末端ライゲーション単離DNAは、ストレプトアビジンなどの第1のアダプター末端の特異的な結合メンバーのための特異的な結合パートナーに結合された固体基質と接触させてもよい。固体基質は磁性ビーズであってもよい。
【0074】
固体基質とアダプター末端ライゲーション単離DNAを接触させる際に、少なくとも1つの第1のアダプター末端を含む単離DNAのみが結合するであろう。1つの第1のアダプター末端のみを含む単離DNAは、単離DNAの一方の末端で結合するであろうが、2つの第1のアダプター末端を含む単離DNAは、2つの末端で結合するであろう。非結合単離DNAは、固体基質から洗い流することができる。固体基質は、低塩類(「融解」または変性)溶液にさらされてもよく、これは、1つの第1のアダプター末端のみを含む単離DNAのみを放出してもよい。2つの第1のアダプター末端を含む単離DNAは、固体基質に結合したままであろう。固体基質から放出される単離一本鎖DNAは、PCR増幅および配列決定においてなどのように、さらなる使用のために収集することができる。
【0075】
3.パッケージDNAの増殖
カプシド形成後に、クローニングされた核酸はまた、適切な宿主細胞に感染させるために使用することができる。宿主細胞において、クローニングされた核酸は、環状にされ、増殖することができる。次いで、クローニングされた核酸は、DNAの単離のために標準的な手法を使用して単離することができる。クローニングされた核酸のIS混入を予防するために、宿主株は、ISエレメントを含んでいなくてもよい。
【0076】
クローニングされた核酸が環状ではない場合、それは、カプシドから単離され、in vitroにおいて環状にすることができる。環状化は、線状DNAの末端と適合性の1つもしくは複数のリンカーのライゲーションまたは組換えを含むが、これらに限定されない方法によって行なわれてもよい。
【0077】
環状核酸は、oriおよび選択可能マーカーまたは可視マーカーを含んでもよい。環状核酸は、細菌などの宿主細胞中で増殖させることができる核酸であってもよく、細菌人工染色体(BAC)であってもよい。環状核酸は、宿主細胞の中に形質転換することができる。
【0078】
4.単離DNAの増幅
単離DNAは、PCRなどによって増幅することができる。単離DNAが環状にされている場合、それは、アダプタープライマー結合部位の間に位置する部位で、DNAを切断することができる制限酵素を用いてそれを消化することによって線状にすることができる。
【0079】
a.エマルションPCR
アダプタープライマー結合部位を含む単離DNAは、ビーズエマルションPCR増幅(emPCR)の使用などによってPCR増幅することができる。emPCRは、米国特許第7,323,305号および国際公開第2007/145612号において記載されるように行なわれてもよく、これらの内容は、参照によって本明細書において組み込まれる。単離DNAは、一本鎖であってもよく、アダプタープライマーにアニールすることができる。アダプタープライマーは、固体支持体に付着させてもよく、これは、球状ビーズであってもよい。固体支持体は、複数の結合アダプタープライマーを含んでもよい。固体支持体は、水性反応混合物中に懸濁され、次いで、油中水型エマルション中にカプセル化することができる。エマルションは、分散した水相微小滴を含んでもよく、これは、60〜100μmの直径であってもよく、耐熱性の油相によって包まれてもよい。それぞれの微小滴は、増幅反応溶液(つまり核酸増幅のために必要な試薬)を含有してもよい。増幅は、PCR反応ミックス(ポリメラーゼ、塩類、dNTPs)ならびに1対のアダプタープライマー(プライマーAおよびプライマーB)を含んでもよい。微小滴集団のサブセットはまた、DNA鋳型を含むDNAビーズを含有してもよい。微小滴のこのサブセットは、増幅のための基本となってもよい。このサブセット内にないマイクロカプセルは、鋳型DNAを有していなくてもよく、増幅に参加しなくてもよい。増幅技術は、PCRであってもよく、PCRプライマーは、非対称PCRを行なうために8:1または16:1の比で存在する(つまり、1つの第2のプライマーに対して8または16の第1のプライマー)。
【0080】
単離DNAは、ビーズに固定することができるオリゴヌクレオチド(プライマーB)にアニールすることができる。サーモサイクリングの間に、一本鎖DNA鋳型およびビーズ上の固定Bプライマーの間の結合は切断される可能性があり、これは、周囲のマイクロカプセル化溶液の中に鋳型を放出する可能性がある。増幅溶液は、追加の溶液相プライマーAおよびプライマーBを含有してもよい。結合反応速度が、固定プライマーよりも溶液相プライマーについてより速いので、溶液相Bプライマーは、鋳型の相補的b’領域に容易に結合してもよい。初期PCRにおいて、A鎖およびB鎖の両方は、等しく十分に増幅することができる。
【0081】
中期増幅(つまり、サイクル10および30の間)によって、Bプライマーは、消耗されてもよく、これは、指数関数的増幅を停止させてもよい。次いで、反応は、非対称増幅に入ってもよく、単位複製配列集団は、A鎖が優位を占めるようになってもよい。後期相増幅において、30〜40のサイクル後に、非対称増幅は、溶液中でA鎖の濃度を増加させてもよい。過剰A鎖は、ビーズ固定Bプライマーにアニールし始める。次いで、耐熱性ポリメラーゼは、単位複製配列の固定ビーズ結合B鎖を合成するために、鋳型としてA鎖を用いる。
【0082】
終期増幅において、熱サイクリングを続けると、ビーズ結合プライマーへのさらなるアニーリングが強いられる可能性がある。溶液相増幅はこの段階で最小限となる可能性があるが、固定B鎖の濃度は増加する可能性がある。次いで、エマルションは、破壊されてもよく、固定産物は、変性によって(熱、pHなどによって)一本鎖にされてもよく、これは、相補的A鎖を取り出してもよい。Aプライマーは、固定鎖のA’領域にアニールされてもよく、固定鎖に、配列決定酵素および任意の必要な補助タンパク質が載せられてもよい。次いで、ビーズは、米国特許第6,274,320号、第6258,568号、および第6,210,891号において記載されるものなどのように、認識されるピロリン酸技術を使用して配列決定されてもよく、これらの内容は、参照によって本明細書において組み込まれる。
【0083】
(1)捕捉ビーズへのアダプタープライマーの結合
アダプタープライマーは、捕捉ビーズに付着することができる。プライマーは、当技術分野において知られている任意の方式において固体支持体捕捉ビーズに付着することができる。多数の方法は、微視的ビーズなどの固体支持体にDNAを付着させるために当技術分野において存在する。ビーズへのDNAの共有化学結合は、ホスホアミデート結合を通してDNA上の5’−リン酸をアミノコート捕捉ビーズに結合するために、水溶性カルボジイミドなどの標準的なカップリング剤の使用によって成し遂げることができる。他の代案は、同様の化学作用を使用して、第1に特異的なオリゴヌクレオチドリンカーをビーズにカップルし、次いで、DNAをビーズ上のリンカーに結合するためにDNAリガーゼを使用することである。ビーズにオリゴヌクレオチドをつなぐための他の結合化学作用は、N−ヒドロキシスクシンアミド(NHS)およびその誘導体の使用を含む。そのような方法において、オリゴヌクレオチドの一方の末端は、固体支持体と共有結合を形成する反応基(アミド基など)を含有してもよく、リンカーの他方の末端は、固定されるオリゴヌクレオチドと結合することができる第2の反応基を含有する。オリゴヌクレオチドは、共有結合によってDNA捕捉ビーズに結合することができる。しかしながら、キレート化または抗原抗体複合体などの非共有結合もまた、ビーズにオリゴヌクレオチドをつなぐために使用することができる。
【0084】
制限酵素部位からのオーバーラップ末端またはバクテリオファージラムダベースのクローニングベクターの「付着末端」などのように、DNA断片の末端でユニークな配列に特異的にハイブリダイズするオリゴヌクレオチドリンカーが、利用することができるが、平滑末端ライゲーションもまた有益に使用することができる。これらの方法は、米国特許第5,674,743号において詳細に記載される。ビーズを固定するために使用される方法は、本発明の方法におけるステップの全体にわたって、固定オリゴヌクレオチドを結合させるために続けてもよい。
【0085】
それぞれの捕捉ビーズは、単離DNAの一部を認識する(つまり、相補的である)複数のプライマーを有するように設計されてもよく、単離DNAは、したがって、捕捉ビーズにハイブリダイズされる。単離DNAのクローン増幅を成し遂げるために、1つのユニークな単離DNAのみがいずれか1つの捕捉ビーズに付着してもよい。
【0086】
本明細書において使用されるビーズは、任意の好都合なサイズであってもよく、任意の数の知られている材料から製造することができる。そのような材料の例は、無機物、天然ポリマー、および合成ポリマーを含む。これらの材料の特定の例は、セルロース、セルロース誘導体、アクリル樹脂、グラス、シリカゲル、ポリスチレン、ゼラチン、ポリビニルピロリドン、ビニールおよびアクリルアミドのコポリマー、ジビニルベンゼンと架橋されたポリスチレン、またはその他同種のもの(たとえばMerrifield、Biochemistry 1964年、3巻、1385〜1390頁において記載される)、ポリアクリルアミド、ラテックスゲル、ポリスチレン、デキストラン、ゴム、シリコン、プラスチック、ニトロセルロース、天然スポンジ、シリカゲル、制御孔ガラス、金属、架橋デキストラン(たとえばSephadex(商標))アガロースゲル(Sepharose(商標))、および当業者らに知られている固相支持体を含む。捕捉ビーズは、約25〜40μmの直径のSepharoseビーズであってもよい。
【0087】
(2)乳化
付着した一本鎖鋳型核酸を有する捕捉ビーズは、熱安定性の油中水型エマルションとして乳化することができる。エマルションは、当技術分野において知られている任意の適した方法によって形成することができる。エマルションを作り出すための1つの方法が下記に記載されるが、エマルションを作製するための任意の方法が使用することができる。これらの方法は、当技術分野において知られており、補助剤法、向流法、逆流法、回転ドラム法、および膜法を含む。そのうえ、マイクロカプセルのサイズは、構成要素の流量および速度を変動させることによって調整することができる。たとえば、液滴追加において、滴のサイズおよび送達の総時間は変動させてもよい。エマルションは、マイクロリットル当たり約3,000ビーズの密度で、ある程度の密度のビーズ「マイクロリアクター」を含有してもよい。
【0088】
エマルションは、増幅溶液中で鋳型付着ビーズを懸濁することによって生成することができる。本明細書において使用されるように、「増幅溶液」という用語は、鋳型DNAの増幅を行なうのに必要な、試薬の十分な混合物を意味し得る。
【0089】
ビーズ/増幅溶液混合物は、液滴に追加して、生物学的適合油(たとえば軽鉱物油、Sigma)の回転混合物にし、乳化させてもよい。使用される油は、1つまたは複数の生物学的適合乳化安定剤で補足することができる。これらの乳化安定剤はAtlox 4912、Span 80、および他の認識され、市販で入手可能な適した安定剤を含んでもよい。形成される液滴は、5ミクロン〜500ミクロン、約50〜300ミクロン、または100〜150ミクロンのサイズに及んでもよい。
【0090】
マイクロリアクターのサイズの制限はない。マイクロリアクターは、必要とされる増幅の程度にとって十分な増幅試薬を包含するのに十分に大きいものであってもよい。マイクロリアクターはまた、それぞれが、DNAライブラリーのメンバーを含有するマイクロリアクターの集団が、従来の実験装置(たとえばPCRサーモサイクリング装置、試験管、インキュベーター、およびその他同種のもの)によって増幅することができるように、十分に小さいものであってもよい。
【0091】
マイクロリアクターの最適サイズは、100〜200ミクロンの直径としてもよい。このサイズのマイクロリアクターは、10ml未満の容量のマイクロリアクターの懸濁液中で、約600,000メンバーを含むDNAライブラリーの増幅を可能にすることができる。たとえば、PCRが選ばれた増幅方法である場合、10mlは、96本の管の容量を有する一般的なサーモサイクラーの96本の管に一致するであろう。600,000マイクロリアクターの懸濁液は、1ml未満の容量を有していてもよい。1ml未満の懸濁液は、従来のPCRサーモサイクラーの約10本の管において増幅することができる。600,000マイクロリアクターの懸濁液はまた、0.5ml未満の容量を有していてもよい。
【0092】
(3)増幅
カプセル化後に、鋳型核酸は、転写ベースの増幅システム(Kwoh D.ら、Proc.Natl. Acad. Sci. (U.S.A.)86巻:1173頁(1989年);Gingeras T. R.ら、PCT特許出願国際公開第88/10315号;Davey,C.ら、欧州特許出願公開第329,822号;Miller, H. I.ら、PCT特許出願国際公開第89/06700号および「race」(Frohman, M.A.、In: PCR Protocols: A Guide to Methods and Applications、Academic Press、NY(1990年))および「片側PCR」(Ohara,O.ら、Proc. Natl. Acad. Sci. (U.S.A.)86巻、5673〜5677頁(1989年))を含むDNA増幅の任意の適した方法によって増幅することができる。なお、「ジオリゴヌクレオチド」増幅、等温増幅(Walker,G. T.ら、Proc. Natl. Acad. Sci. (U.S.A.)89巻:392〜396頁(1992年))、およびローリングサークル増幅(米国特許第5,714,320号において考察される)などのそれほど一般的ではない方法が、本発明において使用することができる。
【0093】
DNA増幅は、PCRによって行なわれてもよい。PCRは、PCRに必要な試薬をすべて含むPCR溶液を用いて、ビーズに結合された単離DNAをカプセル化することによって行なわれてもよい。次いで、PCRは、当技術分野において知られている任意の適したサーモサイクリングレジメンにエマルションをさらすことによって成し遂げることができる。30および50サイクルの間のまたは約40サイクルの増幅が行なわれてもよい。増幅手順に続いて、1つまたは複数のハイブリダイゼーションおよび伸長のサイクルが増幅サイクルに続いてあってもよい。10および30サイクルの間のまたは約25サイクルのハイブリダイゼーションおよび伸長が行なわれてもよい。鋳型DNAは、典型的に、鋳型DNAの少なくとも200万〜5000万のコピーまたは約1000万〜3000万のコピーがビーズ当たりに固定されるまで増幅することができる。
【0094】
(4)エマルションの破壊およびビーズ回収
単離DNAの増幅に続いて、エマルションは、「破壊」することができる(「解乳化」とも呼ばれる)。米国特許第5,989,892号においてなどのように、エマルションを破壊するための多くの方法があり、これらの内容は、参照によって本明細書において組み込まれる。エマルションは、さらなる油を追加して、エマルションを2つの相に分離することによって破壊することができる。次いで、油相は、除去されてもよく、適した有機溶媒(たとえばヘキサン)が追加することができる。混合後に、油/有機溶媒相は除去される。このステップは数回繰り返することができる。最後に、ビーズ上の水層は除去することができる。次いで、ビーズは、有機溶媒/アニーリング緩衝混合液を用いて洗浄してもよく、次いで、アニーリング緩衝液中で再び洗浄してもよい。適した有機溶媒は、メタノール、エタノール、およびその他同種のものなどのアルコールを含む。
【0095】
次いで、増幅された単離DNA含有ビーズは、たとえば知られている技術による配列決定反応における使用のために水溶液中に再懸濁することができる(Sanger, F.ら、Proc. Natl. Acad. Sci. U.S.A.75巻、5463〜5467頁(1977年);Maxam,A. M. & Gilbert、W. Proc Natl Acad Sci USA74巻、560〜564頁(1977年);Ronaghi, M.ら、Science281巻、363巻、365巻(1998年);Lysov,I.ら、Dokl Akad Nauk SSSR303巻、1508〜1511頁(1988年);Bains W. & Smith G. C. J.TheorBiol135巻、303〜307頁(1988年);Drnanac, R.ら、Genomics4巻、114〜128頁(1989年);Khrapko,K. R.ら、FEBS Lett256巻、118〜122頁(1989年);Pevzner P. A. J Biomol Struct Dyn7巻、63〜73頁(1989年);Southern,E. M.ら、Genomics13巻、1008〜1017頁(1992年)を参照されたい)。ビーズは、ピロリン酸ベースの配列決定反応において使用されてもよく(たとえば米国特許第6,274,320号、第6258,568号、および第6,210,891号において記載される、これらの内容は、参照によって本明細書において組み込まれる)、PCR産物の第2の鎖は除去されてもよく、配列決定プライマーは、ビーズに結合された一本鎖単離DNAにアニールすることができる。
【0096】
第2の鎖は、NaOH、低イオン(たとえば塩)強度、または熱処理などの一般的に知られている任意の数の方法を使用して融解することができる。この融解ステップに続いて、ビーズはペレットにされてもよく、上清は捨てられてもよい。ビーズは、アニーリング緩衝液中に再懸濁されてもよく、配列決定プライマーは、追加されてもよく、標準的なアニーリングサイクルを使用して、ビーズ付着一本鎖鋳型にアニールすることができる。
【0097】
(5)ビーズの精製
ビーズ上の増幅DNAはまた、直接ビーズ上でまたは異なる反応槽中で配列決定することができる。DNAは、反応槽にビーズを移し、DNAを配列決定反応にかけることによって、直接ビーズ上で配列決定することができる(たとえばピロリン酸配列決定またはSanger配列決定)。その代わりに、ビーズは、単離することができ、DNAは、それぞれの各ビーズから取り出され、配列決定することができる。いずれの場合においても、配列決定ステップは、それぞれの個々のビーズについて行なわれてもよい。ビーズはまた、米国特許第7,323,305号において記載される方法によって精製されてもよく、これらの内容は、参照によって本明細書において組み込まれる。
【0098】
b.ブリッジングPCR
環状であってもよく、アダプター挿入断片を含んでもよい単離DNAは、「ブリッジング」PCRを使用してPCR増幅することができる。ブリッジングPCRは、米国特許第7,115,400号において記載される方法によって行なわれてもよく、これらの内容は、参照によって本明細書において組み込まれる。増幅に先立って、単離DNAは、2つのプライマー結合部位の間の部位を切断する制限酵素を用いてそれを消化することによって線状にすることができる。単離DNAの一方の末端(5’末端)の配列は、オリゴヌクレオチド配列Yを有していてもよく、「コロニー」プライマーXの配列と同一の配列を含んでもよい。オリゴヌクレオチド配列Yは、知られている配列であってもよく、可変長であってもよい。オリゴヌクレオチド配列Yは、少なくとも5ヌクレオチド長、5〜100ヌクレオチド長、または約20ヌクレオチド長であってもよい。自然発生ヌクレオチドまたは非自然発生ヌクレオチドはオリゴヌクレオチド配列Y中に存在してもよい。
【0099】
配列Yの反対側の単離DNAの末端(3’末端)に含有されるオリゴヌクレオチド配列は、オリゴヌクレオチド配列Zを有していてもよい。オリゴヌクレオチド配列Zは、知られている配列であってもよく、可変長であってもよい。オリゴヌクレオチド配列Zは、少なくとも5ヌクレオチド長、5〜100ヌクレオチド長、または約20ヌクレオチド長であってもよい。自然発生ヌクレオチドまたは非自然発生ヌクレオチドはオリゴヌクレオチド配列Z中に存在してもよい。オリゴヌクレオチド配列Zは、それがコロニープライマーXとハイブリダイズするように設計されてもよく、それが、本明細書においてY’と呼ばれるオリゴヌクレオチド配列Yに相補的となるように設計することができる。核酸鋳型のそれぞれ5’および3’末端に含有されるオリゴヌクレオチド配列YおよびZは、鋳型の末端の先端に位置する必要はない。たとえば、オリゴヌクレオチド配列YおよびZは、核酸鋳型のそれぞれ5’および3’末端(もしくは末端)にまたはその近くに位置してもよいが(たとえば、5’および3’末端の0〜100ヌクレオチド以内)、それらは、核酸鋳型の5’または3’末端からさらに離れて(たとえば100ヌクレオチドを超えて)位置してもよい。したがって、配列YおよびZが、どちらかの側にある、つまり、増幅される核酸配列の側面に位置するという条件で、オリゴヌクレオチド配列YおよびZは核酸鋳型内の任意の位置に位置してもよい。
【0100】
本明細書において使用されるような「核酸鋳型」はまた、二本鎖形態の、増幅され、配列決定される核酸を含む実体を含む。核酸鋳型が二本鎖形態をしている場合、オリゴヌクレオチド配列YおよびZは、鎖のうち1つのそれぞれ5’および3’末端に含有される。他の鎖は、DNAの塩基対合則により、オリゴヌクレオチド配列YおよびZを含有する鎖に相補的であり、したがって、5’末端にオリゴヌクレオチド配列Z’および3’末端にオリゴヌクレオチド配列Y’を含有する。
【0101】
本明細書において使用されるような「コロニープライマー」は、相補的配列にハイブリダイズし、特異的なポリメラーゼ反応を開始することができるオリゴヌクレオチド配列を含む実体を指し得る。コロニープライマーを含む配列は、それが、その相補的配列との最大のハイブリダイズ活性およびあらゆる他の配列への非常に低い非特異的ハイブリダイズ活性を有するように選ばれる。コロニープライマーとして使用される配列は、任意の配列を含むことができるが、5'-AGAAGGAGAAGGAAAGGGAAAGGG(配列番号1)または5'-CACCAACCCAAACCAACCCAAACC(配列番号2)を含んでもよい。コロニープライマーは、5〜100塩基長または15〜25塩基長とすることができる。自然発生ヌクレオチドまたは非自然発生ヌクレオチドがプライマー中に存在してもよい。1つまたは2つの異なるコロニープライマーは、本発明の方法において核酸コロニーを生成するために使用することができる。
【0102】
本明細書において使用されるような「縮重プライマー配列」は、上述の核酸断片の配列と無関係の任意の核酸断片にハイブリダイズすることができる、短いオリゴヌクレオチド配列を指し得る。したがって、そのような縮重プライマーは、鋳型へのハイブリダイゼーションが生じるのに(1つまたは複数の)核酸鋳型におけるオリゴヌクレオチド配列YまたはZの存在を必要としないが、オリゴヌクレオチド配列XまたはYを含有する鋳型にハイブリダイズするための縮重プライマーの使用は除外されない。しかしながら、明白に、本発明の増幅方法における使用のために、縮重プライマーは、どちらかの側のまたは増幅される核酸配列の側面に位置する部位で鋳型中の核酸配列にハイブリダイズしなければならない。
【0103】
本明細書において使用されるような「固体支持体」は、たとえばラテックスビーズ、デキストランビーズ、ポリスチレン、ポリプロピレン表面、ポリアクリルアミドゲル、金表面、ガラス表面、およびシリコンウェーハなどの、核酸が共有結合することができる任意の固体表面を指し得る。
【0104】
本明細書において使用されるような「固体支持体に核酸を付着させるための手段」は、化学的に修飾可能な官能基を含む任意の化学的または非化学的な付着方法を指し得る。「付着」は、共有結合によるまたは不可逆的受動的吸着を介してのまたは分子の間のアフィニティーを介しての、固体支持体上の核酸の固定に関する(たとえばビオチン化分子によるアビジンコート表面上の固定)。付着は、それが、DNA変性条件下で水または水性緩衝液を用いて洗浄することによって除去され得ない十分な強度のものでなければならない。
【0105】
本明細書において使用されるような「化学的に修飾可能な官能基」は、たとえばリン酸基、カルボキシ部分もしくはアルデヒド部分、チオール基、またはアミノ基などの基を指し得る。
【0106】
本明細書において使用されるような「核酸コロニー」は、核酸鎖の多数のコピーを含む分散したエリアを指し得る。核酸鎖に対する相補鎖の多数のコピーもまた、同じコロニー中に存在してもよい。コロニーを構成する核酸鎖の多数のコピーは、固体支持体上に一般に固定され、一本鎖または二本鎖の形態をしていてもよい。本発明の核酸コロニーは、使用される条件に依存して異なるサイズおよび密度で生成することができる。コロニーのサイズは、0.2μm〜6μmまたは0.3μm〜4μmであってもよい。本発明の方法における使用のための核酸コロニーの密度は、10,000/mm2〜100,000/mm2であってもよい。より高い密度、たとえば、100,000/mm2〜1,000,000/mm2および1,000,000/mm2〜10,000,000/mm2が達成することができると考えられる。
【0107】
核酸コロニーは、単離DNAから生成することができる。それぞれが複数の異なる単離DNAのうち1つを表わす複数のコロニーもまた生成することができる。
【0108】
増幅される核酸を含む複数の単離DNAが生成することができ、核酸はそれらの5’末端にオリゴヌクレオチド配列Yおよび3’末端にオリゴヌクレオチド配列Zを含有し、さらに、(1つまたは複数の)核酸は、5’末端に、固体支持体に(1つまたは複数の)核酸を付着させるための手段を有する。複数の単離DNAは、オリゴヌクレオチド配列Zにハイブリダイズし、5’末端に、固体支持体にコロニープライマーを付着させるための手段を持っていてもよい複数のコロニープライマーXと混合される。複数の単離DNAおよびコロニープライマーは、固体支持体に共有結合してもよい。
【0109】
多数の2つの異なるコロニープライマーXは、複数の核酸鋳型と混合することができる。コロニープライマーXの配列は、オリゴヌクレオチド配列Zが、コロニープライマーXのうち1つにハイブリダイズすることができ、オリゴヌクレオチド配列Yが、コロニープライマーXのうち1つの配列と同じであるようなものであってもよい。
【0110】
オリゴヌクレオチド配列Zはまた、オリゴヌクレオチド配列Y(Y’)に相補的であってもよく、複数のコロニープライマーXは、オリゴヌクレオチド配列Yと同じ配列であってもよい。
【0111】
複数のコロニープライマーXは、縮重プライマー配列を含んでもよく、複数の単離DNAは、増幅される核酸を含んでもよく、それぞれ5’および3’末端にオリゴヌクレオチド配列YまたはZを含有していなくてもよい。
【0112】
単離核酸の5’末端に含有されるオリゴヌクレオチド配列は、任意の配列および任意の長さであってもよく、配列Yとして本明細書において示される。オリゴヌクレオチドの適した長さおよび配列は、十分に知られている方法を使用して、選択することができ、当技術分野をにおいて立証されている。たとえば、増幅される核酸のそれぞれの末端に付着されるオリゴヌクレオチド配列は、5および100ヌクレオチドの間の長さの通常、比較的短いヌクレオチド配列である。核酸の3’末端に含有されるオリゴヌクレオチド配列は、任意の配列および任意の長さとすることができ、配列Zとして本明細書において示される。オリゴヌクレオチドの適した長さおよび配列は、十分に知られており、当技術分野において立証されている方法を使用して、選択することができる。たとえば、増幅される核酸のそれぞれの末端に含有されるオリゴヌクレオチド配列は、5および100ヌクレオチドの間の長さの通常、比較的短いヌクレオチド配列である。
【0113】
オリゴヌクレオチド配列Zの配列は、それがコロニープライマーXのうち1つにハイブリダイズすることができるようなものである。オリゴヌクレオチド配列Yの配列は、それがコロニープライマーXのうち1つと同じであるようなものとしてもよい。オリゴヌクレオチド配列Zは、オリゴヌクレオチド配列Y(Y’)に相補的であってもよく、コロニープライマーXは、オリゴヌクレオチド配列Yと同じ配列である。
【0114】
オリゴヌクレオチド配列YおよびZは、当技術分野において標準的もしくは従来のものである技術を使用して調製されてもよく、または商業的供給源から購入することができる。
【0115】
本発明の単離DNAを産生する場合、さらなる望ましい配列は、十分に知られており、当技術分野において立証されている方法によって導入することができる。そのようなさらなる配列は、たとえば、制限酵素部位またはある核酸タグを含み、所与の核酸鋳型配列の増幅産物を同定することを可能にする。他の望ましい配列は、フォールドバックDNA配列(一本鎖にされた場合にヘアピンループまたは他の第2の構造を形成する)、たとえば、核酸ポリメラーゼによって認識されるプロモーターDNA配列などの、タンパク質/DNA相互作用を指示する「コントロール」DNA配列、またはDNA結合タンパク質によって認識されるオペレーターDNA配列を含む。
【0116】
増幅される複数の核酸配列がある場合、オリゴヌクレオチドYおよびZの付着は、同じまたは異なる反応において実行することができる。
【0117】
単離DNAが一度調製されたら、それは、本明細書において記載される方法において使用される前に増幅することができる。そのような増幅は、たとえば、発現ベクターの中に鋳型核酸を挿入し、かつ適した生物学的宿主においてそれを増幅するまたはPCRによってそれを増幅することによって、十分に知られており、当技術分野をにおいて立証されている方法を使用して実行することができる。しかしながら、本発明の方法は、核酸鋳型の多数のコピーが、核酸鋳型の単一コピーから生成される核酸コロニーにおいて産生されるのを可能にするので、この増幅ステップは、必須ではない。
【0118】
単離DNAの5’末端は、固体支持体に核酸鋳型を共有結合させるための手段を有するように修飾することができる。そのような手段は、たとえば、たとえばリン酸基、カルボキシ部分もしくはアルデヒド部分、チオール基、またはアミノ基などの化学的に修飾可能な官能基とすることができる。チオール基、リン酸基、またはアミノ基は、核酸の5’−修飾のために使用することができる。
【0119】
コロニープライマーは、当技術分野において標準的なまたは従来のものである技術を使用して調製することができる。一般に、本発明のコロニープライマーは、十分に知られており、当技術分野において立証されている方法によって生成される合成オリゴヌクレオチドであり、または商業的供給源から購入することができる。
【0120】
1つまたは2つの異なるコロニープライマーXは、任意の核酸配列を増幅するために使用することができる。コロニープライマーXの5’末端は、固体支持体にコロニープライマーを共有結合させるための手段を有するように修飾することができる。共有結合は、上記に記載されるように、化学的に修飾可能な官能基であってもよい。コロニープライマーは、たとえば制限エンドヌクレアーゼ部位またはそれぞれリボザイム切断部位のような他のタイプの切断部位などのさらなる所望の配列を含むように設計することができる。さらなる配列は、フォールドバックDNA配列(一本鎖にされた場合にヘアピンループまたは他の第2の構造を形成する)、たとえば、核酸ポリメラーゼによって認識されるプロモーターDNA配列などの、タンパク質/DNA相互作用を指示する「コントロール」DNA配列、またはDNA結合タンパク質によって認識されるオペレーターDNA配列を含む。
【0121】
5’末端による支持体へのコロニープライマーXの固定は、単離DNAの3’末端に含有される相補的なオリゴヌクレオチド配列とのハイブリダイゼーションが一度起こったら、コロニープライマーがポリメラーゼによる鎖伸長のために利用可能となるように、その3’末端を支持体から遠くにしてもよい。
【0122】
一度、本発明の単離DNAおよびコロニープライマーの両方が合成されたら、それらは、それらが、固体支持体に付着している場合に、付着された単離DNAおよびコロニープライマーの適切な密度が得られるように、適切な割合で共に混合される。混合物中のコロニープライマーの割合は、単離DNAの割合よりも高くてもよい。単離DNAに対するコロニープライマーの比は、コロニープライマーおよび単離DNAが固体支持体に固定される場合に、固体支持体の全体または明示されるエリアにほぼ一定の密度で位置する複数のコロニープライマーを含むコロニープライマーの「ローン(lawn)」が、コロニープライマーのローン内に間隔を置いて個々に固定される1つまたは複数の単離DNAと共に、形成されるようなものであってもよい。
【0123】
単離DNAは、一本鎖形態で提供することができる。しかしながら、それはまた、全体としてまたは部分的に、支持体への付着を可能にするように一方の5’末端または両方の5’末端が修飾された二本鎖形態で提供することができる。その場合、付着プロセスが完了した後に、たとえば放出される鎖を洗浄する前に94℃まで加熱することによって、当技術分野において知られている手段によって鎖を分離することが望まれる。二本鎖分子の両方の鎖が表面と反応し、両方とも付着する場合、結果は、一方の鎖のみが付着し、1回の増幅ステップが行なわれる場合と同じである可能性がある。言いかえれば、二本鎖単離DNAの両方の鎖が付着した場合において、両方の鎖は、必然的に、互いに近くに付着し、一方の鎖のみを付着させ、1回の増幅ステップを行なう結果と区別不能である。したがって、一本鎖および二本鎖単離DNAは、表面に付着され、コロニー生成のために適した鋳型核酸を提供するために使用することができる。
【0124】
個々のコロニープライマーおよび個々の単離DNAの間の距離(したがって、コロニープライマーおよび単離DNAの密度)は、コロニープライマーおよび支持体に固定される単離DNAの濃度を変えることによってコントロールすることができる。コロニープライマーの密度は、少なくとも1fmol/mm2、少なくとも10fmol/mm2、30〜60fmol/mm2としてもよい。単離DNAの密度は、10,000/mm2〜100,000/mm2であってもよい。100,000/mm2〜1,000,000/mm2および1,000,000/mm2〜10,000,000/mm2のより高い密度が達成することができる。
【0125】
付着した単離DNAおよびコロニープライマーの密度のコントロールは、支持体の表面上の核酸コロニーの最終密度をコントロールすることを可能にすることができる。これは、本発明の方法によれば、1つの核酸コロニーが、コロニープライマーが、固体支持体上の適した位置に存在するという条件で、1つの単離DNAの付着の結果として生じ得るといった事実によるものである。単一コロニー内の単離DNAの密度もまた、付着したコロニープライマーの密度をコントロールすることによってコントロールすることができる。
【0126】
コロニープライマーおよび単離DNAが一度、適切な密度で固体支持体上に固定されたら、核酸コロニーは、それぞれのコロニーが、もとの固定された単離DNAおよびその相補的配列の多数のコピーを含むように、共有結合した単離DNA上で適切な数の増幅のサイクルを実行することによって生成することができる。増幅の1回のサイクルは、ハイブリダイゼーション、伸長、および変性のステップからなり、これらのステップは、PCRについて当技術分野において十分に知られている試薬および条件を使用して、一般に行なわれる。
【0127】
典型的な増幅反応は、固体支持体、付着した単離DNA、およびコロニープライマーをプライマーハイブリダイゼーションを誘発する条件にかけること、たとえば、それらを約65℃の温度にかけることを含む。これらの条件下で、単離DNAの3’末端のオリゴヌクレオチド配列Zは、固定されたコロニープライマーXにハイブリダイズするであろう、またヌクレオシド三リン酸分子または任意の他のヌクレオチド前駆体、たとえば修飾ヌクレオシド三リン酸分子の供給を加えての、プライマー伸長を支援するための条件および試薬、たとえば、約72℃の温度、核酸ポリメラーゼ、たとえばDNA依存性DNAポリメラーゼ分子もしくは逆転写酵素分子(つまりRNA依存性DNAポリメラーゼ)またはRNAポリメラーゼの存在下において、コロニープライマーは、単離DNAに相補的なヌクレオチドの追加によって伸長するであろう。
【0128】
核酸ポリメラーゼは、DNAポリメラーゼ(Klenow fragment、T4 DNAポリメラーゼ)、耐熱性細菌からの熱安定性DNAポリメラーゼ(Taq、VENT、Pfu、Tfl DNAポリメラーゼなど)、またはその遺伝子改変誘導体(TaqGold、VENTexo、Pfu exo)であってもよい。RNAポリメラーゼおよび逆転写酵素の組み合わせを使用して、DNAコロニーの増幅を生じさせることもできる。コロニープライマー伸長のために使用される核酸ポリメラーゼは、PCR反応条件、つまり、加熱および冷却のサイクルの繰り返し下で安定性である可能性があり、使用される変性温度、普通、約94℃で安定性である可能性がある。
【0129】
ヌクレオシド三リン酸分子は、デオキシリボヌクレオチド三リン酸、たとえばDATP、dTTP、dCTP、dGTPであってもよく、またはリボヌクレオシド三リン酸、たとえばdATP、dUTP、dCTP、dGTPであってもよい。ヌクレオシド三リン酸分子は、自然発生または非自然発生のものであってもよい。
【0130】
ハイブリダイゼーションおよび伸長のステップの後に、支持体および付着した核酸を変性条件にさらす上で、2つの固定された核酸が存在するであろう、第1は、最初の固定された単離DNAであり、第2は、それに相補的な核酸であり、固定されたコロニープライマーXのうち1つから伸長する。もとの固定された単離DNAおよび形成された固定された伸長コロニープライマーの両方は、次いで、ハイブリダイゼーション、伸長、および変性のさらなるサイクルに支持体をかける上で、さらなる回の増幅を開始することができる。そのようなさらなる回の増幅は、単離DNAの多数の固定されたコピーおよびその相補的配列を含む核酸コロニーをもたらすであろう。
【0131】
単離DNAの最初の固定は、鋳型核酸が、全長の単離DNA内で離れて位置するコロニープライマーとハイブリダイズすることができるのみであることを意味する。したがって、形成される核酸コロニーの境界は、最初の単離DNAが固定されるエリアに対して、比較的局所的なエリアに制限されている。明らかに、単離DNAおよびその相補体のより多くのコピーが、増幅のさらなる回、つまりハイブリダイゼーション、伸長、および変性のさらなる回を実行することによって、一度、合成されたら、次いで、形成されるコロニーの境界が、最初の単離DNAが固定されるエリアに対して、なお、比較的局所的なエリアに制限される可能性があるが、生成されている核酸コロニーの境界は、さらに拡張することができるであろう。
【0132】
単一の固定された単離DNAからの核酸コロニーを生成することができ、これらのコロニーのサイズは、単離DNAがかけられる増幅の回数を変えることによってコントロールすることができる。したがって、それぞれの固定された単離DNAの場所内に十分な数の固定されたコロニープライマーがあるという条件で、固体支持体の表面上に形成される核酸コロニーの数は、支持体に最初に固定される単離DNAの数に依存する。コロニープライマーおよび単離DNAが固定された固体支持体が、プライマーのローン内に間隔を置いて固定された単離DNAと共に適切な密度で、固定されたコロニープライマーのローンを含んでいる可能性があるのは、この理由のためである。
【0133】
核酸コロニーのそのようないわゆる「オートパターニング」は、単離DNAがもともと固定される密度を調節することによって密度をコントロールすることができるという事実によりより高い密度の核酸コロニーが得ることができるという点において、他の方法に対して利点を有している可能性がある。そのような方法は、したがって、たとえば、支持体の特定の局所的なエリア上に特異的なプライマーを特異的に整列させ、次いで、プライマーの同じ局所的なエリア上に単離DNAを含有する特定のサンプルを配置することによってコロニー形成を開始することによって制限されない。先行技術方法を使用して整列することができるコロニーの数、たとえば国際公開第96/04404号(Mosaic Technologies,Inc.)において開示されるものは、したがって、特異的なプライマーエリアが最初のステップにおいて整列され得る密度/間隔によって制限される。
【0134】
単離DNAの最初の密度、したがって、単離DNAから結果として生じる核酸コロニーの密度をコントロールすることができることによって、形成される核酸コロニーのサイズ、さらに個々のコロニー内の単離DNAの密度をコントロールすることができることと共に、高密度の個々の核酸コロニーを十分に大きなサイズの、十分に多くの増幅配列を含有する固体支持体上で産生することができ、核酸コロニーについて行なわれる後の分析またはモニタリングを可能にする、最適の状況に達することができる。
【0135】
一度、核酸コロニーが生成されたら、たとえばコロニー視覚化または配列決定などの、さらなるステップを行なうことが望ましい可能性がある。たとえば、特定の核酸断片の一部または全部の存在または不在について、生成されるコロニーをスクリーニングすることが必要である場合、たとえば、コロニー視覚化は、必要とされる可能性がある。この場合、特定の核酸断片を含有する1つまたは複数のコロニーは、対象とする核酸断片に特異的にハイブリダイズする核酸プローブを設計することによって検出することができる。
【0136】
そのような核酸プローブは、蛍光基、ビオチンを含有する実体(たとえば、蛍光基を用いて標識されたストレプトアビジンとのインキュベーションによって検出することができる)、放射標識(当技術分野において十分に知られており、立証されている方法によって核酸プローブの中に取り込むことができ、たとえばシンチレーション液とのインキュベーションによって放射活性を検出することによって検出することができる)、もしくは染料または他の染色剤などの検出可能な実体を用いて標識してもよい。
【0137】
核酸プローブはまた、非標識であってもよく、核酸ポリメラーゼを用いる多くの標識ヌクレオチドの取り込みのためのプライマーとして作用するように設計することができる。次いで、取り込まれた標識、したがって核酸コロニーの検出を実行することができる。
【0138】
次いで、核酸コロニーは、ハイブリダイゼーションのために調製することができる。そのような調製は、コロニーを構成する核酸鋳型のすべてまたは一部分が、一本鎖形態で存在するように、コロニーの処理を含んでもよい。これは、たとえば、コロニー中の任意の二本鎖DNAの熱変性によって達成することができる。その代わりに、コロニーは、鋳型核酸中の配列の二本鎖形態に特異的な制限エンドヌクレアーゼを用いて処理することができる。したがって、エンドヌクレアーゼは、オリゴヌクレオチド配列YまたはZ中に含有される配列または単離DNA中に存在する他の配列に特異的であってもよい。消化後に、コロニーは、二本鎖DNA分子が分離されるように、加熱され、コロニーは、固定されていない鎖を除去するために洗浄されて、したがって、コロニー中に付着した一本鎖DNAを残す。
【0139】
ハイブリダイゼーションのためのコロニーの調製の後に、標識プローブまたは非標識プローブは、次いで、その特異的なDNA配列とのプローブのハイブリダイゼーションのための適切な条件下でコロニーに追加してもよい。
【0140】
次いで、プローブは、熱変性によって除去されてもよく、所望の場合、第2の核酸のための特異的なプローブが、ハイブリダイズされ、検出することができる。これらのステップは、所望されるだけ繰り返することができる。
【0141】
次いで、核酸コロニーにハイブリダイズされる標識プローブは、適切な検出デバイスを含む機器を使用して検出することができる。蛍光標識のための検出システムは、電荷結合素子(CCD)カメラであってもよく、これは、拡大デバイス、たとえば顕微鏡に任意選択で連結することができる。そのような技術を使用して、並行して、多くのコロニーを同時にモニターすることが可能である。たとえばCCDカメラおよび10×または20×対物レンズを有する顕微鏡を使用すると、1mm2および4mm2の間の表面に対してコロニーを観察することが可能であり、これは、10000および200000コロニーの間のコロニーを並行してモニターすることに対応する。
【0142】
生成されるコロニーをモニターするための代替方法は、コロニーで覆われる表面をスキャンすることである。たとえば、100000000個までのコロニーを同時に整列することができ、全表面に対してCCDカメラを用いて写真を撮ることによってモニターすることができるシステムを使用することができる。このように、100000000個までのコロニーを短時間でモニターすることができることを理解することができる。
【0143】
表面上の蛍光の検出および定量化を可能にする任意の他のデバイスもまた、本発明の核酸コロニーをモニターするために使用することができる。たとえば、蛍光画像装置または共焦点顕微鏡を使用することができる。標識が放射性である場合、放射活性検出システムが必要とすることができる。
【0144】
単離DNAの配列は、任意の適切な固相配列決定技術を使用することによって決定することができる。たとえば、本発明において使用することができる配列決定の1つの技術は、「配列決定プライマー」と本明細書において時に呼ばれる適切なプライマーを、配列決定される核酸鋳型とハイブリダイズすること、プライマーを伸長させること、およびプライマーを伸長させるために使用されるヌクレオチドを検出することを含む。プライマーを伸長させるために使用される核酸は、さらなるヌクレオチドが、成長している核酸鎖に追加される前に、検出されてもよく、したがって、1塩基ずつのin situ核酸配列決定を可能にする。
【0145】
取り込まれるヌクレオチドの検出は、プライマー伸長反応において1つまたは複数の標識ヌクレオチドを含むことによって容易にすることができる。任意の適切な検出可能な標識、たとえば蛍光体、放射標識などが使用することができる。蛍光標識が使用することができる。同じまたは異なる標識が、それぞれの異なるタイプのヌクレオチドについて使用することができる。標識は蛍光体であり、同じ標識が、それぞれの異なるタイプのヌクレオチドについて使用される場合、それぞれのヌクレオチド取り込みは、特定の波長で検出されるシグナルの累積的な増加を提供してもよい。異なる標識が使用される場合、これらのシグナルは、異なる適切な波長で検出することができる。所望の場合、標識ヌクレオチドおよび非標識ヌクレオチドの混合物が提供される。
【0146】
配列決定される単離DNAへの適切な配列決定プライマーのハイブリダイゼーションを可能にするために、核酸鋳型は、一本鎖形態をしていてもよい。核酸コロニーを構成する核酸鋳型が二本鎖形態で存在する場合、これらは、当技術分野において十分に知られている方法を使用して、たとえば変性、切断などによって、一本鎖単離DNAを提供するために処理することができる。
【0147】
単離DNAにハイブリダイズされ、プライマー伸長に使用される配列決定プライマーは、たとえば15〜25ヌクレオチド長の短いオリゴヌクレオチドであってもよい。プライマーの配列は、それらが、配列決定される単離DNAの一部にハイブリダイズするように設計されてもよく、ストリンジェントな条件下であってもよい。配列決定のために使用されるプライマーの配列は、核酸コロニーを生成するために使用されるコロニープライマーの配列と同じまたは同様の配列を有していてもよい。配列決定プライマーは、溶液または固定された形態で提供することができる。
【0148】
一度、配列決定プライマーが、単離DNAおよび配列決定プライマーを、当技術分野において十分に知られている方法によって決定される適切な条件にかけることによって、配列決定される単離DNAにアニールされたら、プライマー伸長は、たとえば、核酸ポリメラーゼおよび少なくともいくつかが、標識形態で提供されるヌクレオチドの供給および適したヌクレオチドが提供される場合に、プライマー伸長に適した条件を使用して、行なわれてもよい。
【0149】
それぞれのプライマー伸長ステップ後に、後のステップに干渉する可能性がある、取り込まれていないヌクレオチドを除去するために、洗浄ステップが含まれていてもよい。一度、プライマー伸長ステップが行なわれたら、核酸コロニーは、標識ヌクレオチドが伸長プライマーの中に取り込まれたかどうかを決定するために、モニターすることができる。次いで、プライマー伸長ステップは、伸長プライマーの中に取り込まれる次のおよび後のヌクレオチドを決定するために、繰り返することができる。
【0150】
適切な標識、たとえば蛍光または放射活性の検出および定量化を可能にする任意のデバイスが配列決定に使用することができる。標識が蛍光である場合、拡大デバイスに任意選択で付着されたCCDカメラが使用することができる。実際に、配列決定のために使用されるデバイスは、増幅核酸コロニーをモニターするための、上記に記載されるデバイスと同じであってもよい。
【0151】
検出システムは、プライマー伸長のそれぞれのステップの後にそれぞれのコロニーに取り込まれるヌクレオチドの数および性質を決定するために、分析システムと組み合わせて使用することができる。この分析は、それぞれのプライマー伸長ステップ直後に実行されてもよくまたは記録されたデータを使用して後に実行されてもよく、所与のコロニー内の核酸鋳型の配列を決定することを可能にする。
【0152】
決定されている配列が未知の場合、所与のコロニーに適用されるヌクレオチドは、次いで、分析の全体にわたって繰り返される選ばれた順、たとえばdATP、dTTP、dCTP、dGTPで適用することができる。しかしながら、決定されている配列が知られており、たとえば、知られている配列からの配列中に小さな差異が存在するかどうかを分析するために、再度配列決定されている場合、配列決定プロセスは、知られている配列に従って選ばれる適切な順で、それぞれのステップで、ヌクレオチドを追加することによって、より速く成することができる。所与の配列との差異は、したがって、プライマー伸長の特定の段階での、あるヌクレオチドの取り込みの欠如によって検出される。
【0153】
固体支持体へのコロニープライマーおよび核酸鋳型の付着は、支持体が、核酸増幅反応の間にかけられてもよい温度、たとえば、約100℃までの温度、たとえば約94℃で耐熱性であってもよい。付着は、共有結合であってもよく、米国特許第7,115,400号において記載されるように成し遂げることができ、これらの内容は、参照によって本明細書において組み込まれる。
【0154】
5.単離DNAの配列決定
配列決定は、国際公開第2007/145612号もしくは国際公開第05003375または米国特許第7,323,305号もしくは第7,115,400号において記載される方法によって行なわれてもよく、これらの内容は、参照によって本明細書において組み込まれる。機械は、Roche(454)GS FLX(454 Life Systems、Branford、CT)、Illuminaゲノム分析機器配列決定プライマー(Illumina Inc.、San Diego、CA)であってもよい。
【0155】
たとえば、ピロリン酸配列決定が使用することができる。この技術は、Hyman、1988年Anew method of sequencing DNA. Anal Biochem.174巻:423〜36頁およびRonaghi、2001年Pyrosequencingsheds light on DNA sequencing. Genome Res11巻:3〜11頁において記載されるように、DNA合成の間に放出されるピロリン酸(Ppi)の検出に基づく。
【0156】
酵素反応のカスケードにおいて、可視光は、取り込まれるヌクレオチドの数に比例して生成し得る。カスケードは、無機Ppiが、ポリメラーゼによるヌクレオチド取り込みと共に放出することができる核酸重合反応を用いて始まってもよい。放出されるPpiは、ATPスルフリラーゼによってATPに変換されてもよく、これは、ルシフェラーゼにエネルギーを提供して、ルシフェリンを酸化してもよく、光を生成してもよい。追加されるヌクレオチドが知られているので、鋳型の配列は決定される可能性がある。固相ピロリン酸配列決定は、3つの酵素システムにおいて固定されたDNAを用いる。シグナル対ノイズ比を増加させるために、自然dATPは、dATPαSと交換することができる。dATPαSは、2つの異性体(SpおよびRp)の混合物であってもよい。ピロリン酸中の純粋2’−デオキシアデノシン−5’−O’−(1−チオ三リン酸)Sp−異性体は、読み取り長の2倍まで、実質的により長い読み取りを可能にするために、配列決定において使用することができる。
【0157】
ピロリン酸ベースの配列決定は、ヌクレオチド三リン酸の存在下において、単離DNAおよび伸長プライマーをポリメラーゼ反応にかけることによって行なわれてもよく、それによって、ヌクレオチド三リン酸は、それが、標的位置における塩基に相補的である場合、取り込まれるようになり、ピロリン酸(PPi)を放出するにすぎないであろう。ヌクレオチド三リン酸は、サンプル−プライマー混合物の別々の一定分量にまたは同じサンプル−プライマー混合物に引き続いて追加される。PPiの放出は、次いで、どのヌクレオチドが取り込まれるかを示すために検出される。
【0158】
配列産物の領域は、単離DNAの領域に配列決定プライマーをアニールすることおよびDNAポリメラーゼおよび知られているヌクレオチド三リン酸、つまり、dATP、dCTP、dGTP、dTTP、またはこれらのヌクレオチドのうち1つの類似体と配列決定プライマーを接触させることによって、決定することができる。配列は、下記に記載されるように、配列反応副産物を検出することによって決定することができる。
【0159】
配列プライマーは、それが、増幅核酸鋳型の領域に特異的にアニールすることができる限り、任意の長さまたは塩基組成とすることができる。配列決定プライマーのための特定の構造は、それが、増幅鋳型核酸上の領域に特異的にプライムすることができる限り、必要とされない。配列決定プライマーは、特徴づけられる配列およびアンカープライマーにハイブリダイズすることができる配列の間にある鋳型の領域に相補的であってもよい。配列決定プライマーは、配列産物を形成するためにDNAポリメラーゼを用いて伸長される。伸長は、1つまたは複数のタイプのヌクレオチド三リン酸および所望の場合、補助結合タンパク質の存在下において行なわれる。
【0160】
dNTPの取り込みは、配列決定副産物の存在についてアッセイすることによって決定することができる。配列決定産物のヌクレオチド配列はまた、dNMPが、伸長配列プライマーの中に取り込まれる時に、ヌクレオチド三リン酸(dNTP)から放たれる無機ピロリン酸(PPi)を測定することによって決定することができる。Pyrosequencing(商標)技術(Pyrosequencing AB、Stockholm、Sweden)と称される配列決定のこの方法は、溶液(液相)中でまたは固相技術として行なわれてもよい。PPiベースの配列決定方法は、たとえば国際公開第WO9813523A1号,Ronaghiら、1996年Anal. Biochem.242巻:84〜89頁、Ronaghiら、1998年Science281巻:363〜365頁(1998年)、および米国特許出願第2001/0024790号において一般に記載され、これらの内容は、参照によって本明細書において組み込まれる。たとえば米国特許第6,210,891号および第6,258,568号もまた参照されたい。これらの内容は、参照によって本明細書において組み込まれる。
【0161】
これらの条件下で放出されるピロリン酸は、酵素で(たとえばルシフェラーゼ−ルシフェリン反応における光の生成によって)検出することができる。そのような方法は、電気泳動の必要性および可能性として危険な放射標識の使用を回避しながら、ヌクレオチドを所与の標的位置において同定することおよびDNAを簡単にかつ急速に配列決定することを可能にする。
【0162】
PPiは、多くの異なる手法によって検出されてもよく、様々な酵素法は、以前に記載されている(たとえばReevesら、1969年Anal. Biochem.28巻:282〜287頁;Guilloryら、1971年Anal. Biochem.39巻:170〜180頁;Johnsonら、1968年Anal.Biochem. 15:273頁;Cookら、1978年Anal. Biochem. 91:557〜565頁、およびDrakeら、1979年Anal.Biochem. 94:117〜120頁を参照されたい)。
【0163】
ポリメラーゼによるdNTPの取り込みの結果として放たれるPPiは、たとえばATPスルフリラーゼを使用して、ATPに変換することができる。この酵素は、硫黄代謝に関与するとして同定された。硫黄は、還元形態および酸化形態の両方において、植物および動物の成長のための、必須の無機栄養素である(たとえば、SchmidtおよびJager、1992年Ann. Rev. Plant Physiol. Plant Mol. Biol.43巻:325〜349頁を参照されたい)。植物および微生物の両方において、硫酸の活発な取り込みの後に硫化物への還元が続く。硫酸は、利用可能な細胞の還元剤に比べて非常に低い酸化/還元電位を有するので、同化における最初のステップは、ATP依存性反応を介してのその活性化を必要とする(たとえばLeyh、1993年Crit.Rev. Biochem. Mol. Biol. 28:515〜542頁を参照されたい)。ATPスルフリラーゼ(ATP:硫酸アデニリルトランスフェラーゼ;EC 2.7.7.4)は、無機硫酸(SO4−2)の代謝における最初の反応を触媒する。たとえば、RobbinsおよびLipmann、1958年J.Biol. Chem.233巻:686〜690頁;HawesおよびNicholas、1973年Biochem. J.133巻:541〜550頁を参照されたい)。この反応において、SO4−2は、アデノシン5’−ホスホ硫酸(APS)に活性化される。
【0164】
ATPスルフリラーゼは、Saccharomyces cerevisiae(たとえばHawesおよびNicholas、1973年Biochem.J.133巻:541〜550頁を参照されたい)、Penicillium chrysogenum(たとえばRenostoら、1990年J. Biol.Chem.265巻:10300〜10308頁を参照されたい)、ラット肝臓(たとえばYuら、1989年Arch. Biochem. Biophys.269巻:165〜174頁を参照されたい)、および植物(たとえばShawおよびAnderson、1972年Biochem.J.127巻:237〜247頁;Osslundら、1982年Plant Physiol.70巻:39〜45頁を参照されたい)などのいくつかの供給源から高度に精製されてきた。そのうえ、ATPスルフリラーゼ遺伝子は、原核生物(たとえばLeyhら、1992年J.Biol. Chem.267巻:10405〜10410頁;SchwedockおよびLong、1989年Mol. Plant Microbe Interaction2巻:181〜194頁;LaueおよびNelson、1994年J.Bacteriol.176巻:3723〜3729頁を参照されたい)、真核生物(たとえばCherestら、1987年Mol. Gen. Genet.210巻:307〜313頁;MountainおよびKorch、1991年Yeast7巻:873〜880頁;Fosterら、1994年J.Biol. Chem.269巻:19777〜19786頁を参照されたい)、植物(たとえばLeustekら、1994年Plant Physiol.105巻:897〜90216頁を参照されたい)、および動物(たとえばLiら、1995年J.Biol. Chem.270巻:29453〜29459頁を参照されたい)からクローニングされてきた。酵素は、特定の供給源に依存して、ホモオリゴマーまたはヘテロダイマーである(たとえばLeyhおよびSuo、1992年J.Biol. Chem.267巻:542〜545頁を参照されたい)。
【0165】
耐熱性スルフリラーゼを使用することができる。耐熱性スルフリラーゼは、たとえばArchaeoglobus種またはPyrococcus種から得られてもよく、耐熱性スルフリラーゼの配列は、データベース受入番号028606、受入番号Q9YCR4、および受入番号P56863で入手可能である。
【0166】
ATPスルフリラーゼは、多くの異なる適用、たとえば高濃度のATPでのADPのバイオルミノメトリック検出(たとえばSchultzら、1993年Anal. Biochem.215巻:302〜304頁を参照されたい)、DNAポリメラーゼ活性の継続的なモニタリング(たとえばNyrbn、1987年Anal.Biochem.167巻:235〜238頁を参照されたい)、およびDNA配列決定(たとえばRonaghiら、1996年Anal. Biochem.242巻:84〜89頁;Ronaghiら、1998年Science281巻:363〜365頁;Ronaghiら、1998年Anal.Biochem.267巻:65〜71頁を参照されたい)に使用されてきた。
【0167】
いくつかのアッセイが、前のATPスルフリラーゼ反応の検出のために開発されてきた。比色モリブドリシスアッセイは、リン酸検出に基づくものであってもよいのに対して(たとえばWilsonおよびBandurski、1958年J. Biol. Chem.233巻:975〜981頁を参照されたい)、継続的な分光光度モリブドリシスアッセイは、NADH酸化の検出に基づくものであってもよい(たとえばSeubertら、1983年Arch.Biochem. Biophys.225巻:679〜691頁;Seubertら、1985年Arch. Biochem. Biophys.240巻:509〜523頁を参照されたい)。後のアッセイは、いくつかの検出酵素の存在を必要としてもよい。さらに、いくつかの放射性アッセイもまた文献において記載されている(たとえばDaleyら、1986年Anal.Biochem.157巻:385〜395頁を参照されたい)。たとえば、あるアッセイは、32P標識ATPから放出される32PPiの検出に基づくものであり(たとえばSeubertら、1985年Arch.Biochem. Biophys.240巻:509〜523頁を参照されたい)、他のアッセイは、[35S]標識APSへの35Sの取り込みに基づき(このアッセイはまた、カップリング酵素として、精製APSキナーゼをも必要とする;たとえばSeubertら、1983年Arch.Biochem. Biophys.225巻:679〜691頁を参照されたい)、第3の反応は、35標識APSからの35SO4−2の放出に依存する(たとえばDaleyら、1986年Anal.Biochem.157巻:385〜395頁を参照されたい)。
【0168】
逆ATPスルフリラーゼ反応の検出については、継続的な分光光度アッセイ(たとえばSegelら、1987年MethodsEnzymol.143巻:334〜349頁を参照されたい)、バイオルミノメトリックアッセイ(たとえばBalharryおよびNicholas、1971年Anal.Biochem.40巻:1〜17頁を参照されたい)、35SO4−2放出アッセイ(たとえばSeubertら、1985年Arch.Biochem. Biophys.240巻:509〜523頁を参照されたい)、または32PPi取り込みアッセイ(たとえばOsslundら、1982年PlantPhysiol.70巻:39〜45頁を参照されたい)が使用することができる。
【0169】
ATPスルフリラーゼによって産生されるATPは、光を生成するために、酵素反応を使用して加水分解することができる。光放出化学反応(つまり化学発光)および生物学的反応(つまり生物発光)は、様々な代謝物質の高感度測定のために分析生化学において広く使用される。生物発光反応において、光の放出をもたらす化学反応は、酵素触媒することができる。たとえば、ルシフェリン−ルシフェラーゼシステムは、ATPの特異的なアッセイを可能にし、細菌ルシフェラーゼ−オキシドリダクターゼシステムは、NAD(P)Hをモニタリングするために使用することができる。両システムは、ATPまたはNAD(P)Hの産生または利用を含むカップル反応の手段によって、多数の物質の分析まで拡張されてきた(たとえばKricka、1991年Chemiluminescent and bioluminescent techniques. Clin.Chem.37巻:1472〜1281頁を参照されたい)。
【0170】
新しい試薬の開発は、ATP(たとえばLundin、1982年Applicationsof firefly luciferase In; Luminescent Assays(Raven Press、New Yorkを参照されたい)またはNAD(P)H(たとえばLovgrenら、Continuousmonitoring of NADH-converting reactions by bacterial luminescence. J. Appl.Biochem.4巻:103〜111頁を参照されたい)の濃度に比例した、安定性の光放出を得ることを可能にしてきた。そのような安定性の光放出試薬を用いると、知られている量のATPまたはNAD(P)Hの追加によって終点アッセイをし、個々のアッセイを較正することが可能である。さらに、安定性の光放出システムはまたATPまたはNAD(P)H変換システムの継続的なモニタリングを可能にする。
【0171】
ATPを光に変換するための適した酵素は、ルシフェラーゼ、たとえば昆虫ルシフェラーゼを含む。ルシフェラーゼは、触媒作用の最終産物として光を産生する。最もよく知られている光放出酵素は、ホタル、Photinus pyralis(甲虫目)の酵素である。対応する遺伝子は、クローニングされ、細菌(たとえばde Wetら、1985年Proc.Natl. Acad. Sci. USA80巻:7870〜7873頁を参照されたい)および植物において(たとえばOwら、1986年Science234巻:856〜859頁を参照されたい)ならびに昆虫(たとえばJhaら、1990年FEBSLett.274巻:24〜26頁)および哺乳動物細胞において(たとえばde Wetら、1987年Mol. Cell. Biol.7巻:725〜7373頁;Kellerら、1987年Proc.Natl. Acad. Sci. USA82巻:3264〜3268頁を参照されたい)発現されてきた。さらに、ジャマイカコメツキムシ、Pyroplorusplagiophihalamus(甲虫目)からの多くのルシフェラーゼ遺伝子は、最近、クローニングされ、部分的に特徴づけられてきた(たとえばWoodら、1989年J.Biolumin. Chemilumin.4巻:289〜301頁;Woodら、1989年Science244巻:700〜702頁を参照されたい)。別のルシフェラーゼは、時に、異なる波長の光を産生することができ、これにより、異なる波長での光放出の同時のモニタリングが可能となる可能性がある。したがって、これらの前述の特徴は、ユニークであり、現在のレポーターシステムの利用に関して新しい特質を追加する。
【0172】
ホタルルシフェラーゼは、ルシフェリン、アデノシン5’−3リン酸(ATP)、マグネシウムイオン、および酸素の存在下において生物発光を触媒し、0.88の量子収量をもたらしてもよい(たとえばMcElroyおよびSelinger、1960年Arch. Biochem. Biophys.88巻:136〜145頁を参照されたい)。ホタルルシフェラーゼ生物発光反応は、約1×10−13Mの検出限界で、ATPの検出のためのアッセイとして用いることができる(たとえばLeach、1981年J.Appl. Biochem.3巻:473〜517頁を参照されたい)。さらに、ルシフェラーゼ媒介検出システムの感度の全体的な程度および利便性は、ホタルルシフェラーゼベースのバイオセンサーの開発への相当な関心を引き起こした(たとえばGreenおよびKricka、1984年Talanta31巻:173〜176頁;Blumら、1989年J.Biolumin. Chemilumin.4巻:543〜550頁を参照されたい)。
【0173】
上記の酵素を使用して、配列プライマーを、ポリメラーゼおよび知られているdNTPにさらすことができる。dNTPが、プライマー配列の3’末端上に取り込まれる場合、dNTPが切断されてもよく、PPi分子が放たれてもよい。次いで、PPiは、ATPスルフリラーゼを用いてATPに変換することができる。ATPスルフリラーゼは、PPiの変換がPPiに関しての一次速度式で進む十分に高い濃度で存在してもよい。ルシフェラーゼの存在下において、ATPは、加水分解されて、光子を生成する。反応は、反応、ATP−−>DP+PO43−+光子(光)が、ATPに関しての一次速度式で進むように、反応混合物内に存在する十分な濃度のルシフェラーゼを有していてもよい。光子は、下記に記載される方法および器具を使用して測定することができる。PPiおよびカップルスルフリラーゼ/ルシフェラーゼ反応は、検出のための光を生成するために使用することができる。スルフリラーゼおよびルシフェラーゼのどちらかまたは両方は、それぞれの反応部位に配置される1つまたは複数の可動性固体支持体上に固定することができる。
【0174】
PPiは、リアルタイムシグナルを発するポリメラーゼ反応の間に検出されるように放出することができる。配列決定反応は、リアルタイムで継続的にモニターすることができる。反応は、2秒未満で起こってもよい(NyrenおよびLundin、前掲)。律速段階は、ATPスルフリラーゼによるATPへのPPiの変換であってもよいが、ルシフェラーゼ反応は速く、0.2秒未満を要すると推測された。ポリメラーゼの取り込み速度もまた、様々な方法によって推測され、たとえば、Klenowポリメラーゼの場合において、1つの塩基の完全な取り込みは、0.5秒未満を要する可能性があることが分かった。したがって、1つの塩基の取り込みおよびこの酵素アッセイによる検出のための推測される総時間は、約3秒である。そのため、非常に速い反応時間が可能であり、リアルタイム検出を可能にすることが分かるであろう。反応時間は、より耐熱性のルシフェラーゼを使用することによってさらに減少し得る。
【0175】
ほとんどの適用については、ATPおよびPPiのような混入物がない試薬が使用することができる。これらの混入物は、樹脂に結合されたアピラーゼおよび/またはピロホスファターゼを含有するプレカラムに試薬を流すことによって除去することができる。その代わりに、アピラーゼまたはピロホスファターゼは、磁性ビーズに結合されてもよく、試薬中に存在する混入しているATPおよびPPiを除去するために使用することができる。さらに、拡散性の配列決定試薬、たとえば取り込まれていないdNTPsは、洗浄緩衝液を用いて洗い流することができる。ピロリン酸配列決定において使用される任意の洗浄緩衝液が使用することができる。
【0176】
配列決定反応における反応物の濃度は、0.2ml緩衝液中に1pmol DNA、3pmolポリメラーゼ、40pmol dNTPを含んでもよい。Ronaghiら、Anal. Biochem.242巻:84〜89頁(1996年)を参照されたい。
【0177】
所望の場合、配列決定反応は、それぞれの4つの所定のヌクレオチドを用いて行なわれてもよい。「完全」サイクルは、所定の順において、ヌクレオチドDATP、dGTP、dCTP、およびdTTP(またはdUTP)のそれぞれについての配列決定試薬を順次、与えることを含んでもよい。取り込まれていないdNTPsは、それぞれのヌクレオチド追加の間に洗い流することができる。その代わりに、取り込まれていないdNTPsは、アピラーゼによって分解することができる。サイクルは、配列産物の所望の量の配列が得られるまで、所望されるように繰り返することができる。約10〜1000、10〜100、10〜75、20〜50、または約30ヌクレオチドの配列情報が、1つのアニールされた配列決定プライマーの伸長から得られてもよい。
【0178】
ヌクレオチドは、ビオチンなどのハプテンのジスルフィド誘導体を含有するように修飾することができる。アンカー基質にアニールした新生プライマーへの修飾ヌクレオチドの追加は、i)修飾がビオチンである例において、酵素分子に結合されたアビジンコンジュゲート部分またはストレプトアビジンコンジュゲート部分の順次結合、ii)過剰なアビジン結合酵素またはストレプトアビジン結合酵素の洗い流し、iii)酵素活性に適用可能な条件下での適した酵素基質の流入、およびiv)1つまたは複数の酵素基質反応産物の検出を含む重合後ステップによって分析することができる。ハプテンは還元剤の追加を通じて除去することができる。そのような方法は、電気泳動の必要性および可能性として危険な放射標識の使用を回避しながら、ヌクレオチドを所与の標的位置において同定することおよびDNAを簡単にかつ急速に配列決定することを可能にする。
【0179】
ハプテンを検出するための酵素は、ホースラディッシュペルオキシダーゼであってもよい。洗浄緩衝液は、本明細書において、様々な反応物の追加の間に使用することができる。アピラーゼは、配列決定プライマーを伸長させるために使用される、未反応dNTPを除去するために使用することができる。洗浄緩衝液は、アピラーゼを含んでもよい。
【0180】
例としてのハプテン、たとえばビオチン、ジゴキシゲニン、蛍光染料分子cy3およびcy5、ならびにフルオレスセインは、伸長DNA分子の中に様々な効率で取り込まれてもよい。ハプテンの付着は、糖、塩基を介しての、およびヌクレオチド上のリン酸部分を介しての結合を通して生じてもよい。シグナル増幅のための例としての手段は、蛍光手段、電気化学手段、および酵素手段を含む。酵素増幅が使用される場合、酵素、たとえばアルカリホスファターゼ(AP)、ホースラディッシュペルオキシダーゼ(HRP)、ベータ−ガラクトシダーゼ、ルシフェラーゼは、光生成基質が知られているものを含んでもよく、光生成(化学発光)基質の検出のための手段はCCDカメラを含んでもよい。
【0181】
修飾塩基は、追加されてもよく、検出が生じてもよく、ハプテンコンジュゲート部分は、除去されてもよく、または切断剤もしくは不活性化剤の使用によって不活性化することができる。たとえば、切断可能なリンカーがジスルフィドである場合、切断剤は、還元剤、たとえばジチオスレイトール(DTT)、ベータ−メルカプトエタノールなどであってもよい。不活性化はまた、熱、冷却、化学的変性剤、界面活性剤、疎水性試薬、または自殺型阻害剤によって成し遂げることができる。
【0182】
ルシフェラーゼは、光子の同時に起こる放出を伴ってdATPを直接、加水分解する可能性がある。加水分解は、伸長配列決定プライマーの中へのdATPの取り込みと無関係に生じるので、これは、偽陽性シグナルをもたらす可能性がある。この問題を回避するために、DNAの中に取り込まれるdATP類似体が使用することができる、つまり、それは、DNAポリメラーゼの基質であるが、ルシフェラーゼの基質ではない。1つのそのような類似体は、α−チオ−dATPである。したがって、α−チオ−dATPの使用により、成長している核酸鎖の中に取り込まれることなく、dATPが加水分解される場合に生じ得る偽性の光子生成を回避してもよい。
【0183】
PPiベースの検出は、配列決定プライマーの追加直後の、配列決定反応混合物へのコントロールヌクレオチドの追加に続いて放出される光の測定によって較正することができる。これにより、反応条件の標準化を可能にすることができる。2つ以上の連続した同一のヌクレオチドの取り込みは、放出される光の量における対応する増加によって明らかにすることができる。したがって、コントロールヌクレオチドと比較した、放出される光の2倍の増加により、伸長プライマーの中への2つの連続のdNTPsの取り込みを明らかにしてもよい。
【0184】
アピラーゼは、配列決定反応混合物内で、あらゆる残りの取り込まれなかったdNTPsの分解を容易にするために、固体支持体の表面に「薄く塗られてもよい」または「流することができる」。アピラーゼはまた、生成されるATPをも分解し、したがって、反応から生成される光を「消す」。アピラーゼを用いる処理の際に、あらゆる残りの反応物は、次に続くdNTPインキュベーションおよび光子検出のステップの調製中に洗い流することができる。その代わりに、アピラーゼは、固体支持体または可動性固体支持体に結合することができる。
【0185】
a.配列決定反応の検出
固体支持体は、画像システム230に光学的に結合されてもよく、これは、従来の光学機械または光ファイバー束と連携してCCDシステムを含んでもよい。灌流チャンバー基質は、水性インターフェースの近くで生成される光が、基質またはチャンバーの外部に光ファイバーを通して直接伝達することができるように、光ファイバーアレイウェーハを含んでもよい。CCDシステムが光ファイバーコネクターを含む場合、画像化は、灌流チャンバー基質をコネクターと直接接触させて配置することによって成し遂げることができる。その代わりに、従来の光学機械は、たとえば、光ファイバー基質の外部から直接、CCDセンサー上への1−1倍率高開口数レンズシステムを使用することによって、光を画像化するために使用することができる。基質が、光ファイバーカップリングを提供しない場合、レンズシステムはまた、上記に記載されるように使用されてもよく、この場合において、基質または灌流チャンバーカバーは光学的に透明である。
【0186】
画像システム230は、基質表面上で反応器から光を収集するために使用することができる。光は、たとえば、当技術分野において知られている高感度低ノイズ器具を使用してCCD上に画像化することができる。光ファイバーベースの画像化については、光ファイバーは、カバースリップの中に直接またはFORAについては、同様に、検出器に光を伝える光ファイバーであるマイクロウェルを形成する光ファイバーを有するように取り込まれてもよい。
【0187】
画像システムは、コンピューター制御およびデータ収集システム240に結合することができる。任意の一般的に入手可能なハードウェアおよびソフトウェアパッケージが使用することができる。コンピューター制御およびデータ収集システムはまた、試薬送達をコントロールするためにコンジット200に結合することができる。
【0188】
ピロリン酸配列決定反応によって生成される光子は、それらが焦点調整デバイス(たとえば光学用レンズまたは光ファイバー)を通過し、CCDエレメント上に集中する場合のみ、CCDによって捕捉することができる。しかしながら、放出される光子は、全方向に等しく流出する可能性がある。平面状のアレイ(たとえばDNAチップ)を用いる場合に、それらの、後の「捕捉」および定量化を最大限にするために、光子は、それらが生成されるポイントにできるだけ近くに、たとえば、平面状の固体支持体の直近に収集することができる。これは、(i)カバースリップおよび従来の光学用レンズもしくは光ファイバー束の間に光学用液浸油を用いることまたは(ii)カバースリップ自体の中に光ファイバーを直接、取り込むことによって成し遂げることができる。同様に、薄く、光学的に透明な平面状の表面が使用される場合、光ファイバー束はまた、その背面に対して配置され、全反応/灌流チャンバーの奥行きを通して「画像化する」必要性を排除してもよい。
【0189】
反応イベント、たとえば、ルシフェラーゼによって生成される光子は、多種多様の検出器具、たとえば、光電子増倍管、CCD、CMOS、吸光度光度計、ルミノメーター、電荷注入デバイス(CID)、または他の固体検出器および本明細書において記載される器具を使用して検出され、定量化することができる。放出される光子の定量化は、溶融光ファイバー束が取り付けられたCCDカメラの使用によって成し遂げることができる。放出される光子の定量化はまた、マイクロチャネルプレート増幅装置が取り付けられたCCDカメラの使用によって成し遂げることができる。背面薄化CCDは、感度を増加させるために使用することができる。CCD検出器は、たとえばBronksら、1995年Anal. Chem.65巻:2750〜2757頁において記載される。
【0190】
CCDシステムは、Lockheed−Martin LM485 CCDチップおよび6−8μmの個々のファイバー直径を有する1−1光ファイバーコネクター(束)を有するSpectral Instruments,Inc.(Tucson、Ariz.)シリーズ600 4ポートカメラであってもよい。このシステムは、4096×4096または1600万超のピクセルを有していてもよく、10%〜>40%に及ぶ量子効率を有する。したがって、波長に依存して、CCDセンサー上に画像化された40%もの光子は、検出可能な電子に変換される可能性がある。
【0191】
蛍光部分は、標識として使用されてもよく、反応イベントの検出は、レーザーを用いてアレイの表面をスキャンするために、共焦点走査型顕微鏡を使用して実行されてもよく、またはより小さな光学解像度が可能な走査型近接場光学顕微鏡(SNOM)などの他の技術が入手可能であり、それによって、「より密な」アレイの使用を可能にする。たとえば、SNOMを使用して、個々のポリヌクレオチドは、100nm、たとえば10nm×10nm未満の距離、離れている場合、識別される可能性がある。さらに、走査トンネル顕微鏡(Binningら、Helvetica Physica Acta、55巻:726〜735頁、1982年)および原子間力顕微鏡(Hanswaら、AnnuRev Biophys Biomol Struct、23巻:115〜139頁、1994年)が使用することができる。
【0192】
本発明は、次に続く非限定的な実施例によって示される、多数の態様を有する。
【実施例】
【0193】
(実施例1)
PACクローニングベクターの中にDNAをクローニングするためのλdoc粒子の使用
Tn7ドナープラスミドpGPS3(New England Biolabs)を使用して、pPAC/oriVベクターの完全なコピーを加えた、λcos部位を含有する転位性カセットを構築する(図1−1)。次いで、転位性カセットは、標的DNAの中に転位する(図1−1)。転位は、サザンブロッティングによって確認することができる。
【0194】
標的DNAの中への転位性エレメントの転位後に、それは、λ抽出物を用いてin vitroにおいてパッケージする(図1−2)。プロヘッドは満たされるであろうが、パッケージングは、第2のcos部位の欠如により完了しなくてもよい。欠けているcos部位を提供するために、頭部からはみ出たあらゆるDNAを除去するために、調製物は、Sau3Aを用いて消化する。次に、ファージ尾部を追加して、ヘッドフルDNAを含有するλdocLビリオンを産生する。ビリオンDNAの一方の末端は、cosLで終了し、他方は、Sau3A突出で終了する。
【0195】
パッケージDNA分子を環状にし、安定性のプラスミドを形成するために、第2のcos部位を、図3において示されるように、リンカーをアニールすることによってSau3Aに追加する。リンカーによって追加したcos部位は、DNAが環状になるまたはコンカテマーを形成することを可能にする。次いで、第2回のin vitroパッケージングは、それぞれの末端にcos突出を有するDNA分子を産生する。DNA分子は、環状になり、両端にλcos部位を有するカプシド形成DNAを有する完全に感染性の形態で再度パッケージされる。これらの調製物は、次いで、高効率でかつ好都合に、細胞の中にPACを導入するために使用される。
【0196】
(実施例2)
PACクローニングベクターの中にDNAをクローニングするためのλdoc粒子のさらなる使用
誘発性複製開始点を含むベクター構築物と一致する、ファージベースのサイズ選択およびランダムクローニングのためのバクテリオファージλ in vitroパッケージングを使用するクローニングシステムを設計した。このクローニングシステムは、バクテリオファージP1またはP7 in vitroパッケージングおよびより大きな(ヘッドフル容量)バクテリオファージを取り込むように拡張することができる。バクテリオファージin vitroパッケージングを使用するためのそのような手法を図2において示す。
【0197】
ファージパッケージング開始認識部位は、in vitro転位によって標的DNAの中に導入する。トランスポゾンは、ファージ特異的パッケージング開始部位、転位のための19bp Tn5モザイク末端、クローニングDNAの増幅のためのoriV、およびプラスミド選択のための抗生物質抵抗性を付与する遺伝子を含む。転位に続いて、バクテリオファージin vitroパッケージングシステムは、クローニングDNAをパッケージするために使用される。パッケージングは、ファージ特異的パッケージング部位で開始され、ファージカプシドのヘッドフル容量に達するまで続く。この容量、およびしたがってクローン挿入断片サイズは、使用する特異的なファージに基づくものであってもよい。
【0198】
ファージ頭部からはみ出たDNAおよびあらゆる非パッケージDNAは、DNアーゼIまたは4塩基認識制限エンドヌクレアーゼによって消化する。パッケージDNAは、ヌクレアーゼ消化から保護される。次いで、クローンの、後の環状化を可能にするDNAリンカーを、パッケージDNAの末端の末端にライゲーションする。その代わりに、パッケージDNAは、ファージ頭部から抽出し、その後、リンカーライゲーションおよび再度パッケージングを続けてもよい。次いで、ファージ尾部を追加し、クローニングDNAを含有するビリオンは、E.coliを形質移入し、その後、抗生物質抵抗性の選択を続けるために使用する。
【0199】
(実施例3)
PACクローニングベクターの中にDNAをクローニングするためのλdoc粒子の代替の使用
実施例1の手順を、プロヘッドがDNAで満たされるポイントまで行ない、次いで、Sau3Aを用いて切断する。ファージ尾部を直接、追加する代わりに、図3のリンカーを使用して、DNAがカプシドからはみ出ている一方で、cosR部位をλdoc粒子にライゲーションする。リンカーをライゲーションした後に、実施例1におけるように、尾部を追加する。正常なcosRおよびcosL末端を有するファージ粒子は、カプシドを破壊して開け、DNAを環状にし、再度パッケージすることが必要ではなく、細胞の中にPACを導入するために直接、使用されるので、これは、全体的なプロセスを単純化する。
【0200】
(実施例4)
in vivoトランスポゾン組換えクローニング
実施例1および3において、かなりの量のクローニング容量がベクターによって取られる。これは、PACベクターが、相当な長さを有し、したがって、そうでなければクローニングDNAに当てることができる、カプシドにおける空間を占めるという事実によって引き起こされる。この制限について検討するために、cos部位の側面に位置する修飾Tn5転位性末端、組換えのためのattP部位、および選択可能マーカーとしてのKanRを含む転位カセットを調製する。トランスポゾンは、ゲノムDNAの中にランダムにin vitroにおいて導入し、cos部位からパッケージし、Sau3A消化し、cosRリンカーを追加し、結果として生じた断片を精製し、再度パッケージする。次いで、挿入断片は、attP部位を含有する修飾PACベクターを含む株の中に導入する。これは、attBに対する同起源の組換え部位である。株は、IPTG誘発性Ptacプロモーター下でpHS3−1プラスミドからIntタンパク質を発現する(Leeら1990年)。宿主はまた、組換えがプラスミドとのみ生じることを確実にするためにIS−ともする。一度、細胞において、挿入断片がcos突出を介して環化したら、それらは、Int媒介性の組換えを通してPACの中に組換えられる。組換え体は、ベクター(CamR)および挿入断片(KanR)の両方に対する抗生物質を含有する培地上で平板培養することによって選択する。
【0201】
(実施例5)
転位性クローニングベクター
一連の転位性クローニングベクターは、バクテリオファージin vitroパッケージングを使用するクローニングのための形質を含むように構築した。これらの形質は、1)バクテリオファージλのファージ特異的パッケージング開始部位(cos)および162bp P1パッケージング開始部位(pac)、2)誘発性複製開始点、oriV、ならびに3)転位のための19bpモザイク末端配列が側面に位置するカナマイシン抵抗性遺伝子を含む。この構築物におけるモザイク末端は、高活性Tn5 in vitro転位システムに基づくものであり、トランスポゾンの発生を可能にするように設計し、パッケージ部位、CAT(クロラムフェニコールアセチルトランスフェラーゼ)遺伝子、oriV、およびPvuII制限酵素を用いる消化後のモザイク末端を含む。標的DNAの中へのトランスポゾンの組み込みは、クロラムフェニコール(Camr)についての選択によって確認することができ、カナマイシン(Kans)に対する感度についてさらにスクリーニングすることができる。cosファージパッケージング開始部位およびpacファージパッケージング開始部位の位置は、組み込みに続いて、パッケージングが、これらの部位で始まり、最終的に隣接標的DNAまで、ベクターを通して時計回りに続く(図4を参照)ように設計する。これは、クローンが、安定して選択され、パッケージング、形質移入、および環状化に続いてプラスミドとして維持されることを可能にする。
【0202】
cos、pac、ならびにcosおよびpacの両方を含有する一連の3つのプラスミドを構築した。最後に、Kanr遺伝子の側面に位置する19bpモザイク末端配列を、最終産物の3つのベクターに追加した。ベクターシリーズのそれぞれの構成要素を個々に試験し、予想通りに機能することを示した。3つのベクターはすべて、in vivoにおいて効率的に転位し、DH10Bゲノムの中に組み込まれることが示された。転位に続いて分析した形質転換体の約98%が、Camr Kansであり、組み込みが確認された。候補からのゲノムDNAのサザンブロッティングにより、ゲノムの中へのランダムな組み込みを確認した。
【0203】
cos機能性を2つの実験によって確認した。精製λターミナーゼ(Epicentre)を用いる単純な消化により、ベクターのcos切断を確認した。さらに、プラスミドマルチマーから構成されるコンカテマーが、λプロファージによってin vivoにおいてパッケージされ得るかどうかを決定するために、in vivoコスミドパッケージングアッセイを行なった。cosを含有する2つのベクターから産生されるコンカテマーは、Camr形質導入粒子の数によって測定されるように、効率的にパッケージされたが、pacのみを有するベクターは、in vivoにおいてλによってパッケージすることができなかった。
【0204】
(実施例6)
ファージパッケージング部位のライゲーション
パッケージ部位は、線状転位性クローニングベクターを、部分的に消化したゲノムDNAに直接、ライゲーションすることによって、標的DNAの中に導入する。EcoRI、BamHI、およびHindIII部位(または全多重クローニング部位)を、tL3ターミネーターおよびモザイク末端の間に位置するユニークなPmeI部位で、転位性クローニングベクターの中に導入する。EcoRI部分的消化物を使用する場合、転位性クローニングベクターは、EcoRIおよびPvuIIによって消化し、精製し、一方の末端に平滑末端化PvuIIおよびpac/cos部位ならびに反対の末端にEcoRI付着末端を有する線状転位性クローニングベクターがもたらされる。EcoRI/PvuII二重消化物は、ベクターエレメントを含有するクローンを用いる一方向のパッケージングをもたらす。
【0205】
線状転位性クローニングベクターを、部分的に消化したゲノムDNAにライゲーションする。次いで、in vitroパッケージング反応を行ない、転位性クローニングベクターの構成要素を通して、転位性クローニングベクター上のパッケージ部位でパッケージングを開始し、ファージ頭部のヘッドフル容量に達するまで、ライゲーションゲノムDNAまで続ける。転位によるパッケージング開始部位の導入と同様に、はみ出たDNAは消化し、適切なリンカーは、DNAの環状化のためにライゲーションし、ファージ尾部を追加し、使用したクローニングDNAを含有するビリオンをE.coliに形質移入する。
【0206】
(実施例7)
λまたはP1を用いるin vitroパッケージング
市販で入手可能なλ in vitroパッケージング抽出物は、シングルチューブシステムとして、段階1および段階2のパッケージング抽出物を組み合わせたものである。クローニングシステムは、順次、2つのパッケージ段階を順次使用してもよい。2つの従来の相補的な溶原性E.coli株BHB2690(段階1)およびBHB2688(段階2)からの抽出物を生成し、試験した。2段階パッケージングシステムの効率は、市販で入手可能なシングルチューブシステムに匹敵した。さらに、発明者らは、段階1抽出物を使用して、パッケージベクターのcos切断を示し、実施例5において記載されるベクターの機能性を確認することができた。バクテリオファージP1のin vitroパッケージングシステムについては、発明者らは、それぞれ、株NS3208およびNS3210から、P1溶原菌の段階1および段階2の抽出物を生成した(Corenら、J Mol Biol、249巻:176〜84頁、1995)。発明者らはまた、段階1(pacase)抽出物を使用して、パッケージベクターのpac部位の切断をも示した。
【0207】
(実施例8)
λファージパッケージングを使用するゲノムDNAのクローニングおよびファージカプシドのアフィニティー精製
本実施例は、λファージパッケージングを使用するDNAのクローニングおよびファージカプシドのアフィニティー精製について記載する。cos部位、複製開始点、および薬剤抵抗性マーカーを含有する核酸は、たとえばin vitro転位によって、ゲノムDNAの中にランダムに挿入する。λターミナーゼ(Nu1およびAの遺伝子の産物を含有する)は、cos部位におよびλプロヘッドに結合し、次いで、cos部位でDNAを切断する(図6)。次いで、ATP駆動モーターは、プロヘッドの中にDNAを詰め込む。ウイルスカプシドは、カプシドへのDタンパク質の追加によって、DNAがその中に挿入されるとともに拡張する。約50kbのDNAがカプシドを満たすまで、このプロセスは続く。カプシドからぶら下がったままのDNAは、頻繁に切断する制限部位を用いて切断することができる。カプシドの内部にあるDNAは保護されるので、カプシドの外部にあるDNAのみを制限酵素によって切断することができる。
【0208】
DNAの制限に続いて、ファージカプシドは、カラムに結合した抗Dタンパク質抗体を使用してアフィニティー精製する。精製カプシドからのDNAは、フェノール抽出によって単離することができる。次いで、DNAは環状にし、細菌の中に形質転換する。
【0209】
(実施例9)
P22HTファージパッケージングを使用するゲノムDNAのクローニング
本実施例は、P22HTファージパッケージングを使用するゲノムDNAのクローニングについて記載する。Gp2およびGp3のタンパク質を含有するP22HTターミナーゼは、ランダムな部位でゲノムDNAに結合し、それを切断する(図7)。ターミナーゼは、DNAに結合したままであり、プロヘッドがいっぱいになるまで、P22プロヘッドの中にそれを詰め込む。カプシドが拡張するとともに、それは、広がり、破壊されたDNAになお付着しているターミナーゼを放出する立体構造変化を受ける。次いで、ターミナーゼは、新しいプロヘッドの中にDNAを挿入し、DNAパッケージングの新しいサイクルを始めることができる。ターミナーゼは、隣接ゲノムDNAセグメントの連続パッケージングの多くのサイクルを続けることができる。
【0210】
パッケージDNAをクローニングするために、ファージカプシドは、拡張されたカプシドの外部に存在するエピトープに結合することができる抗体を含有するカラムを使用してアフィニティー精製することができる。カプシドはまた、差次的沈降または等密度遠心分離によって単離することもできる。これらの方法は、DNアーゼ消化が伴い得る。DNAは、上記に記載されるように、さらにそれを抽出し、環状にし、細菌の中に形質転換することによってクローニングすることができる。
【0211】
(実施例10)
ファージパッケージングによってクローニングされたゲノムDNAの末端の配列の特徴づけ
本実施例は、ファージベースのシステムを使用してパッケージされたゲノムDNAの末端の配列をどのように特徴づけることができるかについて記載する。ゲノムDNAは、ファージカプシドから最初に単離される。次に、EcoP15I部位が側面に位置する2つの外向きリーチングプライマー配列を含有する核酸を、単離DNA断片の末端にライゲーションする(図8)。図9Bは、リーチングプライマー対核酸の構造を示す。リーチングプライマー対断片は、2つの異なるゲノムDNA分子へのリーチングプライマー対断片のライゲーションを回避するのに十分に低い稀釈度で単離DNAにライゲーションする。リーチングプライマー対断片の末端は、それとモノマー環を形成することを回避するためにリン酸化されなくてもよい。
【0212】
ライゲーションに続いて、DNAをEcoP15Iを用いて消化し、これにより、EcoP15I認識部位から27bp離れたところでDNAを切断する。この制限酵素を用いるEcoR消化は、2つのリーチングプライマー配列、側面に位置するEcoP15I部位、およびそれぞれの末端に27bpのゲノムDNAを含有するDNA断片を放出する。放出断片におけるゲノムDNAは、パッケージゲノムDNAのそれぞれの末端を表わす。放出断片は、フィルインすることができる。図9Aは、平滑末端を作り出すために、放出断片のEcoP15I切断末端をどのようにフィルインすることができるかを示す。次いで、放出平滑断片は、ライゲーションして、対になった末端環DNAを作り出すことができる。次いで、このDNAは、2つのリーチングプライマーを使用して、PCR増幅することができ(図10)、次いで、増幅産物は、配列決定することができる。リーチングプライマー対部位の間に制限部位がある場合、対になった末端環は、PCR増幅の前に適切な制限酵素を用いて切断することによって線状にしてもよい。DNAはまた、固相核酸増幅を使用して増幅を行なうことによって、PCR増幅し、直接配列決定してもよい。上記に記載されるアプローチはまた、ビーズエマルションPCR増幅および配列決定における使用のために、磁性ビーズに結合させたリーチングプライマーのうち1つに適応することができる(図11)。
【特許請求の範囲】
【請求項1】
核酸を配列決定する方法であって、
(a)核酸を含むカプシドを提供するステップと、
(b)前記核酸を単離するステップと、
(c)前記核酸を配列決定するステップと
を含む方法。
【請求項2】
前記配列決定が、
(a)水性マイクロリアクターが、前記核酸の単一コピー、前記核酸に結合することができる単一ビーズ、および核酸増幅を行なうために必要な試薬を含有する増幅反応溶液を含むように、油中水型エマルション中で前記水性マイクロリアクターの中に前記核酸を送達するステップと、
(b)前記マイクロリアクター中で前記核酸を増幅して前記核酸の増幅コピーを形成し、前記マイクロリアクター中で前記増幅コピーを前記ビーズに結合させるステップと、
(c)平面表面上にある少なくとも10,000個の反応チャンバーのアレイに前記ビーズを送達するステップであって、複数の前記反応チャンバーが単一核酸結合ビーズしか含まないステップと、
(d)複数の前記反応チャンバー上で配列決定反応を同時に行なうステップと
を含む、請求項1に記載の方法。
【請求項3】
前記ビーズが、少なくとも10,000個の増幅コピーに結合する、請求項2に記載の方法。
【請求項4】
ステップ(b)が、ポリメラーゼ連鎖反応を使用することによって達成される、請求項2に記載の方法。
【請求項5】
前記配列決定反応が、ピロリン酸ベースの配列決定反応である、請求項2に記載の方法。
【請求項6】
前記配列決定反応が、
(a)有効量の配列決定プライマーを前記核酸の前記増幅コピーにアニールし、ポリメラーゼおよび所定のヌクレオチド三リン酸を用いて前記配列決定プライマーを伸長させて、配列決定産物、および前記所定のヌクレオチド三リン酸が前記配列決定プライマーの3’末端上に取り込まれる場合には配列決定反応副産物を得るステップと、
(b)前記配列決定反応副産物を同定し、それによって、反応チャンバー中で前記核酸の配列を決定するステップと
を含む、請求項2に記載の方法。
【請求項7】
複数の核酸を配列決定する方法であって、
(a)それぞれが核酸を含む複数のカプシドを提供するステップと、
(b)前記核酸を単離するステップと、
(c)前記核酸を配列決定するステップと
を含む方法。
【請求項8】
前記配列決定が、
(a)複数の水性マイクロリアクターが、核酸の単一コピー、前記核酸に結合することができる単一ビーズ、および核酸増幅を行なうために必要な試薬を含有する増幅反応溶液を含むように、油中水型エマルション中で前記水性マイクロリアクターの中に前記核酸を送達するステップと、
(b)前記マイクロリアクター中で前記核酸を増幅して前記核酸の増幅コピーを形成し、前記マイクロリアクター中で前記増幅コピーを前記ビーズに結合させるステップと、
(c)平面表面上にある少なくとも10,000個の反応チャンバーのアレイに前記ビーズを送達するステップであって、複数の前記反応チャンバーが単一核酸結合ビーズしか含まないステップと、
(d)複数の前記反応チャンバー上で配列決定反応を同時に行なうステップと
を含む、請求項7に記載の方法。
【請求項9】
前記ビーズが、少なくとも10,000個の増幅コピーに結合する、請求項2に記載の方法。
【請求項10】
ステップ(b)が、ポリメラーゼ連鎖反応を使用することによって達成される、請求項2に記載の方法。
【請求項11】
前記配列決定反応が、ピロリン酸ベースの配列決定反応である、請求項2に記載の方法。
【請求項12】
前記配列決定反応が、
(a)有効量の配列決定プライマーを前記核酸の前記増幅コピーにアニールし、ポリメラーゼおよび所定のヌクレオチド三リン酸を用いて前記配列決定プライマーを伸長させて、配列決定産物、および前記所定のヌクレオチド三リン酸が前記配列決定プライマーの3’末端上に取り込まれる場合には配列決定反応副産物を得るステップと、
(b)前記配列決定反応副産物を同定し、それによって、反応チャンバー中で前記核酸の配列を決定するステップと
を含む、請求項2に記載の方法。
【請求項13】
ステップ(b)における前記核酸を単離するステップが、
(a)第1のアダプター末端および第2のアダプター末端を前記核酸にライゲーションするステップであって、前記第1のアダプター末端が、オリゴヌクレオチド配列Yを含み、前記核酸の5’末端にライゲーションし、前記第2のアダプター末端が、オリゴヌクレオチド配列Zを含み、前記核酸の3’末端にライゲーションし、第1のアダプターが、5’末端で固体支持体に前記核酸を固定するための手段を有するステップと、
(b)ステップ(a)の前記核酸を、前記固体支持体の存在下において、1つまたは複数のコロニープライマーXと混合するステップであって、それぞれのコロニープライマーXが、前記オリゴヌクレオチド配列Zにハイブリダイズすることができ、5’末端で前記固体支持体に前記コロニープライマーを固定するための手段を有し、それによって、前記核酸および前記コロニープライマーの両方の5’末端が、前記固体支持体に固定され、前記核酸および前記コロニープライマーの両方の前記5’末端が、DNA変性条件下での水または水性緩衝液を用いる洗浄によって、それらを除去することができないように、前記固体支持体に固定されるステップと、
(c)核酸コロニーが生成されるように、前記固定された核酸上で1つまたは複数の核酸増幅反応を行なうステップと
を含む、請求項1に記載の方法。
【請求項14】
前記オリゴヌクレオチド配列Zが、オリゴヌクレオチド配列Yに相補的であり、コロニープライマーXが、オリゴヌクレオチド配列Yと同じ配列である、請求項13に記載の方法。
【請求項15】
2つの異なるコロニープライマーXを、請求項13に記載のステップ(a)における前記核酸と混合し、前記オリゴヌクレオチド配列Zが、前記コロニープライマーXのうち1つにハイブリダイズすることができ、前記オリゴヌクレオチドYが、前記コロニープライマーXのうち1つの配列と同じである、請求項13に記載の方法。
【請求項16】
1つまたは複数の前記核酸コロニーにおける前記核酸を配列決定するステップをさらに含む、請求項13に記載の方法。
【請求項17】
前記配列決定するステップが、標識ヌクレオチドの取り込みおよび検出を含む、請求項16に記載の方法。
【請求項18】
前記核酸コロニーを視覚化する追加のステップをさらに含む、請求項16に記載の方法。
【請求項19】
前記視覚化ステップが、標識核酸プローブまたは非標識核酸プローブの使用を含む、請求項18に記載の方法。
【請求項20】
前記核酸および前記コロニープライマーを前記固体支持体に固定するための前記手段が、前記核酸および前記コロニープライマーを前記支持体に共有結合で固定するための手段を含む、請求項13に記載の方法。
【請求項21】
前記核酸および前記コロニープライマーを前記固体支持体に共有結合で固定するための前記手段が、化学的に修飾された基である、請求項20に記載の方法。
【請求項22】
前記核酸および前記コロニープライマーを前記固体支持体に固定するための前記手段が、前記核酸および前記コロニープライマーを前記支持体に共有結合で固定するための手段を含む、請求項21に記載の方法。
【請求項23】
前記化学的に修飾可能な官能基が、アミノ基である、請求項22に記載の方法。
【請求項24】
少なくとも1つの前記核酸および前記コロニープライマーの両方の前記5’末端が固定される前記固体支持体が、ラテックスビーズ、デキストランビーズ、ポリスチレンおよびポリプロピレン表面、ポリアクリルアミドゲル、金表面、ガラス表面、ならびにシリコンウェーハからなる群から選択される、請求項13に記載の方法。
【請求項25】
前記固体支持体が、ガラスである、請求項24に記載の方法。
【請求項26】
前記固体支持体上の前記核酸コロニーの密度が、10,000/mm2〜100,000/mm2である、請求項13に記載の方法。
【請求項27】
前記固体支持体に付着した前記コロニープライマーXの密度が、少なくとも1fmol/mm2である、請求項13に記載の方法。
【請求項28】
前記核酸の密度が、10,000/mm2〜100,000/mm2である、請求項13に記載の方法。
【請求項29】
前記核酸および前記コロニープライマーの両方の前記5’末端が、共有結合を介して前記固体支持体に固定される、請求項13に記載の方法。
【請求項30】
ステップ(b)における前記核酸を単離するステップが、
(a)第1のアダプター末端および第2のアダプター末端を前記核酸にライゲーションするステップであって、前記第1のアダプター末端が、オリゴヌクレオチド配列Yを含み、前記核酸の5’末端にライゲーションし、前記第2のアダプター末端が、オリゴヌクレオチド配列Zを含み、前記核酸の3’末端にライゲーションし、第1のアダプターが、5’末端で固体支持体に前記核酸を固定するための手段を有するステップと、
(b)ステップ(a)の前記核酸を、前記固体支持体の存在下において、1つまたは複数の縮重コロニープライマーXと混合するステップであって、それぞれのコロニープライマーXが、前記オリゴヌクレオチド配列Zにハイブリダイズすることができ、5’末端で前記固体支持体に前記コロニープライマーを固定するための手段を有し、それによって、核酸鋳型および前記コロニープライマーの両方の5’末端が、前記固体支持体に固定され、核酸および前記コロニープライマーの両方の前記5’末端が、DNA変性条件下での水または水性緩衝液を用いる洗浄によって、それらを除去することができないように、前記固体支持体に固定されるステップと、
(c)核酸コロニーが生成されるように、前記固定された核酸上で1つまたは複数の核酸増幅反応を行なうステップと
を含む、請求項1に記載の方法。
【請求項31】
ステップ(a)の前記核酸および前記コロニープライマーの両方の前記5’末端が、共有結合を介して前記固体支持体に固定される、請求項30に記載の方法。
【請求項32】
前記核酸を単離するステップが、
(a)第1のアダプター末端および第2のアダプター末端を前記核酸にライゲーションするステップであって、前記第1のアダプター末端が、オリゴヌクレオチド配列Yを含み、前記核酸の5’末端にライゲーションし、前記第2のアダプター末端が、オリゴヌクレオチド配列Zを含み、前記核酸の3’末端にライゲーションし、第1のアダプターが、5’末端で固体支持体に前記核酸を固定するための手段を有するステップと、
(b)ステップ(a)の前記核酸を、前記固体支持体の存在下において、1つまたは複数のコロニープライマーXと混合するステップであって、それぞれのコロニープライマーXが、前記オリゴヌクレオチド配列Zにハイブリダイズすることができ、5’末端で前記固体支持体に前記コロニープライマーを固定するための手段を有し、それによって、前記核酸および前記コロニープライマーの両方の5’末端が、前記固体支持体に固定され、前記核酸および前記コロニープライマーの両方の前記5’末端が、DNA変性条件下での水または水性緩衝液を用いる洗浄によって、それらを除去することができないように、前記固体支持体に固定されるステップと、
(c)核酸コロニーが生成されるように、前記固定された核酸上で1つまたは複数の核酸増幅反応を行なうステップと
を含む、請求項7に記載の方法。
【請求項33】
前記オリゴヌクレオチド配列Zが、オリゴヌクレオチド配列Yに相補的であり、コロニープライマーXは、オリゴヌクレオチド配列Yと同じ配列である、請求項32に記載の方法。
【請求項34】
2つの異なるコロニープライマーXを、請求項32に記載のステップ(a)における前記核酸と混合し、前記オリゴヌクレオチド配列Zが、前記コロニープライマーXのうち1つにハイブリダイズすることができ、前記オリゴヌクレオチドYが、前記コロニープライマーXのうち1つの配列と同じである、請求項32に記載の方法。
【請求項35】
核酸コロニーにおける前記核酸を配列決定するステップをさらに含む、請求項32に記載の方法。
【請求項36】
前記配列決定するステップが、標識ヌクレオチドの取り込みおよび検出を含む、請求項35に記載の方法。
【請求項37】
前記核酸コロニーを視覚化する追加のステップをさらに含む、請求項35に記載の方法。
【請求項38】
前記視覚化ステップが、標識核酸プローブまたは非標識核酸プローブの使用を含む、請求項37に記載の方法。
【請求項39】
1つより多い核酸コロニー中に存在する前記核酸の完全なまたは部分的な配列が同時に決定される、請求項35に記載の方法。
【請求項40】
前記核酸および前記コロニープライマーを前記固体支持体に固定するための前記手段が、核酸および前記コロニープライマーを前記支持体に共有結合で固定するための手段を含む、請求項32に記載の方法。
【請求項41】
前記核酸および前記コロニープライマーを前記固体支持体に共有結合で固定するための前記手段が、化学的に修飾された基である、請求項40に記載の方法。
【請求項42】
前記核酸および前記コロニープライマーを前記固体支持体に固定するための前記手段が、前記核酸および前記コロニープライマーを前記支持体に共有結合で固定するための手段を含む、請求項41に記載の方法。
【請求項43】
前記化学的に修飾可能な官能基が、アミノ基である、請求項42に記載の方法。
【請求項44】
少なくとも1つの前記核酸および前記コロニープライマーの両方の前記5’末端が固定される前記固体支持体が、ラテックスビーズ、デキストランビーズ、ポリスチレンおよびポリプロピレン表面、ポリアクリルアミドゲル、金表面、ガラス表面、ならびにシリコンウェーハからなる群から選択される、請求項32に記載の方法。
【請求項45】
前記固体支持体が、ガラスである、請求項44に記載の方法。
【請求項46】
前記固体支持体上の前記核酸コロニーの密度が、10,000/mm2〜100,000/mm2である、請求項32に記載の方法。
【請求項47】
前記固体支持体に付着した前記コロニープライマーXの密度が、少なくとも1fmol/mm2である、請求項32に記載の方法。
【請求項48】
それぞれの核酸の密度が、10,000/mm2〜100,000/mm2である、請求項32に記載の方法。
【請求項49】
前記核酸および前記コロニープライマーの両方の前記5’末端が、共有結合を介して前記固体支持体に固定される、請求項32に記載の方法。
【請求項50】
ステップ(b)における前記核酸を単離するステップが、
(a)第1のアダプター末端および第2のアダプター末端を前記核酸にライゲーションするステップであって、前記第1のアダプター末端が、オリゴヌクレオチド配列Yを含み、前記核酸の5’末端にライゲーションし、前記第2のアダプター末端が、オリゴヌクレオチド配列Zを含み、前記核酸の3’末端にライゲーションし、第1のアダプターが、5’末端で固体支持体に前記核酸を固定するための手段を有するステップと、
(b)ステップ(a)の前記核酸を、前記固体支持体の存在下において、1つまたは複数の縮重コロニープライマーXと混合するステップであって、それぞれのコロニープライマーXが、前記オリゴヌクレオチド配列Zにハイブリダイズすることができ、5’末端で前記固体支持体に前記コロニープライマーを固定するための手段を有し、それによって、前記核酸および前記コロニープライマーの両方の5’末端が、前記固体支持体に固定され、核酸および前記コロニープライマーの両方の前記5’末端が、DNA変性条件下での水または水性緩衝液を用いる洗浄によって、それらを除去することができないように、前記固体支持体に固定されるステップと、
(c)核酸コロニーが生成されるように、前記固定された核酸上で核酸増幅反応を行なうステップと
をさらに含む、請求項32に記載の方法。
【請求項51】
ステップ(a)の前記核酸および前記コロニープライマーの両方の前記5’末端が、共有結合を介して前記固体支持体に固定される、請求項50に記載の方法。
【請求項52】
標的核酸から核酸をクローニングする方法であって、
(a)標的核酸の中にパッケージング開始部位を導入するステップであって、前記標的核酸からクローニングされる前記核酸が、前記パッケージング開始部位の下流にあるステップと、
(b)前記パッケージング開始部位でパッケージングを開始し、それによって、前記標的核酸からクローニングされる前記核酸がパッケージされるステップと、
(c)前記パッケージされた核酸を単離するステップと
を含む、方法。
【請求項53】
前記パッケージング開始部位が、λ、P1、P7、またはT4パッケージング開始部位である、請求項52に記載の方法。
【請求項54】
前記標的核酸がベクターである、請求項52に記載の方法。
【請求項55】
前記標的核酸が染色体DNAである、請求項52に記載の方法。
【請求項56】
前記標的核酸が全ゲノムである、請求項52に記載の方法。
【請求項57】
前記パッケージング開始部位が、前記核酸の中にランダムに導入される、請求項52に記載の方法。
【請求項58】
前記パッケージング開始部位が、前記標的核酸の中への第1の核酸の転位によって前記標的核酸の中に導入され、前記第1の核酸が、転位性末端および前記パッケージング開始部位を含む、請求項52に記載の方法。
【請求項59】
前記転位性末端が、Tn7、Tn5、Tn10、Mu、およびMarinerからなる群から選択される、請求項58に記載の方法。
【請求項60】
前記パッケージング開始部位が、第1の核酸および前記標的核酸をライゲーションすることによって、前記標的核酸の中に導入され、前記第1の核酸が、前記パッケージング開始部位を含む、請求項52に記載の方法。
【請求項61】
前記パッケージされた核酸が、カプシド中にパッケージされる、請求項52に記載の方法。
【請求項62】
パッケージングが、in vitroにおいて生じる、請求項61に記載の方法。
【請求項63】
パッケージングは、in vivoにおいて生じる、請求項61に記載の方法。
【請求項64】
前記第1の核酸が、組み込み部位をさらに含み、前記パッケージされた核酸が、前記組み込み部位を含む、請求項58または60に記載の方法。
【請求項65】
相補的な組み込み部位でベクターの中に前記パッケージされた核酸を組み込むステップをさらに含む、請求項64に記載の方法。
【請求項66】
前記第1の核酸の前記組み込み部位が、attBであり、前記ベクターの前記組み込み部位が、attPである、請求項65に記載の方法。
【請求項67】
組み込みは、in vitroにおいて生じる、請求項66に記載の方法。
【請求項68】
組み込みは、in vivoにおいて生じる、請求項66に記載の方法。
【請求項69】
前記第1の核酸が、複製開始点を含む、請求項58または60に記載の方法。
【請求項70】
前記複製開始点が、低コピー複製開始点である、請求項69に記載の方法。
【請求項71】
前記複製開始点が、高コピー複製開始点である、請求項69に記載の方法。
【請求項72】
前記第1の核酸が、低コピー複製開始点および高コピー複製開始点を含む、請求項69に記載の方法。
【請求項73】
前記高コピー複製開始点が、複製誘発剤に条件付きで応答性である、請求項72に記載の方法。
【請求項74】
前記第1の核酸が、前記複製誘発剤をコードする核酸をさらに含む、請求項73に記載の方法。
【請求項75】
前記複製誘発剤が、TrfAである、請求項74に記載の方法。
【請求項76】
請求項56に記載の方法に従ってゲノムから複数の核酸をクローニングするステップを含む、ゲノムライブラリーを産生する方法。
【請求項77】
標的核酸から核酸をクローニングする方法であって、
(a)標的核酸をP22HTターミナーゼと接触させるステップと、
(b)パッケージングを開始し、それによって、前記標的核酸からクローニングされる前記核酸がパッケージされるステップと、
(c)前記パッケージされた核酸を単離するステップと
を含む方法。
【請求項78】
前記標的核酸が全ゲノムである、請求項77に記載の方法。
【請求項79】
前記パッケージされた核酸がカプシド中にパッケージされる、請求項77に記載の方法。
【請求項80】
パッケージングが、in vitroにおいて生じる、請求項79に記載の方法。
【請求項81】
パッケージングが、in vivoにおいて生じる、請求項79に記載の方法。
【請求項1】
核酸を配列決定する方法であって、
(a)核酸を含むカプシドを提供するステップと、
(b)前記核酸を単離するステップと、
(c)前記核酸を配列決定するステップと
を含む方法。
【請求項2】
前記配列決定が、
(a)水性マイクロリアクターが、前記核酸の単一コピー、前記核酸に結合することができる単一ビーズ、および核酸増幅を行なうために必要な試薬を含有する増幅反応溶液を含むように、油中水型エマルション中で前記水性マイクロリアクターの中に前記核酸を送達するステップと、
(b)前記マイクロリアクター中で前記核酸を増幅して前記核酸の増幅コピーを形成し、前記マイクロリアクター中で前記増幅コピーを前記ビーズに結合させるステップと、
(c)平面表面上にある少なくとも10,000個の反応チャンバーのアレイに前記ビーズを送達するステップであって、複数の前記反応チャンバーが単一核酸結合ビーズしか含まないステップと、
(d)複数の前記反応チャンバー上で配列決定反応を同時に行なうステップと
を含む、請求項1に記載の方法。
【請求項3】
前記ビーズが、少なくとも10,000個の増幅コピーに結合する、請求項2に記載の方法。
【請求項4】
ステップ(b)が、ポリメラーゼ連鎖反応を使用することによって達成される、請求項2に記載の方法。
【請求項5】
前記配列決定反応が、ピロリン酸ベースの配列決定反応である、請求項2に記載の方法。
【請求項6】
前記配列決定反応が、
(a)有効量の配列決定プライマーを前記核酸の前記増幅コピーにアニールし、ポリメラーゼおよび所定のヌクレオチド三リン酸を用いて前記配列決定プライマーを伸長させて、配列決定産物、および前記所定のヌクレオチド三リン酸が前記配列決定プライマーの3’末端上に取り込まれる場合には配列決定反応副産物を得るステップと、
(b)前記配列決定反応副産物を同定し、それによって、反応チャンバー中で前記核酸の配列を決定するステップと
を含む、請求項2に記載の方法。
【請求項7】
複数の核酸を配列決定する方法であって、
(a)それぞれが核酸を含む複数のカプシドを提供するステップと、
(b)前記核酸を単離するステップと、
(c)前記核酸を配列決定するステップと
を含む方法。
【請求項8】
前記配列決定が、
(a)複数の水性マイクロリアクターが、核酸の単一コピー、前記核酸に結合することができる単一ビーズ、および核酸増幅を行なうために必要な試薬を含有する増幅反応溶液を含むように、油中水型エマルション中で前記水性マイクロリアクターの中に前記核酸を送達するステップと、
(b)前記マイクロリアクター中で前記核酸を増幅して前記核酸の増幅コピーを形成し、前記マイクロリアクター中で前記増幅コピーを前記ビーズに結合させるステップと、
(c)平面表面上にある少なくとも10,000個の反応チャンバーのアレイに前記ビーズを送達するステップであって、複数の前記反応チャンバーが単一核酸結合ビーズしか含まないステップと、
(d)複数の前記反応チャンバー上で配列決定反応を同時に行なうステップと
を含む、請求項7に記載の方法。
【請求項9】
前記ビーズが、少なくとも10,000個の増幅コピーに結合する、請求項2に記載の方法。
【請求項10】
ステップ(b)が、ポリメラーゼ連鎖反応を使用することによって達成される、請求項2に記載の方法。
【請求項11】
前記配列決定反応が、ピロリン酸ベースの配列決定反応である、請求項2に記載の方法。
【請求項12】
前記配列決定反応が、
(a)有効量の配列決定プライマーを前記核酸の前記増幅コピーにアニールし、ポリメラーゼおよび所定のヌクレオチド三リン酸を用いて前記配列決定プライマーを伸長させて、配列決定産物、および前記所定のヌクレオチド三リン酸が前記配列決定プライマーの3’末端上に取り込まれる場合には配列決定反応副産物を得るステップと、
(b)前記配列決定反応副産物を同定し、それによって、反応チャンバー中で前記核酸の配列を決定するステップと
を含む、請求項2に記載の方法。
【請求項13】
ステップ(b)における前記核酸を単離するステップが、
(a)第1のアダプター末端および第2のアダプター末端を前記核酸にライゲーションするステップであって、前記第1のアダプター末端が、オリゴヌクレオチド配列Yを含み、前記核酸の5’末端にライゲーションし、前記第2のアダプター末端が、オリゴヌクレオチド配列Zを含み、前記核酸の3’末端にライゲーションし、第1のアダプターが、5’末端で固体支持体に前記核酸を固定するための手段を有するステップと、
(b)ステップ(a)の前記核酸を、前記固体支持体の存在下において、1つまたは複数のコロニープライマーXと混合するステップであって、それぞれのコロニープライマーXが、前記オリゴヌクレオチド配列Zにハイブリダイズすることができ、5’末端で前記固体支持体に前記コロニープライマーを固定するための手段を有し、それによって、前記核酸および前記コロニープライマーの両方の5’末端が、前記固体支持体に固定され、前記核酸および前記コロニープライマーの両方の前記5’末端が、DNA変性条件下での水または水性緩衝液を用いる洗浄によって、それらを除去することができないように、前記固体支持体に固定されるステップと、
(c)核酸コロニーが生成されるように、前記固定された核酸上で1つまたは複数の核酸増幅反応を行なうステップと
を含む、請求項1に記載の方法。
【請求項14】
前記オリゴヌクレオチド配列Zが、オリゴヌクレオチド配列Yに相補的であり、コロニープライマーXが、オリゴヌクレオチド配列Yと同じ配列である、請求項13に記載の方法。
【請求項15】
2つの異なるコロニープライマーXを、請求項13に記載のステップ(a)における前記核酸と混合し、前記オリゴヌクレオチド配列Zが、前記コロニープライマーXのうち1つにハイブリダイズすることができ、前記オリゴヌクレオチドYが、前記コロニープライマーXのうち1つの配列と同じである、請求項13に記載の方法。
【請求項16】
1つまたは複数の前記核酸コロニーにおける前記核酸を配列決定するステップをさらに含む、請求項13に記載の方法。
【請求項17】
前記配列決定するステップが、標識ヌクレオチドの取り込みおよび検出を含む、請求項16に記載の方法。
【請求項18】
前記核酸コロニーを視覚化する追加のステップをさらに含む、請求項16に記載の方法。
【請求項19】
前記視覚化ステップが、標識核酸プローブまたは非標識核酸プローブの使用を含む、請求項18に記載の方法。
【請求項20】
前記核酸および前記コロニープライマーを前記固体支持体に固定するための前記手段が、前記核酸および前記コロニープライマーを前記支持体に共有結合で固定するための手段を含む、請求項13に記載の方法。
【請求項21】
前記核酸および前記コロニープライマーを前記固体支持体に共有結合で固定するための前記手段が、化学的に修飾された基である、請求項20に記載の方法。
【請求項22】
前記核酸および前記コロニープライマーを前記固体支持体に固定するための前記手段が、前記核酸および前記コロニープライマーを前記支持体に共有結合で固定するための手段を含む、請求項21に記載の方法。
【請求項23】
前記化学的に修飾可能な官能基が、アミノ基である、請求項22に記載の方法。
【請求項24】
少なくとも1つの前記核酸および前記コロニープライマーの両方の前記5’末端が固定される前記固体支持体が、ラテックスビーズ、デキストランビーズ、ポリスチレンおよびポリプロピレン表面、ポリアクリルアミドゲル、金表面、ガラス表面、ならびにシリコンウェーハからなる群から選択される、請求項13に記載の方法。
【請求項25】
前記固体支持体が、ガラスである、請求項24に記載の方法。
【請求項26】
前記固体支持体上の前記核酸コロニーの密度が、10,000/mm2〜100,000/mm2である、請求項13に記載の方法。
【請求項27】
前記固体支持体に付着した前記コロニープライマーXの密度が、少なくとも1fmol/mm2である、請求項13に記載の方法。
【請求項28】
前記核酸の密度が、10,000/mm2〜100,000/mm2である、請求項13に記載の方法。
【請求項29】
前記核酸および前記コロニープライマーの両方の前記5’末端が、共有結合を介して前記固体支持体に固定される、請求項13に記載の方法。
【請求項30】
ステップ(b)における前記核酸を単離するステップが、
(a)第1のアダプター末端および第2のアダプター末端を前記核酸にライゲーションするステップであって、前記第1のアダプター末端が、オリゴヌクレオチド配列Yを含み、前記核酸の5’末端にライゲーションし、前記第2のアダプター末端が、オリゴヌクレオチド配列Zを含み、前記核酸の3’末端にライゲーションし、第1のアダプターが、5’末端で固体支持体に前記核酸を固定するための手段を有するステップと、
(b)ステップ(a)の前記核酸を、前記固体支持体の存在下において、1つまたは複数の縮重コロニープライマーXと混合するステップであって、それぞれのコロニープライマーXが、前記オリゴヌクレオチド配列Zにハイブリダイズすることができ、5’末端で前記固体支持体に前記コロニープライマーを固定するための手段を有し、それによって、核酸鋳型および前記コロニープライマーの両方の5’末端が、前記固体支持体に固定され、核酸および前記コロニープライマーの両方の前記5’末端が、DNA変性条件下での水または水性緩衝液を用いる洗浄によって、それらを除去することができないように、前記固体支持体に固定されるステップと、
(c)核酸コロニーが生成されるように、前記固定された核酸上で1つまたは複数の核酸増幅反応を行なうステップと
を含む、請求項1に記載の方法。
【請求項31】
ステップ(a)の前記核酸および前記コロニープライマーの両方の前記5’末端が、共有結合を介して前記固体支持体に固定される、請求項30に記載の方法。
【請求項32】
前記核酸を単離するステップが、
(a)第1のアダプター末端および第2のアダプター末端を前記核酸にライゲーションするステップであって、前記第1のアダプター末端が、オリゴヌクレオチド配列Yを含み、前記核酸の5’末端にライゲーションし、前記第2のアダプター末端が、オリゴヌクレオチド配列Zを含み、前記核酸の3’末端にライゲーションし、第1のアダプターが、5’末端で固体支持体に前記核酸を固定するための手段を有するステップと、
(b)ステップ(a)の前記核酸を、前記固体支持体の存在下において、1つまたは複数のコロニープライマーXと混合するステップであって、それぞれのコロニープライマーXが、前記オリゴヌクレオチド配列Zにハイブリダイズすることができ、5’末端で前記固体支持体に前記コロニープライマーを固定するための手段を有し、それによって、前記核酸および前記コロニープライマーの両方の5’末端が、前記固体支持体に固定され、前記核酸および前記コロニープライマーの両方の前記5’末端が、DNA変性条件下での水または水性緩衝液を用いる洗浄によって、それらを除去することができないように、前記固体支持体に固定されるステップと、
(c)核酸コロニーが生成されるように、前記固定された核酸上で1つまたは複数の核酸増幅反応を行なうステップと
を含む、請求項7に記載の方法。
【請求項33】
前記オリゴヌクレオチド配列Zが、オリゴヌクレオチド配列Yに相補的であり、コロニープライマーXは、オリゴヌクレオチド配列Yと同じ配列である、請求項32に記載の方法。
【請求項34】
2つの異なるコロニープライマーXを、請求項32に記載のステップ(a)における前記核酸と混合し、前記オリゴヌクレオチド配列Zが、前記コロニープライマーXのうち1つにハイブリダイズすることができ、前記オリゴヌクレオチドYが、前記コロニープライマーXのうち1つの配列と同じである、請求項32に記載の方法。
【請求項35】
核酸コロニーにおける前記核酸を配列決定するステップをさらに含む、請求項32に記載の方法。
【請求項36】
前記配列決定するステップが、標識ヌクレオチドの取り込みおよび検出を含む、請求項35に記載の方法。
【請求項37】
前記核酸コロニーを視覚化する追加のステップをさらに含む、請求項35に記載の方法。
【請求項38】
前記視覚化ステップが、標識核酸プローブまたは非標識核酸プローブの使用を含む、請求項37に記載の方法。
【請求項39】
1つより多い核酸コロニー中に存在する前記核酸の完全なまたは部分的な配列が同時に決定される、請求項35に記載の方法。
【請求項40】
前記核酸および前記コロニープライマーを前記固体支持体に固定するための前記手段が、核酸および前記コロニープライマーを前記支持体に共有結合で固定するための手段を含む、請求項32に記載の方法。
【請求項41】
前記核酸および前記コロニープライマーを前記固体支持体に共有結合で固定するための前記手段が、化学的に修飾された基である、請求項40に記載の方法。
【請求項42】
前記核酸および前記コロニープライマーを前記固体支持体に固定するための前記手段が、前記核酸および前記コロニープライマーを前記支持体に共有結合で固定するための手段を含む、請求項41に記載の方法。
【請求項43】
前記化学的に修飾可能な官能基が、アミノ基である、請求項42に記載の方法。
【請求項44】
少なくとも1つの前記核酸および前記コロニープライマーの両方の前記5’末端が固定される前記固体支持体が、ラテックスビーズ、デキストランビーズ、ポリスチレンおよびポリプロピレン表面、ポリアクリルアミドゲル、金表面、ガラス表面、ならびにシリコンウェーハからなる群から選択される、請求項32に記載の方法。
【請求項45】
前記固体支持体が、ガラスである、請求項44に記載の方法。
【請求項46】
前記固体支持体上の前記核酸コロニーの密度が、10,000/mm2〜100,000/mm2である、請求項32に記載の方法。
【請求項47】
前記固体支持体に付着した前記コロニープライマーXの密度が、少なくとも1fmol/mm2である、請求項32に記載の方法。
【請求項48】
それぞれの核酸の密度が、10,000/mm2〜100,000/mm2である、請求項32に記載の方法。
【請求項49】
前記核酸および前記コロニープライマーの両方の前記5’末端が、共有結合を介して前記固体支持体に固定される、請求項32に記載の方法。
【請求項50】
ステップ(b)における前記核酸を単離するステップが、
(a)第1のアダプター末端および第2のアダプター末端を前記核酸にライゲーションするステップであって、前記第1のアダプター末端が、オリゴヌクレオチド配列Yを含み、前記核酸の5’末端にライゲーションし、前記第2のアダプター末端が、オリゴヌクレオチド配列Zを含み、前記核酸の3’末端にライゲーションし、第1のアダプターが、5’末端で固体支持体に前記核酸を固定するための手段を有するステップと、
(b)ステップ(a)の前記核酸を、前記固体支持体の存在下において、1つまたは複数の縮重コロニープライマーXと混合するステップであって、それぞれのコロニープライマーXが、前記オリゴヌクレオチド配列Zにハイブリダイズすることができ、5’末端で前記固体支持体に前記コロニープライマーを固定するための手段を有し、それによって、前記核酸および前記コロニープライマーの両方の5’末端が、前記固体支持体に固定され、核酸および前記コロニープライマーの両方の前記5’末端が、DNA変性条件下での水または水性緩衝液を用いる洗浄によって、それらを除去することができないように、前記固体支持体に固定されるステップと、
(c)核酸コロニーが生成されるように、前記固定された核酸上で核酸増幅反応を行なうステップと
をさらに含む、請求項32に記載の方法。
【請求項51】
ステップ(a)の前記核酸および前記コロニープライマーの両方の前記5’末端が、共有結合を介して前記固体支持体に固定される、請求項50に記載の方法。
【請求項52】
標的核酸から核酸をクローニングする方法であって、
(a)標的核酸の中にパッケージング開始部位を導入するステップであって、前記標的核酸からクローニングされる前記核酸が、前記パッケージング開始部位の下流にあるステップと、
(b)前記パッケージング開始部位でパッケージングを開始し、それによって、前記標的核酸からクローニングされる前記核酸がパッケージされるステップと、
(c)前記パッケージされた核酸を単離するステップと
を含む、方法。
【請求項53】
前記パッケージング開始部位が、λ、P1、P7、またはT4パッケージング開始部位である、請求項52に記載の方法。
【請求項54】
前記標的核酸がベクターである、請求項52に記載の方法。
【請求項55】
前記標的核酸が染色体DNAである、請求項52に記載の方法。
【請求項56】
前記標的核酸が全ゲノムである、請求項52に記載の方法。
【請求項57】
前記パッケージング開始部位が、前記核酸の中にランダムに導入される、請求項52に記載の方法。
【請求項58】
前記パッケージング開始部位が、前記標的核酸の中への第1の核酸の転位によって前記標的核酸の中に導入され、前記第1の核酸が、転位性末端および前記パッケージング開始部位を含む、請求項52に記載の方法。
【請求項59】
前記転位性末端が、Tn7、Tn5、Tn10、Mu、およびMarinerからなる群から選択される、請求項58に記載の方法。
【請求項60】
前記パッケージング開始部位が、第1の核酸および前記標的核酸をライゲーションすることによって、前記標的核酸の中に導入され、前記第1の核酸が、前記パッケージング開始部位を含む、請求項52に記載の方法。
【請求項61】
前記パッケージされた核酸が、カプシド中にパッケージされる、請求項52に記載の方法。
【請求項62】
パッケージングが、in vitroにおいて生じる、請求項61に記載の方法。
【請求項63】
パッケージングは、in vivoにおいて生じる、請求項61に記載の方法。
【請求項64】
前記第1の核酸が、組み込み部位をさらに含み、前記パッケージされた核酸が、前記組み込み部位を含む、請求項58または60に記載の方法。
【請求項65】
相補的な組み込み部位でベクターの中に前記パッケージされた核酸を組み込むステップをさらに含む、請求項64に記載の方法。
【請求項66】
前記第1の核酸の前記組み込み部位が、attBであり、前記ベクターの前記組み込み部位が、attPである、請求項65に記載の方法。
【請求項67】
組み込みは、in vitroにおいて生じる、請求項66に記載の方法。
【請求項68】
組み込みは、in vivoにおいて生じる、請求項66に記載の方法。
【請求項69】
前記第1の核酸が、複製開始点を含む、請求項58または60に記載の方法。
【請求項70】
前記複製開始点が、低コピー複製開始点である、請求項69に記載の方法。
【請求項71】
前記複製開始点が、高コピー複製開始点である、請求項69に記載の方法。
【請求項72】
前記第1の核酸が、低コピー複製開始点および高コピー複製開始点を含む、請求項69に記載の方法。
【請求項73】
前記高コピー複製開始点が、複製誘発剤に条件付きで応答性である、請求項72に記載の方法。
【請求項74】
前記第1の核酸が、前記複製誘発剤をコードする核酸をさらに含む、請求項73に記載の方法。
【請求項75】
前記複製誘発剤が、TrfAである、請求項74に記載の方法。
【請求項76】
請求項56に記載の方法に従ってゲノムから複数の核酸をクローニングするステップを含む、ゲノムライブラリーを産生する方法。
【請求項77】
標的核酸から核酸をクローニングする方法であって、
(a)標的核酸をP22HTターミナーゼと接触させるステップと、
(b)パッケージングを開始し、それによって、前記標的核酸からクローニングされる前記核酸がパッケージされるステップと、
(c)前記パッケージされた核酸を単離するステップと
を含む方法。
【請求項78】
前記標的核酸が全ゲノムである、請求項77に記載の方法。
【請求項79】
前記パッケージされた核酸がカプシド中にパッケージされる、請求項77に記載の方法。
【請求項80】
パッケージングが、in vitroにおいて生じる、請求項79に記載の方法。
【請求項81】
パッケージングが、in vivoにおいて生じる、請求項79に記載の方法。
【図1】

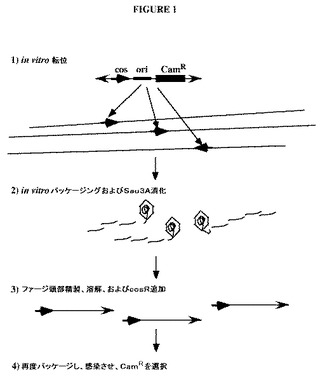
【図3】

【図4】
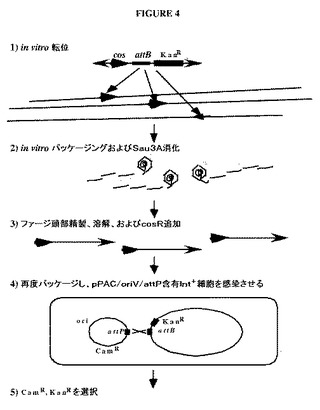
【図2】
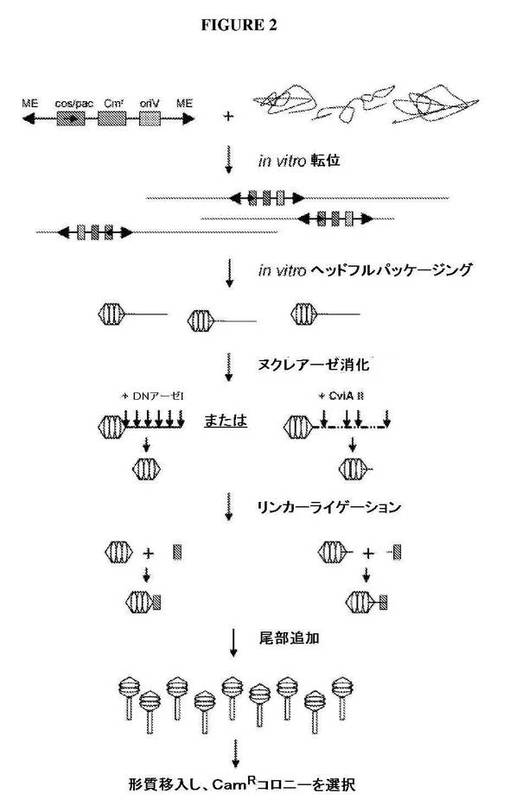
【図5】
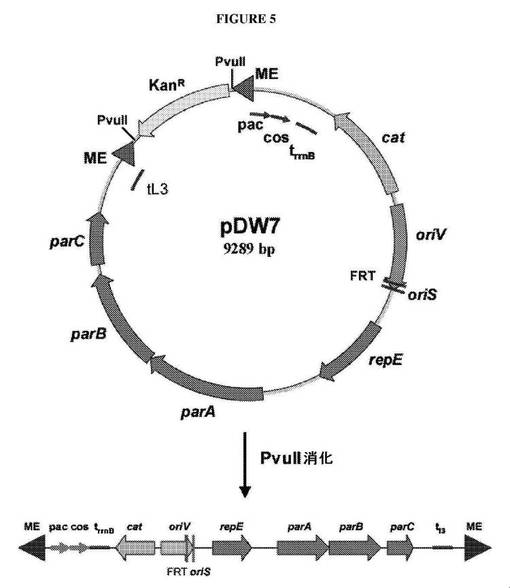
【図6】
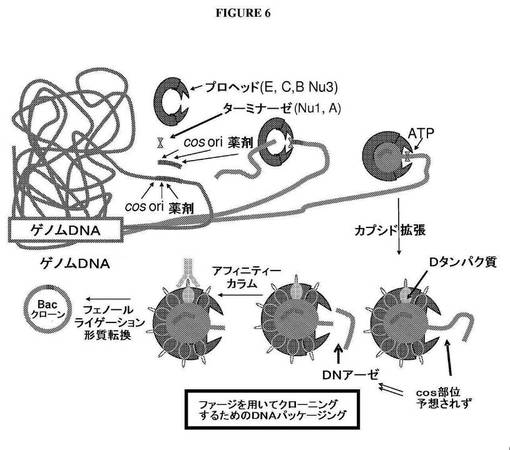
【図7】
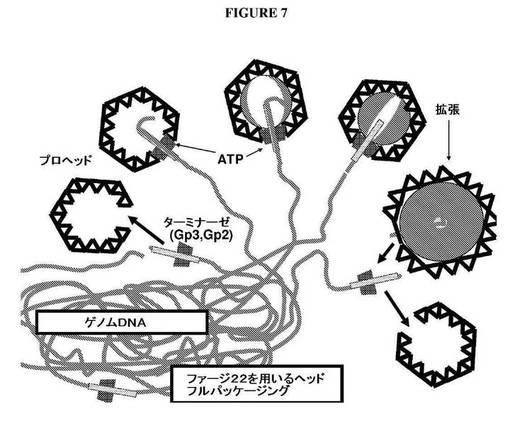
【図8】
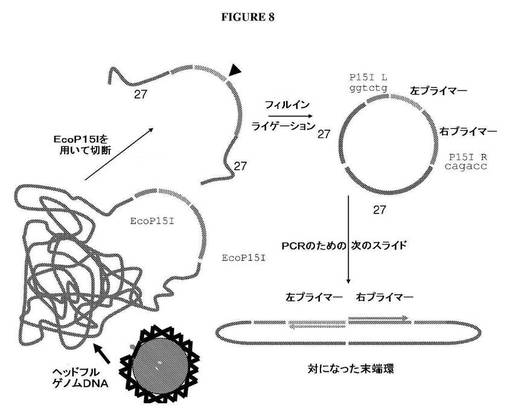
【図9】

【図10】
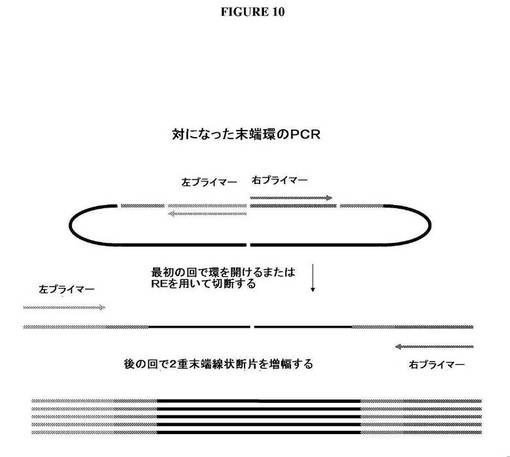
【図11】
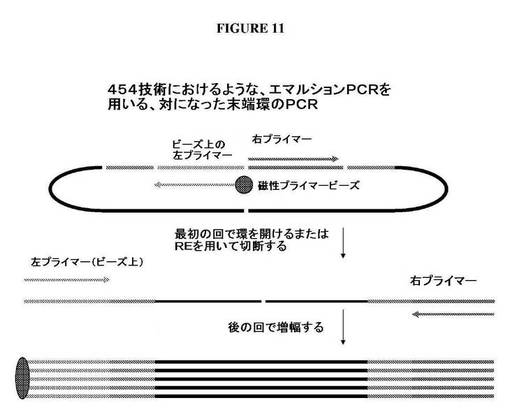
【図3】
【図4】
【図2】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2010−529862(P2010−529862A)
【公表日】平成22年9月2日(2010.9.2)
【国際特許分類】
【出願番号】特願2010−513352(P2010−513352)
【出願日】平成20年6月16日(2008.6.16)
【国際出願番号】PCT/US2008/067160
【国際公開番号】WO2008/157515
【国際公開日】平成20年12月24日(2008.12.24)
【出願人】(508314607)スカラブ ゲノミクス, エルエルシー (4)
【Fターム(参考)】
【公表日】平成22年9月2日(2010.9.2)
【国際特許分類】
【出願日】平成20年6月16日(2008.6.16)
【国際出願番号】PCT/US2008/067160
【国際公開番号】WO2008/157515
【国際公開日】平成20年12月24日(2008.12.24)
【出願人】(508314607)スカラブ ゲノミクス, エルエルシー (4)
【Fターム(参考)】
[ Back to top ]