
核酸精製方法、核酸抽出方法、及び核酸精製用キット
【課題】操作が簡便であり、短時間で高効率に核酸を精製することができる核酸精製方法の提供。
【解決手段】
核酸を含有するサンプルに含まれる物質をイオン交換樹脂10により吸着する手順を含み、上記イオン交換樹脂10には、陽イオン交換樹脂及び陰イオン交換樹脂を用いる核酸精製方法。上記陽イオン交換樹脂には、第1の陽イオン交換樹脂と、当該第1の陽イオン交換樹脂よりも排除限界分子量が小さな第2の陽イオン交換樹脂とを用いてもよい。
【解決手段】
核酸を含有するサンプルに含まれる物質をイオン交換樹脂10により吸着する手順を含み、上記イオン交換樹脂10には、陽イオン交換樹脂及び陰イオン交換樹脂を用いる核酸精製方法。上記陽イオン交換樹脂には、第1の陽イオン交換樹脂と、当該第1の陽イオン交換樹脂よりも排除限界分子量が小さな第2の陽イオン交換樹脂とを用いてもよい。
【発明の詳細な説明】
【技術分野】
【0001】
本技術は、核酸精製方法、核酸抽出方法、及び核酸精製用キットに関する。より詳しくは、イオン交換樹脂により夾雑物を吸着することで核酸を精製する方法等に関する。
【背景技術】
【0002】
PCR(Polymerase Chain Reaction)法やLAMP(Loop-Mediated Isothermal Amplification)法等の核酸増幅反応はバイオテクノロジーにおける様々な分野で応用されている。例えば、医学分野では、DNAやRNAの塩基配列に基づいた診断が行われており、農業分野では遺伝子組み換え作物の判定等でDNA鑑定が活用されている。
【0003】
核酸増幅反応では、微量サンプル中の核酸を高効率に増幅して検出できる。しかし、サンプルに含まれる核酸が極めて微量である場合には、検出下限量未満となる場合がある。さらに、サンプル中の核酸濃度が極めて低い場合には、反応場へ導入可能な容積のサンプル中に増幅対象核酸が含まれなかったために検出が不能となる場合がある。これらの場合には、予め精製や濃縮等をして抽出した核酸を反応場に導入することが有効となる。
【0004】
ここで、核酸の精製という点に着目すると、従来、フェノール/クロロホルム/エタノールを用いた方法が知られている。また、核酸吸着能を有する多孔質担体を用いた核酸の精製方法も知られている(特許文献1参照)。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2005−080555号公報
【特許文献2】国際公開第2009/060847号
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、従来のフェノール/クロロホルム/エタノールを用いた方法は、有害な有機溶媒を使用する必要があり、遠心分離操作等に手間がかかっていた。また、核酸吸着能を有する多孔質担体を用いた核酸の精製方法においては、複数の工程を行う必要があり、操作の簡便性からも課題があった。そのため、操作がより簡便であり、短時間で高効率に核酸を精製することができる方法が希求されていた。
【0007】
そこで、本技術は、操作が簡便であり、短時間で高効率に核酸を精製することができる核酸精製方法を提供することを主な目的とする。
【課題を解決するための手段】
【0008】
上記課題解決のため、本技術は、核酸を含有するサンプルに含まれる物質をイオン交換樹脂により吸着する手順を含み、上記イオン交換樹脂には、陽イオン交換樹脂及び陰イオン交換樹脂を用いる核酸精製方法を提供する。上記陽イオン交換樹脂により、サンプルに含まれる正電荷を有するタンパク質や金属塩等のカチオンを吸着することが可能になる。また、上記陰イオン交換樹脂により、サンプルに含まれる主に核酸以外の負電荷を有するアニオンを吸着することが可能になる。
また、上記陽イオン交換樹脂には、第1の陽イオン交換樹脂と、当該第1の陽イオン交換樹脂よりも排除限界分子量が低い第2の陽イオン交換樹脂とを用いることが好ましい。ここで、排除限界分子量とは、イオン交換樹脂に吸着させようとした種々の化合物において、イオン交換樹脂の細孔内に侵入しづらくなる化合物、すなわちイオン交換樹脂の細孔内に吸着されづらくなる化合物のうち、最も低い分子量をもつものの分子量をいう。換言すれば、当該排除限界分子量より高い分子量を有する化合物については、イオン交換樹脂に吸着されづらくなる。
上記陽イオン交換樹脂のうち、より排除限界分子量が高い上記第1の陽イオン交換樹脂が、上記タンパク質を選択的に吸着しやすくなる。また、上記陽イオン交換樹脂のうち、より排除限界分子量が低い上記第2の陽イオン交換樹脂が、主に金属塩等のカチオンを選択的に吸着しやすくなる。
また、上記第1の陽イオン交換樹脂により上記物質を吸着してから、上記陰イオン交換樹脂と第2の陽イオン交換樹脂とにより上記物質を吸着することが好ましい。この場合、例えば、上層に上記第1の陽イオン交換樹脂を有し、下層に上記陰イオン交換樹脂と上記第2の陽イオン交換樹脂とを有するカラムの上記上層側から上記サンプルを流入してもよい。このように、サンプル中の物質について、まず第1の陽イオン交換樹脂によりサンプル中のタンパク質を主に選択的に吸着し、次いで第2の陽イオン交換樹脂及び陰イオン交換樹脂によりサンプル中の金属塩を主に選択的に吸着することで、よりサンプル中の物質の吸着効率は向上する。
また、上記手順は、緩衝液で希釈された前記サンプルに含まれる物質を前記イオン交換樹脂により吸着する手順であってもよく、上記緩衝液のpHは4.0〜8.0であることが好ましい。
また、上記陽イオン交換樹脂は、強酸性陽イオン交換樹脂であることが好ましい。また、上記第1の陽イオン交換樹脂の対イオンは、Na+(ナトリウムイオン)であることが好ましい。また、上記第2の陽イオン交換樹脂の対イオンは、H+(プロトン)であることが好ましい。
また、上記陰イオン交換樹脂は、強塩基性陰イオン交換樹脂であることが好ましい。また、上記陰イオン交換樹脂の対イオンは、OH−(水酸化物イオン)であることが好ましい。
また、上記第2の陽イオン交換樹脂に対する上記陰イオン交換樹脂のイオン交換容量の割合は、50%〜150%であることが好ましい。ここで、イオン交換容量とは、イオン交換樹脂の単位量当たりの交換基の総数のことを指す。例えば、第2の陽イオン交換樹脂の対イオンがH+である場合、イオン交換容量とは、第2の陽イオン交換樹脂1ml中に含まれるH+の総数のことを指す。上記第2の陽イオン交換樹脂に対する上記陰イオン交換樹脂のイオン交換容量の割合が、50%〜150%であることで、イオン交換樹脂によるサンプルに含まれる物質の吸着をする前とその後のpHの変動をより安定に抑制することが可能になる。
また、上記核酸精製方法では、サンプルに非イオン性界面活性剤及び/又は非イオン性親水性高分子化合物を添加して上記物質を吸着することがより好ましい。サンプルに非イオン性界面活性剤及び/又は非イオン性親水性高分子化合物を添加することで、核酸の樹脂に対する物理的な吸着を抑制することが可能になる。
上記物質は、例えば、夾雑物である。当該夾雑物は、例えば、タンパク質と金属塩とを含有する。また、夾雑物は、タンパク質や金属塩の他にサンプル中の核酸の分析において不要な、種々のペプチド、糖、塩、低分子アニオン(例えば、フマル酸、フタル酸、フミン酸、フルボ酸)等の物質を含有していてもよい。
【0009】
また、上記課題解決のため、本技術は、核酸を含有するサンプルの超音波処理をする手順、陽イオン交換樹脂及び陰イオン交換樹脂により前記サンプルに含まれる物質を吸着する手順、及び電気泳動により移動する前記核酸を堰き止めることで前記核酸を濃縮する手順の各手順を行う核酸抽出方法を提供する。
上記電気泳動は、アニオン性官能基を有するインターカレーターを挿入した前記核酸について行われてもよい。また、上記電気泳動は、上記サンプルと、上記サンプル中に含まれる物質が含有するカルボキシル基と脱水縮合反応をする官能基を有する化合物と、上記脱水縮合反応の縮合剤と、を混合して、上記核酸について行われてもよい。
【0010】
また、本技術は、陽イオン交換樹脂と、陰イオン交換樹脂と、前記陽イオン交換樹脂及び前記陰イオン交換樹脂を内部に保持し、核酸を含有するサンプルを流通可能な核酸精製器具と、を含む核酸精製用キットを提供する。
【発明の効果】
【0011】
本技術により、操作が簡便であり、短時間で高効率に核酸を抽出することができる核酸精製方法が提供される。
【図面の簡単な説明】
【0012】
【図1】本技術の第1実施形態に係る核酸精製方法の手順を説明するための模式図である。
【図2】本技術の第1実施形態に係る第1の陽イオン交換樹脂により夾雑物を吸着する状態を概念的に説明した模式図である。
【図3】本技術の第1実施形態に係る第2の陽イオン交換樹脂及び陰イオン交換樹脂により夾雑物を吸着する状態を概念的に説明した模式図である。
【図4】本技術の第2実施形態に係る核酸精製方法の手順を説明するための模式図である。
【図5】陽イオン交換樹脂による吸着処理後の核酸回収率を示すグラフである(試験例1)。
【図6】陽イオン交換樹脂による吸着処理後のサンプルのLAMP反応の結果を示すグラフである(試験例1)。
【図7】陽イオン交換樹脂の対イオン種(Na+、H+)の核酸回収率に対する影響を検討した結果を示すグラフである(試験例2)。
【図8】イオン交換樹脂による吸着処理後のサンプルのLAMP反応の結果を示すグラフである(実施例4、5)
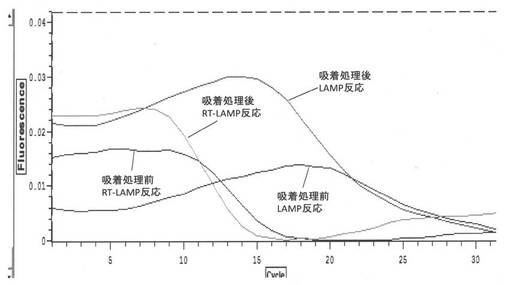
【図9】イオン交換樹脂による吸着処理後のサンプルのLAMP反応及びRT−LAMP反応の結果(Tt値)を示すグラフである(実施例4、5)
【図10】イオン交換樹脂による吸着処理後のLAMP反応及びRT−LAMP反応の結果(Tt値)を示すグラフである(実施例6)
【図11】イオン交換樹脂による吸着処理後のLAMP反応及びRT−LAMP反応の結果(Tt値)を示すグラフである(実施例7)
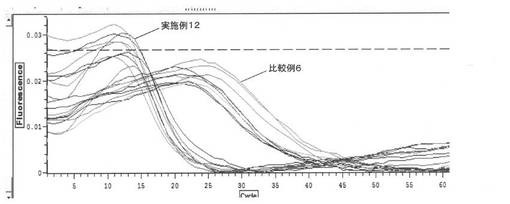
【図12】サンプルのLAMP反応の結果を示すグラフである(実施例12)。
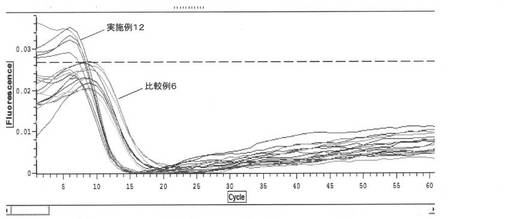
【図13】サンプルのRT−LAMP反応の結果を示すグラフである(実施例12)。
【発明を実施するための形態】
【0013】
以下、本技術を実施するための好適な形態について説明する。なお、以下に説明する実施形態は、本技術の代表的な実施形態の一例を示したものであり、これにより本技術の範囲が狭く解釈されることはない。説明は以下の順序で行う。
1.本技術の第1実施形態に係る核酸精製用キット及び核酸精製方法
(1)核酸精製用キット
(2)核酸精製方法
2.本技術の第2実施形態に係る核酸精製用キット及び核酸精製方法
3.核酸抽出方法
【0014】
1.本技術の第1実施形態に係る核酸精製用キット及び核酸精製方法
(1)核酸精製用キット
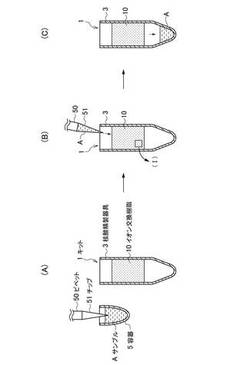
まず、本技術の第1実施形態に係る核酸精製方法に用いる核酸精製用キットについて、図1(A)を参照しながら説明する。図1(A)〜(C)は、本技術の第1実施形態に係る核酸精製方法の手順を説明するための模式図である。
【0015】
図1(A)中、符号1で示す核酸精製用キットは、サンプルに含まれる核酸以外の物質(以下、夾雑物と記す)を吸着し、核酸を精製する。核酸精製用キット1は、イオン交換樹脂10と、イオン交換樹脂10を内部に保持し、核酸を含有するサンプルを流通可能な核酸精製器具3とを主に有する。なお、本技術で用いるサンプルとしては、特に限定されるものではないが、例えば、ぬぐい液、口腔スワブ、唾液、血清、血漿、末梢血単球細胞、脳脊髄液、糞、尿、培養細胞、生検組織等の生体試料が挙げられる。また、サンプルとしては、生体試料の他に、河川水、海水、土壌等も挙げられる。
【0016】
核酸精製器具3は、イオン交換樹脂10を内部に保持し、サンプルを流通させる。核酸精製器具3については、イオン交換樹脂10を内部に保持し、サンプルを流通可能であれば、形状や材質等は特に限定されるものではない。例えば、核酸精製器具3としては、図1(A)に示すように、上部が開口した形状を有する市販のサンプル管やチップ等を用いることが可能である。また、核酸精製器具3は、例えば、上部及び下部が開口した筒状に形成されていてもよい。この場合には、サンプルを核酸精製器具3に流通し、イオン交換樹脂10によりサンプルを吸着した後に、下部から流れ出るサンプルを回収するために、別途チップ等を用いることが可能である。
【0017】
本技術に係るイオン交換樹脂10は、サンプルに含まれる夾雑物を吸着する。イオン交換樹脂10は、陽イオン交換樹脂と、陰イオン交換樹脂とを含む。なお、上記夾雑物は、例えば、タンパク質と金属塩とを含有する。また、夾雑物は、タンパク質や金属塩の他にサンプル中の核酸の分析において不要な、種々のペプチド、糖、塩、低分子アニオン(例えば、フマル酸、フタル酸、フミン酸、フルボ酸)等の物質を含有していてもよい。
【0018】
上記陽イオン交換樹脂は、カチオン性の夾雑物を主に吸着する。陽イオン交換樹脂は、第1の陽イオン交換樹脂と、当該第1の陽イオン交換樹脂よりも排除限界分子量が低い第2の陽イオン交換樹脂とを含むことが好ましい。第1の陽イオン交換樹脂は、サンプルに含まれるタンパク質を主に吸着するために用いられる。第2の陽イオン交換樹脂は、サンプルに含まれる金属イオン等の陽イオン(カチオン)を主に吸着するために用いられる。
【0019】
第1の陽イオン交換樹脂としては、サンプルに含まれるタンパク質を吸着することが可能であれば、特に限定されるものではないが、強酸性陽イオン交換樹脂であることが好ましい。このとき、第1の陽イオン交換樹脂は、SO3−を固定イオンとすることが好ましい。また、対イオンとしては、特に限定されるものではなく、カルシウムイオン(Ca2+)、銅イオン(Cu2+)、亜鉛イオン(Zn2+)、マグネシウムイオン(Mg2+)、カリウムイオン(K+)、アンモニウムイオン(NH4+)、ナトリウムイオン(Na+)、プロトン(H+)等が挙げられる。この点、これらのイオンを用いた場合のイオンを交換する強さ(イオンの選択性)の順序は、Ca2+>Cu2+>Zn2+>Mg2+>K+>NH4+>Na+>H+である。従って、イオン交換する強さを考慮すると、対イオンは、H+であることが好ましい。一方で、対イオンがH+である場合、サンプルのpHが酸性側にシフトしやすい。これに対し、対イオンがNa+である場合、対イオンがH+である場合よりも、金属塩を吸着する機能は弱くなるものの、サンプルのpHの変動を抑制することが可能である。また、対イオンがNa+である場合であっても、第1の陽イオン交換樹脂はタンパク質を十分に吸着することができる。そのため、第1の陽イオン交換樹脂は、Na+を対イオンとして有する強酸性陽イオン交換樹脂(Na+型強酸性陽イオン交換樹脂)であることが好ましい。
【0020】
第2の陽イオン交換樹脂としては、サンプルに含まれる金属塩等のカチオンを吸着することが可能であれば、特に限定されるものではないが、強酸性陽イオン交換樹脂であることが好ましい。このとき、第2の陽イオン交換樹脂は、SO3−を固定イオンとすることが好ましい。また、対イオンとしては、特に限定されるものではないが、上述したイオンを交換する強さ(イオンの選択性)の順序を考慮すると、第2の陽イオン交換樹脂の対イオンはプロトン(H+)であることがより好ましい。
【0021】
陰イオン交換樹脂は、サンプルに含まれる陰イオン(アニオン)を吸着する。陰イオン交換樹脂としては、サンプルに含まれる金属塩等のアニオンを吸着することが可能であれば、特に限定されるものではないが、強塩基性陰イオン交換樹脂であることが好ましい。また、陰イオン交換樹脂は、例えば、トリメチルアンモニウム基を持つI型及びジメチルエタノールアンモニウム基を持つII型の強塩基性陰イオン交換樹脂のうちのいずれとすることも可能である。ここで、I型においては、陰イオンを交換吸着する強さ(イオンの選択性)の順序は、HSO4−>NO3−>Br−>Cl−>HCO3−>HCOO−>CH3COO−>F−>OH−となる。また、II型においては、陰イオンを交換吸着する強さ(イオンの選択性)の順序は、HSO4−>NO3−>Br−>Cl−>HCO3−>OH−>HCOO−>CH3COO−>F−となる。そして、上記I型及びII型の陰イオン交換樹脂のうち、I型の方が塩化物イオン(Cl−)等をより精度良く吸着することが可能である。そのため、陰イオン交換樹脂は、I型であり、対イオンとして水酸化物イオン(OH−)を有することが好ましい。
【0022】
また、核酸精製用キット1には、夾雑物の吸着と共に核酸を吸着しないように、非イオン性界面活性剤や非イオン性親水性高分子化合物等の非イオン性化合物等の添加剤が含まれていてもよい。非イオン性化合物としては、例えば、Brij35、Tween20、又はTritonX100等の非イオン性界面活性剤が挙げられる。また、ポリエチレングリコールやポリヒドロキシエチルセルロース等の非イオン性親水性高分子化合物も挙げられる。これらの例示した非イオン性化合物は、単独で用いてもよいし、複数種組み合わせて用いてもよい。更に、核酸精製用キット1には、EDTA等のキレート添加剤が含まれていてもよい。
【0023】
また、核酸精製用キット1には、緩衝液が含まれていてもよい。かかる緩衝液用の緩衝剤としては、例えば、HomoPIPES(pH:3.9〜5.1、pKa:4.55)、MES(pH:5.5〜7.0、pKa:6.15)、Bis−Tris(pH:5.7〜7.3、pKa:6.46)、ADA(pH:5.8〜7.4、pKa:6.60)、PIPES(pH:6.1〜7.5、pKa:6.80)、ACES(pH:6.0〜7.5、pKa:6.90)、MOPSO(pH:6.2〜7.4、pKa:6.95)、BES(pH:6.6〜8.0、pKa:7.15)、MOPS(pH:6.5〜7.9,pKa:7.20)、TES(pH:6.8〜8.2、pKa:7.50)、HEPES(pH:6.8〜8.2、pKa:7.55)、DIPSO(pH:6.9〜8.1、pKa:7.6)、TAPSO(pH:7.0〜8.2、pKa:7.7)、POPSO(pH:7.2〜8.5、pKa:7.85)、HEPPSO(pH:7.4〜8.6、pKa:7.9)、EPPS(pH:7.5〜8.5、pKa:8.0)、Tricine(pH:7.8〜8.8、pKa:8.15)、Bicine(pH:7.7〜9.1、pKa:8.35)、TAPS(pH:7.7〜9.1、pKa:8.4)、CHES(pH:8.6〜10.0、pKa:9.5)、CAPSO(pH:9.3〜10.7、pKa:10.0)、CAPS(pH:9.7〜11.1、pKa:10.40)等が挙げられる。なお、上記例示した各緩衝剤のpHの範囲は、緩衝液を添加したサンプルのpHとして適当な範囲を指す。また、上記例示した各緩衝剤のpKaは、HomoPIPESを除いて20℃におけるpKaを指す(HomoPIPESについては、37℃におけるpKaを示す。)。サンプルを希釈した緩衝液のpHは、4〜8であることが好ましい。この点、本実施形態において上記例示した緩衝剤の中では、HomoPIPES、MES、Bis−Tris、ADAが好適に用いられる。
【0024】
(2)核酸精製方法
次に、本技術の第1実施形態に係る核酸精製方法の手順について、図1(A)〜(C)を参照しながら説明する。
【0025】
ここで、本技術の第1実施形態に係る核酸精製方法を説明する前に、まず、本技術の関連技術に係る核酸精製方法について説明する。関連技術に係る核酸精製方法としては、ゼオライトを用いてサンプルの夾雑物を吸着する方法を挙げることができる(例えば、特許文献2参照)。
【0026】
関連技術に係る核酸精製方法では、生体試料等のサンプルに含まれるアニオン及びカチオンのうち、カチオンのみの除去が可能である。そのため、関連技術に係る核酸精製方法では、サンプルの精製により、プロトンが放出されることになり、サンプルを含有する液のpHが低下する(より酸性側にシフトする)。この点、サンプルの精製後、核酸増幅反応はpH7〜9で実行される場合があるため、ゼオライトと接触させるサンプルを含有する液のpHを予め高めに設定する(アルカリ側に設定する)必要がある。そのため、上記関連技術に係る核酸精製方法では、操作が煩雑になる場合がある。また、サンプルにRNAが含まれていると、当該RNAの分解をもたらす可能性もある。
【0027】
また、上記関連技術に係る核酸精製方法は、サンプルをアルカリ性の化合物と界面活性剤(例えば、SDS等)との混合試薬で希釈した後に、高温で加熱する工程を含む。そのため、この点からも、上記関連技術に係る核酸精製方法では、操作が煩雑になる場合がある。また、サンプルにRNAが含まれていると、当該RNAの分解をもたらす可能性もある。
【0028】
また、上記関連技術に係る核酸精製方法では、サンプルの精製後、サンプル中にアニオンが残留することがある。更に、サンプル中には、アニオン性界面活性剤が含まれていることもある。そのため、サンプルの精製後、核酸を電気泳動させる際に、サンプル中の電解質濃度が高くなり、サンプルが発熱しやすくなる。従って、この点からも、サンプルにRNAが含まれていると、当該RNAの分解をもたらす可能性がある。更に、発熱によるサンプルの対流の発生により、核酸の電気泳動濃縮の効率が低下する場合もある。
【0029】
この点、以下に詳述する本技術に係る核酸精製方法は、本願発明者らの鋭意検討の結果見出されたものであり、煩雑な操作を必要とせず、操作を非常に簡便にし、短時間に高効率で核酸を精製可能にした方法である。
【0030】
本技術の第1実施形態に係る核酸精製方法では、まず、容器5に収容された核酸を含有するサンプルA及びサンプルAを希釈した緩衝液が、ピペット50に取り付けたピペットチップ51に充填される(図1(A)参照)。次に、ピペットチップ51に充填されたサンプルA及びサンプルAを希釈した緩衝液は、核酸精製器具3に注入される(図1(B)参照)。このとき、核酸精製器具3は、イオン交換樹脂10を内部に保持している。そのため、サンプルAに含まれる夾雑物は、イオン交換樹脂10により吸着される。
【0031】
最後に、夾雑物がイオン交換樹脂10により吸着されることで精製されたサンプルAは、イオン交換樹脂10を通過して核酸精製器具3の下部に貯留される(図1(C)参照)。
【0032】
図1(A)〜(C)に示した工程後、サンプルAについては、核酸増幅反応をすることが可能である。例えば、サンプルAを固相化した核酸増幅試薬に添加して核酸増幅反応をすることが可能である。また、本実施形態では、サンプルをイオン交換樹脂10により脱塩することにより、二重らせんDNAを一本鎖DNAにし、核酸増幅反応前にDNAの変性を適宜行うことも可能である。
【0033】
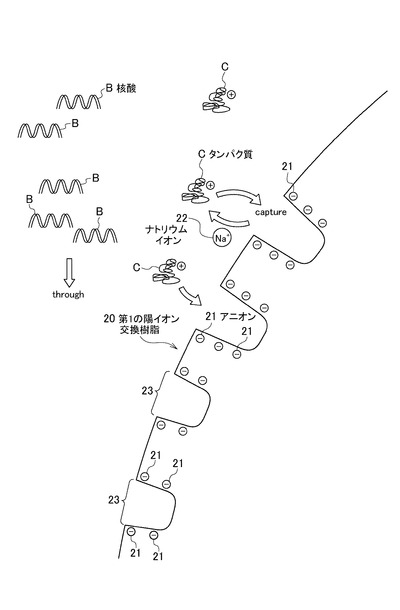
ここで、図2及び図3を参照しながら、サンプルAに含まれる夾雑物がイオン交換樹脂10により吸着される状態(図1(B)中、(I)で示す部分)について、より詳細に説明する。図2は、図1(B)において、特に第1の陽イオン交換樹脂が夾雑物を吸着する状態を概念的に説明した模式図である。図3は、図1(B)において、特に第2の陽イオン交換樹脂及び陰イオン交換樹脂が夾雑物を吸着する状態を概念的に説明した模式図である。
【0034】
まず、図2を参照しながら、イオン交換樹脂10に含まれる第1の陽イオン交換樹脂20が、サンプルAに含まれる夾雑物を吸着する状態を説明する。なお、図2において、符号Bで示されるものは、サンプルAに含まれる核酸であり、符号Cで示されるものは、サンプルAに含まれる夾雑物の一例であるタンパク質である。また、ここでは、第1の陽イオン交換樹脂20については、対イオンとしてナトリウムイオン(Na+)を有する強酸性陽イオン交換樹脂とする。
【0035】
第1の陽イオン交換樹脂20は、SO3−等のアニオン21と、対イオンであるNa+22とを有する。また、第1の陽イオン交換樹脂20には、タンパク質Cを吸着する領域23が形成される。
【0036】
図2に示すように、核酸精製器具3内に通流されたサンプルAに含まれるタンパク質Cは、正に帯電しているため、表面にアニオン21を有する第1の陽イオン交換樹脂20に吸着される。例えば、タンパク質Cは、領域23に吸着される。一方、核酸Bは、負に帯電しているため、第1の陽イオン交換樹脂20に吸着されることなく、サンプルAに含まれる緩衝液等の流れに誘導されて流されていく。また、第1の陽イオン交換樹脂20の対イオンはNa+22であるため、対イオンがH+である場合に比べ、金属塩については吸着しづらく(脱塩しづらく)、タンパク質Cを選択的に吸着することが可能である。
【0037】
この際、サンプルAについては、サンプルAを希釈する緩衝液のpHを4〜8にしておくことが好ましい。サンプルAを希釈する緩衝液のpHを4〜8にしておく(酸性にしておく)ことで、より安定に、核酸Bを負に帯電させつつ、夾雑物(主にタンパク質C)のみを正に帯電させておくことが可能になる。そのため、タンパク質Cのみを第1の陽イオン交換樹脂20に安定に吸着させ、核酸Bをより高効率に精製することができる。また、第1の陽イオン交換樹脂20が強酸性陽イオン交換樹脂であることで、第1の陽イオン交換樹脂20は広範囲のpH条件下(例えば、pH:3〜13)で負電荷を有していることが可能である。そのため、上述したように、サンプルを酸性にしておいても、タンパク質Cを第1の陽イオン交換樹脂20に安定に吸着させることが可能になる。更に、サンプルAのpHを4〜8にしておく(酸性にしておく)ことで、RNA分解酵素の活性を抑えることができるため、RNAを好適に精製可能である。
【0038】
また、第1の陽イオン交換樹脂20の排除限界分子量は、サンプルAに含まれるタンパク質Cが領域23に取り込まれる程度に規定されていることが好ましい。より具体的には、第1の陽イオン交換樹脂20の排除限界分子量は、5000以上であることが好ましい。第1の陽イオン交換樹脂20の排除限界分子量が5000以上であることで、夾雑物中、タンパク質Cを特に選択的に吸着し、除去しやすくなる。
【0039】
また、第1の陽イオン交換樹脂20の平均体積粒子径は、1μm以上3000μm以下であることが好ましい。第1の陽イオン交換樹脂20の平均体積粒子径が1μm以上3000μm以下であることでも、夾雑物中、タンパク質Cを特に選択的に吸着し、除去しやすくなる。
【0040】
また、第1の陽イオン交換樹脂20の粒子自体の比重は、0.5以上2.5以下であることが好ましい。第1の陽イオン交換樹脂20の比重が0.5以上2.5以下であることでも、夾雑物中、タンパク質Cを特に選択的に吸着し、除去しやすくなる。さらに、粒子自体の比重は1.0以上2.5以下であることがより好ましい。上記比重が1.0以上2.5以下であることで、粒子が溶液中で沈降しやすくなることにより、容易に溶液から粒子を除去することが可能になる。
【0041】
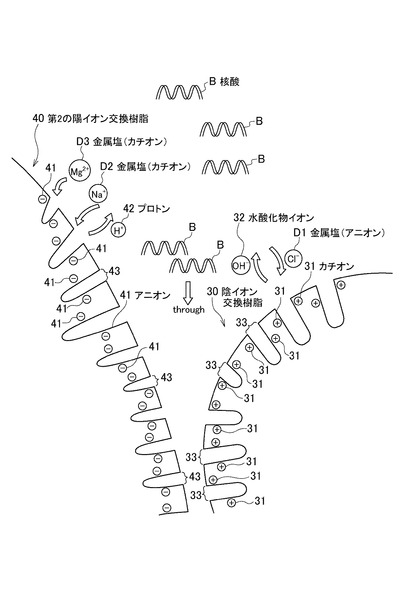
次に、図3を参照しながら、イオン交換樹脂10に含まれる陰イオン交換樹脂30及び第2の陽イオン交換樹脂40によりサンプルAに含まれる夾雑物を吸着する状態を説明する。
【0042】
図3において、符号D1、D2、及びD3で示されるものは、サンプルAに含まれる夾雑物の一例である金属塩が有するカチオン又はアニオンである。本実施形態では、特に限定されるものではないが、ここでは、D1を塩化物イオン等のアニオンとし、D2をナトリウムイオン等のカチオンとし、D3をマグネシウムイオン等のカチオンとする。
【0043】
まず、図3を参照しながら、陰イオン交換樹脂30が夾雑物(主に塩化物イオンD1等のアニオン)を吸着する状態について説明する。本実施形態において、陰イオン交換樹脂30については、対イオンとして水酸化物イオン(OH−)を有するI型の強塩基性陰イオン交換樹脂とする。
【0044】
陰イオン交換樹脂30は、CH2N(CH3)3+等のカチオン31と、対イオンであるOH−32とを有する。また、陰イオン交換樹脂30には、塩化物イオンD1等のアニオンを吸着する領域33が形成される。
【0045】
図3に示すように、核酸精製器具3に通流されたサンプルAに含まれる塩化物イオンD1は、負に帯電しているため、表面にカチオン31を有する陰イオン交換樹脂30に吸着される。例えば、塩化物イオンD1は、領域33に吸着される。一方、核酸Bも負に帯電しているものの、核酸Bは塩化物イオンD1等のアニオンに比べ体積が大きいため、陰イオン交換樹脂30には吸着されにくい。この点、陰イオン交換樹脂30の排除限界分子量は、100以上2000以下であることが好ましい。陰イオン交換樹脂30の排除限界分子量が100以上2000以下であることで、より精度良く、核酸Bの吸着を抑制し、塩化物イオンD1等のカチオンを選択的に吸着し、除去しやすくなる。
【0046】
また、陰イオン交換樹脂30の平均体積粒子径は、1μm以上3000μm以下であることが好ましい。第1の陽イオン交換樹脂20の平均体積粒子径が1μm以上3000μm以下であることでも、より精度良く、塩化物イオンD1等のカチオンを選択的に吸着し、除去しやすくなる。
【0047】
また、陰イオン交換樹脂30の比重は、0.5以上2.5以下であることが好ましい。陰イオン交換樹脂30の比重が0.5以上2.5以下であることでも、より精度良く、塩化物イオンD1等のカチオンを選択的に吸着し、除去しやすくなる。さらに、粒子自体の比重は1.0以上2.5以下であることがより好ましい。上記比重が1.0以上2.5以下であることで、粒子が溶液中で沈降しやすくなることにより、容易に溶液から粒子を除去することが可能になる。
【0048】
次に、図3を参照しながら、第2の陽イオン交換樹脂40が夾雑物(主にナトリウムイオンD2及びマグネシウムイオンD3等のカチオン)を吸着する状態について説明する。本実施形態において、第2の陽イオン交換樹脂40については、対イオンとしてH+イオン(プロトン)を有する強酸性陽イオン交換樹脂とする。
【0049】
第2の陽イオン交換樹脂40は、SO3−等のアニオン41と、H+42とを有する。また、第2の陽イオン交換樹脂40には、ナトリウムイオンD2やマグネシウムイオンD3等のカチオンを吸着する領域43が形成される。
【0050】
図3に示すように、核酸精製器具3に通流されたサンプルAに含まれるナトリウムイオンD2やマグネシウムイオンD3等のカチオンは、正に帯電しているため、表面にアニオン41を有する第2の陽イオン交換樹脂40に吸着される。例えば、ナトリウムイオンD2やマグネシウムイオンD3は、領域43に吸着される。一方、核酸Bは、負に帯電しているため、第2の陽イオン交換樹脂40に吸着されることなく、サンプルAに含まれる緩衝液等の流れに誘導されて流されていく。
【0051】
ここで、第2の陽イオン交換樹脂40は、第1の陽イオン交換樹脂20よりも排除限界分子量が低い。そのため、単位体積当たりの表面積が大きくなり、第1の陽イオン交換樹脂20よりも脱塩しやすくなる。また、第2の陽イオン交換樹脂40が対イオンとしてプロトンを有する場合、第1の陽イオン交換樹脂20よりも脱塩しやすくなる。好ましくは、第2の陽イオン交換樹脂40の排除限界分子量は、100以上2000以下である。第2の陽イオン交換樹脂40の排除限界分子量が100以上2000以下であることでナトリウムイオンD2やマグネシウムイオンD3等のカチオンの吸着をより精度良く行うことが可能になる。
【0052】
また、第2の陽イオン交換樹脂40の平均体積粒子径は、1μm以上3000μm以下であることが好ましい。第2の陽イオン交換樹脂40の平均体積粒子径が1μm以上3000μm以下であることでも、より精度良く、ナトリウムイオンD2やマグネシウムイオンD3等のカチオンの吸着を行うことが可能になる。更に、第2の陽イオン交換樹脂40の平均体積粒子径は、1μm以上2000μm以下であることが好ましい。第2の陽イオン交換樹脂40の平均体積粒子径が1μm以上2000μm以下であることで、溶液をイオン交換樹脂に通液させるときの圧力を低く抑えることが可能になる。
【0053】
また、第2の陽イオン交換樹脂40の比重は、0.5以上2.5以下であることが好ましい。第2の陽イオン交換樹脂40の比重は、0.5以上2.5以下であることでも、より精度良く、ナトリウムイオンD2やマグネシウムイオンD3等のカチオンの吸着を行うことが可能になる。さらに、粒子自体の比重は1.0以上2.5以下であることがより好ましい。上記比重が1.0以上2.5以下であることで、粒子が溶液中で沈降しやすくなることにより、容易に溶液から粒子を除去することが可能になる。
【0054】
また、第2の陽イオン交換樹脂40に対する陰イオン交換樹脂30のイオン交換容量の割合は50%〜150%であることが好ましい。上記イオン交換容量の割合が50%〜150%であることで、サンプルのpHの変動をより安定に抑制しつつ脱塩することが可能になる。
【0055】
また、第2の陽イオン交換樹脂40に対する陰イオン交換樹脂30の用量の割合は50%〜150%であることが好ましい。上記用量の割合が50%〜150%であることでも、サンプルのpHの変動をより安定に抑制しつつ脱塩することが可能になる。
【0056】
また、図2及び図3には図示していないが、核酸Bがイオン交換樹脂により吸着されることを抑制するために、Brij35、Tween20、又はTritonX100等の非イオン性界面活性剤がサンプルA中に適宜添加されることが好ましい。また、ポリエチレングリコールやポリヒドロキシエチルセルロース等の非イオン性親水性高分子化合物等の非イオン性化合物もサンプルA中に適宜添加されることが好ましい。
【0057】
以上、説明してきた本技術の第1実施形態に係る核酸精製方法によれば、陽イオン交換樹脂及び陰イオン交換樹脂からなるイオン交換樹脂10を吸着担体として用いることで、サンプルに含まれる夾雑物を吸着することができる。例えば、サンプルとして血液試料等を用いる場合には、複雑な前処理工程を介さず、直接吸着工程を行うことができる。このように、本実施形態に係る核酸精製方法によれば、非常に簡便な操作で、短時間に高効率で核酸を抽出することができる。また、このような核酸精製方法については、数秒間の時間で全工程を行うことが可能であり、短時間で核酸を精製することができる。
【0058】
また、本実施形態に係る核酸精製方法によれば、例えば、シリカ固相抽出のように洗浄工程を含まないものであるため、微量のサンプル量で操作を行うことができ、装置の小型化も実現できる。また、本実施形態に係る核酸精製方法によれば、サンプルを酸性条件下(好ましくは、pH:4〜8)で夾雑物の吸着処理を行うことが可能である。すなわち、本実施形態に係る核酸精製方法によれば、サンプルをアルカリ性にしたり、加熱したりする工程を要しない。そのため、サンプルにRNAが含まれる場合にはRNAの分解を抑制しつつ、サンプルの精製を行うこともできる。また、本実施形態に係る核酸精製方法によれば、酸性領域で超音波破砕を行うことにより、RNaseの働きを抑制することもできる。
【0059】
また、固相化した核酸増幅試薬を用いて核酸増幅反応をする場合にはサンプルを希釈しないため、夾雑物による核酸増幅反応に対する阻害が顕著に表れることになる。しかしながら、本実施形態に係る核酸精製方法により精製したサンプルでは、サンプルに存在していた夾雑物が除去されているため、核酸を直接固相化した核酸増幅試薬に添加して核酸増幅反応を行うことができる。
【0060】
また、本技術の第1実施形態に係る核酸精製方法によれば、陽イオン交換樹脂として、第1の陽イオン交換樹脂と、該第1の陽イオン交換樹脂よりも排除限界分子量が低い第2の陽イオン交換樹脂とを用いることが可能である。そのため、サンプル中の夾雑物に、金属塩及びタンパク質が含まれている場合にも、双方を効率的に除去することができる。
【0061】
本実施形態に係る核酸精製方法によるサンプル中の夾雑物の吸着・除去については、第1の陽イオン交換樹脂及び第2の陽イオン交換樹脂が強酸性陽イオン交換樹脂である場合、より効率的である。また、第1の陽イオン交換樹脂の対イオンがNa+である場合や第2の陽イオン交換樹脂の対イオンがH+である場合には、より効率的に夾雑物を除去できる。
【0062】
また、本実施形態に係る核酸精製方法によれば、陰イオン交換樹脂が強塩基性陽イオン交換樹脂である場合、より効率的に夾雑物を除去できる。また、陰イオン交換樹脂の対イオンがOH−である場合、より効率的に夾雑物を除去できる。
【0063】
また、本実施形態に係る核酸精製方法によれば、第2の陽イオン交換樹脂に対する陰イオン交換樹脂のイオン交換容量の割合を、50%〜150%とすることで、サンプルのpHの変動を抑制することができる。そのため、上述したように、例えば、サンプルにRNAが含まれる場合にはRNAの分解を抑制しつつ、サンプルの精製を行うこともできる。
【0064】
また、本実施形態に係る核酸精製方法によれば、非イオン性界面活性剤や非イオン性親水性高分子化合物等のノニオン性化合物を用いることが可能である。この場合、核酸がイオン交換樹脂のマトリクスに吸着することを抑制し、効率的に夾雑物を除去することができる。
【0065】
2.本技術の第2実施形態に係る核酸精製用キット及び核酸精製方法
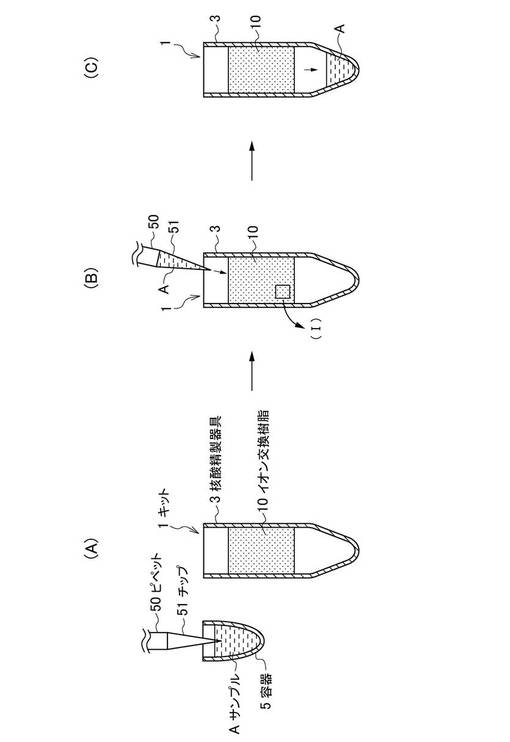
図4(A)〜(C)は、本技術の第2実施形態に係る核酸精製方法の手順を説明するための模式図である。
【0066】
図4(A)中、符号101で示す核酸精製用キットは、イオン交換樹脂10と、イオン交換樹脂10に内部に保持し、核酸を含有するサンプルを流通可能な核酸精製器具3とを含む。イオン交換樹脂10については、陽イオン交換樹脂と、陰イオン交換樹脂とを用いる。そして、本実施形態に係る核酸精製用キット101では、陽イオン交換樹脂に、第1の陽イオン交換樹脂20と、第2の陽イオン交換樹脂40とを用いる。
【0067】
本実施形態に係る核酸精製用キット及び核酸精製方法は、本技術の第1実施形態に係る核酸精製用キット及び核酸精製方法と比し、第1の陽イオン交換樹脂20が陰イオン交換樹脂30及び第2の陽イオン交換樹脂40に対し、上層(サンプルが注入される側)に収容されている状態で核酸の精製が行われるという点が主に異なる。そのため、本実施形態では、第1の陽イオン交換樹脂20が陰イオン交換樹脂30及び第2の陽イオン交換樹脂40に対し、上層に設けられているという点について主に説明する。
【0068】
本実施形態に係る核酸精製方法では、核酸精製器具3に第1の陽イオン交換樹脂20が陰イオン交換樹脂30及び第2の陽イオン交換樹脂40よりも上層側に収容されている。そのため、上層側から注入されるサンプルAは、第1の陽イオン交換樹脂20にまず接触する(図4(B)参照)。
【0069】
次いで、第1の陽イオン交換樹脂20により夾雑物の一部が除去されたサンプルAは、陰イオン交換樹脂30及び第2の陽イオン交換樹脂40に接触する。なお、サンプルAの夾雑物に含まれるタンパク質が第1の陽イオン交換樹脂20により吸着される状態(図4(B)中、(II)で示す部分)については、図2を参照しながら説明した第1の陽イオン交換樹脂20がタンパク質を吸着する状態と同様にして説明できる。そのため、ここではその詳細な説明を省略する。また、サンプルA中の夾雑物に含まれる金属塩が陰イオン交換樹脂30及び第2の陽イオン交換樹脂40により吸着される状態(図4(B)中、(III)で示す部分)についても、図3を参照しながら説明した陰イオン交換樹脂30及び第2の陽イオン交換樹脂40が金属塩を吸着する状態と同様にして説明できる。そのため、ここではその詳細な説明も省略する。なお、陰イオン交換樹脂30及び第2の陽イオン交換樹脂40については、陰イオン交換樹脂30及び第2の陽イオン交換樹脂40の何れが上層に収容されていてもよいし、夫々が混合されて第1の陽イオン交換樹脂20の下層に収容されていてもよい。
【0070】
最後に、イオン交換樹脂10により夾雑物が吸着された後に、精製されたサンプルAは、下層に貯留される(図4(C)参照)。
【0071】
本技術の第2実施形態に係る核酸精製方法によれば、第1の陽イオン交換樹脂20によりサンプルA中の夾雑物を吸着してから、陰イオン交換樹脂30及び第2の陽イオン交換樹脂40により夾雑物を吸着する。そのため、本技術の第2実施形態に係る核酸精製方法によれば、より体積が大きなタンパク質を除去してから、より体積が小さな金属塩を除去することが可能になり、より効率的に夾雑物の除去を行うことができる。
【0072】
3.核酸抽出方法
次に、本技術の各実施形態に係る核酸精製方法を含む核酸抽出方法について説明する。本技術に係る核酸抽出方法は、核酸増幅反応を行うための前処理として、核酸を含有するサンプルの超音波処理をする手順、陽イオン交換樹脂及び陰イオン交換樹脂により前記サンプルに含まれる物質を吸着する手順、及び電気泳動により移動する前記核酸を堰き止めることで前記核酸を濃縮する手順の各手順を行うものである。
【0073】
上記各手順については、特に限定されるものではないが、例えば、同一のセル内で全ての手順を行うことが可能である。同一のセル内で各手順を行う場合、超音波処理を行う超音波発生部をセルに設けることが可能である。また、上記セルに、陽イオン交換樹脂及び陰イオン交換樹脂を収容しておくことが可能である。陽イオン交換樹脂及び陰イオン交換樹脂については、上述した本技術の各実施形態に係る核酸精製方法で用いたイオン交換樹脂10(第1の陽イオン交換樹脂20、陰イオン交換樹脂30、第2の陽イオン交換樹脂40)を用いることが可能である。また、上記セルに、電気泳動用の負極及び正極を設け、電気泳動をした核酸を堰き止める堰止部(例えば、透析膜、高分子ゲル等)を設けることが可能である。
【0074】
また、核酸の電気泳動の際に、核酸にアニオン性官能基を有するインターカレーターを挿入しておくことも可能である。ここで、上記インターカレーターとしては、例えば、スルホ基を官能基として有する化合物が挙げられる。具体的には、上記インターカレーターとしては、例えば、9,10−アントラキノン−2,6−ジスルホン酸、アントラキノン−1−スルホン酸ナトリウム、アントラキノン−2,7−ジスルホン酸二ナトリウム、アントラキノン−1,5−ジスルホン酸二ナトリウム、アントラキノン−2−スルホン酸ナトリウム等が挙げられる。このように、核酸にアニオン性官能基を有するインターカレーターを挿入しておくことで、核酸の電気泳動において、核酸の等電点を調節することができる。すなわち、核酸の電気泳動速度を制御しつつ、核酸を濃縮精製することができる。
【0075】
更に、サンプルに含まれる夾雑物の1つであるタンパク質等のカルボキシル基をN−ヒドロキシスクシンイミド(NHS)、エタノールアミン、エチレンジアミン等の化合物と脱水縮合反応させておくことも可能である。脱水縮合反応においては、縮合剤としてカルボジイミド系化合物を用いることが可能である。カルボジイミド系化合物の具体例としては、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(EDC)、4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルフォリニウムクロリド(DMT−MM)、ジシクロヘキシルカルボジイミド(DCC)、又はジイソピルカルボジイミド(DIC)等が挙げられる。これにより、核酸の電気泳動の際に、サンプル中にタンパク質が存在する場合であっても、タンパク質中のカルボキシル基と脱水縮合反応をする核酸とタンパク質との等電点の差を大きくすることで、核酸とタンパク質とを精度良く分離することができる。
【0076】
このように、上記各手順を行うことで、非常に簡便に、高効率に濃縮した核酸を回収することができる。特に、各手順を同一のセル内で行う場合には、他の装置等にサンプルを移す必要がないため、サンプルとして感染性を有する検体等を扱う場合には、操作者に対する感染のリスクを低減することができる。
【0077】
なお、本技術は以下のような構成も取ることができる。
(1)核酸を含有するサンプルに含まれる物質をイオン交換樹脂により吸着する手順を含み、前記イオン交換樹脂には、陽イオン交換樹脂及び陰イオン交換樹脂を用いる核酸精製方法。
(2)前記陽イオン交換樹脂には、第1の陽イオン交換樹脂と、当該第1の陽イオン交換樹脂よりも排除限界分子量が小さな第2の陽イオン交換樹脂とを用いる、前記(1)記載の核酸精製方法。
(3)前記第1の陽イオン交換樹脂により前記物質を吸着してから、前記陰イオン交換樹脂と第2の陽イオン交換樹脂とにより前記物質を吸着する、前記(2)記載の核酸精製方法。
(4)上層に前記第1の陽イオン交換樹脂を有し、下層に前記陰イオン交換樹脂と前記第2の陽イオン交換樹脂とを有するカラムの前記上層側から前記サンプルを流入する、前記(2)又は(3)に記載の核酸精製方法。
(5)前記手順は、緩衝液で希釈された前記サンプルに含まれる物質を前記イオン交換樹脂により吸着する手順であり、前記緩衝液のpHは4.0〜8.0である、前記(1)〜(4)のいずれか1つに記載の核酸精製方法。
(6)前記陽イオン交換樹脂は、強酸性陽イオン交換樹脂である、前記(1)〜(5)のいずれか1つに記載の核酸精製方法。
(7)前記第1の陽イオン交換樹脂の対イオンは、Na+である、前記(2)〜(6)のいずれか1つに記載の核酸抽出方法。
(8)前記第2の陽イオン交換樹脂の対イオンは、H+である、前記(2)〜(7)のいずれか1つに記載の核酸抽出方法。
(9)前記陰イオン交換樹脂は、強塩基性陰イオン交換樹脂である、前記(1)〜(8)のいずれか1つに記載の核酸精製方法。
(10)前記陰イオン交換樹脂の対イオンは、OH−である、前記(1)〜(9)のいずれか1つに記載の核酸精製方法。
(11)前記第2の陽イオン交換樹脂に対する前記陰イオン交換樹脂のイオン交換容量の割合は、50%〜150%である、前記(2)〜(10)のいずれか1つに記載の核酸精製方法。
(12)非イオン性界面活性剤及び/又は非イオン性親水性高分子化合物を用いて前記物質を吸着する、前記(1)〜(11)のいずれか1つに記載の核酸精製方法。
(13)前記物質は、少なくともタンパク質と金属塩とを含有する夾雑物である、前記(1)〜(12)のいずれか1つに記載の核酸精製方法。
(14)核酸を含有するサンプルの超音波処理をする手順、陽イオン交換樹脂及び陰イオン交換樹脂により前記サンプルに含まれる物質を吸着する手順、及び電気泳動により移動する前記核酸を堰き止めることで前記核酸を濃縮する手順の各手順を行う核酸抽出方法。
(15)アニオン性官能基を有するインターカレーターを挿入した前記核酸について前記電気泳動を行う、前記(14)記載の核酸抽出方法。
(16)前記サンプルと、前記サンプル中に含まれる物質が含有するカルボキシル基と脱水縮合反応をする官能基を有する化合物と、該脱水縮合反応の縮合剤と、を混合し、前記核酸について前記電気泳動を行う、前記(14)又は(15)に記載の核酸抽出方法。
(17)陽イオン交換樹脂と、陰イオン交換樹脂と、前記陽イオン交換樹脂及び前記陰イオン交換樹脂を内部に保持し、核酸を含有するサンプルを流通可能な核酸精製器具と、を含む核酸精製用キット。
【実施例】
【0078】
1.強酸性陽イオン交換樹脂による吸着処理の核酸精製能に対する影響
<試験例1>
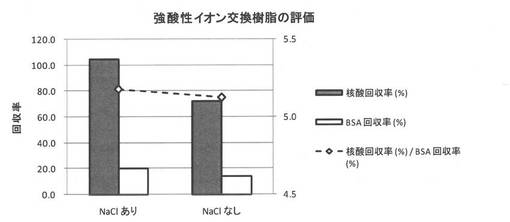
スピンフィルタカラム(Ultrafree-MC,0.45μm, Millipore製)に強酸性陽イオン交換樹脂(Nuvia S Na+型(Bio Rad社製))を100mg計量して入れた。次いで、50mM MESバッファー(pH5)((株)同仁化学研究所製)を調製し、BSA (和光純薬工業(株)製)を0.5質量%となるように加え、さらにCy3修飾した20merオリゴDNA (Sigma Aldrich Co. 製)を5μMとなるように加えた。このタンパク質と核酸の混合液を、常温にて十分に攪拌させた後、強酸性陽イオン交換樹脂封入スピンカラムに200μL滴下し、十分に攪拌させた。その後、12000Gにて2分間遠心し、混合液をスピンダウンした。スピンダウンした液をNanoDrop D-1000 (Thermo Fisher Scientific Inc. 製)にて吸光度測定し、強酸性陽イオン交換樹脂処理前後の吸光度の違いから、核酸精製能を評価した。BSAの吸光度はProtein A280モードで評価し、核酸の吸光度はmicro arrayモードでCy3の吸光度で評価した。さらに、生体環境下に近づけるため、上記タンパク質−核酸混合溶液に最終濃度が0.09質量%となるようにNaCl (和光純薬工業(株)製)を添加し、同様に強酸性イオン交換樹脂処理をおこなった。
【0079】
評価結果を図5に示す。図5に示すように、強酸性陽イオン交換樹脂処理前後のCy3 オリゴ DNAおよびBSA濃度より、核酸の回収率はNaClを含む場合で約100%、NaClを含まない場合で約72%であった。一方、BSAの回収率はNaClを含む場合で約20%、NaClを含まない場合で約14%であった。このことから、強酸性陽イオン交換樹脂処理をすることにより、タンパク質−核酸の混在系においても、DNA存在比を向上させられることを確認できた。
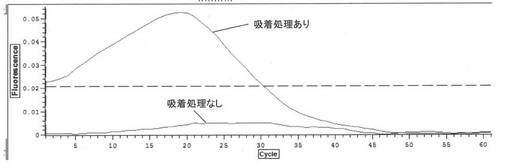
【0080】
また、イオン交換樹脂により夾雑物の吸着処理をしたサンプル、及び当該吸着処理をしていないサンプルの夫々のLAMP反応の結果を図6に示す。LAMP反応については、まず、サンプル、酵素、蛍光色素、核酸モノマー、バッファー、ターゲット核酸鎖増幅用のプライマーセット、リアルタイム測定用のプローブを混合し、LAMP反応液を調製した。各濃度はLoopamp DNA 増幅試薬キット(栄研化学(株)製)およびLoopamp RNA 増幅試薬キット(栄研化学(株)製)に準じて実施した。反応温度は63℃で実施した。LAMP反応は、リアルタイム計測が可能なサーマルサイクラーChromo4(バイオラッド製、米国)を用いて測定を行った。1Cycle を 1min と設定した。プローブは、消光プローブのQPプローブ(J-bio21, 日本: http://www.j-bio21.co.jp/tech/qpmethod.htm([平成23年7月19日]、題目:QP法))を使用しているので、核酸の増幅と共に蛍光強度の低下が観察される。図6に示すように、イオン交換樹脂により夾雑物の吸着処理をしたサンプルでは、核酸増幅反応を検出することができた。なお、以下に記載のLAMP反応は上記と同様の条件で行った。
【0081】
2.陽イオン交換樹脂の対イオンについての検討
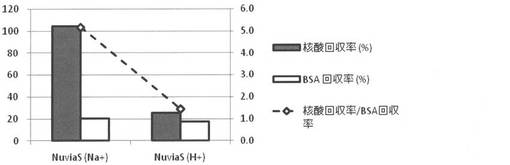
<試験例2>
スピンフィルタカラム(Ultrafree-MC,0.45μm)に陽イオン交換樹脂(Nuvia S Na+型(Bio Rad社製)) を100mg計量して入れた。また、スピンフィルタカラム(Ultrafree-MC,0.45μm)に上記陽イオン交換樹脂(Nuvia S Na+型)を1M HCl溶液に通し、中性になるまで純水で洗浄することで調製した陽イオン交換樹脂(NuviaS H+型)を100mg計量して入れた。次いで、50mM MESバッファー(pH5)を調製し、BSAを0.5質量%となるように加え、Cy3修飾した20merオリゴDNAを4μMとなるように加え、さらにNaClを0.09質量%となるように加えた。このサンプルを、常温にて十分に攪拌させた後、Na+型又はH+型である強酸性陽イオン交換樹脂封入スピンカラムに夫々200μL滴下し、十分に攪拌させた。その後、12000Gにて2分間遠心し、混合液をスピンダウンした。夫々の強酸性陽イオン交換樹脂による核酸精製能については、試験例1と同様にして評価した。Na+の濃度についてはHoriba Cardy Sodium Compact Ion Meter (C-122) ((株)堀場製作所製)により測定した。また、pHについては、ポケットpH計S2K712 (ISFETCOM(株)製)により測定した。評価結果を図7に示す。また、評価結果をまとめたものを表1に示す。
【0082】
【表1】

【0083】
Na+型の陽イオン交換樹脂を用いた場合には、H+型の陽イオン交換樹脂を用いた場合に比べ、核酸の回収率が高く、核酸の精製率も高いことが分かった。一方で、Na+型の陽イオン交換樹脂を用いた場合には、H+型の陽イオン交換樹脂を用いた場合に比べ、Na+の濃度が高く、脱塩しづらいことが分かった。
【0084】
H+型の陽イオン交換樹脂を用いた場合には、Na+型の陽イオン交換樹脂を用いた場合に比べ、吸着処理後のサンプルのpHが吸着処理前のpHに比べ低下する(酸化する)ことが分かった。これは、H+型の陽イオン交換樹脂を用いた場合、Na+型の陽イオン交換樹脂を用いた場合に比べ、核酸の負電荷が弱められ、核酸が陽イオン交換樹脂のH+上に非特異的に保持されやすくなり、核酸の回収率が低くなったものと考えられる。
【0085】
3.塩濃度についての検討
<試験例3>
本試験例では、陽イオン交換樹脂によりサンプル中の夾雑物を吸着する際におけるサンプル中の塩濃度の影響について検討した。
【0086】
スピンフィルタカラム(Ultrafree-MC,0.45μm)に強酸性陽イオン交換樹脂(Nuvia S Na+型(Bio Rad社製))を100mg計量して入れた。次いで、50 mM MESバッファー(pH5)を調製し、BSAを0.5質量%となるように加え、Cy3修飾した20merオリゴDNAを4μMとなるように加え、更にNaClを3.3、0.9、又は0.33mg/mLとなるように加え、3種類のサンプルを用意した。また、NaClを添加していない以外は、上記3種類のサンプルと同様の条件で作製したサンプルも用意した。これらのサンプルを、常温にて十分に攪拌させた後、Na+型又はH+型である強酸性陽イオン交換樹脂封入スピンカラムに夫々200μL滴下し、十分に攪拌させた。その後、12000Gにて2分間遠心し、混合液をスピンダウンした。夫々のサンプルの核酸精製能については、試験例1、2と同様にして評価した。評価結果をまとめたものを表2に示す。
【0087】
【表2】
【0088】
表2に示すように、NaClの濃度が0.33、0.9、3.3mg/mLの場合は、上記濃度が0mg/mLの場合に比べ、核酸の回収率が高くなることが分かった。また、NaClの濃度が0.33、0.9、3.3mg/mLの場合では、核酸回収率はいずれも100%であった。この結果より、血液等のように、内部に塩が存在するサンプルを用いて核酸精製を行う場合には、精製時にサンプルに塩を添加する必要はないものと考えられる。一方、菌等のように、内部に塩が存在しないサンプルを用いて核酸精製を行う場合には、精製時にサンプルに塩を添加することが好ましいものと考えられる。
【0089】
4.イオン交換樹脂種による脱塩に対する影響
以下、実施例1〜3、及び比較例1、2でイオン交換樹脂種による脱塩に対する影響について検討した。
【0090】
<実施例1>
スピンフィルタカラム(Ultrafree-MC,0.45μm)に強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを計量して入れた。吸着処理前のNa+の濃度は、0.45mg/mLであり、pHは4.9であった。次いで、50 mM MESバッファー(pH5)を調製し、ウシ全血を1/10倍に希釈するように加え、サンプルを調製した。このサンプルを、常温にて十分に攪拌させた後、上記イオン交換樹脂封入スピンカラムに200μL滴下し、十分に攪拌させた。その後、12000Gにて2分間遠心し、混合液をスピンダウンした。その後、サンプル中のNa+の濃度及びpHを測定した。
【0091】
<実施例2>
実施例1と比し、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgの代わりに、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いた以外は、実施例1と同様にして評価した。
【0092】
<実施例3>
実施例2と比し、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いる代わりに、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):70mg及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いた以外は、実施例2と同様にして評価した。
【0093】
<比較例1>
実施例1と比し、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いる代わりに、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mgのみを用いた以外は、実施例1と同様にして評価した。
【0094】
<比較例2>
実施例1と比し、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いる代わりに、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mgのみを用いた以外は、実施例1と同様にして評価した。
【0095】
実施例1〜3、及び比較例1、2における評価結果をまとめたものを表3に示す。
【0096】
【表3】
【0097】
実施例1、実施例2、及び実施例3では、H+型の強酸性陽イオン交換樹脂とOH−型の強塩基性陰イオン交換樹脂とを混合したイオン交換樹脂を用いることにより、比較例1及び比較例2に比し、サンプルをより精度良く脱塩できた。また、実施例1、実施例2、及び実施例3では、比較例2に比し、サンプルのpH変化をより精度良く抑制できた。
【0098】
また、実施例2及び実施例3に示すように、強酸性陽イオン交換樹脂と強塩基性陰イオン交換樹脂との用量を等量としても、又は等イオン交換容量としても、脱塩効果及びpH変化の抑制を実現できることが示唆された。
【0099】
5.脱塩によるLAMP反応及びRT-LAMP反応に対する影響
以下、実施例4、5、及び比較例3〜5で脱塩処理によるLAMP反応及びRT-LAMP反応に対する影響について検討した。
【0100】
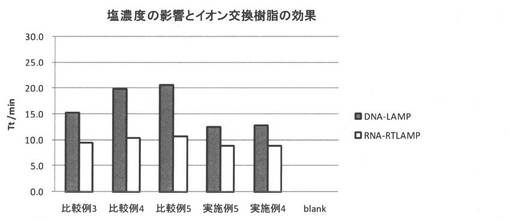
<実施例4>
スピンフィルタカラム(Ultrafree-MC,0.45μm)に強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを計量して入れた。次いで、50 mM MESバッファー(pH5)を調製し、超音波破砕したビフィズス菌を1000copy/uLとなるように加え、更にNaClを0.9mg/mLとなるように加えた。得られたサンプルを、常温にて十分に攪拌させた後、Na+型又はH+型である強酸性陽イオン交換樹脂封入スピンカラムに夫々200μL滴下し、十分に攪拌させた。その後、12000Gにて2分間遠心し、混合液をスピンダウンした。LAMP反応及びRT−LAMP反応については、試験例1と同様の方法にして行った。
【0101】
<実施例5>
実施例4と比し、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgの代わりに、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いた以外は、実施例4と同様にして評価した。
【0102】
<比較例3>
実施例4と比し、サンプルとしてNaClを添加していないものを用い、サンプルをイオン交換樹脂封入スピンカラムに滴下していないという点以外は、実施例4と同様にして評価した。
【0103】
<比較例4>
実施例4と比し、サンプルをイオン交換樹脂封入スピンカラムに滴下していないという点以外は、実施例4と同様にして評価した。
【0104】
<比較例5>
実施例4と比し、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgの代わりに、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mgを用いた以外は、実施例4と同様にして評価した。
【0105】
実施例4、5、及び比較例3〜5における評価結果をまとめたものを図8及び図9に示す。実施例4、実施例5、及び比較例4から、イオン交換樹脂によりサンプルの脱塩を行うことで、LAMP反応及びRT-LAMP反応におけるTt値が小さくなることが分かった。実施例4、実施例5、及び比較例4から、イオン交換樹脂によりサンプルの脱塩を行うことで、LAMP反応の進行が良好になることが示唆された。また、実施例4及び実施例5から、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製))をイオン交換樹脂として用いることによるLAMP反応への影響は小さいことが分かった。これは、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製))をイオン交換樹脂として用いても、サンプルの脱塩に対する影響は小さいことによるものと考えられる。また、比較例4及び比較例5では、LAMP反応の進行を良好にしづらいことが分かった。LAMP反応の進行を良好にするには、サンプルの脱塩を十分に行う必要があることが示唆された。
【0106】
6.添加剤についての検討
以下、実施例6及び実施例7で添加剤(Brij35及びEDTA)の影響について検討した。
<実施例6>
スピンフィルタカラム(Ultrafree-MC,0.45μm)に強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを計量して入れた。次いで、50 mM MESバッファー(pH5)となるように調製し、ウシ全血を10体積%となるように加え、超音波破砕したビフィズス菌を1000copy/uLとなるように加え、更にBrij35を0.5体積%となるように加え、EDTAを1mMとなるように加えた。得られた核酸を含有するサンプルについて、実施例4と同様にし、LAMP反応及びRT-LAMP反応を行い、評価した。
【0107】
<実施例7>
実施例6に比し、Brij35及びEDTAをサンプルに添加していないという点以外は、実施例6と同様にして評価を行った。
【0108】
実施例6及び実施例7における評価結果を図10及び図11に示す。また、評価結果をまとめたものを表4に示す。
【0109】
【表4】
【0110】
実施例6では、イオン交換樹脂によりサンプルの夾雑物の吸着処理を行うことで、LAMP反応及びRT-LAMP反応のいずれにおいても、吸着処理前に比べTt値が小さくなることが分かった。一方、実施例7では、イオン交換樹脂によりサンプルの夾雑物の吸着処理を行うことで、LAMP反応及びRT-LAMP反応のいずれにおいても、吸着処理前に比べTt値が大きくなることが分かった。これは、実施例6では、実施例7に比べ、サンプルにBrij35及び1mM EDTAを添加したことで、核酸の回収率が高くなったことによるものと考えられる。
【0111】
7.界面活性剤/親水性高分子についての検討
以下、実施例8〜11で、界面活性剤/親水性高分子の影響について検討した。
<実施例8>
スピンフィルタカラム(Ultrafree-MC,0.45μm)に強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを計量して入れた。次いで、50 mM MESバッファー(pH5)となるように調製し、ウシ全血を10体積%となるように加え、超音波破砕したビフィズス菌を1000copy/uLとなるように加え、更にBrij35を0.5体積%となるように加えた。実施例7では、サンプルに非イオン界面活性剤又は非イオン性親水性高分子を加えなかった。得られた核酸を含有するサンプルについて、試験例1と同様に、核酸回収率を評価した。
【0112】
<実施例9>
実施例8に比し、イオン交換樹脂による吸着処理の前のサンプルに非イオン性界面活性剤としてBrij35を0.5体積%となるように添加したという点以外は、実施例8と同様にして評価を行った。
【0113】
<実施例10>
実施例9に比し、非イオン性界面活性剤として、Brij35の代わりに、Tween20を用いたという点以外は、実施例9と同様にして評価を行った。
【0114】
<実施例11>
実施例9に比し、Brij35の代わりに、非イオン性親水性高分子であるPEG20000を用いたという点以外は、実施例9と同様にして評価を行った。
【0115】
実施例8〜11における評価結果を表5に示す。
【0116】
【表5】
【0117】
実施例7に比べ、実施例8〜10では核酸の回収率がいずれも高く、非イオン界面活性剤(Brij35、Tween20)又は非イオン性親水性高分子(PEG20000)をサンプルに添加してサンプルの吸着処理を行うことで、核酸の回収率が上がることが実証された。
【0118】
8.本技術に係る吸着処理とゼオライトによる吸着処理との比較
以下、実施例12及び比較例6で本技術に係る吸着処理とゼオライトによる吸着処理との比較実験を行った。
<実施例12>
実施例7で用いたサンプルと同様にして調製したサンプルを準備した。同様の組成の3サンプル夫々について3回ずつ、実施例7と同様にし、LAMP反応及びRT-LAMP反応を行い、評価した。
【0119】
<比較例6>
50 mM MESバッファー(pH5)を調製し、ウシ全血を5体積%加え、超音波破砕したビフィズス菌を1000copy/uLとなるように加え、サンプルを調製した。次いで、サンプルを熱アルカリ処理し、ゼオライトで夾雑物の吸着処理を行った。得られた同様の組成の3サンプル夫々について3回ずつ、実施例4と同様にし、LAMP反応及びRT-LAMP反応を行い、評価した。
【0120】
実施例12及び比較例6における評価結果を図12(LAMP反応)及び図13(RT-LAMP反応)に示す。実施例12及び比較例6から、ゼオライトでサンプルの夾雑物の吸着処理を行った場合よりも、イオン交換樹脂でサンプルの夾雑物の吸着処理を行った場合の方が、核酸増幅反応の進行を良好なものにすることができた。
【産業上の利用可能性】
【0121】
本技術に係る核酸精製方法は、操作が簡便であり、短時間で高効率に核酸を抽出することができる。従って、PCR(Polymerase Chain Reaction)法やLAMP(Loop-Mediated Isothermal Amplification)法等の核酸増幅反応のための核酸精製処理に適用され、核酸が微量あるいは極低濃度でしか含まれないサンプル中の核酸を精製するために用いられ得る。
【符号の説明】
【0122】
1、101:核酸精製用キット、3:核酸精製器具、10:イオン交換樹脂、20:第1の陽イオン交換樹脂、30:陰イオン交換樹脂、40:第2の陽イオン交換樹脂
【技術分野】
【0001】
本技術は、核酸精製方法、核酸抽出方法、及び核酸精製用キットに関する。より詳しくは、イオン交換樹脂により夾雑物を吸着することで核酸を精製する方法等に関する。
【背景技術】
【0002】
PCR(Polymerase Chain Reaction)法やLAMP(Loop-Mediated Isothermal Amplification)法等の核酸増幅反応はバイオテクノロジーにおける様々な分野で応用されている。例えば、医学分野では、DNAやRNAの塩基配列に基づいた診断が行われており、農業分野では遺伝子組み換え作物の判定等でDNA鑑定が活用されている。
【0003】
核酸増幅反応では、微量サンプル中の核酸を高効率に増幅して検出できる。しかし、サンプルに含まれる核酸が極めて微量である場合には、検出下限量未満となる場合がある。さらに、サンプル中の核酸濃度が極めて低い場合には、反応場へ導入可能な容積のサンプル中に増幅対象核酸が含まれなかったために検出が不能となる場合がある。これらの場合には、予め精製や濃縮等をして抽出した核酸を反応場に導入することが有効となる。
【0004】
ここで、核酸の精製という点に着目すると、従来、フェノール/クロロホルム/エタノールを用いた方法が知られている。また、核酸吸着能を有する多孔質担体を用いた核酸の精製方法も知られている(特許文献1参照)。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2005−080555号公報
【特許文献2】国際公開第2009/060847号
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、従来のフェノール/クロロホルム/エタノールを用いた方法は、有害な有機溶媒を使用する必要があり、遠心分離操作等に手間がかかっていた。また、核酸吸着能を有する多孔質担体を用いた核酸の精製方法においては、複数の工程を行う必要があり、操作の簡便性からも課題があった。そのため、操作がより簡便であり、短時間で高効率に核酸を精製することができる方法が希求されていた。
【0007】
そこで、本技術は、操作が簡便であり、短時間で高効率に核酸を精製することができる核酸精製方法を提供することを主な目的とする。
【課題を解決するための手段】
【0008】
上記課題解決のため、本技術は、核酸を含有するサンプルに含まれる物質をイオン交換樹脂により吸着する手順を含み、上記イオン交換樹脂には、陽イオン交換樹脂及び陰イオン交換樹脂を用いる核酸精製方法を提供する。上記陽イオン交換樹脂により、サンプルに含まれる正電荷を有するタンパク質や金属塩等のカチオンを吸着することが可能になる。また、上記陰イオン交換樹脂により、サンプルに含まれる主に核酸以外の負電荷を有するアニオンを吸着することが可能になる。
また、上記陽イオン交換樹脂には、第1の陽イオン交換樹脂と、当該第1の陽イオン交換樹脂よりも排除限界分子量が低い第2の陽イオン交換樹脂とを用いることが好ましい。ここで、排除限界分子量とは、イオン交換樹脂に吸着させようとした種々の化合物において、イオン交換樹脂の細孔内に侵入しづらくなる化合物、すなわちイオン交換樹脂の細孔内に吸着されづらくなる化合物のうち、最も低い分子量をもつものの分子量をいう。換言すれば、当該排除限界分子量より高い分子量を有する化合物については、イオン交換樹脂に吸着されづらくなる。
上記陽イオン交換樹脂のうち、より排除限界分子量が高い上記第1の陽イオン交換樹脂が、上記タンパク質を選択的に吸着しやすくなる。また、上記陽イオン交換樹脂のうち、より排除限界分子量が低い上記第2の陽イオン交換樹脂が、主に金属塩等のカチオンを選択的に吸着しやすくなる。
また、上記第1の陽イオン交換樹脂により上記物質を吸着してから、上記陰イオン交換樹脂と第2の陽イオン交換樹脂とにより上記物質を吸着することが好ましい。この場合、例えば、上層に上記第1の陽イオン交換樹脂を有し、下層に上記陰イオン交換樹脂と上記第2の陽イオン交換樹脂とを有するカラムの上記上層側から上記サンプルを流入してもよい。このように、サンプル中の物質について、まず第1の陽イオン交換樹脂によりサンプル中のタンパク質を主に選択的に吸着し、次いで第2の陽イオン交換樹脂及び陰イオン交換樹脂によりサンプル中の金属塩を主に選択的に吸着することで、よりサンプル中の物質の吸着効率は向上する。
また、上記手順は、緩衝液で希釈された前記サンプルに含まれる物質を前記イオン交換樹脂により吸着する手順であってもよく、上記緩衝液のpHは4.0〜8.0であることが好ましい。
また、上記陽イオン交換樹脂は、強酸性陽イオン交換樹脂であることが好ましい。また、上記第1の陽イオン交換樹脂の対イオンは、Na+(ナトリウムイオン)であることが好ましい。また、上記第2の陽イオン交換樹脂の対イオンは、H+(プロトン)であることが好ましい。
また、上記陰イオン交換樹脂は、強塩基性陰イオン交換樹脂であることが好ましい。また、上記陰イオン交換樹脂の対イオンは、OH−(水酸化物イオン)であることが好ましい。
また、上記第2の陽イオン交換樹脂に対する上記陰イオン交換樹脂のイオン交換容量の割合は、50%〜150%であることが好ましい。ここで、イオン交換容量とは、イオン交換樹脂の単位量当たりの交換基の総数のことを指す。例えば、第2の陽イオン交換樹脂の対イオンがH+である場合、イオン交換容量とは、第2の陽イオン交換樹脂1ml中に含まれるH+の総数のことを指す。上記第2の陽イオン交換樹脂に対する上記陰イオン交換樹脂のイオン交換容量の割合が、50%〜150%であることで、イオン交換樹脂によるサンプルに含まれる物質の吸着をする前とその後のpHの変動をより安定に抑制することが可能になる。
また、上記核酸精製方法では、サンプルに非イオン性界面活性剤及び/又は非イオン性親水性高分子化合物を添加して上記物質を吸着することがより好ましい。サンプルに非イオン性界面活性剤及び/又は非イオン性親水性高分子化合物を添加することで、核酸の樹脂に対する物理的な吸着を抑制することが可能になる。
上記物質は、例えば、夾雑物である。当該夾雑物は、例えば、タンパク質と金属塩とを含有する。また、夾雑物は、タンパク質や金属塩の他にサンプル中の核酸の分析において不要な、種々のペプチド、糖、塩、低分子アニオン(例えば、フマル酸、フタル酸、フミン酸、フルボ酸)等の物質を含有していてもよい。
【0009】
また、上記課題解決のため、本技術は、核酸を含有するサンプルの超音波処理をする手順、陽イオン交換樹脂及び陰イオン交換樹脂により前記サンプルに含まれる物質を吸着する手順、及び電気泳動により移動する前記核酸を堰き止めることで前記核酸を濃縮する手順の各手順を行う核酸抽出方法を提供する。
上記電気泳動は、アニオン性官能基を有するインターカレーターを挿入した前記核酸について行われてもよい。また、上記電気泳動は、上記サンプルと、上記サンプル中に含まれる物質が含有するカルボキシル基と脱水縮合反応をする官能基を有する化合物と、上記脱水縮合反応の縮合剤と、を混合して、上記核酸について行われてもよい。
【0010】
また、本技術は、陽イオン交換樹脂と、陰イオン交換樹脂と、前記陽イオン交換樹脂及び前記陰イオン交換樹脂を内部に保持し、核酸を含有するサンプルを流通可能な核酸精製器具と、を含む核酸精製用キットを提供する。
【発明の効果】
【0011】
本技術により、操作が簡便であり、短時間で高効率に核酸を抽出することができる核酸精製方法が提供される。
【図面の簡単な説明】
【0012】
【図1】本技術の第1実施形態に係る核酸精製方法の手順を説明するための模式図である。
【図2】本技術の第1実施形態に係る第1の陽イオン交換樹脂により夾雑物を吸着する状態を概念的に説明した模式図である。
【図3】本技術の第1実施形態に係る第2の陽イオン交換樹脂及び陰イオン交換樹脂により夾雑物を吸着する状態を概念的に説明した模式図である。
【図4】本技術の第2実施形態に係る核酸精製方法の手順を説明するための模式図である。
【図5】陽イオン交換樹脂による吸着処理後の核酸回収率を示すグラフである(試験例1)。
【図6】陽イオン交換樹脂による吸着処理後のサンプルのLAMP反応の結果を示すグラフである(試験例1)。
【図7】陽イオン交換樹脂の対イオン種(Na+、H+)の核酸回収率に対する影響を検討した結果を示すグラフである(試験例2)。
【図8】イオン交換樹脂による吸着処理後のサンプルのLAMP反応の結果を示すグラフである(実施例4、5)
【図9】イオン交換樹脂による吸着処理後のサンプルのLAMP反応及びRT−LAMP反応の結果(Tt値)を示すグラフである(実施例4、5)
【図10】イオン交換樹脂による吸着処理後のLAMP反応及びRT−LAMP反応の結果(Tt値)を示すグラフである(実施例6)
【図11】イオン交換樹脂による吸着処理後のLAMP反応及びRT−LAMP反応の結果(Tt値)を示すグラフである(実施例7)
【図12】サンプルのLAMP反応の結果を示すグラフである(実施例12)。
【図13】サンプルのRT−LAMP反応の結果を示すグラフである(実施例12)。
【発明を実施するための形態】
【0013】
以下、本技術を実施するための好適な形態について説明する。なお、以下に説明する実施形態は、本技術の代表的な実施形態の一例を示したものであり、これにより本技術の範囲が狭く解釈されることはない。説明は以下の順序で行う。
1.本技術の第1実施形態に係る核酸精製用キット及び核酸精製方法
(1)核酸精製用キット
(2)核酸精製方法
2.本技術の第2実施形態に係る核酸精製用キット及び核酸精製方法
3.核酸抽出方法
【0014】
1.本技術の第1実施形態に係る核酸精製用キット及び核酸精製方法
(1)核酸精製用キット
まず、本技術の第1実施形態に係る核酸精製方法に用いる核酸精製用キットについて、図1(A)を参照しながら説明する。図1(A)〜(C)は、本技術の第1実施形態に係る核酸精製方法の手順を説明するための模式図である。
【0015】
図1(A)中、符号1で示す核酸精製用キットは、サンプルに含まれる核酸以外の物質(以下、夾雑物と記す)を吸着し、核酸を精製する。核酸精製用キット1は、イオン交換樹脂10と、イオン交換樹脂10を内部に保持し、核酸を含有するサンプルを流通可能な核酸精製器具3とを主に有する。なお、本技術で用いるサンプルとしては、特に限定されるものではないが、例えば、ぬぐい液、口腔スワブ、唾液、血清、血漿、末梢血単球細胞、脳脊髄液、糞、尿、培養細胞、生検組織等の生体試料が挙げられる。また、サンプルとしては、生体試料の他に、河川水、海水、土壌等も挙げられる。
【0016】
核酸精製器具3は、イオン交換樹脂10を内部に保持し、サンプルを流通させる。核酸精製器具3については、イオン交換樹脂10を内部に保持し、サンプルを流通可能であれば、形状や材質等は特に限定されるものではない。例えば、核酸精製器具3としては、図1(A)に示すように、上部が開口した形状を有する市販のサンプル管やチップ等を用いることが可能である。また、核酸精製器具3は、例えば、上部及び下部が開口した筒状に形成されていてもよい。この場合には、サンプルを核酸精製器具3に流通し、イオン交換樹脂10によりサンプルを吸着した後に、下部から流れ出るサンプルを回収するために、別途チップ等を用いることが可能である。
【0017】
本技術に係るイオン交換樹脂10は、サンプルに含まれる夾雑物を吸着する。イオン交換樹脂10は、陽イオン交換樹脂と、陰イオン交換樹脂とを含む。なお、上記夾雑物は、例えば、タンパク質と金属塩とを含有する。また、夾雑物は、タンパク質や金属塩の他にサンプル中の核酸の分析において不要な、種々のペプチド、糖、塩、低分子アニオン(例えば、フマル酸、フタル酸、フミン酸、フルボ酸)等の物質を含有していてもよい。
【0018】
上記陽イオン交換樹脂は、カチオン性の夾雑物を主に吸着する。陽イオン交換樹脂は、第1の陽イオン交換樹脂と、当該第1の陽イオン交換樹脂よりも排除限界分子量が低い第2の陽イオン交換樹脂とを含むことが好ましい。第1の陽イオン交換樹脂は、サンプルに含まれるタンパク質を主に吸着するために用いられる。第2の陽イオン交換樹脂は、サンプルに含まれる金属イオン等の陽イオン(カチオン)を主に吸着するために用いられる。
【0019】
第1の陽イオン交換樹脂としては、サンプルに含まれるタンパク質を吸着することが可能であれば、特に限定されるものではないが、強酸性陽イオン交換樹脂であることが好ましい。このとき、第1の陽イオン交換樹脂は、SO3−を固定イオンとすることが好ましい。また、対イオンとしては、特に限定されるものではなく、カルシウムイオン(Ca2+)、銅イオン(Cu2+)、亜鉛イオン(Zn2+)、マグネシウムイオン(Mg2+)、カリウムイオン(K+)、アンモニウムイオン(NH4+)、ナトリウムイオン(Na+)、プロトン(H+)等が挙げられる。この点、これらのイオンを用いた場合のイオンを交換する強さ(イオンの選択性)の順序は、Ca2+>Cu2+>Zn2+>Mg2+>K+>NH4+>Na+>H+である。従って、イオン交換する強さを考慮すると、対イオンは、H+であることが好ましい。一方で、対イオンがH+である場合、サンプルのpHが酸性側にシフトしやすい。これに対し、対イオンがNa+である場合、対イオンがH+である場合よりも、金属塩を吸着する機能は弱くなるものの、サンプルのpHの変動を抑制することが可能である。また、対イオンがNa+である場合であっても、第1の陽イオン交換樹脂はタンパク質を十分に吸着することができる。そのため、第1の陽イオン交換樹脂は、Na+を対イオンとして有する強酸性陽イオン交換樹脂(Na+型強酸性陽イオン交換樹脂)であることが好ましい。
【0020】
第2の陽イオン交換樹脂としては、サンプルに含まれる金属塩等のカチオンを吸着することが可能であれば、特に限定されるものではないが、強酸性陽イオン交換樹脂であることが好ましい。このとき、第2の陽イオン交換樹脂は、SO3−を固定イオンとすることが好ましい。また、対イオンとしては、特に限定されるものではないが、上述したイオンを交換する強さ(イオンの選択性)の順序を考慮すると、第2の陽イオン交換樹脂の対イオンはプロトン(H+)であることがより好ましい。
【0021】
陰イオン交換樹脂は、サンプルに含まれる陰イオン(アニオン)を吸着する。陰イオン交換樹脂としては、サンプルに含まれる金属塩等のアニオンを吸着することが可能であれば、特に限定されるものではないが、強塩基性陰イオン交換樹脂であることが好ましい。また、陰イオン交換樹脂は、例えば、トリメチルアンモニウム基を持つI型及びジメチルエタノールアンモニウム基を持つII型の強塩基性陰イオン交換樹脂のうちのいずれとすることも可能である。ここで、I型においては、陰イオンを交換吸着する強さ(イオンの選択性)の順序は、HSO4−>NO3−>Br−>Cl−>HCO3−>HCOO−>CH3COO−>F−>OH−となる。また、II型においては、陰イオンを交換吸着する強さ(イオンの選択性)の順序は、HSO4−>NO3−>Br−>Cl−>HCO3−>OH−>HCOO−>CH3COO−>F−となる。そして、上記I型及びII型の陰イオン交換樹脂のうち、I型の方が塩化物イオン(Cl−)等をより精度良く吸着することが可能である。そのため、陰イオン交換樹脂は、I型であり、対イオンとして水酸化物イオン(OH−)を有することが好ましい。
【0022】
また、核酸精製用キット1には、夾雑物の吸着と共に核酸を吸着しないように、非イオン性界面活性剤や非イオン性親水性高分子化合物等の非イオン性化合物等の添加剤が含まれていてもよい。非イオン性化合物としては、例えば、Brij35、Tween20、又はTritonX100等の非イオン性界面活性剤が挙げられる。また、ポリエチレングリコールやポリヒドロキシエチルセルロース等の非イオン性親水性高分子化合物も挙げられる。これらの例示した非イオン性化合物は、単独で用いてもよいし、複数種組み合わせて用いてもよい。更に、核酸精製用キット1には、EDTA等のキレート添加剤が含まれていてもよい。
【0023】
また、核酸精製用キット1には、緩衝液が含まれていてもよい。かかる緩衝液用の緩衝剤としては、例えば、HomoPIPES(pH:3.9〜5.1、pKa:4.55)、MES(pH:5.5〜7.0、pKa:6.15)、Bis−Tris(pH:5.7〜7.3、pKa:6.46)、ADA(pH:5.8〜7.4、pKa:6.60)、PIPES(pH:6.1〜7.5、pKa:6.80)、ACES(pH:6.0〜7.5、pKa:6.90)、MOPSO(pH:6.2〜7.4、pKa:6.95)、BES(pH:6.6〜8.0、pKa:7.15)、MOPS(pH:6.5〜7.9,pKa:7.20)、TES(pH:6.8〜8.2、pKa:7.50)、HEPES(pH:6.8〜8.2、pKa:7.55)、DIPSO(pH:6.9〜8.1、pKa:7.6)、TAPSO(pH:7.0〜8.2、pKa:7.7)、POPSO(pH:7.2〜8.5、pKa:7.85)、HEPPSO(pH:7.4〜8.6、pKa:7.9)、EPPS(pH:7.5〜8.5、pKa:8.0)、Tricine(pH:7.8〜8.8、pKa:8.15)、Bicine(pH:7.7〜9.1、pKa:8.35)、TAPS(pH:7.7〜9.1、pKa:8.4)、CHES(pH:8.6〜10.0、pKa:9.5)、CAPSO(pH:9.3〜10.7、pKa:10.0)、CAPS(pH:9.7〜11.1、pKa:10.40)等が挙げられる。なお、上記例示した各緩衝剤のpHの範囲は、緩衝液を添加したサンプルのpHとして適当な範囲を指す。また、上記例示した各緩衝剤のpKaは、HomoPIPESを除いて20℃におけるpKaを指す(HomoPIPESについては、37℃におけるpKaを示す。)。サンプルを希釈した緩衝液のpHは、4〜8であることが好ましい。この点、本実施形態において上記例示した緩衝剤の中では、HomoPIPES、MES、Bis−Tris、ADAが好適に用いられる。
【0024】
(2)核酸精製方法
次に、本技術の第1実施形態に係る核酸精製方法の手順について、図1(A)〜(C)を参照しながら説明する。
【0025】
ここで、本技術の第1実施形態に係る核酸精製方法を説明する前に、まず、本技術の関連技術に係る核酸精製方法について説明する。関連技術に係る核酸精製方法としては、ゼオライトを用いてサンプルの夾雑物を吸着する方法を挙げることができる(例えば、特許文献2参照)。
【0026】
関連技術に係る核酸精製方法では、生体試料等のサンプルに含まれるアニオン及びカチオンのうち、カチオンのみの除去が可能である。そのため、関連技術に係る核酸精製方法では、サンプルの精製により、プロトンが放出されることになり、サンプルを含有する液のpHが低下する(より酸性側にシフトする)。この点、サンプルの精製後、核酸増幅反応はpH7〜9で実行される場合があるため、ゼオライトと接触させるサンプルを含有する液のpHを予め高めに設定する(アルカリ側に設定する)必要がある。そのため、上記関連技術に係る核酸精製方法では、操作が煩雑になる場合がある。また、サンプルにRNAが含まれていると、当該RNAの分解をもたらす可能性もある。
【0027】
また、上記関連技術に係る核酸精製方法は、サンプルをアルカリ性の化合物と界面活性剤(例えば、SDS等)との混合試薬で希釈した後に、高温で加熱する工程を含む。そのため、この点からも、上記関連技術に係る核酸精製方法では、操作が煩雑になる場合がある。また、サンプルにRNAが含まれていると、当該RNAの分解をもたらす可能性もある。
【0028】
また、上記関連技術に係る核酸精製方法では、サンプルの精製後、サンプル中にアニオンが残留することがある。更に、サンプル中には、アニオン性界面活性剤が含まれていることもある。そのため、サンプルの精製後、核酸を電気泳動させる際に、サンプル中の電解質濃度が高くなり、サンプルが発熱しやすくなる。従って、この点からも、サンプルにRNAが含まれていると、当該RNAの分解をもたらす可能性がある。更に、発熱によるサンプルの対流の発生により、核酸の電気泳動濃縮の効率が低下する場合もある。
【0029】
この点、以下に詳述する本技術に係る核酸精製方法は、本願発明者らの鋭意検討の結果見出されたものであり、煩雑な操作を必要とせず、操作を非常に簡便にし、短時間に高効率で核酸を精製可能にした方法である。
【0030】
本技術の第1実施形態に係る核酸精製方法では、まず、容器5に収容された核酸を含有するサンプルA及びサンプルAを希釈した緩衝液が、ピペット50に取り付けたピペットチップ51に充填される(図1(A)参照)。次に、ピペットチップ51に充填されたサンプルA及びサンプルAを希釈した緩衝液は、核酸精製器具3に注入される(図1(B)参照)。このとき、核酸精製器具3は、イオン交換樹脂10を内部に保持している。そのため、サンプルAに含まれる夾雑物は、イオン交換樹脂10により吸着される。
【0031】
最後に、夾雑物がイオン交換樹脂10により吸着されることで精製されたサンプルAは、イオン交換樹脂10を通過して核酸精製器具3の下部に貯留される(図1(C)参照)。
【0032】
図1(A)〜(C)に示した工程後、サンプルAについては、核酸増幅反応をすることが可能である。例えば、サンプルAを固相化した核酸増幅試薬に添加して核酸増幅反応をすることが可能である。また、本実施形態では、サンプルをイオン交換樹脂10により脱塩することにより、二重らせんDNAを一本鎖DNAにし、核酸増幅反応前にDNAの変性を適宜行うことも可能である。
【0033】
ここで、図2及び図3を参照しながら、サンプルAに含まれる夾雑物がイオン交換樹脂10により吸着される状態(図1(B)中、(I)で示す部分)について、より詳細に説明する。図2は、図1(B)において、特に第1の陽イオン交換樹脂が夾雑物を吸着する状態を概念的に説明した模式図である。図3は、図1(B)において、特に第2の陽イオン交換樹脂及び陰イオン交換樹脂が夾雑物を吸着する状態を概念的に説明した模式図である。
【0034】
まず、図2を参照しながら、イオン交換樹脂10に含まれる第1の陽イオン交換樹脂20が、サンプルAに含まれる夾雑物を吸着する状態を説明する。なお、図2において、符号Bで示されるものは、サンプルAに含まれる核酸であり、符号Cで示されるものは、サンプルAに含まれる夾雑物の一例であるタンパク質である。また、ここでは、第1の陽イオン交換樹脂20については、対イオンとしてナトリウムイオン(Na+)を有する強酸性陽イオン交換樹脂とする。
【0035】
第1の陽イオン交換樹脂20は、SO3−等のアニオン21と、対イオンであるNa+22とを有する。また、第1の陽イオン交換樹脂20には、タンパク質Cを吸着する領域23が形成される。
【0036】
図2に示すように、核酸精製器具3内に通流されたサンプルAに含まれるタンパク質Cは、正に帯電しているため、表面にアニオン21を有する第1の陽イオン交換樹脂20に吸着される。例えば、タンパク質Cは、領域23に吸着される。一方、核酸Bは、負に帯電しているため、第1の陽イオン交換樹脂20に吸着されることなく、サンプルAに含まれる緩衝液等の流れに誘導されて流されていく。また、第1の陽イオン交換樹脂20の対イオンはNa+22であるため、対イオンがH+である場合に比べ、金属塩については吸着しづらく(脱塩しづらく)、タンパク質Cを選択的に吸着することが可能である。
【0037】
この際、サンプルAについては、サンプルAを希釈する緩衝液のpHを4〜8にしておくことが好ましい。サンプルAを希釈する緩衝液のpHを4〜8にしておく(酸性にしておく)ことで、より安定に、核酸Bを負に帯電させつつ、夾雑物(主にタンパク質C)のみを正に帯電させておくことが可能になる。そのため、タンパク質Cのみを第1の陽イオン交換樹脂20に安定に吸着させ、核酸Bをより高効率に精製することができる。また、第1の陽イオン交換樹脂20が強酸性陽イオン交換樹脂であることで、第1の陽イオン交換樹脂20は広範囲のpH条件下(例えば、pH:3〜13)で負電荷を有していることが可能である。そのため、上述したように、サンプルを酸性にしておいても、タンパク質Cを第1の陽イオン交換樹脂20に安定に吸着させることが可能になる。更に、サンプルAのpHを4〜8にしておく(酸性にしておく)ことで、RNA分解酵素の活性を抑えることができるため、RNAを好適に精製可能である。
【0038】
また、第1の陽イオン交換樹脂20の排除限界分子量は、サンプルAに含まれるタンパク質Cが領域23に取り込まれる程度に規定されていることが好ましい。より具体的には、第1の陽イオン交換樹脂20の排除限界分子量は、5000以上であることが好ましい。第1の陽イオン交換樹脂20の排除限界分子量が5000以上であることで、夾雑物中、タンパク質Cを特に選択的に吸着し、除去しやすくなる。
【0039】
また、第1の陽イオン交換樹脂20の平均体積粒子径は、1μm以上3000μm以下であることが好ましい。第1の陽イオン交換樹脂20の平均体積粒子径が1μm以上3000μm以下であることでも、夾雑物中、タンパク質Cを特に選択的に吸着し、除去しやすくなる。
【0040】
また、第1の陽イオン交換樹脂20の粒子自体の比重は、0.5以上2.5以下であることが好ましい。第1の陽イオン交換樹脂20の比重が0.5以上2.5以下であることでも、夾雑物中、タンパク質Cを特に選択的に吸着し、除去しやすくなる。さらに、粒子自体の比重は1.0以上2.5以下であることがより好ましい。上記比重が1.0以上2.5以下であることで、粒子が溶液中で沈降しやすくなることにより、容易に溶液から粒子を除去することが可能になる。
【0041】
次に、図3を参照しながら、イオン交換樹脂10に含まれる陰イオン交換樹脂30及び第2の陽イオン交換樹脂40によりサンプルAに含まれる夾雑物を吸着する状態を説明する。
【0042】
図3において、符号D1、D2、及びD3で示されるものは、サンプルAに含まれる夾雑物の一例である金属塩が有するカチオン又はアニオンである。本実施形態では、特に限定されるものではないが、ここでは、D1を塩化物イオン等のアニオンとし、D2をナトリウムイオン等のカチオンとし、D3をマグネシウムイオン等のカチオンとする。
【0043】
まず、図3を参照しながら、陰イオン交換樹脂30が夾雑物(主に塩化物イオンD1等のアニオン)を吸着する状態について説明する。本実施形態において、陰イオン交換樹脂30については、対イオンとして水酸化物イオン(OH−)を有するI型の強塩基性陰イオン交換樹脂とする。
【0044】
陰イオン交換樹脂30は、CH2N(CH3)3+等のカチオン31と、対イオンであるOH−32とを有する。また、陰イオン交換樹脂30には、塩化物イオンD1等のアニオンを吸着する領域33が形成される。
【0045】
図3に示すように、核酸精製器具3に通流されたサンプルAに含まれる塩化物イオンD1は、負に帯電しているため、表面にカチオン31を有する陰イオン交換樹脂30に吸着される。例えば、塩化物イオンD1は、領域33に吸着される。一方、核酸Bも負に帯電しているものの、核酸Bは塩化物イオンD1等のアニオンに比べ体積が大きいため、陰イオン交換樹脂30には吸着されにくい。この点、陰イオン交換樹脂30の排除限界分子量は、100以上2000以下であることが好ましい。陰イオン交換樹脂30の排除限界分子量が100以上2000以下であることで、より精度良く、核酸Bの吸着を抑制し、塩化物イオンD1等のカチオンを選択的に吸着し、除去しやすくなる。
【0046】
また、陰イオン交換樹脂30の平均体積粒子径は、1μm以上3000μm以下であることが好ましい。第1の陽イオン交換樹脂20の平均体積粒子径が1μm以上3000μm以下であることでも、より精度良く、塩化物イオンD1等のカチオンを選択的に吸着し、除去しやすくなる。
【0047】
また、陰イオン交換樹脂30の比重は、0.5以上2.5以下であることが好ましい。陰イオン交換樹脂30の比重が0.5以上2.5以下であることでも、より精度良く、塩化物イオンD1等のカチオンを選択的に吸着し、除去しやすくなる。さらに、粒子自体の比重は1.0以上2.5以下であることがより好ましい。上記比重が1.0以上2.5以下であることで、粒子が溶液中で沈降しやすくなることにより、容易に溶液から粒子を除去することが可能になる。
【0048】
次に、図3を参照しながら、第2の陽イオン交換樹脂40が夾雑物(主にナトリウムイオンD2及びマグネシウムイオンD3等のカチオン)を吸着する状態について説明する。本実施形態において、第2の陽イオン交換樹脂40については、対イオンとしてH+イオン(プロトン)を有する強酸性陽イオン交換樹脂とする。
【0049】
第2の陽イオン交換樹脂40は、SO3−等のアニオン41と、H+42とを有する。また、第2の陽イオン交換樹脂40には、ナトリウムイオンD2やマグネシウムイオンD3等のカチオンを吸着する領域43が形成される。
【0050】
図3に示すように、核酸精製器具3に通流されたサンプルAに含まれるナトリウムイオンD2やマグネシウムイオンD3等のカチオンは、正に帯電しているため、表面にアニオン41を有する第2の陽イオン交換樹脂40に吸着される。例えば、ナトリウムイオンD2やマグネシウムイオンD3は、領域43に吸着される。一方、核酸Bは、負に帯電しているため、第2の陽イオン交換樹脂40に吸着されることなく、サンプルAに含まれる緩衝液等の流れに誘導されて流されていく。
【0051】
ここで、第2の陽イオン交換樹脂40は、第1の陽イオン交換樹脂20よりも排除限界分子量が低い。そのため、単位体積当たりの表面積が大きくなり、第1の陽イオン交換樹脂20よりも脱塩しやすくなる。また、第2の陽イオン交換樹脂40が対イオンとしてプロトンを有する場合、第1の陽イオン交換樹脂20よりも脱塩しやすくなる。好ましくは、第2の陽イオン交換樹脂40の排除限界分子量は、100以上2000以下である。第2の陽イオン交換樹脂40の排除限界分子量が100以上2000以下であることでナトリウムイオンD2やマグネシウムイオンD3等のカチオンの吸着をより精度良く行うことが可能になる。
【0052】
また、第2の陽イオン交換樹脂40の平均体積粒子径は、1μm以上3000μm以下であることが好ましい。第2の陽イオン交換樹脂40の平均体積粒子径が1μm以上3000μm以下であることでも、より精度良く、ナトリウムイオンD2やマグネシウムイオンD3等のカチオンの吸着を行うことが可能になる。更に、第2の陽イオン交換樹脂40の平均体積粒子径は、1μm以上2000μm以下であることが好ましい。第2の陽イオン交換樹脂40の平均体積粒子径が1μm以上2000μm以下であることで、溶液をイオン交換樹脂に通液させるときの圧力を低く抑えることが可能になる。
【0053】
また、第2の陽イオン交換樹脂40の比重は、0.5以上2.5以下であることが好ましい。第2の陽イオン交換樹脂40の比重は、0.5以上2.5以下であることでも、より精度良く、ナトリウムイオンD2やマグネシウムイオンD3等のカチオンの吸着を行うことが可能になる。さらに、粒子自体の比重は1.0以上2.5以下であることがより好ましい。上記比重が1.0以上2.5以下であることで、粒子が溶液中で沈降しやすくなることにより、容易に溶液から粒子を除去することが可能になる。
【0054】
また、第2の陽イオン交換樹脂40に対する陰イオン交換樹脂30のイオン交換容量の割合は50%〜150%であることが好ましい。上記イオン交換容量の割合が50%〜150%であることで、サンプルのpHの変動をより安定に抑制しつつ脱塩することが可能になる。
【0055】
また、第2の陽イオン交換樹脂40に対する陰イオン交換樹脂30の用量の割合は50%〜150%であることが好ましい。上記用量の割合が50%〜150%であることでも、サンプルのpHの変動をより安定に抑制しつつ脱塩することが可能になる。
【0056】
また、図2及び図3には図示していないが、核酸Bがイオン交換樹脂により吸着されることを抑制するために、Brij35、Tween20、又はTritonX100等の非イオン性界面活性剤がサンプルA中に適宜添加されることが好ましい。また、ポリエチレングリコールやポリヒドロキシエチルセルロース等の非イオン性親水性高分子化合物等の非イオン性化合物もサンプルA中に適宜添加されることが好ましい。
【0057】
以上、説明してきた本技術の第1実施形態に係る核酸精製方法によれば、陽イオン交換樹脂及び陰イオン交換樹脂からなるイオン交換樹脂10を吸着担体として用いることで、サンプルに含まれる夾雑物を吸着することができる。例えば、サンプルとして血液試料等を用いる場合には、複雑な前処理工程を介さず、直接吸着工程を行うことができる。このように、本実施形態に係る核酸精製方法によれば、非常に簡便な操作で、短時間に高効率で核酸を抽出することができる。また、このような核酸精製方法については、数秒間の時間で全工程を行うことが可能であり、短時間で核酸を精製することができる。
【0058】
また、本実施形態に係る核酸精製方法によれば、例えば、シリカ固相抽出のように洗浄工程を含まないものであるため、微量のサンプル量で操作を行うことができ、装置の小型化も実現できる。また、本実施形態に係る核酸精製方法によれば、サンプルを酸性条件下(好ましくは、pH:4〜8)で夾雑物の吸着処理を行うことが可能である。すなわち、本実施形態に係る核酸精製方法によれば、サンプルをアルカリ性にしたり、加熱したりする工程を要しない。そのため、サンプルにRNAが含まれる場合にはRNAの分解を抑制しつつ、サンプルの精製を行うこともできる。また、本実施形態に係る核酸精製方法によれば、酸性領域で超音波破砕を行うことにより、RNaseの働きを抑制することもできる。
【0059】
また、固相化した核酸増幅試薬を用いて核酸増幅反応をする場合にはサンプルを希釈しないため、夾雑物による核酸増幅反応に対する阻害が顕著に表れることになる。しかしながら、本実施形態に係る核酸精製方法により精製したサンプルでは、サンプルに存在していた夾雑物が除去されているため、核酸を直接固相化した核酸増幅試薬に添加して核酸増幅反応を行うことができる。
【0060】
また、本技術の第1実施形態に係る核酸精製方法によれば、陽イオン交換樹脂として、第1の陽イオン交換樹脂と、該第1の陽イオン交換樹脂よりも排除限界分子量が低い第2の陽イオン交換樹脂とを用いることが可能である。そのため、サンプル中の夾雑物に、金属塩及びタンパク質が含まれている場合にも、双方を効率的に除去することができる。
【0061】
本実施形態に係る核酸精製方法によるサンプル中の夾雑物の吸着・除去については、第1の陽イオン交換樹脂及び第2の陽イオン交換樹脂が強酸性陽イオン交換樹脂である場合、より効率的である。また、第1の陽イオン交換樹脂の対イオンがNa+である場合や第2の陽イオン交換樹脂の対イオンがH+である場合には、より効率的に夾雑物を除去できる。
【0062】
また、本実施形態に係る核酸精製方法によれば、陰イオン交換樹脂が強塩基性陽イオン交換樹脂である場合、より効率的に夾雑物を除去できる。また、陰イオン交換樹脂の対イオンがOH−である場合、より効率的に夾雑物を除去できる。
【0063】
また、本実施形態に係る核酸精製方法によれば、第2の陽イオン交換樹脂に対する陰イオン交換樹脂のイオン交換容量の割合を、50%〜150%とすることで、サンプルのpHの変動を抑制することができる。そのため、上述したように、例えば、サンプルにRNAが含まれる場合にはRNAの分解を抑制しつつ、サンプルの精製を行うこともできる。
【0064】
また、本実施形態に係る核酸精製方法によれば、非イオン性界面活性剤や非イオン性親水性高分子化合物等のノニオン性化合物を用いることが可能である。この場合、核酸がイオン交換樹脂のマトリクスに吸着することを抑制し、効率的に夾雑物を除去することができる。
【0065】
2.本技術の第2実施形態に係る核酸精製用キット及び核酸精製方法
図4(A)〜(C)は、本技術の第2実施形態に係る核酸精製方法の手順を説明するための模式図である。
【0066】
図4(A)中、符号101で示す核酸精製用キットは、イオン交換樹脂10と、イオン交換樹脂10に内部に保持し、核酸を含有するサンプルを流通可能な核酸精製器具3とを含む。イオン交換樹脂10については、陽イオン交換樹脂と、陰イオン交換樹脂とを用いる。そして、本実施形態に係る核酸精製用キット101では、陽イオン交換樹脂に、第1の陽イオン交換樹脂20と、第2の陽イオン交換樹脂40とを用いる。
【0067】
本実施形態に係る核酸精製用キット及び核酸精製方法は、本技術の第1実施形態に係る核酸精製用キット及び核酸精製方法と比し、第1の陽イオン交換樹脂20が陰イオン交換樹脂30及び第2の陽イオン交換樹脂40に対し、上層(サンプルが注入される側)に収容されている状態で核酸の精製が行われるという点が主に異なる。そのため、本実施形態では、第1の陽イオン交換樹脂20が陰イオン交換樹脂30及び第2の陽イオン交換樹脂40に対し、上層に設けられているという点について主に説明する。
【0068】
本実施形態に係る核酸精製方法では、核酸精製器具3に第1の陽イオン交換樹脂20が陰イオン交換樹脂30及び第2の陽イオン交換樹脂40よりも上層側に収容されている。そのため、上層側から注入されるサンプルAは、第1の陽イオン交換樹脂20にまず接触する(図4(B)参照)。
【0069】
次いで、第1の陽イオン交換樹脂20により夾雑物の一部が除去されたサンプルAは、陰イオン交換樹脂30及び第2の陽イオン交換樹脂40に接触する。なお、サンプルAの夾雑物に含まれるタンパク質が第1の陽イオン交換樹脂20により吸着される状態(図4(B)中、(II)で示す部分)については、図2を参照しながら説明した第1の陽イオン交換樹脂20がタンパク質を吸着する状態と同様にして説明できる。そのため、ここではその詳細な説明を省略する。また、サンプルA中の夾雑物に含まれる金属塩が陰イオン交換樹脂30及び第2の陽イオン交換樹脂40により吸着される状態(図4(B)中、(III)で示す部分)についても、図3を参照しながら説明した陰イオン交換樹脂30及び第2の陽イオン交換樹脂40が金属塩を吸着する状態と同様にして説明できる。そのため、ここではその詳細な説明も省略する。なお、陰イオン交換樹脂30及び第2の陽イオン交換樹脂40については、陰イオン交換樹脂30及び第2の陽イオン交換樹脂40の何れが上層に収容されていてもよいし、夫々が混合されて第1の陽イオン交換樹脂20の下層に収容されていてもよい。
【0070】
最後に、イオン交換樹脂10により夾雑物が吸着された後に、精製されたサンプルAは、下層に貯留される(図4(C)参照)。
【0071】
本技術の第2実施形態に係る核酸精製方法によれば、第1の陽イオン交換樹脂20によりサンプルA中の夾雑物を吸着してから、陰イオン交換樹脂30及び第2の陽イオン交換樹脂40により夾雑物を吸着する。そのため、本技術の第2実施形態に係る核酸精製方法によれば、より体積が大きなタンパク質を除去してから、より体積が小さな金属塩を除去することが可能になり、より効率的に夾雑物の除去を行うことができる。
【0072】
3.核酸抽出方法
次に、本技術の各実施形態に係る核酸精製方法を含む核酸抽出方法について説明する。本技術に係る核酸抽出方法は、核酸増幅反応を行うための前処理として、核酸を含有するサンプルの超音波処理をする手順、陽イオン交換樹脂及び陰イオン交換樹脂により前記サンプルに含まれる物質を吸着する手順、及び電気泳動により移動する前記核酸を堰き止めることで前記核酸を濃縮する手順の各手順を行うものである。
【0073】
上記各手順については、特に限定されるものではないが、例えば、同一のセル内で全ての手順を行うことが可能である。同一のセル内で各手順を行う場合、超音波処理を行う超音波発生部をセルに設けることが可能である。また、上記セルに、陽イオン交換樹脂及び陰イオン交換樹脂を収容しておくことが可能である。陽イオン交換樹脂及び陰イオン交換樹脂については、上述した本技術の各実施形態に係る核酸精製方法で用いたイオン交換樹脂10(第1の陽イオン交換樹脂20、陰イオン交換樹脂30、第2の陽イオン交換樹脂40)を用いることが可能である。また、上記セルに、電気泳動用の負極及び正極を設け、電気泳動をした核酸を堰き止める堰止部(例えば、透析膜、高分子ゲル等)を設けることが可能である。
【0074】
また、核酸の電気泳動の際に、核酸にアニオン性官能基を有するインターカレーターを挿入しておくことも可能である。ここで、上記インターカレーターとしては、例えば、スルホ基を官能基として有する化合物が挙げられる。具体的には、上記インターカレーターとしては、例えば、9,10−アントラキノン−2,6−ジスルホン酸、アントラキノン−1−スルホン酸ナトリウム、アントラキノン−2,7−ジスルホン酸二ナトリウム、アントラキノン−1,5−ジスルホン酸二ナトリウム、アントラキノン−2−スルホン酸ナトリウム等が挙げられる。このように、核酸にアニオン性官能基を有するインターカレーターを挿入しておくことで、核酸の電気泳動において、核酸の等電点を調節することができる。すなわち、核酸の電気泳動速度を制御しつつ、核酸を濃縮精製することができる。
【0075】
更に、サンプルに含まれる夾雑物の1つであるタンパク質等のカルボキシル基をN−ヒドロキシスクシンイミド(NHS)、エタノールアミン、エチレンジアミン等の化合物と脱水縮合反応させておくことも可能である。脱水縮合反応においては、縮合剤としてカルボジイミド系化合物を用いることが可能である。カルボジイミド系化合物の具体例としては、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(EDC)、4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルフォリニウムクロリド(DMT−MM)、ジシクロヘキシルカルボジイミド(DCC)、又はジイソピルカルボジイミド(DIC)等が挙げられる。これにより、核酸の電気泳動の際に、サンプル中にタンパク質が存在する場合であっても、タンパク質中のカルボキシル基と脱水縮合反応をする核酸とタンパク質との等電点の差を大きくすることで、核酸とタンパク質とを精度良く分離することができる。
【0076】
このように、上記各手順を行うことで、非常に簡便に、高効率に濃縮した核酸を回収することができる。特に、各手順を同一のセル内で行う場合には、他の装置等にサンプルを移す必要がないため、サンプルとして感染性を有する検体等を扱う場合には、操作者に対する感染のリスクを低減することができる。
【0077】
なお、本技術は以下のような構成も取ることができる。
(1)核酸を含有するサンプルに含まれる物質をイオン交換樹脂により吸着する手順を含み、前記イオン交換樹脂には、陽イオン交換樹脂及び陰イオン交換樹脂を用いる核酸精製方法。
(2)前記陽イオン交換樹脂には、第1の陽イオン交換樹脂と、当該第1の陽イオン交換樹脂よりも排除限界分子量が小さな第2の陽イオン交換樹脂とを用いる、前記(1)記載の核酸精製方法。
(3)前記第1の陽イオン交換樹脂により前記物質を吸着してから、前記陰イオン交換樹脂と第2の陽イオン交換樹脂とにより前記物質を吸着する、前記(2)記載の核酸精製方法。
(4)上層に前記第1の陽イオン交換樹脂を有し、下層に前記陰イオン交換樹脂と前記第2の陽イオン交換樹脂とを有するカラムの前記上層側から前記サンプルを流入する、前記(2)又は(3)に記載の核酸精製方法。
(5)前記手順は、緩衝液で希釈された前記サンプルに含まれる物質を前記イオン交換樹脂により吸着する手順であり、前記緩衝液のpHは4.0〜8.0である、前記(1)〜(4)のいずれか1つに記載の核酸精製方法。
(6)前記陽イオン交換樹脂は、強酸性陽イオン交換樹脂である、前記(1)〜(5)のいずれか1つに記載の核酸精製方法。
(7)前記第1の陽イオン交換樹脂の対イオンは、Na+である、前記(2)〜(6)のいずれか1つに記載の核酸抽出方法。
(8)前記第2の陽イオン交換樹脂の対イオンは、H+である、前記(2)〜(7)のいずれか1つに記載の核酸抽出方法。
(9)前記陰イオン交換樹脂は、強塩基性陰イオン交換樹脂である、前記(1)〜(8)のいずれか1つに記載の核酸精製方法。
(10)前記陰イオン交換樹脂の対イオンは、OH−である、前記(1)〜(9)のいずれか1つに記載の核酸精製方法。
(11)前記第2の陽イオン交換樹脂に対する前記陰イオン交換樹脂のイオン交換容量の割合は、50%〜150%である、前記(2)〜(10)のいずれか1つに記載の核酸精製方法。
(12)非イオン性界面活性剤及び/又は非イオン性親水性高分子化合物を用いて前記物質を吸着する、前記(1)〜(11)のいずれか1つに記載の核酸精製方法。
(13)前記物質は、少なくともタンパク質と金属塩とを含有する夾雑物である、前記(1)〜(12)のいずれか1つに記載の核酸精製方法。
(14)核酸を含有するサンプルの超音波処理をする手順、陽イオン交換樹脂及び陰イオン交換樹脂により前記サンプルに含まれる物質を吸着する手順、及び電気泳動により移動する前記核酸を堰き止めることで前記核酸を濃縮する手順の各手順を行う核酸抽出方法。
(15)アニオン性官能基を有するインターカレーターを挿入した前記核酸について前記電気泳動を行う、前記(14)記載の核酸抽出方法。
(16)前記サンプルと、前記サンプル中に含まれる物質が含有するカルボキシル基と脱水縮合反応をする官能基を有する化合物と、該脱水縮合反応の縮合剤と、を混合し、前記核酸について前記電気泳動を行う、前記(14)又は(15)に記載の核酸抽出方法。
(17)陽イオン交換樹脂と、陰イオン交換樹脂と、前記陽イオン交換樹脂及び前記陰イオン交換樹脂を内部に保持し、核酸を含有するサンプルを流通可能な核酸精製器具と、を含む核酸精製用キット。
【実施例】
【0078】
1.強酸性陽イオン交換樹脂による吸着処理の核酸精製能に対する影響
<試験例1>
スピンフィルタカラム(Ultrafree-MC,0.45μm, Millipore製)に強酸性陽イオン交換樹脂(Nuvia S Na+型(Bio Rad社製))を100mg計量して入れた。次いで、50mM MESバッファー(pH5)((株)同仁化学研究所製)を調製し、BSA (和光純薬工業(株)製)を0.5質量%となるように加え、さらにCy3修飾した20merオリゴDNA (Sigma Aldrich Co. 製)を5μMとなるように加えた。このタンパク質と核酸の混合液を、常温にて十分に攪拌させた後、強酸性陽イオン交換樹脂封入スピンカラムに200μL滴下し、十分に攪拌させた。その後、12000Gにて2分間遠心し、混合液をスピンダウンした。スピンダウンした液をNanoDrop D-1000 (Thermo Fisher Scientific Inc. 製)にて吸光度測定し、強酸性陽イオン交換樹脂処理前後の吸光度の違いから、核酸精製能を評価した。BSAの吸光度はProtein A280モードで評価し、核酸の吸光度はmicro arrayモードでCy3の吸光度で評価した。さらに、生体環境下に近づけるため、上記タンパク質−核酸混合溶液に最終濃度が0.09質量%となるようにNaCl (和光純薬工業(株)製)を添加し、同様に強酸性イオン交換樹脂処理をおこなった。
【0079】
評価結果を図5に示す。図5に示すように、強酸性陽イオン交換樹脂処理前後のCy3 オリゴ DNAおよびBSA濃度より、核酸の回収率はNaClを含む場合で約100%、NaClを含まない場合で約72%であった。一方、BSAの回収率はNaClを含む場合で約20%、NaClを含まない場合で約14%であった。このことから、強酸性陽イオン交換樹脂処理をすることにより、タンパク質−核酸の混在系においても、DNA存在比を向上させられることを確認できた。
【0080】
また、イオン交換樹脂により夾雑物の吸着処理をしたサンプル、及び当該吸着処理をしていないサンプルの夫々のLAMP反応の結果を図6に示す。LAMP反応については、まず、サンプル、酵素、蛍光色素、核酸モノマー、バッファー、ターゲット核酸鎖増幅用のプライマーセット、リアルタイム測定用のプローブを混合し、LAMP反応液を調製した。各濃度はLoopamp DNA 増幅試薬キット(栄研化学(株)製)およびLoopamp RNA 増幅試薬キット(栄研化学(株)製)に準じて実施した。反応温度は63℃で実施した。LAMP反応は、リアルタイム計測が可能なサーマルサイクラーChromo4(バイオラッド製、米国)を用いて測定を行った。1Cycle を 1min と設定した。プローブは、消光プローブのQPプローブ(J-bio21, 日本: http://www.j-bio21.co.jp/tech/qpmethod.htm([平成23年7月19日]、題目:QP法))を使用しているので、核酸の増幅と共に蛍光強度の低下が観察される。図6に示すように、イオン交換樹脂により夾雑物の吸着処理をしたサンプルでは、核酸増幅反応を検出することができた。なお、以下に記載のLAMP反応は上記と同様の条件で行った。
【0081】
2.陽イオン交換樹脂の対イオンについての検討
<試験例2>
スピンフィルタカラム(Ultrafree-MC,0.45μm)に陽イオン交換樹脂(Nuvia S Na+型(Bio Rad社製)) を100mg計量して入れた。また、スピンフィルタカラム(Ultrafree-MC,0.45μm)に上記陽イオン交換樹脂(Nuvia S Na+型)を1M HCl溶液に通し、中性になるまで純水で洗浄することで調製した陽イオン交換樹脂(NuviaS H+型)を100mg計量して入れた。次いで、50mM MESバッファー(pH5)を調製し、BSAを0.5質量%となるように加え、Cy3修飾した20merオリゴDNAを4μMとなるように加え、さらにNaClを0.09質量%となるように加えた。このサンプルを、常温にて十分に攪拌させた後、Na+型又はH+型である強酸性陽イオン交換樹脂封入スピンカラムに夫々200μL滴下し、十分に攪拌させた。その後、12000Gにて2分間遠心し、混合液をスピンダウンした。夫々の強酸性陽イオン交換樹脂による核酸精製能については、試験例1と同様にして評価した。Na+の濃度についてはHoriba Cardy Sodium Compact Ion Meter (C-122) ((株)堀場製作所製)により測定した。また、pHについては、ポケットpH計S2K712 (ISFETCOM(株)製)により測定した。評価結果を図7に示す。また、評価結果をまとめたものを表1に示す。
【0082】
【表1】
【0083】
Na+型の陽イオン交換樹脂を用いた場合には、H+型の陽イオン交換樹脂を用いた場合に比べ、核酸の回収率が高く、核酸の精製率も高いことが分かった。一方で、Na+型の陽イオン交換樹脂を用いた場合には、H+型の陽イオン交換樹脂を用いた場合に比べ、Na+の濃度が高く、脱塩しづらいことが分かった。
【0084】
H+型の陽イオン交換樹脂を用いた場合には、Na+型の陽イオン交換樹脂を用いた場合に比べ、吸着処理後のサンプルのpHが吸着処理前のpHに比べ低下する(酸化する)ことが分かった。これは、H+型の陽イオン交換樹脂を用いた場合、Na+型の陽イオン交換樹脂を用いた場合に比べ、核酸の負電荷が弱められ、核酸が陽イオン交換樹脂のH+上に非特異的に保持されやすくなり、核酸の回収率が低くなったものと考えられる。
【0085】
3.塩濃度についての検討
<試験例3>
本試験例では、陽イオン交換樹脂によりサンプル中の夾雑物を吸着する際におけるサンプル中の塩濃度の影響について検討した。
【0086】
スピンフィルタカラム(Ultrafree-MC,0.45μm)に強酸性陽イオン交換樹脂(Nuvia S Na+型(Bio Rad社製))を100mg計量して入れた。次いで、50 mM MESバッファー(pH5)を調製し、BSAを0.5質量%となるように加え、Cy3修飾した20merオリゴDNAを4μMとなるように加え、更にNaClを3.3、0.9、又は0.33mg/mLとなるように加え、3種類のサンプルを用意した。また、NaClを添加していない以外は、上記3種類のサンプルと同様の条件で作製したサンプルも用意した。これらのサンプルを、常温にて十分に攪拌させた後、Na+型又はH+型である強酸性陽イオン交換樹脂封入スピンカラムに夫々200μL滴下し、十分に攪拌させた。その後、12000Gにて2分間遠心し、混合液をスピンダウンした。夫々のサンプルの核酸精製能については、試験例1、2と同様にして評価した。評価結果をまとめたものを表2に示す。
【0087】
【表2】
【0088】
表2に示すように、NaClの濃度が0.33、0.9、3.3mg/mLの場合は、上記濃度が0mg/mLの場合に比べ、核酸の回収率が高くなることが分かった。また、NaClの濃度が0.33、0.9、3.3mg/mLの場合では、核酸回収率はいずれも100%であった。この結果より、血液等のように、内部に塩が存在するサンプルを用いて核酸精製を行う場合には、精製時にサンプルに塩を添加する必要はないものと考えられる。一方、菌等のように、内部に塩が存在しないサンプルを用いて核酸精製を行う場合には、精製時にサンプルに塩を添加することが好ましいものと考えられる。
【0089】
4.イオン交換樹脂種による脱塩に対する影響
以下、実施例1〜3、及び比較例1、2でイオン交換樹脂種による脱塩に対する影響について検討した。
【0090】
<実施例1>
スピンフィルタカラム(Ultrafree-MC,0.45μm)に強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを計量して入れた。吸着処理前のNa+の濃度は、0.45mg/mLであり、pHは4.9であった。次いで、50 mM MESバッファー(pH5)を調製し、ウシ全血を1/10倍に希釈するように加え、サンプルを調製した。このサンプルを、常温にて十分に攪拌させた後、上記イオン交換樹脂封入スピンカラムに200μL滴下し、十分に攪拌させた。その後、12000Gにて2分間遠心し、混合液をスピンダウンした。その後、サンプル中のNa+の濃度及びpHを測定した。
【0091】
<実施例2>
実施例1と比し、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgの代わりに、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いた以外は、実施例1と同様にして評価した。
【0092】
<実施例3>
実施例2と比し、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いる代わりに、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):70mg及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いた以外は、実施例2と同様にして評価した。
【0093】
<比較例1>
実施例1と比し、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いる代わりに、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mgのみを用いた以外は、実施例1と同様にして評価した。
【0094】
<比較例2>
実施例1と比し、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いる代わりに、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mgのみを用いた以外は、実施例1と同様にして評価した。
【0095】
実施例1〜3、及び比較例1、2における評価結果をまとめたものを表3に示す。
【0096】
【表3】
【0097】
実施例1、実施例2、及び実施例3では、H+型の強酸性陽イオン交換樹脂とOH−型の強塩基性陰イオン交換樹脂とを混合したイオン交換樹脂を用いることにより、比較例1及び比較例2に比し、サンプルをより精度良く脱塩できた。また、実施例1、実施例2、及び実施例3では、比較例2に比し、サンプルのpH変化をより精度良く抑制できた。
【0098】
また、実施例2及び実施例3に示すように、強酸性陽イオン交換樹脂と強塩基性陰イオン交換樹脂との用量を等量としても、又は等イオン交換容量としても、脱塩効果及びpH変化の抑制を実現できることが示唆された。
【0099】
5.脱塩によるLAMP反応及びRT-LAMP反応に対する影響
以下、実施例4、5、及び比較例3〜5で脱塩処理によるLAMP反応及びRT-LAMP反応に対する影響について検討した。
【0100】
<実施例4>
スピンフィルタカラム(Ultrafree-MC,0.45μm)に強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを計量して入れた。次いで、50 mM MESバッファー(pH5)を調製し、超音波破砕したビフィズス菌を1000copy/uLとなるように加え、更にNaClを0.9mg/mLとなるように加えた。得られたサンプルを、常温にて十分に攪拌させた後、Na+型又はH+型である強酸性陽イオン交換樹脂封入スピンカラムに夫々200μL滴下し、十分に攪拌させた。その後、12000Gにて2分間遠心し、混合液をスピンダウンした。LAMP反応及びRT−LAMP反応については、試験例1と同様の方法にして行った。
【0101】
<実施例5>
実施例4と比し、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgの代わりに、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを用いた以外は、実施例4と同様にして評価した。
【0102】
<比較例3>
実施例4と比し、サンプルとしてNaClを添加していないものを用い、サンプルをイオン交換樹脂封入スピンカラムに滴下していないという点以外は、実施例4と同様にして評価した。
【0103】
<比較例4>
実施例4と比し、サンプルをイオン交換樹脂封入スピンカラムに滴下していないという点以外は、実施例4と同様にして評価した。
【0104】
<比較例5>
実施例4と比し、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgの代わりに、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mgを用いた以外は、実施例4と同様にして評価した。
【0105】
実施例4、5、及び比較例3〜5における評価結果をまとめたものを図8及び図9に示す。実施例4、実施例5、及び比較例4から、イオン交換樹脂によりサンプルの脱塩を行うことで、LAMP反応及びRT-LAMP反応におけるTt値が小さくなることが分かった。実施例4、実施例5、及び比較例4から、イオン交換樹脂によりサンプルの脱塩を行うことで、LAMP反応の進行が良好になることが示唆された。また、実施例4及び実施例5から、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製))をイオン交換樹脂として用いることによるLAMP反応への影響は小さいことが分かった。これは、強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製))をイオン交換樹脂として用いても、サンプルの脱塩に対する影響は小さいことによるものと考えられる。また、比較例4及び比較例5では、LAMP反応の進行を良好にしづらいことが分かった。LAMP反応の進行を良好にするには、サンプルの脱塩を十分に行う必要があることが示唆された。
【0106】
6.添加剤についての検討
以下、実施例6及び実施例7で添加剤(Brij35及びEDTA)の影響について検討した。
<実施例6>
スピンフィルタカラム(Ultrafree-MC,0.45μm)に強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを計量して入れた。次いで、50 mM MESバッファー(pH5)となるように調製し、ウシ全血を10体積%となるように加え、超音波破砕したビフィズス菌を1000copy/uLとなるように加え、更にBrij35を0.5体積%となるように加え、EDTAを1mMとなるように加えた。得られた核酸を含有するサンプルについて、実施例4と同様にし、LAMP反応及びRT-LAMP反応を行い、評価した。
【0107】
<実施例7>
実施例6に比し、Brij35及びEDTAをサンプルに添加していないという点以外は、実施例6と同様にして評価を行った。
【0108】
実施例6及び実施例7における評価結果を図10及び図11に示す。また、評価結果をまとめたものを表4に示す。
【0109】
【表4】
【0110】
実施例6では、イオン交換樹脂によりサンプルの夾雑物の吸着処理を行うことで、LAMP反応及びRT-LAMP反応のいずれにおいても、吸着処理前に比べTt値が小さくなることが分かった。一方、実施例7では、イオン交換樹脂によりサンプルの夾雑物の吸着処理を行うことで、LAMP反応及びRT-LAMP反応のいずれにおいても、吸着処理前に比べTt値が大きくなることが分かった。これは、実施例6では、実施例7に比べ、サンプルにBrij35及び1mM EDTAを添加したことで、核酸の回収率が高くなったことによるものと考えられる。
【0111】
7.界面活性剤/親水性高分子についての検討
以下、実施例8〜11で、界面活性剤/親水性高分子の影響について検討した。
<実施例8>
スピンフィルタカラム(Ultrafree-MC,0.45μm)に強酸性陽イオン交換樹脂(Nuvia S (Na+型)(Bio Rad社製)):100mg、強酸性陽イオン交換樹脂(AG1-X8 (H+型)(Bio Rad社製)):50mg、及び強塩基性陰イオン交換樹脂(AG50W-X8 (OH-型) (Bio Rad社製)):50mgを計量して入れた。次いで、50 mM MESバッファー(pH5)となるように調製し、ウシ全血を10体積%となるように加え、超音波破砕したビフィズス菌を1000copy/uLとなるように加え、更にBrij35を0.5体積%となるように加えた。実施例7では、サンプルに非イオン界面活性剤又は非イオン性親水性高分子を加えなかった。得られた核酸を含有するサンプルについて、試験例1と同様に、核酸回収率を評価した。
【0112】
<実施例9>
実施例8に比し、イオン交換樹脂による吸着処理の前のサンプルに非イオン性界面活性剤としてBrij35を0.5体積%となるように添加したという点以外は、実施例8と同様にして評価を行った。
【0113】
<実施例10>
実施例9に比し、非イオン性界面活性剤として、Brij35の代わりに、Tween20を用いたという点以外は、実施例9と同様にして評価を行った。
【0114】
<実施例11>
実施例9に比し、Brij35の代わりに、非イオン性親水性高分子であるPEG20000を用いたという点以外は、実施例9と同様にして評価を行った。
【0115】
実施例8〜11における評価結果を表5に示す。
【0116】
【表5】
【0117】
実施例7に比べ、実施例8〜10では核酸の回収率がいずれも高く、非イオン界面活性剤(Brij35、Tween20)又は非イオン性親水性高分子(PEG20000)をサンプルに添加してサンプルの吸着処理を行うことで、核酸の回収率が上がることが実証された。
【0118】
8.本技術に係る吸着処理とゼオライトによる吸着処理との比較
以下、実施例12及び比較例6で本技術に係る吸着処理とゼオライトによる吸着処理との比較実験を行った。
<実施例12>
実施例7で用いたサンプルと同様にして調製したサンプルを準備した。同様の組成の3サンプル夫々について3回ずつ、実施例7と同様にし、LAMP反応及びRT-LAMP反応を行い、評価した。
【0119】
<比較例6>
50 mM MESバッファー(pH5)を調製し、ウシ全血を5体積%加え、超音波破砕したビフィズス菌を1000copy/uLとなるように加え、サンプルを調製した。次いで、サンプルを熱アルカリ処理し、ゼオライトで夾雑物の吸着処理を行った。得られた同様の組成の3サンプル夫々について3回ずつ、実施例4と同様にし、LAMP反応及びRT-LAMP反応を行い、評価した。
【0120】
実施例12及び比較例6における評価結果を図12(LAMP反応)及び図13(RT-LAMP反応)に示す。実施例12及び比較例6から、ゼオライトでサンプルの夾雑物の吸着処理を行った場合よりも、イオン交換樹脂でサンプルの夾雑物の吸着処理を行った場合の方が、核酸増幅反応の進行を良好なものにすることができた。
【産業上の利用可能性】
【0121】
本技術に係る核酸精製方法は、操作が簡便であり、短時間で高効率に核酸を抽出することができる。従って、PCR(Polymerase Chain Reaction)法やLAMP(Loop-Mediated Isothermal Amplification)法等の核酸増幅反応のための核酸精製処理に適用され、核酸が微量あるいは極低濃度でしか含まれないサンプル中の核酸を精製するために用いられ得る。
【符号の説明】
【0122】
1、101:核酸精製用キット、3:核酸精製器具、10:イオン交換樹脂、20:第1の陽イオン交換樹脂、30:陰イオン交換樹脂、40:第2の陽イオン交換樹脂
【特許請求の範囲】
【請求項1】
核酸を含有するサンプルに含まれる物質をイオン交換樹脂により吸着する手順を含み、
前記イオン交換樹脂には、陽イオン交換樹脂及び陰イオン交換樹脂を用いる核酸精製方法。
【請求項2】
前記陽イオン交換樹脂には、第1の陽イオン交換樹脂と、当該第1の陽イオン交換樹脂よりも排除限界分子量が低い第2の陽イオン交換樹脂とを用いる、請求項1記載の核酸精製方法。
【請求項3】
前記第1の陽イオン交換樹脂により前記物質を吸着してから、前記陰イオン交換樹脂と第2の陽イオン交換樹脂とにより前記物質を吸着する、請求項2記載の核酸精製方法。
【請求項4】
上層に前記第1の陽イオン交換樹脂を有し、下層に前記陰イオン交換樹脂と前記第2の陽イオン交換樹脂とを有するカラムの前記上層側から前記サンプルを流入する、請求項3記載の核酸精製方法。
【請求項5】
前記手順は、緩衝液で希釈された前記サンプルに含まれる物質を前記イオン交換樹脂により吸着する手順であり、
前記緩衝液のpHは4.0〜8.0である、請求項4記載の核酸精製方法。
【請求項6】
前記陽イオン交換樹脂は、強酸性陽イオン交換樹脂である、請求項5記載の核酸精製方法。
【請求項7】
前記第1の陽イオン交換樹脂の対イオンは、Na+である、請求項6記載の核酸精製方法。
【請求項8】
前記第2の陽イオン交換樹脂の対イオンは、H+である、請求項7記載の核酸精製方法。
【請求項9】
前記陰イオン交換樹脂は、強塩基性陰イオン交換樹脂である、請求項8記載の核酸精製方法。
【請求項10】
前記陰イオン交換樹脂の対イオンは、OH−である、請求項9記載の核酸精製方法。
【請求項11】
前記第2の陽イオン交換樹脂に対する前記陰イオン交換樹脂のイオン交換容量の割合は、50%〜150%である、請求項10記載の核酸精製方法。
【請求項12】
非イオン性界面活性剤及び/又は非イオン性親水性高分子化合物を前記サンプルに混合して前記物質を吸着する、請求項11記載の核酸精製方法。
【請求項13】
前記物質は、少なくともタンパク質と金属塩とを含有する夾雑物である、請求項12記載の核酸精製方法。
【請求項14】
核酸を含有するサンプルの超音波処理をする手順、
陽イオン交換樹脂及び陰イオン交換樹脂により前記サンプルに含まれる物質を吸着する手順、及び
電気泳動により移動する前記核酸を堰き止めることで前記核酸を濃縮する手順の各手順を行う核酸抽出方法。
【請求項15】
アニオン性官能基を有するインターカレーターを挿入した前記核酸について前記電気泳動を行う、請求項14記載の核酸抽出方法。
【請求項16】
前記サンプルと、前記サンプル中に含まれる物質が含有するカルボキシル基と脱水縮合反応をする官能基を有する化合物と、該脱水縮合反応の縮合剤と、を混合し、前記核酸について前記電気泳動を行う、請求項15記載の核酸抽出方法。
【請求項17】
陽イオン交換樹脂と、
陰イオン交換樹脂と、
前記陽イオン交換樹脂及び前記陰イオン交換樹脂を内部に保持し、核酸を含有するサンプルを流通可能な核酸精製器具と、を含む核酸精製用キット。
【請求項1】
核酸を含有するサンプルに含まれる物質をイオン交換樹脂により吸着する手順を含み、
前記イオン交換樹脂には、陽イオン交換樹脂及び陰イオン交換樹脂を用いる核酸精製方法。
【請求項2】
前記陽イオン交換樹脂には、第1の陽イオン交換樹脂と、当該第1の陽イオン交換樹脂よりも排除限界分子量が低い第2の陽イオン交換樹脂とを用いる、請求項1記載の核酸精製方法。
【請求項3】
前記第1の陽イオン交換樹脂により前記物質を吸着してから、前記陰イオン交換樹脂と第2の陽イオン交換樹脂とにより前記物質を吸着する、請求項2記載の核酸精製方法。
【請求項4】
上層に前記第1の陽イオン交換樹脂を有し、下層に前記陰イオン交換樹脂と前記第2の陽イオン交換樹脂とを有するカラムの前記上層側から前記サンプルを流入する、請求項3記載の核酸精製方法。
【請求項5】
前記手順は、緩衝液で希釈された前記サンプルに含まれる物質を前記イオン交換樹脂により吸着する手順であり、
前記緩衝液のpHは4.0〜8.0である、請求項4記載の核酸精製方法。
【請求項6】
前記陽イオン交換樹脂は、強酸性陽イオン交換樹脂である、請求項5記載の核酸精製方法。
【請求項7】
前記第1の陽イオン交換樹脂の対イオンは、Na+である、請求項6記載の核酸精製方法。
【請求項8】
前記第2の陽イオン交換樹脂の対イオンは、H+である、請求項7記載の核酸精製方法。
【請求項9】
前記陰イオン交換樹脂は、強塩基性陰イオン交換樹脂である、請求項8記載の核酸精製方法。
【請求項10】
前記陰イオン交換樹脂の対イオンは、OH−である、請求項9記載の核酸精製方法。
【請求項11】
前記第2の陽イオン交換樹脂に対する前記陰イオン交換樹脂のイオン交換容量の割合は、50%〜150%である、請求項10記載の核酸精製方法。
【請求項12】
非イオン性界面活性剤及び/又は非イオン性親水性高分子化合物を前記サンプルに混合して前記物質を吸着する、請求項11記載の核酸精製方法。
【請求項13】
前記物質は、少なくともタンパク質と金属塩とを含有する夾雑物である、請求項12記載の核酸精製方法。
【請求項14】
核酸を含有するサンプルの超音波処理をする手順、
陽イオン交換樹脂及び陰イオン交換樹脂により前記サンプルに含まれる物質を吸着する手順、及び
電気泳動により移動する前記核酸を堰き止めることで前記核酸を濃縮する手順の各手順を行う核酸抽出方法。
【請求項15】
アニオン性官能基を有するインターカレーターを挿入した前記核酸について前記電気泳動を行う、請求項14記載の核酸抽出方法。
【請求項16】
前記サンプルと、前記サンプル中に含まれる物質が含有するカルボキシル基と脱水縮合反応をする官能基を有する化合物と、該脱水縮合反応の縮合剤と、を混合し、前記核酸について前記電気泳動を行う、請求項15記載の核酸抽出方法。
【請求項17】
陽イオン交換樹脂と、
陰イオン交換樹脂と、
前記陽イオン交換樹脂及び前記陰イオン交換樹脂を内部に保持し、核酸を含有するサンプルを流通可能な核酸精製器具と、を含む核酸精製用キット。
【図2】

【図3】
【図4】

【図1】
【図5】
【図6】
【図7】
【図8】

【図9】
【図10】
【図11】

【図12】
【図13】
【図3】
【図4】
【図1】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公開番号】特開2013−59275(P2013−59275A)
【公開日】平成25年4月4日(2013.4.4)
【国際特許分類】
【出願番号】特願2011−198949(P2011−198949)
【出願日】平成23年9月13日(2011.9.13)
【出願人】(000002185)ソニー株式会社 (34,172)
【Fターム(参考)】
【公開日】平成25年4月4日(2013.4.4)
【国際特許分類】
【出願日】平成23年9月13日(2011.9.13)
【出願人】(000002185)ソニー株式会社 (34,172)
【Fターム(参考)】
[ Back to top ]