
植物でのタンパク質の製造
対象のタンパク質を植物又は植物の部分で合成する方法が提供される。前記方法は、対象のタンパク質をコードする1つ以上の核酸配列であって、植物中で活性な光合成遺伝子から得られる調節領域と機能的に連結されたものを一過性態様で導入する工程を含む。前記植物を続いて、対象のタンパク質をコードする前記核酸配列の前記植物又は植物の部分での発現を可能にする条件下で維持する。前記植物は、1つ以上の核酸配列を導入する前にプルーニングすることができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、植物でタンパク質を製造する方法に関する。本発明はまた、植物でタンパク質を製造するために用い得るヌクレオチド配列を提供する。
【背景技術】
【0002】
免疫グロブリン(IgG)は、多様な性質を有するその特異的抗原対応物に対して特徴的な親和性を有する複雑なヘテロマルチマータンパク質である。今日、IgG産生細胞株の日常的な単離、並びにIgG誘導進化及び分子操作のための技術の出現は、生物性治療薬として及び一般ライフサイエンス市場におけるIgGの展開に重大な影響を及ぼした。治療用モノクローナルIgG(モノクローナル抗体、mAb)は、現在の新規な抗炎症薬及び抗癌薬の市場で優勢であり、さらに何百もの新規な候補物質が、改善された適用又は新規な適用のために現時点で研究及び臨床開発下にある。mAbの市場における年間需要は、数グラム(診断薬)、数キログラム(抗毒素)から百又は数百キログラム(生物防衛、抗癌、抗感染、抗炎症)の範囲である。
CHO細胞株は、今もなお好ましいmAbの産業規模での生成宿主であるが、mAbがライフサイエンス市場でそれらの完璧な効果に達するためには、また別の生産系を開発する必要があることは一般的に理解されている。なぜならば、これらの培養に必要な施設は規模の調節が容易ではなく、それらの構築及び維持コストは極めて高価で、さらに着実に増加しつつあり、GMP基準下でのそれらの実証には構築後なお平均3年を要するからである。初期開発期においてすら、許容可能な収量及び生産性を有するCHO細胞株の選択が、コストのかかる長期に及ぶプロセスであることに変わりはない。上流でのコストを削減させえる新規な生産系(より高収量、より単純な技術及び基盤組織)がリードタイムを短縮し、能力をより柔軟にし、一方で従来の細胞培養系における従来の再現性、品質及び安全性特性を充足させる新規な生産系は、ライフサイエンス市場用のmAb及びワクチンの開発に対し全開発ステージでおそらく大きな影響をもつであろう。
植物は、mAb及びライフサイエンスでこれまで利用されてきたいくつかの他のタンパク質の製造に適した宿主である(最近の概説として以下を参照されたい:Ko and Koprowski 2005;Ma et al. 2005;Yusibov et al. 2006)。mAbは、安定なトランスジェニック植物株で新鮮質量(FW)1kg当たり200mgもの収量で、さらに一過性発現では20mg/kgFWもの割合で生産されている(Kathuria, 2002)。Giritchら(2006)は、IgGについて、葉の質量1kgにつき200−300mgの発現レベルを報告し、多重ウイルス系の一過性発現系を用いた場合の引用最大例は500mg/kgであった。
mAbの合成のために今日まで報告された(例えばKapila et al. 1997;Vaquero et al. 1999;Rodriguez et al. 2004)一過性の系の多くは複雑な方法を含むか、生成物の蓄積レベルが低いか、またはその両方をもたらす。高収量のタンパク質を生じる代替方法を述べる。
【発明の概要】
【発明が解決しようとする課題】
【0003】
本発明は、植物でタンパク質を製造する方法に関する。本発明はまた、植物でタンパク質を製造するために用い得るヌクレオチド配列を提供する。
本発明の目的は、植物でタンパク質を製造するための改善方法を提供することである。
【課題を解決するための手段】
【0004】
本発明は、以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法(A)を提供する:
i)植物又は植物の部分をプルーニングし、プルーニングされた植物又は植物の部分を生産する工程、
ii)植物中で活性な調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を、前記プルーニングされた植物又は植物の部分に一過性態様で導入する工程、及び
iii)前記プルーニングされた植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分中で発現させることを可能にする条件下で維持する工程。
【0005】
対象のタンパク質は抗体、抗原ワクチン又は酵素でありえる。
本発明はまた上記に記載の方法に関し、この場合、導入工程(工程ii)では、2つ以上の核酸配列を植物内に導入し得る。さらにまたこの2つ以上の核酸配列の1つはサイレンシングのサプレッサーをコードし得る。例えば、サイレンシングのサプレッサーは、HcPro、TEV-p1/HC-Pro、BYV-p21、TBSV p19、TCV-CP、CMV-2b、PVX-p25、PVM-p11、PVS-p11、BScV-p16、CTV-p23、GLRaV-2p24、GBV-p14、HLV-p10、GCLV-p16、又はGVA-p10でありえる。
本発明は上記に記載の方法を含み、この場合、導入工程(工程ii)では、1つ以上の核酸配列は、プルーニングした植物又は植物の部分にアグロバクテリア菌を用いて導入し得る。アグロバクテリア菌は、プルーニングした植物又は植物の部分に真空下又は注射筒によって導入し得る。さらにまた、上記に記載した導入工程(工程ii)では、調節領域は光合成遺伝子から得られるプロモーターを含む。例えば、調節領域は、プラストシアニンプロモーター、プラストシアニン3'UTR転写終了配列、又はプラストシアニンプロモーター及びプラストシアニン3'UTR転写終了配列の両方を含み得る。
【0006】
本発明はまた、以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法(B)に関する:
i)植物又は植物の部分中で活性な光合成遺伝子から得られる調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を一過性態様で導入する工程、及び
ii)前記植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分中で発現させることを可能にする条件下で維持する工程。
対象のタンパク質は抗体、抗原ワクチン又は酵素でありえる。
本発明はまた上記に記載の方法(B)に関し、この場合、導入工程(工程i)では、2つ以上の核酸配列が植物に導入される。さらにまたこの2つ以上の核酸配列の1つはサイレンシングのサプレッサーをコードし得る。例えば、サイレンシングのサプレッサーは、HcPro、TEV-p1/HC-Pro、BYV-p21、TBSV p19、TCV-CP、CMV-2b、PVX-p25、PVM-p11、PVS-p11、BScV-p16、CTV-p23、GLRaV-2p24、GBV-p14、HLV-p10、GCLV-p16、又はGVA-p10でありえる。
本発明は上記に記載の方法(B)を含み、この場合、導入工程(工程i)では、1つ以上の核酸配列は、プルーニングした植物又は植物の部分にアグロバクテリア菌を用いて導入し得る。アグロバクテリア菌は、プルーニングした植物又は植物の部分に真空下又は注射筒によって導入し得る。さらにまた、上記に記載した導入工程(工程ii)では、調節領域は光合成遺伝子から得られるプロモーターを含む。例えば、調節領域は、プラストシアニンプロモーター、プラストシアニン3'UTR転写終了配列、又はプラストシアニンプロモーター及びプラストシアニン3'UTR転写終了配列の両方を含み得る。
【0007】
本発明は、以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法(方法C)を提供する:
i)植物又は植物の部分をプルーニングし、プルーニングされた植物又は植物の部分を生産する工程、
ii)植物又は植物の部分中で活性な光合成遺伝子から得られる調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を一過性態様で導入する工程、及び
iii)前記プルーニングされた植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分中で発現させることを可能にする条件下で維持する工程。
【0008】
対象のタンパク質は抗体、抗原ワクチン又は酵素でありえる。
本発明はまた上記に記載の方法(C)に関し、この場合、導入工程(工程ii)では、2つ以上の核酸配列が植物に導入される。さらにまたこの2つ以上の核酸配列の1つはサイレンシングのサプレッサーをコードし得る。例えば、サイレンシングのサプレッサーは、HcPro、TEV-p1/HC-Pro、BYV-p21、TBSV p19、TCV-CP、CMV-2b、PVX-p25、PVM-p11、PVS-p11、BScV-p16、CTV-p23、GLRaV-2p24、GBV-p14、HLV-p10、GCLV-p16、又はGVA-p10でありえる。
本発明は上記に記載の方法(C)を含み、この場合、導入工程(工程ii)では、1つ以上の核酸配列は、プルーニングした植物又は植物の部分にアグロバクテリア菌を用いて導入し得る。アグロバクテリア菌は、プルーニングした植物又は植物の部分に真空下又は注射筒によって導入し得る。さらにまた、上記に記載した導入工程(工程ii)では、調節領域は光合成遺伝子から得られるプロモーターを含む。例えば、調節領域は、プラストシアニンプロモーター、プラストシアニン3'UTR転写終了配列、又はプラストシアニンプロモーター及びプラストシアニン3'UTR転写終了配列の両方を含み得る。
本発明は、一過性発現系を用いて植物で対象のタンパク質の発現を駆動させる、単純化した植物発現系を提供する。本明細書に記載の方法にしたがえば、対象のタンパク質を高収量で生産し得る。本明細書に記載の一過性同時発現系は、極めて長い製造時間、並びに従来技術(例えばBakker, 2005)で記載されたえり抜きの変異体又は糖操作トランスジェニック株の選別プロセス及びそれらの親株としてのその後の使用を回避する。本発現系はまた、変異体又は糖操作植物でしばしば遭遇し、同時に発生する生産性、花粉生産、種子セット(Bakker et al. 2005)及び生存率(Boisson et al. 2005)に関する問題を回避する。本明細書に記載の一過性発現系は、新鮮な葉の質量1kg当たり高品質の抗体1.5gに達する発現レベルをもたらし、他の発現系(多重ウイルスベースの系及びトランスジェニック植物を含む)を有する植物で任意の抗体に関して報告された蓄積レベルを超える。
【0009】
本明細書に記載するように、所望の核酸構築物の浸透(infiltration)前に植物をプルーニングすることによって発現レベル(全合成タンパク質の%として)及び収量(mgタンパク質/kg新鮮質量)の増加が観察された。前記は、注射筒による浸透(syringe-infiltration)又は真空浸透(vacuum-infiltration)を含む(ただしこれらに限定されない)いくつかの浸透方法を用いて観察された。多様なプルーニング方法(例えば機械的プルーニング又は化学的プルーニングを含むが、ただしこれらに限定されない)が、発現レベル及びタンパク質収量を増加させた。
光合成遺伝子由来の調節領域(例えばリブロース1,5-ビスホスフェートカルボキシラーゼ/オキシゲナーゼ(Rubisco)大若しくは小サブユニット又はプラストシアニンをコードする遺伝子から得られるものであるが、ただしこれらに限定されない)の使用、又は光合成遺伝子由来調節領域の使用は、発現レベル及び収量を増加させることが見出された。さらにまた、プルーニングと一緒に光合成遺伝子由来調節領域を使用することによって、発現レベル及び収量が増加することが見出された。
浸透技術は、小さなパイロットユニット内で一日当たりこの抗体のグラム量の生産を可能にする。前記パイロットユニットは、極めて短時間枠内での臨床試験用材料の生産及び年間でキログラムに達するマーケットサイズを有するライセンス製品の供給のために前出のような一過性発現系を使用することを可能にする。高品質の抗体が、ただ1回のアフィニティークロマトグラフィー工程後に浸透リーフから得られた。
本発明の上記要旨は必ずしも本発明のすべての特徴を記述しているわけではない。
本発明のこれらの特徴及び他の特徴は、添付の下記図面に言及する以下の記述からいっそう明白となるであろう。
【図面の簡単な説明】
【0010】
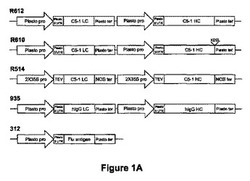
【図1A】図1Aはいくつかのタンパク質の発現のために組み立てた発現カセットの例を示す。R612は、C5-1 LC及びC5-1 HC(各々はプラストシアニンプロモーター及び5'UTR並びにプラストシアニンターミネーターの制御下にある)をコードするヌクレオチド配列を含む。R610は、C5-1 LC及びC5-1 HC-KDEL(各々はプラストシアニンプロモーター及び5'UTR並びにプラストシアニンターミネーターの制御下にある)をコードするヌクレオチド配列を含む。R514は、C5-1 LC及びC5-1 HCをコードするヌクレオチド配列を含む。C5-1 LC:C5-1軽鎖コード配列;C5-1 HC:C5-1重鎖コード配列(各々は2X35Sプロモーター、タバコエッチウイルス(TEV)リーダー配列及びNOSターミネーターの制御下にある)。935は、ヒトIgG-LC及びヒトIgG-HC(各々はプラストシアニンプロモーター及び5'UTR並びにプラストシアニンターミネーターの制御下にある)をコードするヌクレオチド配列を含む。312はfli抗原をコードするヌクレオチド配列を含み、前記はプラストシアニンプロモーター及び5'UTR並びにプラストシアニンターミネーターの制御下にある。
【図1B】図1Bはプラストシアニンプロモーター及び5'UTRのヌクレオチド配列(配列番号:19)を示し、転写開始部位は太字で示され、翻訳開始コドンは下線を付されている。
【図1C】図1Cはプラストシアニン3'UTR及びターミネーターのヌクレオチド配列(配列番号:20)を示し、停止コドンは下線を付されている。
【図1D】図1Dは、R512及びR513アッセンブリに用いられる中継プラスミドの2X35S(配列番号:33)及びNOS(配列番号:34)配列を示す。NOSターミネーター(配列番号:34)はイタリック体で示され、2X35S(配列番号:33)は太字で示されている。制限酵素部位は下線を付されている。
【図2A】図2は、種々の発現カセットを浸透させたニコチアナ・ベンタミアナ(Nicotiana benthamiana)の葉におけるC5-1抗体の蓄積を示す。図2Aは、サイレンシングサプレッサー(例えばHcPro)の存在下又は非存在下での、R514(35Sベースの発現カセット)、R610及びR612(プラストシアニンベースの発現カセット)の注射筒による浸透後に発現されたC5-1抗体の蓄積を示す。
【図2B】図2Bは、サイレンシングサプレッサー(例えばHcPro)を同時発現させ、又は同時発現させずに、R610及びR612(プラストシアニンベースの発現カセット)を用いた、真空浸透又は注射筒浸透葉におけるC5-1抗体の蓄積を示す。提示した値は、3つの植物(注射筒)における6測定値、又は約12の植物(250g)の個々の浸透バッチにおける6測定値から得た平均蓄積レベル及び標準偏差に該当する。
【図3A】図3は、注射筒及び真空浸透させた植物の抽出物における蓄積C5-1のタンパク質ブロット分析を示す。図3Aは、ペルオキシダーゼ結合ヤギ抗マウスIgG(H+L)を、R612(分泌用、レーン1)又はR610(ER保持用、レーン2)を浸透させた植物の抽出物に対して用いたイムノブロッティングを示す。C1:100ngの市販ネズミIgG1(Sigma M9269)、電気泳動の移動度のためのコントロールとしてローディング;C2:擬似浸透バイオマス(空ベクター)から抽出した12μgの全タンパク質;C3:擬似浸透バイオマス(空ベクター)から抽出した12μgの全タンパク質に加えられた100ngの市販ネズミIgG1(Sigma M9269)。
【図3B】図3Bは、ペルオキシダーゼ結合ヒトIgG1を、R612(分泌用、レーン1)又はR610(ER保持用、レーン2)を浸透させた植物の抽出物に対して用いた活性イムノブロッティングを示す。C1:ハイブリドーマから精製した2μgのコントロールC5-1(Khoudi et al. 1999);C2:擬似浸透バイオマス(空ベクター)から抽出した75μgの全タンパク質。
【図4A】図4は、R612(分泌用、レーン1)又はR610(ER保持用、レーン2)のどちらかを浸透させた植物から精製した抗体の分析を示す。図4Aは、非還元条件で実施した粗抽出物及び精製抗体のSDS-PAGEを示す。
【図4B】図4Bは、非還元条件で実施した精製抗体のSDS-PAGEを示す。
【図4C】図4Cは、ペルオキシダーゼ結合ヒトIgG1を用いて実施した精製抗体の活性イムノブロッティングを示す。
【図4D】図4Dは、種々の浸透バッチ由来の精製C5-1の6ロットの夾雑物の比較を示す。C:2.5μgの市販ネズミIgG1(Sigma M9269)、電気泳動の移動度のコントロールとしてローディング。
【図5A】図5Aは、ガラクトシルトランスフェラーゼ発現の天然型(R622)及びハイブリッド型(R621)のために組み立てたカセットの例を示す。GNTI-CTS:N-アセチルグルコース-アミニルトランスフェラーゼIのCTSドメイン;GalT-CAT:ヒトβ1,4ガラクトシルトランスフェラーゼの触媒ドメイン;GalT:ヒトβ1,4ガラクトシルトランスフェラーゼ。
【図5B】図5Bは、GalT(UDP-Gal:ベータGlcNacベータ1,4-ガラクトシルトランスフェラーゼポリペプチド1、ベータ-1,4-ガラクトシルトランスフェラーゼI)のヌクレオチド配列(配列番号:14)を示す。ATG開始部位は下線を付され、トランスメンブレンドメインは下線を付されイタリック体であり、太字の配列はヒトベータ1,4GalTの触媒ドメインに該当し、FLAGエピトープはイタリック体である。
【図5C】図5Cは、GalT(UDP-Gal:ベータGlcNacベータ1,4-ガラクトシルトランスフェラーゼポリペプチド1、ベータ-1,4-ガラクトシルトランスフェラーゼI)のアミノ酸配列(配列番号:15)を示す。トランスメンブレンドメインは下線を付されイタリック体であり、太字の配列はヒトベータ1,4GalTの触媒ドメインに該当し、FLAGエピトープはイタリック体である。
【図5D】図5Dは、GNTIGalTのヌクレオチド配列(配列番号:17)を示す。ATG開始部位は下線を付され、トランスメンブレンドメイン(CTS)は下線を付されイタリック体であり、太字の配列はヒトベータ1,4GalTの触媒ドメインに該当し、FLAGエピトープはイタリック体である。
【図5E】図5EはGNTIGalTのアミノ酸配列(配列番号:18)を示す。トランスメンブレンドメイン(CTS)は下線を付されイタリック体であり、太字の配列はヒトベータ1,4GalTの触媒ドメインに該当し、FLAGエピトープはイタリック体である。
【図5F】図5Fは、N-アセチルグルコサミントランスフェラーゼのCTSドメイン(細胞質テール、トランスメンブレンドメイン、幹領域)(GNT1;配列番号:21)を示す。
【図5G】図5GはCTSのアミノ酸(配列番号:22)を示す。
【図6】図6は、C5-1を発現する植物から得られ、タンパク質のための染色を施されたか、又はウェスタン分析に付された抽出物のプロフィルを示す。一番上のパネルはクマシー染色PAGEゲルを示す。上から二番目のパネルは、β1,4ガラクトースと特異的に結合するエリスリナ・クリスタガリ(Erythrina cristagali)アグルチニン(ECA)を用いた親和性検出を示す。上から三番目のパネルは抗α1,3フコース抗体を用いたウェスタンブロット分析を示す。一番下のパネルは抗β1,2キシロース特異的抗体を用いたウェスタンブロット分析を示す。R612:単独発現C5-1;R612+R622:GalTと同時発現させた(同時浸透させた)C5-1;R612+R621:GNT1-GalTと同時発現させたC5-1。
【図7A】図7は、発現に対する機械的又は化学的プルーニングの影響の例を示す。図7Aは、真空アグロバクテリア浸透(agroinfiltrated)植物における抗原発現(インフルエンザ発現;図1、312参照)に対するプルーニング(機械的プルーニング(浸透の12時間前)及び化学的プルーニング(浸透の7日前)の両プルーニング)の影響を示す。
【図7B】図7Bは、真空アグロバクテリア浸透植物における抗体発現(ヒトIgG、図1、935参照)に対する機械的プルーニング(浸透の12時間前)の影響を示す。
【図7C】図7Cは、注射筒アグロバクテリア浸透植物における抗原発現(インフルエンザ、図1,312参照)に対する機械的プルーニングの影響を示す。条件1:コントロール、非プルーニング植物;条件2:機械的プルーニング植物。
【図8】図8は、真空アグロバクテリア浸透植物における抗原蓄積に対する、プルーニング(機械的プルーニング)の日(形質転換前3、2、又は1日前)、又はプルーニング無し(コントロール)の影響の例を示す。
【図9】図9は、真空浸透された植物における抗体発現(ヒトIgG、図1、935)に対する、サイレンシングのサプレッサー(HcPro)及びプルーニング(浸透の12時間前機械的プルーニング)の併用の影響例を示す。Plasto-HcPro-プルーニング:935単独発現(プルーニング無し、サイレンシングサプレッサーの同時発現無し);Plasto-HcPro+プルーニング:935による形質転換12時間前に植物の機械的プルーニング(サイレンシングサプレッサーの同時発現無し);Plasto+HcPro+プルーニング:935及びHcProの同時発現12時間前に機械的プルーニング。
【発明を実施するための形態】
【0011】
本発明は、植物においてタンパク質を生産する方法に関する。本発明はまた、植物でタンパク質を生産するために用いられ得るヌクレオチド配列を提供する。
植物又は植物の部分内で対象のタンパク質を合成する方法が提供される。その基本的な形態で本方法は以下の工程を含む:植物又は植物の部分中で活性な光合成遺伝子から得られた調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を一過性態様で導入する工程、及び前記対象のタンパク質をコードする前記核酸配列の前記植物または植物の部分での発現を可能にする条件下で前記植物又は植物の部分を維持する工程。
本方法はさらに、先ず初めに、対象のタンパク質をコードする1つ以上の核酸配列を導入する前に植物又は植物の部分をプルーニングする工程を含んでもよい。この方法では、植物又は植物の部分をプルーニングした後で、植物中で活性な調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列が、プルーニングされた植物又は植物の部分に一過性態様で導入される。続いて、前記植物又は植物の部分は、対象のタンパク質をコードする核酸配列の前記植物または植物の部分での発現を可能にする条件下で維持される。
この方法と同様な一過性形質転換プロトコルであって、光合成遺伝子から得られた調節領域の使用、プルーニング工程、又は光合成遺伝子から得られた調節領域とプルーニング工程との両方の使用を含まないものを用いて同じ対象タンパク質を生産する方法とタンパク質生産を比較したとき、この方法を用いて高収量の対象タンパク質が生産された。
【0012】
安定なトランスジェニック発現系で使用するために設計された発現カセットで用いられるプロモーターは、一過性発現系で用いるときは効率が低いことが見出された(Giritch et al. 2006、Fisher, 1999a)。Giritchら(12206)は、各IgGサブユニットのために、1つのリコンビナーゼ及び2つのウイルス性レプリカーゼと一緒に異なるプロベクター(1つはTMV系で他方はPVX系)を同時発現させて、200mg/kgの範囲の発現レベルを達成できたことを示している。本明細書に記載するように、葉の発現で有効性を示したエンハンサー配列を含むプロモーターは、一過性発現で有効であることが見出された。非限定的例示には、プラストシアニンの発現を調節する際に用いられるプロモーターが含まれる(Pwee and Gray 1993、前記文献は参照により本明細書に含まれる)。理論に拘束されないが、核マトリックスへの結合による光合成遺伝子の上流の調節エレメントの結合は、強力な発現を仲介することができる(Sandhu et al. 1998;Chua et al. 2003)。例えば、エンドウのプラストシアニン遺伝子の翻訳開始部位から-784までを用いて、強力なレポーター遺伝子発現を仲介し得る。
光合成遺伝子由来の調節領域(例えばプラストシアニン調節領域であるが、ただしこれらに限定されない(US7,125,978、前記文献は参照により本明細書に含まれる))、又はリブロース1,5-ビスホスフェートカルボキシラーゼ/オキシゲナーゼ(rubisco;US4,962,028、前記文献は参照により本明細書に含まれる)から得られる調節領域、クロロフィルa/b結合タンパク質(CAB;Leutwiler et al. 1986、前記文献は参照により本明細書に含まれる)、ST-LS1(光化学反応系IIの酸素放出複合体と結合する;Stockhaus et al. 1989、前記文献は参照により本明細書に含まれる)を、本発明にしたがって用い得る。
【0013】
リブロース1,5-ビスホスフェートカルボキシラーゼ/オキシゲナーゼ(Rubisco)の大又は小サブユニットをコードする遺伝子若しくはプラストシアニンから得られる調節領域、又は光合成遺伝子から得られる調節領域をプルーニングと併用して使用することによって、発現レベル及び収量が増加することが見出された。例えば、図2Aに示すように、光合成プロモーター(プラストシアニンから得られる;図2A、R610、R612参照)によって駆動される対象のコード領域の浸透後の発現レベルは、35Sによって駆動される同じコード領域(図2A、R514)と比較したとき極めて高い。
したがって、本発明は、以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法を提供する:
i)植物又は植物の部分中で活性な光合成遺伝子から得られる調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を一過性態様で導入する工程、及び
ii)前記植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列の前記植物又は植物の部分での発現を可能にする条件下で維持する工程。
植物又は植物の部分は、1つ以上の核酸配列を導入する工程の前にプルーニングされ得る。所望の核酸構築物の浸透前に植物をプルーニングすることによって、発現レベル(全合成タンパク質の%として)及び収量(mgタンパク質/kg新鮮質量)が増加することが見出された。これは、いくつかの浸透方法(注射筒浸透又は真空浸透が含まれるが、ただしこれらに限定されない)及び多様なプルーニング方法(例えば機械的プルーニング又は化学的プルーニングを含むが、ただしこれらに限定されない)を用いて観察された。理論に拘束されないが、浸透前のプルーニングは、頂芽優勢の低下をもたらすことができ、さらに生長調節物質含有量(例えばジベレリン酸又はエチレン含有量(ただしこれらに限定されない))の低下をもたらしえる。これは、続いて葉の光合成能力の増加を刺激し、さらに光合成遺伝子の転写速度を増加させ得る。したがって、光合成遺伝子から得られる調節領域の使用はより高いタンパク質収量をもたらし得る。さらにまた、光合成遺伝子から得られる調節領域の使用と生長調節物質阻害剤(例えばエチレン又はジベレリン酸)の含有量を低下させる化学的プルーニングの併用はより高いタンパク質収量をもたらし得る。
【0014】
本明細書に記載する方法にしたがって対象タンパク質の収量の増加に対するプルーニングの影響が、機械的又は化学的プルーニング方法及び真空又は注射筒浸透を用いて観察される。プルーニングによる収量の増加は、植物を傷つけたときに、例えば注射筒浸透に続いて観察される(図7C参照)。これは、タンパク質発現の増加は、単なる植物の損傷に対する応答ではないことを示している。
プルーニングとは、1つ以上の腋芽の除去、1つ以上の頂芽の除去、又は1つ以上の腋芽及び1つ以上の頂芽の両方の除去を意味する。プルーニングはまた、枯らすこと(killing)、壊死の誘発、又は植物から芽を除去することなく頂芽及び腋芽の生長を低下させることを含む。芽の生長低下(又は芽の成長を低下させる)とは、処理されていない芽と比較して、芽が、例えば代謝活性の低下、一定期間を超えて約50%から100%又はそれらの間の任意の量のサイズの増加を示すことを意味する。プルーニングはまた、頂芽優勢を低下させる化合物を適用することによって達成され得る。化合物がプルーニングの目的のために適用される場合、用いられる投与量は典型的には化合物の製造業者が推奨する投与量である。
プルーニング(機械的又は化学的プルーニング)を、浸透の約20日前から浸透の約2日後に、又はその間の任意の時点、例えば浸透の7日(168時間)前から浸透の約2日(48時間)後、又はその間の任意の時点、例えば浸透の約48時間(2日)前から浸透の約1日(24時間)後、又はその間の任意の時点、又は浸透の前20日、19日、18日、17日、16日、15日、14日、13日、12日、11日、10日、9日、8日、又は168、144、120、96、72、60、50、40、36、32、30、28、26、24、22、20、18、16、14、12、10、8、6、4、2、1、0時間から浸透の後約1、2、4、6、8、10、12、14、16、18、20、22、24時間、又はそれらの間の任意の時点で実施し得る。プルーニングが浸透の72時間前に又はそれより以前に実施される場合、プルーニング方法は化学的プルーニングであることが好ましい(機械的プルーニングを用いる場合は再生長が生じる可能性があるので)。プルーニング方法が化学的プルーニングである場合は、浸透前により長時間を、例えば浸透前2、3、4、5、6又は7日、又はその間の任意の時点を用い得る。当業者は、プルーニング前の適切な間隔を容易に決定することができる。
【0015】
プルーニングは、当業者に公知の任意の手段によって実施することができ、芽の機械的除去が含まれるが、ただし前記に限定されない。芽の機械的除去は例えば、切断、刈取り、摘取り、例えばトングを用いた圧縮、限局凍結(例えば、液体窒素の限局蒸気を芽に誘導するか、又は液体窒素、ドライアイス、エタノール-ドライアイスなどを含む適切な冷却源を用いて冷却したトング若しくは他の装置を用いて芽を取り囲み、それによって芽の温度を下げて芽の生長を低下させるか又は芽を枯らす(kill)ことによる)が含まれるが、ただしこれらに限定されない。
プルーニングにはまた、化学的プルーニング、例えば、芽を枯らすか若しくは芽の生長を低下させる除草剤(化合物;プルーニング剤)の適用、又は芽を枯らすか若しくは芽の生長を低下させる生長調節剤の適用が含まれる。化学的プルーニングの使用は、噴霧、散布、浸漬、植物上への前記化合物の添加、又は前記化合物を含む溶液への植物のディッピングによって植物を容易に処理することができるので、プルーニングの効率的な処理態様を可能にする。植物は、浸透工程前に1回処理するか、又は浸透工程前に2回以上処理してもよい。使用され得る化合物の例には、除草剤、例えば植物生長調節物質Ethephon(例えばBromeflor、Cerone、Chlorethephon Ethrel、Florel、Prep及びFlordimex)、Daminozide(ブタンジオン酸モノ2,2-ジメチルヒドラジン、-コハク酸2,2-ジメチルヒドラジド;例えばB-9;Alar、Kylar、SADH、B-9、B-995、アミノジド)、Atrimmec(ジケグラクナトリウム)、マレインヒドラジド(1,2-ジヒドロ-3,6-ピリダジンジオン)、2-4-D(2,4,ジクロロフェノキシ酢酸) が含まれ(ただしこれらに限定されない)、さらにジベレリン酸合成の阻害剤、例えばCycocel(クロメクァトクロリド)、A-Rest(アンシミドール)、トリアゾール、例えばBonzi(パクロブトラゾール)、Sumahic(ユニコナゾール)、又は3-アミノ-1,2,4-トリアゾール(3-AT)が含まれる(ただしこれらに限定されない)。これらの化合物は、植物生長調節のために知られている投与量範囲で用いられ得る。例えば用いられる投与量範囲は、前記化合物の製造業者が推奨するものでありえる。これらの化合物はまた、植物生長調節のために知られた投与量範囲未満の投与量範囲で用いてもよい。例えば、用いられる投与量範囲は、前記化合物の製造業者が推奨する投与量範囲の75%、50%、25%で用いられ得る。これらの化合物は約0.2ppmから約5000ppm、及び選択した生長調節物質に応じて前記の間の任意の量で用いられ得る。さらにまた、プルーニング剤(化合物)は1回適用してもよいが、また必要に応じて追加の適用を施してもよい。例えば、化合物は1回適用してもよいが、浸透前又は浸透後の植物の化学的プルーニングを得るために、化合物を2回以上適用してもよい。化学的プルーニングを用いる場合は、化合物は浸透の20日前から浸透の約2日後、又はその間の任意の時間に適用し得る。例えば、浸透の14日、7日又は5日前の化合物の適用が効果的な使用でありえる。
【0016】
図7A、7B、7C、8及び9に示すように、浸透前の植物のプルーニングは、対象タンパク質の発現の増加をもたらす。この効果は機械的又は化学的プルーニング方法を用いたときに観察される。したがって、本発明は、以下の工程を含む、対象のタンパク質を植物又は植物の部分で合成する方法を提供する:
i)植物又は植物の部分をプルーニングし、プルーニングされた植物又は植物の部分を生産する工程、
ii)植物中で活性な調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を、前記プルーニングされた植物又は植物の部分に一過性態様で導入する工程、及び
iii)前記プルーニングされた植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分中で発現させることを可能にする条件下で維持する工程。
対象のタンパク質をコードする核酸配列は、当業者に公知の任意の適切な方法、例えば真空浸透(vacuum infiltration)又は注射筒浸透(syringe infiltration)(前記に限定されると考えるべきではない)によって植物又は植物の部分に導入し得る。真空浸透の方法は当分野では公知であり、Kapilaら(1997、前記文献は参照により本明細書に含まれる)の方法が含まれえるが、ただし前記に限定されない。浸透はまた、注射筒を用いて植物又は植物の部分に対象のタンパク質をコードする核酸配列を導入することにも関連する(Liu and Lomonossoff, 2002、前記文献は参照により本明細書に含まれる)。
本発明で用いる方法及び以前に記載された方法(例えばKapila et al. 1997;又はLiu and Lomonossoff, 2002)は、その使用前に一過性形質転換のためにアセトシリンゴンを含む培養液中でアグロバクテリア菌(Agrobacterium)を培養する。アセトシリンゴン及び他のフェノール系シグナル分子は、アグロバクテリア菌のvir機構を正の方向に調節することが知られている。本明細書に記載の対象タンパク質の発現レベルの増加は、アグロバクテリア菌をアセトシリンゴンの存在下又は非存在下で培養したときに観察される。
【0017】
転写後遺伝子サイレンシング(PTGS)は、植物でのトランスジーンの発現制限に中心的に関与する可能性があり、ジャガイモウイルスY由来のサイレンシングのサプレッサー(HcPro)の同時発現をトランスジーンmRNAの特異的分解に対抗するために用いられ得る(Brigneti et al. 1998)。また別のサイレンシングサプレッサーは当分野で周知であり、本明細書に記載するように用いられ得る(Chiba et al. 2006, Virology 346:7-14、前記文献は参照により本明細書に含まれる)。前記は例えば以下である(ただしこれらに限定されない):TEV-p1/HC-Pro(タバコエッチウイルス-p1/HC-Pro)、トマトブシースタントウイルスのBYV-p21、p19(TBSV p19)、トマトクリンクルウイルスのキャプシドタンパク質(TCV-CP)、キュウリモザイクウイルスの2b(VMC-2b)、ジャガイモウイルスXのp25(PVX-p25)、ジャガイモウイルスMのp11(PVM-p11)、ジャガイモウイルスSのp11(PVS-p11)、ブルーベリースコーチウイルスのp16(BScV-p16)、レモントリステキサウイルスのp23(CTV-p23)、ブドウの木のリーフロール関連ウイルス-2(GLRaV-2p24)、ブドウの木のウイルスAのp10(GVA-p10)、ブドウの木のウイルスB(GVB-p14)、ヘラクレウム潜在ウイルスのp10(HLV-p10)、又はニンニクの共通潜在ウイルス(GCLV-p16)。したがって、サイレンシングのサプレッサー、例えばHcPro、TEV-p1/HC-Pro、BYV-p21、TBS p19、TCV-CP、CMV-2b、PVX-p25、PVM-p11、PVS-p11、BScV-p16、CTV-p23、GLRaV-2 p24、BGV-p14、HLV-p10、GCLV-p16、又はGVA-p10(ただしこれらに限定されない)を、対象のタンパク質をコードする核酸配列と一緒に発現させて、植物における高レベルのタンパク質生産をさらに担保し得る。
図9に示すように、対象のタンパク質をコードする核酸配列とサイレンシングサプレッサーとの同時発現は、対象のタンパク質の収量の顕著な増加をもたらす。この効果はまた、浸透前に植物をプルーニングする場合に観察される。したがって、本明細書に記載する対象のタンパク質を合成する方法は、2つ以上の核酸配列を植物又は植物の部分に導入する工程を含み得る。例えば2つ以上の核酸配列の1つはサイレンシングのサプレッサーをコードし得る。
【0018】
対象のタンパク質を高収量で製造する方法を具体化するために、本発明は、対象のタンパク質、例えば抗体のような複合タンパク質の発現を駆動するための植物発現系を記載する。アグロバクテリア浸透植物、例えばニコチアナ・ベンタミアナ(Nicotiana benthamiana)内での複合タンパク質の発現によって1.5g/kgFW(ほぼ25%TSP)に達するタンパク質レベルが得られた。対象タンパク質の分泌型及びER-保持型について、それぞれ558及び757mg/kg/FWの平均レベルを得た。非限定的な例を提供すれば、この発現レベルは抗体について得られ、これは、マルチウイルス一過性発現系(Giritch et al. 2006)を用いて産生される抗体の3倍高い発現レベルであり、非ウイルス系アグロバクテリア浸透発現系(例えばVaquero et al. 1999)について記載されたレベルを十分に超えている。
本明細書で提供する例(この例に限定されると考えてはならない)では、抗体は改変されたグリコシル化パターンを含み、低フコシル化、低キシロシル化又はフコシル化とキシロシル化がともに低下したN-グリカンを有する。植物及び典型的な哺乳動物のN-グリコシル化の相違の影響は、治療薬の製造に植物を用いるという考えに対する主要な懸念であった。植物特異的グリカンの出現は、植物生成タンパク質の血流中での半減期の短縮の一因となり、あるいは同じグリカンが患者で過敏反応を惹起するかもしれない。本態様では、対象のタンパク質を高収量で製造することが可能で、それらは、過敏反応を惹起する可能性があるか、そうでなければアレルギー反応に関与する可能性があるグリカンを欠く。しかしながら、本明細書に記載の一過性タンパク質生産方法は、改変グリコシル化を含まないタンパク質を含む任意の対象タンパク質のために用いられ得ることは理解されよう。
【0019】
改変グリコシル化パターンを有することを特徴とする、植物内で対象タンパク質を合成する方法が記載される。本方法は、ヒトベータ-1,4ガラクトシルトランスフェラーゼ(hGalT、GaltTとも称される(配列番号:14))を発現するヌクレオチドと一緒に対象タンパク質を同時発現させることを必要とする。hGalTをまたN-アセチルグルコサミントランスフェラーゼ(GNT1;配列番号:21、図5f;アミノ酸配列番号:22、図5g)のCTSドメイン(すなわち、細胞質テール、トランスメンブレンドメイン、幹領域)と融合させて、GNT1-GalTハイブリッド酵素を生成し、このハイブリッド酵素を対象タンパク質と同時発現させ得る。
ハイブリッドGNT1-GalT配列の使用によって、hGalTの触媒ドメインは、複雑なN-グリカン成熟の初期段階が発生するcis-ゴルジ装置に配置される。対象タンパク質はまた、GalTと融合したCTSドメインを含むハイブリッド酵素、例えばGNT1-GalTと同時発現させ得る(R621;図5a;配列番号:18(配列番号:17によってコードされる))。しかしながら、フコシル化レベルが低下し、なおキシロシル化及びガラクトシル化タンパク質を含む対象タンパク質を所望する場合は、GalTを対象タンパク質と同時発現させてもよい。
対象タンパク質の“改変グリコシル化”とは、改変グリコシル化を含む対象タンパク質(例えば本明細書に記載)のN-グリカンプロフィルが、野生型植物で生成される対象タンパク質のN-グリカンプロフィルと異なることを意味する。グリコシル化の改変は、対象タンパク質の1つ以上のグリカンにおける増加又は減少を含み得る。例えば、対象のタンパク質は、キシロシル化の低下、フコシル化の低下、又はキシロシル化の低下及びフコシル化の低下の両低下を示し得る。また別には、対象タンパク質のN-グリカンプロフィルは、ガラクトシル化の量が増加し、さらに場合によってキシロシル化、フコシル化又はその両方が低下するような態様で改変し得る。
【0020】
さらにまた、対象の複合タンパク質を生産するとき、ヌクレオチド配列は、複合タンパク質の2つ以上のペプチド又はドメインをコードし得る。例えば、対象タンパク質が抗体である場合は、ヌクレオチド配列は2つのヌクレオチド配列を含み、その各々は抗体の一部分をコードし得る。例えば一方のヌクレオチド配列は軽鎖をコードし、第二の配列は抗体の重鎖をコードする。そのような構築物の非限定的な例は図1に提供される。図1では、R612及びR610の各構築物は2つのヌクレオチド配列を含み、一方は、植物で活性な調節領域(例えばUS7,125,978(前記文献は参照により本明細書に含まれる)に記載されたプラストシアニンプロモーター(ただし前記に限定されない))と機能的に連結されたC5-1 LC(C5-1の軽鎖)をコードし、第二の配列は、植物で活性な調節領域(例えばプラストシアニンプロモーター(US7,125,978、前記文献は参照により本明細書に含まれる)、ただし前記に限定されない)と機能的に連結されたC5-1の重鎖(C5-1 HC)をコードする。図1に示すように、さらにR610を参照すれば、KDEL配列は、ペプチド2A又は2Bの1つのC-末端領域に融合させ得る。例えば、KDEL配列は抗体の重鎖と融合させて、抗体がERとともに保持されることを担保し得る(前記を限定と捉えるべきではない)。
ヌクレオチド配列によってコードされる各タンパク質はグリコシル化され得る。
一過性発現を用いて製造された精製生成物のクマシー染色によって、少量の多様な夾雑物の存在が示された。これらのフラグメントは関連生成物であるようで、70kDaを越える全ての夾雑物が、活性ブロットによって示されるように、少なくとも1つのFabを含んでいた(図3B)。植物抽出物に存在する生成物関連夾雑物の実体及び量は、哺乳動物細胞の生産系で観察されたものと同様であった。したがって、抗体治療薬の精製に典型的に用いられる精製手順(例えば陰イオン交換、アフィニティー及び陽イオン交換)は、治療薬として使用されるタンパク質の規制機関によって要求される純度を容易に満たす。
【0021】
図6に示すように、本明細書に記載する方法を用いることによって、改変グリコシル化プロフィルを示す対象タンパク質を生産し得る。例えば、免疫遺伝学的に検出不能なフコース又はキシロース残基を有する対象タンパク質は、GNT1-GalTと一緒に対象タンパク質を同時発現させたときに産生された。対象タンパク質のエピトープのMALDI-TOF分析は、改変グリコシル化パターンを有する対象タンパク質は、GalT又はGNT1-GalTのどちらかと一緒に対象タンパク質を同時発現させたときに得られ得ることを示している。
植物、植物部分又は植物材料をフィードとして用いるか、植物又は植物部分に最小限の加工を施すか、或いは対象タンパク質は植物又は植物部分から抽出し得る。所望の場合は、対象タンパク質は標準的な方法を用いて単離及び精製してもよい。
高収量を担保するために、対象タンパク質をコードするヌクレオチド配列に対してまた別の改変を実施し得る。対象タンパク質をコードする核酸配列はまた、前記タンパク質の小胞体(ER)内での保持に活性な配列(例えばKDEL(Lys-Asp-Glu-Leu)又は他の公知のER保持配列(例えばHDEL))をコードする配列と融合させ得る。
本明細書に記載のタンパク質製造方法は、対象タンパク質の製造のための“プラットフォーム”として用いられ得る植物の使用を含み得る。例えば、プラットフォーム植物は典型的には1つ以上のタンパク質を安定的な態様で発現し、このタンパク質は、例えばN-グリコシル化が改変された対象タンパク質を製造するために、いくつかの態様で対象タンパク質の生成を改変する。例えば、プラットフォーム植物は、GalT、GNT1-GalT、又はGalT及びGNT1-GalTの両方をコードする1つ以上の第一のヌクレオチド配列を発現し得る。対象タンパク質を製造するためには、プラットフォーム植物又はプラットフォーム植物の部分をプルーニングした後、対象タンパク質をコードする第二のヌクレオチド配列を一過性形質転換を用いてプラットフォーム植物に導入し、第二のヌクレオチド配列を発現させて対象タンパク質を製造し、この場合、対象タンパク質は、N-グリコシル化が改変されたグリカンを含む。しかしながら、安定的に他のタンパク質を発現するプラットフォーム植物を、所望にしたがって対象タンパク質を改変するために用いてもよいことは理解されよう。植物又は植物の部分をフィードとして用いるか、または植物又は植物部分に最小限の加工を施すか、或いは対象タンパク質を植物又は植物部分から抽出し得る。所望の場合は、対象タンパク質は標準的な方法を用いて単離及び精製してもよい。
【0022】
本発明は、GalT、GNT1-GalT、GalT及びGNT1-GalTの両方、又はその組合せをコードするヌクレオチド配列であって、その各々がプラットフォーム植物中で活性な調節領域と機能的に連結されている前記配列含むプラットフォーム植物又はプラットフォーム植物の部分を用いて、グリコシル化が改変された対象タンパク質を発現させる方法を提供する。続いて前記プラットフォーム植物又はプラットフォーム植物の部分を用いて、1つ以上の対象タンパク質をコードする第二のヌクレオチド配列を発現させ得る。前記第二のヌクレオチド配列は、プラットフォーム植物中で活性な1つ以上の第二の調節領域と機能的に連結されている。第一のヌクレオチド配列、第二のヌクレオチド配列、又は第一及び第二のヌクレオチド配列の両配列は、プラットフォーム植物又はプラットフォーム植物の部分での発現のためにコドンを最適化させ得る。この方法は、先ず初めにプラットフォーム植物又はプラットフォーム植物の部分をプルーニングする工程を含む。プルーニングの後、前記植物中で活性な調節領域と機能的に連結された対象タンパク質をコードする1つ以上の第二の核酸配列を、前記プルーニングした植物又は植物の部分に一過性態様で導入する。続いて、前記植物又は植物の部分を、対象のタンパク質をコードする核酸配列の前記植物又は植物の部分における発現を可能にする条件下で維持する。
対象タンパク質、又は対象のタンパク質のグリコシル化を改変する酵素(例えばGalT、GNT1-GalT、GalT及びGNT1-GalTの両方、又はその組合せ)をコードするヌクレオチド配列は、前記植物での発現レベルを高めるためにコドンを最適化させ得る。コドン最適化とは、植物におけるコドン使用頻度に類似させるために、構造遺伝子又はそのフラグメントのオリゴヌクレオチド構築ブロックの合成及びその後のそれら酵素のアッセンブリに適したDNAヌクレオチドを選択することを意味する。配列は合成配列であってもよく、前記は、Sardanaら(Plant Cell Reports 1996, 15:677-681)が概略した方法と類似する方法を用いて植物のコドン使用頻度について最適化される。双子葉植物で高度に発現される遺伝子から得られるコドン使用頻度表は、Murrayら(Nuc Acids Res 1989, 17:477-498)を含むいくつかの情報源から入手することができる。さらにまた、配列の最適化はまた、コドンタンデムリピート、曖昧なスプライス部位の除去、反復配列の削減(逆リピートを含む)を含んでもよく、さらにLeto1.0(Entelechon, Germany)を用いて決定することができる。
【0023】
“機能的連結される”とは、個々の配列が直接的又は間接的に相互作用して、意図する機能、例えば遺伝子発現の仲介又は調節を達成することを意味する。機能的に連結された配列の相互作用は、例えば前記機能的に連結された配列と相互作用するタンパク質によって仲介されえる。転写調節領域及び対象の配列は、それらの配列が機能的に接続されて、転写調節領域によって仲介又は調節されるべき対象配列の転写が可能となるときに、機能的に連結されている。
“植物の部分”という用語は植物に由来する任意の部分を意味し、全植物、植物から得られる組織、例えば葉、葉及び茎、根、気生部分(植物の葉、茎及び場合によって花の部分を含む)、植物から得られる細胞又はプロトプラストが含まれるが、ただしこれらに限定されない。
“植物材料”という用語は植物に由来する任意の材料を意味する。植物材料は、全植物、組織、細胞又はその任意の部分を含み得る。さらにまた、植物材料は、細胞内の植物成分、細胞外の植物成分、植物の液体若しくは固体抽出物、又は前記の組合せを含み得る。さらにまた、植物材料は、植物、植物細胞、組織、液体抽出物又は前記の組合せであって、植物の葉、茎、果実、根又は前記の組合せに由来するものを含み得る。植物材料は、いずれの加工工程にも付されていない植物又はその部分を含み得る。しかしながら、植物材料は、下記に規定する最小限の加工工程、又はより厳密な加工(部分的又は実質的なタンパク質の精製を含み、前記精製は当分野で一般的に知られている技術(クロマトグラフィー、電気泳動などを含むがただしこれらに限定されない)を用いる)に付すこともまた意図される。
【0024】
“最小限の加工”という用語は、植物材料(例えば対象タンパク質を含む植物又はその部分)が部分的に精製されて、植物抽出物、ホモジネート、植物ホモジネートの分画などを生じること(すなわち最小限の加工)を意味する。部分的精製は、植物の細胞構造を破壊し、それによって可溶性植物成分及び不溶性植物成分を含む組成物を生成する工程を含み得るが、ただしこれらに限定されない(前記成分は、例えば遠心沈殿、ろ過又はその組合せ(ただしこれらに限定されない)によって分離し得る)。前記に関しては、葉又は他の組織の細胞外間隙に分泌されたタンパク質は真空又は遠心抽出を用いて容易に入手することができよう。または、組織は、細胞外間隙からタンパク質を搾り出すか又は遊離させるために、ローラーを通過させるか、又はすり潰しなどにより加圧下で抽出することもできよう。最小限の加工はまた、可溶性タンパク質の粗抽出物の調製を含むことができる。なぜならば、これらの調製物は二次的な植物生成物に由来する夾雑物をほとんど含まないからである。さらにまた、最小限の加工は、葉の可溶性タンパク質の水性抽出物を続いて適切な塩で沈殿させることを含み得る。他の方法には、抽出物の直接的な使用を可能にするための大規模な離解及び絞り汁抽出が含まれ得る。
植物成分又は組織の形態の植物材料は、対象に経口的に送達され得る。植物材料は、食事用サプリメントの部分として他の食物と一緒に又は被包化して投与され得る。植物材料又は組織はまた濃縮して嗜好性を改善又は向上させるか、または要請に応じて他の素材、成分又は医薬賦形剤と一緒に提供してもよい。
【0025】
対象のタンパク質を含む植物は、対象(例えば動物またはヒト)に、必要性及び状況に応じて多様な方法で投与することが意図される。例えば、植物から得られる対象タンパク質は、それを使用する前に、未精製、部分精製又は精製形として抽出され得る。タンパク質を精製することができる場合には、タンパク質は、食用又は非食用植物で生産され得る。さらにまた、タンパク質を経口投与する場合には、植物組織を収穫して対象に直接供給するか、または収穫した組織を供給前に乾燥させるか、または収穫を実施することなく動物に生草を食べさせてもよい。収穫した植物組織を動物の飼料に食物サプリメントとして提供することもまた本発明の範囲内であると考える。植物組織がほとんど又は更なる加工を加えることなく動物に供給される場合は、投与される植物組織が食用であることが好ましい。
実施例でさらに詳細に記載するように、GalT、GNT1-GalT及び対象タンパク質は、一過性態様で植物に導入された。適切な抗体を用いた免疫学的解析によって、MWrが150kDaのタンパク質が形質転換細胞に存在することが明らかにされた(図2、3A及び3B)。さらにまた、GalT又はGNT1-GalTが、どちらかの構築物を発現する植物から得られた抽出物で検出され、さらにGNT1-GalTが植物で発現されたときに、対象タンパク質の改変Nグリコシル化が観察された(図6)。したがって、組換えにより発現されたGalT又はGNT1-GalTはin plantaで生物学的に活性を有する。
“類似体”又は“誘導体”は、GalT(配列番号:14)又はGNT1-GalT(配列番号:17)をコードするヌクレオチド配列への任意の置換、欠失又は付加を含むが、ただし、GalT(配列番号:14)又はGNT1-GalT(配列番号:17)の非存在下で生産された対象タンパク質のグリコシル化プロフィルと比較したとき、前記配列が、対象タンパク質のグリコシル化プロフィルを改変する、例えば対象タンパク質のグリカンのフコシル化、キシロシル化又はその両方の低下、又は対象タンパク質のガラクトシル化の増加をもたらすタンパク質をコードすることを条件とする。例えば、前記配列によってコードされるタンパク質は、Nグリカンの成熟中に末端ガラクトースを付加し得る。核酸配列の誘導体及び類似体は、ある核酸配列と典型的には80%を超える類似性(又は同一性)を示す。
【0026】
2つ以上の核酸又はポリペプチドに関して、“同一”又はパーセント“同一”という用語は、配列比較アルゴリズム(例えばAltschul et al. Nuc Acids Res 1997, 25:3389-3402及びAltschul et al. J Mol Biol 1990, 215:403-410)及びこれらアルゴリズムの任意のアップグレードを用いるか、又は手動アラインメントと目視精査によって測定される比較ウィンドウ又は指定領域にわたって最大一致のために比較及びアラインメントを実施したとき、同じであるか、又は具体的なパーセンテージのアミノ酸残基若しくはヌクレオチドが同じである(すなわち具体的な領域にわたって60%同一、好ましくは65%、70%、75%、80%、85%、90%又は95%同一である)2つ以上の配列又は部分配列に該当する。配列類似性は、BLASTアルゴリズム(GenBank:ncbi.nlm.nih.gov/cgi-bin/BLAST/)により規定値パラメータ(プログラム:blastn;データベース:nr;Expect 10;フィルター:低複雑度;アラインメント:ペア毎;ワードサイズ:11)を用いて決定し得る。
類似体又はその誘導体にはまた、ストリンジェントなハイブリダイゼーション条件下で(以下の文献を参照されたい:Maniatis et al. in Molecular Cloning (A Laboratory Manual), Cold Spring Harbor Laboratory, 1982, p.387-389又はAusubel et al. (eds), 1989, Current Protocols in Molecular Biology, Vol. 1, Green Publishing Associattes, Inc., and John Wiley & Sons, Inc., New York, p.2.10.3)、本明細書に記載のGalT(配列番号:14)配列、GNAT1-GalT(配列番号:17)配列のいずれかとハイブリダイズするヌクレオチド配列が含まれるが、ただし、GalT(配列番号:14)又はGNT1-GalT(配列番号:17)の非存在下で生成された対象タンパク質のグリコシル化プロフィルと比較したとき、前記配列が、対象タンパク質のグリコシル化プロフィルを改変する、例えば対象タンパク質のグリカンのフコシル化、キシロシル化又はその両方の低下、又は対象タンパク質のガラクトシル化の増加をもたらすタンパク質をコードすることを条件とする。例えば、前記配列によってコードされるタンパク質は、Nグリカンの成熟中に末端ガラクトースを付加し得る。そのようなストリンジェントなハイブリダイゼーション条件の例は、適切なプローブ、例えば[ガンマ-32P]dATP標識プローブ(ただし前記に限定されない)との7%SDS、1mM EDTA、0.5M Na2HPO4(pH7.2)中での16−20時間、65℃でのハイブリダイゼーションでありえる。続いて65℃にて5%SDS、1mM EDTA、40mM Na2HPO4(pH7.2)中で30分洗浄し、続いて65℃にて1%SDS、1mM EDTA、40mM Na2HPO4(pH7.2)中で30分洗浄する。この緩衝液での洗浄を繰り返してバックグラウンドを低下させることが得る。
【0027】
“調節領域”、“調節エレメント”又は“プロモーター”とは、核酸の部分、典型的には遺伝子(DNA若しくはRNA又はDNA及びRNAの両方を含み得る)のタンパク質コード領域の上流(ただし常にそうであるとは限らないが)を意味する。調節領域が活性を有し、かつ対象の遺伝子と機能的な結合状態にあるとき、又は機能的に連結されているとき、対象遺伝子の発現がもたらされえる。調節エレメントは器官特異性を仲介するか、又は発生若しくは一過性遺伝子活性化を制御し得る。“調節領域”には、プロモーターエレメント、基礎プロモーター活性を示すコアプロモーターエレメント、外部刺激に応答して誘導されえるエレメント、プロモーター活性を仲介するエレメント、例えば負の調節エレメント又は転写エンハンサーが含まれる。本明細書で用いられる、“調節領域”にはまた転写後に活性を示すエレメントが含まれる。前記は、例えば遺伝子発現を調節する調節エレメント、例えば翻訳及び転写エンハンサー、翻訳及び転写リプレッサー、上流の活性化配列、及びmRNA不安定性決定因子である。これら後者のエレメントのいくつかはコード領域の近位に存在しえる。
本開示に関しては、“調節エレメント”又は“調節領域”は典型的には、構造遺伝子のコード配列の通常は(常にというわけではないが)上流(5')のDNA配列に該当し、前記は、RNAポリメラーゼ及び/又は転写を特定の部位で開始させるために必要な他の因子のための見分けを提供することによってコード領域の発現を制御することは理解されよう。しかしながら、イントロン内又は配列の3'に配置される他のヌクレオチドもまた、対象のコード領域の発現の調節に寄与し得る。特定の部位での開始を担保するためにRNAポリメラーゼ又は他の転写因子のための見分けを提供する調節エレメントはプロモーターエレメントである。ほとんどの(全てではないが)真核細胞性プロモーターエレメントはTATAボックスを含む。前記は保存された核酸配列であって、アデノシン及びチミンヌクレオチド塩基対を含み、通常は転写開始部位のほぼ25塩基対上流に位置している。プロモーターエレメントは基礎プロモーターエレメントを含み、遺伝子の発現を修正する他の調節エレメント(上記に列挙されている)と同様に転写の開始のために極めて重要である。
【0028】
構成的調節領域は、植物の種々の部分にわたって、さらに植物の発育中に持続的に遺伝子の発現を誘導する。公知の構成的調節エレメントの例には、CaMV 35S転写物(Odell et al., 1985, Nature, 313: 810-812)、コメのアクチン1(Zhang et al, 1991, Plant Cell, 3: 1155-1165)、アクチン2(An et al., 1996, Plant J., 10: 107-121)、又はtms2(U.S. 5,428,147、前記文献は参照により本明細書に含まれる)、及びトリオースホスフェートイソメラーゼ1(Xu et. al., 1994, Plant Physiol. 106: 459-467)遺伝子、トウモロコシユビキチン1遺伝子(Cornejo et al, 1993, Plant Mol. Biol. 29: 637-646)、シロイヌナズナ(Arabidopsis)ユビキチン1及び6遺伝子(Holtorf et al, 1995, Plant Mol. Biol. 29: 637-646)、並びにタバコ翻訳開始因子4A遺伝子(Mandel et al, 1995 Plant Mol. Biol. 29: 995-1004)と密接に関連するプロモーターが含まれる。本明細書で用いられる“構成的”という用語は、構成的調節領域の制御下にある遺伝子が全ての細胞タイプで同じレベルで発現されることを必ずしも示しているわけではないが、前記遺伝子は豊富さに変動はしばしば認められるものの広い範囲の細胞タイプで発現される。
光合成遺伝子から得られる調節領域又はプロモーターもまた本発明での使用に適している。例えば、調節領域又はプロモーターは以下の大小のサブユニットをコードする遺伝子から得られ得る:リブロース1,5-ビスホスフェートカルボキシラーゼ/オキシゲナーゼの大若しくは小サブユニット(rubisco;US 4,962,028、前記文献は参照により本明細書に含まれる)、プラストシアニン(US 7,125,978、前記文献は参照により本明細書に含まれる);図1b、配列番号:19)、クロロフィルa/b結合タンパク質(CAB;Leutwiler et al. 1986、前記文献は参照により本明細書に含まれる)、ST-LS1(光化学反応系IIの酸素放出複合体と関連する;Stockhaus et al. 1989、前記文献は参照により本明細書に含まれる)。
【0029】
本発明の1つ以上のヌクレオチド配列は任意の適切な植物宿主において発現させる得る。適切な宿主の例には、シロイヌナズナ、アルファルファ、キャノーラ、ブラシッカ(Brassica)spp.、トウモロコシ、ニコチアナ(Nicotiana)spp.(ニコチアナ・ベンタミアナ(Nicotiana benthamiana)、ニコチアナ・タバクム(Nicotiana tabaccum)を含む)、アルファルファ、ジャガイモ、チョウセンニンジン、エンドウ、エンバク、コメ、ダイズ、コムギ、オオムギ、ヒマワリ、ワタなどが含まれるが、ただしこれらに限定されない。
本発明の1つ以上のキメラ遺伝子構築物はさらに3'非翻訳領域を含み得る。3'非翻訳領域は、ポリアデニル化シグナル及びmRNAプロセッシング又は遺伝子発現を実行することができる他の任意の調節シグナルを収納するDNAセグメントを含む遺伝子の部分に該当する。ポリアデニル化シグナルは、通常mRNA前駆体の3'末端にポリアデニル酸鎖を付加することを特徴とする。ポリアデニル化シグナルは、一般的には正規の形態5'AATAAA-3'(ただし変型は珍しくはないが)との相同性の存在によって認識される。本発明の1つ以上のキメラ遺伝子構築物はさらにまたエンハンサー(翻訳又は転写エンハンサー)を必要に応じて含み得る。これらのエンハンサー領域は当業者には周知であり、ATG開始コドン及び隣接配列を含み得る。開始コドンは、全配列の翻訳を担保するためにコード配列のリーディングフレームと一致しなければならない。
適切な3'領域の非限定的な例は、プラストシアニンの3'UTRを含む3'転写非翻訳領域であり、以下が含まれる:転写終了配列(配列番号:20)、アグロバクテリア菌腫瘍誘発(Ti)遺伝子(当分野では公知)のポリアデニル化シグナル、例えばノパリンシンターゼ(Nos遺伝子)及び植物遺伝子(例えばダイズ貯蔵タンパク質遺伝子)及びリブロース1,5-ビスホスフェートカルボキシラーゼ遺伝子の小サブユニット。
所望の場合には、本発明の構築物はさらに操作して選択可能マーカーを含ませ得る。しかしながら、これは不要な場合もある。有用な選択可能マーカーには、化学物質、例えば抗生物質(例えばゲンタマイシン、ヒグロマイシン、カナマイシン)又は除草剤(例えばホスフィノトリシン、グリホセート、クロロスルフロンなど)に対して耐性を提供する酵素が含まれる。同様に、色の変化によって識別可能な化合物の生成を提供する酵素、例えばGUS(ベータ-グルクロニダーゼ)又はルミネセンス(例えばルシフェラーゼ又はGFP)を用いてもよい。
【0030】
本発明で考慮される部分はまた、本発明のキメラ遺伝子構築物を含むトランスジェニック植物、植物細胞又は種子であって、前記は、本明細書に記載の一過性タンパク質発現に適したプラットフォーム植物として用いられ得る。植物細胞から植物全体を再生する方法もまた当分野では公知である。一般的には、形質転換細胞は適切な培地で培養され、前記培地は選別物質(例えば抗生物質)を含み得る。選別可能マーカーは形質転換植物細胞の識別を容易にするために用いられる。いったんカルスが形成されたら、シュートの形成は、公知の方法にしたがって適切な植物ホルモンを利用することによって促進することができ、このシュートを発根培地に移して植物を再生させる。続いて、この植物を用いて、反復生成を種子から又は栄養繁殖技術を用いて確立し得る。トランスジェニック植物はまた組織培養を用いることなく作製することができる。
安定な形質転換のための方法及びこれら生物の再生は当分野では確立されてあり、当業者には公知である。形質転換及び再生植物を得る方法は、本発明にとって重要ではない。
“形質転換”とは、遺伝子型として、表現型として又は両方において表出される遺伝情報(ヌクレオチド配列)の種間移転を意味する。キメラ構築物から宿主への遺伝情報の種間移転が遺伝可能であって、遺伝情報の移転は安定であるとみなされることもあるが、また移転は一過性であって、遺伝情報の移転は遺伝可能でなくてもよい。
本発明はさらに、安定な発現系又は一過性発現系による使用に適したキメラ構築物を含む適切なベクターを含む。遺伝情報はまた1つ以上の構築物で提供し得る。例えば、対象のタンパク質をコードするヌクレオチド配列は一方の構築物に導入され、対象タンパク質のグリコシル化を改変するタンパク質をコードする第二のヌクレオチド配列は別の構築物を用いて導入されえる。続いてこれらのヌクレオチド配列は、本明細書に記載するように植物内で一過性に同時発現させ得る。対象タンパク質及び対象タンパク質のグリコシル化プロフィルを改変するタンパク質の両タンパク質をコードするヌクレオチド配列を含む構築物もまた用いられ得る。この場合、ヌクレオチド配列は、対象タンパク質をコードする第一の核酸配列(プロモーター又は調節領域と機能的に連結されている)を含む第一の配列、及び対象タンパク質のグリコシル化プロフィルを改変するタンパク質をコードする第二の核酸配列(第二の配列はプロモーター又は調節領域と機能的に連結されている)を含む第二の配列を含むであろう。
【0031】
“同時発現”とは、2つ以上のヌクレオチド配列が、ほぼ同じときに植物内で、及び植物の同じ組織で発現されることを意味する。しかしながら、前記ヌクレオチド配列は、厳密に同じときに発現される必要はない。むしろ、2つ以上のヌクレオチド配列は、コードされた生成物が相互作用する機会を有することができるように発現される。例えば、対象のタンパク質のグリコシル化を改変するタンパク質は、対象タンパク質のグリコシル化の改変が生じるように、対象タンパク質が発現される前又は発現期間中に発現される。2つ以上のヌクレオチドは一過性発現系を用いて同時発現させることができる。この場合、2つ以上の配列は、両配列が発現される条件下でほぼ同じときに植物に導入される。また別には、ヌクレオチド配列の一方(例えば対象タンパク質のグリコシル化プロフィルを改変するタンパク質をコードする配列)を含むプラットフォーム植物は、プラットフォーム植物に一過性態様で導入される対象タンパク質をコードする追加の配列で安定的な態様で形質転換させ得る。この場合、対象タンパク質のグリコシル化プロフィルを改変するタンパク質をコードする配列は、所望の組織で所望の発育段階の間に発現させ得る。あるいは、その発現は誘導性プロモーターを用いて誘導し得、対象タンパク質をコードする追加の配列は、類似の条件下で及び同じ組織で発現させて、ヌクレオチド配列が同時発現されることを担保し得る。
【0032】
本発明の構築物は、Tiプラスミド、Riプラスミド、植物ウイルスベクター、直接DNA形質転換、マイクロインジェクション、エレクトロポレーションなどを用いて植物細胞に導入することができる。そのような技術の検討のためには例えば以下を参照されたい:Weissbach and Weissbach, Methods for Plant Molecular Biology, Academy Press, New York VIII, pp. 421-463 (1988);Geierson and Corey, Plant Molecular Biology, 2d Ed. (1988);及びMiki and Iyer, Fundamentals of Gene Transfer in Plants. ”Plant Metabolism”, 2d Ed. DT. Dennis, DH Turpin, DD Lefebrve, DB Layzell (eds), Addison Wesly, Langmans Ltd. London, pp. 561-579 (1997)。他の方法には、直接DNA取込み、リポソームの使用、エレクトロポレーション(例えばプロトプラストを用いる)、マイクロインジェクション、マイクロプロジェクティル又はウィスカー(whisker)、及び真空浸透が含まれる。例えば以下を参照されたい:Bilang, et al. (Gene 100: 247-250 (1991);Scheid et al. (Mol. Gen. Genet. 228: 104-112, 1991);Guerche et al. (Plant Science 52: 111-116, 1987);Neuhause et al. (Theor. Appl Genet. 75: 30-36, 1987);Klein et al., Nature 327: 70-73 (1987);Howell et al. (Science 208: 1265, 1980);Horsch et al. (Science 227: 1229-1231, 1985);DeBlock et al., (Plant Physiology 91: 694-701, 1989);Methods for Plant Molecular Biology (Weissbach and Weissbach, eds., Academic Press Inc., 1988);Methods in Plant Molecular Biology (Schuler and Zielinski, eds., Academic Press Inc., 1989);Liu and Lomonossoff (J Virol Meth, 105:343-348, 2002,);米国特許4,945,050号、5,036,006号及び5,100,792号;米国特許出願08/438,666号(1995年5月10日出願)及び07/951,715号(1992年9月25日出願)(前記文献はいずれも参照により本明細書に含まれる)。
【0033】
下記で述べるように、一過性発現方法を用いて、本発明の構築物を発現させることができる(以下を参照されたい:Liu and Lomonossoff, 2002, Journal of Virological Methods, 105:343-348、前記文献は参照により本明細書に含まれる)。また別には、Kapilaら(1997)(前記文献は参照により本明細書に含まれる)が記載した真空による一過性発現方法を用いてもよい。これらの方法には、例えばアグロバクテリア接種(Agro-inoculation)若しくはアグロバクテリア浸透(Agro-infiltration)、注射筒浸透が含まれるが(ただしこれらに限定されない)、上記に記載した他の方法もまた用いることができる。アグロバクテリア接種、アグロバクテリア浸透、又は注射筒浸透に関しては、所望の核酸を含むアグロバクテリア菌の混合物が、組織(例えば葉)、植物の気生部分(茎、葉及び花を含む)、植物の他の部分(茎、根、花)又は全植物の細胞間間隙に入り込む。
表皮をクロスさせた後、アグロバクテリア菌が感染してt-DNAコピーが細胞に移される。このt-DNAはエピソーム的に(episomally)転写され、mRNAが翻訳されて感染細胞で対象タンパク質の生成をもたらすが、t-DNAの核内での継代は一過性である。
“対象の遺伝子”、“対象のヌクレオチド配列”、又は“対象のコード領域”(これらの用語は相互に用いられる)とは、植物又は植物の部分で発現されるべき任意の遺伝子、ヌクレオチド配列又はコード領域を意味する。そのような対象ヌクレオチド配列には、その生成物が対象タンパク質である配列又はコード領域が含まれるが、ただしこれらに限定されない。対象タンパク質の例には以下が含まれる(ただしこれらに限定されない):例えば工業用酵素(例えばセルラーゼ、キシラナーゼ、プロテアーゼ、ペルオキシダーゼ)、スブチリシン、タンパク質サプリメント、栄養性医薬(nutraceutical)、付加価値製品、又は前記のフラグメントであって飼料、食品若しくは飼料と食品双方に使用するためのもの、医薬的に活性なタンパク質(例えば成長因子、生長調節物質、抗体、抗原及び前記のフラグメント、又は免疫付与又はワクチン接種のために有用な前記の誘導体が含まれるが、ただしこれらに限定されない)など。さらに追加される対象タンパク質には以下が含まれる(ただしこれらに限定されない):インターロイキン(例えば1つ以上のIL-1からIL-24、IL-26及びIL-27)、サイトカイン、エリスロポエチン(EPO)、インスリン、G-CSF、GM-CSF、hPG-CSF、M-CSF若しくは前記の組合せ、インターフェロン(例えばインターフェロン-アルファ、インターフェロン-ベータ、インターフェロン-ガンマ)、血液凝固因子(例えば第VIII因子、第IX因子)、tPA hGH、レセプター、レセプターアゴニスト、抗体、神経ポリペプチド、インスリン、ワクチン、成長因子(例えば表皮増殖因子、ケラチノサイト増殖因子、形質転換増殖因子、増殖調節因子が含まれるが、ただしこれらに限定されない)、抗原、自己抗原、前記のフラグメント、又は前記の組合せ。
【0034】
対象のヌクレオチド配列が、植物に対して直接的に又は間接的に毒性を有する生成物をコードする場合は、本発明の方法を用いることによって、対象の遺伝子を一過性に発現させて植物全体でそのような毒性を低下させ得る。
下記実施例でより詳細に記載するように、改変N-グリコシル化を有する、対象のタンパク質(例えば抗体、C5-1(ただし前記に限定されない))の合成は、GalT(配列番号:14、図1b)又はGNT1-GalT(配列番号:17、図1c)のどちらかを一過性に同時発現する植物で実施された。
本明細書に記載の一過性発現プロセスの利点は、抗体の一過性発現に用いられるアグロバクテリア菌株数が最小限になり、これはコストを削減し、操作を簡単にし、系の堅実性を高める。Giritchら(2006)によって提唱された一過性発現系は、2つの非競合性ウイルスベクターでの抗体の軽鎖及び重鎖の発現に依存する。この系はまた、プロベクター分子、リコンビナーゼ及び2つのウイルスレプリカーゼの発現のために6つの異なるアグロバクテリア菌培養の同時浸透を必要とする。産業的展望から、6つの接種物の同時調製は、装置における多大なコスト並びに有効性実証及びスケールアップ操作までの期間を必要とする。さらにまた、細菌ベクターの数の増大は、多数の遺伝子の協調的発現に依存するこの発現系の堅実性に影響を与える可能性がある。
比較すれば、本明細書で提唱する系は、ほんの2つの別個のアグロバクテリア菌培養の同時浸透を必要とするだけである。アグロバクテリア菌の培養数は、サイレンシングのサプレッサー(例えばHcPro)をコードする配列、又は対象タンパク質を改変する任意の他の配列を、抗体発現カセットと同じプラスミドに含めることによってただ1つの培養にまで減らすことができる。
【0035】
配列リスト:

【実施例1】
【0036】
発現カセットR610、R612、R514(図1)、R621及びR622(図5)のアッセンブリ
全ての操作は、SambrookとRussel(2001)の一般分子生物学プロトコルを用いて実施した。
使用したオリゴヌクレオチドプライマーを下記に示す:
【0037】
XmaI-pPlas.c: 配列番号:1
5'-AGTTCCCCGGGCTGGTATATTTATATGTTGTC-3' 配列番号:1
SacI-ATG-pPlas.r: 配列番号:2
5'-AATAGAGCTCCATTTTCTCTCAAGATGATTAATTAATTAATTAGTC-3' 配列番号:2
SacI-PlasTer.c: 配列番号:3
5'-AATAGAGCTCGTTAAAATGCTTCTTCGTCTCCTATTTATAATATGG-3' 配列番号:3
EcoRI-PlasTer.r: 配列番号:4
5'-TTACGAATTCTCCTTCCTAATTGGTGTACTATCATTTATCAAAGGGGA-3' 配列番号:4
Plasto-443c: 配列番号:5
5'-GTATTAGTAATTAGAATTTGGTGTC-3' 配列番号:5
Plas+LC-C51.r: 配列番号:6
5'-ATCTGAGGTGTGAAAACCATTTTCTCTCAAGATG-3' 配列番号:6
LC-C51.c: 配列番号:7
5'-ATGGTTTTCACACCTCAGATACTTGG-3' 配列番号:7
LC-C51XhoSac.r: 配列番号:8
5'-ATATGAGCTCCTCGAGCTAACACTCATTCCTGTTGAAGC-3' 配列番号:8
【0038】
Plas+HC-C51.r: 配列番号:9
5'-CAAGGTCCACACCCAAGCCATTTTCTCTCAAGATG-3' 配列番号:9
HC-C51.c: 配列番号:10
5'-ATGGCTTGGGTGTGGACCTTGC-3' 配列番号:10
HC-C51XhoSac.r: 配列番号:11
5'-ATAAGAGCTCCTCGAGTCATTTACCAGGAGAGTGGG-3' 配列番号:11
HC-C51KDEL(SacI).r: 配列番号:12
5'-ATAAGAGCTCTCAAAGTTCATCCTTTTTACCAGGAGAGTGGG-3' 配列番号:12
XhoTEV.c: 配列番号:23
5'-TTTGGAGAGGACCTCGAGAAATAACAAATCTCAACAC-3' 配列番号:23
TEV+LC-C5-1.r: 配列番号:24
5'-ATCTGAGGTGTGAAACCATTGCTATCGTTCGTAAATGGTG-3' 配列番号:24
LC-C5-1.c: 配列番号:25
5'-ATGGTTTTCACACCTCAGATACTTGG-3' 配列番号: 25
LC-C5-1SphSac.r: 配列番号;26
5'-ATATGAGCTGCGATGCCTAACACTCATTCCTGTTGAAGC-3' 配列番号:26
【0039】
第一のクローニング工程は、アルファルファのプラストシアニン遺伝子の上流及び下流の調節エレメントを含むレセプタープラスミドを組み立てることであった。プラストシアニンプロモーター(米国特許7,125,978号、前記文献は参照により本明細書に含まれる)及び5'UTR配列は、オリゴヌクレオチドプライマーXmaI-pPlas.c(配列番号:1)及びSacI-ATG-pPlas.r(配列番号:2)を用いて、アルファルファのゲノムDNAから増幅した。得られた増幅生成物をXmaI及びSacIで消化して、pCAMBIA2300(以前に同じ酵素で消化しておいた)に連結し、pCAMBIA-PromoPlastoを作製した。同様に、プラストシアニン遺伝子の3'UTR配列及びターミネーター(図1c、配列番号:20のヌクレオチド1−399)を、以下のプライマー(SacI-PlasTer.c(配列番号:3)及びEcoRI-PlasTer.r(配列番号:4))を用いアルファルファのゲノム遺伝子から増幅し、生成物をpCAMBIA-PromoPlastoの同じ部位に挿入する前にSacI及びEcoRIで消化してpCAMBIAPlastoを作製した。
プラスミドR610及びR612は、アルファルファのプラストシアニンプロモーター下でC5-1軽鎖及びC5-1重鎖コード配列をタンデム構築物として含むように調製された。R610はアッセンブリされたIgGがERで保持されるように設計され、さらに前記はC5-1の重鎖末端に融合したKDELを含んでいた。R612は分泌が可能なように設計された。
C5-1発現カセットのアッセンブリは、Darveauら(1995)が記載したPCR仲介連結方法を用いて実施した。プラストシアニンの下流の軽鎖コード配列をアッセンブリするために、第一の工程は、開始ATGの下流にあるD'Aoustら(米国特許7,125,978号、前記文献は参照により本明細書に含まれる)が記載したアルファルファプラストシアニンプロモーターの最初の443塩基対(bp)(図1bのヌクレオチド556−999或いは配列番号:19)を、pCAMBIAPlastoを鋳型とし以下のプライマーを用いて増幅させることであった:
Plasto-443c(配列番号:5)及びPlas+LC-C51.r(配列番号:6、オーバーラップには上記で下線が付されている)。
【0040】
並行して、軽鎖コード配列を、プラスミドpGA643-kappa(Khoudi et al. 1999)から以下のプライマーを用いてPCR増幅させた:
LC-C51.c(配列番号:7)及びLC-C51XhoSac.r.(配列番号:8、オーバーラップには下線が付されている)。
得られた2つの増幅生成物を混合して、プライマーPlasto-443c(配列番号:5)及びLC-C51XhoSac.r(配列番号:8)による第三のPCR反応で鋳型として用いた。第一の反応で用いられるプライマーPlas+LC-C51.r(配列番号:6)とLC-C51.c(配列番号:7)との間のオーバーラップは、第三の反応中に増幅生成物のアッセンブリをもたらす。第三のPCR反応から生成されたアッセンブリ生成物をDraIII及びSacIで消化し、DraIII及びSacIで消化したpCAMBIAPlastoに連結してプラスミドR540を作製した。
重鎖コード配列は、プラストシアニンの開始ATGの上流の443 bp、ヌクレオチド556−999(図1b、配列番号:19、)を、pCAMBIAPlastoを鋳型とし以下のプライマーを用いて増幅させることによってプラストシアニンの上流の調節エレメントと融合させた:
Plasto-443c(配列番号:5)及びPlas+HC-C51.r(配列番号:9、オーバーラップには上記で下線が付されている)。
【0041】
これら反応の生成物を混合して、プライマーPlasto-443c(配列番号:5)及びHC-C51XhoSac.r.(配列番号:11)を用い第三のPCR反応でアッセンブリさせた。得られたフラグメントをDraIII及びSacIで消化し、pCAMBIAPlastoのDraIIIとSacI部位の間に連結した。この生成プラスミドをR541と名付けた。
プラスミドR541を鋳型として用い、プライマーPlasto-443c(配列番号:5)及びHC-C51KDEL(SacI).r(配列番号:12)によりPCR増幅によって、重鎖コード配列のC-末端にKDELタグを付加した。生成フラグメントをDraIII及びSacIで消化し、pCAMBIAPlastoの同じ部位にてクローニングし、R550を作製した。
軽鎖及び重鎖発現カセットの同じ二元プラスミドでのアッセンブリは以下のように実施した:R541及びR550をEcoRIで消化して平滑端を得、さらにHindIIIで消化して、R540のHindIII及びSmaI部位に連結してR610(KDELを有する)及びR612(KDELをもたない、図1参照)を作製した。
R514(図5a)
使用したさらに別のオリゴヌクレオチドプライマーは以下に提供される:
【0042】
Tev+HC-C51.2: 配列番号:16
5'-CAAGGTCCACACCCAAGCCATTGCTATCGTTCGTAAATGGTG-3' 配列番号:16
HC-C51SphSac.r 配列番号:32
5'-ATAAGAGCTCGCATGCTCATTTACCAGGAGAGTGGG-3' 配列番号:32
【0043】
完全長のC5-1軽鎖及び重鎖遺伝子(LC及びHC)は、Hema-Quebecから提供され、Darveau(1995)が記載したポリメラーゼ連鎖反応(PCR)仲介方法を用い、植物二元発現ベクターにてインフレーム(in-frame)でクローニングした。先ず初めにタバコエッチウイルス(TEV)エンハンサーを、プライマー”Xho”TEV.c(配列番号:23)及びTEV+LC-C51(配列番号:24)を用いTEVゲノムRNA(Acc. No. NC001555)を基にRT-PCRによって増幅した。並行して、C5-1軽鎖コード配列を、LCのためにプライマーLC-C51.c(配列番号:25)及びLC-C51”SphSac”.r(配列番号:26)を用いプラスミドpGA643(Khoudi et al. 1999)からPCRによって増幅した。続いてTEV及び軽鎖増幅フラグメントを混合し、プライマーとして”Xho”TEV.c(配列番号:23)及びLC-C51”SphSac”.r(配列番号:26)を用いさらにもう1回のPCRによってアッセンブリさせた。生成されたTEV/C5-1LCフラグメントを続いて精製し、中間ベクターにて2X35SプロモーターとNOSターミネーターとの間でXhoI-SacI消化物としてクローニングした。図1dは、使用した2X35Sプロモーター(太字、配列番号:33)及びNOSターミネーター(イタリック体、配列番号:34)並びに制限部位の位置(下線)を示している。続いて、この発現カセットをpCAMBIA2300二元プラスミドにHindIII-EcoRIフラグメントとして移入してプラスミドR512を作製した。
pR513を作製するために、TEVエンハンサーを、プライマー”Xho”TEV.c(配列番号:23)及びTEV+HC-C51.r(配列番号:16)を用いTEVゲノムRNA(Acc. No. NC001555)を基にRT-PCRによって増幅した。並行して、抗体の重鎖のコード配列を、プライマーHC-C51.c(配列番号:10)及びHC-C51”SphSac”.r(配列番号:32)を用いPCRによって増幅した。生成されたTEV及び重鎖増幅フラグメントを混合し、プライマー”Xho”TEV.c(配列番号:23)及びHC-C51”SphSac”.r(配列番号:32)を用いPCRによってアッセンブリさせた。生成されたTEV/C5-1HCフラグメントを精製し、XhoI及びSacIで消化し、中間ベクターにて2X35SプロモーターとNOSターミネーターとの間の同じ部位でクローニングした。図1dは、使用した2X35Sプロモーター(配列番号:33)及び使用したNOSターミネーター(配列番号:34)並びに制限部位の位置を示している。得られた2X35S/TEV/C5-1HC/NOSフラグメントを含むプラスミドをEcoRIで消化し、クレノーフラグメントポリメラーゼを用いて平滑端とし、さらにHindIIIで消化した。続いて、このHindIII-EcoRI(平滑端)フラグメントをR512のHindIII-SmaI消化物に連結してプラスミドR514を作製した。
R621及びR622(図5a)−用いたオリゴヌクレオチドプライマーを下記に示す:
【0044】
FgalT 配列番号:27
5'-GACTCTAGAGCGGGAAGATGAGGCTTCGGGAGCCGCTC-3' 配列番号:27
RgalTFlagStu 配列番号:28
5'- AAGGCCTACG CTACTTGTCAT CGTCATCTTT GTAGTCGCAC GGTGTCCCG AAGTCCAC -3' 配列番号: 28
FGNT 配列番号:29
5'-ATCGAAATCGCACGATGAGAGGGAACAAGTTTTGC-3' 配列番号: 29
RGNTSpe 配列番号:30
5'-CGGGATCCACTAGTCTGACGCTTCATTTGTTCTTC-3' 配列番号: 30
FgalTSpe 配列番号:31
5'-GGACTAGTGCACTGTCGCTGCCCGCCTGC-3' 配列番号: 31
【0045】
GalT及びGNT1GalT発現用プラスミドはpBLTI121(Pagny et al. 2003)からアッセンブリさせた。ヒトβ(1,4)-ガラクトシルトランスフェラーゼ(hGalT)遺伝子(UDPガラクトース:β-N-アセチルグルコサミド:β(1,4)-ガラクトシルトランスフェラーゼ;EC2.41.22)をpUC19-hGalT(Watzele et al. 1991)からEcoRI消化により入手した。クレノーで処理した後、1.2kbのhGalTフラグメントをpBLT221のSmaI部位でクローニングし、プラスミドpBLTI221hGalTを生成した。続いて、プライマーFGalT(配列番号:27)及びRGalTFlagStu(配列番号:28)を増幅のために用い、PCRによって前記コード領域のC-末端にFlagタグを融合させた。続いて、このXbaI-StuIフラグメントを二元ベクターpBLTI121にてクローニングすることによってR622を生成した。トランスメンブレンドメインに対応するN-アセチルグルコサミニルトランスフェラーゼI(GNT1)の最初の77アミノ酸を、鋳型としてN-GNT1をコードするN. tabacumのcDNA(Strasser et al. 1999)並びにプライマーとしてFGNT(配列番号:29)及びRGNTSpe(配列番号:30)を用いてPCRによって増幅させた。増幅生成物を先ず初めにpGEM-Tベクターにてクローニングし、得られたプラスミドをApaI及びBamHIで消化し、pBLTI221に連結し、pBLTI221-GNT1と称されるプラスミドを生成した。hGalTの触媒ドメインを、プライマーFGalTSpe(配列番号:31)及びRGalTFlagStu(配列番号:28)を用いてpBLTI221hGalTを基にPCR増幅によって入手し、SpeI及びStuI部位をそれぞれ5'及び3'末端に作製した。続いて、SpeI/StuI hGalTフラグメントを同じ(SpeI及びStuI)部位を用いてpBLTI221-GNT1にてクローニングし、pBLTI221-GNT1GalTを作製した。最後に、pBLTI221-GNT1GalTをXbaI及びStuIで消化し、GNT1GalTコード配列(図5d、配列番号:17)を単離し、このフラグメントを二元ベクターpBLTI121にてクローニングすることによってR621を作製した。
全てのクローンでシークェンシングを実施して構築物の完全性を確認した。これらのプラスミドを用い、アグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)を大腸菌(E.coli)形質転換の場合のように(W.S. Dower, Electroporation of Bacteria, In “Gentic Engineering”, Volume 12, Plenum Press, New York, 1990, J.K. Setlow eds.)Gene Pulser II装置(Biorad, Hercules, CA, USA)を用いて形質転換した。全てのA.ツメファシエンス株の完全性は制限マッピングによって確認した。
HcPro構築物はHamiltonらの記載にしたがって調製した。
【実施例2】
【0046】
植物バイオマスの調製、接種物、アグロバクテリア浸透及び採集
植物ニコチアナ・ベンタミアナ(Nicotiana benthamiana)を、市販の泥炭ゴケ基質を満たした平箱で種子から育てた。前記植物を16/8の光周期及び25℃日中/20℃夜間の温度スケジュールの下で温室にて生育させた。播種から3週間後に、個々の幼植物を採取してポットに移植し、さらに3週間同じ環境条件下で温室にて生育させた。形質転換の前に、植物から芽を摘み取るか又は植物を化学的に処理することによって、頂芽及び腋芽を下記に示す種々の時期に取り除く。
10mMの2-[N-モルフォリノ]エタンスルホン酸(MES)、20μMアセトシリンゴン、50μg/mLカナマイシン及び25μg/mLのカルベニシリン(pH5.6)を補充したYEB培地で、アグロバクテリア株R612、R610、R621、R622又は35SHcProをOD600が0.6から1.6に達するまで増殖させた。アグロバクテリア菌の懸濁液を使用前に遠心し、浸透培養液(10mMのMgCl2及び10mMのMES(pH5.6))に再懸濁した。
注射筒浸透は、Liu and Lomonossoffの記載(2002、Journal of Virological Methods, 105:343-348)にしたがって実施した。
真空浸透については、A.ツメファシエンス懸濁液を遠心し、浸透培養液に再懸濁して4℃で一晩保存した。浸透の日に、培養バッチを2.5培養体積に希釈し、使用前に温めた。ニコチアナ・ベンタミアナの全植物を気密なスチールタンク中の細菌懸濁液に上下さかさまにして20−40トルの真空下に2分静置した。注射筒又は真空浸透に続いて、植物を温室に戻し、採集まで4−5日間インキュベートした。
【0047】
葉のサンプリング及び全タンパク質の抽出:
インキュベーションに続いて、植物の気生部分を採集し、-80℃で凍結し、小片に粉砕して1.5又は7.5gのサブサンプルに分けた。凍結粉砕した植物材料の各サブサンプルを、冷却した50mMトリス(pH7.4)、0.15MのNaCl、0.1%のトリトンX-100、1mMフェニルメタンスルフォニルフロリド及び10μMキモスタチンの3体積中で均質化することによって(Polytron)、全可溶性タンパク質を抽出した。均質化後に、スラリーを4℃で20分、20,000gで遠心し、これらの清澄粗抽出物(上清)を分析のために維持した。参照標準物としてウシ血清アルブミンを用い、Bradfordアッセイ(Bio-Rad, Hercules, CA)によって、清澄粗抽出物の全タンパク質含有量を決定した。
【実施例3】
【0048】
タンパク質分析、免疫ブロッティング及びELISA
C5-1は抗ヒトネズミIgGであり、したがって検出及び定量は、ヒトIgGに対するその特徴的な親和性により(活性ブロット)または抗マウスIgG対するその免疫反応性によって実施することができる。
全粗抽出物又は精製抗体のタンパク質をSDS-PAGEによって分離し、クマシーブルーR-250若しくはG-250で染色するか、又は免疫的検出のためにポリビニレンジフロリドメンブレン(Roche Giagnostics Corporation, Indianapolis, IN)に電気的に移した。イムノブロッティングの前に、このメンブレンをトリス緩衝食塩水(TBS−T)中の5%脱脂乳及び0.1%トゥイーン20で4℃にて16−18時間ブロッキングした。
イムノブロッティングは以下の抗体とともにインキュベートすることにより実施した:ペルオキシダーゼ結合ヤギ抗マウスIgG(H+L)抗体(Jackson ImmunoResearch, West Grove, PA, Cat#115-035-146)(TBS-T中の2%脱脂乳にて0.04μg/mL)、ペルオキシダーゼ結合ヒトIgG抗体(Gamunex(商標)Bayer Corp., Elkhart, IN)(TBS-T中の2%脱脂乳にて0.2μg/mL)、又はポリクローナルヤギ抗マウスIgG抗体(重鎖特異的)(Sigma-Aldrich, St-Louis, MO)(TBS-T中の2%脱脂乳にて0.25μg/mL)。ペルオキシダーゼ結合ロバ抗ヤギIgG抗体(Jackson ImmunoResearch)(TBS-T中の2%脱脂乳にて0.04μg/mL)は、重鎖特異的抗体で処理したメンブレンについて二次抗体として用いた。免疫反応性複合体は、基質としてルミノール(Roche Giagnostics Corporation)を用いケミルミネセンスによって検出した。ヒトIgG抗体のセイヨウワサビペルオキシダーゼ酵素結合は、EZ-Link Plus(商標)活性化ペルオキシダーゼ結合キット(Pierce, Rockford, IL)を用いて実施した。
【0049】
ELISA定量アッセイ
マルチウェルプレート(Immulon 2HB, ThermoLab System, Franklin, MA)を、IgG1重鎖特異的ヤギ抗マウス抗体(Sigma M8770)(50mMの炭酸緩衝液(pH9.0)中に2.5μg/mL)で4℃にて16−18時間コーティングした。続いてマルチウェルプレートを、リン酸緩衝食塩水(PBS)(Pierce Biotechnology, Rockford, IL)中の1%カゼインで37℃にて1時間ブロッキングした。標準曲線は精製マウスIgG1コントロール(Sigma M9269)の希釈物を用いて作成した。イムノアッセイを実施しているときは、全ての希釈(コントロール及びサンプル)は、一切のマトリックスの作用を排除できるように、模擬接種物を浸透させインキュベートした植物組織から得た植物抽出物中で実施した。プレートをタンパク質サンプル及び標準曲線希釈物とともに1時間37℃でインキュベートした。PBS中の0.1%トゥイーン20で3回洗浄した後、このプレートをペルオキシダーゼ結合ヤギ抗マウスIgG(H+L)抗体(ブロッキング溶液中で0.04μg/mL)(Jackson ImmunoResearch 115-035-146)とともに37℃で1時間インキュベートした。PBS−Tでの洗浄を繰り返し、プレートを3,3',5,5'-テトラメチルベンジジン(TMB)Sure Blueペルオキシダーゼ基質(KPL, Gaithersburg, MD)とともにインキュベートした。反応を1NのHClを添加して停止させ、吸収を450nmで読み取った。各サンプルをトリプリケートでアッセイし、標準曲線の直線部分で濃度を内挿した。
【実施例4】
【0050】
IgG精製
材料の葉からのC5-1の精製は、N.ベンタミアナ(100−150g)の凍結葉を採取する工程、20mMリン酸ナトリウム、150mMのNaCl及び2mMメタ重亜硫酸ナトリウム(pH5.8−6.0)を添加する工程、及び市販のブレンダーを用いて室温で2−3分混ぜ合わせる工程を必要とした。不溶性線維はMiraclothTM(Calbiochem, San Diego, CA)での粗いろ過によって除去し、10mMフェニルメタンスルホニルフルオリド(PMSF)をろ液に添加した。1MのHClで抽出物をpH4.8±0.1に調整し、18000gで15分、2−8分遠心して清澄にした。この清澄にした上清を2MのTRISでpH8.0±0.1に調整し、再度18000gで15分、2−8分遠心して清澄にし、0.8及び0.2μmのメンブレン(Pall Corporation, Canada)で連続してろ過した。ろ過した材料は、有効面積が0.2ft2の100kDa分子量カットオフ限外ろ過膜(GE Healthcare Biosciences, Canada)を用いて接線流ろ過によって濃縮し、前記清澄材料の体積を5−10倍減少させた。続いて、この濃縮サンプルを、組換えプロテインG-セファロースファストフロー(Sigma-Aldrich, St-Louis, MO, Cat.#P4691)の5mmx5cmカラム(カラム体積1mL)に適用した。このカラムを5カラム体積の20mM TRIS-HCl、150mM NaCl(pH7.5)で洗浄した。抗体は100mMのグリシン(pH2.9−3.0)で溶出させ、計算した体積の1M TRIS-HCl(pH7.5)を含むチューブに採取することによって直ちにpHを中性にした。プールした溶出抗体分画を21000gで15分、2−8℃で遠心し、分析まで-80℃で保存した。精製後、前記アフィニティカラムを清浄にし、製造業者の指示にしたがって保存した。同じクロマトグラフィー媒体は、精製性能を顕著に変更することなく数回の精製に再使用することができる(10サイクルまで試験済み)。
【実施例5】
【0051】
N-グリコシル化分析
C5-1(50μg)を含むサンプルを15%SDS/PAGEで泳動した。重鎖及び軽鎖はクマシーブルーで明らかにし、重鎖に対応するタンパク質バンドを切り出し、小断片に切断した。断片を0.1M NH4HCO3/CH3CN(1/1)の溶液600μLで3回、各回15分間洗浄して乾燥させた。
ジスルフィド架橋の還元は、ゲル断片を0.1M NH4HCO3中の0.1MのDTT溶液600μLで56℃にて45分間インキュベートすることにより得られた。アルキル化は、0.1M NH4HCO3中のヨードアセトアミド55mMの溶液600μLを室温で30分添加することによって実施した。上清を廃棄し、ポリアクリルアミド断片を0.1M NH4HCO3/CH3CN(1/1)中でもう1回洗浄した。
続いてタンパク質を、0.05M NH4HCO3 600μL中の7.5μgトリプシン(Promega)で37℃にて16時間消化した。200μLのCH3CNを添加し、上清を収集した。続いて、ゲル断片を0.1M NH4HCO3 200μLで、その後200μLのCH3CNで再度、最後に200μLのギ酸(5%)で洗浄した。全ての上清をプールし凍結乾燥させた。
HPLCによるペプチドの分離は、C18逆相カラム(4.5x250mm)で0.1%のTFA中のCH3CNの直線勾配で実施した。分画を収集して凍結乾燥させ、337-nm窒素レーザーを搭載したVoyager DE-Pro MALDI-TOF装置(Applied Biosystems, USA)でMALDI-TOF-MSによって分析した。質量分析は、マトリックスとしてアルファ-シアノ-4-ヒドロキシ桂皮酸(Sigma-Aldrich)を用い、リフレクター遅延抽出モードで実施した。
【実施例6】
【0052】
アグロバクテリア浸透ニコチアナ・ベンタミアナの葉における一過性IgG発現の定量
強力なプラストシアニン系発現カセットが完全にアッセンブリされたIgGの高度な蓄積を駆動させることができるか否かを試験するために、pCambia二元プラスミドの同じT-DNAセグメント上で、C5-1(ネズミ抗ヒトIgG)(Khoudi et al. 1997)の軽鎖及び重鎖のコード配列をプラストシアニンプロモーター及び5'非翻訳配列の下流でタンデム構築物として、実施例1に記載するように(さらに図1にも提示されている)アッセンブリした。前記をさらにプラストシアニン3'非翻訳配列及び転写終了配列とフランキングさせた。
R612及びR610の両発現カセット(実施例1参照)では、軽鎖及び重鎖コード配列は、C5-1(Khoudi et al. 1999)由来の天然のシグナルペプチドを含んでいたが、R610では、アッセンブリされたIgGのゴルジ装置への移動を抑制するために、KDELペプチドのコード配列を重鎖のC-末端に付加した。
クローニング工程及びアグロバクテリウム・ツメファシエンス(AGL1)のプラスミドの移入に続いて、ニコチアナ・ベンタミアナの3つの植物の全ての葉に、プラスミドR612、R610又はR514(図1)で形質転換したアグロバクテリウム株を注射筒浸透させ、実施例2に記載したように分析を実施する前に、温室の条件下で6日間インキュベートした。インキュベーション期間の後、各植物の葉(ほぼ20gのバイオマス)を凍結し、粉砕し、さらに凍結粉末を混合して均質なサンプルを作製し、前記サンプルから1.5gの2つのサブサンプル(各植物から、実施例3参照)を抽出のために採取した。ポリクローナルヤギ抗マウスIgG1重鎖を捕捉用に、さらにペルオキシダーゼ結合ヤギ抗マウスIgG(H+L)を検出用に用いて、各サンプルの全タンパク抽出物中のC5-1含有量を酵素結合免疫吸着アッセイ(ELISA)によって定量した(実施例3参照)。
図2Aに示すように、R610又はR612(両構築物はプラストシアニンプロモーターを含む)は、サイレンシングサプレッサー(HcPro)の非存在下ではR514(2X35sプロモーターを含む)と比較したとき、極めて高レベルのタンパク質蓄積をもたらす。HcProの存在下では、R610及びR612の両方について大きく増加した発現レベルが観察された。図2Bに示すように、R612のアグロバクテリア浸透は、新鮮質量1kg当たり106mgの抗体の蓄積をもたらし、一方、抗体のER保持型(R610)では同じ条件で211mg/kgFWに達した。
【0053】
転写後遺伝子サイレンシング(PTGS)はアグロバクテリア浸透ニコチアナ・ベンタミアナ植物のトランスジーンの発現を制限し、ジャガイモウイルスYのサイレンシングサプレッサー(HcPro)の同時発現は、トランスジーンのmRNAの特異的分解を妨害することが示されたので(Brigneti et al. 1998)、HcPro構築物(Hamilton et al. 2002)の同時浸透をC5-1の発現増加における有効性について試験した。R612及びR610とHcProとの同時発現は、HcProの非存在下で観察された抗体蓄積と比較して、その蓄積をそれぞれ5.3倍及び3.6倍増加させた。HcProの存在下では、プラストシアニン制御C5-1発現は、R612では558mg/kgFW、R610では757mg/kgFWの平均値に達した(図2A)。C5-1発現の最大レベルは、R612及びR610の両浸透葉由来のいくつかの抽出物で1.5g/kgFW(全可溶性タンパク質の25%)を超えた。
アグロバクテリア浸透発現系の計測能を判定するために、Kapilaら(1997)の記載した方法から応用した真空浸透方法に続いて、C5-1の蓄積を定量した。この実験シリーズでは、全植物の気生部分にR612+HcPro又はR610+HcProを用いて真空浸透を実施し、採集前の6日間温室に戻した。大規模生産系に典型的なデータを提供するために、数植物から得られる葉/葉柄のほぼ250g量のバッチを凍結し、すり潰して均質なサンプルにし、さらにバッチ当たり7.5g量を含む3つのサブサンプルを分析用に収集した。ELISAによる定量で示されたように、平均的なC5-1蓄積レベルは、R612とR610浸透についてそれぞれ238及び328mg/kgFWに達した(図2B)。
【0054】
プルーニングの影響:
適切なプラスミドで形質転換したアグロバクテリア株を用いて葉に真空浸透を施す1、2又は3日前に、3つのニコチアナ・ベンタミアナ植物の頂芽及び腋芽を鋏で植物から機械的に除去するか、又はEthrel、B-9(500ppm)若しくはA-rest(4ppm)を用いて化学的にプルーニングした。
続いて、植物にインフルエンザ抗原(構築物312、図1)、ヒトIgG(構築物935、図1)を浸透させ、実施例2に記載した分析の前に温室条件下で6日間インキュベートした。コントロール植物はプルーニングしなかった。インキュベーション期間の後で、各植物の葉(ほぼ20gのバイオマス)を凍結してすり潰し、さらに凍結粉末を混合して均質なサンプルを作製し、それらサンプルから1.5グラムの2つのサブサンプル(各植物から、実施例3参照)を抽出用に採取した。ポリクローナルヤギ抗マウスIgG1重鎖を捕捉用に、さらにペルオキシダーゼ結合ヤギ抗マウスIgG(H+L)を検出用に用いて、各サンプルの全タンパク抽出物中のC5-1含有量を酵素結合免疫吸着アッセイ(ELISA)によって定量した(実施例3参照)。
図7Aに示すように、312(インフルエンザ抗原)のアグロバクテリア浸透12時間前に機械的プルーニングを実施して頂芽及び腋芽を除去することによって、コントロール処理(プルーニング実施せず)と比較して、抗原の蓄積の増加がもたらされた(150%)。頂芽優勢を阻害することが知られているいくつかの成長調節物質(Ethrel、500ppm;B-9、2500ppm;又はA-rest、4.0ppm)による植物の化学的プルーニング処理とそれに続く512のアグロバクテリアによる浸透を用いて、200%までの発現レベルの増加もまた観察された。同様な結果がまた、これらの成長調節化合物及び他の成長調節化合物を製造業者の推奨した適用率で浸透7日前に用いたときに観察された。
【0055】
機械的プルーニングもまた、植物をアグロバクテリア浸透12時間前に真空浸透(図7B)又は注射筒浸透(図7C)によってプルーニングしたとき、免疫グロブリン935(hIgG、図1)の発現レベルの増加をもたらした。
アグロバクテリア浸透の1から3日前の植物プルーニングは、図8に示すように(機械的プルーニング植物)、タンパク質(インフルエンザ抗原;312、図1)の蓄積の更なる増加をもたらした。発現の極めて大きな増加が、浸透の1から2日目に植物を機械的にプルーニングしたときに観察される。浸透の3日前又は7日前の植物の化学的プルーニングもまた、非プルーニング植物を超えるタンパク質蓄積増加がもたらすことが見出された。
図9に示すように、対象のコード領域が光合成プロモータープラストシアニンによって駆動され、プルーニング(真空浸透の12時間前に機械的プルーニング)とサイレンシングサプレッサーの同時発現とが併用されたとき、抗体(935、図1)の浸透に続いて発現レベルの増加が観察される。プルーニングに続く935とサイレンシングサプレッサーHcProとの同時発現は、HcProの非存在下で観察される抗体蓄積レベルと比較して3−8倍の抗体蓄積レベルの増加をもたらした。HcProと同時発現させたとき、プラストシアニンにより制御される発現は、プルーニングに続いて280mg/kgFWの平均値に達した。
【実施例7】
【0056】
生成抗体の性状決定
タンパク質ブロット分析(実施例3参照)を用いて、注射筒浸透及び真空浸透実験の両実験の後、分泌型(R612)及びER保持型(610)タンパク質を生成する植物でC5-1 IgGのアッセンブリ及び断片化レベルを明らかにした。H+Lペルオキシダーゼ結合ヤギ抗マウスIgGをプローブに用いたウェスタンブロットを先ず初めに用いて、C5-1分子におけるその由来とは無関係に、最大量の抗体フラグメントの存在を明瞭に示した。図3Aに示すように、全てのタンパク質抽出物は、使用した分子内標的誘導方法又は浸透方法とは無関係に、類似の分子サイズのフラグメントを類似の相対的豊富さで含んでいた。各事例で、完全な抗体に一致する約150kDaの主要(≧85%)バンドが、約135kDa及び約100kDaのマイナーバンドとともに明示され、抗体は主としてその完全なアッセンブリ型(H2L2)として蓄積されることが示された。興味深いことに、同様な電気泳動移動度を有するフラグメントがまた、ネズミ腫瘍細胞株(MOPC-21;Sigma #M9269)から生成されたコントロールIgGにも存在し、植物及び哺乳動物細胞株で生じる断片化は類似し、おそらく共通のタンパク質分解活性から生じることが示唆された。同様な結果がまた、検出用抗マウス重鎖特異的抗体を用いて得られた。
抽出物中に存在する抗体フラグメントの実体を調べるために、ブロットタンパク質をペルオキシダーゼ結合ヒトIgG1(C5-1の抗原)プローブを用いて試験する活性ブロットを利用した。約150kDaの完全にアッセンブリした抗体の実体は図3Bで知ることができる。さらにまた、ウェスタンブロットで観察される断片化パターンは活性ブロットで知ることができるが(図3B参照)、ただし100kDaバンドは例外である(図3A参照)。理論に拘束されないが、この結果は、100kDaフラグメントはC5-1抗体のFab領域を含まず、少なくとも部分的には重鎖ダイマー(抗体アッセンブリーの中間物)から成るかもしれない。
【実施例8】
【0057】
抗体精製及び精製生成物の性状決定
プロテインGアフィニティークロマトグラフィー単工程を用いてバイオマスから抗体を精製し、得られた生成物をSDS-PAGEによって分析した(実施例4参照)。図4に提示したクマシーブルー染色ゲルは、プロテインGから溶出した分画中の150kDaの主要バンドを示している。このバンドは、分泌型及びER保持型の両型で精製生成物の85%を占め、夾雑物の含有量は両型について同一である(図4A、レーン4及び5)。ポリクローナル抗マウスIgGプローブで調べたウェスタンブロット分析は、精製C5-1分画中の主要夾雑物がネズミIgG起源であることを示した(データは示されていない)。還元条件下では、2つの主要な生成物が約26kDa及び約55kDaで検出され、これらは、それぞれ軽鎖及び重鎖の分子量と一致している(図4B、レーン2)。ER保持抗体の重鎖は、アポプラスト抗体の重鎖より高い電気泳動移動度を示し(図4B、レーン3)、これは、C-末端に存在する付加KDELアミノ酸とER保持によるN-グリコシル化の相違とが重なった結果と解釈される。図4Cは、精製抗体(150kDa)が、75、90、100及び120kDaの夾雑フラグメントと同様にヒトIgG1と結合したことを示し、このフラグメントに少なくとも1つのFabセグメントが存在することを明瞭に示している。100kDaフラグメントのFabの存在は、粗抽出物から得られた結果と対照的で、後者では100kDaバンドはヒトIgG1と結合しなかった。粗抽出物において100kDaで泳動するFab-含有フラグメントの量はこの活性ブロットによる検出のためには低すぎたか、又は100kDaで泳動するフラグメントは2つの異なる分子(一方は重鎖ダイマー(Fab無し)及び他方は抗原結合領域を含む)から成っていたという仮説が成り立つ。
抗体生成についてこの系の再現性は、2つの異なる浸透バッチから精製した生成物及び各バッチ由来の3つの別個の精製ロットを並べて比較して判定された。精製ロットのクマシー染色SDS-PAGE分析によって、全てのロットで同一バンドが高度に類似する相対的豊富さで存在することが示された(図4D)。
【実施例9】
【0058】
ヒトガラクトシルトランスフェラーゼの同時発現による抗体のN-グリコシル化の改変
一過性同時発現を用いて、一過性発現中に生成されつつあるタンパク質のグリコシル化を制御することができるか否かを調べるために、天然のヒトβ1,4ガラクトシルトランスフェラーゼ(GalT)を含む35S系発現カセットを調製した。R622はGalTを含み(図5B)、R621は、N-アセチルグルコサミニルトランスフェラーゼ(GNTI)のCTSドメインと融合させたGalT触媒ドメインを含んでいた(GNT1GalT、図5A)。N-アセチルグルコサミニルトランスフェラーゼ(GNTI)のCTSドメインは、GNT1が複雑なN-グリカン合成の初期にER及びcis-ゴルジ装置で機能するので、ヒトGalT触媒ドメインのために膜足場として選択した(Saint-Jore-Dupas et al. 2006)。理論に拘束されないが、タンパク質成熟の初期にGalT活性を隔離することによって、成熟グリカンのβ1,4ガラクトース付加及びコアのフコシル化及びキシロシル化の効率的な阻害がもたらされるのかもしれない。これらの構築物をC5-1とともに植物に同時浸透させた。
ニコチアナ・ベンタミアナの植物に、HcProの存在下でR612(C5-1の分泌型)、R612+R621(GNT1GalT)又はR612+R622(GalT)を浸透させた。図6は、これらのバイオマスサンプルから精製したC5-1の免疫学的分析を示す。
抗体のガラクトシル化は、β1,4ガラクトースと特異的に結合するエリトリナ・クリスタガリ(Erythrina cristagali)アグルチニン(ECA)を用いる親和検出によって概算した。期待したように、C5-1を単独で発現させたとき(R612、図6)ガラクトースは検出されなかった。ガラクトシル化は、R512+R622(GalT)による同時浸透から精製したC5-1で認められたが、R612+R621(GNT1GalT、図6)による同時浸透から得たものでは認められなかった。抗α1,3フコース抗体を用いて実施したウェスタンブロットは、ガラクトシルトランスフェラーゼ無しに発現させたコントロールC5-1のN-グリカンのフコシル化を明示した。N-グリカンのフコシル化は、アグロバクテリア浸透の方法に関係なくGNT1GalTを同時発現させた抗体で検出されなかったが、一方、天然のGalTの同時発現は抗体のフコシル化の検出可能な低下をもたらさなかった(図6)。同様な結果が抗β1,2キシロース特異的抗体を用いて得られた。前記の結果では、GNT1GalT同時発現C5-1におけるキシロース特異的免疫シグナルの完全な欠如、及びGalT同時発現C5-1におけるキシロース特異的免疫シグナルの存在が示された。
【0059】
完全にアッセンブリされたIgGの直接視覚的概算のためのクマシーゲル染色、並びにウェスタン及び活性ブロットを同じ抽出物について実施した。このデータを基にすれば、記載した抗体発現系は1.5g/kg新鮮質量の収量を達成し、粗抽出物中の生成物の85%を越えるものが、約150kDaの完全サイズのテトラマーIgGから成っていた。
重鎖のC-末端へのKDELペプチドの付加が以前に用いられ、ゴルジからERへの抗体の回収を仲介することにより抗体蓄積を増加させた(2−10倍)(Schillberg et al. 2003)。本明細書に記載の発現系を用いたとき、C5-1の重鎖へのKDELペプチドの付加は、HcProサイレンシングサプレッサーを使用しないときC5-1の収量を倍増させた。KDELの存在下又は非存在下におけるC5-1の収量の相違は、HcProを使用してサイレンシングを低下させたとき顕著に減少した。ER保持は生成物の品質に影響を与えなかった。なぜならば、ER保持型抗体及び分泌型抗体を産生する植物から得られる粗抽出物で観察されるフラグメントでは、サイズ及び相対的豊富さが同一であったからである。
全ての引用文献は参照により本明細書に含まれる。
本発明は1つ以上の実施態様に関して記載してきた。しかしながら、多数の変型及び改変が、特許請求の範囲で規定した本発明の範囲から逸脱することなく実施されえることは当業者には明白であろう。
【0060】
参考文献
【技術分野】
【0001】
本発明は、植物でタンパク質を製造する方法に関する。本発明はまた、植物でタンパク質を製造するために用い得るヌクレオチド配列を提供する。
【背景技術】
【0002】
免疫グロブリン(IgG)は、多様な性質を有するその特異的抗原対応物に対して特徴的な親和性を有する複雑なヘテロマルチマータンパク質である。今日、IgG産生細胞株の日常的な単離、並びにIgG誘導進化及び分子操作のための技術の出現は、生物性治療薬として及び一般ライフサイエンス市場におけるIgGの展開に重大な影響を及ぼした。治療用モノクローナルIgG(モノクローナル抗体、mAb)は、現在の新規な抗炎症薬及び抗癌薬の市場で優勢であり、さらに何百もの新規な候補物質が、改善された適用又は新規な適用のために現時点で研究及び臨床開発下にある。mAbの市場における年間需要は、数グラム(診断薬)、数キログラム(抗毒素)から百又は数百キログラム(生物防衛、抗癌、抗感染、抗炎症)の範囲である。
CHO細胞株は、今もなお好ましいmAbの産業規模での生成宿主であるが、mAbがライフサイエンス市場でそれらの完璧な効果に達するためには、また別の生産系を開発する必要があることは一般的に理解されている。なぜならば、これらの培養に必要な施設は規模の調節が容易ではなく、それらの構築及び維持コストは極めて高価で、さらに着実に増加しつつあり、GMP基準下でのそれらの実証には構築後なお平均3年を要するからである。初期開発期においてすら、許容可能な収量及び生産性を有するCHO細胞株の選択が、コストのかかる長期に及ぶプロセスであることに変わりはない。上流でのコストを削減させえる新規な生産系(より高収量、より単純な技術及び基盤組織)がリードタイムを短縮し、能力をより柔軟にし、一方で従来の細胞培養系における従来の再現性、品質及び安全性特性を充足させる新規な生産系は、ライフサイエンス市場用のmAb及びワクチンの開発に対し全開発ステージでおそらく大きな影響をもつであろう。
植物は、mAb及びライフサイエンスでこれまで利用されてきたいくつかの他のタンパク質の製造に適した宿主である(最近の概説として以下を参照されたい:Ko and Koprowski 2005;Ma et al. 2005;Yusibov et al. 2006)。mAbは、安定なトランスジェニック植物株で新鮮質量(FW)1kg当たり200mgもの収量で、さらに一過性発現では20mg/kgFWもの割合で生産されている(Kathuria, 2002)。Giritchら(2006)は、IgGについて、葉の質量1kgにつき200−300mgの発現レベルを報告し、多重ウイルス系の一過性発現系を用いた場合の引用最大例は500mg/kgであった。
mAbの合成のために今日まで報告された(例えばKapila et al. 1997;Vaquero et al. 1999;Rodriguez et al. 2004)一過性の系の多くは複雑な方法を含むか、生成物の蓄積レベルが低いか、またはその両方をもたらす。高収量のタンパク質を生じる代替方法を述べる。
【発明の概要】
【発明が解決しようとする課題】
【0003】
本発明は、植物でタンパク質を製造する方法に関する。本発明はまた、植物でタンパク質を製造するために用い得るヌクレオチド配列を提供する。
本発明の目的は、植物でタンパク質を製造するための改善方法を提供することである。
【課題を解決するための手段】
【0004】
本発明は、以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法(A)を提供する:
i)植物又は植物の部分をプルーニングし、プルーニングされた植物又は植物の部分を生産する工程、
ii)植物中で活性な調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を、前記プルーニングされた植物又は植物の部分に一過性態様で導入する工程、及び
iii)前記プルーニングされた植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分中で発現させることを可能にする条件下で維持する工程。
【0005】
対象のタンパク質は抗体、抗原ワクチン又は酵素でありえる。
本発明はまた上記に記載の方法に関し、この場合、導入工程(工程ii)では、2つ以上の核酸配列を植物内に導入し得る。さらにまたこの2つ以上の核酸配列の1つはサイレンシングのサプレッサーをコードし得る。例えば、サイレンシングのサプレッサーは、HcPro、TEV-p1/HC-Pro、BYV-p21、TBSV p19、TCV-CP、CMV-2b、PVX-p25、PVM-p11、PVS-p11、BScV-p16、CTV-p23、GLRaV-2p24、GBV-p14、HLV-p10、GCLV-p16、又はGVA-p10でありえる。
本発明は上記に記載の方法を含み、この場合、導入工程(工程ii)では、1つ以上の核酸配列は、プルーニングした植物又は植物の部分にアグロバクテリア菌を用いて導入し得る。アグロバクテリア菌は、プルーニングした植物又は植物の部分に真空下又は注射筒によって導入し得る。さらにまた、上記に記載した導入工程(工程ii)では、調節領域は光合成遺伝子から得られるプロモーターを含む。例えば、調節領域は、プラストシアニンプロモーター、プラストシアニン3'UTR転写終了配列、又はプラストシアニンプロモーター及びプラストシアニン3'UTR転写終了配列の両方を含み得る。
【0006】
本発明はまた、以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法(B)に関する:
i)植物又は植物の部分中で活性な光合成遺伝子から得られる調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を一過性態様で導入する工程、及び
ii)前記植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分中で発現させることを可能にする条件下で維持する工程。
対象のタンパク質は抗体、抗原ワクチン又は酵素でありえる。
本発明はまた上記に記載の方法(B)に関し、この場合、導入工程(工程i)では、2つ以上の核酸配列が植物に導入される。さらにまたこの2つ以上の核酸配列の1つはサイレンシングのサプレッサーをコードし得る。例えば、サイレンシングのサプレッサーは、HcPro、TEV-p1/HC-Pro、BYV-p21、TBSV p19、TCV-CP、CMV-2b、PVX-p25、PVM-p11、PVS-p11、BScV-p16、CTV-p23、GLRaV-2p24、GBV-p14、HLV-p10、GCLV-p16、又はGVA-p10でありえる。
本発明は上記に記載の方法(B)を含み、この場合、導入工程(工程i)では、1つ以上の核酸配列は、プルーニングした植物又は植物の部分にアグロバクテリア菌を用いて導入し得る。アグロバクテリア菌は、プルーニングした植物又は植物の部分に真空下又は注射筒によって導入し得る。さらにまた、上記に記載した導入工程(工程ii)では、調節領域は光合成遺伝子から得られるプロモーターを含む。例えば、調節領域は、プラストシアニンプロモーター、プラストシアニン3'UTR転写終了配列、又はプラストシアニンプロモーター及びプラストシアニン3'UTR転写終了配列の両方を含み得る。
【0007】
本発明は、以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法(方法C)を提供する:
i)植物又は植物の部分をプルーニングし、プルーニングされた植物又は植物の部分を生産する工程、
ii)植物又は植物の部分中で活性な光合成遺伝子から得られる調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を一過性態様で導入する工程、及び
iii)前記プルーニングされた植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分中で発現させることを可能にする条件下で維持する工程。
【0008】
対象のタンパク質は抗体、抗原ワクチン又は酵素でありえる。
本発明はまた上記に記載の方法(C)に関し、この場合、導入工程(工程ii)では、2つ以上の核酸配列が植物に導入される。さらにまたこの2つ以上の核酸配列の1つはサイレンシングのサプレッサーをコードし得る。例えば、サイレンシングのサプレッサーは、HcPro、TEV-p1/HC-Pro、BYV-p21、TBSV p19、TCV-CP、CMV-2b、PVX-p25、PVM-p11、PVS-p11、BScV-p16、CTV-p23、GLRaV-2p24、GBV-p14、HLV-p10、GCLV-p16、又はGVA-p10でありえる。
本発明は上記に記載の方法(C)を含み、この場合、導入工程(工程ii)では、1つ以上の核酸配列は、プルーニングした植物又は植物の部分にアグロバクテリア菌を用いて導入し得る。アグロバクテリア菌は、プルーニングした植物又は植物の部分に真空下又は注射筒によって導入し得る。さらにまた、上記に記載した導入工程(工程ii)では、調節領域は光合成遺伝子から得られるプロモーターを含む。例えば、調節領域は、プラストシアニンプロモーター、プラストシアニン3'UTR転写終了配列、又はプラストシアニンプロモーター及びプラストシアニン3'UTR転写終了配列の両方を含み得る。
本発明は、一過性発現系を用いて植物で対象のタンパク質の発現を駆動させる、単純化した植物発現系を提供する。本明細書に記載の方法にしたがえば、対象のタンパク質を高収量で生産し得る。本明細書に記載の一過性同時発現系は、極めて長い製造時間、並びに従来技術(例えばBakker, 2005)で記載されたえり抜きの変異体又は糖操作トランスジェニック株の選別プロセス及びそれらの親株としてのその後の使用を回避する。本発現系はまた、変異体又は糖操作植物でしばしば遭遇し、同時に発生する生産性、花粉生産、種子セット(Bakker et al. 2005)及び生存率(Boisson et al. 2005)に関する問題を回避する。本明細書に記載の一過性発現系は、新鮮な葉の質量1kg当たり高品質の抗体1.5gに達する発現レベルをもたらし、他の発現系(多重ウイルスベースの系及びトランスジェニック植物を含む)を有する植物で任意の抗体に関して報告された蓄積レベルを超える。
【0009】
本明細書に記載するように、所望の核酸構築物の浸透(infiltration)前に植物をプルーニングすることによって発現レベル(全合成タンパク質の%として)及び収量(mgタンパク質/kg新鮮質量)の増加が観察された。前記は、注射筒による浸透(syringe-infiltration)又は真空浸透(vacuum-infiltration)を含む(ただしこれらに限定されない)いくつかの浸透方法を用いて観察された。多様なプルーニング方法(例えば機械的プルーニング又は化学的プルーニングを含むが、ただしこれらに限定されない)が、発現レベル及びタンパク質収量を増加させた。
光合成遺伝子由来の調節領域(例えばリブロース1,5-ビスホスフェートカルボキシラーゼ/オキシゲナーゼ(Rubisco)大若しくは小サブユニット又はプラストシアニンをコードする遺伝子から得られるものであるが、ただしこれらに限定されない)の使用、又は光合成遺伝子由来調節領域の使用は、発現レベル及び収量を増加させることが見出された。さらにまた、プルーニングと一緒に光合成遺伝子由来調節領域を使用することによって、発現レベル及び収量が増加することが見出された。
浸透技術は、小さなパイロットユニット内で一日当たりこの抗体のグラム量の生産を可能にする。前記パイロットユニットは、極めて短時間枠内での臨床試験用材料の生産及び年間でキログラムに達するマーケットサイズを有するライセンス製品の供給のために前出のような一過性発現系を使用することを可能にする。高品質の抗体が、ただ1回のアフィニティークロマトグラフィー工程後に浸透リーフから得られた。
本発明の上記要旨は必ずしも本発明のすべての特徴を記述しているわけではない。
本発明のこれらの特徴及び他の特徴は、添付の下記図面に言及する以下の記述からいっそう明白となるであろう。
【図面の簡単な説明】
【0010】
【図1A】図1Aはいくつかのタンパク質の発現のために組み立てた発現カセットの例を示す。R612は、C5-1 LC及びC5-1 HC(各々はプラストシアニンプロモーター及び5'UTR並びにプラストシアニンターミネーターの制御下にある)をコードするヌクレオチド配列を含む。R610は、C5-1 LC及びC5-1 HC-KDEL(各々はプラストシアニンプロモーター及び5'UTR並びにプラストシアニンターミネーターの制御下にある)をコードするヌクレオチド配列を含む。R514は、C5-1 LC及びC5-1 HCをコードするヌクレオチド配列を含む。C5-1 LC:C5-1軽鎖コード配列;C5-1 HC:C5-1重鎖コード配列(各々は2X35Sプロモーター、タバコエッチウイルス(TEV)リーダー配列及びNOSターミネーターの制御下にある)。935は、ヒトIgG-LC及びヒトIgG-HC(各々はプラストシアニンプロモーター及び5'UTR並びにプラストシアニンターミネーターの制御下にある)をコードするヌクレオチド配列を含む。312はfli抗原をコードするヌクレオチド配列を含み、前記はプラストシアニンプロモーター及び5'UTR並びにプラストシアニンターミネーターの制御下にある。
【図1B】図1Bはプラストシアニンプロモーター及び5'UTRのヌクレオチド配列(配列番号:19)を示し、転写開始部位は太字で示され、翻訳開始コドンは下線を付されている。
【図1C】図1Cはプラストシアニン3'UTR及びターミネーターのヌクレオチド配列(配列番号:20)を示し、停止コドンは下線を付されている。
【図1D】図1Dは、R512及びR513アッセンブリに用いられる中継プラスミドの2X35S(配列番号:33)及びNOS(配列番号:34)配列を示す。NOSターミネーター(配列番号:34)はイタリック体で示され、2X35S(配列番号:33)は太字で示されている。制限酵素部位は下線を付されている。
【図2A】図2は、種々の発現カセットを浸透させたニコチアナ・ベンタミアナ(Nicotiana benthamiana)の葉におけるC5-1抗体の蓄積を示す。図2Aは、サイレンシングサプレッサー(例えばHcPro)の存在下又は非存在下での、R514(35Sベースの発現カセット)、R610及びR612(プラストシアニンベースの発現カセット)の注射筒による浸透後に発現されたC5-1抗体の蓄積を示す。
【図2B】図2Bは、サイレンシングサプレッサー(例えばHcPro)を同時発現させ、又は同時発現させずに、R610及びR612(プラストシアニンベースの発現カセット)を用いた、真空浸透又は注射筒浸透葉におけるC5-1抗体の蓄積を示す。提示した値は、3つの植物(注射筒)における6測定値、又は約12の植物(250g)の個々の浸透バッチにおける6測定値から得た平均蓄積レベル及び標準偏差に該当する。
【図3A】図3は、注射筒及び真空浸透させた植物の抽出物における蓄積C5-1のタンパク質ブロット分析を示す。図3Aは、ペルオキシダーゼ結合ヤギ抗マウスIgG(H+L)を、R612(分泌用、レーン1)又はR610(ER保持用、レーン2)を浸透させた植物の抽出物に対して用いたイムノブロッティングを示す。C1:100ngの市販ネズミIgG1(Sigma M9269)、電気泳動の移動度のためのコントロールとしてローディング;C2:擬似浸透バイオマス(空ベクター)から抽出した12μgの全タンパク質;C3:擬似浸透バイオマス(空ベクター)から抽出した12μgの全タンパク質に加えられた100ngの市販ネズミIgG1(Sigma M9269)。
【図3B】図3Bは、ペルオキシダーゼ結合ヒトIgG1を、R612(分泌用、レーン1)又はR610(ER保持用、レーン2)を浸透させた植物の抽出物に対して用いた活性イムノブロッティングを示す。C1:ハイブリドーマから精製した2μgのコントロールC5-1(Khoudi et al. 1999);C2:擬似浸透バイオマス(空ベクター)から抽出した75μgの全タンパク質。
【図4A】図4は、R612(分泌用、レーン1)又はR610(ER保持用、レーン2)のどちらかを浸透させた植物から精製した抗体の分析を示す。図4Aは、非還元条件で実施した粗抽出物及び精製抗体のSDS-PAGEを示す。
【図4B】図4Bは、非還元条件で実施した精製抗体のSDS-PAGEを示す。
【図4C】図4Cは、ペルオキシダーゼ結合ヒトIgG1を用いて実施した精製抗体の活性イムノブロッティングを示す。
【図4D】図4Dは、種々の浸透バッチ由来の精製C5-1の6ロットの夾雑物の比較を示す。C:2.5μgの市販ネズミIgG1(Sigma M9269)、電気泳動の移動度のコントロールとしてローディング。
【図5A】図5Aは、ガラクトシルトランスフェラーゼ発現の天然型(R622)及びハイブリッド型(R621)のために組み立てたカセットの例を示す。GNTI-CTS:N-アセチルグルコース-アミニルトランスフェラーゼIのCTSドメイン;GalT-CAT:ヒトβ1,4ガラクトシルトランスフェラーゼの触媒ドメイン;GalT:ヒトβ1,4ガラクトシルトランスフェラーゼ。
【図5B】図5Bは、GalT(UDP-Gal:ベータGlcNacベータ1,4-ガラクトシルトランスフェラーゼポリペプチド1、ベータ-1,4-ガラクトシルトランスフェラーゼI)のヌクレオチド配列(配列番号:14)を示す。ATG開始部位は下線を付され、トランスメンブレンドメインは下線を付されイタリック体であり、太字の配列はヒトベータ1,4GalTの触媒ドメインに該当し、FLAGエピトープはイタリック体である。
【図5C】図5Cは、GalT(UDP-Gal:ベータGlcNacベータ1,4-ガラクトシルトランスフェラーゼポリペプチド1、ベータ-1,4-ガラクトシルトランスフェラーゼI)のアミノ酸配列(配列番号:15)を示す。トランスメンブレンドメインは下線を付されイタリック体であり、太字の配列はヒトベータ1,4GalTの触媒ドメインに該当し、FLAGエピトープはイタリック体である。
【図5D】図5Dは、GNTIGalTのヌクレオチド配列(配列番号:17)を示す。ATG開始部位は下線を付され、トランスメンブレンドメイン(CTS)は下線を付されイタリック体であり、太字の配列はヒトベータ1,4GalTの触媒ドメインに該当し、FLAGエピトープはイタリック体である。
【図5E】図5EはGNTIGalTのアミノ酸配列(配列番号:18)を示す。トランスメンブレンドメイン(CTS)は下線を付されイタリック体であり、太字の配列はヒトベータ1,4GalTの触媒ドメインに該当し、FLAGエピトープはイタリック体である。
【図5F】図5Fは、N-アセチルグルコサミントランスフェラーゼのCTSドメイン(細胞質テール、トランスメンブレンドメイン、幹領域)(GNT1;配列番号:21)を示す。
【図5G】図5GはCTSのアミノ酸(配列番号:22)を示す。
【図6】図6は、C5-1を発現する植物から得られ、タンパク質のための染色を施されたか、又はウェスタン分析に付された抽出物のプロフィルを示す。一番上のパネルはクマシー染色PAGEゲルを示す。上から二番目のパネルは、β1,4ガラクトースと特異的に結合するエリスリナ・クリスタガリ(Erythrina cristagali)アグルチニン(ECA)を用いた親和性検出を示す。上から三番目のパネルは抗α1,3フコース抗体を用いたウェスタンブロット分析を示す。一番下のパネルは抗β1,2キシロース特異的抗体を用いたウェスタンブロット分析を示す。R612:単独発現C5-1;R612+R622:GalTと同時発現させた(同時浸透させた)C5-1;R612+R621:GNT1-GalTと同時発現させたC5-1。
【図7A】図7は、発現に対する機械的又は化学的プルーニングの影響の例を示す。図7Aは、真空アグロバクテリア浸透(agroinfiltrated)植物における抗原発現(インフルエンザ発現;図1、312参照)に対するプルーニング(機械的プルーニング(浸透の12時間前)及び化学的プルーニング(浸透の7日前)の両プルーニング)の影響を示す。
【図7B】図7Bは、真空アグロバクテリア浸透植物における抗体発現(ヒトIgG、図1、935参照)に対する機械的プルーニング(浸透の12時間前)の影響を示す。
【図7C】図7Cは、注射筒アグロバクテリア浸透植物における抗原発現(インフルエンザ、図1,312参照)に対する機械的プルーニングの影響を示す。条件1:コントロール、非プルーニング植物;条件2:機械的プルーニング植物。
【図8】図8は、真空アグロバクテリア浸透植物における抗原蓄積に対する、プルーニング(機械的プルーニング)の日(形質転換前3、2、又は1日前)、又はプルーニング無し(コントロール)の影響の例を示す。
【図9】図9は、真空浸透された植物における抗体発現(ヒトIgG、図1、935)に対する、サイレンシングのサプレッサー(HcPro)及びプルーニング(浸透の12時間前機械的プルーニング)の併用の影響例を示す。Plasto-HcPro-プルーニング:935単独発現(プルーニング無し、サイレンシングサプレッサーの同時発現無し);Plasto-HcPro+プルーニング:935による形質転換12時間前に植物の機械的プルーニング(サイレンシングサプレッサーの同時発現無し);Plasto+HcPro+プルーニング:935及びHcProの同時発現12時間前に機械的プルーニング。
【発明を実施するための形態】
【0011】
本発明は、植物においてタンパク質を生産する方法に関する。本発明はまた、植物でタンパク質を生産するために用いられ得るヌクレオチド配列を提供する。
植物又は植物の部分内で対象のタンパク質を合成する方法が提供される。その基本的な形態で本方法は以下の工程を含む:植物又は植物の部分中で活性な光合成遺伝子から得られた調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を一過性態様で導入する工程、及び前記対象のタンパク質をコードする前記核酸配列の前記植物または植物の部分での発現を可能にする条件下で前記植物又は植物の部分を維持する工程。
本方法はさらに、先ず初めに、対象のタンパク質をコードする1つ以上の核酸配列を導入する前に植物又は植物の部分をプルーニングする工程を含んでもよい。この方法では、植物又は植物の部分をプルーニングした後で、植物中で活性な調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列が、プルーニングされた植物又は植物の部分に一過性態様で導入される。続いて、前記植物又は植物の部分は、対象のタンパク質をコードする核酸配列の前記植物または植物の部分での発現を可能にする条件下で維持される。
この方法と同様な一過性形質転換プロトコルであって、光合成遺伝子から得られた調節領域の使用、プルーニング工程、又は光合成遺伝子から得られた調節領域とプルーニング工程との両方の使用を含まないものを用いて同じ対象タンパク質を生産する方法とタンパク質生産を比較したとき、この方法を用いて高収量の対象タンパク質が生産された。
【0012】
安定なトランスジェニック発現系で使用するために設計された発現カセットで用いられるプロモーターは、一過性発現系で用いるときは効率が低いことが見出された(Giritch et al. 2006、Fisher, 1999a)。Giritchら(12206)は、各IgGサブユニットのために、1つのリコンビナーゼ及び2つのウイルス性レプリカーゼと一緒に異なるプロベクター(1つはTMV系で他方はPVX系)を同時発現させて、200mg/kgの範囲の発現レベルを達成できたことを示している。本明細書に記載するように、葉の発現で有効性を示したエンハンサー配列を含むプロモーターは、一過性発現で有効であることが見出された。非限定的例示には、プラストシアニンの発現を調節する際に用いられるプロモーターが含まれる(Pwee and Gray 1993、前記文献は参照により本明細書に含まれる)。理論に拘束されないが、核マトリックスへの結合による光合成遺伝子の上流の調節エレメントの結合は、強力な発現を仲介することができる(Sandhu et al. 1998;Chua et al. 2003)。例えば、エンドウのプラストシアニン遺伝子の翻訳開始部位から-784までを用いて、強力なレポーター遺伝子発現を仲介し得る。
光合成遺伝子由来の調節領域(例えばプラストシアニン調節領域であるが、ただしこれらに限定されない(US7,125,978、前記文献は参照により本明細書に含まれる))、又はリブロース1,5-ビスホスフェートカルボキシラーゼ/オキシゲナーゼ(rubisco;US4,962,028、前記文献は参照により本明細書に含まれる)から得られる調節領域、クロロフィルa/b結合タンパク質(CAB;Leutwiler et al. 1986、前記文献は参照により本明細書に含まれる)、ST-LS1(光化学反応系IIの酸素放出複合体と結合する;Stockhaus et al. 1989、前記文献は参照により本明細書に含まれる)を、本発明にしたがって用い得る。
【0013】
リブロース1,5-ビスホスフェートカルボキシラーゼ/オキシゲナーゼ(Rubisco)の大又は小サブユニットをコードする遺伝子若しくはプラストシアニンから得られる調節領域、又は光合成遺伝子から得られる調節領域をプルーニングと併用して使用することによって、発現レベル及び収量が増加することが見出された。例えば、図2Aに示すように、光合成プロモーター(プラストシアニンから得られる;図2A、R610、R612参照)によって駆動される対象のコード領域の浸透後の発現レベルは、35Sによって駆動される同じコード領域(図2A、R514)と比較したとき極めて高い。
したがって、本発明は、以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法を提供する:
i)植物又は植物の部分中で活性な光合成遺伝子から得られる調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を一過性態様で導入する工程、及び
ii)前記植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列の前記植物又は植物の部分での発現を可能にする条件下で維持する工程。
植物又は植物の部分は、1つ以上の核酸配列を導入する工程の前にプルーニングされ得る。所望の核酸構築物の浸透前に植物をプルーニングすることによって、発現レベル(全合成タンパク質の%として)及び収量(mgタンパク質/kg新鮮質量)が増加することが見出された。これは、いくつかの浸透方法(注射筒浸透又は真空浸透が含まれるが、ただしこれらに限定されない)及び多様なプルーニング方法(例えば機械的プルーニング又は化学的プルーニングを含むが、ただしこれらに限定されない)を用いて観察された。理論に拘束されないが、浸透前のプルーニングは、頂芽優勢の低下をもたらすことができ、さらに生長調節物質含有量(例えばジベレリン酸又はエチレン含有量(ただしこれらに限定されない))の低下をもたらしえる。これは、続いて葉の光合成能力の増加を刺激し、さらに光合成遺伝子の転写速度を増加させ得る。したがって、光合成遺伝子から得られる調節領域の使用はより高いタンパク質収量をもたらし得る。さらにまた、光合成遺伝子から得られる調節領域の使用と生長調節物質阻害剤(例えばエチレン又はジベレリン酸)の含有量を低下させる化学的プルーニングの併用はより高いタンパク質収量をもたらし得る。
【0014】
本明細書に記載する方法にしたがって対象タンパク質の収量の増加に対するプルーニングの影響が、機械的又は化学的プルーニング方法及び真空又は注射筒浸透を用いて観察される。プルーニングによる収量の増加は、植物を傷つけたときに、例えば注射筒浸透に続いて観察される(図7C参照)。これは、タンパク質発現の増加は、単なる植物の損傷に対する応答ではないことを示している。
プルーニングとは、1つ以上の腋芽の除去、1つ以上の頂芽の除去、又は1つ以上の腋芽及び1つ以上の頂芽の両方の除去を意味する。プルーニングはまた、枯らすこと(killing)、壊死の誘発、又は植物から芽を除去することなく頂芽及び腋芽の生長を低下させることを含む。芽の生長低下(又は芽の成長を低下させる)とは、処理されていない芽と比較して、芽が、例えば代謝活性の低下、一定期間を超えて約50%から100%又はそれらの間の任意の量のサイズの増加を示すことを意味する。プルーニングはまた、頂芽優勢を低下させる化合物を適用することによって達成され得る。化合物がプルーニングの目的のために適用される場合、用いられる投与量は典型的には化合物の製造業者が推奨する投与量である。
プルーニング(機械的又は化学的プルーニング)を、浸透の約20日前から浸透の約2日後に、又はその間の任意の時点、例えば浸透の7日(168時間)前から浸透の約2日(48時間)後、又はその間の任意の時点、例えば浸透の約48時間(2日)前から浸透の約1日(24時間)後、又はその間の任意の時点、又は浸透の前20日、19日、18日、17日、16日、15日、14日、13日、12日、11日、10日、9日、8日、又は168、144、120、96、72、60、50、40、36、32、30、28、26、24、22、20、18、16、14、12、10、8、6、4、2、1、0時間から浸透の後約1、2、4、6、8、10、12、14、16、18、20、22、24時間、又はそれらの間の任意の時点で実施し得る。プルーニングが浸透の72時間前に又はそれより以前に実施される場合、プルーニング方法は化学的プルーニングであることが好ましい(機械的プルーニングを用いる場合は再生長が生じる可能性があるので)。プルーニング方法が化学的プルーニングである場合は、浸透前により長時間を、例えば浸透前2、3、4、5、6又は7日、又はその間の任意の時点を用い得る。当業者は、プルーニング前の適切な間隔を容易に決定することができる。
【0015】
プルーニングは、当業者に公知の任意の手段によって実施することができ、芽の機械的除去が含まれるが、ただし前記に限定されない。芽の機械的除去は例えば、切断、刈取り、摘取り、例えばトングを用いた圧縮、限局凍結(例えば、液体窒素の限局蒸気を芽に誘導するか、又は液体窒素、ドライアイス、エタノール-ドライアイスなどを含む適切な冷却源を用いて冷却したトング若しくは他の装置を用いて芽を取り囲み、それによって芽の温度を下げて芽の生長を低下させるか又は芽を枯らす(kill)ことによる)が含まれるが、ただしこれらに限定されない。
プルーニングにはまた、化学的プルーニング、例えば、芽を枯らすか若しくは芽の生長を低下させる除草剤(化合物;プルーニング剤)の適用、又は芽を枯らすか若しくは芽の生長を低下させる生長調節剤の適用が含まれる。化学的プルーニングの使用は、噴霧、散布、浸漬、植物上への前記化合物の添加、又は前記化合物を含む溶液への植物のディッピングによって植物を容易に処理することができるので、プルーニングの効率的な処理態様を可能にする。植物は、浸透工程前に1回処理するか、又は浸透工程前に2回以上処理してもよい。使用され得る化合物の例には、除草剤、例えば植物生長調節物質Ethephon(例えばBromeflor、Cerone、Chlorethephon Ethrel、Florel、Prep及びFlordimex)、Daminozide(ブタンジオン酸モノ2,2-ジメチルヒドラジン、-コハク酸2,2-ジメチルヒドラジド;例えばB-9;Alar、Kylar、SADH、B-9、B-995、アミノジド)、Atrimmec(ジケグラクナトリウム)、マレインヒドラジド(1,2-ジヒドロ-3,6-ピリダジンジオン)、2-4-D(2,4,ジクロロフェノキシ酢酸) が含まれ(ただしこれらに限定されない)、さらにジベレリン酸合成の阻害剤、例えばCycocel(クロメクァトクロリド)、A-Rest(アンシミドール)、トリアゾール、例えばBonzi(パクロブトラゾール)、Sumahic(ユニコナゾール)、又は3-アミノ-1,2,4-トリアゾール(3-AT)が含まれる(ただしこれらに限定されない)。これらの化合物は、植物生長調節のために知られている投与量範囲で用いられ得る。例えば用いられる投与量範囲は、前記化合物の製造業者が推奨するものでありえる。これらの化合物はまた、植物生長調節のために知られた投与量範囲未満の投与量範囲で用いてもよい。例えば、用いられる投与量範囲は、前記化合物の製造業者が推奨する投与量範囲の75%、50%、25%で用いられ得る。これらの化合物は約0.2ppmから約5000ppm、及び選択した生長調節物質に応じて前記の間の任意の量で用いられ得る。さらにまた、プルーニング剤(化合物)は1回適用してもよいが、また必要に応じて追加の適用を施してもよい。例えば、化合物は1回適用してもよいが、浸透前又は浸透後の植物の化学的プルーニングを得るために、化合物を2回以上適用してもよい。化学的プルーニングを用いる場合は、化合物は浸透の20日前から浸透の約2日後、又はその間の任意の時間に適用し得る。例えば、浸透の14日、7日又は5日前の化合物の適用が効果的な使用でありえる。
【0016】
図7A、7B、7C、8及び9に示すように、浸透前の植物のプルーニングは、対象タンパク質の発現の増加をもたらす。この効果は機械的又は化学的プルーニング方法を用いたときに観察される。したがって、本発明は、以下の工程を含む、対象のタンパク質を植物又は植物の部分で合成する方法を提供する:
i)植物又は植物の部分をプルーニングし、プルーニングされた植物又は植物の部分を生産する工程、
ii)植物中で活性な調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を、前記プルーニングされた植物又は植物の部分に一過性態様で導入する工程、及び
iii)前記プルーニングされた植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分中で発現させることを可能にする条件下で維持する工程。
対象のタンパク質をコードする核酸配列は、当業者に公知の任意の適切な方法、例えば真空浸透(vacuum infiltration)又は注射筒浸透(syringe infiltration)(前記に限定されると考えるべきではない)によって植物又は植物の部分に導入し得る。真空浸透の方法は当分野では公知であり、Kapilaら(1997、前記文献は参照により本明細書に含まれる)の方法が含まれえるが、ただし前記に限定されない。浸透はまた、注射筒を用いて植物又は植物の部分に対象のタンパク質をコードする核酸配列を導入することにも関連する(Liu and Lomonossoff, 2002、前記文献は参照により本明細書に含まれる)。
本発明で用いる方法及び以前に記載された方法(例えばKapila et al. 1997;又はLiu and Lomonossoff, 2002)は、その使用前に一過性形質転換のためにアセトシリンゴンを含む培養液中でアグロバクテリア菌(Agrobacterium)を培養する。アセトシリンゴン及び他のフェノール系シグナル分子は、アグロバクテリア菌のvir機構を正の方向に調節することが知られている。本明細書に記載の対象タンパク質の発現レベルの増加は、アグロバクテリア菌をアセトシリンゴンの存在下又は非存在下で培養したときに観察される。
【0017】
転写後遺伝子サイレンシング(PTGS)は、植物でのトランスジーンの発現制限に中心的に関与する可能性があり、ジャガイモウイルスY由来のサイレンシングのサプレッサー(HcPro)の同時発現をトランスジーンmRNAの特異的分解に対抗するために用いられ得る(Brigneti et al. 1998)。また別のサイレンシングサプレッサーは当分野で周知であり、本明細書に記載するように用いられ得る(Chiba et al. 2006, Virology 346:7-14、前記文献は参照により本明細書に含まれる)。前記は例えば以下である(ただしこれらに限定されない):TEV-p1/HC-Pro(タバコエッチウイルス-p1/HC-Pro)、トマトブシースタントウイルスのBYV-p21、p19(TBSV p19)、トマトクリンクルウイルスのキャプシドタンパク質(TCV-CP)、キュウリモザイクウイルスの2b(VMC-2b)、ジャガイモウイルスXのp25(PVX-p25)、ジャガイモウイルスMのp11(PVM-p11)、ジャガイモウイルスSのp11(PVS-p11)、ブルーベリースコーチウイルスのp16(BScV-p16)、レモントリステキサウイルスのp23(CTV-p23)、ブドウの木のリーフロール関連ウイルス-2(GLRaV-2p24)、ブドウの木のウイルスAのp10(GVA-p10)、ブドウの木のウイルスB(GVB-p14)、ヘラクレウム潜在ウイルスのp10(HLV-p10)、又はニンニクの共通潜在ウイルス(GCLV-p16)。したがって、サイレンシングのサプレッサー、例えばHcPro、TEV-p1/HC-Pro、BYV-p21、TBS p19、TCV-CP、CMV-2b、PVX-p25、PVM-p11、PVS-p11、BScV-p16、CTV-p23、GLRaV-2 p24、BGV-p14、HLV-p10、GCLV-p16、又はGVA-p10(ただしこれらに限定されない)を、対象のタンパク質をコードする核酸配列と一緒に発現させて、植物における高レベルのタンパク質生産をさらに担保し得る。
図9に示すように、対象のタンパク質をコードする核酸配列とサイレンシングサプレッサーとの同時発現は、対象のタンパク質の収量の顕著な増加をもたらす。この効果はまた、浸透前に植物をプルーニングする場合に観察される。したがって、本明細書に記載する対象のタンパク質を合成する方法は、2つ以上の核酸配列を植物又は植物の部分に導入する工程を含み得る。例えば2つ以上の核酸配列の1つはサイレンシングのサプレッサーをコードし得る。
【0018】
対象のタンパク質を高収量で製造する方法を具体化するために、本発明は、対象のタンパク質、例えば抗体のような複合タンパク質の発現を駆動するための植物発現系を記載する。アグロバクテリア浸透植物、例えばニコチアナ・ベンタミアナ(Nicotiana benthamiana)内での複合タンパク質の発現によって1.5g/kgFW(ほぼ25%TSP)に達するタンパク質レベルが得られた。対象タンパク質の分泌型及びER-保持型について、それぞれ558及び757mg/kg/FWの平均レベルを得た。非限定的な例を提供すれば、この発現レベルは抗体について得られ、これは、マルチウイルス一過性発現系(Giritch et al. 2006)を用いて産生される抗体の3倍高い発現レベルであり、非ウイルス系アグロバクテリア浸透発現系(例えばVaquero et al. 1999)について記載されたレベルを十分に超えている。
本明細書で提供する例(この例に限定されると考えてはならない)では、抗体は改変されたグリコシル化パターンを含み、低フコシル化、低キシロシル化又はフコシル化とキシロシル化がともに低下したN-グリカンを有する。植物及び典型的な哺乳動物のN-グリコシル化の相違の影響は、治療薬の製造に植物を用いるという考えに対する主要な懸念であった。植物特異的グリカンの出現は、植物生成タンパク質の血流中での半減期の短縮の一因となり、あるいは同じグリカンが患者で過敏反応を惹起するかもしれない。本態様では、対象のタンパク質を高収量で製造することが可能で、それらは、過敏反応を惹起する可能性があるか、そうでなければアレルギー反応に関与する可能性があるグリカンを欠く。しかしながら、本明細書に記載の一過性タンパク質生産方法は、改変グリコシル化を含まないタンパク質を含む任意の対象タンパク質のために用いられ得ることは理解されよう。
【0019】
改変グリコシル化パターンを有することを特徴とする、植物内で対象タンパク質を合成する方法が記載される。本方法は、ヒトベータ-1,4ガラクトシルトランスフェラーゼ(hGalT、GaltTとも称される(配列番号:14))を発現するヌクレオチドと一緒に対象タンパク質を同時発現させることを必要とする。hGalTをまたN-アセチルグルコサミントランスフェラーゼ(GNT1;配列番号:21、図5f;アミノ酸配列番号:22、図5g)のCTSドメイン(すなわち、細胞質テール、トランスメンブレンドメイン、幹領域)と融合させて、GNT1-GalTハイブリッド酵素を生成し、このハイブリッド酵素を対象タンパク質と同時発現させ得る。
ハイブリッドGNT1-GalT配列の使用によって、hGalTの触媒ドメインは、複雑なN-グリカン成熟の初期段階が発生するcis-ゴルジ装置に配置される。対象タンパク質はまた、GalTと融合したCTSドメインを含むハイブリッド酵素、例えばGNT1-GalTと同時発現させ得る(R621;図5a;配列番号:18(配列番号:17によってコードされる))。しかしながら、フコシル化レベルが低下し、なおキシロシル化及びガラクトシル化タンパク質を含む対象タンパク質を所望する場合は、GalTを対象タンパク質と同時発現させてもよい。
対象タンパク質の“改変グリコシル化”とは、改変グリコシル化を含む対象タンパク質(例えば本明細書に記載)のN-グリカンプロフィルが、野生型植物で生成される対象タンパク質のN-グリカンプロフィルと異なることを意味する。グリコシル化の改変は、対象タンパク質の1つ以上のグリカンにおける増加又は減少を含み得る。例えば、対象のタンパク質は、キシロシル化の低下、フコシル化の低下、又はキシロシル化の低下及びフコシル化の低下の両低下を示し得る。また別には、対象タンパク質のN-グリカンプロフィルは、ガラクトシル化の量が増加し、さらに場合によってキシロシル化、フコシル化又はその両方が低下するような態様で改変し得る。
【0020】
さらにまた、対象の複合タンパク質を生産するとき、ヌクレオチド配列は、複合タンパク質の2つ以上のペプチド又はドメインをコードし得る。例えば、対象タンパク質が抗体である場合は、ヌクレオチド配列は2つのヌクレオチド配列を含み、その各々は抗体の一部分をコードし得る。例えば一方のヌクレオチド配列は軽鎖をコードし、第二の配列は抗体の重鎖をコードする。そのような構築物の非限定的な例は図1に提供される。図1では、R612及びR610の各構築物は2つのヌクレオチド配列を含み、一方は、植物で活性な調節領域(例えばUS7,125,978(前記文献は参照により本明細書に含まれる)に記載されたプラストシアニンプロモーター(ただし前記に限定されない))と機能的に連結されたC5-1 LC(C5-1の軽鎖)をコードし、第二の配列は、植物で活性な調節領域(例えばプラストシアニンプロモーター(US7,125,978、前記文献は参照により本明細書に含まれる)、ただし前記に限定されない)と機能的に連結されたC5-1の重鎖(C5-1 HC)をコードする。図1に示すように、さらにR610を参照すれば、KDEL配列は、ペプチド2A又は2Bの1つのC-末端領域に融合させ得る。例えば、KDEL配列は抗体の重鎖と融合させて、抗体がERとともに保持されることを担保し得る(前記を限定と捉えるべきではない)。
ヌクレオチド配列によってコードされる各タンパク質はグリコシル化され得る。
一過性発現を用いて製造された精製生成物のクマシー染色によって、少量の多様な夾雑物の存在が示された。これらのフラグメントは関連生成物であるようで、70kDaを越える全ての夾雑物が、活性ブロットによって示されるように、少なくとも1つのFabを含んでいた(図3B)。植物抽出物に存在する生成物関連夾雑物の実体及び量は、哺乳動物細胞の生産系で観察されたものと同様であった。したがって、抗体治療薬の精製に典型的に用いられる精製手順(例えば陰イオン交換、アフィニティー及び陽イオン交換)は、治療薬として使用されるタンパク質の規制機関によって要求される純度を容易に満たす。
【0021】
図6に示すように、本明細書に記載する方法を用いることによって、改変グリコシル化プロフィルを示す対象タンパク質を生産し得る。例えば、免疫遺伝学的に検出不能なフコース又はキシロース残基を有する対象タンパク質は、GNT1-GalTと一緒に対象タンパク質を同時発現させたときに産生された。対象タンパク質のエピトープのMALDI-TOF分析は、改変グリコシル化パターンを有する対象タンパク質は、GalT又はGNT1-GalTのどちらかと一緒に対象タンパク質を同時発現させたときに得られ得ることを示している。
植物、植物部分又は植物材料をフィードとして用いるか、植物又は植物部分に最小限の加工を施すか、或いは対象タンパク質は植物又は植物部分から抽出し得る。所望の場合は、対象タンパク質は標準的な方法を用いて単離及び精製してもよい。
高収量を担保するために、対象タンパク質をコードするヌクレオチド配列に対してまた別の改変を実施し得る。対象タンパク質をコードする核酸配列はまた、前記タンパク質の小胞体(ER)内での保持に活性な配列(例えばKDEL(Lys-Asp-Glu-Leu)又は他の公知のER保持配列(例えばHDEL))をコードする配列と融合させ得る。
本明細書に記載のタンパク質製造方法は、対象タンパク質の製造のための“プラットフォーム”として用いられ得る植物の使用を含み得る。例えば、プラットフォーム植物は典型的には1つ以上のタンパク質を安定的な態様で発現し、このタンパク質は、例えばN-グリコシル化が改変された対象タンパク質を製造するために、いくつかの態様で対象タンパク質の生成を改変する。例えば、プラットフォーム植物は、GalT、GNT1-GalT、又はGalT及びGNT1-GalTの両方をコードする1つ以上の第一のヌクレオチド配列を発現し得る。対象タンパク質を製造するためには、プラットフォーム植物又はプラットフォーム植物の部分をプルーニングした後、対象タンパク質をコードする第二のヌクレオチド配列を一過性形質転換を用いてプラットフォーム植物に導入し、第二のヌクレオチド配列を発現させて対象タンパク質を製造し、この場合、対象タンパク質は、N-グリコシル化が改変されたグリカンを含む。しかしながら、安定的に他のタンパク質を発現するプラットフォーム植物を、所望にしたがって対象タンパク質を改変するために用いてもよいことは理解されよう。植物又は植物の部分をフィードとして用いるか、または植物又は植物部分に最小限の加工を施すか、或いは対象タンパク質を植物又は植物部分から抽出し得る。所望の場合は、対象タンパク質は標準的な方法を用いて単離及び精製してもよい。
【0022】
本発明は、GalT、GNT1-GalT、GalT及びGNT1-GalTの両方、又はその組合せをコードするヌクレオチド配列であって、その各々がプラットフォーム植物中で活性な調節領域と機能的に連結されている前記配列含むプラットフォーム植物又はプラットフォーム植物の部分を用いて、グリコシル化が改変された対象タンパク質を発現させる方法を提供する。続いて前記プラットフォーム植物又はプラットフォーム植物の部分を用いて、1つ以上の対象タンパク質をコードする第二のヌクレオチド配列を発現させ得る。前記第二のヌクレオチド配列は、プラットフォーム植物中で活性な1つ以上の第二の調節領域と機能的に連結されている。第一のヌクレオチド配列、第二のヌクレオチド配列、又は第一及び第二のヌクレオチド配列の両配列は、プラットフォーム植物又はプラットフォーム植物の部分での発現のためにコドンを最適化させ得る。この方法は、先ず初めにプラットフォーム植物又はプラットフォーム植物の部分をプルーニングする工程を含む。プルーニングの後、前記植物中で活性な調節領域と機能的に連結された対象タンパク質をコードする1つ以上の第二の核酸配列を、前記プルーニングした植物又は植物の部分に一過性態様で導入する。続いて、前記植物又は植物の部分を、対象のタンパク質をコードする核酸配列の前記植物又は植物の部分における発現を可能にする条件下で維持する。
対象タンパク質、又は対象のタンパク質のグリコシル化を改変する酵素(例えばGalT、GNT1-GalT、GalT及びGNT1-GalTの両方、又はその組合せ)をコードするヌクレオチド配列は、前記植物での発現レベルを高めるためにコドンを最適化させ得る。コドン最適化とは、植物におけるコドン使用頻度に類似させるために、構造遺伝子又はそのフラグメントのオリゴヌクレオチド構築ブロックの合成及びその後のそれら酵素のアッセンブリに適したDNAヌクレオチドを選択することを意味する。配列は合成配列であってもよく、前記は、Sardanaら(Plant Cell Reports 1996, 15:677-681)が概略した方法と類似する方法を用いて植物のコドン使用頻度について最適化される。双子葉植物で高度に発現される遺伝子から得られるコドン使用頻度表は、Murrayら(Nuc Acids Res 1989, 17:477-498)を含むいくつかの情報源から入手することができる。さらにまた、配列の最適化はまた、コドンタンデムリピート、曖昧なスプライス部位の除去、反復配列の削減(逆リピートを含む)を含んでもよく、さらにLeto1.0(Entelechon, Germany)を用いて決定することができる。
【0023】
“機能的連結される”とは、個々の配列が直接的又は間接的に相互作用して、意図する機能、例えば遺伝子発現の仲介又は調節を達成することを意味する。機能的に連結された配列の相互作用は、例えば前記機能的に連結された配列と相互作用するタンパク質によって仲介されえる。転写調節領域及び対象の配列は、それらの配列が機能的に接続されて、転写調節領域によって仲介又は調節されるべき対象配列の転写が可能となるときに、機能的に連結されている。
“植物の部分”という用語は植物に由来する任意の部分を意味し、全植物、植物から得られる組織、例えば葉、葉及び茎、根、気生部分(植物の葉、茎及び場合によって花の部分を含む)、植物から得られる細胞又はプロトプラストが含まれるが、ただしこれらに限定されない。
“植物材料”という用語は植物に由来する任意の材料を意味する。植物材料は、全植物、組織、細胞又はその任意の部分を含み得る。さらにまた、植物材料は、細胞内の植物成分、細胞外の植物成分、植物の液体若しくは固体抽出物、又は前記の組合せを含み得る。さらにまた、植物材料は、植物、植物細胞、組織、液体抽出物又は前記の組合せであって、植物の葉、茎、果実、根又は前記の組合せに由来するものを含み得る。植物材料は、いずれの加工工程にも付されていない植物又はその部分を含み得る。しかしながら、植物材料は、下記に規定する最小限の加工工程、又はより厳密な加工(部分的又は実質的なタンパク質の精製を含み、前記精製は当分野で一般的に知られている技術(クロマトグラフィー、電気泳動などを含むがただしこれらに限定されない)を用いる)に付すこともまた意図される。
【0024】
“最小限の加工”という用語は、植物材料(例えば対象タンパク質を含む植物又はその部分)が部分的に精製されて、植物抽出物、ホモジネート、植物ホモジネートの分画などを生じること(すなわち最小限の加工)を意味する。部分的精製は、植物の細胞構造を破壊し、それによって可溶性植物成分及び不溶性植物成分を含む組成物を生成する工程を含み得るが、ただしこれらに限定されない(前記成分は、例えば遠心沈殿、ろ過又はその組合せ(ただしこれらに限定されない)によって分離し得る)。前記に関しては、葉又は他の組織の細胞外間隙に分泌されたタンパク質は真空又は遠心抽出を用いて容易に入手することができよう。または、組織は、細胞外間隙からタンパク質を搾り出すか又は遊離させるために、ローラーを通過させるか、又はすり潰しなどにより加圧下で抽出することもできよう。最小限の加工はまた、可溶性タンパク質の粗抽出物の調製を含むことができる。なぜならば、これらの調製物は二次的な植物生成物に由来する夾雑物をほとんど含まないからである。さらにまた、最小限の加工は、葉の可溶性タンパク質の水性抽出物を続いて適切な塩で沈殿させることを含み得る。他の方法には、抽出物の直接的な使用を可能にするための大規模な離解及び絞り汁抽出が含まれ得る。
植物成分又は組織の形態の植物材料は、対象に経口的に送達され得る。植物材料は、食事用サプリメントの部分として他の食物と一緒に又は被包化して投与され得る。植物材料又は組織はまた濃縮して嗜好性を改善又は向上させるか、または要請に応じて他の素材、成分又は医薬賦形剤と一緒に提供してもよい。
【0025】
対象のタンパク質を含む植物は、対象(例えば動物またはヒト)に、必要性及び状況に応じて多様な方法で投与することが意図される。例えば、植物から得られる対象タンパク質は、それを使用する前に、未精製、部分精製又は精製形として抽出され得る。タンパク質を精製することができる場合には、タンパク質は、食用又は非食用植物で生産され得る。さらにまた、タンパク質を経口投与する場合には、植物組織を収穫して対象に直接供給するか、または収穫した組織を供給前に乾燥させるか、または収穫を実施することなく動物に生草を食べさせてもよい。収穫した植物組織を動物の飼料に食物サプリメントとして提供することもまた本発明の範囲内であると考える。植物組織がほとんど又は更なる加工を加えることなく動物に供給される場合は、投与される植物組織が食用であることが好ましい。
実施例でさらに詳細に記載するように、GalT、GNT1-GalT及び対象タンパク質は、一過性態様で植物に導入された。適切な抗体を用いた免疫学的解析によって、MWrが150kDaのタンパク質が形質転換細胞に存在することが明らかにされた(図2、3A及び3B)。さらにまた、GalT又はGNT1-GalTが、どちらかの構築物を発現する植物から得られた抽出物で検出され、さらにGNT1-GalTが植物で発現されたときに、対象タンパク質の改変Nグリコシル化が観察された(図6)。したがって、組換えにより発現されたGalT又はGNT1-GalTはin plantaで生物学的に活性を有する。
“類似体”又は“誘導体”は、GalT(配列番号:14)又はGNT1-GalT(配列番号:17)をコードするヌクレオチド配列への任意の置換、欠失又は付加を含むが、ただし、GalT(配列番号:14)又はGNT1-GalT(配列番号:17)の非存在下で生産された対象タンパク質のグリコシル化プロフィルと比較したとき、前記配列が、対象タンパク質のグリコシル化プロフィルを改変する、例えば対象タンパク質のグリカンのフコシル化、キシロシル化又はその両方の低下、又は対象タンパク質のガラクトシル化の増加をもたらすタンパク質をコードすることを条件とする。例えば、前記配列によってコードされるタンパク質は、Nグリカンの成熟中に末端ガラクトースを付加し得る。核酸配列の誘導体及び類似体は、ある核酸配列と典型的には80%を超える類似性(又は同一性)を示す。
【0026】
2つ以上の核酸又はポリペプチドに関して、“同一”又はパーセント“同一”という用語は、配列比較アルゴリズム(例えばAltschul et al. Nuc Acids Res 1997, 25:3389-3402及びAltschul et al. J Mol Biol 1990, 215:403-410)及びこれらアルゴリズムの任意のアップグレードを用いるか、又は手動アラインメントと目視精査によって測定される比較ウィンドウ又は指定領域にわたって最大一致のために比較及びアラインメントを実施したとき、同じであるか、又は具体的なパーセンテージのアミノ酸残基若しくはヌクレオチドが同じである(すなわち具体的な領域にわたって60%同一、好ましくは65%、70%、75%、80%、85%、90%又は95%同一である)2つ以上の配列又は部分配列に該当する。配列類似性は、BLASTアルゴリズム(GenBank:ncbi.nlm.nih.gov/cgi-bin/BLAST/)により規定値パラメータ(プログラム:blastn;データベース:nr;Expect 10;フィルター:低複雑度;アラインメント:ペア毎;ワードサイズ:11)を用いて決定し得る。
類似体又はその誘導体にはまた、ストリンジェントなハイブリダイゼーション条件下で(以下の文献を参照されたい:Maniatis et al. in Molecular Cloning (A Laboratory Manual), Cold Spring Harbor Laboratory, 1982, p.387-389又はAusubel et al. (eds), 1989, Current Protocols in Molecular Biology, Vol. 1, Green Publishing Associattes, Inc., and John Wiley & Sons, Inc., New York, p.2.10.3)、本明細書に記載のGalT(配列番号:14)配列、GNAT1-GalT(配列番号:17)配列のいずれかとハイブリダイズするヌクレオチド配列が含まれるが、ただし、GalT(配列番号:14)又はGNT1-GalT(配列番号:17)の非存在下で生成された対象タンパク質のグリコシル化プロフィルと比較したとき、前記配列が、対象タンパク質のグリコシル化プロフィルを改変する、例えば対象タンパク質のグリカンのフコシル化、キシロシル化又はその両方の低下、又は対象タンパク質のガラクトシル化の増加をもたらすタンパク質をコードすることを条件とする。例えば、前記配列によってコードされるタンパク質は、Nグリカンの成熟中に末端ガラクトースを付加し得る。そのようなストリンジェントなハイブリダイゼーション条件の例は、適切なプローブ、例えば[ガンマ-32P]dATP標識プローブ(ただし前記に限定されない)との7%SDS、1mM EDTA、0.5M Na2HPO4(pH7.2)中での16−20時間、65℃でのハイブリダイゼーションでありえる。続いて65℃にて5%SDS、1mM EDTA、40mM Na2HPO4(pH7.2)中で30分洗浄し、続いて65℃にて1%SDS、1mM EDTA、40mM Na2HPO4(pH7.2)中で30分洗浄する。この緩衝液での洗浄を繰り返してバックグラウンドを低下させることが得る。
【0027】
“調節領域”、“調節エレメント”又は“プロモーター”とは、核酸の部分、典型的には遺伝子(DNA若しくはRNA又はDNA及びRNAの両方を含み得る)のタンパク質コード領域の上流(ただし常にそうであるとは限らないが)を意味する。調節領域が活性を有し、かつ対象の遺伝子と機能的な結合状態にあるとき、又は機能的に連結されているとき、対象遺伝子の発現がもたらされえる。調節エレメントは器官特異性を仲介するか、又は発生若しくは一過性遺伝子活性化を制御し得る。“調節領域”には、プロモーターエレメント、基礎プロモーター活性を示すコアプロモーターエレメント、外部刺激に応答して誘導されえるエレメント、プロモーター活性を仲介するエレメント、例えば負の調節エレメント又は転写エンハンサーが含まれる。本明細書で用いられる、“調節領域”にはまた転写後に活性を示すエレメントが含まれる。前記は、例えば遺伝子発現を調節する調節エレメント、例えば翻訳及び転写エンハンサー、翻訳及び転写リプレッサー、上流の活性化配列、及びmRNA不安定性決定因子である。これら後者のエレメントのいくつかはコード領域の近位に存在しえる。
本開示に関しては、“調節エレメント”又は“調節領域”は典型的には、構造遺伝子のコード配列の通常は(常にというわけではないが)上流(5')のDNA配列に該当し、前記は、RNAポリメラーゼ及び/又は転写を特定の部位で開始させるために必要な他の因子のための見分けを提供することによってコード領域の発現を制御することは理解されよう。しかしながら、イントロン内又は配列の3'に配置される他のヌクレオチドもまた、対象のコード領域の発現の調節に寄与し得る。特定の部位での開始を担保するためにRNAポリメラーゼ又は他の転写因子のための見分けを提供する調節エレメントはプロモーターエレメントである。ほとんどの(全てではないが)真核細胞性プロモーターエレメントはTATAボックスを含む。前記は保存された核酸配列であって、アデノシン及びチミンヌクレオチド塩基対を含み、通常は転写開始部位のほぼ25塩基対上流に位置している。プロモーターエレメントは基礎プロモーターエレメントを含み、遺伝子の発現を修正する他の調節エレメント(上記に列挙されている)と同様に転写の開始のために極めて重要である。
【0028】
構成的調節領域は、植物の種々の部分にわたって、さらに植物の発育中に持続的に遺伝子の発現を誘導する。公知の構成的調節エレメントの例には、CaMV 35S転写物(Odell et al., 1985, Nature, 313: 810-812)、コメのアクチン1(Zhang et al, 1991, Plant Cell, 3: 1155-1165)、アクチン2(An et al., 1996, Plant J., 10: 107-121)、又はtms2(U.S. 5,428,147、前記文献は参照により本明細書に含まれる)、及びトリオースホスフェートイソメラーゼ1(Xu et. al., 1994, Plant Physiol. 106: 459-467)遺伝子、トウモロコシユビキチン1遺伝子(Cornejo et al, 1993, Plant Mol. Biol. 29: 637-646)、シロイヌナズナ(Arabidopsis)ユビキチン1及び6遺伝子(Holtorf et al, 1995, Plant Mol. Biol. 29: 637-646)、並びにタバコ翻訳開始因子4A遺伝子(Mandel et al, 1995 Plant Mol. Biol. 29: 995-1004)と密接に関連するプロモーターが含まれる。本明細書で用いられる“構成的”という用語は、構成的調節領域の制御下にある遺伝子が全ての細胞タイプで同じレベルで発現されることを必ずしも示しているわけではないが、前記遺伝子は豊富さに変動はしばしば認められるものの広い範囲の細胞タイプで発現される。
光合成遺伝子から得られる調節領域又はプロモーターもまた本発明での使用に適している。例えば、調節領域又はプロモーターは以下の大小のサブユニットをコードする遺伝子から得られ得る:リブロース1,5-ビスホスフェートカルボキシラーゼ/オキシゲナーゼの大若しくは小サブユニット(rubisco;US 4,962,028、前記文献は参照により本明細書に含まれる)、プラストシアニン(US 7,125,978、前記文献は参照により本明細書に含まれる);図1b、配列番号:19)、クロロフィルa/b結合タンパク質(CAB;Leutwiler et al. 1986、前記文献は参照により本明細書に含まれる)、ST-LS1(光化学反応系IIの酸素放出複合体と関連する;Stockhaus et al. 1989、前記文献は参照により本明細書に含まれる)。
【0029】
本発明の1つ以上のヌクレオチド配列は任意の適切な植物宿主において発現させる得る。適切な宿主の例には、シロイヌナズナ、アルファルファ、キャノーラ、ブラシッカ(Brassica)spp.、トウモロコシ、ニコチアナ(Nicotiana)spp.(ニコチアナ・ベンタミアナ(Nicotiana benthamiana)、ニコチアナ・タバクム(Nicotiana tabaccum)を含む)、アルファルファ、ジャガイモ、チョウセンニンジン、エンドウ、エンバク、コメ、ダイズ、コムギ、オオムギ、ヒマワリ、ワタなどが含まれるが、ただしこれらに限定されない。
本発明の1つ以上のキメラ遺伝子構築物はさらに3'非翻訳領域を含み得る。3'非翻訳領域は、ポリアデニル化シグナル及びmRNAプロセッシング又は遺伝子発現を実行することができる他の任意の調節シグナルを収納するDNAセグメントを含む遺伝子の部分に該当する。ポリアデニル化シグナルは、通常mRNA前駆体の3'末端にポリアデニル酸鎖を付加することを特徴とする。ポリアデニル化シグナルは、一般的には正規の形態5'AATAAA-3'(ただし変型は珍しくはないが)との相同性の存在によって認識される。本発明の1つ以上のキメラ遺伝子構築物はさらにまたエンハンサー(翻訳又は転写エンハンサー)を必要に応じて含み得る。これらのエンハンサー領域は当業者には周知であり、ATG開始コドン及び隣接配列を含み得る。開始コドンは、全配列の翻訳を担保するためにコード配列のリーディングフレームと一致しなければならない。
適切な3'領域の非限定的な例は、プラストシアニンの3'UTRを含む3'転写非翻訳領域であり、以下が含まれる:転写終了配列(配列番号:20)、アグロバクテリア菌腫瘍誘発(Ti)遺伝子(当分野では公知)のポリアデニル化シグナル、例えばノパリンシンターゼ(Nos遺伝子)及び植物遺伝子(例えばダイズ貯蔵タンパク質遺伝子)及びリブロース1,5-ビスホスフェートカルボキシラーゼ遺伝子の小サブユニット。
所望の場合には、本発明の構築物はさらに操作して選択可能マーカーを含ませ得る。しかしながら、これは不要な場合もある。有用な選択可能マーカーには、化学物質、例えば抗生物質(例えばゲンタマイシン、ヒグロマイシン、カナマイシン)又は除草剤(例えばホスフィノトリシン、グリホセート、クロロスルフロンなど)に対して耐性を提供する酵素が含まれる。同様に、色の変化によって識別可能な化合物の生成を提供する酵素、例えばGUS(ベータ-グルクロニダーゼ)又はルミネセンス(例えばルシフェラーゼ又はGFP)を用いてもよい。
【0030】
本発明で考慮される部分はまた、本発明のキメラ遺伝子構築物を含むトランスジェニック植物、植物細胞又は種子であって、前記は、本明細書に記載の一過性タンパク質発現に適したプラットフォーム植物として用いられ得る。植物細胞から植物全体を再生する方法もまた当分野では公知である。一般的には、形質転換細胞は適切な培地で培養され、前記培地は選別物質(例えば抗生物質)を含み得る。選別可能マーカーは形質転換植物細胞の識別を容易にするために用いられる。いったんカルスが形成されたら、シュートの形成は、公知の方法にしたがって適切な植物ホルモンを利用することによって促進することができ、このシュートを発根培地に移して植物を再生させる。続いて、この植物を用いて、反復生成を種子から又は栄養繁殖技術を用いて確立し得る。トランスジェニック植物はまた組織培養を用いることなく作製することができる。
安定な形質転換のための方法及びこれら生物の再生は当分野では確立されてあり、当業者には公知である。形質転換及び再生植物を得る方法は、本発明にとって重要ではない。
“形質転換”とは、遺伝子型として、表現型として又は両方において表出される遺伝情報(ヌクレオチド配列)の種間移転を意味する。キメラ構築物から宿主への遺伝情報の種間移転が遺伝可能であって、遺伝情報の移転は安定であるとみなされることもあるが、また移転は一過性であって、遺伝情報の移転は遺伝可能でなくてもよい。
本発明はさらに、安定な発現系又は一過性発現系による使用に適したキメラ構築物を含む適切なベクターを含む。遺伝情報はまた1つ以上の構築物で提供し得る。例えば、対象のタンパク質をコードするヌクレオチド配列は一方の構築物に導入され、対象タンパク質のグリコシル化を改変するタンパク質をコードする第二のヌクレオチド配列は別の構築物を用いて導入されえる。続いてこれらのヌクレオチド配列は、本明細書に記載するように植物内で一過性に同時発現させ得る。対象タンパク質及び対象タンパク質のグリコシル化プロフィルを改変するタンパク質の両タンパク質をコードするヌクレオチド配列を含む構築物もまた用いられ得る。この場合、ヌクレオチド配列は、対象タンパク質をコードする第一の核酸配列(プロモーター又は調節領域と機能的に連結されている)を含む第一の配列、及び対象タンパク質のグリコシル化プロフィルを改変するタンパク質をコードする第二の核酸配列(第二の配列はプロモーター又は調節領域と機能的に連結されている)を含む第二の配列を含むであろう。
【0031】
“同時発現”とは、2つ以上のヌクレオチド配列が、ほぼ同じときに植物内で、及び植物の同じ組織で発現されることを意味する。しかしながら、前記ヌクレオチド配列は、厳密に同じときに発現される必要はない。むしろ、2つ以上のヌクレオチド配列は、コードされた生成物が相互作用する機会を有することができるように発現される。例えば、対象のタンパク質のグリコシル化を改変するタンパク質は、対象タンパク質のグリコシル化の改変が生じるように、対象タンパク質が発現される前又は発現期間中に発現される。2つ以上のヌクレオチドは一過性発現系を用いて同時発現させることができる。この場合、2つ以上の配列は、両配列が発現される条件下でほぼ同じときに植物に導入される。また別には、ヌクレオチド配列の一方(例えば対象タンパク質のグリコシル化プロフィルを改変するタンパク質をコードする配列)を含むプラットフォーム植物は、プラットフォーム植物に一過性態様で導入される対象タンパク質をコードする追加の配列で安定的な態様で形質転換させ得る。この場合、対象タンパク質のグリコシル化プロフィルを改変するタンパク質をコードする配列は、所望の組織で所望の発育段階の間に発現させ得る。あるいは、その発現は誘導性プロモーターを用いて誘導し得、対象タンパク質をコードする追加の配列は、類似の条件下で及び同じ組織で発現させて、ヌクレオチド配列が同時発現されることを担保し得る。
【0032】
本発明の構築物は、Tiプラスミド、Riプラスミド、植物ウイルスベクター、直接DNA形質転換、マイクロインジェクション、エレクトロポレーションなどを用いて植物細胞に導入することができる。そのような技術の検討のためには例えば以下を参照されたい:Weissbach and Weissbach, Methods for Plant Molecular Biology, Academy Press, New York VIII, pp. 421-463 (1988);Geierson and Corey, Plant Molecular Biology, 2d Ed. (1988);及びMiki and Iyer, Fundamentals of Gene Transfer in Plants. ”Plant Metabolism”, 2d Ed. DT. Dennis, DH Turpin, DD Lefebrve, DB Layzell (eds), Addison Wesly, Langmans Ltd. London, pp. 561-579 (1997)。他の方法には、直接DNA取込み、リポソームの使用、エレクトロポレーション(例えばプロトプラストを用いる)、マイクロインジェクション、マイクロプロジェクティル又はウィスカー(whisker)、及び真空浸透が含まれる。例えば以下を参照されたい:Bilang, et al. (Gene 100: 247-250 (1991);Scheid et al. (Mol. Gen. Genet. 228: 104-112, 1991);Guerche et al. (Plant Science 52: 111-116, 1987);Neuhause et al. (Theor. Appl Genet. 75: 30-36, 1987);Klein et al., Nature 327: 70-73 (1987);Howell et al. (Science 208: 1265, 1980);Horsch et al. (Science 227: 1229-1231, 1985);DeBlock et al., (Plant Physiology 91: 694-701, 1989);Methods for Plant Molecular Biology (Weissbach and Weissbach, eds., Academic Press Inc., 1988);Methods in Plant Molecular Biology (Schuler and Zielinski, eds., Academic Press Inc., 1989);Liu and Lomonossoff (J Virol Meth, 105:343-348, 2002,);米国特許4,945,050号、5,036,006号及び5,100,792号;米国特許出願08/438,666号(1995年5月10日出願)及び07/951,715号(1992年9月25日出願)(前記文献はいずれも参照により本明細書に含まれる)。
【0033】
下記で述べるように、一過性発現方法を用いて、本発明の構築物を発現させることができる(以下を参照されたい:Liu and Lomonossoff, 2002, Journal of Virological Methods, 105:343-348、前記文献は参照により本明細書に含まれる)。また別には、Kapilaら(1997)(前記文献は参照により本明細書に含まれる)が記載した真空による一過性発現方法を用いてもよい。これらの方法には、例えばアグロバクテリア接種(Agro-inoculation)若しくはアグロバクテリア浸透(Agro-infiltration)、注射筒浸透が含まれるが(ただしこれらに限定されない)、上記に記載した他の方法もまた用いることができる。アグロバクテリア接種、アグロバクテリア浸透、又は注射筒浸透に関しては、所望の核酸を含むアグロバクテリア菌の混合物が、組織(例えば葉)、植物の気生部分(茎、葉及び花を含む)、植物の他の部分(茎、根、花)又は全植物の細胞間間隙に入り込む。
表皮をクロスさせた後、アグロバクテリア菌が感染してt-DNAコピーが細胞に移される。このt-DNAはエピソーム的に(episomally)転写され、mRNAが翻訳されて感染細胞で対象タンパク質の生成をもたらすが、t-DNAの核内での継代は一過性である。
“対象の遺伝子”、“対象のヌクレオチド配列”、又は“対象のコード領域”(これらの用語は相互に用いられる)とは、植物又は植物の部分で発現されるべき任意の遺伝子、ヌクレオチド配列又はコード領域を意味する。そのような対象ヌクレオチド配列には、その生成物が対象タンパク質である配列又はコード領域が含まれるが、ただしこれらに限定されない。対象タンパク質の例には以下が含まれる(ただしこれらに限定されない):例えば工業用酵素(例えばセルラーゼ、キシラナーゼ、プロテアーゼ、ペルオキシダーゼ)、スブチリシン、タンパク質サプリメント、栄養性医薬(nutraceutical)、付加価値製品、又は前記のフラグメントであって飼料、食品若しくは飼料と食品双方に使用するためのもの、医薬的に活性なタンパク質(例えば成長因子、生長調節物質、抗体、抗原及び前記のフラグメント、又は免疫付与又はワクチン接種のために有用な前記の誘導体が含まれるが、ただしこれらに限定されない)など。さらに追加される対象タンパク質には以下が含まれる(ただしこれらに限定されない):インターロイキン(例えば1つ以上のIL-1からIL-24、IL-26及びIL-27)、サイトカイン、エリスロポエチン(EPO)、インスリン、G-CSF、GM-CSF、hPG-CSF、M-CSF若しくは前記の組合せ、インターフェロン(例えばインターフェロン-アルファ、インターフェロン-ベータ、インターフェロン-ガンマ)、血液凝固因子(例えば第VIII因子、第IX因子)、tPA hGH、レセプター、レセプターアゴニスト、抗体、神経ポリペプチド、インスリン、ワクチン、成長因子(例えば表皮増殖因子、ケラチノサイト増殖因子、形質転換増殖因子、増殖調節因子が含まれるが、ただしこれらに限定されない)、抗原、自己抗原、前記のフラグメント、又は前記の組合せ。
【0034】
対象のヌクレオチド配列が、植物に対して直接的に又は間接的に毒性を有する生成物をコードする場合は、本発明の方法を用いることによって、対象の遺伝子を一過性に発現させて植物全体でそのような毒性を低下させ得る。
下記実施例でより詳細に記載するように、改変N-グリコシル化を有する、対象のタンパク質(例えば抗体、C5-1(ただし前記に限定されない))の合成は、GalT(配列番号:14、図1b)又はGNT1-GalT(配列番号:17、図1c)のどちらかを一過性に同時発現する植物で実施された。
本明細書に記載の一過性発現プロセスの利点は、抗体の一過性発現に用いられるアグロバクテリア菌株数が最小限になり、これはコストを削減し、操作を簡単にし、系の堅実性を高める。Giritchら(2006)によって提唱された一過性発現系は、2つの非競合性ウイルスベクターでの抗体の軽鎖及び重鎖の発現に依存する。この系はまた、プロベクター分子、リコンビナーゼ及び2つのウイルスレプリカーゼの発現のために6つの異なるアグロバクテリア菌培養の同時浸透を必要とする。産業的展望から、6つの接種物の同時調製は、装置における多大なコスト並びに有効性実証及びスケールアップ操作までの期間を必要とする。さらにまた、細菌ベクターの数の増大は、多数の遺伝子の協調的発現に依存するこの発現系の堅実性に影響を与える可能性がある。
比較すれば、本明細書で提唱する系は、ほんの2つの別個のアグロバクテリア菌培養の同時浸透を必要とするだけである。アグロバクテリア菌の培養数は、サイレンシングのサプレッサー(例えばHcPro)をコードする配列、又は対象タンパク質を改変する任意の他の配列を、抗体発現カセットと同じプラスミドに含めることによってただ1つの培養にまで減らすことができる。
【0035】
配列リスト:
【実施例1】
【0036】
発現カセットR610、R612、R514(図1)、R621及びR622(図5)のアッセンブリ
全ての操作は、SambrookとRussel(2001)の一般分子生物学プロトコルを用いて実施した。
使用したオリゴヌクレオチドプライマーを下記に示す:
【0037】
XmaI-pPlas.c: 配列番号:1
5'-AGTTCCCCGGGCTGGTATATTTATATGTTGTC-3' 配列番号:1
SacI-ATG-pPlas.r: 配列番号:2
5'-AATAGAGCTCCATTTTCTCTCAAGATGATTAATTAATTAATTAGTC-3' 配列番号:2
SacI-PlasTer.c: 配列番号:3
5'-AATAGAGCTCGTTAAAATGCTTCTTCGTCTCCTATTTATAATATGG-3' 配列番号:3
EcoRI-PlasTer.r: 配列番号:4
5'-TTACGAATTCTCCTTCCTAATTGGTGTACTATCATTTATCAAAGGGGA-3' 配列番号:4
Plasto-443c: 配列番号:5
5'-GTATTAGTAATTAGAATTTGGTGTC-3' 配列番号:5
Plas+LC-C51.r: 配列番号:6
5'-ATCTGAGGTGTGAAAACCATTTTCTCTCAAGATG-3' 配列番号:6
LC-C51.c: 配列番号:7
5'-ATGGTTTTCACACCTCAGATACTTGG-3' 配列番号:7
LC-C51XhoSac.r: 配列番号:8
5'-ATATGAGCTCCTCGAGCTAACACTCATTCCTGTTGAAGC-3' 配列番号:8
【0038】
Plas+HC-C51.r: 配列番号:9
5'-CAAGGTCCACACCCAAGCCATTTTCTCTCAAGATG-3' 配列番号:9
HC-C51.c: 配列番号:10
5'-ATGGCTTGGGTGTGGACCTTGC-3' 配列番号:10
HC-C51XhoSac.r: 配列番号:11
5'-ATAAGAGCTCCTCGAGTCATTTACCAGGAGAGTGGG-3' 配列番号:11
HC-C51KDEL(SacI).r: 配列番号:12
5'-ATAAGAGCTCTCAAAGTTCATCCTTTTTACCAGGAGAGTGGG-3' 配列番号:12
XhoTEV.c: 配列番号:23
5'-TTTGGAGAGGACCTCGAGAAATAACAAATCTCAACAC-3' 配列番号:23
TEV+LC-C5-1.r: 配列番号:24
5'-ATCTGAGGTGTGAAACCATTGCTATCGTTCGTAAATGGTG-3' 配列番号:24
LC-C5-1.c: 配列番号:25
5'-ATGGTTTTCACACCTCAGATACTTGG-3' 配列番号: 25
LC-C5-1SphSac.r: 配列番号;26
5'-ATATGAGCTGCGATGCCTAACACTCATTCCTGTTGAAGC-3' 配列番号:26
【0039】
第一のクローニング工程は、アルファルファのプラストシアニン遺伝子の上流及び下流の調節エレメントを含むレセプタープラスミドを組み立てることであった。プラストシアニンプロモーター(米国特許7,125,978号、前記文献は参照により本明細書に含まれる)及び5'UTR配列は、オリゴヌクレオチドプライマーXmaI-pPlas.c(配列番号:1)及びSacI-ATG-pPlas.r(配列番号:2)を用いて、アルファルファのゲノムDNAから増幅した。得られた増幅生成物をXmaI及びSacIで消化して、pCAMBIA2300(以前に同じ酵素で消化しておいた)に連結し、pCAMBIA-PromoPlastoを作製した。同様に、プラストシアニン遺伝子の3'UTR配列及びターミネーター(図1c、配列番号:20のヌクレオチド1−399)を、以下のプライマー(SacI-PlasTer.c(配列番号:3)及びEcoRI-PlasTer.r(配列番号:4))を用いアルファルファのゲノム遺伝子から増幅し、生成物をpCAMBIA-PromoPlastoの同じ部位に挿入する前にSacI及びEcoRIで消化してpCAMBIAPlastoを作製した。
プラスミドR610及びR612は、アルファルファのプラストシアニンプロモーター下でC5-1軽鎖及びC5-1重鎖コード配列をタンデム構築物として含むように調製された。R610はアッセンブリされたIgGがERで保持されるように設計され、さらに前記はC5-1の重鎖末端に融合したKDELを含んでいた。R612は分泌が可能なように設計された。
C5-1発現カセットのアッセンブリは、Darveauら(1995)が記載したPCR仲介連結方法を用いて実施した。プラストシアニンの下流の軽鎖コード配列をアッセンブリするために、第一の工程は、開始ATGの下流にあるD'Aoustら(米国特許7,125,978号、前記文献は参照により本明細書に含まれる)が記載したアルファルファプラストシアニンプロモーターの最初の443塩基対(bp)(図1bのヌクレオチド556−999或いは配列番号:19)を、pCAMBIAPlastoを鋳型とし以下のプライマーを用いて増幅させることであった:
Plasto-443c(配列番号:5)及びPlas+LC-C51.r(配列番号:6、オーバーラップには上記で下線が付されている)。
【0040】
並行して、軽鎖コード配列を、プラスミドpGA643-kappa(Khoudi et al. 1999)から以下のプライマーを用いてPCR増幅させた:
LC-C51.c(配列番号:7)及びLC-C51XhoSac.r.(配列番号:8、オーバーラップには下線が付されている)。
得られた2つの増幅生成物を混合して、プライマーPlasto-443c(配列番号:5)及びLC-C51XhoSac.r(配列番号:8)による第三のPCR反応で鋳型として用いた。第一の反応で用いられるプライマーPlas+LC-C51.r(配列番号:6)とLC-C51.c(配列番号:7)との間のオーバーラップは、第三の反応中に増幅生成物のアッセンブリをもたらす。第三のPCR反応から生成されたアッセンブリ生成物をDraIII及びSacIで消化し、DraIII及びSacIで消化したpCAMBIAPlastoに連結してプラスミドR540を作製した。
重鎖コード配列は、プラストシアニンの開始ATGの上流の443 bp、ヌクレオチド556−999(図1b、配列番号:19、)を、pCAMBIAPlastoを鋳型とし以下のプライマーを用いて増幅させることによってプラストシアニンの上流の調節エレメントと融合させた:
Plasto-443c(配列番号:5)及びPlas+HC-C51.r(配列番号:9、オーバーラップには上記で下線が付されている)。
【0041】
これら反応の生成物を混合して、プライマーPlasto-443c(配列番号:5)及びHC-C51XhoSac.r.(配列番号:11)を用い第三のPCR反応でアッセンブリさせた。得られたフラグメントをDraIII及びSacIで消化し、pCAMBIAPlastoのDraIIIとSacI部位の間に連結した。この生成プラスミドをR541と名付けた。
プラスミドR541を鋳型として用い、プライマーPlasto-443c(配列番号:5)及びHC-C51KDEL(SacI).r(配列番号:12)によりPCR増幅によって、重鎖コード配列のC-末端にKDELタグを付加した。生成フラグメントをDraIII及びSacIで消化し、pCAMBIAPlastoの同じ部位にてクローニングし、R550を作製した。
軽鎖及び重鎖発現カセットの同じ二元プラスミドでのアッセンブリは以下のように実施した:R541及びR550をEcoRIで消化して平滑端を得、さらにHindIIIで消化して、R540のHindIII及びSmaI部位に連結してR610(KDELを有する)及びR612(KDELをもたない、図1参照)を作製した。
R514(図5a)
使用したさらに別のオリゴヌクレオチドプライマーは以下に提供される:
【0042】
Tev+HC-C51.2: 配列番号:16
5'-CAAGGTCCACACCCAAGCCATTGCTATCGTTCGTAAATGGTG-3' 配列番号:16
HC-C51SphSac.r 配列番号:32
5'-ATAAGAGCTCGCATGCTCATTTACCAGGAGAGTGGG-3' 配列番号:32
【0043】
完全長のC5-1軽鎖及び重鎖遺伝子(LC及びHC)は、Hema-Quebecから提供され、Darveau(1995)が記載したポリメラーゼ連鎖反応(PCR)仲介方法を用い、植物二元発現ベクターにてインフレーム(in-frame)でクローニングした。先ず初めにタバコエッチウイルス(TEV)エンハンサーを、プライマー”Xho”TEV.c(配列番号:23)及びTEV+LC-C51(配列番号:24)を用いTEVゲノムRNA(Acc. No. NC001555)を基にRT-PCRによって増幅した。並行して、C5-1軽鎖コード配列を、LCのためにプライマーLC-C51.c(配列番号:25)及びLC-C51”SphSac”.r(配列番号:26)を用いプラスミドpGA643(Khoudi et al. 1999)からPCRによって増幅した。続いてTEV及び軽鎖増幅フラグメントを混合し、プライマーとして”Xho”TEV.c(配列番号:23)及びLC-C51”SphSac”.r(配列番号:26)を用いさらにもう1回のPCRによってアッセンブリさせた。生成されたTEV/C5-1LCフラグメントを続いて精製し、中間ベクターにて2X35SプロモーターとNOSターミネーターとの間でXhoI-SacI消化物としてクローニングした。図1dは、使用した2X35Sプロモーター(太字、配列番号:33)及びNOSターミネーター(イタリック体、配列番号:34)並びに制限部位の位置(下線)を示している。続いて、この発現カセットをpCAMBIA2300二元プラスミドにHindIII-EcoRIフラグメントとして移入してプラスミドR512を作製した。
pR513を作製するために、TEVエンハンサーを、プライマー”Xho”TEV.c(配列番号:23)及びTEV+HC-C51.r(配列番号:16)を用いTEVゲノムRNA(Acc. No. NC001555)を基にRT-PCRによって増幅した。並行して、抗体の重鎖のコード配列を、プライマーHC-C51.c(配列番号:10)及びHC-C51”SphSac”.r(配列番号:32)を用いPCRによって増幅した。生成されたTEV及び重鎖増幅フラグメントを混合し、プライマー”Xho”TEV.c(配列番号:23)及びHC-C51”SphSac”.r(配列番号:32)を用いPCRによってアッセンブリさせた。生成されたTEV/C5-1HCフラグメントを精製し、XhoI及びSacIで消化し、中間ベクターにて2X35SプロモーターとNOSターミネーターとの間の同じ部位でクローニングした。図1dは、使用した2X35Sプロモーター(配列番号:33)及び使用したNOSターミネーター(配列番号:34)並びに制限部位の位置を示している。得られた2X35S/TEV/C5-1HC/NOSフラグメントを含むプラスミドをEcoRIで消化し、クレノーフラグメントポリメラーゼを用いて平滑端とし、さらにHindIIIで消化した。続いて、このHindIII-EcoRI(平滑端)フラグメントをR512のHindIII-SmaI消化物に連結してプラスミドR514を作製した。
R621及びR622(図5a)−用いたオリゴヌクレオチドプライマーを下記に示す:
【0044】
FgalT 配列番号:27
5'-GACTCTAGAGCGGGAAGATGAGGCTTCGGGAGCCGCTC-3' 配列番号:27
RgalTFlagStu 配列番号:28
5'- AAGGCCTACG CTACTTGTCAT CGTCATCTTT GTAGTCGCAC GGTGTCCCG AAGTCCAC -3' 配列番号: 28
FGNT 配列番号:29
5'-ATCGAAATCGCACGATGAGAGGGAACAAGTTTTGC-3' 配列番号: 29
RGNTSpe 配列番号:30
5'-CGGGATCCACTAGTCTGACGCTTCATTTGTTCTTC-3' 配列番号: 30
FgalTSpe 配列番号:31
5'-GGACTAGTGCACTGTCGCTGCCCGCCTGC-3' 配列番号: 31
【0045】
GalT及びGNT1GalT発現用プラスミドはpBLTI121(Pagny et al. 2003)からアッセンブリさせた。ヒトβ(1,4)-ガラクトシルトランスフェラーゼ(hGalT)遺伝子(UDPガラクトース:β-N-アセチルグルコサミド:β(1,4)-ガラクトシルトランスフェラーゼ;EC2.41.22)をpUC19-hGalT(Watzele et al. 1991)からEcoRI消化により入手した。クレノーで処理した後、1.2kbのhGalTフラグメントをpBLT221のSmaI部位でクローニングし、プラスミドpBLTI221hGalTを生成した。続いて、プライマーFGalT(配列番号:27)及びRGalTFlagStu(配列番号:28)を増幅のために用い、PCRによって前記コード領域のC-末端にFlagタグを融合させた。続いて、このXbaI-StuIフラグメントを二元ベクターpBLTI121にてクローニングすることによってR622を生成した。トランスメンブレンドメインに対応するN-アセチルグルコサミニルトランスフェラーゼI(GNT1)の最初の77アミノ酸を、鋳型としてN-GNT1をコードするN. tabacumのcDNA(Strasser et al. 1999)並びにプライマーとしてFGNT(配列番号:29)及びRGNTSpe(配列番号:30)を用いてPCRによって増幅させた。増幅生成物を先ず初めにpGEM-Tベクターにてクローニングし、得られたプラスミドをApaI及びBamHIで消化し、pBLTI221に連結し、pBLTI221-GNT1と称されるプラスミドを生成した。hGalTの触媒ドメインを、プライマーFGalTSpe(配列番号:31)及びRGalTFlagStu(配列番号:28)を用いてpBLTI221hGalTを基にPCR増幅によって入手し、SpeI及びStuI部位をそれぞれ5'及び3'末端に作製した。続いて、SpeI/StuI hGalTフラグメントを同じ(SpeI及びStuI)部位を用いてpBLTI221-GNT1にてクローニングし、pBLTI221-GNT1GalTを作製した。最後に、pBLTI221-GNT1GalTをXbaI及びStuIで消化し、GNT1GalTコード配列(図5d、配列番号:17)を単離し、このフラグメントを二元ベクターpBLTI121にてクローニングすることによってR621を作製した。
全てのクローンでシークェンシングを実施して構築物の完全性を確認した。これらのプラスミドを用い、アグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)を大腸菌(E.coli)形質転換の場合のように(W.S. Dower, Electroporation of Bacteria, In “Gentic Engineering”, Volume 12, Plenum Press, New York, 1990, J.K. Setlow eds.)Gene Pulser II装置(Biorad, Hercules, CA, USA)を用いて形質転換した。全てのA.ツメファシエンス株の完全性は制限マッピングによって確認した。
HcPro構築物はHamiltonらの記載にしたがって調製した。
【実施例2】
【0046】
植物バイオマスの調製、接種物、アグロバクテリア浸透及び採集
植物ニコチアナ・ベンタミアナ(Nicotiana benthamiana)を、市販の泥炭ゴケ基質を満たした平箱で種子から育てた。前記植物を16/8の光周期及び25℃日中/20℃夜間の温度スケジュールの下で温室にて生育させた。播種から3週間後に、個々の幼植物を採取してポットに移植し、さらに3週間同じ環境条件下で温室にて生育させた。形質転換の前に、植物から芽を摘み取るか又は植物を化学的に処理することによって、頂芽及び腋芽を下記に示す種々の時期に取り除く。
10mMの2-[N-モルフォリノ]エタンスルホン酸(MES)、20μMアセトシリンゴン、50μg/mLカナマイシン及び25μg/mLのカルベニシリン(pH5.6)を補充したYEB培地で、アグロバクテリア株R612、R610、R621、R622又は35SHcProをOD600が0.6から1.6に達するまで増殖させた。アグロバクテリア菌の懸濁液を使用前に遠心し、浸透培養液(10mMのMgCl2及び10mMのMES(pH5.6))に再懸濁した。
注射筒浸透は、Liu and Lomonossoffの記載(2002、Journal of Virological Methods, 105:343-348)にしたがって実施した。
真空浸透については、A.ツメファシエンス懸濁液を遠心し、浸透培養液に再懸濁して4℃で一晩保存した。浸透の日に、培養バッチを2.5培養体積に希釈し、使用前に温めた。ニコチアナ・ベンタミアナの全植物を気密なスチールタンク中の細菌懸濁液に上下さかさまにして20−40トルの真空下に2分静置した。注射筒又は真空浸透に続いて、植物を温室に戻し、採集まで4−5日間インキュベートした。
【0047】
葉のサンプリング及び全タンパク質の抽出:
インキュベーションに続いて、植物の気生部分を採集し、-80℃で凍結し、小片に粉砕して1.5又は7.5gのサブサンプルに分けた。凍結粉砕した植物材料の各サブサンプルを、冷却した50mMトリス(pH7.4)、0.15MのNaCl、0.1%のトリトンX-100、1mMフェニルメタンスルフォニルフロリド及び10μMキモスタチンの3体積中で均質化することによって(Polytron)、全可溶性タンパク質を抽出した。均質化後に、スラリーを4℃で20分、20,000gで遠心し、これらの清澄粗抽出物(上清)を分析のために維持した。参照標準物としてウシ血清アルブミンを用い、Bradfordアッセイ(Bio-Rad, Hercules, CA)によって、清澄粗抽出物の全タンパク質含有量を決定した。
【実施例3】
【0048】
タンパク質分析、免疫ブロッティング及びELISA
C5-1は抗ヒトネズミIgGであり、したがって検出及び定量は、ヒトIgGに対するその特徴的な親和性により(活性ブロット)または抗マウスIgG対するその免疫反応性によって実施することができる。
全粗抽出物又は精製抗体のタンパク質をSDS-PAGEによって分離し、クマシーブルーR-250若しくはG-250で染色するか、又は免疫的検出のためにポリビニレンジフロリドメンブレン(Roche Giagnostics Corporation, Indianapolis, IN)に電気的に移した。イムノブロッティングの前に、このメンブレンをトリス緩衝食塩水(TBS−T)中の5%脱脂乳及び0.1%トゥイーン20で4℃にて16−18時間ブロッキングした。
イムノブロッティングは以下の抗体とともにインキュベートすることにより実施した:ペルオキシダーゼ結合ヤギ抗マウスIgG(H+L)抗体(Jackson ImmunoResearch, West Grove, PA, Cat#115-035-146)(TBS-T中の2%脱脂乳にて0.04μg/mL)、ペルオキシダーゼ結合ヒトIgG抗体(Gamunex(商標)Bayer Corp., Elkhart, IN)(TBS-T中の2%脱脂乳にて0.2μg/mL)、又はポリクローナルヤギ抗マウスIgG抗体(重鎖特異的)(Sigma-Aldrich, St-Louis, MO)(TBS-T中の2%脱脂乳にて0.25μg/mL)。ペルオキシダーゼ結合ロバ抗ヤギIgG抗体(Jackson ImmunoResearch)(TBS-T中の2%脱脂乳にて0.04μg/mL)は、重鎖特異的抗体で処理したメンブレンについて二次抗体として用いた。免疫反応性複合体は、基質としてルミノール(Roche Giagnostics Corporation)を用いケミルミネセンスによって検出した。ヒトIgG抗体のセイヨウワサビペルオキシダーゼ酵素結合は、EZ-Link Plus(商標)活性化ペルオキシダーゼ結合キット(Pierce, Rockford, IL)を用いて実施した。
【0049】
ELISA定量アッセイ
マルチウェルプレート(Immulon 2HB, ThermoLab System, Franklin, MA)を、IgG1重鎖特異的ヤギ抗マウス抗体(Sigma M8770)(50mMの炭酸緩衝液(pH9.0)中に2.5μg/mL)で4℃にて16−18時間コーティングした。続いてマルチウェルプレートを、リン酸緩衝食塩水(PBS)(Pierce Biotechnology, Rockford, IL)中の1%カゼインで37℃にて1時間ブロッキングした。標準曲線は精製マウスIgG1コントロール(Sigma M9269)の希釈物を用いて作成した。イムノアッセイを実施しているときは、全ての希釈(コントロール及びサンプル)は、一切のマトリックスの作用を排除できるように、模擬接種物を浸透させインキュベートした植物組織から得た植物抽出物中で実施した。プレートをタンパク質サンプル及び標準曲線希釈物とともに1時間37℃でインキュベートした。PBS中の0.1%トゥイーン20で3回洗浄した後、このプレートをペルオキシダーゼ結合ヤギ抗マウスIgG(H+L)抗体(ブロッキング溶液中で0.04μg/mL)(Jackson ImmunoResearch 115-035-146)とともに37℃で1時間インキュベートした。PBS−Tでの洗浄を繰り返し、プレートを3,3',5,5'-テトラメチルベンジジン(TMB)Sure Blueペルオキシダーゼ基質(KPL, Gaithersburg, MD)とともにインキュベートした。反応を1NのHClを添加して停止させ、吸収を450nmで読み取った。各サンプルをトリプリケートでアッセイし、標準曲線の直線部分で濃度を内挿した。
【実施例4】
【0050】
IgG精製
材料の葉からのC5-1の精製は、N.ベンタミアナ(100−150g)の凍結葉を採取する工程、20mMリン酸ナトリウム、150mMのNaCl及び2mMメタ重亜硫酸ナトリウム(pH5.8−6.0)を添加する工程、及び市販のブレンダーを用いて室温で2−3分混ぜ合わせる工程を必要とした。不溶性線維はMiraclothTM(Calbiochem, San Diego, CA)での粗いろ過によって除去し、10mMフェニルメタンスルホニルフルオリド(PMSF)をろ液に添加した。1MのHClで抽出物をpH4.8±0.1に調整し、18000gで15分、2−8分遠心して清澄にした。この清澄にした上清を2MのTRISでpH8.0±0.1に調整し、再度18000gで15分、2−8分遠心して清澄にし、0.8及び0.2μmのメンブレン(Pall Corporation, Canada)で連続してろ過した。ろ過した材料は、有効面積が0.2ft2の100kDa分子量カットオフ限外ろ過膜(GE Healthcare Biosciences, Canada)を用いて接線流ろ過によって濃縮し、前記清澄材料の体積を5−10倍減少させた。続いて、この濃縮サンプルを、組換えプロテインG-セファロースファストフロー(Sigma-Aldrich, St-Louis, MO, Cat.#P4691)の5mmx5cmカラム(カラム体積1mL)に適用した。このカラムを5カラム体積の20mM TRIS-HCl、150mM NaCl(pH7.5)で洗浄した。抗体は100mMのグリシン(pH2.9−3.0)で溶出させ、計算した体積の1M TRIS-HCl(pH7.5)を含むチューブに採取することによって直ちにpHを中性にした。プールした溶出抗体分画を21000gで15分、2−8℃で遠心し、分析まで-80℃で保存した。精製後、前記アフィニティカラムを清浄にし、製造業者の指示にしたがって保存した。同じクロマトグラフィー媒体は、精製性能を顕著に変更することなく数回の精製に再使用することができる(10サイクルまで試験済み)。
【実施例5】
【0051】
N-グリコシル化分析
C5-1(50μg)を含むサンプルを15%SDS/PAGEで泳動した。重鎖及び軽鎖はクマシーブルーで明らかにし、重鎖に対応するタンパク質バンドを切り出し、小断片に切断した。断片を0.1M NH4HCO3/CH3CN(1/1)の溶液600μLで3回、各回15分間洗浄して乾燥させた。
ジスルフィド架橋の還元は、ゲル断片を0.1M NH4HCO3中の0.1MのDTT溶液600μLで56℃にて45分間インキュベートすることにより得られた。アルキル化は、0.1M NH4HCO3中のヨードアセトアミド55mMの溶液600μLを室温で30分添加することによって実施した。上清を廃棄し、ポリアクリルアミド断片を0.1M NH4HCO3/CH3CN(1/1)中でもう1回洗浄した。
続いてタンパク質を、0.05M NH4HCO3 600μL中の7.5μgトリプシン(Promega)で37℃にて16時間消化した。200μLのCH3CNを添加し、上清を収集した。続いて、ゲル断片を0.1M NH4HCO3 200μLで、その後200μLのCH3CNで再度、最後に200μLのギ酸(5%)で洗浄した。全ての上清をプールし凍結乾燥させた。
HPLCによるペプチドの分離は、C18逆相カラム(4.5x250mm)で0.1%のTFA中のCH3CNの直線勾配で実施した。分画を収集して凍結乾燥させ、337-nm窒素レーザーを搭載したVoyager DE-Pro MALDI-TOF装置(Applied Biosystems, USA)でMALDI-TOF-MSによって分析した。質量分析は、マトリックスとしてアルファ-シアノ-4-ヒドロキシ桂皮酸(Sigma-Aldrich)を用い、リフレクター遅延抽出モードで実施した。
【実施例6】
【0052】
アグロバクテリア浸透ニコチアナ・ベンタミアナの葉における一過性IgG発現の定量
強力なプラストシアニン系発現カセットが完全にアッセンブリされたIgGの高度な蓄積を駆動させることができるか否かを試験するために、pCambia二元プラスミドの同じT-DNAセグメント上で、C5-1(ネズミ抗ヒトIgG)(Khoudi et al. 1997)の軽鎖及び重鎖のコード配列をプラストシアニンプロモーター及び5'非翻訳配列の下流でタンデム構築物として、実施例1に記載するように(さらに図1にも提示されている)アッセンブリした。前記をさらにプラストシアニン3'非翻訳配列及び転写終了配列とフランキングさせた。
R612及びR610の両発現カセット(実施例1参照)では、軽鎖及び重鎖コード配列は、C5-1(Khoudi et al. 1999)由来の天然のシグナルペプチドを含んでいたが、R610では、アッセンブリされたIgGのゴルジ装置への移動を抑制するために、KDELペプチドのコード配列を重鎖のC-末端に付加した。
クローニング工程及びアグロバクテリウム・ツメファシエンス(AGL1)のプラスミドの移入に続いて、ニコチアナ・ベンタミアナの3つの植物の全ての葉に、プラスミドR612、R610又はR514(図1)で形質転換したアグロバクテリウム株を注射筒浸透させ、実施例2に記載したように分析を実施する前に、温室の条件下で6日間インキュベートした。インキュベーション期間の後、各植物の葉(ほぼ20gのバイオマス)を凍結し、粉砕し、さらに凍結粉末を混合して均質なサンプルを作製し、前記サンプルから1.5gの2つのサブサンプル(各植物から、実施例3参照)を抽出のために採取した。ポリクローナルヤギ抗マウスIgG1重鎖を捕捉用に、さらにペルオキシダーゼ結合ヤギ抗マウスIgG(H+L)を検出用に用いて、各サンプルの全タンパク抽出物中のC5-1含有量を酵素結合免疫吸着アッセイ(ELISA)によって定量した(実施例3参照)。
図2Aに示すように、R610又はR612(両構築物はプラストシアニンプロモーターを含む)は、サイレンシングサプレッサー(HcPro)の非存在下ではR514(2X35sプロモーターを含む)と比較したとき、極めて高レベルのタンパク質蓄積をもたらす。HcProの存在下では、R610及びR612の両方について大きく増加した発現レベルが観察された。図2Bに示すように、R612のアグロバクテリア浸透は、新鮮質量1kg当たり106mgの抗体の蓄積をもたらし、一方、抗体のER保持型(R610)では同じ条件で211mg/kgFWに達した。
【0053】
転写後遺伝子サイレンシング(PTGS)はアグロバクテリア浸透ニコチアナ・ベンタミアナ植物のトランスジーンの発現を制限し、ジャガイモウイルスYのサイレンシングサプレッサー(HcPro)の同時発現は、トランスジーンのmRNAの特異的分解を妨害することが示されたので(Brigneti et al. 1998)、HcPro構築物(Hamilton et al. 2002)の同時浸透をC5-1の発現増加における有効性について試験した。R612及びR610とHcProとの同時発現は、HcProの非存在下で観察された抗体蓄積と比較して、その蓄積をそれぞれ5.3倍及び3.6倍増加させた。HcProの存在下では、プラストシアニン制御C5-1発現は、R612では558mg/kgFW、R610では757mg/kgFWの平均値に達した(図2A)。C5-1発現の最大レベルは、R612及びR610の両浸透葉由来のいくつかの抽出物で1.5g/kgFW(全可溶性タンパク質の25%)を超えた。
アグロバクテリア浸透発現系の計測能を判定するために、Kapilaら(1997)の記載した方法から応用した真空浸透方法に続いて、C5-1の蓄積を定量した。この実験シリーズでは、全植物の気生部分にR612+HcPro又はR610+HcProを用いて真空浸透を実施し、採集前の6日間温室に戻した。大規模生産系に典型的なデータを提供するために、数植物から得られる葉/葉柄のほぼ250g量のバッチを凍結し、すり潰して均質なサンプルにし、さらにバッチ当たり7.5g量を含む3つのサブサンプルを分析用に収集した。ELISAによる定量で示されたように、平均的なC5-1蓄積レベルは、R612とR610浸透についてそれぞれ238及び328mg/kgFWに達した(図2B)。
【0054】
プルーニングの影響:
適切なプラスミドで形質転換したアグロバクテリア株を用いて葉に真空浸透を施す1、2又は3日前に、3つのニコチアナ・ベンタミアナ植物の頂芽及び腋芽を鋏で植物から機械的に除去するか、又はEthrel、B-9(500ppm)若しくはA-rest(4ppm)を用いて化学的にプルーニングした。
続いて、植物にインフルエンザ抗原(構築物312、図1)、ヒトIgG(構築物935、図1)を浸透させ、実施例2に記載した分析の前に温室条件下で6日間インキュベートした。コントロール植物はプルーニングしなかった。インキュベーション期間の後で、各植物の葉(ほぼ20gのバイオマス)を凍結してすり潰し、さらに凍結粉末を混合して均質なサンプルを作製し、それらサンプルから1.5グラムの2つのサブサンプル(各植物から、実施例3参照)を抽出用に採取した。ポリクローナルヤギ抗マウスIgG1重鎖を捕捉用に、さらにペルオキシダーゼ結合ヤギ抗マウスIgG(H+L)を検出用に用いて、各サンプルの全タンパク抽出物中のC5-1含有量を酵素結合免疫吸着アッセイ(ELISA)によって定量した(実施例3参照)。
図7Aに示すように、312(インフルエンザ抗原)のアグロバクテリア浸透12時間前に機械的プルーニングを実施して頂芽及び腋芽を除去することによって、コントロール処理(プルーニング実施せず)と比較して、抗原の蓄積の増加がもたらされた(150%)。頂芽優勢を阻害することが知られているいくつかの成長調節物質(Ethrel、500ppm;B-9、2500ppm;又はA-rest、4.0ppm)による植物の化学的プルーニング処理とそれに続く512のアグロバクテリアによる浸透を用いて、200%までの発現レベルの増加もまた観察された。同様な結果がまた、これらの成長調節化合物及び他の成長調節化合物を製造業者の推奨した適用率で浸透7日前に用いたときに観察された。
【0055】
機械的プルーニングもまた、植物をアグロバクテリア浸透12時間前に真空浸透(図7B)又は注射筒浸透(図7C)によってプルーニングしたとき、免疫グロブリン935(hIgG、図1)の発現レベルの増加をもたらした。
アグロバクテリア浸透の1から3日前の植物プルーニングは、図8に示すように(機械的プルーニング植物)、タンパク質(インフルエンザ抗原;312、図1)の蓄積の更なる増加をもたらした。発現の極めて大きな増加が、浸透の1から2日目に植物を機械的にプルーニングしたときに観察される。浸透の3日前又は7日前の植物の化学的プルーニングもまた、非プルーニング植物を超えるタンパク質蓄積増加がもたらすことが見出された。
図9に示すように、対象のコード領域が光合成プロモータープラストシアニンによって駆動され、プルーニング(真空浸透の12時間前に機械的プルーニング)とサイレンシングサプレッサーの同時発現とが併用されたとき、抗体(935、図1)の浸透に続いて発現レベルの増加が観察される。プルーニングに続く935とサイレンシングサプレッサーHcProとの同時発現は、HcProの非存在下で観察される抗体蓄積レベルと比較して3−8倍の抗体蓄積レベルの増加をもたらした。HcProと同時発現させたとき、プラストシアニンにより制御される発現は、プルーニングに続いて280mg/kgFWの平均値に達した。
【実施例7】
【0056】
生成抗体の性状決定
タンパク質ブロット分析(実施例3参照)を用いて、注射筒浸透及び真空浸透実験の両実験の後、分泌型(R612)及びER保持型(610)タンパク質を生成する植物でC5-1 IgGのアッセンブリ及び断片化レベルを明らかにした。H+Lペルオキシダーゼ結合ヤギ抗マウスIgGをプローブに用いたウェスタンブロットを先ず初めに用いて、C5-1分子におけるその由来とは無関係に、最大量の抗体フラグメントの存在を明瞭に示した。図3Aに示すように、全てのタンパク質抽出物は、使用した分子内標的誘導方法又は浸透方法とは無関係に、類似の分子サイズのフラグメントを類似の相対的豊富さで含んでいた。各事例で、完全な抗体に一致する約150kDaの主要(≧85%)バンドが、約135kDa及び約100kDaのマイナーバンドとともに明示され、抗体は主としてその完全なアッセンブリ型(H2L2)として蓄積されることが示された。興味深いことに、同様な電気泳動移動度を有するフラグメントがまた、ネズミ腫瘍細胞株(MOPC-21;Sigma #M9269)から生成されたコントロールIgGにも存在し、植物及び哺乳動物細胞株で生じる断片化は類似し、おそらく共通のタンパク質分解活性から生じることが示唆された。同様な結果がまた、検出用抗マウス重鎖特異的抗体を用いて得られた。
抽出物中に存在する抗体フラグメントの実体を調べるために、ブロットタンパク質をペルオキシダーゼ結合ヒトIgG1(C5-1の抗原)プローブを用いて試験する活性ブロットを利用した。約150kDaの完全にアッセンブリした抗体の実体は図3Bで知ることができる。さらにまた、ウェスタンブロットで観察される断片化パターンは活性ブロットで知ることができるが(図3B参照)、ただし100kDaバンドは例外である(図3A参照)。理論に拘束されないが、この結果は、100kDaフラグメントはC5-1抗体のFab領域を含まず、少なくとも部分的には重鎖ダイマー(抗体アッセンブリーの中間物)から成るかもしれない。
【実施例8】
【0057】
抗体精製及び精製生成物の性状決定
プロテインGアフィニティークロマトグラフィー単工程を用いてバイオマスから抗体を精製し、得られた生成物をSDS-PAGEによって分析した(実施例4参照)。図4に提示したクマシーブルー染色ゲルは、プロテインGから溶出した分画中の150kDaの主要バンドを示している。このバンドは、分泌型及びER保持型の両型で精製生成物の85%を占め、夾雑物の含有量は両型について同一である(図4A、レーン4及び5)。ポリクローナル抗マウスIgGプローブで調べたウェスタンブロット分析は、精製C5-1分画中の主要夾雑物がネズミIgG起源であることを示した(データは示されていない)。還元条件下では、2つの主要な生成物が約26kDa及び約55kDaで検出され、これらは、それぞれ軽鎖及び重鎖の分子量と一致している(図4B、レーン2)。ER保持抗体の重鎖は、アポプラスト抗体の重鎖より高い電気泳動移動度を示し(図4B、レーン3)、これは、C-末端に存在する付加KDELアミノ酸とER保持によるN-グリコシル化の相違とが重なった結果と解釈される。図4Cは、精製抗体(150kDa)が、75、90、100及び120kDaの夾雑フラグメントと同様にヒトIgG1と結合したことを示し、このフラグメントに少なくとも1つのFabセグメントが存在することを明瞭に示している。100kDaフラグメントのFabの存在は、粗抽出物から得られた結果と対照的で、後者では100kDaバンドはヒトIgG1と結合しなかった。粗抽出物において100kDaで泳動するFab-含有フラグメントの量はこの活性ブロットによる検出のためには低すぎたか、又は100kDaで泳動するフラグメントは2つの異なる分子(一方は重鎖ダイマー(Fab無し)及び他方は抗原結合領域を含む)から成っていたという仮説が成り立つ。
抗体生成についてこの系の再現性は、2つの異なる浸透バッチから精製した生成物及び各バッチ由来の3つの別個の精製ロットを並べて比較して判定された。精製ロットのクマシー染色SDS-PAGE分析によって、全てのロットで同一バンドが高度に類似する相対的豊富さで存在することが示された(図4D)。
【実施例9】
【0058】
ヒトガラクトシルトランスフェラーゼの同時発現による抗体のN-グリコシル化の改変
一過性同時発現を用いて、一過性発現中に生成されつつあるタンパク質のグリコシル化を制御することができるか否かを調べるために、天然のヒトβ1,4ガラクトシルトランスフェラーゼ(GalT)を含む35S系発現カセットを調製した。R622はGalTを含み(図5B)、R621は、N-アセチルグルコサミニルトランスフェラーゼ(GNTI)のCTSドメインと融合させたGalT触媒ドメインを含んでいた(GNT1GalT、図5A)。N-アセチルグルコサミニルトランスフェラーゼ(GNTI)のCTSドメインは、GNT1が複雑なN-グリカン合成の初期にER及びcis-ゴルジ装置で機能するので、ヒトGalT触媒ドメインのために膜足場として選択した(Saint-Jore-Dupas et al. 2006)。理論に拘束されないが、タンパク質成熟の初期にGalT活性を隔離することによって、成熟グリカンのβ1,4ガラクトース付加及びコアのフコシル化及びキシロシル化の効率的な阻害がもたらされるのかもしれない。これらの構築物をC5-1とともに植物に同時浸透させた。
ニコチアナ・ベンタミアナの植物に、HcProの存在下でR612(C5-1の分泌型)、R612+R621(GNT1GalT)又はR612+R622(GalT)を浸透させた。図6は、これらのバイオマスサンプルから精製したC5-1の免疫学的分析を示す。
抗体のガラクトシル化は、β1,4ガラクトースと特異的に結合するエリトリナ・クリスタガリ(Erythrina cristagali)アグルチニン(ECA)を用いる親和検出によって概算した。期待したように、C5-1を単独で発現させたとき(R612、図6)ガラクトースは検出されなかった。ガラクトシル化は、R512+R622(GalT)による同時浸透から精製したC5-1で認められたが、R612+R621(GNT1GalT、図6)による同時浸透から得たものでは認められなかった。抗α1,3フコース抗体を用いて実施したウェスタンブロットは、ガラクトシルトランスフェラーゼ無しに発現させたコントロールC5-1のN-グリカンのフコシル化を明示した。N-グリカンのフコシル化は、アグロバクテリア浸透の方法に関係なくGNT1GalTを同時発現させた抗体で検出されなかったが、一方、天然のGalTの同時発現は抗体のフコシル化の検出可能な低下をもたらさなかった(図6)。同様な結果が抗β1,2キシロース特異的抗体を用いて得られた。前記の結果では、GNT1GalT同時発現C5-1におけるキシロース特異的免疫シグナルの完全な欠如、及びGalT同時発現C5-1におけるキシロース特異的免疫シグナルの存在が示された。
【0059】
完全にアッセンブリされたIgGの直接視覚的概算のためのクマシーゲル染色、並びにウェスタン及び活性ブロットを同じ抽出物について実施した。このデータを基にすれば、記載した抗体発現系は1.5g/kg新鮮質量の収量を達成し、粗抽出物中の生成物の85%を越えるものが、約150kDaの完全サイズのテトラマーIgGから成っていた。
重鎖のC-末端へのKDELペプチドの付加が以前に用いられ、ゴルジからERへの抗体の回収を仲介することにより抗体蓄積を増加させた(2−10倍)(Schillberg et al. 2003)。本明細書に記載の発現系を用いたとき、C5-1の重鎖へのKDELペプチドの付加は、HcProサイレンシングサプレッサーを使用しないときC5-1の収量を倍増させた。KDELの存在下又は非存在下におけるC5-1の収量の相違は、HcProを使用してサイレンシングを低下させたとき顕著に減少した。ER保持は生成物の品質に影響を与えなかった。なぜならば、ER保持型抗体及び分泌型抗体を産生する植物から得られる粗抽出物で観察されるフラグメントでは、サイズ及び相対的豊富さが同一であったからである。
全ての引用文献は参照により本明細書に含まれる。
本発明は1つ以上の実施態様に関して記載してきた。しかしながら、多数の変型及び改変が、特許請求の範囲で規定した本発明の範囲から逸脱することなく実施されえることは当業者には明白であろう。
【0060】
参考文献
【特許請求の範囲】
【請求項1】
以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法:
i)前記植物又は植物の部分をプルーニングし、プルーニングされた植物又は植物の部分を生産する工程、
ii)前記植物中で活性な調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を、前記プルーニングされた植物又は植物の部分に一過性態様で導入する工程、及び
iii)前記プルーニングされた植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分において発現させることを可能にする条件下で維持する工程。
【請求項2】
前記導入工程(工程ii)において、2つ以上の核酸配列が植物に導入される、請求項1に記載の方法。
【請求項3】
前記導入工程(工程ii)において、前記1つ以上の核酸配列が、前記プルーニングされた植物又は植物の部分に真空下で導入される、請求項1に記載の方法。
【請求項4】
前記導入工程(工程ii)において、前記1つ以上の核酸配列が、前記プルーニングされた植物又は植物の部分に注射筒による浸透を用いて導入される、請求項1に記載の方法。
【請求項5】
前記プルーニング工程(工程i)において、前記プルーニングされた植物が、機械的プルーニングを用いて生産される、請求項1に記載の方法。
【請求項6】
前記プルーニング工程(工程i)において、前記プルーニングされた植物が、化学的プルーニングを用いて生産される、請求項1に記載の方法。
【請求項7】
前記導入工程(工程ii)において、前記調節領域が、光合成遺伝子から得られるプロモーターである、請求項1に記載の方法。
【請求項8】
前記調節領域が、プラストシアニンプロモーター、プラストシアニン3'UTR及びターミネーター、又は両方である、請求項7に記載の方法。
【請求項9】
前記2つ以上の核酸配列のうちの1つが、サイレンシングのサプレッサーをコードする、請求項2に記載の方法。
【請求項10】
前記サイレンシングのサプレッサーがHcProである、請求項9に記載の方法。
【請求項11】
対象のタンパク質が抗体、抗原又はワクチンである、請求項1に記載の方法。
【請求項12】
以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法:
i)前記植物又は植物の部分中で活性な光合成遺伝子から得られる調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を一過性態様で導入する工程、及び
ii)前記植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分において発現させることを可能にする条件下で維持する工程。
【請求項13】
前記導入工程(工程i)において、2つ以上の核酸配列が植物に導入される、請求項12に記載の方法。
【請求項14】
前記導入工程(工程i)において、前記1つ以上の核酸配列が、前記植物又は植物の部分に真空下で導入される、請求項12に記載の方法。
【請求項15】
前記導入工程(工程i)において、前記1つ以上の核酸配列が、前記植物又は植物の部分に注射筒による浸透を用いて導入される、請求項12に記載の方法。
【請求項16】
前記調節領域が、プラストシアニン遺伝子から得られる、請求項12に記載の方法。
【請求項17】
前記2つ以上の核酸配列のうちの1つが、サイレンシングのサプレッサーをコードする、請求項13に記載の方法。
【請求項18】
前記サイレンシングのサプレッサーがHcProである、請求項17に記載の方法。
【請求項19】
対象のタンパク質が抗体、抗原又はワクチンである、請求項10に記載の方法。
【請求項20】
以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法:
i)前記植物又は植物の部分をプルーニングし、プルーニングされた植物又は植物の部分を生産する工程、
ii)前記植物又は植物の部分中で活性な光合成遺伝子から得られる調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を、一過性態様で導入する工程、及び
iii)前記プルーニングされた植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分において発現させることを可能にする条件下で維持する工程。
【請求項1】
以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法:
i)前記植物又は植物の部分をプルーニングし、プルーニングされた植物又は植物の部分を生産する工程、
ii)前記植物中で活性な調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を、前記プルーニングされた植物又は植物の部分に一過性態様で導入する工程、及び
iii)前記プルーニングされた植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分において発現させることを可能にする条件下で維持する工程。
【請求項2】
前記導入工程(工程ii)において、2つ以上の核酸配列が植物に導入される、請求項1に記載の方法。
【請求項3】
前記導入工程(工程ii)において、前記1つ以上の核酸配列が、前記プルーニングされた植物又は植物の部分に真空下で導入される、請求項1に記載の方法。
【請求項4】
前記導入工程(工程ii)において、前記1つ以上の核酸配列が、前記プルーニングされた植物又は植物の部分に注射筒による浸透を用いて導入される、請求項1に記載の方法。
【請求項5】
前記プルーニング工程(工程i)において、前記プルーニングされた植物が、機械的プルーニングを用いて生産される、請求項1に記載の方法。
【請求項6】
前記プルーニング工程(工程i)において、前記プルーニングされた植物が、化学的プルーニングを用いて生産される、請求項1に記載の方法。
【請求項7】
前記導入工程(工程ii)において、前記調節領域が、光合成遺伝子から得られるプロモーターである、請求項1に記載の方法。
【請求項8】
前記調節領域が、プラストシアニンプロモーター、プラストシアニン3'UTR及びターミネーター、又は両方である、請求項7に記載の方法。
【請求項9】
前記2つ以上の核酸配列のうちの1つが、サイレンシングのサプレッサーをコードする、請求項2に記載の方法。
【請求項10】
前記サイレンシングのサプレッサーがHcProである、請求項9に記載の方法。
【請求項11】
対象のタンパク質が抗体、抗原又はワクチンである、請求項1に記載の方法。
【請求項12】
以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法:
i)前記植物又は植物の部分中で活性な光合成遺伝子から得られる調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を一過性態様で導入する工程、及び
ii)前記植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分において発現させることを可能にする条件下で維持する工程。
【請求項13】
前記導入工程(工程i)において、2つ以上の核酸配列が植物に導入される、請求項12に記載の方法。
【請求項14】
前記導入工程(工程i)において、前記1つ以上の核酸配列が、前記植物又は植物の部分に真空下で導入される、請求項12に記載の方法。
【請求項15】
前記導入工程(工程i)において、前記1つ以上の核酸配列が、前記植物又は植物の部分に注射筒による浸透を用いて導入される、請求項12に記載の方法。
【請求項16】
前記調節領域が、プラストシアニン遺伝子から得られる、請求項12に記載の方法。
【請求項17】
前記2つ以上の核酸配列のうちの1つが、サイレンシングのサプレッサーをコードする、請求項13に記載の方法。
【請求項18】
前記サイレンシングのサプレッサーがHcProである、請求項17に記載の方法。
【請求項19】
対象のタンパク質が抗体、抗原又はワクチンである、請求項10に記載の方法。
【請求項20】
以下の工程を含む、植物又は植物の部分内で対象のタンパク質を合成する方法:
i)前記植物又は植物の部分をプルーニングし、プルーニングされた植物又は植物の部分を生産する工程、
ii)前記植物又は植物の部分中で活性な光合成遺伝子から得られる調節領域と機能的に連結された、対象のタンパク質をコードする1つ以上の核酸配列を、一過性態様で導入する工程、及び
iii)前記プルーニングされた植物又は植物の部分を、対象のタンパク質をコードする前記核酸配列を前記植物又は植物の部分において発現させることを可能にする条件下で維持する工程。
【図1A】

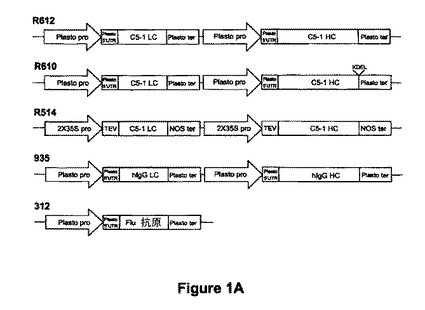
【図1B】

【図1C】

【図1D】

【図2A】
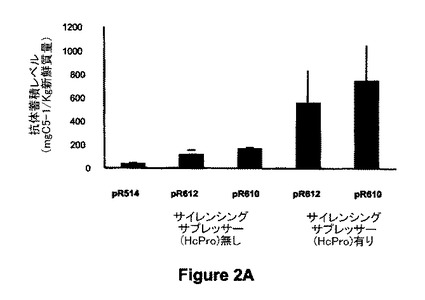
【図2B】
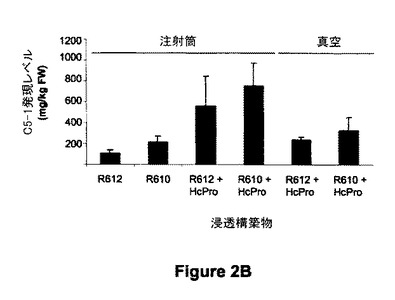
【図3A】
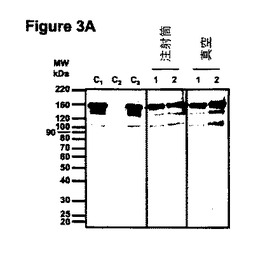
【図3B】

【図4A】
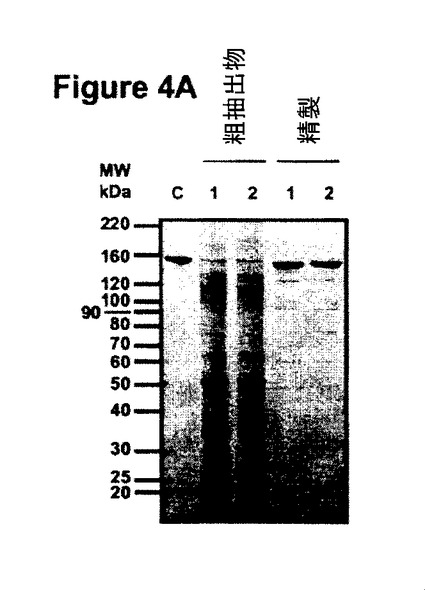
【図4B】
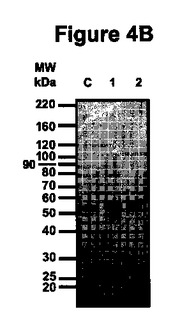
【図4C】
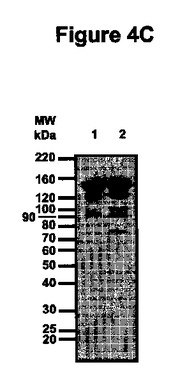
【図4D】
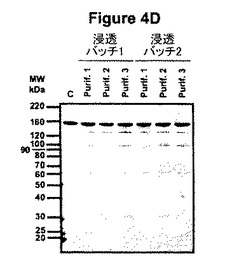
【図5A】
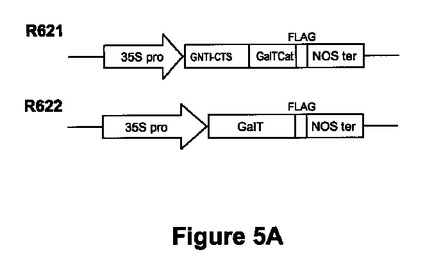
【図5B】
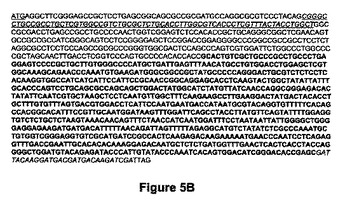
【図5C】

【図5D】
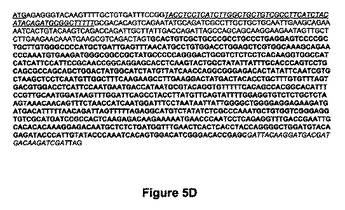
【図5E】

【図5F】

【図5G】

【図6】
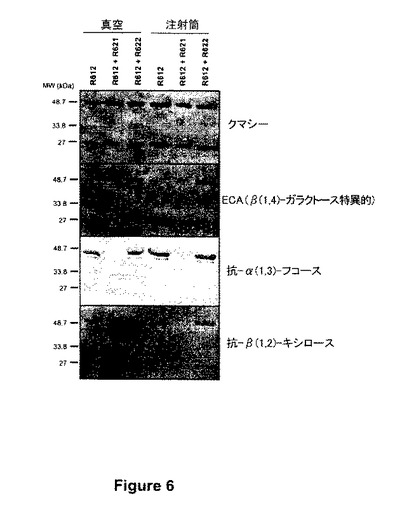
【図7A】
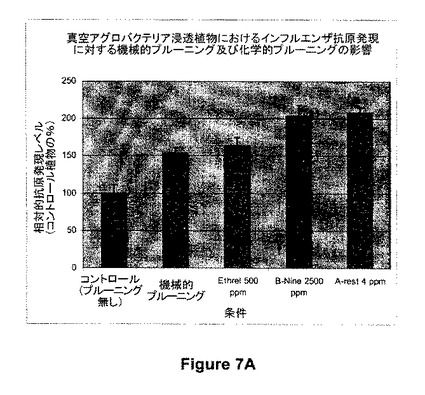
【図7B】
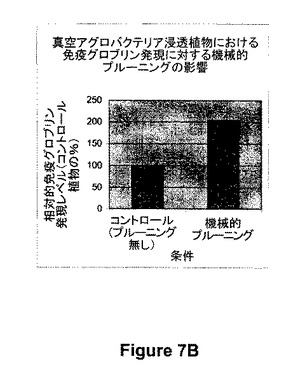
【図7C】
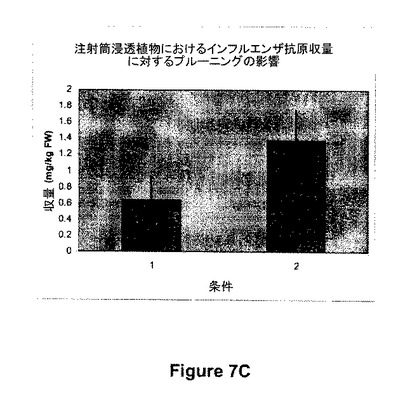
【図8】
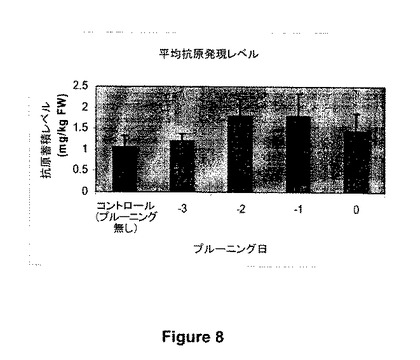
【図9】
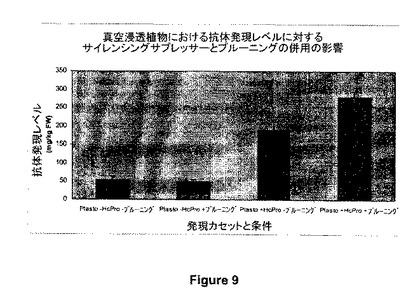
【図1B】
【図1C】
【図1D】
【図2A】
【図2B】
【図3A】
【図3B】
【図4A】
【図4B】
【図4C】
【図4D】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】
【図5G】
【図6】
【図7A】
【図7B】
【図7C】
【図8】
【図9】
【公表番号】特表2010−529845(P2010−529845A)
【公表日】平成22年9月2日(2010.9.2)
【国際特許分類】
【出願番号】特願2010−511463(P2010−511463)
【出願日】平成20年6月13日(2008.6.13)
【国際出願番号】PCT/CA2008/001146
【国際公開番号】WO2008/151444
【国際公開日】平成20年12月18日(2008.12.18)
【出願人】(502121395)メディカゴ インコーポレイテッド (13)
【Fターム(参考)】
【公表日】平成22年9月2日(2010.9.2)
【国際特許分類】
【出願日】平成20年6月13日(2008.6.13)
【国際出願番号】PCT/CA2008/001146
【国際公開番号】WO2008/151444
【国際公開日】平成20年12月18日(2008.12.18)
【出願人】(502121395)メディカゴ インコーポレイテッド (13)
【Fターム(参考)】
[ Back to top ]