
植物における、化学物質による外来遺伝子の誘導発現方法
【課題】銅イオン誘導性システムについて、目的とする外来遺伝子の植物における誘導発現における誘導倍率の向上を達成できる方法等を提供すること。
【解決手段】植物における、化学物質による目的とする外来遺伝子の誘導発現方法であって、前記の目的とする外来遺伝子が含むプロモーター機能を有する領域の下流に任意の外来遺伝子の5'−非翻訳領域を有するように構築されてなる遺伝子構築物に含まれる転写因子であり、前記の目的とする外来遺伝子とは異なる他の外来遺伝子によりコードされる転写因子を銅イオンにより活性化することにより、前記の目的とする外来遺伝子が含むプロモーター機能を有する領域による前記の目的とする外来遺伝子の転写活性化を誘導する工程を有することを特徴とする方法等。
【解決手段】植物における、化学物質による目的とする外来遺伝子の誘導発現方法であって、前記の目的とする外来遺伝子が含むプロモーター機能を有する領域の下流に任意の外来遺伝子の5'−非翻訳領域を有するように構築されてなる遺伝子構築物に含まれる転写因子であり、前記の目的とする外来遺伝子とは異なる他の外来遺伝子によりコードされる転写因子を銅イオンにより活性化することにより、前記の目的とする外来遺伝子が含むプロモーター機能を有する領域による前記の目的とする外来遺伝子の転写活性化を誘導する工程を有することを特徴とする方法等。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、植物における、化学物質による外来遺伝子の誘導発現方法等に関するものである。
【背景技術】
【0002】
目的とする外来遺伝子を植物に導入し発現させる場合には、通常、例えば、カリフラワーモザイクウイルス(CaMV)35Sプロモーター(例えば、非特許文献1参照)のような常に発現する構成的プロモーターを用いることが多い。しかしながら、このような構成的プロモーターを利用する場合には、植物の転写系・翻訳系に多大な負荷をかけることになるうえ、目的とする外来遺伝子の種類によっては、発芽や生育の阻害等植物に悪影響を与えることがある。このような悪影響を回避する一つの方法として、目的とする外来遺伝子を所望の時期に誘導発現させる方法を挙げることができる。そして、このような誘導発現方法を利用することによって、目的とする外来遺伝子の一層効率的な発現、ひいては当該外来遺伝子が含む構造遺伝子領域によりコードされる目的とする外来蛋白質等の高生産が実現できるうえ、植物の生長や生理を自在に制御できるようになり、その産業的利用価値は計りしれない。
【0003】
目的とする外来遺伝子を所望の時期に誘導発現させる方法は、その誘導条件の差異に基づき、2つに大別することができる。即ち、温度や光、植物病原体等の非化学物質により誘導発現させるものと、化学物質により誘導発現させるものとである。
前者の例としては、具体的には例えば、熱誘導性システム(例えば、特許文献1参照)、低温誘導性システム(例えば、特許文献2参照)、植物病原体の攻撃により誘導されるシステム(例えば、特許文献3参照)等が挙げられる。これらは、誘導性プロモーターを目的とする外来遺伝子の上流に連結してなる遺伝子構造体を構築し当該遺伝子構造体を利用するだけでよく、当該遺伝子構造体の構造は極めて単純である。しかしながら、温度や光、植物病原体等の非化学物質により誘導発現させるものは、予期せぬ環境の変化や植物病原体の襲来等で突発的に目的とする外来遺伝子が発現してしまう危険性がある。
一方、後者の例としては、具体的には例えば、銅イオン誘導性システム(例えば、非特許文献2参照)、ステロイドホルモン誘導性システム(例えば、非特許文献3参照)、エタノール誘導性システム(例えば、非特許文献4参照)、テトラサイクリン誘導性システム(例えば、非特許文献5参照)等が挙げられる。これらは、Moore I et al (例えば、非特許文献6参照)、Padidam M (例えば、非特許文献7参照)等により詳細に解説されており、特定の化学物質の濃度に依存して目的とする外来遺伝子の発現を制御することができる。即ち、所望の時期に必要な分だけ目的とする外来遺伝子の発現を誘導することができる。
【0004】
従来、目的とする外来遺伝子が含む構造遺伝子領域によりコードされる目的とする外来蛋白質等を組換え植物にて生産(植物内に蓄積)させるために、目的とする外来遺伝子が含む構造遺伝子領域の上流に位置させるタバコモザイクウイルス(TMV)のΩ配列(例えば、非特許文献8参照)やタバコアルコールデヒドロゲナーゼ遺伝子の5’−非翻訳領域配列(例えば、非特許文献9参照)を利用した例が存在しているが、一方で、Ω配列が連結されているmRNAはヌクレアーゼにより分解されやすいと報告する例も存在しており(例えば、非特許文献10参照)、5’−非翻訳領域配列の利用が如何なる結果をもたらすかを容易に想像できる技術状況にはない。まして、特定な誘導性システム(例えば、銅イオン誘導性システム等)における化学物質による目的とする外来遺伝子の誘導発現にどのような影響を与えるものであるか等は全く不明であった。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Benfey PN & Chua NH、1990、Science 250、959-966
【非特許文献2】Mett VL et al、1993、Proc Natl Acad Sci 90、4567-4571
【非特許文献3】Aoyama T & Chua NH、1997、Plant J.11、605-612; 米国特許第6063985号
【非特許文献4】Caddick MX et al、1998、Nature Biotech. 16、177-180; 国際公開第93/21334号パンフレット
【非特許文献5】Weinmann P et al、1994、Plant J. 5、559-569
【非特許文献6】Moore I et al 、2006、Plant J 45、651-683
【非特許文献7】Padidam M、2003、Current Opin Plant Biol 6、169-177
【非特許文献8】Gallie D et al、1987、Nucleic Acid Res 15、3257-3273; 米国特許第5489527号
【非特許文献9】Satoh J et al、2004、J Biosci Bioeng 98(1):1-8; 特開2003-079372号公報
【非特許文献10】Gallie D et al、1988、Nucleic Acid Res 16、8675-8694
【特許文献1】米国特許第5447858号
【特許文献2】米国特許第5847102号
【特許文献3】米国特許第5942662号
【発明の概要】
【発明が解決しようとする課題】
【0006】
理想的な、植物における、化学物質による目的とする外来遺伝子の誘導発現方法は、非誘導時の目的とする外来遺伝子の発現レベルが非常に低く、誘導時の目的とする外来遺伝子の発現レベルが非常に高いものである。非誘導時の目的とする外来遺伝子の発現レベルが低ければ低いほど、宿主植物への負荷が少なくなるうえ、微量で機能するシグナル因子等をも制御できるようになる。また、誘導時の目的とする外来遺伝子の発現レベルが高ければ高いほど、目的とする外来遺伝子が含む構造遺伝子領域によりコードされる目的とする外来蛋白質等を効率的に生産可能となるうえ、誘導に必要な刺激が少量で済むからである。
上述のような誘導性システムにおける化学物質による目的とする外来遺伝子の誘導発現のうち、銅イオン誘導性システムについては、当該システムが有する誘導発現の低い誘導倍率が問題となっており(例えば、非特許文献7参照)、本発明では、より理想に近づけることによって、当該銅イオン誘導性システムにおいて、目的とする外来遺伝子の誘導発現での誘導倍率の向上を達成できる方法を提供することを課題としている。
【課題を解決するための手段】
【0007】
本発明者らは、かかる状況下鋭意検討した結果、本発明に至った。
即ち、本発明は、
1.植物における、化学物質による目的とする外来遺伝子の誘導発現方法であって、前記の目的とする外来遺伝子が含むプロモーター機能を有する領域の下流に任意の外来遺伝子の5'−非翻訳領域を有するように構築されてなる遺伝子構築物に含まれる転写因子であり、前記の目的とする外来遺伝子とは異なる他の外来遺伝子によりコードされる転写因子を、銅イオンにより活性化することにより、前記の目的とする外来遺伝子が含むプロモーター機能を有する領域による前記の目的とする外来遺伝子の転写活性化を誘導する工程を有することを特徴とする方法(以下、本発明誘導発現方法と記すこともある。);
2.前記の5'−非翻訳領域の塩基配列が、下記の5'−非翻訳領域に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項1記載の方法;
<5'−非翻訳領域に係る塩基配列群>
(1)ウイルスの遺伝子又は植物の誘導性遺伝子由来の塩基配列
(2)トバモウイルス属に属するウイルスの遺伝子由来の塩基配列
(3)トマトモザイクウイルスの遺伝子由来の塩基配列
(4)トマトモザイクウイルスの130k/180k遺伝子由来の塩基配列
3.前記の転写因子の塩基配列が、下記の転写因子に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項1又は2記載の方法;
<転写因子に係る塩基配列群>
(1)真核生物由来の塩基配列
(2)酵母由来の塩基配列
(3)酵母ACE1由来の塩基配列
4.前記の遺伝子構築物が、前記の転写因子とは異なる他の転写因子の転写活性化領域であり、前記の転写因子の下流に位置する転写活性化領域を、さらに追加的に有するように構築されてなる遺伝子構築物であることを特徴とする前項1、2又は3記載の方法;
5.前記の転写活性化領域が、下記の転写活性化領域に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項4記載の方法;
<転写活性化領域に係る塩基配列群>
(1)ウイルスの転写因子由来の塩基配列
(2)シンプレックスウイルス属に属するウイルスの転写因子由来の塩基配列
(3)単純ヘルペスウイルスの転写因子由来の塩基配列
(4)単純ヘルペスウイルスのVP16転写因子由来の塩基配列
6.前記の遺伝子構築物が、任意のプロモーターを前記の転写因子の上流に追加的に有するように構築されてなる遺伝子構築物であることを特徴とする前項1、2、3、4又は5記載の方法;
7.植物において、化学物質により目的とする外来遺伝子を誘導発現させるための遺伝子構築物であって、
(a)プロモーター機能を有する領域と目的とする外来蛋白質に係る構造遺伝子領域とを含む、目的とする外来遺伝子と、
(b)前記の目的とする外来遺伝子とは異なる他の外来遺伝子によりコードされ且つ銅イオンにより活性化される転写因子と、
(c)前記の目的とする外来遺伝子が含むプロモーター機能を有する領域(以下、本プロモーター領域と記すこともある。)の下流に位置する任意の外来遺伝子の5'−非翻訳領域と
を有するように構築可能に調製されてなることを特徴とする遺伝子構築物(以下、本発明遺伝子構築物と記すこともある。);
8.前記の5'−非翻訳領域の塩基配列が、下記の5'−非翻訳領域に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項7記載の遺伝子構築物;
<5'−非翻訳領域に係る塩基配列群>
(1)ウイルスの遺伝子又は植物の誘導性遺伝子由来の塩基配列
(2)トバモウイルス属に属するウイルスの遺伝子由来の塩基配列
(3)トマトモザイクウイルスの遺伝子由来の塩基配列
(4)トマトモザイクウイルスの130k/180k遺伝子由来の塩基配列
9.前記の転写因子の塩基配列が、下記の転写因子に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項7又は8記載の遺伝子構築物;
<転写因子に係る塩基配列群>
(1)真核生物由来の塩基配列
(2)酵母由来の塩基配列
(3)酵母ACE1由来の塩基配列
10.前記の転写因子とは異なる他の転写因子の転写活性化領域であり、前記の転写因子の下流に位置する転写活性化領域を、さらに追加的に有するように構築されてなることを特徴とする前項7、8又は9記載の遺伝子構築物;
11.前記の転写活性化領域が、下記の転写活性化領域に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項10記載の遺伝子構築物;
<転写活性化領域に係る塩基配列群>
(1)ウイルスの転写因子由来の塩基配列
(2)シンプレックスウイルス属に属するウイルスの転写因子由来の塩基配列
(3)単純ヘルペスウイルスの転写因子由来の塩基配列
(4)単純ヘルペスウイルスのVP16転写因子由来の塩基配列
12.任意のプロモーターを前記の転写因子の上流に追加的に有するように構築されてなる遺伝子構築物であることを特徴とする前項7、8、9、10又は11記載の遺伝子構築物;
13.前項7〜12のいずれかの前項記載の遺伝子構築物が導入されてなることを特徴とする形質転換体植物(以下、本発明形質転換体植物と記すこともある。);
14.前項13記載の形質転換体植物から目的とする外来蛋白質を回収することを特徴とする外来蛋白質の取得方法(以下、本発明外来蛋白質取得方法と記すこともある。);
等を提供するものである。
【発明の効果】
【0008】
本発明により、銅イオン誘導性システムについて、目的とする外来遺伝子の植物における誘導発現における誘導倍率の向上を達成できる方法等を提供することが可能となる。
【図面の簡単な説明】
【0009】
【図1】図1は、銅イオン誘導性sGFP遺伝子発現ベクターのT-DNA領域の構造を示す模式図である。図中の*印は、as-1サイトへの変異導入部位を示している。
【図2】図2は、銅イオン誘導性sGFP遺伝子発現ベクターが導入されてなる組換えシロイヌナズナにおける、sGFP遺伝子のmRNA量をリアルタイムPCRにより定量した結果を示す図である。グラフ内の数値は、非誘導発現処理区でのsGFP遺伝子のmRNA量に対する、誘導発現処理区でのsGFP遺伝子のmRNA量の割合を示している。
【図3】図3は、銅イオン誘導性sGFP遺伝子発現ベクターが導入されてなる組換えタバコにおける、sGFP遺伝子のmRNA量をリアルタイムPCRにより定量した結果を示す図である。グラフ内の数値は、非誘導発現処理区でのsGFP遺伝子のmRNA量に対する、誘導発現処理区でのsGFP遺伝子のmRNA量の割合を示している。
【図4】図4は、銅イオン誘導性sGFP遺伝子発現ベクターが導入されてなる組換えタバコ培養細胞における、GFPの蓄積量(GFPの蛍光強度から算出したGFP換算量)の定量の結果を示す図である。グラフ内の数値は、非誘導発現処理区でのGFPの蓄積量に対する、誘導発現処理区でのGFPの蓄積量の割合を示している。
【図5】図5は、銅イオン誘導性FT遺伝子発現ベクター及び銅イオン誘導性rSt-1609soy遺伝子発現ベクターのT-DNA領域の構造を示す模式図である。図中の*印は、as-1サイトへの変異導入部位を示している。
【図6】図6は、銅イオン誘導性FT遺伝子発現ベクターが導入されてなる組換えシロイヌナズナにおける、開花時期の相違を示す図である。
【図7】図7は、銅イオン誘導性sGFP遺伝子発現ベクターのT-DNA領域の構造を示す模式図である。
【図8】図8は、銅イオン誘導性sGFP遺伝子発現ベクターが導入されてなる組換えシロイヌナズナにおける、GFPの蛍光発光状態を示す図である。夫々、誘導発現処理0日目を左に、誘導発現処理3日目を右に示している。
【発明を実施するための形態】
【0010】
以下に本発明を詳細に説明する。
本発明遺伝子構築物は、植物において、化学物質により目的とする外来遺伝子を誘導発現させるために利用することができる。当該遺伝子構築物は、
(a)プロモーター機能を有する領域と目的とする外来蛋白質に係る構造遺伝子領域とを含む、目的とする外来遺伝子と、
(b)前記の目的とする外来遺伝子とは異なる他の外来遺伝子によりコードされ且つ銅イオンにより活性化される転写因子(以下、本転写因子と記すことがある。)と、
(c)前記の目的とする外来遺伝子が含むプロモーター機能を有する領域(以下、本プロモーター領域と記すこともある。)の下流に位置する任意の外来遺伝子の5'−非翻訳領域(以下、本5'−非翻訳領域と記すことがある。)と
を有するように構築可能に調製すればよい。このようにして、本発明遺伝子構築物を得ることができる。
【0011】
好ましい本発明遺伝子構築物としては、例えば、本転写因子とは異なる他の転写因子の転写活性化領域であり、本転写因子の下流に位置する転写活性化領域(以下、本転写活性化領域と記すことがある。)を、さらに追加的に有するように構築されてなる遺伝子構築物を挙げることができる。
【0012】
本発明遺伝子構築物は、前記(a)、前記(b)及び前記(c)の各要素を有するように構築可能に調製すればよい。即ち、1つの発現カセットとして前記(a)、前記(b)及び前記(c)の各要素を有するように構築してもよいし、また2つの発現カセットのうち、1つの発現カセットとして前記(a)及び前記(c)の要素を有するように構築し、且つ、他の1つの発現カセットとして前記(b)の要素を有するように構築してもよい。
このような発現カセットは、通常の遺伝子工学的手法を用いて行えばよい。尚、2つの発現カセットとして構築する場合には、例えば、前記(a)及び(c)を第一発現カセットとして構築し、また前記(b)を第二発現カセットとして構築してもよい。
【0013】
本発明遺伝子構築物としては、好ましくは、任意のプロモーターを前記の転写因子の上流に追加的に有するように構築されてなる遺伝子構築物等を挙げることができる。
また更に、本発明遺伝子構築物としては、好ましくは、任意のターミネーターを前記の転写因子の下流に追加的に有するように構築されてなる遺伝子構築物等を挙げることができる。当該ターミネーターとしては、例えば、NOSターミネーター、CR16ターミネーター(特開2000-166577)、ダイズ種子グリシニンターミネーター(特開06-189777)等を挙げることができる。
【0014】
本発明遺伝子構築物を、例えば、植物体、培養細胞等の宿主植物へ導入し、当該遺伝子構築物の誘導発現を行う。本発明遺伝子構築物の宿主植物への導入は、宿主植物夫々に適用可能な方法に応じて、通常の遺伝子工学的手法を用いて行えばよい。
具体的には例えば、第一発現カセットと第二発現カセットとをベクター上で連結してなる遺伝子構築物に含まれる目的とする外来遺伝子を導入してもよいし、またこれらの発現カセットを連結せずに混合して導入してもよい。また、第一発現カセット又は第二発現カセットのいずれかを導入した宿主植物に、再度、他方の発現カセットを導入してもよい。
さらに、第一発現カセットを導入した個体と第二発現カセットを導入した個体とを交配してもよい。第一発現カセットを複数種類導入することで、複数種類の外来遺伝子を同時に誘導発現することも可能である。
本発明遺伝子構築物の導入方法としては、アグロバクテリウム法、パーティクルガン法、エレクトロポレーション法、リン酸カルシウム法、ウイルスベクター法等の各種の公知方法を用いることができる。
宿主植物には、農業上及び園芸上、重要な種又はゲノム解析等遺伝学上有用な種が挙げられるが、さらに、任意の植物種が含まれる。例えば、ダイズ、エンドウ、インゲン、アルファルファ、ミヤコグサ、クローバ、ピーナッツ、スイートピー、クルミ、チャ、ワタ、コショウ、キュウリ、スイカ、カボチャ、メロン、ダイコン、ナタネ、キャノーラ、テンサイ、レタス、キャベツ、ブロッコリー、カリフラワー、シロイヌナズナ、タバコ、ナス、ジャガイモ、サツマイモ、サトイモ、キクイモ、トマト、ホウレンソウ、アスパラガス、ニンジン、ゴマ、エンダイブ、キク、フウロウソウ、キンギョソウ、カーネーション、ナデシコ、ニチニチソウ、ブバルディア、カスミソウ、ガーベラ、トルコキキョウ、チューリップ、ストック、スターチス、シクラメン、ユキノシタ、ノースポール、スミレ、バラ、サクラ、リンゴ、イチゴ、ウメ、ミカン、ボケ、サツキ、ツツジ、ナンヨウアブラギリ、リンドウ、コスモス、アサガオ、ヒマワリ、イチョウ、スギ、ヒノキ、ポプラ、マツ、セコイア、オーク、スイレン、トチュウ、ブナ、イネ、コムギ、オオムギ、ライムギ、エンバク、トウモロコシ、ネギ、タマネギ、ニンニク、ユリ、オニユリ、ラン、グラジオラス、パイナップルである。
【0015】
本発明誘導発現方法では、例えば、銅イオンを本発明形質転換体植物に接触させたり、施用したりすること等が必要である。当該接触・施用方法等としては、本発明形質転換体植物が培養細胞である場合には、例えば、培地や栄養溶液への銅イオンの混合(具体的には例えば、銅イオン濃度1μM〜24mM)等の方法を挙げることができる。また、本発明形質転換体植物が植物体である場合には、土壌や植物体の茎葉への銅イオンの処理・散布(具体的には例えば、銅イオン量1nmol〜2.4mmol、銅イオン濃度1μM〜24mM)等の方法が挙げられる。宿主植物の種類や使用場面に応じて適宜選択することが望ましい。本発明誘導発現方法では、銅イオンを目的とする外来遺伝子を誘導発現させたい植物細胞の中に浸透させればよく、施用する銅イオンを錯体にする等製剤を工夫したり、ボルドー(住友化学)、ジーファイン(八洲化学)、トモノZボルドー(トモノアグリカ)、リドミルプラス(日本農薬)、ハイカッパー(住化武田農薬)、クプラビットホルテ(バイエルクロップサイエンス)、コサイドボルドー(デュポン)、キノンドー(アグロカネショウ)、ヨネポン(米澤化学)等、農業用途に使用される銅剤や、グルコン酸銅(和光純薬)等、食品添加物用途に使用される銅剤を所望の濃度に調整して用いてもよい。また施用に際して展着剤等を混合してもよい。
【0016】
使用される本転写因子は、銅イオンにより活性化され、目的とする外来遺伝子に含まれるプロモーター機能を有する領域(即ち、本プロモーター領域)の転写活性を誘導する。
当該転写因子は、銅イオンが存在しない状態においては不活性型でありプロモーター機能を有する領域(即ち、本プロモーター領域)の転写活性を誘導できないが、銅イオンが存在する状態においては活性型に変化しプロモーター機能を有する領域(即ち、本プロモーター領域)の転写活性を誘導することができる。因みに、転写因子は、通常、DNA結合領域と転写活性化領域とを有している。このとき、当該転写因子に本転写因子とは異なる他の転写因子の転写活性化領域を連結したり、元の転写因子の転写活性化領域と当該転写因子とは異なる他の転写因子の転写活性化領域を置換することもできる。尚、本転写活性化領域は宿主植物の細胞内において、RNAポリメラーゼ複合体のメディエーターをリクルートし、転写活性化能力を増強する働きを有する。
【0017】
本転写因子の好ましい塩基配列としては、前述のように、例えば、下記の転写因子に係る塩基配列群から選ばれてなる塩基配列を挙げることができる。
<転写因子に係る塩基配列群>
(1)真核生物由来の塩基配列
(2)酵母由来の塩基配列
(3)酵母ACE1由来の塩基配列
また例えば、candida glabrataのAMT1(Thorvaldsen JL et al、1993、J Biol Chem 268、12512-12518)由来の塩基配列、Yarrowia lipolyticaのCRF1(Garcia S、2002、J Biol Chem 277、37359-37368)由来の塩基配列、トウモロコシのSOD遺伝子群を銅イオン依存的に誘導発現させる転写因子(Ruzsa SM et al、2003、Biochemistry 42、1508-1516)由来の塩基配列等を挙げることができる。尚、由来の塩基配列には、当該塩基配列の一部使用や他の塩基配列とのキメラ、欠失及び挿入、置換等の変異を導入した塩基配列も含まれる。
【0018】
使用される本プロモーター領域は、前記の本転写因子が銅イオン存在下において転写活性を誘導しうる塩基配列からなる。誘導条件下の宿主細胞中で、目的とする外来遺伝子が発現可能であればよい。好ましくは、例えば、酵母のMRE配列(Mett VL et al、1993、Proc Natl Acad Sci 90、4567-4571)等を含む配列を挙げることができる。具体的には、通常使用されるプロモーター機能を有する領域に存在するTATA配列の上流に、酵母のMRE配列等を繰返し配置してなるものが挙げられる。
尚、前記の通常使用されるプロモーター機能を有する領域は、構成的プロモーターでもよいし、組織特異的プロモーターや、ある刺激により転写活性が誘導される誘導性プロモーターでもよい。使用場面に応じて適宜選択することが望ましい。
構成的プロモーターとしては、例えば、CaMV 35Sプロモーター、PG10-90(特開09-131187)、ユビキチンプロモーター(国際公開01/094394)、アクチンプロモーター(国際公開00/070067)等を挙げることができる。また、組織特異的プロモーターとしては、例えば、ダイズ種子グリシニンプロモーター(特開06-189777)、プロラミンプロモーター(国際公開2004/056993)、インゲンマメ種子ファゼオリンプロモーター(国際公開91/013993)、ナタネ種子ナピンプロモーター(国際公開91/013972)、シロイヌナズナSultr2;2プロモーター(Takahashi H et al、2000、Plant J 23、171-82)、アグロバクテリウムrolCプロモーター(Almon E et al、1997、Physiol 115、1599-1607)等を挙げることができる。
【0019】
使用される本5'−非翻訳領域は、本プロモーターの下流に位置し、転写されるが翻訳はされない塩基配列からなる領域である。但し、目的とする外来遺伝子が、アンチセンスRNAをコードする場合やRNAiを誘発するRNAをコードする場合等は、転写開始点から、当該RNAをコードする領域の直前までを5'−非翻訳領域とする。当該5'−非翻訳領域配列は、ウイルスの遺伝子由来であっても植物の誘導性遺伝子由来であってもよい。ウイルスの遺伝子由来の5'−非翻訳領域配列としては、例えば、タバコモザイクウイルス(TMV)のΩ配列(例えば、Gallie D et al、1987、Nucleic Acid Res 15、3257-3273; 米国特許5489527参照)やタバコエッチウイルス(TEV)のL配列(例えば、Lindbo JA、2007、BMC Biotechnol 7、52参照)、イネ縞葉枯ウイルス(RSV)のΦ配列(例えば、Mori M et al、2006、Plant Biotechnology 23(1)、55-61参照)等を挙げることができる。植物の誘導性遺伝子とは、ある刺激によってその発現量が増大する遺伝子を意味し、例えば、タバコのアルコールデヒドロゲナーゼ遺伝子(Satoh J et al、2004、J Biosci Bioeng 98(1):1-8; 特開2003-79372)やPR1a遺伝子(Ohshima M et al、1990、Plant Cell 2(2)、95-106)、トウモロコシのIn2-1遺伝子又はIn2-2遺伝子(国際公開90/11361; De Veylder L et al、1997、Plant Cell Physiol 38(5)、568-577)やGST27遺伝子(Jepson I et al、1994、Plant Mol Biol 26(6)、1855-1866; 国際公開97/11189)、シロイヌナズナのRD29遺伝子(Yamaguchi-Shinozaki K、1993、Mol Gen Genet 236(2-3)、331-340)、シロイヌナズナ又はダイズ、ヒマワリのヒートショック蛋白質遺伝子(Yoshida K et al、1995、Appl Microbiol Biotechnol 44(3-4):466-472; Nagao RT et al、1985、Mol Cell Biol 5(12)、3417-3428; Almoguera C et al、2002、J Biol Chem、277(46):43866-43872)等を挙げることができる。
【0020】
本5'−非翻訳領域の好ましい塩基配列としては、例えば、下記の5'−非翻訳領域に係る塩基配列群から選ばれてなる塩基配列を挙げることができる。
<5'−非翻訳領域に係る塩基配列群>
(1)ウイルスの遺伝子又は植物の誘導性遺伝子由来の塩基配列
(2)トバモウイルス属に属するウイルスの遺伝子由来の塩基配列(例えば、Gallie D et al、1987、Nucleic Acid Res 15、3257-3273; 米国特許5489527参照)
(3)トマトモザイクウイルスの遺伝子由来の塩基配列
(4)トマトモザイクウイルスの130k/180k遺伝子由来の塩基配列
尚、由来の塩基配列には、当該塩基配列の一部使用や他の塩基配列とのキメラ、欠失及び挿入、置換等の変異を導入した塩基配列も含まれる。また、5'−非翻訳領域の塩基配列数が48 bp以上の塩基配列、又は、二次構造予測プログラム「mfold (version 2.3)」(http://frontend.bioinfo.rpi.edu/applications/mfold/cgi-bin/rna-form1-2.3.cgi、The Bioinformatics Center at Rensselaer and Wadsworth提供; 例えば、Zuker M、2003、Nucleic Acids Res 31(13)、3406-3415参照)を用いて25℃の条件下で二次構造を予測した場合に、分子内で相互作用が予測されない連続した最長の塩基数が11 bp以上である塩基配列を挙げることもできる。
【0021】
本転写活性化領域の好ましい塩基配列としては、前述のように、例えば、下記の転写活性化領域に係る塩基配列群から選ばれてなる塩基配列を挙げることができる。
<転写活性化領域に係る塩基配列群>
(1)ウイルスの転写因子由来の塩基配列
(2)シンプレックスウイルス属に属するウイルスの転写因子由来の塩基配列
(3)単純ヘルペスウイルスの転写因子由来の塩基配列
(4)単純ヘルペスウイルスのVP16転写因子由来の塩基配列(例えば、Triezenberg SJ et al、1988、Genes Dev 2(6)、718-29参照)
また例えば、GAL4転写因子(Gill G、Ptashne M、1987、Cell 51(1)、121-126)由来の塩基配列、転写活性化能力を有する人工的に合成されたペプチドAH(Ansari AZ et al、2001、Chem Biol 8(6)、583-592)由来の塩基配列等を挙げることもできる。尚、由来の塩基配列には、当該塩基配列の一部使用や他の塩基配列とのキメラ、欠失及び挿入、置換等の変異を導入した塩基配列も含まれる。
【0022】
本発明誘導発現方法で使用される転写因子は、前述のような本転写因子であり、目的とする外来遺伝子とは異なる他の外来遺伝子によりコードされる転写因子である。そして当該転写因子は、銅イオンにより活性化するものである。また、本発明誘導発現方法で使用される遺伝子構築物は、例えば、前述のような本発明遺伝子構築物である。また、本発明誘導発現方法で使用される外来遺伝子の5'−非翻訳領域は、前述のような本5'−非翻訳領域であり、任意の外来遺伝子の5'−非翻訳領域である。また、本発明誘導発現方法で使用されるプロモーター機能を有する領域は、前述のような本プロモーター領域であり、目的とする外来遺伝子が含む、プロモーター機能を有する領域である。
【0023】
本発明形質転換体植物は、上述のように、本発明遺伝子構築物が導入されてなる形質転換体植物である。本発明外来蛋白質取得方法では、当該形質転換体植物から目的とする外来蛋白質を通常の蛋白質工学的な手法に準じて回収すればよい。
【0024】
本発明の応用例として、以下のようなことを挙げることができるが、特にこれらに限定されるものではない。
【0025】
FT遺伝子を過剰発現させたシロイヌナズナは、花成時期が早まる(例えば、特開2000-139250参照)。本発明誘導発現方法における目的とする外来遺伝子がFT遺伝子である
場合には、所望する時期に花成を誘導することができる。花成促進遺伝子への変異導入やRNAi、花成遅延遺伝子の過剰発現等により花成が起こり難くした植物に本FT遺伝子を誘導発現させることにより、より厳密に花成を制御することが可能となる。
因みに、FT遺伝子とは、Flowering Locus T 遺伝子の略称であり、花成制御に関して正に働く因子をコードする遺伝子を意味する。FT遺伝子は、日長に応答して維管束師部での発現が増大し、茎頂に移動して、茎頂で発現しているFDと呼ばれるbZIP型転写因子と相互作用することにより花成を誘導する作用を有する(例えば、Kobayashi Y and Weigel D、2007、Genes Dev 21、2371-2384参照)。
【0026】
コラーゲン遺伝子を導入したタバコは、コラーゲンを生産する(例えば、米国特許6617431参照)。本発明誘導発現方法における目的とする外来遺伝子がコラーゲン遺伝子である場合には、高いレベルでの生産が可能となる。
【0027】
PPO遺伝子を導入したトウモロコシは、除草剤耐性となる(例えば、米国特許6307129参照)。本発明誘導発現方法における目的とする外来遺伝子がPPO遺伝子である場合には、所望の時期にのみ除草剤に耐性を示すようになる。栽培の必要がなくなった時には、同じ除草剤を用いて枯死させることが可能となる。
【0028】
EPSPS遺伝子を導入したダイズは、除草剤耐性となる(例えば、国際公開92/00377参照)。本発明誘導発現方法における目的とする外来遺伝子がEPSPS遺伝子である場合には、所望の時期にのみ除草剤に耐性を示すようになる。栽培の必要がなくなった時には、同じ除草剤を用いて枯死させることが可能となる。
【0029】
Δ15脂肪酸不飽和化酵素遺伝子を導入したダイズは、高度不飽和脂肪酸含量が上昇する(例えば、国際公開2005/047479参照)。本発明誘導発現方法における目的とする外来遺伝子がΔ15脂肪酸不飽和化酵素遺伝子である場合には、所望の時期に油脂中の脂肪酸組成を制御することが可能となる。
【0030】
ポリプレニル2リン酸合成酵素遺伝子を導入したイネは、CoQ10含量が上昇する(例えば、特開2006-212019参照)。本発明誘導発現方法における目的とする外来遺伝子がポリプレニル2リン酸合成酵素遺伝子である場合には、所望の時期にCoQ10含量を上昇させることが可能となる。
【0031】
テオブロミン合成酵素遺伝子の発現を抑制したコーヒーは、カフェイン含量が減少する(例えば、特開2002-112785; Ogita S et al、2004、Plant Mol Biol 54(6)、931-941参照)。本発明誘導発現方法における目的とする外来遺伝子がテオブロミン合成酵素遺伝子のアンチセンス遺伝子等である場合には、所望の時期に種子中のカフェイン含量を制御することが可能となる。
【0032】
リボヌクレアーゼ遺伝子を葯にて発現させたアブラナは、雄性不稔となる(例えば、Mariani C et al、1990、Nature 347、737-741参照)。本発明誘導発現方法における目的とする外来遺伝子がリボヌクレアーゼ遺伝子である場合には、所望の個体のみを雄性不稔にすることが可能となる。一方、S糖蛋白質遺伝子の発現を抑制すると、自家不和合性が打破される(例えば、特開08-322412参照)。本発明誘導発現方法における目的とする外来遺伝子がS糖蛋白質遺伝子のアンチセンス遺伝子等である場合には、所望の個体のみを自家和合性に変換することが可能となる。これにより、効率的な雑種種子採取ができる。
【0033】
またステロイドホルモン誘導性システムについては、以下のような報告(例えば、Zuo J et al、2006、Methods Mol Biol 323、329-342参照)がある。
(1)CRE DNAリコンビナーゼ遺伝子を誘導発現させることにより、2つのloxPサイトに挟まれた薬剤耐性遺伝子発現カセット等を切り出す。
(2)RNAiを誘発するRNAを誘導発現させることにより、所望の遺伝子の発現を抑制する。
(3)イントロンのドナーサイトとアクセプターサイトを含むmRNAを誘導発現させることにより、所望のcDNAを単離したり、無作為にcDNAを単離する。
(4)第一発現カセットの5’−非翻訳領域下流に遺伝子やターミネーターを配置せず、偶発的に5’−非翻訳領域下流に組み込まれた染色体上の遺伝子を誘導発現させることにより、機能的な遺伝子を単離する。
以上、同様なコンストラクトを作製することにより、これらの技術の全てに対して、本発明誘導発現方法を適用させることも可能である。
【0034】
外被蛋白質をコードする領域を目的とする外来蛋白質に係る構造遺伝子領域に置換したブロモモザイクウイルスcDNAやトマトモザイクウイルスcDNAをステロイドホルモンにより誘導発現させ、外来蛋白質を高いレベルで生産したという報告がある(例えば、特開2005-102652; Mori M et al、2001、Plant Journal 27(1)、79-86; Dohi K et al、2006、Arc
h Virol 151、1075-1084参照)。本発明誘導発現方法における目的とする外来遺伝子が、外被蛋白質等の構造遺伝子領域を置換したウイルスcDNAの場合においても、置換した構造遺伝子を高いレベルで転写誘導することが可能となる。
【0035】
本発明は上述した各実施形態に限定されるものではなく、請求項に示した範囲で種々の変更が可能であり、異なる実施形態に夫々開示された技術的手段を適宜組み合わせて得られる実施形態についても本発明の技術的範囲に含まれる。
【実施例】
【0036】
以下に実施例により本発明を詳細に説明するが、本発明はこれらに限定されるものではない。
【0037】
実施例1 (導入ベクターの作製)
(1)転写因子遺伝子発現カセットの構築
YPD培地(1% 酵母エキス、2% ポリペプトン、2% グルコース)中、30℃で2日間振とう培養した出芽酵母(Saccharomyces cerevisiae strain AH22)から、ゲノムDNA抽出キット「Genとるくん」(タカラバイオ)を用いてゲノムDNAを抽出した。抽出されたゲノムDNAを鋳型に、2種の特異的プライマー(ACE1-1F、ACE1-1RC)を用いてPCRを行うことにより、ACE1転写因子遺伝子を増幅した。増幅されたACE1転写因子遺伝子を、pBI221(Clontech)のGUS遺伝子と置換し、p35S-ACE1-NOSを作製した。次に、p35S-ACE1-NOSに含まれるNOSターミネーターを、CR16ターミネーター(特開2000-166577)に置換し、p35S-ACE1-CRを作製した。
【0038】
NASC(Nottingham Arabidopsis Stock Centre)より購入した組換えシロイヌナズナ種子(No.N70016)を改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種した。23℃で3週間生育させた組換えシロイヌナズナの本葉から、植物ゲノムDNA抽出キット「DNeasy Plant mini kit」(QIAGEN)を用いてゲノムDNAを抽出した。
抽出されたゲノムDNAを鋳型に、2種の特異的プライマー(VP16-1F、VP16-1RC)を用いてPCRを行うことにより、単純ヘルペスウイルスのVP16転写因子の転写活性化ドメイン(VP16AD)遺伝子を増幅した。増幅されたVP16AD遺伝子を、pCR2.1(Invitrogen)にTAクローニングし、pCR2.1-VP16ADを作製した。pCR2.1-VP16ADを鋳型に、2種の特異的プライマー(VP16-2F、VP16-2RC)を用いてPCRを行い、VP16AD遺伝子内のSacIサイトに変異を導入し、pCR2.1-VP16AD(dSacI)を作製した。その後、pCR2.1-VP16AD(dSacI)を鋳型に、2種の特異的プライマー(VP16-3F、VP16-3RC)を用いてPCRを行うことにより、VP16AD(dSacI)遺伝子の5’末端にXhoIサイトを付加し3’末端にSacIサイトを付加した。
【0039】
p35S-ACE1-CRを鋳型に、2種の特異的プライマー(ACE1-1F、ACE1-2RC)を用いてPCRを行うことにより、ACE1転写因子遺伝子の終始コドンを除去しXhoIサイトを付加した。
3’末端にXhoIサイトを付加したACE1転写因子遺伝子の下流に、VP16AD(dSacI)遺伝子を読み枠が合うように連結し、p35S-ACE1/VP16AD-CRを作製した。
【0040】
ACE1-1F:5'-atggatccatggtcgtaattaacggg-3'(配列番号1)
ACE1-1RC:5'-tggagctcttattgtgaatgtgagttatg-3'(配列番号2)
ACE1-2RC:5'-aactcgagttgtgaatgtgagttatgcg-3’(配列番号3)
VP16-1F:5'-acggctccaccgaccgacgtc-3'(配列番号4)
VP16-1RC:5'-ctacccaccgtactcgtcaattc-3'(配列番号5)
VP16-2F:5'-ggacgaactccacttagacgg-3'(配列番号6)
VP16-2RC:5'-ccgtctaagtggagttcgtcc-3'(配列番号7)
VP16-3F:5'-tactcgagtcaacggctccaccgaccgacgt-3'(配列番号8)
VP16-3RC:5'-aagagctcttacccaccgtactcgtcaattccaag-3'(配列番号9)
【0041】
(2)sGFP遺伝子発現カセットの構築
プラスミドCaMV35S-sGFP(S65T)-NOS3'(「植物の細胞を観る実験プロトコール」 福田裕穂他監修、1997年、秀潤社、ISBN 4-87962-170-6)からsGFP遺伝子を切り出し、アダプター(NS-1F、NS-1RC)を使用して、pBI221のGUS遺伝子と置換、p35S-sGFPを作製した。p35S-sGFPに含まれるCaMV35Sプロモーターの-830bpから-91bpまでの領域を、合成オリゴヌクレオチド(MRE-1F、MRE-1RC)に置換し、pMRE/35S(-90)-sGFPを作製した。
pMRE/35S(-90)-sGFPを制限酵素EcoRVで処理した後、「Calf intestine Alkaline Phosphatase」(タカラバイオ)で脱リン酸化した。そこへ、「Blunting Kination Ligation kit」(タカラバイオ)を用いて平滑末端化及びリン酸化した合成オリゴヌクレオチド(MRE-1F、MRE-1RC)を挿入することで、MRE配列を順向きに2回繰返して配置させた。得られたMRE配列の2回繰返し部分を切り出し、上記と同様の方法で平滑末端化し、再度EcoRVサイトに挿入した。これにより、MRE配列を順向きに4回繰返して配置させたpMRE4/35S(-90)-sGFPを作製した。
さらに、pBI221を鋳型に、2種の特異的プライマー(46bp-1F、46bp-1RC)を用いてPCRを行うことにより、CaMV35Sプロモーターの-46bpから-1bpまでの領域を含むDNA断片を増幅した。増幅されたDNA断片を、pMRE4/35S(-90)-sGFPの4回繰返し配置させたMRE配列の下流に位置するCaMV35Sプロモーターの-90bpから-1bpまでの領域と置換し、pMRE4/35S(-46)-sGFPを作製した。
他方では、CaMV35Sプロモーターの-90bpから-1bpまでの領域に存在するas-1サイト(非特許文献1)に変異が導入された配列とも置換した。
まず、NASCより購入した組換えシロイヌナズナ(No.N70016)からゲノムDNAを抽出した。抽出されたゲノムDNAを鋳型に、2種の特異的プライマー(90m-1F、90m-1RC)を用いてPCRを行うことにより、as-1サイトに変異を持つ35S(-90m)配列をpCR2.1(Invitrogen)にTAクローニングし、pCR2.1-35S(-90m)を作製した。pCR2.1-35S(-90m)を鋳型に、2種の特異的プライマー(90m-2F、90m-2RC)を用いてPCRを行うことにより、5’末端にEcoRVサイトを付加し3’末端にXbaIサイトを付加した。得られたDNA断片を、pMRE4/35S(-90)-sGFPの4回繰返し配置させたMRE配列の下流に位置するCaMV35Sプロモーターの-90bpから-1bpまでの領域と置換し、pMRE4/35S(-90m)-sGFPを作製した。
加えて、プラスミドpiL.erG3(Tamai A et al、2001、Mol Plant Microbe Interact 14(2)、126-134)を鋳型に、2種の特異的プライマー(71bp-1F、71bp-1RC)を用いてPCRを行うことにより、トマトモザイクウイルスの130k/180k遺伝子の5’−非翻訳領域配列(To71配列)を増幅した。増幅された5’−非翻訳領域配列をpMRE4/35S(-46)-sGFP及びpMRE4/35S(-90m)-sGFPのsGFP遺伝子の上流に挿入し、pMRE4/35S(-46)-To71sGFP及びpMRE4/35S(-90m)-To71sGFPを作製した。
【0042】
NS-1F:5'-ggccgcgagctcagt-3'(配列番号10)
NS-1RC:5'-gactgagctcgc-3'(配列番号11)
MRE-1F:5'-agcttagcgatgcgtcttttccgctgaaccgttccagcaaaaaagactagat-3'(配列番号12)
MRE-1RC:5'-atctagtcttttttgctggaacggttcagcggaaaagacgcatcgcta-3'(配列番号13)46bp-1F:5'-tagatatcgcaagacccttcctctatataagg-3'(配列番号14)
46bp-1RC:5'-atcctctagagtcccccgtgttc-3'(配列番号15)
90m-1F:5'-gctatgaccatgattacgccaagcttg-3'(配列番号16)
90m-1RC:5'-cattgttatatctccttggatccgtcg-3'(配列番号17)
90m-2F:5'-tagatatctccacgtccataagggac-3'(配列番号18)
90m-2RC:5'-aatctagactgcaggtcgtcctctcca-3'(配列番号19)
71bp-1F:5'-tgtctagagtatttttacaacaattaccaacaac-3'(配列番号20)
71bp-1RC:5'-aaggatcctgtagttgtagaatgtaaaatgtaatg-3'(配列番号21)
【0043】
(3)転写因子遺伝子発現カセットとsGFP遺伝子発現カセットとの連結
転写因子遺伝子発現カセットを含むp35S-ACE1-CR及びp35S-ACE1/VP16AD-CRを制限酵素HindIII及びEcoRIで処理し、夫々転写因子遺伝子発現カセットを切り出した。
sGFP遺伝子発現カセットを含むpMRE/35S(-90)-sGFP及びpMRE4/35S(-46)-sGFP、pMRE4/35S(-46)-To71sGFP、pMRE4/35S(-90m)-To71sGFPを制限酵素HindIIIで処理した後、「Blunting Kination Ligation kit」を用いて平滑末端化及びリン酸化し、そこへ合成オリゴヌクレオチド(KXS-1F、KXS-1RC)を挿入、pKXS-MRE/35S(-90)-sGFP及びpKXS-MRE4/35S(-46)-sGFP、pKXS-MRE4/35S(-46)-To71sGFP、pKXS-MRE4/35S(-90m)-To71sGFPを作製した。作製したこれらのプラスミドを制限酵素KpnI及びEcoRIで処理し、夫々sGFP遺伝子発現カセットを切り出した。
一方、pBI121(Clontech)に含まれるGUS遺伝子発現カセットを、合成オリゴヌクレオチド(HEK-1F、HEK-1RC)に置換し、pBI121-HEKを作製した。pBI121-HEKを制限酵素HindIII及びKpnIで処理した。
制限酵素HindIII及びKpnIで処理したpBI121-HEKに、切り出した転写因子遺伝子発現カセットとsGFP遺伝子発現カセットをライゲーションすることにより、転写因子遺伝子発現カセットとsGFP遺伝子発現カセットとが、夫々のターミネーター側で連結したベクターを得た(図1参照)。p35S-ACE1-CRとpKXS-MRE/35S(-90)-sGFPを由来とするものをベクターA、p35S-ACE1-CRとpKXS-MRE4/35S(-46)-sGFPを由来とするものをベクターB、p35S-ACE1/VP16AD-CRとpKXS-MRE4/35S(-46)-sGFPを由来とするものをベクターC、p35S-ACE1/VP16AD-CRとpKXS-MRE4/35S(-46)-To71sGFPを由来とするものをベクターD、p35S-ACE1/VP16AD-CRとpKXS-MRE4/35S(-90m)-To71sGFPを由来とするものをベクターEとした。
【0044】
KXS-1F:5'-ggtacctcgagtcgac-3'(配列番号22)
KXS-1RC:5'-gtcgactcgaggtacc-3'(配列番号23)
HEK-1F:5'-agcttgaattcgtcgacggtacctaggacgagctc-3'(配列番号24)
HEK-1RC:5'-aattgagctcgtcctaggtaccgtcgacgaattca-3'(配列番号25)
【0045】
実施例2 (組換えシロイヌナズナにおけるsGFP遺伝子の発現量解析)
(1)組換えシロイヌナズナの作製及び選抜
実施例1で作製されたベクターAからEをそれぞれアグロバクテリウム(Agrobacterium tumefaciens strain C58C1)に導入した。アグロバクテリウムを50 mg/L カナマイシン、100 mg/L アンピシリン、100 mg/L リファンピシンを含むLB寒天培地(0.5% 酵母エキス、1.0% バクトトリプトン、0.5% 食塩、1% 寒天)で培養して薬剤耐性コロニーを選抜することにより、組換えアグロバクテリウムを得た。得られた組換えアグロバクテリウムを、モデル植物ラボマニュアル(岩渕雅樹他編集、2000年、シュプリンガー・フェアラーク東京株式会社、ISBN 4-431-70881-2 C3045)に記載される方法に準じて、シロイヌナズナ(Arabidopsis thaliana ecotype Columbia)に感染させることにより、遺伝子導入を行った。遺伝子導入されたシロイヌナズナから採取されたT1種子を、20 mg/L ベンレート、200 mg/L クラフォラン、25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、1% ショ糖、0.8 % 寒天)に播種し生育した後、カナマイシンに耐性を示す植物個体を選抜した。選抜された植物個体を予め培土が入れられたポットに移植し、人工気象器内で生育させることにより、T2種子を得た。得られたT2種子を25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種し生育した後、χ2検定に基づいた5%の有意水準において、カナマイシンに耐性を示す植物個体が3:1の割合で出現するラインを選抜した。尚、植物個体の生育のための培養条件は、明期23時間、暗期1時間、23〜25℃とした。
【0046】
(2)リアルタイムPCRによるsGFP遺伝子の発現量解析
選抜されたラインについて、播種後10日目の植物個体を、誘導発現処理区として100μM CuSO4を含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8 % 寒天)に6個体ずつ移植することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った(尚、非誘導発現処理区として CuSO4を含まない改変MS寒天培地を用いた。)。
その後、当該誘導発現処理6時間後の植物個体から、植物RNA抽出キット「RNeasy Plant Mini Kit」(QIAGEN)を用いて全RNAを抽出した。抽出された全RNAから、cDNA合成キット「ReverTra Ace」(TOYOBO)を用いてcDNAを合成した。合成されたcDNAを鋳型に、7500 Fast Real-time PCR装置(Applied Biosystems)を用いてリアルタイムPCRを行うことにより、mRNA量の定量を行った。sGFP遺伝子のmRNA量の定量には、2種の特異的プライマー(S01F、S01R)及びTaqManプローブ(S01)を用いた。内部標準としては、シロイヌナズナアクチン遺伝子(AtACT2、GenBank Accession Number NM180280)を用いた。AtACT遺伝子のmRNA量の定量には、2種の特異的プライマー(S03F、S03R)及びTaqManプローブ(S03)を用いた。
【0047】
S01F:5'-tccgccctgagcaaagac-3'(配列番号26)
S01R:5'-gaactccagcaggaccatgtg-3'(配列番号27)
S01:5'-FAM-ccaacgagaagcgcga-MGB-3'(配列番号28)
S03F:5'-cggtggttccattcttgctt-3'(配列番号29)
S03R:5'-cggccttggagatccacat-3'(配列番号30)
S03:5'-VIC-cctcagcacattcc-MGB-3'(配列番号31)
【0048】
その結果、ベクターAが導入されてなる組換えシロイヌナズナでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して4.6倍(以下、ベクターAが導入されてなる組換えシロイヌナズナにおける誘導倍率と記すこともある。)であった。MRE配列が4回繰返し配置され且つCaMV35Sプロモーターが-90bpから-46bpまでの領域に短縮されたベクターBが導入されてなる組換えシロイヌナズナでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して8.6倍(以下、ベクターBが導入されてなる組換えシロイヌナズナにおける誘導倍率と記すこともある。)であり、またベクターAが導入されてなる組換えシロイヌナズナにおける誘導倍率と比較して1.9倍の改善が認められた。
一方、ACE1転写因子にVP16ADが付加されたベクターCが導入されてなる組換えシロイヌナズナでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して273.0倍(以下、ベクターCが導入されてなる組換えシロイヌナズナにおける誘導倍率と記すこともある。)であり、またベクターBが導入されてなる組換えシロイヌナズナにおける誘導倍率と比較して31.7倍の改善が認められた。
さらに、トマトモザイクウイルスの130k/180k遺伝子の5’−非翻訳領域配列が挿入されたベクターDが導入されてなる組換えシロイヌナズナでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して628.6倍(以下、ベクターDが導入されてなる組換えシロイヌナズナにおける誘導倍率と記すこともある。)であり、またベクターCが導入されてなる組換えシロイヌナズナにおける誘導倍率と比較して2.3倍の改善が認められた。
また、as-1サイトに変異を持つ35S(-90m)配列が用いられたベクターEが導入されてなる組換えシロイヌナズナでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して325.6倍(以下、ベクターEが導入されてなる組換えシロイヌナズナにおける誘導倍率と記すこともある。)であった(図2参照)。
【0049】
実施例3 (組換えタバコにおけるsGFP遺伝子の発現量解析)
(1)組換えタバコの作製及び選抜
実施例1で作製されたベクターB及びEをそれぞれアグロバクテリウム(Agrobacterium tumefaciens strain LBA4404)に導入した。アグロバクテリウムを50 mg/L カナマイシン、300 mg/L ストレプトマイシン、100 mg/L リファンピシンを含むLB寒天培地(0.5% 酵母エキス、1.0% バクトトリプトン、0.5% 食塩、1% 寒天)で培養して薬剤耐性コロニーを選抜することにより、組換えアグロバクテリウムを得た。得られた組換えアグロバクテリウムを、植物遺伝子操作マニュアル(内宮博文著、1992年、講談社サイエンティフィック)に記載される方法に準じて、タバコ(Nicotiana tabacum strain SR1)に感染させることにより、遺伝子導入を行った。遺伝子導入されたタバコの葉片から100 mg/L カナマイシンに耐性を示す不定芽を選抜し、選抜された不定芽から植物個体を再生させた。得られた植物個体から採取されたT1種子を50 mg/L カナマイシンを含むMS寒天培地(MS無機塩類、MSビタミン、3% ショ糖、0.8% 寒天)に播種し生育した後、χ2検定に基づいた5%の有意水準において、カナマイシンに耐性を示す植物個体が3:1の割合で出現するラインを選抜した。尚、植物個体の生育のための培養条件は、明期23時間、暗期1時間、23〜25℃とした。
【0050】
(2)リアルタイムPCRによるsGFP遺伝子の発現量解析
選抜されたラインについて、播種後12日目の植物個体を、誘導発現処理区として100μM CuSO4を含むMS寒天培地(MS無機塩類、MSビタミン、3% ショ糖、0.8% 寒天)に3個体ずつ移植することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った(尚、非誘導発現処理区として CuSO4を含まないMS寒天培地を用いた。)。
その後、当該誘導発現処理6時間後の植物個体から、植物RNA抽出キット「RNeasy Plant Mini Kit」(QIAGEN)を用いて全RNAを抽出した。抽出された全RNAから、cDNA合成キット「ReverTra Ace」(TOYOBO)を用いてcDNAを合成した。合成されたcDNAを鋳型に、7500 Fast Real-time PCR装置(Applied Biosystems)を用いてリアルタイムPCRを行うことにより、mRNA量の定量を行った。sGFP遺伝子のmRNA量の定量には、2種の特異的プライマー(S01F、S01R)及びTaqManプローブ(S01)を用いた。内部標準としては、タバコユビキチン遺伝子(NtUBI、GenBank Accession Number U66264)を用いた。NtUBI遺伝子のmRNA量の定量には、2種の特異的プライマー(S06F、S06R)及びTaqManプローブ(S06)を用いた。
【0051】
S06F:5'-gaagcagctcgaggatggaa-3'(配列番号32)
S06R:5'-gacgggttgactctttctggat-3'(配列番号33)
S06:5'-VIC-accttggctgactacaa-MGB-3'(配列番号34)
【0052】
その結果、ベクターBが導入されてなる組換えタバコでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して9.6倍(以下、ベクターBが導入されてなる組換えタバコにおける誘導倍率と記すこともある。)であった。
一方、ACE1転写因子にVP16ADが付加され且つsGFP遺伝子の上流にトマトモザイクウイルスの130k/180k遺伝子の5’−非翻訳領域配列が挿入されたベクターEが導入されてなる組換えタバコでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して40.8倍(以下、ベクターEが導入されてなる組換えタバコにおける誘導倍率と記すこともある。)であり、またベクターBが導入されてなる組換えタバコにおける誘導倍率と比較して4.2倍の改善が認められた(図3参照)。
【0053】
実施例4 (組換えタバコ培養細胞におけるGFPの蓄積量解析)
(1)組換えタバコ培養細胞の作製及び選抜
実施例1で作製されたベクターA及びB、Eがコーティングされた直径1.0μmの金粒子を用いたパーティクルガン法(森川弘道ら著、1992年、植物細胞工学 Vol.4 No.1 p.47-52、秀潤社)により、前記ベクターをそれぞれタバコ培養細胞(BY-2)に導入した。金粒子1mg当たりのDNA量は0.1μgとした。遺伝子導入されたタバコ培養細胞の細胞懸濁液を、遺伝子導入操作後3〜5日目に、30 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、3% ショ糖、1μM 2,4-D、1 mg/L thiamin・HCl、100 mg/L myo-inositol、200 mg/L KH2PO4、0.8 % 寒天)に広げた。培養1ヵ月間後に、30 mg/L カナマイシンに耐性を示す細胞塊を選抜し、選抜された細胞塊をその後1〜2週間毎に新しい寒天培地に植え継ぎながら4〜8週間培養した。培養条件は、暗下、23〜25℃とした。
【0054】
(2)蛍光プレートリーダーによるGFPの蓄積量解析
得られた細胞塊を、誘導発現処理区として100μM CuSO4を含む改変MS寒天培地(MS無機塩類、3% ショ糖、1μM 2,4-D、1 mg/L thiamin・HCl、100 mg/L myo-inositol、200 mg/L KH2PO4、0.8 % 寒天)に移植することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った(尚、非誘導発現処理区として CuSO4を含まない改変MS寒天培地を用いた。)。
その後、当該誘導発現処理3日間後の細胞塊について、蛍光顕微鏡(Nikon)にて、GFPの蛍光発光状態を調べた。
【0055】
GFPの蛍光発光の増大が観察された細胞塊を液体窒素で凍結した後、当該凍結物にガラスビーズ(0.25〜0.5 mm)及び抽出バッファー(1×PBS(-)、5 mM DTT、1 mM PMSF、0.1 % protease inhibitor cocktail)を加えて、破砕装置「ミキサーミル」(QIAGEN)にて破砕処理した。得られた破砕物を卓上遠心機にて15000 rpm、4℃で5分間遠心することにより上清を回収した後、得られた上清を蛋白質抽出液とした。次いで、当該蛋白質抽出液を適宜、1×PBS(-) で希釈し、得られた希釈液におけるGFPの蛍光強度を、マルチラベルカウンタ「wallac 1420 ARVOMX」(パーキンエルマー)を用いて測定した。尚、標準サンプルとしては、リコンビナントGFP(コスモバイオ)を1×PBS(-)で希釈して得られる希釈液を用いた。測定された蛍光強度から可溶性蛋白質量当たりのGFP換算量を算出し、対照となる野生型の細胞塊での対応値をバックグランドとして差し引くことにより、GFPの蓄積量を算出した。尚、蛋白質抽出液の中の蛋白質濃度はBradford法により定量した。
【0056】
その結果、ベクターAが導入されてなる組換えタバコ培養細胞では、誘導発現処理区でのGFPの蓄積量が非誘導発現処理区でのGFPの蓄積量と比較して1.6倍(以下、ベクターAが導入されてなる組換えタバコ培養細胞における誘導倍率と記すこともある。)であった。MRE配列が4回繰返し配置され且つCaMV35Sプロモーターが-90bpから-46bpまでの領域に短縮されたベクターBが導入されてなる組換えタバコ培養細胞では、誘導発現処理区でのGFPの蓄積量が非誘導発現処理区でのGFPの蓄積量と比較して1.9倍(以下、ベクターBが導入されてなる組換えタバコ培養細胞における誘導倍率と記すこともある。)であった。
一方、ACE1転写因子にVP16ADが付加され且つsGFP遺伝子の上流にトマトモザイクウイルスの130k/180k遺伝子の5’−非翻訳領域配列が挿入されたベクターEが導入されてなる組換えタバコ培養細胞では、誘導発現処理区でのGFPの蓄積量が非誘導発現処理区でのGFPの蓄積量と比較して21.9倍(以下、ベクターEが導入されてなる組換えタバコ培養細胞における誘導倍率と記すこともある。)であり、またベクターBが導入されてなる組換えタバコ培養細胞における誘導倍率と比較して11.5倍の改善が認められた(図4参照)。
【0057】
実施例5 (誘導発現システムのトランジェントアッセイ)
(1)試料の準備及びパーティクルガンによる遺伝子導入
シロイヌナズナ(Arabidopsis thaliana)、タバコ(Nicotiana tabacum)、ダイズ(Glycine max 'Jack')、カーネーション(Dianthus caryophyllus 'True Love'、キリン グリーン アンド フラワー)、イネ(Oryza sativa ssp. japonica cv. Nipponbare)、オニユリ(Liliunm lancifolium)の夫々の葉を切り取り、切り取られた葉(葉片)を誘導発現処理区として100μM CuSO4を含む0.6% 寒天培地上に静置することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った(尚、非誘導発現処理区として CuSO4を含まない寒天培地を用いた。
)。
その後、当該誘導発現処理1日間後の葉片に、実施例1で作製されたベクターEとpBI221(Clontech)とを等量混合してなる組成物がコーティングされた直径1.0μmの金粒子を用いたパーティクルガン法により、前記組成物を導入した。金粒子1mg当たりのDNA量は0.1μgとした。pBI221は、遺伝子導入効率を補正する目的で混合導入した。対照として、前記組成物に代えて実施例1で作製されたp35S-sGFPとpBI221とを等量混合してなる組成物が用いられた。
【0058】
(2)レポーター遺伝子のトランジェントな発現解析
遺伝子導入された葉片について、遺伝子導入後1日目に、蛍光顕微鏡(Nikon)にてGFPの蛍光発光が観察されるスポット数をカウントした。その後、遺伝子導入された葉片をGUS染色液(0.5 mg/ml X-Gluc、0.5 mM K3Fe(CN)6、0.5 mM K4Fe(CN)6、0.01% Triton X-100、100 mM リン酸ナトリウム)に浸し、これを400 mmHgで10分間、減圧処理した後、37℃で1日間、染色した。染色された葉片について、70% エタノールで脱色処理した後、光学顕微鏡(Nikon)にてGUS染色が観察されるスポット数をカウントした。
【0059】
その結果、p35S-sGFPが導入されてなる葉片では、誘導発現処理区及び非誘導発現処理区におけるGFP蛍光によるスポット数に有意な差は認められなかった。一方、ベクターEが導入されてなる葉片では、誘導発現処理区におけるGFP蛍光によるスポット数が、非誘導発現処理区におけるGFP蛍光によるスポット数と比較して、多く認められた。尚、GUS染色によるスポット数はいずれの処理区においても安定的に観察され、遺伝子導入効率に大きな差異が存在しないことが確認された(表1参照)。
【0060】
【表1】

【0061】
実施例6 (組換えシロイヌナズナの開花誘導)
(1)導入ベクターの作製
シロイヌナズナ(Arabidopsis thaliana ecotype Columbia)を改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種し、23℃で3週間生育させた。得られた植物を、予め培土が入れられたポットに移植し、人工気象器内で生育させた。移植後2週間目の植物の花芽及び本葉から、植物RNA抽出キット「RNeasy Plant Mini Kit」(QIAGEN)を用いて全RNAを抽出した。抽出された全RNAから、cDNA合成キット「ReverTra Ace」(TOYOBO)を用いてcDNAを合成した。合成されたcDNAを鋳型に、2種の特異的プライマー(FT-1F、FT-1RC)を用いてPCRを行うことにより、FT遺伝子(GenBank Accession Number AB027504)を増幅した。増幅されたFT遺伝子を、pBI221(Clontech)のGUS遺伝子と置換した。このようにして得られたプラスミドを鋳型に、6種の特異的プライマー(FT-2F、FT-2RC、FT-3F、FT-3RC、FT-4F、FT-1RC)を用いてfusion PCRを行うことにより、FT遺伝子内部に存在するBamHIサイト及びEcoRIサイトにアミノ酸置換を伴わない変異を導入し且つ5’末端のXbaIサイトをBamHIサイトに変更した。このようにして得られた改変FT遺伝子を、実施例1で作製されたpKXS-MRE4/35S(-90m)-To71sGFPのsGFP遺伝子と置換することにより、pKXS-MRE4/35S(-90m)-To71FTを作製した。pKXS-MRE4/35S(-90m)-To71FTに含まれるFT遺伝子発現カセットを実施例1に記載された方法と同様の方法に準じて、p35S-ACE1/VP16AD-CRに含まれる転写因子遺伝子発現カセットとターミネーター側で連結したベクターFを得た(図5参照)。
【0062】
FT-1F:5'-taatctagaatgtctataaatataagagaccctc-3'(配列番号35)
FT-1RC:5'-atagagctcctaaagtcttcttcctccg-3'(配列番号36)
FT-2F:5'-taaggatccatgtctataaatataagagaccctc-3'(配列番号37)
FT-2RC:5'-gaacatctggatcgaccataaccaaagta-3'(配列番号38)
FT-3F:5'-tactttggttatggtcgatccagatgttc-3'(配列番号39)
FT-3RC:5'-gacacgatgaatacctgcagtggga-3'(配列番号40)
FT-4F:5'-tcccactgcaggtattcatcgtgtc-3'(配列番号41)
【0063】
(2)組換えシロイヌナズナの作製と選抜及び開花誘導
作製されたベクターFを、アグロバクテリウム(Agrobacterium tumefaciens strain C58C1)に導入した。アグロバクテリウムを50 mg/L カナマイシン、100 mg/L アンピシリン、100 mg/L リファンピシンを含むLB寒天培地(0.5% 酵母エキス、1.0% バクトトリプトン、0.5% 食塩、1% 寒天)で培養して薬剤耐性コロニーを選抜することにより、組換えアグロバクテリウムを得た。得られた組換えアグロバクテリウムを、モデル植物ラボマニュアル(岩渕雅樹他編集、2000年、シュプリンガー・フェアラーク東京株式会社、ISBN 4-431-70881-2 C3045)に記載される方法に準じて、シロイヌナズナ(Arabidopsis thalia
na ecotype Columbia)に感染させることにより、遺伝子導入を行った。遺伝子導入されたシロイヌナズナから採取されたT1種子を、20 mg/L ベンレート、200 mg/L クラフォラン、25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、1% ショ糖、0.8 % 寒天)に播種し生育した後、カナマイシンに耐性を示す植物個体を選抜した。選抜された植物個体を予め培土が入れられたポットに移植し、人工気象器内で生育させることにより、T2種子を得た。得られたT2種子を25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種し生育した後、χ2検定に基づいた5%の有意水準において、カナマイシンに耐性を示す植物個体が3:1の割合で出現するラインを選抜した。選抜されたラインの植物数個体を予め培土が入れられたポットに移植し、人工気象器内で生育させることにより、T3種子を得た。得られたT3種子を、25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種し生育した後、全てがカナマイシンに耐性を示すホモ接合体を選抜した。尚、植物個体の生育のための培養条件は、明期23時間、暗期1時間、23〜25℃とした。
選抜されたホモ接合体について、T3種子を、改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種した。明期23時間、暗期1時間、23〜25℃の条件下で3日間生育させた後、明期12時間、暗期12時間、23〜25℃の条件下で生育させた。播種後10日経過した植物個体を誘導発現処理区として100μM CuSO4を含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8 % 寒天)に移植することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った(尚、非誘導発現処理区として CuSO4を含まない改変MS寒天培地を用いた。)。
【0064】
当該誘導発現処理3日後(播種後13日目)の植物個体を、CuSO4を含まない改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8 % 寒天)に移植した。当該誘導発現処理後11日目(播種後24日目)の植物個体を観察した。その結果、誘導発現処理区では、非誘導発現処理区と比較して、早期に開花が誘導された(図6参照)。
【0065】
実施例7 (組換えタバコの除草剤耐性誘導)
(1)導入ベクターの作製
pSUM-35S-rSt-1609soy(特開2006-001921)から、制限酵素BamHI及びSacIを用いて、rSt-1609soy遺伝子を切り出した。切り出しされたrSt-1609soy遺伝子を、実施例1で作製されたpKXS-MRE4/35S(-90m)-To71sGFPのsGFP遺伝子と置換することにより、pKXS-MRE4/35S(-90m)-To71rSt-1609soyを作製した。pKXS-MRE4/35S(-90m)-To71rSt-1609soyに含まれるrS
t-1609soy遺伝子発現カセットを実施例1に記載された方法と同様の方法に準じて、p35S-ACE1/VP16AD-CRに含まれる転写因子遺伝子発現カセットとターミネーター側で連結したベクターGを得た(図5参照)。
【0066】
(2)組換えタバコの作製と選抜及び除草剤耐性誘導
作製されたベクターGをアグロバクテリウム(Agrobacterium tumefaciens strain LBA4404)に導入した。アグロバクテリウムを50 mg/L カナマイシン、300 mg/L ストレプトマイシン、100 mg/L リファンピシンを含むLB寒天培地(0.5% 酵母エキス、1.0% バクトトリプトン、0.5% 食塩、1% 寒天)で培養して薬剤耐性コロニーを選抜することにより、組換えアグロバクテリウムを得た。得られた組換えアグロバクテリウムを、植物遺伝子操作マニュアル(内宮博文著、1992年、講談社サイエンティフィック)に記載される方法に準じて、タバコ(Nicotiana tabacum strain SR1)に感染させることにより、遺伝子導入を行った。遺伝子導入されたタバコの葉片から100 mg/L カナマイシンに耐性を示す不定芽を選抜し、選抜された不定芽から植物個体を再生させた。得られた植物個体から採取されたT1種子を50 mg/L カナマイシンを含むMS寒天培地(MS無機塩類、MSビタミン、3% ショ糖、0.8% 寒天)に播種し生育した後、χ2検定に基づいた5%の有意水準において、カナマイシンに耐性を示す植物個体が3:1の割合で出現するラインを選抜した。尚、植物個体の生育のための培養条件は、明期23時間、暗期1時間、23〜25℃とした。
選抜されたラインについて、播種後数日経過した植物個体を、予め培土が入れられたポットに移植した後、当該植物個体に、誘導発現処理区としてCuSO4を含む水溶液を散布する(尚、非誘導発現処理区として CuSO4を含まない水溶液を用いた。)。さらに、前記植物個体に、PPO阻害型除草剤を散布する。
誘導発現処理区では、非誘導発現処理区と比較して、より高濃度の除草剤に耐性を示す結果となる。
【0067】
実施例8 (他の5’−非翻訳領域配列の利用)
(1)導入ベクターの作製
実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPを鋳型に2種の特異的プライマー(EV46-1F、o46-1RC)を用いてPCRを行うことにより、46o断片を増幅した。また、プラスミドCaMV35S-sGFP(S65T)-NOS3'(「植物の細胞を観る実験プロトコール」 福田裕穂他監修、1997年、秀潤社、ISBN 4-87962-170-6)を鋳型に2種の特異的プライマー(omg-1F、omg-1RC)を用いてPCRを行うことにより、Ω配列断片を増幅した。さらに、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPを鋳型に2種の特異的プライマー(oGFP-1F、SIGFP-1RC)を用いてPCRを行うことにより、oGFP断片を増幅した。得られた3種のDNA断片(46o、Ω配列、oGFP)の混合物を鋳型に、2種の特異的プライマー(EV46-1F、SIGFP-1RC)を用いてPCRを行うことにより、ΩsGFP断片を増幅した。得られたΩsGFP断片を、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPのEcoRV及びSacIサイトで切り出される断片と置換することにより、pKXS-MRE4/35S(-46)-ΩsGFPを作製した。作製されたpKXS-MRE4/35S(-46)-ΩsGFPを鋳型に2種の特異的プライマー(Xo-1F、SIGFP-1RC)を用いてPCRを行うことにより、xΩsGFP断片を増幅した。得られたxΩsGFP断片を、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPのXbaI及びSacIサイトで切り出される断片と置換することにより、pKXS-MRE4/35S(-46)-xΩsGFPを作製した。
【0068】
実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPを鋳型に2種の特異的プライマー(EV46-1F、a46-1RC)を用いてPCRを行うことにより、46a断片を増幅した。また、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPを鋳型に2種の特異的プライマー(aGFP-1F、SIGFP-1RC)を用いてPCRを行うことにより、aGFP断片を増幅した。得られた2種のDNA断片(46a、aGFP)と合成オリゴヌクレオチド(A94-1F、A94-1RC)を混合した。この混合物を鋳型に、2種の特異的プライマー(EV46-1F、SIGFP-1RC)を用いてPCRを行うことにより、タバコのアルコールデヒドロゲナーゼ遺伝子由来の5'−非翻訳領域配列を有するA94sGFP断片を増幅した。得られたA94sGFP断片を、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPのEcoRV及びSacIサイトで切り出される断片と置換することにより、pKXS-MRE4/35S(-46)-A94sGFPを作製した。作製されたpKXS-MRE4/35S(-46)-A94sGFPを鋳型に2種の特異的プライマー(Xa-1F、SIGFP-1RC)を用いてPCRを行うことにより、xA94sGFP断片を増幅した。得られたxA94sGFP断片を、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPのXbaI及びSacIサイトで切り出される断片と置換することにより、pKXS-MRE4/35S(-46)-xA94sGFPを作製した。
作製された4種のsGFP遺伝子発現カセットを、実施例1に記載された方法と同様の方法に準じて、p35S-ACE1/VP16AD-CRに含まれる転写因子遺伝子発現カセットとターミネーター側で連結し、ベクターを得た(図7参照)。pKXS-MRE4/35S(-46)-ΩsGFPを由来とするものをベクターH、pKXS-MRE4/35S(-46)-xΩsGFPを由来とするものをベクターI、pKXS-MRE4/35S(-46)-A94sGFPを由来とするものをベクターJ、pKXS-MRE4/35S(-46)-xA94sGFPを由来とするものをベクターKとした。
【0069】
EV46-1F:5'-ctagatatcgcaagacccttcctct-3'(配列番号42)
o46-1RC:5'-gtaaaaataccctctccaaatgaaatgaacttc-3'(配列番号43)
omg-1F:5'-ggtatttttacaacaattaccaacaacaac-3'(配列番号44)
omg-1RC:5'-cattgtaattgtaaatagtaattgtaatgt-3'(配列番号45)
oGFP-1F:5'-acaattacaatggtgagcaagggcga-3'(配列番号46)
SIGFP-1RC:5'-ttagagctcttacttgtacagctcgtcc-3'(配列番号47)
Xo-1F:5'-gactctagagtatttttacaacaattaccaac-3'(配列番号48)
a46-1RC:5'-gttaaatagaccctctccaaatgaaatgaacttc-3'(配列番号49)
aGFP-1F:5'-gaaaaataaatggtgagcaagggcgag-3'(配列番号50)
A94-1F:5'-gtctatttaactcagtattcagaaacaacaaaagttcttctctacataaaattttcctattttagtgatcagtgaaggaaatcaagaaaaataaatg-3'(配列番号51)
A94-1RC:5'-catttatttttcttgatttccttcactgatcactaaaataggaaaattttatgtagagaagaacttttgttgtttctgaatactgagttaaatagac-3'(配列番号52)
Xa-1F:5'-actctagagtctatttaactcagtattcag-3'(配列番号53)
【0070】
(2)組換えシロイヌナズナの作製と選抜及び発現解析
作製されたベクターHからKをそれぞれアグロバクテリウム(Agrobacterium tumefaciens strain C58C1)に導入した。アグロバクテリウムを50 mg/L カナマイシン、100 mg/L アンピシリン、100 mg/L リファンピシンを含むLB寒天培地(0.5% 酵母エキス、1.0% バクトトリプトン、0.5% 食塩、1% 寒天)で培養して薬剤耐性コロニーを選抜することにより、組換えアグロバクテリウムを得た。得られた組換えアグロバクテリウムを、モデル植物ラボマニュアル(岩渕雅樹他編集、2000年、シュプリンガー・フェアラーク東京株式会社、ISBN 4-431-70881-2 C3045)に記載される方法に準じて、シロイヌナズナ(Arabidopsis thaliana ecotype Columbia)に感染させることにより、遺伝子導入を行った。遺伝子導入されたシロイヌナズナから採取されたT1種子を、20 mg/L ベンレート、200 mg/L クラフォラン、25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、1% ショ糖、0.8 % 寒天)に播種し11日間生育させた後、カナマイシンに耐性を示す植物個体を選抜した。選抜された植物個体を20 mg/L ベンレート、200 mg/L クラフォラン、25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、1% ショ糖、0.8 % 寒天)に移植し、さらに6日間生育させた。播種後17日経過した植物個体を100μM CuSO4を含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8 % 寒天)に移植することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った。当該誘導発現処理1時間以内(0日目)に、植物個体におけるGFPの蛍光発光状態をマクロ蛍光顕微鏡VB-G05(キーエンス)にて観察し、高感度冷却CCDカメラVB-7010(キーエンス)を用いて蛍光画像を取得した。その後、当該誘導発現処理3日目に、同様の観察をし、蛍光画像を取得した。その結果、ベクターHからKが導入されてなる組換えシロイヌナズナは、誘導発現処理0日目ではGFP蛍光が観察されないのに対して、誘導発現処理3日目では強いGFP蛍光が観察された(図8参照)。
尚、植物個体の生育のための培養条件は、播種後11日目までは明期23時間、暗期1時間、23〜25℃とし、播種後11日目以降は明期12時間、暗期12時間、23〜25℃とした。
【産業上の利用可能性】
【0071】
本発明により、銅イオン誘導性システムについて、目的とする外来遺伝子の植物における誘導発現における誘導倍率の向上を達成できる方法等を提供することが可能となる。
【配列表フリーテキスト】
【0072】
配列番号1
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号2
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号3
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号4
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号5
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号6
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号7
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号8
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号9
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号10
アダプターのために設計されたオリゴヌクレオチド
配列番号11
アダプターのために設計されたオリゴヌクレオチド
配列番号12
置換DNA断片のために設計されたオリゴヌクレオチド
配列番号13
置換DNA断片のために設計されたオリゴヌクレオチド
配列番号14
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号15
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号16
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号17
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号18
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号19
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号20
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号21
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号22
挿入DNA断片のために設計されたオリゴヌクレオチド
配列番号23
挿入DNA断片のために設計されたオリゴヌクレオチド
配列番号24
置換DNA断片のために設計されたオリゴヌクレオチド
配列番号25
置換DNA断片のために設計されたオリゴヌクレオチド
配列番号26
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号27
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号28
TaqManプローブのために設計されたオリゴヌクレオチド
配列番号29
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号30
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号31
TaqManプローブのために設計されたオリゴヌクレオチド
配列番号32
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号33
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号34
TaqManプローブのために設計されたオリゴヌクレオチド
配列番号35
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号36
TaqManプローブのために設計されたオリゴヌクレオチド
配列番号37
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号38
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号39
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号40
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号41
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号42
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号43
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号44
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号45
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号46
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号47
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号48
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号49
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号50
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号51
PCRのために設計されたオリゴヌクレオチド
配列番号52
PCRのために設計されたオリゴヌクレオチド
配列番号53
PCRのために設計されたオリゴヌクレオチドプライマー
【技術分野】
【0001】
本発明は、植物における、化学物質による外来遺伝子の誘導発現方法等に関するものである。
【背景技術】
【0002】
目的とする外来遺伝子を植物に導入し発現させる場合には、通常、例えば、カリフラワーモザイクウイルス(CaMV)35Sプロモーター(例えば、非特許文献1参照)のような常に発現する構成的プロモーターを用いることが多い。しかしながら、このような構成的プロモーターを利用する場合には、植物の転写系・翻訳系に多大な負荷をかけることになるうえ、目的とする外来遺伝子の種類によっては、発芽や生育の阻害等植物に悪影響を与えることがある。このような悪影響を回避する一つの方法として、目的とする外来遺伝子を所望の時期に誘導発現させる方法を挙げることができる。そして、このような誘導発現方法を利用することによって、目的とする外来遺伝子の一層効率的な発現、ひいては当該外来遺伝子が含む構造遺伝子領域によりコードされる目的とする外来蛋白質等の高生産が実現できるうえ、植物の生長や生理を自在に制御できるようになり、その産業的利用価値は計りしれない。
【0003】
目的とする外来遺伝子を所望の時期に誘導発現させる方法は、その誘導条件の差異に基づき、2つに大別することができる。即ち、温度や光、植物病原体等の非化学物質により誘導発現させるものと、化学物質により誘導発現させるものとである。
前者の例としては、具体的には例えば、熱誘導性システム(例えば、特許文献1参照)、低温誘導性システム(例えば、特許文献2参照)、植物病原体の攻撃により誘導されるシステム(例えば、特許文献3参照)等が挙げられる。これらは、誘導性プロモーターを目的とする外来遺伝子の上流に連結してなる遺伝子構造体を構築し当該遺伝子構造体を利用するだけでよく、当該遺伝子構造体の構造は極めて単純である。しかしながら、温度や光、植物病原体等の非化学物質により誘導発現させるものは、予期せぬ環境の変化や植物病原体の襲来等で突発的に目的とする外来遺伝子が発現してしまう危険性がある。
一方、後者の例としては、具体的には例えば、銅イオン誘導性システム(例えば、非特許文献2参照)、ステロイドホルモン誘導性システム(例えば、非特許文献3参照)、エタノール誘導性システム(例えば、非特許文献4参照)、テトラサイクリン誘導性システム(例えば、非特許文献5参照)等が挙げられる。これらは、Moore I et al (例えば、非特許文献6参照)、Padidam M (例えば、非特許文献7参照)等により詳細に解説されており、特定の化学物質の濃度に依存して目的とする外来遺伝子の発現を制御することができる。即ち、所望の時期に必要な分だけ目的とする外来遺伝子の発現を誘導することができる。
【0004】
従来、目的とする外来遺伝子が含む構造遺伝子領域によりコードされる目的とする外来蛋白質等を組換え植物にて生産(植物内に蓄積)させるために、目的とする外来遺伝子が含む構造遺伝子領域の上流に位置させるタバコモザイクウイルス(TMV)のΩ配列(例えば、非特許文献8参照)やタバコアルコールデヒドロゲナーゼ遺伝子の5’−非翻訳領域配列(例えば、非特許文献9参照)を利用した例が存在しているが、一方で、Ω配列が連結されているmRNAはヌクレアーゼにより分解されやすいと報告する例も存在しており(例えば、非特許文献10参照)、5’−非翻訳領域配列の利用が如何なる結果をもたらすかを容易に想像できる技術状況にはない。まして、特定な誘導性システム(例えば、銅イオン誘導性システム等)における化学物質による目的とする外来遺伝子の誘導発現にどのような影響を与えるものであるか等は全く不明であった。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Benfey PN & Chua NH、1990、Science 250、959-966
【非特許文献2】Mett VL et al、1993、Proc Natl Acad Sci 90、4567-4571
【非特許文献3】Aoyama T & Chua NH、1997、Plant J.11、605-612; 米国特許第6063985号
【非特許文献4】Caddick MX et al、1998、Nature Biotech. 16、177-180; 国際公開第93/21334号パンフレット
【非特許文献5】Weinmann P et al、1994、Plant J. 5、559-569
【非特許文献6】Moore I et al 、2006、Plant J 45、651-683
【非特許文献7】Padidam M、2003、Current Opin Plant Biol 6、169-177
【非特許文献8】Gallie D et al、1987、Nucleic Acid Res 15、3257-3273; 米国特許第5489527号
【非特許文献9】Satoh J et al、2004、J Biosci Bioeng 98(1):1-8; 特開2003-079372号公報
【非特許文献10】Gallie D et al、1988、Nucleic Acid Res 16、8675-8694
【特許文献1】米国特許第5447858号
【特許文献2】米国特許第5847102号
【特許文献3】米国特許第5942662号
【発明の概要】
【発明が解決しようとする課題】
【0006】
理想的な、植物における、化学物質による目的とする外来遺伝子の誘導発現方法は、非誘導時の目的とする外来遺伝子の発現レベルが非常に低く、誘導時の目的とする外来遺伝子の発現レベルが非常に高いものである。非誘導時の目的とする外来遺伝子の発現レベルが低ければ低いほど、宿主植物への負荷が少なくなるうえ、微量で機能するシグナル因子等をも制御できるようになる。また、誘導時の目的とする外来遺伝子の発現レベルが高ければ高いほど、目的とする外来遺伝子が含む構造遺伝子領域によりコードされる目的とする外来蛋白質等を効率的に生産可能となるうえ、誘導に必要な刺激が少量で済むからである。
上述のような誘導性システムにおける化学物質による目的とする外来遺伝子の誘導発現のうち、銅イオン誘導性システムについては、当該システムが有する誘導発現の低い誘導倍率が問題となっており(例えば、非特許文献7参照)、本発明では、より理想に近づけることによって、当該銅イオン誘導性システムにおいて、目的とする外来遺伝子の誘導発現での誘導倍率の向上を達成できる方法を提供することを課題としている。
【課題を解決するための手段】
【0007】
本発明者らは、かかる状況下鋭意検討した結果、本発明に至った。
即ち、本発明は、
1.植物における、化学物質による目的とする外来遺伝子の誘導発現方法であって、前記の目的とする外来遺伝子が含むプロモーター機能を有する領域の下流に任意の外来遺伝子の5'−非翻訳領域を有するように構築されてなる遺伝子構築物に含まれる転写因子であり、前記の目的とする外来遺伝子とは異なる他の外来遺伝子によりコードされる転写因子を、銅イオンにより活性化することにより、前記の目的とする外来遺伝子が含むプロモーター機能を有する領域による前記の目的とする外来遺伝子の転写活性化を誘導する工程を有することを特徴とする方法(以下、本発明誘導発現方法と記すこともある。);
2.前記の5'−非翻訳領域の塩基配列が、下記の5'−非翻訳領域に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項1記載の方法;
<5'−非翻訳領域に係る塩基配列群>
(1)ウイルスの遺伝子又は植物の誘導性遺伝子由来の塩基配列
(2)トバモウイルス属に属するウイルスの遺伝子由来の塩基配列
(3)トマトモザイクウイルスの遺伝子由来の塩基配列
(4)トマトモザイクウイルスの130k/180k遺伝子由来の塩基配列
3.前記の転写因子の塩基配列が、下記の転写因子に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項1又は2記載の方法;
<転写因子に係る塩基配列群>
(1)真核生物由来の塩基配列
(2)酵母由来の塩基配列
(3)酵母ACE1由来の塩基配列
4.前記の遺伝子構築物が、前記の転写因子とは異なる他の転写因子の転写活性化領域であり、前記の転写因子の下流に位置する転写活性化領域を、さらに追加的に有するように構築されてなる遺伝子構築物であることを特徴とする前項1、2又は3記載の方法;
5.前記の転写活性化領域が、下記の転写活性化領域に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項4記載の方法;
<転写活性化領域に係る塩基配列群>
(1)ウイルスの転写因子由来の塩基配列
(2)シンプレックスウイルス属に属するウイルスの転写因子由来の塩基配列
(3)単純ヘルペスウイルスの転写因子由来の塩基配列
(4)単純ヘルペスウイルスのVP16転写因子由来の塩基配列
6.前記の遺伝子構築物が、任意のプロモーターを前記の転写因子の上流に追加的に有するように構築されてなる遺伝子構築物であることを特徴とする前項1、2、3、4又は5記載の方法;
7.植物において、化学物質により目的とする外来遺伝子を誘導発現させるための遺伝子構築物であって、
(a)プロモーター機能を有する領域と目的とする外来蛋白質に係る構造遺伝子領域とを含む、目的とする外来遺伝子と、
(b)前記の目的とする外来遺伝子とは異なる他の外来遺伝子によりコードされ且つ銅イオンにより活性化される転写因子と、
(c)前記の目的とする外来遺伝子が含むプロモーター機能を有する領域(以下、本プロモーター領域と記すこともある。)の下流に位置する任意の外来遺伝子の5'−非翻訳領域と
を有するように構築可能に調製されてなることを特徴とする遺伝子構築物(以下、本発明遺伝子構築物と記すこともある。);
8.前記の5'−非翻訳領域の塩基配列が、下記の5'−非翻訳領域に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項7記載の遺伝子構築物;
<5'−非翻訳領域に係る塩基配列群>
(1)ウイルスの遺伝子又は植物の誘導性遺伝子由来の塩基配列
(2)トバモウイルス属に属するウイルスの遺伝子由来の塩基配列
(3)トマトモザイクウイルスの遺伝子由来の塩基配列
(4)トマトモザイクウイルスの130k/180k遺伝子由来の塩基配列
9.前記の転写因子の塩基配列が、下記の転写因子に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項7又は8記載の遺伝子構築物;
<転写因子に係る塩基配列群>
(1)真核生物由来の塩基配列
(2)酵母由来の塩基配列
(3)酵母ACE1由来の塩基配列
10.前記の転写因子とは異なる他の転写因子の転写活性化領域であり、前記の転写因子の下流に位置する転写活性化領域を、さらに追加的に有するように構築されてなることを特徴とする前項7、8又は9記載の遺伝子構築物;
11.前記の転写活性化領域が、下記の転写活性化領域に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする前項10記載の遺伝子構築物;
<転写活性化領域に係る塩基配列群>
(1)ウイルスの転写因子由来の塩基配列
(2)シンプレックスウイルス属に属するウイルスの転写因子由来の塩基配列
(3)単純ヘルペスウイルスの転写因子由来の塩基配列
(4)単純ヘルペスウイルスのVP16転写因子由来の塩基配列
12.任意のプロモーターを前記の転写因子の上流に追加的に有するように構築されてなる遺伝子構築物であることを特徴とする前項7、8、9、10又は11記載の遺伝子構築物;
13.前項7〜12のいずれかの前項記載の遺伝子構築物が導入されてなることを特徴とする形質転換体植物(以下、本発明形質転換体植物と記すこともある。);
14.前項13記載の形質転換体植物から目的とする外来蛋白質を回収することを特徴とする外来蛋白質の取得方法(以下、本発明外来蛋白質取得方法と記すこともある。);
等を提供するものである。
【発明の効果】
【0008】
本発明により、銅イオン誘導性システムについて、目的とする外来遺伝子の植物における誘導発現における誘導倍率の向上を達成できる方法等を提供することが可能となる。
【図面の簡単な説明】
【0009】
【図1】図1は、銅イオン誘導性sGFP遺伝子発現ベクターのT-DNA領域の構造を示す模式図である。図中の*印は、as-1サイトへの変異導入部位を示している。
【図2】図2は、銅イオン誘導性sGFP遺伝子発現ベクターが導入されてなる組換えシロイヌナズナにおける、sGFP遺伝子のmRNA量をリアルタイムPCRにより定量した結果を示す図である。グラフ内の数値は、非誘導発現処理区でのsGFP遺伝子のmRNA量に対する、誘導発現処理区でのsGFP遺伝子のmRNA量の割合を示している。
【図3】図3は、銅イオン誘導性sGFP遺伝子発現ベクターが導入されてなる組換えタバコにおける、sGFP遺伝子のmRNA量をリアルタイムPCRにより定量した結果を示す図である。グラフ内の数値は、非誘導発現処理区でのsGFP遺伝子のmRNA量に対する、誘導発現処理区でのsGFP遺伝子のmRNA量の割合を示している。
【図4】図4は、銅イオン誘導性sGFP遺伝子発現ベクターが導入されてなる組換えタバコ培養細胞における、GFPの蓄積量(GFPの蛍光強度から算出したGFP換算量)の定量の結果を示す図である。グラフ内の数値は、非誘導発現処理区でのGFPの蓄積量に対する、誘導発現処理区でのGFPの蓄積量の割合を示している。
【図5】図5は、銅イオン誘導性FT遺伝子発現ベクター及び銅イオン誘導性rSt-1609soy遺伝子発現ベクターのT-DNA領域の構造を示す模式図である。図中の*印は、as-1サイトへの変異導入部位を示している。
【図6】図6は、銅イオン誘導性FT遺伝子発現ベクターが導入されてなる組換えシロイヌナズナにおける、開花時期の相違を示す図である。
【図7】図7は、銅イオン誘導性sGFP遺伝子発現ベクターのT-DNA領域の構造を示す模式図である。
【図8】図8は、銅イオン誘導性sGFP遺伝子発現ベクターが導入されてなる組換えシロイヌナズナにおける、GFPの蛍光発光状態を示す図である。夫々、誘導発現処理0日目を左に、誘導発現処理3日目を右に示している。
【発明を実施するための形態】
【0010】
以下に本発明を詳細に説明する。
本発明遺伝子構築物は、植物において、化学物質により目的とする外来遺伝子を誘導発現させるために利用することができる。当該遺伝子構築物は、
(a)プロモーター機能を有する領域と目的とする外来蛋白質に係る構造遺伝子領域とを含む、目的とする外来遺伝子と、
(b)前記の目的とする外来遺伝子とは異なる他の外来遺伝子によりコードされ且つ銅イオンにより活性化される転写因子(以下、本転写因子と記すことがある。)と、
(c)前記の目的とする外来遺伝子が含むプロモーター機能を有する領域(以下、本プロモーター領域と記すこともある。)の下流に位置する任意の外来遺伝子の5'−非翻訳領域(以下、本5'−非翻訳領域と記すことがある。)と
を有するように構築可能に調製すればよい。このようにして、本発明遺伝子構築物を得ることができる。
【0011】
好ましい本発明遺伝子構築物としては、例えば、本転写因子とは異なる他の転写因子の転写活性化領域であり、本転写因子の下流に位置する転写活性化領域(以下、本転写活性化領域と記すことがある。)を、さらに追加的に有するように構築されてなる遺伝子構築物を挙げることができる。
【0012】
本発明遺伝子構築物は、前記(a)、前記(b)及び前記(c)の各要素を有するように構築可能に調製すればよい。即ち、1つの発現カセットとして前記(a)、前記(b)及び前記(c)の各要素を有するように構築してもよいし、また2つの発現カセットのうち、1つの発現カセットとして前記(a)及び前記(c)の要素を有するように構築し、且つ、他の1つの発現カセットとして前記(b)の要素を有するように構築してもよい。
このような発現カセットは、通常の遺伝子工学的手法を用いて行えばよい。尚、2つの発現カセットとして構築する場合には、例えば、前記(a)及び(c)を第一発現カセットとして構築し、また前記(b)を第二発現カセットとして構築してもよい。
【0013】
本発明遺伝子構築物としては、好ましくは、任意のプロモーターを前記の転写因子の上流に追加的に有するように構築されてなる遺伝子構築物等を挙げることができる。
また更に、本発明遺伝子構築物としては、好ましくは、任意のターミネーターを前記の転写因子の下流に追加的に有するように構築されてなる遺伝子構築物等を挙げることができる。当該ターミネーターとしては、例えば、NOSターミネーター、CR16ターミネーター(特開2000-166577)、ダイズ種子グリシニンターミネーター(特開06-189777)等を挙げることができる。
【0014】
本発明遺伝子構築物を、例えば、植物体、培養細胞等の宿主植物へ導入し、当該遺伝子構築物の誘導発現を行う。本発明遺伝子構築物の宿主植物への導入は、宿主植物夫々に適用可能な方法に応じて、通常の遺伝子工学的手法を用いて行えばよい。
具体的には例えば、第一発現カセットと第二発現カセットとをベクター上で連結してなる遺伝子構築物に含まれる目的とする外来遺伝子を導入してもよいし、またこれらの発現カセットを連結せずに混合して導入してもよい。また、第一発現カセット又は第二発現カセットのいずれかを導入した宿主植物に、再度、他方の発現カセットを導入してもよい。
さらに、第一発現カセットを導入した個体と第二発現カセットを導入した個体とを交配してもよい。第一発現カセットを複数種類導入することで、複数種類の外来遺伝子を同時に誘導発現することも可能である。
本発明遺伝子構築物の導入方法としては、アグロバクテリウム法、パーティクルガン法、エレクトロポレーション法、リン酸カルシウム法、ウイルスベクター法等の各種の公知方法を用いることができる。
宿主植物には、農業上及び園芸上、重要な種又はゲノム解析等遺伝学上有用な種が挙げられるが、さらに、任意の植物種が含まれる。例えば、ダイズ、エンドウ、インゲン、アルファルファ、ミヤコグサ、クローバ、ピーナッツ、スイートピー、クルミ、チャ、ワタ、コショウ、キュウリ、スイカ、カボチャ、メロン、ダイコン、ナタネ、キャノーラ、テンサイ、レタス、キャベツ、ブロッコリー、カリフラワー、シロイヌナズナ、タバコ、ナス、ジャガイモ、サツマイモ、サトイモ、キクイモ、トマト、ホウレンソウ、アスパラガス、ニンジン、ゴマ、エンダイブ、キク、フウロウソウ、キンギョソウ、カーネーション、ナデシコ、ニチニチソウ、ブバルディア、カスミソウ、ガーベラ、トルコキキョウ、チューリップ、ストック、スターチス、シクラメン、ユキノシタ、ノースポール、スミレ、バラ、サクラ、リンゴ、イチゴ、ウメ、ミカン、ボケ、サツキ、ツツジ、ナンヨウアブラギリ、リンドウ、コスモス、アサガオ、ヒマワリ、イチョウ、スギ、ヒノキ、ポプラ、マツ、セコイア、オーク、スイレン、トチュウ、ブナ、イネ、コムギ、オオムギ、ライムギ、エンバク、トウモロコシ、ネギ、タマネギ、ニンニク、ユリ、オニユリ、ラン、グラジオラス、パイナップルである。
【0015】
本発明誘導発現方法では、例えば、銅イオンを本発明形質転換体植物に接触させたり、施用したりすること等が必要である。当該接触・施用方法等としては、本発明形質転換体植物が培養細胞である場合には、例えば、培地や栄養溶液への銅イオンの混合(具体的には例えば、銅イオン濃度1μM〜24mM)等の方法を挙げることができる。また、本発明形質転換体植物が植物体である場合には、土壌や植物体の茎葉への銅イオンの処理・散布(具体的には例えば、銅イオン量1nmol〜2.4mmol、銅イオン濃度1μM〜24mM)等の方法が挙げられる。宿主植物の種類や使用場面に応じて適宜選択することが望ましい。本発明誘導発現方法では、銅イオンを目的とする外来遺伝子を誘導発現させたい植物細胞の中に浸透させればよく、施用する銅イオンを錯体にする等製剤を工夫したり、ボルドー(住友化学)、ジーファイン(八洲化学)、トモノZボルドー(トモノアグリカ)、リドミルプラス(日本農薬)、ハイカッパー(住化武田農薬)、クプラビットホルテ(バイエルクロップサイエンス)、コサイドボルドー(デュポン)、キノンドー(アグロカネショウ)、ヨネポン(米澤化学)等、農業用途に使用される銅剤や、グルコン酸銅(和光純薬)等、食品添加物用途に使用される銅剤を所望の濃度に調整して用いてもよい。また施用に際して展着剤等を混合してもよい。
【0016】
使用される本転写因子は、銅イオンにより活性化され、目的とする外来遺伝子に含まれるプロモーター機能を有する領域(即ち、本プロモーター領域)の転写活性を誘導する。
当該転写因子は、銅イオンが存在しない状態においては不活性型でありプロモーター機能を有する領域(即ち、本プロモーター領域)の転写活性を誘導できないが、銅イオンが存在する状態においては活性型に変化しプロモーター機能を有する領域(即ち、本プロモーター領域)の転写活性を誘導することができる。因みに、転写因子は、通常、DNA結合領域と転写活性化領域とを有している。このとき、当該転写因子に本転写因子とは異なる他の転写因子の転写活性化領域を連結したり、元の転写因子の転写活性化領域と当該転写因子とは異なる他の転写因子の転写活性化領域を置換することもできる。尚、本転写活性化領域は宿主植物の細胞内において、RNAポリメラーゼ複合体のメディエーターをリクルートし、転写活性化能力を増強する働きを有する。
【0017】
本転写因子の好ましい塩基配列としては、前述のように、例えば、下記の転写因子に係る塩基配列群から選ばれてなる塩基配列を挙げることができる。
<転写因子に係る塩基配列群>
(1)真核生物由来の塩基配列
(2)酵母由来の塩基配列
(3)酵母ACE1由来の塩基配列
また例えば、candida glabrataのAMT1(Thorvaldsen JL et al、1993、J Biol Chem 268、12512-12518)由来の塩基配列、Yarrowia lipolyticaのCRF1(Garcia S、2002、J Biol Chem 277、37359-37368)由来の塩基配列、トウモロコシのSOD遺伝子群を銅イオン依存的に誘導発現させる転写因子(Ruzsa SM et al、2003、Biochemistry 42、1508-1516)由来の塩基配列等を挙げることができる。尚、由来の塩基配列には、当該塩基配列の一部使用や他の塩基配列とのキメラ、欠失及び挿入、置換等の変異を導入した塩基配列も含まれる。
【0018】
使用される本プロモーター領域は、前記の本転写因子が銅イオン存在下において転写活性を誘導しうる塩基配列からなる。誘導条件下の宿主細胞中で、目的とする外来遺伝子が発現可能であればよい。好ましくは、例えば、酵母のMRE配列(Mett VL et al、1993、Proc Natl Acad Sci 90、4567-4571)等を含む配列を挙げることができる。具体的には、通常使用されるプロモーター機能を有する領域に存在するTATA配列の上流に、酵母のMRE配列等を繰返し配置してなるものが挙げられる。
尚、前記の通常使用されるプロモーター機能を有する領域は、構成的プロモーターでもよいし、組織特異的プロモーターや、ある刺激により転写活性が誘導される誘導性プロモーターでもよい。使用場面に応じて適宜選択することが望ましい。
構成的プロモーターとしては、例えば、CaMV 35Sプロモーター、PG10-90(特開09-131187)、ユビキチンプロモーター(国際公開01/094394)、アクチンプロモーター(国際公開00/070067)等を挙げることができる。また、組織特異的プロモーターとしては、例えば、ダイズ種子グリシニンプロモーター(特開06-189777)、プロラミンプロモーター(国際公開2004/056993)、インゲンマメ種子ファゼオリンプロモーター(国際公開91/013993)、ナタネ種子ナピンプロモーター(国際公開91/013972)、シロイヌナズナSultr2;2プロモーター(Takahashi H et al、2000、Plant J 23、171-82)、アグロバクテリウムrolCプロモーター(Almon E et al、1997、Physiol 115、1599-1607)等を挙げることができる。
【0019】
使用される本5'−非翻訳領域は、本プロモーターの下流に位置し、転写されるが翻訳はされない塩基配列からなる領域である。但し、目的とする外来遺伝子が、アンチセンスRNAをコードする場合やRNAiを誘発するRNAをコードする場合等は、転写開始点から、当該RNAをコードする領域の直前までを5'−非翻訳領域とする。当該5'−非翻訳領域配列は、ウイルスの遺伝子由来であっても植物の誘導性遺伝子由来であってもよい。ウイルスの遺伝子由来の5'−非翻訳領域配列としては、例えば、タバコモザイクウイルス(TMV)のΩ配列(例えば、Gallie D et al、1987、Nucleic Acid Res 15、3257-3273; 米国特許5489527参照)やタバコエッチウイルス(TEV)のL配列(例えば、Lindbo JA、2007、BMC Biotechnol 7、52参照)、イネ縞葉枯ウイルス(RSV)のΦ配列(例えば、Mori M et al、2006、Plant Biotechnology 23(1)、55-61参照)等を挙げることができる。植物の誘導性遺伝子とは、ある刺激によってその発現量が増大する遺伝子を意味し、例えば、タバコのアルコールデヒドロゲナーゼ遺伝子(Satoh J et al、2004、J Biosci Bioeng 98(1):1-8; 特開2003-79372)やPR1a遺伝子(Ohshima M et al、1990、Plant Cell 2(2)、95-106)、トウモロコシのIn2-1遺伝子又はIn2-2遺伝子(国際公開90/11361; De Veylder L et al、1997、Plant Cell Physiol 38(5)、568-577)やGST27遺伝子(Jepson I et al、1994、Plant Mol Biol 26(6)、1855-1866; 国際公開97/11189)、シロイヌナズナのRD29遺伝子(Yamaguchi-Shinozaki K、1993、Mol Gen Genet 236(2-3)、331-340)、シロイヌナズナ又はダイズ、ヒマワリのヒートショック蛋白質遺伝子(Yoshida K et al、1995、Appl Microbiol Biotechnol 44(3-4):466-472; Nagao RT et al、1985、Mol Cell Biol 5(12)、3417-3428; Almoguera C et al、2002、J Biol Chem、277(46):43866-43872)等を挙げることができる。
【0020】
本5'−非翻訳領域の好ましい塩基配列としては、例えば、下記の5'−非翻訳領域に係る塩基配列群から選ばれてなる塩基配列を挙げることができる。
<5'−非翻訳領域に係る塩基配列群>
(1)ウイルスの遺伝子又は植物の誘導性遺伝子由来の塩基配列
(2)トバモウイルス属に属するウイルスの遺伝子由来の塩基配列(例えば、Gallie D et al、1987、Nucleic Acid Res 15、3257-3273; 米国特許5489527参照)
(3)トマトモザイクウイルスの遺伝子由来の塩基配列
(4)トマトモザイクウイルスの130k/180k遺伝子由来の塩基配列
尚、由来の塩基配列には、当該塩基配列の一部使用や他の塩基配列とのキメラ、欠失及び挿入、置換等の変異を導入した塩基配列も含まれる。また、5'−非翻訳領域の塩基配列数が48 bp以上の塩基配列、又は、二次構造予測プログラム「mfold (version 2.3)」(http://frontend.bioinfo.rpi.edu/applications/mfold/cgi-bin/rna-form1-2.3.cgi、The Bioinformatics Center at Rensselaer and Wadsworth提供; 例えば、Zuker M、2003、Nucleic Acids Res 31(13)、3406-3415参照)を用いて25℃の条件下で二次構造を予測した場合に、分子内で相互作用が予測されない連続した最長の塩基数が11 bp以上である塩基配列を挙げることもできる。
【0021】
本転写活性化領域の好ましい塩基配列としては、前述のように、例えば、下記の転写活性化領域に係る塩基配列群から選ばれてなる塩基配列を挙げることができる。
<転写活性化領域に係る塩基配列群>
(1)ウイルスの転写因子由来の塩基配列
(2)シンプレックスウイルス属に属するウイルスの転写因子由来の塩基配列
(3)単純ヘルペスウイルスの転写因子由来の塩基配列
(4)単純ヘルペスウイルスのVP16転写因子由来の塩基配列(例えば、Triezenberg SJ et al、1988、Genes Dev 2(6)、718-29参照)
また例えば、GAL4転写因子(Gill G、Ptashne M、1987、Cell 51(1)、121-126)由来の塩基配列、転写活性化能力を有する人工的に合成されたペプチドAH(Ansari AZ et al、2001、Chem Biol 8(6)、583-592)由来の塩基配列等を挙げることもできる。尚、由来の塩基配列には、当該塩基配列の一部使用や他の塩基配列とのキメラ、欠失及び挿入、置換等の変異を導入した塩基配列も含まれる。
【0022】
本発明誘導発現方法で使用される転写因子は、前述のような本転写因子であり、目的とする外来遺伝子とは異なる他の外来遺伝子によりコードされる転写因子である。そして当該転写因子は、銅イオンにより活性化するものである。また、本発明誘導発現方法で使用される遺伝子構築物は、例えば、前述のような本発明遺伝子構築物である。また、本発明誘導発現方法で使用される外来遺伝子の5'−非翻訳領域は、前述のような本5'−非翻訳領域であり、任意の外来遺伝子の5'−非翻訳領域である。また、本発明誘導発現方法で使用されるプロモーター機能を有する領域は、前述のような本プロモーター領域であり、目的とする外来遺伝子が含む、プロモーター機能を有する領域である。
【0023】
本発明形質転換体植物は、上述のように、本発明遺伝子構築物が導入されてなる形質転換体植物である。本発明外来蛋白質取得方法では、当該形質転換体植物から目的とする外来蛋白質を通常の蛋白質工学的な手法に準じて回収すればよい。
【0024】
本発明の応用例として、以下のようなことを挙げることができるが、特にこれらに限定されるものではない。
【0025】
FT遺伝子を過剰発現させたシロイヌナズナは、花成時期が早まる(例えば、特開2000-139250参照)。本発明誘導発現方法における目的とする外来遺伝子がFT遺伝子である
場合には、所望する時期に花成を誘導することができる。花成促進遺伝子への変異導入やRNAi、花成遅延遺伝子の過剰発現等により花成が起こり難くした植物に本FT遺伝子を誘導発現させることにより、より厳密に花成を制御することが可能となる。
因みに、FT遺伝子とは、Flowering Locus T 遺伝子の略称であり、花成制御に関して正に働く因子をコードする遺伝子を意味する。FT遺伝子は、日長に応答して維管束師部での発現が増大し、茎頂に移動して、茎頂で発現しているFDと呼ばれるbZIP型転写因子と相互作用することにより花成を誘導する作用を有する(例えば、Kobayashi Y and Weigel D、2007、Genes Dev 21、2371-2384参照)。
【0026】
コラーゲン遺伝子を導入したタバコは、コラーゲンを生産する(例えば、米国特許6617431参照)。本発明誘導発現方法における目的とする外来遺伝子がコラーゲン遺伝子である場合には、高いレベルでの生産が可能となる。
【0027】
PPO遺伝子を導入したトウモロコシは、除草剤耐性となる(例えば、米国特許6307129参照)。本発明誘導発現方法における目的とする外来遺伝子がPPO遺伝子である場合には、所望の時期にのみ除草剤に耐性を示すようになる。栽培の必要がなくなった時には、同じ除草剤を用いて枯死させることが可能となる。
【0028】
EPSPS遺伝子を導入したダイズは、除草剤耐性となる(例えば、国際公開92/00377参照)。本発明誘導発現方法における目的とする外来遺伝子がEPSPS遺伝子である場合には、所望の時期にのみ除草剤に耐性を示すようになる。栽培の必要がなくなった時には、同じ除草剤を用いて枯死させることが可能となる。
【0029】
Δ15脂肪酸不飽和化酵素遺伝子を導入したダイズは、高度不飽和脂肪酸含量が上昇する(例えば、国際公開2005/047479参照)。本発明誘導発現方法における目的とする外来遺伝子がΔ15脂肪酸不飽和化酵素遺伝子である場合には、所望の時期に油脂中の脂肪酸組成を制御することが可能となる。
【0030】
ポリプレニル2リン酸合成酵素遺伝子を導入したイネは、CoQ10含量が上昇する(例えば、特開2006-212019参照)。本発明誘導発現方法における目的とする外来遺伝子がポリプレニル2リン酸合成酵素遺伝子である場合には、所望の時期にCoQ10含量を上昇させることが可能となる。
【0031】
テオブロミン合成酵素遺伝子の発現を抑制したコーヒーは、カフェイン含量が減少する(例えば、特開2002-112785; Ogita S et al、2004、Plant Mol Biol 54(6)、931-941参照)。本発明誘導発現方法における目的とする外来遺伝子がテオブロミン合成酵素遺伝子のアンチセンス遺伝子等である場合には、所望の時期に種子中のカフェイン含量を制御することが可能となる。
【0032】
リボヌクレアーゼ遺伝子を葯にて発現させたアブラナは、雄性不稔となる(例えば、Mariani C et al、1990、Nature 347、737-741参照)。本発明誘導発現方法における目的とする外来遺伝子がリボヌクレアーゼ遺伝子である場合には、所望の個体のみを雄性不稔にすることが可能となる。一方、S糖蛋白質遺伝子の発現を抑制すると、自家不和合性が打破される(例えば、特開08-322412参照)。本発明誘導発現方法における目的とする外来遺伝子がS糖蛋白質遺伝子のアンチセンス遺伝子等である場合には、所望の個体のみを自家和合性に変換することが可能となる。これにより、効率的な雑種種子採取ができる。
【0033】
またステロイドホルモン誘導性システムについては、以下のような報告(例えば、Zuo J et al、2006、Methods Mol Biol 323、329-342参照)がある。
(1)CRE DNAリコンビナーゼ遺伝子を誘導発現させることにより、2つのloxPサイトに挟まれた薬剤耐性遺伝子発現カセット等を切り出す。
(2)RNAiを誘発するRNAを誘導発現させることにより、所望の遺伝子の発現を抑制する。
(3)イントロンのドナーサイトとアクセプターサイトを含むmRNAを誘導発現させることにより、所望のcDNAを単離したり、無作為にcDNAを単離する。
(4)第一発現カセットの5’−非翻訳領域下流に遺伝子やターミネーターを配置せず、偶発的に5’−非翻訳領域下流に組み込まれた染色体上の遺伝子を誘導発現させることにより、機能的な遺伝子を単離する。
以上、同様なコンストラクトを作製することにより、これらの技術の全てに対して、本発明誘導発現方法を適用させることも可能である。
【0034】
外被蛋白質をコードする領域を目的とする外来蛋白質に係る構造遺伝子領域に置換したブロモモザイクウイルスcDNAやトマトモザイクウイルスcDNAをステロイドホルモンにより誘導発現させ、外来蛋白質を高いレベルで生産したという報告がある(例えば、特開2005-102652; Mori M et al、2001、Plant Journal 27(1)、79-86; Dohi K et al、2006、Arc
h Virol 151、1075-1084参照)。本発明誘導発現方法における目的とする外来遺伝子が、外被蛋白質等の構造遺伝子領域を置換したウイルスcDNAの場合においても、置換した構造遺伝子を高いレベルで転写誘導することが可能となる。
【0035】
本発明は上述した各実施形態に限定されるものではなく、請求項に示した範囲で種々の変更が可能であり、異なる実施形態に夫々開示された技術的手段を適宜組み合わせて得られる実施形態についても本発明の技術的範囲に含まれる。
【実施例】
【0036】
以下に実施例により本発明を詳細に説明するが、本発明はこれらに限定されるものではない。
【0037】
実施例1 (導入ベクターの作製)
(1)転写因子遺伝子発現カセットの構築
YPD培地(1% 酵母エキス、2% ポリペプトン、2% グルコース)中、30℃で2日間振とう培養した出芽酵母(Saccharomyces cerevisiae strain AH22)から、ゲノムDNA抽出キット「Genとるくん」(タカラバイオ)を用いてゲノムDNAを抽出した。抽出されたゲノムDNAを鋳型に、2種の特異的プライマー(ACE1-1F、ACE1-1RC)を用いてPCRを行うことにより、ACE1転写因子遺伝子を増幅した。増幅されたACE1転写因子遺伝子を、pBI221(Clontech)のGUS遺伝子と置換し、p35S-ACE1-NOSを作製した。次に、p35S-ACE1-NOSに含まれるNOSターミネーターを、CR16ターミネーター(特開2000-166577)に置換し、p35S-ACE1-CRを作製した。
【0038】
NASC(Nottingham Arabidopsis Stock Centre)より購入した組換えシロイヌナズナ種子(No.N70016)を改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種した。23℃で3週間生育させた組換えシロイヌナズナの本葉から、植物ゲノムDNA抽出キット「DNeasy Plant mini kit」(QIAGEN)を用いてゲノムDNAを抽出した。
抽出されたゲノムDNAを鋳型に、2種の特異的プライマー(VP16-1F、VP16-1RC)を用いてPCRを行うことにより、単純ヘルペスウイルスのVP16転写因子の転写活性化ドメイン(VP16AD)遺伝子を増幅した。増幅されたVP16AD遺伝子を、pCR2.1(Invitrogen)にTAクローニングし、pCR2.1-VP16ADを作製した。pCR2.1-VP16ADを鋳型に、2種の特異的プライマー(VP16-2F、VP16-2RC)を用いてPCRを行い、VP16AD遺伝子内のSacIサイトに変異を導入し、pCR2.1-VP16AD(dSacI)を作製した。その後、pCR2.1-VP16AD(dSacI)を鋳型に、2種の特異的プライマー(VP16-3F、VP16-3RC)を用いてPCRを行うことにより、VP16AD(dSacI)遺伝子の5’末端にXhoIサイトを付加し3’末端にSacIサイトを付加した。
【0039】
p35S-ACE1-CRを鋳型に、2種の特異的プライマー(ACE1-1F、ACE1-2RC)を用いてPCRを行うことにより、ACE1転写因子遺伝子の終始コドンを除去しXhoIサイトを付加した。
3’末端にXhoIサイトを付加したACE1転写因子遺伝子の下流に、VP16AD(dSacI)遺伝子を読み枠が合うように連結し、p35S-ACE1/VP16AD-CRを作製した。
【0040】
ACE1-1F:5'-atggatccatggtcgtaattaacggg-3'(配列番号1)
ACE1-1RC:5'-tggagctcttattgtgaatgtgagttatg-3'(配列番号2)
ACE1-2RC:5'-aactcgagttgtgaatgtgagttatgcg-3’(配列番号3)
VP16-1F:5'-acggctccaccgaccgacgtc-3'(配列番号4)
VP16-1RC:5'-ctacccaccgtactcgtcaattc-3'(配列番号5)
VP16-2F:5'-ggacgaactccacttagacgg-3'(配列番号6)
VP16-2RC:5'-ccgtctaagtggagttcgtcc-3'(配列番号7)
VP16-3F:5'-tactcgagtcaacggctccaccgaccgacgt-3'(配列番号8)
VP16-3RC:5'-aagagctcttacccaccgtactcgtcaattccaag-3'(配列番号9)
【0041】
(2)sGFP遺伝子発現カセットの構築
プラスミドCaMV35S-sGFP(S65T)-NOS3'(「植物の細胞を観る実験プロトコール」 福田裕穂他監修、1997年、秀潤社、ISBN 4-87962-170-6)からsGFP遺伝子を切り出し、アダプター(NS-1F、NS-1RC)を使用して、pBI221のGUS遺伝子と置換、p35S-sGFPを作製した。p35S-sGFPに含まれるCaMV35Sプロモーターの-830bpから-91bpまでの領域を、合成オリゴヌクレオチド(MRE-1F、MRE-1RC)に置換し、pMRE/35S(-90)-sGFPを作製した。
pMRE/35S(-90)-sGFPを制限酵素EcoRVで処理した後、「Calf intestine Alkaline Phosphatase」(タカラバイオ)で脱リン酸化した。そこへ、「Blunting Kination Ligation kit」(タカラバイオ)を用いて平滑末端化及びリン酸化した合成オリゴヌクレオチド(MRE-1F、MRE-1RC)を挿入することで、MRE配列を順向きに2回繰返して配置させた。得られたMRE配列の2回繰返し部分を切り出し、上記と同様の方法で平滑末端化し、再度EcoRVサイトに挿入した。これにより、MRE配列を順向きに4回繰返して配置させたpMRE4/35S(-90)-sGFPを作製した。
さらに、pBI221を鋳型に、2種の特異的プライマー(46bp-1F、46bp-1RC)を用いてPCRを行うことにより、CaMV35Sプロモーターの-46bpから-1bpまでの領域を含むDNA断片を増幅した。増幅されたDNA断片を、pMRE4/35S(-90)-sGFPの4回繰返し配置させたMRE配列の下流に位置するCaMV35Sプロモーターの-90bpから-1bpまでの領域と置換し、pMRE4/35S(-46)-sGFPを作製した。
他方では、CaMV35Sプロモーターの-90bpから-1bpまでの領域に存在するas-1サイト(非特許文献1)に変異が導入された配列とも置換した。
まず、NASCより購入した組換えシロイヌナズナ(No.N70016)からゲノムDNAを抽出した。抽出されたゲノムDNAを鋳型に、2種の特異的プライマー(90m-1F、90m-1RC)を用いてPCRを行うことにより、as-1サイトに変異を持つ35S(-90m)配列をpCR2.1(Invitrogen)にTAクローニングし、pCR2.1-35S(-90m)を作製した。pCR2.1-35S(-90m)を鋳型に、2種の特異的プライマー(90m-2F、90m-2RC)を用いてPCRを行うことにより、5’末端にEcoRVサイトを付加し3’末端にXbaIサイトを付加した。得られたDNA断片を、pMRE4/35S(-90)-sGFPの4回繰返し配置させたMRE配列の下流に位置するCaMV35Sプロモーターの-90bpから-1bpまでの領域と置換し、pMRE4/35S(-90m)-sGFPを作製した。
加えて、プラスミドpiL.erG3(Tamai A et al、2001、Mol Plant Microbe Interact 14(2)、126-134)を鋳型に、2種の特異的プライマー(71bp-1F、71bp-1RC)を用いてPCRを行うことにより、トマトモザイクウイルスの130k/180k遺伝子の5’−非翻訳領域配列(To71配列)を増幅した。増幅された5’−非翻訳領域配列をpMRE4/35S(-46)-sGFP及びpMRE4/35S(-90m)-sGFPのsGFP遺伝子の上流に挿入し、pMRE4/35S(-46)-To71sGFP及びpMRE4/35S(-90m)-To71sGFPを作製した。
【0042】
NS-1F:5'-ggccgcgagctcagt-3'(配列番号10)
NS-1RC:5'-gactgagctcgc-3'(配列番号11)
MRE-1F:5'-agcttagcgatgcgtcttttccgctgaaccgttccagcaaaaaagactagat-3'(配列番号12)
MRE-1RC:5'-atctagtcttttttgctggaacggttcagcggaaaagacgcatcgcta-3'(配列番号13)46bp-1F:5'-tagatatcgcaagacccttcctctatataagg-3'(配列番号14)
46bp-1RC:5'-atcctctagagtcccccgtgttc-3'(配列番号15)
90m-1F:5'-gctatgaccatgattacgccaagcttg-3'(配列番号16)
90m-1RC:5'-cattgttatatctccttggatccgtcg-3'(配列番号17)
90m-2F:5'-tagatatctccacgtccataagggac-3'(配列番号18)
90m-2RC:5'-aatctagactgcaggtcgtcctctcca-3'(配列番号19)
71bp-1F:5'-tgtctagagtatttttacaacaattaccaacaac-3'(配列番号20)
71bp-1RC:5'-aaggatcctgtagttgtagaatgtaaaatgtaatg-3'(配列番号21)
【0043】
(3)転写因子遺伝子発現カセットとsGFP遺伝子発現カセットとの連結
転写因子遺伝子発現カセットを含むp35S-ACE1-CR及びp35S-ACE1/VP16AD-CRを制限酵素HindIII及びEcoRIで処理し、夫々転写因子遺伝子発現カセットを切り出した。
sGFP遺伝子発現カセットを含むpMRE/35S(-90)-sGFP及びpMRE4/35S(-46)-sGFP、pMRE4/35S(-46)-To71sGFP、pMRE4/35S(-90m)-To71sGFPを制限酵素HindIIIで処理した後、「Blunting Kination Ligation kit」を用いて平滑末端化及びリン酸化し、そこへ合成オリゴヌクレオチド(KXS-1F、KXS-1RC)を挿入、pKXS-MRE/35S(-90)-sGFP及びpKXS-MRE4/35S(-46)-sGFP、pKXS-MRE4/35S(-46)-To71sGFP、pKXS-MRE4/35S(-90m)-To71sGFPを作製した。作製したこれらのプラスミドを制限酵素KpnI及びEcoRIで処理し、夫々sGFP遺伝子発現カセットを切り出した。
一方、pBI121(Clontech)に含まれるGUS遺伝子発現カセットを、合成オリゴヌクレオチド(HEK-1F、HEK-1RC)に置換し、pBI121-HEKを作製した。pBI121-HEKを制限酵素HindIII及びKpnIで処理した。
制限酵素HindIII及びKpnIで処理したpBI121-HEKに、切り出した転写因子遺伝子発現カセットとsGFP遺伝子発現カセットをライゲーションすることにより、転写因子遺伝子発現カセットとsGFP遺伝子発現カセットとが、夫々のターミネーター側で連結したベクターを得た(図1参照)。p35S-ACE1-CRとpKXS-MRE/35S(-90)-sGFPを由来とするものをベクターA、p35S-ACE1-CRとpKXS-MRE4/35S(-46)-sGFPを由来とするものをベクターB、p35S-ACE1/VP16AD-CRとpKXS-MRE4/35S(-46)-sGFPを由来とするものをベクターC、p35S-ACE1/VP16AD-CRとpKXS-MRE4/35S(-46)-To71sGFPを由来とするものをベクターD、p35S-ACE1/VP16AD-CRとpKXS-MRE4/35S(-90m)-To71sGFPを由来とするものをベクターEとした。
【0044】
KXS-1F:5'-ggtacctcgagtcgac-3'(配列番号22)
KXS-1RC:5'-gtcgactcgaggtacc-3'(配列番号23)
HEK-1F:5'-agcttgaattcgtcgacggtacctaggacgagctc-3'(配列番号24)
HEK-1RC:5'-aattgagctcgtcctaggtaccgtcgacgaattca-3'(配列番号25)
【0045】
実施例2 (組換えシロイヌナズナにおけるsGFP遺伝子の発現量解析)
(1)組換えシロイヌナズナの作製及び選抜
実施例1で作製されたベクターAからEをそれぞれアグロバクテリウム(Agrobacterium tumefaciens strain C58C1)に導入した。アグロバクテリウムを50 mg/L カナマイシン、100 mg/L アンピシリン、100 mg/L リファンピシンを含むLB寒天培地(0.5% 酵母エキス、1.0% バクトトリプトン、0.5% 食塩、1% 寒天)で培養して薬剤耐性コロニーを選抜することにより、組換えアグロバクテリウムを得た。得られた組換えアグロバクテリウムを、モデル植物ラボマニュアル(岩渕雅樹他編集、2000年、シュプリンガー・フェアラーク東京株式会社、ISBN 4-431-70881-2 C3045)に記載される方法に準じて、シロイヌナズナ(Arabidopsis thaliana ecotype Columbia)に感染させることにより、遺伝子導入を行った。遺伝子導入されたシロイヌナズナから採取されたT1種子を、20 mg/L ベンレート、200 mg/L クラフォラン、25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、1% ショ糖、0.8 % 寒天)に播種し生育した後、カナマイシンに耐性を示す植物個体を選抜した。選抜された植物個体を予め培土が入れられたポットに移植し、人工気象器内で生育させることにより、T2種子を得た。得られたT2種子を25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種し生育した後、χ2検定に基づいた5%の有意水準において、カナマイシンに耐性を示す植物個体が3:1の割合で出現するラインを選抜した。尚、植物個体の生育のための培養条件は、明期23時間、暗期1時間、23〜25℃とした。
【0046】
(2)リアルタイムPCRによるsGFP遺伝子の発現量解析
選抜されたラインについて、播種後10日目の植物個体を、誘導発現処理区として100μM CuSO4を含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8 % 寒天)に6個体ずつ移植することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った(尚、非誘導発現処理区として CuSO4を含まない改変MS寒天培地を用いた。)。
その後、当該誘導発現処理6時間後の植物個体から、植物RNA抽出キット「RNeasy Plant Mini Kit」(QIAGEN)を用いて全RNAを抽出した。抽出された全RNAから、cDNA合成キット「ReverTra Ace」(TOYOBO)を用いてcDNAを合成した。合成されたcDNAを鋳型に、7500 Fast Real-time PCR装置(Applied Biosystems)を用いてリアルタイムPCRを行うことにより、mRNA量の定量を行った。sGFP遺伝子のmRNA量の定量には、2種の特異的プライマー(S01F、S01R)及びTaqManプローブ(S01)を用いた。内部標準としては、シロイヌナズナアクチン遺伝子(AtACT2、GenBank Accession Number NM180280)を用いた。AtACT遺伝子のmRNA量の定量には、2種の特異的プライマー(S03F、S03R)及びTaqManプローブ(S03)を用いた。
【0047】
S01F:5'-tccgccctgagcaaagac-3'(配列番号26)
S01R:5'-gaactccagcaggaccatgtg-3'(配列番号27)
S01:5'-FAM-ccaacgagaagcgcga-MGB-3'(配列番号28)
S03F:5'-cggtggttccattcttgctt-3'(配列番号29)
S03R:5'-cggccttggagatccacat-3'(配列番号30)
S03:5'-VIC-cctcagcacattcc-MGB-3'(配列番号31)
【0048】
その結果、ベクターAが導入されてなる組換えシロイヌナズナでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して4.6倍(以下、ベクターAが導入されてなる組換えシロイヌナズナにおける誘導倍率と記すこともある。)であった。MRE配列が4回繰返し配置され且つCaMV35Sプロモーターが-90bpから-46bpまでの領域に短縮されたベクターBが導入されてなる組換えシロイヌナズナでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して8.6倍(以下、ベクターBが導入されてなる組換えシロイヌナズナにおける誘導倍率と記すこともある。)であり、またベクターAが導入されてなる組換えシロイヌナズナにおける誘導倍率と比較して1.9倍の改善が認められた。
一方、ACE1転写因子にVP16ADが付加されたベクターCが導入されてなる組換えシロイヌナズナでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して273.0倍(以下、ベクターCが導入されてなる組換えシロイヌナズナにおける誘導倍率と記すこともある。)であり、またベクターBが導入されてなる組換えシロイヌナズナにおける誘導倍率と比較して31.7倍の改善が認められた。
さらに、トマトモザイクウイルスの130k/180k遺伝子の5’−非翻訳領域配列が挿入されたベクターDが導入されてなる組換えシロイヌナズナでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して628.6倍(以下、ベクターDが導入されてなる組換えシロイヌナズナにおける誘導倍率と記すこともある。)であり、またベクターCが導入されてなる組換えシロイヌナズナにおける誘導倍率と比較して2.3倍の改善が認められた。
また、as-1サイトに変異を持つ35S(-90m)配列が用いられたベクターEが導入されてなる組換えシロイヌナズナでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して325.6倍(以下、ベクターEが導入されてなる組換えシロイヌナズナにおける誘導倍率と記すこともある。)であった(図2参照)。
【0049】
実施例3 (組換えタバコにおけるsGFP遺伝子の発現量解析)
(1)組換えタバコの作製及び選抜
実施例1で作製されたベクターB及びEをそれぞれアグロバクテリウム(Agrobacterium tumefaciens strain LBA4404)に導入した。アグロバクテリウムを50 mg/L カナマイシン、300 mg/L ストレプトマイシン、100 mg/L リファンピシンを含むLB寒天培地(0.5% 酵母エキス、1.0% バクトトリプトン、0.5% 食塩、1% 寒天)で培養して薬剤耐性コロニーを選抜することにより、組換えアグロバクテリウムを得た。得られた組換えアグロバクテリウムを、植物遺伝子操作マニュアル(内宮博文著、1992年、講談社サイエンティフィック)に記載される方法に準じて、タバコ(Nicotiana tabacum strain SR1)に感染させることにより、遺伝子導入を行った。遺伝子導入されたタバコの葉片から100 mg/L カナマイシンに耐性を示す不定芽を選抜し、選抜された不定芽から植物個体を再生させた。得られた植物個体から採取されたT1種子を50 mg/L カナマイシンを含むMS寒天培地(MS無機塩類、MSビタミン、3% ショ糖、0.8% 寒天)に播種し生育した後、χ2検定に基づいた5%の有意水準において、カナマイシンに耐性を示す植物個体が3:1の割合で出現するラインを選抜した。尚、植物個体の生育のための培養条件は、明期23時間、暗期1時間、23〜25℃とした。
【0050】
(2)リアルタイムPCRによるsGFP遺伝子の発現量解析
選抜されたラインについて、播種後12日目の植物個体を、誘導発現処理区として100μM CuSO4を含むMS寒天培地(MS無機塩類、MSビタミン、3% ショ糖、0.8% 寒天)に3個体ずつ移植することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った(尚、非誘導発現処理区として CuSO4を含まないMS寒天培地を用いた。)。
その後、当該誘導発現処理6時間後の植物個体から、植物RNA抽出キット「RNeasy Plant Mini Kit」(QIAGEN)を用いて全RNAを抽出した。抽出された全RNAから、cDNA合成キット「ReverTra Ace」(TOYOBO)を用いてcDNAを合成した。合成されたcDNAを鋳型に、7500 Fast Real-time PCR装置(Applied Biosystems)を用いてリアルタイムPCRを行うことにより、mRNA量の定量を行った。sGFP遺伝子のmRNA量の定量には、2種の特異的プライマー(S01F、S01R)及びTaqManプローブ(S01)を用いた。内部標準としては、タバコユビキチン遺伝子(NtUBI、GenBank Accession Number U66264)を用いた。NtUBI遺伝子のmRNA量の定量には、2種の特異的プライマー(S06F、S06R)及びTaqManプローブ(S06)を用いた。
【0051】
S06F:5'-gaagcagctcgaggatggaa-3'(配列番号32)
S06R:5'-gacgggttgactctttctggat-3'(配列番号33)
S06:5'-VIC-accttggctgactacaa-MGB-3'(配列番号34)
【0052】
その結果、ベクターBが導入されてなる組換えタバコでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して9.6倍(以下、ベクターBが導入されてなる組換えタバコにおける誘導倍率と記すこともある。)であった。
一方、ACE1転写因子にVP16ADが付加され且つsGFP遺伝子の上流にトマトモザイクウイルスの130k/180k遺伝子の5’−非翻訳領域配列が挿入されたベクターEが導入されてなる組換えタバコでは、誘導発現処理区でのsGFP遺伝子のmRNA量が非誘導発現処理区でのsGFP遺伝子のmRNA量と比較して40.8倍(以下、ベクターEが導入されてなる組換えタバコにおける誘導倍率と記すこともある。)であり、またベクターBが導入されてなる組換えタバコにおける誘導倍率と比較して4.2倍の改善が認められた(図3参照)。
【0053】
実施例4 (組換えタバコ培養細胞におけるGFPの蓄積量解析)
(1)組換えタバコ培養細胞の作製及び選抜
実施例1で作製されたベクターA及びB、Eがコーティングされた直径1.0μmの金粒子を用いたパーティクルガン法(森川弘道ら著、1992年、植物細胞工学 Vol.4 No.1 p.47-52、秀潤社)により、前記ベクターをそれぞれタバコ培養細胞(BY-2)に導入した。金粒子1mg当たりのDNA量は0.1μgとした。遺伝子導入されたタバコ培養細胞の細胞懸濁液を、遺伝子導入操作後3〜5日目に、30 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、3% ショ糖、1μM 2,4-D、1 mg/L thiamin・HCl、100 mg/L myo-inositol、200 mg/L KH2PO4、0.8 % 寒天)に広げた。培養1ヵ月間後に、30 mg/L カナマイシンに耐性を示す細胞塊を選抜し、選抜された細胞塊をその後1〜2週間毎に新しい寒天培地に植え継ぎながら4〜8週間培養した。培養条件は、暗下、23〜25℃とした。
【0054】
(2)蛍光プレートリーダーによるGFPの蓄積量解析
得られた細胞塊を、誘導発現処理区として100μM CuSO4を含む改変MS寒天培地(MS無機塩類、3% ショ糖、1μM 2,4-D、1 mg/L thiamin・HCl、100 mg/L myo-inositol、200 mg/L KH2PO4、0.8 % 寒天)に移植することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った(尚、非誘導発現処理区として CuSO4を含まない改変MS寒天培地を用いた。)。
その後、当該誘導発現処理3日間後の細胞塊について、蛍光顕微鏡(Nikon)にて、GFPの蛍光発光状態を調べた。
【0055】
GFPの蛍光発光の増大が観察された細胞塊を液体窒素で凍結した後、当該凍結物にガラスビーズ(0.25〜0.5 mm)及び抽出バッファー(1×PBS(-)、5 mM DTT、1 mM PMSF、0.1 % protease inhibitor cocktail)を加えて、破砕装置「ミキサーミル」(QIAGEN)にて破砕処理した。得られた破砕物を卓上遠心機にて15000 rpm、4℃で5分間遠心することにより上清を回収した後、得られた上清を蛋白質抽出液とした。次いで、当該蛋白質抽出液を適宜、1×PBS(-) で希釈し、得られた希釈液におけるGFPの蛍光強度を、マルチラベルカウンタ「wallac 1420 ARVOMX」(パーキンエルマー)を用いて測定した。尚、標準サンプルとしては、リコンビナントGFP(コスモバイオ)を1×PBS(-)で希釈して得られる希釈液を用いた。測定された蛍光強度から可溶性蛋白質量当たりのGFP換算量を算出し、対照となる野生型の細胞塊での対応値をバックグランドとして差し引くことにより、GFPの蓄積量を算出した。尚、蛋白質抽出液の中の蛋白質濃度はBradford法により定量した。
【0056】
その結果、ベクターAが導入されてなる組換えタバコ培養細胞では、誘導発現処理区でのGFPの蓄積量が非誘導発現処理区でのGFPの蓄積量と比較して1.6倍(以下、ベクターAが導入されてなる組換えタバコ培養細胞における誘導倍率と記すこともある。)であった。MRE配列が4回繰返し配置され且つCaMV35Sプロモーターが-90bpから-46bpまでの領域に短縮されたベクターBが導入されてなる組換えタバコ培養細胞では、誘導発現処理区でのGFPの蓄積量が非誘導発現処理区でのGFPの蓄積量と比較して1.9倍(以下、ベクターBが導入されてなる組換えタバコ培養細胞における誘導倍率と記すこともある。)であった。
一方、ACE1転写因子にVP16ADが付加され且つsGFP遺伝子の上流にトマトモザイクウイルスの130k/180k遺伝子の5’−非翻訳領域配列が挿入されたベクターEが導入されてなる組換えタバコ培養細胞では、誘導発現処理区でのGFPの蓄積量が非誘導発現処理区でのGFPの蓄積量と比較して21.9倍(以下、ベクターEが導入されてなる組換えタバコ培養細胞における誘導倍率と記すこともある。)であり、またベクターBが導入されてなる組換えタバコ培養細胞における誘導倍率と比較して11.5倍の改善が認められた(図4参照)。
【0057】
実施例5 (誘導発現システムのトランジェントアッセイ)
(1)試料の準備及びパーティクルガンによる遺伝子導入
シロイヌナズナ(Arabidopsis thaliana)、タバコ(Nicotiana tabacum)、ダイズ(Glycine max 'Jack')、カーネーション(Dianthus caryophyllus 'True Love'、キリン グリーン アンド フラワー)、イネ(Oryza sativa ssp. japonica cv. Nipponbare)、オニユリ(Liliunm lancifolium)の夫々の葉を切り取り、切り取られた葉(葉片)を誘導発現処理区として100μM CuSO4を含む0.6% 寒天培地上に静置することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った(尚、非誘導発現処理区として CuSO4を含まない寒天培地を用いた。
)。
その後、当該誘導発現処理1日間後の葉片に、実施例1で作製されたベクターEとpBI221(Clontech)とを等量混合してなる組成物がコーティングされた直径1.0μmの金粒子を用いたパーティクルガン法により、前記組成物を導入した。金粒子1mg当たりのDNA量は0.1μgとした。pBI221は、遺伝子導入効率を補正する目的で混合導入した。対照として、前記組成物に代えて実施例1で作製されたp35S-sGFPとpBI221とを等量混合してなる組成物が用いられた。
【0058】
(2)レポーター遺伝子のトランジェントな発現解析
遺伝子導入された葉片について、遺伝子導入後1日目に、蛍光顕微鏡(Nikon)にてGFPの蛍光発光が観察されるスポット数をカウントした。その後、遺伝子導入された葉片をGUS染色液(0.5 mg/ml X-Gluc、0.5 mM K3Fe(CN)6、0.5 mM K4Fe(CN)6、0.01% Triton X-100、100 mM リン酸ナトリウム)に浸し、これを400 mmHgで10分間、減圧処理した後、37℃で1日間、染色した。染色された葉片について、70% エタノールで脱色処理した後、光学顕微鏡(Nikon)にてGUS染色が観察されるスポット数をカウントした。
【0059】
その結果、p35S-sGFPが導入されてなる葉片では、誘導発現処理区及び非誘導発現処理区におけるGFP蛍光によるスポット数に有意な差は認められなかった。一方、ベクターEが導入されてなる葉片では、誘導発現処理区におけるGFP蛍光によるスポット数が、非誘導発現処理区におけるGFP蛍光によるスポット数と比較して、多く認められた。尚、GUS染色によるスポット数はいずれの処理区においても安定的に観察され、遺伝子導入効率に大きな差異が存在しないことが確認された(表1参照)。
【0060】
【表1】
【0061】
実施例6 (組換えシロイヌナズナの開花誘導)
(1)導入ベクターの作製
シロイヌナズナ(Arabidopsis thaliana ecotype Columbia)を改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種し、23℃で3週間生育させた。得られた植物を、予め培土が入れられたポットに移植し、人工気象器内で生育させた。移植後2週間目の植物の花芽及び本葉から、植物RNA抽出キット「RNeasy Plant Mini Kit」(QIAGEN)を用いて全RNAを抽出した。抽出された全RNAから、cDNA合成キット「ReverTra Ace」(TOYOBO)を用いてcDNAを合成した。合成されたcDNAを鋳型に、2種の特異的プライマー(FT-1F、FT-1RC)を用いてPCRを行うことにより、FT遺伝子(GenBank Accession Number AB027504)を増幅した。増幅されたFT遺伝子を、pBI221(Clontech)のGUS遺伝子と置換した。このようにして得られたプラスミドを鋳型に、6種の特異的プライマー(FT-2F、FT-2RC、FT-3F、FT-3RC、FT-4F、FT-1RC)を用いてfusion PCRを行うことにより、FT遺伝子内部に存在するBamHIサイト及びEcoRIサイトにアミノ酸置換を伴わない変異を導入し且つ5’末端のXbaIサイトをBamHIサイトに変更した。このようにして得られた改変FT遺伝子を、実施例1で作製されたpKXS-MRE4/35S(-90m)-To71sGFPのsGFP遺伝子と置換することにより、pKXS-MRE4/35S(-90m)-To71FTを作製した。pKXS-MRE4/35S(-90m)-To71FTに含まれるFT遺伝子発現カセットを実施例1に記載された方法と同様の方法に準じて、p35S-ACE1/VP16AD-CRに含まれる転写因子遺伝子発現カセットとターミネーター側で連結したベクターFを得た(図5参照)。
【0062】
FT-1F:5'-taatctagaatgtctataaatataagagaccctc-3'(配列番号35)
FT-1RC:5'-atagagctcctaaagtcttcttcctccg-3'(配列番号36)
FT-2F:5'-taaggatccatgtctataaatataagagaccctc-3'(配列番号37)
FT-2RC:5'-gaacatctggatcgaccataaccaaagta-3'(配列番号38)
FT-3F:5'-tactttggttatggtcgatccagatgttc-3'(配列番号39)
FT-3RC:5'-gacacgatgaatacctgcagtggga-3'(配列番号40)
FT-4F:5'-tcccactgcaggtattcatcgtgtc-3'(配列番号41)
【0063】
(2)組換えシロイヌナズナの作製と選抜及び開花誘導
作製されたベクターFを、アグロバクテリウム(Agrobacterium tumefaciens strain C58C1)に導入した。アグロバクテリウムを50 mg/L カナマイシン、100 mg/L アンピシリン、100 mg/L リファンピシンを含むLB寒天培地(0.5% 酵母エキス、1.0% バクトトリプトン、0.5% 食塩、1% 寒天)で培養して薬剤耐性コロニーを選抜することにより、組換えアグロバクテリウムを得た。得られた組換えアグロバクテリウムを、モデル植物ラボマニュアル(岩渕雅樹他編集、2000年、シュプリンガー・フェアラーク東京株式会社、ISBN 4-431-70881-2 C3045)に記載される方法に準じて、シロイヌナズナ(Arabidopsis thalia
na ecotype Columbia)に感染させることにより、遺伝子導入を行った。遺伝子導入されたシロイヌナズナから採取されたT1種子を、20 mg/L ベンレート、200 mg/L クラフォラン、25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、1% ショ糖、0.8 % 寒天)に播種し生育した後、カナマイシンに耐性を示す植物個体を選抜した。選抜された植物個体を予め培土が入れられたポットに移植し、人工気象器内で生育させることにより、T2種子を得た。得られたT2種子を25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種し生育した後、χ2検定に基づいた5%の有意水準において、カナマイシンに耐性を示す植物個体が3:1の割合で出現するラインを選抜した。選抜されたラインの植物数個体を予め培土が入れられたポットに移植し、人工気象器内で生育させることにより、T3種子を得た。得られたT3種子を、25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種し生育した後、全てがカナマイシンに耐性を示すホモ接合体を選抜した。尚、植物個体の生育のための培養条件は、明期23時間、暗期1時間、23〜25℃とした。
選抜されたホモ接合体について、T3種子を、改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8% 寒天)に播種した。明期23時間、暗期1時間、23〜25℃の条件下で3日間生育させた後、明期12時間、暗期12時間、23〜25℃の条件下で生育させた。播種後10日経過した植物個体を誘導発現処理区として100μM CuSO4を含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8 % 寒天)に移植することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った(尚、非誘導発現処理区として CuSO4を含まない改変MS寒天培地を用いた。)。
【0064】
当該誘導発現処理3日後(播種後13日目)の植物個体を、CuSO4を含まない改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8 % 寒天)に移植した。当該誘導発現処理後11日目(播種後24日目)の植物個体を観察した。その結果、誘導発現処理区では、非誘導発現処理区と比較して、早期に開花が誘導された(図6参照)。
【0065】
実施例7 (組換えタバコの除草剤耐性誘導)
(1)導入ベクターの作製
pSUM-35S-rSt-1609soy(特開2006-001921)から、制限酵素BamHI及びSacIを用いて、rSt-1609soy遺伝子を切り出した。切り出しされたrSt-1609soy遺伝子を、実施例1で作製されたpKXS-MRE4/35S(-90m)-To71sGFPのsGFP遺伝子と置換することにより、pKXS-MRE4/35S(-90m)-To71rSt-1609soyを作製した。pKXS-MRE4/35S(-90m)-To71rSt-1609soyに含まれるrS
t-1609soy遺伝子発現カセットを実施例1に記載された方法と同様の方法に準じて、p35S-ACE1/VP16AD-CRに含まれる転写因子遺伝子発現カセットとターミネーター側で連結したベクターGを得た(図5参照)。
【0066】
(2)組換えタバコの作製と選抜及び除草剤耐性誘導
作製されたベクターGをアグロバクテリウム(Agrobacterium tumefaciens strain LBA4404)に導入した。アグロバクテリウムを50 mg/L カナマイシン、300 mg/L ストレプトマイシン、100 mg/L リファンピシンを含むLB寒天培地(0.5% 酵母エキス、1.0% バクトトリプトン、0.5% 食塩、1% 寒天)で培養して薬剤耐性コロニーを選抜することにより、組換えアグロバクテリウムを得た。得られた組換えアグロバクテリウムを、植物遺伝子操作マニュアル(内宮博文著、1992年、講談社サイエンティフィック)に記載される方法に準じて、タバコ(Nicotiana tabacum strain SR1)に感染させることにより、遺伝子導入を行った。遺伝子導入されたタバコの葉片から100 mg/L カナマイシンに耐性を示す不定芽を選抜し、選抜された不定芽から植物個体を再生させた。得られた植物個体から採取されたT1種子を50 mg/L カナマイシンを含むMS寒天培地(MS無機塩類、MSビタミン、3% ショ糖、0.8% 寒天)に播種し生育した後、χ2検定に基づいた5%の有意水準において、カナマイシンに耐性を示す植物個体が3:1の割合で出現するラインを選抜した。尚、植物個体の生育のための培養条件は、明期23時間、暗期1時間、23〜25℃とした。
選抜されたラインについて、播種後数日経過した植物個体を、予め培土が入れられたポットに移植した後、当該植物個体に、誘導発現処理区としてCuSO4を含む水溶液を散布する(尚、非誘導発現処理区として CuSO4を含まない水溶液を用いた。)。さらに、前記植物個体に、PPO阻害型除草剤を散布する。
誘導発現処理区では、非誘導発現処理区と比較して、より高濃度の除草剤に耐性を示す結果となる。
【0067】
実施例8 (他の5’−非翻訳領域配列の利用)
(1)導入ベクターの作製
実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPを鋳型に2種の特異的プライマー(EV46-1F、o46-1RC)を用いてPCRを行うことにより、46o断片を増幅した。また、プラスミドCaMV35S-sGFP(S65T)-NOS3'(「植物の細胞を観る実験プロトコール」 福田裕穂他監修、1997年、秀潤社、ISBN 4-87962-170-6)を鋳型に2種の特異的プライマー(omg-1F、omg-1RC)を用いてPCRを行うことにより、Ω配列断片を増幅した。さらに、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPを鋳型に2種の特異的プライマー(oGFP-1F、SIGFP-1RC)を用いてPCRを行うことにより、oGFP断片を増幅した。得られた3種のDNA断片(46o、Ω配列、oGFP)の混合物を鋳型に、2種の特異的プライマー(EV46-1F、SIGFP-1RC)を用いてPCRを行うことにより、ΩsGFP断片を増幅した。得られたΩsGFP断片を、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPのEcoRV及びSacIサイトで切り出される断片と置換することにより、pKXS-MRE4/35S(-46)-ΩsGFPを作製した。作製されたpKXS-MRE4/35S(-46)-ΩsGFPを鋳型に2種の特異的プライマー(Xo-1F、SIGFP-1RC)を用いてPCRを行うことにより、xΩsGFP断片を増幅した。得られたxΩsGFP断片を、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPのXbaI及びSacIサイトで切り出される断片と置換することにより、pKXS-MRE4/35S(-46)-xΩsGFPを作製した。
【0068】
実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPを鋳型に2種の特異的プライマー(EV46-1F、a46-1RC)を用いてPCRを行うことにより、46a断片を増幅した。また、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPを鋳型に2種の特異的プライマー(aGFP-1F、SIGFP-1RC)を用いてPCRを行うことにより、aGFP断片を増幅した。得られた2種のDNA断片(46a、aGFP)と合成オリゴヌクレオチド(A94-1F、A94-1RC)を混合した。この混合物を鋳型に、2種の特異的プライマー(EV46-1F、SIGFP-1RC)を用いてPCRを行うことにより、タバコのアルコールデヒドロゲナーゼ遺伝子由来の5'−非翻訳領域配列を有するA94sGFP断片を増幅した。得られたA94sGFP断片を、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPのEcoRV及びSacIサイトで切り出される断片と置換することにより、pKXS-MRE4/35S(-46)-A94sGFPを作製した。作製されたpKXS-MRE4/35S(-46)-A94sGFPを鋳型に2種の特異的プライマー(Xa-1F、SIGFP-1RC)を用いてPCRを行うことにより、xA94sGFP断片を増幅した。得られたxA94sGFP断片を、実施例1で作製されたpKXS-MRE4/35S(-46)-To71sGFPのXbaI及びSacIサイトで切り出される断片と置換することにより、pKXS-MRE4/35S(-46)-xA94sGFPを作製した。
作製された4種のsGFP遺伝子発現カセットを、実施例1に記載された方法と同様の方法に準じて、p35S-ACE1/VP16AD-CRに含まれる転写因子遺伝子発現カセットとターミネーター側で連結し、ベクターを得た(図7参照)。pKXS-MRE4/35S(-46)-ΩsGFPを由来とするものをベクターH、pKXS-MRE4/35S(-46)-xΩsGFPを由来とするものをベクターI、pKXS-MRE4/35S(-46)-A94sGFPを由来とするものをベクターJ、pKXS-MRE4/35S(-46)-xA94sGFPを由来とするものをベクターKとした。
【0069】
EV46-1F:5'-ctagatatcgcaagacccttcctct-3'(配列番号42)
o46-1RC:5'-gtaaaaataccctctccaaatgaaatgaacttc-3'(配列番号43)
omg-1F:5'-ggtatttttacaacaattaccaacaacaac-3'(配列番号44)
omg-1RC:5'-cattgtaattgtaaatagtaattgtaatgt-3'(配列番号45)
oGFP-1F:5'-acaattacaatggtgagcaagggcga-3'(配列番号46)
SIGFP-1RC:5'-ttagagctcttacttgtacagctcgtcc-3'(配列番号47)
Xo-1F:5'-gactctagagtatttttacaacaattaccaac-3'(配列番号48)
a46-1RC:5'-gttaaatagaccctctccaaatgaaatgaacttc-3'(配列番号49)
aGFP-1F:5'-gaaaaataaatggtgagcaagggcgag-3'(配列番号50)
A94-1F:5'-gtctatttaactcagtattcagaaacaacaaaagttcttctctacataaaattttcctattttagtgatcagtgaaggaaatcaagaaaaataaatg-3'(配列番号51)
A94-1RC:5'-catttatttttcttgatttccttcactgatcactaaaataggaaaattttatgtagagaagaacttttgttgtttctgaatactgagttaaatagac-3'(配列番号52)
Xa-1F:5'-actctagagtctatttaactcagtattcag-3'(配列番号53)
【0070】
(2)組換えシロイヌナズナの作製と選抜及び発現解析
作製されたベクターHからKをそれぞれアグロバクテリウム(Agrobacterium tumefaciens strain C58C1)に導入した。アグロバクテリウムを50 mg/L カナマイシン、100 mg/L アンピシリン、100 mg/L リファンピシンを含むLB寒天培地(0.5% 酵母エキス、1.0% バクトトリプトン、0.5% 食塩、1% 寒天)で培養して薬剤耐性コロニーを選抜することにより、組換えアグロバクテリウムを得た。得られた組換えアグロバクテリウムを、モデル植物ラボマニュアル(岩渕雅樹他編集、2000年、シュプリンガー・フェアラーク東京株式会社、ISBN 4-431-70881-2 C3045)に記載される方法に準じて、シロイヌナズナ(Arabidopsis thaliana ecotype Columbia)に感染させることにより、遺伝子導入を行った。遺伝子導入されたシロイヌナズナから採取されたT1種子を、20 mg/L ベンレート、200 mg/L クラフォラン、25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、1% ショ糖、0.8 % 寒天)に播種し11日間生育させた後、カナマイシンに耐性を示す植物個体を選抜した。選抜された植物個体を20 mg/L ベンレート、200 mg/L クラフォラン、25 mg/L カナマイシンを含む改変MS寒天培地(MS無機塩類、B5ビタミン、1% ショ糖、0.8 % 寒天)に移植し、さらに6日間生育させた。播種後17日経過した植物個体を100μM CuSO4を含む改変MS寒天培地(MS無機塩類、B5ビタミン、2% ショ糖、0.8 % 寒天)に移植することにより、銅イオンによる目的とする外来遺伝子の誘導発現のための処理(以下、誘導発現処理と記すこともある。)を行った。当該誘導発現処理1時間以内(0日目)に、植物個体におけるGFPの蛍光発光状態をマクロ蛍光顕微鏡VB-G05(キーエンス)にて観察し、高感度冷却CCDカメラVB-7010(キーエンス)を用いて蛍光画像を取得した。その後、当該誘導発現処理3日目に、同様の観察をし、蛍光画像を取得した。その結果、ベクターHからKが導入されてなる組換えシロイヌナズナは、誘導発現処理0日目ではGFP蛍光が観察されないのに対して、誘導発現処理3日目では強いGFP蛍光が観察された(図8参照)。
尚、植物個体の生育のための培養条件は、播種後11日目までは明期23時間、暗期1時間、23〜25℃とし、播種後11日目以降は明期12時間、暗期12時間、23〜25℃とした。
【産業上の利用可能性】
【0071】
本発明により、銅イオン誘導性システムについて、目的とする外来遺伝子の植物における誘導発現における誘導倍率の向上を達成できる方法等を提供することが可能となる。
【配列表フリーテキスト】
【0072】
配列番号1
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号2
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号3
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号4
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号5
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号6
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号7
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号8
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号9
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号10
アダプターのために設計されたオリゴヌクレオチド
配列番号11
アダプターのために設計されたオリゴヌクレオチド
配列番号12
置換DNA断片のために設計されたオリゴヌクレオチド
配列番号13
置換DNA断片のために設計されたオリゴヌクレオチド
配列番号14
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号15
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号16
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号17
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号18
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号19
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号20
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号21
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号22
挿入DNA断片のために設計されたオリゴヌクレオチド
配列番号23
挿入DNA断片のために設計されたオリゴヌクレオチド
配列番号24
置換DNA断片のために設計されたオリゴヌクレオチド
配列番号25
置換DNA断片のために設計されたオリゴヌクレオチド
配列番号26
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号27
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号28
TaqManプローブのために設計されたオリゴヌクレオチド
配列番号29
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号30
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号31
TaqManプローブのために設計されたオリゴヌクレオチド
配列番号32
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号33
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号34
TaqManプローブのために設計されたオリゴヌクレオチド
配列番号35
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号36
TaqManプローブのために設計されたオリゴヌクレオチド
配列番号37
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号38
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号39
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号40
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号41
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号42
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号43
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号44
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号45
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号46
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号47
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号48
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号49
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号50
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号51
PCRのために設計されたオリゴヌクレオチド
配列番号52
PCRのために設計されたオリゴヌクレオチド
配列番号53
PCRのために設計されたオリゴヌクレオチドプライマー
【特許請求の範囲】
【請求項1】
植物において、化学物質により目的遺伝子を誘導発現させるための遺伝子構築物であって、
(a)プロモーター機能を有する領域と目的とする外来蛋白質に係る構造遺伝子領域とを含む、目的遺伝子と、
(b)前記の目的遺伝子とは異なる他の外来遺伝子によりコードされ且つ銅イオンにより活性化される転写因子と、
(c)前記の目的遺伝子が含むプロモーター機能を有する領域の下流に位置する任意の外来遺伝子の5’−非翻訳領域と、
を有するように構築可能に調製されてなり、前記の転写因子とは異なる他の転写因子の転写活性化領域であり、前記の転写因子の下流に位置する転写活性化領域を、さらに追加的に有するように構築されてなることを特徴とする遺伝子構築物、ここで
前記の転写因子の塩基配列は、酵母ACE1由来の塩基配列である、および
前記の転写活性化領域の塩基配列は、単純ヘルペスウイルスのVP16転写因子由来の塩基配列である。
【請求項2】
前記の5’−非翻訳領域の塩基配列が、下記の5’−非翻訳領域に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする請求項1記載の遺伝子構築物。
<5’−非翻訳領域に係る塩基配列群>
(1)タバコモザイクウイルスのΩ配列からなる塩基配列
(2)タバコのアルコールデヒドロゲナーゼ遺伝子の5’−非翻訳領域配列からなる塩基配列
【請求項3】
任意のプロモーターを前記の転写因子の上流に追加的に有するように構築されてなる遺伝子構築物であることを特徴とする請求項1または2記載の遺伝子構築物。
【請求項4】
請求項1〜3のいずれかに記載の遺伝子構築物が導入されてなることを特徴とする形質転換体植物。
【請求項5】
請求項4記載の形質転換体植物から目的とする外来蛋白質を回収することを特徴とする外来蛋白質の取得方法。
【請求項1】
植物において、化学物質により目的遺伝子を誘導発現させるための遺伝子構築物であって、
(a)プロモーター機能を有する領域と目的とする外来蛋白質に係る構造遺伝子領域とを含む、目的遺伝子と、
(b)前記の目的遺伝子とは異なる他の外来遺伝子によりコードされ且つ銅イオンにより活性化される転写因子と、
(c)前記の目的遺伝子が含むプロモーター機能を有する領域の下流に位置する任意の外来遺伝子の5’−非翻訳領域と、
を有するように構築可能に調製されてなり、前記の転写因子とは異なる他の転写因子の転写活性化領域であり、前記の転写因子の下流に位置する転写活性化領域を、さらに追加的に有するように構築されてなることを特徴とする遺伝子構築物、ここで
前記の転写因子の塩基配列は、酵母ACE1由来の塩基配列である、および
前記の転写活性化領域の塩基配列は、単純ヘルペスウイルスのVP16転写因子由来の塩基配列である。
【請求項2】
前記の5’−非翻訳領域の塩基配列が、下記の5’−非翻訳領域に係る塩基配列群から選ばれてなる塩基配列であることを特徴とする請求項1記載の遺伝子構築物。
<5’−非翻訳領域に係る塩基配列群>
(1)タバコモザイクウイルスのΩ配列からなる塩基配列
(2)タバコのアルコールデヒドロゲナーゼ遺伝子の5’−非翻訳領域配列からなる塩基配列
【請求項3】
任意のプロモーターを前記の転写因子の上流に追加的に有するように構築されてなる遺伝子構築物であることを特徴とする請求項1または2記載の遺伝子構築物。
【請求項4】
請求項1〜3のいずれかに記載の遺伝子構築物が導入されてなることを特徴とする形質転換体植物。
【請求項5】
請求項4記載の形質転換体植物から目的とする外来蛋白質を回収することを特徴とする外来蛋白質の取得方法。
【図1】

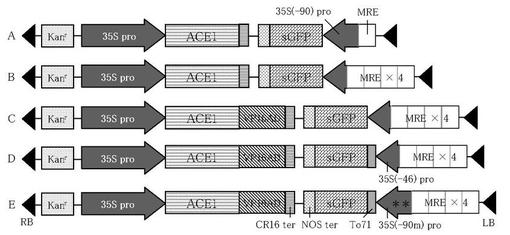
【図2】
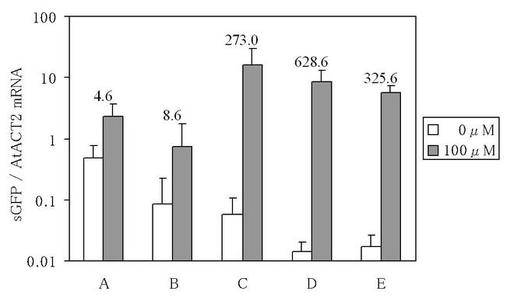
【図3】
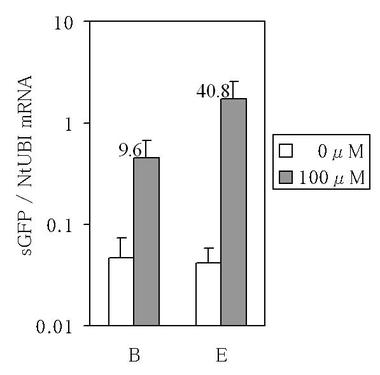
【図4】
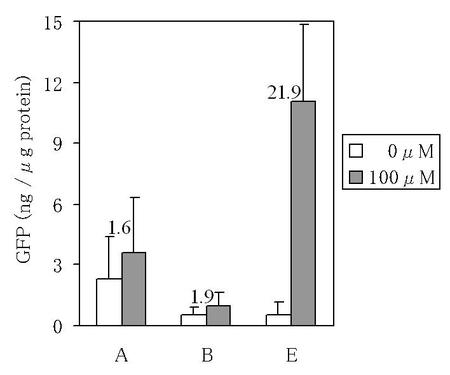
【図5】
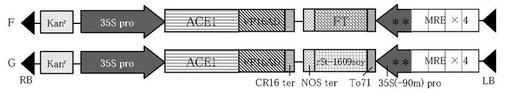
【図6】

【図7】
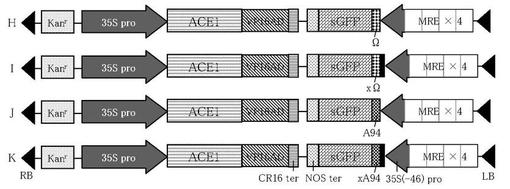
【図8】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2012−191941(P2012−191941A)
【公開日】平成24年10月11日(2012.10.11)
【国際特許分類】
【出願番号】特願2012−133673(P2012−133673)
【出願日】平成24年6月13日(2012.6.13)
【分割の表示】特願2008−1027(P2008−1027)の分割
【原出願日】平成20年1月8日(2008.1.8)
【出願人】(000002093)住友化学株式会社 (8,981)
【Fターム(参考)】
【公開日】平成24年10月11日(2012.10.11)
【国際特許分類】
【出願日】平成24年6月13日(2012.6.13)
【分割の表示】特願2008−1027(P2008−1027)の分割
【原出願日】平成20年1月8日(2008.1.8)
【出願人】(000002093)住友化学株式会社 (8,981)
【Fターム(参考)】
[ Back to top ]