
標的分子−ポリペプチド間相互作用解析方法
【課題】医薬リード化合物の設計などにおいて重要な、標的分子と相互作用するファーマコフォアを明らかにするための、ポリペプチドを効率良く取得する方法を提供する。
【解決手段】変異を導入した構造モチーフ、あるいは変異を導入した構造モチーフとランダムペプチドを標的分子と接触させ、標的分子に対し高親和性を示すポリペプチドを選択する。
【解決手段】変異を導入した構造モチーフ、あるいは変異を導入した構造モチーフとランダムペプチドを標的分子と接触させ、標的分子に対し高親和性を示すポリペプチドを選択する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、標的分子と相互作用するポリペプチドの選択方法、および目的蛋白質のファーマコフォアを明らかにするポリペプチドの取得方法等に関する。
【背景技術】
【0002】
従来の医薬品は各種抗生物質のような天然物、あるいはステロイドやカテコールアミンのようなヒト内在性物質から誘導された低分子化合物をベースに開発されているものが多い。一方で人工的に合成されたベンゼン置換体、複素環化合物誘導体など比較的シンプルな化合物についても医薬品としての成功例がしばしば見られる。これら従来の医薬品の開発においては限られた数の候補化合物を合成し、活性の確認を経たのち実用性の向上を目指した誘導体の合成展開が行なわれるのが一般的であり、合成と活性測定の対象となる化合物の数は限られていた。しかし医薬品開発の初期段階における合理性が追求された結果、多数の化合物に対して並行して活性試験を行なうハイスループットスクリーニングシステム(HTS)がリード化合物取得のために用いられるようになり、特に90年代初期に進展したコンビナトリアルケミストリーの技術は多検体アッセイ系の進歩とあいまって数百から数十万の低分子化合物ライブラリーを短期間でスクリーニングすることを可能にしている。こうしたシステムは天然の化合物に依存しないリード化合物開発の成功確率を高める一方、アッセイ系でヒットした化合物が医薬として不適である場合が少なくないという問題も生み出している。
【0003】
しばしば医薬リードとして重要視される生理活性ペプチドはアミノ酸重合体としてコンビナトリアルケミストリーの概念が適用されやすい対象であり、天然のペプチド性リガンドの活性増強のみならず、新規なリガンドの開発も数多く試みられている。化学合成の手法においては非天然型アミノ酸も導入可能な固相合成法がアレイ技術などとともに用いられることが多い。遺伝子工学的な手法も応用可能であり、特に80年代後半に実用化されたファージディスプレー法はペプチドなどの発現物を増幅可能な遺伝子と対応付けさせることで数百万から数億の多様性を有するライブラリーをスクリーニング対象とすることを可能にした。
【0004】
ペプチドあるいはその類縁体に属する従来の医薬品はアンギオテンシン、エンドセリン、ソマトスタチン、バソプレッシンなどヒトに内在する生理活性ペプチドリガンドに由来するアゴニストまたはアンタゴニスト、あるいはプロテアーゼの活性を抑えるための基質配列に由来する阻害剤であることが多い。こうした内在性のリガンドや基質に由来する医薬品の有用性は明らかであるが、創薬リードに成り得るそれらのペプチドの数には限界があり、疾患に対して理想的な創薬を可能にするには新たな相互作用の組合せを開拓する必要がある。ファージディスプレー法のように人工的なライブラリーを提示してスクリーニングをする技術はそれに適していると考えられ、蛋白質―蛋白質相互作用を阻害するペプチドの取得などが実際に試みられている。
【0005】
しかし医薬のリードとなるペプチドがこの方法により取得される可能性は極めて限られていると予想される。その理由の第一は、ペプチドによる蛋白質―蛋白質相互作用阻害といった人工的な作用機序が受容体へのリガンド結合による応答という自然な機序と同等な薬理活性を生み出し得る場合が限られること、第二は受容体などの相互作用対象と共進化の結果生まれた内在性アゴニストと同じような高い親和性を標的蛋白質と人工的ペプチドの間に持たせることが原理的に難しい場合が多いこと、第三は化学的手法に比べて多様性が高いとは言えファージディスプレー法でスクリーニング可能なライブラリーにも限界が見られること、であり、技術的な解決が期待できる第三の問題において改善を試みない限り人工的な相互作用の同定に基づく医薬リードの創製は効率の悪い方法となる。
【0006】
ファージディスプレー法において1度のスクリーニングにより探索可能な多様性の範囲は10の6乗から9乗程度であり、これは20種類のアミノ酸の組合せの数として5残基から7残基分に相当する。構造活性相関研究により標的分子との相互作用において特に重要な役割を果たしているとされるアミノ酸の数は一般に3から8程度であることが多いが、これは標的分子と直接相互作用しているアミノ酸に注目した場合の数であり、標的分子側が連続した数残基を特異的にリガンドとして認識するような深いキャビティを有していない限り安定な主鎖構造を持たない数残基のペプチドが高い親和性と特異性をもって標的分子に結合することはほとんど起こらない。
【0007】
ペプチドと標的分子の相互作用の強さは両者の結合に伴う自由エネルギーの変化により決まるものであり、深いキャビティを持たない標的分子に対して強い相互作用がはたらくためには、結合に適した主鎖構造や側鎖の空間配置をペプチド側が持つことにより結合に伴うエントロピー的な不利が少なくなること、あるいは広い結合界面により局所的な相互作用の足し合わせがエネルギー的に有利であることが求められる。前者の条件に関しては、ジスルフィド結合で環状化しているような場合を除いて10残基以下のペプチドが特定の安定した構造をとることは少なく、後者についても広い結合界面を形成するには10残基以上が必要となり、いずれにしてもファージディスプレー法で網羅できる多様性の範囲を超えている。
【0008】
医薬開発におけるペプチドスクリーニングは相互作用様式の解明による低分子化を目的として行なわれることが多い。ペプチドが内在性リガンドである場合、それが得られていることは標的分子の機能解明とリード化合物のスクリーニングに貢献するだけでなく、低分子リガンド設計にも直接的な情報を与える。人工的な相互作用様式の創出を伴うランダムペプチドライブラリーからのスクリーニングもファーマコフォアの同定による低分子化を目的の一つとして行われることが多い。この場合、特定の構造を持たずに標的分子と広い結合界面を形成することによって全体の親和性を獲得している比較的大きなペプチドは低分子でそれを再現することが原理的に困難である。したがって低分子化を目的として行なわれる人工的ペプチドリガンドのスクリーニングにおいては比較的限られた空間内において相互作用界面を抽出すること、そしてその成功確率を高めるためにはペプチド全体から生み出される構造の安定性が結合に伴うエントロピー的な不利を少なくするようなライブラリーを用いること、が重要となる。
【0009】
独立して安定に存在できる蛋白質の3次構造は一般に百残基以上によって形成され、しばしば機能の単位と重複する。一方で比較的独立した小さな構造が蛋白質の中に含まれていることもあり、核内受容体のDNA結合ドメインに含まれるジンクフィンガーモチーフ、カルモジュリンなどにおいてカルシウム結合部位となるEFハンドモチーフなどはその代表例である。さらに十数残基から50残基程度の大きさで3次構造を形成するペプチド・小型蛋白質も存在し、その中で複数のジスルフィド結合が構造形成の中心になっているものは特にシスチンノットモチーフと呼ばれる。これらの構造モチーフは構造の安定性を持ったペプチドライブラリーの土台として有用であり、ファージディスプレー法において応用されているものも多い(例えば、非特許文献1および2を参照)。
【0010】
しかしファージディスプレー法でスクリーニング可能なペプチドの数を考えた場合、こうした構造モチーフの導入はむしろライブラリーの多様性を自ら限定することにつながり易いだけでなく、構造維持に耐えられない変異が導入されたものやフォールディングの問題からファージ上で提示不可能なものなどが想定の範囲を超えて含まれているという理由により目的の質を持ったライブラリーの多様性が期待したスケールよりも低いという問題ももたらす。限られた空間内においてファーマコフォアを抽出し、医薬開発に有益な情報を効率良く得るためには投入できるライブラリーのスケールにおいてファージディスプレー法を上回るスクリーニング技術が求められ、さらにそのスケールの大きなライブラリーは質の点においても優れていることが望まれる。
【0011】
スクリーニング可能なライブラリーのスケールがファージディスプレー法を上回り、遺伝子工学的手法に特有な問題点に対してもある程度解決が期待できる方法としてリボソームディスプレー法と無細胞蛋白質ディスプレー法(例えば、特許文献1を参照)がある。いずれも細胞を介さずに遺伝情報と発現物を対応付けさせる技術であるが、前者は対応付け分子複合体の作製が容易である反面、その安定性維持のためスクリーニング条件に制約があり柔軟性に乏しい。後者は遺伝情報(mRNA)と発現物(蛋白質あるいはポリペプチド)をピューロマイシンリンカーを介して共有結合させることにより対応付けする技術であり、様々な条件下でスクリーニングできるだけでなく対応付け分子として前者よりはるかに小さいという利点も持つ(例えば、特許文献1あるいは非特許文献3を参照)。
【0012】
しかしmRNAが溶媒中に露出しているというリボソームディスプレー法と共通した特徴がリボヌクレアーゼなどの混入する系での安定性という面で問題となるだけでなく、発現物と同等あるいはそれ以上の多様性を有するmRNAがアプタマーとして標的分子その他と無視できない親和性で結合するという問題も生じさせ、特にランダムペプチドライブラリーのような多様性が大きい系ではそれが無視できなくなることが推測されていた。我々は新たなピューロマイシンリンカーの開発などにより、遺伝情報側を効率良く2本鎖核酸とすることでmRNAのアプタマーとしての性質を抑える実用的なスクリーニング系の開発に成功し、無細胞蛋白質ディスプレー法によってファージディスプレー法では対処できない多様性の大きさを持ったライブラリーから有用ペプチド・蛋白質をスクリーニングすることを可能にしていた(例えば、特許文献2を参照)。
【0013】
これらの技術の応用としてはこれまでのところ、天然に得られる遺伝子からライブラリー化されたポリペプチドから標的分子と相互作用する配列を選抜取得するスクリーニングのみしか行われておらず、取得される標的分子と相互作用するポリペプチドの機能にも限界があった。
【特許文献1】WO98/16636号公報
【特許文献2】特願2003−315385号明細書
【非特許文献1】Science,263,671−673(1994)
【非特許文献2】J.Mol.Biol.277,317−332(1998)
【非特許文献3】Nature,410,715−718(2001)
【発明の開示】
【発明が解決しようとする課題】
【0014】
本発明は、ポリペプチドあるいは蛋白質が持ち得る多様性の大きさと比較した場合に、実際の溶液中において行なわれる従来のペプチドあるいは蛋白質のスクリーニング系が、網羅性に著しく欠けていた点を改善し、医薬リード化合物の設計などにおいて重要な標的分子とのファーマコフォアを明らかにするポリペプチドを効率良く取得する方法を提供するためになされたものである。
【課題を解決するための手段】
【0015】
本発明者は、上記課題を達成するために鋭意検討を進めた結果、比較的安定な2次ないし3次の構造を有する天然に由来する構造モチーフあるいはそれらの構造を持つことが期待される人工的な構造モチーフを複数選び出し、それぞれに4ヶ所以上の変異を導入したライブラリーを構築し、さらに通常のランダムペプチドライブラリーもこれに加えて、無細胞蛋白質ディスプレー法を用いて標的蛋白質と接触させ、該蛋白質と相互作用するポリペプチドを選択する工程を繰り返すことにより、より特異的に、また高い親和性で標的蛋白質と相互作用をするポリペプチドを取得することに成功した。本発明は、これらの知見に基づいてなされたものである。
【0016】
すなわち、本発明によれば、
(1)変異を導入した構造モチーフを標的分子と接触させ、標的分子と相互作用するポリペプチドを選択することを特徴とする標的分子−ポリペプチド間相互作用解析方法、
(2)変異を導入した構造モチーフおよびランダムペプチドを標的分子と接触させ、標的分子と相互作用するポリペプチドを選択することを特徴とする標的分子−ポリペプチド間相互作用解析方法、
(3)標的分子と相互作用するポリペプチドを選択する方法が、無細胞蛋白質ディスプレー法である上記(1)または(2)に記載の方法、
(4)変異を導入した構造モチーフを標的分子と接触させ、標的分子に対し高親和性を示すポリペプチドを選択することを特徴とするファーマコフォアを明らかにするポリペプチドの取得方法、
(5)変異を導入した構造モチーフおよびランダムペプチドを標的分子と接触させ、標的分子に対し高親和性を示すポリペプチドを選択することを特徴とするファーマコフォアを明らかにするポリペプチドの取得方法、
(6)標的分子に対し高親和性を示すポリペプチドを選択する方法が無細胞蛋白質ディスプレー法である上記(5)に記載の方法、
(7)上記(4)〜(6)のいずれかに記載の方法により得られたポリペプチドのアミノ酸配列からアミノ酸の存在頻度および/または出現頻度を指標として共通配列を選択する工程を含むことを特徴とする医薬的活性を有するポリペプチドの取得方法、
(8)上記(4)〜(7)のいずれかに記載の方法により得られたポリペプチドを化学的に改変して医薬的活性を向上させることを特徴とするペプチド誘導体の取得方法、
(9)上記(4)〜(8)のいずれかに記載の方法により得られたポリペプチドあるいはペプチド誘導体と標的分子との複合体を作製する工程、該複合体の立体構造解析を行い、標的分子と該ポリペプチドあるいは該ペプチド誘導体との結合部位を特定する工程、同定された結合部位から標的分子のファーマコフォアを明らかにして、該ファーマコフォアに合う低分子化合物を設計する工程を含むことを特徴とする標的分子と相互作用する化合物の分子設計方法、
が提供される。
【発明の効果】
【0017】
本発明によれば、従来の高次構造解析の結果からは予測が困難な標的蛋白質のファーマコフォアを明らかにするポリペプチド、サブタイプなど類似の標的分子を選択的に認識するポリペプチド等を溶液中での実際のスクリーニングにより効率良く取得する方法が提供される。該方法は非天然型アミノ酸を変異部位に導入するスクリーニングにも対応しやすく、得られたポリペプチドのC末端にPEGなどの高分子を導入して安定化を図ることにも適する。該方法により得られたポリペプチドは標的分子と複合体を形成した状態での構造解析、低分子医薬の設計、ペプチド誘導体あるいは蛋白性医薬の開発、医薬リードスクリーニング系におけるコントロール用リガンドの取得、標的分子の検出あるいはイメージング、等において有用である。さらに、安定な構造を土台に持つポリペプチドライブラリーを複数使用してファーマコフォア同定のためにスクリーニングするという方法は実際の溶液中でのスクリーニングのみならず、コンピューターを用いた仮想結合実験においても応用できる。
【発明を実施するための最良の形態】
【0018】
以下、本発明を更に詳細に説明するが、以下の構成要件の説明は、本発明の実施態様の代表例であり、本発明はこれらの内容のみに特定されるものではない。
(1)ライブラリーの設計と構築
本発明の1つは、変異構造モチーフ、あるいは変異構造モチーフとランダムペプチドからなるライブラリー(以下、これを「ライブラリー」と称することがある)を標的分子と接触させ、標的分子と相互作用するポリペプチドを選択することを特徴とする標的分子−ポリペプチド間相互作用解析方法である。また、標的分子が蛋白質である場合、上記ライブラリーと標的蛋白質とを接触させ、標的分子と高い親和性を示すポリペプチドを選択することで得られるポリペプチド(本明細書中ではこれを「ファーマコフォアを明らかにするポリペプチド」と称する)は該蛋白質のファーマコフォアを明らかにするものであり、医薬として実用化が可能な低分子化合物あるいはポリペプチド誘導体の開発において有用である。
【0019】
そこで、本発明の方法に適したポリペプチドのライブラリーを考えると、高い親和性と特異性をもたらすファーマコフォアが選択されるポリペプチドと標的分子との間の比較的限られた空間に存在することが望ましい。そのようなファーマコフォアを効率良く同定するためのポリペプチドを選択するためには限られた空間におけるポリペプチドの多様性を可能な限り大きくすることが必要であり、標的分子と直接相互作用するアミノ酸残基のみならず、それらの配向や揺らぎを規定する他の残基においても多様性が求められる。
【0020】
ペプチド鎖は側鎖だけでなくアルファ炭素の両側においても比較的高い回転の自由度を持つために仮想的には一定の空間内で様々な構造をとり得るが、実際の標的分子との相互作用においては結合に伴う内部エネルギー的な有利性がエントロピー的な不利を相殺できないような主鎖構造は生じ得ない。選択されるポリペプチドと標的分子との高い親和性は、標的分子側が選択されるポリペプチドの主鎖構造を安定化させるような明確なキャビティを持っていること、あるいは選択されるポリペプチド側が標的分子との結合に適した主鎖構造をとり易いこと、のいずれかによって可能になっていることがほとんどであり、特に20残基を超えるようなポリペプチドに関しては後者の例が多い。したがって、深いキャビティを持たない標的分子に対してファーマコフォアを特定するためには、主鎖構造が二次ないし三次構造のレベルで安定になりやすいポリペプチドをライブラリー化してスクリーニング対象とすることが合理的と考えられる。
【0021】
直接的に医薬リード化合物となるようなポリペプチドをスクリーニングにより効率良く得る技術を提供することも本方法の目的の一つであるが、安定な構造をとり易いポリペプチドはペプチダーゼやプロテアーゼに対する感受性が一般に低くなることが期待でき、血中での安定性という点で有利になりやすいと予想される。本発明において使用されるライブラリーは以上のことを踏まえて選定と構築がなされる。
【0022】
ここで、「ポリペプチド」とは、残基数において6残基から10残基程度の一般にオリゴペプチドと呼ばれるものから、構造安定性の高い120残基程度の大きさを持った蛋白質までを含むものとする。また、「相互作用する」とは、標的分子に結合して該物質の機能を制御することを意味する。さらに、「ファーマコフォア」とは、標的分子と相互作用する分子と標的分子の結合界面に存在して、親和性と特異性の獲得に寄与しているアミノ酸残基や官能基などの複数の部分構造の空間配置を意味する。また、本発明の方法で用いる「標的分子」とは、蛋白質、ポリペプチド、核酸、糖質、脂質、低分子化合物等のいずれのものでもよいが、特定の疾患と関連し、該分子の機能の制御が、該疾患の治療に有用であることがわかっている物質が特に好ましく用いられる。
【0023】
本発明において、用いられる「構造モチーフ」とは、天然の蛋白質あるいはポリペプチドの全体ないし一部であってもよく、天然の構造からの類推または論理的設計によって得られた人工的なものでもよい。天然のポリペプチドの例としては、コノトキシン、アガトキシン、デンドロトキシン、チャリブドトキシン、アパミンなどのイオンチャネル阻害ペプチド、テッポウウリ由来トリプシンインヒビターやアプロチニンなどのプロテアーゼ阻害ペプチド、ディフェンシンやヘプシジンなどの抗菌ペプチド、エンドセリンやインシュリンなどのペプチド性ホルモンが挙げられ、これらの多くにおいては二次以上の高次構造が複数のジスルフィド結合によって安定化されている。バソプレッシン、ウロテンシン、オキシトシン、ソマトスタチンのように一つのジスルフィド結合を有する重要な生理活性ペプチドもあり、二つのシステインの間に4残基から12残基のアミノ酸を含むライブラリーも主鎖構造がある程度制限されたライブラリーとして利用される。
【0024】
蛋白質の一部ないし全体に由来するものとしては、ジンクフィンガーモチーフ、EFハンドモチーフ、アンキリンリピート、4本ヘリックスバンドル、免疫グロブリンフォールドのように多くの蛋白質に共通して現れる構造が有用である。人工的に設計される構造としては、ヘリックスターンヘリックスのようなアルファヘリックス構造を中心とした配列のライブラリー化が比較的容易である。構造モチーフという言葉は、βターン構造のような定義が明確なものを除けば、二次構造が連結して生じる部分構造や金属イオンへの配位結合で生じる小さな構造など十数残基から数十残基程度の特徴的な構造を意味することが多いが、ここでは一つのジスルフィド結合で生じる数残基から成るループ構造や、一般的な蛋白質のドメイン構造に近い大きさを持つ三次構造、あるいは標的分子との相互作用により初めて安定化される柔軟な構造も含めて「構造モチーフ」と定義する。
【0025】
このような構造モチーフの中で、本発明の方法に用いるライブラリーとしての配列の選定は、選定された構造モチーフの組合わせによって一次構造上の多様性だけでなくファーマコフォアを探索する空間におけるライブラリーの多様性が大きくなることを目的として行なわれる。例えば一本のαヘリックス、βストランド、あるいはループ上において空間的に近い数残基はその組合せが同じであっても異なるものと見なすことができ、特にループ部分はその配列や周辺の構造によって異なる主鎖構造をとり易い。さらに三次構造的にはそれらの残基が一定の空間内で組合されることになり、主鎖構造及び側鎖の配向と種類が生み出す多様性が著しく大きくなる。
【0026】
このような多様性を確保するには異なる構造モチーフライブラリーを多種類併用することが望ましいが、実際に用いるスクリーニング系が扱うことの可能なライブラリーのスケールには限界があるため、併用する構造モチーフライブラリーの数と種類、各ライブラリーにおける変異導入箇所の選択は制約の下で有効な多様性を生み出すように行なわれる必要がある。具体的には、用いる構造モチーフのもととなるポリペプチドはその構造が比較的複雑である場合に、立体構造がX線結晶構造解析などにより解明され実験的な証明が得られていることが望ましい。立体構造上の情報は相互作用クラスターとして複数の変異を導入する空間を決定する目的で求められるだけでなく、構造維持に著しく不利な変異の導入を避けるためにも重要となる。
【0027】
例えばβターン構造上に存在するグリシン残基の変異はしばしば分子全体のフォールディングを著しく阻害することが知られており、あるいはプロリン残基は立体構造の中心となる二次構造の形成に不利になることが多い。立体構造が既知であればフォールディングにおいて脱落するポリペプチドを少なくしながら構造モチーフライブラリーの多様性の維持を図ることが比較的容易になる。
【0028】
変異の導入がフォールディングに著しく不利にならない範囲では、二つ以上の相互作用クラスターを一つの分子に導入することも可能である。無細胞蛋白質ディスプレー法をスクリーニングに用いる場合、4〜25の変異部位を導入した構造モチーフ、および完全ランダムライブラリーとともに4〜50種類まとめてスクリーニングすることが好ましい。
スクリーニング技術に由来する制約もいくつか存在し、中でもフォールディングに関する問題はライブラリーの選択に大きな影響を与える。酵母ツーハイブリッド法、ファージディスプレー法、リボソームディスプレー法、無細胞蛋白質ディスプレー法は、フォールディングも含めた発現の効率において一長一短を持ち、翻訳される蛋白質あるいはポリペプチドごとに適不適がある。生物系に依存しない上に比較的安定な対応付け分子を形成する無細胞蛋白質ディスプレー法は翻訳後の巻き戻し操作などにおいて有利な点を持っていると考えられ、複数のジスルフィド結合を持つシスチンノットモチーフ等に対しては特にこの利点を応用しやすいが、すべての構造モチーフのフォールディングに適した条件を確立することは困難であり、さらに提示されるポリペプチドのC末端がピューロマイシンを介したmRNAとの連結のために塞がれているということも時として問題になると予想される。このため構造モチーフの選択に際しては、化学合成物や組換発現物などにおいてフォールディングの容易さが確認されているものを優先することが望ましく、さらにそれらの中ではC末端が溶液中に露出してリンカー配列等の付加によるフォールディングの阻害が予想されないものが望ましい。
【0029】
ライブラリーの変異箇所に導入されるアミノ酸残基の出現確率は、構造モチーフに合わせて可能な範囲で変えることが必要となる。例えば、ジスルフィド結合によって形成される構造モチーフをライブラリーとして用い、あるいは標的分子が細胞外の蛋白質でジスルフィド結合が形成され易い条件でスクリーニングを行なう場合、ランダム部位に出現したシステインは期待した構造モチーフの形成を阻害し、あるいは他の分子との間で架橋される可能性があるため、ランダム化した部位のシステイン出現頻度は低いことが望まれる。さらにαヘリックスやβストランドの変異による不安定化が構造維持を阻害する可能性が高いと考えられる部位においては、それらの二次構造に不利にならないセミランダム化を行なうなどの対応も必要となる。加えて、トリプトファンやチロシンなど一分子に数多く出現した場合に非特異結合が増えるような残基は出現頻度を高くしないこと、最初に選択するライブラリーのスケールを落とすことになる終止コドンの出現頻度も制限すること、なども望まれる。これらのアミノ酸残基出現頻度の制御は、主にDNA化学合成の段階で塩基の混合比を設定することにより行なわれる。
【0030】
ライブラリーの構築は、主に化学的に合成した複数のDNAのオーバーラップ伸長法により行なわれる。オーバーラップさせる末端の相補鎖形成配列は、ランダム化部位を含まないほうが望ましい。ライブラリーの多様性の維持と各分子のコピー数格差抑制のため、オーバーラップ伸長反応のサイクルは1〜10回程度に抑え、増幅のためのPCR反応は行なわないことが望ましい。
【0031】
(2) ファーマコフォアを明らかにするポリペプチドおよびそれをコードする核酸の選抜取得方法
本発明のファーマコフォアを明らかにするポリペプチドあるいは後述するドメインポリペプチドの選抜取得方法として、酵母ツーハイブリッド法、ファージディスプレー法、リボソームディスプレー法、無細胞蛋白質ディスプレー法などが用いられるが、より大きなスケールのライブラリーの中からポリペプチドを柔軟な条件で選択できる無細胞蛋白質ディスプレー法が特に好ましく用いられる。
【0032】
無細胞蛋白質ディスプレー法によるポリペプチドの選抜取得方法は、 (1)ポリペプチド部とそれをコードする核酸部が、核酸部の3’末端に結合した核酸構築物を介して直接結合したポリペプチド−核酸連結体群と標的分子とを接触させる工程、(2)標的分子に特異的に結合した該ポリペプチド−核酸連結体を取得し、該連結体の核酸部を増幅し、これを鋳型の一部として用いてポリペプチド部とそれをコードする核酸部が、核酸部の3’末端に結合した核酸構築物を介して直接結合したポリペプチド−核酸連結体群を調製する工程、(3)上記(1)および(2)の工程を、標的分子と相互作用させたポリペプチド−核酸連結体の核酸部が、標的分子に特異的に結合するポリペプチドをコードすることが確認されるまで繰り返す工程を含むことを特徴とするものである。具体的には、以下のとおりである。
【0033】
(i)ポリペプチド−核酸連結体および該連結体と標的分子を接触させる工程
本発明の方法で用いられる「ポリペプチド−核酸連結体」は、例えば、WO98/16636号公報に記載のもの等のように、ポリペプチド部とそれをコードする核酸部が、核酸部の3’末端に結合した核酸構築物を介して直接結合したものであり、蛋白質の相互作用解析等の強力なツールとなり得る分子である。
【0034】
ここで、「ポリペプチド部」とは、核酸部に含まれるコーディング配列によりコードされるポリペプチドを含み、ポリペプチドは上記ライブラリーである。また、ポリペプチド−核酸連結体を精製するためのタグと上記蛋白質との融合ポリペプチドも含まれる。「核酸部」とは、ポリペプチド部のポリペプチドをコードするコーディング配列を含む核酸であり、さらに、用いるポリペプチド合成系においてポリペプチドの翻訳が開始されるための配列も含むことが多い。核酸部は一般にmRNAであり、その鋳型となるDNAを「核酸部前駆体」とする。
【0035】
核酸部前駆体は、具体的には、コーディング領域と発現制御領域を含むものである。発現制御配列とは、(1)プロモーター配列、(2)翻訳の際にリボソームによって認識される配列等が挙げられる。プロモーター配列の種類は、適用する発現系に適したものを適宜選択すればよく、特に限定されない。例えば、大腸菌ウイルスT7のRNAポリメラーゼによって認識されるT7プロモーター配列、SP6 RNAポリメラーゼにより認識されるSP6プロモーター配列などが挙げられる。また、翻訳の際にリボソームによって認識される配列としては、翻訳の際に真核細胞のリボソームによって認識されるRNA配列(Kozak配列)に対応するDNA配列や、原核細胞のリボソームによって認識されるシャイン・ダルガノ配列(Shine-Dalgarno)、5’キャップ構造(Shatkin,Cell,9,645−(1976))、オメガ配列等のtabacco mosaic virusのリボソームによって認識される配列、WO03/56009号公報に記載の配列、rabbitβ−globlin、Xenopusβ−globlin あるいはbromo mosaic virusのリボゾーム認識領域などが挙げられる。また、ポリペプチド−核酸連結体の核酸部を増幅するのに、PCRを用いる場合、PCRプライマー用配列を含むものも挙げられる。また、核酸部前駆体を増幅するためにPCRを用いる場合には、PCRプライマー結合用の共通配列を含むものも挙げられる。
【0036】
コーディング配列とは、ポリペプチド−核酸連結体の蛋白質部に含まれるポリペプチドをコードする配列であり、この種類は特に限定されず、目的に応じて適宜選択できる。また、該核酸部の3’末端に結合した「核酸構築物」とは、その末端に「核酸誘導体」を結合しており、該核酸部を鋳型として無細胞蛋白質翻訳系又は生細胞中で蛋白質(ポリペプチド)の翻訳を行った場合、mRNAの末端付近まで翻訳が進んだ後、該核酸誘導体(例えば、ピューロマイシンなど)がリボソームのAサイトに入ることにより蛋白質(ポリペプチド)と結合させる機能を有するものを意味する。具体的には、スペーサー、該ポリペプチド−核酸連結体を標識する標識部、また該連結体を固相に結合するための構造、核酸部を逆転写するためのプライマー部等を含むものが好ましい。
【0037】
「核酸誘導体」としては、無細胞蛋白質翻訳系又は生細胞中で蛋白質(ポリペプチド)の翻訳が行われた時に、合成された蛋白質(ポリペプチド)のC末端に結合する能力を有する化合物である限り限定されないが、その3’末端がアミノアシルtRNAに化学構造骨格が類似しているものを選択することができる。代表的な化合物として、ピューロマイシン(Puromycin)と3’−N−アミノアシルピューロマイシンアミノヌクレオシド(3'-N-Aminoacylpuromycinaminonucleoside 、 PANS-アミノ酸)、すなわち、アミノ酸部がグリシンのPANS−Gly、アミノ酸部がバリンのPANS−Val、アミノ酸部がアラニンのPANS−Ala、その他、アミノ酸部が全ての各アミノ酸に対応するPANS−アミノ酸化合物が挙げられる。
【0038】
また、3’−アミノアデノシンのアミノ基とアミノ酸のカルボキシル基が脱水縮合して連結した3’−N−アミノアシルアデノシンアミノヌクレオシド(3'-Aminoacyladenosine aminonucleoside, AANS-アミノ酸)、すなわち、アミノ酸部がグリシンのAANS−Gly、アミノ酸部がバリンのAANS−Val、アミノ酸部がアラニンのAANS−Ala、その他、アミノ酸部が全アミノ酸の各アミノ酸に対応するAANS−アミノ酸化合物を使用できる。また、核酸あるいは核酸とアミノ酸のエステル結合したものなども使用できる。さらにまた、核酸あるいは核酸に類似した化学構造骨格及び塩基を有する物質と、アミノ酸に類似した化学構造骨格を有する物質とを化学的に結合した化合物は、すべて本発明で用いられる核酸誘導体に含まれる。核酸誘導体としては、ピューロマイシン、PANS−アミノ酸もしくはAANS−アミノ酸がリン酸基を介してヌクレオシドと結合している化合物がより好ましい。これらの化合物の中でピューロマイシン、リボシチジルピューロマイシン、デオキシシチジルピューロマイシン、デオキシシチジルデオキシシチジルピューロマイシン、デオキシウリジルピューロマイシンなどのピューロマイシン誘導体が特に好ましい。
【0039】
このような核酸誘導体は、それ自体既知の化学結合方法によって製造することができる。具体的には、リン酸ジエステル結合で合成ユニットを結合させる場合は、DNA合成機に一般的に用いられているホスホアミダイド法などにより固相合成で合成することが可能である。ペプチド結合を導入する場合は、活性エステル法などにより合成ユニットを結合させるが、DNAとの複合体を合成する場合は、両方の合成法に対応が可能な保護基が必要になる。
【0040】
「スペーサー」は、ポリエチレン又はポリエチレングリコールあるいはその誘導体などの高分子物質や、オリゴヌクレオチドやペプチドあるいはその誘導体などの生体高分子物質等が用いられ、好ましくはポリエチレングリコールが用いられる。スペーサーの長さは特に限定されないが、好ましくは、分子量150〜6000であるか、または主鎖の原子数は10原子から400原子であり、さらに好ましくは、分子量600〜3000であるか、または主鎖の原子数が40原子から200原子である。また、標識部を有しててもよい。
【0041】
「標識部」は、親和性物質、共有結合性物質、蛍光物質、分解性物質等が挙げられる。これらは、「該連結物を固相に結合するための構造」としても用いられるし、また「該連結物を精製するための構造」としても用いられる。親和性物質としては、ポリA配列、ポリT配列、ビオチン、FLAG等の各種抗原又は抗体、Hisタグ、NTA等の配位子、受容体リガンド等が挙げられる。また、共有結合性物質としては、デオキシリボヌクレオチド、リボヌクレオチド等の核酸末端部分、ヒドラジド、ケトン、チオエステル等の官能基、ソラレン等の架橋性物質が挙げられる。蛍光物質としては、フルオレセイン、オレゴングリーン、ローダミン、テトラメチルローダミン、テキサスレッド、Cy3、Cy5、Alexa488等が挙げられる。分解性物質としては、光反応で分解する1-(2-ニトロフェニル)-エチル基を有する誘導体や、プロテアーゼやペプチダーゼに認識されるアミノ酸配列等が挙げられる。これらの標識物質は、それ自体既知の通常用いられるものであり、容易に入手可能であり、また常法により核酸等に結合して標識することができる。
【0042】
好ましい核酸部とその3’末端側に結合した核酸構築物、核酸誘導体(以下、これを「ポリペプチド−核酸連結体鋳型」と称することがある)の好ましい構造としては、核酸部であるRNAと、核酸誘導体を末端に有するスペーサーが枝分かれした状態で結合している1本鎖核酸(以下、これを「スペーサー鎖」と称することがある)がアニーリングした構造を有し、さらに該スペーサー鎖と標識部を含む1本鎖核酸と核酸部がそれぞれ結合しているものが挙げられる。具体的には、PCT/JP2004/013399号明細書に記載のもので、製造法も該明細書に記載された方法が好ましく用いられる。このような構造を有することにより、核酸部と核酸構築物のRNAリガーセ等による結合効率が高く、またポリペプチド−核酸連結体を標識部により精製でき、核酸部のRNAを逆転写することができ、さらに核酸部を2本鎖DNAとすることもできる。
【0043】
かくして構築されたポリペプチド−核酸連結体鋳型を蛋白質翻訳系に導入することによりポリペプチド−核酸連結体を製造することができる。核酸からそれがコードする蛋白質を人工的に生成させるための翻訳系は当業者に公知である。具体的には、適当な細胞より蛋白質合成能を有する成分を抽出し、その抽出液を用いて目的の蛋白質を合成させる無細胞蛋白質合成系が挙げられる。このような無細胞蛋白質合成系には、リボゾーム、開始因子、伸長因子及びtRNA等の翻訳に必要な要素が含まれている。
【0044】
このような無細胞蛋白質合成系としては、例えば、真核生物の無細胞蛋白質合成系が用いられ、より具体的には、ウサギ網状赤血球抽出液やコムギ胚芽抽出液などが挙げられるが、これらに限られるものではない。無細胞蛋白質合成系は、キットとして市販されているものを使用することができる。例えば、ウサギ網状赤血球抽出液のキットとしては、Rabbit Reticulocyte Lysate Systems, Nuclease Treated(Promega社製)等が用いられ、またコムギ胚芽抽出液としては、ゾイジーン無細胞蛋白質合成システム(ゾイジーン社製)や、PROTEIOSTM Wheat germ cell−free protein synthesis core kit(TOYOBO社製)等が挙げられる。蛋白質翻訳系としては、生細胞を使用してもよく、具体的には、原核又は真核生物、例えば大腸菌の細胞等を用いることができる。無細胞蛋白質翻訳系又は生細胞などは、その中に蛋白質をコードする核酸を添加するか又は導入することによって蛋白質合成が行われるものである限り特に制限はない。本発明の核酸構築物を無細胞蛋白質合成系に導入する直前に、60〜90℃で加熱した後急冷する工程を行うと、蛋白質−核酸連結体の合成効率が高くなるため好ましい。
【0045】
上記翻訳反応液から、ポリペプチド−核酸連結体を精製する場合、標識鎖に親和性物質あるいは共有結合物質が結合している場合には、該親和性物質あるいは共有結合物質を介して精製を行うことができる。精製の方法は、用いる親和性物質および共有結合物質に応じて適宜選択してそれ自体既知の定法を用いることができる。
上記で製造されたポリペプチド−核酸連結体は、上記標的分子と接触させ、該物質と相互作用する連結体を選択取得する工程に供する。この選択方法は、ポリペプチド−核酸連結体中のポリペプチドが有する機能(生物活性)を用いて標的分子と相互作用するポリペプチドを該連結体として選択することを意味する。このような相互作用の解析方法としては、例えばWO98/16636号公報に記載の方法を用いることができる。また、標的分子と接触させるポリペプチド−核酸連結体は、上記ライブラリーを有する複数の連結体群である。
【0046】
また、本発明のポリペプチド−核酸連結体および標的分子は、固相(支持体)に結合させて用いることもできる。ポリペプチド−核酸連結体の固相への結合は、上記標識部に親和性物質や共有結合性物質が結合している場合には、これらを用いて行うことができる。具体的には、親和性物質又は共有結合性物質が親和性を有するまたは結合する物質を予め固定化した固相に、上記ポリペプチド−核酸連結体を接触させることにより、当該ポリペプチド−核酸連結体を固相に容易に固定化することができる。固相への標的分子の結合は、例えば、Scott,J.K.&Smith,G.P.(1990)Science,249,386−390;Devlin,P.E.et al.(1990)Science,249,404−406;Mattheakis,L.C.et al.(1994)Proc.Natl.Acad.Sci.USA,91,9022−9026等に記載されている方法等により行うことができる。
【0047】
固相(支持体)としては、通常の核酸、ポリペプチド、糖質、脂質、化合物等の固定化に用いることができる支持体であれば特に限定されない。支持体としては、親和性物質や共有結合性物質どうしの結合形成、あるいは標的分子の結合に悪影響を及ぼさないものであればその形状は特に限定されず、例えば、平板、マイクロウェル、ビーズ等の任意の形態をとることができる。支持体の材質としては、例えば、ガラス、セメント、陶磁器等のセラミックス、アガロース、ポリエチレンテレフタレート、酢酸セルロース、ビスフェノールAのポリカーボネート、ポリスチレン、ポリメチルメタクリレート等のポリマー類、シリコン、活性炭、多孔質ガラス、多孔質セラミックス、多孔質シリコン、多孔質活性炭、織編物、不織布、濾紙、短繊維、メンブランフィルター等の多孔質物質を挙げることができる。
【0048】
上記選択方法に付するポリペプチド−核酸連結体は、核酸部がRNAのものでもよいし、mRNA鎖をDNAに逆転写したポリペプチド−逆転写核酸連結体でもよい。逆転写反応に必要な試薬及び反応条件は当業者に周知であり、必要に応じて適宜選択することができる。さらに得られたポリペプチド−逆転写核酸連結体のRNAをRNA分解酵素などを用いて分解し、DNAを鋳型にポリメラーゼ反応をすることにより2本鎖DNA−蛋白質連結体を作製して用いることもできる。このような、ポリペプチド−逆転写核酸連結体、あるいは2本鎖DNA−ポリペプチド連結体を用いれば、核酸部分の安定性がよいこと、また1本鎖RNAの非特異的吸着がないため好ましい。
【0049】
(ii)標的分子に特異的に結合したポリペプチド−核酸連結体の取得、核酸部の増幅、該核酸部を鋳型の一部としたポリペプチド−核酸連結体の調製工程
上記選択方法で選択されたポリペプチド−核酸連結体は、これを再度標的分子との相互作用に基づいて選択する工程に供する。一度選択されたポリペプチド−核酸連結体の核酸部を増幅して、さらにポリペプチド−核酸連結体とするためには、(a)選択取得されたポリペプチド−核酸連結体の1本鎖RNA部分を必要に応じて分解する等した後に、これをPCRなどで増幅し(増幅工程)、(b)増幅されたDNA鎖をもとにmRNA鎖を製造し、さらに上記のポリペプチド−核酸連結体鋳型を製造し、これを翻訳してポリペプチド−核酸連結体を調製することにより行なうことができる。
【0050】
(a)増幅工程は、PCRを用いて例えば以下のように行うことも好ましい。ポリペプチド−核酸連結体の核酸中、増幅するのは少なくともコーディング配列を含む領域である。このように増幅されたDNAについて、それ自体既知の定法により塩基配列を解析することにより、上記選択方法で選択された蛋白質をコードするDNA配列を同定できるので、該配列をもとにDNAまたはRNAも取得することができる。該領域を増幅するのに用いられるPCRプライマーとしては、特に制限はないが、全てのポリペプチド−核酸連結体に共通に用いられる配列として、5’側のプライマーは、コーディング配列の5’上流側に連結されている配列が好ましく用いられる。具体的には、上記したポリペプチド−核酸連結体の場合、5’側のプライマーは、翻訳の際にリボソームによって認識されるDNA配列などが好ましく用いられ、3’側のプライマーは、タグ配列や共通配列が好ましく用いられる。
かくして調製されたポリペプチド−核酸連結体鋳型は、上記(i)に記載の方法により翻訳され、ポリペプチド−核酸連結体が調製される。
【0051】
(iii)濃縮操作
本発明の方法では、上記(i)および(ii)に記載の工程を、標的分子に特異的に結合したポリペプチド−核酸連結体が全体として標的分子に対して高い親和性と特異性を示すまで繰り返す(本明細書では、これを「濃縮操作」と称することがある)。求められる親和性は目的により異なるが、高い親和性を有するポリペプチドの取得を目的とする場合はライブラリーと接触させる標的分子の濃度を低くすることが必要であり、期待する解離定数の近辺でその値を設定する。
【0052】
本発明の方法では、一般にこの濃縮操作を2〜15回行なうことによって、ファーマコフォアを明らかにするポリペプチドと、それをコードする核酸の連結体が取得される。取得された連結体の核酸部を上記方法により増幅して常法により塩基配列を解析することにより、該ポリペプチドをコードする塩基配列を同定することができる。また、該塩基配列を有する核酸を常法により合成すればポリペプチドをコードする核酸を取得することができ、またこの核酸を公知の、例えば、上記した蛋白質合成方法等により翻訳合成することにより、ファーマコフォアを明らかにするポリペプチドを取得することができる。
【0053】
さらには、多様性の大きなライブラリーから濃縮操作の結果として同定される上記塩基配列は、一般に複数であるので、上記の標的分子への親和性に基づいて選択されたポリペプチドが、ファーマコフォアを明らかにするポリペプチドであることを確認するためには、標的分子に作用してその機能に影響するかどうかを調べることが好ましい。具体的方法としては、まず上記で取得された塩基配列がコードするポリペプチドを常法により化学合成し、あるいは該塩基配列を有する核酸を常法により合成して、大腸菌あるいは無細胞翻訳系で翻訳合成する等の方法により、活性測定用のポリペプチドを調製する。生体内で標的分子と相互作用する蛋白質等と標的分子の結合に該ペプチドが与える影響、あるいは標的分子の機能を測定可能な細胞実験系における該ペプチドの薬理作用等を調べることにより、ファーマコフォアの解明に有用なポリペプチドを選択することができる。
【0054】
また、上記のようにして取得したポリペプチドのアミノ酸配列からアミノ酸の存在頻度および/または出現頻度を指標として共通配列を選択する工程を含むことを特徴とする医薬的活性を有するポリペプチド(本明細書中では、これを「ドメインポリペプチド」と称することがある)を取得することもできる。具体的には、上記で取得されたポリペプチドをコードする核酸からドメインポリペプチドをコードするクラスターを取得することにより行う。「ドメインポリペプチドをコードするクラスター」(以下、「クラスター」と称することがある)とは、上記ドメインポリペプチドをコードする核酸を共通配列として含むほぼ同じ長さの核酸のかたまりを意味する。共通配列を含むほぼ同じ長さのかたまりとは、上記で取得されるポリペプチド−核酸連結体群の50%以上が該共通配列を含み、その鎖長が最も短い核酸と長い核酸が1倍〜10倍の範囲に入ってくることを意味する。このようなクラスターが形成されたことは、形成を確認し得る方法であれば特に制限はないが、例えば、標的分子と相互作用することで選択取得されたポリペプチド−核酸連結体の核酸部を、RNAのまま、又はDNAとしてアガロースゲルまたはSDS−ポリアクリルアミドゲル中を電気泳動等で分離した場合にバンドが観察されることにより確認することができる。
【0055】
(3)ファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドを用いた医薬開発
本発明に従ったライブラリーのスクリーニングで取得されたポリペプチドに対して解析実験を行なうことにより、医薬リード開発に必要な情報がもたらされる。相同性の高い配列が複数得られている場合にはそのアライメントから構造活性相関についての情報が少なからず得られることがある。配列のアライメントから必要な情報が充分得られない場合は、取得された配列をもとにそれをコードするDNAの再ランダム化を行い、無細胞蛋白質ディスプレー法またはその他の方法でスクリーニングするか、あるいは置換した部位を有するポリペプチドを無細胞翻訳、大腸菌発現、化学合成などにより1つないし数百種合成して活性測定をすることで構造活性相関についての情報を得ることができる。
【0056】
得られたファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドが明確に区別され得る複数の立体構造あるいはジスルフィド架橋をとる場合には、実際に該ポリペプチドを合成して活性に必要な立体構造あるいはジスルフィド架橋を確認する必要があり、立体構造の確認方法としてはX線結晶構造解析、NMRによる構造解析、円偏光二色性解析などが挙げられ、ジスルフィド架橋の確認方法としては酵素による限定加水分解法と質量分析法を合わせた方法がある。
【0057】
得られたファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドと標的分子の間でファーマコフォアを抽出する方法としては、両者が結合した共結晶を形成させ、X線結晶構造解析により両者の座標を得ることがもっとも望ましい。得られた空間座標をもとに上記ポリペプチド側を改変する、低分子をドッキングさせる、あるいは新規に原子を発生させてde novoで分子を形成する等の手法により上記ポリペプチドと同様の活性を持った低分子化合物を開発することが可能となる。共結晶が得られない時でも、上記ポリペプチド側の立体構造が土台となる構造モチーフ等から推測できる場合、あるいはMD等の手法により限られた主鎖構造が候補となる場合には標的分子側の立体構造情報の有無に関わらず上記ポリペプチドの低分子化合物への改変ができることもある。さらに一般的に、上記ポリペプチド側についての詳細な構造情報はNMRによる溶液中での立体構造解析により取得することが比較的容易である。上記ポリペプチド側あるいは標的分子側において相互作用に関わっている部位についての情報が著しく不明である場合にはHSQC(Heteronuclear single quantum correlation)等の手法が有用である。これらの構造情報とペプチドミメティクスの手法の組み合わせによっても新たな低分子化合物が創出される。
【0058】
かくして設計された低分子化合物は、標的分子が疾病に関連するものである場合、該疾病治療薬として用いることができる。この場合、上記工程により医薬組成物を製造する方法も本発明に含まれる。
取得されたファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドをそのまま、あるいは改変を加えてペプチド医薬として応用することも可能である。親和性と選択性の向上を目的とした改変には通常のアミノ酸あるいは非天然型アミノ酸による置換等が有効である。また、ヒトに対する抗原性を低下させることを目的とした改変が行なわれる場合もある。安定性を目的とした改変においては、該ポリペプチドの主鎖あるいは側鎖の置換の他にPEGなどの高分子との融合が行なわれることもあり、上述した無細胞蛋白質ディスプレー法で選択されたポリペプチドは特にC末端とその周辺への導入に対して許容性が高いと予想される。
【0059】
取得されたファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドはそれ自体あるいはその構造情報から医薬リードを開発する目的以外にも応用でき、例えば蛍光プローブをポリペプチドに導入することでHTSシステムにおいて多数の低分子化合物ライブラリーの活性を調べるための競合リガンド(Surrogate ligand)として用いることも可能である。蛍光プローブを導入した該ポリペプチドはイメージング等にも応用できる。
【0060】
さらに本発明で取得されたファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドを固相化することにより標的分子を検出するチップ等を作製することもできる。固相化の方法としては、ビオチン化、ポリペプチドの官能基とオルトゴナルな官能基の導入等が挙げられる。
【実施例】
【0061】
以下、実施例を挙げて本発明を詳細に説明するが、本発明の範囲はこれらの実施例により限定されるものではない。
実施例1 ライブラリーDNAの配列とその構築
ライブラリー化する配列として、3本のジスルフィド結合を持つ構造モチーフを10種、2本のジスルフィド結合を持つ構造モチーフを4種、1本のジスルフィド結合を持つループ状のモチーフを5種、ジスルフィド結合を持たない構造モチーフを3種、15残基のランダムライブラリーを1種、合計23種選択した。これらを発現させるために用いられるDNAの原料として、配列番号1〜51のDNAをDNA合成機により合成した。このうち配列番号1〜33はランダムあるいはセミランダム化する残基に相当する部位に混合塩基が導入されており、そのA:T:G:C比がKにおいて0:50:50:0、Mにおいて50:0:0:50となるようにホスホアミダイトを混合した。Nは合計4種類の混合比でホスホアミダイトを混合し、そのA:T:G:C比は混合比1が30:15:30:25、混合比2が30:30:30:10、混合比3が15:30:25:30、混合比4が30:30:10:30となるようにした。基本的に配列番号1〜14、16〜22においてKの前に位置するNが混合比2、Kの2つ前に位置するNが混合比1であり、配列番号15、23〜33においてはMの後に位置するNが混合比4、Mの2つ後に位置するNが混合比3である。この基本則に優先してNの混合比を指定した部位もあり、Gの次で後にKが続くNは混合比1、TとTに挟まれたNは混合比3、Mの次で後にCが続くNは混合比3とした。さらに配列番号1の37番目、配列番号2の72番目、配列番号6の61番目、配列番号8の57番目および配列番号10の57番目のNは混合比1、配列番号5の72番目および配列番号10の39番目のNは混合比2、配列番号28の49番目、配列番号29の20番目、配列番号31の42番目および配列番号33の24番目のNは混合比3、配列番号31の21番目および配列番号33の23番目のNは混合比4とした。
【0062】
化学合成したDNAをオーバーラップ伸長反応により連結した。反応系としてはTOYOBO社製KOD plusを用い、連結する2種のDNAの濃度を0.5Mとし、PCR装置中で94℃で2分反応後、98℃で10秒、60℃で30秒、68℃で5分の反応を5回ないし10回繰り返し、さらに68℃で5分反応させた。配列番号1と23、配列番号2と24、配列番号3と35、配列番号4と25、配列番号5と26、配列番号6と27、配列番号7と28、配列番号8と29、配列番号9と30、配列番号10と31、配列番号11と36、配列番号12と41、配列番号13と42、配列番号14と36、配列番号15と50、配列番号16と36、配列番号17と36、配列番号18と36、配列番号19と36、配列番号20と43、配列番号20と32、配列番号21と32、配列番号22と33、これらの組合わせで連結を行なうことによりそれぞれ生成物1〜23を得た。
【0063】
目的物の純度が低い場合は8M尿素変性ポリアクリルアミドゲル電気泳動(PAGE)で分離し、抽出濃縮操作によって次の反応に用いた。続いて生成物1と配列番号34、生成物2と配列番号34、生成物4と配列番号36、生成物5と配列番号36、生成物6と配列番号37、生成物7と配列番号38、生成物8と配列番号39、生成物9と配列番号37、生成物10と配列番号40、生成物21と配列番号44、生成物22と配列番号44、生成物23と配列番号36のオーバーラップ伸長反応によりそれぞれ生成物24〜35を得た。目的物の純度が低い場合は8M尿素変性PAGEで精製した。さらに生成物24と配列番号45、生成物25と配列番号46、生成物3と配列番号47、生成物26と配列番号45、生成物27と配列番号46、生成物28と配列番号48、生成物29と配列番号47、生成物30と配列番号45、生成物31と配列番号47、生成物32と配列番号47、生成物11と配列番号47、生成物12と配列番号47、生成物13と配列番号47、生成物14と配列番号49、生成物15と配列番号34、生成物16と配列番号47、生成物17と配列番号47、生成物18と配列番号47、生成物19と配列番号47、生成物20と配列番号47、生成物33と配列番号47、生成物34と配列番号51、生成物35と配列番号47、の組合せでオーバーラップ伸長反応を行い、それぞれ生成物36〜58を得た。これらはすべて8M尿素変性PAGEで精製し、得られたDNAを定量して転写用の鋳型とした。
【0064】
連結されたDNA配列は転写プロモーター配列のあとにmRNAに転写される配列として翻訳エンハンサー配列、ライブラリー配列、リンカー配列、FLAG配列、リンカー配列、ピューロマイシンリンカー結合配列を有している。得られたDNAのうち生成物39と41の配列解析を行なったところ、上記のコンストラクトを完全に含む配列は生成物39で19/31(61%)、生成物41で18/32(56%)であり、このうちランダム化部位に終止コドンを含まないものはそれぞれ15(48%)、14(44%)であった。この配列解析の結果得られたランダム化部位におけるアミノ酸出現頻度(観測値)をその期待値とともに図1に示した。
【0065】
実施例2 ライブラリーDNAの転写と転写産物のピューロマイシンリンカーとの連結
生成物36〜58のDNA分子を大きさなどを基準として9つのグループに分類し、Promega社製Ribomax Large Scale RNA Production Systemを用いて定法に従い転写反応を行なった。翻訳時におけるmRNAの安定性向上を目的として転写反応液にはキャップアナログ(7.2mM、RNA Capping Analog;Gibco BRL社製)を加え、mRNAの5’側を修飾した。9つのグループの組み合わせとDNA量(括弧内pmol)、転写反応液の量(括弧内・μl)は次の通りである。生成物36(4.2pmol)(50μl)、生成物37
(1.7pmol)と生成物49(6.7pmol)(50μl)、生成物38(5.8pmol)と生成物47(3.3pmol)と生成物48(4.2pmol)(70μl)、生成物39(7.5pmol)と生成物40(7.5pmol)(75μl)、生成物41(7.5 pmol)と生成物43(5.8pmol)と生成物44(5.8pmol)(100μl)、生成物42(6.7pmol)と生成物45(3.3pmol)(50μl)、生成物46(2.5pmol)と生成物55(3.3pmol)(50μl)、生成物50〜54(各3.3pmol)(80μl)、生成物56(1.7pmol)と生成物57(3.3pmol)と生成物58(3.3pmol)(60μl)。得られた生成物をこの順に従って生成物59〜67とする。分析のために各DNAを鋳型に小スケールで転写反応を行い、生成物36〜58から生成物68〜90を得た。
【0066】
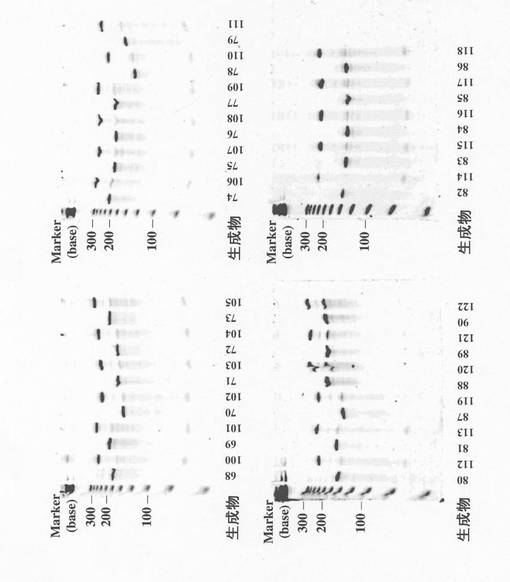
続いてこれらのmRNAを特願2003−315385号明細書に記載のピューロマイシンリンカー(T−splint5.9FA)と連結した。ピューロマイシンとmRNAの濃度がそれぞれ0.75μM、0.5μMになるように5%ジメチルスルホキシドを含むT4 RNAリガーゼ緩衝液(50mM Tris−HCl、pH7.5、10mM MgCl2、10mM DTT、1mM ATP)に溶解し、72℃で150秒加熱したのち室温で12分放置し、さらに15℃まで冷却したのち100分の1量(体積比)のT4 RNAリガーゼ(Takara社製)を加えて15℃で約2時間反応させた。実際に反応させたmRNAの量は生成物59〜67それぞれに対し100pmol、200pmol、320pmol、360pmol、460pmol、240pmol、140pmol、380pmol、200pmolであり、反応後に得られた産物をそれぞれ生成物91〜99とする。生成物68〜90それぞれに対しても20〜150 pmol相当のmRNAをピューロマイシンリンカーと反応させ、生成物100〜122を得た。生成物68〜90と生成物100〜122を8M尿素変性7%ポリアクリルアミドゲル電気泳動で分析した結果を図2に示す。
【0067】
実施例3 ポリペプチド−核酸連結分子の調製
上記の生成物91〜99を小麦胚芽無細胞蛋白質合成系(ゾイジーン社製)に6pmol/24μlの割合で加えて75分間翻訳させ、ポリペプチドと核酸の連結した対応分子が形成されるかどうかを確認した。合成系に加えた直後と75分後の混合液の一部(各生成物0.75pmol相当)を8M尿素変性7.5%SDS−PAGEで分離し、蛍光で検出した結果を図3に示す。さらに実際の選択実験に用いる対応付け分子を調製するため、生成物91〜99をそれぞれ40pmol、65pmol、65pmol、65pmol、80pmol、65pmol、30pmol、88pmol、102pmol、生成物100〜122を13.5pmolずつ同様の条件で翻訳した。
【0068】
翻訳後の溶液合計約3650μlに5M NaClを988μl、1M Tris−HCl(pH8.0)を49.4μl、0.5M EDTA−Naを148μl、10% Triton−X100を49.4μl、0.1M DTTを57μl加え、Biotinylated Oligo dTが結合したMAGNOTEX−SA(Takara社製)600μlと4℃で約2時間反応させた。上清を除き、緩衝液A(1M NaCl、50mM Tris−HCl(pH8.0)、0.1% Triton−X100、1mM DTT)で3回、緩衝液B(0.5M NaCl、50mM Tris−HCl(pH8.0)、0.1% Triton−X100、1mM DTT)で1回、さらに緩衝液C(0.25M NaCl、50mM Tris−HCl(pH8.0)、0.1% Triton−X100、1mM DTT)で1回洗浄した後、5mM DTT水溶液330μlで3回溶出した。
【0069】
この溶出画分に324μlの5×RT Buffer(Invitrogen社製)、162μlの10mM dNTP、60μlのRNase Inhibitor(WAKO社製;40U/μl)、84μlのSuperScript III Reverse Transcriptase(Invitrogen社製;200U/μl)を添加し、50℃で3分間加熱後、室温で12分放置した。この溶液に水94.5μl、5M NaClを54μl、0.1M リン酸水素2カリウムを324μl、10% Tween 20を22.5μl、50μg/ml tRNAを22.5μl、1mg/ml BSAを112.5μl加え、150μlの抗FLAG M2抗体アガロースビーズ(Sigma社製)と室温で1時間反応させた。
【0070】
上清を除き、0.1%のTween 20を含むpH 7.4のPBS(リン酸緩衝生理食塩水)360μlで3回、0.1%のTween 20を含むpH8.0のPBS360μlで3回洗浄し、100μMの3×FLAG peptideを溶かしたPBS(pH8.0)96μlで2回、37μMの3×FLAG peptideと0.1%のTween 20を溶かしたPBS(pH8.0)257μlで1回溶出した。合わせた溶出液にPBS(pH8.0)を16.8μl、0.5M EDTA−Naを2.4μl、10% Tween 20を2.4μl、2mM 酸化型グルタチオンを4.8μl、2mM 還元型グルタチオンを4.8μl加えて4℃で15時間反応させた後、約60μlずつをPBS(pH7.4)に置換したゲルろ過カラム(Bio−Gel P−6;BIO−RAD社製)に通して対応付け分子を精製した。これらを再び合わせた約480mlの溶液に10倍濃度のPBS(pH7.4)を12μl、4.8% BSAを78μl、2.5mg/ml tRNAを24μl、10% Tween 20を6μl加えて選択実験に用いる対応付け分子の溶液(約600μl)とした。
【0071】
実施例4 標的蛋白質結合担体の調製と対応付け分子の選択
スクリーニングの標的蛋白質の調製については、まず常法に従ってヒトインターロイキン6(interluekin 6;IL6)あるいはヒト腫瘍壊死因子α(tumor necrosis factor;TNF)のN末側にグルタチオンS−トランスフェラーゼ(gulutathione S−transferase;GST)を融合した蛋白質をコードするDNAが挿入された大腸菌発現用ベクターを調製した。両者の間にはprecision proteaseで切断されるペプチド鎖をコードした配列を挿入した。これを大腸菌(BL21(DE3)pLysS)で発現させ、目的とする融合蛋白質をGlutathione Sepharose4Bビーズ(アマシャムファルマシア社製)を用いて精製し、溶出に使用したグルタチオンは脱塩カラムにより除去した。またネガティブコントロール用としてGST蛋白質のみを合成し、同様の方法で精製した。もう一つの標的蛋白質としてgp130のFcキメラ(R&D SYSTEMS社製)を購入し、精製せずにそのまま用いた。この融合蛋白質はgp130とFcの間にFactor Xaで切断可能な配列を有する。
【0072】
GST(100μg)、GST−IL6(35μg)、GST−TNF(130μg)をそれぞれ250μlのPBS(pH7.4)−0.1% Tween 20溶液(以下、「PBST」と称することがある)に溶解し、30μlのGlutathione Sepharose4Bビーズと合わせて15時間静かに混合した後、上清を除いてPBSTで4回洗浄し、GSTビーズ、GST−IL6ビーズ、GST−TNFビーズを得た。プロテインGビーズ(Protein G Sepharose 4 Fast Flow;アマシャムバイオサイエンス社製)30μlをPBSTで洗浄し、gp130/Fc 18μgを溶かした0.2% BSAを含むPBST、200μlと合わせて1時間静かに混合し、上清を除いてPBSTで4回洗浄し、gp130/Fcビーズを得た。
【0073】
対応付け分子の溶液(約600μl)をGSTビーズと混合し、室温で1時間反応させた。上清を続いてGST−IL6ビーズと混合して反応を行い、その上清をとってGST−TNFビーズと反応させ、さらにその上清をプロテインGビーズ70μlに加えて反応を行い、最後にその上清をgp130/Fcビーズと混合して反応させた。反応はすべて室温で約1時間行なった。上清を除いた後のビーズはただちに150μlのPBSTで6回洗浄し、gp130/Fcビーズはさらに50mM Tris−HCl(pH7.5)、150mM NaCl、0.1% Tween 20の溶液(以下、これを「TBST」と称することがある)150μlで2回洗浄した。洗浄後のGSTビーズ、GST−IL6ビーズ、GST−TNFビーズには50mM Tris−HCl(pH8.0)、150mM NaCl、1mM DTTの溶液200μlとprecision protease溶液2μlを加えて室温で1時間反応させ、上清をとりビーズを同溶液200μlで洗った溶液と合わせて酵素溶出液とした。
【0074】
洗浄後のgp130/Fcビーズには50mM Tris−HCl(pH7.5)、150mM NaCl、0.1% Tween 20の溶液42μlとFactor Xa溶液(BioLabs社製)8μlを加えて室温で2時間反応させ、上清をとり、50mM Tris−HCl(pH7.5)、150mM NaCl、0.1% Tween 20、1mM EDTAの溶液175μlでビーズを2回洗って上清と合わせ酵素溶出液とした。各酵素溶出液にQuick precip Plusを30μl、イソプロパノールを430μl加えて遠心し、対応付け分子を沈殿物として回収して70%エタノール水溶液で洗い、風乾後、0.1%のTriton X−100を含む100mMの水酸化カリウム水溶液25μlに溶解した。室温で1時間反応させた後、0.1% Triton X−100を含む100mMの酢酸カリウム水溶液をほぼ等量加えて中和を確認し、PCR反応の鋳型溶液とした。
【0075】
PCRの反応サイクル数を決めるため、鋳型溶液の一部(4μl)で条件検討を行なった。プライマーとして配列番号52と53のDNA(それぞれ40nM)、反応系としてTOYOBO社製KOD plusを用い(鋳型溶液1μlに対し10μl)、PCR装置上94℃で2分反応後、98℃で10秒、60℃で30秒、68℃で90秒の反応サイクルを8、12、16、あるいは20回実施後、さらに68℃で5分反応させた。アガロースゲル電気泳動による解析結果より、GSTとGST−TNFに対しては反応サイクルを10回、GST−IL6とgp130/Fcに対しては12回として残りの鋳型溶液を用いてPCR反応を行なった。精製して得られたそれぞれのDNA溶液のうち約半分(DNAとして480 ng)を配列番号54と55のプライマーDNA(それぞれ40nM)とともにKOD plus反応液に加え(計480μl)、反応サイクル8回で再びPCR反応を行い、転写プロモーター配列からピューロマイシンリンカー結合配列までを含む最初のライブラリーと同じコンストラクトを有するDNAを得た。
【0076】
実施例5 ポリペプチド−核酸連結分子の濃縮操作
実施例2から実施例4の操作を第1ラウンドとし、GST、GST−IL6、GST−TNF、gp130/Fcのそれぞれに対して選択されたDNAをもとに次の選択を繰り返す操作を第10ラウンドまで実施した。具体的には、選択後にPCR反応を行なって得られた4種のDNA混合物のそれぞれを鋳型に実施例2の方法に基づいて転写反応を行い、得られたそれぞれのmRNA混合物をピューロマイシンリンカーと連結させた。この核酸複合体を実施例3の方法に基づいてそれぞれ翻訳し、mRNA/cDNAを遺伝情報として有する対応付け分子を調製した。実施例2と実施例3の方法に準じた第2ラウンド以降の操作は4種のライブラリー由来混合物を対象に並行して行い、反応のスケールを徐々に減らして実施した。実施例4に準じた選択実験も4種を並行して行い、GSTに対して選択された対応付け分子はGSTビーズと反応させ、GST−IL6あるいはGST−TNF に対して選択された対応付け分子はGSTビーズと混合した後に上清画分をGST−IL6ビーズあるいはGST−TNFビーズと反応させた。
【0077】
gp130/Fcに対して選択された対応付け分子はプロテインGビーズとまず反応させた後に上清画分をgp130/Fcビーズと反応させた。反応はいずれも第1ラウンドと同様に室温約1時間とし、ビーズに固定する蛋白量は段階的に減らして、最後の第10ラウンドにおいては対応付け分子と反応させる時の溶液量に対し、GSTが20μg/250μl、GST−IL6が10μg/250μl、GST−TNFが5μg/250μl、gp130/Fcが1.5μg/250μlであった。反応後の洗浄液の量と洗浄回数は段階的に増やし、第10ラウンドにおいてはGSTビーズを250μlのPBSTで8回、GST−IL6ビーズを250μlのPBSTで10回、GST−TNFビーズを250μlのPBSTで10回、gp130/Fcビーズを250μlのPBSTで4回、さらに250μlのTBSTで8回洗浄した。
【0078】
酵素溶出とPCR鋳型溶液の調製は実施例4とほぼ同様の条件で行い、配列番号52と53のDNAをプライマーとしたPCR反応は実施例4と同様に一部で条件検討を行い反応サイクル数を決定してから実施した。このPCR産物を鋳型とし、配列番号54と55のDNAをプライマーとしたPCRは鋳型DNA、150ng/KOD plus反応液300μl、反応サイクル8回を基本として実施した。
【0079】
実施例6 選択されたポリペプチドの結合試験
第10ラウンド選択後のPCRで得られたDNAをTaqポリメラーゼ(Takara社製)によりA付加し、pGEM T easy(プロメガ社製)を用いて常法によりプラスミドに挿入した。これを大腸菌JM109にトランスフェクションして増幅し、選択されたDNA配列を含むプラスミドを回収した。配列番号54と55のDNAをプライマーとしてPCR反応を行い、得られたDNAを鋳型に転写反応を実施してmRNAを得た。このmRNAを実施例3で用いた無細胞翻訳系で翻訳し、抗FLAG M2抗体アガロースビーズで精製した。DTTを含まない溶出液(TBST)中で15時間放置した後、1%のBSAを含むPBSTで約5倍に希釈してポリペプチド溶液とした。
【0080】
96ウェルのポリスチレンプレート(COSTAR社製;9018)の各ウェルにGST、GST−IL6、TNFのそれぞれを1μg/100μlの濃度でPBSに溶かした溶液を加えて4℃で一晩静置した。GSTとGST−IL6は実施例4で用いたものを使用し、TNFはGST−TNFをPrecision proteaseで消化してGlutathione Sepharose4Bビーズ上でGSTを除くことにより得た。
【0081】
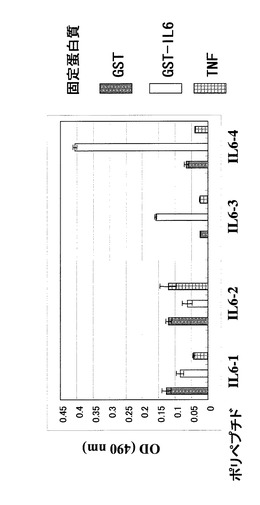
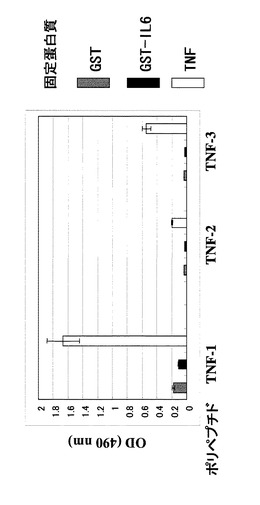
ウェルをPBSで2回洗浄し、1%のBSAを含むPBSを200μl加えて37℃で2時間振とうした後に溶液を除き、上記のポリペプチド溶液を加えて室温で1時間振とうした。溶液を除いてPBSTで3回ウェルを洗浄し、1mg/mlの抗FLAG M2抗体−HRP(SIGMA社製;A8592)を1%BSA/PBSで1万倍に希釈した溶液100μlを各ウェルに添加して室温で1時間静置した。溶液を除いてPBSTで3回ウェルを洗浄し、10mg/25mlのo−フェニレンジアミンと10μl/25mlの30%過酸化水素水を含むクエン酸緩衝液を各ウェルに100μlずつ加えて室温で10分静置した後に1規定の硫酸水溶液を等量加えて酵素反応を停止し、490nmの吸収をプレートリーダーで測定した。GST−IL6に対して選択された4種のポリペプチドに対する結果を図4に、GST−TNFに対して選択された3種のポリペプチドに対する結果を図5にそれぞれ示した。GST−IL6に対して選択されたもののうち、2種がGST−IL6に対して選択性を示し、GST−TNFに対して選択されたものは3種すべてTNFに対する選択性を示した。
【図面の簡単な説明】
【0082】
【図1】生成物39と41の配列解析の結果得られたランダム化部位におけるアミノ酸出現頻度(観測値)をその期待値とともに示したグラフである。
【図2】mRNA(生成物68〜90)とそれをピューロマイシンリンカーと連結させた反応物(生成物100〜122)を8M尿素変性7%ポリアクリルアミドゲル電気泳動で解析した写真である。
【図3】mRNA−ピューロマイシンリンカー複合体(生成物91〜99)を無細胞翻訳系に加えた反応液をSDS−PAGEで分離し蛍光で検出した写真である。
【図4】GST−IL6ビーズに対して選択されたポリペプチドの結合活性をプレートアッセイで測定した結果を示すグラフである。
【図5】GST−TNFビーズに対して選択されたポリペプチドの結合活性をプレートアッセイで測定した結果を示すグラフである。
【技術分野】
【0001】
本発明は、標的分子と相互作用するポリペプチドの選択方法、および目的蛋白質のファーマコフォアを明らかにするポリペプチドの取得方法等に関する。
【背景技術】
【0002】
従来の医薬品は各種抗生物質のような天然物、あるいはステロイドやカテコールアミンのようなヒト内在性物質から誘導された低分子化合物をベースに開発されているものが多い。一方で人工的に合成されたベンゼン置換体、複素環化合物誘導体など比較的シンプルな化合物についても医薬品としての成功例がしばしば見られる。これら従来の医薬品の開発においては限られた数の候補化合物を合成し、活性の確認を経たのち実用性の向上を目指した誘導体の合成展開が行なわれるのが一般的であり、合成と活性測定の対象となる化合物の数は限られていた。しかし医薬品開発の初期段階における合理性が追求された結果、多数の化合物に対して並行して活性試験を行なうハイスループットスクリーニングシステム(HTS)がリード化合物取得のために用いられるようになり、特に90年代初期に進展したコンビナトリアルケミストリーの技術は多検体アッセイ系の進歩とあいまって数百から数十万の低分子化合物ライブラリーを短期間でスクリーニングすることを可能にしている。こうしたシステムは天然の化合物に依存しないリード化合物開発の成功確率を高める一方、アッセイ系でヒットした化合物が医薬として不適である場合が少なくないという問題も生み出している。
【0003】
しばしば医薬リードとして重要視される生理活性ペプチドはアミノ酸重合体としてコンビナトリアルケミストリーの概念が適用されやすい対象であり、天然のペプチド性リガンドの活性増強のみならず、新規なリガンドの開発も数多く試みられている。化学合成の手法においては非天然型アミノ酸も導入可能な固相合成法がアレイ技術などとともに用いられることが多い。遺伝子工学的な手法も応用可能であり、特に80年代後半に実用化されたファージディスプレー法はペプチドなどの発現物を増幅可能な遺伝子と対応付けさせることで数百万から数億の多様性を有するライブラリーをスクリーニング対象とすることを可能にした。
【0004】
ペプチドあるいはその類縁体に属する従来の医薬品はアンギオテンシン、エンドセリン、ソマトスタチン、バソプレッシンなどヒトに内在する生理活性ペプチドリガンドに由来するアゴニストまたはアンタゴニスト、あるいはプロテアーゼの活性を抑えるための基質配列に由来する阻害剤であることが多い。こうした内在性のリガンドや基質に由来する医薬品の有用性は明らかであるが、創薬リードに成り得るそれらのペプチドの数には限界があり、疾患に対して理想的な創薬を可能にするには新たな相互作用の組合せを開拓する必要がある。ファージディスプレー法のように人工的なライブラリーを提示してスクリーニングをする技術はそれに適していると考えられ、蛋白質―蛋白質相互作用を阻害するペプチドの取得などが実際に試みられている。
【0005】
しかし医薬のリードとなるペプチドがこの方法により取得される可能性は極めて限られていると予想される。その理由の第一は、ペプチドによる蛋白質―蛋白質相互作用阻害といった人工的な作用機序が受容体へのリガンド結合による応答という自然な機序と同等な薬理活性を生み出し得る場合が限られること、第二は受容体などの相互作用対象と共進化の結果生まれた内在性アゴニストと同じような高い親和性を標的蛋白質と人工的ペプチドの間に持たせることが原理的に難しい場合が多いこと、第三は化学的手法に比べて多様性が高いとは言えファージディスプレー法でスクリーニング可能なライブラリーにも限界が見られること、であり、技術的な解決が期待できる第三の問題において改善を試みない限り人工的な相互作用の同定に基づく医薬リードの創製は効率の悪い方法となる。
【0006】
ファージディスプレー法において1度のスクリーニングにより探索可能な多様性の範囲は10の6乗から9乗程度であり、これは20種類のアミノ酸の組合せの数として5残基から7残基分に相当する。構造活性相関研究により標的分子との相互作用において特に重要な役割を果たしているとされるアミノ酸の数は一般に3から8程度であることが多いが、これは標的分子と直接相互作用しているアミノ酸に注目した場合の数であり、標的分子側が連続した数残基を特異的にリガンドとして認識するような深いキャビティを有していない限り安定な主鎖構造を持たない数残基のペプチドが高い親和性と特異性をもって標的分子に結合することはほとんど起こらない。
【0007】
ペプチドと標的分子の相互作用の強さは両者の結合に伴う自由エネルギーの変化により決まるものであり、深いキャビティを持たない標的分子に対して強い相互作用がはたらくためには、結合に適した主鎖構造や側鎖の空間配置をペプチド側が持つことにより結合に伴うエントロピー的な不利が少なくなること、あるいは広い結合界面により局所的な相互作用の足し合わせがエネルギー的に有利であることが求められる。前者の条件に関しては、ジスルフィド結合で環状化しているような場合を除いて10残基以下のペプチドが特定の安定した構造をとることは少なく、後者についても広い結合界面を形成するには10残基以上が必要となり、いずれにしてもファージディスプレー法で網羅できる多様性の範囲を超えている。
【0008】
医薬開発におけるペプチドスクリーニングは相互作用様式の解明による低分子化を目的として行なわれることが多い。ペプチドが内在性リガンドである場合、それが得られていることは標的分子の機能解明とリード化合物のスクリーニングに貢献するだけでなく、低分子リガンド設計にも直接的な情報を与える。人工的な相互作用様式の創出を伴うランダムペプチドライブラリーからのスクリーニングもファーマコフォアの同定による低分子化を目的の一つとして行われることが多い。この場合、特定の構造を持たずに標的分子と広い結合界面を形成することによって全体の親和性を獲得している比較的大きなペプチドは低分子でそれを再現することが原理的に困難である。したがって低分子化を目的として行なわれる人工的ペプチドリガンドのスクリーニングにおいては比較的限られた空間内において相互作用界面を抽出すること、そしてその成功確率を高めるためにはペプチド全体から生み出される構造の安定性が結合に伴うエントロピー的な不利を少なくするようなライブラリーを用いること、が重要となる。
【0009】
独立して安定に存在できる蛋白質の3次構造は一般に百残基以上によって形成され、しばしば機能の単位と重複する。一方で比較的独立した小さな構造が蛋白質の中に含まれていることもあり、核内受容体のDNA結合ドメインに含まれるジンクフィンガーモチーフ、カルモジュリンなどにおいてカルシウム結合部位となるEFハンドモチーフなどはその代表例である。さらに十数残基から50残基程度の大きさで3次構造を形成するペプチド・小型蛋白質も存在し、その中で複数のジスルフィド結合が構造形成の中心になっているものは特にシスチンノットモチーフと呼ばれる。これらの構造モチーフは構造の安定性を持ったペプチドライブラリーの土台として有用であり、ファージディスプレー法において応用されているものも多い(例えば、非特許文献1および2を参照)。
【0010】
しかしファージディスプレー法でスクリーニング可能なペプチドの数を考えた場合、こうした構造モチーフの導入はむしろライブラリーの多様性を自ら限定することにつながり易いだけでなく、構造維持に耐えられない変異が導入されたものやフォールディングの問題からファージ上で提示不可能なものなどが想定の範囲を超えて含まれているという理由により目的の質を持ったライブラリーの多様性が期待したスケールよりも低いという問題ももたらす。限られた空間内においてファーマコフォアを抽出し、医薬開発に有益な情報を効率良く得るためには投入できるライブラリーのスケールにおいてファージディスプレー法を上回るスクリーニング技術が求められ、さらにそのスケールの大きなライブラリーは質の点においても優れていることが望まれる。
【0011】
スクリーニング可能なライブラリーのスケールがファージディスプレー法を上回り、遺伝子工学的手法に特有な問題点に対してもある程度解決が期待できる方法としてリボソームディスプレー法と無細胞蛋白質ディスプレー法(例えば、特許文献1を参照)がある。いずれも細胞を介さずに遺伝情報と発現物を対応付けさせる技術であるが、前者は対応付け分子複合体の作製が容易である反面、その安定性維持のためスクリーニング条件に制約があり柔軟性に乏しい。後者は遺伝情報(mRNA)と発現物(蛋白質あるいはポリペプチド)をピューロマイシンリンカーを介して共有結合させることにより対応付けする技術であり、様々な条件下でスクリーニングできるだけでなく対応付け分子として前者よりはるかに小さいという利点も持つ(例えば、特許文献1あるいは非特許文献3を参照)。
【0012】
しかしmRNAが溶媒中に露出しているというリボソームディスプレー法と共通した特徴がリボヌクレアーゼなどの混入する系での安定性という面で問題となるだけでなく、発現物と同等あるいはそれ以上の多様性を有するmRNAがアプタマーとして標的分子その他と無視できない親和性で結合するという問題も生じさせ、特にランダムペプチドライブラリーのような多様性が大きい系ではそれが無視できなくなることが推測されていた。我々は新たなピューロマイシンリンカーの開発などにより、遺伝情報側を効率良く2本鎖核酸とすることでmRNAのアプタマーとしての性質を抑える実用的なスクリーニング系の開発に成功し、無細胞蛋白質ディスプレー法によってファージディスプレー法では対処できない多様性の大きさを持ったライブラリーから有用ペプチド・蛋白質をスクリーニングすることを可能にしていた(例えば、特許文献2を参照)。
【0013】
これらの技術の応用としてはこれまでのところ、天然に得られる遺伝子からライブラリー化されたポリペプチドから標的分子と相互作用する配列を選抜取得するスクリーニングのみしか行われておらず、取得される標的分子と相互作用するポリペプチドの機能にも限界があった。
【特許文献1】WO98/16636号公報
【特許文献2】特願2003−315385号明細書
【非特許文献1】Science,263,671−673(1994)
【非特許文献2】J.Mol.Biol.277,317−332(1998)
【非特許文献3】Nature,410,715−718(2001)
【発明の開示】
【発明が解決しようとする課題】
【0014】
本発明は、ポリペプチドあるいは蛋白質が持ち得る多様性の大きさと比較した場合に、実際の溶液中において行なわれる従来のペプチドあるいは蛋白質のスクリーニング系が、網羅性に著しく欠けていた点を改善し、医薬リード化合物の設計などにおいて重要な標的分子とのファーマコフォアを明らかにするポリペプチドを効率良く取得する方法を提供するためになされたものである。
【課題を解決するための手段】
【0015】
本発明者は、上記課題を達成するために鋭意検討を進めた結果、比較的安定な2次ないし3次の構造を有する天然に由来する構造モチーフあるいはそれらの構造を持つことが期待される人工的な構造モチーフを複数選び出し、それぞれに4ヶ所以上の変異を導入したライブラリーを構築し、さらに通常のランダムペプチドライブラリーもこれに加えて、無細胞蛋白質ディスプレー法を用いて標的蛋白質と接触させ、該蛋白質と相互作用するポリペプチドを選択する工程を繰り返すことにより、より特異的に、また高い親和性で標的蛋白質と相互作用をするポリペプチドを取得することに成功した。本発明は、これらの知見に基づいてなされたものである。
【0016】
すなわち、本発明によれば、
(1)変異を導入した構造モチーフを標的分子と接触させ、標的分子と相互作用するポリペプチドを選択することを特徴とする標的分子−ポリペプチド間相互作用解析方法、
(2)変異を導入した構造モチーフおよびランダムペプチドを標的分子と接触させ、標的分子と相互作用するポリペプチドを選択することを特徴とする標的分子−ポリペプチド間相互作用解析方法、
(3)標的分子と相互作用するポリペプチドを選択する方法が、無細胞蛋白質ディスプレー法である上記(1)または(2)に記載の方法、
(4)変異を導入した構造モチーフを標的分子と接触させ、標的分子に対し高親和性を示すポリペプチドを選択することを特徴とするファーマコフォアを明らかにするポリペプチドの取得方法、
(5)変異を導入した構造モチーフおよびランダムペプチドを標的分子と接触させ、標的分子に対し高親和性を示すポリペプチドを選択することを特徴とするファーマコフォアを明らかにするポリペプチドの取得方法、
(6)標的分子に対し高親和性を示すポリペプチドを選択する方法が無細胞蛋白質ディスプレー法である上記(5)に記載の方法、
(7)上記(4)〜(6)のいずれかに記載の方法により得られたポリペプチドのアミノ酸配列からアミノ酸の存在頻度および/または出現頻度を指標として共通配列を選択する工程を含むことを特徴とする医薬的活性を有するポリペプチドの取得方法、
(8)上記(4)〜(7)のいずれかに記載の方法により得られたポリペプチドを化学的に改変して医薬的活性を向上させることを特徴とするペプチド誘導体の取得方法、
(9)上記(4)〜(8)のいずれかに記載の方法により得られたポリペプチドあるいはペプチド誘導体と標的分子との複合体を作製する工程、該複合体の立体構造解析を行い、標的分子と該ポリペプチドあるいは該ペプチド誘導体との結合部位を特定する工程、同定された結合部位から標的分子のファーマコフォアを明らかにして、該ファーマコフォアに合う低分子化合物を設計する工程を含むことを特徴とする標的分子と相互作用する化合物の分子設計方法、
が提供される。
【発明の効果】
【0017】
本発明によれば、従来の高次構造解析の結果からは予測が困難な標的蛋白質のファーマコフォアを明らかにするポリペプチド、サブタイプなど類似の標的分子を選択的に認識するポリペプチド等を溶液中での実際のスクリーニングにより効率良く取得する方法が提供される。該方法は非天然型アミノ酸を変異部位に導入するスクリーニングにも対応しやすく、得られたポリペプチドのC末端にPEGなどの高分子を導入して安定化を図ることにも適する。該方法により得られたポリペプチドは標的分子と複合体を形成した状態での構造解析、低分子医薬の設計、ペプチド誘導体あるいは蛋白性医薬の開発、医薬リードスクリーニング系におけるコントロール用リガンドの取得、標的分子の検出あるいはイメージング、等において有用である。さらに、安定な構造を土台に持つポリペプチドライブラリーを複数使用してファーマコフォア同定のためにスクリーニングするという方法は実際の溶液中でのスクリーニングのみならず、コンピューターを用いた仮想結合実験においても応用できる。
【発明を実施するための最良の形態】
【0018】
以下、本発明を更に詳細に説明するが、以下の構成要件の説明は、本発明の実施態様の代表例であり、本発明はこれらの内容のみに特定されるものではない。
(1)ライブラリーの設計と構築
本発明の1つは、変異構造モチーフ、あるいは変異構造モチーフとランダムペプチドからなるライブラリー(以下、これを「ライブラリー」と称することがある)を標的分子と接触させ、標的分子と相互作用するポリペプチドを選択することを特徴とする標的分子−ポリペプチド間相互作用解析方法である。また、標的分子が蛋白質である場合、上記ライブラリーと標的蛋白質とを接触させ、標的分子と高い親和性を示すポリペプチドを選択することで得られるポリペプチド(本明細書中ではこれを「ファーマコフォアを明らかにするポリペプチド」と称する)は該蛋白質のファーマコフォアを明らかにするものであり、医薬として実用化が可能な低分子化合物あるいはポリペプチド誘導体の開発において有用である。
【0019】
そこで、本発明の方法に適したポリペプチドのライブラリーを考えると、高い親和性と特異性をもたらすファーマコフォアが選択されるポリペプチドと標的分子との間の比較的限られた空間に存在することが望ましい。そのようなファーマコフォアを効率良く同定するためのポリペプチドを選択するためには限られた空間におけるポリペプチドの多様性を可能な限り大きくすることが必要であり、標的分子と直接相互作用するアミノ酸残基のみならず、それらの配向や揺らぎを規定する他の残基においても多様性が求められる。
【0020】
ペプチド鎖は側鎖だけでなくアルファ炭素の両側においても比較的高い回転の自由度を持つために仮想的には一定の空間内で様々な構造をとり得るが、実際の標的分子との相互作用においては結合に伴う内部エネルギー的な有利性がエントロピー的な不利を相殺できないような主鎖構造は生じ得ない。選択されるポリペプチドと標的分子との高い親和性は、標的分子側が選択されるポリペプチドの主鎖構造を安定化させるような明確なキャビティを持っていること、あるいは選択されるポリペプチド側が標的分子との結合に適した主鎖構造をとり易いこと、のいずれかによって可能になっていることがほとんどであり、特に20残基を超えるようなポリペプチドに関しては後者の例が多い。したがって、深いキャビティを持たない標的分子に対してファーマコフォアを特定するためには、主鎖構造が二次ないし三次構造のレベルで安定になりやすいポリペプチドをライブラリー化してスクリーニング対象とすることが合理的と考えられる。
【0021】
直接的に医薬リード化合物となるようなポリペプチドをスクリーニングにより効率良く得る技術を提供することも本方法の目的の一つであるが、安定な構造をとり易いポリペプチドはペプチダーゼやプロテアーゼに対する感受性が一般に低くなることが期待でき、血中での安定性という点で有利になりやすいと予想される。本発明において使用されるライブラリーは以上のことを踏まえて選定と構築がなされる。
【0022】
ここで、「ポリペプチド」とは、残基数において6残基から10残基程度の一般にオリゴペプチドと呼ばれるものから、構造安定性の高い120残基程度の大きさを持った蛋白質までを含むものとする。また、「相互作用する」とは、標的分子に結合して該物質の機能を制御することを意味する。さらに、「ファーマコフォア」とは、標的分子と相互作用する分子と標的分子の結合界面に存在して、親和性と特異性の獲得に寄与しているアミノ酸残基や官能基などの複数の部分構造の空間配置を意味する。また、本発明の方法で用いる「標的分子」とは、蛋白質、ポリペプチド、核酸、糖質、脂質、低分子化合物等のいずれのものでもよいが、特定の疾患と関連し、該分子の機能の制御が、該疾患の治療に有用であることがわかっている物質が特に好ましく用いられる。
【0023】
本発明において、用いられる「構造モチーフ」とは、天然の蛋白質あるいはポリペプチドの全体ないし一部であってもよく、天然の構造からの類推または論理的設計によって得られた人工的なものでもよい。天然のポリペプチドの例としては、コノトキシン、アガトキシン、デンドロトキシン、チャリブドトキシン、アパミンなどのイオンチャネル阻害ペプチド、テッポウウリ由来トリプシンインヒビターやアプロチニンなどのプロテアーゼ阻害ペプチド、ディフェンシンやヘプシジンなどの抗菌ペプチド、エンドセリンやインシュリンなどのペプチド性ホルモンが挙げられ、これらの多くにおいては二次以上の高次構造が複数のジスルフィド結合によって安定化されている。バソプレッシン、ウロテンシン、オキシトシン、ソマトスタチンのように一つのジスルフィド結合を有する重要な生理活性ペプチドもあり、二つのシステインの間に4残基から12残基のアミノ酸を含むライブラリーも主鎖構造がある程度制限されたライブラリーとして利用される。
【0024】
蛋白質の一部ないし全体に由来するものとしては、ジンクフィンガーモチーフ、EFハンドモチーフ、アンキリンリピート、4本ヘリックスバンドル、免疫グロブリンフォールドのように多くの蛋白質に共通して現れる構造が有用である。人工的に設計される構造としては、ヘリックスターンヘリックスのようなアルファヘリックス構造を中心とした配列のライブラリー化が比較的容易である。構造モチーフという言葉は、βターン構造のような定義が明確なものを除けば、二次構造が連結して生じる部分構造や金属イオンへの配位結合で生じる小さな構造など十数残基から数十残基程度の特徴的な構造を意味することが多いが、ここでは一つのジスルフィド結合で生じる数残基から成るループ構造や、一般的な蛋白質のドメイン構造に近い大きさを持つ三次構造、あるいは標的分子との相互作用により初めて安定化される柔軟な構造も含めて「構造モチーフ」と定義する。
【0025】
このような構造モチーフの中で、本発明の方法に用いるライブラリーとしての配列の選定は、選定された構造モチーフの組合わせによって一次構造上の多様性だけでなくファーマコフォアを探索する空間におけるライブラリーの多様性が大きくなることを目的として行なわれる。例えば一本のαヘリックス、βストランド、あるいはループ上において空間的に近い数残基はその組合せが同じであっても異なるものと見なすことができ、特にループ部分はその配列や周辺の構造によって異なる主鎖構造をとり易い。さらに三次構造的にはそれらの残基が一定の空間内で組合されることになり、主鎖構造及び側鎖の配向と種類が生み出す多様性が著しく大きくなる。
【0026】
このような多様性を確保するには異なる構造モチーフライブラリーを多種類併用することが望ましいが、実際に用いるスクリーニング系が扱うことの可能なライブラリーのスケールには限界があるため、併用する構造モチーフライブラリーの数と種類、各ライブラリーにおける変異導入箇所の選択は制約の下で有効な多様性を生み出すように行なわれる必要がある。具体的には、用いる構造モチーフのもととなるポリペプチドはその構造が比較的複雑である場合に、立体構造がX線結晶構造解析などにより解明され実験的な証明が得られていることが望ましい。立体構造上の情報は相互作用クラスターとして複数の変異を導入する空間を決定する目的で求められるだけでなく、構造維持に著しく不利な変異の導入を避けるためにも重要となる。
【0027】
例えばβターン構造上に存在するグリシン残基の変異はしばしば分子全体のフォールディングを著しく阻害することが知られており、あるいはプロリン残基は立体構造の中心となる二次構造の形成に不利になることが多い。立体構造が既知であればフォールディングにおいて脱落するポリペプチドを少なくしながら構造モチーフライブラリーの多様性の維持を図ることが比較的容易になる。
【0028】
変異の導入がフォールディングに著しく不利にならない範囲では、二つ以上の相互作用クラスターを一つの分子に導入することも可能である。無細胞蛋白質ディスプレー法をスクリーニングに用いる場合、4〜25の変異部位を導入した構造モチーフ、および完全ランダムライブラリーとともに4〜50種類まとめてスクリーニングすることが好ましい。
スクリーニング技術に由来する制約もいくつか存在し、中でもフォールディングに関する問題はライブラリーの選択に大きな影響を与える。酵母ツーハイブリッド法、ファージディスプレー法、リボソームディスプレー法、無細胞蛋白質ディスプレー法は、フォールディングも含めた発現の効率において一長一短を持ち、翻訳される蛋白質あるいはポリペプチドごとに適不適がある。生物系に依存しない上に比較的安定な対応付け分子を形成する無細胞蛋白質ディスプレー法は翻訳後の巻き戻し操作などにおいて有利な点を持っていると考えられ、複数のジスルフィド結合を持つシスチンノットモチーフ等に対しては特にこの利点を応用しやすいが、すべての構造モチーフのフォールディングに適した条件を確立することは困難であり、さらに提示されるポリペプチドのC末端がピューロマイシンを介したmRNAとの連結のために塞がれているということも時として問題になると予想される。このため構造モチーフの選択に際しては、化学合成物や組換発現物などにおいてフォールディングの容易さが確認されているものを優先することが望ましく、さらにそれらの中ではC末端が溶液中に露出してリンカー配列等の付加によるフォールディングの阻害が予想されないものが望ましい。
【0029】
ライブラリーの変異箇所に導入されるアミノ酸残基の出現確率は、構造モチーフに合わせて可能な範囲で変えることが必要となる。例えば、ジスルフィド結合によって形成される構造モチーフをライブラリーとして用い、あるいは標的分子が細胞外の蛋白質でジスルフィド結合が形成され易い条件でスクリーニングを行なう場合、ランダム部位に出現したシステインは期待した構造モチーフの形成を阻害し、あるいは他の分子との間で架橋される可能性があるため、ランダム化した部位のシステイン出現頻度は低いことが望まれる。さらにαヘリックスやβストランドの変異による不安定化が構造維持を阻害する可能性が高いと考えられる部位においては、それらの二次構造に不利にならないセミランダム化を行なうなどの対応も必要となる。加えて、トリプトファンやチロシンなど一分子に数多く出現した場合に非特異結合が増えるような残基は出現頻度を高くしないこと、最初に選択するライブラリーのスケールを落とすことになる終止コドンの出現頻度も制限すること、なども望まれる。これらのアミノ酸残基出現頻度の制御は、主にDNA化学合成の段階で塩基の混合比を設定することにより行なわれる。
【0030】
ライブラリーの構築は、主に化学的に合成した複数のDNAのオーバーラップ伸長法により行なわれる。オーバーラップさせる末端の相補鎖形成配列は、ランダム化部位を含まないほうが望ましい。ライブラリーの多様性の維持と各分子のコピー数格差抑制のため、オーバーラップ伸長反応のサイクルは1〜10回程度に抑え、増幅のためのPCR反応は行なわないことが望ましい。
【0031】
(2) ファーマコフォアを明らかにするポリペプチドおよびそれをコードする核酸の選抜取得方法
本発明のファーマコフォアを明らかにするポリペプチドあるいは後述するドメインポリペプチドの選抜取得方法として、酵母ツーハイブリッド法、ファージディスプレー法、リボソームディスプレー法、無細胞蛋白質ディスプレー法などが用いられるが、より大きなスケールのライブラリーの中からポリペプチドを柔軟な条件で選択できる無細胞蛋白質ディスプレー法が特に好ましく用いられる。
【0032】
無細胞蛋白質ディスプレー法によるポリペプチドの選抜取得方法は、 (1)ポリペプチド部とそれをコードする核酸部が、核酸部の3’末端に結合した核酸構築物を介して直接結合したポリペプチド−核酸連結体群と標的分子とを接触させる工程、(2)標的分子に特異的に結合した該ポリペプチド−核酸連結体を取得し、該連結体の核酸部を増幅し、これを鋳型の一部として用いてポリペプチド部とそれをコードする核酸部が、核酸部の3’末端に結合した核酸構築物を介して直接結合したポリペプチド−核酸連結体群を調製する工程、(3)上記(1)および(2)の工程を、標的分子と相互作用させたポリペプチド−核酸連結体の核酸部が、標的分子に特異的に結合するポリペプチドをコードすることが確認されるまで繰り返す工程を含むことを特徴とするものである。具体的には、以下のとおりである。
【0033】
(i)ポリペプチド−核酸連結体および該連結体と標的分子を接触させる工程
本発明の方法で用いられる「ポリペプチド−核酸連結体」は、例えば、WO98/16636号公報に記載のもの等のように、ポリペプチド部とそれをコードする核酸部が、核酸部の3’末端に結合した核酸構築物を介して直接結合したものであり、蛋白質の相互作用解析等の強力なツールとなり得る分子である。
【0034】
ここで、「ポリペプチド部」とは、核酸部に含まれるコーディング配列によりコードされるポリペプチドを含み、ポリペプチドは上記ライブラリーである。また、ポリペプチド−核酸連結体を精製するためのタグと上記蛋白質との融合ポリペプチドも含まれる。「核酸部」とは、ポリペプチド部のポリペプチドをコードするコーディング配列を含む核酸であり、さらに、用いるポリペプチド合成系においてポリペプチドの翻訳が開始されるための配列も含むことが多い。核酸部は一般にmRNAであり、その鋳型となるDNAを「核酸部前駆体」とする。
【0035】
核酸部前駆体は、具体的には、コーディング領域と発現制御領域を含むものである。発現制御配列とは、(1)プロモーター配列、(2)翻訳の際にリボソームによって認識される配列等が挙げられる。プロモーター配列の種類は、適用する発現系に適したものを適宜選択すればよく、特に限定されない。例えば、大腸菌ウイルスT7のRNAポリメラーゼによって認識されるT7プロモーター配列、SP6 RNAポリメラーゼにより認識されるSP6プロモーター配列などが挙げられる。また、翻訳の際にリボソームによって認識される配列としては、翻訳の際に真核細胞のリボソームによって認識されるRNA配列(Kozak配列)に対応するDNA配列や、原核細胞のリボソームによって認識されるシャイン・ダルガノ配列(Shine-Dalgarno)、5’キャップ構造(Shatkin,Cell,9,645−(1976))、オメガ配列等のtabacco mosaic virusのリボソームによって認識される配列、WO03/56009号公報に記載の配列、rabbitβ−globlin、Xenopusβ−globlin あるいはbromo mosaic virusのリボゾーム認識領域などが挙げられる。また、ポリペプチド−核酸連結体の核酸部を増幅するのに、PCRを用いる場合、PCRプライマー用配列を含むものも挙げられる。また、核酸部前駆体を増幅するためにPCRを用いる場合には、PCRプライマー結合用の共通配列を含むものも挙げられる。
【0036】
コーディング配列とは、ポリペプチド−核酸連結体の蛋白質部に含まれるポリペプチドをコードする配列であり、この種類は特に限定されず、目的に応じて適宜選択できる。また、該核酸部の3’末端に結合した「核酸構築物」とは、その末端に「核酸誘導体」を結合しており、該核酸部を鋳型として無細胞蛋白質翻訳系又は生細胞中で蛋白質(ポリペプチド)の翻訳を行った場合、mRNAの末端付近まで翻訳が進んだ後、該核酸誘導体(例えば、ピューロマイシンなど)がリボソームのAサイトに入ることにより蛋白質(ポリペプチド)と結合させる機能を有するものを意味する。具体的には、スペーサー、該ポリペプチド−核酸連結体を標識する標識部、また該連結体を固相に結合するための構造、核酸部を逆転写するためのプライマー部等を含むものが好ましい。
【0037】
「核酸誘導体」としては、無細胞蛋白質翻訳系又は生細胞中で蛋白質(ポリペプチド)の翻訳が行われた時に、合成された蛋白質(ポリペプチド)のC末端に結合する能力を有する化合物である限り限定されないが、その3’末端がアミノアシルtRNAに化学構造骨格が類似しているものを選択することができる。代表的な化合物として、ピューロマイシン(Puromycin)と3’−N−アミノアシルピューロマイシンアミノヌクレオシド(3'-N-Aminoacylpuromycinaminonucleoside 、 PANS-アミノ酸)、すなわち、アミノ酸部がグリシンのPANS−Gly、アミノ酸部がバリンのPANS−Val、アミノ酸部がアラニンのPANS−Ala、その他、アミノ酸部が全ての各アミノ酸に対応するPANS−アミノ酸化合物が挙げられる。
【0038】
また、3’−アミノアデノシンのアミノ基とアミノ酸のカルボキシル基が脱水縮合して連結した3’−N−アミノアシルアデノシンアミノヌクレオシド(3'-Aminoacyladenosine aminonucleoside, AANS-アミノ酸)、すなわち、アミノ酸部がグリシンのAANS−Gly、アミノ酸部がバリンのAANS−Val、アミノ酸部がアラニンのAANS−Ala、その他、アミノ酸部が全アミノ酸の各アミノ酸に対応するAANS−アミノ酸化合物を使用できる。また、核酸あるいは核酸とアミノ酸のエステル結合したものなども使用できる。さらにまた、核酸あるいは核酸に類似した化学構造骨格及び塩基を有する物質と、アミノ酸に類似した化学構造骨格を有する物質とを化学的に結合した化合物は、すべて本発明で用いられる核酸誘導体に含まれる。核酸誘導体としては、ピューロマイシン、PANS−アミノ酸もしくはAANS−アミノ酸がリン酸基を介してヌクレオシドと結合している化合物がより好ましい。これらの化合物の中でピューロマイシン、リボシチジルピューロマイシン、デオキシシチジルピューロマイシン、デオキシシチジルデオキシシチジルピューロマイシン、デオキシウリジルピューロマイシンなどのピューロマイシン誘導体が特に好ましい。
【0039】
このような核酸誘導体は、それ自体既知の化学結合方法によって製造することができる。具体的には、リン酸ジエステル結合で合成ユニットを結合させる場合は、DNA合成機に一般的に用いられているホスホアミダイド法などにより固相合成で合成することが可能である。ペプチド結合を導入する場合は、活性エステル法などにより合成ユニットを結合させるが、DNAとの複合体を合成する場合は、両方の合成法に対応が可能な保護基が必要になる。
【0040】
「スペーサー」は、ポリエチレン又はポリエチレングリコールあるいはその誘導体などの高分子物質や、オリゴヌクレオチドやペプチドあるいはその誘導体などの生体高分子物質等が用いられ、好ましくはポリエチレングリコールが用いられる。スペーサーの長さは特に限定されないが、好ましくは、分子量150〜6000であるか、または主鎖の原子数は10原子から400原子であり、さらに好ましくは、分子量600〜3000であるか、または主鎖の原子数が40原子から200原子である。また、標識部を有しててもよい。
【0041】
「標識部」は、親和性物質、共有結合性物質、蛍光物質、分解性物質等が挙げられる。これらは、「該連結物を固相に結合するための構造」としても用いられるし、また「該連結物を精製するための構造」としても用いられる。親和性物質としては、ポリA配列、ポリT配列、ビオチン、FLAG等の各種抗原又は抗体、Hisタグ、NTA等の配位子、受容体リガンド等が挙げられる。また、共有結合性物質としては、デオキシリボヌクレオチド、リボヌクレオチド等の核酸末端部分、ヒドラジド、ケトン、チオエステル等の官能基、ソラレン等の架橋性物質が挙げられる。蛍光物質としては、フルオレセイン、オレゴングリーン、ローダミン、テトラメチルローダミン、テキサスレッド、Cy3、Cy5、Alexa488等が挙げられる。分解性物質としては、光反応で分解する1-(2-ニトロフェニル)-エチル基を有する誘導体や、プロテアーゼやペプチダーゼに認識されるアミノ酸配列等が挙げられる。これらの標識物質は、それ自体既知の通常用いられるものであり、容易に入手可能であり、また常法により核酸等に結合して標識することができる。
【0042】
好ましい核酸部とその3’末端側に結合した核酸構築物、核酸誘導体(以下、これを「ポリペプチド−核酸連結体鋳型」と称することがある)の好ましい構造としては、核酸部であるRNAと、核酸誘導体を末端に有するスペーサーが枝分かれした状態で結合している1本鎖核酸(以下、これを「スペーサー鎖」と称することがある)がアニーリングした構造を有し、さらに該スペーサー鎖と標識部を含む1本鎖核酸と核酸部がそれぞれ結合しているものが挙げられる。具体的には、PCT/JP2004/013399号明細書に記載のもので、製造法も該明細書に記載された方法が好ましく用いられる。このような構造を有することにより、核酸部と核酸構築物のRNAリガーセ等による結合効率が高く、またポリペプチド−核酸連結体を標識部により精製でき、核酸部のRNAを逆転写することができ、さらに核酸部を2本鎖DNAとすることもできる。
【0043】
かくして構築されたポリペプチド−核酸連結体鋳型を蛋白質翻訳系に導入することによりポリペプチド−核酸連結体を製造することができる。核酸からそれがコードする蛋白質を人工的に生成させるための翻訳系は当業者に公知である。具体的には、適当な細胞より蛋白質合成能を有する成分を抽出し、その抽出液を用いて目的の蛋白質を合成させる無細胞蛋白質合成系が挙げられる。このような無細胞蛋白質合成系には、リボゾーム、開始因子、伸長因子及びtRNA等の翻訳に必要な要素が含まれている。
【0044】
このような無細胞蛋白質合成系としては、例えば、真核生物の無細胞蛋白質合成系が用いられ、より具体的には、ウサギ網状赤血球抽出液やコムギ胚芽抽出液などが挙げられるが、これらに限られるものではない。無細胞蛋白質合成系は、キットとして市販されているものを使用することができる。例えば、ウサギ網状赤血球抽出液のキットとしては、Rabbit Reticulocyte Lysate Systems, Nuclease Treated(Promega社製)等が用いられ、またコムギ胚芽抽出液としては、ゾイジーン無細胞蛋白質合成システム(ゾイジーン社製)や、PROTEIOSTM Wheat germ cell−free protein synthesis core kit(TOYOBO社製)等が挙げられる。蛋白質翻訳系としては、生細胞を使用してもよく、具体的には、原核又は真核生物、例えば大腸菌の細胞等を用いることができる。無細胞蛋白質翻訳系又は生細胞などは、その中に蛋白質をコードする核酸を添加するか又は導入することによって蛋白質合成が行われるものである限り特に制限はない。本発明の核酸構築物を無細胞蛋白質合成系に導入する直前に、60〜90℃で加熱した後急冷する工程を行うと、蛋白質−核酸連結体の合成効率が高くなるため好ましい。
【0045】
上記翻訳反応液から、ポリペプチド−核酸連結体を精製する場合、標識鎖に親和性物質あるいは共有結合物質が結合している場合には、該親和性物質あるいは共有結合物質を介して精製を行うことができる。精製の方法は、用いる親和性物質および共有結合物質に応じて適宜選択してそれ自体既知の定法を用いることができる。
上記で製造されたポリペプチド−核酸連結体は、上記標的分子と接触させ、該物質と相互作用する連結体を選択取得する工程に供する。この選択方法は、ポリペプチド−核酸連結体中のポリペプチドが有する機能(生物活性)を用いて標的分子と相互作用するポリペプチドを該連結体として選択することを意味する。このような相互作用の解析方法としては、例えばWO98/16636号公報に記載の方法を用いることができる。また、標的分子と接触させるポリペプチド−核酸連結体は、上記ライブラリーを有する複数の連結体群である。
【0046】
また、本発明のポリペプチド−核酸連結体および標的分子は、固相(支持体)に結合させて用いることもできる。ポリペプチド−核酸連結体の固相への結合は、上記標識部に親和性物質や共有結合性物質が結合している場合には、これらを用いて行うことができる。具体的には、親和性物質又は共有結合性物質が親和性を有するまたは結合する物質を予め固定化した固相に、上記ポリペプチド−核酸連結体を接触させることにより、当該ポリペプチド−核酸連結体を固相に容易に固定化することができる。固相への標的分子の結合は、例えば、Scott,J.K.&Smith,G.P.(1990)Science,249,386−390;Devlin,P.E.et al.(1990)Science,249,404−406;Mattheakis,L.C.et al.(1994)Proc.Natl.Acad.Sci.USA,91,9022−9026等に記載されている方法等により行うことができる。
【0047】
固相(支持体)としては、通常の核酸、ポリペプチド、糖質、脂質、化合物等の固定化に用いることができる支持体であれば特に限定されない。支持体としては、親和性物質や共有結合性物質どうしの結合形成、あるいは標的分子の結合に悪影響を及ぼさないものであればその形状は特に限定されず、例えば、平板、マイクロウェル、ビーズ等の任意の形態をとることができる。支持体の材質としては、例えば、ガラス、セメント、陶磁器等のセラミックス、アガロース、ポリエチレンテレフタレート、酢酸セルロース、ビスフェノールAのポリカーボネート、ポリスチレン、ポリメチルメタクリレート等のポリマー類、シリコン、活性炭、多孔質ガラス、多孔質セラミックス、多孔質シリコン、多孔質活性炭、織編物、不織布、濾紙、短繊維、メンブランフィルター等の多孔質物質を挙げることができる。
【0048】
上記選択方法に付するポリペプチド−核酸連結体は、核酸部がRNAのものでもよいし、mRNA鎖をDNAに逆転写したポリペプチド−逆転写核酸連結体でもよい。逆転写反応に必要な試薬及び反応条件は当業者に周知であり、必要に応じて適宜選択することができる。さらに得られたポリペプチド−逆転写核酸連結体のRNAをRNA分解酵素などを用いて分解し、DNAを鋳型にポリメラーゼ反応をすることにより2本鎖DNA−蛋白質連結体を作製して用いることもできる。このような、ポリペプチド−逆転写核酸連結体、あるいは2本鎖DNA−ポリペプチド連結体を用いれば、核酸部分の安定性がよいこと、また1本鎖RNAの非特異的吸着がないため好ましい。
【0049】
(ii)標的分子に特異的に結合したポリペプチド−核酸連結体の取得、核酸部の増幅、該核酸部を鋳型の一部としたポリペプチド−核酸連結体の調製工程
上記選択方法で選択されたポリペプチド−核酸連結体は、これを再度標的分子との相互作用に基づいて選択する工程に供する。一度選択されたポリペプチド−核酸連結体の核酸部を増幅して、さらにポリペプチド−核酸連結体とするためには、(a)選択取得されたポリペプチド−核酸連結体の1本鎖RNA部分を必要に応じて分解する等した後に、これをPCRなどで増幅し(増幅工程)、(b)増幅されたDNA鎖をもとにmRNA鎖を製造し、さらに上記のポリペプチド−核酸連結体鋳型を製造し、これを翻訳してポリペプチド−核酸連結体を調製することにより行なうことができる。
【0050】
(a)増幅工程は、PCRを用いて例えば以下のように行うことも好ましい。ポリペプチド−核酸連結体の核酸中、増幅するのは少なくともコーディング配列を含む領域である。このように増幅されたDNAについて、それ自体既知の定法により塩基配列を解析することにより、上記選択方法で選択された蛋白質をコードするDNA配列を同定できるので、該配列をもとにDNAまたはRNAも取得することができる。該領域を増幅するのに用いられるPCRプライマーとしては、特に制限はないが、全てのポリペプチド−核酸連結体に共通に用いられる配列として、5’側のプライマーは、コーディング配列の5’上流側に連結されている配列が好ましく用いられる。具体的には、上記したポリペプチド−核酸連結体の場合、5’側のプライマーは、翻訳の際にリボソームによって認識されるDNA配列などが好ましく用いられ、3’側のプライマーは、タグ配列や共通配列が好ましく用いられる。
かくして調製されたポリペプチド−核酸連結体鋳型は、上記(i)に記載の方法により翻訳され、ポリペプチド−核酸連結体が調製される。
【0051】
(iii)濃縮操作
本発明の方法では、上記(i)および(ii)に記載の工程を、標的分子に特異的に結合したポリペプチド−核酸連結体が全体として標的分子に対して高い親和性と特異性を示すまで繰り返す(本明細書では、これを「濃縮操作」と称することがある)。求められる親和性は目的により異なるが、高い親和性を有するポリペプチドの取得を目的とする場合はライブラリーと接触させる標的分子の濃度を低くすることが必要であり、期待する解離定数の近辺でその値を設定する。
【0052】
本発明の方法では、一般にこの濃縮操作を2〜15回行なうことによって、ファーマコフォアを明らかにするポリペプチドと、それをコードする核酸の連結体が取得される。取得された連結体の核酸部を上記方法により増幅して常法により塩基配列を解析することにより、該ポリペプチドをコードする塩基配列を同定することができる。また、該塩基配列を有する核酸を常法により合成すればポリペプチドをコードする核酸を取得することができ、またこの核酸を公知の、例えば、上記した蛋白質合成方法等により翻訳合成することにより、ファーマコフォアを明らかにするポリペプチドを取得することができる。
【0053】
さらには、多様性の大きなライブラリーから濃縮操作の結果として同定される上記塩基配列は、一般に複数であるので、上記の標的分子への親和性に基づいて選択されたポリペプチドが、ファーマコフォアを明らかにするポリペプチドであることを確認するためには、標的分子に作用してその機能に影響するかどうかを調べることが好ましい。具体的方法としては、まず上記で取得された塩基配列がコードするポリペプチドを常法により化学合成し、あるいは該塩基配列を有する核酸を常法により合成して、大腸菌あるいは無細胞翻訳系で翻訳合成する等の方法により、活性測定用のポリペプチドを調製する。生体内で標的分子と相互作用する蛋白質等と標的分子の結合に該ペプチドが与える影響、あるいは標的分子の機能を測定可能な細胞実験系における該ペプチドの薬理作用等を調べることにより、ファーマコフォアの解明に有用なポリペプチドを選択することができる。
【0054】
また、上記のようにして取得したポリペプチドのアミノ酸配列からアミノ酸の存在頻度および/または出現頻度を指標として共通配列を選択する工程を含むことを特徴とする医薬的活性を有するポリペプチド(本明細書中では、これを「ドメインポリペプチド」と称することがある)を取得することもできる。具体的には、上記で取得されたポリペプチドをコードする核酸からドメインポリペプチドをコードするクラスターを取得することにより行う。「ドメインポリペプチドをコードするクラスター」(以下、「クラスター」と称することがある)とは、上記ドメインポリペプチドをコードする核酸を共通配列として含むほぼ同じ長さの核酸のかたまりを意味する。共通配列を含むほぼ同じ長さのかたまりとは、上記で取得されるポリペプチド−核酸連結体群の50%以上が該共通配列を含み、その鎖長が最も短い核酸と長い核酸が1倍〜10倍の範囲に入ってくることを意味する。このようなクラスターが形成されたことは、形成を確認し得る方法であれば特に制限はないが、例えば、標的分子と相互作用することで選択取得されたポリペプチド−核酸連結体の核酸部を、RNAのまま、又はDNAとしてアガロースゲルまたはSDS−ポリアクリルアミドゲル中を電気泳動等で分離した場合にバンドが観察されることにより確認することができる。
【0055】
(3)ファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドを用いた医薬開発
本発明に従ったライブラリーのスクリーニングで取得されたポリペプチドに対して解析実験を行なうことにより、医薬リード開発に必要な情報がもたらされる。相同性の高い配列が複数得られている場合にはそのアライメントから構造活性相関についての情報が少なからず得られることがある。配列のアライメントから必要な情報が充分得られない場合は、取得された配列をもとにそれをコードするDNAの再ランダム化を行い、無細胞蛋白質ディスプレー法またはその他の方法でスクリーニングするか、あるいは置換した部位を有するポリペプチドを無細胞翻訳、大腸菌発現、化学合成などにより1つないし数百種合成して活性測定をすることで構造活性相関についての情報を得ることができる。
【0056】
得られたファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドが明確に区別され得る複数の立体構造あるいはジスルフィド架橋をとる場合には、実際に該ポリペプチドを合成して活性に必要な立体構造あるいはジスルフィド架橋を確認する必要があり、立体構造の確認方法としてはX線結晶構造解析、NMRによる構造解析、円偏光二色性解析などが挙げられ、ジスルフィド架橋の確認方法としては酵素による限定加水分解法と質量分析法を合わせた方法がある。
【0057】
得られたファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドと標的分子の間でファーマコフォアを抽出する方法としては、両者が結合した共結晶を形成させ、X線結晶構造解析により両者の座標を得ることがもっとも望ましい。得られた空間座標をもとに上記ポリペプチド側を改変する、低分子をドッキングさせる、あるいは新規に原子を発生させてde novoで分子を形成する等の手法により上記ポリペプチドと同様の活性を持った低分子化合物を開発することが可能となる。共結晶が得られない時でも、上記ポリペプチド側の立体構造が土台となる構造モチーフ等から推測できる場合、あるいはMD等の手法により限られた主鎖構造が候補となる場合には標的分子側の立体構造情報の有無に関わらず上記ポリペプチドの低分子化合物への改変ができることもある。さらに一般的に、上記ポリペプチド側についての詳細な構造情報はNMRによる溶液中での立体構造解析により取得することが比較的容易である。上記ポリペプチド側あるいは標的分子側において相互作用に関わっている部位についての情報が著しく不明である場合にはHSQC(Heteronuclear single quantum correlation)等の手法が有用である。これらの構造情報とペプチドミメティクスの手法の組み合わせによっても新たな低分子化合物が創出される。
【0058】
かくして設計された低分子化合物は、標的分子が疾病に関連するものである場合、該疾病治療薬として用いることができる。この場合、上記工程により医薬組成物を製造する方法も本発明に含まれる。
取得されたファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドをそのまま、あるいは改変を加えてペプチド医薬として応用することも可能である。親和性と選択性の向上を目的とした改変には通常のアミノ酸あるいは非天然型アミノ酸による置換等が有効である。また、ヒトに対する抗原性を低下させることを目的とした改変が行なわれる場合もある。安定性を目的とした改変においては、該ポリペプチドの主鎖あるいは側鎖の置換の他にPEGなどの高分子との融合が行なわれることもあり、上述した無細胞蛋白質ディスプレー法で選択されたポリペプチドは特にC末端とその周辺への導入に対して許容性が高いと予想される。
【0059】
取得されたファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドはそれ自体あるいはその構造情報から医薬リードを開発する目的以外にも応用でき、例えば蛍光プローブをポリペプチドに導入することでHTSシステムにおいて多数の低分子化合物ライブラリーの活性を調べるための競合リガンド(Surrogate ligand)として用いることも可能である。蛍光プローブを導入した該ポリペプチドはイメージング等にも応用できる。
【0060】
さらに本発明で取得されたファーマコフォアを明らかにするポリペプチドあるいはドメインポリペプチドを固相化することにより標的分子を検出するチップ等を作製することもできる。固相化の方法としては、ビオチン化、ポリペプチドの官能基とオルトゴナルな官能基の導入等が挙げられる。
【実施例】
【0061】
以下、実施例を挙げて本発明を詳細に説明するが、本発明の範囲はこれらの実施例により限定されるものではない。
実施例1 ライブラリーDNAの配列とその構築
ライブラリー化する配列として、3本のジスルフィド結合を持つ構造モチーフを10種、2本のジスルフィド結合を持つ構造モチーフを4種、1本のジスルフィド結合を持つループ状のモチーフを5種、ジスルフィド結合を持たない構造モチーフを3種、15残基のランダムライブラリーを1種、合計23種選択した。これらを発現させるために用いられるDNAの原料として、配列番号1〜51のDNAをDNA合成機により合成した。このうち配列番号1〜33はランダムあるいはセミランダム化する残基に相当する部位に混合塩基が導入されており、そのA:T:G:C比がKにおいて0:50:50:0、Mにおいて50:0:0:50となるようにホスホアミダイトを混合した。Nは合計4種類の混合比でホスホアミダイトを混合し、そのA:T:G:C比は混合比1が30:15:30:25、混合比2が30:30:30:10、混合比3が15:30:25:30、混合比4が30:30:10:30となるようにした。基本的に配列番号1〜14、16〜22においてKの前に位置するNが混合比2、Kの2つ前に位置するNが混合比1であり、配列番号15、23〜33においてはMの後に位置するNが混合比4、Mの2つ後に位置するNが混合比3である。この基本則に優先してNの混合比を指定した部位もあり、Gの次で後にKが続くNは混合比1、TとTに挟まれたNは混合比3、Mの次で後にCが続くNは混合比3とした。さらに配列番号1の37番目、配列番号2の72番目、配列番号6の61番目、配列番号8の57番目および配列番号10の57番目のNは混合比1、配列番号5の72番目および配列番号10の39番目のNは混合比2、配列番号28の49番目、配列番号29の20番目、配列番号31の42番目および配列番号33の24番目のNは混合比3、配列番号31の21番目および配列番号33の23番目のNは混合比4とした。
【0062】
化学合成したDNAをオーバーラップ伸長反応により連結した。反応系としてはTOYOBO社製KOD plusを用い、連結する2種のDNAの濃度を0.5Mとし、PCR装置中で94℃で2分反応後、98℃で10秒、60℃で30秒、68℃で5分の反応を5回ないし10回繰り返し、さらに68℃で5分反応させた。配列番号1と23、配列番号2と24、配列番号3と35、配列番号4と25、配列番号5と26、配列番号6と27、配列番号7と28、配列番号8と29、配列番号9と30、配列番号10と31、配列番号11と36、配列番号12と41、配列番号13と42、配列番号14と36、配列番号15と50、配列番号16と36、配列番号17と36、配列番号18と36、配列番号19と36、配列番号20と43、配列番号20と32、配列番号21と32、配列番号22と33、これらの組合わせで連結を行なうことによりそれぞれ生成物1〜23を得た。
【0063】
目的物の純度が低い場合は8M尿素変性ポリアクリルアミドゲル電気泳動(PAGE)で分離し、抽出濃縮操作によって次の反応に用いた。続いて生成物1と配列番号34、生成物2と配列番号34、生成物4と配列番号36、生成物5と配列番号36、生成物6と配列番号37、生成物7と配列番号38、生成物8と配列番号39、生成物9と配列番号37、生成物10と配列番号40、生成物21と配列番号44、生成物22と配列番号44、生成物23と配列番号36のオーバーラップ伸長反応によりそれぞれ生成物24〜35を得た。目的物の純度が低い場合は8M尿素変性PAGEで精製した。さらに生成物24と配列番号45、生成物25と配列番号46、生成物3と配列番号47、生成物26と配列番号45、生成物27と配列番号46、生成物28と配列番号48、生成物29と配列番号47、生成物30と配列番号45、生成物31と配列番号47、生成物32と配列番号47、生成物11と配列番号47、生成物12と配列番号47、生成物13と配列番号47、生成物14と配列番号49、生成物15と配列番号34、生成物16と配列番号47、生成物17と配列番号47、生成物18と配列番号47、生成物19と配列番号47、生成物20と配列番号47、生成物33と配列番号47、生成物34と配列番号51、生成物35と配列番号47、の組合せでオーバーラップ伸長反応を行い、それぞれ生成物36〜58を得た。これらはすべて8M尿素変性PAGEで精製し、得られたDNAを定量して転写用の鋳型とした。
【0064】
連結されたDNA配列は転写プロモーター配列のあとにmRNAに転写される配列として翻訳エンハンサー配列、ライブラリー配列、リンカー配列、FLAG配列、リンカー配列、ピューロマイシンリンカー結合配列を有している。得られたDNAのうち生成物39と41の配列解析を行なったところ、上記のコンストラクトを完全に含む配列は生成物39で19/31(61%)、生成物41で18/32(56%)であり、このうちランダム化部位に終止コドンを含まないものはそれぞれ15(48%)、14(44%)であった。この配列解析の結果得られたランダム化部位におけるアミノ酸出現頻度(観測値)をその期待値とともに図1に示した。
【0065】
実施例2 ライブラリーDNAの転写と転写産物のピューロマイシンリンカーとの連結
生成物36〜58のDNA分子を大きさなどを基準として9つのグループに分類し、Promega社製Ribomax Large Scale RNA Production Systemを用いて定法に従い転写反応を行なった。翻訳時におけるmRNAの安定性向上を目的として転写反応液にはキャップアナログ(7.2mM、RNA Capping Analog;Gibco BRL社製)を加え、mRNAの5’側を修飾した。9つのグループの組み合わせとDNA量(括弧内pmol)、転写反応液の量(括弧内・μl)は次の通りである。生成物36(4.2pmol)(50μl)、生成物37
(1.7pmol)と生成物49(6.7pmol)(50μl)、生成物38(5.8pmol)と生成物47(3.3pmol)と生成物48(4.2pmol)(70μl)、生成物39(7.5pmol)と生成物40(7.5pmol)(75μl)、生成物41(7.5 pmol)と生成物43(5.8pmol)と生成物44(5.8pmol)(100μl)、生成物42(6.7pmol)と生成物45(3.3pmol)(50μl)、生成物46(2.5pmol)と生成物55(3.3pmol)(50μl)、生成物50〜54(各3.3pmol)(80μl)、生成物56(1.7pmol)と生成物57(3.3pmol)と生成物58(3.3pmol)(60μl)。得られた生成物をこの順に従って生成物59〜67とする。分析のために各DNAを鋳型に小スケールで転写反応を行い、生成物36〜58から生成物68〜90を得た。
【0066】
続いてこれらのmRNAを特願2003−315385号明細書に記載のピューロマイシンリンカー(T−splint5.9FA)と連結した。ピューロマイシンとmRNAの濃度がそれぞれ0.75μM、0.5μMになるように5%ジメチルスルホキシドを含むT4 RNAリガーゼ緩衝液(50mM Tris−HCl、pH7.5、10mM MgCl2、10mM DTT、1mM ATP)に溶解し、72℃で150秒加熱したのち室温で12分放置し、さらに15℃まで冷却したのち100分の1量(体積比)のT4 RNAリガーゼ(Takara社製)を加えて15℃で約2時間反応させた。実際に反応させたmRNAの量は生成物59〜67それぞれに対し100pmol、200pmol、320pmol、360pmol、460pmol、240pmol、140pmol、380pmol、200pmolであり、反応後に得られた産物をそれぞれ生成物91〜99とする。生成物68〜90それぞれに対しても20〜150 pmol相当のmRNAをピューロマイシンリンカーと反応させ、生成物100〜122を得た。生成物68〜90と生成物100〜122を8M尿素変性7%ポリアクリルアミドゲル電気泳動で分析した結果を図2に示す。
【0067】
実施例3 ポリペプチド−核酸連結分子の調製
上記の生成物91〜99を小麦胚芽無細胞蛋白質合成系(ゾイジーン社製)に6pmol/24μlの割合で加えて75分間翻訳させ、ポリペプチドと核酸の連結した対応分子が形成されるかどうかを確認した。合成系に加えた直後と75分後の混合液の一部(各生成物0.75pmol相当)を8M尿素変性7.5%SDS−PAGEで分離し、蛍光で検出した結果を図3に示す。さらに実際の選択実験に用いる対応付け分子を調製するため、生成物91〜99をそれぞれ40pmol、65pmol、65pmol、65pmol、80pmol、65pmol、30pmol、88pmol、102pmol、生成物100〜122を13.5pmolずつ同様の条件で翻訳した。
【0068】
翻訳後の溶液合計約3650μlに5M NaClを988μl、1M Tris−HCl(pH8.0)を49.4μl、0.5M EDTA−Naを148μl、10% Triton−X100を49.4μl、0.1M DTTを57μl加え、Biotinylated Oligo dTが結合したMAGNOTEX−SA(Takara社製)600μlと4℃で約2時間反応させた。上清を除き、緩衝液A(1M NaCl、50mM Tris−HCl(pH8.0)、0.1% Triton−X100、1mM DTT)で3回、緩衝液B(0.5M NaCl、50mM Tris−HCl(pH8.0)、0.1% Triton−X100、1mM DTT)で1回、さらに緩衝液C(0.25M NaCl、50mM Tris−HCl(pH8.0)、0.1% Triton−X100、1mM DTT)で1回洗浄した後、5mM DTT水溶液330μlで3回溶出した。
【0069】
この溶出画分に324μlの5×RT Buffer(Invitrogen社製)、162μlの10mM dNTP、60μlのRNase Inhibitor(WAKO社製;40U/μl)、84μlのSuperScript III Reverse Transcriptase(Invitrogen社製;200U/μl)を添加し、50℃で3分間加熱後、室温で12分放置した。この溶液に水94.5μl、5M NaClを54μl、0.1M リン酸水素2カリウムを324μl、10% Tween 20を22.5μl、50μg/ml tRNAを22.5μl、1mg/ml BSAを112.5μl加え、150μlの抗FLAG M2抗体アガロースビーズ(Sigma社製)と室温で1時間反応させた。
【0070】
上清を除き、0.1%のTween 20を含むpH 7.4のPBS(リン酸緩衝生理食塩水)360μlで3回、0.1%のTween 20を含むpH8.0のPBS360μlで3回洗浄し、100μMの3×FLAG peptideを溶かしたPBS(pH8.0)96μlで2回、37μMの3×FLAG peptideと0.1%のTween 20を溶かしたPBS(pH8.0)257μlで1回溶出した。合わせた溶出液にPBS(pH8.0)を16.8μl、0.5M EDTA−Naを2.4μl、10% Tween 20を2.4μl、2mM 酸化型グルタチオンを4.8μl、2mM 還元型グルタチオンを4.8μl加えて4℃で15時間反応させた後、約60μlずつをPBS(pH7.4)に置換したゲルろ過カラム(Bio−Gel P−6;BIO−RAD社製)に通して対応付け分子を精製した。これらを再び合わせた約480mlの溶液に10倍濃度のPBS(pH7.4)を12μl、4.8% BSAを78μl、2.5mg/ml tRNAを24μl、10% Tween 20を6μl加えて選択実験に用いる対応付け分子の溶液(約600μl)とした。
【0071】
実施例4 標的蛋白質結合担体の調製と対応付け分子の選択
スクリーニングの標的蛋白質の調製については、まず常法に従ってヒトインターロイキン6(interluekin 6;IL6)あるいはヒト腫瘍壊死因子α(tumor necrosis factor;TNF)のN末側にグルタチオンS−トランスフェラーゼ(gulutathione S−transferase;GST)を融合した蛋白質をコードするDNAが挿入された大腸菌発現用ベクターを調製した。両者の間にはprecision proteaseで切断されるペプチド鎖をコードした配列を挿入した。これを大腸菌(BL21(DE3)pLysS)で発現させ、目的とする融合蛋白質をGlutathione Sepharose4Bビーズ(アマシャムファルマシア社製)を用いて精製し、溶出に使用したグルタチオンは脱塩カラムにより除去した。またネガティブコントロール用としてGST蛋白質のみを合成し、同様の方法で精製した。もう一つの標的蛋白質としてgp130のFcキメラ(R&D SYSTEMS社製)を購入し、精製せずにそのまま用いた。この融合蛋白質はgp130とFcの間にFactor Xaで切断可能な配列を有する。
【0072】
GST(100μg)、GST−IL6(35μg)、GST−TNF(130μg)をそれぞれ250μlのPBS(pH7.4)−0.1% Tween 20溶液(以下、「PBST」と称することがある)に溶解し、30μlのGlutathione Sepharose4Bビーズと合わせて15時間静かに混合した後、上清を除いてPBSTで4回洗浄し、GSTビーズ、GST−IL6ビーズ、GST−TNFビーズを得た。プロテインGビーズ(Protein G Sepharose 4 Fast Flow;アマシャムバイオサイエンス社製)30μlをPBSTで洗浄し、gp130/Fc 18μgを溶かした0.2% BSAを含むPBST、200μlと合わせて1時間静かに混合し、上清を除いてPBSTで4回洗浄し、gp130/Fcビーズを得た。
【0073】
対応付け分子の溶液(約600μl)をGSTビーズと混合し、室温で1時間反応させた。上清を続いてGST−IL6ビーズと混合して反応を行い、その上清をとってGST−TNFビーズと反応させ、さらにその上清をプロテインGビーズ70μlに加えて反応を行い、最後にその上清をgp130/Fcビーズと混合して反応させた。反応はすべて室温で約1時間行なった。上清を除いた後のビーズはただちに150μlのPBSTで6回洗浄し、gp130/Fcビーズはさらに50mM Tris−HCl(pH7.5)、150mM NaCl、0.1% Tween 20の溶液(以下、これを「TBST」と称することがある)150μlで2回洗浄した。洗浄後のGSTビーズ、GST−IL6ビーズ、GST−TNFビーズには50mM Tris−HCl(pH8.0)、150mM NaCl、1mM DTTの溶液200μlとprecision protease溶液2μlを加えて室温で1時間反応させ、上清をとりビーズを同溶液200μlで洗った溶液と合わせて酵素溶出液とした。
【0074】
洗浄後のgp130/Fcビーズには50mM Tris−HCl(pH7.5)、150mM NaCl、0.1% Tween 20の溶液42μlとFactor Xa溶液(BioLabs社製)8μlを加えて室温で2時間反応させ、上清をとり、50mM Tris−HCl(pH7.5)、150mM NaCl、0.1% Tween 20、1mM EDTAの溶液175μlでビーズを2回洗って上清と合わせ酵素溶出液とした。各酵素溶出液にQuick precip Plusを30μl、イソプロパノールを430μl加えて遠心し、対応付け分子を沈殿物として回収して70%エタノール水溶液で洗い、風乾後、0.1%のTriton X−100を含む100mMの水酸化カリウム水溶液25μlに溶解した。室温で1時間反応させた後、0.1% Triton X−100を含む100mMの酢酸カリウム水溶液をほぼ等量加えて中和を確認し、PCR反応の鋳型溶液とした。
【0075】
PCRの反応サイクル数を決めるため、鋳型溶液の一部(4μl)で条件検討を行なった。プライマーとして配列番号52と53のDNA(それぞれ40nM)、反応系としてTOYOBO社製KOD plusを用い(鋳型溶液1μlに対し10μl)、PCR装置上94℃で2分反応後、98℃で10秒、60℃で30秒、68℃で90秒の反応サイクルを8、12、16、あるいは20回実施後、さらに68℃で5分反応させた。アガロースゲル電気泳動による解析結果より、GSTとGST−TNFに対しては反応サイクルを10回、GST−IL6とgp130/Fcに対しては12回として残りの鋳型溶液を用いてPCR反応を行なった。精製して得られたそれぞれのDNA溶液のうち約半分(DNAとして480 ng)を配列番号54と55のプライマーDNA(それぞれ40nM)とともにKOD plus反応液に加え(計480μl)、反応サイクル8回で再びPCR反応を行い、転写プロモーター配列からピューロマイシンリンカー結合配列までを含む最初のライブラリーと同じコンストラクトを有するDNAを得た。
【0076】
実施例5 ポリペプチド−核酸連結分子の濃縮操作
実施例2から実施例4の操作を第1ラウンドとし、GST、GST−IL6、GST−TNF、gp130/Fcのそれぞれに対して選択されたDNAをもとに次の選択を繰り返す操作を第10ラウンドまで実施した。具体的には、選択後にPCR反応を行なって得られた4種のDNA混合物のそれぞれを鋳型に実施例2の方法に基づいて転写反応を行い、得られたそれぞれのmRNA混合物をピューロマイシンリンカーと連結させた。この核酸複合体を実施例3の方法に基づいてそれぞれ翻訳し、mRNA/cDNAを遺伝情報として有する対応付け分子を調製した。実施例2と実施例3の方法に準じた第2ラウンド以降の操作は4種のライブラリー由来混合物を対象に並行して行い、反応のスケールを徐々に減らして実施した。実施例4に準じた選択実験も4種を並行して行い、GSTに対して選択された対応付け分子はGSTビーズと反応させ、GST−IL6あるいはGST−TNF に対して選択された対応付け分子はGSTビーズと混合した後に上清画分をGST−IL6ビーズあるいはGST−TNFビーズと反応させた。
【0077】
gp130/Fcに対して選択された対応付け分子はプロテインGビーズとまず反応させた後に上清画分をgp130/Fcビーズと反応させた。反応はいずれも第1ラウンドと同様に室温約1時間とし、ビーズに固定する蛋白量は段階的に減らして、最後の第10ラウンドにおいては対応付け分子と反応させる時の溶液量に対し、GSTが20μg/250μl、GST−IL6が10μg/250μl、GST−TNFが5μg/250μl、gp130/Fcが1.5μg/250μlであった。反応後の洗浄液の量と洗浄回数は段階的に増やし、第10ラウンドにおいてはGSTビーズを250μlのPBSTで8回、GST−IL6ビーズを250μlのPBSTで10回、GST−TNFビーズを250μlのPBSTで10回、gp130/Fcビーズを250μlのPBSTで4回、さらに250μlのTBSTで8回洗浄した。
【0078】
酵素溶出とPCR鋳型溶液の調製は実施例4とほぼ同様の条件で行い、配列番号52と53のDNAをプライマーとしたPCR反応は実施例4と同様に一部で条件検討を行い反応サイクル数を決定してから実施した。このPCR産物を鋳型とし、配列番号54と55のDNAをプライマーとしたPCRは鋳型DNA、150ng/KOD plus反応液300μl、反応サイクル8回を基本として実施した。
【0079】
実施例6 選択されたポリペプチドの結合試験
第10ラウンド選択後のPCRで得られたDNAをTaqポリメラーゼ(Takara社製)によりA付加し、pGEM T easy(プロメガ社製)を用いて常法によりプラスミドに挿入した。これを大腸菌JM109にトランスフェクションして増幅し、選択されたDNA配列を含むプラスミドを回収した。配列番号54と55のDNAをプライマーとしてPCR反応を行い、得られたDNAを鋳型に転写反応を実施してmRNAを得た。このmRNAを実施例3で用いた無細胞翻訳系で翻訳し、抗FLAG M2抗体アガロースビーズで精製した。DTTを含まない溶出液(TBST)中で15時間放置した後、1%のBSAを含むPBSTで約5倍に希釈してポリペプチド溶液とした。
【0080】
96ウェルのポリスチレンプレート(COSTAR社製;9018)の各ウェルにGST、GST−IL6、TNFのそれぞれを1μg/100μlの濃度でPBSに溶かした溶液を加えて4℃で一晩静置した。GSTとGST−IL6は実施例4で用いたものを使用し、TNFはGST−TNFをPrecision proteaseで消化してGlutathione Sepharose4Bビーズ上でGSTを除くことにより得た。
【0081】
ウェルをPBSで2回洗浄し、1%のBSAを含むPBSを200μl加えて37℃で2時間振とうした後に溶液を除き、上記のポリペプチド溶液を加えて室温で1時間振とうした。溶液を除いてPBSTで3回ウェルを洗浄し、1mg/mlの抗FLAG M2抗体−HRP(SIGMA社製;A8592)を1%BSA/PBSで1万倍に希釈した溶液100μlを各ウェルに添加して室温で1時間静置した。溶液を除いてPBSTで3回ウェルを洗浄し、10mg/25mlのo−フェニレンジアミンと10μl/25mlの30%過酸化水素水を含むクエン酸緩衝液を各ウェルに100μlずつ加えて室温で10分静置した後に1規定の硫酸水溶液を等量加えて酵素反応を停止し、490nmの吸収をプレートリーダーで測定した。GST−IL6に対して選択された4種のポリペプチドに対する結果を図4に、GST−TNFに対して選択された3種のポリペプチドに対する結果を図5にそれぞれ示した。GST−IL6に対して選択されたもののうち、2種がGST−IL6に対して選択性を示し、GST−TNFに対して選択されたものは3種すべてTNFに対する選択性を示した。
【図面の簡単な説明】
【0082】
【図1】生成物39と41の配列解析の結果得られたランダム化部位におけるアミノ酸出現頻度(観測値)をその期待値とともに示したグラフである。
【図2】mRNA(生成物68〜90)とそれをピューロマイシンリンカーと連結させた反応物(生成物100〜122)を8M尿素変性7%ポリアクリルアミドゲル電気泳動で解析した写真である。
【図3】mRNA−ピューロマイシンリンカー複合体(生成物91〜99)を無細胞翻訳系に加えた反応液をSDS−PAGEで分離し蛍光で検出した写真である。
【図4】GST−IL6ビーズに対して選択されたポリペプチドの結合活性をプレートアッセイで測定した結果を示すグラフである。
【図5】GST−TNFビーズに対して選択されたポリペプチドの結合活性をプレートアッセイで測定した結果を示すグラフである。
【特許請求の範囲】
【請求項1】
変異を導入した構造モチーフを標的分子と接触させ、標的分子と相互作用するポリペプチドを選択することを特徴とする標的分子−ポリペプチド間相互作用解析方法。
【請求項2】
変異を導入した構造モチーフおよびランダムペプチドを標的分子と接触させ、標的分子と相互作用するポリペプチドを選択することを特徴とする標的分子−ポリペプチド間相互作用解析方法。
【請求項3】
標的分子と相互作用するポリペプチドを選択する方法が、無細胞蛋白質ディスプレー法である請求項1または2に記載の方法。
【請求項4】
変異を導入した構造モチーフを標的分子と接触させ、標的分子に対し高親和性を示すポリペプチドを選択することを特徴とするファーマコフォアを明らかにするポリペプチドの取得方法。
【請求項5】
変異を導入した構造モチーフおよびランダムペプチドを標的分子と接触させ、標的分子に対し高親和性を示すポリペプチドを選択することを特徴とするファーマコフォアを明らかにするポリペプチドの取得方法。
【請求項6】
標的分子に対し高親和性を示すポリペプチドを選択する方法が無細胞蛋白質ディスプレー法である請求項5に記載の方法。
【請求項7】
請求項4〜6のいずれか1項に記載の方法により得られたポリペプチドのアミノ酸配列からアミノ酸の存在頻度および/または出現頻度を指標として共通配列を選択する工程を含むことを特徴とする医薬的活性を有するポリペプチドの取得方法。
【請求項8】
請求項4〜7のいずれか1項に記載の方法により得られたポリペプチドを化学的に改変して医薬的活性を向上させることを特徴とするペプチド誘導体の取得方法。
【請求項9】
請求項4〜8のいずれか1項に記載の方法により得られたポリペプチドあるいはペプチド誘導体と標的分子との複合体を作製する工程、該複合体の立体構造解析を行い、標的分子と該ポリペプチドあるいは該ペプチド誘導体との結合部位を特定する工程、同定された結合部位から標的分子のファーマコフォアを明らかにして、該ファーマコフォアに合う低分子化合物を設計する工程を含むことを特徴とする標的分子と相互作用する化合物の分子設計方法。
【請求項1】
変異を導入した構造モチーフを標的分子と接触させ、標的分子と相互作用するポリペプチドを選択することを特徴とする標的分子−ポリペプチド間相互作用解析方法。
【請求項2】
変異を導入した構造モチーフおよびランダムペプチドを標的分子と接触させ、標的分子と相互作用するポリペプチドを選択することを特徴とする標的分子−ポリペプチド間相互作用解析方法。
【請求項3】
標的分子と相互作用するポリペプチドを選択する方法が、無細胞蛋白質ディスプレー法である請求項1または2に記載の方法。
【請求項4】
変異を導入した構造モチーフを標的分子と接触させ、標的分子に対し高親和性を示すポリペプチドを選択することを特徴とするファーマコフォアを明らかにするポリペプチドの取得方法。
【請求項5】
変異を導入した構造モチーフおよびランダムペプチドを標的分子と接触させ、標的分子に対し高親和性を示すポリペプチドを選択することを特徴とするファーマコフォアを明らかにするポリペプチドの取得方法。
【請求項6】
標的分子に対し高親和性を示すポリペプチドを選択する方法が無細胞蛋白質ディスプレー法である請求項5に記載の方法。
【請求項7】
請求項4〜6のいずれか1項に記載の方法により得られたポリペプチドのアミノ酸配列からアミノ酸の存在頻度および/または出現頻度を指標として共通配列を選択する工程を含むことを特徴とする医薬的活性を有するポリペプチドの取得方法。
【請求項8】
請求項4〜7のいずれか1項に記載の方法により得られたポリペプチドを化学的に改変して医薬的活性を向上させることを特徴とするペプチド誘導体の取得方法。
【請求項9】
請求項4〜8のいずれか1項に記載の方法により得られたポリペプチドあるいはペプチド誘導体と標的分子との複合体を作製する工程、該複合体の立体構造解析を行い、標的分子と該ポリペプチドあるいは該ペプチド誘導体との結合部位を特定する工程、同定された結合部位から標的分子のファーマコフォアを明らかにして、該ファーマコフォアに合う低分子化合物を設計する工程を含むことを特徴とする標的分子と相互作用する化合物の分子設計方法。
【図1】



【図4】
【図5】
【図2】
【図3】

【図4】
【図5】
【図2】
【図3】
【公開番号】特開2007−143423(P2007−143423A)
【公開日】平成19年6月14日(2007.6.14)
【国際特許分類】
【出願番号】特願2005−338881(P2005−338881)
【出願日】平成17年11月24日(2005.11.24)
【出願人】(502262469)ゾイジーン株式会社 (8)
【Fターム(参考)】
【公開日】平成19年6月14日(2007.6.14)
【国際特許分類】
【出願日】平成17年11月24日(2005.11.24)
【出願人】(502262469)ゾイジーン株式会社 (8)
【Fターム(参考)】
[ Back to top ]