
標的化された細胞死
【課題】ニューロン障害のための治療剤に対するスクリーニングを行うための組成物および方法を提供する。
【解決手段】神経障害の病因および、特に、ニューロン脱髄の解明は、効果的な動物モデルの継続的欠如によって妨げられてきた。かくして、ニューロン障害のための治療剤に対するスクリーニングを行うための組成物および方法に対する緊急の要望が存在する。本発明は、神経障害を研究するための組成物および方法を提供する。本明細書中に提供される組成物および方法は、治療および/または診断潜在能力の薬剤をスクリーニングするのに特に有用である。
【解決手段】神経障害の病因および、特に、ニューロン脱髄の解明は、効果的な動物モデルの継続的欠如によって妨げられてきた。かくして、ニューロン障害のための治療剤に対するスクリーニングを行うための組成物および方法に対する緊急の要望が存在する。本発明は、神経障害を研究するための組成物および方法を提供する。本明細書中に提供される組成物および方法は、治療および/または診断潜在能力の薬剤をスクリーニングするのに特に有用である。
【発明の詳細な説明】
【技術分野】
【0001】
本願は、2007年6月12日に出願された米国仮特許出願第60/943,448号の利益を主張する。米国仮特許出願第60/943,448号は、その全体が参考として本明細書中に援用される。
【背景技術】
【0002】
ニューロン脱髄は、神経系におけるミエリンの低下によって特徴付けられる有害な状態である。ミエリンは、中枢(CNS)および末梢(PNS)神経系の双方に対して非常に重要であり、ニューロンの軸索を包み、ミエリン鞘として知られた絶縁性層を形成する。ミエリン鞘の存在は、神経軸索を下って伝達する電位の形態の神経シグナルの速度および完全性を増強させる。ミエリン鞘の喪失は、感覚、運動および他のタイプの機能における重要な損傷を生じさせる。というのは、神経シグナルはあまりにも遅く、(例えば、神経中のいくつかの軸索が他の軸索よりも速く伝わる場合には)非同期的に、(例えば、伝達が高い周波数においてのみ損なわれる場合には)間歇的に、のいずれかでそれらの標的に到達し、あるいは全く到達しないからである。
【0003】
ニューロン組織は、一般に、ニューロンおよび支持する神経膠細胞を含む。神経膠細胞は、哺乳動物の脳においては、約10:1でニューロンよりも数が多い。神経膠細胞は4つのタイプ:神経膠星状細胞、乏突起神経膠細胞、シュワン細胞および小神経膠細胞に分けることができる。ミエリン鞘は神経膠細胞(CNSにおいては乏突起神経膠細胞およびPNSにおいてはシュワン細胞)の原形質膜または細胞膜によって形成される。髄鞘形成の活性相の間に、CNS中の各乏突起神経膠細胞は、典型的には、1日当たりミエリン表面の約5000μm2、および1分当たり約105ミエリン蛋白質分子も多くを生じる(非特許文献1)。髄鞘形成乏突起神経膠細胞は、脱髄された病巣において同定されており、脱髄された軸索は新たに合成されたミエリンで修復できることを示す。
【0004】
ニューロン脱髄は、CNSおよびPNSの非常に多数の遺伝的および後天的障害で発現される。これらの障害は、例えば、多発性硬化症(MS)、進行性多病巣性白質脳障害(PML)、脳脊髄炎、中枢橋ミエリン分解(CPM)、抗MAG病、白質萎縮症:副腎脳白質ジストロフィー(ALD)、アレクサンダー病、キャナヴァン病、クラッベ病、異染性白質萎縮症(MLD)、ペリツェーウス・メルツバッヒャー病、レフスム病、コケーン症候群、ファン・デル・クナップ症候群、およびツェルヴェーガー症候群、ギラン・バレー症候群(GBS)、慢性炎症性脱髄多発性神経障害(CIDP)、および多病巣性運動神経障害(MMN)を含む。これらの障害のうちほとんど大部分では、治癒はなく、および効果的な療法はほとんどない。
【0005】
パーキンソン病(パーキンソン症候群または振せん麻痺)の間には、脳の細胞は未知の理由で劣化するように見える。しかしながら、炎症性反応についての役割は、パーキンソン病の病因において役割を演じると仮定されている。パーキンソン病は、振せん、および歩行、運動、および協調に伴う困難によって特徴付けられる脳の障害である。前記障害は、1,000人の人々のうち約2人が罹り、最もしばしば、50歳後に発症する。それは男性および女性の双方に罹り、老人の最も一般的な神経学的障害の1つである。パーキンソン病は、筋肉の運動を制御する脳の部分(基底神経節および錐体外路領域)の神経細胞
の進行性劣化によって引き起こされる。
【0006】
筋肉制御の喪失に加えて、パーキンソン病を持つ一部の人々はひどく鬱になる。知的能力の初期の喪失は一般的ではないが、重度のパーキンソン病の人々は(認知症、幻覚等を含めた)総じての精神的劣化を呈し得る。認知症は、該障害を処置するのに用いる投薬のいくらかの副作用でもあり得る。
【0007】
筋委縮性側索硬化症(ALS)は、脳および脊髄における神経細胞の破壊により随意筋の神経制御の喪失を引き起こす迅速に進行する不変に致命的な障害であり、筋肉の使用および制御の喪失をもたらす。これらの筋肉を制御する神経は収縮し、消失し、その結果、神経刺激の欠如による筋肉組織の喪失がもたらされる。筋肉の強度および協調は減少し、随意筋(例えば、意識制御下にあるもの)で開始する。筋肉制御の喪失の程度は継続的に進行し、より大きな筋肉の群が関与するようになる。呼吸および嚥下を制御する筋肉のような、半随意筋に対する神経刺激の喪失があろう。結局は、随意制御の下にある全ての筋肉は侵され、患者は、腕、脚および身体を動かす力および能力を喪失する。
【0008】
脳、脳幹、および脊髄に位置する運動ニューロンは制御ユニットとして働き、重要な通信は神経系および身体の随意筋の間をリンクさせる。脳内の運動ニューロン(上位運動ニューロン)からのメッセージは脊髄中の運動ニューロン(下位運動ニューロン)に伝達され、そこから特定の筋肉に伝達される。ALSにおいては、上位運動ニューロンおよび下位運動ニューロンは変性するか、あるいは死滅し、メッセージを筋肉に送るのを止める。筋肉は機能できず、除々に弱まり、段々減り(萎縮)、およびひきつる(束性攣縮)。結局は、随意運動を開始し、制御する脳の能力は失われる。原因は知られていない。
【0009】
MSは若い成人において非外傷性CNS罹患率の主な原因である。該疾患の若い年齢での開始および進行性性質は、社会に対して多大の経済的および社会的負荷を課す。典型的な再発軽減MSにおける急性増悪は、CNSにおける急性および病巣性炎症および脱髄の発現であり、長い間、MSの一次的疾患原因と考えられてきた。これらの事象は、現在認可されている治療剤の標的である。しかしながら、磁気共鳴(MR)イメージについてのT2炎症シグナル、および疾患の進行の相関は、再発軽減(RR)相および不能の引き続いての進行の間における臨床的特徴がそうであるように、弱い。さらに、いったん不可逆的不能に到達すれば、さらなる不能への進行は、不可逆的負傷の開始の前または後に起こるものを含めて、再発によって影響されない。
【0010】
軸索喪失による永久的神経学的不能に加えて、炎症性脱髄はMS病因において役割を演じる。炎症とは対照的に、軸索喪失は、典型的には、T1黒色穴、磁気共鳴分光測定(MRS)による減少したアスパラギン酸N−アセチル(NAA)、および脊髄萎縮の程度に相関し、これは、患者における臨床的不能と相関し得る。これらの変化は、診断から6カ月後と初期の患者において認められているが、ほとんどの患者においては、慢性、およびおそらくは全体的な軸索負傷は、疾患の二次的進行相の開始において臨床的な閾値を破る。
【0011】
(再発/軽減MS、またはRRMSと臨床的に定義される)疾患の急性炎症段階の間に、炎症メディエーターは軸索負傷に寄与するようである。CD8+T細胞の数、および軸索損傷の程度の間で関連付けがなされており、動物モデルは、これが、軸索破壊のCD8−MHCクラスI経路に関与することを支持する傾向がある(非特許文献2)。さらなる裏付けは、脱髄された軸索に向かって分極された細胞傷害性顆粒を含有する活性化されたCD8T細胞が直接的なCD8+T細胞毒性を示唆するという病理学的研究からくるものである。マクロファージおよび小神経膠細胞は、変性する軸索と近接して見出されている。これらの細胞型は、CNSからデブリスを除去するホメオスタシスメカニズムにおいて
役割を演じているが、それらはプロテアーゼ、サイトカイン、および酸化窒素(NO)のようなフリーラジカルを含めた炎症メディエーターも放出する。最後に、抗体および補体もまた、急性炎症の間に軸索損傷において役割を演じ得る。抗ガングリオシド抗体のレベルは、二次的な進行性またはRRMSにおけるよりも一次的進行性MS(PPMS)において有意に高いことが判明し、脱髄後に補体に暴露された軸索は、補体カスケードを直接的に活性化し得る。
【0012】
MSにおける炎症およびニューロン喪失の間の関係は十分に明らかにされていない。乏突起神経膠細胞の喪失、および全身的に特異的CNS抗原に対する炎症の誘導に依拠せず、または効力ある系アジュバントの使用を必要としない、引き続いての脱髄のモデルを確立する必要性がある。抗原および炎症を利用するそのようなモデルは、典型的には、MS患者において同定された脱髄およびニューロン喪失を反復しない。
【0013】
軸索横断およびニューロン喪失の正確な動物モデルは、MS脳検体において同定された病理学的特徴を模倣すべきである。これは、急性皮質病巣における横断された軸索、横断された樹状突起およびニューロンアポトーシスの同定を含む。急性事象は、また、MS病理学検体において同定されているように、Na+チャネルの再分布によって、程度は変わるが、回復される測定可能な損傷神経伝達をもたらすべきである。慢性病巣は、種々の程度の髄鞘再形成を示すべきであり、内向する持続性の軸索喪失が明らかでなければならない。最後に、動物モデルにおけるニューロン喪失が、解剖学的に区別され、かつNAWMに対する疾患の影響を模倣する元来の脱髄病巣から一時的に区別される領域で同定されなければならない。これらの特徴は、MS患者からの病理学検体において同定されたニューロンに対する影響の重要かつ正確な描写を提供するであろう。
【0014】
正確な分子メカニズム、および神経障害の病因および、特に、ニューロン脱髄の解明は、効果的な動物モデルの継続的欠如によって妨げられてきた。かくして、ニューロン障害のための治療剤に対する頑強なスクリーニングを行うための組成物および方法に対する緊急の要望が存在する。
【先行技術文献】
【非特許文献】
【0015】
【非特許文献1】Pfeifferら(1993)Trends Cell Biol.3:191−197
【非特許文献2】Rivera−Quinonesら、(1998)Nat Med.4:187−193
【発明の概要】
【課題を解決するための手段】
【0016】
本発明は、神経障害のプロセスを理解するための組成物および方法を提供し、およびニューロン脱髄の処置のための生物活性剤を同定しおよび開発する。本発明のトランスジェニック動物/細胞は、髄鞘再形成についての、および候補生物活性剤をスクリーニングするためのメカニズムを解明するのに利用することができるモデル系を提供する。マーカーおよび他の手段の髄鞘再形成特異的発現を利用することによって、本発明は、効果が肯定的(潜在的に治療的)であるか、または効果が否定的(潜在的に有害)であるかを問わず、髄鞘再形成に対する効果についてテスト剤をアッセイするのに利用することができる。さらに、髄鞘再形成に関連する現象に対するテスト剤の効果を決定することは、限定されるものではないが、細胞ベースのアッセイまたは技術を利用するものを含めた、当該分野で公知のいずれの適当な方法も含むことができる。
【0017】
本発明は、細胞死メディエーター蛋白質(CDMP)をコードする核酸配列を含む組換
え核酸分子を提供し、ここに、前記核酸配列は神経細胞特異的調節エレメントに作動可能に連結されている。また、核酸配列は、GFPまたは他の蛍光マーカーのようなマーカー蛋白質をコードする第2の核酸配列に作動可能に連結できる。また、本発明は、細胞死メディエーター蛋白質(CDMP)をコードする核酸配列を含む組換え核酸分子を含む宿主細胞も提供し、ここに、前記核酸配列は神経細胞特異的調節エレメントに作動可能に連結されている。前記宿主細胞は神経細胞または壁細胞であり得、例えば、それはニューロン細胞または神経膠細胞で有り得る。例えば、神経膠細胞は、乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞またはシュワン細胞であり得る。ニューロン細胞は頸部神経節ニューロン、皮質ニューロン、セロトニンニューロン、後根神経節、結節性神経節ニューロン、脊髄運動ニューロン、中脳ドーパミン作動性ニューロン、中枢ノルアドレナリン作動性ニューロンまたは腸ニューロンであり得る。壁細胞は内皮細胞、血管周囲細胞または平滑筋細胞であり得る。いくつかの実施形態において宿主細胞はBまたはTリンパ球のような免疫細胞であり得る。
【0018】
また、細胞型特異的発現調節エレメントに作動可能に連結された細胞死メディエーター蛋白質(CDMP)をコードするヌクレオチド配列を含むトランスジェニック動物も提供され、ここに、前記動物は、前記ヌクレオチド配列を含まない動物に対してより大きな程度の神経障害を呈する。動物は哺乳動物、霊長類、またはマウス、ラット、モルモットのようなげっ歯類、イヌ、ネコ、ウサギ、ブタ、チンパンジーまたはサルであり得る。神経障害は多発性硬化症のようなニューロン脱髄を含み得る。前記動物は対照動物のそれに対してアポトーシス神経稀突起膠細胞の増加を呈することができる。CDMPは中枢神経系に対して異所性的に制限され得る。
【0019】
CDMPをコードする核酸配列は宿主細胞または動物のゲノムに組み込むことができる。別法として、CDMPをコードする核酸配列はエピソームのものとすることができる。さらに、組換え核酸分子を宿主細胞または動物に送達して、レンチウイルスベクターのようなウイルスベクターによってトランスジェニック動物を作成することができる。
【0020】
1つの態様において、CDMPはカスパーゼ2、カスパーゼ5、カスパーゼ8、カスパーゼ9、カスパーゼ10、またはカスパーゼ11であり得る。CDMPは、FK506型リガンド、FKBP12型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドについての結合ドメインを含むキメラ蛋白質であり得る。CDMPの発現および/またはアポトーシス促進活性は、例えば、誘導性カスパーゼ9(iCP9)のように、誘導性であり得る。例えば、発現調節エレメントは誘導性、構成的、および/または細胞型または組織特異的であり得る。いくつかの実施形態において、発現および/または活性は動物におけるもののように神経細胞に特異的である。CDMPの活性は、AP20187のような二量体化化学誘導因子(CID)によって誘導することができる。
【0021】
(CDMP)をコードする核酸配列を含む核酸分子は、壁細胞または神経細胞特異的調節エレメントに作動可能に連結され得る。例えば、調節エレメントはニューロン細胞または神経膠細胞特異的調節エレメントであり得る。核酸配列によってコードされるCDMPの発現または活性は、乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、またはシュワン細胞のようなニューロン細胞または神経膠細胞におけるものであり得る。神経膠細胞特異的調節エレメントはCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、プロテオリピド蛋白質(PLT)、乏突起神経膠細胞ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFR−α、PDGF−α、RG5、p糖蛋白質、ニュールツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、P
DGFR−β、またはスルファチド遺伝子からのものであり得る。
【0022】
本発明のもう1つの態様は、生物学的に活性な剤、または生物活性剤をスクリーニングする方法である。本発明は、候補剤を、細胞死メディエーター蛋白質(CDMP)をコードする核酸を含む細胞と接触させる工程であって、ここに、前記核酸は細胞型特異的発現調節エレメントに作動可能に連結されている、工程;脱髄障害に関連する現象に対する効果を検出する工程;前記CDMPの活性のレベルが対照細胞に対して調節されているならば、前記薬剤を前記現象を調節するのに効果的であるとして選択する工程を含む、前記現象を調節する生物学的に活性な剤についてスクリーニングする方法を提供する。前記細胞は神経膠細胞、または壁細胞のようなニューロン細胞であり得る。神経膠細胞は乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、またはシュワン細胞であり得る。
【0023】
また、本明細書において、非ヒトトランスジェニック動物に候補剤を投与する工程であって、ここに、細胞死メディエーター蛋白質(CDMP)をコードする核酸配列の発現に際して脱髄が前記動物で起こり;ここに、前記核酸配列の発現は細胞特異的発現調節エレメントによって調節される、工程;前記CDMPを活性化して、前記動物における少なくとも1つの細胞でアポトーシスをもたらす工程であって、ここに、前記細胞は脱髄障害に関連している、工程;前記脱髄障害に関連する現象に対する前記薬剤の効果を検出する工程を含む、脱髄障害に関連する現象を調節する生物学的に活性な剤についてスクリーニングする方法も提供される。いくつかの実施形態において、動物は候補剤の投与に先立って脱髄から回復させる。脱髄障害は動物における乏突起神経膠細胞、神経膠星状細胞またはシュワン細胞の喪失によって特徴付けることができ、脱髄障害に関連する現象は髄鞘を有する軸索の減少によって特徴付けることができる。いくつかの実施形態において、脱髄障害は多発性硬化症である。もう1つの態様において、薬剤の効果の決定はPCR、イムノアッセイ、ハイブリダイゼーションアッセイまたはその組合せを含むことができる。候補剤はアンチセンスオリゴヌクレオチド、ペプチド、抗体、リポソーム、低分子干渉RNA、低分子有機化合物、または無機化合物であり得る。
【0024】
本発明のなおもう1つの態様において、細胞死メディエーター蛋白質(CDMP)をコードする核酸を含むトランスジェニック動物または細胞を供する工程であって、ここで前記核酸は神経細胞または神経膠細胞特異的発現調節エレメントに作動可能に連結されている、工程;CDMPを活性化し、それにより、アポトーシスを誘導する工程;活性化に続いて少なくとも1つの生存するニューロン細胞または神経膠細胞を得る工程;前記生存する神経膠細胞または神経細胞においてRNA転写物および/またはコードされた産物をプロファイルする工程;それにより、多発性硬化症に関連する現象、または多発性硬化症に関連するMS関連状態を特徴付けるプロファイルデータセットを編集する工程を含む、多発性硬化症(MS)に関連する現象、または多発性硬化症に関連するMS関連状態を特徴付けるプロファイルデータセットを編集する方法が提供される。
引用による援用
本明細書中で言及された全ての刊行物および特許出願は、各個々の刊行物または特許出願が引用により援用されることが具体的かつ個々に意図されているのと同等の程度まで引用によりここに援用される。
本発明の好ましい実施形態では、例えば以下が提供される:
(項目1)
細胞死メディエーター蛋白質(CDMP)をコードする核酸配列を含む組換え核酸分子であって、ここで、前記核酸配列は神経細胞特異的調節エレメントに作動可能に連結されている、組換え核酸分子。
(項目2)
前記CDMPがカスパーゼ2、カスパーゼ5、カスパーゼ8、カスパーゼ9、カスパーゼ10、またはカスパーゼ11である、項目1記載の組換え核酸分子。
(項目3)
前記CDMPが、FK506型リガンド、FKBP12型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドについての結合ドメインを含むキメラ蛋白質である、項目1記載の組換え核酸分子。
(項目4)
前記CDMPの発現が誘導性である、項目1記載の組換え核酸分子。
(項目5)
前記CDMPのアポトーシス促進活性が誘導性である、項目1記載の組換え核酸分子。(項目6)
前記活性が二量体化化学誘導因子(CID)によって誘導される、項目5記載の組換え核酸分子。
(項目7)
前記CDMPが誘導性カスパーゼ9(iCP9)である、項目5記載の組換え核酸分子。
(項目8)
前記CIDがAP20187である、項目6記載の組換え核酸分子。
(項目9)
前記神経細胞特異的調節エレメントが神経膠細胞特異的調節エレメントである、項目1記載の組換え核酸分子。
(項目10)
前記神経膠細胞が乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、またはシュワン細胞である、項目1記載の組換え核酸分子。
(項目11)
前記神経膠細胞特異的調節エレメントがCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、プロテオリピド蛋白質(PLP)、乏突起神経膠細胞−ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFR−α、PDGF−α、RG5、P糖蛋白質、ニュールツリン(neurturin)(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、PDGFR−β、またはスルファチド遺伝子からのものである、項目9記載の組換え核酸分子。
(項目12)
細胞死メディエーター蛋白質(CDMP)をコードする核酸配列を含む組換え核酸分子を含む宿主細胞であって、ここで、前記核酸配列は神経細胞特異的調節エレメントに作動可能に連結されている、宿主細胞。
(項目13)
前記CDMPがカスパーゼ2、カスパーゼ5、カスパーゼ8、カスパーゼ9、カスパーゼ10、またはカスパーゼ11である、項目12記載の宿主細胞。
(項目14)
前記CDMPがFK506型リガンド、FKBP12型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドについての結合ドメインを含むキメラ蛋白質である、項目12記載の宿主細胞。
(項目15)
前記CDMPの発現が誘導性である、項目12記載の宿主細胞。
(項目16)
前記CDMPのアポトーシス促進活性が誘導性である、項目12記載の宿主細胞。
(項目17)
前記活性が二量体化化学誘導因子(CID)によって誘導される、項目16記載の宿主細胞。
(項目18)
前記CDMPが誘導性カスパーゼ9(iCP9)である、項目12記載の宿主細胞。
(項目19)
前記CIDがAP20187である、項目17記載の宿主細胞。
(項目20)
前記神経細胞特異的調節エレメントが神経膠細胞特異的調節エレメントである、項目12記載の宿主細胞。
(項目21)
前記神経膠細胞が乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、またはシュワン細胞である、項目20記載の宿主細胞。
(項目22)
前記神経膠細胞特異的調節エレメントがCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、プロテオリピド蛋白質(PLP)、乏突起神経膠細胞−ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGF−α、RG5、P糖蛋白質、ニュールツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、PDGFR−β、PDGFR−α、またはスルファチドから選択される遺伝子からのものである、項目20記載の宿主細胞。
(項目23)
前記宿主細胞が神経細胞または壁細胞である、項目12記載の宿主細胞。
(項目24)
前記宿主細胞が神経膠細胞である、項目23記載の宿主細胞。
(項目25)
前記神経膠細胞が乏突起神経膠細胞、神経膠細胞星状細胞、小神経膠細胞、またはシュワン細胞である、項目24記載の宿主細胞。
(項目26)
CDMPをコードする前記核酸配列が前記宿主細胞のゲノムに組み込まれる、項目12記載の宿主細胞。
(項目27)
CDMPをコードする前記核酸配列がエピソームのものである、項目12記載の宿主細胞。
(項目28)
前記組換え核酸分子がウイルスベクターによって前記宿主細胞に送達される、項目12記載の宿主細胞。
(項目29)
前記ウイルスベクターがレンチウイルスベクターである、項目28記載の宿主細胞。
(項目30)
細胞型特異的発現調節エレメントに作動可能に連結された細胞死メディエーター蛋白質(CDMP)をコードするヌクレオチド配列を含むトランスジェニック動物であって、ここで、前記動物は前記ヌクレオチド配列を含まない動物と比較してより大きな程度の神経障害を呈する、トランスジェニック動物。
(項目31)
前記動物が哺乳動物、霊長類、またはげっ歯類である、項目30記載の動物。
(項目32)
前記動物がマウス、ラット、モルモット、イヌ、ネコ、ウサギ、ブタ、チンパンジーまたはサルである、項目30記載の動物。
(項目33)
前記神経障害がニューロン脱髄および/または血液脳関門における欠陥を含む、項目30記載の動物。
(項目34)
前記動物が対照動物の場合と比較してアポトーシス乏突起神経膠細胞または血管周囲細胞の増加を呈する、項目30記載の動物。
(項目35)
前記CDMPがカスパーゼ2、カスパーゼ5、カスパーゼ8、カスパーゼ9、カスパーゼ10またはカスパーゼ11である、項目30記載の動物。
(項目36)
前記CDMPがFK506型リガンド、FKBP12型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドについての結合ドメインを含むキメラ蛋白質である、項目30記載の動物。
(項目37)
前記CDMPの発現が誘導性である、項目30記載の動物。
(項目38)
前記CDMPの発現が中枢神経系に異所的に制限される、項目30記載の動物。
(項目39)
前記CDMPのアポトーシス促進活性が誘導性である、項目30記載の動物。
(項目40)
前記アポトーシス促進活性が、特異的に、前記動物の神経細胞におけるものである、項目39記載の動物。
(項目41)
前記活性が二量体化化学誘導因子(CID)によって誘導される、項目39記載の動物。
(項目42)
前記CDMPが誘導性カスパーゼ9(iCP9)である、項目39記載の動物。
(項目43)
前記CIDがAP20187である、項目41記載の動物。
(項目44)
前記細胞型特異的発現調節エレメントが神経または壁細胞特異的調節エレメントである、項目30記載の動物。
(項目45)
前記神経細胞特異的調節エレメントが神経膠細胞特異的調節エレメントである、項目44記載の動物。
(項目46)
前記神経膠細胞が乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、またはシュワン細胞である項目45記載の動物。
(項目47)
前記神経膠細胞特異的調節エレメントがCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、プロテオリピド蛋白質(PLP)、乏突起神経膠細胞−ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFα、RG5、P糖蛋白質、ニュールツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、PDGFR−α、PDGFR−β、またはスルファチドから選択される遺伝子からのものである、項目45記載の動物。
(項目48)
前記核酸配列がマーカー蛋白質をコードする第2の核酸配列に作動可能に連結されている、項目30記載の動物。
(項目49)
脱髄または血液脳関門障害に関連する現象を調節する生物活性剤についてスクリーニングする方法であって;
a)候補剤を、細胞死メディエーター蛋白質(CDMP)をコードする核酸を含む細胞
と接触させる工程であって、ここで、前記核酸は細胞型特異的発現調節エレメントに作動可能に連結されている工程;
b)前記現象に対する効果を検出する工程;
c)前記CDMPの活性のレベルが対照細胞と比べて調節されている場合、前記薬剤を前記現象を調節するのに効果的であるとして選択する工程
を含む、方法。
(項目50)
前記細胞が神経細胞、神経膠細胞または壁細胞である、項目49記載の方法。
(項目51)
前記細胞が乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、血管周囲細胞、またはシュワン細胞である、項目49記載の方法。
(項目52)
脱髄または血液脳関門障害に関連する現象を調節する生物活性剤についてスクリーニングする方法であって;
a)非−ヒトトランスジェニック動物に候補剤を投与する工程であって、ここで、前記現象は、細胞死メディエーター蛋白質(CDMP)をコードする核酸配列の発現に際して前記動物で起こり;ここで、前記核酸配列の発現は細胞特異的発現調節エレメントによって調節される工程;
b)前記CDMPを活性化して、前記動物における少なくとも1つの細胞においてアポトーシスをもたらす工程であって、ここで、前記細胞は脱髄または血液脳関門障害に関連する工程;
c)前記現象における前記薬剤の効果を検出する工程
を含む、方法。
(項目53)
工程b)後に、前記動物を前記現象から回復させる、項目52記載の方法。
(項目54)
前記現象が、前記動物における乏突起神経膠細胞、神経膠星状細胞、血管周囲細胞、シュワン細胞の喪失によって特徴付けられる、項目52記載の方法。
(項目55)
前記現象が髄鞘形成軸索の減少、または血液脳関門透過性の増加によって特徴付けられる、項目52記載の方法。
(項目56)
前記調節エレメントがCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、乏突起神経膠細胞−ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFα、RG5、P糖蛋白質、ニュールツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、スルファチド、PDGFR−β、PDGFR−α、またはプロテオリピド蛋白質(PLP)遺伝子から選択される遺伝子からのものである、項目49または52記載の方法。
(項目57)
前記脱髄障害が多発性硬化症である、項目52記載の方法。
(項目58)
前記効果を決定することがPCR、イムノアッセイ、ハイブリダイゼーションアッセイまたはそれらの組合せを含む、項目49または52記載の方法。
(項目59)
前記候補剤がアンチセンスオリゴヌクレオチド、ペプチド、抗体、リポソーム、低分子干渉RNA、低分子有機化合物または無機化合物である、項目49または52記載の方法。
(項目60)
前記少なくとも1つの細胞が、ニューロン細胞、神経膠細胞または壁細胞である、項目52記載の方法。
(項目61)
前記少なくとも1つの細胞が乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、血管周囲細胞、またはシュワン細胞である、項目60記載の方法。
(項目62)
前記CDMPがカスパーゼ2、ガスパーゼ5、ガスパーゼ8、カスパーゼ9、カスパーゼ10またはカスパーゼ11である、項目49または52記載の方法。
(項目63)
前記CDMPがFK506型リガンド、FKBP12型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドについての結合ドメインを含むキメラ蛋白質である、項目49または52記載の方法。
(項目64)
前記CDMPの発現が誘導性である、項目49または52記載の方法。
(項目65)
前記CDMPのアポトーシス促進活性が誘導性である、項目49または52記載の方法。
(項目66)
前記活性が二量体化化学誘導因子(CID)によって誘導される、項目65記載の方法。
(項目67)
前記CDMPが誘導性カスパーゼ9(iCP9)である、項目49または52記載の方法。
(項目68)
前記CIDがAP20187である、項目67記載の方法。
(項目69)
多発性硬化症(MS)または多発性硬化症に関連するMS関連疾患に関連する現象を特徴付けるためのプロファイルデータセットを編集する方法であって:
a)細胞死メディエーター蛋白質(CDMP)をコードする核酸を含むトランスジェニック動物または細胞を供する工程であって、ここで、前記核酸はニューロンまたは神経膠細胞特異的発現調節エレメントに作動可能に連結されている工程;
b)前記CDMPを活性化し、それによりアポトーシスを誘導する工程;
c)前記活性化に続いて少なくとも1つの生存するニューロン細胞または神経膠細胞を得る工程;
d)前記生存する神経膠細胞または神経細胞におけるRNA転写物および/またはコードされる産物をプロファイルし、それにより多発性硬化症または多発性硬化症に関連するMS−関連障害に関連する現象を特徴付けるプロファイルデータセットを編集する工程
を含む、方法。
(項目70)
前記CDMPがカスパーゼ9またはカスパーゼ11である、項目69記載の方法。
(項目71)
前記CDMPのアポトーシス誘導活性が誘導性である、項目69記載の方法。
(項目72)
前記神経膠細胞特異的発現調節エレメントがCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、およびプロテオリピド蛋白質(PLP)、CC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、乏突起神経膠細胞−ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFα、PDGFR−β、PDGFR−α、RG5、P糖蛋白質、ニュールツリン(NRTN)
、アルテミン(ARTN)、ペルセフィン(PSPN)、スルファチド、またはプロテオリピド蛋白質(PLP)遺伝子からなる群のものである、項目69記載の方法。
【図面の簡単な説明】
【0025】
本発明の新規な特徴は、特に、添付の請求の範囲に記載されている。本発明の特徴および利点は、本発明の原理が利用される例示としての実施形態を説明する以下の詳細な記載、および添付の図面を参照することによってより良く理解されるであろう。
【図1】操作されたSINレンチウイルスベクター(pΔcmvICP9/mEGFP(図1B)およびpΔmbpICP9/mEGFP)を示す。図1AはpΔcmv/mEGFPベクター構築体を提供する。第2世代のpΔmbpICP9/mEGFP(図1C)は、遺伝子発現を感染した乏突起神経膠細胞に制限するMBP特異的プロモーターを有し、その細胞特異的発現は乏突起神経膠細胞特異的細胞死を引き起こす。デュアルプロモーターの特徴は、感染した細胞が蛍光顕微鏡検査法を用いて容易に同定することができるような、上流遺伝子から独立したGFPの同時発現を可能とする。LTR=末端反復配列;pCMV=サイトメガロウイルスプロモーター配列;iCP9=操作された誘導性カスパーゼ−9遺伝子配列;pMND=修飾されたLTRプロモーター配列;EGFP=増強された緑色蛍光蛋白質遺伝子配列;pMBP=ミエリン塩基性蛋白質プロモーター配列。
【図2】細胞型選択的にアポトーシスを誘導するための種々の異なる組織特異的プロモーターを使用して作製されたレンチウイルスベクターを示す。前記ウイルスベクターは、典型的には、感染した細胞においてEGFP発現をもたらす。LRT=末端反復配列;pMND=ウイルスプロモーター配列;GFP=緑色蛍光蛋白質;pCMV=サイトメガロウイルスプロモーター配列;iCP9=誘導性カスパーゼ遺伝子配列。
【図3】濃縮されたレンチウイルスを直接的にCNSに適用することができ感染の程度および領域は、犠牲後に検出されたGFP陽性細胞の数、および脳または脊髄の薄切片に基づいて決定することができることを示す写真を提供する。このデータは、インビトロおよびインビボにおける特異的細胞死についての導入メカニズムを確立する。
【図4】本発明の特定の実施形態のグラフ表示である。該図は、本発明の1つの方法、および該方法についての特定の適用の一般的概略を示す。
【図5】GFP+細胞の持続性によって示されるように、CIDを培養物に加えた後に生存したままである対照ウイルスで感染させた細胞(上段)を示す写真を提供する。iCP9を発現するウイルスで感染させた細胞は、GFP+細胞の不存在によって示されるように、CID暴露後にアポトーシスを受ける(下段)。明視野における細胞の持続性は、感染した細胞のみに対するアポトーシスの特異性を示す。BF=明視野。
【図6】iCP9遺伝子をコードするウイルスの種々の希釈物に暴露され、次いで、CIDを培養基に加えた細胞を示すグラフである。4時間後に、アポトーシスELISAを行った。アポトーシスのレベルは、ウイルスの希釈に伴って減少した。というのは、より少数の細胞が感染し、iCP9を発現したからである。かくして、アポトーシスはiCP9遺伝子発現に相関した。X軸はウイルス希釈である。Y軸は405nmにおけるELISA吸収である。
【図7】pLpMBP(iCP9)MGに感染され、次いで、CIDに暴露されたラットの子に由来する混合皮質培養物を示す。4時間後に、細胞を染色して、細胞型を同定した。CID暴露(+CID)後におけるMBP+細胞の形態は、アポトーシスを示唆する。他の細胞型は、生きた細胞と合致する形態を維持した。これは、MBP発現細胞においてウイルスが細胞死を誘導することを示す。
【図8】pLpGFAP(iCP9)MGで感染させ、CIDに暴露した(+CID)パン精製GFAP+細胞(神経膠星状細胞)を示す。GFP+細胞は、CIDへの暴露から4時間後にEGFP+細胞の不存在によって示されるアポトーシスを受けた(上段顕微鏡写真)。CIDに暴露されなかった(−CID)並行培養物は、多数のEGFP+細胞を維持した(下段顕微鏡写真)。
【図9】混合皮質培養物をpLpPDGFR(iCP9)MGウイルスに感染させ、CIDに暴露した(+CID)ことを示す。4時間後に、A2B5+細胞は、生きた細胞の形態の喪失に基づいてアポトーシスを受けていると同定された。
【図10】ラットの脳梁へのpLpGFAP(iCP9)MGウイルスの注射からの結果を示す。CIDへの暴露から24時間後に、アポトーシスは、GFAP+細胞の喪失を伴う感染の部位において同定される(右側パネル)。しかしながら、感染、その後のビヒクルへの暴露は、アポトーシスまたはGFAP+細胞の喪失を生じさせなかった(左側パネル)。これは、ウイルスの結果、アポトーシスを介するGFAP+細胞(神経膠星状細胞)の特異的喪失をもたらすことを示唆する。
【図11】特異的に乏突起神経膠細胞を除去するためにラットの脳梁に注射されたpLpMBP(iCP9)MGウイルスを示す。CIDへの暴露(CID+)から24時間後に、アポトーシスは、TUNEL染色(最上段パネル)およびMBP染色細胞の不存在(第2段)を介して感染の部位において同定された。抗MBP抗体で染色されない領域は、抗GFAP抗体で陽性に染色され、これは、細胞がその領域に持続することを示す。黒色矢印によって示される領域は双方について陰性に染色され、針挿入によって生じた組織欠陥を表す。V=脳室。最下段パネルは、全ての感染した細胞はGFP+であるので感染の程度を示す。
【発明を実施するための形態】
【0026】
本発明において、トランスジェニック動物または細胞は、神経障害を研究するためのモデル系を提供する。前記モデル系は、ニューロンの健康および/または脱髄/髄鞘再形成を同定し、評価し、および/または定量するために使用することができる。さらに、本発明の組成物および方法は、ニューロンまたは髄鞘形成の健康または維持を調節し、促進し、または低下させる生物学的に活性な剤の同定および開発のための種々の実施形態において利用される。
【0027】
本発明の1つの態様は、ニューロン脱髄および/または髄鞘再形成に対する効果を評価するための特定の細胞型における細胞死の選択的誘導を含む。これにより、脱髄に対するニューロンの応答はインビボおよびインビトロにて人工的に誘導された炎症なくして定義することができる。
【0028】
ニューロンの喪失は、多発性硬化症に罹った患者での不能の進行における重要な因子である。さらに、ニューロンの喪失は炎症からかなり独立でき、ニューロン細胞およびミエリンを産生する乏突起神経膠細胞の間の共依存性関係の破壊後に起こり得る。
【0029】
したがって、1つの実施形態において、系(例えば、動物)を提供して、その共依存性乏突起神経膠細胞の喪失に対するニューロンの応答を特徴付ける。例えば、修飾されたレトロウイルスを利用して、誘導性自殺因子、または細胞死メディエーター蛋白質をコードする遺伝子を脳内の乏突起神経膠細胞に送達する。誘導性因子を動物に投与すると、自殺遺伝子は乏突起神経膠細胞(ミエリン産生細胞)がアポトーシスを受けるように誘導し、その結果、炎症によって誘導されない病巣脱髄がもたらされる。乏突起神経膠細胞およびミエリンの喪失に対するニューロンの応答を、次いで特徴付ける。脱髄に対するニューロンの応答を理解することによって、この事象後にニューロンを保護する介入を開発することができる。
【0030】
神経細胞のような細胞を、制御された細胞死について、被験体において、または細胞培養物において標的化することができる。いくつかの実施形態において、さらなる研究のために、例えば、エクスビボ技術を含めた、細胞ベースのまたは細胞培養設定で行われるアッセイのために、そのような細胞を本発明のトランスジェニック動物から単離する。
【0031】
本発明の種々の態様は、臨床的に発現する、乏突起神経膠細胞またはニューロン喪失のようなCNS中の病理学的変化を評価するための方法を提供する。
【0032】
本発明のいくつかの態様において、当該分野で公知の方法を用いて非ヒトトランスジェニック動物を操作して、細胞型特異的または誘導性発現調節エレメント(例えば、プロモーター/エンハンサー)に作動可能に連結された細胞死メディエーター蛋白質の発現を供する。いくつかの実施形態において、ヒトまたは非ヒトの細胞を操作して、細胞型特異的または誘導性発現調節エレメント(例えば、プロモーター/エンハンサー)に作動可能に連結された1以上の細胞死メディエーター蛋白質を発現させることができる。本発明の種々の態様において有用なそのような調節エレメント、ならびに本発明の組成物/方法で標的化することができる種々の細胞型は本明細書中により十分に記載されている。
【0033】
1つの実施形態において、本発明の非ヒトトランスジェニック動物(「テスト動物」)における1以上の細胞死メディエーター蛋白質の発現の結果、そのような動物において神経障害または増悪した神経障害がもたらされる。
【0034】
なおもう1つの実施形態において、1以上の細胞死メディエーター蛋白質の発現は、テスト動物の中枢神経系または末梢神経系におけるものである。
【0035】
本発明の1つの態様において、1以上の細胞死メディエーター蛋白質(CDMP)の発現の結果、テスト動物においてニューロンの変性がもたらされる。いくつかの実施形態において、そのようなニューロンは頸部神経節ニューロン、皮質ニューロン、セロトニンニューロン、後根神経節、結節性神経節ニューロン、脊髄運動ニューロン、中脳ドーパミン作動性ニューロン、中枢ノルアドレナリン作動性ニューロンまたは腸ニューロンを含む。
【0036】
種々の実施形態において、本発明の細胞死メディエーター核酸構築体を含む細胞は、限定されるものではないが、神経細胞または壁細胞である。
【0037】
いくつかの実施形態において、そのような細胞は神経膠細胞であり、限定されるものではないが、乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞および/またはシュワン細胞を含む。いくつかの実施形態において、そのような細胞は、限定されるものではないが、血管周囲細胞、内皮細胞および/または平滑筋細胞を含む。
【0038】
本発明のもう1つの態様は、選択的および限定された細胞死後に、インビボにて、ニューロンおよび乏突起神経膠細胞の応答を決定する方法を対象とする。例えば、本発明の核酸構築体(例えば、細胞/組織特異的発現調節エレメントに作動可能に連結された細胞死メディエーター蛋白質をコードする核酸構築体)を、成体ラットに適用することができ、その結果、抗原/アジュバント刺激炎症によって合併されないCNS中の病巣に対するニューロンの応答を評価するためのモデル系がもたらされる。さらに、本発明は、病巣のサイズ、病巣の位置、病巣の数、および病巣の間の一時的な関係の制御を可能とする組成物および方法を提供する。
一般的な技術
本発明の実施は、特に断りのない限り、当業者の技量内にある、免疫学、生化学、化学、分子生物学、微生物学、細胞生物学、ゲノミックスおよび組換えDNAの慣用的な技術を使用する。Sambrook,Fritsch and Maniatis,MOLECULAR CLONING:A LABORATORY MANUAL,第2版(1989);CURRENT PROTOCOLS IN MOLECULAR BIOLOGY(F.M.Ausubel,ら編,(1987));METHODS IN ENZYMOLOGYのシリーズ(Academic Press Inc.);PCR2:A
PRACTICAL APPROARCH(M.J.MacPherson,B.D.
Hames and G.R.Taylor編(1995)),Harlow and Lane編(1988)ANTIBODIES,A LABORATORY MANUAL,and ANIMAL CELL CULTURE (R.I.Freshney編(1987))参照。
定義
明細書および特許請求の範囲で用いるように、単数形「ある」、および「前記」は文脈が明瞭にそうでないことを指示するのでなければ、複数への言及を含む。例えば、用語「ある細胞」はその混合物を含めた複数の細胞を含む。
【0039】
用語「ポリヌクレオチド」、「ヌクレオチド」、「ヌクレオチド配列」、「核酸」および「オリゴヌクレオチド」は相互交換可能に用いる。それらは、デオキシリボヌクレオチドまたはリボヌクレオチド、またはそのアナログいずれかである、いずれかの長さのヌクレオチドのポリマー形態をいう。ポリヌクレオチドはいずれかの三次元構造を有することができ、公知または未知のいずれかの機能を行うことができる。以下に示すのはポリヌクレオチドの非限定的例である:遺伝子または遺伝子断片のコーディング、または非コーディング領域、連鎖分析から定義された複数の遺伝子座(遺伝子座)、エクソン、イントロン、メッセンジャーRNA(mRNA)、トランスファーRNA、リボソームRNA、リボザイム、cDNA、組換えポリヌクレオチド、分岐ポリヌクレオチド、プラスミド、ベクター、いずれかの配列の単離されたDNA、いずれかの配列の単離されたRNA、核酸プローブ、およびプライマー。ポリヌクレオチドはメチル化ヌクレオチドおよびヌクレオチドアナログのような修飾されたヌクレオチドを含むことができる。もし存在すれば、ヌクレオチド構造に対する修飾は、ポリマーの組立前にまたは後に付与することができる。ヌクレオチド配列は非ヌクレオチド成分によって中断してもよい。ポリヌクレオチドは、さらに、標識成分でのコンジュゲーションなどによって、重合の後に修飾することができる。
【0040】
「ヌクレオチドプローブ」または「プローブ」とは、ハイブリダイゼーション反応においてその対応する標的ポリヌクレオチドを検出し、または同定するのに用いるポリヌクレオチドをいう。
【0041】
「ハイブリダイゼーション」とは、1以上のポリヌクレオチドが反応して複合体を形成し、これはヌクレオチド残基の塩基の間の水素結合を介して安定化される反応をいう。水素結合はワトソン・クリック塩基対合、フーグステン結合によって、あるいはいずれかの他の配列特異的様式で起こり得る。複合体はデュプレックス構造を形成する2つのストランド、マルチストランデッド複合体を形成する3以上のストランド、単一の自己ハイブリダイジングストランド、またはこれらのいずれかの組合せを含むことができる。ハイブリダイゼーション反応はPCRの開始、またはリボザイムによるポリヌクレオチドの酵素切断のようなより広汎なプロセスにおいて工程を構成することができる。
【0042】
ポリヌクレオチドに適用される用語「ハイブリダイズされた」とは、ヌクレオチド残基の塩基の間の水素結合を介して安定化される複合体を形成するポリヌクレオチドの能力をいう。水素結合はワトソン・クリック塩基対合、フーグステン結合によって、またはいずれかの他の配列特異的様式で起こり得る。前記複合体はデュプレックス構造を形成する2つのストランド、マルチストランデッド複合体を形成する3以上のストランド、単一の自己ハイブリダイジングストランド、またはこれらのいずれかの組合せを含むことができる。ハイブリダイゼーション反応はPCRの開始、またはリボザイムによるポリヌクレオチドの酵素切断のようなより広汎なプロセスにおいて工程を構成することができる。
【0043】
本明細書中で用いるように、「発現」とは、ポリヌクレオチドがmRNAに転写されるプロセスおよび/または前記転写されたmRNA(「転写物」ともいう)が引き続いて、
ペプチド、ポリペプチドまたは蛋白質に翻訳されているプロセスをいう。転写物およびコードされたポリペプチドは、集合的に、「遺伝子産物」といわれる。もしポリヌクレオチドがゲノムDNAに由来するならば、発現は、真核生物細胞におけるmRNAのスプライシングを含むことができる。
【0044】
被験体におけるヌクレオチド配列またはポリペプチド配列に適用される「異なって発現される」とは、対照において検出されるのと比較した場合のその配列の過剰発現または過小発現をいう。過小発現は、対照と比較した場合に、テスト被験体における検出可能な発現の不存在によって証明される、特定の配列の発現の不存在も含む。
【0045】
用語「ポリペプチド」、「ペプチド」および「蛋白質」は、本明細書中において相互交換可能に用いられて、いずれかの長さのアミノ酸のポリマーをいう。ポリマーは直線または分岐であってよく、それは修飾されたアミノ酸を含んでもよく、およびそれは非アミノ酸によって中断されていてもよい。前記用語は修飾(例えば、ジスルフィド結合形成、グリコシル化、脂質化、アセチル化、リン酸化、または標識成分とのコンジュゲーションのようないずれかの他の操作)されているアミノ酸ポリマーも含む。本明細書中で用いるように、用語「アミノ酸」とは、グリシン、およびDまたはL双方の光学異性体、およびアミノ酸アナログおよびペプチドミメティクスを含めた、天然および/または非天然または合成アミノ酸のいずれかをいう。
【0046】
本明細書中で用いるように、「髄鞘形成細胞」とは、神経系において軸索を絶縁するミエリンを産生することができる細胞をいう。例示的な髄鞘形成細胞は中枢神経系においてミエリンを産生することを担当する乏突起神経膠細胞、および末梢神経系においてミエリンを産生することを担当するシュワン細胞である。
【0047】
用語「髄鞘再形成する」または「髄鞘再形成」とは、例えば、脱髄傷害に応答してのミエリンの再生をいう。
【0048】
「被験体」、「個体」または「患者」は本明細書中においては相互交換的に用いられ、これは、脊椎動物、好ましくは哺乳動物、より好ましくはヒトをいう。哺乳動物は、限定されるものではないが、ネズミ、サル、ヒト、農場動物、競技動物およびペットを含む。組織、細胞およびそれらの子孫またはインビボで得られたかもしくはインビトロで培養された生物学的実体も含まれる。
【0049】
本明細書中に記載される動物モデルまたは細胞培養アッセイで使用される「生物学的に活性な剤」または「生物活性剤」は、単純なまたは複雑な有機または無機分子、ペプチド、ペプチドミメティック、蛋白質(例えば、抗体)、リボソーム、低分子干渉RNA、またはポリヌクレオチド(例えば、アンチセンス)のような生物学的または化学的化合物よりなる群から選択することができる。さらに、そのような薬剤は複雑な有機または無機分子を含み、粗製または精製された植物抽出物のような化合物の不均一混合物を含むことができる。
【0050】
「プロモーターエレメント」はそのような配列に連結された遺伝子の転写を促進する調節配列である。調節配列はエンハンサー配列またはその機能的部分を含むことができる。
【0051】
「対照」は比較目的にて実験で用いる代替被験体、細胞または試料である。
細胞死メディエーター蛋白質(CDMP)
本発明は、細胞または組織特異的に発現された細胞死メディエーター蛋白質(CDMP)を含む方法および組成物を提供する。例えば、発現はCNSまたはPNSに対して、あるいは特異的神経細胞または壁細胞に対して特異的であってよい。発現は動物におけるも
のであり得るが、動物におけるそのような発現は、例えば、髄鞘形成または髄鞘再形成において役割を持つ細胞において特異的に細胞死を誘導することによって脱髄を引き起こすことができる。発現はインビトロでもあり得る。細胞死メディエーター蛋白質の発現は、インビボまたはインビトロにて誘導性であり得る。
【0052】
細胞死メディエーター蛋白質は細胞死またはアポトーシスの媒介において役割を有する。CDMPは、例えば、アポトーシスに直接的に影響する蛋白質の活性を調節することによって、直接的にまたは間接的にアポトーシスに影響し得る。例えば、CDMPはSMAC(カスパーゼの第2のミトコンドリア由来アクチベーター)、IAP(アポトーシス蛋白質の阻害剤)、カスパーゼ、またはそれらのモジュレーターであり得る。他の実施形態において、CDMPはTNF(腫瘍壊死因子)受容体、およびFas受容体、およびTRAIL受容体経路のような他の死滅受容体シグナリング経路のモジュレーターであり得る。CDMPは、グランザイムB、またはグランザイムBのモジュレーターを含めたカスパーゼのアクチベーターでもあり得る。
【0053】
種々の実施形態において、細胞死メディエーター蛋白質は核酸構築体によってコードされる。いくつかの実施形態において、核酸構築体は1以上のCDMPをコードすることができる。CDMPは同一または異なることができる。例えば、単一の核酸は、タンデムにカスパーゼ9をコードする2つの配列のような上記の蛋白質の2つをコードすることができ、あるいはカスパーゼ9およびカスパーゼ3をコードすることができる。本発明の核酸構築体はカスパーゼ2、5、8、9、10または11、それらのプロ酵素形態、またはその誘導体をコードすることができる。さらなる実施形態において、そのようなカスパーゼ蛋白質をコードする配列を、架橋剤化合物と反応性である二量体化ドメインを含むように修飾する。かくして、いくつかの実施形態において、野生型カスパーゼ配列を修飾して、架橋剤化合物に対して選択的に反応性である二量体化ドメインを含むキメラ配列を産生する。そのような二量体化ドメインの例は、その関連部分をここに引用してその全体を援用する、米国特許出願第20050187177号および米国特許第6,984,635号に開示されているものを含む。
二量体化
いくつかの実施形態において、二量体化は生物学的プロセス(例えば、カスパーゼ9の活性化を介するアポトーシス)を活性化し、種々のキメラ蛋白質を利用することができる。キメラ蛋白質は、種々のドメインが(例えば、天然では一緒に連結しては見出されない異なる源に由来する)相互に対して異種である点で組換体である。天然では直接的に相互に連結しては見出されない、例えば、特定のドメインまたは発現制御配列をコードする異種成分を含む組換えDNA構築体は、インビトロまたはインビボにて標的宿主細胞を遺伝子工学的に作成するのに用いられる。そのように操作された細胞は、少なくとも1つのそのようなキメラ蛋白質、またはキメラ蛋白質をコードする遺伝子構築体の第1のシリーズを含有する。1つのそのようなDNA構築体は、(b)異種のさらなる(「作用」)蛋白質ドメインに融合した(a)(選択されたリガンドに結合することができる)少なくとも1つの受容体ドメインを含むキメラ蛋白質をコードする。前記リガンドは、好ましくは、ほぼ10−6から<10−9までの範囲のKd値でもって、キメラ蛋白質内の2(またはそれ以上)の受容体ドメインに結合することができ、好ましくは、ほぼ1kDa、5kDa、10kDa、15kDaまたは20kDa未満の分子量を有する非蛋白質の化合物である。そのようにオリゴマー化されたキメラ蛋白質受容体のドメインは同一または異なってもよい(すなわち、ホモ二量体化またはヘテロ二量体化)。リガンドへの暴露および受容体のオリゴマー化に際して、キメラ蛋白質は生物学的プロセス(例えば、補体カスケード)を開始することができる。コードされたキメラ蛋白質は、さらに、キメラ蛋白質を所望の細胞区画に向けることができる細胞内標的化ドメイン(例えば、蛋白質に指令して核と会合させる配列)を含むことができる。
【0054】
キメラ蛋白質が結合することができるリガンドのタイプの例は、FK506タイプのリガンド、サイクロフィリンタイプのリガンド(例えば、サイクロスポリンAタイプのリガンド)、テトラサイクリンまたはステロイドリガンドを含む。そのような結合はホモタイプ(同一)またはヘテロタイプ(異なる)のキメラ蛋白質分子のオリゴマー化を引き起こす。そのようなリガンドおよび/または受容体ドメインの例は、その各々についての開示をここに引用してその全体を援用する、米国特許第5534418号、同第5002753号、同第5298429号、同第6235872号、同第6656971号、同第7196182号、同第7101357号、同第7109317号、同第7153685号および同第7169564号に開示されている;また、Straathofら、Blood,(2005)105:4247−4254;Belshawら、Proc Natl Acad Sci.(1996)93:4604−4607も参照。
【0055】
かくして、本明細書中に記載された方法を利用し、標的細胞は、(i)選択されたオリゴマー化リガンドに結合することができる少なくとも1つの受容体ドメイン、および(ii)カスパーゼ9をコードする、受容体ドメインに対して異種であるもう1つの蛋白質ドメインを含むキメラ蛋白質をコードするDNA構築体を含むことができる。よって、選択されたリガンドへの暴露に続き、そのような標的細胞において発現されたカスパーゼ9のオリゴマー化は、アポトーシスプログラムを誘導し、前記DNA構築体を含む細胞を殺傷する。
【0056】
カスパーゼ9は、典型的には、アポトーシスプロテアーゼ活性化因子1(Apaf−1)とのチトクロームCおよびATP依存性相互作用後にダイマーとして機能する蛋白質である。カスパーゼ9の二量体化は、このポリペプチドが下流エフェクター分子を活性化するのを可能とし、最終的に、当該細胞のアポトーシスをもたらす(Springer,J
Biochem.Mol.Biol.(2002)35:94−105)。
【0057】
かくして、種々の実施形態において、細胞死メディエーター蛋白質は、FK506タイプのリガンド、サイクロスポリンAタイプのリガンド、テトラサイクリンまたはステロイドリガンドのための結合ドメインを含むカスパーゼ9キメラ蛋白質である。1つの実施形態において、カスパーゼ9はFKBP12結合ドメインを含む。さらなる実施形態において、キメラ構築体(例えば、カスパーゼ9)で利用される結合ドメインは、リガンド(例えば、二量体化化学誘導因子)に結合するように最適化することができる。
【0058】
誘導性カスパーゼ9 cDNA(iCP9)は、カスパーゼ動員ドメイン(CARD)の除去後に、カスパーゼ9 cDNA配列(GenBank AH002 818)をFK506結合蛋白質(FKBP)配列(GenBank AH002818)に連結させることによって操作した。CARD配列の不存在は蛋白質の生理学的二量体化を妨げ、それにより、カスパーゼ細胞死カスケードの自発的な開始を妨げる。FKBP配列への融合は、二量体化化学誘導因子(CID)の投与後に化学的に誘導された凝集を可能とする。CID、AP20187(ARIAD Pharmaceuticals,Cambridge,MA)は、内因性FKBPとの相互作用を妨げるように改変されている非毒性合成FK506アナログである。CIDシステムは、従前に、インビトロおよびインビボの両方で無関係なシステムにおいて利用されている(Straathofら、Blood.(2005)105:4247−4254)。
【0059】
本発明の種々の実施形態において、本発明の核酸構築体は、限定されるものではないが、米国特許第6,982,082号に開示されたもののような、アデノウイルス、アデノウイルス関連ウイルス、ネズミ白血病ウイルス、レンチウイルス、泡沫状ウイルス、狂犬病ウイルスまたは当該分野で公知の他のウイルスベクターを含めた、ウイルスベクターを介して(培養中のまたはインビボで)細胞に送達される。
調節された発現
本発明の種々の態様において、細胞または組織特異的および/または誘導性発現調節エレメントは細胞死メディエーター蛋白質に作動可能に連結されて、発現に際して選択的な細胞死をもたらす。前記したように、組織特異的および細胞特異的調節配列が、中枢神経系において導入遺伝子を発現させるために入手可能である。調節配列は、特定の細胞型において中枢神経系または末梢神経系での導入遺伝子の異所性発現を可能とする。例えば、選択的死滅は、限定されるものではないが、乏突起神経膠細胞、小神経膠細胞、シュワン細胞または神経膠星状細胞のような細胞において達成することができる。
【0060】
調節配列の例示的な発現は、限定されるものではないが、CC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、ミエリン関連糖蛋白質(MAG)、ミエリン乏突起神経膠細胞糖蛋白質(MOG)、乏突起神経膠細胞ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2),GFAP、AQP4、PDGFR−α、PDGF−α、RG5、p糖蛋白質、ノイルツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、スルファチド、2(VEGFR2)、スーパーオキシドジスムターゼ(SOD1)、チロシンヒドロキシラーゼ、ニューロン特異的エノラーゼ、パルキン遺伝子(PARK2)、パルキン共調節遺伝子(PAGRG)、ニューロン特異的Tα1 α−チューブリン(Tα1)、小胞モノアミントランスポーター(VMAT2)、およびα−シヌクレイン(SNCA)、PDGFR−β、またはプロテオリピド蛋白質(PLP)を含めた遺伝子から選択される調節配列を含む。
【0061】
神経細胞特異的プロモーターのさらなる例は、その関連開示をここに引用して援用する、米国特許出願公開番号2003/0110524;2003/0199022;2006/0052327、2006/0193841、2006/0040386、2006/0034767、2006/0030541;米国特許第6472520号、同第6245330号、同第7022319号、および同第7033595号に開示されているように、当該分野で公知である。また、ウェブサイト<chinook.uoregon.edu/promoters.html>または<tiprod.cbi.pku.edu.cn:8080/index>(ある細胞/組織に対して特異的な遺伝子のプロモーターをリストする);およびPattersonら、J.Biol.Chem.(1995)270:23111−23118も参照。
【0062】
かくして、本発明の1つの態様は、細胞死の一時的制御のための誘導性/細胞型特異的発現調節エレメントの利用であり、これは今度は、MS、ALSおよびパーキンソン病のような神経障害に関連するものとして、ニューロンの健康/維持に対する特定の細胞の効果を評価し、またはニューロンの健康/維持に対する候補分子の効果を評価するのに利用される。
【0063】
細胞死メディエーター蛋白質の発現は、(例えば、エフェクター分子を局所投与する場合)細胞/組織特異的プロモーターを利用するもの以外の発現系を利用することによって一時的に調節することもできる。したがって、いくつかの実施形態において、細胞死メディエーター蛋白質をコードする遺伝子は、tet応答性プロモーターのような制御可能なプロモーターエレメントに作動可能に連結させることができる。例えば、望まれる場所および時期に、誘導性剤(例えば、テトラサイクリンまたはそのアナログ)を細胞または被験体に投与して、細胞/組織特異的に細胞死メディエーター蛋白質の発現を誘導することができる(例えば、単にテトラサイクリンが局所化された/限定された様式で送達される)。そのような系は、テトラサイクリンの存在が発現を阻害する「オフスイッチ」系、またはテトラサイクリンの存在が発現を誘導するようにE.coli TetRの変異体が
用いられる「可逆性」Tet系を含めることによって、真核生物細胞における遺伝子発現の密接な制御を提供することができる。これらの系は、例えば、Gossen and Bujard(Proc.Natl.Acad.Sci.U.S.A.(1992)89:5547)において、および、Pujardらによる米国特許第5,464,758号;同第5,650,298号;および同第5,589,362号に開示されている。
【0064】
誘導性プロモーターのさらなる例は、限定されるものではないが、MMTV、ヒートショック70プロモーター、GAL1−GAL10プロモーター、メタロチオネイン誘導性プロモーター(例えば、銅誘導性ACE1;他の金属イオン)、ホルモン応答エレメント(例えば、グルココルチコイド、エストロゲン、プロゲストロゲン)、フォルボールエステル(TREエレメント)、カルシウムイオノフォア応答性エレメント、または非カップリング蛋白質3、αヒト葉酸受容体、ホエイ酸性蛋白質、前立腺特異的プロモーター、ならびに米国特許第6,313,373号に開示されたものを含む;また、(遺伝子/活性によって分類された15,000を超える異なるプロモーター配列を含むデータベースを提供する)オンライン上での<biobase/de/pages/products/transpor.html>;およびChenら、Nuc.Acids.Res.2006,34:Database issue,D104−107も参照。
【0065】
なお他の誘導性プロモーターは、成長ホルモンプロモーター;アデノウイルスE1A蛋白質によって誘導可能なアデノウイルス初期遺伝子プロモーター、またはアデノウイルス主要後期プロモーターのようなヘルパーウイルスによって誘導可能なプロモーター;VP16または1CP4のようなヘルペスウイルスタンパク質によって誘導可能なヘルペスウイルスプロモーター;ワクシニアまたはポックスウイルスRNAポリメラーゼによって誘導可能なプロモーター;または、各々、T7、T3、またはSP6 RNAポリメラーゼによって誘導可能な、T7、T3およびSP6のようなバクテリオファージプロモーターを含む。
【0066】
他の実施形態において、構成的プロモーターが望ましいであろう。例えば、アデノウイルス主要後期プロモーター、サイトメガロウイルス即時型初期プロモーター、βアクチンプロモーター、またはβグロビンプロモーターを含めた、本発明で用いるのに適した多くの構成的プロモーターがある。多くの他のものが当該分野で知られており、本発明で用いることができる。なおさらなる実施形態において、調節エレメントを改変し、または修飾して、発現を増強させる(すなわち、プロモーターの強度を増加させる)ことができる。例えば、エンハンサー機能を含むイントロン性配列を利用して、プロモーターの機能を増加させることができる。ミエリンプロテオリピド蛋白質(PLP)遺伝子はエンハンサーエレメントとして機能するイントロン性配列を含む。この調節エレメント/領域ASE(アンチサイレンサー/エンハンサー)は、エクソン1 DNAからほぼ1kb下流に位置し、ほぼ100bpを含む。Mengら、J Neurosci Res.(2005)82:346−356参照。
【0067】
さらに、特定の細胞内位置における導入遺伝子の発現が望まれる場合、導入遺伝子は、当該分野で広く実施されている組み換えDNA技術によって対応する細胞内局所化配列に作動可能に連結させることができる。例示的な細胞内局所化配列は、限定されるものではないが、(a)細胞の外側へ遺伝子産物の分泌を指令するシグナル配列;(b)原形質膜、または細胞の他の膜区画への蛋白質の付着を可能とする膜係留ドメイン;(c)コードされた蛋白質の核へのトランスロケーションを媒介する核局所化配列;(d)主としてコードされた蛋白質をERに制限する小胞体滞留配列、(例えば、KDEL配列);(e)蛋白質は、蛋白質を細胞膜に会合させるようにファルネシル化されるように設計することができる;または(f)コードされた蛋白質産物の異なる細胞内分布において役割を演じるいずれかの他の配列を含む。
【0068】
いくつかの実施形態において、遺伝子的に修飾された細胞を区別するためのマーカーを検出することができる。そのようなマーカーは、限定されるものではないが、CC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、ミエリン関連糖蛋白質(MAG)、ミエリン乏突起神経膠細胞糖蛋白質(MOG)、乏突起神経膠細胞ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、ガラクトセレブロシド(GalC)、スルファチド、PDGFR−β、PDGFR−α、PDGF−α、およびプロテオリピド蛋白質(PLP)を含む。
【0069】
種々の実施形態において、乏突起神経膠細胞の選択的アポトーシスのような選択的細胞死が誘導される動物もまた、脱髄/髄鞘再形成状態に対する効果についてアッセイすることもできる。例えば、脱髄/髄鞘再形成現象は、当該分野で知られているように免疫組織化学手段または蛋白質分析によって観察することができる。例えば、テスト動物の脳の切片を、乏突起神経膠細胞マーカーを特異的に認識する抗体で染色することができる。もう1つの態様において、乏突起神経膠細胞マーカーの発現レベルは、免疫ブロッティング、ハイブリダイゼーション手段および増幅手法ならびに当該分野でよく確立されたいずれかの他の方法によって定量することができる。例えば、その各々の開示をここに引用して援用する、Mukouyamaら、Proc.Natl.Acad.Sci.(2006)103:1551−1556;Zhangら(2003)、supra.;Girardら、J.Neuroscience.(2005)25:7924−7933;および米国特許第6,909,031号;同第6,891,081号;同第6,903,244号;同第6,905,823号;同第6,781,029号;および同第6,753,456号。
【0070】
なお他の実施形態において、選択的細胞死が血管周囲細胞のような血液脳関門の維持で重要である細胞で起こる動物。例えば、動物における、PDGFR−βプロモーターの制御下でのカスパーゼ9またはiCP9のようなカスパーゼの発現を用いて、その浸透性、維持または完全性のように、血液脳関門(BBB)に対する効果についてアッセイすることができる。例えば、テスト動物の脳の切片を、接着結合蛋白質のようなBBBマーカーを特異的に認識する抗体で染色することができる。例えば、検出することができるマーカーはオクルジン、およびクラウジン2、クラウジン5、クラウジン6、クラウジン7、クラウジン10、クラウジン12、クラウジン15および/またはクラウジン19のようなクラウジンを含む。もう1つの実施形態において、接着結合蛋白質マーカーの発現レベルは、イムノブロッティング、ハイブリダイゼーション手段、および増幅手法ならびに当該分野で確立された他のいずれかの方法によって定量することができる。
【0071】
BBBの完全性または透過性は、血液−脳関門透過性を決定し、可視化し、測定し、同定し、または定量するのに利用される、当該分野で公知のいずれかの色素、マーカー、またはトレーサーのようなインジケーターを用いてアッセイすることもできる。非限定的な例として、エバンスブルーおよびナトリウムフルオレセインが挙げられる。そのようなインジケーターの例は当業者に明らかであり、無傷のBBBを横切ることができないが、より透過性のBBBを横切ることができ、ならびに同定し、測定し、または定量することができる実質的にいずれの化合物も含む。
【0072】
インジケーターは、BBB透過性の変化を決定するのに利用される酵素、トレーサーまたはマーカーであり得るが、非限定的な例は以下の通りである:
【0073】
【化1】

【0074】
【化2】
色素、トレーサーまたはマーカーのさらなる例は、デキストラン、ビオチン、フィブリノーゲン、アルブミン、クーンス(Coons)反応を用いる血中グロブリン、テキサスレッド結合デキストラン(70,000daMW)、Na(+)−フルオレセイン(MW376)またはフルオレセインイソチオシアネート(FITC)標識デキストラン(MW62,000または145,000)、または分子質量10,000DaのFITC−標識デキストラン(FITC−デキストラン−10K)を含む。
トランスジェニック動物
本発明の1つの態様において、目的の蛋白質をコードする遺伝子に作動可能に連結された神経細胞特異的調節エレメントをコードするトランスジェニックヌクレオチド配列がゲノムに安定に組み込まれたトランスジェニック動物が生成される。前記遺伝子の発現は誘導性プロモーターの制御下とすることができる。いくつかの実施形態において、細胞特異性は神経細胞(例えば、神経膠細胞、好ましくは神経膠星状細胞、乏突起神経膠細胞および/またはシュワン細胞;頸部神経節ニューロン、後根神経節細胞、結節性神経節ニューロン、脊髄運動ニューロン、中脳ドーパミン作動性ニューロン、中枢ノルアドレナリン作動性ニューロンおよび腸ニューロンのようなニューロン細胞)に対するものである。
【0075】
好ましい実施形態において、目的の蛋白質をコードする遺伝子は細胞死メディエーター蛋白質をコードする。かくして、本発明のトランスジェニック動物は、細胞型特異的発現
調節エレメントに作動可能に連結された細胞死メディエーター蛋白質(CDMP)をコードするヌクレオチド配列を含むことができ;ここで、動物は、細胞型特異的発現調節エレメントに作動可能に連結されたCDMPをコードするヌクレオチド配列がない動物に対してより大きな程度の神経障害を呈する。神経障害は多発性硬化症のようなニューロン脱髄であり得る。トランスジェニック動物は、対照動物のそれに対してアポトーシス乏突起神経膠細胞の増加を呈することができ、CDMPの発現は中枢神経系に異所的に制限することができる。いくつかの実施形態において、神経障害は、血液脳関門の欠陥によるものであり、またはBBBおよび脱髄双方の欠陥によるものである。例えば、神経障害は筋委縮性側索硬化症(ALS)、多発性硬化症(MS)、免疫機能不全筋肉中枢神経系破壊、筋ジストロフィ(MD)、アルツハイマー病、パーキンソン病、ハンチントン病、脳虚血症、脳性麻痺、皮質基底神経節変性、クロイツェルフェルト・ヤコブ症候群、ダンディー・ウォーカー症候群、認知症、血管性脳炎、脳脊髄炎、癲癇、本態性振せん、クル・ランダウ・クレッフナー症候群、レーヴィ体病、マチャド・ジョセフ病、メージ症候群、偏頭痛障害、ポリオ、多系統萎縮症、髄膜炎、ドラガー症候群、チューレット症候群、ハレルフォルデン・シュパッツ症候群、水頭症、オリーブ橋小脳萎縮、核上麻痺、または脊髄空洞症であり得る。
【0076】
1つの実施形態において、細胞死メディエーター蛋白質(CDMP)はカスパーゼ2、5、8、9、10または11である。もう1つの実施形態において、そのような細胞死メディエーター蛋白質の1以上は神経細胞または壁細胞のような細胞で発現される。例えば、CDMPは、神経膠細胞のような、動物の神経細胞に標的化され得る。神経細胞は、限定されるものではないが、乏突起神経膠細胞、神経膠星状細胞、シュワン細胞、または小神経膠細胞であり得る。標的化は、動物の壁細胞、例えば、限定されるものではないが、血管周囲細胞、内皮細胞または平滑筋細胞のような壁細胞に対するものであり得る。いくつかの実施形態において、1以上の標的細胞の組合せが選択される(例えば、神経細胞および壁細胞、または異なる神経細胞、または異なる壁細胞)。
【0077】
いくつかの実施形態において、CDMPの発現は誘導性である。例えば、CDMPは、誘導性プロモーターを持つ調節エレメントに作動可能に連結していてもよい。アポトーシスの促進のようなCDMPの活性もまた誘導性であってよい。例えば、CIDによって誘導されるCDMPをコードするウイルスベクターは、非ヒト成体動物(例えば、ラット)の脊髄または脳に直接的に注射することができ、CIDは投与される。CIDはウイルスベクターの投与に同時に、またはそれに引き続いて投与することができる。
【0078】
病巣感染に由来する、乏突起神経膠細胞の喪失の病巣面積の程度は、投与されるウイルスの量によって制御することができる。乏突起神経膠細胞の選択的喪失に対する生存細胞の応答は、当業者に明らかである、組織学、分子および電気生理学アッセイを用いて評価することができる。(例えば、乏突起神経膠細胞の死滅によって測定される)病巣サイズの程度は、病巣部位における軸索喪失と相関させることができる。同様に、その軸索が細胞喪失の領域を通過するニューロンを評価することによって、一旦超過したら、離れたニューロンの喪失をもたらす脱髄閾値を存在するか否かを決定することができる。従って、本発明の方法は、CNS乏突起神経膠細胞の喪失に続いての分子応答に対する洞察を提供し、候補剤の同定が、多発性硬化症においてニューロン保護に対する時間的および環境ベースのアプローチを行うのを可能とする。1つの実施形態において、ウイルスベクターはレンチウイルスベクターである。さらなる実施形態において、ウイルスベクターは(後にさらに記載する)iCP9レンチウイルスである。
【0079】
本発明の動物モデルは、中枢および末梢神経系を含めた動物の神経系においてニューロン脱髄状態を生じさせる手法に使用できるいずれの非ヒト脊髄動物を含む。好ましいモデル生物は、限定されるものではないが、哺乳動物、霊長類、およびげっ歯類を含む。好ま
しいモデルの非限定的例は、ラット、マウス、モルモット、ネコ、イヌ、ウサギ、ブタ、チンパンジー、およびサルである。テスト動物は野生型またはトランスジェニックであり得る。1つの実施形態において、動物はげっ歯類である。なおさらなる実施形態において、動物はマウスである。もう1つの実施形態において、動物はサル種である。なおさらなる実施形態において、動物は、神経学的疾患を調べるのに通常利用されるキヌザルである(例えば、Eslamboi,A.Brain Res Bull.(2005)68,140−149;Kirikら、Proc.Natl.Acad.Sci.(2004)100:2884−89)。
【0080】
トランスジェニック動物は広く2つのタイプ:「ノックアウト」および「ノックイン」にカテゴリー化することができる。「ノックアウト」は、好ましくは、例えば、標的遺伝子の一部を標的遺伝子に無関係な配列で置き換えることによって、標的遺伝子の発現が微々たるものとなり、または検出できないものとなるように、標的遺伝子の機能の減少をもたらすトランスジェニック配列の導入を介する標的遺伝子の改変を有する。「ノックイン」は、例えば、標的遺伝子の調節配列を作動可能に挿入することによる、標的遺伝子の改変された発現を有する動物であり;または例えば、標的遺伝子を修飾されたコピーで置き換えることによる、標的遺伝子の修飾されたコピーを発現する動物である。修飾は標的遺伝子の失欠または変異であり得る。ノックインまたはノックアウト動物は標的遺伝子に対してヘテロ接合性またはホモ接合性であり得る。ノックアウトおよびノックインの双方は、ダブルノックインまたはダブルノックアウトとしても知られた「バイジェニック」であり得る。バイジェニック動物は、改変されている少なくとも2つの宿主細胞遺伝子を有する。1つの実施形態において、バイジェニック動物は、細胞特異的細胞死メディエーター蛋白質をコードする導入遺伝子、および細胞特異的マーカー遺伝子をコードするもう1つのトランスジェニック配列を保持する。本発明のトランスジェニック動物は広くノックインとして分類することができる。いくつかの実施形態において、動物の特異的細胞型を標的としてもよい。例えば、標的細胞は神経細胞または壁細胞であり得る。種々の実施形態において、神経細胞は乏突起神経膠細胞、シュワン細胞、小神経膠細胞または神経膠星状細胞を含み、壁細胞は内皮細胞、血管周囲細胞または平滑筋細胞を含む。
【0081】
胚顕微操作のための技術の進歩は、いまや、同様に、受精した哺乳動物卵子への異種DNAの導入を可能とする。例えば、全能性または多能性幹細胞は、マイクロインジェクション、リン酸カルシウム媒介沈殿、リポソーム融合、レトロウイルス感染または他の手段によって形質転換することができる。次いで、形質転換された細胞を胚に導入し、胚は発生してトランスジェニック動物となる。好ましい実施形態において、発生する胚を、導入遺伝子を発現するトランスジェニック動物を感染した胚から生じさせることができるように、所望の導入遺伝子を含有するウイルスベクターで感染させる。もう1つの好ましい実施形態において、所望の導入遺伝子を、好ましくは、単一細胞段階で、胚の前核または細胞質に共注射し、胚は発生して成熟トランスジェニック動物となる。トランスジェニック動物を生成させるためのこれらおよび他の変種の方法は当該分野において確立されており、本明細書中においては詳細に記載しない。例えば、米国特許第5,175,385号および同第5,175,384号参照。
【0082】
従って、本発明は、細胞特異的に髄鞘再形成を検出し、および定量するために動物モデルを用いる方法を提供する。そのような実施形態において、前記方法は:(a)細胞における細胞死メディエーター蛋白質の発現によって細胞型特異的に細胞死を誘導する工程;(b)細胞死を起こすために時間をおく工程;(c)動物において髄鞘形成/髄鞘再形成の調節を決定する工程を含む。生物活性剤をスクリーニングし、生物活性剤の髄鞘形成または髄鞘細形成の調節を決定するためにトランスジェニック動物を用いてもよい。
動物実験
動物モデルを、神経障害現象(例えば、脱髄障害)に関連する標的細胞において選択的
または制御された細胞死をアッセイする本発明の1以上の方法で利用する。前記現象は、髄鞘を有する軸索の減少、乏突起神経膠細胞マーカー、神経膠星状細胞マーカーまたはシュワン細胞マーカーの低下によって特徴付けられる脱髄障害と関連付けることができる。脱髄障害は遺伝的、または病原体またはウイルスによって罹るものであり得る。
【0083】
動物モデルを用いて、神経障害を調節する生物活性剤についてスクリーニングすることができる。例えば、本明細書中に開示されたモデル系の適用は、ラットCNSに適用した場合、インビボでの脱髄に対するニューロン応答の研究、および脱髄障害の処置の開発を可能とするより正確なモデルを提供する。
【0084】
本発明は、動物に投与されて、細胞死メディエーター蛋白質の発現および、よって、標的細胞の選択的除去を達成する細胞/組織特異的または誘導性プロモーターに作動可能に連結された細胞死メディエーター蛋白質をコードする核酸構築体を提供する。そのような発現は、異所的に維持された導入遺伝子送達ビヒクルを介して達成することができるか、あるいはそのような導入遺伝子は、当該分野で公知の方法を用いて動物のゲノムに組み込むことができる。例えば、発現は、エピソームにより、またはCDMPをコードする核酸の安定な組込みを介して達成することができよう。
【0085】
種々の実施形態において、細胞死メディエーター蛋白質をコードする遺伝子(「自殺遺伝子」)を含む核酸構築体は、細胞/組織特異的な、または誘導性である発現調節エレメントに作動可能に連結される。種々の実施形態において、標的細胞は神経細胞または壁細胞である。神経細胞は、限定されるものではないが、乏突起神経膠細胞、神経膠星状細胞、シュワン細胞、または小神経膠細胞であり得る。壁細胞は、限定されるものではないが、血管周囲細胞、内皮細胞および平滑筋細胞を含む。いくつかの実施形態において、1以上の標的細胞の組合せが選択される(例えば、神経細胞および壁細胞、または異なる神経細胞、または異なる壁細胞)。
【0086】
なお他の実施形態において、標的細胞は、限定されるものではないが、Bリンパ球またはTリンパ球細胞のような免疫細胞である。なおさらなる実施形態において、Bリンパ球および/またはTリンパ球は標的細胞ではない。
【0087】
本発明の1つの態様において、細胞死メディエーター蛋白質は、限定されるものではないが、カスパーゼ2、5、8、9、10または11を含めたカスパーゼ蛋白質である。1つの実施形態において、本発明の核酸構築体は、標的細胞において発現させることができる、少なくとも2つの異なるカスパーゼ蛋白質を含む。CDMPは、例えば、FK506型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドに対する結合ドメインを有するキメラのCDMPであってよい。1つの実施形態において、カスパーゼ9は、FKBP12結合ドメインを含む。さらなる実施形態において、CDMPのアポトーシス促進活性は誘導性であってよく、例えば、キメラ構築体(例えば、カスパーゼ9)で利用される結合ドメインは、カスパーゼ9活性および、かくして、アポトーシスを促進する、二量体化化学誘導因子に結合するように最適化することができる。
【0088】
本発明のもう1つの態様において、髄鞘形成/髄鞘再形成調節活性について生物学的に活性な剤をテストする方法が提供される。
【0089】
1つの実施形態において、神経障害関連現象の調節について候補剤をテストする方法は、細胞死メディエーター蛋白質の発現によってテスト動物において細胞死を誘導する工程、髄鞘形成/髄鞘再形成に対する効果を評価するのに十分な時間おいておく工程、テスト生物活性剤を投与する工程、ならびに、投与無しと比較した髄鞘形成/髄鞘再形成に対する効果を決定し、かくして、テスト剤が髄鞘形成/髄鞘再形成を増強し/低下させるか否
かを決定するする工程を含む。
【0090】
かくして、いくつかの実施形態において、前記方法は:(a)細胞型特異的に細胞死を誘導する工程;(b)本発明のトランスジェニック動物において脱髄傷害を評価する工程;(c)テスト剤を該動物に投与する工程;(d)必要に応じて、工程(c)の前または後に細胞特異的マーカー遺伝子の発現を検出し、および/または定量する工程;(e)髄鞘再形成が工程(d)において起こったか、およびどれくらい多くの髄鞘再形成が工程(d)において起こったかを検出する工程;(f)(例えば、組織学、組織化学、生化学アッセイによって、またはテスト剤の投与に応答してアップレギュレートまたはダウンレギュレートされ得る髄鞘再形成特異的マーカー蛋白質の発現を測定することによって)もし髄鞘再形成が増強され、または減少したならば、テスト剤が髄鞘再形成調節活性を有すると決定する工程を含む。種々の実施形態において、検出は、当該分野で公知の組織化学または生化学アッセイを含む。いくつかの実施形態において、検出は種々の時点で行い、テスト剤の投与は、アッセイの過程の間に、ならびに異なる投与養生法を用いて反復することができる。
【0091】
もう1つの実施形態において、神経障害に関連する現象を調節する化合物についてテストする方法は、候補剤を本明細書中に記載したようにテスト動物に投与する工程、細胞死メディエーター蛋白質の発現を誘導することによってテスト動物における脱髄を増強させる工程、候補剤の投与が増強されたまたは低下した髄鞘再形成をもたらすか否かを決定し、かくして、もし髄鞘再形成が増強され、または低下されたならば、テスト剤が神経障害に関連する現象を調節すると決定する工程を含む。1つの実施形態において、前記方法の実施は、候補剤が細胞死媒介脱髄/髄鞘再形成を調節するか否かを決定する。
【0092】
例えば、1つのそのような実施形態において、神経障害に関連する現象を調節する生物学的に活性な剤をテストする方法は、(a)誘導性/細胞特異的発現調節エレメントに作動可能に連結された死滅メディエーター蛋白質をコードする導入遺伝子を含む動物に候補剤を投与する工程;(b)細胞死メディエーター蛋白質の発現を誘導し、かくして、細胞死をもたらす工程;ならびに(c)候補剤が髄鞘形成または髄鞘再形成を増強させるかを決定する工程を含む。いくつかの実施形態において、検出は種々の時点でなされ、テスト剤の投与はアッセイの過程の間、ならびに異なる投与養生法を用いて反復することができる。1つの実施形態において、神経障害は脱髄障害(例えば、MS)である。髄鞘形成または髄鞘再形成のレベルは、限定されるものではないが、組織学、組織化学、生化学アッセイを含めた方法によって、あるいはテスト剤の投与に応答してアップレギュレートまたはダウンレギュレートされ得る髄鞘再形成特異的マーカー蛋白質の発現を測定することによって、対照動物と比較し、決定することができる。例えば、脱髄障害に関連する現象に対する薬剤の効果はイムノアッセイ、ハイブリダイゼーションアッセイまたはPCRアッセイを含むことができる。
【0093】
種々の実施形態において、検出は、当該分野で公知の組織化学または生化学アッセイを含む。いくつかの実施形態において、検出は種々の時点でなされ、テスト剤の投与はアッセイの過程の間に、ならびに異なる投与養生法を用いて反復することができる。
【0094】
また、本発明は、ニューロンの維持、ニューロンの死滅、またはニューロンの成長または神経膠細胞の維持、神経膠細胞の死滅、または神経膠細胞の成長に対する効果について候補剤をテストする方法も提供する。
【0095】
免疫細胞学および組織学的実験を用いて、ニューロンまたは神経膠の維持、成長または死滅に対する効果を決定することができる。いくつかの実施形態において、テスト動物における髄鞘再形成特異的マーカー蛋白質の発現を対照または参照動物と比較することがで
きる。他の実施形態において、テスト動物における細胞特異的マーカー蛋白質の発現を同一動物において種々の時点になした測定値と比較して、ニューロンの死滅または成長の開始または進行を決定する。
【0096】
本発明の候補剤は、髄鞘再形成促進活性、あるいは逆に、髄鞘再形成阻害活性または髄鞘再形成低下活性についての本明細書中に記載された方法によってテストすることができる。例えば、前記方法は(a)1以上の特定の細胞型において、細胞死メディエーター蛋白質の発現を通じて本発明のトランスジェニック動物において脱髄傷害を誘導する工程であって、ここに、前記1以上の細胞型が髄鞘形成またはニューロン支持に影響する、工程;(b)ミエリン細胞特異的マーカー遺伝子の発現によって証明されるように、ミエリン修復を行うのに十分な時間おいておく工程;(c)候補剤を工程(a)および/または(b)の前、その間および/またはその後に前記動物に投与する工程;(d)もしあれば、髄鞘再形成に対する投与された候補の効果を検出する工程を含むことができる。
【0097】
いくつかの実施形態において、候補剤は、脱髄傷害工程の誘導、例えば、1以上のCDMPの発現および/または活性を誘導することによる傷害の誘導の前、間または後に投与される。1つの実施形態において、候補剤は脱髄傷害の誘導前に投与される。もう1つの実施形態において、候補剤は脱髄傷害の誘導の間に投与される。なおもう1つの実施形態において、候補剤は脱髄傷害の誘導後に投与される。
【0098】
いくつかの実施形態において、候補剤はミエリン修復をもたらすのに十分な見込み時間より前、その間またはそれより後に投与される。1つの実施形態において、候補剤は傷害直後に投与される。もう1つの実施形態において、候補剤は、ミエリン修復が起こることができる時間の間に投与される。なおさらなる実施形態において、候補剤はミエリン修復が起こった後に投与される。
【0099】
いくつかの実施形態において、候補剤は傷害後約1から約24時間に投与される。いくつかの実施形態において、候補剤は傷害後約1から約30日に投与される。種々の実施形態において、候補剤は傷害後約1、2、3、4、5、6、7、8、9、10,11、12、13、14、15、16、17、18、19、20、21、22、23または24時間に投与される。種々の実施形態において、候補剤は約1、2、3、4、5、6、7、8、9、10,11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29または30日目から投与される。なおさらなる実施形態において、候補剤は約1から約12ヶ月まで投与される。
【0100】
髄鞘再形成された軸索を発生させるのに必要な時間の量は異なる動物の間で変化するが、それは、一般に、少なくとも約1週間を必要とし、よりしばしば、少なくとも約2から10週間必要とし、およびよりしばしば、約4から約10週間必要とする。
【0101】
候補剤をスクリーニングすることに関する方法のいずれかにおいて、1以上の候補剤は同時にスクリーニングすることができるのは理解されるべきである。種々の実施形態において、候補剤は髄鞘再形成を促進するものとして同定され、ここに、髄鞘再形成および/またはミエリン特異的マーカー蛋白質の発現は増強され、または増加する。いくつかの実施形態において、テスト動物における細胞特異的マーカー蛋白質の発現は対照または参照動物と比較することができる。他の実施形態において、テスト動物における細胞特異的マーカー蛋白質の発現は同一動物において種々の時点になした測定値と比較され、ここに、より早い時点を参照または対照時点として用いることができる。なお他の実施形態において、髄鞘再形成特異的マーカー蛋白質の発現は、候補剤が髄鞘再形成阻害活性または髄鞘再形成低下活性を有するか否かを決定することにおいて、テスト動物、および対照または参照動物における尺度である。そのような薬剤は髄鞘再形成阻害剤または髄鞘再形成トキ
シンとしてカテゴリー化することができる。
【0102】
髄鞘再形成は、マーカー蛋白質(例えば、EGFP)の発現によるなどして、(例えば、中枢または末梢神経系における)マーカー遺伝子/遺伝子産物の細胞特異的発現の増加を観察することによって確認することができる。脱髄または髄鞘形成が同定されることが求められる1以上の本明細書中における方法において、種々のマーカーが当該分野で入手可能である。髄鞘形成細胞を同定するための例示的なマーカーは、限定されるものではないが、CC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、ミエリン関連糖蛋白質(MAG)、ミエリン乏突起神経膠細胞糖蛋白質(MOG)、乏突起神経膠細胞ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、ガラクトセレブロシド(GalC)、スルファチド、PDGFRβ、PDGFRα、PDGFα、およびプロテオリピド蛋白質(PLP)を含む。
【0103】
髄鞘形成細胞におけるアポトーシスの誘導の後、および髄鞘再形成が起こるのに十分な時間の後のような傷害に続いて、マーカー蛋白質の蛍光は、小さな動物における蛍光の検出のための当該分野で知られたインビトロまたはインビボ方法を用いて検出することができる。インビボ蛍光は、関連分野において入手可能なデバイスを利用して検出し、および/または定量することができる。例えば、パルスレーザーダイオードおよび可視化システムにカップリングされた時間相関単一フォトン計数検出システムを用いて、組織からの蛍光の発光のレベルを検出することができる(Gallantら、Annual Conference of the Optical Society of America(2004);Contagら、Mol.Microbiol.18:593−603(1995);Schindehutteら、Stem Cells 23:10−15(2005))。組織空気界面における背面反射したフォトンからの大きなシグナルを回避するために、検出点は、典型的には、源点の右側3mmに位置させる。レーザーおよびフィルター双方の波長選択は、選択の蛍光マーカーに依存する。生物学的組織の吸収が低い場合、より深い組織深さ(例えば、レーザー出力に依存して数センチから5または6センチメートル)からの蛍光シグナルをインビボイメージングのために検出することができる。
【0104】
イメージングすべきマウスをイソフルラン/オキシテンで麻酔し、イメージングステージ上に置くことができる。腹側および背側イメージを、関連分野で入手可能なイメージングシステム(例えば、IVISイメージングシステム、Xenogen Corp.,Alameda,CA)を用いて種々の時点について収集することができる。種々の標的組織からの蛍光をイメージし、定量することができる。例えば、シグナル強度は、平均+/−平均の周りの標準偏差として本文または図面中に示すことができる。蛍光シグナルを事後のt検定を使用した分散分析によって分析して、時間ゼロおよび引き続いての各時点における所定のマーカーに対する蛍光シグナルの間の差を評価することができる。
【0105】
蛍光可視化、イメージングまたは検出は当該分野で公知の、および前記した本明細書中で記載された方法を用いて行うことができる。可視化、イメージングまたは検出は侵襲性、最小限に侵襲性または非侵襲性技術を通じてなすことができる。典型的には、顕微鏡検査技術を利用して、トランスジェニック動物から、生きた細胞から得られた細胞/組織からの蛍光、またはインビボイメージング技術を介する蛍光を検出し、またはイメージする。前記、「一般的方法」。
【0106】
ルミネセンス、蛍光または生物ルミネセンスシグナルは、蛍光マルチ・ウエルプレートリーダー、蛍光細胞分析分離装置(FACS)およびシグナルの空間的分解能を供する自
動細胞ベースのイメージングシステムを含めた、種々の自動/または高スループット機器システムのいずれか1つで容易に検出し、定量される。種々の機器システムが、検出を自動化するために開発されており、Cellomics、Amersham、TTP、Q3DM、Evotec、Universal ImagingおよびZeissによって開発された自動蛍光イメージング、および自動顕微鏡検査システムを含む。光漂白後の蛍光回復(FRAP)および微速度蛍光顕微鏡法は、生きた細胞における蛋白質の移動度を研究するためにやはり用いられてきた。
【0107】
蛍光の可視化(例えば、蛍光蛋白質をコードするマーカー遺伝子)は、(例えば、共焦点顕微鏡を用いるセクショニングおよびイメージングを通じての)動物から得られた細胞/組織試料の調査、生きた細胞の調査、またはインビボでの蛍光の検出いずれかを通じて、顕微鏡検査技術で行うことができる。可視化技術は、限定されるものではないが、当該分野で公知の共焦点顕微鏡検査法または光−光学スキャンニング技術の利用を含む。一般に、近赤外において発光波長を持つ蛍光標識が深い組織のイメージングにより使用できる。なぜならば、バックグラウンドノイズを増加させる散乱および自家蛍光の双方は波長が増加するにつれて低下するからである。インビボイメージングの例は、その各開示をここに引用して援用する、Mansfieldら、J.Biomed.Opt.10:41207(2005);Zhangら、Drug Met.Disp.31:1054−1064(2003);Flusbergら、Nat.Meth.2:941−950(2005);Mehthaら、Curr Opin Neurobiol.14:617−628(2004);Jungら、J.Neurophysiol.92:3121−3133(2004);米国特許第6977733号および同第6839586号によって開示されているように当該分野で公知である。
【0108】
インビボイメージングプロセスの1つの例は、インビボイメージング実験から1週間前を含み、休止期の背側毛髪は脱毛剤(Nair,Carter−Wallace Inc.)を用いて脱毛される(約2.5cm×2.5cm面積)。イメージング実験の日に、マウスを麻酔し、顕微鏡ステージ上の顕微鏡カバーグラス上にその背側皮膚を乗せて置く。表皮の脱毛した領域に50W水銀ランプによって照射し、×10対物レンズおよびLP520発光フィルター(Zeiss)を備えた倒立レーザー走査共焦点蛍光顕微鏡(Zeiss LSM510)を用いてスキャンする。アルゴンレーザー(488nm)のようなレーザー、および×10対物レンズは、深い組織から表皮細胞まで徐々により効果的に蛍光発光をイメージすることができる。増強されたエミッターまたはより長い波長のエミッターを利用することによって、より深い組織のイメージングについての感度を高めることができる。別法として、マウスのような小さな動物は種々の異なる位置(例えば、背側、腹側など)を利用して容易にスキャン/イメージすることができる。インビボイメージングは、肝臓のような深い組織領域にさえ効果的であった(例えば、Zhangら、supra)。
【0109】
DAPIを含むVectashield封入剤(Vector Laboratories)でマウントした細胞/組織切片は、Zeiss Axioplan蛍光顕微鏡で可視化することができる。イメージは、Open Labソフトウエアスイートを用いてApple Macintoshコンピューターに連結されたPhotometrics PXL CCDカメラを用いて捕獲することができる。異なる波長の蛍光は、ほぼ0.04mm2の領域に制限された脳梁の中央内の陽性細胞をカウントすることによって検出され、定量される。蛍光または共焦点顕微鏡検査法を利用してインビトロにて組織/細胞からの蛍光のレベルを検出し、測定するさらなる方法は当該分野で公知であり、前記にて本明細書中で開示された1以上のマーカー蛋白質からの蛍光を検出し、または測定するのに利用することができる。
【0110】
もう1つの例において、神経細胞は、エピ蛍光励起および発光経路においてLudlフィルターホイール(CarlZeiss,Thornwood,NY,USA)が嵌合したAxiovert S100 TV倒立顕微鏡でイメージすることができ、冷却された電荷結合素子(CCD)カメラ(Micro−MAXO;Roper Scientific,Trenton,NJ,USA)を用いてイメージを収集することができる。特定の励起および発光フィルター、および通常の二色エレメントを用いて、2つの異なる蛍光蛋白質のシグナルを単離することができる(HQFITCおよびテキサスレッド励起および発光フィルター、およびFITC/テキサスレッドV3二色;Chroma Technology,Brattleboro,VT,USA)。フィルターホイールおよびカメラはソフトウエア(例えば、IPLabsソフトウエア,Scanalytics,Fairfax,VA,USA)によって制御することができる。赤色および緑色蛍光イメージの組を収集して、赤色または緑色蛍光を有する細胞の相対的パーセンテージを分析することができる。イメージを分析し、IPLabsおよびAdobe InDesignソフトウエアでの公表のために調製することができる。イメージの操作を、赤色および緑色蛍光蛋白質のグレースケールイメージの合併に制限して、RGBカラーファイルを作り出し、最終プリントアウトの明るさ/コントラストを調整して、顕微鏡を通じて観察されたものに最も密接にマッチさせ、レタリングおよびスケールバーを加えることができる。蛍光顕微鏡検査装置は当該分野で公知であり、商業的に入手可能である。例えば、<confocal−microscopy.com/website/sc llt.nsf.>におけるウェブサイト参照。
【0111】
代替実施形態において、蛍光検出は網膜または角膜からの直接的なものである。網膜部位は全身毒性の研究のための非侵襲性箇所である。角膜は、身体に侵入することなく神経膠細胞を含有する器官に直接的に適用された物質の毒性を評価するのによく適合している。従って、マーカー蛋白質を異なって発現する神経細胞から発せられた蛍光は、所望の領域へレーザービームを向け、発せられた蛍光のレベルを検出することにより、網膜または角膜の共焦点顕微鏡検査法を用いることによって検出することができる。
【0112】
さらに、脱髄/髄鞘再形成現象は当該分野で公知の免疫組織化学的手段または蛋白質分析によって観察することができる。例えば、テスト動物の脳の切片を、乏突起神経膠細胞マーカーを特異的に認識する抗体で染色することができる。もう1つの態様において、乏突起神経膠細胞マーカーの発現レベルは、イムノブロッティング、ハイブリダイゼーション手段、および増幅手法ならびに当該分野でよく確立されたいずれかの他の方法によって定量することができる。例えば、その各々の開示をここに引用して援用する、Mukouyamaら、Proc.Natl.Acad.Sci.103:1551−1556(2006);Zhangら、supra;Girardら、J.Neurosci.25:7924−7933(2005);および米国特許第6,909,031号;同第6,891,081号;同第6,903,244号;同第6,905,823号;同第6,781,029号;および同第6,753,456号。
【0113】
もう1つの態様において、中枢または末梢神経系からの細胞/組織を切り出し、蛋白質のために処理することができ、例えば、組織をホモジナイズし、蛋白質をSDS−10%ポリアクリルアミドゲルで分離し、次いで、ニトロセルロース膜に移して、マーカー蛋白質を検出する。蛍光蛋白質のレベルは、一次抗体/抗血清(例えば、特定のマーカー蛋白質に対して生起させたヤギポリクローナル;BD Gentest,Woburn,MA)、およびペルオキシダーゼ結合二次抗体(例えば、ウサギ抗ヤギIgG,Sigma Aldrich)を利用して検出することができる。化学発光は、当該分野で入手可能な標準試薬を用いて検出して、組織試料中の蛍光マーカー蛋白質のレベルを検出し、決定する。
【0114】
従って、もし候補治療剤/薬物またはテスト生物活性剤が本発明の1以上の方法でアッセイされるならば、異なる時点で比較して、ならびに参照または対照と比較して薬物に対する応答の総じての差があるか否かを決定することができる。
脱髄
本発明の1つの態様において、本発明の組成物および方法を利用して、全身抗原送達、または免疫応答を開始させるためのアジュバントプライミングの必要性なくして、病巣脱髄を達成する。本発明の種々の実施形態において、細胞特異的な様式での細胞死メディエーター蛋白質の発現は、標的細胞における細胞死および/またはニューロン喪失を誘導するのに利用される。細胞死メディエーター蛋白質の発現および/または活性もまた誘導性であり得る。
【0115】
CDMPはSMAC(カスパーゼの第2のミトコンドリア由来アクチベーター)、IAP(アポトーシス蛋白質の阻害剤)、カスパーゼおよびそれらのモジュレーターであり得る。他の実施形態において、CDMPはTNF(腫瘍壊死因子)受容体、およびFas受容体、およびTRAIL受容体経路のような他の死滅受容体シグナリング経路のモジュレーターであり得る。また、CDMPは、グランザイムB、またはグランザイムBのモジュレーターを含めたカスパーゼのアクチベーターでもあり得る。
【0116】
種々の実施形態において、細胞死メディエーター蛋白質は核酸構築体によってコードされる。いくつかの実施形態において、核酸構築体は1以上のCDMPをコードすることができる。CDMPは同一または異なり得る。例えば、単一の核酸は、タンデムにカスパーゼ9をコードする2つの配列のような蛋白質の2つをコードすることができるか、あるいはカスパーゼ9およびカスパーゼ3をコードすることができる。本発明の核酸構築体はカスパーゼ2、5、8、9、10または11、それらのプロ酵素形態、またはその誘導体をコードすることができる。さらなる実施形態において、そのようなカスパーゼ蛋白質をコードする配列を、架橋剤化合物と反応性である二量体化ドメインを含むように修飾する。かくして、いくつかの実施形態において、野生型カスパーゼ配列を、架橋剤化合物に対して選択的に反応性である二量体化ドメインを含むキメラ配列を生じるように修飾する。そのような二量体化ドメインの例は、その関連部分をここに引用してその全体を援用する、米国特許出願第20050187177号および米国特許第6,984,635号に開示されたものを含む。
【0117】
さらに、インビボまたはインビトロでの選択的細胞死は、デュアルプロモーター、自殺遺伝子またはCDMPの組織特異的発現を可能とするように構築された自己不活性化(SIN)レンチウイルスベクター、およびマーカー遺伝子(例えば、eGFP)の使用によって達成することができる。1つの実施形態において、乏突起神経膠細胞のような細胞を、図1に示すように、本発明の核酸構築体を用いて標的化する。得られたベクターは、自殺遺伝子およびマーカー遺伝子eGFPの組織特異的発現を可能とする。従って、前記ベクターは、乏突起神経膠細胞のような有糸分裂後細胞の感染を可能とする。さらに、前記ベクターを利用して、高ウイルス力価を生じさせ、インビボにて有効な適用を促進することができる。かくして、1つの実施形態において、そのような細胞特異的発現の結果は乏突起神経膠細胞特異的細胞死である。
【0118】
デュアルプロモーターの特徴は、感染された細胞を蛍光顕微鏡検査法を用いて、容易に同定できるように、上流遺伝子から独立したGFPの共発現を可能とする。当該分野で公知の他の蛍光マーカーをGFPの代わりに用いることができる。
【0119】
1つの実施形態において、核酸送達ビヒクルを用いて、細胞死メディエーター蛋白質(または自殺蛋白質)をコードする配列をインビトロにて細胞に送達する。送達は、病巣パターンの、CNSに対するものでもあり得る。このシステムに対して数個の利点がある。
【0120】
まず、病巣の位置は予め決定することができ、関連するニューロンおよび細胞応答の正確な検出を可能とする。第2に、生じた病巣のサイズを、引き続いての実験において制御し、および改変することができる。これは重要である。というのは、関連するニューロンの死滅を広めるために破らなければならない乏突起神経膠細胞の喪失の閾値があり得るからである。第3に、前記モデルは、脱髄の正確なタイミングを指定する能力を可能とする。
【0121】
従って、細胞および分子レベルにおけるニューロン応答の一時的記載を達成することができる。さらに、CNS中の異なる位置で生じた第2の病巣に対する1つの病巣の一時的関係を調査して、脱髄病巣の間の時間がCNS応答のタイプを決定するにおいて重要な因子であるかを決定することができる。加えて、送達ベクターは細胞特異的発現調節配列を組み込み、その結果、細胞死メディエーター蛋白質の細胞型特異的(例えば、乏突起神経膠細胞特異的)発現がもたらされる。この特徴は、細胞の実験的に課された死滅が最小の傍観者効果でもって望まれる細胞型集団に制限されることを確実とする。最後に、前記モデルは修飾および増大レベルの複雑性に適合している。
【0122】
例えば、1つの実施形態において、iCP9をレンチウイルスプラスミドにクローン化し、MBPプロモーター配列をiCP9遺伝子の上流にクローン化する。得られたレンチウイルスベクター、pΔmbpICP9/mEGFPを用いて、複製能力がないウイルスを作り出す(実施例1参照)。かくして、前記ベクターを利用して、細胞/組織特異的発現調節配列または誘導性発現調節配列の選択によって、1以上の特定の細胞型(例えば、所望により神経細胞および/または壁細胞)の結果として、時間をわたっての、あるいは特定の時点におけるニューロン応答を規定することができる。
【0123】
種々の実施形態において、本発明の方法を利用し、乏突起神経膠細胞の喪失に対するニューロンの応答を経時的に定義することができる。これは、公知の生存、栄養およびアポトーシス経路における変化を観察することによって、ならびにそのような事象に対する新規な応答を同定するためのマイクロアレイ分析を通じて達成することができる。さらに、環境ストレスに対する規定されたニューロン応答の分類は、アポトーシス誘導因子、抗アポトーシス因子、神経栄養因子および神経栄養関係転写因子の発現を含む。
【0124】
従って、種々の実施形態において、本発明の動物/細胞を利用して、種々の因子/化合物をスクリーニングして、そのような因子/化合物がニューロンの維持または健康を増強させ/減少させるかを決定することができる。例えば、多数の転写因子は外部刺激に応答してニューロンで発現される。2つの高度に保存されかつよく規定された転写因子であるNF−κBおよびcAMP応答エレメント結合蛋白質(CREB)は、ニューロンがストレスを受け、引き続いて、部分的に規定された様式で、広い範囲の下流標的を活性化する場合に発現される。NF−κBは、卒中、心臓停止および全体的虚血症、発作およびグルタメート、グルコース剥奪およびβ−アミロイドペプチドへの実験的暴露を含めた生理学的および病理学的状態によって調節される。可変傷害に対するこの高度に保存された応答は、ニューロンの生存およびアポトーシスの制御に関連する基本的な細胞応答を示唆する。同様に、CREBは生理学的刺激の膨大なアレイに対して応答して活性化される。最初のインビトロ研究およびCREBナルマウスを利用するその後のインビボの研究は、CREBが多数のニューロンサブタイプの生存に必要であることを示唆する。CREBは多数のげっ歯類モデルにおいて低酸素症、虚血症および酸化的ストレスに応答して活性化され、これらのストレッサーの間におけるCREBの不活性化は、典型的には、ニューロン細胞死を増悪させる。
【0125】
CREBおよびNF−κB誘導細胞応答の双方は、第1に、ニューロンの生存を究極的
には支持する様式で作用するように見える。同様に、神経栄養因子はニューロンの成長および生存を支持する。この群の分子は、限定されるものではないが:神経膠由来神経栄養因子(GDNF)、毛様体神経栄養因子(CNTF)、脳由来神経栄養因子(BNDF)、神経成長因子(NGF)、NT−3およびNT−4/5を含む。これらの因子は、急性CNS傷害後にアップレギュレートされると考えられている。さらに、乏突起神経膠細胞はニューロンに対する栄養支持を供すると考えられている。PLPを欠如するトランスジェニックマウスにおける、および誕生時における乏突起神経膠細胞前駆体の照射後に、急性MS病巣における乏突起神経膠細胞の喪失を記載するインビボの観察はインビトロ実験によって確証されている。具体的には、乏突起神経膠細胞前駆体細胞またはそれらの馴化培地の、黒質への添加は、ニューロンの生存を有意に増強させた。同様に、視神経乏突起神経膠細胞前駆体細胞またはそれらの培養された培地は、網膜神経節細胞の生存を有意に増強させた。NGF、BDNF、GDNF、ニューレグリンおよびNT−3は、全て、乏突起神経膠細胞の培養した培地において同定されている。
【0126】
ニューロンの生存とは対照的に、ニューロンのプログラムされた細胞死は急性乏突起神経膠細胞の喪失に対する重要な応答であろう。死滅受容体ファミリーのメンバーであるFasは、カスパーゼカスケードの活性化によってそのリガンドFasLによって結合された場合にアポトーシスを誘導する。脳においては、皮質ニューロンはFasを発現し、およびインビトロにて、これらの細胞はFas活性化の後に迅速にアポトーシスを受ける。卒中のモデルにおいて、Fas発現は、カスパーゼ8の発現と共に局所化されるニューロンにおいてアップレギュレートされる。さらに、もしFasLが不存在であれば、卒中誘導脳損傷が低下する。運動ニューロンのインビトロ培養物は、プログラムされた細胞死の開始についてFasの活性化に対する同様な依拠を示している。というのは、インビトロにて神経栄養支持が奪われた運動ニューロンは、Fas/FasL相互作用がなくなった後にのみ培養で維持されたからである。
【0127】
最後に、抗アポトーシス因子の発現は、急性脱髄に対する応答の重要な態様であり得る。天然で生じる細胞死のアポトーシスプロセスは種の間で高度に保存されており、線虫C.elegansにおける実験は、最初に、このプロセスに対して必須の分子の同定に導き、引き続いて、哺乳動物ホモログE4BP4の同定を可能とした。この分子は、発達する脳において、天然に生じる細胞死の時において運動ニューロンによって発現されることが示されている。さらに、インビボ過剰発現は、標的を刺激するニューロンの数を増加させ、該分子が天然で生じるアポトーシスのプロセスを妨げることを示唆する。
【0128】
従って、種々の実施形態において、抗アポトーシス化合物を種々の時点で本発明の細胞または動物に投与して、ニューロン維持に対する抗アポトーシス化合物の効果を決定することができる。抗アポトーシス化合物は細胞死メディエーター蛋白質(例えば、抗カスパーゼ9)に対する公知の活性を有することができるか、あるいはそれは、抗アポトーシス活性を決定するためにスクリーニングされる候補剤であり得る。
【0129】
かくして、乏突起神経膠細胞の喪失に対する経時的なニューロン応答を規定することによって、いずれの化合物が、最終的に、ニューロンの喪失、または細胞死からのニューロンの救済に導く事象のカスケードに影響するかを決定することができる。分子の空間的および時間的発現パターンを規定することから得られた情報は、適当な機能の実験を設計し、負傷に対する神経応答の根底にある細胞相互作用を解明することにおいて重要である。
【0130】
1つの実施形態において、本発明の方法を利用して、全身合併症がない乏突起神経膠細胞−ニューロン相互作用をアッセイして、生存するニューロンの分子応答を規定する(例えば、実施例2)。急性および慢性MS病巣における、ならびにNAWM(正常に見える白色物質)における軸索の横断および引き続いてのニューロンの喪失は、この疾患の比較
的最近に規定された態様である。証拠は、ニューロンの継続的喪失が患者における不能の蓄積を説明することを示唆する。
細胞ベースのスクリーニングアッセイ
本発明のいくつかの態様において、細胞培養物は、本発明の1以上の方法で利用される。標的細胞は被験体に由来することができ、かつ形質転換され得る(例えば、遺伝子的に修飾された)か、または本発明のトランスジェニック動物は細胞/組織培養物のための源となり得る。もう1つの態様において、本発明の細胞は細胞系に由来する細胞であってよい。
【0131】
種々の実施形態において、本発明の実施は、対照細胞に対する、テスト細胞(例えば、トランスジェニック乏突起神経膠細胞またはシュワン細胞)における遺伝子または遺伝子産物の発現、または遺伝子産物の活性の比較を供するための細胞ベースのアッセイを含むことができる。本発明で用いるテスト細胞は中枢または末梢神経系から単離することができ、それに由来する細胞培養物、およびその子孫、および源から調製された切片またはスミアー、または例えば、乏突起神経膠細胞またはシュワン細胞またはそれらの先祖を含有する脳のいずれかの他の試料を含む。望まれる場合、限定されるものではないが、ニューロン、神経膠細胞、小神経膠細胞、および神経膠星状細胞のような他のニューロン細胞型が実質的に含まれない富化された細胞培養物を用いるように選択することができる。成熟した乏突起神経膠細胞およびシュワン細胞を単離し、生成させ、または維持する種々の方法は当該分野で公知であって、本明細書中に例示される。
【0132】
1つの実施形態において、細胞死メディエーター蛋白質をコードする導入遺伝子を含む非ヒトトランスジェニック動物または細胞を供する工程であって、ここに、前記導入遺伝子はニューロン細胞または神経膠細胞特異的発現調節エレメントに作動可能に連結されている、工程;前記細胞死メディエーター蛋白質を活性化し、それにより、アポトーシスを誘導する工程;前記活性化に続いて少なくとも1つの生存するニューロン細胞または神経膠細胞を得る工程;ならびに、生存する神経膠細胞またはニューロン細胞においてRNA転写物および/またはコードされた産物をプロファイルする工程;それにより、MSまたはMS関連状態に関連する現象を特徴付けるプロファイルデータセットを編集する工程を含む、MSまたはMS関連状態に関連する現象を特徴付けるためのプロファイルデータセットを編集する方法が提供される。他の実施形態において、この方法は、ALSまたはパーキンソン病に関連する現象を特徴付けるためのプロファイルデータセットの編集を提供する。
【0133】
いくつかの実施形態において、細胞死メディエーター蛋白質はカスパーゼ−9またはカスパーゼ−11である。1つの実施形態において、カスパーゼ−9の活性化は、カスパーゼ−9の二量体化を誘導することを含む。もう1つの実施形態において、カスパーゼ−11の活性化は自己活性化を含む。
【0134】
種々の実施形態において、細胞型特異的発現調節エレメントは、CC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、ミエリン関連糖蛋白質(MAG)、ミエリン乏突起神経膠細胞糖蛋白質(MOG)、乏突起神経膠細胞ミエリン糖蛋白質(OMGP)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFα、RG5、p糖蛋白質、ニュールツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、スルファチド、2(VEGFR2)、スーパーオキシドジスムターゼ(SOD1)、チロシンヒドロキシラーゼ、ニューロン特異的エノラーゼ、パルキン遺伝子(PARK2)、パルキン共調節遺伝子(PACRG)、ニューロン特異的Tα1 α―チューブリン(Tα1)、小胞モノアミントランスポーター(VMAT2)、α−シノクレイン(SNCA)、PDGFR
−α、PDGFR−β、またはプロテオリピド蛋白質(PLP)の1以上を含めた群から選択される遺伝子からのものである。
【0135】
1つの実施形態において、本発明は、髄鞘再形成を調節する候補の生物学的に活性な剤を同定する方法を提供する。前記方法は、細胞死メディエーター蛋白質の細胞ごとに異なっての発現が可能な本発明のトランスジェニック動物からトランスジェニック細胞を入手または単離し、そのような細胞を培養する工程;候補剤を培養した細胞と接触させる工程;対照細胞に対する、遺伝子または遺伝子産物の改変された発現、または遺伝子産物の改変された活性を検出する工程であって、前記遺伝子または遺伝子産物は細胞死メディエーター蛋白質による細胞死の調節に相関している、工程;ならびに、もし前記遺伝子または遺伝子産物の発現のレベルが前記対照細胞に対して調節されているならば、前記薬剤を候補として選択する工程を含む。
【0136】
もう1つの実施形態において、もし細胞死を受けている標的細胞の数が、対照細胞と比較して候補剤の添加によって調節されるならば、薬剤は候補剤であると決定される。
【0137】
もう1つの実施形態において、本発明は、髄鞘再形成を促進する生物学的に活性な剤を同定する方法を提供する。前記方法は、本発明のトランスジェニック動物に存在する脱髄病巣からの標的細胞を入手し、単離し、および培養する工程;培養した細胞を候補の生物学的に活性な剤と接触させる工程;ならびに、細胞死メディエーター蛋白質をコードする導入遺伝子の改変された発現または改変された活性を検出する工程;ならびに、もし遺伝子または遺伝子産物の発現のレベル、または遺伝子産物の活性のレベルが対照細胞に対して増加するならば、前記薬剤を候補として選択する工程を含む。例えば、もし候補剤が細胞死メディエーター蛋白質の細胞死媒介活性を増強させるならば、前記候補は脱髄を促進することができ、他方、もし前記候補剤が細胞死媒介活性を低下させるならば、前記薬剤は髄鞘再形成を促進することができる。
【0138】
ある実施形態において、その神経系が活動的に髄鞘形成を受けている若い被験体からの髄鞘形成細胞を使用するのが好ましいであろう。他の実施形態において、限定されるものではないが、病原体または物理的負傷によって冒された病巣、およびクプリゾンのような毒性剤によって引き起こされた病巣を含めた、脱髄病巣における成体乏突起神経膠細胞前駆体に由来する髄鞘再形成細胞を用いるのが好ましいであろう。
【0139】
1つの実施形態において、高密度の皮質培養物を、iCP9を発現する複製欠損レンチウイルスでトランスフェクトする。CID(二量体化化学誘導因子)の添加、および乏突起神経膠細胞の引き続いての死滅の後、遺伝子転写のマイクロアレイ分析を用いて、生存する培養細胞における新規な因子の発現を評価することができる。これらの培養物における乏突起神経膠細胞の細胞死の程度は、細胞破壊のひどさに対する分子応答を同定するために系統的に変化されるであろう。従って、種々の実施形態において、細胞死を調節する1以上の分子標的が同定される。
【0140】
本発明に適した種々の遺伝子ビヒクルは当該分野で入手可能である。それらはウイルスおよび非ウイルス発現ベクターを共に含む。非限定的例示的なウイルス発現ベクターは、レトロウイルスのようなRNAウイルス、およびアデノウイルス、泡沫状ウイルス、狂犬病ウイルスおよびアデノ関連ウイルスのようなDNAウイルスに由来するベクターである。非ウイルス発現ベクターは、限定されるものでなはいが、プラスミド、コスミド、およびDNA/リポソーム複合体を含む。遺伝子ビヒクルは、その中に運ばれる外因性遺伝子の組織特異的、細胞特異的、またはオルガネラ特異的でさえある発現を指令する調節配列を運ぶように作成することができる。
【0141】
いくつかの実施形態において、標的細胞を多数の遺伝子ビヒクル(例えば、その各々が、所望の産物をコードする遺伝子構築体、および1以上のレポーター遺伝子をコードする遺伝子構築体を含む2つのベクター)で共トランスフェクトすることができる。
【0142】
さらに、所望であれば、広く種々の細胞内局所化配列またはシグナルが特徴付けられ、導入遺伝子のオルガネラ特異的発現を指令するために適用可能である。例えば、細胞内局所化配列は以下の:(a)細胞の外部への遺伝子産物の分泌を指令するシグナル配列;(b)細胞の原形質膜または他の膜区画への蛋白質の付着を可能とする膜係留ドメイン;(c)コードされた蛋白質の核へのトランスロケーションを媒介する核局所化配列;(d)コードされた蛋白質を主としてERに制限する小胞体滞留配列(例えば、KDEL配列);(e)目的の蛋白質が、該蛋白質が膜に結合するように、ファルネシル化することができ;または(f)コードされた蛋白質産物の異なる細胞内分布において役割を演じるいずれかの他の配列のうちのいずれか1つであり得る。
【0143】
所望であれば、遺伝子ビヒクルを当該分野で公知のいずれかの方法によって宿主細胞(例えば、乏突起神経膠細胞またはシュワン細胞のような髄鞘形成細胞)に挿入することができる。適当な方法はリン酸カルシウム沈殿、DEAE−デキストラン、エレクトロポレーション、またはマイクロインジェクションを用いるトランスフェクションを含むことができる。
【0144】
適当な対照細胞または組織の選択は、最初に選択されたテスト細胞または組織、および調査下にあるその表現型または遺伝子型の特徴に依存する。テスト髄鞘再形成細胞をテスト化合物に接触させるが、対照細胞または組織は非処理の対応物であってよい。テスト髄鞘再形成細胞は脱髄後に検出されたテスト細胞であるが、対照細胞は非処理の対応物であってよい。一般には、テスト細胞および対照を並行して分析するのが好ましい。
【0145】
先のセクションで議論したように、これらの細胞は、ニューロン髄鞘再形成状態の分子基礎を解明するための細胞ベースのアッセイを行うのに、およびニューロン脱髄を阻害し、または髄鞘再形成を促進するのに有効な薬剤をアッセイするのに有用である。
【0146】
本発明のいくつかの態様において、トランスジェニック細胞は本発明のトランスジェニック細胞から入手し、培養し、拡大培養し、標的蛋白質をコードする遺伝子で形質導入し、そして、源動物またはいくつかの他の動物に移植し、または再度導入することができる。そのようなエクスビボ方法において、トランスジェニック細胞は、例えば、マーカー遺伝子発現/産生の調節用のテスト遺伝子/蛋白質をアッセイするために、誘導により生成され得る生物学的に活性な剤をコードする遺伝子(例えば、テスト産物をコードする遺伝子)でトランスフェクトすることができる。そのような調節は、本明細書中において前記したように細胞培養物においてアッセイすることができる。別法として、形質導入された細胞は被験体の動物に再度導入され、マーカー遺伝子発現をアッセイし、対照または参照と比較できる場合、移植された前記細胞が形質導入されていない場合、目的のベクターで生じる産物を発現せず、時間制御様式で目的のベクターで生じる産物を発現せず(例えば、誘導性発現)、または目的の産物を構成的に発現しない(例えば、CMVプロモーター)。
【0147】
例えば、神経膠細胞(例えば、乏突起神経膠細胞またはシュワン細胞)は神経バイオプシーに由来することができる。細胞を(例えば、10%熱不活化胎児ウシ血清(FBS)を含有し、抗生物質、組換えNeu分化因子(LDF)、インスリンおよびフォルスコリン(1ng/ml)を補足した、DMEMよりなる増殖培地を利用して)培養において拡大することができる。さらに、細胞は導入遺伝子によって供された蛍光標識を利用して非トランスジェニック神経細胞からソーティングする(例えば、FACS)ことができる。
形質導入のために、細胞を、目的の産物を送達する当該分野で公知の種々のベクタービヒクルでトランスフェクトすることができる。従って、種々の実施形態において、本発明の核酸構築体は、やはり、マーカー蛋白質をコードする1以上の配列に作動可能に連結される。
【0148】
本発明の1以上の組成物または方法で利用することができるベクターは、SV−40、泡沫状ウイルス、狂犬病ウイルス、アデノウイルス、レンチウイルス、レトロウイルス由来DNA配列の誘導体、および機能的哺乳動物ベクターの組合せに由来するシャトルベクター、ならびに機能的プラスミドおよびファージDNAを含む。真核生物発現ベクターは、例えば、ここに引用して援用する、P J Southern and P Berg,J MoL Appl Genet 1:327−341(1982);Subraminiら、Mol Cell.Biol.1:854−864(1981),Kaufinann and Sharp,J Mol.Biol.159:601−621(1982);Scahillら、PNAS USA 80:4654−4659(1983)およびUrlaub and Chasin PNAS USA 77:4216−4220(1980)によって記載されているように、よく知られている。本発明の方法で用いるベクターは、複製欠損アデノウイルスのような、レトロウイルスベクターのごときウイルスベクターであってよい。例えば、レトロウイルスの構造遺伝子が、末端反復配列に含有されたウイルス調節配列の制御下にある、目的の単一遺伝子によって置き換えられた「単一遺伝子ベクター」を用いることができる。例えば、ここに引用して援用する、Eglitis and Andersen,BioTechniques 6(7):608−614(1988)によって記載されているような、モロニーネズミ白血病ウイルス(MoMulV)、ハーベイネズミ肉腫ウイルス(HaMuSV)、マウス乳腺癌ウイルス(MuMTV)、およびネズミ骨髄増殖性肉腫ウイルス(MuMPSV)、ならびに細網内皮症ウイルス(Rev)およびラウス肉腫ウイルス(RSV)のような鳥類レトロウイルス。好ましくは、本発明のベクターはレンチウイルスベクターである。
【0149】
1つの実施形態において、操作された誘導性カスパーゼ9(iCP9)cDNA配列は、ミエリン塩基性蛋白質遺伝子プロモーター配列、図1Cの制御下にあるレンチウイルスベクターにクローン化する。引き続いて、複製能力がないレンチウイルスはこのプラスミドから生じさせることができ、細胞および動物系に適用することができる。前記ウイルスは、暴露された全ての細胞型の感染をもたらすがiCP9遺伝子の発現は、細胞特異的プロモーター系のため乏突起神経膠細胞に制限される。二量体化化学誘導因子(CID)の添加の結果、乏突起神経膠細胞のみ細胞死がもたらされるであろう。このアプローチは、インビトロおよびインビボ双方において、乏突起神経膠細胞を特異的に殺傷することにおいてかなり有効であろう。当業者であれば、他のカスパーゼまたはその誘導体のような他のCDMPをiCP9の代りに用いることができること、他のプロモーター配列をMBPプロモーターの代りに用いることができることを認識する。本発明のもう1つの態様において、当該分野で公知のマイクロアレイまたは他の発現プロファイリングプロセスを利用して、細胞死に応答してアップレギュレートまたはダウンレギュレートされる遺伝子または遺伝子の組を同定する。種々の実施形態において、発現データセットを、細胞死の誘導前、その間およびその後を含めた、種々のおよび特定の時点について編集することができる(例えば、実施例5)。
生物活性剤
ニューロン髄鞘再形成を調節するのに有効な生物学的に活性な剤または生物活性剤は、限定されるものではないが、単純なまたは複雑な有機または無機分子、ペプチド、ペプチドミメティック、蛋白質(例えば、抗体)、リポソーム、低分子干渉RNA、またはポリヌクレオチド(例えば、アンチセンス)のような生物学的または化学的化合物を含むことが意図される。
【0150】
化合物、例えば、ポリペプチドおよびポリヌクレオチドのようなポリマー、および種々のコア構造に基づく合成有機化合物の膨大なアレイを合成することができ、これらは本明細書中においても考えられる。加えて、植物または動物抽出物などのような種々の天然源はスクリーニングのための化合物を提供することができる。常には明確に述べられないが、活性剤は、単独で、あるいは主題のスクリーニング方法によって同定された薬剤と同一または異なる生物学的活性を有するもう1つのモジュレーターと組み合わせて用いることができるのは理解されるべきである。
【0151】
生物学的に活性な剤が裸のRNA以外の組成物である場合、前記薬剤を細胞培養物に直接的に加えてもよく、あるいは添加のための培養基に加えてもよい。当業者に明らかなように、経験的に決定することができる「有効」量が加えられなければならない。前記薬剤がポリヌクレオチドである場合、それはトランスフェクションまたはエレクトロポレーションによって直接的に細胞に導入することができる。別法として、それは、遺伝子送達ビヒクル、または前記した他の方法を用いて細胞に挿入することができる。
【0152】
蛋白質レベルを検出するのに適した広く種々の標識は当該分野で公知である。非限定的例は放射性同位体、酵素、コロイド状金属、蛍光化合物、生物ルミネセント化合物、および化学発光化合物を含む。
【0153】
主題の方法によって同定された候補の生物学的に活性な剤は以下の2つのクラスに広くカテゴリー化することができる。最初のクラスは、細胞または被験体に投与した場合に、細胞死メディエーター蛋白質の発現または活性のレベルを低下させる薬剤を含む。第2のクラスは、細胞死メディエーター蛋白質の発現または活性のレベルを増大させる薬剤を含む。
医薬組成物
本発明の方法を用いて、脱髄障害の処置で引き続いて実施できる生物学的に活性な剤を選択することができる。髄鞘再形成を調節するのに有効な選択された生物学的に活性な剤は、脱髄障害を処置するための医薬の調製で用いることができる。1つの態様において、本発明の同定された/選択された生物学的に活性な剤を投与して、細菌およびウイルスのような病原体によって冒されたニューロン脱髄を処置することができる。もう1つの態様において、選択された薬剤を用いて、例えば、橋中央ミエリン溶解およびビタミン欠乏症におけるように、毒性物質によって引き起こされたニューロン脱髄、または身体中への毒性代謝産物の蓄積を処置することができる。なおもう1つの態様において、前記薬剤を用いて、脊髄負傷のような物理的負傷によって引き起こされた脱髄を処置することができる。さらになおもう1つの態様において、前記薬剤を投与して、遺伝的特質を有する障害、限定されるものではないが、白質萎縮症、副腎脳白質ジストロフィー、変性多系統萎縮症、ビンスヴァンガー脳障害、中枢神経系における腫瘍および多発性硬化症を含む遺伝的障害において発現される脱髄を処置することができる。
【0154】
種々の送達系は公知であり、それを用いて、本発明の生物活性剤を投与することができる(例えば、リポソーム、マイクロ粒子、マイクロカプセルでのカプセル化、組換え細胞による発現、受容体媒介エンドサイトーシス(例えば、Wu and Wu,(1987),J.Biol.Chem.262:4429−4432)、レトロウイルスまたは他のベクターの一部としての治療核酸の構築など)。送達の方法は、限定されるものではないが、動脈内、筋肉内、静脈内、鼻内および経口経路を含む。特定の実施形態においては、本発明の医薬組成物を処置を必要とする領域に局所投与するのが望ましいであろう;これは、例えば、限定されるものではないが、手術の間における局所的注入によって、注射によって、またはカテーテルによって達成することができる。ある実施形態において、前記薬剤は被験体の神経系、好ましくは、中枢神経系に送達される。もう1つの実施形態において、前記薬剤は髄鞘再形成を受けている神経組織に投与される。
【0155】
選択された薬剤の投与は、処置の過程全体にわたって継続的にまたは間歇的に、1用量で行うことができる。投与の最も有効な手段および投与量を決定する方法は当業者によく知られており、治療で用いる組成物、治療の目的、処置されている標的細胞、および処置されている被験体で変化するであろう。単一または複数の投与を、処置している医師によって選択されている用量レベルおよびパターンで行うことができる。
【0156】
本発明の医薬組成物の調製は、医薬製剤の調製のための一般的に許容される手法に従って行われる。例えば、Remington’s Pharmaceutical Sciences 第18版(1990),E.W.Martin編,Mack Publishing Co.,PA参照。意図する使用および投与の態様に依存して、有効成分を、さらに、医薬組成物の調製において加工するのが望ましいであろう。適当な加工は、適当な非毒性および非干渉性成分との混合、滅菌、用量単位への分割、および送達デバイスへの封入を含むことができる。
【0157】
経口、鼻内、または局所投与用の医薬組成物は、錠剤、カプセル、粉末、液体、および懸濁液を含めた固体、半固体または液体形態で供することができる。注射用の組成物は液体溶液または懸濁液として、エマルジョンとして、または注射に先立っての液体中の溶解または懸濁に適した固体形態として供することができる。気道を介する投与のために、好ましい組成物は、適当なエアロゾライザーデバイスで用いる場合に固体、粉末、またはエアロゾルを供するものである。
【0158】
液体の医薬上許容される組成物は、例えば、本明細書中に具体化されたポリペプチドを、水、生理食塩水、水性デキストロース、グリセロール、またはエタノールのような液体賦形剤に溶解または分散させることによって調製することができる。組成物は他の薬効のある薬剤、医薬剤、補助薬、担体、および湿潤剤または乳化剤、およびpH緩衝剤のような補助的物質を含有することもできる。
【実施例】
【0159】
実施例1:iCP9ウイルスの生産
複製不能のウイルスを生産すること
図1および2は、本明細書中で用いる導入ベクターについての一般的な模式図を描く。第1世代自己不活性化レンチウイルスベクターpLpMGは、サイトメガロウイルスプロモーター配列(pCMV)、ラットミエリン塩基性蛋白質プロモーター(pMBP)の断片、ラット神経膠細胞繊維状酸性蛋白質プロモーター(pGFAP)の断片、または血小板由来成長因子受容体(PDGFR)−アルファプロモーター(pPDGFR)の断片いずれかの挿入によって修飾した。得られたベクター(pLpCMVXMG、pLpMBPXMG、pLpGFAPXMG、pLpPDGFRXMG)は、さらに、鋳型として元来のベクター(David Spencer博士,Baylor Medical Centerの親切な贈り物)を用いてPCRを介して得られたiCP9 cDNA配列の連結によって修飾し、新しいプロモーター配列の下流にクローン化した。
【0160】
次いで、3つのベクター、pLpCMV(iCP9)MG、pLpMBP(iCP9)MG、pLpGFAP(iCP9)MG、pLpPDGFR(iCP9)MGを用いて、高力価の複製不能レンチウイルスを得た。ウイルス力価は293T細胞に対して決定し、続いて、全てのベクターがEGFPを共発現するので、EGFP発現についてのFACS分析を行った。イン・ビボで用いた全ての力価は、ml当たり少なくとも109コロニー形成単位であった。この結果、EGFP発現を通じての感染した細胞の同定を可能としつつ、誘導性自殺遺伝子(iCP9)の発現のイン・ビトロおよびイン・ビボでの細胞への導入のためのビヒクルが得られた。というのは、それは同一ベクター内のウイルスプロモ
ーター配列から駆動されるだろうからである。また、それは、いずれのプロモーターを用いたかに基づいて遺伝子発現を特定の細胞型に限定し、これは、時間制御様式で系の細胞型特異的除去を与える。
【0161】
293T細胞を、リポフェクタミン(Invitrogen,Carlsbad,CA)を用い、gag−polおよびRD114エンベロープ蛋白質をコードするプラスミドと共にpΔmbpICP9/mEGFPベクターでトランスフェクトした。トランスフェクションから48時間後に、ウイルス上清を収穫し、標的細胞に直接的に適用するか、あるいはスナップ凍結させ、−80℃で貯蔵した。
【0162】
適時に細胞死を誘導すること。収集されたウイルスを用いて、樹立されている初代乏突起神経膠細胞培養物を形質導入した。細胞を培養し、GFPの発現について蛍光顕微鏡検査法を用いて評価した。GFP陽性細胞の程度はウイルス感染効率に相関した。細胞蛋白質の単離を行い、蛋白質単離体をウエスタンブロット導入に付して、iCP9蛋白質の安定な発現を検出した。得られた膜を抗−カスパーゼ9抗体(R&D Systems Inc.#AF8301)でプローブした。一旦ウエスタンブロット技術で蛋白質レベルにおいてiCP9の安定な発現が確認されたならば、CIDを10nM濃度にて培養物中のこれらの細胞に適用した。これは感染した細胞の細胞死を誘導した。細胞死は、蛍光顕微鏡下で培養物を観察することによって、およびCIDへの暴露の後に培養物中の細胞のメチレンブルー(Sigma)生存性染色を用いることによって検出した。全ての実験は、陰性対照として神経膠星状細胞−由来U87細胞系を用いて並行して行った。
【0163】
CIDの存在下でアポトーシスを誘導し、および細胞型選択性を示すために、レンチウイルスをイン・ビトロ培養物に適用した。アポトーシスは、最初にiCP9の構成的発現を生じるpLpCMV(iCP9)MGウイルスを用いて293T細胞において誘導した。EGFPは同一のウイルスプラスミドおよびプロモーターからやはり生産される。CIDの添加後に、(ウイルスでトランスフェクトした細胞に対応する)EGFP−陽性細胞は迅速にアポトーシスを受け、培養物中に同定されなかった。しかしながら、ウイルスでトランスフェクトされていない細胞(EGFP陰性)は培養物に持続した。対照ウイルス、pLpCMVXMGでトランスフェクトされた同一培養物において、EGFP−陽性細胞はCIDへの暴露の後に持続した(図5)。また、細胞死検出(Cell Death
Detection) ELISA Plus(Roche Inc)を行って、ウイルスの種々の希釈物で感染させた細胞をCIDに暴露した後に、培養物におけるアポトーシスのレベルを決定した。該ELISAシステムは、アポトーシスを受けている細胞によって生産されるヒストン−複合体化DNA断片を検出する。該ELISAのデータは、感染、引き続いての遺伝子発現および細胞死の間の相関を示す(図6)。
【0164】
可能な代替細胞死メディエーターシステムは、モノクローナルキメラ抗−CD20抗体によって活性化されて、アポトーシスを誘導することができる、E.coli−由来サイトシンデアミナーゼ遺伝子、HSV−tkシステムおよびトランスジェニックCD20の使用を含む。
【0165】
結果は、pΔcmvICP9/mEGFP由来レンチウイルスでの標的細胞(例えば、初代乏突起神経膠細胞)の効果的感染を示した。感染率は、予備的データに基づき、培養物中の標的細胞のほぼ100%に近付くはずである。さらに、遺伝子の乏突起神経膠細胞特異的発現を駆動するためにMBPプロモーター配列を用い、GFPは乏突起神経膠細胞において検出されるがU87対照細胞系(または他の対照細胞系、例えば、NIH 3T3)においては検出されない。CIDを含むiCP9システムを用い、CIDの培養物への投与は、乏突起神経膠細胞腫細胞系において迅速かつ有効な細胞死を生じると期待されるがU87細胞系においてはそうでない。
【0166】
CIDのイン・ビトロ系への添加。CIDを、50ng/ml NGFを含むNeurobasal培地中で10nmの最終濃度で前記した混合皮質培養物に加えた。乏突起神経膠細胞死への最初の時間は、かくして、前記したように確立され、ニューロン応答の分析についての最初の時点を表す。第2の時点は、24および72時間分析における亜急性応答を表わした。最後に、乏突起神経膠細胞の喪失に対する慢性応答はCID投与から1週間、2週間および2カ月後に分析した。
実施例2:乏突起神経膠細胞の喪失
iCP細胞死系を乏突起神経膠細胞初代細胞培養物に適用し、細胞死の有効性、ならびに時間経過を規定した。第2に、該系を高密度皮質培養物に適用して、MSで起こる乏突起神経膠細胞の急性喪失をモデル化した。
【0167】
この系を用い、乏突起神経膠細胞の喪失に対するニューロンの応答を経時的に規定した。これは、公知の生存、栄養およびアポトーシス経路の変化を観察することによって、ならびにそのような事象に対する新規な応答を同定するためのマイクロアレイ分析を通じて達成された。
【0168】
初代乏突起神経膠細胞培養物の樹立。乏突起神経膠細胞の富化された集団をFischer P2ラットから単離した。前脳をハンクスの緩衝塩溶液中で切開した。ポリL−リシン−被覆25cm2フラスコ中で組織をほぼ1−mm片に切断し、次いで、0.01%トリプシンおよび10μg/ml DNaseにて、湿潤化5%CO2部屋空気インキュベーター中で37℃で15分間インキュベートした。このインキュベーション時間に続き、20%胎児ウシ血清(FCS)を補足した、DMEM培地を組織混合物に加え、次いで、湿潤化5%CO2部屋空気インキュベーター中で10日間増殖させた。10日後に、乏突起神経膠細胞前駆体細胞(A2B5+)を、フラスコを200rpm、37℃にて一晩振盪することによって収集した。フラスコに接着したままの細胞を、線維芽細胞成長因子(FGF)および血小板由来成長因子(PDGF)を10ng/mlの最終濃度まで補足した高−グルコースDMEM中で1週間培養した。その後、FGFおよびPDGFを培養基から取り出し、A2B5+細胞を3ないし7日間にわたってO4+前髄鞘形成乏突起神経膠細胞に、次いで、7ないし10日間後にO4+およびMBP+成熟乏突起神経膠細胞に分化させた。細胞分化の状態は、培養物における細胞の形態の変化をみることによってモニターした。成熟MBP+乏突起神経膠細胞への分化が、顕微鏡調査で容易に同定される。分化を確認するために、パターンを細胞形態の変化として光学顕微鏡検査法で観察し、抗−A2B5,抗−O4および抗−MBP抗体を、分化経路における種々の時点において、培養された細胞のアリコットを用いる基本的免疫組織化学染色プロトコルで利用した。
【0169】
一旦この系および分化パターンを確認したならば、A2B5+初代細胞培養物を、pΔmbpICP9/mEGFPウイルス上澄み中で8時間培養した。8時間後に、上清を取り出し、24時間、DMEM/FGF PDGF培地に交換した。次いで、細胞を蛍光顕微鏡検査法下で可視化して、感染の有効性を決定した。感染された細胞はEGFPを発現し、適当な蛍光フィルター系を用いて容易に同定された。
【0170】
pΔmbpICP9/mEGFPでの有効な感染を確認した後、培養されたGFP+、A2B5+細胞のアリコットを前記したように培養物中で分化させる。成熟MBP+乏突起神経膠細胞が培養物で同定された後、CIDをDMEM/20%FCS中10nMの最終濃度まで加える。多数の培養物の維持は、CIDの添加の後の種々の時点におけるアポトーシスの分析を可能とした。CIDを培養上清に加えてから1、4、12、および24時間後に、培養物のメチレンブルー染色を行った。メチレンブルー陽性細胞がかくして同定可能であり、生きた細胞を表す。この実験はpΔmbpICP9/mEGFPウイルス
上清に暴露されていない(従って、アポトーシスを受けないはずである)A2B5+細胞を用いて二連で行った。従って、細胞の100%についての死滅までの時間はCIDの添加の後に確立された。
実施例3:高密度混合皮質培養物
高密度混合皮質培養物を、従前に記載されているように、3匹のFischerラットからの大脳皮質の単離後に樹立した。大脳皮質を取り出し、氷冷ハンクスの平衡塩溶液に入れ、遠心し、トリプシンで37°で10分間消化した。組織を遠心し、加熱−不活化胎児ウシ血清およびウマ血清を含有するEarleの塩(Invitrogen)を含む最小必須培地に再懸濁させた。細胞懸濁液を細胞ストレイナーに通し、平板培養した。3時間後に、培地を、B27 Minus AO(Invitrogen)を補足したNeurobasal培地に代える。培養物を7日間維持し、次いで、免疫組織化学染色を行って、培養物の組成を確認した。
【0171】
(全てのCNS細胞型よりなる)混合初代皮質培養物をpLpMBP(iCP9)MGで感染させ、CIDに暴露し、次いで、乏突起神経膠細胞の細胞死について分析した(MBP+細胞)。MBPプロモーターは、その大部分が成熟乏突起神経膠細胞であるMBP+細胞中でのみ細胞死を誘導するように設計した。CID発現に先立って、細胞は形態学的に正常に見え、対照培養物およびウイルスによりトランスフェクトされた培養物の細胞カウントは、細胞型の間の有意な差を示さず、これは、ウイルストランスフェクションが培養物の構成を有意に改変しなかったことを示す。CID暴露の後MBP−陽性、EGFP−陽性細胞はCIDに暴露しなかったMBP−陽性、EGFP−陽性細胞に対して破壊されているように見えた(図7)。さらに、A2B5(乏突起神経膠細胞前駆体細胞)/EGFP陽性細胞、GFAP(神経膠星状細胞)/EGFP陽性細胞およびO4陽性細胞(乏突起神経膠細胞前駆体細胞)/EGFP陽性細胞はCID暴露の前および後に形態学的に正常に見えた。
【0172】
GFAPプロモーター系の特異性をテストするために、GFAP+パン精製培養物(精製された神経膠星状細胞の培養物)をpLpGFAP(iCP9)MGでトランスフェクトし、CIDに暴露した。pLpGFAP(iCP9)MGで感染させたパン精製GFAP細胞をCIDに暴露し、4時間後に、EGFP陽性細胞の減少が示され、他方、CIDに暴露されなかった同様な培養物はEGFP陽性細胞の高いレベルを維持した(図8)。これらのデータは、GFAPプロモーターがiCP9発現を駆動し、その結果、CID暴露の後に神経膠星状細胞のアポトーシスがもたさられることを確認した。
【0173】
PDGFR−αプロモーター系の細胞特異性を確認するためにラット−由来初代皮質培養物をpLpPDGFR(iCP9)MGウイルスで感染させた。混合初代皮質培養物(全てのCNS細胞型を含有する培養物)において、A2B5−陽性細胞(A2B5発現はPDGFR発現と重複し、抗−PDDGFR抗体の代わりに用いられる、なぜなら、それはより労働集約的ではないからである)は、改変された形態を伴うアポトーシスを受けていると同定され、他方、他の細胞型は影響されていないように見える(図9)。これは、PDGFR−αプロモーター系のオリゴ前駆体−特異性を支持するデータを提供する。
実施例4:急性傷害に対するニューロン応答の分析
応答の4つの主なクラス:アポトーシス誘導因子、抗−アポトーシス因子、神経栄養因子および神経栄養関連転写因子を分析する。神経栄養因子の分析は、製造業者のプロトコルに従い、GDNF、CNTF、BNDF、NGF、NT−3およびNT4/5についての市販のELISAアッセイを用いて行う。混合皮質培養物からの24時間上清を記載された時点においてアポトーシス後に収集し、そしてCID投与されていない混合皮質培養物からの上清を収集し、それは対照として役立つ。全ての試料は二連で行い、最終のデータは2つの値の平均を表す。
【0174】
神経栄養関連転写因子NF−κBおよびCREB活性化の分析は、製造業者の指示に従い、Trans AMP−CREBおよびNF−κB p65キット(Active Motif Europe,Rixensart,Belgium)を用いて行う。該Trans−Amアッセイは、96−ウエルプレートに被覆された環状AMP−応答エレメントおよびNF−κBコンセンサス部位(5’−GGGACTTTCC−3’)を含有するオリゴヌクレオチドに特異的に結合することができる細胞抽出物に含まれるリン酸化CREBおよびNF−κBの活性形態のレベルを測定する。二次ホースラディッシュペルオキシダーゼ−コンジュゲーテッド抗体は、450nmにおいてELISAプレ−トリーダー(BIORAD)を用いて定量されるであろう感度が良い比色の読みを提供する。全ての試料は二連で行う。細胞抽出物は、従前に規定された時点において誘導アポトーシス後の混合皮質培養物から由来する。対照細胞抽出物は、CIDに暴露されていない混合皮質培養物よりなる。
【0175】
混合皮質培養物中での乏突起神経膠細胞の喪失に続いてアポトーシスを受けているニューロンの数は、製造業者のプロトコルに従い、TdT−媒介dUTPニック末端標識アッセイ(TUNELアッセイ,Roche,Indianapolis,IN)を用いて定量する。陽性細胞の数を、顕微鏡検査法を用いて定量し、CIDに暴露されていない培養物中の陽性細胞の数と比較する。抗−FAS抗体および抗−NOS抗体を用いて免疫組織化学(IHC)染色を行って、乏突起神経膠細胞の喪失後における混合皮質培養物からのニューロンでの発現を定量し、CIDに暴露されていなかった培養物と比較する。染色は、プロトコル当たり、VECTASTAIN ABCキット(Vector Laboratories Inc.,Burlingame,CA)を用いて行う。
【0176】
抗アポトーシス分子はニューロン生存において役割を演じることができるか、あるいは、逆に、発現の減少はニューロン喪失に寄与することができる。新規な因子E4BP4は、CNSにおけるニューロンで重要な抗アポトーシス機能を演じるように見える。従って、E4BP4の発現のレベルは、混合皮質培養物中での乏突起神経膠細胞の喪失に続いてアポトーシスを受けているニューロンにおける免疫組織化学(IHC)染色を介して調べ、CIDに暴露されていなかった培養物と比較する。IHC染色は前記したように行う。
実施例5:急性乏突起神経膠細胞によって調節されたユニークな分子の同定
乏突起神経膠細胞の急性喪失および慢性不存在は、中枢神経系におけるユニークな病理学的状態を表し、従って、新規な遺伝子はこれらの事象に応答してニューロンでアップレギュレートされ、同定される。これらの遺伝子を検出するためには、マイクロアレイ分析を行う。傷害後種々の時点において、乏突起神経膠細胞の誘導されたアポトーシス後に残る培養物から、RNeasy Miniキット(Qiagen,Valencia,CA)を用いて全RNAを単離する。対照アレイを生じさせるには、CIDを差し控えることによってイン・ビトロにて、乏突起神経膠細胞死に暴露されていない培養物から全RNAを同様に単離する。RNAプロセッシングおよび分析は、標準的なプロトコルを用い、Case Western Medical Schoolのコア施設によって行う。走査された出力ファイルはハイブリダイゼーションアーティファクトについて視覚的に調べられるであろう。アレイを平均強度に対して一定の基準で決め、次いで、Affymetrix Microarray5.0ソフトウエアを用いて分析する。もし発現が対照RNAに対して<1.5倍変化すれば、遺伝子はアップレギュレートされていると考えられる。
【0177】
マイクロアレイの結果を培養物の免疫組織化学染色で確認して、もし抗体が入手可能であれば、増加発現レベルを示す。抗体が入手可能でない場合、遺伝子配列に基づいて定量的RT−PCRを行って遺伝子発現のアップレギュレーションを確認する。簡単に述べれば、β―アクチンはベースライン遺伝子発現として働く。プライマーはcDNA配列分析に基づいて購入する。BioRad iCyclerを用いてリアル−タイムPCRを行
い、コンピューターは閾値サイクルについての標準曲線を計算する。平均閾値サイクルは各試料について3つのウエルから計算され、平均TCおよび標準曲線を用いて、試料のmRNAの量を外挿する。各細胞培養物において、mRNAはβ―アクチンmRNAの割合として定量され、対照培養物からの平均割合を比較する。
【0178】
pΔmbpICP9/mEGFPウイルスの乏突起神経膠細胞先駆体細胞への投与の結果、ほとんど100%のGFP陽性細胞がもたらされる。乏突起神経膠細胞が成熟するまでiCPの発現は起こらず、CIDの投与の結果、他の細胞型における公開されたデータに基づき、4ないし6時間にわたる乏突起神経膠細胞の細胞死がもたらされる(Straathofら、2005)。虚血症または低酸素症に対するもののような予め規定された経路の最初の分析は、乏突起神経膠細胞の喪失後の急性期において培養ニューロンで明らかであろう。なぜなら、ニューロンにおける有害事象に対する通常の応答が最も可能性があるからである。しかしながら、乏突起神経膠細胞喪失から過ぎた時間が長いほど、応答は公知の負傷応答から隔たり得る。さらに、乏突起神経膠細胞の喪失に続いてのニューロンのマイクロアレイ分析の結果、対照ニューロンと比較して、2ないし4、2ないし6、または5ないし10の新規かつ関連する遺伝子のアップレギュレーションおよび同定がもたらされる。そのようなプロファイルは、急性期におけるよりはむしろ、乏突起神経膠細胞喪失から72時間ないし1週間後に同定される。
実施例6−動物モデル
誘導性カスパーゼ9配列を含有する前記したウイルスpΔmbpICP9m/EGFPを成体ラットのCNSに適用して、CNS脱髄のイン・ビボモデルを作成した。ウイルスベクターの投与は、投与の速度(対流性分布)およびウイルス力価に基づく、実質の可変領域における遺伝的情報の有効な導入を可能とした。データは、CNSの領域がレンチウイルスで効果的に感染されることを示した。まず、成体Fischerラットをケタミン塩酸塩(80mg/kg)およびキシラジン(4mg/ml)の腹腔内注射によって麻酔し、定位フレームに入れた。中央頭皮切開を行う。脳実質へのアクセスは、病巣の所望の位置に依存し、種々の所定の座標において、頭蓋を通って右側穿頭孔に設置することによって達成された。標的領域サイズに依存した速度にてNo.32S−ゲージ針を備えた滅菌10μlハミルトンシリンジを用いてレンチウイルスを無血清培地で投与した(ウイルスの対流性分布は0.1ないし0.5μl/分の速度で最も有効である)。送達されたウイルスベクター(例えば、レンチウイルス)の容量は、本明細書中で決定される感染有効性および標的領域のサイズに依存する。注射の後に、針を5分間所定の場所に残し、次いで、次の4分間にわたってゆっくりと引き出す。皮膚を縫合糸を用いて閉じた。
【0179】
脊髄へのウイルス投与では、脊柱胸部において切開を行い、続いて、ラメネクトミーを行って、脊髄を露出させた。No.32S−ゲージ針を所定の座標において後柱に通した。ウイルスの注射後種々の時点において、CIDを腹腔内注射を介して投与して、CNSの感染した領域のアポトーシスをもたらした。
【0180】
急性脱髄後の可逆的生理学的機能不全の確認。データに基づき、ウイルスベクターを利用して、CNSの領域を首尾よく感染させ、導入遺伝子を有効に発現させた。急性乏突起神経膠細胞の死滅の生理学的発現は、まず、脳病巣を持つラットにおいて経時的に観察された。脊髄における病巣では、より正確な記録メカニズムを使用した。後柱における軸索の機能的完全性は、体性感覚惹起電位記録(SSEP)を用いてイン・ビボで調べる。
【0181】
CIDの投与後種々の時点において、ラットをSSEPの記録のために従前に記載されたように麻酔した。SSEPは、ハードプレート下に置かれたAg/AgClディスク電極を参照して、右側体性感覚皮質上のスクリュー電極から記録し、他方、対側性坐骨神経は1Hzで刺激する(200掃引の平均についての0.2msパルス持続、および40mA一定電流強度)。接地電極は頭皮に経皮的に置いた。SSEPの振幅は正ピークに対す
る最初の負ピークから測定される。応答潜時は、刺激の開始および最初のピークの間の時間として測定される。振幅および潜時の値は3つの独立した測定の平均として記録した。これらの測定を、CIDの投与から1、2、7および14日後に反復して、CNS損傷および修復の測定可能なパターンを確立した。急性病巣の免疫組織化学分析。iCP9乏突起神経膠細胞死に関連する細胞変化を評価するために、動物を、CIDの投与後種々の時点で犠牲にした。
【0182】
ラットの脳または脊髄を、イソペンタン中で20秒間スナップ凍結させ、次いで、セクションニングまで―80℃で貯蔵した。10μm厚さの脳の薄い冠状切片および脊髄の軸方向の切片をクリオスタット(−20℃)を用いて作成した。まず、これを死亡後1日において行って、CID誘導細胞死の成功を決定した。その後、CID後1、2、7および14日動物を犠牲にし、蛍光顕微鏡検査法、ルクソール速青色染色およびヘマトキシリンおよびエオシン(H&E)染色を用いて調べた。脳および脊髄の薄い切片を種々の抗体を用いる免疫組織化学染色を介して調べて、起こった、炎症(抗−LCA抗体、抗−ED1)および神経膠症(抗−GFAP抗体)の程度を決定した。
【0183】
図2中の写真は、濃縮されたレンチウイルスがCNSに直接的に適用でき、感染の程度および領域は、脳または脊髄の薄い切片における犠牲後に検出されたGFP陽性細胞の数に基づいて決定できることを示す。このデータは、本明細書中で提案されたモデルの基礎であるイン・ビトロおよびイン・ビボでの特異的細胞死についての導入メカニズムを確立する。
実施例7:急性病巣における軸索相互作用およびニューロンアポトーシスの同定
薄い切片を用いて、横断軸索またはアポトーシスニューロンの形態の病巣の部位における病巣ニューロン損傷を検出する。免疫組織化学染色を、抗−アミロイド前駆体蛋白質(APP)抗体を用い、脱髄病巣を含む薄い切片で行う。これは、APPは当該軸索の末端で蓄積するので、軸索の輸送および横断の乱れを同定する。同様に、Bielschowskys銀含浸染色を用いて、ニューロンおよび横断軸索を検出する。固定された切片を予め温められた(40℃)10%硝酸銀で10分間染色し、次いでPBS中で洗浄する。水酸化アンモニウムを硝酸銀溶液に加え、スライドを40℃にて30分間インキュベートし、その時点の後、スライドを発色剤作用溶液(40%ホルムアルデヒド、クエン酸、硝酸溶液)に直接的に1分間入れる。
【0184】
反応を1%水酸化アンモニウムで停止し、PBS中で洗浄し、次いで、5%チオ硫酸ナトリウム中で5分間インキュベートする。最後に、スライドを脱水し、マウントする。この染色技術を用い、横断軸索を同定することができ、軸索の数をカウントし、ウイルスで感染されているが、CIDを投与されておらず、従って、脱髄の病巣領域を有しない対照動物と比較する。ニューロンを細胞死後種々の時点で急性病巣中で、および急性病巣の周りでカウントし、ウイルスを受けているが、CIDを受けておらず、従って、脱髄の病巣領域を有しない動物に由来する薄い切片と比較する。各病巣領域または離れた正常領域の中央を通じて取られた、目での形態計測グリットによって規定された0.01mm2視野を調査のために選択する。この視野において、APP陽性線維およびBirelschowskys銀含浸線維を100×対物レンズ下でカウントする。
実施例8:急性脱髄病巣後の離れたニューロン喪失の同定
CNS中の脱髄の結果、病巣を横切る軸索を持つ離れたニューロンの死滅がもたらされるか否かを決定するために、種々の時点において、脊髄の側方束中での脱髄病巣の生成に続いて、対側性赤核においてニューロンを調べる。脊髄における乏突起神経膠細胞死の誘導後におけるラットの赤核中のニューロンの数を、CIDを受けていないウイルスを感染させたラットの赤核における数と比較する。また、TdT−媒介dUTPニック末端−標識アッセイ(TUNELアッセイ,Roche,Indianapolis,IN)を利用して、アポトーシス活性を検出する。これは製造業者のプロトコルに従って薄い切片で
行う。皮質下病巣負荷および脊髄萎縮。広範な脊髄萎縮はMSのよく規定された特徴である。CNSの離れた領域における脱髄事象は脊髄萎縮を誘導し、この誘導は脱髄負荷の程度に関連することを決定する。
【0185】
従って、従前に記載されているように、ウイルスベクター(例えば、iCP9)をCNSの皮質下領域に送達し、CIDの送達の結果、脱髄領域がもたらされる。元来の傷害ラットが犠牲にされてから8カ月後に、脊髄を取り出し、スナップ凍結させ、薄い切片を調製し、H&E染色する。脊髄の直径を測定し、iCP9ウイルスで同様に感染されるが、CIDを受けず、従って、急性脱髄病巣を有しない対照動物と比較する。病巣の数、位置、ならびに病巣の間の時間間隔は、全て、脱髄領域および頻度のより大きな程度を生じさせるために変化させ、脊髄における萎縮を検出する可能性を最大化することができる。
【0186】
最後に、結果は、生存またはアポトーシス因子の変化を同定するか、またはイン・ビボモデルを用いてユニークな遺伝子を確認する。これは免疫組織化学染色を用いて行われる。また、結果はイン・ビボモデルでアッセイすることもでき、これは、脱髄に対するニューロン応答を研究するためのもう1つの生理学的基盤を提供する。脊髄における乏突起神経膠細胞の急性喪失の結果、急性生理学的機能不全およびSSEP記録の測定可能な変化がもたらされ得る。具体的には、振幅の減衰および潜時の増加が同定される。これは決定される時間経過にわたってのベースラインを修正する。というのは、ラットは最初の傷害から回復するからである。細胞死後にルクソール速青色(Luxol Fast Blue)染色を用い、急性脱髄の領域を規定する。炎症性浸潤の程度およびタイプは免疫組織学染色を用いて決定される。
【0187】
かくして、温和な非特異的炎症性浸潤が続いて起き、数週間にわたって消失し得る。MS患者からの病理学的検体において示されているように、横断軸索が急性病巣内に局在化する。最後に、多数の離れた脱髄病巣の誘導の後に赤核中の生きたニューロンの数の減少が観察される。喪失の時間経過および程度は経時的にゆっくりと起こり得るが、例えば、脱髄から約6ないし12カ月の後に同定することができる。
実施例9:カスパーゼ−9を発現するトランスジェニック動物
ミエリン塩基性プロモーターの制御下にあるカスパーゼ−9を発現するトランスジェニックマウスは、まず、トランスジェニック標的化ベクター構築体を生じさせることによって作り出される。種々の核酸エレメントを組み込んで、マウスにおけるカスパーゼ−9の発現を確実とする。合成イントロンエレメントを、ベクターに由来するプレ−mRNAの適切なプロセッシングのためにカスパーゼ−9 cDNAの前面に置く。ポリアデニル化シグナルを、mRNAの適切なプロセッシングのためにカスパーゼ−9 cDNAの末端に組み込む。カスパーゼ−9の誘導性発現を可能とするために、2つのLoxP部位(Cre−レコンビナーゼ認識部位)によってフロックスされた停止コドンをミエリン塩基性プロモーターおよび合成イントロンの間に組み込む。このベクターは、ゲノムへのベクターの有効な組込みのために線状化され、マイクロインジェクションによって多数の前核に注射される。注射された前核を偽妊娠FVB/N株マウスに移植する。動物の子供を、注射されたベクターの宿主ゲノムへの組み込みについてスクリーニングする。スクリーニングにおいて陽性と同定された動物の子供を離乳させ、ER−Cre導入遺伝子を含有するトランスジェニックマウスと交配させる。交配からの動物の子供を、カスパーゼ−9およびER−Cre導入遺伝子双方を保有する動物についてスクリーニングする。タモキシフェンを腹腔内に注射してER−Cre導入遺伝子からのCre蛋白質の発現を誘導する。LoxP部位の切り出し、およびミエリン鞘におけるカスパーゼ−9の結果としての発現は、抗−カスパーゼ−9抗体を用いて免疫蛍光染色で確認される。
実施例10:イン・ビボでのアポトーシスの細胞特異的誘導
該系をイン・ビボでテストして、細胞特異的アポトーシスを誘導することができ、および細胞除去のタイミングはそれがイン・ビトロであるように制御されたことを確認した。
この目的で、ウイルスを成体Fischerラットの脳梁に注射し、次いで、3週間後に、CIDを同側脳室に投与した。それから24時間後にラットを犠牲にし、脳を全部摘出し、固定し、分析のために低温切断した。ラットにpLpGFAP(iCP9)MG(GFAPプロモーターは発現を神経膠星状細胞に制限する)およびその後CIDを注射し、24時間後に犠牲にし、ウイルス感染の部位におけるTUNEL陽性染色を示した(というのは、該ベクターは、EGFPの構成的な発現をもたらすので、EGFP陽性細胞によって境界が画定されるからである)。TUNEL(TdT−媒介dUTP−Xニック末端−標識)系は、アポトーシスカスケードの間に断片化されたDNAにタグを加え、標準的に免疫組織化学を用いる標識を可能とした。CIDよりはむしろグリセロールを受ける対照ラットに由来する薄い切片はTUNEL陰性であった。次いで、連続した薄い切片を抗−GFAP抗体で染色して、神経膠星状細胞を同定した。GFAP+細胞は、グリセロールを受ける対照ラットに由来する切片とは対照的に、CIDが投与されたラットに由来する薄い切片におけるウイルス感染の領域で検出されなかった(図10)。これは、イン・ビボにて、iCP9系がCID投与後のみにアポトーシスを誘導し、GFAP+神経膠星状細胞を効果的に除去することを確認した。
【0188】
同様に、(乏突起神経膠細胞を除去するように設計された)pLpMBP(iCP9)MGを注射し、CIDに暴露されたラットは、感染の部位において抗−MBP抗体で染色されず、他方、pLpMBP(iCP9)MGで感染させ、グリセロールを投与した対照ラットからの薄い切片は、抗−MBP抗体を用いる免疫組織学染色後に正常に見えた。TUNEL陽性細胞はCID暴露後に感染の部位において同定されたが、グリセロールのみに暴露された対照においては同定されなかった(図11)。
【0189】
これらのデータは、iCP9系が時間制御様式でイン・ビボでアポトーシスを誘導でき、利用されたプロモーター配列に基づいて細胞−型に特異的なように見えることを確認した。
実施例11:CNSにおける急性乏突起神経膠細胞アポトーシスに対する細胞応答
脊髄および脳双方におけるラットでの脱髄および修復プロセスは、ケタミン塩酸塩(80mg/kg)、アセプロマジン(2.1mg/kg)、およびキシラジン(4mg/ml)の腹腔内注射によって麻酔され、および定位フレーム(Stoelting Co)に入れられたラットの3日齢子供(P3)を用いることによって規定される。No.26S−ゲージ針を備えた10μlのハミルトンシリンジを、ブレグマの0.5mm前方かつ0.5mm側方の柔軟な頭蓋を0.2mmの深さで通す。無血清培地中の5マイクロリットルのpLpMBP(iCP9)MGウイルス(乏突起神経膠細胞を選択的に除去する)を、No.26S−ケージ針を備えた10μlのハミルトンシリンジを用いて2μl/分の速度で注射する。胸脊髄への注射を受けるラットについては、No.26S−ゲージ針を備えた10μlのハミルトンシリンジを手動で脊髄に通す。次いで、マイクロインジェクションシステム(Harvard Co)によって、脳におけるのと同一速度でウイルスを注射する。注射の後に、針を5分間所定の場所に残し、次いで、次の4分間にわたってゆっくりと引き抜く。
【0190】
ウイルスの注射から3週間後に、5μlのCID(10nm)を同側脳室(3.6mmの深さにてブレグマから−0.8mm前方かつ1.4mm側方)に注射し、その結果、感染した乏突起神経膠細胞のアポトーシスがもたらされる。1日置きに、CID注射の日に開始し、動物は、増殖細胞を取り込み、免疫組織化学を用いるそれらの同定を可能とする100mg/kgのBrdU標識ミックス(Sigma)の腹腔内注射を受ける。ラットをCIDから1,7,14、21および28日後に犠牲にする。
【0191】
対照ラットには、CIDよりはむしろグリセロールの注射を受ける(CIDはグリセロール中に希釈する)以外は実験動物と同様にウイルスを注射する。ラットを深く麻酔し、
まず、150mlの0.9%NaCl生理食塩水で、次に等容量の氷冷4%パラホルムアルデヒドで経心的に灌流する。次いで、全脳および/または脊髄を摘出し、パラホルムアルデヒド中で少なくとも4時間、後固定し、続いて、組織が容器の底に沈むまで、30%スクロース中で低温保護する。次いで、試料をOCT中で凍結させ、スーパーフロスト+スライド上で10ないし20μmにて低温切断する。この実験を同様に脊髄において反復する。
免疫組織化学。CIDから1、7、14、21及び28日後の動物から由来する組織を、蛍光顕微鏡検査法を用いて調べて、感染の領域を同定する。急性脱髄および引き続いての髄鞘再形成の領域を規定するために、ミエリンの黒色金染色を、各時点において急性乏突起神経膠細胞の死滅の誘導後に薄い切片で行う。この実験における脱髄までの時間は、感染の領域が黒色金で染色されない時点と定義され、およびこの実験における髄鞘再形成までの時間は、黒色金が隣接する影響されていないミエリンと同等な強度で感染の領域を染色する脱髄後の時点と定義され、これを各動物について記され、そして平均および標準偏差を計算する。これらのデータを、対照動物についての脱髄までの時間および髄鞘再形成までの時間と比較する。統計学的解析は行わない。というのは、対照は脱髄/髄鞘再形成すると予測されないからである。しかしながら、これらのデータ点はさらなる実験のための対照として働く。
【0192】
脳および/または脊髄の薄い切片を調べて、抗−CD45抗体(TおよびB細胞)、抗−ED1抗体(小神経膠細胞)、抗−CD68抗体(マクロファージ)を用いて炎症の程度、および抗−GFAP抗体(神経膠星状細胞)を用いて神経膠症を決定する。これは、各時点においてなし、同等な対照動物に由来する標識された薄い切片と定量的に比較する。
【0193】
また、切片を抗−ネスチン抗体(多能性幹細胞)、抗−NG2+抗体(神経膠に定められた幹細胞)、抗−PDGFR−アルファ(OPCの全プール)、抗−O4+抗体(O1陰性である場合の初期OPC系)、抗−O1抗体(後期OPC系)および抗−MBP抗体(成熟乏突起神経膠細胞)で染色して、幹細胞および乏突起神経膠細胞前駆体細胞の動員、および動的病巣部位への取り込みを同定する。これらの抗体は、CNS幹細胞から成熟乏突起神経膠細胞への経路に沿って異なる細胞を同定する。次いで、薄い切片を、蛍光顕微鏡検査法を用いて調べ、100×対物レンズ下で、目での形態計測グリットによって規定された0.01mm2の視野において標識された細胞をカウントする。病巣内の4つの異なる位置をランダムに選択し、カウントし、各時点における各切片についての平均細胞数および標準偏差が計算される。これを、脱髄病巣を有しない反対側半球中の対応する部位と、およびウイルスで感染されたが、CIDを受けず、従って、脱髄病巣を有するはずのない対照動物と比較する。対照動物におけるカウンティングのための領域は、EGFP+細胞の存在によって同定されるウイルス注射の部位である。各領域についての平均細胞カウントを対応t−検定を用いて比較する。これを各時点において用いた各抗体について反復して、いずれの乏突起神経膠細胞のサブタイプが病巣に移動するか、およびこの移動の時間的分布を決定する。
【0194】
CID暴露の後に、ラットにBrdU標識ミックスを注射する。分裂の間に、BrdUは細胞のDNAに取り込まれ、抗−BrdU抗体の適用、および標準免疫組織化学によって分裂された細胞の同定を可能とする。細胞型についての染色の完了の後に、薄い切片を、最初の標識手法で用いたものとは異なる二次抗体と共に抗−BrdU抗体で標識する。シグナルの重複は増殖する細胞型を同定する。二重に染色された切片を、同様な時点での対照切片と比較する。もし同一の増殖する細胞型が対照切片で同定されるならば、二重染色細胞を4つの切片の全同側半球でカウントし、平均し、対照切片に由来する平均カウントと比較し、次いで、標準t−検定を用いてこれを統計学的に比較することができる。もし二重標識細胞が対照で不存在であれば、定量的な尺度は二重標識細胞の存在または不存
在である。
【0195】
また、前記実験を、脊髄へのウイルス注射を受けるが、それがCIDではなくビヒクル(グリセロール)である匹敵する対照と共に脊髄において行う。脊髄脱髄についての動物の調製は、CIDが同側脳室よりはむしろ大槽に注射される点で異なる。これは、脊髄ないし頭蓋のベースに沿っての触診、続いての、この空間へのハミルトンシリンジの通過によって達成される。
実施例12:イン・ビボでの乏突起神経膠細胞の急性喪失に対するニューロンの応答
急性病巣における軸索横断の同定。脱髄病巣は、実施例11に記載したように生じさせる。動物を実施例11からのデータによって決定された時点で犠牲にする。対照ラットに、CIDよりはむしろグリセロールの注射(CIDはグリセロール中で希釈する)を受ける以外は、実験動物と同様にウイルスを注射する。ラットを犠牲にし、記載したように調製する。病巣内の軸索の応答を特徴付けるために、薄い切片を抗−ニューロフィラメント(NF)抗体および抗−アミロイド前駆体蛋白質(APP)で標識する。前者は病巣中の軸索を同定し、他方、後者は軸索の輸送および横断の乱れを同定する。NF+軸索およびAPP陽性軸索を、目での形態計測グリットによって規定された0.01mm2視野を用いてカウントし、100×対物レンズ下でカウントする。脱髄された病巣内の4つの区別される領域をカウントし平均する。
【0196】
横断軸索(APP+)のパーセンテージは、平均APP+細胞のカウントを、病巣中の軸索の平均数(NF+)で割ることによって計算する。これらのデータを、比較可能な様式で、感染の部位において行われた対照動物カウントと比較する。ニューロンのカウンティングはCID後1、7、14、21および28日に行う。単純t−検定を用いて、いずれかの与えられた時点において、対照のカウントと実験のカウントとの間の統計学的有意性を決定する。
【0197】
急性脱髄病巣後の離れたニューロン喪失の同定。CNSにおける脱髄の結果、CNSの空間的に異なる領域においてニューロンの喪失がもたらされ得るか否かを決定するために、下部胸脊髄における脱税病巣の誘導に続いて、ニューロンを黒質において調べる。ラットは、病巣の部位が胸脊髄であって、CIDを従前に記載したように大槽に注射する以外は記載したように調製される。対照ラットにウイルスを注射するが、CIDよりはむしろ大槽へのグリセロール注射を受ける。同一の時点(1、7、14、21および28日)においてラットを犠牲にし、全脳および脊髄を摘出し、記載したように調製する。脊髄を切断し、黒色金で染色して、脱髄している病巣の存在を確認する。その解剖学的位置および外観によって同定された黒質を取り込む脳切片を抗−NF1抗体で標識し、ニューロンを前記したようにカウントする。脊髄における乏突起神経膠細胞の死滅の誘導後におけるラットの黒質中のニューロンの平均数を、脱髄病巣を有しない対照ラットの黒質中での平均数と比較する。同様に、TUNEL染色を、脊髄中の脱髄病巣を持つラットに由来する黒質を含む脳の薄い切片で行って、ニューロンの活性なアポトーシスを検出する。TUNEL(TdT−媒介dUTP−Xニック末端−標識)系は、アポトーシスカスケードの間に断片化されているDNAにタグを加え、標準免疫組織化学を用いる標識を可能とする。もしTUNEL陽性細胞が黒質内で同定されれば、それらをカウントする。前記したように、TUNEL染色の結果を、対照ラットの黒質における染色およびカウントと比較し、平均カウントをt−検定を用いて比較する。これらの実験は、乏突起神経膠細胞の離れた喪失がCNSでの無関係なニューロンの生存に影響するかを決定する。
【0198】
脱髄負荷およびニューロン喪失の間の関係の決定。正常に見える脳中の離れた軸索の喪失は、CNSにおける脱髄の程度に依存し得る。これをテストするために脱髄病巣のサイズ、および脱髄病巣の数を改変する。P3ラットに、胸脊髄に、および両側脳梁にウイルスを注射する。ラットを成熟させ、P20でのラットに前記したようにCIDを注射する
。ラットをCID注射から28日後に犠牲にし、脳および脊髄からの組織を前記したように調製する。分析の時点は変化し得るが、病巣が髄鞘を再形成した後には完了する。対照ラットは同一ウイルス注射を受けるが、P20においてはCIDよりはむしろグリセロールを受ける。前記したように、本実施例においては多病巣である病巣誘導の後に、黒質中のニューロンの完全性をTUNELアッセイおよびニューロンカウンティングを介して調べる。100×対物レンズ下で目での形態計測グリットによって規定された0.01mm2視野当たりの生きたニューロンおよびアポトーシス細胞の数を、同一切片における黒質内の4つの別々の視野から平均し、同様にカウントされた対照動物と比較する。細胞カウントを標準t−検定を用いて比較する。
【0199】
急性脱髄後の可逆的生理学的機能不全の確認。後柱における軸索の機能的完全性を、体性感覚惹起電位記録(SSEP)を用いてイン・ビボで調べる。CIDの投与後の種々の時点において、ラットを従前に記載したように麻酔する。ハードプレート下に置かれたAg/AgClディスク電極を参照とした病巣上方の脊髄に挿入された電極からSSPEを記録し、他方、反対側坐骨神経を1Hzで刺激する(200掃引の平均についての0.2msのパルス持続、および40mA一定電流強度)。接地電極を頭皮上に経皮的に置く。SSEP振幅を第1の負ピークから正のピークまで測定する。応答の潜時は、刺激の開始および最初のピークの間の時間として測定される。振幅および潜時の値を3つの独立した測定の平均として記録する。これらのデータをいくつかの時点で収集するが、脱髄に先立ってベースライン、脱髄後のデータ点および修復が組織学的に起こった後の1つのデータ点も含む。対照のデータは、同一ラットにおける反対側後柱からの記録によって得られる。病巣側(実験側)からの平均振幅および潜時の値を、標準t−検定を用いて反対側後柱(対照側)と比較する。
実施例13:成体ラットでの髄鞘再形成プロセスにおける乏突起神経膠細胞前駆体細胞の役割
OPCを選択的に除去するために、新規なウイルスベクターを記載されたベクター戦略を用いて作成した。細胞の形態学変化に基づき、A2B5標識細胞は、前記したように、pLpPDGFR(iCP9)MGでの感染、およびCID暴露の後にアポトーシスを受ける。パン精製A2B5+細胞(抗体で細胞を捕獲することによってA2B5+を富化した培養物)および並行して成長させたP3由来混合皮質培養物(先に記載された培養物を記載するための方法)を感染させるが、CIDに暴露せず、標識およびELISA研究のための対照として供する。培養物の双方の組を抗−A2B5抗体およびTUNEL染色で染色して、細胞除去の特異性を決定する。A2B5発現はPDGFR−α発現と重複し、イン・ビトロで標識するのがより容易であり、従って、PDGFR−α+細胞の代理マーカーとして役立つ。混合皮質培養物を抗−GFAP(神経膠星状細胞標識)および抗−MBP(乏突起神経膠細胞標識)でやはり染色して、他の細胞型はこのウイルス構築体での感染によって影響されないことを確認する。パン−精製A2B5+細胞培養物を、pLpPDGFR(iCP9)MGウイルスの系列希釈物に暴露し、CIDに暴露する。次いで、(予備的実験に記載された)製造業者のプロトコルに従い、培養物を細胞死検出(Cell Death Detection)Elisa Plus(Roche Inc)システムに付して、アポトーシスレベルを定量する。これは、PDGFR+細胞は感染およびCID暴露の後に除去された唯一の細胞であることを確認する。次いで、全ての培養を三連で行って、再現性を確認する。
【0200】
有効性および特異性の確認に際して、pLpPDGFR(iCP9)MGウイルスをP3ラットの脳または胸脊髄に注射する。動物を成熟させ、P30の齢において、リソレシチンを同側脳梁に元来のウイルス感染の部位に注射する。リソレシチン(LPC)は、迅速な細胞膜の破壊、および軸索保持でのミエリンの急性喪失をもたらす洗剤である。LPCをラットの脳に注射してから24時間後に、動物を犠牲にして、実験手法の結果、予測されるイン・ビボでのパラダイムをもたらすことを確認する。元来のpLpPDGFR(
iCP9)MG感染の領域内のLPC誘導脱髄病巣の存在は、蛍光顕微鏡検査法を用いて薄い切片上のウイルス感染の領域を同定することによって確認される。引き続いて、EGFP+細胞を含む切片は黒色金で染色され、重ねられた脱髄病巣の存在が確認される。私は、元来のウイルス感染の領域内でLPC病巣を再現性よく私が作成できることを確認するために4匹の動物の使用を提案する。対照はなく、この実験についての統計学的分析はない。
【0201】
一旦感染および脱髄の位置が確認されれば、LPCの注射から24時間後の、PDGFR+OPCを除去するためのCIDの添加でもって実験を反復する。1、7、14、21および28日に動物を犠牲にして、ミエリン修復に対するOPC除去の効果を決定する。これらの実験についての対照は、P30において重ねられたLPC病巣を伴うP3においてpLpPDGFR(iCP9)MGウイルスで感染させた動物であるが、その後CIDよりもむしろグリセロールを受ける。従って、それらは脱髄LPC誘導病巣を有するはずであるが、OPC除去は有さず、予め規定された様式で修復されるはずである。
【0202】
重ねられたLPC脱髄病巣および引き続いてのOPC除去を持つ動物(実験群)を、黒色金染色で同定されたデータ点で髄鞘再形成するまでの時間を用いて、重ねられたLPC病巣を持つが、OPC除去を持たない動物(対照群)と比較する(実施例11参照)。さらに、LPC病巣の部位までのOPC移動に要する時間もまた記録し、群の間で比較する。髄鞘再形成までの時間、およびOPC移動までの時間を各動物について記録し、標準t−検定分析を用いて各群の統計学的平均を比較する。実施例11に記載されたように、切片を乏突起神経膠細胞の細胞型、神経膠星状細胞および炎症細胞の存在について染色する。細胞を実施例11に記載されたプロトコルに従い、標識された切片についてカウントし、平均を計算し、標準t−検定を用いて対照群における平均細胞カウントと比較する。
【0203】
最後に、CID後の1日毎にBrdUで全ての動物を注射すると、記載したように増殖する細胞の同定が可能となる。細胞型についての染色の完了の後、次いで、薄い切片を、最初の標識手法で用いたものとは異なる二次抗体と共に抗−BrdU抗体で標識する。シグナルの重複は増殖する細胞型を同定する。二重染色切片を、同様な時点において対照切片で比較する。もし同一の増殖する細胞型が対照切片で同定されるならば、二重染色細胞を4つの切片の全同側半球においてカウントし、平均し、対照切片に由来する平均カウントと比較し、次いで、これは標準t−検定を用いて統計学的に比較することができる。もし二重標識細胞が対照動物に置いて同定されたものと異なるならば、定量はない。
【0204】
この実験を脊髄において反復することができる。
【0205】
本発明は前記した実施態様に限定されず、添付の請求の範囲の範囲内で変更することが可能である。当業者であれば、慣例的なものを超えない実験、本明細書中に記載された発明の具体例の多くの同等体の使用を認識し、または確認することができるであろう。
【技術分野】
【0001】
本願は、2007年6月12日に出願された米国仮特許出願第60/943,448号の利益を主張する。米国仮特許出願第60/943,448号は、その全体が参考として本明細書中に援用される。
【背景技術】
【0002】
ニューロン脱髄は、神経系におけるミエリンの低下によって特徴付けられる有害な状態である。ミエリンは、中枢(CNS)および末梢(PNS)神経系の双方に対して非常に重要であり、ニューロンの軸索を包み、ミエリン鞘として知られた絶縁性層を形成する。ミエリン鞘の存在は、神経軸索を下って伝達する電位の形態の神経シグナルの速度および完全性を増強させる。ミエリン鞘の喪失は、感覚、運動および他のタイプの機能における重要な損傷を生じさせる。というのは、神経シグナルはあまりにも遅く、(例えば、神経中のいくつかの軸索が他の軸索よりも速く伝わる場合には)非同期的に、(例えば、伝達が高い周波数においてのみ損なわれる場合には)間歇的に、のいずれかでそれらの標的に到達し、あるいは全く到達しないからである。
【0003】
ニューロン組織は、一般に、ニューロンおよび支持する神経膠細胞を含む。神経膠細胞は、哺乳動物の脳においては、約10:1でニューロンよりも数が多い。神経膠細胞は4つのタイプ:神経膠星状細胞、乏突起神経膠細胞、シュワン細胞および小神経膠細胞に分けることができる。ミエリン鞘は神経膠細胞(CNSにおいては乏突起神経膠細胞およびPNSにおいてはシュワン細胞)の原形質膜または細胞膜によって形成される。髄鞘形成の活性相の間に、CNS中の各乏突起神経膠細胞は、典型的には、1日当たりミエリン表面の約5000μm2、および1分当たり約105ミエリン蛋白質分子も多くを生じる(非特許文献1)。髄鞘形成乏突起神経膠細胞は、脱髄された病巣において同定されており、脱髄された軸索は新たに合成されたミエリンで修復できることを示す。
【0004】
ニューロン脱髄は、CNSおよびPNSの非常に多数の遺伝的および後天的障害で発現される。これらの障害は、例えば、多発性硬化症(MS)、進行性多病巣性白質脳障害(PML)、脳脊髄炎、中枢橋ミエリン分解(CPM)、抗MAG病、白質萎縮症:副腎脳白質ジストロフィー(ALD)、アレクサンダー病、キャナヴァン病、クラッベ病、異染性白質萎縮症(MLD)、ペリツェーウス・メルツバッヒャー病、レフスム病、コケーン症候群、ファン・デル・クナップ症候群、およびツェルヴェーガー症候群、ギラン・バレー症候群(GBS)、慢性炎症性脱髄多発性神経障害(CIDP)、および多病巣性運動神経障害(MMN)を含む。これらの障害のうちほとんど大部分では、治癒はなく、および効果的な療法はほとんどない。
【0005】
パーキンソン病(パーキンソン症候群または振せん麻痺)の間には、脳の細胞は未知の理由で劣化するように見える。しかしながら、炎症性反応についての役割は、パーキンソン病の病因において役割を演じると仮定されている。パーキンソン病は、振せん、および歩行、運動、および協調に伴う困難によって特徴付けられる脳の障害である。前記障害は、1,000人の人々のうち約2人が罹り、最もしばしば、50歳後に発症する。それは男性および女性の双方に罹り、老人の最も一般的な神経学的障害の1つである。パーキンソン病は、筋肉の運動を制御する脳の部分(基底神経節および錐体外路領域)の神経細胞
の進行性劣化によって引き起こされる。
【0006】
筋肉制御の喪失に加えて、パーキンソン病を持つ一部の人々はひどく鬱になる。知的能力の初期の喪失は一般的ではないが、重度のパーキンソン病の人々は(認知症、幻覚等を含めた)総じての精神的劣化を呈し得る。認知症は、該障害を処置するのに用いる投薬のいくらかの副作用でもあり得る。
【0007】
筋委縮性側索硬化症(ALS)は、脳および脊髄における神経細胞の破壊により随意筋の神経制御の喪失を引き起こす迅速に進行する不変に致命的な障害であり、筋肉の使用および制御の喪失をもたらす。これらの筋肉を制御する神経は収縮し、消失し、その結果、神経刺激の欠如による筋肉組織の喪失がもたらされる。筋肉の強度および協調は減少し、随意筋(例えば、意識制御下にあるもの)で開始する。筋肉制御の喪失の程度は継続的に進行し、より大きな筋肉の群が関与するようになる。呼吸および嚥下を制御する筋肉のような、半随意筋に対する神経刺激の喪失があろう。結局は、随意制御の下にある全ての筋肉は侵され、患者は、腕、脚および身体を動かす力および能力を喪失する。
【0008】
脳、脳幹、および脊髄に位置する運動ニューロンは制御ユニットとして働き、重要な通信は神経系および身体の随意筋の間をリンクさせる。脳内の運動ニューロン(上位運動ニューロン)からのメッセージは脊髄中の運動ニューロン(下位運動ニューロン)に伝達され、そこから特定の筋肉に伝達される。ALSにおいては、上位運動ニューロンおよび下位運動ニューロンは変性するか、あるいは死滅し、メッセージを筋肉に送るのを止める。筋肉は機能できず、除々に弱まり、段々減り(萎縮)、およびひきつる(束性攣縮)。結局は、随意運動を開始し、制御する脳の能力は失われる。原因は知られていない。
【0009】
MSは若い成人において非外傷性CNS罹患率の主な原因である。該疾患の若い年齢での開始および進行性性質は、社会に対して多大の経済的および社会的負荷を課す。典型的な再発軽減MSにおける急性増悪は、CNSにおける急性および病巣性炎症および脱髄の発現であり、長い間、MSの一次的疾患原因と考えられてきた。これらの事象は、現在認可されている治療剤の標的である。しかしながら、磁気共鳴(MR)イメージについてのT2炎症シグナル、および疾患の進行の相関は、再発軽減(RR)相および不能の引き続いての進行の間における臨床的特徴がそうであるように、弱い。さらに、いったん不可逆的不能に到達すれば、さらなる不能への進行は、不可逆的負傷の開始の前または後に起こるものを含めて、再発によって影響されない。
【0010】
軸索喪失による永久的神経学的不能に加えて、炎症性脱髄はMS病因において役割を演じる。炎症とは対照的に、軸索喪失は、典型的には、T1黒色穴、磁気共鳴分光測定(MRS)による減少したアスパラギン酸N−アセチル(NAA)、および脊髄萎縮の程度に相関し、これは、患者における臨床的不能と相関し得る。これらの変化は、診断から6カ月後と初期の患者において認められているが、ほとんどの患者においては、慢性、およびおそらくは全体的な軸索負傷は、疾患の二次的進行相の開始において臨床的な閾値を破る。
【0011】
(再発/軽減MS、またはRRMSと臨床的に定義される)疾患の急性炎症段階の間に、炎症メディエーターは軸索負傷に寄与するようである。CD8+T細胞の数、および軸索損傷の程度の間で関連付けがなされており、動物モデルは、これが、軸索破壊のCD8−MHCクラスI経路に関与することを支持する傾向がある(非特許文献2)。さらなる裏付けは、脱髄された軸索に向かって分極された細胞傷害性顆粒を含有する活性化されたCD8T細胞が直接的なCD8+T細胞毒性を示唆するという病理学的研究からくるものである。マクロファージおよび小神経膠細胞は、変性する軸索と近接して見出されている。これらの細胞型は、CNSからデブリスを除去するホメオスタシスメカニズムにおいて
役割を演じているが、それらはプロテアーゼ、サイトカイン、および酸化窒素(NO)のようなフリーラジカルを含めた炎症メディエーターも放出する。最後に、抗体および補体もまた、急性炎症の間に軸索損傷において役割を演じ得る。抗ガングリオシド抗体のレベルは、二次的な進行性またはRRMSにおけるよりも一次的進行性MS(PPMS)において有意に高いことが判明し、脱髄後に補体に暴露された軸索は、補体カスケードを直接的に活性化し得る。
【0012】
MSにおける炎症およびニューロン喪失の間の関係は十分に明らかにされていない。乏突起神経膠細胞の喪失、および全身的に特異的CNS抗原に対する炎症の誘導に依拠せず、または効力ある系アジュバントの使用を必要としない、引き続いての脱髄のモデルを確立する必要性がある。抗原および炎症を利用するそのようなモデルは、典型的には、MS患者において同定された脱髄およびニューロン喪失を反復しない。
【0013】
軸索横断およびニューロン喪失の正確な動物モデルは、MS脳検体において同定された病理学的特徴を模倣すべきである。これは、急性皮質病巣における横断された軸索、横断された樹状突起およびニューロンアポトーシスの同定を含む。急性事象は、また、MS病理学検体において同定されているように、Na+チャネルの再分布によって、程度は変わるが、回復される測定可能な損傷神経伝達をもたらすべきである。慢性病巣は、種々の程度の髄鞘再形成を示すべきであり、内向する持続性の軸索喪失が明らかでなければならない。最後に、動物モデルにおけるニューロン喪失が、解剖学的に区別され、かつNAWMに対する疾患の影響を模倣する元来の脱髄病巣から一時的に区別される領域で同定されなければならない。これらの特徴は、MS患者からの病理学検体において同定されたニューロンに対する影響の重要かつ正確な描写を提供するであろう。
【0014】
正確な分子メカニズム、および神経障害の病因および、特に、ニューロン脱髄の解明は、効果的な動物モデルの継続的欠如によって妨げられてきた。かくして、ニューロン障害のための治療剤に対する頑強なスクリーニングを行うための組成物および方法に対する緊急の要望が存在する。
【先行技術文献】
【非特許文献】
【0015】
【非特許文献1】Pfeifferら(1993)Trends Cell Biol.3:191−197
【非特許文献2】Rivera−Quinonesら、(1998)Nat Med.4:187−193
【発明の概要】
【課題を解決するための手段】
【0016】
本発明は、神経障害のプロセスを理解するための組成物および方法を提供し、およびニューロン脱髄の処置のための生物活性剤を同定しおよび開発する。本発明のトランスジェニック動物/細胞は、髄鞘再形成についての、および候補生物活性剤をスクリーニングするためのメカニズムを解明するのに利用することができるモデル系を提供する。マーカーおよび他の手段の髄鞘再形成特異的発現を利用することによって、本発明は、効果が肯定的(潜在的に治療的)であるか、または効果が否定的(潜在的に有害)であるかを問わず、髄鞘再形成に対する効果についてテスト剤をアッセイするのに利用することができる。さらに、髄鞘再形成に関連する現象に対するテスト剤の効果を決定することは、限定されるものではないが、細胞ベースのアッセイまたは技術を利用するものを含めた、当該分野で公知のいずれの適当な方法も含むことができる。
【0017】
本発明は、細胞死メディエーター蛋白質(CDMP)をコードする核酸配列を含む組換
え核酸分子を提供し、ここに、前記核酸配列は神経細胞特異的調節エレメントに作動可能に連結されている。また、核酸配列は、GFPまたは他の蛍光マーカーのようなマーカー蛋白質をコードする第2の核酸配列に作動可能に連結できる。また、本発明は、細胞死メディエーター蛋白質(CDMP)をコードする核酸配列を含む組換え核酸分子を含む宿主細胞も提供し、ここに、前記核酸配列は神経細胞特異的調節エレメントに作動可能に連結されている。前記宿主細胞は神経細胞または壁細胞であり得、例えば、それはニューロン細胞または神経膠細胞で有り得る。例えば、神経膠細胞は、乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞またはシュワン細胞であり得る。ニューロン細胞は頸部神経節ニューロン、皮質ニューロン、セロトニンニューロン、後根神経節、結節性神経節ニューロン、脊髄運動ニューロン、中脳ドーパミン作動性ニューロン、中枢ノルアドレナリン作動性ニューロンまたは腸ニューロンであり得る。壁細胞は内皮細胞、血管周囲細胞または平滑筋細胞であり得る。いくつかの実施形態において宿主細胞はBまたはTリンパ球のような免疫細胞であり得る。
【0018】
また、細胞型特異的発現調節エレメントに作動可能に連結された細胞死メディエーター蛋白質(CDMP)をコードするヌクレオチド配列を含むトランスジェニック動物も提供され、ここに、前記動物は、前記ヌクレオチド配列を含まない動物に対してより大きな程度の神経障害を呈する。動物は哺乳動物、霊長類、またはマウス、ラット、モルモットのようなげっ歯類、イヌ、ネコ、ウサギ、ブタ、チンパンジーまたはサルであり得る。神経障害は多発性硬化症のようなニューロン脱髄を含み得る。前記動物は対照動物のそれに対してアポトーシス神経稀突起膠細胞の増加を呈することができる。CDMPは中枢神経系に対して異所性的に制限され得る。
【0019】
CDMPをコードする核酸配列は宿主細胞または動物のゲノムに組み込むことができる。別法として、CDMPをコードする核酸配列はエピソームのものとすることができる。さらに、組換え核酸分子を宿主細胞または動物に送達して、レンチウイルスベクターのようなウイルスベクターによってトランスジェニック動物を作成することができる。
【0020】
1つの態様において、CDMPはカスパーゼ2、カスパーゼ5、カスパーゼ8、カスパーゼ9、カスパーゼ10、またはカスパーゼ11であり得る。CDMPは、FK506型リガンド、FKBP12型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドについての結合ドメインを含むキメラ蛋白質であり得る。CDMPの発現および/またはアポトーシス促進活性は、例えば、誘導性カスパーゼ9(iCP9)のように、誘導性であり得る。例えば、発現調節エレメントは誘導性、構成的、および/または細胞型または組織特異的であり得る。いくつかの実施形態において、発現および/または活性は動物におけるもののように神経細胞に特異的である。CDMPの活性は、AP20187のような二量体化化学誘導因子(CID)によって誘導することができる。
【0021】
(CDMP)をコードする核酸配列を含む核酸分子は、壁細胞または神経細胞特異的調節エレメントに作動可能に連結され得る。例えば、調節エレメントはニューロン細胞または神経膠細胞特異的調節エレメントであり得る。核酸配列によってコードされるCDMPの発現または活性は、乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、またはシュワン細胞のようなニューロン細胞または神経膠細胞におけるものであり得る。神経膠細胞特異的調節エレメントはCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、プロテオリピド蛋白質(PLT)、乏突起神経膠細胞ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFR−α、PDGF−α、RG5、p糖蛋白質、ニュールツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、P
DGFR−β、またはスルファチド遺伝子からのものであり得る。
【0022】
本発明のもう1つの態様は、生物学的に活性な剤、または生物活性剤をスクリーニングする方法である。本発明は、候補剤を、細胞死メディエーター蛋白質(CDMP)をコードする核酸を含む細胞と接触させる工程であって、ここに、前記核酸は細胞型特異的発現調節エレメントに作動可能に連結されている、工程;脱髄障害に関連する現象に対する効果を検出する工程;前記CDMPの活性のレベルが対照細胞に対して調節されているならば、前記薬剤を前記現象を調節するのに効果的であるとして選択する工程を含む、前記現象を調節する生物学的に活性な剤についてスクリーニングする方法を提供する。前記細胞は神経膠細胞、または壁細胞のようなニューロン細胞であり得る。神経膠細胞は乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、またはシュワン細胞であり得る。
【0023】
また、本明細書において、非ヒトトランスジェニック動物に候補剤を投与する工程であって、ここに、細胞死メディエーター蛋白質(CDMP)をコードする核酸配列の発現に際して脱髄が前記動物で起こり;ここに、前記核酸配列の発現は細胞特異的発現調節エレメントによって調節される、工程;前記CDMPを活性化して、前記動物における少なくとも1つの細胞でアポトーシスをもたらす工程であって、ここに、前記細胞は脱髄障害に関連している、工程;前記脱髄障害に関連する現象に対する前記薬剤の効果を検出する工程を含む、脱髄障害に関連する現象を調節する生物学的に活性な剤についてスクリーニングする方法も提供される。いくつかの実施形態において、動物は候補剤の投与に先立って脱髄から回復させる。脱髄障害は動物における乏突起神経膠細胞、神経膠星状細胞またはシュワン細胞の喪失によって特徴付けることができ、脱髄障害に関連する現象は髄鞘を有する軸索の減少によって特徴付けることができる。いくつかの実施形態において、脱髄障害は多発性硬化症である。もう1つの態様において、薬剤の効果の決定はPCR、イムノアッセイ、ハイブリダイゼーションアッセイまたはその組合せを含むことができる。候補剤はアンチセンスオリゴヌクレオチド、ペプチド、抗体、リポソーム、低分子干渉RNA、低分子有機化合物、または無機化合物であり得る。
【0024】
本発明のなおもう1つの態様において、細胞死メディエーター蛋白質(CDMP)をコードする核酸を含むトランスジェニック動物または細胞を供する工程であって、ここで前記核酸は神経細胞または神経膠細胞特異的発現調節エレメントに作動可能に連結されている、工程;CDMPを活性化し、それにより、アポトーシスを誘導する工程;活性化に続いて少なくとも1つの生存するニューロン細胞または神経膠細胞を得る工程;前記生存する神経膠細胞または神経細胞においてRNA転写物および/またはコードされた産物をプロファイルする工程;それにより、多発性硬化症に関連する現象、または多発性硬化症に関連するMS関連状態を特徴付けるプロファイルデータセットを編集する工程を含む、多発性硬化症(MS)に関連する現象、または多発性硬化症に関連するMS関連状態を特徴付けるプロファイルデータセットを編集する方法が提供される。
引用による援用
本明細書中で言及された全ての刊行物および特許出願は、各個々の刊行物または特許出願が引用により援用されることが具体的かつ個々に意図されているのと同等の程度まで引用によりここに援用される。
本発明の好ましい実施形態では、例えば以下が提供される:
(項目1)
細胞死メディエーター蛋白質(CDMP)をコードする核酸配列を含む組換え核酸分子であって、ここで、前記核酸配列は神経細胞特異的調節エレメントに作動可能に連結されている、組換え核酸分子。
(項目2)
前記CDMPがカスパーゼ2、カスパーゼ5、カスパーゼ8、カスパーゼ9、カスパーゼ10、またはカスパーゼ11である、項目1記載の組換え核酸分子。
(項目3)
前記CDMPが、FK506型リガンド、FKBP12型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドについての結合ドメインを含むキメラ蛋白質である、項目1記載の組換え核酸分子。
(項目4)
前記CDMPの発現が誘導性である、項目1記載の組換え核酸分子。
(項目5)
前記CDMPのアポトーシス促進活性が誘導性である、項目1記載の組換え核酸分子。(項目6)
前記活性が二量体化化学誘導因子(CID)によって誘導される、項目5記載の組換え核酸分子。
(項目7)
前記CDMPが誘導性カスパーゼ9(iCP9)である、項目5記載の組換え核酸分子。
(項目8)
前記CIDがAP20187である、項目6記載の組換え核酸分子。
(項目9)
前記神経細胞特異的調節エレメントが神経膠細胞特異的調節エレメントである、項目1記載の組換え核酸分子。
(項目10)
前記神経膠細胞が乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、またはシュワン細胞である、項目1記載の組換え核酸分子。
(項目11)
前記神経膠細胞特異的調節エレメントがCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、プロテオリピド蛋白質(PLP)、乏突起神経膠細胞−ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFR−α、PDGF−α、RG5、P糖蛋白質、ニュールツリン(neurturin)(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、PDGFR−β、またはスルファチド遺伝子からのものである、項目9記載の組換え核酸分子。
(項目12)
細胞死メディエーター蛋白質(CDMP)をコードする核酸配列を含む組換え核酸分子を含む宿主細胞であって、ここで、前記核酸配列は神経細胞特異的調節エレメントに作動可能に連結されている、宿主細胞。
(項目13)
前記CDMPがカスパーゼ2、カスパーゼ5、カスパーゼ8、カスパーゼ9、カスパーゼ10、またはカスパーゼ11である、項目12記載の宿主細胞。
(項目14)
前記CDMPがFK506型リガンド、FKBP12型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドについての結合ドメインを含むキメラ蛋白質である、項目12記載の宿主細胞。
(項目15)
前記CDMPの発現が誘導性である、項目12記載の宿主細胞。
(項目16)
前記CDMPのアポトーシス促進活性が誘導性である、項目12記載の宿主細胞。
(項目17)
前記活性が二量体化化学誘導因子(CID)によって誘導される、項目16記載の宿主細胞。
(項目18)
前記CDMPが誘導性カスパーゼ9(iCP9)である、項目12記載の宿主細胞。
(項目19)
前記CIDがAP20187である、項目17記載の宿主細胞。
(項目20)
前記神経細胞特異的調節エレメントが神経膠細胞特異的調節エレメントである、項目12記載の宿主細胞。
(項目21)
前記神経膠細胞が乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、またはシュワン細胞である、項目20記載の宿主細胞。
(項目22)
前記神経膠細胞特異的調節エレメントがCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、プロテオリピド蛋白質(PLP)、乏突起神経膠細胞−ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGF−α、RG5、P糖蛋白質、ニュールツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、PDGFR−β、PDGFR−α、またはスルファチドから選択される遺伝子からのものである、項目20記載の宿主細胞。
(項目23)
前記宿主細胞が神経細胞または壁細胞である、項目12記載の宿主細胞。
(項目24)
前記宿主細胞が神経膠細胞である、項目23記載の宿主細胞。
(項目25)
前記神経膠細胞が乏突起神経膠細胞、神経膠細胞星状細胞、小神経膠細胞、またはシュワン細胞である、項目24記載の宿主細胞。
(項目26)
CDMPをコードする前記核酸配列が前記宿主細胞のゲノムに組み込まれる、項目12記載の宿主細胞。
(項目27)
CDMPをコードする前記核酸配列がエピソームのものである、項目12記載の宿主細胞。
(項目28)
前記組換え核酸分子がウイルスベクターによって前記宿主細胞に送達される、項目12記載の宿主細胞。
(項目29)
前記ウイルスベクターがレンチウイルスベクターである、項目28記載の宿主細胞。
(項目30)
細胞型特異的発現調節エレメントに作動可能に連結された細胞死メディエーター蛋白質(CDMP)をコードするヌクレオチド配列を含むトランスジェニック動物であって、ここで、前記動物は前記ヌクレオチド配列を含まない動物と比較してより大きな程度の神経障害を呈する、トランスジェニック動物。
(項目31)
前記動物が哺乳動物、霊長類、またはげっ歯類である、項目30記載の動物。
(項目32)
前記動物がマウス、ラット、モルモット、イヌ、ネコ、ウサギ、ブタ、チンパンジーまたはサルである、項目30記載の動物。
(項目33)
前記神経障害がニューロン脱髄および/または血液脳関門における欠陥を含む、項目30記載の動物。
(項目34)
前記動物が対照動物の場合と比較してアポトーシス乏突起神経膠細胞または血管周囲細胞の増加を呈する、項目30記載の動物。
(項目35)
前記CDMPがカスパーゼ2、カスパーゼ5、カスパーゼ8、カスパーゼ9、カスパーゼ10またはカスパーゼ11である、項目30記載の動物。
(項目36)
前記CDMPがFK506型リガンド、FKBP12型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドについての結合ドメインを含むキメラ蛋白質である、項目30記載の動物。
(項目37)
前記CDMPの発現が誘導性である、項目30記載の動物。
(項目38)
前記CDMPの発現が中枢神経系に異所的に制限される、項目30記載の動物。
(項目39)
前記CDMPのアポトーシス促進活性が誘導性である、項目30記載の動物。
(項目40)
前記アポトーシス促進活性が、特異的に、前記動物の神経細胞におけるものである、項目39記載の動物。
(項目41)
前記活性が二量体化化学誘導因子(CID)によって誘導される、項目39記載の動物。
(項目42)
前記CDMPが誘導性カスパーゼ9(iCP9)である、項目39記載の動物。
(項目43)
前記CIDがAP20187である、項目41記載の動物。
(項目44)
前記細胞型特異的発現調節エレメントが神経または壁細胞特異的調節エレメントである、項目30記載の動物。
(項目45)
前記神経細胞特異的調節エレメントが神経膠細胞特異的調節エレメントである、項目44記載の動物。
(項目46)
前記神経膠細胞が乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、またはシュワン細胞である項目45記載の動物。
(項目47)
前記神経膠細胞特異的調節エレメントがCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、プロテオリピド蛋白質(PLP)、乏突起神経膠細胞−ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFα、RG5、P糖蛋白質、ニュールツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、PDGFR−α、PDGFR−β、またはスルファチドから選択される遺伝子からのものである、項目45記載の動物。
(項目48)
前記核酸配列がマーカー蛋白質をコードする第2の核酸配列に作動可能に連結されている、項目30記載の動物。
(項目49)
脱髄または血液脳関門障害に関連する現象を調節する生物活性剤についてスクリーニングする方法であって;
a)候補剤を、細胞死メディエーター蛋白質(CDMP)をコードする核酸を含む細胞
と接触させる工程であって、ここで、前記核酸は細胞型特異的発現調節エレメントに作動可能に連結されている工程;
b)前記現象に対する効果を検出する工程;
c)前記CDMPの活性のレベルが対照細胞と比べて調節されている場合、前記薬剤を前記現象を調節するのに効果的であるとして選択する工程
を含む、方法。
(項目50)
前記細胞が神経細胞、神経膠細胞または壁細胞である、項目49記載の方法。
(項目51)
前記細胞が乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、血管周囲細胞、またはシュワン細胞である、項目49記載の方法。
(項目52)
脱髄または血液脳関門障害に関連する現象を調節する生物活性剤についてスクリーニングする方法であって;
a)非−ヒトトランスジェニック動物に候補剤を投与する工程であって、ここで、前記現象は、細胞死メディエーター蛋白質(CDMP)をコードする核酸配列の発現に際して前記動物で起こり;ここで、前記核酸配列の発現は細胞特異的発現調節エレメントによって調節される工程;
b)前記CDMPを活性化して、前記動物における少なくとも1つの細胞においてアポトーシスをもたらす工程であって、ここで、前記細胞は脱髄または血液脳関門障害に関連する工程;
c)前記現象における前記薬剤の効果を検出する工程
を含む、方法。
(項目53)
工程b)後に、前記動物を前記現象から回復させる、項目52記載の方法。
(項目54)
前記現象が、前記動物における乏突起神経膠細胞、神経膠星状細胞、血管周囲細胞、シュワン細胞の喪失によって特徴付けられる、項目52記載の方法。
(項目55)
前記現象が髄鞘形成軸索の減少、または血液脳関門透過性の増加によって特徴付けられる、項目52記載の方法。
(項目56)
前記調節エレメントがCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、乏突起神経膠細胞−ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFα、RG5、P糖蛋白質、ニュールツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、スルファチド、PDGFR−β、PDGFR−α、またはプロテオリピド蛋白質(PLP)遺伝子から選択される遺伝子からのものである、項目49または52記載の方法。
(項目57)
前記脱髄障害が多発性硬化症である、項目52記載の方法。
(項目58)
前記効果を決定することがPCR、イムノアッセイ、ハイブリダイゼーションアッセイまたはそれらの組合せを含む、項目49または52記載の方法。
(項目59)
前記候補剤がアンチセンスオリゴヌクレオチド、ペプチド、抗体、リポソーム、低分子干渉RNA、低分子有機化合物または無機化合物である、項目49または52記載の方法。
(項目60)
前記少なくとも1つの細胞が、ニューロン細胞、神経膠細胞または壁細胞である、項目52記載の方法。
(項目61)
前記少なくとも1つの細胞が乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞、血管周囲細胞、またはシュワン細胞である、項目60記載の方法。
(項目62)
前記CDMPがカスパーゼ2、ガスパーゼ5、ガスパーゼ8、カスパーゼ9、カスパーゼ10またはカスパーゼ11である、項目49または52記載の方法。
(項目63)
前記CDMPがFK506型リガンド、FKBP12型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドについての結合ドメインを含むキメラ蛋白質である、項目49または52記載の方法。
(項目64)
前記CDMPの発現が誘導性である、項目49または52記載の方法。
(項目65)
前記CDMPのアポトーシス促進活性が誘導性である、項目49または52記載の方法。
(項目66)
前記活性が二量体化化学誘導因子(CID)によって誘導される、項目65記載の方法。
(項目67)
前記CDMPが誘導性カスパーゼ9(iCP9)である、項目49または52記載の方法。
(項目68)
前記CIDがAP20187である、項目67記載の方法。
(項目69)
多発性硬化症(MS)または多発性硬化症に関連するMS関連疾患に関連する現象を特徴付けるためのプロファイルデータセットを編集する方法であって:
a)細胞死メディエーター蛋白質(CDMP)をコードする核酸を含むトランスジェニック動物または細胞を供する工程であって、ここで、前記核酸はニューロンまたは神経膠細胞特異的発現調節エレメントに作動可能に連結されている工程;
b)前記CDMPを活性化し、それによりアポトーシスを誘導する工程;
c)前記活性化に続いて少なくとも1つの生存するニューロン細胞または神経膠細胞を得る工程;
d)前記生存する神経膠細胞または神経細胞におけるRNA転写物および/またはコードされる産物をプロファイルし、それにより多発性硬化症または多発性硬化症に関連するMS−関連障害に関連する現象を特徴付けるプロファイルデータセットを編集する工程
を含む、方法。
(項目70)
前記CDMPがカスパーゼ9またはカスパーゼ11である、項目69記載の方法。
(項目71)
前記CDMPのアポトーシス誘導活性が誘導性である、項目69記載の方法。
(項目72)
前記神経膠細胞特異的発現調節エレメントがCC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、およびプロテオリピド蛋白質(PLP)、CC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、乏突起神経膠細胞−ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFα、PDGFR−β、PDGFR−α、RG5、P糖蛋白質、ニュールツリン(NRTN)
、アルテミン(ARTN)、ペルセフィン(PSPN)、スルファチド、またはプロテオリピド蛋白質(PLP)遺伝子からなる群のものである、項目69記載の方法。
【図面の簡単な説明】
【0025】
本発明の新規な特徴は、特に、添付の請求の範囲に記載されている。本発明の特徴および利点は、本発明の原理が利用される例示としての実施形態を説明する以下の詳細な記載、および添付の図面を参照することによってより良く理解されるであろう。
【図1】操作されたSINレンチウイルスベクター(pΔcmvICP9/mEGFP(図1B)およびpΔmbpICP9/mEGFP)を示す。図1AはpΔcmv/mEGFPベクター構築体を提供する。第2世代のpΔmbpICP9/mEGFP(図1C)は、遺伝子発現を感染した乏突起神経膠細胞に制限するMBP特異的プロモーターを有し、その細胞特異的発現は乏突起神経膠細胞特異的細胞死を引き起こす。デュアルプロモーターの特徴は、感染した細胞が蛍光顕微鏡検査法を用いて容易に同定することができるような、上流遺伝子から独立したGFPの同時発現を可能とする。LTR=末端反復配列;pCMV=サイトメガロウイルスプロモーター配列;iCP9=操作された誘導性カスパーゼ−9遺伝子配列;pMND=修飾されたLTRプロモーター配列;EGFP=増強された緑色蛍光蛋白質遺伝子配列;pMBP=ミエリン塩基性蛋白質プロモーター配列。
【図2】細胞型選択的にアポトーシスを誘導するための種々の異なる組織特異的プロモーターを使用して作製されたレンチウイルスベクターを示す。前記ウイルスベクターは、典型的には、感染した細胞においてEGFP発現をもたらす。LRT=末端反復配列;pMND=ウイルスプロモーター配列;GFP=緑色蛍光蛋白質;pCMV=サイトメガロウイルスプロモーター配列;iCP9=誘導性カスパーゼ遺伝子配列。
【図3】濃縮されたレンチウイルスを直接的にCNSに適用することができ感染の程度および領域は、犠牲後に検出されたGFP陽性細胞の数、および脳または脊髄の薄切片に基づいて決定することができることを示す写真を提供する。このデータは、インビトロおよびインビボにおける特異的細胞死についての導入メカニズムを確立する。
【図4】本発明の特定の実施形態のグラフ表示である。該図は、本発明の1つの方法、および該方法についての特定の適用の一般的概略を示す。
【図5】GFP+細胞の持続性によって示されるように、CIDを培養物に加えた後に生存したままである対照ウイルスで感染させた細胞(上段)を示す写真を提供する。iCP9を発現するウイルスで感染させた細胞は、GFP+細胞の不存在によって示されるように、CID暴露後にアポトーシスを受ける(下段)。明視野における細胞の持続性は、感染した細胞のみに対するアポトーシスの特異性を示す。BF=明視野。
【図6】iCP9遺伝子をコードするウイルスの種々の希釈物に暴露され、次いで、CIDを培養基に加えた細胞を示すグラフである。4時間後に、アポトーシスELISAを行った。アポトーシスのレベルは、ウイルスの希釈に伴って減少した。というのは、より少数の細胞が感染し、iCP9を発現したからである。かくして、アポトーシスはiCP9遺伝子発現に相関した。X軸はウイルス希釈である。Y軸は405nmにおけるELISA吸収である。
【図7】pLpMBP(iCP9)MGに感染され、次いで、CIDに暴露されたラットの子に由来する混合皮質培養物を示す。4時間後に、細胞を染色して、細胞型を同定した。CID暴露(+CID)後におけるMBP+細胞の形態は、アポトーシスを示唆する。他の細胞型は、生きた細胞と合致する形態を維持した。これは、MBP発現細胞においてウイルスが細胞死を誘導することを示す。
【図8】pLpGFAP(iCP9)MGで感染させ、CIDに暴露した(+CID)パン精製GFAP+細胞(神経膠星状細胞)を示す。GFP+細胞は、CIDへの暴露から4時間後にEGFP+細胞の不存在によって示されるアポトーシスを受けた(上段顕微鏡写真)。CIDに暴露されなかった(−CID)並行培養物は、多数のEGFP+細胞を維持した(下段顕微鏡写真)。
【図9】混合皮質培養物をpLpPDGFR(iCP9)MGウイルスに感染させ、CIDに暴露した(+CID)ことを示す。4時間後に、A2B5+細胞は、生きた細胞の形態の喪失に基づいてアポトーシスを受けていると同定された。
【図10】ラットの脳梁へのpLpGFAP(iCP9)MGウイルスの注射からの結果を示す。CIDへの暴露から24時間後に、アポトーシスは、GFAP+細胞の喪失を伴う感染の部位において同定される(右側パネル)。しかしながら、感染、その後のビヒクルへの暴露は、アポトーシスまたはGFAP+細胞の喪失を生じさせなかった(左側パネル)。これは、ウイルスの結果、アポトーシスを介するGFAP+細胞(神経膠星状細胞)の特異的喪失をもたらすことを示唆する。
【図11】特異的に乏突起神経膠細胞を除去するためにラットの脳梁に注射されたpLpMBP(iCP9)MGウイルスを示す。CIDへの暴露(CID+)から24時間後に、アポトーシスは、TUNEL染色(最上段パネル)およびMBP染色細胞の不存在(第2段)を介して感染の部位において同定された。抗MBP抗体で染色されない領域は、抗GFAP抗体で陽性に染色され、これは、細胞がその領域に持続することを示す。黒色矢印によって示される領域は双方について陰性に染色され、針挿入によって生じた組織欠陥を表す。V=脳室。最下段パネルは、全ての感染した細胞はGFP+であるので感染の程度を示す。
【発明を実施するための形態】
【0026】
本発明において、トランスジェニック動物または細胞は、神経障害を研究するためのモデル系を提供する。前記モデル系は、ニューロンの健康および/または脱髄/髄鞘再形成を同定し、評価し、および/または定量するために使用することができる。さらに、本発明の組成物および方法は、ニューロンまたは髄鞘形成の健康または維持を調節し、促進し、または低下させる生物学的に活性な剤の同定および開発のための種々の実施形態において利用される。
【0027】
本発明の1つの態様は、ニューロン脱髄および/または髄鞘再形成に対する効果を評価するための特定の細胞型における細胞死の選択的誘導を含む。これにより、脱髄に対するニューロンの応答はインビボおよびインビトロにて人工的に誘導された炎症なくして定義することができる。
【0028】
ニューロンの喪失は、多発性硬化症に罹った患者での不能の進行における重要な因子である。さらに、ニューロンの喪失は炎症からかなり独立でき、ニューロン細胞およびミエリンを産生する乏突起神経膠細胞の間の共依存性関係の破壊後に起こり得る。
【0029】
したがって、1つの実施形態において、系(例えば、動物)を提供して、その共依存性乏突起神経膠細胞の喪失に対するニューロンの応答を特徴付ける。例えば、修飾されたレトロウイルスを利用して、誘導性自殺因子、または細胞死メディエーター蛋白質をコードする遺伝子を脳内の乏突起神経膠細胞に送達する。誘導性因子を動物に投与すると、自殺遺伝子は乏突起神経膠細胞(ミエリン産生細胞)がアポトーシスを受けるように誘導し、その結果、炎症によって誘導されない病巣脱髄がもたらされる。乏突起神経膠細胞およびミエリンの喪失に対するニューロンの応答を、次いで特徴付ける。脱髄に対するニューロンの応答を理解することによって、この事象後にニューロンを保護する介入を開発することができる。
【0030】
神経細胞のような細胞を、制御された細胞死について、被験体において、または細胞培養物において標的化することができる。いくつかの実施形態において、さらなる研究のために、例えば、エクスビボ技術を含めた、細胞ベースのまたは細胞培養設定で行われるアッセイのために、そのような細胞を本発明のトランスジェニック動物から単離する。
【0031】
本発明の種々の態様は、臨床的に発現する、乏突起神経膠細胞またはニューロン喪失のようなCNS中の病理学的変化を評価するための方法を提供する。
【0032】
本発明のいくつかの態様において、当該分野で公知の方法を用いて非ヒトトランスジェニック動物を操作して、細胞型特異的または誘導性発現調節エレメント(例えば、プロモーター/エンハンサー)に作動可能に連結された細胞死メディエーター蛋白質の発現を供する。いくつかの実施形態において、ヒトまたは非ヒトの細胞を操作して、細胞型特異的または誘導性発現調節エレメント(例えば、プロモーター/エンハンサー)に作動可能に連結された1以上の細胞死メディエーター蛋白質を発現させることができる。本発明の種々の態様において有用なそのような調節エレメント、ならびに本発明の組成物/方法で標的化することができる種々の細胞型は本明細書中により十分に記載されている。
【0033】
1つの実施形態において、本発明の非ヒトトランスジェニック動物(「テスト動物」)における1以上の細胞死メディエーター蛋白質の発現の結果、そのような動物において神経障害または増悪した神経障害がもたらされる。
【0034】
なおもう1つの実施形態において、1以上の細胞死メディエーター蛋白質の発現は、テスト動物の中枢神経系または末梢神経系におけるものである。
【0035】
本発明の1つの態様において、1以上の細胞死メディエーター蛋白質(CDMP)の発現の結果、テスト動物においてニューロンの変性がもたらされる。いくつかの実施形態において、そのようなニューロンは頸部神経節ニューロン、皮質ニューロン、セロトニンニューロン、後根神経節、結節性神経節ニューロン、脊髄運動ニューロン、中脳ドーパミン作動性ニューロン、中枢ノルアドレナリン作動性ニューロンまたは腸ニューロンを含む。
【0036】
種々の実施形態において、本発明の細胞死メディエーター核酸構築体を含む細胞は、限定されるものではないが、神経細胞または壁細胞である。
【0037】
いくつかの実施形態において、そのような細胞は神経膠細胞であり、限定されるものではないが、乏突起神経膠細胞、神経膠星状細胞、小神経膠細胞および/またはシュワン細胞を含む。いくつかの実施形態において、そのような細胞は、限定されるものではないが、血管周囲細胞、内皮細胞および/または平滑筋細胞を含む。
【0038】
本発明のもう1つの態様は、選択的および限定された細胞死後に、インビボにて、ニューロンおよび乏突起神経膠細胞の応答を決定する方法を対象とする。例えば、本発明の核酸構築体(例えば、細胞/組織特異的発現調節エレメントに作動可能に連結された細胞死メディエーター蛋白質をコードする核酸構築体)を、成体ラットに適用することができ、その結果、抗原/アジュバント刺激炎症によって合併されないCNS中の病巣に対するニューロンの応答を評価するためのモデル系がもたらされる。さらに、本発明は、病巣のサイズ、病巣の位置、病巣の数、および病巣の間の一時的な関係の制御を可能とする組成物および方法を提供する。
一般的な技術
本発明の実施は、特に断りのない限り、当業者の技量内にある、免疫学、生化学、化学、分子生物学、微生物学、細胞生物学、ゲノミックスおよび組換えDNAの慣用的な技術を使用する。Sambrook,Fritsch and Maniatis,MOLECULAR CLONING:A LABORATORY MANUAL,第2版(1989);CURRENT PROTOCOLS IN MOLECULAR BIOLOGY(F.M.Ausubel,ら編,(1987));METHODS IN ENZYMOLOGYのシリーズ(Academic Press Inc.);PCR2:A
PRACTICAL APPROARCH(M.J.MacPherson,B.D.
Hames and G.R.Taylor編(1995)),Harlow and Lane編(1988)ANTIBODIES,A LABORATORY MANUAL,and ANIMAL CELL CULTURE (R.I.Freshney編(1987))参照。
定義
明細書および特許請求の範囲で用いるように、単数形「ある」、および「前記」は文脈が明瞭にそうでないことを指示するのでなければ、複数への言及を含む。例えば、用語「ある細胞」はその混合物を含めた複数の細胞を含む。
【0039】
用語「ポリヌクレオチド」、「ヌクレオチド」、「ヌクレオチド配列」、「核酸」および「オリゴヌクレオチド」は相互交換可能に用いる。それらは、デオキシリボヌクレオチドまたはリボヌクレオチド、またはそのアナログいずれかである、いずれかの長さのヌクレオチドのポリマー形態をいう。ポリヌクレオチドはいずれかの三次元構造を有することができ、公知または未知のいずれかの機能を行うことができる。以下に示すのはポリヌクレオチドの非限定的例である:遺伝子または遺伝子断片のコーディング、または非コーディング領域、連鎖分析から定義された複数の遺伝子座(遺伝子座)、エクソン、イントロン、メッセンジャーRNA(mRNA)、トランスファーRNA、リボソームRNA、リボザイム、cDNA、組換えポリヌクレオチド、分岐ポリヌクレオチド、プラスミド、ベクター、いずれかの配列の単離されたDNA、いずれかの配列の単離されたRNA、核酸プローブ、およびプライマー。ポリヌクレオチドはメチル化ヌクレオチドおよびヌクレオチドアナログのような修飾されたヌクレオチドを含むことができる。もし存在すれば、ヌクレオチド構造に対する修飾は、ポリマーの組立前にまたは後に付与することができる。ヌクレオチド配列は非ヌクレオチド成分によって中断してもよい。ポリヌクレオチドは、さらに、標識成分でのコンジュゲーションなどによって、重合の後に修飾することができる。
【0040】
「ヌクレオチドプローブ」または「プローブ」とは、ハイブリダイゼーション反応においてその対応する標的ポリヌクレオチドを検出し、または同定するのに用いるポリヌクレオチドをいう。
【0041】
「ハイブリダイゼーション」とは、1以上のポリヌクレオチドが反応して複合体を形成し、これはヌクレオチド残基の塩基の間の水素結合を介して安定化される反応をいう。水素結合はワトソン・クリック塩基対合、フーグステン結合によって、あるいはいずれかの他の配列特異的様式で起こり得る。複合体はデュプレックス構造を形成する2つのストランド、マルチストランデッド複合体を形成する3以上のストランド、単一の自己ハイブリダイジングストランド、またはこれらのいずれかの組合せを含むことができる。ハイブリダイゼーション反応はPCRの開始、またはリボザイムによるポリヌクレオチドの酵素切断のようなより広汎なプロセスにおいて工程を構成することができる。
【0042】
ポリヌクレオチドに適用される用語「ハイブリダイズされた」とは、ヌクレオチド残基の塩基の間の水素結合を介して安定化される複合体を形成するポリヌクレオチドの能力をいう。水素結合はワトソン・クリック塩基対合、フーグステン結合によって、またはいずれかの他の配列特異的様式で起こり得る。前記複合体はデュプレックス構造を形成する2つのストランド、マルチストランデッド複合体を形成する3以上のストランド、単一の自己ハイブリダイジングストランド、またはこれらのいずれかの組合せを含むことができる。ハイブリダイゼーション反応はPCRの開始、またはリボザイムによるポリヌクレオチドの酵素切断のようなより広汎なプロセスにおいて工程を構成することができる。
【0043】
本明細書中で用いるように、「発現」とは、ポリヌクレオチドがmRNAに転写されるプロセスおよび/または前記転写されたmRNA(「転写物」ともいう)が引き続いて、
ペプチド、ポリペプチドまたは蛋白質に翻訳されているプロセスをいう。転写物およびコードされたポリペプチドは、集合的に、「遺伝子産物」といわれる。もしポリヌクレオチドがゲノムDNAに由来するならば、発現は、真核生物細胞におけるmRNAのスプライシングを含むことができる。
【0044】
被験体におけるヌクレオチド配列またはポリペプチド配列に適用される「異なって発現される」とは、対照において検出されるのと比較した場合のその配列の過剰発現または過小発現をいう。過小発現は、対照と比較した場合に、テスト被験体における検出可能な発現の不存在によって証明される、特定の配列の発現の不存在も含む。
【0045】
用語「ポリペプチド」、「ペプチド」および「蛋白質」は、本明細書中において相互交換可能に用いられて、いずれかの長さのアミノ酸のポリマーをいう。ポリマーは直線または分岐であってよく、それは修飾されたアミノ酸を含んでもよく、およびそれは非アミノ酸によって中断されていてもよい。前記用語は修飾(例えば、ジスルフィド結合形成、グリコシル化、脂質化、アセチル化、リン酸化、または標識成分とのコンジュゲーションのようないずれかの他の操作)されているアミノ酸ポリマーも含む。本明細書中で用いるように、用語「アミノ酸」とは、グリシン、およびDまたはL双方の光学異性体、およびアミノ酸アナログおよびペプチドミメティクスを含めた、天然および/または非天然または合成アミノ酸のいずれかをいう。
【0046】
本明細書中で用いるように、「髄鞘形成細胞」とは、神経系において軸索を絶縁するミエリンを産生することができる細胞をいう。例示的な髄鞘形成細胞は中枢神経系においてミエリンを産生することを担当する乏突起神経膠細胞、および末梢神経系においてミエリンを産生することを担当するシュワン細胞である。
【0047】
用語「髄鞘再形成する」または「髄鞘再形成」とは、例えば、脱髄傷害に応答してのミエリンの再生をいう。
【0048】
「被験体」、「個体」または「患者」は本明細書中においては相互交換的に用いられ、これは、脊椎動物、好ましくは哺乳動物、より好ましくはヒトをいう。哺乳動物は、限定されるものではないが、ネズミ、サル、ヒト、農場動物、競技動物およびペットを含む。組織、細胞およびそれらの子孫またはインビボで得られたかもしくはインビトロで培養された生物学的実体も含まれる。
【0049】
本明細書中に記載される動物モデルまたは細胞培養アッセイで使用される「生物学的に活性な剤」または「生物活性剤」は、単純なまたは複雑な有機または無機分子、ペプチド、ペプチドミメティック、蛋白質(例えば、抗体)、リボソーム、低分子干渉RNA、またはポリヌクレオチド(例えば、アンチセンス)のような生物学的または化学的化合物よりなる群から選択することができる。さらに、そのような薬剤は複雑な有機または無機分子を含み、粗製または精製された植物抽出物のような化合物の不均一混合物を含むことができる。
【0050】
「プロモーターエレメント」はそのような配列に連結された遺伝子の転写を促進する調節配列である。調節配列はエンハンサー配列またはその機能的部分を含むことができる。
【0051】
「対照」は比較目的にて実験で用いる代替被験体、細胞または試料である。
細胞死メディエーター蛋白質(CDMP)
本発明は、細胞または組織特異的に発現された細胞死メディエーター蛋白質(CDMP)を含む方法および組成物を提供する。例えば、発現はCNSまたはPNSに対して、あるいは特異的神経細胞または壁細胞に対して特異的であってよい。発現は動物におけるも
のであり得るが、動物におけるそのような発現は、例えば、髄鞘形成または髄鞘再形成において役割を持つ細胞において特異的に細胞死を誘導することによって脱髄を引き起こすことができる。発現はインビトロでもあり得る。細胞死メディエーター蛋白質の発現は、インビボまたはインビトロにて誘導性であり得る。
【0052】
細胞死メディエーター蛋白質は細胞死またはアポトーシスの媒介において役割を有する。CDMPは、例えば、アポトーシスに直接的に影響する蛋白質の活性を調節することによって、直接的にまたは間接的にアポトーシスに影響し得る。例えば、CDMPはSMAC(カスパーゼの第2のミトコンドリア由来アクチベーター)、IAP(アポトーシス蛋白質の阻害剤)、カスパーゼ、またはそれらのモジュレーターであり得る。他の実施形態において、CDMPはTNF(腫瘍壊死因子)受容体、およびFas受容体、およびTRAIL受容体経路のような他の死滅受容体シグナリング経路のモジュレーターであり得る。CDMPは、グランザイムB、またはグランザイムBのモジュレーターを含めたカスパーゼのアクチベーターでもあり得る。
【0053】
種々の実施形態において、細胞死メディエーター蛋白質は核酸構築体によってコードされる。いくつかの実施形態において、核酸構築体は1以上のCDMPをコードすることができる。CDMPは同一または異なることができる。例えば、単一の核酸は、タンデムにカスパーゼ9をコードする2つの配列のような上記の蛋白質の2つをコードすることができ、あるいはカスパーゼ9およびカスパーゼ3をコードすることができる。本発明の核酸構築体はカスパーゼ2、5、8、9、10または11、それらのプロ酵素形態、またはその誘導体をコードすることができる。さらなる実施形態において、そのようなカスパーゼ蛋白質をコードする配列を、架橋剤化合物と反応性である二量体化ドメインを含むように修飾する。かくして、いくつかの実施形態において、野生型カスパーゼ配列を修飾して、架橋剤化合物に対して選択的に反応性である二量体化ドメインを含むキメラ配列を産生する。そのような二量体化ドメインの例は、その関連部分をここに引用してその全体を援用する、米国特許出願第20050187177号および米国特許第6,984,635号に開示されているものを含む。
二量体化
いくつかの実施形態において、二量体化は生物学的プロセス(例えば、カスパーゼ9の活性化を介するアポトーシス)を活性化し、種々のキメラ蛋白質を利用することができる。キメラ蛋白質は、種々のドメインが(例えば、天然では一緒に連結しては見出されない異なる源に由来する)相互に対して異種である点で組換体である。天然では直接的に相互に連結しては見出されない、例えば、特定のドメインまたは発現制御配列をコードする異種成分を含む組換えDNA構築体は、インビトロまたはインビボにて標的宿主細胞を遺伝子工学的に作成するのに用いられる。そのように操作された細胞は、少なくとも1つのそのようなキメラ蛋白質、またはキメラ蛋白質をコードする遺伝子構築体の第1のシリーズを含有する。1つのそのようなDNA構築体は、(b)異種のさらなる(「作用」)蛋白質ドメインに融合した(a)(選択されたリガンドに結合することができる)少なくとも1つの受容体ドメインを含むキメラ蛋白質をコードする。前記リガンドは、好ましくは、ほぼ10−6から<10−9までの範囲のKd値でもって、キメラ蛋白質内の2(またはそれ以上)の受容体ドメインに結合することができ、好ましくは、ほぼ1kDa、5kDa、10kDa、15kDaまたは20kDa未満の分子量を有する非蛋白質の化合物である。そのようにオリゴマー化されたキメラ蛋白質受容体のドメインは同一または異なってもよい(すなわち、ホモ二量体化またはヘテロ二量体化)。リガンドへの暴露および受容体のオリゴマー化に際して、キメラ蛋白質は生物学的プロセス(例えば、補体カスケード)を開始することができる。コードされたキメラ蛋白質は、さらに、キメラ蛋白質を所望の細胞区画に向けることができる細胞内標的化ドメイン(例えば、蛋白質に指令して核と会合させる配列)を含むことができる。
【0054】
キメラ蛋白質が結合することができるリガンドのタイプの例は、FK506タイプのリガンド、サイクロフィリンタイプのリガンド(例えば、サイクロスポリンAタイプのリガンド)、テトラサイクリンまたはステロイドリガンドを含む。そのような結合はホモタイプ(同一)またはヘテロタイプ(異なる)のキメラ蛋白質分子のオリゴマー化を引き起こす。そのようなリガンドおよび/または受容体ドメインの例は、その各々についての開示をここに引用してその全体を援用する、米国特許第5534418号、同第5002753号、同第5298429号、同第6235872号、同第6656971号、同第7196182号、同第7101357号、同第7109317号、同第7153685号および同第7169564号に開示されている;また、Straathofら、Blood,(2005)105:4247−4254;Belshawら、Proc Natl Acad Sci.(1996)93:4604−4607も参照。
【0055】
かくして、本明細書中に記載された方法を利用し、標的細胞は、(i)選択されたオリゴマー化リガンドに結合することができる少なくとも1つの受容体ドメイン、および(ii)カスパーゼ9をコードする、受容体ドメインに対して異種であるもう1つの蛋白質ドメインを含むキメラ蛋白質をコードするDNA構築体を含むことができる。よって、選択されたリガンドへの暴露に続き、そのような標的細胞において発現されたカスパーゼ9のオリゴマー化は、アポトーシスプログラムを誘導し、前記DNA構築体を含む細胞を殺傷する。
【0056】
カスパーゼ9は、典型的には、アポトーシスプロテアーゼ活性化因子1(Apaf−1)とのチトクロームCおよびATP依存性相互作用後にダイマーとして機能する蛋白質である。カスパーゼ9の二量体化は、このポリペプチドが下流エフェクター分子を活性化するのを可能とし、最終的に、当該細胞のアポトーシスをもたらす(Springer,J
Biochem.Mol.Biol.(2002)35:94−105)。
【0057】
かくして、種々の実施形態において、細胞死メディエーター蛋白質は、FK506タイプのリガンド、サイクロスポリンAタイプのリガンド、テトラサイクリンまたはステロイドリガンドのための結合ドメインを含むカスパーゼ9キメラ蛋白質である。1つの実施形態において、カスパーゼ9はFKBP12結合ドメインを含む。さらなる実施形態において、キメラ構築体(例えば、カスパーゼ9)で利用される結合ドメインは、リガンド(例えば、二量体化化学誘導因子)に結合するように最適化することができる。
【0058】
誘導性カスパーゼ9 cDNA(iCP9)は、カスパーゼ動員ドメイン(CARD)の除去後に、カスパーゼ9 cDNA配列(GenBank AH002 818)をFK506結合蛋白質(FKBP)配列(GenBank AH002818)に連結させることによって操作した。CARD配列の不存在は蛋白質の生理学的二量体化を妨げ、それにより、カスパーゼ細胞死カスケードの自発的な開始を妨げる。FKBP配列への融合は、二量体化化学誘導因子(CID)の投与後に化学的に誘導された凝集を可能とする。CID、AP20187(ARIAD Pharmaceuticals,Cambridge,MA)は、内因性FKBPとの相互作用を妨げるように改変されている非毒性合成FK506アナログである。CIDシステムは、従前に、インビトロおよびインビボの両方で無関係なシステムにおいて利用されている(Straathofら、Blood.(2005)105:4247−4254)。
【0059】
本発明の種々の実施形態において、本発明の核酸構築体は、限定されるものではないが、米国特許第6,982,082号に開示されたもののような、アデノウイルス、アデノウイルス関連ウイルス、ネズミ白血病ウイルス、レンチウイルス、泡沫状ウイルス、狂犬病ウイルスまたは当該分野で公知の他のウイルスベクターを含めた、ウイルスベクターを介して(培養中のまたはインビボで)細胞に送達される。
調節された発現
本発明の種々の態様において、細胞または組織特異的および/または誘導性発現調節エレメントは細胞死メディエーター蛋白質に作動可能に連結されて、発現に際して選択的な細胞死をもたらす。前記したように、組織特異的および細胞特異的調節配列が、中枢神経系において導入遺伝子を発現させるために入手可能である。調節配列は、特定の細胞型において中枢神経系または末梢神経系での導入遺伝子の異所性発現を可能とする。例えば、選択的死滅は、限定されるものではないが、乏突起神経膠細胞、小神経膠細胞、シュワン細胞または神経膠星状細胞のような細胞において達成することができる。
【0060】
調節配列の例示的な発現は、限定されるものではないが、CC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、ミエリン関連糖蛋白質(MAG)、ミエリン乏突起神経膠細胞糖蛋白質(MOG)、乏突起神経膠細胞ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2),GFAP、AQP4、PDGFR−α、PDGF−α、RG5、p糖蛋白質、ノイルツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、スルファチド、2(VEGFR2)、スーパーオキシドジスムターゼ(SOD1)、チロシンヒドロキシラーゼ、ニューロン特異的エノラーゼ、パルキン遺伝子(PARK2)、パルキン共調節遺伝子(PAGRG)、ニューロン特異的Tα1 α−チューブリン(Tα1)、小胞モノアミントランスポーター(VMAT2)、およびα−シヌクレイン(SNCA)、PDGFR−β、またはプロテオリピド蛋白質(PLP)を含めた遺伝子から選択される調節配列を含む。
【0061】
神経細胞特異的プロモーターのさらなる例は、その関連開示をここに引用して援用する、米国特許出願公開番号2003/0110524;2003/0199022;2006/0052327、2006/0193841、2006/0040386、2006/0034767、2006/0030541;米国特許第6472520号、同第6245330号、同第7022319号、および同第7033595号に開示されているように、当該分野で公知である。また、ウェブサイト<chinook.uoregon.edu/promoters.html>または<tiprod.cbi.pku.edu.cn:8080/index>(ある細胞/組織に対して特異的な遺伝子のプロモーターをリストする);およびPattersonら、J.Biol.Chem.(1995)270:23111−23118も参照。
【0062】
かくして、本発明の1つの態様は、細胞死の一時的制御のための誘導性/細胞型特異的発現調節エレメントの利用であり、これは今度は、MS、ALSおよびパーキンソン病のような神経障害に関連するものとして、ニューロンの健康/維持に対する特定の細胞の効果を評価し、またはニューロンの健康/維持に対する候補分子の効果を評価するのに利用される。
【0063】
細胞死メディエーター蛋白質の発現は、(例えば、エフェクター分子を局所投与する場合)細胞/組織特異的プロモーターを利用するもの以外の発現系を利用することによって一時的に調節することもできる。したがって、いくつかの実施形態において、細胞死メディエーター蛋白質をコードする遺伝子は、tet応答性プロモーターのような制御可能なプロモーターエレメントに作動可能に連結させることができる。例えば、望まれる場所および時期に、誘導性剤(例えば、テトラサイクリンまたはそのアナログ)を細胞または被験体に投与して、細胞/組織特異的に細胞死メディエーター蛋白質の発現を誘導することができる(例えば、単にテトラサイクリンが局所化された/限定された様式で送達される)。そのような系は、テトラサイクリンの存在が発現を阻害する「オフスイッチ」系、またはテトラサイクリンの存在が発現を誘導するようにE.coli TetRの変異体が
用いられる「可逆性」Tet系を含めることによって、真核生物細胞における遺伝子発現の密接な制御を提供することができる。これらの系は、例えば、Gossen and Bujard(Proc.Natl.Acad.Sci.U.S.A.(1992)89:5547)において、および、Pujardらによる米国特許第5,464,758号;同第5,650,298号;および同第5,589,362号に開示されている。
【0064】
誘導性プロモーターのさらなる例は、限定されるものではないが、MMTV、ヒートショック70プロモーター、GAL1−GAL10プロモーター、メタロチオネイン誘導性プロモーター(例えば、銅誘導性ACE1;他の金属イオン)、ホルモン応答エレメント(例えば、グルココルチコイド、エストロゲン、プロゲストロゲン)、フォルボールエステル(TREエレメント)、カルシウムイオノフォア応答性エレメント、または非カップリング蛋白質3、αヒト葉酸受容体、ホエイ酸性蛋白質、前立腺特異的プロモーター、ならびに米国特許第6,313,373号に開示されたものを含む;また、(遺伝子/活性によって分類された15,000を超える異なるプロモーター配列を含むデータベースを提供する)オンライン上での<biobase/de/pages/products/transpor.html>;およびChenら、Nuc.Acids.Res.2006,34:Database issue,D104−107も参照。
【0065】
なお他の誘導性プロモーターは、成長ホルモンプロモーター;アデノウイルスE1A蛋白質によって誘導可能なアデノウイルス初期遺伝子プロモーター、またはアデノウイルス主要後期プロモーターのようなヘルパーウイルスによって誘導可能なプロモーター;VP16または1CP4のようなヘルペスウイルスタンパク質によって誘導可能なヘルペスウイルスプロモーター;ワクシニアまたはポックスウイルスRNAポリメラーゼによって誘導可能なプロモーター;または、各々、T7、T3、またはSP6 RNAポリメラーゼによって誘導可能な、T7、T3およびSP6のようなバクテリオファージプロモーターを含む。
【0066】
他の実施形態において、構成的プロモーターが望ましいであろう。例えば、アデノウイルス主要後期プロモーター、サイトメガロウイルス即時型初期プロモーター、βアクチンプロモーター、またはβグロビンプロモーターを含めた、本発明で用いるのに適した多くの構成的プロモーターがある。多くの他のものが当該分野で知られており、本発明で用いることができる。なおさらなる実施形態において、調節エレメントを改変し、または修飾して、発現を増強させる(すなわち、プロモーターの強度を増加させる)ことができる。例えば、エンハンサー機能を含むイントロン性配列を利用して、プロモーターの機能を増加させることができる。ミエリンプロテオリピド蛋白質(PLP)遺伝子はエンハンサーエレメントとして機能するイントロン性配列を含む。この調節エレメント/領域ASE(アンチサイレンサー/エンハンサー)は、エクソン1 DNAからほぼ1kb下流に位置し、ほぼ100bpを含む。Mengら、J Neurosci Res.(2005)82:346−356参照。
【0067】
さらに、特定の細胞内位置における導入遺伝子の発現が望まれる場合、導入遺伝子は、当該分野で広く実施されている組み換えDNA技術によって対応する細胞内局所化配列に作動可能に連結させることができる。例示的な細胞内局所化配列は、限定されるものではないが、(a)細胞の外側へ遺伝子産物の分泌を指令するシグナル配列;(b)原形質膜、または細胞の他の膜区画への蛋白質の付着を可能とする膜係留ドメイン;(c)コードされた蛋白質の核へのトランスロケーションを媒介する核局所化配列;(d)主としてコードされた蛋白質をERに制限する小胞体滞留配列、(例えば、KDEL配列);(e)蛋白質は、蛋白質を細胞膜に会合させるようにファルネシル化されるように設計することができる;または(f)コードされた蛋白質産物の異なる細胞内分布において役割を演じるいずれかの他の配列を含む。
【0068】
いくつかの実施形態において、遺伝子的に修飾された細胞を区別するためのマーカーを検出することができる。そのようなマーカーは、限定されるものではないが、CC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、ミエリン関連糖蛋白質(MAG)、ミエリン乏突起神経膠細胞糖蛋白質(MOG)、乏突起神経膠細胞ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、ガラクトセレブロシド(GalC)、スルファチド、PDGFR−β、PDGFR−α、PDGF−α、およびプロテオリピド蛋白質(PLP)を含む。
【0069】
種々の実施形態において、乏突起神経膠細胞の選択的アポトーシスのような選択的細胞死が誘導される動物もまた、脱髄/髄鞘再形成状態に対する効果についてアッセイすることもできる。例えば、脱髄/髄鞘再形成現象は、当該分野で知られているように免疫組織化学手段または蛋白質分析によって観察することができる。例えば、テスト動物の脳の切片を、乏突起神経膠細胞マーカーを特異的に認識する抗体で染色することができる。もう1つの態様において、乏突起神経膠細胞マーカーの発現レベルは、免疫ブロッティング、ハイブリダイゼーション手段および増幅手法ならびに当該分野でよく確立されたいずれかの他の方法によって定量することができる。例えば、その各々の開示をここに引用して援用する、Mukouyamaら、Proc.Natl.Acad.Sci.(2006)103:1551−1556;Zhangら(2003)、supra.;Girardら、J.Neuroscience.(2005)25:7924−7933;および米国特許第6,909,031号;同第6,891,081号;同第6,903,244号;同第6,905,823号;同第6,781,029号;および同第6,753,456号。
【0070】
なお他の実施形態において、選択的細胞死が血管周囲細胞のような血液脳関門の維持で重要である細胞で起こる動物。例えば、動物における、PDGFR−βプロモーターの制御下でのカスパーゼ9またはiCP9のようなカスパーゼの発現を用いて、その浸透性、維持または完全性のように、血液脳関門(BBB)に対する効果についてアッセイすることができる。例えば、テスト動物の脳の切片を、接着結合蛋白質のようなBBBマーカーを特異的に認識する抗体で染色することができる。例えば、検出することができるマーカーはオクルジン、およびクラウジン2、クラウジン5、クラウジン6、クラウジン7、クラウジン10、クラウジン12、クラウジン15および/またはクラウジン19のようなクラウジンを含む。もう1つの実施形態において、接着結合蛋白質マーカーの発現レベルは、イムノブロッティング、ハイブリダイゼーション手段、および増幅手法ならびに当該分野で確立された他のいずれかの方法によって定量することができる。
【0071】
BBBの完全性または透過性は、血液−脳関門透過性を決定し、可視化し、測定し、同定し、または定量するのに利用される、当該分野で公知のいずれかの色素、マーカー、またはトレーサーのようなインジケーターを用いてアッセイすることもできる。非限定的な例として、エバンスブルーおよびナトリウムフルオレセインが挙げられる。そのようなインジケーターの例は当業者に明らかであり、無傷のBBBを横切ることができないが、より透過性のBBBを横切ることができ、ならびに同定し、測定し、または定量することができる実質的にいずれの化合物も含む。
【0072】
インジケーターは、BBB透過性の変化を決定するのに利用される酵素、トレーサーまたはマーカーであり得るが、非限定的な例は以下の通りである:
【0073】
【化1】
【0074】
【化2】
色素、トレーサーまたはマーカーのさらなる例は、デキストラン、ビオチン、フィブリノーゲン、アルブミン、クーンス(Coons)反応を用いる血中グロブリン、テキサスレッド結合デキストラン(70,000daMW)、Na(+)−フルオレセイン(MW376)またはフルオレセインイソチオシアネート(FITC)標識デキストラン(MW62,000または145,000)、または分子質量10,000DaのFITC−標識デキストラン(FITC−デキストラン−10K)を含む。
トランスジェニック動物
本発明の1つの態様において、目的の蛋白質をコードする遺伝子に作動可能に連結された神経細胞特異的調節エレメントをコードするトランスジェニックヌクレオチド配列がゲノムに安定に組み込まれたトランスジェニック動物が生成される。前記遺伝子の発現は誘導性プロモーターの制御下とすることができる。いくつかの実施形態において、細胞特異性は神経細胞(例えば、神経膠細胞、好ましくは神経膠星状細胞、乏突起神経膠細胞および/またはシュワン細胞;頸部神経節ニューロン、後根神経節細胞、結節性神経節ニューロン、脊髄運動ニューロン、中脳ドーパミン作動性ニューロン、中枢ノルアドレナリン作動性ニューロンおよび腸ニューロンのようなニューロン細胞)に対するものである。
【0075】
好ましい実施形態において、目的の蛋白質をコードする遺伝子は細胞死メディエーター蛋白質をコードする。かくして、本発明のトランスジェニック動物は、細胞型特異的発現
調節エレメントに作動可能に連結された細胞死メディエーター蛋白質(CDMP)をコードするヌクレオチド配列を含むことができ;ここで、動物は、細胞型特異的発現調節エレメントに作動可能に連結されたCDMPをコードするヌクレオチド配列がない動物に対してより大きな程度の神経障害を呈する。神経障害は多発性硬化症のようなニューロン脱髄であり得る。トランスジェニック動物は、対照動物のそれに対してアポトーシス乏突起神経膠細胞の増加を呈することができ、CDMPの発現は中枢神経系に異所的に制限することができる。いくつかの実施形態において、神経障害は、血液脳関門の欠陥によるものであり、またはBBBおよび脱髄双方の欠陥によるものである。例えば、神経障害は筋委縮性側索硬化症(ALS)、多発性硬化症(MS)、免疫機能不全筋肉中枢神経系破壊、筋ジストロフィ(MD)、アルツハイマー病、パーキンソン病、ハンチントン病、脳虚血症、脳性麻痺、皮質基底神経節変性、クロイツェルフェルト・ヤコブ症候群、ダンディー・ウォーカー症候群、認知症、血管性脳炎、脳脊髄炎、癲癇、本態性振せん、クル・ランダウ・クレッフナー症候群、レーヴィ体病、マチャド・ジョセフ病、メージ症候群、偏頭痛障害、ポリオ、多系統萎縮症、髄膜炎、ドラガー症候群、チューレット症候群、ハレルフォルデン・シュパッツ症候群、水頭症、オリーブ橋小脳萎縮、核上麻痺、または脊髄空洞症であり得る。
【0076】
1つの実施形態において、細胞死メディエーター蛋白質(CDMP)はカスパーゼ2、5、8、9、10または11である。もう1つの実施形態において、そのような細胞死メディエーター蛋白質の1以上は神経細胞または壁細胞のような細胞で発現される。例えば、CDMPは、神経膠細胞のような、動物の神経細胞に標的化され得る。神経細胞は、限定されるものではないが、乏突起神経膠細胞、神経膠星状細胞、シュワン細胞、または小神経膠細胞であり得る。標的化は、動物の壁細胞、例えば、限定されるものではないが、血管周囲細胞、内皮細胞または平滑筋細胞のような壁細胞に対するものであり得る。いくつかの実施形態において、1以上の標的細胞の組合せが選択される(例えば、神経細胞および壁細胞、または異なる神経細胞、または異なる壁細胞)。
【0077】
いくつかの実施形態において、CDMPの発現は誘導性である。例えば、CDMPは、誘導性プロモーターを持つ調節エレメントに作動可能に連結していてもよい。アポトーシスの促進のようなCDMPの活性もまた誘導性であってよい。例えば、CIDによって誘導されるCDMPをコードするウイルスベクターは、非ヒト成体動物(例えば、ラット)の脊髄または脳に直接的に注射することができ、CIDは投与される。CIDはウイルスベクターの投与に同時に、またはそれに引き続いて投与することができる。
【0078】
病巣感染に由来する、乏突起神経膠細胞の喪失の病巣面積の程度は、投与されるウイルスの量によって制御することができる。乏突起神経膠細胞の選択的喪失に対する生存細胞の応答は、当業者に明らかである、組織学、分子および電気生理学アッセイを用いて評価することができる。(例えば、乏突起神経膠細胞の死滅によって測定される)病巣サイズの程度は、病巣部位における軸索喪失と相関させることができる。同様に、その軸索が細胞喪失の領域を通過するニューロンを評価することによって、一旦超過したら、離れたニューロンの喪失をもたらす脱髄閾値を存在するか否かを決定することができる。従って、本発明の方法は、CNS乏突起神経膠細胞の喪失に続いての分子応答に対する洞察を提供し、候補剤の同定が、多発性硬化症においてニューロン保護に対する時間的および環境ベースのアプローチを行うのを可能とする。1つの実施形態において、ウイルスベクターはレンチウイルスベクターである。さらなる実施形態において、ウイルスベクターは(後にさらに記載する)iCP9レンチウイルスである。
【0079】
本発明の動物モデルは、中枢および末梢神経系を含めた動物の神経系においてニューロン脱髄状態を生じさせる手法に使用できるいずれの非ヒト脊髄動物を含む。好ましいモデル生物は、限定されるものではないが、哺乳動物、霊長類、およびげっ歯類を含む。好ま
しいモデルの非限定的例は、ラット、マウス、モルモット、ネコ、イヌ、ウサギ、ブタ、チンパンジー、およびサルである。テスト動物は野生型またはトランスジェニックであり得る。1つの実施形態において、動物はげっ歯類である。なおさらなる実施形態において、動物はマウスである。もう1つの実施形態において、動物はサル種である。なおさらなる実施形態において、動物は、神経学的疾患を調べるのに通常利用されるキヌザルである(例えば、Eslamboi,A.Brain Res Bull.(2005)68,140−149;Kirikら、Proc.Natl.Acad.Sci.(2004)100:2884−89)。
【0080】
トランスジェニック動物は広く2つのタイプ:「ノックアウト」および「ノックイン」にカテゴリー化することができる。「ノックアウト」は、好ましくは、例えば、標的遺伝子の一部を標的遺伝子に無関係な配列で置き換えることによって、標的遺伝子の発現が微々たるものとなり、または検出できないものとなるように、標的遺伝子の機能の減少をもたらすトランスジェニック配列の導入を介する標的遺伝子の改変を有する。「ノックイン」は、例えば、標的遺伝子の調節配列を作動可能に挿入することによる、標的遺伝子の改変された発現を有する動物であり;または例えば、標的遺伝子を修飾されたコピーで置き換えることによる、標的遺伝子の修飾されたコピーを発現する動物である。修飾は標的遺伝子の失欠または変異であり得る。ノックインまたはノックアウト動物は標的遺伝子に対してヘテロ接合性またはホモ接合性であり得る。ノックアウトおよびノックインの双方は、ダブルノックインまたはダブルノックアウトとしても知られた「バイジェニック」であり得る。バイジェニック動物は、改変されている少なくとも2つの宿主細胞遺伝子を有する。1つの実施形態において、バイジェニック動物は、細胞特異的細胞死メディエーター蛋白質をコードする導入遺伝子、および細胞特異的マーカー遺伝子をコードするもう1つのトランスジェニック配列を保持する。本発明のトランスジェニック動物は広くノックインとして分類することができる。いくつかの実施形態において、動物の特異的細胞型を標的としてもよい。例えば、標的細胞は神経細胞または壁細胞であり得る。種々の実施形態において、神経細胞は乏突起神経膠細胞、シュワン細胞、小神経膠細胞または神経膠星状細胞を含み、壁細胞は内皮細胞、血管周囲細胞または平滑筋細胞を含む。
【0081】
胚顕微操作のための技術の進歩は、いまや、同様に、受精した哺乳動物卵子への異種DNAの導入を可能とする。例えば、全能性または多能性幹細胞は、マイクロインジェクション、リン酸カルシウム媒介沈殿、リポソーム融合、レトロウイルス感染または他の手段によって形質転換することができる。次いで、形質転換された細胞を胚に導入し、胚は発生してトランスジェニック動物となる。好ましい実施形態において、発生する胚を、導入遺伝子を発現するトランスジェニック動物を感染した胚から生じさせることができるように、所望の導入遺伝子を含有するウイルスベクターで感染させる。もう1つの好ましい実施形態において、所望の導入遺伝子を、好ましくは、単一細胞段階で、胚の前核または細胞質に共注射し、胚は発生して成熟トランスジェニック動物となる。トランスジェニック動物を生成させるためのこれらおよび他の変種の方法は当該分野において確立されており、本明細書中においては詳細に記載しない。例えば、米国特許第5,175,385号および同第5,175,384号参照。
【0082】
従って、本発明は、細胞特異的に髄鞘再形成を検出し、および定量するために動物モデルを用いる方法を提供する。そのような実施形態において、前記方法は:(a)細胞における細胞死メディエーター蛋白質の発現によって細胞型特異的に細胞死を誘導する工程;(b)細胞死を起こすために時間をおく工程;(c)動物において髄鞘形成/髄鞘再形成の調節を決定する工程を含む。生物活性剤をスクリーニングし、生物活性剤の髄鞘形成または髄鞘細形成の調節を決定するためにトランスジェニック動物を用いてもよい。
動物実験
動物モデルを、神経障害現象(例えば、脱髄障害)に関連する標的細胞において選択的
または制御された細胞死をアッセイする本発明の1以上の方法で利用する。前記現象は、髄鞘を有する軸索の減少、乏突起神経膠細胞マーカー、神経膠星状細胞マーカーまたはシュワン細胞マーカーの低下によって特徴付けられる脱髄障害と関連付けることができる。脱髄障害は遺伝的、または病原体またはウイルスによって罹るものであり得る。
【0083】
動物モデルを用いて、神経障害を調節する生物活性剤についてスクリーニングすることができる。例えば、本明細書中に開示されたモデル系の適用は、ラットCNSに適用した場合、インビボでの脱髄に対するニューロン応答の研究、および脱髄障害の処置の開発を可能とするより正確なモデルを提供する。
【0084】
本発明は、動物に投与されて、細胞死メディエーター蛋白質の発現および、よって、標的細胞の選択的除去を達成する細胞/組織特異的または誘導性プロモーターに作動可能に連結された細胞死メディエーター蛋白質をコードする核酸構築体を提供する。そのような発現は、異所的に維持された導入遺伝子送達ビヒクルを介して達成することができるか、あるいはそのような導入遺伝子は、当該分野で公知の方法を用いて動物のゲノムに組み込むことができる。例えば、発現は、エピソームにより、またはCDMPをコードする核酸の安定な組込みを介して達成することができよう。
【0085】
種々の実施形態において、細胞死メディエーター蛋白質をコードする遺伝子(「自殺遺伝子」)を含む核酸構築体は、細胞/組織特異的な、または誘導性である発現調節エレメントに作動可能に連結される。種々の実施形態において、標的細胞は神経細胞または壁細胞である。神経細胞は、限定されるものではないが、乏突起神経膠細胞、神経膠星状細胞、シュワン細胞、または小神経膠細胞であり得る。壁細胞は、限定されるものではないが、血管周囲細胞、内皮細胞および平滑筋細胞を含む。いくつかの実施形態において、1以上の標的細胞の組合せが選択される(例えば、神経細胞および壁細胞、または異なる神経細胞、または異なる壁細胞)。
【0086】
なお他の実施形態において、標的細胞は、限定されるものではないが、Bリンパ球またはTリンパ球細胞のような免疫細胞である。なおさらなる実施形態において、Bリンパ球および/またはTリンパ球は標的細胞ではない。
【0087】
本発明の1つの態様において、細胞死メディエーター蛋白質は、限定されるものではないが、カスパーゼ2、5、8、9、10または11を含めたカスパーゼ蛋白質である。1つの実施形態において、本発明の核酸構築体は、標的細胞において発現させることができる、少なくとも2つの異なるカスパーゼ蛋白質を含む。CDMPは、例えば、FK506型リガンド、サイクロスポリンA型リガンド、テトラサイクリンまたはステロイドリガンドに対する結合ドメインを有するキメラのCDMPであってよい。1つの実施形態において、カスパーゼ9は、FKBP12結合ドメインを含む。さらなる実施形態において、CDMPのアポトーシス促進活性は誘導性であってよく、例えば、キメラ構築体(例えば、カスパーゼ9)で利用される結合ドメインは、カスパーゼ9活性および、かくして、アポトーシスを促進する、二量体化化学誘導因子に結合するように最適化することができる。
【0088】
本発明のもう1つの態様において、髄鞘形成/髄鞘再形成調節活性について生物学的に活性な剤をテストする方法が提供される。
【0089】
1つの実施形態において、神経障害関連現象の調節について候補剤をテストする方法は、細胞死メディエーター蛋白質の発現によってテスト動物において細胞死を誘導する工程、髄鞘形成/髄鞘再形成に対する効果を評価するのに十分な時間おいておく工程、テスト生物活性剤を投与する工程、ならびに、投与無しと比較した髄鞘形成/髄鞘再形成に対する効果を決定し、かくして、テスト剤が髄鞘形成/髄鞘再形成を増強し/低下させるか否
かを決定するする工程を含む。
【0090】
かくして、いくつかの実施形態において、前記方法は:(a)細胞型特異的に細胞死を誘導する工程;(b)本発明のトランスジェニック動物において脱髄傷害を評価する工程;(c)テスト剤を該動物に投与する工程;(d)必要に応じて、工程(c)の前または後に細胞特異的マーカー遺伝子の発現を検出し、および/または定量する工程;(e)髄鞘再形成が工程(d)において起こったか、およびどれくらい多くの髄鞘再形成が工程(d)において起こったかを検出する工程;(f)(例えば、組織学、組織化学、生化学アッセイによって、またはテスト剤の投与に応答してアップレギュレートまたはダウンレギュレートされ得る髄鞘再形成特異的マーカー蛋白質の発現を測定することによって)もし髄鞘再形成が増強され、または減少したならば、テスト剤が髄鞘再形成調節活性を有すると決定する工程を含む。種々の実施形態において、検出は、当該分野で公知の組織化学または生化学アッセイを含む。いくつかの実施形態において、検出は種々の時点で行い、テスト剤の投与は、アッセイの過程の間に、ならびに異なる投与養生法を用いて反復することができる。
【0091】
もう1つの実施形態において、神経障害に関連する現象を調節する化合物についてテストする方法は、候補剤を本明細書中に記載したようにテスト動物に投与する工程、細胞死メディエーター蛋白質の発現を誘導することによってテスト動物における脱髄を増強させる工程、候補剤の投与が増強されたまたは低下した髄鞘再形成をもたらすか否かを決定し、かくして、もし髄鞘再形成が増強され、または低下されたならば、テスト剤が神経障害に関連する現象を調節すると決定する工程を含む。1つの実施形態において、前記方法の実施は、候補剤が細胞死媒介脱髄/髄鞘再形成を調節するか否かを決定する。
【0092】
例えば、1つのそのような実施形態において、神経障害に関連する現象を調節する生物学的に活性な剤をテストする方法は、(a)誘導性/細胞特異的発現調節エレメントに作動可能に連結された死滅メディエーター蛋白質をコードする導入遺伝子を含む動物に候補剤を投与する工程;(b)細胞死メディエーター蛋白質の発現を誘導し、かくして、細胞死をもたらす工程;ならびに(c)候補剤が髄鞘形成または髄鞘再形成を増強させるかを決定する工程を含む。いくつかの実施形態において、検出は種々の時点でなされ、テスト剤の投与はアッセイの過程の間、ならびに異なる投与養生法を用いて反復することができる。1つの実施形態において、神経障害は脱髄障害(例えば、MS)である。髄鞘形成または髄鞘再形成のレベルは、限定されるものではないが、組織学、組織化学、生化学アッセイを含めた方法によって、あるいはテスト剤の投与に応答してアップレギュレートまたはダウンレギュレートされ得る髄鞘再形成特異的マーカー蛋白質の発現を測定することによって、対照動物と比較し、決定することができる。例えば、脱髄障害に関連する現象に対する薬剤の効果はイムノアッセイ、ハイブリダイゼーションアッセイまたはPCRアッセイを含むことができる。
【0093】
種々の実施形態において、検出は、当該分野で公知の組織化学または生化学アッセイを含む。いくつかの実施形態において、検出は種々の時点でなされ、テスト剤の投与はアッセイの過程の間に、ならびに異なる投与養生法を用いて反復することができる。
【0094】
また、本発明は、ニューロンの維持、ニューロンの死滅、またはニューロンの成長または神経膠細胞の維持、神経膠細胞の死滅、または神経膠細胞の成長に対する効果について候補剤をテストする方法も提供する。
【0095】
免疫細胞学および組織学的実験を用いて、ニューロンまたは神経膠の維持、成長または死滅に対する効果を決定することができる。いくつかの実施形態において、テスト動物における髄鞘再形成特異的マーカー蛋白質の発現を対照または参照動物と比較することがで
きる。他の実施形態において、テスト動物における細胞特異的マーカー蛋白質の発現を同一動物において種々の時点になした測定値と比較して、ニューロンの死滅または成長の開始または進行を決定する。
【0096】
本発明の候補剤は、髄鞘再形成促進活性、あるいは逆に、髄鞘再形成阻害活性または髄鞘再形成低下活性についての本明細書中に記載された方法によってテストすることができる。例えば、前記方法は(a)1以上の特定の細胞型において、細胞死メディエーター蛋白質の発現を通じて本発明のトランスジェニック動物において脱髄傷害を誘導する工程であって、ここに、前記1以上の細胞型が髄鞘形成またはニューロン支持に影響する、工程;(b)ミエリン細胞特異的マーカー遺伝子の発現によって証明されるように、ミエリン修復を行うのに十分な時間おいておく工程;(c)候補剤を工程(a)および/または(b)の前、その間および/またはその後に前記動物に投与する工程;(d)もしあれば、髄鞘再形成に対する投与された候補の効果を検出する工程を含むことができる。
【0097】
いくつかの実施形態において、候補剤は、脱髄傷害工程の誘導、例えば、1以上のCDMPの発現および/または活性を誘導することによる傷害の誘導の前、間または後に投与される。1つの実施形態において、候補剤は脱髄傷害の誘導前に投与される。もう1つの実施形態において、候補剤は脱髄傷害の誘導の間に投与される。なおもう1つの実施形態において、候補剤は脱髄傷害の誘導後に投与される。
【0098】
いくつかの実施形態において、候補剤はミエリン修復をもたらすのに十分な見込み時間より前、その間またはそれより後に投与される。1つの実施形態において、候補剤は傷害直後に投与される。もう1つの実施形態において、候補剤は、ミエリン修復が起こることができる時間の間に投与される。なおさらなる実施形態において、候補剤はミエリン修復が起こった後に投与される。
【0099】
いくつかの実施形態において、候補剤は傷害後約1から約24時間に投与される。いくつかの実施形態において、候補剤は傷害後約1から約30日に投与される。種々の実施形態において、候補剤は傷害後約1、2、3、4、5、6、7、8、9、10,11、12、13、14、15、16、17、18、19、20、21、22、23または24時間に投与される。種々の実施形態において、候補剤は約1、2、3、4、5、6、7、8、9、10,11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29または30日目から投与される。なおさらなる実施形態において、候補剤は約1から約12ヶ月まで投与される。
【0100】
髄鞘再形成された軸索を発生させるのに必要な時間の量は異なる動物の間で変化するが、それは、一般に、少なくとも約1週間を必要とし、よりしばしば、少なくとも約2から10週間必要とし、およびよりしばしば、約4から約10週間必要とする。
【0101】
候補剤をスクリーニングすることに関する方法のいずれかにおいて、1以上の候補剤は同時にスクリーニングすることができるのは理解されるべきである。種々の実施形態において、候補剤は髄鞘再形成を促進するものとして同定され、ここに、髄鞘再形成および/またはミエリン特異的マーカー蛋白質の発現は増強され、または増加する。いくつかの実施形態において、テスト動物における細胞特異的マーカー蛋白質の発現は対照または参照動物と比較することができる。他の実施形態において、テスト動物における細胞特異的マーカー蛋白質の発現は同一動物において種々の時点になした測定値と比較され、ここに、より早い時点を参照または対照時点として用いることができる。なお他の実施形態において、髄鞘再形成特異的マーカー蛋白質の発現は、候補剤が髄鞘再形成阻害活性または髄鞘再形成低下活性を有するか否かを決定することにおいて、テスト動物、および対照または参照動物における尺度である。そのような薬剤は髄鞘再形成阻害剤または髄鞘再形成トキ
シンとしてカテゴリー化することができる。
【0102】
髄鞘再形成は、マーカー蛋白質(例えば、EGFP)の発現によるなどして、(例えば、中枢または末梢神経系における)マーカー遺伝子/遺伝子産物の細胞特異的発現の増加を観察することによって確認することができる。脱髄または髄鞘形成が同定されることが求められる1以上の本明細書中における方法において、種々のマーカーが当該分野で入手可能である。髄鞘形成細胞を同定するための例示的なマーカーは、限定されるものではないが、CC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、ミエリン関連糖蛋白質(MAG)、ミエリン乏突起神経膠細胞糖蛋白質(MOG)、乏突起神経膠細胞ミエリン糖蛋白質(OMG)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、ガラクトセレブロシド(GalC)、スルファチド、PDGFRβ、PDGFRα、PDGFα、およびプロテオリピド蛋白質(PLP)を含む。
【0103】
髄鞘形成細胞におけるアポトーシスの誘導の後、および髄鞘再形成が起こるのに十分な時間の後のような傷害に続いて、マーカー蛋白質の蛍光は、小さな動物における蛍光の検出のための当該分野で知られたインビトロまたはインビボ方法を用いて検出することができる。インビボ蛍光は、関連分野において入手可能なデバイスを利用して検出し、および/または定量することができる。例えば、パルスレーザーダイオードおよび可視化システムにカップリングされた時間相関単一フォトン計数検出システムを用いて、組織からの蛍光の発光のレベルを検出することができる(Gallantら、Annual Conference of the Optical Society of America(2004);Contagら、Mol.Microbiol.18:593−603(1995);Schindehutteら、Stem Cells 23:10−15(2005))。組織空気界面における背面反射したフォトンからの大きなシグナルを回避するために、検出点は、典型的には、源点の右側3mmに位置させる。レーザーおよびフィルター双方の波長選択は、選択の蛍光マーカーに依存する。生物学的組織の吸収が低い場合、より深い組織深さ(例えば、レーザー出力に依存して数センチから5または6センチメートル)からの蛍光シグナルをインビボイメージングのために検出することができる。
【0104】
イメージングすべきマウスをイソフルラン/オキシテンで麻酔し、イメージングステージ上に置くことができる。腹側および背側イメージを、関連分野で入手可能なイメージングシステム(例えば、IVISイメージングシステム、Xenogen Corp.,Alameda,CA)を用いて種々の時点について収集することができる。種々の標的組織からの蛍光をイメージし、定量することができる。例えば、シグナル強度は、平均+/−平均の周りの標準偏差として本文または図面中に示すことができる。蛍光シグナルを事後のt検定を使用した分散分析によって分析して、時間ゼロおよび引き続いての各時点における所定のマーカーに対する蛍光シグナルの間の差を評価することができる。
【0105】
蛍光可視化、イメージングまたは検出は当該分野で公知の、および前記した本明細書中で記載された方法を用いて行うことができる。可視化、イメージングまたは検出は侵襲性、最小限に侵襲性または非侵襲性技術を通じてなすことができる。典型的には、顕微鏡検査技術を利用して、トランスジェニック動物から、生きた細胞から得られた細胞/組織からの蛍光、またはインビボイメージング技術を介する蛍光を検出し、またはイメージする。前記、「一般的方法」。
【0106】
ルミネセンス、蛍光または生物ルミネセンスシグナルは、蛍光マルチ・ウエルプレートリーダー、蛍光細胞分析分離装置(FACS)およびシグナルの空間的分解能を供する自
動細胞ベースのイメージングシステムを含めた、種々の自動/または高スループット機器システムのいずれか1つで容易に検出し、定量される。種々の機器システムが、検出を自動化するために開発されており、Cellomics、Amersham、TTP、Q3DM、Evotec、Universal ImagingおよびZeissによって開発された自動蛍光イメージング、および自動顕微鏡検査システムを含む。光漂白後の蛍光回復(FRAP)および微速度蛍光顕微鏡法は、生きた細胞における蛋白質の移動度を研究するためにやはり用いられてきた。
【0107】
蛍光の可視化(例えば、蛍光蛋白質をコードするマーカー遺伝子)は、(例えば、共焦点顕微鏡を用いるセクショニングおよびイメージングを通じての)動物から得られた細胞/組織試料の調査、生きた細胞の調査、またはインビボでの蛍光の検出いずれかを通じて、顕微鏡検査技術で行うことができる。可視化技術は、限定されるものではないが、当該分野で公知の共焦点顕微鏡検査法または光−光学スキャンニング技術の利用を含む。一般に、近赤外において発光波長を持つ蛍光標識が深い組織のイメージングにより使用できる。なぜならば、バックグラウンドノイズを増加させる散乱および自家蛍光の双方は波長が増加するにつれて低下するからである。インビボイメージングの例は、その各開示をここに引用して援用する、Mansfieldら、J.Biomed.Opt.10:41207(2005);Zhangら、Drug Met.Disp.31:1054−1064(2003);Flusbergら、Nat.Meth.2:941−950(2005);Mehthaら、Curr Opin Neurobiol.14:617−628(2004);Jungら、J.Neurophysiol.92:3121−3133(2004);米国特許第6977733号および同第6839586号によって開示されているように当該分野で公知である。
【0108】
インビボイメージングプロセスの1つの例は、インビボイメージング実験から1週間前を含み、休止期の背側毛髪は脱毛剤(Nair,Carter−Wallace Inc.)を用いて脱毛される(約2.5cm×2.5cm面積)。イメージング実験の日に、マウスを麻酔し、顕微鏡ステージ上の顕微鏡カバーグラス上にその背側皮膚を乗せて置く。表皮の脱毛した領域に50W水銀ランプによって照射し、×10対物レンズおよびLP520発光フィルター(Zeiss)を備えた倒立レーザー走査共焦点蛍光顕微鏡(Zeiss LSM510)を用いてスキャンする。アルゴンレーザー(488nm)のようなレーザー、および×10対物レンズは、深い組織から表皮細胞まで徐々により効果的に蛍光発光をイメージすることができる。増強されたエミッターまたはより長い波長のエミッターを利用することによって、より深い組織のイメージングについての感度を高めることができる。別法として、マウスのような小さな動物は種々の異なる位置(例えば、背側、腹側など)を利用して容易にスキャン/イメージすることができる。インビボイメージングは、肝臓のような深い組織領域にさえ効果的であった(例えば、Zhangら、supra)。
【0109】
DAPIを含むVectashield封入剤(Vector Laboratories)でマウントした細胞/組織切片は、Zeiss Axioplan蛍光顕微鏡で可視化することができる。イメージは、Open Labソフトウエアスイートを用いてApple Macintoshコンピューターに連結されたPhotometrics PXL CCDカメラを用いて捕獲することができる。異なる波長の蛍光は、ほぼ0.04mm2の領域に制限された脳梁の中央内の陽性細胞をカウントすることによって検出され、定量される。蛍光または共焦点顕微鏡検査法を利用してインビトロにて組織/細胞からの蛍光のレベルを検出し、測定するさらなる方法は当該分野で公知であり、前記にて本明細書中で開示された1以上のマーカー蛋白質からの蛍光を検出し、または測定するのに利用することができる。
【0110】
もう1つの例において、神経細胞は、エピ蛍光励起および発光経路においてLudlフィルターホイール(CarlZeiss,Thornwood,NY,USA)が嵌合したAxiovert S100 TV倒立顕微鏡でイメージすることができ、冷却された電荷結合素子(CCD)カメラ(Micro−MAXO;Roper Scientific,Trenton,NJ,USA)を用いてイメージを収集することができる。特定の励起および発光フィルター、および通常の二色エレメントを用いて、2つの異なる蛍光蛋白質のシグナルを単離することができる(HQFITCおよびテキサスレッド励起および発光フィルター、およびFITC/テキサスレッドV3二色;Chroma Technology,Brattleboro,VT,USA)。フィルターホイールおよびカメラはソフトウエア(例えば、IPLabsソフトウエア,Scanalytics,Fairfax,VA,USA)によって制御することができる。赤色および緑色蛍光イメージの組を収集して、赤色または緑色蛍光を有する細胞の相対的パーセンテージを分析することができる。イメージを分析し、IPLabsおよびAdobe InDesignソフトウエアでの公表のために調製することができる。イメージの操作を、赤色および緑色蛍光蛋白質のグレースケールイメージの合併に制限して、RGBカラーファイルを作り出し、最終プリントアウトの明るさ/コントラストを調整して、顕微鏡を通じて観察されたものに最も密接にマッチさせ、レタリングおよびスケールバーを加えることができる。蛍光顕微鏡検査装置は当該分野で公知であり、商業的に入手可能である。例えば、<confocal−microscopy.com/website/sc llt.nsf.>におけるウェブサイト参照。
【0111】
代替実施形態において、蛍光検出は網膜または角膜からの直接的なものである。網膜部位は全身毒性の研究のための非侵襲性箇所である。角膜は、身体に侵入することなく神経膠細胞を含有する器官に直接的に適用された物質の毒性を評価するのによく適合している。従って、マーカー蛋白質を異なって発現する神経細胞から発せられた蛍光は、所望の領域へレーザービームを向け、発せられた蛍光のレベルを検出することにより、網膜または角膜の共焦点顕微鏡検査法を用いることによって検出することができる。
【0112】
さらに、脱髄/髄鞘再形成現象は当該分野で公知の免疫組織化学的手段または蛋白質分析によって観察することができる。例えば、テスト動物の脳の切片を、乏突起神経膠細胞マーカーを特異的に認識する抗体で染色することができる。もう1つの態様において、乏突起神経膠細胞マーカーの発現レベルは、イムノブロッティング、ハイブリダイゼーション手段、および増幅手法ならびに当該分野でよく確立されたいずれかの他の方法によって定量することができる。例えば、その各々の開示をここに引用して援用する、Mukouyamaら、Proc.Natl.Acad.Sci.103:1551−1556(2006);Zhangら、supra;Girardら、J.Neurosci.25:7924−7933(2005);および米国特許第6,909,031号;同第6,891,081号;同第6,903,244号;同第6,905,823号;同第6,781,029号;および同第6,753,456号。
【0113】
もう1つの態様において、中枢または末梢神経系からの細胞/組織を切り出し、蛋白質のために処理することができ、例えば、組織をホモジナイズし、蛋白質をSDS−10%ポリアクリルアミドゲルで分離し、次いで、ニトロセルロース膜に移して、マーカー蛋白質を検出する。蛍光蛋白質のレベルは、一次抗体/抗血清(例えば、特定のマーカー蛋白質に対して生起させたヤギポリクローナル;BD Gentest,Woburn,MA)、およびペルオキシダーゼ結合二次抗体(例えば、ウサギ抗ヤギIgG,Sigma Aldrich)を利用して検出することができる。化学発光は、当該分野で入手可能な標準試薬を用いて検出して、組織試料中の蛍光マーカー蛋白質のレベルを検出し、決定する。
【0114】
従って、もし候補治療剤/薬物またはテスト生物活性剤が本発明の1以上の方法でアッセイされるならば、異なる時点で比較して、ならびに参照または対照と比較して薬物に対する応答の総じての差があるか否かを決定することができる。
脱髄
本発明の1つの態様において、本発明の組成物および方法を利用して、全身抗原送達、または免疫応答を開始させるためのアジュバントプライミングの必要性なくして、病巣脱髄を達成する。本発明の種々の実施形態において、細胞特異的な様式での細胞死メディエーター蛋白質の発現は、標的細胞における細胞死および/またはニューロン喪失を誘導するのに利用される。細胞死メディエーター蛋白質の発現および/または活性もまた誘導性であり得る。
【0115】
CDMPはSMAC(カスパーゼの第2のミトコンドリア由来アクチベーター)、IAP(アポトーシス蛋白質の阻害剤)、カスパーゼおよびそれらのモジュレーターであり得る。他の実施形態において、CDMPはTNF(腫瘍壊死因子)受容体、およびFas受容体、およびTRAIL受容体経路のような他の死滅受容体シグナリング経路のモジュレーターであり得る。また、CDMPは、グランザイムB、またはグランザイムBのモジュレーターを含めたカスパーゼのアクチベーターでもあり得る。
【0116】
種々の実施形態において、細胞死メディエーター蛋白質は核酸構築体によってコードされる。いくつかの実施形態において、核酸構築体は1以上のCDMPをコードすることができる。CDMPは同一または異なり得る。例えば、単一の核酸は、タンデムにカスパーゼ9をコードする2つの配列のような蛋白質の2つをコードすることができるか、あるいはカスパーゼ9およびカスパーゼ3をコードすることができる。本発明の核酸構築体はカスパーゼ2、5、8、9、10または11、それらのプロ酵素形態、またはその誘導体をコードすることができる。さらなる実施形態において、そのようなカスパーゼ蛋白質をコードする配列を、架橋剤化合物と反応性である二量体化ドメインを含むように修飾する。かくして、いくつかの実施形態において、野生型カスパーゼ配列を、架橋剤化合物に対して選択的に反応性である二量体化ドメインを含むキメラ配列を生じるように修飾する。そのような二量体化ドメインの例は、その関連部分をここに引用してその全体を援用する、米国特許出願第20050187177号および米国特許第6,984,635号に開示されたものを含む。
【0117】
さらに、インビボまたはインビトロでの選択的細胞死は、デュアルプロモーター、自殺遺伝子またはCDMPの組織特異的発現を可能とするように構築された自己不活性化(SIN)レンチウイルスベクター、およびマーカー遺伝子(例えば、eGFP)の使用によって達成することができる。1つの実施形態において、乏突起神経膠細胞のような細胞を、図1に示すように、本発明の核酸構築体を用いて標的化する。得られたベクターは、自殺遺伝子およびマーカー遺伝子eGFPの組織特異的発現を可能とする。従って、前記ベクターは、乏突起神経膠細胞のような有糸分裂後細胞の感染を可能とする。さらに、前記ベクターを利用して、高ウイルス力価を生じさせ、インビボにて有効な適用を促進することができる。かくして、1つの実施形態において、そのような細胞特異的発現の結果は乏突起神経膠細胞特異的細胞死である。
【0118】
デュアルプロモーターの特徴は、感染された細胞を蛍光顕微鏡検査法を用いて、容易に同定できるように、上流遺伝子から独立したGFPの共発現を可能とする。当該分野で公知の他の蛍光マーカーをGFPの代わりに用いることができる。
【0119】
1つの実施形態において、核酸送達ビヒクルを用いて、細胞死メディエーター蛋白質(または自殺蛋白質)をコードする配列をインビトロにて細胞に送達する。送達は、病巣パターンの、CNSに対するものでもあり得る。このシステムに対して数個の利点がある。
【0120】
まず、病巣の位置は予め決定することができ、関連するニューロンおよび細胞応答の正確な検出を可能とする。第2に、生じた病巣のサイズを、引き続いての実験において制御し、および改変することができる。これは重要である。というのは、関連するニューロンの死滅を広めるために破らなければならない乏突起神経膠細胞の喪失の閾値があり得るからである。第3に、前記モデルは、脱髄の正確なタイミングを指定する能力を可能とする。
【0121】
従って、細胞および分子レベルにおけるニューロン応答の一時的記載を達成することができる。さらに、CNS中の異なる位置で生じた第2の病巣に対する1つの病巣の一時的関係を調査して、脱髄病巣の間の時間がCNS応答のタイプを決定するにおいて重要な因子であるかを決定することができる。加えて、送達ベクターは細胞特異的発現調節配列を組み込み、その結果、細胞死メディエーター蛋白質の細胞型特異的(例えば、乏突起神経膠細胞特異的)発現がもたらされる。この特徴は、細胞の実験的に課された死滅が最小の傍観者効果でもって望まれる細胞型集団に制限されることを確実とする。最後に、前記モデルは修飾および増大レベルの複雑性に適合している。
【0122】
例えば、1つの実施形態において、iCP9をレンチウイルスプラスミドにクローン化し、MBPプロモーター配列をiCP9遺伝子の上流にクローン化する。得られたレンチウイルスベクター、pΔmbpICP9/mEGFPを用いて、複製能力がないウイルスを作り出す(実施例1参照)。かくして、前記ベクターを利用して、細胞/組織特異的発現調節配列または誘導性発現調節配列の選択によって、1以上の特定の細胞型(例えば、所望により神経細胞および/または壁細胞)の結果として、時間をわたっての、あるいは特定の時点におけるニューロン応答を規定することができる。
【0123】
種々の実施形態において、本発明の方法を利用し、乏突起神経膠細胞の喪失に対するニューロンの応答を経時的に定義することができる。これは、公知の生存、栄養およびアポトーシス経路における変化を観察することによって、ならびにそのような事象に対する新規な応答を同定するためのマイクロアレイ分析を通じて達成することができる。さらに、環境ストレスに対する規定されたニューロン応答の分類は、アポトーシス誘導因子、抗アポトーシス因子、神経栄養因子および神経栄養関係転写因子の発現を含む。
【0124】
従って、種々の実施形態において、本発明の動物/細胞を利用して、種々の因子/化合物をスクリーニングして、そのような因子/化合物がニューロンの維持または健康を増強させ/減少させるかを決定することができる。例えば、多数の転写因子は外部刺激に応答してニューロンで発現される。2つの高度に保存されかつよく規定された転写因子であるNF−κBおよびcAMP応答エレメント結合蛋白質(CREB)は、ニューロンがストレスを受け、引き続いて、部分的に規定された様式で、広い範囲の下流標的を活性化する場合に発現される。NF−κBは、卒中、心臓停止および全体的虚血症、発作およびグルタメート、グルコース剥奪およびβ−アミロイドペプチドへの実験的暴露を含めた生理学的および病理学的状態によって調節される。可変傷害に対するこの高度に保存された応答は、ニューロンの生存およびアポトーシスの制御に関連する基本的な細胞応答を示唆する。同様に、CREBは生理学的刺激の膨大なアレイに対して応答して活性化される。最初のインビトロ研究およびCREBナルマウスを利用するその後のインビボの研究は、CREBが多数のニューロンサブタイプの生存に必要であることを示唆する。CREBは多数のげっ歯類モデルにおいて低酸素症、虚血症および酸化的ストレスに応答して活性化され、これらのストレッサーの間におけるCREBの不活性化は、典型的には、ニューロン細胞死を増悪させる。
【0125】
CREBおよびNF−κB誘導細胞応答の双方は、第1に、ニューロンの生存を究極的
には支持する様式で作用するように見える。同様に、神経栄養因子はニューロンの成長および生存を支持する。この群の分子は、限定されるものではないが:神経膠由来神経栄養因子(GDNF)、毛様体神経栄養因子(CNTF)、脳由来神経栄養因子(BNDF)、神経成長因子(NGF)、NT−3およびNT−4/5を含む。これらの因子は、急性CNS傷害後にアップレギュレートされると考えられている。さらに、乏突起神経膠細胞はニューロンに対する栄養支持を供すると考えられている。PLPを欠如するトランスジェニックマウスにおける、および誕生時における乏突起神経膠細胞前駆体の照射後に、急性MS病巣における乏突起神経膠細胞の喪失を記載するインビボの観察はインビトロ実験によって確証されている。具体的には、乏突起神経膠細胞前駆体細胞またはそれらの馴化培地の、黒質への添加は、ニューロンの生存を有意に増強させた。同様に、視神経乏突起神経膠細胞前駆体細胞またはそれらの培養された培地は、網膜神経節細胞の生存を有意に増強させた。NGF、BDNF、GDNF、ニューレグリンおよびNT−3は、全て、乏突起神経膠細胞の培養した培地において同定されている。
【0126】
ニューロンの生存とは対照的に、ニューロンのプログラムされた細胞死は急性乏突起神経膠細胞の喪失に対する重要な応答であろう。死滅受容体ファミリーのメンバーであるFasは、カスパーゼカスケードの活性化によってそのリガンドFasLによって結合された場合にアポトーシスを誘導する。脳においては、皮質ニューロンはFasを発現し、およびインビトロにて、これらの細胞はFas活性化の後に迅速にアポトーシスを受ける。卒中のモデルにおいて、Fas発現は、カスパーゼ8の発現と共に局所化されるニューロンにおいてアップレギュレートされる。さらに、もしFasLが不存在であれば、卒中誘導脳損傷が低下する。運動ニューロンのインビトロ培養物は、プログラムされた細胞死の開始についてFasの活性化に対する同様な依拠を示している。というのは、インビトロにて神経栄養支持が奪われた運動ニューロンは、Fas/FasL相互作用がなくなった後にのみ培養で維持されたからである。
【0127】
最後に、抗アポトーシス因子の発現は、急性脱髄に対する応答の重要な態様であり得る。天然で生じる細胞死のアポトーシスプロセスは種の間で高度に保存されており、線虫C.elegansにおける実験は、最初に、このプロセスに対して必須の分子の同定に導き、引き続いて、哺乳動物ホモログE4BP4の同定を可能とした。この分子は、発達する脳において、天然に生じる細胞死の時において運動ニューロンによって発現されることが示されている。さらに、インビボ過剰発現は、標的を刺激するニューロンの数を増加させ、該分子が天然で生じるアポトーシスのプロセスを妨げることを示唆する。
【0128】
従って、種々の実施形態において、抗アポトーシス化合物を種々の時点で本発明の細胞または動物に投与して、ニューロン維持に対する抗アポトーシス化合物の効果を決定することができる。抗アポトーシス化合物は細胞死メディエーター蛋白質(例えば、抗カスパーゼ9)に対する公知の活性を有することができるか、あるいはそれは、抗アポトーシス活性を決定するためにスクリーニングされる候補剤であり得る。
【0129】
かくして、乏突起神経膠細胞の喪失に対する経時的なニューロン応答を規定することによって、いずれの化合物が、最終的に、ニューロンの喪失、または細胞死からのニューロンの救済に導く事象のカスケードに影響するかを決定することができる。分子の空間的および時間的発現パターンを規定することから得られた情報は、適当な機能の実験を設計し、負傷に対する神経応答の根底にある細胞相互作用を解明することにおいて重要である。
【0130】
1つの実施形態において、本発明の方法を利用して、全身合併症がない乏突起神経膠細胞−ニューロン相互作用をアッセイして、生存するニューロンの分子応答を規定する(例えば、実施例2)。急性および慢性MS病巣における、ならびにNAWM(正常に見える白色物質)における軸索の横断および引き続いてのニューロンの喪失は、この疾患の比較
的最近に規定された態様である。証拠は、ニューロンの継続的喪失が患者における不能の蓄積を説明することを示唆する。
細胞ベースのスクリーニングアッセイ
本発明のいくつかの態様において、細胞培養物は、本発明の1以上の方法で利用される。標的細胞は被験体に由来することができ、かつ形質転換され得る(例えば、遺伝子的に修飾された)か、または本発明のトランスジェニック動物は細胞/組織培養物のための源となり得る。もう1つの態様において、本発明の細胞は細胞系に由来する細胞であってよい。
【0131】
種々の実施形態において、本発明の実施は、対照細胞に対する、テスト細胞(例えば、トランスジェニック乏突起神経膠細胞またはシュワン細胞)における遺伝子または遺伝子産物の発現、または遺伝子産物の活性の比較を供するための細胞ベースのアッセイを含むことができる。本発明で用いるテスト細胞は中枢または末梢神経系から単離することができ、それに由来する細胞培養物、およびその子孫、および源から調製された切片またはスミアー、または例えば、乏突起神経膠細胞またはシュワン細胞またはそれらの先祖を含有する脳のいずれかの他の試料を含む。望まれる場合、限定されるものではないが、ニューロン、神経膠細胞、小神経膠細胞、および神経膠星状細胞のような他のニューロン細胞型が実質的に含まれない富化された細胞培養物を用いるように選択することができる。成熟した乏突起神経膠細胞およびシュワン細胞を単離し、生成させ、または維持する種々の方法は当該分野で公知であって、本明細書中に例示される。
【0132】
1つの実施形態において、細胞死メディエーター蛋白質をコードする導入遺伝子を含む非ヒトトランスジェニック動物または細胞を供する工程であって、ここに、前記導入遺伝子はニューロン細胞または神経膠細胞特異的発現調節エレメントに作動可能に連結されている、工程;前記細胞死メディエーター蛋白質を活性化し、それにより、アポトーシスを誘導する工程;前記活性化に続いて少なくとも1つの生存するニューロン細胞または神経膠細胞を得る工程;ならびに、生存する神経膠細胞またはニューロン細胞においてRNA転写物および/またはコードされた産物をプロファイルする工程;それにより、MSまたはMS関連状態に関連する現象を特徴付けるプロファイルデータセットを編集する工程を含む、MSまたはMS関連状態に関連する現象を特徴付けるためのプロファイルデータセットを編集する方法が提供される。他の実施形態において、この方法は、ALSまたはパーキンソン病に関連する現象を特徴付けるためのプロファイルデータセットの編集を提供する。
【0133】
いくつかの実施形態において、細胞死メディエーター蛋白質はカスパーゼ−9またはカスパーゼ−11である。1つの実施形態において、カスパーゼ−9の活性化は、カスパーゼ−9の二量体化を誘導することを含む。もう1つの実施形態において、カスパーゼ−11の活性化は自己活性化を含む。
【0134】
種々の実施形態において、細胞型特異的発現調節エレメントは、CC1、ミエリン塩基性蛋白質(MBP)、セラミドガラクトシルトランスフェラーゼ(CGT)、ミエリン関連糖蛋白質(MAG)、ミエリン乏突起神経膠細胞糖蛋白質(MOG)、乏突起神経膠細胞ミエリン糖蛋白質(OMGP)、環状ヌクレオチドホスホジエステラーゼ(CNP)、NOGO、ミエリン蛋白質ゼロ(MPZ)、末梢ミエリン蛋白質22(PMP22)、蛋白質2(P2)、GFAP、AQP4、PDGFα、RG5、p糖蛋白質、ニュールツリン(NRTN)、アルテミン(ARTN)、ペルセフィン(PSPN)、スルファチド、2(VEGFR2)、スーパーオキシドジスムターゼ(SOD1)、チロシンヒドロキシラーゼ、ニューロン特異的エノラーゼ、パルキン遺伝子(PARK2)、パルキン共調節遺伝子(PACRG)、ニューロン特異的Tα1 α―チューブリン(Tα1)、小胞モノアミントランスポーター(VMAT2)、α−シノクレイン(SNCA)、PDGFR
−α、PDGFR−β、またはプロテオリピド蛋白質(PLP)の1以上を含めた群から選択される遺伝子からのものである。
【0135】
1つの実施形態において、本発明は、髄鞘再形成を調節する候補の生物学的に活性な剤を同定する方法を提供する。前記方法は、細胞死メディエーター蛋白質の細胞ごとに異なっての発現が可能な本発明のトランスジェニック動物からトランスジェニック細胞を入手または単離し、そのような細胞を培養する工程;候補剤を培養した細胞と接触させる工程;対照細胞に対する、遺伝子または遺伝子産物の改変された発現、または遺伝子産物の改変された活性を検出する工程であって、前記遺伝子または遺伝子産物は細胞死メディエーター蛋白質による細胞死の調節に相関している、工程;ならびに、もし前記遺伝子または遺伝子産物の発現のレベルが前記対照細胞に対して調節されているならば、前記薬剤を候補として選択する工程を含む。
【0136】
もう1つの実施形態において、もし細胞死を受けている標的細胞の数が、対照細胞と比較して候補剤の添加によって調節されるならば、薬剤は候補剤であると決定される。
【0137】
もう1つの実施形態において、本発明は、髄鞘再形成を促進する生物学的に活性な剤を同定する方法を提供する。前記方法は、本発明のトランスジェニック動物に存在する脱髄病巣からの標的細胞を入手し、単離し、および培養する工程;培養した細胞を候補の生物学的に活性な剤と接触させる工程;ならびに、細胞死メディエーター蛋白質をコードする導入遺伝子の改変された発現または改変された活性を検出する工程;ならびに、もし遺伝子または遺伝子産物の発現のレベル、または遺伝子産物の活性のレベルが対照細胞に対して増加するならば、前記薬剤を候補として選択する工程を含む。例えば、もし候補剤が細胞死メディエーター蛋白質の細胞死媒介活性を増強させるならば、前記候補は脱髄を促進することができ、他方、もし前記候補剤が細胞死媒介活性を低下させるならば、前記薬剤は髄鞘再形成を促進することができる。
【0138】
ある実施形態において、その神経系が活動的に髄鞘形成を受けている若い被験体からの髄鞘形成細胞を使用するのが好ましいであろう。他の実施形態において、限定されるものではないが、病原体または物理的負傷によって冒された病巣、およびクプリゾンのような毒性剤によって引き起こされた病巣を含めた、脱髄病巣における成体乏突起神経膠細胞前駆体に由来する髄鞘再形成細胞を用いるのが好ましいであろう。
【0139】
1つの実施形態において、高密度の皮質培養物を、iCP9を発現する複製欠損レンチウイルスでトランスフェクトする。CID(二量体化化学誘導因子)の添加、および乏突起神経膠細胞の引き続いての死滅の後、遺伝子転写のマイクロアレイ分析を用いて、生存する培養細胞における新規な因子の発現を評価することができる。これらの培養物における乏突起神経膠細胞の細胞死の程度は、細胞破壊のひどさに対する分子応答を同定するために系統的に変化されるであろう。従って、種々の実施形態において、細胞死を調節する1以上の分子標的が同定される。
【0140】
本発明に適した種々の遺伝子ビヒクルは当該分野で入手可能である。それらはウイルスおよび非ウイルス発現ベクターを共に含む。非限定的例示的なウイルス発現ベクターは、レトロウイルスのようなRNAウイルス、およびアデノウイルス、泡沫状ウイルス、狂犬病ウイルスおよびアデノ関連ウイルスのようなDNAウイルスに由来するベクターである。非ウイルス発現ベクターは、限定されるものでなはいが、プラスミド、コスミド、およびDNA/リポソーム複合体を含む。遺伝子ビヒクルは、その中に運ばれる外因性遺伝子の組織特異的、細胞特異的、またはオルガネラ特異的でさえある発現を指令する調節配列を運ぶように作成することができる。
【0141】
いくつかの実施形態において、標的細胞を多数の遺伝子ビヒクル(例えば、その各々が、所望の産物をコードする遺伝子構築体、および1以上のレポーター遺伝子をコードする遺伝子構築体を含む2つのベクター)で共トランスフェクトすることができる。
【0142】
さらに、所望であれば、広く種々の細胞内局所化配列またはシグナルが特徴付けられ、導入遺伝子のオルガネラ特異的発現を指令するために適用可能である。例えば、細胞内局所化配列は以下の:(a)細胞の外部への遺伝子産物の分泌を指令するシグナル配列;(b)細胞の原形質膜または他の膜区画への蛋白質の付着を可能とする膜係留ドメイン;(c)コードされた蛋白質の核へのトランスロケーションを媒介する核局所化配列;(d)コードされた蛋白質を主としてERに制限する小胞体滞留配列(例えば、KDEL配列);(e)目的の蛋白質が、該蛋白質が膜に結合するように、ファルネシル化することができ;または(f)コードされた蛋白質産物の異なる細胞内分布において役割を演じるいずれかの他の配列のうちのいずれか1つであり得る。
【0143】
所望であれば、遺伝子ビヒクルを当該分野で公知のいずれかの方法によって宿主細胞(例えば、乏突起神経膠細胞またはシュワン細胞のような髄鞘形成細胞)に挿入することができる。適当な方法はリン酸カルシウム沈殿、DEAE−デキストラン、エレクトロポレーション、またはマイクロインジェクションを用いるトランスフェクションを含むことができる。
【0144】
適当な対照細胞または組織の選択は、最初に選択されたテスト細胞または組織、および調査下にあるその表現型または遺伝子型の特徴に依存する。テスト髄鞘再形成細胞をテスト化合物に接触させるが、対照細胞または組織は非処理の対応物であってよい。テスト髄鞘再形成細胞は脱髄後に検出されたテスト細胞であるが、対照細胞は非処理の対応物であってよい。一般には、テスト細胞および対照を並行して分析するのが好ましい。
【0145】
先のセクションで議論したように、これらの細胞は、ニューロン髄鞘再形成状態の分子基礎を解明するための細胞ベースのアッセイを行うのに、およびニューロン脱髄を阻害し、または髄鞘再形成を促進するのに有効な薬剤をアッセイするのに有用である。
【0146】
本発明のいくつかの態様において、トランスジェニック細胞は本発明のトランスジェニック細胞から入手し、培養し、拡大培養し、標的蛋白質をコードする遺伝子で形質導入し、そして、源動物またはいくつかの他の動物に移植し、または再度導入することができる。そのようなエクスビボ方法において、トランスジェニック細胞は、例えば、マーカー遺伝子発現/産生の調節用のテスト遺伝子/蛋白質をアッセイするために、誘導により生成され得る生物学的に活性な剤をコードする遺伝子(例えば、テスト産物をコードする遺伝子)でトランスフェクトすることができる。そのような調節は、本明細書中において前記したように細胞培養物においてアッセイすることができる。別法として、形質導入された細胞は被験体の動物に再度導入され、マーカー遺伝子発現をアッセイし、対照または参照と比較できる場合、移植された前記細胞が形質導入されていない場合、目的のベクターで生じる産物を発現せず、時間制御様式で目的のベクターで生じる産物を発現せず(例えば、誘導性発現)、または目的の産物を構成的に発現しない(例えば、CMVプロモーター)。
【0147】
例えば、神経膠細胞(例えば、乏突起神経膠細胞またはシュワン細胞)は神経バイオプシーに由来することができる。細胞を(例えば、10%熱不活化胎児ウシ血清(FBS)を含有し、抗生物質、組換えNeu分化因子(LDF)、インスリンおよびフォルスコリン(1ng/ml)を補足した、DMEMよりなる増殖培地を利用して)培養において拡大することができる。さらに、細胞は導入遺伝子によって供された蛍光標識を利用して非トランスジェニック神経細胞からソーティングする(例えば、FACS)ことができる。
形質導入のために、細胞を、目的の産物を送達する当該分野で公知の種々のベクタービヒクルでトランスフェクトすることができる。従って、種々の実施形態において、本発明の核酸構築体は、やはり、マーカー蛋白質をコードする1以上の配列に作動可能に連結される。
【0148】
本発明の1以上の組成物または方法で利用することができるベクターは、SV−40、泡沫状ウイルス、狂犬病ウイルス、アデノウイルス、レンチウイルス、レトロウイルス由来DNA配列の誘導体、および機能的哺乳動物ベクターの組合せに由来するシャトルベクター、ならびに機能的プラスミドおよびファージDNAを含む。真核生物発現ベクターは、例えば、ここに引用して援用する、P J Southern and P Berg,J MoL Appl Genet 1:327−341(1982);Subraminiら、Mol Cell.Biol.1:854−864(1981),Kaufinann and Sharp,J Mol.Biol.159:601−621(1982);Scahillら、PNAS USA 80:4654−4659(1983)およびUrlaub and Chasin PNAS USA 77:4216−4220(1980)によって記載されているように、よく知られている。本発明の方法で用いるベクターは、複製欠損アデノウイルスのような、レトロウイルスベクターのごときウイルスベクターであってよい。例えば、レトロウイルスの構造遺伝子が、末端反復配列に含有されたウイルス調節配列の制御下にある、目的の単一遺伝子によって置き換えられた「単一遺伝子ベクター」を用いることができる。例えば、ここに引用して援用する、Eglitis and Andersen,BioTechniques 6(7):608−614(1988)によって記載されているような、モロニーネズミ白血病ウイルス(MoMulV)、ハーベイネズミ肉腫ウイルス(HaMuSV)、マウス乳腺癌ウイルス(MuMTV)、およびネズミ骨髄増殖性肉腫ウイルス(MuMPSV)、ならびに細網内皮症ウイルス(Rev)およびラウス肉腫ウイルス(RSV)のような鳥類レトロウイルス。好ましくは、本発明のベクターはレンチウイルスベクターである。
【0149】
1つの実施形態において、操作された誘導性カスパーゼ9(iCP9)cDNA配列は、ミエリン塩基性蛋白質遺伝子プロモーター配列、図1Cの制御下にあるレンチウイルスベクターにクローン化する。引き続いて、複製能力がないレンチウイルスはこのプラスミドから生じさせることができ、細胞および動物系に適用することができる。前記ウイルスは、暴露された全ての細胞型の感染をもたらすがiCP9遺伝子の発現は、細胞特異的プロモーター系のため乏突起神経膠細胞に制限される。二量体化化学誘導因子(CID)の添加の結果、乏突起神経膠細胞のみ細胞死がもたらされるであろう。このアプローチは、インビトロおよびインビボ双方において、乏突起神経膠細胞を特異的に殺傷することにおいてかなり有効であろう。当業者であれば、他のカスパーゼまたはその誘導体のような他のCDMPをiCP9の代りに用いることができること、他のプロモーター配列をMBPプロモーターの代りに用いることができることを認識する。本発明のもう1つの態様において、当該分野で公知のマイクロアレイまたは他の発現プロファイリングプロセスを利用して、細胞死に応答してアップレギュレートまたはダウンレギュレートされる遺伝子または遺伝子の組を同定する。種々の実施形態において、発現データセットを、細胞死の誘導前、その間およびその後を含めた、種々のおよび特定の時点について編集することができる(例えば、実施例5)。
生物活性剤
ニューロン髄鞘再形成を調節するのに有効な生物学的に活性な剤または生物活性剤は、限定されるものではないが、単純なまたは複雑な有機または無機分子、ペプチド、ペプチドミメティック、蛋白質(例えば、抗体)、リポソーム、低分子干渉RNA、またはポリヌクレオチド(例えば、アンチセンス)のような生物学的または化学的化合物を含むことが意図される。
【0150】
化合物、例えば、ポリペプチドおよびポリヌクレオチドのようなポリマー、および種々のコア構造に基づく合成有機化合物の膨大なアレイを合成することができ、これらは本明細書中においても考えられる。加えて、植物または動物抽出物などのような種々の天然源はスクリーニングのための化合物を提供することができる。常には明確に述べられないが、活性剤は、単独で、あるいは主題のスクリーニング方法によって同定された薬剤と同一または異なる生物学的活性を有するもう1つのモジュレーターと組み合わせて用いることができるのは理解されるべきである。
【0151】
生物学的に活性な剤が裸のRNA以外の組成物である場合、前記薬剤を細胞培養物に直接的に加えてもよく、あるいは添加のための培養基に加えてもよい。当業者に明らかなように、経験的に決定することができる「有効」量が加えられなければならない。前記薬剤がポリヌクレオチドである場合、それはトランスフェクションまたはエレクトロポレーションによって直接的に細胞に導入することができる。別法として、それは、遺伝子送達ビヒクル、または前記した他の方法を用いて細胞に挿入することができる。
【0152】
蛋白質レベルを検出するのに適した広く種々の標識は当該分野で公知である。非限定的例は放射性同位体、酵素、コロイド状金属、蛍光化合物、生物ルミネセント化合物、および化学発光化合物を含む。
【0153】
主題の方法によって同定された候補の生物学的に活性な剤は以下の2つのクラスに広くカテゴリー化することができる。最初のクラスは、細胞または被験体に投与した場合に、細胞死メディエーター蛋白質の発現または活性のレベルを低下させる薬剤を含む。第2のクラスは、細胞死メディエーター蛋白質の発現または活性のレベルを増大させる薬剤を含む。
医薬組成物
本発明の方法を用いて、脱髄障害の処置で引き続いて実施できる生物学的に活性な剤を選択することができる。髄鞘再形成を調節するのに有効な選択された生物学的に活性な剤は、脱髄障害を処置するための医薬の調製で用いることができる。1つの態様において、本発明の同定された/選択された生物学的に活性な剤を投与して、細菌およびウイルスのような病原体によって冒されたニューロン脱髄を処置することができる。もう1つの態様において、選択された薬剤を用いて、例えば、橋中央ミエリン溶解およびビタミン欠乏症におけるように、毒性物質によって引き起こされたニューロン脱髄、または身体中への毒性代謝産物の蓄積を処置することができる。なおもう1つの態様において、前記薬剤を用いて、脊髄負傷のような物理的負傷によって引き起こされた脱髄を処置することができる。さらになおもう1つの態様において、前記薬剤を投与して、遺伝的特質を有する障害、限定されるものではないが、白質萎縮症、副腎脳白質ジストロフィー、変性多系統萎縮症、ビンスヴァンガー脳障害、中枢神経系における腫瘍および多発性硬化症を含む遺伝的障害において発現される脱髄を処置することができる。
【0154】
種々の送達系は公知であり、それを用いて、本発明の生物活性剤を投与することができる(例えば、リポソーム、マイクロ粒子、マイクロカプセルでのカプセル化、組換え細胞による発現、受容体媒介エンドサイトーシス(例えば、Wu and Wu,(1987),J.Biol.Chem.262:4429−4432)、レトロウイルスまたは他のベクターの一部としての治療核酸の構築など)。送達の方法は、限定されるものではないが、動脈内、筋肉内、静脈内、鼻内および経口経路を含む。特定の実施形態においては、本発明の医薬組成物を処置を必要とする領域に局所投与するのが望ましいであろう;これは、例えば、限定されるものではないが、手術の間における局所的注入によって、注射によって、またはカテーテルによって達成することができる。ある実施形態において、前記薬剤は被験体の神経系、好ましくは、中枢神経系に送達される。もう1つの実施形態において、前記薬剤は髄鞘再形成を受けている神経組織に投与される。
【0155】
選択された薬剤の投与は、処置の過程全体にわたって継続的にまたは間歇的に、1用量で行うことができる。投与の最も有効な手段および投与量を決定する方法は当業者によく知られており、治療で用いる組成物、治療の目的、処置されている標的細胞、および処置されている被験体で変化するであろう。単一または複数の投与を、処置している医師によって選択されている用量レベルおよびパターンで行うことができる。
【0156】
本発明の医薬組成物の調製は、医薬製剤の調製のための一般的に許容される手法に従って行われる。例えば、Remington’s Pharmaceutical Sciences 第18版(1990),E.W.Martin編,Mack Publishing Co.,PA参照。意図する使用および投与の態様に依存して、有効成分を、さらに、医薬組成物の調製において加工するのが望ましいであろう。適当な加工は、適当な非毒性および非干渉性成分との混合、滅菌、用量単位への分割、および送達デバイスへの封入を含むことができる。
【0157】
経口、鼻内、または局所投与用の医薬組成物は、錠剤、カプセル、粉末、液体、および懸濁液を含めた固体、半固体または液体形態で供することができる。注射用の組成物は液体溶液または懸濁液として、エマルジョンとして、または注射に先立っての液体中の溶解または懸濁に適した固体形態として供することができる。気道を介する投与のために、好ましい組成物は、適当なエアロゾライザーデバイスで用いる場合に固体、粉末、またはエアロゾルを供するものである。
【0158】
液体の医薬上許容される組成物は、例えば、本明細書中に具体化されたポリペプチドを、水、生理食塩水、水性デキストロース、グリセロール、またはエタノールのような液体賦形剤に溶解または分散させることによって調製することができる。組成物は他の薬効のある薬剤、医薬剤、補助薬、担体、および湿潤剤または乳化剤、およびpH緩衝剤のような補助的物質を含有することもできる。
【実施例】
【0159】
実施例1:iCP9ウイルスの生産
複製不能のウイルスを生産すること
図1および2は、本明細書中で用いる導入ベクターについての一般的な模式図を描く。第1世代自己不活性化レンチウイルスベクターpLpMGは、サイトメガロウイルスプロモーター配列(pCMV)、ラットミエリン塩基性蛋白質プロモーター(pMBP)の断片、ラット神経膠細胞繊維状酸性蛋白質プロモーター(pGFAP)の断片、または血小板由来成長因子受容体(PDGFR)−アルファプロモーター(pPDGFR)の断片いずれかの挿入によって修飾した。得られたベクター(pLpCMVXMG、pLpMBPXMG、pLpGFAPXMG、pLpPDGFRXMG)は、さらに、鋳型として元来のベクター(David Spencer博士,Baylor Medical Centerの親切な贈り物)を用いてPCRを介して得られたiCP9 cDNA配列の連結によって修飾し、新しいプロモーター配列の下流にクローン化した。
【0160】
次いで、3つのベクター、pLpCMV(iCP9)MG、pLpMBP(iCP9)MG、pLpGFAP(iCP9)MG、pLpPDGFR(iCP9)MGを用いて、高力価の複製不能レンチウイルスを得た。ウイルス力価は293T細胞に対して決定し、続いて、全てのベクターがEGFPを共発現するので、EGFP発現についてのFACS分析を行った。イン・ビボで用いた全ての力価は、ml当たり少なくとも109コロニー形成単位であった。この結果、EGFP発現を通じての感染した細胞の同定を可能としつつ、誘導性自殺遺伝子(iCP9)の発現のイン・ビトロおよびイン・ビボでの細胞への導入のためのビヒクルが得られた。というのは、それは同一ベクター内のウイルスプロモ
ーター配列から駆動されるだろうからである。また、それは、いずれのプロモーターを用いたかに基づいて遺伝子発現を特定の細胞型に限定し、これは、時間制御様式で系の細胞型特異的除去を与える。
【0161】
293T細胞を、リポフェクタミン(Invitrogen,Carlsbad,CA)を用い、gag−polおよびRD114エンベロープ蛋白質をコードするプラスミドと共にpΔmbpICP9/mEGFPベクターでトランスフェクトした。トランスフェクションから48時間後に、ウイルス上清を収穫し、標的細胞に直接的に適用するか、あるいはスナップ凍結させ、−80℃で貯蔵した。
【0162】
適時に細胞死を誘導すること。収集されたウイルスを用いて、樹立されている初代乏突起神経膠細胞培養物を形質導入した。細胞を培養し、GFPの発現について蛍光顕微鏡検査法を用いて評価した。GFP陽性細胞の程度はウイルス感染効率に相関した。細胞蛋白質の単離を行い、蛋白質単離体をウエスタンブロット導入に付して、iCP9蛋白質の安定な発現を検出した。得られた膜を抗−カスパーゼ9抗体(R&D Systems Inc.#AF8301)でプローブした。一旦ウエスタンブロット技術で蛋白質レベルにおいてiCP9の安定な発現が確認されたならば、CIDを10nM濃度にて培養物中のこれらの細胞に適用した。これは感染した細胞の細胞死を誘導した。細胞死は、蛍光顕微鏡下で培養物を観察することによって、およびCIDへの暴露の後に培養物中の細胞のメチレンブルー(Sigma)生存性染色を用いることによって検出した。全ての実験は、陰性対照として神経膠星状細胞−由来U87細胞系を用いて並行して行った。
【0163】
CIDの存在下でアポトーシスを誘導し、および細胞型選択性を示すために、レンチウイルスをイン・ビトロ培養物に適用した。アポトーシスは、最初にiCP9の構成的発現を生じるpLpCMV(iCP9)MGウイルスを用いて293T細胞において誘導した。EGFPは同一のウイルスプラスミドおよびプロモーターからやはり生産される。CIDの添加後に、(ウイルスでトランスフェクトした細胞に対応する)EGFP−陽性細胞は迅速にアポトーシスを受け、培養物中に同定されなかった。しかしながら、ウイルスでトランスフェクトされていない細胞(EGFP陰性)は培養物に持続した。対照ウイルス、pLpCMVXMGでトランスフェクトされた同一培養物において、EGFP−陽性細胞はCIDへの暴露の後に持続した(図5)。また、細胞死検出(Cell Death
Detection) ELISA Plus(Roche Inc)を行って、ウイルスの種々の希釈物で感染させた細胞をCIDに暴露した後に、培養物におけるアポトーシスのレベルを決定した。該ELISAシステムは、アポトーシスを受けている細胞によって生産されるヒストン−複合体化DNA断片を検出する。該ELISAのデータは、感染、引き続いての遺伝子発現および細胞死の間の相関を示す(図6)。
【0164】
可能な代替細胞死メディエーターシステムは、モノクローナルキメラ抗−CD20抗体によって活性化されて、アポトーシスを誘導することができる、E.coli−由来サイトシンデアミナーゼ遺伝子、HSV−tkシステムおよびトランスジェニックCD20の使用を含む。
【0165】
結果は、pΔcmvICP9/mEGFP由来レンチウイルスでの標的細胞(例えば、初代乏突起神経膠細胞)の効果的感染を示した。感染率は、予備的データに基づき、培養物中の標的細胞のほぼ100%に近付くはずである。さらに、遺伝子の乏突起神経膠細胞特異的発現を駆動するためにMBPプロモーター配列を用い、GFPは乏突起神経膠細胞において検出されるがU87対照細胞系(または他の対照細胞系、例えば、NIH 3T3)においては検出されない。CIDを含むiCP9システムを用い、CIDの培養物への投与は、乏突起神経膠細胞腫細胞系において迅速かつ有効な細胞死を生じると期待されるがU87細胞系においてはそうでない。
【0166】
CIDのイン・ビトロ系への添加。CIDを、50ng/ml NGFを含むNeurobasal培地中で10nmの最終濃度で前記した混合皮質培養物に加えた。乏突起神経膠細胞死への最初の時間は、かくして、前記したように確立され、ニューロン応答の分析についての最初の時点を表す。第2の時点は、24および72時間分析における亜急性応答を表わした。最後に、乏突起神経膠細胞の喪失に対する慢性応答はCID投与から1週間、2週間および2カ月後に分析した。
実施例2:乏突起神経膠細胞の喪失
iCP細胞死系を乏突起神経膠細胞初代細胞培養物に適用し、細胞死の有効性、ならびに時間経過を規定した。第2に、該系を高密度皮質培養物に適用して、MSで起こる乏突起神経膠細胞の急性喪失をモデル化した。
【0167】
この系を用い、乏突起神経膠細胞の喪失に対するニューロンの応答を経時的に規定した。これは、公知の生存、栄養およびアポトーシス経路の変化を観察することによって、ならびにそのような事象に対する新規な応答を同定するためのマイクロアレイ分析を通じて達成された。
【0168】
初代乏突起神経膠細胞培養物の樹立。乏突起神経膠細胞の富化された集団をFischer P2ラットから単離した。前脳をハンクスの緩衝塩溶液中で切開した。ポリL−リシン−被覆25cm2フラスコ中で組織をほぼ1−mm片に切断し、次いで、0.01%トリプシンおよび10μg/ml DNaseにて、湿潤化5%CO2部屋空気インキュベーター中で37℃で15分間インキュベートした。このインキュベーション時間に続き、20%胎児ウシ血清(FCS)を補足した、DMEM培地を組織混合物に加え、次いで、湿潤化5%CO2部屋空気インキュベーター中で10日間増殖させた。10日後に、乏突起神経膠細胞前駆体細胞(A2B5+)を、フラスコを200rpm、37℃にて一晩振盪することによって収集した。フラスコに接着したままの細胞を、線維芽細胞成長因子(FGF)および血小板由来成長因子(PDGF)を10ng/mlの最終濃度まで補足した高−グルコースDMEM中で1週間培養した。その後、FGFおよびPDGFを培養基から取り出し、A2B5+細胞を3ないし7日間にわたってO4+前髄鞘形成乏突起神経膠細胞に、次いで、7ないし10日間後にO4+およびMBP+成熟乏突起神経膠細胞に分化させた。細胞分化の状態は、培養物における細胞の形態の変化をみることによってモニターした。成熟MBP+乏突起神経膠細胞への分化が、顕微鏡調査で容易に同定される。分化を確認するために、パターンを細胞形態の変化として光学顕微鏡検査法で観察し、抗−A2B5,抗−O4および抗−MBP抗体を、分化経路における種々の時点において、培養された細胞のアリコットを用いる基本的免疫組織化学染色プロトコルで利用した。
【0169】
一旦この系および分化パターンを確認したならば、A2B5+初代細胞培養物を、pΔmbpICP9/mEGFPウイルス上澄み中で8時間培養した。8時間後に、上清を取り出し、24時間、DMEM/FGF PDGF培地に交換した。次いで、細胞を蛍光顕微鏡検査法下で可視化して、感染の有効性を決定した。感染された細胞はEGFPを発現し、適当な蛍光フィルター系を用いて容易に同定された。
【0170】
pΔmbpICP9/mEGFPでの有効な感染を確認した後、培養されたGFP+、A2B5+細胞のアリコットを前記したように培養物中で分化させる。成熟MBP+乏突起神経膠細胞が培養物で同定された後、CIDをDMEM/20%FCS中10nMの最終濃度まで加える。多数の培養物の維持は、CIDの添加の後の種々の時点におけるアポトーシスの分析を可能とした。CIDを培養上清に加えてから1、4、12、および24時間後に、培養物のメチレンブルー染色を行った。メチレンブルー陽性細胞がかくして同定可能であり、生きた細胞を表す。この実験はpΔmbpICP9/mEGFPウイルス
上清に暴露されていない(従って、アポトーシスを受けないはずである)A2B5+細胞を用いて二連で行った。従って、細胞の100%についての死滅までの時間はCIDの添加の後に確立された。
実施例3:高密度混合皮質培養物
高密度混合皮質培養物を、従前に記載されているように、3匹のFischerラットからの大脳皮質の単離後に樹立した。大脳皮質を取り出し、氷冷ハンクスの平衡塩溶液に入れ、遠心し、トリプシンで37°で10分間消化した。組織を遠心し、加熱−不活化胎児ウシ血清およびウマ血清を含有するEarleの塩(Invitrogen)を含む最小必須培地に再懸濁させた。細胞懸濁液を細胞ストレイナーに通し、平板培養した。3時間後に、培地を、B27 Minus AO(Invitrogen)を補足したNeurobasal培地に代える。培養物を7日間維持し、次いで、免疫組織化学染色を行って、培養物の組成を確認した。
【0171】
(全てのCNS細胞型よりなる)混合初代皮質培養物をpLpMBP(iCP9)MGで感染させ、CIDに暴露し、次いで、乏突起神経膠細胞の細胞死について分析した(MBP+細胞)。MBPプロモーターは、その大部分が成熟乏突起神経膠細胞であるMBP+細胞中でのみ細胞死を誘導するように設計した。CID発現に先立って、細胞は形態学的に正常に見え、対照培養物およびウイルスによりトランスフェクトされた培養物の細胞カウントは、細胞型の間の有意な差を示さず、これは、ウイルストランスフェクションが培養物の構成を有意に改変しなかったことを示す。CID暴露の後MBP−陽性、EGFP−陽性細胞はCIDに暴露しなかったMBP−陽性、EGFP−陽性細胞に対して破壊されているように見えた(図7)。さらに、A2B5(乏突起神経膠細胞前駆体細胞)/EGFP陽性細胞、GFAP(神経膠星状細胞)/EGFP陽性細胞およびO4陽性細胞(乏突起神経膠細胞前駆体細胞)/EGFP陽性細胞はCID暴露の前および後に形態学的に正常に見えた。
【0172】
GFAPプロモーター系の特異性をテストするために、GFAP+パン精製培養物(精製された神経膠星状細胞の培養物)をpLpGFAP(iCP9)MGでトランスフェクトし、CIDに暴露した。pLpGFAP(iCP9)MGで感染させたパン精製GFAP細胞をCIDに暴露し、4時間後に、EGFP陽性細胞の減少が示され、他方、CIDに暴露されなかった同様な培養物はEGFP陽性細胞の高いレベルを維持した(図8)。これらのデータは、GFAPプロモーターがiCP9発現を駆動し、その結果、CID暴露の後に神経膠星状細胞のアポトーシスがもたさられることを確認した。
【0173】
PDGFR−αプロモーター系の細胞特異性を確認するためにラット−由来初代皮質培養物をpLpPDGFR(iCP9)MGウイルスで感染させた。混合初代皮質培養物(全てのCNS細胞型を含有する培養物)において、A2B5−陽性細胞(A2B5発現はPDGFR発現と重複し、抗−PDDGFR抗体の代わりに用いられる、なぜなら、それはより労働集約的ではないからである)は、改変された形態を伴うアポトーシスを受けていると同定され、他方、他の細胞型は影響されていないように見える(図9)。これは、PDGFR−αプロモーター系のオリゴ前駆体−特異性を支持するデータを提供する。
実施例4:急性傷害に対するニューロン応答の分析
応答の4つの主なクラス:アポトーシス誘導因子、抗−アポトーシス因子、神経栄養因子および神経栄養関連転写因子を分析する。神経栄養因子の分析は、製造業者のプロトコルに従い、GDNF、CNTF、BNDF、NGF、NT−3およびNT4/5についての市販のELISAアッセイを用いて行う。混合皮質培養物からの24時間上清を記載された時点においてアポトーシス後に収集し、そしてCID投与されていない混合皮質培養物からの上清を収集し、それは対照として役立つ。全ての試料は二連で行い、最終のデータは2つの値の平均を表す。
【0174】
神経栄養関連転写因子NF−κBおよびCREB活性化の分析は、製造業者の指示に従い、Trans AMP−CREBおよびNF−κB p65キット(Active Motif Europe,Rixensart,Belgium)を用いて行う。該Trans−Amアッセイは、96−ウエルプレートに被覆された環状AMP−応答エレメントおよびNF−κBコンセンサス部位(5’−GGGACTTTCC−3’)を含有するオリゴヌクレオチドに特異的に結合することができる細胞抽出物に含まれるリン酸化CREBおよびNF−κBの活性形態のレベルを測定する。二次ホースラディッシュペルオキシダーゼ−コンジュゲーテッド抗体は、450nmにおいてELISAプレ−トリーダー(BIORAD)を用いて定量されるであろう感度が良い比色の読みを提供する。全ての試料は二連で行う。細胞抽出物は、従前に規定された時点において誘導アポトーシス後の混合皮質培養物から由来する。対照細胞抽出物は、CIDに暴露されていない混合皮質培養物よりなる。
【0175】
混合皮質培養物中での乏突起神経膠細胞の喪失に続いてアポトーシスを受けているニューロンの数は、製造業者のプロトコルに従い、TdT−媒介dUTPニック末端標識アッセイ(TUNELアッセイ,Roche,Indianapolis,IN)を用いて定量する。陽性細胞の数を、顕微鏡検査法を用いて定量し、CIDに暴露されていない培養物中の陽性細胞の数と比較する。抗−FAS抗体および抗−NOS抗体を用いて免疫組織化学(IHC)染色を行って、乏突起神経膠細胞の喪失後における混合皮質培養物からのニューロンでの発現を定量し、CIDに暴露されていなかった培養物と比較する。染色は、プロトコル当たり、VECTASTAIN ABCキット(Vector Laboratories Inc.,Burlingame,CA)を用いて行う。
【0176】
抗アポトーシス分子はニューロン生存において役割を演じることができるか、あるいは、逆に、発現の減少はニューロン喪失に寄与することができる。新規な因子E4BP4は、CNSにおけるニューロンで重要な抗アポトーシス機能を演じるように見える。従って、E4BP4の発現のレベルは、混合皮質培養物中での乏突起神経膠細胞の喪失に続いてアポトーシスを受けているニューロンにおける免疫組織化学(IHC)染色を介して調べ、CIDに暴露されていなかった培養物と比較する。IHC染色は前記したように行う。
実施例5:急性乏突起神経膠細胞によって調節されたユニークな分子の同定
乏突起神経膠細胞の急性喪失および慢性不存在は、中枢神経系におけるユニークな病理学的状態を表し、従って、新規な遺伝子はこれらの事象に応答してニューロンでアップレギュレートされ、同定される。これらの遺伝子を検出するためには、マイクロアレイ分析を行う。傷害後種々の時点において、乏突起神経膠細胞の誘導されたアポトーシス後に残る培養物から、RNeasy Miniキット(Qiagen,Valencia,CA)を用いて全RNAを単離する。対照アレイを生じさせるには、CIDを差し控えることによってイン・ビトロにて、乏突起神経膠細胞死に暴露されていない培養物から全RNAを同様に単離する。RNAプロセッシングおよび分析は、標準的なプロトコルを用い、Case Western Medical Schoolのコア施設によって行う。走査された出力ファイルはハイブリダイゼーションアーティファクトについて視覚的に調べられるであろう。アレイを平均強度に対して一定の基準で決め、次いで、Affymetrix Microarray5.0ソフトウエアを用いて分析する。もし発現が対照RNAに対して<1.5倍変化すれば、遺伝子はアップレギュレートされていると考えられる。
【0177】
マイクロアレイの結果を培養物の免疫組織化学染色で確認して、もし抗体が入手可能であれば、増加発現レベルを示す。抗体が入手可能でない場合、遺伝子配列に基づいて定量的RT−PCRを行って遺伝子発現のアップレギュレーションを確認する。簡単に述べれば、β―アクチンはベースライン遺伝子発現として働く。プライマーはcDNA配列分析に基づいて購入する。BioRad iCyclerを用いてリアル−タイムPCRを行
い、コンピューターは閾値サイクルについての標準曲線を計算する。平均閾値サイクルは各試料について3つのウエルから計算され、平均TCおよび標準曲線を用いて、試料のmRNAの量を外挿する。各細胞培養物において、mRNAはβ―アクチンmRNAの割合として定量され、対照培養物からの平均割合を比較する。
【0178】
pΔmbpICP9/mEGFPウイルスの乏突起神経膠細胞先駆体細胞への投与の結果、ほとんど100%のGFP陽性細胞がもたらされる。乏突起神経膠細胞が成熟するまでiCPの発現は起こらず、CIDの投与の結果、他の細胞型における公開されたデータに基づき、4ないし6時間にわたる乏突起神経膠細胞の細胞死がもたらされる(Straathofら、2005)。虚血症または低酸素症に対するもののような予め規定された経路の最初の分析は、乏突起神経膠細胞の喪失後の急性期において培養ニューロンで明らかであろう。なぜなら、ニューロンにおける有害事象に対する通常の応答が最も可能性があるからである。しかしながら、乏突起神経膠細胞喪失から過ぎた時間が長いほど、応答は公知の負傷応答から隔たり得る。さらに、乏突起神経膠細胞の喪失に続いてのニューロンのマイクロアレイ分析の結果、対照ニューロンと比較して、2ないし4、2ないし6、または5ないし10の新規かつ関連する遺伝子のアップレギュレーションおよび同定がもたらされる。そのようなプロファイルは、急性期におけるよりはむしろ、乏突起神経膠細胞喪失から72時間ないし1週間後に同定される。
実施例6−動物モデル
誘導性カスパーゼ9配列を含有する前記したウイルスpΔmbpICP9m/EGFPを成体ラットのCNSに適用して、CNS脱髄のイン・ビボモデルを作成した。ウイルスベクターの投与は、投与の速度(対流性分布)およびウイルス力価に基づく、実質の可変領域における遺伝的情報の有効な導入を可能とした。データは、CNSの領域がレンチウイルスで効果的に感染されることを示した。まず、成体Fischerラットをケタミン塩酸塩(80mg/kg)およびキシラジン(4mg/ml)の腹腔内注射によって麻酔し、定位フレームに入れた。中央頭皮切開を行う。脳実質へのアクセスは、病巣の所望の位置に依存し、種々の所定の座標において、頭蓋を通って右側穿頭孔に設置することによって達成された。標的領域サイズに依存した速度にてNo.32S−ゲージ針を備えた滅菌10μlハミルトンシリンジを用いてレンチウイルスを無血清培地で投与した(ウイルスの対流性分布は0.1ないし0.5μl/分の速度で最も有効である)。送達されたウイルスベクター(例えば、レンチウイルス)の容量は、本明細書中で決定される感染有効性および標的領域のサイズに依存する。注射の後に、針を5分間所定の場所に残し、次いで、次の4分間にわたってゆっくりと引き出す。皮膚を縫合糸を用いて閉じた。
【0179】
脊髄へのウイルス投与では、脊柱胸部において切開を行い、続いて、ラメネクトミーを行って、脊髄を露出させた。No.32S−ゲージ針を所定の座標において後柱に通した。ウイルスの注射後種々の時点において、CIDを腹腔内注射を介して投与して、CNSの感染した領域のアポトーシスをもたらした。
【0180】
急性脱髄後の可逆的生理学的機能不全の確認。データに基づき、ウイルスベクターを利用して、CNSの領域を首尾よく感染させ、導入遺伝子を有効に発現させた。急性乏突起神経膠細胞の死滅の生理学的発現は、まず、脳病巣を持つラットにおいて経時的に観察された。脊髄における病巣では、より正確な記録メカニズムを使用した。後柱における軸索の機能的完全性は、体性感覚惹起電位記録(SSEP)を用いてイン・ビボで調べる。
【0181】
CIDの投与後種々の時点において、ラットをSSEPの記録のために従前に記載されたように麻酔した。SSEPは、ハードプレート下に置かれたAg/AgClディスク電極を参照して、右側体性感覚皮質上のスクリュー電極から記録し、他方、対側性坐骨神経は1Hzで刺激する(200掃引の平均についての0.2msパルス持続、および40mA一定電流強度)。接地電極は頭皮に経皮的に置いた。SSEPの振幅は正ピークに対す
る最初の負ピークから測定される。応答潜時は、刺激の開始および最初のピークの間の時間として測定される。振幅および潜時の値は3つの独立した測定の平均として記録した。これらの測定を、CIDの投与から1、2、7および14日後に反復して、CNS損傷および修復の測定可能なパターンを確立した。急性病巣の免疫組織化学分析。iCP9乏突起神経膠細胞死に関連する細胞変化を評価するために、動物を、CIDの投与後種々の時点で犠牲にした。
【0182】
ラットの脳または脊髄を、イソペンタン中で20秒間スナップ凍結させ、次いで、セクションニングまで―80℃で貯蔵した。10μm厚さの脳の薄い冠状切片および脊髄の軸方向の切片をクリオスタット(−20℃)を用いて作成した。まず、これを死亡後1日において行って、CID誘導細胞死の成功を決定した。その後、CID後1、2、7および14日動物を犠牲にし、蛍光顕微鏡検査法、ルクソール速青色染色およびヘマトキシリンおよびエオシン(H&E)染色を用いて調べた。脳および脊髄の薄い切片を種々の抗体を用いる免疫組織化学染色を介して調べて、起こった、炎症(抗−LCA抗体、抗−ED1)および神経膠症(抗−GFAP抗体)の程度を決定した。
【0183】
図2中の写真は、濃縮されたレンチウイルスがCNSに直接的に適用でき、感染の程度および領域は、脳または脊髄の薄い切片における犠牲後に検出されたGFP陽性細胞の数に基づいて決定できることを示す。このデータは、本明細書中で提案されたモデルの基礎であるイン・ビトロおよびイン・ビボでの特異的細胞死についての導入メカニズムを確立する。
実施例7:急性病巣における軸索相互作用およびニューロンアポトーシスの同定
薄い切片を用いて、横断軸索またはアポトーシスニューロンの形態の病巣の部位における病巣ニューロン損傷を検出する。免疫組織化学染色を、抗−アミロイド前駆体蛋白質(APP)抗体を用い、脱髄病巣を含む薄い切片で行う。これは、APPは当該軸索の末端で蓄積するので、軸索の輸送および横断の乱れを同定する。同様に、Bielschowskys銀含浸染色を用いて、ニューロンおよび横断軸索を検出する。固定された切片を予め温められた(40℃)10%硝酸銀で10分間染色し、次いでPBS中で洗浄する。水酸化アンモニウムを硝酸銀溶液に加え、スライドを40℃にて30分間インキュベートし、その時点の後、スライドを発色剤作用溶液(40%ホルムアルデヒド、クエン酸、硝酸溶液)に直接的に1分間入れる。
【0184】
反応を1%水酸化アンモニウムで停止し、PBS中で洗浄し、次いで、5%チオ硫酸ナトリウム中で5分間インキュベートする。最後に、スライドを脱水し、マウントする。この染色技術を用い、横断軸索を同定することができ、軸索の数をカウントし、ウイルスで感染されているが、CIDを投与されておらず、従って、脱髄の病巣領域を有しない対照動物と比較する。ニューロンを細胞死後種々の時点で急性病巣中で、および急性病巣の周りでカウントし、ウイルスを受けているが、CIDを受けておらず、従って、脱髄の病巣領域を有しない動物に由来する薄い切片と比較する。各病巣領域または離れた正常領域の中央を通じて取られた、目での形態計測グリットによって規定された0.01mm2視野を調査のために選択する。この視野において、APP陽性線維およびBirelschowskys銀含浸線維を100×対物レンズ下でカウントする。
実施例8:急性脱髄病巣後の離れたニューロン喪失の同定
CNS中の脱髄の結果、病巣を横切る軸索を持つ離れたニューロンの死滅がもたらされるか否かを決定するために、種々の時点において、脊髄の側方束中での脱髄病巣の生成に続いて、対側性赤核においてニューロンを調べる。脊髄における乏突起神経膠細胞死の誘導後におけるラットの赤核中のニューロンの数を、CIDを受けていないウイルスを感染させたラットの赤核における数と比較する。また、TdT−媒介dUTPニック末端−標識アッセイ(TUNELアッセイ,Roche,Indianapolis,IN)を利用して、アポトーシス活性を検出する。これは製造業者のプロトコルに従って薄い切片で
行う。皮質下病巣負荷および脊髄萎縮。広範な脊髄萎縮はMSのよく規定された特徴である。CNSの離れた領域における脱髄事象は脊髄萎縮を誘導し、この誘導は脱髄負荷の程度に関連することを決定する。
【0185】
従って、従前に記載されているように、ウイルスベクター(例えば、iCP9)をCNSの皮質下領域に送達し、CIDの送達の結果、脱髄領域がもたらされる。元来の傷害ラットが犠牲にされてから8カ月後に、脊髄を取り出し、スナップ凍結させ、薄い切片を調製し、H&E染色する。脊髄の直径を測定し、iCP9ウイルスで同様に感染されるが、CIDを受けず、従って、急性脱髄病巣を有しない対照動物と比較する。病巣の数、位置、ならびに病巣の間の時間間隔は、全て、脱髄領域および頻度のより大きな程度を生じさせるために変化させ、脊髄における萎縮を検出する可能性を最大化することができる。
【0186】
最後に、結果は、生存またはアポトーシス因子の変化を同定するか、またはイン・ビボモデルを用いてユニークな遺伝子を確認する。これは免疫組織化学染色を用いて行われる。また、結果はイン・ビボモデルでアッセイすることもでき、これは、脱髄に対するニューロン応答を研究するためのもう1つの生理学的基盤を提供する。脊髄における乏突起神経膠細胞の急性喪失の結果、急性生理学的機能不全およびSSEP記録の測定可能な変化がもたらされ得る。具体的には、振幅の減衰および潜時の増加が同定される。これは決定される時間経過にわたってのベースラインを修正する。というのは、ラットは最初の傷害から回復するからである。細胞死後にルクソール速青色(Luxol Fast Blue)染色を用い、急性脱髄の領域を規定する。炎症性浸潤の程度およびタイプは免疫組織学染色を用いて決定される。
【0187】
かくして、温和な非特異的炎症性浸潤が続いて起き、数週間にわたって消失し得る。MS患者からの病理学的検体において示されているように、横断軸索が急性病巣内に局在化する。最後に、多数の離れた脱髄病巣の誘導の後に赤核中の生きたニューロンの数の減少が観察される。喪失の時間経過および程度は経時的にゆっくりと起こり得るが、例えば、脱髄から約6ないし12カ月の後に同定することができる。
実施例9:カスパーゼ−9を発現するトランスジェニック動物
ミエリン塩基性プロモーターの制御下にあるカスパーゼ−9を発現するトランスジェニックマウスは、まず、トランスジェニック標的化ベクター構築体を生じさせることによって作り出される。種々の核酸エレメントを組み込んで、マウスにおけるカスパーゼ−9の発現を確実とする。合成イントロンエレメントを、ベクターに由来するプレ−mRNAの適切なプロセッシングのためにカスパーゼ−9 cDNAの前面に置く。ポリアデニル化シグナルを、mRNAの適切なプロセッシングのためにカスパーゼ−9 cDNAの末端に組み込む。カスパーゼ−9の誘導性発現を可能とするために、2つのLoxP部位(Cre−レコンビナーゼ認識部位)によってフロックスされた停止コドンをミエリン塩基性プロモーターおよび合成イントロンの間に組み込む。このベクターは、ゲノムへのベクターの有効な組込みのために線状化され、マイクロインジェクションによって多数の前核に注射される。注射された前核を偽妊娠FVB/N株マウスに移植する。動物の子供を、注射されたベクターの宿主ゲノムへの組み込みについてスクリーニングする。スクリーニングにおいて陽性と同定された動物の子供を離乳させ、ER−Cre導入遺伝子を含有するトランスジェニックマウスと交配させる。交配からの動物の子供を、カスパーゼ−9およびER−Cre導入遺伝子双方を保有する動物についてスクリーニングする。タモキシフェンを腹腔内に注射してER−Cre導入遺伝子からのCre蛋白質の発現を誘導する。LoxP部位の切り出し、およびミエリン鞘におけるカスパーゼ−9の結果としての発現は、抗−カスパーゼ−9抗体を用いて免疫蛍光染色で確認される。
実施例10:イン・ビボでのアポトーシスの細胞特異的誘導
該系をイン・ビボでテストして、細胞特異的アポトーシスを誘導することができ、および細胞除去のタイミングはそれがイン・ビトロであるように制御されたことを確認した。
この目的で、ウイルスを成体Fischerラットの脳梁に注射し、次いで、3週間後に、CIDを同側脳室に投与した。それから24時間後にラットを犠牲にし、脳を全部摘出し、固定し、分析のために低温切断した。ラットにpLpGFAP(iCP9)MG(GFAPプロモーターは発現を神経膠星状細胞に制限する)およびその後CIDを注射し、24時間後に犠牲にし、ウイルス感染の部位におけるTUNEL陽性染色を示した(というのは、該ベクターは、EGFPの構成的な発現をもたらすので、EGFP陽性細胞によって境界が画定されるからである)。TUNEL(TdT−媒介dUTP−Xニック末端−標識)系は、アポトーシスカスケードの間に断片化されたDNAにタグを加え、標準的に免疫組織化学を用いる標識を可能とした。CIDよりはむしろグリセロールを受ける対照ラットに由来する薄い切片はTUNEL陰性であった。次いで、連続した薄い切片を抗−GFAP抗体で染色して、神経膠星状細胞を同定した。GFAP+細胞は、グリセロールを受ける対照ラットに由来する切片とは対照的に、CIDが投与されたラットに由来する薄い切片におけるウイルス感染の領域で検出されなかった(図10)。これは、イン・ビボにて、iCP9系がCID投与後のみにアポトーシスを誘導し、GFAP+神経膠星状細胞を効果的に除去することを確認した。
【0188】
同様に、(乏突起神経膠細胞を除去するように設計された)pLpMBP(iCP9)MGを注射し、CIDに暴露されたラットは、感染の部位において抗−MBP抗体で染色されず、他方、pLpMBP(iCP9)MGで感染させ、グリセロールを投与した対照ラットからの薄い切片は、抗−MBP抗体を用いる免疫組織学染色後に正常に見えた。TUNEL陽性細胞はCID暴露後に感染の部位において同定されたが、グリセロールのみに暴露された対照においては同定されなかった(図11)。
【0189】
これらのデータは、iCP9系が時間制御様式でイン・ビボでアポトーシスを誘導でき、利用されたプロモーター配列に基づいて細胞−型に特異的なように見えることを確認した。
実施例11:CNSにおける急性乏突起神経膠細胞アポトーシスに対する細胞応答
脊髄および脳双方におけるラットでの脱髄および修復プロセスは、ケタミン塩酸塩(80mg/kg)、アセプロマジン(2.1mg/kg)、およびキシラジン(4mg/ml)の腹腔内注射によって麻酔され、および定位フレーム(Stoelting Co)に入れられたラットの3日齢子供(P3)を用いることによって規定される。No.26S−ゲージ針を備えた10μlのハミルトンシリンジを、ブレグマの0.5mm前方かつ0.5mm側方の柔軟な頭蓋を0.2mmの深さで通す。無血清培地中の5マイクロリットルのpLpMBP(iCP9)MGウイルス(乏突起神経膠細胞を選択的に除去する)を、No.26S−ケージ針を備えた10μlのハミルトンシリンジを用いて2μl/分の速度で注射する。胸脊髄への注射を受けるラットについては、No.26S−ゲージ針を備えた10μlのハミルトンシリンジを手動で脊髄に通す。次いで、マイクロインジェクションシステム(Harvard Co)によって、脳におけるのと同一速度でウイルスを注射する。注射の後に、針を5分間所定の場所に残し、次いで、次の4分間にわたってゆっくりと引き抜く。
【0190】
ウイルスの注射から3週間後に、5μlのCID(10nm)を同側脳室(3.6mmの深さにてブレグマから−0.8mm前方かつ1.4mm側方)に注射し、その結果、感染した乏突起神経膠細胞のアポトーシスがもたらされる。1日置きに、CID注射の日に開始し、動物は、増殖細胞を取り込み、免疫組織化学を用いるそれらの同定を可能とする100mg/kgのBrdU標識ミックス(Sigma)の腹腔内注射を受ける。ラットをCIDから1,7,14、21および28日後に犠牲にする。
【0191】
対照ラットには、CIDよりはむしろグリセロールの注射を受ける(CIDはグリセロール中に希釈する)以外は実験動物と同様にウイルスを注射する。ラットを深く麻酔し、
まず、150mlの0.9%NaCl生理食塩水で、次に等容量の氷冷4%パラホルムアルデヒドで経心的に灌流する。次いで、全脳および/または脊髄を摘出し、パラホルムアルデヒド中で少なくとも4時間、後固定し、続いて、組織が容器の底に沈むまで、30%スクロース中で低温保護する。次いで、試料をOCT中で凍結させ、スーパーフロスト+スライド上で10ないし20μmにて低温切断する。この実験を同様に脊髄において反復する。
免疫組織化学。CIDから1、7、14、21及び28日後の動物から由来する組織を、蛍光顕微鏡検査法を用いて調べて、感染の領域を同定する。急性脱髄および引き続いての髄鞘再形成の領域を規定するために、ミエリンの黒色金染色を、各時点において急性乏突起神経膠細胞の死滅の誘導後に薄い切片で行う。この実験における脱髄までの時間は、感染の領域が黒色金で染色されない時点と定義され、およびこの実験における髄鞘再形成までの時間は、黒色金が隣接する影響されていないミエリンと同等な強度で感染の領域を染色する脱髄後の時点と定義され、これを各動物について記され、そして平均および標準偏差を計算する。これらのデータを、対照動物についての脱髄までの時間および髄鞘再形成までの時間と比較する。統計学的解析は行わない。というのは、対照は脱髄/髄鞘再形成すると予測されないからである。しかしながら、これらのデータ点はさらなる実験のための対照として働く。
【0192】
脳および/または脊髄の薄い切片を調べて、抗−CD45抗体(TおよびB細胞)、抗−ED1抗体(小神経膠細胞)、抗−CD68抗体(マクロファージ)を用いて炎症の程度、および抗−GFAP抗体(神経膠星状細胞)を用いて神経膠症を決定する。これは、各時点においてなし、同等な対照動物に由来する標識された薄い切片と定量的に比較する。
【0193】
また、切片を抗−ネスチン抗体(多能性幹細胞)、抗−NG2+抗体(神経膠に定められた幹細胞)、抗−PDGFR−アルファ(OPCの全プール)、抗−O4+抗体(O1陰性である場合の初期OPC系)、抗−O1抗体(後期OPC系)および抗−MBP抗体(成熟乏突起神経膠細胞)で染色して、幹細胞および乏突起神経膠細胞前駆体細胞の動員、および動的病巣部位への取り込みを同定する。これらの抗体は、CNS幹細胞から成熟乏突起神経膠細胞への経路に沿って異なる細胞を同定する。次いで、薄い切片を、蛍光顕微鏡検査法を用いて調べ、100×対物レンズ下で、目での形態計測グリットによって規定された0.01mm2の視野において標識された細胞をカウントする。病巣内の4つの異なる位置をランダムに選択し、カウントし、各時点における各切片についての平均細胞数および標準偏差が計算される。これを、脱髄病巣を有しない反対側半球中の対応する部位と、およびウイルスで感染されたが、CIDを受けず、従って、脱髄病巣を有するはずのない対照動物と比較する。対照動物におけるカウンティングのための領域は、EGFP+細胞の存在によって同定されるウイルス注射の部位である。各領域についての平均細胞カウントを対応t−検定を用いて比較する。これを各時点において用いた各抗体について反復して、いずれの乏突起神経膠細胞のサブタイプが病巣に移動するか、およびこの移動の時間的分布を決定する。
【0194】
CID暴露の後に、ラットにBrdU標識ミックスを注射する。分裂の間に、BrdUは細胞のDNAに取り込まれ、抗−BrdU抗体の適用、および標準免疫組織化学によって分裂された細胞の同定を可能とする。細胞型についての染色の完了の後に、薄い切片を、最初の標識手法で用いたものとは異なる二次抗体と共に抗−BrdU抗体で標識する。シグナルの重複は増殖する細胞型を同定する。二重に染色された切片を、同様な時点での対照切片と比較する。もし同一の増殖する細胞型が対照切片で同定されるならば、二重染色細胞を4つの切片の全同側半球でカウントし、平均し、対照切片に由来する平均カウントと比較し、次いで、標準t−検定を用いてこれを統計学的に比較することができる。もし二重標識細胞が対照で不存在であれば、定量的な尺度は二重標識細胞の存在または不存
在である。
【0195】
また、前記実験を、脊髄へのウイルス注射を受けるが、それがCIDではなくビヒクル(グリセロール)である匹敵する対照と共に脊髄において行う。脊髄脱髄についての動物の調製は、CIDが同側脳室よりはむしろ大槽に注射される点で異なる。これは、脊髄ないし頭蓋のベースに沿っての触診、続いての、この空間へのハミルトンシリンジの通過によって達成される。
実施例12:イン・ビボでの乏突起神経膠細胞の急性喪失に対するニューロンの応答
急性病巣における軸索横断の同定。脱髄病巣は、実施例11に記載したように生じさせる。動物を実施例11からのデータによって決定された時点で犠牲にする。対照ラットに、CIDよりはむしろグリセロールの注射(CIDはグリセロール中で希釈する)を受ける以外は、実験動物と同様にウイルスを注射する。ラットを犠牲にし、記載したように調製する。病巣内の軸索の応答を特徴付けるために、薄い切片を抗−ニューロフィラメント(NF)抗体および抗−アミロイド前駆体蛋白質(APP)で標識する。前者は病巣中の軸索を同定し、他方、後者は軸索の輸送および横断の乱れを同定する。NF+軸索およびAPP陽性軸索を、目での形態計測グリットによって規定された0.01mm2視野を用いてカウントし、100×対物レンズ下でカウントする。脱髄された病巣内の4つの区別される領域をカウントし平均する。
【0196】
横断軸索(APP+)のパーセンテージは、平均APP+細胞のカウントを、病巣中の軸索の平均数(NF+)で割ることによって計算する。これらのデータを、比較可能な様式で、感染の部位において行われた対照動物カウントと比較する。ニューロンのカウンティングはCID後1、7、14、21および28日に行う。単純t−検定を用いて、いずれかの与えられた時点において、対照のカウントと実験のカウントとの間の統計学的有意性を決定する。
【0197】
急性脱髄病巣後の離れたニューロン喪失の同定。CNSにおける脱髄の結果、CNSの空間的に異なる領域においてニューロンの喪失がもたらされ得るか否かを決定するために、下部胸脊髄における脱税病巣の誘導に続いて、ニューロンを黒質において調べる。ラットは、病巣の部位が胸脊髄であって、CIDを従前に記載したように大槽に注射する以外は記載したように調製される。対照ラットにウイルスを注射するが、CIDよりはむしろ大槽へのグリセロール注射を受ける。同一の時点(1、7、14、21および28日)においてラットを犠牲にし、全脳および脊髄を摘出し、記載したように調製する。脊髄を切断し、黒色金で染色して、脱髄している病巣の存在を確認する。その解剖学的位置および外観によって同定された黒質を取り込む脳切片を抗−NF1抗体で標識し、ニューロンを前記したようにカウントする。脊髄における乏突起神経膠細胞の死滅の誘導後におけるラットの黒質中のニューロンの平均数を、脱髄病巣を有しない対照ラットの黒質中での平均数と比較する。同様に、TUNEL染色を、脊髄中の脱髄病巣を持つラットに由来する黒質を含む脳の薄い切片で行って、ニューロンの活性なアポトーシスを検出する。TUNEL(TdT−媒介dUTP−Xニック末端−標識)系は、アポトーシスカスケードの間に断片化されているDNAにタグを加え、標準免疫組織化学を用いる標識を可能とする。もしTUNEL陽性細胞が黒質内で同定されれば、それらをカウントする。前記したように、TUNEL染色の結果を、対照ラットの黒質における染色およびカウントと比較し、平均カウントをt−検定を用いて比較する。これらの実験は、乏突起神経膠細胞の離れた喪失がCNSでの無関係なニューロンの生存に影響するかを決定する。
【0198】
脱髄負荷およびニューロン喪失の間の関係の決定。正常に見える脳中の離れた軸索の喪失は、CNSにおける脱髄の程度に依存し得る。これをテストするために脱髄病巣のサイズ、および脱髄病巣の数を改変する。P3ラットに、胸脊髄に、および両側脳梁にウイルスを注射する。ラットを成熟させ、P20でのラットに前記したようにCIDを注射する
。ラットをCID注射から28日後に犠牲にし、脳および脊髄からの組織を前記したように調製する。分析の時点は変化し得るが、病巣が髄鞘を再形成した後には完了する。対照ラットは同一ウイルス注射を受けるが、P20においてはCIDよりはむしろグリセロールを受ける。前記したように、本実施例においては多病巣である病巣誘導の後に、黒質中のニューロンの完全性をTUNELアッセイおよびニューロンカウンティングを介して調べる。100×対物レンズ下で目での形態計測グリットによって規定された0.01mm2視野当たりの生きたニューロンおよびアポトーシス細胞の数を、同一切片における黒質内の4つの別々の視野から平均し、同様にカウントされた対照動物と比較する。細胞カウントを標準t−検定を用いて比較する。
【0199】
急性脱髄後の可逆的生理学的機能不全の確認。後柱における軸索の機能的完全性を、体性感覚惹起電位記録(SSEP)を用いてイン・ビボで調べる。CIDの投与後の種々の時点において、ラットを従前に記載したように麻酔する。ハードプレート下に置かれたAg/AgClディスク電極を参照とした病巣上方の脊髄に挿入された電極からSSPEを記録し、他方、反対側坐骨神経を1Hzで刺激する(200掃引の平均についての0.2msのパルス持続、および40mA一定電流強度)。接地電極を頭皮上に経皮的に置く。SSEP振幅を第1の負ピークから正のピークまで測定する。応答の潜時は、刺激の開始および最初のピークの間の時間として測定される。振幅および潜時の値を3つの独立した測定の平均として記録する。これらのデータをいくつかの時点で収集するが、脱髄に先立ってベースライン、脱髄後のデータ点および修復が組織学的に起こった後の1つのデータ点も含む。対照のデータは、同一ラットにおける反対側後柱からの記録によって得られる。病巣側(実験側)からの平均振幅および潜時の値を、標準t−検定を用いて反対側後柱(対照側)と比較する。
実施例13:成体ラットでの髄鞘再形成プロセスにおける乏突起神経膠細胞前駆体細胞の役割
OPCを選択的に除去するために、新規なウイルスベクターを記載されたベクター戦略を用いて作成した。細胞の形態学変化に基づき、A2B5標識細胞は、前記したように、pLpPDGFR(iCP9)MGでの感染、およびCID暴露の後にアポトーシスを受ける。パン精製A2B5+細胞(抗体で細胞を捕獲することによってA2B5+を富化した培養物)および並行して成長させたP3由来混合皮質培養物(先に記載された培養物を記載するための方法)を感染させるが、CIDに暴露せず、標識およびELISA研究のための対照として供する。培養物の双方の組を抗−A2B5抗体およびTUNEL染色で染色して、細胞除去の特異性を決定する。A2B5発現はPDGFR−α発現と重複し、イン・ビトロで標識するのがより容易であり、従って、PDGFR−α+細胞の代理マーカーとして役立つ。混合皮質培養物を抗−GFAP(神経膠星状細胞標識)および抗−MBP(乏突起神経膠細胞標識)でやはり染色して、他の細胞型はこのウイルス構築体での感染によって影響されないことを確認する。パン−精製A2B5+細胞培養物を、pLpPDGFR(iCP9)MGウイルスの系列希釈物に暴露し、CIDに暴露する。次いで、(予備的実験に記載された)製造業者のプロトコルに従い、培養物を細胞死検出(Cell Death Detection)Elisa Plus(Roche Inc)システムに付して、アポトーシスレベルを定量する。これは、PDGFR+細胞は感染およびCID暴露の後に除去された唯一の細胞であることを確認する。次いで、全ての培養を三連で行って、再現性を確認する。
【0200】
有効性および特異性の確認に際して、pLpPDGFR(iCP9)MGウイルスをP3ラットの脳または胸脊髄に注射する。動物を成熟させ、P30の齢において、リソレシチンを同側脳梁に元来のウイルス感染の部位に注射する。リソレシチン(LPC)は、迅速な細胞膜の破壊、および軸索保持でのミエリンの急性喪失をもたらす洗剤である。LPCをラットの脳に注射してから24時間後に、動物を犠牲にして、実験手法の結果、予測されるイン・ビボでのパラダイムをもたらすことを確認する。元来のpLpPDGFR(
iCP9)MG感染の領域内のLPC誘導脱髄病巣の存在は、蛍光顕微鏡検査法を用いて薄い切片上のウイルス感染の領域を同定することによって確認される。引き続いて、EGFP+細胞を含む切片は黒色金で染色され、重ねられた脱髄病巣の存在が確認される。私は、元来のウイルス感染の領域内でLPC病巣を再現性よく私が作成できることを確認するために4匹の動物の使用を提案する。対照はなく、この実験についての統計学的分析はない。
【0201】
一旦感染および脱髄の位置が確認されれば、LPCの注射から24時間後の、PDGFR+OPCを除去するためのCIDの添加でもって実験を反復する。1、7、14、21および28日に動物を犠牲にして、ミエリン修復に対するOPC除去の効果を決定する。これらの実験についての対照は、P30において重ねられたLPC病巣を伴うP3においてpLpPDGFR(iCP9)MGウイルスで感染させた動物であるが、その後CIDよりもむしろグリセロールを受ける。従って、それらは脱髄LPC誘導病巣を有するはずであるが、OPC除去は有さず、予め規定された様式で修復されるはずである。
【0202】
重ねられたLPC脱髄病巣および引き続いてのOPC除去を持つ動物(実験群)を、黒色金染色で同定されたデータ点で髄鞘再形成するまでの時間を用いて、重ねられたLPC病巣を持つが、OPC除去を持たない動物(対照群)と比較する(実施例11参照)。さらに、LPC病巣の部位までのOPC移動に要する時間もまた記録し、群の間で比較する。髄鞘再形成までの時間、およびOPC移動までの時間を各動物について記録し、標準t−検定分析を用いて各群の統計学的平均を比較する。実施例11に記載されたように、切片を乏突起神経膠細胞の細胞型、神経膠星状細胞および炎症細胞の存在について染色する。細胞を実施例11に記載されたプロトコルに従い、標識された切片についてカウントし、平均を計算し、標準t−検定を用いて対照群における平均細胞カウントと比較する。
【0203】
最後に、CID後の1日毎にBrdUで全ての動物を注射すると、記載したように増殖する細胞の同定が可能となる。細胞型についての染色の完了の後、次いで、薄い切片を、最初の標識手法で用いたものとは異なる二次抗体と共に抗−BrdU抗体で標識する。シグナルの重複は増殖する細胞型を同定する。二重染色切片を、同様な時点において対照切片で比較する。もし同一の増殖する細胞型が対照切片で同定されるならば、二重染色細胞を4つの切片の全同側半球においてカウントし、平均し、対照切片に由来する平均カウントと比較し、次いで、これは標準t−検定を用いて統計学的に比較することができる。もし二重標識細胞が対照動物に置いて同定されたものと異なるならば、定量はない。
【0204】
この実験を脊髄において反復することができる。
【0205】
本発明は前記した実施態様に限定されず、添付の請求の範囲の範囲内で変更することが可能である。当業者であれば、慣例的なものを超えない実験、本明細書中に記載された発明の具体例の多くの同等体の使用を認識し、または確認することができるであろう。
【特許請求の範囲】
【請求項1】
本明細書の一部に記載の発明。
【請求項1】
本明細書の一部に記載の発明。
【図1】

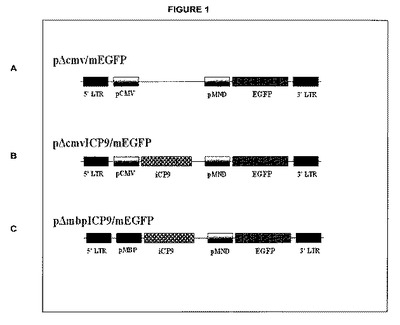
【図2】
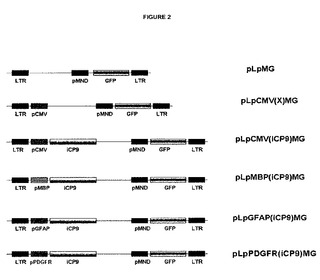
【図3】

【図4】
【図5】

【図6】
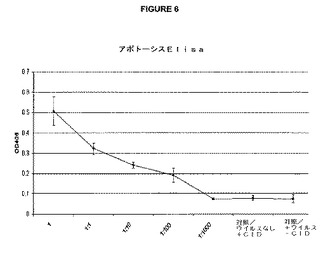
【図7】

【図8】

【図9】

【図10】

【図11】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公開番号】特開2013−81480(P2013−81480A)
【公開日】平成25年5月9日(2013.5.9)
【国際特許分類】
【外国語出願】
【出願番号】特願2013−15116(P2013−15116)
【出願日】平成25年1月30日(2013.1.30)
【分割の表示】特願2010−512360(P2010−512360)の分割
【原出願日】平成20年6月12日(2008.6.12)
【出願人】(597138069)ケース ウエスタン リザーブ ユニバーシティ (16)
【Fターム(参考)】
【公開日】平成25年5月9日(2013.5.9)
【国際特許分類】
【出願番号】特願2013−15116(P2013−15116)
【出願日】平成25年1月30日(2013.1.30)
【分割の表示】特願2010−512360(P2010−512360)の分割
【原出願日】平成20年6月12日(2008.6.12)
【出願人】(597138069)ケース ウエスタン リザーブ ユニバーシティ (16)
【Fターム(参考)】
[ Back to top ]