
標的化ゲノム変更
例えば、1つ以上の目的のポリペプチドの発現のための、1つ以上の配列の、細胞への標的組み込みおよび/または標的切除のための方法および組成物が本明細書に開示される。

【発明の詳細な説明】
【技術分野】
【0001】
技術分野
本開示は、ゲノム工学の分野、特に、細胞のゲノムへの、1つ以上の外来性配列の標的化組み込みおよび/または標的化切除に関する。
【背景技術】
【0002】
関連出願の相互参照
本出願は、2010年1月22日に出願された米国仮出願第61/336,457号の利益を主張し、その開示は、参照によりその全体が本明細書に組み込まれる。
【0003】
連邦支援による研究下でなされた発明に対する権利の記載
該当なし。
【0004】
背景
生物工学は、食物生産に対する増加し続ける世界的需要の課題に対応する努力における必須のツールとして出現した。農業生産性の改善、例えば、収率の向上または病虫害抵抗性の改変のための従来のアプローチは、突然変異育種、または形質転換による作物種のゲノムへの新規遺伝子の導入のいずれかに依存する。両プロセスは、本質的に非特異的であり相対的に非効率的である。例えば、従来の植物の形質転換方法は、無作為位置におけるゲノムに組み込む、外来性DNAを送達する。故に、望ましい属性を有する遺伝子導入系を特定および単離するために、数千の固有の無作為組み込み事象を生成し、その後、所望の個体のためにスクリーニングすることが必要である。結果として、従来の植物形質改変は手間がかかり、時間を浪費し、かつ予測不可能な事業である。更に、これらの組み込みの無作為的性質は、意図されないゲノム破壊に起因する多面効果が生じているかどうかを予測することを困難にする。結果として、改変された遺伝子または形質を有する植物株の生成、単離、および特性評価は、成功確立の低い、非常に労働集約的およびコスト集約的プロセスとなっている。
【0005】
標的化遺伝子修飾は、植物系における従来の実務の事業計画上の課題を克服するものであり、したがって、基本的な植物生物学研究および農業生物工学の両方における長年の目標でありながら、定義し難い目標であった。しかしながら、イネにおける陽性−陰性薬物選択を介した「遺伝子標的法」、または事前に改変された制限部位の使用を除いて、全ての植物種のモデルおよび作物の両方における標的化ゲノム修飾は、最近まで非常に困難であることが確認されていた。Terada et al.(2002)Nat Biotechnol 20(10):1030、Terada et al.(2007)Plant Physiol 144(2):846、D’Halluin et al.(2008)Plant Biotechnology J.6(1):93。
【0006】
哺乳類細胞において、安定な遺伝子組み換えおよび標的化遺伝子挿入は、遺伝子療法および細胞改変の両方において、多くの潜在的な用途を有する。しかしながら、現在の戦略は、しばしば非効率的であり、導入遺伝子をゲノムDNAに非特異的に挿入する。ゲノム挿入の位置を制御する能力の欠如は、ゲノム内の位置効果に起因して、集団全体にわたる導入遺伝子発現レベルの高い変動性をもたらす可能性がある。更に、安定な遺伝子組み換えおよび導入遺伝子の増幅の現在の方法は、しばしば導入遺伝子の物理的損失、経時的な導入遺伝子サイレンシング、内在性遺伝子の内側もしくはそれに隣接した遺伝子および自律的プロモーターの組み込みによる挿入変異、染色体異常の作成および再配列遺伝子産物の発現(内在性遺伝子、挿入された導入遺伝子、または両方からなる)、ならびに/または安定な導入遺伝子発現を提供するためのベクターの長期持続性の必要性に起因して永続的に発現される、ベクター由来遺伝子からのインビボのベクター関連毒性もしくは免疫原性の作成をもたらす。
【0007】
最近、ゲノムDNAの標的化開裂のための方法および組成物が記載されている。かかる標的化開裂事象を使用して、例えば、標的突然変異誘発を誘導し、細胞DNA配列の標的欠失を誘導し、かつ所定の染色体座における標的組み換えを促進することができる。例えば、米国特許公開第20030232410号、同第20050208489号、同第20050026157号、同第20050064474号、および同第20060188987号、ならびに国際公開第WO2007/014275号を参照されたく、それらの開示は、参照によりそれらの全体が全目的で組み込まれる。米国特許公開第20080182332号は、植物ゲノムの標的化修飾のための非標準的な亜鉛フィンガーヌクレアーゼ(ZFN)の使用を記載し、米国特許公開第20090205083号は、植物EPSPS遺伝子座のZFN媒介型標的修飾を記載する。加えて、Moehle et al.(2007)Proc.Natl.Acad,Sci.USA 104(9):3055−3060)は、特異的遺伝子座における標的化遺伝子付加のために設計されたZFNを使用することを記載する。
【0008】
しかしながら、植物およびその子孫における安定な遺伝性の遺伝子修飾を確立するための植物への標的化組み込みのため、ならびに遺伝子療法および細胞株育成目的の哺乳類細胞への標的組み込みのためを含む、標的化組み込みのための組成物および方法に対する必要性が依然として存在する。
【発明の概要】
【0009】
概要
本開示は、細胞ゲノムに組み込まれる、多重挿入部位に組み込まれた外来性核酸配列(すなわち、タンパク質またはRNA分子)の1つ以上の産物を発現させるための方法および組成物を提供する。細胞は、真核細胞、例えば、植物、酵母、または哺乳類細胞であり得る。
【0010】
外来性核酸配列の組み込みは、1つ以上のヌクレアーゼ、例えば、亜鉛フィンガーヌクレアーゼ(ZFN)に対する多重標的部位を含むポリヌクレオチド配列の、細胞のゲノムへのゲノム組み込みによって促進される。ポリヌクレオチド(本明細書で多重挿入部位とも称される)は、細胞のゲノム内での特異的な標的化二本鎖開裂を可能にし、その二本鎖開裂は次に、相同性依存性および相同性非依存性機構の両方を通じた外来性配列の組み込みをもたらす。
【0011】
故に、一態様では、亜鉛フィンガーヌクレアーゼ(ZFN)等のヌクレアーゼに対する1つ以上の標的部位を含む、多重挿入部位としても知られる核酸分子が本明細書に開示される。ある実施形態では、標的部位は、多重挿入部位が組み込まれる内在性ゲノム中に存在しない。多重挿入部位は、ヌクレアーゼに対する1つ、2つ、3つ、4つ、5つ、6つ、7つ、またはそれを超える標的部位を含んでもよい。ある実施形態では、隣接した標的部位(対形成した標的部位)に結合する2つの結合DNA結合タンパク質の開裂片側ドメインの二量体化が、開裂に必要とされる(例えば、各部位に対のそれぞれが結合する、一対のヌクレアーゼが開裂に必要とされる)。本明細書に記載される多重挿入部位のいずれかにおいて、各対の標的部位のうち一方の標的部位は、同じ配列を含んでもよい。例えば、図1を参照されたい。ある実施形態では、少なくとも一つの対は標的部位が同じである。他の実施形態では、少なくとも1対の標的部位は、異なる標的からの個々の標的配列(例えば、異なる遺伝子および/または異なる生物からの遺伝子)を含む。ある実施形態では、対形成した標的部位のうちの少なくとも1つは、配列番号1〜20からなる群から選択される配列を含む。ある実施形態では、多重挿入部位は、更に1つのコード配列、例えば、ホスフィノスリシンアセチルトランスフェラーゼ(PAT)コード配列を含む植物転写単位(PTU)、または哺乳類細胞と共に使用するためのスクリーニングマーカーを含んでもよい。
【0012】
多重挿入部位は、ヌクレアーゼ(例えば、ZFN)に対するゲノム標的を提供するために細胞(例えば、植物または哺乳類細胞)のゲノムに組み込まれる。ある実施形態では、標的部位は、亜鉛フィンガーヌクレアーゼの1つ以上の対がホモ二量体として結合し、開裂するように位置する。他の実施形態では、標的部位は、亜鉛フィンガーヌクレアーゼの1つ以上の対がヘテロ二量体として結合し、開裂するように位置する。
【0013】
別の態様では、本明細書に記載される1つ以上の多重挿入部位および/または多重挿入部位に組み込まれる1つ以上の外来性配列を含む、植物または種子が本明細書に開示される。ある実施形態では、多重挿入部位および/または外来性配列は、トウモロコシ植物の配偶体に組み込まれる。
【0014】
ある態様では、本明細書に記載される1つ以上の多重挿入部位および/または多重挿入部位に組み込まれる1つ以上の外来性配列を含む、修飾された哺乳類細胞株、修飾された初代細胞、修飾された幹細胞、および/または遺伝子導入動物が本明細書に提供される。
【0015】
別の態様では、外来性配列を、細胞(例えば、植物または哺乳類細胞)のゲノムに組み込まれた多重挿入部位に組み込むための方法が本明細書に提供され、本方法は、(a)ヌクレアーゼに対する1つ以上の標的部位を含む多重挿入部位ポリヌクレオチドを、細胞のゲノムに組み込むことと、(b)ヌクレアーゼのそれらの標的部位への結合が細胞のゲノムを開裂させるように、多重挿入部位ポリヌクレオチドにおける第1の標的部位に結合する1つ以上のヌクレアーゼを提供および/または発現させることと、(c)細胞を、外来性核酸配列を含むポリヌクレオチドと接触させ、それによって外来性配列の、多重挿入部位ポリヌクレオチド内の細胞のゲノムへの相同性依存性組み込みをもたらすことと、を含む。
【0016】
別の態様では、多重外来性配列を細胞(例えば、植物または哺乳類細胞)のゲノムに組み込むための方法が本明細書に提供され、本方法は、(a)第1の多重挿入部位ポリヌクレオチドが、第1および第2のヌクレアーゼに対する標的部位が側面に位置する少なくとも1つの第1の遺伝子を含む、ヌクレアーゼに対する1つ以上の標的部位を含む第1の多重挿入部位ポリヌクレオチドを、細胞のゲノムに組み込むことと、(b)第3および第4のヌクレアーゼに対する標的部位が側面に位置する少なくとも1つの第2の遺伝子を含む第2の多重挿入部位ポリヌクレオチドの存在下で、細胞中の第1または第2のヌクレアーゼを発現させ、それによって第1および第2の遺伝子の細胞のゲノムへの組み込みをもたらすことと、を含む。ある実施形態では、本方法は更に、コード配列および/またはヌクレアーゼ部位を含む、追加的な外来性配列を組み込むために、挿入された多重挿入部位上に存在する適切なヌクレアーゼを発現させるステップを一回以上反復することを含む。ヌクレアーゼは、ヘテロ二量体ZFNであってもよく、ヌクレアーゼのうちの1つ以上の間にあるものと共通の1つの単量体が存在してもよい。幾つかの実施形態では、挿入のための外来性DNA配列は、外来性配列の組み込み時に、ドナーDNAに関連する片側標的部位、およびゲノムDNAに関連する片側標的部位を含む新規ZFN標的部位が作り出されるように、ZFN片側標的部位を含んでもよい。この新規ZFN標的部位は、類似の新規ヘテロ二量体ZFNに対する標的部位としての役割を果たすことができる。
【0017】
別の態様では、細胞(例えば、植物または哺乳類細胞)中で1つ以上の外来性核酸配列の産物を発現させるための方法が本明細書に開示され、本方法は、外来性配列が、組み込まれた核酸分子中の細胞のゲノムに組み込まれ、外来性配列の産物が発現されるように、本明細書に記載される方法のうちのいずれかに従って1つ以上の外来性核酸配列を組み込むことを含む。
【0018】
細胞のゲノムに挿入された1つ以上の遺伝子を欠失させる方法もまた提供され、本方法は、複数の外来性配列を、本明細書に記載される方法のうちのいずれかによって組み込むことと、外来性配列のうちの1つ以上がゲノムから欠失させられるように、細胞中で適切なヌクレアーゼを発現させることと、を含む。ある実施形態では、欠失させられる外来性配列は、マーカー遺伝子である。ある実施形態では、外来性配列の欠失およびその後のゲノム内の末端部の再結合は、ゲノム位置における機能性遺伝子または配列を作り出し、例えば、発現可能なスクリーニングマーカーを作り出す。
【0019】
なおも別の態様では、ゲノムが変更された細胞を提供する方法が提供され、本方法は、本明細書に記載される方法のうちのいずれかに従って、第1の細胞中の1つ以上の外来性核酸配列を組み込むおよび/または切除することと、第1の細胞を第1の性的に成熟した生物に発達させることと、生物を、対立位置におけるゲノム変更を含む第2の生物と交配させて、第1および第2の生物のゲノム変更を有する第2の細胞を生成することと、を含む。ある実施形態では、生物は、植物である。他の実施形態では、生物は、遺伝子導入動物である。
【0020】
本明細書に記載される方法のうちのいずれかにおいて、本方法は、1つ以上の内在性遺伝子座における標的化組み込みおよび/または標的化不活性化を含む、ゲノム変更の他の方法と組み合わせて使用されてもよい。更に、本明細書に記載される方法のうちのいずれかにおいて、ヌクレアーゼは、亜鉛フィンガー結合ドメインおよび開裂片側ドメインを含む1つ以上の融合タンパク質を含んでもよく、ここで亜鉛フィンガー結合ドメインは、多重挿入部位において標的部位に結合するように改変されている。更に、これらの方法のうちのいずれにおいても、外来性核酸配列は、多重挿入部位における配列、および/または多重挿入部位が組み込まれる領域における、内在性配列と相同である1つ以上の配列を含む。
【0021】
本明細書に記載される方法のうちのいずれかにおいて、1つ以上の多重挿入部位は、任意の好適な方法によって、例えば、挿入が所望される内在性遺伝子を標的とするZFNを使用する、ヌクレアーゼ(例えば、ZFN)を介した標的化組み込みによって、ゲノムに組み込まれてもよい。代替的に、1つ以上の多重挿入部位は、標準技術を使用して細胞のゲノムに無作為に組み込まれてもよい。
【0022】
外来性核酸配列は、1つ以上のプロモーターを含むもしくは含まない、1つ以上の機能性ポリペプチド(例えば、cDNA)をコードする配列を含んでもよく、かつ/または望ましい形質を生物に付与する、1つ以上のRNA配列(例えば、1つ以上のshRNA発現カセットを介して)を産生してもよい。植物におけるかかる形質には、除草剤抵抗性または耐性;虫害抵抗性または耐性;病害抵抗性または耐性(ウイルス、細菌、真菌、線虫);干ばつ、暑気、冷気、凍結、過度の湿気、塩ストレスに対する抵抗性または耐性によって例示されるようなストレス耐性および/または抵抗性;酸化ストレス;増加した収率;食物含量および組成(food content and makeup);外見;雄性不稔性;ドライダウン;標準性(standability);多産性;デンプンの量および質;油の量および質;タンパク質の量および質;アミノ酸組成等が含まれるが、これらに限定されない。当然のことながら、除草剤、虫害、病害(ウイルス、細菌、真菌、線虫)、もしくは干ばつ抵抗性、雄性不稔性、ドライダウン、標準性、多産性、デンプン特性、油の量および質を付与するもの、または収率もしくは栄養の質を増加させるもの等の、任意の説明の任意の2つ以上の外来性核酸が、所望に応じて用いられてもよい。ある実施形態では、外来性核酸配列は、除草剤抵抗性タンパク質(例えば、AAD(アリールオキシアルカノエートジオキシゲナーゼ)遺伝子)および/またはその機能的断片をコードする配列を含む。組み込まれる配列の発現は、組み込まれる配列に操作可能に連結されたプロモーターによって駆動することができる。代替的には、組み込まれる配列は、プロモーターを含まず、転写は、多重挿入部位ポリヌクレオチドの挿入の領域における内在性プロモーターによって駆動される。他の実施形態では、結合部位の開裂および不正確な修復は、目的の遺伝子を不活性化または活性化する可能性がある。ある実施形態では、ポリヌクレオチドは、プラスミドである。他の実施形態では、ポリヌクレオチドは、線状DNA分子である。
【0023】
哺乳類細胞中で、本発明の方法および組成物は、細胞株構築のため、例えば、抗体等の多量体ポリペプチドを発現する細胞株の構築のために使用されてもよい。幾つかの実施形態では、細胞株は、研究目的のため、例えば、目的の経路のメンバーを発現する細胞株の構築のために使用されてもよい。幾つかの実施形態では、初代細胞または幹細胞を使用して、細胞治療目的のために目的の多量体タンパク質を発現させてもよい。
【0024】
別の態様では、亜鉛フィンガーヌクレアーゼ活性を測定する方法が本明細書に提供される。ある実施形態では、本方法は、(a)対形成した標的部位の各々が、亜鉛フィンガーヌクレアーゼが結合する2つの亜鉛フィンガーヌクレアーゼ片側標的部位を含む、本明細書に記載される少なくとも1つの亜鉛フィンガーヌクレアーゼおよび核酸分子、ならびに片側標的部位の間に挿まれる切断部位である、結合した亜鉛フィンガーヌクレアーゼによって切断される切断部位を提供することと、(b)亜鉛フィンガーヌクレアーゼが、少なくとも切断部位内で対形成した標的部位を開裂させるように、亜鉛フィンガーヌクレアーゼを核酸と組み合わせることと、(c)少なくとも切断部位の配列を決定して、配列データを生成することと、(d)配列データにおいて、切断部位内の塩基対欠失の数および長さを、亜鉛フィンガーヌクレアーゼの不在下での切断部位内の塩基対欠失の数および長さと比較して、それによって対形成した標的部位における亜鉛フィンガーヌクレアーゼ活性を測定することと、を含む。ある実施形態では、1つを超える塩基対の欠失は、亜鉛フィンガーヌクレアーゼの増加した活性を示す。
【0025】
なおも他の実施形態では、対形成した標的部位における亜鉛フィンガーヌクレアーゼ活性を最適化するための方法が本明細書に提供される。ある実施形態では、本方法は、(a)対形成した標的部位の各々が、亜鉛フィンガーヌクレアーゼが結合する2つの亜鉛フィンガーヌクレアーゼ片側標的部位を含む、本明細書に記載される少なくとも1つの亜鉛フィンガーヌクレアーゼおよび核酸分子、ならびに片側標的部位の間に挿まれる切断部位である、結合した亜鉛フィンガーヌクレアーゼによって切断される切断部位を提供することと、(b)亜鉛フィンガーヌクレアーゼが、少なくとも切断部位内で対形成した標的部位を開裂させるように、亜鉛フィンガーヌクレアーゼを核酸と組み合わせることと、(c)切断部位における亜鉛フィンガーヌクレアーゼ活性レベルを決定することと、(d)切断部位における塩基対の数に変化を与えることと、(e)ステップ(b)〜(d)を数回反復することと、(f)最高レベルの亜鉛フィンガーヌクレアーゼ活性を提供する塩基対の数を含む、核酸に組み込むための切断部位を選択し、それによって対形成した標的部位における亜鉛フィンガーヌクレアーゼ活性を最適化することと、を含む。
【0026】
亜鉛フィンガーヌクレアーゼを伴う本明細書に記載される方法のうちのいずれかにおいて、第1および第2の開裂片側ドメインは、Type IIS制限エンドヌクレアーゼ、例えば、FokIまたはStsIからのものである。更に、本明細書に記載される方法のうちのいずれかにおいて、融合タンパク質のうちの少なくとも1つは、例えば、開裂片側ドメインの偏性ヘテロ二量体が形成されるように、開裂片側ドメインの二量体化成面のアミノ酸配列における変更を含んでもよい。
【0027】
本明細書に記載される方法のうちのいずれかにおいて、植物細胞は、単子葉または双子葉植物細胞を含む可能性がある。ある実施形態では、植物細胞は、作物植物、例えば、トウモロコシである。ある実施形態では、細胞は、初代細胞、細胞株、または幹細胞等の哺乳類細胞を含む可能性がある。幾つかの実施形態では、哺乳類細胞株は、目的のポリペプチドの産生のために使用することができる。
【図面の簡単な説明】
【0028】
【図1】本明細書に記載される例示的な多重挿入部位を示す概略図。図1は、7つのZFN標的部位から成り立つ多重挿入部位を示す。標的部位に結合するZFN対は、幾何学的図として示される。「ブロック1」は、ZFN標的部位(影付きおよび格子模様の三角)を維持しながら、適切なZFN対の存在下で多重挿入部位に組み込まれる外来性配列である。図1は、ZFN標的部位の代わりの、適切なZFN対の存在下での「ブロック1」の多重挿入部位への組み込みを示す。
【図2】「ブロック2」が、適切なZFN対の存在下で多重挿入部位に組み込まれる外来性配列である、図1に示される例示的な多重挿入部位を示す概略図。
【図3】ZFNによって強化された対立遺伝子間組み換えの概略図。同一のゲノム位置にあるが、互いから移動させられる2つの挿入物は、ZFNによる二本鎖開裂後に、相同組み換えまたは鎖交換を経る可能性がある。ZFN対(このうち両方のZFN単量体が一緒に発現される)は、ZFN対を発現する植物を、両方の対立遺伝子を含む植物と一緒に交配させることによって、または単一の対立遺伝子を含有する植物との交配の両側からの2つのZFN単量体を導入することによって、提供することができる。
【図4】ヘテロ二量体ZFN「左」および「右」標的ドメインの使用を示す概略図。上部の線は、左および右標的ZFNドメインを有するゲノムを示す(影付き三角および碁盤格子三角)。異なるヘテロ二量体対が側面に位置する遺伝子を含む外来性分子の存在下で、適切なZFN対が付加されるとき、遺伝子および隣接するヌクレアーゼ部位は、示されるようにゲノムに挿入される。
【図5】ゲノムが組み込まれた配列のいずれかの側上での、外来性配列(「遺伝子」として示される)の組み込みおよび切除を示す概略図。付加された遺伝子の側面には、遺伝子カセットを適切な部位内に導くための相同性領域が位置する。挿入のために使用されるZFN標的部位の2つの片側は、挿入されるDNAにおいて2つの新たな組み合わせを作り出すことによって組み換えられる。遺伝子カセットの切除は、隣接するZFN標的部位において開裂するための適切なZFN対を結合させることによって遂行される。切除は、所望のDNA配列の欠失を防止するために、相同性アームを含有するテンプレートを必要とする可能性がある。各「遺伝子」は、1つ以上の配列、例えば、1つ以上のコード配列を含む可能性がある。
【図6】ZFNヘテロ二量体(異なる陰影を有する三角として示される)を使用する、挿入されたマーカー遺伝子の切除および「再利用」を示す概略図。
【図7】pDAB105900のプラスミドマップ。
【図8】pDAB105908のプラスミドマップ。
【図9】亜鉛フィンガーヌクレアーゼホモ二量体発現カセットの線図。
【図10】亜鉛フィンガーヌクレアーゼヘテロ二量体発現カセットの線図。
【図11】開裂後の非相同末端結合からもたらされる欠失の頻度によって決定される、トウモロコシにおけるeZFN開裂活性を示す。
【図12】開裂後の非相同末端結合からもたらされる欠失の頻度によって決定される、タバコにおけるeZFN開裂活性を示す。
【図13】同じ遺伝子座への2つの遺伝子導入挿入物の概略図。上部の線は、遺伝子座における相同組み換えに必要とされるeZFN結合部位を含有する、多重挿入部位(本明細書でランディングパッドとも称される)を表すMISと標識された無作為配列、ならびにカナマイシン選択可能マーカー遺伝子およびGUSスクリーニング可能マーカー遺伝子を含むブロック1を示す。中間の線は、上部のDNAにあるものと同じ多重挿入部位(MIS)と共に、ハイグロマイシン抵抗性選択可能マーカー遺伝子および黄色蛍光タンパク質スクリーン可能マーカー遺伝子(HPT/YFP)を含むブロック2を示す。下部の線は、組み換えに続く遺伝子座を示す。
【図14】ZFNによる対立位置における相同組み換え、および図13に説明されるものと同じ遺伝子座における2つの異なるDNA挿入物の生成を示す。ブロック1(カナマイシンおよびGUCマーカーを含む、GUS/NPT)、多重挿入部位(MISまたはランディングパッド)、およびブロック2(ハイグロマイシン(hygomycin)および黄色蛍光マーカーを含む、HPT/YFP)を含む構築物が、アラビドプシスに形質転換される。各ブロックのみを別個の植物における多重挿入部位と一緒に生成するために、ブロック2は、ブロック1のみの構成を生成するために組み込まれる部位から切除されるか、またはブロック1は、ブロック2のみの構成を生成するために組み込まれる部位から切除される。遺伝子ブロックの除去は、元の遺伝子導入事象を含有する植物を、遺伝子ブロックの各々に隣接するeZFN結合部位において開裂するZFNを発現する植物と交配させることによって遂行される。回収された単一のブロック植物は交配されて、2つの構成物を単一の植物中に一緒にまとめ、その植物が減数分裂特異的プロモーターを発現する植物と交配されて、2つのブロック1とブロック2対立遺伝子との間のDNAの交換に影響を及ぼす。
【図15】2つの配列の間の中間部位におけるZFN開裂によって、同じ遺伝子座に位置する2つのDNA配列の間の組み換えを得るためのステップを示す概略フローチャート。図16に説明される構築物は、アラビドプシスに形質転換される。2つの遺伝子ブロック(図14に説明される)の1つは、結合部位がブロックに隣接するeZFNを発現する植物と交配させることによって除去され、ブロック1またはブロック2のいずれかを含有する植物をもたらす。
【図16】交換遺伝子座をアラビドプシスに導入するために使用されるプラスミドの概略図。それは、図14に説明されるブロック1および2、ならびに多重挿入部位配列を含有する。eZFN結合部位が示され、ブロック1および2(ブロック1:eZFN1および8、ブロック2:eZFN3および6)に隣接するか、または相同組み換えを促進するために多重挿入部位(eZFN4および7)における中心に位置する。
【発明を実施するための形態】
【0029】
詳細な説明
本開示は、ゲノム、例えば、トウモロコシ等の作物植物または哺乳類細胞への標的化組み込み(TI)のための方法および組成物に関する。1つ以上のヌクレアーゼ(例えば、ZFN)に対する多重標的部位を含有する多重挿入部位が、ゲノムに組み込まれる。多重挿入部位のゲノムへの組み込みに続いて、適切なヌクレアーゼが、挿入対象の外来性配列と共に細胞に導入される。
【0030】
ある実施形態では、ヌクレアーゼは、1つ以上のZFNを含む。ZFNは典型的には、開裂ドメイン(または開裂片側ドメイン)および亜鉛フィンガー結合ドメインを含み、タンパク質として、これらのタンパク質をコードするポリヌクレオチドとして、またはポリペプチドおよびポリペプチドをコードするポリヌクレオチドの組み合わせとして導入されてもよい。亜鉛フィンガーヌクレアーゼは典型的には、開裂片側ドメインの二量体化に続いて、二量体タンパク質として機能する。「左」および「右」認識ドメインに結合するZFN単量体が会合して活性ヌクレアーゼを形成することができる、偏性ヘテロ二量体ZFNが記載されている。例えば、米国特許公開第2008/0131962号を参照されたい。故に、適切な標的部位が与えられると、「左」単量体は、任意の「右」単量体を有する活性ZFヌクレアーゼを形成し得る。これは、様々な組み合わせで使用することができる、立証された左および右ドメインに基づいて有用なヌクレアーゼ部位の数を有意に増加させる。例えば、4個のホモ二量体ZFヌクレアーゼの結合部位を組み換えることは、追加的な12個のヘテロ二量体ZFヌクレアーゼを生み出す。より重要なことに、あらゆる新たな導入された配列の側面に、遺伝子を再び切除するため、またはそれの隣の追加的な遺伝子を標的とするために使用することができる固有のZFN部位が位置することになるような、遺伝子導入設計に対する系統的な手法を可能にする。更に、この方法は、ZFN依存性二本鎖切断によって駆動される、単一の遺伝子座へのスタッキングの戦略を単純化することができる。
【0031】
亜鉛フィンガー結合ドメインは、標準(C2H2)亜鉛フィンガーまたは非標準(例えば、C3H)亜鉛フィンガーであり得る。更に、亜鉛フィンガー結合ドメインは、1つ以上の亜鉛フィンガー(例えば、2つ、3つ、4つ、5つ、6つ、7つ、8つ、9つ、またはそれを超える亜鉛フィンガー)を含む可能性があり、多重挿入部位内の任意の配列に結合するように改変することができる。細胞中のかかる融合タンパク質(複数可)の存在は、融合タンパク質(複数可)のその(それらの)結合部位への結合および多重挿入部位内の開裂をもたらし、それは外来性配列の組み込みをもたらす。
【0032】
概論
本明細書で開示される方法の実践、ならびに組成物の調製と使用は、特に指示がない限り、分子生物学、生化学、クロマチン構造および分析、計算化学、細胞培養、組換えDNA、および関連分野における従来の技術を採用しており、当技術分野の技術範囲内である。これらの技術は、文献で完全に説明される。例えば、Sambrook et al.MOLECULAR CLONING:A LABORATORY MANUAL,Second edition,Cold Spring Harbor Laboratory Press,1989 and Third edition,2001、Ausubel et al.,CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John Wiley&Sons,New York,1987 and periodic updates、the series METHODS IN ENZYMOLOGY,Academic Press,San Diego、Wolffe,CHROMATIN STRUCTURE AND FUNCTION,Third edition,Academic Press,San Diego,1998、METHODS IN ENZYMOLOGY,Vol.304,“Chromatin”(P.M.Wassarman and A.P.Wolffe,eds.),Academic Press,San Diego,1999、およびMETHODS IN MOLECULAR BIOLOGY,Vol.119,“Chromatin Protocols”(P.B.Becker,ed.)Humana Press,Totowa,1999を参照されたい。
【0033】
定義
「核酸」、「ポリヌクレオチド」、および「オリゴヌクレオチド」という用語は、交換可能に使用され、線状または環状コンフォメーションで、一本鎖または二本鎖形態の、デオキシリボヌクレオチドポリマーまたはリボヌクレオチドポリマーを指す。本開示については、これらの用語をポリマーの長さに関する限定と解釈すべきではない。これらの用語は、天然ヌクレオチドの既知の類似体、ならびに塩基、糖、および/またはリン酸(ホスホロチオエート主鎖)部分内で修飾されるヌクレオチドを包含することができる。一般に、特定のヌクレオチドの類似体は、同一の塩基対形成特異性を有し、すなわち、Aの類似体は、Tと塩基対を形成する。
【0034】
「ポリペプチド」、「ペプチド」、および「タンパク質」という用語は、アミノ酸残基のポリマーを指すために交換可能に使用される。この用語は、1つ以上のアミノ酸が、対応する天然に存在するアミノ酸の化学的類似体または修飾誘導体である、アミノ酸ポリマーにも適用される。
【0035】
「結合」は、高分子間(例えば、タンパク質と核酸との間)の配列特異的で非共有結合的な相互作用を指す。結合相互作用の全ての構成要素は、相互作用が全体として配列特異的である限り(例えば、DNA主鎖中のリン酸残基との接触)、配列特異的である必要はない。かかる相互作用は、一般に、10-6M-1以下の解離定数(Kd)によって特徴付けられる。「親和性」は、結合の強さを指す。高い結合親和性は、低いKdと関連している。
【0036】
「結合タンパク質」は、別の分子に結合することができるタンパク質である。結合タンパク質は、例えば、DNA分子(DNA結合タンパク質)、RNA分子(RNA結合タンパク質)、および/またはタンパク質分子(タンパク質結合タンパク質)に結合することができる。タンパク質結合タンパク質の場合においては、それは、それ自体に結合する(その結果、ホモ二量体、ホモ三量体等を形成する)ことができる、および/または異なるタンパク質または複数のタンパク質の1つ以上の分子に結合することができる。結合タンパク質は、2種類以上の結合活性を有することができる。例えば、亜鉛フィンガータンパク質は、DNA結合、RNA結合、およびタンパク質結合活性を有する。
【0037】
「亜鉛フィンガーDNA結合タンパク質」(または結合ドメイン)は、タンパク質、または構造が亜鉛イオンの配位によって安定化される結合ドメイン内のアミノ酸配列の領域である、1つ以上の亜鉛フィンガーによって配列特異的な様式でDNAを結合するより大きなタンパク質内のドメインである。亜鉛フィンガーDNA結合タンパク質という用語は、しばしば、亜鉛フィンガータンパク質またはZFPとして略記される。
【0038】
亜鉛フィンガー結合ドメインは、所定のヌクレオチド配列に結合するように、「改変」することができる。亜鉛フィンガータンパク質を改変するための方法の非制限的な例は、設計および選択である。設計された亜鉛フィンガータンパク質は、天然に存在しないタンパク質であり、その設計/組成は、主として合理的基準によってもたらされる。設計のための合理的基準には、置換規則の適用、ならびに既存のZFP設計および結合データの情報を格納したデータベース中の情報を処理するためのコンピュータアルゴリズムの適用が含まれる。例えば、米国特許第6,140,081号、同第6,453,242号、および同第6,534,261号を参照されたく、また、国際公開第98/53058号、国際公開第98/53059号、国際公開第98/53060号、国際公開第02/016536号、および国際公開第03/016496号も参照されたい。
【0039】
「選択された」亜鉛フィンガータンパク質は、天然には見出されないタンパク質であり、その産生は、ファージディスプレイ、相互作用トラップ、またはハイブリッド選択等の実験的プロセスによって主にもたらされる。例えば、米国特許第5,789,538号、同第5,925,523号、同第6,007,988号、同第6,013,453号、同第6,200,759号、国際公開第95/19431号、国際公開第96/06166号、国際公開第98/53057号、国際公開第98/54311号、国際公開第00/27878号、国際公開第01/60970号、国際公開第01/88197号、および国際公開第02/099084号を参照されたい。
【0040】
「配列」という用語は、任意の長さのヌクレオチド配列を指し、それは、DNAまたはRNAであってもよく、線状、環状、または分岐状であってもよく、一本鎖または二本鎖のいずれかであってもよい。「ドナー配列」という用語は、ゲノム内に挿入されるヌクレオチド配列を指す。ドナー配列は、任意の長さ、例えば、2〜10,000(またはそれらの間もしくはそれらを超える任意の整数)ヌクレオチド長、好ましくは約100〜1,000(またはそれらの間の任意の整数)ヌクレオチド長、より好ましくは約200〜500ヌクレオチド長であってもよい。
【0041】
「相同非同一配列」は、第2の配列とある程度の配列同一性を共有するが、その配列は、第2の配列と同一ではない、第1の配列を指す。例えば、突然変異遺伝子の野生型配列を含むポリヌクレオチドは、突然変異遺伝子の配列に対して相同的かつ非同一である。ある実施形態では、2つの配列の間の相同性の度合いは、通常の細胞機構を利用して、それらの間で相同組み換えを可能にするために十分である。2つの相同非同一配列は、任意の長さである可能性があり、非相同性の度合いは、1ヌクレオチドほど小さいか(例えば、標的化相同組み換えによるゲノム点突然変異の補正のため)、または10キロベース以上ほど大きい(例えば、染色体内の所定の異所性部位における遺伝子の挿入のため)可能性がある。相同非同一配列を含む2つのポリヌクレオチドは、同一の長さである必要はない。例えば、20〜10,000ヌクレオチドまたはヌクレオチド対の外因性ポリヌクレオチド(すなわち、ドナーポリヌクレオチド)を使用することができる。
【0042】
核酸およびアミノ酸配列同一性を判定するための技術は、当該技術分野において既知である。典型的には、かかる技術は、遺伝子のためのmRNAのヌクレオチド配列を判定すること、および/またはそれによってコードされたアミノ酸配列を判定すること、ならびにそれらの配列を、第2のヌクレオチドまたはアミノ酸配列と比較することを含む。ゲノム配列もまた、この方法で決定し、比較することができる。一般に、同一性は、それぞれ、2つのポリヌクレオチドまたはポリペプチド配列のヌクレオチド対ヌクレオチド、またはアミノ酸対アミノ酸の対応が完全であることを指す。2つ以上の配列(ポリヌクレオチドまたはアミノ酸)は、それらの同一性百分率を判定することによって比較することができる。核酸配列であれアミノ酸配列であれ、2つの配列の同一性百分率は、短い方の配列の長さで割り、100を乗じた、2つの整合配列の間の厳密な合致数である。核酸配列の近似配列は、Smith and Waterman,Advances in Applied Mathematics 2:482−489(1981)の局所性相同性アルゴリズムによって提供される。このアルゴリズムは、Dayhoff,Atlas of Protein Sequences and Structure,M.O.Dayhoff ed.,5 suppl.3:353−358,National Biomedical Research Foundation,Washington,D.C.,USAによって開発され、Gribskov,Nucl.Acids Res.14(6):6745−6763(1986)によって正規化されたスコアリングマトリックスを使用することによって、アミノ酸配列に適用することができる。配列の同一性百分率を判定するためのこのアルゴリズムの例示的実施は、「BestFit」実用用途において、Genetics Computer Group(Madison,WI)によって提供される。配列の間の同一性百分率または類似性を計算するための好適なプログラムは、当該技術分野において一般に既知であり、例えば、別の整合プログラムは、デフォルトパラメータと共に使用されるBLASTである。例えば、BLASTNおよびBLASTPは、次のデフォルトパラメータを使用して、使用することができる:遺伝コード=標準、フィルター=なし、鎖=両方、カットオフ=60、期待数=10、マトリックス=BLOSUM62、表示=50配列、分類=高スコア、データベース=非重複、GenBank+EMBL+DDBJ+PDB+GenBank CDS tranlsations+Swiss protein+Spupdate+PIR。これらのプログラムの詳細は、インターネット上で見出すことができる。本明細書に説明する配列に関して、望ましい配列同一性の度合いの範囲は、約80%〜100%、およびそれらの間の任意の整数値である。典型的には、配列の間の同一性百分率は、少なくとも70〜75%、好ましくは、80〜82%、より好ましくは85〜90%、更に好ましくは92%、より一層好ましくは95%、最も好ましくは98%の配列同一性である。
【0043】
代替的には、ポリヌクレオチド間の配列類似性の度合いは、相同領域間で安定した二重鎖の形成を可能にする条件下でのポリヌクレオチドのハイブリダイゼーション、続いて一本鎖特異的ヌクレアーゼでの消化、および消化された断片の寸法決定によって決定することができる。2つの核酸または2つのポリペプチド配列は、配列が、上記の方法を使用して判定されるように、分子の定義された長さにわたって、少なくとも70%〜75%、好ましくは80%〜82%、より好ましくは85%〜90%、更に好ましくは92%、より一層好ましくは95%、および最も好ましくは98%の配列同一性を示すとき、互いに実質的に相同的である。本明細書に使用するとき、実質的に相同的はまた、指定されるDNAまたはポリペプチド配列に完全な同一性を示す配列を指す。実質的に相同的であるDNA配列は、例えば、サザンハイブリダイゼーション実験において、その特定の体系のために定義されるストリンジェントな条件下で特定することができる。適切なハイブリダイゼーション条件を定義することは、当該技術分野の技術の範囲内である。例えば、Sambrook et al.,supra;Nucleic Acid Hybridization:A Practical Approach,editors B.D.Hames and S.J.Higgins,(1985)Oxford;Washington,DC;IRL Press)を参照されたい。
【0044】
2つの核酸断片の選択的ハイブリダイゼーションは、次のように決定することができる。2つの核酸分子の間の配列同一性の度合いは、かかる分子の間のハイブリダイゼーション事象の効率および強度に影響する。部分的に同一な核酸配列は、完全に同一な配列の標的分子へのハイブリダイゼーションを少なくとも部分的に阻害する。完全に同一な配列のハイブリダイゼーションの阻害は、当該技術分野において周知である、ハイブリダイゼーションアッセイ(例えば、サザン(DNA)ブロット、ノーザン(RNA)ブロット、溶液ハイブリダイゼーション、または同等物、Sambrook,et al.,Molecular Cloning:A Laboratory Manual,Second Edition,(1989)Cold Spring Harbor,N.Y.を参照されたい)を使用して評価することができる。かかるアッセイは、異なる度合いの選択性を使用して、例えば、低ストリンジェンシーから高いストリンジェンシーまで異なる条件を使用して、実施することができる。低ストリンジェンシーの条件を使用する場合では、非特異的結合の不在は、非特異的結合事象の不在下で二次プローブが標的にハイブリダイズしないように、部分的な配列同一性の度合いさえ持たない二次プローブ(例えば、標的分子との配列同一性が約30%未満であるプローブ)を使用し、評価することができる。
【0045】
ハイブリダイゼーションベースの検出系を利用する時、参照核酸配列に相補的である核酸プローブが選択され、次いで、適切な条件の選択によって、プローブおよび参照配列が二重鎖分子を形成するように、互いに選択的にハイブリダイズするか、または結合する。中等度にストリンジェントなハイブリダイゼーション条件下で、参照配列に選択的にハイブリダイズする能力を有する核酸分子は、典型的には、選択された核酸プローブの配列と少なくとも約70%の配列同一性を有する、少なくとも約10〜14ヌクレオチド長の標的核酸配列の検出を可能にするような条件下でハイブリダイズする。ストリンジェントなハイブリダイゼーション条件は、典型的には、選択された核酸プローブの配列と少なくとも約90〜95%を上回る配列同一性を有する、少なくとも約10〜14ヌクレオチド長の標的核酸配列の検出を可能にする。プローブおよび参照配列が、特異的な度合いの配列同一性を有する、プローブ/参照配列ハイブリダイゼーションに有用なハイブリダイゼーション条件は、当該技術分野において既知のとおり決定することができる(例えば、Nucleic Acid Hybridization:A Practical Approach,editors B.D.Hames and S.J.Higgins,(1985)Oxford;Washington,DC;IRL Pressを参照されたい)。
【0046】
ハイブリダイゼーションの条件は、当業者に周知である。ハイブリダイゼーションストリンジェンシーは、ハイブリダイゼーション条件が、ミスマッチしたヌクレオチドを含むハイブリッドの形成を嫌う度合いを指し、より高いストリンジェンシーは、ミスマッチしたハイブリッドに対する許容度の低さに相関する。ハイブリダイゼーションのストリンジェンシーに影響を及ぼす因子は、当業者に周知であり、温度、pH、イオン強度、ならびに例えば、ホルムアミドおよびジメチルスルホキシド等の有機溶媒の濃度を含むが、これらに限定されない。当業者には既知であるが、ハイブリダイゼーションのストリンジェンシーは、より高い温度、より低いイオン強度、およびより低い溶媒濃度によって増加する。
【0047】
ハイブリダイゼーションのストリンジェンシー条件に関して、数々の等価条件は、例えば、次の因子:配列の長さおよび性質、様々な配列の塩基組成、塩および他のハイブリダイゼーション溶液成分の濃度、ハイブリダイゼーション溶液中の遮断薬(例えば、硫酸デキストランおよびポリエチレングリコール)の存否、ハイブリダイゼーション反応温度および時間パラメータ、ならびに洗浄条件を変化させることによって、特定のストリンジェンシーを確立するために使用することができることは、当該技術分野において周知である。特定の一連のハイブリダイゼーション条件の選択は、当該技術分野における標準方法に従い選択される(例えば、Sambrook et al.,Molecular Cloning:A Laboratory Manual,Second Edition,(1989)Cold Spring Harbor,N.Y.を参照されたい)。
【0048】
「組み換え」は、2つのポリヌクレオチドの間の遺伝子情報の交換のプロセスを指す。この開示に関して、「相同組み換え(HR)」は、例えば、細胞中の二本鎖切断の修復中に行われるような交換の特殊な形態を指す。このプロセスは、ヌクレオチド配列相同性を必要とし、「標的」分子(すなわち、二本鎖切断を経験するもの)のテンプレート修復に「ドナー」分子を使用し、かつ、それがドナーから標的への遺伝子情報の移動をもたらすため、「非交差遺伝子変換」または「ショートトラクト遺伝子変換」として多様に知られる。いかなる特定の理論にも拘束されることを望むものではないが、かかる移動は、切断された標的とドナーとの間に形成されるヘテロ二重鎖DNAのミスマッチ修正、および/または標的の一部になる遺伝子情報を再合成するためにドナーが使用される、「合成依存性鎖アニーリング」、および/または関連プロセスを伴う可能性がある。かかる特殊なHRは、しばしば、ドナーポリヌクレオチドの配列の一部または全てが、標的ポリヌクレオチド内に組み込まれるような、標的分子の配列の変化をもたらす。
【0049】
「開裂」は、DNA分子の共有主鎖の切断を指す。開裂は、ホスホジエステル結合の酵素的または化学的加水分解を含むが、これらに限定されない多様な方法によって開始することができる。一本鎖開裂および二本鎖開裂の両方が可能であり、二本鎖開裂は、2つの別々の一本鎖開裂事象の結果として生じ得る。DNA開裂は、平滑末端または付着末端のいずれかの産生をもたらす可能性がある。ある実施形態では、融合ポリペプチドは、標的化二本鎖DNA開裂のために使用される。
【0050】
「開裂ドメイン」は、DNA開裂に対する触媒活性を有する1つ以上のポリペプチド配列を含む。開裂ドメインは、単一のポリペプチド鎖内に含有され得るか、または開裂活性は、2つ(またはそれを超える数)のポリペプチドの会合からもたらされ得る。
【0051】
「開裂片側ドメイン」は、第2のポリペプチド(同一のまたは異なるいずれかの)と併せて、開裂活性(好ましくは二本鎖開裂活性)を有する複合体を形成するポリペプチド配列である。
【0052】
「クロマチン」は、細胞ゲノムを含む核タンパク質構造である。細胞クロマチンは、核酸、主にDNA、ならびにヒストンおよび非ヒストン染色体タンパク質を含むタンパク質を含む。真核細胞クロマチンの大半は、ヌクレオソームの形態で存在し、そこでは、ヌクレオソームコアが、それぞれ2つのヒストンH2A、H2B、H3、およびH4を含む八量体と会合した約150塩基対のDNAを含み、リンカーDNA(生物に応じて変化する長さ)がヌクレオソームコアの間に延在する。1分子のヒストンH1は、一般にリンカーDNAと会合する。本開示に関して、「クロマチン」という用語は、原核性および真核性の両方の全ての種類の細胞核タンパク質を包含するように意図されている。細胞クロマチンは、染色体クロマチンおよびエピソームクロマチンの両方を含む。
【0053】
「染色体」は、細胞のゲノムの全てまたは一部を含むクロマチン複合体である。細胞のゲノムは、しばしば、その細胞のゲノムを含む全染色体の集合である、その核型によって特徴付けられる。細胞のゲノムは、1つ以上の染色体を含むことができる。
【0054】
「エピソーム」は、細胞の染色体核型の一部ではない核酸を含む、複製する核酸、核タンパク質複合体、または他の構造である。エピソームの例としては、プラスミドおよびある種のウイルスゲノムが挙げられる。
【0055】
「到達可能領域」は、核酸内に存在する標的部位が標的部位を認識する外来性分子によって結合され得る、細胞クロマチン内の部位である。いかなる特定の理論にも拘束されることを望むものではないが、到達可能領域は、ヌクレオソーム構造内にパッケージングされない領域であると考えられる。到達可能領域の別々の構造は、しばしば、化学的および酵素的プローブ、例えば、ヌクレアーゼに対する、その感受性によって検出することができる。
【0056】
「標的部位」または「標的配列」は、結合にとって十分な条件が存在すれば、結合分子が結合する核酸の一部を規定する核酸配列である。例えば、配列5’−GAATTC−3’は、EcoRI制限エンドヌクレアーゼに対する標的部位である。
【0057】
「外来性」分子は、通常は細胞中に存在しないが、1つ以上の遺伝学的、生化学的、または他の方法によって細胞中に導入することができる分子である。「通常は細胞中に存在」は、細胞の特定の発達段階および環境条件に対して決定される。したがって、例えば、筋の胚発達中にのみ存在する分子は、成体筋細胞に対しては外来性分子である。同様に、熱ショックによって誘導される分子は、非熱ショック細胞に対しては外来性分子である。外来性分子は、例えば、任意のポリペプチドもしくはその断片に対するコード配列、機能不全型内在性分子の機能型、または正常機能型内在性分子の機能不全型を含む可能性がある。更に、外来性分子は、宿主細胞中の内在性遺伝子のオルソログである別の種からのコード配列を含む可能性がある。
【0058】
外来性分子は、とりわけ、コンビナトリアルケミストリープロセスによって生成されるもの等の小分子、または、タンパク質、核酸、糖質、脂質、糖タンパク質、リポタンパク質、多糖、上記の分子の任意の修飾誘導体、または上記の分子の1つ以上を含む任意の複合体等の高分子であってもよい。核酸にはDNAおよびRNAが含まれ、それは一本鎖または二本鎖であってもよく、線状、分岐状、または環状であってもよく、任意の長さであってもよい。核酸には、二重鎖を形成する能力のあるもの、ならびに三重鎖形成核酸が含まれる。例えば、米国特許第5,176,996号および同第5,422,251号明細書を参照されたい。タンパク質には、DNA結合タンパク質、転写因子、クロマチンリモデリング因子、メチル化DNA結合タンパク質、ポリメラーゼ、メチラーゼ、デメチラーゼ、アセチラーゼ、デアセチラーゼ、キナーゼ、ホスファターゼ、インテグラーゼ、リコンビナーゼ、リガーゼ、トポイソメラーゼ、ジャイレース、およびヘリカーゼが含まれるが、これらに限定されない。
【0059】
外来性分子は、内在性分子と同一の種類の分子、例えば、外来性タンパク質または核酸であってもよい。例えば、外来性核酸は、感染ウイルスゲノム、細胞中に導入されたプラスミドもしくはエピソーム、または細胞内に通常存在しない染色体を含むことができる。細胞中に外来性分子を導入するための方法は当業者に既知であり、脂質媒介導入(すなわち、中性およびカチオン性脂質を含むリポソーム)、エレクトロポレーション、直接注入、細胞融合、マイクロプロジェクタイルボンバードメント、リン酸カルシウム共沈、DEAE−デキストラン媒介導入、およびウイルスベクター媒介導入を含むが、これらに限定されない。
【0060】
対照的に、「内在性」分子は、特定の環境条件下で特定の発達段階にある特定の細胞中に通常存在するものである。例えば、内在性核酸は、染色体、ミトコンドリア、クロロプラストもしくは他の細胞小器官のゲノム、または天然に存在するエピソーム核酸を含むことができる。更なる内在性分子には、タンパク質、例えば、転写因子および酵素が含まれてもよい。
【0061】
本明細書で使用されるとき、「外来性核酸の産物」という用語は、ポリヌクレオチドおよびポリペプチド産物の両方、例えば、転写産物(RNA等のポリヌクレオチド)および翻訳産物(ポリペプチド)を含む。
【0062】
「融合」分子は、2つ以上のサブユニット分子が、好ましくは共有結合的に、連結された分子である。サブユニット分子は、同一の化学種類の分子であってもよいか、または異なる化学種類の分子であってもよい。第1の種類の融合分子の例としては、融合タンパク質(例えば、ZFP DNA結合ドメインと開裂ドメインとの間の融合物)および融合核酸(例えば、上述の融合タンパク質をコードする核酸)が挙げられるが、これらに限定されない。第2の種類の融合分子の例としては、三重鎖形成核酸とポリペプチドとの間の融合物、および副溝結合剤と核酸との間の融合が挙げられるが、これらに限定されない。
【0063】
細胞中の融合タンパク質の発現は、細胞に融合タンパク質を送達すること、または細胞に融合タンパク質をコードするポリヌクレオチドを送達することによってもたらされてもよく、ポリヌクレオチドが転写され、転写物が翻訳されて、融合タンパク質を生成する。トランススプライシング、ポリペプチド開裂、およびポリペプチド連結はまた、細胞中のタンパク質の発現に関与することができる。細胞へのポリヌクレオチドおよびポリペプチドの送達の方法が本開示の他所で提示される。
【0064】
本開示に関して、「遺伝子」は、かかる調節配列がコードおよび/または転写配列に隣接しているか否かに関わらず、遺伝子産物(以下を参照)をコードするDNA領域、ならびに遺伝子産物の産生を調節する全てのDNA領域を含む。したがって、遺伝子には、プロモーター配列、ターミネータ、リポソーム結合部位および内部リポソーム侵入部位等の翻訳調節配列、エンハンサー、サイレンサー、インスレーター、境界エレメント、複製起点、マトリックス付着部位、ならびに遺伝子座制御領域が含まれるが、これらに限定されない。
【0065】
「遺伝子発現」は、遺伝子に含有される情報の、遺伝子産物への変換を指す。遺伝子産物は、遺伝子の直接転写産物(例えば、mRNA、tRNA、rRNA、アンチセンスRNA、リボザイム、構造RNA、または任意の他の種類のRNA)、またはmRNAの翻訳によって産生されるタンパク質である可能性がある。遺伝子産物には、キャッピング、ポリアデニル化、メチル化、および編集等のプロセスによって修飾されたRNA、および、例えば、メチル化、アセチル化、リン酸化、ユビキチン化、ADPリボシル化、ミリスチリル化、およびグリコシル化によって修飾されたタンパク質も含まれる。
【0066】
遺伝子発現の「調節」は、遺伝子の活性の変化を指す。発現の調節には、遺伝子活性化および遺伝子抑制が含まれてもよいが、これらに限定されない。
【0067】
「植物」細胞には、単子葉(単子葉類)または双子葉(双子葉類)植物の細胞が含まれるが、これらに限定されない。単子葉類の非限定的例としては、トウモロコシ、イネ、オオムギ、オートムギ、コムギ、ソルガム、ライムギ、サトウキビ、パイナップル、タマネギ、バナナ、およびココナッツ等の穀物植物が挙げられる。双子葉類の非限定的例としては、タバコ、トマト、ヒマワリ、綿花、テンサイ、ジャガイモ、レタス、メロン、ダイズ、キャノーラ(ナタネ)、およびアルファルファが挙げられる。植物細胞は、植物の任意の部分からの、および/または植物発達の任意の段階からのものであってもよい。
【0068】
「目的の領域」は、例えば、領域内で外因性分子を結合させることが望まれる、遺伝子、または遺伝子内のもしくはそれに隣接する非コード配列等の細胞クロマチンの任意の領域である。結合の目的は、標的化DNA開裂および/または標的化組み換えであり得る。目的の領域は、例えば、染色体、エピソーム、細胞小器官ゲノム(例えば、ミトコンドリア、クロロプラスト)、または感染ウイルスゲノム内に存在することができる。目的の領域は、遺伝子のコード領域内、例えば、リーダー配列、トレーラー配列、もしくはイントロン等の転写された非コード領域内、またはコード領域の上流もしくは下流のいずれかの非転写領域内である可能性がある。目的の領域は、単一のヌクレオチド対ほど小さいか、または最大2,000ヌクレオチド対長、またはヌクレオチド対の任意の整数値である可能性がある。
【0069】
「作動的連結」および「作動的に連結」(または「作動可能に連結」)という用語は、2つ以上の構成要素(配列要素等)の並列に関連して交換可能に使用され、その中で両方の構成要素が正常に機能し、かつ構成要素の少なくとも1つが、他の構成要素の少なくとも1つに対して発揮される機能を媒介することができる可能性を許容するように各構成要素が配置される。例証として、プロモーター等の転写調節配列は、転写調節配列が1つ以上の転写調節因子の存在または不在に応じてコード配列の転写レベルを制御する場合に、コード配列に作動的に連結される。転写調節配列は、一般的に、コード配列とシスで作動的に連結されるが、それに直接隣接している必要はない。例えば、エンハンサーは、隣接していなくても、コード配列に作動的に連結される転写調節配列である。
【0070】
融合ポリペプチドに関して、「作動的に連結」という用語は、構成要素の各々が連動して機能し、そのように連結されていない場合にそれが果たす機能と同じ機能を実行するという事実を指すことができる。例えば、ZFP DNA結合ドメインが開裂ドメインに融合されている融合ポリペプチドに関しては、ZFP DNA結合ドメインおよび開裂ドメインは、融合ポリペプチドにおいて、ZFP DNA結合ドメイン部分が、その標的部位および/またはその結合部位に結合することが可能であり、開裂ドメインが、標的部位の近傍でDNAを開裂することが可能である場合に、作動的連結状態にある。
【0071】
タンパク質、ポリペプチド、または核酸の「機能的断片」は、その配列が、完全長タンパク質、ポリペプチド、または核酸と同一でないが、完全長タンパク質、ポリペプチド、または核酸と同一の機能を保持するタンパク質、ポリペプチド、または核酸である。機能的断片は、対応する天然分子より多い、少ない、または同一の残基数を有することができ、かつ/または1つ以上のアミノ酸またはヌクレオチド置換を含有することができる。核酸の機能(例えば、コード機能、別の核酸とハイブリッド形成する能力)を決定するための方法は、当技術分野で周知である。同様に、タンパク質機能を決定するための方法は周知である。例えば、ポリペプチドのDNA結合機能は、例えば、フィルター結合、電気泳動移動度シフト、または免疫沈降アッセイによって決定することができる。DNA開裂は、ゲル電気泳動によってアッセイすることができる。上記のAusubelらを参照されたい。別のタンパク質と相互作用するタンパク質の能力は、例えば、共免疫沈降、ツーハイブリッドアッセイ、または遺伝的相補性および生化学的相補性の両方の相補性によって決定することができる。例えば、Fields et al.(1989)Nature 340:245−246、米国特許第5,585,245号、およびPCT国際公開第98/44350号を参照されたい。
【0072】
多重挿入部位
多重挿入部位、つまり、適切なZFN対の結合時に、多重挿入部位がZFN対の標的部位の間で開裂されるように複数の亜鉛フィンガーヌクレアーゼ(ZFN)結合部位を含むポリヌクレオチドが、本明細書に開示される。
【0073】
多重挿入部位上に含まれる標的部位は、好ましくは、それが組み込まれる細胞のゲノム内には見出されない。したがって、ゲノム内の望まれない開裂の発生は、低減または排除される。任意の数、例えば、1〜50(またはその間の任意の数)、好ましくは2〜30(またはその間の任意の数)、および更により好ましくは5〜20(またはその間の任意の数)の標的部位が、多重挿入部位ポリヌクレオチドに含まれてもよい。亜鉛フィンガーヌクレアーゼについて、標的部位は、典型的には、亜鉛フィンガーヌクレアーゼが、ホモ二量体またはヘテロ二量体を形成して、適切な部位で開裂するような対になっている。
【0074】
更に、図1に示されるように、標的部位(図1の影付き三角)の各対の1つの標的部位は、多重挿入部位全体にわたって同じであってもよい。代替的には、ヘテロ二量体対は、部位の間で異なってもよい。
【0075】
多重挿入部位は、ホモ二量体のみが結合する標的部位、ヘテロ二量体のみが結合する標的部位、またはホモ二量体またはヘテロ二量体が結合する標的部位の組み合わせを含んでもよい。場合によっては、次の1つ以上の理由で、ホモ二量体が結合する標的部位が好ましい可能性がある:1つのZFNの送達が2つのそれよりも効率的であり得ること、ホモ二量体化が、ZFNの不均等な発現に起因する不均等な化学量論の問題を低減すること;オフターゲット部位における開裂からの毒性が低減され得ること;CCHC(非標準)亜鉛フィンガードメインを使用するとき、それによってホモ二量体が破壊される可能性が半減すること;および/または固有の標的可能部位の総数が拡大され得ること。代替的に、他の場合には、ヘテロ二量体は、異なる標的部位の混合およびマッチング、故にZFN対に対する標的可能部位の増加の可能性を許容するため、好ましい可能性がある。また、ヘテロ二量体は、実行者の必要に応じて、ドナーの逐次的付加も許容する。ヘテロ二量体組み合わせはまた、新規ZFN対の使用を通じて、ドナーの任意の所望の部分の特異的欠失も許容し得る。
【0076】
亜鉛フィンガーヌクレアーゼに対する標的部位が3ヌクレオチドの倍数である必要がないことは明白であろう。例えば、交差鎖相互作用が生じる場合(例えば、米国特許第6,453,242号および国際公開第02/077227号を参照されたい)、マルチフィンガー結合ドメインの個々の亜鉛フィンガーのうちの1つ以上は、重複する四重亜部位に結合することができる。結果として、3フィンガータンパク質は、10ヌクレオチド配列に結合することができ、ここで10番目のヌクレオチドは、末端フィンガーが結合する四重の一部であり、4フィンガータンパク質は、13ヌクレオチド配列に結合することができ、ここで13番目のヌクレオチドは、末端フィンガーが結合する四重の一部である、等である。
【0077】
マルチフィンガー結合ドメインにおける個々の亜鉛フィンガーの間のアミノ酸リンカー配列の長さおよび性質もまた、標的配列への結合に影響を及ぼす。例えば、マルチフィンガー結合ドメインにおける隣接した亜鉛フィンガーの間の、いわゆる「非標準的なリンカー」、「長いリンカー」、または「構造化リンカー」の存在は、それらのフィンガーが直接隣接していない亜部位に結合することを可能にし得る。かかるリンカーの非限定的例は、例えば、米国特許第6,479,626号および国際公開第01/53480号に記載される。したがって、亜鉛フィンガー結合ドメインに対する標的部位における1つ以上の亜部位は、互いから1、2、3、4、5、またはそれを超えるヌクレオチド分だけ隔てられてもよい。一例を提供するにすぎないが、4フィンガー結合ドメインは、順番に、2つの隣接する3ヌクレオチド亜部位、介入するヌクレオチド、および2つの隣接する三重亜部位を含む、13ヌクレオチド標的部位に結合することができる。
【0078】
配列(例えば、標的部位)の間の距離は、互いに最も近接した配列の端から測定した、2つの配列の間に介入するヌクレオチドまたはヌクレオチド対の数を指す。
【0079】
開裂が2つの亜鉛フィンガードメイン/開裂片側ドメイン融合分子の、別個の標的部位への結合に依存するある実施形態では、2つの標的部位は、反対側のDNA鎖上にあり得る。他の実施形態では、両方の標的部位は、同じDNA鎖上にある。
【0080】
多重挿入部位は、植物ゲノム内のいずれの場所にも組み込むことができる。ある実施形態では、多重挿入部位は、トウモロコシゲノムにおけるZp15に組み込まれ、それは米国出願第12/653,735号に記載されるように、外来性配列の標的の組み込みのために望ましい部位である。
【0081】
DNA結合ドメイン
任意のDNA結合ドメインを、本明細書に開示される方法で使用することができる。ある実施形態では、DNA結合ドメインは、亜鉛フィンガータンパク質を含む。亜鉛フィンガー結合ドメインは、1つ以上の亜鉛フィンガーを含む。Miller et al.(1985)EMBO J.4:1609−1614、Rhodes(1993)Scientific American Feb.:56−65、米国特許第6,453,242号。本明細書に記載される亜鉛フィンガー結合ドメインは、一般に、2つ、3つ、4つ、5つ、6つ、または更にそれを超える亜鉛フィンガーを含む。
【0082】
典型的には、単一の亜鉛フィンガードメインは、約30アミノ酸長である。構造的研究は、各亜鉛フィンガードメイン(モチーフ)が、2つのベータシート(2つの不変のシステイン残基を含有するベータターンにおいて保有される)およびアルファヘリックス(2つの不変のヒスチジン残基を含有する)を含有し、それらが2つのシステインおよび2つのヒスチジンによる亜鉛原子の配位を通じて、特定のコンフォメーションにおいて保有されることを実証している。
【0083】
亜鉛フィンガーは、標準的なC2H2亜鉛フィンガー(すなわち、亜鉛イオンが2つのシステインおよび2つのヒスチジン残基によって配位されるもの)、ならびに例えば、C3H亜鉛フィンガー(亜鉛イオンが3つのシステイン残基および1つのヒスチジン残基によって配位されるもの)、およびC4亜鉛フィンガー(亜鉛イオンが4つのシステイン残基によって配位されるもの)等の非標準的な亜鉛フィンガーの両方を含む。植物における使用のための非標準的なZFPに関してはまた、国際公開第02/057293号、およびまた米国特許公開第20080182332号も参照されたい。
【0084】
改変された亜鉛フィンガー結合ドメインは、天然に存在する亜鉛フィンガータンパク質と比較して、新規の結合特異性を有することができる。改変方法には、合理的設計および様々な種類の選択が含まれるが、これらに限定されない。合理的設計には、例えば、三重(または四重)ヌクレオチド配列および個々の亜鉛フィンガーアミノ酸配列を含むデータベースを使用することが含まれ、ここで各三重または四重ヌクレオチド配列は、特定の三重または四重配列に結合する亜鉛フィンガーの1つ以上のアミノ酸配列に関連する。
【0085】
ファージディスプレイおよび2ハイブリッド系を含む例示的な選択方法は、米国特許第5,789,538号、同第5,925,523号、同第6,007,988号、同第6,013,453号、同第6,410,248号、同第6,140,466号、同第6,200,759号、および同第6,242,568号、ならびに国際公開第98/37186号、国際公開第98/53057号、国際公開第00/27878号、国際公開第01/88197号、および英国特許第2,338,237号に開示される。
【0086】
亜鉛フィンガー結合ドメインに対する結合特異性の強化は、例えば、譲受人共通の国際公開第02/077227号に記載されている。
【0087】
個々の亜鉛フィンガーは、3ヌクレオチド(すなわち、三重)配列(または隣接した亜鉛フィンガーの4ヌクレオチド結合部位により、1ヌクレオチド分だけ重複し得る4ヌクレオチド配列)に結合するため、亜鉛フィンガー結合ドメインが改変されて結合する配列(例えば、標的配列)の長さは、改変亜鉛フィンガー結合ドメインにおける亜鉛フィンガーの数を決定するであろう。例えば、フィンガーモチーフが重複する亜部位に結合しないZFPについて、6ヌクレオチド標的配列には、2フィンガー結合ドメインが結合し、9ヌクレオチド標的配列には、3フィンガー結合ドメインが結合する、等である。本明細書に記載されるように、標的部位における個々の亜鉛フィンガーに対する結合部位(すなわち、亜部位)は、隣接している必要はないが、マルチフィンガー結合ドメインにおける亜鉛フィンガーの間のアミノ酸配列(すなわち、フィンガー間リンカー)の長さおよび性質に依存して、1つ以上のヌクレオチド分だけ隔てられる可能性がある。
【0088】
マルチフィンガー亜鉛フィンガー結合ドメインにおいて、隣接した亜鉛フィンガーは、およそ5アミノ酸(いわゆる「標準」フィンガー間リンカー)のアミノ酸リンカー配列分、または代替的に、1つ以上の非標準的なリンカー分だけ隔てられる可能性がある。例えば、譲受人共通の米国特許第6,453,242号および同第6,534,261号を参照されたい。3つを超えるフィンガーを含む改変された亜鉛フィンガー結合ドメインについて、場合によっては、ある種の亜鉛フィンガー間のより長い(「非標準」)フィンガー間リンカーの挿入は、それが結合ドメインによる結合の親和性および/または特異性を増加させ得るため、望ましい可能性がある。例えば、米国特許第6,479,626号および国際公開第01/53480号を参照されたい。したがって、マルチフィンガー亜鉛フィンガー結合ドメインはまた、非標準的なフィンガー間リンカーの存在および位置に関して特徴付けることができる。例えば、3フィンガー(2つの標準的なフィンガー間リンカーによって結合される)、長いリンカー、および3つの追加的なフィンガー(2つの標準的なフィンガー間リンカーによって結合される)を含む6フィンガー亜鉛フィンガー結合ドメインは、2×3構成と表示される。同様に、2つのフィンガー(その間に標準的なリンカーを有する)、長いリンカー、および2つの追加的なフィンガー(標準的なリンカーによって結合される)を含む結合ドメインは、2×2構成と表示される。3つの2フィンガー単位(その各々において2つのフィンガーは、標準的なリンカーによって結合される)を含み、その中で各2つのフィンガー単位が長いリンカーによって隣接した2つのフィンガー単位に結合されるタンパク質は、3×2構成と称される。
【0089】
マルチフィンガー結合ドメインにおける2つの隣接した亜鉛フィンガーの間の、長いまたは非標準的なフィンガー間リンカーの存在は、しばしば、2つのフィンガーが、標的配列内で直接隣接していない亜部位に結合することを可能にする。したがって、標的部位における亜部位の間に1つ以上のヌクレオチドのギャップが存在し得、すなわち、標的部位は、亜鉛フィンガーが接触していない1つ以上のヌクレオチドを含有し得る。例えば、2×2亜鉛フィンガー結合ドメインは、1ヌクレオチド分だけ隔てられた2つの6ヌクレオチド配列に結合することができ、すなわち、それは13ヌクレオチド標的部位に結合する。また、Moore et al.(2001a)Proc.Natl.Acad.Sci.USA 98:1432−1436、Moore et al.(2001b)Proc.Natl.Acad.Sci.USA 98:1437−1441、および国際公開第01/53480号も参照されたい。
【0090】
前述のように、標的亜部位は、単一の亜鉛フィンガーが結合する、3ヌクレオチドまたは4ヌクレオチド配列である。ある目的に関して、2フィンガー単位は、「結合モジュール」と表示される。結合モジュールは、例えば、特定の6ヌクレオチド標的配列に結合するマルチフィンガータンパク質(一般に、3フィンガー)の文脈において、2つの隣接したフィンガーについて選択することによって得ることができる。代替的に、モジュールは、個々の亜鉛フィンガーの組立によって構築することができる。また、国際公開第98/53057号および国際公開第01/53480号も参照されたい。
【0091】
代替的に、DNA結合ドメインは、ヌクレアーゼに由来し得る。例えば、I−SceI、I−CeuI、PI−PspI,PI−Sce、I−SceIV、I−CsmI、I−PanI、I−SceII、I−PpoI、I−SceIII、I−CreI、I−TevI、I−TevII、およびI−TevIII等のホーミングエンドヌクレアーゼおよびメガヌクレアーゼの認識配列が知られている。また、米国特許第5,420,032号、米国特許第6,833,252号、Belfort et al.(1997)Nucleic Acids Res.25:3379−3388、Dujon et al.(1989)Gene 82:115−118;Perler et al.(1994)Nucleic Acids Res.22,1125−1127、Jasin(1996)Trends Genet.12:224−228、Gimble et al.(1996)J.Mol.Biol.263:163−180、Argast et al.(1998)J.Mol.Biol.280:345−353、およびNew England Biolabsカタログも参照されたい。加えて、ホーミングエンドヌクレアーゼおよびメガヌクレアーゼのDNA結合特異性を、非天然の標的部位に結合するよう改変することができる。例えば、Chevalier et al.(2002)Molec.Cell 10:895−905、Epinat et al.(2003)Nucleic Acids Res.31:2952−2962、Ashworth et al.(2006)Nature 441:656−659、Paques et al.(2007)Current Gene Therapy 7:49−66、米国特許公開第20070117128号を参照されたい。
【0092】
別の代替として、DNA結合ドメインは、ロイシンジッパータンパク質に由来し得る。ロイシンジッパーは、遺伝子発現に関連する重要な転写因子である多くの真核性調節タンパク質におけるタンパク質−タンパク質相互作用に関与する、タンパク質のクラスである。ロイシンジッパーは、動物、植物、酵母等を含む複数の界にわたって、これらの転写因子において共有される共通の構造モチーフを指す。ロイシンジッパーは、2つのポリペプチドのロイシン残基がヘリックスの同じ面上で終了するように、ロイシン残基がα−ヘリックスを通じて均等に間隔をあけられる様態で、特異的DNA配列に結合する2つのポリペプチド(ホモ二量体またはヘテロ二量体)によって形成される。ロイシンジッパーのDNA結合特異性は、本明細書に開示されるDNA結合ドメインにおいて利用することができる。
【0093】
幾つかの実施形態では、DNA結合ドメインは、植物病原菌キサントモナスに由来するTALエフェクターからの改変ドメインである(Miller et al.(2010)Nature Biotechnology,Dec 22 [Epub ahead of print]、Boch et al,(2009)Science 29 Oct 2009(10.1126/science.117881)、およびMoscou and Bogdanove,(2009)Science 29 Oct 2009(10.1126/science.1178817)を参照されたい。
【0094】
開裂ドメイン
上記のように、DNA結合ドメインは、開裂(ヌクレアーゼ)ドメインに関連し得る。例えば、ホーミングエンドヌクレアーゼは、ヌクレアーゼ機能を保持しながら、それらのDNA結合特異性において修飾されてもよい。加えて、亜鉛フィンガータンパク質を、開裂ドメインに融合して、亜鉛フィンガーヌクレアーゼ(ZFN)を形成してもよい。本明細書に開示される融合タンパク質の開裂ドメイン部分は、任意のエンドヌクレアーゼまたはエキソヌクレアーゼから得ることができる。開裂ドメインの由来であり得る例示的なエンドヌクレアーゼには、制限エンドヌクレアーゼおよびホーミングエンドヌクレアーゼが含まれるが、これらに限定されない。例えば、2002−2003 Catalogue,New England Biolabs,Beverly,MA;and Belfort et al.(1997)Nucleic Acids Res.25:3379−3388を参照されたい。DNAを開裂する追加的な酵素は既知である(例えば、S1ヌクレアーゼ;マングビーンヌクレアーゼ;膵臓DNase I;ミクロコッカルヌクレアーゼ;酵母HOエンドヌクレアーゼ、また、Linn et al.(eds.)Nucleases,Cold Spring Harbor Laboratory Press,1993も参照されたい)。ホーミングエンドヌクレアーゼおよびメガヌクレアーゼの非限定的例としては、I−SceI、I−CeuI、PI−PspI、PI−Sce、I−SceIV、I−CsmI、I−PanI、I−SceII、I−PpoI、I−SceIII、I−CreI、I−TevI、I−TevII、およびI−TevIIIが既知である。また、米国特許第5,420,032号、米国特許第6,833,252号、Belfort et al.(1997)Nucleic Acids Res.25:3379−3388、Dujon et al.(1989)Gene 82:115−118、Perler et al.(1994)Nucleic Acids Res.22,1125−1127、Jasin(1996)Trends Genet.12:224−228、Gimble et al.(1996)J.Mol.Biol.263:163−180、Argast et al.(1998)J.Mol.Biol.280:345−353、およびNew England Biolabsカタログも参照されたい。これらの酵素(またはその機能的断片)のうちの1つ以上を、開裂ドメインおよび開裂片側ドメインの源として使用することができる。
【0095】
制限エンドヌクレアーゼ(制限酵素)は、多くの種に存在し、DNA(認識部位における)に配列特異的に結合すること、また結合の部位におけるまたはそれに近接するDNAを開裂することができる。ある種の制限酵素(例えば、IIS型)は、認識部位から除去された部位におけるDNAを開裂し、分離可能な結合および開裂ドメインを有する。例えば、IIS型酵素FokIは、1つの鎖上のその認識部位から9ヌクレオチド、およびもう1つの鎖上のその認識部位から13ヌクレオチドにおいて、DNAの2本鎖切断を触媒する。例えば、米国特許第5,356,802号、同第5,436,150号、および同第5,487,994号、ならびにLi et al.(1992)Proc.Natl.Acad.Sci.USA 89:4275−4279、Li et al.(1993)Proc.Natl.Acad.Sci.USA 90:2764−2768、Kim et al.(1994a)Proc.Natl.Acad.Sci.USA 91:883−887、Kim et al.(1994b)J.Biol.Chem.269:31,978−31,982を参照されたい。故に、一実施形態では、融合タンパク質は、少なくとも1つのIIS型制限酵素からの開裂ドメイン(または開裂片側ドメイン)および1つ以上の亜鉛フィンガー結合ドメインを含み、それは改変されてもよく、されなくてもよい。
【0096】
開裂ドメインが結合ドメインから分離可能な、例示的なIIS型制限酵素は、FokIである。この特定の酵素は、二量体として活性である。Bitinaite et al.(1998)Proc.Natl.Acad.Sci.USA 95:10,570−10,575。したがって、本開示に関して、開示される融合タンパク質に使用されるFokI酵素の部分は、開裂片側ドメインとみなされる。故に、亜鉛フィンガー−FokI融合を使用する細胞配列の標的化二本鎖開裂および/または標的化置換のために、各々がFokI開裂片側ドメインを含む2つの融合タンパク質を使用して、触媒的に活性な開裂ドメインを再構成することができる。代替的に、亜鉛フィンガー結合ドメインおよび2つのFokI開裂片側ドメインを含有する単一のポリペプチド分子を使用することもできる。亜鉛フィンガー−FokI融合を使用する標的化開裂および標的化配列変更についてのパラメータは、本開示の他の箇所に提供される。
【0097】
開裂ドメインまたは開裂片側ドメインは、開裂活性を保持するか、または多量体化(例えば、二量体化)して機能性開裂ドメインを形成する能力を保持するタンパク質の任意の部分であってもよい。
【0098】
例示的なIIS型制限酵素は、譲受人共通の国際公開第WO2007/014275号に記載され、参照によりその全体が本明細書に組み込まれる。
【0099】
開裂特異性を強化するために、開裂ドメインがまた修飾されてもよい。ある実施形態では、開裂片側ドメインの変異体が用いられ、これらの変異体は、開裂片側ドメインのホモ二量体化を最小化または防止する。かかる修飾された開裂片側ドメインの非限定的例は、国際公開第2007/014275号に詳細に開示され、参照によりその全体が本明細書に組み込まれる。また、実施例も参照されたい。ある実施形態では、ホモ二量体化を最小化または防止する改変開裂片側ドメイン(二量体化ドメイン突然変異体とも称される)を含む開裂ドメインは、当業者に既知であり、例えば、米国特許公開第20050064474号および同第20060188987号に記載され、参照によりそれらの全体が本明細書に組み込まれる。FokIの446、447、479、483、484、486、487、490、491、496、498、499、500、531、534、537、および538位におけるアミノ酸残基は全てFokI開裂片側ドメインの二量体化に影響を及ぼすための標的である。例えば、米国特許公開第20050064474号および同第20060188987号、国際特許公開第WO07/139898号、Miller et al.(2007)Nat.Biotechnol.25(7):778−785、およびDoyon et al(2011)Nature Methods 8(1):74−79を参照されたい。
【0100】
偏性ヘテロ二量体を形成するFokIの追加的な改変された開裂片側ドメインを、本明細書に記載されるZFNにおいて使用することもできる。一実施形態では、第1の開裂片側ドメインは、FokIの490および538位におけるアミノ酸残基における突然変異を含み、第2の開裂片側ドメインは、アミノ酸残基486および499における突然変異を含む。
【0101】
ある実施形態では、開裂ドメインは、2つの開裂片側ドメインを含み、それらの両方が、結合ドメイン、第1の開裂片側ドメイン、および第2の開裂片側ドメインを含む単一のポリペプチドの一部である。開裂片側ドメインは、それらがDNAを開裂させるように機能する限り、同じアミノ酸配列または異なるアミノ酸配列を有することができる。
【0102】
一般に、融合タンパク質が、開裂片側ドメインを含む場合、2つの融合タンパク質が、開裂に必要とされる。代替的に、2つの開裂片側ドメインを含む単一のタンパク質を使用することができる。2つの開裂片側ドメインは、同じエンドヌクレアーゼ(またはその機能的断片)に由来し得るか、または各開裂片側ドメインが、異なるエンドヌクレアーゼ(またはその機能的断片)に由来し得る。加えて、2つの融合タンパク質に対する標的部位は、好ましくは、互いに対して、2つの融合タンパク質のそれらの対応する標的部位への結合が、例えば、二量体化によって、開裂片側ドメインが機能性開裂ドメインを形成することを可能にする相互に対する空間的配向で開裂片側ドメインを配置するように、配置される。故に、ある実施形態では、標的部位の近端は、5〜8ヌクレオチド分または15〜18ヌクレオチド分、隔てられる。しかしながら、任意の整数の数のヌクレオチドまたはヌクレオチド対が、2つの標的部位(例えば、2〜50ヌクレオチド以上)の間に介入してもよい。一般に、開裂の点は、標的部位の間にある。
【0103】
融合タンパク質
融合タンパク質(およびそれをコードするポリヌクレオチド)の設計および構築のための方法は、当業者に既知である。例えば、DNA結合ドメイン(例えば、亜鉛フィンガードメイン)および調節または開裂ドメイン(または開裂片側ドメイン)を含む融合タンパク質、ならびにかかる融合タンパク質をコードするポリヌクレオチドの設計および構築のための方法は、譲受人共通の米国特許第6,453,242号および同第6,534,261号、ならびに米国特許出願公開第2007/0134796号および同第2005/0064474号に記載され、参照によりそれらの全体が本明細書に組み込まれる。ある実施形態では、融合タンパク質をコードするポリヌクレオチドが構築される。これらのポリヌクレオチドは、ベクターに挿入することができ、ベクターは、細胞に導入することができる(ポリヌクレオチドを細胞に導入するためのベクターおよび方法に関する追加的な開示については下記を参照されたい)。
【0104】
本明細書に記載される方法のある実施形態では、亜鉛フィンガーヌクレアーゼは、FokI制限酵素からの亜鉛フィンガー結合ドメインおよび開裂片側ドメインを含む融合タンパク質を含み、2つのかかる融合タンパク質は、細胞中で発現される。2つの融合タンパク質の細胞中の発現は、2つのタンパク質の細胞中への送達、1つのタンパク質およびタンパク質の1つをコードする1つの核酸の、細胞中への送達、各々がタンパク質の1つをコードする2つの核酸の、細胞中への送達から、または両方のタンパク質をコードする単一の核酸の、細胞中への送達によって、もたらされ得る。追加的な実施形態では、融合タンパク質は、2つの開裂片側ドメインおよび亜鉛フィンガー結合ドメインを含む、単一のポリペプチド鎖を含む。この場合、単一の融合タンパク質が細胞中で発現され、理論に拘束されることを望むものではないが、開裂片側ドメインの分子内二量体の形成の結果としてDNAを開裂させると考えられる。
【0105】
ある実施形態では、融合タンパク質(例えば、ZFP−FokI融合)の構成要素は、亜鉛フィンガードメインが融合タンパク質のアミノ末端に最近接し、開裂片側ドメインがカルボキシ末端に最近接するように配列される。これは、FokI酵素に由来するもの等の天然に存在する二量体化開裂ドメインにおける開裂ドメインの相対的配向を反映し、ここでDNA結合ドメインは、アミノ末端に最近接し、開裂片側ドメインは、カルボキシ末端に最近接する。これらの実施形態では、機能性ヌクレアーゼを形成するための開裂片側ドメインの二量体化は、融合タンパク質の、反対側のDNA鎖上の部位への結合によって引き起こされ、このうち結合部位の5’末端は、互いに近位にある。
【0106】
追加的な実施形態では、融合タンパク質(例えば、ZFP−FokI融合)の構成要素は、開裂片側ドメインが融合タンパク質のアミノ末端に最近接し、亜鉛フィンガードメインがカルボキシ末端に最近接するように配列される。これらの実施形態では、機能性ヌクレアーゼを形成するための開裂片側ドメインの二量体化は、融合タンパク質の、反対側のDNA鎖上の部位への結合によって引き起こされ、このうち結合部位の3’末端は、互いに近位にある。
【0107】
なおも追加的な実施形態では、第1の融合タンパク質は、融合タンパク質のアミノ末端に最近接した開裂片側ドメイン、およびカルボキシ末端に最近接した亜鉛フィンガードメインを含有し、第2の融合タンパク質は、亜鉛フィンガードメインが融合タンパク質のアミノ末端に最近接し、開裂片側ドメインがカルボキシ末端に最近接するように配列される。これらの実施形態では、両方の融合タンパク質が同じDNA鎖に結合し、このうち第1の融合タンパク質の結合部位は、アミノ末端に最近接した亜鉛フィンガードメインを含有する第2の融合タンパク質の結合部位の5’側に対して位置する、カルボキシ末端に最近接した亜鉛フィンガードメインを含有する。
【0108】
開示される融合タンパク質のある実施形態では、亜鉛フィンガードメインと開裂ドメイン(または開裂片側ドメイン)との間のアミノ酸配列は、「ZCリンカー」と表示される。ZCリンカーは、上述のフィンガー間リンカーと区別されるべきである。開裂を最適化するZCリンカーを得ることに関する詳細については、例えば、米国特許公開第20050064474A1号および同第20030232410号、ならびに国際特許公開第WO05/084190号を参照されたい。
【0109】
一実施形態では、本開示は、表1に示される認識ヘリックスアミノ酸配列のうちの1つ以上を有する亜鉛フィンガータンパク質を含むZFNを提供する。別の実施形態では、表1に示される1つ以上の認識ヘリックスを有するZFPをコードするヌクレオチド配列を含む、ZFP発現ベクターが本明細書に提供される。
【0110】
標的化組込み
開示される方法および組成物を使用して、多重挿入部位が組み込まれている任意の細胞ゲノム中のDNAを開裂することができ、それは適切なZFN対の存在下での、外来性配列の多重挿入部位への安定な標的化組み込みおよび/または外来性配列の切除を促進する。図1および2を参照されたい。
【0111】
ZFN挿入部位が外来性配列の一部として細胞のゲノムに連続して導入される方法もまた、本明細書に記載される。図4および5を参照されたい。例えば、ヘテロ二量体ヌクレアーゼ部位の異なる組み合わせが側面に位置する外来性配列が、ゲノムに挿入される。その後、適切な隣接するZFN部位の1つにおいて開裂するZFN対が、再びヘテロ二量体ヌクレアーゼ部位の異なる組み合わせを含む別の外来性配列の存在下で、細胞に導入される。このプロセスは、外来性配列を挿入するために所望に応じて反復することができる。加えて、適切なZFN対の存在下で、1つ以上の外来性配列がゲノムから切除されてもよい。
【0112】
図6は、外来性配列がマーカー遺伝子および目的の遺伝子を含む、別の実施形態を示す。マーカー遺伝子および目的の遺伝子の両方の側面には、適切な場合、例えば、追加的な遺伝子を挿入するときに、マーカー遺伝子が欠失させられ得るように、異なるZFN結合部位(異なる陰影を有する三角として示される)が位置する。選択可能マーカーの数が限定される植物等の生物において、これはわずか1つにすぎない選択可能マーカー遺伝子の使用を可能にし、目的の遺伝子をスタッキングする可能性を大幅に促進する。ある実施形態では、例えば、相同性指向性DNA修復の効率に依存して、「分裂した」選択可能マーカーが使用されてもよい。分裂した選択可能マーカーを使用するドナーDNA配列の正しい組み込みにより、発現可能な選択可能マーカー遺伝子が作り出される。選択可能マーカーは、組み込まれたDNA配列から切除することができ、したがって再利用することができる。別の実施形態では、除去のための外来性配列には、ゲノムにおいて分裂したマーカー遺伝子の部分配列が隣接する。切除されると、マーカー遺伝子は再構築され、機能性マーカー遺伝子の作成をもたらす。選択可能マーカー切除の使用は、必要とされる選択可能マーカーの数を2つまたは場合によっては1つのみに限定する。
【0113】
本明細書に記載されるように組み込まれた多重挿入部位への標的化組み込みについて、1つ以上のDNA結合ドメイン(例えば、ZFP)は、所定の開裂部位における、またはそれに近接した標的部位に結合するように改変され、改変DNA結合ドメインおよび開裂ドメインを含む融合タンパク質が細胞中で発現される。融合タンパク質のDNA結合(例えば、亜鉛フィンガー)部分が標的部位に結合すると、DNAは、好ましくは二本鎖切断を介して、開裂ドメインによって標的部位に近隣して開裂される。
【0114】
多重挿入部位における二本鎖切断の存在は、相同組み換えを介した外来性配列の組み込みを促進する。ある実施形態では、多重挿入部位への挿入対象の外来性配列を含むポリヌクレオチドは、相同組み換えを促進するために、多重挿入部位ポリヌクレオチドおよび/または周辺のゲノムと相同性の1つ以上の領域を含むであろう。ドナー配列とゲノム配列との間の、およそ25、50、100、200、500、750、1,000、1,500、2,000ヌクレオチドまたはそれを超える配列相同性(または10〜2,000ヌクレオチドもしくはそれを超える任意の整数値)がその間の相同組み換えを支持するであろう。ある実施形態では、相同性アームは、1,000塩基対長未満である。他の実施形態では、相同性アームは、750塩基対長未満である。また、米国仮特許出願第61/124,047号も参照されたく、それは参照により本明細書に組み込まれる。ドナー分子(外来性配列)は、細胞クロマチンに対して相同性の、複数の不連続の領域を含有し得る。例えば、目的の領域に通常存在しない配列の標的化挿入のために、該配列は、ドナー核酸分子に存在し、目的の領域における遺伝子配列に対して相同性の領域が側面に位置する可能性がある。
【0115】
目的の任意の配列(外来性配列)は、本明細書に記載されるように多重挿入部位に導入、またはそこから切除することができる。例示的な外来性配列には、任意のポリペプチドコード配列(例えば、cDNA)、プロモーター、エンハンサー、および他の調節配列(例えば、干渉RNA配列、shRNA発現カセット、エピトープタグ、マーカー遺伝子、開裂酵素認識部位および様々な種類の発現構築物が含まれるが、これらに限定されない。かかる配列は、標準的な分子生物学技術(クローニング、合成等)を使用して容易に得ることができ、かつ/または市販されている。外来性配列は、融合タンパク質の発現の前に、それと同時に、またはその後に細胞に導入することができる。
【0116】
ドナーポリヌクレオチドは、DNAまたはRNAであっても、一本鎖または二本鎖のいずれか一方であってもよく、線状または環状形態で細胞に導入されてもよい。線状形態で導入される場合、ドナー配列の末端部は、当業者に既知の方法によって保護(例えば、エキソヌクレアーゼ分解から)することができる。例えば、1つ以上のジデオキシヌクレオチド残基が線状分子の3’末端に付加され、かつ/または自己相補性オリゴヌクレオチドは、1つまたは両方の末端部に結合される。例えば、Chang et al.(1987)Proc.Natl.Acad.Sci.USA 84:4959−4963、Nehls et al.(1996)Science 272:886−889を参照されたい。外来性ポリヌクレオチドを分解から保護するための追加的な方法には、末端アミノ基の付加、ならびに例えば、ホスホロチオエート、ホスホロアミデート、およびO−メチルリボースまたはデオキシリボース残基等の修飾されたヌクレオチド間結合の使用が含まれるが、これらに限定されない。
【0117】
ポリヌクレオチドは、例えば、複製起点、プロモーター、および抗体抵抗性をコードする遺伝子等の追加的な配列を有するベクター分子の一部として細胞に導入することができる。更に、ドナーポリヌクレオチドは、ネイキッド核酸として、ナノ粒子、リポソーム、もしくはポロキサマー等の薬剤と複合体化した核酸として、導入することができるか、または細菌もしくはウイルスによって植物細胞に送達することができる(例えば、アグロバクテリウム、リゾビウム属NGR234、シノリゾビウム・メリロティ(Sinorhizoboium meliloti)、メソリゾビウム・ロティ、タバコモザイクウイルス、ジャガイモXウイルス、カリフラワーモザイクウイルス、およびカッサバ葉脈モザイクウイルス。例えば、Chung et al.(2006)Trends Plant Sci.11(1):1−4を参照されたい。
【0118】
上に詳述されるように、各々が亜鉛フィンガー結合ドメインおよび開裂片側ドメインを含む、2つの融合タンパク質(ホモ二量体またはヘテロ二量体)に対する多重挿入部位上の結合部位は、他の結合部位に最近接した各結合部位の端から測定して5〜8または15〜18ヌクレオチド離れて位置し得、開裂は、結合部位の間で生じる。開裂が単一の部位においてまたは結合部位の間の多重部位において生じるかは、開裂されたゲノム配列がドナー配列によって置換されるため、重要でない。故に、標的化組み換えによる単一のヌクレオチド対の配列の効率的変更のために、結合部位の間の領域の中点は、そのヌクレオチド対の10,000ヌクレオチド内、好ましくは1,000ヌクレオチド、または500ヌクレオチド、または200ヌクレオチド、または100ヌクレオチド、または50ヌクレオチド、または20ヌクレオチド、または10ヌクレオチド、または5ヌクレオチド、または2ヌクレオチド、または1ヌクレオチド内であるか、または目的のヌクレオチド対における。
【0119】
例えば、RAD54エピスタシスグループ(例えば、AtRad54、AtRad51)の植物遺伝子、遺伝子の産物が前述の遺伝子産物と相互作用する遺伝子等の相同組み換えに関与する遺伝子の発現を活性化するための、追加的なZFP機能性ドメイン融合の使用を含むが、これらに限定されない、標的化組み換えのレベルを強化し得る方法および組成物もまた、提供される。例えば、Klutstein et al.Genetics.2008 Apr;178(4):2389−97を参照されたい。
【0120】
同様に、ZFP機能性ドメイン融合は、非相同末端結合(例えば、Ku70/80、XRCC4、ポリ(ADPリボース)ポリメラーゼ、DNAリガーゼ4)に関与する遺伝子の発現を抑制するために、本明細書に開示される方法および組成物と組み合わせて使用することができる。例えば、Riha et al.(2002)EMBO 21:2819−2826、Freisner et al.(2003)Plant J.34:427−440、Chen et al.(1994)European Journal of Biochemistry 224:135−142を参照されたい。亜鉛フィンガー結合ドメインと機能性ドメインとの間の融合を使用した、遺伝子発現の活性化および抑制のための方法は、例えば、譲受人共通の米国特許第6,534,261号、同第6,824,978号、および同第6,933,113号に開示される。追加的な抑制方法には、アンチセンスオリゴヌクレオチドおよび/または抑制対象の遺伝子の配列を標的とする低分子干渉RNA(siRNAまたはRNAi)もしくはshRNAの使用が含まれる。
【0121】
亜鉛フィンガー/ヌクレアーゼ融合分子およびドナーDNA分子を含む細胞中の標的化組み換えの効率における更なる増加は、相同性駆動型修復プロセスが最大限に活性である細胞周期のG2期において細胞を遮断することによって達成される。かかる停止は、幾つかの方法で達成することができる。例えば、細胞は、G2期の細胞を停止させるために、例えば、細胞周期進行に影響を及ぼす薬物、化合物および/または小分子で処置することができる。この種類の例示的な分子には、微小管重合に影響を及ぼす化合物(例えば、ビンブラスチン、ノコダゾール、タキソール)、DNAと相互作用する化合物(例えば、シス−プラチナム(II)ジアミンジクロリド、シスプラチン、ドキソルビシン)、および/またはDNA合成に影響を及ぼす化合物(例えば、チミジン、ヒドロキシ尿素、L−ミモシン、エトポシド、5−フルオロウラシル)が含まれるが、これらに限定されない。組み換え効率における追加的な増加は、クロマチン構造を変化させて、ゲノムDNAを細胞組み換え機構へとより到達可能にする、ヒストンデアセチラーゼ(HDAC)阻害剤(例えば、酪酸ナトリウム、トリコスタチンA)の使用によって達成される。
【0122】
細胞周期停止のための追加的な方法には、例えば、タンパク質をコードするcDNAを細胞中に導入することによって、または細胞中にタンパク質をコードする遺伝子の発現を活性化する改変されたZFPを導入することによって、CDK細胞周期キナーゼの活性を阻害するタンパク質を過剰発現させることが含まれる。細胞周期停止はまた、例えば、RNAi法を使用して(例えば、米国特許第6,506,559号)サイクリンおよびCDKの活性を阻害することによって、または細胞中に、例えば、サイクリンおよび/またはCDK遺伝子等の、細胞周期進行に関与する1つ以上の遺伝子の発現を抑制する改変されたZFPを導入することによっても達成される。遺伝子発現の調節のために改変された亜鉛フィンガータンパク質の合成のための方法については、例えば、譲受人共通の米国特許第6,534,261号を参照されたい。
【0123】
代替的に、ある場合には、ドナーポリヌクレオチドの不在下で(好ましくはSまたはG2期において)標的化開裂が実施され、組み換えが相同染色体の間で生じる。
【0124】
発現ベクター
本明細書に記載される1つ以上の融合タンパク質(例えば、ZFN)をコードする核酸は、複製および/または発現のための原核細胞または真核細胞への形質転換のために、ベクターにクローン化することができる。ベクターは、原核性ベクター、例えば、プラスミド、またはシャトルベクター、昆虫ベクター、または真核性ベクターであり得る。融合タンパク質をコードする核酸もまた、細胞への投与のために発現ベクターにクローン化することができる。
【0125】
融合タンパク質(例えば、ZFN)を発現させるために、融合タンパク質をコードする配列は、典型的には、転写を導くためのプロモーターを含有する発現ベクターにサブクローン化される。好適な細菌および真核性プロモーターは、当該技術分野において周知であり、例えば、Sambrook et al.,Molecular Cloning,A Laboratory Manual(2nd ed.1989;3rd ed.,2001)、Kriegler,Gene Transfer and Expression:A Laboratory Manual(1990);and Current Protocols in Molecular Biologyに記載される(上記のAusubelらを参照されたい。ZFPを発現させるための細菌発現系は、例えば、大腸菌、バチルス属、およびサルモネラで入手可能である(Palva et al.,Gene 22:229−235(1983))。かかる発現系のためのキットが市販されている。哺乳類細胞、酵母、および昆虫細胞のための真核性発現系は、当業者に周知であり、また市販されている。
【0126】
融合タンパク質コード核酸の発現を導くために使用されるプロモーターは、特定の用途に依存する。例えば、宿主細胞に適合させられた強力な構成的プロモーターは、典型的には融合タンパク質の発現および精製のために使用される。
【0127】
対照的に、植物遺伝子の調節のために融合タンパク質がインビボ投与されるとき(下記の「植物細胞への核酸送達」節を参照されたい)、融合タンパク質の特定の使用に依存して、構成的プロモーター、調節されたプロモーター(例えば、発達中に、組織または細胞型によって、または環境によって)、または誘導性プロモーターのいずれかが、使用される。植物プロモーターの非限定的例としては、シロイヌナズナ・ユビキチン−3(ubi−3)(Callis,et al.,1990,J.Biol.Chem.265−12486−12493)、アグロバクター・ツメファシエンス(Δmas)(Petolinoら、米国特許第6,730,824号)、および/またはカッサバ葉脈モザイクウイルス(CsVMV)(Verdaguer et al.,1996,Plant Molecular Biology 31:1129−1139)に由来するプロモーター配列が挙げられる。また、実施例も参照されたい。
【0128】
プロモーターに加えて、発現ベクターは、典型的には、原核性または真核性のいずれかの宿主細胞中での核酸の発現に必要とされる全ての追加的な要素を含有する、転写単位または発現カセットを含有する。典型的な発現カセットは故に、例えば、融合タンパク質をコードする核酸配列に作動可能に連結されたプロモーター、および例えば、転写物の効率的なポリアデニル化、転写終結、リボソーム結合部位、または翻訳終結のために必要とされるシグナルを含有する。カセットの追加的な要素には、例えば、エンハンサー、異種スプライシングシグナル、および/または核局在化シグナル(NLS)が含まれてもよい。
【0129】
遺伝子情報を細胞中に移入するために使用される特定の発現ベクターは、融合タンパク質の意図される使用、例えば、植物、動物、細菌、真菌、原虫等における発現に関連して選択される(後述の発現ベクターを参照されたい)。標準的な細菌および動物発現ベクターは、当該技術分野において既知であり、例えば、米国特許公開第20050064474A1号、ならびに国際特許公開第WO05/084190号、同第WO05/014791号、および同第WO03/080809号に詳述される。
【0130】
標準的なトランスフェクション方法を使用して、大量のタンパク質を発現する細菌、哺乳類、酵母、または昆虫細胞株を産生することができ、それは次いで標準技術を使用して精製することができる(例えば、Colley et al.,J.Biol.Chem.264:17619−17622(1989)、Guide to Protein Purification,in Methods in Enzymology,vol.182(Deutscher,ed.,1990)を参照されたい)。真核細胞および原核細胞の形質転換は、標準技術に従って実施される(例えば、Morrison,J.Bact.132:349−351(1977)、Clark−Curtiss&Curtiss,Methods in Enzymology 101:347−362(Wu et al.,eds.,1983)を参照されたい。
【0131】
外来ヌクレオチド配列をかかる宿主細胞に導入するための周知の手順のうちのいずれが使用されてもよい。これらには、リン酸カルシウムトランスフェクション、ポリブレン、プロトプラスト融合、エレクトロポレーション、超音波法(例えば、ソノポレーション)、リポソーム、マイクロインジェクション、ネイキッドDNA、プラスミドベクター、エピソーム型および組み込み型の両方のウイルスベクターの使用、ならびにクローン化ゲノムDNA、cDNA、合成DNA、または他の外来遺伝材料を、宿主細胞に導入するための他の周知の方法のいずれの使用も含まれる(例えば、上記のSambrookらを参照されたい)。使用される特定の遺伝子改変手順が、少なくとも1つの遺伝子を、選択のタンパク質を発現することができる宿主細胞に首尾よく導入できれば十分である。
【0132】
植物細胞への核酸送達
上記のように、DNA構築物は、多様な従来の技術によって所望の植物宿主に(例えば、そのゲノムに)導入されてもよい。かかる技術の概説については、例えば、Weissbach&Weissbach Methods for Plant Molecular Biology(1988,Academic Press,N.Y.)Section VIII,pp.421−463、およびGrierson&Corey,Plant Molecular Biology(1988,2d Ed.),Blackie,London,Ch.7−9を参照されたい。
【0133】
例えば、DNA構築物は、植物細胞プロトプラストのエレクトロポレーションおよびマイクロインジェクション等の技術を使用して植物細胞のゲノムDNAに直接導入されてもよく、またはDNA構築物は、DNAマイクロプロジェクタイルボンバードメント等のバイオリスティック法を使用して植物組織に直接導入することができる(例えば、Klein et al.(1987)Nature 327:70−73を参照されたい)。代替的に、DNA構築物は、ナノ粒子形質転換を介して植物細胞に導入することができる(例えば、米国特許出願第12/245,685号を参照されたく、それは参照によりその全体が本明細書に組み込まれる)。代替的に、DNA構築物は、好適なT−DNA境界/隣接領域と組み合わされ、従来のアグロバクテリウム・ツメファシエンス宿主ベクターに導入されてもよい。アグロバクテリウム・ツメファシエンス媒介型形質転換技術は、無害化(disarming)およびバイナリーベクターの使用を含めて、科学文献に詳細に記載される。例えば、Horsch et al.(1984)Science 233:496−498、およびFraley et al.(1983)Proc.Nat’l.Acad.Sci.USA 80:4803を参照されたい。
【0134】
加えて、遺伝子移入は、リゾビウム属NGR234、シノリゾビウム・メリロティ(Sinorhizoboium meliloti)、メソリゾビウム・ロティ、ジャガイモXウイルス、カリフラワーモザイクウイルス、およびカッサバ葉脈モザイクウイルスおよび/またはタバコモザイクウイルス等の、非アグロバクテリウム細菌またはウイルスを使用して達成してもよく、例えば、Chung et al.(2006)Trends Plant Sci.11(1):1−4を参照されたい。
【0135】
アグロバクテリウム・ツメファシエンス宿主のビルレンス機能は、バイナリーT DNAベクター(Bevan(1984)Nuc.Acid Res.12:8711−8721)または共培養手順(Horsch et al.(1985)Science 227:1229−1231)を使用して、細胞をこの細菌に感染させるとき、構築物および隣接マーカーを含有するT鎖の、植物細胞DNAへの挿入を導く。一般に、アグロバクテリウム形質転換系は、双子葉植物を改変するために使用される(Bevan et al.(1982)Ann.Rev.Genet 16:357−384、Rogers et al.(1986)Methods Enzymol.118:627−641)。アグロバクテリウム形質転換系は、単子葉植物および植物細胞にDNAを形質転換ならびに移入するためにも使用することができる。米国特許第5、591,616号、Hernalsteen et al.(1984)EMBO J 3:3039−3041、Hooykass−Van Slogteren et al.(1984)Nature 311:763−764、Grimsley et al.(1987)Nature 325:1677−179、Boulton et al.(1989)Plant Mol.Biol.12:31−40、およびGould et al.(1991)Plant Physiol.95:426−434を参照されたい。
【0136】
代替的な遺伝子移入および形質転換方法には、カルシウム、ポリエチレングリコール(PEG)、またはエレクトロポレーション媒介型ネイキッドDNAの取り込みを通じたプロトプラスト形質転換(Paszkowski et al.(1984)EMBO J 3:2717−2722、Potrykus et al.(1985)Molec.Gen.Genet.199:169−177、Fromm et al.(1985)Proc.Nat.Acad.Sci.USA 82:5824−5828、およびShimamoto(1989)Nature 338:274−276を参照されたい)、および植物組織のエレクトロポレーション(D’Halluin et al.(1992)Plant Cell 4:1495−1505)が含まれるが、これらに限定されない。植物細胞形質転換のための追加的な方法には、マイクロインジェクション、炭化ケイ素媒介型DNA取り込み(Kaeppler et al.(1990)Plant Cell Reporter 9:415−418)、およびマイクロプロジェクタイルボンバードメント(Klein et al.(1988)Proc.Nat.Acad.Sci.USA 85:4305−4309、およびGordon−Kamm et al.(1990)Plant Cell 2:603−618を参照されたい)が含まれる。
【0137】
開示される方法および組成物を使用して、外来配列を、植物細胞のゲノムに挿入されている多重挿入部位に挿入することができる。植物ゲノムに導入された導入遺伝子の発現は、その組み込み部位に決定的に依存するため、これは有用である。例えば、除草剤耐性、虫害抵抗性、栄養素、抗体または治療用分子をコードする遺伝子は、標的化組み換えによって、それらの発現に好ましい植物ゲノムの領域に挿入することができる。
【0138】
上記の形質転換技術のいずれかによって産生される形質転換された植物細胞を培養して、形質転換された遺伝子型を有し、故に所望の表現型を有する、全植物体を再生することができる。かかる再生技術は、組織培養成長培地中のある種の植物ホルモンの操作に依存し、典型的には、所望のヌクレオチド配列と一緒に導入されている殺生物剤および/または除草剤マーカーに依存する。培養プロトプラストからの植物再生は、Evans,et al.,”Protoplasts Isolation and Culture” in Handbook of Plant Cell Culture,pp.124−176、Macmillian Publishing Company,New York,1983、およびBinding,Regeneration of Plants,Plant Protoplasts,pp.21−73,CRC Press,Boca Raton,1985に記載される。再生はまた、植物のカルス、外植片、器官、花粉、胚、またはそれらの一部から得ることもできる。かかる再生技法は、一般に、Klee et al.(1987)Ann.Rev.of Plant Phys.38:467−486に記載される。
【0139】
植物細胞に導入された核酸を使用して、本質的にいずれの植物にも、所望の形質を付与することができる。本開示の核酸構築物および上述の様々な形質転換方法を使用して、本明細書に記載される所望の生理学的および作物栽培学的特徴のために、多種多様な植物および植物細胞系が改変されてもよい。好ましい実施形態では、改変用の標的植物および植物細胞には、穀物作物(例えばコムギ、トウモロコシ、イネ、アワ(millet)、オオムギ)、果実作物(例えばトマト、リンゴ、ナシ、イチゴ、オレンジ)、飼料作物(例えばアルファルファ)、根菜作物(例えばニンジン、ジャガイモ、サトウダイコン、ヤムイモ)、葉菜作物(例えばレタス、ホウレンソウ);顕花植物(例えばペチュニア、バラ、キク)、針葉樹およびマツ(例えばマツ、モミ、トウヒ)、ファイトレメディエーションに使用される植物(例えば重金属蓄積植物);油料作物(例えば、ヒマワリ、ナタネ)および実験目的のために使用される植物(例えば、アラビドプシス)を含む作物等の単子葉植物および双子葉植物が含まれるが、これらに限定されない。故に、開示される方法および組成物は、例えば、限定するわけではないが、アスパラガス属、カラスムギ属、アブラナ属、ミカン属、スイカ属、トウガラシ属、カボチャ属、ニンジン属、ダイズ属、ワタ属、オオムギ属、アキノノゲシ属、トマト属、リンゴ属、マニホット属、タバコ属、イネ属、ワニナシ属、エンドウ属、ナシ属、サクラ属、ダイコン属、ライムギ属、ナス属、モロコシ属、コムギ属、ブドウ属、ササゲ属、およびトウモロコシ属に属する種を含むが、これらに限定されない、広範囲にわたる植物に使用することができる。
【0140】
当業者であれば、現カセットがトランスジェニック植物に安定に組み込まれ、作動可能であることが確認された後、それを有性交配によって他の植物体に導入できることを認識するであろう。幾つかの標準的育種技術のうちのいずれも、交配させる種に依存して使用することができる。
【0141】
形質転換された植物細胞、カルス、組織または植物体は、形質転換DNA上に存在するマーカー遺伝子によってコードされる形質に関して、改変された植物材料を選択またはスクリーニングすることによって特定し、単離することができる。例えば、選択は、形質転換遺伝子構築物によって耐性を付与される、阻害量の抗生物質または除草剤を含有する培地上で、改変された植物材料を成長させることよって行うことができる。更に、形質転換された植物および植物細胞はまた、組み換え核酸構築物上に存在し得る任意の可視マーカー遺伝子(例えば、β−グルクロニダーゼ、ルシフェラーゼ、BまたはC1遺伝子)の活性についてスクリーニングすることによって特定することもできる。かかる選択およびスクリーニング手法は、当業者に周知である。
【0142】
また、物理的および生化学的方法を使用して、挿入された遺伝子構築物を含有する植物形質転換体または植物細胞形質転換体を特定してもよい。これらの方法には、1)組み換えDNA挿入を検出し、その構造を決定するためのサザン分析またはPCR増幅、2)遺伝子構築物のRNA転写物を検出し、検査するためのノーザンブロット、S1 RNase保護、プライマー伸長、または逆転写酵素−PCR増幅、3)かかる遺伝子産物が遺伝子構築物によってコードされる場合、酵素活性またはリボザイム活性を検出するための酵素アッセイ、4)遺伝子構築物産物がタンパク質である場合、タンパク質ゲル電気泳動、ウェスタンブロット技術、免疫沈降、または酵素結合免疫測定法(ELISA)が含まれるが、これらに限定されない。また、インサイツハイブリダイゼーション、酵素染色、および免疫染色等の追加的な技術を使用して、特定の植物器官または植物組織における組み換え構築物の存在または発現を検出することもできる。全てのこれらのアッセイを行うための方法は、当業者に周知である。
【0143】
本明細書に開示される方法を使用する遺伝子操作の効果は、例えば、目的の組織から単離されるRNA(例えば、mRNA)のノーザンブロットよって観察することができる。典型的には、mRNAが存在するか、またはmRNAの量が増加している場合、対応する導入遺伝子が発現されていると想定することができる。遺伝子および/またはコードされたポリペプチド活性を測定する他の方法を使用することができる。使用される基質および反応産物または副産物の増減を検出する方法に依存して、異なる種類の酵素アッセイを使用することができる。加えて、発現されるポリペプチドのレベルは、免疫化学的に、すなわち、ELISA、RIA、EIA、および電気泳動検出アッセイ(染色またはウェスタンブロッティングのいずれかによる)等による当業者に周知の他の抗体に基づくアッセイで測定することができる。1つの非限定的例として、ELISAアッセイを使用するAAD−1およびPATタンパク質の検出は、米国特許出願第11/587,893号に記載され、この参考文献は、本明細書での参照によりその全体が本明細書に組み込まれる。導入遺伝子は、植物体の一部の組織において、または一部の発達段階で、選択的に発現させてもよく、または導入遺伝子は、実質的に全ての植物組織において、実質的にそのライフサイクル全体にわたって発現させてもよい。しかしながら、任意の組み合わせ発現(combinatorial expression)様式もまた適用可能である。
【0144】
本開示はまた、上述の遺伝子導入植物の種子も包含し、ここで種子は、導入遺伝子または遺伝子構築物を有する。本開示は更に、上述の遺伝子導入植物の子孫、クローン、細胞株もしくは細胞を包含し、該子孫、クローン、細胞株もしくは細胞は、導入遺伝子または遺伝子構築物を有する。
【0145】
融合タンパク質(例えば、ZFN)および融合タンパク質をコードする発現ベクターは、遺伝子調節、標的化開裂、および/または組み換えのために、植物に直接投与することができる。ある実施形態では、植物は、多重パラロガス標的遺伝子を含有する。故に、1つ以上の異なる融合タンパク質または融合タンパク質をコードする発現ベクターは、植物におけるこれらのパラロガス遺伝子(例えば、Zp15、PCT特許公開第WO2010077319号を参照されたい)遺伝子のうちの1つ以上を標的とするために、植物に投与されてもよい。
【0146】
有効量の投与は、融合タンパク質を導入して、処置対象の植物細胞と最終接触させるために通常使用される経路のうちのいずれによっても行われる。ZFPは、好ましくは許容される担体と共に、任意の好適な様態で投与される。かかるモジュレータを投与する好適な方法が利用可能であり、かつ当業者に周知であり、1つ以上の経路を使用して特定の組成物を投与することができるが、特定の経路は、しばしば、別の経路よりも即時かつ有効な反応を提供することができる。
【0147】
また、担体が使用されてもよく、それは一部には、投与されている特定の組成物によって、ならびに組成物を投与するために使用される特定の方法によって決定される。したがって、多種多様の好適な担体の製剤が利用可能である。
【0148】
哺乳類細胞への送達
本明細書に記載されるZFNは、任意の好適な手段によって、例えば、ZFN mRNAの注入によって、標的哺乳類細胞に送達されてもよい。Hammerschmidt et al.(1999)Methods Cell Biol.59:87−115を参照されたい。
【0149】
亜鉛フィンガーを含むタンパク質を送達する方法は、例えば、米国特許第6,453,242号、同第6,503,717号、同第6,534,261号、同第6,599,692号、同第6,607,882号、同第6,689,558号、同第6,824,978号、同第6,933,113号、同第6,979,539号、同第7,013,219号、および同第7,163,824号に記載され、これらの全ての開示は、参照によりそれらの全体が本明細書に組み込まれる。
【0150】
本明細書に記載されるZFNはまた、ZFNのうちの1つ以上をコードする配列を含有するベクターを使用して送達されてもよい。プラスミドベクター、レトロウイルスベクター、レンチウイルスベクター、アデノウイルスベクター、ポックスウイルスベクター;ヘルペスウイルスベクター、およびアデノ関連ウイルスベクター等を含むが、これらに限定されない、任意のベクター系が使用されてもよい。また、米国特許第6,534,261号、同第6,607,882号、同第6,824,978号、同第6,933,113号、同第6,979,539号、同第7,013,219号、および同第7,163,824号も参照されたく、参照によりそれらの全体が本明細書に組み込まれる。更に、これらのベクターのうちのいずれも、ZFNをコードする1つ以上の配列を含み得ることは明白であろう。故に、ZFNの1つ以上の対が細胞に導入される時、ZFNは、同じベクター上または異なるベクター上で担持され得る。複数のベクターが使用されるとき、各ベクターは、1つまたは複数のZFNをコードする配列を含み得る。
【0151】
従来のウイルスおよび非ウイルスに基づく遺伝子移入方法を使用して、改変されたZFPをコードする核酸を、哺乳類細胞に導入することができる。また、かかる方法を使用して、ZFPをコードする核酸を、哺乳類細胞にインビトロ投与することもできる。ある実施形態では、ZFPをコードする核酸は、インビボまたはエクスビボ用途のために投与される。
【0152】
非ウイルスベクター送達系には、エレクトロポレーション、リポフェクション、マイクロインジェクション、バイオリスティック、ビロソーム、リポソーム、免疫リポソーム、ポリカチオン、または脂質:核酸共役体、ネイキッドDNA、人工ビリオン、および薬剤により強化されたDNAの取り込みが含まれる。例えば、Sonitron 2000 system(Rich−Mar)を使用するソノポレーションも、核酸の送達に使用することができる。ウイルスベクター送達系には、細胞への送達後にエピソームゲノムまたは組み込まれたゲノムのいずれかを有する、DNAおよびRNAウイルスが含まれる。追加的な例示的核酸送達系には、Amaxa Biosystems(Cologne,Germany)、Maxcyte,Inc.(Rockville,Maryland)、BTX Molecular Delivery Systems(Holliston,MA)、およびCopernicus Therapeutics Inc,(例えば、米国特許第6008336号を参照されたい)によって提供されるものが含まれる。リポフェクションは、例えば、米国特許第5,049,386号、米国特許第4,946,787号、および米国特許第4,897,355号に記載され、リポフェクション試薬は市販されている(例えば、TRANSFECTAM(商標)およびLIPOFECTIN(商標))。ポリヌクレオチドの効率的な受容体認識リポフェクションに好適なカチオン性脂肪および中性脂肪には、Felgner、国際公開第91/17424号、国際公開第91/16024号のものが含まれる。送達は、細胞(エクスビボ投与)または標的組織(インビボ投与)に行うことができる。免疫脂質複合体等の標的リポソームを含む脂質:核酸複合体の調製は、当業者に周知である(例えば、Crystal,Science 270:404−410(1995)、Blaese et al.,Cancer Gene Ther.2:291−297(1995)、Behr et al.,Bioconjugate Chem.5:382−389(1994)、Remy et al.,Bioconjugate Chem.5:647−654(1994)、Gao et al.,Gene Therapy 2:710−722(1995)、Ahmad et al.,Cancer Res.52:4817−4820(1992)、米国特許第4,186,183号、同第4,217,344号、同第4,235,871号、同第4,261,975号、同第4,485,054号、同第4,501,728号、同第4,774,085号、同第4,837,028号、および同第4,946,787号を参照されたい)。
【0153】
上記のように、開示される方法および組成物は、任意の種類の哺乳類細胞において使用することができる。タンパク質(例えば、ZFP)、それをコードするヌクレオチド、ならびに本明細書に記載されるタンパク質および/またはポリヌクレオチドを含む組成物は、任意の好適な手段によって標的細胞に送達され得る。好適な細胞には、真核細胞および/または細胞株ならびに原核細胞および/または細胞株が含まれるが、これらに限定されない。かかる細胞、またはかかる細胞から生成された細胞株の非制限的な例には、COS、CHO(例えば、CHO−S、CHO−K1、CHO−DG44、CHO−DUXB11、CHO−DUKX、CHOK1SV)、VERO、MDCK、WI38、V79、B14AF28−G3、BHK、HaK、NS0、SP2/0−Ag14、HeLa、HEK293(例えば、HEK293−F、HEK293−H、HEK293−T)、およびperC6細胞、ならびにツマジロクサヨトウ(Sf)等の昆虫細胞、またはサッカロミセス、ピチア、およびシゾサッカロミセス等の真菌細胞が挙げられるが、これらに限定されない。ある特定の実施形態では、細胞株は、CHO−K1、MDCK、またはHEK293細胞株である。好適な初代細胞には、抹消血液単核細胞(PBMC)、およびCD4+T細胞またはCD8+T細胞等であるが、これらに限定されない他の血液細胞サブセットが含まれる。好適な細胞にはまた、例として、胚性幹細胞、人口多能性幹細胞、造血幹細胞、神経幹細胞、および間葉系幹細胞等の幹細胞も含まれる。
【実施例】
【0154】
実施例1:プラスミド設計
実施例1.1:eZFN結合部位
【0155】
8つの改変亜鉛フィンガーヌクレアーゼ(eZFN)結合部位(CL:AR−配列番号1、RL:PR−配列番号2、AL:PR−配列番号3、PL:AR−配列番号4、CL:RR−配列番号5、RL:CR−配列番号6、CL:PR−配列番号7、RL:AR−配列番号8)を、eZFN結合部位の各々に対して固有の、隣接するPCRプライマー部位を有する単一のDNA断片(多重eZFN結合部位)中に組み合わせた。加えて、他のeZFN結合部位を設計し、酵母において高レベルで開裂することを示したが(例えば、米国特許公開第2009/0111119号を参照されたい)、それは次のものを含む:PL:RR−配列番号9、AL:RR−配列番号10、AL:CR−配列番号11、PL:CR−配列番号12、ならびにホモ二量体eZFNのRR:RR−配列番号13、RL:RL−配列番号14、PR:PR−配列番号15、PL:PL−配列番号16、CL:CL−配列番号17、CR:CR−配列番号18、AR:AR−配列番号19、およびAL:AL−配列番号20。「CL」および「CR」は、それぞれ、8266および8196と指定される、CCR5受容体に対する「左」および「右」手亜鉛フィンガー設計を指し、それは米国特許出願第2008/0159996号に示される配列を有し、そこで示される標的部位に結合する。「AL」および「AR」は、それぞれ、15556および15590と指定される、AAVS1遺伝子座に対する「左」および「右」手亜鉛フィンガー設計を指し、米国特許出願第2008/0299580号に示される認識ヘリックス配列を有し、そこで示される標的部位に結合する。「PL」および「PR」設計ならびに「RL」および「RR」設計に対する認識ヘリックス配列および標的部位は、下記の表1および2に列挙される。PLおよびPRの両方は、ヒトPRMT1遺伝子に特異的なZFNに対する「左」および「右」手亜鉛フィンガー設計を指す一方で、「RL」および「RR」は、マウスRosa26遺伝子座に特異的なZFNに対する「左」および「右」手亜鉛フィンガー設計を指す。
【0156】
これらの標的部位のうちのいずれも、バイオインフォマティクス分析によって計測するとき、トウモロコシゲノム中に存在しない。PCRプライマー部位を、eZFNによる染色体局在性DNA断片の二本鎖開裂からもたらされるNHEJの評価のために含めた。
【0157】
【表1】
【0158】
【表2】
【0159】
Att部位を、合成したDNA断片中に含めて、断片を、TOPOクローニング(Invitrogen,Carlsbad,CA)を使用して、プラスミド中にクローン化した。Gateway LR CLONASE(商標)(Invitrogen)反応を使用して、この断片をpDAB101834およびpDAB101849に移入した。これらのベクターは、それぞれタバコおよびトウモロコシに好適な選択可能マーカーを含有する。pDAB101834は、カッサバ葉脈モザイクウイルスプロモーター(CsVMV、カッサバ葉脈モザイクウイルスに由来するプロモーターおよび5’非翻訳領域、Verdaguer et al.,(1996)Plant Molecular Biology,31(6)1129−1139)、ホスフィノスリシンアセチルトランスフェラーゼ遺伝子(PAT、Wohlleben et al.,(1988)Gene 70(1),25−37)、およびAtuORF1 3’ UTR(アグロバクテリウム・ツメファシエンスpTi15955のオープンリーディングフレーム1(ORF1)の転写終結因子およびポリアデニル化部位を含む3’非翻訳領域(UTR)、Barker et al.,(1983)Plant Molecular Biology,2(6),335−50)からなる。トウモロコシpDAB101849ベクターは、イネ(rice)アクチン1遺伝子プロモーター(OsAct1、イネ(Oryza sativa)アクチン1(Act1)遺伝子に由来するプロモーター、5’非翻訳領域(UTR)、およびイントロン、McElroy et al.,(1990)Plant Cell 2(2):163−71)およびZmLip 3’ UTR(トウモロコシLIP遺伝子の転写終結因子およびポリアデニル化部位を含む3’非翻訳領域(UTR)、GenBank受入L35913)を含む選択可能マーカーカセットを含有する。
【0160】
結果として得られたタバコベクター、pDAB105900(図7)を、エレクトロポレーションを使用してアグロバクテリウム・ツメファシエンスに移入した。制限酵素検証後、アグロバクテリウムを、使用するまでグリセロールストックとして保管した。トウモロコシベクター、pDAB105908(図8)を一括にして、製造業者のプロトコルに従ってQiagen QIAfilter Plasmid Giga kit(Qiagen,Valencia,CA)を使用して精製した。
【0161】
実施例1.2:eZFNを発現させるためのベクター
標準(C2H2)主鎖または非標準(C3H)主鎖のいずれかにおいて適切な認識ヘリックスを発現するZFNベクターを、本質的に、米国特許出願第2008/0182332号および同第2008/0159996号に記載されるように調製した。
【0162】
ZFNの機能を、酵母ZFNスクリーニング系に挿入した、実施例1.1に記載されるeZFN多重挿入部位に対して試験した(米国特許公開第2009/0111119号を参照されたい)。試験した全てのZFN対は、酵母系において活性であった。
【0163】
8つのeZFNを、植物細胞中の発現に必要な調節配列を含有するベクター中にクローン化した。構築用に配備したクローニング戦略は、米国特許出願第2009/0111188A1号および同第20100199389号に本質的に記載される通りである。図9および10は、一般化したeZFN発現カセットの概略図を示す。
【0164】
実施例2:トウモロコシにおけるeZFNの評価
実施例2.1:WHISKERS(商標)媒介型DNA送達
トウモロコシの胚形成Hi−II細胞培養物を産生し、内部への組み込みが実証された生存植物細胞の源として使用した。当業者は、多様な植物種に由来する細胞培養物、または多様な植物種に由来する分化した植物組織を、内部への組み込みが実証された生存植物細胞の源として利用することを想定し得る。
【0165】
この実施例において、PAT植物選択可能マーカーカセットおよび多重eZFN結合部位挿入配列を含有するプラスミド(pDAB105908)を使用して、遺伝子導入事象を得た。遺伝子導入単離株を、eZFNにより形質転換して、二本鎖開裂を評価した。
【0166】
特に、事前に凍結保存した細胞株からの12mL血中血球容積(PCV)、加えて28mLの条件培地を、500mLエルレンマイヤーフラスコ中、80mLのGN6液体培地(N6培地(Chu et al.(1975)Scientia Sin 18:659−668)、2.0mg/L 2,4−D、30g/Lスクロース、pH5.8)中に継代培養し、125rpmで、28℃でシェーカー上に配置した。このステップを、同じ細胞株を使用して2回反復して、総計36mL PCVが3つのフラスコにわたって分配されるようにした。24時間後、GN6液体培地を除去し、72mL GN6 S/M浸透培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、45.5g/Lソルビトール、45.5g/Lマンニトール、100mg/Lミオイノシトール、pH6.0)と交換した。フラスコを暗所で30〜35分間、28℃で適度に撹拌しながらインキュベートした(125rpm)。インキュベーション時間中、炭化ケイ素ウィスカ(Advanced Composite Materials,LLC,Greer,SC)の50mg/mL懸濁液を、8.1mLのGN6 S/M液体培地を405mgの滅菌炭化ケイ素ウィスカに添加することによって調製した。
【0167】
GN6 S/M浸透培地中でのインキュベーションに続いて、各フラスコの内容物を250mL遠心瓶中にプールした。フラスコ中の全ての細胞が底に沈殿した後、およそ14mLを超える内容積のGN6 S/M液体を引き出し、後の使用のために滅菌1−Lフラスコ中に収集した。事前に湿潤させたウィスカの懸濁液を、ボルテックス上で、最大速度で60秒間混合し、次いで遠心瓶に添加した。
【0168】
この実施例において、pDAB105908プラスミドDNAからの170μgの精製した断片を各瓶に添加した。一旦、DNAを添加すると、瓶を、改変したRed Devil 5400商業用塗料ミキサ(Red Devil Equipment Co.,Plymouth,MN)中に即座に配置し、10秒間撹拌した。撹拌に続いて、細胞、培地、ウィスカ、およびDNAのカクテルを、125mLの新鮮なGN6液体培地と共に1−Lフラスコの内容物に添加して、浸透圧溶質を減少させた。細胞を、125rpmに設定したシェーカー上で2時間回収させた。6mLの分散した懸濁液を、ハウス真空管路(house vacuum line)に接続したガラス細胞収集ユニットを使用して、Whatman第4濾紙(5.5cm)上で濾過して、1瓶当たり60のフィルターを得るようにした。フィルターをGN6固体培地(2.5g/Lゲルライトゲル化剤を用いたことを除いてGN6液体培地と同じ)の60×20mmプレート上に配置し、暗所条件下で、28℃で1週間培養した。
【0169】
実施例2.2:推定遺伝子導入事象の特定および単離
DNA送達の1週間後、濾紙を、選択剤を含有するGN6(1H)選択培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、100mg/Lミオイノシトール、2.5g/Lゲルライト、pH5.8)の60×20mmプレートに移した。これらの選択プレートを、暗所で、28℃で1週間インキュベートした。暗所での1週間の選択後、細胞の半分を各プレートから、37〜38℃で保持された3.0mLのGN6アガロース培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、100mg/Lミオイノシトール、7g/L SeaPlaqueアガロース、pH5.8、121度でわずか10分間オートクレーブ処理)を含有する管にこすり落とすことによって、組織を新鮮な培地上に埋め込んだ。
【0170】
アガロース/組織混合物をへらで粉砕し、その後、3mLのアガロース/組織混合物を、GN6(1H)培地を含有する100×15mmペトリ皿の表面上に均等に注いだ。このプロセスを、各プレートの両方の片について反復した。一旦、全ての組織を埋め込むと、プレートをNESCOFILM(登録商標)またはPARAFILM M(登録商標)で個々に密閉し、暗所条件下で、28℃で最長10週間培養した。
【0171】
これらの選択条件下で増殖する推定的に形質転換した単離株を、埋め込まれたプレートから除去し、60×20mmプレート中の新鮮な選択培地に移した。およそ2週間後に持続的な増殖が明白であった場合、事象を、適用された除草剤(選択剤)に対して抵抗性であるとみなし、細胞のアリコートをその後、遺伝子型分析のために採取した。
【0172】
実施例2.3:ゲノムDNA抽出
ゲノムDNA(gDNA)を、実施例2.2に記載される単離したトウモロコシ細胞から抽出し、PCR遺伝子型同定実験のためのテンプレートとして利用した。gDNAを、DNeasy 96 Plant Kit(QIAGEN Inc.,Valencia,CA)に詳述される製造業者のプロトコルに従って上述のように単離した、およそ100〜300μL血中血球容積(PCV)のHi−IIカルスから抽出した。ゲノムDNAを、100μLのキット提供の溶出緩衝剤中に溶出して20〜200ng/μLの最終濃度を得、その後、下に概説されるPCRに基づく遺伝子型同定法を介して分析した。
【0173】
実施例2.4:コピー数の分子分析
TAQMAN(登録商標)アッセイを行って、除草剤抵抗性カルスの試料をスクリーニングして、pDAB105908導入遺伝子の単一コピー組み込みを含有したものを特定した。遺伝子発現カセットに特異的なプライマーおよびプローブを使用して、詳細な分析を行った。単一コピー事象を追加的な分析のために特定した。
【0174】
カスタムTAQMAN(登録商標)アッセイを、Third Wave Technologies(Madison,WI)によるHi−IIカルスにおけるPAT遺伝子分析のために開発した。ゲノムDNA試料を、96ウェルプレートフォーマットで95℃でのインキュベーションによって最初に変性させ、次いで周囲温度まで冷却した。次に、マスターミックス(緩衝剤に加えてPATのためのプローブミックスおよび内部参照遺伝子を含有する)を各ウェルに添加し、試料を鉱物油で上塗りした。プレートを密閉し、BioRad TETRAD(登録商標)サーモサイクラー中でインキュベートした。プレートを周囲温度まで冷却した後、蛍光プレートリーダー上で読み取った。全てのプレートは、1コピー、2コピー、および4コピー標準物、ならびに野生型対照試料および試料を含有しないブランクウェルを含有した。読み取りを収集し、各チャネルについてのフォールドオーバーゼロ(fold over zero)(すなわち、バックグラウンド)との比較は、試料の原シグナルを、テンプレートを含有しない原シグナルで割ることによって、各試料について決定した。
【0175】
このデータから標準的な曲線を構築し、最良適合を線形回帰分析によって決定した。この適合から特定したパラメータを使用して、次いで見かけのPATコピー数を各試料について推定した。
【0176】
実施例2.5:PCR遺伝子型同定のためのプライマー設計
この実施例において、PCR遺伝子型同定は、ゲノムに埋め込まれたドナーDNAを含有することが予測される単離したトウモロコシカルス組織に由来するゲノムDNAのポリメラーゼ鎖反応(PCR)増幅、続いてPCR増幅産物の標準的なクローニングおよび配列分析を含むが、これらに限定されないものとして理解した。PCR遺伝子型同定の方法は、詳細に記載されており(例えば、Rios,G.et al.(2002)Plant J.32:243−253)、細胞培養物を含む任意の植物種または組織型に由来するゲノムDNAに適用されてもよい。
【0177】
当業者は、植物ゲノムにおける特異的配列の増幅、植物ゲノムにおける多重特異的配列の増幅、植物ゲノムにおける非特異的配列の増幅、またはそれらの組み合わせを含む(しかしこれらに限定されない)、PCR遺伝子型同定のための戦略を考案し得る。増幅には、この実施例に記載されるように、クローニングおよび配列決定が続いてもよく、または増幅産物の直接配列分析が続いてもよい。当業者は、本明細書で生成される増幅産物の分析のための代替的な方法を想定することもあり得る。本明細書に記載される一実施形態では、遺伝子標的に特異的なオリゴヌクレオチドプライマーが、PCR増幅において用いられる。
【0178】
本明細書に提示される実施例において、オリゴヌクレオチドプライマーは、例えば、Integrated DNA Technologies,Inc.(Coralville,IA)によって、標準的な脱塩の条件下で、水で100μMの濃度まで希釈して合成される。オリゴヌクレオチドプライマーを、DNA挿入の隣接する領域とアニーリングするように設計した。プライマーを、プラスミドDNAの希釈を使用して、非遺伝子導入植物から単離したDNAの存在下で試験した。pDAB105908導入遺伝子を、プライマーを使用して推定事象のゲノムDNAからPCR増幅した。結果として得られた断片をプラスミドベクター中にクローン化し、配列決定して、多重eZFN結合部位配列が形質転換中に植物ゲノムに完全に組み込まれたことを確認した。
【0179】
実施例2.6:標的DNAを有する遺伝子導入事象の選択
低コピー(1〜2)事象をPCRによって、無傷の多重eZFN結合部位配列について、およびPAT遺伝子について、スクリーニングした。コピー数を、サザン分析によって、PAT遺伝子プローブによる標準的な方法を使用して確認した。単一コピーの無傷の挿入物を有する選択された遺伝子導入事象からのカルスを、一過性発現eZFNによるその後の評価のために維持した。
【0180】
実施例3:植物細胞中へのeZFN DNA送達
eZFN媒介型二本鎖開裂を可能にするために、DNAをコードするeZFNの送達、続いて植物細胞中の機能性eZFNタンパク質の発現が必要であることを理解する。当業者は、機能性ZFNタンパク質の発現が、ZFNをコードする構築物の遺伝子組み換え、またはZFNをコードする構築物の一過性発現を含むが、これらに限定されない複数の方法によって達成され得ることを想定し得る。
【0181】
本明細書に引用される実施例において、eZFNをコードするDNAの植物細胞中への送達のための方法が記載される。当業者は、アグロバクテリウム媒介型形質転換、バイオリスティックに基づくDNA送達、またはWHISKERS(商標)媒介型DNA送達を含むが、これらに限定されない、植物細胞に適切な多様なDNA送達方法のうちのいずれを使用することもできる。本明細書に記載される一実施形態では、バイオリスティック媒介型DNA送達実験を、様々なeZFNをコードするDNA構築物を使用して行った。
【0182】
実施例3.1:バイオリスティック媒介型DNA送達
上述のように、トウモロコシの胚形成Hi−II細胞培養物を産生し、eZFN機能を評価するための生存植物の源として使用した。当業者は、多様な植物種に由来する細胞培養物、または多様な植物種に由来する分化した植物組織を、内部への標的化組み込みが実証される生存植物細胞の源として利用することを想定し得る。
【0183】
多重eZFN結合部位上の特異的標的配列において結合する8つのeZFNのうちの1つを発現するプラスミドを、内部対照(IPK−1)と一緒に、5〜10個の遺伝子導入単離株からのカルスのプール中に衝撃した。
【0184】
遺伝子導入Hi−IIトウモロコシカルス事象を、GN6(1H)培地上で毎週継代培養した。培養の7日後、およそ400mgの細胞を、2.5g/Lゲルライトで凝固させたGN6 S/M培地を含有する100×15mmペトリ皿の中心上で、直径2.5cmの円形に薄く広げた。細胞を暗所条件下で4時間培養した。バイオリスティック粒子をDNAでコーティングするために、3mgの0.6ミクロン直径の金粒子を100%エタノールで1回、滅菌蒸留水で2回洗浄し、シリコン処理したエッペンドルフ管中の50μL水中に再懸濁させた。総計5μgのプラスミドDNA、20μLスペルミジン(0.1M)、および50μL塩化カルシウム(2.5M)を金懸濁液に別個に添加し、ボルテックス上で穏やかに混合した。混合物を室温で10分間インキュベートし、ベンチトップ微量遠心機において10,000rpmで10秒間ペレット化し、60μL冷100%エタノール中に再懸濁させ、8〜9μLを各マクロ担体上に分配した。
【0185】
Biolistic PDS−1000/HE(商標)系(Bio−Rad Laboratories,Hercules,CA)を使用して衝撃を行った。細胞を含有するプレートを、1100psiおよび27インチのHg真空の条件下で棚中段に配置し、作動マニュアルに従って衝撃した。衝撃の24時間後、組織を小さい塊でGN6固体培地に移した。
【0186】
実施例4:Solexa配列決定および分析
実施例4.1:試料調製
eZFNおよび対照IPK1−ZFN(Shukla et al.(1990)Nature 459,437−441)による衝撃の72時間後、組織を2mL微量遠心管中に収集し、少なくとも48時間凍結乾燥させた。ゲノムDNAを、製造業者の仕様書に従ってQIAGEN(登録商標)gDNA抽出キットを使用して、凍結乾燥させた組織から抽出した。最後に、DNAを200μLの水中に再懸濁させ、濃度を、Nanodrop分光光度計(Thermo Scientific,Wilmington,DE)を使用して決定した。DNAの無傷を、全ての試料を0.8%アガロースEゲル(Invitrogen,Carlsbad,CA)上で泳動させることによって推定した。全ての試料をPCR増幅に対して正規化して(25ng/μL)、Solexa配列決定に対するアンプリコンを生成した。
【0187】
eZFN開裂部位の各々を包含する領域の増幅のためのPCRプライマー、ならびに標的(ZFN処置された)試料および対照試料からのIPK1−ZFN標的部位を、IDT(Integrated DNA Technologies,San Jose,CA)から購入した。これらのプライマーのために最適な増幅条件を、勾配PCRによって、23.5μL反応中、0.2μMの適切なプライマー、Accuprime Pfx Supermix(1.1X,Invitrogen,Carlsbad,CA)、および100ngのテンプレートゲノムDNAを使用して特定した。循環パラメータには、95℃(5分間)での最初の変性、続いて35周期の変性(95℃、15秒間)、アニーリング[55−72℃、30秒間]、伸張(68℃、1分間)、および最終伸張(72℃、7分間)が含まれる。増幅産物を3.5%TAEアガロースゲル上で分析した。最適なアニーリング温度を特定した後、分取PCR反応を行って、各々一連のPCRプライマーを検証し、またSolexaアンプリコンを生成した。トウモロコシおよびタバコにおけるeZFN標的領域の増幅のために使用されるオリゴヌクレオチドを、下記の表3に示す。IPK1標的領域は、プライマー(配列番号27 GCAGTGCATGTTATGAGC(順方向プライマー)および配列番号28 CAGGACATAAATGAACTGAATC(逆方向プライマー))を使用して増幅した。
【0188】
【表3】
【0189】
分取PCRのために、上述の条件を使用して各テンプレートについて8つの個々の小規模PCR反応を完了し、産物を一緒にプールし、Qiagen MinElute(商標)ゲル精製キットを使用して3.5%アガロースゲル上でゲル精製した。ゲル精製したアンプリコンの濃度を、Nanodrop分光光度計を使用して決定し、eZFN標的化および対応する野生型対照ならびに正規化IPK−1標的化および野生型対照からのおよそ100ngのアンプリコンをプールすることによって、Solexa試料を調製した。eZFN+IPK−1標的化試料、IPK−1標的化試料、および野生型対照から、アンプリコンを含む4つの最終Solexa試料を生成し、配列決定した。アンプリコンをPCR−Blunt II−TOPO(Invitrogen)中にクローン化し、配列決定に供してプライマーを検証した後、Solexa配列決定を行った。
【0190】
実施例4.2:Solexa配列決定および分析
Solexa配列決定は、数千の配列の産生をもたらした。DAS Next Generation Sequence(NGS)分析スクリプトを使用して配列を分析した。低質の配列(品質スコアカットオフ<5を有する配列)を取り除いた。次いで配列を参照配列と整列させ、しばしばZFN活性の指標であるインデルを引き起こす、ZFN媒介型開裂およびNHEJ媒介型修復によって引き起こされた、ZFN開裂部位における挿入/欠失(インデル)についてスコア化した。バックグラウンド活性を差し引いた後の、ZFNタンパク質に対する結合部位の間の「ギャップ」配列内での1bpを超える欠失の数によって、編集活性を決定した。本研究における各eZFNに対する活性を、野生型対照と比較して算出し、IPK−1 ZFN活性に対して正規化した。次いで各eZFNについて正規化した活性を比較して、研究に使用したeZFNをランク付けした。また、活性を、eZFN開裂部位におけるインデルの存在によって、配列整列レベル(Solexa出力と比較した参照)において査定した。
【0191】
図11に示すように、8つのうち7つのeZFNは、トウモロコシにおける編集活性を示す。
【0192】
実施例5:タバコにおけるeZFNの評価
実施例5.1:多重eZFN結合部位配列の安定な組み込み
本明細書に上述される多重eZFN結合部位配列の組み込まれたコピーを有する遺伝子導入植物事象を作製するために、PhytaTrays(Sigma,St.Louis,MO)中のMS培地(Phytotechnology Labs,Shawnee Mission,KS)および30g/Lスクロース上に無菌で増殖させた、Petit Havanaタバコ植物から切り取った葉ディスク(1cm2)(例えば、事前に組み込まれたZFN−IL1結合部位を含有する事象1585−10)を、OD600約1.2まで増殖させたアグロバクテリウムLBA4404含有プラスミドpDAB105900の一晩の培養物上に浮遊させ、滅菌濾紙上でブロッティング乾燥させ、次いで60×20mm皿(1皿当たり5ディスク)中、1mg/Lインドール酢酸および1mg/Lベンジアミノ(benzyamino)プリンを添加した同じ培地上に配置した。72時間の共培養後、葉ディスクを、250mg/Lセフォタキシムおよび5mg/L BASTA(登録商標)を有する同じ培地に移した。3〜4週間後、小植物を、PhytaTrays中、250mg/Lセフォタキシムおよび10mg/L BASTA(登録商標)を有するMS培地に更に2〜3週間移した後、葉の採取および分子分析を行った。
【0193】
実施例5.2:多重eZFN結合部位配列遺伝子導入事象のコピー数およびPTU分析
DNA単離。遺伝子導入タバコ植物組織をBASTA(登録商標)抵抗性小植物から採取し、96ウェル収集プレート中で少なくとも2日間凍結乾燥させた。次いでDNAを、製造業者の指示に従って、DNEASY(商標)96ウェル抽出キット(Qiagen,Valencia,CA)を使用して単離した。Model 2−96A Kleco組織粉砕機(Garcia Manufacturing,Visalia CA)を使用して組織破壊を行った。
【0194】
DNA定量化。結果として得られたゲノムDNAを、QUANT−IT(登録商標)Pico Green DNAアッセイキット(Molecular Probes,Invitrogen,Carlsbad,CA)を使用して定量化した。20ng/μL〜1.25ng/μL(段階希釈した)の範囲の、5つの事前に定量化したDNA標準物を使用して標準曲線生成を行った。未知の試料を、アッセイの線形範囲内に入るように、最初に1:10または1:20希釈に希釈した。5μLの希釈した試料および標準物を、100μLの希釈したPico Green基質(1:200)と混合し、暗所で10分間インキュベートした。次いで蛍光を、Synergy2 プレートリーダー(Biotek,Winooski,VT)を使用して記録した。ゲノムDNA濃度を、バックグラウンド蛍光補正後に算定された標準曲線から推定した。TEまたは水を使用して、DNAを次いで、Biorobot3000自動液体処理機を使用して(Qiagen)10ng/μLの一般的な濃度まで希釈した。
【0195】
コピー数推定。推定遺伝子導入事象を、TAQMAN(登録商標)アッセイに類似した多重化DNA加水分解プローブアッセイを使用して、組み込み複雑性について分析した。多重部位構築物のコピー数を、PAT導入遺伝子および内在性タバコ参照遺伝子、PALの両方に対する、配列特異的プライマーおよびプローブを使用して推定した。両方の遺伝子についてのアッセイを、LIGHTCYCLER(登録商標)プローブ設計ソフトウェア2.0リアルタイムPCRを使用して設計し、両方の遺伝子について、LIGHTCYCLER(登録商標)480系(Roche Applied Science,Indianapolis,IN)を使用して評価した。増幅のために、LIGHTCYCLER(登録商標)480プローブマスターミックスを、0.4μMの各プライマーおよび0.2μMの各プローブを含有する10μL容量の多重化反応物中、1倍の最終濃度で調製した(下記の表4)。2ステップ増幅反応を、蛍光取得により58℃で38秒間の伸張により行った。全ての試料を3系列で実験し、平均Ct値を各試料の分析のために使用した。リアルタイムPCRデータの分析を、相対定量モジュール(relative quant module)を使用するLIGHTCYCLER(登録商標)ソフトウェアを使用して行い、それはΔΔCt法に基づいた。このために、単一コピーキャリブレーターからのDNAの試料を含めて、結果を正規化した。単一コピーキャリブレーター事象をサザン分析によって特定し、それがPAT遺伝子の単一の挿入物を有することを確認した。
【0196】
【表4】
【0197】
PCR。低コピー(1〜2)事象をその後、PAT遺伝子に対する無傷の植物転写単位(PTU)、および無傷の多重eZFN結合部位についてPCRによってスクリーニングした。
【0198】
実施例6:多重eZFN結合部位配列におけるeZFN開裂の試験
組み込まれた多重eZFN結合部位配列における標的開裂を促進する、eZFNの能力を試験するために、遺伝子導入タバコ葉ディスクのアグロバクテリウム共培養を介したeZFN構築物の一過性発現に基づいて、一過性アッセイを使用した。多重eZFN結合部位配列含有構築物の単一の完全長コピー(ならびにZFN−IL1構築物の単一の完全長コピー)を含有する、遺伝子導入事象から切り取った葉ディスク(1cm2)を、OD600約1.2まで増殖させたアグロバクテリウムの一晩の培養物上に浮遊させ、滅菌濾紙上でブロッティング乾燥させ、次いで1mg/Lインドール酢酸および1mg/Lベンジアミノ(benzyamino)プリンを添加した同じ培地上に配置した。試験した各eZFNに対して、次の3つの処置を使用した:1処置当たり20個の葉ディスクを用いて、pDAB1601(陰性対照PATのみ)、pDAB4346のみ(陽性対照ZFN−IL1のみ)、およびpDAB4346+pDABeZFN−X(試験対象のZFN−IL1+eZFN)。
【0199】
実施例6.1:配列分析
ゲノムDNAを、Qiagen DNA抽出キットを使用して、アグロバクテリウム処置した遺伝子導入タバコ葉ディスク物から単離した。全ての処置を2系列で行い、全ての試料からのゲノムDNAを100μLの水中に再懸濁させ、濃度をNanodropによって決定した。個々の処置に対する各複製物からの等量のゲノムDNAを一緒にプールし、Solexaアンプリコン生成のための出発テンプレートとして使用した。
【0200】
標的(eZFN処置された)試料および対照試料からの、多重eZFN結合部位配列および開裂部位を包含する領域の増幅のためのPCRプライマーは、Integrated DNA Technologies(Coralville,IA)からのものであり、それをHPLC精製した。最適な増幅条件を、勾配PCRによって、23.5μL反応中、0.2μMの適切なプライマー、Accuprime Pfx Supermix(1.1X,Invitrogen,Carlsbad,CA)、および100ngのテンプレートゲノムDNAを使用して特定した。循環パラメータは、95℃(5分間)での最初の変性、続いて35周期の変性(95℃、15秒間)、アニーリング[55〜72℃、30秒間]、伸張(68℃、1分間)、および最終伸張(72℃、7分間)であった。増幅産物を3.5%TAEアガロースゲル上で分析した。最適なアニーリング温度(56.1℃)を特定した後、分取PCR反応を行って、各々一連のPCRプライマーを検証し、またSolexaアンプリコンを生成した。
【0201】
分取PCRのために、上述の条件を使用して各テンプレートについて8つの個々の小規模PCR反応を行い、産物を一緒にプールし、Qiagen MinEluteゲル精製キットを使用して3.5%アガロースゲル上でゲル精製した。ゲル精製したアンプリコンの濃度を、Nanodrop分光光度計を使用することによって決定し、およそ200ngの各アンプリコンを一緒にプールして、最終Solexa配列決定試料(800ng総試料)を調製した。また、アンプリコンをPCR−Blunt II−TOPO中にクローン化し、通常の配列決定に供してプライマーを検証した後、Solexa配列決定を行った。Solexa分析(Shendure et al.(2008)Nat.Biotechnology,26:1135−1145)を行い、配列を分析した。
【0202】
実施例6.2:Solexa配列決定および分析
Solexa配列決定を行って、数千の配列の産生がもたらされた。DAS NGS分析スクリプトを使用して配列を分析した。低質の配列(品質スコアカットオフ<5を有する配列)を取り除いた。次いで配列を参照配列(多重eZFN結合部位を含有するpDAB105900)と整列させ、開裂部位における挿入/欠失(インデル)についてスコア化した。各eZFNおよび無処置対照に対する編集活性(NHEJ%)を算出し(インデルを有する高質配列の数/高質配列の総数×100)、下記の図12に示す。2つの遺伝子導入タバコ事象(105900/第33および105900/第45)における8−eZFNの活性を実証した(図12)。8つのうちの3つのeZFNが、試験した2つの遺伝子導入タバコ事象において活性であった。活性をまた、eZFN処置試料中のeZFN開裂部位におけるインデルの存在によって、配列整列レベル(参照対solexa出力)において査定した。
【0203】
ZFN単量体(「右」および「左」)片側の全ての組み合わせが、酵母アッセイにおいて活性であった。トウモロコシおよびタバコ実験について記載されるデータは、組み合わせのうちの幾つかまたはほとんどが植物において活性であることを実証し、研究のために選択された4つの元のZFNからの2つのZFN単量体の、有意な数の順列を使用する可能性を支持している。
【0204】
実施例7:対立遺伝子内組み換え
対立遺伝子内組み換えは、導入遺伝子の2つの独立したブロックの発達および最適化を可能にし、次いでそれを組み換えによって1つの遺伝子座において一緒にスタッキングすることができる。2つのブロックの間の組み換えのレベルを強化するために、二本鎖開裂は、遺伝子変換または染色分体交換によってDNA交換を開始する。
【0205】
植物におけるこの概念を実証するために、図13に例証する遺伝子導入挿入物を、アラビドプシス・タリアナ中で作製する。構築物には、選択可能マーカー(ネオマイシンホスホトランスフェラーゼ(NPTII)またはハイグロマイシンホスホトランスフェラーゼ(HPT)、およびにスコア化可能なマーカー(β−グルクロニダーゼ(GUS)および黄色蛍光タンパク質(YFP))を含有する遺伝子ブロックが含まれる。これらの遺伝子ブロックは、同一のゲノム位置にあるが、互いからおよそ2kbが置換される。2つのブロックの間の組み換えは、子孫を、2つのブロックの間の中心の位置において開裂するZFNを発現する植物と交配させ、次いでそれらを再交配させることによって、2つのブロックの各々を担持する染色体を、単一の植物中に組み合わせることによって遂行する(図13MIS上部の黒色バー)。ZFNは、減数分裂特異的/選好的プロモーターを使用して発現させる。使用されるランディングパッド配列には、参照により本明細書に組み込まれる米国特許出願第61/297,641号に記載されるものが含まれる。
【0206】
同一のゲノム位置における独立したブロックを生成するために、隣接する配列において両方のブロックを含む構築物を作製した(図14)。独立したブロックのみを担持する植物を作り出すために、対応するZFN結合部位におけるそれぞれのブロックのいずれかの側上で、DNAを切断するように設計したZFNを使用して、別個の交配において各ブロックを切除する(赤色および青色バー)。図15は、適切な株(ZFNを発現するアラビドプシス)と交配させた後、ブロックが切断されて、単一のブロック挿入物を生成することを例証する。これらの株は、選択可能マーカーとしてPAT遺伝子を担持する。予想される表現型(HygR+、KanR−、PAT+、YFP+or Ka nR+、HygR−、PAT+、GUS+)を有する植物の回収を、表現型スクリーニング(HygR、KanR、およびPAT遺伝子に対する除草剤抵抗性、またはGUSおよびYFPのスコア化可能なマーカー遺伝子発現)を介して、またはPCRおよびサザン分析等の分子分析によって確認する。2つの異なるブロックの1つを担持する植物を交配させて、HygR+、KanR+、PAT−、GUS+、YFP+子孫を生成する。
【0207】
結果として得られた植物の分子特性評価後、確認された挿入物を有する植物を、減数分裂特異的プロモーターを使用して、結合部位が2つのブロックの間に位置するZFNを発現する株と交配させて、DNAの交換を達成する。これは、1つのDNA位置における2つのブロックのスタッキングをもたらす。最終のスタッキングされた遺伝子植物は、単一の分離遺伝子座として、HygR+、KanR+、GUS+、YFP+構成を担持する。代替的には、ブロックのうちの1つを含有する植物を、減数分裂プロモーター/ZFN構築物を含む2つの単量体のうちの1つと交配させ、2つの挿入物に対してホモ接合型である植物を得、次いで一緒に交配させる。
【0208】
実施例7.1:DNA構築
ZFN構築物の構築用に配備したクローニング戦略は、米国特許出願第2009/0111188A1号および同第2010/0199389号に本質的に記載される通りである。図9は、例示的なeZFN発現カセットを示す。ZmUbi1プロモーター(トウモロコシユビキチン1(Ubi−1)遺伝子に由来するプロモーター、5’非翻訳領域(UTR)、およびイントロン、Christensen et al.(1992)Plant Molec.Biol.18(4),675−89)を使用して、ZFNコード配列を発現させる。これらをその後、PAT遺伝子の発現を駆動するイネアクチン1プロモーターを含有する、バイナリーGATEWAY(商標)デスティネーションベクター中にクローン化した。それぞれブロック1 Excisor(eZFN1,8)またはブロック2 Excisor(eZFN3,6)構築物として指定する、結果として得られたプラスミドpDAB105951(ZFN1、CL:AR)、105954(ZFN8、RL:AR)、105952(ZFN3、AL:PR)、105953(ZFN6、CL:RR)を、アグロバクテリウム株DA2552recAに移した。
【0209】
アグロバクテリウムDA2552株を、DA2552株をグリセロールストックから、スペクチノマイシン(spec)(100μg/mL)およびエリスロマイシン(ery)(150μg/mL)を含有する10mLのYEP中に植菌することによって種培養を調製することによって、エレクトロポレーションのためにコンピテントにした。10mL培養物を、200rpmで、28℃で一晩インキュベートした。5ミリリットルの種培養を使用して、適切に標識化した1.5Lエルレンマイヤーフラスコ中、適切な抗生物質を有する500mLのYEPを植菌した。培養を、200rpmで、28℃で一晩インキュベートした。一晩のインキュベーション後、培養を、湿潤の氷水浴中に配置し、穏やかに旋回させることによって冷却した。細胞を、全ての更なるステップのために4℃で維持した。細胞を、事前冷却したローター内で標識化した滅菌遠心瓶中、4000xgで、4℃で10分間、遠心分離機にかけることによってペレット化した。上清を注ぎ出して破棄し、次いで5〜10mLの氷冷滅菌再蒸留水を添加し、塊が残らなくなるまで、細胞を上下に穏やかにピペット操作した。懸濁液容量を、氷冷滅菌再蒸留水でおよそ500mLまで調整した。細胞を、事前冷却したローター内で、4000xgで、4℃で10分間、遠心分離機にかけることによってペレット化した。上清を破棄し、5〜10mLの氷冷滅菌再蒸留水を添加し、次いで塊が残らなくなるまで、滅菌ワイドボアピペットを使用して細胞を上下に穏やかにピペット操作した。懸濁液容量を、氷冷滅菌再蒸留水でおよそ250mLまで調整し、細胞を、事前冷却したローター内で、4000xgで、4℃で10分間、遠心分離機にかけることによってペレット化した。上清を破棄し、5〜10mLの氷冷滅菌再蒸留水を添加し、ペレットを穏やかに再懸濁させ、最終容量を氷冷滅菌再蒸留水で50mLまで調整した。細胞を、事前冷却したローター内で、4000xgで、4℃で10分間、遠心分離機にかけることによってペレット化した。細胞を、5mLの10%(v/v)氷冷の滅菌グリセロールの最終容量中に再懸濁させた。細胞を滅菌0.5mL微量遠心管中の50μLアリコートに分与し、液体窒素中で凍結させた。
【0210】
20マイクロリットルのコンピテントDA2552細胞を、製造業者のプリセット設定およびアグロバクテリウムエレクトロポレーションのためのプロトコルに従って、GENE PULSER(登録商標)XCELL(登録商標)Electroporation System(BioRad Hercules,CA.)を使用して、50ngのプラスミドDNAで電気穿孔処理した。細胞をSOC中、28℃で2時間回収し、次いでYEP spec/ery寒天プレート上にプレートし、28℃で48時間増殖させた。
【0211】
実施例7.2:交換遺伝子座構築物
交換遺伝子座DNA構築物を、ベクター1:AtAct2プロモーター(AtAct2プロモーターv2(アラビドプシス・タリアナアクチン遺伝子(ACT2)からのプロモーター、5’非翻訳領域、およびイントロン、An et al.(1996)Plant J.10,107−121))/GUS(Jefferson,(1987)EMBO J.6,3901−3907)/AtuORF23 3’ UTR(アグロバクテリウム・ツメファシエンスpTi15955のオープンリーディングフレーム23(ORF23)の転写終結因子およびポリアデニル化部位を含む、3’非翻訳領域(UTR)、Barker et al.,(1983)Plant Molec.Biol.2(6):335−50)::AtAct2プロモーター/NPTII(Bevan et al.(1983)Nature 304,184−187)/ eZFN 1および8が側面に位置するAtuORF23 3’ UTR、ベクター2:配列の中心にeZFN 4および7を有する合成2kb領域、およびベクター3:CsVMVプロモーター/HPT(Kaster et al.(1983)Nucleic Acids Res.11(19),6895−6911(1983))/AtuORF23 3’ UTR::AtUbi10プロモーター(アラビドプシス・タリアナポリユビキチン10(UBQ10)遺伝子からのプロモーター、5’非翻訳領域、およびイントロン、Norris et al.(1993)Plant Molecular Biology 21(5):895−906)/PhiYFP(Shagin et al.,(2004)Molecular Biol.Evol.21:841−850)/ eZFN 3および6が側面に位置するAtuORF23 3’ UTR、を含むGATEWAY(商標)エントリーベクターから調製した。デスティネーションベクターを、2つの1kb無作為化合成DNA配列を、アグロバクテリウムバイナリーベクター主鎖に挿入することによって調製し、このうち制限部位をそれらの間に含めて、GATEWAY(商標)ccdB陰性選択可能マーカーカセットをクローン化した。エントリーベクターを、LR Clonase反応によってデスティネーションベクター中にクローン化した。結果として得られたベクター、pDAB100646(図16)を、上述のアグロバクテリウムに移した。
【0212】
実施例7.3:アラビドプシス形質転換
Clough&Bent(1998 Plant J.,16,735−743)によって記載される方法に従って、アラビドプシスへの全ての形質転換を行った。
【0213】
切除因子株
「切除因子」株構築物は、グルフォシネートに対する抵抗性を伝達するホスフィノトリシンアセチルトランスフェラーゼ(PAT)遺伝子をプロセスする。植付けの7、10、および13日後、T1植物に、Liberty除草剤(グルフォシネート1リットル当たり200グラムの活性成分(g ai/L)、Bayer Crop Sciences,Kansas City,MO)の284mg/L溶液を、10mL/トレー(703L/ha)の噴霧容量で、DeVilbiss圧縮空気ノズルチップを使用して噴霧して、1回の塗布当たり200g ai/haグルフォシネートの有効率を送達した。最終噴霧の4〜7日後、生存物(活発に成長している植物)を特定し、培養土(Metro Mix 360)で調製した3インチポットに個々に移植した。
【0214】
切除因子事象におけるeZFNの発現を逆転写酵素PCR(RT PCR)によって決定し、コピー数を、PAT遺伝子について本明細書に記載されるようにqPCRによって決定し、サザン分析によって確認する。ZFNを高レベルで発現する3つの低コピー事象を、交換遺伝子座(Exchange Locus)事象と交配させる。
【0215】
交換遺伝子座株
交換遺伝子座株を、ハイグロマイシンまたはカナマイシンを含有する培地についての選択を含む、Clough&Bent(1998 Plant J.,16,735−743)によって記載される方法に従って、アラビドプシスにおいて生成する。
【0216】
実施例7.4:アラビドプシス交配および子孫回収
交換遺伝子座事象の、2つの一連のブロック1およびブロック2切除因子株との交配を、標準的な方法を使用して行う。
【0217】
交配からの種子をハイグロマイシン(ブロック1欠失)またはカナマイシン(ブロック2欠失)上で増殖させ、抵抗性植物をGUS発現(ブロック1欠失)またはYFP発現(ブロック2欠失)について分析する。GUS活性を、組織化学的アッセイ(Jefferson et al.(1987)Plant Mol.Biol.Rep 5,387−405)を用いて、またYFPを、蛍光顕微鏡検査法を使用して、決定する。所望の表現型(ブロック1陽性:GUS+、NPT+、HPT−、YFP−、ブロック2陽性:GUS−、NPT−、HPT+、YFP+)を有する植物を、PCRおよびサザン法によって分析して、所望の遺伝子構成を確認する。選択された植物からの葉を、ビアラホス溶液で塗布して、どれがPAT+であるかを査定する。
【0218】
ブロック1およびブロック2遺伝子カセットを含有する植物を交配させ、子孫をハイグロマイシン/カナマイシンプレート上で選択する。HygR/KanR植物を、PCRおよび表現型スクリーニングによって、全ての遺伝子の存在について分析する。所望の表現型を有するF1植物を増殖させ、減数分裂プロモーター/ZFN植物と交配させて、ブロック1およびブロック2の間の組み換えを達成する。結果として得られた子孫を、ハイグロマイシン/カナマイシンプレート上で増殖させる。選択後に生存した植物を、GUSおよびYFPについてスクリーニングする。PCR、サザン、配列決定、および分離比分析を使用して、組み換えの確認および特性評価を行う。
【0219】
実施例8:eZFN部位における遺伝子スタッキング
図1、2、4、5、および6に示す戦略は、次の方法を使用して遂行することができる。
【0220】
構築物設計
ヘテロ二量体eZFN部位の様々な組み合わせは、植物形質転換に好適なプラスミドベクター中のコンカテマーとして組み立てることができる。図1、図2、および図4は、ベクターに組み入れて、植物の染色体に形質転換することができるヘテロ二量体eZFN部位の様々なバージョンを例証する。
【0221】
WHISKERS(商標)形質転換
トウモロコシの胚形成Hi−II細胞培養物を、米国特許第7,179,902号に記載されるように産生し、標的組み込みが例示される生存植物細胞の源として使用する。植物選択可能マーカーカセットに結合されたヘテロ二量体eZFN部位を含有するDNA断片を使用して、遺伝子導入事象を生成する。遺伝子導入事象を、単離し、特徴付ける。
【0222】
事前に凍結保存した細胞株からの12mLの血中血球容積(PCV)、加えて28mLの条件培地を、500mLエルレンマイヤーフラスコ中、80mLのGN6液体培地(N6培地(Chu et al.,(1975)Sci Sin.18:659−668)、2.0mg/L 2,4−D、30g/Lスクロース、pH5.8)中に継代培養し、シェーカー上に125rpmで、28℃で配置する。同じ細胞株を使用してこのステップを2回反復して、総計36mLPCVが3フラスコにわたって分配されるようにする。
【0223】
24時間後、GN6液体培地を除去し、72mL GN6 S/M浸透培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、45.5g/Lソルビトール、45.5g/Lマンニトール、100mg/Lミオイノシトール、pH6.0)と交換する。フラスコを暗所で30〜35分間、28℃で適度に撹拌しながらインキュベートする(125rpm)。インキュベーション時間中、炭化ケイ素ウィスカ(Advanced Composite Materials,LLC,Greer,SC)の50mg/mL(w/v)懸濁液を、8.1mLのGN6 S/M液体培地を405mgの滅菌炭化ケイ素ウィスカに添加することによって調製する。GN6 S/M浸透培地中でのインキュベーションに続いて、各フラスコの内容物を250mL遠心瓶中にプールする。フラスコ中の全ての細胞が底に沈殿した後、およそ14mLを超える内容積のGN6 S/M液体を引き出し、後の使用のために滅菌1−Lフラスコ中に収集する。事前に湿潤させたウィスカの懸濁液を、ボルテックス上で、最大速度で60秒間混合し、次いで遠心瓶に添加する。
【0224】
85μgの精製したDNA断片のアリコートを各瓶に添加する。一旦、DNAを添加すると、瓶を、改変したRed Devil 5400商業用塗料ミキサ(Red Devil Equipment Co.,Plymouth,MN)中に即座に配置し、10秒間撹拌する。撹拌に続いて、細胞、培地、ウィスカ、およびDNAのカクテルを、125mLの新鮮なGN6液体培地と共に1−Lフラスコの内容物に添加して、浸透圧溶質を減少させる。細胞を、125rpmに設定したシェーカー上に2時間回収させる。6mLの分散懸濁液を、ハウス真空管路(house vacuum line)に接続したガラス細胞収集ユニットを使用して、Whatman第4濾紙(5.5cm)上で濾過して、1瓶当たり60のフィルターを得るようにする。フィルターをGN6固体培地(2.5g/L Gelriteゲル化剤を用いることを除いてGN6液体培地と同じ)の60×20mmプレート上に配置し、暗所条件下で、28℃で1週間培養する。
【0225】
推定標的組み込み遺伝子導入事象の特定および単離
DNA送達の1週間後、濾紙を、選択剤を含有するGN6(1H)選択培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、100mg/Lミオイノシトール、2.5g/Lゲルライト、pH5.8)の60×20mmプレートに移す。これらの選択プレートを、暗所で、28℃で1週間インキュベートする。暗所での1週間の選択後、細胞の1/2を各プレートから、37〜38℃で保持される3.0mLのGN6アガロース培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、100mg/Lミオイノシトール、7g/L SeaPlaque(登録商標)アガロース、pH5.8、121度で10分間オートクレーブ処理)を含有する管にこすり落とすことによって、組織を新鮮な培地上に埋め込む。
【0226】
アガロース/組織混合物をへらで粉砕し、その後、3mLのアガロース/組織混合物を、GN6(1H)培地を含有する100×25mm皿の表面上に均等に注ぐ。このプロセスを、各プレートの両方の片について反復する。一旦、全ての組織を埋め込むと、プレートを暗所条件下で、28℃で最長10週間培養する。これらの選択条件下で増殖した推定的に形質転換した単離株を、埋め込まれたプレートから除去し、60×20mmプレート中の新鮮な選択培地に移す。およそ2週間後に持続的な増殖が明白である場合、事象を、適用された除草剤(選択剤)に対して抵抗性であるとみなし、細胞のアリコートをその後、遺伝子型分析のために採取する。安定な植物形質転換は、スタッキング実験のために使用される単一コピー組み込み体を産生する。
【0227】
事象の分子特性評価
ゲノムDNA(gDNA)を、記載される単離されたトウモロコシ細胞から抽出し、PCR遺伝子型同定実験のためのテンプレートとして利用する。gDNAを、DNEASY(登録商標)96 Plant Kit(QIAGEN Inc.,Valencia,CA)に詳述される製造業者のプロトコルに従って単離する、およそ100〜300μL血中血球容積(PCV)のHi−IIカルスから抽出する。ゲノムDNAを、100μLのキット提供緩衝剤中に溶出して20〜200ng/μLの最終濃度を得、その後、PCRに基づく遺伝子型同定法を介して分析する。
【0228】
コピー数の分子分析
INVADER(登録商標)または加水分解プローブアッセイを行って、除草剤抵抗性カルスの試料をスクリーニングして、T鎖DNAの単一コピー組み込みを含有するものを特定する。遺伝子発現カセットに特異的なプライマーおよびプローブを使用して、詳細な分析を行う。単一コピー事象を追加的な分析のために特定する。
【0229】
カスタムINVADER(登録商標)アッセイを、Third Wave Technologies(Madison,WI)によるHi−IIカルスにおける選択可能マーカー遺伝子分析のために開発する。ゲノム試料を、INVADER(登録商標)アッセイキットを使用して増幅し、読み取りを収集する。これらの読み取りから、各チャネルについてのフォールドオーバーゼロ(fold−over zero)(すなわち、バックグラウンド)を、試料の原シグナルを、テンプレートを含有しない原シグナルで割ることによって、各試料について決定する。このデータから標準的な曲線を構築し、最良適合を線形回帰分析によって決定する。この適合から特定したパラメータを使用して、次いで見かけの選択可能マーカーコピー数を各試料について推定する。
【0230】
標的DNAを有する遺伝子導入事象の選択
低コピー(1〜2コピーの導入遺伝子)事象を、上述のように、選択可能マーカー遺伝子カセットおよび無傷のeZFN部位を含有する無傷の植物転写単位(PTU)について、PCRによってスクリーニングする。コピー数を、サザン分析によって、選択可能マーカー遺伝子による標準的な方法を使用して更に確認する。単一コピーの無傷の挿入を有する選択された遺伝子導入事象からのカルスを維持する。
【0231】
eZFNを含有する植物細胞中へのバイオリスティック媒介型DNA送達
上述のように、トウモロコシの胚形成Hi−II細胞培養物を産生し、内部への標的組み込みが実証される生存植物細胞の源として使用する。トウモロコシの胚形成懸濁液を、GN6液体培地中におよそ24時間継代培養し、その後、上述される実験を行う。およそ0.4mL PCVの細胞を、2.5g/Lゲルライトで凝固させたGN6 S/M培地を含有する100×15mmペトリ皿の中心上で、直径2.5cmの円形に薄く広げる。
【0232】
細胞を暗所条件下で4時間培養する。バイオリスティック粒子を、ドナーDNA(図1のブロック1、図2のブロック2、または図4の遺伝子1)を含有するDNAでコーティングするために、3mgの1.0ミクロン直径の金粒子を100%エタノールで1回、滅菌蒸留水で2回洗浄し、シリコン処理したエッペンドルフ管中の50μL水中に再懸濁させる。総計5μgのプラスミドDNA(単一のベクター中または別個のベクター中に、eZFNをコードする核酸分子およびドナーDNA断片を含有する)、20μLスペルミジン(0.1M)、および50μL塩化カルシウム(2.5M)を金懸濁液に別個に添加し、ボルテックス上で混合する。混合物を室温で10分間インキュベートし、ベンチトップ微量遠心機において10,000rpmで10秒間ペレット化し、60μL冷100%エタノール中に再懸濁させ、8〜9μLを各マクロ担体上に分配する。
【0233】
Biolistic PDS−1000/HE(商標)系(Bio−Rad Laboratories,Hercules,CA)を使用して衝撃を行う。細胞を含有するプレートを、1,100psiおよび27インチのHg真空の条件下で棚中段に配置し、作動マニュアルに従って衝撃する。衝撃の16時間後、組織を小さい塊でGN6(1H)培地に移し、暗所条件下で、28℃で2〜3週間培養する。ドナーDNAの組み込みからもたらされる推定遺伝子導入単離株が現れ始めるまで、移入を2〜4週間毎に継続する。ビアラホス抵抗性コロニーを、一般に、PCRおよびサザンブロッティングによって、標的配列を含有する単離株を生成するために上に詳述される方法を使用して分析する。
【0234】
PCR遺伝子型同定を介する標的組み込み事象についてのスクリーニング
PCR反応を行って、ドナーDNAの無傷のコピーの存在を調査する。追加的な反応は、標的とドナーとの間の5’境界およびドナーと標的との間の3’境界に焦点を当てる。増幅した断片をゲル切除し、標準的なプロトコルに従って精製する。精製した断片をその後、製造業者のプロトコルに従って、TOPO TA CLONING(登録商標)Kit(pCR2.1ベクターを有する)およびONE SHOT(登録商標)TOP10 Chemicallyコンピテント大腸菌細胞(Invitrogen Life Technologies,Carlsbad,CA)を使用してpCR2.1プラスミド中にクローン化する。
【0235】
個々のコロニーを選択し、増幅したPCR断片を含有することを確認する。プラスミドクローンの二本鎖配列決定反応を行って、PCR増幅したゲノム配列が組み込まれたドナーを含有することを確認する。ドナー断片を含有することが特定された事象は、ZFN媒介型二本鎖切断の相同性駆動型修復、およびドナーDNAの特異的遺伝子標的における標的組み込みが存在する標的を表す。
【0236】
eZFN部位を使用する遺伝子スタッキングの特異的用途
図1は、植物の染色体に安定に形質転換される7つの(7)eZFN標的部位から成り立つ多重挿入部位の変形を示す。標的部位に結合するeZFN対を幾何学的図として示す。「ブロック1」は、特異的eZFN部位を開裂するように設計されたeZFNにより形質転換されるとき、適切なeZFN対の多重挿入部位に組み込まれ得る外来性ポリヌクレオチド配列である。eZFNおよび「ブロック1」ドナーDNA配列の共形質転換は、前述のバイオリスティック形質転換方法を使用して達成することができる。様々な他のeZFN部位の忠実度は、植物細胞中に形質転換されるeZFNがこれらの他の部位を開裂しないため、維持される。「ブロック1」は、相同組み換えを介して植物染色体に組み込み、「ブロック1」の配列を含有する植物細胞をもたらす。結果として得られた植物細胞は、成熟植物に成長させられ、サザンブロッティング、Taqmanアッセイ、またはInvaderアッセイ等の当該技術分野において既知である分析分子生物学的方法を使用して「ブロック1」の存在についてスクリーニングされ得る。
【0237】
図2は、図1の別の変形を例証し、ここで異なるeZFN結合部位は、ポリヌクレオチドドナー配列「ブロック2」により標的化される。DNA断片の結果として得られた組み込みは、染色体内に「ブロック2」を含有する安定な植物を産生する。
【0238】
図4は、eZFN「左」および「右」ドメインの使用を例証する。上部の線は、左および右eZFNドメインにより形質転換された植物のゲノムを示す(影付き三角および碁盤格子三角)。新たなおよび異なるヘテロ二量体eZFN部位が側面に位置する「遺伝子1」を含む外来分子の存在下で、適切なeZFNが付加されるとき、「遺伝子1」および隣接するeZFN部位がゲノムに挿入される。「遺伝子1」および隣接するeZFN部位を含有する、結果として得られた子孫が特定され、これらの植物を、その後、親植物内には存在しなかった新たなヘテロ二量体eZFN部位を使用して、再標的化することができる(すなわち、影付き三角および碁盤格子三角を含有するeZFN部位)。
【0239】
図5および図6は、eZFN部位を使用して、新たな導入遺伝子を染色体位置中にスタッキングすることができる様態を例証する。更に、この戦略は、他の遺伝子発現カセットの切除を可能にする。場合によっては、遺伝子発現カセットを完全に除去することができ(図5)、他のシナリオでは、遺伝子発現カセットを植物の特異的生成において除去し、最終的にそれらの植物の子孫に再導入することができ、それによって遺伝子発現カセットの再利用が可能となる。欠失したマーカー(図6)配列は、上述のプロトコルを使用して相同組み換え媒介型遺伝子標的法を介して再導入することができる。ヘテロ二量体eZFN部位への遺伝子標的法は、上述のプロトコルを使用して完了する。この実施例において、eZFN結合部位は、形質転換植物からの、選択可能マーカー遺伝子を含む、植物体での任意の導入遺伝子の欠失を可能にするために使用される。2010年1月22日に出願された米国仮特許出願第61/297628号を参照されたく、それは参照により本明細書に組み込まれる。
【0240】
本明細書で言及した全ての特許、特許出願、および刊行物は、参照によりそれらの全体が全目的で本明細書に組み込まれる。
【0241】
理解の明瞭化を目的とした図示および例を用いて、本開示を多少詳しく提供したが、本開示の趣旨または範囲から逸脱することなく、様々な変更および修正を実施することができることは当業者には明らかであろう。したがって、前述の説明および例を限定的であると解釈すべきではない。
【技術分野】
【0001】
技術分野
本開示は、ゲノム工学の分野、特に、細胞のゲノムへの、1つ以上の外来性配列の標的化組み込みおよび/または標的化切除に関する。
【背景技術】
【0002】
関連出願の相互参照
本出願は、2010年1月22日に出願された米国仮出願第61/336,457号の利益を主張し、その開示は、参照によりその全体が本明細書に組み込まれる。
【0003】
連邦支援による研究下でなされた発明に対する権利の記載
該当なし。
【0004】
背景
生物工学は、食物生産に対する増加し続ける世界的需要の課題に対応する努力における必須のツールとして出現した。農業生産性の改善、例えば、収率の向上または病虫害抵抗性の改変のための従来のアプローチは、突然変異育種、または形質転換による作物種のゲノムへの新規遺伝子の導入のいずれかに依存する。両プロセスは、本質的に非特異的であり相対的に非効率的である。例えば、従来の植物の形質転換方法は、無作為位置におけるゲノムに組み込む、外来性DNAを送達する。故に、望ましい属性を有する遺伝子導入系を特定および単離するために、数千の固有の無作為組み込み事象を生成し、その後、所望の個体のためにスクリーニングすることが必要である。結果として、従来の植物形質改変は手間がかかり、時間を浪費し、かつ予測不可能な事業である。更に、これらの組み込みの無作為的性質は、意図されないゲノム破壊に起因する多面効果が生じているかどうかを予測することを困難にする。結果として、改変された遺伝子または形質を有する植物株の生成、単離、および特性評価は、成功確立の低い、非常に労働集約的およびコスト集約的プロセスとなっている。
【0005】
標的化遺伝子修飾は、植物系における従来の実務の事業計画上の課題を克服するものであり、したがって、基本的な植物生物学研究および農業生物工学の両方における長年の目標でありながら、定義し難い目標であった。しかしながら、イネにおける陽性−陰性薬物選択を介した「遺伝子標的法」、または事前に改変された制限部位の使用を除いて、全ての植物種のモデルおよび作物の両方における標的化ゲノム修飾は、最近まで非常に困難であることが確認されていた。Terada et al.(2002)Nat Biotechnol 20(10):1030、Terada et al.(2007)Plant Physiol 144(2):846、D’Halluin et al.(2008)Plant Biotechnology J.6(1):93。
【0006】
哺乳類細胞において、安定な遺伝子組み換えおよび標的化遺伝子挿入は、遺伝子療法および細胞改変の両方において、多くの潜在的な用途を有する。しかしながら、現在の戦略は、しばしば非効率的であり、導入遺伝子をゲノムDNAに非特異的に挿入する。ゲノム挿入の位置を制御する能力の欠如は、ゲノム内の位置効果に起因して、集団全体にわたる導入遺伝子発現レベルの高い変動性をもたらす可能性がある。更に、安定な遺伝子組み換えおよび導入遺伝子の増幅の現在の方法は、しばしば導入遺伝子の物理的損失、経時的な導入遺伝子サイレンシング、内在性遺伝子の内側もしくはそれに隣接した遺伝子および自律的プロモーターの組み込みによる挿入変異、染色体異常の作成および再配列遺伝子産物の発現(内在性遺伝子、挿入された導入遺伝子、または両方からなる)、ならびに/または安定な導入遺伝子発現を提供するためのベクターの長期持続性の必要性に起因して永続的に発現される、ベクター由来遺伝子からのインビボのベクター関連毒性もしくは免疫原性の作成をもたらす。
【0007】
最近、ゲノムDNAの標的化開裂のための方法および組成物が記載されている。かかる標的化開裂事象を使用して、例えば、標的突然変異誘発を誘導し、細胞DNA配列の標的欠失を誘導し、かつ所定の染色体座における標的組み換えを促進することができる。例えば、米国特許公開第20030232410号、同第20050208489号、同第20050026157号、同第20050064474号、および同第20060188987号、ならびに国際公開第WO2007/014275号を参照されたく、それらの開示は、参照によりそれらの全体が全目的で組み込まれる。米国特許公開第20080182332号は、植物ゲノムの標的化修飾のための非標準的な亜鉛フィンガーヌクレアーゼ(ZFN)の使用を記載し、米国特許公開第20090205083号は、植物EPSPS遺伝子座のZFN媒介型標的修飾を記載する。加えて、Moehle et al.(2007)Proc.Natl.Acad,Sci.USA 104(9):3055−3060)は、特異的遺伝子座における標的化遺伝子付加のために設計されたZFNを使用することを記載する。
【0008】
しかしながら、植物およびその子孫における安定な遺伝性の遺伝子修飾を確立するための植物への標的化組み込みのため、ならびに遺伝子療法および細胞株育成目的の哺乳類細胞への標的組み込みのためを含む、標的化組み込みのための組成物および方法に対する必要性が依然として存在する。
【発明の概要】
【0009】
概要
本開示は、細胞ゲノムに組み込まれる、多重挿入部位に組み込まれた外来性核酸配列(すなわち、タンパク質またはRNA分子)の1つ以上の産物を発現させるための方法および組成物を提供する。細胞は、真核細胞、例えば、植物、酵母、または哺乳類細胞であり得る。
【0010】
外来性核酸配列の組み込みは、1つ以上のヌクレアーゼ、例えば、亜鉛フィンガーヌクレアーゼ(ZFN)に対する多重標的部位を含むポリヌクレオチド配列の、細胞のゲノムへのゲノム組み込みによって促進される。ポリヌクレオチド(本明細書で多重挿入部位とも称される)は、細胞のゲノム内での特異的な標的化二本鎖開裂を可能にし、その二本鎖開裂は次に、相同性依存性および相同性非依存性機構の両方を通じた外来性配列の組み込みをもたらす。
【0011】
故に、一態様では、亜鉛フィンガーヌクレアーゼ(ZFN)等のヌクレアーゼに対する1つ以上の標的部位を含む、多重挿入部位としても知られる核酸分子が本明細書に開示される。ある実施形態では、標的部位は、多重挿入部位が組み込まれる内在性ゲノム中に存在しない。多重挿入部位は、ヌクレアーゼに対する1つ、2つ、3つ、4つ、5つ、6つ、7つ、またはそれを超える標的部位を含んでもよい。ある実施形態では、隣接した標的部位(対形成した標的部位)に結合する2つの結合DNA結合タンパク質の開裂片側ドメインの二量体化が、開裂に必要とされる(例えば、各部位に対のそれぞれが結合する、一対のヌクレアーゼが開裂に必要とされる)。本明細書に記載される多重挿入部位のいずれかにおいて、各対の標的部位のうち一方の標的部位は、同じ配列を含んでもよい。例えば、図1を参照されたい。ある実施形態では、少なくとも一つの対は標的部位が同じである。他の実施形態では、少なくとも1対の標的部位は、異なる標的からの個々の標的配列(例えば、異なる遺伝子および/または異なる生物からの遺伝子)を含む。ある実施形態では、対形成した標的部位のうちの少なくとも1つは、配列番号1〜20からなる群から選択される配列を含む。ある実施形態では、多重挿入部位は、更に1つのコード配列、例えば、ホスフィノスリシンアセチルトランスフェラーゼ(PAT)コード配列を含む植物転写単位(PTU)、または哺乳類細胞と共に使用するためのスクリーニングマーカーを含んでもよい。
【0012】
多重挿入部位は、ヌクレアーゼ(例えば、ZFN)に対するゲノム標的を提供するために細胞(例えば、植物または哺乳類細胞)のゲノムに組み込まれる。ある実施形態では、標的部位は、亜鉛フィンガーヌクレアーゼの1つ以上の対がホモ二量体として結合し、開裂するように位置する。他の実施形態では、標的部位は、亜鉛フィンガーヌクレアーゼの1つ以上の対がヘテロ二量体として結合し、開裂するように位置する。
【0013】
別の態様では、本明細書に記載される1つ以上の多重挿入部位および/または多重挿入部位に組み込まれる1つ以上の外来性配列を含む、植物または種子が本明細書に開示される。ある実施形態では、多重挿入部位および/または外来性配列は、トウモロコシ植物の配偶体に組み込まれる。
【0014】
ある態様では、本明細書に記載される1つ以上の多重挿入部位および/または多重挿入部位に組み込まれる1つ以上の外来性配列を含む、修飾された哺乳類細胞株、修飾された初代細胞、修飾された幹細胞、および/または遺伝子導入動物が本明細書に提供される。
【0015】
別の態様では、外来性配列を、細胞(例えば、植物または哺乳類細胞)のゲノムに組み込まれた多重挿入部位に組み込むための方法が本明細書に提供され、本方法は、(a)ヌクレアーゼに対する1つ以上の標的部位を含む多重挿入部位ポリヌクレオチドを、細胞のゲノムに組み込むことと、(b)ヌクレアーゼのそれらの標的部位への結合が細胞のゲノムを開裂させるように、多重挿入部位ポリヌクレオチドにおける第1の標的部位に結合する1つ以上のヌクレアーゼを提供および/または発現させることと、(c)細胞を、外来性核酸配列を含むポリヌクレオチドと接触させ、それによって外来性配列の、多重挿入部位ポリヌクレオチド内の細胞のゲノムへの相同性依存性組み込みをもたらすことと、を含む。
【0016】
別の態様では、多重外来性配列を細胞(例えば、植物または哺乳類細胞)のゲノムに組み込むための方法が本明細書に提供され、本方法は、(a)第1の多重挿入部位ポリヌクレオチドが、第1および第2のヌクレアーゼに対する標的部位が側面に位置する少なくとも1つの第1の遺伝子を含む、ヌクレアーゼに対する1つ以上の標的部位を含む第1の多重挿入部位ポリヌクレオチドを、細胞のゲノムに組み込むことと、(b)第3および第4のヌクレアーゼに対する標的部位が側面に位置する少なくとも1つの第2の遺伝子を含む第2の多重挿入部位ポリヌクレオチドの存在下で、細胞中の第1または第2のヌクレアーゼを発現させ、それによって第1および第2の遺伝子の細胞のゲノムへの組み込みをもたらすことと、を含む。ある実施形態では、本方法は更に、コード配列および/またはヌクレアーゼ部位を含む、追加的な外来性配列を組み込むために、挿入された多重挿入部位上に存在する適切なヌクレアーゼを発現させるステップを一回以上反復することを含む。ヌクレアーゼは、ヘテロ二量体ZFNであってもよく、ヌクレアーゼのうちの1つ以上の間にあるものと共通の1つの単量体が存在してもよい。幾つかの実施形態では、挿入のための外来性DNA配列は、外来性配列の組み込み時に、ドナーDNAに関連する片側標的部位、およびゲノムDNAに関連する片側標的部位を含む新規ZFN標的部位が作り出されるように、ZFN片側標的部位を含んでもよい。この新規ZFN標的部位は、類似の新規ヘテロ二量体ZFNに対する標的部位としての役割を果たすことができる。
【0017】
別の態様では、細胞(例えば、植物または哺乳類細胞)中で1つ以上の外来性核酸配列の産物を発現させるための方法が本明細書に開示され、本方法は、外来性配列が、組み込まれた核酸分子中の細胞のゲノムに組み込まれ、外来性配列の産物が発現されるように、本明細書に記載される方法のうちのいずれかに従って1つ以上の外来性核酸配列を組み込むことを含む。
【0018】
細胞のゲノムに挿入された1つ以上の遺伝子を欠失させる方法もまた提供され、本方法は、複数の外来性配列を、本明細書に記載される方法のうちのいずれかによって組み込むことと、外来性配列のうちの1つ以上がゲノムから欠失させられるように、細胞中で適切なヌクレアーゼを発現させることと、を含む。ある実施形態では、欠失させられる外来性配列は、マーカー遺伝子である。ある実施形態では、外来性配列の欠失およびその後のゲノム内の末端部の再結合は、ゲノム位置における機能性遺伝子または配列を作り出し、例えば、発現可能なスクリーニングマーカーを作り出す。
【0019】
なおも別の態様では、ゲノムが変更された細胞を提供する方法が提供され、本方法は、本明細書に記載される方法のうちのいずれかに従って、第1の細胞中の1つ以上の外来性核酸配列を組み込むおよび/または切除することと、第1の細胞を第1の性的に成熟した生物に発達させることと、生物を、対立位置におけるゲノム変更を含む第2の生物と交配させて、第1および第2の生物のゲノム変更を有する第2の細胞を生成することと、を含む。ある実施形態では、生物は、植物である。他の実施形態では、生物は、遺伝子導入動物である。
【0020】
本明細書に記載される方法のうちのいずれかにおいて、本方法は、1つ以上の内在性遺伝子座における標的化組み込みおよび/または標的化不活性化を含む、ゲノム変更の他の方法と組み合わせて使用されてもよい。更に、本明細書に記載される方法のうちのいずれかにおいて、ヌクレアーゼは、亜鉛フィンガー結合ドメインおよび開裂片側ドメインを含む1つ以上の融合タンパク質を含んでもよく、ここで亜鉛フィンガー結合ドメインは、多重挿入部位において標的部位に結合するように改変されている。更に、これらの方法のうちのいずれにおいても、外来性核酸配列は、多重挿入部位における配列、および/または多重挿入部位が組み込まれる領域における、内在性配列と相同である1つ以上の配列を含む。
【0021】
本明細書に記載される方法のうちのいずれかにおいて、1つ以上の多重挿入部位は、任意の好適な方法によって、例えば、挿入が所望される内在性遺伝子を標的とするZFNを使用する、ヌクレアーゼ(例えば、ZFN)を介した標的化組み込みによって、ゲノムに組み込まれてもよい。代替的に、1つ以上の多重挿入部位は、標準技術を使用して細胞のゲノムに無作為に組み込まれてもよい。
【0022】
外来性核酸配列は、1つ以上のプロモーターを含むもしくは含まない、1つ以上の機能性ポリペプチド(例えば、cDNA)をコードする配列を含んでもよく、かつ/または望ましい形質を生物に付与する、1つ以上のRNA配列(例えば、1つ以上のshRNA発現カセットを介して)を産生してもよい。植物におけるかかる形質には、除草剤抵抗性または耐性;虫害抵抗性または耐性;病害抵抗性または耐性(ウイルス、細菌、真菌、線虫);干ばつ、暑気、冷気、凍結、過度の湿気、塩ストレスに対する抵抗性または耐性によって例示されるようなストレス耐性および/または抵抗性;酸化ストレス;増加した収率;食物含量および組成(food content and makeup);外見;雄性不稔性;ドライダウン;標準性(standability);多産性;デンプンの量および質;油の量および質;タンパク質の量および質;アミノ酸組成等が含まれるが、これらに限定されない。当然のことながら、除草剤、虫害、病害(ウイルス、細菌、真菌、線虫)、もしくは干ばつ抵抗性、雄性不稔性、ドライダウン、標準性、多産性、デンプン特性、油の量および質を付与するもの、または収率もしくは栄養の質を増加させるもの等の、任意の説明の任意の2つ以上の外来性核酸が、所望に応じて用いられてもよい。ある実施形態では、外来性核酸配列は、除草剤抵抗性タンパク質(例えば、AAD(アリールオキシアルカノエートジオキシゲナーゼ)遺伝子)および/またはその機能的断片をコードする配列を含む。組み込まれる配列の発現は、組み込まれる配列に操作可能に連結されたプロモーターによって駆動することができる。代替的には、組み込まれる配列は、プロモーターを含まず、転写は、多重挿入部位ポリヌクレオチドの挿入の領域における内在性プロモーターによって駆動される。他の実施形態では、結合部位の開裂および不正確な修復は、目的の遺伝子を不活性化または活性化する可能性がある。ある実施形態では、ポリヌクレオチドは、プラスミドである。他の実施形態では、ポリヌクレオチドは、線状DNA分子である。
【0023】
哺乳類細胞中で、本発明の方法および組成物は、細胞株構築のため、例えば、抗体等の多量体ポリペプチドを発現する細胞株の構築のために使用されてもよい。幾つかの実施形態では、細胞株は、研究目的のため、例えば、目的の経路のメンバーを発現する細胞株の構築のために使用されてもよい。幾つかの実施形態では、初代細胞または幹細胞を使用して、細胞治療目的のために目的の多量体タンパク質を発現させてもよい。
【0024】
別の態様では、亜鉛フィンガーヌクレアーゼ活性を測定する方法が本明細書に提供される。ある実施形態では、本方法は、(a)対形成した標的部位の各々が、亜鉛フィンガーヌクレアーゼが結合する2つの亜鉛フィンガーヌクレアーゼ片側標的部位を含む、本明細書に記載される少なくとも1つの亜鉛フィンガーヌクレアーゼおよび核酸分子、ならびに片側標的部位の間に挿まれる切断部位である、結合した亜鉛フィンガーヌクレアーゼによって切断される切断部位を提供することと、(b)亜鉛フィンガーヌクレアーゼが、少なくとも切断部位内で対形成した標的部位を開裂させるように、亜鉛フィンガーヌクレアーゼを核酸と組み合わせることと、(c)少なくとも切断部位の配列を決定して、配列データを生成することと、(d)配列データにおいて、切断部位内の塩基対欠失の数および長さを、亜鉛フィンガーヌクレアーゼの不在下での切断部位内の塩基対欠失の数および長さと比較して、それによって対形成した標的部位における亜鉛フィンガーヌクレアーゼ活性を測定することと、を含む。ある実施形態では、1つを超える塩基対の欠失は、亜鉛フィンガーヌクレアーゼの増加した活性を示す。
【0025】
なおも他の実施形態では、対形成した標的部位における亜鉛フィンガーヌクレアーゼ活性を最適化するための方法が本明細書に提供される。ある実施形態では、本方法は、(a)対形成した標的部位の各々が、亜鉛フィンガーヌクレアーゼが結合する2つの亜鉛フィンガーヌクレアーゼ片側標的部位を含む、本明細書に記載される少なくとも1つの亜鉛フィンガーヌクレアーゼおよび核酸分子、ならびに片側標的部位の間に挿まれる切断部位である、結合した亜鉛フィンガーヌクレアーゼによって切断される切断部位を提供することと、(b)亜鉛フィンガーヌクレアーゼが、少なくとも切断部位内で対形成した標的部位を開裂させるように、亜鉛フィンガーヌクレアーゼを核酸と組み合わせることと、(c)切断部位における亜鉛フィンガーヌクレアーゼ活性レベルを決定することと、(d)切断部位における塩基対の数に変化を与えることと、(e)ステップ(b)〜(d)を数回反復することと、(f)最高レベルの亜鉛フィンガーヌクレアーゼ活性を提供する塩基対の数を含む、核酸に組み込むための切断部位を選択し、それによって対形成した標的部位における亜鉛フィンガーヌクレアーゼ活性を最適化することと、を含む。
【0026】
亜鉛フィンガーヌクレアーゼを伴う本明細書に記載される方法のうちのいずれかにおいて、第1および第2の開裂片側ドメインは、Type IIS制限エンドヌクレアーゼ、例えば、FokIまたはStsIからのものである。更に、本明細書に記載される方法のうちのいずれかにおいて、融合タンパク質のうちの少なくとも1つは、例えば、開裂片側ドメインの偏性ヘテロ二量体が形成されるように、開裂片側ドメインの二量体化成面のアミノ酸配列における変更を含んでもよい。
【0027】
本明細書に記載される方法のうちのいずれかにおいて、植物細胞は、単子葉または双子葉植物細胞を含む可能性がある。ある実施形態では、植物細胞は、作物植物、例えば、トウモロコシである。ある実施形態では、細胞は、初代細胞、細胞株、または幹細胞等の哺乳類細胞を含む可能性がある。幾つかの実施形態では、哺乳類細胞株は、目的のポリペプチドの産生のために使用することができる。
【図面の簡単な説明】
【0028】
【図1】本明細書に記載される例示的な多重挿入部位を示す概略図。図1は、7つのZFN標的部位から成り立つ多重挿入部位を示す。標的部位に結合するZFN対は、幾何学的図として示される。「ブロック1」は、ZFN標的部位(影付きおよび格子模様の三角)を維持しながら、適切なZFN対の存在下で多重挿入部位に組み込まれる外来性配列である。図1は、ZFN標的部位の代わりの、適切なZFN対の存在下での「ブロック1」の多重挿入部位への組み込みを示す。
【図2】「ブロック2」が、適切なZFN対の存在下で多重挿入部位に組み込まれる外来性配列である、図1に示される例示的な多重挿入部位を示す概略図。
【図3】ZFNによって強化された対立遺伝子間組み換えの概略図。同一のゲノム位置にあるが、互いから移動させられる2つの挿入物は、ZFNによる二本鎖開裂後に、相同組み換えまたは鎖交換を経る可能性がある。ZFN対(このうち両方のZFN単量体が一緒に発現される)は、ZFN対を発現する植物を、両方の対立遺伝子を含む植物と一緒に交配させることによって、または単一の対立遺伝子を含有する植物との交配の両側からの2つのZFN単量体を導入することによって、提供することができる。
【図4】ヘテロ二量体ZFN「左」および「右」標的ドメインの使用を示す概略図。上部の線は、左および右標的ZFNドメインを有するゲノムを示す(影付き三角および碁盤格子三角)。異なるヘテロ二量体対が側面に位置する遺伝子を含む外来性分子の存在下で、適切なZFN対が付加されるとき、遺伝子および隣接するヌクレアーゼ部位は、示されるようにゲノムに挿入される。
【図5】ゲノムが組み込まれた配列のいずれかの側上での、外来性配列(「遺伝子」として示される)の組み込みおよび切除を示す概略図。付加された遺伝子の側面には、遺伝子カセットを適切な部位内に導くための相同性領域が位置する。挿入のために使用されるZFN標的部位の2つの片側は、挿入されるDNAにおいて2つの新たな組み合わせを作り出すことによって組み換えられる。遺伝子カセットの切除は、隣接するZFN標的部位において開裂するための適切なZFN対を結合させることによって遂行される。切除は、所望のDNA配列の欠失を防止するために、相同性アームを含有するテンプレートを必要とする可能性がある。各「遺伝子」は、1つ以上の配列、例えば、1つ以上のコード配列を含む可能性がある。
【図6】ZFNヘテロ二量体(異なる陰影を有する三角として示される)を使用する、挿入されたマーカー遺伝子の切除および「再利用」を示す概略図。
【図7】pDAB105900のプラスミドマップ。
【図8】pDAB105908のプラスミドマップ。
【図9】亜鉛フィンガーヌクレアーゼホモ二量体発現カセットの線図。
【図10】亜鉛フィンガーヌクレアーゼヘテロ二量体発現カセットの線図。
【図11】開裂後の非相同末端結合からもたらされる欠失の頻度によって決定される、トウモロコシにおけるeZFN開裂活性を示す。
【図12】開裂後の非相同末端結合からもたらされる欠失の頻度によって決定される、タバコにおけるeZFN開裂活性を示す。
【図13】同じ遺伝子座への2つの遺伝子導入挿入物の概略図。上部の線は、遺伝子座における相同組み換えに必要とされるeZFN結合部位を含有する、多重挿入部位(本明細書でランディングパッドとも称される)を表すMISと標識された無作為配列、ならびにカナマイシン選択可能マーカー遺伝子およびGUSスクリーニング可能マーカー遺伝子を含むブロック1を示す。中間の線は、上部のDNAにあるものと同じ多重挿入部位(MIS)と共に、ハイグロマイシン抵抗性選択可能マーカー遺伝子および黄色蛍光タンパク質スクリーン可能マーカー遺伝子(HPT/YFP)を含むブロック2を示す。下部の線は、組み換えに続く遺伝子座を示す。
【図14】ZFNによる対立位置における相同組み換え、および図13に説明されるものと同じ遺伝子座における2つの異なるDNA挿入物の生成を示す。ブロック1(カナマイシンおよびGUCマーカーを含む、GUS/NPT)、多重挿入部位(MISまたはランディングパッド)、およびブロック2(ハイグロマイシン(hygomycin)および黄色蛍光マーカーを含む、HPT/YFP)を含む構築物が、アラビドプシスに形質転換される。各ブロックのみを別個の植物における多重挿入部位と一緒に生成するために、ブロック2は、ブロック1のみの構成を生成するために組み込まれる部位から切除されるか、またはブロック1は、ブロック2のみの構成を生成するために組み込まれる部位から切除される。遺伝子ブロックの除去は、元の遺伝子導入事象を含有する植物を、遺伝子ブロックの各々に隣接するeZFN結合部位において開裂するZFNを発現する植物と交配させることによって遂行される。回収された単一のブロック植物は交配されて、2つの構成物を単一の植物中に一緒にまとめ、その植物が減数分裂特異的プロモーターを発現する植物と交配されて、2つのブロック1とブロック2対立遺伝子との間のDNAの交換に影響を及ぼす。
【図15】2つの配列の間の中間部位におけるZFN開裂によって、同じ遺伝子座に位置する2つのDNA配列の間の組み換えを得るためのステップを示す概略フローチャート。図16に説明される構築物は、アラビドプシスに形質転換される。2つの遺伝子ブロック(図14に説明される)の1つは、結合部位がブロックに隣接するeZFNを発現する植物と交配させることによって除去され、ブロック1またはブロック2のいずれかを含有する植物をもたらす。
【図16】交換遺伝子座をアラビドプシスに導入するために使用されるプラスミドの概略図。それは、図14に説明されるブロック1および2、ならびに多重挿入部位配列を含有する。eZFN結合部位が示され、ブロック1および2(ブロック1:eZFN1および8、ブロック2:eZFN3および6)に隣接するか、または相同組み換えを促進するために多重挿入部位(eZFN4および7)における中心に位置する。
【発明を実施するための形態】
【0029】
詳細な説明
本開示は、ゲノム、例えば、トウモロコシ等の作物植物または哺乳類細胞への標的化組み込み(TI)のための方法および組成物に関する。1つ以上のヌクレアーゼ(例えば、ZFN)に対する多重標的部位を含有する多重挿入部位が、ゲノムに組み込まれる。多重挿入部位のゲノムへの組み込みに続いて、適切なヌクレアーゼが、挿入対象の外来性配列と共に細胞に導入される。
【0030】
ある実施形態では、ヌクレアーゼは、1つ以上のZFNを含む。ZFNは典型的には、開裂ドメイン(または開裂片側ドメイン)および亜鉛フィンガー結合ドメインを含み、タンパク質として、これらのタンパク質をコードするポリヌクレオチドとして、またはポリペプチドおよびポリペプチドをコードするポリヌクレオチドの組み合わせとして導入されてもよい。亜鉛フィンガーヌクレアーゼは典型的には、開裂片側ドメインの二量体化に続いて、二量体タンパク質として機能する。「左」および「右」認識ドメインに結合するZFN単量体が会合して活性ヌクレアーゼを形成することができる、偏性ヘテロ二量体ZFNが記載されている。例えば、米国特許公開第2008/0131962号を参照されたい。故に、適切な標的部位が与えられると、「左」単量体は、任意の「右」単量体を有する活性ZFヌクレアーゼを形成し得る。これは、様々な組み合わせで使用することができる、立証された左および右ドメインに基づいて有用なヌクレアーゼ部位の数を有意に増加させる。例えば、4個のホモ二量体ZFヌクレアーゼの結合部位を組み換えることは、追加的な12個のヘテロ二量体ZFヌクレアーゼを生み出す。より重要なことに、あらゆる新たな導入された配列の側面に、遺伝子を再び切除するため、またはそれの隣の追加的な遺伝子を標的とするために使用することができる固有のZFN部位が位置することになるような、遺伝子導入設計に対する系統的な手法を可能にする。更に、この方法は、ZFN依存性二本鎖切断によって駆動される、単一の遺伝子座へのスタッキングの戦略を単純化することができる。
【0031】
亜鉛フィンガー結合ドメインは、標準(C2H2)亜鉛フィンガーまたは非標準(例えば、C3H)亜鉛フィンガーであり得る。更に、亜鉛フィンガー結合ドメインは、1つ以上の亜鉛フィンガー(例えば、2つ、3つ、4つ、5つ、6つ、7つ、8つ、9つ、またはそれを超える亜鉛フィンガー)を含む可能性があり、多重挿入部位内の任意の配列に結合するように改変することができる。細胞中のかかる融合タンパク質(複数可)の存在は、融合タンパク質(複数可)のその(それらの)結合部位への結合および多重挿入部位内の開裂をもたらし、それは外来性配列の組み込みをもたらす。
【0032】
概論
本明細書で開示される方法の実践、ならびに組成物の調製と使用は、特に指示がない限り、分子生物学、生化学、クロマチン構造および分析、計算化学、細胞培養、組換えDNA、および関連分野における従来の技術を採用しており、当技術分野の技術範囲内である。これらの技術は、文献で完全に説明される。例えば、Sambrook et al.MOLECULAR CLONING:A LABORATORY MANUAL,Second edition,Cold Spring Harbor Laboratory Press,1989 and Third edition,2001、Ausubel et al.,CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John Wiley&Sons,New York,1987 and periodic updates、the series METHODS IN ENZYMOLOGY,Academic Press,San Diego、Wolffe,CHROMATIN STRUCTURE AND FUNCTION,Third edition,Academic Press,San Diego,1998、METHODS IN ENZYMOLOGY,Vol.304,“Chromatin”(P.M.Wassarman and A.P.Wolffe,eds.),Academic Press,San Diego,1999、およびMETHODS IN MOLECULAR BIOLOGY,Vol.119,“Chromatin Protocols”(P.B.Becker,ed.)Humana Press,Totowa,1999を参照されたい。
【0033】
定義
「核酸」、「ポリヌクレオチド」、および「オリゴヌクレオチド」という用語は、交換可能に使用され、線状または環状コンフォメーションで、一本鎖または二本鎖形態の、デオキシリボヌクレオチドポリマーまたはリボヌクレオチドポリマーを指す。本開示については、これらの用語をポリマーの長さに関する限定と解釈すべきではない。これらの用語は、天然ヌクレオチドの既知の類似体、ならびに塩基、糖、および/またはリン酸(ホスホロチオエート主鎖)部分内で修飾されるヌクレオチドを包含することができる。一般に、特定のヌクレオチドの類似体は、同一の塩基対形成特異性を有し、すなわち、Aの類似体は、Tと塩基対を形成する。
【0034】
「ポリペプチド」、「ペプチド」、および「タンパク質」という用語は、アミノ酸残基のポリマーを指すために交換可能に使用される。この用語は、1つ以上のアミノ酸が、対応する天然に存在するアミノ酸の化学的類似体または修飾誘導体である、アミノ酸ポリマーにも適用される。
【0035】
「結合」は、高分子間(例えば、タンパク質と核酸との間)の配列特異的で非共有結合的な相互作用を指す。結合相互作用の全ての構成要素は、相互作用が全体として配列特異的である限り(例えば、DNA主鎖中のリン酸残基との接触)、配列特異的である必要はない。かかる相互作用は、一般に、10-6M-1以下の解離定数(Kd)によって特徴付けられる。「親和性」は、結合の強さを指す。高い結合親和性は、低いKdと関連している。
【0036】
「結合タンパク質」は、別の分子に結合することができるタンパク質である。結合タンパク質は、例えば、DNA分子(DNA結合タンパク質)、RNA分子(RNA結合タンパク質)、および/またはタンパク質分子(タンパク質結合タンパク質)に結合することができる。タンパク質結合タンパク質の場合においては、それは、それ自体に結合する(その結果、ホモ二量体、ホモ三量体等を形成する)ことができる、および/または異なるタンパク質または複数のタンパク質の1つ以上の分子に結合することができる。結合タンパク質は、2種類以上の結合活性を有することができる。例えば、亜鉛フィンガータンパク質は、DNA結合、RNA結合、およびタンパク質結合活性を有する。
【0037】
「亜鉛フィンガーDNA結合タンパク質」(または結合ドメイン)は、タンパク質、または構造が亜鉛イオンの配位によって安定化される結合ドメイン内のアミノ酸配列の領域である、1つ以上の亜鉛フィンガーによって配列特異的な様式でDNAを結合するより大きなタンパク質内のドメインである。亜鉛フィンガーDNA結合タンパク質という用語は、しばしば、亜鉛フィンガータンパク質またはZFPとして略記される。
【0038】
亜鉛フィンガー結合ドメインは、所定のヌクレオチド配列に結合するように、「改変」することができる。亜鉛フィンガータンパク質を改変するための方法の非制限的な例は、設計および選択である。設計された亜鉛フィンガータンパク質は、天然に存在しないタンパク質であり、その設計/組成は、主として合理的基準によってもたらされる。設計のための合理的基準には、置換規則の適用、ならびに既存のZFP設計および結合データの情報を格納したデータベース中の情報を処理するためのコンピュータアルゴリズムの適用が含まれる。例えば、米国特許第6,140,081号、同第6,453,242号、および同第6,534,261号を参照されたく、また、国際公開第98/53058号、国際公開第98/53059号、国際公開第98/53060号、国際公開第02/016536号、および国際公開第03/016496号も参照されたい。
【0039】
「選択された」亜鉛フィンガータンパク質は、天然には見出されないタンパク質であり、その産生は、ファージディスプレイ、相互作用トラップ、またはハイブリッド選択等の実験的プロセスによって主にもたらされる。例えば、米国特許第5,789,538号、同第5,925,523号、同第6,007,988号、同第6,013,453号、同第6,200,759号、国際公開第95/19431号、国際公開第96/06166号、国際公開第98/53057号、国際公開第98/54311号、国際公開第00/27878号、国際公開第01/60970号、国際公開第01/88197号、および国際公開第02/099084号を参照されたい。
【0040】
「配列」という用語は、任意の長さのヌクレオチド配列を指し、それは、DNAまたはRNAであってもよく、線状、環状、または分岐状であってもよく、一本鎖または二本鎖のいずれかであってもよい。「ドナー配列」という用語は、ゲノム内に挿入されるヌクレオチド配列を指す。ドナー配列は、任意の長さ、例えば、2〜10,000(またはそれらの間もしくはそれらを超える任意の整数)ヌクレオチド長、好ましくは約100〜1,000(またはそれらの間の任意の整数)ヌクレオチド長、より好ましくは約200〜500ヌクレオチド長であってもよい。
【0041】
「相同非同一配列」は、第2の配列とある程度の配列同一性を共有するが、その配列は、第2の配列と同一ではない、第1の配列を指す。例えば、突然変異遺伝子の野生型配列を含むポリヌクレオチドは、突然変異遺伝子の配列に対して相同的かつ非同一である。ある実施形態では、2つの配列の間の相同性の度合いは、通常の細胞機構を利用して、それらの間で相同組み換えを可能にするために十分である。2つの相同非同一配列は、任意の長さである可能性があり、非相同性の度合いは、1ヌクレオチドほど小さいか(例えば、標的化相同組み換えによるゲノム点突然変異の補正のため)、または10キロベース以上ほど大きい(例えば、染色体内の所定の異所性部位における遺伝子の挿入のため)可能性がある。相同非同一配列を含む2つのポリヌクレオチドは、同一の長さである必要はない。例えば、20〜10,000ヌクレオチドまたはヌクレオチド対の外因性ポリヌクレオチド(すなわち、ドナーポリヌクレオチド)を使用することができる。
【0042】
核酸およびアミノ酸配列同一性を判定するための技術は、当該技術分野において既知である。典型的には、かかる技術は、遺伝子のためのmRNAのヌクレオチド配列を判定すること、および/またはそれによってコードされたアミノ酸配列を判定すること、ならびにそれらの配列を、第2のヌクレオチドまたはアミノ酸配列と比較することを含む。ゲノム配列もまた、この方法で決定し、比較することができる。一般に、同一性は、それぞれ、2つのポリヌクレオチドまたはポリペプチド配列のヌクレオチド対ヌクレオチド、またはアミノ酸対アミノ酸の対応が完全であることを指す。2つ以上の配列(ポリヌクレオチドまたはアミノ酸)は、それらの同一性百分率を判定することによって比較することができる。核酸配列であれアミノ酸配列であれ、2つの配列の同一性百分率は、短い方の配列の長さで割り、100を乗じた、2つの整合配列の間の厳密な合致数である。核酸配列の近似配列は、Smith and Waterman,Advances in Applied Mathematics 2:482−489(1981)の局所性相同性アルゴリズムによって提供される。このアルゴリズムは、Dayhoff,Atlas of Protein Sequences and Structure,M.O.Dayhoff ed.,5 suppl.3:353−358,National Biomedical Research Foundation,Washington,D.C.,USAによって開発され、Gribskov,Nucl.Acids Res.14(6):6745−6763(1986)によって正規化されたスコアリングマトリックスを使用することによって、アミノ酸配列に適用することができる。配列の同一性百分率を判定するためのこのアルゴリズムの例示的実施は、「BestFit」実用用途において、Genetics Computer Group(Madison,WI)によって提供される。配列の間の同一性百分率または類似性を計算するための好適なプログラムは、当該技術分野において一般に既知であり、例えば、別の整合プログラムは、デフォルトパラメータと共に使用されるBLASTである。例えば、BLASTNおよびBLASTPは、次のデフォルトパラメータを使用して、使用することができる:遺伝コード=標準、フィルター=なし、鎖=両方、カットオフ=60、期待数=10、マトリックス=BLOSUM62、表示=50配列、分類=高スコア、データベース=非重複、GenBank+EMBL+DDBJ+PDB+GenBank CDS tranlsations+Swiss protein+Spupdate+PIR。これらのプログラムの詳細は、インターネット上で見出すことができる。本明細書に説明する配列に関して、望ましい配列同一性の度合いの範囲は、約80%〜100%、およびそれらの間の任意の整数値である。典型的には、配列の間の同一性百分率は、少なくとも70〜75%、好ましくは、80〜82%、より好ましくは85〜90%、更に好ましくは92%、より一層好ましくは95%、最も好ましくは98%の配列同一性である。
【0043】
代替的には、ポリヌクレオチド間の配列類似性の度合いは、相同領域間で安定した二重鎖の形成を可能にする条件下でのポリヌクレオチドのハイブリダイゼーション、続いて一本鎖特異的ヌクレアーゼでの消化、および消化された断片の寸法決定によって決定することができる。2つの核酸または2つのポリペプチド配列は、配列が、上記の方法を使用して判定されるように、分子の定義された長さにわたって、少なくとも70%〜75%、好ましくは80%〜82%、より好ましくは85%〜90%、更に好ましくは92%、より一層好ましくは95%、および最も好ましくは98%の配列同一性を示すとき、互いに実質的に相同的である。本明細書に使用するとき、実質的に相同的はまた、指定されるDNAまたはポリペプチド配列に完全な同一性を示す配列を指す。実質的に相同的であるDNA配列は、例えば、サザンハイブリダイゼーション実験において、その特定の体系のために定義されるストリンジェントな条件下で特定することができる。適切なハイブリダイゼーション条件を定義することは、当該技術分野の技術の範囲内である。例えば、Sambrook et al.,supra;Nucleic Acid Hybridization:A Practical Approach,editors B.D.Hames and S.J.Higgins,(1985)Oxford;Washington,DC;IRL Press)を参照されたい。
【0044】
2つの核酸断片の選択的ハイブリダイゼーションは、次のように決定することができる。2つの核酸分子の間の配列同一性の度合いは、かかる分子の間のハイブリダイゼーション事象の効率および強度に影響する。部分的に同一な核酸配列は、完全に同一な配列の標的分子へのハイブリダイゼーションを少なくとも部分的に阻害する。完全に同一な配列のハイブリダイゼーションの阻害は、当該技術分野において周知である、ハイブリダイゼーションアッセイ(例えば、サザン(DNA)ブロット、ノーザン(RNA)ブロット、溶液ハイブリダイゼーション、または同等物、Sambrook,et al.,Molecular Cloning:A Laboratory Manual,Second Edition,(1989)Cold Spring Harbor,N.Y.を参照されたい)を使用して評価することができる。かかるアッセイは、異なる度合いの選択性を使用して、例えば、低ストリンジェンシーから高いストリンジェンシーまで異なる条件を使用して、実施することができる。低ストリンジェンシーの条件を使用する場合では、非特異的結合の不在は、非特異的結合事象の不在下で二次プローブが標的にハイブリダイズしないように、部分的な配列同一性の度合いさえ持たない二次プローブ(例えば、標的分子との配列同一性が約30%未満であるプローブ)を使用し、評価することができる。
【0045】
ハイブリダイゼーションベースの検出系を利用する時、参照核酸配列に相補的である核酸プローブが選択され、次いで、適切な条件の選択によって、プローブおよび参照配列が二重鎖分子を形成するように、互いに選択的にハイブリダイズするか、または結合する。中等度にストリンジェントなハイブリダイゼーション条件下で、参照配列に選択的にハイブリダイズする能力を有する核酸分子は、典型的には、選択された核酸プローブの配列と少なくとも約70%の配列同一性を有する、少なくとも約10〜14ヌクレオチド長の標的核酸配列の検出を可能にするような条件下でハイブリダイズする。ストリンジェントなハイブリダイゼーション条件は、典型的には、選択された核酸プローブの配列と少なくとも約90〜95%を上回る配列同一性を有する、少なくとも約10〜14ヌクレオチド長の標的核酸配列の検出を可能にする。プローブおよび参照配列が、特異的な度合いの配列同一性を有する、プローブ/参照配列ハイブリダイゼーションに有用なハイブリダイゼーション条件は、当該技術分野において既知のとおり決定することができる(例えば、Nucleic Acid Hybridization:A Practical Approach,editors B.D.Hames and S.J.Higgins,(1985)Oxford;Washington,DC;IRL Pressを参照されたい)。
【0046】
ハイブリダイゼーションの条件は、当業者に周知である。ハイブリダイゼーションストリンジェンシーは、ハイブリダイゼーション条件が、ミスマッチしたヌクレオチドを含むハイブリッドの形成を嫌う度合いを指し、より高いストリンジェンシーは、ミスマッチしたハイブリッドに対する許容度の低さに相関する。ハイブリダイゼーションのストリンジェンシーに影響を及ぼす因子は、当業者に周知であり、温度、pH、イオン強度、ならびに例えば、ホルムアミドおよびジメチルスルホキシド等の有機溶媒の濃度を含むが、これらに限定されない。当業者には既知であるが、ハイブリダイゼーションのストリンジェンシーは、より高い温度、より低いイオン強度、およびより低い溶媒濃度によって増加する。
【0047】
ハイブリダイゼーションのストリンジェンシー条件に関して、数々の等価条件は、例えば、次の因子:配列の長さおよび性質、様々な配列の塩基組成、塩および他のハイブリダイゼーション溶液成分の濃度、ハイブリダイゼーション溶液中の遮断薬(例えば、硫酸デキストランおよびポリエチレングリコール)の存否、ハイブリダイゼーション反応温度および時間パラメータ、ならびに洗浄条件を変化させることによって、特定のストリンジェンシーを確立するために使用することができることは、当該技術分野において周知である。特定の一連のハイブリダイゼーション条件の選択は、当該技術分野における標準方法に従い選択される(例えば、Sambrook et al.,Molecular Cloning:A Laboratory Manual,Second Edition,(1989)Cold Spring Harbor,N.Y.を参照されたい)。
【0048】
「組み換え」は、2つのポリヌクレオチドの間の遺伝子情報の交換のプロセスを指す。この開示に関して、「相同組み換え(HR)」は、例えば、細胞中の二本鎖切断の修復中に行われるような交換の特殊な形態を指す。このプロセスは、ヌクレオチド配列相同性を必要とし、「標的」分子(すなわち、二本鎖切断を経験するもの)のテンプレート修復に「ドナー」分子を使用し、かつ、それがドナーから標的への遺伝子情報の移動をもたらすため、「非交差遺伝子変換」または「ショートトラクト遺伝子変換」として多様に知られる。いかなる特定の理論にも拘束されることを望むものではないが、かかる移動は、切断された標的とドナーとの間に形成されるヘテロ二重鎖DNAのミスマッチ修正、および/または標的の一部になる遺伝子情報を再合成するためにドナーが使用される、「合成依存性鎖アニーリング」、および/または関連プロセスを伴う可能性がある。かかる特殊なHRは、しばしば、ドナーポリヌクレオチドの配列の一部または全てが、標的ポリヌクレオチド内に組み込まれるような、標的分子の配列の変化をもたらす。
【0049】
「開裂」は、DNA分子の共有主鎖の切断を指す。開裂は、ホスホジエステル結合の酵素的または化学的加水分解を含むが、これらに限定されない多様な方法によって開始することができる。一本鎖開裂および二本鎖開裂の両方が可能であり、二本鎖開裂は、2つの別々の一本鎖開裂事象の結果として生じ得る。DNA開裂は、平滑末端または付着末端のいずれかの産生をもたらす可能性がある。ある実施形態では、融合ポリペプチドは、標的化二本鎖DNA開裂のために使用される。
【0050】
「開裂ドメイン」は、DNA開裂に対する触媒活性を有する1つ以上のポリペプチド配列を含む。開裂ドメインは、単一のポリペプチド鎖内に含有され得るか、または開裂活性は、2つ(またはそれを超える数)のポリペプチドの会合からもたらされ得る。
【0051】
「開裂片側ドメイン」は、第2のポリペプチド(同一のまたは異なるいずれかの)と併せて、開裂活性(好ましくは二本鎖開裂活性)を有する複合体を形成するポリペプチド配列である。
【0052】
「クロマチン」は、細胞ゲノムを含む核タンパク質構造である。細胞クロマチンは、核酸、主にDNA、ならびにヒストンおよび非ヒストン染色体タンパク質を含むタンパク質を含む。真核細胞クロマチンの大半は、ヌクレオソームの形態で存在し、そこでは、ヌクレオソームコアが、それぞれ2つのヒストンH2A、H2B、H3、およびH4を含む八量体と会合した約150塩基対のDNAを含み、リンカーDNA(生物に応じて変化する長さ)がヌクレオソームコアの間に延在する。1分子のヒストンH1は、一般にリンカーDNAと会合する。本開示に関して、「クロマチン」という用語は、原核性および真核性の両方の全ての種類の細胞核タンパク質を包含するように意図されている。細胞クロマチンは、染色体クロマチンおよびエピソームクロマチンの両方を含む。
【0053】
「染色体」は、細胞のゲノムの全てまたは一部を含むクロマチン複合体である。細胞のゲノムは、しばしば、その細胞のゲノムを含む全染色体の集合である、その核型によって特徴付けられる。細胞のゲノムは、1つ以上の染色体を含むことができる。
【0054】
「エピソーム」は、細胞の染色体核型の一部ではない核酸を含む、複製する核酸、核タンパク質複合体、または他の構造である。エピソームの例としては、プラスミドおよびある種のウイルスゲノムが挙げられる。
【0055】
「到達可能領域」は、核酸内に存在する標的部位が標的部位を認識する外来性分子によって結合され得る、細胞クロマチン内の部位である。いかなる特定の理論にも拘束されることを望むものではないが、到達可能領域は、ヌクレオソーム構造内にパッケージングされない領域であると考えられる。到達可能領域の別々の構造は、しばしば、化学的および酵素的プローブ、例えば、ヌクレアーゼに対する、その感受性によって検出することができる。
【0056】
「標的部位」または「標的配列」は、結合にとって十分な条件が存在すれば、結合分子が結合する核酸の一部を規定する核酸配列である。例えば、配列5’−GAATTC−3’は、EcoRI制限エンドヌクレアーゼに対する標的部位である。
【0057】
「外来性」分子は、通常は細胞中に存在しないが、1つ以上の遺伝学的、生化学的、または他の方法によって細胞中に導入することができる分子である。「通常は細胞中に存在」は、細胞の特定の発達段階および環境条件に対して決定される。したがって、例えば、筋の胚発達中にのみ存在する分子は、成体筋細胞に対しては外来性分子である。同様に、熱ショックによって誘導される分子は、非熱ショック細胞に対しては外来性分子である。外来性分子は、例えば、任意のポリペプチドもしくはその断片に対するコード配列、機能不全型内在性分子の機能型、または正常機能型内在性分子の機能不全型を含む可能性がある。更に、外来性分子は、宿主細胞中の内在性遺伝子のオルソログである別の種からのコード配列を含む可能性がある。
【0058】
外来性分子は、とりわけ、コンビナトリアルケミストリープロセスによって生成されるもの等の小分子、または、タンパク質、核酸、糖質、脂質、糖タンパク質、リポタンパク質、多糖、上記の分子の任意の修飾誘導体、または上記の分子の1つ以上を含む任意の複合体等の高分子であってもよい。核酸にはDNAおよびRNAが含まれ、それは一本鎖または二本鎖であってもよく、線状、分岐状、または環状であってもよく、任意の長さであってもよい。核酸には、二重鎖を形成する能力のあるもの、ならびに三重鎖形成核酸が含まれる。例えば、米国特許第5,176,996号および同第5,422,251号明細書を参照されたい。タンパク質には、DNA結合タンパク質、転写因子、クロマチンリモデリング因子、メチル化DNA結合タンパク質、ポリメラーゼ、メチラーゼ、デメチラーゼ、アセチラーゼ、デアセチラーゼ、キナーゼ、ホスファターゼ、インテグラーゼ、リコンビナーゼ、リガーゼ、トポイソメラーゼ、ジャイレース、およびヘリカーゼが含まれるが、これらに限定されない。
【0059】
外来性分子は、内在性分子と同一の種類の分子、例えば、外来性タンパク質または核酸であってもよい。例えば、外来性核酸は、感染ウイルスゲノム、細胞中に導入されたプラスミドもしくはエピソーム、または細胞内に通常存在しない染色体を含むことができる。細胞中に外来性分子を導入するための方法は当業者に既知であり、脂質媒介導入(すなわち、中性およびカチオン性脂質を含むリポソーム)、エレクトロポレーション、直接注入、細胞融合、マイクロプロジェクタイルボンバードメント、リン酸カルシウム共沈、DEAE−デキストラン媒介導入、およびウイルスベクター媒介導入を含むが、これらに限定されない。
【0060】
対照的に、「内在性」分子は、特定の環境条件下で特定の発達段階にある特定の細胞中に通常存在するものである。例えば、内在性核酸は、染色体、ミトコンドリア、クロロプラストもしくは他の細胞小器官のゲノム、または天然に存在するエピソーム核酸を含むことができる。更なる内在性分子には、タンパク質、例えば、転写因子および酵素が含まれてもよい。
【0061】
本明細書で使用されるとき、「外来性核酸の産物」という用語は、ポリヌクレオチドおよびポリペプチド産物の両方、例えば、転写産物(RNA等のポリヌクレオチド)および翻訳産物(ポリペプチド)を含む。
【0062】
「融合」分子は、2つ以上のサブユニット分子が、好ましくは共有結合的に、連結された分子である。サブユニット分子は、同一の化学種類の分子であってもよいか、または異なる化学種類の分子であってもよい。第1の種類の融合分子の例としては、融合タンパク質(例えば、ZFP DNA結合ドメインと開裂ドメインとの間の融合物)および融合核酸(例えば、上述の融合タンパク質をコードする核酸)が挙げられるが、これらに限定されない。第2の種類の融合分子の例としては、三重鎖形成核酸とポリペプチドとの間の融合物、および副溝結合剤と核酸との間の融合が挙げられるが、これらに限定されない。
【0063】
細胞中の融合タンパク質の発現は、細胞に融合タンパク質を送達すること、または細胞に融合タンパク質をコードするポリヌクレオチドを送達することによってもたらされてもよく、ポリヌクレオチドが転写され、転写物が翻訳されて、融合タンパク質を生成する。トランススプライシング、ポリペプチド開裂、およびポリペプチド連結はまた、細胞中のタンパク質の発現に関与することができる。細胞へのポリヌクレオチドおよびポリペプチドの送達の方法が本開示の他所で提示される。
【0064】
本開示に関して、「遺伝子」は、かかる調節配列がコードおよび/または転写配列に隣接しているか否かに関わらず、遺伝子産物(以下を参照)をコードするDNA領域、ならびに遺伝子産物の産生を調節する全てのDNA領域を含む。したがって、遺伝子には、プロモーター配列、ターミネータ、リポソーム結合部位および内部リポソーム侵入部位等の翻訳調節配列、エンハンサー、サイレンサー、インスレーター、境界エレメント、複製起点、マトリックス付着部位、ならびに遺伝子座制御領域が含まれるが、これらに限定されない。
【0065】
「遺伝子発現」は、遺伝子に含有される情報の、遺伝子産物への変換を指す。遺伝子産物は、遺伝子の直接転写産物(例えば、mRNA、tRNA、rRNA、アンチセンスRNA、リボザイム、構造RNA、または任意の他の種類のRNA)、またはmRNAの翻訳によって産生されるタンパク質である可能性がある。遺伝子産物には、キャッピング、ポリアデニル化、メチル化、および編集等のプロセスによって修飾されたRNA、および、例えば、メチル化、アセチル化、リン酸化、ユビキチン化、ADPリボシル化、ミリスチリル化、およびグリコシル化によって修飾されたタンパク質も含まれる。
【0066】
遺伝子発現の「調節」は、遺伝子の活性の変化を指す。発現の調節には、遺伝子活性化および遺伝子抑制が含まれてもよいが、これらに限定されない。
【0067】
「植物」細胞には、単子葉(単子葉類)または双子葉(双子葉類)植物の細胞が含まれるが、これらに限定されない。単子葉類の非限定的例としては、トウモロコシ、イネ、オオムギ、オートムギ、コムギ、ソルガム、ライムギ、サトウキビ、パイナップル、タマネギ、バナナ、およびココナッツ等の穀物植物が挙げられる。双子葉類の非限定的例としては、タバコ、トマト、ヒマワリ、綿花、テンサイ、ジャガイモ、レタス、メロン、ダイズ、キャノーラ(ナタネ)、およびアルファルファが挙げられる。植物細胞は、植物の任意の部分からの、および/または植物発達の任意の段階からのものであってもよい。
【0068】
「目的の領域」は、例えば、領域内で外因性分子を結合させることが望まれる、遺伝子、または遺伝子内のもしくはそれに隣接する非コード配列等の細胞クロマチンの任意の領域である。結合の目的は、標的化DNA開裂および/または標的化組み換えであり得る。目的の領域は、例えば、染色体、エピソーム、細胞小器官ゲノム(例えば、ミトコンドリア、クロロプラスト)、または感染ウイルスゲノム内に存在することができる。目的の領域は、遺伝子のコード領域内、例えば、リーダー配列、トレーラー配列、もしくはイントロン等の転写された非コード領域内、またはコード領域の上流もしくは下流のいずれかの非転写領域内である可能性がある。目的の領域は、単一のヌクレオチド対ほど小さいか、または最大2,000ヌクレオチド対長、またはヌクレオチド対の任意の整数値である可能性がある。
【0069】
「作動的連結」および「作動的に連結」(または「作動可能に連結」)という用語は、2つ以上の構成要素(配列要素等)の並列に関連して交換可能に使用され、その中で両方の構成要素が正常に機能し、かつ構成要素の少なくとも1つが、他の構成要素の少なくとも1つに対して発揮される機能を媒介することができる可能性を許容するように各構成要素が配置される。例証として、プロモーター等の転写調節配列は、転写調節配列が1つ以上の転写調節因子の存在または不在に応じてコード配列の転写レベルを制御する場合に、コード配列に作動的に連結される。転写調節配列は、一般的に、コード配列とシスで作動的に連結されるが、それに直接隣接している必要はない。例えば、エンハンサーは、隣接していなくても、コード配列に作動的に連結される転写調節配列である。
【0070】
融合ポリペプチドに関して、「作動的に連結」という用語は、構成要素の各々が連動して機能し、そのように連結されていない場合にそれが果たす機能と同じ機能を実行するという事実を指すことができる。例えば、ZFP DNA結合ドメインが開裂ドメインに融合されている融合ポリペプチドに関しては、ZFP DNA結合ドメインおよび開裂ドメインは、融合ポリペプチドにおいて、ZFP DNA結合ドメイン部分が、その標的部位および/またはその結合部位に結合することが可能であり、開裂ドメインが、標的部位の近傍でDNAを開裂することが可能である場合に、作動的連結状態にある。
【0071】
タンパク質、ポリペプチド、または核酸の「機能的断片」は、その配列が、完全長タンパク質、ポリペプチド、または核酸と同一でないが、完全長タンパク質、ポリペプチド、または核酸と同一の機能を保持するタンパク質、ポリペプチド、または核酸である。機能的断片は、対応する天然分子より多い、少ない、または同一の残基数を有することができ、かつ/または1つ以上のアミノ酸またはヌクレオチド置換を含有することができる。核酸の機能(例えば、コード機能、別の核酸とハイブリッド形成する能力)を決定するための方法は、当技術分野で周知である。同様に、タンパク質機能を決定するための方法は周知である。例えば、ポリペプチドのDNA結合機能は、例えば、フィルター結合、電気泳動移動度シフト、または免疫沈降アッセイによって決定することができる。DNA開裂は、ゲル電気泳動によってアッセイすることができる。上記のAusubelらを参照されたい。別のタンパク質と相互作用するタンパク質の能力は、例えば、共免疫沈降、ツーハイブリッドアッセイ、または遺伝的相補性および生化学的相補性の両方の相補性によって決定することができる。例えば、Fields et al.(1989)Nature 340:245−246、米国特許第5,585,245号、およびPCT国際公開第98/44350号を参照されたい。
【0072】
多重挿入部位
多重挿入部位、つまり、適切なZFN対の結合時に、多重挿入部位がZFN対の標的部位の間で開裂されるように複数の亜鉛フィンガーヌクレアーゼ(ZFN)結合部位を含むポリヌクレオチドが、本明細書に開示される。
【0073】
多重挿入部位上に含まれる標的部位は、好ましくは、それが組み込まれる細胞のゲノム内には見出されない。したがって、ゲノム内の望まれない開裂の発生は、低減または排除される。任意の数、例えば、1〜50(またはその間の任意の数)、好ましくは2〜30(またはその間の任意の数)、および更により好ましくは5〜20(またはその間の任意の数)の標的部位が、多重挿入部位ポリヌクレオチドに含まれてもよい。亜鉛フィンガーヌクレアーゼについて、標的部位は、典型的には、亜鉛フィンガーヌクレアーゼが、ホモ二量体またはヘテロ二量体を形成して、適切な部位で開裂するような対になっている。
【0074】
更に、図1に示されるように、標的部位(図1の影付き三角)の各対の1つの標的部位は、多重挿入部位全体にわたって同じであってもよい。代替的には、ヘテロ二量体対は、部位の間で異なってもよい。
【0075】
多重挿入部位は、ホモ二量体のみが結合する標的部位、ヘテロ二量体のみが結合する標的部位、またはホモ二量体またはヘテロ二量体が結合する標的部位の組み合わせを含んでもよい。場合によっては、次の1つ以上の理由で、ホモ二量体が結合する標的部位が好ましい可能性がある:1つのZFNの送達が2つのそれよりも効率的であり得ること、ホモ二量体化が、ZFNの不均等な発現に起因する不均等な化学量論の問題を低減すること;オフターゲット部位における開裂からの毒性が低減され得ること;CCHC(非標準)亜鉛フィンガードメインを使用するとき、それによってホモ二量体が破壊される可能性が半減すること;および/または固有の標的可能部位の総数が拡大され得ること。代替的に、他の場合には、ヘテロ二量体は、異なる標的部位の混合およびマッチング、故にZFN対に対する標的可能部位の増加の可能性を許容するため、好ましい可能性がある。また、ヘテロ二量体は、実行者の必要に応じて、ドナーの逐次的付加も許容する。ヘテロ二量体組み合わせはまた、新規ZFN対の使用を通じて、ドナーの任意の所望の部分の特異的欠失も許容し得る。
【0076】
亜鉛フィンガーヌクレアーゼに対する標的部位が3ヌクレオチドの倍数である必要がないことは明白であろう。例えば、交差鎖相互作用が生じる場合(例えば、米国特許第6,453,242号および国際公開第02/077227号を参照されたい)、マルチフィンガー結合ドメインの個々の亜鉛フィンガーのうちの1つ以上は、重複する四重亜部位に結合することができる。結果として、3フィンガータンパク質は、10ヌクレオチド配列に結合することができ、ここで10番目のヌクレオチドは、末端フィンガーが結合する四重の一部であり、4フィンガータンパク質は、13ヌクレオチド配列に結合することができ、ここで13番目のヌクレオチドは、末端フィンガーが結合する四重の一部である、等である。
【0077】
マルチフィンガー結合ドメインにおける個々の亜鉛フィンガーの間のアミノ酸リンカー配列の長さおよび性質もまた、標的配列への結合に影響を及ぼす。例えば、マルチフィンガー結合ドメインにおける隣接した亜鉛フィンガーの間の、いわゆる「非標準的なリンカー」、「長いリンカー」、または「構造化リンカー」の存在は、それらのフィンガーが直接隣接していない亜部位に結合することを可能にし得る。かかるリンカーの非限定的例は、例えば、米国特許第6,479,626号および国際公開第01/53480号に記載される。したがって、亜鉛フィンガー結合ドメインに対する標的部位における1つ以上の亜部位は、互いから1、2、3、4、5、またはそれを超えるヌクレオチド分だけ隔てられてもよい。一例を提供するにすぎないが、4フィンガー結合ドメインは、順番に、2つの隣接する3ヌクレオチド亜部位、介入するヌクレオチド、および2つの隣接する三重亜部位を含む、13ヌクレオチド標的部位に結合することができる。
【0078】
配列(例えば、標的部位)の間の距離は、互いに最も近接した配列の端から測定した、2つの配列の間に介入するヌクレオチドまたはヌクレオチド対の数を指す。
【0079】
開裂が2つの亜鉛フィンガードメイン/開裂片側ドメイン融合分子の、別個の標的部位への結合に依存するある実施形態では、2つの標的部位は、反対側のDNA鎖上にあり得る。他の実施形態では、両方の標的部位は、同じDNA鎖上にある。
【0080】
多重挿入部位は、植物ゲノム内のいずれの場所にも組み込むことができる。ある実施形態では、多重挿入部位は、トウモロコシゲノムにおけるZp15に組み込まれ、それは米国出願第12/653,735号に記載されるように、外来性配列の標的の組み込みのために望ましい部位である。
【0081】
DNA結合ドメイン
任意のDNA結合ドメインを、本明細書に開示される方法で使用することができる。ある実施形態では、DNA結合ドメインは、亜鉛フィンガータンパク質を含む。亜鉛フィンガー結合ドメインは、1つ以上の亜鉛フィンガーを含む。Miller et al.(1985)EMBO J.4:1609−1614、Rhodes(1993)Scientific American Feb.:56−65、米国特許第6,453,242号。本明細書に記載される亜鉛フィンガー結合ドメインは、一般に、2つ、3つ、4つ、5つ、6つ、または更にそれを超える亜鉛フィンガーを含む。
【0082】
典型的には、単一の亜鉛フィンガードメインは、約30アミノ酸長である。構造的研究は、各亜鉛フィンガードメイン(モチーフ)が、2つのベータシート(2つの不変のシステイン残基を含有するベータターンにおいて保有される)およびアルファヘリックス(2つの不変のヒスチジン残基を含有する)を含有し、それらが2つのシステインおよび2つのヒスチジンによる亜鉛原子の配位を通じて、特定のコンフォメーションにおいて保有されることを実証している。
【0083】
亜鉛フィンガーは、標準的なC2H2亜鉛フィンガー(すなわち、亜鉛イオンが2つのシステインおよび2つのヒスチジン残基によって配位されるもの)、ならびに例えば、C3H亜鉛フィンガー(亜鉛イオンが3つのシステイン残基および1つのヒスチジン残基によって配位されるもの)、およびC4亜鉛フィンガー(亜鉛イオンが4つのシステイン残基によって配位されるもの)等の非標準的な亜鉛フィンガーの両方を含む。植物における使用のための非標準的なZFPに関してはまた、国際公開第02/057293号、およびまた米国特許公開第20080182332号も参照されたい。
【0084】
改変された亜鉛フィンガー結合ドメインは、天然に存在する亜鉛フィンガータンパク質と比較して、新規の結合特異性を有することができる。改変方法には、合理的設計および様々な種類の選択が含まれるが、これらに限定されない。合理的設計には、例えば、三重(または四重)ヌクレオチド配列および個々の亜鉛フィンガーアミノ酸配列を含むデータベースを使用することが含まれ、ここで各三重または四重ヌクレオチド配列は、特定の三重または四重配列に結合する亜鉛フィンガーの1つ以上のアミノ酸配列に関連する。
【0085】
ファージディスプレイおよび2ハイブリッド系を含む例示的な選択方法は、米国特許第5,789,538号、同第5,925,523号、同第6,007,988号、同第6,013,453号、同第6,410,248号、同第6,140,466号、同第6,200,759号、および同第6,242,568号、ならびに国際公開第98/37186号、国際公開第98/53057号、国際公開第00/27878号、国際公開第01/88197号、および英国特許第2,338,237号に開示される。
【0086】
亜鉛フィンガー結合ドメインに対する結合特異性の強化は、例えば、譲受人共通の国際公開第02/077227号に記載されている。
【0087】
個々の亜鉛フィンガーは、3ヌクレオチド(すなわち、三重)配列(または隣接した亜鉛フィンガーの4ヌクレオチド結合部位により、1ヌクレオチド分だけ重複し得る4ヌクレオチド配列)に結合するため、亜鉛フィンガー結合ドメインが改変されて結合する配列(例えば、標的配列)の長さは、改変亜鉛フィンガー結合ドメインにおける亜鉛フィンガーの数を決定するであろう。例えば、フィンガーモチーフが重複する亜部位に結合しないZFPについて、6ヌクレオチド標的配列には、2フィンガー結合ドメインが結合し、9ヌクレオチド標的配列には、3フィンガー結合ドメインが結合する、等である。本明細書に記載されるように、標的部位における個々の亜鉛フィンガーに対する結合部位(すなわち、亜部位)は、隣接している必要はないが、マルチフィンガー結合ドメインにおける亜鉛フィンガーの間のアミノ酸配列(すなわち、フィンガー間リンカー)の長さおよび性質に依存して、1つ以上のヌクレオチド分だけ隔てられる可能性がある。
【0088】
マルチフィンガー亜鉛フィンガー結合ドメインにおいて、隣接した亜鉛フィンガーは、およそ5アミノ酸(いわゆる「標準」フィンガー間リンカー)のアミノ酸リンカー配列分、または代替的に、1つ以上の非標準的なリンカー分だけ隔てられる可能性がある。例えば、譲受人共通の米国特許第6,453,242号および同第6,534,261号を参照されたい。3つを超えるフィンガーを含む改変された亜鉛フィンガー結合ドメインについて、場合によっては、ある種の亜鉛フィンガー間のより長い(「非標準」)フィンガー間リンカーの挿入は、それが結合ドメインによる結合の親和性および/または特異性を増加させ得るため、望ましい可能性がある。例えば、米国特許第6,479,626号および国際公開第01/53480号を参照されたい。したがって、マルチフィンガー亜鉛フィンガー結合ドメインはまた、非標準的なフィンガー間リンカーの存在および位置に関して特徴付けることができる。例えば、3フィンガー(2つの標準的なフィンガー間リンカーによって結合される)、長いリンカー、および3つの追加的なフィンガー(2つの標準的なフィンガー間リンカーによって結合される)を含む6フィンガー亜鉛フィンガー結合ドメインは、2×3構成と表示される。同様に、2つのフィンガー(その間に標準的なリンカーを有する)、長いリンカー、および2つの追加的なフィンガー(標準的なリンカーによって結合される)を含む結合ドメインは、2×2構成と表示される。3つの2フィンガー単位(その各々において2つのフィンガーは、標準的なリンカーによって結合される)を含み、その中で各2つのフィンガー単位が長いリンカーによって隣接した2つのフィンガー単位に結合されるタンパク質は、3×2構成と称される。
【0089】
マルチフィンガー結合ドメインにおける2つの隣接した亜鉛フィンガーの間の、長いまたは非標準的なフィンガー間リンカーの存在は、しばしば、2つのフィンガーが、標的配列内で直接隣接していない亜部位に結合することを可能にする。したがって、標的部位における亜部位の間に1つ以上のヌクレオチドのギャップが存在し得、すなわち、標的部位は、亜鉛フィンガーが接触していない1つ以上のヌクレオチドを含有し得る。例えば、2×2亜鉛フィンガー結合ドメインは、1ヌクレオチド分だけ隔てられた2つの6ヌクレオチド配列に結合することができ、すなわち、それは13ヌクレオチド標的部位に結合する。また、Moore et al.(2001a)Proc.Natl.Acad.Sci.USA 98:1432−1436、Moore et al.(2001b)Proc.Natl.Acad.Sci.USA 98:1437−1441、および国際公開第01/53480号も参照されたい。
【0090】
前述のように、標的亜部位は、単一の亜鉛フィンガーが結合する、3ヌクレオチドまたは4ヌクレオチド配列である。ある目的に関して、2フィンガー単位は、「結合モジュール」と表示される。結合モジュールは、例えば、特定の6ヌクレオチド標的配列に結合するマルチフィンガータンパク質(一般に、3フィンガー)の文脈において、2つの隣接したフィンガーについて選択することによって得ることができる。代替的に、モジュールは、個々の亜鉛フィンガーの組立によって構築することができる。また、国際公開第98/53057号および国際公開第01/53480号も参照されたい。
【0091】
代替的に、DNA結合ドメインは、ヌクレアーゼに由来し得る。例えば、I−SceI、I−CeuI、PI−PspI,PI−Sce、I−SceIV、I−CsmI、I−PanI、I−SceII、I−PpoI、I−SceIII、I−CreI、I−TevI、I−TevII、およびI−TevIII等のホーミングエンドヌクレアーゼおよびメガヌクレアーゼの認識配列が知られている。また、米国特許第5,420,032号、米国特許第6,833,252号、Belfort et al.(1997)Nucleic Acids Res.25:3379−3388、Dujon et al.(1989)Gene 82:115−118;Perler et al.(1994)Nucleic Acids Res.22,1125−1127、Jasin(1996)Trends Genet.12:224−228、Gimble et al.(1996)J.Mol.Biol.263:163−180、Argast et al.(1998)J.Mol.Biol.280:345−353、およびNew England Biolabsカタログも参照されたい。加えて、ホーミングエンドヌクレアーゼおよびメガヌクレアーゼのDNA結合特異性を、非天然の標的部位に結合するよう改変することができる。例えば、Chevalier et al.(2002)Molec.Cell 10:895−905、Epinat et al.(2003)Nucleic Acids Res.31:2952−2962、Ashworth et al.(2006)Nature 441:656−659、Paques et al.(2007)Current Gene Therapy 7:49−66、米国特許公開第20070117128号を参照されたい。
【0092】
別の代替として、DNA結合ドメインは、ロイシンジッパータンパク質に由来し得る。ロイシンジッパーは、遺伝子発現に関連する重要な転写因子である多くの真核性調節タンパク質におけるタンパク質−タンパク質相互作用に関与する、タンパク質のクラスである。ロイシンジッパーは、動物、植物、酵母等を含む複数の界にわたって、これらの転写因子において共有される共通の構造モチーフを指す。ロイシンジッパーは、2つのポリペプチドのロイシン残基がヘリックスの同じ面上で終了するように、ロイシン残基がα−ヘリックスを通じて均等に間隔をあけられる様態で、特異的DNA配列に結合する2つのポリペプチド(ホモ二量体またはヘテロ二量体)によって形成される。ロイシンジッパーのDNA結合特異性は、本明細書に開示されるDNA結合ドメインにおいて利用することができる。
【0093】
幾つかの実施形態では、DNA結合ドメインは、植物病原菌キサントモナスに由来するTALエフェクターからの改変ドメインである(Miller et al.(2010)Nature Biotechnology,Dec 22 [Epub ahead of print]、Boch et al,(2009)Science 29 Oct 2009(10.1126/science.117881)、およびMoscou and Bogdanove,(2009)Science 29 Oct 2009(10.1126/science.1178817)を参照されたい。
【0094】
開裂ドメイン
上記のように、DNA結合ドメインは、開裂(ヌクレアーゼ)ドメインに関連し得る。例えば、ホーミングエンドヌクレアーゼは、ヌクレアーゼ機能を保持しながら、それらのDNA結合特異性において修飾されてもよい。加えて、亜鉛フィンガータンパク質を、開裂ドメインに融合して、亜鉛フィンガーヌクレアーゼ(ZFN)を形成してもよい。本明細書に開示される融合タンパク質の開裂ドメイン部分は、任意のエンドヌクレアーゼまたはエキソヌクレアーゼから得ることができる。開裂ドメインの由来であり得る例示的なエンドヌクレアーゼには、制限エンドヌクレアーゼおよびホーミングエンドヌクレアーゼが含まれるが、これらに限定されない。例えば、2002−2003 Catalogue,New England Biolabs,Beverly,MA;and Belfort et al.(1997)Nucleic Acids Res.25:3379−3388を参照されたい。DNAを開裂する追加的な酵素は既知である(例えば、S1ヌクレアーゼ;マングビーンヌクレアーゼ;膵臓DNase I;ミクロコッカルヌクレアーゼ;酵母HOエンドヌクレアーゼ、また、Linn et al.(eds.)Nucleases,Cold Spring Harbor Laboratory Press,1993も参照されたい)。ホーミングエンドヌクレアーゼおよびメガヌクレアーゼの非限定的例としては、I−SceI、I−CeuI、PI−PspI、PI−Sce、I−SceIV、I−CsmI、I−PanI、I−SceII、I−PpoI、I−SceIII、I−CreI、I−TevI、I−TevII、およびI−TevIIIが既知である。また、米国特許第5,420,032号、米国特許第6,833,252号、Belfort et al.(1997)Nucleic Acids Res.25:3379−3388、Dujon et al.(1989)Gene 82:115−118、Perler et al.(1994)Nucleic Acids Res.22,1125−1127、Jasin(1996)Trends Genet.12:224−228、Gimble et al.(1996)J.Mol.Biol.263:163−180、Argast et al.(1998)J.Mol.Biol.280:345−353、およびNew England Biolabsカタログも参照されたい。これらの酵素(またはその機能的断片)のうちの1つ以上を、開裂ドメインおよび開裂片側ドメインの源として使用することができる。
【0095】
制限エンドヌクレアーゼ(制限酵素)は、多くの種に存在し、DNA(認識部位における)に配列特異的に結合すること、また結合の部位におけるまたはそれに近接するDNAを開裂することができる。ある種の制限酵素(例えば、IIS型)は、認識部位から除去された部位におけるDNAを開裂し、分離可能な結合および開裂ドメインを有する。例えば、IIS型酵素FokIは、1つの鎖上のその認識部位から9ヌクレオチド、およびもう1つの鎖上のその認識部位から13ヌクレオチドにおいて、DNAの2本鎖切断を触媒する。例えば、米国特許第5,356,802号、同第5,436,150号、および同第5,487,994号、ならびにLi et al.(1992)Proc.Natl.Acad.Sci.USA 89:4275−4279、Li et al.(1993)Proc.Natl.Acad.Sci.USA 90:2764−2768、Kim et al.(1994a)Proc.Natl.Acad.Sci.USA 91:883−887、Kim et al.(1994b)J.Biol.Chem.269:31,978−31,982を参照されたい。故に、一実施形態では、融合タンパク質は、少なくとも1つのIIS型制限酵素からの開裂ドメイン(または開裂片側ドメイン)および1つ以上の亜鉛フィンガー結合ドメインを含み、それは改変されてもよく、されなくてもよい。
【0096】
開裂ドメインが結合ドメインから分離可能な、例示的なIIS型制限酵素は、FokIである。この特定の酵素は、二量体として活性である。Bitinaite et al.(1998)Proc.Natl.Acad.Sci.USA 95:10,570−10,575。したがって、本開示に関して、開示される融合タンパク質に使用されるFokI酵素の部分は、開裂片側ドメインとみなされる。故に、亜鉛フィンガー−FokI融合を使用する細胞配列の標的化二本鎖開裂および/または標的化置換のために、各々がFokI開裂片側ドメインを含む2つの融合タンパク質を使用して、触媒的に活性な開裂ドメインを再構成することができる。代替的に、亜鉛フィンガー結合ドメインおよび2つのFokI開裂片側ドメインを含有する単一のポリペプチド分子を使用することもできる。亜鉛フィンガー−FokI融合を使用する標的化開裂および標的化配列変更についてのパラメータは、本開示の他の箇所に提供される。
【0097】
開裂ドメインまたは開裂片側ドメインは、開裂活性を保持するか、または多量体化(例えば、二量体化)して機能性開裂ドメインを形成する能力を保持するタンパク質の任意の部分であってもよい。
【0098】
例示的なIIS型制限酵素は、譲受人共通の国際公開第WO2007/014275号に記載され、参照によりその全体が本明細書に組み込まれる。
【0099】
開裂特異性を強化するために、開裂ドメインがまた修飾されてもよい。ある実施形態では、開裂片側ドメインの変異体が用いられ、これらの変異体は、開裂片側ドメインのホモ二量体化を最小化または防止する。かかる修飾された開裂片側ドメインの非限定的例は、国際公開第2007/014275号に詳細に開示され、参照によりその全体が本明細書に組み込まれる。また、実施例も参照されたい。ある実施形態では、ホモ二量体化を最小化または防止する改変開裂片側ドメイン(二量体化ドメイン突然変異体とも称される)を含む開裂ドメインは、当業者に既知であり、例えば、米国特許公開第20050064474号および同第20060188987号に記載され、参照によりそれらの全体が本明細書に組み込まれる。FokIの446、447、479、483、484、486、487、490、491、496、498、499、500、531、534、537、および538位におけるアミノ酸残基は全てFokI開裂片側ドメインの二量体化に影響を及ぼすための標的である。例えば、米国特許公開第20050064474号および同第20060188987号、国際特許公開第WO07/139898号、Miller et al.(2007)Nat.Biotechnol.25(7):778−785、およびDoyon et al(2011)Nature Methods 8(1):74−79を参照されたい。
【0100】
偏性ヘテロ二量体を形成するFokIの追加的な改変された開裂片側ドメインを、本明細書に記載されるZFNにおいて使用することもできる。一実施形態では、第1の開裂片側ドメインは、FokIの490および538位におけるアミノ酸残基における突然変異を含み、第2の開裂片側ドメインは、アミノ酸残基486および499における突然変異を含む。
【0101】
ある実施形態では、開裂ドメインは、2つの開裂片側ドメインを含み、それらの両方が、結合ドメイン、第1の開裂片側ドメイン、および第2の開裂片側ドメインを含む単一のポリペプチドの一部である。開裂片側ドメインは、それらがDNAを開裂させるように機能する限り、同じアミノ酸配列または異なるアミノ酸配列を有することができる。
【0102】
一般に、融合タンパク質が、開裂片側ドメインを含む場合、2つの融合タンパク質が、開裂に必要とされる。代替的に、2つの開裂片側ドメインを含む単一のタンパク質を使用することができる。2つの開裂片側ドメインは、同じエンドヌクレアーゼ(またはその機能的断片)に由来し得るか、または各開裂片側ドメインが、異なるエンドヌクレアーゼ(またはその機能的断片)に由来し得る。加えて、2つの融合タンパク質に対する標的部位は、好ましくは、互いに対して、2つの融合タンパク質のそれらの対応する標的部位への結合が、例えば、二量体化によって、開裂片側ドメインが機能性開裂ドメインを形成することを可能にする相互に対する空間的配向で開裂片側ドメインを配置するように、配置される。故に、ある実施形態では、標的部位の近端は、5〜8ヌクレオチド分または15〜18ヌクレオチド分、隔てられる。しかしながら、任意の整数の数のヌクレオチドまたはヌクレオチド対が、2つの標的部位(例えば、2〜50ヌクレオチド以上)の間に介入してもよい。一般に、開裂の点は、標的部位の間にある。
【0103】
融合タンパク質
融合タンパク質(およびそれをコードするポリヌクレオチド)の設計および構築のための方法は、当業者に既知である。例えば、DNA結合ドメイン(例えば、亜鉛フィンガードメイン)および調節または開裂ドメイン(または開裂片側ドメイン)を含む融合タンパク質、ならびにかかる融合タンパク質をコードするポリヌクレオチドの設計および構築のための方法は、譲受人共通の米国特許第6,453,242号および同第6,534,261号、ならびに米国特許出願公開第2007/0134796号および同第2005/0064474号に記載され、参照によりそれらの全体が本明細書に組み込まれる。ある実施形態では、融合タンパク質をコードするポリヌクレオチドが構築される。これらのポリヌクレオチドは、ベクターに挿入することができ、ベクターは、細胞に導入することができる(ポリヌクレオチドを細胞に導入するためのベクターおよび方法に関する追加的な開示については下記を参照されたい)。
【0104】
本明細書に記載される方法のある実施形態では、亜鉛フィンガーヌクレアーゼは、FokI制限酵素からの亜鉛フィンガー結合ドメインおよび開裂片側ドメインを含む融合タンパク質を含み、2つのかかる融合タンパク質は、細胞中で発現される。2つの融合タンパク質の細胞中の発現は、2つのタンパク質の細胞中への送達、1つのタンパク質およびタンパク質の1つをコードする1つの核酸の、細胞中への送達、各々がタンパク質の1つをコードする2つの核酸の、細胞中への送達から、または両方のタンパク質をコードする単一の核酸の、細胞中への送達によって、もたらされ得る。追加的な実施形態では、融合タンパク質は、2つの開裂片側ドメインおよび亜鉛フィンガー結合ドメインを含む、単一のポリペプチド鎖を含む。この場合、単一の融合タンパク質が細胞中で発現され、理論に拘束されることを望むものではないが、開裂片側ドメインの分子内二量体の形成の結果としてDNAを開裂させると考えられる。
【0105】
ある実施形態では、融合タンパク質(例えば、ZFP−FokI融合)の構成要素は、亜鉛フィンガードメインが融合タンパク質のアミノ末端に最近接し、開裂片側ドメインがカルボキシ末端に最近接するように配列される。これは、FokI酵素に由来するもの等の天然に存在する二量体化開裂ドメインにおける開裂ドメインの相対的配向を反映し、ここでDNA結合ドメインは、アミノ末端に最近接し、開裂片側ドメインは、カルボキシ末端に最近接する。これらの実施形態では、機能性ヌクレアーゼを形成するための開裂片側ドメインの二量体化は、融合タンパク質の、反対側のDNA鎖上の部位への結合によって引き起こされ、このうち結合部位の5’末端は、互いに近位にある。
【0106】
追加的な実施形態では、融合タンパク質(例えば、ZFP−FokI融合)の構成要素は、開裂片側ドメインが融合タンパク質のアミノ末端に最近接し、亜鉛フィンガードメインがカルボキシ末端に最近接するように配列される。これらの実施形態では、機能性ヌクレアーゼを形成するための開裂片側ドメインの二量体化は、融合タンパク質の、反対側のDNA鎖上の部位への結合によって引き起こされ、このうち結合部位の3’末端は、互いに近位にある。
【0107】
なおも追加的な実施形態では、第1の融合タンパク質は、融合タンパク質のアミノ末端に最近接した開裂片側ドメイン、およびカルボキシ末端に最近接した亜鉛フィンガードメインを含有し、第2の融合タンパク質は、亜鉛フィンガードメインが融合タンパク質のアミノ末端に最近接し、開裂片側ドメインがカルボキシ末端に最近接するように配列される。これらの実施形態では、両方の融合タンパク質が同じDNA鎖に結合し、このうち第1の融合タンパク質の結合部位は、アミノ末端に最近接した亜鉛フィンガードメインを含有する第2の融合タンパク質の結合部位の5’側に対して位置する、カルボキシ末端に最近接した亜鉛フィンガードメインを含有する。
【0108】
開示される融合タンパク質のある実施形態では、亜鉛フィンガードメインと開裂ドメイン(または開裂片側ドメイン)との間のアミノ酸配列は、「ZCリンカー」と表示される。ZCリンカーは、上述のフィンガー間リンカーと区別されるべきである。開裂を最適化するZCリンカーを得ることに関する詳細については、例えば、米国特許公開第20050064474A1号および同第20030232410号、ならびに国際特許公開第WO05/084190号を参照されたい。
【0109】
一実施形態では、本開示は、表1に示される認識ヘリックスアミノ酸配列のうちの1つ以上を有する亜鉛フィンガータンパク質を含むZFNを提供する。別の実施形態では、表1に示される1つ以上の認識ヘリックスを有するZFPをコードするヌクレオチド配列を含む、ZFP発現ベクターが本明細書に提供される。
【0110】
標的化組込み
開示される方法および組成物を使用して、多重挿入部位が組み込まれている任意の細胞ゲノム中のDNAを開裂することができ、それは適切なZFN対の存在下での、外来性配列の多重挿入部位への安定な標的化組み込みおよび/または外来性配列の切除を促進する。図1および2を参照されたい。
【0111】
ZFN挿入部位が外来性配列の一部として細胞のゲノムに連続して導入される方法もまた、本明細書に記載される。図4および5を参照されたい。例えば、ヘテロ二量体ヌクレアーゼ部位の異なる組み合わせが側面に位置する外来性配列が、ゲノムに挿入される。その後、適切な隣接するZFN部位の1つにおいて開裂するZFN対が、再びヘテロ二量体ヌクレアーゼ部位の異なる組み合わせを含む別の外来性配列の存在下で、細胞に導入される。このプロセスは、外来性配列を挿入するために所望に応じて反復することができる。加えて、適切なZFN対の存在下で、1つ以上の外来性配列がゲノムから切除されてもよい。
【0112】
図6は、外来性配列がマーカー遺伝子および目的の遺伝子を含む、別の実施形態を示す。マーカー遺伝子および目的の遺伝子の両方の側面には、適切な場合、例えば、追加的な遺伝子を挿入するときに、マーカー遺伝子が欠失させられ得るように、異なるZFN結合部位(異なる陰影を有する三角として示される)が位置する。選択可能マーカーの数が限定される植物等の生物において、これはわずか1つにすぎない選択可能マーカー遺伝子の使用を可能にし、目的の遺伝子をスタッキングする可能性を大幅に促進する。ある実施形態では、例えば、相同性指向性DNA修復の効率に依存して、「分裂した」選択可能マーカーが使用されてもよい。分裂した選択可能マーカーを使用するドナーDNA配列の正しい組み込みにより、発現可能な選択可能マーカー遺伝子が作り出される。選択可能マーカーは、組み込まれたDNA配列から切除することができ、したがって再利用することができる。別の実施形態では、除去のための外来性配列には、ゲノムにおいて分裂したマーカー遺伝子の部分配列が隣接する。切除されると、マーカー遺伝子は再構築され、機能性マーカー遺伝子の作成をもたらす。選択可能マーカー切除の使用は、必要とされる選択可能マーカーの数を2つまたは場合によっては1つのみに限定する。
【0113】
本明細書に記載されるように組み込まれた多重挿入部位への標的化組み込みについて、1つ以上のDNA結合ドメイン(例えば、ZFP)は、所定の開裂部位における、またはそれに近接した標的部位に結合するように改変され、改変DNA結合ドメインおよび開裂ドメインを含む融合タンパク質が細胞中で発現される。融合タンパク質のDNA結合(例えば、亜鉛フィンガー)部分が標的部位に結合すると、DNAは、好ましくは二本鎖切断を介して、開裂ドメインによって標的部位に近隣して開裂される。
【0114】
多重挿入部位における二本鎖切断の存在は、相同組み換えを介した外来性配列の組み込みを促進する。ある実施形態では、多重挿入部位への挿入対象の外来性配列を含むポリヌクレオチドは、相同組み換えを促進するために、多重挿入部位ポリヌクレオチドおよび/または周辺のゲノムと相同性の1つ以上の領域を含むであろう。ドナー配列とゲノム配列との間の、およそ25、50、100、200、500、750、1,000、1,500、2,000ヌクレオチドまたはそれを超える配列相同性(または10〜2,000ヌクレオチドもしくはそれを超える任意の整数値)がその間の相同組み換えを支持するであろう。ある実施形態では、相同性アームは、1,000塩基対長未満である。他の実施形態では、相同性アームは、750塩基対長未満である。また、米国仮特許出願第61/124,047号も参照されたく、それは参照により本明細書に組み込まれる。ドナー分子(外来性配列)は、細胞クロマチンに対して相同性の、複数の不連続の領域を含有し得る。例えば、目的の領域に通常存在しない配列の標的化挿入のために、該配列は、ドナー核酸分子に存在し、目的の領域における遺伝子配列に対して相同性の領域が側面に位置する可能性がある。
【0115】
目的の任意の配列(外来性配列)は、本明細書に記載されるように多重挿入部位に導入、またはそこから切除することができる。例示的な外来性配列には、任意のポリペプチドコード配列(例えば、cDNA)、プロモーター、エンハンサー、および他の調節配列(例えば、干渉RNA配列、shRNA発現カセット、エピトープタグ、マーカー遺伝子、開裂酵素認識部位および様々な種類の発現構築物が含まれるが、これらに限定されない。かかる配列は、標準的な分子生物学技術(クローニング、合成等)を使用して容易に得ることができ、かつ/または市販されている。外来性配列は、融合タンパク質の発現の前に、それと同時に、またはその後に細胞に導入することができる。
【0116】
ドナーポリヌクレオチドは、DNAまたはRNAであっても、一本鎖または二本鎖のいずれか一方であってもよく、線状または環状形態で細胞に導入されてもよい。線状形態で導入される場合、ドナー配列の末端部は、当業者に既知の方法によって保護(例えば、エキソヌクレアーゼ分解から)することができる。例えば、1つ以上のジデオキシヌクレオチド残基が線状分子の3’末端に付加され、かつ/または自己相補性オリゴヌクレオチドは、1つまたは両方の末端部に結合される。例えば、Chang et al.(1987)Proc.Natl.Acad.Sci.USA 84:4959−4963、Nehls et al.(1996)Science 272:886−889を参照されたい。外来性ポリヌクレオチドを分解から保護するための追加的な方法には、末端アミノ基の付加、ならびに例えば、ホスホロチオエート、ホスホロアミデート、およびO−メチルリボースまたはデオキシリボース残基等の修飾されたヌクレオチド間結合の使用が含まれるが、これらに限定されない。
【0117】
ポリヌクレオチドは、例えば、複製起点、プロモーター、および抗体抵抗性をコードする遺伝子等の追加的な配列を有するベクター分子の一部として細胞に導入することができる。更に、ドナーポリヌクレオチドは、ネイキッド核酸として、ナノ粒子、リポソーム、もしくはポロキサマー等の薬剤と複合体化した核酸として、導入することができるか、または細菌もしくはウイルスによって植物細胞に送達することができる(例えば、アグロバクテリウム、リゾビウム属NGR234、シノリゾビウム・メリロティ(Sinorhizoboium meliloti)、メソリゾビウム・ロティ、タバコモザイクウイルス、ジャガイモXウイルス、カリフラワーモザイクウイルス、およびカッサバ葉脈モザイクウイルス。例えば、Chung et al.(2006)Trends Plant Sci.11(1):1−4を参照されたい。
【0118】
上に詳述されるように、各々が亜鉛フィンガー結合ドメインおよび開裂片側ドメインを含む、2つの融合タンパク質(ホモ二量体またはヘテロ二量体)に対する多重挿入部位上の結合部位は、他の結合部位に最近接した各結合部位の端から測定して5〜8または15〜18ヌクレオチド離れて位置し得、開裂は、結合部位の間で生じる。開裂が単一の部位においてまたは結合部位の間の多重部位において生じるかは、開裂されたゲノム配列がドナー配列によって置換されるため、重要でない。故に、標的化組み換えによる単一のヌクレオチド対の配列の効率的変更のために、結合部位の間の領域の中点は、そのヌクレオチド対の10,000ヌクレオチド内、好ましくは1,000ヌクレオチド、または500ヌクレオチド、または200ヌクレオチド、または100ヌクレオチド、または50ヌクレオチド、または20ヌクレオチド、または10ヌクレオチド、または5ヌクレオチド、または2ヌクレオチド、または1ヌクレオチド内であるか、または目的のヌクレオチド対における。
【0119】
例えば、RAD54エピスタシスグループ(例えば、AtRad54、AtRad51)の植物遺伝子、遺伝子の産物が前述の遺伝子産物と相互作用する遺伝子等の相同組み換えに関与する遺伝子の発現を活性化するための、追加的なZFP機能性ドメイン融合の使用を含むが、これらに限定されない、標的化組み換えのレベルを強化し得る方法および組成物もまた、提供される。例えば、Klutstein et al.Genetics.2008 Apr;178(4):2389−97を参照されたい。
【0120】
同様に、ZFP機能性ドメイン融合は、非相同末端結合(例えば、Ku70/80、XRCC4、ポリ(ADPリボース)ポリメラーゼ、DNAリガーゼ4)に関与する遺伝子の発現を抑制するために、本明細書に開示される方法および組成物と組み合わせて使用することができる。例えば、Riha et al.(2002)EMBO 21:2819−2826、Freisner et al.(2003)Plant J.34:427−440、Chen et al.(1994)European Journal of Biochemistry 224:135−142を参照されたい。亜鉛フィンガー結合ドメインと機能性ドメインとの間の融合を使用した、遺伝子発現の活性化および抑制のための方法は、例えば、譲受人共通の米国特許第6,534,261号、同第6,824,978号、および同第6,933,113号に開示される。追加的な抑制方法には、アンチセンスオリゴヌクレオチドおよび/または抑制対象の遺伝子の配列を標的とする低分子干渉RNA(siRNAまたはRNAi)もしくはshRNAの使用が含まれる。
【0121】
亜鉛フィンガー/ヌクレアーゼ融合分子およびドナーDNA分子を含む細胞中の標的化組み換えの効率における更なる増加は、相同性駆動型修復プロセスが最大限に活性である細胞周期のG2期において細胞を遮断することによって達成される。かかる停止は、幾つかの方法で達成することができる。例えば、細胞は、G2期の細胞を停止させるために、例えば、細胞周期進行に影響を及ぼす薬物、化合物および/または小分子で処置することができる。この種類の例示的な分子には、微小管重合に影響を及ぼす化合物(例えば、ビンブラスチン、ノコダゾール、タキソール)、DNAと相互作用する化合物(例えば、シス−プラチナム(II)ジアミンジクロリド、シスプラチン、ドキソルビシン)、および/またはDNA合成に影響を及ぼす化合物(例えば、チミジン、ヒドロキシ尿素、L−ミモシン、エトポシド、5−フルオロウラシル)が含まれるが、これらに限定されない。組み換え効率における追加的な増加は、クロマチン構造を変化させて、ゲノムDNAを細胞組み換え機構へとより到達可能にする、ヒストンデアセチラーゼ(HDAC)阻害剤(例えば、酪酸ナトリウム、トリコスタチンA)の使用によって達成される。
【0122】
細胞周期停止のための追加的な方法には、例えば、タンパク質をコードするcDNAを細胞中に導入することによって、または細胞中にタンパク質をコードする遺伝子の発現を活性化する改変されたZFPを導入することによって、CDK細胞周期キナーゼの活性を阻害するタンパク質を過剰発現させることが含まれる。細胞周期停止はまた、例えば、RNAi法を使用して(例えば、米国特許第6,506,559号)サイクリンおよびCDKの活性を阻害することによって、または細胞中に、例えば、サイクリンおよび/またはCDK遺伝子等の、細胞周期進行に関与する1つ以上の遺伝子の発現を抑制する改変されたZFPを導入することによっても達成される。遺伝子発現の調節のために改変された亜鉛フィンガータンパク質の合成のための方法については、例えば、譲受人共通の米国特許第6,534,261号を参照されたい。
【0123】
代替的に、ある場合には、ドナーポリヌクレオチドの不在下で(好ましくはSまたはG2期において)標的化開裂が実施され、組み換えが相同染色体の間で生じる。
【0124】
発現ベクター
本明細書に記載される1つ以上の融合タンパク質(例えば、ZFN)をコードする核酸は、複製および/または発現のための原核細胞または真核細胞への形質転換のために、ベクターにクローン化することができる。ベクターは、原核性ベクター、例えば、プラスミド、またはシャトルベクター、昆虫ベクター、または真核性ベクターであり得る。融合タンパク質をコードする核酸もまた、細胞への投与のために発現ベクターにクローン化することができる。
【0125】
融合タンパク質(例えば、ZFN)を発現させるために、融合タンパク質をコードする配列は、典型的には、転写を導くためのプロモーターを含有する発現ベクターにサブクローン化される。好適な細菌および真核性プロモーターは、当該技術分野において周知であり、例えば、Sambrook et al.,Molecular Cloning,A Laboratory Manual(2nd ed.1989;3rd ed.,2001)、Kriegler,Gene Transfer and Expression:A Laboratory Manual(1990);and Current Protocols in Molecular Biologyに記載される(上記のAusubelらを参照されたい。ZFPを発現させるための細菌発現系は、例えば、大腸菌、バチルス属、およびサルモネラで入手可能である(Palva et al.,Gene 22:229−235(1983))。かかる発現系のためのキットが市販されている。哺乳類細胞、酵母、および昆虫細胞のための真核性発現系は、当業者に周知であり、また市販されている。
【0126】
融合タンパク質コード核酸の発現を導くために使用されるプロモーターは、特定の用途に依存する。例えば、宿主細胞に適合させられた強力な構成的プロモーターは、典型的には融合タンパク質の発現および精製のために使用される。
【0127】
対照的に、植物遺伝子の調節のために融合タンパク質がインビボ投与されるとき(下記の「植物細胞への核酸送達」節を参照されたい)、融合タンパク質の特定の使用に依存して、構成的プロモーター、調節されたプロモーター(例えば、発達中に、組織または細胞型によって、または環境によって)、または誘導性プロモーターのいずれかが、使用される。植物プロモーターの非限定的例としては、シロイヌナズナ・ユビキチン−3(ubi−3)(Callis,et al.,1990,J.Biol.Chem.265−12486−12493)、アグロバクター・ツメファシエンス(Δmas)(Petolinoら、米国特許第6,730,824号)、および/またはカッサバ葉脈モザイクウイルス(CsVMV)(Verdaguer et al.,1996,Plant Molecular Biology 31:1129−1139)に由来するプロモーター配列が挙げられる。また、実施例も参照されたい。
【0128】
プロモーターに加えて、発現ベクターは、典型的には、原核性または真核性のいずれかの宿主細胞中での核酸の発現に必要とされる全ての追加的な要素を含有する、転写単位または発現カセットを含有する。典型的な発現カセットは故に、例えば、融合タンパク質をコードする核酸配列に作動可能に連結されたプロモーター、および例えば、転写物の効率的なポリアデニル化、転写終結、リボソーム結合部位、または翻訳終結のために必要とされるシグナルを含有する。カセットの追加的な要素には、例えば、エンハンサー、異種スプライシングシグナル、および/または核局在化シグナル(NLS)が含まれてもよい。
【0129】
遺伝子情報を細胞中に移入するために使用される特定の発現ベクターは、融合タンパク質の意図される使用、例えば、植物、動物、細菌、真菌、原虫等における発現に関連して選択される(後述の発現ベクターを参照されたい)。標準的な細菌および動物発現ベクターは、当該技術分野において既知であり、例えば、米国特許公開第20050064474A1号、ならびに国際特許公開第WO05/084190号、同第WO05/014791号、および同第WO03/080809号に詳述される。
【0130】
標準的なトランスフェクション方法を使用して、大量のタンパク質を発現する細菌、哺乳類、酵母、または昆虫細胞株を産生することができ、それは次いで標準技術を使用して精製することができる(例えば、Colley et al.,J.Biol.Chem.264:17619−17622(1989)、Guide to Protein Purification,in Methods in Enzymology,vol.182(Deutscher,ed.,1990)を参照されたい)。真核細胞および原核細胞の形質転換は、標準技術に従って実施される(例えば、Morrison,J.Bact.132:349−351(1977)、Clark−Curtiss&Curtiss,Methods in Enzymology 101:347−362(Wu et al.,eds.,1983)を参照されたい。
【0131】
外来ヌクレオチド配列をかかる宿主細胞に導入するための周知の手順のうちのいずれが使用されてもよい。これらには、リン酸カルシウムトランスフェクション、ポリブレン、プロトプラスト融合、エレクトロポレーション、超音波法(例えば、ソノポレーション)、リポソーム、マイクロインジェクション、ネイキッドDNA、プラスミドベクター、エピソーム型および組み込み型の両方のウイルスベクターの使用、ならびにクローン化ゲノムDNA、cDNA、合成DNA、または他の外来遺伝材料を、宿主細胞に導入するための他の周知の方法のいずれの使用も含まれる(例えば、上記のSambrookらを参照されたい)。使用される特定の遺伝子改変手順が、少なくとも1つの遺伝子を、選択のタンパク質を発現することができる宿主細胞に首尾よく導入できれば十分である。
【0132】
植物細胞への核酸送達
上記のように、DNA構築物は、多様な従来の技術によって所望の植物宿主に(例えば、そのゲノムに)導入されてもよい。かかる技術の概説については、例えば、Weissbach&Weissbach Methods for Plant Molecular Biology(1988,Academic Press,N.Y.)Section VIII,pp.421−463、およびGrierson&Corey,Plant Molecular Biology(1988,2d Ed.),Blackie,London,Ch.7−9を参照されたい。
【0133】
例えば、DNA構築物は、植物細胞プロトプラストのエレクトロポレーションおよびマイクロインジェクション等の技術を使用して植物細胞のゲノムDNAに直接導入されてもよく、またはDNA構築物は、DNAマイクロプロジェクタイルボンバードメント等のバイオリスティック法を使用して植物組織に直接導入することができる(例えば、Klein et al.(1987)Nature 327:70−73を参照されたい)。代替的に、DNA構築物は、ナノ粒子形質転換を介して植物細胞に導入することができる(例えば、米国特許出願第12/245,685号を参照されたく、それは参照によりその全体が本明細書に組み込まれる)。代替的に、DNA構築物は、好適なT−DNA境界/隣接領域と組み合わされ、従来のアグロバクテリウム・ツメファシエンス宿主ベクターに導入されてもよい。アグロバクテリウム・ツメファシエンス媒介型形質転換技術は、無害化(disarming)およびバイナリーベクターの使用を含めて、科学文献に詳細に記載される。例えば、Horsch et al.(1984)Science 233:496−498、およびFraley et al.(1983)Proc.Nat’l.Acad.Sci.USA 80:4803を参照されたい。
【0134】
加えて、遺伝子移入は、リゾビウム属NGR234、シノリゾビウム・メリロティ(Sinorhizoboium meliloti)、メソリゾビウム・ロティ、ジャガイモXウイルス、カリフラワーモザイクウイルス、およびカッサバ葉脈モザイクウイルスおよび/またはタバコモザイクウイルス等の、非アグロバクテリウム細菌またはウイルスを使用して達成してもよく、例えば、Chung et al.(2006)Trends Plant Sci.11(1):1−4を参照されたい。
【0135】
アグロバクテリウム・ツメファシエンス宿主のビルレンス機能は、バイナリーT DNAベクター(Bevan(1984)Nuc.Acid Res.12:8711−8721)または共培養手順(Horsch et al.(1985)Science 227:1229−1231)を使用して、細胞をこの細菌に感染させるとき、構築物および隣接マーカーを含有するT鎖の、植物細胞DNAへの挿入を導く。一般に、アグロバクテリウム形質転換系は、双子葉植物を改変するために使用される(Bevan et al.(1982)Ann.Rev.Genet 16:357−384、Rogers et al.(1986)Methods Enzymol.118:627−641)。アグロバクテリウム形質転換系は、単子葉植物および植物細胞にDNAを形質転換ならびに移入するためにも使用することができる。米国特許第5、591,616号、Hernalsteen et al.(1984)EMBO J 3:3039−3041、Hooykass−Van Slogteren et al.(1984)Nature 311:763−764、Grimsley et al.(1987)Nature 325:1677−179、Boulton et al.(1989)Plant Mol.Biol.12:31−40、およびGould et al.(1991)Plant Physiol.95:426−434を参照されたい。
【0136】
代替的な遺伝子移入および形質転換方法には、カルシウム、ポリエチレングリコール(PEG)、またはエレクトロポレーション媒介型ネイキッドDNAの取り込みを通じたプロトプラスト形質転換(Paszkowski et al.(1984)EMBO J 3:2717−2722、Potrykus et al.(1985)Molec.Gen.Genet.199:169−177、Fromm et al.(1985)Proc.Nat.Acad.Sci.USA 82:5824−5828、およびShimamoto(1989)Nature 338:274−276を参照されたい)、および植物組織のエレクトロポレーション(D’Halluin et al.(1992)Plant Cell 4:1495−1505)が含まれるが、これらに限定されない。植物細胞形質転換のための追加的な方法には、マイクロインジェクション、炭化ケイ素媒介型DNA取り込み(Kaeppler et al.(1990)Plant Cell Reporter 9:415−418)、およびマイクロプロジェクタイルボンバードメント(Klein et al.(1988)Proc.Nat.Acad.Sci.USA 85:4305−4309、およびGordon−Kamm et al.(1990)Plant Cell 2:603−618を参照されたい)が含まれる。
【0137】
開示される方法および組成物を使用して、外来配列を、植物細胞のゲノムに挿入されている多重挿入部位に挿入することができる。植物ゲノムに導入された導入遺伝子の発現は、その組み込み部位に決定的に依存するため、これは有用である。例えば、除草剤耐性、虫害抵抗性、栄養素、抗体または治療用分子をコードする遺伝子は、標的化組み換えによって、それらの発現に好ましい植物ゲノムの領域に挿入することができる。
【0138】
上記の形質転換技術のいずれかによって産生される形質転換された植物細胞を培養して、形質転換された遺伝子型を有し、故に所望の表現型を有する、全植物体を再生することができる。かかる再生技術は、組織培養成長培地中のある種の植物ホルモンの操作に依存し、典型的には、所望のヌクレオチド配列と一緒に導入されている殺生物剤および/または除草剤マーカーに依存する。培養プロトプラストからの植物再生は、Evans,et al.,”Protoplasts Isolation and Culture” in Handbook of Plant Cell Culture,pp.124−176、Macmillian Publishing Company,New York,1983、およびBinding,Regeneration of Plants,Plant Protoplasts,pp.21−73,CRC Press,Boca Raton,1985に記載される。再生はまた、植物のカルス、外植片、器官、花粉、胚、またはそれらの一部から得ることもできる。かかる再生技法は、一般に、Klee et al.(1987)Ann.Rev.of Plant Phys.38:467−486に記載される。
【0139】
植物細胞に導入された核酸を使用して、本質的にいずれの植物にも、所望の形質を付与することができる。本開示の核酸構築物および上述の様々な形質転換方法を使用して、本明細書に記載される所望の生理学的および作物栽培学的特徴のために、多種多様な植物および植物細胞系が改変されてもよい。好ましい実施形態では、改変用の標的植物および植物細胞には、穀物作物(例えばコムギ、トウモロコシ、イネ、アワ(millet)、オオムギ)、果実作物(例えばトマト、リンゴ、ナシ、イチゴ、オレンジ)、飼料作物(例えばアルファルファ)、根菜作物(例えばニンジン、ジャガイモ、サトウダイコン、ヤムイモ)、葉菜作物(例えばレタス、ホウレンソウ);顕花植物(例えばペチュニア、バラ、キク)、針葉樹およびマツ(例えばマツ、モミ、トウヒ)、ファイトレメディエーションに使用される植物(例えば重金属蓄積植物);油料作物(例えば、ヒマワリ、ナタネ)および実験目的のために使用される植物(例えば、アラビドプシス)を含む作物等の単子葉植物および双子葉植物が含まれるが、これらに限定されない。故に、開示される方法および組成物は、例えば、限定するわけではないが、アスパラガス属、カラスムギ属、アブラナ属、ミカン属、スイカ属、トウガラシ属、カボチャ属、ニンジン属、ダイズ属、ワタ属、オオムギ属、アキノノゲシ属、トマト属、リンゴ属、マニホット属、タバコ属、イネ属、ワニナシ属、エンドウ属、ナシ属、サクラ属、ダイコン属、ライムギ属、ナス属、モロコシ属、コムギ属、ブドウ属、ササゲ属、およびトウモロコシ属に属する種を含むが、これらに限定されない、広範囲にわたる植物に使用することができる。
【0140】
当業者であれば、現カセットがトランスジェニック植物に安定に組み込まれ、作動可能であることが確認された後、それを有性交配によって他の植物体に導入できることを認識するであろう。幾つかの標準的育種技術のうちのいずれも、交配させる種に依存して使用することができる。
【0141】
形質転換された植物細胞、カルス、組織または植物体は、形質転換DNA上に存在するマーカー遺伝子によってコードされる形質に関して、改変された植物材料を選択またはスクリーニングすることによって特定し、単離することができる。例えば、選択は、形質転換遺伝子構築物によって耐性を付与される、阻害量の抗生物質または除草剤を含有する培地上で、改変された植物材料を成長させることよって行うことができる。更に、形質転換された植物および植物細胞はまた、組み換え核酸構築物上に存在し得る任意の可視マーカー遺伝子(例えば、β−グルクロニダーゼ、ルシフェラーゼ、BまたはC1遺伝子)の活性についてスクリーニングすることによって特定することもできる。かかる選択およびスクリーニング手法は、当業者に周知である。
【0142】
また、物理的および生化学的方法を使用して、挿入された遺伝子構築物を含有する植物形質転換体または植物細胞形質転換体を特定してもよい。これらの方法には、1)組み換えDNA挿入を検出し、その構造を決定するためのサザン分析またはPCR増幅、2)遺伝子構築物のRNA転写物を検出し、検査するためのノーザンブロット、S1 RNase保護、プライマー伸長、または逆転写酵素−PCR増幅、3)かかる遺伝子産物が遺伝子構築物によってコードされる場合、酵素活性またはリボザイム活性を検出するための酵素アッセイ、4)遺伝子構築物産物がタンパク質である場合、タンパク質ゲル電気泳動、ウェスタンブロット技術、免疫沈降、または酵素結合免疫測定法(ELISA)が含まれるが、これらに限定されない。また、インサイツハイブリダイゼーション、酵素染色、および免疫染色等の追加的な技術を使用して、特定の植物器官または植物組織における組み換え構築物の存在または発現を検出することもできる。全てのこれらのアッセイを行うための方法は、当業者に周知である。
【0143】
本明細書に開示される方法を使用する遺伝子操作の効果は、例えば、目的の組織から単離されるRNA(例えば、mRNA)のノーザンブロットよって観察することができる。典型的には、mRNAが存在するか、またはmRNAの量が増加している場合、対応する導入遺伝子が発現されていると想定することができる。遺伝子および/またはコードされたポリペプチド活性を測定する他の方法を使用することができる。使用される基質および反応産物または副産物の増減を検出する方法に依存して、異なる種類の酵素アッセイを使用することができる。加えて、発現されるポリペプチドのレベルは、免疫化学的に、すなわち、ELISA、RIA、EIA、および電気泳動検出アッセイ(染色またはウェスタンブロッティングのいずれかによる)等による当業者に周知の他の抗体に基づくアッセイで測定することができる。1つの非限定的例として、ELISAアッセイを使用するAAD−1およびPATタンパク質の検出は、米国特許出願第11/587,893号に記載され、この参考文献は、本明細書での参照によりその全体が本明細書に組み込まれる。導入遺伝子は、植物体の一部の組織において、または一部の発達段階で、選択的に発現させてもよく、または導入遺伝子は、実質的に全ての植物組織において、実質的にそのライフサイクル全体にわたって発現させてもよい。しかしながら、任意の組み合わせ発現(combinatorial expression)様式もまた適用可能である。
【0144】
本開示はまた、上述の遺伝子導入植物の種子も包含し、ここで種子は、導入遺伝子または遺伝子構築物を有する。本開示は更に、上述の遺伝子導入植物の子孫、クローン、細胞株もしくは細胞を包含し、該子孫、クローン、細胞株もしくは細胞は、導入遺伝子または遺伝子構築物を有する。
【0145】
融合タンパク質(例えば、ZFN)および融合タンパク質をコードする発現ベクターは、遺伝子調節、標的化開裂、および/または組み換えのために、植物に直接投与することができる。ある実施形態では、植物は、多重パラロガス標的遺伝子を含有する。故に、1つ以上の異なる融合タンパク質または融合タンパク質をコードする発現ベクターは、植物におけるこれらのパラロガス遺伝子(例えば、Zp15、PCT特許公開第WO2010077319号を参照されたい)遺伝子のうちの1つ以上を標的とするために、植物に投与されてもよい。
【0146】
有効量の投与は、融合タンパク質を導入して、処置対象の植物細胞と最終接触させるために通常使用される経路のうちのいずれによっても行われる。ZFPは、好ましくは許容される担体と共に、任意の好適な様態で投与される。かかるモジュレータを投与する好適な方法が利用可能であり、かつ当業者に周知であり、1つ以上の経路を使用して特定の組成物を投与することができるが、特定の経路は、しばしば、別の経路よりも即時かつ有効な反応を提供することができる。
【0147】
また、担体が使用されてもよく、それは一部には、投与されている特定の組成物によって、ならびに組成物を投与するために使用される特定の方法によって決定される。したがって、多種多様の好適な担体の製剤が利用可能である。
【0148】
哺乳類細胞への送達
本明細書に記載されるZFNは、任意の好適な手段によって、例えば、ZFN mRNAの注入によって、標的哺乳類細胞に送達されてもよい。Hammerschmidt et al.(1999)Methods Cell Biol.59:87−115を参照されたい。
【0149】
亜鉛フィンガーを含むタンパク質を送達する方法は、例えば、米国特許第6,453,242号、同第6,503,717号、同第6,534,261号、同第6,599,692号、同第6,607,882号、同第6,689,558号、同第6,824,978号、同第6,933,113号、同第6,979,539号、同第7,013,219号、および同第7,163,824号に記載され、これらの全ての開示は、参照によりそれらの全体が本明細書に組み込まれる。
【0150】
本明細書に記載されるZFNはまた、ZFNのうちの1つ以上をコードする配列を含有するベクターを使用して送達されてもよい。プラスミドベクター、レトロウイルスベクター、レンチウイルスベクター、アデノウイルスベクター、ポックスウイルスベクター;ヘルペスウイルスベクター、およびアデノ関連ウイルスベクター等を含むが、これらに限定されない、任意のベクター系が使用されてもよい。また、米国特許第6,534,261号、同第6,607,882号、同第6,824,978号、同第6,933,113号、同第6,979,539号、同第7,013,219号、および同第7,163,824号も参照されたく、参照によりそれらの全体が本明細書に組み込まれる。更に、これらのベクターのうちのいずれも、ZFNをコードする1つ以上の配列を含み得ることは明白であろう。故に、ZFNの1つ以上の対が細胞に導入される時、ZFNは、同じベクター上または異なるベクター上で担持され得る。複数のベクターが使用されるとき、各ベクターは、1つまたは複数のZFNをコードする配列を含み得る。
【0151】
従来のウイルスおよび非ウイルスに基づく遺伝子移入方法を使用して、改変されたZFPをコードする核酸を、哺乳類細胞に導入することができる。また、かかる方法を使用して、ZFPをコードする核酸を、哺乳類細胞にインビトロ投与することもできる。ある実施形態では、ZFPをコードする核酸は、インビボまたはエクスビボ用途のために投与される。
【0152】
非ウイルスベクター送達系には、エレクトロポレーション、リポフェクション、マイクロインジェクション、バイオリスティック、ビロソーム、リポソーム、免疫リポソーム、ポリカチオン、または脂質:核酸共役体、ネイキッドDNA、人工ビリオン、および薬剤により強化されたDNAの取り込みが含まれる。例えば、Sonitron 2000 system(Rich−Mar)を使用するソノポレーションも、核酸の送達に使用することができる。ウイルスベクター送達系には、細胞への送達後にエピソームゲノムまたは組み込まれたゲノムのいずれかを有する、DNAおよびRNAウイルスが含まれる。追加的な例示的核酸送達系には、Amaxa Biosystems(Cologne,Germany)、Maxcyte,Inc.(Rockville,Maryland)、BTX Molecular Delivery Systems(Holliston,MA)、およびCopernicus Therapeutics Inc,(例えば、米国特許第6008336号を参照されたい)によって提供されるものが含まれる。リポフェクションは、例えば、米国特許第5,049,386号、米国特許第4,946,787号、および米国特許第4,897,355号に記載され、リポフェクション試薬は市販されている(例えば、TRANSFECTAM(商標)およびLIPOFECTIN(商標))。ポリヌクレオチドの効率的な受容体認識リポフェクションに好適なカチオン性脂肪および中性脂肪には、Felgner、国際公開第91/17424号、国際公開第91/16024号のものが含まれる。送達は、細胞(エクスビボ投与)または標的組織(インビボ投与)に行うことができる。免疫脂質複合体等の標的リポソームを含む脂質:核酸複合体の調製は、当業者に周知である(例えば、Crystal,Science 270:404−410(1995)、Blaese et al.,Cancer Gene Ther.2:291−297(1995)、Behr et al.,Bioconjugate Chem.5:382−389(1994)、Remy et al.,Bioconjugate Chem.5:647−654(1994)、Gao et al.,Gene Therapy 2:710−722(1995)、Ahmad et al.,Cancer Res.52:4817−4820(1992)、米国特許第4,186,183号、同第4,217,344号、同第4,235,871号、同第4,261,975号、同第4,485,054号、同第4,501,728号、同第4,774,085号、同第4,837,028号、および同第4,946,787号を参照されたい)。
【0153】
上記のように、開示される方法および組成物は、任意の種類の哺乳類細胞において使用することができる。タンパク質(例えば、ZFP)、それをコードするヌクレオチド、ならびに本明細書に記載されるタンパク質および/またはポリヌクレオチドを含む組成物は、任意の好適な手段によって標的細胞に送達され得る。好適な細胞には、真核細胞および/または細胞株ならびに原核細胞および/または細胞株が含まれるが、これらに限定されない。かかる細胞、またはかかる細胞から生成された細胞株の非制限的な例には、COS、CHO(例えば、CHO−S、CHO−K1、CHO−DG44、CHO−DUXB11、CHO−DUKX、CHOK1SV)、VERO、MDCK、WI38、V79、B14AF28−G3、BHK、HaK、NS0、SP2/0−Ag14、HeLa、HEK293(例えば、HEK293−F、HEK293−H、HEK293−T)、およびperC6細胞、ならびにツマジロクサヨトウ(Sf)等の昆虫細胞、またはサッカロミセス、ピチア、およびシゾサッカロミセス等の真菌細胞が挙げられるが、これらに限定されない。ある特定の実施形態では、細胞株は、CHO−K1、MDCK、またはHEK293細胞株である。好適な初代細胞には、抹消血液単核細胞(PBMC)、およびCD4+T細胞またはCD8+T細胞等であるが、これらに限定されない他の血液細胞サブセットが含まれる。好適な細胞にはまた、例として、胚性幹細胞、人口多能性幹細胞、造血幹細胞、神経幹細胞、および間葉系幹細胞等の幹細胞も含まれる。
【実施例】
【0154】
実施例1:プラスミド設計
実施例1.1:eZFN結合部位
【0155】
8つの改変亜鉛フィンガーヌクレアーゼ(eZFN)結合部位(CL:AR−配列番号1、RL:PR−配列番号2、AL:PR−配列番号3、PL:AR−配列番号4、CL:RR−配列番号5、RL:CR−配列番号6、CL:PR−配列番号7、RL:AR−配列番号8)を、eZFN結合部位の各々に対して固有の、隣接するPCRプライマー部位を有する単一のDNA断片(多重eZFN結合部位)中に組み合わせた。加えて、他のeZFN結合部位を設計し、酵母において高レベルで開裂することを示したが(例えば、米国特許公開第2009/0111119号を参照されたい)、それは次のものを含む:PL:RR−配列番号9、AL:RR−配列番号10、AL:CR−配列番号11、PL:CR−配列番号12、ならびにホモ二量体eZFNのRR:RR−配列番号13、RL:RL−配列番号14、PR:PR−配列番号15、PL:PL−配列番号16、CL:CL−配列番号17、CR:CR−配列番号18、AR:AR−配列番号19、およびAL:AL−配列番号20。「CL」および「CR」は、それぞれ、8266および8196と指定される、CCR5受容体に対する「左」および「右」手亜鉛フィンガー設計を指し、それは米国特許出願第2008/0159996号に示される配列を有し、そこで示される標的部位に結合する。「AL」および「AR」は、それぞれ、15556および15590と指定される、AAVS1遺伝子座に対する「左」および「右」手亜鉛フィンガー設計を指し、米国特許出願第2008/0299580号に示される認識ヘリックス配列を有し、そこで示される標的部位に結合する。「PL」および「PR」設計ならびに「RL」および「RR」設計に対する認識ヘリックス配列および標的部位は、下記の表1および2に列挙される。PLおよびPRの両方は、ヒトPRMT1遺伝子に特異的なZFNに対する「左」および「右」手亜鉛フィンガー設計を指す一方で、「RL」および「RR」は、マウスRosa26遺伝子座に特異的なZFNに対する「左」および「右」手亜鉛フィンガー設計を指す。
【0156】
これらの標的部位のうちのいずれも、バイオインフォマティクス分析によって計測するとき、トウモロコシゲノム中に存在しない。PCRプライマー部位を、eZFNによる染色体局在性DNA断片の二本鎖開裂からもたらされるNHEJの評価のために含めた。
【0157】
【表1】
【0158】
【表2】
【0159】
Att部位を、合成したDNA断片中に含めて、断片を、TOPOクローニング(Invitrogen,Carlsbad,CA)を使用して、プラスミド中にクローン化した。Gateway LR CLONASE(商標)(Invitrogen)反応を使用して、この断片をpDAB101834およびpDAB101849に移入した。これらのベクターは、それぞれタバコおよびトウモロコシに好適な選択可能マーカーを含有する。pDAB101834は、カッサバ葉脈モザイクウイルスプロモーター(CsVMV、カッサバ葉脈モザイクウイルスに由来するプロモーターおよび5’非翻訳領域、Verdaguer et al.,(1996)Plant Molecular Biology,31(6)1129−1139)、ホスフィノスリシンアセチルトランスフェラーゼ遺伝子(PAT、Wohlleben et al.,(1988)Gene 70(1),25−37)、およびAtuORF1 3’ UTR(アグロバクテリウム・ツメファシエンスpTi15955のオープンリーディングフレーム1(ORF1)の転写終結因子およびポリアデニル化部位を含む3’非翻訳領域(UTR)、Barker et al.,(1983)Plant Molecular Biology,2(6),335−50)からなる。トウモロコシpDAB101849ベクターは、イネ(rice)アクチン1遺伝子プロモーター(OsAct1、イネ(Oryza sativa)アクチン1(Act1)遺伝子に由来するプロモーター、5’非翻訳領域(UTR)、およびイントロン、McElroy et al.,(1990)Plant Cell 2(2):163−71)およびZmLip 3’ UTR(トウモロコシLIP遺伝子の転写終結因子およびポリアデニル化部位を含む3’非翻訳領域(UTR)、GenBank受入L35913)を含む選択可能マーカーカセットを含有する。
【0160】
結果として得られたタバコベクター、pDAB105900(図7)を、エレクトロポレーションを使用してアグロバクテリウム・ツメファシエンスに移入した。制限酵素検証後、アグロバクテリウムを、使用するまでグリセロールストックとして保管した。トウモロコシベクター、pDAB105908(図8)を一括にして、製造業者のプロトコルに従ってQiagen QIAfilter Plasmid Giga kit(Qiagen,Valencia,CA)を使用して精製した。
【0161】
実施例1.2:eZFNを発現させるためのベクター
標準(C2H2)主鎖または非標準(C3H)主鎖のいずれかにおいて適切な認識ヘリックスを発現するZFNベクターを、本質的に、米国特許出願第2008/0182332号および同第2008/0159996号に記載されるように調製した。
【0162】
ZFNの機能を、酵母ZFNスクリーニング系に挿入した、実施例1.1に記載されるeZFN多重挿入部位に対して試験した(米国特許公開第2009/0111119号を参照されたい)。試験した全てのZFN対は、酵母系において活性であった。
【0163】
8つのeZFNを、植物細胞中の発現に必要な調節配列を含有するベクター中にクローン化した。構築用に配備したクローニング戦略は、米国特許出願第2009/0111188A1号および同第20100199389号に本質的に記載される通りである。図9および10は、一般化したeZFN発現カセットの概略図を示す。
【0164】
実施例2:トウモロコシにおけるeZFNの評価
実施例2.1:WHISKERS(商標)媒介型DNA送達
トウモロコシの胚形成Hi−II細胞培養物を産生し、内部への組み込みが実証された生存植物細胞の源として使用した。当業者は、多様な植物種に由来する細胞培養物、または多様な植物種に由来する分化した植物組織を、内部への組み込みが実証された生存植物細胞の源として利用することを想定し得る。
【0165】
この実施例において、PAT植物選択可能マーカーカセットおよび多重eZFN結合部位挿入配列を含有するプラスミド(pDAB105908)を使用して、遺伝子導入事象を得た。遺伝子導入単離株を、eZFNにより形質転換して、二本鎖開裂を評価した。
【0166】
特に、事前に凍結保存した細胞株からの12mL血中血球容積(PCV)、加えて28mLの条件培地を、500mLエルレンマイヤーフラスコ中、80mLのGN6液体培地(N6培地(Chu et al.(1975)Scientia Sin 18:659−668)、2.0mg/L 2,4−D、30g/Lスクロース、pH5.8)中に継代培養し、125rpmで、28℃でシェーカー上に配置した。このステップを、同じ細胞株を使用して2回反復して、総計36mL PCVが3つのフラスコにわたって分配されるようにした。24時間後、GN6液体培地を除去し、72mL GN6 S/M浸透培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、45.5g/Lソルビトール、45.5g/Lマンニトール、100mg/Lミオイノシトール、pH6.0)と交換した。フラスコを暗所で30〜35分間、28℃で適度に撹拌しながらインキュベートした(125rpm)。インキュベーション時間中、炭化ケイ素ウィスカ(Advanced Composite Materials,LLC,Greer,SC)の50mg/mL懸濁液を、8.1mLのGN6 S/M液体培地を405mgの滅菌炭化ケイ素ウィスカに添加することによって調製した。
【0167】
GN6 S/M浸透培地中でのインキュベーションに続いて、各フラスコの内容物を250mL遠心瓶中にプールした。フラスコ中の全ての細胞が底に沈殿した後、およそ14mLを超える内容積のGN6 S/M液体を引き出し、後の使用のために滅菌1−Lフラスコ中に収集した。事前に湿潤させたウィスカの懸濁液を、ボルテックス上で、最大速度で60秒間混合し、次いで遠心瓶に添加した。
【0168】
この実施例において、pDAB105908プラスミドDNAからの170μgの精製した断片を各瓶に添加した。一旦、DNAを添加すると、瓶を、改変したRed Devil 5400商業用塗料ミキサ(Red Devil Equipment Co.,Plymouth,MN)中に即座に配置し、10秒間撹拌した。撹拌に続いて、細胞、培地、ウィスカ、およびDNAのカクテルを、125mLの新鮮なGN6液体培地と共に1−Lフラスコの内容物に添加して、浸透圧溶質を減少させた。細胞を、125rpmに設定したシェーカー上で2時間回収させた。6mLの分散した懸濁液を、ハウス真空管路(house vacuum line)に接続したガラス細胞収集ユニットを使用して、Whatman第4濾紙(5.5cm)上で濾過して、1瓶当たり60のフィルターを得るようにした。フィルターをGN6固体培地(2.5g/Lゲルライトゲル化剤を用いたことを除いてGN6液体培地と同じ)の60×20mmプレート上に配置し、暗所条件下で、28℃で1週間培養した。
【0169】
実施例2.2:推定遺伝子導入事象の特定および単離
DNA送達の1週間後、濾紙を、選択剤を含有するGN6(1H)選択培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、100mg/Lミオイノシトール、2.5g/Lゲルライト、pH5.8)の60×20mmプレートに移した。これらの選択プレートを、暗所で、28℃で1週間インキュベートした。暗所での1週間の選択後、細胞の半分を各プレートから、37〜38℃で保持された3.0mLのGN6アガロース培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、100mg/Lミオイノシトール、7g/L SeaPlaqueアガロース、pH5.8、121度でわずか10分間オートクレーブ処理)を含有する管にこすり落とすことによって、組織を新鮮な培地上に埋め込んだ。
【0170】
アガロース/組織混合物をへらで粉砕し、その後、3mLのアガロース/組織混合物を、GN6(1H)培地を含有する100×15mmペトリ皿の表面上に均等に注いだ。このプロセスを、各プレートの両方の片について反復した。一旦、全ての組織を埋め込むと、プレートをNESCOFILM(登録商標)またはPARAFILM M(登録商標)で個々に密閉し、暗所条件下で、28℃で最長10週間培養した。
【0171】
これらの選択条件下で増殖する推定的に形質転換した単離株を、埋め込まれたプレートから除去し、60×20mmプレート中の新鮮な選択培地に移した。およそ2週間後に持続的な増殖が明白であった場合、事象を、適用された除草剤(選択剤)に対して抵抗性であるとみなし、細胞のアリコートをその後、遺伝子型分析のために採取した。
【0172】
実施例2.3:ゲノムDNA抽出
ゲノムDNA(gDNA)を、実施例2.2に記載される単離したトウモロコシ細胞から抽出し、PCR遺伝子型同定実験のためのテンプレートとして利用した。gDNAを、DNeasy 96 Plant Kit(QIAGEN Inc.,Valencia,CA)に詳述される製造業者のプロトコルに従って上述のように単離した、およそ100〜300μL血中血球容積(PCV)のHi−IIカルスから抽出した。ゲノムDNAを、100μLのキット提供の溶出緩衝剤中に溶出して20〜200ng/μLの最終濃度を得、その後、下に概説されるPCRに基づく遺伝子型同定法を介して分析した。
【0173】
実施例2.4:コピー数の分子分析
TAQMAN(登録商標)アッセイを行って、除草剤抵抗性カルスの試料をスクリーニングして、pDAB105908導入遺伝子の単一コピー組み込みを含有したものを特定した。遺伝子発現カセットに特異的なプライマーおよびプローブを使用して、詳細な分析を行った。単一コピー事象を追加的な分析のために特定した。
【0174】
カスタムTAQMAN(登録商標)アッセイを、Third Wave Technologies(Madison,WI)によるHi−IIカルスにおけるPAT遺伝子分析のために開発した。ゲノムDNA試料を、96ウェルプレートフォーマットで95℃でのインキュベーションによって最初に変性させ、次いで周囲温度まで冷却した。次に、マスターミックス(緩衝剤に加えてPATのためのプローブミックスおよび内部参照遺伝子を含有する)を各ウェルに添加し、試料を鉱物油で上塗りした。プレートを密閉し、BioRad TETRAD(登録商標)サーモサイクラー中でインキュベートした。プレートを周囲温度まで冷却した後、蛍光プレートリーダー上で読み取った。全てのプレートは、1コピー、2コピー、および4コピー標準物、ならびに野生型対照試料および試料を含有しないブランクウェルを含有した。読み取りを収集し、各チャネルについてのフォールドオーバーゼロ(fold over zero)(すなわち、バックグラウンド)との比較は、試料の原シグナルを、テンプレートを含有しない原シグナルで割ることによって、各試料について決定した。
【0175】
このデータから標準的な曲線を構築し、最良適合を線形回帰分析によって決定した。この適合から特定したパラメータを使用して、次いで見かけのPATコピー数を各試料について推定した。
【0176】
実施例2.5:PCR遺伝子型同定のためのプライマー設計
この実施例において、PCR遺伝子型同定は、ゲノムに埋め込まれたドナーDNAを含有することが予測される単離したトウモロコシカルス組織に由来するゲノムDNAのポリメラーゼ鎖反応(PCR)増幅、続いてPCR増幅産物の標準的なクローニングおよび配列分析を含むが、これらに限定されないものとして理解した。PCR遺伝子型同定の方法は、詳細に記載されており(例えば、Rios,G.et al.(2002)Plant J.32:243−253)、細胞培養物を含む任意の植物種または組織型に由来するゲノムDNAに適用されてもよい。
【0177】
当業者は、植物ゲノムにおける特異的配列の増幅、植物ゲノムにおける多重特異的配列の増幅、植物ゲノムにおける非特異的配列の増幅、またはそれらの組み合わせを含む(しかしこれらに限定されない)、PCR遺伝子型同定のための戦略を考案し得る。増幅には、この実施例に記載されるように、クローニングおよび配列決定が続いてもよく、または増幅産物の直接配列分析が続いてもよい。当業者は、本明細書で生成される増幅産物の分析のための代替的な方法を想定することもあり得る。本明細書に記載される一実施形態では、遺伝子標的に特異的なオリゴヌクレオチドプライマーが、PCR増幅において用いられる。
【0178】
本明細書に提示される実施例において、オリゴヌクレオチドプライマーは、例えば、Integrated DNA Technologies,Inc.(Coralville,IA)によって、標準的な脱塩の条件下で、水で100μMの濃度まで希釈して合成される。オリゴヌクレオチドプライマーを、DNA挿入の隣接する領域とアニーリングするように設計した。プライマーを、プラスミドDNAの希釈を使用して、非遺伝子導入植物から単離したDNAの存在下で試験した。pDAB105908導入遺伝子を、プライマーを使用して推定事象のゲノムDNAからPCR増幅した。結果として得られた断片をプラスミドベクター中にクローン化し、配列決定して、多重eZFN結合部位配列が形質転換中に植物ゲノムに完全に組み込まれたことを確認した。
【0179】
実施例2.6:標的DNAを有する遺伝子導入事象の選択
低コピー(1〜2)事象をPCRによって、無傷の多重eZFN結合部位配列について、およびPAT遺伝子について、スクリーニングした。コピー数を、サザン分析によって、PAT遺伝子プローブによる標準的な方法を使用して確認した。単一コピーの無傷の挿入物を有する選択された遺伝子導入事象からのカルスを、一過性発現eZFNによるその後の評価のために維持した。
【0180】
実施例3:植物細胞中へのeZFN DNA送達
eZFN媒介型二本鎖開裂を可能にするために、DNAをコードするeZFNの送達、続いて植物細胞中の機能性eZFNタンパク質の発現が必要であることを理解する。当業者は、機能性ZFNタンパク質の発現が、ZFNをコードする構築物の遺伝子組み換え、またはZFNをコードする構築物の一過性発現を含むが、これらに限定されない複数の方法によって達成され得ることを想定し得る。
【0181】
本明細書に引用される実施例において、eZFNをコードするDNAの植物細胞中への送達のための方法が記載される。当業者は、アグロバクテリウム媒介型形質転換、バイオリスティックに基づくDNA送達、またはWHISKERS(商標)媒介型DNA送達を含むが、これらに限定されない、植物細胞に適切な多様なDNA送達方法のうちのいずれを使用することもできる。本明細書に記載される一実施形態では、バイオリスティック媒介型DNA送達実験を、様々なeZFNをコードするDNA構築物を使用して行った。
【0182】
実施例3.1:バイオリスティック媒介型DNA送達
上述のように、トウモロコシの胚形成Hi−II細胞培養物を産生し、eZFN機能を評価するための生存植物の源として使用した。当業者は、多様な植物種に由来する細胞培養物、または多様な植物種に由来する分化した植物組織を、内部への標的化組み込みが実証される生存植物細胞の源として利用することを想定し得る。
【0183】
多重eZFN結合部位上の特異的標的配列において結合する8つのeZFNのうちの1つを発現するプラスミドを、内部対照(IPK−1)と一緒に、5〜10個の遺伝子導入単離株からのカルスのプール中に衝撃した。
【0184】
遺伝子導入Hi−IIトウモロコシカルス事象を、GN6(1H)培地上で毎週継代培養した。培養の7日後、およそ400mgの細胞を、2.5g/Lゲルライトで凝固させたGN6 S/M培地を含有する100×15mmペトリ皿の中心上で、直径2.5cmの円形に薄く広げた。細胞を暗所条件下で4時間培養した。バイオリスティック粒子をDNAでコーティングするために、3mgの0.6ミクロン直径の金粒子を100%エタノールで1回、滅菌蒸留水で2回洗浄し、シリコン処理したエッペンドルフ管中の50μL水中に再懸濁させた。総計5μgのプラスミドDNA、20μLスペルミジン(0.1M)、および50μL塩化カルシウム(2.5M)を金懸濁液に別個に添加し、ボルテックス上で穏やかに混合した。混合物を室温で10分間インキュベートし、ベンチトップ微量遠心機において10,000rpmで10秒間ペレット化し、60μL冷100%エタノール中に再懸濁させ、8〜9μLを各マクロ担体上に分配した。
【0185】
Biolistic PDS−1000/HE(商標)系(Bio−Rad Laboratories,Hercules,CA)を使用して衝撃を行った。細胞を含有するプレートを、1100psiおよび27インチのHg真空の条件下で棚中段に配置し、作動マニュアルに従って衝撃した。衝撃の24時間後、組織を小さい塊でGN6固体培地に移した。
【0186】
実施例4:Solexa配列決定および分析
実施例4.1:試料調製
eZFNおよび対照IPK1−ZFN(Shukla et al.(1990)Nature 459,437−441)による衝撃の72時間後、組織を2mL微量遠心管中に収集し、少なくとも48時間凍結乾燥させた。ゲノムDNAを、製造業者の仕様書に従ってQIAGEN(登録商標)gDNA抽出キットを使用して、凍結乾燥させた組織から抽出した。最後に、DNAを200μLの水中に再懸濁させ、濃度を、Nanodrop分光光度計(Thermo Scientific,Wilmington,DE)を使用して決定した。DNAの無傷を、全ての試料を0.8%アガロースEゲル(Invitrogen,Carlsbad,CA)上で泳動させることによって推定した。全ての試料をPCR増幅に対して正規化して(25ng/μL)、Solexa配列決定に対するアンプリコンを生成した。
【0187】
eZFN開裂部位の各々を包含する領域の増幅のためのPCRプライマー、ならびに標的(ZFN処置された)試料および対照試料からのIPK1−ZFN標的部位を、IDT(Integrated DNA Technologies,San Jose,CA)から購入した。これらのプライマーのために最適な増幅条件を、勾配PCRによって、23.5μL反応中、0.2μMの適切なプライマー、Accuprime Pfx Supermix(1.1X,Invitrogen,Carlsbad,CA)、および100ngのテンプレートゲノムDNAを使用して特定した。循環パラメータには、95℃(5分間)での最初の変性、続いて35周期の変性(95℃、15秒間)、アニーリング[55−72℃、30秒間]、伸張(68℃、1分間)、および最終伸張(72℃、7分間)が含まれる。増幅産物を3.5%TAEアガロースゲル上で分析した。最適なアニーリング温度を特定した後、分取PCR反応を行って、各々一連のPCRプライマーを検証し、またSolexaアンプリコンを生成した。トウモロコシおよびタバコにおけるeZFN標的領域の増幅のために使用されるオリゴヌクレオチドを、下記の表3に示す。IPK1標的領域は、プライマー(配列番号27 GCAGTGCATGTTATGAGC(順方向プライマー)および配列番号28 CAGGACATAAATGAACTGAATC(逆方向プライマー))を使用して増幅した。
【0188】
【表3】
【0189】
分取PCRのために、上述の条件を使用して各テンプレートについて8つの個々の小規模PCR反応を完了し、産物を一緒にプールし、Qiagen MinElute(商標)ゲル精製キットを使用して3.5%アガロースゲル上でゲル精製した。ゲル精製したアンプリコンの濃度を、Nanodrop分光光度計を使用して決定し、eZFN標的化および対応する野生型対照ならびに正規化IPK−1標的化および野生型対照からのおよそ100ngのアンプリコンをプールすることによって、Solexa試料を調製した。eZFN+IPK−1標的化試料、IPK−1標的化試料、および野生型対照から、アンプリコンを含む4つの最終Solexa試料を生成し、配列決定した。アンプリコンをPCR−Blunt II−TOPO(Invitrogen)中にクローン化し、配列決定に供してプライマーを検証した後、Solexa配列決定を行った。
【0190】
実施例4.2:Solexa配列決定および分析
Solexa配列決定は、数千の配列の産生をもたらした。DAS Next Generation Sequence(NGS)分析スクリプトを使用して配列を分析した。低質の配列(品質スコアカットオフ<5を有する配列)を取り除いた。次いで配列を参照配列と整列させ、しばしばZFN活性の指標であるインデルを引き起こす、ZFN媒介型開裂およびNHEJ媒介型修復によって引き起こされた、ZFN開裂部位における挿入/欠失(インデル)についてスコア化した。バックグラウンド活性を差し引いた後の、ZFNタンパク質に対する結合部位の間の「ギャップ」配列内での1bpを超える欠失の数によって、編集活性を決定した。本研究における各eZFNに対する活性を、野生型対照と比較して算出し、IPK−1 ZFN活性に対して正規化した。次いで各eZFNについて正規化した活性を比較して、研究に使用したeZFNをランク付けした。また、活性を、eZFN開裂部位におけるインデルの存在によって、配列整列レベル(Solexa出力と比較した参照)において査定した。
【0191】
図11に示すように、8つのうち7つのeZFNは、トウモロコシにおける編集活性を示す。
【0192】
実施例5:タバコにおけるeZFNの評価
実施例5.1:多重eZFN結合部位配列の安定な組み込み
本明細書に上述される多重eZFN結合部位配列の組み込まれたコピーを有する遺伝子導入植物事象を作製するために、PhytaTrays(Sigma,St.Louis,MO)中のMS培地(Phytotechnology Labs,Shawnee Mission,KS)および30g/Lスクロース上に無菌で増殖させた、Petit Havanaタバコ植物から切り取った葉ディスク(1cm2)(例えば、事前に組み込まれたZFN−IL1結合部位を含有する事象1585−10)を、OD600約1.2まで増殖させたアグロバクテリウムLBA4404含有プラスミドpDAB105900の一晩の培養物上に浮遊させ、滅菌濾紙上でブロッティング乾燥させ、次いで60×20mm皿(1皿当たり5ディスク)中、1mg/Lインドール酢酸および1mg/Lベンジアミノ(benzyamino)プリンを添加した同じ培地上に配置した。72時間の共培養後、葉ディスクを、250mg/Lセフォタキシムおよび5mg/L BASTA(登録商標)を有する同じ培地に移した。3〜4週間後、小植物を、PhytaTrays中、250mg/Lセフォタキシムおよび10mg/L BASTA(登録商標)を有するMS培地に更に2〜3週間移した後、葉の採取および分子分析を行った。
【0193】
実施例5.2:多重eZFN結合部位配列遺伝子導入事象のコピー数およびPTU分析
DNA単離。遺伝子導入タバコ植物組織をBASTA(登録商標)抵抗性小植物から採取し、96ウェル収集プレート中で少なくとも2日間凍結乾燥させた。次いでDNAを、製造業者の指示に従って、DNEASY(商標)96ウェル抽出キット(Qiagen,Valencia,CA)を使用して単離した。Model 2−96A Kleco組織粉砕機(Garcia Manufacturing,Visalia CA)を使用して組織破壊を行った。
【0194】
DNA定量化。結果として得られたゲノムDNAを、QUANT−IT(登録商標)Pico Green DNAアッセイキット(Molecular Probes,Invitrogen,Carlsbad,CA)を使用して定量化した。20ng/μL〜1.25ng/μL(段階希釈した)の範囲の、5つの事前に定量化したDNA標準物を使用して標準曲線生成を行った。未知の試料を、アッセイの線形範囲内に入るように、最初に1:10または1:20希釈に希釈した。5μLの希釈した試料および標準物を、100μLの希釈したPico Green基質(1:200)と混合し、暗所で10分間インキュベートした。次いで蛍光を、Synergy2 プレートリーダー(Biotek,Winooski,VT)を使用して記録した。ゲノムDNA濃度を、バックグラウンド蛍光補正後に算定された標準曲線から推定した。TEまたは水を使用して、DNAを次いで、Biorobot3000自動液体処理機を使用して(Qiagen)10ng/μLの一般的な濃度まで希釈した。
【0195】
コピー数推定。推定遺伝子導入事象を、TAQMAN(登録商標)アッセイに類似した多重化DNA加水分解プローブアッセイを使用して、組み込み複雑性について分析した。多重部位構築物のコピー数を、PAT導入遺伝子および内在性タバコ参照遺伝子、PALの両方に対する、配列特異的プライマーおよびプローブを使用して推定した。両方の遺伝子についてのアッセイを、LIGHTCYCLER(登録商標)プローブ設計ソフトウェア2.0リアルタイムPCRを使用して設計し、両方の遺伝子について、LIGHTCYCLER(登録商標)480系(Roche Applied Science,Indianapolis,IN)を使用して評価した。増幅のために、LIGHTCYCLER(登録商標)480プローブマスターミックスを、0.4μMの各プライマーおよび0.2μMの各プローブを含有する10μL容量の多重化反応物中、1倍の最終濃度で調製した(下記の表4)。2ステップ増幅反応を、蛍光取得により58℃で38秒間の伸張により行った。全ての試料を3系列で実験し、平均Ct値を各試料の分析のために使用した。リアルタイムPCRデータの分析を、相対定量モジュール(relative quant module)を使用するLIGHTCYCLER(登録商標)ソフトウェアを使用して行い、それはΔΔCt法に基づいた。このために、単一コピーキャリブレーターからのDNAの試料を含めて、結果を正規化した。単一コピーキャリブレーター事象をサザン分析によって特定し、それがPAT遺伝子の単一の挿入物を有することを確認した。
【0196】
【表4】
【0197】
PCR。低コピー(1〜2)事象をその後、PAT遺伝子に対する無傷の植物転写単位(PTU)、および無傷の多重eZFN結合部位についてPCRによってスクリーニングした。
【0198】
実施例6:多重eZFN結合部位配列におけるeZFN開裂の試験
組み込まれた多重eZFN結合部位配列における標的開裂を促進する、eZFNの能力を試験するために、遺伝子導入タバコ葉ディスクのアグロバクテリウム共培養を介したeZFN構築物の一過性発現に基づいて、一過性アッセイを使用した。多重eZFN結合部位配列含有構築物の単一の完全長コピー(ならびにZFN−IL1構築物の単一の完全長コピー)を含有する、遺伝子導入事象から切り取った葉ディスク(1cm2)を、OD600約1.2まで増殖させたアグロバクテリウムの一晩の培養物上に浮遊させ、滅菌濾紙上でブロッティング乾燥させ、次いで1mg/Lインドール酢酸および1mg/Lベンジアミノ(benzyamino)プリンを添加した同じ培地上に配置した。試験した各eZFNに対して、次の3つの処置を使用した:1処置当たり20個の葉ディスクを用いて、pDAB1601(陰性対照PATのみ)、pDAB4346のみ(陽性対照ZFN−IL1のみ)、およびpDAB4346+pDABeZFN−X(試験対象のZFN−IL1+eZFN)。
【0199】
実施例6.1:配列分析
ゲノムDNAを、Qiagen DNA抽出キットを使用して、アグロバクテリウム処置した遺伝子導入タバコ葉ディスク物から単離した。全ての処置を2系列で行い、全ての試料からのゲノムDNAを100μLの水中に再懸濁させ、濃度をNanodropによって決定した。個々の処置に対する各複製物からの等量のゲノムDNAを一緒にプールし、Solexaアンプリコン生成のための出発テンプレートとして使用した。
【0200】
標的(eZFN処置された)試料および対照試料からの、多重eZFN結合部位配列および開裂部位を包含する領域の増幅のためのPCRプライマーは、Integrated DNA Technologies(Coralville,IA)からのものであり、それをHPLC精製した。最適な増幅条件を、勾配PCRによって、23.5μL反応中、0.2μMの適切なプライマー、Accuprime Pfx Supermix(1.1X,Invitrogen,Carlsbad,CA)、および100ngのテンプレートゲノムDNAを使用して特定した。循環パラメータは、95℃(5分間)での最初の変性、続いて35周期の変性(95℃、15秒間)、アニーリング[55〜72℃、30秒間]、伸張(68℃、1分間)、および最終伸張(72℃、7分間)であった。増幅産物を3.5%TAEアガロースゲル上で分析した。最適なアニーリング温度(56.1℃)を特定した後、分取PCR反応を行って、各々一連のPCRプライマーを検証し、またSolexaアンプリコンを生成した。
【0201】
分取PCRのために、上述の条件を使用して各テンプレートについて8つの個々の小規模PCR反応を行い、産物を一緒にプールし、Qiagen MinEluteゲル精製キットを使用して3.5%アガロースゲル上でゲル精製した。ゲル精製したアンプリコンの濃度を、Nanodrop分光光度計を使用することによって決定し、およそ200ngの各アンプリコンを一緒にプールして、最終Solexa配列決定試料(800ng総試料)を調製した。また、アンプリコンをPCR−Blunt II−TOPO中にクローン化し、通常の配列決定に供してプライマーを検証した後、Solexa配列決定を行った。Solexa分析(Shendure et al.(2008)Nat.Biotechnology,26:1135−1145)を行い、配列を分析した。
【0202】
実施例6.2:Solexa配列決定および分析
Solexa配列決定を行って、数千の配列の産生がもたらされた。DAS NGS分析スクリプトを使用して配列を分析した。低質の配列(品質スコアカットオフ<5を有する配列)を取り除いた。次いで配列を参照配列(多重eZFN結合部位を含有するpDAB105900)と整列させ、開裂部位における挿入/欠失(インデル)についてスコア化した。各eZFNおよび無処置対照に対する編集活性(NHEJ%)を算出し(インデルを有する高質配列の数/高質配列の総数×100)、下記の図12に示す。2つの遺伝子導入タバコ事象(105900/第33および105900/第45)における8−eZFNの活性を実証した(図12)。8つのうちの3つのeZFNが、試験した2つの遺伝子導入タバコ事象において活性であった。活性をまた、eZFN処置試料中のeZFN開裂部位におけるインデルの存在によって、配列整列レベル(参照対solexa出力)において査定した。
【0203】
ZFN単量体(「右」および「左」)片側の全ての組み合わせが、酵母アッセイにおいて活性であった。トウモロコシおよびタバコ実験について記載されるデータは、組み合わせのうちの幾つかまたはほとんどが植物において活性であることを実証し、研究のために選択された4つの元のZFNからの2つのZFN単量体の、有意な数の順列を使用する可能性を支持している。
【0204】
実施例7:対立遺伝子内組み換え
対立遺伝子内組み換えは、導入遺伝子の2つの独立したブロックの発達および最適化を可能にし、次いでそれを組み換えによって1つの遺伝子座において一緒にスタッキングすることができる。2つのブロックの間の組み換えのレベルを強化するために、二本鎖開裂は、遺伝子変換または染色分体交換によってDNA交換を開始する。
【0205】
植物におけるこの概念を実証するために、図13に例証する遺伝子導入挿入物を、アラビドプシス・タリアナ中で作製する。構築物には、選択可能マーカー(ネオマイシンホスホトランスフェラーゼ(NPTII)またはハイグロマイシンホスホトランスフェラーゼ(HPT)、およびにスコア化可能なマーカー(β−グルクロニダーゼ(GUS)および黄色蛍光タンパク質(YFP))を含有する遺伝子ブロックが含まれる。これらの遺伝子ブロックは、同一のゲノム位置にあるが、互いからおよそ2kbが置換される。2つのブロックの間の組み換えは、子孫を、2つのブロックの間の中心の位置において開裂するZFNを発現する植物と交配させ、次いでそれらを再交配させることによって、2つのブロックの各々を担持する染色体を、単一の植物中に組み合わせることによって遂行する(図13MIS上部の黒色バー)。ZFNは、減数分裂特異的/選好的プロモーターを使用して発現させる。使用されるランディングパッド配列には、参照により本明細書に組み込まれる米国特許出願第61/297,641号に記載されるものが含まれる。
【0206】
同一のゲノム位置における独立したブロックを生成するために、隣接する配列において両方のブロックを含む構築物を作製した(図14)。独立したブロックのみを担持する植物を作り出すために、対応するZFN結合部位におけるそれぞれのブロックのいずれかの側上で、DNAを切断するように設計したZFNを使用して、別個の交配において各ブロックを切除する(赤色および青色バー)。図15は、適切な株(ZFNを発現するアラビドプシス)と交配させた後、ブロックが切断されて、単一のブロック挿入物を生成することを例証する。これらの株は、選択可能マーカーとしてPAT遺伝子を担持する。予想される表現型(HygR+、KanR−、PAT+、YFP+or Ka nR+、HygR−、PAT+、GUS+)を有する植物の回収を、表現型スクリーニング(HygR、KanR、およびPAT遺伝子に対する除草剤抵抗性、またはGUSおよびYFPのスコア化可能なマーカー遺伝子発現)を介して、またはPCRおよびサザン分析等の分子分析によって確認する。2つの異なるブロックの1つを担持する植物を交配させて、HygR+、KanR+、PAT−、GUS+、YFP+子孫を生成する。
【0207】
結果として得られた植物の分子特性評価後、確認された挿入物を有する植物を、減数分裂特異的プロモーターを使用して、結合部位が2つのブロックの間に位置するZFNを発現する株と交配させて、DNAの交換を達成する。これは、1つのDNA位置における2つのブロックのスタッキングをもたらす。最終のスタッキングされた遺伝子植物は、単一の分離遺伝子座として、HygR+、KanR+、GUS+、YFP+構成を担持する。代替的には、ブロックのうちの1つを含有する植物を、減数分裂プロモーター/ZFN構築物を含む2つの単量体のうちの1つと交配させ、2つの挿入物に対してホモ接合型である植物を得、次いで一緒に交配させる。
【0208】
実施例7.1:DNA構築
ZFN構築物の構築用に配備したクローニング戦略は、米国特許出願第2009/0111188A1号および同第2010/0199389号に本質的に記載される通りである。図9は、例示的なeZFN発現カセットを示す。ZmUbi1プロモーター(トウモロコシユビキチン1(Ubi−1)遺伝子に由来するプロモーター、5’非翻訳領域(UTR)、およびイントロン、Christensen et al.(1992)Plant Molec.Biol.18(4),675−89)を使用して、ZFNコード配列を発現させる。これらをその後、PAT遺伝子の発現を駆動するイネアクチン1プロモーターを含有する、バイナリーGATEWAY(商標)デスティネーションベクター中にクローン化した。それぞれブロック1 Excisor(eZFN1,8)またはブロック2 Excisor(eZFN3,6)構築物として指定する、結果として得られたプラスミドpDAB105951(ZFN1、CL:AR)、105954(ZFN8、RL:AR)、105952(ZFN3、AL:PR)、105953(ZFN6、CL:RR)を、アグロバクテリウム株DA2552recAに移した。
【0209】
アグロバクテリウムDA2552株を、DA2552株をグリセロールストックから、スペクチノマイシン(spec)(100μg/mL)およびエリスロマイシン(ery)(150μg/mL)を含有する10mLのYEP中に植菌することによって種培養を調製することによって、エレクトロポレーションのためにコンピテントにした。10mL培養物を、200rpmで、28℃で一晩インキュベートした。5ミリリットルの種培養を使用して、適切に標識化した1.5Lエルレンマイヤーフラスコ中、適切な抗生物質を有する500mLのYEPを植菌した。培養を、200rpmで、28℃で一晩インキュベートした。一晩のインキュベーション後、培養を、湿潤の氷水浴中に配置し、穏やかに旋回させることによって冷却した。細胞を、全ての更なるステップのために4℃で維持した。細胞を、事前冷却したローター内で標識化した滅菌遠心瓶中、4000xgで、4℃で10分間、遠心分離機にかけることによってペレット化した。上清を注ぎ出して破棄し、次いで5〜10mLの氷冷滅菌再蒸留水を添加し、塊が残らなくなるまで、細胞を上下に穏やかにピペット操作した。懸濁液容量を、氷冷滅菌再蒸留水でおよそ500mLまで調整した。細胞を、事前冷却したローター内で、4000xgで、4℃で10分間、遠心分離機にかけることによってペレット化した。上清を破棄し、5〜10mLの氷冷滅菌再蒸留水を添加し、次いで塊が残らなくなるまで、滅菌ワイドボアピペットを使用して細胞を上下に穏やかにピペット操作した。懸濁液容量を、氷冷滅菌再蒸留水でおよそ250mLまで調整し、細胞を、事前冷却したローター内で、4000xgで、4℃で10分間、遠心分離機にかけることによってペレット化した。上清を破棄し、5〜10mLの氷冷滅菌再蒸留水を添加し、ペレットを穏やかに再懸濁させ、最終容量を氷冷滅菌再蒸留水で50mLまで調整した。細胞を、事前冷却したローター内で、4000xgで、4℃で10分間、遠心分離機にかけることによってペレット化した。細胞を、5mLの10%(v/v)氷冷の滅菌グリセロールの最終容量中に再懸濁させた。細胞を滅菌0.5mL微量遠心管中の50μLアリコートに分与し、液体窒素中で凍結させた。
【0210】
20マイクロリットルのコンピテントDA2552細胞を、製造業者のプリセット設定およびアグロバクテリウムエレクトロポレーションのためのプロトコルに従って、GENE PULSER(登録商標)XCELL(登録商標)Electroporation System(BioRad Hercules,CA.)を使用して、50ngのプラスミドDNAで電気穿孔処理した。細胞をSOC中、28℃で2時間回収し、次いでYEP spec/ery寒天プレート上にプレートし、28℃で48時間増殖させた。
【0211】
実施例7.2:交換遺伝子座構築物
交換遺伝子座DNA構築物を、ベクター1:AtAct2プロモーター(AtAct2プロモーターv2(アラビドプシス・タリアナアクチン遺伝子(ACT2)からのプロモーター、5’非翻訳領域、およびイントロン、An et al.(1996)Plant J.10,107−121))/GUS(Jefferson,(1987)EMBO J.6,3901−3907)/AtuORF23 3’ UTR(アグロバクテリウム・ツメファシエンスpTi15955のオープンリーディングフレーム23(ORF23)の転写終結因子およびポリアデニル化部位を含む、3’非翻訳領域(UTR)、Barker et al.,(1983)Plant Molec.Biol.2(6):335−50)::AtAct2プロモーター/NPTII(Bevan et al.(1983)Nature 304,184−187)/ eZFN 1および8が側面に位置するAtuORF23 3’ UTR、ベクター2:配列の中心にeZFN 4および7を有する合成2kb領域、およびベクター3:CsVMVプロモーター/HPT(Kaster et al.(1983)Nucleic Acids Res.11(19),6895−6911(1983))/AtuORF23 3’ UTR::AtUbi10プロモーター(アラビドプシス・タリアナポリユビキチン10(UBQ10)遺伝子からのプロモーター、5’非翻訳領域、およびイントロン、Norris et al.(1993)Plant Molecular Biology 21(5):895−906)/PhiYFP(Shagin et al.,(2004)Molecular Biol.Evol.21:841−850)/ eZFN 3および6が側面に位置するAtuORF23 3’ UTR、を含むGATEWAY(商標)エントリーベクターから調製した。デスティネーションベクターを、2つの1kb無作為化合成DNA配列を、アグロバクテリウムバイナリーベクター主鎖に挿入することによって調製し、このうち制限部位をそれらの間に含めて、GATEWAY(商標)ccdB陰性選択可能マーカーカセットをクローン化した。エントリーベクターを、LR Clonase反応によってデスティネーションベクター中にクローン化した。結果として得られたベクター、pDAB100646(図16)を、上述のアグロバクテリウムに移した。
【0212】
実施例7.3:アラビドプシス形質転換
Clough&Bent(1998 Plant J.,16,735−743)によって記載される方法に従って、アラビドプシスへの全ての形質転換を行った。
【0213】
切除因子株
「切除因子」株構築物は、グルフォシネートに対する抵抗性を伝達するホスフィノトリシンアセチルトランスフェラーゼ(PAT)遺伝子をプロセスする。植付けの7、10、および13日後、T1植物に、Liberty除草剤(グルフォシネート1リットル当たり200グラムの活性成分(g ai/L)、Bayer Crop Sciences,Kansas City,MO)の284mg/L溶液を、10mL/トレー(703L/ha)の噴霧容量で、DeVilbiss圧縮空気ノズルチップを使用して噴霧して、1回の塗布当たり200g ai/haグルフォシネートの有効率を送達した。最終噴霧の4〜7日後、生存物(活発に成長している植物)を特定し、培養土(Metro Mix 360)で調製した3インチポットに個々に移植した。
【0214】
切除因子事象におけるeZFNの発現を逆転写酵素PCR(RT PCR)によって決定し、コピー数を、PAT遺伝子について本明細書に記載されるようにqPCRによって決定し、サザン分析によって確認する。ZFNを高レベルで発現する3つの低コピー事象を、交換遺伝子座(Exchange Locus)事象と交配させる。
【0215】
交換遺伝子座株
交換遺伝子座株を、ハイグロマイシンまたはカナマイシンを含有する培地についての選択を含む、Clough&Bent(1998 Plant J.,16,735−743)によって記載される方法に従って、アラビドプシスにおいて生成する。
【0216】
実施例7.4:アラビドプシス交配および子孫回収
交換遺伝子座事象の、2つの一連のブロック1およびブロック2切除因子株との交配を、標準的な方法を使用して行う。
【0217】
交配からの種子をハイグロマイシン(ブロック1欠失)またはカナマイシン(ブロック2欠失)上で増殖させ、抵抗性植物をGUS発現(ブロック1欠失)またはYFP発現(ブロック2欠失)について分析する。GUS活性を、組織化学的アッセイ(Jefferson et al.(1987)Plant Mol.Biol.Rep 5,387−405)を用いて、またYFPを、蛍光顕微鏡検査法を使用して、決定する。所望の表現型(ブロック1陽性:GUS+、NPT+、HPT−、YFP−、ブロック2陽性:GUS−、NPT−、HPT+、YFP+)を有する植物を、PCRおよびサザン法によって分析して、所望の遺伝子構成を確認する。選択された植物からの葉を、ビアラホス溶液で塗布して、どれがPAT+であるかを査定する。
【0218】
ブロック1およびブロック2遺伝子カセットを含有する植物を交配させ、子孫をハイグロマイシン/カナマイシンプレート上で選択する。HygR/KanR植物を、PCRおよび表現型スクリーニングによって、全ての遺伝子の存在について分析する。所望の表現型を有するF1植物を増殖させ、減数分裂プロモーター/ZFN植物と交配させて、ブロック1およびブロック2の間の組み換えを達成する。結果として得られた子孫を、ハイグロマイシン/カナマイシンプレート上で増殖させる。選択後に生存した植物を、GUSおよびYFPについてスクリーニングする。PCR、サザン、配列決定、および分離比分析を使用して、組み換えの確認および特性評価を行う。
【0219】
実施例8:eZFN部位における遺伝子スタッキング
図1、2、4、5、および6に示す戦略は、次の方法を使用して遂行することができる。
【0220】
構築物設計
ヘテロ二量体eZFN部位の様々な組み合わせは、植物形質転換に好適なプラスミドベクター中のコンカテマーとして組み立てることができる。図1、図2、および図4は、ベクターに組み入れて、植物の染色体に形質転換することができるヘテロ二量体eZFN部位の様々なバージョンを例証する。
【0221】
WHISKERS(商標)形質転換
トウモロコシの胚形成Hi−II細胞培養物を、米国特許第7,179,902号に記載されるように産生し、標的組み込みが例示される生存植物細胞の源として使用する。植物選択可能マーカーカセットに結合されたヘテロ二量体eZFN部位を含有するDNA断片を使用して、遺伝子導入事象を生成する。遺伝子導入事象を、単離し、特徴付ける。
【0222】
事前に凍結保存した細胞株からの12mLの血中血球容積(PCV)、加えて28mLの条件培地を、500mLエルレンマイヤーフラスコ中、80mLのGN6液体培地(N6培地(Chu et al.,(1975)Sci Sin.18:659−668)、2.0mg/L 2,4−D、30g/Lスクロース、pH5.8)中に継代培養し、シェーカー上に125rpmで、28℃で配置する。同じ細胞株を使用してこのステップを2回反復して、総計36mLPCVが3フラスコにわたって分配されるようにする。
【0223】
24時間後、GN6液体培地を除去し、72mL GN6 S/M浸透培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、45.5g/Lソルビトール、45.5g/Lマンニトール、100mg/Lミオイノシトール、pH6.0)と交換する。フラスコを暗所で30〜35分間、28℃で適度に撹拌しながらインキュベートする(125rpm)。インキュベーション時間中、炭化ケイ素ウィスカ(Advanced Composite Materials,LLC,Greer,SC)の50mg/mL(w/v)懸濁液を、8.1mLのGN6 S/M液体培地を405mgの滅菌炭化ケイ素ウィスカに添加することによって調製する。GN6 S/M浸透培地中でのインキュベーションに続いて、各フラスコの内容物を250mL遠心瓶中にプールする。フラスコ中の全ての細胞が底に沈殿した後、およそ14mLを超える内容積のGN6 S/M液体を引き出し、後の使用のために滅菌1−Lフラスコ中に収集する。事前に湿潤させたウィスカの懸濁液を、ボルテックス上で、最大速度で60秒間混合し、次いで遠心瓶に添加する。
【0224】
85μgの精製したDNA断片のアリコートを各瓶に添加する。一旦、DNAを添加すると、瓶を、改変したRed Devil 5400商業用塗料ミキサ(Red Devil Equipment Co.,Plymouth,MN)中に即座に配置し、10秒間撹拌する。撹拌に続いて、細胞、培地、ウィスカ、およびDNAのカクテルを、125mLの新鮮なGN6液体培地と共に1−Lフラスコの内容物に添加して、浸透圧溶質を減少させる。細胞を、125rpmに設定したシェーカー上に2時間回収させる。6mLの分散懸濁液を、ハウス真空管路(house vacuum line)に接続したガラス細胞収集ユニットを使用して、Whatman第4濾紙(5.5cm)上で濾過して、1瓶当たり60のフィルターを得るようにする。フィルターをGN6固体培地(2.5g/L Gelriteゲル化剤を用いることを除いてGN6液体培地と同じ)の60×20mmプレート上に配置し、暗所条件下で、28℃で1週間培養する。
【0225】
推定標的組み込み遺伝子導入事象の特定および単離
DNA送達の1週間後、濾紙を、選択剤を含有するGN6(1H)選択培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、100mg/Lミオイノシトール、2.5g/Lゲルライト、pH5.8)の60×20mmプレートに移す。これらの選択プレートを、暗所で、28℃で1週間インキュベートする。暗所での1週間の選択後、細胞の1/2を各プレートから、37〜38℃で保持される3.0mLのGN6アガロース培地(N6培地、2.0mg/L 2,4−D、30g/Lスクロース、100mg/Lミオイノシトール、7g/L SeaPlaque(登録商標)アガロース、pH5.8、121度で10分間オートクレーブ処理)を含有する管にこすり落とすことによって、組織を新鮮な培地上に埋め込む。
【0226】
アガロース/組織混合物をへらで粉砕し、その後、3mLのアガロース/組織混合物を、GN6(1H)培地を含有する100×25mm皿の表面上に均等に注ぐ。このプロセスを、各プレートの両方の片について反復する。一旦、全ての組織を埋め込むと、プレートを暗所条件下で、28℃で最長10週間培養する。これらの選択条件下で増殖した推定的に形質転換した単離株を、埋め込まれたプレートから除去し、60×20mmプレート中の新鮮な選択培地に移す。およそ2週間後に持続的な増殖が明白である場合、事象を、適用された除草剤(選択剤)に対して抵抗性であるとみなし、細胞のアリコートをその後、遺伝子型分析のために採取する。安定な植物形質転換は、スタッキング実験のために使用される単一コピー組み込み体を産生する。
【0227】
事象の分子特性評価
ゲノムDNA(gDNA)を、記載される単離されたトウモロコシ細胞から抽出し、PCR遺伝子型同定実験のためのテンプレートとして利用する。gDNAを、DNEASY(登録商標)96 Plant Kit(QIAGEN Inc.,Valencia,CA)に詳述される製造業者のプロトコルに従って単離する、およそ100〜300μL血中血球容積(PCV)のHi−IIカルスから抽出する。ゲノムDNAを、100μLのキット提供緩衝剤中に溶出して20〜200ng/μLの最終濃度を得、その後、PCRに基づく遺伝子型同定法を介して分析する。
【0228】
コピー数の分子分析
INVADER(登録商標)または加水分解プローブアッセイを行って、除草剤抵抗性カルスの試料をスクリーニングして、T鎖DNAの単一コピー組み込みを含有するものを特定する。遺伝子発現カセットに特異的なプライマーおよびプローブを使用して、詳細な分析を行う。単一コピー事象を追加的な分析のために特定する。
【0229】
カスタムINVADER(登録商標)アッセイを、Third Wave Technologies(Madison,WI)によるHi−IIカルスにおける選択可能マーカー遺伝子分析のために開発する。ゲノム試料を、INVADER(登録商標)アッセイキットを使用して増幅し、読み取りを収集する。これらの読み取りから、各チャネルについてのフォールドオーバーゼロ(fold−over zero)(すなわち、バックグラウンド)を、試料の原シグナルを、テンプレートを含有しない原シグナルで割ることによって、各試料について決定する。このデータから標準的な曲線を構築し、最良適合を線形回帰分析によって決定する。この適合から特定したパラメータを使用して、次いで見かけの選択可能マーカーコピー数を各試料について推定する。
【0230】
標的DNAを有する遺伝子導入事象の選択
低コピー(1〜2コピーの導入遺伝子)事象を、上述のように、選択可能マーカー遺伝子カセットおよび無傷のeZFN部位を含有する無傷の植物転写単位(PTU)について、PCRによってスクリーニングする。コピー数を、サザン分析によって、選択可能マーカー遺伝子による標準的な方法を使用して更に確認する。単一コピーの無傷の挿入を有する選択された遺伝子導入事象からのカルスを維持する。
【0231】
eZFNを含有する植物細胞中へのバイオリスティック媒介型DNA送達
上述のように、トウモロコシの胚形成Hi−II細胞培養物を産生し、内部への標的組み込みが実証される生存植物細胞の源として使用する。トウモロコシの胚形成懸濁液を、GN6液体培地中におよそ24時間継代培養し、その後、上述される実験を行う。およそ0.4mL PCVの細胞を、2.5g/Lゲルライトで凝固させたGN6 S/M培地を含有する100×15mmペトリ皿の中心上で、直径2.5cmの円形に薄く広げる。
【0232】
細胞を暗所条件下で4時間培養する。バイオリスティック粒子を、ドナーDNA(図1のブロック1、図2のブロック2、または図4の遺伝子1)を含有するDNAでコーティングするために、3mgの1.0ミクロン直径の金粒子を100%エタノールで1回、滅菌蒸留水で2回洗浄し、シリコン処理したエッペンドルフ管中の50μL水中に再懸濁させる。総計5μgのプラスミドDNA(単一のベクター中または別個のベクター中に、eZFNをコードする核酸分子およびドナーDNA断片を含有する)、20μLスペルミジン(0.1M)、および50μL塩化カルシウム(2.5M)を金懸濁液に別個に添加し、ボルテックス上で混合する。混合物を室温で10分間インキュベートし、ベンチトップ微量遠心機において10,000rpmで10秒間ペレット化し、60μL冷100%エタノール中に再懸濁させ、8〜9μLを各マクロ担体上に分配する。
【0233】
Biolistic PDS−1000/HE(商標)系(Bio−Rad Laboratories,Hercules,CA)を使用して衝撃を行う。細胞を含有するプレートを、1,100psiおよび27インチのHg真空の条件下で棚中段に配置し、作動マニュアルに従って衝撃する。衝撃の16時間後、組織を小さい塊でGN6(1H)培地に移し、暗所条件下で、28℃で2〜3週間培養する。ドナーDNAの組み込みからもたらされる推定遺伝子導入単離株が現れ始めるまで、移入を2〜4週間毎に継続する。ビアラホス抵抗性コロニーを、一般に、PCRおよびサザンブロッティングによって、標的配列を含有する単離株を生成するために上に詳述される方法を使用して分析する。
【0234】
PCR遺伝子型同定を介する標的組み込み事象についてのスクリーニング
PCR反応を行って、ドナーDNAの無傷のコピーの存在を調査する。追加的な反応は、標的とドナーとの間の5’境界およびドナーと標的との間の3’境界に焦点を当てる。増幅した断片をゲル切除し、標準的なプロトコルに従って精製する。精製した断片をその後、製造業者のプロトコルに従って、TOPO TA CLONING(登録商標)Kit(pCR2.1ベクターを有する)およびONE SHOT(登録商標)TOP10 Chemicallyコンピテント大腸菌細胞(Invitrogen Life Technologies,Carlsbad,CA)を使用してpCR2.1プラスミド中にクローン化する。
【0235】
個々のコロニーを選択し、増幅したPCR断片を含有することを確認する。プラスミドクローンの二本鎖配列決定反応を行って、PCR増幅したゲノム配列が組み込まれたドナーを含有することを確認する。ドナー断片を含有することが特定された事象は、ZFN媒介型二本鎖切断の相同性駆動型修復、およびドナーDNAの特異的遺伝子標的における標的組み込みが存在する標的を表す。
【0236】
eZFN部位を使用する遺伝子スタッキングの特異的用途
図1は、植物の染色体に安定に形質転換される7つの(7)eZFN標的部位から成り立つ多重挿入部位の変形を示す。標的部位に結合するeZFN対を幾何学的図として示す。「ブロック1」は、特異的eZFN部位を開裂するように設計されたeZFNにより形質転換されるとき、適切なeZFN対の多重挿入部位に組み込まれ得る外来性ポリヌクレオチド配列である。eZFNおよび「ブロック1」ドナーDNA配列の共形質転換は、前述のバイオリスティック形質転換方法を使用して達成することができる。様々な他のeZFN部位の忠実度は、植物細胞中に形質転換されるeZFNがこれらの他の部位を開裂しないため、維持される。「ブロック1」は、相同組み換えを介して植物染色体に組み込み、「ブロック1」の配列を含有する植物細胞をもたらす。結果として得られた植物細胞は、成熟植物に成長させられ、サザンブロッティング、Taqmanアッセイ、またはInvaderアッセイ等の当該技術分野において既知である分析分子生物学的方法を使用して「ブロック1」の存在についてスクリーニングされ得る。
【0237】
図2は、図1の別の変形を例証し、ここで異なるeZFN結合部位は、ポリヌクレオチドドナー配列「ブロック2」により標的化される。DNA断片の結果として得られた組み込みは、染色体内に「ブロック2」を含有する安定な植物を産生する。
【0238】
図4は、eZFN「左」および「右」ドメインの使用を例証する。上部の線は、左および右eZFNドメインにより形質転換された植物のゲノムを示す(影付き三角および碁盤格子三角)。新たなおよび異なるヘテロ二量体eZFN部位が側面に位置する「遺伝子1」を含む外来分子の存在下で、適切なeZFNが付加されるとき、「遺伝子1」および隣接するeZFN部位がゲノムに挿入される。「遺伝子1」および隣接するeZFN部位を含有する、結果として得られた子孫が特定され、これらの植物を、その後、親植物内には存在しなかった新たなヘテロ二量体eZFN部位を使用して、再標的化することができる(すなわち、影付き三角および碁盤格子三角を含有するeZFN部位)。
【0239】
図5および図6は、eZFN部位を使用して、新たな導入遺伝子を染色体位置中にスタッキングすることができる様態を例証する。更に、この戦略は、他の遺伝子発現カセットの切除を可能にする。場合によっては、遺伝子発現カセットを完全に除去することができ(図5)、他のシナリオでは、遺伝子発現カセットを植物の特異的生成において除去し、最終的にそれらの植物の子孫に再導入することができ、それによって遺伝子発現カセットの再利用が可能となる。欠失したマーカー(図6)配列は、上述のプロトコルを使用して相同組み換え媒介型遺伝子標的法を介して再導入することができる。ヘテロ二量体eZFN部位への遺伝子標的法は、上述のプロトコルを使用して完了する。この実施例において、eZFN結合部位は、形質転換植物からの、選択可能マーカー遺伝子を含む、植物体での任意の導入遺伝子の欠失を可能にするために使用される。2010年1月22日に出願された米国仮特許出願第61/297628号を参照されたく、それは参照により本明細書に組み込まれる。
【0240】
本明細書で言及した全ての特許、特許出願、および刊行物は、参照によりそれらの全体が全目的で本明細書に組み込まれる。
【0241】
理解の明瞭化を目的とした図示および例を用いて、本開示を多少詳しく提供したが、本開示の趣旨または範囲から逸脱することなく、様々な変更および修正を実施することができることは当業者には明らかであろう。したがって、前述の説明および例を限定的であると解釈すべきではない。
【特許請求の範囲】
【請求項1】
亜鉛フィンガーヌクレアーゼの1つ以上の対に対する複数の対形成した標的部位を含む、単離した核酸。
【請求項2】
各対形成した標的部位からの1つの標的部位が、同じ配列を含む、請求項1に記載の核酸分子。
【請求項3】
1つ以上のコード配列を更に含む、請求項1または2に記載の核酸分子。
【請求項4】
請求項1〜3のいずれか1項に記載の核酸分子を含む、細胞または細胞株。
【請求項5】
前記細胞は、植物細胞または哺乳類細胞もしくは哺乳類細胞株等の真核細胞である、請求項1に記載の細胞。
【請求項6】
前記核酸分子は、前記細胞のゲノムに組み込まれる、請求項4または請求項5に記載の細胞。
【請求項7】
1つ以上の外来性配列を細胞のゲノムに組み込むための方法であって、
(a)亜鉛フィンガーヌクレアーゼの1つ以上の対を、請求項6に記載の細胞に提供することであって、前記亜鉛フィンガーヌクレアーゼは、前記組み込まれた核酸分子中の標的部位に結合し、その結果、前記ヌクレアーゼのそれらの標的部位への結合が、前記細胞のゲノムを開裂させる、提供すること、
(b)前記細胞を、外来性配列を含むポリヌクレオチドと接触させることであって、前記外来性配列は、前記組み込まれた核酸内の細胞のゲノムに組み込まれる、接触させること、
を含む、方法。
【請求項8】
追加的な外来性配列の存在下で、前記組み込まれた核酸分子中の追加的な標的部位を開裂させる追加的な亜鉛フィンガーヌクレアーゼを用いて、ステップ(a)および(b)を反復し、それによって前記追加的な外来性配列を、前記細胞のゲノムに挿入することを更に含む、請求項7に記載の方法。
【請求項9】
前記外来性配列のうちの1つ以上は、亜鉛フィンガーヌクレアーゼに対する1つ以上の標的部位を含む、請求項8に記載の方法。
【請求項10】
前記標的部位は、亜鉛フィンガーヌクレアーゼの片側標的部位であり、前記片側標的部位の組み込み時に標的部位が形成される、請求項9に記載の方法。
【請求項11】
前記外来性配列のうちの1つ以上は、コード配列を含み、前記細胞は、前記コード配列の産物を発現する、請求項7〜10のいずれかに記載の方法。
【請求項12】
細胞のゲノムに挿入される1つ以上の配列を欠失させる方法であって、
(a)請求項7〜11のいずれかに記載の複数の外来性配列を組み込むこと、
(b)前記外来性配列のうちの1つ以上がゲノムから欠失させられるように、前記細胞中に適切なヌクレアーゼを発現させること、
を含む、方法。
【請求項13】
ゲノムが変更された細胞を提供する方法であって、
(a)請求項7〜12のいずれかに記載の方法に従って、少なくとも第1の細胞中の1つ以上の外来性配列を組み込むか、または欠失させること、
(b)前記第1の細胞を性的に成熟した生物に発達させること、
(c)前記生物を、ゲノム変更を含む第2の生物と交配させて、前記第1および第2の生物の前記ゲノム変更のうちの少なくとも1つを有する、第2の細胞を生成すること、
を含む、方法。
【請求項14】
前記第2の細胞のゲノム変更は、前記第2の細胞中の単一のゲノム位置における複数の異種遺伝子を含む、請求項13に記載の方法。
【請求項15】
前記細胞は更に、前記組み込まれた核酸分子を含む領域の外側で、そのゲノムの修飾を含む、請求項7〜14のいずれかに記載の方法。
【請求項16】
前記細胞は、植物細胞である、請求項7〜14のいずれかに記載の方法。
【請求項1】
亜鉛フィンガーヌクレアーゼの1つ以上の対に対する複数の対形成した標的部位を含む、単離した核酸。
【請求項2】
各対形成した標的部位からの1つの標的部位が、同じ配列を含む、請求項1に記載の核酸分子。
【請求項3】
1つ以上のコード配列を更に含む、請求項1または2に記載の核酸分子。
【請求項4】
請求項1〜3のいずれか1項に記載の核酸分子を含む、細胞または細胞株。
【請求項5】
前記細胞は、植物細胞または哺乳類細胞もしくは哺乳類細胞株等の真核細胞である、請求項1に記載の細胞。
【請求項6】
前記核酸分子は、前記細胞のゲノムに組み込まれる、請求項4または請求項5に記載の細胞。
【請求項7】
1つ以上の外来性配列を細胞のゲノムに組み込むための方法であって、
(a)亜鉛フィンガーヌクレアーゼの1つ以上の対を、請求項6に記載の細胞に提供することであって、前記亜鉛フィンガーヌクレアーゼは、前記組み込まれた核酸分子中の標的部位に結合し、その結果、前記ヌクレアーゼのそれらの標的部位への結合が、前記細胞のゲノムを開裂させる、提供すること、
(b)前記細胞を、外来性配列を含むポリヌクレオチドと接触させることであって、前記外来性配列は、前記組み込まれた核酸内の細胞のゲノムに組み込まれる、接触させること、
を含む、方法。
【請求項8】
追加的な外来性配列の存在下で、前記組み込まれた核酸分子中の追加的な標的部位を開裂させる追加的な亜鉛フィンガーヌクレアーゼを用いて、ステップ(a)および(b)を反復し、それによって前記追加的な外来性配列を、前記細胞のゲノムに挿入することを更に含む、請求項7に記載の方法。
【請求項9】
前記外来性配列のうちの1つ以上は、亜鉛フィンガーヌクレアーゼに対する1つ以上の標的部位を含む、請求項8に記載の方法。
【請求項10】
前記標的部位は、亜鉛フィンガーヌクレアーゼの片側標的部位であり、前記片側標的部位の組み込み時に標的部位が形成される、請求項9に記載の方法。
【請求項11】
前記外来性配列のうちの1つ以上は、コード配列を含み、前記細胞は、前記コード配列の産物を発現する、請求項7〜10のいずれかに記載の方法。
【請求項12】
細胞のゲノムに挿入される1つ以上の配列を欠失させる方法であって、
(a)請求項7〜11のいずれかに記載の複数の外来性配列を組み込むこと、
(b)前記外来性配列のうちの1つ以上がゲノムから欠失させられるように、前記細胞中に適切なヌクレアーゼを発現させること、
を含む、方法。
【請求項13】
ゲノムが変更された細胞を提供する方法であって、
(a)請求項7〜12のいずれかに記載の方法に従って、少なくとも第1の細胞中の1つ以上の外来性配列を組み込むか、または欠失させること、
(b)前記第1の細胞を性的に成熟した生物に発達させること、
(c)前記生物を、ゲノム変更を含む第2の生物と交配させて、前記第1および第2の生物の前記ゲノム変更のうちの少なくとも1つを有する、第2の細胞を生成すること、
を含む、方法。
【請求項14】
前記第2の細胞のゲノム変更は、前記第2の細胞中の単一のゲノム位置における複数の異種遺伝子を含む、請求項13に記載の方法。
【請求項15】
前記細胞は更に、前記組み込まれた核酸分子を含む領域の外側で、そのゲノムの修飾を含む、請求項7〜14のいずれかに記載の方法。
【請求項16】
前記細胞は、植物細胞である、請求項7〜14のいずれかに記載の方法。
【図1】

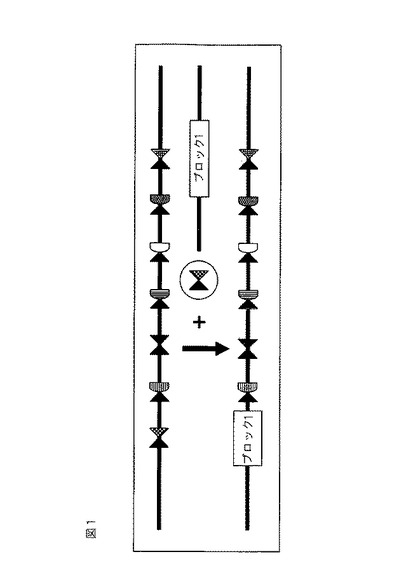
【図2】
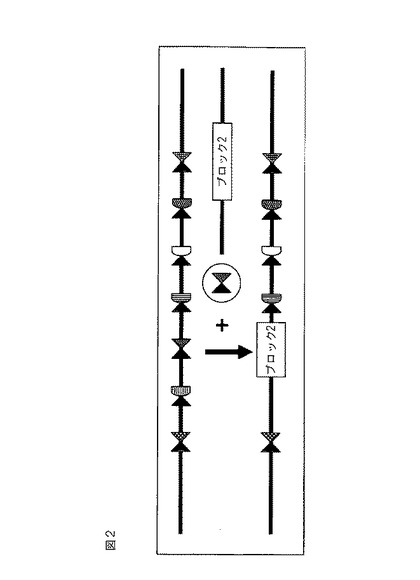
【図3】
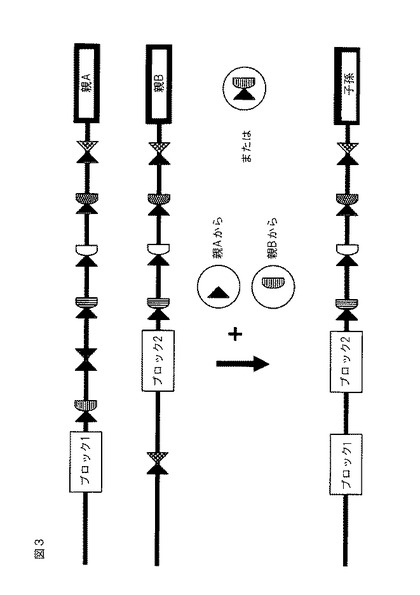
【図4】
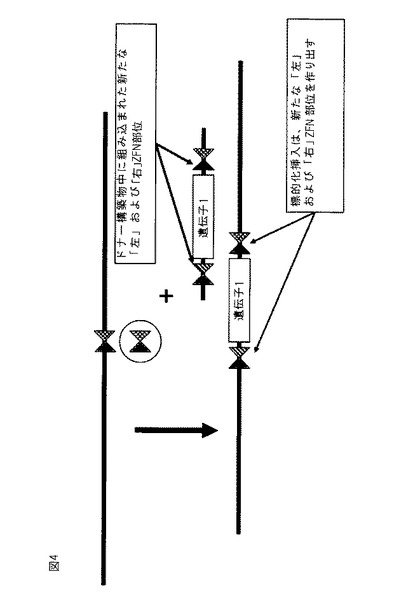
【図5】
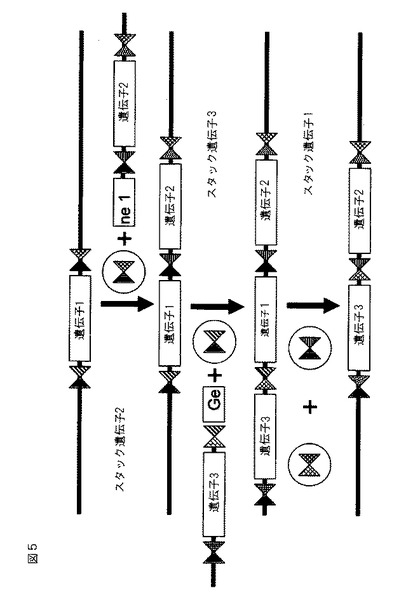
【図6】
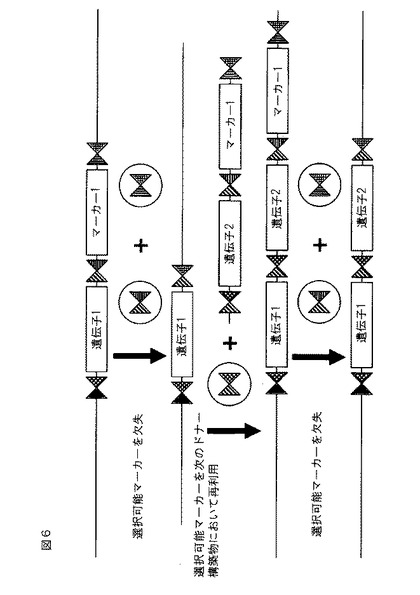
【図7】
【図8】

【図9】
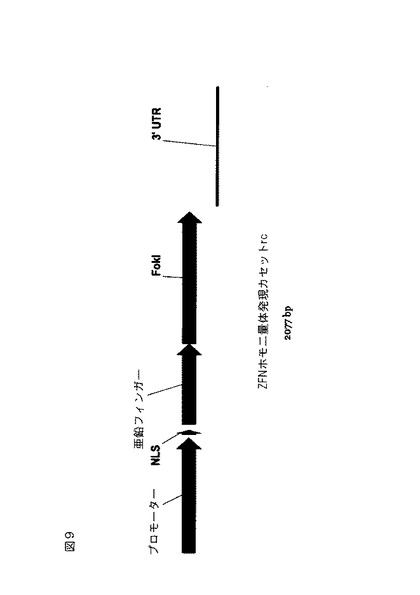
【図10】
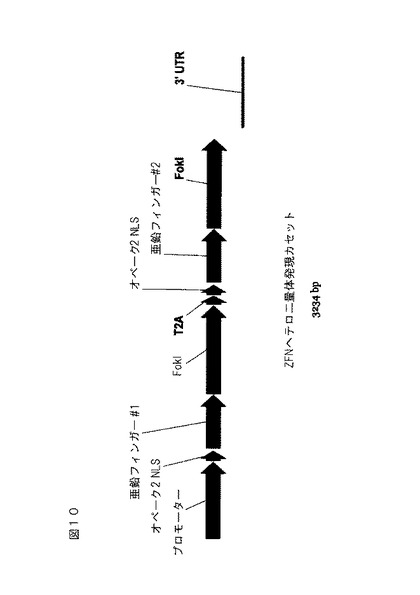
【図11】
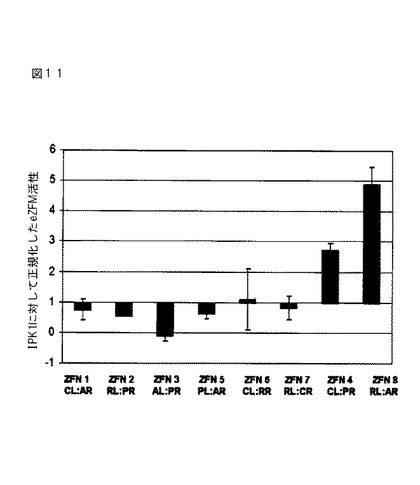
【図12】
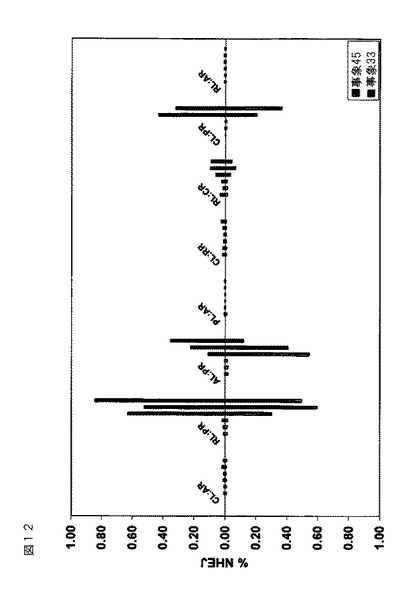
【図13】
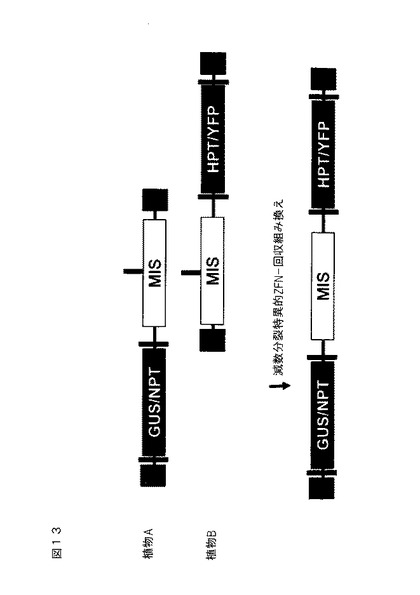
【図14】
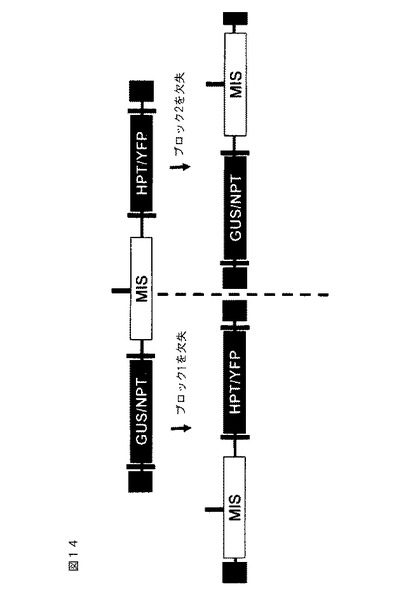
【図15】

【図16】
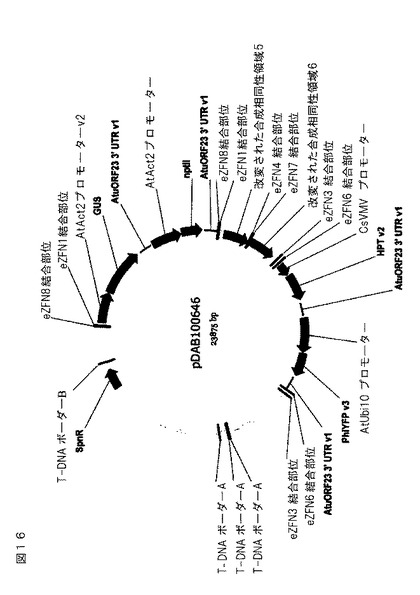
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公表番号】特表2013−517770(P2013−517770A)
【公表日】平成25年5月20日(2013.5.20)
【国際特許分類】
【出願番号】特願2012−550018(P2012−550018)
【出願日】平成23年1月24日(2011.1.24)
【国際出願番号】PCT/US2011/000125
【国際公開番号】WO2011/090804
【国際公開日】平成23年7月28日(2011.7.28)
【出願人】(390039192)ダウ・アグロサイエンス・エル・エル・シー (20)
【氏名又は名称原語表記】Dow AgroSciences LLC
【出願人】(508241200)サンガモ バイオサイエンシーズ, インコーポレイテッド (28)
【Fターム(参考)】
【公表日】平成25年5月20日(2013.5.20)
【国際特許分類】
【出願日】平成23年1月24日(2011.1.24)
【国際出願番号】PCT/US2011/000125
【国際公開番号】WO2011/090804
【国際公開日】平成23年7月28日(2011.7.28)
【出願人】(390039192)ダウ・アグロサイエンス・エル・エル・シー (20)
【氏名又は名称原語表記】Dow AgroSciences LLC
【出願人】(508241200)サンガモ バイオサイエンシーズ, インコーポレイテッド (28)
【Fターム(参考)】
[ Back to top ]