
樹状細胞補助刺激分子
【課題】T細胞活性化についての新規な樹状細胞(DC)特異的補助刺激分子をコードする遺伝子を提供する。
【解決手段】補助刺激タンパク質分子であるB7−DCは、B7ファミリーのメンバーであり、これをコードするDNAおよびこのDNAを含む発現ベクターが記載される。B7−DCタンパク質、フラグメント、融合ポリペプチド/タンパク質および他の機能的誘導体、ならびにB7−DCを発現する形質転換された細胞は、ワクチン組成物および方法に有用である。抗腫瘍性免疫および抗ウイルス性免疫に利用され得る、強力なT細胞媒介性応答を誘導するための組成物および方法。
【解決手段】補助刺激タンパク質分子であるB7−DCは、B7ファミリーのメンバーであり、これをコードするDNAおよびこのDNAを含む発現ベクターが記載される。B7−DCタンパク質、フラグメント、融合ポリペプチド/タンパク質および他の機能的誘導体、ならびにB7−DCを発現する形質転換された細胞は、ワクチン組成物および方法に有用である。抗腫瘍性免疫および抗ウイルス性免疫に利用され得る、強力なT細胞媒介性応答を誘導するための組成物および方法。
【発明の詳細な説明】
【技術分野】
【0001】
生化学および医薬の分野における本発明は、樹状細胞の表面上で選択的に発現され、そして免疫応答を刺激するために細胞表面分子として用いられ得るか、またはワクチン組成物において可溶性形態で用いられ得る新規なタンパク質に関する。
【背景技術】
【0002】
Tリンパ球応答の生成は、細胞間相互作用および可溶性メディエーター(サイトカインまたはリンホカイン)の生成に関与する複雑なプロセスである。この応答は、「レセプター」として働くいくつかのT細胞表面分子によって調節される。このT細胞表面分子としては、T細胞レセプター(TCR)複合体、および他の「補助的な」表面分子(その多くはモノクローナル抗体によって最初に規定された細胞表面の「ディファレンシャル抗原」(「CD分子」)である)が挙げられる。
【0003】
全てのリンパ球の至適な活性化には、以下の2つのシグナルを要すると考えられる:抗原特異的シグナルまたはクローン(clonal)シグナル、および第二の抗原非特異的シグナル(Janeway,C.,Cold Spring Harbor Symp.Quant.Biol.54:1〜14(1989))。リンパ球が、いわゆる補助刺激分子(例えば、以下に記載のB7)による補助刺激なしに、単独の抗原に接触する場合、リンパ球は、「アネルギー」とも呼ばれるクローン性不活性(Schwartz,R,Science 248:1349(1990))、またはアポトーシス(プログラムされた細胞死)のいずれかに応答する;補助刺激シグナルが提供される場合、それは刺激抗原に特異的なクローン性増殖に応答する。所定の抗原に対する免疫応答の有意な増大は、補助刺激なしには生じない(Juneら(Immunology Today 15:321〜331,1994);Chenら(Immunology Today 14:483〜486);Townsend,SEおよびAllison,JP(1993)Science 259:368〜370)。
【0004】
免疫応答の質および力価は、大部分は、T細胞に対して抗原を処理して提示する抗原提示細胞(APC)の型に依存する。TCRの係合に利用可能なペプチド抗原/MHCリガンドの密度、ならびにT細胞の係合および活性化の時点でのAPCによる可溶性のおよび/または膜結合型の補助刺激シグナルの提供が重要である。これらの理由によって、免疫治療ストラテジーは、(a)標的抗原を適切な型のAPCに提供すること、そして(b)適切な補助刺激分子を提供してT細胞活性化を増強することに、集中し始めてきている。
【0005】
T細胞の活性化に必要なシグナルを提供するAPCとしては、単球/マクロファージ、Bリンパ球、そして最も重要である樹状細胞(DC)が挙げられる。過去には、活性化されたマクロファージは、インビボにおいてT細胞応答を開始する重要なAPCであると考えられた。この知見はマクロファージが抗原を効率的に食菌し、そして表面ディスプレイおよび提示のために抗原を処理する能力に基づく。より最近では、抗原特異的T細胞応答のインビボにおける主なイニシエータとして、DCに注意が移ってきた。DCは、活性化マクロファージ由来の異なる表現型を有し、そして異なる免疫応答を開始し得る異なるサブタイプに分類される。DCの機能的な欠点は、インビトロにおいてナイーブなT細胞を活性化する能力がマクロファージよりも約100倍大きいことである。今日まで、この力価の解釈は、抗原提示に重要であると考えられる分子の量的な差異に基づいていた。本発明は、重要な量的差異の発見に基づく。
【0006】
抗原提示における最初のシグナルは、APC上におけるクラスII主要組織適合性複合体(MHC)分子の状況で提示された抗原とTCRとの相互作用によって開始される(Allen、Immunol.Today 8:270(1987))。補助刺激シグナルは、他の分子に由来する。その分子のうちの最も特徴づけされたものはB7ファミリー(すなわち、B7.1、B7.2、およびおそらくB7.3)であり、これはAPC上にも存在する。
【0007】
T細胞の表面上に発現した2つのタンパク質は、B7のような補助刺激分子の最も特徴付けられたリガンドまたは対応レセプターである。CD28は、T細胞活性化において機能する最も成熟したヒトT細胞で見出された免疫グロブリン(Ig)スーパーファミリー(AruffoおよびSeed,Proc.Natl.Acad.Sci.84:8573〜8577(1987))のホモダイマー糖タンパク質である。CD28は、休止しているT細胞で構成的に発現され、そして活性化後に増大する。T細胞レセプターを通じたシグナル伝達後、CD28の連結は、T細胞が増殖してIL−2を分泌するように誘導する(Linsley,PSら(1991)J.Exp.Med.173、721〜730;Gimmi,CDら(1991)Proc.Natl.Acad.Sci.USA.88,6575〜6579;Thompson,C.B.ら(1989)Proc.Natl.Acad.Sci.USA.86,1333〜1337;June,C.H.ら(1990)Immunol.Today.11,211〜6;Harding,F.A.ら(1992)Nature.356,607〜609)。CD28は、免疫応答に必須である抗原依存性の細胞間相互作用の形態である、細胞間接触(「細胞間接着」)を媒介する(Springerら、Ann.Rev.Immunol.5:223〜252(1987))。
【0008】
CTLA4は、CD28に非常に相同であるT細胞表面分子であるが、休止しているT細胞では発現されず、そしてT細胞活性化後に生じる(Brunet,J.Fら(1987)Nature 328,267〜270)。CTLA−4は、マウス細胞傷害性T細胞cDNAライブラリーの示差的スクリーニングによってもともと同定された(Brunetら、前出)。B7の第二のレセプターとしてのCTLA−4の役割は、Linsleyら(1991)J.Exp.Med.174:561〜569(これはまた、B7がCD28についてよりもCTLA4について高い親和性を有することを注記している)に考察されている。Freemanら(1993)Science 262:907〜909は、B7欠損マウスにおけるCTLA−4を考察した。CTLA−4のリガンドは、Lenschowら(1993)Proc.Nat’l.Acad.Sci.90:11054〜11058に記載されている。
【0009】
Th細胞は、Th細胞に対して抗原を提示するB細胞のみの活性化を確実にするように働き、そしてバイスタンダーB細胞の活性化を回避する、Th細胞−B細胞接触の領域において、おそらく集中的な様式で、IL−2、IL−4、およびIL−6のような増殖および分化誘導のサイトカインを分泌する。
【0010】
CD28およびCTLA−4は、B7として一般に公知の補助刺激分子と相互作用する。B7は、B細胞活性化抗原としてもともと記載されている。なぜなら、B7はB細胞上で見出され、そしてB7/BB−1と名づけられたからである(Linsleyら、Proc.Natl.Acad.Sci.USA 87:5031〜5035(1990))。本明細書中において以降は、この分子をB7、B7−1、またはB7.1と呼ぶ。B7(より詳細には、B7ホモログと呼ばれる)はまた、Igスーパーファミリーのメンバーである。CD28およびCTLA−4と比較して、B7は、2つの細胞外Igドメイン(N末端可変(V)様ドメイン、それに続く定常(C)様ドメイン)を含む。
【0011】
B7ファミリーメンバーは、一般にAPC上で発現され、そして注記されるように、ナイーブT細胞の活性化に非常に重要なものである。これらのファミリーのマンバーとしては、B7−1(=B7、CD80とも呼ばれる)、およびB7−2(CD86とも呼ばれる)が挙げられる。B7−1を記載する参考文献としては、Schwartz,R.H.Cell 71:1065〜1068,1992;Chen,Lら、Cell 71:1093〜1102、1992;Freeman,G.J.ら、J.Immunol 143:2714〜2722,1989;およびFreeman,G.J.ら、J.Exp.Med.174:625〜631,1991が挙げられる。B7−2を記載する参考文献としては、Freeman,G.J.ら、Science 262:909〜911 813〜960、1993が挙げられる。現在まで、マウスB7−1およびB7−2ならびにヒトB7−1およびB7−2の両方が記載されている(Freemanら、1989、前出;1991、前出;および1993、前出)。活性化ヒトBリンパ球は、CTLA4/CD28結合カウンターレセプターであるB7−2およびB7−3を発現する。これは両方ともCD28またはCTLA4のいずれかを介してT細胞に補助刺激シグナルを送達し得る。
【0012】
B7−2は、抗IgクラスIImAbまたは抗MHCクラスIImAbのいずれかでの刺激後約24時間でB細胞によって発現される。B7−2は、検出可能IL−2分泌およびT細胞増殖を誘導する。活性化後約48時間〜72時間で、B細胞は、B7−1およびB7−3と名づけられた第三のCTLA4対応レセプター(mAb BB−1によって同定された)(Yokochi,Tら(1982)J.Immunol.128,823〜827)の両方を発現する。B7−3はまた、B7陰性活性化B細胞で発現され、そして検出可能なIL−2産生なしにT細胞増殖を補助刺激し得、このことは、B7−1分子およびB7−3分子が異なることを示す。B7−3は、活性化B細胞、活性化単球、樹状細胞、ランゲルハンス細胞、およびケラチノサイトを含む広範な種々の細胞で発現される。B細胞活性化後72時間で、B7−1およびB7−3の発現が低下し始める。活性化Bリンパ球の表面でのこれらのCTLA4/CD28結合対応レセプターの存在によって、T細胞補助刺激が、部分的には、B細胞活性化後のこれらの分子の一時的発現によって調節されることが示される。
【0013】
B7:CD28/CTLA4補助刺激経路の重要性は、インビトロおよびインビボで実証されている。T細胞活性化の増大とB7発現の増大との間に直接的な関係が存在する(Razi−Wolfら,Proc.Natl.Acad.Sci.USA,89:4210〜4214(1992))。T細胞は、それが補助刺激リガンド(この補助刺激経路のCD28遮断に結合する)を欠く細胞上のペプチド抗原に接触した場合、アネルギーにされ、マウスおよびヒトの系において抗原特異的寛容の発生を生じる(Hardingら、前出;Lenschow,D.J.ら(1992)Science.257,789〜792;Turka、LAら(1992)Proc.Natl.Acad.Sci.USA.89,11102〜11105;Gimmi,CDら(1993)Proc.Natl.Acad.Sci.USA 90、6586〜6590;Boussiotis,V.ら(1993)J.Exp.Med.178、1753〜1763)。逆に、B7−陰性マウス腫瘍細胞によるB7の発現は、腫瘍拒絶によって付随するT細胞媒介特異的免疫、および腫瘍チャレンジに対する長期防御を誘導する(Chen,Lら(1992)Cell 71:1093〜1102;Townsendら、前出;Baskar、Sら(1993)Proc.Natl.Acad.Sci.90,5687〜5690)。従って、B7:CD28/CTLA4経路の操作によって、ヒトにおいて免疫応答を刺激または抑制する大きな能力が得られる。
【0014】
CD28とB7との間の相互作用は、B7またはCD28の細胞外部分とIg Cγ1鎖の遺伝子融合を用いて特徴づけされた(Linsleyら、J.Exp.Med.173:721〜730(1991))。B7Ig融合タンパク質を固定するか、またはB7を細胞(例えば、トランスフェクトされたCHO細胞)の表面で発現する場合、それらはT細胞増殖を補助刺激する。B7+CHO細胞でのT細胞刺激はまた、IL−2の転写物のレベルの増大を特異的に刺激する。
【0015】
米国特許第5,521,288号は、CD28陽性T細胞を、B7をコードするDNAの一部によってコードされるフラグメント(B7の細胞外ドメイン(ECD)に主に対応する)と接触させることによって、免疫応答を調節するための方法を記載する。免疫応答はまた、B7 ECDの少なくとも一部および別のタンパク質(例えば、B7の可溶性、結合親和性、および/または原子価を変化させるヒトIgCγ1ドメイン)を含む融合タンパク質構築物であるB7の誘導体によって調節された。例えば、B7 ECDの位置1〜215に由来するアミノ酸残基をコードするDNAは、ヒトIgCγ1のヒンジ領域、CH2領域およびCH3領域に対応する配列のアミノ酸残基をコードするDNAに連結され、B7Ig融合タンパク質をコードしたDNA融合産物を形成する。また、CD28レセプターを結合することによって、T細胞と反応するB7またはB7Ig融合タンパク質を投与することにより、T細胞によって媒介される免疫系疾患を処置するための方法が開示されている。対宿主性移植片病におけるT細胞増殖を、CD28+T細胞を、B7抗原またはB7Ig融合タンパク質と、免疫抑制剤と組み合わせて、反応させることによって阻害した。
【0016】
米国特許第5,861,310号は、1つ以上のT細胞補助刺激分子(B7−2およびB7−3を含む)を発現するように改変された腫瘍細胞を開示する。1つの実施形態は、B7のさらなる発現を含む。改変は、B7−2タンパク質、B7−3タンパク質またはB7タンパク質をコードする核酸でのトランスフェクションによるものであった。腫瘍細胞はまたインビボで遺伝的に改変され得る。このような改変腫瘍細胞は、腫瘍を有する患者の処置に有用であり、腫瘍の転移性伝播を防止もしくは阻害するために、または腫瘍の再発を阻害するために有用であると言われている。本明細書は、腫瘍に対するCD4+T細胞応答を特異的に誘導するための方法を開示している。
【0017】
米国特許第5,942,607号は、T細胞活性化を補助刺激する新規なCTLA4/CD28リガンドをコードする単離された核酸を開示する。1つの実施形態において、単離された核酸は、B7−2をコードした。また、開示された全長B7−2配列の少なくとも一部を含む核酸を開示している、本明細書に従って、核酸配列は、哺乳動物細胞および昆虫細胞を含む種々の宿主細胞においてタンパク質またはペプチドに対応する合成を指向し得る種々の発現ベクター中に組み込まれ得る。これらの核酸配列によってコードされるタンパク質またはペプチド、および単離されたタンパク質およびペプチド(B7−2配列の少なくとも一部を含む)を生成するように形質転換された宿主細胞がまた開示されている。
【0018】
Dong Hら、Nat Med 1999 5:1365〜1399は、CD28、CTLA4、またはICOS(誘導性補助刺激因子)に結合しないB7−H1と名付けられたB7ファミリーの第三のメンバーを記載している。B7−H1の連結は、T細胞応答をポリクローナル刺激およびアロ抗原に対して補助刺激し、そして優先的にインターロイキン10の生成を刺激した。少量生成されたIL−2は、B7−H1補助刺激の効果を必要とした。この研究によって、細胞媒介性免疫応答の負の調節に関与し得る、以前には未知であった補助刺激分子が規定された。同じ研究室(Wang Sら、Blood.2000;96:2808〜2813)は、B7−H2と名付けられた新しいヒトB7様遺伝子を記載している。この遺伝子の発現は単球由来の未成熟DCの表面で検出された。可溶性のB7−H2およびIgの融合タンパク質である、B7−H2Igは、活性化T細胞には結合したが休止T細胞には結合しない。この結合は、ICOSの可溶性形態(ICOSIg)により阻害されたが、CTLA4Igによっては阻害されなかった。ICOSIgは、B7−H2遺伝子でトランスフェクトされたCHO細胞を染色した。刺激因子としてCD3の最適以下の架橋を用いて、B7−H2IgによるT細胞増殖の補助刺激は、用量依存性でありそしてIL−2の分泌と関連しており、一方至適CD3連結は好ましくはIL−10生成を刺激したことが見出された。著者らは、B7−H2がICOS T細胞分子の推定リガンドであると結論した。
【0019】
Swallow MMら、Immunity,1999,11:423〜432は、APC上で発現されるB7分子の近接するホモログ(相同体)である、新規な遺伝子b7hのクローニングを報告した。B7hは、CD28またはCTLA−4由来の異なるレセプター上で作用することにより精製したT細胞の増殖を補助刺激した。驚くべきことに、B7hは、刺激されていないB細胞で発現されたが、その発現は、TNFαで処理した非リンパ球性細胞(3T3細胞;胎児性線維芽細胞)で誘導され、そしてLPS(TNFαの強力なアクチベータ)で処理されたマウスの非リンパ球性組織で上方制御された。これらの研究は、T細胞の新規な補助刺激リガンドを規定し、そしてTNFαによるB7hの誘導は、炎症中の自己の認識を直接増強し得る。
【0020】
Yoshinaga SKら、Nature,1999、402:827〜832は、新しいマウス補助刺激レセプターリガンド対を記載している。このレセプター(CD28に関連する)は、ヒトタンパク質ICOSのマウスホモログであり、そして活性化T細胞および休止している記憶T細胞で発現された。B7分子に相同であるリガンドは、B7関連タンパク質−1(B7−related protein−1)(B7RP−1)と呼ばれる。B7RP−1は、それぞれマウスB7.1(CD80)およびB7.2(CD86)に対して、20%アミノ酸同一性および19%アミノ酸同一性である1型膜貫通タンパク質である。この相同性は、27%アミノ酸同一性しか共有しないB7.1およびB7.2として有意である(Freeman,GJら、J.Exp.Med.178:2185〜2192(1993))。この相同性は、保存位置でIgループ形成に重要であるシステインを含む(開始メチオニンから残基62、138、185および242)。B7RP−1の膜貫通ドメインの全体的長さおよび相対的位置は、B7分子の長さおよび位置と類似している(Greenfield,EAら、Crit.Rev.Immunol.18:389〜418(1998))。B7RP−1は、B細胞およびマクロファージで発現されることが示された。ICOSおよびB7RP−Iは、CD28−B7経路においてタンパク質と相互作用せず、そしてB7RP−1は、CD28と独立してT細胞を補助刺激した。B7RP−1とIgのFc部分との間の融合タンパク質(「B7−RP1−Fc」)を発現するトランスジェニックマウスは、脾臓においてリンパ過形成、リンパ節およびパイアー斑を有した。インビボにおけるB7RP−1の補助刺激活性は、抗原チャレンジの時点でB7RP−1−Fcで処理された抗原提示化マウスにおいて、増強した遅延型過敏症を実証することによって見出された。著者らは、ICOSおよびB7RP−1が、CD28−B7に構造的に関連し、そして適応的免疫反応に関与する異なる新しいレセプター−リガンド対を規定すると結論した。
【0021】
Yoshinaga SKら、Int Immunol,2000 Oct,12:1439〜1447は、ヒトB7RP−1およびICOS相互作用を通じたヒトT細胞の補助刺激を報告した。このリガンド−レセプター対は、約33nMのKDおよび10分より長いt(1/2)を有するオフレート(off−rate)で相互作用した。TNFαは、B細胞、単球、およびDC上のヒトB7RP−1の発現を示差的に調節した。TNFαは、B細胞および単球上のB7RP−1発現を増強したが、DCの発現は阻害した。ヒトB7RP−1−Fcタンパク質または膜結合したB7RP−1を発現した細胞は、インビトロにおいてT細胞増殖を補助刺激した。特定のサイトカイン(例えば、IFNγおよびIL−10)は、B7RP−1補助刺激によって誘導された。IL−2レベルは有意に増大しなかったが、B7RP−1が誘導した補助刺激は、IL−2に依存した。これらの研究により、マウスB7RP−1に対するヒトオルソログが規定され、そしてヒトICOSとの相互作用が特徴付けられた。
【0022】
PD−1は、活性化T細胞、B細胞および骨髄細胞によって発現された免疫阻害レセプターである。PD−1欠損マウスは、末梢寛容の損失に起因して自己免疫の複数の形態を示した。Freeman、GJら、J.Exp.Med.192:1027〜1034(2000)は、PD−1(PD−L1)のリガンドが、B7遺伝子ファミリーのメンバーであることを報告した。PD−L1によるPD−1の係合によって、TCR媒介リンパ球活性化(増殖、サイトカイン分泌)の阻害が生じた。さらに、PD−1シグナル伝達は、CD28媒介補助刺激の至適以下のレベルを阻害した。PD−L1は、APC(IFNγで刺激されたヒト単球、活性化ヒトDC)により発現される。さらに、PD−L1は、心臓および肺において発現されることが示された。著者らは、APC上の阻害性PD−L1シグナルおよび補助刺激B7−1/B7−2シグナルの相対的な大きさが、T細胞活性化の程度および寛容と自己免疫との間の閾値を決定し得ることを推測した。非リンパ性組織でのPD−L1の存在は、炎症の部位での免疫応答の大きさに寄与し得る。
【0023】
上記の文献の引用は、前述のいずれの文献も先行技術に関連するという承認とは解釈されない。これらの文献の内容についての、今日までの全ての記述または説明は、本出願人に対して利用可能な情報に基づき、そしてこれらの文献の日付または内容の正確さに関してなんらの承認をも構成しない。
【発明の概要】
【発明が解決しようとする課題】
【0024】
T細胞活性化についての新規な樹状細胞(DC)特異的補助刺激分子をコードする遺伝子を同定するために、本発明者らは、DCと活性化マクロファージとの間のサブトラクトcDNAライブラリーをスクリーニングした。このcDNAのサブトラクションアプローチによって、DCによって発現されたが、活性化マクロファージによって発現されない遺伝子が規定される。このアプローチを用いて、T細胞の活性化に依存するワクチンの力価を増強するのに有用ないくつかの新規なDC特異的遺伝子の発見がもたらされた。本出願はこのような遺伝子の1つに焦点をあてる。
【課題を解決するための手段】
【0025】
DCライブラリーにおいて存在し、活性化マクロファージライブラリーから非存在であることに基づいて、新規なコード配列(「B7−DC」と名付けられた)を同定した。B7−DC遺伝子は、補助刺激分子をコードする遺伝子のB7ファミリーのメンバーである。B7−DCは、DC特異的発現および異なるレセプター特異性を有する第一のB7ファミリーメンバーである。この遺伝子の産物は、DCがT細胞を刺激する特有の能力を媒介するのにおいて重要な役割を有する。機能的分析によって、T細胞によるIFNγ産生を刺激する際に、B7−1よりもB7−DCがさらに活性であることが示された。従って、B7−DC DNAおよびポリペプチドは、抗原特異的であろうとなかろうと、細胞性および分子性のワクチン組成物の効力を増強するための組成物および方法において有用である。
【0026】
1つの実施形態において、本発明は、活性化マクロファージと比べて樹状細胞上で選択的に発現されるB7−DCと名付けられた哺乳動物タンパク質をコードする単離された核酸分子を提供する。核酸分子は好ましくは、配列番号1(ヒト起源)、または配列番号5(マウス起源)から選択されたヌクレオチド配列を含む。本発明はまた、ストリンジェントなハイブリダイゼーション条件下で上記の核酸分子にハイブリダイズする単離された核酸に関する。好ましいストリンジェントな条件は、約45℃での6×塩化ナトリウム/クエン酸ナトリウム(SSC)中でのインキュベーション、続いて約50℃の温度での約0.2×SSC中での洗浄を含む。好ましくは、上記の核酸分子は、配列番号1のヌクレオチド配列を含む。上記のような好ましい核酸分子は、配列番号2および配列番号4から選択されたアミノ酸配列を有するタンパク質をコードするか、またはこのタンパク質の生物学的に活性なフラグメント、ホモログ、もしくは他の機能的誘導体をコードする。好ましくは、この核酸分子は、配列番号2(ヒト起源のB7−DC)の配列を有するタンパク質をコードするか、または配列番号2の生物学的に活性なフラグメント、ホモログ、もしくは他の機能的誘導体をコードする。
【0027】
好ましい実施形態において、核酸分子は、B7−DCタンパク質の細胞外ドメイン(残基26〜221を含む)をコードする。この核酸分子は、その補助刺激ホモログ、フラグメントまたは他の機能的誘導体をコードする。
【0028】
別の実施形態において、上記の核酸分子は、B7−DC融合タンパク質をコードし、この核酸分子は以下:
(a)B7−DCタンパク質(好ましくは、配列番号2または配列番号4)の全てまたは一部である第一のポリペプチドをコードする第一の核酸配列;
(b)必要に応じて、この第一の核酸配列とインフレームで融合した、リンカーペプチドをコードするリンカー核酸;および
(c)この第一の核酸配列またはこのリンカー核酸配列にインフレームで結合し、そして第二のポリペプチドをコードする第二の核酸配列、
を含む。この第二のポリペプチドは、好ましくは、Igの重鎖定常領域の1つ以上のドメイン、好ましくはヒトIgG(好ましくはIgG1)の2つのCドメイン、から構成される。
【0029】
発現ベクターであって、以下:
(a)プロモーター、および
(b)必要に応じて、真核生物細胞において核酸の発現を調節するさらなる調節配列、
に作動可能に連結された上記の任意の核酸分子、
を含む発現ベクターも提供される。
【0030】
上記の発現ベクターは、プラスミドまたはウイルスベクターであり得る。これらのベクターとしては、自己複製するRNAレプリコン(DNA−ランチャー、またはRNA)、自殺RNAベクター、DNAウイルス(例えば、アデノウイルス、ワクシニアウイルスなど)、およびRNAビリオン(パッケージング細胞株で増殖する)が挙げられる。
【0031】
ベクターDNAまたはRNAは、例えば、所望の標的細胞および組織への送達を増強する、徐放性処方物中で、宿主への遺伝子銃媒介導入のための金粒子に複合体化されてもよいし、または他のポリマーと複合体化されてもよい。
【0032】
また、以下:
(a)第一の組み換え発現ベクターであって、該ベクターは、免疫応答が誘導される目的の抗原をコードするヌクレオチド配列を、その配列中に組み込んでいる、ベクター;および
(b)第二の組み換え発現ベクターであって、該ベクターは、補助刺激ポリペプチド(このポリペプチドの少なくとも1つは、B7−DCであるか、またはその生物学的に活性なフラグメント、ホモログ、もしくは他の機能的誘導体である)をコードする1つ以上のヌクレオチド配列をその核酸配列中に組み込んでおり、ここで該発現ベクターは、宿主細胞に同時感染または同時トランスフェクトし得、抗原および補助刺激ポリペプチド、複合体、ホモログもしくは誘導体の同時発現を生じる、ベクター、
を含むベクター組成物も含まれる。
【0033】
上記の実施形態の改変において、本発明は、以下:(i)細胞間(好ましくは、APC)で発現された産物(抗原)の伝播を促進し、(ii)核酸が発現されるAPCでの抗原の提示を増大し、そして/または(iii)ベクターが導入されている宿主のAPCにおける抗原の再提示(交差プライミング)および提示を促進する、標的タンパク質をコードする第三の核酸配列を提供する。この標的化タンパク質コード核酸は、抗原または補助刺激因子またはその両方をコードする核酸に融合され得る。第一または第二のベクターは、核酸を含む。1つの実施形態において、このベクター組成物は、これらの抗原コード核酸、補助刺激コード核酸(好ましくはB7−DC)、および「標的化」タンパク質コード核酸を、単一の融合構築物中に組みあわせる。
【0034】
本発明は、上記の任意の核酸分子または発現ベクターで形質転換またはトランスフェクトされた細胞を含む。この細胞は、好ましくは真核生物細胞、より好ましくは哺乳動物細胞、最も好ましくはヒト細胞である。この細胞は、樹状細胞またはその前駆体であり得る。別の実施形態において、この細胞は、腫瘍細胞、好ましくは抗原(宿主における腫瘍上の抗原(それに対して免疫応答が所望される)と同じであるか、またはその抗原と交差反応性である抗原)を保有する腫瘍細胞である。
【0035】
好ましい実施形態は、哺乳動物B7−DCタンパク質(好ましくは配列番号2または配列番号4)、またはその生物学的に活性なフラグメント、ホモログ、もしくは他の機能的誘導体をコードする外因性核酸分子でトランスフェクトされた単離された哺乳動物腫瘍細胞であり、その結果、この腫瘍細胞によって、このタンパク質、フラグメント、ホモログ、または誘導体が発現される場合、そしてこの腫瘍細胞がT細胞と接触する場合、
(i)B7−DCタンパク質、フラグメント、ホモログ、または誘導体が、T細胞に結合し;そして
(ii)この腫瘍細胞は、T細胞を補助刺激して増殖し、そして/またはサイトカインを産生し、そして分泌する。
【0036】
本発明はまた、活性化マクロファージと比べて樹状細胞で選択的に発現され、そして以下:
(a)T細胞上の結合パートナーに結合する;
(b)T細胞を補助刺激して増殖し、そして/またはサイトカインを産生し、そして分泌する、
という機能的特性を有するポリペプチドに関連する。
【0037】
このポリペプチドの生物学的に活性なフラグメント、ホモログまたは他の機能的誘導体もまた含まれる。
【0038】
このポリペプチド、フラグメント、ホモログまたは機能的誘導体は、好ましくは、配列番号1または配列番号5の配列を有する核酸分子、またはこの核酸分子のフラグメント、ホモログもしくは等価物によってコードされる。好ましいポリペプチドは、配列番号2または配列番号4のアミノ酸配列を有する。
【0039】
このポリペプチドまたはこのポリペプチドの生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体は、上記核酸の一つの組換え発現によって、生成され得る。
【0040】
好ましいポリペプチドは、B7−DCタンパク質の細胞外ドメインを含み、好ましくは、以下:
(a)配列番号2(ヒト)のアミノ酸残基26〜221または
(b)配列番号4(マウス)のアミノ酸残基26〜221
である。上記ポリペプチドは、B7−DCの細胞外ドメインから「本質的になり」得る。
【0041】
(i)第二のポリペプチドに直接的に融合しているか、または
(ii)必要に応じて、この第二のポリペプチドに融合したリンカーペプチド配列に融合している、
B7−DCタンパク質の全てまたは一部を含む、第一の融合パートナーを有するB7−DC融合ポリペプチドもまた、提供される。
【0042】
上記A B7−DC融合タンパク質はまた、第二のポリペプチドに融合し得、この第二のポリペプチドは、好ましくは、Ig重鎖定常領域の1つ以上のドメインであり、好ましくは、ヒト免疫グロブリンCγ1鎖のヒンジ領域、CH2領域およびCH3領域に対応するアミノ酸配列を有する。
【0043】
上記融合タンパク質の1つの実施形態において、第一の融合パートナーは、B7−DCタンパク質の細胞外ドメインであり、これの全長配列は、配列番号2または配列番号4である。
【0044】
この融合タンパク質は、好ましくは、T細胞上の結合パートナーに結合し、そしてT細胞レセプターに対する十分な刺激の存在下で、T細胞を補助刺激する。
【0045】
上記融合タンパク質のダイマーまたはトリマーである、ダイマーまたはトリマーの融合タンパク質もまた提供される。好ましくは、これらの鎖は、ジスルフィド結合または他の鎖間共有結合を介して、縦列で連結される。
【0046】
好ましいダイマー融合タンパク質において、このダイマーは、Ig重鎖の2つのCH領域のCys残基の共有結合から生じ、これらのCys残基は、二量化した通常のIg H鎖において、ジスルフィド結合しているものと同じCys残基である。
【0047】
本発明の融合タンパク質は、直接的にかまたは1つ以上の単量体間のリンカー配列を伴って、末端同士で結合した第一の融合パートナーの2つ以上の繰返し体のマルチマーを含み得る。
【0048】
本発明はまた、B7−DCタンパク質のエピトープに対して特異的な抗体を提供し、このエピトープは、B7ファミリータンパク質の公知のメンバーには存在しない。このエピトープは、配列番号2または配列番号4のポリペプチドの「直鎖またはコンホメーション」エピトープであり得る。この抗体は、好ましくは、モノクローナル抗体であり、より好ましくは、ヒトまたはヒト化(操作による)モノクローナル抗体である。
【0049】
上記抗体を使用して、細胞集団においてB7−DCポリペプチドをその表面において発現する細胞を同定または定量する方法もまた提供され、この方法は、以下の工程を包含する:
(a)この抗体がこのエピトープを発現する細胞に結合するように、この集団の細胞を、上記抗体と接触させる工程;
(b)この抗体が結合する細胞の数の存在を評価するかまたはその細胞の数を定量する工程。
【0050】
細胞集団から、その表面でB7−DCポリペプチドを発現する細胞を単離するための、別の方法が提供され、この方法は、以下の工程を包含する:
(a)この抗体がこのエピトープを発現する細胞に結合するように、この集団を、上記抗体に接触させる工程;
(b)この抗体が結合する細胞をポジティブに選択するか、またはこの抗体が結合しない細胞をネガティブに選択する工程。
【0051】
サンプル中のB7−DCポリペプチド、フラグメントまたはホモログの存在を検出するかまたは定量する方法もまた提供され、この方法は、以下の工程を包含する:
(a)この抗体が、このエピトープを保有する任意のポリペプチドまたはフラグメントに結合するように、このサンプルを請求項43の抗体と接触させる工程;
(b)この抗体に結合するポリペプチドまたはフラグメントの存在を検出するか、または定量する工程。
【0052】
本発明はまた、抗原提示細胞またはその前駆細胞におけるB7−DCポリペプチドの発現を誘導または増加して、この細胞が、インビトロまたはインビボで、T細胞レセプターに対する十分な刺激の存在下で、T細胞を補助刺激する能力を増加させる方法に関し、この方法は、B7−DCポリペプチドの発現がその細胞において誘導または増加されるように、抗原提示細胞または前駆細胞を上記のような発現ベクターで形質転換またはトランスフェクトする工程を包含する。この抗原提示細胞は、好ましくは、樹状細胞であり、そして前駆細胞は、樹状細胞前駆細胞である。
【0053】
本発明は、細胞補助刺激組成物およびポリペプチド補助刺激剤を使用して、免疫応答を刺激するための方法を提供する。抗原性刺激に対する哺乳動物被験体のT細胞応答を増加させるための1つの方法は、この被験体に、有効量の上記細胞(好ましくは、腫瘍細胞)を、抗原性刺激と組み合わせて投与する工程を包含し、ここで、これらの細胞は、この抗原性刺激に対するこの被験体のT細胞応答を増加するために効果的である。上記のことは、好ましくは、抗原と補助刺激組成物との同時注入によって、達成される。
【0054】
抗原性刺激に対する哺乳動物被験体のT細胞応答を、腫瘍関連抗原で増加させるための方法は、この被験体に、有効量の上記のような腫瘍細胞を投与する工程を包含し、ここで、これらの腫瘍細胞は、この抗原を発現し、この腫瘍細胞の投与は、腫瘍抗原刺激に対するこの被験体のT細胞応答を増加させるために効果的である。
【0055】
抗原性刺激に対する哺乳動物被験体のT細胞応答を増加させるための方法は、この被験体に、有効量のポリペプチド、上記のようなフラグメント、ホモログもしくは機能的誘導体、または上記のような融合ポリペプチドもしくはタンパク質を、抗原性刺激と組み合わせて投与する工程を包含し、ここで、このポリペプチドの投与は、抗原性刺激に対するこの被験体のT細胞応答を増加させるために効果的である。
【0056】
本発明はまた、抗原性刺激に対する哺乳動物被験体のT細胞応答を阻害するための方法を提供し、この方法は、この被験体に、有効量の上記のような「抗体」を投与する工程を包含し、ここで、この抗体の投与は、T細胞の刺激をブロックするかまたは抗原反応性T細胞を排除するために効果的であり、これによって、T細胞応答を阻害する。これらの方法は、組織または器官の移植を有する被験体を処置して、移植片の拒絶を阻害するためおよび/または移植を促進するために、特に有用である。自己抗原の場合には、この方法は、自己免疫応答およびこれらの病理学的続発症をブロックまたは低下させる。
【0057】
本発明は、本発明の組成物でのエキソビボ刺激を受けたT細胞を使用する、治療方法を提供する。抗原性刺激に対する哺乳動物被験体の免疫応答を増加させるための1つの方法は、以下:
(a)この被験体から、この被験体に免疫学的に適合性のドナーから、または免疫学的に受容可能な培養細胞株から、T細胞を得る工程;
(b)このT細胞を、エキソビボで、有効量の上記のような細胞と接触させる工程であって、ここで、この接触は、抗原性刺激に対するT細胞の応答を増加させるために効果的である、工程;および
(c)工程(b)のT細胞を、この被験体に投与する工程、
を包含し、これによって、この被験体の免疫応答を増加させる。
【0058】
別の実施形態において、抗原性刺激に対する哺乳動物被験体の免疫応答を増加させるための方法は、以下の工程:
(a)被験体から、この被験体に対して免疫学的に適合性のドナーから、または免疫学的に受容可能な培養細胞株から、T細胞を得る工程;
(b)このT細胞をエキソビボで有効量の以下:(i)上記のようなポリペプチド、フラグメント、ホモログまたは機能的誘導体、あるいは(ii)上記のような融合ポリペプチドと接触させる工程であって、ここで、この接触は、抗原性刺激に対するT細胞の応答を増加させるために効果的である、工程;ならびに
(c)工程(b)のT細胞をこの被験体に投与する工程、
を包含し、これによって、この被験体の免疫応答を増加(または発生)させる。
【0059】
本明細書中にはまた、ワクチン組成物もまた提供され、このワクチン組成物は、以下を含有する:
(a)(i)B7−DC構築物を発現する、上記のような細胞、(ii)B7−DCポリペプチド、フラグメント、ホモログまたは機能的誘導体、(iii)B7−DC融合ポリペプチドまたはタンパク質;
(b)一般に、免疫応答が所望される抗原のさらなる供給源(これは、細胞自体がこの抗原を発現する、細胞に基づくワクチンの場合には、必要とされないかもしれないが(腫瘍抗原を保有する腫瘍細胞の場合のように));
(c)必要に応じて、一般的な免疫刺激因子またはアジュバント;ならびに
(d)(a)、(b)および(c)のための薬学的かつ免疫学的に受容可能な賦形剤またはキャリア。
【0060】
哺乳動物被験体において抗原に対する免疫応答を増加または増強するための方法は、この被験体に、有効量の上記ワクチン組成物を投与する工程を包含する。
【0061】
抗原またはワクチンと共に使用するための、補助刺激組成物もまた提供され、この組成物は、以下を含有する:
(a)B7−DCポリペプチド(好ましくは、配列番号2または配列番号4)、そのフラグメント、ホモログもしくは機能的誘導体、あるいはB7−DC融合ポリペプチド、ならびに
(b)薬学的かつ免疫学的に受容可能な賦形剤またはキャリア。
【0062】
哺乳動物被験体において、抗原またはワクチンに対する免疫応答を相乗するための方法は、この被験体に、この抗原またはワクチンと組み合わせて、上記補助刺激組成物を投与する工程を包含する。
【0063】
被験体において、腫瘍に対する全身性免疫応答を刺激する方法は、この被験体に、遺伝子改変された腫瘍細胞を投与する工程を包含し、この細胞は:
(a)この被験体中の腫瘍細胞由来であり、そして
(b)上記のようなB7−DC核酸のエキソビボでの導入によって遺伝子改変されており、
この核酸の発現は、この被験体において補助刺激シグナルを提供し、ここで、この投与は、この腫瘍に対する全身性免疫応答の刺激を生じる。
【0064】
これらの腫瘍細胞は、これらが投与された後のこれらの増殖が防止されるように、好ましくは照射によって、好ましくは処理されている。
【0065】
この被験体は、上記治療組成物の投与の前に、化学療法、照射または外科的切除の腫瘍減少レジメンに供され得る。
【0066】
抗原陽性腫瘍を有する哺乳動物において、抗腫瘍応答を誘導する方法もまた提供され、この方法は、以下:
(a)(i)この哺乳動物の腫瘍と共通する抗原を発現し;
(ii)上記のようにB7−DCコード核酸ベクターでトランスフェクトされ、これは、B7−DC分子として発現される場合に、この細胞に、この腫瘍の抗原に対するT細胞応答を補助刺激させ;
(iii)必要に応じて、工程(b)の前に照射される、腫瘍または腫瘍細胞株の細胞を提供する工程;
(b)有効数の細胞をこの哺乳動物に投与する工程であって、この細胞は、この抗原およびB7−DC分子を発現する、工程;
を包含し、これによって、抗腫瘍応答を誘導する。
【0067】
上記方法において、抗腫瘍応答は、以下によって特徴付けられる:
(A)全ての測定可能な病巣の最大垂直直径の積の合計の、少なくとも50%の減少;
(B)新たな病巣の証拠がないこと、および
(C)任意の先在病巣の進行がないこと。
【0068】
腫瘍を保有する哺乳動物において、腫瘍の一次増殖または再増殖の抑制または弱化を誘導する方法もまた提供され、この方法は、以下:
(a)(i)この哺乳動物の腫瘍と共通する抗原を発現し;
(ii)上記のようにB7−DCコード核酸ベクターでトランスフェクトされ、これは、B7−DC分子として発現される場合に、この細胞に、この腫瘍の抗原に対するT細胞応答を補助刺激させ;
(iii)必要に応じて、工程(b)の前に照射される、腫瘍または腫瘍細胞株の細胞を提供する工程;
(b)有効数の細胞をこの哺乳動物に投与する工程であって、この細胞は、この抗原およびB7−DC分子を発現する、工程;
を包含し、これによって、黒色腫の腫瘍細胞に対して特異的な全身性免疫応答を誘導し、これによって、この抑制または弱化を誘導する。
【0069】
哺乳動物における抗原陽性腫瘍の反復性の増殖を阻害する方法は、以下の工程を包含する:
(a)(i)この哺乳動物の腫瘍と共通する抗原を発現し;
(ii)上記のようにB7−DCコード核酸ベクターでトランスフェクトされ、これは、発現される場合に、この細胞に、この腫瘍の抗原に対するT細胞応答を補助刺激させ;
(iii)必要に応じて、工程(b)の前に照射される、腫瘍または腫瘍細胞株の細胞を提供する工程;
(b)有効数の細胞をこの哺乳動物に投与する工程であって、この細胞は、この抗原およびB7−DC分子を発現する、工程;
を包含し、これによって、この哺乳動物において腫瘍抗原に対して特異的な全身性免疫応答を誘導し、この免疫応答が、腫瘍の反復性の増殖を阻害する。
【0070】
別の実施形態は、哺乳動物被験体に局所的に投与された抗原の近隣に、補助刺激シグナルを提供して、この抗原に対する全身性免疫の状態を生じる炎症応答および免疫応答の局所的発生を促進する方法に関し、この方法は、被験体における局所部位に、以下:
(a)補助刺激に有効な量の、上記のようなB7−DCポリペプチド、フラグメント、ホモログまたは機能的誘導体を発現する細胞、および
(b)抗原
を投与する工程を包含し、その結果、この抗原に物理的に近い位置での補助刺激が、この応答の局所的発生を促進し、そして全身性免疫の状態を生じる。
【0071】
上記方法において、抗原は、好ましくは、工程(b)において、腫瘍細胞または非細胞抗原性物質の形態で投与される、腫瘍抗原である。この腫瘍細胞はまた、工程(a)において、B7−DCポリペプチド、フラグメント、ホモログまたは誘導体を発現する細胞であり得る。
【図面の簡単な説明】
【0072】
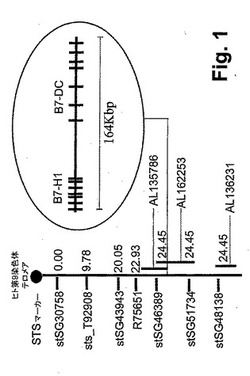
【図1】図1は、ヒト染色体9p24に局在したhB7−DCのマップを示す、ダイヤグラムである。hB7−DCは、BACクローンRPCI−11.2にマッピングされる。
【図2】図2は、B7−DCが、DCとマクロファージとの間で差示的に発現されることを示す。骨髄DC、脾性DC、マクロファージ、マクロファージ株および組織におけるB7−DC mRNAの分布を、0.5μg/レーンの精製したDNAを使用して、1%アガロースゲルで泳動する、バーチャルノーザンブロット分析によって評価した。G3PDHを、コントロールとして使用した。J774A1細胞、Raw264.7細胞、Pu5−1.8細胞およびWEHI細胞は、マクロファージ細胞株である。BM:骨髄。
【図3】図3は、ヒトDCにおけるB7−DC発現のバーチャルノーザンブロットを示す。レーン1は、GM−CSFおよびFlt−3Lと共に培養されたヒトDCを示し、レーン2は、ヒト胎盤を示し、そしてレーン3は、GM−CSFおよびIL4と共に培養されたヒトDCを示す。ヒトB7−DCの5’および3’UTR由来のオリゴヌクレオチドを使用して、ヒトDCの全RNAのバーチャルノーザン分析のためのPCR DNAプローブを作製した。β−アクチンをコントロールとして使用して、mRNAの品質を確認した。
【図4】図4は、成熟BM−DCにおけるB7−DCの表面発現を示す、フローサイトメトリー分析を代表する。9日目に、マウスBM−DCをFcブロックし、そしてコントロール抗体またはB7−DC抗血清を用いて染色した。結合の特異性を、DCの表面への抗B7−DCの結合に対して競合するためのB7−DC−Igを添加することによって、実証した。
【図5】図5は、B7−DCがPD−1に結合するが、CTLA−4にもCD28にも結合しないことを示す。293T細胞を、pCAGGS−B7.1 o pCAGGS−B7−DCを用いて、一時的にトランスフェクトした。トランスフェクト体を、PD−1−Ig融合分子、28−Ig融合分子およびCTLA−4−Ig融合分子で染色し、続いてPE標識した二次抗体で染色した。pCAGGS(空のベクター)トランスフェクト体の染色は、ネガティブであった(示さず)。
【図6】図6(左右のパネル)は、抗CD3およびB7−DC−IgによるT細胞増殖の補助刺激を示す。左側のグラフ:精製T細胞(CD4+CD8)を、次第に増加する濃度の抗CD3(mAb 2C11)および一定濃度(0.1μg/ml)の固定したB7.1−Ig(黒菱形)、B7−DC−Ig(黒丸)またはアイソタイプコントロール(黒三角)で予めコートしたウェル内で培養した。結果は、3つのうちの1つの代表的な実験を示す。細胞を、72時間インキュベートし、そして3H−チミジンで標識した。CPMとは、1分間あたりの計数である。右側のグラフ:精製CD8 T細胞を、次第に増加する濃度の抗CD3および一定濃度の固定したB7.1−Ig(黒菱形)、B7−DC−Ig(黒丸)またはアイソタイプコントロール(黒三角)で(a)においてと同様に予めコートしたウェル内で、培養した。結果は、2つのうちの1つの代表的な実験のものである。細胞を72時間インキュベートし、そして3H−チミジンで標識した。CPMとは、1分間あたりの計数である。
【図7】図7は、抗原特異的T細胞増殖応答の補助刺激を示す。RENCA細胞をIFNγで72時間処理して、MHCクラスIIの発現を誘導し、そして12.5μg/mlのHA110−120ペプチドとともにインキュベートした。精製したHAおよびI−Ed特異的トランスジェニックT細胞を、次第に増加する濃度のB7.1−Ig(黒菱形)、B7−DC−Ig(黒丸)またはアイソタイプコントロール(黒三角)のいずれかと共に、可溶化形態で添加した。細胞を48時間インキュベートし、そして3H−チミジンで標識した。CPMとは、1分間あたりの計数である。結果は、3つのうちの1つの代表的な実験である。
【図8】図8は、B7−DCによって補助刺激されたT細胞のサイトカイン分泌を示す。上のパネル:精製したT細胞を、抗CD3(0.12μg/ml)および0.1μg/mlの固定化B7.1−Ig(黒菱形)、B7−DC−Ig(黒丸)またはアイソタイプコントロール(黒三角)で、図6(左)と同様に予めコートしたウェル内で培養した。結果は、3つのうちの1つの代表的な実験を示す。下のパネル:12.5μg/ml HA(110−120)ペプチドで負荷したγ−IFN処理したRENCA細胞を、精製したHAおよびI−Ed特異的トランスジェニックT細胞と共に、2μg/mlの可溶性B7.1−Ig、B7−DC−Igまたはアイソタイプコントロール(記号は上記のとおり)と一緒にインキュベートした。結果は、2つのうちの1つの代表的な実験を示す。上清を、24時間および48時間の培養の後に収集し、そしてELISAを使用して、示されるリンホカインについてアッセイした。
【図9】図9は、B7−DC−Igが、インビボ補助刺激の後に、抗原特異的増殖を大いに増強することを示す。HAに対して特異的な2.5×106のTCRトランスジェニック細胞の養子移入の後に、3群のマウスを、HAペプチド(110−120)、不完全フロイントアジュバント(IFA)のいずれかで、単独でかまたはB7−DC−IgおよびIFAまたはIFAを含むアイソタイプコントロール抗体のいずれかとの組合せで、後肢裏において皮下で免疫した。リンパ節の排液を、7日目に獲得した。1.5×105のLN細胞を、HAペプチドと共に48時間インキュベートし、1μCi[3H]チミジンでパルス化し、そして12時間後に組み込んだ放射能を、決定した。
【発明を実施するための形態】
【0073】
本発明者らは、ここで、改善された免疫治療組成物および方法のための基礎として働く、新たなタンパク質および核酸を同定した。ヒトおよびマウス形態の、新規補助刺激タンパク質(B7−DCと命名された)が発見され、そして本明細書中に開示される。
【0074】
ヒトB7−DCをコードするDNAは、以下に示す配列番号1のヌクレオチド配列を有する:
【0075】
【化1】

ヒトB7−DCタンパク質は、以下に示す配列番号2のアミノ酸配列を有する(リーダー配列、膜貫通ドメインおよび細胞質テイルが注釈を付けられている):
【化2】
このタンパク質の細胞外ドメインは、残基P26〜残基W221由来である。
【0076】
マウスB7−DCをコードするコード配列を含むDNAクローンは、以下に示す配列番号3のヌクレオチド配列を有する。このコード配列(下線を引かれた、三つ組みで示される)は、ヌクレオチド210で開始するメチオニンコドンatg(太字)から開始し、そしてtag終止コドン(太字)(ヌクレオチド951〜953)で終止する。
【化3】
配列番号5は、配列番号3のコード配列部分である。
【0077】
配列番号3のコード領域によって(すなわち、配列番号5によって)コードされるマウスB7−DCタンパク質は、以下に示す配列番号4のアミノ酸配列を有する(リーダー配列、膜貫通ドメインおよび細胞質テイルが注釈をつけられている):
【化4】
このタンパク質の細胞外ドメインは、残基P26〜残基W221由来である。
マウスB7−DCの完全DNA配列(もともとは「ブチロフィリン様タンパク質」または「Btdc」と称された)は、Genbank登録番号AF142780.2を有する。
【0078】
(基本的な分子アプローチ)
このアプローチは、実施例により詳細に記載される。本発明者らは、2つの重要な特徴を組み込むPCR Selectアプローチを利用した。第1に、ハイブリダイゼーション工程前の最初のPCR反応は、少量のRNAのみを必要とする。この技術は、夾雑するマクロファージ、前駆体細胞または他の潜在的な夾雑細胞を実質的に含まないようになった高度に精製された成熟DCの使用を可能にする。このような高度に精製されたDCを非常に多数で得ることが困難であることは公知である。第2に、PCR Select手順は、低いコピー数の差次的に発現される遺伝子のクローニングを可能にする。
【0079】
その細胞性の対応物である活性化マクロファージと比較して、DCによって差次的に発現される遺伝子を同定するために、ならびにDC特異的機能と関連する遺伝子を同定するために、本発明者らは、cDNAサブトラクションアプローチを適用した。これは、抑制PCR(PCR SelectTM)と組み合わせた、改変型PCRベースの「代表差次的分析(representative differential analysis)」(RDA)を使用した(Diatchenko,L.ら,Proc.Natl.Acad.Sci USA 93:66025−6030(1996))。
【0080】
(一般的な組み換えDNA方法)
分子生物学の一般的な方法を開示する基本書(それらの全てが、参考として援用される)としては、以下が挙げられる:Sambrook,Jら,Molecular Cloning:A Laboratory Manual,第2版,Cold Spring Harbor Press,Cold Spring Harbor,NY,1989;Ausubel,FMら、Current Protocols in Molecular Biology,Vol.2,Wiley−Interscience,New York(最新版);Kriegler,Gene Transfer and Expression:A Laboratory Manual(1990);Glover,DM,編,DNA Cloning:A Practical Approach,vol.I & II,IRL Press,1985;Albers,B.ら,Molecular Biology of the Cell,第2版,Garland Publishing,Inc.,New York,NY(1989);Watson,JDら,Recombinant DNA,第2版,Scientific American Books,New York,1992;ならびにOld,RWら,Principles of Gene Manipulation:An Introduction to Genetic Engineering,第2版,University of California Press,Berkeley,CA(1981)。
【0081】
他に示されない限り、特定の核酸配列は、その保存的置換(例えば、縮重コドン置換)改変体および相補配列を含むことが意図される。用語「核酸」は、「ポリヌクレオチド」と同義であり、そして遺伝子、cDNA分子、mRNA分子、ならびにこれらのいずれかのフラグメント(例えば、オリゴヌクレオチド)を含み、そしてさらに、これらの等価物を含むことが意図される(より完全に以下に説明される)。核酸のサイズは、キロベース(kb)または塩基対(bp)のいずれかで示される。これらは、アガロースまたはポリアクリルアミドゲル電気泳動(PAGE)から、使用者によって決定される核酸配列から、または公開される核酸配列から誘導される推定値である。タンパク質サイズは、キロダルトン(kDa)の分子量または長さ(アミノ酸残基の数)で示される。タンパク質サイズは、PAGEから、配列決定から、コード核酸配列に基づく推定アミノ酸配列から、または公開されるアミノ酸配列から概算される。
【0082】
詳細には、B7−DCまたはそのフラグメントもしくは誘導体に対応するアミノ酸配列をコードするcDNA分子は、本明細書中に開示されるタンパク質の配列由来のプライマーを用いてポリメラーゼ連鎖反応(PCR)(例えば、米国特許第4,683,202号を参照のこと)によって合成され得る。次いでこれらのcDNA配列は、真核生物発現ベクターまたは原核生物発現ベクターに組み立てられ得、そして生じるベクターが使用されて、適切な宿主細胞(例えば、COS細胞またはCHO細胞)によってB7−DCまたはそのフラグメントもしくは誘導体の合成を指向し得る。
【0083】
本発明は、新規B7−DCをコードするヌクレオチド配列を有する単離された核酸、そのフラグメントまたはその等価物を含む。用語、核酸は、本明細書中に使用される場合、そのようなフラグメントまたは等価物を含むことが意図される。本発明の核酸配列は、DNAであってもRNAであってもよい。好ましい核酸は、配列番号1の配列を有するヒトB7−DCをコードするcDNAまたはその等価物である。
【0084】
好ましくは、本発明の核酸は、B7−DCの少なくとも一部をコードするcDNA分子である。このcDNAは、成熟DCまたはこのタンパク質を天然に発現する他の細胞から抽出されたmRNAから作製され得る。B7−DCをコードする核酸配列は、DCゲノムDNAから入手可能である。従って、B7−DCをコードするDNAは、公知のプロトコールに従ってcDNAライブラリーまたはゲノムライブラリーからクローン化され得る。
【0085】
B7−DCをコードするcDNAヌクレオチド配列は、適切な細胞株から総mRNAを単離することによって入手され得る。二本鎖cDNAが総mRNAから調製される。cDNAが、多数の公知の技術のうちのいずれか1つを用いて、適切なプラスミド、バクテリオファージまたはウイルスベクターに挿入され得る。
【0086】
ヌクレオチド配列に関して、用語「等価」は、構造的に相同なタンパク質および/または機能的に等価なタンパク質をコードする配列を含むことが意図される。例えば、B7−DCヌクレオチド配列の天然の多型(特にコドンの第3の塩基において)は、アミノ酸配列を変更しない「サイレント」な変異として公知であり得る。しかし、B7−DC中のアミノ酸配列変化を含む多型が、ヒト(または他の哺乳動物)集団中に存在し得る。1つ以上のヌクレオチド中に変化を有するこれらの対立遺伝子改変体(コード配列の総数のうちの約3〜4%まで)が、天然の対立遺伝子バリエーションに起因してヒト集団中におそらく見出されるということを、当業者は認識する。B7−DCをコードするDNAにおける、これらの対立遺伝子改変体ならびに生じる核酸多型およびポリペプチド多型のいずれかおよび全てが、本発明の範囲内である。
【0087】
さらに、本明細書中に記載されるB7−DCタンパク質の、1つ以上の天然に存在するアイソフォームまたは関連する免疫学的に交差反応性のファミリーのメンバーが存在し得る。このようなアイソフォームまたはファミリーのメンバーは、たとえそれらが異なる遺伝子座の遺伝子によってコードされるとしても、B7−DCに対して機能アミノ酸配列の類似性を共有するタンパク質として規定される。
【0088】
(核酸のフラグメント)
核酸配列のフラグメントは、全長B7−DCタンパク質をコードするヌクレオチド配列よりも少ないヌクレオチドを有するヌクレオチド配列として規定される。本発明は、(1)T細胞上でその天然のリガンドに結合するB7−DCの能力を保持し、かつ(2)活性化T細胞媒介免疫応答(一次活性化シグナルを受けたT細胞によるサイトカイン産生および/またはT細胞増殖として測定される)を(それらがどのように提示されるかに依存して)増強または阻害するポリペプチドをコードする、核酸フラグメントを含む。
【0089】
例えば、本明細書中に意図されるような核酸フラグメントは、未だ同定されていない(しかし、CD28またはCTLA−4ではないようである)レセプターに対してT細胞表面に結合し、そして補助刺激シグナルをTリンパ球に送達する能力を保持するB7−DCポリペプチドをコードする。別の基準によって、本発明の核酸フラグメントは、別の動物種由来の核酸とハイブリダイズする核酸フラグメントであり、ゆえに、B7−DCと「交差反応性」である新規タンパク質を検出するためのスクリーニングアッセイにおいて有用である。
【0090】
一般的に、B7−DCポリペプチドのフラグメントをコードする核酸配列は、成熟タンパク質をコードする配列由来のヌクレオチドを含む。しかし、いくつかの場合、核酸のリーダー配列部分の全てまたは一部を含むことが、望ましくあり得る。本発明の核酸配列はまた、リンカー配列、天然または改変された制限エンドヌクレアーゼ部位、およびコードされるタンパク質もしくはフラグメントのクローニング、発現または精製に関連する操作のために有用である他の配列を含み得る。核酸配列のこれらおよび他の改変は、本明細書中に記載されるか、または当該分野で周知である。
【0091】
1つの実施形態において、B7−DCのECDに対応するアミノ酸配列(おおよそ26〜221位のアミノ酸を含む)をコードするDNAは、PCRを用いて、ヒトIgCγ1のヒンジ領域、CH2領域、CH3領域に対応するアミノ酸配列をコードするDNAに連結されて、B7−DC−Ig融合タンパク質として発現される構築物を形成する。
【0092】
B7−Ig融合タンパク質をコードする類似のDNA分子は、米国特許第5,521,288号に開示され、そして登録番号68627の下でAmerican Type Culture Collection(Rockville,Md.)に寄託されている。
【0093】
B7−DCおよび可溶性B7−DC融合タンパク質をコードするDNAを組み立て、そして発現させるための技術(例えば、オリゴヌクレオチド合成、PCR、細胞の形質転換、ベクターの構築、発現系など)は、当該分野で十分に確立されている。当業者は、特定の条件および手順のための標準的な供給原材料に精通している。
【0094】
他の実施形態において、B7−DCのドメインまたはフラグメントをコードするDNAは、別のB7ファミリータンパク質(例えば、B7.1、B7.2、またはB7.3)の残りの部分のほとんどまたは全てをコードする核酸と融合される。ヒトB7.1(CD80)の完全DNA配列は、Genbank登録番号X60958を有し;マウス配列についての登録番号は、X60958であり;ラット配列についての登録番号は、U05593である。ヒトB7.2(CD86)の完全cDNA配列は、Genbank登録番号L25259を有し;マウス配列についての登録番号は、L25606である。
【0095】
(発現ベクターおよび宿主細胞)
本発明は、少なくとも1つの調節配列に作動可能に連結されたB7−DCポリペプチドをコードする核酸配列を含む発現ベクターを含む。「作動可能に連結された」は、コード配列の発現を可能にする条件下で、コード配列が調節配列に連結されていることを意味する。既知の調節配列が選択されて、適切な宿主細胞中での所望のタンパク質の発現を指向する。従って、用語「調節配列」は、プロモーター、エンハンサーおよび他の発現制御エレメントを含む。このような調節配列は、例えば、Goeddel,Gene Expression Technology.Methods in Enzymology,vol.185,Academic Press,San Diego,Calif.(1990)に記載される。
【0096】
当業者は、本発明の発現ベクターの特定の設計が、考慮(例えば、トランスフェクトされる宿主細胞および/または発現されるタンパク質の型)に依存することを、認識する。
【0097】
本発明の発現ベクターは、B7−DCの種々の実施形態(全長タンパク質およびその機能的誘導体(本明細書中に規定される)(ポリペプチドフラグメント、改変体、融合タンパク質などを含む))をコードする核酸分子の完全な範囲を含む。従って、1つの実施形態において、発現ベクターは、単独または別のタンパク質に融合した、B7−DCタンパク質の少なくとも一部(例えば、ECD)をコードする核酸を含む。
【0098】
このような発現ベクターが使用されて、DNAの発現のためおよび融合タンパク質またはペプチドを含むコードされるタンパク質の産生のために、宿主細胞にトランスフェクトされる。B7−DCポリペプチドを発現する遺伝学的に改変された細胞は、細胞が示された目的のために有用であるために十分な時間、外因性DNAを一過性に発現し得ることが理解される。従って、細胞が、インビボまたはエキソビボにおいて増大した補助刺激能力を有する免疫源として作用する場合、発現が必要とされる時間の長さまたは細胞が生存したままである長さは、細胞がその免疫原性機能および/または補助刺激機能と発揮するために必要とされる時間である。例えば、本発明のB7−DCを発現する形質導入された腫瘍細胞の場合、B7−DCの発現は、少なくとも6時間、好ましくは24時間、より好ましくは少なくとも2〜4日間であり得る。当然ながら、発現はまた、安定(すなわち、その細胞の寿命の間、安定)であり得る。以下に考察される適切な発現ベクターおよび調節エレメント(例えば、誘導性プロモーターまたは構成性プロモーター)は、所望または必要とされる発現の安定性に従って選択される。
【0099】
本発明において、本発明は、B7−DCタンパク質、フラグメント、および誘導体を産生するための方法を提供する。例えば、B7−DCタンパク質の少なくとも一部をコードする核酸ベクターを用いてトランスフェクトされた宿主細胞は、B7−DCポリペプチドの発現を可能にする適切な条件下で培養される。
【0100】
宿主細胞はまた、B7−DCタンパク質の少なくとも一部をコードするDNAおよび第2タンパク質の少なくとも一部をコードするDNAを、単独でかまたは組み合わせて含む1つ以上の発現ベクターを用いてトランスフェクトされ得、その結果、この宿主細胞は、両方の部分を含む融合ポリペプチドを産生する。
【0101】
組み換え発現ベクターが、B7−DCの一部をコードするDNAおよび別のタンパク質(例えば、ヒトIgCγ1)をコードするDNAを含む場合、生じる融合タンパク質は、変更された可溶性、結合親和性および/または結合価を有し得る。例えば、B7−DC Ig融合タンパク質は、好ましくは、培養物においてトランスフェクトされた宿主細胞によって分泌され、ゆえに、この培養培地から単離される。あるいは、タンパク質が細胞質に保持される場合、細胞が収集され、溶解され、そしてタンパク質がこの溶解物から単離される。
【0102】
培養は、代表的に、宿主細胞、適切な増殖培地、および他の副生成物を含む。適切な培養培地は、当該分野で周知である。B7−DCタンパク質は、タンパク質およびペプチドを精製するための従来技術を用いて培地または細胞溶解物から単離され得、これには、硫酸アンモニウム沈澱、分画カラムクロマトグラフィー(例えば、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィー、アフィニティークロマトグラフィーなど)、および/または電気泳動が挙げられる(一般的に、「Enzyme Purification and Related Techniques」Methods in Enzymology,22:233−577(1971)を参照のこと)。一旦、部分的または均質的にまで精製されると、本発明の組み換えB7−DCタンパク質は、本明細書中により詳細に記載されるように薬学的組成物中に使用され得る。
【0103】
形質転換またはトランスフェクトされてB7−DCまたはそのホモログもしくは機能的誘導体を発現する、原核生物宿主細胞または真核生物宿主細胞は、本発明の範囲内である。例えば、B7−DCは、E.coliのような細菌細胞、昆虫細胞(バキュロウイルス)、酵母、またはチャイニーズハムスター卵巣細胞(CHO)もしくはヒト細胞のような哺乳動物細胞中で発現され得る。他の適切な宿主細胞は、Goeddel(1990)(前出)に見出され得るか、さもなくば当業者に公知である。
【0104】
真核生物細胞における発現は、部分的もしくは完全なグリコシル化および/または組み換えタンパク質の関連する鎖間ジスルフィド結合もしくは鎖内ジスルフィド結合の形成を導く。
【0105】
酵母、S.cerevisiaeにおける発現のためのベクターの例としては、pYepSec1(Baldariら(1987)EMBO J.6:229−234)、pMFa(Kurjanら(1982)Cell 30:933−943)、pJRY88(Schultzら(1987)Gene 54:113−123)、およびpYES2(Invitrogen Corporation,San Diego,Calif.)が挙げられる。培養される昆虫細胞(SF9細胞)におけるタンパク質発現のために利用可能なバキュロウイルスベクターとしては、pAcシリーズ(Smithら(1983)Mol.Cell Biol.3:2156−2165)およびpVLシリーズ(Lucklow,V.A.,およびSummers,M.D.(1989)Virology 170:31−39)が挙げられる。一般的に、COS細胞(Gluzman,Y.(1981)Cell 23:175−182)が、哺乳動物細胞における一過性の増殖/発現のためにpCDM8のようなベクター(Aruffo A.およびSeed,B.(前出))と組み合わせて使用され、一方、CHO(dhfr−ネガティブCHO)細胞が、哺乳動物細胞における安定な増殖/発現のためにpMT2PCのようなベクター(Kaufmanら(1987)EMBO J.6:187−195)と共に使用される。NS0骨髄腫細胞株(グルタミンシンセターゼ発現系)が、Celltech Ltd.から利用可能である。
【0106】
しばしば、融合発現ベクターにおいて、タンパク質分解性切断部位が、レポーター基および標的タンパク質の連結部に導入されて、融合タンパク質の精製に続く、標的タンパク質のレポーター基からの分離を可能にする。このような切断およびその認識配列に対するタンパク質分解性酵素としては、第Xa因子、トロンビンおよびエンテロキナーゼが挙げられる。
【0107】
代表的な融合発現ベクターとしては、pGEX(Amrad Corp.,Melbourne,Australia)、pMAL(New England Biolabs,Beverly,Mass.)およびpRIT5(Pharmacia,Piscataway,N.J.)が挙げられ、これらはそれぞれ、グルタチオンS−トランスフェラーゼ、マルトースE結合タンパク質、プロテインAを標的組み換えタンパク質に融合する。
【0108】
誘導可能な非融合発現ベクターとしては、pTrc(Amannら(1988)Gene 69:301−315)およびpET 11d(Studierら,Gene Expression Technology:Methods in Enzymology 185,Academic Press,San Diego,Calif.(1990)60−89)が挙げられる。標的遺伝子発現は、pTrcにおいてハイブリッドtrp−lac融合プロモーターからの宿主RNAポリメラーゼ転写に依存するが、pET11dに挿入された標的遺伝子の発現は、同時発現されるウイルスRNAポリメラーゼ(T7gn1)によって媒介されるT7 gn10−lacO融合プロモーターからの転写に依存する。これは、ウイルスポリメラーゼであり、lacUV5プロモーターの転写制御下で、T7gn1を保有するレジデントλプロファージから宿主株BL21(DE3)またはHMS174(DE3)によって供給される。
【0109】
本発明の1つの実施形態は、新規B7−DCを新規に発現するトランスフェクトされた細胞である。すでにB7−DCを発現する細胞(例えば、成熟DC)の場合、トランスフェクトされた細胞は、細胞表面上に増加した量のB7−DCタンパク質またはそのフラグメントを発現させる。
【0110】
例えば、腫瘍細胞(例えば、肉腫、黒色腫、白血病、リンパ球、癌腫または神経芽細胞腫)は、腫瘍細胞表面上でのB7−DCの発現を指向する発現ベクターでトランスフェクトされる。このようなトランスフェクトされた腫瘍細胞は、本明細書中に記載されるように治療的抗腫瘍免疫を誘導するための免疫原として使用され得る。
【0111】
(ベクター構築)
所望されるコード配列および制御配列を含む適切なベクターの構築は、当該分野で十分に理解された標準的な連結技術および制限技術を使用する。単離されたプラスミド、DNA配列、または合成オリゴヌクレオチドは、所望される形態に、切断され、調整され、そして連結され得る。
【0112】
ベクターを形成するDNA配列は、多数の供給源から利用可能である。骨格のベクターおよびコントロール系は、一般的に構築において大量の配列のために使用される利用可能な「宿主」ベクター上に見出される。適切なコード配列について、最初の構築物は、適切な配列をcDNAライブラリーまたはゲノムDNAライブラリーから回収することが問題であるかもしれず、通常はこれが問題である。しかし、一旦配列が開示されると、個々のヌクレオチド誘導体から開始して、全遺伝子配列をインビトロで合成することが可能である。かなり大きいサイズ(例えば、500〜1000bp)の長さの遺伝子についての全遺伝子配列は、個々の重複する相補オリゴヌクレオチドを合成し、そして一本鎖の非重複部分をデオキシリボヌクレオチド三リン酸の存在下でDNAポリメラーゼを用いて充填することによって、調製され得る。このアプローチは、既知配列のいくつかの遺伝子の構築の場合に好首尾に使用されている。例えば、Edge,M.D.,Nature(1981)292:756;Nambair,K.P.ら,Science(1984)223:1299;およびJay,E.,J Biol Chem(1984)259:6311を参照のこと。
【0113】
合成オリゴヌクレオチドは、上記に引用される参考文献によって記載されるようなホスホトリエステル方法によってか、またはBeaucage,S.L.,およびCaruthers,M.H.,Tet Lett(1981)22:1859;ならびにMatteucci,M.D.,およびCaruthers,M.H.,J Am Chem Soc(1981)103:3185によって記載されるようなホスホロアミダイト方法のいずれかによって調製され、そして市販の自動化オリゴヌクレオチド合成機を用いて調製され得る。アニーリング前または標識のための一本鎖のキナーゼ処理は、50mM Tris(pH7.6)、10mM MgCl2、5mM ジチオスレイトール、1〜2mM ATP、1.7pmole γ−32P−ATP(2.9mCi/mmole)、0.1mM スペルミジン、0.1mM EDTAの存在下、過剰のポリヌクレオチドキナーゼ(例えば、1nmoleの基質に対して約10ユニットのポリヌクレオチドキナーゼ)を使用することによって達成される。
【0114】
一旦所望されるベクターの成分がこのように利用可能になると、これらは、標準的な制限手順および連結手順を用いて、切り出され、そして連結され得る。部位特異的DNA切断は、当該分野で一般的に理解される条件下で適切な制限酵素(単数または複数)を用いて処理することによって実行され、そしてこの詳細は、市販の制限酵素の製造業者らによって特定される。例えば、New England Biolabsの製品カタログを参照のこと。一般的に、約1mgのプラスミドまたはDNA配列が、約20mlの緩衝溶液中で1ユニットの酵素によって切断される;本明細書中の実施例において、代表的に、過剰の制限酵素が、DNA基質の完全な消化を確実にするために使用される。37℃で約1時間〜2時間のインキュベーション時間が、実行可能であるが、バリエーションが許容され得る。各インキュベーション後、タンパク質が、フェノール/クロロホルムを用いた抽出によって除去され、そしてエーテル抽出が続き得、そして核酸が、エタノールを用いる沈澱によって水性画分から回収される。所望される場合、切断されたフラグメントのサイズ分画は、標準的な技術を用いてポリアクリルアミドゲルまたはアガロースゲル電気泳動によって実行され得る。サイズ分画の一般的な説明は、Methods in Enzymology(1980)65:499−560に見出される。
【0115】
制限切断されたフラグメントは、4つのデオキシヌクレオチド三リン酸(dNTP)の存在下、20℃〜25℃で約15〜25分間のインキュベーション時間を用いて、50mM Tris(pH7.6)、50mM NaCl、6mM MgCl2、6mM DTTおよび0.1〜1.0mM dNTP中でE.coli DNAポリメラーゼI(Klenow)の大きなフラグメントを用いて処理することによって、平滑末端化処理し得る。Klenowフラグメントは、5’一本鎖オーバーハングを充填するが、たとえ4つのdNTPが存在していても突出している(protruding)3’一本鎖を破壊する。所望される場合、選択的な修復が、オーバーハングの性質によって規定される制限内で、1種類のdNTPだけ、または選択されたdNTPを供給することによって、実行され得る。Klenowを用いた処理後、混合物を、フェノール/クロロホルムを用いて抽出し、そしてエタノール沈澱する。S1ヌクレアーゼまたはBAL−31を用いての適切な条件下での処理は、いずれかの一本鎖部分の加水分解をもたらす。
【0116】
連結は、代表的に、例えば、以下の標準的な条件および温度の下で、15〜50mlの容量で実行される:20mM Tris−HCl(pH7.5)、10mM MgCl2、10mM DTT、33μg/ml BSA、10〜50mM NaCl、40μM ATP、0.01〜0.02(Weiss)ユニットのT4 DNAリガーゼ、0℃(「粘着性末端」連結について)または1mM ATP、0.3〜0.6(Weiss)ユニットのT4 DNAリガーゼ、14℃(「平滑末端」連結について)のいずれか。分子間「粘着末端」連結は、通常、33〜100μg/mlの総DNA濃度(5〜100nMの末端濃度)で実行される。分子間平滑末端連結は、1mMの総末端濃度で実行される。
【0117】
「ベクターフラグメント」を使用するベクター構築の際、フラグメントは、一般的に、5’ホスフェートを除去し、そして自己連結を回避するために、細菌アルカリホスファターゼ(BAP)またはウシ腸アルカリホスファターゼ(CIAP)を用いて処理される。消化は、おおよそ10mM Tris−HCl、1mM EDTA中、pH8、60°で、約1時間かけて約1ユニット/mgベクターでBAPまたはCIAPを用いて実行される。調製物は、フェノール/クロロホルムを用いて抽出され、そしてエタノール沈澱される。あるいは、さらなる制限酵素および所望されないフラグメントの分離によって二重に消化されたベクターにおいて、再連結が回避され得る。
【0118】
多数の方法のうちのいずれかが使用されて、コード配列中に変異を誘導して、本発明の改変体を作製する。これらの変異としては、単純な欠失または挿入、体系的な欠失、挿入または塩基クラスターの置換あるいは1塩基の置換が挙げられる。
【0119】
例えば、B7−DC DNA配列(cDNAまたはゲノムDNA)の改変は、プロトコールおよび試薬が市販されている周知技術である部位指向型変異誘発によって作製される(Zoller,MJら,Nucleic Acids Res(1982)10:6487−6500およびAdelman,JPら,DNA(1983)2:183−193))。プラスミド構築のための正確な連結が、例えば、最初にE.coli株MC1061(Casadaban,M.ら,J Mol Biol(1980)138:179−207)、または他の適切な宿主を、連結混合物を用いて形質転換することによって確立される。従来方法を用いて、形質転換体は、プラスミド構築の様式に依存して、アンピシリン耐性遺伝子、テトラサイクリン耐性遺伝子、または他の抗生物質耐性遺伝子(または他の選択マーカー)の存在に基づいて選択される。次いで、プラスミドは、任意のクロラムフェニコール増幅(必要に応じてクロラムフェニコール増幅が続く)を用いて形質転換体から調製される(Clewell,DBら,Proc Natl Acad Sci USA(1969)62:1159;Clewell,D.B.,J Bacteriol(1972)110:667)。いくつかのミニDNAプレップが、一般的に使用される。例えば、Holmes,DSら,Anal Biochem(1981)114:193−197;Birnboim,HCら,Nucleic Acids Res(1979)7:1513−1523を参照のこと。単離されたDNAは、制限によって分析され、そして/またはMessingら,Nucleic Acids Res(1981)9:309によってさらに記載されるようなSanger(Proc Natl Acad Sci USA(1977)74:5463)のジデオキシヌクレオチド方法、またはMaxamら、Methods in Enzymology(1980)65:499の方法によって配列決定される。
【0120】
ベクターDNAは、従来の技術、例えば、リン酸カルシウムまたは塩化カルシウム共沈澱、DEAE−デキストラン媒介トランスフェクション、リポフェクション、またはエレクトロポレーションを介して、哺乳動物細胞に導入され得る。宿主細胞を形質転換するための適切な方法は、Sambrookら(前出)および他の標準的な文書に見出され得る。
【0121】
しばしば、融合発現ベクターにおいて、タンパク質分解性切断部位が、レポーター基および標的タンパク質の連結部に導入されて、融合タンパク質の精製に続く、標的タンパク質のレポーター基からの分離を可能にする。このような切断およびその認識配列に対するタンパク質分解性酵素としては、第Xa因子、トロンビンおよびエンテロキナーゼが挙げられる。
【0122】
代表的な融合発現ベクターとしては、pGEX(Amrad Corp.,Melbourne,Australia)、pMAL(New England Biolabs,Beverly,Mass.)およびpRIT5(Pharmacia,Piscataway,N.J.)が挙げられ、これらはそれぞれ、グルタチオンS−トランスフェラーゼ、マルトースE結合タンパク質、プロテインAを標的組み換えタンパク質に融合する。
【0123】
誘導可能な非融合発現ベクターとしては、pTrc(Amannら(1988)Gene 69:301−315)およびpET 11d(Studierら,Gene Expression Technology:Methods in Enzymology 185,Academic Press,San Diego,Calif.(1990)60−89)が挙げられる。標的遺伝子発現は、pTrcにおいてハイブリッドtrp−lac融合プロモーターからの宿主RNAポリメラーゼ転写に依存するが、pET11dに挿入された標的遺伝子の発現は、同時発現されるウイルスRNAポリメラーゼ(T7gn1)によって媒介されるT7 gn10−lacO融合プロモーターからの転写に依存する。これは、ウイルスポリメラーゼであり、lacUV5プロモーターの転写制御下で、T7gn1を保有するレジデントλプロファージから宿主株BL21(DE3)またはHMS174(DE3)によって供給される。
【0124】
(プロモーターおよびエンハンサー)
DNA分子またはRNA分子のプロモーター領域は、RNAポリメラーゼに結合し、そして「作動可能に連結された」核酸配列の転写を促進する。本明細書中に使用される場合、「プロモーター配列」は、RNAポリメラーゼによって転写されるDNAまたはRNAの鎖の上に見出されるプロモーターのヌクレオチド配列である。核酸分子の2つの配列(例えば、プロモーター配列およびコード配列)は、両方の配列が同じRNA転写物上に転写されることを可能にするか、または1つの配列において開始するRNA転写物が第2配列に伸長されることを可能にする様式で、これらが互いに連結している場合、「作動可能に連結されている」。従って、2つの配列(例えば、DNAまたはRNAのプロモーター配列およびコード配列)は、プロモーター配列から始まる転写物が作動可能に連結されたコード配列のRNA転写物を産生する場合、作動可能に連結されている。「作動可能に連結された」状態であるためには、2つの配列が、直線配列中で互いに直ぐに隣接する必要はない。
【0125】
本発明の好ましいプロモーター配列は、哺乳動物細胞において作動可能でなければならず、そして真核生物プロモーターまたはウイルス性プロモーターのいずれかであり得る。適切なプロモーターは、誘導可能、抑制可能または構成的であり得る。構成的プロモーターの例は、ウイルス性プロモーターMSV−LTRであり、これは、種々の細胞において有効であり、かつ活性であり、そしてほとんどの他のプロモーターとは対照的に、停止細胞および増殖中の細胞において同じ高い活性を有する。他の好ましいウイルス性プロモーターとしては、CMV−LTR(サイトメガロウイルス由来)(Bashart,M.ら、Cell 41:521(1985))またはRSV−LTR(ラウス肉腫ウイルス由来)(Gorman,C.M.,Proc.Natl.Acad.Sci.USA 79:6777(1982))に存在するウイルス性プロモーターが挙げられる。マウスメタロチオネインI遺伝子のプロモーター(Hamer,D.ら、J.Mol.Appl.Gen.1:273−288(1982));ヘルペスウイルスのTKプロモーター(McKnight,S.,Cell 31:355−365(1982));SV40初期プロモーター(Benoist,C.ら、Nature 290:304−310(1981));および酵母gal4遺伝子プロモーター(Johnston,S.A.ら、Proc.Natl.Acad.Sci.(USA)79:6971−6975(1982);Silver,P.A.ら、Proc.Natl.Acad.Sci(USA)81:5951−5955(1984))もまた、有用である。プロモーター領域に関連する転写因子および分離活性化および転写因子のDNA結合の他の例示的記載としては、以下が挙げられる:Keeganら、Nature(1986)231:699;Fieldsら、Nature(1989)340:245;Jones,Cell(1990)61:9;Lewin,Cell(1990)61:1161;Ptashneら、Nature(1990)346:329;Adamsら、Cell(1993)72:306。これらの上記の参考文献全ての関連する開示は、参考として本明細書に援用される。
【0126】
プロモーター領域はさらに、特異的な組織に見出される特定のタンパク質との相互作用によって組織特異的エンハンサーとして機能もし得る八量体領域を含み得る。本発明のDNA構築物のエンハンサードメインは、トランスフェクトされるべき標的細胞に特異的かまたは、このような標的細胞の細胞性因子によって高く活性化されるものである。ベクター(プラスミドまたはレトロウイルス)の例は、(Roy−Burmanら、米国特許第5,112,767号)に開示される。エンハンサーおよび転写におけるその作用の一般的な議論については、Lewin,B.M.,Genes IV,Oxford University Press,Oxford、(1990)、pp.552−576を参照のこと。特に有用なものは、レトロウイルスエンハンサー(例えば、ウイルス性LTRである)。エンハンサーは、好ましくは、プロモーターから上流に配置され、相互作用して遺伝子発現を刺激する。レトロウイルスベクターの使用のために、内因性ウイルスLTRは、エンハンサーがなくされ、そして組織特異性または他の所望の特性(例えば、本発明のB7−DCコードDNA分子に対する転写効果)を与える他の所望のエンハンサー配列で置換され得る。
【0127】
本発明の核酸配列はまた、標準的技術を用いて化学的に合成され得る。ポリデオキシヌクレオチドを化学的に合成する種々の方法は公知であり、市販されるDNA合成装置で完全に自動化されている固相合成(ペプチド合成など)を含む(例えば、本明細書中に援用されるItakuraら、米国特許第4,598,049号;Caruthersら、米国特許第4,458,066号;ならびにItakura 米国特許第4,401,796号および同第4,373,071号を参照のこと)。
【0128】
(タンパク質およびポリペプチド)
本発明は、配列番号2または配列番号4の配列を有する「単離された」B7−DCタンパク質を含む。本開示は、全長ヒトおよびマウスB7−DCタンパク質(およびDNA)を例示するが、本明細書中に開示される特徴を所有する他の哺乳動物種由来のB7−DCの相同体およびその変異体が、本発明の範囲内であることを意図されることは理解されるべきである。
【0129】
含まれるものはまた、アミノ酸置換改変体を意味するB7−DCの「機能的な誘導体」、「フラグメント」、またはB7−DCの「化学誘導体」である。これらの用語は、下で定義される。機能的誘導体は、測定可能なB7−DC活性、好ましくはT細胞上のレセプターに結合する活性および本発明に従ってその有用性を可能にするT細胞活性を補助刺激する活性を保持する。「機能的誘導体」は、この用語が本明細書中に接続語でまたは選択肢で使用されるか否かによらず「改変体」および「フラグメント」を含む。
【0130】
機能的相同体は、上の生化学的活性および生物学的活性を保有しなければならない。この機能的特徴付けを考慮して、他の種由来の相同体タンパク質B7−DC(まだ発見されていないタンパク質を含む)の使用は、これらのタンパク質が配列類似性ならびに上記の生化学的活性および生物学的活性を有する場合、本発明の範囲内に含まれる。
【0131】
2つのアミノ酸配列のまたは、2つの核酸配列のパーセント同一性を測定するために、この配列は、最適な比較目的のためにアラインメントされる(例えば、ギャップが、最適なアラインメントのために第1のアミノ酸および第2のアミノ酸の1つまたは両方、あるいは核酸配列において導入され得、そして非相同配列は比較目的のために無視され得る)。好ましいアラインメントの方法において、Cys残基がアラインメントされる。
【0132】
好ましい実施形態において、比較されるべき配列の長さは、参照配列の長さの少なくとも30%、好ましくは少なくとも40%、より好ましくは少なくとも50%、さらにより好ましくは少なくとも60%、そしてより好ましくは少なくとも70%、80%、または90%である。例えば、276アミノ酸残基を有するヒトB7−DCタンパク質アミノ酸配列(配列番号2)に対して第2の配列をアラインメントする場合、少なくとも83アミノ酸残基、好ましくは少なくとも110アミノ酸残基、より好ましくは少なくとも138アミノ酸残基、さらにより好ましくは少なくとも166アミノ酸残基、そしてより好ましくは193アミノ酸残基、221アミノ酸残基または248アミノ酸残基が、アラインメントされる。次いで、対応するアミノ酸位置(またはヌクレオチド位置)におけるアミノ酸残基(またはヌクレオチド)が、比較される。次いで第1の配列における位置が、第2の配列における対応位置と同じアミノ酸残基(またはヌクレオチド)によって占有される場合、この分子は、その位置で同一性である(本明細書で使用されるアミノ酸または核酸「同一性」は、アミノ酸または核酸「相同」と等価である)。2つの配列間のパーセント同一性は、この2つの配列の最適なアラインメントのために誘導される必要があるギャップの数、および各ギャップの長さを考慮に入れて、配列によって共有される同一位置の数の関数である。
【0133】
2つの配列間の配列の比較およびパーセント同一性の決定は、数学的アルゴリズムを使用して達成され得る。好ましい実施形態において、2つのアミノ酸配列間のパーセント同一性は、NeedlemanおよびWunsch(J.Mol.Biol.48:444−453(1970))アルゴリズムを使用して測定される。このアルゴリズムは、GCGソフトウェアパッケージ(http://www.gcg.comから入手可能)中のGAPプログラムに組み込まれている。これは、Blossom62マトリクスまたはPAM250マトリクスのいずれか、および16、14、12、10、8、6、もしくは4のgap weightおよび1、2、3、4、5、もしくは6のlength weightを使用する。さらに別の好ましい実施形態において、2つのヌクレオチド配列間のパーセント同一性は、GCGソフトウェアパッケージ(http://www.gcg.comから入手可能)中のGAPプログラムを使用して測定される。これは、NMSgapdna.CMPマトリクスおよび40、50、60、70、もしくは80のgap weightおよび1、2、3、4、5、もしくは6のlength weightを使用する。別の実施形態において、2つのアミノ酸または2つのヌクレオチド配列間のパーセント同一性は、E.MeyersおよびW.Miller(CABIOS,4:11−17(1989))のアルゴリズムを使用して測定される。これは、PAM120 weight residue table、12のgap length penaltyおよび4のgap penaltyを使用するALIGNプログラム(バージョン2.0)に組み込まれている。
【0134】
本発明の核酸およびタンパク質配列は、さらに「クエリー配列」として使用されて、公開されているデータベースに対する探索を実行(例えば、他のファミリーメンバーまたは関連配列を同定する)し得る。このような探索は、Altshulら(1990)J.Mol.Biol.215:403−10のNBLASTおよびXBLASTプログラム(バージョン2.0)を使用して実行され得る。BLASTヌクレオチド探索は、NBLASTプログラム(score=100、wordlength=12)を用いて実行されて、ヒトまたはマウスB7−DC核酸分子に対するヌクレオチド配列相同性を獲得し得る。BLASTタンパク質探索は、XBLASTプログラム(score=50、wordlength=3)を用いて実行されて、本発明のヒトまたはマウスB7−DCタンパク質分子に対するアミノ酸配列相同性を獲得し得る。比較目的のためのギャップを挿入したアラインメントを獲得するために、Gapped BLASTは、Altshulら(1997)Nucleic Acids Res.25:3389−3402に記載されるように利用され得る。BLASTおよびGapped BLASTプログラムを利用する場合、各々のプログラムのデフォルトパラメータ(例えば、XBLASTおよびNBLAST)が使用され得る。http://www.ncbi.nlm.nih.gov.を参照のこと。
【0135】
従って、上記のB7−DCタンパク質の相同体は、(a)ネイティブのB7−DC機能的活性、および(b)ネイティブのB7−DCタンパク質(例えば、上で測定された配列番号2または配列番号4)に対する配列類似性(少なくとも約30%(アミノ酸レベルで)、好ましくは少なくとも約50%、より好ましくは少なくとも約70%、さらにより好ましくは少なくとも約90%)を有すると特徴付けられる。
【0136】
B7−DCの開示された配列に基づくDNAプローブを使用してこのようなタンパク質を得、そして発現することは当該分野の技術範囲内である。次いで、このタンパク質の生化学的活性および生物学的活性は、本明細書中に記載されるような当該分野で認識される方法(例えば、標準的なT細胞増殖またはサイトカイン分泌アッセイ)を使用して、容易に試験され得る。T細胞補助刺激の生物学的アッセイは、相同体が「機能的」相同体として適切であるような必須な活性を有するかどうかを示す。
【0137】
好ましいアッセイは、TCR(「一次活性化シグナル」)の結合または架橋、および補助刺激シグナルの送達に依存するB7−DCの機能的特徴(例えば、サイトカインのT細胞合成の刺激)を測定する。T細胞上のその天然のリガンドへのB7−DCの結合は、増加したサイトカイン産生(例えば、IL−2)を誘導するシグナルを伝達し、IL−2は、慣用的に測定もされ得る増殖を順番に刺激する。
【0138】
B6−DCの「改変体」とは、1つ以上のアミノ酸残基が置換されている(置換改変体)または1つもしくはいくつかの欠失した残基(欠失改変体)もしくは付加された残基(付加改変体)を有する、全タンパク質あるいはそのフラグメントのいずれかに実質的に同一な分子をいう。B6−DCの「フラグメント」とは、分子の任意のサブセット、好ましくはECDを含むサブセット(すなわち、全長タンパク質のより短いポリペプチド)をいう。
【0139】
多数のプロセスが使用されて、単離されたDNA配列のフラグメント、変異体および改変体を産生し得る。B7−DCタンパク質をコードする核酸の小さなサブ領域またはフラグメント(例えば、1〜30塩基長)は、標準的な、化学的な合成によって調製され得る。より大きな合成フラグメントの産生において使用するためのアンチセンスオリゴヌクレオチドおよびプライマー。
【0140】
好ましい機能的誘導体は、融合タンパク質(B7−DCの機能的フラグメントを含むポリペプチド)である。例えば、B7の有用な誘導体は、B7−DCのECDおよびIg C領域に対応するポリペプチドを含むB7−DC−Ig融合タンパク質である。融合パートナーの存在は、B7−DCタンパク質の可溶性、親和性および/または結合価(ここでは1分子あたりの利用可能な結合部位の数として規定される)を変更し得る。可溶性B7−DC融合タンパク質(T細胞上のレセプターにまだ結合している間)は、APC上に発現されるネイティブタンパク質の効果とは異なる生物学的効果(すなわち、補助刺激よりも競合結合によるT細胞刺激の阻害)を有し得る。
【0141】
本明細書中に使用される場合、B7−DCの細胞外ドメイン(ECD)は、PD−1またはCD28もしくはCTLA−4ではないT細胞上の別のレセプターを認識し、そして結合するタンパク質またはその任意のフラグメントの全体の細胞外部分である。好ましくは、B7−DCのECDは、配列番号2または配列番号4の約26位〜約221位のアミノ酸残基によりコードされる部分である。
【0142】
「可溶性B7−DC」によって、産生細胞から流れるか、分泌されるか、または、さもなければ抽出され得るB7−DCの無細胞形態が意図される。可溶性B7−DCとしては、可溶性融合タンパク質(例えば、生物学的に活性な分子に(遺伝的にまたは化学的に)融合されるB7−DC−Ig、B7−DCの遊離ECD、またはB7−DC ECD)が挙げられるが、それらに限定されない。
【0143】
初期に示されたように、本発明はまた、B7−DCドメインと別のB7ファミリータンパク質のドメインまたはフラグメント(好ましくは補助刺激形態で細胞表面に発現される)との間のハイブリッド融合タンパク質を含む。
【0144】
B7−DC改変体の好ましい群は、少なくとも1つのアミノ酸残基および好ましくは、たった1つのアミノ酸残基が異なる残基によって置換されている群である。タンパク質化学およびタンパク質構造の詳細な記載について、Schulz,GEら、Principles of Protein Structure,Springer−Verlag,New York,1978,およびCreighton,T.E.,Proteins:Structure and Molecular Properties,W.H.Freeman & Co.,San Francisco,1983(本明細書中に参考として援用される)を参照のこと。タンパク質分子において作製され得る置換の型は、異なる種の相同性タンパク質(例えば、Schulzら(上記)の表1〜2およびCreighton(上記)の表3〜9に示されるタンパク質)間のアミノ酸変化の頻度の分析に基づき得る。このような分析に基づいて、保存的置換は、以下の5つの群のうちの1つにおける交換として本明細書中に定義される。
【0145】
【化5】
上の括弧の3つのアミノ酸残基は、タンパク質構造に特別な役割を有する。Glyは、側鎖を欠く唯一の残基であり、従って鎖に可撓性を与える。その通常ではないジオメトリーのため、Proは、鎖をきつく拘束する。Cysは、タンパク質折り畳みに重要なジスルフィド結合形成に関係し得る。
【0146】
生物学的、機能的(または免疫学的)特性におけるより実質的な変化は、例えば、上の5つの群の中よりも間でほとんど保存されていない置換を選択することによって作製される。このような変化は、(a)置換の領域におけるペプチド骨格の構造(例えば、シートまたはヘリックス構造)、(b)標的部位での分子の荷電もしくは疎水性、または(c)側鎖の大きさを維持することに対する効果においてより有意に異なる。このような置換の例は、(i)別のアミノ酸によるGlyおよび/もしくはProの置換またはGlyもしくはProの欠失または挿入;(ii)疎水性残基(例えば、Leu、Lle、Phe、ValもしくはAla)による親水性残基(例えば、SerもしくはThr)の置換;(iii)任意のほかの残基によるCys残基の置換;(iv)電気陰性の電荷を有する残基(例えば、GluもしくはAsp)による電気陽性の側鎖を有する残基(例えば、Lys、ArgもしくはHis)の置換;あるいは(v)巨大な側鎖を有さない残基(例えば、Gly)によるこのような側鎖を有する残基(例えば、Phe)の置換である。
【0147】
本発明に従う大部分の受容可能な欠失、挿入および置換は、そのT細胞補助刺激活性に関するB7−DCタンパク質の特徴においてラジカル変化を産生しないものである。しかし、そのようにするより前に、置換、欠失または挿入の確かな効果を予測することが困難な場合、当業者は、この効果が慣習的なスクリーニングアッセイ(例えば、本明細書に記載される)によって過度の実験を必要とせず評価され得ることを正しく認識する。
【0148】
より短い鎖の改変体が化学合成によって作製され得るが、本発明について、好ましいより長い鎖の改変体は、B7−DCポリペプチドをコードする核酸の部位特異的変異誘発、細胞培養における改変体核酸の発現、および必要に応じて細胞培養物からのポリペプチドの精製によって(例えば、(少なくとも1つのエピトープに結合することによって改変体を吸収するため)カラムに固定された特異的な抗体を使用する免疫親和性クロマトグラフィーによって)典型的に作製される。
【0149】
(B7−DCの化学的誘導体)
B7−DCの「化学的誘導体」は、通常はタンパク質の一部分ではないさらなる化学的部分を含む。ポリペプチドの共有結合修飾は、本発明の範囲内に含まれる。このような誘導された部分は、可溶性、吸収性、生物学的半減期などを改良し得る。このような効果を媒介し得る部分は、例えば、Remington’s Pharmaceutical Sciences,第16版、Mack Publishing Co.,Easton,PA(1980)に開示される。
【0150】
このような修飾は、ポリペプチドの標的化アミノ酸残基を、選択された側鎖または末端残基と反応し得る有機誘導化因子と反応することによって分子に導入され得る。
【0151】
ポリペプチドの化学的誘導体の例は、以下である。
【0152】
リジニル(lysinyl)およびアミノ末端残基は、コハク酸または他のカルボキシル酸無水物で誘導される。環式カルボキシル無水物を用いる誘導は、リジニル残基の電荷を逆転する効果を有する。アミノ含有残基を誘導するための他の適切な試薬としては、イミドエステル(例えば、メチルピコリンイミデート;ピリドキサールリン酸;ピリドキサール;クロロ水素化ホウ素(chlorobrohydride);トリニトロベンゼンスルホン酸;O−メチルイソ尿素;2,4ペンタンジオン;およびグリオキシレートを用いるトランスアミラーゼ触媒反応)が挙げられる。
【0153】
カルボキシル側鎖(アスパルチルまたはグルタミル)は、カルボジイミド(R−N=C=N−R’)(例えば、1−シクロヘキシル−3−(2−モルホリニル−(4−エチル)カルボジイミドまたは1−エチル−3−(4−アゾニア−4,4−ジメチルペンチル)カルボジイミド)との反応によって選択的に修飾され得る。さらに、アスパルチル残基およびグルタミル残基は、アンモニアとの反応によってアスパラギニル残基グルタミニル残基へ変換され得る。
【0154】
他の修飾としては、プロリンおよびリジンのヒドロキシル化、セリル残基またはスレオニル残基のヒドロキシル基のリン酸化、リジンのアミノ基のメチル化(Creighton、上記、pp.79−86)、N末端アミンのアセチル化、ならびに、C末端カルボキシル基のアミド化が挙げられる。
【0155】
含まれるものはまた、1つ以上のD−アミノ酸が1つ以上のL−アミノ酸の代わりに置換されたペプチドである。
【0156】
(マルチマーペプチド)
本発明はまた、B7−DCの配列から得られた塩基性ペプチド配列が、介在スペーサ−またはリンカーを用いてか、または用いずに約2〜約100回繰り返される、より長いポリペプチドを含む。このようなマルチマーは、本明細書中に定義される任意のペプチド改変体から構築され得ることが理解される。さらに、ペプチドマルチマーは、ペプチドモノマーおよびその開示された置換改変体の異なる組合せを含み得る。このようなオリゴマーまたはマルチマーペプチドは、化学合成によってか、または本明細書中で考察されるような組換えDNA技術によって作製され得る。化学的に産生される場合、オリゴマーは、好ましくは、塩基性ペプチド配列の2〜8回繰り返しを有する。組換え的に産生される場合、マルチマーは、例えば、発現系が2〜約100回繰り返すことを可能にするのと同じくらい多くの繰り返しを有し得る。
【0157】
B7−DCペプチドまたはポリペプチドのタンデムマルチマー、好ましくはダイマーおよびトリマーにおいて、鎖は、鎖間ジスルフィド結合または鎖間の他の「構造的」共有結合によって結合され、その結果、この鎖は、「末端から末端」よりも「横 対 横」である。好ましいダイマーおよびトリマーは、本明細書に記載されるようなB7−DCの融合タンパク質(例えば、B7−DC−Ig)間のものである。
【0158】
(B7−DCのエピトープに対して特異的な抗体)
以下の記載において、免疫学、細胞生物学、および分子生物学の当業者に公知の種々の方法論について言及がなされる。刊行物および言及されるこのような公知の方法論を示す他の材料は、まるで全てを示すようにその全体において参考として本明細書中に援用される。免疫学の一般的な原理を記載する標準的な参考著述としては、A.K.Abbasら、Cellular and Molecular Immunology(第4版),W.B.Saunders Co.,Philadelphia,2000;C.A.Janewayら、Immunobiology.The Immune System in Health and Disease,第4版,Garland Publishing Co.,New York,1999;Roitt,I.ら、Immunology,(最新版)C.V.Mosby Co.,St.Louis,MO(1999);Klein,J.,Immunology,Blackwell Scientific Publications,Inc.,Cambridge,MA,(1990)が挙げられる。
【0159】
モノクローナル抗体(mAb)およびその産生および使用のための方法は、KohlerおよびMilstein,Nature 256:495−497(1975);米国特許第4,376,110号;Hartlow,E.ら、Antibodies:A Laboratory Manual,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY,1988);Monoclonal Antibodies and Hybridomas:A New Dimension in Biological Analyses,Plenum Press,New York,NY(1980);H.Zolaら、Monoclonal Hybridoma Antibodies:Techniques and Applications,CRC Press,1982)に記載される。
【0160】
免疫アッセイはまた、Coligan,J.E.ら、編、Current Protocols in Immunology,Wiley−Interscience,New York 1991(または最新版);Butt,W.R.(編)Practical Immunoassay:The State of the Art,Dekker,New York,1984;Bizollon,Ch.A.編、Monoclonal Antibodies and New Trends in Immunoassays,Elsevier,New York,1984;Butler,J.E.,ELISA(29章),In:van Oss,C.J.ら、(編),IMMUNOCHEMISTRY,Marcel Dekker,Inc.,New York,1994,pp.759−803;Butler,J.E.(編),Immunochemistry of Solid−Phase Immunoassay,CRC Press,Boca Raton,1991;Weintraub,B.,Principles of Radioimmunoassays,Seventh Training Course on Radioligand Assay Techniques,The Endocrine Society,March,1986;Work,T.S.ら、Laboratory Techniques and Biochemistry in Molecular Biology,North Holland Publishing Company,NY,(1978)(Chard,T.による章,「An Introduction to Radioimmune Assay and Related Techniques」)に記載される。
【0161】
抗イディオタイプ抗体は、例えば、Idiotypy in Biology and Medicine,Academic Press,New York,1984;Immunological Reviews Volume 79,1984;Immunological Reviews Volume 90,1986,Curr.Top.Microbiol.,Immunol.Volume 119,1985;Bona,C.ら、CRC Crit.Rev.Immunol.,pp.33−81(1981);Jerne,NK,Ann.Immunol.125C:373−389(1974);Jerne,NK,In:Idiotypes−Antigens on the Inside,Westen−Schnurr,I.編,Editiones Roche,Basel,1982,Urbain,Jら、Ann.Immunol.133D:179−(1982);Rajewsky,K.ら、Ann.Rev.Immunol.1:569−607(1983)に記載される。
【0162】
本発明は、公知のB7ファミリータンパク質にないB7−DCの新規なエピトープを反応する抗体(ポリクローナルおよびモノクローナルの両方)を提供する。抗体は、異種の、同種異系の、同系のまたはそれらの修飾形態(例えば、ヒト化抗体またはキメラ抗体)であり得る。抗B7−DC抗体のイディオタイプに対して特異的な抗イディオタイプ抗体もまた、含まれる。用語「抗体」はまた、抗原結合部位を含み、そしてB7−DCエピトープに結合し得るインタクトな分子およびそのフラグメントの両方を含むことが意味される。これらは、インタクトな抗体のFcフラグメントを欠くFabおよびF(ab’)2フラグメント(循環からより速やかに除去され、そしてインタクトな抗体よりも少ない非特異的な組織結合を有し得る)を含む(Wahlら、J.Nucl.Med.24:316−325(1983))。含まれるものはまた、Fvフラグメントである(Hochman,J.ら、(1973)Biochemistry 12:1130−1135;Sharon,J.ら、(1976)Biochemistry 15:1591−1594))。これらの種々のフラグメントは、従来の技術(例えば、プロテアーゼ切断または化学的切断)を使用して産生される(例えば、Rousseauxら、Meth.Enzymol.,121:663−69(1986)を参照のこと)。
【0163】
ポリクローナル抗体は、免疫化動物(ウサギ、ヤギ、げっ歯類など)由来の血清として得られ、そしてさらなる処理をせずに直接使用され得るか、または、慣習的な富化または精製方法(例えば、硫酸アンモニウム沈殿、イオン交換クロマトグラフィー、および親和性クロマトグラフィー)に供され得る(Zolaら、前記を参照のこと)。
【0164】
免疫原は、完全B7−D6タンパク質、あるいはそのフラグメントまたは誘導体を含み得る。好ましい免疫原は、ヒトB7−DCのECD(アミノ酸残基26−221)の全てまたは一部を含み、ここでこれらの残基は、ネイティブなB7−DC上に見出される翻訳後修飾(例えば、グリコシル化)を含む。細胞外ドメインを含む免疫原は、当該分野で公知の種々の方法(例えば、従来の組換え方法を使用するクローニングされた遺伝子の発現、起源の細胞からの単離、高いレベルのB7−DCを発現する細胞集団など)において産生される。
【0165】
mAbは、従来のハイブリドーマ技術(例えば、KohlerおよびMilstein(Nature,256:495−97(1975))、ならびにその改変(上の参考文献を参照のこと)によって導入された手順)を使用して産生され得る。動物、好ましくはマウスは、上のような免疫原を用いる免疫化によってプライムされて、プライムされた動物において所望の抗体応答を誘導する。
【0166】
プライムされた動物のリンパ節、脾臓または末梢血由来のBリンパ球は、一般的に、融合促進剤(例えば、ポリエチレングリコール(PEG))の存在化で骨髄腫細胞と融合させる。任意の多くのマウス骨髄腫細胞株は、このような使用のために入手可能である:P3−NS1/1−Ag4−1,P3−x63−kOAg8.653,Sp2/0−Agl4,またはHL1−653骨髄腫株(ATCC,Rockville,MDから入手可能)。次の工程は、選択培地における増殖を含み、その結果、融合されていない親骨髄腫細胞およびドナーリンパ球細胞は、最終的に死亡するが、ハイブリドーマ細胞のみが生存する。これらは、クローニングされ、そして増殖させて、そしてその上清は、例えば、B7−DC−Ig融合タンパク質を使用する免疫アッセイ技術によって、所望の特異性の抗体の存在についてスクリーニングされる。陽性クローンは、例えば、限界希釈によってサブクローニングされ、そしてmAbが単離される。
【0167】
これらの方法に従って産生されたハイブリドーマは、当該分野で公知の技術を使用してインビトロまたはインビボで(腹水中で)増殖され得る(一般的に、Finkら、Prog.Clin.Pathol.,9:121−33(1984)を参照のこと)。一般的に、個々の細胞株は、培養において増殖され、そして高い濃度の単一のmAbを含有する培養培地は、デカンテーション、濾過、または遠心沈殿法によって収穫され得る。
【0168】
この抗体は、通常のマルチマー構造の代わりに、単鎖抗体またはscFvとして生成され得る。単鎖抗体は、目的のIg由来の超可変領域を含み、未知量の標識された抗体(これは、「リポーター分子」として機能する)を含む溶液と接触させた場合、インタクトなIgの大きさの一部ではあるが、ネイティブなIgの抗原結合部位を再現する(Skerra,A.ら(1988)Science、240:1038−1041;Pluckthun,A.ら(1989)Methods Enzymol.178:497−515;Winter,G.ら(1991)Nature、349:293−299);Birdら(1988)Science 242:423;Hustonら(1988)Proc.Natl.Acad.Sci.USA 85:5879;Jost CRら、J Biol Chem.(1994)269:26267−26273;米国特許第4,704,692号、同第4,853,871号、同第4,946,778号、同第5,260,203号、同第5,455,0Kn)。標識抗体が、非標識抗体を介して固体支持体に結合した抗原と複合体化するのを可能にするための、二回目のインキュベーション期間の後、この固体支持体は2回目に洗浄され、未反応の標識抗体が除去される。この型の正のサンドイッチアッセイは、抗原が存在するか否かを決定するための単純な「あり/なし」のアッセイであり得るか、または標識抗体の程度と、既知量の抗原を含む標準サンプルについて得られた程度を比較することによって定量的にされ得る。
【0169】
別の型の「サンドイッチ」アッセイでは、いわゆる「同時」アッセイおよび「逆」アッセイが使用される。同時アッセイは、固体支持体に結合した抗体および標識抗体の両方が、試験されるサンプルに同時に添加されるので、1回のインキュベーション工程を包含する。このインキュベーションが完了した後、この固体支持体を洗浄して、残りの液体サンプルおよび複合体化されていない標識抗体を除去する。次いで、固体支持体に結合した標識抗体の存在は、従来の「正」サンドイッチアッセイにおけるのと同じようにして決定される。
【0170】
「逆」アッセイにおいて、標識抗体溶液の流体サンプルへの最初の添加、続いて適切なインキュベーション期間後の、固体支持体に結合した非標識抗体の添加という段階的添加が利用される。2回目のインキュベーション後、この固相を従来の様式で洗浄し、試験される残りのサンプルおよび未反応の標識抗体の溶液が固相にない状態にする。次いで、固体支持体に結合した標識抗体の決定は、「同時」アッセイおよび「正」アッセイのように決定される。
【0171】
上記の抗体は、T細胞刺激を阻害するための方法および所望されないT細胞の活性化に関連する疾患(例えば、移植片拒絶および自己免疫)を処置するための方法において有用である。この方法は、このような処置が必要な被験体にB7−DCの補助刺激エピトープに特異的な有効量の抗体、好ましくはmAb、より好ましくはヒトmAbまたはヒト化mAbを、投与する工程を包含する。抗体の投与は、T細胞刺激をブロックするか、または抗原反応性T細胞を排除し、これによって、標的T細胞応答を阻害するのに効果的でなければならない。関連する用量範囲を以下に記載する。
【0172】
(B7−DCタンパク質をコードする核酸の使用)
本発明の核酸は、生物学的サンプル由来の細胞中のB7−DCの発現を測定することによって、疾患の進行をモニターするために診断的に使用されるかまたはB7−DCの発現に対する薬剤の影響をアッセイするために使用される。好ましくは、これは、細胞性mRNAレベルの測定によって達成される。このような診断方法での使用のために、この核酸配列は、例えば、放射性標識もしくは蛍光標識またはビオチンで検出可能に標識され、従来のドットブロットまたはノザン−ハイブリダイゼーション手順において使用され、例えば、生物学的サンプル由来の総またはポリ(A+)RNAの調製物中に存在するmRNA分子がプローブされる。
【0173】
(治療的組成物およびその投与)
B6−DCポリペプチドまたはこのポリペプチドを発現する細胞(例えば、DCまたは腫瘍細胞)は、哺乳動物被験体に、好ましくはヒトに投与される。細胞に結合した形態、固定化された形態、またそうでなければ凝集された形態のポリペプチドを使用して、Tリンパ球の反応性および得られる免疫を増強する。B6−DC−Ig融合タンパク質はダイマーとして組み立てられる、実施例に示されるようにT細胞を補助刺激する。B6−DCポリペプチドの可溶性単量体形態は、活性を刺激することなくT細胞上のレセプターに結合し得、従って、これは、この分子の刺激性形態によるT細胞補助刺激の競合的インヒビターまたはアンタゴニストと考えられ得る。このようなB6−DCアンタゴニストの結合は、進行中のT細胞反応性を抑制し得るか、または内因性のB6−DCまたはさらにこれらのレセプター(例えば、CD28またはCTLA−4)を介して作用するB7ファミリーの他のメンバーによって提示される補助刺激シグナルの効果を妨害し得る。
【0174】
本明細書中に記載のようなB7−DC活性を有する組成物は、生物学的に有効な量または治療的に有効な量で、薬学的に受容可能なキャリア中で投与される。B7−DCポリペプチド(またはそのポリペプチドを発現する細胞)は、単独で、またはB7ファミリーの別のメンバーもしくは別の免疫刺激分子の活性を有するタンパク質もしくはペプチドのような別のタンパク質もしくはペプチドと組合せて、投与され得る。処置はアジュバント(アジュバントの最も広い意味で使用され、インターフェロンのような任意の非特異的免疫刺激化合物を含む)の投与を含み得る。本明細書中で意図されるアジュバントとして、レゾルシノール、非イオン性界面活性剤(例えば、ポリオキシエチレンオレイルエーテルおよびn−ヘキサデシルポリエチレンエーテル)が挙げられる。
【0175】
また、被験体に投与される場合に、以下の用量および量が、本発明の抗体に関連する。
【0176】
治療的に有効な量は、効果的な時期に投与される場合に、所望の免疫学的効果または臨床的効果を達成する投与量である。
【0177】
B7−DC活性を有するポリペプチド(または抗B7−DC抗体)の治療的に活性な量は、因子(例えば、個体の疾患状態、年齢、性別および体重)、ならびにペプチドが個体において所望の応答を惹起するための能力によって変化し得る。投与量のレジメは、最適な治療応答を提供するように調整され得る。例えば、いくつかに分けられた用量を毎日投与してもよいし、この用量を治療状態の緊急性によって示されるように比例して減少させてもよい。細胞に結合した形態の治療的に有効な量のタンパク質は、タンパク質当量または細胞当量で記述され得る。
【0178】
従って、有効な量は、レシピエントの体重1kg当たり約1ngと約1gとの間、より好ましくは約1μgと100mg/kgとの間、より好ましくは約100μgと100mg/kgとの間である。内部投与に適切な投薬形態は、好ましくは、(後者の容量範囲について)1単位当たり約0.1mg〜500mgの活性成分のを含む。活性成分は、組成物の総重量に基づいて、0.5重量%〜95重量%まで変化し得る。あるいは、B7−DCを発現する細胞(DCまたは不活化された腫瘍細胞のような、好ましく形質導入された細胞)の有効な用量は、好ましくは分割用量で、被験体当たり、約104個との細胞と約109個の細胞との間、より好ましくは約106個の細胞と108個の細胞との間である。免疫療法の当業者は、過度の実験なしにこれらの用量を調整し得る。
【0179】
活性な化合物(例えば、B6−DCポリペプチドまたはB6−DC DNAで形質導入された細胞)は、便利な様式(例えば、便利でかつ有効な経路による注射)で投与され得る。好ましい経路として、皮下経路、皮内経路、静脈内経路および筋肉内経路が挙げられる。他の可能性のある経路としては、経口投与、くも膜下腔内適用、吸入適用、経皮適用、または直腸投与が挙げられる。完全には除去されていない腫瘍の処置については、腫瘍内への直接注入もまた、意図される。
【0180】
投与経路に依存して、活性な化合物は、酵素、酸、およびこの化合物を不活化し得る他の天然の条件の作用からこの化合物を保護するための材料でコーティングされ得る。従って、B7−DC活性を有するポリペプチドまたはペプチドを、経腸経路によって投与するために、B7−DC活性の不活化を防止するための材料で組成物をコーティングするか、またはそのような材料と組成物を同時投与する必要があり得る。例えば、ペプチドは、適切なキャリア、希釈液またはアジュバント中で個体に投与され得るか、酵素インヒビター(例えば、膵臓トリプシンインヒビター、ジイソプロピルフルオロホスフェート(DEP)およびトラシロール(trasylol))と共に同時投与され得るか、またはリポソーム(水中油中水エマルジョンおよび従来のリポソーム(Strejanら(1984)J.Neuroimmunol 7:27)を含む)のような適切なキャリア中で投与され得る。
【0181】
本明細書中で使用される場合、「薬学的に受容可能なキャリア」として、任意のおよび全ての溶媒、分散媒体、コーティング剤、抗細菌剤および抗真菌剤、等張剤および吸収遅延剤などが挙げられる。薬学的に活性な物質についてこのような媒体および薬剤の使用は、当該分野で周知である。任意の従来の媒体または薬剤が、この活性な化合物と不適合である範囲を除いて、治療組成物中でその使用が意図される。補助的な活性化合物もまた、この組成物中に組込まれ得る。
【0182】
好ましい薬学的に受容可能な希釈液として、生理食塩液および水性緩衝溶液が挙げられる。注射のために適切な薬学的組成物としては、滅菌水性溶液(水溶性である場合)または滅菌水性分散剤および滅菌注射用溶液または滅菌注射用分散液の即時調製のための滅菌粉末が挙げられる。等張剤(例えば、糖、ポリアルコール(例えば、マンニトール、ソルビトール)、塩化ナトリウム)は、薬学的組成物中に含まれ得る。全ての場合において、組成物は無菌でありかつ流体であるべきである。組成物は、製造および保存の条件下で安定であるべきであり、そして細菌および真菌のような微生物による汚染を予防する保存剤を含まなければならない。分散液はまた、グリセロール、液体ポリエチレングリコール、およびそれらの混合物中に、ならびに油中に調製され得る。保存および使用の通常の条件下で、これらの調製物は、微生物の増殖を防ぐための保存剤を含み得る。
【0183】
キャリアは、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、およびそれらの適切な混合物を含む、溶媒または分散媒体であり得る。適切な流動性は、例えば、レシチンのようなコーティングの使用によって、必要とされる粒子サイズの維持によって(分散液の場合)、および界面活性剤の使用によって、維持され得る。
【0184】
微生物作用の防止は、種々の抗細菌剤および抗真菌剤(例えば、パラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサールなど)によって達成され得る。
【0185】
注射可能な組成物の長期にわたる吸収は、吸収を遅延させる薬剤(例えば、モノステアリン酸アルミニウムおよびゼラチン)を組成物中に含ませることによってもたらされ得る。
【0186】
非経口組成物は、好ましくは、投与の容易さ、および投薬量の均一性のために投薬単位形態で処方される。投薬単位形態は、哺乳動物の被験体のための投薬単位として適切な物理学的に別個の単位を言い;各単位は、必要とされる薬学的キャリアと結合して所望の治療的効果を生成するように計算された予め決定された量の活性化合物を含む。本発明の投薬単位形態のための詳細は、(a)活性化合物の固有の特徴および達成されるべき特定の治療効果、ならびに(b)個体の感受性を処置するためのこのような活性化合物を配合する分野に固有の制限によって、決定され、そしてこれらに直接的に依存する。
【0187】
肺への注入のために、エアゾール化された溶液が使用される。噴霧可能なエアゾール調製物において、活性なタンパク質は、固体または液体の不活性キャリア材料との組合せで存在し得る。これはまた、スクイーズボトルにパッケージングされ得るか、加圧された揮発性(通常は気体の)プロペラントと混合してパッケージングされ得る。エアゾール調製物は、本発明のタンパク質に加えて溶媒、緩衝剤、界面活性剤、および抗酸化剤を含み得る。
【0188】
局所適用のために、本発明のタンパク質は、軟膏剤(salve)または軟膏剤(ointment)(これは、皮膚への平滑効果および罹患領域への活性成分の直接的な投与のための手段の両方を有する)のような局所適用ビヒクルに組込まれ得る。
【0189】
活性成分のためのキャリアは、噴霧可能な形態または噴霧不可能な形態のいずれであってもよい。噴霧不可能な形態は、局所適用に固有のキャリアを含み、かつ、好ましくは水の動的粘度よりも大きな動的粘度を有する半固形または固形であり得る。適切な処方物として、液剤、懸濁剤、乳剤、クリーム剤、軟膏剤(ointment)、散剤、リニメント剤、軟膏剤(salve)などが挙げられるが、これらに限定されない。所望である場合には、これらは、滅菌しても、補助剤(例えば、保存剤、安定化剤、湿潤剤、緩衝剤、または浸透圧に影響を与えるための塩など)と混合してもよい。噴霧不可能な局所的調製物のための好ましいビヒクルの例として、軟膏基剤(例えば、ポリエチレングリコール−1000(PEG−1000);HEBクリームのような従来のクリーム;ゲル;ならびにワセリンなどが挙げられる。
【0190】
本発明によるB7−DCポリペプチドのための他の薬学的に受容可能なキャリアは、リポソームであり、これは、活性なタンパク質が、脂質層に接着した水性の同心層からなる小体に分散しているか、またはその中に多様に存在しているかのいずれかで含まれる、の薬学的組成物である。活性なタンパク質は、好ましくは、水性層および脂質層の内側または外側(すなわち、いずれの事象においても、リポソーム懸濁液として一般的に公知の不均一な系)に存在する。疎水性の層、または脂質層は、一般的には(しかし、排他的にではない)、レシチンおよびスフィンゴミエリンのようなリン脂質、コレステロールのようなステロイド、多少イオン性の界面活性剤(例えば、ジセチルホスフェート、ステアリルアミンまたはホスファチジン酸)、および/または疎水性の性質の他の物質を含む。
【0191】
(B7−DCおよび複数の補助刺激分子を発現させるための腫瘍細胞の改変)
本発明の別の局面は、複数の補助刺激分子を発現するように改変された細胞(例えば、腫瘍細胞)である。活性化B細胞での補助刺激分子の時間的な発現は、B7、B7−2、およびB7−3で異なる。例えば、B7−2は、B細胞の活性化に続いて初期に発現されるが、B7−3は、後期に発現される。従って、この異なる補助刺激分子は、免疫応答の過程の間に、別個の機能を果たし得る。効果的なT細胞応答は、T細胞が複数の補助刺激分子から補助刺激シグナルを受けることを必要とし得る。
【0192】
従って、本発明は、遺伝的に改変されるか、または1つより多くの補助刺激分子を発現する腫瘍細胞を含む。例えば、腫瘍細胞を改変して、B7−DCと、B7、B7−2、およびB7−3のうちの1つ以上とを発現させ得る。
【0193】
改変の前に、細胞(例えば、腫瘍細胞)は、いずれかの補助刺激分子を発現しないかもしれず、または特定の補助刺激分子は発現するが、他の分子は発現しないかもしれない。本明細書中で記載されるように、腫瘍細胞は、B7−DCのみをコードする核酸または別の補助刺激分子とともにB7−DCをコードする核酸を用いるトランスフェクションによって改変され得る。例えば、B7−DCをコードする核酸でトランスフェトされた腫瘍細胞は、B7をコードする核酸でさらにトランスフェクトされ得る。ヒトB7−DCタンパク質またはマウスB7−DCタンパク質をコードするcDNA分子の配列は、それぞれ、配列番号1および配列番号3のコード部分である。あるいは、1より多くの型の改変が使用され得る。例えば、B7−DCをコードする核酸でトランスフェクトされた腫瘍細胞は、B7−1、B7−2、またはB7−3の発現を誘導する薬剤で刺激され得る。
【0194】
(病原体に関連する抗原)
本発明についての主な有用性は、癌および世界中で罹患率および死亡率を引き起こしている主な慢性的ウイルス感染症についての治療用ワクチンの本願組成物の使用である。このようなワクチンは、感染した細胞を排除する(これは、抗体が無効であるためにT細胞応答を必要とする)ように設計される。本発明のワクチンは、抗原性エピトープそれ自身に加えて:
(a)ベクター(例えば、裸の(naked)DNA、裸のRNA、自己複製RNAレプリコン)ならびにウイルス(ワクシニアウイルス、アデノウイルス、アデノ随伴ウイルス(AAV)、レンチウイルスおよびRNAアルファウイルスを含む);
(b)抗原標的化シグナルまたはプロセシングシグナル(例えば、HSP70)、カルレティキュリン、Flt−3リガンドの細胞外ドメイン、Pseudomonasの菌体外毒素ETAのドメインII、単純ヘルペスのVP22標的化タンパク質など(同一人に譲渡された米国特許出願第09/421,608号;同第09/501,097号;同第09/693,450号;同第60/222,9002号;同第60/222,985号;同第60/268,575号およびChang,W−Fら、J.Virol.75:2368−2376(2001)(これらは、その全体が参考として本明細書中で援用される);ならびに
(c)補助刺激シグナル、好ましくは本発明のB7−DCタンパク質またはその融合タンパク質、フラグメントもしくは機能的誘導体(単独でまたはB7.1、B7.2、可溶性CD40などのような他の公知の補助刺激タンパク質との組み合わせ)
を含む。
【0195】
腫瘍細胞または他の型の宿主細胞(APCを含む)は、所望の免疫応答に対する抗原をコードする核酸で形質転換されるか、トランスフェクトされるか、またはそうでなければ、形質導入される。このような抗原は、好ましくは、病原性微生物のエピトープであり、これに対して宿主は、エフェクターT細胞応答(細胞障害性Tリンパ球(CTL)および遅延型過敏症を含む)によって防御されている。代表的には、これらとして、ウイルス、マラリアのような細胞内寄生生物、およびミコバクテリアおよびリステリアのような細胞内で増殖する細菌が挙げられる。従って、本発明のワクチン組成物に含まれるこの型の抗原は、(もちろん、腫瘍特異性抗原に加えて)このような病原体に関連する抗原のいずれかである。いくつかのウイルス抗原がまた、ウイルスが癌の原因となる因子である場合の腫瘍抗原であることは注目すべきである。
【0196】
実際に、世界中で最も一般的な2つの癌、肝癌および子宮頚癌は、ウイルス感染と関連する。B型肝炎ウイルス(HBV)は、肝癌の病因となる因子として関連付けられてきた(Beasley,R.P.ら、Lancet 2、1129−1133(1981))。80〜90%の子宮頚癌は、以下の4つの「高度に危険な(ハイリスク)」ヒトパピローマウイルス型のうちの1つに由来するE6抗原およびE7抗原を発現する:HPV−16、HPV−18、HPV−31およびHPV−45(Gissmann,L.ら、Ciba Found Symp.120、190−207(1986);Beaudenon,S.ら、Nature 321、246−249(1986))。HPV E6抗原およびE7抗原は、免疫適格性個体におけるウイルス関連の癌のための最も有望な標的である。なぜなら、これらが、子宮頚癌において遍在的に発現されるからである。治療的癌ワクチンの標的としてのこれらの重要性に加えて、ウイルス関連腫瘍抗原はまた、予防的ワクチンのための理想的な候補である。実際に、アジアにおける予防的HBVワクチンの導入は、肝癌の発生率を減少させ、癌予防に対する大きな影響を示している(Chang,M.H.ら、New Engl.J.Med.336、1855−1859(1997))。
【0197】
慢性的なヒトウイルス感染において、とりわけ最も重要なウイルスは、ヒトパピローマウイルス(HPV)、B型肝炎ウイルス(HBV)、C型肝炎ウイルス(HCV)、ヒト免疫不全ウイルス(HIV)、エプスタイン−バーウイルス(EBV)および単純ヘルペスウイルス(HSV)である。
【0198】
ヒトの癌および感染性疾患へのその適用性に加えて、本発明はまた、獣医学的医薬の状況において動物の疾患の処置に使用することが意図される。従って、本明細書中で記載されるこのアプローチは、当業者によって容易に適用されて、獣医学的なヘルペスウイルス感染(ウマのヘルペスウイルス、ウシのヘルペスウイルス、ニワトリおよび他の家禽におけるマレク病ウイルスを含む);動物のレトロウイルス疾患;仮性狂犬病および狂犬病などを処置し得る。
【0199】
以下の参考文献は、基礎的な医学的および獣医学的なウイルス学の分野における原理ならびに現在の情報を示し、これらは、参考として援用される:Fields Virology、Fields,BNら編、Lippincott Williams & Wilkins、NY、1996;
Principles of Virology:Molecular Biology,Pathogenesis,and Control、Flint,S.J.ら編、Amer Society for Microbiology、Washington、1999;Principles and Practice of Clinical Virology,第4版、Zuckerman.A.J.ら編、John Wiley & Sons、NY、1999;The Hepatitis C Viruses、Hagedorn,CHら編、Springer Verlag、1999;Hepatitis B Virus:Molecular Mechanisms in Disease and Novel Strategies for Therapy、
Koshy,R.ら編、World Scientific Pub Co、1998;Veterinary Virology、Murphy,F.A.ら編、Academic Press、NY、1999;Avian Viruses:Function and Control、Ritchie,B.W.、Iowa State University Press、Ames、2000;Virus Taxonomy:Classification and Nomenclature of Viruses:Seventh Report of the International Committee on Taxonomy of Viruses、M.H.V.Van Regenmortel,MHVら編、Academic Press;NY、2000。
【0200】
(標的化分子)
種々の作用様式を有する多数のタンパク質が「標的化」分子として関連付けられてきた。この「標的化」分子は、抗原と共に(好ましくは、融合ポリペプチドとして)使用されて、細胞および細胞下区画へ抗原を標的化し、このことは、より強力かつ効果的な様式でT細胞に対する抗原提示を促進する。
【0201】
熱ショックタンパク質(HSP)への抗原の結合は、核酸に基づく(および他の)ワクチンの能力を増強するための潜在的アプローチを示す。HSPは、癌性ワクチン接種およびウイルス性ワクチン接種における天然の生物学的アジュバントとして作用するらしい。小胞体(ER)に常在するgp96 HSPおよび細胞質Hsp70は両方とも、免疫学的アジュバントとして作用する(Srivastava,PKら、Semin.Immunol.3、57−64(1991);Udono,Hら、Proc.Natl.Acad.Sci.USA 91、3077−3081(1994))。これらのHSPまたはシャペロニンは、広範なペプチドアレイに結合する(Lammert,E.ら、Eur.J.Immunol.27、923−927(1997))。Hsp70は、会合したタンパク質をプロテオソーム(MHCクラスI分子と会合するためのペプチドを生成する主な細胞性プロテアーゼ複合体)に標的化し得るシャペロニンである。従って、Hsp70に直接結合する抗原は、MHCクラスIによってより効率的に提示される(特に、CTL応答を生じる)。2つの特徴が、HSPのアジュバント活性の原因であるらしい:(1)インビトロでペプチドをロードされたgp96は、MHCクラスIプロセシング経路に抗原を効果的に導入する;(2)gp96のマクロファージへの結合は、炎症誘発性サイトカインの分泌を誘導し、従って、ペプチド性抗原が標的化されている細胞の機能を増強する。
【0202】
腫瘍またはウイルスに感染した細胞から単離されたHSP複合体での免疫は、強力な抗腫瘍免疫(Srivastava,PKら、Int J Cancer.33:417−22、1984;Srivastava,PKら、Proc Natl Acad Sci USA.83:3407−11、1986;Udono,Hら、J Immunol.152:5398−5403、1994;Blachere,NEら、J Immunother.14:352−6、1993;Udono,Hら(前出);Tamura,Yら、Science.278:117−20、1997;Janetzki,Sら、J Immunother.21:269−76、1998)、または抗ウイルス免疫(Heikema,Aら、Immunol Lett.57:69−74、1997;Suto,Rら、Science.269:1585−8、1995)を誘導する。ペプチドとHSPとをインビトロで混合することにより、免疫原性HSP−ペプチド複合体が生成された(Ciupitu,AMら、J Exp Med.187:685−91、1998;Blachere,NEら、J Exp Med.186:1315−22、1997)。HSPに基づくいくつかのタンパク質ワクチンは、HSPへの抗原の融合物を含んだ(Suzue,Kら、J Immunol.156:873−9、1996;Suzue,K.ら、Proc Natl Acad Sci USA 94:13146−51、1997)。より最近では、本発明者およびその同僚は、キメラDNAまたはRNAレプリコンワクチンの形態でHSPを使用した(例えば、Chen,C−Hら、Canc.Res.60:1035−1042(2000))。彼らは、Mycobacterium tuberculosis HSP70に融合させる抗原としてHPV−16 E7を使用し、そしてE7特異的CD8+T細胞の増加した増殖および活性化(これは、確立された腫瘍に対する強力な抗腫瘍免疫を生じた)を示した(Lin,K.−Y.ら、Cancer Res.56:21−26.、1996)。
【0203】
別の有用な標的化分子は、Pseudomonas菌体外毒素A(ETA)(例えば、ETAのドメインII(dII)(残基253〜364にわたる))のトランスロケーションドメインである。トランスロケーションドメインは、細胞の細胞質ゾルに結合するタンパク質またはポリペプチドのトランスロケーションを誘導するポリペプチドである。例えば、同様に適用可能なポリペプチドは、ジフテリア毒素、クロストリジウム属(botulinum、tetani)毒素、炭疽毒素、エルジニア属毒素、Vibrio choleraeの毒素、またはBordetella pertussis毒素由来である。毒素をコードするDNAの毒素ドメインは、好ましくは、このような組成物の調製物において変異または欠失されている。
【0204】
カルレティキュリン(CRT)は、レクチン活性を示し、そして新生糖タンパク質の折り畳みおよび組立てに関連することが公知である、小胞体(ER)腔に豊富に存在する46kDaのタンパク質である(Nash(1994)Mol.Cell.Biochem.135:71−78;Hebert(1997)J.Cell Biol.139:613−623;Vassilakos(1998)Biochemistry 37:3480−3490;Spiro(1996)J.Biol.Chem.271:11588−11594)。CRTは、抗原プロセシングに関連するトランスポーター(例えば、TAP−1およびTAP−2)によってERに輸送されたペプチドと会合する(Spee(1997)Eur.J.Immunol.27:2441−2449)。CRTは、インビトロでペプチドと複合体を形成する。これらの複合体は、マウスに投与された場合に、ペプチド特異的CD8+T細胞応答を惹起した(Basu(1999)J.Exp.Med.189:797−802;Nair(1999)J.Immunol.162:6426−6432)。マウス腫瘍から精製されたCTRは、CRTの供給源として使用される腫瘍に対して特異的な免疫を惹起したが、抗原的に別個の腫瘍に対しては免疫を惹起しなかった(Basu、前出)。ペプチドに結合するCRTを用いて、DCをインビトロでパルスすることによって、このペプチドは、DCクラスI分子の状況において再提示され、そしてペプチド特異的CTLを刺激した(Nair、前出)。
【0205】
Flt−3リガンドは、DC前駆体の増殖を刺激し、そしてインビボで多数のDCの生成を促進し得る(Maraskovsky,E.ら、J Exp Med.184:1953−62、1996;Shurin,MR.ら、Cell Immunol.179:174−84、1997)。Flt3は、マウスチロシンキナーゼレセプターであり(Rosnet,O.ら、Oncogene 6:1641−50、1991)、これは、第IIIレセプターキナーゼファミリーのメンバーである(概説については、Lyman,SD、Curr Opin Hematol.5:192−6、1998を参照のこと)。造血組織において、Flt3の発現は、CD34ポジティブ前駆体に制限される。Flt3を使用して、対応するリガンドであるFlt3−リガンドを同定し、そして引続いてクローン化した(Lyman,SDら、Cell 75:1157−67、1993;Hannum,Cら、Nature 368:643−8、1994)。Flt3−リガンドの主な形態は、膜貫通タンパク質として合成され、機能的に類似の可溶性ECDは、このタンパク質からタンパク質分解切断によって生成される(Lymanら、前出)。これらのタンパク質は、固有のチロシンキナーゼレセプターに結合し、そして活性化する。造血細胞の中で、Flt3レセプターの発現は、DC前駆体を含む最も未分化の前駆体細胞に主に限定される。Flt3−リガンドのECDは、いくつかのマウスモデル腫瘍(線維肉腫、乳癌、肝癌、肺癌、黒色腫およびリンパ腫を含む)に対する強力な抗腫瘍効果を生成した(Lynch,DHら、Nat Med.3:625−631、1997;Chen,Kら、Cancer Res.57:3511−3516、1997;Braun,SEら、Hum Gene Ther.10:2141−2151、1999;Peron,JMら、J Immunol.161:6164−6170、1998;Chakravarty,PKら、Cancer Res.59:6028−6032、1999;Esche,Cら、Cancer Res.58:380−383、1998)(19)。本発明者らの同僚らは、HPV Ek7タンパク質をコードするDNAを、Flt3−リガンドECDをコードするDNAに連結した。この構築物での免疫は、E7抗原特異的CD8+T細胞の増殖および活性化を劇的に増強し、確立されたE7発現転移性腫瘍に対する強力な抗腫瘍免疫を生じた。
【0206】
HSV−1タンパク質VP22は、数ある中で、その顕著な細胞内輸送特性が使用され得る(Elliott,G.およびP.O’Hare.1997.Cell 88:223−33)ことに起因して、抗原の伝播の増強に寄与するプロトタイプタンパク質である。例えば、p53に連結されたVP22(Phelan,A.ら,1998,Nat Biotechnol 16:440−443)またはチミジンキナーゼに連結されたVP22(Dilber,MSら,1999,Gene Ther 6:12−21)は、連結したタンパク質の、インビトロでの周囲の細胞への伝播およびモデル腫瘍の処置を容易にした。DNAワクチンの状況でHPV−16 E7抗原に連結されたVP22は、ワクチン接種したマウスにおいてE7特異的CD8+T細胞前駆体の数における劇的な増加(約50倍)を導き、そしてあまり有効でないDNAワクチンをE7発現腫瘍に対して顕著な効力を有するDNAワクチンへと変換させた。非伝播性VP22変異体は、ワクチン効力を増強することができなかった。VP22および同様の作用形態を有し得るタンパク質は、以下のいくつかの方法でワクチン効力の増強に寄与する:(1)トランスフェクトされた細胞から周囲のAPCへの抗原の伝播を容易にし、それによってMHCクラスI経路を通して抗原を提示するAPCの数を増加させる;(2)抗原を、トランスフェクトされた細胞において、より効率的に提示する;(3)「交叉プライム(crosspriming)」を行い、それによってVP22/抗原融合タンパク質の放出が、MHC−I拘束経路を介した提示のための、DC(または他のAPC)による取り込みおよびプロセシングを導く(Huang,AYら,1994,Science 264:961−965)。
【0207】
当業者は、本発明に従って使用するための病原体由来の関連タンパク質の適切なエピトープ(例えば、CTLエピトープ)をどのようにして同定するかがわかる。
【0208】
(細胞および動物へのB7−DC DNAの送達)
DNA送達(例えば、一般的に「遺伝子治療」として公知であるものをもたらすためのDNA送達)は、細胞への、そして最終的には生存動物への「外来」DNAの導入を含む。遺伝子治療についてのいくつかの一般的ストラテジーが研究されており、そして広範に概説されている(Yang,N−S.,Crit.Rev.Biotechnol.12:335−356(1992);Anderson,W.F.,Science 256:808−813(1992);Miller,A.S.,Nature 357:455−460(1992);Crystal,R.G.,Amer.J.Med.92(補遺6A):44S−52S(1992);Zwiebel,J.A.ら,Ann.N.Y.Acad.Sci.618:394−404(1991);McLachlin,J.R.ら,Prog.Nucl.Acid Res.Molec.Biol.38:91−135(1990);Kohn,D.B.ら,Cancer Invest.7:179−192(1989)(これらの参考文献は、その全体が、本明細書中に参考として援用される)。
【0209】
1つのアプローチは、培養中の初代細胞への核酸移入、続いてエキソビボで形質転換された細胞の、全身的または特定の器官もしくは組織へかのいずれかの、宿主への自己移植を含む。
【0210】
本発明の目的を達成するために、核酸治療は、インビボでの哺乳動物の体細胞組織または器官への機能的に活性なDNAの直接的移入によって達成される。DNA移入は、以下に記載の多数のアプローチを用いて達成され得る。これらの系は、選択マーカー(例えば、G418耐性)の使用によってインビトロでの好首尾の発現について試験されて、DNAを発現するトランスフェクトされたクローンが選択され得、続いて適切な免疫アッセイにおいてB7−DC発現産物に対する抗体を用いてのこの産物の存在が(誘導性系での場合はインデューサーでの処置後に)検出され得る。この手順(DNA取り込み、プラスミド組み込みおよび組み込まれたプラスミドの安定性を含む)の効率は、公知の方法を用いたプラスミドDNAの直鎖化および高分子量哺乳動物DNAを「キャリア」として用いた同時トランスフェクションによって改善され得る。
【0211】
当該分野で報告される好首尾の「遺伝子移入」の例としては、以下が挙げられる:(a)マウス筋肉組織へのプラスミドDNAの直接注入(これは、無期限にわたってマーカー遺伝子の発現を導いた)(Wolff,J.A.ら,Science 247:1465(1990);Acsadi,G.ら,The New Biologist 3:71(1991));(b)レトロウイルスベクターは、インビボおよびインサイチュでの血管組織の感染に有効である;(c)レトロウイルス調製物の門脈注射および肝臓への直接注射は、遺伝子移入およびインビボでの発現をもたらした(Horzaglou,M.ら,J.Biol.Chem.265:17285(1990);Koleko,M.ら,Human Gene Therapy 2:27(1991);Ferry,N.ら,Proc.Natl.Acad.Sci.USA 88:8387(1991));(d)肺組織への組換えアデノウイルスの気管内注入は、肺の気道上皮における外来遺伝子のインビボでの移入および長期発現に有効であった(Rosenfeld,M.A.ら,Science 252:431(1991);(e)単純ヘルペスウイルスベクターは、脳組織へのインビボでの遺伝子移入を達成した(Ahmad,F.ら編,Miami Short Reports−Advances in Gene Technology:The Molecular Biology of Human Genetic Disease,第1巻,Boerringer Manneheim Biochemicals,USA,1991)。
【0212】
レトロウイルス媒介ヒト治療は、両栄養性の複製欠損レトロウイルス系を利用する(Temin,H.M.,Human Gene Therapy 1:111(1990);Teminら,米国特許4,980,289;Teminら,米国特許4,650,764;Teminら,米国特許第5,124,263号;Wills,J.W.米国特許5,175,099;Miller,A.D.,米国特許第4,861,719号)。このようなベクターは、機能的DNAをヒトの細胞または組織へと(例えば、アデノシンデアミナーゼ遺伝子をリンパ球へと、NPT−II遺伝子および腫瘍壊死因子遺伝子を腫瘍浸潤性リンパ球へと)導入するために用いられている。レトロウイルス媒介遺伝子送達は一般に、遺伝子移入のために標的細胞の増殖を必要とする(Miller,D.G.ら,Mol.Cell.Biol.10:4239(1990))。この条件は、本発明のDNA分子が導入されるべき特定の好ましい標的細胞(すなわち、活発に増殖する腫瘍細胞)によって満たされる。多数の方法のうちのいずれかを用いたプラスミドによる、およびレトロウイルスベクターによる、トランスフェクションを用いた嚢胞性線維症の遺伝子治療は、Collinsら,米国特許5,240,846によって記載されている。
【0213】
B7−DC配列をコードするDNA分子は、当該分野において周知であるように、複製欠損レトロウイルスを産生するパッケージング細胞株を用いてレトロウイルスベクター中にパッケージングされ得る(例えば、Cone,R.D.ら,Proc.Natl.Acad.Sci.USA 81:6349−6353(1984);Mann,R.F.ら,Cell 33:153−159(1983);Miller,A.D.ら,Molec.Cell.Biol.5:431−437(1985);Sorge,J.ら,Molec.Cell.Biol.4:1730−1737(1984);Hock,R.A.ら,Nature 320:257(1986);Miller,A.D.ら,Molec.Cell.Biol.6:2895−2902(1986)を参照のこと)。遺伝子移入について効率的でかつ安全な、より新しいパッケージング細胞株もまた記載されている(Bankら,U.S.5,278,056)。
【0214】
このアプローチは、レトロウイルスベクターを選り抜きの組織または器官へと送達するために部位特異的様式で利用され得る。従って、例えば、カテーテル送達系が使用され得る(Nabel,EGら,Science 244:1342(1989))。レトロウイルスベクターまたはリポソームベクターのいずれかを使用するこのような方法は、発現されるべき核酸を血管壁へと、または腫瘍の血液循環物へと送達するために特に有用である。
【0215】
以下を含めた他のウイルスベクターもまた使用され得る:組換えアデノウイルス(Horowitz,M.S.,In:Virology,Fields,BNら編,Raven Press,New York,1990,1679頁;Berkner,K.L.,Biotechniques 6:616−919,1988),Strauss,S.E.,In:The Adenoviruses,Ginsberg,HS編,Plenum Press,New York,1984,第11章)、ニューロン特異的送達および持続性のための単純ヘルペスウイルス(HSV)。ヒトの遺伝子治療のためのアデノウイルスベクターの利点としては、組換えは稀であり、このようなウイルスと関連するヒトの悪性疾患は知られておらず、アデノウイルスゲノムは7.5kbの大きさまでの外来遺伝子を受け入れるように操作され得る二本鎖DNAであり、そして生のアデノウイルスは、安全なヒトワクチン生物であるという事実が挙げられる。アデノ随伴ウイルスもまた、本発明によるヒトの治療のために有用である(Samulski,R.J.ら,EMBO J.10:3941(1991)。
【0216】
本発明のDNA分子を発現し得、そして(特にヒトにおける)本発明の治療設定において有用である別のベクターは、ワクシニアウイルスであり、ワクシニアウイルスは、非複製性にされ得る(米国特許5,225,336号;同5,204,243号;同5,155,020号;同4,769,330号;Sutter,Gら,Proc.Natl.Acad.Sci.USA(1992)89:10847−10851;Fuerst,T.R.ら,Proc.Natl.Acad.Sci.USA(1989)86:2549−2553;Falkner F.G.ら;Nucl.Acids Res(1987)15:7192;Chakrabarti,Sら,Molec.Cell.Biol.(1985)5:3403−3409)。組換えワクシニアウイルスおよび異種DNAを含む他のウイルス、ならびに免疫およびDNA治療におけるそれらの使用の説明は、以下に概説されている:Moss,B.,Curr.Opin.Genet.Dev.(1993)3:86−90;Moss,B.Biotechnology(1992)20:345−362;Moss,B.,Curr Top Microbiol Immunol(1992)158:25−38;Moss,B.,Science(1991)252:1662−1667;Piccini,Aら,Adv.Virus Res.(1988)34:43−64;Moss,B.ら,Gene Amplif Anal(1983)3:201−213。
【0217】
裸のDNAもしくはRNAまたはウイルスベクターに加えて、操作された細菌はベクターとして使用され得る。以下を含む多数の細菌株:Salmonella、BCGおよびListeria monocytogenes(LM)(HoisethおよびStocker,Nature 291,238−239(1981);Poirier,TPらJ:Exp.Med.168,25−32(1988);(Sadoff,J.C.ら,Science 240,336−338(1988);Stover,C.K.ら,Nature 351,456−460(1991);Aldovini,A.ら,Nature 351,479−482(1991);Schafer,R.ら,J.Immunol.149,53−59(1992);Ikonomidis,G.ら,J.Exp.Med.180,2209−2218(1994))。これらの生物は、ワクチンベクターとしての使用のために有望な以下の2つの特徴を提示する:(1)経口ワクチン送達の可能性を提供する、腸感染経路;および(2)単球/マクロファージの感染(それによってプロフェッショナルAPCへと抗原を標的化する)。
【0218】
インビボでのウイルス媒介遺伝子移入に加えて、以下を含めて、当該分野で周知の物理的手段は、DNAの直接移入のために用いられ得る:プラスミドDNAの投与(Wolffら,1990,前出)およびパーティクル−ボンバードメント媒介遺伝子移入(Yang,N.−S.ら,Proc.Natl.Acad.Sci.USA 87:9568(1990);Williams,R.S.ら,Proc.Natl.Acad.Sci.USA 88:2726(1991);Zelenin,A.V.ら,FEBS Lett.280:94(1991);Zelenin,A.V.ら,FEBS Lett.244:65(1989);Johnston,S.A.ら,In Vitro Cell.Dev.Biol.27:11(1991))。さらに、インビトロで細胞へと遺伝子を移入するための周知の手段であるエレクトロポレーションを用いて、本発明によるDNA分子をインビボで組織へと移入し得る(Titomirov,A.V.ら,Biochim.Biophys.Acta 1088:131((1991))。
【0219】
「キャリア媒介遺伝子移入」もまた記載されている(Wu,C.H.ら,J.Biol.Chem.264:16985(1989);Wu,G.Y.ら,J.Biol.Chem.263:14621(1988);Soriano,P.ら,Proc.Natl.Acad.Sci.USA 80:7128(1983);Wang,C−Y.ら,Proc.Natl.Acad.Sci.USA 84:7851(1982);Wilson,J.M.ら,J:Biol.Chem.267:963(1992))。好ましいキャリアは、標的化されたリポソーム(Nicolau,C.ら,Proc.Natl.Acad.Sci.USA 80:1068(1983);Sorianoら,前出)(例えば、免疫リポソーム(これは、アシル化mAbを脂質二重層へと取り込み得る(Wangら,前出)))である。アシアロ糖タンパク質/ポリリジン(Wuら,1989,前出)のようなポリカチオンが用いられ得、ここで、結合体は、標的組織を認識する分子(例えば、肝臓についてはアシアロオロソムコイド)およびトランスフェクトされるべきDNAに結合するDNA結合化合物を含む。ポリリジンは、DNAに損傷を与えることなくDNAを結合するDNA結合分子の一例である。次いで、この結合体は、移入のために本発明に従ってプラスミドDNAと複合体化される。
【0220】
トランスフェクションまたはマイクロインジェクションのために用いられるプラスミドDNAは、当該分野で周知の方法を用いて(例えば、Quiagen手順(Quiagen)を用いて)調製され得、続いてDNAが、公知の方法(例えば、本明細書中に例示される方法)を用いて精製され得る。
【0221】
さらに、上記のように、本発明による形質導入されたB7−DC分子の有用性については、安定な発現を必要としないかもしれない。むしろ、ポリペプチドの一過性の発現は、形質導入された細胞がその免疫原性機能および/または補助刺激機能を果たすために充分であり得る。
【0222】
ここで、本発明を一般的に記載してきたが、本発明は、以下の実施例を参照してより容易に理解される。以下の実施例は、例示のために提供され、明記しない限り、本発明を限定することを意図しない。
【実施例】
【0223】
(実施例I)
(材料および方法)
(細胞の調製および培養)
6〜12週齢の雌性BALB/cマウスをNCIから購入し、そしてDCおよびマクロファージの調製のために用いた。
【0224】
骨髄由来DCを、以前に記載された(26)ように、5%ウシ胎仔血清(FCS)(Hyclone)、ペニシリン/ストレプトマイシン(JRH Biosciences)、ゲンタマイシン(Sigma)、非必須アミノ酸(JRH Biosciences)、L−グルタメート(JRH Biosciences)、ピルビン酸ナトリウム(Sigma)、2メルカプトエタノール(Sigma)および1000単位/ml組換えマウスGM−CSF(Immunex)を補充したRPMI1640(Gibco BRL)培地中で培養した。8日目の骨髄由来DCを、従来法によってモノクローナル抗体で染色した。MHCクラスIIに対するモノクローナル抗体14−4−4を、ハイブリドーマ上清から精製した。Johns Hopkins UniversityのWilliam Baldwin博士は親切なことに、CTLA4−Ig融合分子を供給した。MHCクラスIについての抗体(28−14−8)、F4/80についての抗体(Cl.A3−1)、B7.1についての抗体(1G10)、B7.2についての抗体(GL1)、FcγRII/IIIについての抗体(2.4G2)およびMac−1についての抗体(M1/70)を、PharMingenから購入した。テスターcDNA調製のために、8日目の細胞を、Johns Hopkins University Oncology Centerで14−4−4sおよびCTLA4−Igを用いてセルソーターによって精製した。選別後のMHCクラスIIhiおよびB7hi集団の純度は93%〜98%であった。
【0225】
骨髄由来マクロファージを、10% FCS、ペニシリン/ストレプトマイシン、非必須アミノ酸、ピルビン酸ナトリウム、Lグルタミン、2−メルカプトエタノールおよび250単位/ml組換えマウスM−CSFを補充したRPMI−1640培地で培養し、そして以前に記載された(27)通りにこれらを500単位/mlのγ−IFN(Pharmingen)および5μg/ml LPS(Sigma)で処理した。刺激後、MHCクラスIIおよびB7の細胞表面発現を、培養10日目のフローサイトメトリー分析を用いて確認した。
【0226】
マクロファージ細胞株WEHI−3、RAW264.7、J774.A.1、PU5−1.8は、親切なことに、NIAID,National Institutes of HealthのJoshua Farber博士によって提供された。これらを、ATCCの推奨する培地で培養した。
【0227】
(同種混合リンパ球反応)
MHCクラスIIhiおよびB7hiと特徴付けされた8日目のBM由来DCを、MLC中の同種T細胞を刺激する能力について試験した。MLC反応を、3×105同種C57BL/6リンパ球へ漸増数のBALB/c刺激細胞を添加することによって、96ウェル平底マイクロプレートにおいて行った。3日間の培養後、T細胞増殖を、最後の18時間の培養にわたる、各ウェルへの1μCiの[3H]−メチル−チミジン(Amersham)の添加によって評価した。次いで、細胞を収集し、そして放射能の取り込みを、β計数管(Packard 96)を用いて決定した。
【0228】
(cDNA差引きハイブリダイゼーション)
選別したDCおよび活性化したマクロファージからの総RNAを、TRIZOL(Gibco BRL)を用いて抽出した。メッセンジャーRNAを、Oligotex mRNA精製キット(Qiagen)によって精製した。本発明者らは、PCRベースのSMART cDNA合成系(Clonetech)を用いてcDNAを増幅し、続いてPCRベースの差引き系PCR Select(Clonetech)を用いた。差引きを、製造業者のプロトコルに従って行った。最後の差引きPCR後、DNAフラグメントを、プラスミドベクターpCR2.1(Invitrogen)またはpCR Blunt(Invitrogen)に連結した。形質転換後、各クローンを、プラスミドDNA増幅およびミニプレップDNAのために増殖させ、次いでEcoRIで消化して挿入物の存在を確認した。次いで、プラスミドドットブロットを行って、このcDNAクローンが樹状細胞特異的であることを確認した。アルカリ変性ミニプレップDNAをHybond N+メンブレン(Amersham)にスポットし、そして選別されたDCまたは活性化されたマクロファージ由来のSMART cDNAプローブとハイブリダイズさせた。これらのcDNAプローブを、ランダムプライマー標識法(Stratagene Prime−It II)を用いて32P標識した。ハイブリダイゼーションおよび洗浄を、以前に記載された(28)通りに行った。メンブレンをフィルム(Amersham)に1日間〜2日間にわたって暴露し、そして現像した。
【0229】
(プラスミドドットブロット分析)
アルカリ変性ミニプレップDNAサンプルを、Hybond N+メンブレン(Amersham)にスポッティングし、選別されたDCまたは活性化したマクロファージから誘導されたSMART(登録商標)cDNAプローブとハイブリダイズさせた。これらのcDNAプローブを、ランダムプライマー標識法(Stratagene Prime−It II)を用いて32P標識した。ハイブリダイゼーションおよび洗浄を、以前に記載された通りに行った。オートラジオグラフィーについては、メンブレンを、フィルム(Amersham)に1〜2日間にわたって暴露し、そして現像した。
【0230】
(cDNAライブラリーの構築およびスクリーニング−B7−DCのクローニング)
骨髄由来DCを8日目に選別をせずに収集した。これらの細胞の約20%〜40%は、高いMHCクラスIIおよびB7を発現した。総RNA抽出、続いてポリA RNA精製を上記に記載された通りに行った。オリゴdTプライムDCライブラリー構築については、本発明者らは、λZAP Express cDNA合成系(Stratagene)を用いた。B7−DCのPCR DNAフラグメントをプローブし、そしてスクリーニングのために用いた。メンブレン転移、変性、再生を、Stratageneのプロトコルを用いて行った。プローブの放射性標識、ハイブリダイゼーション、洗浄およびオートラジオグラフィーを、上記に記載の通りに行った。陽性クローンを単離し、そして2回目のスクリーニングを行った。2回目のスクリーニングの後、プラスミドをインビボ切り出しによって切り出し、そしてドットブロッティングおよび配列決定によって試験した。配列決定を、Johns Hopkins University School of MedicineのCore Facilityによって行った。BLASTプログラムを用いて、以前に報告された遺伝子に対する類似性について、このヌクレオチド配列の、Genbank(NCBI)に対する相同性検索を行った。全長のB7−DC cDNAクローンを、DC cDNAライブラリーから取り出した。5’RACEを、SMART RACE cDNA増幅キット(Clontech)を用いて行った。5’−RACE産物をpCR2.1ベクター中にクローニングし、そして配列決定した。さらに2つの全長B7−DCクローンをRT−PCRによって入手し、そしてそれらの配列を比較して、配列の誤りを回避した。
【0231】
ヒトB7−DCを以下の通りにクローニングした:ヒトDCを、以前に記載(29)の通りのGM−CSF+IL−4またはGM−CSF+Flt−3Lのいずれかにおける培養によって正常末梢血単核細胞から入手した。RNAを上記の通りに抽出した。BLAST検索は、マウスB7−DCに対する相同性を有する、GenBank登録番号AK001879の重複するESTクローンを同定した。5’RACEを、上記の通りに行った。本発明者らは、5’−RACE PCRフラグメントを配列決定し、そしてヒトB7DCの5’−UTRに対応するプライマーを設計した。B7−DCの5’−UTRおよび3’−UTRにおける以下のプライマーを用いて、全長ヒトB7−DCを増幅した:
【化6】
ヒトおよびマウスのB7−DC cDNAの全長cDNA配列は、EMBL/GenBank/DDBSに登録番号AF329193およびAF142780の下で寄託された。
【0232】
(BAC(129SVJ)ライブラリーのスクリーニング/ゲノムクローニングおよびマッピング)
BACライブラリーのスクリーニングは、製造業者(Genome Systems,Inc.)のプロトコルに従った。以下のプライマーを用いた:
【化7】
BACライブラリーのスクリーニングによって、3つの陽性クローンが得られた。染色体位置決定マッピングを、蛍光インサイチュハイブリダイゼーション(Genome Systems Inc.)によって行った。合計80個の分裂中期細胞を分析し、79個が特異的標識を示した。ヒトB7−DCマッピングを、入手可能なバイオインフォーマティックツールであるNCBIのBLASTプログラムおよびInternational RH Mapping Consortiumを用いることによって行った。hB7−DCの配列をhtsgにおいて検索し、そして第9染色体に局在する2つのBACクローンRP11−574F11(AL162253)およびRp11−635N21(AL354744)にマッピングされることが見い出された。
【0233】
(仮想ノーザンブロッティング)
4〜6週齢の雌性Balb/cマウスをNCIから購入し、そして組織RNA調製のために用いた。組織、選別されたDCおよび活性化されたマクロファージについてのRNA抽出およびSMART cDNA合成を、上記の通りに行った。SMART PCR cDNAを、PCR精製キット(Qiagen)によって精製した。0.5μg/レーンの精製DNAを1%アガロースゲルに泳動し、そしてNytranナイロンメンブレン(Schleier and Schuell)に移した。放射性プローブを作製するために、本発明者らは、差引きしたライブラリーから誘導されたプラスミドDNAをテンプレートとして増幅した。本発明者らは、プラスミドDNAのクローニング部位にすぐ隣接したプライマーセットを用いるPCRによってDNAを増幅し、そしてクローンの各々の精製したPCR DNAをハイブリダイゼーションプローブのために用いた。これらのプライマーのヌクレオチド配列は以下の通りである。
【化8】
ヒトDCおよびコントロールの胎盤の総RNAの仮想のノーザン分析もまた行った。用いたプローブおよびRNA調製は、上に記載された。プローブの放射性標識、ハイブリダイゼーション、洗浄およびオートラジオグラフィーを、上記の通りに行った。
【0234】
(ハムスター抗mB7−DC Ab産生)
DC2.4細胞株、RAW246.7細胞株およびRENCA細胞株中のB7−DCの安定なトランスフェクタントを用いて、Armenian Hamsterを免疫した。B7−DCを、改変したpCAGGSベクター(30)中にクローニングした。このハムスターを、B7−DC(Rockland)を含むプラスミドで3回追加免疫した。この研究において用いた抗B7−DC抗体は、免疫した3匹のハムスターのうちの1匹の血清由来であった。
【0235】
(CD28−Ig、CTLA4−IgおよびPD−1−Igの結合アッセイ)
293T細胞を、Lipofectamine 2000(Gibco BRL)を用いてB7.1−pCAGGS、B7−DC−pCAGG、PD−1−pCAGGSまたはベクター単独でトランスフェクトした。24時間後、細胞をFACS緩衝液(1×HBSS、2%仔ウシ血清、10M HEPESおよび0.1% NaN3)中に再懸濁し、そして1000rpmで5分間、4℃で回転させた。次いで、この緩衝液を棄て、抗体をチューブに添加し、4℃で20分間インキュベートし、FACS緩衝液で2回洗浄し、そしてこれを二次抗体について繰り返した。サンプルをFACScanに泳動した。B7.1抗体を、1:5希釈、10μl/サンプル(Gal−Tag)で用いた。組換えCD28−Ig、CTLA−4−IgおよびPD−1−Igキメラを、2μg/ml、10μl/サンプル(R&D System,Inc)で用いた。ヤギF(ab’)2抗ヒトIgG−PEを、1:20希釈(Southern Biotechnology Associates,Inc.)で用いた。
【0236】
(B7−DC−Igダイマー合成)
B7−DC−Ig構築物を、膜貫通ドメインを含まないB7−DCのN末端アミノ酸をコードする配列を、pIg−Tail Plusベクター(R&D systems)中のヒトIgG1 FcのC末端アミノ酸をコードする配列に対してインフレームで融合することによって作製した。COS−7細胞を、LipofectAMINE 2000(GIBCO BRL)またはGINE JAMMER(Stratagene)を用いてpIg/B7−DCで一過性にトランスフェクトした。B7−DC−Ig融合タンパク質を、飽和硫酸アンモニウム沈澱を用いて無血清上清から精製した。SDS−PAGEおよび銀染色は、90%を超える純度を実証した。
【0237】
(T細胞増殖およびサイトカインアッセイ)
抗CD3を用いた補助刺激アッセイについて、96ウェル平底プレート(Dynex製のImmulon 4)を、1×PBS(Gibco)pH7.4中に希釈した100ng/mlの抗CD3抗体(2C11,Pharmingen)およびB7.1−Ig(R&D System)、B7−DC−IgまたはIsotypeコントロール(Sigma)で37℃で2時間にわたって予めコーティングした。次いで、これらのプレートを1×PBSで3回洗浄し、そして10% FCS、ペニシリン/ストレプトマイシン、非必須アミノ酸、ピルビン酸ナトリウム、Lグルタミン、2−メルカプトエタノールを補充したRPMI1640培地で30分間にわたってブロッキングし、その後、T細胞を添加した。脾臓およびリンパ節を、6〜10週齢のBALB/cマウスから入手した。RBCをACK緩衝液を用いて溶解し、そしてT細胞を、間接的方法で、ダイナビーズ(dynabeads)M−280(Dynex)を用いて精製した。このビーズをPBS(pH7.4)+1% FCSで2回洗浄し、その後、この細胞を添加し、そして抗IEdおよびB220/CD45ROまたはCD8α(Pharmingen)から構成される抗体カクテルを、この細胞に添加し、そして4℃で30分間にわたって二方向性混合を用いてインキュベートした。この細胞を、Dynal MPC中のチューブに5分間配置することによって単離し、1500rpmで5分間遠心分離し、そしてPBS(pH7.4)+1% FCSで2回洗浄して未結合のAbを除去した。同じ手順を15分間のインキュベーションを用いて繰り返し、そしてこの細胞を2×105細胞/ウェルでプレーティングした。72時間のインキュベーション後、10μlの3H−チミジン(1μCi/ウェル)を各ウェルに添加し、そして18時間にわたってインキュベートした。細胞をPackard Micromate Cell Harvesterを用いて収集し、そしてフィルターをPackard Matrix 96直接β計数管で読み取った。
【0238】
HA抗原を提示するためのRENCA系を用いた補助刺激アッセイについては、RENCA細胞を、10% FCS、ペニシリン/ストレプトマイシン、非必須アミノ酸、ピルビン酸ナトリウムを補充したRPMI−1640培地で培養した。これを、MHCクラスII発現について、IFN−γ(75U/ml)を用いて72時間にわたって誘導した。次いで、これらを13,200Radで照射し、そして2×104細胞/ウェル(96ウェル平底プレート)でプレーティングした。次いで、HA110−120ペプチドを2.5μg/ウェルで添加し、そして種々の濃度のIg−融合分子を添加した。トランスジェニックI−Ed+HA特異的T細胞(H.von Boehmer,Harvard Universityの親切な贈与物)を上記の通りに単離し、そして4×105細胞/ウェルでプレーティングした。48時間のインキュベーション後、10μlの3Hチミジン(1μCi/ウェル)を各ウェルに添加し、そして18時間にわたってインキュベートした。細胞を、Packard Micromate Cell Harvesterを用いて収集し、そしてフィルターをPackard Matrix 96 Directβ計数管で読み取った。
【0239】
ELISAによるサイトカイン産生の分析について、培養物を上記の通りに設定し、そして上清を指定の時間で収集した。IL−2、IL−10およびIFN−γの濃度を、市販のELISAキット(Endogen)ならびにIL−4およびIL−6(R&D System)を用いて決定した。
【0240】
(インビボでの補助刺激)
B10.D2遺伝子バックグランド上のI−Ed拘束HAエピトープ(110SFERFEIFPKE120[配列番号12])を認識するTCRを発現するTCRトランスジェニックマウス系統6.5由来のプールされた腋窩、鼡径、頚部、および腸間膜のLNを、RPMI培地(GIBCO BRL)中で解離し、100μmのナイロン細胞ストレイナーを通過させ、そして滅菌ハンクス緩衝液(GIBCO BRL)中で洗浄した。FACS(登録商標)染色して、クローン型(clonotypic)CD4細胞の割合を決定した後、0.2mlの滅菌ハンクス緩衝液中に2.5×106クローン型細胞を含む細胞調製物を、レシピエントB10.D2マウスの尾静脈に静脈内(i.v.)注射した。この養子移入から3日後、この動物を、後足蹠(footpad)に皮下(s.c.)注射することによりワクチン接種した。各マウスに、以下の3つの調製物のうちの1つを両側に注射した:
(A)10μgの合成HA(足蹠1つにつき)(HAペプチド(110−120)を1:1のv/v比で不完全フロイントアジュバント(IFA)(Sigma)と合わせた)
(B)HA−IFAと25μgのB7−DC−Igとの混合物、または
(C)HA−IFAと25μgのアイソタイプコントロール抗体との混合物。7日後、排出LN節を採取し;1.5×105LN細胞を丸底96ウェル組織培養プレート中で示された濃度の合成HAペプチドとともにインキュベートした。48時間培養物を1μCi[3H]チミジンでパルスし、そしてさらに12時間インキュベートした後採取し、そして組み込まれた放射活性の量を決定することにより増殖アッセイを行った。
【0241】
(実施例II:B7−DCの同定および特徴づけ)
B7−DCを、DCと活性化マクロファージとの間の差引きライブラリーから単離した。cDNA差引きに用いた2つの集団は、「テスター」集団としてのGM−CSFと培養した骨髄由来DC、および「ドライバー」集団としてのγ−インターフェロン+LPS活性化した接着性骨髄由来M−CSFマクロファージであった。8日目に、MHCクラスIIhiB7hi「成熟」DCを、テスターcDNAの供給源として、>93%純度までソートした。DCを、フローサイトメトリーにより、マクロファージより約50倍MHCクラスIIレベルが高いと特徴付けた。両方の集団がB7.1およびB7.2を発現したが、B7.2レベルはDCにおいて有意に高かった。F4/80およびCD16は、マクロファージ集団で高レベルで発現された。2つの集団の機能的比較により、DC集団は、同種異系の混合したリンパ球反応を刺激することにおいてマクロファージ集団より約100倍強力であることが実証された。両方の集団からRNAを抽出した後、本発明者らは、PCRベースのcDNA合成システム、続いてPCRベースの差引き手順であるPCR Selectを用いた。差示的に発現されるクローンのうちの1つが、新規な免疫グロブリンスーパー遺伝子ファミリーのメンバー(本発明者らは、これをB7−DCと名付けた)をコードした。マウスB7−DC cDNAは、長さが約1.7kbであり、これは、23アミノ酸(aa)のN末端シグナルペプチドを有し、そして推定分子量が約25kdの247アミノ酸(aa)の前駆体タンパク質をコードする(表1)。推定リーダー配列および膜貫通ドメインを、SOSUIプログラム(31)を用いて同定した。2つの荷電aaは、mB7−DCの23aa膜貫通ドメイン内に見出される。このことは、あり得る結合パートナーを示唆する。aaレベルで、マウスB7−DCは、ヒトB7−DCと70%同一であり、このことは、それらがオルソログであることを示す(以下の表を参照のこと)。hB7−DCは、より長い細胞質テイルを有するという点で、マウスB7−DCとはわずかに異なる。
【0242】
相同性検索を介して、B7−DCがB7−H1に対して有意な相同性を有し(34%同一性、48%類似性)(表2)、より低い程度には、ブチロフィリン(butyrophilin)に対して(30%同一性、45%類似性)、そしてB7.1およびB7.2に対して20%未満の同一性を有することが見出された(表3)。系統発生的な研究により、エキソンシャッフリング(32,33)を介して、ブチロフィリンがB7スーパーファミリーに関連する可能性が高いことが示される。これらは、各々、標準的な(canonical)IgV−IgC構造および膜貫通ドメインを有する。他のB7ファミリーのメンバーとは対照的に、マウスB7−DCは、非常に短い細胞質テイル(4aa)を有する。
【0243】
(表1)
マウスB7−DCおよびヒトB7−DCのアミノ酸配列比較。mB7−DCの推定リーダードメインおよび膜貫通ドメインは、上に線を引いた。アラインメントは、Clustalw−Gonnet Pam250マトリクスを用いて行った。[*]は、同一アミノ酸を示し、[:]は、保存的置換を示す。免疫グロブリンVまたはCドメイン内部のジスルフィド結合の形式に重要であり得るシステイン残基は、斜体にされている。
【表1】
(表2:mB7−DCおよびmB7−H1のアミノ酸配列比較)
【表2】
(表3:B7−DCとB7ファミリーメンバーとのアミノ酸配列比較)
【表3】
NCBI blast2検索を用いて比較を行った(マトリクスBLOSUM62)。
1.対応する位置での同一アミノ酸
2.対応する位置での類似アミノ酸−以下のようにグループ分けされる:(A,G);(S,T);(E,D);(R,K,H);(Q,N);(V,I,L,M);(Y,F);(W);(C);(P)。
3.マトリクスBLOSUM62を用いて有意な類似性は見出されなかった。
4.BT=ブチロフィリン5.=PDL−1。
【0244】
mB7−DCのゲノム構造を決定するために、本発明者らは、5’および3’UTRに由来するプローブを用いてプールされたBacterial Artificial Chromosome(BAC)ライブラリーをスクリーニングすることによりゲノムクローンを単離した。染色体位置のマッピングを、BACクローンを用いて行った。B7−DCの染色体位置は、蛍光インサイチュハイブリダイゼーション(FISH)を用いて行った。10の特異的に染色した第19染色体の測定により、mB7−DCが、第19染色体の異質染色質−真性染色質境界からテロメアまでの距離(19C2と19C3のバンドの間の境界に対応する領域)の47%である位置に位置することが実証された。ハイブリダイズしたスライドをフルオレセイン化抗ジゴキシゲニン抗体中でインキュベートし、続いてDAPIで対比染色することにより、特異的ハイブリダイゼーションシグナルを検出した。この遺伝子座は、ヒト第9染色体の領域に対応し、この領域においてhB7−H1がマッピングされている。
【0245】
hB7−DCは2つの第9染色体のBACクローンに位置することが見出された。さらに、hB7−DCおよびhB7−H1はともに、約164kbの挿入物サイズを有する、単一の第9染色体のBACクローンに位置することが見出された(図1)。B7−DCおよびB7−H1がゲノム上で近位にあることは、B7.1/B7.2対を暗示し、この対は、互いに1メガ塩基以内にマッピングされる。
【0246】
(実施例III:B7−DCは樹上細胞において選択的に発現される)
B7−DCの発現パターンを決定するために、複数の組織、マクロファージ細胞株、マクロファージ培養物、ならびに骨髄および脾臓の両方から得られた樹上細胞から抽出したRNAを用いて、事実上のノザンブロッティングを行った。未熟(4日目および6日目)および成熟(8日目およびソートされたMHC IIhiB7hi)の骨髄由来DCおよび脾臓由来DCにおいてB7−DCプローブを用いて強いハイブリダイゼーションが検出されたが、4つのマクロファージ細胞株、活性化BMマクロファージ、腹膜マクロファージのいずれにおいてもシグナルは検出されなかった(図2)。GM−CSF+IL−4またはGM−CSF+Flt−3Lのいずれかを用いて末梢血単球から増殖させたヒトDCにおいてhB7−DCの強い発現が検出された(図3)。B7−DCタンパク質の細胞表面発現を確認するために、抗mB7−DC抗体を用いて、DCを染色した。可溶性B7−DC−Igを用いた染色および遮断が、DCで観察された(図4)。
【0247】
(B7−DCは、CD28にもCTLA−4にも結合しないが、PD−1には結合する)
B7−DCはB7ファミリーに対して構造的相同性および配列相同性を有するが、推定CD28/CTLA−4結合配列、SQDXXXELY(配列番号13)も、XXXYXXRT(配列番号14)も含まない(34)(ここで、X=任意のアミノ酸)。結合を直接評価するために、2量体CD28−IgおよびCTLA−4−Igが、B7−DCまたはB7.1のいずれかでトランスフェクトされた293T細胞を染色する能力を調査した。B7.1トランスフェクタントで強い結合が観察された一方、B7−DCトランスフェクタントに対する結合は観察されなかった(図5)。B7−DCとB7−H1/PD−L1との間の相同性およびゲノム上で近位であることに基づいて、B7−DCについての候補結合パートナーとしてPD−1を試験するために実験を行った。実際に、PD−100IGは、B7−DCトランスフェクタントに結合したが、B7.1トランスフェクタントには結合しなかった。PD−1−IgのB7−DCトランスフェクタントへの結合は、CTLA−IgおよびCD28−IgのB7.1トランスフェクタントへの結合より低かったが、特異的であった。PD−1のB7−DCへの結合のさらなる確認を、本発明者らは、PD−1−Igとの安定なB7−DC−GFPトランスフェクタントのポジティブな染色から得た。B7−H1およびB7h/B7RP−1と同様に、B7−DCは、レセプターとしてCD28もCTLA−4も用いないことが結論付けられた。むしろ、PD−1は、B7−DCについてのレセプターであるようである。
【0248】
(実施例IV:B7−DCは、T細胞についての補助刺激分子として機能する)
T細胞刺激アッセイに添加され得る可溶性B7−DC−Ig融合タンパク質を、B7−DCが補助刺激活性を有するか否かを試験することにおける使用のために生成した。T細胞の増殖応答を、B7−DC−Ig、B7.1−Igまたはアイソタイプコントロールいずれかの存在下で、漸増量の固定化抗CD3による刺激に対して測定した。図6(左)は、最適未満の抗CD3濃度の存在下で、B7−DCがB7.1より大きなT細胞増殖応答を補助刺激したことを示す。さらに、B7−DCにより補助刺激された増殖応答は、CD8細胞よりCD4細胞において高かった(図6、右)。B7−DCは、TCRに集中した刺激がない場合にはT細胞を刺激できなかった。このことは、B7−DCが、真の補助刺激シグナルを提供することを示す。
【0249】
B7−DCはまた、「シグナル1」がMHCペプチド複合体により提供された場合に増殖応答を補助刺激した。RENCA細胞(RT−PCR分析により内因性B7.1、B7.2、またはB7−DCを発現しない)を、γ−IFNで処理して、MHCクラスII発現を誘導した。これらの細胞に、I−Ed拘束HA110−120ペプチド(FERFEIFPKE)(35)(配列番号15)を負荷した。I−Ed+HA110−120特異的TCRトランスジェニックマウス系統から精製した脾臓T細胞を添加し、そして増殖応答をB7−DC−Ig、B7.1−Igまたはアイソタイプコントロールいずれかの存在下で測定した。図7は、B7−DCがB7.1より大きな補助刺激活性を有したことを示す。
【0250】
(B7−DCにより補助刺激されたリンホカイン生成のパターン)
B7ファミリー分子による補助刺激に対して最もよく特徴付けられたT細胞応答は、リンホカイン生成である。これらのリンホカインは、T細胞効果の重要なメディエーターである。抗CD3またはHA抗原で刺激し、B7−DC−Ig、B7.1−Igまたはアイソタイプコントロールいずれかで補助刺激したT細胞による多くの異なるリンホカイン生成を分析するための研究を行った(図8)。リンホカイン補助刺激のパターンは、抗CD3またはMHCペプチド複合体が「シグナル1」として利用されるか否かとかなり一致した。重大なことには、B7−DCは、B7.1より大きなレベルのγ−IFNを補助刺激した。B7−DCはまた、有意な量のIL−6生成を補助刺激したのに対し、B7.1は、実質的に補助刺激しなかった。両方の分子が、IL−2生成を補助刺激したが、B7.1は、B7−DCより非常に効率的に刺激した。従って、B7−DCおよびB7.1の補助刺激のパターンは異なり、B7−DCは重要な前炎症性リンホカインを補助刺激することにおいて効率的である。
【0251】
(実施例V:B7−DCは、インビボ免疫応答を増強する)
B7−DCがインビボで生物学的活性を有するか否かを決定するために、本発明者らは、B7−DC−Igがペプチドワクチンに対する免疫応答を増強するか否かを調べた。B7−DC−Igまたはアイソタイプコントロール抗体を、HA110−120ペプチドおよびIFAの免疫原性カクテルに添加した。HA特異的CD4T細胞のインビボでの計数を可能にするために、2.5×106抗HA6.5T細胞を免疫の3日前にマウスに移入した。免疫して7日後、排出LN細胞を採取し、そしてこの細胞をインビトロで2日間、種々の量のHA110−120ペプチドで刺激した。図9は、B7−DC−Igの添加が、実際にHAに対する増殖応答を劇的に増強したことを示す。排出LNにおけるHA特異的T細胞の総数は、アイソタイプ抗体コントロールに対して、B7−DC−Igを受けた群において約2倍増加した。従って、B7−DCが1細胞あたりですら抗原特異的応答を増強する能力を有することが結論付けられた。
【0252】
(実施例VI:考察および結論)
本発明者らは、DCに高度に拘束された発現を伴い、T細胞について独特な補助刺激特性を有する新たなB7ファミリーメンバーを発見し、そして特徴づけた。B7−DCのヒトオルソログもまた、DCにおいて発現される。
【0253】
この拘束された発現パターンは、先に記載されたB7ファミリーメンバーと対照をなし、このことは、B7−DCが、公知のB7.1/2経路とは異なる免疫応答に関与することを示唆する。弱いB7−DCシグナルが、活性化マクロファージにおいてRT−PCRにより検出されたが、予備のリアルタイムRT−PCR分析により、DCにおけるB7−DC mRNA発現が、活性化マクロファージより15倍高いことが示された。同様に、抗体染色により、活性化マクロファージの表面で非常に低いレベルのB7−DCが検出された。このことが、有意なT細胞活性化に十分か否かは明瞭ではない。
【0254】
B7−DCが補助刺激するリンホカイン生成の異常なパターンは、他のB7ファミリーメンバーと比較して、独特の生物学的役割を示す。サイトカインの伝統的な分類は、以下のとおりである:Th1サイトカインは、IL−2、γ−IFNおよびリンホトキシンを含む;Th2サイトカインは、IL−4、IL−5、IL−6およびIL−13を含む(36)。B7−DCは、いずれの古典的なTh1リンホカインプロフィールもTh2リンホカインプロフィールも誘導しない。B7−DCは、非常にわずかなIL−4を誘導し、IL−10を誘導しない。しかし、IL−6は、Th2サイトカインと考えられる。B7.1に対して、B7−DCにより補助刺激されたより低いIL−2およびより高いγ−IFNは、古典的なTh1パターンに合わない。にもかかわらず、高いγ−IFN生成は、B7−DCが重要なT細胞エフェクター機能を誘起することを示唆する。
【0255】
B7−DCは、IL−6を補助刺激する能力において著しい。B7−DCにより誘導されたT細胞の活発な増殖性応答は、B7.1では観察されないIL−6生成の強い補助刺激により一部説明される。IL−6は、(他の増殖刺激と関連して)T細胞増殖の強力な増幅因子(amplifier)である(37,38)。IL−6は、T細胞機能のみならず、前炎症性応答、単球分化、B細胞分化、血小板新生、骨の吸収、および特定の造血性腫瘍の増殖もまた調節する多機能性サイトカインである(39,40)。IL−6は、ケモカインおよび白血球動員の誘導において、可溶性IL6レセプター(sIL−6R)と協同して機能し得る(41)。これは、Stat−3活性化を介して強力な抗アポトーシス効果を媒介し得る。T細胞におけるIL−6依存性Stat−3活性化は、活性化T細胞の生存についての重要な経路であると報告されている(42,43)が、他の報告では、Stat−3が休止T細胞に対してその効果を発揮することが示唆されている。
【0256】
B7−DCは、CD28もCTLA−4も結合できないが、PD−1(B7−H1/PD−L1についてのレセプター)を結合する(22,47,48)。B7−DCが、ICOS(B7h/B7RP−1についてのレセプター)を結合するか否かは未だ決定されていない(23−25,44−46)。B7−DCとB7−H1/PDL−1との間の顕著な相同性(B7.1とB7.2との間の相同性より大きい)、hB7−H1/PD−L1とhB7−DCとの密接な物理的連鎖、およびそれらの共通するレセプターへの結合により、これらが比較的最近の遺伝子重複事象に関連することが示唆される。このことは、B7.1とB7.2との間の関係と同様であり、B7.1およびB7.2の両方が、マウス第16染色体およびヒト第3染色体上の1メガ塩基内にマッピングする(49)。
【0257】
B7−H1/PD−L1に対するB7−DCの相対的生物学的役割が、PD−1および他の推定レセプターにより媒介されると認識することは重要である。PD−1は、T細胞活性化に続いて発現され、そしてT細胞活性化を阻害するようである。PD−1は、高濃度の抗CD−3でのT細胞刺激条件下でアポトーシスを誘導する。PD−1ノックアウトマウスは、肥大型心筋症の臨床的発現により特徴付けられる自己免疫症候群を発症する(22)。対照的に、Dongら(21)は、B7−H1/PD−L1が、低濃度の抗CD−3でT細胞増殖およびサイトカイン増殖を補助刺激することを報告した。CD28/CTLA−4の関係に類推して、PD−L1は、未だ同定されていない活性化レセプターについての対レセプター(counterreceptor)であり得る。PD−1に結合するという特性を共有しているにもかかわらず、B7−DCおよびB7−H1は、それらのリンホカイン補助刺激パターンが異なる;B7−H1は、T細胞のIL−10生成を補助刺激するのに対し、B7−DCはIL−10生成を補助刺激しない。B7−DCの異なる細胞発現パターンおよび補助刺激機能は、免疫機能における独特の役割を示唆する。
【0258】
上記および以下に引用された参考文献は、これらが具体的に援用されているか否かにかかわらず、全て本明細書中に参考として援用される。
【0259】
今や本発明を十分に記載してきたが、本発明が、広範な等価なパラメーター、濃度、および条件の範囲内で、本発明の趣旨および範囲から逸脱することなく、そして過度な実験なくして実施され得ることは当業者に明らかである。
【0260】
本発明は、その具体的な実施形態と関連付けて記載してきたが、さらなる改変が行われ得ることは理解される。本出願は、概して、本発明の原理に従って、そして本発明が属する分野において公知または慣用的な実施の範囲内であり、および添付の特許請求の範囲の範囲に入るように本明細書中で先に記載された本質的特徴に適用され得る、本開示からのこのような逸脱を含む、本発明の任意のバリエーション、使用または適合を含むことが意図される。
【0261】
(引用文献)
本願明細書に完全に列挙される文書に加えて、特定の文書は番号のみによって列挙(挿入)され、以下に列挙される。
配列表
【技術分野】
【0001】
生化学および医薬の分野における本発明は、樹状細胞の表面上で選択的に発現され、そして免疫応答を刺激するために細胞表面分子として用いられ得るか、またはワクチン組成物において可溶性形態で用いられ得る新規なタンパク質に関する。
【背景技術】
【0002】
Tリンパ球応答の生成は、細胞間相互作用および可溶性メディエーター(サイトカインまたはリンホカイン)の生成に関与する複雑なプロセスである。この応答は、「レセプター」として働くいくつかのT細胞表面分子によって調節される。このT細胞表面分子としては、T細胞レセプター(TCR)複合体、および他の「補助的な」表面分子(その多くはモノクローナル抗体によって最初に規定された細胞表面の「ディファレンシャル抗原」(「CD分子」)である)が挙げられる。
【0003】
全てのリンパ球の至適な活性化には、以下の2つのシグナルを要すると考えられる:抗原特異的シグナルまたはクローン(clonal)シグナル、および第二の抗原非特異的シグナル(Janeway,C.,Cold Spring Harbor Symp.Quant.Biol.54:1〜14(1989))。リンパ球が、いわゆる補助刺激分子(例えば、以下に記載のB7)による補助刺激なしに、単独の抗原に接触する場合、リンパ球は、「アネルギー」とも呼ばれるクローン性不活性(Schwartz,R,Science 248:1349(1990))、またはアポトーシス(プログラムされた細胞死)のいずれかに応答する;補助刺激シグナルが提供される場合、それは刺激抗原に特異的なクローン性増殖に応答する。所定の抗原に対する免疫応答の有意な増大は、補助刺激なしには生じない(Juneら(Immunology Today 15:321〜331,1994);Chenら(Immunology Today 14:483〜486);Townsend,SEおよびAllison,JP(1993)Science 259:368〜370)。
【0004】
免疫応答の質および力価は、大部分は、T細胞に対して抗原を処理して提示する抗原提示細胞(APC)の型に依存する。TCRの係合に利用可能なペプチド抗原/MHCリガンドの密度、ならびにT細胞の係合および活性化の時点でのAPCによる可溶性のおよび/または膜結合型の補助刺激シグナルの提供が重要である。これらの理由によって、免疫治療ストラテジーは、(a)標的抗原を適切な型のAPCに提供すること、そして(b)適切な補助刺激分子を提供してT細胞活性化を増強することに、集中し始めてきている。
【0005】
T細胞の活性化に必要なシグナルを提供するAPCとしては、単球/マクロファージ、Bリンパ球、そして最も重要である樹状細胞(DC)が挙げられる。過去には、活性化されたマクロファージは、インビボにおいてT細胞応答を開始する重要なAPCであると考えられた。この知見はマクロファージが抗原を効率的に食菌し、そして表面ディスプレイおよび提示のために抗原を処理する能力に基づく。より最近では、抗原特異的T細胞応答のインビボにおける主なイニシエータとして、DCに注意が移ってきた。DCは、活性化マクロファージ由来の異なる表現型を有し、そして異なる免疫応答を開始し得る異なるサブタイプに分類される。DCの機能的な欠点は、インビトロにおいてナイーブなT細胞を活性化する能力がマクロファージよりも約100倍大きいことである。今日まで、この力価の解釈は、抗原提示に重要であると考えられる分子の量的な差異に基づいていた。本発明は、重要な量的差異の発見に基づく。
【0006】
抗原提示における最初のシグナルは、APC上におけるクラスII主要組織適合性複合体(MHC)分子の状況で提示された抗原とTCRとの相互作用によって開始される(Allen、Immunol.Today 8:270(1987))。補助刺激シグナルは、他の分子に由来する。その分子のうちの最も特徴づけされたものはB7ファミリー(すなわち、B7.1、B7.2、およびおそらくB7.3)であり、これはAPC上にも存在する。
【0007】
T細胞の表面上に発現した2つのタンパク質は、B7のような補助刺激分子の最も特徴付けられたリガンドまたは対応レセプターである。CD28は、T細胞活性化において機能する最も成熟したヒトT細胞で見出された免疫グロブリン(Ig)スーパーファミリー(AruffoおよびSeed,Proc.Natl.Acad.Sci.84:8573〜8577(1987))のホモダイマー糖タンパク質である。CD28は、休止しているT細胞で構成的に発現され、そして活性化後に増大する。T細胞レセプターを通じたシグナル伝達後、CD28の連結は、T細胞が増殖してIL−2を分泌するように誘導する(Linsley,PSら(1991)J.Exp.Med.173、721〜730;Gimmi,CDら(1991)Proc.Natl.Acad.Sci.USA.88,6575〜6579;Thompson,C.B.ら(1989)Proc.Natl.Acad.Sci.USA.86,1333〜1337;June,C.H.ら(1990)Immunol.Today.11,211〜6;Harding,F.A.ら(1992)Nature.356,607〜609)。CD28は、免疫応答に必須である抗原依存性の細胞間相互作用の形態である、細胞間接触(「細胞間接着」)を媒介する(Springerら、Ann.Rev.Immunol.5:223〜252(1987))。
【0008】
CTLA4は、CD28に非常に相同であるT細胞表面分子であるが、休止しているT細胞では発現されず、そしてT細胞活性化後に生じる(Brunet,J.Fら(1987)Nature 328,267〜270)。CTLA−4は、マウス細胞傷害性T細胞cDNAライブラリーの示差的スクリーニングによってもともと同定された(Brunetら、前出)。B7の第二のレセプターとしてのCTLA−4の役割は、Linsleyら(1991)J.Exp.Med.174:561〜569(これはまた、B7がCD28についてよりもCTLA4について高い親和性を有することを注記している)に考察されている。Freemanら(1993)Science 262:907〜909は、B7欠損マウスにおけるCTLA−4を考察した。CTLA−4のリガンドは、Lenschowら(1993)Proc.Nat’l.Acad.Sci.90:11054〜11058に記載されている。
【0009】
Th細胞は、Th細胞に対して抗原を提示するB細胞のみの活性化を確実にするように働き、そしてバイスタンダーB細胞の活性化を回避する、Th細胞−B細胞接触の領域において、おそらく集中的な様式で、IL−2、IL−4、およびIL−6のような増殖および分化誘導のサイトカインを分泌する。
【0010】
CD28およびCTLA−4は、B7として一般に公知の補助刺激分子と相互作用する。B7は、B細胞活性化抗原としてもともと記載されている。なぜなら、B7はB細胞上で見出され、そしてB7/BB−1と名づけられたからである(Linsleyら、Proc.Natl.Acad.Sci.USA 87:5031〜5035(1990))。本明細書中において以降は、この分子をB7、B7−1、またはB7.1と呼ぶ。B7(より詳細には、B7ホモログと呼ばれる)はまた、Igスーパーファミリーのメンバーである。CD28およびCTLA−4と比較して、B7は、2つの細胞外Igドメイン(N末端可変(V)様ドメイン、それに続く定常(C)様ドメイン)を含む。
【0011】
B7ファミリーメンバーは、一般にAPC上で発現され、そして注記されるように、ナイーブT細胞の活性化に非常に重要なものである。これらのファミリーのマンバーとしては、B7−1(=B7、CD80とも呼ばれる)、およびB7−2(CD86とも呼ばれる)が挙げられる。B7−1を記載する参考文献としては、Schwartz,R.H.Cell 71:1065〜1068,1992;Chen,Lら、Cell 71:1093〜1102、1992;Freeman,G.J.ら、J.Immunol 143:2714〜2722,1989;およびFreeman,G.J.ら、J.Exp.Med.174:625〜631,1991が挙げられる。B7−2を記載する参考文献としては、Freeman,G.J.ら、Science 262:909〜911 813〜960、1993が挙げられる。現在まで、マウスB7−1およびB7−2ならびにヒトB7−1およびB7−2の両方が記載されている(Freemanら、1989、前出;1991、前出;および1993、前出)。活性化ヒトBリンパ球は、CTLA4/CD28結合カウンターレセプターであるB7−2およびB7−3を発現する。これは両方ともCD28またはCTLA4のいずれかを介してT細胞に補助刺激シグナルを送達し得る。
【0012】
B7−2は、抗IgクラスIImAbまたは抗MHCクラスIImAbのいずれかでの刺激後約24時間でB細胞によって発現される。B7−2は、検出可能IL−2分泌およびT細胞増殖を誘導する。活性化後約48時間〜72時間で、B細胞は、B7−1およびB7−3と名づけられた第三のCTLA4対応レセプター(mAb BB−1によって同定された)(Yokochi,Tら(1982)J.Immunol.128,823〜827)の両方を発現する。B7−3はまた、B7陰性活性化B細胞で発現され、そして検出可能なIL−2産生なしにT細胞増殖を補助刺激し得、このことは、B7−1分子およびB7−3分子が異なることを示す。B7−3は、活性化B細胞、活性化単球、樹状細胞、ランゲルハンス細胞、およびケラチノサイトを含む広範な種々の細胞で発現される。B細胞活性化後72時間で、B7−1およびB7−3の発現が低下し始める。活性化Bリンパ球の表面でのこれらのCTLA4/CD28結合対応レセプターの存在によって、T細胞補助刺激が、部分的には、B細胞活性化後のこれらの分子の一時的発現によって調節されることが示される。
【0013】
B7:CD28/CTLA4補助刺激経路の重要性は、インビトロおよびインビボで実証されている。T細胞活性化の増大とB7発現の増大との間に直接的な関係が存在する(Razi−Wolfら,Proc.Natl.Acad.Sci.USA,89:4210〜4214(1992))。T細胞は、それが補助刺激リガンド(この補助刺激経路のCD28遮断に結合する)を欠く細胞上のペプチド抗原に接触した場合、アネルギーにされ、マウスおよびヒトの系において抗原特異的寛容の発生を生じる(Hardingら、前出;Lenschow,D.J.ら(1992)Science.257,789〜792;Turka、LAら(1992)Proc.Natl.Acad.Sci.USA.89,11102〜11105;Gimmi,CDら(1993)Proc.Natl.Acad.Sci.USA 90、6586〜6590;Boussiotis,V.ら(1993)J.Exp.Med.178、1753〜1763)。逆に、B7−陰性マウス腫瘍細胞によるB7の発現は、腫瘍拒絶によって付随するT細胞媒介特異的免疫、および腫瘍チャレンジに対する長期防御を誘導する(Chen,Lら(1992)Cell 71:1093〜1102;Townsendら、前出;Baskar、Sら(1993)Proc.Natl.Acad.Sci.90,5687〜5690)。従って、B7:CD28/CTLA4経路の操作によって、ヒトにおいて免疫応答を刺激または抑制する大きな能力が得られる。
【0014】
CD28とB7との間の相互作用は、B7またはCD28の細胞外部分とIg Cγ1鎖の遺伝子融合を用いて特徴づけされた(Linsleyら、J.Exp.Med.173:721〜730(1991))。B7Ig融合タンパク質を固定するか、またはB7を細胞(例えば、トランスフェクトされたCHO細胞)の表面で発現する場合、それらはT細胞増殖を補助刺激する。B7+CHO細胞でのT細胞刺激はまた、IL−2の転写物のレベルの増大を特異的に刺激する。
【0015】
米国特許第5,521,288号は、CD28陽性T細胞を、B7をコードするDNAの一部によってコードされるフラグメント(B7の細胞外ドメイン(ECD)に主に対応する)と接触させることによって、免疫応答を調節するための方法を記載する。免疫応答はまた、B7 ECDの少なくとも一部および別のタンパク質(例えば、B7の可溶性、結合親和性、および/または原子価を変化させるヒトIgCγ1ドメイン)を含む融合タンパク質構築物であるB7の誘導体によって調節された。例えば、B7 ECDの位置1〜215に由来するアミノ酸残基をコードするDNAは、ヒトIgCγ1のヒンジ領域、CH2領域およびCH3領域に対応する配列のアミノ酸残基をコードするDNAに連結され、B7Ig融合タンパク質をコードしたDNA融合産物を形成する。また、CD28レセプターを結合することによって、T細胞と反応するB7またはB7Ig融合タンパク質を投与することにより、T細胞によって媒介される免疫系疾患を処置するための方法が開示されている。対宿主性移植片病におけるT細胞増殖を、CD28+T細胞を、B7抗原またはB7Ig融合タンパク質と、免疫抑制剤と組み合わせて、反応させることによって阻害した。
【0016】
米国特許第5,861,310号は、1つ以上のT細胞補助刺激分子(B7−2およびB7−3を含む)を発現するように改変された腫瘍細胞を開示する。1つの実施形態は、B7のさらなる発現を含む。改変は、B7−2タンパク質、B7−3タンパク質またはB7タンパク質をコードする核酸でのトランスフェクションによるものであった。腫瘍細胞はまたインビボで遺伝的に改変され得る。このような改変腫瘍細胞は、腫瘍を有する患者の処置に有用であり、腫瘍の転移性伝播を防止もしくは阻害するために、または腫瘍の再発を阻害するために有用であると言われている。本明細書は、腫瘍に対するCD4+T細胞応答を特異的に誘導するための方法を開示している。
【0017】
米国特許第5,942,607号は、T細胞活性化を補助刺激する新規なCTLA4/CD28リガンドをコードする単離された核酸を開示する。1つの実施形態において、単離された核酸は、B7−2をコードした。また、開示された全長B7−2配列の少なくとも一部を含む核酸を開示している、本明細書に従って、核酸配列は、哺乳動物細胞および昆虫細胞を含む種々の宿主細胞においてタンパク質またはペプチドに対応する合成を指向し得る種々の発現ベクター中に組み込まれ得る。これらの核酸配列によってコードされるタンパク質またはペプチド、および単離されたタンパク質およびペプチド(B7−2配列の少なくとも一部を含む)を生成するように形質転換された宿主細胞がまた開示されている。
【0018】
Dong Hら、Nat Med 1999 5:1365〜1399は、CD28、CTLA4、またはICOS(誘導性補助刺激因子)に結合しないB7−H1と名付けられたB7ファミリーの第三のメンバーを記載している。B7−H1の連結は、T細胞応答をポリクローナル刺激およびアロ抗原に対して補助刺激し、そして優先的にインターロイキン10の生成を刺激した。少量生成されたIL−2は、B7−H1補助刺激の効果を必要とした。この研究によって、細胞媒介性免疫応答の負の調節に関与し得る、以前には未知であった補助刺激分子が規定された。同じ研究室(Wang Sら、Blood.2000;96:2808〜2813)は、B7−H2と名付けられた新しいヒトB7様遺伝子を記載している。この遺伝子の発現は単球由来の未成熟DCの表面で検出された。可溶性のB7−H2およびIgの融合タンパク質である、B7−H2Igは、活性化T細胞には結合したが休止T細胞には結合しない。この結合は、ICOSの可溶性形態(ICOSIg)により阻害されたが、CTLA4Igによっては阻害されなかった。ICOSIgは、B7−H2遺伝子でトランスフェクトされたCHO細胞を染色した。刺激因子としてCD3の最適以下の架橋を用いて、B7−H2IgによるT細胞増殖の補助刺激は、用量依存性でありそしてIL−2の分泌と関連しており、一方至適CD3連結は好ましくはIL−10生成を刺激したことが見出された。著者らは、B7−H2がICOS T細胞分子の推定リガンドであると結論した。
【0019】
Swallow MMら、Immunity,1999,11:423〜432は、APC上で発現されるB7分子の近接するホモログ(相同体)である、新規な遺伝子b7hのクローニングを報告した。B7hは、CD28またはCTLA−4由来の異なるレセプター上で作用することにより精製したT細胞の増殖を補助刺激した。驚くべきことに、B7hは、刺激されていないB細胞で発現されたが、その発現は、TNFαで処理した非リンパ球性細胞(3T3細胞;胎児性線維芽細胞)で誘導され、そしてLPS(TNFαの強力なアクチベータ)で処理されたマウスの非リンパ球性組織で上方制御された。これらの研究は、T細胞の新規な補助刺激リガンドを規定し、そしてTNFαによるB7hの誘導は、炎症中の自己の認識を直接増強し得る。
【0020】
Yoshinaga SKら、Nature,1999、402:827〜832は、新しいマウス補助刺激レセプターリガンド対を記載している。このレセプター(CD28に関連する)は、ヒトタンパク質ICOSのマウスホモログであり、そして活性化T細胞および休止している記憶T細胞で発現された。B7分子に相同であるリガンドは、B7関連タンパク質−1(B7−related protein−1)(B7RP−1)と呼ばれる。B7RP−1は、それぞれマウスB7.1(CD80)およびB7.2(CD86)に対して、20%アミノ酸同一性および19%アミノ酸同一性である1型膜貫通タンパク質である。この相同性は、27%アミノ酸同一性しか共有しないB7.1およびB7.2として有意である(Freeman,GJら、J.Exp.Med.178:2185〜2192(1993))。この相同性は、保存位置でIgループ形成に重要であるシステインを含む(開始メチオニンから残基62、138、185および242)。B7RP−1の膜貫通ドメインの全体的長さおよび相対的位置は、B7分子の長さおよび位置と類似している(Greenfield,EAら、Crit.Rev.Immunol.18:389〜418(1998))。B7RP−1は、B細胞およびマクロファージで発現されることが示された。ICOSおよびB7RP−Iは、CD28−B7経路においてタンパク質と相互作用せず、そしてB7RP−1は、CD28と独立してT細胞を補助刺激した。B7RP−1とIgのFc部分との間の融合タンパク質(「B7−RP1−Fc」)を発現するトランスジェニックマウスは、脾臓においてリンパ過形成、リンパ節およびパイアー斑を有した。インビボにおけるB7RP−1の補助刺激活性は、抗原チャレンジの時点でB7RP−1−Fcで処理された抗原提示化マウスにおいて、増強した遅延型過敏症を実証することによって見出された。著者らは、ICOSおよびB7RP−1が、CD28−B7に構造的に関連し、そして適応的免疫反応に関与する異なる新しいレセプター−リガンド対を規定すると結論した。
【0021】
Yoshinaga SKら、Int Immunol,2000 Oct,12:1439〜1447は、ヒトB7RP−1およびICOS相互作用を通じたヒトT細胞の補助刺激を報告した。このリガンド−レセプター対は、約33nMのKDおよび10分より長いt(1/2)を有するオフレート(off−rate)で相互作用した。TNFαは、B細胞、単球、およびDC上のヒトB7RP−1の発現を示差的に調節した。TNFαは、B細胞および単球上のB7RP−1発現を増強したが、DCの発現は阻害した。ヒトB7RP−1−Fcタンパク質または膜結合したB7RP−1を発現した細胞は、インビトロにおいてT細胞増殖を補助刺激した。特定のサイトカイン(例えば、IFNγおよびIL−10)は、B7RP−1補助刺激によって誘導された。IL−2レベルは有意に増大しなかったが、B7RP−1が誘導した補助刺激は、IL−2に依存した。これらの研究により、マウスB7RP−1に対するヒトオルソログが規定され、そしてヒトICOSとの相互作用が特徴付けられた。
【0022】
PD−1は、活性化T細胞、B細胞および骨髄細胞によって発現された免疫阻害レセプターである。PD−1欠損マウスは、末梢寛容の損失に起因して自己免疫の複数の形態を示した。Freeman、GJら、J.Exp.Med.192:1027〜1034(2000)は、PD−1(PD−L1)のリガンドが、B7遺伝子ファミリーのメンバーであることを報告した。PD−L1によるPD−1の係合によって、TCR媒介リンパ球活性化(増殖、サイトカイン分泌)の阻害が生じた。さらに、PD−1シグナル伝達は、CD28媒介補助刺激の至適以下のレベルを阻害した。PD−L1は、APC(IFNγで刺激されたヒト単球、活性化ヒトDC)により発現される。さらに、PD−L1は、心臓および肺において発現されることが示された。著者らは、APC上の阻害性PD−L1シグナルおよび補助刺激B7−1/B7−2シグナルの相対的な大きさが、T細胞活性化の程度および寛容と自己免疫との間の閾値を決定し得ることを推測した。非リンパ性組織でのPD−L1の存在は、炎症の部位での免疫応答の大きさに寄与し得る。
【0023】
上記の文献の引用は、前述のいずれの文献も先行技術に関連するという承認とは解釈されない。これらの文献の内容についての、今日までの全ての記述または説明は、本出願人に対して利用可能な情報に基づき、そしてこれらの文献の日付または内容の正確さに関してなんらの承認をも構成しない。
【発明の概要】
【発明が解決しようとする課題】
【0024】
T細胞活性化についての新規な樹状細胞(DC)特異的補助刺激分子をコードする遺伝子を同定するために、本発明者らは、DCと活性化マクロファージとの間のサブトラクトcDNAライブラリーをスクリーニングした。このcDNAのサブトラクションアプローチによって、DCによって発現されたが、活性化マクロファージによって発現されない遺伝子が規定される。このアプローチを用いて、T細胞の活性化に依存するワクチンの力価を増強するのに有用ないくつかの新規なDC特異的遺伝子の発見がもたらされた。本出願はこのような遺伝子の1つに焦点をあてる。
【課題を解決するための手段】
【0025】
DCライブラリーにおいて存在し、活性化マクロファージライブラリーから非存在であることに基づいて、新規なコード配列(「B7−DC」と名付けられた)を同定した。B7−DC遺伝子は、補助刺激分子をコードする遺伝子のB7ファミリーのメンバーである。B7−DCは、DC特異的発現および異なるレセプター特異性を有する第一のB7ファミリーメンバーである。この遺伝子の産物は、DCがT細胞を刺激する特有の能力を媒介するのにおいて重要な役割を有する。機能的分析によって、T細胞によるIFNγ産生を刺激する際に、B7−1よりもB7−DCがさらに活性であることが示された。従って、B7−DC DNAおよびポリペプチドは、抗原特異的であろうとなかろうと、細胞性および分子性のワクチン組成物の効力を増強するための組成物および方法において有用である。
【0026】
1つの実施形態において、本発明は、活性化マクロファージと比べて樹状細胞上で選択的に発現されるB7−DCと名付けられた哺乳動物タンパク質をコードする単離された核酸分子を提供する。核酸分子は好ましくは、配列番号1(ヒト起源)、または配列番号5(マウス起源)から選択されたヌクレオチド配列を含む。本発明はまた、ストリンジェントなハイブリダイゼーション条件下で上記の核酸分子にハイブリダイズする単離された核酸に関する。好ましいストリンジェントな条件は、約45℃での6×塩化ナトリウム/クエン酸ナトリウム(SSC)中でのインキュベーション、続いて約50℃の温度での約0.2×SSC中での洗浄を含む。好ましくは、上記の核酸分子は、配列番号1のヌクレオチド配列を含む。上記のような好ましい核酸分子は、配列番号2および配列番号4から選択されたアミノ酸配列を有するタンパク質をコードするか、またはこのタンパク質の生物学的に活性なフラグメント、ホモログ、もしくは他の機能的誘導体をコードする。好ましくは、この核酸分子は、配列番号2(ヒト起源のB7−DC)の配列を有するタンパク質をコードするか、または配列番号2の生物学的に活性なフラグメント、ホモログ、もしくは他の機能的誘導体をコードする。
【0027】
好ましい実施形態において、核酸分子は、B7−DCタンパク質の細胞外ドメイン(残基26〜221を含む)をコードする。この核酸分子は、その補助刺激ホモログ、フラグメントまたは他の機能的誘導体をコードする。
【0028】
別の実施形態において、上記の核酸分子は、B7−DC融合タンパク質をコードし、この核酸分子は以下:
(a)B7−DCタンパク質(好ましくは、配列番号2または配列番号4)の全てまたは一部である第一のポリペプチドをコードする第一の核酸配列;
(b)必要に応じて、この第一の核酸配列とインフレームで融合した、リンカーペプチドをコードするリンカー核酸;および
(c)この第一の核酸配列またはこのリンカー核酸配列にインフレームで結合し、そして第二のポリペプチドをコードする第二の核酸配列、
を含む。この第二のポリペプチドは、好ましくは、Igの重鎖定常領域の1つ以上のドメイン、好ましくはヒトIgG(好ましくはIgG1)の2つのCドメイン、から構成される。
【0029】
発現ベクターであって、以下:
(a)プロモーター、および
(b)必要に応じて、真核生物細胞において核酸の発現を調節するさらなる調節配列、
に作動可能に連結された上記の任意の核酸分子、
を含む発現ベクターも提供される。
【0030】
上記の発現ベクターは、プラスミドまたはウイルスベクターであり得る。これらのベクターとしては、自己複製するRNAレプリコン(DNA−ランチャー、またはRNA)、自殺RNAベクター、DNAウイルス(例えば、アデノウイルス、ワクシニアウイルスなど)、およびRNAビリオン(パッケージング細胞株で増殖する)が挙げられる。
【0031】
ベクターDNAまたはRNAは、例えば、所望の標的細胞および組織への送達を増強する、徐放性処方物中で、宿主への遺伝子銃媒介導入のための金粒子に複合体化されてもよいし、または他のポリマーと複合体化されてもよい。
【0032】
また、以下:
(a)第一の組み換え発現ベクターであって、該ベクターは、免疫応答が誘導される目的の抗原をコードするヌクレオチド配列を、その配列中に組み込んでいる、ベクター;および
(b)第二の組み換え発現ベクターであって、該ベクターは、補助刺激ポリペプチド(このポリペプチドの少なくとも1つは、B7−DCであるか、またはその生物学的に活性なフラグメント、ホモログ、もしくは他の機能的誘導体である)をコードする1つ以上のヌクレオチド配列をその核酸配列中に組み込んでおり、ここで該発現ベクターは、宿主細胞に同時感染または同時トランスフェクトし得、抗原および補助刺激ポリペプチド、複合体、ホモログもしくは誘導体の同時発現を生じる、ベクター、
を含むベクター組成物も含まれる。
【0033】
上記の実施形態の改変において、本発明は、以下:(i)細胞間(好ましくは、APC)で発現された産物(抗原)の伝播を促進し、(ii)核酸が発現されるAPCでの抗原の提示を増大し、そして/または(iii)ベクターが導入されている宿主のAPCにおける抗原の再提示(交差プライミング)および提示を促進する、標的タンパク質をコードする第三の核酸配列を提供する。この標的化タンパク質コード核酸は、抗原または補助刺激因子またはその両方をコードする核酸に融合され得る。第一または第二のベクターは、核酸を含む。1つの実施形態において、このベクター組成物は、これらの抗原コード核酸、補助刺激コード核酸(好ましくはB7−DC)、および「標的化」タンパク質コード核酸を、単一の融合構築物中に組みあわせる。
【0034】
本発明は、上記の任意の核酸分子または発現ベクターで形質転換またはトランスフェクトされた細胞を含む。この細胞は、好ましくは真核生物細胞、より好ましくは哺乳動物細胞、最も好ましくはヒト細胞である。この細胞は、樹状細胞またはその前駆体であり得る。別の実施形態において、この細胞は、腫瘍細胞、好ましくは抗原(宿主における腫瘍上の抗原(それに対して免疫応答が所望される)と同じであるか、またはその抗原と交差反応性である抗原)を保有する腫瘍細胞である。
【0035】
好ましい実施形態は、哺乳動物B7−DCタンパク質(好ましくは配列番号2または配列番号4)、またはその生物学的に活性なフラグメント、ホモログ、もしくは他の機能的誘導体をコードする外因性核酸分子でトランスフェクトされた単離された哺乳動物腫瘍細胞であり、その結果、この腫瘍細胞によって、このタンパク質、フラグメント、ホモログ、または誘導体が発現される場合、そしてこの腫瘍細胞がT細胞と接触する場合、
(i)B7−DCタンパク質、フラグメント、ホモログ、または誘導体が、T細胞に結合し;そして
(ii)この腫瘍細胞は、T細胞を補助刺激して増殖し、そして/またはサイトカインを産生し、そして分泌する。
【0036】
本発明はまた、活性化マクロファージと比べて樹状細胞で選択的に発現され、そして以下:
(a)T細胞上の結合パートナーに結合する;
(b)T細胞を補助刺激して増殖し、そして/またはサイトカインを産生し、そして分泌する、
という機能的特性を有するポリペプチドに関連する。
【0037】
このポリペプチドの生物学的に活性なフラグメント、ホモログまたは他の機能的誘導体もまた含まれる。
【0038】
このポリペプチド、フラグメント、ホモログまたは機能的誘導体は、好ましくは、配列番号1または配列番号5の配列を有する核酸分子、またはこの核酸分子のフラグメント、ホモログもしくは等価物によってコードされる。好ましいポリペプチドは、配列番号2または配列番号4のアミノ酸配列を有する。
【0039】
このポリペプチドまたはこのポリペプチドの生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体は、上記核酸の一つの組換え発現によって、生成され得る。
【0040】
好ましいポリペプチドは、B7−DCタンパク質の細胞外ドメインを含み、好ましくは、以下:
(a)配列番号2(ヒト)のアミノ酸残基26〜221または
(b)配列番号4(マウス)のアミノ酸残基26〜221
である。上記ポリペプチドは、B7−DCの細胞外ドメインから「本質的になり」得る。
【0041】
(i)第二のポリペプチドに直接的に融合しているか、または
(ii)必要に応じて、この第二のポリペプチドに融合したリンカーペプチド配列に融合している、
B7−DCタンパク質の全てまたは一部を含む、第一の融合パートナーを有するB7−DC融合ポリペプチドもまた、提供される。
【0042】
上記A B7−DC融合タンパク質はまた、第二のポリペプチドに融合し得、この第二のポリペプチドは、好ましくは、Ig重鎖定常領域の1つ以上のドメインであり、好ましくは、ヒト免疫グロブリンCγ1鎖のヒンジ領域、CH2領域およびCH3領域に対応するアミノ酸配列を有する。
【0043】
上記融合タンパク質の1つの実施形態において、第一の融合パートナーは、B7−DCタンパク質の細胞外ドメインであり、これの全長配列は、配列番号2または配列番号4である。
【0044】
この融合タンパク質は、好ましくは、T細胞上の結合パートナーに結合し、そしてT細胞レセプターに対する十分な刺激の存在下で、T細胞を補助刺激する。
【0045】
上記融合タンパク質のダイマーまたはトリマーである、ダイマーまたはトリマーの融合タンパク質もまた提供される。好ましくは、これらの鎖は、ジスルフィド結合または他の鎖間共有結合を介して、縦列で連結される。
【0046】
好ましいダイマー融合タンパク質において、このダイマーは、Ig重鎖の2つのCH領域のCys残基の共有結合から生じ、これらのCys残基は、二量化した通常のIg H鎖において、ジスルフィド結合しているものと同じCys残基である。
【0047】
本発明の融合タンパク質は、直接的にかまたは1つ以上の単量体間のリンカー配列を伴って、末端同士で結合した第一の融合パートナーの2つ以上の繰返し体のマルチマーを含み得る。
【0048】
本発明はまた、B7−DCタンパク質のエピトープに対して特異的な抗体を提供し、このエピトープは、B7ファミリータンパク質の公知のメンバーには存在しない。このエピトープは、配列番号2または配列番号4のポリペプチドの「直鎖またはコンホメーション」エピトープであり得る。この抗体は、好ましくは、モノクローナル抗体であり、より好ましくは、ヒトまたはヒト化(操作による)モノクローナル抗体である。
【0049】
上記抗体を使用して、細胞集団においてB7−DCポリペプチドをその表面において発現する細胞を同定または定量する方法もまた提供され、この方法は、以下の工程を包含する:
(a)この抗体がこのエピトープを発現する細胞に結合するように、この集団の細胞を、上記抗体と接触させる工程;
(b)この抗体が結合する細胞の数の存在を評価するかまたはその細胞の数を定量する工程。
【0050】
細胞集団から、その表面でB7−DCポリペプチドを発現する細胞を単離するための、別の方法が提供され、この方法は、以下の工程を包含する:
(a)この抗体がこのエピトープを発現する細胞に結合するように、この集団を、上記抗体に接触させる工程;
(b)この抗体が結合する細胞をポジティブに選択するか、またはこの抗体が結合しない細胞をネガティブに選択する工程。
【0051】
サンプル中のB7−DCポリペプチド、フラグメントまたはホモログの存在を検出するかまたは定量する方法もまた提供され、この方法は、以下の工程を包含する:
(a)この抗体が、このエピトープを保有する任意のポリペプチドまたはフラグメントに結合するように、このサンプルを請求項43の抗体と接触させる工程;
(b)この抗体に結合するポリペプチドまたはフラグメントの存在を検出するか、または定量する工程。
【0052】
本発明はまた、抗原提示細胞またはその前駆細胞におけるB7−DCポリペプチドの発現を誘導または増加して、この細胞が、インビトロまたはインビボで、T細胞レセプターに対する十分な刺激の存在下で、T細胞を補助刺激する能力を増加させる方法に関し、この方法は、B7−DCポリペプチドの発現がその細胞において誘導または増加されるように、抗原提示細胞または前駆細胞を上記のような発現ベクターで形質転換またはトランスフェクトする工程を包含する。この抗原提示細胞は、好ましくは、樹状細胞であり、そして前駆細胞は、樹状細胞前駆細胞である。
【0053】
本発明は、細胞補助刺激組成物およびポリペプチド補助刺激剤を使用して、免疫応答を刺激するための方法を提供する。抗原性刺激に対する哺乳動物被験体のT細胞応答を増加させるための1つの方法は、この被験体に、有効量の上記細胞(好ましくは、腫瘍細胞)を、抗原性刺激と組み合わせて投与する工程を包含し、ここで、これらの細胞は、この抗原性刺激に対するこの被験体のT細胞応答を増加するために効果的である。上記のことは、好ましくは、抗原と補助刺激組成物との同時注入によって、達成される。
【0054】
抗原性刺激に対する哺乳動物被験体のT細胞応答を、腫瘍関連抗原で増加させるための方法は、この被験体に、有効量の上記のような腫瘍細胞を投与する工程を包含し、ここで、これらの腫瘍細胞は、この抗原を発現し、この腫瘍細胞の投与は、腫瘍抗原刺激に対するこの被験体のT細胞応答を増加させるために効果的である。
【0055】
抗原性刺激に対する哺乳動物被験体のT細胞応答を増加させるための方法は、この被験体に、有効量のポリペプチド、上記のようなフラグメント、ホモログもしくは機能的誘導体、または上記のような融合ポリペプチドもしくはタンパク質を、抗原性刺激と組み合わせて投与する工程を包含し、ここで、このポリペプチドの投与は、抗原性刺激に対するこの被験体のT細胞応答を増加させるために効果的である。
【0056】
本発明はまた、抗原性刺激に対する哺乳動物被験体のT細胞応答を阻害するための方法を提供し、この方法は、この被験体に、有効量の上記のような「抗体」を投与する工程を包含し、ここで、この抗体の投与は、T細胞の刺激をブロックするかまたは抗原反応性T細胞を排除するために効果的であり、これによって、T細胞応答を阻害する。これらの方法は、組織または器官の移植を有する被験体を処置して、移植片の拒絶を阻害するためおよび/または移植を促進するために、特に有用である。自己抗原の場合には、この方法は、自己免疫応答およびこれらの病理学的続発症をブロックまたは低下させる。
【0057】
本発明は、本発明の組成物でのエキソビボ刺激を受けたT細胞を使用する、治療方法を提供する。抗原性刺激に対する哺乳動物被験体の免疫応答を増加させるための1つの方法は、以下:
(a)この被験体から、この被験体に免疫学的に適合性のドナーから、または免疫学的に受容可能な培養細胞株から、T細胞を得る工程;
(b)このT細胞を、エキソビボで、有効量の上記のような細胞と接触させる工程であって、ここで、この接触は、抗原性刺激に対するT細胞の応答を増加させるために効果的である、工程;および
(c)工程(b)のT細胞を、この被験体に投与する工程、
を包含し、これによって、この被験体の免疫応答を増加させる。
【0058】
別の実施形態において、抗原性刺激に対する哺乳動物被験体の免疫応答を増加させるための方法は、以下の工程:
(a)被験体から、この被験体に対して免疫学的に適合性のドナーから、または免疫学的に受容可能な培養細胞株から、T細胞を得る工程;
(b)このT細胞をエキソビボで有効量の以下:(i)上記のようなポリペプチド、フラグメント、ホモログまたは機能的誘導体、あるいは(ii)上記のような融合ポリペプチドと接触させる工程であって、ここで、この接触は、抗原性刺激に対するT細胞の応答を増加させるために効果的である、工程;ならびに
(c)工程(b)のT細胞をこの被験体に投与する工程、
を包含し、これによって、この被験体の免疫応答を増加(または発生)させる。
【0059】
本明細書中にはまた、ワクチン組成物もまた提供され、このワクチン組成物は、以下を含有する:
(a)(i)B7−DC構築物を発現する、上記のような細胞、(ii)B7−DCポリペプチド、フラグメント、ホモログまたは機能的誘導体、(iii)B7−DC融合ポリペプチドまたはタンパク質;
(b)一般に、免疫応答が所望される抗原のさらなる供給源(これは、細胞自体がこの抗原を発現する、細胞に基づくワクチンの場合には、必要とされないかもしれないが(腫瘍抗原を保有する腫瘍細胞の場合のように));
(c)必要に応じて、一般的な免疫刺激因子またはアジュバント;ならびに
(d)(a)、(b)および(c)のための薬学的かつ免疫学的に受容可能な賦形剤またはキャリア。
【0060】
哺乳動物被験体において抗原に対する免疫応答を増加または増強するための方法は、この被験体に、有効量の上記ワクチン組成物を投与する工程を包含する。
【0061】
抗原またはワクチンと共に使用するための、補助刺激組成物もまた提供され、この組成物は、以下を含有する:
(a)B7−DCポリペプチド(好ましくは、配列番号2または配列番号4)、そのフラグメント、ホモログもしくは機能的誘導体、あるいはB7−DC融合ポリペプチド、ならびに
(b)薬学的かつ免疫学的に受容可能な賦形剤またはキャリア。
【0062】
哺乳動物被験体において、抗原またはワクチンに対する免疫応答を相乗するための方法は、この被験体に、この抗原またはワクチンと組み合わせて、上記補助刺激組成物を投与する工程を包含する。
【0063】
被験体において、腫瘍に対する全身性免疫応答を刺激する方法は、この被験体に、遺伝子改変された腫瘍細胞を投与する工程を包含し、この細胞は:
(a)この被験体中の腫瘍細胞由来であり、そして
(b)上記のようなB7−DC核酸のエキソビボでの導入によって遺伝子改変されており、
この核酸の発現は、この被験体において補助刺激シグナルを提供し、ここで、この投与は、この腫瘍に対する全身性免疫応答の刺激を生じる。
【0064】
これらの腫瘍細胞は、これらが投与された後のこれらの増殖が防止されるように、好ましくは照射によって、好ましくは処理されている。
【0065】
この被験体は、上記治療組成物の投与の前に、化学療法、照射または外科的切除の腫瘍減少レジメンに供され得る。
【0066】
抗原陽性腫瘍を有する哺乳動物において、抗腫瘍応答を誘導する方法もまた提供され、この方法は、以下:
(a)(i)この哺乳動物の腫瘍と共通する抗原を発現し;
(ii)上記のようにB7−DCコード核酸ベクターでトランスフェクトされ、これは、B7−DC分子として発現される場合に、この細胞に、この腫瘍の抗原に対するT細胞応答を補助刺激させ;
(iii)必要に応じて、工程(b)の前に照射される、腫瘍または腫瘍細胞株の細胞を提供する工程;
(b)有効数の細胞をこの哺乳動物に投与する工程であって、この細胞は、この抗原およびB7−DC分子を発現する、工程;
を包含し、これによって、抗腫瘍応答を誘導する。
【0067】
上記方法において、抗腫瘍応答は、以下によって特徴付けられる:
(A)全ての測定可能な病巣の最大垂直直径の積の合計の、少なくとも50%の減少;
(B)新たな病巣の証拠がないこと、および
(C)任意の先在病巣の進行がないこと。
【0068】
腫瘍を保有する哺乳動物において、腫瘍の一次増殖または再増殖の抑制または弱化を誘導する方法もまた提供され、この方法は、以下:
(a)(i)この哺乳動物の腫瘍と共通する抗原を発現し;
(ii)上記のようにB7−DCコード核酸ベクターでトランスフェクトされ、これは、B7−DC分子として発現される場合に、この細胞に、この腫瘍の抗原に対するT細胞応答を補助刺激させ;
(iii)必要に応じて、工程(b)の前に照射される、腫瘍または腫瘍細胞株の細胞を提供する工程;
(b)有効数の細胞をこの哺乳動物に投与する工程であって、この細胞は、この抗原およびB7−DC分子を発現する、工程;
を包含し、これによって、黒色腫の腫瘍細胞に対して特異的な全身性免疫応答を誘導し、これによって、この抑制または弱化を誘導する。
【0069】
哺乳動物における抗原陽性腫瘍の反復性の増殖を阻害する方法は、以下の工程を包含する:
(a)(i)この哺乳動物の腫瘍と共通する抗原を発現し;
(ii)上記のようにB7−DCコード核酸ベクターでトランスフェクトされ、これは、発現される場合に、この細胞に、この腫瘍の抗原に対するT細胞応答を補助刺激させ;
(iii)必要に応じて、工程(b)の前に照射される、腫瘍または腫瘍細胞株の細胞を提供する工程;
(b)有効数の細胞をこの哺乳動物に投与する工程であって、この細胞は、この抗原およびB7−DC分子を発現する、工程;
を包含し、これによって、この哺乳動物において腫瘍抗原に対して特異的な全身性免疫応答を誘導し、この免疫応答が、腫瘍の反復性の増殖を阻害する。
【0070】
別の実施形態は、哺乳動物被験体に局所的に投与された抗原の近隣に、補助刺激シグナルを提供して、この抗原に対する全身性免疫の状態を生じる炎症応答および免疫応答の局所的発生を促進する方法に関し、この方法は、被験体における局所部位に、以下:
(a)補助刺激に有効な量の、上記のようなB7−DCポリペプチド、フラグメント、ホモログまたは機能的誘導体を発現する細胞、および
(b)抗原
を投与する工程を包含し、その結果、この抗原に物理的に近い位置での補助刺激が、この応答の局所的発生を促進し、そして全身性免疫の状態を生じる。
【0071】
上記方法において、抗原は、好ましくは、工程(b)において、腫瘍細胞または非細胞抗原性物質の形態で投与される、腫瘍抗原である。この腫瘍細胞はまた、工程(a)において、B7−DCポリペプチド、フラグメント、ホモログまたは誘導体を発現する細胞であり得る。
【図面の簡単な説明】
【0072】
【図1】図1は、ヒト染色体9p24に局在したhB7−DCのマップを示す、ダイヤグラムである。hB7−DCは、BACクローンRPCI−11.2にマッピングされる。
【図2】図2は、B7−DCが、DCとマクロファージとの間で差示的に発現されることを示す。骨髄DC、脾性DC、マクロファージ、マクロファージ株および組織におけるB7−DC mRNAの分布を、0.5μg/レーンの精製したDNAを使用して、1%アガロースゲルで泳動する、バーチャルノーザンブロット分析によって評価した。G3PDHを、コントロールとして使用した。J774A1細胞、Raw264.7細胞、Pu5−1.8細胞およびWEHI細胞は、マクロファージ細胞株である。BM:骨髄。
【図3】図3は、ヒトDCにおけるB7−DC発現のバーチャルノーザンブロットを示す。レーン1は、GM−CSFおよびFlt−3Lと共に培養されたヒトDCを示し、レーン2は、ヒト胎盤を示し、そしてレーン3は、GM−CSFおよびIL4と共に培養されたヒトDCを示す。ヒトB7−DCの5’および3’UTR由来のオリゴヌクレオチドを使用して、ヒトDCの全RNAのバーチャルノーザン分析のためのPCR DNAプローブを作製した。β−アクチンをコントロールとして使用して、mRNAの品質を確認した。
【図4】図4は、成熟BM−DCにおけるB7−DCの表面発現を示す、フローサイトメトリー分析を代表する。9日目に、マウスBM−DCをFcブロックし、そしてコントロール抗体またはB7−DC抗血清を用いて染色した。結合の特異性を、DCの表面への抗B7−DCの結合に対して競合するためのB7−DC−Igを添加することによって、実証した。
【図5】図5は、B7−DCがPD−1に結合するが、CTLA−4にもCD28にも結合しないことを示す。293T細胞を、pCAGGS−B7.1 o pCAGGS−B7−DCを用いて、一時的にトランスフェクトした。トランスフェクト体を、PD−1−Ig融合分子、28−Ig融合分子およびCTLA−4−Ig融合分子で染色し、続いてPE標識した二次抗体で染色した。pCAGGS(空のベクター)トランスフェクト体の染色は、ネガティブであった(示さず)。
【図6】図6(左右のパネル)は、抗CD3およびB7−DC−IgによるT細胞増殖の補助刺激を示す。左側のグラフ:精製T細胞(CD4+CD8)を、次第に増加する濃度の抗CD3(mAb 2C11)および一定濃度(0.1μg/ml)の固定したB7.1−Ig(黒菱形)、B7−DC−Ig(黒丸)またはアイソタイプコントロール(黒三角)で予めコートしたウェル内で培養した。結果は、3つのうちの1つの代表的な実験を示す。細胞を、72時間インキュベートし、そして3H−チミジンで標識した。CPMとは、1分間あたりの計数である。右側のグラフ:精製CD8 T細胞を、次第に増加する濃度の抗CD3および一定濃度の固定したB7.1−Ig(黒菱形)、B7−DC−Ig(黒丸)またはアイソタイプコントロール(黒三角)で(a)においてと同様に予めコートしたウェル内で、培養した。結果は、2つのうちの1つの代表的な実験のものである。細胞を72時間インキュベートし、そして3H−チミジンで標識した。CPMとは、1分間あたりの計数である。
【図7】図7は、抗原特異的T細胞増殖応答の補助刺激を示す。RENCA細胞をIFNγで72時間処理して、MHCクラスIIの発現を誘導し、そして12.5μg/mlのHA110−120ペプチドとともにインキュベートした。精製したHAおよびI−Ed特異的トランスジェニックT細胞を、次第に増加する濃度のB7.1−Ig(黒菱形)、B7−DC−Ig(黒丸)またはアイソタイプコントロール(黒三角)のいずれかと共に、可溶化形態で添加した。細胞を48時間インキュベートし、そして3H−チミジンで標識した。CPMとは、1分間あたりの計数である。結果は、3つのうちの1つの代表的な実験である。
【図8】図8は、B7−DCによって補助刺激されたT細胞のサイトカイン分泌を示す。上のパネル:精製したT細胞を、抗CD3(0.12μg/ml)および0.1μg/mlの固定化B7.1−Ig(黒菱形)、B7−DC−Ig(黒丸)またはアイソタイプコントロール(黒三角)で、図6(左)と同様に予めコートしたウェル内で培養した。結果は、3つのうちの1つの代表的な実験を示す。下のパネル:12.5μg/ml HA(110−120)ペプチドで負荷したγ−IFN処理したRENCA細胞を、精製したHAおよびI−Ed特異的トランスジェニックT細胞と共に、2μg/mlの可溶性B7.1−Ig、B7−DC−Igまたはアイソタイプコントロール(記号は上記のとおり)と一緒にインキュベートした。結果は、2つのうちの1つの代表的な実験を示す。上清を、24時間および48時間の培養の後に収集し、そしてELISAを使用して、示されるリンホカインについてアッセイした。
【図9】図9は、B7−DC−Igが、インビボ補助刺激の後に、抗原特異的増殖を大いに増強することを示す。HAに対して特異的な2.5×106のTCRトランスジェニック細胞の養子移入の後に、3群のマウスを、HAペプチド(110−120)、不完全フロイントアジュバント(IFA)のいずれかで、単独でかまたはB7−DC−IgおよびIFAまたはIFAを含むアイソタイプコントロール抗体のいずれかとの組合せで、後肢裏において皮下で免疫した。リンパ節の排液を、7日目に獲得した。1.5×105のLN細胞を、HAペプチドと共に48時間インキュベートし、1μCi[3H]チミジンでパルス化し、そして12時間後に組み込んだ放射能を、決定した。
【発明を実施するための形態】
【0073】
本発明者らは、ここで、改善された免疫治療組成物および方法のための基礎として働く、新たなタンパク質および核酸を同定した。ヒトおよびマウス形態の、新規補助刺激タンパク質(B7−DCと命名された)が発見され、そして本明細書中に開示される。
【0074】
ヒトB7−DCをコードするDNAは、以下に示す配列番号1のヌクレオチド配列を有する:
【0075】
【化1】
ヒトB7−DCタンパク質は、以下に示す配列番号2のアミノ酸配列を有する(リーダー配列、膜貫通ドメインおよび細胞質テイルが注釈を付けられている):
【化2】
このタンパク質の細胞外ドメインは、残基P26〜残基W221由来である。
【0076】
マウスB7−DCをコードするコード配列を含むDNAクローンは、以下に示す配列番号3のヌクレオチド配列を有する。このコード配列(下線を引かれた、三つ組みで示される)は、ヌクレオチド210で開始するメチオニンコドンatg(太字)から開始し、そしてtag終止コドン(太字)(ヌクレオチド951〜953)で終止する。
【化3】
配列番号5は、配列番号3のコード配列部分である。
【0077】
配列番号3のコード領域によって(すなわち、配列番号5によって)コードされるマウスB7−DCタンパク質は、以下に示す配列番号4のアミノ酸配列を有する(リーダー配列、膜貫通ドメインおよび細胞質テイルが注釈をつけられている):
【化4】
このタンパク質の細胞外ドメインは、残基P26〜残基W221由来である。
マウスB7−DCの完全DNA配列(もともとは「ブチロフィリン様タンパク質」または「Btdc」と称された)は、Genbank登録番号AF142780.2を有する。
【0078】
(基本的な分子アプローチ)
このアプローチは、実施例により詳細に記載される。本発明者らは、2つの重要な特徴を組み込むPCR Selectアプローチを利用した。第1に、ハイブリダイゼーション工程前の最初のPCR反応は、少量のRNAのみを必要とする。この技術は、夾雑するマクロファージ、前駆体細胞または他の潜在的な夾雑細胞を実質的に含まないようになった高度に精製された成熟DCの使用を可能にする。このような高度に精製されたDCを非常に多数で得ることが困難であることは公知である。第2に、PCR Select手順は、低いコピー数の差次的に発現される遺伝子のクローニングを可能にする。
【0079】
その細胞性の対応物である活性化マクロファージと比較して、DCによって差次的に発現される遺伝子を同定するために、ならびにDC特異的機能と関連する遺伝子を同定するために、本発明者らは、cDNAサブトラクションアプローチを適用した。これは、抑制PCR(PCR SelectTM)と組み合わせた、改変型PCRベースの「代表差次的分析(representative differential analysis)」(RDA)を使用した(Diatchenko,L.ら,Proc.Natl.Acad.Sci USA 93:66025−6030(1996))。
【0080】
(一般的な組み換えDNA方法)
分子生物学の一般的な方法を開示する基本書(それらの全てが、参考として援用される)としては、以下が挙げられる:Sambrook,Jら,Molecular Cloning:A Laboratory Manual,第2版,Cold Spring Harbor Press,Cold Spring Harbor,NY,1989;Ausubel,FMら、Current Protocols in Molecular Biology,Vol.2,Wiley−Interscience,New York(最新版);Kriegler,Gene Transfer and Expression:A Laboratory Manual(1990);Glover,DM,編,DNA Cloning:A Practical Approach,vol.I & II,IRL Press,1985;Albers,B.ら,Molecular Biology of the Cell,第2版,Garland Publishing,Inc.,New York,NY(1989);Watson,JDら,Recombinant DNA,第2版,Scientific American Books,New York,1992;ならびにOld,RWら,Principles of Gene Manipulation:An Introduction to Genetic Engineering,第2版,University of California Press,Berkeley,CA(1981)。
【0081】
他に示されない限り、特定の核酸配列は、その保存的置換(例えば、縮重コドン置換)改変体および相補配列を含むことが意図される。用語「核酸」は、「ポリヌクレオチド」と同義であり、そして遺伝子、cDNA分子、mRNA分子、ならびにこれらのいずれかのフラグメント(例えば、オリゴヌクレオチド)を含み、そしてさらに、これらの等価物を含むことが意図される(より完全に以下に説明される)。核酸のサイズは、キロベース(kb)または塩基対(bp)のいずれかで示される。これらは、アガロースまたはポリアクリルアミドゲル電気泳動(PAGE)から、使用者によって決定される核酸配列から、または公開される核酸配列から誘導される推定値である。タンパク質サイズは、キロダルトン(kDa)の分子量または長さ(アミノ酸残基の数)で示される。タンパク質サイズは、PAGEから、配列決定から、コード核酸配列に基づく推定アミノ酸配列から、または公開されるアミノ酸配列から概算される。
【0082】
詳細には、B7−DCまたはそのフラグメントもしくは誘導体に対応するアミノ酸配列をコードするcDNA分子は、本明細書中に開示されるタンパク質の配列由来のプライマーを用いてポリメラーゼ連鎖反応(PCR)(例えば、米国特許第4,683,202号を参照のこと)によって合成され得る。次いでこれらのcDNA配列は、真核生物発現ベクターまたは原核生物発現ベクターに組み立てられ得、そして生じるベクターが使用されて、適切な宿主細胞(例えば、COS細胞またはCHO細胞)によってB7−DCまたはそのフラグメントもしくは誘導体の合成を指向し得る。
【0083】
本発明は、新規B7−DCをコードするヌクレオチド配列を有する単離された核酸、そのフラグメントまたはその等価物を含む。用語、核酸は、本明細書中に使用される場合、そのようなフラグメントまたは等価物を含むことが意図される。本発明の核酸配列は、DNAであってもRNAであってもよい。好ましい核酸は、配列番号1の配列を有するヒトB7−DCをコードするcDNAまたはその等価物である。
【0084】
好ましくは、本発明の核酸は、B7−DCの少なくとも一部をコードするcDNA分子である。このcDNAは、成熟DCまたはこのタンパク質を天然に発現する他の細胞から抽出されたmRNAから作製され得る。B7−DCをコードする核酸配列は、DCゲノムDNAから入手可能である。従って、B7−DCをコードするDNAは、公知のプロトコールに従ってcDNAライブラリーまたはゲノムライブラリーからクローン化され得る。
【0085】
B7−DCをコードするcDNAヌクレオチド配列は、適切な細胞株から総mRNAを単離することによって入手され得る。二本鎖cDNAが総mRNAから調製される。cDNAが、多数の公知の技術のうちのいずれか1つを用いて、適切なプラスミド、バクテリオファージまたはウイルスベクターに挿入され得る。
【0086】
ヌクレオチド配列に関して、用語「等価」は、構造的に相同なタンパク質および/または機能的に等価なタンパク質をコードする配列を含むことが意図される。例えば、B7−DCヌクレオチド配列の天然の多型(特にコドンの第3の塩基において)は、アミノ酸配列を変更しない「サイレント」な変異として公知であり得る。しかし、B7−DC中のアミノ酸配列変化を含む多型が、ヒト(または他の哺乳動物)集団中に存在し得る。1つ以上のヌクレオチド中に変化を有するこれらの対立遺伝子改変体(コード配列の総数のうちの約3〜4%まで)が、天然の対立遺伝子バリエーションに起因してヒト集団中におそらく見出されるということを、当業者は認識する。B7−DCをコードするDNAにおける、これらの対立遺伝子改変体ならびに生じる核酸多型およびポリペプチド多型のいずれかおよび全てが、本発明の範囲内である。
【0087】
さらに、本明細書中に記載されるB7−DCタンパク質の、1つ以上の天然に存在するアイソフォームまたは関連する免疫学的に交差反応性のファミリーのメンバーが存在し得る。このようなアイソフォームまたはファミリーのメンバーは、たとえそれらが異なる遺伝子座の遺伝子によってコードされるとしても、B7−DCに対して機能アミノ酸配列の類似性を共有するタンパク質として規定される。
【0088】
(核酸のフラグメント)
核酸配列のフラグメントは、全長B7−DCタンパク質をコードするヌクレオチド配列よりも少ないヌクレオチドを有するヌクレオチド配列として規定される。本発明は、(1)T細胞上でその天然のリガンドに結合するB7−DCの能力を保持し、かつ(2)活性化T細胞媒介免疫応答(一次活性化シグナルを受けたT細胞によるサイトカイン産生および/またはT細胞増殖として測定される)を(それらがどのように提示されるかに依存して)増強または阻害するポリペプチドをコードする、核酸フラグメントを含む。
【0089】
例えば、本明細書中に意図されるような核酸フラグメントは、未だ同定されていない(しかし、CD28またはCTLA−4ではないようである)レセプターに対してT細胞表面に結合し、そして補助刺激シグナルをTリンパ球に送達する能力を保持するB7−DCポリペプチドをコードする。別の基準によって、本発明の核酸フラグメントは、別の動物種由来の核酸とハイブリダイズする核酸フラグメントであり、ゆえに、B7−DCと「交差反応性」である新規タンパク質を検出するためのスクリーニングアッセイにおいて有用である。
【0090】
一般的に、B7−DCポリペプチドのフラグメントをコードする核酸配列は、成熟タンパク質をコードする配列由来のヌクレオチドを含む。しかし、いくつかの場合、核酸のリーダー配列部分の全てまたは一部を含むことが、望ましくあり得る。本発明の核酸配列はまた、リンカー配列、天然または改変された制限エンドヌクレアーゼ部位、およびコードされるタンパク質もしくはフラグメントのクローニング、発現または精製に関連する操作のために有用である他の配列を含み得る。核酸配列のこれらおよび他の改変は、本明細書中に記載されるか、または当該分野で周知である。
【0091】
1つの実施形態において、B7−DCのECDに対応するアミノ酸配列(おおよそ26〜221位のアミノ酸を含む)をコードするDNAは、PCRを用いて、ヒトIgCγ1のヒンジ領域、CH2領域、CH3領域に対応するアミノ酸配列をコードするDNAに連結されて、B7−DC−Ig融合タンパク質として発現される構築物を形成する。
【0092】
B7−Ig融合タンパク質をコードする類似のDNA分子は、米国特許第5,521,288号に開示され、そして登録番号68627の下でAmerican Type Culture Collection(Rockville,Md.)に寄託されている。
【0093】
B7−DCおよび可溶性B7−DC融合タンパク質をコードするDNAを組み立て、そして発現させるための技術(例えば、オリゴヌクレオチド合成、PCR、細胞の形質転換、ベクターの構築、発現系など)は、当該分野で十分に確立されている。当業者は、特定の条件および手順のための標準的な供給原材料に精通している。
【0094】
他の実施形態において、B7−DCのドメインまたはフラグメントをコードするDNAは、別のB7ファミリータンパク質(例えば、B7.1、B7.2、またはB7.3)の残りの部分のほとんどまたは全てをコードする核酸と融合される。ヒトB7.1(CD80)の完全DNA配列は、Genbank登録番号X60958を有し;マウス配列についての登録番号は、X60958であり;ラット配列についての登録番号は、U05593である。ヒトB7.2(CD86)の完全cDNA配列は、Genbank登録番号L25259を有し;マウス配列についての登録番号は、L25606である。
【0095】
(発現ベクターおよび宿主細胞)
本発明は、少なくとも1つの調節配列に作動可能に連結されたB7−DCポリペプチドをコードする核酸配列を含む発現ベクターを含む。「作動可能に連結された」は、コード配列の発現を可能にする条件下で、コード配列が調節配列に連結されていることを意味する。既知の調節配列が選択されて、適切な宿主細胞中での所望のタンパク質の発現を指向する。従って、用語「調節配列」は、プロモーター、エンハンサーおよび他の発現制御エレメントを含む。このような調節配列は、例えば、Goeddel,Gene Expression Technology.Methods in Enzymology,vol.185,Academic Press,San Diego,Calif.(1990)に記載される。
【0096】
当業者は、本発明の発現ベクターの特定の設計が、考慮(例えば、トランスフェクトされる宿主細胞および/または発現されるタンパク質の型)に依存することを、認識する。
【0097】
本発明の発現ベクターは、B7−DCの種々の実施形態(全長タンパク質およびその機能的誘導体(本明細書中に規定される)(ポリペプチドフラグメント、改変体、融合タンパク質などを含む))をコードする核酸分子の完全な範囲を含む。従って、1つの実施形態において、発現ベクターは、単独または別のタンパク質に融合した、B7−DCタンパク質の少なくとも一部(例えば、ECD)をコードする核酸を含む。
【0098】
このような発現ベクターが使用されて、DNAの発現のためおよび融合タンパク質またはペプチドを含むコードされるタンパク質の産生のために、宿主細胞にトランスフェクトされる。B7−DCポリペプチドを発現する遺伝学的に改変された細胞は、細胞が示された目的のために有用であるために十分な時間、外因性DNAを一過性に発現し得ることが理解される。従って、細胞が、インビボまたはエキソビボにおいて増大した補助刺激能力を有する免疫源として作用する場合、発現が必要とされる時間の長さまたは細胞が生存したままである長さは、細胞がその免疫原性機能および/または補助刺激機能と発揮するために必要とされる時間である。例えば、本発明のB7−DCを発現する形質導入された腫瘍細胞の場合、B7−DCの発現は、少なくとも6時間、好ましくは24時間、より好ましくは少なくとも2〜4日間であり得る。当然ながら、発現はまた、安定(すなわち、その細胞の寿命の間、安定)であり得る。以下に考察される適切な発現ベクターおよび調節エレメント(例えば、誘導性プロモーターまたは構成性プロモーター)は、所望または必要とされる発現の安定性に従って選択される。
【0099】
本発明において、本発明は、B7−DCタンパク質、フラグメント、および誘導体を産生するための方法を提供する。例えば、B7−DCタンパク質の少なくとも一部をコードする核酸ベクターを用いてトランスフェクトされた宿主細胞は、B7−DCポリペプチドの発現を可能にする適切な条件下で培養される。
【0100】
宿主細胞はまた、B7−DCタンパク質の少なくとも一部をコードするDNAおよび第2タンパク質の少なくとも一部をコードするDNAを、単独でかまたは組み合わせて含む1つ以上の発現ベクターを用いてトランスフェクトされ得、その結果、この宿主細胞は、両方の部分を含む融合ポリペプチドを産生する。
【0101】
組み換え発現ベクターが、B7−DCの一部をコードするDNAおよび別のタンパク質(例えば、ヒトIgCγ1)をコードするDNAを含む場合、生じる融合タンパク質は、変更された可溶性、結合親和性および/または結合価を有し得る。例えば、B7−DC Ig融合タンパク質は、好ましくは、培養物においてトランスフェクトされた宿主細胞によって分泌され、ゆえに、この培養培地から単離される。あるいは、タンパク質が細胞質に保持される場合、細胞が収集され、溶解され、そしてタンパク質がこの溶解物から単離される。
【0102】
培養は、代表的に、宿主細胞、適切な増殖培地、および他の副生成物を含む。適切な培養培地は、当該分野で周知である。B7−DCタンパク質は、タンパク質およびペプチドを精製するための従来技術を用いて培地または細胞溶解物から単離され得、これには、硫酸アンモニウム沈澱、分画カラムクロマトグラフィー(例えば、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィー、アフィニティークロマトグラフィーなど)、および/または電気泳動が挙げられる(一般的に、「Enzyme Purification and Related Techniques」Methods in Enzymology,22:233−577(1971)を参照のこと)。一旦、部分的または均質的にまで精製されると、本発明の組み換えB7−DCタンパク質は、本明細書中により詳細に記載されるように薬学的組成物中に使用され得る。
【0103】
形質転換またはトランスフェクトされてB7−DCまたはそのホモログもしくは機能的誘導体を発現する、原核生物宿主細胞または真核生物宿主細胞は、本発明の範囲内である。例えば、B7−DCは、E.coliのような細菌細胞、昆虫細胞(バキュロウイルス)、酵母、またはチャイニーズハムスター卵巣細胞(CHO)もしくはヒト細胞のような哺乳動物細胞中で発現され得る。他の適切な宿主細胞は、Goeddel(1990)(前出)に見出され得るか、さもなくば当業者に公知である。
【0104】
真核生物細胞における発現は、部分的もしくは完全なグリコシル化および/または組み換えタンパク質の関連する鎖間ジスルフィド結合もしくは鎖内ジスルフィド結合の形成を導く。
【0105】
酵母、S.cerevisiaeにおける発現のためのベクターの例としては、pYepSec1(Baldariら(1987)EMBO J.6:229−234)、pMFa(Kurjanら(1982)Cell 30:933−943)、pJRY88(Schultzら(1987)Gene 54:113−123)、およびpYES2(Invitrogen Corporation,San Diego,Calif.)が挙げられる。培養される昆虫細胞(SF9細胞)におけるタンパク質発現のために利用可能なバキュロウイルスベクターとしては、pAcシリーズ(Smithら(1983)Mol.Cell Biol.3:2156−2165)およびpVLシリーズ(Lucklow,V.A.,およびSummers,M.D.(1989)Virology 170:31−39)が挙げられる。一般的に、COS細胞(Gluzman,Y.(1981)Cell 23:175−182)が、哺乳動物細胞における一過性の増殖/発現のためにpCDM8のようなベクター(Aruffo A.およびSeed,B.(前出))と組み合わせて使用され、一方、CHO(dhfr−ネガティブCHO)細胞が、哺乳動物細胞における安定な増殖/発現のためにpMT2PCのようなベクター(Kaufmanら(1987)EMBO J.6:187−195)と共に使用される。NS0骨髄腫細胞株(グルタミンシンセターゼ発現系)が、Celltech Ltd.から利用可能である。
【0106】
しばしば、融合発現ベクターにおいて、タンパク質分解性切断部位が、レポーター基および標的タンパク質の連結部に導入されて、融合タンパク質の精製に続く、標的タンパク質のレポーター基からの分離を可能にする。このような切断およびその認識配列に対するタンパク質分解性酵素としては、第Xa因子、トロンビンおよびエンテロキナーゼが挙げられる。
【0107】
代表的な融合発現ベクターとしては、pGEX(Amrad Corp.,Melbourne,Australia)、pMAL(New England Biolabs,Beverly,Mass.)およびpRIT5(Pharmacia,Piscataway,N.J.)が挙げられ、これらはそれぞれ、グルタチオンS−トランスフェラーゼ、マルトースE結合タンパク質、プロテインAを標的組み換えタンパク質に融合する。
【0108】
誘導可能な非融合発現ベクターとしては、pTrc(Amannら(1988)Gene 69:301−315)およびpET 11d(Studierら,Gene Expression Technology:Methods in Enzymology 185,Academic Press,San Diego,Calif.(1990)60−89)が挙げられる。標的遺伝子発現は、pTrcにおいてハイブリッドtrp−lac融合プロモーターからの宿主RNAポリメラーゼ転写に依存するが、pET11dに挿入された標的遺伝子の発現は、同時発現されるウイルスRNAポリメラーゼ(T7gn1)によって媒介されるT7 gn10−lacO融合プロモーターからの転写に依存する。これは、ウイルスポリメラーゼであり、lacUV5プロモーターの転写制御下で、T7gn1を保有するレジデントλプロファージから宿主株BL21(DE3)またはHMS174(DE3)によって供給される。
【0109】
本発明の1つの実施形態は、新規B7−DCを新規に発現するトランスフェクトされた細胞である。すでにB7−DCを発現する細胞(例えば、成熟DC)の場合、トランスフェクトされた細胞は、細胞表面上に増加した量のB7−DCタンパク質またはそのフラグメントを発現させる。
【0110】
例えば、腫瘍細胞(例えば、肉腫、黒色腫、白血病、リンパ球、癌腫または神経芽細胞腫)は、腫瘍細胞表面上でのB7−DCの発現を指向する発現ベクターでトランスフェクトされる。このようなトランスフェクトされた腫瘍細胞は、本明細書中に記載されるように治療的抗腫瘍免疫を誘導するための免疫原として使用され得る。
【0111】
(ベクター構築)
所望されるコード配列および制御配列を含む適切なベクターの構築は、当該分野で十分に理解された標準的な連結技術および制限技術を使用する。単離されたプラスミド、DNA配列、または合成オリゴヌクレオチドは、所望される形態に、切断され、調整され、そして連結され得る。
【0112】
ベクターを形成するDNA配列は、多数の供給源から利用可能である。骨格のベクターおよびコントロール系は、一般的に構築において大量の配列のために使用される利用可能な「宿主」ベクター上に見出される。適切なコード配列について、最初の構築物は、適切な配列をcDNAライブラリーまたはゲノムDNAライブラリーから回収することが問題であるかもしれず、通常はこれが問題である。しかし、一旦配列が開示されると、個々のヌクレオチド誘導体から開始して、全遺伝子配列をインビトロで合成することが可能である。かなり大きいサイズ(例えば、500〜1000bp)の長さの遺伝子についての全遺伝子配列は、個々の重複する相補オリゴヌクレオチドを合成し、そして一本鎖の非重複部分をデオキシリボヌクレオチド三リン酸の存在下でDNAポリメラーゼを用いて充填することによって、調製され得る。このアプローチは、既知配列のいくつかの遺伝子の構築の場合に好首尾に使用されている。例えば、Edge,M.D.,Nature(1981)292:756;Nambair,K.P.ら,Science(1984)223:1299;およびJay,E.,J Biol Chem(1984)259:6311を参照のこと。
【0113】
合成オリゴヌクレオチドは、上記に引用される参考文献によって記載されるようなホスホトリエステル方法によってか、またはBeaucage,S.L.,およびCaruthers,M.H.,Tet Lett(1981)22:1859;ならびにMatteucci,M.D.,およびCaruthers,M.H.,J Am Chem Soc(1981)103:3185によって記載されるようなホスホロアミダイト方法のいずれかによって調製され、そして市販の自動化オリゴヌクレオチド合成機を用いて調製され得る。アニーリング前または標識のための一本鎖のキナーゼ処理は、50mM Tris(pH7.6)、10mM MgCl2、5mM ジチオスレイトール、1〜2mM ATP、1.7pmole γ−32P−ATP(2.9mCi/mmole)、0.1mM スペルミジン、0.1mM EDTAの存在下、過剰のポリヌクレオチドキナーゼ(例えば、1nmoleの基質に対して約10ユニットのポリヌクレオチドキナーゼ)を使用することによって達成される。
【0114】
一旦所望されるベクターの成分がこのように利用可能になると、これらは、標準的な制限手順および連結手順を用いて、切り出され、そして連結され得る。部位特異的DNA切断は、当該分野で一般的に理解される条件下で適切な制限酵素(単数または複数)を用いて処理することによって実行され、そしてこの詳細は、市販の制限酵素の製造業者らによって特定される。例えば、New England Biolabsの製品カタログを参照のこと。一般的に、約1mgのプラスミドまたはDNA配列が、約20mlの緩衝溶液中で1ユニットの酵素によって切断される;本明細書中の実施例において、代表的に、過剰の制限酵素が、DNA基質の完全な消化を確実にするために使用される。37℃で約1時間〜2時間のインキュベーション時間が、実行可能であるが、バリエーションが許容され得る。各インキュベーション後、タンパク質が、フェノール/クロロホルムを用いた抽出によって除去され、そしてエーテル抽出が続き得、そして核酸が、エタノールを用いる沈澱によって水性画分から回収される。所望される場合、切断されたフラグメントのサイズ分画は、標準的な技術を用いてポリアクリルアミドゲルまたはアガロースゲル電気泳動によって実行され得る。サイズ分画の一般的な説明は、Methods in Enzymology(1980)65:499−560に見出される。
【0115】
制限切断されたフラグメントは、4つのデオキシヌクレオチド三リン酸(dNTP)の存在下、20℃〜25℃で約15〜25分間のインキュベーション時間を用いて、50mM Tris(pH7.6)、50mM NaCl、6mM MgCl2、6mM DTTおよび0.1〜1.0mM dNTP中でE.coli DNAポリメラーゼI(Klenow)の大きなフラグメントを用いて処理することによって、平滑末端化処理し得る。Klenowフラグメントは、5’一本鎖オーバーハングを充填するが、たとえ4つのdNTPが存在していても突出している(protruding)3’一本鎖を破壊する。所望される場合、選択的な修復が、オーバーハングの性質によって規定される制限内で、1種類のdNTPだけ、または選択されたdNTPを供給することによって、実行され得る。Klenowを用いた処理後、混合物を、フェノール/クロロホルムを用いて抽出し、そしてエタノール沈澱する。S1ヌクレアーゼまたはBAL−31を用いての適切な条件下での処理は、いずれかの一本鎖部分の加水分解をもたらす。
【0116】
連結は、代表的に、例えば、以下の標準的な条件および温度の下で、15〜50mlの容量で実行される:20mM Tris−HCl(pH7.5)、10mM MgCl2、10mM DTT、33μg/ml BSA、10〜50mM NaCl、40μM ATP、0.01〜0.02(Weiss)ユニットのT4 DNAリガーゼ、0℃(「粘着性末端」連結について)または1mM ATP、0.3〜0.6(Weiss)ユニットのT4 DNAリガーゼ、14℃(「平滑末端」連結について)のいずれか。分子間「粘着末端」連結は、通常、33〜100μg/mlの総DNA濃度(5〜100nMの末端濃度)で実行される。分子間平滑末端連結は、1mMの総末端濃度で実行される。
【0117】
「ベクターフラグメント」を使用するベクター構築の際、フラグメントは、一般的に、5’ホスフェートを除去し、そして自己連結を回避するために、細菌アルカリホスファターゼ(BAP)またはウシ腸アルカリホスファターゼ(CIAP)を用いて処理される。消化は、おおよそ10mM Tris−HCl、1mM EDTA中、pH8、60°で、約1時間かけて約1ユニット/mgベクターでBAPまたはCIAPを用いて実行される。調製物は、フェノール/クロロホルムを用いて抽出され、そしてエタノール沈澱される。あるいは、さらなる制限酵素および所望されないフラグメントの分離によって二重に消化されたベクターにおいて、再連結が回避され得る。
【0118】
多数の方法のうちのいずれかが使用されて、コード配列中に変異を誘導して、本発明の改変体を作製する。これらの変異としては、単純な欠失または挿入、体系的な欠失、挿入または塩基クラスターの置換あるいは1塩基の置換が挙げられる。
【0119】
例えば、B7−DC DNA配列(cDNAまたはゲノムDNA)の改変は、プロトコールおよび試薬が市販されている周知技術である部位指向型変異誘発によって作製される(Zoller,MJら,Nucleic Acids Res(1982)10:6487−6500およびAdelman,JPら,DNA(1983)2:183−193))。プラスミド構築のための正確な連結が、例えば、最初にE.coli株MC1061(Casadaban,M.ら,J Mol Biol(1980)138:179−207)、または他の適切な宿主を、連結混合物を用いて形質転換することによって確立される。従来方法を用いて、形質転換体は、プラスミド構築の様式に依存して、アンピシリン耐性遺伝子、テトラサイクリン耐性遺伝子、または他の抗生物質耐性遺伝子(または他の選択マーカー)の存在に基づいて選択される。次いで、プラスミドは、任意のクロラムフェニコール増幅(必要に応じてクロラムフェニコール増幅が続く)を用いて形質転換体から調製される(Clewell,DBら,Proc Natl Acad Sci USA(1969)62:1159;Clewell,D.B.,J Bacteriol(1972)110:667)。いくつかのミニDNAプレップが、一般的に使用される。例えば、Holmes,DSら,Anal Biochem(1981)114:193−197;Birnboim,HCら,Nucleic Acids Res(1979)7:1513−1523を参照のこと。単離されたDNAは、制限によって分析され、そして/またはMessingら,Nucleic Acids Res(1981)9:309によってさらに記載されるようなSanger(Proc Natl Acad Sci USA(1977)74:5463)のジデオキシヌクレオチド方法、またはMaxamら、Methods in Enzymology(1980)65:499の方法によって配列決定される。
【0120】
ベクターDNAは、従来の技術、例えば、リン酸カルシウムまたは塩化カルシウム共沈澱、DEAE−デキストラン媒介トランスフェクション、リポフェクション、またはエレクトロポレーションを介して、哺乳動物細胞に導入され得る。宿主細胞を形質転換するための適切な方法は、Sambrookら(前出)および他の標準的な文書に見出され得る。
【0121】
しばしば、融合発現ベクターにおいて、タンパク質分解性切断部位が、レポーター基および標的タンパク質の連結部に導入されて、融合タンパク質の精製に続く、標的タンパク質のレポーター基からの分離を可能にする。このような切断およびその認識配列に対するタンパク質分解性酵素としては、第Xa因子、トロンビンおよびエンテロキナーゼが挙げられる。
【0122】
代表的な融合発現ベクターとしては、pGEX(Amrad Corp.,Melbourne,Australia)、pMAL(New England Biolabs,Beverly,Mass.)およびpRIT5(Pharmacia,Piscataway,N.J.)が挙げられ、これらはそれぞれ、グルタチオンS−トランスフェラーゼ、マルトースE結合タンパク質、プロテインAを標的組み換えタンパク質に融合する。
【0123】
誘導可能な非融合発現ベクターとしては、pTrc(Amannら(1988)Gene 69:301−315)およびpET 11d(Studierら,Gene Expression Technology:Methods in Enzymology 185,Academic Press,San Diego,Calif.(1990)60−89)が挙げられる。標的遺伝子発現は、pTrcにおいてハイブリッドtrp−lac融合プロモーターからの宿主RNAポリメラーゼ転写に依存するが、pET11dに挿入された標的遺伝子の発現は、同時発現されるウイルスRNAポリメラーゼ(T7gn1)によって媒介されるT7 gn10−lacO融合プロモーターからの転写に依存する。これは、ウイルスポリメラーゼであり、lacUV5プロモーターの転写制御下で、T7gn1を保有するレジデントλプロファージから宿主株BL21(DE3)またはHMS174(DE3)によって供給される。
【0124】
(プロモーターおよびエンハンサー)
DNA分子またはRNA分子のプロモーター領域は、RNAポリメラーゼに結合し、そして「作動可能に連結された」核酸配列の転写を促進する。本明細書中に使用される場合、「プロモーター配列」は、RNAポリメラーゼによって転写されるDNAまたはRNAの鎖の上に見出されるプロモーターのヌクレオチド配列である。核酸分子の2つの配列(例えば、プロモーター配列およびコード配列)は、両方の配列が同じRNA転写物上に転写されることを可能にするか、または1つの配列において開始するRNA転写物が第2配列に伸長されることを可能にする様式で、これらが互いに連結している場合、「作動可能に連結されている」。従って、2つの配列(例えば、DNAまたはRNAのプロモーター配列およびコード配列)は、プロモーター配列から始まる転写物が作動可能に連結されたコード配列のRNA転写物を産生する場合、作動可能に連結されている。「作動可能に連結された」状態であるためには、2つの配列が、直線配列中で互いに直ぐに隣接する必要はない。
【0125】
本発明の好ましいプロモーター配列は、哺乳動物細胞において作動可能でなければならず、そして真核生物プロモーターまたはウイルス性プロモーターのいずれかであり得る。適切なプロモーターは、誘導可能、抑制可能または構成的であり得る。構成的プロモーターの例は、ウイルス性プロモーターMSV−LTRであり、これは、種々の細胞において有効であり、かつ活性であり、そしてほとんどの他のプロモーターとは対照的に、停止細胞および増殖中の細胞において同じ高い活性を有する。他の好ましいウイルス性プロモーターとしては、CMV−LTR(サイトメガロウイルス由来)(Bashart,M.ら、Cell 41:521(1985))またはRSV−LTR(ラウス肉腫ウイルス由来)(Gorman,C.M.,Proc.Natl.Acad.Sci.USA 79:6777(1982))に存在するウイルス性プロモーターが挙げられる。マウスメタロチオネインI遺伝子のプロモーター(Hamer,D.ら、J.Mol.Appl.Gen.1:273−288(1982));ヘルペスウイルスのTKプロモーター(McKnight,S.,Cell 31:355−365(1982));SV40初期プロモーター(Benoist,C.ら、Nature 290:304−310(1981));および酵母gal4遺伝子プロモーター(Johnston,S.A.ら、Proc.Natl.Acad.Sci.(USA)79:6971−6975(1982);Silver,P.A.ら、Proc.Natl.Acad.Sci(USA)81:5951−5955(1984))もまた、有用である。プロモーター領域に関連する転写因子および分離活性化および転写因子のDNA結合の他の例示的記載としては、以下が挙げられる:Keeganら、Nature(1986)231:699;Fieldsら、Nature(1989)340:245;Jones,Cell(1990)61:9;Lewin,Cell(1990)61:1161;Ptashneら、Nature(1990)346:329;Adamsら、Cell(1993)72:306。これらの上記の参考文献全ての関連する開示は、参考として本明細書に援用される。
【0126】
プロモーター領域はさらに、特異的な組織に見出される特定のタンパク質との相互作用によって組織特異的エンハンサーとして機能もし得る八量体領域を含み得る。本発明のDNA構築物のエンハンサードメインは、トランスフェクトされるべき標的細胞に特異的かまたは、このような標的細胞の細胞性因子によって高く活性化されるものである。ベクター(プラスミドまたはレトロウイルス)の例は、(Roy−Burmanら、米国特許第5,112,767号)に開示される。エンハンサーおよび転写におけるその作用の一般的な議論については、Lewin,B.M.,Genes IV,Oxford University Press,Oxford、(1990)、pp.552−576を参照のこと。特に有用なものは、レトロウイルスエンハンサー(例えば、ウイルス性LTRである)。エンハンサーは、好ましくは、プロモーターから上流に配置され、相互作用して遺伝子発現を刺激する。レトロウイルスベクターの使用のために、内因性ウイルスLTRは、エンハンサーがなくされ、そして組織特異性または他の所望の特性(例えば、本発明のB7−DCコードDNA分子に対する転写効果)を与える他の所望のエンハンサー配列で置換され得る。
【0127】
本発明の核酸配列はまた、標準的技術を用いて化学的に合成され得る。ポリデオキシヌクレオチドを化学的に合成する種々の方法は公知であり、市販されるDNA合成装置で完全に自動化されている固相合成(ペプチド合成など)を含む(例えば、本明細書中に援用されるItakuraら、米国特許第4,598,049号;Caruthersら、米国特許第4,458,066号;ならびにItakura 米国特許第4,401,796号および同第4,373,071号を参照のこと)。
【0128】
(タンパク質およびポリペプチド)
本発明は、配列番号2または配列番号4の配列を有する「単離された」B7−DCタンパク質を含む。本開示は、全長ヒトおよびマウスB7−DCタンパク質(およびDNA)を例示するが、本明細書中に開示される特徴を所有する他の哺乳動物種由来のB7−DCの相同体およびその変異体が、本発明の範囲内であることを意図されることは理解されるべきである。
【0129】
含まれるものはまた、アミノ酸置換改変体を意味するB7−DCの「機能的な誘導体」、「フラグメント」、またはB7−DCの「化学誘導体」である。これらの用語は、下で定義される。機能的誘導体は、測定可能なB7−DC活性、好ましくはT細胞上のレセプターに結合する活性および本発明に従ってその有用性を可能にするT細胞活性を補助刺激する活性を保持する。「機能的誘導体」は、この用語が本明細書中に接続語でまたは選択肢で使用されるか否かによらず「改変体」および「フラグメント」を含む。
【0130】
機能的相同体は、上の生化学的活性および生物学的活性を保有しなければならない。この機能的特徴付けを考慮して、他の種由来の相同体タンパク質B7−DC(まだ発見されていないタンパク質を含む)の使用は、これらのタンパク質が配列類似性ならびに上記の生化学的活性および生物学的活性を有する場合、本発明の範囲内に含まれる。
【0131】
2つのアミノ酸配列のまたは、2つの核酸配列のパーセント同一性を測定するために、この配列は、最適な比較目的のためにアラインメントされる(例えば、ギャップが、最適なアラインメントのために第1のアミノ酸および第2のアミノ酸の1つまたは両方、あるいは核酸配列において導入され得、そして非相同配列は比較目的のために無視され得る)。好ましいアラインメントの方法において、Cys残基がアラインメントされる。
【0132】
好ましい実施形態において、比較されるべき配列の長さは、参照配列の長さの少なくとも30%、好ましくは少なくとも40%、より好ましくは少なくとも50%、さらにより好ましくは少なくとも60%、そしてより好ましくは少なくとも70%、80%、または90%である。例えば、276アミノ酸残基を有するヒトB7−DCタンパク質アミノ酸配列(配列番号2)に対して第2の配列をアラインメントする場合、少なくとも83アミノ酸残基、好ましくは少なくとも110アミノ酸残基、より好ましくは少なくとも138アミノ酸残基、さらにより好ましくは少なくとも166アミノ酸残基、そしてより好ましくは193アミノ酸残基、221アミノ酸残基または248アミノ酸残基が、アラインメントされる。次いで、対応するアミノ酸位置(またはヌクレオチド位置)におけるアミノ酸残基(またはヌクレオチド)が、比較される。次いで第1の配列における位置が、第2の配列における対応位置と同じアミノ酸残基(またはヌクレオチド)によって占有される場合、この分子は、その位置で同一性である(本明細書で使用されるアミノ酸または核酸「同一性」は、アミノ酸または核酸「相同」と等価である)。2つの配列間のパーセント同一性は、この2つの配列の最適なアラインメントのために誘導される必要があるギャップの数、および各ギャップの長さを考慮に入れて、配列によって共有される同一位置の数の関数である。
【0133】
2つの配列間の配列の比較およびパーセント同一性の決定は、数学的アルゴリズムを使用して達成され得る。好ましい実施形態において、2つのアミノ酸配列間のパーセント同一性は、NeedlemanおよびWunsch(J.Mol.Biol.48:444−453(1970))アルゴリズムを使用して測定される。このアルゴリズムは、GCGソフトウェアパッケージ(http://www.gcg.comから入手可能)中のGAPプログラムに組み込まれている。これは、Blossom62マトリクスまたはPAM250マトリクスのいずれか、および16、14、12、10、8、6、もしくは4のgap weightおよび1、2、3、4、5、もしくは6のlength weightを使用する。さらに別の好ましい実施形態において、2つのヌクレオチド配列間のパーセント同一性は、GCGソフトウェアパッケージ(http://www.gcg.comから入手可能)中のGAPプログラムを使用して測定される。これは、NMSgapdna.CMPマトリクスおよび40、50、60、70、もしくは80のgap weightおよび1、2、3、4、5、もしくは6のlength weightを使用する。別の実施形態において、2つのアミノ酸または2つのヌクレオチド配列間のパーセント同一性は、E.MeyersおよびW.Miller(CABIOS,4:11−17(1989))のアルゴリズムを使用して測定される。これは、PAM120 weight residue table、12のgap length penaltyおよび4のgap penaltyを使用するALIGNプログラム(バージョン2.0)に組み込まれている。
【0134】
本発明の核酸およびタンパク質配列は、さらに「クエリー配列」として使用されて、公開されているデータベースに対する探索を実行(例えば、他のファミリーメンバーまたは関連配列を同定する)し得る。このような探索は、Altshulら(1990)J.Mol.Biol.215:403−10のNBLASTおよびXBLASTプログラム(バージョン2.0)を使用して実行され得る。BLASTヌクレオチド探索は、NBLASTプログラム(score=100、wordlength=12)を用いて実行されて、ヒトまたはマウスB7−DC核酸分子に対するヌクレオチド配列相同性を獲得し得る。BLASTタンパク質探索は、XBLASTプログラム(score=50、wordlength=3)を用いて実行されて、本発明のヒトまたはマウスB7−DCタンパク質分子に対するアミノ酸配列相同性を獲得し得る。比較目的のためのギャップを挿入したアラインメントを獲得するために、Gapped BLASTは、Altshulら(1997)Nucleic Acids Res.25:3389−3402に記載されるように利用され得る。BLASTおよびGapped BLASTプログラムを利用する場合、各々のプログラムのデフォルトパラメータ(例えば、XBLASTおよびNBLAST)が使用され得る。http://www.ncbi.nlm.nih.gov.を参照のこと。
【0135】
従って、上記のB7−DCタンパク質の相同体は、(a)ネイティブのB7−DC機能的活性、および(b)ネイティブのB7−DCタンパク質(例えば、上で測定された配列番号2または配列番号4)に対する配列類似性(少なくとも約30%(アミノ酸レベルで)、好ましくは少なくとも約50%、より好ましくは少なくとも約70%、さらにより好ましくは少なくとも約90%)を有すると特徴付けられる。
【0136】
B7−DCの開示された配列に基づくDNAプローブを使用してこのようなタンパク質を得、そして発現することは当該分野の技術範囲内である。次いで、このタンパク質の生化学的活性および生物学的活性は、本明細書中に記載されるような当該分野で認識される方法(例えば、標準的なT細胞増殖またはサイトカイン分泌アッセイ)を使用して、容易に試験され得る。T細胞補助刺激の生物学的アッセイは、相同体が「機能的」相同体として適切であるような必須な活性を有するかどうかを示す。
【0137】
好ましいアッセイは、TCR(「一次活性化シグナル」)の結合または架橋、および補助刺激シグナルの送達に依存するB7−DCの機能的特徴(例えば、サイトカインのT細胞合成の刺激)を測定する。T細胞上のその天然のリガンドへのB7−DCの結合は、増加したサイトカイン産生(例えば、IL−2)を誘導するシグナルを伝達し、IL−2は、慣用的に測定もされ得る増殖を順番に刺激する。
【0138】
B6−DCの「改変体」とは、1つ以上のアミノ酸残基が置換されている(置換改変体)または1つもしくはいくつかの欠失した残基(欠失改変体)もしくは付加された残基(付加改変体)を有する、全タンパク質あるいはそのフラグメントのいずれかに実質的に同一な分子をいう。B6−DCの「フラグメント」とは、分子の任意のサブセット、好ましくはECDを含むサブセット(すなわち、全長タンパク質のより短いポリペプチド)をいう。
【0139】
多数のプロセスが使用されて、単離されたDNA配列のフラグメント、変異体および改変体を産生し得る。B7−DCタンパク質をコードする核酸の小さなサブ領域またはフラグメント(例えば、1〜30塩基長)は、標準的な、化学的な合成によって調製され得る。より大きな合成フラグメントの産生において使用するためのアンチセンスオリゴヌクレオチドおよびプライマー。
【0140】
好ましい機能的誘導体は、融合タンパク質(B7−DCの機能的フラグメントを含むポリペプチド)である。例えば、B7の有用な誘導体は、B7−DCのECDおよびIg C領域に対応するポリペプチドを含むB7−DC−Ig融合タンパク質である。融合パートナーの存在は、B7−DCタンパク質の可溶性、親和性および/または結合価(ここでは1分子あたりの利用可能な結合部位の数として規定される)を変更し得る。可溶性B7−DC融合タンパク質(T細胞上のレセプターにまだ結合している間)は、APC上に発現されるネイティブタンパク質の効果とは異なる生物学的効果(すなわち、補助刺激よりも競合結合によるT細胞刺激の阻害)を有し得る。
【0141】
本明細書中に使用される場合、B7−DCの細胞外ドメイン(ECD)は、PD−1またはCD28もしくはCTLA−4ではないT細胞上の別のレセプターを認識し、そして結合するタンパク質またはその任意のフラグメントの全体の細胞外部分である。好ましくは、B7−DCのECDは、配列番号2または配列番号4の約26位〜約221位のアミノ酸残基によりコードされる部分である。
【0142】
「可溶性B7−DC」によって、産生細胞から流れるか、分泌されるか、または、さもなければ抽出され得るB7−DCの無細胞形態が意図される。可溶性B7−DCとしては、可溶性融合タンパク質(例えば、生物学的に活性な分子に(遺伝的にまたは化学的に)融合されるB7−DC−Ig、B7−DCの遊離ECD、またはB7−DC ECD)が挙げられるが、それらに限定されない。
【0143】
初期に示されたように、本発明はまた、B7−DCドメインと別のB7ファミリータンパク質のドメインまたはフラグメント(好ましくは補助刺激形態で細胞表面に発現される)との間のハイブリッド融合タンパク質を含む。
【0144】
B7−DC改変体の好ましい群は、少なくとも1つのアミノ酸残基および好ましくは、たった1つのアミノ酸残基が異なる残基によって置換されている群である。タンパク質化学およびタンパク質構造の詳細な記載について、Schulz,GEら、Principles of Protein Structure,Springer−Verlag,New York,1978,およびCreighton,T.E.,Proteins:Structure and Molecular Properties,W.H.Freeman & Co.,San Francisco,1983(本明細書中に参考として援用される)を参照のこと。タンパク質分子において作製され得る置換の型は、異なる種の相同性タンパク質(例えば、Schulzら(上記)の表1〜2およびCreighton(上記)の表3〜9に示されるタンパク質)間のアミノ酸変化の頻度の分析に基づき得る。このような分析に基づいて、保存的置換は、以下の5つの群のうちの1つにおける交換として本明細書中に定義される。
【0145】
【化5】
上の括弧の3つのアミノ酸残基は、タンパク質構造に特別な役割を有する。Glyは、側鎖を欠く唯一の残基であり、従って鎖に可撓性を与える。その通常ではないジオメトリーのため、Proは、鎖をきつく拘束する。Cysは、タンパク質折り畳みに重要なジスルフィド結合形成に関係し得る。
【0146】
生物学的、機能的(または免疫学的)特性におけるより実質的な変化は、例えば、上の5つの群の中よりも間でほとんど保存されていない置換を選択することによって作製される。このような変化は、(a)置換の領域におけるペプチド骨格の構造(例えば、シートまたはヘリックス構造)、(b)標的部位での分子の荷電もしくは疎水性、または(c)側鎖の大きさを維持することに対する効果においてより有意に異なる。このような置換の例は、(i)別のアミノ酸によるGlyおよび/もしくはProの置換またはGlyもしくはProの欠失または挿入;(ii)疎水性残基(例えば、Leu、Lle、Phe、ValもしくはAla)による親水性残基(例えば、SerもしくはThr)の置換;(iii)任意のほかの残基によるCys残基の置換;(iv)電気陰性の電荷を有する残基(例えば、GluもしくはAsp)による電気陽性の側鎖を有する残基(例えば、Lys、ArgもしくはHis)の置換;あるいは(v)巨大な側鎖を有さない残基(例えば、Gly)によるこのような側鎖を有する残基(例えば、Phe)の置換である。
【0147】
本発明に従う大部分の受容可能な欠失、挿入および置換は、そのT細胞補助刺激活性に関するB7−DCタンパク質の特徴においてラジカル変化を産生しないものである。しかし、そのようにするより前に、置換、欠失または挿入の確かな効果を予測することが困難な場合、当業者は、この効果が慣習的なスクリーニングアッセイ(例えば、本明細書に記載される)によって過度の実験を必要とせず評価され得ることを正しく認識する。
【0148】
より短い鎖の改変体が化学合成によって作製され得るが、本発明について、好ましいより長い鎖の改変体は、B7−DCポリペプチドをコードする核酸の部位特異的変異誘発、細胞培養における改変体核酸の発現、および必要に応じて細胞培養物からのポリペプチドの精製によって(例えば、(少なくとも1つのエピトープに結合することによって改変体を吸収するため)カラムに固定された特異的な抗体を使用する免疫親和性クロマトグラフィーによって)典型的に作製される。
【0149】
(B7−DCの化学的誘導体)
B7−DCの「化学的誘導体」は、通常はタンパク質の一部分ではないさらなる化学的部分を含む。ポリペプチドの共有結合修飾は、本発明の範囲内に含まれる。このような誘導された部分は、可溶性、吸収性、生物学的半減期などを改良し得る。このような効果を媒介し得る部分は、例えば、Remington’s Pharmaceutical Sciences,第16版、Mack Publishing Co.,Easton,PA(1980)に開示される。
【0150】
このような修飾は、ポリペプチドの標的化アミノ酸残基を、選択された側鎖または末端残基と反応し得る有機誘導化因子と反応することによって分子に導入され得る。
【0151】
ポリペプチドの化学的誘導体の例は、以下である。
【0152】
リジニル(lysinyl)およびアミノ末端残基は、コハク酸または他のカルボキシル酸無水物で誘導される。環式カルボキシル無水物を用いる誘導は、リジニル残基の電荷を逆転する効果を有する。アミノ含有残基を誘導するための他の適切な試薬としては、イミドエステル(例えば、メチルピコリンイミデート;ピリドキサールリン酸;ピリドキサール;クロロ水素化ホウ素(chlorobrohydride);トリニトロベンゼンスルホン酸;O−メチルイソ尿素;2,4ペンタンジオン;およびグリオキシレートを用いるトランスアミラーゼ触媒反応)が挙げられる。
【0153】
カルボキシル側鎖(アスパルチルまたはグルタミル)は、カルボジイミド(R−N=C=N−R’)(例えば、1−シクロヘキシル−3−(2−モルホリニル−(4−エチル)カルボジイミドまたは1−エチル−3−(4−アゾニア−4,4−ジメチルペンチル)カルボジイミド)との反応によって選択的に修飾され得る。さらに、アスパルチル残基およびグルタミル残基は、アンモニアとの反応によってアスパラギニル残基グルタミニル残基へ変換され得る。
【0154】
他の修飾としては、プロリンおよびリジンのヒドロキシル化、セリル残基またはスレオニル残基のヒドロキシル基のリン酸化、リジンのアミノ基のメチル化(Creighton、上記、pp.79−86)、N末端アミンのアセチル化、ならびに、C末端カルボキシル基のアミド化が挙げられる。
【0155】
含まれるものはまた、1つ以上のD−アミノ酸が1つ以上のL−アミノ酸の代わりに置換されたペプチドである。
【0156】
(マルチマーペプチド)
本発明はまた、B7−DCの配列から得られた塩基性ペプチド配列が、介在スペーサ−またはリンカーを用いてか、または用いずに約2〜約100回繰り返される、より長いポリペプチドを含む。このようなマルチマーは、本明細書中に定義される任意のペプチド改変体から構築され得ることが理解される。さらに、ペプチドマルチマーは、ペプチドモノマーおよびその開示された置換改変体の異なる組合せを含み得る。このようなオリゴマーまたはマルチマーペプチドは、化学合成によってか、または本明細書中で考察されるような組換えDNA技術によって作製され得る。化学的に産生される場合、オリゴマーは、好ましくは、塩基性ペプチド配列の2〜8回繰り返しを有する。組換え的に産生される場合、マルチマーは、例えば、発現系が2〜約100回繰り返すことを可能にするのと同じくらい多くの繰り返しを有し得る。
【0157】
B7−DCペプチドまたはポリペプチドのタンデムマルチマー、好ましくはダイマーおよびトリマーにおいて、鎖は、鎖間ジスルフィド結合または鎖間の他の「構造的」共有結合によって結合され、その結果、この鎖は、「末端から末端」よりも「横 対 横」である。好ましいダイマーおよびトリマーは、本明細書に記載されるようなB7−DCの融合タンパク質(例えば、B7−DC−Ig)間のものである。
【0158】
(B7−DCのエピトープに対して特異的な抗体)
以下の記載において、免疫学、細胞生物学、および分子生物学の当業者に公知の種々の方法論について言及がなされる。刊行物および言及されるこのような公知の方法論を示す他の材料は、まるで全てを示すようにその全体において参考として本明細書中に援用される。免疫学の一般的な原理を記載する標準的な参考著述としては、A.K.Abbasら、Cellular and Molecular Immunology(第4版),W.B.Saunders Co.,Philadelphia,2000;C.A.Janewayら、Immunobiology.The Immune System in Health and Disease,第4版,Garland Publishing Co.,New York,1999;Roitt,I.ら、Immunology,(最新版)C.V.Mosby Co.,St.Louis,MO(1999);Klein,J.,Immunology,Blackwell Scientific Publications,Inc.,Cambridge,MA,(1990)が挙げられる。
【0159】
モノクローナル抗体(mAb)およびその産生および使用のための方法は、KohlerおよびMilstein,Nature 256:495−497(1975);米国特許第4,376,110号;Hartlow,E.ら、Antibodies:A Laboratory Manual,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY,1988);Monoclonal Antibodies and Hybridomas:A New Dimension in Biological Analyses,Plenum Press,New York,NY(1980);H.Zolaら、Monoclonal Hybridoma Antibodies:Techniques and Applications,CRC Press,1982)に記載される。
【0160】
免疫アッセイはまた、Coligan,J.E.ら、編、Current Protocols in Immunology,Wiley−Interscience,New York 1991(または最新版);Butt,W.R.(編)Practical Immunoassay:The State of the Art,Dekker,New York,1984;Bizollon,Ch.A.編、Monoclonal Antibodies and New Trends in Immunoassays,Elsevier,New York,1984;Butler,J.E.,ELISA(29章),In:van Oss,C.J.ら、(編),IMMUNOCHEMISTRY,Marcel Dekker,Inc.,New York,1994,pp.759−803;Butler,J.E.(編),Immunochemistry of Solid−Phase Immunoassay,CRC Press,Boca Raton,1991;Weintraub,B.,Principles of Radioimmunoassays,Seventh Training Course on Radioligand Assay Techniques,The Endocrine Society,March,1986;Work,T.S.ら、Laboratory Techniques and Biochemistry in Molecular Biology,North Holland Publishing Company,NY,(1978)(Chard,T.による章,「An Introduction to Radioimmune Assay and Related Techniques」)に記載される。
【0161】
抗イディオタイプ抗体は、例えば、Idiotypy in Biology and Medicine,Academic Press,New York,1984;Immunological Reviews Volume 79,1984;Immunological Reviews Volume 90,1986,Curr.Top.Microbiol.,Immunol.Volume 119,1985;Bona,C.ら、CRC Crit.Rev.Immunol.,pp.33−81(1981);Jerne,NK,Ann.Immunol.125C:373−389(1974);Jerne,NK,In:Idiotypes−Antigens on the Inside,Westen−Schnurr,I.編,Editiones Roche,Basel,1982,Urbain,Jら、Ann.Immunol.133D:179−(1982);Rajewsky,K.ら、Ann.Rev.Immunol.1:569−607(1983)に記載される。
【0162】
本発明は、公知のB7ファミリータンパク質にないB7−DCの新規なエピトープを反応する抗体(ポリクローナルおよびモノクローナルの両方)を提供する。抗体は、異種の、同種異系の、同系のまたはそれらの修飾形態(例えば、ヒト化抗体またはキメラ抗体)であり得る。抗B7−DC抗体のイディオタイプに対して特異的な抗イディオタイプ抗体もまた、含まれる。用語「抗体」はまた、抗原結合部位を含み、そしてB7−DCエピトープに結合し得るインタクトな分子およびそのフラグメントの両方を含むことが意味される。これらは、インタクトな抗体のFcフラグメントを欠くFabおよびF(ab’)2フラグメント(循環からより速やかに除去され、そしてインタクトな抗体よりも少ない非特異的な組織結合を有し得る)を含む(Wahlら、J.Nucl.Med.24:316−325(1983))。含まれるものはまた、Fvフラグメントである(Hochman,J.ら、(1973)Biochemistry 12:1130−1135;Sharon,J.ら、(1976)Biochemistry 15:1591−1594))。これらの種々のフラグメントは、従来の技術(例えば、プロテアーゼ切断または化学的切断)を使用して産生される(例えば、Rousseauxら、Meth.Enzymol.,121:663−69(1986)を参照のこと)。
【0163】
ポリクローナル抗体は、免疫化動物(ウサギ、ヤギ、げっ歯類など)由来の血清として得られ、そしてさらなる処理をせずに直接使用され得るか、または、慣習的な富化または精製方法(例えば、硫酸アンモニウム沈殿、イオン交換クロマトグラフィー、および親和性クロマトグラフィー)に供され得る(Zolaら、前記を参照のこと)。
【0164】
免疫原は、完全B7−D6タンパク質、あるいはそのフラグメントまたは誘導体を含み得る。好ましい免疫原は、ヒトB7−DCのECD(アミノ酸残基26−221)の全てまたは一部を含み、ここでこれらの残基は、ネイティブなB7−DC上に見出される翻訳後修飾(例えば、グリコシル化)を含む。細胞外ドメインを含む免疫原は、当該分野で公知の種々の方法(例えば、従来の組換え方法を使用するクローニングされた遺伝子の発現、起源の細胞からの単離、高いレベルのB7−DCを発現する細胞集団など)において産生される。
【0165】
mAbは、従来のハイブリドーマ技術(例えば、KohlerおよびMilstein(Nature,256:495−97(1975))、ならびにその改変(上の参考文献を参照のこと)によって導入された手順)を使用して産生され得る。動物、好ましくはマウスは、上のような免疫原を用いる免疫化によってプライムされて、プライムされた動物において所望の抗体応答を誘導する。
【0166】
プライムされた動物のリンパ節、脾臓または末梢血由来のBリンパ球は、一般的に、融合促進剤(例えば、ポリエチレングリコール(PEG))の存在化で骨髄腫細胞と融合させる。任意の多くのマウス骨髄腫細胞株は、このような使用のために入手可能である:P3−NS1/1−Ag4−1,P3−x63−kOAg8.653,Sp2/0−Agl4,またはHL1−653骨髄腫株(ATCC,Rockville,MDから入手可能)。次の工程は、選択培地における増殖を含み、その結果、融合されていない親骨髄腫細胞およびドナーリンパ球細胞は、最終的に死亡するが、ハイブリドーマ細胞のみが生存する。これらは、クローニングされ、そして増殖させて、そしてその上清は、例えば、B7−DC−Ig融合タンパク質を使用する免疫アッセイ技術によって、所望の特異性の抗体の存在についてスクリーニングされる。陽性クローンは、例えば、限界希釈によってサブクローニングされ、そしてmAbが単離される。
【0167】
これらの方法に従って産生されたハイブリドーマは、当該分野で公知の技術を使用してインビトロまたはインビボで(腹水中で)増殖され得る(一般的に、Finkら、Prog.Clin.Pathol.,9:121−33(1984)を参照のこと)。一般的に、個々の細胞株は、培養において増殖され、そして高い濃度の単一のmAbを含有する培養培地は、デカンテーション、濾過、または遠心沈殿法によって収穫され得る。
【0168】
この抗体は、通常のマルチマー構造の代わりに、単鎖抗体またはscFvとして生成され得る。単鎖抗体は、目的のIg由来の超可変領域を含み、未知量の標識された抗体(これは、「リポーター分子」として機能する)を含む溶液と接触させた場合、インタクトなIgの大きさの一部ではあるが、ネイティブなIgの抗原結合部位を再現する(Skerra,A.ら(1988)Science、240:1038−1041;Pluckthun,A.ら(1989)Methods Enzymol.178:497−515;Winter,G.ら(1991)Nature、349:293−299);Birdら(1988)Science 242:423;Hustonら(1988)Proc.Natl.Acad.Sci.USA 85:5879;Jost CRら、J Biol Chem.(1994)269:26267−26273;米国特許第4,704,692号、同第4,853,871号、同第4,946,778号、同第5,260,203号、同第5,455,0Kn)。標識抗体が、非標識抗体を介して固体支持体に結合した抗原と複合体化するのを可能にするための、二回目のインキュベーション期間の後、この固体支持体は2回目に洗浄され、未反応の標識抗体が除去される。この型の正のサンドイッチアッセイは、抗原が存在するか否かを決定するための単純な「あり/なし」のアッセイであり得るか、または標識抗体の程度と、既知量の抗原を含む標準サンプルについて得られた程度を比較することによって定量的にされ得る。
【0169】
別の型の「サンドイッチ」アッセイでは、いわゆる「同時」アッセイおよび「逆」アッセイが使用される。同時アッセイは、固体支持体に結合した抗体および標識抗体の両方が、試験されるサンプルに同時に添加されるので、1回のインキュベーション工程を包含する。このインキュベーションが完了した後、この固体支持体を洗浄して、残りの液体サンプルおよび複合体化されていない標識抗体を除去する。次いで、固体支持体に結合した標識抗体の存在は、従来の「正」サンドイッチアッセイにおけるのと同じようにして決定される。
【0170】
「逆」アッセイにおいて、標識抗体溶液の流体サンプルへの最初の添加、続いて適切なインキュベーション期間後の、固体支持体に結合した非標識抗体の添加という段階的添加が利用される。2回目のインキュベーション後、この固相を従来の様式で洗浄し、試験される残りのサンプルおよび未反応の標識抗体の溶液が固相にない状態にする。次いで、固体支持体に結合した標識抗体の決定は、「同時」アッセイおよび「正」アッセイのように決定される。
【0171】
上記の抗体は、T細胞刺激を阻害するための方法および所望されないT細胞の活性化に関連する疾患(例えば、移植片拒絶および自己免疫)を処置するための方法において有用である。この方法は、このような処置が必要な被験体にB7−DCの補助刺激エピトープに特異的な有効量の抗体、好ましくはmAb、より好ましくはヒトmAbまたはヒト化mAbを、投与する工程を包含する。抗体の投与は、T細胞刺激をブロックするか、または抗原反応性T細胞を排除し、これによって、標的T細胞応答を阻害するのに効果的でなければならない。関連する用量範囲を以下に記載する。
【0172】
(B7−DCタンパク質をコードする核酸の使用)
本発明の核酸は、生物学的サンプル由来の細胞中のB7−DCの発現を測定することによって、疾患の進行をモニターするために診断的に使用されるかまたはB7−DCの発現に対する薬剤の影響をアッセイするために使用される。好ましくは、これは、細胞性mRNAレベルの測定によって達成される。このような診断方法での使用のために、この核酸配列は、例えば、放射性標識もしくは蛍光標識またはビオチンで検出可能に標識され、従来のドットブロットまたはノザン−ハイブリダイゼーション手順において使用され、例えば、生物学的サンプル由来の総またはポリ(A+)RNAの調製物中に存在するmRNA分子がプローブされる。
【0173】
(治療的組成物およびその投与)
B6−DCポリペプチドまたはこのポリペプチドを発現する細胞(例えば、DCまたは腫瘍細胞)は、哺乳動物被験体に、好ましくはヒトに投与される。細胞に結合した形態、固定化された形態、またそうでなければ凝集された形態のポリペプチドを使用して、Tリンパ球の反応性および得られる免疫を増強する。B6−DC−Ig融合タンパク質はダイマーとして組み立てられる、実施例に示されるようにT細胞を補助刺激する。B6−DCポリペプチドの可溶性単量体形態は、活性を刺激することなくT細胞上のレセプターに結合し得、従って、これは、この分子の刺激性形態によるT細胞補助刺激の競合的インヒビターまたはアンタゴニストと考えられ得る。このようなB6−DCアンタゴニストの結合は、進行中のT細胞反応性を抑制し得るか、または内因性のB6−DCまたはさらにこれらのレセプター(例えば、CD28またはCTLA−4)を介して作用するB7ファミリーの他のメンバーによって提示される補助刺激シグナルの効果を妨害し得る。
【0174】
本明細書中に記載のようなB7−DC活性を有する組成物は、生物学的に有効な量または治療的に有効な量で、薬学的に受容可能なキャリア中で投与される。B7−DCポリペプチド(またはそのポリペプチドを発現する細胞)は、単独で、またはB7ファミリーの別のメンバーもしくは別の免疫刺激分子の活性を有するタンパク質もしくはペプチドのような別のタンパク質もしくはペプチドと組合せて、投与され得る。処置はアジュバント(アジュバントの最も広い意味で使用され、インターフェロンのような任意の非特異的免疫刺激化合物を含む)の投与を含み得る。本明細書中で意図されるアジュバントとして、レゾルシノール、非イオン性界面活性剤(例えば、ポリオキシエチレンオレイルエーテルおよびn−ヘキサデシルポリエチレンエーテル)が挙げられる。
【0175】
また、被験体に投与される場合に、以下の用量および量が、本発明の抗体に関連する。
【0176】
治療的に有効な量は、効果的な時期に投与される場合に、所望の免疫学的効果または臨床的効果を達成する投与量である。
【0177】
B7−DC活性を有するポリペプチド(または抗B7−DC抗体)の治療的に活性な量は、因子(例えば、個体の疾患状態、年齢、性別および体重)、ならびにペプチドが個体において所望の応答を惹起するための能力によって変化し得る。投与量のレジメは、最適な治療応答を提供するように調整され得る。例えば、いくつかに分けられた用量を毎日投与してもよいし、この用量を治療状態の緊急性によって示されるように比例して減少させてもよい。細胞に結合した形態の治療的に有効な量のタンパク質は、タンパク質当量または細胞当量で記述され得る。
【0178】
従って、有効な量は、レシピエントの体重1kg当たり約1ngと約1gとの間、より好ましくは約1μgと100mg/kgとの間、より好ましくは約100μgと100mg/kgとの間である。内部投与に適切な投薬形態は、好ましくは、(後者の容量範囲について)1単位当たり約0.1mg〜500mgの活性成分のを含む。活性成分は、組成物の総重量に基づいて、0.5重量%〜95重量%まで変化し得る。あるいは、B7−DCを発現する細胞(DCまたは不活化された腫瘍細胞のような、好ましく形質導入された細胞)の有効な用量は、好ましくは分割用量で、被験体当たり、約104個との細胞と約109個の細胞との間、より好ましくは約106個の細胞と108個の細胞との間である。免疫療法の当業者は、過度の実験なしにこれらの用量を調整し得る。
【0179】
活性な化合物(例えば、B6−DCポリペプチドまたはB6−DC DNAで形質導入された細胞)は、便利な様式(例えば、便利でかつ有効な経路による注射)で投与され得る。好ましい経路として、皮下経路、皮内経路、静脈内経路および筋肉内経路が挙げられる。他の可能性のある経路としては、経口投与、くも膜下腔内適用、吸入適用、経皮適用、または直腸投与が挙げられる。完全には除去されていない腫瘍の処置については、腫瘍内への直接注入もまた、意図される。
【0180】
投与経路に依存して、活性な化合物は、酵素、酸、およびこの化合物を不活化し得る他の天然の条件の作用からこの化合物を保護するための材料でコーティングされ得る。従って、B7−DC活性を有するポリペプチドまたはペプチドを、経腸経路によって投与するために、B7−DC活性の不活化を防止するための材料で組成物をコーティングするか、またはそのような材料と組成物を同時投与する必要があり得る。例えば、ペプチドは、適切なキャリア、希釈液またはアジュバント中で個体に投与され得るか、酵素インヒビター(例えば、膵臓トリプシンインヒビター、ジイソプロピルフルオロホスフェート(DEP)およびトラシロール(trasylol))と共に同時投与され得るか、またはリポソーム(水中油中水エマルジョンおよび従来のリポソーム(Strejanら(1984)J.Neuroimmunol 7:27)を含む)のような適切なキャリア中で投与され得る。
【0181】
本明細書中で使用される場合、「薬学的に受容可能なキャリア」として、任意のおよび全ての溶媒、分散媒体、コーティング剤、抗細菌剤および抗真菌剤、等張剤および吸収遅延剤などが挙げられる。薬学的に活性な物質についてこのような媒体および薬剤の使用は、当該分野で周知である。任意の従来の媒体または薬剤が、この活性な化合物と不適合である範囲を除いて、治療組成物中でその使用が意図される。補助的な活性化合物もまた、この組成物中に組込まれ得る。
【0182】
好ましい薬学的に受容可能な希釈液として、生理食塩液および水性緩衝溶液が挙げられる。注射のために適切な薬学的組成物としては、滅菌水性溶液(水溶性である場合)または滅菌水性分散剤および滅菌注射用溶液または滅菌注射用分散液の即時調製のための滅菌粉末が挙げられる。等張剤(例えば、糖、ポリアルコール(例えば、マンニトール、ソルビトール)、塩化ナトリウム)は、薬学的組成物中に含まれ得る。全ての場合において、組成物は無菌でありかつ流体であるべきである。組成物は、製造および保存の条件下で安定であるべきであり、そして細菌および真菌のような微生物による汚染を予防する保存剤を含まなければならない。分散液はまた、グリセロール、液体ポリエチレングリコール、およびそれらの混合物中に、ならびに油中に調製され得る。保存および使用の通常の条件下で、これらの調製物は、微生物の増殖を防ぐための保存剤を含み得る。
【0183】
キャリアは、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、およびそれらの適切な混合物を含む、溶媒または分散媒体であり得る。適切な流動性は、例えば、レシチンのようなコーティングの使用によって、必要とされる粒子サイズの維持によって(分散液の場合)、および界面活性剤の使用によって、維持され得る。
【0184】
微生物作用の防止は、種々の抗細菌剤および抗真菌剤(例えば、パラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサールなど)によって達成され得る。
【0185】
注射可能な組成物の長期にわたる吸収は、吸収を遅延させる薬剤(例えば、モノステアリン酸アルミニウムおよびゼラチン)を組成物中に含ませることによってもたらされ得る。
【0186】
非経口組成物は、好ましくは、投与の容易さ、および投薬量の均一性のために投薬単位形態で処方される。投薬単位形態は、哺乳動物の被験体のための投薬単位として適切な物理学的に別個の単位を言い;各単位は、必要とされる薬学的キャリアと結合して所望の治療的効果を生成するように計算された予め決定された量の活性化合物を含む。本発明の投薬単位形態のための詳細は、(a)活性化合物の固有の特徴および達成されるべき特定の治療効果、ならびに(b)個体の感受性を処置するためのこのような活性化合物を配合する分野に固有の制限によって、決定され、そしてこれらに直接的に依存する。
【0187】
肺への注入のために、エアゾール化された溶液が使用される。噴霧可能なエアゾール調製物において、活性なタンパク質は、固体または液体の不活性キャリア材料との組合せで存在し得る。これはまた、スクイーズボトルにパッケージングされ得るか、加圧された揮発性(通常は気体の)プロペラントと混合してパッケージングされ得る。エアゾール調製物は、本発明のタンパク質に加えて溶媒、緩衝剤、界面活性剤、および抗酸化剤を含み得る。
【0188】
局所適用のために、本発明のタンパク質は、軟膏剤(salve)または軟膏剤(ointment)(これは、皮膚への平滑効果および罹患領域への活性成分の直接的な投与のための手段の両方を有する)のような局所適用ビヒクルに組込まれ得る。
【0189】
活性成分のためのキャリアは、噴霧可能な形態または噴霧不可能な形態のいずれであってもよい。噴霧不可能な形態は、局所適用に固有のキャリアを含み、かつ、好ましくは水の動的粘度よりも大きな動的粘度を有する半固形または固形であり得る。適切な処方物として、液剤、懸濁剤、乳剤、クリーム剤、軟膏剤(ointment)、散剤、リニメント剤、軟膏剤(salve)などが挙げられるが、これらに限定されない。所望である場合には、これらは、滅菌しても、補助剤(例えば、保存剤、安定化剤、湿潤剤、緩衝剤、または浸透圧に影響を与えるための塩など)と混合してもよい。噴霧不可能な局所的調製物のための好ましいビヒクルの例として、軟膏基剤(例えば、ポリエチレングリコール−1000(PEG−1000);HEBクリームのような従来のクリーム;ゲル;ならびにワセリンなどが挙げられる。
【0190】
本発明によるB7−DCポリペプチドのための他の薬学的に受容可能なキャリアは、リポソームであり、これは、活性なタンパク質が、脂質層に接着した水性の同心層からなる小体に分散しているか、またはその中に多様に存在しているかのいずれかで含まれる、の薬学的組成物である。活性なタンパク質は、好ましくは、水性層および脂質層の内側または外側(すなわち、いずれの事象においても、リポソーム懸濁液として一般的に公知の不均一な系)に存在する。疎水性の層、または脂質層は、一般的には(しかし、排他的にではない)、レシチンおよびスフィンゴミエリンのようなリン脂質、コレステロールのようなステロイド、多少イオン性の界面活性剤(例えば、ジセチルホスフェート、ステアリルアミンまたはホスファチジン酸)、および/または疎水性の性質の他の物質を含む。
【0191】
(B7−DCおよび複数の補助刺激分子を発現させるための腫瘍細胞の改変)
本発明の別の局面は、複数の補助刺激分子を発現するように改変された細胞(例えば、腫瘍細胞)である。活性化B細胞での補助刺激分子の時間的な発現は、B7、B7−2、およびB7−3で異なる。例えば、B7−2は、B細胞の活性化に続いて初期に発現されるが、B7−3は、後期に発現される。従って、この異なる補助刺激分子は、免疫応答の過程の間に、別個の機能を果たし得る。効果的なT細胞応答は、T細胞が複数の補助刺激分子から補助刺激シグナルを受けることを必要とし得る。
【0192】
従って、本発明は、遺伝的に改変されるか、または1つより多くの補助刺激分子を発現する腫瘍細胞を含む。例えば、腫瘍細胞を改変して、B7−DCと、B7、B7−2、およびB7−3のうちの1つ以上とを発現させ得る。
【0193】
改変の前に、細胞(例えば、腫瘍細胞)は、いずれかの補助刺激分子を発現しないかもしれず、または特定の補助刺激分子は発現するが、他の分子は発現しないかもしれない。本明細書中で記載されるように、腫瘍細胞は、B7−DCのみをコードする核酸または別の補助刺激分子とともにB7−DCをコードする核酸を用いるトランスフェクションによって改変され得る。例えば、B7−DCをコードする核酸でトランスフェトされた腫瘍細胞は、B7をコードする核酸でさらにトランスフェクトされ得る。ヒトB7−DCタンパク質またはマウスB7−DCタンパク質をコードするcDNA分子の配列は、それぞれ、配列番号1および配列番号3のコード部分である。あるいは、1より多くの型の改変が使用され得る。例えば、B7−DCをコードする核酸でトランスフェクトされた腫瘍細胞は、B7−1、B7−2、またはB7−3の発現を誘導する薬剤で刺激され得る。
【0194】
(病原体に関連する抗原)
本発明についての主な有用性は、癌および世界中で罹患率および死亡率を引き起こしている主な慢性的ウイルス感染症についての治療用ワクチンの本願組成物の使用である。このようなワクチンは、感染した細胞を排除する(これは、抗体が無効であるためにT細胞応答を必要とする)ように設計される。本発明のワクチンは、抗原性エピトープそれ自身に加えて:
(a)ベクター(例えば、裸の(naked)DNA、裸のRNA、自己複製RNAレプリコン)ならびにウイルス(ワクシニアウイルス、アデノウイルス、アデノ随伴ウイルス(AAV)、レンチウイルスおよびRNAアルファウイルスを含む);
(b)抗原標的化シグナルまたはプロセシングシグナル(例えば、HSP70)、カルレティキュリン、Flt−3リガンドの細胞外ドメイン、Pseudomonasの菌体外毒素ETAのドメインII、単純ヘルペスのVP22標的化タンパク質など(同一人に譲渡された米国特許出願第09/421,608号;同第09/501,097号;同第09/693,450号;同第60/222,9002号;同第60/222,985号;同第60/268,575号およびChang,W−Fら、J.Virol.75:2368−2376(2001)(これらは、その全体が参考として本明細書中で援用される);ならびに
(c)補助刺激シグナル、好ましくは本発明のB7−DCタンパク質またはその融合タンパク質、フラグメントもしくは機能的誘導体(単独でまたはB7.1、B7.2、可溶性CD40などのような他の公知の補助刺激タンパク質との組み合わせ)
を含む。
【0195】
腫瘍細胞または他の型の宿主細胞(APCを含む)は、所望の免疫応答に対する抗原をコードする核酸で形質転換されるか、トランスフェクトされるか、またはそうでなければ、形質導入される。このような抗原は、好ましくは、病原性微生物のエピトープであり、これに対して宿主は、エフェクターT細胞応答(細胞障害性Tリンパ球(CTL)および遅延型過敏症を含む)によって防御されている。代表的には、これらとして、ウイルス、マラリアのような細胞内寄生生物、およびミコバクテリアおよびリステリアのような細胞内で増殖する細菌が挙げられる。従って、本発明のワクチン組成物に含まれるこの型の抗原は、(もちろん、腫瘍特異性抗原に加えて)このような病原体に関連する抗原のいずれかである。いくつかのウイルス抗原がまた、ウイルスが癌の原因となる因子である場合の腫瘍抗原であることは注目すべきである。
【0196】
実際に、世界中で最も一般的な2つの癌、肝癌および子宮頚癌は、ウイルス感染と関連する。B型肝炎ウイルス(HBV)は、肝癌の病因となる因子として関連付けられてきた(Beasley,R.P.ら、Lancet 2、1129−1133(1981))。80〜90%の子宮頚癌は、以下の4つの「高度に危険な(ハイリスク)」ヒトパピローマウイルス型のうちの1つに由来するE6抗原およびE7抗原を発現する:HPV−16、HPV−18、HPV−31およびHPV−45(Gissmann,L.ら、Ciba Found Symp.120、190−207(1986);Beaudenon,S.ら、Nature 321、246−249(1986))。HPV E6抗原およびE7抗原は、免疫適格性個体におけるウイルス関連の癌のための最も有望な標的である。なぜなら、これらが、子宮頚癌において遍在的に発現されるからである。治療的癌ワクチンの標的としてのこれらの重要性に加えて、ウイルス関連腫瘍抗原はまた、予防的ワクチンのための理想的な候補である。実際に、アジアにおける予防的HBVワクチンの導入は、肝癌の発生率を減少させ、癌予防に対する大きな影響を示している(Chang,M.H.ら、New Engl.J.Med.336、1855−1859(1997))。
【0197】
慢性的なヒトウイルス感染において、とりわけ最も重要なウイルスは、ヒトパピローマウイルス(HPV)、B型肝炎ウイルス(HBV)、C型肝炎ウイルス(HCV)、ヒト免疫不全ウイルス(HIV)、エプスタイン−バーウイルス(EBV)および単純ヘルペスウイルス(HSV)である。
【0198】
ヒトの癌および感染性疾患へのその適用性に加えて、本発明はまた、獣医学的医薬の状況において動物の疾患の処置に使用することが意図される。従って、本明細書中で記載されるこのアプローチは、当業者によって容易に適用されて、獣医学的なヘルペスウイルス感染(ウマのヘルペスウイルス、ウシのヘルペスウイルス、ニワトリおよび他の家禽におけるマレク病ウイルスを含む);動物のレトロウイルス疾患;仮性狂犬病および狂犬病などを処置し得る。
【0199】
以下の参考文献は、基礎的な医学的および獣医学的なウイルス学の分野における原理ならびに現在の情報を示し、これらは、参考として援用される:Fields Virology、Fields,BNら編、Lippincott Williams & Wilkins、NY、1996;
Principles of Virology:Molecular Biology,Pathogenesis,and Control、Flint,S.J.ら編、Amer Society for Microbiology、Washington、1999;Principles and Practice of Clinical Virology,第4版、Zuckerman.A.J.ら編、John Wiley & Sons、NY、1999;The Hepatitis C Viruses、Hagedorn,CHら編、Springer Verlag、1999;Hepatitis B Virus:Molecular Mechanisms in Disease and Novel Strategies for Therapy、
Koshy,R.ら編、World Scientific Pub Co、1998;Veterinary Virology、Murphy,F.A.ら編、Academic Press、NY、1999;Avian Viruses:Function and Control、Ritchie,B.W.、Iowa State University Press、Ames、2000;Virus Taxonomy:Classification and Nomenclature of Viruses:Seventh Report of the International Committee on Taxonomy of Viruses、M.H.V.Van Regenmortel,MHVら編、Academic Press;NY、2000。
【0200】
(標的化分子)
種々の作用様式を有する多数のタンパク質が「標的化」分子として関連付けられてきた。この「標的化」分子は、抗原と共に(好ましくは、融合ポリペプチドとして)使用されて、細胞および細胞下区画へ抗原を標的化し、このことは、より強力かつ効果的な様式でT細胞に対する抗原提示を促進する。
【0201】
熱ショックタンパク質(HSP)への抗原の結合は、核酸に基づく(および他の)ワクチンの能力を増強するための潜在的アプローチを示す。HSPは、癌性ワクチン接種およびウイルス性ワクチン接種における天然の生物学的アジュバントとして作用するらしい。小胞体(ER)に常在するgp96 HSPおよび細胞質Hsp70は両方とも、免疫学的アジュバントとして作用する(Srivastava,PKら、Semin.Immunol.3、57−64(1991);Udono,Hら、Proc.Natl.Acad.Sci.USA 91、3077−3081(1994))。これらのHSPまたはシャペロニンは、広範なペプチドアレイに結合する(Lammert,E.ら、Eur.J.Immunol.27、923−927(1997))。Hsp70は、会合したタンパク質をプロテオソーム(MHCクラスI分子と会合するためのペプチドを生成する主な細胞性プロテアーゼ複合体)に標的化し得るシャペロニンである。従って、Hsp70に直接結合する抗原は、MHCクラスIによってより効率的に提示される(特に、CTL応答を生じる)。2つの特徴が、HSPのアジュバント活性の原因であるらしい:(1)インビトロでペプチドをロードされたgp96は、MHCクラスIプロセシング経路に抗原を効果的に導入する;(2)gp96のマクロファージへの結合は、炎症誘発性サイトカインの分泌を誘導し、従って、ペプチド性抗原が標的化されている細胞の機能を増強する。
【0202】
腫瘍またはウイルスに感染した細胞から単離されたHSP複合体での免疫は、強力な抗腫瘍免疫(Srivastava,PKら、Int J Cancer.33:417−22、1984;Srivastava,PKら、Proc Natl Acad Sci USA.83:3407−11、1986;Udono,Hら、J Immunol.152:5398−5403、1994;Blachere,NEら、J Immunother.14:352−6、1993;Udono,Hら(前出);Tamura,Yら、Science.278:117−20、1997;Janetzki,Sら、J Immunother.21:269−76、1998)、または抗ウイルス免疫(Heikema,Aら、Immunol Lett.57:69−74、1997;Suto,Rら、Science.269:1585−8、1995)を誘導する。ペプチドとHSPとをインビトロで混合することにより、免疫原性HSP−ペプチド複合体が生成された(Ciupitu,AMら、J Exp Med.187:685−91、1998;Blachere,NEら、J Exp Med.186:1315−22、1997)。HSPに基づくいくつかのタンパク質ワクチンは、HSPへの抗原の融合物を含んだ(Suzue,Kら、J Immunol.156:873−9、1996;Suzue,K.ら、Proc Natl Acad Sci USA 94:13146−51、1997)。より最近では、本発明者およびその同僚は、キメラDNAまたはRNAレプリコンワクチンの形態でHSPを使用した(例えば、Chen,C−Hら、Canc.Res.60:1035−1042(2000))。彼らは、Mycobacterium tuberculosis HSP70に融合させる抗原としてHPV−16 E7を使用し、そしてE7特異的CD8+T細胞の増加した増殖および活性化(これは、確立された腫瘍に対する強力な抗腫瘍免疫を生じた)を示した(Lin,K.−Y.ら、Cancer Res.56:21−26.、1996)。
【0203】
別の有用な標的化分子は、Pseudomonas菌体外毒素A(ETA)(例えば、ETAのドメインII(dII)(残基253〜364にわたる))のトランスロケーションドメインである。トランスロケーションドメインは、細胞の細胞質ゾルに結合するタンパク質またはポリペプチドのトランスロケーションを誘導するポリペプチドである。例えば、同様に適用可能なポリペプチドは、ジフテリア毒素、クロストリジウム属(botulinum、tetani)毒素、炭疽毒素、エルジニア属毒素、Vibrio choleraeの毒素、またはBordetella pertussis毒素由来である。毒素をコードするDNAの毒素ドメインは、好ましくは、このような組成物の調製物において変異または欠失されている。
【0204】
カルレティキュリン(CRT)は、レクチン活性を示し、そして新生糖タンパク質の折り畳みおよび組立てに関連することが公知である、小胞体(ER)腔に豊富に存在する46kDaのタンパク質である(Nash(1994)Mol.Cell.Biochem.135:71−78;Hebert(1997)J.Cell Biol.139:613−623;Vassilakos(1998)Biochemistry 37:3480−3490;Spiro(1996)J.Biol.Chem.271:11588−11594)。CRTは、抗原プロセシングに関連するトランスポーター(例えば、TAP−1およびTAP−2)によってERに輸送されたペプチドと会合する(Spee(1997)Eur.J.Immunol.27:2441−2449)。CRTは、インビトロでペプチドと複合体を形成する。これらの複合体は、マウスに投与された場合に、ペプチド特異的CD8+T細胞応答を惹起した(Basu(1999)J.Exp.Med.189:797−802;Nair(1999)J.Immunol.162:6426−6432)。マウス腫瘍から精製されたCTRは、CRTの供給源として使用される腫瘍に対して特異的な免疫を惹起したが、抗原的に別個の腫瘍に対しては免疫を惹起しなかった(Basu、前出)。ペプチドに結合するCRTを用いて、DCをインビトロでパルスすることによって、このペプチドは、DCクラスI分子の状況において再提示され、そしてペプチド特異的CTLを刺激した(Nair、前出)。
【0205】
Flt−3リガンドは、DC前駆体の増殖を刺激し、そしてインビボで多数のDCの生成を促進し得る(Maraskovsky,E.ら、J Exp Med.184:1953−62、1996;Shurin,MR.ら、Cell Immunol.179:174−84、1997)。Flt3は、マウスチロシンキナーゼレセプターであり(Rosnet,O.ら、Oncogene 6:1641−50、1991)、これは、第IIIレセプターキナーゼファミリーのメンバーである(概説については、Lyman,SD、Curr Opin Hematol.5:192−6、1998を参照のこと)。造血組織において、Flt3の発現は、CD34ポジティブ前駆体に制限される。Flt3を使用して、対応するリガンドであるFlt3−リガンドを同定し、そして引続いてクローン化した(Lyman,SDら、Cell 75:1157−67、1993;Hannum,Cら、Nature 368:643−8、1994)。Flt3−リガンドの主な形態は、膜貫通タンパク質として合成され、機能的に類似の可溶性ECDは、このタンパク質からタンパク質分解切断によって生成される(Lymanら、前出)。これらのタンパク質は、固有のチロシンキナーゼレセプターに結合し、そして活性化する。造血細胞の中で、Flt3レセプターの発現は、DC前駆体を含む最も未分化の前駆体細胞に主に限定される。Flt3−リガンドのECDは、いくつかのマウスモデル腫瘍(線維肉腫、乳癌、肝癌、肺癌、黒色腫およびリンパ腫を含む)に対する強力な抗腫瘍効果を生成した(Lynch,DHら、Nat Med.3:625−631、1997;Chen,Kら、Cancer Res.57:3511−3516、1997;Braun,SEら、Hum Gene Ther.10:2141−2151、1999;Peron,JMら、J Immunol.161:6164−6170、1998;Chakravarty,PKら、Cancer Res.59:6028−6032、1999;Esche,Cら、Cancer Res.58:380−383、1998)(19)。本発明者らの同僚らは、HPV Ek7タンパク質をコードするDNAを、Flt3−リガンドECDをコードするDNAに連結した。この構築物での免疫は、E7抗原特異的CD8+T細胞の増殖および活性化を劇的に増強し、確立されたE7発現転移性腫瘍に対する強力な抗腫瘍免疫を生じた。
【0206】
HSV−1タンパク質VP22は、数ある中で、その顕著な細胞内輸送特性が使用され得る(Elliott,G.およびP.O’Hare.1997.Cell 88:223−33)ことに起因して、抗原の伝播の増強に寄与するプロトタイプタンパク質である。例えば、p53に連結されたVP22(Phelan,A.ら,1998,Nat Biotechnol 16:440−443)またはチミジンキナーゼに連結されたVP22(Dilber,MSら,1999,Gene Ther 6:12−21)は、連結したタンパク質の、インビトロでの周囲の細胞への伝播およびモデル腫瘍の処置を容易にした。DNAワクチンの状況でHPV−16 E7抗原に連結されたVP22は、ワクチン接種したマウスにおいてE7特異的CD8+T細胞前駆体の数における劇的な増加(約50倍)を導き、そしてあまり有効でないDNAワクチンをE7発現腫瘍に対して顕著な効力を有するDNAワクチンへと変換させた。非伝播性VP22変異体は、ワクチン効力を増強することができなかった。VP22および同様の作用形態を有し得るタンパク質は、以下のいくつかの方法でワクチン効力の増強に寄与する:(1)トランスフェクトされた細胞から周囲のAPCへの抗原の伝播を容易にし、それによってMHCクラスI経路を通して抗原を提示するAPCの数を増加させる;(2)抗原を、トランスフェクトされた細胞において、より効率的に提示する;(3)「交叉プライム(crosspriming)」を行い、それによってVP22/抗原融合タンパク質の放出が、MHC−I拘束経路を介した提示のための、DC(または他のAPC)による取り込みおよびプロセシングを導く(Huang,AYら,1994,Science 264:961−965)。
【0207】
当業者は、本発明に従って使用するための病原体由来の関連タンパク質の適切なエピトープ(例えば、CTLエピトープ)をどのようにして同定するかがわかる。
【0208】
(細胞および動物へのB7−DC DNAの送達)
DNA送達(例えば、一般的に「遺伝子治療」として公知であるものをもたらすためのDNA送達)は、細胞への、そして最終的には生存動物への「外来」DNAの導入を含む。遺伝子治療についてのいくつかの一般的ストラテジーが研究されており、そして広範に概説されている(Yang,N−S.,Crit.Rev.Biotechnol.12:335−356(1992);Anderson,W.F.,Science 256:808−813(1992);Miller,A.S.,Nature 357:455−460(1992);Crystal,R.G.,Amer.J.Med.92(補遺6A):44S−52S(1992);Zwiebel,J.A.ら,Ann.N.Y.Acad.Sci.618:394−404(1991);McLachlin,J.R.ら,Prog.Nucl.Acid Res.Molec.Biol.38:91−135(1990);Kohn,D.B.ら,Cancer Invest.7:179−192(1989)(これらの参考文献は、その全体が、本明細書中に参考として援用される)。
【0209】
1つのアプローチは、培養中の初代細胞への核酸移入、続いてエキソビボで形質転換された細胞の、全身的または特定の器官もしくは組織へかのいずれかの、宿主への自己移植を含む。
【0210】
本発明の目的を達成するために、核酸治療は、インビボでの哺乳動物の体細胞組織または器官への機能的に活性なDNAの直接的移入によって達成される。DNA移入は、以下に記載の多数のアプローチを用いて達成され得る。これらの系は、選択マーカー(例えば、G418耐性)の使用によってインビトロでの好首尾の発現について試験されて、DNAを発現するトランスフェクトされたクローンが選択され得、続いて適切な免疫アッセイにおいてB7−DC発現産物に対する抗体を用いてのこの産物の存在が(誘導性系での場合はインデューサーでの処置後に)検出され得る。この手順(DNA取り込み、プラスミド組み込みおよび組み込まれたプラスミドの安定性を含む)の効率は、公知の方法を用いたプラスミドDNAの直鎖化および高分子量哺乳動物DNAを「キャリア」として用いた同時トランスフェクションによって改善され得る。
【0211】
当該分野で報告される好首尾の「遺伝子移入」の例としては、以下が挙げられる:(a)マウス筋肉組織へのプラスミドDNAの直接注入(これは、無期限にわたってマーカー遺伝子の発現を導いた)(Wolff,J.A.ら,Science 247:1465(1990);Acsadi,G.ら,The New Biologist 3:71(1991));(b)レトロウイルスベクターは、インビボおよびインサイチュでの血管組織の感染に有効である;(c)レトロウイルス調製物の門脈注射および肝臓への直接注射は、遺伝子移入およびインビボでの発現をもたらした(Horzaglou,M.ら,J.Biol.Chem.265:17285(1990);Koleko,M.ら,Human Gene Therapy 2:27(1991);Ferry,N.ら,Proc.Natl.Acad.Sci.USA 88:8387(1991));(d)肺組織への組換えアデノウイルスの気管内注入は、肺の気道上皮における外来遺伝子のインビボでの移入および長期発現に有効であった(Rosenfeld,M.A.ら,Science 252:431(1991);(e)単純ヘルペスウイルスベクターは、脳組織へのインビボでの遺伝子移入を達成した(Ahmad,F.ら編,Miami Short Reports−Advances in Gene Technology:The Molecular Biology of Human Genetic Disease,第1巻,Boerringer Manneheim Biochemicals,USA,1991)。
【0212】
レトロウイルス媒介ヒト治療は、両栄養性の複製欠損レトロウイルス系を利用する(Temin,H.M.,Human Gene Therapy 1:111(1990);Teminら,米国特許4,980,289;Teminら,米国特許4,650,764;Teminら,米国特許第5,124,263号;Wills,J.W.米国特許5,175,099;Miller,A.D.,米国特許第4,861,719号)。このようなベクターは、機能的DNAをヒトの細胞または組織へと(例えば、アデノシンデアミナーゼ遺伝子をリンパ球へと、NPT−II遺伝子および腫瘍壊死因子遺伝子を腫瘍浸潤性リンパ球へと)導入するために用いられている。レトロウイルス媒介遺伝子送達は一般に、遺伝子移入のために標的細胞の増殖を必要とする(Miller,D.G.ら,Mol.Cell.Biol.10:4239(1990))。この条件は、本発明のDNA分子が導入されるべき特定の好ましい標的細胞(すなわち、活発に増殖する腫瘍細胞)によって満たされる。多数の方法のうちのいずれかを用いたプラスミドによる、およびレトロウイルスベクターによる、トランスフェクションを用いた嚢胞性線維症の遺伝子治療は、Collinsら,米国特許5,240,846によって記載されている。
【0213】
B7−DC配列をコードするDNA分子は、当該分野において周知であるように、複製欠損レトロウイルスを産生するパッケージング細胞株を用いてレトロウイルスベクター中にパッケージングされ得る(例えば、Cone,R.D.ら,Proc.Natl.Acad.Sci.USA 81:6349−6353(1984);Mann,R.F.ら,Cell 33:153−159(1983);Miller,A.D.ら,Molec.Cell.Biol.5:431−437(1985);Sorge,J.ら,Molec.Cell.Biol.4:1730−1737(1984);Hock,R.A.ら,Nature 320:257(1986);Miller,A.D.ら,Molec.Cell.Biol.6:2895−2902(1986)を参照のこと)。遺伝子移入について効率的でかつ安全な、より新しいパッケージング細胞株もまた記載されている(Bankら,U.S.5,278,056)。
【0214】
このアプローチは、レトロウイルスベクターを選り抜きの組織または器官へと送達するために部位特異的様式で利用され得る。従って、例えば、カテーテル送達系が使用され得る(Nabel,EGら,Science 244:1342(1989))。レトロウイルスベクターまたはリポソームベクターのいずれかを使用するこのような方法は、発現されるべき核酸を血管壁へと、または腫瘍の血液循環物へと送達するために特に有用である。
【0215】
以下を含めた他のウイルスベクターもまた使用され得る:組換えアデノウイルス(Horowitz,M.S.,In:Virology,Fields,BNら編,Raven Press,New York,1990,1679頁;Berkner,K.L.,Biotechniques 6:616−919,1988),Strauss,S.E.,In:The Adenoviruses,Ginsberg,HS編,Plenum Press,New York,1984,第11章)、ニューロン特異的送達および持続性のための単純ヘルペスウイルス(HSV)。ヒトの遺伝子治療のためのアデノウイルスベクターの利点としては、組換えは稀であり、このようなウイルスと関連するヒトの悪性疾患は知られておらず、アデノウイルスゲノムは7.5kbの大きさまでの外来遺伝子を受け入れるように操作され得る二本鎖DNAであり、そして生のアデノウイルスは、安全なヒトワクチン生物であるという事実が挙げられる。アデノ随伴ウイルスもまた、本発明によるヒトの治療のために有用である(Samulski,R.J.ら,EMBO J.10:3941(1991)。
【0216】
本発明のDNA分子を発現し得、そして(特にヒトにおける)本発明の治療設定において有用である別のベクターは、ワクシニアウイルスであり、ワクシニアウイルスは、非複製性にされ得る(米国特許5,225,336号;同5,204,243号;同5,155,020号;同4,769,330号;Sutter,Gら,Proc.Natl.Acad.Sci.USA(1992)89:10847−10851;Fuerst,T.R.ら,Proc.Natl.Acad.Sci.USA(1989)86:2549−2553;Falkner F.G.ら;Nucl.Acids Res(1987)15:7192;Chakrabarti,Sら,Molec.Cell.Biol.(1985)5:3403−3409)。組換えワクシニアウイルスおよび異種DNAを含む他のウイルス、ならびに免疫およびDNA治療におけるそれらの使用の説明は、以下に概説されている:Moss,B.,Curr.Opin.Genet.Dev.(1993)3:86−90;Moss,B.Biotechnology(1992)20:345−362;Moss,B.,Curr Top Microbiol Immunol(1992)158:25−38;Moss,B.,Science(1991)252:1662−1667;Piccini,Aら,Adv.Virus Res.(1988)34:43−64;Moss,B.ら,Gene Amplif Anal(1983)3:201−213。
【0217】
裸のDNAもしくはRNAまたはウイルスベクターに加えて、操作された細菌はベクターとして使用され得る。以下を含む多数の細菌株:Salmonella、BCGおよびListeria monocytogenes(LM)(HoisethおよびStocker,Nature 291,238−239(1981);Poirier,TPらJ:Exp.Med.168,25−32(1988);(Sadoff,J.C.ら,Science 240,336−338(1988);Stover,C.K.ら,Nature 351,456−460(1991);Aldovini,A.ら,Nature 351,479−482(1991);Schafer,R.ら,J.Immunol.149,53−59(1992);Ikonomidis,G.ら,J.Exp.Med.180,2209−2218(1994))。これらの生物は、ワクチンベクターとしての使用のために有望な以下の2つの特徴を提示する:(1)経口ワクチン送達の可能性を提供する、腸感染経路;および(2)単球/マクロファージの感染(それによってプロフェッショナルAPCへと抗原を標的化する)。
【0218】
インビボでのウイルス媒介遺伝子移入に加えて、以下を含めて、当該分野で周知の物理的手段は、DNAの直接移入のために用いられ得る:プラスミドDNAの投与(Wolffら,1990,前出)およびパーティクル−ボンバードメント媒介遺伝子移入(Yang,N.−S.ら,Proc.Natl.Acad.Sci.USA 87:9568(1990);Williams,R.S.ら,Proc.Natl.Acad.Sci.USA 88:2726(1991);Zelenin,A.V.ら,FEBS Lett.280:94(1991);Zelenin,A.V.ら,FEBS Lett.244:65(1989);Johnston,S.A.ら,In Vitro Cell.Dev.Biol.27:11(1991))。さらに、インビトロで細胞へと遺伝子を移入するための周知の手段であるエレクトロポレーションを用いて、本発明によるDNA分子をインビボで組織へと移入し得る(Titomirov,A.V.ら,Biochim.Biophys.Acta 1088:131((1991))。
【0219】
「キャリア媒介遺伝子移入」もまた記載されている(Wu,C.H.ら,J.Biol.Chem.264:16985(1989);Wu,G.Y.ら,J.Biol.Chem.263:14621(1988);Soriano,P.ら,Proc.Natl.Acad.Sci.USA 80:7128(1983);Wang,C−Y.ら,Proc.Natl.Acad.Sci.USA 84:7851(1982);Wilson,J.M.ら,J:Biol.Chem.267:963(1992))。好ましいキャリアは、標的化されたリポソーム(Nicolau,C.ら,Proc.Natl.Acad.Sci.USA 80:1068(1983);Sorianoら,前出)(例えば、免疫リポソーム(これは、アシル化mAbを脂質二重層へと取り込み得る(Wangら,前出)))である。アシアロ糖タンパク質/ポリリジン(Wuら,1989,前出)のようなポリカチオンが用いられ得、ここで、結合体は、標的組織を認識する分子(例えば、肝臓についてはアシアロオロソムコイド)およびトランスフェクトされるべきDNAに結合するDNA結合化合物を含む。ポリリジンは、DNAに損傷を与えることなくDNAを結合するDNA結合分子の一例である。次いで、この結合体は、移入のために本発明に従ってプラスミドDNAと複合体化される。
【0220】
トランスフェクションまたはマイクロインジェクションのために用いられるプラスミドDNAは、当該分野で周知の方法を用いて(例えば、Quiagen手順(Quiagen)を用いて)調製され得、続いてDNAが、公知の方法(例えば、本明細書中に例示される方法)を用いて精製され得る。
【0221】
さらに、上記のように、本発明による形質導入されたB7−DC分子の有用性については、安定な発現を必要としないかもしれない。むしろ、ポリペプチドの一過性の発現は、形質導入された細胞がその免疫原性機能および/または補助刺激機能を果たすために充分であり得る。
【0222】
ここで、本発明を一般的に記載してきたが、本発明は、以下の実施例を参照してより容易に理解される。以下の実施例は、例示のために提供され、明記しない限り、本発明を限定することを意図しない。
【実施例】
【0223】
(実施例I)
(材料および方法)
(細胞の調製および培養)
6〜12週齢の雌性BALB/cマウスをNCIから購入し、そしてDCおよびマクロファージの調製のために用いた。
【0224】
骨髄由来DCを、以前に記載された(26)ように、5%ウシ胎仔血清(FCS)(Hyclone)、ペニシリン/ストレプトマイシン(JRH Biosciences)、ゲンタマイシン(Sigma)、非必須アミノ酸(JRH Biosciences)、L−グルタメート(JRH Biosciences)、ピルビン酸ナトリウム(Sigma)、2メルカプトエタノール(Sigma)および1000単位/ml組換えマウスGM−CSF(Immunex)を補充したRPMI1640(Gibco BRL)培地中で培養した。8日目の骨髄由来DCを、従来法によってモノクローナル抗体で染色した。MHCクラスIIに対するモノクローナル抗体14−4−4を、ハイブリドーマ上清から精製した。Johns Hopkins UniversityのWilliam Baldwin博士は親切なことに、CTLA4−Ig融合分子を供給した。MHCクラスIについての抗体(28−14−8)、F4/80についての抗体(Cl.A3−1)、B7.1についての抗体(1G10)、B7.2についての抗体(GL1)、FcγRII/IIIについての抗体(2.4G2)およびMac−1についての抗体(M1/70)を、PharMingenから購入した。テスターcDNA調製のために、8日目の細胞を、Johns Hopkins University Oncology Centerで14−4−4sおよびCTLA4−Igを用いてセルソーターによって精製した。選別後のMHCクラスIIhiおよびB7hi集団の純度は93%〜98%であった。
【0225】
骨髄由来マクロファージを、10% FCS、ペニシリン/ストレプトマイシン、非必須アミノ酸、ピルビン酸ナトリウム、Lグルタミン、2−メルカプトエタノールおよび250単位/ml組換えマウスM−CSFを補充したRPMI−1640培地で培養し、そして以前に記載された(27)通りにこれらを500単位/mlのγ−IFN(Pharmingen)および5μg/ml LPS(Sigma)で処理した。刺激後、MHCクラスIIおよびB7の細胞表面発現を、培養10日目のフローサイトメトリー分析を用いて確認した。
【0226】
マクロファージ細胞株WEHI−3、RAW264.7、J774.A.1、PU5−1.8は、親切なことに、NIAID,National Institutes of HealthのJoshua Farber博士によって提供された。これらを、ATCCの推奨する培地で培養した。
【0227】
(同種混合リンパ球反応)
MHCクラスIIhiおよびB7hiと特徴付けされた8日目のBM由来DCを、MLC中の同種T細胞を刺激する能力について試験した。MLC反応を、3×105同種C57BL/6リンパ球へ漸増数のBALB/c刺激細胞を添加することによって、96ウェル平底マイクロプレートにおいて行った。3日間の培養後、T細胞増殖を、最後の18時間の培養にわたる、各ウェルへの1μCiの[3H]−メチル−チミジン(Amersham)の添加によって評価した。次いで、細胞を収集し、そして放射能の取り込みを、β計数管(Packard 96)を用いて決定した。
【0228】
(cDNA差引きハイブリダイゼーション)
選別したDCおよび活性化したマクロファージからの総RNAを、TRIZOL(Gibco BRL)を用いて抽出した。メッセンジャーRNAを、Oligotex mRNA精製キット(Qiagen)によって精製した。本発明者らは、PCRベースのSMART cDNA合成系(Clonetech)を用いてcDNAを増幅し、続いてPCRベースの差引き系PCR Select(Clonetech)を用いた。差引きを、製造業者のプロトコルに従って行った。最後の差引きPCR後、DNAフラグメントを、プラスミドベクターpCR2.1(Invitrogen)またはpCR Blunt(Invitrogen)に連結した。形質転換後、各クローンを、プラスミドDNA増幅およびミニプレップDNAのために増殖させ、次いでEcoRIで消化して挿入物の存在を確認した。次いで、プラスミドドットブロットを行って、このcDNAクローンが樹状細胞特異的であることを確認した。アルカリ変性ミニプレップDNAをHybond N+メンブレン(Amersham)にスポットし、そして選別されたDCまたは活性化されたマクロファージ由来のSMART cDNAプローブとハイブリダイズさせた。これらのcDNAプローブを、ランダムプライマー標識法(Stratagene Prime−It II)を用いて32P標識した。ハイブリダイゼーションおよび洗浄を、以前に記載された(28)通りに行った。メンブレンをフィルム(Amersham)に1日間〜2日間にわたって暴露し、そして現像した。
【0229】
(プラスミドドットブロット分析)
アルカリ変性ミニプレップDNAサンプルを、Hybond N+メンブレン(Amersham)にスポッティングし、選別されたDCまたは活性化したマクロファージから誘導されたSMART(登録商標)cDNAプローブとハイブリダイズさせた。これらのcDNAプローブを、ランダムプライマー標識法(Stratagene Prime−It II)を用いて32P標識した。ハイブリダイゼーションおよび洗浄を、以前に記載された通りに行った。オートラジオグラフィーについては、メンブレンを、フィルム(Amersham)に1〜2日間にわたって暴露し、そして現像した。
【0230】
(cDNAライブラリーの構築およびスクリーニング−B7−DCのクローニング)
骨髄由来DCを8日目に選別をせずに収集した。これらの細胞の約20%〜40%は、高いMHCクラスIIおよびB7を発現した。総RNA抽出、続いてポリA RNA精製を上記に記載された通りに行った。オリゴdTプライムDCライブラリー構築については、本発明者らは、λZAP Express cDNA合成系(Stratagene)を用いた。B7−DCのPCR DNAフラグメントをプローブし、そしてスクリーニングのために用いた。メンブレン転移、変性、再生を、Stratageneのプロトコルを用いて行った。プローブの放射性標識、ハイブリダイゼーション、洗浄およびオートラジオグラフィーを、上記に記載の通りに行った。陽性クローンを単離し、そして2回目のスクリーニングを行った。2回目のスクリーニングの後、プラスミドをインビボ切り出しによって切り出し、そしてドットブロッティングおよび配列決定によって試験した。配列決定を、Johns Hopkins University School of MedicineのCore Facilityによって行った。BLASTプログラムを用いて、以前に報告された遺伝子に対する類似性について、このヌクレオチド配列の、Genbank(NCBI)に対する相同性検索を行った。全長のB7−DC cDNAクローンを、DC cDNAライブラリーから取り出した。5’RACEを、SMART RACE cDNA増幅キット(Clontech)を用いて行った。5’−RACE産物をpCR2.1ベクター中にクローニングし、そして配列決定した。さらに2つの全長B7−DCクローンをRT−PCRによって入手し、そしてそれらの配列を比較して、配列の誤りを回避した。
【0231】
ヒトB7−DCを以下の通りにクローニングした:ヒトDCを、以前に記載(29)の通りのGM−CSF+IL−4またはGM−CSF+Flt−3Lのいずれかにおける培養によって正常末梢血単核細胞から入手した。RNAを上記の通りに抽出した。BLAST検索は、マウスB7−DCに対する相同性を有する、GenBank登録番号AK001879の重複するESTクローンを同定した。5’RACEを、上記の通りに行った。本発明者らは、5’−RACE PCRフラグメントを配列決定し、そしてヒトB7DCの5’−UTRに対応するプライマーを設計した。B7−DCの5’−UTRおよび3’−UTRにおける以下のプライマーを用いて、全長ヒトB7−DCを増幅した:
【化6】
ヒトおよびマウスのB7−DC cDNAの全長cDNA配列は、EMBL/GenBank/DDBSに登録番号AF329193およびAF142780の下で寄託された。
【0232】
(BAC(129SVJ)ライブラリーのスクリーニング/ゲノムクローニングおよびマッピング)
BACライブラリーのスクリーニングは、製造業者(Genome Systems,Inc.)のプロトコルに従った。以下のプライマーを用いた:
【化7】
BACライブラリーのスクリーニングによって、3つの陽性クローンが得られた。染色体位置決定マッピングを、蛍光インサイチュハイブリダイゼーション(Genome Systems Inc.)によって行った。合計80個の分裂中期細胞を分析し、79個が特異的標識を示した。ヒトB7−DCマッピングを、入手可能なバイオインフォーマティックツールであるNCBIのBLASTプログラムおよびInternational RH Mapping Consortiumを用いることによって行った。hB7−DCの配列をhtsgにおいて検索し、そして第9染色体に局在する2つのBACクローンRP11−574F11(AL162253)およびRp11−635N21(AL354744)にマッピングされることが見い出された。
【0233】
(仮想ノーザンブロッティング)
4〜6週齢の雌性Balb/cマウスをNCIから購入し、そして組織RNA調製のために用いた。組織、選別されたDCおよび活性化されたマクロファージについてのRNA抽出およびSMART cDNA合成を、上記の通りに行った。SMART PCR cDNAを、PCR精製キット(Qiagen)によって精製した。0.5μg/レーンの精製DNAを1%アガロースゲルに泳動し、そしてNytranナイロンメンブレン(Schleier and Schuell)に移した。放射性プローブを作製するために、本発明者らは、差引きしたライブラリーから誘導されたプラスミドDNAをテンプレートとして増幅した。本発明者らは、プラスミドDNAのクローニング部位にすぐ隣接したプライマーセットを用いるPCRによってDNAを増幅し、そしてクローンの各々の精製したPCR DNAをハイブリダイゼーションプローブのために用いた。これらのプライマーのヌクレオチド配列は以下の通りである。
【化8】
ヒトDCおよびコントロールの胎盤の総RNAの仮想のノーザン分析もまた行った。用いたプローブおよびRNA調製は、上に記載された。プローブの放射性標識、ハイブリダイゼーション、洗浄およびオートラジオグラフィーを、上記の通りに行った。
【0234】
(ハムスター抗mB7−DC Ab産生)
DC2.4細胞株、RAW246.7細胞株およびRENCA細胞株中のB7−DCの安定なトランスフェクタントを用いて、Armenian Hamsterを免疫した。B7−DCを、改変したpCAGGSベクター(30)中にクローニングした。このハムスターを、B7−DC(Rockland)を含むプラスミドで3回追加免疫した。この研究において用いた抗B7−DC抗体は、免疫した3匹のハムスターのうちの1匹の血清由来であった。
【0235】
(CD28−Ig、CTLA4−IgおよびPD−1−Igの結合アッセイ)
293T細胞を、Lipofectamine 2000(Gibco BRL)を用いてB7.1−pCAGGS、B7−DC−pCAGG、PD−1−pCAGGSまたはベクター単独でトランスフェクトした。24時間後、細胞をFACS緩衝液(1×HBSS、2%仔ウシ血清、10M HEPESおよび0.1% NaN3)中に再懸濁し、そして1000rpmで5分間、4℃で回転させた。次いで、この緩衝液を棄て、抗体をチューブに添加し、4℃で20分間インキュベートし、FACS緩衝液で2回洗浄し、そしてこれを二次抗体について繰り返した。サンプルをFACScanに泳動した。B7.1抗体を、1:5希釈、10μl/サンプル(Gal−Tag)で用いた。組換えCD28−Ig、CTLA−4−IgおよびPD−1−Igキメラを、2μg/ml、10μl/サンプル(R&D System,Inc)で用いた。ヤギF(ab’)2抗ヒトIgG−PEを、1:20希釈(Southern Biotechnology Associates,Inc.)で用いた。
【0236】
(B7−DC−Igダイマー合成)
B7−DC−Ig構築物を、膜貫通ドメインを含まないB7−DCのN末端アミノ酸をコードする配列を、pIg−Tail Plusベクター(R&D systems)中のヒトIgG1 FcのC末端アミノ酸をコードする配列に対してインフレームで融合することによって作製した。COS−7細胞を、LipofectAMINE 2000(GIBCO BRL)またはGINE JAMMER(Stratagene)を用いてpIg/B7−DCで一過性にトランスフェクトした。B7−DC−Ig融合タンパク質を、飽和硫酸アンモニウム沈澱を用いて無血清上清から精製した。SDS−PAGEおよび銀染色は、90%を超える純度を実証した。
【0237】
(T細胞増殖およびサイトカインアッセイ)
抗CD3を用いた補助刺激アッセイについて、96ウェル平底プレート(Dynex製のImmulon 4)を、1×PBS(Gibco)pH7.4中に希釈した100ng/mlの抗CD3抗体(2C11,Pharmingen)およびB7.1−Ig(R&D System)、B7−DC−IgまたはIsotypeコントロール(Sigma)で37℃で2時間にわたって予めコーティングした。次いで、これらのプレートを1×PBSで3回洗浄し、そして10% FCS、ペニシリン/ストレプトマイシン、非必須アミノ酸、ピルビン酸ナトリウム、Lグルタミン、2−メルカプトエタノールを補充したRPMI1640培地で30分間にわたってブロッキングし、その後、T細胞を添加した。脾臓およびリンパ節を、6〜10週齢のBALB/cマウスから入手した。RBCをACK緩衝液を用いて溶解し、そしてT細胞を、間接的方法で、ダイナビーズ(dynabeads)M−280(Dynex)を用いて精製した。このビーズをPBS(pH7.4)+1% FCSで2回洗浄し、その後、この細胞を添加し、そして抗IEdおよびB220/CD45ROまたはCD8α(Pharmingen)から構成される抗体カクテルを、この細胞に添加し、そして4℃で30分間にわたって二方向性混合を用いてインキュベートした。この細胞を、Dynal MPC中のチューブに5分間配置することによって単離し、1500rpmで5分間遠心分離し、そしてPBS(pH7.4)+1% FCSで2回洗浄して未結合のAbを除去した。同じ手順を15分間のインキュベーションを用いて繰り返し、そしてこの細胞を2×105細胞/ウェルでプレーティングした。72時間のインキュベーション後、10μlの3H−チミジン(1μCi/ウェル)を各ウェルに添加し、そして18時間にわたってインキュベートした。細胞をPackard Micromate Cell Harvesterを用いて収集し、そしてフィルターをPackard Matrix 96直接β計数管で読み取った。
【0238】
HA抗原を提示するためのRENCA系を用いた補助刺激アッセイについては、RENCA細胞を、10% FCS、ペニシリン/ストレプトマイシン、非必須アミノ酸、ピルビン酸ナトリウムを補充したRPMI−1640培地で培養した。これを、MHCクラスII発現について、IFN−γ(75U/ml)を用いて72時間にわたって誘導した。次いで、これらを13,200Radで照射し、そして2×104細胞/ウェル(96ウェル平底プレート)でプレーティングした。次いで、HA110−120ペプチドを2.5μg/ウェルで添加し、そして種々の濃度のIg−融合分子を添加した。トランスジェニックI−Ed+HA特異的T細胞(H.von Boehmer,Harvard Universityの親切な贈与物)を上記の通りに単離し、そして4×105細胞/ウェルでプレーティングした。48時間のインキュベーション後、10μlの3Hチミジン(1μCi/ウェル)を各ウェルに添加し、そして18時間にわたってインキュベートした。細胞を、Packard Micromate Cell Harvesterを用いて収集し、そしてフィルターをPackard Matrix 96 Directβ計数管で読み取った。
【0239】
ELISAによるサイトカイン産生の分析について、培養物を上記の通りに設定し、そして上清を指定の時間で収集した。IL−2、IL−10およびIFN−γの濃度を、市販のELISAキット(Endogen)ならびにIL−4およびIL−6(R&D System)を用いて決定した。
【0240】
(インビボでの補助刺激)
B10.D2遺伝子バックグランド上のI−Ed拘束HAエピトープ(110SFERFEIFPKE120[配列番号12])を認識するTCRを発現するTCRトランスジェニックマウス系統6.5由来のプールされた腋窩、鼡径、頚部、および腸間膜のLNを、RPMI培地(GIBCO BRL)中で解離し、100μmのナイロン細胞ストレイナーを通過させ、そして滅菌ハンクス緩衝液(GIBCO BRL)中で洗浄した。FACS(登録商標)染色して、クローン型(clonotypic)CD4細胞の割合を決定した後、0.2mlの滅菌ハンクス緩衝液中に2.5×106クローン型細胞を含む細胞調製物を、レシピエントB10.D2マウスの尾静脈に静脈内(i.v.)注射した。この養子移入から3日後、この動物を、後足蹠(footpad)に皮下(s.c.)注射することによりワクチン接種した。各マウスに、以下の3つの調製物のうちの1つを両側に注射した:
(A)10μgの合成HA(足蹠1つにつき)(HAペプチド(110−120)を1:1のv/v比で不完全フロイントアジュバント(IFA)(Sigma)と合わせた)
(B)HA−IFAと25μgのB7−DC−Igとの混合物、または
(C)HA−IFAと25μgのアイソタイプコントロール抗体との混合物。7日後、排出LN節を採取し;1.5×105LN細胞を丸底96ウェル組織培養プレート中で示された濃度の合成HAペプチドとともにインキュベートした。48時間培養物を1μCi[3H]チミジンでパルスし、そしてさらに12時間インキュベートした後採取し、そして組み込まれた放射活性の量を決定することにより増殖アッセイを行った。
【0241】
(実施例II:B7−DCの同定および特徴づけ)
B7−DCを、DCと活性化マクロファージとの間の差引きライブラリーから単離した。cDNA差引きに用いた2つの集団は、「テスター」集団としてのGM−CSFと培養した骨髄由来DC、および「ドライバー」集団としてのγ−インターフェロン+LPS活性化した接着性骨髄由来M−CSFマクロファージであった。8日目に、MHCクラスIIhiB7hi「成熟」DCを、テスターcDNAの供給源として、>93%純度までソートした。DCを、フローサイトメトリーにより、マクロファージより約50倍MHCクラスIIレベルが高いと特徴付けた。両方の集団がB7.1およびB7.2を発現したが、B7.2レベルはDCにおいて有意に高かった。F4/80およびCD16は、マクロファージ集団で高レベルで発現された。2つの集団の機能的比較により、DC集団は、同種異系の混合したリンパ球反応を刺激することにおいてマクロファージ集団より約100倍強力であることが実証された。両方の集団からRNAを抽出した後、本発明者らは、PCRベースのcDNA合成システム、続いてPCRベースの差引き手順であるPCR Selectを用いた。差示的に発現されるクローンのうちの1つが、新規な免疫グロブリンスーパー遺伝子ファミリーのメンバー(本発明者らは、これをB7−DCと名付けた)をコードした。マウスB7−DC cDNAは、長さが約1.7kbであり、これは、23アミノ酸(aa)のN末端シグナルペプチドを有し、そして推定分子量が約25kdの247アミノ酸(aa)の前駆体タンパク質をコードする(表1)。推定リーダー配列および膜貫通ドメインを、SOSUIプログラム(31)を用いて同定した。2つの荷電aaは、mB7−DCの23aa膜貫通ドメイン内に見出される。このことは、あり得る結合パートナーを示唆する。aaレベルで、マウスB7−DCは、ヒトB7−DCと70%同一であり、このことは、それらがオルソログであることを示す(以下の表を参照のこと)。hB7−DCは、より長い細胞質テイルを有するという点で、マウスB7−DCとはわずかに異なる。
【0242】
相同性検索を介して、B7−DCがB7−H1に対して有意な相同性を有し(34%同一性、48%類似性)(表2)、より低い程度には、ブチロフィリン(butyrophilin)に対して(30%同一性、45%類似性)、そしてB7.1およびB7.2に対して20%未満の同一性を有することが見出された(表3)。系統発生的な研究により、エキソンシャッフリング(32,33)を介して、ブチロフィリンがB7スーパーファミリーに関連する可能性が高いことが示される。これらは、各々、標準的な(canonical)IgV−IgC構造および膜貫通ドメインを有する。他のB7ファミリーのメンバーとは対照的に、マウスB7−DCは、非常に短い細胞質テイル(4aa)を有する。
【0243】
(表1)
マウスB7−DCおよびヒトB7−DCのアミノ酸配列比較。mB7−DCの推定リーダードメインおよび膜貫通ドメインは、上に線を引いた。アラインメントは、Clustalw−Gonnet Pam250マトリクスを用いて行った。[*]は、同一アミノ酸を示し、[:]は、保存的置換を示す。免疫グロブリンVまたはCドメイン内部のジスルフィド結合の形式に重要であり得るシステイン残基は、斜体にされている。
【表1】
(表2:mB7−DCおよびmB7−H1のアミノ酸配列比較)
【表2】
(表3:B7−DCとB7ファミリーメンバーとのアミノ酸配列比較)
【表3】
NCBI blast2検索を用いて比較を行った(マトリクスBLOSUM62)。
1.対応する位置での同一アミノ酸
2.対応する位置での類似アミノ酸−以下のようにグループ分けされる:(A,G);(S,T);(E,D);(R,K,H);(Q,N);(V,I,L,M);(Y,F);(W);(C);(P)。
3.マトリクスBLOSUM62を用いて有意な類似性は見出されなかった。
4.BT=ブチロフィリン5.=PDL−1。
【0244】
mB7−DCのゲノム構造を決定するために、本発明者らは、5’および3’UTRに由来するプローブを用いてプールされたBacterial Artificial Chromosome(BAC)ライブラリーをスクリーニングすることによりゲノムクローンを単離した。染色体位置のマッピングを、BACクローンを用いて行った。B7−DCの染色体位置は、蛍光インサイチュハイブリダイゼーション(FISH)を用いて行った。10の特異的に染色した第19染色体の測定により、mB7−DCが、第19染色体の異質染色質−真性染色質境界からテロメアまでの距離(19C2と19C3のバンドの間の境界に対応する領域)の47%である位置に位置することが実証された。ハイブリダイズしたスライドをフルオレセイン化抗ジゴキシゲニン抗体中でインキュベートし、続いてDAPIで対比染色することにより、特異的ハイブリダイゼーションシグナルを検出した。この遺伝子座は、ヒト第9染色体の領域に対応し、この領域においてhB7−H1がマッピングされている。
【0245】
hB7−DCは2つの第9染色体のBACクローンに位置することが見出された。さらに、hB7−DCおよびhB7−H1はともに、約164kbの挿入物サイズを有する、単一の第9染色体のBACクローンに位置することが見出された(図1)。B7−DCおよびB7−H1がゲノム上で近位にあることは、B7.1/B7.2対を暗示し、この対は、互いに1メガ塩基以内にマッピングされる。
【0246】
(実施例III:B7−DCは樹上細胞において選択的に発現される)
B7−DCの発現パターンを決定するために、複数の組織、マクロファージ細胞株、マクロファージ培養物、ならびに骨髄および脾臓の両方から得られた樹上細胞から抽出したRNAを用いて、事実上のノザンブロッティングを行った。未熟(4日目および6日目)および成熟(8日目およびソートされたMHC IIhiB7hi)の骨髄由来DCおよび脾臓由来DCにおいてB7−DCプローブを用いて強いハイブリダイゼーションが検出されたが、4つのマクロファージ細胞株、活性化BMマクロファージ、腹膜マクロファージのいずれにおいてもシグナルは検出されなかった(図2)。GM−CSF+IL−4またはGM−CSF+Flt−3Lのいずれかを用いて末梢血単球から増殖させたヒトDCにおいてhB7−DCの強い発現が検出された(図3)。B7−DCタンパク質の細胞表面発現を確認するために、抗mB7−DC抗体を用いて、DCを染色した。可溶性B7−DC−Igを用いた染色および遮断が、DCで観察された(図4)。
【0247】
(B7−DCは、CD28にもCTLA−4にも結合しないが、PD−1には結合する)
B7−DCはB7ファミリーに対して構造的相同性および配列相同性を有するが、推定CD28/CTLA−4結合配列、SQDXXXELY(配列番号13)も、XXXYXXRT(配列番号14)も含まない(34)(ここで、X=任意のアミノ酸)。結合を直接評価するために、2量体CD28−IgおよびCTLA−4−Igが、B7−DCまたはB7.1のいずれかでトランスフェクトされた293T細胞を染色する能力を調査した。B7.1トランスフェクタントで強い結合が観察された一方、B7−DCトランスフェクタントに対する結合は観察されなかった(図5)。B7−DCとB7−H1/PD−L1との間の相同性およびゲノム上で近位であることに基づいて、B7−DCについての候補結合パートナーとしてPD−1を試験するために実験を行った。実際に、PD−100IGは、B7−DCトランスフェクタントに結合したが、B7.1トランスフェクタントには結合しなかった。PD−1−IgのB7−DCトランスフェクタントへの結合は、CTLA−IgおよびCD28−IgのB7.1トランスフェクタントへの結合より低かったが、特異的であった。PD−1のB7−DCへの結合のさらなる確認を、本発明者らは、PD−1−Igとの安定なB7−DC−GFPトランスフェクタントのポジティブな染色から得た。B7−H1およびB7h/B7RP−1と同様に、B7−DCは、レセプターとしてCD28もCTLA−4も用いないことが結論付けられた。むしろ、PD−1は、B7−DCについてのレセプターであるようである。
【0248】
(実施例IV:B7−DCは、T細胞についての補助刺激分子として機能する)
T細胞刺激アッセイに添加され得る可溶性B7−DC−Ig融合タンパク質を、B7−DCが補助刺激活性を有するか否かを試験することにおける使用のために生成した。T細胞の増殖応答を、B7−DC−Ig、B7.1−Igまたはアイソタイプコントロールいずれかの存在下で、漸増量の固定化抗CD3による刺激に対して測定した。図6(左)は、最適未満の抗CD3濃度の存在下で、B7−DCがB7.1より大きなT細胞増殖応答を補助刺激したことを示す。さらに、B7−DCにより補助刺激された増殖応答は、CD8細胞よりCD4細胞において高かった(図6、右)。B7−DCは、TCRに集中した刺激がない場合にはT細胞を刺激できなかった。このことは、B7−DCが、真の補助刺激シグナルを提供することを示す。
【0249】
B7−DCはまた、「シグナル1」がMHCペプチド複合体により提供された場合に増殖応答を補助刺激した。RENCA細胞(RT−PCR分析により内因性B7.1、B7.2、またはB7−DCを発現しない)を、γ−IFNで処理して、MHCクラスII発現を誘導した。これらの細胞に、I−Ed拘束HA110−120ペプチド(FERFEIFPKE)(35)(配列番号15)を負荷した。I−Ed+HA110−120特異的TCRトランスジェニックマウス系統から精製した脾臓T細胞を添加し、そして増殖応答をB7−DC−Ig、B7.1−Igまたはアイソタイプコントロールいずれかの存在下で測定した。図7は、B7−DCがB7.1より大きな補助刺激活性を有したことを示す。
【0250】
(B7−DCにより補助刺激されたリンホカイン生成のパターン)
B7ファミリー分子による補助刺激に対して最もよく特徴付けられたT細胞応答は、リンホカイン生成である。これらのリンホカインは、T細胞効果の重要なメディエーターである。抗CD3またはHA抗原で刺激し、B7−DC−Ig、B7.1−Igまたはアイソタイプコントロールいずれかで補助刺激したT細胞による多くの異なるリンホカイン生成を分析するための研究を行った(図8)。リンホカイン補助刺激のパターンは、抗CD3またはMHCペプチド複合体が「シグナル1」として利用されるか否かとかなり一致した。重大なことには、B7−DCは、B7.1より大きなレベルのγ−IFNを補助刺激した。B7−DCはまた、有意な量のIL−6生成を補助刺激したのに対し、B7.1は、実質的に補助刺激しなかった。両方の分子が、IL−2生成を補助刺激したが、B7.1は、B7−DCより非常に効率的に刺激した。従って、B7−DCおよびB7.1の補助刺激のパターンは異なり、B7−DCは重要な前炎症性リンホカインを補助刺激することにおいて効率的である。
【0251】
(実施例V:B7−DCは、インビボ免疫応答を増強する)
B7−DCがインビボで生物学的活性を有するか否かを決定するために、本発明者らは、B7−DC−Igがペプチドワクチンに対する免疫応答を増強するか否かを調べた。B7−DC−Igまたはアイソタイプコントロール抗体を、HA110−120ペプチドおよびIFAの免疫原性カクテルに添加した。HA特異的CD4T細胞のインビボでの計数を可能にするために、2.5×106抗HA6.5T細胞を免疫の3日前にマウスに移入した。免疫して7日後、排出LN細胞を採取し、そしてこの細胞をインビトロで2日間、種々の量のHA110−120ペプチドで刺激した。図9は、B7−DC−Igの添加が、実際にHAに対する増殖応答を劇的に増強したことを示す。排出LNにおけるHA特異的T細胞の総数は、アイソタイプ抗体コントロールに対して、B7−DC−Igを受けた群において約2倍増加した。従って、B7−DCが1細胞あたりですら抗原特異的応答を増強する能力を有することが結論付けられた。
【0252】
(実施例VI:考察および結論)
本発明者らは、DCに高度に拘束された発現を伴い、T細胞について独特な補助刺激特性を有する新たなB7ファミリーメンバーを発見し、そして特徴づけた。B7−DCのヒトオルソログもまた、DCにおいて発現される。
【0253】
この拘束された発現パターンは、先に記載されたB7ファミリーメンバーと対照をなし、このことは、B7−DCが、公知のB7.1/2経路とは異なる免疫応答に関与することを示唆する。弱いB7−DCシグナルが、活性化マクロファージにおいてRT−PCRにより検出されたが、予備のリアルタイムRT−PCR分析により、DCにおけるB7−DC mRNA発現が、活性化マクロファージより15倍高いことが示された。同様に、抗体染色により、活性化マクロファージの表面で非常に低いレベルのB7−DCが検出された。このことが、有意なT細胞活性化に十分か否かは明瞭ではない。
【0254】
B7−DCが補助刺激するリンホカイン生成の異常なパターンは、他のB7ファミリーメンバーと比較して、独特の生物学的役割を示す。サイトカインの伝統的な分類は、以下のとおりである:Th1サイトカインは、IL−2、γ−IFNおよびリンホトキシンを含む;Th2サイトカインは、IL−4、IL−5、IL−6およびIL−13を含む(36)。B7−DCは、いずれの古典的なTh1リンホカインプロフィールもTh2リンホカインプロフィールも誘導しない。B7−DCは、非常にわずかなIL−4を誘導し、IL−10を誘導しない。しかし、IL−6は、Th2サイトカインと考えられる。B7.1に対して、B7−DCにより補助刺激されたより低いIL−2およびより高いγ−IFNは、古典的なTh1パターンに合わない。にもかかわらず、高いγ−IFN生成は、B7−DCが重要なT細胞エフェクター機能を誘起することを示唆する。
【0255】
B7−DCは、IL−6を補助刺激する能力において著しい。B7−DCにより誘導されたT細胞の活発な増殖性応答は、B7.1では観察されないIL−6生成の強い補助刺激により一部説明される。IL−6は、(他の増殖刺激と関連して)T細胞増殖の強力な増幅因子(amplifier)である(37,38)。IL−6は、T細胞機能のみならず、前炎症性応答、単球分化、B細胞分化、血小板新生、骨の吸収、および特定の造血性腫瘍の増殖もまた調節する多機能性サイトカインである(39,40)。IL−6は、ケモカインおよび白血球動員の誘導において、可溶性IL6レセプター(sIL−6R)と協同して機能し得る(41)。これは、Stat−3活性化を介して強力な抗アポトーシス効果を媒介し得る。T細胞におけるIL−6依存性Stat−3活性化は、活性化T細胞の生存についての重要な経路であると報告されている(42,43)が、他の報告では、Stat−3が休止T細胞に対してその効果を発揮することが示唆されている。
【0256】
B7−DCは、CD28もCTLA−4も結合できないが、PD−1(B7−H1/PD−L1についてのレセプター)を結合する(22,47,48)。B7−DCが、ICOS(B7h/B7RP−1についてのレセプター)を結合するか否かは未だ決定されていない(23−25,44−46)。B7−DCとB7−H1/PDL−1との間の顕著な相同性(B7.1とB7.2との間の相同性より大きい)、hB7−H1/PD−L1とhB7−DCとの密接な物理的連鎖、およびそれらの共通するレセプターへの結合により、これらが比較的最近の遺伝子重複事象に関連することが示唆される。このことは、B7.1とB7.2との間の関係と同様であり、B7.1およびB7.2の両方が、マウス第16染色体およびヒト第3染色体上の1メガ塩基内にマッピングする(49)。
【0257】
B7−H1/PD−L1に対するB7−DCの相対的生物学的役割が、PD−1および他の推定レセプターにより媒介されると認識することは重要である。PD−1は、T細胞活性化に続いて発現され、そしてT細胞活性化を阻害するようである。PD−1は、高濃度の抗CD−3でのT細胞刺激条件下でアポトーシスを誘導する。PD−1ノックアウトマウスは、肥大型心筋症の臨床的発現により特徴付けられる自己免疫症候群を発症する(22)。対照的に、Dongら(21)は、B7−H1/PD−L1が、低濃度の抗CD−3でT細胞増殖およびサイトカイン増殖を補助刺激することを報告した。CD28/CTLA−4の関係に類推して、PD−L1は、未だ同定されていない活性化レセプターについての対レセプター(counterreceptor)であり得る。PD−1に結合するという特性を共有しているにもかかわらず、B7−DCおよびB7−H1は、それらのリンホカイン補助刺激パターンが異なる;B7−H1は、T細胞のIL−10生成を補助刺激するのに対し、B7−DCはIL−10生成を補助刺激しない。B7−DCの異なる細胞発現パターンおよび補助刺激機能は、免疫機能における独特の役割を示唆する。
【0258】
上記および以下に引用された参考文献は、これらが具体的に援用されているか否かにかかわらず、全て本明細書中に参考として援用される。
【0259】
今や本発明を十分に記載してきたが、本発明が、広範な等価なパラメーター、濃度、および条件の範囲内で、本発明の趣旨および範囲から逸脱することなく、そして過度な実験なくして実施され得ることは当業者に明らかである。
【0260】
本発明は、その具体的な実施形態と関連付けて記載してきたが、さらなる改変が行われ得ることは理解される。本出願は、概して、本発明の原理に従って、そして本発明が属する分野において公知または慣用的な実施の範囲内であり、および添付の特許請求の範囲の範囲に入るように本明細書中で先に記載された本質的特徴に適用され得る、本開示からのこのような逸脱を含む、本発明の任意のバリエーション、使用または適合を含むことが意図される。
【0261】
(引用文献)
本願明細書に完全に列挙される文書に加えて、特定の文書は番号のみによって列挙(挿入)され、以下に列挙される。
配列表
【特許請求の範囲】
【請求項1】
B7−DCと称される哺乳動物タンパク質をコードする単離された核酸分子であって、該タンパク質は、活性化したマクロファージと比較して樹状細胞上に選択的に発現される、核酸分子。
【請求項2】
配列番号1または配列番号5より選択されるヌクレオチド配列を含む、請求項1に記載の核酸分子。
【請求項3】
ストリンジェントなハイブリダイゼーション条件下で、請求項1または2に記載の核酸分子とハイブリダイズする、単離された核酸分子。
【請求項4】
配列番号1のヌクレオチド配列を含む、請求項2に記載の核酸分子。
【請求項5】
請求項1〜4のいずれか1項に記載の核酸分子であって、該核酸分子は、配列番号2および配列番号4より選択されるアミノ酸配列を有するタンパク質をコードするか、または該タンパク質の生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体をコードする、核酸分子。
【請求項6】
請求項5に記載の核酸分子であって、該核酸分子は、配列番号2の配列を有するタンパク質をコードするか、または配列番号2の生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体をコードする、核酸分子。
【請求項7】
前記B7−DCタンパク質の細胞外ドメインの全てまたは一部をコードする、請求項5または6に記載の核酸分子の、フラグメント。
【請求項8】
B7−DC融合ポリペプチドをコードする、請求項5に記載の核酸分子であって、該分子は、以下:
(a)配列番号2または配列番号4のB7−DCタンパク質の全てまたは一部である、第一のポリペプチドをコードする、第一の核酸配列;
(b)必要に応じて、該第一の核酸配列とインフレームで融合される、リンカーポリペプチドをコードする、リンカー核酸配列;および
(c)該第一の核酸配列または該リンカー核酸配列にインフレームで連結されかつ第二のポリペプチドをコードする、第二の核酸配列、
を含む、核酸分子。
【請求項9】
前記第二のポリペプチドがIg重鎖定常領域の1以上のドメインからなる、請求項8に記載の核酸分子。
【請求項10】
前記第二のポリペプチドがヒトIgG定常領域の2つのドメインからなる、請求項8に記載の核酸分子。
【請求項11】
請求項1〜10のいずれか1項に記載の核酸を含む発現ベクターであって、以下:
(a)プロモーター、および
(b)必要に応じて、真核生物細胞において該核酸の発現を調節する、さらなる調節配列、
に作動可能に連結された、発現ベクター。
【請求項12】
プラスミドである、請求項11に記載の発現ベクター。
【請求項13】
ウイルスベクターである、請求項11に記載の発現ベクター。
【請求項14】
ベクター組成物であって、以下:
(a)第一の組み換え体発現ベクターであって、その核酸中に、免疫応答が誘導される目的の抗原をコードするヌクレオチド配列が組み込まれている、ベクター;および
(b)第二の組み換え体発現ベクターであって、その核酸中に、補助刺激ポリペプチドをコードする1以上のヌクレオチド配列が組み込まれており、ここで、該補助刺激ポリペプチドの少なくとも1つは、B7−DCであるか、またはその生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体である、ベクター、
を含み、ここで、該発現ベクターは、抗原と、該補助刺激ポリペプチド、フラグメント、ホモログもしくは誘導体との同時発現を生じる宿主細胞に同時感染され得るかまたは同時トランスフェクトされ得る、ベクター組成物。
【請求項15】
前記第二のベクターが配列番号1または配列番号5を含む、請求項14に記載のベクター組成物。
【請求項16】
前記第二のベクターが配列番号1を含む、請求項14または15に記載のベクター組成物。
【請求項17】
請求項1〜6のいずれか1項に記載の核酸分子を用いて形質転換またはトランスフェクトされる、細胞。
【請求項18】
請求項11〜16のいずれか1項に記載の発現ベクターを用いて形質転換またはトランスフェクトされる、細胞。
【請求項19】
哺乳動物細胞である、請求項18に記載の細胞。
【請求項20】
ヒト細胞である、請求項19に記載の細胞。
【請求項21】
樹状細胞またはその前駆体である、請求項19に記載の細胞。
【請求項22】
腫瘍細胞である、請求項19に記載の細胞。
【請求項23】
外因性核酸分子でトランスフェクトされた、単離された哺乳動物腫瘍細胞であって、該核酸分子は、哺乳動物B7−DCタンパク質またはその生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体をコードし、その結果、該タンパク質、フラグメント、ホモログまたは誘導体は、該腫瘍細胞によって発現され、そして該腫瘍細胞は、T細胞と接触され、
(i)該B7−DCタンパク質、フラグメント、ホモログまたは誘導体は、該T細胞に結合し;そして
(ii)該腫瘍細胞は、該T細胞を補助刺激して増殖ならびに/またはサイトカインを生成および分泌する、
腫瘍細胞。
【請求項24】
前記外因性核酸分子は、配列番号1もしくは配列番号5の配列、または該配列のフラグメントを含む、請求項23に記載の腫瘍細胞。
【請求項25】
ポリペプチドであって、活性化されたマクロファージと比較して樹状細胞上に選択的に発現されかつ以下の機能的特性:
(a)T細胞上の結合パートナーに結合すること;および
(b)増殖ならびに/またはサイトカインを生成および分泌するようにT細胞を補助刺激すること、
を有する、ポリペプチド。
【請求項26】
配列番号1もしくは配列番号5の配列を有する核酸分子、または該核酸分子のフラグメント、ホモログもしくは等価物によってコードされる、請求項25に記載のポリペプチド、フラグメント、ホモログまたは機能的誘導体。
【請求項27】
請求項26に記載のポリペプチドであって、該ポリペプチドは、配列番号2もしくは配列番号4のアミノ酸配列、または該ポリペプチドのフラグメント、ホモログもしくは等価物を有する、ポリペプチド。
【請求項28】
請求項1〜10のいずれか1項に記載の核酸の組み換え体発現によって生成される、ポリペプチド、または該ポリペプチドの生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体。
【請求項29】
B7−DCタンパク質の細胞外ドメインを含む、請求項25〜28のいずれか1項に記載のポリペプチドであって、該ドメインは、配列番号2もしくは配列番号4のアミノ酸残基26〜221、または該ドメインの補助刺激フラグメント、ホモログもしくは他の機能的誘導体を含む、ポリペプチド。
【請求項30】
B7−DCの細胞外ドメインを実質的に含む、請求項25〜28のいずれか1項に記載のポリペプチドであって、該ドメインは、配列番号2もしくは配列番号4のアミノ酸残基26〜221、または残基26〜221の補助刺激フラグメント、ホモログもしくは他の機能的誘導体を含む、ポリペプチド。
【請求項31】
B7−DCタンパク質の全てまたは一部を含む第一の融合パートナーを有する、B7−DC融合ポリペプチドであって、該B7−DCタンパク質は:
(i)第二のポリペプチドに直接融合されるか、または
(ii)必要に応じて、該第二のポリペプチドに融合されるリンカーペプチド配列に融合される、
B7−DC融合ポリペプチド。
【請求項32】
第二のポリペプチドに融合される請求項25〜30のいずれか1項に記載のポリペプチドを含む、B7−DC融合ポリペプチド。
【請求項33】
請求項32に記載の融合ポリペプチドであって、T細胞上の結合パートナー分子に結合し、そしてT細胞レセプターに対する十分な刺激の存在下で該T細胞を補助刺激する、融合ポリペプチド。
【請求項34】
前記結合パートナー分子がPD−1である、請求項33に記載の融合ポリペプチド。
【請求項35】
前記第二のポリペプチドがIg重鎖定常領域の1以上のドメインである、請求項31〜33のいずれか1項に記載の融合ポリペプチド。
【請求項36】
請求項31〜35のいずれか1項に記載の融合ポリペプチドであって、前記第一の融合パートナーは、配列番号2もしくは配列番号4のアミノ酸26〜221、またはその補助刺激フラグメント、ホモログもしくは他の機能的誘導体を含む、融合ポリペプチド。
【請求項37】
請求項36に記載の融合ポリペプチドであって、前記第一の融合パートナーは、配列番号2のアミノ酸26〜221、またはその補助刺激フラグメント、ホモログもしくは他の機能的誘導体を含む、融合ポリペプチド。
【請求項38】
ヒト免疫グロブリンCγ1鎖のヒンジ領域、CH2領域およびCH3領域に対応するアミノ酸配列を有する、請求項35に記載の融合ポリペプチド。
【請求項39】
請求項31〜38のいずれか1項に記載の融合ポリペプチドの直列に連結されたダイマーまたはトリマーである、ダイマー融合ポリペプチドまたはトリマー融合ポリペプチド。
【請求項40】
末端と末端とで、直接にか、または前記モノマーの反復の間に存在するリンカー配列を有して連結された、前記第一の融合パートナーのモノマーが2以上反復する直鎖状マルチマーを含む、請求項31〜38のいずれか1項に記載の融合ポリペプチド。
【請求項41】
B7−DCタンパク質のエピトープに特異的である抗体であって、該エピトープは、B7ファミリータンパク質の既知のメンバー中には存在しない、抗体。
【請求項42】
前記エピトープは、配列番号2または配列番号4のポリペプチドの線状エピトープまたは立体的エピトープである、請求項41に記載の抗体。
【請求項43】
前記エピトープは、配列番号2のポリペプチドの線状エピトープまたは立体的エピトープである、請求項42に記載の抗体。
【請求項44】
モノクローナル抗体である、請求項41または42に記載の抗体。
【請求項45】
ヒトモノクローナル抗体またはヒト化モノクローナル抗体である、請求項43に記載の抗体。
【請求項46】
細胞集団中の、その表面上にB7−DCポリペプチドを発現する細胞を同定または定量化する方法であって、該方法は、以下の工程:
(a)該集団の細胞を、請求項41〜45のいずれか1項に記載の抗体と接触させて、その結果、該抗体が該エピトープを発現する細胞に結合する、工程;および
(b)該抗体が結合する細胞の存在を評価する工程、または該抗体が結合する細胞の数を定量化する工程、
を包含する、方法。
【請求項47】
その表面上にB7−DCポリペプチドを発現する細胞を細胞集団から単離する方法であって、該方法は、以下の工程:
(a)該集団の細胞を、請求項41〜45のいずれか1項に記載の抗体と接触させて、その結果、該抗体が該エピトープを発現する細胞に結合する、工程;
(b)該抗体が結合している細胞をポジティブに選択する工程、または該抗体が結合していない細胞をネガティブに選択する工程、
を包含し、これにより該細胞を単離する、方法。
【請求項48】
サンプル中の、B7−DCポリペプチド、フラグメントもしくはホモログの存在を検出するか、またはB7−DCポリペプチド、フラグメントもしくはホモログを定量化する方法であって、該方法は、以下の工程:
(a)該サンプルを、請求項41〜45のいずれか1項に記載の抗体と接触させて、その結果、該抗体が該エピトープを保有する任意のポリペプチドまたはフラグメントに結合する、工程;および
(b)該抗体に結合する該ポリペプチドもしくはフラグメントの存在を検出する工程、または該抗体に結合する該ポリペプチドもしくはフラグメントを定量化する工程、
を包含する、方法。
【請求項49】
T細胞レセプターに対する十分な刺激の存在下にて、インビトロまたはインビボでT細胞を補助刺激する抗原提示細胞またはその前駆体の能力を増加するために、該抗原提示細胞またはその前駆体中にB7−DCポリペプチドの発現を誘導するかまたは増加する方法であって、該方法は、該抗原提示細胞またはその前駆体細胞を請求項11〜13のいずれか1項に記載の発現ベクターを用いて形質転換またはトランスフェクトし、その結果、該B7−DCポリペプチドの発現が該細胞上で誘導または増加される工程を包含する、方法。
【請求項50】
T細胞レセプターに対する十分な刺激の存在下にて、インビトロまたはインビボでT細胞を補助刺激する抗原提示細胞またはその前駆体の能力を増加するために、該抗原提示細胞またはその前駆体中にB7−DCポリペプチドの発現を誘導するかまたは増加する方法であって、該方法は、該抗原提示細胞またはその前駆体細胞を請求項11〜13のいずれか1項に記載の発現ベクターを用いて形質転換またはトランスフェクトし、その結果、該B7−DCポリペプチドの発現が該細胞上で誘導または増加される工程を包含する、方法。
【請求項51】
前記抗原提示細胞は樹状細胞であり、そして前記前駆体は樹状細胞前駆体である、請求項49または50に記載の方法。
【請求項52】
抗原性刺激に対する哺乳動物被験体のT細胞応答を増加する方法であって、該方法は、請求項17〜24にいずれか1項に記載の、有効量の細胞を、抗原とともに該被験体に投与する工程を包含し、ここで、該細胞は、該抗原性刺激に対する該被験体のT細胞応答を増加するために効果的である、方法。
【請求項53】
腫瘍関連抗原を用いる抗原性刺激に対する哺乳動物被験体のT細胞応答を増加する方法であって、該方法は、請求項22〜24にいずれか1項に記載の、有効量の腫瘍細胞を、該被験体に投与する工程を包含し、ここで、該腫瘍細胞は、該抗原を発現し、該腫瘍細胞の投与は、該腫瘍抗原刺激に対する該被験体のT細胞応答を増加するために効果的である、方法。
【請求項54】
抗原性刺激に対する哺乳動物被験体のT細胞応答を増加する方法であって、該方法は、請求項25〜30にいずれか1項に記載の、有効量のポリペプチド、フラグメント、ホモログまたは機能的誘導体を、抗原とともに該被験体に投与する工程を包含し、ここで、該ポリペプチドの投与は、該抗原性刺激に対する該被験体のT細胞応答を増加するために効果的である、方法。
【請求項55】
抗原性刺激に対する哺乳動物被験体のT細胞応答を増加する方法であって、該方法は、請求項31〜40にいずれか1項に記載の、有効量の融合ポリペプチドを、抗原とともに該被験体に投与する工程を包含し、ここで、該ポリペプチドの投与は、該抗原性刺激に対する該被験体のT細胞応答を増加するために効果的である、方法。
【請求項56】
抗原性刺激に対する哺乳動物被験体のT細胞応答を阻害する方法であって、該方法は、請求項41〜45にいずれか1項に記載の、有効量の抗体を、該被験体に投与する工程を包含し、ここで、該抗体の投与は、T細胞の刺激をブロックするかまたは抗原反応性T細胞を排除して、それによりT細胞応答を阻害するために効果的である、方法。
【請求項57】
前記T細胞応答は、組織移植物または自己抗原に対して指向され、その結果、移植物拒絶または自己免疫反応を阻害する、請求項56に記載の方法。
【請求項58】
抗原性刺激に対する哺乳動物被験体の免疫応答を増加する方法であって、該方法は、以下の工程:
(a)(i)該被験体、(ii)該被験体に対して免疫学的に適合可能なドナー、または(iii)免疫学的に適合可能であるかまたは受容可能な培養された細胞株、よりT細胞を得る工程;
(b)該T細胞を、請求項17〜24のいずれか1項に記載の、有効量の細胞とエキソビボで接触させる工程であって、ここで、該接触させる工程は、抗原性刺激に対する該T細胞の応答を増加するために効果的である、工程;および
(c)該被験体に工程(b)の該T細胞を投与する工程、
を包含し、これにより、該被験体の該免疫応答を増加する、方法。
【請求項59】
抗原性刺激に対する哺乳動物被験体の免疫応答を増加する方法であって、該方法は、以下の工程:
(a)(i)該被験体、(ii)該被験体に対して免疫学的に適合可能なドナー、または(iii)免疫学的に適合可能であるかまたは受容可能な培養された細胞株、よりT細胞を得る工程;
(b)該T細胞を、請求項17〜24のいずれか1項に記載の、有効量の細胞とエキソビボで接触させる工程であって、ここで、該接触させる工程は、抗原性刺激に対する該T細胞の応答を増加するために効果的である、工程;および
(c)該被験体に工程(b)の該T細胞を投与する工程、
を包含し、これにより、該被験体の該免疫応答を増加する、方法。
【請求項60】
抗原性刺激に対する哺乳動物被験体の免疫応答を増加する方法であって、該方法は、以下の工程:
(a)(i)該被験体、(ii)該被験体に対して免疫学的に適合可能なドナー、または(iii)免疫学的に適合可能であるかまたは受容可能な培養された細胞株、よりT細胞を得る工程;
(b)該T細胞を、請求項25〜30のいずれか1項に記載の、有効量のポリペプチド、フラグメント、ホモログまたは機能的誘導体とエキソビボで接触させる工程であって、ここで、該接触させる工程は、抗原性刺激に対する該T細胞の応答を増加するために効果的である、工程;および
(c)該被験体に工程(b)の該T細胞を投与する工程、
を包含し、これにより、該被験体の該免疫応答を増加する、方法。
【請求項61】
抗原性刺激に対する哺乳動物被験体の免疫応答を増加する方法であって、該方法は、以下の工程:
(a)(i)該被験体、(ii)該被験体に対して免疫学的に適合可能なドナー、または(iii)免疫学的に適合可能であるかまたは受容可能な培養された細胞株、よりT細胞を得る工程;
(b)該T細胞を、請求項31〜40のいずれか1項に記載の、有効量の融合ポリペプチドとエキソビボで接触させる工程であって、ここで、該接触させる工程は、抗原性刺激に対する該T細胞の応答を増加するために効果的である、工程;および
(c)該被験体に工程(b)の該T細胞を投与する工程、
を包含し、これにより、該被験体の該免疫応答を増加する、方法。
【請求項62】
病原細胞または病原微生物に関連する抗原に対する保護免疫応答を誘導するために有用なワクチン組成物であって、以下:
(a)必要に応じて該抗原を発現する、請求項17〜24のいずれか1項に記載の形質転換された細胞またはトランスフェクトされた細胞;
(b)(a)の細胞が該抗原を発現しない場合、該抗原のさらなる供給源;
(c)必要に応じて、一般的な免疫刺激剤またはアジュバント;ならびに (d)(a)、(b)および(c)のための薬学的および免疫学的に受容可能な賦形剤またはキャリア、
を含む、ワクチン組成物。
【請求項63】
病原細胞または病原微生物に関連する抗原に対する保護免疫応答を誘導するために有用なワクチン組成物であって、以下:
(a)免疫応答が所望される抗原の供給源;
(b)請求項25〜30のいずれか1項に記載のポリペプチド、フラグメント、ホモログまたは機能的誘導体;
(c)必要に応じて、一般的な免疫刺激剤またはアジュバント;ならびに
(d)(a)、(b)および(c)のための薬学的および免疫学的に受容可能な賦形剤またはキャリア、
を含む、ワクチン組成物。
【請求項64】
病原細胞または病原微生物に関連する抗原に対する保護免疫応答を誘導するために有用なワクチン組成物であって、以下:
(a)免疫応答が所望される抗原の供給源;
(b)請求項31〜40のいずれか1項に記載の融合ポリペプチド;
(c)必要に応じて、一般的な免疫刺激剤またはアジュバント;ならびに
(d)(a)、(b)および(c)のための薬学的および免疫学的に受容可能な賦形剤またはキャリア、
を含む、ワクチン組成物。
【請求項65】
抗原またはワクチンの免疫原性を増加するために、抗原またはワクチンと使用するための補助刺激組成物であって、以下:
(a)請求項25〜30のいずれか1項に記載のポリペプチド、フラグメント、ホモログまたは機能的誘導体;ならびに
(b)薬学的および免疫学的に受容可能な賦形剤またはキャリア、
を含む、補助刺激組成物。
【請求項66】
抗原またはワクチンの免疫原性を増加するために、抗原またはワクチンと使用するための補助刺激組成物であって、以下:
(a)請求項31〜40のいずれか1項に記載の融合ポリペプチド;ならびに
(b)薬学的および免疫学的に受容可能な賦形剤またはキャリア、
を含む、補助刺激組成物。
【請求項67】
請求項62〜64のいずれか1項に記載の、有効量のワクチン組成物を、被験体に投与する工程を包含する、哺乳動物被験体において抗原に対する免疫応答を誘導または増強する方法。
【請求項68】
請求項65または66に記載の補助刺激組成物を、抗原またはワクチンとともに被験体に投与する工程を包含する、哺乳動物被験体において抗原またはワクチンに対する免疫応答を増強する方法。
【請求項1】
B7−DCと称される哺乳動物タンパク質をコードする単離された核酸分子であって、該タンパク質は、活性化したマクロファージと比較して樹状細胞上に選択的に発現される、核酸分子。
【請求項2】
配列番号1または配列番号5より選択されるヌクレオチド配列を含む、請求項1に記載の核酸分子。
【請求項3】
ストリンジェントなハイブリダイゼーション条件下で、請求項1または2に記載の核酸分子とハイブリダイズする、単離された核酸分子。
【請求項4】
配列番号1のヌクレオチド配列を含む、請求項2に記載の核酸分子。
【請求項5】
請求項1〜4のいずれか1項に記載の核酸分子であって、該核酸分子は、配列番号2および配列番号4より選択されるアミノ酸配列を有するタンパク質をコードするか、または該タンパク質の生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体をコードする、核酸分子。
【請求項6】
請求項5に記載の核酸分子であって、該核酸分子は、配列番号2の配列を有するタンパク質をコードするか、または配列番号2の生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体をコードする、核酸分子。
【請求項7】
前記B7−DCタンパク質の細胞外ドメインの全てまたは一部をコードする、請求項5または6に記載の核酸分子の、フラグメント。
【請求項8】
B7−DC融合ポリペプチドをコードする、請求項5に記載の核酸分子であって、該分子は、以下:
(a)配列番号2または配列番号4のB7−DCタンパク質の全てまたは一部である、第一のポリペプチドをコードする、第一の核酸配列;
(b)必要に応じて、該第一の核酸配列とインフレームで融合される、リンカーポリペプチドをコードする、リンカー核酸配列;および
(c)該第一の核酸配列または該リンカー核酸配列にインフレームで連結されかつ第二のポリペプチドをコードする、第二の核酸配列、
を含む、核酸分子。
【請求項9】
前記第二のポリペプチドがIg重鎖定常領域の1以上のドメインからなる、請求項8に記載の核酸分子。
【請求項10】
前記第二のポリペプチドがヒトIgG定常領域の2つのドメインからなる、請求項8に記載の核酸分子。
【請求項11】
請求項1〜10のいずれか1項に記載の核酸を含む発現ベクターであって、以下:
(a)プロモーター、および
(b)必要に応じて、真核生物細胞において該核酸の発現を調節する、さらなる調節配列、
に作動可能に連結された、発現ベクター。
【請求項12】
プラスミドである、請求項11に記載の発現ベクター。
【請求項13】
ウイルスベクターである、請求項11に記載の発現ベクター。
【請求項14】
ベクター組成物であって、以下:
(a)第一の組み換え体発現ベクターであって、その核酸中に、免疫応答が誘導される目的の抗原をコードするヌクレオチド配列が組み込まれている、ベクター;および
(b)第二の組み換え体発現ベクターであって、その核酸中に、補助刺激ポリペプチドをコードする1以上のヌクレオチド配列が組み込まれており、ここで、該補助刺激ポリペプチドの少なくとも1つは、B7−DCであるか、またはその生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体である、ベクター、
を含み、ここで、該発現ベクターは、抗原と、該補助刺激ポリペプチド、フラグメント、ホモログもしくは誘導体との同時発現を生じる宿主細胞に同時感染され得るかまたは同時トランスフェクトされ得る、ベクター組成物。
【請求項15】
前記第二のベクターが配列番号1または配列番号5を含む、請求項14に記載のベクター組成物。
【請求項16】
前記第二のベクターが配列番号1を含む、請求項14または15に記載のベクター組成物。
【請求項17】
請求項1〜6のいずれか1項に記載の核酸分子を用いて形質転換またはトランスフェクトされる、細胞。
【請求項18】
請求項11〜16のいずれか1項に記載の発現ベクターを用いて形質転換またはトランスフェクトされる、細胞。
【請求項19】
哺乳動物細胞である、請求項18に記載の細胞。
【請求項20】
ヒト細胞である、請求項19に記載の細胞。
【請求項21】
樹状細胞またはその前駆体である、請求項19に記載の細胞。
【請求項22】
腫瘍細胞である、請求項19に記載の細胞。
【請求項23】
外因性核酸分子でトランスフェクトされた、単離された哺乳動物腫瘍細胞であって、該核酸分子は、哺乳動物B7−DCタンパク質またはその生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体をコードし、その結果、該タンパク質、フラグメント、ホモログまたは誘導体は、該腫瘍細胞によって発現され、そして該腫瘍細胞は、T細胞と接触され、
(i)該B7−DCタンパク質、フラグメント、ホモログまたは誘導体は、該T細胞に結合し;そして
(ii)該腫瘍細胞は、該T細胞を補助刺激して増殖ならびに/またはサイトカインを生成および分泌する、
腫瘍細胞。
【請求項24】
前記外因性核酸分子は、配列番号1もしくは配列番号5の配列、または該配列のフラグメントを含む、請求項23に記載の腫瘍細胞。
【請求項25】
ポリペプチドであって、活性化されたマクロファージと比較して樹状細胞上に選択的に発現されかつ以下の機能的特性:
(a)T細胞上の結合パートナーに結合すること;および
(b)増殖ならびに/またはサイトカインを生成および分泌するようにT細胞を補助刺激すること、
を有する、ポリペプチド。
【請求項26】
配列番号1もしくは配列番号5の配列を有する核酸分子、または該核酸分子のフラグメント、ホモログもしくは等価物によってコードされる、請求項25に記載のポリペプチド、フラグメント、ホモログまたは機能的誘導体。
【請求項27】
請求項26に記載のポリペプチドであって、該ポリペプチドは、配列番号2もしくは配列番号4のアミノ酸配列、または該ポリペプチドのフラグメント、ホモログもしくは等価物を有する、ポリペプチド。
【請求項28】
請求項1〜10のいずれか1項に記載の核酸の組み換え体発現によって生成される、ポリペプチド、または該ポリペプチドの生物学的に活性なフラグメント、ホモログもしくは他の機能的誘導体。
【請求項29】
B7−DCタンパク質の細胞外ドメインを含む、請求項25〜28のいずれか1項に記載のポリペプチドであって、該ドメインは、配列番号2もしくは配列番号4のアミノ酸残基26〜221、または該ドメインの補助刺激フラグメント、ホモログもしくは他の機能的誘導体を含む、ポリペプチド。
【請求項30】
B7−DCの細胞外ドメインを実質的に含む、請求項25〜28のいずれか1項に記載のポリペプチドであって、該ドメインは、配列番号2もしくは配列番号4のアミノ酸残基26〜221、または残基26〜221の補助刺激フラグメント、ホモログもしくは他の機能的誘導体を含む、ポリペプチド。
【請求項31】
B7−DCタンパク質の全てまたは一部を含む第一の融合パートナーを有する、B7−DC融合ポリペプチドであって、該B7−DCタンパク質は:
(i)第二のポリペプチドに直接融合されるか、または
(ii)必要に応じて、該第二のポリペプチドに融合されるリンカーペプチド配列に融合される、
B7−DC融合ポリペプチド。
【請求項32】
第二のポリペプチドに融合される請求項25〜30のいずれか1項に記載のポリペプチドを含む、B7−DC融合ポリペプチド。
【請求項33】
請求項32に記載の融合ポリペプチドであって、T細胞上の結合パートナー分子に結合し、そしてT細胞レセプターに対する十分な刺激の存在下で該T細胞を補助刺激する、融合ポリペプチド。
【請求項34】
前記結合パートナー分子がPD−1である、請求項33に記載の融合ポリペプチド。
【請求項35】
前記第二のポリペプチドがIg重鎖定常領域の1以上のドメインである、請求項31〜33のいずれか1項に記載の融合ポリペプチド。
【請求項36】
請求項31〜35のいずれか1項に記載の融合ポリペプチドであって、前記第一の融合パートナーは、配列番号2もしくは配列番号4のアミノ酸26〜221、またはその補助刺激フラグメント、ホモログもしくは他の機能的誘導体を含む、融合ポリペプチド。
【請求項37】
請求項36に記載の融合ポリペプチドであって、前記第一の融合パートナーは、配列番号2のアミノ酸26〜221、またはその補助刺激フラグメント、ホモログもしくは他の機能的誘導体を含む、融合ポリペプチド。
【請求項38】
ヒト免疫グロブリンCγ1鎖のヒンジ領域、CH2領域およびCH3領域に対応するアミノ酸配列を有する、請求項35に記載の融合ポリペプチド。
【請求項39】
請求項31〜38のいずれか1項に記載の融合ポリペプチドの直列に連結されたダイマーまたはトリマーである、ダイマー融合ポリペプチドまたはトリマー融合ポリペプチド。
【請求項40】
末端と末端とで、直接にか、または前記モノマーの反復の間に存在するリンカー配列を有して連結された、前記第一の融合パートナーのモノマーが2以上反復する直鎖状マルチマーを含む、請求項31〜38のいずれか1項に記載の融合ポリペプチド。
【請求項41】
B7−DCタンパク質のエピトープに特異的である抗体であって、該エピトープは、B7ファミリータンパク質の既知のメンバー中には存在しない、抗体。
【請求項42】
前記エピトープは、配列番号2または配列番号4のポリペプチドの線状エピトープまたは立体的エピトープである、請求項41に記載の抗体。
【請求項43】
前記エピトープは、配列番号2のポリペプチドの線状エピトープまたは立体的エピトープである、請求項42に記載の抗体。
【請求項44】
モノクローナル抗体である、請求項41または42に記載の抗体。
【請求項45】
ヒトモノクローナル抗体またはヒト化モノクローナル抗体である、請求項43に記載の抗体。
【請求項46】
細胞集団中の、その表面上にB7−DCポリペプチドを発現する細胞を同定または定量化する方法であって、該方法は、以下の工程:
(a)該集団の細胞を、請求項41〜45のいずれか1項に記載の抗体と接触させて、その結果、該抗体が該エピトープを発現する細胞に結合する、工程;および
(b)該抗体が結合する細胞の存在を評価する工程、または該抗体が結合する細胞の数を定量化する工程、
を包含する、方法。
【請求項47】
その表面上にB7−DCポリペプチドを発現する細胞を細胞集団から単離する方法であって、該方法は、以下の工程:
(a)該集団の細胞を、請求項41〜45のいずれか1項に記載の抗体と接触させて、その結果、該抗体が該エピトープを発現する細胞に結合する、工程;
(b)該抗体が結合している細胞をポジティブに選択する工程、または該抗体が結合していない細胞をネガティブに選択する工程、
を包含し、これにより該細胞を単離する、方法。
【請求項48】
サンプル中の、B7−DCポリペプチド、フラグメントもしくはホモログの存在を検出するか、またはB7−DCポリペプチド、フラグメントもしくはホモログを定量化する方法であって、該方法は、以下の工程:
(a)該サンプルを、請求項41〜45のいずれか1項に記載の抗体と接触させて、その結果、該抗体が該エピトープを保有する任意のポリペプチドまたはフラグメントに結合する、工程;および
(b)該抗体に結合する該ポリペプチドもしくはフラグメントの存在を検出する工程、または該抗体に結合する該ポリペプチドもしくはフラグメントを定量化する工程、
を包含する、方法。
【請求項49】
T細胞レセプターに対する十分な刺激の存在下にて、インビトロまたはインビボでT細胞を補助刺激する抗原提示細胞またはその前駆体の能力を増加するために、該抗原提示細胞またはその前駆体中にB7−DCポリペプチドの発現を誘導するかまたは増加する方法であって、該方法は、該抗原提示細胞またはその前駆体細胞を請求項11〜13のいずれか1項に記載の発現ベクターを用いて形質転換またはトランスフェクトし、その結果、該B7−DCポリペプチドの発現が該細胞上で誘導または増加される工程を包含する、方法。
【請求項50】
T細胞レセプターに対する十分な刺激の存在下にて、インビトロまたはインビボでT細胞を補助刺激する抗原提示細胞またはその前駆体の能力を増加するために、該抗原提示細胞またはその前駆体中にB7−DCポリペプチドの発現を誘導するかまたは増加する方法であって、該方法は、該抗原提示細胞またはその前駆体細胞を請求項11〜13のいずれか1項に記載の発現ベクターを用いて形質転換またはトランスフェクトし、その結果、該B7−DCポリペプチドの発現が該細胞上で誘導または増加される工程を包含する、方法。
【請求項51】
前記抗原提示細胞は樹状細胞であり、そして前記前駆体は樹状細胞前駆体である、請求項49または50に記載の方法。
【請求項52】
抗原性刺激に対する哺乳動物被験体のT細胞応答を増加する方法であって、該方法は、請求項17〜24にいずれか1項に記載の、有効量の細胞を、抗原とともに該被験体に投与する工程を包含し、ここで、該細胞は、該抗原性刺激に対する該被験体のT細胞応答を増加するために効果的である、方法。
【請求項53】
腫瘍関連抗原を用いる抗原性刺激に対する哺乳動物被験体のT細胞応答を増加する方法であって、該方法は、請求項22〜24にいずれか1項に記載の、有効量の腫瘍細胞を、該被験体に投与する工程を包含し、ここで、該腫瘍細胞は、該抗原を発現し、該腫瘍細胞の投与は、該腫瘍抗原刺激に対する該被験体のT細胞応答を増加するために効果的である、方法。
【請求項54】
抗原性刺激に対する哺乳動物被験体のT細胞応答を増加する方法であって、該方法は、請求項25〜30にいずれか1項に記載の、有効量のポリペプチド、フラグメント、ホモログまたは機能的誘導体を、抗原とともに該被験体に投与する工程を包含し、ここで、該ポリペプチドの投与は、該抗原性刺激に対する該被験体のT細胞応答を増加するために効果的である、方法。
【請求項55】
抗原性刺激に対する哺乳動物被験体のT細胞応答を増加する方法であって、該方法は、請求項31〜40にいずれか1項に記載の、有効量の融合ポリペプチドを、抗原とともに該被験体に投与する工程を包含し、ここで、該ポリペプチドの投与は、該抗原性刺激に対する該被験体のT細胞応答を増加するために効果的である、方法。
【請求項56】
抗原性刺激に対する哺乳動物被験体のT細胞応答を阻害する方法であって、該方法は、請求項41〜45にいずれか1項に記載の、有効量の抗体を、該被験体に投与する工程を包含し、ここで、該抗体の投与は、T細胞の刺激をブロックするかまたは抗原反応性T細胞を排除して、それによりT細胞応答を阻害するために効果的である、方法。
【請求項57】
前記T細胞応答は、組織移植物または自己抗原に対して指向され、その結果、移植物拒絶または自己免疫反応を阻害する、請求項56に記載の方法。
【請求項58】
抗原性刺激に対する哺乳動物被験体の免疫応答を増加する方法であって、該方法は、以下の工程:
(a)(i)該被験体、(ii)該被験体に対して免疫学的に適合可能なドナー、または(iii)免疫学的に適合可能であるかまたは受容可能な培養された細胞株、よりT細胞を得る工程;
(b)該T細胞を、請求項17〜24のいずれか1項に記載の、有効量の細胞とエキソビボで接触させる工程であって、ここで、該接触させる工程は、抗原性刺激に対する該T細胞の応答を増加するために効果的である、工程;および
(c)該被験体に工程(b)の該T細胞を投与する工程、
を包含し、これにより、該被験体の該免疫応答を増加する、方法。
【請求項59】
抗原性刺激に対する哺乳動物被験体の免疫応答を増加する方法であって、該方法は、以下の工程:
(a)(i)該被験体、(ii)該被験体に対して免疫学的に適合可能なドナー、または(iii)免疫学的に適合可能であるかまたは受容可能な培養された細胞株、よりT細胞を得る工程;
(b)該T細胞を、請求項17〜24のいずれか1項に記載の、有効量の細胞とエキソビボで接触させる工程であって、ここで、該接触させる工程は、抗原性刺激に対する該T細胞の応答を増加するために効果的である、工程;および
(c)該被験体に工程(b)の該T細胞を投与する工程、
を包含し、これにより、該被験体の該免疫応答を増加する、方法。
【請求項60】
抗原性刺激に対する哺乳動物被験体の免疫応答を増加する方法であって、該方法は、以下の工程:
(a)(i)該被験体、(ii)該被験体に対して免疫学的に適合可能なドナー、または(iii)免疫学的に適合可能であるかまたは受容可能な培養された細胞株、よりT細胞を得る工程;
(b)該T細胞を、請求項25〜30のいずれか1項に記載の、有効量のポリペプチド、フラグメント、ホモログまたは機能的誘導体とエキソビボで接触させる工程であって、ここで、該接触させる工程は、抗原性刺激に対する該T細胞の応答を増加するために効果的である、工程;および
(c)該被験体に工程(b)の該T細胞を投与する工程、
を包含し、これにより、該被験体の該免疫応答を増加する、方法。
【請求項61】
抗原性刺激に対する哺乳動物被験体の免疫応答を増加する方法であって、該方法は、以下の工程:
(a)(i)該被験体、(ii)該被験体に対して免疫学的に適合可能なドナー、または(iii)免疫学的に適合可能であるかまたは受容可能な培養された細胞株、よりT細胞を得る工程;
(b)該T細胞を、請求項31〜40のいずれか1項に記載の、有効量の融合ポリペプチドとエキソビボで接触させる工程であって、ここで、該接触させる工程は、抗原性刺激に対する該T細胞の応答を増加するために効果的である、工程;および
(c)該被験体に工程(b)の該T細胞を投与する工程、
を包含し、これにより、該被験体の該免疫応答を増加する、方法。
【請求項62】
病原細胞または病原微生物に関連する抗原に対する保護免疫応答を誘導するために有用なワクチン組成物であって、以下:
(a)必要に応じて該抗原を発現する、請求項17〜24のいずれか1項に記載の形質転換された細胞またはトランスフェクトされた細胞;
(b)(a)の細胞が該抗原を発現しない場合、該抗原のさらなる供給源;
(c)必要に応じて、一般的な免疫刺激剤またはアジュバント;ならびに (d)(a)、(b)および(c)のための薬学的および免疫学的に受容可能な賦形剤またはキャリア、
を含む、ワクチン組成物。
【請求項63】
病原細胞または病原微生物に関連する抗原に対する保護免疫応答を誘導するために有用なワクチン組成物であって、以下:
(a)免疫応答が所望される抗原の供給源;
(b)請求項25〜30のいずれか1項に記載のポリペプチド、フラグメント、ホモログまたは機能的誘導体;
(c)必要に応じて、一般的な免疫刺激剤またはアジュバント;ならびに
(d)(a)、(b)および(c)のための薬学的および免疫学的に受容可能な賦形剤またはキャリア、
を含む、ワクチン組成物。
【請求項64】
病原細胞または病原微生物に関連する抗原に対する保護免疫応答を誘導するために有用なワクチン組成物であって、以下:
(a)免疫応答が所望される抗原の供給源;
(b)請求項31〜40のいずれか1項に記載の融合ポリペプチド;
(c)必要に応じて、一般的な免疫刺激剤またはアジュバント;ならびに
(d)(a)、(b)および(c)のための薬学的および免疫学的に受容可能な賦形剤またはキャリア、
を含む、ワクチン組成物。
【請求項65】
抗原またはワクチンの免疫原性を増加するために、抗原またはワクチンと使用するための補助刺激組成物であって、以下:
(a)請求項25〜30のいずれか1項に記載のポリペプチド、フラグメント、ホモログまたは機能的誘導体;ならびに
(b)薬学的および免疫学的に受容可能な賦形剤またはキャリア、
を含む、補助刺激組成物。
【請求項66】
抗原またはワクチンの免疫原性を増加するために、抗原またはワクチンと使用するための補助刺激組成物であって、以下:
(a)請求項31〜40のいずれか1項に記載の融合ポリペプチド;ならびに
(b)薬学的および免疫学的に受容可能な賦形剤またはキャリア、
を含む、補助刺激組成物。
【請求項67】
請求項62〜64のいずれか1項に記載の、有効量のワクチン組成物を、被験体に投与する工程を包含する、哺乳動物被験体において抗原に対する免疫応答を誘導または増強する方法。
【請求項68】
請求項65または66に記載の補助刺激組成物を、抗原またはワクチンとともに被験体に投与する工程を包含する、哺乳動物被験体において抗原またはワクチンに対する免疫応答を増強する方法。
【図1】

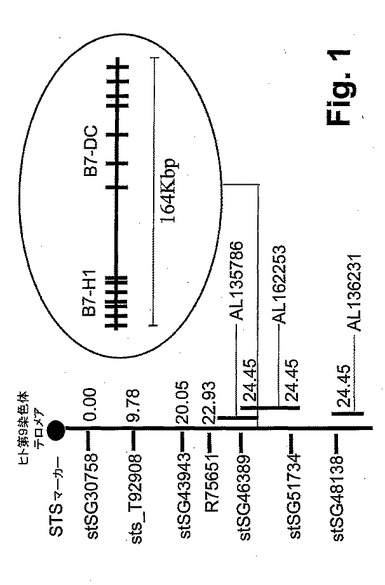
【図2】
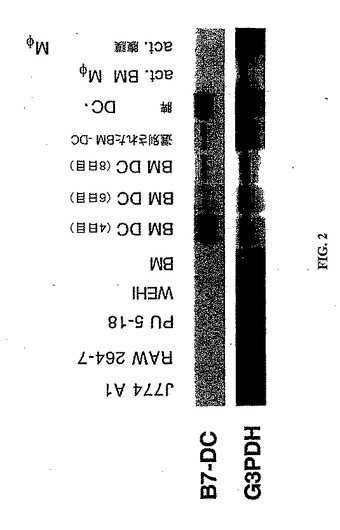
【図3】
【図4】
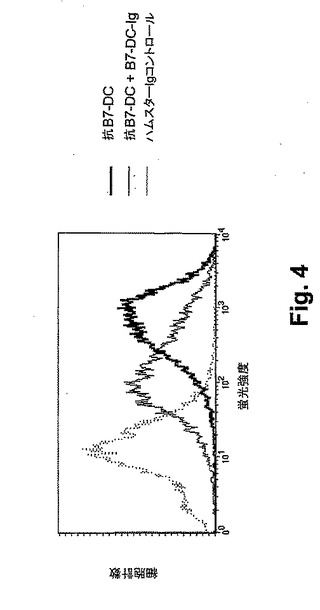
【図5】

【図6】
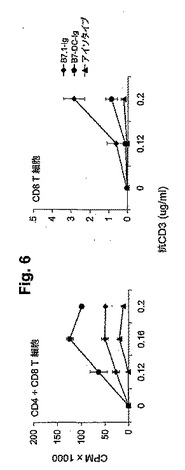
【図7】
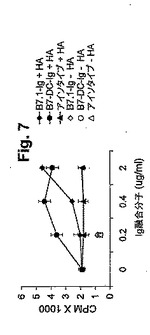
【図8】
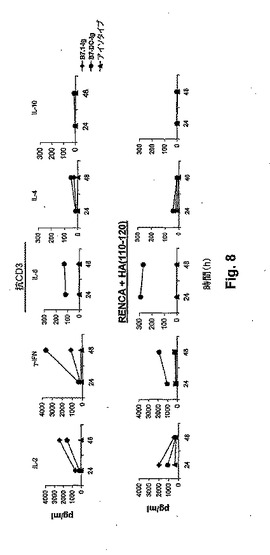
【図9】
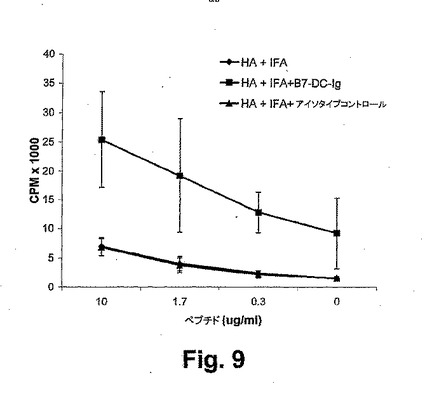
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2011−224008(P2011−224008A)
【公開日】平成23年11月10日(2011.11.10)
【国際特許分類】
【出願番号】特願2011−104379(P2011−104379)
【出願日】平成23年5月9日(2011.5.9)
【分割の表示】特願2001−580357(P2001−580357)の分割
【原出願日】平成13年4月27日(2001.4.27)
【出願人】(398076227)ザ・ジョンズ・ホプキンス・ユニバーシティー (35)
【Fターム(参考)】
【公開日】平成23年11月10日(2011.11.10)
【国際特許分類】
【出願日】平成23年5月9日(2011.5.9)
【分割の表示】特願2001−580357(P2001−580357)の分割
【原出願日】平成13年4月27日(2001.4.27)
【出願人】(398076227)ザ・ジョンズ・ホプキンス・ユニバーシティー (35)
【Fターム(参考)】
[ Back to top ]