
樹脂、低温配合物、およびそれに由来するコーティング
分子構造、したがって本発明の樹脂の特性を制御する一連のバイオベース材料を使用して、一連の樹脂を合成した。これらの樹脂の有用性を、架橋β−ヒドロキシアミドやハイブリッドタイプなどの粉体コーティングの配合物で実証した。一般に、バイオベース樹脂は、加熱時に、通常の石油化学系樹脂より速く流展し、硬化オーブンで通常可能な温度より低い温度の使用が可能になり、特にカルボン酸−エポキシ架橋ハイブリッドコーティング配合物でより活性の高い触媒系が可能になる。

【発明の詳細な説明】
【発明の開示】
【0001】
本願は、2005年3月18日出願の米国特許仮出願第60/663,422号、および2006年1月13日出願の米国特許仮出願第60/758,757号の利益を主張する。
【0002】
該米国特許仮出願2件の内容全体を参照により本明細書に組み込む。
発明の分野
本発明は、基材、具体的には感温性基材用の粉体コーティングを製造するのに有用である。典型的な感温性基材としては、プラスチックなどのポリマーを含むがこれらに限定されない有機基材、および木質およびプラスチックの複合体を含むがこれらに限定されない複合体が挙げられる。
【0003】
現在の粉体コーティング樹脂および配合物には、1つの重要な制限がある。これらは、一般に許容できる性能に必要とされる良好なフローおよび架橋を有するためにかなり高いオーブン温度(通常は177℃超)を必要とする。プラスチック、木質、バイオ複合体など、被覆対象の基材の多くは、まさに感温性であり、現在の粉体コーティング配合物で使用される高温に耐えることができない。このような基材の使用は、ここ数年で大幅に増大し、将来極めて劇的に増大するものと予想される。低温硬化粉体コーティングの領域における最近の研究の例のMuthiah文献を参照のこと。
【0004】
金属などの高温基材上で使用することもでき、耐久性があり、費用効果の高い感温性基材用低温熱硬化粉体コーティングが必要とされている。このような場合、温度がより低くなれば、プロセスにおけるエネルギーコストがより低くなるはずである。コストがより低くなれば、その新技術の受入れが大幅に増加するはずである。
【0005】
石油化学系供給原料の一部を広範囲の応用分野に使用するためのバイオベース供給原料で置換することは非常に興味深い。この興味の証拠は、長年にわたって出版された総説の数に反映されている。ポリエステル樹脂の合成でバイオベース供給原料を利用する試みは、米国特許第6,063,464号、およびトウモロコシバイオマス由来イソソルビドをポリエステル材料の合成で使用するGuoらの論文(下記を参照のこと)に例示されている。
【0006】
現在産業界で使用されるものより低温で流展し硬化する粉体コーティングを製造する必要もある。粉体コーティングは、使用中のVOC放出が非常に低いという点で環境上の利点を与える。残念なことには、その利点の一部は、硬化サイクルにおけるエネルギー需要の高さにより失われ、仕上がりの粗さは、通常はそれらが低温において流展性が不十分であることに由来する。
【0007】
他の関連特許および雑誌記事には、下記が含まれる。
低温硬化(LOW TEMPERATURE CURE):米国特許第6,703,070号、03/2004、Muthiah
合成および加工(SYNTHESIS AND PROCESSING):欧州特許出願公開第1491593号、12/2004、Mons
バイオベース材料総説:
Applied Microbiology and Biotechnology(2001),55(4),387−394.Huttermann,A.;Mai,C.;Kharazipour,A.“Modification of lignin for the production of new compounded materials”;
Biopolymers from Renewable Resources(1998),1−29.Kaplan,David L.“Introduction to biopolymers from renewable resources”;
Bioresource Technology(1994),49(1),1−6.Sharma,D.K.;Tiwari,M.;Behera,B.K.“Review of integrated processes to get value−added chemicals and fuels from petrocrops”;
Applied Biochemistry and Biotechnology(1988),17 7−22.Narayan,Ramani.“Preparation of bio−based polymers for materials applications”.
バイオベース樹脂合成:
Abstracts of Papers,224th ACS National Meeting,Boston,MA,United States,August 18−22,2002(2002).Guo,Yinzhong;Mannari,Vijaykumar M.;Massingill,John L.,Jr.“Hyperbranched bio−based polyols”.
粉体コーティング:
“Powder Coatings Volume 1:The Technology,Formulation,and Application of Powder Coatings”.Howell,David M.John Wiley and Sons,London,2000.
Polymer Preprints 2003,44(1).Gedan−Smolka,Michaela;Lehmann,Dieter;Lehmann,Frank.“Catalysis In Uretdione Powder Coatings Enables Innovative Processing Lines”.
【0008】
粉体コーティングにおいて低温フローおよび硬化が必要であるのに加えて、コーティングタイプに関わらず、コーティングマトリックス内において顔料の良好な分散も必要である。これを実現するために、異なる相溶性の成分を有するポリマーが設計されている。ポリマー分散剤は、塗料、コーティング、およびインク系中の顔料および他の材料を、最も典型的には立体安定化によって安定化する。ポリマー分散剤は、定着基とポリマー鎖からなる2成分構造を有する。最も典型的には、定着基は、粒子表面、およびコーティングの連続相と相溶であるポリマー鎖と相互作用する極性材料である。実際には、該ポリマー基は粒子の周りにコーティングを形成し、粒子同士が接触し、より大きい非相溶な凝集体になるのを防止する。
【0009】
有効なポリマー分散剤を与えるものと予想され得る定着基/ポリマーの配置は多数存在する。本発明の樹脂は、極性カルボン酸定着部位および非極性植物油鎖を有し、したがって分散剤および結合剤として働くことができる。分散剤としても働くことができる硬化結合剤によって、多数の顔料を分散するために別々の添加剤を必要とすることがなくなる可能性がある。関連技術としては、米国特許第5,959,066号、第6,025,061号、第6,063,464号、および第6,107,447号が挙げられる。
発明の簡単な説明
簡潔に言えば、金属などの高温基材上で使用することもでき、耐久性があり、費用効果の高い感温性基材用低温熱硬化粉体コーティングが必要とされている。石油化学系供給原料の置換材料を見い出すことが、特に大量のバイオベース供給原料をこの置換で利用することができる場合にさらに求められている。本明細書で開示するバイオベース粉体コーティング技術は、再生可能なバイオ供給源に由来する新規樹脂、および特許配合物技術、特に低温硬化技術を組み合わせることによってこの必要に応じる。後者の場合、温度がより低くなれば、プロセスにおけるエネルギーコストがより低くなるはずであり、新規バイオベース技術の受入れが大幅に増加するはずである。
【0010】
本発明の一実施形態は、Tgが50℃より高く、バイオベース内容物が少なくとも5%、別の実施形態では少なくとも50%であり、粘度が比較的低いポリエステル樹脂の合成を提供する。
【0011】
広範囲の実施形態では、樹脂をコーティング、特に粉体コーティングの配合物で利用する。
別の実施形態では、樹脂には、二酸およびジオールの反応によるカルボン酸官能性ポリエステルが含まれる。
【0012】
別の実施形態では、ポリエステル樹脂を形成するために利用される酸およびジオールは、得られるコーティングの特性を最大限にし、かつ樹脂中のバイオベース材料の量を最大限にするために、必要に応じてバイオベースまたは石油系である。
【0013】
本発明のさらに別の実施形態では、樹脂を架橋性樹脂と配合して、しばしば比較的低い温度で、良好なフローおよび柔軟性を有する保護コーティング被膜に硬化する。
本発明のさらに別の実施形態では、樹脂をPRIMID樹脂と配合して、良好なフローおよび柔軟性を有する保護コーティング被膜に硬化する。
【0014】
本発明のさらに別の実施形態では、樹脂をアクリル酸エポキシ樹脂と配合して、比較的低い温度で良好なフローおよび柔軟性を有するハイブリッド粉体コーティング被膜に硬化する。
【0015】
別の実施形態では、配合物は、外観、硬化速度、および他の特性を制御するために触媒、フローコントロール剤、硬化改質添加剤などを含む。
別の実施形態では、配合物は、イミダゾールおよび置換イミダゾールからなる触媒を含む。
【0016】
別の実施形態では、配合物は、イミダゾールおよび置換イミダゾール触媒の活性を改質するために酸性添加剤などの硬化改質添加剤を含む。
別の実施形態では、配合物は、色、外観、腐食制御、隠蔽、または他の機能のための顔料を含めて、当技術分野で知られている添加剤および賦形剤を含有することができる。
発明の詳細な説明および最良の態様
概して、本発明は、所望のバイオベース供給原料の使用と、より低い温度の粉体コーティングの必要性を組み合わせる。トウモロコシおよびダイズの供給原料を利用して、粉体コーティング性能に適した特性のバランスを有する樹脂を作製することができる。次いで、これらの樹脂を、様々な粉体コーティング配合物に配合することができる。
【0017】
通常は、本発明による粉体コーティング配合物は、下記によって調製される。主要な樹脂を粉砕し、粉砕硬化剤および選択した粉砕添加剤とドライブレンドし、ドライブレンドを溶融混合し、溶融混合ブレンドを押し出し、続いて急速冷却する。次いで、冷却したブレンドを所望の粒径に粉砕し、最後に、得られた粉末を最終粒径に分類する。
【0018】
本発明のいくつかの実施形態について本明細書で用いるバイオベース供給原料、配合物、生成物、材料、樹脂などは、通常の化学修飾、および/または発酵などの生物プロセスによって加工された農業および森林をベースとする再生可能な資源の変換に少なくとも一部由来する供給原料、配合物、材料、樹脂、および生成物などを意味する。炭素源は、有限で枯渇しつつある通常の化石由来炭素源とは異なる再生可能な植物の作物/樹木資源に由来する。
【0019】
本明細書では、ハイブリッド樹脂は、樹脂が1タイプを超える樹脂、例えばポリエステルとエポキシのブレンドであることを意味する。
本発明による特に有用な樹脂は、明らかに矛盾する2つの特性が良好なバランスをとり、
(1)非晶質樹脂の特徴である、適用中の良好な流展性のための、溶融状態における低粘度を有するが、
(2)結晶質樹脂の特徴である、良好な貯蔵安定性のための、比較的高いガラス転移温度(Tg)も有さなければならない。Tgが低すぎる場合、粉末粒子は「軟質」であり、特に高い貯蔵温度で貯蔵中に使用不可能な塊に合体する。通常は、これらの特性は、結晶質および非晶質の樹脂を、実際に半結晶質な樹脂ブレンドにブレンドすることによってバランスがとられている。通常は、本発明に従って得られた樹脂はこれらの所望の特性を提供する。
【0020】
注:別段の指定のない限り、材料の量を意味する場合の%は、重量パーセント(重量%)を意味する。
本明細書で開示する樹脂合成の一般的な手法は4つある。
【0021】
1.ダイマージオール、イソソルビド由来のジオール、および/またはダイマー酸をベースとするヒドロキシル官能性ポリエステル。通常は、ポリエステルのカルボキシルまたはヒドロキシル官能性は、二酸またはジオール基のモル過剰の比によって決定される。ポリエステルは、通常は正味のバイオベース内容物が少なくとも約5重量%であるが、最も典型的には約20〜約50重量%である。
【0022】
2.ダイマージオール、イソソルビド由来のジオール、および/またはダイマー酸をベースとするカルボキシル官能性ポリエステル。通常は、ポリエステルのカルボキシルまたはヒドロキシル官能性は、二酸またはジオール基のモル過剰の比によって決定される。ポリエステルは、通常は正味のバイオベース内容物が少なくとも約5重量%であるが、最も典型的には約50〜約70重量%である。
【0023】
3.ダイマー酸、および/またはダイマージオールをベースとするヒドロキシル、カルボキシル、またはイソシアナート官能性ポリウレタン。通常は、過剰のイソソルビドおよび/またはダイマージオールがヒドロキシル官能性を生成し、過剰のダイマー酸がカルボキシル官能性を生成し、過剰のポリイソシアナートがイソシアナート官能性を生成する。ポリウレタンは、通常は正味のバイオベース内容物が少なくとも約5重量%であるが、最も典型的には約20〜約50重量%である。
【0024】
4.米国を指定国とする2004年2月2日出願の国際公開第2004/077169号の易脱墨性トナー(Readily Deinkable Toners)に開示されるアミド−アミン官能性樹脂。アミド−アミン樹脂は、参照により内容が本明細書に組み込まれる該特許出願に記載されるダイマー酸とジアミンの反応生成物である。本発明のいくつかの実施形態では、典型的なアミドアミン官能性樹脂のTgは約80℃未満である。本発明の他の実施形態では、アミド−アミンのTgは約70℃未満である。正味のバイオベース内容物は、典型的には少なくとも5重量%であるが、より典型的には約40〜約60重量%である。
【0025】
本発明による樹脂は、イソソルビド(通常はトウモロコシ供給原料由来)など、硬質化効果を与える傾向がある共反応成分、およびダイマー酸またはダイマージオール(通常は植物油供給原料由来)など、柔軟化効果を与える成分からなり得る。これらの成分を適切に樹脂に共反応させることによって、樹脂の流展性と貯蔵安定性とを制御することができる。一般に、硬質化成分は、環状構造に結合し可動性が限定されたアルコール、エステル、カルボン酸、または酸塩化物などの官能性化学基を含み、柔軟化成分は、脂肪族炭素鎖に結合した官能性化学基を含む。イソソルビドは、縮合環状エーテル環からなるジオールであり、一般にジアンヒドロヘキシトールと呼ばれる、バイオベース糖誘導体のより大きいファミリーのメンバーである。ダイマー酸およびダイマージオールはそれぞれ、性質が極めて脂肪族であるバイオベース脂肪酸に由来するジカルボン酸およびジアルコールである。同様に、これらの硬質化および柔軟化の効果を、図3に示すポリウレタンに適用することもできる。
【0026】
通常は、触媒および/または熱を用いて架橋することによって、樹脂を硬化する。典型的な硬化温度は最高125℃である。
本明細書で開示するポリエステルポリオール樹脂は、イソシアナート、エポキシ、メラミンホルムアルデヒド、尿素ホルムアルデヒドなどを有する反応性配合物において、コーティング、接着剤、シーラント、および他の用途として有用である。
【0027】
本明細書で開示するポリカルボン酸樹脂は、β−ヒドロキシルアミド、エポキシなどを有する反応性配合物において、コーティング、接着剤、シーラント、および他の用途として有用である。
【0028】
アミド−アミン官能性樹脂は、イソシアナート、エポキシ、メラミンホルムアルデヒド、尿素ホルムアルデヒドなどを有する反応性配合物において、コーティング、接着剤、シーラント、および他の用途として有用である。
【0029】
粉体コーティング配合物では、開示するバイオ由来樹脂が特に有用である。実施例4には、粉体コーティング配合物中のβ−ヒドロキシルアミドとエステル交換方式で硬化して、透明なコーティングを形成するバイオ由来のカルボン酸官能性樹脂が記載される。実施例4Aには、実施例4の樹脂と同様の樹脂が記載されるが、より大きな規模で樹脂が作製されている。得られた樹脂のTgはわずかに高い。実施例5には、粉体コーティング配合物中のアクリル酸エポキシ樹脂で硬化して、透明なコーティングを形成するバイオ由来のカルボン酸官能性樹脂が記載される。実施例6には、顔料を含む粉体コーティング配合物中の市販のエポキシ架橋性樹脂で硬化して、黒色コーティングを形成するバイオ由来のカルボン酸官能性樹脂が記載される。実施例6Aには、市販のカルボン酸官能性樹脂およびカーボンブラックに比べて、バイオ由来のカルボン酸官能性樹脂およびカーボンブラックからなる顔料分散体、ならびに白色粉体コーティング配合物に添加された場合に、色に及ぼすそれらの効果が記載される。実施例6Bには、イソシアヌル酸トリグリシジル(TGIC)架橋剤で硬化されたバイオベースのカルボン酸官能性樹脂が記載される。
【0030】
実施例7には、粉体コーティング配合物中の市販のエポキシ架橋性樹脂で硬化して、透明なコーティングを形成するバイオ由来のアミド−アミン官能性樹脂が記載される。
実施例8には、フロープロモーターとしてバイオベースポリエステルを使用して、粉体コーティングの生成が例示されている。ポリエステル樹脂は、実施例3Bに記載されている。
【0031】
最後の実施例である実施例9には、実施例3Fに従って調製された樹脂の顔料分散体特性が例示されている。
本発明の一実施形態は、最小量から最大量のバイオベース材料の粉体コーティング用途向け樹脂の製造方法に関する。本発明の別の実施形態の樹脂は、少なくとも1つの飽和または不飽和のバイオベースポリエステルを含む。
【0032】
本発明は、コーティング、粉体コーティング、接着剤、トナー、インク、シーラント、ポリマー添加剤などを含むが、これらに限定されない様々な用途における、これらのバイオベース材料の1つまたは複数の使用にも関する。一実施形態では、ガラス転移温度(Tg)が約80℃未満である樹脂、他の実施形態では、ガラス転移温度が約70℃未満である樹脂、さらに別の実施形態では、約60℃未満で適切な溶融レオロジーの樹脂が設計された。本発明の広範囲の一般的な実施形態による樹脂は、ガラス転移温度の最小限が少なくとも約20℃であり、最大限が約80℃で、適切な溶融レオロジーを有する。フローコントロールに有用な樹脂は、通常はガラス転移温度範囲の下端である(例えば、Tgが約28.4℃である実施例3B)が、約20℃〜約80℃とすることができ、いくつかの実施形態では、通常は約25℃〜約60℃とすることができる。
【0033】
50%超のバイオベースカルボキシ官能基を含有する本発明の樹脂からなるハイブリッド粉体コーティング樹脂は、粉体コーティングに配合した。本明細書に記載する本発明の樹脂は、イソソルビド(通常は、トウモロコシ供給原料由来)など、硬質で非常に官能性である傾向がある共反応成分、およびダイマー酸(通常は、ダイズ供給原料由来)など、軟質で柔軟である傾向がある成分からなる。これらの成分を適切に樹脂に共反応させることによって、流展性と貯蔵安定性とを制御することができる。
【0034】
本発明は、本発明の1つまたは複数の樹脂による粉体コーティングの配合物にも関する。この粉体コーティングの著しい特徴は、このコーティングが、粉体コーティング操作に典型的な温度より低い温度で連続被膜に流展し硬化する能力である。低温硬化能力は、その組成物で利用するバイオベース樹脂の低粘度性、および本発明の樹脂性成分の有利なフロー特性を活用する配合物に由来する。本発明の樹脂から得られた利点は、ほぼ等価な市販の樹脂に比べて、所与の温度において低粘度である。
【0035】
粉体コーティング配合物で使用される樹脂の重要な特徴は、最終的な粉体コーティング粉末の貯蔵安定性には通常は少なくとも約50℃であり、好ましくは少なくとも約60℃であるガラス転移温度(Tg)である。表1は、いくつかのダイズベース樹脂、その官能性、およびそのTgの一覧を示す。この表は、低粘度のダイズベースモノマーを含む材料から、許容できるTgを有する樹脂を生成することが困難であることを例証している。
【0036】
【表1】
【0037】
樹脂1−1しか、Tgの基準を満たさなかった。ダイズベース材料の存在下でより高いTgを実現し、樹脂中バイオベース材料の負荷を高く維持するために、別のバイオベース材料であるが、高い固有Tgに寄与するものであるイソソルビドを利用した。
【0038】
Tgがより高いバイオベース材料(トウモロコシ供給原料由来のイソソルビド)は、ダイズベース材料と共反応して、粉体コーティング配合物のバイオベース含有量が高く、Tgが十分に高い樹脂を与えることができることが確認された。その後の合成によって、ダイズ、イソソルビド、および他の成分のバランスをとる試みを行って、樹脂、最終的には粉体コーティングにおいて特性の適切なバランスを実現した。
樹脂合成(実施例1および2を参照のこと):
コーティングの生成におけるバイオベース材料の使用は、以下の通り記述することができる。
【0039】
ポリエステルポリマーは、(1)反応器中で、イソソルビド(トウモロコシ供給原料由来);脂肪ダイマージオールおよび/またはダイマー二酸(ダイズ供給原料由来);二酸、ジエステル、または二酸塩化物;任意選択のコジオール;および任意選択のコ二酸、コジエステル、またはコ二酸塩化物を、芳香族二酸およびジオールを重合するのに適した縮合触媒と混合し、(2)モノマーおよび触媒を加熱して、モノマーを重合し、ポリエステルを得ることによって調製される。(図1を参照のこと)
カルボキシル官能性ポリエステル樹脂は、(1)反応器中で、イソソルビド;脂肪ダイマー二酸;任意選択のコ二酸、コジエステル、またはコ二酸塩化物;および任意選択のコジオールを縮合触媒と混合し、(2)モノマーおよび触媒を加熱して、モノマーを重合し、カルボキシル官能性ポリエステル樹脂を得ることによって調製される。(図2を参照のこと)
ヒドロキシル、カルボキシル、またはイソシアナート官能性ポリウレタンは、(1)反応器中で、イソソルビド;脂肪ダイマー二酸および/またはダイマージオール、ポリイソシアナート;任意選択のコジオール;および任意選択のコ二酸、コジエステル、またはコ二酸塩化物を、ジオールおよび二酸をポリイソシアナートと共に重合するのに適した触媒を使用して、あるいは使用せずに混合し、(2)モノマーおよび任意選択の触媒を加熱して、モノマーを重合し、ポリウレタンを得ることによって調製される。(図3を参照のこと)
次に、本明細書の実施形態に有用な様々な反応物質を開示する図1、2、および3を参照する。本発明は広範囲の実施形態によれば、開示するダイマージオールおよびダイマー酸に加えて、通常は約4〜約20個の炭素原子を有する脂肪族鎖を含む。より好ましくは、脂肪族鎖は約6〜約16個の炭素原子を有する。
【0040】
開示した追加のダイマージオールおよびダイマー酸は、アルコールまたはカルボン酸の官能基と共に、約4〜20個の炭素原子の脂肪族側鎖である2つの側鎖、および約8〜12個の炭素原子の他の2つの側鎖を有する6員環を含む。
【0041】
さらに、ジエステル、二酸、コ二酸、およびコジエステルは、式R2−CO−R1−CO−R2(式中、R2=−OH、−OR3、または−Cl(式中、R3=1〜4個の炭素原子を有する脂肪族鎖))を有することができる。R1は、2〜12個の炭素原子を有する芳香族または脂肪族の基である。
【0042】
特定の理論に拘泥するものではないが、現時点ではダイマー酸およびダイマージオールの脂肪族側鎖が樹脂に低粘度性をもたらすと考えられている。脂肪族側鎖は、低温で軟化し、粘度を低下させ、フローをよりよくさせる傾向がある。鎖が長くなれば、より軟化が見られ、加熱中により速く軟化する。
【0043】
これらは、いくつかの実施形態では、実施例9で例示するような顔料分散体の改善ももたらすと考えられる。よりよいフローがもたらす1つの結果は、顔料の優れた濡れであり、それによって顔料分散体が改善される。
【0044】
さらにかつより広範囲に、ジアンヒドロヘキシトールを本発明で使用することができる。したがって、他の環状ジオールを含む二環式を組み込むことによって硬質化構造を調製する際に、他のジアンヒドロヘキシトールをD−イソソルビドまたはその異性体の代わりとすることができ、本発明で使用することができる。シクロヘキシル、イソホロン、および他の環状構造を組み込むジオールは、イソソルビドと同様な硬質化効果を加えることができる。
【0045】
ダイマー二酸は、通常はC18の不飽和脂肪酸の二量化によって生成された粘性液体である。C18の不飽和脂肪酸のバイオ供給源には、植物、トール油、および動物の3つが存在する。C18単位は、いくつかの方式で連結され得る。優勢な成分であるC36の二酸では、非環式、単環式、二環式、および芳香族の4つの主要な構造タイプが知られている。これらの構造タイプのそれぞれにも、多くの構造異性体が存在する。これらの構造タイプおよび異性体の分布は、二量化に使用する出発物の脂肪酸供給原料の単不飽和/多不飽和の比、およびプロセス条件に依存する。いくつかの実施形態で典型的に使用する最小のダイマー二酸は、C18の二酸である。
【0046】
ダイマー二酸は、(1)約80%のC36の二塩基酸を含有する標準(未蒸留)タイプ、(2)C36の二塩基酸含有量を92〜98%に上昇させた、蒸留を行ったタイプ、(3)色を改善するため、蒸留および部分水素化を行ったタイプ、ならびに(4)安定性を最大限にするため、蒸留および完全水素化を行ったタイプの4タイプが現在市販されている。
【0047】
バイオベースポリエステル樹脂を調製するために使用する典型的なダイマー酸は、Empol 1018(登録商標)(実施例3、3C、および3E)、およびPripol 1013(登録商標)(実施例2、3A、および3D)(両者共、植物系ダイマー酸である)であった。Empol 1018(登録商標)はCognis Corporationが製造し、Pripol 1013(登録商標)は、Uniqemaが製造している。Cognisは、以降トール油系ダイマー酸を選択して、植物系ダイマー酸生産を中止した。表3では、Pripol 1013(登録商標)とEmpol 1018(登録商標)の物理的諸特性および組成物を比較している。Pripol 1013(登録商標)のほうが、明るい色であり、二塩基酸含有量が高い。この2つの異なるダイマー酸を使用して得られたカルボキシル官能性樹脂は、同様な物理的諸特性を有した。
【0048】
【表2】
【0049】
ダイマージオールは、通常はダイマー二酸メチルエステルの高圧水素化によって生成される。バイオベースポリエステル樹脂(実施例1、1A、および3B)を調製するために使用するダイマージオールは、SPEZIOL C36/2 1075(登録商標)ダイマージオールであった。これは、Cognis製の植物系ダイマージオールである。
【0050】
本明細書で開示する樹脂は、一度溶融させた市販の石油化学系樹脂に比べて、低い粘度を有する(実施例を参照のこと)。現在市販されている樹脂粉体コーティング配合物では、硬化サイクル後に得られた被膜の流展性およびレベリング性を良好にするために、フロー性のある材料(フローコントロール添加剤)を添加する必要がある。バイオベース樹脂は、良好な被膜レベリング性および外観を実現するためのこのような添加剤を殆どまたは全く必要としない。組み込むことに成功したバイオベース樹脂は、通常の石油化学系樹脂を含有する配合物中でフロー添加剤として機能することもできる。通常は、コーティング配合物に、他の主要な粉末樹脂を使用すると共に、バイオベース樹脂(含有量約0.1重量%〜約5重量%)を使用して、本発明の樹脂のフローコントロール特性を利用する。
【0051】
本発明のポリエステルポリマーは、イソソルビド、ダイマージオール、および/またはダイマー酸、二酸、ジエステル、または二酸塩化物;任意選択のコジオール;および任意選択のコ二酸、コジエステル、またはコ二酸塩化物の溶融重合によって調製した(図1の方法)。
【0052】
実施例1に、本発明のポリエステルを調製するために使用する典型的な手順を記載する。脂肪族ポリエステルは、軟質で柔軟なゴム状材料である。大部分の芳香族ポリエステルは結晶質である。軟質ダイマージオールを、高官能性イソソルビドおよび結晶質の芳香族二酸とブレンドすることによって、特性の良好なバランスが得られる。しかし、このバランスは、反応においてエチレングリコールなど、他の材料を(すなわち、図1および2の「ジオール」として)含めることによって助長され得る。
【0053】
ガラス転移温度(Tg)が61℃〜165℃(表2Aおよび2B)のポリエステルを調製することによって、様々なモノマーの効果を検討した。表2Aは、本明細書に記載するように合成された樹脂の典型的な特性、およびガラス転移温度(Tg)が61℃〜165℃の様々なモノマーの効果を示す。表2Bは、実施例2〜3Aに記載するように合成されたカルボキシル官能性樹脂の典型的な特性を示す。
【0054】
【表3】
【0055】
【表4】
【0056】
データは、これらのモノマーで可能な広範囲のTgを示す。イソソルビドなしで調製された試験ポリエステルは、イソソルビド含有のもののように非晶質でなかったが、挙動は結晶質であった。
【0057】
D−イソソルビド(1,4:3,6−ジアンヒドロ−D−グルシトール)(1a)もしくはその異性体、および/またはD−イソソルビドを含むすべての異性体の混合物を、D−イソソルビドの代わりに使用することができた。1,4:3,6−ジアンヒドロ−D−マンニトール(1b)および1,4:3,6−ジアンヒドロ−D−イジトール(1c)は、イソソルビドの2つの異性体である。D−イソソルビドを本発明で使用したが、D−イソソルビドの異性体も同様に働くものと予想される。本発明で有用なイソソルビドの異性体を図3Aに例示する。
【0058】
酸官能性ポリエステルを形成するのに適切なポリオールの例としては、1,2−エタンジオール(エチレングリコール)、1,3−プロパンジオール、1,4ブタンジオール、1,6−ヘキサンジオール、1,10−デカンジオール、1,12−ドデカンジオール、1,4−シクロヘキサンジメタノール、ジエチレングリコール、トリエチレングリコール、ネオペンチルグリコール、トリメチロールプロパン、水素化ビスフェノールA(2,2−(ジシクロヘキサノール)プロパン)、2,2,4−トリメチル−1,3−ペンタンジオール、2−メチル−1,3−プロパンジオール、2−メチル−2−ヒドロキシメチル−1,3−プロパンジオール、2−エチル−2−ヒドロキシメチル−1,3−プロパンジオールなど、および前述のポリオールの少なくとも1つを含む組合せが挙げられる。現時点での働きは、バイオベース内容物を最大限にすることを目標としているので、好ましいポリオールは、イソソルビド(トウモロコシ原料由来)およびダイマー酸ジオール(ダイズ原料由来)であるが、必要に応じてエチレングリコールなどを使用して、特性を向上させることができる。
【0059】
適切なポリカルボン酸、酸エステル、および酸塩化物には、コハク酸、アジピン酸、アゼライン酸、セバシン酸、1,12−ドデカン二酸、テレフタル酸、イソフタル酸、トリメシン酸、テトラヒドロフタル酸、ヘキサヒドロフタル酸、1,4−シクロヘキサンジカルボン酸、トリメリト酸、ナフタレンジカルボン酸、ダイマー酸など、および前述のポリカルボン酸の少なくとも1つを含む組合せに由来するものが含まれる。好ましいジエステルは、テレフタル酸のジメチルエステルである。いくつかの配合物では、ドデカン二酸(DDA)を改質剤として使用する。1,4−シクロヘキサンジカルボン酸、Empol 1018(登録商標)、Pripol 1013(登録商標)などの二酸が現時点で好ましい。
【0060】
所望の分子量のカルボキシル官能性ポリエステルを得るために、ポリエステルを形成するために使用するモノマー混合物は、通常はヒドロキシル官能基に対して適切に過剰なカルボキシル官能基を有する(ただし、酸当量に対するヒドロキシル当量の比は通常は0.85〜0.95である)。ポリエステルは、非晶質から結晶質まで多様であり得る。
【0061】
架橋は、カルボキシ官能性のカルボキシル基を、β−ヒドロキシルアミドと自己触媒エステル交換反応(アミドの商標名の後に、PRIMID反応と記載される場合が多い(表3))で反応させ、または市販のポリエポキシ官能性ポリマーと反応させることによって実現される。特に低温硬化組成物に好ましいポリエポキシ化合物は、グリシジルアクリラートやグリシジルメタクリラートのコポリマー(総称して「GMA」)樹脂など、エポキシ官能性アクリル酸またはメタクリル酸の樹脂である。GMA樹脂は、通常は約5〜約30重量%のグリシジルアクリラートまたはグリシジルメタクリラート、および約80〜約95重量%のメチルメタクリラート(ただし、約50重量%までのメチルメタクリラートを別のα,β−不飽和モノマー、例えばスチレン、アクリロニトリルなどで置換することができる)から得られる。適切なGMA樹脂は、エポキシ当量が約200〜約1000、好ましくは約200〜約600であり、Mnがゲルパーミエーションクロマトグラフィーで決定して200〜約2000原子質量単位(AMU)である。これらは室温で固体であり、融点が約40℃超であり、好ましくは軟化点が約50℃〜約75℃であり、Tgが約40℃〜約60℃である(表3)。
【0062】
バイオベースの樹脂性成分で低温流展性を実現できることを考えれば、約115℃〜約140℃の温度で硬化を開始する触媒を利用することが有利であり、これは市販されている多くの中から選択可能である。通常は、触媒を約0.1〜約5部/樹脂100部(phr)、好ましくは約0.2〜2(phr)のレベルで使用して、硬化反応を低温硬化剤で加速することができる。本発明に好ましい触媒は、式1に示す一般式を有するイミダゾール:
【0063】
【化1】
【0064】
(式中、R1、R2、R3、およびR4は独立に、水素、メチル、フェニル、またはベンジルである)、およびその付加物である。
概して、置換基はエポキシ樹脂との反応性を示さないことがある。第三級アミンおよびポリアミン材料も、この反応の触媒として有用である。
【0065】
良好な流動性を維持するためには、付加物を生成し、イミダゾール触媒の反応性を部分ブロックすることによって、その反応性を改変する必要があり得る。これは、イミダゾールとエポキシとの付加物を作製することによって行われることがある(例えば、米国特許第6,703,070号を参照のこと)。本発明の一実施形態では、親イミダゾールが好ましい触媒であり、イミダゾールの反応性を軽減するために酸性材料を配合物に添加した。
【0066】
イミダゾールの反応性を軽減するのに適した酸性材料には、ベンゼンスルホン酸もしくはナフタレンスルホン酸およびその置換変形体などの芳香族スルホナート、またはナフタレンカルボン酸およびその置換変形体などの芳香族カルボキシラートが含まれ、無機物や超酸などの固体酸性材料も使用することもできる。後者の場合、イミダゾール触媒の一部を固体酸性表面に吸着させ、したがって加熱するまで結合剤のバルクに利用させなくすることができる。このような一固体材料は、例えばKing Industriesのブロック超酸NACURE(登録商標)7231(アンチモン酸アンモニウム)である。
【0067】
コーティング粉末は、約0〜約5重量%の範囲のフローコントロール剤を含有することもでき、約0.1重量%〜約2重量%の範囲が最も好ましい。フローコントロール剤の例としては、MODAFLOW(登録商標)ポリ(アルキルアクリラート)(すなわち、MODAFLOW 6000(登録商標))製品、およびサーフィノール(Surfynol)(登録商標)アセチレンジオール(すなわち、P200(登録商標))など、ヒドロキシル、カルボキシル、または他の官能基を含む他のものが挙げられる。官能化フロー添加剤は、粉体コーティングの修正(touch−up)または修復が必要である場合に層間付着力の助けにもなる。フロー添加剤を単独または組み合わせて使用することができる。抗酸化剤を約0.5〜約2.0phrの濃度で使用して、本発明に適した比較的低い硬化温度でさえコーティングの変色を防止することもできる。本明細書の他で記載するように、実施例8の実施例3B樹脂によって例示される本発明の樹脂自体は、フローコントロール剤として働くことができる。
【0068】
二酸化チタンおよび/またはカーボンブラックなどの顔料、炭酸カルシウムなどの充填剤、粒状ゴムなどのテキスチャー付与剤、ベントナイト粘土、商標LANCOWAX(登録商標)で販売されているものなどのポリエチレン粉末含有または不含ポリテトラフルオロエチレン(PTFE)粉末、ならびに他の通常の添加剤も、外観およびコスト削減のために存在させることができる。
【0069】
ベンゾインを、通常はピンホール防止剤として使用する(Howellの参考文献を参照のこと)。
表3は、市販の粉体コーティング樹脂の一覧を示し、その官能性およびTgを記述する。これらの樹脂の一部を本明細書の実施例の調製で使用した。残りは、他の配合物で使用され、本発明の様々な実施形態で有用である。
【0070】
【表5】
【0071】
下記に、本発明による粉体コーティング配合物を調製するために適応させることができる一般的手順を記載する。
手順:粉体コーティング混合プロトコル:
通常は、Brabender(登録商標)混合機を使用するが、手順を他のタイプの混合機向けに適応させることができる。
・120mlのボウルサイズに対して約70〜80gに等しくなるように粉体コーティング配合物を算出する。典型的な小型Sigma Bladeボウルには、顔料不含から低顔料/結合剤(P/B)のコーティング配合物70g、またはより高いP/Bの塗料80gが入る。
・油加熱器を始動させることによって、Brabender(登録商標)混合機または同様な混合機を99℃に予熱する。30分間予熱しておく。
・予熱が完了したとき、ローターを始動させ、ボウルが装置に対して安全であるか試験する。
・トルクセンサーのスイッチを入れる。これは、ミックスが前進するためのガイドとして働く。
・約30gの一次樹脂をボウルに徐々に添加する。
・樹脂を、トルクセンサーが定常値を示すまで(約5分間)混合し、溶融させ、次いで一次樹脂の残部を混合ボウルに添加し、混合し、溶融させる。
・添加剤があればそれ/全部を、ローター間の混合ゾーン中心部に添加する。
・トルク値が定常になるまで混合しておく(通常は、約10分間)。
・架橋性樹脂を全部、ボウルに徐々に添加する。少なくとも3分間混合し、架橋が開始する(トルク示度が急速に上昇し始める)といけないので、トルク示度が定常のままであることを確認する。
・最後に、触媒が(必要である場合には)それを、トルク示度を綿密に見ながら添加する。トルクは上昇するはずであり、粘度(トルク)が10%上昇した後、バッチを止めるべきである。
・混合ボウルから、生成物を濃厚な溶融材料としてすぐに取り出し、硬質になる(通常は、硬質で脆弱な光沢のある材料になる)まで所望の温度(通常は、室温)に冷却する。
・所望の温度に冷却した後、生成物を小チップに分解する。
・次いで、チップをボールミルにかけ、またはその他の(例えば、塗料チップを10mm〜15mmのスチールメディアの存在下で16時間ボールミルする)方法で微粉砕して、細粉末を得る。
・粉末を適切なスクリーンに通してふるいにかけて、大きな断片、通常は約105ミクロンより大きい断片を除去する。
【0072】
記載された方法を利用して、いくつかのクラスの粉体コーティング配合物を調製することができる。
本発明に従って仕上げ粉体コーティングを製造するための一般的手順は、以下の通りである。
・コーティングするために、基材に適した溶媒(例えば、水、メチルエチルケトン、イソプロピルアルコール)で、基材をきれいに拭くことによって調製する。
・基材を下塗りする。
・粉体コーティングを、Nordson Corporationから供給されるVersa−Spray(登録商標)などの粉末スプレーガンの試料リザーバーに注ぎ込む。
・電圧コントロールをスプレーガン制御装置で調整して、適切な電荷を粉末に加えるようにする。
・粉末を、標準的な粉体コーティング技法で塗布して、乾燥被膜厚が約50.8〜76.2ミクロン(2〜3ミル)の所望の被膜厚を実現する。
・次いで、基材をオーブンに適切な時間および温度で配置する。
【0073】
通常は、β−ヒドロキシアミド系粉体コーティングは、ジ−N−β−ヒドロキシアミド架橋剤でカルボキシル官能性をエステル交換架橋することに基づく。これらのタイプの粉末は、市販のPRIMID(登録商標)タイプの粉末によって例示される。実施例4は、バイオベース樹脂対通常の石油化学系樹脂の配合物および硬化の詳細を示す。このタイプの化学は、触媒作用の影響を受けない。したがって、硬化速度には有意差は期待されず、実際、いずれの樹脂についても硬化速度の利点がこの場合には検出されなかった。
【0074】
2つのコーティングを、121℃と147℃の2つの異なる温度で30分間硬化した。最大の差は、より高い温度での光沢であり、バイオベース樹脂配合物は対照より約50ユニット高かった。しかし、耐溶媒性については、バイオベース配合物のほうがわずかに低かった。明らかに、バイオベース樹脂は、β−ヒドロキシアミド(登録商標)架橋配合物において市販の対照と比較したとき、全体的に性能が劣らない。
ハイブリッド粉体コーティング:カルボン酸−エポキシ架橋
カルボン酸−エポキシ架橋粉体コーティングは、ハイブリッドコーティングの最も一般的なものである。通常は、これらは、アクリル酸エポキシ架橋剤と共に配合された石油由来のポリエステル酸からなる。本発明のカルボン酸官能性バイオベース樹脂を合成し、典型的な配合物において市販の石油化学系ポリエステル酸と比較して試験した。
【0075】
図4Aは、本開発によるバイオベース樹脂と典型的な市販の樹脂(FINE−CLAD 8400)について、121℃におけるせん断速度に対する粘度(単位:ポアズ)の比較を示す。バイオベース樹脂(下部のデータ点群)は、その対照物より粘度が低いことに留意されたい。
【0076】
粘度の差に基づいて、バイオベース材料はより低い温度でより高いフローをもたらし、低温硬化コーティングの外観を全般的に改善できるという可能性がある。様々な工業製造方法による粗さ平均測定値を用いて、改善の影響を概算測定することができる。
【0077】
実施例4Aでは、対照の石油化学系透明コートの表面粗さ(Ra)は4.2と評価し、バイオベース透明コートは1.3と評価される。石油由来のパネルの粗さは、典型的な鋸引き操作に等価であり、バイオベースのパネルは、典型的な電子ビームまたはレーザー操作に等価であった。バイオベース配合物は、対照よりはるかに「クラスA」仕上げに近い。
【0078】
実施例4Aのバイオベース粉体コーティングおよび市販の石油由来の低温硬化粉体コーティング(Forrest Powder Low Temperature Cure(登録商標))のパネルの比較を行った。両パネルは、約63.5ミクロン(約2.5ミル)の被膜厚でスプレーし、次いで121℃で30分間熱硬化した。バイオベース粉末材料は、比較の粉体コーティングよりかなり少ないゆず肌(または表面粗さ)しか示さなかった。さらに、バイオベース粉末で被覆されたパネルのほうは、60℃ではるかに高い光沢を示した(72点対50点)。配合物の詳細については実施例4Aを参照のこと。
【0079】
バイオ由来配合物の改善されたメルトフローを、応力制御レオメータで測定した。粉末の試料を、100℃に加熱されたプラテン間に配置し、典型的な粉体コーティング被膜の厚さ(50.8ミクロン(約2ミル))に圧縮した。温度を121℃に上げ、試料が硬化するまで粘度の変化(単位:ポアズ)を測定する。粘度データを図5に示す。
【0080】
上部の曲線データ点は、比較対照の粉末試料を示し、下部の曲線データ点は、バイオベース粉末試料を示す。バイオベース配合物の初期粘度は、対照試料より有意に低い(3694ポアズ対11980ポアズ)。時間が経過するにつれて、試料が硬化するので試料は両方とも粘度が上昇した。硬化時間の残りの間、バイオベース配合物のほうの粘度は低いままだったので、この粉末は、被膜が架橋し硬化する前に溶融し流展する機会が多かった。
【0081】
柔軟性(靭性の尺度)は、最終生成物が、被覆された物体の使用に伴う日常的な突起および凹みに耐えることを可能にする重要なコーティング属性である。柔軟性が十分でないと、コーティングのひび割れが起こり、衝撃が起きたときに基材の剥離が起こることもある。基材を戸外で使用する場合、水、紫外線、酸化、および酸性雨など、大気中の化学物質は、被膜を劣化させ、脆化させる恐れがある。これらの要因の多くは腐食の原因ともなり、錆、および不十分な外観という損失、ならびに不十分な柔軟性がもたらされる。
【0082】
対照試料と真上で述べたバイオベース配合物を、マンドレル屈曲と呼ばれる柔軟性試験(ASTM D522)で比較した。この試験では、被覆された基材を万力にクランプで留め、円錐形マンドレル上を回転させた。領域のテープ接着性試験によって、コーティングの最終性能が決定された。テープをコーティング上の屈曲領域に貼り付け、剥がして、コーティングがまだパネルに接着しているかどうかを決定する。
【0083】
円錐形マンドレルは、その長さの全域で、試験に利用可能な最小のサイズであり、かつコーティングの剥離またはひび割れがなく通過するのに最も靭性の高いサイズである3.18mm(1/8インチ)まで様々な幅を有した。
【0084】
バイオベースのコーティングは、わずかなひび割れしか明らかでなく、かつ剥離の徴候は明らかでなかったので、良好な柔軟性を有した。対照の石油由来のコーティングは、パネルの長さ全体にわたってひび割れが起こり、屈曲の長さの約40%でコーティングが剥がれた。配合物の詳細については実施例5を参照のこと。
【0085】
使用温度で低粘度であることが顔料表面の良好な濡れにとって好ましい場合、顔料を含む粉体コーティングはいくつかの利点がバイオベース樹脂配合物に由来することもある。実施例6では、2つの黒色配合物(対照配合物1つおよびバイオベース配合物1つ)を記載する。
【0086】
バイオベース粉体コーティングは、石油化学製品由来のコーティングより、60℃で光沢がはるかに高かった(85点対44点)。これもやはり、おそらく熱硬化中、配合物のメルトフローがよりよいことに起因した。
【0087】
黒色顔料の発色/漆黒度(jetness)を、バイオベース配合物で改善した。漆黒度は、コーティングのLおよびb色成分を測定することによって決定することができる。(Hunter Color Scaleの説明については、Wicks,Z.W.ら、「Organic Coatings:Science and Technology」、第2版、特に351〜355頁、Wiley Interscience,NY,NY.ISBN 0−471−24507−0 1999を参照のこと)。
【0088】
2つの黒色パネルの全デルタE、すなわち色差は0.52であり、バイオベースのほうが、発色が強かった(漆黒)。対照パネル(左)は、黒色顔料がバイオベース配合物ほどコーティング系にうまく分散されていなかったので、バイオベース配合物より灰色に見えた。これは、おそらくバイオベース樹脂の低粘度に起因する。
【0089】
次に図6を参照して、この図は、本発明に従って樹脂を作製するのに使用された装置100の様々な要素を示す概略図である。加熱マントル102は、反応器101を少なくとも一部包囲し、反応混合物104を含む反応器101の温度を制御するのに使用される。反応器101は、反応容器106およびトップ部108からなる。トップ部108は、様々な器具に連結するための複数の頸部110、112、114、116を有する。撹拌は、撹拌軸122(例えば、ステンレス鋼)の端部にあるパドル120(例えば、通常は45°の角度の羽根)によって行われる。撹拌軸122は頸部116を貫通している。コネクタ131を介して熱電対132に連結された熱電対コントローラ130は、シールした装置のガス入口コネクタ111で頸部110を貫通して、反応混合物104に到達する。Vigreauxカラム140を、シールした関係で頸部114に取り付ける。温度計141または他の温度測定装置を、Vigreauxカラム140の塔頂部(蒸留ヘッド)142に取り付ける。コネクタ146を用い、冷却器入口152を介して、冷却器150をVigreauxカラム140の頸部144に取り付ける。Vigreauxカラム140は、ジャケットで包囲された独立型装置とすることができ、またはジャケットおよびカラムは一体型とすることができる。排気出口164および頸部出口166を有する頸部160の頸部入口162に、冷却器出口154を連結する。受けフラスコ170は、頸部出口166に連結させた入口172を有する。冷却液体155は、冷却器150に入口156から入り、出口158から出る。
【0090】
操作の際には、アルゴンガス111−1は、ガス入口コネクタ111から入って、反応混合物104をブランケットし、ガス出口164から流出する。材料は、装置を閉じる前に、または頸部112のシールしたコネクタ118を通して、添加することができる。図6では、頸部112は、頸部116の真後ろに配置されていることに留意されたい。頸部112を反応器101の中心軸190上に配置する。留出液178を受けフラスコ170に回収する。
【0091】
下記の実施例は、本発明の様々な態様を例示するものであり、決して本発明の範囲を限定するものではない。
【0092】
[実施例1]
樹脂生成(実施例1〜実施例3F)
この実施例は、ヒドロキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、ジメチルテレフタラート(DMT)(228.30g、1.1757モル)、Speziol C36/2 1075(登録商標)ダイマージオール(バッチ#415252)(77.61g、0.1411モル)、D−イソソルビド(123.90g、0.84785モル)、およびエチレングリコール(EG)(102.81、1.6563モル)と、続いて酢酸マンガン(II)四水和物(0.0917g)、酢酸コバルト(II)四水和物(0.0618g)、および酸化アンチモン(III)(0.103g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を30分間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約150℃超で加熱したとき、メタノールを連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これはメタノールが除去されたことを示唆する。約95mlのメタノールを留去した。引き続いて、ポリリン酸(0.0634g)のEG(1g)溶液を反応混合物に添加した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、エチレングリコールが留出し(91g)、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分間維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0093】
下記の特性を有する樹脂を生成した。
溶液固有粘度:0.29(溶媒はo−クロロフェノールである。92%のみ可溶)
Tg=61℃
ヒドロキシル値=24.3
酸価=8.0
分子量(MW)=3470(酸およびヒドロキシル値から算出)
ポリマー特性:
色:茶色
粘着性:非粘着性
清澄性:わずかに半透明
柔軟性:脆い
固体
【0094】
[実施例1A]
この実施例は、バイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、ジメチルテレフタラート(DMT)(197.74g、1.0183モル)、D−イソソルビド(119.05g、0.81463モル)、およびSpeziol C36/2 1075(登録商標)ダイマージオール(バッチ# 415252)(112.06g、0.20371モル)と、続いて1,2,3,4−テトラヒドロナフタレン(2ml)および酸化アンチモン(III)(0.089g)を加えた。反応器をアルゴンでブランケットした。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を12分間維持した。反応混合物を、20分間かけて250℃に徐々に加熱した(2.5℃/分)。この温度を8分間維持した。反応を約150℃超で加熱したとき、メタノールを連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これはメタノールが除去されたことを示唆する。約83mlのメタノールを留去した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、13分かけて280℃に徐々に加熱した(2.3℃/分)。次いで、反応混合物を260℃まで放冷した。追加のD−イソソルビド(14.87g、0.1018モル)を反応混合物に加えた。反応混合物を280°に加熱した。この温度を30分間維持した。留出液受器を真空受器に換え、徐々に真空にした(≦1199.898Pa(9トル))。この間に、低分子量ポリマーが形成した。反応混合物温度を280℃で2時間40分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0095】
下記の特性を有する樹脂を生成した。
溶液固有粘度:0.10(溶媒はo−クロロフェノールである)
Tg=165℃
ヒドロキシル値=45.0
酸価=2.3
分子量(MW)=2372(酸およびヒドロキシル値から算出)
ポリマー特性:
色:明茶色
粘着性:粘着性
清澄性:半透明
柔軟性:幾分脆い
固体
【0096】
[実施例2]
この実施例は、カルボキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)。
4頸トップを備えた5リットルの丸底ガラス反応容器、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、D−イソソルビド(1337.0g、9.1490モル)(受理したまま)、Pripol 1013(登録商標)ダイマー酸(バッチ# 091687)(699.1g、1.215モル)、および1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(1563.8g、9.0826モル)と、続いて酸化アンチモン(III)(1.231g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、47分間かけて250℃に徐々に加熱した(1.1℃/分)。この温度を3.1時間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約329mlの水が留出した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0097】
下記の特性を有する樹脂を生成した。Tg=64.2℃
酸価=34.8
分子量(MW)
GPC(ポリスチレン標準)Mn=1689
GPC(ポリスチレン標準)Mw=11681
多分散性(Mw/Mn)=6.91
ポリマー特性:
色:明コハク色
粘着性:非粘着性
清澄性:半透明
柔軟性:脆い
固体
【0098】
[実施例3]
この実施例は、カルボキシル官能性バイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと).
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(204.66g、1.1886モル)、Empol 1018(登録商標)ダイマー酸(バッチ# U42G151910)(72.54g、0.1251モル)、およびD−イソソルビド(172.80g、1.1824モル)と、続いて酸化アンチモン(III)(0.1594g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を30分間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約47mlの水が留出した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0099】
下記の特性を有する樹脂を生成した。
溶液固有粘度=0.25dl/g(溶媒はo−クロロフェノールである):
Tg=66.9℃
ヒドロキシル値=13.0
酸価=36.3
分子量(MW)
GPC(ポリスチレン標準)Mn=2995
GPC(ポリスチレン標準)Mw=9560
多分散性(Mw/Mn)=3.19
ポリマー特性:
色:明茶色
粘着性:非粘着性
清澄性:殆ど半透明
柔軟性:脆いが硬質
固体
【0100】
[実施例3A]
この実施例は、カルボキシル官能性バイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
4頸トップを備えた5リットルの丸底ガラス反応容器、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(1570.3g、9.1202モル)、Pripol 1013(登録商標)ダイマー酸(バッチ# 091687)(675.7g、1.174モル)、D−イソソルビド(得たまま)(1354.0g、9.2648モル)と、続いて酸化アンチモン(III)(1.247g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、51分間かけて250℃に徐々に加熱した(1.0℃/分)。この温度を3.1時間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約334mlの水を留去した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0101】
下記の特性を有する樹脂を生成した。
Tg=65.3
酸価=29.0
分子量(MW)
GPC(ポリスチレン標準)Mn=2162
GPC(ポリスチレン標準)Mw=11872
多分散性(MW/Mn)=5.49
ポリマー特性:
色:黄色/明コハク色
粘着性:非粘着性
清澄性:半透明
柔軟性:脆い
固体
【0102】
[実施例3B]
この実施例は、ヒドロキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、ジメチルテレフタラート(DMT)(228.30g、1.1757モル)、Speziol C36/2 1075(登録商標)ダイマージオール(バッチ# 415252)(129.40g、0.23523モル)、D−イソソルビド(123.90g、0.84785モル)、およびエチレングリコール(EG)(89.66g、1.444モル)と、続いて酢酸マンガン(II)四水和物(0.0917g)、酢酸コバルト(II)四水和物(0.0618g)、および酸化アンチモン(III)(0.103g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を30分間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約150℃超で加熱したとき、メタノールを連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これはメタノールが除去されたことを示唆する。約95mlのメタノールを留去した。引き続いて、ポリリン酸(0.0634g)のEG(1g)溶液を反応混合物に添加した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、30分間かけて280℃に徐々に加熱した(1℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、エチレングリコールが留出し(84g)、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0103】
下記の特性を有する樹脂を生成した。
溶液固有粘度:0.19(溶媒はo−クロロフェノールである)
Tg=28.4℃
ヒドロキシル値=35.4
酸価=6.1
分子量(MW)=2700(酸およびヒドロキシル値から算出)
ポリマー特性:
色:茶色
粘着性:非粘着性
清澄性:殆ど半透明
柔軟性:脆い
固体
【0104】
[実施例3C]
この実施例は、ヒドロキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、ジメチルテレフタラート(DMT)(213.96g、1.1018モル)、Empol 1018(登録商標)ダイマー酸(バッチ# U42G151910)(71.02g、0.1225モル)、D−イソソルビド(128.79g、0.88128モル)、およびエチレングリコール(EG)(116.28g、1.8734モル)と、続いて酢酸マンガン(II)四水和物(0.0859g)、酢酸コバルト(II)四水和物(0.0579g)、および酸化アンチモン(III)(0.0965g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を30分間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約150℃超で加熱したとき、メタノールを連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これはメタノール/水の混合物が除去されたことを示唆する。約93mlのメタノール/水の混合物を留去した。引き続いて、ポリリン酸(0.0594g)のEG(1g)溶液を反応混合物に添加した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、エチレングリコールが留出し(95g)、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0105】
下記の特性を有する樹脂を生成した。
溶液固有粘度:0.23(溶媒はo−クロロフェノールである)
Tg=58.8℃
ヒドロキシル値=23.7
酸価=1.4
分子量(MW)=4470(酸およびヒドロキシル値から算出)
ポリマー特性:
色:明茶色
粘着性:非粘着性
清澄性:幾分半透明、わずかな曇り
柔軟性:脆い
固体
【0106】
[実施例3D]
この実施例は、カルボキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
2リットルの4頸円柱壁付き丸底ガラス反応容器、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(610.68g、3.5468モル)、Pripol 1013(登録商標)ダイマー酸(バッチ# 091687)(262.78g、0.45670モル)、D−イソソルビド(アセトンで再結晶)(526.54g、3.6030モル)と、続いて酸化アンチモン(III)(0.4849g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中で反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を2.1時間維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約129mlの水を留去した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0107】
下記の特性を有する樹脂を生成した。
Tg=62.3
酸価=34.7
分子量(MW)
GPC(ポリスチレン標準)Mn=3517
GPC(ポリスチレン標準)Mw=12753
多分散性(Mw/Mn)=3.63
ポリマー特性:
色:コハク色/橙色
粘着性:非粘着性
清澄性:半透明
柔軟性:脆い
固体
【0108】
[実施例3E]
この実施例は、カルボキシル官能性バイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと).
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(318.36g、1.8490モル)、Empol 1018(登録商標)ダイマー酸(バッチ# U42G151910)(112.84g、0.1946モル)、およびD−イソソルビド(268.80g、1.8393モル)と、続いて酸化アンチモン(III)(0.2479g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を2.3時間維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約74mlの水が留出した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0109】
下記の特性を有する樹脂を生成した。
溶液固有粘度=0.24dl/g(溶媒はo−クロロフェノールである):
Tg=72.3℃
ヒドロキシル値=0.0
酸価=32.8
分子量(MW)
GPC(ポリスチレン標準)Mn=4027
GPC(ポリスチレン標準)MW=15756
多分散性(MW/Mn)=3.91
ポリマー特性:
色:黄色−茶色
粘着性:非粘着性
清澄性:殆ど半透明
柔軟性:脆い
固体
下記の実施例4〜8は、本発明によるいくつかの典型的な粉末配合物、および仕上げコーティングを例示する。
【0110】
[実施例3F]
(顔料分散剤)
この実施例は、顔料の存在下で使用した場合に改善された分散剤特性を有するカルボキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
4頸トップを備えた2リットルの丸底ガラス反応容器、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、D−イソソルビド(545.35g、3.7317モル)(受理したまま)、Pripol 1013(登録商標)ダイマー酸(バッチ# 091687)(272.17g、0.47302モル)、および1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(632.49g、3.6734モル)と、続いて酸化アンチモン(III)(0.498g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を2.1時間維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約134mlの水が留出した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で30分間維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0111】
下記の特性を有する樹脂を生成した。
Tg=52.9℃
酸価=47.7
120℃における粘度=7772ポアズ
160℃における粘度=247ポアズ
ポリマー特性:
色:黄色/明コハク色
粘着性:非粘着性
清澄性:半透明
柔軟性:脆い
固体
【0112】
[実施例4]
この実施例は、実施例3のカルボキシル官能性ポリエステル樹脂を使用したβ−ヒドロキシアミド型架橋による粉体コーティング配合物の調製を例示する。次いで、粉末配合物を基材に塗布する。
【0113】
上記に記載するように混合し、β−ヒドロキシアミドエステル交換で架橋した典型的な粉体コーティング配合物の並列比較で、カルボキシル官能性ポリエステル(実施例3の生成物)を市販のポリエステルと比較した。下記の表4は、これらの配合物を重量百分率で示す。
【0114】
【表6】
【0115】
FINE−CLAD M8930(登録商標)は、比較のために使用されたポリエステル酸の例である。
手順:粉体コーティング混合プロトコル(Brabender(登録商標)混合機による99℃での混合):
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。約30分間予熱させた。予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで、30gの一次樹脂をボウルに徐々に添加し、溶融するまで混合した。次いで、一次樹脂の残部35.2gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。次いで、1.3gの添加剤(0.9gのベンゾインおよび0.4gのModaflow 6000(登録商標))をローター間の混合ゾーン中心部に添加した。
【0116】
混合を10分間続行し(トルク値を安定性について監視した)、次いで3.4gの架橋性樹脂(Primid XL−552(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0117】
混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0118】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧した。パネルを121℃または147℃で30分間硬化した(試験結果については、表6を参照のこと)。
【0119】
上記の手順を繰り返して、対照材料を得た。
バイオベースのコーティングは、光沢に関して様々な温度でロバスト性がはるかに高く、より低い温度では等価な光沢を有し、より高い温度ではるかによりよい光沢を有した。
【0120】
示差走査熱分析(DSC)の結果は、実験した樹脂が、硬化温度、硬化の大きさ、またはコーティングの最終Tgに、ポジティブにもまたはネガティブにも影響しないことを示した。これは、外的影響に対するβ−ヒドロキシアミド硬化速度の非感受性(Howellの参考文献を参照のこと)に一致した。
【0121】
【表7】
【0122】
バイオベースのコーティングも、両方の硬化温度において市販対照と同様な最終特性を有した。
【0123】
【表8】
【0124】
表5および6のデータは、両方のコーティングが性能全体において本質的に等価であることを示す。バイオベースのコーティングは、より高い硬化温度における光沢でわずかな利点を有し、市販対照は、耐溶媒性でわずかな利点を有した。先に述べたように、多大な温度ロバスト性を必要とする配合物において、より高い温度での光沢は顕著な利点となり得る。
【0125】
したがって、このタイプのバイオベース樹脂は、β−ヒドロキシアルキルアミドを用いたエステル交換架橋反応として有用である。
【0126】
[実施例4A]
この実施例は、カルボキシル官能性樹脂を使用したβ−ヒドロキシアミド型架橋による粉体コーティング配合物の調製を例示する。
上記に記載するように混合し、β−ヒドロキシアミドエステル交換で架橋した典型的な粉体コーティング配合物の並列比較で、カルボン酸官能性ポリエステル(実施例3E)を市販のポリエステルと比較した。表4Aは、これらの配合物を重量百分率で示す。
【0127】
【表9】
【0128】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約85gの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。約30分間予熱させた。予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂をボウルに徐々に添加し、溶融するまで混合した。次いで、一次樹脂の残部37.8gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させ(約5分間)、次いで3.6gの添加剤(1.1gのベンゾインおよび2.5gのFine Clad A241(登録商標))をローター間の混合ゾーン中心部に添加し、混合を10分間続行し(トルク値を安定性について監視した)、次いで4.1gの架橋性樹脂(Primid XL−552(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0129】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧した。パネルを121℃または147℃で30分間硬化した(試験結果については、表6を参照のこと)。実施例4Aの上記の手順を繰り返して、対照材料を得た。
【0130】
バイオベースのコーティングは、光沢に関して様々な温度でロバスト性がはるかに高く、より低い温度では等価な光沢を有し、より高い温度ではるかによりよい光沢を有した。
示差走査熱分析(DSC)の結果は、実験したコーティングが、硬化温度、硬化の大きさ、またはコーティングの最終Tgに、ポジティブにもまたはネガティブにも影響しないことを示した。これは、外的影響に対するβ−ヒドロキシアミド硬化速度の非感受性(Howellの参考文献を参照のこと)に一致した。
【0131】
【表10】
【0132】
下記の表6Aからわかるように、バイオベースのコーティングも、両方の硬化温度において市販対照と同様な最終特性を有した。
【0133】
【表11】
【0134】
表5Aおよび6Aのデータは、両方のコーティングが性能全体において本質的に等価であることを示す。バイオベースは、より低い硬化温度でより硬質の被膜の利点を有した(#H鉛筆対#HB鉛筆)。バイオベースはまた、より高い硬化温度における光沢で利点を有し、市販対照は、耐溶媒性で利点を有した。先に述べたように、多大な温度ロバスト性を必要とする配合物において、より高い温度での光沢は顕著な利点となり得る。
【0135】
したがって、このタイプのバイオベース樹脂は、β−ヒドロキシアルキルアミドでのエステル交換架橋反応として有用である。
【0136】
[実施例5]
この実施例は、カルボキシル官能性ポリエステル樹脂(実施例2の生成物)およびアクリル酸エポキシ架橋剤と配合されたハイブリッドコーティングのための配合物の調製を例示する。
手順:バイオベースのハイブリッド粉体コーティングのための粉体コーティング混合プロトコル(Brabender(登録商標)混合機による99℃での混合):
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働く。次いで、30gの一次樹脂(実施例2に記載した樹脂)をボウルに徐々に添加し、溶融するまで混合した。次いで、一次樹脂の残部21.5gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。混合を10分間続行した(トルク値を安定性について監視した)。16.9gの架橋性樹脂(Fine Clad A229−30A(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒を細粉に粉砕し、最後に添加した(0.4gのイミダゾールおよび1.1gのドデカン二酸)。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0137】
混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0138】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、約63.5ミクロン(約2.5ミル)の乾燥被膜厚にした。パネルを121℃で30分間硬化した。
手順:対照ポリエステルハイブリッド粉体コーティングのための粉体コーティング混合プロトコル(Brabender(登録商標)混合機による99℃での混合):
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。
【0139】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(Fine−Clad M8400(登録商標))をボウルに徐々に添加し、溶融するまで混合した。一次樹脂の残部25.8gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させ(約5分間)、混合を10分間続行した(トルク値を安定性について監視した)。12.7gの架橋性樹脂(Fine Clad A229−30A(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒(0.4gのイミダゾールおよび1.1gのドデカン二酸)を最後に添加し、トルク示度を綿密に監視した。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0140】
混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0141】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、約63.5ミクロン(約2.5ミル)の乾燥被膜厚にした。パネルを121℃で30分間硬化した。
【0142】
図4に記載された粘度を有する、この実施例に記載した2つのポリエステル樹脂を、これらの手順に従って透明コートに配合し、下記の表7に示す。表7は、様々な材料の量を重量百分率で示す。
【0143】
【表12】
【0144】
バイオベース材料と対照系について、硬化の外観に及ぼす影響を比較した。低温硬化型粉体コーティングを対照として選択した。被験製品を1PC−306−0040(F−0040)S−9 Clear Gloss(登録商標)と称する。硬化スケジュールは、145℃で15分間、または162℃で10分間であった。
【0145】
コーティングの表面粗さを測面計で定量化した。この試験の最中に、細針がコーティングの表面上を通過し、同時に表面の山と谷を記録した。谷をRv(nm)として記録し、山をRp(nm)として記録した。これらの2つの値から、平均粗さ(Ra)を算出する。より低いR値は、より平坦またはより滑らかな表面を示唆する。被覆パネルのR値については、表8を参照のこと。
【0146】
【表13】
【0147】
一般的な生成方法で生成された表面粗さについての典型的なR値(ASME B46.1−1995に列挙)を、表8の値と比較することができる。バイオベースのパネルの表面粗さは、グライディング、ホーニング、および電解研摩で生成された表面と同様である。対照パネルの表面粗さは、スナッギング、平削り、および形削り操作で生成された表面と同様である。
【0148】
[実施例6]
この実施例は、実施例3Aのバイオベースのカルボキシル官能性ポリエステルおよびエポキシ架橋剤と配合された、顔料を含むハイブリッド粉体コーティングを例示する。
バイオベース樹脂が、顔料を分散し、顔料を発色させる優れた能力を有する場合に、顔料を含む粉体コーティングは利益を得ることができる。黒色粉体コーティング配合物(下記)の例は、市販対照に対して作製した。表9を参照のこと。表9の配合物は、様々な材料の量を重量百分率で示す。
【0149】
【表14】
【0150】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。
【0151】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(実施例3Aに記載)をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部23.1gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。1.7gの添加剤(0.8gの黒色顔料および0.9gのベンゾイン)をローター間の混合ゾーン中心部に添加した。混合を10分間続行した(トルク値を安定性について監視した)。架橋性樹脂としてエポキシ官能基を含むアクリル(FineClad A257(登録商標))11.3gを先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒を細粉に粉砕し、最後に添加した(0.5gのイミダゾール、1.7gのドデカン二酸、および1.7gのNacure XC−7231(登録商標))。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0152】
混合ボウルから、滑らかで堅い光沢のある黒色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0153】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0154】
対照のポリエステル顔料を含む粉体コーティングでは、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。
【0155】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(Fine−Clad M8400(登録商標))をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部21.9gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。1.7gの添加剤(0.8gの黒色顔料および0.9gのベンゾイン)をローター間の混合ゾーン中心部に添加した。混合を10分間続行した(トルク値を安定性について監視した)。12.5gの架橋性樹脂(FlneClad A257(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒(0.5gのイミダゾール、1.7gのドデカン二酸、および1.7gのNacure XC−7231(登録商標))を最後に添加し、トルク示度を綿密に監視した。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0156】
混合ボウルから、滑らかで堅い光沢のある黒色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0157】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0158】
バイオベース粉体コーティングは、石油化学製品由来のコーティングより、はるかに高い60度光沢値を有した(85点対44点)。これは、おそらく熱硬化中、配合物のメルトフローがよりよいことに起因した。
【0159】
バイオベース配合物については、黒色顔料の発色/漆黒度が改善される。コーティングのLおよびbの色成分を測定することによって、漆黒度を決定することができる。(Hunter color Scaleの説明については、上記に引用したWicks,Z.W.らを参照のこと)。
【0160】
極黒色は低いL値で決まり、藍色のアンダートーンは低いb値で決まる。Lおよびbの値が低くなると、コーティングの漆黒度が高くなる。漆黒度は、よりよい顔料分散および発色で改善される。下記の表10は、被覆パネルの色測定値を示す。
【0161】
【表15】
【0162】
2つの黒色パネルの全体のデルタE、すなわち色差は、0.52である。石油由来のパネルは、黒色顔料が、バイオベース配合物ほどよくコーティング系に分散していないので、バイオベース配合物のパネルより灰色に見える。
【0163】
[実施例6A]
粉体コーティングにおいて低温フローおよび硬化が必要であるのに加えて、コーティングタイプに関わらず、コーティングマトリックス内において顔料の良好な分散も必要である。これを実現するために、異なる相溶性の成分を有するポリマーが設計されている。ポリマー分散剤は、塗料、コーティング、およびインク系中の顔料および他の材料を、最も典型的には立体安定化によって安定化する。ポリマー分散剤は、定着基とポリマー鎖からなる2成分構造を有する。最も典型的には、定着基は、粒子表面、およびコーティングの連続相と相溶であるポリマー鎖と相互作用する極性材料である。実際には、該ポリマー基は粒子の周りにコーティングを形成し、粒子同士が接触し、より大きい非相溶な凝集体になるのを防止する。
【0164】
有効なポリマー分散剤を与えるものと予想され得る定着基/ポリマーの立体配置は多数存在する。本発明の樹脂は、極性カルボン酸定着部位および非極性植物油鎖を有し、したがって分散剤および結合剤として働くことができる。分散剤としても働くことができる硬化結合剤によって、多数の顔料を分散するために別々の添加剤を必要とすることがなくなる可能性がある。
【0165】
表10−6A−1の配合物は、様々な材料の量を重量百分率で示す。
【0166】
【表16】
【0167】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を110℃に予備加熱した(ボウルは約110℃に上昇した)。30分間予熱させた。
【0168】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30.0gの一次樹脂(実施例3に記載)をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部33.0gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させ(約5分間)、混合羽根の速度を40回転/分に設定した。7.0gの黒色顔料をローター間の混合ゾーン中心部に添加した。混合を15分間続行した。混合ボウルから、滑らかで堅い光沢のある黒色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。
【0169】
対照ポリエステル黒色分散液では、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を110℃に予備加熱した(ボウルは約110℃に上昇した)。30分間予熱させた。
【0170】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30.0gの対照ポリエステル(FineClad M8400)をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部33.0gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させ(約5分間)、混合羽根の速度を40回転/分に設定した。7.0gの黒色顔料をローター間の混合ゾーン中心部に添加した。混合を15分間続行した。混合ボウルから、滑らかで堅い光沢のある黒色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。
【0171】
これらの材料をその後の配合物で使用して、カーボンブラック顔料の分散度を決定した。黒色顔料の分散がよくなると、最終配合物の得られる色が濃く(黒く)なる。
表10−6A−2 の配合物は、様々な材料の量を重量百分率で示す。
【0172】
【表17】
【0173】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。
【0174】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで34.9gの一次樹脂(FINE−CLAD M8400(登録商標))をボウルに徐々に添加し、溶融するまで混合させた。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。22.1gの添加剤(21.0gの白色顔料(Kronos 2310)および1.1gのベンゾイン)をローター間の混合ゾーン中心部に添加し、ローターの速度を60回転/分に設定し、混合を15分間続行した。バイオベースポリエステル樹脂に事前に分散させた黒色顔料(表10−6A−2の実施例A)3.5gを添加し、ローターの速度を40回転/分に下げ、混合を5分間続行した。7.2gの架橋性樹脂(FineClad A257(登録商標))を先の混合物に添加し、混合を少なくとも2分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒を細粉に粉砕し、最後に添加した(0.5gのイミダゾールおよび1.9gのドデカン二酸)。混合を2分間続行した。
【0175】
混合ボウルから、滑らかで堅い光沢のある灰色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0176】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0177】
120mlのボウルサイズに対して、対照ポリエステル粉体コーティング全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。
【0178】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで34.9gの一次樹脂(FineClad M8400)をボウルに徐々に添加し、溶融するまで混合させた。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。22.1gの添加剤(21.0gの白色顔料(Kronos 2310)および1.1gのベンゾイン)をローター間の混合ゾーン中心部に添加し、ローターの速度を60回転/分に設定し、混合を15分間続行した。バイオベースポリエステル樹脂に事前に分散させた黒色顔料(表10−6a−2の実施例B)3.5gを添加し、ローターの速度を40回転/分に下げ、混合を5分間続行した。7.2gの架橋性樹脂(FineCiad A257(登録商標))を先の混合物に添加し、混合を少なくとも2分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒を細粉に粉砕し、最後に添加した(0.5gのイミダゾールおよび1.9gのドデカン二酸)。混合を2分間続行した。
【0179】
混合ボウルから、滑らかで堅い光沢のある灰色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0180】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0181】
バイオベース配合物については、黒色顔料の発色および比着色力が改善される。比着色力は、カーボンブラックが二酸化チタンなど他の顔料の存在する配合物を暗色化/左右する能力である。コーティングのLおよびbの色成分を測定することによって、比着色力を決定することができる。(Hunter color Scaleの説明については、上記に引用したWicks,Z.W.らを参照のこと)。
【0182】
より高い比着色力は低いL値で決まる。L値が低くなると、カーボンブラックの分散がよくなり、コーティングでの発色がよくなる。バイオベースポリエステルは、カーボンブラックの分散をよりよくし、カーボンブラックの発色をより完全にすることができて、比着色力がより高くなる。下記の表10−6A−3は、被覆パネルの色測定値を示す。
【0183】
【表18】
【0184】
2つの黒色パネルの全体のデルタE、すなわち色差は、2.53である。石油由来のパネルは、黒色顔料が、バイオベース配合物ほどよくコーティング系に分散していないので、バイオベース配合物のパネルより明るく見える。
【0185】
[実施例6B]
この実施例は、実施例3Aのバイオベースのカルボキシル官能性ポリエステル、およびイソシアヌル酸トリグリシジル(TGIC)架橋剤と配合された、顔料を含むハイブリッド粉体コーティングを例示する。
【0186】
バイオベース樹脂が、フィッシュアイ、すなわち被膜の欠点の出現なしにコーティングのフローおよびレベリング性を促進する優れた能力を有する場合に、粉体コーティングは利益を得ることもできる。白色粉体コーティング配合物(下記)の例は、市販対照に対して作製した。表10−6B−1を参照のこと。表10−6B−1の配合物は、様々な材料の量を重量百分率で示す。
【0187】
【表19】
【0188】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を110℃に予備加熱した(ボウルは約110℃に上昇した)。30分間予熱させた。
【0189】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(実施例3に記載)をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部27.2gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。42.8gの添加剤(4.4gのTGIC、37.9gの二酸化チタン(Kronos CR2310)、および0.5gのベンゾイン)をローター間の混合ゾーン中心部に添加した。
【0190】
混合を10分間続行した(トルク値を安定性について監視した)。
混合ボウルから、滑らかで堅い光沢のある白色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0191】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0192】
対照のポリエステル顔料を含む粉体コーティングでは、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を110℃に予備加熱した(ボウルは約110℃に上昇した)。30分間予熱させた。
【0193】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(Albester 5140)をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部27.2gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。42.8gの添加剤(4.4gのTGIC、37.9gの二酸化チタン(Kronos CR2310)、および0.5gのベンゾイン)をローター間の混合ゾーン中心部に添加した。混合を10分間続行した(トルク値を安定性について監視した)。
【0194】
混合ボウルから、滑らかで堅い光沢のある白色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0195】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0196】
【表20】
【0197】
バイオベース配合物は、対照配合物よりよい耐溶媒性、高い光沢、および高い鉛筆硬度を有する。バイオベース配合物の全体の外観は、「フィッシュアイ」と呼ばれる被膜の欠点を有する対照配合物よりはるかによい。コーティング中のフィッシュアイの存在は単に外観の問題であるだけでなく、基材が環境に曝露されるので、これらの領域はさびおよび腐食の影響を受けやすい。
【0198】
本発明の別の実施形態による樹脂を添加顔料として使用して、より効率的に顔料を分散することができる。例えば、顔料を添加して、発色を助けることができる。
【0199】
[実施例7]
この実施例は、アミド−アミン官能性ポリエステル、およびエポキシ架橋剤と配合されたハイブリッド粉体コーティングの生成を例示する。
アミド−アミン官能性ポリエステル粉体コーティングをバイオベース樹脂と配合することもできる。粉体コーティング配合物の例を下記の表11に示し、材料の量を重量百分率で示す。利用可能な市販のアミドアミン官能性ポリエステル樹脂はないので、したがって対照は用いなかった。
【0200】
【表21】
【0201】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全粉体コーティングを算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。約30分間予熱させた。予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(49251−23−22)をボウルに徐々に添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。次いで、一次樹脂の残部3.7gを添加し、約5分間混合させた。次いで、0.7gの添加剤(Modaflow 6000(登録商標))をローター間の混合ゾーン中心部に添加した。混合を10分間続行した(トルク値を安定性について監視した)。次いで、30.6gの架橋性樹脂(FineClad A249A(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒(0.7gのイミダゾール、2.1gのドデカン二酸、および2.1gのNacure XC−7231(登録商標))を最後に添加し、トルク示度を綿密に監視した。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0202】
混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0203】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、95℃、107℃、または121℃で30分間硬化した(試験結果については、表13を参照のこと)。
【0204】
実施例7からの試料をDSCで分析した。結果を下記の表12に示す。
【0205】
【表22】
【0206】
どのパネルも非常に高い光沢を有していないが、被膜特性は、硬化温度107℃で正当であった。表13を参照のこと。硬度は、耐溶媒性(MEK二重摩擦)と同様に劇的に改善した。マンドレル屈曲の結果も、接着性および柔軟性の改善を示した。
【0207】
【表23】
【0208】
[実施例8]
この実施例は、バイオベースポリエステルをフロープロモーターとして使用する粉体コーティングの生成を例示する。ポリエステルポリマーを実施例3Bに記載する。
まず、対照配合物を下記の通り調製した。
【0209】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全粉体コーティングを算出した。Brabender(登録商標)混合機を93℃に予備加熱した(ボウルは約99℃に上昇した)。約30分間予熱させた。予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働いた。次いで、30gの一次樹脂(Fine−Clad M8710(登録商標))をボウルに徐々に添加し、トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。次いで、一次樹脂の残部31.8gを添加し、約5分間混合させた。次いで、0.44gの添加剤(ベンゾイン)をローター間の混合ゾーン中心部に添加した。混合を10分間続行した(トルク値を安定性について監視した)。
【0210】
17.7gの架橋性樹脂(FineClad A249A(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。
【0211】
混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。
上記の手順を6回以上繰り返して、3つのフロープロモーターをそれぞれ1%(重量)または3%(重量)組み込んだ。フロープロモーターは、ベンゾイン添加時に添加した。バイオベースのフロープロモーター(実施例3B)、および2つの市販フロープロモーター(Fine−Clad A241(登録商標)および Additol VXL9820(登録商標))を使用して、配合物B〜Gを調製した(表14を参照のこと)。
【0212】
【表24】
【0213】
低い硬化温度における粉体コーティングの粘度を下げる際のフロープロモーターの有効性を決定するために、表14の配合物の粘度を試験した。粘度が低くなれば、ベーキング中、最終被膜の流展性がよくなり、滑らかになることを示唆するはずである。表15は、90℃〜140℃における粘度(単位、ポアズ)を示す。
【0214】
【表25】
【0215】
他の被検フロープロモーターより低い温度(90℃および100℃)における粉体コーティングの粘度を下げる際に、バイオベースポリエステルフロープロモーター(試料B)の1%添加が最も有効である。90℃における3%添加(試料C)は、市販材料よりさらに低い。これは、図7にグラフで示している。
【0216】
この一連のDSC結果は、バイオベースのフロープロモーターの添加によって、90℃および100℃における溶融粘度が下がっても、粉体コーティング配合物全体のTgは下がらず、粉末安定性は損なわれないことを示す。粉体コーティングの硬化ピーク温度および反応エンタルピー(ΔH)は、フロープロモーターから悪影響を受けなかった。配合物A〜Gについて、結果を表16に示す。
【0217】
【表26】
【0218】
[実施例9]
実施例3Fの樹脂を、その顔料分散体特性についてカラーコンセントレートで評価した。
材料:
2つのカラーコンセントレート処方を選択した。一方は、ポリスチレン担体樹脂にPB 15:3(フタロブルー)10%添加であり、他方は、アクリロニトリル・ブタジエン・スチレンコポリマー(ABS)系担体樹脂にカスタムグリーンであった。カスタムグリーンは有機および無機顔料のブレンドからなり、約18%加えた。対照試料は、ステアリン酸亜鉛、およびステアリン酸亜鉛とエチレンビステアラミド分散剤の組合せなどの典型的な分散剤を用いて実施され、試料は、実施例3Fからの分散剤助剤を用いて実施された。
混合:
同方向回転の直径18mmのLeistritz二軸押出機で、混合を行った。
試験:
フィルター試験で、分散試験を行い、圧力上昇(単位、バール/顔料1グラム)を報告した。これは、分散の定量試験であり、値が低くなれば、分散がよくなることを示唆する。
【0219】
表17は、試験に使用した配合物、および結果を示す。
【0220】
【表27】
【0221】
市販の分散剤であるステアリン酸亜鉛、およびEBSとステアリン酸亜鉛の混合物との比較は、良好な結果を示した。結果は、2つの異なるポリマー系で一定の優れた発色を示した。
【0222】
本明細書に開示する本発明の形態は、現在好ましい実施形態を構成するが、他の多くの形態が可能である。本明細書では、本発明の可能な等価の形態または細分化形態をすべて記述するように意図されていない。本明細書で使用する用語は、説明するものにすぎず、限定するものでないこと、本発明の範囲の趣旨から逸脱することなく、様々な変更を行うことができることを理解されたい。
【図面の簡単な説明】
【0223】
【図1】硬質の結晶質イソソルビドを非晶質ダイマージオール、芳香族ジエステル、および他の材料とブレンドして、ポリエステル材料を合成する経路を示す概略フローチャートである。
【図2】硬質の結晶質イソソルビドを非晶質ダイマー二酸および他の材料とブレンドして、ポリエステル酸を合成する経路を示す概略フローチャートである。これらを、具体的にはハイブリッド粉体コーティング配合物で利用して、エポキシ官能性部位を架橋することができる。
【図3】硬質の結晶質イソソルビドをダイマージオール、非晶質ダイマー二酸、ポリイソシアナート(例えば、ジイソシアナート)、および他の材料とブレンドして、ポリウレタンを合成する経路を示す概略フローチャートである。
【図3A】本発明で有用なイソソルビドの典型的な異性体1a、1b、および1cを示す図である。
【図4】バイオベース樹脂(実施例2)および典型的な市販樹脂(FINE−CLAD 8400(登録商標))のレオロジー曲線を示すグラフである。
【図5】バイオベース配合物(実施例4A)および市販の対照配合物について、約121℃における粘度プロファイルを示すグラフである。
【図6】実施例1〜3Fの樹脂を作製するために使用した装置の様々な構成要素を示す概略図である。
【図7】実施例8の配合物A〜Gについて、90℃と100℃における粘度を示す棒グラフである。
【発明の開示】
【0001】
本願は、2005年3月18日出願の米国特許仮出願第60/663,422号、および2006年1月13日出願の米国特許仮出願第60/758,757号の利益を主張する。
【0002】
該米国特許仮出願2件の内容全体を参照により本明細書に組み込む。
発明の分野
本発明は、基材、具体的には感温性基材用の粉体コーティングを製造するのに有用である。典型的な感温性基材としては、プラスチックなどのポリマーを含むがこれらに限定されない有機基材、および木質およびプラスチックの複合体を含むがこれらに限定されない複合体が挙げられる。
【0003】
現在の粉体コーティング樹脂および配合物には、1つの重要な制限がある。これらは、一般に許容できる性能に必要とされる良好なフローおよび架橋を有するためにかなり高いオーブン温度(通常は177℃超)を必要とする。プラスチック、木質、バイオ複合体など、被覆対象の基材の多くは、まさに感温性であり、現在の粉体コーティング配合物で使用される高温に耐えることができない。このような基材の使用は、ここ数年で大幅に増大し、将来極めて劇的に増大するものと予想される。低温硬化粉体コーティングの領域における最近の研究の例のMuthiah文献を参照のこと。
【0004】
金属などの高温基材上で使用することもでき、耐久性があり、費用効果の高い感温性基材用低温熱硬化粉体コーティングが必要とされている。このような場合、温度がより低くなれば、プロセスにおけるエネルギーコストがより低くなるはずである。コストがより低くなれば、その新技術の受入れが大幅に増加するはずである。
【0005】
石油化学系供給原料の一部を広範囲の応用分野に使用するためのバイオベース供給原料で置換することは非常に興味深い。この興味の証拠は、長年にわたって出版された総説の数に反映されている。ポリエステル樹脂の合成でバイオベース供給原料を利用する試みは、米国特許第6,063,464号、およびトウモロコシバイオマス由来イソソルビドをポリエステル材料の合成で使用するGuoらの論文(下記を参照のこと)に例示されている。
【0006】
現在産業界で使用されるものより低温で流展し硬化する粉体コーティングを製造する必要もある。粉体コーティングは、使用中のVOC放出が非常に低いという点で環境上の利点を与える。残念なことには、その利点の一部は、硬化サイクルにおけるエネルギー需要の高さにより失われ、仕上がりの粗さは、通常はそれらが低温において流展性が不十分であることに由来する。
【0007】
他の関連特許および雑誌記事には、下記が含まれる。
低温硬化(LOW TEMPERATURE CURE):米国特許第6,703,070号、03/2004、Muthiah
合成および加工(SYNTHESIS AND PROCESSING):欧州特許出願公開第1491593号、12/2004、Mons
バイオベース材料総説:
Applied Microbiology and Biotechnology(2001),55(4),387−394.Huttermann,A.;Mai,C.;Kharazipour,A.“Modification of lignin for the production of new compounded materials”;
Biopolymers from Renewable Resources(1998),1−29.Kaplan,David L.“Introduction to biopolymers from renewable resources”;
Bioresource Technology(1994),49(1),1−6.Sharma,D.K.;Tiwari,M.;Behera,B.K.“Review of integrated processes to get value−added chemicals and fuels from petrocrops”;
Applied Biochemistry and Biotechnology(1988),17 7−22.Narayan,Ramani.“Preparation of bio−based polymers for materials applications”.
バイオベース樹脂合成:
Abstracts of Papers,224th ACS National Meeting,Boston,MA,United States,August 18−22,2002(2002).Guo,Yinzhong;Mannari,Vijaykumar M.;Massingill,John L.,Jr.“Hyperbranched bio−based polyols”.
粉体コーティング:
“Powder Coatings Volume 1:The Technology,Formulation,and Application of Powder Coatings”.Howell,David M.John Wiley and Sons,London,2000.
Polymer Preprints 2003,44(1).Gedan−Smolka,Michaela;Lehmann,Dieter;Lehmann,Frank.“Catalysis In Uretdione Powder Coatings Enables Innovative Processing Lines”.
【0008】
粉体コーティングにおいて低温フローおよび硬化が必要であるのに加えて、コーティングタイプに関わらず、コーティングマトリックス内において顔料の良好な分散も必要である。これを実現するために、異なる相溶性の成分を有するポリマーが設計されている。ポリマー分散剤は、塗料、コーティング、およびインク系中の顔料および他の材料を、最も典型的には立体安定化によって安定化する。ポリマー分散剤は、定着基とポリマー鎖からなる2成分構造を有する。最も典型的には、定着基は、粒子表面、およびコーティングの連続相と相溶であるポリマー鎖と相互作用する極性材料である。実際には、該ポリマー基は粒子の周りにコーティングを形成し、粒子同士が接触し、より大きい非相溶な凝集体になるのを防止する。
【0009】
有効なポリマー分散剤を与えるものと予想され得る定着基/ポリマーの配置は多数存在する。本発明の樹脂は、極性カルボン酸定着部位および非極性植物油鎖を有し、したがって分散剤および結合剤として働くことができる。分散剤としても働くことができる硬化結合剤によって、多数の顔料を分散するために別々の添加剤を必要とすることがなくなる可能性がある。関連技術としては、米国特許第5,959,066号、第6,025,061号、第6,063,464号、および第6,107,447号が挙げられる。
発明の簡単な説明
簡潔に言えば、金属などの高温基材上で使用することもでき、耐久性があり、費用効果の高い感温性基材用低温熱硬化粉体コーティングが必要とされている。石油化学系供給原料の置換材料を見い出すことが、特に大量のバイオベース供給原料をこの置換で利用することができる場合にさらに求められている。本明細書で開示するバイオベース粉体コーティング技術は、再生可能なバイオ供給源に由来する新規樹脂、および特許配合物技術、特に低温硬化技術を組み合わせることによってこの必要に応じる。後者の場合、温度がより低くなれば、プロセスにおけるエネルギーコストがより低くなるはずであり、新規バイオベース技術の受入れが大幅に増加するはずである。
【0010】
本発明の一実施形態は、Tgが50℃より高く、バイオベース内容物が少なくとも5%、別の実施形態では少なくとも50%であり、粘度が比較的低いポリエステル樹脂の合成を提供する。
【0011】
広範囲の実施形態では、樹脂をコーティング、特に粉体コーティングの配合物で利用する。
別の実施形態では、樹脂には、二酸およびジオールの反応によるカルボン酸官能性ポリエステルが含まれる。
【0012】
別の実施形態では、ポリエステル樹脂を形成するために利用される酸およびジオールは、得られるコーティングの特性を最大限にし、かつ樹脂中のバイオベース材料の量を最大限にするために、必要に応じてバイオベースまたは石油系である。
【0013】
本発明のさらに別の実施形態では、樹脂を架橋性樹脂と配合して、しばしば比較的低い温度で、良好なフローおよび柔軟性を有する保護コーティング被膜に硬化する。
本発明のさらに別の実施形態では、樹脂をPRIMID樹脂と配合して、良好なフローおよび柔軟性を有する保護コーティング被膜に硬化する。
【0014】
本発明のさらに別の実施形態では、樹脂をアクリル酸エポキシ樹脂と配合して、比較的低い温度で良好なフローおよび柔軟性を有するハイブリッド粉体コーティング被膜に硬化する。
【0015】
別の実施形態では、配合物は、外観、硬化速度、および他の特性を制御するために触媒、フローコントロール剤、硬化改質添加剤などを含む。
別の実施形態では、配合物は、イミダゾールおよび置換イミダゾールからなる触媒を含む。
【0016】
別の実施形態では、配合物は、イミダゾールおよび置換イミダゾール触媒の活性を改質するために酸性添加剤などの硬化改質添加剤を含む。
別の実施形態では、配合物は、色、外観、腐食制御、隠蔽、または他の機能のための顔料を含めて、当技術分野で知られている添加剤および賦形剤を含有することができる。
発明の詳細な説明および最良の態様
概して、本発明は、所望のバイオベース供給原料の使用と、より低い温度の粉体コーティングの必要性を組み合わせる。トウモロコシおよびダイズの供給原料を利用して、粉体コーティング性能に適した特性のバランスを有する樹脂を作製することができる。次いで、これらの樹脂を、様々な粉体コーティング配合物に配合することができる。
【0017】
通常は、本発明による粉体コーティング配合物は、下記によって調製される。主要な樹脂を粉砕し、粉砕硬化剤および選択した粉砕添加剤とドライブレンドし、ドライブレンドを溶融混合し、溶融混合ブレンドを押し出し、続いて急速冷却する。次いで、冷却したブレンドを所望の粒径に粉砕し、最後に、得られた粉末を最終粒径に分類する。
【0018】
本発明のいくつかの実施形態について本明細書で用いるバイオベース供給原料、配合物、生成物、材料、樹脂などは、通常の化学修飾、および/または発酵などの生物プロセスによって加工された農業および森林をベースとする再生可能な資源の変換に少なくとも一部由来する供給原料、配合物、材料、樹脂、および生成物などを意味する。炭素源は、有限で枯渇しつつある通常の化石由来炭素源とは異なる再生可能な植物の作物/樹木資源に由来する。
【0019】
本明細書では、ハイブリッド樹脂は、樹脂が1タイプを超える樹脂、例えばポリエステルとエポキシのブレンドであることを意味する。
本発明による特に有用な樹脂は、明らかに矛盾する2つの特性が良好なバランスをとり、
(1)非晶質樹脂の特徴である、適用中の良好な流展性のための、溶融状態における低粘度を有するが、
(2)結晶質樹脂の特徴である、良好な貯蔵安定性のための、比較的高いガラス転移温度(Tg)も有さなければならない。Tgが低すぎる場合、粉末粒子は「軟質」であり、特に高い貯蔵温度で貯蔵中に使用不可能な塊に合体する。通常は、これらの特性は、結晶質および非晶質の樹脂を、実際に半結晶質な樹脂ブレンドにブレンドすることによってバランスがとられている。通常は、本発明に従って得られた樹脂はこれらの所望の特性を提供する。
【0020】
注:別段の指定のない限り、材料の量を意味する場合の%は、重量パーセント(重量%)を意味する。
本明細書で開示する樹脂合成の一般的な手法は4つある。
【0021】
1.ダイマージオール、イソソルビド由来のジオール、および/またはダイマー酸をベースとするヒドロキシル官能性ポリエステル。通常は、ポリエステルのカルボキシルまたはヒドロキシル官能性は、二酸またはジオール基のモル過剰の比によって決定される。ポリエステルは、通常は正味のバイオベース内容物が少なくとも約5重量%であるが、最も典型的には約20〜約50重量%である。
【0022】
2.ダイマージオール、イソソルビド由来のジオール、および/またはダイマー酸をベースとするカルボキシル官能性ポリエステル。通常は、ポリエステルのカルボキシルまたはヒドロキシル官能性は、二酸またはジオール基のモル過剰の比によって決定される。ポリエステルは、通常は正味のバイオベース内容物が少なくとも約5重量%であるが、最も典型的には約50〜約70重量%である。
【0023】
3.ダイマー酸、および/またはダイマージオールをベースとするヒドロキシル、カルボキシル、またはイソシアナート官能性ポリウレタン。通常は、過剰のイソソルビドおよび/またはダイマージオールがヒドロキシル官能性を生成し、過剰のダイマー酸がカルボキシル官能性を生成し、過剰のポリイソシアナートがイソシアナート官能性を生成する。ポリウレタンは、通常は正味のバイオベース内容物が少なくとも約5重量%であるが、最も典型的には約20〜約50重量%である。
【0024】
4.米国を指定国とする2004年2月2日出願の国際公開第2004/077169号の易脱墨性トナー(Readily Deinkable Toners)に開示されるアミド−アミン官能性樹脂。アミド−アミン樹脂は、参照により内容が本明細書に組み込まれる該特許出願に記載されるダイマー酸とジアミンの反応生成物である。本発明のいくつかの実施形態では、典型的なアミドアミン官能性樹脂のTgは約80℃未満である。本発明の他の実施形態では、アミド−アミンのTgは約70℃未満である。正味のバイオベース内容物は、典型的には少なくとも5重量%であるが、より典型的には約40〜約60重量%である。
【0025】
本発明による樹脂は、イソソルビド(通常はトウモロコシ供給原料由来)など、硬質化効果を与える傾向がある共反応成分、およびダイマー酸またはダイマージオール(通常は植物油供給原料由来)など、柔軟化効果を与える成分からなり得る。これらの成分を適切に樹脂に共反応させることによって、樹脂の流展性と貯蔵安定性とを制御することができる。一般に、硬質化成分は、環状構造に結合し可動性が限定されたアルコール、エステル、カルボン酸、または酸塩化物などの官能性化学基を含み、柔軟化成分は、脂肪族炭素鎖に結合した官能性化学基を含む。イソソルビドは、縮合環状エーテル環からなるジオールであり、一般にジアンヒドロヘキシトールと呼ばれる、バイオベース糖誘導体のより大きいファミリーのメンバーである。ダイマー酸およびダイマージオールはそれぞれ、性質が極めて脂肪族であるバイオベース脂肪酸に由来するジカルボン酸およびジアルコールである。同様に、これらの硬質化および柔軟化の効果を、図3に示すポリウレタンに適用することもできる。
【0026】
通常は、触媒および/または熱を用いて架橋することによって、樹脂を硬化する。典型的な硬化温度は最高125℃である。
本明細書で開示するポリエステルポリオール樹脂は、イソシアナート、エポキシ、メラミンホルムアルデヒド、尿素ホルムアルデヒドなどを有する反応性配合物において、コーティング、接着剤、シーラント、および他の用途として有用である。
【0027】
本明細書で開示するポリカルボン酸樹脂は、β−ヒドロキシルアミド、エポキシなどを有する反応性配合物において、コーティング、接着剤、シーラント、および他の用途として有用である。
【0028】
アミド−アミン官能性樹脂は、イソシアナート、エポキシ、メラミンホルムアルデヒド、尿素ホルムアルデヒドなどを有する反応性配合物において、コーティング、接着剤、シーラント、および他の用途として有用である。
【0029】
粉体コーティング配合物では、開示するバイオ由来樹脂が特に有用である。実施例4には、粉体コーティング配合物中のβ−ヒドロキシルアミドとエステル交換方式で硬化して、透明なコーティングを形成するバイオ由来のカルボン酸官能性樹脂が記載される。実施例4Aには、実施例4の樹脂と同様の樹脂が記載されるが、より大きな規模で樹脂が作製されている。得られた樹脂のTgはわずかに高い。実施例5には、粉体コーティング配合物中のアクリル酸エポキシ樹脂で硬化して、透明なコーティングを形成するバイオ由来のカルボン酸官能性樹脂が記載される。実施例6には、顔料を含む粉体コーティング配合物中の市販のエポキシ架橋性樹脂で硬化して、黒色コーティングを形成するバイオ由来のカルボン酸官能性樹脂が記載される。実施例6Aには、市販のカルボン酸官能性樹脂およびカーボンブラックに比べて、バイオ由来のカルボン酸官能性樹脂およびカーボンブラックからなる顔料分散体、ならびに白色粉体コーティング配合物に添加された場合に、色に及ぼすそれらの効果が記載される。実施例6Bには、イソシアヌル酸トリグリシジル(TGIC)架橋剤で硬化されたバイオベースのカルボン酸官能性樹脂が記載される。
【0030】
実施例7には、粉体コーティング配合物中の市販のエポキシ架橋性樹脂で硬化して、透明なコーティングを形成するバイオ由来のアミド−アミン官能性樹脂が記載される。
実施例8には、フロープロモーターとしてバイオベースポリエステルを使用して、粉体コーティングの生成が例示されている。ポリエステル樹脂は、実施例3Bに記載されている。
【0031】
最後の実施例である実施例9には、実施例3Fに従って調製された樹脂の顔料分散体特性が例示されている。
本発明の一実施形態は、最小量から最大量のバイオベース材料の粉体コーティング用途向け樹脂の製造方法に関する。本発明の別の実施形態の樹脂は、少なくとも1つの飽和または不飽和のバイオベースポリエステルを含む。
【0032】
本発明は、コーティング、粉体コーティング、接着剤、トナー、インク、シーラント、ポリマー添加剤などを含むが、これらに限定されない様々な用途における、これらのバイオベース材料の1つまたは複数の使用にも関する。一実施形態では、ガラス転移温度(Tg)が約80℃未満である樹脂、他の実施形態では、ガラス転移温度が約70℃未満である樹脂、さらに別の実施形態では、約60℃未満で適切な溶融レオロジーの樹脂が設計された。本発明の広範囲の一般的な実施形態による樹脂は、ガラス転移温度の最小限が少なくとも約20℃であり、最大限が約80℃で、適切な溶融レオロジーを有する。フローコントロールに有用な樹脂は、通常はガラス転移温度範囲の下端である(例えば、Tgが約28.4℃である実施例3B)が、約20℃〜約80℃とすることができ、いくつかの実施形態では、通常は約25℃〜約60℃とすることができる。
【0033】
50%超のバイオベースカルボキシ官能基を含有する本発明の樹脂からなるハイブリッド粉体コーティング樹脂は、粉体コーティングに配合した。本明細書に記載する本発明の樹脂は、イソソルビド(通常は、トウモロコシ供給原料由来)など、硬質で非常に官能性である傾向がある共反応成分、およびダイマー酸(通常は、ダイズ供給原料由来)など、軟質で柔軟である傾向がある成分からなる。これらの成分を適切に樹脂に共反応させることによって、流展性と貯蔵安定性とを制御することができる。
【0034】
本発明は、本発明の1つまたは複数の樹脂による粉体コーティングの配合物にも関する。この粉体コーティングの著しい特徴は、このコーティングが、粉体コーティング操作に典型的な温度より低い温度で連続被膜に流展し硬化する能力である。低温硬化能力は、その組成物で利用するバイオベース樹脂の低粘度性、および本発明の樹脂性成分の有利なフロー特性を活用する配合物に由来する。本発明の樹脂から得られた利点は、ほぼ等価な市販の樹脂に比べて、所与の温度において低粘度である。
【0035】
粉体コーティング配合物で使用される樹脂の重要な特徴は、最終的な粉体コーティング粉末の貯蔵安定性には通常は少なくとも約50℃であり、好ましくは少なくとも約60℃であるガラス転移温度(Tg)である。表1は、いくつかのダイズベース樹脂、その官能性、およびそのTgの一覧を示す。この表は、低粘度のダイズベースモノマーを含む材料から、許容できるTgを有する樹脂を生成することが困難であることを例証している。
【0036】
【表1】
【0037】
樹脂1−1しか、Tgの基準を満たさなかった。ダイズベース材料の存在下でより高いTgを実現し、樹脂中バイオベース材料の負荷を高く維持するために、別のバイオベース材料であるが、高い固有Tgに寄与するものであるイソソルビドを利用した。
【0038】
Tgがより高いバイオベース材料(トウモロコシ供給原料由来のイソソルビド)は、ダイズベース材料と共反応して、粉体コーティング配合物のバイオベース含有量が高く、Tgが十分に高い樹脂を与えることができることが確認された。その後の合成によって、ダイズ、イソソルビド、および他の成分のバランスをとる試みを行って、樹脂、最終的には粉体コーティングにおいて特性の適切なバランスを実現した。
樹脂合成(実施例1および2を参照のこと):
コーティングの生成におけるバイオベース材料の使用は、以下の通り記述することができる。
【0039】
ポリエステルポリマーは、(1)反応器中で、イソソルビド(トウモロコシ供給原料由来);脂肪ダイマージオールおよび/またはダイマー二酸(ダイズ供給原料由来);二酸、ジエステル、または二酸塩化物;任意選択のコジオール;および任意選択のコ二酸、コジエステル、またはコ二酸塩化物を、芳香族二酸およびジオールを重合するのに適した縮合触媒と混合し、(2)モノマーおよび触媒を加熱して、モノマーを重合し、ポリエステルを得ることによって調製される。(図1を参照のこと)
カルボキシル官能性ポリエステル樹脂は、(1)反応器中で、イソソルビド;脂肪ダイマー二酸;任意選択のコ二酸、コジエステル、またはコ二酸塩化物;および任意選択のコジオールを縮合触媒と混合し、(2)モノマーおよび触媒を加熱して、モノマーを重合し、カルボキシル官能性ポリエステル樹脂を得ることによって調製される。(図2を参照のこと)
ヒドロキシル、カルボキシル、またはイソシアナート官能性ポリウレタンは、(1)反応器中で、イソソルビド;脂肪ダイマー二酸および/またはダイマージオール、ポリイソシアナート;任意選択のコジオール;および任意選択のコ二酸、コジエステル、またはコ二酸塩化物を、ジオールおよび二酸をポリイソシアナートと共に重合するのに適した触媒を使用して、あるいは使用せずに混合し、(2)モノマーおよび任意選択の触媒を加熱して、モノマーを重合し、ポリウレタンを得ることによって調製される。(図3を参照のこと)
次に、本明細書の実施形態に有用な様々な反応物質を開示する図1、2、および3を参照する。本発明は広範囲の実施形態によれば、開示するダイマージオールおよびダイマー酸に加えて、通常は約4〜約20個の炭素原子を有する脂肪族鎖を含む。より好ましくは、脂肪族鎖は約6〜約16個の炭素原子を有する。
【0040】
開示した追加のダイマージオールおよびダイマー酸は、アルコールまたはカルボン酸の官能基と共に、約4〜20個の炭素原子の脂肪族側鎖である2つの側鎖、および約8〜12個の炭素原子の他の2つの側鎖を有する6員環を含む。
【0041】
さらに、ジエステル、二酸、コ二酸、およびコジエステルは、式R2−CO−R1−CO−R2(式中、R2=−OH、−OR3、または−Cl(式中、R3=1〜4個の炭素原子を有する脂肪族鎖))を有することができる。R1は、2〜12個の炭素原子を有する芳香族または脂肪族の基である。
【0042】
特定の理論に拘泥するものではないが、現時点ではダイマー酸およびダイマージオールの脂肪族側鎖が樹脂に低粘度性をもたらすと考えられている。脂肪族側鎖は、低温で軟化し、粘度を低下させ、フローをよりよくさせる傾向がある。鎖が長くなれば、より軟化が見られ、加熱中により速く軟化する。
【0043】
これらは、いくつかの実施形態では、実施例9で例示するような顔料分散体の改善ももたらすと考えられる。よりよいフローがもたらす1つの結果は、顔料の優れた濡れであり、それによって顔料分散体が改善される。
【0044】
さらにかつより広範囲に、ジアンヒドロヘキシトールを本発明で使用することができる。したがって、他の環状ジオールを含む二環式を組み込むことによって硬質化構造を調製する際に、他のジアンヒドロヘキシトールをD−イソソルビドまたはその異性体の代わりとすることができ、本発明で使用することができる。シクロヘキシル、イソホロン、および他の環状構造を組み込むジオールは、イソソルビドと同様な硬質化効果を加えることができる。
【0045】
ダイマー二酸は、通常はC18の不飽和脂肪酸の二量化によって生成された粘性液体である。C18の不飽和脂肪酸のバイオ供給源には、植物、トール油、および動物の3つが存在する。C18単位は、いくつかの方式で連結され得る。優勢な成分であるC36の二酸では、非環式、単環式、二環式、および芳香族の4つの主要な構造タイプが知られている。これらの構造タイプのそれぞれにも、多くの構造異性体が存在する。これらの構造タイプおよび異性体の分布は、二量化に使用する出発物の脂肪酸供給原料の単不飽和/多不飽和の比、およびプロセス条件に依存する。いくつかの実施形態で典型的に使用する最小のダイマー二酸は、C18の二酸である。
【0046】
ダイマー二酸は、(1)約80%のC36の二塩基酸を含有する標準(未蒸留)タイプ、(2)C36の二塩基酸含有量を92〜98%に上昇させた、蒸留を行ったタイプ、(3)色を改善するため、蒸留および部分水素化を行ったタイプ、ならびに(4)安定性を最大限にするため、蒸留および完全水素化を行ったタイプの4タイプが現在市販されている。
【0047】
バイオベースポリエステル樹脂を調製するために使用する典型的なダイマー酸は、Empol 1018(登録商標)(実施例3、3C、および3E)、およびPripol 1013(登録商標)(実施例2、3A、および3D)(両者共、植物系ダイマー酸である)であった。Empol 1018(登録商標)はCognis Corporationが製造し、Pripol 1013(登録商標)は、Uniqemaが製造している。Cognisは、以降トール油系ダイマー酸を選択して、植物系ダイマー酸生産を中止した。表3では、Pripol 1013(登録商標)とEmpol 1018(登録商標)の物理的諸特性および組成物を比較している。Pripol 1013(登録商標)のほうが、明るい色であり、二塩基酸含有量が高い。この2つの異なるダイマー酸を使用して得られたカルボキシル官能性樹脂は、同様な物理的諸特性を有した。
【0048】
【表2】
【0049】
ダイマージオールは、通常はダイマー二酸メチルエステルの高圧水素化によって生成される。バイオベースポリエステル樹脂(実施例1、1A、および3B)を調製するために使用するダイマージオールは、SPEZIOL C36/2 1075(登録商標)ダイマージオールであった。これは、Cognis製の植物系ダイマージオールである。
【0050】
本明細書で開示する樹脂は、一度溶融させた市販の石油化学系樹脂に比べて、低い粘度を有する(実施例を参照のこと)。現在市販されている樹脂粉体コーティング配合物では、硬化サイクル後に得られた被膜の流展性およびレベリング性を良好にするために、フロー性のある材料(フローコントロール添加剤)を添加する必要がある。バイオベース樹脂は、良好な被膜レベリング性および外観を実現するためのこのような添加剤を殆どまたは全く必要としない。組み込むことに成功したバイオベース樹脂は、通常の石油化学系樹脂を含有する配合物中でフロー添加剤として機能することもできる。通常は、コーティング配合物に、他の主要な粉末樹脂を使用すると共に、バイオベース樹脂(含有量約0.1重量%〜約5重量%)を使用して、本発明の樹脂のフローコントロール特性を利用する。
【0051】
本発明のポリエステルポリマーは、イソソルビド、ダイマージオール、および/またはダイマー酸、二酸、ジエステル、または二酸塩化物;任意選択のコジオール;および任意選択のコ二酸、コジエステル、またはコ二酸塩化物の溶融重合によって調製した(図1の方法)。
【0052】
実施例1に、本発明のポリエステルを調製するために使用する典型的な手順を記載する。脂肪族ポリエステルは、軟質で柔軟なゴム状材料である。大部分の芳香族ポリエステルは結晶質である。軟質ダイマージオールを、高官能性イソソルビドおよび結晶質の芳香族二酸とブレンドすることによって、特性の良好なバランスが得られる。しかし、このバランスは、反応においてエチレングリコールなど、他の材料を(すなわち、図1および2の「ジオール」として)含めることによって助長され得る。
【0053】
ガラス転移温度(Tg)が61℃〜165℃(表2Aおよび2B)のポリエステルを調製することによって、様々なモノマーの効果を検討した。表2Aは、本明細書に記載するように合成された樹脂の典型的な特性、およびガラス転移温度(Tg)が61℃〜165℃の様々なモノマーの効果を示す。表2Bは、実施例2〜3Aに記載するように合成されたカルボキシル官能性樹脂の典型的な特性を示す。
【0054】
【表3】
【0055】
【表4】
【0056】
データは、これらのモノマーで可能な広範囲のTgを示す。イソソルビドなしで調製された試験ポリエステルは、イソソルビド含有のもののように非晶質でなかったが、挙動は結晶質であった。
【0057】
D−イソソルビド(1,4:3,6−ジアンヒドロ−D−グルシトール)(1a)もしくはその異性体、および/またはD−イソソルビドを含むすべての異性体の混合物を、D−イソソルビドの代わりに使用することができた。1,4:3,6−ジアンヒドロ−D−マンニトール(1b)および1,4:3,6−ジアンヒドロ−D−イジトール(1c)は、イソソルビドの2つの異性体である。D−イソソルビドを本発明で使用したが、D−イソソルビドの異性体も同様に働くものと予想される。本発明で有用なイソソルビドの異性体を図3Aに例示する。
【0058】
酸官能性ポリエステルを形成するのに適切なポリオールの例としては、1,2−エタンジオール(エチレングリコール)、1,3−プロパンジオール、1,4ブタンジオール、1,6−ヘキサンジオール、1,10−デカンジオール、1,12−ドデカンジオール、1,4−シクロヘキサンジメタノール、ジエチレングリコール、トリエチレングリコール、ネオペンチルグリコール、トリメチロールプロパン、水素化ビスフェノールA(2,2−(ジシクロヘキサノール)プロパン)、2,2,4−トリメチル−1,3−ペンタンジオール、2−メチル−1,3−プロパンジオール、2−メチル−2−ヒドロキシメチル−1,3−プロパンジオール、2−エチル−2−ヒドロキシメチル−1,3−プロパンジオールなど、および前述のポリオールの少なくとも1つを含む組合せが挙げられる。現時点での働きは、バイオベース内容物を最大限にすることを目標としているので、好ましいポリオールは、イソソルビド(トウモロコシ原料由来)およびダイマー酸ジオール(ダイズ原料由来)であるが、必要に応じてエチレングリコールなどを使用して、特性を向上させることができる。
【0059】
適切なポリカルボン酸、酸エステル、および酸塩化物には、コハク酸、アジピン酸、アゼライン酸、セバシン酸、1,12−ドデカン二酸、テレフタル酸、イソフタル酸、トリメシン酸、テトラヒドロフタル酸、ヘキサヒドロフタル酸、1,4−シクロヘキサンジカルボン酸、トリメリト酸、ナフタレンジカルボン酸、ダイマー酸など、および前述のポリカルボン酸の少なくとも1つを含む組合せに由来するものが含まれる。好ましいジエステルは、テレフタル酸のジメチルエステルである。いくつかの配合物では、ドデカン二酸(DDA)を改質剤として使用する。1,4−シクロヘキサンジカルボン酸、Empol 1018(登録商標)、Pripol 1013(登録商標)などの二酸が現時点で好ましい。
【0060】
所望の分子量のカルボキシル官能性ポリエステルを得るために、ポリエステルを形成するために使用するモノマー混合物は、通常はヒドロキシル官能基に対して適切に過剰なカルボキシル官能基を有する(ただし、酸当量に対するヒドロキシル当量の比は通常は0.85〜0.95である)。ポリエステルは、非晶質から結晶質まで多様であり得る。
【0061】
架橋は、カルボキシ官能性のカルボキシル基を、β−ヒドロキシルアミドと自己触媒エステル交換反応(アミドの商標名の後に、PRIMID反応と記載される場合が多い(表3))で反応させ、または市販のポリエポキシ官能性ポリマーと反応させることによって実現される。特に低温硬化組成物に好ましいポリエポキシ化合物は、グリシジルアクリラートやグリシジルメタクリラートのコポリマー(総称して「GMA」)樹脂など、エポキシ官能性アクリル酸またはメタクリル酸の樹脂である。GMA樹脂は、通常は約5〜約30重量%のグリシジルアクリラートまたはグリシジルメタクリラート、および約80〜約95重量%のメチルメタクリラート(ただし、約50重量%までのメチルメタクリラートを別のα,β−不飽和モノマー、例えばスチレン、アクリロニトリルなどで置換することができる)から得られる。適切なGMA樹脂は、エポキシ当量が約200〜約1000、好ましくは約200〜約600であり、Mnがゲルパーミエーションクロマトグラフィーで決定して200〜約2000原子質量単位(AMU)である。これらは室温で固体であり、融点が約40℃超であり、好ましくは軟化点が約50℃〜約75℃であり、Tgが約40℃〜約60℃である(表3)。
【0062】
バイオベースの樹脂性成分で低温流展性を実現できることを考えれば、約115℃〜約140℃の温度で硬化を開始する触媒を利用することが有利であり、これは市販されている多くの中から選択可能である。通常は、触媒を約0.1〜約5部/樹脂100部(phr)、好ましくは約0.2〜2(phr)のレベルで使用して、硬化反応を低温硬化剤で加速することができる。本発明に好ましい触媒は、式1に示す一般式を有するイミダゾール:
【0063】
【化1】
【0064】
(式中、R1、R2、R3、およびR4は独立に、水素、メチル、フェニル、またはベンジルである)、およびその付加物である。
概して、置換基はエポキシ樹脂との反応性を示さないことがある。第三級アミンおよびポリアミン材料も、この反応の触媒として有用である。
【0065】
良好な流動性を維持するためには、付加物を生成し、イミダゾール触媒の反応性を部分ブロックすることによって、その反応性を改変する必要があり得る。これは、イミダゾールとエポキシとの付加物を作製することによって行われることがある(例えば、米国特許第6,703,070号を参照のこと)。本発明の一実施形態では、親イミダゾールが好ましい触媒であり、イミダゾールの反応性を軽減するために酸性材料を配合物に添加した。
【0066】
イミダゾールの反応性を軽減するのに適した酸性材料には、ベンゼンスルホン酸もしくはナフタレンスルホン酸およびその置換変形体などの芳香族スルホナート、またはナフタレンカルボン酸およびその置換変形体などの芳香族カルボキシラートが含まれ、無機物や超酸などの固体酸性材料も使用することもできる。後者の場合、イミダゾール触媒の一部を固体酸性表面に吸着させ、したがって加熱するまで結合剤のバルクに利用させなくすることができる。このような一固体材料は、例えばKing Industriesのブロック超酸NACURE(登録商標)7231(アンチモン酸アンモニウム)である。
【0067】
コーティング粉末は、約0〜約5重量%の範囲のフローコントロール剤を含有することもでき、約0.1重量%〜約2重量%の範囲が最も好ましい。フローコントロール剤の例としては、MODAFLOW(登録商標)ポリ(アルキルアクリラート)(すなわち、MODAFLOW 6000(登録商標))製品、およびサーフィノール(Surfynol)(登録商標)アセチレンジオール(すなわち、P200(登録商標))など、ヒドロキシル、カルボキシル、または他の官能基を含む他のものが挙げられる。官能化フロー添加剤は、粉体コーティングの修正(touch−up)または修復が必要である場合に層間付着力の助けにもなる。フロー添加剤を単独または組み合わせて使用することができる。抗酸化剤を約0.5〜約2.0phrの濃度で使用して、本発明に適した比較的低い硬化温度でさえコーティングの変色を防止することもできる。本明細書の他で記載するように、実施例8の実施例3B樹脂によって例示される本発明の樹脂自体は、フローコントロール剤として働くことができる。
【0068】
二酸化チタンおよび/またはカーボンブラックなどの顔料、炭酸カルシウムなどの充填剤、粒状ゴムなどのテキスチャー付与剤、ベントナイト粘土、商標LANCOWAX(登録商標)で販売されているものなどのポリエチレン粉末含有または不含ポリテトラフルオロエチレン(PTFE)粉末、ならびに他の通常の添加剤も、外観およびコスト削減のために存在させることができる。
【0069】
ベンゾインを、通常はピンホール防止剤として使用する(Howellの参考文献を参照のこと)。
表3は、市販の粉体コーティング樹脂の一覧を示し、その官能性およびTgを記述する。これらの樹脂の一部を本明細書の実施例の調製で使用した。残りは、他の配合物で使用され、本発明の様々な実施形態で有用である。
【0070】
【表5】
【0071】
下記に、本発明による粉体コーティング配合物を調製するために適応させることができる一般的手順を記載する。
手順:粉体コーティング混合プロトコル:
通常は、Brabender(登録商標)混合機を使用するが、手順を他のタイプの混合機向けに適応させることができる。
・120mlのボウルサイズに対して約70〜80gに等しくなるように粉体コーティング配合物を算出する。典型的な小型Sigma Bladeボウルには、顔料不含から低顔料/結合剤(P/B)のコーティング配合物70g、またはより高いP/Bの塗料80gが入る。
・油加熱器を始動させることによって、Brabender(登録商標)混合機または同様な混合機を99℃に予熱する。30分間予熱しておく。
・予熱が完了したとき、ローターを始動させ、ボウルが装置に対して安全であるか試験する。
・トルクセンサーのスイッチを入れる。これは、ミックスが前進するためのガイドとして働く。
・約30gの一次樹脂をボウルに徐々に添加する。
・樹脂を、トルクセンサーが定常値を示すまで(約5分間)混合し、溶融させ、次いで一次樹脂の残部を混合ボウルに添加し、混合し、溶融させる。
・添加剤があればそれ/全部を、ローター間の混合ゾーン中心部に添加する。
・トルク値が定常になるまで混合しておく(通常は、約10分間)。
・架橋性樹脂を全部、ボウルに徐々に添加する。少なくとも3分間混合し、架橋が開始する(トルク示度が急速に上昇し始める)といけないので、トルク示度が定常のままであることを確認する。
・最後に、触媒が(必要である場合には)それを、トルク示度を綿密に見ながら添加する。トルクは上昇するはずであり、粘度(トルク)が10%上昇した後、バッチを止めるべきである。
・混合ボウルから、生成物を濃厚な溶融材料としてすぐに取り出し、硬質になる(通常は、硬質で脆弱な光沢のある材料になる)まで所望の温度(通常は、室温)に冷却する。
・所望の温度に冷却した後、生成物を小チップに分解する。
・次いで、チップをボールミルにかけ、またはその他の(例えば、塗料チップを10mm〜15mmのスチールメディアの存在下で16時間ボールミルする)方法で微粉砕して、細粉末を得る。
・粉末を適切なスクリーンに通してふるいにかけて、大きな断片、通常は約105ミクロンより大きい断片を除去する。
【0072】
記載された方法を利用して、いくつかのクラスの粉体コーティング配合物を調製することができる。
本発明に従って仕上げ粉体コーティングを製造するための一般的手順は、以下の通りである。
・コーティングするために、基材に適した溶媒(例えば、水、メチルエチルケトン、イソプロピルアルコール)で、基材をきれいに拭くことによって調製する。
・基材を下塗りする。
・粉体コーティングを、Nordson Corporationから供給されるVersa−Spray(登録商標)などの粉末スプレーガンの試料リザーバーに注ぎ込む。
・電圧コントロールをスプレーガン制御装置で調整して、適切な電荷を粉末に加えるようにする。
・粉末を、標準的な粉体コーティング技法で塗布して、乾燥被膜厚が約50.8〜76.2ミクロン(2〜3ミル)の所望の被膜厚を実現する。
・次いで、基材をオーブンに適切な時間および温度で配置する。
【0073】
通常は、β−ヒドロキシアミド系粉体コーティングは、ジ−N−β−ヒドロキシアミド架橋剤でカルボキシル官能性をエステル交換架橋することに基づく。これらのタイプの粉末は、市販のPRIMID(登録商標)タイプの粉末によって例示される。実施例4は、バイオベース樹脂対通常の石油化学系樹脂の配合物および硬化の詳細を示す。このタイプの化学は、触媒作用の影響を受けない。したがって、硬化速度には有意差は期待されず、実際、いずれの樹脂についても硬化速度の利点がこの場合には検出されなかった。
【0074】
2つのコーティングを、121℃と147℃の2つの異なる温度で30分間硬化した。最大の差は、より高い温度での光沢であり、バイオベース樹脂配合物は対照より約50ユニット高かった。しかし、耐溶媒性については、バイオベース配合物のほうがわずかに低かった。明らかに、バイオベース樹脂は、β−ヒドロキシアミド(登録商標)架橋配合物において市販の対照と比較したとき、全体的に性能が劣らない。
ハイブリッド粉体コーティング:カルボン酸−エポキシ架橋
カルボン酸−エポキシ架橋粉体コーティングは、ハイブリッドコーティングの最も一般的なものである。通常は、これらは、アクリル酸エポキシ架橋剤と共に配合された石油由来のポリエステル酸からなる。本発明のカルボン酸官能性バイオベース樹脂を合成し、典型的な配合物において市販の石油化学系ポリエステル酸と比較して試験した。
【0075】
図4Aは、本開発によるバイオベース樹脂と典型的な市販の樹脂(FINE−CLAD 8400)について、121℃におけるせん断速度に対する粘度(単位:ポアズ)の比較を示す。バイオベース樹脂(下部のデータ点群)は、その対照物より粘度が低いことに留意されたい。
【0076】
粘度の差に基づいて、バイオベース材料はより低い温度でより高いフローをもたらし、低温硬化コーティングの外観を全般的に改善できるという可能性がある。様々な工業製造方法による粗さ平均測定値を用いて、改善の影響を概算測定することができる。
【0077】
実施例4Aでは、対照の石油化学系透明コートの表面粗さ(Ra)は4.2と評価し、バイオベース透明コートは1.3と評価される。石油由来のパネルの粗さは、典型的な鋸引き操作に等価であり、バイオベースのパネルは、典型的な電子ビームまたはレーザー操作に等価であった。バイオベース配合物は、対照よりはるかに「クラスA」仕上げに近い。
【0078】
実施例4Aのバイオベース粉体コーティングおよび市販の石油由来の低温硬化粉体コーティング(Forrest Powder Low Temperature Cure(登録商標))のパネルの比較を行った。両パネルは、約63.5ミクロン(約2.5ミル)の被膜厚でスプレーし、次いで121℃で30分間熱硬化した。バイオベース粉末材料は、比較の粉体コーティングよりかなり少ないゆず肌(または表面粗さ)しか示さなかった。さらに、バイオベース粉末で被覆されたパネルのほうは、60℃ではるかに高い光沢を示した(72点対50点)。配合物の詳細については実施例4Aを参照のこと。
【0079】
バイオ由来配合物の改善されたメルトフローを、応力制御レオメータで測定した。粉末の試料を、100℃に加熱されたプラテン間に配置し、典型的な粉体コーティング被膜の厚さ(50.8ミクロン(約2ミル))に圧縮した。温度を121℃に上げ、試料が硬化するまで粘度の変化(単位:ポアズ)を測定する。粘度データを図5に示す。
【0080】
上部の曲線データ点は、比較対照の粉末試料を示し、下部の曲線データ点は、バイオベース粉末試料を示す。バイオベース配合物の初期粘度は、対照試料より有意に低い(3694ポアズ対11980ポアズ)。時間が経過するにつれて、試料が硬化するので試料は両方とも粘度が上昇した。硬化時間の残りの間、バイオベース配合物のほうの粘度は低いままだったので、この粉末は、被膜が架橋し硬化する前に溶融し流展する機会が多かった。
【0081】
柔軟性(靭性の尺度)は、最終生成物が、被覆された物体の使用に伴う日常的な突起および凹みに耐えることを可能にする重要なコーティング属性である。柔軟性が十分でないと、コーティングのひび割れが起こり、衝撃が起きたときに基材の剥離が起こることもある。基材を戸外で使用する場合、水、紫外線、酸化、および酸性雨など、大気中の化学物質は、被膜を劣化させ、脆化させる恐れがある。これらの要因の多くは腐食の原因ともなり、錆、および不十分な外観という損失、ならびに不十分な柔軟性がもたらされる。
【0082】
対照試料と真上で述べたバイオベース配合物を、マンドレル屈曲と呼ばれる柔軟性試験(ASTM D522)で比較した。この試験では、被覆された基材を万力にクランプで留め、円錐形マンドレル上を回転させた。領域のテープ接着性試験によって、コーティングの最終性能が決定された。テープをコーティング上の屈曲領域に貼り付け、剥がして、コーティングがまだパネルに接着しているかどうかを決定する。
【0083】
円錐形マンドレルは、その長さの全域で、試験に利用可能な最小のサイズであり、かつコーティングの剥離またはひび割れがなく通過するのに最も靭性の高いサイズである3.18mm(1/8インチ)まで様々な幅を有した。
【0084】
バイオベースのコーティングは、わずかなひび割れしか明らかでなく、かつ剥離の徴候は明らかでなかったので、良好な柔軟性を有した。対照の石油由来のコーティングは、パネルの長さ全体にわたってひび割れが起こり、屈曲の長さの約40%でコーティングが剥がれた。配合物の詳細については実施例5を参照のこと。
【0085】
使用温度で低粘度であることが顔料表面の良好な濡れにとって好ましい場合、顔料を含む粉体コーティングはいくつかの利点がバイオベース樹脂配合物に由来することもある。実施例6では、2つの黒色配合物(対照配合物1つおよびバイオベース配合物1つ)を記載する。
【0086】
バイオベース粉体コーティングは、石油化学製品由来のコーティングより、60℃で光沢がはるかに高かった(85点対44点)。これもやはり、おそらく熱硬化中、配合物のメルトフローがよりよいことに起因した。
【0087】
黒色顔料の発色/漆黒度(jetness)を、バイオベース配合物で改善した。漆黒度は、コーティングのLおよびb色成分を測定することによって決定することができる。(Hunter Color Scaleの説明については、Wicks,Z.W.ら、「Organic Coatings:Science and Technology」、第2版、特に351〜355頁、Wiley Interscience,NY,NY.ISBN 0−471−24507−0 1999を参照のこと)。
【0088】
2つの黒色パネルの全デルタE、すなわち色差は0.52であり、バイオベースのほうが、発色が強かった(漆黒)。対照パネル(左)は、黒色顔料がバイオベース配合物ほどコーティング系にうまく分散されていなかったので、バイオベース配合物より灰色に見えた。これは、おそらくバイオベース樹脂の低粘度に起因する。
【0089】
次に図6を参照して、この図は、本発明に従って樹脂を作製するのに使用された装置100の様々な要素を示す概略図である。加熱マントル102は、反応器101を少なくとも一部包囲し、反応混合物104を含む反応器101の温度を制御するのに使用される。反応器101は、反応容器106およびトップ部108からなる。トップ部108は、様々な器具に連結するための複数の頸部110、112、114、116を有する。撹拌は、撹拌軸122(例えば、ステンレス鋼)の端部にあるパドル120(例えば、通常は45°の角度の羽根)によって行われる。撹拌軸122は頸部116を貫通している。コネクタ131を介して熱電対132に連結された熱電対コントローラ130は、シールした装置のガス入口コネクタ111で頸部110を貫通して、反応混合物104に到達する。Vigreauxカラム140を、シールした関係で頸部114に取り付ける。温度計141または他の温度測定装置を、Vigreauxカラム140の塔頂部(蒸留ヘッド)142に取り付ける。コネクタ146を用い、冷却器入口152を介して、冷却器150をVigreauxカラム140の頸部144に取り付ける。Vigreauxカラム140は、ジャケットで包囲された独立型装置とすることができ、またはジャケットおよびカラムは一体型とすることができる。排気出口164および頸部出口166を有する頸部160の頸部入口162に、冷却器出口154を連結する。受けフラスコ170は、頸部出口166に連結させた入口172を有する。冷却液体155は、冷却器150に入口156から入り、出口158から出る。
【0090】
操作の際には、アルゴンガス111−1は、ガス入口コネクタ111から入って、反応混合物104をブランケットし、ガス出口164から流出する。材料は、装置を閉じる前に、または頸部112のシールしたコネクタ118を通して、添加することができる。図6では、頸部112は、頸部116の真後ろに配置されていることに留意されたい。頸部112を反応器101の中心軸190上に配置する。留出液178を受けフラスコ170に回収する。
【0091】
下記の実施例は、本発明の様々な態様を例示するものであり、決して本発明の範囲を限定するものではない。
【0092】
[実施例1]
樹脂生成(実施例1〜実施例3F)
この実施例は、ヒドロキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、ジメチルテレフタラート(DMT)(228.30g、1.1757モル)、Speziol C36/2 1075(登録商標)ダイマージオール(バッチ#415252)(77.61g、0.1411モル)、D−イソソルビド(123.90g、0.84785モル)、およびエチレングリコール(EG)(102.81、1.6563モル)と、続いて酢酸マンガン(II)四水和物(0.0917g)、酢酸コバルト(II)四水和物(0.0618g)、および酸化アンチモン(III)(0.103g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を30分間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約150℃超で加熱したとき、メタノールを連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これはメタノールが除去されたことを示唆する。約95mlのメタノールを留去した。引き続いて、ポリリン酸(0.0634g)のEG(1g)溶液を反応混合物に添加した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、エチレングリコールが留出し(91g)、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分間維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0093】
下記の特性を有する樹脂を生成した。
溶液固有粘度:0.29(溶媒はo−クロロフェノールである。92%のみ可溶)
Tg=61℃
ヒドロキシル値=24.3
酸価=8.0
分子量(MW)=3470(酸およびヒドロキシル値から算出)
ポリマー特性:
色:茶色
粘着性:非粘着性
清澄性:わずかに半透明
柔軟性:脆い
固体
【0094】
[実施例1A]
この実施例は、バイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、ジメチルテレフタラート(DMT)(197.74g、1.0183モル)、D−イソソルビド(119.05g、0.81463モル)、およびSpeziol C36/2 1075(登録商標)ダイマージオール(バッチ# 415252)(112.06g、0.20371モル)と、続いて1,2,3,4−テトラヒドロナフタレン(2ml)および酸化アンチモン(III)(0.089g)を加えた。反応器をアルゴンでブランケットした。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を12分間維持した。反応混合物を、20分間かけて250℃に徐々に加熱した(2.5℃/分)。この温度を8分間維持した。反応を約150℃超で加熱したとき、メタノールを連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これはメタノールが除去されたことを示唆する。約83mlのメタノールを留去した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、13分かけて280℃に徐々に加熱した(2.3℃/分)。次いで、反応混合物を260℃まで放冷した。追加のD−イソソルビド(14.87g、0.1018モル)を反応混合物に加えた。反応混合物を280°に加熱した。この温度を30分間維持した。留出液受器を真空受器に換え、徐々に真空にした(≦1199.898Pa(9トル))。この間に、低分子量ポリマーが形成した。反応混合物温度を280℃で2時間40分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0095】
下記の特性を有する樹脂を生成した。
溶液固有粘度:0.10(溶媒はo−クロロフェノールである)
Tg=165℃
ヒドロキシル値=45.0
酸価=2.3
分子量(MW)=2372(酸およびヒドロキシル値から算出)
ポリマー特性:
色:明茶色
粘着性:粘着性
清澄性:半透明
柔軟性:幾分脆い
固体
【0096】
[実施例2]
この実施例は、カルボキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)。
4頸トップを備えた5リットルの丸底ガラス反応容器、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、D−イソソルビド(1337.0g、9.1490モル)(受理したまま)、Pripol 1013(登録商標)ダイマー酸(バッチ# 091687)(699.1g、1.215モル)、および1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(1563.8g、9.0826モル)と、続いて酸化アンチモン(III)(1.231g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、47分間かけて250℃に徐々に加熱した(1.1℃/分)。この温度を3.1時間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約329mlの水が留出した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0097】
下記の特性を有する樹脂を生成した。Tg=64.2℃
酸価=34.8
分子量(MW)
GPC(ポリスチレン標準)Mn=1689
GPC(ポリスチレン標準)Mw=11681
多分散性(Mw/Mn)=6.91
ポリマー特性:
色:明コハク色
粘着性:非粘着性
清澄性:半透明
柔軟性:脆い
固体
【0098】
[実施例3]
この実施例は、カルボキシル官能性バイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと).
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(204.66g、1.1886モル)、Empol 1018(登録商標)ダイマー酸(バッチ# U42G151910)(72.54g、0.1251モル)、およびD−イソソルビド(172.80g、1.1824モル)と、続いて酸化アンチモン(III)(0.1594g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を30分間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約47mlの水が留出した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0099】
下記の特性を有する樹脂を生成した。
溶液固有粘度=0.25dl/g(溶媒はo−クロロフェノールである):
Tg=66.9℃
ヒドロキシル値=13.0
酸価=36.3
分子量(MW)
GPC(ポリスチレン標準)Mn=2995
GPC(ポリスチレン標準)Mw=9560
多分散性(Mw/Mn)=3.19
ポリマー特性:
色:明茶色
粘着性:非粘着性
清澄性:殆ど半透明
柔軟性:脆いが硬質
固体
【0100】
[実施例3A]
この実施例は、カルボキシル官能性バイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
4頸トップを備えた5リットルの丸底ガラス反応容器、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(1570.3g、9.1202モル)、Pripol 1013(登録商標)ダイマー酸(バッチ# 091687)(675.7g、1.174モル)、D−イソソルビド(得たまま)(1354.0g、9.2648モル)と、続いて酸化アンチモン(III)(1.247g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、51分間かけて250℃に徐々に加熱した(1.0℃/分)。この温度を3.1時間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約334mlの水を留去した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0101】
下記の特性を有する樹脂を生成した。
Tg=65.3
酸価=29.0
分子量(MW)
GPC(ポリスチレン標準)Mn=2162
GPC(ポリスチレン標準)Mw=11872
多分散性(MW/Mn)=5.49
ポリマー特性:
色:黄色/明コハク色
粘着性:非粘着性
清澄性:半透明
柔軟性:脆い
固体
【0102】
[実施例3B]
この実施例は、ヒドロキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、ジメチルテレフタラート(DMT)(228.30g、1.1757モル)、Speziol C36/2 1075(登録商標)ダイマージオール(バッチ# 415252)(129.40g、0.23523モル)、D−イソソルビド(123.90g、0.84785モル)、およびエチレングリコール(EG)(89.66g、1.444モル)と、続いて酢酸マンガン(II)四水和物(0.0917g)、酢酸コバルト(II)四水和物(0.0618g)、および酸化アンチモン(III)(0.103g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を30分間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約150℃超で加熱したとき、メタノールを連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これはメタノールが除去されたことを示唆する。約95mlのメタノールを留去した。引き続いて、ポリリン酸(0.0634g)のEG(1g)溶液を反応混合物に添加した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、30分間かけて280℃に徐々に加熱した(1℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、エチレングリコールが留出し(84g)、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0103】
下記の特性を有する樹脂を生成した。
溶液固有粘度:0.19(溶媒はo−クロロフェノールである)
Tg=28.4℃
ヒドロキシル値=35.4
酸価=6.1
分子量(MW)=2700(酸およびヒドロキシル値から算出)
ポリマー特性:
色:茶色
粘着性:非粘着性
清澄性:殆ど半透明
柔軟性:脆い
固体
【0104】
[実施例3C]
この実施例は、ヒドロキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、ジメチルテレフタラート(DMT)(213.96g、1.1018モル)、Empol 1018(登録商標)ダイマー酸(バッチ# U42G151910)(71.02g、0.1225モル)、D−イソソルビド(128.79g、0.88128モル)、およびエチレングリコール(EG)(116.28g、1.8734モル)と、続いて酢酸マンガン(II)四水和物(0.0859g)、酢酸コバルト(II)四水和物(0.0579g)、および酸化アンチモン(III)(0.0965g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を30分間、またはVigreauxカラムの塔頂部の温度が30℃以下に下がるまで維持した。反応を約150℃超で加熱したとき、メタノールを連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これはメタノール/水の混合物が除去されたことを示唆する。約93mlのメタノール/水の混合物を留去した。引き続いて、ポリリン酸(0.0594g)のEG(1g)溶液を反応混合物に添加した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、エチレングリコールが留出し(95g)、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0105】
下記の特性を有する樹脂を生成した。
溶液固有粘度:0.23(溶媒はo−クロロフェノールである)
Tg=58.8℃
ヒドロキシル値=23.7
酸価=1.4
分子量(MW)=4470(酸およびヒドロキシル値から算出)
ポリマー特性:
色:明茶色
粘着性:非粘着性
清澄性:幾分半透明、わずかな曇り
柔軟性:脆い
固体
【0106】
[実施例3D]
この実施例は、カルボキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
2リットルの4頸円柱壁付き丸底ガラス反応容器、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(610.68g、3.5468モル)、Pripol 1013(登録商標)ダイマー酸(バッチ# 091687)(262.78g、0.45670モル)、D−イソソルビド(アセトンで再結晶)(526.54g、3.6030モル)と、続いて酸化アンチモン(III)(0.4849g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中で反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を2.1時間維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約129mlの水を留去した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0107】
下記の特性を有する樹脂を生成した。
Tg=62.3
酸価=34.7
分子量(MW)
GPC(ポリスチレン標準)Mn=3517
GPC(ポリスチレン標準)Mw=12753
多分散性(Mw/Mn)=3.63
ポリマー特性:
色:コハク色/橙色
粘着性:非粘着性
清澄性:半透明
柔軟性:脆い
固体
【0108】
[実施例3E]
この実施例は、カルボキシル官能性バイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと).
1リットルの4頸円柱壁付き丸底ガラスフラスコ、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(318.36g、1.8490モル)、Empol 1018(登録商標)ダイマー酸(バッチ# U42G151910)(112.84g、0.1946モル)、およびD−イソソルビド(268.80g、1.8393モル)と、続いて酸化アンチモン(III)(0.2479g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を2.3時間維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約74mlの水が留出した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で3時間10分維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0109】
下記の特性を有する樹脂を生成した。
溶液固有粘度=0.24dl/g(溶媒はo−クロロフェノールである):
Tg=72.3℃
ヒドロキシル値=0.0
酸価=32.8
分子量(MW)
GPC(ポリスチレン標準)Mn=4027
GPC(ポリスチレン標準)MW=15756
多分散性(MW/Mn)=3.91
ポリマー特性:
色:黄色−茶色
粘着性:非粘着性
清澄性:殆ど半透明
柔軟性:脆い
固体
下記の実施例4〜8は、本発明によるいくつかの典型的な粉末配合物、および仕上げコーティングを例示する。
【0110】
[実施例3F]
(顔料分散剤)
この実施例は、顔料の存在下で使用した場合に改善された分散剤特性を有するカルボキシル官能性のバイオベースポリエステル樹脂の生成を例示する。
装置(図6を参照のこと)
4頸トップを備えた2リットルの丸底ガラス反応容器、ジャケット付きVigreauxカラム、蒸留ヘッド、ガス入口および出口アダプター、ステンレス鋼撹拌軸および4枚羽根(45°の角度)パドル、冷却器、ならびに受けフラスコ。
手順
反応器に、D−イソソルビド(545.35g、3.7317モル)(受理したまま)、Pripol 1013(登録商標)ダイマー酸(バッチ# 091687)(272.17g、0.47302モル)、および1,4−シクロヘキサンジカルボン酸(1,4−CHDA)(632.49g、3.6734モル)と、続いて酸化アンチモン(III)(0.498g)を加えた。反応器をアルゴンでブランケットした。次いで、1,2,3,4−テトラヒドロナフタレン(2ml)を、アルゴン中、反応混合物に添加した。アルゴン中、(固体物が溶融した後)撹拌しながら、反応器内容物の温度を200℃に上げた。この温度を30分間維持した。反応混合物を、30分間かけて250℃に徐々に加熱した(1.6℃/分)。この温度を2.1時間維持した。反応を約180℃超で加熱したとき、水を連続的に回収した。Vigreauxカラムの塔頂部の温度が下がったとき、これは大部分の水が除去されたことを示唆する。約134mlの水が留出した。イソソルビドが蒸留されるのを回避するために、反応混合物上のアルゴン流量をチェックし、必要な場合は低速にした。反応混合物を、2時間かけて280℃に徐々に加熱した(0.25℃/分)。留出液受器を真空受器に換え、徐々に真空にした(<133.322Pa(1トル))。この間に、残留水が留出し、低分子量ポリマーが形成した。反応混合物温度を280℃で30分間維持した。反応混合物をアルゴンでブランケットして、大気圧を得ることによって、反応を終結した。次いで、反応混合物を≦250℃に冷却し、フッ素化ファイバーグラスシートに注いだ。
【0111】
下記の特性を有する樹脂を生成した。
Tg=52.9℃
酸価=47.7
120℃における粘度=7772ポアズ
160℃における粘度=247ポアズ
ポリマー特性:
色:黄色/明コハク色
粘着性:非粘着性
清澄性:半透明
柔軟性:脆い
固体
【0112】
[実施例4]
この実施例は、実施例3のカルボキシル官能性ポリエステル樹脂を使用したβ−ヒドロキシアミド型架橋による粉体コーティング配合物の調製を例示する。次いで、粉末配合物を基材に塗布する。
【0113】
上記に記載するように混合し、β−ヒドロキシアミドエステル交換で架橋した典型的な粉体コーティング配合物の並列比較で、カルボキシル官能性ポリエステル(実施例3の生成物)を市販のポリエステルと比較した。下記の表4は、これらの配合物を重量百分率で示す。
【0114】
【表6】
【0115】
FINE−CLAD M8930(登録商標)は、比較のために使用されたポリエステル酸の例である。
手順:粉体コーティング混合プロトコル(Brabender(登録商標)混合機による99℃での混合):
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。約30分間予熱させた。予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで、30gの一次樹脂をボウルに徐々に添加し、溶融するまで混合した。次いで、一次樹脂の残部35.2gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。次いで、1.3gの添加剤(0.9gのベンゾインおよび0.4gのModaflow 6000(登録商標))をローター間の混合ゾーン中心部に添加した。
【0116】
混合を10分間続行し(トルク値を安定性について監視した)、次いで3.4gの架橋性樹脂(Primid XL−552(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0117】
混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0118】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧した。パネルを121℃または147℃で30分間硬化した(試験結果については、表6を参照のこと)。
【0119】
上記の手順を繰り返して、対照材料を得た。
バイオベースのコーティングは、光沢に関して様々な温度でロバスト性がはるかに高く、より低い温度では等価な光沢を有し、より高い温度ではるかによりよい光沢を有した。
【0120】
示差走査熱分析(DSC)の結果は、実験した樹脂が、硬化温度、硬化の大きさ、またはコーティングの最終Tgに、ポジティブにもまたはネガティブにも影響しないことを示した。これは、外的影響に対するβ−ヒドロキシアミド硬化速度の非感受性(Howellの参考文献を参照のこと)に一致した。
【0121】
【表7】
【0122】
バイオベースのコーティングも、両方の硬化温度において市販対照と同様な最終特性を有した。
【0123】
【表8】
【0124】
表5および6のデータは、両方のコーティングが性能全体において本質的に等価であることを示す。バイオベースのコーティングは、より高い硬化温度における光沢でわずかな利点を有し、市販対照は、耐溶媒性でわずかな利点を有した。先に述べたように、多大な温度ロバスト性を必要とする配合物において、より高い温度での光沢は顕著な利点となり得る。
【0125】
したがって、このタイプのバイオベース樹脂は、β−ヒドロキシアルキルアミドを用いたエステル交換架橋反応として有用である。
【0126】
[実施例4A]
この実施例は、カルボキシル官能性樹脂を使用したβ−ヒドロキシアミド型架橋による粉体コーティング配合物の調製を例示する。
上記に記載するように混合し、β−ヒドロキシアミドエステル交換で架橋した典型的な粉体コーティング配合物の並列比較で、カルボン酸官能性ポリエステル(実施例3E)を市販のポリエステルと比較した。表4Aは、これらの配合物を重量百分率で示す。
【0127】
【表9】
【0128】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約85gの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。約30分間予熱させた。予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂をボウルに徐々に添加し、溶融するまで混合した。次いで、一次樹脂の残部37.8gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させ(約5分間)、次いで3.6gの添加剤(1.1gのベンゾインおよび2.5gのFine Clad A241(登録商標))をローター間の混合ゾーン中心部に添加し、混合を10分間続行し(トルク値を安定性について監視した)、次いで4.1gの架橋性樹脂(Primid XL−552(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0129】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧した。パネルを121℃または147℃で30分間硬化した(試験結果については、表6を参照のこと)。実施例4Aの上記の手順を繰り返して、対照材料を得た。
【0130】
バイオベースのコーティングは、光沢に関して様々な温度でロバスト性がはるかに高く、より低い温度では等価な光沢を有し、より高い温度ではるかによりよい光沢を有した。
示差走査熱分析(DSC)の結果は、実験したコーティングが、硬化温度、硬化の大きさ、またはコーティングの最終Tgに、ポジティブにもまたはネガティブにも影響しないことを示した。これは、外的影響に対するβ−ヒドロキシアミド硬化速度の非感受性(Howellの参考文献を参照のこと)に一致した。
【0131】
【表10】
【0132】
下記の表6Aからわかるように、バイオベースのコーティングも、両方の硬化温度において市販対照と同様な最終特性を有した。
【0133】
【表11】
【0134】
表5Aおよび6Aのデータは、両方のコーティングが性能全体において本質的に等価であることを示す。バイオベースは、より低い硬化温度でより硬質の被膜の利点を有した(#H鉛筆対#HB鉛筆)。バイオベースはまた、より高い硬化温度における光沢で利点を有し、市販対照は、耐溶媒性で利点を有した。先に述べたように、多大な温度ロバスト性を必要とする配合物において、より高い温度での光沢は顕著な利点となり得る。
【0135】
したがって、このタイプのバイオベース樹脂は、β−ヒドロキシアルキルアミドでのエステル交換架橋反応として有用である。
【0136】
[実施例5]
この実施例は、カルボキシル官能性ポリエステル樹脂(実施例2の生成物)およびアクリル酸エポキシ架橋剤と配合されたハイブリッドコーティングのための配合物の調製を例示する。
手順:バイオベースのハイブリッド粉体コーティングのための粉体コーティング混合プロトコル(Brabender(登録商標)混合機による99℃での混合):
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働く。次いで、30gの一次樹脂(実施例2に記載した樹脂)をボウルに徐々に添加し、溶融するまで混合した。次いで、一次樹脂の残部21.5gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。混合を10分間続行した(トルク値を安定性について監視した)。16.9gの架橋性樹脂(Fine Clad A229−30A(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒を細粉に粉砕し、最後に添加した(0.4gのイミダゾールおよび1.1gのドデカン二酸)。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0137】
混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0138】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、約63.5ミクロン(約2.5ミル)の乾燥被膜厚にした。パネルを121℃で30分間硬化した。
手順:対照ポリエステルハイブリッド粉体コーティングのための粉体コーティング混合プロトコル(Brabender(登録商標)混合機による99℃での混合):
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。
【0139】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(Fine−Clad M8400(登録商標))をボウルに徐々に添加し、溶融するまで混合した。一次樹脂の残部25.8gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させ(約5分間)、混合を10分間続行した(トルク値を安定性について監視した)。12.7gの架橋性樹脂(Fine Clad A229−30A(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒(0.4gのイミダゾールおよび1.1gのドデカン二酸)を最後に添加し、トルク示度を綿密に監視した。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0140】
混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0141】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、約63.5ミクロン(約2.5ミル)の乾燥被膜厚にした。パネルを121℃で30分間硬化した。
【0142】
図4に記載された粘度を有する、この実施例に記載した2つのポリエステル樹脂を、これらの手順に従って透明コートに配合し、下記の表7に示す。表7は、様々な材料の量を重量百分率で示す。
【0143】
【表12】
【0144】
バイオベース材料と対照系について、硬化の外観に及ぼす影響を比較した。低温硬化型粉体コーティングを対照として選択した。被験製品を1PC−306−0040(F−0040)S−9 Clear Gloss(登録商標)と称する。硬化スケジュールは、145℃で15分間、または162℃で10分間であった。
【0145】
コーティングの表面粗さを測面計で定量化した。この試験の最中に、細針がコーティングの表面上を通過し、同時に表面の山と谷を記録した。谷をRv(nm)として記録し、山をRp(nm)として記録した。これらの2つの値から、平均粗さ(Ra)を算出する。より低いR値は、より平坦またはより滑らかな表面を示唆する。被覆パネルのR値については、表8を参照のこと。
【0146】
【表13】
【0147】
一般的な生成方法で生成された表面粗さについての典型的なR値(ASME B46.1−1995に列挙)を、表8の値と比較することができる。バイオベースのパネルの表面粗さは、グライディング、ホーニング、および電解研摩で生成された表面と同様である。対照パネルの表面粗さは、スナッギング、平削り、および形削り操作で生成された表面と同様である。
【0148】
[実施例6]
この実施例は、実施例3Aのバイオベースのカルボキシル官能性ポリエステルおよびエポキシ架橋剤と配合された、顔料を含むハイブリッド粉体コーティングを例示する。
バイオベース樹脂が、顔料を分散し、顔料を発色させる優れた能力を有する場合に、顔料を含む粉体コーティングは利益を得ることができる。黒色粉体コーティング配合物(下記)の例は、市販対照に対して作製した。表9を参照のこと。表9の配合物は、様々な材料の量を重量百分率で示す。
【0149】
【表14】
【0150】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。
【0151】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(実施例3Aに記載)をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部23.1gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。1.7gの添加剤(0.8gの黒色顔料および0.9gのベンゾイン)をローター間の混合ゾーン中心部に添加した。混合を10分間続行した(トルク値を安定性について監視した)。架橋性樹脂としてエポキシ官能基を含むアクリル(FineClad A257(登録商標))11.3gを先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒を細粉に粉砕し、最後に添加した(0.5gのイミダゾール、1.7gのドデカン二酸、および1.7gのNacure XC−7231(登録商標))。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0152】
混合ボウルから、滑らかで堅い光沢のある黒色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0153】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0154】
対照のポリエステル顔料を含む粉体コーティングでは、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。
【0155】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(Fine−Clad M8400(登録商標))をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部21.9gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。1.7gの添加剤(0.8gの黒色顔料および0.9gのベンゾイン)をローター間の混合ゾーン中心部に添加した。混合を10分間続行した(トルク値を安定性について監視した)。12.5gの架橋性樹脂(FlneClad A257(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒(0.5gのイミダゾール、1.7gのドデカン二酸、および1.7gのNacure XC−7231(登録商標))を最後に添加し、トルク示度を綿密に監視した。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0156】
混合ボウルから、滑らかで堅い光沢のある黒色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0157】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0158】
バイオベース粉体コーティングは、石油化学製品由来のコーティングより、はるかに高い60度光沢値を有した(85点対44点)。これは、おそらく熱硬化中、配合物のメルトフローがよりよいことに起因した。
【0159】
バイオベース配合物については、黒色顔料の発色/漆黒度が改善される。コーティングのLおよびbの色成分を測定することによって、漆黒度を決定することができる。(Hunter color Scaleの説明については、上記に引用したWicks,Z.W.らを参照のこと)。
【0160】
極黒色は低いL値で決まり、藍色のアンダートーンは低いb値で決まる。Lおよびbの値が低くなると、コーティングの漆黒度が高くなる。漆黒度は、よりよい顔料分散および発色で改善される。下記の表10は、被覆パネルの色測定値を示す。
【0161】
【表15】
【0162】
2つの黒色パネルの全体のデルタE、すなわち色差は、0.52である。石油由来のパネルは、黒色顔料が、バイオベース配合物ほどよくコーティング系に分散していないので、バイオベース配合物のパネルより灰色に見える。
【0163】
[実施例6A]
粉体コーティングにおいて低温フローおよび硬化が必要であるのに加えて、コーティングタイプに関わらず、コーティングマトリックス内において顔料の良好な分散も必要である。これを実現するために、異なる相溶性の成分を有するポリマーが設計されている。ポリマー分散剤は、塗料、コーティング、およびインク系中の顔料および他の材料を、最も典型的には立体安定化によって安定化する。ポリマー分散剤は、定着基とポリマー鎖からなる2成分構造を有する。最も典型的には、定着基は、粒子表面、およびコーティングの連続相と相溶であるポリマー鎖と相互作用する極性材料である。実際には、該ポリマー基は粒子の周りにコーティングを形成し、粒子同士が接触し、より大きい非相溶な凝集体になるのを防止する。
【0164】
有効なポリマー分散剤を与えるものと予想され得る定着基/ポリマーの立体配置は多数存在する。本発明の樹脂は、極性カルボン酸定着部位および非極性植物油鎖を有し、したがって分散剤および結合剤として働くことができる。分散剤としても働くことができる硬化結合剤によって、多数の顔料を分散するために別々の添加剤を必要とすることがなくなる可能性がある。
【0165】
表10−6A−1の配合物は、様々な材料の量を重量百分率で示す。
【0166】
【表16】
【0167】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を110℃に予備加熱した(ボウルは約110℃に上昇した)。30分間予熱させた。
【0168】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30.0gの一次樹脂(実施例3に記載)をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部33.0gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させ(約5分間)、混合羽根の速度を40回転/分に設定した。7.0gの黒色顔料をローター間の混合ゾーン中心部に添加した。混合を15分間続行した。混合ボウルから、滑らかで堅い光沢のある黒色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。
【0169】
対照ポリエステル黒色分散液では、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を110℃に予備加熱した(ボウルは約110℃に上昇した)。30分間予熱させた。
【0170】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30.0gの対照ポリエステル(FineClad M8400)をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部33.0gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させ(約5分間)、混合羽根の速度を40回転/分に設定した。7.0gの黒色顔料をローター間の混合ゾーン中心部に添加した。混合を15分間続行した。混合ボウルから、滑らかで堅い光沢のある黒色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。
【0171】
これらの材料をその後の配合物で使用して、カーボンブラック顔料の分散度を決定した。黒色顔料の分散がよくなると、最終配合物の得られる色が濃く(黒く)なる。
表10−6A−2 の配合物は、様々な材料の量を重量百分率で示す。
【0172】
【表17】
【0173】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。
【0174】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで34.9gの一次樹脂(FINE−CLAD M8400(登録商標))をボウルに徐々に添加し、溶融するまで混合させた。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。22.1gの添加剤(21.0gの白色顔料(Kronos 2310)および1.1gのベンゾイン)をローター間の混合ゾーン中心部に添加し、ローターの速度を60回転/分に設定し、混合を15分間続行した。バイオベースポリエステル樹脂に事前に分散させた黒色顔料(表10−6A−2の実施例A)3.5gを添加し、ローターの速度を40回転/分に下げ、混合を5分間続行した。7.2gの架橋性樹脂(FineClad A257(登録商標))を先の混合物に添加し、混合を少なくとも2分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒を細粉に粉砕し、最後に添加した(0.5gのイミダゾールおよび1.9gのドデカン二酸)。混合を2分間続行した。
【0175】
混合ボウルから、滑らかで堅い光沢のある灰色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0176】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0177】
120mlのボウルサイズに対して、対照ポリエステル粉体コーティング全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。30分間予熱させた。
【0178】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで34.9gの一次樹脂(FineClad M8400)をボウルに徐々に添加し、溶融するまで混合させた。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。22.1gの添加剤(21.0gの白色顔料(Kronos 2310)および1.1gのベンゾイン)をローター間の混合ゾーン中心部に添加し、ローターの速度を60回転/分に設定し、混合を15分間続行した。バイオベースポリエステル樹脂に事前に分散させた黒色顔料(表10−6a−2の実施例B)3.5gを添加し、ローターの速度を40回転/分に下げ、混合を5分間続行した。7.2gの架橋性樹脂(FineCiad A257(登録商標))を先の混合物に添加し、混合を少なくとも2分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒を細粉に粉砕し、最後に添加した(0.5gのイミダゾールおよび1.9gのドデカン二酸)。混合を2分間続行した。
【0179】
混合ボウルから、滑らかで堅い光沢のある灰色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0180】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0181】
バイオベース配合物については、黒色顔料の発色および比着色力が改善される。比着色力は、カーボンブラックが二酸化チタンなど他の顔料の存在する配合物を暗色化/左右する能力である。コーティングのLおよびbの色成分を測定することによって、比着色力を決定することができる。(Hunter color Scaleの説明については、上記に引用したWicks,Z.W.らを参照のこと)。
【0182】
より高い比着色力は低いL値で決まる。L値が低くなると、カーボンブラックの分散がよくなり、コーティングでの発色がよくなる。バイオベースポリエステルは、カーボンブラックの分散をよりよくし、カーボンブラックの発色をより完全にすることができて、比着色力がより高くなる。下記の表10−6A−3は、被覆パネルの色測定値を示す。
【0183】
【表18】
【0184】
2つの黒色パネルの全体のデルタE、すなわち色差は、2.53である。石油由来のパネルは、黒色顔料が、バイオベース配合物ほどよくコーティング系に分散していないので、バイオベース配合物のパネルより明るく見える。
【0185】
[実施例6B]
この実施例は、実施例3Aのバイオベースのカルボキシル官能性ポリエステル、およびイソシアヌル酸トリグリシジル(TGIC)架橋剤と配合された、顔料を含むハイブリッド粉体コーティングを例示する。
【0186】
バイオベース樹脂が、フィッシュアイ、すなわち被膜の欠点の出現なしにコーティングのフローおよびレベリング性を促進する優れた能力を有する場合に、粉体コーティングは利益を得ることもできる。白色粉体コーティング配合物(下記)の例は、市販対照に対して作製した。表10−6B−1を参照のこと。表10−6B−1の配合物は、様々な材料の量を重量百分率で示す。
【0187】
【表19】
【0188】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を110℃に予備加熱した(ボウルは約110℃に上昇した)。30分間予熱させた。
【0189】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(実施例3に記載)をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部27.2gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。42.8gの添加剤(4.4gのTGIC、37.9gの二酸化チタン(Kronos CR2310)、および0.5gのベンゾイン)をローター間の混合ゾーン中心部に添加した。
【0190】
混合を10分間続行した(トルク値を安定性について監視した)。
混合ボウルから、滑らかで堅い光沢のある白色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0191】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0192】
対照のポリエステル顔料を含む粉体コーティングでは、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全配合物重量を算出した。Brabender(登録商標)混合機を110℃に予備加熱した(ボウルは約110℃に上昇した)。30分間予熱させた。
【0193】
予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(Albester 5140)をボウルに徐々に添加し、溶融するまで混合させ、樹脂の残部27.2gを添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。42.8gの添加剤(4.4gのTGIC、37.9gの二酸化チタン(Kronos CR2310)、および0.5gのベンゾイン)をローター間の混合ゾーン中心部に添加した。混合を10分間続行した(トルク値を安定性について監視した)。
【0194】
混合ボウルから、滑らかで堅い光沢のある白色材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0195】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、121℃で30分間硬化した。
【0196】
【表20】
【0197】
バイオベース配合物は、対照配合物よりよい耐溶媒性、高い光沢、および高い鉛筆硬度を有する。バイオベース配合物の全体の外観は、「フィッシュアイ」と呼ばれる被膜の欠点を有する対照配合物よりはるかによい。コーティング中のフィッシュアイの存在は単に外観の問題であるだけでなく、基材が環境に曝露されるので、これらの領域はさびおよび腐食の影響を受けやすい。
【0198】
本発明の別の実施形態による樹脂を添加顔料として使用して、より効率的に顔料を分散することができる。例えば、顔料を添加して、発色を助けることができる。
【0199】
[実施例7]
この実施例は、アミド−アミン官能性ポリエステル、およびエポキシ架橋剤と配合されたハイブリッド粉体コーティングの生成を例示する。
アミド−アミン官能性ポリエステル粉体コーティングをバイオベース樹脂と配合することもできる。粉体コーティング配合物の例を下記の表11に示し、材料の量を重量百分率で示す。利用可能な市販のアミドアミン官能性ポリエステル樹脂はないので、したがって対照は用いなかった。
【0200】
【表21】
【0201】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全粉体コーティングを算出した。Brabender(登録商標)混合機を99℃に予備加熱した(ボウルは約99℃に上昇した)。約30分間予熱させた。予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働き、次いで30gの一次樹脂(49251−23−22)をボウルに徐々に添加した。トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。次いで、一次樹脂の残部3.7gを添加し、約5分間混合させた。次いで、0.7gの添加剤(Modaflow 6000(登録商標))をローター間の混合ゾーン中心部に添加した。混合を10分間続行した(トルク値を安定性について監視した)。次いで、30.6gの架橋性樹脂(FineClad A249A(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。触媒(0.7gのイミダゾール、2.1gのドデカン二酸、および2.1gのNacure XC−7231(登録商標))を最後に添加し、トルク示度を綿密に監視した。トルクは上昇し、粘度(トルク)が10%上昇した後、バッチを止めた。
【0202】
混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。最後に、生成物を、10mm〜15mmのスチールメディアの存在下、ボールミルで16時間微粉化した。最終粉末が得られた。これをふるいにかけて、150ミクロンを超える粒子を除去した。
【0203】
Nordson Corporationから供給されたVersa−Spray(登録商標)マニュアルスプレーガンを使用して、粉末を10.16cm(4インチ)×15.24cm(6インチ)のベア鋼パネルに静電噴霧し、乾燥被膜厚が約76.2ミクロン(3ミル)の被膜構造にした。パネルを、対流式オーブン中、95℃、107℃、または121℃で30分間硬化した(試験結果については、表13を参照のこと)。
【0204】
実施例7からの試料をDSCで分析した。結果を下記の表12に示す。
【0205】
【表22】
【0206】
どのパネルも非常に高い光沢を有していないが、被膜特性は、硬化温度107℃で正当であった。表13を参照のこと。硬度は、耐溶媒性(MEK二重摩擦)と同様に劇的に改善した。マンドレル屈曲の結果も、接着性および柔軟性の改善を示した。
【0207】
【表23】
【0208】
[実施例8]
この実施例は、バイオベースポリエステルをフロープロモーターとして使用する粉体コーティングの生成を例示する。ポリエステルポリマーを実施例3Bに記載する。
まず、対照配合物を下記の通り調製した。
【0209】
まず、120mlのボウルサイズに対して、全配合物重量、すなわち約70グラムの全粉体コーティングを算出した。Brabender(登録商標)混合機を93℃に予備加熱した(ボウルは約99℃に上昇した)。約30分間予熱させた。予熱が完了したとき、混合羽根を始動し、トルクセンサーを作動させた。これは、混合の進行状況のためのガイドとして働いた。次いで、30gの一次樹脂(Fine−Clad M8710(登録商標))をボウルに徐々に添加し、トルクセンサーが定常値を示すまで、樹脂を混合溶融させた(約5分間)。次いで、一次樹脂の残部31.8gを添加し、約5分間混合させた。次いで、0.44gの添加剤(ベンゾイン)をローター間の混合ゾーン中心部に添加した。混合を10分間続行した(トルク値を安定性について監視した)。
【0210】
17.7gの架橋性樹脂(FineClad A249A(登録商標))を先の混合物に添加し、混合を少なくとも3分間続行した。架橋が開始する可能性がある(トルク示度は急速に上昇し始める)ので、トルク示度が安定なままであるように監視し、トルク示度を綿密に監視した。
【0211】
混合ボウルから、滑らかで堅い光沢のある材料として生成物を取り出し、室温まで放冷した。材料をハンマーで小チップに砕いた。
上記の手順を6回以上繰り返して、3つのフロープロモーターをそれぞれ1%(重量)または3%(重量)組み込んだ。フロープロモーターは、ベンゾイン添加時に添加した。バイオベースのフロープロモーター(実施例3B)、および2つの市販フロープロモーター(Fine−Clad A241(登録商標)および Additol VXL9820(登録商標))を使用して、配合物B〜Gを調製した(表14を参照のこと)。
【0212】
【表24】
【0213】
低い硬化温度における粉体コーティングの粘度を下げる際のフロープロモーターの有効性を決定するために、表14の配合物の粘度を試験した。粘度が低くなれば、ベーキング中、最終被膜の流展性がよくなり、滑らかになることを示唆するはずである。表15は、90℃〜140℃における粘度(単位、ポアズ)を示す。
【0214】
【表25】
【0215】
他の被検フロープロモーターより低い温度(90℃および100℃)における粉体コーティングの粘度を下げる際に、バイオベースポリエステルフロープロモーター(試料B)の1%添加が最も有効である。90℃における3%添加(試料C)は、市販材料よりさらに低い。これは、図7にグラフで示している。
【0216】
この一連のDSC結果は、バイオベースのフロープロモーターの添加によって、90℃および100℃における溶融粘度が下がっても、粉体コーティング配合物全体のTgは下がらず、粉末安定性は損なわれないことを示す。粉体コーティングの硬化ピーク温度および反応エンタルピー(ΔH)は、フロープロモーターから悪影響を受けなかった。配合物A〜Gについて、結果を表16に示す。
【0217】
【表26】
【0218】
[実施例9]
実施例3Fの樹脂を、その顔料分散体特性についてカラーコンセントレートで評価した。
材料:
2つのカラーコンセントレート処方を選択した。一方は、ポリスチレン担体樹脂にPB 15:3(フタロブルー)10%添加であり、他方は、アクリロニトリル・ブタジエン・スチレンコポリマー(ABS)系担体樹脂にカスタムグリーンであった。カスタムグリーンは有機および無機顔料のブレンドからなり、約18%加えた。対照試料は、ステアリン酸亜鉛、およびステアリン酸亜鉛とエチレンビステアラミド分散剤の組合せなどの典型的な分散剤を用いて実施され、試料は、実施例3Fからの分散剤助剤を用いて実施された。
混合:
同方向回転の直径18mmのLeistritz二軸押出機で、混合を行った。
試験:
フィルター試験で、分散試験を行い、圧力上昇(単位、バール/顔料1グラム)を報告した。これは、分散の定量試験であり、値が低くなれば、分散がよくなることを示唆する。
【0219】
表17は、試験に使用した配合物、および結果を示す。
【0220】
【表27】
【0221】
市販の分散剤であるステアリン酸亜鉛、およびEBSとステアリン酸亜鉛の混合物との比較は、良好な結果を示した。結果は、2つの異なるポリマー系で一定の優れた発色を示した。
【0222】
本明細書に開示する本発明の形態は、現在好ましい実施形態を構成するが、他の多くの形態が可能である。本明細書では、本発明の可能な等価の形態または細分化形態をすべて記述するように意図されていない。本明細書で使用する用語は、説明するものにすぎず、限定するものでないこと、本発明の範囲の趣旨から逸脱することなく、様々な変更を行うことができることを理解されたい。
【図面の簡単な説明】
【0223】
【図1】硬質の結晶質イソソルビドを非晶質ダイマージオール、芳香族ジエステル、および他の材料とブレンドして、ポリエステル材料を合成する経路を示す概略フローチャートである。
【図2】硬質の結晶質イソソルビドを非晶質ダイマー二酸および他の材料とブレンドして、ポリエステル酸を合成する経路を示す概略フローチャートである。これらを、具体的にはハイブリッド粉体コーティング配合物で利用して、エポキシ官能性部位を架橋することができる。
【図3】硬質の結晶質イソソルビドをダイマージオール、非晶質ダイマー二酸、ポリイソシアナート(例えば、ジイソシアナート)、および他の材料とブレンドして、ポリウレタンを合成する経路を示す概略フローチャートである。
【図3A】本発明で有用なイソソルビドの典型的な異性体1a、1b、および1cを示す図である。
【図4】バイオベース樹脂(実施例2)および典型的な市販樹脂(FINE−CLAD 8400(登録商標))のレオロジー曲線を示すグラフである。
【図5】バイオベース配合物(実施例4A)および市販の対照配合物について、約121℃における粘度プロファイルを示すグラフである。
【図6】実施例1〜3Fの樹脂を作製するために使用した装置の様々な構成要素を示す概略図である。
【図7】実施例8の配合物A〜Gについて、90℃と100℃における粘度を示す棒グラフである。
【特許請求の範囲】
【請求項1】
a.ヒドロキシル、カルボキシル、またはイソシイソシアナート基で官能化され、Tgが約80℃未満の樹脂と、
b.前記樹脂の少なくとも5重量%のバイオベース内容物と
c.架橋剤と
の反応生成物を含む粉体コーティング。
【請求項2】
樹脂が、ポリエステル樹脂、ポリアミド樹脂、またはポリウレタン樹脂である、請求項1に記載の粉体コーティング。
【請求項3】
粉体コーティングが、約125℃以下で硬化する、請求項1または2に記載の粉体コーティング。
【請求項4】
A.ジアンヒドロヘキシトールと、
B.ダイマージオールおよび/またはダイマー二酸と、
C.二酸、ジエステル、または二酸塩化物と
D.触媒と
の反応生成物を含むカルボキシルまたはヒドロキシル官能性ポリエステル樹脂。
【請求項5】
エチレングリコールでない任意選択のジオールをさらに含む、請求項4に記載のポリエステル樹脂。
【請求項6】
任意選択の第2の二酸、ジエステル、または二酸塩化物を含む、請求項4に記載の樹脂。
【請求項7】
ジアンヒドロヘキシトールがイソソルビドを含む、請求項4に記載の樹脂。
【請求項8】
ヒドロキシル、カルボキシル、またはイソシアナート官能基を有するポリウレタン樹脂であって、
A.ジアンヒドロヘキシトールと、
B.ダイマージオールおよび/またはダイマー二酸と、
C.ポリイソシアナートと、
D.触媒と
の反応生成物を含む樹脂。
【請求項9】
ジアンヒドロヘキシトールがイソソルビドである、請求項4に記載の樹脂。
【請求項10】
A.請求項4または請求項8に記載の樹脂、アミド−アミン樹脂、およびその混合物からなる群から選択された樹脂と、
B.架橋剤と
の反応生成物を含む粉末配合物。
【請求項11】
架橋剤がエポキシ官能基を有する、請求項10に記載の粉末配合物。
【請求項12】
架橋剤がβ−ヒドロキシアミド官能性を有する、請求項10に記載の粉末配合物。
【請求項13】
A.基材と、
B.前記基材上で硬化させた粉末と
を含み、前記硬化させた粉末が、
a.請求項4または請求項8に記載の樹脂、アミド−アミン樹脂、およびその混合物からなる群から選択された樹脂と、
b.架橋剤と
の反応生成物である粉体コーティング。
【請求項14】
架橋剤がエポキシ官能性を有する、請求項13に記載の粉体コーティング。
【請求項15】
架橋剤がβ−ヒドロキシアミド官能性を有する、請求項13に記載の粉体コーティング。
【請求項16】
粉体コーティングがクラスAの仕上がりを提供する、請求項13に記載の粉体コーティング。
【請求項17】
基材が感温性である、請求項13に記載の粉体コーティング。
【請求項18】
粉体コーティングがクラスAの仕上がりを提供する、請求項13に記載の粉体コーティング。
【請求項19】
樹脂のTgが約40℃から80℃までの間である、請求項13に記載の粉体コーティング。
【請求項20】
a.ジアンヒドロヘキシトールの靭性結晶質バイオベースモノマーを選択する工程と、
b.フロー性のある非晶質バイオベース樹脂と共反応させて、樹脂を形成する工程
とを含み、前記樹脂が
a.約40℃より高く、約80℃より低いTgと、
b.前記樹脂組成物中に少なくとも5重量%のバイオマスと、
c.125℃以下、30分間の低温ベーキング条件での硬化、柔軟性、および流展性と
を含む方法。
【請求項21】
樹脂が、ヒドロキシル、カルボキシル、および/または窒素含有基(アミン、イソシアナート、またはアミド基)で官能化されている、請求項20に記載の方法。
【請求項22】
樹脂が、第2の硬化機序で利用可能な過剰の不飽和を有する、請求項20に記載の方法。
【請求項23】
ポリエステル酸樹脂のカルボキシル基と硬化反応するβ−ヒドロキシルアミドの反応であるβ−ヒドロキシアミドエステル交換硬化を利用して、請求項2に記載の樹脂をコーティング組成物に配合する、請求項20に記載の方法。
【請求項24】
樹脂をエポキシ−アクリルなどのハイブリッド粉体コーティングになる材料と配合する請求項21に記載の方法。
【請求項25】
ハイブリッドコーティング組成物が、次の一般式を有する触媒:
【化1】
[式中、R1、R2、R3、およびR4は独立に、水素、またはエポキシ樹脂と反応しない任意の置換基である]
と反応する、請求項20に記載の方法。
【請求項26】
靭性結晶質バイオベースモノマー(イソソルビドなど)を、フロー性のある非晶質バイオベース樹脂と共反応させて、コーティング、接着剤、インク、シーラント、ポリマー用途、および他の市販の材料を製造するために使用することができる樹脂を形成する組成物。
【請求項27】
靭性結晶質バイオベースモノマー(イソソルビドなど)をフロー性のある非晶質バイオベース樹脂と共反応させて、新規クラスの樹脂を形成し、
前記樹脂が
a.約40℃〜約80℃のTgと、
b.前記樹脂組成物中に少なくとも約5重量%のバイオマスと、
c.硬化温度125℃で30分間
とを有する組成物。
【請求項28】
樹脂が、カルボキシル基、ヒドロキシル基、および/または窒素含有基(アミン、イソシアナート、および/またはアミド)で官能化されている、請求項27に記載の組成物。
【請求項29】
樹脂が過剰の不飽和を有する、請求項27に記載の組成物。
【請求項30】
樹脂がカルボキシル基で官能化されている、請求項27に記載の組成物。
【請求項31】
樹脂がヒドロキシル基で官能化されている、請求項27に記載の組成物。
【請求項32】
樹脂が窒素含有基(アミン、イソシアナート、アミドなど)で官能化されている、請求項27に記載の組成物。
【請求項33】
樹脂のカルボキシル基と硬化反応するβ−ヒドロキシルアミドの反応であるβ−ヒドロキシアミドエステル交換硬化を利用して、請求項30に記載の樹脂をコーティング組成物に配合する、請求項27に記載の組成物。
【請求項34】
樹脂が、≧40℃のTg、および≧20%のバイオベース含有量を有し、粉体コーティングに混合され、これらの粉体コーティングを、
a.β−ヒドロキシルアミド
b.グリシジルメタクリレートアクリルコポリマー樹脂を含むが、これに限定されないエポキシ含有樹脂
などの架橋性樹脂で硬化することができる、請求項27に記載の組成物。
【請求項35】
A.樹脂と、
B.架橋剤と、
C.任意選択の顔料
とを含み、前記樹脂が、
a.ジアンヒドロヘキシトールと、
b.ダイマー酸および/またはダイマージオールと、
c.二酸、ジエステル、または二酸塩化物と
の反応生成物を含む粉体コーティング。
【請求項36】
反応のための触媒を含む、請求項35に記載の粉体コーティング。
【請求項37】
顔料分散助剤を含む、請求項35に記載の粉末。
【請求項38】
樹脂がカルボキシル基を有する場合、カルボキシ官能性のカルボキシル基を自己触媒エステル交換反応でβ−ヒドロキシルアミドと反応させ、またはポリエポキシ官能性ポリマーと反応させる工程を含む、請求項35に記載の粉体コーティング。
【請求項39】
樹脂がヒドロキシル官能基を有する場合、前記ヒドロキシル官能性の前記ヒドロキシル基をエポキシ官能性アクリルと反応させる工程を含む、請求項35に記載の粉体コーティング。
【請求項40】
任意選択の賦形剤を含む、請求項35に記載の粉体コーティング。
【請求項41】
本明細書で開示されたすべての新規で非自明の組成物、方法、および使用。
【請求項1】
a.ヒドロキシル、カルボキシル、またはイソシイソシアナート基で官能化され、Tgが約80℃未満の樹脂と、
b.前記樹脂の少なくとも5重量%のバイオベース内容物と
c.架橋剤と
の反応生成物を含む粉体コーティング。
【請求項2】
樹脂が、ポリエステル樹脂、ポリアミド樹脂、またはポリウレタン樹脂である、請求項1に記載の粉体コーティング。
【請求項3】
粉体コーティングが、約125℃以下で硬化する、請求項1または2に記載の粉体コーティング。
【請求項4】
A.ジアンヒドロヘキシトールと、
B.ダイマージオールおよび/またはダイマー二酸と、
C.二酸、ジエステル、または二酸塩化物と
D.触媒と
の反応生成物を含むカルボキシルまたはヒドロキシル官能性ポリエステル樹脂。
【請求項5】
エチレングリコールでない任意選択のジオールをさらに含む、請求項4に記載のポリエステル樹脂。
【請求項6】
任意選択の第2の二酸、ジエステル、または二酸塩化物を含む、請求項4に記載の樹脂。
【請求項7】
ジアンヒドロヘキシトールがイソソルビドを含む、請求項4に記載の樹脂。
【請求項8】
ヒドロキシル、カルボキシル、またはイソシアナート官能基を有するポリウレタン樹脂であって、
A.ジアンヒドロヘキシトールと、
B.ダイマージオールおよび/またはダイマー二酸と、
C.ポリイソシアナートと、
D.触媒と
の反応生成物を含む樹脂。
【請求項9】
ジアンヒドロヘキシトールがイソソルビドである、請求項4に記載の樹脂。
【請求項10】
A.請求項4または請求項8に記載の樹脂、アミド−アミン樹脂、およびその混合物からなる群から選択された樹脂と、
B.架橋剤と
の反応生成物を含む粉末配合物。
【請求項11】
架橋剤がエポキシ官能基を有する、請求項10に記載の粉末配合物。
【請求項12】
架橋剤がβ−ヒドロキシアミド官能性を有する、請求項10に記載の粉末配合物。
【請求項13】
A.基材と、
B.前記基材上で硬化させた粉末と
を含み、前記硬化させた粉末が、
a.請求項4または請求項8に記載の樹脂、アミド−アミン樹脂、およびその混合物からなる群から選択された樹脂と、
b.架橋剤と
の反応生成物である粉体コーティング。
【請求項14】
架橋剤がエポキシ官能性を有する、請求項13に記載の粉体コーティング。
【請求項15】
架橋剤がβ−ヒドロキシアミド官能性を有する、請求項13に記載の粉体コーティング。
【請求項16】
粉体コーティングがクラスAの仕上がりを提供する、請求項13に記載の粉体コーティング。
【請求項17】
基材が感温性である、請求項13に記載の粉体コーティング。
【請求項18】
粉体コーティングがクラスAの仕上がりを提供する、請求項13に記載の粉体コーティング。
【請求項19】
樹脂のTgが約40℃から80℃までの間である、請求項13に記載の粉体コーティング。
【請求項20】
a.ジアンヒドロヘキシトールの靭性結晶質バイオベースモノマーを選択する工程と、
b.フロー性のある非晶質バイオベース樹脂と共反応させて、樹脂を形成する工程
とを含み、前記樹脂が
a.約40℃より高く、約80℃より低いTgと、
b.前記樹脂組成物中に少なくとも5重量%のバイオマスと、
c.125℃以下、30分間の低温ベーキング条件での硬化、柔軟性、および流展性と
を含む方法。
【請求項21】
樹脂が、ヒドロキシル、カルボキシル、および/または窒素含有基(アミン、イソシアナート、またはアミド基)で官能化されている、請求項20に記載の方法。
【請求項22】
樹脂が、第2の硬化機序で利用可能な過剰の不飽和を有する、請求項20に記載の方法。
【請求項23】
ポリエステル酸樹脂のカルボキシル基と硬化反応するβ−ヒドロキシルアミドの反応であるβ−ヒドロキシアミドエステル交換硬化を利用して、請求項2に記載の樹脂をコーティング組成物に配合する、請求項20に記載の方法。
【請求項24】
樹脂をエポキシ−アクリルなどのハイブリッド粉体コーティングになる材料と配合する請求項21に記載の方法。
【請求項25】
ハイブリッドコーティング組成物が、次の一般式を有する触媒:
【化1】
[式中、R1、R2、R3、およびR4は独立に、水素、またはエポキシ樹脂と反応しない任意の置換基である]
と反応する、請求項20に記載の方法。
【請求項26】
靭性結晶質バイオベースモノマー(イソソルビドなど)を、フロー性のある非晶質バイオベース樹脂と共反応させて、コーティング、接着剤、インク、シーラント、ポリマー用途、および他の市販の材料を製造するために使用することができる樹脂を形成する組成物。
【請求項27】
靭性結晶質バイオベースモノマー(イソソルビドなど)をフロー性のある非晶質バイオベース樹脂と共反応させて、新規クラスの樹脂を形成し、
前記樹脂が
a.約40℃〜約80℃のTgと、
b.前記樹脂組成物中に少なくとも約5重量%のバイオマスと、
c.硬化温度125℃で30分間
とを有する組成物。
【請求項28】
樹脂が、カルボキシル基、ヒドロキシル基、および/または窒素含有基(アミン、イソシアナート、および/またはアミド)で官能化されている、請求項27に記載の組成物。
【請求項29】
樹脂が過剰の不飽和を有する、請求項27に記載の組成物。
【請求項30】
樹脂がカルボキシル基で官能化されている、請求項27に記載の組成物。
【請求項31】
樹脂がヒドロキシル基で官能化されている、請求項27に記載の組成物。
【請求項32】
樹脂が窒素含有基(アミン、イソシアナート、アミドなど)で官能化されている、請求項27に記載の組成物。
【請求項33】
樹脂のカルボキシル基と硬化反応するβ−ヒドロキシルアミドの反応であるβ−ヒドロキシアミドエステル交換硬化を利用して、請求項30に記載の樹脂をコーティング組成物に配合する、請求項27に記載の組成物。
【請求項34】
樹脂が、≧40℃のTg、および≧20%のバイオベース含有量を有し、粉体コーティングに混合され、これらの粉体コーティングを、
a.β−ヒドロキシルアミド
b.グリシジルメタクリレートアクリルコポリマー樹脂を含むが、これに限定されないエポキシ含有樹脂
などの架橋性樹脂で硬化することができる、請求項27に記載の組成物。
【請求項35】
A.樹脂と、
B.架橋剤と、
C.任意選択の顔料
とを含み、前記樹脂が、
a.ジアンヒドロヘキシトールと、
b.ダイマー酸および/またはダイマージオールと、
c.二酸、ジエステル、または二酸塩化物と
の反応生成物を含む粉体コーティング。
【請求項36】
反応のための触媒を含む、請求項35に記載の粉体コーティング。
【請求項37】
顔料分散助剤を含む、請求項35に記載の粉末。
【請求項38】
樹脂がカルボキシル基を有する場合、カルボキシ官能性のカルボキシル基を自己触媒エステル交換反応でβ−ヒドロキシルアミドと反応させ、またはポリエポキシ官能性ポリマーと反応させる工程を含む、請求項35に記載の粉体コーティング。
【請求項39】
樹脂がヒドロキシル官能基を有する場合、前記ヒドロキシル官能性の前記ヒドロキシル基をエポキシ官能性アクリルと反応させる工程を含む、請求項35に記載の粉体コーティング。
【請求項40】
任意選択の賦形剤を含む、請求項35に記載の粉体コーティング。
【請求項41】
本明細書で開示されたすべての新規で非自明の組成物、方法、および使用。
【図1】


【図2】

【図3】
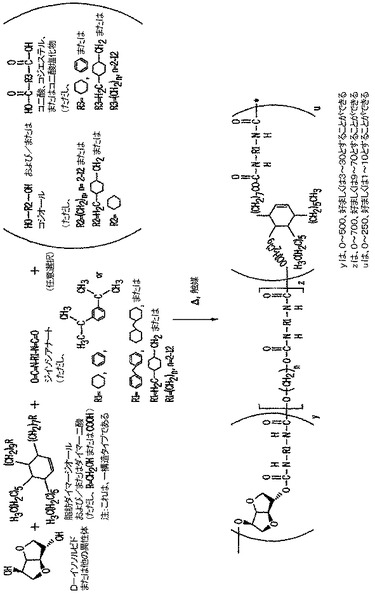
【図3A】

【図4】

【図5】
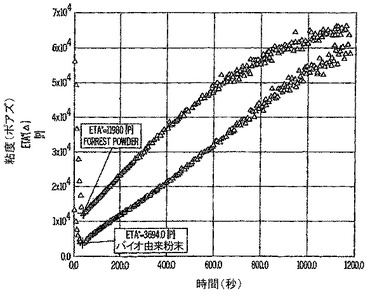
【図6】
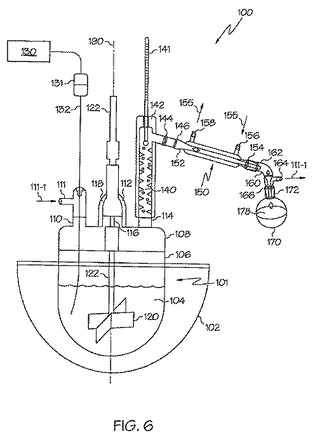
【図7】
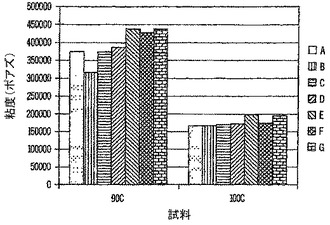
【図2】
【図3】
【図3A】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2008−533290(P2008−533290A)
【公表日】平成20年8月21日(2008.8.21)
【国際特許分類】
【出願番号】特願2008−502152(P2008−502152)
【出願日】平成18年3月20日(2006.3.20)
【国際出願番号】PCT/US2006/010135
【国際公開番号】WO2006/102279
【国際公開日】平成18年9月28日(2006.9.28)
【出願人】(504306714)バッテル メモリアル インスティテュート (26)
【氏名又は名称原語表記】BATTELLE MEMORIAL INSTITUTE
【住所又は居所原語表記】505 King Avenue, Columbus, OH 43201−2693 (US)
【Fターム(参考)】
【公表日】平成20年8月21日(2008.8.21)
【国際特許分類】
【出願日】平成18年3月20日(2006.3.20)
【国際出願番号】PCT/US2006/010135
【国際公開番号】WO2006/102279
【国際公開日】平成18年9月28日(2006.9.28)
【出願人】(504306714)バッテル メモリアル インスティテュート (26)
【氏名又は名称原語表記】BATTELLE MEMORIAL INSTITUTE
【住所又は居所原語表記】505 King Avenue, Columbus, OH 43201−2693 (US)
【Fターム(参考)】
[ Back to top ]