
樹脂成形体の製造方法、樹脂成形体、光学部品および光学デバイス
【課題】 樹脂成形体の内部での屈折率のバラつきの小さい樹脂成形体を製造する方法を提供すること。また、樹脂成形体の内部でのバラつきの小さい樹脂成形体およびそれを用いて性能に優れた光学部品、光学デバイスを提供すること。
【解決手段】 本発明の樹脂成形体の製造方法は、屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を所望の形状の形成し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体の製造方法であって、前記第1加熱工程において樹脂成形体の屈折率が第1屈折率(A1)になるまで加熱し、前記第2加熱工程において樹脂成形体の屈折率が第2屈折率(A2)になるまで加熱したときに、前記樹脂成形体の屈折率の差(A2−A1)が0.03以上となるような条件で加熱する。
【解決手段】 本発明の樹脂成形体の製造方法は、屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を所望の形状の形成し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体の製造方法であって、前記第1加熱工程において樹脂成形体の屈折率が第1屈折率(A1)になるまで加熱し、前記第2加熱工程において樹脂成形体の屈折率が第2屈折率(A2)になるまで加熱したときに、前記樹脂成形体の屈折率の差(A2−A1)が0.03以上となるような条件で加熱する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、樹脂成形体の製造方法、樹脂成形体、光学部品および光学デバイスに関する。
【背景技術】
【0002】
近年、軽量でかつ加工性に富み、各種の光学部材に好適な樹脂材料として、1.8〜2.2程度の高い屈折率を有する透明樹脂材料が要求されている。この要求に対して、ポリチオール化合物とポリイソシアネート化合物から得られるチオウレタン(例えば、特許文献1参照)、エポキシ樹脂またはエピスルフィド樹脂から得られる重合体(例えば、特許文献2参照)などが検討されている。しかし、これらの材料が有するような屈折率では、依然として不十分であった。
【0003】
また、透明性を維持したまま、高い屈折率を与えるために、樹脂中に酸化チタンや酸化亜鉛などの高屈折率金属酸化物微粒子を分散させる技術が提案されているが(例えば、特許文献3参照)、光散乱を引き起こさないようにこれらの微粒子を分散させるには、分散剤が必須であったが、これによっても透明材料中で屈折率にバラつきがあった。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特公平4−58489号公報
【特許文献2】特開平3−81320号公報
【特許文献3】特開2002−277609号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明の目的は、樹脂成形体の内部での屈折率のバラつきの小さい樹脂成形体を製造する方法を提供することにある。
また、本発明の別の目的は、樹脂成形体の内部でのバラつきの小さい樹脂成形体およびそれを用いて性能に優れた光学部品、光学デバイスを提供することにある。
【課題を解決するための手段】
【0006】
このような目的は、下記(1)〜(15)に記載の本発明により達成される。
(1)屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を所望の形状に形成し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体の製造方法であって、前記第1加熱工程において樹脂成形体の屈折率が第1屈折率(A1)になるまで加熱し、前記第2加熱工程において樹脂成形体の屈折率が第2屈折率(A2)になるまで加熱したときに、前記樹脂成形体の屈折率の差(A2−A1)が0.03以上となるような条件で加熱することを特徴とする樹脂成形体の製造方法。
(2)前記第1加熱工程は、一定温度で所定時間加熱することにより樹脂成形体の屈折率を、第1屈折率(A1)にするものである上記(1)に記載の樹脂成形体の製造方法。
(3)前記第1加熱工程で樹脂成形体の屈折率が、1.70以上となるまでに加熱するものである上記(1)または(2)に記載の樹脂成形体の製造方法。
(4)前記第2加熱工程は、前記第1加熱工程の加熱温度よりも高い温度で、所定時間加熱することにより樹脂成形体の屈折率を、第2屈折率(A2)にするものである上記(1)ないし(3)のいずれかに記載の樹脂成形体の製造方法。
(5)前記第2加熱工程は、前記分散剤が分解または揮発するような温度で加熱するものである上記(1)ないし(4)のいずれかに記載の樹脂成形体の製造方法。
(6)前記屈折率が1.6以上の樹脂は、加熱により脱水閉環反応または脱アルコール閉環反応する樹脂である上記(1)ないし(5)のいずれかに記載の樹脂成形体の製造方法。
(7)前記屈折率が1.6以上の樹脂は、ポリベンゾオキサゾール前駆体を含むものである上記(1)ないし(6)のいずれかに記載の樹脂成形体の製造方法。
(8)前記ポリベンゾオキサゾール前駆体は、ジカルボン酸化合物をビスアミノフェノール化合物よりも過剰に反応させてなるものである上記(1)ないし(7)のいずれかに記載の樹脂成形体の製造方法。
(9)前記樹脂成形体は、直径0.4〜1.0μm、深さ0.5〜2.5μmの凹部に埋め込まれるものである上記(1)ないし(8)のいずれかに記載の樹脂成形体の製造方法。
(10)屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を成膜し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体であって、前記第1加熱工程後における樹脂成形体の第1屈折率(A1)と、前記第2加熱工程後における樹脂成形体の第2屈折率(A2)との差(A2−A1)が、0.03以上であることを特徴とする樹脂成形体。
(11)前記第1屈折率が、1.70以上である上記(10)に記載の樹脂成形体。
(12)前記屈折率が1.6以上の樹脂は、加熱により脱水閉環反応または脱アルコール閉環反応する樹脂である上記(10)または(11)に記載の樹脂成形体。
(13)前記樹脂成形体は、直径0.4〜1.0μm、深さ0.5〜2.5μmの凹部を埋め込むために用いるものである上記(10)ないし(12)のいずれかに記載の樹脂成形体。
(14)上記(10)ないし(13)のいずれかに記載の樹脂成形体を用いてなることを特徴とする光学部品。
(15)上記(14)に記載の光学部品を有することを特徴とする光学デバイス。
【発明の効果】
【0007】
本発明によれば、樹脂成形体の内部での屈折率のバラつきの小さい樹脂成形体を製造する方法を提供することができる。
また、本発明の別の目的は、樹脂成形体の内部でのバラつきの小さい樹脂成形体およびそれを用いて性能に優れた光学部品、光学デバイスを得ることができる。
【図面の簡単な説明】
【0008】
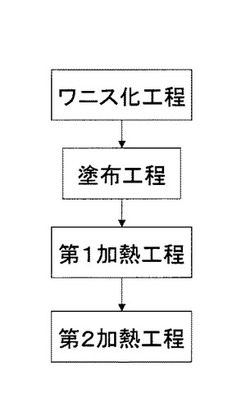
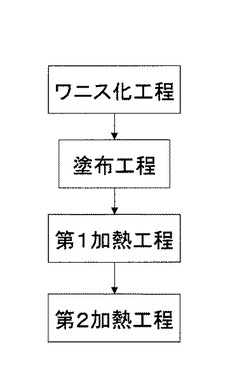
【図1】樹脂成形体の製造方法の主なプロセスを記載した工程フロー図である。
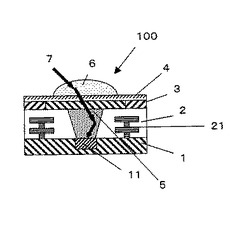
【図2】光学部品の一例を示す断面図である。
【発明を実施するための形態】
【0009】
以下、本発明の樹脂成形体の製造方法、樹脂成形体、光学部品および光学デバイスについて説明する。
本発明の樹脂成形体の製造方法は、屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を所望の形状の形成し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体の製造方法であって、前記第1加熱工程において樹脂成形体の屈折率が第1屈折率(A1)になるまで加熱し、前記第2加熱工程において樹脂成形体の屈折率が第2屈折率(A2)になるまで加熱したときに、前記樹脂成形体の屈折率の差(A2−A1)が0.03以上となるような条件で加熱することを特徴とする。
また、本発明の樹脂成形体は、屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を成膜し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体であって、前記第1加熱工程後における樹脂成形体の第1屈折率(A1)と、前記第2加熱工程後における樹脂成形体の第2屈折率(A2)との差(A2−A1)が、0.03以上であることを特徴とする。
また、本発明の光学部品は、上記に記載の樹脂成形体を用いてなることを特徴とする。
また、本発明の光学デバイスは、上記に記載の光学部品を有することを特徴とする。
【0010】
まず、樹脂成形体の製造方法および樹脂成形体について、所望の形状として膜(フィルム)状の成形体を形成する場合について説明する。
図1に示すように、本発明の樹脂成形体の製造方法は、ワニス化工程、塗布工程、第1加熱工程および第2加熱工程を有している。
【0011】
<ワニス化工程>
ワニス化工程では、屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤とを、溶媒に溶解して樹脂組成物をワニスの状態とする。これにより、前記塗布工程を容易にする。
前記樹脂組成物は、屈折率が1.6以上の樹脂を含む。前記屈折率が1.6以上の樹脂としては、例えばポリアミド系樹脂、ポリエステル系樹脂、エポキシ系樹脂、ポリウレタン系樹脂、ポリスルフィド系樹脂、加熱により脱水閉環反応または脱アルコール閉環反応する樹脂等が挙げられる。
これらの中でも加熱により脱水閉環反応または脱アルコール閉環反応する樹脂が好ましい。これにより、耐熱性をより向上することができる。
【0012】
前記加熱により脱水閉環反応または脱アルコール閉環反応する樹脂としては、具体的にはポリベンゾオキサゾール前駆体、ポリイミド前駆体、ポリベンゾイミダゾール前駆体およびポリベンゾチアゾール前駆体等が挙げられ、これらのうちの1種または2種以上を組み合わせて用いることができる。これらの中でもポリベンゾオキサゾール前駆体およびポリイミド前駆体のうちの少なくとも1種を用いるのが好ましい。これにより、脱水閉環による硬化物の可視光域での透過性および耐熱性を向上することができる。
【0013】
前記ポリベンゾオキサゾール前駆体としては、特に限定されないが、例えば、下記一般式(A)で表わされるヒドロキシアミド重合体が用いられる。
【0014】
このヒドロキシアミド重合体は、加熱により脱水する閉環反応(縮合反応)により、ポリベンゾオキサゾール(硬化物)となるものである。
【0015】
【化1】

【0016】
【化2】
【0017】
【化3】
【0018】
【化4】
【0019】
【化5】
【0020】
【化6】
【0021】
【化7】
【0022】
また、式(B)、式(C)、式(D)、式(E)、式(F)および式(G)で表される基における環構造上の水素原子は、炭素数1〜4のアルキル基の中から選ばれる少なくとも1個の基で置換されていてもよい。
【0023】
このような一般式(A)で表されるヒドロキシアミド重合体は、前記式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物の少なくとも1種と、前記式(C)および前記式(D)で表される基の中から選ばれる基を有するジカルボン酸化合物の少なくとも1種とを用いて反応させて合成することができる。
【0024】
また、ジカルボン酸化合物として、前記式(C)および前記式(D)で表される基の中から選ばれる基を有するジカルボン酸化合物の少なくとも1種と、式(E)および式(F)で表される基の中から選ばれる基を有するジカルボン酸化合物とを用いて反応させることもできる。
【0025】
上記ヒドロキシアミド重合体の合成において、ビスアミノフェノール化合物と、ジカルボン酸化合物とを反応させる方法としては、従来の酸クロライド法、活性化エステル法、ポリリン酸やジシクロヘキシルカルボジイミド等の脱水縮合剤の存在下での縮合反応等の方法を挙げることができる。
【0026】
なお、前記ビスアミノフェノール化合物における基とは、アミノ基およびフェノール性水酸基と結合し得る結合手を四つ有する基を意味する。また、前記ジカルボン酸化合物における基とは、カルボキシル基と結合し得る結合手を2つ有する基を意味する。
【0027】
なお、式(B)で表される基を有するビスアミノフェノール化合物の具体例としては、2,5−ジアミノヒドロキノン、2,3−ジアミノヒドロキノン、3,6−ジアミノカテコール、4,6−ジアミノレゾルシノール、ビス(3−アミノ−4−ヒドロキシフェニル)メタン、ビス(4−アミノ−3−ヒドロキシフェニル)メタン、2,2−ビス(3−アミノ−4−ヒドロキシフェニル)プロパン、2,2−ビス(4−アミノ−3−ヒドロキシフェニル)プロパン、2,2−(2,3’−アミノ−3,4’−ヒドロキシフェニル)プロパン、2,2−(3,4’−アミノ−2,3’−ヒドロキシフェニル)プロパン、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルスルホン、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルスルホン、2,3’−ジアミノ−3,4’−ジヒドロキシジフェニルスルホン、3,4’−ジアミノ−2,3’−ジヒドロキシジフェニルスルホン、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルエーテル、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルエーテル、2,3’−ジアミノ−3,4’−ジヒドロキシジフェニルエーテル、3,4’−ジアミノ−2,3’−ジヒドロキシジフェニルエーテル、1,3−ビス(3−アミノ−4ヒドロキシフェノキシ)ベンゼン、1,3−ビス(4−アミノ−3ヒドロキシフェノキシ)ベンゼン、1,4−ビス(3−アミノ−4ヒドロキシフェノキシ)ベンゼン、1,4−ビス(4−アミノ−3ヒドロキシフェノキシ)ベンゼン、1,3−ビス(3−アミノ−4ヒドロキシフェニルスルファニル)ベンゼン、1,3−ビス(4−アミノ−3ヒドロキシフェニルスルファニル)ベンゼン、1,4−ビス(3−アミノ−4ヒドロキシフェニルスルファニル)ベンゼン、1,4−ビス(4−アミノ−3ヒドロキシフェニルスルファニル)ベンゼン、3,3’−ジアミノ−4,4’−ジヒドロキシビフェニル、4,4’−ジアミノ−3,3’−ジヒドロキシビフェニル、3,4’−ジアミノ−2,3’−ジヒドロキシビフェニル、2,3’−ジアミノ−3,4’−ジヒドロキシビフェニル、3,3’−ジアミノ−2,2’−ジヒドロキシビナフチル、2,2’−ジアミノ−3,3’−ジヒドロキシビナフチル、2,3’−ジアミノ−3,2’−ジヒドロキシビナフチル、9,9−ビス(4−アミノ−3−ヒドロキシフェニル)フルオレン、9,9−ビス(3−アミノ−4−ヒドロキシフェニル)フルオレン、9,9−ビス(4−(3−アミノ−4−ヒドロキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−アミノ−3−ヒドロキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−アミノ−3−ヒドロキシフェノキシ)フェニル)フルオレンおよび9,9−ビス(4−(3−アミノ−4−ヒドロキシフェノキシ)フェニル)フルオレン等が挙げられる。
【0028】
これらの中でも、溶媒に対する溶解性と、耐熱性の上で、2,5−ジアミノヒドロキノン、4,6−ジアミノレゾルシノール、ビス(3−アミノ−4−ヒドロキシフェニル)メタン、ビス(4−アミノ−3−ヒドロキシフェニル)メタン、2,2−ビス(3−アミノ−4−ヒドロキシフェニル)プロパン、2,2−ビス(4−アミノ−3−ヒドロキシフェニル)プロパン、2,2−(2,3’−アミノ−3,4’−ヒドロキシフェニル)プロパン、2,2−(3,4’−アミノ−2,3’−ヒドロキシフェニル)プロパン、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルスルホン、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルスルホン、2,3’−ジアミノ−3,4’−ジヒドロキシジフェニルスルホン、3,4’−ジアミノ−2,3’−ジヒドロキシジフェニルスルホン、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルエーテル、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルエーテル、2,3’−ジアミノ−3,4’−ジヒドロキシジフェニルエーテル、3,4’−ジアミノ−2,3’−ジヒドロキシジフェニルエーテル、1,3−ビス(3−アミノ−4ヒドロキシフェノキシ)ベンゼン、1,3−ビス(4−アミノ−3ヒドロキシフェノキシ)ベンゼン、1,4−ビス(3−アミノ−4ヒドロキシフェノキシ)ベンゼン、1,4−ビス(4−アミノ−3ヒドロキシフェノキシ)ベンゼン、1,3−ビス(3−アミノ−4ヒドロキシフェニルスルファニル)ベンゼン、1,3−ビス(4−アミノ−3ヒドロキシフェニルスルファニル)ベンゼン、1,4−ビス(3−アミノ−4ヒドロキシフェニルスルファニル)ベンゼン、1,4−ビス(4−アミノ−3ヒドロキシフェニルスルファニル)ベンゼン、9,9−ビス(4−アミノ−3−ヒドロキシフェニル)フルオレン、9,9−ビス(3−アミノ−4−ヒドロキシフェニル)フルオレン、9,9−ビス(4−(4−アミノ−3−ヒドロキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−アミノ−3−ヒドロキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−アミノ−3−ヒドロキシフェノキシ)フェニル)フルオレンおよび9,9−ビス(4−(3−アミノ−4−ヒドロキシフェノキシ)フェニル)フルオレンが好ましく、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルスルホン、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルスルホン、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルエーテル、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルエーテル、1,3−ビス(3−アミノ−4ヒドロキシフェノキシ)ベンゼン、1,3−ビス(4−アミノ−3ヒドロキシフェノキシ)ベンゼン、9,9−ビス(4−アミノ−3−ヒドロキシフェニル)フルオレンおよび9,9−ビス(3−アミノ−4−ヒドロキシフェニル)フルオレン、がより好ましい。これらのビスアミノフェノール化合物は単独で用いてもよく、また2種類以上を組み合わせて用いることができる。
【0029】
また、式(C)で表される基を有するジカルボン酸化合物の具体例としては、特に限定されないが、イソフタル酸、テレフタル酸、4,4’−ビフェニルジカルボン酸、3,3’−ビフェニルジカルボン酸、1,4−ナフタレンジカルボン酸、2,3−ナフタレンジカルボン酸、2,6−ナフタレンジカルボン酸、4,4’−スルホニルビス安息香酸、3,3’−スルホニルビス安息香酸、4,4’−チオビス安息香酸、3,3’−チオビス安息香酸、4,4’−オキシビス安息香酸、3,3’−オキシビス安息香酸、ビス(4−カルボキシフェニル)メタン、ビス(3−カルボキシフェニル)メタン、2,2−ビス(4−カルボキシフェニル)プロパン、2,2−ビス(3−カルボキシフェニル)プロパン、2,2’−ジメチル−4,4’−ビフェニルジカルボン酸、3,3’−ジメチル−4,4’−ビフェニルジカルボン酸、2,2’−ジメチル−3,3’−ビフェニルジカルボン酸、1,3−ビス(4−カルボキシフェノキシ)ベンゼン、1,3−ビス(3−カルボキシフェノキシ)ベンゼン、1,4−ビス(4−カルボキシフェノキシ)ベンゼン、1,4−ビス(3−カルボキシフェノキシ)ベンゼン、1,3−ビス(4−カルボキシフェニルスルファニル)ベンゼン、1,3−ビス(3−カルボキシフェニルスルファニル)ベンゼン、1,4−ビス(4−カルボキシフェニルスルファニル)ベンゼン、1,4−ビス(3−カルボキシフェニルスルファニル)ベンゼン、4,4’−ビス(4−カルボキシフェノキシ)ビフェニル、4,4’−ビス(3−カルボキシフェノキシ)ビフェニル、3,4’−ビス(4−カルボキシフェノキシ)ビフェニル、3,4’−ビス(3−カルボキシフェノキシ)ビフェニル、3,3’−ビス(4−カルボキシフェノキシ)ビフェニル、3,3’−ビス(3−カルボキシフェノキシ)ビフェニル、9,9−ビス(3−カルボキシフェニル)フルオレン、9,9−ビス(4−カルボキシフェニル)フルオレン、9,9−ビス(4−(3−カルボキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−カルボキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−カルボキシフェノキシフェニル)フルオレンおよび9,9−ビス(3−カルボキシフェノキシフェニル)フルオレン等が挙げられる。
【0030】
これらの中でも、溶媒に対する溶解性の上で、イソフタル酸、テレフタル酸、4,4’−スルホニルビス安息香酸、3,3’−スルホニルビス安息香酸、4,4’−チオビス安息香酸、3,3’−チオビス安息香酸、4,4’−オキシビス安息香酸、3,3’−オキシビス安息香酸、ビス(4−カルボキシフェニル)メタン、ビス(3−カルボキシフェニル)メタン、2,2−ビス(4−カルボキシフェニル)プロパン、2,2−ビス(3−カルボキシフェニル)プロパン、1,3−ビス(4−カルボキシフェノキシ)ベンゼン、1,3−ビス(3−カルボキシフェノキシ)ベンゼン、1,4−ビス(4−カルボキシフェノキシ)ベンゼン、1,4−ビス(3−カルボキシフェノキシ)ベンゼン、1,3−ビス(4−カルボキシフェニルスルファニル)ベンゼン、1,3−ビス(3−カルボキシフェニルスルファニル)ベンゼン、1,4−ビス(4−カルボキシフェニルスルファニル)ベンゼン、1,4−ビス(3−カルボキシフェニルスルファニル)ベンゼン、9,9−ビス(3−カルボキシフェニル)フルオレン、9,9−ビス(4−カルボキシフェニル)フルオレン、9,9−ビス(4−(3−カルボキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−カルボキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−カルボキシフェノキシフェニル)フルオレンおよび9,9−ビス(3−カルボキシフェノキシフェニル)フルオレンが好ましく、イソフタル酸、4,4’−スルホニルビス安息香酸、3,3’−スルホニルビス安息香酸、4,4’−チオビス安息香酸、3,3’−チオビス安息香酸、4,4’−オキシビス安息香酸、3,3’−オキシビス安息香酸、1,3−ビス(4−カルボキシフェノキシ)ベンゼン、1,3−ビス(3−カルボキシフェノキシ)ベンゼン、1,4−ビス(4−カルボキシフェノキシ)ベンゼン、1,4−ビス(3−カルボキシフェノキシ)ベンゼン、9,9−ビス(3−カルボキシフェニル)フルオレンおよび9,9−ビス(4−カルボキシフェニル)フルオレンがより好ましい。これらのジカルボン酸化合物は単独で用いてもよく、また2種類以上を組み合わせて用いることができる。
【0031】
式(D)で表される基を有するジカルボン酸化合物の具体例としては、特に限定されないが、1,3−シクロヘキサンジカルボン酸、cis−1,3−シクロヘキサンジカルボン酸、1,4−シクロヘキサンジカルボン酸、cis−1,4−シクロヘキサンジカルボン酸、trans−1,4−シクロヘキサンジカルボン酸、1,3−デカリンジカルボン酸、1,4−デカリンジカルボン酸、2,6−デカリンジカルボン酸、1,3−アダマンタンジカルボン酸、5−メチル−1,3−アダマンタンジカルボン酸、2,2−アダマンタンジカルボン酸、3,3’−(2,2−アダマンチル)フェニルジカルボン酸、4,4’−(2,2−アダマンチル)フェニルジカルボン酸、3,3’−(2,2−アダマンチルオキシ)フェニルジカルボン酸、4,4’−(2,2−アダマンチルオキシ)フェニルジカルボン酸、3,3’−(1,3−アダマンチル)フェニルジカルボン酸、4,4’−(1,3−アダマンチル)フェニルジカルボン酸、3,3’−(1,1’−ビアダマンタン)ジカルボン酸、3,5−(1,1’−ビアダマンタン)ジカルボン酸、3,3’−(1,1’−ビアダマンタン)ジカルボン酸、3,5’−(1,1’−ビアダマンタン)ジカルボン酸、3’,5’,7,7’−テトラメチル−1,1’−ビアダマンタン−3,5−ジカルボン酸、5,5’,7,7’−テトラメチル−1,1’−ビアダマンタン−3,3’ジカルボン酸および3’,5,7,7’−テトラメチル−1,1’−ビアダマンタン−3,5’−ジカルボン酸等が挙げられる。
【0032】
これらの中でも、溶媒に対する溶解性と、耐熱性の上で、1,4−シクロヘキサンジカルボン酸、cis−1,4−シクロヘキサンジカルボン酸、trans−1,4−シクロヘキサンジカルボン酸、1,3−アダマンタンジカルボン酸、5−メチル−1,3−アダマンタンジカルボン酸、3,3’−(1,1’−ビアダマンタン)ジカルボン酸、3,5−(1,1’−ビアダマンタン)ジカルボン酸、3,3’−(1,1’−ビアダマンタン)ジカルボン酸、3,5’−(1,1’−ビアダマンタン)ジカルボン酸、3’,5’,7,7’−テトラメチル−1,1’−ビアダマンタン−3,5−ジカルボン酸、5,5’,7,7’−テトラメチル−1,1’−ビアダマンタン−3,3’ジカルボン酸および3’,5,7,7’−テトラメチル−1,1’−ビアダマンタン−3,5’−ジカルボン酸が好ましい。これらのジカルボン酸化合物は単独で用いてもよく、また2種類以上を組み合わせて用いることができる。
【0033】
式(E)で表される基を有するジカルボン酸化合物において、R置換基は、アリール基または炭素数1〜10のアルキル基である。これらのうち、前記アリール基としては、具体的には、フェニル基、ベンジル基、トリル基、o−キシリル基、m−キシリル基およびp−キシリル基等が挙げられる。また、前記アルキル基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、t−ブチル基、シクロヘキシル基およびアダマンチル基等が挙げられる。これらのなかでも、フェニル基、トリル基、o−キシリル基、m−キシリル基、p−キシリル基、メチル基、イソブチル基およびt−ブチル基が好ましく、フェニル基、トリル基、o−キシリル基、m−キシリル基およびp−キシリル基がより好ましい。
【0034】
このような式(E)で表される基を有するジカルボン酸化合物のうち、R置換基がフェニル基であるフェニルエチニル基を有するジカルボン酸化合物の具体例としては、特に限定されないが、4−フェニルエチニルイソフタル酸、5−フェニルエチニルイソフタル酸、2−フェニルエチニルテレフタル酸、3−フェニルエチニルテレフタル酸、2−フェニルエチニル−1,5−ナフタレンジカルボン酸、3−フェニルエチニル−1,5−ナフタレンジカルボン酸、4−フェニルエチニル−1,5−ナフタレンジカルボン酸、1−フェニルエチニル−2,6−ナフタレンジカルボン酸、3−フェニルエチニル−2,6−ナフタレンジカルボン酸、4−フェニルエチニル−2,6−ナフタレンジカルボン酸、3,7−ビスフェニルエチニル−1,5−ナフタレンジカルボン酸、4,8−ビスフェニルエチニル−2,6−ナフタレンジカルボン酸、4−フェニルエチニル−2,2'−ビフェニルジカルボン酸、6−フェニルエチニル−2,2'−ビフェニルジカルボン酸、2−フェニルエチニル−4,4'−ビフェニルジカルボン酸、4,4'−ビスフェニルエチニル−2,2'−ビフェニルジカルボン酸、6,6'−ビスフェニルエチニル−2,2'−ビフェニルジカルボン酸、2,2'−ビスフェニルエチニル−4,4'−ビフェニルジカルボン酸、2,2−ビス(3−カルボキシ−4−フェニルエチニルフェニル)プロパン、2,2−ビス(3−カルボキシ−5−フェニルエチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−2−フェニルエチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−3−フェニルエチニルフェニル)プロパン、ビス(3−カルボキシ−4−フェニルエチニルフェニル)スルホン、ビス(3−カルボキシ−5−フェニルエチニルフェニル)スルホン、ビス(4−カルボキシ−2−フェニルエチニルフェニル)スルホン、ビス(4−カルボキシ−3−フェニルエチニルフェニル)スルホン、ビス(3−カルボキシ−4−フェニルエチニルフェニル)エーテル、ビス(3−カルボキシ−5−フェニルエチニルフェニル)エーテル、ビス(4−カルボキシ−2−フェニルエチニルフェニル)エーテル、ビス(4−カルボキシ−3−フェニルエチニルフェニル)エーテル等が挙げられる。
【0035】
これらの中でも、溶媒に対する溶解性の上で、4−フェニルエチニルイソフタル酸、5−フェニルエチニルイソフタル酸、2−フェニルエチニルテレフタル酸、3−フェニルエチニルテレフタル酸、2,2−ビス(3−カルボキシ−4−フェニルエチニルフェニル)プロパン、2,2−ビス(3−カルボキシ−5−フェニルエチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−2−フェニルエチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−3−フェニルエチニルフェニル)プロパン、ビス(3−カルボキシ−4−フェニルエチニルフェニル)スルホン、ビス(3−カルボキシ−5−フェニルエチニルフェニル)スルホン、ビス(4−カルボキシ−2−フェニルエチニルフェニル)スルホン、ビス(4−カルボキシ−3−フェニルエチニルフェニル)スルホン、ビス(3−カルボキシ−4−フェニルエチニルフェニル)エーテル、ビス(3−カルボキシ−5−フェニルエチニルフェニル)エーテル、ビス(4−カルボキシ−2−フェニルエチニルフェニル)エーテル、ビス(4−カルボキシ−3−フェニルエチニルフェニル)エーテルが好ましく、4−フェニルエチニルイソフタル酸、5−フェニルエチニルイソフタル酸、2−フェニルエチニルテレフタル酸、3−フェニルエチニルテレフタル酸がより好ましい。これらのジカルボン酸化合物は単独で用いてもよく、また2種類以上を組み合わせて用いることができる。
【0036】
式(F)で表される基を有するジカルボン酸化合物の具体例としては、特に限定されないが、4−エチニルイソフタル酸、5−エチニルイソフタル酸、2−エチニルテレフタル酸、3−エチニルテレフタル酸、2−エチニル−1,5−ナフタレンジカルボン酸、3−エチニル−1,5−ナフタレンジカルボン酸、4−エチニル−1,5−ナフタレンジカルボン酸、1−エチニル−2,6−ナフタレンジカルボン酸、3−エチニル−2,6−ナフタレンジカルボン酸、4−エチニル−2,6−ナフタレンジカルボン酸、3,7−ジエチニル−1,5−ナフタレンジカルボン酸、4,8−ジエチニル−2,6−ナフタレンジカルボン酸、4−エチニル−2,2'−ビフェニルジカルボン酸、6−エチニル−2,2'−ビフェニルジカルボン酸、2−エチニル−4,4'−ビフェニルジカルボン酸、4,4'−ジエチニル−2,2'−ビフェニルジカルボン酸、6,6'−ジエチニル−2,2'−ビフェニルジカルボン酸、2,2'−ジエチニル−4,4'−ビフェニルジカルボン酸、2,2−ビス(3−カルボキシ−4−エチニルフェニル)プロパン、2,2−ビス(3−カルボキシ−5−エチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−2−エチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−3−エチニルフェニル)プロパン、ビス(3−カルボキシ−4−エチニルフェニル)スルホン、ビス(3−カルボキシ−5−エチニルフェニル)スルホン、ビス(4−カルボキシ−2−エチニルフェニル)スルホン、ビス(4−カルボキシ−3−エチニルフェニル)スルホン、ビス(3−カルボキシ−4−エチニルフェニル)エーテル、ビス(3−カルボキシ−5−エチニルフェニル)エーテル、ビス(4−カルボキシ−2−エチニルフェニル)エーテル、ビス(4−カルボキシ−3−エチニルフェニル)エーテル等が挙げられる。
【0037】
これらの中でも、溶媒に対する溶解性の上で、4−エチニルイソフタル酸、5−エチニルイソフタル酸、2−エチニルテレフタル酸、3−エチニルテレフタル酸、2,2−ビス(3−カルボキシ−4−エチニルフェニル)プロパン、2,2−ビス(3−カルボキシ−5−エチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−2−エチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−3−エチニルフェニル)プロパン、ビス(3−カルボキシ−4−エチニルフェニル)スルホン、ビス(3−カルボキシ−5−エチニルフェニル)スルホン、ビス(4−カルボキシ−2−エチニルフェニル)スルホン、ビス(4−カルボキシ−3−エチニルフェニル)スルホン、ビス(3−カルボキシ−4−エチニルフェニル)エーテル、ビス(3−カルボキシ−5−エチニルフェニル)エーテル、ビス(4−カルボキシ−2−エチニルフェニル)エーテル、ビス(4−カルボキシ−3−エチニルフェニル)エーテルが好ましく、4−エチニルイソフタル酸、5−エチニルイソフタル酸、2−エチニルテレフタル酸、3−エチニルテレフタル酸がより好ましい。これらのジカルボン酸化合物は単独で用いてもよく、また2種類以上を組み合わせて用いることができる。
【0038】
なお、式(B)、式(C)、式(D)、式(E)、式(F)および式(G)で表される基における環構造上の水素原子は、炭素数1〜4のアルキル基の中から選ばれる少なくとも1個の基で置換されていてもよい。上記炭素数1〜4のアルキル基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基およびt−ブチル基等が挙げられる。
【0039】
なお、下記一般式(A)で表わされるヒドロキシアミド重合体の製造方法としては、例えば、酸クロライド法では、使用する酸クロライドは、まず、N,N’−ジメチルホルムアミド等の触媒存在下で、前記ジカルボン酸化合物と、過剰量の塩化チオニルとを、室温ないし100℃程度の温度で反応させ、過剰の塩化チオニルを加熱および減圧により留去した後、残渣をヘキサン等の溶媒で再結晶することにより得ることができる。このようにして製造したジカルボン酸クロライド化合物を、前記ビスアミノフェノール化合物と共に、通常、N−メチル−2−ピロリドンおよびN,N’−ジメチルアセトアミド等の極性溶媒に溶解し、ピリジン等の酸受容剤存在下で、室温ないし−30℃程度の温度で反応させることにより、ヒドロキシアミド重合体を得ることができる。このようにして得られるヒドロキシアミド重合体が共重合体である場合の繰り返し単位の配列は、ブロック的であっても、ランダム的であっても良い。
【0040】
例えば、ブロック的な繰り返し単位の前記ヒドロキシアミド重合体の製造方法としては、酸クロライド法による場合、式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物と、式(C)および式(D)で表される基の中から選ばれる基を有するジカルボン酸クロライド化合物の少なくとも1種とを、予め反応させて分子量を上げた後、更に式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物と、式(E)および式(F)で表される基の中から選ばれる基を有するジカルボン酸クロライドの少なくとも1種とを反応させることにより得ることができる。
【0041】
また、逆に、式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物と、式(E)および式(F)で表される基の中から選ばれる基を有するジカルボン酸クロライド化合物の少なくとも1種とを、予め反応させて、重合体の分子量を上げた後、更に式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物と式(C)または式(D)で表される基の中から選ばれる基を有するジカルボン酸クロライド化合物の少なくとも1種とを反応させてもよい。
【0042】
ランダム的な繰り返し単位の前記ヒドロキシアミド重合体を製造する場合は、式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物と式(C)または式(D)で表される基の中から選ばれる基を有するジカルボン酸クロライド化合物の少なくとも1種と、式(E)および式(F)で表される基の中から選ばれる基を有するジカルボン酸クロライド化合物の少なくとも1種とを、同時に反応させることにより得ることができる。
【0043】
また、前記一般式(A)で表されるヒドロキシアミド重合体の構造中のlおよびmは、lが4以上の整数、mが0以上の整数、かつl+mが4以上の整数であり、上記ヒドロキシアミド重合体に用いるビスアミノフェノール化合物およびジカルボン酸化合物の組み合わせにより異なるが、一般的には、l+mが4〜100の範囲である。
【0044】
このようにして得られる一般式(A)で表されるヒドロキシアミド重合体は、溶媒に対する溶解性、および耐熱性の上で、下記一般式(H)で表される構造を有するヒドロキシアミド重合体であることが好ましく、下記一般式(N)で表される構造を有するヒドロキシアミド重合体であることがより好ましい。
【0045】
【化8】
【0046】
【化9】
【0047】
【化10】
【0048】
【化11】
【0049】
【化12】
【0050】
【化13】
【0051】
【化14】
【0052】
【化15】
【0053】
【化16】
【0054】
【化17】
【0055】
【化18】
【0056】
また、上記一般式(A)で表されるヒドロキシアミド重合体は、標準ポリスチレン換算の重量平均分子量が、一般的には3,000以上、100,000以下であるが、5,000以上、30,000以下が好ましく、より好ましくは6,000以上、15,000以下である。重量平均分子量は上記範囲外でも使用できるが、下限値より小さいと成膜後のプロセス中に耐熱性が劣化するおそれがあり、上限値より大きいと、溶媒への溶解性が低下するおそれがあり、また、高屈折率樹脂組成物における溶液粘度が上昇することにより、塗膜形成時に基板への濡れ性が低下して、膜厚均一性の低下が起こるおそれがある。
【0057】
上記重量平均分子量は、高速液体クロマトグラフを用い、標準ポリスチレンで検量線を作成し、ポリスチレン換算で求めたものをいう。例えば、装置として、東ソー株式会社製高速液体クロマトグラフSC−8020システムに、TSKgelGMH−HRH高速SEC用カラム、UV(λ=270nm)検出器を用い、移動相としてLiBr0.5%を添加したN−メチル−2−ピロリドン液を用いて40℃で測定し、標準ポリスチレンとして、東ソー製PS−オリゴマーキットにより、リテンションタイムと分子量の検量線を作製し、ヒドロキシアミド重合体のポリスチレン換算の重量平均分子量を求めることができる。
【0058】
また、ヒドロキシアミド重合体の標準ポリスチレン換算の重量平均分子量を前記の範囲にするための反応方法としては、例えば、上記製造方法において、ジカルボン酸クロライド化合物とビスアミノフェノール化合物の反応モル比を調整して分子量を制御する方法、ジカルボン酸クロライド化合物とビスアミノフェノール化合物に、カルボン酸クロライド化合物またはアミノフェノール化合物を用いて、反応を停止させ、任意の分子量に調整する方法等を例示することができる。上記カルボン酸クロライド化合物およびアミノフェノール化合物は、ヒドロキシアミド重合体の末端基として反応を終結させ、それ以上分子量が大きくならないようにするために用い、例えば、安息香酸クロライド、2−アミノフェノール等を例示することができる。
【0059】
前記ジカルボン酸化合物とビスアミノフェノール化合物の反応モル比を調整することにより分子量を制御する方法においては、上記の標準ポリスチレン換算の重量平均分子量とも関係し、上記で用いるジカルボン酸化合物およびビスアミノフェノール化合物の構造によりモル比が異なるが、一方の化合物のモル数を過剰に、例えば、反応モル比が0.7以上、0.9以下に調整されることが好ましく、0.75以上、0.90以下がより好ましく、さらに好ましくは、0.8以上、0.90以下とするのが良い。過剰にする一方の化合物は、ビスアミノフェノール化合物でもジカルボン酸化合物のどちらでも構わないが、硬化物であるポリベンゾオキサゾールの透明性の観点から、ジカルボン酸化合物が過剰であることが望ましい。反応モル比は、前記範囲外でも使用できるが、前記下限値より低いと比較的低分子の成分が多く生じるため、その除去等により、工程の複雑化する恐れがある。また、前記上限値を超える場合、標準ポリスチレン換算の重量平均分子量が目的とする範囲よりも大きくなる恐れがある。
【0060】
また、前記ポリイミド前駆体としては、特に限定されないが、例えば、下記一般式(S)で表わされるアミド酸重合体が用いられる。
【0061】
【化19】
【0062】
このアミド酸重合体は、加熱により脱水する閉環反応(縮合反応)により、ポリイミド(硬化物)となるものである。
【0063】
このような一般式(S)で表わされるアミド酸重合体は、前記式(C)および前記式(D)で表わされる基の中から選ばれる基を有するジアミン化合物の少なくとも1種と、ジフェニルエーテルテトラカルボン酸二無水物とを用いて反応させて合成することができる。
【0064】
なお、前記ジアミン化合物における基とは、アミノ基と結合し得る結合手を2つ有する基を意味する。
【0065】
また、前記式(C)または式(D)で表わされる基を有するジアミン化合物の具体例としては、例えば、3,3'−ジアミノジフェニルエーテル、3,4'−ジアミノジフェニルエーテル、1,3−ビス(3−アミノフェノキシ)ベンゼン、1,3−ビス(4−アミノフェノキシ)ベンゼン、2,6−ビス(3−アミノフェノキシ)ベンゾニトリル、1,3−ビス(3−アミノフェノキシ)−5−クロロベンゼン、4,4'−ビス(3−アミノフェノキシ)ジフェニルスルフィド、4,4'−ビス(4−アミノフェノキシ)ジフェニルスルフィド、4,4'−ビス(3−アミノフェノキシ)ビフェニル、4,4'−ビス(4−アミノフェノキシ)ビフェニル等が挙げられる。
【0066】
なお、一般式(S)で表わされるアミド酸重合体において、前記式(C)または式(D)で表わされる基における環構造上の水素原子は、一般式(A)で表わされるヒドロキシアミド重合体と同様に、炭素数1〜4のアルキル基の中から選ばれる少なくとも1個の基で置換されていてもよい。
【0067】
また、このような一般式(S)で表わされるアミド酸重合体を生成するための、ジアミン化合物とテトラカルボン酸二無水物との反応は、一般的に有機溶媒中において実施される。
【0068】
かかる反応に用いられる有機溶媒として、例えば、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド、N−メチル−2−ピロリドン、1,3−ジメチル−2−イミダゾリジノン、N−メチルカプロラクタム、1,2−ジメトキシエタン、ビス(2−メトキシエチル)エーテル、1,2−ビス(2−メトキシエトキシ)エタン、ビス{2−(2−メトキシエトキシ)エチル}エーテル、テトラヒドロフラン、1,3−ジオキサン、1,4−ジオキサン、ピリジン、ピコリン、ジメチルスルホキシド、ジメチルスルホン、テトラメチル尿素、ヘキサメチルホスホルアミド、m−クレゾール、p−クレゾール、クレゾールサン、p−クロロフェノール、o−クロロフェノール、フェノール、アニソール等が挙げられ、単独またはこれらの混合溶媒が用いられる。
【0069】
また、これらジアミン化合物とテトラカルボン酸二無水物との反応温度は、通常、200℃以下、好ましくは30〜50℃程度に設定される。さらに、反応時間は、通常、4〜24時間程度、好ましくは10〜15時間程度に設定される。
【0070】
なお、加熱により脱水または脱アルコール反応する樹脂としては、ポリベンゾオキサゾール前駆体を用いるのが好ましい。ポリベンゾオキサゾール前駆体の硬化物であるポリベンゾオキサゾールには、カルボニル基のような極性基が存在しない。そのため、高屈折率樹脂組成物の硬化物の吸水率および誘電率を効果的に低減させることができる。
【0071】
上述したような樹脂の屈折率は、1.6以上であるが、より好ましくは1.65以上であることが好ましく、最も1.7以上であることが好ましい。ベース樹脂の屈折率が高いと、最終的に得られる樹脂成形体の屈折率もより高くすることができる。
【0072】
前記屈折率が1.6以上の樹脂の含有量は、特に限定されないが、前記樹脂組成物全体の1〜25重量%が好ましく、特に5〜20重量%が好ましい。含有量が前記範囲内であると、高屈折率と埋め込み性との両方に優れる。
【0073】
前記樹脂組成物は、前記樹脂よりも屈折率が高い高屈折率粒子を含む。これにより、最終的に得られる樹脂成形体をより高屈折率にすることができる。前記高屈折率粒子としては、有機樹脂からなる微粒子でも無機微粒子でも構わないが、無機微粒子が好ましく用いられる。
前記無機微粒子としては、可視光線の長波長帯域において吸収の少ないもの(具体的には透過率が50〜100%程度のもの)が好適に用いられ、例えば、酸化アルミニウム粒子、酸化ジルコニウム粒子、酸化チタン粒子、酸化亜鉛粒子、硫化鉛粒子、硫化亜鉛粒子、チタン酸バリウム、チタン酸カリウム、窒化ガリウム等が挙げられる。これらの中でも、屈折率および透明性等の観点から、特に、酸化チタン粒子および酸化ジルコニウム粒子が好ましい。
【0074】
なお、これらの無機粒子は、それ自身の触媒活性を抑制するためや、樹脂や分散媒との分散性を向上させるために、粒子表面がシリカやアルミナ等で被覆されていてもよい。
【0075】
これらの無機粒子は、単独で用いてもよく、また、2種類以上を混合して使用することもできる。
【0076】
これらの高屈折率粒子(無機粒子)の比表面積は、特に限定されないが、好ましくは30〜220m2/g程度、より好ましくは60〜200m2/g程度に設定される。比表面積が前記下限値より小さいと、高屈折率粒子(無機粒子)の種類によっては、光の散乱により透明性が低下するおそれがある。また、比表面積が前記上限値より大きいと、高屈折率粒子(無機粒子)の種類によっては、表面自由エネルギーが大きくなり、凝集を起こし易くなるため、樹脂組成物の硬化物(膜)の均一性が低下するおそれがある。なお、上記比表面積は、一般的なBET法により求めることができる。
【0077】
高屈折率粒子(無機粒子)の平均粒子径としては、1〜60nmが好ましく、1〜40nmがより好ましい。なお、上記平均粒子径は、動的光散乱法による有効径として測定することができる。
【0078】
前記高屈折率粒子(無機粒子)の含有量は、特に限定されないが、前記樹脂100重量部に対して20質量部〜400質量部程度が好ましく、50質量部〜200質量部程度がより好ましい。含有量が前記範囲内であると、高屈折率化に加え、高屈折率粒子(無機粒子)の分散状態の均一性にも優れる。
【0079】
前記樹脂組成物は、分散剤を含む。これにより、樹脂組成物中での高屈折率粒子の分散性を向上させ、高屈折率粒子同士が凝集した凝集体(二次粒子)が生じることに起因する透明性の低下を防止または抑制できる。
【0080】
前記分散剤としては、50%重量減少温度が400℃以下であるものを好ましく用いることができる。なお、本発明において、分散剤として、50%重量減少温度が400℃以下のものを用いることが好ましい理由は、樹脂組成物の硬化物中での分散剤の残存率を低下させることができ、それによって屈折率を向上することができるからである。
【0081】
ここで、分散剤の50%重量減少温度とは、分散剤を、示差熱熱重量同時測定装置を用いて、50℃から600℃まで10℃/分昇温させたときの重量減少率が、50%に達したときの温度である。また、70%重量減少温度とは、上記測定方法で測定した分散剤の重量減少率が、70%に達したときの温度である。同様に、90%重量減少温度とは、上記測定方法で測定した分散剤の重量減少率が、90%に達したときの温度である。
【0082】
この50%重量減少温度が400℃以下である分散剤としては、例えば、ポリオキシアルキルエーテル系のものでは、ポリオキシエチレン(5)ドコシルエーテル、ポリオキシエチレンドデシルエーテル、ポリオキシエチレンアルキレンアミン等が挙げられ、ポリオキシアリールエーテル系のものでは、ポリオキシエチレン(2)ノニルフェニルエーテル等が挙げられ、ポリエチレングリコール(PEG)系のものでは、PEG400、PEG600、PEG04モノステアリレート等が挙げられ、ポリプロピレングリコール(PPG)系のものでは、PPG400、PPG700、アミン末端PPGであるPPGビス−2−アミノプロピルエーテル400、ポリウレタン系のものでは、ポリブチレンヘキサメチレンジイソシアネート、ポリエステル系のものでは、ポリブチレンアジペート、シラン系カップリング剤のものでは、ビニルトリメトキシシラン縮合物、3−メタクリロキシプロピルトリメトキシシラン縮合物、アルミニウム系カップリング剤のものでは、アセトアルコキシアルミニウムイソプロポキシド縮合物、チタン系カップリング剤のものでは、チタンテトラブトキシド縮合物等が挙げられる。
【0083】
なお、上記分散剤の縮合物は、純水または純水/アルコール溶液中において、酸や塩基の添加により緩やかに加水分解を進行させて縮合物とする、一般的なゾル−ゲル法で合成することができる。
【0084】
なお、PEGおよびPPGの後に記載している数字は、それぞれ、PEGおよびPPGの平均分子量を示す。
【0085】
また、樹脂組成物の硬化物中での分散剤の残存率を低下させるという観点からは、50%重量減少温度が400℃以下である分散剤の他、70%重量減少温度が420℃以下である分散剤および90%重量減少温度が440℃以下である分散剤も、50%重量減少温度が400℃以下である分散剤と同様に用いることができる。したがって、50%重量減少温度が400℃以下であることの他、70%重量減少温度が420℃以下であることおよび90%重量減少温度が440℃以下であることのいずれか一方を満足する分散剤が好ましく用いられ、これらの双方を満足する分散剤がより好ましく用いられることは言うまでもない。
【0086】
なお、本発明では、後述するように、溶媒としては、その沸点が、70〜210℃程度のものが好ましく用いられ、かかる沸点を有する溶媒が樹脂組成物中に存在している際には、分散剤も樹脂組成物中に存在しているのが好ましいため、分散剤は、その10%重量減少温度が210℃以上であるものが好ましく用いられる。
【0087】
また、上述した50%重量減少温度が400℃以下である分散剤の、50%重量減少温度、70%重量減少温度および90%重量減少温度は、表1に示す通りである。
【0088】
【表1】
【0089】
また、分散剤としては、溶媒中においてミセルを形成し、このミセル中に高屈折率粒子を取り込むことにより、高屈折率粒子の溶媒中での分散性を向上させるものであってもよいし、高屈折率粒子の表面を修飾することにより、高屈折率粒子の溶媒中での分散性を向上させるものの何れであってもよい。
【0090】
なお、分散剤として、溶媒中でミセルを形成するものを用いた場合、溶媒中の高屈折率粒子は、その全てがミセル中に取り込まれる必要はなく、その少なくとも一部がミセル中に取り込まれることにより、高屈折率粒子全体としての溶媒中での分散性が向上していれば良い。
【0091】
上述した分散剤のうち、ミセルを形成する分散剤としては、例えば、前述した分散剤のうち、ポリオキシアルキルエーテル系、ポリオキシアリールエーテル系、ポリエチレングリコール(PEG)系、ポリプロピレングリコール(PPG)系、ポリエステル系およびポリウレタン系のものが挙げられる。また、高屈折率粒子の表面を修飾する分散剤としては、例えば、シラン系カップリング剤、アルミニウム系カップリング剤、チタン系カップリング剤のものが挙げられる。
【0092】
前記分散剤の含有量は、特に限定されないが、前記樹脂100質量部に対して、5質量部〜100質量部が好ましく、10質量部〜50質量部がより好ましい。含有量が前記範囲内であると、高屈折率粒子の分散状態の均一性に特に優れる。
【0093】
前記樹脂組成物は、溶媒を含む。溶媒は、屈折率が1.6以上の樹脂、高屈折率粒子および分散剤の種類に応じて選択され、これらの組み合わせによってそれぞれ異なるが、ケトン系分散媒、エーテル系分散媒、エステル系分散媒および非プロトン極性分散媒等が好適に用いられる。
【0094】
具体的には、例えば、ケトン系溶媒として、シクロペンタノン、シクロヘキサノン、シクロヘプタノン、4−メチル−シクロヘキサノン、3,3,5−トリメチルシクロヘキサノン、メチルエチルケトン、メチルイソブチルケトン、炭酸プロピレン、ジアセトンアルコールおよびγ−ブチロラクトン等;エーテル系溶媒として、ジエチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテル、ジエチレングリコールジブチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノプロピルエーテル、プロピレングリコール1−モノ−n−ブチルエーテル、ジプロピレングリコールモノメチルエーテルおよび1,3−ブチレングリコール−3−モノメチルエーテル等;エステル系溶媒として、プロピレングリコールジアセテート、プロピレングリコールモノメチルエーテルアセテート、乳酸メチル、乳酸エチル、乳酸ブチル、メチル−1,3−ブチレングリコールアセテート、ピルビン酸メチル、ピルビン酸エチルおよびメチル−3−メトキシプロピオネート等;非プロトン系極性溶媒として、N−メチル−2−ピロリドン、N,N−ジメチルアセトアミドおよびジメチルスルホキシド等;等を挙げることができる。これらは1種または2種以上を混合して用いることができる。これらの中で、加熱により脱水または脱アルコール反応する樹脂(特に、ヒドロキシアミド重合体)の構造により異なるが、シクロペンタノンと上記シクロペンタノン以外の溶媒の混合物、シクロヘキサノンと上記シクロヘキサノン以外の溶媒の混合物が、好適に使用することができる。
【0095】
これらの中でも、溶媒としては、その沸点が好ましくは70〜210℃程度のもの、より好ましくは120〜170℃程度のものが用いられる。沸点が前記下限値より低いと、樹脂組成物で構成される液状被膜を基板上に形成する際に、液状被膜中から早期に脱溶媒して、均一な膜厚の液状被膜が形成されないおそれがある。また、沸点が前記上限値より高いと、加熱により脱水または脱アルコール反応する樹脂を、脱水または脱アルコール反応させて、樹脂組成物の硬化物を得る際に、この硬化物中に溶媒が残存し、これに起因して、硬化物の屈折率および透明性が低下するおそれがあり好ましくない。
【0096】
なお、このような溶媒としては、上述したもののうち、例えば、シクロペンタノン(沸点:131℃)およびシクロヘキサノン(沸点:156℃)等が好ましく用いられる。
【0097】
前記溶媒の含有量は、特に限定されないが、前記樹脂100質量部に対して、400質量部〜3,000質量部が好ましく、900質量部〜2,000質量部がより好ましい。含有量が前記範囲内であると、特に塗布性に優れる。
【0098】
なお、この樹脂組成物には、前記屈折率が1.6以上の樹脂、高屈折率粒子、分散剤および溶媒以外の成分として、酸素ラジカルやイオウラジカルを加熱により発生するラジカル開始剤等の添加剤を添加することができる。また、前記屈折率が1.6以上の樹脂に、感光剤としての例えばナフトキノンジアジド化合物と一緒に用いることで、樹脂組成物を感光性樹脂組成物として用いることが可能である。
【0099】
このような、屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤とを、溶媒に溶解して樹脂組成物をワニスの状態とするが、これらの高屈折率粒子を溶媒中に、より均一に分散させる方法としては、特に限定されないが、例えば、溶媒と、高屈折率粒子と、必要に応じて表面処理剤や分散剤とを混合し、高周波超音波ホモジナイザーや、微小ビーズを用いたビーズミル処理を行うことにより、無機粒子が均一に分散された分散液を得て、それと屈折率が1.6の樹脂とを混合してワニス化する方法が好ましい。
【0100】
<塗布工程>
塗布工程では上述したような樹脂組成物のワニスは、スピンナーを用いた回転塗布、スプレーコーターを用いた噴霧塗布、浸漬、印刷、ロールコーティング等の方法により成膜され、所望の形状に形成される。これらの中でもスピンコート法が好ましい。これにより、成膜上面のコプラナリティが良好な形状を得ることができる。
なお、本実施形態においては、膜(フィルム)状の成形体を形成する場合について説明したが、これに限定されない。
【0101】
この塗布工程では、上述の樹脂組成物のワニスを塗布することで、直径0.4〜1.0μmで、深さ0.5〜2.5μmの凹部を埋め込むことが好ましい。これにより、後述するような光学部品として好適に用いることができる。
【0102】
この塗布工程では、膜状体の成形体を形成すると共に、直径0.4〜1.0μm、深さ0.5〜2.5μmの凹部を埋め込むことが好ましい。このような凹部に前記樹脂組成物を埋め込み、光学部品として用いることができるからである。
【0103】
<第1加熱工程および第2加熱工程>
上述したような塗布工程を経て得られた膜状体に、第1加熱工程を実施して膜状体の屈折率が第1屈折率(A1)となるまで加熱し、さらに第2加熱工程を実施して膜状体の屈折率が第2屈折率(A2)になるまで加熱したときに、前記膜状体の屈折率の差(A2−A1)が0.03以上となるように加熱することを特徴とする。これにより、膜状体の内部での屈折率のバラつきを小さくすることができる。前記屈折率の差は、0.03以上であることがより好ましく、もっとも好ましくは0.04以上である
【0104】
このように前記屈折率の差(A2−A1)が前記下限値以上であると、膜状体の内部での屈折率のバラつきを小さくすることができる。その理由は、次のように考えられる。
屈折率の差(A2−A1)が大きくなるためには、A2値が大きいまたはA1値が小さいことが必要である。第1加熱工程は、膜状体の屈折率が第1屈折率(A1)となるまで加熱するものであるが、この工程の目的は、主として溶媒を除去し膜状体を得るために行われるものであり、第2加熱工程の加熱より低温で行われる。この第1加熱工程後の膜状体には、主として屈折率が1.6以上の樹脂、高屈折率粒子、分散剤が含まれるが、低屈折率の分散剤が含まれているため、膜状体の第1屈折率(A1)は、第2屈折率(A2)より小さな値を示す。これに対して、第2加熱工程は、膜状体の屈折率が第2屈折率(A2)となるまで加熱するものであるが、この工程では、分解または揮発による分散剤の除去が行われるので、樹脂組成物の硬化物中での分散剤の残存率は小さくなり、それによって第2屈折率(A2)を大きくすることができる。さらに、屈折率が1.6以上の樹脂が脱水閉環反応または脱アルコール閉環反応する樹脂である場合、この第2加熱工程で、脱水閉環反応または脱アルコール閉環反応が起こり、これらの閉環反応前よりも屈折率は高くなる。
【0105】
このような第1加熱工程および第2加熱工程での目的を達成するために、上述のような屈折率を示すような条件で加熱することにより、溶媒の除去、分散剤の除去が適宜実施されることになる。このように、溶媒、分散剤の除去にそれぞれ適した加熱温度で除去が行われることで膜状体の内部の屈折率のバラつきを低減することができる。
【0106】
さらには、屈折率が1.6以上の樹脂が脱水閉環反応または脱アルコール閉環反応する樹脂である場合には、第2加熱工程において、分散剤の除去等に加えて樹脂の脱水閉環反応等も適切に実施されることになるので、これによって膜状体の内部の屈折率のバラつきが抑制される。
例えば、第1加熱工程を行わないで、第2加熱工程のみとした場合には、溶媒の除去、分散剤の除去、屈折率が1.6以上の樹脂が脱水閉環反応または脱アルコール閉環反応する樹脂である場合には脱水分及び脱アルコール分の除去が、同時に一気に起こることになり、膜状体の内部での屈折率のバラつきが生じたり、膜厚の不均一による外観の悪化が生じたりしてしまう。
【0107】
前記第1加熱工程では、膜状体の屈折率が第1屈折率(A1)となるまで加熱するが、具体的には膜状体の第1屈折率が1.70以上となるまで加熱することが好ましく、特に1.75以上となるまで加熱することが好ましい。第1屈折率が前記下限値未満である場合は、膜状体内部に溶媒が残存している可能性が高く、その後の第2工程で残存した溶媒が一気に除去されることにより膜厚の均一性に問題が生じる場合がある。
【0108】
前記第1加熱工程では、一定温度で所定時間加熱する工程が好ましく、より具体的には、80〜230℃×30秒〜5分間が好ましく、より好ましくは90〜200℃×30秒〜2分間が好ましい。前記第1加熱条件であると、特に膜厚の均一性を向上することができる。
なお、具体的な加熱条件は、樹脂組成および分子量等により異なる。
【0109】
前記第2加熱工程は、前記第加熱1工程よりも高い温度で前記膜状体を加熱して、前記膜状体が第2屈折率となるまで加熱するが、具体的には膜状体の第2屈折率が1.75以上となるまで加熱することが好ましく、特に1.8〜1.9となるまで加熱することが好ましい。第2屈折率が前記下限値未満である場合は、膜状体内部に分散剤が残存しているということに他ならず、その残存分散剤の偏在により膜状体の内部での屈折率がバラつく原因となる。
【0110】
前記第2加熱工程では、前記第1加熱工程よりも高い一定温度で所定時間加熱する工程が好ましく、より具体的には、250〜450℃×1〜10分間が好ましく、より好ましくは250〜400℃×1〜5分間が好ましい。前記第2加熱条件であると、特に膜状体の内部での屈折率のバラつきを低減することができる。
なお、具体的な加熱条件は、樹脂組成および分子量等により異なる。
【0111】
また、前記第2加熱工程では、前記分散剤が分解または揮発するような温度で加熱することが好ましい。これにより、膜状体(樹脂成形体)中に残留する低屈折率の分散剤が低減され、それによって膜状体を高屈折率化することができる。
【0112】
このような工程を経ることにより、樹脂成形体を得ることができる。このような工程で得られる樹脂成形体は、内部での屈折率のバラつきが低減されているものである。
前記屈折率は、例えばプリズムカップリング法を用いた屈折率測定装置、プリズムカプラー(メトリコン社製2010プリズムカプラー)を用いて評価することができる。その際の測定には、直径2インチ、厚み1mmの石英基板上に本発明の樹脂組成物の膜状体を作成し、それぞれ所定の加熱・乾燥させたものを用いることができる。
【0113】
次に、光学部品について、好適な実施形態に基づいて説明する。
光学部品100は、基板1と、基板1に埋め込まれているフォトダイオード11と、フォトダイオード11を覆うように設けられている樹脂成形体5と、樹脂成形体5の側面を囲むように設けられた絶縁材2と、絶縁材2中に設けられた回路配線21と、樹脂成形体5および絶縁材2の上面(図2中の上側)に設けられたカラーフィルター3と、カラーフィルター3の上面に設けられた平坦化膜4と、樹脂成形体5の上方に相当する、平坦化膜4の上側の位置に設けられたレンズ6とで構成されている。このような構成により、レンズ6から入射した光信号7が、平坦化膜4およびカラーフィルター3を透過して、樹脂成形体5に入射される。
ここで、レンズ6に垂直方向に入射する光は、光漏れ等を特に生じることなくフォトダイオード11に入射される。これに対して、レンズ6に対して入射角度θが大きい光は、樹脂成形体5で光漏れ等が生じる場合があった。本発明では、樹脂成形体5は前述したように高屈折率の光学部品用樹脂組成物の硬化物で構成されているので、入射角度θが大きい光信号7に対しても光信号7を外部に漏らすことなく、フォトダイオード11に誘導することができる。
【0114】
ここで、樹脂成形体5は、樹脂組成物を上述したような第1加熱工程、第2加熱工程を経て得られるものである。そのため、樹脂成形体5の屈折率は高く、またその内部での屈折率のバラつきが小さいものである。そのため、フォトダイオードでの集光効率を向上することができる。
【0115】
さらに、樹脂成形体5が、高屈折率を有する樹脂組成物の硬化物で構成されている場合、光漏れ等を低減することができるため、フォトダイオードの受光量を向上することができる。
【0116】
また、このような樹脂成形体5を形成する場合、予め樹脂成形体5に相当する部分が凹部となっており、そこに前述の樹脂組成物を埋め込んだ後に、硬化させる手法が取られている。
このような凹部の大きさとしては、例えば径0.4〜1μm、深さ0.5〜2μmの場合が挙げられ、非常に小さい空間である。このような凹部に前述した樹脂組成物を埋め込むためには、樹脂組成物の室温での溶液粘度を低いものとして流動性を十分に確保する必要がある。そのような室温での溶液粘度としては、例えば3〜11mPa・sであることが好ましく、特に3〜8mPa・sであることが好ましい。溶液粘度が前記範囲内であると、凹部への埋込み性に優れる。
【0117】
具体的には、上述したような凹部に前記樹脂組成物を埋め込み、前記第1加熱工程および前記第2加熱工程を経て樹脂成形体5を得て、最終的な光学部品100を得る。
【0118】
樹脂成形体5の633nmでの屈折率は、特に限定されないが、1.75以上であることが好ましく、特に1.8〜1.9であることが好ましい。屈折率が前記範囲内であると、光信号7が樹脂成形体5より漏れるのを防止する効果が特に優れる。
前記屈折率は、例えばプリズムカップリング法を用いた屈折率測定装置、プリズムカプラー(メトリコン社製2010プリズムカプラー)を用いて評価することができる。その際の測定には、直径2インチ、厚み1mmの石英基板上に前記樹脂組成物の液状被膜を作成し、加熱・乾燥させたものを用いる。
【0119】
樹脂成形体5は、特に限定されないが、波長400nmにおける厚み1.5μmでの光線透過率が85%以上であることが好ましく、特に90%以上であることが好ましい。光線透過率が前記範囲内であると、特に透明性に優れる。
【0120】
このような光学部品100の具体例としては、例えば光学レンズ、光学フィルター、光スイッチ、光導波路、光ファイバー、集光レンズ等が挙げられる。
また、このような光学部品100は、上述したような樹脂成形体5で光学素子が被覆されているので、光漏れが小さく、光学素子の受光特性に優れる。
【0121】
また、上述したような光学部品100を有する光学デバイスとしては、例えば光電集積回路、光集積回路、CCDセンサ、CMOSセンサ等の光学デバイス等が挙げられる。
【実施例】
【0122】
以下、本発明を実施例および比較例に基づいて詳細に説明するが、本発明はこれに限定されるものではない。
【0123】
<実施例1>
1−1.4,4’−オキシビス安息香酸ジクロライドの合成
温度計、ジムロート冷却管、撹拌機を備えた300mLの4つ口フラスコに、東京化成工業(株)製4,4’−オキシビス安息香酸10.33g(0.04mol)、ベンジルトリエチルアンモニウムクロライド0.08g(0.0004mol)および塩化チオニル50g(0.42mol)を加え、3時間還流した。100℃で塩化チオニルを留去し、残留物を60℃で12時間真空乾燥し、10.9gの4,4’−オキシビス安息香酸ジクロライドを得た(収率92%)。
【0124】
1−2.ポリアミド系樹脂(屈折率1.6以上の樹脂)の合成
窒素ガスフロー下で、ビスアミノフェノール化合物(A)として9,9−ビス(3−アミノ−4−ヒドロキシフェニル)フルオレン30.44g(0.08mol)を、乾燥したN−メチル−2−ピロリドン200gに溶解し、ピリジン17.4g(0.22mol)を添加した後、−15℃に冷却し、ジカルボン酸化合物(B)として上述の4,4’−オキシビス安息香酸ジクロリド29.5g(0.1mol)を、少しずつ添加した。滴下終了後、−15℃で、1時間撹拌後、室温まで3時間かけて戻し、さらに室温で2時間撹拌した(反応モル比A/B=0.8)。その後、反応液を50%メタノール水溶液4リットルに小さな液滴で滴下し、沈殿物を集めた。さらに、50%メタノール水溶液4リットル中での攪拌、沈殿物の回収を3回繰り返した。その後、沈殿物を乾燥することにより、ポリアミド系樹脂(P)を得た。得られたポリアミド系樹脂の末端は、電位差滴定を用いた中和滴定により、90%以上がカルボン酸であった。このポリアミド系樹脂(P)の屈折率をプリズムカップリング法で求めたところ、1.67であった。
【0125】
1−3.酸化チタン粒子シクロヘキサノン分散液の調製
無機粒子として酸化チタン粒子(堺化学工業(株)製SSP−20、比表面積170m2/g)20重量部、分散媒としてシクロヘキサノン74重量部、および、分散剤としてポリオキシエチレン(2)ノニルフェニルエーテル(和光純薬工業(株)製)10重量部をそれぞれ混合し、微粉砕・分散機(寿工業(株)製ウルトラアペックスミルUAM−015)を用いて、ビーズミル分散処理を行い、酸化チタン粒子シクロヘキサノン分散液を得た。
【0126】
2.樹脂組成物の調製
得られたポリアミド系樹脂(P)100重量部にシクロヘキサノン330重量部を加え溶解させた後、酸化チタン粒子シクロヘキサノン分散液520重量部を混合し30分間攪拌を行った後、超音波ホモジナイザー処理を行い、樹脂組成物(A)を得た。
【0127】
3.樹脂成形体の製造(塗布工程、第1加熱工程および第2加熱工程)
次に、上述の樹脂組成物(A)をシリコン基板上に、スピンコートにより、塗膜を形成した。次に、第1加熱工程として、第1屈折率が1.77となるまで、該塗膜を大気雰囲気下で、200℃で、60秒間加熱した。さらに、第2加熱工程として、第2屈折率が1.84となるまで、N2ガス雰囲気下で、380℃で、250秒間加熱し樹脂成形体を得た。この樹脂成形体の屈折率の差は0.07であった。また、下記に記載の方法で測定した膜質均一性は0.5%、凹部埋込性は良好であった。
【0128】
(実施例2)
第1加熱工程および第2加熱工程の条件を下記のようにした以外は、実施例1と同様にした。
第1加熱工程として、第1屈折率が1.77となるまで、90℃で、120秒間加熱し、第2加熱工程として、第2屈折率が1.83となるまで、350℃、250秒間加熱し樹脂成形体を得た。この樹脂成形体の屈折率の差は、0.06であり、膜質均一性は0.6%、凹部埋込性は良好であった。
【0129】
(実施例3)
第1加熱工程および第2加熱工程の条件を下記のようにした以外は、実施例1と同様にした。
第1加熱工程として、第1屈折率が1.80となるまで220℃で、120秒間加熱し、第2加熱工程として、第2屈折率が1.84となるまで、400℃、250秒間加熱し樹脂成形体を得た。この樹脂成形体の屈折率の差は、0.04であり、膜質均一性は0.7%、凹部埋込性は良好であった。
【0130】
(実施例4)
樹脂組成物として下記に記載の樹脂組成物を用い、第1加熱条件および第2加熱条件を下記のようにした以外は、実施例1と同様にした。
まず、無機微粒子として酸化ジルコニウム粒子(日本稀元素化学工業(株)製UEP−100、比表面積90m2/g)20重量部、シクロヘキサノン74重量部及びポリプロピレングリコール400(和光純薬(株)製)6重量部を混合し、実施例1と同様にして、酸化ジルコニウム粒子シクロヘキサノン分散液を得た。
つぎに、ポリアミド系樹脂(P)100重量部にシクロヘキサノン300重量部を加え溶解させた後、この酸化ジルコニウム粒子シクロヘキサノン分散液750重量部を混合し30分間攪拌を行った後、超音波ホモジナイザー処理を行い、樹脂組成物(B)を得た。
また、第1加熱工程として、第1屈折率が1.76となるまで、該塗膜を大気雰囲気下で、200℃で、60秒間加熱した。さらに、第2加熱工程として、第2屈折率が1.80となるまで、N2ガス雰囲気下で、380℃で、250秒間加熱し樹脂成形体を得た。この樹脂成形体の屈折率の差は0.04であり、膜質均一性は0.5%、凹部埋込性は良好であった。
【0131】
(比較例1)
第1加熱工程および第2加熱工程を以下のようにした以外は、実施例1と同様にした。
実施例1と同様にして、第1加熱工程として、第1屈折率が1.77となるまで、200℃で、60秒間加熱し、第2加熱工程として、第2屈折率が1.79となるまで、250℃、250秒間加熱し樹脂成形体を得た。この樹脂成形体の屈折率の差は、0.02であり、膜質均一性は2.8%であった。
なお、第2加熱工程後の樹脂成形体のIR測定を実施したところ、分散剤であるポリオキシエチレン(2)ノニルフェニルエーテル樹脂のピークが観察されたことから分散剤が残存していること、またアミド結合のピークが観察されたことから閉環脱水反応が完了していないことが判明した。また閉環脱水反応が完了していないため、実施例1と比較し、ガラス転移温度が245℃と低かった。
【0132】
(比較例2)
ポリアミド系樹脂として、以下のものを用いた以外は、実施例1と同様にした。
9,9−ビス(3−アミノ−4−ヒドロキシフェニル)フルオレン30.44g(0.08mol)の代わりに、2,2−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパン29.3g(0.08mol)を用いた以外は、全て実施例1と同様にして、ポリアミド系樹脂(Q)を得た。このポリアミド系樹脂(Q)の屈折率は、1.58であった。
このポリアミド系樹脂(Q)100重量部にシクロヘキサノン330重量部を加え溶解させた後、製造例2−1の酸化チタン粒子シクロヘキサノン分散液520重量部を混合し30分間攪拌を行った後、超音波ホモジナイザー処理を行い、樹脂組成物(C)を得た。
【0133】
そして、実施例1と同様に第1加熱工程として、第1屈折率が1.70となるまで、該塗膜を大気雰囲気下で、200℃で、60秒間加熱した。さらに、第2加熱工程として、N2ガス雰囲気下で、350℃で、250秒間加熱したところ、第2屈折率としては1.74であった。第2屈折率を高めるために、450℃、250秒間とより高い温度にて加熱する条件でも第2工程を行ったが、1.74のままであった。屈折率の差は0.04であり、膜質均一性も0.7%と悪くないが、使用したポリアミド系樹脂(Q)の屈折率値が低いために、第2屈折率としては1.75以上という望ましい値が得られていなかった。
【0134】
(比較例3)
第1加熱工程のみを行い、第2加熱工程を行わないこと以外は、実施例1と同様にした。得られた樹脂成形体の膜質均一性は2.0%であった。樹脂成形体のIR測定を実施したところ、分散剤であるポリオキシエチレン(2)ノニルフェニルエーテル樹脂のピークが観察されたことから分散剤が残存していること、またアミド結合のピークが観察されたことから閉環脱水反応が完了していないことが判明した。閉環脱水反応が完了していないため、実施例1と比較し、ガラス転移温度が242℃と低かった。
【0135】
(比較例4)
第1加熱工程を行わず、第2加熱工程のみを行った以外は、実施例1と同様に行った。得られた樹脂成形体の膜質均一性は2.3%であった。第1加熱工程がなく、膜状体内部に溶媒が多く残存している状態で、第2加熱工程を行ったため、残存した溶媒、分散剤及び脱水閉環反応による脱水が一気に除去されることにより膜厚の均一性に問題が生じた結果、バラつき度が大きくなったと予想される。凹部埋込性は、FIB−SEMによる観察では、断面部に気泡が見られ不良であった。
【0136】
各実施例および比較例で得られた樹脂成形体について、以下の評価を行った。評価項目を評価内容と共に示す。
【0137】
1.ガラス転移温度
上記で得た塗膜を削り落とした粉末について、MDSC(温度サイクルモード示差操作熱量計:ティー・エイ・インスツルメント社製2910MDSC)により、N2ガスを30mL/分の流量で流しながら、昇温速度2℃/分、温度振幅±2℃/分の条件で昇温しながら、40℃から420℃までの温度範囲で測定を行い、リバース曲線の変移点から算出を行った。
【0138】
2.膜質均一性
上記で得た加熱後の塗膜において、ウエハ面内をXY軸それぞれ等間隔で19ポイント(合計37ポイント)をn&k Technology Inc.社製n&kアナライザー1500を用いて屈折率を測定し、その相対標準偏差値を膜質均一性とした。
【0139】
3.凹部への埋込性
凹部への埋込性は、直径0.5μmで、深さ1.5μmの凹部を加工したSiO2加工ウエハを用い、上述の樹脂組成物をスピンコートして塗膜を形成し、上述の条件加熱・乾燥させたものを用い、FIB−SEMによる断面観察を行うことで、良好か不良かを判断した。良好とはボイドや基板との剥離などなく均一であることが観察されることとした。
【0140】
4.樹脂成形体の簡易評価
各実施例および比較例で用いた樹脂組成物を、石英基板上にスピンコートにより、熱処理後の最終膜厚が1.5μmとなるように塗膜を形成した。この塗膜を用いて、下記のように光線透過率および屈折率を評価した。
【0141】
4−1.光線透過率
上記で得た樹脂膜を用いて、株式会社島津製作所製分光光度計UV−3100を用いて波長400nmで測定した。
【0142】
4−2.屈折率
上記で得た樹脂膜を用いて、Metricon製プリズムカプラーを用いて、20℃での633nm(He−Neレーザーを使用)での膜面に対して垂直方向の屈折率(TM)を測定した。
【0143】
【表2】
【0144】
表2から明らかなように、実施例1〜4の樹脂成形体は、樹脂成形体の内部での屈折率のバラつきを小さくすることができた。したがって、この樹脂成形体を用いた光学部品、光学デバイスは性能に優れていることが示唆された。
また、実施例1〜4で用いている樹脂成形体は、ガラス転移温度が高く、耐熱性に優れていた。
また、実施例1〜4で用いている樹脂組成物を使用した凹部への埋込性の評価より、凹部への埋め込み性に優れていることが示された。
また、実施例1〜4で用いている樹脂成形体は、屈折率も高く、光学部品として用いた際の性能に優れることが示唆された。
【符号の説明】
【0145】
1 基板
11 フォトダイオード
2 絶縁材
21 回路配線
3 カラーフィルター
4 平坦化膜
5 樹脂成形体
6 レンズ
7 光信号
100 光学部品
【技術分野】
【0001】
本発明は、樹脂成形体の製造方法、樹脂成形体、光学部品および光学デバイスに関する。
【背景技術】
【0002】
近年、軽量でかつ加工性に富み、各種の光学部材に好適な樹脂材料として、1.8〜2.2程度の高い屈折率を有する透明樹脂材料が要求されている。この要求に対して、ポリチオール化合物とポリイソシアネート化合物から得られるチオウレタン(例えば、特許文献1参照)、エポキシ樹脂またはエピスルフィド樹脂から得られる重合体(例えば、特許文献2参照)などが検討されている。しかし、これらの材料が有するような屈折率では、依然として不十分であった。
【0003】
また、透明性を維持したまま、高い屈折率を与えるために、樹脂中に酸化チタンや酸化亜鉛などの高屈折率金属酸化物微粒子を分散させる技術が提案されているが(例えば、特許文献3参照)、光散乱を引き起こさないようにこれらの微粒子を分散させるには、分散剤が必須であったが、これによっても透明材料中で屈折率にバラつきがあった。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特公平4−58489号公報
【特許文献2】特開平3−81320号公報
【特許文献3】特開2002−277609号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明の目的は、樹脂成形体の内部での屈折率のバラつきの小さい樹脂成形体を製造する方法を提供することにある。
また、本発明の別の目的は、樹脂成形体の内部でのバラつきの小さい樹脂成形体およびそれを用いて性能に優れた光学部品、光学デバイスを提供することにある。
【課題を解決するための手段】
【0006】
このような目的は、下記(1)〜(15)に記載の本発明により達成される。
(1)屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を所望の形状に形成し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体の製造方法であって、前記第1加熱工程において樹脂成形体の屈折率が第1屈折率(A1)になるまで加熱し、前記第2加熱工程において樹脂成形体の屈折率が第2屈折率(A2)になるまで加熱したときに、前記樹脂成形体の屈折率の差(A2−A1)が0.03以上となるような条件で加熱することを特徴とする樹脂成形体の製造方法。
(2)前記第1加熱工程は、一定温度で所定時間加熱することにより樹脂成形体の屈折率を、第1屈折率(A1)にするものである上記(1)に記載の樹脂成形体の製造方法。
(3)前記第1加熱工程で樹脂成形体の屈折率が、1.70以上となるまでに加熱するものである上記(1)または(2)に記載の樹脂成形体の製造方法。
(4)前記第2加熱工程は、前記第1加熱工程の加熱温度よりも高い温度で、所定時間加熱することにより樹脂成形体の屈折率を、第2屈折率(A2)にするものである上記(1)ないし(3)のいずれかに記載の樹脂成形体の製造方法。
(5)前記第2加熱工程は、前記分散剤が分解または揮発するような温度で加熱するものである上記(1)ないし(4)のいずれかに記載の樹脂成形体の製造方法。
(6)前記屈折率が1.6以上の樹脂は、加熱により脱水閉環反応または脱アルコール閉環反応する樹脂である上記(1)ないし(5)のいずれかに記載の樹脂成形体の製造方法。
(7)前記屈折率が1.6以上の樹脂は、ポリベンゾオキサゾール前駆体を含むものである上記(1)ないし(6)のいずれかに記載の樹脂成形体の製造方法。
(8)前記ポリベンゾオキサゾール前駆体は、ジカルボン酸化合物をビスアミノフェノール化合物よりも過剰に反応させてなるものである上記(1)ないし(7)のいずれかに記載の樹脂成形体の製造方法。
(9)前記樹脂成形体は、直径0.4〜1.0μm、深さ0.5〜2.5μmの凹部に埋め込まれるものである上記(1)ないし(8)のいずれかに記載の樹脂成形体の製造方法。
(10)屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を成膜し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体であって、前記第1加熱工程後における樹脂成形体の第1屈折率(A1)と、前記第2加熱工程後における樹脂成形体の第2屈折率(A2)との差(A2−A1)が、0.03以上であることを特徴とする樹脂成形体。
(11)前記第1屈折率が、1.70以上である上記(10)に記載の樹脂成形体。
(12)前記屈折率が1.6以上の樹脂は、加熱により脱水閉環反応または脱アルコール閉環反応する樹脂である上記(10)または(11)に記載の樹脂成形体。
(13)前記樹脂成形体は、直径0.4〜1.0μm、深さ0.5〜2.5μmの凹部を埋め込むために用いるものである上記(10)ないし(12)のいずれかに記載の樹脂成形体。
(14)上記(10)ないし(13)のいずれかに記載の樹脂成形体を用いてなることを特徴とする光学部品。
(15)上記(14)に記載の光学部品を有することを特徴とする光学デバイス。
【発明の効果】
【0007】
本発明によれば、樹脂成形体の内部での屈折率のバラつきの小さい樹脂成形体を製造する方法を提供することができる。
また、本発明の別の目的は、樹脂成形体の内部でのバラつきの小さい樹脂成形体およびそれを用いて性能に優れた光学部品、光学デバイスを得ることができる。
【図面の簡単な説明】
【0008】
【図1】樹脂成形体の製造方法の主なプロセスを記載した工程フロー図である。
【図2】光学部品の一例を示す断面図である。
【発明を実施するための形態】
【0009】
以下、本発明の樹脂成形体の製造方法、樹脂成形体、光学部品および光学デバイスについて説明する。
本発明の樹脂成形体の製造方法は、屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を所望の形状の形成し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体の製造方法であって、前記第1加熱工程において樹脂成形体の屈折率が第1屈折率(A1)になるまで加熱し、前記第2加熱工程において樹脂成形体の屈折率が第2屈折率(A2)になるまで加熱したときに、前記樹脂成形体の屈折率の差(A2−A1)が0.03以上となるような条件で加熱することを特徴とする。
また、本発明の樹脂成形体は、屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を成膜し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体であって、前記第1加熱工程後における樹脂成形体の第1屈折率(A1)と、前記第2加熱工程後における樹脂成形体の第2屈折率(A2)との差(A2−A1)が、0.03以上であることを特徴とする。
また、本発明の光学部品は、上記に記載の樹脂成形体を用いてなることを特徴とする。
また、本発明の光学デバイスは、上記に記載の光学部品を有することを特徴とする。
【0010】
まず、樹脂成形体の製造方法および樹脂成形体について、所望の形状として膜(フィルム)状の成形体を形成する場合について説明する。
図1に示すように、本発明の樹脂成形体の製造方法は、ワニス化工程、塗布工程、第1加熱工程および第2加熱工程を有している。
【0011】
<ワニス化工程>
ワニス化工程では、屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤とを、溶媒に溶解して樹脂組成物をワニスの状態とする。これにより、前記塗布工程を容易にする。
前記樹脂組成物は、屈折率が1.6以上の樹脂を含む。前記屈折率が1.6以上の樹脂としては、例えばポリアミド系樹脂、ポリエステル系樹脂、エポキシ系樹脂、ポリウレタン系樹脂、ポリスルフィド系樹脂、加熱により脱水閉環反応または脱アルコール閉環反応する樹脂等が挙げられる。
これらの中でも加熱により脱水閉環反応または脱アルコール閉環反応する樹脂が好ましい。これにより、耐熱性をより向上することができる。
【0012】
前記加熱により脱水閉環反応または脱アルコール閉環反応する樹脂としては、具体的にはポリベンゾオキサゾール前駆体、ポリイミド前駆体、ポリベンゾイミダゾール前駆体およびポリベンゾチアゾール前駆体等が挙げられ、これらのうちの1種または2種以上を組み合わせて用いることができる。これらの中でもポリベンゾオキサゾール前駆体およびポリイミド前駆体のうちの少なくとも1種を用いるのが好ましい。これにより、脱水閉環による硬化物の可視光域での透過性および耐熱性を向上することができる。
【0013】
前記ポリベンゾオキサゾール前駆体としては、特に限定されないが、例えば、下記一般式(A)で表わされるヒドロキシアミド重合体が用いられる。
【0014】
このヒドロキシアミド重合体は、加熱により脱水する閉環反応(縮合反応)により、ポリベンゾオキサゾール(硬化物)となるものである。
【0015】
【化1】
【0016】
【化2】
【0017】
【化3】
【0018】
【化4】
【0019】
【化5】
【0020】
【化6】
【0021】
【化7】
【0022】
また、式(B)、式(C)、式(D)、式(E)、式(F)および式(G)で表される基における環構造上の水素原子は、炭素数1〜4のアルキル基の中から選ばれる少なくとも1個の基で置換されていてもよい。
【0023】
このような一般式(A)で表されるヒドロキシアミド重合体は、前記式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物の少なくとも1種と、前記式(C)および前記式(D)で表される基の中から選ばれる基を有するジカルボン酸化合物の少なくとも1種とを用いて反応させて合成することができる。
【0024】
また、ジカルボン酸化合物として、前記式(C)および前記式(D)で表される基の中から選ばれる基を有するジカルボン酸化合物の少なくとも1種と、式(E)および式(F)で表される基の中から選ばれる基を有するジカルボン酸化合物とを用いて反応させることもできる。
【0025】
上記ヒドロキシアミド重合体の合成において、ビスアミノフェノール化合物と、ジカルボン酸化合物とを反応させる方法としては、従来の酸クロライド法、活性化エステル法、ポリリン酸やジシクロヘキシルカルボジイミド等の脱水縮合剤の存在下での縮合反応等の方法を挙げることができる。
【0026】
なお、前記ビスアミノフェノール化合物における基とは、アミノ基およびフェノール性水酸基と結合し得る結合手を四つ有する基を意味する。また、前記ジカルボン酸化合物における基とは、カルボキシル基と結合し得る結合手を2つ有する基を意味する。
【0027】
なお、式(B)で表される基を有するビスアミノフェノール化合物の具体例としては、2,5−ジアミノヒドロキノン、2,3−ジアミノヒドロキノン、3,6−ジアミノカテコール、4,6−ジアミノレゾルシノール、ビス(3−アミノ−4−ヒドロキシフェニル)メタン、ビス(4−アミノ−3−ヒドロキシフェニル)メタン、2,2−ビス(3−アミノ−4−ヒドロキシフェニル)プロパン、2,2−ビス(4−アミノ−3−ヒドロキシフェニル)プロパン、2,2−(2,3’−アミノ−3,4’−ヒドロキシフェニル)プロパン、2,2−(3,4’−アミノ−2,3’−ヒドロキシフェニル)プロパン、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルスルホン、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルスルホン、2,3’−ジアミノ−3,4’−ジヒドロキシジフェニルスルホン、3,4’−ジアミノ−2,3’−ジヒドロキシジフェニルスルホン、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルエーテル、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルエーテル、2,3’−ジアミノ−3,4’−ジヒドロキシジフェニルエーテル、3,4’−ジアミノ−2,3’−ジヒドロキシジフェニルエーテル、1,3−ビス(3−アミノ−4ヒドロキシフェノキシ)ベンゼン、1,3−ビス(4−アミノ−3ヒドロキシフェノキシ)ベンゼン、1,4−ビス(3−アミノ−4ヒドロキシフェノキシ)ベンゼン、1,4−ビス(4−アミノ−3ヒドロキシフェノキシ)ベンゼン、1,3−ビス(3−アミノ−4ヒドロキシフェニルスルファニル)ベンゼン、1,3−ビス(4−アミノ−3ヒドロキシフェニルスルファニル)ベンゼン、1,4−ビス(3−アミノ−4ヒドロキシフェニルスルファニル)ベンゼン、1,4−ビス(4−アミノ−3ヒドロキシフェニルスルファニル)ベンゼン、3,3’−ジアミノ−4,4’−ジヒドロキシビフェニル、4,4’−ジアミノ−3,3’−ジヒドロキシビフェニル、3,4’−ジアミノ−2,3’−ジヒドロキシビフェニル、2,3’−ジアミノ−3,4’−ジヒドロキシビフェニル、3,3’−ジアミノ−2,2’−ジヒドロキシビナフチル、2,2’−ジアミノ−3,3’−ジヒドロキシビナフチル、2,3’−ジアミノ−3,2’−ジヒドロキシビナフチル、9,9−ビス(4−アミノ−3−ヒドロキシフェニル)フルオレン、9,9−ビス(3−アミノ−4−ヒドロキシフェニル)フルオレン、9,9−ビス(4−(3−アミノ−4−ヒドロキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−アミノ−3−ヒドロキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−アミノ−3−ヒドロキシフェノキシ)フェニル)フルオレンおよび9,9−ビス(4−(3−アミノ−4−ヒドロキシフェノキシ)フェニル)フルオレン等が挙げられる。
【0028】
これらの中でも、溶媒に対する溶解性と、耐熱性の上で、2,5−ジアミノヒドロキノン、4,6−ジアミノレゾルシノール、ビス(3−アミノ−4−ヒドロキシフェニル)メタン、ビス(4−アミノ−3−ヒドロキシフェニル)メタン、2,2−ビス(3−アミノ−4−ヒドロキシフェニル)プロパン、2,2−ビス(4−アミノ−3−ヒドロキシフェニル)プロパン、2,2−(2,3’−アミノ−3,4’−ヒドロキシフェニル)プロパン、2,2−(3,4’−アミノ−2,3’−ヒドロキシフェニル)プロパン、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルスルホン、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルスルホン、2,3’−ジアミノ−3,4’−ジヒドロキシジフェニルスルホン、3,4’−ジアミノ−2,3’−ジヒドロキシジフェニルスルホン、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルエーテル、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルエーテル、2,3’−ジアミノ−3,4’−ジヒドロキシジフェニルエーテル、3,4’−ジアミノ−2,3’−ジヒドロキシジフェニルエーテル、1,3−ビス(3−アミノ−4ヒドロキシフェノキシ)ベンゼン、1,3−ビス(4−アミノ−3ヒドロキシフェノキシ)ベンゼン、1,4−ビス(3−アミノ−4ヒドロキシフェノキシ)ベンゼン、1,4−ビス(4−アミノ−3ヒドロキシフェノキシ)ベンゼン、1,3−ビス(3−アミノ−4ヒドロキシフェニルスルファニル)ベンゼン、1,3−ビス(4−アミノ−3ヒドロキシフェニルスルファニル)ベンゼン、1,4−ビス(3−アミノ−4ヒドロキシフェニルスルファニル)ベンゼン、1,4−ビス(4−アミノ−3ヒドロキシフェニルスルファニル)ベンゼン、9,9−ビス(4−アミノ−3−ヒドロキシフェニル)フルオレン、9,9−ビス(3−アミノ−4−ヒドロキシフェニル)フルオレン、9,9−ビス(4−(4−アミノ−3−ヒドロキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−アミノ−3−ヒドロキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−アミノ−3−ヒドロキシフェノキシ)フェニル)フルオレンおよび9,9−ビス(4−(3−アミノ−4−ヒドロキシフェノキシ)フェニル)フルオレンが好ましく、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルスルホン、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルスルホン、3,3’−ジアミノ−4,4’−ジヒドロキシジフェニルエーテル、4,4’−ジアミノ−3,3’−ジヒドロキシジフェニルエーテル、1,3−ビス(3−アミノ−4ヒドロキシフェノキシ)ベンゼン、1,3−ビス(4−アミノ−3ヒドロキシフェノキシ)ベンゼン、9,9−ビス(4−アミノ−3−ヒドロキシフェニル)フルオレンおよび9,9−ビス(3−アミノ−4−ヒドロキシフェニル)フルオレン、がより好ましい。これらのビスアミノフェノール化合物は単独で用いてもよく、また2種類以上を組み合わせて用いることができる。
【0029】
また、式(C)で表される基を有するジカルボン酸化合物の具体例としては、特に限定されないが、イソフタル酸、テレフタル酸、4,4’−ビフェニルジカルボン酸、3,3’−ビフェニルジカルボン酸、1,4−ナフタレンジカルボン酸、2,3−ナフタレンジカルボン酸、2,6−ナフタレンジカルボン酸、4,4’−スルホニルビス安息香酸、3,3’−スルホニルビス安息香酸、4,4’−チオビス安息香酸、3,3’−チオビス安息香酸、4,4’−オキシビス安息香酸、3,3’−オキシビス安息香酸、ビス(4−カルボキシフェニル)メタン、ビス(3−カルボキシフェニル)メタン、2,2−ビス(4−カルボキシフェニル)プロパン、2,2−ビス(3−カルボキシフェニル)プロパン、2,2’−ジメチル−4,4’−ビフェニルジカルボン酸、3,3’−ジメチル−4,4’−ビフェニルジカルボン酸、2,2’−ジメチル−3,3’−ビフェニルジカルボン酸、1,3−ビス(4−カルボキシフェノキシ)ベンゼン、1,3−ビス(3−カルボキシフェノキシ)ベンゼン、1,4−ビス(4−カルボキシフェノキシ)ベンゼン、1,4−ビス(3−カルボキシフェノキシ)ベンゼン、1,3−ビス(4−カルボキシフェニルスルファニル)ベンゼン、1,3−ビス(3−カルボキシフェニルスルファニル)ベンゼン、1,4−ビス(4−カルボキシフェニルスルファニル)ベンゼン、1,4−ビス(3−カルボキシフェニルスルファニル)ベンゼン、4,4’−ビス(4−カルボキシフェノキシ)ビフェニル、4,4’−ビス(3−カルボキシフェノキシ)ビフェニル、3,4’−ビス(4−カルボキシフェノキシ)ビフェニル、3,4’−ビス(3−カルボキシフェノキシ)ビフェニル、3,3’−ビス(4−カルボキシフェノキシ)ビフェニル、3,3’−ビス(3−カルボキシフェノキシ)ビフェニル、9,9−ビス(3−カルボキシフェニル)フルオレン、9,9−ビス(4−カルボキシフェニル)フルオレン、9,9−ビス(4−(3−カルボキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−カルボキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−カルボキシフェノキシフェニル)フルオレンおよび9,9−ビス(3−カルボキシフェノキシフェニル)フルオレン等が挙げられる。
【0030】
これらの中でも、溶媒に対する溶解性の上で、イソフタル酸、テレフタル酸、4,4’−スルホニルビス安息香酸、3,3’−スルホニルビス安息香酸、4,4’−チオビス安息香酸、3,3’−チオビス安息香酸、4,4’−オキシビス安息香酸、3,3’−オキシビス安息香酸、ビス(4−カルボキシフェニル)メタン、ビス(3−カルボキシフェニル)メタン、2,2−ビス(4−カルボキシフェニル)プロパン、2,2−ビス(3−カルボキシフェニル)プロパン、1,3−ビス(4−カルボキシフェノキシ)ベンゼン、1,3−ビス(3−カルボキシフェノキシ)ベンゼン、1,4−ビス(4−カルボキシフェノキシ)ベンゼン、1,4−ビス(3−カルボキシフェノキシ)ベンゼン、1,3−ビス(4−カルボキシフェニルスルファニル)ベンゼン、1,3−ビス(3−カルボキシフェニルスルファニル)ベンゼン、1,4−ビス(4−カルボキシフェニルスルファニル)ベンゼン、1,4−ビス(3−カルボキシフェニルスルファニル)ベンゼン、9,9−ビス(3−カルボキシフェニル)フルオレン、9,9−ビス(4−カルボキシフェニル)フルオレン、9,9−ビス(4−(3−カルボキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−(4−カルボキシフェニル)スルファニルフェニル)フルオレン、9,9−ビス(4−カルボキシフェノキシフェニル)フルオレンおよび9,9−ビス(3−カルボキシフェノキシフェニル)フルオレンが好ましく、イソフタル酸、4,4’−スルホニルビス安息香酸、3,3’−スルホニルビス安息香酸、4,4’−チオビス安息香酸、3,3’−チオビス安息香酸、4,4’−オキシビス安息香酸、3,3’−オキシビス安息香酸、1,3−ビス(4−カルボキシフェノキシ)ベンゼン、1,3−ビス(3−カルボキシフェノキシ)ベンゼン、1,4−ビス(4−カルボキシフェノキシ)ベンゼン、1,4−ビス(3−カルボキシフェノキシ)ベンゼン、9,9−ビス(3−カルボキシフェニル)フルオレンおよび9,9−ビス(4−カルボキシフェニル)フルオレンがより好ましい。これらのジカルボン酸化合物は単独で用いてもよく、また2種類以上を組み合わせて用いることができる。
【0031】
式(D)で表される基を有するジカルボン酸化合物の具体例としては、特に限定されないが、1,3−シクロヘキサンジカルボン酸、cis−1,3−シクロヘキサンジカルボン酸、1,4−シクロヘキサンジカルボン酸、cis−1,4−シクロヘキサンジカルボン酸、trans−1,4−シクロヘキサンジカルボン酸、1,3−デカリンジカルボン酸、1,4−デカリンジカルボン酸、2,6−デカリンジカルボン酸、1,3−アダマンタンジカルボン酸、5−メチル−1,3−アダマンタンジカルボン酸、2,2−アダマンタンジカルボン酸、3,3’−(2,2−アダマンチル)フェニルジカルボン酸、4,4’−(2,2−アダマンチル)フェニルジカルボン酸、3,3’−(2,2−アダマンチルオキシ)フェニルジカルボン酸、4,4’−(2,2−アダマンチルオキシ)フェニルジカルボン酸、3,3’−(1,3−アダマンチル)フェニルジカルボン酸、4,4’−(1,3−アダマンチル)フェニルジカルボン酸、3,3’−(1,1’−ビアダマンタン)ジカルボン酸、3,5−(1,1’−ビアダマンタン)ジカルボン酸、3,3’−(1,1’−ビアダマンタン)ジカルボン酸、3,5’−(1,1’−ビアダマンタン)ジカルボン酸、3’,5’,7,7’−テトラメチル−1,1’−ビアダマンタン−3,5−ジカルボン酸、5,5’,7,7’−テトラメチル−1,1’−ビアダマンタン−3,3’ジカルボン酸および3’,5,7,7’−テトラメチル−1,1’−ビアダマンタン−3,5’−ジカルボン酸等が挙げられる。
【0032】
これらの中でも、溶媒に対する溶解性と、耐熱性の上で、1,4−シクロヘキサンジカルボン酸、cis−1,4−シクロヘキサンジカルボン酸、trans−1,4−シクロヘキサンジカルボン酸、1,3−アダマンタンジカルボン酸、5−メチル−1,3−アダマンタンジカルボン酸、3,3’−(1,1’−ビアダマンタン)ジカルボン酸、3,5−(1,1’−ビアダマンタン)ジカルボン酸、3,3’−(1,1’−ビアダマンタン)ジカルボン酸、3,5’−(1,1’−ビアダマンタン)ジカルボン酸、3’,5’,7,7’−テトラメチル−1,1’−ビアダマンタン−3,5−ジカルボン酸、5,5’,7,7’−テトラメチル−1,1’−ビアダマンタン−3,3’ジカルボン酸および3’,5,7,7’−テトラメチル−1,1’−ビアダマンタン−3,5’−ジカルボン酸が好ましい。これらのジカルボン酸化合物は単独で用いてもよく、また2種類以上を組み合わせて用いることができる。
【0033】
式(E)で表される基を有するジカルボン酸化合物において、R置換基は、アリール基または炭素数1〜10のアルキル基である。これらのうち、前記アリール基としては、具体的には、フェニル基、ベンジル基、トリル基、o−キシリル基、m−キシリル基およびp−キシリル基等が挙げられる。また、前記アルキル基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、t−ブチル基、シクロヘキシル基およびアダマンチル基等が挙げられる。これらのなかでも、フェニル基、トリル基、o−キシリル基、m−キシリル基、p−キシリル基、メチル基、イソブチル基およびt−ブチル基が好ましく、フェニル基、トリル基、o−キシリル基、m−キシリル基およびp−キシリル基がより好ましい。
【0034】
このような式(E)で表される基を有するジカルボン酸化合物のうち、R置換基がフェニル基であるフェニルエチニル基を有するジカルボン酸化合物の具体例としては、特に限定されないが、4−フェニルエチニルイソフタル酸、5−フェニルエチニルイソフタル酸、2−フェニルエチニルテレフタル酸、3−フェニルエチニルテレフタル酸、2−フェニルエチニル−1,5−ナフタレンジカルボン酸、3−フェニルエチニル−1,5−ナフタレンジカルボン酸、4−フェニルエチニル−1,5−ナフタレンジカルボン酸、1−フェニルエチニル−2,6−ナフタレンジカルボン酸、3−フェニルエチニル−2,6−ナフタレンジカルボン酸、4−フェニルエチニル−2,6−ナフタレンジカルボン酸、3,7−ビスフェニルエチニル−1,5−ナフタレンジカルボン酸、4,8−ビスフェニルエチニル−2,6−ナフタレンジカルボン酸、4−フェニルエチニル−2,2'−ビフェニルジカルボン酸、6−フェニルエチニル−2,2'−ビフェニルジカルボン酸、2−フェニルエチニル−4,4'−ビフェニルジカルボン酸、4,4'−ビスフェニルエチニル−2,2'−ビフェニルジカルボン酸、6,6'−ビスフェニルエチニル−2,2'−ビフェニルジカルボン酸、2,2'−ビスフェニルエチニル−4,4'−ビフェニルジカルボン酸、2,2−ビス(3−カルボキシ−4−フェニルエチニルフェニル)プロパン、2,2−ビス(3−カルボキシ−5−フェニルエチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−2−フェニルエチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−3−フェニルエチニルフェニル)プロパン、ビス(3−カルボキシ−4−フェニルエチニルフェニル)スルホン、ビス(3−カルボキシ−5−フェニルエチニルフェニル)スルホン、ビス(4−カルボキシ−2−フェニルエチニルフェニル)スルホン、ビス(4−カルボキシ−3−フェニルエチニルフェニル)スルホン、ビス(3−カルボキシ−4−フェニルエチニルフェニル)エーテル、ビス(3−カルボキシ−5−フェニルエチニルフェニル)エーテル、ビス(4−カルボキシ−2−フェニルエチニルフェニル)エーテル、ビス(4−カルボキシ−3−フェニルエチニルフェニル)エーテル等が挙げられる。
【0035】
これらの中でも、溶媒に対する溶解性の上で、4−フェニルエチニルイソフタル酸、5−フェニルエチニルイソフタル酸、2−フェニルエチニルテレフタル酸、3−フェニルエチニルテレフタル酸、2,2−ビス(3−カルボキシ−4−フェニルエチニルフェニル)プロパン、2,2−ビス(3−カルボキシ−5−フェニルエチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−2−フェニルエチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−3−フェニルエチニルフェニル)プロパン、ビス(3−カルボキシ−4−フェニルエチニルフェニル)スルホン、ビス(3−カルボキシ−5−フェニルエチニルフェニル)スルホン、ビス(4−カルボキシ−2−フェニルエチニルフェニル)スルホン、ビス(4−カルボキシ−3−フェニルエチニルフェニル)スルホン、ビス(3−カルボキシ−4−フェニルエチニルフェニル)エーテル、ビス(3−カルボキシ−5−フェニルエチニルフェニル)エーテル、ビス(4−カルボキシ−2−フェニルエチニルフェニル)エーテル、ビス(4−カルボキシ−3−フェニルエチニルフェニル)エーテルが好ましく、4−フェニルエチニルイソフタル酸、5−フェニルエチニルイソフタル酸、2−フェニルエチニルテレフタル酸、3−フェニルエチニルテレフタル酸がより好ましい。これらのジカルボン酸化合物は単独で用いてもよく、また2種類以上を組み合わせて用いることができる。
【0036】
式(F)で表される基を有するジカルボン酸化合物の具体例としては、特に限定されないが、4−エチニルイソフタル酸、5−エチニルイソフタル酸、2−エチニルテレフタル酸、3−エチニルテレフタル酸、2−エチニル−1,5−ナフタレンジカルボン酸、3−エチニル−1,5−ナフタレンジカルボン酸、4−エチニル−1,5−ナフタレンジカルボン酸、1−エチニル−2,6−ナフタレンジカルボン酸、3−エチニル−2,6−ナフタレンジカルボン酸、4−エチニル−2,6−ナフタレンジカルボン酸、3,7−ジエチニル−1,5−ナフタレンジカルボン酸、4,8−ジエチニル−2,6−ナフタレンジカルボン酸、4−エチニル−2,2'−ビフェニルジカルボン酸、6−エチニル−2,2'−ビフェニルジカルボン酸、2−エチニル−4,4'−ビフェニルジカルボン酸、4,4'−ジエチニル−2,2'−ビフェニルジカルボン酸、6,6'−ジエチニル−2,2'−ビフェニルジカルボン酸、2,2'−ジエチニル−4,4'−ビフェニルジカルボン酸、2,2−ビス(3−カルボキシ−4−エチニルフェニル)プロパン、2,2−ビス(3−カルボキシ−5−エチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−2−エチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−3−エチニルフェニル)プロパン、ビス(3−カルボキシ−4−エチニルフェニル)スルホン、ビス(3−カルボキシ−5−エチニルフェニル)スルホン、ビス(4−カルボキシ−2−エチニルフェニル)スルホン、ビス(4−カルボキシ−3−エチニルフェニル)スルホン、ビス(3−カルボキシ−4−エチニルフェニル)エーテル、ビス(3−カルボキシ−5−エチニルフェニル)エーテル、ビス(4−カルボキシ−2−エチニルフェニル)エーテル、ビス(4−カルボキシ−3−エチニルフェニル)エーテル等が挙げられる。
【0037】
これらの中でも、溶媒に対する溶解性の上で、4−エチニルイソフタル酸、5−エチニルイソフタル酸、2−エチニルテレフタル酸、3−エチニルテレフタル酸、2,2−ビス(3−カルボキシ−4−エチニルフェニル)プロパン、2,2−ビス(3−カルボキシ−5−エチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−2−エチニルフェニル)プロパン、2,2−ビス(4−カルボキシ−3−エチニルフェニル)プロパン、ビス(3−カルボキシ−4−エチニルフェニル)スルホン、ビス(3−カルボキシ−5−エチニルフェニル)スルホン、ビス(4−カルボキシ−2−エチニルフェニル)スルホン、ビス(4−カルボキシ−3−エチニルフェニル)スルホン、ビス(3−カルボキシ−4−エチニルフェニル)エーテル、ビス(3−カルボキシ−5−エチニルフェニル)エーテル、ビス(4−カルボキシ−2−エチニルフェニル)エーテル、ビス(4−カルボキシ−3−エチニルフェニル)エーテルが好ましく、4−エチニルイソフタル酸、5−エチニルイソフタル酸、2−エチニルテレフタル酸、3−エチニルテレフタル酸がより好ましい。これらのジカルボン酸化合物は単独で用いてもよく、また2種類以上を組み合わせて用いることができる。
【0038】
なお、式(B)、式(C)、式(D)、式(E)、式(F)および式(G)で表される基における環構造上の水素原子は、炭素数1〜4のアルキル基の中から選ばれる少なくとも1個の基で置換されていてもよい。上記炭素数1〜4のアルキル基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基およびt−ブチル基等が挙げられる。
【0039】
なお、下記一般式(A)で表わされるヒドロキシアミド重合体の製造方法としては、例えば、酸クロライド法では、使用する酸クロライドは、まず、N,N’−ジメチルホルムアミド等の触媒存在下で、前記ジカルボン酸化合物と、過剰量の塩化チオニルとを、室温ないし100℃程度の温度で反応させ、過剰の塩化チオニルを加熱および減圧により留去した後、残渣をヘキサン等の溶媒で再結晶することにより得ることができる。このようにして製造したジカルボン酸クロライド化合物を、前記ビスアミノフェノール化合物と共に、通常、N−メチル−2−ピロリドンおよびN,N’−ジメチルアセトアミド等の極性溶媒に溶解し、ピリジン等の酸受容剤存在下で、室温ないし−30℃程度の温度で反応させることにより、ヒドロキシアミド重合体を得ることができる。このようにして得られるヒドロキシアミド重合体が共重合体である場合の繰り返し単位の配列は、ブロック的であっても、ランダム的であっても良い。
【0040】
例えば、ブロック的な繰り返し単位の前記ヒドロキシアミド重合体の製造方法としては、酸クロライド法による場合、式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物と、式(C)および式(D)で表される基の中から選ばれる基を有するジカルボン酸クロライド化合物の少なくとも1種とを、予め反応させて分子量を上げた後、更に式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物と、式(E)および式(F)で表される基の中から選ばれる基を有するジカルボン酸クロライドの少なくとも1種とを反応させることにより得ることができる。
【0041】
また、逆に、式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物と、式(E)および式(F)で表される基の中から選ばれる基を有するジカルボン酸クロライド化合物の少なくとも1種とを、予め反応させて、重合体の分子量を上げた後、更に式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物と式(C)または式(D)で表される基の中から選ばれる基を有するジカルボン酸クロライド化合物の少なくとも1種とを反応させてもよい。
【0042】
ランダム的な繰り返し単位の前記ヒドロキシアミド重合体を製造する場合は、式(B)で表される基の中から選ばれる基を有するビスアミノフェノール化合物と式(C)または式(D)で表される基の中から選ばれる基を有するジカルボン酸クロライド化合物の少なくとも1種と、式(E)および式(F)で表される基の中から選ばれる基を有するジカルボン酸クロライド化合物の少なくとも1種とを、同時に反応させることにより得ることができる。
【0043】
また、前記一般式(A)で表されるヒドロキシアミド重合体の構造中のlおよびmは、lが4以上の整数、mが0以上の整数、かつl+mが4以上の整数であり、上記ヒドロキシアミド重合体に用いるビスアミノフェノール化合物およびジカルボン酸化合物の組み合わせにより異なるが、一般的には、l+mが4〜100の範囲である。
【0044】
このようにして得られる一般式(A)で表されるヒドロキシアミド重合体は、溶媒に対する溶解性、および耐熱性の上で、下記一般式(H)で表される構造を有するヒドロキシアミド重合体であることが好ましく、下記一般式(N)で表される構造を有するヒドロキシアミド重合体であることがより好ましい。
【0045】
【化8】
【0046】
【化9】
【0047】
【化10】
【0048】
【化11】
【0049】
【化12】
【0050】
【化13】
【0051】
【化14】
【0052】
【化15】
【0053】
【化16】
【0054】
【化17】
【0055】
【化18】
【0056】
また、上記一般式(A)で表されるヒドロキシアミド重合体は、標準ポリスチレン換算の重量平均分子量が、一般的には3,000以上、100,000以下であるが、5,000以上、30,000以下が好ましく、より好ましくは6,000以上、15,000以下である。重量平均分子量は上記範囲外でも使用できるが、下限値より小さいと成膜後のプロセス中に耐熱性が劣化するおそれがあり、上限値より大きいと、溶媒への溶解性が低下するおそれがあり、また、高屈折率樹脂組成物における溶液粘度が上昇することにより、塗膜形成時に基板への濡れ性が低下して、膜厚均一性の低下が起こるおそれがある。
【0057】
上記重量平均分子量は、高速液体クロマトグラフを用い、標準ポリスチレンで検量線を作成し、ポリスチレン換算で求めたものをいう。例えば、装置として、東ソー株式会社製高速液体クロマトグラフSC−8020システムに、TSKgelGMH−HRH高速SEC用カラム、UV(λ=270nm)検出器を用い、移動相としてLiBr0.5%を添加したN−メチル−2−ピロリドン液を用いて40℃で測定し、標準ポリスチレンとして、東ソー製PS−オリゴマーキットにより、リテンションタイムと分子量の検量線を作製し、ヒドロキシアミド重合体のポリスチレン換算の重量平均分子量を求めることができる。
【0058】
また、ヒドロキシアミド重合体の標準ポリスチレン換算の重量平均分子量を前記の範囲にするための反応方法としては、例えば、上記製造方法において、ジカルボン酸クロライド化合物とビスアミノフェノール化合物の反応モル比を調整して分子量を制御する方法、ジカルボン酸クロライド化合物とビスアミノフェノール化合物に、カルボン酸クロライド化合物またはアミノフェノール化合物を用いて、反応を停止させ、任意の分子量に調整する方法等を例示することができる。上記カルボン酸クロライド化合物およびアミノフェノール化合物は、ヒドロキシアミド重合体の末端基として反応を終結させ、それ以上分子量が大きくならないようにするために用い、例えば、安息香酸クロライド、2−アミノフェノール等を例示することができる。
【0059】
前記ジカルボン酸化合物とビスアミノフェノール化合物の反応モル比を調整することにより分子量を制御する方法においては、上記の標準ポリスチレン換算の重量平均分子量とも関係し、上記で用いるジカルボン酸化合物およびビスアミノフェノール化合物の構造によりモル比が異なるが、一方の化合物のモル数を過剰に、例えば、反応モル比が0.7以上、0.9以下に調整されることが好ましく、0.75以上、0.90以下がより好ましく、さらに好ましくは、0.8以上、0.90以下とするのが良い。過剰にする一方の化合物は、ビスアミノフェノール化合物でもジカルボン酸化合物のどちらでも構わないが、硬化物であるポリベンゾオキサゾールの透明性の観点から、ジカルボン酸化合物が過剰であることが望ましい。反応モル比は、前記範囲外でも使用できるが、前記下限値より低いと比較的低分子の成分が多く生じるため、その除去等により、工程の複雑化する恐れがある。また、前記上限値を超える場合、標準ポリスチレン換算の重量平均分子量が目的とする範囲よりも大きくなる恐れがある。
【0060】
また、前記ポリイミド前駆体としては、特に限定されないが、例えば、下記一般式(S)で表わされるアミド酸重合体が用いられる。
【0061】
【化19】
【0062】
このアミド酸重合体は、加熱により脱水する閉環反応(縮合反応)により、ポリイミド(硬化物)となるものである。
【0063】
このような一般式(S)で表わされるアミド酸重合体は、前記式(C)および前記式(D)で表わされる基の中から選ばれる基を有するジアミン化合物の少なくとも1種と、ジフェニルエーテルテトラカルボン酸二無水物とを用いて反応させて合成することができる。
【0064】
なお、前記ジアミン化合物における基とは、アミノ基と結合し得る結合手を2つ有する基を意味する。
【0065】
また、前記式(C)または式(D)で表わされる基を有するジアミン化合物の具体例としては、例えば、3,3'−ジアミノジフェニルエーテル、3,4'−ジアミノジフェニルエーテル、1,3−ビス(3−アミノフェノキシ)ベンゼン、1,3−ビス(4−アミノフェノキシ)ベンゼン、2,6−ビス(3−アミノフェノキシ)ベンゾニトリル、1,3−ビス(3−アミノフェノキシ)−5−クロロベンゼン、4,4'−ビス(3−アミノフェノキシ)ジフェニルスルフィド、4,4'−ビス(4−アミノフェノキシ)ジフェニルスルフィド、4,4'−ビス(3−アミノフェノキシ)ビフェニル、4,4'−ビス(4−アミノフェノキシ)ビフェニル等が挙げられる。
【0066】
なお、一般式(S)で表わされるアミド酸重合体において、前記式(C)または式(D)で表わされる基における環構造上の水素原子は、一般式(A)で表わされるヒドロキシアミド重合体と同様に、炭素数1〜4のアルキル基の中から選ばれる少なくとも1個の基で置換されていてもよい。
【0067】
また、このような一般式(S)で表わされるアミド酸重合体を生成するための、ジアミン化合物とテトラカルボン酸二無水物との反応は、一般的に有機溶媒中において実施される。
【0068】
かかる反応に用いられる有機溶媒として、例えば、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド、N−メチル−2−ピロリドン、1,3−ジメチル−2−イミダゾリジノン、N−メチルカプロラクタム、1,2−ジメトキシエタン、ビス(2−メトキシエチル)エーテル、1,2−ビス(2−メトキシエトキシ)エタン、ビス{2−(2−メトキシエトキシ)エチル}エーテル、テトラヒドロフラン、1,3−ジオキサン、1,4−ジオキサン、ピリジン、ピコリン、ジメチルスルホキシド、ジメチルスルホン、テトラメチル尿素、ヘキサメチルホスホルアミド、m−クレゾール、p−クレゾール、クレゾールサン、p−クロロフェノール、o−クロロフェノール、フェノール、アニソール等が挙げられ、単独またはこれらの混合溶媒が用いられる。
【0069】
また、これらジアミン化合物とテトラカルボン酸二無水物との反応温度は、通常、200℃以下、好ましくは30〜50℃程度に設定される。さらに、反応時間は、通常、4〜24時間程度、好ましくは10〜15時間程度に設定される。
【0070】
なお、加熱により脱水または脱アルコール反応する樹脂としては、ポリベンゾオキサゾール前駆体を用いるのが好ましい。ポリベンゾオキサゾール前駆体の硬化物であるポリベンゾオキサゾールには、カルボニル基のような極性基が存在しない。そのため、高屈折率樹脂組成物の硬化物の吸水率および誘電率を効果的に低減させることができる。
【0071】
上述したような樹脂の屈折率は、1.6以上であるが、より好ましくは1.65以上であることが好ましく、最も1.7以上であることが好ましい。ベース樹脂の屈折率が高いと、最終的に得られる樹脂成形体の屈折率もより高くすることができる。
【0072】
前記屈折率が1.6以上の樹脂の含有量は、特に限定されないが、前記樹脂組成物全体の1〜25重量%が好ましく、特に5〜20重量%が好ましい。含有量が前記範囲内であると、高屈折率と埋め込み性との両方に優れる。
【0073】
前記樹脂組成物は、前記樹脂よりも屈折率が高い高屈折率粒子を含む。これにより、最終的に得られる樹脂成形体をより高屈折率にすることができる。前記高屈折率粒子としては、有機樹脂からなる微粒子でも無機微粒子でも構わないが、無機微粒子が好ましく用いられる。
前記無機微粒子としては、可視光線の長波長帯域において吸収の少ないもの(具体的には透過率が50〜100%程度のもの)が好適に用いられ、例えば、酸化アルミニウム粒子、酸化ジルコニウム粒子、酸化チタン粒子、酸化亜鉛粒子、硫化鉛粒子、硫化亜鉛粒子、チタン酸バリウム、チタン酸カリウム、窒化ガリウム等が挙げられる。これらの中でも、屈折率および透明性等の観点から、特に、酸化チタン粒子および酸化ジルコニウム粒子が好ましい。
【0074】
なお、これらの無機粒子は、それ自身の触媒活性を抑制するためや、樹脂や分散媒との分散性を向上させるために、粒子表面がシリカやアルミナ等で被覆されていてもよい。
【0075】
これらの無機粒子は、単独で用いてもよく、また、2種類以上を混合して使用することもできる。
【0076】
これらの高屈折率粒子(無機粒子)の比表面積は、特に限定されないが、好ましくは30〜220m2/g程度、より好ましくは60〜200m2/g程度に設定される。比表面積が前記下限値より小さいと、高屈折率粒子(無機粒子)の種類によっては、光の散乱により透明性が低下するおそれがある。また、比表面積が前記上限値より大きいと、高屈折率粒子(無機粒子)の種類によっては、表面自由エネルギーが大きくなり、凝集を起こし易くなるため、樹脂組成物の硬化物(膜)の均一性が低下するおそれがある。なお、上記比表面積は、一般的なBET法により求めることができる。
【0077】
高屈折率粒子(無機粒子)の平均粒子径としては、1〜60nmが好ましく、1〜40nmがより好ましい。なお、上記平均粒子径は、動的光散乱法による有効径として測定することができる。
【0078】
前記高屈折率粒子(無機粒子)の含有量は、特に限定されないが、前記樹脂100重量部に対して20質量部〜400質量部程度が好ましく、50質量部〜200質量部程度がより好ましい。含有量が前記範囲内であると、高屈折率化に加え、高屈折率粒子(無機粒子)の分散状態の均一性にも優れる。
【0079】
前記樹脂組成物は、分散剤を含む。これにより、樹脂組成物中での高屈折率粒子の分散性を向上させ、高屈折率粒子同士が凝集した凝集体(二次粒子)が生じることに起因する透明性の低下を防止または抑制できる。
【0080】
前記分散剤としては、50%重量減少温度が400℃以下であるものを好ましく用いることができる。なお、本発明において、分散剤として、50%重量減少温度が400℃以下のものを用いることが好ましい理由は、樹脂組成物の硬化物中での分散剤の残存率を低下させることができ、それによって屈折率を向上することができるからである。
【0081】
ここで、分散剤の50%重量減少温度とは、分散剤を、示差熱熱重量同時測定装置を用いて、50℃から600℃まで10℃/分昇温させたときの重量減少率が、50%に達したときの温度である。また、70%重量減少温度とは、上記測定方法で測定した分散剤の重量減少率が、70%に達したときの温度である。同様に、90%重量減少温度とは、上記測定方法で測定した分散剤の重量減少率が、90%に達したときの温度である。
【0082】
この50%重量減少温度が400℃以下である分散剤としては、例えば、ポリオキシアルキルエーテル系のものでは、ポリオキシエチレン(5)ドコシルエーテル、ポリオキシエチレンドデシルエーテル、ポリオキシエチレンアルキレンアミン等が挙げられ、ポリオキシアリールエーテル系のものでは、ポリオキシエチレン(2)ノニルフェニルエーテル等が挙げられ、ポリエチレングリコール(PEG)系のものでは、PEG400、PEG600、PEG04モノステアリレート等が挙げられ、ポリプロピレングリコール(PPG)系のものでは、PPG400、PPG700、アミン末端PPGであるPPGビス−2−アミノプロピルエーテル400、ポリウレタン系のものでは、ポリブチレンヘキサメチレンジイソシアネート、ポリエステル系のものでは、ポリブチレンアジペート、シラン系カップリング剤のものでは、ビニルトリメトキシシラン縮合物、3−メタクリロキシプロピルトリメトキシシラン縮合物、アルミニウム系カップリング剤のものでは、アセトアルコキシアルミニウムイソプロポキシド縮合物、チタン系カップリング剤のものでは、チタンテトラブトキシド縮合物等が挙げられる。
【0083】
なお、上記分散剤の縮合物は、純水または純水/アルコール溶液中において、酸や塩基の添加により緩やかに加水分解を進行させて縮合物とする、一般的なゾル−ゲル法で合成することができる。
【0084】
なお、PEGおよびPPGの後に記載している数字は、それぞれ、PEGおよびPPGの平均分子量を示す。
【0085】
また、樹脂組成物の硬化物中での分散剤の残存率を低下させるという観点からは、50%重量減少温度が400℃以下である分散剤の他、70%重量減少温度が420℃以下である分散剤および90%重量減少温度が440℃以下である分散剤も、50%重量減少温度が400℃以下である分散剤と同様に用いることができる。したがって、50%重量減少温度が400℃以下であることの他、70%重量減少温度が420℃以下であることおよび90%重量減少温度が440℃以下であることのいずれか一方を満足する分散剤が好ましく用いられ、これらの双方を満足する分散剤がより好ましく用いられることは言うまでもない。
【0086】
なお、本発明では、後述するように、溶媒としては、その沸点が、70〜210℃程度のものが好ましく用いられ、かかる沸点を有する溶媒が樹脂組成物中に存在している際には、分散剤も樹脂組成物中に存在しているのが好ましいため、分散剤は、その10%重量減少温度が210℃以上であるものが好ましく用いられる。
【0087】
また、上述した50%重量減少温度が400℃以下である分散剤の、50%重量減少温度、70%重量減少温度および90%重量減少温度は、表1に示す通りである。
【0088】
【表1】
【0089】
また、分散剤としては、溶媒中においてミセルを形成し、このミセル中に高屈折率粒子を取り込むことにより、高屈折率粒子の溶媒中での分散性を向上させるものであってもよいし、高屈折率粒子の表面を修飾することにより、高屈折率粒子の溶媒中での分散性を向上させるものの何れであってもよい。
【0090】
なお、分散剤として、溶媒中でミセルを形成するものを用いた場合、溶媒中の高屈折率粒子は、その全てがミセル中に取り込まれる必要はなく、その少なくとも一部がミセル中に取り込まれることにより、高屈折率粒子全体としての溶媒中での分散性が向上していれば良い。
【0091】
上述した分散剤のうち、ミセルを形成する分散剤としては、例えば、前述した分散剤のうち、ポリオキシアルキルエーテル系、ポリオキシアリールエーテル系、ポリエチレングリコール(PEG)系、ポリプロピレングリコール(PPG)系、ポリエステル系およびポリウレタン系のものが挙げられる。また、高屈折率粒子の表面を修飾する分散剤としては、例えば、シラン系カップリング剤、アルミニウム系カップリング剤、チタン系カップリング剤のものが挙げられる。
【0092】
前記分散剤の含有量は、特に限定されないが、前記樹脂100質量部に対して、5質量部〜100質量部が好ましく、10質量部〜50質量部がより好ましい。含有量が前記範囲内であると、高屈折率粒子の分散状態の均一性に特に優れる。
【0093】
前記樹脂組成物は、溶媒を含む。溶媒は、屈折率が1.6以上の樹脂、高屈折率粒子および分散剤の種類に応じて選択され、これらの組み合わせによってそれぞれ異なるが、ケトン系分散媒、エーテル系分散媒、エステル系分散媒および非プロトン極性分散媒等が好適に用いられる。
【0094】
具体的には、例えば、ケトン系溶媒として、シクロペンタノン、シクロヘキサノン、シクロヘプタノン、4−メチル−シクロヘキサノン、3,3,5−トリメチルシクロヘキサノン、メチルエチルケトン、メチルイソブチルケトン、炭酸プロピレン、ジアセトンアルコールおよびγ−ブチロラクトン等;エーテル系溶媒として、ジエチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテル、ジエチレングリコールジブチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノプロピルエーテル、プロピレングリコール1−モノ−n−ブチルエーテル、ジプロピレングリコールモノメチルエーテルおよび1,3−ブチレングリコール−3−モノメチルエーテル等;エステル系溶媒として、プロピレングリコールジアセテート、プロピレングリコールモノメチルエーテルアセテート、乳酸メチル、乳酸エチル、乳酸ブチル、メチル−1,3−ブチレングリコールアセテート、ピルビン酸メチル、ピルビン酸エチルおよびメチル−3−メトキシプロピオネート等;非プロトン系極性溶媒として、N−メチル−2−ピロリドン、N,N−ジメチルアセトアミドおよびジメチルスルホキシド等;等を挙げることができる。これらは1種または2種以上を混合して用いることができる。これらの中で、加熱により脱水または脱アルコール反応する樹脂(特に、ヒドロキシアミド重合体)の構造により異なるが、シクロペンタノンと上記シクロペンタノン以外の溶媒の混合物、シクロヘキサノンと上記シクロヘキサノン以外の溶媒の混合物が、好適に使用することができる。
【0095】
これらの中でも、溶媒としては、その沸点が好ましくは70〜210℃程度のもの、より好ましくは120〜170℃程度のものが用いられる。沸点が前記下限値より低いと、樹脂組成物で構成される液状被膜を基板上に形成する際に、液状被膜中から早期に脱溶媒して、均一な膜厚の液状被膜が形成されないおそれがある。また、沸点が前記上限値より高いと、加熱により脱水または脱アルコール反応する樹脂を、脱水または脱アルコール反応させて、樹脂組成物の硬化物を得る際に、この硬化物中に溶媒が残存し、これに起因して、硬化物の屈折率および透明性が低下するおそれがあり好ましくない。
【0096】
なお、このような溶媒としては、上述したもののうち、例えば、シクロペンタノン(沸点:131℃)およびシクロヘキサノン(沸点:156℃)等が好ましく用いられる。
【0097】
前記溶媒の含有量は、特に限定されないが、前記樹脂100質量部に対して、400質量部〜3,000質量部が好ましく、900質量部〜2,000質量部がより好ましい。含有量が前記範囲内であると、特に塗布性に優れる。
【0098】
なお、この樹脂組成物には、前記屈折率が1.6以上の樹脂、高屈折率粒子、分散剤および溶媒以外の成分として、酸素ラジカルやイオウラジカルを加熱により発生するラジカル開始剤等の添加剤を添加することができる。また、前記屈折率が1.6以上の樹脂に、感光剤としての例えばナフトキノンジアジド化合物と一緒に用いることで、樹脂組成物を感光性樹脂組成物として用いることが可能である。
【0099】
このような、屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤とを、溶媒に溶解して樹脂組成物をワニスの状態とするが、これらの高屈折率粒子を溶媒中に、より均一に分散させる方法としては、特に限定されないが、例えば、溶媒と、高屈折率粒子と、必要に応じて表面処理剤や分散剤とを混合し、高周波超音波ホモジナイザーや、微小ビーズを用いたビーズミル処理を行うことにより、無機粒子が均一に分散された分散液を得て、それと屈折率が1.6の樹脂とを混合してワニス化する方法が好ましい。
【0100】
<塗布工程>
塗布工程では上述したような樹脂組成物のワニスは、スピンナーを用いた回転塗布、スプレーコーターを用いた噴霧塗布、浸漬、印刷、ロールコーティング等の方法により成膜され、所望の形状に形成される。これらの中でもスピンコート法が好ましい。これにより、成膜上面のコプラナリティが良好な形状を得ることができる。
なお、本実施形態においては、膜(フィルム)状の成形体を形成する場合について説明したが、これに限定されない。
【0101】
この塗布工程では、上述の樹脂組成物のワニスを塗布することで、直径0.4〜1.0μmで、深さ0.5〜2.5μmの凹部を埋め込むことが好ましい。これにより、後述するような光学部品として好適に用いることができる。
【0102】
この塗布工程では、膜状体の成形体を形成すると共に、直径0.4〜1.0μm、深さ0.5〜2.5μmの凹部を埋め込むことが好ましい。このような凹部に前記樹脂組成物を埋め込み、光学部品として用いることができるからである。
【0103】
<第1加熱工程および第2加熱工程>
上述したような塗布工程を経て得られた膜状体に、第1加熱工程を実施して膜状体の屈折率が第1屈折率(A1)となるまで加熱し、さらに第2加熱工程を実施して膜状体の屈折率が第2屈折率(A2)になるまで加熱したときに、前記膜状体の屈折率の差(A2−A1)が0.03以上となるように加熱することを特徴とする。これにより、膜状体の内部での屈折率のバラつきを小さくすることができる。前記屈折率の差は、0.03以上であることがより好ましく、もっとも好ましくは0.04以上である
【0104】
このように前記屈折率の差(A2−A1)が前記下限値以上であると、膜状体の内部での屈折率のバラつきを小さくすることができる。その理由は、次のように考えられる。
屈折率の差(A2−A1)が大きくなるためには、A2値が大きいまたはA1値が小さいことが必要である。第1加熱工程は、膜状体の屈折率が第1屈折率(A1)となるまで加熱するものであるが、この工程の目的は、主として溶媒を除去し膜状体を得るために行われるものであり、第2加熱工程の加熱より低温で行われる。この第1加熱工程後の膜状体には、主として屈折率が1.6以上の樹脂、高屈折率粒子、分散剤が含まれるが、低屈折率の分散剤が含まれているため、膜状体の第1屈折率(A1)は、第2屈折率(A2)より小さな値を示す。これに対して、第2加熱工程は、膜状体の屈折率が第2屈折率(A2)となるまで加熱するものであるが、この工程では、分解または揮発による分散剤の除去が行われるので、樹脂組成物の硬化物中での分散剤の残存率は小さくなり、それによって第2屈折率(A2)を大きくすることができる。さらに、屈折率が1.6以上の樹脂が脱水閉環反応または脱アルコール閉環反応する樹脂である場合、この第2加熱工程で、脱水閉環反応または脱アルコール閉環反応が起こり、これらの閉環反応前よりも屈折率は高くなる。
【0105】
このような第1加熱工程および第2加熱工程での目的を達成するために、上述のような屈折率を示すような条件で加熱することにより、溶媒の除去、分散剤の除去が適宜実施されることになる。このように、溶媒、分散剤の除去にそれぞれ適した加熱温度で除去が行われることで膜状体の内部の屈折率のバラつきを低減することができる。
【0106】
さらには、屈折率が1.6以上の樹脂が脱水閉環反応または脱アルコール閉環反応する樹脂である場合には、第2加熱工程において、分散剤の除去等に加えて樹脂の脱水閉環反応等も適切に実施されることになるので、これによって膜状体の内部の屈折率のバラつきが抑制される。
例えば、第1加熱工程を行わないで、第2加熱工程のみとした場合には、溶媒の除去、分散剤の除去、屈折率が1.6以上の樹脂が脱水閉環反応または脱アルコール閉環反応する樹脂である場合には脱水分及び脱アルコール分の除去が、同時に一気に起こることになり、膜状体の内部での屈折率のバラつきが生じたり、膜厚の不均一による外観の悪化が生じたりしてしまう。
【0107】
前記第1加熱工程では、膜状体の屈折率が第1屈折率(A1)となるまで加熱するが、具体的には膜状体の第1屈折率が1.70以上となるまで加熱することが好ましく、特に1.75以上となるまで加熱することが好ましい。第1屈折率が前記下限値未満である場合は、膜状体内部に溶媒が残存している可能性が高く、その後の第2工程で残存した溶媒が一気に除去されることにより膜厚の均一性に問題が生じる場合がある。
【0108】
前記第1加熱工程では、一定温度で所定時間加熱する工程が好ましく、より具体的には、80〜230℃×30秒〜5分間が好ましく、より好ましくは90〜200℃×30秒〜2分間が好ましい。前記第1加熱条件であると、特に膜厚の均一性を向上することができる。
なお、具体的な加熱条件は、樹脂組成および分子量等により異なる。
【0109】
前記第2加熱工程は、前記第加熱1工程よりも高い温度で前記膜状体を加熱して、前記膜状体が第2屈折率となるまで加熱するが、具体的には膜状体の第2屈折率が1.75以上となるまで加熱することが好ましく、特に1.8〜1.9となるまで加熱することが好ましい。第2屈折率が前記下限値未満である場合は、膜状体内部に分散剤が残存しているということに他ならず、その残存分散剤の偏在により膜状体の内部での屈折率がバラつく原因となる。
【0110】
前記第2加熱工程では、前記第1加熱工程よりも高い一定温度で所定時間加熱する工程が好ましく、より具体的には、250〜450℃×1〜10分間が好ましく、より好ましくは250〜400℃×1〜5分間が好ましい。前記第2加熱条件であると、特に膜状体の内部での屈折率のバラつきを低減することができる。
なお、具体的な加熱条件は、樹脂組成および分子量等により異なる。
【0111】
また、前記第2加熱工程では、前記分散剤が分解または揮発するような温度で加熱することが好ましい。これにより、膜状体(樹脂成形体)中に残留する低屈折率の分散剤が低減され、それによって膜状体を高屈折率化することができる。
【0112】
このような工程を経ることにより、樹脂成形体を得ることができる。このような工程で得られる樹脂成形体は、内部での屈折率のバラつきが低減されているものである。
前記屈折率は、例えばプリズムカップリング法を用いた屈折率測定装置、プリズムカプラー(メトリコン社製2010プリズムカプラー)を用いて評価することができる。その際の測定には、直径2インチ、厚み1mmの石英基板上に本発明の樹脂組成物の膜状体を作成し、それぞれ所定の加熱・乾燥させたものを用いることができる。
【0113】
次に、光学部品について、好適な実施形態に基づいて説明する。
光学部品100は、基板1と、基板1に埋め込まれているフォトダイオード11と、フォトダイオード11を覆うように設けられている樹脂成形体5と、樹脂成形体5の側面を囲むように設けられた絶縁材2と、絶縁材2中に設けられた回路配線21と、樹脂成形体5および絶縁材2の上面(図2中の上側)に設けられたカラーフィルター3と、カラーフィルター3の上面に設けられた平坦化膜4と、樹脂成形体5の上方に相当する、平坦化膜4の上側の位置に設けられたレンズ6とで構成されている。このような構成により、レンズ6から入射した光信号7が、平坦化膜4およびカラーフィルター3を透過して、樹脂成形体5に入射される。
ここで、レンズ6に垂直方向に入射する光は、光漏れ等を特に生じることなくフォトダイオード11に入射される。これに対して、レンズ6に対して入射角度θが大きい光は、樹脂成形体5で光漏れ等が生じる場合があった。本発明では、樹脂成形体5は前述したように高屈折率の光学部品用樹脂組成物の硬化物で構成されているので、入射角度θが大きい光信号7に対しても光信号7を外部に漏らすことなく、フォトダイオード11に誘導することができる。
【0114】
ここで、樹脂成形体5は、樹脂組成物を上述したような第1加熱工程、第2加熱工程を経て得られるものである。そのため、樹脂成形体5の屈折率は高く、またその内部での屈折率のバラつきが小さいものである。そのため、フォトダイオードでの集光効率を向上することができる。
【0115】
さらに、樹脂成形体5が、高屈折率を有する樹脂組成物の硬化物で構成されている場合、光漏れ等を低減することができるため、フォトダイオードの受光量を向上することができる。
【0116】
また、このような樹脂成形体5を形成する場合、予め樹脂成形体5に相当する部分が凹部となっており、そこに前述の樹脂組成物を埋め込んだ後に、硬化させる手法が取られている。
このような凹部の大きさとしては、例えば径0.4〜1μm、深さ0.5〜2μmの場合が挙げられ、非常に小さい空間である。このような凹部に前述した樹脂組成物を埋め込むためには、樹脂組成物の室温での溶液粘度を低いものとして流動性を十分に確保する必要がある。そのような室温での溶液粘度としては、例えば3〜11mPa・sであることが好ましく、特に3〜8mPa・sであることが好ましい。溶液粘度が前記範囲内であると、凹部への埋込み性に優れる。
【0117】
具体的には、上述したような凹部に前記樹脂組成物を埋め込み、前記第1加熱工程および前記第2加熱工程を経て樹脂成形体5を得て、最終的な光学部品100を得る。
【0118】
樹脂成形体5の633nmでの屈折率は、特に限定されないが、1.75以上であることが好ましく、特に1.8〜1.9であることが好ましい。屈折率が前記範囲内であると、光信号7が樹脂成形体5より漏れるのを防止する効果が特に優れる。
前記屈折率は、例えばプリズムカップリング法を用いた屈折率測定装置、プリズムカプラー(メトリコン社製2010プリズムカプラー)を用いて評価することができる。その際の測定には、直径2インチ、厚み1mmの石英基板上に前記樹脂組成物の液状被膜を作成し、加熱・乾燥させたものを用いる。
【0119】
樹脂成形体5は、特に限定されないが、波長400nmにおける厚み1.5μmでの光線透過率が85%以上であることが好ましく、特に90%以上であることが好ましい。光線透過率が前記範囲内であると、特に透明性に優れる。
【0120】
このような光学部品100の具体例としては、例えば光学レンズ、光学フィルター、光スイッチ、光導波路、光ファイバー、集光レンズ等が挙げられる。
また、このような光学部品100は、上述したような樹脂成形体5で光学素子が被覆されているので、光漏れが小さく、光学素子の受光特性に優れる。
【0121】
また、上述したような光学部品100を有する光学デバイスとしては、例えば光電集積回路、光集積回路、CCDセンサ、CMOSセンサ等の光学デバイス等が挙げられる。
【実施例】
【0122】
以下、本発明を実施例および比較例に基づいて詳細に説明するが、本発明はこれに限定されるものではない。
【0123】
<実施例1>
1−1.4,4’−オキシビス安息香酸ジクロライドの合成
温度計、ジムロート冷却管、撹拌機を備えた300mLの4つ口フラスコに、東京化成工業(株)製4,4’−オキシビス安息香酸10.33g(0.04mol)、ベンジルトリエチルアンモニウムクロライド0.08g(0.0004mol)および塩化チオニル50g(0.42mol)を加え、3時間還流した。100℃で塩化チオニルを留去し、残留物を60℃で12時間真空乾燥し、10.9gの4,4’−オキシビス安息香酸ジクロライドを得た(収率92%)。
【0124】
1−2.ポリアミド系樹脂(屈折率1.6以上の樹脂)の合成
窒素ガスフロー下で、ビスアミノフェノール化合物(A)として9,9−ビス(3−アミノ−4−ヒドロキシフェニル)フルオレン30.44g(0.08mol)を、乾燥したN−メチル−2−ピロリドン200gに溶解し、ピリジン17.4g(0.22mol)を添加した後、−15℃に冷却し、ジカルボン酸化合物(B)として上述の4,4’−オキシビス安息香酸ジクロリド29.5g(0.1mol)を、少しずつ添加した。滴下終了後、−15℃で、1時間撹拌後、室温まで3時間かけて戻し、さらに室温で2時間撹拌した(反応モル比A/B=0.8)。その後、反応液を50%メタノール水溶液4リットルに小さな液滴で滴下し、沈殿物を集めた。さらに、50%メタノール水溶液4リットル中での攪拌、沈殿物の回収を3回繰り返した。その後、沈殿物を乾燥することにより、ポリアミド系樹脂(P)を得た。得られたポリアミド系樹脂の末端は、電位差滴定を用いた中和滴定により、90%以上がカルボン酸であった。このポリアミド系樹脂(P)の屈折率をプリズムカップリング法で求めたところ、1.67であった。
【0125】
1−3.酸化チタン粒子シクロヘキサノン分散液の調製
無機粒子として酸化チタン粒子(堺化学工業(株)製SSP−20、比表面積170m2/g)20重量部、分散媒としてシクロヘキサノン74重量部、および、分散剤としてポリオキシエチレン(2)ノニルフェニルエーテル(和光純薬工業(株)製)10重量部をそれぞれ混合し、微粉砕・分散機(寿工業(株)製ウルトラアペックスミルUAM−015)を用いて、ビーズミル分散処理を行い、酸化チタン粒子シクロヘキサノン分散液を得た。
【0126】
2.樹脂組成物の調製
得られたポリアミド系樹脂(P)100重量部にシクロヘキサノン330重量部を加え溶解させた後、酸化チタン粒子シクロヘキサノン分散液520重量部を混合し30分間攪拌を行った後、超音波ホモジナイザー処理を行い、樹脂組成物(A)を得た。
【0127】
3.樹脂成形体の製造(塗布工程、第1加熱工程および第2加熱工程)
次に、上述の樹脂組成物(A)をシリコン基板上に、スピンコートにより、塗膜を形成した。次に、第1加熱工程として、第1屈折率が1.77となるまで、該塗膜を大気雰囲気下で、200℃で、60秒間加熱した。さらに、第2加熱工程として、第2屈折率が1.84となるまで、N2ガス雰囲気下で、380℃で、250秒間加熱し樹脂成形体を得た。この樹脂成形体の屈折率の差は0.07であった。また、下記に記載の方法で測定した膜質均一性は0.5%、凹部埋込性は良好であった。
【0128】
(実施例2)
第1加熱工程および第2加熱工程の条件を下記のようにした以外は、実施例1と同様にした。
第1加熱工程として、第1屈折率が1.77となるまで、90℃で、120秒間加熱し、第2加熱工程として、第2屈折率が1.83となるまで、350℃、250秒間加熱し樹脂成形体を得た。この樹脂成形体の屈折率の差は、0.06であり、膜質均一性は0.6%、凹部埋込性は良好であった。
【0129】
(実施例3)
第1加熱工程および第2加熱工程の条件を下記のようにした以外は、実施例1と同様にした。
第1加熱工程として、第1屈折率が1.80となるまで220℃で、120秒間加熱し、第2加熱工程として、第2屈折率が1.84となるまで、400℃、250秒間加熱し樹脂成形体を得た。この樹脂成形体の屈折率の差は、0.04であり、膜質均一性は0.7%、凹部埋込性は良好であった。
【0130】
(実施例4)
樹脂組成物として下記に記載の樹脂組成物を用い、第1加熱条件および第2加熱条件を下記のようにした以外は、実施例1と同様にした。
まず、無機微粒子として酸化ジルコニウム粒子(日本稀元素化学工業(株)製UEP−100、比表面積90m2/g)20重量部、シクロヘキサノン74重量部及びポリプロピレングリコール400(和光純薬(株)製)6重量部を混合し、実施例1と同様にして、酸化ジルコニウム粒子シクロヘキサノン分散液を得た。
つぎに、ポリアミド系樹脂(P)100重量部にシクロヘキサノン300重量部を加え溶解させた後、この酸化ジルコニウム粒子シクロヘキサノン分散液750重量部を混合し30分間攪拌を行った後、超音波ホモジナイザー処理を行い、樹脂組成物(B)を得た。
また、第1加熱工程として、第1屈折率が1.76となるまで、該塗膜を大気雰囲気下で、200℃で、60秒間加熱した。さらに、第2加熱工程として、第2屈折率が1.80となるまで、N2ガス雰囲気下で、380℃で、250秒間加熱し樹脂成形体を得た。この樹脂成形体の屈折率の差は0.04であり、膜質均一性は0.5%、凹部埋込性は良好であった。
【0131】
(比較例1)
第1加熱工程および第2加熱工程を以下のようにした以外は、実施例1と同様にした。
実施例1と同様にして、第1加熱工程として、第1屈折率が1.77となるまで、200℃で、60秒間加熱し、第2加熱工程として、第2屈折率が1.79となるまで、250℃、250秒間加熱し樹脂成形体を得た。この樹脂成形体の屈折率の差は、0.02であり、膜質均一性は2.8%であった。
なお、第2加熱工程後の樹脂成形体のIR測定を実施したところ、分散剤であるポリオキシエチレン(2)ノニルフェニルエーテル樹脂のピークが観察されたことから分散剤が残存していること、またアミド結合のピークが観察されたことから閉環脱水反応が完了していないことが判明した。また閉環脱水反応が完了していないため、実施例1と比較し、ガラス転移温度が245℃と低かった。
【0132】
(比較例2)
ポリアミド系樹脂として、以下のものを用いた以外は、実施例1と同様にした。
9,9−ビス(3−アミノ−4−ヒドロキシフェニル)フルオレン30.44g(0.08mol)の代わりに、2,2−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパン29.3g(0.08mol)を用いた以外は、全て実施例1と同様にして、ポリアミド系樹脂(Q)を得た。このポリアミド系樹脂(Q)の屈折率は、1.58であった。
このポリアミド系樹脂(Q)100重量部にシクロヘキサノン330重量部を加え溶解させた後、製造例2−1の酸化チタン粒子シクロヘキサノン分散液520重量部を混合し30分間攪拌を行った後、超音波ホモジナイザー処理を行い、樹脂組成物(C)を得た。
【0133】
そして、実施例1と同様に第1加熱工程として、第1屈折率が1.70となるまで、該塗膜を大気雰囲気下で、200℃で、60秒間加熱した。さらに、第2加熱工程として、N2ガス雰囲気下で、350℃で、250秒間加熱したところ、第2屈折率としては1.74であった。第2屈折率を高めるために、450℃、250秒間とより高い温度にて加熱する条件でも第2工程を行ったが、1.74のままであった。屈折率の差は0.04であり、膜質均一性も0.7%と悪くないが、使用したポリアミド系樹脂(Q)の屈折率値が低いために、第2屈折率としては1.75以上という望ましい値が得られていなかった。
【0134】
(比較例3)
第1加熱工程のみを行い、第2加熱工程を行わないこと以外は、実施例1と同様にした。得られた樹脂成形体の膜質均一性は2.0%であった。樹脂成形体のIR測定を実施したところ、分散剤であるポリオキシエチレン(2)ノニルフェニルエーテル樹脂のピークが観察されたことから分散剤が残存していること、またアミド結合のピークが観察されたことから閉環脱水反応が完了していないことが判明した。閉環脱水反応が完了していないため、実施例1と比較し、ガラス転移温度が242℃と低かった。
【0135】
(比較例4)
第1加熱工程を行わず、第2加熱工程のみを行った以外は、実施例1と同様に行った。得られた樹脂成形体の膜質均一性は2.3%であった。第1加熱工程がなく、膜状体内部に溶媒が多く残存している状態で、第2加熱工程を行ったため、残存した溶媒、分散剤及び脱水閉環反応による脱水が一気に除去されることにより膜厚の均一性に問題が生じた結果、バラつき度が大きくなったと予想される。凹部埋込性は、FIB−SEMによる観察では、断面部に気泡が見られ不良であった。
【0136】
各実施例および比較例で得られた樹脂成形体について、以下の評価を行った。評価項目を評価内容と共に示す。
【0137】
1.ガラス転移温度
上記で得た塗膜を削り落とした粉末について、MDSC(温度サイクルモード示差操作熱量計:ティー・エイ・インスツルメント社製2910MDSC)により、N2ガスを30mL/分の流量で流しながら、昇温速度2℃/分、温度振幅±2℃/分の条件で昇温しながら、40℃から420℃までの温度範囲で測定を行い、リバース曲線の変移点から算出を行った。
【0138】
2.膜質均一性
上記で得た加熱後の塗膜において、ウエハ面内をXY軸それぞれ等間隔で19ポイント(合計37ポイント)をn&k Technology Inc.社製n&kアナライザー1500を用いて屈折率を測定し、その相対標準偏差値を膜質均一性とした。
【0139】
3.凹部への埋込性
凹部への埋込性は、直径0.5μmで、深さ1.5μmの凹部を加工したSiO2加工ウエハを用い、上述の樹脂組成物をスピンコートして塗膜を形成し、上述の条件加熱・乾燥させたものを用い、FIB−SEMによる断面観察を行うことで、良好か不良かを判断した。良好とはボイドや基板との剥離などなく均一であることが観察されることとした。
【0140】
4.樹脂成形体の簡易評価
各実施例および比較例で用いた樹脂組成物を、石英基板上にスピンコートにより、熱処理後の最終膜厚が1.5μmとなるように塗膜を形成した。この塗膜を用いて、下記のように光線透過率および屈折率を評価した。
【0141】
4−1.光線透過率
上記で得た樹脂膜を用いて、株式会社島津製作所製分光光度計UV−3100を用いて波長400nmで測定した。
【0142】
4−2.屈折率
上記で得た樹脂膜を用いて、Metricon製プリズムカプラーを用いて、20℃での633nm(He−Neレーザーを使用)での膜面に対して垂直方向の屈折率(TM)を測定した。
【0143】
【表2】
【0144】
表2から明らかなように、実施例1〜4の樹脂成形体は、樹脂成形体の内部での屈折率のバラつきを小さくすることができた。したがって、この樹脂成形体を用いた光学部品、光学デバイスは性能に優れていることが示唆された。
また、実施例1〜4で用いている樹脂成形体は、ガラス転移温度が高く、耐熱性に優れていた。
また、実施例1〜4で用いている樹脂組成物を使用した凹部への埋込性の評価より、凹部への埋め込み性に優れていることが示された。
また、実施例1〜4で用いている樹脂成形体は、屈折率も高く、光学部品として用いた際の性能に優れることが示唆された。
【符号の説明】
【0145】
1 基板
11 フォトダイオード
2 絶縁材
21 回路配線
3 カラーフィルター
4 平坦化膜
5 樹脂成形体
6 レンズ
7 光信号
100 光学部品
【特許請求の範囲】
【請求項1】
屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を所望の形状に形成し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体の製造方法であって、
前記第1加熱工程において樹脂成形体の屈折率が第1屈折率(A1)になるまで加熱し、前記第2加熱工程において樹脂成形体の屈折率が第2屈折率(A2)になるまで加熱したときに、前記樹脂成形体の屈折率の差(A2−A1)が0.03以上となるような条件で加熱することを特徴とする樹脂成形体の製造方法。
【請求項2】
前記第1加熱工程は、一定温度で所定時間加熱することにより樹脂成形体の屈折率を、第1屈折率(A1)にするものである請求項1に記載の樹脂成形体の製造方法。
【請求項3】
前記第1加熱工程で樹脂成形体の屈折率が、1.70以上となるまでに加熱するものである請求項1または2に記載の樹脂成形体の製造方法。
【請求項4】
前記第2加熱工程は、前記第1加熱工程の加熱温度よりも高い温度で、所定時間加熱することにより樹脂成形体の屈折率を、第2屈折率(A2)にするものである請求項1ないし3のいずれかに記載の樹脂成形体の製造方法。
【請求項5】
前記第2加熱工程は、前記分散剤が分解または揮発するような温度で加熱するものである請求項1ないし4のいずれかに記載の樹脂成形体の製造方法。
【請求項6】
前記屈折率が1.6以上の樹脂は、加熱により脱水閉環反応または脱アルコール閉環反応する樹脂である請求項1ないし5のいずれかに記載の樹脂成形体の製造方法。
【請求項7】
前記屈折率が1.6以上の樹脂は、ポリベンゾオキサゾール前駆体を含むものである請求項1ないし6のいずれかに記載の樹脂成形体の製造方法。
【請求項8】
前記ポリベンゾオキサゾール前駆体は、ジカルボン酸化合物をビスアミノフェノール化合物よりも過剰に反応させてなるものである請求項1ないし7のいずれかに記載の樹脂成形体の製造方法。
【請求項9】
前記樹脂成形体は、直径0.4〜1.0μm、深さ0.5〜2.5μmの凹部に埋め込まれるものである請求項1ないし8のいずれかに記載の樹脂成形体の製造方法。
【請求項10】
屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を成膜し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体であって、
前記第1加熱工程後における樹脂成形体の第1屈折率(A1)と、前記第2加熱工程後における樹脂成形体の第2屈折率(A2)との差(A2−A1)が、0.03以上であることを特徴とする樹脂成形体。
【請求項11】
前記第1屈折率が、1.70以上である請求項10に記載の樹脂成形体。
【請求項12】
前記屈折率が1.6以上の樹脂は、加熱により脱水閉環反応または脱アルコール閉環反応する樹脂である請求項10または11に記載の樹脂成形体。
【請求項13】
前記樹脂成形体は、直径0.4〜1.0μm、深さ0.5〜2.5μmの凹部を埋め込むために用いるものである請求項10ないし12のいずれかに記載の樹脂成形体。
【請求項14】
請求項10ないし13のいずれかに記載の樹脂成形体を用いてなることを特徴とする光学部品。
【請求項15】
請求項14に記載の光学部品を有することを特徴とする光学デバイス。
【請求項1】
屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を所望の形状に形成し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体の製造方法であって、
前記第1加熱工程において樹脂成形体の屈折率が第1屈折率(A1)になるまで加熱し、前記第2加熱工程において樹脂成形体の屈折率が第2屈折率(A2)になるまで加熱したときに、前記樹脂成形体の屈折率の差(A2−A1)が0.03以上となるような条件で加熱することを特徴とする樹脂成形体の製造方法。
【請求項2】
前記第1加熱工程は、一定温度で所定時間加熱することにより樹脂成形体の屈折率を、第1屈折率(A1)にするものである請求項1に記載の樹脂成形体の製造方法。
【請求項3】
前記第1加熱工程で樹脂成形体の屈折率が、1.70以上となるまでに加熱するものである請求項1または2に記載の樹脂成形体の製造方法。
【請求項4】
前記第2加熱工程は、前記第1加熱工程の加熱温度よりも高い温度で、所定時間加熱することにより樹脂成形体の屈折率を、第2屈折率(A2)にするものである請求項1ないし3のいずれかに記載の樹脂成形体の製造方法。
【請求項5】
前記第2加熱工程は、前記分散剤が分解または揮発するような温度で加熱するものである請求項1ないし4のいずれかに記載の樹脂成形体の製造方法。
【請求項6】
前記屈折率が1.6以上の樹脂は、加熱により脱水閉環反応または脱アルコール閉環反応する樹脂である請求項1ないし5のいずれかに記載の樹脂成形体の製造方法。
【請求項7】
前記屈折率が1.6以上の樹脂は、ポリベンゾオキサゾール前駆体を含むものである請求項1ないし6のいずれかに記載の樹脂成形体の製造方法。
【請求項8】
前記ポリベンゾオキサゾール前駆体は、ジカルボン酸化合物をビスアミノフェノール化合物よりも過剰に反応させてなるものである請求項1ないし7のいずれかに記載の樹脂成形体の製造方法。
【請求項9】
前記樹脂成形体は、直径0.4〜1.0μm、深さ0.5〜2.5μmの凹部に埋め込まれるものである請求項1ないし8のいずれかに記載の樹脂成形体の製造方法。
【請求項10】
屈折率が1.6以上の樹脂と、前記樹脂よりも屈折率が高い高屈折率粒子と、分散剤と、溶媒とを含む樹脂組成物を成膜し、第1加熱工程および前記第1加熱工程よりも高い温度で加熱する第2加熱工程を経てなる樹脂成形体であって、
前記第1加熱工程後における樹脂成形体の第1屈折率(A1)と、前記第2加熱工程後における樹脂成形体の第2屈折率(A2)との差(A2−A1)が、0.03以上であることを特徴とする樹脂成形体。
【請求項11】
前記第1屈折率が、1.70以上である請求項10に記載の樹脂成形体。
【請求項12】
前記屈折率が1.6以上の樹脂は、加熱により脱水閉環反応または脱アルコール閉環反応する樹脂である請求項10または11に記載の樹脂成形体。
【請求項13】
前記樹脂成形体は、直径0.4〜1.0μm、深さ0.5〜2.5μmの凹部を埋め込むために用いるものである請求項10ないし12のいずれかに記載の樹脂成形体。
【請求項14】
請求項10ないし13のいずれかに記載の樹脂成形体を用いてなることを特徴とする光学部品。
【請求項15】
請求項14に記載の光学部品を有することを特徴とする光学デバイス。
【図1】

【図2】
【図2】
【公開番号】特開2011−26365(P2011−26365A)
【公開日】平成23年2月10日(2011.2.10)
【国際特許分類】
【出願番号】特願2009−170130(P2009−170130)
【出願日】平成21年7月21日(2009.7.21)
【出願人】(000002141)住友ベークライト株式会社 (2,927)
【Fターム(参考)】
【公開日】平成23年2月10日(2011.2.10)
【国際特許分類】
【出願日】平成21年7月21日(2009.7.21)
【出願人】(000002141)住友ベークライト株式会社 (2,927)
【Fターム(参考)】
[ Back to top ]